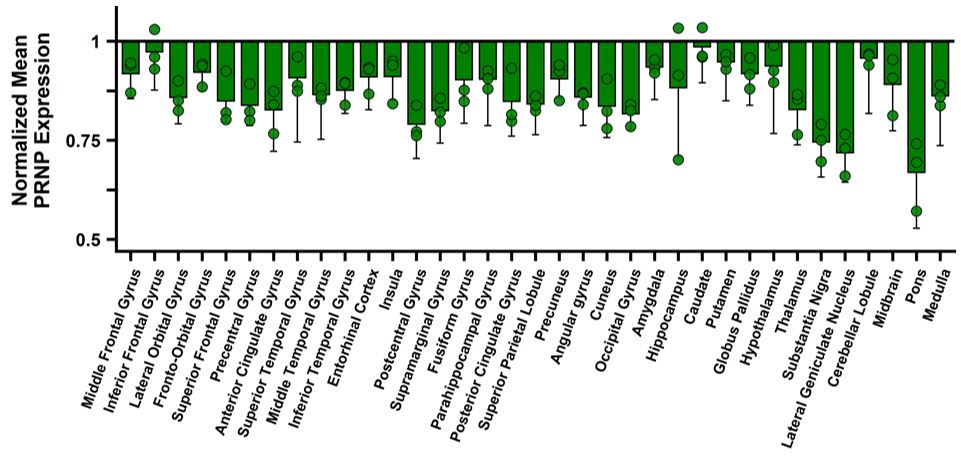
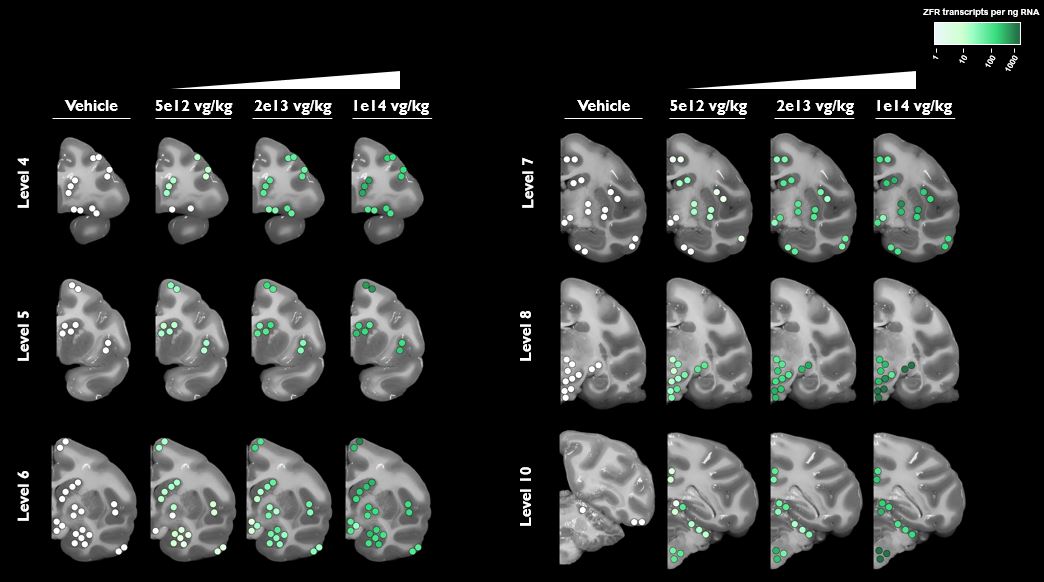
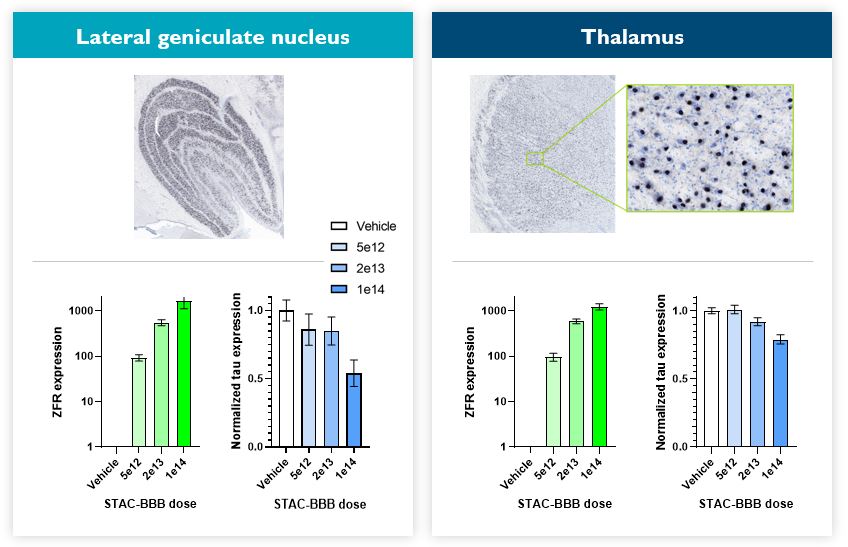
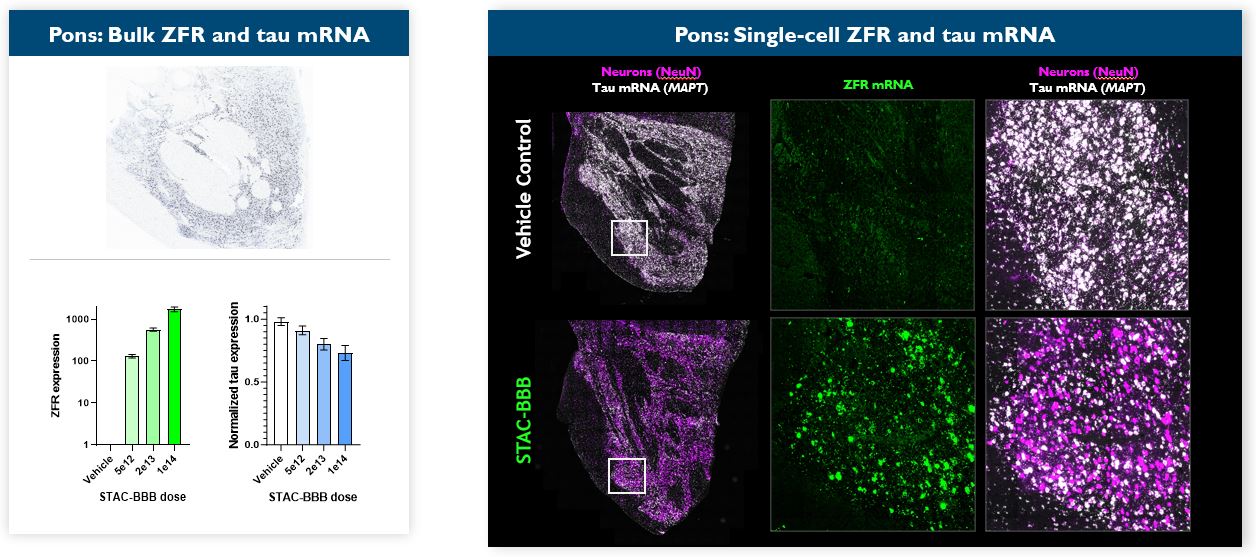
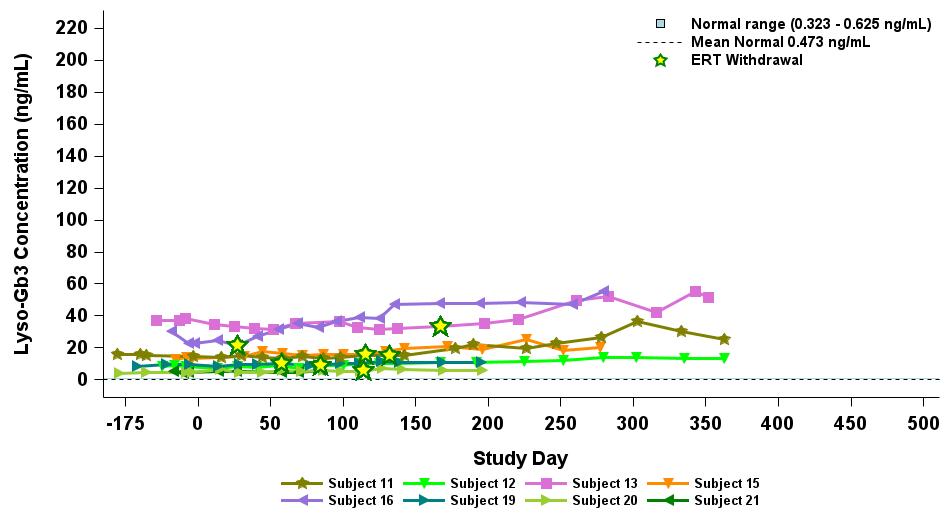
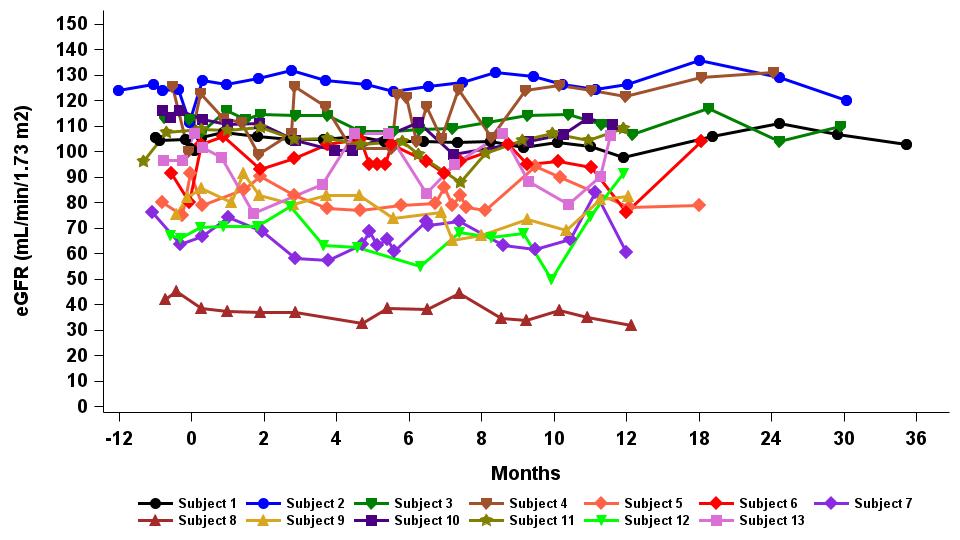
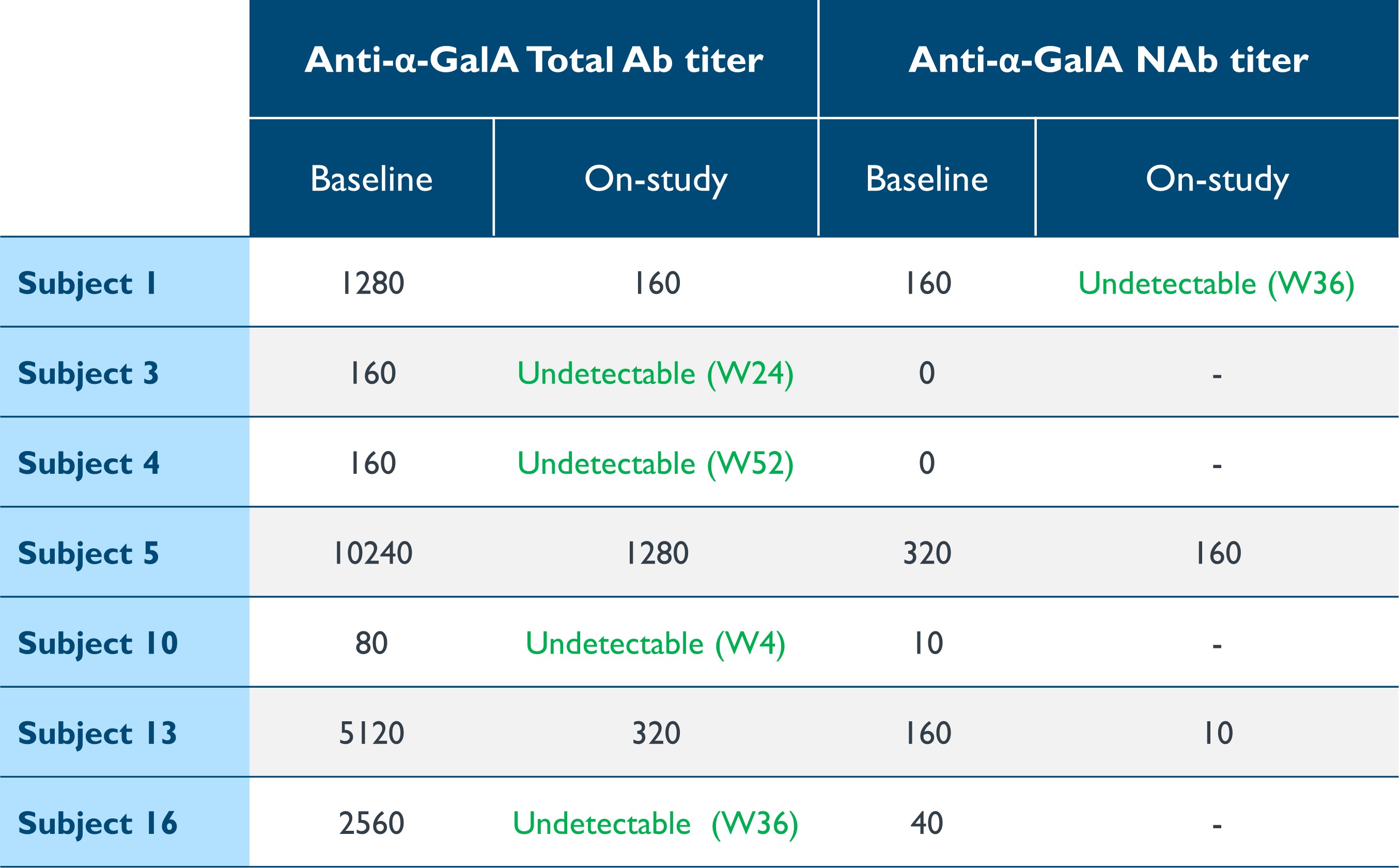
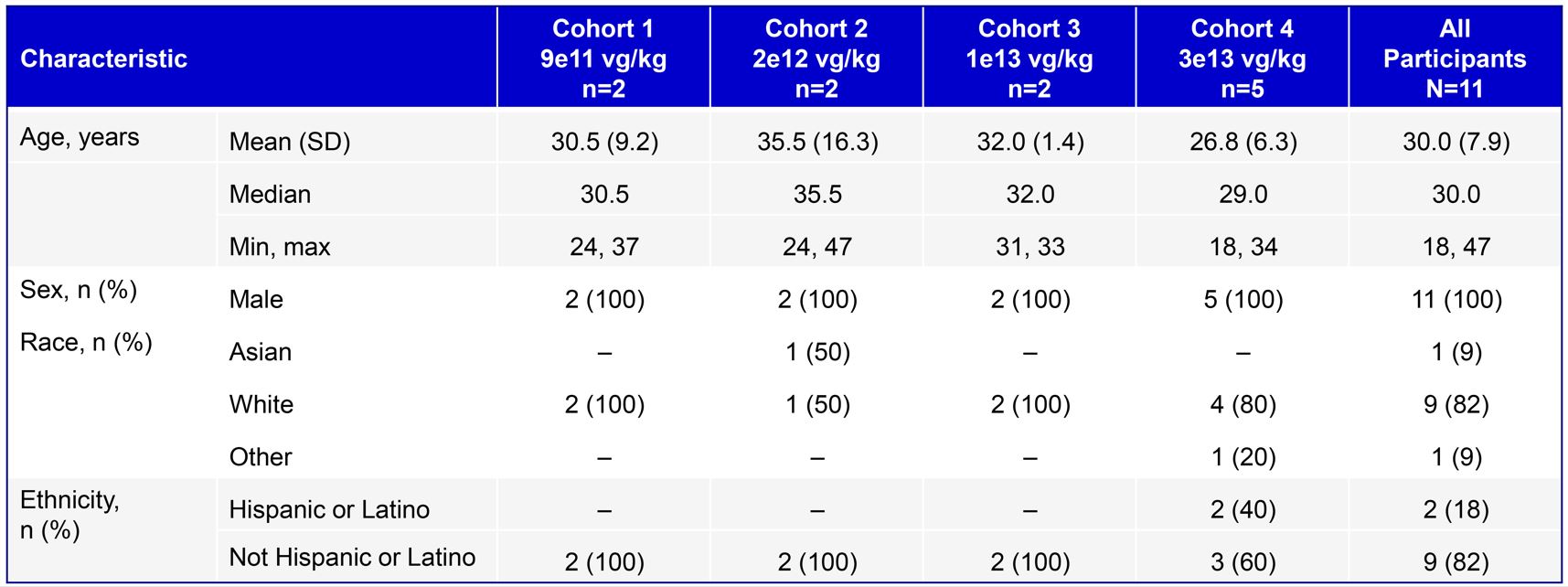
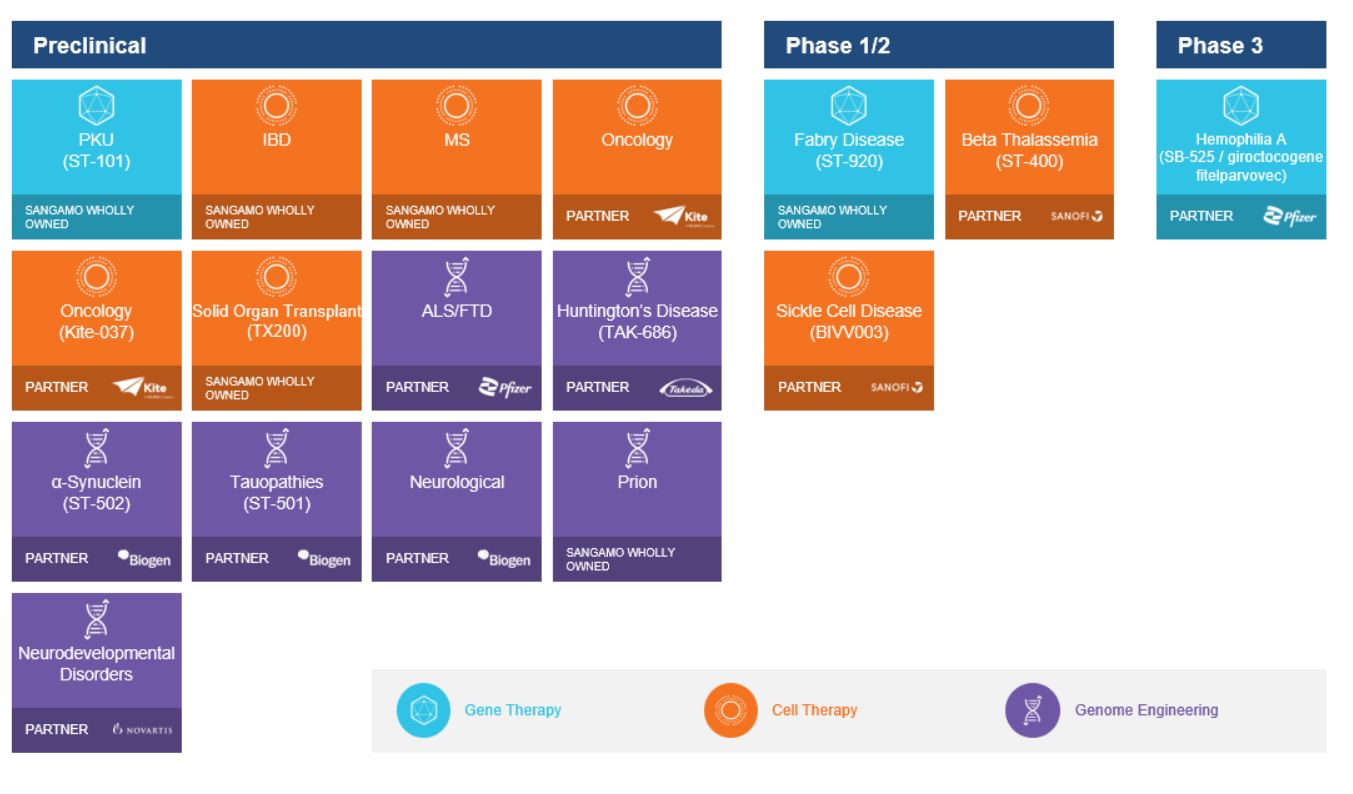
Under the amended and restated agreement, we have full control over, and full responsibility for the costs of, the hemophilia A and B programs returned to us by Takeda, subject to certain diligence obligations. We also granted Takeda a right of first negotiation to obtain a license to such programs under certain circumstances. We areShould we proceed to commercialize the specific hemophilia A and B programs returned to us by Takeda, we will be required to pay single digit percentage royalties to Takeda, up to a specified maximum cap, on commercial sales of therapeutic products from the programs returned to us byus. We do not have any obligations under the amended and restated agreement to make milestone payments to Takeda.
The amended and restated agreement may be terminated by (i) us or Takeda, in whole or in part, for the uncured material breach of the other party, (ii) us or Takeda for the bankruptcy or other insolvency proceeding of the other party and (iii) Takeda, in its entirety, effective upon at least 90 days’ advance written notice.
In addition to our partnerships for the development of human therapeutic applications, we have also licensed our technology in several other areas, such as plant agriculture and research reagents, including the production of transgenic animals and cell-line engineering. These license partners include Corteva AgriScience, formerly known as Dow AgroSciences LLC, or DAS, Sigma-Aldrich Corporation Genentech, Inc.(now MilliporeSigma in the United States and Merck KGaA outside the United States), and Open Monoclonal Technology, Inc. and F. Hoffmann-La Roche Ltd and Hoffmann-La Roche(now Ligand Pharmaceuticals Inc.
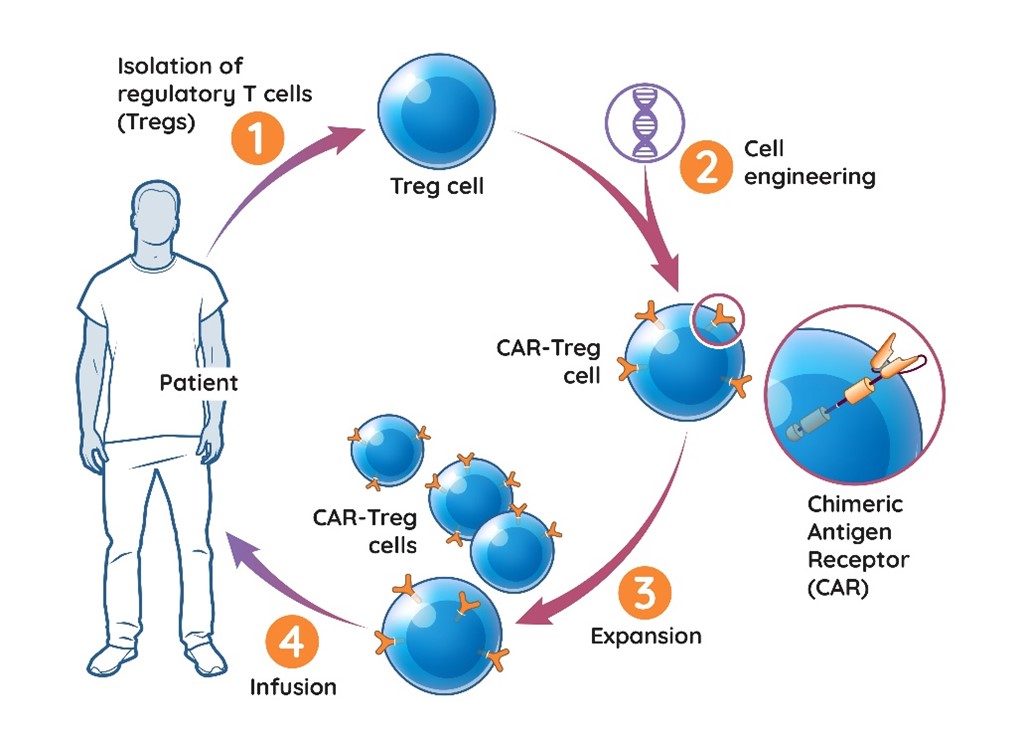
In addition, with respect to our cell therapy products, our subsidiary, Sangamo France, has a license agreement with the University of British Columbia pursuant to which it exclusively licensed in relevant fields the right to the CAR for use in our TX200 product candidate. This license includes one patent family, which expiresis expected to expire in September 2038, absent any patent term adjustment, or PTA, patent term extension, or terminalPTE, or disclaimers.
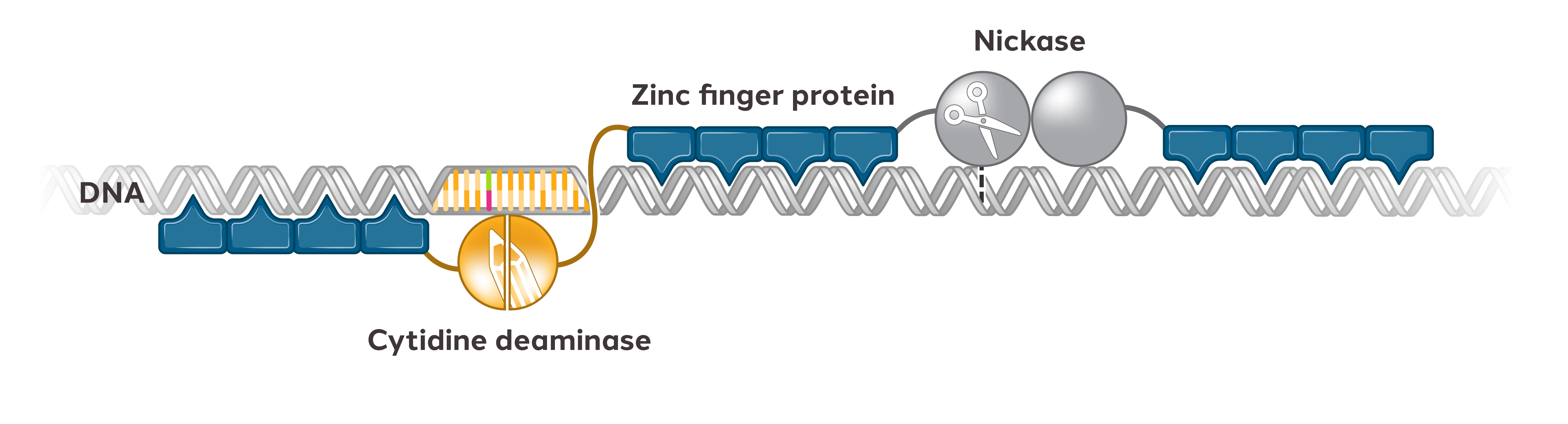
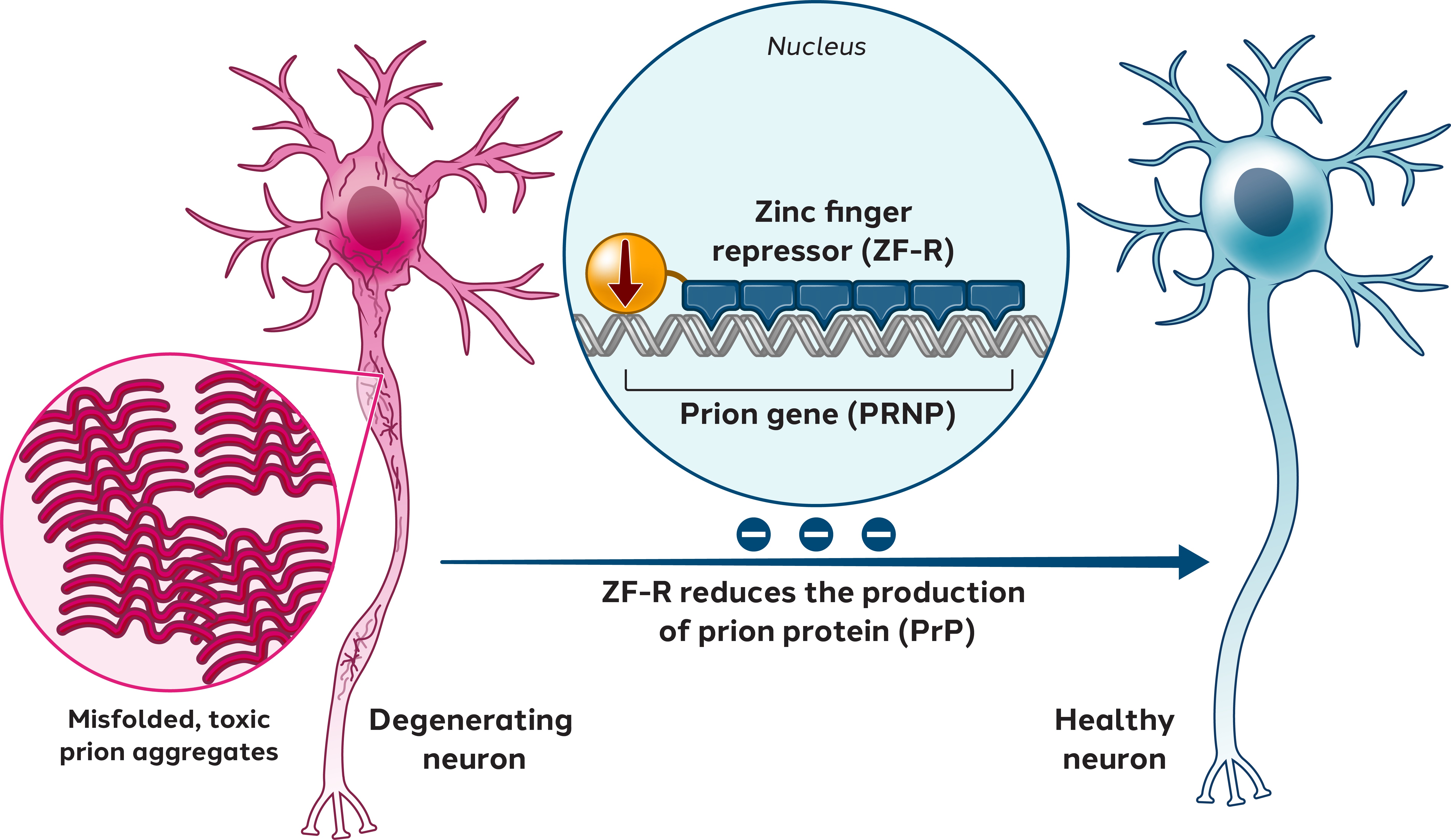
In addition to our in-licensed patent portfolio, we have numerous issued patents and pending patent filingsapplications comprising approximately 170 patent families that are directed to the design, compositioncompositions and useuses of ZFPs, ZFNs, ZFP-TFs,ZF-transcriptional repressors, TALE proteins and CRISPR/Cas editing systems, Treg cell therapy platforms, viral vector delivery platforms, and other technologies related to our program.
development of cell lines for improved protein production, methods of transgenic animal development, engineering of stem cells, methods of genome editing (see, e.g., US9890395).
The patent positions of biopharmaceutical companies, including our patent position, are uncertain and involve complex legal and factual questions for which important legal tenets are largely unresolved and are subject to administrative, judicial, and regulatory interpretation and refinement byrefinement. Obtaining, maintaining, and enforcing patent protection in the court system.United States and other countries remains uncertain and depends, in part, upon decisions of the patent offices, courts, administrative bodies and lawmakers in these countries. It is also possible that we may develop proprietary products or technologies in the future that are not patentable. Patent applications may not result in the issuance of patents and the coverage claimed in a patent application may be significantly reduced before a patent is issued. It is possible that, under certain circumstances, patent applications will be rejected and we subsequently abandon them. It is possible that we may decide that an issued patent or pending patent application may provide us with little or no competitive advantage in view of its associated costs, in which case we may abandon or allow to lapse such patent or patent applications. Although we have filed for patents on some aspects of our technology, we cannot provide assurances that patents will be issued as a result of these pending applications or that any patent that has been or may be issued will be upheld. It is possible that our current patents, or patents which we may later acquire, may be successfully challenged, invalidated in whole or in part, or deemed unenforceable. The laws of some foreign countries may not protect our proprietary rights to the same extent as do the laws of the United States.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements. Periodic maintenance fees, renewal fees, annuity fees and various other governmental fees on patents and/or applications will be due to be paid to the U.S. PTOUSPTO and various governmental patent agencies outside of the United States in several stages over the lifetime of the patents and/or applications. We have systems in place to remind us to pay these fees, and we employ an outside firm and rely on our outside counsel to pay these fees due to non-U.S. patent agencies. The U.S. PTOUSPTO and various non-U.S. governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. We employ professionals to help us comply, and in many cases, an inadvertent lapse can be cured by payment of a late fee or by other means in accordance with the applicable rules. However, there are situations in which non-compliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, our competitors might be able to enter the market and this circumstance would have a material adverse effect on our business. We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting and defending patents on product candidates in all countries throughout the world would be prohibitively expensive, and our intellectual property rights in some countries outside the United States can be less extensive than those in the United States. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as federal and state laws in the United States. Competitors may use our technologies in jurisdictions where we have
not obtained patent protection to develop their own products and further, may export otherwise infringing products to territories where we have patent protection, but enforcement is not as strong as that in the United States.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents, trade secrets and other intellectual property protection, particularly those relating to biotechnology products, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly and our patent applications at risk of not issuing and could provoke third parties to assert claims against us. We may not be able to prevent, alone or with our licensors, misappropriation of our intellectual property rights, particularly in countries where the laws may not protect those rights as fully as in the United States.
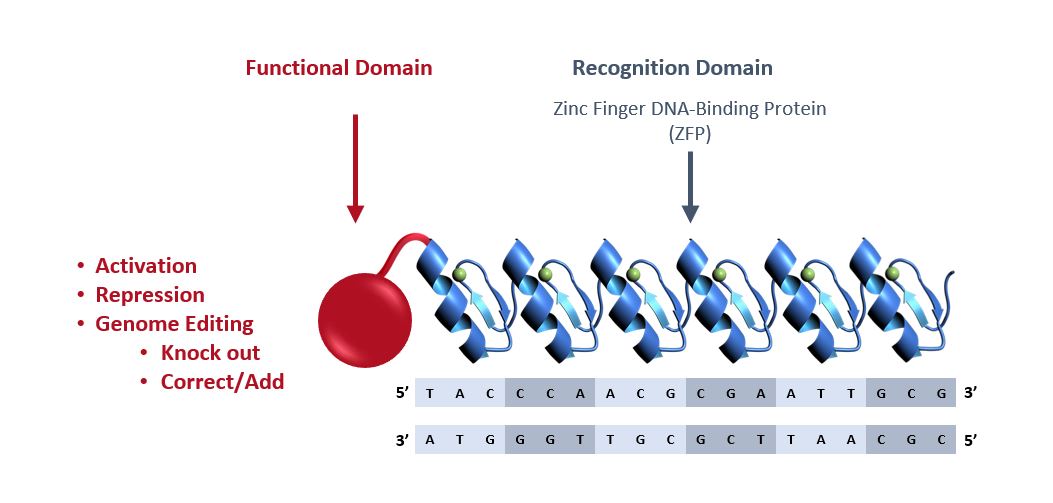
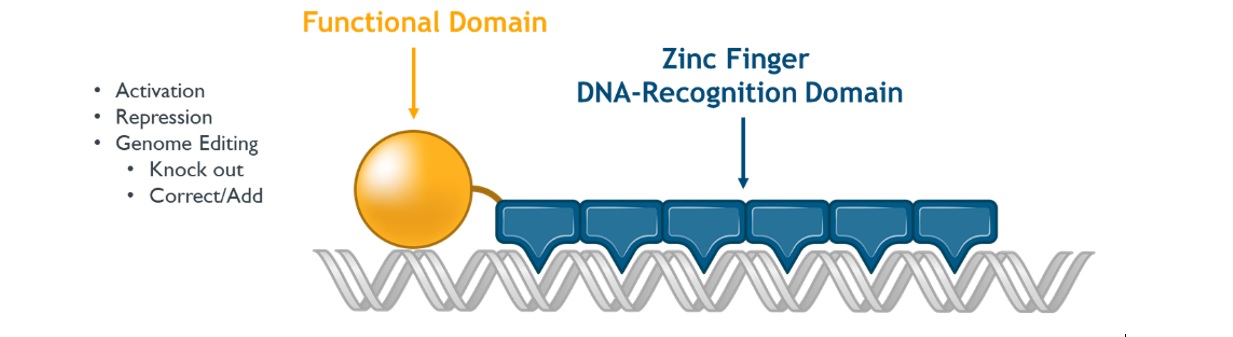
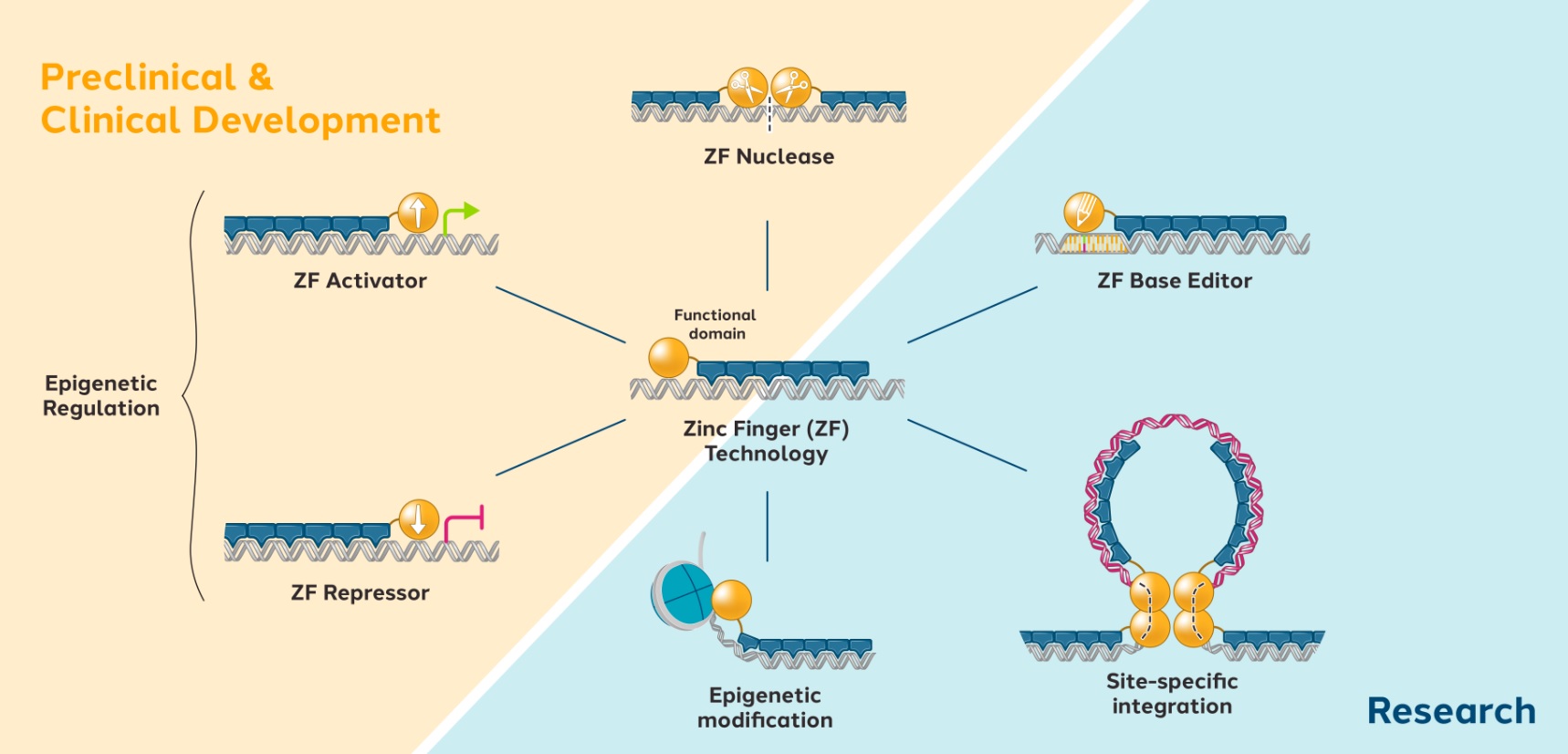
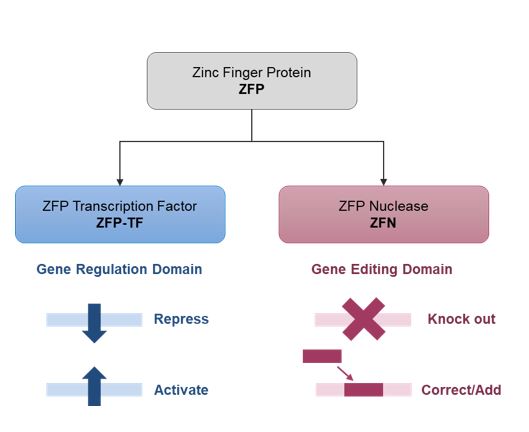
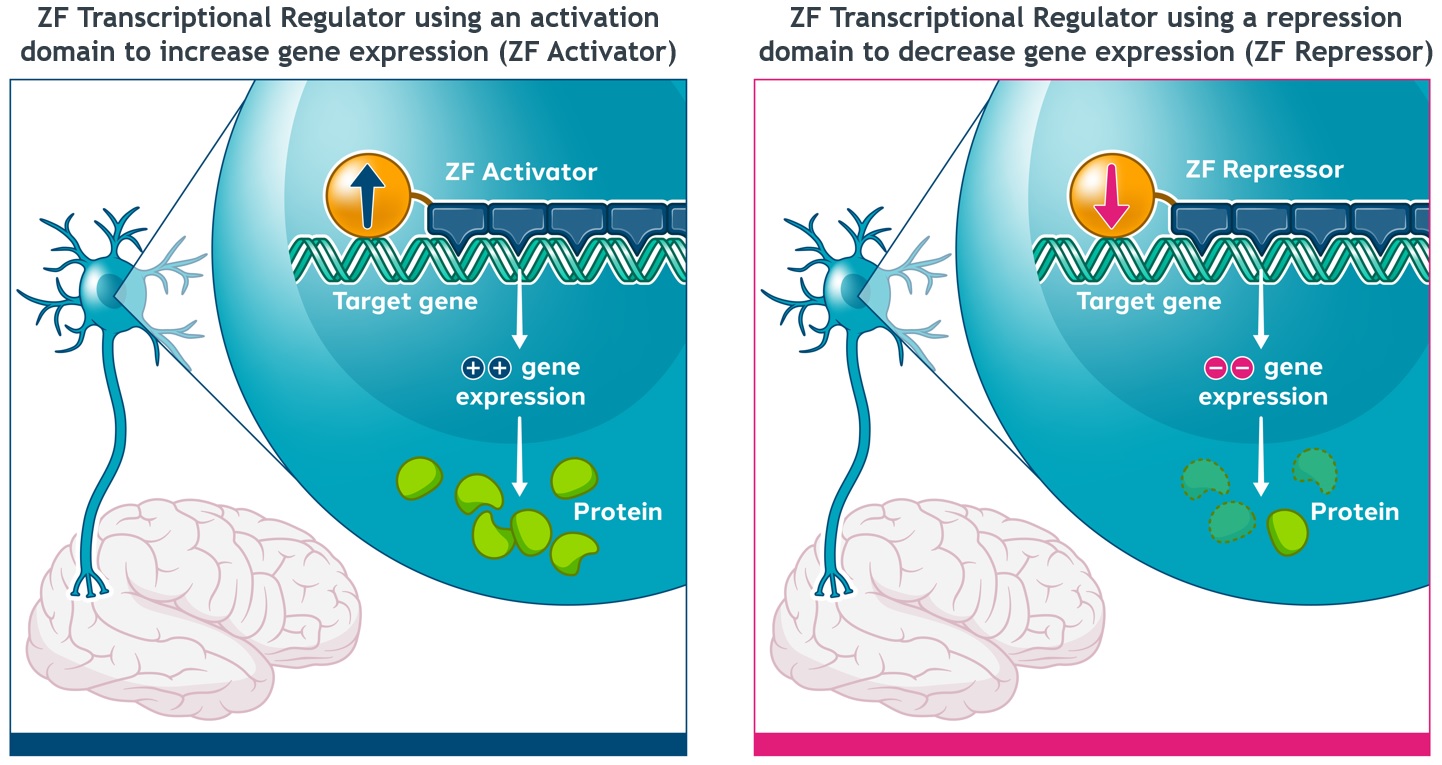
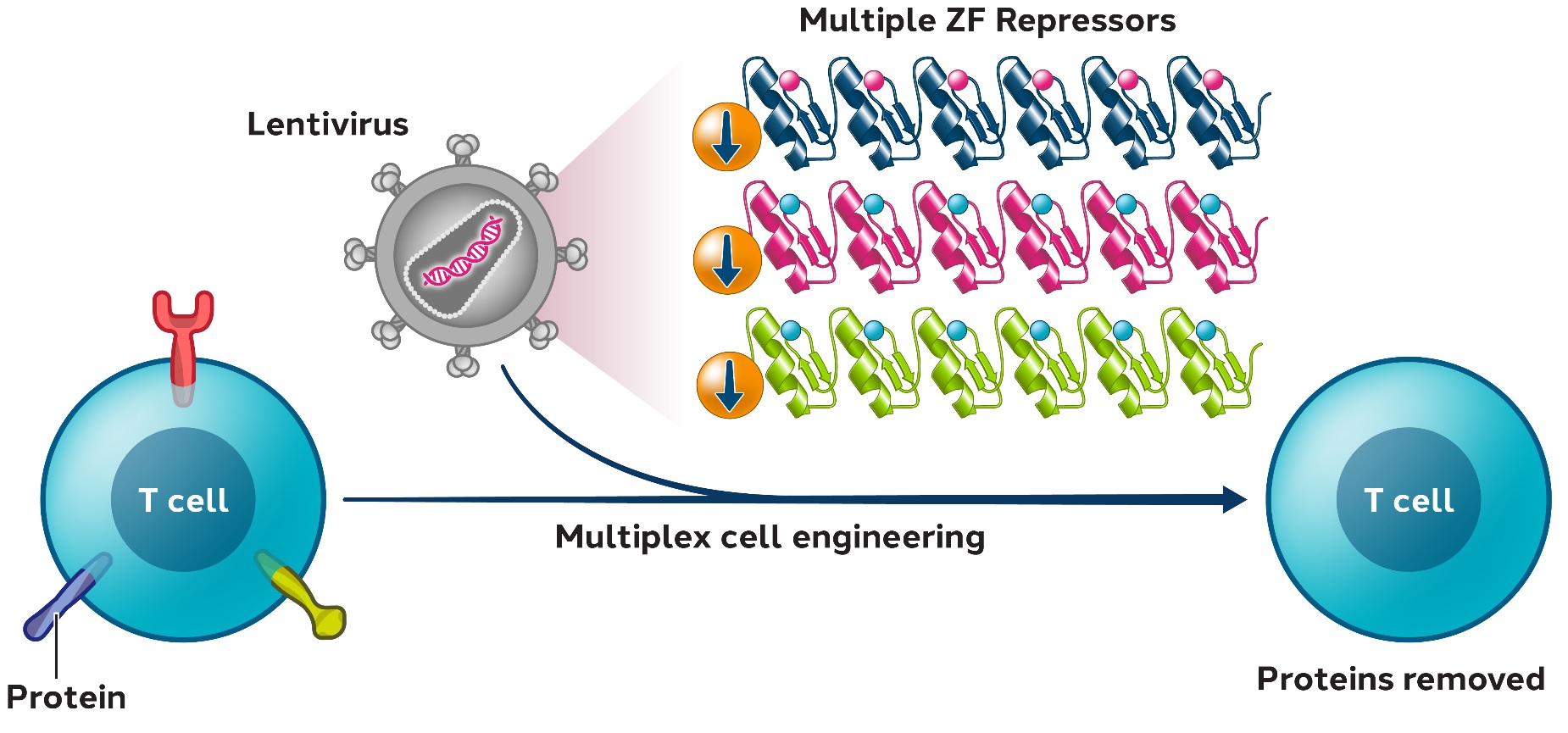
We and our biopharmaceutical collaborators are leaders in the research and development of gene therapies, cell therapies and genome engineering therapies using ZFPZF DNA-binding proteins.
We are aware of several other companies focused on other methods for editing genes and regulating gene expression and a limited number of commercial and academic groups pursuing the development of ZFPZF genome engineering technologies. The fields of gene therapy, cell therapy and genome engineering are highly competitive, and we expect competition to persist and intensify in the future from a number of different sources, including other biopharmaceutical companies; academic and research institutions; and government agencies that will seek to develop ZFPsZFs as well as technologies that will compete with our ZFPZF technology platform, such as TALE proteins and the CRISPR-Cas editing system.
Accordingly, our competitors may succeed in obtaining patent protection, receiving FDA approval or commercializing competitive products before us.we do. If we commence commercial product sales, we may be competing against companies with greater marketing, sales, distribution and manufacturing capabilities, areas in which we have limited or no experience. In addition, any product candidate that we successfully develop may compete with existing products that have long histories of safe and effective use.
Although we are in the clinical development phase of operations and have no current therapeutic product sales, we believe the following companies, products and/or technologies may potentially be competitive with our technology or our product candidates under development:
We expect to face intense competition from other companies for collaborative arrangements with biopharmaceutical companies, for establishing relationships with academic and research institutions, for licenses to proprietary technology and for subjects in our clinical trials of treatments for rare diseases. These competitors, either alone or with their collaborative partners, may succeed in developing technologies or products that are more effective or less costly than ours.
GOVERNMENT REGULATION
We operate within the heavily regulated biopharmaceutical industry and much of our operations, including nonclinical and clinical trials, development, manufacturing, commercialization, marketing and reimbursement are subject to regulatory approvals. Relevant regulatory authorities include, but are not limited to, the FDA, the EMA, the European Commission, national competent authorities of the European Union, or EU, Member State agencies, includingStates and the UKU.K. Medicines and Healthcare Products Regulatory Agency, or MHRA.
Product Regulation
In the United States, the FDA regulates biologic products including gene therapy and human cellular therapy products under the Federal Food, Drug, and Cosmetic Act, or the FDCA, the Public Health Service Act, or the PHSA, and regulations and guidance implementing these laws. The FDCA, PHSA and their corresponding regulations govern, among other things, the testing, manufacturing, safety, efficacy, labeling, packaging, storage, record keeping, distribution, reporting, advertising and other promotional practices involving biologic products. Applications to the FDA are required before conducting human clinical testing of biologic products and in the EU, approval must be obtained from the EMA.products. FDA approval also must be obtained before marketing of biologic products. In the EU, approval from the competent authorities of EU Member States must be obtained before commencing clinical trials. In addition, medicinal products can only be marketed if a marketing authorization, or MA, from the competent regulatory agencies has been obtained.
The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes, regulations and regulationsapplicable guidance require the expenditure of substantial time and financial resources and we may not be able to obtain the required regulatory approvals.
Accelerated Assessment
A number of agencies, including the FDA and the EMA, have expedited development programs, including for innovative products and in areas of high unmet medical need, such as PRIME in the EU. These programs require a certain level of evidence demonstrating safety and efficacy in patients from early-stage clinical trials. Entry into one of these expedited programs may result in assistance with the scientific opinion and faster approval timelines. Some of these programs may offer joint approval and reimbursement advice. It is noted that even applications in an expedited development program may be assessed under standard timelines, where the regulatory authority deems that the program may no longer meet the requirements for priority review.
U.S. Biologic Products Development Process
Our product candidates must be approved by the FDA before they may be legally marketed in the United States. The process required by the FDA before a biologic product candidate may be marketed in the United States generally involves the following:
•completion of preclinical laboratory tests and in vivo studies in accordance with the FDA’s current Good Laboratory Practice, or GLP, regulations and applicable requirements for the humane use of laboratory animals or other applicable regulations;
•submission to the FDA of an IND application, which allows human clinical trials to begin unless FDA objects within 30 days;
•approval by an independent institutional review board, or IRB, reviewing each clinical site before each clinical trial may be initiated;
•performance of adequate and well-controlled human clinical trials according to the FDA’s Good Clinical Practice, or GCP, regulations, and any additional requirements for the protection of human research subjects and their health information, to establish the safety and efficacy of the proposed biologic product candidate for its intended use;
•preparation and submission to the FDA of a biologics license application, or BLA for marketing approval that includes substantial evidence of safety and efficacy from results of nonclinical testing and clinical trials;trials and payment of user fees, if applicable;
•review of the product by an FDA advisory committee, if applicable;
•satisfactory completion of an FDA inspection of the manufacturing facility or facilities where the biologic product candidate is produced to assess compliance with cGMP requirements and to assure that the facilities, methods and controls are adequate to preserve the biologic product candidate’s identity, safety, strength, quality, potency and purity;
•potential FDA inspection of the nonclinical and clinical trial sites that generated the data in support of the BLA; and
•payment of user fees, if applicable, and FDA review and approval, or licensure, of the BLA.
Before testing any biologic product candidate in humans, including a gene therapy product candidate, the product candidate must undergo preclinical testing. Preclinical tests, also referred to as nonclinical studies, include laboratory evaluations of product chemistry, toxicity and formulation, as well as in vivo studies to assess the potential safety and activity of the product candidate and to establish a rationale for therapeutic use. The conduct of the preclinical tests must comply with federal regulations and requirements including GLPs.
Concurrent with clinical trials, companies usually must complete additional preclinical testing, that may include animal tests of reproductive adverse events and carcinogenicity, and must also develop additional information about the chemistry and physical characteristics of the drug and finalize a process for manufacturing the drug in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the drug candidate and, among other things, the manufacturer must develop methods for testing the identity, strength, quality and purity of the final drug product. Additionally, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate that the drug candidate does not undergo unacceptable deterioration over its shelf life.
Human gene transfer protocols are subject to the FDA’s oversight and other clinical trial regulations, and oversight at the local level as set forth in National Institutes of Health, or NIH, Guidelines. Specifically, under the NIH Guidelines, supervision of human gene transfer trials includes evaluation and assessment by an institutional biosafety committee, or IBC, a local institutional committee that reviews and oversees research utilizing recombinant or synthetic nucleic acid molecules at that institution. The IBC assesses the safety of the research and identifies any potential risk to public health or the environment, and such review may result in some delay before initiation of a clinical trial. Compliance with the NIH Guidelines is mandatory for investigators at institutions receiving NIH funds for research involving recombinant DNA. However, many companies and other institutions, not otherwise subject to the NIH Guidelines, voluntarily follow them.
The clinical trial sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and a proposed clinical protocol, to the FDA as part of the IND. Some preclinical testing may continue even after the IND is submitted. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA places the clinical trial on a clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. The FDA also may impose clinical holds on a biologic product candidate at any time before or during clinical trials due to safety concerns or non-compliance. If the FDA imposes a clinical hold, trials may not recommence without FDA authorization and then only under terms authorized by the FDA. Accordingly, we cannot be sure that submission of an IND will result in the FDA allowing clinical studies to begin, or that, once begun, issues will not arise that suspend or terminate such studies.
EU Drug Development Process
Similar to the United States, the various phases of preclinical and clinical research in the EU are subject to significant regulatory framework sets both EU-wide and national, Member State-specific requirements for the development and approval of medicinal products. Article 8(3) of Directive 2001/83/EC sets out the contents of a marketing authorization, or MA, application and all the information that must be submitted for the evaluation of a medicinal product.controls. Certain preclinical (also termed “non-clinical”) data is required in order to enable clinical trials and later be used in dossier for a marketing authorization application.MAA. All studies should take placebe conducted in accordance with GLP and all applicable EMA, European Commission and European Pharmacopoeia guidelines onrelated to preclinical studies, including guidance on quality, non-clinical and clinical aspects of medicinal products containing genetically modified cells.
The requisite amount of preclinical data enables the design of a clinical trial, from Phase 1 (first-in-human clinical trials) through to Phases 2 and 3, which are quality, safety and efficacy studies. Similar restrictions and requirements apply as in the United States regarding preclinical data to support trials using viral vectors. The preclinical tests should establish parameters such as toxicity, pharmacodynamics and pharmacokinetic properties, as well as the quality of the gene therapy medicinal products. Due to the particular nature of gene therapy medicinal products, it is recognized that it may not always be possible for the non-clinical safety studies to be in conformity with the principles of GLP and a proper justification should be submitted where a pivotal non-clinical safety study has not been conducted under GLP rules.
Clinical studies are crucial to obtaining the required data and the requirements governing the conduct of clinical trials are further analyzed below.
All medicinal products and advanced therapy medicinal products, or ATMPs, must be manufactured in accordance with the guidelines on Good Manufacturing Practice, or GMP and in a GMP licensed facility, which can be subject to GMP inspections.
Human Clinical Trials Under an IND
Clinical trials involve the administration of the biologic product candidate to patients under the supervision of qualified investigators which generally are physicians not employed by, or under, the control of the trial sponsor. Clinical trials are conducted under written study protocols detailing, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria and the parameters to be used to monitor subject safety, including stopping rules that assure a clinical trial will be stopped if certain adverse events should occur. Each protocol and any amendments to the protocol must be submitted to the FDA as part of the IND. Clinical trials must be conducted and monitored in accordance with the FDA’s regulations comprising the GCP requirements, including the requirement that all research subjects provide informed consent.
Further, each clinical trial must be reviewed and approved by an IRB at or servicing each institution at which the clinical trial will be conducted. An IRB is charged with protecting the welfare and rights of trial participants and considers items such as whether the risks to individuals participating in the clinical trials are minimized and are reasonable in relation to
anticipated benefits. The IRB also approves the form and content of the informed consent that must be signed by each clinical trial subject, or their legal representative, reviews and approves the study protocol, and must monitor the clinical trial until completed.
Human clinical trials typically are conducted in three sequential phases that may overlap or be combined:
•Phase 1. The biologic product candidate initially is introduced into a small number of human subjects and tested for safety, dosage tolerance, absorption, metabolism, distribution, excretion and, if possible, to gain an early understanding of its effectiveness. Phase 1 clinical trials of gene and cell therapies are typically conducted in patients rather than healthy volunteers.
•Phase 2. The biologic product candidate is evaluated in a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product candidate for specific targeted diseases and to determine dosage tolerance, optimal dosage and dosing schedule.
•Phase 3. Phase 3 clinical trials are commonly referred to as “pivotal” studies, which typically denotes a study which presents the data that the FDA or other relevant regulatory agency will use to determine whether or not to approve a biologic product. In Phase 3 studies, the biologic product candidate is administered to an expanded patient population, generally at multiple geographically dispersed clinical trial sites in adequate and well-controlled clinical trials to generate sufficient data to statistically confirm the efficacy and safety of the product for approval. These clinical trials are intended to establish the overall risk/benefit ratio of the product candidate and provide an adequate basis for product labeling.
Post-approval clinical trials, sometimes referred to as Phase 4 clinical trials, may be conducted after initial approval. These clinical trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication, particularly for long-term safety follow-up. Sometimes approval for a product is conditional upon the completion of post-marketing clinical studies.
During all phases of clinical development, regulatory agencies (such as the FDA, the EMA, national competent authorities of EU Member States and other comparable regulatory agencies) require extensive monitoring and auditing of all clinical activities, clinical data and clinical trial investigators. Annual progress reports detailing the results of the clinical trials must be submitted to the FDA.
Written IND safety reports must be promptly submitted to the FDA and the investigators for: serious and unexpected adverse events; any findings from other trials, in vivo laboratory tests or in vitro testing that suggest a significant risk for human subjects; or any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigator brochure. The sponsor must submit an IND safety report within 15 calendar days after the sponsor determines that the information qualifies for reporting. The sponsor also must notify the FDA of any unexpected fatal or life-threateninglife‑threatening suspected adverse reaction within seven calendar days after the sponsor’s initial receipt of the information.
The FDA and comparable foreign regulatory authorities or the sponsor or its data safety monitoring board may suspend a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable safety risk. Similarly, an IRB and comparable foreign regulatory authorities can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s or comparable foreign regulatory authority’s requirements or if the biologic product candidate has been associated with unexpected serious harm to patients.
The FDA and comparable regulatory authorities in the EU usually recommends that sponsors observe subjects for potential gene therapy-related delayed adverse events for up to a 15-year period.
In the EU, clinical trials almost always require approval fromare governed by the Clinical Trials Regulation (EU) No 536/2014, or the CTR, which entered into application on January 31, 2022, repealing and replacing the former Clinical Trials Directive 2001/20, or CTD. The CTR is intended to harmonize and streamline CTAs, simplify adverse-event reporting procedures, improve the supervision of clinical trials and increase clinical trial transparency. Specifically, the CTR, which is directly applicable in all EU Member States, introduces a nationalstreamlined application procedure through a single-entry point, the Clinical Trials Information System, or CTIS, which is a single set of documents to be prepared and submitted for the application, as well as simplified reporting procedures for clinical trial sponsors. A harmonized procedure for the assessment of applications for clinical trials has been introduced and is divided into two parts. Part I assessment is led by the competent authorityauthorities of a reference Member State selected by the trial sponsor and relates to clinical trial aspects that are considered to be scientifically harmonized across EU Member States. This assessment is then submitted to the competent authorities of all the concerned Member States in which the trial is to be conducted for their review. Part II is assessed separately by the competent authorities and ethics committees in each concerned EU Member State. Individual EU Member States retain the power to authorize the conduct of clinical trials in their territory. The extent to which ongoing clinical trials will be governed by the CTR will depend on the duration of the relevant Member State and anindividual clinical trial. For clinical trials in relation to which application for approval was made on the basis of the CTD before January 31, 2023,
the CTD will continue to apply on a transitional basis for three years until January 31, 2025. By that date, all ongoing trials will become subject to the provisions of the CTR. The CTR will apply to clinical trials from an Ethics Committee. earlier date if the related clinical trial application was made on the basis of the CTR or if the clinical trial has already transitioned to the CTR framework before January 31, 2025.
If the medicinal product is considered to be a genetically modified organism, or GMO, then GMO approval mustmay also be obtained.required from the national GMO competent authorities of EU Member States. There is no harmonization between EU Member States regarding the approach to and timelines of GMO approval, which may result in diverging requirements between EU Member States. In addition, the submission of additional information, whichapplications for approval of GMOs to national competent authorities of EU Member States is not made in tandem with applications for the approval of clinical trials that must be submitted via CTIS. As a result, sponsors of clinical trials that include GMOs requiring separate approval cannot benefit from submission of a single application dossier for the approval of a clinical trial and the subsequent synchronized response from EU Member States. This may impact study initiation in a given country.
The conduct of clinical trials should follow the approved clinical trial protocol, informed consents requirements, including patient informed consents, procedures and controls designed and approved for such studies, accepted standard medical and scientific research procedures and be conducted in accordance with the relevant principles of GCP.GCP and all applicable laws and regulations. Gene therapy medicinal products are in addition subject to the rules of GCP for ATMPs, which outline specific additional safeguards and requirements. Record retention requirements are increased for ATMPs as there are relevant long-term follow-up and human safety and traceability requirements.
Compliance with cGMP Requirements
Manufacturers of biologics must comply with applicable current Good Manufacturing Practices, or cGMP regulations, including quality control and quality assurance and maintenance of records and documentation. Manufacturers and others involved in the manufacture and distribution of such products also must register their establishments with the FDA and certain state agencies.agencies, as well as foreign authorities including the competent authorities of the EU Member States. Both domestic and foreign manufacturing establishments must register and provide additional information to the FDA, as well as foreign authorities including the competent authorities of the EU Member States, upon their initial participation in the manufacturing process. Any material changes to the manufacturing equipment, process or location of the approved manufacturing site must be reported to the relevant agency/authority. Establishments may be subject to periodic, unannounced inspections by government authorities (including regulatory agencies) to ensure compliance with cGMP requirements and other laws. Discovery of problems may result in a government entity placing restrictions on a product, manufacturer or holder of an approved BLA or authorization for clinical trial, and may extend to requiring withdrawal of the product from the market, issue warning or similar letters or seeking civil, criminal or administrative sanctions against the company. The
FDAthe EU Member States will not approve a BLA unless it determinesthey determine that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specification.
Concurrent with clinical trials, companies develop additional information about the physical and biological characteristics of the product candidate as well as finalize a process for manufacturing the product candidate in commercial quantities in accordance with cGMP requirements. To help reduce the risk of the introduction of adventitious agents or of causing other adverse events with the use of biologic products, the PHSA emphasizes the importance of manufacturing control for products whose attributes cannot be precisely defined. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other requirements, the sponsor must develop methods for testing the identity, strength, quality, potency and purity of the final biologic product. Additionally, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate that the biologic product candidate does not undergo unacceptable deterioration over its shelf life.
For a product candidate that is also a human cellular or tissue product, the FDA also requires compliance with current Good Tissue Practices, or cGTPs. These are FDA and EU regulations that govern the methods used in, and the facilities and controls used for, the manufacture of human cells, tissues and cellular and tissue-based products, or HCT/Ps, which are human cells or tissue intended for implantation, transplant, infusion, or transfer into a human recipient. The primary intent of the GTP requirements is to ensure that cell and tissue-based products are manufactured in a manner designed to prevent the introduction, transmission and spread of communicable disease. FDA and EU regulations also require tissue establishments to register and list their HCT/Ps with the FDA or the competent authorities of the EU Member States and, when applicable, to evaluate donors through screening and testing.
U.S. Review and Approval Processes
The results of the preclinical tests and clinical trials, together with detailed information relating to the product’s Chemistry, Manufacturing and Controls, or CMC, and proposed labeling, among other things, are submitted to the FDA as part of a BLA requesting approval to market the product for one or more indications.
Under the Prescription Drug User Fee Act, or PDUFA, as amended, each BLA must be accompanied by a significant user fee. The FDA adjusts the PDUFA user fees on an annual basis. The PDUFA also imposes an annual program fee for approved biologics. Fee waivers or reductions are available in certain circumstances, including a waiver of the application fee for the first application filed by a small business or for a product indication for orphan diseases.
The FDA reviews a BLA within 60 days of submission to determine if it is substantially complete before the agency accepts it for filing. The FDA may refuse to file any BLA that it deems incomplete or not properly reviewable at the time of submission and may request additional information. In that event, the BLA must be resubmitted with the additional information. The resubmitted application also is subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth, substantive review of the BLA.
The FDA reviews the BLA to determine, among other things, whether the proposed product candidate is safe and effective, for its intended use and whether the product candidate is being manufactured in accordance with cGMP to assure and preserve the product candidate’s identity, safety, strength, quality, potency and purity. The FDA may refer applications for novel biologic products or biologic products that present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions. During the product approval process, the FDA also will determine whether a risk evaluation and mitigation strategy, or REMS, is necessary to assure the safe use of the product candidate. REMS use risk minimization strategies beyond the professional labeling to ensure that the benefits of the product outweigh the potential risks. To determine whether a REMS is needed, the FDA will consider the size of the population likely to use the product, seriousness of the disease, expected benefit of the product, expected duration of treatment, seriousness of known or potential adverse events, and whether the product is a new molecular entity. A REMS could include medication guides, physician communication plans and elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. If the FDA concludes a REMS is needed, the sponsor of the BLA must submit a proposed REMS; the FDA will not approve the BLA without a REMS, if required.
Before approving a BLA, the FDA will inspect the facilities at which the product candidate is manufactured. The FDA will not approve the product candidate unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product candidate within required specifications. Additionally, before approving a BLA, the FDA typically will inspect one or more clinical sites to assure that the clinical trials were conducted in compliance with IND trial requirements and GCP requirements.
On the basis of the BLA and accompanying information, including the results of the inspection of the manufacturing facilities, the FDA may issue an approval letter or a complete response letter. An approval letter authorizes commercial marketing of the biologic product with specific prescribing information for specific indications. A complete response letter generally outlines the deficiencies in the submission and may require substantial additional testing or information in order for the FDA to reconsider the application. If and when those deficiencies have been addressed to the FDA’s satisfaction in a resubmission of the BLA, the FDA will issue an approval letter.
If a product candidate receives regulatory approval, the approval may be significantly limited to specific diseases and dosages or the indications for use may otherwise be limited. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling. The FDA may impose restrictions and conditions on product distribution, prescribing or dispensing in the form of a REMS, or otherwise limit the scope of any approval. In addition, the FDA may require post-marketing clinical trials, sometimes referred to as Phase 4 clinical trials, designed to further assess a biologic product’s safety and effectiveness, and testing and surveillance programs to monitor the safety of approved products that have been commercialized.
The FDA has agreed to specified performance goals in the review of BLAs under the PDUFA. One such goal is to review standard BLAs in 10ten months after the FDA accepts the BLA for filing, and priority BLAs in six months, whereupon a review decision is to be made. The FDA does not always meet its PDUFA goal dates for standard and priority BLAs and its review goals are subject to change from time to time. The review process and the PDUFA goal date may be extended by three months if the FDA requests or the BLA sponsor otherwise provides additional information or clarification regarding information already provided in the submission within the last three months before the PDUFA goal date.
EU Review and Approval Process
Before a medicinal product can be placed on the market inIn the EU, it must have received an MA. This could eithermedicinal products can only be at national or EU level undercommercialized after a mutual recognition, decentralized or centralized procedure. Our product candidates are innovative treatments, which will bear the classification of ATMP. As such, the appropriate authorization procedure is the centralized procedure, which involvesrelated MA has been granted. To obtain an MA being granted by the European Commission followingfor a positive opinion by the EMA. A centralized MA is simultaneously validproduct in all EU Member States and the European Economic Area, or EEA (Iceland, Liechtenstein(which is comprised of the 27 Member States of the EU plus Norway, Iceland and Norway)Liechtenstein), an applicant must submit a MAA, either under a centralized procedure administered by the EMA or one of the procedures administered by competent authorities in the EU Member States (decentralized procedure, national procedure or mutual recognition procedure). AAn MA may be granted only to an applicant established in the EU.
The centralized MA also results inprocedure provides for the grant of a single set of product information (patient information leaflet, labelling and summary of product characteristics)MA by the European Commission that is valid for all EU Member States.
The timeline Pursuant to Regulation (EC) No 726/2004, the centralized procedure is compulsory for specific products, including for (i) medicinal products derived from biotechnological processes, (ii) products designated as orphan medicinal products, (iii) ATMPs and (iv) products with a new active substance indicated for the granttreatment of HIV/AIDS, cancer, neurodegenerative diseases, diabetes, autoimmune and other immune dysfunctions and viral diseases. For products with a new active substance indicated for the treatment of other diseases and products that are highly innovative or for which a centralized MA sinceprocess is in the timeinterest of patients, authorization through the applicationcentralized procedure is 210 days foroptional on related approval.
Under the assessment ofcentralized procedure, the application (including “clock stops” for the applicant to prepare answers to the questions from the EMA). TheEMA’s Committee for Medicinal Products for Human Use, or CHMP, conducts the initial assessment of a product. The CHMP is also responsible for several post-authorization and maintenance activities, such as the assessment of modifications or extensions to an existing MA. The maximum timeframe for the evaluation of an MAA is 210 days, excluding clock stops when additional information or written or oral explanation is to be provided by the applicant in response to questions of the CHMP. Accelerated assessment may be granted by the CHMP in exceptional cases. If the CHMP accepts a request for accelerated assessment, the time limit of 210 days will be reduced to 150 days (excluding clock stops). The CHMP can, however, revert to the standard time limit for the centralized procedure if it considers that it is no longer appropriate to conduct an accelerated assessment.
Unlike the centralized authorization procedure, the decentralized MA procedure requires a separate application to, and leads to separate approval by, the competent authorities of each EU Member State in which the product is to be marketed. This application is identical to the application that would be submitted to the EMA for authorization through the centralized procedure. The reference EU Member State prepares a draft assessment and drafts of the related materials within 120 days after receipt of a valid application. The resulting assessment report is submitted to the concerned EU Member States that, within 90 days of receipt, must decide whether to approve the assessment report and related materials. If a concerned EU Member State cannot approve the assessment report and related materials due to concerns relating to a potential serious risk to public health, disputed elements may either provide a positive or negative opinion. Following a positive opinion,be referred to the Heads of Medicines Agencies’ Coordination Group for Mutual Recognition and Decentralised Procedures – Human for review. The subsequent decision of the European Commission will usually issue its legallyis binding on all EU Member States.
The mutual recognition procedure allows companies that have a medicinal product already authorized in one EU Member State to apply for this authorization to be recognized by the competent authorities in other EU Member States. Like the decentralized procedure, the mutual recognition procedure is based on the acceptance by the competent authorities of the EU Member States of the MA after 67 days. A negative opinionof a medicinal product by the competent authorities of other EU Member States. The holder of a national MA may submit an application to the competent authority of an EU Member State requesting that this authority recognize the MA delivered by the competent authority of another EU Member State.
An MA has, in principle, an initial validity of five years. The MA may be appealedrenewed after five years on the basis of a reevaluation of the risk-benefit balance by the applicant who must submit a request for re-examination within 60 days. There isEMA or by the possibility for accelerated timelinescompetent authority of drug applications for eligible applicants,the EU Member State in which can reduce the timeline to 150 days, iforiginal MA was granted. To support the applicant can produce sufficient justification.
Ifapplication, the MA application contains less comprehensive thanholder must provide the required standard as atEMA or the timecompetent authority with a consolidated version of the application, when there are public health groundselectronic Common Technical Document providing up-to-date data concerning the quality, safety and often inefficacy of the case of orphan medicinal products,product, including all variations introduced since the EMA may recommendMA was granted, at least nine months before the MA ceases to thebe valid. The European Commission that it issues a different typeor the competent authorities of the EU Member States may decide on justified grounds relating to pharmacovigilance to proceed with one further five-year renewal period for the MA. Once subsequently definitively renewed, the MA shall be valid for an MA, as follows: (a) a Conditional MA (valid for one year and renewable), whenunlimited period. Any authorization which is not followed by the actual placing of the medicinal product showson the EU market (for a positive benefit-risk balance and targetscentralized MA) or on the market of the authorizing EU Member State (for a decentralized MA) within three years after authorization ceases to be valid (the so-called sunset clause).
Innovative products that target an unmet medical need and are expected to be of major public health interest may be eligible for a number of expedited development and review programs, such as PRIME designation. Products eligible for PRIME must target conditions for which there is an unmet medical need and demonstrate the potential to address the unmet medical need by introducing new methods of therapy or improving existing ones. Benefits accrue to sponsors of product candidates with PRIME designation, including but not limited to, early and proactive regulatory dialogue with the EMA, frequent discussions on clinical trial designs and other development program elements and potentially accelerated MAA assessment once a dossier has been submitted.
In the EU, a “conditional” MA may be granted in cases where all the required safety and efficacy data are not yet available. The European Commission may grant a conditional MA for a medicinal product if it is expecteddemonstrated that all of the following criteria are met: (i) the benefit-risk balance of the medicinal product is positive, (ii) it is likely that the applicant will be able to provide comprehensive data post-authorization, (iii) the medicinal product fulfils an unmet medical need and (iv) the benefit of the immediate availability to patients of the medicinal product is greater than the risk inherent in due course;the fact that additional data are still required. The conditional MA is subject to conditions to be fulfilled for generating the missing data or (b) anensuring increased safety measures. It is valid for one year and must be renewed annually until all related conditions have been fulfilled. Once any pending studies are provided, the conditional MA can be converted into a traditional MA. However, if the conditions are not fulfilled within the timeframe set by the EMA and approved by the European Commission, the MA will cease to be renewed.
An MA may also be granted “under exceptional circumstances” where the applicant can show that it is unable to provide comprehensive data on efficacy and safety under ‘exceptional circumstances’,normal conditions of use even after the product has been authorized and subject to specific procedures being introduced. These circumstances may arise in particular when the intended indications are very rare and, in the state of scientific knowledge at that time, it is not expected that the applicant will be ablepossible to provide comprehensive efficacyinformation, or when generating data may be contrary to generally accepted ethical principles. Like a conditional MA, an MA granted in exceptional circumstances is reserved to medicinal products intended to be authorized for treatment of rare diseases or unmet medical needs for which the applicant does not hold a complete data set that is required for the grant of a standard MA. However, unlike the conditional MA, an applicant for authorization in exceptional circumstances is not subsequently required to provide the missing data. Although the MA “under exceptional circumstances” is granted definitively, the risk-benefit balance of the medicinal product is reviewed annually, and safety data (often for very rare indications).
Manufacturing RegulationRegulations in Europe
Various requirements apply to the manufacturing and placing on the EU market of medicinal products. The manufacturing of medicinal products in the EU requires a manufacturing authorization, and import of medicinal products into the EU requires a manufacturing authorization allowing for import. The manufacturing authorization holder must comply with various requirements set out in the applicable EU laws, regulations and guidance, including EU cGMP standards. Similarly, the distribution of medicinal products into and within the EU is subject to compliance with the applicable EU laws, regulations and guidelines, including the requirement to hold appropriate authorizations for distribution granted by the competent authorities of the EU member states.Member States. Marketing authorization holders and/or manufacturing and import authorization, or MA holders and/or distribution authorization holders may be subject to civil, criminal or administrative sanctions, including suspension of manufacturing authorization, in case of non-compliance with the EU or EU member states’Member States’ requirements applicable to the manufacturing of medicinal products.
Post-approval Requirements
Rigorous and extensive FDA and EU regulation of biologic products continues after approval, particularly with respect to cGMP requirements. Manufacturers are required to comply with applicable requirements in the cGMP regulations, including quality control and quality assurance and maintenance of records and documentation. Other post-approval requirements applicable to biologic products include reporting of cGMP deviations that may affect the identity, potency, purity and overall safety of a distributed product, record-keeping requirements, reporting of adverse effects, reporting updated safety and efficacy information and complying with electronic record and signature requirements. After a BLA is approved, the product also may be subject to official lot release. If the product is subject to official release by the FDA, the manufacturer submits samples of each lot of product to the FDA, together with a release protocol, showing a summary of the history of manufacture of the lot and the results of all tests performed on the lot. The FDA also may perform certain confirmatory tests on lots of some products before releasing the lots for distribution. In addition, the FDA conducts laboratory research related to the regulatory standards on the safety, purity, potency and effectiveness of biologic products. Failure to comply with the FDA’s post-approval regulations can result in withdrawal of product approval and licensure.
A sponsor also must comply with the FDA’s or appropriate national authority’sEU and/or the applicable EU Member States’ laws and requirements governing advertising and promotion requirements, such as the prohibition on promoting products for uses or in patient populations that are not described in the product’s approved labeling (or Summary of Product Characteristics in the EU) (known as “off-label use”). Discovery of previously unknown problems or the failure to comply with the applicable regulatory requirements may result in restrictions on the marketing of a product or withdrawal of the product from the market as well as possible civil or criminal sanctions. In addition, changes to the manufacturing process or facility generally require prior approval by the FDA approvalor competent foreign regulatory authority before being implemented and other types of changes to the approved product, such as adding new indications and additional labeling claims, are also subject to further FDA review and approval.
Orphan and RMAT designation
Products that are intended for treating rare conditions that affect fewer than 200,000 people in the U.S.,United States, or that affect more than 200,000 persons but are not expected to recover the costs of developing and marketing a treatment drug, may qualify for orphan designation. In the EU, these rarea medicinal product can be designated as an orphan medicinal product by the European Commission if the product sponsor can establish that: (i) the product is intended for the diagnosis, prevention or treatment of life-threatening or chronically debilitating conditions; (ii) either (a) such conditions are defined as having a prevalence of noaffect not more than five in every 10,000 peoplepersons in the EU. EU when the application is made, or (b) the product without the benefits derived from orphan status, would not generate sufficient return in the EU to justify the necessary investment in developing the medicinal product; and (iii) there exists no satisfactory authorized method of diagnosis, prevention or treatment of the condition that has been authorized in the EU, or even if such method exists, the product will be of significant benefit to those affected by that condition.
Once marketing authorization has been granted in relation to a medicinal product with orphan designation, obtains a marketing approval, itthe product can benefit from a marketingmarket exclusivity period in respect of the specific orphan indication for which the drug has been approved for a period of seven years in the U.S.United States and for up to 10ten years in the EU. If the manufacturer is no longer able to assert that the product meets the orphan designation criteria or is not able to providesupply sufficient quantities of the product, it may lose the orphan market exclusivity. In the EU, the period of market exclusivity may be reduced to six years if, at the end of the fifth year, it is established that the product no longer meets the criteria on the basis of which it received orphan medicinal product destination, including where it can be demonstrated on the basis of available evidence that the original orphan medicinal product is sufficiently profitable not to justify maintenance of market exclusivity or where the prevalence of the condition has increased above the threshold. Additionally, an MA may be granted to a similar medicinal product with the same orphan indication during the ten year period if: (i) if the applicant consents to a second original orphan medicinal product application, (ii) if the manufacturer of the original orphan medicinal product is unable to supply sufficient quantities; or (iii) if the second applicant can establish that its product, although similar, is safer, more effective or otherwise clinically superior to the original orphan medicinal product. A company may voluntarily remove a product from the register of orphan products.
Regenerative medicine advanced therapy, or RMAT designation is intended to expedite review of a cell therapy, therapeutic tissue engineering product, human cell and tissue product, or any combination product using such therapies or products, intended to treat, modify, reverse, or cure a serious or life-threateninglife‑threatening disease or condition and for which preliminary clinical evidence indicates the potential to address unmet medical needs for such a disease or condition.
RMAT designation provides potential benefits that include more frequent meetings with the FDA to discuss the development plan for the product candidate, and eligibility for rolling review and priority review of the related BLA. Products granted RMAT designation may also be eligible for accelerated approval on the basis of a surrogate or intermediate endpoint reasonably likely to predict long-term clinical benefit, or reliance upon data obtained from a meaningful number of sites, including through expansion to additional sites. However, RMAT designation does not change the FDA’s standards for product approval. Additionally, RMAT designation can be revoked if the criteria for eligibility cease to be met as clinical data emerges.
Clinical Trial Data Disclosure
Many jurisdictions have mandatory clinical trial information obligations incumbent on sponsors. In the EU, thistransparency requirements relating to clinical trial information are established in the CTR, which establishes a general principle according to which information contained in CTIS shall be made publicly accessible unless confidentiality is underjustified on grounds of protecting personal data or commercially confidential information, protecting confidential communications between EU Member States in relation to the Transparencypreparation of an assessment report or ensuring effective supervision of the conduct of a clinical trial by EU Member States. This confidentiality exception may be overruled if there is an overriding public interest in disclosure. The publication of data and documents in relation to the conduct of a clinical trial will take place in accordance with specific timelines. The timelines are established by the EMA and are determined based on the documents and the categorization of the clinical trial.
In addition, Regulation No. 1049/2001 on access to documents, or the ATD Regulation, and the related EMA Policypolicy 0043 EMA Policy 0070, as well as the new Clinical Trials Regulation No. 536/2014, all of which impose on sponsors the obligationaccess to make publicly available certain information stemming from clinical studies. In the EU, the transparency framework providesdocuments provide for a wide right for (EU-based at the moment)EU-based interested parties to submit an access to documents request to the EMA forto access certain information included inheld by the marketing authorization application dossier for approved medicinal products.EMA. Only very limited information is exempted from disclosure (i.e., commercially confidential information, which is construed increasingly narrowly and protected personal data). It is possible for competitors to access and use this data in their own research and development programs anywhere in the world, once these data are in the public domain.
Regulation of Our Operations
Although we currently do not have any products on the market, we may be subject to additional healthcare regulation and enforcement by the federal government and by authorities in the states and foreign jurisdictions in which we conduct our business. Such laws include, without limitation:
•the federal healthcare Anti-Kickback Statute, which prohibits, among other things, persons and entities from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, overtly or covertly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made under a federal healthcare program such as Medicare and Medicaid;
•federal civil and criminal false claims laws, including the federal False Claims Act, and civil monetary penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, to the federal government, including the Medicare and Medicaid programs, claims for payment or approval that are false or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government;
•the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which imposes criminal and civil liability for executing a scheme to defraud any healthcare benefit program and also created federal criminal laws that prohibit, among other things, knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statements in connection with the delivery of or payment for healthcare benefits, items or services;
•HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH, and their implementing regulations, which impose obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information held by certain healthcare providers, health plans and healthcare clearinghouses, known as covered entities, and individuals and entities that perform services for them that involve individually identifiable health information, known as business associates as well as covered subcontractors;
•the federal Physician Payments Sunshine Act created under the Patient Protection and Affordable Care Act of 2010, as amended by the Health Care and Education Reconciliation Act of 2010, or collectively, the ACA, which requires certain manufacturers of drugs, devices, biologics and medical supplies to report annually to the Centers for Medicare and Medicaid Services, or CMS, information related to payments and other transfers of value to physicians currently(currently defined to includedinclude doctors, dentists, optometrists, podiatrists and chiropractors,chiropractors), other healthcare professionals (such as physician assistants and nurse practitioners) and teaching hospitals, and ownership and investment interests held by physicians and their immediate family members. Effective January 1, 2022, these reporting obligations will extend to include transfers of value made to physician assistants, nurse practitioners, clinical nurse specialists, anesthesiologist assistants, certified registered nurse anesthetists and certified nurse midwives during the previous year;members;
•analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers; some state laws require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, require drug manufacturers to report information related to payments and other transfers of value to other healthcare providers and healthcare entities, marketing expenditures; or drug pricing; and/or ensure the registration of sales personnel; and
•state and foreign laws govern the privacy and security of health information in specified circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts.
If our operations are found to be in violation of any of such laws or any other governmental regulations that apply to us, we may be subject to significant penalties, including, without limitation, administrative, civil and criminal penalties, damages, fines, disgorgement, the curtailment or restructuring of our operations, exclusion from participation in federal and state healthcare programs, imprisonment, suspension or withdrawal of our marketing and commercialization in respect of our commercially approved products, and additional reporting requirements and oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations of non-compliance with these laws, any of which could adversely affect our ability to operate our business and our financial results. Responding to investigations can be time-and resource-consuming and can divert management’s attention from the business. Any such investigation or settlement could increase our costs or otherwise have an adverse effect on our business. See “Risk Factors—Our current and future relationships with healthcare providers, customers and third-party payors subject us to applicable anti-kickback, fraud and abuse, privacy, data security and other healthcare laws and regulations. If we fail to comply with such regulations, we could face substantial penalties and our business, results of operations, financial condition and prospects could be adversely affected.”
Healthcare Reform
The U.S. and some foreign jurisdictions are considering enacting or have enacted a number of additional legislative and regulatory proposals to change the healthcare system in ways that could affect our ability to sell our product candidates profitably, if approved. Among policy makers and payors in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality and expanding access. In the United States, the pharmaceutical industry has been a particular focus of these efforts, which include major legislative initiatives, such as the ACA, to reduce the cost of care through changes in the healthcare system, including limits on the pricing, coverage, and reimbursement of pharmaceutical and biopharmaceutical products, especially under government-funded health care programs, and increased governmental control of drug pricing. The ACA and its implementing regulations, among other things, addressed a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for certain drugs and biologics, including products similar to our product candidates, that are inhaled, infused, instilled, implanted or injected, increased the minimum Medicaid rebates owed by manufacturers under the Medicaid Drug Rebate Program, extended the Medicaid Drug Rebate Program to utilization of prescriptions of individuals enrolled in Medicaid managed care organizations, subjected manufacturers to new annual fees and taxes for certain branded prescription drugs, created a new Patient Centered Outcomes Research Institute, which provides incentives to programs that increase the federal government’s comparative effectiveness research, established a new Medicare Part D coverage gap discount program, in which manufacturers must now agree to offer 70% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D, and created a licensure framework for follow-on biologic products.
There remainhave been legal and political challenges to certain aspects of the ACA, as well as efforts to repeal or replace certain aspects of the ACA. Several bills affectingFor example, on June 17, 2021, the implementation of certain taxes under the ACA have been signed into law. The Tax Cuts and Jobs Act of 2017, or Tax Act, includedU.S. Supreme Court dismissed a provisionchallenge on procedural grounds that repealed, effective January 1, 2019, the tax-based shared responsibility payment imposed by the ACA on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate”. In addition, the 2020 federal spending package permanently eliminated, effective January 1, 2020, the ACA-mandated “Cadillac” tax on high-cost employer-sponsored health coverage and medical device tax and, effective January 1, 2021, also eliminates the health insurer tax. In addition, the Bipartisan Budget Act of 2018, or the BBA, among other things, amended the ACA, effective January 1, 2019, to close the coverage gap in most Medicare drug plans, commonly referred to as the “donut hole.”
On December 14, 2018, a Texas U.S. District Court Judge ruled thatargued the ACA is unconstitutional in its entirety because the “individual mandate” was repealed by Congress as partCongress. Prior to the U.S. Supreme Court ruling, on January 28, 2021, President Biden issued an executive order that initiated a special enrollment period for purposes of obtaining health insurance coverage through the ACA marketplace. The executive order also instructed certain governmental agencies to review and reconsider their existing policies and rules that limit access to healthcare, including among others, reexamining Medicaid demonstration projects and waiver programs that include work requirements, and policies that create unnecessary barriers to obtaining access to health insurance coverage through Medicaid or the ACA. Further, on August 16, 2022, President Biden signed the Inflation Reduction Act of 2022, or the IRA, into law, which among other things, extends enhanced subsidies for individuals purchasing health insurance coverage in ACA marketplaces through plan year 2025. The IRA also eliminates the “donut hole” under the Medicare Part D program beginning in 2025 by significantly lowering the beneficiary maximum out-of-pocket cost through a newly established manufacturer discount program. It is possible that the ACA will be subject to judicial or Congressional challenges in the future. It is unclear how additional challenges and the healthcare reform measures of the Tax Act. Additionally, on December 18, 2019,Biden administration will impact the U.S. Court of Appeals for the 5th Circuit upheld the District Court ruling that the individual mandate was unconstitutional and remanded the case back to the District Court to determine whether the remaining provisions of the ACA are invalid as well. The United States Supreme Court is currently reviewing this case, although it is unclear when a decision will be made.
Other legislative changes have been proposed and adopted in the United States since the ACA was enacted. In August 2011, the Budget Control Act of 2011, among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions of Medicare payments to providers of 2% per fiscal year, which went into effect in April 2013, and, due to subsequent legislative amendments to the statute, including the Bipartisan Budget Act of 2018 and the Consolidated Appropriations Act of 2023, will remain in effect through 20292032 unless additional Congressional action is taken. However, pursuant to COVID-19 pandemic relief legislation, these Medicare sequester reductions are suspended from MayAdditionally, on March 11, 2021, President Biden signed the American Rescue Plan Act of 2021 into law, which eliminates the statutory Medicaid drug rebate cap, currently set at 100% of a drug’s average manufacturer price, for single source and innovator multiple source drugs, beginning January 1, 2020 through March 31, 2021 due to the COVID-19 pandemic.2024. In January 2013, the American Taxpayer Relief Act of 2012, or the ATRA, was signed into law, which, among other things, further reduced Medicare payments to several providers, including hospitals and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
Also, there has been heightened governmental scrutiny recently over pharmaceutical pricing practices in light of the rising cost of prescription drugs and biologics. Such scrutiny has resulted in several recent Presidential executive orders, Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for pharmaceutical products. AtAdditionally, in July 2021, the federal level, the Trump administration’s budget proposal for fiscal year 2021 included a $135 billion allowance to support legislative proposals seeking to reduce drug prices, increase competition, lower out-of-pocket drug costs for patients, and increase patient access to lower-cost generic and biosimilar drugs. On March 10, 2020, the Trump administration sent “principles” for drug pricing to Congress, calling for legislation that would, among other things, cap Medicare Part D beneficiary out-of-pocket pharmacy expenses, provide an option to cap Medicare Part D
beneficiary monthly out-of-pocket expenses, and place limits on pharmaceutical price increases. Further, the TrumpBiden administration released a “Blueprint”an executive order, “Promoting Competition in the American Economy,” with multiple provisions aimed at prescription drugs. In response to lower drug prices and reduce out-of-pocket costs of drugs that contained proposals to increase drug manufacturer competition, increase the negotiating power of certain federal healthcare programs, incentivize manufacturers to lower the list price of their products, and reduce the out-of-pocket costs of drug products paid by consumers. On July 24, 2020 and September 13, 2020, the Trump administration announced severalBiden’s executive orders related to prescription drug pricing that seek to implement several of the administration’s proposals. As a result, the FDA released a final ruleorder, on September 24, 2020, effective November 30, 2020, providing guidance for states to build and submit importation plans for drugs from Canada. Further, on November 20, 2020,9, 2021, the U.S. Department of Health and Human Services, or HHS, finalizedreleased a regulation removing safe harbor protectionComprehensive Plan for price reductions from pharmaceutical manufacturers to plan sponsors under Part D, either directly or through pharmacy benefit managers, unless the price reduction is required by law. The rule also createsAddressing High Drug Prices that outlines principles for drug pricing reform and sets out a new safe harbor for price reductions reflected at the point-of-sale,variety of potential legislative policies that Congress could pursue as well as a safe harbor forpotential administrative actions HHS can take to advance these principles. It is unclear whether these or similar measures will be implemented in the future. In addition, the IRA, among other things, (1) directs HHS to negotiate the price of certain fixed fee arrangements between pharmacy benefit managerssingle-
source drugs and manufacturers. On November 20, 2020, CMS issued an interim final rule implementing President Trump’s Most Favored Nation executive order, which would tiebiologics covered under Medicare and (2) imposes rebates under Medicare Part B payments for certain physician-administeredand Medicare Part D to penalize price increases that outpace inflation. These provisions take effect progressively starting in fiscal year 2023. On August 29, 2023, HHS announced the list of the first ten drugs that will be subject to price negotiations, although the Medicare drug price negotiation program is currently subject to legal challenges. It is currently unclear how the IRA will be implemented but is likely to have a significant impact on the pharmaceutical industry. Further, in response to the lowest price paid in other economically advanced countries, effective January 1, 2021. On December 28, 2020,Biden administration’s October 2022 executive order, on February 14, 2023, HHS released a report outlining three new models for testing by the United States District Court in Northern California issued a nationwide preliminary injunction against implementationCMS Innovation Center which will be evaluated on their ability to lower the cost of the interim final rule.drugs, promote accessibility, and improve quality of care. It is unclear whether the Biden administrationmodels will work to reverse thesebe utilized in any health reform measures or pursue similar policy initiatives.in the future. At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing.
In the United States, the EU and other potentially significant markets for our product candidates, government authorities and third-party payors are increasingly attempting to limit or regulate the price of medical products and services, particularly for new and innovative products and therapies, which has resulted in lower average selling prices. Furthermore, the increased emphasis on managed healthcare in the United States and on country and regional pricing and reimbursement controls in the EU will put additional pressure on product pricing, reimbursement and usage, which may adversely affect our future product sales and results of operations. These pressures can arise from rules and practices of managed care groups, judicial decisions and governmental laws and regulations related to Medicare, Medicaid and healthcare reform, pharmaceutical reimbursement policies and pricing in general.
See “Risk Factors—Recently enacted and future legislation, including potentially unfavorable pricing regulations or other healthcare reform initiatives, may increase the difficulty and cost for us to obtain regulatory approval of and commercialize our product candidates and affect the prices we may obtain.”
Pricing, Coverage and Reimbursement
Pricing and reimbursement of a therapeutic product will largely determine the affordability of the product, and whether the product is prescribed and supplied to patients and private insurance companies may take into account government reimbursement methodologies. Due to these proposed and enacted laws, as well as other actions, significant uncertainty exists as to the coverage and reimbursement status of any product candidates for which we obtain regulatory approval, particularly for novel products. In both domestic and foreign markets, sales and reimbursement of any approved products will depend, in part, on the extent to which third-party payors, such as government health programs, commercial insurance and managed healthcare organizations provide coverage, and establish adequate reimbursement levels, for such products. Third-party payors are increasingly challenging the prices charged for medical products and services and imposing controls to manage costs. Third-party payors may limit coverage to specific products on an approved list, also known as a formulary, which might not include all of the FDA-approved products for a particular indication. Additionally, we may need to conduct expensive pharmacoeconomic studies in order to demonstrate the cost-effectiveness of our products. If third-party payors do not consider our products to be cost-effective compared to other therapies, these payors may not cover our products after approved as a benefit under their plans or, if they do, the level of reimbursement may not be sufficient to allow us to sell our products on a profitable basis.
In the EU, pricing and reimbursement schemes vary widely from country to country. The EU provides options for its Member States to restrict the range of products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. EU Member States may approve a specific price for a product, or they may instead adopt a system of direct or indirect controls on the profitability of the company placing the product on the market. Other EU Member States allow companies to fix their own prices for products but monitor and control prescription volumes and issue guidance to physicians to limit prescriptions. Recently, many countries in the EU have increased the amount of discounts required on pharmaceuticals and these efforts could continue as countries attempt to manage healthcare expenditures. Some countries may also require the completion of additional studies that compare the cost-effectiveness of a particular product candidate to currently available therapies (so called health technology assessments) in order to obtain reimbursement or pricing approval. The Health Technology Assessment, or HTA, process is the procedure according to which the assessment of the public health impact, therapeutic impact and the economic and societal impact of use of a given medicinal product in the national healthcare systems of the individual country is conducted. The outcome of HTA regarding specific medicinal products will often influence the pricing and reimbursement status granted to these medicinal products by the competent authorities of individual EU Member States. In December 2021, the EU HTA Regulation was adopted. Although the HTA Regulation entered into force in January 2022, it will only begin to apply from January 12, 2025, with preparatory and implementation-related steps to take place in the interim. The HTA Regulation is intended to boost cooperation among EU Member States in assessing health technologies and providing the basis for cooperation at EU level for joint clinical assessments in these areas. The results of assessments conducted on the basis of the HTA may result in increased parity of reimbursement levels for medicinal products between EU Member States. See “Risk Factors—Even if we are able to commercialize our product candidates, theany approved products, such products may not receive coverage and adequate reimbursement from third-party payors in the United States and in other countries in which we seek to commercialize our products,them, which could harm our business.”
In the EU, pricing and reimbursement are the prerogative of Member States. Therefore, the requirements around reimbursement of medicinal products can vary widely. Each Member State can follow its own approach, subject to common rules of transparency, competition, and freedom of trade and movement in the EU. Many Member States, including France, Germany and the United Kingdom, follow a health technology assessment, or HTA, procedure for medicinal products in order to assess the cost-effectiveness of a product which could then be recommended for reimbursement under the national health services. There is increasingly exchange of information concerning HTAs on a voluntary basis among EU Member States. In
the United Kingdom, the National Institute for Health and Care Excellence is the body which conducts HTAs and issues guidance to be followed by the regional health bodies called clinical commissioning groups.
Environmental Regulation
U.S. federal and state laws regarding safe working conditions, environmental protection and hazardous substances, including the Occupational Safety and Health Act, the Resource Conservancy and Recovery Act and the Toxic Substances Control Act, affect our business. These and other laws govern our use, handling and disposal of various biological, chemical and radioactive substances used in, and wastes generated by, our operations. We may incur significant costs to comply with such laws and regulations now or in the future. If our operations result in contamination of the environment or expose individuals to hazardous substances, we could be liable for damages and governmental fines. We believe that we are in material compliance with applicable environmental laws and regulations that continued compliance therewith will not have a material effect on our business. We cannot predict, however, how changes in these laws and regulations may affect our future operations.
Privacy Regulation
We are, requiredor may become, subject to comply withnumerous privacy and data security laws and regulations in the United States and in other foreign jurisdiction in which we operate, suchjurisdictions, including, as applicable, the Federal Trade Commission Act, the EU General Data Protection Regulation, or EU GDPR, the EU GDPR as it forms part of the United Kingdom’s law by virtue of Section 3 of the European Union (Withdrawal) Act 2018, as amended, or U.K. GDPR, and the California Consumer Privacy Act of 2018, as amended by the California Privacy Rights Act of 2020, or CCPA, which apply tocollectively the collection, use, disclosure, transfer, or other processing of personal data.
The collection, use, disclosure, transfer or other processing of personal data regarding individuals in the EU,EEA and U.K., including personal health data, is subject to the EU GDPR, which became effective on May 25, 2018.and U.K. GDPR, or collectively the GDPR, as applicable. The GDPR, which is wide-ranging in scope, imposes several requirements on us relating to, among other things, the control over personal data by individuals to whom the personal data relates;relates, notice we must provide to individuals regarding our processing of their personal data;data, the documentation we must maintain;maintain, the security and confidentiality of the personal data;data, data breach notification;notification, and the use of third-party processors in connection with the processing of personal data. The GDPR also imposes strict rules on the transfer of personal data outto countries that the European Commission does not consider to provide an adequate level of the EU. The post-Brexit tradeprivacy and cooperation agreement betweendata security (including the United Kingdom and the EU provides that transfers of personal data from the EU to the United Kingdom will not be treated as restricted transfers to a non-EU country for a period of up to six months from January 1, 2021. However, unless the EU Commission makes an “adequacy finding” with respect to the United Kingdom before the end of that transition period, from that date the United Kingdom will be a “third country” under the GDPR, and transfers of personal data from the European Economic Area to the United Kingdom will require an “adequacy mechanism,” such as the Standard Contractual Clauses.States). The GDPR authorizes the imposition of large penalties and other corrective actions for noncompliance, including the potential for fines of up to €20 million (£7.5 million) or 4% of the annual global revenue of the noncompliant company, whichever is greater.greater, definitive bans on data processing or private litigation related to processing of personal data brought by classes of data subjects or consumer protection organizations authorized at law to represent their interests. The GDPR requirements related to international data transfers apply not only to third-party transactions, but also to transfers of information between us and our subsidiaries such as Sangamo France, including employee information. The GDPR has increased our responsibility and potential liability in relation to personal data that we process, compared to prior EU law, particularly in light of our acquisition of Sangamo France, and we may be required to put in place additional mechanisms to ensure compliance with the GDPR, which could divert management’s attention and increase our cost of doing business.
Despite our ongoing efforts to bring our practices into compliance with the GDPR, we may not be successful either due to various factors within our control or other factors outside our control. It is possible that local data protection authorities may interpret the GDPR in different ways, leading to potential inconsistencies in the requirements we must meet in different EU member states. Any failure or alleged failure (including as a result of deficiencies in our policies, procedures or measures relating to privacy, data security, marketing or communications) by us to comply with laws, regulations, policies, legal or contractual obligations, industry standards or regulatory guidance relating to privacy or data security may result in governmental investigations and enforcement actions, litigation, fines and penalties or adverse publicity.
In the United States, California adoptedfederal, state and local governments have enacted numerous privacy and data security laws, including laws on data breach notification, personal data privacy and consumer protection. For example, the CCPA which became effective in January 2020. The CCPA establishes a privacy framework for covered businesses, including an expansive definitionapplies to personal data of personal informationconsumers, business representatives and data privacy rights foremployees who are California residents. The CCPA includes a framework with potentially severe statutory damagesresidents and private rights of action. The CCPA requires covered businesses to provide newdetailed disclosures in privacy notices and honor requests of California residents to California consumers (as that word is broadly definedexercise certain privacy rights related to their personal data. The CCPA provides for civil penalties of up to $7,500 per intentional violation and allows private litigants affected by certain data breaches to recover significant statutory damages. Although the CCPA (like other U.S. comprehensive privacy laws) exempts some data processed in the CCPA), provide such consumers new ways to opt-outcontext of certain sales of their personal information and allow for a new cause of action for data breaches. It remains unclear how the CCPA will be interpreted, but as currently written, it will likely impact our business activities and exemplifies the vulnerability of our business not only to cyber threats but also to the evolving regulatory environment related to personal data. As we expand our operations,clinical trials, the CCPA may increase our compliance costs and potential liability.
The CCPA itself will be expanded substantially effective January 1, 2023, as a result ofliability with respect to other personal data we may maintain about California voters approving the California Privacy Rights Act of 2020,residents. Similar laws have been enacted or CPRA, on the November 2020 ballot, which will, amongproposed by several other things, establish the California Privacy Protection Agency to implement and enforce the new law,states, as well as impose administrative fines. Some observers have noted thatat the CCPAfederal and CPRA could marklocal levels. These laws may further complicate compliance efforts, and may increase legal risk and compliance costs for us and the beginning of a trend toward more stringent privacy legislation in the United States. Other states are beginning to pass similar laws.
Compliance with these and any other applicable privacy and data security laws and regulations is a rigorous and time-intensive process, and we may be required to put in place additional mechanisms ensuring compliance with the new data protection rules. If we fail to comply with any such laws or regulations, we may face significant fines and penalties that could adversely affect our business, financial condition and results of operations. Furthermore, the laws are not consistent, and compliance in the event of a widespread data breach is costly. See “Risk FactorsFactors——Our current and future relationships with healthcare providers, customers and third-party payors subject us to applicable anti-kickback, fraud and abuse, privacy, data security and other healthcare laws and regulations. If we fail to comply with federal, state and foreign laws and regulations, including healthcare, privacy and data security laws andsuch regulations, we could face regulatory investigations or actions, litigation, and substantial fines and penalties, and our business, reputation, results of operations, financial condition and prospects could be adversely affected.”
HUMAN CAPITAL MANAGEMENT
Our Mission &and Our Employees
At Sangamo, we are committed to translating ground-breaking science into genomic medicines that transform patients’ lives. We are a passionate group of biotechnology professionals based in the United States, France and the United Kingdom with years of experience and technical expertise, committed to developing best-in-class genomic medicines. We embrace collaboration, discipline and efficiency while welcoming fresh ideas and stimulating personal development. We encourage and embrace diversity, equity and inclusion, and believe it enhances our work towards one common goal: to transform the lives of the patients we aim to serve.
We view our employees as one of our most valuable assets in serving our mission. We compete in the highly competitive biotechnology industry, and attracting, retaining and developing a diverse group of talented employees is crucial to our strategy and our ability to compete effectively. We needare committed to grow the sizedevelopment and retention of our organization in orderworkforce. There continues to support our current research, product development, manufacturing and regulatory efforts and our plans for commercializing our wholly-owned product candidates when approved. This growth is critical to our success. There currently isbe a shortage of skilled individuals with substantial experience discovering, developing and manufacturing genomic medicines, which is likely to continue. As a result, there continues to be competition for these individuals is intense and the turnover rate can be high. We face substantial competition among numerousbetween biopharmaceutical companies and academic institutions for individuals with these skills.
Our Values
We believe success comes when we and our employees align our core values with our mission to translate our ground-breaking science intodeliver genomic medicines that replace today’s symptomatic treatments and transform patients’ lives. Our core values are:
◦We collaborate with purpose and are driven by results that benefit patients.
◦We strive to put patient safety and quality of care first.
◦Patient needs drive our sense of urgency to deliver medicines.
◦We embrace our responsibility to pioneer the field of genomic medicine bioethically.
◦We take an inclusive approach to guide our development efforts.
•Patient CentricSucceeding through teamwork: Patient welfare is the rule against which we measure our actions and decisions. Before making decisions affecting patients, we work to understand their needs and experiences. Our focus on patients is a source of competitive advantage: it sharpens our research and development efforts and guides us to develop important medicines.
•◦Teamwork & Accountability: We seek appropriate input acrossare driven by our company to make better decisionsshared vision that genomic medicine will transform the lives of patients and achieve our goals. We treat one another with respect and expect a great dealthe field of each other. We honor our commitments.healthcare.
•◦Innovation & Excellence: The bold pursuit of technological advancement and scientific breakthrough is our foundation. We are also a company wherepassionate and dedicated group of individuals can flourish, growwho collaborate proactively and develop their expertise. We celebrateopenly to execute and progress our successes and the personal and professional accomplishments of our colleagues.business forward.
•◦Urgency & Efficiency: We make decisions quickly and thoughtfully; we decentralize decision making authority appropriately to foster ownership and action. We define our priorities clearly, communicate them, and move forward decisively. take collective accountability to deliver results for all stakeholders.
◦We commitare resilient and determined to achieving or surpassingsucceed together because patients are depending on us.
•Innovating through smart decisions:
◦We courageously, relentlessly, and urgently pursue the journey of innovation to succeed in the field of genomic medicine.
◦We mine scientific possibilities with the goal of unlocking new treatment solutions for serious diseases.
◦We strive to achieve our business goals through agile, inclusive and efficient decision making.
◦We learn and grow from decades of scientific experience to develop therapies at the cutting edge of medicine.
◦We learn from failure, and seek to continuously improve performance, objectives,as part of the journey to achieve breakthroughs.
•Fostering belonging:
◦We develop shared goals that create a sense of belonging.
◦We are a company where diverse individuals can flourish, grow and develop their expertise while bringing their authentic selves to work.
◦We feel connected to our local communities, the environment in which we celebrate exceedinglive and the expectationspatient communities we serve.
◦We come together to understand our scientific learnings and progress the evolution of our colleagues and stakeholders.
◦We embrace diversity, equity and inclusion.
◦We are committed to nurturing diverse and inclusive environments to advance healthcare equity.
Our Management of Human Capital
We recently hired our first Chief People Officer in September 2020 to lead ourOur human resources function focuses on the attraction and to expand our programs to recruit, retainrecruitment of candidates, leadership training and develop our employees.development, diversity and inclusion efforts, total rewards packages consisting of compensation and benefits, and employee engagement and retention. As of December 31, 2020,2023, our global human resources function was comprised of nineeight full time human resources professionals.
Of the 405 full time employees, 180 were primarily engaged in research and development activities, 138 were primarily engaged in technical operations and manufacturing and 87 were primarily engaged in general and administrative activities. We also engage the services of independent contractors and consultants as needed for special or temporary projects or specific expertise.
To manage our human resources, we track and report internally on key talent metrics including headcount by business unit and country, historical headcount growth, turnover, new hires and terminations, open roles and employee demographics including gender, race and ethnicity. Our senior executives use these metrics to assist with resource planning, recruitment and retention initiatives and the design of our compensation and benefits programs. We share these metrics quarterly with the Compensation Committee of our Board of Directors to assist it in fulfilling its duties to (a) establish our enterprise compensation philosophy, (b) administer our compensation and benefit plans, (c) evaluate the performance of our executive officers and key employees and (d) review and monitor management development and succession plans.
AsOur employees participate in employee engagement surveys throughout the year. The results of December 31, 2020, we had 413 full timethis survey continue to help us better understand the culture, work dynamics and overall commitment of our employees located in the United States, France and the United Kingdom. Of these employees, 349 are located in the United States, primarily in the San Francisco Bay Area, 57 are located in Valbonne, France and the remaining 7 are located near London, United Kingdom. Of these employees, 185 were primarily engaged in research and development activities, 140 were primarily engaged in technical operations and manufacturing and 88 were primarily engaged in general and administrative activities. Weto also engage the servicesidentify areas of independent contractors and consultants as needed for special or temporary projects or specific expertise.
Our Commitment to Diversity, Equity &and Inclusion
We strongly believe in a diverse workplace where all Sangamo employees can thrive in an inclusive environment free from discrimination, harassment, bias and prejudice. We aim to treat all individuals with respect and dignity and to provide all Sangamo employees with equal opportunity and fair treatment based on merit. By embracing diversity and inclusion, we strive to create an organization committed to working together to develop innovative solutions in support of the Sangamo mission consistent with our values. At Sangamo, we cultivate a culture and environment where different backgrounds and perspectives are not only respected and heard, but also embraced and celebrated. Not only is a diverse, equitable and inclusive mindset and culture critical to an engaged and committed workplace, but it is also imperative to understanding and meeting the needsin delivering innovative solutions for our patients.
The following depicts Sangamo’s United States employee demographics, based on self-identification, as of December 31, 2023:
In 2020, with the support
We foster Diversity, Equity, and Inclusion, or DEI, working group comprised ofthrough a diverse groupcollaborative approach led by three dedicated groups of employees taskedfrom across the organization: the DEI Committee, DEI Advocates and DEI Champions. These three groups work together to identify key areas of focus for the company and implement strategic and impactful DEI initiatives that reinforce Sangamo’s culture and build community. We continue to partner with designing and implementing specific initiatives to promote greater diversity, equity and inclusion at Sangamo worldwide, including potential amendments to our company values to reflect our commitment to these ideals andLife Science Cares, a non-profit organization with a mission of leveraging the launchresources of a corporate social responsibility scorecard tracking our DEI initiatives. We also have engaged an experienced DEI consultantlife science companies to help survey our employee population and advise us onreduce the design and planningeffects of new DEI initiatives based on this input and external benchmarking. In 2021, we expect to implement these measures, institute new DEI tracking and benchmarking and increase our focus on DEI in recruitment and retention initiatives.
As of December 31, 2020, women accounted for the following approximate percentages of our full-time employees: 51% globally; 48%poverty. We have also participated in the United States; 65% in FranceBloomberg Gender Equality Index to better align our investments and 71% in the United Kingdom. As of December 31, 2020,initiatives with our employee records indicate that approximately 57% of our full-time U.S. employees identify as non-white.
Our Compensation &and Benefits
Given the highly competitive nature of our industry and the importance of recruitment and retention to our success, we strive to furnishprovide our employees with what we believe is a very competitive and comprehensive total rewards package of compensation, benefits and services.development opportunities. This package includes at or above-market pay; healthcare benefits for employees and family members; a health savings account for eligible U.S. employees with above market employer contributions; generous paid time off benefits; family leave; bereavement leave; flexible work schedules; contributions to retirement and/or pension plans; a supplemental long term disability plan; mental health benefits and onsite services.gym access. In addition, we offer a monthly stipend for employees to spend on health and well-being. We also offer every full-time employee globally the benefit of equity ownership in the companySangamo through stock option grants and/or restricted stock units. Our U.S. employees are also eligible to participate in an employee stock purchase plan, which offers the opportunity to purchase our common stock at a discount of at least 15%.
Our Efforts to Address the COVID-19 Pandemic
Employee safetySangamo is headquartered in Richmond, California, with additional facilities in Brisbane, California and wellbeing is of paramount importance to us in any year and was of particular focus in 2020 in light of the evolving COVID-19 pandemic. In response to the pandemic, we have supported our employees and government efforts to curb the COVID-19 pandemic through safety and communication efforts and investments, which include:
•Creating a COVID-19 task force responsible for establishing COVID-19, safety protocols and regularly communicating updates to all employees;
•A strict working from home policyValbonne, France. We are in the U.S., in which all work that can be done from home, must be done from home;
•Decreasing density and increasing physical distancing inprocess of closing our facilities for employees working onsite using scheduling adjustmentsin Brisbane, California and flexibility;
•Mandatory weekly COVID-19 testing for all onsite employeesValbonne, France in our U.S. facilities;
•Robust cleaning protocols across all locations;
•Provision of masks to all onsite employees and strict masking requirements;
•Rigorous procedures to address actual and suspected COVID-19 cases and potential exposure; and
•Prohibition of all domestic and international non-essential travel for all employees.
Additionally, from time to time we have instituted additional programs during the pandemic to support our employees, including monthly subsidies to all employees for home office expenses and upgrades, mental well-being private coaching and therapy services.
Trademarks &and Tradenames
SANGAMO®, Better Therapeutics By Design®, ZFP Therapeutic® and Engineering Genetic Cures® are ourBETTER THERAPEUTICS BY DESIGN is a registered trademarkstrademark in the United States, EXPRESSING LIFE is a registered trademark in the European Union and Sangamo Therapeutics™the United Kingdom, and Pioneering Genetic Cures™SANGAMO is a registered trademark in Australia, Canada, the European Union and the United Kingdom. SANGAMO THERAPEUTICS and the SANGAMO THERAPEUTICS Design are our trademarks.registered trademarks in Australia, Canada, the European Union, Japan and the United Kingdom. They have been allowed, but are not yet registered, in the United States. The trademarks UNIVERSAL GENE RECOGNITION, UNIVERSAL GENETOOLS and ZFP THERAPEUTIC are registered in Canada. All other trademarks or trade names referred to in this Annual Report on Form 10-K are the property of their respective owners.
Available Information
Our website is located at www.sangamo.com. This Annual Report on Form 10-K, our Quarterly Reports on Form 10-Q and our Current Reports on Form 8-K and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act are available free of charge on our website as soon as reasonably practicable after we electronically file this material with, or furnish it to, the Securities and Exchange Commission, or SEC. Information found on, or accessible through, our website is not a part of, and is not incorporated into, this Annual Report on Form 10-K. In addition, the SEC maintains a website at www.sec.gov that contains reports, proxy and information statements and other information regarding issuers that file electronically with the SEC.
ITEM 1A – RISK FACTORS
Our business involves significantmaterial risks, some of which are described below. Before making investment decisions regarding our common stock, you should carefully consider these risks, as well as the other information in this Annual Report on Form 10-K, including our financial statements and the related notes and “Management’s Discussion and Analysis of Financial Condition and Results of Operations.” The occurrence of any of the events or developments described below could have a material adverse effect on our business, results of operations, financial condition prospects and stock price.prospects. In such event, the market price of our common stock could decline, and you could lose all or part of your investment. In addition, there are also additional risks not described below that either are either not presently known to us or that we currently deem immaterial, and these additional risks could also materially impair our business, operations or market price of our common stock.
Risks Relating to Research, Development, Regulatory Approval and Commercialization of Our Product Candidates and Technologies
Our success depends substantially on clinical trial results demonstrating safety and efficacy of our product candidates to the satisfaction of regulatory authorities. We may be unable to obtain positive clinical trial results and regulatory approvals for any of our product candidates.
We are a clinical-stage biotechnology company with no approved products and no product revenues. We have ongoing clinical trials evaluating product candidates that use our platform technologies in gene therapy and cell therapy and we
anticipate initiating additional clinical trials in the future on other product candidates. We are substantially dependent on the results of these clinical trials, and there is no guarantee that final results of clinical trials conducted on our product candidates now or in the future will demonstrate the safety and efficacy of any of our product candidates. In addition, none of our product candidates have obtained regulatory approval. Obtaining positive clinical trial results and regulatory approvals is expensive, lengthy, challenging and unpredictable and may never occur for any of our product candidates. If we fail to obtain positive clinical trial results and regulatory approvals for our product candidates, our anticipated revenues from our product candidates and our prospects for profitability would be adversely affected, which would likely cause the market price of our common stock to significantly decline.
Conducting clinical trials and obtaining regulatory approvals is complex and exposes our business to numerous risks, including potential unexpected costs and delays.
We must conduct extensive clinical trials to demonstrate the safety and efficacy of our product candidates to the satisfaction of regulatory authorities in order to obtain regulatory approvals necessary for commercialization. We have limited experience in conducting later stage clinical trials and may not possess the necessary resources and expertise to complete such trials. Clinical trials are expensive, lengthy and unpredictable. We cannot guarantee that any clinical trials will be conducted as planned or completed on schedule, if at all. A failure of one or more clinical trials can occur at any stage. Events that may prevent successful or timely completion of clinical development and regulatory approval include, among others:
•delays in reaching a consensus with regulatory authorities on clinical trial design;
•delays in reaching agreement on acceptable terms with prospective clinical research organizations, or CROs, and clinical trial sites;
•delays in opening clinical trial sites or obtaining required institutional review board, or IRB, or independent ethics committee approval at each clinical trial site;
•delays in recruiting and enrolling suitable patients to participate in our clinical trials;
•delays in clinical trial activities due to the evolving COVID-19 global pandemic and the diversion of healthcare resources to fight the pandemic, such as the delays that have previously impacted clinical trial timelines for our Fabry and TX200 programs;
•imposition of clinical holds by regulatory authorities as a result of serious adverse events or after an inspection of clinical trial operations or trial sites;
•failure by us, any CROs we engage or any other third parties to adhere to clinical trial requirements;
•failure to perform in accordance with the Good Clinical Practice regulations of the U.S. FDA, or applicable regulatory guidelines in the EU and other countries;
•delays in the testing, validation, manufacturing and delivery of our product candidates to the clinical sites, including delays by third parties with whom we have contracted to perform certain of those functions, or as a result of manufacturing or formulation changes to our product candidates;
•delays in having patients complete participation in a trial or return for post-treatment follow-up;
•clinical trial sites or patients dropping out of a trial;
•selections of clinical endpoints that require prolonged periods of clinical observation or analysis of the resulting data;
•occurrences of serious adverse events or other safety concerns associated with product candidates that are viewed to outweigh their potential benefits, result in approval delays or other regulatory restrictions, or harm our reputation;
•occurrences of serious adverse events or other safety concerns in clinical trials of the same class of agents conducted by other sponsors;
•failures to demonstrate that product candidates are safe and effective for their proposed indication;
•changes in regulatory requirements and guidance that require amending or submitting new clinical protocols;
•unexpected costs and expenses and lack of sufficient funding to develop our product candidates; and
•losses of licenses to critical intellectual properties.
We have not yet reached agreement with regulatory authorities on the complete development pathway for certain product candidates, and such authorities have the ability to change decisions or guidance with respect to approvable endpoints, particularly as the technology continues to develop in these areas. For example, we are aware of another company developing a gene therapy to treat hemophilia A that the FDA recommended complete its Phase 3 study and submit two-year follow-up safety and efficacy data on all study participants notwithstanding the company’s contention that it and the FDA had previously agreed on the extent of data necessary to support a BLA.
Due to the novelty of certain product candidates and their technologies, the endpoints needed to support regulatory approvals will likely be different from those originally anticipated. Any inability to successfully complete preclinical and clinical development of our product candidates, or complete such trials in the time frames anticipated, could result in additional costs to us or impair our ability to generate revenues from product sales or achieve regulatory and commercialization milestones and royalties, or shorten any periods during which we may have exclusivity.
Even if a product candidate successfully obtains approval from the FDA and comparable foreign regulatory authorities, any approval might contain significant limitations related to use restrictions for specified age groups, warnings, precautions or contraindications, or may be subject to burdensome post-approval study or risk management requirements. Also, any regulatory approval of our product candidates, once obtained, may be withdrawn. If we are unable to obtain and maintain regulatory approvals for our product candidates in one or more jurisdictions, or if any approval contains significant limitations, we would not be able to generate anticipated revenues and may struggle to become profitable, which would have an adverse effect on our business operations and financial conditions.
Success in research and preclinical studies or early clinical trial results may not be indicative of results obtained in later trials. Likewise, preliminary, initial or interim data from clinical trials may be materially different from final data.
Results from research and preclinical studies or early clinical trials are not necessarily predictive of future clinical trial results, and interim results of a clinical trial are not necessarily indicative of final results. Our product candidates may fail to show the desired safety and efficacy in clinical trials despite demonstrating positive results in preclinical studies or having successfully advanced through initial clinical trials or preliminary stages of clinical trials. From time to time, we have and may in the future publish or report preliminary, initial or interim data. Preliminary, initial or interim data from our clinical trials and those of our collaborators may not be indicative of the final results of the trial and are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues and/or more patient data become available. In this regard, such data may show initial evidence of clinical benefit, but as patients continue to be followed and more patient data becomes available, there is a risk that any therapeutic effects will not be durable in patients and/or will decrease over time, or cease entirely. Preliminary, initial or interim data also remain subject to audit and verification procedures that may result in the final data being materially different from such preliminary, initial or interim data. As a result, preliminary, initial or interim data should be considered carefully and with caution until the final data are available. For example, there can be no assurance that the steady-state FVIII levels shown in the preliminary follow-up data presented in December 2020 by Pfizer and us from the Phase 1/2 Alta study of giroctocogene fitelparvovec will persist in future follow-up or any other data from the Alta study, or that giroctocogene fitelparvovec will ultimately demonstrate a clinical benefit in the final results of the Alta study or in the Phase 3 AFFINE clinical trial of giroctocogene fitelparvovec.
There is no guarantee that any of our pending clinical trials will be successful. Many of our product candidates currently use our ZFP technology platform, including ZFN and ZPT-TF technologies, which has not yet yielded any approved therapeutic products. Moreover, many of our product candidates are preclinical and have never demonstrated any clinical benefit. In addition, our viral delivery systems continue to evolve and have not been used in any approved products. If our product candidates using our ZFP technology platform and viral delivery systems are not able to demonstrate the safe, effective and durable results we are hoping to see in clinical trials, we may be forced to suspend or terminate development of some or all of our product candidates or seek alternative technologies to develop or deliver product candidates.
In addition, there is a high failure rate for product candidates proceeding through clinical trials. Many companies in the biopharmaceutical industries have suffered significant setbacks in late-stage clinical trials even after achieving promising results in preclinical testing and earlier-stage clinical trials. Data obtained from preclinical and clinical activities are subject to varying interpretations, which may delay, limit or prevent regulatory approval. Any such setbacks could adversely affect our business, financial condition, results of operations and prospects.
Our product candidates are subject to a lengthy and unpredictable regulatory approval process in each jurisdiction where approval is sought.
A regulatory authority such as the FDA or the EMA must approve any human therapeutic product before it can be marketed in the jurisdiction it governs. The process for receiving regulatory approval is lengthy and unpredictable, and a product candidate may not withstand the rigors of testing under the process. Before commencing clinical trials in humans in the United States, we must submit an IND, to the FDA. Certain countries outside of the United States have a similar process that requires the submission of a clinical trial application much like the IND prior to the commencement of human clinical trials. In the EU, for example, a clinical trial authorization, or CTA, must be submitted for each clinical protocol to each country’s national health authority and an independent ethics committee. Only after an IND becomes effective and/or the applicable CTA has been accepted may clinical trials begin. See “Business—Government Regulation” for details regarding the regulatory approval processes applicable to our product candidates. While there is some overlap, the regulatory requirements to conduct clinical trials and seek marketing approval vary by jurisdiction. There is no guarantee that the safety studies and other data generated will be sufficient to permit us to conduct clinical trials in all jurisdictions where planned, or once generated, that such clinical trial data will be sufficient to obtain marketing approval in all jurisdictions in which we intend to seek such approval. If we are not able to obtain the necessary regulatory approvals to conduct our clinical trials and commercialize our product
candidates, or if such approvals are delayed or suspended, our business, prospects and market price of our common stock would be adversely affected.
We may not be able to identify, qualify and enroll sufficient patients for our clinical trials or complete our clinical trials in a timely manner, which could delay or prevent us from proceeding with the development of our product candidates.
Identifying, qualifying and enrolling patients in clinical trials of our product candidates, and completing these clinical trials, is critical to our success. Patient enrollment and trial completion is affected by factors including:
•size of the patient population and process for identifying patients;
•design of the trial protocol;
•eligibility and exclusion criteria;
•perceived risks and benefits of the product candidate under study;
•perceived risks and benefits of genomic approaches to treatment of diseases;
•availability of competing therapies and clinical trials;
•potential delays related to the evolving COVID-19 global pandemic and the diversion of healthcare resources to fight the pandemic;
•severity of the disease under investigation;
•availability of genetic testing for potential patients;
•proximity and availability of clinical trial sites for prospective patients;
•required and desired characteristics of patients;
•ability to obtain and maintain patient consent;
•risk that enrolled patients will drop out before completion of the trial;
•patient referral practices of physicians; and
•ability to monitor patients adequately during and after treatment.
The timing of our clinical trials depends on our ability to recruit patients to participate as well as completion of required follow-up periods. There are also a number of other product candidates in development by our competitors, who compete for the same limited patient populations. If we are not able to enroll the necessary number of patients in a timely manner, we may not be able to complete our clinical trials on our desired timelines or at all. If fewer patients are willing to participate in our clinical trials because of negative publicity from adverse events related to genomic medicines, competitive clinical trials for similar patient populations or for other reasons, the timelines for conducting clinical trials of our product candidates may be delayed. These delays could result in increased costs, limitation or termination of clinical trials, and delays in product development timelines. If we are forced to expand to additional jurisdictions to address these challenges, it could impose additional costs, delays and risks. If we are not successful in conducting our clinical trials as planned, it would have an adverse effect on our business, financial condition, results of operations, prospects and market price of our common stock.
We may encounter difficulties in advancing product candidates from research programs to preclinical and clinical development.
We intend to advance our product candidates from research programs through preclinical development and to submit new INDs, CTAs and equivalent filings in other jurisdictions necessary to conduct human clinical trials evaluating our product candidates. The preparation and submission of applications to conduct clinical trials requires us to conduct rigorous and time-consuming preclinical testing and studies and prepare documentation relating to, among other things, the toxicity, safety, manufacturing, chemistry and clinical protocols of our product candidates. We may experience unforeseen difficulties that could delay or otherwise prevent us from executing this strategy successfully. For example, we may encounter problems in the manufacturing of a product candidates and may fail to demonstrate consistency in the formulation of a product candidate. Our preclinical tests may produce negative or inconclusive results, which may lead us to decide, or which may lead regulators to require us, to conduct additional preclinical testing. If we cannot obtain positive results in preclinical testing, we may decide to abandon a product candidate altogether. In addition, our ability to complete and submit such applications to conduct clinical trials may depend on the support of our collaborators and the timely performance of their obligations under relevant collaboration agreements. If our collaborators are not able to perform such obligations or if they choose to slow down or delay the development of a product candidate, we may not be able to submit the clinical trial applications on a timely basis or at all. Furthermore, the submission of applications to conduct clinical trials involves significant cost and labor, and we may not have sufficient resources and personnel to complete the filing of all intended applications, which may force us to scale back the number of applications or forego potential applications that we believe are promising. Any delay, suspension or reduction of our efforts to pursue our preclinical and clinical development strategy could have an adverse effect on our business and cause the market price of our common stock to decline.
Special regulatory designations, such as RMAT, or orphan drug designations, may not be available for our product candidates or may not lead to a faster development or regulatory review or approval process.
We have received RMAT, designation for our product candidate to treat severe hemophilia A. Additionally, some of our product candidates, including our product candidate to treat Fabry disease, have also been granted Orphan Drug Designation by the FDA, and some have also been designated Orphan Medicinal Products by the EMA. Regulatory authorities in some jurisdictions, including the United States and the EU, may designate drugs for relatively small patient populations as orphan drugs. For additional information regarding these special regulatory designations, see “Business—Government Regulation.”
If we request such designations for our other current or future product candidates, there can be no assurances that the FDA or the EMA will grant any of our product candidates such designations. Additionally, such designations do not guarantee that any regulatory agency will accelerate regulatory review of, or ultimately approve, those product candidates, nor does it limit the ability of any regulatory agency to grant such designations to product candidates of other companies that treat the same indications as our product candidates prior to our product candidates receiving exclusive marketing approval. Such designations can also be revoked. RMAT designation can be revoked if the criteria for eligibility cease to be met as clinical data emerges. Orphan drug exclusivity may be revoked if any regulatory agency determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantity of the product to meet the needs of patients with the rare disease or condition.
Our product candidates may cause undesirable side effects or have other properties that could delay or prevent their regulatory approval, limit the approved indications or commercial potential, or result in significant negative consequences following any potential marketing approval.
During the conduct of clinical trials, patients report changes in their health, including illnesses, injuries and discomforts, to their study doctor. Often, it is not possible to determine whether or not the product candidate being studied caused these conditions, particularly as many of the diseases we are studying have complex comorbidities. If clinical experience indicates that a product candidate has side effects or cause serious or life-threatening side effects, the development of the product candidate may fail or be delayed, or, if the product candidate has received regulatory approval, such approval may be revoked, which would severely harm our business, prospects, operating results and financial condition.
There have been several significant adverse side effects in gene therapy treatments in the past, including reported cases of leukemia and death seen in other trials using other genomic therapies. Gene therapy is still a relatively new approach to disease treatment and additional adverse side effects could develop. There also is the potential risk of significantly delayed adverse events following exposure to gene therapy products due to persistent biologic activity of the genetic material or other components of products used to carry the genetic material. Possible adverse side effects that could occur with treatment with gene therapy products include an immunologic reaction early after administration that, while not necessarily adverse to the patient’s health, could substantially limit the effectiveness of the treatment.
Even if our product development efforts are successful and even if the requisite regulatory approvals are obtained, our products may not gain market acceptance among physicians, patients, healthcare payors and the medical community.
Even if we obtain regulatory approval for any of our product candidates that we may develop or acquire in the future, the approved product may not gain market acceptance among physicians, healthcare payors, patients or the medical community. Market acceptance of approved products depends on a number of factors, including:
•the efficacy and safety of the product as demonstrated in clinical trials;
•the clinical indications and patient populations for which the product is approved;
•acceptance by physicians, treatment centers and patients of the product as a safe and effective treatment;
•the adoption of novel genomic therapies by physicians, hospitals and third-party payors;
•the potential and perceived advantages of the product over alternative treatments;
•the safety of the product seen in a broader patient group, including its use outside the approved indications;
•any restrictions on product use together with other medications;
•the prevalence and severity of any side effects;
•product labeling or product insert requirements of the FDA or other regulatory authorities;
•the timing of market introduction of the product as well as competitive products;
•the development of manufacturing and distribution processes for the product;
•the cost of treatment in relation to alternative treatments;
•the availability of coverage and adequate reimbursement and the willingness of patients to pay out-of-pocket in the absence of coverage or inadequacy of reimbursement by third-party payors and government authorities;
•relative convenience and ease of administration; and
•the effectiveness of our sales and marketing efforts and those of our collaborators.
If any of our product candidates are approved but fail to achieve market acceptance among physicians, patients, healthcare payors or treatment centers, we will not be able to generate significant revenues from the approved product, which would compromise our ability to become profitable.
Even if we are able to commercialize any approved products, such products may not receive coverage and adequate reimbursement from third-party payors in the United States and in other countries in which we seek to commercialize them, which could harm our business.
Our ability to commercialize any product successfully will depend, in part, on the extent to which coverage and adequate reimbursement for these products and related treatments will be available. Government authorities and third-party payors, such as private health insurers and health maintenance organizations, determine which medications they will cover and establish reimbursement levels, which can affect demand for, or the price of, any approved product. Given the nature of the product candidates that we are developing, some patients may require treatment only one-time (e.g., single dose administration), and there is substantial uncertainty about the pricing structure for such products, and the level of coverage and reimbursement that will be available for a shift to single-dose treatment as compared to chronic therapy over a patient’s lifetime. If other companies establish a new pricing structure or business model, including payment based on demonstration of long-term efficacy, our ability to price or obtain reimbursement for our products may be adversely affected. If such pricing structure or business model do not adequately fund the costs of our research and development, manufacturing and commercialization efforts, our business may be adversely affected.
In addition to uncertainty about the potential pricing structure for certain of our product candidates, cost containment is a recurrent trend in the healthcare industry. Government authorities and third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for certain medications. We cannot be sure that coverage and adequate reimbursement will be available for any product that we commercialize and, if reimbursement is available, what the level of reimbursement will be. If reimbursement is not available or is available only at limited levels, we may be unable to successfully commercialize any product candidate for which we obtain regulatory approval. Our inability to promptly obtain coverage and profitable reimbursement rates from both government-funded and private payors for any approved products that we develop could have an adverse effect on our operating results, our ability to raise capital needed to commercialize products and our overall financial condition.
Recently enacted and future legislation, including potentially unfavorable pricing regulations or other healthcare reform initiatives, may increase the difficulty and cost for us to obtain regulatory approval of and commercialize our product candidates and affect the prices we may obtain.
The regulations that govern, among other things, regulatory approvals, coverage, pricing and reimbursement for new drug products vary widely from country to country. In the United States and some foreign jurisdictions, there have been a number of legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay regulatory approval of our product candidates, restrict or regulate post-approval activities and affect our ability to successfully sell any product candidates for which we obtain regulatory approval. Also, there has been heightened governmental scrutiny recently over biopharmaceutical pricing practices in light of the rising cost of prescription drugs and biologics. Such scrutiny has resulted in several recent Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for biopharmaceutical products. At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, have been designed to encourage importation from other countries and bulk purchasing. For a discussion of health reform activity and the current pricing framework, see “Business—Government Regulation—Healthcare Reform” and “—Pricing, Coverage and Reimbursement.”
The continuing efforts of the government, insurance companies, managed care organizations and other payors of healthcare services to contain or reduce costs of healthcare and/or impose price controls may adversely affect:
•the demand for our product candidates, if we obtain regulatory approval;
•our ability to set a price that we believe is fair for our products;
•our ability to generate revenue and achieve or maintain profitability;
•the level of taxes that we are required to pay; and
•the availability of capital.
Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors, which may adversely affect our future profitability.
Even if we obtain regulatory approval for a product candidate, our products will remain subject to regulatory scrutiny.
Even if we obtain regulatory approval in a jurisdiction, the regulatory authority may still impose significant restrictions on the indicated uses or marketing of our product candidates or impose ongoing requirements for potentially costly post-approval studies, post-market surveillance or patient or drug restrictions. For example, the FDA typically advises that patients treated with gene therapy undergo follow-up observations for potential adverse events for a 15-year period. Additionally, the holder of an approved BLA is required to comply with FDA rules and is subject to FDA review and periodic inspections, in addition to other potentially applicable federal and state laws, to ensure compliance with cGMP and adherence to commitments made in the BLA.
If we or a regulatory agency discovers previously unknown problems with a product such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, a regulatory agency may impose restrictions relative to that product or the manufacturing facility, including requiring recall or withdrawal of the product from the market or suspension of manufacturing. Moreover, product labeling, advertising and promotion for any approved product will be subject to regulatory requirements and continuing regulatory review. Failure to comply with such requirements, when and if applicable, could subject us to a number of actions ranging from warning letters to product seizures or significant fines, among other actions. See “Business—Government Regulation—U.S. Review and Approval Processes” for more information.
Any government investigation of alleged violations of laws or regulations could require us to expend significant time and resources in response and could generate negative publicity. The occurrence of any event or penalty described above may inhibit our ability to commercialize our product candidates and generate revenues.
Our employees or contractors may engage in misconduct or other improper activities, including noncompliance with research, development, manufacturing or regulatory standards and requirements, which could cause significant liability for us and harm our reputation.
We are exposed to the risk of fraud or other misconduct by our employees and contractors, including intentional failures to comply with FDA regulations or similar regulations of comparable foreign regulatory authorities, provide accurate information to the FDA or comparable foreign regulatory authorities, comply with manufacturing standards we have established, comply with federal and state healthcare fraud and abuse laws and regulations and similar laws and regulations established and enforced by comparable foreign regulatory authorities, report financial information or data accurately or disclose unauthorized activities to us. Misconduct by our employees and contractors could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. It is not always possible to identify and deter such misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business and results of operations, including the imposition of significant civil, criminal and administrative penalties, damages, fines, disgorgement, personal imprisonment, exclusion from government funded healthcare programs, such as Medicare and Medicaid, additional reporting requirements and oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations of non-compliance with these laws, and the curtailment or restructuring of our operations.
We may use our financial and human resources to pursue a particular research program or product candidate and fail to capitalize on other programs or product candidates that may be more profitable or for which there is a greater likelihood of success.
We have limited resources and may forego or delay pursuit of certain research programs or product candidates that later prove to have greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities or pursue collaborations rather than retain sole responsibility for development. Our current and future research and development programs for product candidates may not yield any commercially viable products. The evaluation of the commercial potential or target market for a particular product candidate is forward-looking and based upon assumptions involving, for example and not limited to, market evolution, advances in disease
standard of care, competition and reimbursement. This reliance on assumptions means that, if our assumptions prove to be inaccurate or incomplete, we may pursue opportunities that end up having a number of competitors that are more advanced than our product candidates, or we may relinquish valuable rights to a product candidate through strategic collaboration, licensing or other royalty arrangements in cases where it would have been more advantageous for us to retain sole development and commercialization rights to such product candidate. We may also allocate internal resources to a product candidate in a therapeutic area in which it would have been more advantageous to enter into a collaboration or that does not prove to have viable commercial opportunities. Any failure to use our financial and human resources efficiently could harm our business and operations.
ZFP technology is novel and has never been used to develop any approved, commercially viable therapeutic products.
Our ZFP technology is a novel technology which to date has not yielded any approved commercially viable therapeutic products, and there can be no guarantee that our product development efforts using ZFP technology will be fruitful. We have invested heavily in development of this technology, and our failure to develop approved, commercially viable products using ZFP technology would significantly limit our business and prospects and would adversely impact the market value of our common stock.
Risks Relating to Manufacturing
We are building manufacturing facilities for clinical trial supplies. We have limited experience manufacturing biopharmaceutical products, and there can be no assurance that we will be able to maintain compliant manufacturing facilities, build additional facilities and manufacture our product candidates as intended.
We expect to use both contract manufacturing organizations, or CMOs, and our own facilities to meet our projected needs for clinical trial supply. In 2020, we completed the construction of an AAV manufacturing facility in our Brisbane, California headquarters building to manufacture Phase 1/2 clinical study supplies for our gene therapy product candidates and we are building additional facilities in our headquarters building and in Valbonne, France to manufacture supplies for our cell therapy product candidates. We expect the cell therapy facilities to be operational by the end of 2021. Building these facilities requires us to transition manufacturing processes and know-how of our product candidates from our CMOs to our own facilities. Transferring manufacturing processes and know-how is complex and involves review and incorporation of both documented and undocumented processes that may have evolved over time. In addition, transferring production to different facilities may require utilization of new or different processes to meet the specific requirements of a given facility. Additional studies may also need to be conducted to support the transfer of certain manufacturing processes and process improvements. We cannot be certain that all relevant know-how and data has been adequately incorporated into the manufacturing process until the completion of studies and evaluations intended to demonstrate the comparability of material previously produced by CMOs with that generated by our facilities. Although some of our employees have experience in the manufacturing of biopharmaceutical products from prior employment at other companies, we, as a company, have no prior experience in biopharmaceutical product manufacturing, and operating these facilities will require us to comply with complex regulations and to continue to hire and retain experienced scientific, quality control, quality assurance and manufacturing personnel. Designing and building manufacturing facilities has been and will continue to be time-consuming and expensive, and we may experience delays or cost overruns. In addition, government approvals are required for us to operate manufacturing facilities and are time-consuming to obtain and maintain. As a manufacturer of biopharmaceutical products, we also will be required to demonstrate and maintain cGMP compliance. These requirements include, among other things, quality control, quality assurance and the maintenance of records and documentation. Furthermore, establishing manufacturing operations will require a reallocation of other resources, particularly the time and attention of our senior management. Even if we are able to establish our own manufacturing capabilities, we could encounter challenges in operating the manufacturing facilities in compliance with cGMP, regulatory or other applicable requirements, resulting in potential negative consequences, including regulatory actions, which could undermine our ability to use these facilities for our own manufacturing needs. Any failure or delay in the development of our manufacturing capabilities could adversely impact the development of our product candidates.
The manufacture, storage and transport of our product candidates is complex, expensive, highly regulated and risky, which could hamper their commercial viability.
There are significant risks associated with manufacturing, storing and transporting our product candidates including, among others, cGMP compliance, cost overruns, technical problems with process scale-up, specialized facilities, process reproducibility, stability issues, lot consistency, yields and timely availability of highly specific raw materials. Even though product batches released for use in clinical trials undergo sample testing, some defects may only be identified following release. In addition, process deviations or unanticipated effects of approved process changes may result in these intermediate products
not complying with stability requirements or specifications. Also, our product candidates must be stored and transported at temperatures within a certain range. If these environmental conditions deviate, our product candidates’ remaining shelf-lives could be impaired or their efficacy and safety could be adversely affected, making them no longer suitable for use. Moreover, product candidates that are biologics involving complex processes, including the development of cell lines or cell systems to produce the biologic, with the challenge of significant variability. There are difficulties in growing large quantities of such cells, consistently and sufficiently isolating certain types of cells and harvesting and purifying the biologic produced by them. The cost to manufacture biologics is generally far higher than traditional small molecule chemical compounds, and the manufacturing process can be difficult to reproduce.
Moreover, manufacturing, storing and transporting our product candidates is subject to strict regulatory standards, which adds additional production risk. Even if efficacy and safety data from our clinical trials would otherwise support regulatory approval of a product candidate, there is no assurance that we or our CMOs will be able to manufacture our product candidates to specifications at levels necessary to support or maintain regulatory approval by the FDA or other regulatory authorities.
Thus, there is no guarantee we will be successful in establishing a larger-scale commercial manufacturing process for our product candidates or obtaining the needed manufacturing capacity. Due to these manufacturing challenges, there is risk that some of our product candidates could be subject to inventory outages, reputational damage and product liability risks, and result in additional expense and delays to clinical trials and commercialization. Supply interruptions or shortages could result in potential negative impacts to our business, prospects and market price of our common stock.
If we use chemical, biological or hazardous materials in a manner that causes injury or violates laws, we may be liable for damages.
Our research and development activities involve the controlled use of potentially harmful biological materials as well as hazardous materials, chemicals, and various radioactive compounds typically employed in the study of molecular and cellular biology. We routinely use cells in culture and gene delivery vectors, and we employ small amounts of radioisotopes in trace experiments. Although we maintain up-to-date licensing and training programs, we cannot completely eliminate the risk of accidental contamination or injury from the use, storage, handling, or disposal of these materials. In the event of contamination or injury, we could be held liable for damages that result, and any liability could exceed our resources. We currently carry insurance covering certain claims arising from our use of these materials. However, if we are unable to maintain our insurance coverage at a reasonable cost and with adequate coverage, our insurance may not cover any liability that may arise. We are subject to federal, state, and local laws and regulations governing the use, storage, handling, and disposal of these materials and specified waste products. Failure to comply with these laws and regulations could result in fines, penalties and additional liabilities and restrictions on our operations.
We currently rely on third parties to conduct some or all aspects of manufacturing of our product candidates for preclinical and clinical development. If one of our third-party manufacturers fails to perform adequately or fulfill our needs, we may be required to incur significant costs and devote significant efforts, to find new suppliers or manufacturers.
We currently have limited experience in clinical-scale manufacturing of our product candidates, and we rely in large part upon third-party CMOs to manufacture and supply drug product for our preclinical studies and clinical trials. Although we are in the process of establishing manufacturing facilities in Brisbane, California and Valbonne, France, these facilities will only manufacture limited quantities of our product candidates for our early-stage clinical trials. We intend to continue to rely on third parties for the manufacture of product candidates for later stage clinical trials, and for commercial-scale manufacturing for any approved product. The manufacture of biopharmaceutical products in compliance with the FDA’s cGMP requires significant expertise and capital investment, including the development of advanced manufacturing techniques and process controls. Manufacturers of biopharmaceutical products often encounter difficulties in production, including difficulties with production costs and yields, quality control, including stability of the product candidate and quality assurance testing, shortages of qualified personnel, as well as compliance with strictly enforced cGMP requirements, other federal and state regulatory requirements and foreign regulations. If our manufacturers were to encounter any of these difficulties or otherwise fail to comply with their obligations to us or under applicable regulations, our ability to conduct later-stage clinical trials could be jeopardized. Any delay or interruption in the supply of clinical trial materials could delay the completion of our clinical trials, increase the costs associated with developing our product candidates and, depending upon the period of delay, require us to commence new clinical trials at significant additional expense or terminate the clinical trials completely.
We and our CMOs must comply with cGMP requirements enforced by the FDA through its facilities inspection program. These requirements include, among other things, quality control, quality assurance and the maintenance of records and documentation. We and our CMOs may be unable to comply with these cGMP requirements and with other FDA, state and
foreign regulatory requirements. The FDA or similar foreign regulatory agencies may also implement new standards at any time or change their interpretation and enforcement of existing standards for manufacture, packaging or testing of products. We have limited control over our manufacturers’ compliance with these regulations and standards. Failure to comply with these requirements may result in fines and civil penalties, suspension of production, suspension or delay in product approval, product seizure or recall or withdrawal of product approval. If the safety of any product supplied is compromised due to our manufacturers’ failure to adhere to applicable laws or for other reasons, we may not be able to obtain regulatory approval for or successfully commercialize our products and we may be held liable for any injuries sustained as a result. Any of these factors could cause a delay of clinical trials, regulatory submissions, approvals or commercialization of our product candidates, entail higher costs or impair our reputation.
Our current agreements with our CMOs do not provide for the entire supply of the drug product necessary for all anticipated clinical trials or for full scale commercialization. If we and our CMOs cannot agree to the terms and conditions for them to provide the drug product necessary for our clinical and commercial supply needs, we may not be able to manufacture the product candidate until a qualified alternative manufacturer is identified, which could also delay the development of, and impair our ability to commercialize our product candidates.
The number of third-party CMOs with the necessary manufacturing and regulatory expertise and facilities is limited, and it could be expensive and take a significant amount of time to arrange for alternative CMOs, which could have an adverse effect on our business. New manufacturers of any product candidate would be required to qualify under applicable regulatory requirements and would need to have sufficient rights under applicable intellectual property laws to the method of manufacturing the product candidate. Obtaining the necessary FDA approvals or other qualifications under applicable regulatory requirements and ensuring non-infringement of third-party intellectual property rights could result in a significant interruption of supply and could require the new manufacturer to bear significant additional costs which may be passed on to us.
We and third parties on which we rely may be adversely affected by natural disasters and catastrophic or other events outside of our control, and our business continuity and disaster recovery plans may not adequately protect us from a serious disaster or event.
Natural disasters could severely disrupt our operations and our facilities, including our current and planned manufacturing facilities in Brisbane, California and Valbonne, France and the manufacturing facilities of our CMOs, and any disruption would likely have a negative impact on our business, financial condition, results of operations and prospects. If a natural disaster, pandemic or epidemic, including the evolving COVID-19 pandemic, political crisis, power outage or any other event that is out of our control occurred that prevented us or third parties on which we rely from using all or a significant portion of our or their facilities, that damaged critical infrastructure or that otherwise disrupted our or their operations, it may be difficult or, in certain cases, impossible for us to continue our business and operations for a substantial period of time. The disaster recovery and business continuity plans we have in place currently are limited and may not prove adequate in the event of a serious disaster or similar event. We may incur substantial expenses as a result of the limited nature of our disaster recovery and business continuity plans, which could have an adverse effect on our business, financial condition, results of operations and prospects. Such disasters or events occurring at facilities of third parties on which we rely could also negatively impact our business and operations.
Risks Relating to our Industry
Our product candidates are based on novel genomic medicine technologies, which makes it difficult to predict the timing and costs of development and of subsequently obtaining regulatory approval.
We have concentrated our research and development efforts on genomic medicine, consisting of gene therapy, gene-edited cell therapy and genome engineering. The regulatory approval process for novel product candidates such as ours is unclear and may be lengthier and more expensive than the process for other, better-known or more extensively studied product candidates.
Adverse developments in clinical trials of genomic medicines conducted by others may cause the FDA or other oversight bodies to change the requirements for approval of our product candidates.
These regulatory review committees and advisory groups, and any new guidelines they promulgate, may lengthen the regulatory review process, require us to perform additional preclinical studies or clinical trials, increase our development costs, lead to changes in regulatory positions and interpretations, delay or prevent approval and commercialization of our current or future product candidates or lead to significant post-approval limitations or restrictions. As we advance our product candidates,
we will be required to consult with these regulatory and advisory groups and comply with applicable guidelines. If we fail to do so, we may be required to delay or discontinue development of our product candidates. These additional processes may result in a review and approval process that is longer than we otherwise would have expected. Delay or failure to obtain, or unexpected costs in obtaining, the regulatory approval necessary to bring a potential product to market could decrease our ability to generate sufficient product revenue, and our business, financial condition, results of operations and prospects would be harmed. Even if our product candidates are approved, we expect that the FDA will require us to submit follow-up data regarding our clinical trial patients for a number of years after any approval. If this follow-up data shows negative long-term safety or efficacy outcomes for these patients, the FDA may revoke its approval or change the label of our products in a manner that could have an adverse impact on our business.
In addition, adverse developments in clinical trials of genomic medicines conducted by others may cause the FDA or other oversight bodies to change the requirements for approval of our product candidates. The FDA and EMA have only very recent and limited experience in the approval of in vivo gene therapy products. As a result, it is difficult to determine how long it will take or how much it will cost to obtain regulatory approvals for our product candidates.
If we or our competitors develop, acquire, or market technologies or products that are more effective than ours, our financial condition and ability to successfully market or commercialize our product candidates or be profitable would be adversely affected.
The biopharmaceutical industry is highly competitive and subject to significant and rapid technological change. We are aware of several companies focused on other methods for editing cells, editing genes and regulating gene expression and a growing number of commercial and academic groups pursuing the development of genome engineering technology. The field of genomic medicine is highly competitive, and we expect competition to persist and intensify in the future from a number of different sources, including biopharmaceutical companies, academic and research institutions, and government agencies that will seek to develop competing products as well as technologies that will compete with our ZFP technology platform. For example, in genome engineering and gene therapy products, competing proprietary technologies with our product development focus include but are not limited to, recombinant proteins, other gene therapy/cDNAs, antisense, siRNA and microRNA approaches, exon skipping, small molecule drugs, monoclonal antibodies, CRISPR/Cas technology and TALE proteins, meganucleases, and MegaTALs. See “Business—Competition” for more information on the competition we may face.
Any products that we or our collaborators or strategic partners develop will enter into highly competitive markets. Even if we are able to generate products that are safe and effective for their intended use, competing technologies may prove to be more effective or less expensive, which, to the extent these competing technologies achieve market acceptance, will limit our revenue opportunities. In some cases, competing technologies have proven to be effective and less expensive. Competing technologies may include other methods of regulating gene expression or modifying genes. ZFNs and ZFP-TFs have broad application in the life sciences industry and compete with a broad array of new technologies and approaches being applied to genetic research by many companies.
In addition to possessing competing technologies, our competitors include biopharmaceutical companies with:
•substantially greater capital resources than ours;
•larger research and development staffs and facilities than ours; and
•greater experience in product development and in obtaining regulatory approvals and patent protection.
These organizations also compete with us to attract qualified personnel, attract parties for acquisitions, joint ventures or other collaborations and license the proprietary technologies of academic and research institutions that are competitive with our technology, which may preclude us from pursuing similar opportunities. Accordingly, our competitors may succeed in obtaining patent protection or commercializing products before us. Even if our product candidate is more effective, it may be disadvantaged if it is not first to market. In addition, any products that we develop may compete with existing products or services that are well established in the marketplace. Further, some of our product candidates in development are designed for use once. Any success in developing single-dose therapeutics could cause us to lose potential recurring revenues from therapeutics that are designed to be taken over a patient’s lifetime.
The evolving COVID-19 pandemic could continue to adversely impact our business and operations and the business and operations of our collaborators, manufacturers and other business partners.
In March 2020, the World Health Organization declared the novel coronavirus, or COVID-19, outbreak a pandemic,
and since such time, actions taken around the world to help mitigate the spread of COVID-19 have included varying restrictions on travel, quarantines in certain areas, and forced closures for certain types of public places and businesses, including in the
three countries where we have most of our day-to-day operations, the United States, France and the United Kingdom. Our business has been directly impacted by pandemic restrictions aimed at reducing the spread of the disease, including multiple California executive orders, several semi-coordinated San Francisco Bay Area orders, several other state and additional local orders across the country and similar orders outside the United States, which, among other things, direct individuals to shelter at their places of residence, direct businesses and governmental agencies to cease non-essential operations at physical locations, prohibit certain non-essential gatherings, and order cessation of non-essential travel.
To comply with these orders, we implemented an operating plan to continue business operations during the ongoing COVID-19 pandemic, including enhanced workplace safety protocols and modified working schedules in our laboratories. In the United States, employees who are able to fulfill their job duties working from home are required to work from home, and have been doing so since March 2020. These protocols and modifications have slowed our productivity and disrupted our business to a moderate degree and are likely to continue doing so through 2021. For example, we have experienced periodic short-term disruptions to our onsite laboratory and manufacturing operations while addressing positive cases of COVID-19 by onsite workers, and our laboratory and manufacturing operations could experience longer term disruptions in the future in the event of a significant outbreak of COVID-19 among our onsite workers. Moreover, from time to time, we have been required to reorganize and prioritize our research resources to mitigate moderate COVID-19 impacts arising from travel restrictions, laboratory density restrictions and laboratory supply constraints. If our research programs encounter longer-term disruptions, it could impact our ability to support our biopharmaceutical partners as contemplated in our collaboration agreements and could result in adjustments to our research timelines, although we do not believe that the short-term disruptions to date have resulted in any such impacts.
Additionally, our Phase 1/2 STAAR clinical study evaluating ST-920, our wholly-owned gene therapy product candidate for the treatment of Fabry disease, has experienced delays in its timeline due to COVID-19 impacts and the diversion of healthcare resources to fight the pandemic. For example, the clinical trial site in the UK for this study has not been able to open due to the significant prevalence of COVID-19 in the UK. Additionally, we have experienced delays in recruiting, enrolling and dosing patients for this study at our US trial sites, due in some part to the understandable hesitation of patients to travel by plane to trial sites not within driving distance and to enter medical facilities during the pandemic and in other part to trial sites prioritizing COVID-19 clinical care over research activities such as the STAAR study. Moreover, we have experienced some short-term delays in sourcing the necessary raw materials to manufacture supplies for the STAAR study due to COVID-19 impacts. We estimate that these challenges have set back our STAAR study timelines three to six months. While we currently still expect to share initial clinical study data by the end of 2021, this timeline could be revised if COVID-19 impacts to our enrollment and dosing of patients and to our sourcing of raw materials for this study intensify because of vaccination delays, new COVID-19 variants or unexpected events.
In addition, our STEADFAST clinical study evaluating TX200, our wholly-owned CAR-Treg cell therapy product candidate for the treatment of kidney transplant rejection, has experienced delays in its commencement timeline due to COVID-19 impacts related to manufacturing and technology transfer challenges with our CMOs. We estimate that these challenges have set back our commencement timeline by approximately three months. While we currently still expect to initiate this clinical study by the end of 2021, this timeline could be revised if COVID-19 impacts result in additional delays.
With respect to our partnered programs, the timelines for the studies and trials managed by our collaborators are also subject to potential delay in the future if these studies and trials experience similar challenges that we have experienced in our STAAR and STEADFAST studies.
While we have been working with our collaborators, clinical trial sites and CMOs to minimize any impact of COVID-19 on clinical trials and research and development operations conducted by us and our collaborators, we do expect that at least some of our programs will nonetheless experience delays and disruptions in the future due to COVID-19 impacts on the operations of us and our business partners. These delays and disruptions have in the past and could in the future relate to clinical site initiation, patient recruitment and enrollment or dosing of patients. Some patients and clinical trial staff may not be able or willing to comply with clinical trial protocols if quarantines impede movement or interrupt healthcare services.
It is uncertain when restrictions will be fully lifted, and if so, when we will be able to resume pre-pandemic work routines. Imposition of government orders, including quarantine and shelter-in-place orders related to COVID-19 or other infectious diseases, is expected to continue to impact personnel at our laboratories and our third-party manufacturing facilities in the United States and other countries, for the foreseeable future, and could impact the availability or cost of materials, which would disrupt our supply chain. Many of our third-party manufacturers which we use for the supply of materials for product candidates or other materials necessary to manufacture product to conduct preclinical tests and clinical trials are located in countries heavily affected by the COVID-19 pandemic, and should they experience disruptions in the future, such as temporary closures or suspension of services, we would likely experience delays in advancing these tests and trials.
While the potential economic impact brought by, and the duration of, the COVID-19 pandemic may be difficult to assess or predict, it could continue to result in significant disruption of global financial markets, impairing our ability to access capital, which could in the future negatively affect our liquidity. In addition, a recession or market correction resulting from the COVID-19 pandemic could materially affect our business and the value of our common stock.
The extent to which the COVID-19 pandemic impacts our business, our clinical development and regulatory efforts will depend on future developments that remain highly uncertain and cannot be predicted with confidence at this time, such as the ultimate duration and severity of the pandemic, travel restrictions, quarantines and social distancing requirements in the United States, France, United Kingdom and other countries, business closures or business disruptions and the effectiveness and timeliness of actions taken in the United States, France, United Kingdom and other countries to contain and treat the disease, including the effectiveness and timing of vaccination programs in the United States and worldwide. Accordingly, we do not yet know the full extent of potential delays or impacts on our business, sales of our products, our clinical and regulatory activities, healthcare systems or the global economy as a whole. However, these effects could have material adverse impacts on our business, financial condition, results of operations and growth prospects.
In addition, to the extent the evolving COVID-19 pandemic continues to adversely affect our business and results of operations, it may also have the effect of heightening many of the other risks and uncertainties described in this “Risk Factors” section.
Negative public opinion and increased regulatory scrutiny of genomic medicines may damage public perception of the safety of our product candidates and adversely affect our ability to conduct our business or obtain regulatory approvals for our product candidates.
Genetically modified products are currently subject to public debate and heightened regulatory scrutiny. Gene therapy remains a novel technology, with only two in vivo gene therapy products approved for a genetic disease to date in the United States and only a few in vivo gene therapy products for genetic diseases approved to date in the EU. Public perception may be influenced by claims that gene therapy is unsafe, and gene therapy may not gain the acceptance of the public or the medical community. For example, reports of serious adverse events in a retroviral gene transfer trial for infants with X-linked severe combined immunodeficiency (X-linked SCID) in France and subsequent FDA actions putting related trials on hold in the United States had a significant negative impact on the public perception and stock price of certain companies involved in gene therapy, whether or not the specific company was involved with retroviral gene transfer, or whether the specific company’s clinical trials were placed on hold in connection with these events. Other adverse events could occur in the field of genomic medicine that could result in increased regulatory scrutiny, potential regulatory delays or negative impact on public perception genomic medicines, which could cause our stock price to decline.
In particular, our success will depend upon physicians who specialize in the treatment of genetic diseases targeted by our product candidates, prescribing treatments that involve the use of our product candidates in lieu of, or in addition to, existing treatments with which they are familiar and for which greater clinical data may be available.
Even if the regulatory approval for genetically modified products developed using our technology is obtained, our success will also depend on public acceptance of the use of genetically modified products including medicines, plants and plant products. Claims that genetically modified products are unsafe for consumption or pose a danger to the environment may influence public attitudes. Our genetically modified products may not gain public acceptance. More restrictive government regulations or negative public opinion would have an adverse effect on our business, financial condition, results of operations and prospects and may delay or impair the development and commercialization of our product candidates or demand for any products we may develop. For example, earlier gene therapy trials led to several well-publicized adverse events, including cases of leukemia and death seen in other trials using other vectors. Serious adverse events in our clinical trials, or other clinical trials involving gene therapy products or our competitors’ products, even if not ultimately attributable to the relevant product candidates, and the resulting publicity, could result in increased government regulation, unfavorable public perception, potential regulatory delays in the testing or approval of our product candidates, stricter labeling requirements for those product candidates that are approved and a decrease in demand for any such product candidates.
Our current and future relationships with healthcare providers, customers and third-party payors subject us to applicable anti-kickback, fraud and abuse, privacy, data security and other healthcare laws and regulations. If we fail to comply with such regulations, we could face substantial penalties and our business, results of operations, financial condition and prospects could be adversely affected.
Healthcare providers, physicians and third-party payors will play a primary role in the recommendation and prescription of any product candidates for which we obtain regulatory approval. Arrangements with healthcare providers, third-party payors and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we would market, sell and distribute our products. As a biotechnology company, even though we will not control referrals of healthcare services or bill directly to Medicare, Medicaid or other third-party payors, federal and state healthcare laws and regulations pertaining to fraud and abuse, transparency, health privacy and security and patients’ rights are and will be applicable to our business. For details regarding the restrictions under applicable federal and state healthcare laws and regulations that may affect our ability to operate see “Business—Government Regulation—Additional Regulation”.
The scope and enforcement of each of these laws is uncertain and subject to rapid change in the current environment of healthcare reform, especially in light of the lack of applicable precedent and regulations. Scrutiny has also increased, which has led to a number of investigations, prosecutions, convictions and settlements in the healthcare industry. Responding to investigations can be time-and resource-consuming and can divert management’s attention from the business. Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations will involve substantial costs. If our operations or if any physicians or other healthcare providers or entities with whom we expect to do business are found to not be in compliance with applicable laws or applicable regulations, we and they could be subjected to significant civil, criminal and administrative enforcement actions, see “Business—Government Regulation—Additional Regulation”.
Further, we are required to comply with privacy and data security laws, such as the GDPR and the CCPA, and after January 1, 2023, the California Privacy Rights Act, which apply to the collection, use, disclosure, transfer, or other processing of personal data. For more information regarding these regulations, see “Business—Government Regulation—Privacy Regulation”. To comply with the GDPR restrictions on the transfer of personal data from Europe, we have relied on Standard Contractual Clauses. However, a July 2020 decision of the EU’s highest court has called into question this practice, and UK authorities may similarly question the viability of the Standard Contractual Clauses as a mechanism for the lawful transfer of personal data outside of that country. Moreover, there is uncertainty regarding how ‘Brexit’ will impact these practices. If we are unable to implement safeguards necessary to ensure that our transfers of personal data from and within Europe are lawful, we will face increased exposure to regulatory actions, substantial fines, and injunctions against processing personal data from Europe, and could be required to increase our data processing capabilities in Europe at significant expense. Restrictions on our ability to transfer personal data from Europe could impact our clinical trial activities in Europe and limit our ability to collaborate with CROs and other third parties subject to European data protection laws. Other countries may adopt restrictions similar to the GDPR, which could further impact our operations.
Any failure or alleged failure (including as a result of deficiencies in our policies, procedures or measures relating to privacy, data security, marketing or communications) by us to comply with laws, regulations, policies, legal or contractual obligations, industry standards or regulatory guidance relating to privacy or data security, may result in governmental investigations and enforcement actions, litigation, fines and penalties or adverse publicity. In addition, new regulation, legislative actions or changes in interpretation of existing laws or regulations regarding privacy and data security (together with applicable industry standards) may increase our costs of doing business. In this regard, we expect that there will continue to be new proposed laws, regulations and industry standards relating to privacy and data security in the United States, the EU and other jurisdictions, and we cannot determine the impact such future laws, regulations and standards will have on our business.
Product liability lawsuits against us could cause us to incur substantial liabilities and to limit commercialization of any products that we may develop.
We face inherent risks of product liability exposure related to the testing of our product candidates in human clinical trials and will face even greater product liability risks if we commercially sell any approved products. Product liability claims may be brought against us by subjects enrolled in our clinical trials, patients, healthcare providers or others using, administering or selling our products. If we cannot successfully defend ourselves against claims that our product candidates or products caused injuries, we could incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
•decreased demand for any product candidates or products that we may develop;
•termination of clinical trial sites or entire trial programs;
•injury to our reputation and significant negative media attention;
•withdrawal of clinical trial participants;
•significant costs to defend the related litigation;
•substantial monetary awards to clinical trial patients;
•loss of revenue;
•diversion of management and scientific resources from our business operations; and
•the inability to commercialize any products that we may develop.
We currently hold product liability insurance coverage at a level that we believe is customary for similarly situated companies and adequate to provide us with insurance coverage for foreseeable risks, but which may not be adequate to cover all liabilities that we may incur. Insurance coverage is increasingly expensive. We may not be able to maintain insurance coverage at a reasonable cost or in an amount adequate to satisfy any liability that may arise. We intend to expand our insurance coverage for products to include the sale of commercial products if we obtain regulatory approval for our product candidates in development, but we may be unable to obtain commercially reasonable product liability insurance for any products that receive regulatory approval. Large judgments have been awarded in class action lawsuits based on drugs that had unanticipated side effects. A successful product liability claim or series of claims brought against us, particularly if judgments exceed our insurance coverage, could decrease our cash and adversely affect our business.
Risks Relating to our Finances
We have incurred significant operating losses since inception and anticipate that we will incur continued losses for the foreseeable future.
We have a history of recurring net losses, including $257.8 million and $192.3 million for the years ended December 31, 2023 and 2022, respectively, and we have otherwise generated operating losses since we began operations in 1995. The
extent of our future losses and the timing of profitability are uncertain, and we expect to incur losses for the foreseeable future. We have been engaged in developing our ZFPZF technology since inception, which has and will continue to require significant research and development expenditures. To date, we have generated our funding from issuance of equity securities, revenues derived from collaboration agreements, other strategic partnerships in non-therapeutic applications of our technology, federal government research grants and grants awarded by research foundations. We expect to continue to incur additional operating losses for the next several years as we continue to develop our product candidates.preclinical core neurology therapeutic programs and capsid engineering platform. If the time required to generate significant product revenues and achieve profitability is longer than we currently anticipate or if we are unable to generate liquidity through equity financing or other sources of funding, we may be forced to further curtail or suspend, or entirely cease, our operations.
We may be unable to raise additional capital on favorable terms, if at all, which would harmThere is substantial doubt about our ability to developcontinue to operate as a going concern. We need substantial additional funding to execute our technologyoperating plan and product candidatesto continue to operate as a going concern. If adequate funds are not available to us on a timely basis, or at all, we will be required to take additional actions to address our liquidity needs, including additional cost reduction measures such as further reducing operating expenses and could delaydelaying, reducing the scope of, discontinuing or terminate somealtering our research and development activities, which would have a material adverse effect on our business and prospects, or we may be required to cease operations entirely, liquidate all or a portion of our programs.assets, and/or seek protection under the U.S. Bankruptcy Code, and you may lose all or part of your investment. Future sales and issuances of equity securities couldwould also result in substantial dilution to our stockholders.
We have incurred significant operating losses and negative operating cash flows since inception and have not achieved profitability. We expect capital outlays andBased on our current operating expenditures to increase over the next several years asplan, we expand our infrastructure and research and product development activities. While we believeestimate that our available cash, cash equivalents and marketable securities as of December 31, 2020, when combined2023, in combination with additional capital raises and expected revenues from collaborations, strategic partners and research grants,potential future cost reductions, will be adequatesufficient to fund our currently planned operations through at leastonly into the next 12 months from the date thethird quarter of 2024. Our financial statements in this Annual Report on Form 10-K are issued, we will needposition raises substantial doubt about our ability to continue to operate as a going concern. Our ability to continue to operate as a going concern is dependent upon our ability to raise substantial additional capital to fund our operations and support our research and development endeavors, including to progress our preclinical and clinical programs as described in this Annual Report on Form 10-K. In this regard, we have been seeking, and continue to actively seek substantial additional capital, including through public or private equity or debt financing, royalty financing or other sources, such as strategic collaborations and other direct investments in our programs. We have been unsuccessful in securing such additional capital to date. If we are unable to secure additional funding in the very near term, we will likely seek protection under the U.S. Bankruptcy Code. We have explored, and continue to explore, whether filing for bankruptcy protection is in the best interest of our Company and our stakeholders. Additional capital may not be available on acceptable terms or at all. In particular, the perception of our ability to continue to operate as a going concern may make it more difficult to obtain financing for the continuation of our operations, particularly in light of currently challenging macroeconomic and market conditions. Further, we may be unable to attract new investments as a result of the speculative nature of our newly reprioritized core neurology preclinical programs. If adequate funds are not available to us on a timely basis, or at all, we will be required to take additional actions to address our liquidity needs, including additional cost reduction measures such as further reducing operating expenses and delaying, reducing the scope of, discontinuing or altering our research and development activities, which would have a material adverse effect on our business and prospects, or we may be required to cease operations entirely, liquidate all or a portion of our assets, and/or or seek protection under the U.S. Bankruptcy Code, and you may lose all or part of your investment.
In this regard, in April 2023, we announced a restructuring of operations and a reduction in force and a significant reduction in our internal manufacturing and potential commercializationallogeneic research footprints in California, or the April Restructuring, and in November 2023, we announced a further restructuring of operations and reduction in force, or the November Restructuring, including a strategic transformation to focus resources on our proprietary neurology-focused epigenetic regulation programs and AAV capsid delivery technology and move all U.S. operations, including our headquarters, to our Richmond, California facility. On March 1, 2024, our board of directors approved the wind-down of our product candidates. We regularly consider fund raising opportunitiesoperations in France and closure of our facility in Valbonne France by the end of 2024, or the France Restructuring. While we expect the April, November and France Restructurings to be complete by the third quarter of 2024, the second quarter of 2024 and the fourth quarter of 2024, respectively, we may also incur other charges or cash expenditures not currently contemplated due to events that may occur as a result of, or associated with, each of the restructurings. In addition, we may not achieve the expected benefits of these cost reduction measures and other cost reduction plans on the anticipated timeline, or at all, or we may use our available capital more quickly than we expect, which could otherwise accelerate our liquidity needs and could force us to further curtail or suspend, or entirely cease, our operations. Moreover, we have historically relied in part on collaboration partners to provide funding for and otherwise advance our preclinical and clinical programs. However, in June 2022, our collaboration agreement with Sanofi terminated, in June 2023 our collaboration agreements with Biogen and Novartis terminated, and our collaboration agreement with Kite expires by its terms in April 2024, and we do not expect such agreement to be extended. While we may identify new collaboration partners who can progress some of the programs that were the subject of these collaborations as well as our Fabry disease gene therapy program and our CAR-Treg cell therapy programs, we have not yet been, and may decide, from time to time,never be, successful in doing so in a timely manner, on acceptable terms or at all, and we may otherwise fail to raise sufficient additional
capital based on various factors, including market conditionsin order to progress these and our plansother programs ourselves, in which case, we will not receive any return on our investments in these programs. In any event, we need substantial additional funding in order to progress the programs that were the subject of operation. these collaborations as well as our Fabry disease and CAR-Treg cell therapy programs, and to advance our core neurology programs and otherwise execute on our current operating plan.
If we raise additional capital through public or private equity offerings, including sales pursuant to our at-the-market offering program with Jefferies LLC, the ownership interest of our existing stockholders will be diluted, and such dilution may be substantial given our current stock price decline, and the terms of any new equity securities may have a preference over, and include rights superior to, our common stock. If we raise additional capital through royalty financings or other collaborations, strategic alliances or licensing arrangements with third parties, we may need to relinquish certain valuable rights to our product candidates, technologies, future revenue streams or research programs or grant licenses on terms that may not be favorable. If we raise additional capital through debt financing, we may be subject to specified financial covenants or covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or pursuing certain transactions, any of which could restrict our ability to commercialize our product candidates or operate as a business.
In addition, as we focus our efforts on proprietary human therapeutics, we will need to seek FDAregulatory approvals of our product candidates from the FDA or other comparable foreign regulatory authorities, a process that could cost in excess of hundreds of millions of dollars per product. We may experience difficulties in accessing the capital markets due to external factors beyond our control, such as volatility in the equity markets for emerging biotechnology companies and general economic and market conditions both in the United States and abroad. For example,In particular, our ability to raise the substantial additional capital we need in order to fund our business may be adversely impacted by global economic conditions and disruptions to and volatility in the credit and financial markets in the United States and worldwide, which could be impacted by the evolving COVID-19 pandemic.such as has been experienced recently. We cannot be certain that we will be able to obtain financing on terms acceptable to us, or at all. Our failure to obtain adequate and timely funding will adversely affect our businessability to continue to operate as a going concern and our ability to develop our technology and products candidates.
ToBased on our current operating plan, our cash, cash equivalents and marketable securities as of December 31, 2023 are expected to allow us to meet our liquidity requirements only into the extentthird quarter of 2024. Although we raisehave been actively seeking, and continue to actively seek, substantial additional capital, by issuingincluding through public or private equity securities,or debt financing, royalty financing or other sources, such as strategic collaborations and other direct investments in our programs, we have been unsuccessful in securing any such additional capital to date. As a result, we have explored, and continue to explore, whether filing for bankruptcy protection is in the best interest of our Company and our stakeholders. In the event we file for relief under the U.S. Bankruptcy Code, our operations, our ability to develop our product candidates and execute on our operating plan, and our ability to continue as a going concern will be subject to the risks and uncertainties associated with bankruptcy proceedings, including, sales pursuantamong others: our ability to execute, confirm and consummate a plan of reorganization; the additional, significant costs of bankruptcy proceedings and related fees; our ability to obtain sufficient financing to allow us to emerge from bankruptcy and execute our business plan thereafter, and our ability to comply with terms and conditions of any such financing; our ability to continue our operations in the ordinary course; our ability to maintain our relationships with our collaborators, counterparties, employees and other third parties; our ability to obtain, maintain or renew contracts that are critical to our at-the-market offering programoperations on reasonably acceptable terms and conditions or at all; our ability to attract, motivate and retain key employees; the ability of third parties to use certain provisions of the U.S. Bankruptcy Code to terminate contracts without first seeking Bankruptcy Court approval; the ability of third parties to seek and obtain court approval to terminate or shorten the exclusivity period for us to propose and confirm a plan of reorganization, to appoint a trustee, or to convert a proceeding under Chapter 11 of the U.S. Bankruptcy Code to a proceeding under Chapter 7 of the U.S. Bankruptcy Code; and the actions and decisions of our stakeholders and other third parties who have interests in our bankruptcy proceedings that may be inconsistent with Jefferies LLC, our stockholdersoperational and strategic plans. Any delays in our bankruptcy proceedings would increase the risks that we may experience substantial dilution. Wenot be able to reorganize our business and emerge from bankruptcy proceedings and may issueincrease our costs associated with the bankruptcy process or result in prolonged operational disruption. In addition, we would need the prior approval of the Bankruptcy Court for transactions outside the ordinary course of business during the course of any bankruptcy proceedings, which may limit our ability to respond timely to certain events or take advantage of certain opportunities. Because of the risks and uncertainties associated with any bankruptcy proceedings, we cannot accurately predict or quantify the ultimate impact of events that could occur during any such proceedings. There can be no guarantees that if we seek protection under the U.S. Bankruptcy Code, we will emerge from any such proceedings as a going concern or that holders of our common stock convertible securitieswill receive any recovery from any bankruptcy proceedings.
In the event we are unable to pursue protection under Chapter 11 of the U.S. Bankruptcy Code, or, other equity securities in one or more transactions at prices and in a manner we determineif pursued, successfully emerge from timesuch proceedings, it may be necessary for us to time. New investors could gain rights superior to our existing stockholders.pursue protection under Chapter 7 of the U.S.
Bankruptcy Code for all or a part of our businesses. In such event, a Chapter 7 trustee would be appointed or elected to liquidate our assets for distribution in accordance with the priorities established by the U.S. Bankruptcy Code. We believe that liquidation under Chapter 7 would result in significantly smaller distributions being made to our stakeholders than those we might obtain under Chapter 11, or no distribution at all, primarily because of the likelihood that the assets would have to be sold or otherwise disposed of in a distressed fashion over a short period of time rather than in a controlled manner and as a going concern. In such event, you may lose part or all of your investment.
We have fully impaired our goodwill and indefinite-lived intangible assets, have recorded significant impairment of our long-lived assets, and may be required to record significant additional charges if our long-lived assets become further impaired in the future.
It is possible that changes in circumstances, many of which are outside of our control, or in the numerous variables associated with the assumptions and estimates used in assessing the appropriate valuation of our long-lived assets, could in the future result in significant additional impairment charges to our long-lived assets, which could adversely affect our results of operations.
Our ability to use net operating losses to offset future taxable income may be subject to limitations.
Although a certain amount of our federal net operating loss carryforwards carry forward indefinitely (but are subject to a percentage limitation), a significant amount of our federal and all of our state net operating loss carryforwards will begin to expire, if not utilized, beginning in 2024 and 2029, respectively. The net operating loss carryforwards subject to expiration could expire unused and be unavailable to offset future income tax liabilities. In addition, under Sections 382 and 383 of the Internal Revenue Code of 1986, as amended, and corresponding provisions of state law, if a corporation undergoes an “ownership change,” which is generally defined as a greater than 50 percentage point change in its equity ownership value over a three-year period, the corporation’s ability to use its pre-change net operating loss carryforwards and other pre-change tax attributes to offset its post-change income or taxes may be limited. We have experienced an ownership change in the past and we may also experience additional ownership changes in the future as a result of subsequent shifts in our stock ownership, some of which may be outside of our control. If an ownership change occurs and our ability to use our net operating loss carryforwards is materially limited, it would harm our future operating results by effectively increasing our future tax obligations. In addition, at the state level, there may be periods during which the use of net operating loss carryforwards is suspended or otherwise limited, which could accelerate or permanently increase state taxes owed. For example, California has imposed limits on the usability of California state net operating losses to offset California taxable income in tax years beginning after 2019 and before 2023. As a result, if we earn net taxable income, we may be unable to use all or a material portion of our net operating loss carryforwards and other tax attributes, which could potentially result in increased future tax liability to us and adversely affect our future cash flows.
Risks Relating to Research, Development, Regulatory Approval and Commercialization of Our Product Candidates and Technologies
We are a biotechnology company with a reprioritized preclinical focus and no approved products or product revenues. Our success depends substantially on preclinical studies supporting advancement of product candidates into the clinic and subsequent clinical trial results demonstrating safety, efficacy and durability of our product candidates to the satisfaction of regulatory authorities. Obtaining positive clinical trial results and regulatory approvals is expensive, lengthy, challenging and unpredictable and may never occur for any product candidates.
We are a biotechnology company with no approved products or product revenues. Following our strategic reprioritization in 2023, we expect to focus substantially all of our efforts on our core preclinical neurology programs. As a result, we may find that we reduce spending and resources on product candidates or indications that later prove to have greater commercial potential than our core preclinical neurology programs. Our spending on current and future research and development programs may not yield any commercially viable products.
Should we be successful in raising additional funds necessary to execute our operating plan and to continue to operate as a going concern, we anticipate initiating clinical trials in the future on our product candidates if our preclinical studies are supportive. We are and will be substantially dependent on the results of our preclinical studies and subsequent clinical trials, and there is no guarantee that final results of clinical trials conducted on our product candidates now or in the future will
demonstrate the safety and efficacy of any of our product candidates. In addition, none of our product candidates has obtained regulatory approval. Obtaining positive clinical trial results and regulatory approvals is expensive, lengthy, challenging and unpredictable and may never occur for any of our product candidates. If securities or industry analysts do not publish research or publish inaccurate or unfavorable research aboutwe fail to obtain positive results from our business,preclinical studies and subsequent clinical trial and regulatory approvals for our stock priceproduct candidates, our anticipated revenues from our product candidates and trading volume could decline.
Theour prospects for profitability would be adversely affected, which would likely cause the market price of our common stock to significantly decline.
Conducting clinical trials and obtaining regulatory approvals is complex and exposes our business to numerous risks, including potential unexpected costs and delays.
We must conduct extensive clinical trials to demonstrate the safety and efficacy of our product candidates to the satisfaction of regulatory authorities in order to obtain regulatory approvals necessary for commercialization. We have limited experience in conducting later stage clinical trials and may not possess the necessary resources and expertise to complete such trials. Clinical trials are expensive, lengthy and unpredictable. We cannot guarantee that any clinical trials will be conducted as planned or completed on schedule, if at all. A failure of one or more clinical trials can occur at any stage. Events that may delay or prevent successful or timely completion of clinical development and regulatory approval include, among others:
•delays in reaching a consensus with regulatory authorities on clinical trial design;
•delays in reaching agreement on acceptable terms with prospective clinical research organizations, or CROs, and clinical trial sites;
•delays in opening clinical trial sites or obtaining required IRB ethics committee or national competent authority approval at each clinical trial site;
•delays or interruptions in recruiting, screening and enrolling suitable patients to participate in our clinical trials and dosing enrolled patients, such as the pause in dosing of additional patients in the Phase 3 AFFINE trial of giroctocogene fitelparvovec implemented by Pfizer in March 2022 and lifted in September 2022;
•the imposition of clinical holds by regulatory authorities on our clinical trials or those of our collaborators, such as the clinical hold imposed by the FDA on the Phase 3 AFFINE trial of giroctocogene fitelparvovec imposed in November 2021 and lifted in March 2022;
•failure by us, any CROs we engage or any other third parties to adhere to clinical trial requirements;
•failure to perform in accordance with the Good Clinical Practice and Good Laboratory Practice regulations of the FDA, or applicable comparable foreign regulations in the EU and other countries;
•delays in the testing, validation, manufacturing and delivery of our product candidates to the clinical sites, including delays by third parties with whom we have contracted to perform certain of those functions, or as a result of manufacturing or formulation changes to our product candidates;
•delays in having patients complete participation in a trial or return for post-treatment follow-up;
•clinical trial sites or patients dropping out of a trial;
•selections of clinical endpoints that require prolonged periods of clinical observation or analysis of the resulting data;
•occurrences of serious adverse events or other safety concerns associated with product candidates that are viewed to outweigh their potential benefits, result in approval delays or other regulatory restrictions, or harm our reputation;
•occurrences of serious adverse events or other safety concerns in clinical trials of the same class of agents conducted by other sponsors;
•failures to demonstrate that product candidates are safe and effective for their proposed indication;
•changes in regulatory requirements and guidance that require amending or submitting new clinical protocols;
•unexpected costs and expenses and lack of sufficient funding to develop our product candidates; and
•losses of licenses to critical intellectual properties.
We have not yet reached agreement with regulatory authorities on the complete development pathway for certain product candidates, and such authorities have the ability to change decisions or guidance with respect to approvable endpoints, particularly as the technology continues to develop in these areas. For example, we are aware of another company developing a gene therapy to treat hemophilia A that the FDA recommended complete its Phase 3 study and submit two-year follow-up safety and efficacy data on all study participants notwithstanding the company’s contention that it and the FDA had previously
agreed on the extent of data necessary to support a BLA. While we and Pfizer anticipate pivotal data readouts for our Phase 3 AFFINE trial to be based on full analyses of all study participants, when the first 50 patients are twelve months past reaching a steady-state of FVIII expression, the FDA or other comparable foreign regulatory authorities could determine that we need to treat more patients in this trial than expected or follow patients for longer than expected to generate the required data, or that we need to make other modifications to the trial, any of which could negatively impact the ability to complete the trial and seek regulatory approvals for giroctocogene fitelparvovec, which could in turn materially and adversely affect its competitive position and commercial viability and therefore our business, prospects and market price of our stock.
Due to the novelty of certain product candidates and their technologies, the endpoints needed to support regulatory approvals will likely be different from those originally anticipated. Any inability to successfully complete preclinical and clinical development of our product candidates, or complete such trials in the timeframes anticipated, could result in additional costs to us or impair our ability to generate revenues from product sales or achieve regulatory and commercialization milestones and royalties, or shorten any periods during which we may have exclusivity.
Even if a product candidate successfully obtains approval from the FDA and comparable foreign regulatory authorities, any approval might contain significant limitations related to use restrictions for specified age groups, warnings, precautions or contraindications, or may be subject to burdensome post-approval study or risk management requirements. Also, any regulatory approval of our product candidates, once obtained, may be withdrawn, varied or suspended. If we are unable to obtain and maintain regulatory approvals for our product candidates in one or more jurisdictions, or if any approval contains significant limitations, we would not be able to generate anticipated revenues and may struggle to become profitable, which would have an adverse effect on our business operations and financial condition.
We are early in our research and development efforts for our core preclinical neurology programs that are the current focus of our research and development efforts. We may encounter difficulties in advancing product candidates from research programs to preclinical and clinical development and may fail to capitalize on product candidates with a greater commercial opportunity or for which there is a greater likelihood of success.
We are early in our research and development efforts for our core preclinical neurology programs that are the current focus of our research and development efforts. We have not yet demonstrated our ability to successfully commence any clinical trials of any product candidates from our core preclinical neurology programs. We intend to advance our core neurology program product candidates from research programs through preclinical development and to submit new INDs, applications for clinical trial approval and equivalent filings in other jurisdictions necessary to conduct human clinical trials evaluating our product candidates. The preparation and submission of applications to conduct clinical trials require us to conduct rigorous and time-consuming preclinical testing and studies and prepare documentation relating to, among other things, the toxicity, safety, manufacturing, chemistry and clinical protocols of our product candidates. We may experience unforeseen difficulties that could delay or otherwise prevent us from executing this strategy successfully. For example, we may encounter problems in the manufacturing of a product candidate and may fail to demonstrate consistency in the formulation of a product candidate. Our preclinical tests may produce negative or inconclusive results, which may lead us to decide, or which may lead regulators to require us, to conduct additional preclinical testing. If we cannot obtain positive results in preclinical testing, we may decide to abandon a product candidate altogether. In addition, our ability to complete and submit such applications to conduct clinical trials may depend on the support of collaborators and the timely performance of their obligations under relevant collaboration agreements. If our collaborators are not able to perform such obligations or if they choose to slow down or delay the development of a product candidate, we may not be able to submit the clinical trial applications on a timely basis or at all. Furthermore, the submission of applications to conduct clinical trials involves significant cost and labor, and we may not have sufficient resources and personnel to complete the filing of all intended applications, which may force us to scale back the number of applications or forego potential applications that we believe are promising. Any delay, suspension or reduction of our efforts to pursue our preclinical and clinical development strategy could have an adverse effect on our business and cause the market price of our common stock to decline.
Furthermore, if our existing product candidates do not receive regulatory approval or are not successfully commercialized, then the success of our business will depend on our ability to continue to expand our product pipeline through discovery, in-licensing or acquisitions. We may be unable to do so. If we do identify potential product candidates for licensing or acquisition, we may be unable to reach acceptable terms with the licensors or sellers. Further, there may be risks and liabilities associated with the product candidates which our due diligence efforts fail to discover, that are not disclosed to us, that we inadequately assess, or that we are unable to manage effectively. Additionally, we may not realize the anticipated benefits of such licenses or acquisitions for a variety of reasons, including the possibility that the product candidates prove not to be safe or effective in clinical trials, that we are unable to successfully integrate the product candidate into our operations, or that the anticipated benefits will not otherwise be realized within the expected timeframe.
Success in research and preclinical studies or early clinical trial results may not be indicative of results obtained in later trials. Likewise, preliminary, initial or interim data from clinical trials may be materially different from final data.
Results from research and preclinical studies or early clinical trials are not necessarily predictive of future clinical trial results, and preliminary, initial and interim results of a clinical trial are not necessarily indicative of final results. Our product candidates may fail to show the desired safety and efficacy in clinical trials despite demonstrating positive results in preclinical studies or having successfully advanced through initial clinical trials or preliminary stages of clinical trials. For example, there can be no assurance that the effects demonstrated by the STAC-BBB capsid variant in our recent preclinical study in NHPs will translate into similar results in any clinical trial of human subjects. Our inability to demonstrate positive results in clinical trials using the STAC-BBB capsid could result in delays and difficulties in furthering development of our capsid platform and the epigenetic regulation therapies that incorporate or depend on the use of the STAC-BBB capsid or other capsids we may discover, or may require us to cease development of such therapies entirely.
From time to time, we have and may in the future publish or report preliminary, initial or interim data. Preliminary, initial or interim data from our clinical trials and those of our collaborators may not be indicative of the final results of the trial and are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues and/or more patient data become available. In this regard, such data may show initial evidence of clinical benefit, but as patients continue to be followed and more patient data becomes available, there is a risk that any therapeutic effects will not be durable in patients and/or will decrease over time, or cease entirely. Preliminary, initial or interim data also remain subject to audit and verification procedures that may result in the final data being materially different from such preliminary, initial or interim data. As a result, preliminary, initial or interim data should be considered carefully and with caution until the final data are available. For example, there can be no assurance that the FVIII levels shown in the updated data announced in December 2023 by Pfizer and us from the Phase 1/2 Alta study of giroctocogene fitelparvovec will persist in future follow-up or any other data from the Alta study or the Phase 3 AFFINE trial. Mean FVIII levels shown in the Alta study, after an initial peak, have tended to fall to lower levels post peak and then stabilize. We cannot anticipate whether and to what extent this trend will continue downward over time. For this reason and potentially other reasons, giroctocogene fitelparvovec may not ultimately demonstrate a durable, safe and effective clinical benefit to the satisfaction of regulatory authorities in the final results of the Alta study and the Phase 3 AFFINE clinical trial, as applicable, and even if satisfactory to regulatory authorities, such benefit may not be sufficient to yield a commercially-viable product.
There is no guarantee that any of our pending clinical trials will be successful. Many of our product candidates currently use our ZF technology platform, including ZFN and ZF-transcriptional regulator-technologies, which has not yet yielded any approved therapeutic products. Moreover, most of our product candidates are still in preclinical development and have never demonstrated any clinical benefit. In addition, our engineered capsids, including STAC-BBB, continue to evolve and have not been used in any approved products. If our product candidates using our ZF technology platform and viral delivery systems are not able to demonstrate safe, effective and durable results, we may be forced to suspend or terminate development of some or all of our product candidates or seek alternative technologies to develop or deliver product candidates.
In addition, there is a high failure rate for product candidates proceeding through clinical trials. Many companies in the biopharmaceutical industry have suffered significant setbacks in late-stage clinical trials even after achieving promising results in preclinical testing and earlier-stage clinical trials. Data obtained from preclinical and clinical activities are subject to varying interpretations, which may delay, limit or prevent regulatory approval. Any such setbacks could adversely affect our business, financial condition, results of operations and prospects.
Our product candidates are subject to a lengthy and unpredictable regulatory approval process in each jurisdiction where approval is sought.
A regulatory authority such as the FDA, the European Commission or comparable foreign regulatory authorities must approve any human therapeutic product before it can be marketed in the jurisdiction it governs. The process for receiving regulatory approval is lengthy and unpredictable, and a product candidate may not withstand the rigors of testing under the process. Before commencing clinical trials in humans in the United States, we must submit an IND to the FDA. Certain countries outside of the United States have a similar process that requires the submission of a clinical trial application much like the IND prior to the commencement of human clinical trials. In the EU, for example, an application for the approval of a clinical trial must be submitted for each clinical trial to each national competent authority and relevant ethics committee of EU Member States in which sponsor wishes to conduct the clinical trial. Only after an IND becomes effective and/or the CTA has been obtained may clinical trials begin. See “Business—Government Regulation” for details regarding the regulatory approval processes applicable to our product candidates. While there is some overlap, the regulatory requirements to conduct clinical trials and seek marketing approval vary by jurisdiction. There is no guarantee that the safety studies and other data generated will be sufficient to permit us to conduct clinical trials in all jurisdictions where planned, or once generated, that such clinical trial data will be sufficient to obtain marketing approval in all jurisdictions in which we intend to seek such approval. If we are not able to obtain the necessary regulatory approvals to conduct our clinical trials and commercialize our product candidates, or
if such approvals are delayed or suspended, our business, prospects and market price of our common stock would be adversely affected.
We may not be able to identify, qualify and enroll sufficient patients for our clinical trials or complete our clinical trials in a timely manner, which could delay or prevent us from proceeding with the development of our product candidates.
Identifying, qualifying and enrolling patients in clinical trials of our product candidates, and completing these clinical trials, is critical to our success. Patient enrollment and trial completion is affected by factors including:
•size of the patient population and process for identifying patients;
•design of the trial protocol;
•eligibility and exclusion criteria;
•perceived risks and benefits of the product candidate under study;
•perceived risks and benefits of genomic approaches to treatment of diseases;
•availability of competing therapies and clinical trials;
•delays or interruptions related to voluntary pauses of our clinical trials or those of our collaborators, such as the prior voluntary pause in March 2022 in enrolling and dosing additional patients in the Phase 3 AFFINE trial of giroctocogene fitelparvovec, which pause was lifted in September 2022, and the activation of trial sites;
•the imposition of clinical holds by regulatory authorities on our clinical trials or those of our collaborators, such as the prior clinical hold imposed by the FDA on the Phase 3 AFFINE trial of giroctocogene fitelparvovec, which hold has since been lifted, and the potential inability of Sangamo and our collaborators to lift clinical holds imposed by regulatory authorities in a timely manner or on acceptable terms, or at all;
•severity of the disease under investigation;
•availability of genetic testing for potential patients;
•proximity and availability of clinical trial sites for prospective patients;
•required and desired characteristics of patients;
•ability to obtain and maintain patient consent;
•risk that enrolled patients will drop out before completion of the trial;
•patient referral practices of physicians; and
•ability to monitor patients adequately during and after treatment.
The timing of our clinical trials depends on our ability to recruit patients to participate as well as completion of required follow-up periods. There are also a number of other product candidates in development by our competitors, who compete for the same limited patient populations. If we are not able to enroll the necessary number of patients in a timely manner, we may not be able to complete our clinical trials on our desired timelines or at all, which could negatively impact the competitive position and commercial viability of our product candidates or delay or reduce the product revenues, milestone payments or royalty payments we expect to earn from our product candidates.
In addition, we and Pfizer previously announced that some of the patients treated in the Phase 3 AFFINE trial of giroctocogene fitelparvovec have experienced FVIII activity greater than 150% following treatment, and that Pfizer had decided to voluntarily pause screening and dosing of additional patients in this trial to implement a proposed protocol amendment intended to provide guidelines for the clinical management of elevated FVIII levels. Subsequent to the voluntary pause, the FDA put this trial on clinical hold, which was subsequently lifted in March 2022. While the voluntary pause initiated by Pfizer was lifted, the trial re-opened, and recruitment, enrollment and dosing was completed, we cannot assure you that the presentation of data from such trial will be published in a timely manner, if at all. Continued delays or additional pauses to the Phase 3 AFFINE trial could negatively impact the projected timelines for conducting and completing the trial and seeking regulatory approvals for giroctocogene fitelparvovec, which could in turn materially and adversely affect giroctocogene fitelparvovec’s competitive position and commercial viability and therefore our business, prospects and market price of our common stock.
In addition, if fewer patients are willing to participate in our clinical trials because of negative publicity from adverse events related to genomic medicines, competitive clinical trials for similar patient populations or for other reasons, the timelines for conducting clinical trials of our product candidates and presenting clinical data may be delayed. These delays could result in increased costs, limitation or termination of clinical trials, and delays in product development timelines. If we are forced to expand to additional jurisdictions to address these challenges, it could impose additional costs, delays and risks. If we are not successful in conducting our clinical trials as planned, it would have an adverse effect on our business, financial condition, results of operations, prospects and market price of our common stock.
Special regulatory designations, such as RMAT, orphan drug designations, fast track designation, or PRIME may not be available for our product candidates or may not lead to a faster development or regulatory review or approval process.
We have received RMAT designation for our product candidates to treat severe hemophilia A and Fabry disease. Additionally, some of our product candidates, including our product candidate to treat Fabry disease, have also been granted Orphan Drug Designation by the FDA and PRIME eligibility by the EMA, and some have also been designated Orphan Medicinal Products by the EMA. Regulatory authorities in some jurisdictions, including the United States and the EU, may designate drugs for relatively small patient populations as orphan drugs. In addition, our product candidate to treat Fabry disease was granted FDA Fast Track Designation in May 2023. For additional information regarding these special regulatory designations, see “Business—Government Regulation.”
If we request such designations for our other current or future product candidates, there can be no assurances that the FDA, the European Commission or comparable foreign regulatory authorities will grant any of our product candidates such designations. Additionally, such designations do not guarantee that any regulatory agency will accelerate regulatory review of, or ultimately approve, those product candidates, nor does it limit the ability of any regulatory agency to grant such designations to product candidates of other companies that treat the same indications as our product candidates prior to our product candidates receiving marketing approval. Such designations can also be revoked. RMAT designation can be revoked if the criteria for eligibility cease to be met as clinical data emerges. Orphan drug exclusivity may be revoked if any regulatory authority determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantity of the product to meet the needs of patients with the rare disease or condition. In addition, the FDA may withdraw Fast Track designation if it believes that the designation is no longer supported by data from our clinical development program. Fast Track designation alone does not guarantee qualification for the FDA’s priority review procedures.
Our product candidates may cause undesirable side effects or have other properties that could delay or prevent their regulatory approval, limit the approved indications or commercial potential, or result in significant negative consequences following any potential marketing approval.
During the conduct of clinical trials, patients report changes in their health, including illnesses, injuries and discomforts, to their study doctor. Often, it is not possible to determine whether or not the product candidate being studied caused these conditions, particularly as many of the diseases we are studying have complex comorbidities. If clinical experience indicates that a product candidate has side effects or causes serious or life-threatening side effects, the development of the product candidate may fail or be delayed, or, if the product candidate has received regulatory approval, such approval may be revoked, which would severely harm our business, prospects, operating results and financial condition.
There have been several significant adverse side effects in gene therapy treatments in the past, including reported cases of leukemia and death seen in other trials using other genomic therapies. Gene therapy is still a relatively new approach to disease treatment and additional adverse side effects could develop. There also is the potential risk of significantly delayed adverse events following exposure to gene therapy products due to persistent biologic activity of the genetic material or other components of products used to carry the genetic material. Possible adverse side effects that could occur with treatment with gene therapy products include an immunologic reaction early after administration that, while not necessarily adverse to the patient’s health, could substantially limit the effectiveness of the treatment. Possible adverse side effects, including SAEs, could develop in the future, which could delay or halt any further development or potential commercialization of the applicable product candidate.
Even if our product development efforts are successful and even if the requisite regulatory approvals are obtained, our products may not gain market acceptance among physicians, patients, healthcare payors and the medical community.
Even if we obtain regulatory approval for any of our product candidates that we may develop or acquire in the future, the approved product may not gain market acceptance among physicians, healthcare payors, patients or the medical community. Market acceptance of approved products depends on a number of factors, including:
•the efficacy and safety of the product as demonstrated in clinical trials;
•the clinical indications and patient populations for which the product is approved;
•acceptance by physicians, treatment centers and patients of the product as a safe and effective treatment;
•the adoption of novel genomic therapies by physicians, hospitals and third-party payors;
•the potential and perceived advantages of the product over alternative treatments;
•the safety of the product seen in a broader patient group, including its use outside the approved indications;
•any restrictions on product use together with other medications;
•the prevalence and severity of any side effects;
•product labeling or product insert requirements of the FDA or other comparable foreign regulatory authorities;
•the timing of market introduction of the product as well as competitive products;
•the development of manufacturing and distribution processes for the product;
•the cost of treatment in relation to alternative treatments;
•the availability of coverage and adequate reimbursement and the willingness of patients to pay out-of-pocket in the absence of coverage or inadequacy of reimbursement by third-party payors and government authorities;
•relative convenience and ease of administration; and
•the effectiveness of our sales and marketing efforts and those of our collaborators.
If any of our product candidates are approved but fail to achieve market acceptance among physicians, patients, healthcare payors or treatment centers, we will not be able to generate significant revenues from the approved product, which would compromise our ability to become profitable.
Even if we are able to commercialize any approved products, such products may not receive coverage and adequate reimbursement from third-party payors in the United States and in other countries in which we seek to commercialize them, which could harm our business.
Our ability to commercialize any product successfully will depend, in part, on the extent to which coverage and adequate reimbursement for these products and related treatments will be available. Government authorities and third-party payors, such as private health insurers and health maintenance organizations, determine which medications they will cover and establish reimbursement levels, which can affect demand for, or the price of, any approved product. Given the nature of the product candidates that we are developing, some patients may require treatment only one-time (e.g., single dose administration), and there is substantial uncertainty about the pricing structure for such products, and the level of coverage and reimbursement that will be available for a shift to single-dose treatment as compared to chronic therapy over a patient’s lifetime. If other companies establish a new pricing structure or business model, including payment based on demonstration of long-term efficacy, our ability to price or obtain reimbursement for our products may be adversely affected. If such pricing structure or business model do not adequately fund the costs of our research and reportsdevelopment, manufacturing and commercialization efforts, our business may be adversely affected.
In addition to uncertainty about the potential pricing structure for certain of our product candidates, cost containment is a recurrent trend in the healthcare industry. Government authorities and third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for certain medications. We cannot be sure that securitiescoverage and adequate reimbursement will be available for any product that we commercialize and, if reimbursement is available, what the level of reimbursement will be. If reimbursement is not available or industry analysts publish aboutis available only at limited levels, we may be unable to successfully commercialize any product candidate for which we obtain regulatory approval. Our inability to promptly obtain coverage and profitable reimbursement rates from both government-funded and private payors for any approved products that we develop could have an adverse effect on our operating results, our ability to raise capital needed to commercialize products and our overall financial condition.
Many EU Member States periodically review their reimbursement procedures for medicinal products, which could have an adverse impact on reimbursement status. We expect that legislators, policymakers and healthcare insurance funds in the EU Member States will continue to propose and implement cost-containing measures, such as lower maximum prices, lower or lack of reimbursement coverage and incentives to use cheaper, usually generic, products as an alternative to branded products, and/or branded products available through parallel import to keep healthcare costs down. These measures could include limitations on the prices we would be able to charge for product candidates that we may successfully develop and for which we may obtain regulatory approval or the level of reimbursement available for these products from governmental authorities or third-party payors. Further, an increasing number of EU and other foreign countries use prices for medicinal products established in other countries as “reference prices” to help determine the price of the product in their own territory. Consequently, a downward trend in prices of medicinal products in some countries could contribute to similar downward trends elsewhere.
Moreover, in order to obtain reimbursement for our products in some European countries, including some EU Member States, we may be required to compile additional data comparing the cost-effectiveness of our products to other available therapies. HTA of medicinal products is becoming an increasingly common part of the pricing and reimbursement procedures in some EU Member States, including those representing the larger markets. The outcome of an HTA will often influence the pricing and reimbursement status granted to these medicinal products by the competent authorities of individual EU Member States. The extent to which pricing and reimbursement decisions are influenced by the HTA of the specific medicinal product currently varies between EU Member States. In December 2021, the EU HTA Regulation was adopted, which will enter into application in 2025 and is intended to harmonize the clinical benefit assessment of HTA across the EU. However, individual EU Member States will continue to be responsible for assessing non-clinical (e.g., economic, social and ethical) aspects of health technologies and making decisions on pricing and reimbursement. If we are unable to maintain favorable pricing and reimbursement status in EU Member States for product candidates that we may successfully develop and for which we may obtain regulatory approval, any anticipated revenue from and growth prospects for those products in the EU could be negatively affected.
Recently enacted and future legislation, including potentially unfavorable pricing regulations or other healthcare reform initiatives, may increase the difficulty and cost for us to obtain regulatory approval of and commercialize our product candidates and affect the prices we may obtain.
The regulations that govern, among other things, regulatory approvals, coverage, pricing and reimbursement for new drug products vary widely from country to country. In the United States and some foreign jurisdictions, there have been a number of legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay regulatory approval of our product candidates, restrict or regulate post-approval activities and affect our ability to successfully sell any product candidates for which we obtain regulatory approval. Also, there has been heightened governmental scrutiny recently over biopharmaceutical pricing practices in light of the rising cost of prescription drugs and biologics. Such scrutiny has resulted in several recent Presidential executive orders, Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for biopharmaceutical products. At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, have been designed to encourage importation from other countries and bulk purchasing. For a discussion of health reform activity and the current pricing framework, see “Business—Government Regulation—Healthcare Reform” and “Business—Government Regulation—Pricing, Coverage and Reimbursement.”
The continuing efforts of the government, insurance companies, managed care organizations and other payors of healthcare services to contain or reduce costs of healthcare and/or impose price controls may adversely affect:
•the demand for our product candidates, if we obtain regulatory approval;
•our ability to set a price that we believe is fair for our products;
•our ability to generate revenue and achieve or maintain profitability;
•the level of taxes that we are required to pay; and
•the availability of capital.
Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors, which may adversely affect our future profitability.
In addition, the policies of the FDA, the competent authorities of the EU Member States, the EMA, the European Commission and other comparable regulatory authorities with respect to clinical trials may change, and additional government regulations may be enacted. For instance, the regulatory landscape related to clinical trials in the EU recently evolved. The CTR, which was adopted in April 2014 and repeals the EU Clinical Trials Directive, became applicable on January 31, 2022. The CTR allows sponsors to make a single submission to both the competent authority and an ethics committee in each EU Member State along with a harmonized assessment procedure, leading to a single decision for each EU Member State. Compliance with the CTR requirements by us and our third-party service providers, such as CROs, may impact our developments plans. It is currently unclear to what extent the U.K. will seek to align its regulations with the EU in the future. A decision by the U.K. not to closely align its regulations with the new approach that will be adopted in the EU may have an effect on the cost of conducting clinical trials in the U.K. as opposed to other countries and/or make it harder to seek a marketing authorization in the EU for our product candidates on the basis of clinical trials conducted in the U.K.
If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies governing clinical trials, our development plans may be impacted.
Even if we obtain regulatory approval for a product candidate, our products will remain subject to regulatory scrutiny.
Even if we obtain regulatory approval in a jurisdiction, the competent regulatory authority may still impose significant restrictions on the indicated uses or marketing of our product candidates or impose ongoing requirements for potentially costly post-approval studies, post-market surveillance or patient or drug restrictions. For example, the FDA typically advises that patients treated with gene therapy undergo follow-up observations for potential adverse events for a 15-year period. Additionally, the holder of an approved BLA is required to comply with FDA rules and is subject to FDA review and periodic inspections, in addition to other potentially applicable federal and state laws, to ensure compliance with cGMP and adherence to commitments made in the BLA.
If we or a regulatory authority discovers previously unknown problems with a product such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, a regulatory agency may impose restrictions relative to that product or the manufacturing facility, including requiring recall or withdrawal of the product from the market or suspension of manufacturing. Moreover, product labeling, advertising and promotion for any approved product will be subject to regulatory requirements and continuing regulatory review. Failure to comply with such requirements, when and if applicable, could subject us to a number of actions ranging from warning letters to product seizures or significant fines, among other actions.
Any government investigation of alleged violations of laws or regulations could require us to expend significant time and resources in response and could generate negative publicity. The occurrence of any event or penalty described above may inhibit our ability to commercialize our product candidates and generate revenues.
Failure to comply with EU and EU Member State laws that apply to the conduct of clinical trials, manufacturing approval, marketing authorization of medicinal products and marketing of such products, both before and after grant of the marketing authorization, or with other applicable regulatory requirements may result in administrative, civil or criminal penalties. These penalties could include delays or refusal to authorize the conduct of clinical trials, or to grant marketing authorization, product withdrawals and recalls, product seizures, suspension, withdrawal or variation of the marketing authorization, total or partial suspension of production, distribution, manufacturing or clinical trials, operating restrictions, injunctions, suspension of licenses, fines and criminal penalties.
See “Business—Government Regulation—Post-approval Requirements” for more information.
Our employees or contractors may engage in misconduct or other improper activities, including noncompliance with research, development, manufacturing or regulatory standards and requirements, which could cause significant liability for us and harm our reputation.
We are exposed to the risk of fraud or other misconduct by our employees and contractors, including intentional failures to comply with FDA regulations or similar regulations of comparable foreign regulatory authorities, provide accurate information to the FDA or comparable foreign regulatory authorities, comply with manufacturing standards we have established, comply with federal and state healthcare fraud and abuse laws and regulations and similar laws and regulations established and enforced by comparable foreign regulatory authorities, report financial information or data accurately or disclose unauthorized activities to us. Misconduct by our employees and contractors could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. It is not always possible to identify and deter such misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business and results of operations, including the imposition of significant civil, criminal and administrative penalties, damages, fines, disgorgement, personal imprisonment, exclusion from government funded healthcare programs, such as Medicare and Medicaid, or comparable foreign programs, additional reporting requirements and oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations of non-compliance with these laws, and the curtailment or restructuring of our operations.
We may use our financial and human resources to pursue a particular research program or product candidate and fail to capitalize on other programs or product candidates that may be more profitable or for which there is a greater likelihood of success.
We have limited resources and may forego or delay pursuit of certain research programs or product candidates that later prove to have greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities or pursue collaborations rather than retain sole responsibility for development. Our current and future research and development programs for core neurology program product candidates may not yield any commercially viable products. The evaluation of the commercial potential or target market for a particular product
candidate is forward-looking and based upon assumptions involving, for example and not limited to, market evolution, advances in disease standard of care, competition and reimbursement. This reliance on assumptions means that, if our assumptions prove to be inaccurate or incomplete, we may pursue opportunities that end up having a number of competitors that are more advanced than our product candidates, or we may relinquish valuable rights to a product candidate through strategic collaboration, licensing or other royalty arrangements in cases where it would have been more advantageous for us to retain sole development and commercialization rights to such product candidate.
We have implemented several strategic decisions to reallocate resources among our various clinical and preclinical development programs, including most recently our restructurings, including decisions to defer new investments in our Fabry disease gene therapy program and our CAR-Treg cell therapy programs unless and until we are able to successfully secure a collaboration partner or external investment in these programs. Although we are actively seeking collaboration partners or a direct external investment, as applicable, to progress our Fabry disease and CAR-Treg cell therapy programs, there can be no assurance that such efforts will be successful in a timely manner, or at all, in which case, we will not receive any return on our investments in these programs. As part of our restructurings and the related strategic reprioritization, we have determined to focus substantially all of our efforts on our core preclinical neurology programs. Investment in preclinical programs is highly speculative, as it entails substantial upfront capital expenditures and significant risk that any potential product candidate will fail to demonstrate adequate effect and/or an acceptable safety profile. In addition, we are developing, and may in the future develop certain product candidates designed to treat neurological diseases using our novel capsid delivery technology. If our capsid development efforts are not successful, we may be required to defer, or cease entirely, development of such product candidates. In the last several years, we have also paused further investment in our BIVV003 SCD program beyond completion of the Phase 1/2 PRECIZN-1 study, as well as our programs previously the subject of collaborations with Biogen and Novartis. While we may potentially identify new collaboration partners who can progress some of the programs that were the subject of these collaborations as well as our Fabry disease and CAR-Treg cell therapy programs, we may not be successful in doing so in a timely manner, on acceptable terms or at all, and we may otherwise fail to raise sufficient additional capital in order to progress these programs ourselves. As a result of these strategic decisions, we could miss valuable opportunities to capitalize on the potential of our discontinued and halted programs. We may also allocate internal resources to a product candidate in a therapeutic area in which it would have been more advantageous to enter into a collaboration or that does not prove to have viable commercial opportunities. Any failure to use our financial and human resources efficiently could harm our business and operations.
ZF technology is novel and has never been used to develop any approved, commercially viable therapeutic products.
Our ZF technology is a novel technology which to date has not yielded any approved commercially viable therapeutic products, and there can be no guarantee that our product development efforts using ZF technology will be fruitful. We have invested heavily in development of this technology, and our failure to develop approved, commercially viable products using ZF technology would significantly limit our business and prospects and would adversely impact the market value of our common stock.
Risks Relating to Manufacturing
We have limited experience manufacturing biopharmaceutical products, and there can be no assurance that we will be able to maintain compliant manufacturing facilities and manufacture our product candidates as intended.
In connection with our restructurings and the anticipated closure of our Brisbane, California and Valbonne, France facilities in 2024, we expect to rely solely on CMOs to manufacture clinical supply. Transferring manufacturing processes and know-how is complex and involves review and incorporation of both documented and undocumented processes that may have evolved over time. In addition, transferring production to different facilities may require utilization of new or different processes and equipment to meet the specific requirements of a given facility. Additional studies may also need to be conducted to support the transfer of certain manufacturing processes and process improvements. We cannot be certain that all relevant know-how and data have been adequately incorporated into the manufacturing process until the completion of studies and evaluations intended to demonstrate the comparability of material previously produced by our facilities with that generated by our CMOs.
The manufacture, storage and transport of our product candidates is complex, expensive, highly regulated and risky, which could hamper their commercial viability.
There are significant risks associated with manufacturing, storing and transporting our product candidates including, among others, cGMP compliance, cost overruns, technical problems with process scale-up, specialized facilities, process reproducibility, stability issues, lot consistency, yields and timely availability of highly specific raw materials. Even though product batches released for use in clinical trials undergo sample testing, some defects may only be identified following release. In addition, process deviations or unanticipated effects of approved process changes may result in these intermediate products not complying with stability requirements or specifications. Also, our product candidates must be stored and transported at
temperatures within a certain range. If these environmental conditions deviate, our product candidates’ remaining shelf-lives could be impaired or their efficacy and safety could be adversely affected, making them no longer suitable for use. Moreover, product candidates that are biologics involve complex processes, including the development of cell lines or cell systems to produce the biologic, with the challenge of significant variability. There are difficulties in growing large quantities of such cells, consistently and sufficiently isolating certain types of cells and harvesting and purifying the biologic produced by them. The cost to manufacture biologics is generally far higher than traditional small molecule chemical compounds, and the manufacturing process can be difficult to reproduce.
Moreover, manufacturing, storing and transporting our product candidates is subject to strict regulatory standards, which adds additional production risk. Even if efficacy and safety data from our clinical trials would otherwise support regulatory approval of a product candidate, there is no assurance that we or our business.CMOs will be able to manufacture our product candidates to specifications at levels necessary to support or maintain regulatory approval by the FDA or other comparable foreign regulatory authorities.
Thus, there is no guarantee we will be successful in establishing a larger-scale commercial manufacturing process for our product candidates or obtaining the needed manufacturing capacity. Due to these manufacturing challenges, there is risk that some of our product candidates could be subject to inventory outages, reputational damage and product liability risks, and result in additional expense and delays to clinical trials and commercialization. Supply interruptions or shortages could result in potential negative impacts to our business, prospects and market price of our common stock.
If we use chemical, biological or hazardous materials in a manner that causes injury or violates laws, we may be liable for damages.
Our research and development activities involve the controlled use of potentially harmful biological materials as well as hazardous materials, chemicals, and various radioactive compounds typically employed in the study of molecular and cellular biology. We routinely use cells in culture and gene delivery vectors, and we employ small amounts of radioisotopes in trace experiments. Although we maintain up-to-date licensing and training programs, we cannot completely eliminate the risk of accidental contamination or injury from the use, storage, handling, or disposal of these materials. In the event securitiesof contamination or industry analysts whoinjury, we could be held liable for damages that result, and any liability could exceed our resources. We currently carry insurance covering certain claims arising from our use of these materials. However, if we are unable to maintain our insurance coverage at a reasonable cost and with adequate coverage, our insurance may not cover any liability that may arise. We are subject to federal, state, and local laws and regulations governing the use, storage, handling, and disposal of these materials and specified waste products. Failure to comply with these laws and regulations could result in fines, penalties and additional liabilities and restrictions on our operations.
We are reliant on third parties for the manufacture of our product candidates for preclinical and clinical development. If one of our third-party manufacturers fails to perform adequately or fulfill our needs, we may be required to incur significant costs and devote significant efforts to find new suppliers or manufacturers.
In connection with our restructurings and the closure of our Brisbane, California facility in 2024, we now rely solely on CMOs to manufacture preclinical and clinical supply. We intend to continue to rely on third parties for the manufacture of product candidates for later stage clinical trials, and for commercial-scale manufacturing for any approved product. The manufacture of biopharmaceutical products in compliance with the FDA’s cGMP, or comparable foreign GMP regulations, requires significant expertise and capital investment, including the development of advanced manufacturing techniques and process controls. Manufacturers of biopharmaceutical products often encounter difficulties in production, including difficulties with production costs and yields, quality control, including stability of the product candidate and quality assurance testing, shortages of qualified personnel, as well as compliance with strictly enforced cGMP requirements, other federal and state regulatory requirements and foreign regulations. If our manufacturers were to encounter any of these difficulties or otherwise fail to comply with their obligations to us downgradeor under applicable regulations, our stockability to conduct clinical trials could be jeopardized. Any delay or publish inaccurateinterruption in the supply of clinical trial materials could delay the completion of our clinical trials, increase the costs associated with developing our product candidates and, depending upon the period of delay, require us to commence new clinical trials at significant additional expense or unfavorable research aboutterminate the clinical trials completely.
We and our CMOs must comply with cGMP requirements enforced by the FDA through its facilities inspection program and comparable foreign regulatory authorities. These requirements include, among other things, quality control, quality assurance and the maintenance of records and documentation. We and our CMOs may be unable to comply with these cGMP requirements and with other FDA, state and comparable foreign regulatory requirements. The FDA or similar foreign regulatory agencies may also implement new standards at any time or change their interpretation and enforcement of existing standards for manufacture, packaging or testing of products. We have limited control over our manufacturers’ compliance with these regulations and standards. Failure to comply with these requirements may result in fines and civil penalties, suspension of production, suspension, variation or delay in product approval, product seizure or recall or withdrawal of product approval. If the safety of any product supplied is compromised due to our manufacturers’ failure to adhere to applicable laws or for other
reasons, we may not be able to obtain regulatory approval for or successfully commercialize our products and we may be held liable for any injuries sustained as a result. Any of these factors could cause a delay of clinical trials, regulatory submissions, approvals or commercialization of our product candidates, entail higher costs or impair our reputation.
Our current agreements with our CMOs do not provide for the entire supply of the drug product necessary for all anticipated clinical trials or for full scale commercialization. If we and our CMOs cannot agree to the terms and conditions for them to provide the drug product necessary for our clinical and commercial supply needs, we may not be able to manufacture the product candidate until a qualified alternative manufacturer is identified, which could also delay the development of, and impair our ability to commercialize our product candidates.
The number of third-party CMOs with the necessary manufacturing and regulatory expertise and facilities is limited, and it could be expensive and take a significant amount of time to arrange for alternative CMOs, which could have an adverse effect on our business. New manufacturers of any product candidate would be required to qualify under applicable regulatory requirements and would need to have sufficient rights under applicable intellectual property laws to the method of manufacturing the product candidate. Obtaining the necessary approvals or other qualifications under applicable regulatory requirements and ensuring non-infringement of third-party intellectual property rights could result in a significant interruption of supply and could require the new manufacturer to bear significant additional costs which may be passed on to us.
We and third parties on which we rely may be adversely affected by natural disasters and catastrophic or other events outside of our control, and our business continuity and disaster recovery plans may not adequately protect us from a serious disaster or event.
Natural disasters could severely disrupt our operations and our facilities and the manufacturing facilities of our CMOs, and any disruption would likely have a negative impact on our business, financial condition, results of operations and prospects. If a natural disaster, pandemic or epidemic, geopolitical crisis, including the ongoing conflict between Russia and Ukraine and conflicts in the Middle East, power outage or any other event that is out of our control occurred that prevented us or third parties on which we rely from using all or a significant portion of our or their facilities, that damaged critical infrastructure or that otherwise disrupted our or their operations, it may be difficult or, in certain cases, impossible for us to continue our business and operations for a substantial period of time. The disaster recovery and business continuity plans we have in place currently are limited and may not prove adequate in the event of a serious disaster or similar event. We may incur substantial expenses as a result of the limited nature of our disaster recovery and business continuity plans, which could have an adverse effect on our business, financial condition, results of operations and prospects. Such disasters or events occurring at facilities of third parties on which we rely could also negatively impact our business and operations.
Risks Relating to our Industry
Our product candidates are based on novel genomic medicine technologies, which makes it difficult to predict the timing and costs of development and of subsequently obtaining regulatory approval.
We have concentrated our research and development efforts on genomic medicine, consisting of gene therapy, gene-edited cell therapy and genome engineering. The regulatory approval process for novel product candidates such as ours is unclear and may be lengthier and more expensive than the process for other, better-known or more extensively studied product candidates.
Regulatory review committees and advisory groups, and any new guidelines they promulgate, may lengthen the regulatory review process, require us to perform additional preclinical studies or clinical trials, increase our development costs, lead to changes in regulatory positions and interpretations, delay or prevent approval and commercialization of our current or future product candidates or lead to significant post-approval limitations or restrictions. As we advance our product candidates, we will be required to consult with these regulatory and advisory groups and comply with applicable guidelines. If we fail to do so, we may be required to delay or discontinue development of our product candidates. These additional processes may result in a review and approval process that is longer than we otherwise would have expected. Delay or failure to obtain, or unexpected costs in obtaining, the regulatory approval necessary to bring a potential product to market could decrease our ability to generate sufficient product revenue, and our business, financial condition, results of operations and prospects would be harmed. Even if our product candidates are approved, we expect that the FDA, or comparable foreign regulatory authorities, will require us to submit follow-up data regarding our clinical trial patients for a number of years after any approval. If this follow-up data shows negative long-term safety or efficacy outcomes for these patients, the FDA, or comparable foreign regulatory authorities, may revoke their approval or change the label of our products in a manner that could have an adverse impact on our business.
In addition, adverse developments in clinical trials of genomic medicines conducted by others may cause the FDA or other comparable foreign regulatory authorities to change the requirements for approval of our product candidates. The FDA and European Commission have only very recent and limited experience in the approval of in vivo gene therapy products. As a result, it is difficult to determine how long it will take or how much it will cost to obtain regulatory approvals for our product candidates.
If we or our competitors develop, acquire, or market technologies or products that are more effective than ours, our financial condition and ability to successfully market or commercialize our product candidates or be profitable would be adversely affected.
The biopharmaceutical industry is highly competitive and subject to significant and rapid technological change. We are aware of several companies focused on other methods for editing cells, editing genes and regulating gene expression and a growing number of commercial and academic groups pursuing the development of genome engineering technology. The field of genomic medicine is highly competitive, and we expect competition to persist and intensify in the future from a number of different sources, including biopharmaceutical companies, academic and research institutions, and government agencies that will seek to develop competing products as well as technologies that will compete with our ZF technology platform. For example, in genome engineering and gene therapy products, competing proprietary technologies with our product development focus include but are not limited to, recombinant proteins, other gene therapy/cDNAs, nuclease and base editing technologies, antisense therapeutics and RNA interference technologies, siRNA, RNAi and microRNA approaches, exon skipping, small molecule drugs, monoclonal antibodies, CRISPR/Cas technology and TALE proteins, meganucleases, and MegaTALs. A growing number of companies are also developing rival cell therapy technologies and product candidates. See “Business—Competition” for more information on the competition we may face.
Any products that we or our collaborators or strategic partners develop will enter into highly competitive markets. Even if we are able to generate products that are safe and effective for their intended use, competing technologies may prove to be more effective or less expensive, which, to the extent these competing technologies achieve market acceptance, will limit our revenue opportunities. In some cases, competing technologies have proven to be effective and less expensive. Competing technologies may include other methods of regulating gene expression or modifying genes. ZFNs and ZF-transcriptional regulators have broad application in the life sciences industry and compete with a broad array of new technologies and approaches being applied to genetic research by many companies.
In addition to possessing competing technologies, our competitors include biopharmaceutical companies with:
•substantially greater capital resources than ours;
•larger research and development staffs and facilities than ours; and
•greater experience in product development and in obtaining regulatory approvals and patent protection.
These organizations also compete with us to attract qualified personnel, attract parties for acquisitions, joint ventures or other collaborations and license the proprietary technologies of academic and research institutions that are competitive with our technology, which may preclude us from pursuing similar opportunities. Accordingly, our competitors may succeed in obtaining patent protection or commercializing products before we do. Even if our product candidate is more effective, it may be disadvantaged if it is not first to market. In addition, any products that we develop may compete with existing products or services that are well established in the marketplace. Further, some of our product candidates in development are designed for use once. Any success in developing one-time use therapeutics could cause us to lose potential recurring revenues from therapeutics that are designed to be taken over a patient’s lifetime.
Negative public opinion and increased regulatory scrutiny of genomic medicines may damage public perception of the safety of our product candidates and adversely affect our ability to conduct our business or obtain regulatory approvals for our product candidates.
Genetically modified products are currently subject to public debate and heightened regulatory scrutiny. Gene therapy remains a novel technology, with only two in vivo gene therapy products approved for a genetic disease to date in the United States and only a few in vivo gene therapy products for genetic diseases approved to date in the EU. Public perception may be influenced by claims that gene therapy is unsafe, and gene therapy may not gain the acceptance of the public or the medical community. For example, reports of serious adverse events in a retroviral gene transfer trial for infants with X-linked severe combined immunodeficiency, or X-linked SCID, in France and subsequent FDA actions putting related trials on hold in the United States had a significant negative impact on the public perception and stock price would likely decline. If oneof certain companies involved in gene therapy, whether or morenot the specific company was involved with retroviral gene transfer, or whether the specific company’s clinical trials were placed on hold in connection with these events. Other adverse events could occur in the field of these analysts cease coverage of our companygenomic medicine that could result in increased regulatory scrutiny, potential regulatory delays or fail to publish reportsnegative impact on us regularly, demand for our stockpublic perception genomic medicines, which could decrease, which might cause our stock price and trading volume to decline.
In particular, our success will depend upon physicians who specialize in the treatment of genetic diseases targeted by our product candidates, prescribing treatments that involve the use of our product candidates in lieu of, or in addition to, existing treatments with which they are familiar and for which greater clinical data may be available.
Even if the regulatory approval for genetically modified products developed using our technology is obtained, our success will also depend on public acceptance of the use of genetically modified products including medicines, plants and plant
products. Claims that genetically modified products are unsafe for consumption or pose a danger to the environment may influence public attitudes. Our genetically modified products may not gain public acceptance. More restrictive government regulations or negative public opinion would have an adverse effect on our business, financial condition, results of operations and prospects and may delay or impair the development and commercialization of our product candidates or demand for any products we may develop. For example, earlier gene therapy trials led to several well-publicized adverse events, including cases of leukemia and death seen in other trials using other vectors. Serious adverse events in our clinical trials, or other clinical trials involving gene therapy products or our competitors’ products, even if not ultimately attributable to the relevant product candidates, and the resulting publicity, could result in increased government regulation, unfavorable public perception, potential regulatory delays in the testing or approval of our product candidates, stricter labeling requirements for those product candidates that are approved and a decrease in demand for any such product candidates.
Our current and future relationships with healthcare providers, customers and third-party payors subject us to applicable anti-kickback, fraud and abuse, privacy, data security and other healthcare laws and regulations. If we fail to comply with such regulations, we could face regulatory investigations or actions, litigation (including class claims) and mass arbitration demands, and substantial fines and penalties, and our business, reputation, results of operations, financial condition and prospects could be adversely affected.
Healthcare providers, including physicians, and third-party payors will play a primary role in the recommendation and prescription of any product candidates for which we obtain regulatory approval. Arrangements with healthcare providers, third-party payors and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we would market, sell and distribute our products. As a biotechnology company, even though we will not control referrals of healthcare services or bill directly to Medicare, Medicaid or other third-party payors, federal and state healthcare laws and regulations pertaining to fraud and abuse, transparency, health privacy and security and patients’ rights and comparable foreign legislation are and will be applicable to our business. Outside the United States, interactions between pharmaceutical companies and health care professionals are also governed by strict laws, such as national anti-bribery laws of EU Member States, national sunshine rules, regulations, industry self-regulation codes of conduct and physicians’ codes of professional conduct. If we fail to comply with these, or to comply with these adequately or appropriately, we could be subject to significant penalties.
For details regarding the restrictions under applicable federal and state healthcare laws and regulations that may affect our ability to operate see “Business—Government Regulation—Additional Regulation.”
The scope and enforcement of each of these laws is uncertain and subject to rapid change in the current environment of healthcare reform, especially in light of the lack of applicable precedent and regulations. Scrutiny has also increased, which has led to a number of investigations, prosecutions, convictions and settlements in the healthcare industry. Responding to investigations can be time-and resource-consuming and can divert management’s attention from the business. Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations will involve substantial costs. If our operations or if any physicians or other healthcare providers or entities with whom we expect to do business are found to not be in compliance with applicable laws or applicable regulations, we and they could be subjected to significant civil, criminal and administrative enforcement actions, see “Business—Government Regulation—Additional Regulation.”
Further, we are required to comply with domestic and international privacy and data security laws, such as the EU GDPR and the CCPA, which apply to the collection, use, disclosure, transfer, or other processing of personal data, including data we collect about trial participants in connection with clinical trials. In the past few years, numerous U.S. states – including California, Virginia, Colorado, Connecticut and Utah – have enacted comprehensive privacy and data security laws that impose certain obligations on covered businesses, including providing specific disclosures in privacy notices and affording residents with certain rights concerning their personal data. As applicable, such rights may include the right to access, correct or delete certain personal data, and to opt-out of certain data processing activities, such as targeted advertising, profiling and automated decision-making. The exercise of these rights may impact our business and ability to provide our products and services. Certain states also impose stricter requirements for processing certain personal data, including sensitive information, such as conducting data privacy impact assessments. These state laws allow for statutory fines for noncompliance.
Certain jurisdictions have enacted data localization and cross-border data transfer laws, which could make it more difficult to transfer information across jurisdictions. In particular, the EEA and the U.K. have significantly restricted the transfer of personal data to the United States and other countries whose privacy and data security laws they believe to be inadequate. Although there are currently various mechanisms that may be used to transfer personal data from the EEA and United Kingdom to the United States in compliance with law, such as the EEA standard contractual clauses, the U.K.’s International Data Transfer Agreement / Addendum and the EU-U.S. Data Privacy Framework and the U.K. extension thereto (which allows for transfers to relevant U.S.-based organizations who self-certify compliance and participate in the Framework), these mechanisms are subject to legal challenges, and there is no assurance that we can satisfy or rely on these measures to lawfully transfer personal data to the United States. If we are unable to implement a legal mechanism to ensure that our transfers of personal data
from the EEA or the U.K. are lawful, we could face adverse consequences, including increased exposure to regulatory actions, substantial fines and injunctions against processing or transferring personal data, and could be required to increase our data processing capabilities in the EEA, the U.K. or elsewhere at significant expense. Restrictions on our ability to transfer personal data from the EEA, the U.K. or elsewhere could impact our clinical trial activities in the EEA or the U.K. and limit our ability to collaborate with CROs and other third parties. For more information regarding these regulations, see “Business—Government Regulation—Privacy Regulation.”
We are also bound by contractual obligations related to data privacy and security, and our efforts to comply with such obligations may not be successful. For example, certain privacy laws, such as the GDPR and the CCPA, require our customers to impose specific contractual restrictions on their service providers. We publish privacy policies, marketing materials and other statements, such as compliance with certain certifications or self-regulatory principles, regarding data privacy and security. If these policies, materials or statements are found to be deficient, lacking in transparency, deceptive, unfair or misrepresentative of our practices, we may be subject to investigation, enforcement actions by regulators or other adverse consequences. In addition, privacy advocates and industry groups have proposed, and may propose, standards with which we are legally or contractually bound to comply, or may become subject to in the future.
Our obligations related to privacy and data security (and consumers’ expectations regarding them) are quickly changing and becoming increasingly stringent, creating uncertainty. These obligations may be subject to differing applications and interpretations, which may be inconsistent or in conflict among jurisdictions. Preparing for and complying with these obligations requires us to devote significant resources. These obligations may also necessitate changes to our information technologies, systems and practices and those of third parties upon which we rely. Moreover, despite our efforts, our personnel or third parties upon which we rely may fail to comply with such obligations, which could negatively impact our business operations and compliance posture.
Any failure or alleged failure (including as a result of deficiencies in our policies, procedures or measures relating to privacy, data security, marketing or communications) by us or our third-party partners to comply with laws, regulations, policies, legal or contractual obligations, industry standards or regulatory guidance relating to privacy or data security, may result in significant consequences. These consequences may include, but are not limited to, governmental enforcement actions (e.g., investigations, fines, penalties, audits, inspections, and similar), litigation (including class-related claims) and mass arbitration demands, additional reporting requirements and/or oversight, bans on processing personal data, orders to destroy or not use personal data, civil and criminal liability and imprisonment of company officials. In particular, plaintiffs have become increasingly more active in bringing privacy-related claims against companies, including class claims and mass arbitration demands. Some of these claims allow for the recovery of statutory damages on a per violation basis, and, if viable, carry the potential for monumental statutory damages, depending on the volume of data and the number of violations. Any of these events could have a material adverse effect on our reputation, business or financial condition, including but not limited to interruptions or stoppages in business operations (including clinical trials), inability to process personal data or to operate in certain jurisdictions, limited ability to develop or commercialize our products, expenditure of time and resources to defend any claim or inquiry or revision or restructuring of our operations.
Product liability lawsuits against us could cause us to incur substantial liabilities and to limit commercialization of any products that we may develop.
We face inherent risks of product liability exposure related to the testing of our product candidates in human clinical trials and will face even greater product liability risks if we commercially sell any approved products. Product liability claims may be brought against us by patients enrolled in our clinical trials, patients, healthcare providers or others using, administering or selling our products. If we cannot successfully defend ourselves against claims that our product candidates or products caused injuries, we could incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
•termination of clinical trial sites or entire trial programs;
•injury to our reputation and significant negative media attention;
•withdrawal of clinical trial participants;
•significant costs to defend the related litigation;
•substantial monetary awards to clinical trial patients;
•loss of revenue;
•diversion of management and scientific resources from our business operations; and
•the inability to commercialize any products that we may develop.
We currently hold product liability insurance coverage at a level that we believe is customary for similarly situated companies and adequate to provide us with insurance coverage for foreseeable risks, but which may not be adequate to cover all liabilities that we may incur. Insurance coverage is increasingly expensive. We may not be able to maintain insurance coverage at a reasonable cost or in an amount adequate to satisfy any liability that may arise. We intend to expand our insurance coverage for products to include the sale of commercial products if we obtain regulatory approval for our product candidates in development, but we may be unable to obtain commercially reasonable product liability insurance for any products that receive regulatory approval. Large judgments have been awarded in class action lawsuits based on drugs that had unanticipated side effects. A successful product liability claim or series of claims brought against us, particularly if judgments exceed our insurance coverage, could decrease our cash and adversely affect our business.
Unfavorable global economic conditions could have a negative impact on our operations, which could materially and adversely affect our ability to continue to operate as a going concern and otherwise have a material adverse effect on our business, financial condition, results of operations, prospects and market price of our common stock.
Financial instability and a general decline in economic conditions in the United States and other countries caused by political instability and conflict, including the ongoing conflict between Russia and Ukraine and conflicts in the Middle East, and economic or financial challenges caused by recent and potential future bank failures or by general health crises, have led to market disruptions, including significant volatility in commodity prices, credit and capital markets instability, including disruptions in access to bank deposits and lending commitments, supply chain interruptions, rising interest rates and global inflationary pressures. These macroeconomic factors could materially and adversely affect our ability to continue to operate as a going concern and could otherwise have a material adverse effect on our business, operations, operating results and financial condition as well as the price of our common stock. For example, the recent closures of Silicon Valley Bank, or SVB, Signature Bank and First Republic Bank have resulted in broader financial institution liquidity risk and concerns. Although we were able to access all of the funds we had in deposit with SVB, future adverse developments with respect to specific financial institutions or the broader financial services industry may lead to market-wide liquidity shortages. The failure of any bank in which we deposit our funds could reduce the amount of cash we have available for our operations or delay our ability to access such funds. Any such failure may increase the possibility of a sustained deterioration of financial market liquidity, or illiquidity at clearing, cash management and/or custodial financial institutions. In the event we have a commercial relationship with a bank that has failed or is otherwise distressed, we may experience delays or other issues in meeting our financial obligations. If other banks and financial institutions fail or become insolvent in the future in response to financial conditions affecting the banking system and financial markets, our ability to access our cash and cash equivalents and investments may be threatened and our ability to raise additional capital when needed could be substantially impaired, which could have a material adverse effect on our business, operations, operating results and financial condition as well as the price of our common stock. In particular, failure to secure any necessary financing in a timely manner and on favorable terms could require us to delay or abandon clinical development plans or we may be forced to further curtail or suspend, or entirely cease, our operations. In addition, any or all of these factors could disrupt our and our collaborators’ supply chains and adversely affect our and our collaborators’ ability to conduct ongoing and future clinical trials of our product candidates.
Risks Relating to our Reliance on Third Parties
If conflicts arise with our contractors, collaborators or other business partners, these conflicts may limit our ability to implement our strategies and may harm our business and prospects.
If conflicts arise with our contractors, collaborators or other business partners, the other party will likely act in its self-interest, which may limit our ability to implement our strategies. For example, some of our collaborators are conducting multiple product development efforts within each area that is the subject of their collaboration with us. Our collaborators may develop, either alone or with others, product candidates in related fields that are competitive with the product candidates that are the subject of their collaborations with us. Competing products, either developed by the collaborators or to which the collaborators or have rights, may result in the withdrawal of their support for our product candidates.
Some of our collaborators could also become our competitors in the future. Our collaborators could develop or invest in competing products, preclude us from entering into collaborations with their competitors, fail to obtain timely regulatory approvals, terminate or breach their agreements with us unexpectedly or prematurely, or fail to devote sufficient resources to the development and commercialization of product candidates covered by the collaboration.
In addition, conflicts could arise between us and our collaborators resulting from disputes regarding our or our collaborators’ or strategic partners’ performance under the applicable agreement, including disputes arising from alleged breaches of our agreements with our collaborators.
Any of these conflicts could harm our product development efforts and otherwise adversely affect our business and prospects.
Our collaborators control certain aspects of our product development efforts, including certain of oura clinical trials,trial, which could result in unanticipated delays and other obstacles in the commercialization of our product candidates.
We depend on collaborators to design and conduct certain of our clinical trials for some of our product candidates. As a result, these clinical trials may not be conducted in the manner or on the timeline we desire, which may negatively impact our
product development efforts. In addition, if any of these collaborators withdraws support for our product candidates or otherwise impairs their development, our business could be negatively affected.
Our lack of control over aspects of product development in our agreements with Novartis, Biogen, Kite, Sanofi, Takeda and Pfizercollaborations could cause delays or other difficulties in the development and commercialization of our product candidates, which may prevent us from completing the intended IND filings in a timely fashion and receiving any milestone, royalty payments and other benefits under the agreement. For example, Pfizer is the trial sponsor of the Phase 3 AFFINE trial of giroctocogene fitelparvovec and we depended on the efforts of Pfizer to diligently seek to lift the clinical hold on the Phase 3 AFFINE trial and resume the trial. Although dosing in the AFFINE trial is now complete, we cannot guarantee that we will not experience future delays in this trial or that the trial will be completed on the anticipated timeframe or at all.
In addition, under their respective agreements, our third-party collaborators have certain rights to terminate the agreements by providing us with advance notices, therefore, the actual milestone payments that we may receive under these agreements may be substantially lower than the full amounts provided for under these agreements.
Our collaborators licensing our ZFPZF technologies or AAV capsid technologies may decide to adopt alternative technologies or products or may be unable or unwilling to develop commercially viable products with our ZFPZF technologies or AAV capsid technologies, which would negatively impact our revenues and our strategy to develop product candidates using ZFPZF technologies or AAV capsid technologies.
SeveralSome of our ongoing collaborations leverage our ZFPZF technology and AAV capsid technology platform. These collaborators may elect to adopt alternative technologies in the future, which could decrease the value of either or both of our ZFPZF technology platform and AAV capsid technology platform and impede the development of product candidates using the platform.these platforms. Additionally, because many of our collaborators are likely to be working on more than one development project, they could choose to shift their resources to projects other than those they are working on with us. If they do so, this would delay our ability to test and develop our ZFPZF technology platform and AAV capsid technology platform and would delay or terminate the development of our product candidates using thesuch platform. Further, our collaborators may elect not to develop product candidates arising out of our collaborations or not to devote sufficient resources to the development, manufacturing, marketing or sale of these product candidates. If they terminate the collaborations with us or allow them to expire, such as the terminations for convenience of our collaboration agreements with Biogen and Novartis and the anticipated expiration of our collaboration agreement with Kite by its terms in April 2024, and we wish to continue developing the product candidates, we will be required to seek the support of other collaborators or develop the products ourselves. We mayParticularly as a result of the November Restructuring, we do not be ableexpect to identify a suitable partner or negotiate a favorable collaboration agreement, and we may not have sufficient resources and expertise internally to allow us to continue the development of these product candidates and we may not be able to identify a suitable partner or negotiate a favorable collaboration agreement to allow us to continue the development of these product candidates.
Commercialization of our technologies will depend, in part, on collaborations with other companies. If we are not able to find collaborators in the future or if our collaborators do not diligently pursue product development efforts, we may not be able to develop our technologies or product candidates, which could slow our growth and decrease the market value of our common stock.
We do not have financial resources ourselves to fully develop, obtain regulatory approval for and commercialize our product candidates. We have relied, and expect to continue to rely, significantly on our collaborations with other biopharmaceutical companies to provide funding for our research and development efforts, including preclinical studies and clinical tests, and expect to rely significantly on such collaborations to provide funding for the lengthy regulatory approval processes required to commercialize our product candidates.
For example, we have collaborationsWe were party to collaboration agreements with Novartis to develop product candidates to treat certain neurodevelopment disorders, including autism and intellectual disability;disability and with Biogen to develop product candidates to treat tauopathies including Alzheimer’s disease, alpha-synuclein related diseases including Parkinson’s disease and other neurological diseases;diseases. In June 2023, our collaboration agreements with Novartis and Biogen terminated. We are also party to a collaboration agreement with Kite to develop product candidatesengineered cell therapies for cancer, which we expect to treat cancer; with Pfizerexpire by its terms and without an extension in April 2024. As a result of these terminations and expirations, we are no longer entitled to any milestone payments or royalties from Novartis, Biogen or Kite, and such counterparties have no further obligations to develop product candidatesor to treat hemophilia A and amyotrophic lateral sclerosis and frontotemporal lobar degeneration linked to mutationsreimburse the costs of any of the C9ORF72 gene; andprograms under the applicable agreement. In connection with Sanofithe Restructurings, we made the strategic decision to develop product candidatespause further development of the programs that were the subject of these collaborations. In the future, we may identify alternative options to treat beta thalassemia and sickle cell disease.advance some of the programs that were subject to such agreements, including potential development internally or with a collaboration partner. However, we cannot guarantee that we will be able to successfully secure any such options, including identifying an alternative suitable collaboration partner or negotiate a favorable alternative
If we are unable to secure additional collaborations or if our collaborators are unable or unwilling to diligently advance the development, regulatory approval and commercialization of our product candidates, our growth may slow and adversely affect our ability to generate funding for development of our technologies and product candidates.candidates as well as our ability to continue to operate as a going concern, and we may be required to cease operations. For example, although we have decided to defer new investments in our Fabry disease gene therapy program and our CAR-Treg cell therapy programs unless and until we are able to successfully secure a collaboration partner or external investment in these programs, there can be no assurance that such efforts will be successful in a timely manner, or at all, in which case, we will not receive any return on our investments in these programs and our ability to continue to operate as a going concern may be materially and adversely affected. In addition, our ongoing collaborators may sublicense or abandon development programs with little advance notice, or we may have disagreements or disputes with our collaborators, which would cause associated product development to slow or cease. In addition, the business or operations of our collaborators may change significantly through restructurings, acquisitions, other strategic transactions that may negatively impact their ability to advance our programs. The evolving COVID-19 pandemic could similarly impact our ability to realize the expected benefits of our collaborations due to the impacts of the pandemic on our collaborators and their business and operations.
Under typical collaborations, we expect to receive revenue for the research and development of our product candidates based on achievement of specific milestones, as well as royalties based on a percentage of sales of any commercialized products. Achieving these milestones will depend, in part, on the efforts of our collaborators, which we have no control over, as well as our own efforts. IfIn addition, business combinations, changes in a collaborator’s business strategy and financial difficulties or other factors could result in that collaborator abandoning or delaying development of any product candidates covered by our collaboration agreement with that collaborator. For example, the transition back to us of the rights and obligations of Sanofi related to BIVV003 and the related termination for convenience by Sanofi of our prior collaboration agreement followed a change in Sanofi’s strategic direction to focus on allogeneic universal genomic medicine approaches rather than autologous personalized cell therapies. In addition, Novartis’s and Biogen’s decisions to terminate their respective collaboration agreements with us each related to a recent strategic review. Further, if we fail or any collaboration partner fails to meet specific milestones, then the collaboration agreement may be terminated, which could
would preclude our ability to earn any additional milestone payments under that collaboration agreement and would reduce our revenues. In addition, even if a collaboration product candidate is successfully developed and approved for marketing by relevant regulatory authorities, if sales of the commercialized products failproduct fails to meet expectations, we could receive lower royalties than expected.
Risks Relating to our Intellectual Property
Because it is difficult, time consuming and costly to obtain, maintain and enforce patent protections for our technologies and product candidates, and because third parties may have made inventions that are similar to ours, we may not be able to secure optimal patent protections of our technologies and product candidates.
Our commercial success may depend in part on obtaining, maintaining and enforcing patent protection for our technologies and product candidates and successfully defending any of our patents that may be challenged. Obtaining, maintaining and enforcing biopharmaceutical patents is costly, time consuming and complex, and we may not be able to file and prosecute all necessary or desirable patent applications in all desired jurisdictions, or maintain, enforce and license any patents that may issue from such patent applications, at a reasonable cost or in a timely manner or at all. It is also possible that we will fail to identify patentable aspects of our research and development output before it is too late to obtain patent protection. The patent positions of biopharmaceutical companies can be highly uncertain and can involve complex legal and factual questions. No consistent policy regarding the breadth of claims allowed in biotechnology patents has emerged to date. In addition, future patent laws, regulations, rules, and court decisions may affect the scope, validity, enforceability, and associated remedies of our current and future patent claims. Accordingly, we cannot predict the breadth of claims that may issue from any patent applications that we own or license, nor are we able to predict whether any third-party patents might issue with claims that are relevant to our product candidates or technologies. Even if patents do successfully issue and even if such patents cover our technologies and product candidates, third parties may challenge their validity, enforceability or scope, which may result in such patents being narrowed, invalidated or invalidated.deemed unenforceable. There is no assurance that all of the potentially relevant prior art relating to our patents and patent applications has been found, the existence of which cancould invalidate a patent or prevent a patent from issuing from a pending patent application. Furthermore, if third parties have made similar inventions, there are multiple ways they could impact the coverage of our own applications.
We are a party to various license agreements that grant us rights under specified patents and patent applications. We are also party to various license agreements by which we grant third parties rights under specified patents and patent
applications. Our current licenses contain performance obligations. If we fail to meet those obligations, the licenses could be terminated. If we are unable to continue to license these technologies on commercially reasonable terms, or at all, we may be forced to delay or terminate aspects of our product development and research activities.
We are unable to exercise the same degree of control over intellectual property that we license from third parties as we exercise over our internally developed intellectual property. We do not control the prosecution of certain of the patent applications that we license from third parties; therefore, the patent applications may not be prosecuted as we desire or in a timely manner.
The degree of future protection for our proprietary rights is uncertain, and we cannot ensure that:
•we or our licensors were the first to makeconceive and/or reduce to practice the inventions covered by each of our pending patent applications;
•we or our licensors were the first to file patent applications for these inventions;
•the patents of others will not have an adverse effect on our ability to do business;
•others will not independently develop similar or alternative technologies or reverse engineer any of our products, processes or technologies;
•any of our pending patent applications will result in issued patents;
•any patents issued or licensed to us, our collaborators or strategic partners will provide a basis for commercially viable products or will provide us with any competitive advantages;
•any patents issued or licensed to us will not be challenged and invalidated by third parties;
•the laws, regulations, rules, or court decisions in the U.S.United States and foreign countries will not change or be interpreted in a way that modifies our patent rights or impacts our ability to enforce or maintain our patent rights; or
•we will develop additional products, processes or technologies that are patentable.
Others have filed and in the future are likely to file patent applications that are similar to ours. We are aware that there are academic groups and other companies that are attempting to develop technology that is based on the use of zinc finger, TALE, CRISPR/Cas and other DNA-binding proteins, and that these groups and companies have filed patent applications. Several patents with claims directed to this technologythese technologies have issued, although we have no current plans to use the claimed inventions. If these or other patent applications issue as patents, it is possible that the holder of any patent or patents granted on these applications may bring an infringement action against us, our collaborators, or strategic partners claiming damages and seeking to enjoin research, development or commercial activities relating to the affected products and processes. The costs of litigating the claim could be substantial regardless of outcome. Moreover, we cannot predict whether we, our collaborators, or strategic partners would
prevail in any actions. In addition, if the relevant patent claims were upheld as valid and enforceable and our products or processes were found to infringe a patent or patents, we or our collaborators may have to pay substantial damages, including treble damages and attorneys’ fees for willful infringement, and we may be prevented from making, using, selling, offering to sell, or sellingimporting into the United States the relevant product or process unless we or our collaborators could obtain a license or were able to design around the patent claims. We can give no assurance that such a license would be available to us or our collaborators on commercially reasonable terms, or at all, or that we would be able to successfully design around the relevant patent claims. There may be significant litigation in the genomics or cell therapy industry regarding patent and other intellectual property rights, which could subject us to costly, lengthy and distracting litigation with unpredictable results.
We rely on trade secrets to protect technology where we believe patent protection is not appropriate or obtainable. Trade secrets, however, are difficult to protect. While we require employees, academic collaborators and consultants to enter into confidentiality agreements, we may not be able to adequately protect our trade secrets or other proprietary information or enforce these confidentiality agreements.
Our collaborators, strategic partners, and scientific advisors have rights to publish data and information in which we may have rights. If we cannot maintain the confidentiality of our technology and other confidential information in connection with our collaborations and strategic partnerships, then we may not be able to receive patent protection or protect our proprietary information.
Patent terms may be inadequate to protect our competitive position on our product candidates for an adequate amount of time and may vary based on jurisdiction.
Patents have a limited lifespan. In the United States, the natural expiration of a patent is generally 20 years from its earliest U.S. non-provisional filing date or from the filing date of the corresponding international application. Various extensionsmeans to
extend this expected expiration date may be available. However,Regardless, the life of a patent, and the protection it affords, is limited. Even if patents covering our product candidates are obtained, once the patent life has expired for a product, we may be open to competition from generic medications. Given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. As a result, our owned and licensed patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours.
If we are unable to protect the confidentiality of our trade secrets, the value of our technology could be adversely affected, and our business would be harmed.
We rely on trade secret protection and confidentiality agreements to protect proprietary know-how that is not patentable or that we elect not to patent, processes for which patents are difficult to enforce and any other elements of our product candidate discovery and development processes that involve proprietary know-how, information or technology that is not covered by patents. However, trade secrets can be difficult to protect. We seek to protect our proprietary technology and processes, in part, by entering into confidentiality agreements with our employees, consultants, scientific advisors, collaborators, partners and contractors. We also seek to preserve the integrity and confidentiality of our data and trade secrets by maintaining physical security of our premises and physical and electronic security of our information technology systems. While we have confidence in these individuals, organizations and systems, agreements or security measures have been and may in the future be breached, and we may not have adequate remedies for any breach. See also the risk factor titled “Significant disruptions of“If our information technology systems or data, security incidentsor those of third parties upon which we rely, are or were compromised, we could result in significant financial, legal,experience adverse consequences, including but not limited to regulatory investigations or actions, litigation, fines and penalties, disruptions of our business operations and reputational harm to us.harm.” In addition, our trade secrets may otherwise become known or be independently discovered by competitors.
Although we expect all of our employees and consultants to assign their inventions to us, and all of our employees, consultants, advisors, collaborators, partners and any third parties who have access to our proprietary know-how, information or technology to enter into confidentiality agreements, we cannot provide any assurances that all such agreements have been duly executed or that our trade secrets and other confidential proprietary information will not be disclosed or that competitors will not otherwise gain access to our trade secrets or independently develop substantially equivalent information and techniques. Misappropriation or unauthorized disclosure of our trade secrets could impair our competitive position and may have an adverse effect on our business. Additionally, if the steps taken to maintain our trade secrets are deemed inadequate, we may have insufficient recourse against third parties for misappropriating the trade secret. In addition, others may independently discover our trade secrets and proprietary information.
Further, the laws of some foreign countries do not protect proprietary rights to the same extent or in the same manner as the laws of the United States. As a result, we may encounter significant problems in protecting and defending our intellectual
property both in the United States and abroad. If we are unable to prevent material disclosure of the non-patented intellectual property related to our technologies to third parties, and there is no guarantee that we will have any such enforceable trade secret protection, we may not be able to establish or maintain a competitive advantage in our market, which could adversely affect our business, results of operations and financial condition.
We may not be successful in obtaining or maintaining necessary rights to product components, platforms and processes for our development pipeline through acquisitions and in-licenses.
Presently, we believe we have rights to the intellectual property, through licenses from third parties and under patents that we own, to develop our gene and cell therapy product candidates. Because our programs may involve additional product candidates, such as TX200 and potential future CAR-Treg therapies that may require the use of proprietary rights held by third parties, the growth of our business will likely depend in part on our ability to acquire, in-license or use these proprietary rights. In addition, our product candidates may require specific formulations to work effectively and efficiently and these rights may be held by others. We may be unable to acquire or in-license any compositions, methods of use, processes or other third-party intellectual property rights from third parties that we identify on commercially reasonable terms, if at all. The licensing and acquisition of third-party intellectual property rights is a competitive area, and a number of more established companies are also pursuing strategies to license or acquire third-party intellectual property rights, including from other companies and academic institutions, that we may consider attractive. Other companies may have a competitive advantage over us due to their size, cash resources and greater clinical development and commercialization capabilities.
In addition, companies that perceive us to be a competitor may be unwilling to assign or license rights to us. We also may be unable to license or acquire third-party intellectual property rights on terms that would allow us to make an appropriate
return on our investment. If we are unable to successfully obtain rights to required third-party intellectual property rights, our business, financial condition and prospects for growth could suffer.
If we fail to comply with our obligations in the agreements under which we license intellectual property rights from third parties or otherwise experience disruptions to our business relationships with our licensors, we could lose license rights that are important to our business.
We are a party to a number of intellectual property license agreements that are important to our business and expect to enter into additional license agreements in the future. Our existing license agreements impose, and we expect that future license agreements will impose, various diligence, milestone, royalty and other obligations on us. If we fail to comply with our obligations under these agreements, or we are subject to a bankruptcy, the licensor may have the right to terminate the license, in which event we would not be able to market products covered by the license.
We may need to obtain licenses from third parties to advance our research or allow commercialization of our product candidates, and we have done so from time to time. We may fail to obtain any of these licenses at a reasonable cost or on reasonable terms, if at all. In that event, we may be required to expend significant time and resources to develop or license replacement technology. If we are unable to do so, we may be unable to develop or commercialize the affected product candidates, which could harm our business significantly. We cannot provide any assurances that third-party patents do not exist that might be enforced against our current product candidates or future products, resulting in either an injunction prohibiting our sales, or, with respect to our sales, an obligation on our part to pay royalties and/or other forms of compensation to third parties.
In many cases, patent prosecution of our in-licensed technology is controlled solely by the licensor. If our licensors fail to obtain and maintain patent or other protection for the proprietary intellectual property we license from them, we could lose our rights to the intellectual property or our exclusivity with respect to those rights, and our competitors could market competing products using the intellectual property. In certain cases, we control the prosecution of patents resulting from licensed technology. In the event we breach any of our obligations related to such prosecution, we may incur significant liability to our licensing partners. Licensing of intellectual property is of critical importance to our business and involves complex legal, business and scientific issues and is complicated by the rapid pace of scientific discovery in our industry.
The agreements under which we currently license intellectual property or technology from third parties are complex, and certain provisions in such agreements may be susceptible to multiple interpretations. The resolution of any contract interpretation disagreement that may arise could narrow what we believe to be the scope of our rights to the relevant intellectual property or technology, or increase what we believe to be our financial or other obligations under the relevant agreement, either of which could have an adverse effect on our business, financial condition, results of operations and prospects. If disputes over intellectual property that we have licensed prevent or impair our ability to maintain our current licensing arrangements on acceptable terms, we may be unable to successfully develop and commercialize the affected product candidates, which could
have an adverse effect on our business, financial conditions,condition, results of operations and prospects. As an example, Sangamo France has exclusively licensed the right to the CAR for use in TX200 from the University of British Columbia, or UBC. Should UBC terminate this license agreement, we may have to develop or acquire the appropriate CAR which would extend our anticipated development timeline and add expense, and which could result in our failure to realize the anticipated benefits of the acquisition of Sangamo France.
We may be involved in patent or intellectual property lawsuits or similar disputes involving patents under our control or patents of third parties claiming infringement, which lawsuits could be expensive, time-consuming and impair or prevent development and commercialization activities.
Our commercial success depends in part on our avoiding infringement of the patents and proprietary rights of third parties. There is a substantial amount of litigation, both within and outside the United States, involving patent and other intellectual property rights in the biotechnology and pharmaceutical industries, including patent infringement lawsuits, declaratory judgment lawsuits, invalidity proceedings, interferences, oppositions, ex parte or inter partes reexaminations, post-grant reviewreviews and inter partes review proceedings before the U.S. PTO, and corresponding foreign patent offices. Numerous U.S. and foreign issued patents and pending patent applications, which are owned by third parties, exist in the fields in which we are pursuing development candidates. As the biotechnology and pharmaceutical industries expand and more patents are issued, the risk increases that our product candidates may be subject to claims of infringement of the patent rights of third parties.
Third parties may assert that we are employing their proprietary technology without authorization, and such parties may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. There may be third-party patents or patent applications with claims to materials, formulations, methods of manufacture or methods for treatment related to the use or manufacture of our product candidates. For example, we are aware of certain patents held by third parties related to certain vector and vector manufacturing methods that are related to certain of our product candidates. We have not yet finalized the commercial scale manufacturing process for any of our product candidates. If our commercial scale manufacturing process utilizes these vector manufacturing methods, and if these third-party
patents are valid and in force at the time of commercialization, we may need to challenge these patents, use or develop non-infringing alternatives or seek a license to these patents. In any event, if any third-party patents were held by a court of competent jurisdiction to cover our product candidates, any molecules formed during the manufacturing process or any final product itself, the holders of any such patents may be able to block or hinder our ability to commercialize such product candidate unless we obtained a license under the applicable patents, or until such patents expire. Similarly, if any third-party patents were held by a court of competent jurisdiction to cover aspects of our formulations or processes for manufacture or methods of use, the holders of any such patents may be able to block our ability to develop and commercialize the applicable product candidate unless we obtained a license, or until such patents expires. Moreover, because patent applications can take many years to issue, there may be currently pending patent applications that may later result in issued patents that our product candidates may infringe.
Defense of these claims, regardless of their merit, would involve substantial litigation expense, could expose proprietary information and would be a substantial diversion of employee resources from our business. In the event of a successful claim of infringement against us, we may have to pay substantial damages, including treble damages and attorneys’ fees for willful infringement, pay royalties, redesign our infringing products or obtain one or more licenses from third parties, which may be impossible or require substantial time and monetary expenditure.
Competitors may also infringe our patents or the patents of our licensors. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time-consuming. In addition, in an infringement proceeding, a court may decide that a patent of ours or our licensors is not valid, is unenforceable, in whole or in part, or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation or defense proceedings could put one or more of our patents at risk of being invalidated, held unenforceable or interpreted narrowly and could put our patent applications at risk of not issuing. Moreover, if we or one of our licensing partners initiated legal proceedings against a third party to enforce a patent covering one of our product candidates, the defendant could counterclaim that the patent covering our product candidate is invalid and/or unenforceable. Such proceedings could result in revocation or amendment to our patents in such a way that they no longer cover our product candidate. The outcome following legal assertions of invalidity and unenforceability is unpredictable. With respect to the validity question, for example, we cannot be certain that there is no invalidating prior art, of which we and the patent examiner were unaware during prosecution. If a defendant were to prevail on a legal assertion of invalidity and/or unenforceability, we would lose at least part, and perhaps all, of the patent protection on our product candidates. Such a loss of patent protection could have an adverse impact on our business.
Interference or derivation proceedings provoked by third parties or brought by us or declared by the U.S. PTO may be necessary to determine the priority of inventions or other matters of inventorship with respect to our patents or patent applications or those of our licensors. An unfavorable outcome could expose us to significant monetary damages, result in the loss of valuable intellectual property, require us to cease using the related technology or to attempt to license rights to it from the prevailing party. Our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms or at all, or if a non-exclusive license is offered and our competitors gain access to the same technology. Our defense of litigation, interference, derivation or other proceedings may fail and, even if successful, may result in substantial costs and distract our management and other employees. We may not be able to prevent, alone or with our licensors, misappropriation of our intellectual property rights, particularly in countries where the laws may not protect those rights as fully as in the United States.
We may be subject to claims that our employees, consultants or independent contractors have wrongfully used or disclosed confidential information of third parties or that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
We employ individuals who were previously employed at universities or other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although we try to ensure that our employees, consultants and independent contractors do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or our employees, consultants or independent contractors have inadvertently or otherwise used or disclosed intellectual property, including trade secrets or other proprietary information, of any of our employee’s former employer or other third parties. Litigation may be necessary to defend against these claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel, which could adversely impact our business. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.
We may be unable to license gene transfer technologies that we may need to commercialize our ZFPZF technology and potential products, if approved.
In order to regulate or modify a gene in a cell, the ZFP must be efficiently delivered to the cell. We have licensed certain gene transfer technologies for our ZFP in research, including AAV and mRNA technology, and we are evaluating these systems and other technologies that may need to be used in the delivery of ZFP into cells for in vitro and in vivo applications. We have not fully developed our own gene transfer technologies, and we rely on our ability to enter into license agreements to provide us with rights to the necessary gene transfer technology. Our approach has been to license appropriate technology as required. For example, in addition to our own vector manufacturing methods currently being used in our product candidates, we are aware of certain patents held by a third party related to certain vector manufacturing methods that are currently being used in certain of our product candidates. We have not yet finalized the commercial scale manufacturing process for any of our product candidates. If our commercial scale manufacturing process utilizes these vector manufacturing methods, and if these third-party patents are in force at the time of commercialization, we may need to use or develop a non-infringing manufacturing method or seek a license to these patents. However, we may not be able to license the gene transfer technologies on reasonable terms, if at all, required to develop and commercialize our product candidates. The inability to obtain a license to use gene transfer technologies with entities that own such technology on reasonable commercial terms, if at all, could delay or prevent the preclinical evaluation, drug development collaborations, clinical testing and/or commercialization of our therapeutic product candidates.
We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting and defending all current and future patents and patent applications in all countries throughout the world would be prohibitively expensive, and our intellectual property rights in some countries outside the United States can be less extensive or more difficult to enforce than those in the United States. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as federal and state laws in the United States. Consequently, we may not be able to prevent third parties from practicing our inventions in all countries outside the United States, or from selling or importing products made using our inventions in and into the United States or other jurisdictions. Competitors may use, make, sell, or import our technologies in or into jurisdictions where we have not obtained patent protection to develop their own products and, further, may export otherwise infringing products to territories where we have patent protection but enforcement is not as strong as that in the United States. These products may compete with our product candidates, and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
The legal systems of many foreign countries do not favor the enforcement of patents and other intellectual property protections, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights. For example, some foreign countries have compulsory licensing laws under which a patent owner must grant licenses to third parties. In addition, some countries limit the enforceability of patents against third parties, including government agencies or government contractors. In these countries, patents may provide limited or no benefit. Further, the complexity and uncertainty of European patent laws have increased in recent years. In Europe, the new unitary patent system that came into effect in June 2023 would significantly impact European patents, including those granted before the introduction of such a system. Under the unitary patent system, European applications will have the option, upon grant of a patent, of becoming a Unitary Patent which will be subject to the jurisdiction of the newly formed Unified Patent Court, or UPC. As the UPC is a new court system, there is no precedent or established body of case law on which the court can base its decisions, thus increasing the uncertainty of any litigation before the UPC. Patents granted before the implementation of the UPC will have the option of opting out of the jurisdiction of the UPC and remaining as national patents in the UPC countries. Patents that remain under the jurisdiction of the UPC will be potentially vulnerable to a single UPC-based revocation challenge that, if successful, could invalidate the patent in all countries who are signatories to the UPC. We cannot predict with certainty the long-term effects of any potential changes. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly and our patent applications at risk of not issuing and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate, and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
We may become subject to claims challenging the inventorship or ownership of our patents and other intellectual property.
We, our licensors and collaborators may be subject to claims that former employees, consultants, independent contractors, collaborators or other third parties have an interest in our patents or other intellectual property as an owner, co-owner, inventor or co-inventor. The failure to name the proper inventors on a patent application can result in the patents issuing thereon being unenforceable or invalid. Inventorship disputes may arise from conflicting views regarding the contributions of different individuals named as inventors, the effects of foreign laws where foreign nationals are involved in the development of the subject matter of the patent, conflicting obligations of third parties involved in developing our product candidates or as a
Risks Relating to our Business Operations
SignificantOur recent restructurings are designed to reduce costs and increase focus on our key strategic priorities. We may incur additional expenses not currently contemplated due to events associated with the reduction in force, and our restructuring activities may subject us to reputational risks and litigation risks and expenses. We may not fully realize the anticipated benefits and savings from these restructurings due to unforeseen difficulties, disruptions, delays or unexpected costs, which could adversely affect our financial condition. In addition, we may need to undertake additional workforce reductions or restructuring activities in the future.
Furthermore, our restructurings may be disruptive to our operations. For example, in connection with the November Restructuring, we expect to close our Brisbane, California facility in 2024 and move all U.S. operations, including our headquarters, to our Richmond, California facility, and in connection with the France Restructuring, we expect to close our Valbonne, France facility and eliminate all 93 roles in France in 2024. We may experience delays or other difficulties in effectuating the transition of certain research and other operations, which could result in significant disruptions to our business and delays in our development efforts and related timelines. In addition, our workforce reductions could yield unanticipated consequences, such as attrition beyond planned staff reductions, increased difficulties in our day-to-day operations, loss of institutional knowledge and expertise and increased risk to our internal controls and disclosure controls. Our workforce reductions could also harm our ability to attract and retain qualified personnel who are critical to our operations.
If our information technology systems or data, security incidentsor those of third parties upon which we rely, are or were compromised, we could result in significant financial, legal,experience adverse consequences resulting from such compromise, including but not limited to regulatory investigations or actions, litigation, fines and penalties, disruptions of our business andoperations, reputational harm to us.
We are increasingly dependent on information technology systems and infrastructure to operate our business, which are large and complex. In the ordinary course of our business, we and the third parties upon which we rely, may collect, receive, store, process, generate, use, transfer, disclose, make accessible, protect, secure, dispose of, share and transmit large amounts of proprietary, confidential and sensitive information, including intellectual property, proprietary business information,trade secrets and personal data (including personal health(such as health-related information) and other confidential information.. It is critical that we do so in a secure manner to maintain the confidentiality, integrity and availability of such sensitive information. We have also outsourced elements of our operations (including elements of our information technology infrastructure) to third parties, and as a result, we manage a number of third-party vendors who may or could have access to our computer networks or our confidential information. Many of those third parties in turn subcontract or outsource some of their responsibilities to other third parties. Our ability to monitor third parties’ information security practices is limited, and these third parties may not have adequate information security measures in place.
While all information technology operations are inherently vulnerable to inadvertent or intentional security breaches, incidents, attacks and exposures, the size, complexity, accessibility and distributed nature of our information technology systems, and the large amounts of sensitive information stored on those systems, make such systems potentially vulnerable to unintentional or malicious, internal and external attacks
on our technology environment. Attacks of this nature are increasing in their frequency, levels of persistence, sophistication and intensity. Threats to information systems and data are increasingly difficult to detect and come from a variety of sources, including traditional computer “hackers,” threat actors, “hacktivists,” organized criminal threat actors, personnel (such as through theft or misuse), sophisticated nation-states and nation-state-supported actors. Some actors now engage and are expected to continue to engage in cyber-attacks, including nation-state actors for geopolitical reasons and in conjunction with military conflicts and defense activities. During times of war and other major
conflicts, we and the third parties on which we rely may be vulnerable to a heightened risk of these attacks, including retaliatory cyber-attacks, that could materially disrupt our systems, operations and supply chain. We and the third parties upon which we rely may be subject to a variety of evolving threats, including but not limited to social-engineering attacks (including through phishing attacks), malicious code (such as viruses and worms), malware (including as a result of advanced persistent threat intrusions), denial-of-service attacks (such as credential stuffing), credential harvesting, personnel misconduct or error, ransomware attacks, supply-chain attacks, software bugs, server malfunctions, software or hardware failures, loss of data or other information technology assets, adware, telecommunications failures, natural disasters (such as earthquakes, fires, floods), war, terrorism, attacks enhanced or facilitated by artificial intelligence, or AI, and other similar threats. Ransomware attacks are becoming increasingly prevalent and severe and can lead to significant interruptions in our operations, loss of data and income, reputational harm and diversions of funds. Extortion payments may alleviate the negative impact of a ransomware attack, but we may be unwilling or unable to make such payments due to, for example, applicable laws or regulations prohibiting such payments. In addition, the effects of the COVID-19 pandemicour updated work from home policies have intensified our dependence on information technology systems and could increase our cybersecurity risk as many of our critical business activities are currently being conducted remotely utilizing network connections, computers and devices outside our premises or network and our increased reliance on personnel working from home, could increase our cybersecurity risk.while in transit and in public locations.
Significant disruptions of our, our third-party vendors’ and/or business partners’ information technology systems or other similarWhile we have implemented security measures designed to protect against data security incidents, could adversely affect our business operations and/or resultthere can be no assurance that these measures will be effective. We have not always been able in the loss, misappropriation,past and may be unable in the future to detect vulnerabilities in our information technology systems. We take steps designed to detect, mitigate and remediate vulnerabilities in our information security systems (such as our hardware and/or unauthorized access, use or disclosuresoftware, including that of or the prevention of accessthird parties upon which we rely), but we may not be able to sensitive information, which could result in financialdetect, mitigate and reputational harm to us.remediate all such vulnerabilities on a timely basis. For example, in April 2018, we announced a data security incident involving the compromise of a senior executive’s company email account. Our investigation of the incident did not reveal any evidence that our systems were otherwise compromised in connection with the incident or that personal data about patients or other individuals besides the executive were accessed or disclosed. However, proprietary, confidential and other sensitive information of ours and that of other entities was accessed and may have been compromised as a result of the incident. Unforeseen developments related to this incident could occur, which could have a further adverse impact on us. Any litigation or regulatory review or investigation arising from this incident could result in significant legal exposure to us. In addition, information technology system disruptions, whether from attacks on our technology environmentA security incident or from computer viruses, natural disasters, terrorism, war and telecommunication and electrical failures,other interruption could also result in a material disruption of our facility, development programs and our business operations. For example, the loss of clinical trial data from completed or future clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data.
While we are aware of the company email incident described above, there is no way of knowing with certainty whether we have experienced any other data security incidents that have not been discovered. While we have no reason to believe this to be the case, attackers have become very sophisticated in the way they conceal access to systems, and many companies that have been attacked are not aware that they have been attacked. Any delay in the discovery of an attack may result in increased expense and may harm our reputation. Any event, includingsecurity incident or interruption that we, or a third-party upon which we rely, experience (including the company email incident described above, that leadsabove) could lead to unauthorized access, use adverse consequences, including government enforcement actions (for example, investigations, fines, penalties, audits and inspections), additional reporting requirements and/or disclosure ofoversight, restrictions on processing sensitive data (including personal data could, among other consequences, disrupt our business,data), litigation (including class claims), indemnification obligations, harm to our reputation, compelmonetary fund diversions, diversion of management attention, interruptions in our operations (including availability of data) and financial loss. Applicable data privacy and security obligations may require us to notify relevant stakeholders, including affected individuals, customers, regulators and investors, of security incidents. Such disclosures are costly, and the disclosures or the failure to comply with applicable federal and/or state data breach notification laws and foreign law equivalents.such requirements could lead to adverse consequences. In addition, failure to maintain effective internal accounting controls related to security breaches and cybersecurity in general could impact our ability to produce timely and accurate financial statements and subject us to regulatory scrutiny. While we have implemented security measures intendedWe may expend significant resources or modify our business activities in an effort to protect our information technology systemsagainst security incidents or other interruptions. Further, we may experience delays in developing and infrastructure,deploying remedial measures designed to address any such identified vulnerabilities. Our contracts may not contain limitations of liability, and even where they do, there can be no assurance that limitations of liability in our contracts are sufficient to protect us from liabilities, damages or claims related to our data privacy and security obligations. While we may be entitled to damages if our third-party partners fail to satisfy their privacy or data security-related obligations to us, any award may be insufficient to cover our damages, or we may be unable to recover such measuresaward. Additionally, we cannot be sure that our insurance coverage, if any, will successfully prevent service interruptionsbe adequate or furthersufficient to protect us from or mitigate liabilities arising out of our privacy and security incidents.practices, that such coverage will continue to be available on commercially reasonable terms, or at all, or that such coverage will pay future claims.
In addition to experiencing a security incident, third parties may gather, collect or infer sensitive information about us from public sources, data brokers or other means that reveals competitively sensitive details about our organization and could be used to undermine our competitive advantage or market position. Additionally, our sensitive information could be leaked,
disclosed or revealed as a result of or in connection with our employees’, personnel’s or vendors’ use of generative AI technologies.
We have business operations in France and the United Kingdom, which exposes us to additional costs and risks.
Our business operations in France and the United Kingdom subject us to certain additional costs and risks associated with doing business outside the United States, including:
•the increased complexity and costs inherent in managing international operations in geographically disparate locations;
•challenges of complying with diverse regulatory, financial and legal requirements, which are subject to change at any time;
•potentially adverse tax consequences, including changes in applicable tax laws and regulations;
•potentially costly trade laws, tariffs, export quotas, custom duties or other trade restrictions, and any changes to them;
•compliance with tax, employment, immigration and labor laws for employees living or traveling abroad;
•liabilities for activities of, or related to, our international operations;
•challenges inherent in efficiently managing employees in diverse geographies, including the need to adapt systems, policies, benefits and compliance programs to differing labor and other regulations;
•natural disasters, political and economic instability, including wars, terrorism and political unrest, including the conflicts between Russia and Ukraine and in the Middle East, outbreak of health epidemics including the evolving COVID-19 pandemic, and the resulting global economic and social impacts;
•workforce uncertainty in countries where labor unrest is more common than in the United States; and
•differing laws and regulations relating to data security and the unauthorized use of, or access to, commercial and personal information.
In addition, our international operations in France and the United Kingdom expose us to fluctuations in currency exchange rates between the Euro and the U.S. dollar and between the Pound Sterling and the U.S. dollar. Given the volatility of currency exchange rates, there is no assurance that we will be able to effectively manage currency transaction and/or conversion risks. To date, we have not entered into derivative instruments to offset the impact of foreign exchange fluctuations, which fluctuations could have an adverse effect on our financial condition and results of operations. In any event, difficulties resulting
from these and other risks related to our operations outside of the United States could expose us to increased expenses, impair our development efforts, adversely affect our financial condition and results of operations and harm our competitive position.
We are in the process of growing the size of our organization globally,have experienced and we may continue to experience difficulties in hiring, integrating and retaining qualified skilled employees.
We need to grow the sizeThe stability and potential growth of our organization in order to support our research, product development, manufacturing and regulatory efforts, and such growth is critical to our success.ability to successfully achieve our strategic objectives. We may not be able to hire, integrate and retain a sufficient number of qualified employees with the appropriate levels of experience and skills to accomplish our growth objectives.
There currently is a shortage of skilled individuals with substantial experience discovering, developing and manufacturing genomic medicines, which is likely to continue. As a result, competition for these individuals is intense and the turnover rate can be high. We have experienced, and may not be ablecontinue to hire, integrateexperience, difficulty hiring, integrating and retainretaining employees with these skills on acceptable terms given the uncertainty regarding our ability to obtain sufficient additional funding and to continue to operate as a going concern as well as the competition among numerous biopharmaceutical companies and academic institutions for individuals with these skills. In this regard, as a result of the April and November Restructurings, approximately 272 roles at our Company were eliminated, and as a result of the France Restructuring, 93 roles will be eliminated. Accordingly, we have been and are operating with a shortage of resources and may not be able to effectively conduct our operations with our substantially reduced number of employees. In addition, our history of implementing significant workforce reductions, along with the potential for future workforce reductions, may negatively affect our ability to retain or attract talented employees. Moreover, any negative or unexpected results in our preclinical studies or clinical trials or applications for marketing approval would make it more challenging to hire and retain qualified skilled employees. Moreover, the evolving COVID-19 pandemic has further challenged our ability to hire skilled employees. If we do not obtain sufficient additional funding in the near term so that we can continue to operate as a going concern and to potentially
achieve our growth objectives, the progress of our research, development, manufacturing and regulatory efforts will slow down or halt altogether, which willwould materially and adversely impactaffect our business, financial condition, results of operations and prospects.
We are dependent on certain key members of our executive team and certain of our scientific, clinical development and manufacturing personnel, the loss of whose services may impede the progress of our research, development manufacturing and regulatory efforts. For example, in 2024, our former Senior Vice President and Chief People Officer and our former Chief Medical Officer resigned from Sangamo, and in connection with our Restructurings, the employment of each of our former Executive Vice President, Technical Operations, Executive Vice President, Chief Operating Officer, and Senior Vice President, Chief Scientific Officer, was terminated. We could experience resignations of other executives and employees in the future given the uncertainty regarding our ability to obtain sufficient additional funding and to continue to operate as a going concern as well as the intensity of the competition for talent in the biotechnology industry, particularly in the San Francisco Bay Area. Additional resignations or workforce reductions could result in more significant disruptions and threats to our stability and potential growth. While we have entered into employment agreements with each of our executive officers, any of them could leave our employment at any time, as all of our employees are “at will” employees. We do not have “key person” insurance on any of our employees.
We may not be successful in our efforts to discover, license or acquire new potential product candidates and may fail to capitalize on product candidates with a greater commercial opportunity or for which there is a greater likelihood of success.
If our existing product candidates do not receive regulatory approval or are not successfully commercialized, then the success of our business will depend on our ability to continue to expand our product pipeline through discovery, in-licensing or acquisitions. We may be unable to do so. If we do identify potential product candidates for licensing or acquisition, we may be unable to reach acceptable terms with the licensors or sellers. Further, there may be risks and liabilities associated with the product candidates which our due diligence efforts fail to discover, that are not disclosed to us, that we inadequately assess, or that we are unable to manage effectively. Additionally, we may not realize the anticipated benefits of such licenses or acquisitions for a variety of reasons, including the possibility that the product candidates prove not to be safe or effective in clinical trials, that we are unable to successfully integrate the product candidate into our operations, or that the anticipated benefits will not otherwise be realized within the expected timeframe.
Additionally, because we have limited resources, we may forego or delay pursuit of opportunities with certain product candidates or indications that later prove to have greater commercial potential. Our spending on current and future research and development programs may not yield any commercially viable products. If we do not accurately evaluate the commercial potential for a particular product candidate, we may relinquish valuable rights to that product candidate through strategic collaboration, licensing or other arrangements in cases in which it would have been more advantageous for us to retain sole development and commercialization rights to such product candidate. Alternatively, we may allocate internal resources to a product candidate in a therapeutic area in which it would have been more advantageous to enter into a collaboration arrangement.
Risks Relating to our Common Stock and Corporate Organization
We have in the past, and may in the future, be unable to comply with the listing standards of the Nasdaq Stock Market LLC, or Nasdaq. If we fail to comply with listing standards in the future, our common stock may be delisted. Delisting could adversely affect the liquidity of our common stock and the market price of our common stock could decrease, and our ability to obtain sufficient additional capital to fund our operations and to continue to operate as a going concern would be substantially impaired.
Our common stock is currently listed on the Nasdaq Global Select Market, which has minimum requirements that a company must meet in order to remain listed. These requirements include maintaining a minimum closing bid price of $1.00 per share, which closing bid price cannot fall below $1.00 per share for a period of more than 30 consecutive trading days, or the Bid Price Requirement. On October 27, 2023, we received a deficiency notice from the Listing Qualifications Staff, or the Staff of Nasdaq, notifying us that, for the last 30 consecutive business days, the bid price of our common stock had closed below $1.00 per share, thereby failing to satisfy the Bid Price Requirement set forth in the continued listing requirements of Nasdaq Listing Rule 5450(a)(1). On March 1, 2024, we received a notice from the Staff of Nasdaq notifying us that, for the prior 10 consecutive business days, the bid price of our common stock had closed at $1.00 per share or greater, thereby satisfying the Bid Price Requirement set forth in the continued listing requirements of Nasdaq Listing Rule 5450(a)(1). However, since receiving such notice of compliance, closing prices of our common stock on Nasdaq have fluctuated to below $1.00 and, accordingly, there can be no assurance that we will be able to maintain compliance with the Bid Price Requirement. In the event we fail to comply with listing standards in the future, and we do not regain compliance with the Bid Price Requirement prior to the expiration of the compliance period, unless Nasdaq exercises its discretion to extend this period, our common stock may be subject to a delisting action by Nasdaq.
If we again fail to satisfy the Bid Price Requirement, a reverse stock split may allow us to meet the Bid Price Requirement, but we cannot assure you that a reverse stock split will be approved by our stockholders or that any reverse stock split, if implemented, will be sufficient to enable us to maintain our Nasdaq listing. Additionally, if a reverse stock split is implemented, there can be no assurance that the market price per new share of our common stock following the reverse stock split will remain unchanged or will increase in proportion to the reduction in the number of shares of our common stock outstanding before the reverse stock split. The liquidity of the shares of our common stock may be affected adversely by any reverse stock split given the reduced number of shares of our common stock that will be outstanding following such reverse stock split. Furthermore, following any reverse stock split, the resulting market price of our common stock may not attract new investors and may not satisfy the investing requirements of those investors.
In the event that our common stock is delisted from Nasdaq as a result of our failure to comply with the Bid Price Requirement, as a result of Nasdaq not granting us an extension or the panel not granting us a favorable decision or due to our failure to continue to comply with any other requirement for continued listing on Nasdaq, trading of our common stock could be conducted in the over-the-counter market or on an electronic bulletin board established for unlisted securities such as the Pink Sheets or the OTC Bulletin Board, but there can be no assurance that our common stock will be eligible for trading on such alternative exchange or market.
Additionally, if our common stock is delisted from Nasdaq, the liquidity of our common stock would be adversely affected, the market price of our common stock could decrease, our ability to obtain sufficient additional capital to fund our operations and to continue to operate as a going concern would be substantially impaired and transactions in our common stock could lose federal preemption of state securities laws. Furthermore, there could also be a further reduction in our coverage by securities analysts and the news media and broker-dealers may be deterred from making a market in or otherwise seeking or
generating interest in our common stock, which could cause the price of our common stock to decline further. Moreover, delisting may also negatively affect our collaborators’, vendors’, suppliers’ and employees’ confidence in us and employee morale.
Our stock price has been volatile and will likely continue to be volatile, which could result in substantial losses for investors and potentially class action securities litigation against us, and could be influenced by public perception of genomic medicines and the biotechnology sector.
Our stock price has been volatile and may continue to be volatile, which could cause stockholders to incur substantial losses. An active public market for our common stock may not be sustained, and the market price of our common stock may continue to be volatile. The market price of our common stock has fluctuated significantly in response to various factors, some of which are beyond our control, including but not limited to the following:
•announcements by us or collaborators providing updates on the progress or development status of product candidates or data from clinical trials;
•initiation or termination of clinical trials;
•changes in market valuations of similar companies;
•overall market and economic conditions, including the equity markets for emerging biotechnology companies;
•deviations in our results of operations from the guidance given by us;
•announcements by us or our competitors of new or enhanced products or technologies or significant contracts, acquisitions, strategic relationships, joint ventures or capital commitments;
•announcement of changes in business and operations by our collaborators, or changes to our existing collaboration agreements;
•changes in public opinions of genomic medicines;
•regulatory developments, including increased regulatory scrutiny of genomic medicines;
•changes by one or more of our securities analysts in recommendations, ratings or coverage of our stock;
•additions or departures of key personnel; and
•sales of our common stock or other securities by us, officers or directors, liquidation of institutional funds that comprised large holdings of our stock and decreases in our cash balances.
In addition, the stock marketsemerging biotechnology stocks have recently experienced extreme price and volume fluctuations that have affected and continue to affect the market prices of equity securities of many biotechnology companies, including very recently in connection with the evolving COVID-19 pandemic, which has resulted in decreased stock prices for many emerging biotechnology companies notwithstanding the lack of a fundamental change in their underlying business models or prospects. These fluctuations have often been unrelated or disproportionate to the operating performance of those biotechnology companies. Broad market and industry factors, including potentially worsening economicmacroeconomic conditions and other adverse effects or developments relating to the evolving COVID-19 pandemic, andcurrent political, geopolitical, regulatory and other market conditions, may negatively affect the market price of shares of our common stock, regardless of our actual operating performance.
Actual or potential sales of significant amounts of shares of our common stock into the market could cause the market price of our common stock to fall or prevent it from increasing for numerous reasons.
Sales of a substantial number of shares of our common stock in the public market could occur at any time. These sales, or the perception in the market that holders of a large number of shares intend to sell shares, could reduce the market price of our common stock. Our outstanding shares of common stock generally may be freely sold in the public market at any time to the extent permitted by Rules 144 and 701 under the Securities Act of 1933, as amended, or the Securities Act, or to the extent the issuance of such shares has already been registered under the Securities Act and are held by non-affiliates of ours. While Biogen agreed notIn 2022, the restrictions applicable to sell anythe sale of the shares that we issued to Biogen lapsed, and accordingly, may be sold in April 2020 until the first anniversary of the effectiveness of the Biogen collaboration, and to limit resales through the second anniversary, such restrictions are only temporary. Further, we alsopublic market without restriction. We agreed, subject to certain limitations, to register for resale under the Securities Act any of the shares we issued to Biogen.Biogen in connection with our prior collaboration. We have also filed registration statements registering allthe shares of common stock that we may issue under our equity compensation plans. Such shares can be freely sold in the public market upon issuance, subject to volume limitations and black-out periods applicable to affiliates. Additionally, we are party to
a sales agreement with Jefferies LLC which permits us from time to time at our discretion to sell up to $150.0$325.0 million of shares of our common stock in the public markets at prevailing market prices. As of February 19, 2021, we have sold 1,034,762 shares of our common stock under the sales agreement for net proceeds of approximately $15.7 million.
In addition, in accordance with the guidelines specified under Rule 10b5-1 of the Securities Exchange Act of 1934, as amended, or the Exchange Act, and our policies regarding stock transactions, certaincertain of our employees, executive officers and directors have adopted, and may continue to adopt, stock trading plans pursuant to which they have arranged to sell shares of our common stock from time to time in the future. Generally, sales under such plans by our executive officers and directors require public filings.disclosure. Our employees, executive officers, directors and affiliated stockholders also may buy or sell additional shares outside of a Rule 10b5-1 plan when they are not in possession of material, nonpublic information. Actual or potential sales of our common stock by such persons could be viewed negatively by other investors and could cause the price of our common stock to fall or prevent it from increasing.
We do not intend to pay dividends on our common stock so any returns will be limited to the value of our stock.
We have never declared or paid any cash dividends on our common stock. We currently anticipate that we will retain future earnings for the development, operation and expansion of our business and do not anticipate declaring or paying any cash dividends for the foreseeable future. Any return to stockholders will therefore be limited to the appreciation of their stock.
our common stock will depend in part on the research and reports that securities or industry analysts publish about us or our business. In the event securities or industry analysts who cover us downgrade our stock or publish inaccurate or unfavorable research about our business, our stock price would likely decline. If one or more of these analysts cease coverage of our company or fail to publish reports on us regularly, demand for our stock could decrease, which might cause our stock price and trading volume to decline. Anti-takeover provisions in our certificate of incorporation, Delaware law and our bylaws could make an acquisition of our company more difficult and could prevent attempts by our stockholders to remove or replace current management.
Anti-takeover provisions of Delaware law and in our certificate of incorporation and our bylaws may discourage, delay or prevent a change in control of our company, even if a change in control would be beneficial to our stockholders. In addition, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our board of directors. In particular, under our certificate of incorporation our board of directors may issue up to 5,000,000 shares of preferred stock with rights and privileges that might be senior to our common stock, without the consent of the holders of the common stock. Moreover, without any further vote or action on the part of the stockholders, the board of directors would have the authority to determine the price, rights, preferences, privileges, and restrictions of the preferred stock. This preferred stock, if it is ever issued, may have preference over, and harm the rights of, the holders of common stock. Although the issuance of this preferred stock would provide us with flexibility in connection with possible acquisitions and other corporate purposes, this issuance may make it more difficult for a third party to acquire a majority of our outstanding voting stock.
Similarly, our authorized but unissued common stock is available for future issuance without stockholder approval. Our certificate of incorporation further provides that stockholders may not take action by written consent.
In addition, our amended and restated bylaws:
•establish advance notice requirements for nominations for election to the board of directors or proposing matters that can be acted upon at stockholders’ meetings; and
•prohibit stockholders from calling a special meeting of stockholders.
We are also subject to Section 203 of the General Corporation Law of the State of Delaware, which provides, subject to certain exceptions, that if a person acquires 15% of our voting stock, the person is an “interested stockholder” and may not engage in “business combinations” with us for a period of three years from the time the person acquired 15% or more or our voting stock. The application of Section 203 may, in some circumstances, deter or prevent a change in control of our company even when such change may be beneficial to our stockholders.
Our amended and restated bylaws designate exclusive forums for the adjudication of certain disputes, which could limit our stockholders’ ability to bring claims in a judicial forum it finds favorable for disputes with us or our directors, officers, or employees.
Our amended and restated bylaws provide that, a state or federal court located withinunless we consent in writing to the selection of an alternative forum, the Court of Chancery of the State of Delaware isor, if such court does not have subject matter jurisdiction, the federal district court of the State of Delaware, will be the sole and exclusive forum for:
•any derivative action or proceeding brought on our behalf;
•any action asserting a claim of breach of a fiduciary duty owed by any director, officer or other employee or stockholder of Sangamo to us or our stockholders;
•any action asserting a claim arising pursuant to any provision of the General Corporation Law of the State of Delaware, our charter or our bylaws, as to which the General Corporation Law of the State of Delaware confers jurisdiction on the Court of Chancery of the State of Delaware; and
•any action asserting a claim governed by the internal affairs doctrine.
Our amended and restated bylaws further provide that a federal district court of the United State is the sole and exclusive forum for any complaint asserting a cause of action arising under the Securities Act of 1933, as amended. These provisions further provide that any person or entity that acquires any interest in shares of our capital stock will be deemed to have notice of and consented to these provisions.
These provisions may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, officers, or other employees, which may discourage lawsuits against us and our directors, officers, and other employees. If a court were to find any of these provisions to be inapplicable or unenforceable in an action, we may incur additional costs associated with resolving the dispute in other jurisdictions, which could seriously harm our business.
ITEM 1B – UNRESOLVED STAFF COMMENTS
None.
ITEM 1C – CYBERSECURITY
Risk Management and Strategy
We have implemented and maintain various information security processes designed to identify, assess and manage material risks from cybersecurity threats to our critical computer networks, third party hosted services, communications systems, hardware and software, and our critical data, including intellectual property, confidential information that is proprietary, strategic or competitive in nature (“Information Systems and Data”).
Our cybersecurity risk management program includes a risk assessment methodology designed to escalate cybersecurity risks to the appropriate channels within our organization in order to help identify material cybersecurity risks to our critical systems, information, products, services and our broader enterprise IT environment. The information technology and legal departments help identify, assess and manage Sangamo’s cybersecurity threats and risks. The information technology department, in coordination with the legal department, identifies and assesses risks from cybersecurity threats by monitoring and evaluating our threat environment and Sangamo’s risk profile using various methods including, for example, evaluating threats reported to us, conducting audits, performing threat assessments, and conducting vulnerability assessments to identify vulnerabilities.
Depending on the environment, we implement and maintain various technical, physical, and organizational measures, processes, standards and policies designed to manage and mitigate material risks from cybersecurity threats to our Information Systems and Data, including, for example: an incident response plan that includes procedures for responding to cybersecurity incidents and escalating cybersecurity incidents to cross-functional teams, management and our board of directors, business continuity plans, encryption of data, network security controls, systems monitoring, employee training, and cybersecurity insurance.
Our assessment and management of material risks from cybersecurity threats are integrated into our risk management protocols. Our cybersecurity risk management program shares common methodologies, reporting channels and governance processes that apply across the enterprise risk management program to other legal, compliance, strategic, operational and financial risk areas, including the involvement of cross-functional teams and, depending on the nature and severity of an incident, an escalation path to notify our executive and senior management teams and our board of directors. For example, the
information technology department works with management to prioritize our risk management processes and mitigate cybersecurity threats that are more likely to lead to a material impact to our business.
We use third-party service providers to assist us from time to time to identify, assess, and manage material risks from cybersecurity threats, including for example: professional service firms, including legal counsel, cybersecurity software providers and managed cybersecurity service providers.
We use third-party service providers to perform a variety of functions throughout our business, such as application providers, hosting companies, contract research organizations, contract manufacturing organizations and supply chain resources. Depending on the nature of the services provided, the sensitivity of the Information Systems and Data at issue, and the identity of the provider, our vendor management process may involve different levels of assessment designed to help identify cybersecurity risks associated with a provider and impose contractual obligations related to cybersecurity on the provider.
For a description of the risks from cybersecurity threats that may materially affect us and how they may do so, see our risk factors included in Part I, Item 1A. “Risk Factors” of this Annual Report on Form 10-K, including “Risk Factors—If our information technology systems or data, or those of third parties upon which we rely, are or were compromised, we could experience adverse consequences resulting from such compromise, including but not limited to regulatory investigations or actions, litigation, fines and penalties, disruptions of our business operations, reputational harm and other adverse consequences.”
Governance
Our board of directors addresses Sangamo’s cybersecurity risk management as part of its general oversight function. The board of directors’ audit committee is responsible for overseeing Company’s cybersecurity risk management processes, including oversight of mitigation of risks from cybersecurity threats.
Our cybersecurity risk assessment and management processes are implemented and maintained by certain Company management, including our Vice President, Information Technology, who has over 20 years of technology experience which includes extensive cybersecurity implementation and oversight.
Our Vice President, Information Technology, is responsible for hiring appropriate personnel, helping to integrate cybersecurity risk considerations into our overall risk management strategy, and communicating key priorities to relevant personnel. Our Vice President, Information Technology is responsible for helping prepare for cybersecurity incidents, approving cybersecurity processes, and reviewing security assessments and other security-related reports. Our Chief Financial Officer is responsible for approving budgets related to the foregoing.
Our cybersecurity incident response and vulnerability management protocol is designed to escalate certain cybersecurity incidents to members of management depending on the circumstances, including the executive leadership team. The executive leadership team works with the incident response team to help us mitigate and remediate cybersecurity incidents of which they are notified. In addition, our incident response and vulnerability management protocol includes reporting to the Audit Committee of the Board of Directors for certain cybersecurity incidents.
The Audit Committee of our Board of Directors receives periodic reports from the Vice President, Information Technology, concerning the Sangamo’s significant cybersecurity threats and risk and the processes we have implemented to address them. The Board of Directors also has access to various reports, summaries or presentations related to cybersecurity threats, risk and mitigation.
ITEM 2 – PROPERTIES
Our corporate headquarters occupies approximately 87,70059,485 square feet of research and office space, pursuant to a lease that expires in August 2031, and approximately 7,700 of office space, pursuant to a lease that expires in August 2026, in Richmond, California. We also lease approximately 103,089 square feet of office and research and development laboratory facilities in Brisbane, California, pursuant to a lease that expires in May 2029. We also lease approximately 54,20028,070 square feet of researchoffice and office space in Richmond, California, subject to leases that expire in August 2026. We also lease approximately 20,800 square feet of office, research and development space in Valbonne, France, subjectpursuant to leases that expire beginning in June 2025 through March 2028.January 2030. We believe that our facilities are currently adequate to meet our needs. As we continueWe expect to expandclose our operations, we may need to lease or purchase additional facilities.facilities in Brisbane, California and Valbonne, France in 2024.
ITEM 3 – LEGAL PROCEEDINGS
We are not a party to any material pending legal proceeding. From time to time, we may be involved in legal proceedings arising in the ordinary course of business.
ITEM 4 – MINE SAFETY DISCLOSURES
Not Applicable.
PART II
ITEM 5 – MARKET FOR THE REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information
Our common stock trades on the Nasdaq Global Select Market under the symbol “SGMO.”
Holders
As of February 19, 2021,March 8, 2024, there were 53 holders of record of our common stock. This number does not include “street name,” or beneficial holders, whose shares are held of record by banks, brokers, financial institutions and other nominees.
Dividends
We have not paid dividends on our common stock, and currently do not plan to pay any cash dividends in the foreseeable future.
Stock Performance Graph
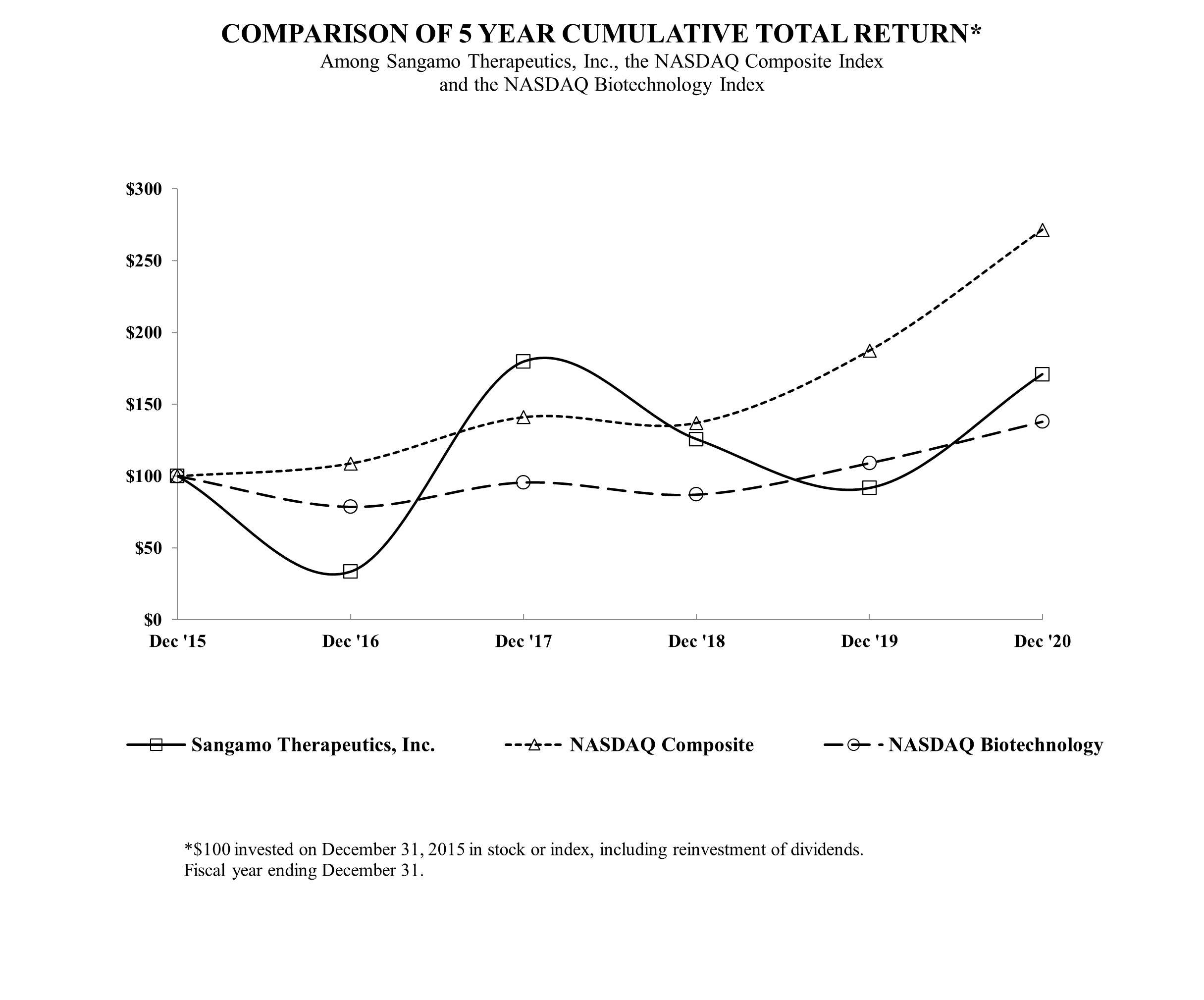
The above Stock Performance Graph and related information shall not be deemed “soliciting material” or to be “filed” with the SEC nor shall such information be incorporated by reference into any future filing under the Securities Act or the Exchange Act, each as amended, except to the extent that the Companywe specifically incorporatesincorporate it by reference into such filing.
ITEM 6 – SELECTED FINANCIAL DATA[RESERVED]
The following Selected Financial Data should be read in conjunction with “Item 7 – Management’s Discussion and Analysis of Financial Condition and Results of Operations” and “Item 8 – Financial Statements and Supplementary Data” included elsewhere in this Annual Report on Form 10-K. Historical results areresponsive to Item 6 have not necessarily indicative of future results.
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2020 | | 2019 | | 2018 | | 2017 | | 2016 |
| (In thousands, except per share data) |
| Consolidated Statement of Operations Data: | | | | | | | | | |
| Revenues | $ | 118,192 | | | $ | 102,428 | | | $ | 84,452 | | | $ | 36,567 | | | $ | 19,389 | |
| Operating expenses: | | | | | | | | | |
| Research and development | 180,647 | | | 145,922 | | | 114,866 | | | 65,728 | | | 65,618 | |
| General and administrative | 67,097 | | | 61,686 | | | 46,736 | | | 27,200 | | | 26,330 | |
| Total operating expenses | 247,744 | | | 207,608 | | | 161,602 | | | 92,928 | | | 91,948 | |
| Loss from operations | (129,552) | | | (105,180) | | | (77,150) | | | (56,361) | | | (72,559) | |
| Interest and other income, net | 8,775 | | | 9,761 | | | 8,261 | | | 1,793 | | | 887 | |
| Loss before income taxes | (120,777) | | | (95,419) | | | (68,889) | | | (54,568) | | | (71,672) | |
| Income tax expense (benefit) | 345 | | | — | | | — | | | — | | | (14) | |
| Net loss | (121,122) | | | (95,419) | | | (68,889) | | | (54,568) | | | (71,658) | |
| Net loss attributable to non-controlling interest | (126) | | | (233) | | | (555) | | | — | | | — | |
| Net loss attributable to Sangamo Therapeutics, Inc. stockholders | $ | (120,996) | | | $ | (95,186) | | | $ | (68,334) | | | $ | (54,568) | | | $ | (71,658) | |
| Basic and diluted net loss per share attributable to Sangamo Therapeutics, Inc. stockholders | $ | (0.90) | | | $ | (0.85) | | | $ | (0.70) | | | $ | (0.70) | | | $ | (1.02) | |
| Shares used in computing basic and diluted net loss per share attributable to Sangamo Therapeutics, Inc. stockholders | 134,449 | | | 112,114 | | | 96,941 | | | 78,084 | | | 70,553 | |
| | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| As of December 31, |
| 2020 | | 2019 | | 2018 | | 2017 | | 2016 |
| (In thousands) |
| Consolidated Balance Sheet Data: | | | | | | | | | |
| Cash, cash equivalents, marketable securities, and interest receivable | $ | 692,988 | | | $ | 384,988 | | | $ | 400,508 | | | $ | 244,560 | | | $ | 142,759 | |
| Working capital | $ | 516,121 | | | $ | 335,601 | | | $ | 332,010 | | | $ | 203,538 | | | $ | 136,289 | |
| Total assets | $ | 938,550 | | | $ | 637,516 | | | $ | 590,395 | | | $ | 286,741 | | | $ | 157,891 | |
Long-term portion of lease liabilities (¹) | $ | 38,396 | | | $ | 41,192 | | | $ | 27,689 | | | $ | 24,738 | | | $ | 3,945 | |
| Accumulated deficit | $ | (777,981) | | | $ | (656,985) | | | $ | (562,696) | | | $ | (495,479) | | | $ | (440,911) | |
| Total stockholders’ equity | $ | 497,366 | | | $ | 432,739 | | | $ | 367,257 | | | $ | 187,900 | | | $ | 136,195 | |
(1) In 2019, we adopted Accounting Standards Update No. 2016-02 (Topic 842) “Leases,” which requires lessees to recognize right-of-use assets and lease liabilities for operating leases with a lease term greater than one year. We adopted Topic 842 using the modified retrospective method. As such, results for reporting periods beginning after January 1, 2019 arebeen presented under Topic 842, while prior period amounts are not adjusted and continue to be reported in accordance with our historical accounting under Topic 840 “Leases.”
(2) Sangamo France was acquiredamendments to Item 301 of Regulation S-K contained in October 2018 and the results of operations have been included in the selected consolidated financial data since the date of acquisition (see Note 5 – Acquisition of Sangamo France in our Consolidated Financial Statements).
SEC Release No. 33-10890.
ITEM 7 – MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The discussion in “Management’s Discussion and Analysis of Financial Condition and Results of Operations” contains trend analysis, estimates and other forward-looking statements within the meaning of Section 27A of the Securities Act, as amended, and Section 21E of the Exchange Act, as amended. These forward-looking statements include, without limitation, statements containing the words “anticipates,” “believes,” “continues,” “could,” “estimates,” “expects,” “intends,” “may,” “plans,
“plans,” “seeks,” “should,” “will,” and other words of similar import or the negative of those terms or expressions. Such forward-looking statements are subject to known and unknown risks, uncertainties, estimates and other factors that may cause our actual results, performance or achievements, or industry results, to be materially different from any future results, performance or achievements expressed or implied by such forward-looking statements. Actual results could differ materially from those set forth in such forward-looking statements as a result of, but not limited to, the “Risk Factors” described in Part I, Item 1A of this Annual Report on Form 10-K. You should read the following discussion and analysis along with the “Selected Financial Data” and the Consolidated Financial Statements and notes attached to those statements included elsewhere in this report.
In addition, the “Results of Operations” section of this “Management’s Discussion and Analysis of Financial Condition and Results of Operations” generally discusses 20202023 and 20192022 items and year-to-year comparisons between 20202023 and 2019.2022. Discussions of 20182021 items and year-to-year comparisons between 20192022 and 20182021 are not included in this Annual Report on Form 10-K and can be found in “Management’s Discussion and Analysis of Financial Condition and Results of Operations” in Part II, Item 7 of our Annual Report on Form 10-K for the fiscal year ended December 31, 2019,2022, filed with the SEC on February 28, 2020.23, 2023. Overview
We are a clinical-stage genomic medicine company committed to translating ground-breaking science into medicines that transform the lives of patients and families afflicted with serious neurological diseases. We plan to deliver on this mission through (i) development of our clinical and preclinical product candidates, (ii) our novel science and (iii) our in-house manufacturing capabilities.
Our current clinical-stage product candidates are:
•Giroctocogene fitelparvovec, also known as SB-525, our lead product candidate, is a gene therapy for the treatment of hemophilia A and is currently being evaluated in the registrational Phase 3 AFFINE (efficAcy and saFety Factor vIii geNe thErapy) clinical trial. We are developing giroctocogene fitelparvovec with our collaborator Pfizer Inc., or Pfizer;
•ST-920, our wholly-owned gene therapy product candidate for the treatment of Fabry disease, is currently being evaluated in our Phase 1/2 STAAR clinical study;
•BIVV003, our cell therapy product candidate for the treatment of sickle cell disease, or SCD, is currently being evaluated in our Phase 1/2 PRECIZN-1 clinical study. We are developing BIVV003 with our collaborator Sanofi S.A., or Sanofi; and
•ST-400, our cell therapy product candidate for the treatment of transfusion dependent beta thalassemia, is currently being evaluated in our Phase 1/2 Thales clinical study. We are developing ST-400 with our collaborator Sanofi.
In addition, we expect to initiate clinical studies for two additional product candidates in 2021:
•TX200, our wholly-owned Chimeric Antigen Receptor, or CAR, engineered regulatory T cell, or CAR-Treg, cell therapy product candidate for the treatment of HLA-A2 mismatched kidney transplant rejection; and
•KITE-037, our allogeneic anti-CD19 CAR-T cell therapy product candidate for the treatment of cancer. We are developing KITE-037 with our collaborator Kite Pharma Inc., or Kite, a wholly-owned subsidiary of Gilead Sciences, Inc.
Moreover, we are focusing our preclinical development in emerging areas for us including CAR-Treg cell therapies for autoimmune disorders and genome engineering for neurological diseases. Indications for our other preclinical programs include neurodevelopmental disorders, cancer, inflammatory bowel disease, or IBD, tauopathies such as Alzheimer’s and neurodegenerative diseases such as amyotrophic lateral sclerosis, or ALS, multiple sclerosis, or MS, and Huntington’s disease, some of which we are developing with our collaborators Biogen MA, Inc. and Biogen International GmbH, which we refer to together as Biogen, Kite, Novartis Institutes for BioMedical Research, Inc., or Novartis, Pfizer and Takeda Pharmaceutical Company Limited, or Takeda.
Our multiple collaborations with biopharmaceutical companies bring us important financial and strategic benefits and reinforce the potential of our research and development efforts andbelieve our zinc finger protein,epigenetic regulators are ideally suited to potentially address devastating neurology disorders and our capsid engineering platform has demonstrated the ability to expand delivery beyond currently available intrathecal delivery capsids, including in the central nervous system, or ZFP, technology platform. They leverage our collaborators’ therapeutic and clinical expertise and commercial resources with the goal to bring our medicines more rapidly to patients. We believe these collaborations reflect of the value of our ZFP technology platform and will potentially expand the addressable markets of our product candidates. To date, we have received approximately $815.0 millionCNS, in upfront licensing fees, milestone payments, and proceeds from sale of our common stock to collaborators and have the right to earn up to $7.0 billion in future milestone payments from our collaborations, in addition to potential product royalties.
preclinical studies. For additional information regarding our business, see “Business” in Part I, Item 1 of this Annual Report on
Form 10-K.
Recent Business Highlights
Corporate Updates
France Restructuring
Financial Position
Based on our current operating plan, our cash, cash equivalents and marketable securities as of December 31, 2023 are expected to allow us to meet our liquidity requirements only into the third quarter of 2024. Our ability to continue to operate as a going concern is dependent upon our ability to raise substantial additional capital to fund our operations and support our research and development endeavors, including to progress our preclinical and clinical programs as described in this Annual Report on Form 10-K. We have been actively seeking, and continue to actively seek, substantial additional capital, including through public or private equity or debt financing, royalty financing or other sources, such as strategic collaborations and other direct investments in our programs. However, we have been unsuccessful in securing any such additional capital to date. If we are unable to secure additional funding in the very near term, we will likely seek protection under the U.S. Bankruptcy Code. We have explored, and continue to explore, whether filing for bankruptcy protection is in the best interest of our Company and our stakeholders.
Core Preclinical Neurology Programs and Technology
Novel AAV Capsid Delivery Technology
•In December 2020,March 2024, we and Pfizer jointly announced updated follow-upthe first data from the Phase 1/2 Alta study of giroctocogene fitelparvovec, an investigational gene therapy for the treatment of severe hemophilia A.
•In October 2020, weour novel proprietary neurotropic adeno-associated virus, or AAV, capsid demonstrating industry-leading blood-brain barrier, or BBB, penetration and Pfizer jointly announced that we dosed the first participantbrain transduction in the registrational Phase 3 AFFINE clinical trial of giroctocogene fitelparvovec.
•In August and September 2020, we dosed the first two patients in our Phase 1/2 STAAR study evaluating our ST-920 gene therapy product candidate to treat Fabry disease, a rare inherited metabolic disease. In February 2021, we dosed the first patient in the second cohort.
•In July 2020, we entered into a collaboration agreement with Novartis to develop and commercialize gene regulation therapies to treat three neurodevelopmental targets, including genes linked to autism spectrum disorder and intellectual disability.
•In February 2020, we entered into a collaboration agreement with Biogen to develop gene regulation therapies for Alzheimer’s, Parkinson’s, neuromuscular and other neurological diseases.
•In 2020, we brought in-house AAV manufacturing capabilities online in our Brisbane, California headquarters.
Estimated Impacts of Evolving COVID-19 Pandemic
In March 2020, the World Health Organization declared the novel coronavirus disease,non-human primates, or COVID-19, outbreak a global pandemic, and since such time, actions taken around the world to help mitigate the spread of COVID-19 have included varying restrictions on travel, quarantines in certain areas, and forced closures for certain types of public places and businesses, including in the three countries where we have most of our day-to-day operations, the United States, France and the United Kingdom. Our business has been directly impacted by pandemic restrictions aimed at reducing the spread of the disease, including multiple California executive orders, several semi-coordinated San Francisco Bay Area orders, several other state and additional local orders across the country and similar orders outside the United States, which, among other things, direct individuals to shelter at their places of residence, direct businesses and governmental agencies to cease non-essential operations at physical locations, prohibit certain non-essential gatherings, and order cessation of non-essential travel.
To comply with these orders, we implemented an operating plan to continue business operations during the ongoing COVID-19 pandemic, including enhanced workplace safety protocols and modified working schedules in our laboratories. Employees who are able to fulfill their job duties working from home are required to work from home, and have been doing so since March 2020. These protocols and modifications have slowed our productivity and disrupted our business to a moderate degree and are likely to continue doing so through 2021. For example, we have experienced periodic short-term disruptions to our onsite laboratory and manufacturing operations while addressing positive cases of COVID-19 by onsite workers, and our laboratory and manufacturing operations could experience longer term disruptions in the future in the event of a significant outbreak of COVID-19 among our onsite workers. Moreover, from time to time, we have been required to reorganize and prioritize our research resources to mitigate moderate COVID-19 impacts arising from travel restrictions, laboratory density restrictions and laboratory supply constraints. If our research programs encounter longer-term disruptions, it could impact our ability to support our biopharmaceutical partners as contemplated in our collaboration agreements and could result in adjustments to our research timelines, although we do not believe that the short-term disruptions to date have resulted in any such impacts.
Additionally, our Phase 1/2 STAAR clinical study evaluating ST-920, our wholly-owned gene therapy product candidate for the treatment of Fabry disease, has experienced delays in its timeline due to COVID-19 impacts and the diversion of healthcare resources to fight the pandemic. For example, the clinical trial sites in the UK for this study have not been able to open due to the significant prevalence of COVID-19 in the UK. Additionally, we have experienced delays in recruiting, enrolling and dosing patients for this study at our US trial sites, due in some part to the understandable hesitation of patients to travel by plane to trial sites not within driving distance and to enter medical facilities during the pandemic and in other part to
trial sites prioritizing COVID-19 clinical care over research•The novel STAC-BBB capsid demonstrated robust penetration of BBB in NHPs with 700-fold higher transgene expression in neurons compared to the benchmark capsid AAV9 and outperformed all other known published capsid variants evaluated in our study.
•The STAC-BBB capsid variant mediated robust expression of zinc finger cargo in neurons of NHPs, with potent and widespread repression of prion and tau genes observed across key brain regions, demonstrating the potential for modification of disease progression in prion disease and various tauopathies in human patients.
•Visualization of gene expression in individual brain cells by RNAscope revealed highly potent repression of tau in neurons expressing the zinc finger cargo, across multiple brain regions.
•We believe the capsid biodistribution profile of STAC-BBB is optimal for the treatment of neurological diseases with AAV-based treatments, highlighted by the observed enrichment in the CNS and de-targeting from the liver, dorsal root ganglia, or DRG, and other peripheral organs.
•STAC-BBB was generally well tolerated in NHPs, with no notable treatment related pathological findings in brain, spinal cord and peripheral tissues.
•We believe STAC-BBB is potentially manufacturable at commercial scale using standard cell culture and purification processes, is soluble using known excipients, and can be characterized using available analytics.
Chronic Neuropathic Pain – Nav1.7
•Since our last update in November 2023, investigational new drug application, or IND, enabling Toxicology studies are nearing completion in our Nav1.7 program to treat chronic neuropathic pain.
•An IND submission is expected for this program in the fourth quarter of 2024, subject to our ability to secure adequate funding.
Prion Disease
•CTA-enabling activities suchcontinue to advance for our epigenetic regulation program in prion disease.
•We demonstrated that our prion-targeted zinc finger repressor, delivered via an intravenous administration of our novel STAC-BBB capsid, resulted in a dose-dependent repression of prion genes in NHPs.
•A CTA submission is expected for this program in the fourth quarter of 2025, subject to our ability to secure adequate funding.
Tauopathies
•We intend to resume development of our previously paused tau program, developing epigenetic regulation therapies addressing tauopathies, leveraging our newly identified STAC-BBB capsid variant.
•We demonstrated that our tau clinical-lead zinc finger repressor, delivered via an intravenous administration of the STAC-BBB novel capsid, resulted in a dose-dependent repression of tau genes in NHPs. Visualization of gene expression in individual brain cells by RNAscope revealed highly potent repression of tau in neurons expressing the zinc finger cargo across multiple brain regions.
•The IND submission for this program could occur as early as the fourth quarter of 2025, subject to our ability to secure adequate funding.
Clinical Programs
Fabry Disease
•Since our last update in November 2023, an additional seven patients have been dosed in the Phase 1/2 STAAR study. Moreover,study of isaralgagene civaparvovec, our investigational gene therapy for the treatment of Fabry disease, resulting in a total of 32 patients dosed to date. One additional patient has been withdrawn from Enzyme Replacement Therapy, or ERT, resulting in a total of 13 patients withdrawn from ERT to date. All patients withdrawn from ERT remain off ERT as of March 12, 2024.
•On February 5, 2024, we announced updated preliminary clinical data from our Phase 1/2 STAAR study in advance of our presentation at the 20th Annual WORLDSymposium on February 7, 2024. The data showed that as of the September 19, 2023 data cutoff date, sustained elevated expression of α-Gal A activity was maintained for up to three years for the longest-treated patient. All 12 patients withdrawn from ERT remained off ERT, with sustained elevated expression of α-Gal A activity observed for up to 19 months as of the September 19, 2023 data cutoff date. Total antibody or neutralizing antibody titers against α-Gal A decreased markedly in all seven patients with antibodies
associated with ERT at baseline, and became undetectable in five. In the 13 patients followed for 12-months or more after treatment, renal function remained stable, and significant improvements in overall disease severity, quality of life, and gastrointestinal symptoms compared to baseline were reported. ST-920 continued to demonstrate a favorable safety profile, with no liver function test elevations requiring steroids post-treatment.
•Screening and enrollment are complete, and we expect to complete the dosing of remaining enrolled patients in the Phase 1/2 study in the first half of 2024.
•In February 2024, we aligned with the U.S. Food and Drug Administration, or FDA, on an abbreviated pathway to potential approval of isaralgagene civaparvovec. The FDA agreed in a Type D meeting that data from a single, adequate, and well-controlled study may form the primary basis of approval of a Biologics License Application, or BLA, for isaralgagene civaparvovec. The proposed study would enroll up to 25 patients, both male and female, without the need for a control arm. A head-to-head comparison with ERT is not part of the proposed study design deemed acceptable by the FDA. This approach enables a potentially more rapid, efficient and cost-effective pathway to BLA submission than originally anticipated.
•In addition, the European Medicine Agency, or EMA, granted priority medicines, or PRIME, eligibility to isaralgagene civaparvovec. PRIME is a scheme designed to enhance support for the development of medicines that target an unmet medical need and is intended to optimize development plans and expedite the review and approval process so that these medicines may reach patients as early as possible.
•The U.K. Medicines and Healthcare products Regulatory Agency also granted Innovative Licensing and Access Pathway to isaralgagene civaparvovec, which aims to accelerate time to market and facilitate access to medicines. Isaralgagene civaparvovec has already received Orphan Medicinal Product designation from the EMA as well as Orphan Drug, Fast Track and regenerative medicine advanced therapy, or RMAT, designations from the U.S. FDA.
•We are actively seeking a potential collaboration partner for our Fabry disease program. We are deferring additional investments in planning for a registrational trial until a collaboration partnership or financing for this program is secured.
Renal Transplant Rejection
•Since our last update in November 2023, we have experienced some short-term delaysdosed two additional patients in sourcing the necessary raw materials to manufacture supplies for the STAAR study due to COVID-19 impacts. We estimate that these challenges have set back our STAAR study timelines three to six months. While we currently still expect to share initial clinical study data by the end of 2021, this timeline could be revised if COVID-19 impacts to our enrollment and dosing of patients and to our sourcing of raw materials for this study intensify because of vaccination delays, new COVID-19 variants or unexpected events.
In addition, ourPhase 1/2 STEADFAST clinical study evaluating TX200, our wholly-owned autologous CAR-Treg cell therapy candidate for renal transplant rejection, to achieve a total of six patients dosed. In 2023, we received all necessary regulatory and ethics approvals for an accelerated dose escalation protocol from European regulatory authorities that allows dosing to potentially advance more quickly through the cohorts, and which also allows for a new and highest fourth dose cohort, compared to the three cohorts in the previously approved study protocol. The new, fourth cohort dose is 18-fold higher than the first cohort starting dose. As a result, in total three patients have been dosed at dose level 1, one patient at dose level 2, one patient at dose level 3 and the first patient at the new, and highest, dose level 4. The product candidate continues to be generally well tolerated in all six patients dosed to date.
•We expect to dose up to an additional two patients in the Phase 1/2 STEADFAST study and expect to complete dosing in the study in the first half of 2024. We plan to continue seeking a potential collaboration partner or external investment in our autologous CAR-Treg cell therapy programs.
Partnered Program
Hemophilia A
•The Phase 3 AFFINE trial of giroctocogene fitelparvovec, an investigational gene therapy that we are developing with Pfizer Inc., or Pfizer, for patients with moderately severe to severe hemophilia A, continues to progress. Dosing of all patients in the treatmenttrial is now complete.
•A pivotal readout is expected in the middle of kidney transplant rejection, has experienced delays2024, with Pfizer anticipating submitting a BLA and a marketing authorization application, or MAA, in its commencement timelineearly 2025 if the pivotal readout is supportive.
•We and Pfizer presented updated data from the Phase 1/2 ALTA study of giroctocogene fitelparvovec via an oral presentation at the 65th American Society for Hematology Annual Meeting and Exposition on December 11, 2023. These data are described in detail in “Overview” in Part I, Item 1 of this Annual Report on Form 10-K
•We are eligible to earn from Pfizer up to $220.0 million in milestone payments upon the achievement of certain regulatory and commercial milestones for giroctocogene fitelparvovec and product royalties of 14% - 20% if giroctocogene fitelparvovec is approved and commercialized, subject to reduction due to COVID-19 impacts relatedpatent expiration, entry of biosimilar products to manufacturingthe market and technology transfer challenges with our CMOs. We estimate that these challenges have set back our commencement timeline by approximately three months. While we currently still expect to initiate this clinical study by the endpayment made under certain licenses for third-party intellectual property.
With respect to our partnered programs, the timelines for the studies and trials managed by our collaborators are also subject to potential delay in the future if these studies and trials experience similar challenges that we have experienced in our STAAR and STEADFAST studies.
Going forward, we will continue to monitor the impact of COVID-19Concern
Based on our operations, research commitments and clinical trials and those ofcurrent operating plan, our collaborators, clinical trial sites and CMOs. The magnitude of these impacts will depend, in part, on the length and severity of the COVID-19 pandemic and related government orders and restrictions, and how the pandemic limits the ability of us and our business partners to operate business in the ordinary course. Disruptions to these operations, and possibly more severe disruptions in the future that could arise due to the extension of government orders or new government orders applicable in the places we operate or our industry generally or to us and our facilities specifically, could impede our ability to conduct research in a timely manner, comply with our research obligations to our collaborators and advance the development of our therapeutic programs. These delays and disruptions could result in adverse material impacts to our business, operating results and financial condition.
We do not anticipate any material negative impact on our financial condition in 2021 as a result of the COVID-19 pandemic. We believe we are well positioned financially in the near term to execute on our wholly-owned and partnered research and clinical programs. We ended 2020 with $692.0 million in cash, cash equivalents and marketable securities as of December 31, 2023 are expected to allow us to meet our liquidity requirements only into the third quarter of 2024. Our history of significant losses, negative cash flows from operations, limited liquidity resources currently on hand and have raised an additional $15.7 million in cash through February 19, 2021 through sales of our common stock under our at-the-market offering program. Although we believe we are well capitalized currently, the effects of the evolving pandemic could result in disruption of global financial markets, impairingdependence on our ability to access capital, which couldobtain additional financing to fund our operations have resulted in the future negatively affectmanagement’s assessment that there is substantial doubt about our liquidity. We do not currently anticipate any material impairmentsability to the valuation of the financial assets or goodwill on our balance sheetcontinue as a result of COVID-19. We do not believe thatgoing concern for at least the remote workplace arrangements we have implemented for our office-based employees have affected our financial reporting or control systems.
The extent to whichnext 12 months from the COVID-19 pandemic will impact our business, operations and financial condition, either directly or indirectly, will depend on future developments that remain highly uncertain atdate the present time. These developments include the ultimate duration and severity of the pandemic, the impacts of new COVID-19 variants, travel restrictions, quarantines and social distancing requirements in the United States, France, United Kingdom and other countries, business closures or business disruptions and the effectiveness and timeliness of actions taken in the United States, France, United Kingdom and other countries to contain and treat the disease, including the effectiveness and timing of vaccination programs. Although certain government orders and restrictions have eased, and phased re-openings are underway, it is not certain when such restrictions and orders will be fully lifted, and recent resurgences in number and rates of infections and the surge of new variants of the virus may result in the return of prior orders and restrictions or new quarantine and shelter-in-place orders or other restrictions. As our understanding of events evolves and additional information becomes available, we may materially change our guidance relating to our revenues, expenses and timelines for manufacturing, clinical trials and research and development.
See also the section titled “Risk Factors”Consolidated Financial Statements included in Part I, Item 1A of this Annual Report on Form 10-K are issued. Our ability to continue to operate as a going concern is dependent upon our ability to raise substantial additional capital to fund our operations and support our research and development endeavors, including to progress our preclinical and clinical programs as described in this Annual Report on Form 10-K. In this regard, we have been actively seeking, and continue to actively seek, substantial additional capital, including through public or private equity or debt financing, royalty financing or other sources, such as strategic collaborations and other direct investments in our programs. We may be unable to attract new investments as a result of the speculative nature of our newly reprioritized core neurology preclinical programs. We have been unsuccessful in securing such additional capital to date. If we are unable to secure additional funding in the very near term, we will likely seek protection under the U.S. Bankruptcy Code. We have explored, and continue to explore, whether filing for bankruptcy protection is in the best interest of our Company and our stakeholders. Additional capital may not be available on acceptable terms or at all. If adequate funds are not available to us on a timely basis, or at all, we will be required to take additional informationactions to address our liquidity needs, including additional cost reduction measures such as further reducing operating expenses and delaying, reducing the scope of, discontinuing or altering our research and development activities, which would have a material adverse effect on risksour business and uncertainties relatedprospects, or we may be required to cease operations entirely, liquidate all or a portion of our assets, and/or seek protection under the evolving COVID-19 pandemic.
Certain Components of Results of Operations
Our revenues have consisted primarily of revenues from ongoing collaboration agreements, which includes upfront licensing fees, reimbursements for research services, milestonesand milestone achievements, and research grant funding. In 2022, our collaboration agreement with Sanofi S.A., or Sanofi, terminated. In 2023, our collaboration agreements with Biogen MA, Inc. and Biogen International GmbH, which we refer to together as Biogen, and Novartis Institutes for BioMedical Research, Inc., or Novartis, were terminated, and the Kite Pharma, Inc., a Gilead Sciences, Inc. subsidiary, or Kite, collaboration agreement will expire pursuant to its terms on April 4, 2024. We expect revenues to continue to fluctuate from period to period and there can be no assurance that new collaborations or partner reimbursements will continue beyond their initial terms or that we are able to meet the milestones specified in these agreements.
Major Customers, Partnerships and Strategic Alliances in the accompanying notes to the Consolidated Financial Statements included in Part II, Item 8, “Financial Statements and Supplementary Data” of this Annual Report on Form 10-K. We have incurred net losses since inception and expect to incur losses for at least the next several years as we continue our research and development activities. To date, we have funded our operations primarily through the issuance of equity securities and revenues from collaborations and research grants.
WeAlthough we expect research and development expenses to decrease in the near-term in connection with the restructuring of operations and reduction in workforce and significant reduction in our internal manufacturing and allogeneic research footprints in California announced in April, or the April Restructuring, the further restructuring of operations and corresponding reduction in workforce announced in November, or the November Restructuring, and the France Restructuring, we expect to continue to devote substantial resources to research and development in the future and expect research and development expenses to increase in the next several years if we are successful in advancing our product candidates from research stage through clinical trials. Pursuant to the terms of our agreementstermination and transition agreement with Biogen, Kite, Novartis, Pfizer and Sanofi certain expenses related to research and development activities willmay be reimbursed to us. TheAny reimbursement funds to be received from Biogen, Kite, Novartis, Pfizer and Sanofi will be recognized as revenue as the related costs are incurreddecrease our research and collection is reasonably assured.development expense.
General and administrative expenses consist primarily of salaries and personnel related expenses for executive, finance and administrative personnel, stock-based compensation expenses,expense, professional fees, allocated facilities and information technology expenses, patent prosecution expenses and other general corporate expenses. AsAlthough we continueexpect general and administrative expenses to advance our product candidates intodecrease in the near-term in connection with the April, November, and through the clinic,France Restructurings, we expect the growth of our business to require increased general and administrative expenses.expenses as we continue to advance our product candidates into and through the clinic.
Critical Accounting Policies and Estimates
The accompanying discussion and analysis of our financial condition and results of operations are based upon ourOur Consolidated Financial Statements and the related disclosures which have been prepared in accordance with generally accepted accounting principles generally accepted in the United States. The preparation of these Consolidated Financial Statements requires us to
make estimates, assumptions and judgments that affect the reported amounts in our Consolidated Financial Statements and accompanying notes. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions. We believe the following policies to be the most critical to an understanding of our financial condition and results of operations because they require us to make estimates, assumptions and judgments about matters that are inherently uncertain.
We believe our critical accounting policies and estimates relating to revenue recognition including the estimated fair value of common shares issued as part of the transaction price and valuation of long-liveslong-lived assets including goodwill and intangible assets are the moremost significant estimates and assumptions used in the preparation of our Consolidated Financial Statements.
For a complete description of our significant accounting policies, see Note 1 – Organization, Basis of Presentation and Summary of Significant Accounting Policies in the accompanying notes to the Consolidated Financial Statements included in Part II, Item 8, "Financial“Financial Statements and Supplementary Data"Data” of this Annual Report on Form 10-K.
Revenue Recognition
Our revenues are primarily derived from collaboration arrangements which primarily include licensing intellectual property and providing research and development services. We recognize revenuesrevenue when our customers obtain control of promised goods or services in a contract for an amount that reflects the consideration we expect to receive in exchange for those goods or services.
For most of our arrangements, the licenses granted to our intellectual property are not distinct from providing related research and development services and such combined performance obligations are satisfied over time. Such agreements may also contain options for additional goods and services that are considered to be material rights. For these agreements, we are required to estimate a transaction price and then allocate such transaction price based on the estimated standalone selling price of each distinct performance obligation. Most of our performance obligations are delivered over time. We generally as services are provided while revenues from non-refundable upfront fees are recognized overrecognize revenue using measure of progress based on an input method (e.g., cumulative actual level of effort, which includes the value of actual time eitherincurred by measuring progress towardsour researchers plus third-party cost reimbursements, relative to the total estimated level of effort to be incurred, or cumulative actual hours incurred relative to total estimated hours to be incurred) which we believe best depicts our satisfaction of the relevant performance obligation usingobligation. We evaluate the input method (i.e., cumulative actual costs incurred relative to total estimated costs) or on a straight-line basis when a performance obligation is expected to be satisfied evenly over a period of time (when there is a stand-ready obligation).
The estimation of measure of progress each reporting period and, if necessary, adjust the measure of performance and related revenue recognition.
Estimating the standalone selling price of material rights including their likelihood of exercise requires significant judgment. Estimating the measure of progress is also complex, involves significant judgment, and is affected by our estimates of the total costs required to complete the performance obligations including the total internal personnel costs and external costs to be incurred as well as, in certain cases,to satisfy the estimated stand-ready obligation period.respective performance obligation. Changes in these estimates can have a material effect on our revenue recognition.
For a further description of our revenue recognition, see Note 4 – Major Customers, Partnerships and Strategic Alliances in the accompanying notes to the Consolidated Financial Statements included in Part II, Item 8, "Financial“Financial Statements and Supplementary Data"Data” of this Annual Report on Form 10-K.
Valuation of Long-lived Assets including Goodwill and Intangible Assets
We allocate the fair value of purchase consideration in a business combination to the tangible assets acquired, liabilities assumed, and intangible assets acquired based on their estimated fair values. Any excess of purchase consideration over the fair value of net assets acquired is recorded as goodwill. Such valuations require management to make significant estimates and assumptions, especially with respect to intangible assets. Significant estimates in valuing certain intangible assets include, but are not limited to, future expected cash flows from acquired in-process research and development, or IPR&D, projects. Management’s estimates of fair value are based upon assumptions believed to be reasonable, but which are inherently uncertain and unpredictable and, as a result, actual results may differ from estimates. Allocation of purchase consideration to
identifiable assets and liabilities affects our amortization expense, as acquired finite-lived intangible assets are amortized over the useful lives, whereas any indefinite lived intangible assets, including goodwill, are not amortized. During the measurement period, which is not to exceed one year from the acquisition date, we may record adjustments to the assets acquired and liabilities assumed, with the corresponding offset to goodwill. Upon the conclusion of the measurement period, any subsequent adjustments are recorded to earnings.
We review goodwill and indefinite-lived intangible assets for impairment at least annually or more frequently if events or changes in circumstances would more likely than not reduce the fair value these assets below their carrying values. As of December 31, 2020, no impairment of2023, we have fully impaired our goodwill orand our indefinite-lived intangible assets was identified.asset. For a further description of the impairment, see Note 6 – Impairment of Goodwill, Indefinite-Lived Intangible Assets and Other Long-Lived Assets in theaccompanying notes to the Consolidated Financial Statements included in Part II, Item 8, “Financial Statements and Supplementary Data” of this Annual Report on Form 10-K.
Long-lived assets, including property and equipment and finite-lived intangible assets, are reviewed for possible impairment whenever events or circumstances indicate that the carrying amount of such assets may not be recoverable. The evaluation is performed at the lowest level for which identifiable cash flows are largely independent of the cash flows of other assets and liabilities. Recoverability of these assets is measured by a comparison of the carrying amounts to the future undiscounted cash flows the assets are expected to generate from the use and eventual disposition. If such review indicates that the carrying amount of property and equipment and intangiblethe long-lived assets is not recoverable, the carrying amount of such assets is reduced to fair value. We have not recorded any significant impairment charges during the years presented.year ended December 31, 2023. For a further description of the impairment, see Note 6 – Impairment of Goodwill, Indefinite-Lived Intangible Assets and Other Long-Lived Assets in theaccompanying notes to the Consolidated Financial Statements included in Part II, Item 8, “Financial Statements and Supplementary Data” of this Annual Report on Form 10-K.
Recent Accounting Pronouncements
For a summary of recent accounting pronouncements and the anticipated effects on our Consolidated Financial Statements, see Note 1 – Organization, Basis of Presentation and Summary of Significant Accounting Policies in theaccompanying notes to the Consolidated Financial Statements included in Part II, Item 8, “Financial Statements and Supplementary Data” of this Annual Report on Form 10-K.
Results of Operations
Years Ended December 31, 2020, 20192023, 2022 and 20182021
Revenues | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| (in thousands, except percentage values) |
| 2023 | | 2022 | | Change | | % | | 2022 | | 2021 | | Change | | % |
| | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | |
| Revenues | $ | 176,232 | | | $ | 111,299 | | | $ | 64,933 | | | 58 | % | | $ | 111,299 | | | $ | 110,701 | | | $ | 598 | | | 1 | % |
Revenues
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| (In thousands, except percentage values) |
| 2020 | | 2019 | | Change | | % | | 2019 | | 2018 | | Change | | % |
| | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | |
| Revenues | $ | 118,192 | | | $ | 102,428 | | | $ | 15,764 | | | 15 | % | | $ | 102,428 | | | $ | 84,452 | | | $ | 17,976 | | | 21 | % |
Total revenues primarily consisted of revenuesamounts earned from our collaboration agreements and research grants.agreements. We anticipate revenues overin the next several yearsfuture will be derived primarily from our collaboration agreements with Biogen, Kite, Novartis, Pfizer and Sanofi as we continue to recognize upfront and milestone payments received under such agreements over time.
agreements. The increaseterminations of $15.8 million in revenues in 2020 compared to 2019 was primarily due to the recognition of upfront license fees of $21.4 million and $4.1 million under our collaboration agreements with Biogen and Novartis entered intobecame effective in 2020, respectively. In addition,June 2023, following which we are not entitled to any further milestone payments or royalties from either Biogen or Novartis, nor does either Biogen or Novartis have any further obligations to develop or to reimburse us the costs of any of the programs previously subject to the Biogen and Novartis collaborations.
The increase of $64.9 million in revenues in 2023 compared to 2022, was primarily attributed to an increase of $6.2$106.4 million in recognition of upfront license fee relatedrevenue relating to ourC9ORF72 collaboration agreement with PfizerBiogen, primarily due to advancementthe impact of termination of the program.collaboration agreement, which resulted in an increase in the measure of proportional cumulative performance, an increase of $3.8 million in revenue relating to our license agreement with Sigma-Aldrich Corporation, and an increase of $3.6 million in revenues from other licensing agreements.
These increases were partially offset by a decrease of $12.6$27.5 million in revenues relatedrevenue relating to our hemophilia A collaboration agreement with PfizerNovartis due to the termination of the collaboration agreement in June 2023, a decrease of $18.1 million in activities followingrevenue relating to our collaboration agreement with Kite due to reductions in the IND transfer in December 2019,estimated future level of our research and development services, and a decrease of $3.2$3.3 million in license revenue fromrelating to our collaboration agreement with Sanofi primarily due to a changethe termination of the collaboration agreement in estimate driven by a change in project scope and related project cost.June 2022.
Operating Expenses
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| (In thousands, except percentage values) |
| 2020 | | 2019 | | Change | | % | | 2019 | | 2018 | | Change | | % |
| Operating expenses: | | | | | | | | | | | | | | | |
| Research and development | $ | 180,647 | | | $ | 145,922 | | | $ | 34,725 | | | 24 | % | | $ | 145,922 | | | $ | 114,866 | | | $ | 31,056 | | | 27 | % |
| General and administrative | 67,097 | | | 61,686 | | | 5,411 | | | 9 | % | | 61,686 | | | 46,736 | | | 14,950 | | | 32 | % |
| Total operating expenses | $ | 247,744 | | | $ | 207,608 | | | $ | 40,136 | | | 19 | % | | $ | 207,608 | | | $ | 161,602 | | | $ | 46,006 | | | 28 | % |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| (in thousands, except percentage values) |
| 2023 | | 2022 | | Change | | % | | 2022 | | 2021 | | Change | | % |
| Operating expenses: | | | | | | | | | | | | | | | |
| Research and development | $ | 234,057 | | | $ | 249,898 | | | $ | (15,841) | | | (6) | % | | $ | 249,898 | | | $ | 230,819 | | | $ | 19,079 | | | 8 | % |
| General and administrative | 61,167 | | | 62,682 | | | (1,515) | | | (2) | % | | 62,682 | | | 63,219 | | | (537) | | | (1) | % |
| Impairment of goodwill and indefinite-lived intangible assets | 89,485 | | | — | | | 89,485 | | | 100 | % | | — | | | — | | | — | | | — | |
| Impairment of long-lived assets | 65,528 | | | — | | | 65,528 | | | 100 | % | | — | | | — | | | — | | | — | |
| Total operating expenses | $ | 450,237 | | | $ | 312,580 | | | $ | 137,657 | | | 44 | % | | $ | 312,580 | | | $ | 294,038 | | | $ | 18,542 | | | 6 | % |
Research and Development Expenses
Research and development expenses consisted primarily of compensation related expenses including restructuring charges and stock-based compensation, laboratory supplies, preclinical and clinical studies, manufacturing clinical supply, contracted research, and allocated facilities and information technology expenses.
The decrease of $15.8 million in research and development expenses in 2023 compared to 2022 was primarily attributable to lower compensation and other personnel costs of $16.6 million mainly due to a reduction in the bonus expense and lower headcount as a result of the April Restructuring, lower preclinical and clinical expenses of $14.1 million primarily related to the termination of collaboration agreements with Biogen and Novartis, and deferral and reprioritization of certain programs. These decreases were partially offset by restructuring charges of $12.9 million and higher depreciation expense of
The increase of $34.7 million in research and development expenses in 2020 compared to 2019 was primarily driven by a $20.0 million increase in compensation expense as a result of increased headcount to support our programs, clinical trials and start-up of our internal manufacturing operations, a $14.2 million increase in allocated facility overhead costs as we ramped up our internal manufacturing operations, and a $2.1 million increase in preclinical, clinical, research and manufacturing supply expenses. These increases were partially offset by a decrease of $1.7 million in travel and corporate costs due to COVID-19 travel restrictions.$2.5 million. Stock-based compensation expense included in research and development expenses was $13.5$15.2 million and $10.1$18.4 million for the years ended December 31, 20202023 and 2019,2022, respectively.
The table below shows research and development expenses related to our clinical, preclinical and preclinicalother research and development programs. As shown in the table below, preclinical and researchclinical programs contributed $50.7$18.4 million of the increasedecrease in our research and development expenses in 2023 as compared to 2022, primarily driven by deferral and reprioritization of certain programs. Preclinical and research programs decreased by $0.2 million as our CNS partner programs decreased by $19.2 million, primarily driven by the termination of our collaboration agreements with Biogen and Novartis, and decreases in our oncology partner programs by $0.2 million, offset by an increase of $19.2 million in our wholly-owned programs and early research activities due to advancement of our technical platformpreclinical pipeline related to our strategic transformation into a genomic medicine company focused on epigenetic regulation programs addressing serious neurological diseases and our wholly-owned preclinicalnovel AAV capsid delivery technology. Decreases in clinical programs and our CNSpreclinical and research programs partnered with Biogen and Novartis in 2020,were partially offset by a decreasean increase of $16.0$2.8 million in our other research and development programs. | | | | | | | | | | | | | | | | |
| | Year Ended December 31, |
| | (in thousands) |
| Programs | | 2023 | | 2022 | | |
| Clinical programs: | | | | | | |
| Fabry clinical programs | | $ | 46,938 | | | $ | 67,351 | | | |
| | | | | | |
| TX200 clinical programs | | 28,179 | | | 26,185 | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| Subtotal | | 75,117 | | | 93,536 | | | |
| Preclinical and research programs: | | | | | | |
| Wholly-owned programs and early research activities | | 122,352 | | | 103,160 | | | |
| CNS partner programs | | 9,052 | | | 28,247 | | | |
| Oncology partner programs | | 2,038 | | | 2,222 | | | |
| | | | | | |
| Subtotal | | 133,442 | | | 133,629 | | | |
| Other research and development programs | | 25,498 | | | 22,733 | | | |
| Total research and development expenses | | $ | 234,057 | | | $ | 249,898 | | | |
We expect to continue to devote substantial resources to research and development in the future. While we anticipate that our research and development expenses will decrease in the near-term in connection with the April, November, and France Restructurings and the related reprioritization of certain programs and deferral of certain new investments, we ultimately expect research and development expenses to increase in the next several years if we are successful in advancing our clinical programs and if we are able to progress our preclinical product candidates into clinical trials and/or if we are successful in 2020 as comparedsecuring new collaborations or other capital necessary to 2019, primarily driven by a decrease in activities following the IND transfer foradvance our giroctocogene fitelparvovec program in 2019.
| | | | | | | | | | | | | | | | | | | | |
| | Year Ended December 31, |
| | (In thousands) |
| Programs | | 2020 | | 2019 | | 2018 |
| Clinical Programs: | | | | | | |
| Inherited metabolic disorders clinical programs | | $ | 59,030 | | | $ | 61,845 | | | $ | 37,668 | |
| Beta thalassemia clinical program | | 8,672 | | | 13,634 | | | 12,317 | |
| HIV clinical programs | | 3,289 | | | 1,762 | | | 1,612 | |
| Hemophilia clinical programs | | 3,108 | | | 12,805 | | | 23,006 | |
| Subtotal | | 74,099 | | | 90,046 | | | 74,603 | |
| Preclinical Programs: | | | | | | |
| Wholly-owned programs and early research activities | | 79,193 | | | 37,760 | | | 28,018 | |
| CNS programs | | 21,073 | | | 5,721 | | | 3,916 | |
| Oncology programs | | 6,214 | | | 12,281 | | | 7,845 | |
| Other | | 68 | | | 114 | | | 484 | |
| Subtotal | | 106,548 | | | 55,876 | | | 40,263 | |
| Total research and development expenses | | $ | 180,647 | | | $ | 145,922 | | | $ | 114,866 | |
The length of time required to complete our development programs and our development costs for those programs may be impacted by the results of preclinical testing, scope and timing of enrollment in clinical trials for our product candidates, our decisions to pursue development programs in other therapeutic areas, and whether we pursue development of our product candidates with a partner or collaborator or independently.independently and our ability to secure the necessary funding to progress the development of our programs. For example, our product candidates are being developed in multiple therapeutic areas,current focus is on our core neurology preclinical program, and we do not yet know-how many of those therapeutic areasknow whether and to what extent we will continueprogress any resulting product candidates from our preclinical program into the clinic and in what therapeutic areas. In this regard, in connection with the April and November Restructurings, we have paused further development of certain preclinical programs following conclusion of collaborations with Biogen and Novartis and are deferring new investments in registrational trial planning activities for our Fabry disease gene therapy program and in our CAR-Treg cell therapy programs until we secure a collaboration partner or external investment in these programs. We are actively seeking collaboration partners or a direct external investment, as applicable, to pursue.progress our Fabry and CAR-Treg cell therapy programs. Furthermore, the scope and number of clinical trials required to obtain regulatory approval for each pursued therapeutic area is subject to the input of the applicable regulatory authorities, and we have not yet sought such input for all potential therapeutic areas that we may elect to pursue, and even after having given such input, applicable regulatory authorities may subsequently require additional clinical studies prior to granting regulatory approval based on new data generated by us or other companies, or for other reasons outside of our control. As a condition to any regulatory approval, we may also be subject to post-marketing development commitments, including additional clinical trial requirements. As a result of the uncertainties discussed above, we are unable to determine the duration of or completioncomplete costs associated with our development programs.
In any event, ourOur potential therapeutic products are subject to a lengthy and uncertain regulatory process that may not result in our receipt of any necessary regulatory approvals. Failure to receive the necessary regulatory approvals would prevent us from commercializing the product candidates affected. In addition, clinical trials of our product candidates may fail to demonstrate
safety and efficacy, which could prevent or significantly delay regulatory approval. A discussion of the risks and uncertainties with respect to our research and development activities, including completing the development of our product candidates, and the consequences to our business, financial position and growth prospects can be found in “Risk Factors” in Part I, Item 1A of this Annual Report on Form 10-K.
General and Administrative Expenses
General and administrative expenses consist primarily of compensation related expenses including restructuring charges and stock-based compensation for executive, legal, finance and administrative personnel, professional fees, allocated facilities and information technology expenses, and other general corporate expenses.
The increasedecrease of $5.4$1.5 million in general and administrative expenses in 20202023 compared to 20192022 was primarily attributable to lower compensation and other personnel costs of $7.9 million mainly due to an increasea reduction in the bonus expense and lower headcount as a result of $7.3 million in headcount driven compensation costs, and an increase of $6.3 million in professional fees associated
with our 2020 business activities. These increases were partiallythe April Restructuring. The decrease was offset by a decreaserestructuring charges of $7.4$3.8 million, in allocated facility overhead costs, and a decreaseBiogen contract cost asset amortization of $1.1$1.8 million in travel related expenses due to COVID-19 travel restrictions.the termination of the collaboration agreement, and $0.8 million increase in external professional services. Stock-based compensation expense included in general and administrative expenses was $12.2 million and $9.2$13.2 million for the years ended December 31, 20202023 and 2019,2022, respectively.
Restructuring Charges
In 2023, we executed a series of restructurings of operations and corresponding reductions in workforce announced in April and November, and we initiated an information and consultation procedure with the Works Council for our Valbonne, France workforce related to a planned wind-down of our French operations and a corresponding workforce reduction. The information and consultation procedure with the Works Council concluded in the first quarter of 2024, and our Board of Directors approved the France Restructuring on March 1, 2024. These restructurings were designed to reduce overall costs and advance our strategic transformation into a neurology focused genomic medicine company focused on epigenetic regulation programs addressing serious neurological diseases and novel AAV capsid delivery technology. In connection with the April, November, and France Restructurings, we incurred approximately $16.4 million in expenses related to employee severance and notice period payments, benefits, and related restructuring charges, of which $12.5 million is included in research and development expense and $4.0 million is included in general and administrative expense in the accompanying Consolidated Statements of Operations.
Impairment of Goodwill, Indefinite-lived Intangible Assets and Other Long-lived Assets
During the year ended December 31, 2023, we recognized impairment charges of $155.0 million due to identified impairment indicators, including sustained decline in our stock price and related market capitalization, termination of our collaboration agreements with Biogen and Novartis, and actions we initiated including deferral and reprioritization of certain research and development programs. As a result of these impairment indicators, we performed interim impairment tests and concluded our goodwill, indefinite-lived intangible asset, and long-lived assets, primarily comprising right-of-use assets, related leasehold improvements and construction-in-progress, and manufacturing and laboratory equipment, were impaired. Based on our analyses, we recognized a pre-tax goodwill impairment charge of $38.1 million, a pre-tax indefinite-lived intangible asset impairment charge of $51.4 million along with the income tax benefit from the reduction of the associated deferred tax liability of $6.3 million, and a pre-tax long-lived assets impairment charge of $65.5 million during the year ended December 31, 2023.
For more information see Note 6 – Impairment of Goodwill, Indefinite-lived Intangible Assets and Other Long-lived Assets in the accompanying notes to the Consolidated Financial Statements included in Part II, Item 8, “Financial Statements and Supplementary Data” of this Annual Report on Form 10-K.
Interest and other income, net
The decrease of $1.0 million in interestInterest and other income, net was $11.1 million and $9.4 million for the years ended December 31, 2023 and 2022, respectively. The increase of $1.7 million in 20202023 compared to 20192022 was primarily due to a decreasedriven by an increase of $5.1$2.3 million in interest income reflecting the declineincrease in market interest rates. This decrease was partially offset byrates, an increase of $3.1$1.9 million related to fluctuations in foreign exchange gains as a result of favorable foreigncurrency exchange rates, and an increase of $1.1$1.0 million in other income.research tax credits. These increases were partially offset by a $3.0 million benefit received in 2022 from Employee Retention Credit under the Coronavirus Aid, Relief, and Economic Security Act.
Income tax expense
ProvisionThe income tax benefit was $5.1 million for 2023. The provision for income taxes was $0.4 million and $0.3 million zero,for 2022 and zero2021, respectively. The income tax benefit for 2020, 2019the period-ended December 31, 2023 was primarily driven by the reduction of the foreign deferred tax liability due to the impairment of the indefinite-lived intangible asset and 2018, respectively.was offset by the
setup of the valuation allowance for the Company’s United Kingdom subsidiary. The income tax expense for 2020the years ended December 31, 2022 and December 31, 2021 was primarily due to an increase in the long-term liability associated with uncertain tax positions, foreign income taxes and state income taxes. This expense was partially offset by a foreign deferred tax benefit.
Beginning in 2022, the 2017 Tax Cuts and Jobs Act amended Section 174 to eliminate current-year deductibility of research and experimentation, or R&E, expenditures and software development costs, collectively, R&E expenditures, and instead require taxpayers to charge their R&E expenditures to a capital account amortized over five years (15 years for expenditures attributable R&E activity performed outside the United States). We generated a deferred tax asset for capitalized R&E expenditures for the years ended December 31, 2023 and December 31, 2022, which is fully offset with a valuation allowance.
As of December 31, 2020,2023, we had net operating loss carryforwards for federal and state income tax purposes of approximately $622.6$764.6 million and $261.7$338.9 million, respectively. The federal net operating loss generated before 2018 will begin to expire in 2024 and will keep expiring through 2037, if not utilized. Federal net operating losses generated infrom 2018 will carry forward indefinitely. If not utilized, the state net operating loss carryforwards will begin to expire in 2029. We also have federal and state research tax credit carryforwards of $21.9$45.3 million and $18.3$31.3 million, respectively. The federal research credits will begin to expire in 2021,2024, while the state research credits have no expiration date. Utilization of our net operating loss carryforwards and research tax credit carryforwards may be subject to substantial annual limitations due to the ownership change limitations provided by the Internal Revenue Code and similar state provisions. The annual limitation could result in the expiration of the net operating loss carryforwards and research tax credit carryforwards before use. Due to the carryforwards related to the net operating losses and research and development tax credits, we do not expect to pay any U.S. federal taxes related to income in the near future.
Liquidity and Capital Resources
Liquidity
Since inception, we have incurred significant net losses, and we have funded our operations primarily through the issuance of equity securities, payments from corporate collaborators and strategic partners and research grants.
As of December 31, 2020,2023, we had cash, cash equivalents, and marketable securities totaling $692.0$81.0 million compared to $384.3$307.5 million as of December 31, 2019 with the increase primarily attributable to our collaboration with and sale of our common stock to Biogen, which became effective and closed in April 2020, and our collaboration with Novartis, which became effective in July 2020 and included an upfront fee.2022. Our most significant use of capital during the year was for employee compensation and external research and development expenses, such as manufacturing, clinical trials and preclinical activity related to our therapeutic programs. Our cash and investment balances are held in a variety of interest-bearing instruments, including U.S. government-sponsored entity debt securities, commercial paper securities, money market funds, corporate debt securities, certificates of deposit, asset-backed securities and U.S. government-sponsored entity debt securities.certificates of deposit. Cash in excess of immediate requirements is invested in accordance with our investment policy with a view toward capital preservation and liquidity.
Since the beginning of 2017, we have received significant amounts of capital as upfront payments under our collaboration agreements. In addition, our collaboration agreements provide for the payment of development, regulatory, and commercial milestones. For example, in May 2020, we received an upfront license fee of $125.0 million from Biogen upon the effectiveness of our collaboration and license agreement for the research, development and commercialization of gene regulation therapies for the treatment of neurological diseases. And in August 2020, we received an upfront license fee of $75.0 million from Novartis upon the effectiveness of our collaboration and license agreement for the development and commercialization of gene regulation therapies for the treatment of neurodevelopment disorders. For more information, see “Business – Collaborations” in Part I, Item 1 of this Annual Report on Form 10-K.
In August 2020, we entered into an Open Market Sale AgreementAgreement℠, or the sales agreement, with Jefferies LLC, or Jefferies, providing for the sale of up to $150.0 million of our common stock from time to time in ‘at-the-market’“at-the-market” offerings under ouran existing shelf registration statement. AsIn December 2022, we entered into Amendment No. 2 to the Open Market Sale Agreement℠, which increased the aggregate offering price under the sales agreement by an additional $175.0 million. Approximately $194.5 million remained available under the sales agreement as of February 19, 2021, we haveDecember 31, 2023. We sold 1,034,7628,249,261 shares of our common stock under the sales agreement for net proceeds of approximately $15.7 million.$15.1 million during the year ended December 31, 2023.
WhileUnder Accounting Standard Codification Topic 205-40, Presentation of Financial Statements—Going Concern, or ASC Topic 205-40, we expecthave the responsibility to evaluate whether conditions and/or events raise substantial doubt about our rateability to meet our future financial obligations as they become due within one year after the date that the Consolidated Financial Statements included in this Annual Report on Form 10-K are issued. As required under ASC Topic 205-40, management’s evaluation should initially not take into consideration the potential mitigating effects of cash usagemanagement’s plans that have not been fully implemented as of the date the Consolidated Financial Statements are issued. When substantial doubt exists, management evaluates whether the mitigating effects of its plans sufficiently alleviate the substantial doubt about the company’s ability to increase incontinue as a going concern. The mitigating effects of management’s plans, however, are only considered if both (i) it is probable that the future, in particularplans will be effectively implemented within one year after the date that the financial statements are issued, and (ii) it is probable that the plans, when implemented, will mitigate the relevant conditions or events that raise substantial doubt about the entity’s ability to supportcontinue as a going concern within one year after the date that the financial statements are issued. Generally, to be considered probable of being effectively implemented, the plans must have been approved by the company’s board of directors before the date that the financial statements are issued.
Based on our product development endeavors, we currently believe thatcurrent operating plan, our available cash, cash equivalents and marketable securities when combined withas of December 31, 2023 are expected to allow us to meet our liquidity requirements only into the third quarter of 2024. Our history of significant losses, negative cash flows from operations, limited liquidity resources currently on hand and dependence on our ability to obtain
additional capital raises and expected revenues from collaborations, strategic partnerships and research grants, will be adequatefinancing to fund our currently planned operations throughhave resulted in management’s assessment that there is substantial doubt about our ability to continue as a going concern for at least the next 12 months from the date the financial statementsConsolidated Financial Statements included in this Annual Report on Form 10-K are issued. DuringOur ability to continue to operate as a going concern is dependent upon our ability to raise substantial additional capital to fund our operations and support our research and development endeavors, including to progress our preclinical and clinical programs as described in this periodAnnual Report on Form 10-K. In this regard, we have been actively seeking, and continue to actively seek, substantial additional capital, including through public or private equity or debt financing, royalty financing or other sources, such as strategic collaborations and other direct investments in our programs. We may be unable to attract new investments as a result of uncertaintythe speculative nature of our newly reprioritized core neurology preclinical programs. We have been unsuccessful in securing such additional capital to date. If we are unable to secure additional funding in the very near term, we will likely seek protection under the U.S. Bankruptcy Code. We have explored, and continue to explore, whether filing for bankruptcy protection is in the best interest of our Company and our stakeholders. Additional captial may not be available on acceptable terms or at all. If adequate funds are not available to us on a timely basis, or at all, we will be required to take additional actions to address our liquidity needs, including additional cost reduction measures such as further reducing operating expenses and delaying, reducing the scope of, discontinuing or altering our research and development activities, which would have a material adverse effect on our business and prospects, or we may be required to cease operations entirely, liquidate all or a portion of our assets, and/or seek protection under the U.S. Bankruptcy Code, and you may lose all or part of your investment.
While we expect the April, November, and France Restructurings to be complete by the third quarter of 2024, the second quarter of 2024, and fourth quarter of 2024, respectively, we may also incur other charges or cash expenditures not currently contemplated due to events that may occur as a result of, or associated with, each of the restructurings. In addition, we may not achieve the expected benefits of these cost reduction measures and other cost reduction plans on the anticipated timeline, or at all, or we may use our available capital more quickly than we expect, which could otherwise accelerate our liquidity needs and could force us to further curtail or suspend, or entirely cease, our operations. Moreover, we rely in part on our collaboration partners to provide funding for and otherwise advance our preclinical and clinical programs. However, in June 2022, our collaboration agreement with Sanofi terminated, and in June 2023 our collaboration agreements with Biogen and Novartis terminated. While we may identify new collaboration partners who can progress some of the programs that were the subject of these collaborations as well as our Fabry disease gene therapy program and our CAR-Treg cell therapy programs, we have not yet been, and may never be, successful in doing so in a timely manner, on acceptable terms or at all, and we may otherwise fail to raise sufficient additional capital in order to progress these programs ourselves, in which case, we will not receive any return on our investments in these programs. In any event, we need substantial additional funding in order to progress the programs that were the subject of these collaborations as well as our Fabry disease and CAR-Treg cell therapy programs, and to otherwise execute on our current operating plan. If we raise additional capital through public or private equity offerings, including sales pursuant to our at-the-market offering program with Jefferies LLC, the ownership interest of our existing stockholders will be diluted, and such dilution may be substantial given our current stock price decline, and the terms of any new equity securities may have a preference over, and include rights superior to, our common stock. If we raise additional capital through royalty financings or other collaborations, strategic alliances or licensing arrangements with third parties, we may need to relinquish certain valuable rights to our product candidates, technologies, future revenue streams or research programs or grant licenses on terms that may not be favorable. If we raise additional capital through debt financing, we may be subject to specified financial covenants or covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or pursuing certain transactions, any of which could restrict our ability to commercialize our product candidates or operate as a business.
In addition, as we focus our efforts on proprietary human therapeutics, we will need to seek regulatory approvals of our product candidates from the FDA or other comparable foreign regulatory authorities, a process that could cost in excess of hundreds of millions of dollars per product. We may experience difficulties in accessing the capital markets due to external factors beyond our control, such as volatility in the equity markets for emerging biotechnology companies and general economic and market conditions both in the United States and abroad. In particular, our ability to raise the substantial additional capital we need in order to fund our business may be adversely impacted by global economic conditions and disruptions to and volatility relatedin the credit and financial markets in the United States and worldwide, such as has been experienced recently due in part to, among other things, the evolving COVID-19 pandemic,ongoing conflict between Russia and Ukraine and conflicts in the Middle East and disruptions in access to bank deposits and lending commitments due to bank failure. We cannot be certain that we will continuebe able to monitor our liquidity.obtain financing on terms acceptable to us, or at all.
Cash Flows
Operating activities
Net cash provided by operating activities was $169.9 million in 2020 compared to net cash used in operating activities was $224.8 million in 2023, primarily due to:
•a net loss of $257.8 million, adjusted for non-cash goodwill, indefinite-lived intangible assets, and long-lived asset impairment charges of $155.0 million, other non-cash expenses related to stock-based compensation of $27.4 million, depreciation and amortization of $15.1 million, amortization of operating lease right-of-use assets of $7.1 million, and other non-cash adjustments of $1.1 million, offset by income tax benefit of $6.3 million related to reversal of the deferred tax liability as a result of impairment of the associated indefinite-lived intangible assets, accretion of discounts and impairment of marketable securities of $2.3 million; and
•$144.4a decrease in deferred revenues of $161.2 million, in 2019. The increase was primarily attributable mainly attributed to favorablethe impact of the termination and related contract modification of our collaboration agreement with Biogen and changes in working capital of $325.6estimate for our collaboration agreement with Kite, a decrease in accrued compensation and employee benefits by $12.7 million, drivenand decrease in lease liabilities by $5.0 million. These were partially offset by an increase in deferred revenues of $252.2accounts payable and other accrued liabilities by $5.6 million, reflecting the cash received under the Biogendecrease in prepaid expenses and Novartis collaboration agreements,other assets by $6.1 million, and a decrease in accounts receivable of $63.9by $2.8 million. This increase
Net cash used in operating activities was partially offset by an increase$223.6 million in 2022, primarily reflecting our net loss of $25.7$192.3 million, a decrease in deferred revenues of $91.3 million, an increase in prepaid expenses and other assets of $4.9 million, and a decrease in lease liabilities by $2.2 million. These decreases were partially offset by $51.0 million of non-cash expenses related to stock-based compensation, depreciation and amortization, amortization of operating lease right-of-use assets, and net amortization of premium (discount) on marketable securities, an increase in accounts payable and other accrued liabilities of $13.3 million, and an increase in accounts receivable of $2.3 million.
Investing activities
Net cash used inprovided by investing activities was $271.6$153.5 million in 2020, and $59.8 million in 2019. The increase was2023, primarily duerelated to a net increase of $217.3 million in the purchasematurities of marketable securities of $214.5 million and sales of marketable securities of $19.7 million, partially offset by a decreasepurchases of $6.0marketable securities of $59.6 million inand purchases of property and equipment.equipment of $21.2 million.
Net cash provided by investing activities was $59.3 million in 2022, primarily related to maturities of marketable securities of $354.6 million and sales of marketable securities of $2.3 million, partially offset by purchases of marketable securities of $277.4 million and purchases of property and equipment of $20.2 million.
Financing activities
Net cash provided by financing activities was $153.1$14.6 million in2020, and $142.0 2023, primarily related to $15.1 million in2019.The increase was primarily attributed toof proceeds from the $145.4 million estimated fair value of the Biogen Shares issued in April 2020,at-the-market offering, net of $2.9offering expenses of $0.4 million, and proceeds from purchases of common stock under the employee stock purchase plan of $0.9 million, offset by a decrease of $1.5 million for taxes paid related to net share settlement of equity awards.
Net cash provided by financing activities was $84.7 million in 2022, primarily related to $87.1 million of proceeds from the at-the-market offering, net of offering expenses of $2.2 million, and an increase of $1.8 million related to proceeds from the issuance costs, partiallyof common stock under our employee stock purchase plan, offset by $136.3a decrease of $2.1 million received in 2019for taxes paid related to our April 2019 underwritten public offering.net share settlement of equity awards.
Operating Capital and Capital Expenditure Requirements
We anticipate continuing to incur operating losses for at least the next several years. While we expect our rateyears and need to raise substantial additional capital. The effects of cash usage to increase in the future, in particular to support our product development endeavors, we currently believe that our available cash resources, when combined with additional capital raises and expected revenues from collaborations, strategic partners and research grants, will be adequate to fund our currently planned operations through at least the next 12 months from the date the financial statements are issued. Although we believe we are well capitalized currently,current macroeconomic environment, including the effects of war in Ukraine and conflicts in the ongoing COVID-19 pandemic couldMiddle East, financial and liquidity challenges associated with current and potential future bank failures, inflation, climate change, rising interest rates and other economic uncertainty and volatility, has resulted and may continue to result in significant disruption of global financial markets, impairingwhich could impair our ability to access capital whichon terms that are acceptable or at all, and in turn could in the future negatively affect our liquidity.liquidity and our ability to continue to operate as a going concern. Future capital requirements beyond the next 12 monthsthird quarter of 2024, the period until which we expect our existing cash and cash equivalents, in combination with potential future cost reductions, will be sufficient to fund our planned operations, will be substantial, and we will need to raise substantial additional capital to continue to operate as a going concern and to fund the development, manufacturing and potential commercialization of our product candidatescandidates. In this regard, we have been actively seeking, and continue to actively seek, substantial additional capital, including through public or private equity or debt financing. In addition,financings, royalty financings or other sources, such as strategic collaborations and other direct investments in our programs. We have been unsuccessful in securing such additional capital to date. If we are unable to secure additional funding in the very near term, we
will likely seek protection under the U.S. Bankruptcy Code.We have explored, and continue to explore, whether filing for bankruptcy protection is in the best interest of our Company and our stakeholders. Additional capital may not be available on acceptable terms or at all. If adequate funds are not available to us on a timely basis, or at all, we will be required to take additional actions to address our liquidity needs, including additional cost reduction measures such as further reducing operating expenses and delaying, reducing the scope of, discontinuing or altering our research and development activities, which would have a material adverse effect on our business and prospects, or we may be required to cease operations entirely, liquidate all or a portion of our assets, and/or seek protection under the U.S. Bankruptcy Code, and you may lose all or part of your investment.
As we focus our efforts on proprietary human therapeutics, we will need to seek FDA approvals of our product candidates, a process that could cost in excess of hundreds of millions of dollars per product. We regularly consider fund-raising opportunities and may decide, from time to time, to raise capital based on various factors, including market conditions and our plans of operation. Additional capital may not be available on terms acceptable to us, or at all. If adequate funds are not available, or if the terms of potential funding sources are unfavorable, our business and our ability to advance our product candidate pipeline would be harmed. Furthermore, any sales of additional equity securities, including sales pursuant to our at-the-market financing offering programs, may result in dilution to our stockholders, and any debt financing may include covenants that restrict our business.
Our future capital requirements will depend on many forward-looking factors, including the following:
•the results of preclinical testing of our early-stage core neurology program product candidates;
•the initiation, progress, timing and completion of clinical trials for our product candidates and potential product candidates;
•the outcome, timing and cost of regulatory approvals;
•the success of our collaboration agreements;
•delays that may be caused by changing regulatory requirements;
•the number of product candidates that we pursue;
•the costs involved in filing and prosecuting patent applications and enforcing and defending patent claims;
•the timing and terms of future in-licensing and out-licensing transactions;
•the cost and timing of establishing sales, marketing, manufacturing and distribution capabilities;
•the cost of procuring clinical and commercial supplies of our product candidates;
•the extent to which we acquire or invest in businesses, products or technologies, including the costs associated with such acquisitions and investments; and
•the costs of potential disputes and litigation.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements as defined in Item 303(a)(4)(ii) of Regulation S-K.
Contractual Obligations
AsOur contractual obligations as of December 31, 2020, we had contractual obligations and commercial commitments as follows (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Payments Due by Period |
| Contractual Obligations | | Total | | Less Than
1 Year | | 1-3
Years | | 4-5
Years | | More Than
5 Years |
| Operating leases | | $ | 53,422 | | | $ | 6,191 | | | $ | 13,607 | | | $ | 14,053 | | | $ | 19,571 | |
| License obligations | | 958 | | | 178 | | | 290 | | | 280 | | | 210 | |
| Manufacturing obligations | | 21,507 | | | 14,605 | | | 6,902 | | | — | | | — | |
| Total contractual obligations | | $ | 75,887 | | | $ | 20,974 | | | $ | 20,799 | | | $ | 14,333 | | | $ | 19,781 | |
Operating2023 relate primarily to (i) operating leases consistconsisting of base rents for facilities we occupy in Brisbane, California; Richmond, California; and Valbonne, France.
LicenseFrance, (ii) purchase obligations includes anrelated to manufacturing, facilities, and equipment, and (iii) license obligations for ongoing license maintenance fee associated with cancellable in-licensed patent agreements.
Manufacturing These agreements are enforceable and legally binding and specify all significant terms, including fixed or minimum services to be used, fixed, minimum or variable price, and the approximate timing of the actions under the contracts. For more information regarding our contractual obligations include the following non-cancelable material contractualand commitments under manufacturing-related supplier arrangements as of December 31, 2020 (in thousands):
| | | | | | | | | | | | | | |
Party2023, see Note 8 – | | Total | | Expiry date |
Brammer | | $ | 7,736 | | | December 2022 |
Lonza Netherlands, B.V. | | 13,771 | | | December 2022 |
Total manufacturing obligations | | $ | 21,507 | | | |
ITEM 7A – QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Our exposure to market risk relates to our cash, cash equivalents, and marketable securities. The goals of our investment policy are preservation of capital, fulfillment of liquidity needs and capturing a market rate of return based on our investment policy parameters and market conditions. We select investments that maximize interest income to the extent possible within these guidelines. To achieve our goals, we maintain a portfolio of cash equivalents and investments in securities of high credit quality and with varying maturities to match projected cash needs.
The securities in our investment portfolio are not leveraged and are classified as available-for-sale. The majority of these available-for-sale securities are short-term in nature and subject to minimal interest rate risk. Our investments currently consist of commercial paper, money market funds, corporate debt securities, certificates of deposit, asset-backed securities and U.S. government-sponsored entity debt securities.securities, commercial paper securities, corporate debt securities, asset-backed securities, U.S. treasury bills, and certificates of deposit. Our investment policy, approved by our Board of Directors, limits the amount we may invest in any one type of investment issuer, thereby reducing credit risk concentrations. All investments have a fixed interest rate and are carried at market value, which approximates cost. We do not use derivative financial instruments in our investment portfolio. We do not believe that a change inIf market interest rates would have a material negative impact onwere to increase or decrease by one hundred basis points, the fair value of our investment portfolio.portfolio would increase or decrease by an immaterial amount.
Foreign Currency Exchange Risk
We have operations in the United States as well as in Europe. The functional currency of each foreign subsidiary is the local currency. We are exposed to foreign currency risk, primarily through operations of our subsidiaries in Europe which conduct business primarily in Euros. We record gains and losses within our stockholders’ equity due to the translation of our subsidiaries’ financial statements into U.S. dollars.
A hypothetical 10% strengthening/(weakening) in the rates used to translate the results of our foreign subsidiaries would have increased/(decreased) net loss for the year ended December 31, 20202023 by approximately $2.1$12.6 million and wouldmay have resulted in a material impact on our Consolidated Financial Statements. To date, we have not have materially impactedhad a formal hedging program with respect to foreign currencies, but we may do so in the future if our operating loss.exposure to foreign currencies should become more significant.
Additionally, we incur foreign currency transaction gains and losses related to the level of activity between the United States and Europe. In 2020,2023, we incurred foreign currency transaction gainslosses of $2.2$1.2 million. A 10% unfavorable change in the Euro and U.S. dollar exchange rate on December 31, 20202023 would have had an immaterial impact on foreign currency transaction gainslosses for 2020.2023.
As of December 31, 2020 and 2019, we maintainedWe did not maintain any cash balances of approximately $16.8 million and $22.6 million, respectively, denominated in a foreign currency in the United States. A hypothetical 10% change in foreign exchange rates would have increased/(decreased) net loss for the year endedStates as of December 31, 2020 by approximately $1.7 million2023 and would not have materially impacted our operating loss.2022.
ITEM 8 – FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
SANGAMO THERAPEUTICS, INC.
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Stockholders and the Board of Directors of Sangamo Therapeutics, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Sangamo Therapeutics, Inc. (the Company) as of December 31, 20202023 and 2019,2022, the related consolidated statements of operations, comprehensive loss, stockholders'stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2020,2023, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 20202023 and 2019,2022, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2020,2023, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company'sCompany’s internal control over financial reporting as of December 31, 2020,2023, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) and our report dated February 24, 2021March 13, 2024 expressed an unqualified opinion thereon.
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the financial statements, the Company has suffered recurring losses from operations and has stated that substantial doubt exists about the Company’s ability to continue as a going concern. Management’s evaluation of the events and conditions and management’s plans regarding these matters are also described in Note 1. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company'sCompany’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matter
The critical audit mattermatters communicated below is a matterare matters arising from the current period audit of the financial statements that was communicated or required to be communicated to the audit committee and that: (1) relatesrelate to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of the critical audit mattermatters does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit mattermatters below, providing a separate opinionopinions on the critical audit mattermatters or on the accounts or disclosures to which it relates.
| | | | | | | | |
| | Impairment of goodwill, intangibles and other long-lived assets |
| | | Estimation of Fair value of shares sold to Biogen
| | |
| | As describeddiscussed in Note 4 to1 of the consolidated financial statements, in 2020,goodwill and the Company entered into a collaborationin-process research and license agreement with Biogen MA, Inc.development (“IPR&D”) intangible asset are tested for impairment at least annually and Biogen International GmbH (together, “Biogen”) for the research, development and commercializationmore frequently when indicators of gene regulation therapies for the treatment of neurological diseases. Concurrent with the execution of the Biogen collaboration agreement, the Company entered into a stock purchase agreement pursuant to which Biogen agreed to purchase 24.4 million shares of the Company's common stock for an aggregate purchase price of approximately $225.0 million, a price which exceededimpairment exist. Management estimated the fair value of the shares at the time of the purchase. Since the purchase price exceededIPR&D intangible asset and estimated the fair value of the common shares issuedreporting unit for purposes of testing goodwill. As discussed in this transaction, management hadNote 1 of the consolidated financial statements, the Company also reviews its other long-lived assets, including right-of-use assets (“RoU”) and property, plant and equipment for impairment whenever events or changes in circumstances indicate that the carrying amounts of the assets may not be recoverable. For the purposes of such review of recoverability, the Company determined all of its long-lived assets represent one entity-wide asset group. The Company compares expected future cash flows of the single asset group to estimatethe carrying value of the assets. If the expected future cash flows (undiscounted) are less than the carrying amount of such assets, the Company recognizes an impairment loss for the difference between the carrying value of the asset group and its estimated fair value, which is allocated to long-lived assets of the group on a pro rata basis using the relative carrying amounts of those assets. The carrying amount of a long-lived asset of the group must not be reduced below its fair value. During the year ended December 31, 2023, the Company recorded impairment of $155.0 million, consisting of $38.1 million related to goodwill, $51.4 million related to IPR&D, and $65.5 million related to other long-lived assets.
Auditing the Company's impairment tests of goodwill, indefinite-lived intangible assets and long-lived assets was complex and required a high degree of auditor judgment when performing procedures due to the significant estimation uncertainty in determining the fair value of these shares using an option pricing model to reflect certain holding period restrictionsgoodwill, IPR&D, RoU and other unique termslong-lived assets. Significant judgments made by management related to valuation of the agreement. The fair valuegoodwill included estimating a reasonable range of these shares was estimated to be $145.4 million, and the $79.6 million excess consideration was included in the transaction price of the collaboration and license agreement.
Auditing the estimated fair value of the shares issued in this transaction was complex as management had to use significant judgment in selecting the valuation methodology and estimating the significant unobservable inputs used in the valuation model. The usevalues of a different valuation methodology and inputs could significantly affect the fair value assignedcontrol premium applied to the common shares thereby impactingCompany’s implied business enterprise value. Significant judgements made by management related to valuation of IPR&D included certain assumptions, including weighted average cost of capital, forecasted net sales, the total transaction priceprobability of clinical success of research and development programs and of obtaining regulatory approvals. Significant judgements made by management related to valuation of RoU assets included determining discount rate, the collaborationlength of time to enter into a sublease and license agreementmarket rental rates. Management also made significant judgments in estimating market-based pricing for its machinery and ultimately the amount of revenue that is recognized under this agreement.equipment.
|
| | We obtained an understanding, evaluated the design and tested the operating effectiveness of controls over the Company’s accounting for this new collaboration agreement, including management’s reviewevaluation of impairment of goodwill, IPR&D, RoU and other long-lived asset. For example, we tested management’s controls over developmentthe determination of the significant unobservable inputs that affectedcontrol premium, weighted average cost of capital, forecasted net sales, the probability of clinical success of research and development programs and obtaining regulatory approvals, the discount rate, the length of time to enter into sublease, market rental rates and other key assumptions used in determination of the fair value of the common shares issued to Biogen.goodwill, IPR&D, RoU assets and other long-lived assets.
To test the fair value of the common shares, ourOur audit procedures included, among others, evaluating the Company’s valuation methodology and the significant unobservable inputsvaluation models used and testing the mathematical accuracykey inputs and significant assumptions discussed above. We evaluated the significant assumptions described above comparing to observable industry data, external data sources, probability of success benchmarks and regulatory factors for IPR&D intangible assets, and external market data and comparable executed transactions and listings for right-of-use assets and machinery and equipment. Our procedures included evaluating the Company’s valuation calculations.data sources used by management in determining its significant assumptions and included an evaluation of available information that either corroborated or contradicted management’s conclusions. We involved our valuation specialistsprofessionals to assist us with evaluatingassess the Company’s valuation methodology and assessingvaluation of the IPR&D intangible assets and other long-lived assets including right-of-use and machinery and equipment, including evaluating the reasonableness of the inputs by comparing them to information available from third-party sources and market data. We also independently developed fair value estimates with the assistance of our valuation specialists and compared them to the Company’s valuation results.certain significant assumptions.
|
| | | | | | | | |
| | Termination of Biogen collaboration agreement and termination of Kite research plans |
| | |
| Description of the Matter | | The Company recorded revenue from collaboration agreements of $176.2 million for the year ended December 31, 2023. The Company’s revenues are derived from collaboration arrangements which primarily include licensing intellectual property and providing research and development services. Revenues are recognized over time by measuring progress towards satisfaction of the relevant performance obligation, using the input method (i.e., cumulative actual costs incurred relative to total estimated costs). As described in Note 4, during the first quarter of 2023, the Company received a contract termination notice from Biogen MA, Inc., (“Biogen”). The termination resulted in a reduction to the scope and price of the Company’s ongoing performance obligations under the collaboration agreement. The termination provisions of the Biogen collaboration required the Company to perform services for certain targets beyond the notice date, some of which were distinct, while some were not distinct from prior services performed. The Company updated the transaction price and allocated it to the remaining performance obligations and adjusted revenue previously recognized based on the updated measure of progress for remaining targets. The Company updated the transaction price and calculated total revenue to be recognized to date, including a cumulative catch-up adjustment of $127.1 million, by using the reassessed measure of progress taking into account the revised transaction price and the revised scope of services. During the year ended December 31, 2023, the Company received letters from Kite Pharma, Inc., (“Kite”), terminating two research plans. The termination of the research plans changed the estimate of required effort to complete the performance obligation under the Kite collaboration resulting in recognition of revenue through a total cumulative catch-up adjustment of $13.9 million.
Auditing the Company’s accounting for revenue pertaining to the modification of the existing collaboration agreements affected the amount and timing of revenue recognition and required an increased extent of effort and a high degree of auditor judgment, due to the complex and judgmental nature of evaluating the terms and assumptions of the related modified agreements. |
| How We Addressed the Matter in Our Audit | | We obtained an understanding, evaluated the design, and tested the operating effectiveness of controls addressing the risk of material misstatement relating to accounting for modification to its existing collaboration agreements. For example, we tested management’s controls over the determination of the remaining performance obligations, including any material rights and the likelihood of exercise as well as the Company’s estimates of the total costs expected to complete the performance obligations. We also tested management’s controls over the completeness and accuracy of the data used in the underlying calculations.
To test the Company’s application of the contract modification guidance under ASC 606 to the amended collaboration contracts, our procedures included, among others, inspecting the termination letters and obtaining information from the Company’s personnel that oversee the collaboration activities to confirm our understanding of the nature of the amendments. We inspected the minutes of the relevant joint steering committee meetings and the updated budgets by project, to evaluate management’s assumptions used in the Company’s estimates of total expected costs by project and evaluated changes in the total expected costs due to the modification or change in estimate. We also assessed whether the termination provisions were appropriately accounted for and that the recognition of revenue for the remaining performance obligations was appropriate by inspecting the associated model used to determine the amount of revenue to be recognized through cumulative catch-up adjustments. |
/s/ ERNST & YOUNG LLP
We have served as the Company’s auditor since 1997.
Redwood City,San Mateo, California
February 24, 2021March 13, 2024
SANGAMO THERAPEUTICS, INC.
CONSOLIDATED BALANCE SHEETS
(in thousands, except share and per share amounts)
| | | | | | | | | | | |
| December 31,
2020 | | December 31,
2019 |
| ASSETS | | | |
| Current assets: | | | |
| Cash and cash equivalents | $ | 131,329 | | | $ | 80,428 | |
| | | |
| Marketable securities | 510,094 | | | 282,046 | |
| Interest receivable | 1,035 | | | 682 | |
| Accounts receivable | 5,224 | | | 36,909 | |
| Prepaid expenses and other current assets | 11,986 | | | 5,408 | |
| Total current assets | 659,668 | | | 405,473 | |
| Marketable securities, non-current | 50,530 | | | 21,832 | |
| Property and equipment, net | 41,324 | | | 29,926 | |
| Intangible assets | 58,128 | | | 53,156 | |
| Goodwill | 42,798 | | | 39,273 | |
| Operating lease right-of-use assets | 71,045 | | | 77,289 | |
| Other non-current assets | 13,557 | | | 9,067 | |
| Restricted cash | 1,500 | | | 1,500 | |
| Total assets | $ | 938,550 | | | $ | 637,516 | |
| LIABILITIES AND STOCKHOLDERS’ EQUITY | | | |
| Current liabilities: | | | |
| Accounts payable | $ | 12,553 | | | $ | 6,671 | |
| Other accrued liabilities | 18,612 | | | 10,885 | |
| Accrued compensation and employee benefits | 20,738 | | | 13,605 | |
| Deferred revenues | 91,644 | | | 38,711 | |
| Total current liabilities | 143,547 | | | 69,872 | |
| Deferred revenues, non-current | 245,045 | | | 81,432 | |
| Long-term portion of lease liabilities | 38,396 | | | 41,192 | |
| Deferred income tax | 7,185 | | | 6,570 | |
| Other non-current liabilities | 7,011 | | | 5,711 | |
| Total liabilities | 441,184 | | | 204,777 | |
| Commitments and contingencies | 0 | | 0 |
| Stockholders’ equity: | | | |
| Preferred stock, $0.01 par value, 5,000,000 shares authorized, and 0 shares issued or outstanding | 0 | | | 0 | |
| Common stock, $0.01 par value; 320,000,000 shares authorized, 142,063,203 and 115,972,708 shares issued and outstanding at December 31, 2020 and 2019, respectively | 1,421 | | | 1,160 | |
| Additional paid-in capital | 1,269,375 | | | 1,090,828 | |
| Accumulated deficit | (777,981) | | | (656,985) | |
| Accumulated other comprehensive income (loss) | 5,419 | | | (2,449) | |
| Total Sangamo Therapeutics, Inc. stockholders’ equity | 498,234 | | | 432,554 | |
| Non-controlling interest | (868) | | | 185 | |
| Total stockholders’ equity | 497,366 | | | 432,739 | |
| Total liabilities and stockholders’ equity | $ | 938,550 | | | $ | 637,516 | |
| | | | | | | | | | | |
| December 31, |
| 2023 | | 2022 |
| ASSETS | | | |
| Current assets: | | | |
| Cash and cash equivalents | $ | 45,204 | | | $ | 100,444 | |
| | | |
| Marketable securities | 35,798 | | | 177,188 | |
| Interest receivable | 403 | | | 794 | |
| Accounts receivable | 923 | | | 3,678 | |
| Prepaid expenses and other current assets | 12,000 | | | 18,223 | |
| Total current assets | 94,328 | | | 300,327 | |
| Marketable securities, non-current | — | | | 29,845 | |
| Property and equipment, net | 26,874 | | | 63,531 | |
| Intangible assets | — | | | 50,729 | |
| Goodwill | — | | | 37,552 | |
| Operating lease right-of-use assets | 25,991 | | | 62,002 | |
| Other non-current assets | 16,627 | | | 17,023 | |
| Restricted cash | 1,500 | | | 1,500 | |
| Total assets | $ | 165,320 | | | $ | 562,509 | |
| LIABILITIES AND STOCKHOLDERS’ EQUITY | | | |
| Current liabilities: | | | |
| Accounts payable | $ | 15,259 | | | $ | 22,418 | |
| Accrued compensation and employee benefits | 8,918 | | | 21,506 | |
| Other accrued liabilities | 23,554 | | | 16,007 | |
| Deferred revenues | — | | | 51,780 | |
| Total current liabilities | 47,731 | | | 111,711 | |
| Deferred revenues, non-current | — | | | 109,377 | |
| Long-term portion of lease liabilities | 33,515 | | | 38,986 | |
| Deferred income tax | — | | | 6,270 | |
| Other non-current liabilities | 1,187 | | | 1,207 | |
| Total liabilities | 82,433 | | | 267,551 | |
| Commitments and contingencies | | | |
| Stockholders’ equity: | | | |
| Preferred stock, $0.01 par value, 5,000,000 shares authorized, and no shares issued or outstanding | — | | | — | |
| Common stock, $0.01 par value; 640,000,000 shares authorized; 178,133,548 and 166,793,320 shares issued and outstanding at December 31, 2023 and 2022, respectively | 1,781 | | | 1,668 | |
| Additional paid-in capital | 1,492,077 | | | 1,450,239 | |
| Accumulated deficit | (1,406,376) | | | (1,148,545) | |
| Accumulated other comprehensive loss | (4,595) | | | (8,404) | |
| | | |
| | | |
| Total stockholders’ equity | 82,887 | | | 294,958 | |
| Total liabilities and stockholders’ equity | $ | 165,320 | | | $ | 562,509 | |
See accompanying Notes to Consolidated Financial Statements.
SANGAMO THERAPEUTICS, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
(in thousands, except per share amounts)
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2020 | | 2019 | | 2018 |
| | | | | |
| | | | | |
| | | | | |
| Revenues | $ | 118,192 | | | $ | 102,428 | | | $ | 84,452 | |
| Operating expenses: | | | | | |
| Research and development | 180,647 | | | 145,922 | | | 114,866 | |
| General and administrative | 67,097 | | | 61,686 | | | 46,736 | |
| Total operating expenses | 247,744 | | | 207,608 | | | 161,602 | |
| Loss from operations | (129,552) | | | (105,180) | | | (77,150) | |
| Interest and other income, net | 8,775 | | | 9,761 | | | 8,261 | |
| Loss before income taxes | (120,777) | | | (95,419) | | | (68,889) | |
| Income tax expense | 345 | | | 0 | | | 0 | |
| Net loss | (121,122) | | | (95,419) | | | (68,889) | |
| Net loss attributable to non-controlling interest | (126) | | | (233) | | | (555) | |
| Net loss attributable to Sangamo Therapeutics, Inc. stockholders | $ | (120,996) | | | $ | (95,186) | | | $ | (68,334) | |
| Basic and diluted net loss per share attributable to Sangamo Therapeutics, Inc. stockholders | $ | (0.90) | | | $ | (0.85) | | | $ | (0.70) | |
| Shares used in computing basic and diluted net loss per share attributable to Sangamo Therapeutics, Inc. stockholders | 134,449 | | | 112,114 | | | 96,941 | |
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2023 | | 2022 | | 2021 |
| | | | | |
| | | | | |
| | | | | |
| Revenues | $ | 176,232 | | | $ | 111,299 | | | $ | 110,701 | |
| Operating expenses: | | | | | |
| Research and development | 234,057 | | | 249,898 | | | 230,819 | |
| General and administrative | 61,167 | | | 62,682 | | | 63,219 | |
| Impairment of goodwill and indefinite-lived intangible assets | 89,485 | | | — | | | — | |
| Impairment of long-lived assets | 65,528 | | | — | | | — | |
| Total operating expenses | 450,237 | | | 312,580 | | | 294,038 | |
| Loss from operations | (274,005) | | | (201,281) | | | (183,337) | |
| Interest and other income, net | 11,102 | | | 9,432 | | | 5,346 | |
| Loss before income taxes | (262,903) | | | (191,849) | | | (177,991) | |
| Income tax (benefit) expense | (5,072) | | | 429 | | | 306 | |
| Net loss | (257,831) | | | (192,278) | | | (178,297) | |
| Net loss attributable to non-controlling interest | — | | | — | | | (11) | |
| Net loss attributable to Sangamo Therapeutics, Inc. stockholders | $ | (257,831) | | | $ | (192,278) | | | $ | (178,286) | |
| Basic and diluted net loss per share attributable to Sangamo Therapeutics, Inc. stockholders | $ | (1.48) | | | $ | (1.25) | | | $ | (1.23) | |
| Shares used in computing basic and diluted net loss per share attributable to Sangamo Therapeutics, Inc. stockholders | 174,444 | | | 154,345 | | | 144,568 | |
See accompanying Notes to Consolidated Financial Statements.
SANGAMO THERAPEUTICS, INC.
CONSOLIDATED STATEMENTS OF COMPREHENSIVE LOSS
(in thousands)
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2020 | | 2019 | | 2018 |
| Net loss | $ | (121,122) | | | $ | (95,419) | | | $ | (68,889) | |
| Foreign currency translation adjustment | 8,345 | | | (1,573) | | | (1,148) | |
| Net pension losses | (193) | | | (28) | | | (21) | |
| Change in unrealized (loss) gain on marketable securities, net of tax | (284) | | | 592 | | | (4) | |
| Comprehensive loss | (113,254) | | | (96,428) | | | (70,062) | |
| Comprehensive loss attributable to non-controlling interest | (126) | | | (233) | | | (574) | |
| Comprehensive loss attributable to Sangamo Therapeutics, Inc. | $ | (113,128) | | | $ | (96,195) | | | $ | (69,488) | |
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2023 | | 2022 | | 2021 |
| Net loss | $ | (257,831) | | | $ | (192,278) | | | $ | (178,297) | |
| Foreign currency translation adjustment | 2,848 | | | (4,606) | | | (8,351) | |
| Net pension gain (loss) | (163) | | | 786 | | | (716) | |
| Unrealized gain (loss) on marketable securities, net of tax | 1,124 | | | (597) | | | (339) | |
| Comprehensive loss | (254,022) | | | (196,695) | | | (187,703) | |
| Comprehensive loss attributable to non-controlling interest | — | | | — | | | (11) | |
| Comprehensive loss attributable to Sangamo Therapeutics, Inc. | $ | (254,022) | | | $ | (196,695) | | | $ | (187,692) | |
See accompanying Notes to Consolidated Financial Statements.
SANGAMO THERAPEUTICS, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
(in thousands, except share amounts)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Common Stock | | Additional
Paid-in
Capital | | Accumulated
Deficit | | Accumulated
Other
Comprehensive
Income (Loss) | | Non-
Controlling
Interest | | Total
Stockholders’
Equity |
| Shares | | Amount | | | | | |
| Balances at December 31, 2017 | 85,598,534 | | | $ | 856 | | | $ | 682,809 | | | $ | (495,479) | | | $ | (286) | | | $ | 0 | | | $ | 187,900 | |
Cumulative-effect adjustment of ASC Topic 606 on January 1, 2018 | — | | | — | | | — | | | 1,117 | | | — | | | — | | | 1,117 | |
| Issuance of common stock upon exercise of stock options and in connection with restricted stock units, net of tax | 2,103,727 | | | 20 | | | 14,447 | | | — | | | — | | | — | | | 14,467 | |
| Issuance of common stock under employee stock purchase plan | 328,710 | | | 4 | | | 1,480 | | | — | | | — | | | — | | | 1,484 | |
| Issuance of common stock under public offering, net of issuance costs | 14,156,500 | | | 142 | | | 215,616 | | | — | | | — | | | — | | | 215,758 | |
| Stock-based compensation | — | | | — | | | 14,677 | | | — | | | — | | | — | | | 14,677 | |
| Additional paid-in capital for acquisition of Sangamo France | — | | | — | | | 603 | | | — | | | — | | | — | | | 603 | |
| Non-controlling interest upon acquisition of Sangamo France | — | | | — | | | — | | | — | | | — | | | 1,313 | | | 1,313 | |
| Foreign currency translation adjustment | — | | | — | | | — | | | — | | | (1,129) | | | (19) | | | (1,148) | |
| Net pension losses | — | | | — | | | — | | | — | | | (21) | | | — | | | (21) | |
| Net unrealized loss on marketable securities, net of tax | — | | | — | | | — | | | — | | | (4) | | | — | | | (4) | |
| Net loss | — | | | — | | | — | | | (68,334) | | | — | | | (555) | | | (68,889) | |
| Balances at December 31, 2018 | 102,187,471 | | | 1,022 | | | 929,632 | | | (562,696) | | | (1,440) | | | 739 | | | 367,257 | |
Cumulative-effect adjustment of ASC Topic 842 on January 1, 2019 | — | | | — | | | — | | | 897 | | | — | | | — | | | 897 | |
| Issuance of common stock upon exercise of stock options and in connection with restricted stock units, net of tax | 885,873 | | | 9 | | | 3,668 | | | — | | | — | | | — | | | 3,677 | |
| Issuance of common stock under employee stock purchase plan | 249,364 | | | 2 | | | 2,042 | | | — | | | — | | | — | | | 2,044 | |
| Issuance of common stock under public offering, net of issuance costs | 12,650,000 | | | 127 | | | 136,181 | | | — | | | — | | | — | | | 136,308 | |
| Stock-based compensation | — | | | — | | | 19,330 | | | — | | | — | | | — | | | 19,330 | |
Acquisition of additional shares of Sangamo
France | — | | | — | | | — | | | — | | | — | | | (321) | | | (321) | |
| Issuance costs related to Sangamo France acquisition | — | | | — | | | (25) | | | — | | | — | | | — | | | (25) | |
| Foreign currency translation adjustment | — | | | — | | | — | | | — | | | (1,573) | | | 0 | | (1,573) | |
| Net pension losses | — | | | — | | | — | | | — | | | (28) | | | — | | | (28) | |
| Net unrealized gain on marketable securities, net of tax | — | | | — | | | — | | | — | | | 592 | | | — | | | 592 | |
| Net loss | — | | | — | | | — | | | (95,186) | | | — | | | (233) | | | (95,419) | |
| Balances at December 31, 2019 | 115,972,708 | | | 1,160 | | | 1,090,828 | | | (656,985) | | | (2,449) | | | 185 | | | 432,739 | |
| Issuance of common stock upon exercise of stock options and in connection with restricted stock units, net of tax | 1,395,956 | | | 14 | | | 8,545 | | | — | | | — | | | — | | | 8,559 | |
| Issuance of common stock under employee stock purchase plan | 274,382 | | | 3 | | | 2,012 | | | — | | | — | | | — | | | 2,015 | |
| Issuance of common stock in connection with the Biogen collaboration agreement, net of issuance costs | 24,420,157 | | | 244 | | | 142,282 | | | — | | | — | | | — | | | 142,526 | |
| Stock-based compensation | — | | | — | | | 25,708 | | | — | | | — | | | — | | | 25,708 | |
Acquisition of additional shares of Sangamo
France | — | | | — | | | — | | | — | | | — | | | (927) | | | (927) | |
| Foreign currency translation adjustment | — | | | — | | | — | | | — | | | 8,345 | | | — | | | 8,345 | |
| Net pension losses | — | | | — | | | — | | | — | | | (193) | | | — | | | (193) | |
| Net unrealized loss on marketable securities, net of tax | — | | | — | | | — | | | — | | | (284) | | | — | | | (284) | |
| Net loss | — | | | — | | | — | | | (120,996) | | | — | | | (126) | | | (121,122) | |
| Balances at December 31, 2020 | 142,063,203 | | | $ | 1,421 | | | $ | 1,269,375 | | | $ | (777,981) | | | $ | 5,419 | | | $ | (868) | | | $ | 497,366 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Common Stock | | Additional
Paid-in
Capital | | Accumulated
Deficit | | Accumulated
Other
Comprehensive (Loss) Income | | Non-
Controlling
Interest | | Total
Stockholders’
Equity |
| Shares | | Amount | | | | | |
| Balances at December 31, 2020 | 142,063,203 | | | $ | 1,421 | | | $ | 1,269,375 | | | $ | (777,981) | | | $ | 5,419 | | | $ | (868) | | | $ | 497,366 | |
| | | | | | | | | | | | | |
| Issuance of common stock in at-the-market offering, net of offering expenses | 2,007,932 | | | 20 | | | 27,079 | | | — | | | — | | | — | | | 27,099 | |
| Issuance of common stock upon exercise of stock options and vesting of restricted stock units, net of tax | 1,417,288 | | | 14 | | | 2,375 | | | — | | | — | | | — | | | 2,389 | |
| Issuance of common stock under employee stock purchase plan | 433,107 | | | 4 | | | 3,366 | | | — | | | — | | | — | | | 3,370 | |
| | | | | | | | | | | | | |
| Stock-based compensation | — | | | — | | | 32,956 | | | — | | | — | | | — | | | 32,956 | |
Acquisition of additional shares of Sangamo
France | — | | | — | | | (70) | | | — | | | — | | | (64) | | | (134) | |
| | | | | | | | | | | | | |
| Foreign currency translation adjustment | — | | | — | | | — | | | — | | | (8,351) | | | | | (8,351) | |
| Net pension losses | — | | | — | | | — | | | — | | | (716) | | | — | | | (716) | |
| Net unrealized loss on marketable securities, net of tax | — | | | — | | | — | | | — | | | (339) | | | — | | | (339) | |
| Buy-out of non-controlling interest | — | | | — | | | (943) | | | — | | | — | | | 943 | | | — | |
| Net loss | — | | | — | | | — | | | (178,286) | | | — | | | (11) | | | (178,297) | |
| Balances at December 31, 2021 | 145,921,530 | | | 1,459 | | | 1,334,138 | | | (956,267) | | | (3,987) | | | — | | | 375,343 | |
| Issuance of common stock in at-the-market offering, net of offering expenses | 19,300,743 | | | 193 | | | 84,676 | | | — | | | — | | | — | | | 84,869 | |
| Issuance of common stock upon exercise of stock options and vesting of restricted stock units, net of tax | 994,097 | | | 10 | | | (1,990) | | | — | | | — | | | — | | | (1,980) | |
| Issuance of common stock under employee stock purchase plan | 576,950 | | | 6 | | | 1,765 | | | — | | | — | | | — | | | 1,771 | |
| Stock-based compensation | — | | | — | | | 31,650 | | | — | | | — | | | — | | | 31,650 | |
| | | | | | | | | | | | | |
| Foreign currency translation adjustment | — | | | — | | | — | | | — | | | (4,606) | | | — | | | (4,606) | |
| Net pension gains | — | | | — | | | — | | | — | | | 786 | | | — | | | 786 | |
| Net unrealized loss on marketable securities, net of tax | — | | | — | | | — | | | — | | | (597) | | | — | | | (597) | |
| | | | | | | | | | | | | |
| Net loss | — | | | — | | | — | | | (192,278) | | | — | | | — | | | (192,278) | |
| Balances at December 31, 2022 | 166,793,320 | | | 1,668 | | | 1,450,239 | | | (1,148,545) | | | (8,404) | | | — | | | 294,958 | |
| Issuance of common stock in at-the-market offering, net of offering expenses | 8,249,261 | | | 82 | | | 15,024 | | | — | | | — | | | — | | | 15,106 | |
| Issuance of common stock upon exercise of stock options and vesting of restricted stock units, net of tax | 1,744,118 | | | 18 | | | (1,471) | | | — | | | — | | | — | | | (1,453) | |
| Issuance of common stock under employee stock purchase plan | 1,346,849 | | | 13 | | | 922 | | | — | | | — | | | — | | | 935 | |
| Stock-based compensation | — | | | — | | | 27,363 | | | — | | | — | | | — | | | 27,363 | |
| | | | | | | | | | | | | |
| Foreign currency translation adjustment | — | | | — | | | — | | | — | | | 2,848 | | | — | | | 2,848 | |
| Net pension losses | — | | | — | | | — | | | — | | | (163) | | | — | | | (163) | |
| Net unrealized gain on marketable securities, net of tax | — | | | — | | | — | | | — | | | 1,124 | | | — | | | 1,124 | |
| | | | | | | | | | | | | |
| Net loss | — | | | — | | | — | | | (257,831) | | | — | | | — | | | (257,831) | |
| Balances at December 31, 2023 | 178,133,548 | | | $ | 1,781 | | | $ | 1,492,077 | | | $ | (1,406,376) | | | $ | (4,595) | | | $ | — | | | $ | 82,887 | |
See accompanying Notes to Consolidated Financial Statements.
SANGAMO THERAPEUTICS, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(in thousands)
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2020 | | 2019 | | 2018 |
| Operating Activities: | | | | | |
| Net loss | $ | (121,122) | | | $ | (95,419) | | | $ | (68,889) | |
| Adjustments to reconcile net loss to net cash provided by (used in) operating activities: | | | | | |
| Depreciation and amortization | 5,682 | | | 3,930 | | | 2,359 | |
| Amortization of discount on marketable securities | (825) | | | (4,708) | | | (5,829) | |
| Amortization and other changes in right-of-use assets | 7,687 | | | 5,677 | | | 0 | |
| Gain on free shares | (63) | | | (488) | | | 0 | |
| | | | | |
| Net loss on disposal of property and equipment | 222 | | | 68 | | | 0 | |
| Stock-based compensation | 25,708 | | | 19,330 | | | 14,677 | |
| | | | | |
| Net loss on lease termination | 0 | | | 218 | | | 0 | |
| Build-to-suit leases | — | | | 0 | | | 966 | |
| | | | | |
| Net changes in operating assets and liabilities: | | | | | |
| Interest receivable | (353) | | | (307) | | | (135) | |
| Accounts receivable | 31,685 | | | (32,236) | | | (1,330) | |
| Prepaid expenses and other assets | (10,411) | | | (6,660) | | | (2,828) | |
| | | | | |
| Accounts payable and other accrued liabilities | 10,703 | | | (4,192) | | | (6,372) | |
| Accrued compensation and employee benefits | 6,877 | | | 4,129 | | | 2,604 | |
| Deferred revenues | 216,546 | | | (35,693) | | | 99,364 | |
| Long-term portion of lease liabilities | (3,761) | | | (1,800) | | | 0 | |
| Other non-current liabilities | 1,300 | | | 3,749 | | | 1,963 | |
| Net cash provided by (used in) operating activities | 169,875 | | | (144,402) | | | 36,550 | |
| Investing Activities: | | | | | |
| Acquisition, net of cash acquired | 0 | | | 0 | | | (75,647) | |
| Purchases of marketable securities | (570,779) | | | (443,711) | | | (451,239) | |
| Maturities of marketable securities | 314,570 | | | 404,847 | | | 391,845 | |
| Purchases of property and equipment | (14,714) | | | (20,675) | | | (43,065) | |
| Purchase of additional Sangamo France shares | (704) | | | (262) | | | 0 | |
| | | | | |
| | | | | |
| Net cash used in investing activities | (271,627) | | | (59,801) | | | (178,106) | |
| Financing Activities: | | | | | |
| Proceeds from public offering of common stock, net of issuance costs | 0 | | | 136,308 | | | 215,758 | |
| Proceeds from issuance of common stock in connection with the Biogen collaboration agreement, net of issuance costs | 142,526 | | | 0 | | | 0 | |
| Taxes paid related to net share settlement of equity awards | (765) | | | (422) | | | (254) | |
| Proceeds from issuance of common stock under employee stock purchase plan | 2,015 | | | 2,044 | | | 1,484 | |
| Proceeds from exercise of stock options and restricted stock units | 9,324 | | | 4,099 | | | 14,721 | |
| Net cash provided by financing activities | 153,100 | | | 142,029 | | | 231,709 | |
| Effect of exchange rate changes on cash and cash equivalents | (447) | | | 184 | | | 439 | |
| Net increase (decrease) in cash, cash equivalents, and restricted cash | 50,901 | | | (61,990) | | | 90,592 | |
| Cash, cash equivalents, and restricted cash, beginning of period | 81,928 | | | 143,918 | | | 53,326 | |
| Cash, cash equivalents, and restricted cash, end of period | $ | 132,829 | | | $ | 81,928 | | | $ | 143,918 | |
| Supplemental cash flow disclosures: | | | | | |
| Non-controlling interest for acquisition | $ | 0 | | | $ | 0 | | | $ | 1,313 | |
| Property and equipment included in unpaid liabilities | $ | 4,569 | | | $ | 2,114 | | | $ | 4,953 | |
| Build-to-suit leases included in build-to-suit liabilities | $ | 0 | | | $ | 0 | | | $ | 2,950 | |
| Right-of-use assets obtained in exchange for lease obligations | $ | 1,333 | | | $ | 31,291 | | | $ | 0 | |
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2023 | | 2022 | | 2021 |
| Operating Activities: | | | | | |
| Net loss | $ | (257,831) | | | $ | (192,278) | | | $ | (178,297) | |
| Adjustments to reconcile net loss to net cash used in operating activities: | | | | | |
| Impairment of goodwill and indefinite-lived intangible assets | 89,485 | | | — | | | — | |
| Impairment of long-lived assets | 65,528 | | | — | | | — | |
| Depreciation and amortization | 15,065 | | | 12,108 | | | 9,439 | |
| (Accretion of discount) amortization of premium on marketable securities | (2,329) | | | (1,242) | | | 2,844 | |
| Amortization in operating lease right-of-use assets | 7,127 | | | 8,454 | | | 8,199 | |
| Deferred income tax benefit | (6,335) | | | — | | | — | |
| Stock-based compensation | 27,363 | | | 31,650 | | | 32,956 | |
| Other | 1,119 | | | — | | | (70) | |
| | | | | |
| | | | | |
| | | | | |
| Adjustment of CIRM award liability related to termination of the grant | — | | | — | | | (6,427) | |
| | | | | |
| | | | | |
| Net changes in operating assets and liabilities: | | | | | |
| Interest receivable | 392 | | | (445) | | | 686 | |
| Accounts receivable | 2,755 | | | 2,335 | | | (789) | |
| Prepaid expenses and other assets | 6,133 | | | (4,909) | | | (7,175) | |
| | | | | |
| Accounts payable and other accrued liabilities | 5,589 | | | 13,348 | | | (7,664) | |
| Accrued compensation and employee benefits | (12,690) | | | 941 | | | 373 | |
| Deferred revenues | (161,156) | | | (91,331) | | | (84,202) | |
| Lease liabilities | (5,037) | | | (2,249) | | | (4,340) | |
| | | | | |
| Other non-current liabilities | (20) | | | (9) | | | 1,216 | |
| Net cash used in by operating activities | (224,842) | | | (223,627) | | | (233,251) | |
| Investing Activities: | | | | | |
| | | | | |
| Purchases of marketable securities | (59,551) | | | (277,391) | | | (338,159) | |
| Maturities of marketable securities | 214,500 | | | 354,587 | | | 602,885 | |
| Sales of marketable securities | 19,737 | | | 2,260 | | | 6,870 | |
| Purchases of property and equipment | (21,155) | | | (20,171) | | | (23,278) | |
| Purchase of additional Sangamo France shares | — | | | — | | | (119) | |
| | | | | |
| | | | | |
| Net cash provided by investing activities | 153,531 | | | 59,285 | | | 248,199 | |
| Financing Activities: | | | | | |
| Proceeds from at-the-market offering, net of offering expenses | 15,106 | | | 84,869 | | | 27,099 | |
| | | | | |
| | | | | |
| Taxes paid related to net share settlement of equity awards | (1,453) | | | (2,104) | | | (3,258) | |
| Proceeds from issuance of common stock under employee stock purchase plan | 935 | | | 1,771 | | | 3,369 | |
| Proceeds from exercise of stock options | — | | | 124 | | | 5,648 | |
| Net cash provided by financing activities | 14,588 | | | 84,660 | | | 32,858 | |
| Effect of exchange rate changes on cash and cash equivalents, and restricted cash | 1,483 | | | 1,254 | | | (263) | |
| Net (decrease) increase in cash, cash equivalents, and restricted cash | (55,240) | | | (78,428) | | | 47,543 | |
| Cash, cash equivalents, and restricted cash, beginning of period | 101,944 | | | 180,372 | | | 132,829 | |
| Cash, cash equivalents, and restricted cash, end of period | $ | 46,704 | | | $ | 101,944 | | | $ | 180,372 | |
| Supplemental cash flow disclosures: | | | | | |
| | | | | |
| Property and equipment included in unpaid liabilities | $ | 447 | | | $ | 6,539 | | | $ | 1,535 | |
| Tenant improvement allowance included in contra-lease liability | $ | — | | | $ | 243 | | | $ | — | |
| Buy-out of non-controlling interest | $ | — | | | $ | — | | | $ | 943 | |
| Right-of-use assets obtained in exchange for lease obligations | $ | — | | | $ | — | | | $ | 10,418 | |
See accompanying Notes to Consolidated Financial Statements
SANGAMO THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 1 – ORGANIZATION, BASIS OF PRESENTATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Organization and Description of Business Overview
Sangamo Therapeutics, Inc. (“Sangamo” or “the Company”) was incorporated in the State of Delaware in June 1995 and changed its name from Sangamo Biosciences, Inc. in January 2017. Sangamo is a clinical-stage genomic medicine company committed to translating ground-breaking science into medicines that transform the lives of patients and families afflicted with serious neurological diseases. The Company believes its zinc finger (“ZF”) epigenetic regulators are ideally suited to potentially address devastating neurology disorders and its capsid engineering platform has demonstrated the ability to expand delivery beyond currently available intrathecal delivery capsids, including in the central nervous system (“CNS”) in preclinical studies.
In 2023, the Company announced its strategic transformation into a neurology-focused genomic medicine company focused on developing epigenetic regulation therapies designed to address serious neurological diseases using its platform technologies in gene therapy, cell therapy and genome engineering.novel adeno-associated virus (“AAV”) capsid delivery technology.
Basis of Presentation
The accompanying Consolidated Financial Statements include the accounts of the Company and its subsidiaries and have been prepared in conformity with generally accepted accounting principles in the United States of America (“U.S. GAAP”) and includepursuant to the accountsrules and regulations of the CompanyUnited States Securities and its subsidiaries.Exchange Commission (“SEC”). All intercompany balances and transactions have been eliminated in the Consolidated Financial Statements. For consolidated entities where the Company owns or is exposed to less than 100% of the economics, the Company records net loss attributable to non-controlling interests on its Consolidated Statements of Operations equal to the percentage of the economic or ownership interest retained in such entities by the respective non-controlling parties.
Liquidity, Going Concern, and Management’s PlanCapital Resources
Sangamo is currently working on a number of long-term development projects that involve experimental technologies. The projects maywill require several years and substantial expenditures to complete and ultimately may be unsuccessful. The Company plans to financeIn recent years, the Company’s operations with available cash resources,have been funded primarily through collaborations and strategic partnerships, funds, research grants and from the issuance of equity securities. As of December 31, 2023, the Company had capital resources of $81.0 million consisting of cash, cash equivalents, and marketable securities.
Under Accounting Standard Codification (“ASC”) Topic 205-40, Presentation of Financial Statements—Going Concern (“ASC Topic 205-40”), the Company has the responsibility to evaluate whether conditions and/or debt securities. Sangamo believesevents raise substantial doubt about its ability to meet its future financial obligations as they become due within one year after the date that the Consolidated Financial Statements are issued. As required under ASC Topic 205-40, management’s evaluation should initially not take into consideration the potential mitigating effects of management’s plans that have not been fully implemented as of the date the Consolidated Financial Statements are issued. When substantial doubt exists, management evaluates whether the mitigating effects of its availableplans sufficiently alleviates the substantial doubt about the Company’s ability to continue as a going concern. The mitigating effects of management’s plans, however, are only considered if both (i) it is probable that the plans will be effectively implemented within one year after the date that the financial statements are issued, and (ii) it is probable that the plans, when implemented, will mitigate the relevant conditions or events that raise substantial doubt about the entity’s ability to continue as a going concern within one year after the date that the financial statements are issued. Generally, to be considered probable of being effectively implemented, the plans must have been approved by the Company’s board of directors before the date that the financial statements are issued.
The Company’s history of significant losses, its negative cash flows from operations, its limited liquidity resources currently on hand, and its dependence on additional financing to fund its operations after the current resources are exhausted raise substantial doubt about its ability to continue to operate as a going concern within one year after the date that the Consolidated Financial Statements are issued. The Company’s current operating plan, its cash, cash equivalents, and marketable securities as of December 31, 2020, when combined with additional capital raises and2023 are expected revenues from collaborations, strategic partnerships and research grants, will be adequate to fundallow the Company to meet its operations at least throughliquidity requirements only into the next 12 months fromthird quarter of 2024, which is less than one year following the date these Consolidated Financial Statements are issued. Sangamo will require substantial
Successful completion of the Company’s development programs and, ultimately, the attainment of profitable operations are dependent upon future events, including obtaining adequate financing to support the Company’s cost structure
and operating plan. Management’s plans include, among other things, pursuing one or more of the following steps to raise additional financial resourcescapital, none of which can be guaranteed or are entirely within the Company’s control:
•raise funding through the sale of the Company’s common stock;
•raise funding through debt or royalty financing; and
•establish collaborations with potential partners to completeadvance the development and commercializationCompany’s product pipeline.
If the Company is unable to raise capital on acceptable terms, or at all, or if it is unable to procure collaboration arrangements or external direct investments to advance its programs, the Company would be required to discontinue some or all of its product candidates.operations or develop and implement a plan to further extend payables, reduce overhead or scale back its current operating plan until sufficient additional capital is raised to support further operations. There can be no assurance that such a plan would be successful. Additional capital may not be available on terms acceptable to the Company on a timely basis, on terms that are acceptable or at all. In particular, the perception of the Company’s ability to continue to operate as a going concern may make it more difficult to obtain financing for the continuation of its operations, particularly in light of currently challenging macroeconomic and market conditions. Further, the Company may be unable to attract new investments as a result of the speculative nature of its newly reprioritized core neurology preclinical programs. If adequate funds are not available to the Company on a timely basis, or ifat all, the termsCompany will be required to take additional actions to address its liquidity needs, including additional cost reduction measures such as further reducing operating expenses and delaying, reducing the scope of, potential funding sources are unfavorable, the Company’sdiscontinuing or altering its research and development activities, which would have a material adverse effect on its business and abilityprospects, or the Company may be required to developcease operations entirely, liquidate all or a portion of its technologyassets, and/or seek protection under the U.S. Bankruptcy Code.
The accompanying Consolidated Financial Statements have been prepared assuming the Company will continue to operate as a going concern, which contemplates the realization of assets and therapeutic products would be harmed. Furthermore,the settlement of liabilities in the normal course of business. The Consolidated Financial Statements do not include any salesadjustments to reflect the possible future effects on the recoverability and classification of additional equity securitiesassets or the amounts of liabilities that may result in dilutionfrom uncertainty related to the Company’s stockholders, and any debt financing may include covenants that restrict the Company’s business.ability to continue as a going concern.
Summary of Significant Accounting Policies
Use of Estimates
The preparation of the accompanying Consolidated Financial Statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the amounts reported in the Consolidated Financial Statements and the accompanying notes. On an ongoing basis, management evaluates its estimates including critical accounting policies or estimates related to revenue recognition, including the estimated fair value of common shares issued as part of the transaction price, clinical trial accruals, income taxes, fair value of assets and liabilities, including from acquisitions, useful lives and impairment of long-lived assets, and stock-based compensation. Estimates are based on historical experience and on various other market specific and other relevant assumptions that the Company believes to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results could differ from those estimates.
During the year ended December 31, 2020,2023, the Company recorded additional revenue related to changes in estimates in connection with the collaboration agreement with Kite Pharma, Inc., a Gilead Sciences, Inc. subsidiary (“Kite”). These changes in estimates were driven by reductions in the estimated future level of the Company’s research and development services in March and September 2023, and as a result, future project costs. These changes resulted in an increase in proportional cumulative performance on this collaboration and increased revenue by $13.9 million, decreased net loss by $13.9 million, and decreased the Company’s basic and diluted net loss per share by $0.08 for the year ended December 31, 2023.
During the year ended December 31, 2021, the Company recorded adjustments to revenue related to changes in estimates in connection with the collaboration agreementsagreement with Sanofi GenzymeS.A. (“Sanofi”) and Pfizer Inc. (“Pfizer”). These changes in estimates were driven by changesa change in project scope and related project costs in September 2021 and subsequent notification of termination of the collaboration agreement, effective June 28, 2022, which resulted in changes to the measure of proportional cumulative performance. These adjustments increaseddecreased revenue by $8.9$1.6 million, decreasedincreased net loss by $8.9$1.6 million and decreasedincreased the Company’s basic and diluted net loss per share by $0.06$0.01 for the year ended December 31, 2020.
During the year ended December 31, 2019, the Company recorded adjustments to revenue related to a change in estimate in connection with the giroctocogene fitelparvovec collaboration agreement with Pfizer Inc. (“Pfizer”). This change in estimate was driven by changes in project scope and related project costs which resulted in changes to the measure of proportional cumulative performance. These adjustments increased revenue by $5.7 million, decreased net loss by $5.7 million and decreased the Company’s basic net loss per share by $0.05 for the year ended December 31, 2019.
Revenue Recognition
The Company accounts for its revenues pursuant to the provisions of Accounting Standards Codification (“ASC”)ASC Topic 606, Revenue from Contracts with Customers (“ASC Topic 606”). The Company’s contract revenues are derived from collaboration agreements including licensing arrangements and research activity grants.services. Research and licensing agreements typically include nonrefundable upfront signing or license fees, payments at negotiated rates for time incurred by Company researchers, third-party cost reimbursements, for research services, minimumadditional target selection fees, sublicense fees, milestone payments tied to ongoing development and product
commercialization, and royalties on future licensee’slicensees’ product sales. The Company has agreements with both fixed and variable consideration.All funds received from the Company’s collaboration partners are generally not refundable. Non-refundable upfront fees and funding of research and development activities are considered fixed while milestone payments are generally identified as variable consideration. Sangamo’s research grants are typically multi-year agreements and provide forat the reimbursement of qualified expenses for research and development as defined under the termscommencement of the grant agreement. Revenues under research grant agreements are generally recognized whencontract. All other fees represent variable consideration in contracts. For contracts that contain a provision where the related qualified research expenses are incurred.Company reimburses its customer for certain costs they incur and where the Company does not acquire any distinct goods or services in exchange for such payments, the Company accounts for it as a reduction to the contract transaction price. Deferred revenue primarily represents the portion of researchnonrefundable upfront fees or licensemilestone payments received but not earned.
In determining the appropriate amount of revenue to be recognized as the Company fulfills its obligations under its agreements, the Company performs the following steps: (i) identification of the promised goods or services in the contract; (ii) determination of whether the promised goods or services are performance obligations, including whether they are distinct in the context of the contract; (iii) measurement of the transaction price, including the constraint on variable consideration; (iv) allocation of the transaction price to the performance obligations based on estimated selling prices; and (v) recognition of revenue when (or as) the Company satisfies each performance obligation.
A performance obligation is a promise in a contract to transfer a distinct good or service toMost of the customer and is the unit of account in ASC Topic 606. The Company’s performance obligations include license rights,in its collaboration agreements represent distinct bundles of licenses of intellectual property and research and development services, with these components being individually non-distinct. Options to license the Company’s intellectual property and/or acquire research and development services associated with regulatory submission and approval processes. also represent performance obligations when they grant customers a material right, e.g. a right to a discount the customer would not have received if they did not purchase the Company’s services under the existing contract.
Revenues from bundles of licenses of intellectual property and research and development services earned under collaboration agreements are generally recognized as revenue as the related services are provided. Revenues from non-refundable upfront fees are recognized over time eitherusing a proportional performance method. Under this method, revenue is recognized by measuring progress towards satisfaction of the relevant performance obligation using a measure that best depicts the progress towards satisfaction of the relevant performance obligation. For most of the Company’s agreements the measure of progress is an input method (i.e., cumulative actual costs incurred relative to total estimated costs) ormeasure based on a straight-line basis when alevel of effort incurred, which includes the value of actual time by Company researchers plus third-party cost reimbursements.
Consideration allocated to options that include material rights is deferred until the options are exercised or expire. The exercise of such options is accounted for as contract continuation, with target selection fees and estimated variable consideration included in the transaction price at that time and allocated specifically to the respective target’s performance obligation is expected to be satisfied evenly over a period of time (or when the entity has a stand-ready obligation). obligation.
Significant management judgment is required to determine the level of effort required under an arrangement, and the period over which the Company expects to complete its performance obligations under the arrangement, which may include total internal personnel costs and external costs to be incurred as well as, in certain cases, the estimated stand-ready obligation period.arrangement. Changes in these estimates can have a material effect on revenue recognized. If the Company cannot reasonably estimate when its performance obligations either are completed or become inconsequential, then revenue recognition is deferred until the Company can reasonably make such estimates. The Company includes the unconstrained amount of estimatedFor variable consideration, in the transaction price. The amount included in the transaction price is constrained to the amount for which it is probable that a significant reversal of cumulative revenue recognized will not occur. At the end of each subsequent reporting period, the Company re-evaluates the estimated variable consideration included in the transaction price and any related constraint and, if necessary, adjusts its estimate of the overall transaction price. RevenueA cumulative catch-up is then recognized overrecorded in the remaining estimatedcurrent period to reflect the updated transaction price and the updated measure of performance using the cumulative catch-up method.progress. The estimated period of performance and projectlevel of effort, including the value of Company researchers’ time and third-party costs, such as personnel and manufacturing cost, are reviewed quarterly and adjusted, as needed, to reflect the Company’s current assumptions regarding the timing of its deliverables.expectations.
As part of the accounting for these arrangements, the Company must develop assumptions that require judgment to determine the stand-alone selling price of each performance obligation identified in the contract. The Company uses key assumptions to determine the stand-alone selling price, which may include forecasted revenues, development timelines, reimbursement rates for personnel costs, discount rates and probabilities of exercise of technical and regulatory success. Related costssuccess, and expenses underthe expected level of effort for research and development services.
Contract modifications occur when the price and/or scope of an arrangement changes. If the modification consists of adding new distinct goods or services in exchange for consideration that reflects standalone selling prices of these arrangements have historically approximatedgoods and services, the revenues recognized.modification is accounted for as a separate contract with the customer. Otherwise, if the remaining goods and services are distinct from those previously provided, the existing contract is considered terminated, and the remaining consideration is allocated to the remaining goods and services as if this was a newly signed contract. If the remaining goods and services are not distinct from those previously provided, the effects of the modification are accounted for in a manner similar to the effect of a change in the estimated measure of progress, with cumulative catch-up in revenue recorded at the time of the modification. If some of the remaining goods and services are distinct from those previously provided and others are not, to account for the effects of the modification the Company applies principles consistent with the objectives of the modification accounting.
Revenues from major collaboration agreements and research activity grantslicensing agreements as a percentage of total revenues were as follows: | | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2023 | | 2022 | | 2021 |
| | | | | |
| Biogen MA, Inc. | 77 | % | | 26 | % | | 38 | % |
| Kite Pharma, Inc. | 12 | % | | 35 | % | | 23 | % |
| Novartis Institutes for BioMedical Research, Inc. | 7 | % | | 36 | % | | 34 | % |
| | | | | |
| | | | | |
| Other licensing agreements | 4 | % | | 3 | % | | 5 | % |
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2020 | | 2019 | | 2018 |
| Pfizer Inc. | 40 | % | | 40 | % | | 47 | % |
| Kite Pharma, Inc. | 24 | % | | 34 | % | | 30 | % |
| Biogen MA, Inc. | 24 | % | | 0 | | | 0 | |
| Sanofi Genzyme | 5 | % | | 22 | % | | 16 | % |
| Novartis Institutes for BioMedical Research, Inc. | 4 | % | | 0 | | | 0 | |
amounts billed to the Company’s collaboration partners for cost reimbursements for research services, sublicensing revenue, and royalty payments. Receivables from collaborations are typically unsecured and are concentrated in the biopharmaceutical industry. Accordingly, the Company may be exposed to credit risk generally associated with biopharmaceutical companies or specific to its collaboration agreements. To date,The Company records trade receivables net of allowances for credit losses. The Company applies an aging method to estimate credit losses and considers its historical loss information, adjusted to account for current conditions, and reasonable and supportable forecasts of future economic conditions affecting its customers. Accounts receivable as of December 31, 2023 and 2022 were $0.9 million and $3.7 million, respectively, and the Company hashad not experiencedincurred any losses related to these receivables.
Funds received fromaccounts receivable. As of December 31, 2023 and 2022, the Company’spercentage of accounts receivable by collaboration partners are generally not refundablewho individually accounted for 10% or more of accounts receivable were as follows:
| | | | | | | | | | | |
| As of December 31, |
| 2023 | | 2022 |
| Sigma-Aldrich Corporation | 78 | % | | — | % |
| Kite Pharma, Inc. | 18 | % | | 19 | % |
| Novartis Institutes for BioMedical Research, Inc. | — | % | | 59 | % |
| Biogen MA, Inc. | — | % | | 14 | % |
| | | |
| | | |
| | | |
| | | |
Impairment of Goodwill, Indefinite-lived Intangible Assets and are recorded as revenue as it fulfills its performance obligations. The Company’s performance obligations are satisfied over time (i.e., stand ready obligations) or using the input method (i.e., cumulative actual costs incurred relative to total estimated costs). Revenue is also recognized when the Company has incurred qualified research and development costs that are reimbursable from its collaboration partners and when there is reasonable assurance that such costs will be reimbursed. Any payments received from a collaboration partner in advance of the completion of the relevant performance obligation are recorded as deferred revenue.
Business Combinations
The Company accounts for acquisitions using the acquisition method of accounting, which requires that assets acquired, including in-process research and development (“IPR&D”) projects, liabilities assumed and any non-controlling interests in the acquired target in an acquisition be recorded at their fair values as of the acquisition date on the Company’s Consolidated Balance Sheets. Any excess of purchase consideration over the fair value of net assets acquired is recorded as goodwill. The determination of estimated fair value requires the Company to make significant estimates and assumptions. As a result, the Company may record adjustments to the fair values of assets acquired and liabilities assumed within the measurement period (up to one year from the acquisition date) with the corresponding offset to goodwill. Transaction costs associated with business combinations are expensed as they are incurred.
Goodwill and IntangibleLong-lived Assets
Goodwill represents the excess of the purchase consideration transferred over the estimated fair values of assets acquired and liabilities assumed in a business combination. Intangible assets with indefinite useful lives are related to purchased acquired in-process research and development (“IPR&D&D”) projects and are initially measured at their respective fair values as of the acquisition date. Goodwill and indefinite-lived intangible assets with indefinite useful lives are not amortized. Intangible assets related to IPR&D projects are considered to be indefinite-lived until the completion or abandonment of the associated research and development efforts. If and when development is complete, which generally occurs if and when regulatory approval to market a product is obtained, the associated assets would be deemed finite-lived and would then be amortized based on their respective estimated useful lives at that point in time. The Company tests goodwill
Goodwill and indefinite-lived intangible assets are assessed for impairment on an annual basis and between annual tests ifwhenever events and circumstances indicate that these assets may be impaired. The Company evaluates the Company becomes awarecarrying value of anylong-lived assets, which include property and equipment, leasehold improvements and right-of-use assets, for impairment whenever events occurring or changes in circumstances indicate that would indicatethe carrying amounts of the asset may not be fully recoverable.
In testing for goodwill impairment, the Company has the option of first performing a qualitative assessment to determine whether it is more likely than not that the fair valuesvalue of the assets are below their respectivereporting unit is less than its carrying amounts. As of December 31, 2020, 0 impairment of goodwillamount. If the Company elects to bypass the qualitative assessment, or indefinite-lived intangible assets was identified.
Valuation of Long-Lived Assets
Long-lived assets, including property and equipment and finite-lived intangible assets, are reviewed for impairment whenever facts or circumstances either internally or externally may suggestif a qualitative assessment indicates it is more likely than not that the carrying value exceeds its fair value, the Company performs a quantitative goodwill impairment test to compare the fair value of its reporting unit to its carrying value, including goodwill. If the carrying value, including goodwill, exceeds the reporting unit’s fair value, the Company will recognize an asset mayimpairment loss for the amount by which the carrying amount exceeds the reporting unit’s fair value (but not be recoverable. Recoverability of these assets is measured by comparisonin excess of the carrying value of goodwill).
In performing each annual impairment assessment and any interim impairment assessment for its indefinite-lived intangible assets, the Company determines if it should qualitatively assess whether it is more likely than not the fair value of its IPR&D asset is less than its carrying amount of each asset(the qualitative impairment test). If the Company concludes that is the case, or elects not to use the future undiscounted cash flows expected to result fromqualitative impairment test, the useCompany will proceed with quantitatively determining the fair value of the IPR&D asset and comparing its eventual disposition. If the asset is consideredfair value to be impaired,its carrying value to determine the amount of impairment, if any (the quantitative impairment test).
In performing the qualitative impairment test, the Company considers the results of the most recent quantitative impairment test and identifies the most relevant drivers of the fair value for the IPR&D asset. The most relevant drivers of fair value identified are consistent with the assumptions used in the quantitative estimate of the IPR&D asset. Using these drivers of fair value, the Company identifies events and circumstances which may have an effect on the fair value of the IPR&D asset since the last time the IPR&D’s fair value was quantitatively determined. The Company then weighs these factors to determine and conclude if it is measurednot more likely than not the IPR&D asset is impaired. If it is more likely than not the IPR&D asset is impaired, the Company proceeds with quantitatively determining the fair value of the IPR&D asset.
When performing the quantitative impairment test, the Company uses the income approach to determine the fair value of its IPR&D asset. This approach calculates fair value by estimating the after-tax cash flows attributable to an in-process project over its useful life and then discounting these after-tax cash flows back to a present value. This estimate includes judgmental assumptions regarding the estimates that market participants would make in evaluating the IPR&D asset, including the probability of successfully completing clinical trials and obtaining regulatory approval, the timing of and the expected costs to complete IPR&D projects, future net cash flows from potential drug sales, which are based on estimates of the sales price of the drug, the size of the patient population and cure rate, its competitive position in the marketplace, and appropriate discount and tax rates. Any impairment to be recorded is calculated as the difference between the estimated fair value and the carrying value of the IPR&D asset on the Company’s Consolidated Balance Sheet.
If a change in circumstance occurs that indicates long-lived assets may be impaired, the Company performs a test of recoverability by comparing the carrying value of the asset or asset group to its undiscounted expected future cash flows. The long-lived asset evaluation is performed at the asset group level, i.e., the lowest level for which identifiable cash flows are largely independent of the cash flows of other assets and liabilities. If this review indicates that the carrying amount of the asset group is not recoverable, an impairment loss is measured as the amount by which the carrying amount of an asset group exceeds its fair value. Any impairment loss is allocated to the long-lived assets of the group on a pro rata basis using the relative carrying amounts of those assets, except that the carrying amount of an individual asset shall not be reduced below its fair value.
Factors that may indicate potential impairment and trigger an impairment test include, but are not limited to, general macroeconomic conditions, conditions specific to the industry and market, an adverse change in legal factors, impairment of indefinite-lived intangible assets, business climate or operational performance of the business, and sustained decline in the stock price and market capitalization compared to the net book value.
Calculating the fair value of a reporting unit, an asset group and an individual asset involves significant estimates and assumptions. These estimates and assumptions include, among others, projected future cash flows, risk-adjusted discount rates, future economic and market conditions, and the impaired asset. Asdetermination of December 31, 2020, 0appropriate market comparables. Changes in these factors and assumptions used can materially affect the amount of impairment of any long-lived assetsloss recognized in the period the asset was identified.considered impaired.
Fair Value Measurements
The carrying amounts for financial instruments consisting of cash and cash equivalents, accounts receivable, accounts payable and other accrued liabilities approximate fair value due to their shortshort-term maturities. Marketable securities are stated at their estimated fair values. The free shares asset or liability is measured using a binomial-lattice pricing model and is reviewed each reporting period and adjusted, as needed, and is expected to approximate fair value.
Cash, Cash Equivalents, and Restricted Cash
Sangamo considers all highly-liquidhighly liquid investments purchased with original maturities of three months or less at the purchase date to be cash equivalents. Cash and cash equivalents consist of cash, deposits in demand money market accounts and commercial paper.U.S. government-sponsored entity debt securities. Restricted cash consists of a letter of credit for $1.5 million, asrepresenting a deposit for the lease of office and research and development laboratory facilities in Brisbane, lease.California.
A reconciliation of cash, cash equivalents, and restricted cash reported within the accompanying Consolidated Balance Sheets to the amounts reported within the accompanying Consolidated Statements of Cash Flows is as follows (in thousands):
| | | | | | | | | | | | | | | | | | | |
| As of December 31, |
| 2023 | | 2022 | | 2021 | | |
| Cash and cash equivalents | $ | 45,204 | | | $ | 100,444 | | | $ | 178,872 | | | |
| | | | | | | |
| Non-current restricted cash | 1,500 | | | 1,500 | | | 1,500 | | | |
| Cash, cash equivalents, and restricted cash as reported within the Consolidated Statements of Cash Flows | $ | 46,704 | | | $ | 101,944 | | | $ | 180,372 | | | |
| | | | | | | | | | | | | | | | | | | |
| As of December 31, |
| 2020 | | 2019 | | 2018 | | |
| Cash and cash equivalents | $ | 131,329 | | | $ | 80,428 | | | $ | 140,418 | | | |
| | | | | | | |
| Non-current restricted cash | 1,500 | | | 1,500 | | | 3,500 | | | |
| Cash, cash equivalents and restricted cash as reported within the Consolidated Statements of Cash Flows | $ | 132,829 | | | $ | 81,928 | | | $ | 143,918 | | | |
Marketable Securities
Sangamo classifies its marketable securities as available-for-sale and records its investments at estimated fair value based on quoted market prices or observable market inputs of almost identical assets, with the unrealized holding gains and losses included in accumulated other comprehensive lossincome (loss) (“AOCI”). The Company classifies those investments that are not required for use in current operations and that mature in more than 12 months as non-current marketable securities in the accompanying Consolidated Balance Sheets.
The Company’s investments are subject to a periodic impairment review. The Company recognizes an impairment charge, if material, when a decline in the fair value of its investments below the cost basis is judged to be other-than-temporary. The Company considers various factors in determining whether to recognize an impairment charge, including the length of time and extent to which the fair value has been less than the Company’s cost basis, the Company’s financial condition and near-term prospects of the investee and the Company’sits intent and ability to hold the investment for a period of time sufficient to allow for any anticipated recovery in the market value. Realized gains and losses on marketable securities are included in interest and other income, net, which are determined using the specific identification method. Credit losses related to the marketable securities are recorded in interest and other income, net in the Consolidated Statements of Operations through an allowance for credit losses rather than as a reduction in the amortized cost basis of the securities.
If the Company intends to sell, or if it is more likely than not that the Company will be required to sell, a security before recovery of its amortized cost basis, the allowance for credit losses is written off, and the amortized cost of the security is written down to its fair value, with any incremental impairment charge recognized in earnings. This also results in a reversal of any unrealized gains and losses for this security that were previously included in AOCI. Impairment charges are included in interest and other income, net in the Consolidated Statements of Operations.
Concentrations of Credit Risk and Other Risks
Cash, cash equivalents, and marketable securities consist of financial instruments that potentially subject the Company to a concentration of credit risk to the extent of the fair value recorded in the Consolidated Balance Sheets. The Company invests cash that is not required for immediate operating needs primarily in highly liquid instruments with high credit ratings which are expected tothat bear minimal risk. The Company has established policies relating to the quality, diversification, and maturities of securities to enable the Company to manage its credit risk. The Company is exposed to credit risk in the event of a default by the financial institutions or issuers of investments holding its cash, cash equivalents, and marketable securities and issuers of investments to the extent recorded on the Consolidated Balance Sheets.
Certain materials and key components that the Company utilizes in its operations are obtained through single suppliers. Since the suppliers of key components and materials must be named in an investigational new drug (“IND”) application filed with the U.S. Food and Drug Administration (“FDA”) for a product, significant delays can occur if the qualification of a new supplier is required. If delivery of material from the Company’s suppliers waswere interrupted for any reason, the Company may be unable to supply any of its product candidates for clinical trials.
Property and Equipment
Property and equipment are stated at cost, less accumulated depreciation and amortization. Depreciation is calculated using the straight-line method based on the estimated useful lives of the related assets which is generally three to five years. For leasehold improvements, amortization is calculated using the straight-line method based on the shorter of the useful life or the lease term. The Company reviews its property and equipment for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable.
Research and Development Expenses
Research and development expenses consist primarily of personnel costs, including salaries, benefits and stock-based compensation, restructuring charges, clinical studies performed by contract research organizations, materials and supplies and overhead allocations consisting of various support and facility-related costs.Research and development costs are expensed as incurred.
General and Administrative Expenses
General and administrative expenses consist of finance, human resources, legal and other administrative activities. These expenses consist primarily of personnel costs, including salaries, benefits and stock-based compensation, restructuring charges, facilities and overhead costs, legal expenses, and other general and administrative costs.
Stock-based Compensation
The Company measures and recognizes compensation expense for all stock-based payment awards made to Sangamo employees and directors, including employee share options, restricted stock units (“RSUs”) and employee stock purchases related to the Employee Stock Purchase Plan (“ESPP”) based on estimated fair values at the award grant date. The fair value of stock-based awards is amortized over the vesting period of the award using a straight-line method.
To estimate the fair value of an award, the Company uses the Black-Scholes option pricing model. This model requires inputs such as expected life, expected volatility, expected dividend yield of stock and risk-free interest rate. These inputs are subjective and generally require significant analysis and judgment to develop. While estimates of expected life and volatility are derived primarily from the Company’s historical data, the risk-free rate is based on the U.S. Treasury yield curve in effect at the time of grant commensurate with the expected life assumption. The Company accounts for forfeitures in the period they occur.
Income Taxes
Income tax expense has been providedcalculated using the liability method. Deferred tax assets and liabilities are determined based on the difference between the financial statement and tax bases of assets and liabilities as measured by the enacted tax rates that will be in effect when these differences reverse. The Company provides a valuation allowance against net deferred tax assets if, based upon the available evidence, it is not more likely than not that the deferred tax assets will be realized.
The Company recognizes a tax benefit from an uncertain tax position only if it is more likely than not that the tax position will be sustained on examination by the taxing authorities, based on the technical merits of the position. The tax benefits recognized in the Company’s Consolidated Financial Statements from such positions are measured based on the largest benefit that has a greater than 50% likelihood of being realized. The Company recognizes interest and penalties associated with tax matters as part of the income tax provision and includes accrued interest and penalties with the related income tax liability within other accrued liabilities on its Consolidated Balance Sheets. The Company evaluates uncertain tax positions on a regular basis and makes adjustments to these accruals when facts and circumstances change, such as the closing of a tax audit or the refinement of an estimate.
Leases
The Company adopted the Accounting Standard Update (“ASU”) No. 2016-02, Leases (Topic 842) on January 1, 2019, using the modified retrospective approach with a cumulative-effect adjustment. The Company determines if an arrangement is or contains a lease at inception by assessing whether the arrangement contains an identified asset and whether it has the right to control the identified asset. Right-of-use (“ROU”) assets represent the Company’s right to use an underlying asset for the lease term and lease liabilities represent the Company’s obligation to make lease payments arising from the lease. Lease liabilities are recognized at the lease commencement date based on the present value of future lease payments over the lease term. ROURight-of-use assets are based on the measurement of the lease liability and also include any lease payments made prior to or on lease commencement and exclude lease incentives and initial direct costs incurred, as applicable.
As the implicit rate in the Company’s leases is generally unknown, the Company uses its incremental borrowing rate based on the information available at the lease commencement date in determining the present value of remaining lease payments. The incremental borrowing rate represents an estimate of the interest rate the Company would incur at lease commencement to borrow an amount equal to the lease payments on a collateralized basis over the term of a lease in a similar economic environment. The Company considers its credit risk, term of the lease, and total lease payments and adjusts for the impacts of collateral, as necessary, when calculating its incremental borrowing rates. The lease terms may include options to extend or terminate the lease when it is reasonably certain the Company will exercise any such options. Rent expense for the Company’s operating leases is recognized on a straight-line basis over the lease term. The Company will evaluate the lease arrangement for impairment whenever events or changes in circumstances indicate that the carrying amounts of the right-of-use asset may not be fully recoverable. To the extent an impairment of the right-of-use asset is identified, the Company will recognize lease impairment and subsequently amortize the remaining lease asset on a straight-line basis (unless another systematic basis is more representative of the pattern in which the Company expects to consume the future economic benefits from the asset) from the date of impairment to the earlier of the end of the right-of-use asset’s useful life or the end of the lease term.
The Company has elected not to not separate lease and non-lease components for its real estate and copier leases and, as a result, accounts for any lease and non-lease components as a single lease component. The Company has also elected not to not apply the recognition requirement to any leases with a term of 12 months or less and does not include an option to purchase the underlying asset that the Company is reasonably certain to exercise.
Foreign Currency Translation
The functional currency of the Company’s foreign subsidiaries is primarily the Euro. Assets and liabilities denominated in foreign currencies are translated to U.S. dollars using the exchange rates at the balance sheet date. Foreign currency translation adjustments are recorded as a component of AOCI within stockholders’ equity. Revenues and expenses from the Company’s foreign subsidiaries are translated using the monthly average exchange rates in effect during the period in which the transactions occur. Foreign currency transaction gains and losses are recorded in interest and other income, net, on the Company’s Consolidated Statements of Operations.
Net Loss Per Share
Basic net loss per share attributable to Sangamo Therapeutics, Inc. stockholders has been computed by dividing net loss attributable to Sangamo Therapeutics, Inc. stockholders by the weighted-average number of shares of common stock outstanding during the period. Diluted net loss per share attributable to Sangamo Therapeutics, Inc. stockholders is calculated by dividing net loss attributable to Sangamo Therapeutics, Inc. stockholders by the weighted-average number of shares of common stock plus potentially dilutive securities outstanding during the period.
The total number of shares subject to stock options and RSUs outstanding and the ESPP shares reserved for issuance, which are all anti-dilutive, were excluded from consideration in the calculation of diluted net loss per share attributable to Sangamo Therapeutics, Inc. stockholders. Stock options and RSUs outstanding and ESPP shares reserved for issuance as of December 31, 2020, 20192023, 2022 and 20182021 were 14,237,871, 10,750,550,21,254,556, 18,560,755, and 9,048,793,15,159,908, respectively.
Segments
The Company operates in 1one segment. Management uses one measure of profitability and does not segregate its business for internal reporting. As of December 31, 20202023 and 2019, substantially all2022, the majority of the Company’s assetsproperty and equipment were maintained in the United States. For the years ended December 31, 2020, 20192023, 2022 and 2018, substantially2021, all of the Company’s revenues and operating expenses were generated and incurred in the United States.
Restructuring
The Company records employee severance costs based on whether the termination benefits are provided under an on-going benefit arrangement or under a one-time benefit arrangement. The Company accounts for on-going termination benefit arrangements, such as those arising from employment agreements, applicable regulations or past practices, in accordance with ASC Topic 712, Compensation—Nonretirement Postemployment Benefits (“ASC Topic 712”). Under ASC 712, liabilities for post-employment benefits related to past services and that vest or are accumulated over time are recorded at the time the obligations are probable of being incurred and can be reasonably estimated. The Company accounts for one-time employment benefit arrangements in accordance with ASC Topic 420, Exit or Disposal Cost Obligations (“ASC Topic 420”). One-time termination benefits are expensed at the date the entity notifies the employee, unless the employee must provide future service over a period extending past the minimum notification period, in which case the benefits are expensed ratably over the future service period. Other associated costs are recognized in the period in which the liability is incurred.
Costs incurred to terminate contracts are recognized upon their termination, e.g., when notice of termination is provided to the counterparty. Costs related to contracts without future benefit are recognized at the cease-use date. Other exit-related costs are recognized as incurred.
Recent Accounting Pronouncements
Recently Adopted
Collaborative ArrangementsNone.
Not Yet Adopted
In November 2018,2023, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) 2023-07, Segment Reporting (Topic 280): Improvements to Reportable Segment Disclosures (“ASU 2023-07”), which requires public entities to disclose information about their reportable segments’ significant expenses and other segment items on an interim and annual basis. Public entities with a single reportable segment are required to apply the disclosure requirements in ASU 2023-07, as well as all existing segment disclosures and reconciliation requirements in ASC Topic 280, Segment Reporting on an interim and annual basis. ASU 2023-07 is effective for fiscal years beginning after December 15, 2023, and for interim periods within fiscal years beginning after December 15, 2024, with early adoption permitted. The Company is currently evaluating the impact of adopting ASU 2023-07.
In December 2023, the FASB issued ASU No. 2018-18, Collaborative Arrangements (ASC Topic 808): Clarifying the Interaction between Topic 808 and Topic 6062023-09, (“Income Taxes (Topic 740): Improvements to Income Tax Disclosures (“ASU 2018-18”2023-09”), which clarifies that certain transactions between participantsrequires public entities, on an annual basis, to provide disclosure of specific categories in a collaborative arrangement should be accounted for under ASC Topic 606 when the counterparty is a customer. In addition,rate reconciliation, as well as disclosure of income taxes paid disaggregated by jurisdiction. ASU 2018-18 precludes an entity from presenting consideration from a transaction in a collaborative arrangement as revenue from contracts with customers if the counterparty is not a customer for that transaction. ASU 2018-182023-09 is effective for all interim and annual reporting periodsfiscal years beginning after December 15, 2019. On January 1, 2020, the Company adopted ASU 2018-18. The adoption of ASU 2018-18 did not have a material impact on the Company’s Consolidated Financial Statements.
Goodwill Impairment Testing
In January 2017, the FASB issued ASU No. 2017-04, Intangibles – Goodwill and Other (Topic 350): Simplifying the Test of Goodwill Impairment (“ASU 2017-04”). The new guidance simplifies the subsequent measurement of goodwill by eliminating Step 2 from the goodwill impairment test. ASU 2017-04 requires goodwill impairment to be measured as the amount by which a reporting unit’s carrying amount exceeds its fair value, not to exceed the carrying amount of its goodwill. ASU 2017-04 requires prospective application and is effective for annual periods beginning after December 15, 2019. ASU 2017-04 required the Company to amend its methodology for determining any goodwill impairment beginning in 2020. On January 1, 2020, the Company adopted ASU 2017-04. The adoption of ASU 2017-04 did not have a material impact on the Company’s Consolidated Financial Statements.
Credit Losses
In June 2016, the FASB issued ASU No. 2016-13, Financial Instruments – Credit Losses (Topic 326) (“ASU 2016-13”). ASU 2016-13 implements an impairment model, known as the current expected credit loss model, that is based on expected losses rather than incurred losses. Under the new guidance, an entity will recognize as an allowance its estimate of expected credit losses. ASU 2016-13 is effective for all interim and annual reporting periods beginning after December 15, 2019 and must be adopted using a modified retrospective approach, with certain exceptions. Early adoption is permitted. On January 1, 2020, the Company adopted ASU 2016-13 by using a modified retrospective approach. The adoption of ASU 2016-13 did not have a material impact on the Company’s Consolidated Financial Statements.
Income Taxes
In December 2019, the FASB issued ASU No. 2019-12, Income Taxes – Simplifying the Accounting for Income Taxes (“ASU 2019-12”). The guidance removes exceptions to the general principles in Income Taxes (Topic 740) for allocating tax expense between financial statement components, accounting basis differences stemming from an ownership change in foreign investments and interim period income tax accounting for year-to-date losses that exceed projected losses. The guidance becomes effective for annual reporting periods beginning after December 15, 2020 and interim periods within those fiscal years2024, with early adoption permitted. On January 1, 2020,The Company is currently evaluating the Company early adoptedimpact of adopting ASU 2019-12. The adoption of ASU 2019-12 did not have a material impact on the Company's Consolidated Financial Statements.2023-09.
Cloud Computing Arrangements
In August 2018, the FASB issued ASU 2018-15, Intangibles-Goodwill and Other-Internal Use Software: Customer’s Accounting for Implementation Costs Incurred in a Cloud Computing Arrangement That Is a Service Contract (“ASU 2018-15”). ASU 2018-15 aligns the requirements for capitalizing implementation costs incurred in a cloud computing arrangement that is a service contract with the requirements for capitalizing implementation costs incurred to develop or obtain internal-use software. The guidance becomes effective for annual reporting periods beginning after December 15, 2019 and interim periods within those fiscal years with early adoption permitted. The Company adopted this standard on January 1, 2020 using the prospective method. The adoption of ASU 2018-15 did not have a material impact on the Company’s Consolidated Financial Statements.
NOTE 2 – FAIR VALUE MEASUREMENTMEASUREMENTS
The Company measures certain financial assets and liabilities at fair value on a recurring basis, including cash equivalents and marketable securities and the free shares asset. The accounting guidance establishessecurities. Fair value is determined based on a three-tier hierarchy whichunder the authoritative guidance for fair value measurements and disclosures that prioritizes the inputs used in the valuation methodologies in measuring fair value as follows:
Level 1: Unadjusted quoted prices in active markets that are accessible at the measurement date for identical, unrestricted assets or liabilities;
Level 2: Quoted prices in markets that are not active or inputs which are observable, either directly or indirectly, for substantially the full term of the asset or liability; and
Level 3: Prices or valuation techniques that require inputs that are both significant to the fair value measurementmeasurements and unobservable (i.e., supported by little or no market activity).
The fair value measurements of the Company’s cash equivalents and marketable securities and the free shares asset are identified at the following levels within the fair value hierarchy (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | |
| December 31, 2020 |
| Fair Value Measurement |
| Total | | Level 1 | | Level 2 | | Level 3 |
| Assets: | | | | | | | |
| Cash equivalents: | | | | | | | |
| Money market funds | $ | 53,165 | | | $ | 53,165 | | | $ | 0 | | | $ | 0 | |
| | | | | | | |
| Total | 53,165 | | | 53,165 | | | 0 | | | 0 | |
| Marketable securities: | | | | | | | |
| Commercial paper | 213,533 | | | 0 | | | 213,533 | | | 0 | |
| Corporate debt securities | 59,574 | | | 0 | | | 59,574 | | | 0 | |
| Certificates of deposit | 12,311 | | | 0 | | | 12,311 | | | 0 | |
| Asset-backed securities | 17,908 | | | 0 | | | 17,908 | | | 0 | |
| U.S. government-sponsored entity debt securities | 257,298 | | | 0 | | | 257,298 | | | 0 | |
| Total | 560,624 | | | 0 | | | 560,624 | | | 0 | |
| Total cash equivalents and marketable securities | $ | 613,789 | | | $ | 53,165 | | | $ | 560,624 | | | $ | 0 | |
| | | | | | | |
| Free shares asset | $ | 70 | | | $ | 0 | | | $ | 0 | | | $ | 70 | |
| | | | | | | | | | | | | | | | | | | | | | | |
| December 31, 2023 |
| Fair Value Measurements |
| Total | | Level 1 | | Level 2 | | Level 3 |
| Assets: | | | | | | | |
| Cash equivalents: | | | | | | | |
| Money market funds | $ | 2,508 | | | $ | 2,508 | | | $ | — | | | $ | — | |
| | | | | | | |
| Total | 2,508 | | | 2,508 | | | — | | | — | |
| Marketable securities: | | | | | | | |
| U.S. government-sponsored entity debt securities | 22,566 | | | — | | | 22,566 | | | — | |
| Commercial paper securities | 2,826 | | | — | | | 2,826 | | | — | |
| Corporate debt securities | 1,405 | | | — | | | 1,405 | | | — | |
| Asset-backed securities | 2,377 | | | — | | | 2,377 | | | — | |
| U.S. treasury bills | 5,593 | | | | | 5,593 | | | |
| Certificates of deposit | 1,031 | | | — | | | 1,031 | | | — | |
| | | | | | | |
| Total | 35,798 | | | — | | | 35,798 | | | — | |
| Total cash equivalents and marketable securities | $ | 38,306 | | | $ | 2,508 | | | $ | 35,798 | | | $ | — | |
| | | | | | | |
| | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | |
| December 31, 2022 |
| Fair Value Measurements |
| Total | | Level 1 | | Level 2 | | Level 3 |
| Assets: | | | | | | | |
| Cash equivalents: | | | | | | | |
| Money market funds | $ | 50,820 | | | $ | 50,820 | | | $ | — | | | $ | — | |
| | | | | | | |
| Total | 50,820 | | | 50,820 | | | — | | | — | |
| Marketable securities: | | | | | | | |
| U.S. government-sponsored entity debt securities | 18,417 | | | — | | | 18,417 | | | — | |
| Commercial paper securities | 101,165 | | | — | | | 101,165 | | | — | |
| Corporate debt securities | 11,670 | | | — | | | 11,670 | | | — | |
| Asset-backed securities | 24,792 | | | — | | | 24,792 | | | — | |
| U.S. treasury bills | 7,938 | | | — | | | 7,938 | | | — | |
| Certificates of deposit | 37,461 | | | — | | | 37,461 | | | — | |
| Agency bonds | 5,590 | | | — | | | 5,590 | | | — | |
| Total | 207,033 | | | — | | | 207,033 | | | — | |
| Total cash equivalents and marketable securities | $ | 257,853 | | | $ | 50,820 | | | $ | 207,033 | | | $ | — | |
| | | | | | | |
| | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | |
| December 31, 2019 |
| Fair Value Measurement |
| Total | | Level 1 | | Level 2 | | Level 3 |
| Assets: | | | | | | | |
| Cash equivalents: | | | | | | | |
| Money market funds | $ | 30,496 | | | $ | 30,496 | | | $ | 0 | | | $ | 0 | |
| Commercial paper | 2,999 | | | 0 | | | 2,999 | | | 0 | |
| Total | 33,495 | | | 30,496 | | | 2,999 | | | 0 | |
| Marketable securities: | | | | | | | |
| Commercial paper | 155,368 | | | 0 | | | 155,368 | | | 0 | |
| Corporate debt securities | 95,017 | | | 0 | | | 95,017 | | | 0 | |
| U.S. government-sponsored entity debt securities | 53,493 | | | 0 | | | 53,493 | | | 0 | |
| Total | 303,878 | | | 0 | | | 303,878 | | | 0 | |
| Total cash equivalents and marketable securities | $ | 337,373 | | | $ | 30,496 | | | $ | 306,877 | | | $ | 0 | |
| | | | | | | |
| Free shares asset | $ | 236 | | | $ | 0 | | | $ | 0 | | | $ | 236 | |
Cash Equivalents and Marketable Securities
The Company generally classifies its marketable securities and some cash equivalents as Level 2. Instruments are classified as Level 2 when observable market prices for identical securities that are traded in less active markets are used. When observable market prices for identical securities are not available, such instruments are priced using benchmark curves, benchmarking of like securities, sector groupings, matrix pricing and valuation models. These valuation models are proprietary to the pricing providers or brokers and incorporate a number of inputs, including in approximate order of priority: benchmark yields, reported trades, broker/dealer quotes, issuer spreads, two-sided markets, benchmark securities, bids, offers and reference data including market research publications. For certain security types, additional inputs may be used, or some of the standard inputs may not be applicable. Evaluators may prioritize inputs differently on any given day for any security based on market conditions, and not all inputs listed are available for use in the evaluation process for each security evaluation on any given day.
Free Shares Asset
As a result of the July 20, 2018 Share Purchase Agreement (“Sangamo France SPA”) to acquire Sangamo France (see Note 5 – Acquisition of Sangamo France), the Company entered into arrangements with the holders of approximately 477,000 “free shares” of Sangamo France pursuant to which the Company has the right to purchase such shares from the holders (a call option) and such holders have the right to sell to the Company such shares from time to time through mid-2021 (a put option). The Company initially recorded a liability of $0.2 million on the acquisition date. The put options were classified within Level 3 of the fair value hierarchy as the Company utilized a binomial-lattice pricing model (the “Monte Carlo simulation model”) that involved certain market conditions to estimate the fair value of the options. The assumptions used in this simulation model are reviewed on a quarterly basis and adjusted, as needed. Subsequent changes in the fair value of the free shares are recorded in general and administrative expenses in the Consolidated Statements of Operations. The Company purchased approximately 111,000 and 322,000 shares during 2019 and 2020 respectively, of the 477,000 total free shares for a cash payment of approximately $0.3 million and $0.7 million respectively, upon exercise of the put options. As of December 31, 2020, approximately 44,000 free shares remain outstanding and subject to purchase by the Company.
The fair value of the free shares’ asset was approximately $0.2 million at December 31, 2019. The Company recognized a gain due to an increase in the fair value of the free shares of approximately $0.1 million for the year ended December 31, 2020, offset by approximately $0.2 million for the shares purchased during the year, resulting in an asset balance of approximately $0.1 million at December 31, 2020.
| | | | | | | | | | | |
| December 31, |
| Free Shares valuation assumptions: | 2020 | | 2019 |
| Sangamo stock price (USD) | $ | 15.61 | | | $ | 8.68 | |
| Sangamo France stock price (EUR) | € | 3.85 | | | € | 2.14 | |
| EUR/ USD exchange rate | 0.82 | | | 0.91 | |
| Estimated correlation Sangamo and Sangamo France stock prices | 100.0 | % | | 100.0 | % |
| Sangamo stock price (USD) volatility estimate | 88.9 | % | | 72.5 | % |
| Sangamo France stock price (EUR) volatility estimate | 88.9 | % | | 72.5 | % |
| EUR/ USD exchange rate volatility estimate | 6.3 | % | | 6.6 | % |
| Risk free rate and cost of debt by expected exercise date | Varies | | Varies |
NOTE 3 – CASH EQUIVALENTS AND MARKETABLE SECURITIES
The table below summarizes the Company’s cash equivalents and marketable securities (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | |
| Amortized
Cost | | Gross
Unrealized
Gains | | Gross
Unrealized
Losses | | Estimated
Fair Value |
| December 31, 2020 | | | | | | | |
| Cash equivalents: | | | | | | | |
| Money market funds | $ | 53,165 | | | $ | 0 | | | $ | 0 | | | $ | 53,165 | |
| | | | | | | |
| Total | 53,165 | | | 0 | | | 0 | | | 53,165 | |
| Marketable securities: | | | | | | | |
| Commercial paper | 213,500 | | | 41 | | | (8) | | | 213,533 | |
| Corporate debt securities | 59,575 | | | 16 | | | (17) | | | 59,574 | |
| Certificates of deposit | 12,311 | | | 0 | | | 0 | | | 12,311 | |
| Asset-backed securities | 17,905 | | | 10 | | | (7) | | | 17,908 | |
| U.S. government-sponsored entity debt securities | 257,284 | | | 19 | | | (5) | | | 257,298 | |
| Total | 560,575 | | | 86 | | | (37) | | | 560,624 | |
| Total cash equivalents and marketable securities | $ | 613,740 | | | $ | 86 | | | $ | (37) | | | $ | 613,789 | |
| | | | | | | |
| December 31, 2019 | | | | | | | |
| Cash equivalents: | | | | | | | |
| Money market funds | $ | 30,496 | | | $ | 0 | | | $ | 0 | | | $ | 30,496 | |
| Commercial paper | 2,998 | | | 1 | | | 0 | | | 2,999 | |
| Total | 33,494 | | | 1 | | | 0 | | | 33,495 | |
| Marketable securities: | | | | | | | |
| Commercial paper | 155,230 | | | 145 | | | (7) | | | 155,368 | |
| Corporate debt securities | 94,905 | | | 115 | | | (3) | | | 95,017 | |
| U.S. government-sponsored entity debt securities | 53,411 | | | 91 | | | (9) | | | 53,493 | |
| Total | 303,546 | | | 351 | | | (19) | | | 303,878 | |
| Total cash equivalents and marketable securities | $ | 337,040 | | | $ | 352 | | | $ | (19) | | | $ | 337,373 | |
| | | | | | | | | | | | | | | | | | | | | | | |
| Amortized
Cost | | Gross
Unrealized
Gains | | Gross
Unrealized
Losses | | Estimated
Fair Value |
| December 31, 2023 | | | | | | | |
| Assets | | | | | | | |
| Cash equivalents: | | | | | | | |
| Money market funds | $ | 2,508 | | | $ | — | | | $ | — | | | $ | 2,508 | |
| | | | | | | |
| Total | 2,508 | | | — | | | — | | | 2,508 | |
| Marketable securities: | | | | | | | |
| U.S. government-sponsored entity debt securities | 22,347 | | | 219 | | | — | | | 22,566 | |
| Commercial paper securities | 2,825 | | | 2 | | | (1) | | | 2,826 | |
| Corporate debt securities | 1,399 | | | 6 | | | — | | | 1,405 | |
| Asset-backed securities | 2,368 | | | 9 | | | — | | | 2,377 | |
| U.S. treasury bills | 5,599 | | | — | | | (6) | | | 5,593 | |
| Certificates of deposit | 1,026 | | | 5 | | | — | | | 1,031 | |
| | | | | | | |
| Total | 35,564 | | | 241 | | | (7) | | | 35,798 | |
| Total cash equivalents and marketable securities | $ | 38,072 | | | $ | 241 | | | $ | (7) | | | $ | 38,306 | |
| | | | | | | |
| December 31, 2022 | | | | | | | |
| Assets | | | | | | | |
| Cash equivalents: | | | | | | | |
| Money market funds | $ | 50,820 | | | $ | — | | | $ | — | | | $ | 50,820 | |
| | | | | | | |
| Total | 50,820 | | | — | | | — | | | 50,820 | |
| Marketable securities: | | | | | | | |
| U.S. government-sponsored entity debt securities | 18,710 | | | — | | | (293) | | | 18,417 | |
| Commercial paper securities | 101,336 | | | 22 | | | (193) | | | 101,165 | |
| Corporate debt securities | 11,760 | | | — | | | (90) | | | 11,670 | |
| Asset-backed securities | 24,970 | | | 2 | | | (180) | | | 24,792 | |
| U.S. treasury bills | 7,950 | | | — | | | (12) | | | 7,938 | |
| Certificates of deposit | 37,599 | | | 4 | | | (142) | | | 37,461 | |
| Agency bonds | 5,598 | | | — | | | (8) | | | 5,590 | |
| Total | 207,923 | | | 28 | | | (918) | | | 207,033 | |
| Total cash equivalents and marketable securities | $ | 258,743 | | | $ | 28 | | | $ | (918) | | | $ | 257,853 | |
The fair value of marketable securities by contractual maturity were as follows (in thousands):
| | | | | | | | | | | |
| December 31, |
| 2023 | | 2022 |
| Maturing in one year or less | $ | 10,855 | | | $ | 177,188 | |
| Maturing after one year through five years | 24,943 | | | 29,845 | |
| Total | $ | 35,798 | | | $ | 207,033 | |
| | | | | | | | | | | |
| December 31, |
| 2020 | | 2019 |
| Maturing in one year or less | $ | 510,094 | | | $ | 282,046 | |
| Maturing after one year through five years | 50,530 | | | 21,832 | |
| Total | $ | 560,624 | | | $ | 303,878 | |
The Company had 0 realizedmanages credit risk associated with its investment portfolio through its investment policy, which limits purchases to high-quality issuers and also limits the amount of its portfolio that can be invested in a single issuer. The Company
did not record an allowance for credit losses from the sale ofrelated to its marketable securities for the years ended December 31, 2020, 20192023, 2022, or 2018. NaN investments were other-than-temporarily impaired at either2021.
The Company had unrealized losses related to its marketable securities for the years ended December 31, 2020 or 2019.2023, 2022 and 2021. The Company had no material unrealized losses, individually and in the aggregate, for marketable securities that are in a continuous unrealized loss position for greater than 12 months as of December 31, 2023, 2022 and 2021. These unrealized losses were not attributed to credit risk and were associated with changes in market conditions. The Company periodically reviews its marketable securities for indications of credit losses. The Company considers factors such as the duration, the magnitude and the reason for the decline in value, the potential recovery period, creditworthiness of the issuers of the securities and its intent to sell. For marketable securities, it also considers whether (i) it is more likely than not that the Company will be required to sell the debt securities before recovery of their amortized cost basis, and (ii) the amortized cost basis cannot be recovered as a result of credit losses. No significant facts or circumstances have arisen to indicate that there has been any significant deterioration in the creditworthiness of the issuers of the securities held by the Company. Based on the Company’s review of these securities, including the assessmentCompany determined that no allowance for credit losses related to its marketable securities was required at either December 31, 2023 or 2022.
The Company also considers whether it is more likely than not that the Company will be required to sell the debt securities before recovery of their amortized cost basis. Based on the durationscheduled maturities of its investments and severityprojection of its cash flows in accordance with the unrealizedcurrent operating plan, the Company determined that it was more likely than not that it will be required to sell various securities before recovery of their amortized cost basis during the year ended December 31, 2023. As a result, the Company had reclassified certain non-current marketable securities investments of $34.4 million as current during the year ended December 31, 2023 and recorded an impairment charge of $0.4 million related to those securities. Realized gains and losses andon the Company’ssubsequent sale of these securities during the year ended December 31, 2023 were not material. No impairment charges were recorded during the year ended December 31, 2022, as the Company concluded at that time it had the ability and intent to hold the long-term investments until maturity, there were no other-than-temporary impairments for these marketable securities at December 31, 2020.
NOTE 4 – MAJOR CUSTOMERS, PARTNERSHIPS AND STRATEGIC ALLIANCES
Collaboration Agreements
Novartis Institutes for BioMedical Research, Inc.
On July 27, 2020, the Company entered into a collaboration and license agreement with Novartis Institutes for BioMedical Research, Inc. (“Novartis”) for the research, development and commercialization of gene regulation therapies to treat three neurodevelopmental disorders. Under the agreement, which was effective upon execution, the Company granted Novartis an exclusive, royalty bearing and worldwide license, under its relevant patents and know-how, to develop, manufacture and commercialize certain of its zinc finger protein (“ZFP”) transcription factors (“ZFP-TFs”) targeted to three undisclosed genes that are associated with certain neurodevelopmental disorders, including autism spectrum disorder and intellectual disability. The Company performs early research activities over the collaboration period for each gene target and manufacture the ZPF-TFs required for such research, costs of which will be funded by Novartis. Novartis is responsible for additional research activities, investigational new drug-enabling studies, clinical development, regulatory approvals, manufacturing of preclinical, clinical and approved products, and global commercialization. Subject to certain exceptions set forth in the agreement, the Company is prohibited from developing, manufacturing or commercializing any therapeutic product targeting any of the three genes that are the subject of the collaboration. Novartis also has the option to license certain of the Company’s proprietary adeno-associated viruses (“AAVs”) for the sole purpose of developing, manufacturing and commercializing licensed products arising from the collaboration.
Under the agreement, Novartis paid the Company a $75.0 million upfront license fee in August 2020. In addition to this fee and the cost reimbursements for early research activities, the Company is eligible to earn from Novartis up to $420.0 million in development milestones and up to $300.0 million in commercial milestones. The Company is also eligible to earn from Novartis tiered high single-digit to sub-teen double-digit royalties on potential net commercial sales of licensed products arising from the collaboration. These royalty payments will be subject to reduction due to patent expiration, loss of market exclusivity and payments made under certain licenses for third-party intellectual property. The agreement will continue, on a product-by-product and country-by-country basis, until the expiration of the applicable royalty term. Novartis has the right to terminate the agreement, in its entirety or on a target-by-target basis, for any reason after a specified notice period. Each party also has the right to terminate the agreement on account of the other party’s bankruptcy or material, uncured breach.
All payments received under the agreement, when earned, are non-refundable and non-creditable. The transaction price of $95.1 million includes the upfront license fee of $75.0 million and estimated research costs of $20.1 million for identified research projects over the estimated research period. None of the development and commercial milestones have been included in the transaction price, as all such amounts are fully constrained. As part of its evaluation of the constraint, the Company considered numerous factors, including the fact that achievement of the milestones at this time is uncertain and contingent upon future periods when the uncertainty related to the variable consideration is resolved. The Company will re-evaluate the transaction price, including the estimated variable consideration included in the transaction price and all constrained amounts, in each reporting period and as uncertain events are resolved or other changes in circumstances occur.
The Company assessed the agreement with Novartis in accordance with ASC Topic 606 and concluded that Novartis is a customer. The Company has identified a single performance obligation within this arrangement as a license to the technology and ongoing research services. The Company concluded that the license is not discrete as it does not have stand-alone value to Novartis apart from the research services to be performed pursuant to the agreement. As a result, the Company recognizes revenue from the upfront payment based on proportional performance of the ongoing research services through the estimated research period. The estimation of progress towards the satisfaction of performance obligation and project cost is reviewed quarterly and adjusted, as needed, to reflect the Company’s current assumptions regarding the timing of its performance obligation. As of December 31, 2020, the Company had deferred revenue of $70.9 million related to the upfront license fee received.
The Company recognized $4.1 million of upfront license fee and $1.1 million research reimbursement costs as revenue related to the Novartis agreement during the year ended December 31, 2020.
The Company paid $1.5 million for financial advisory fees related to the Novartis collaboration and license agreement during the year ended December 31, 2020, equal to 2% of $75.0 million received for the upfront license fee related to the collaboration and license agreement with Novartis. The Company recognized this $1.5 million as a contract asset as such amount represents a cost of obtaining the agreement. This balance will be amortized and included in general and administrative costs on a systematic basis consistent with the transfer of the services to Novartis in accordance with ASC Topic 340, Other Assets and Deferred Costs. The Company amortized $0.1 million during the year ended December 31, 2020.
Biogen MA, Inc.
In February 2020, the Company entered into a collaboration and license agreement with Biogen MA, Inc. (“BIMA”) and Biogen International GmbH (together with BIMA, “Biogen”) for the research, development and commercialization of gene
regulation therapies for the treatment of neurological diseases. The Company and Biogen plan to leverage the Company’s proprietary ZFP technology delivered via AAV to modulate expression of key genes involved in neurological diseases. Concurrently with the execution of the collaboration agreement, the Company entered into a stock purchase agreement with BIMA, pursuant to which BIMA agreed to purchase 24,420,157 shares of the Company’s common stock (the “Biogen Shares”), at a price per share of $9.2137, for an aggregate purchase price of approximately $225.0 million.
The collaboration agreement became effective in April 2020 following the termination of the waiting period under the Hart-Scott-Rodino Antitrust Improvements Act of 1976, as amended, and satisfaction of other customary closing conditions, including the payment of $225.0 million for the purchase of the Biogen Shares.
Under the collaboration agreement, Biogen paid the Company an upfront license fee of $125.0 million in May 2020. The Company is also eligible to receive research, development, regulatory and commercial milestone payments that could total up to approximately $2.37 billion if Biogen selects all of the targets allowed under the agreement and all the specified milestones set forth in the agreement are achieved, which includes up to $925.0 million in pre-approval milestone payments and up to $1.45 billion in first commercial sale and other sales-based milestone payments. In addition, the Company is also eligible to receive tiered high single-digit to sub-teen royalties on potential net commercial sales of licensed products arising from the collaboration. These royalty payments are subject to reduction due to patent expiration, entry of biosimilar products to the market and payments made under certain licenses for third-party intellectual property.
Under the collaboration agreement, the Company granted to Biogen an exclusive, royalty bearing and worldwide license, under its relevant patents and know-how, to develop, manufacture and commercialize certain ZFP and/or AAV-based products directed to up to 12 neurological disease gene targets selected by Biogen. Biogen has already selected 3 of these: ST-501 for tauopathies including Alzheimer’s disease, ST-502 for synucleinopathies including Parkinson’s disease, and a third undisclosed neuromuscular disease target. Biogen has exclusive rights to nominate up to 9 additional targets over a target selection period of five years. For each gene target selected by Biogen, the Company performs early research activities, costs for which are shared by the companies, aimed at the development of the combination of proprietary central nervous system delivery vectors and ZFP-TFs (or potential other ZFP products) targeting therapeutically relevant genes. Biogen has assumed responsibility and costs for the IND enabling studies, clinical development, related regulatory interactions, and global commercialization. The Company is responsible for manufacturing activities for the initial clinical trials for the first 3 products of the collaboration and plans to leverage its in-house manufacturing capacity, where appropriate, which is currently in development. Biogen is responsible for manufacturing activities beyond the first clinical trial for each of the first 3 products. The Company’s research activities for any targets will be performed over the period not to exceed seven years from the effective date of the agreement (i.e., through April 2027). Subject to certain exceptions set forth in the collaboration agreement, the Company is prohibited from developing, manufacturing or commercializing any therapeutic product directed to the targets selected by Biogen.
The collaboration agreement continues on a product-by-product and country-by-country basis until the expiration of all applicable royalty terms. Biogen has the right to terminate the collaboration agreement, in its entirety or on target-by-target basis, for any reason after a specified notice period, and also has the right to replace up to 10 targets. Each party has the right to terminate this agreement on account of the other party’s bankruptcy or material, uncured breach. In addition, the Company may terminate the collaboration agreement if Biogen challenges any patents licensed by the Company to Biogen.
Pursuant to the terms of the stock purchase agreement, Biogen has agreed not to, without the Company’s prior written consent and subject to specified conditions and exceptions, directly or indirectly acquire shares of the Company’s outstanding common stock, seek or propose a tender or exchange offer or merger between the parties, solicit proxies or consents with respect to any matter, or undertake other specified actions related to the potential acquisition of additional equity interests in the Company. Such standstill restrictions expire on the earlier of the three-year anniversary of the effectiveness of the collaboration agreement and the date that Biogen beneficially owns less than 5% of the Company’s common stock.
The stock purchase agreement also provides that until the first anniversary of the effectiveness of the collaboration agreement, Biogen will hold and not sell any of the Biogen Shares and from the first anniversary through the second anniversary, Biogen will hold and not sell at least 50% of the Biogen Shares, in addition to being subject to certain volume limitations. The stock purchase agreement further provides that, subject to certain limitations, until such time as all remaining Biogen Shares may be sold pursuant to Rule 144 promulgated under the Securities Exchange Act of 1933, as amended, within a 90-day period, Biogen may request the Company to register for resale any of the Biogen Shares on a registration statement to be filed with the Securities and Exchange Commission.
In addition, Biogen has agreed that, excluding specified extraordinary matters, it will vote the Biogen Shares in accordance with the Company’s recommendation and has granted the Company an irrevocable proxy with respect to the foregoing. Such voting provisions expire on the earlier of (i) the two-year anniversary of the effectiveness of the collaboration agreement, (ii) the date that Biogen beneficially owns less than 5% of the Company’s common stock and (iii) the date the
collaboration agreement is terminated; provided, however, that in no event shall such expiration date be prior to the one-year anniversary of the effectiveness of the collaboration agreement.
The Company assessed the collaboration agreement with Biogen in accordance with ASC Topic 606 and concluded that Biogen is a customer. As of December 31, 2020, the transaction price includes the upfront license fee of $125.0 million and the excess consideration from the stock purchase of $79.6 million, which represents the difference between the $225.0 million received for the purchase of the Biogen Shares and the $145.4 million estimated fair value of the equity issued. The equity issued to Biogen was valued using an option pricing model to reflect certain holding period restrictions. None of the target selection fees and clinical or regulatory milestones have been included in the transaction price, as all such amounts are fully constrained. As part of its evaluation of the constraint, the Company considered numerous factors, including the fact that nomination of additional targets and achievement of the milestones at this time is uncertain and contingent upon future periods when the uncertainty related to the variable consideration is resolved. The Company will re-evaluate the transaction price as uncertain events are resolved or other changes in circumstances occur.
The Company has identified a single performance obligation within the Biogen collaboration agreement, which is a stand-ready obligation consisting of a series of distinct days of research services, during which Biogen obtains access to the Company’s license and research resources. Revenue from the upfront license fee relates to access to the license and Company’s obligation to stand-ready to perform such research services corresponding to the targets selected by Biogen. As a result of this obligation to perform research services when and if requested throughout the duration of the contract, the upfront license fee and the excess consideration from the stock purchase will be recognized over time on a straight-line basis consistent with the resources expected to be dedicated to providing the research services through April 2027, the estimated period of the obligation. The estimated period of performance is reviewed quarterly and adjusted, as needed, to reflect the Company’s current assumptions regarding the timing of its deliverable. Revenue from the reimbursement by Biogen of shared costs of early research activities performed by Sangamo is recognized as the research services are performed. As of December 31, 2020, the Company had deferred revenue of $183.2 million related to this agreement.
The Company recognized $21.4 million of upfront license fee and the excess consideration from the stock purchase, and $6.5 million research reimbursement costs as revenue related to the Biogen agreement during the year ended December 31, 2020.
The Company paid $7.0 million for financial advisory fees during the year ended December 31, 2020, equal to 2% of $225.0 million received for the sale of shares and 2% of $125.0 million received for the upfront fee. The fees incurred related to both the collaboration agreement with Biogen and to the stock purchase agreement for the sale of shares. The Company believes that the allocation of fees on a relative fair value basis between the two agreements is reasonable. The Company recognized $4.1 million, which represents 2% of the upfront license fee of $125.0 million and 2% of the excess consideration from the stock purchase of $79.6 million, as a contract asset. This balance will be amortized and included in general and administrative costs on a systematic basis consistent with the transfer of the services to Biogen in accordance with ASC Topic 340, Other Assets and Deferred Costs. The Company amortized $0.4 million during the year ended December 31, 2020. The Company recognized $2.9 million, which represents 2% of the $145.4 million estimated fair value of the equity issued, as a share issuance cost and recorded this amount in equity as reduction in proceeds during the year ended December 31, 2020.
Kite Pharma, Inc.
In February 2018, the Company entered into a global collaboration and license agreement with Kite Pharma, Inc. (“Kite”), which became effective on April 5, 2018, and was amended and restated in September 2019, for the research, development and commercialization of potential engineered cell therapies for cancer. In this collaboration, Sangamo is working together with Kite on a research program under which the companies are designing zinc finger nucleases (“ZFNs”) and viral vectors to disrupt and insert certain genes in T cells and natural killer cells (“NK-cells”) including the insertion of genes that encode chimeric antigen receptors (“CARs”), T cell receptors (“TCRs”), and NK-cell receptors (“NKRs”) directed to mutually agreed targets. Kite is responsible for all clinical development and commercialization of any resulting products.
Subject to the terms of this agreement, the Company granted Kite an exclusive, royalty-bearing, worldwide sublicensable license under the Company’s relevant patents and know-how to develop, manufacture and commercialize, for the purpose of treating cancer, specific cell therapy products that may result from the research program and that are engineered ex vivo using selected ZFNs and viral vectors developed under the research program to express CARs, TCRs or NKRs directed to candidate targets.
During the research program term and subject to certain exceptions, except pursuant to this agreement, the Company is prohibited from researching, developing, manufacturing and commercializing, for the purpose of treating cancer, any cell therapy product that, as a result of ex vivo genome editing, expresses a CAR, TCR or NKR that is directed to a target expressed on or in a human cancer cell. After the research program term concludes and subject to certain exceptions, except pursuant to this agreement, the Company will be prohibited from developing, manufacturing and commercializing, for the purpose of
treating cancer, any cell therapy product that, as a result of ex vivo genome editing, expresses a CAR, TCR or NKR that is directed to a candidate target.
Following the effective date, in April 2018, the Company received a $150.0 million upfront payment from Kite. In addition, Kite will reimburse the Company’s direct costs to conduct service under the joint research program provisions of the agreement, and Kite will be responsible for all subsequent development, manufacturing and commercialization of any licensed products. Sangamo is also eligible to receive contingent development- and sales-based milestone payments that could total up to $3.01 billion if all of the specified milestones in this agreement are achieved. Of this amount, approximately $1.26 billion relates to the achievement of specified research, clinical development, regulatory and first commercial sale milestones, and approximately $1.75 billion relates to the achievement of specified sales-based milestones if annual worldwide net sales of licensed products reach specified levels. Each development- and sales-based milestone payment is payable (i) only once for each licensed product, regardless of the number of times that the associated milestone event is achieved by such licensed product, and (ii) only for the first 10 times that the associated milestone event is achieved, regardless of the number of licensed products that may achieve such milestone event. In addition, the Company will be entitled to receive escalating, tiered royalty payments with a percentage in the single digits based on potential future annual worldwide net sales of licensed products. These royalty payments will be subject to reduction due to patent expiration, entry of biosimilar products to the market and payments made under certain licenses for third-party intellectual property.
The initial research term of the agreement is six years. Kite has an option to extend the research term for up to 2 additional one-year periods for a separate fee of $10.0 million per year. All contingent payments under the agreement, when earned, will be non-refundable and non-creditable. In connection with the amendment and restatement of the agreement in September 2019, the Company entered into a new research plan with Kite, with estimated reimbursable service costs of approximately $3.4 million. The Company concluded the transaction price under this agreement is $189.3 million and includes the upfront license fee of $150.0 million and $39.3 million estimated reimbursable service costs for identified research projects over the estimated performance period. The Company concluded that the estimated fees for the presumed exercise of the research term extension options and all milestone amounts are fully constrained. As part of its evaluation of the constraint, the Company considered numerous factors, including the fact that achievement of the milestones at this time is uncertain and contingent upon future periods when the uncertainty related to the variable consideration is resolved. The Company will re-evaluate the transaction price, including the estimated variable consideration included in the transaction price and all constrained amounts, in each reporting period and as uncertain events are resolved or other changes in circumstances occur. None of the development and sales-based milestone payments have been included in the transaction price.
The Company assessed the agreement with Kite in accordance with ASC Topic 606 and concluded that Kite is a customer. Kite has the right to terminate this agreement, in its entirety or on a per licensed product or per candidate target basis, for any reason after a specified notice period. Each party has the right to terminate this agreement on account of the other party’s bankruptcy or material, uncured breach.
The Company has identified the primary performance obligations within the Kite agreement as: (1) a license to the technology along with the stand-ready obligation to perform research services, and (2) the ongoing research services. Revenue from the upfront license fee relates to access to the license and Company’s obligation to stand-ready to perform such research services as additional targets are selected by Kite. As a result of this obligation to perform research services when and if requested throughout the duration of the contract, the fee for the license and the stand-ready obligation will be recognized over time on a straight-line basis through June 2024, the estimated period of the stand-ready obligation. Revenue from the reimbursable costs related to the integrated service deliverable is recognized as the research services are performed. Related costs and expenses under these arrangements have historically approximated the revenues recognized. The estimated period of performance is reviewed quarterly and adjusted, as needed, to reflect the Company’s current assumptions regarding the timing of its deliverables. As of December 31, 2020, and 2019 the Company had deferred revenue of $81.4 million and $106.5 million, respectively, related to this agreement.
Revenues recognized under the agreement were as follows (in thousands):
| | | | | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2020 | | 2019 | | 2018 | | | |
| Revenue related to Kite agreement: | | | | | | | | |
| Recognition of license and stand-ready fee | $ | 25,046 | | | $ | 24,977 | | | $ | 18,545 | | | | |
| Research services | 3,562 | | | 9,373 | | | 6,972 | | | | |
| Total | $ | 28,608 | | | $ | 34,350 | | | $ | 25,517 | | | | |
Pfizer Inc.
Giroctocogene Fitelparvovec Global Collaboration and License Agreement
In May 2017, the Company entered into an exclusive global collaboration and license agreement with Pfizer Inc. (“Pfizer”), pursuant to which it established a collaboration for the research, development and commercialization of giroctocogene fitelparvovec, its gene therapy product candidate for hemophilia A, and closely related products.
Under this agreement, the Company is responsible for conducting the Phase 1/2 clinical trial and for certain manufacturing activities for giroctocogene fitelparvovec, while Pfizer is responsible for subsequent worldwide development, manufacturing, marketing and commercialization of giroctocogene fitelparvovec. Sangamo may also collaborate in the research and development of additional AAV basedAAV-based gene therapy products for hemophilia A.
The Company originally received an upfront fee of $70.0 million and is eligible to receive up to $208.5 million in payments upon the achievement of specified clinical development, intellectual property and regulatory milestones and up to $266.5 million in payments upon first commercial sale milestones for giroctocogene fitelparvovec and potentially other products. The total amount of potential clinical development, intellectual property, regulatory, and first commercial sale milestone payments, assuming the achievement of all specified milestones in the giroctocogene fitelparvovec Pfizer agreement, is up to $475.0 million, which includes up to $300.0 million for giroctocogene fitelparvovec and up to $175.0 million for other products that may be developed under the agreement, subject to reduction on account of payments made under certain licenses for third party intellectual property. In addition, Pfizer agreed to pay the Company royalties for each potential licensed product developed under the agreement based on an escalating tiered, double-digit percentage of the annual net sales of such product. These royalties are subject to reduction due to patent expiration, entry of biosimilar products to the market and payment made under certain licenses for third-party intellectual property. To date, two milestones of $55.0 million in aggregate have been achieved and paid, however 0 products have been approved and therefore 0 royalty fees have been earned under the giroctocogene fitelparvovec Pfizer agreement.
The Company assessed the agreement with Pfizer in accordance with ASC Topic 606 and concluded that Pfizer is a customer. As of December 31, 2020, the total transaction price under this agreement is $134.0 million, which represents the upfront and research services fees of $79.0 million and two unconstrained milestones achieved of an aggregate amount of $55.0 million. Sangamo is responsible for internal and external research costs as part of the upfront fee and has the ability to request additional reimbursement from Pfizer if certain conditions are met. None of the constrained clinical or regulatory milestones have been included in the transaction price, as all such milestone amounts are fully constrained. As part of its evaluation of the constraint, the Company considered numerous factors, including the fact that achievement of the milestones at this time is uncertain and contingent upon future periods when the uncertainty related to the variable consideration is resolved. The Company will re-evaluate the transaction price, including its estimated variable consideration included in the transaction price and all constrained amounts in each reporting period and as uncertain events are resolved or other changes in circumstances occur.
Subject to the terms of the agreement, the Company granted Pfizer an exclusive worldwide royalty-bearing license, with the right to grant sublicenses, to use certain technology controlled by the Company for the purpose of developing, manufacturing and commercializing giroctocogene fitelparvovec and related products. Pfizer granted the Company a non-exclusive, worldwide, royalty free,royalty-free, fully paid license, with the right to grant sublicenses, to use certain manufacturing technology developed under the agreement and controlled by Pfizer to manufacture the Company’s products that utilize the AAV delivery system. During a specified period, neither the Company nor Pfizer will beis permitted to clinically develop or commercialize, outside of the collaboration, certain AAV-based gene therapy products for hemophilia A.
Unless earlier terminated, the agreement has a term that continues on a per product and per country basis until the later of (i) the expiration of patent claims that cover the product in a country, (ii) the expiration of regulatory exclusivity for a product in a country, and (iii) 15 years after the first commercial sale of a product in a country. Pfizer has the right to terminate the agreement without cause in its entirety or on a per product or per country basis. The agreement may also be terminated by either party based on an uncured material breach by the other party or the bankruptcy of the other party. Upon termination for any reason, the license granted by the Company to Pfizer to develop, manufacture and commercialize giroctocogene fitelparvovec and related products will automatically terminate. Upon termination by the Company for cause or by Pfizer in any country or countries, Pfizer will automatically grant the Company an exclusive, royalty-bearing license under certain technology controlled by Pfizer to develop, manufacture and commercialize giroctocogene fitelparvovec in the terminated country or countries.
Upon execution of the agreement, the Company received an upfront fee of $70.0 million and was eligible to receive up to $208.5 million in payments upon the achievement of specified clinical development, intellectual property and regulatory milestones and up to $266.5 million in payments upon first commercial sale milestones for giroctocogene fitelparvovec and potentially other products. To date, two milestones of $55.0 million in aggregate have been achieved and paid. The Company is eligible to earn from Pfizer up to $220.0 million in remaining milestone payments for giroctocogene fitelparvovec and up to
$175.0 million for other products that may be developed under the agreement, subject to reduction on account of payments made under certain licenses for third-party intellectual property. In addition, Pfizer agreed to pay the Company royalties for each potential licensed product developed under the agreement that are 14% - 20% of the annual worldwide net sales of such product and are subject to reduction due to patent expiration, entry of biosimilar products to the market and payment made under certain licenses for third-party intellectual property.
The Company assessed the agreement with Pfizer in accordance with ASC Topic 606 and concluded that Pfizer was a customer. The total transaction price under this agreement was $134.0 million, which represented the upfront fee and research services fees of $79.0 million and fees related to two achieved milestones in an aggregate amount of $55.0 million. Sangamo was responsible for internal and external research costs as part of the upfront fee and had the ability to request additional reimbursement from Pfizer if certain conditions were met. None of the constrained clinical or regulatory milestones were included in the transaction price. As part of its evaluation of the constraint, the Company considered numerous factors, including the fact that achievement of the milestones at the time was uncertain and contingent upon future periods when the uncertainty related to the variable consideration is resolved.
The Company has identified onethe performance obligationobligations within the giroctocogene fitelparvovec Pfizer agreement as a license to the technology and ongoing research services. The Company concluded that the license iswas not discrete as it doesdid not have stand-alone value to Pfizer apart from the research services to be performed by the Company pursuant to the agreement. As a result, the Company recognizesrecognized revenue from the upfront payment based on proportional performance of the ongoing
research services through 2020, the estimated period the Company will perform research services. The estimate of progress towards the satisfaction of its performance obligation and project cost is reviewed quarterly and adjusted, as needed, to reflect the Company’s current assumptions regarding the timing of its deliverables. The Company satisfied the deliverables and research services responsibilities within the arrangement which were completed in December 2020. As a result, the Company recognized the remaining deferred revenue from the upfront payment in December 2020. As of December 31, 2019, the Company had deferred revenue of $4.0 million related to this agreement.
In December 2019, the Company entered into an amendment to the collaboration agreement, pursuant toduring which the Company transferred the IND for giroctocogene fitelparvovec to Pfizer. Upon this transfer the Company achieved a $25.0 million milestone as the conditions for achieving the milestone were met. The cumulative revenue recognized in connection with this milestone was $25.0 million as of December 31, 2020 and included $1.3 million recognized during the year ended December 31, 2020.
In September 2020, the Company determined that there was a high probability of achievement of a $30.0 million milestone with Pfizer for giroctocogene fitelparvovec. The milestone was subsequently achieved upon dosing of the first subject in a Phase 3 clinical trial in early October 2020. The cumulative revenue recognized in connection with this milestone was $30.0 million during the year ended December 31, 2020.
Revenues recognized under the agreement were as follows (in thousands):
| | | | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2020 | | 2019 | | 2018 | | |
| Revenue related to Pfizer giroctocogene fitelparvovec agreement: | | | | | | | |
| Recognition of upfront fee and research services | $ | 3,111 | | | $ | 15,697 | | | $ | 37,810 | | | |
| Milestone achievement | 31,338 | | | 23,662 | | | 0 | | | |
| Total | $ | 34,449 | | | $ | 39,359 | | | $ | 37,810 | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
In March 2019, the Company updated its estimated project cost and related revenues under this program. This adjustment was a direct result of the increase in project scope during the first quarter of 2019 and the corresponding costs, which resulted in a decrease in the measure of proportional performance. In December 2019, the Company updated its estimated project cost and related revenues upon transfer of the IND for giroctocogene fitelparvovec to Pfizer. This adjustment was a direct result of the decrease in project scope during the fourth quarter of 2019 and the corresponding costs, which resulted in an increase in the measure of proportional performance. During the year ended December 31, 2019, the Company recognized $15.7 million in revenues related to the Pfizer giroctocogene fitelparvovec agreement, which included approximately $8.7 million acceleration in revenues recorded in the three months ended December 31, 2019 related to the updated estimated project cost, offset by approximately $3.0 million reduction in revenues recorded in the three months ended March 31, 2019 related to the updated estimated project cost.
In March 2020, the Company recorded an adjustment to revenue related to a change in estimate in connection with the giroctocogene fitelparvovec collaboration agreement with Pfizer. This adjustment was a direct result of the decision to decrease the project scope and the corresponding costs, after the successful IND transfer of the giroctocogene fitelparvovec product candidate to Pfizer, both of which resulted in an increase in the measure of proportional cumulative performance. This adjustment increased revenue by $2.4 million, decreased net loss by $2.4 million and decreased the Company’s basic net loss per share by $0.02 for year ended December 31, 2020.performed research services.
C9ORF72 Research Collaboration and License Agreement
In December 2017, the Company entered into a separate exclusive, global collaboration and license agreement with Pfizer for the development and commercialization of potential gene therapy products that use ZFP-TFszinc finger transcriptional regulators (“ZF-transcriptional regulators”) to treat amyotrophic lateral sclerosis (“ALS”) and frontotemporal lobar degeneration (“FTLD”) linked to mutations of the C9ORF72 gene. Pursuant to this agreement, the Company agreed to work with Pfizer on a research program to identify, characterize and preclinically develop ZFP-TFsZF-transcriptional regulators that bind to and specifically reduce expression of the mutant form of the C9ORF72 gene.
Subject to the terms of this agreement, the Company granted Pfizer an exclusive, royalty-bearing, worldwide license under the Company’s relevant patents and know-how to develop, manufacture and commercialize gene therapy products that use resulting ZFP-TFsZF-transcriptional regulators that satisfy pre-agreed criteria. During a specified period, neither the Company nor Pfizer will be permitted to research, develop, manufacture or commercialize outside of the collaboration any ZFPszinc finger proteins (“ZFPs”) that specifically bind to the C9ORF7C9ORF722 gene.
Unless earlier terminated, the agreement has a term that continues on a per licensed product and per country basis until the later of (i) the expiration of patent claims that cover the licensed product in a country, (ii) the expiration of regulatory exclusivity for a licensed product in a country, and (iii) 15 years after the first commercial sale of a licensed product in a major
market country. Pfizer also has the right to terminate the agreement without cause in its entirety or on a per product or per country basis. The agreement may also be terminated by either party based on an uncured material breach by the other party or the bankruptcy of the other party. The agreement will also terminate if the Company is unable to identify any lead candidates for development within a specified period of time or if Pfizer elects not to advance a lead candidate beyond a certain development milestone within a specified period of time. Upon termination for any reason, the license granted by the Company to Pfizer to develop, manufacture and commercialize licensed products under the agreement will automatically terminate. Upon termination by the Company for cause or by Pfizer without cause for any licensed product or licensed products in any country or countries, the Company will have the right to negotiate with Pfizer to obtain a non-exclusive, royalty-bearing license under certain technology controlled by Pfizer to develop, manufacture and commercialize the licensed product or licensed products in the terminated country or countries.
Following termination by the Company for Pfizer’s material breach, Pfizer will not be permitted to research, develop, manufacture or commercialize ZFPs that specifically bind to the C9ORF72gene for a period of time. Following termination by Pfizer for the Company’s material breach, the Company will not be permitted to research, develop, manufacture or commercialize ZFPs that specifically bind to the C9ORF72 gene for a period of time.
The Company received a $12.0 million upfront payment from Pfizer and is eligible to receive up to $60.0 million in development milestone payments from Pfizer contingent on the achievement of specified preclinical development, clinical development and first commercial sale milestones, and up to $90.0 million in commercial milestone payments if annual worldwide net sales of the licensed products reach specified levels. In addition, Pfizer will pay the Company royalties based on an escalating tiered, mid- to high-single digit percentageof 14% - 20% of the annual worldwide net sales of the licensed products. These royalty payments are subject to reduction due to patent expiration, entry of biosimilar products to the market and payments made under certain licenses for third partythird-party intellectual property. Each party will be responsible for the cost of its performance of the research program. Pfizer will be operationally and
financially responsible for subsequent development, manufacturing and commercialization of the licensed products. To date, a milestone of $5.0 million has been achieved and paid, however 0no products have been approved and therefore 0no royalty fees have been earned under the C9ORF72 Pfizer agreement.
The Company assessed the agreement with Pfizer in accordance with ASC Topic 606 and concluded that Pfizer iswas a customer. The Company concluded the total transaction price under this agreement iswas $17.0 million, which representsrepresented the upfront feefees of $12.0 million and fees related to achievement of one unconstrained milestone in the amount of $5.0 million. None of the constrained clinical or regulatory milestones have beenwere included in the transaction price. As part of its evaluation of the constraint, the Company considered numerous factors, including the fact that achievement of the milestones at thisthe time iswas uncertain and contingent upon future periods when the uncertainty related to the variable consideration is resolved. The Company will re-evaluate the transaction price, including its estimated variable consideration included in the transaction price and all constrained amounts, in each reporting period and as uncertain events are resolved or other changes in circumstances occur.
The Company hashad identified the performance obligationobligations within this agreement as a license to the technology and ongoing research services. The Company concluded that the license is not discrete as it does not have stand-alone value to Pfizer apart from the services to be performed by the Company pursuant to the agreement. As a result, the Company recognizesrecognized revenue from the upfront payment based on proportional performance of the ongoingresearch services overthrough 2020, the estimated period the Company will performperformed research services. The estimation of progress towards the satisfaction of its performance obligation and project cost is reviewed quarterly and adjusted, as needed, to reflect the Company’s current assumptions regarding the timing of its deliverables. The Company satisfied the deliverables and research services responsibilities within the arrangement which were completed in September 2020. As a result, the Company recognized the remaining deferred revenue from the upfront payment in September 2020. As of December 31, 2019, the Company had deferred revenue of $8.0 million related to this agreement.
In September 2020, the Company earned a $5.0 million milestone associated with the completion of the Company’s research activities in its collaboration withOctober 2023, Pfizer to develop genome regulation therapies using ZFP-TFs for the treatment of C9ORF72-related ALS and frontotemporal lobar degeneration. This milestone was achieved upon Pfizer’s notification tonotified the Company of its election to pay the first development milestone payment underPfizer’s assignment of the collaboration agreement. This milestone payment is non-refundable, and the Company recognizedlicense agreement to Alexion, AstraZeneca Rare Disease, pursuant to a definitive purchase and license agreement for preclinical gene therapy assets and enabling technologies that closed on a cumulative basis $5.0 million for the year ended December 31, 2020.September 20, 2023.
Revenues recognized under the agreement were as follows (in thousands):
| | | | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2020 | | 2019 | | 2018 | | |
Revenue related to Pfizer C9ORF72 agreement: | | | | | | | |
| Recognition of upfront fee | $ | 7,985 | | | $ | 1,827 | | | $ | 2,188 | | | |
| Milestone achievement | 5,000 | | | 0 | | | 0 | | | |
| Total | $ | 12,985 | | | $ | 1,827 | | | $ | 2,188 | | | |
During the year ended December 31, 2020, the Company recorded adjustments to revenue related to changes in estimate in connection with the C9ORF72 collaboration agreement with Pfizer. These adjustments were a direct result of the decision to decrease the project scope and the corresponding costs due to advancement of the program, which resulted in an increase in the measure of proportional cumulative performance. These adjustments increased revenue by $8.8 million, decreased net loss by $8.8 million and decreased the Company’s basic net loss per share by $0.06 for the year ended December 31, 2020.
Sanofi GenzymeKite Pharma, Inc.
In January 2014,February 2018, the Company entered into an exclusive worldwidea global collaboration and license agreement with Bioverativ Inc., (now Sanofi Genzyme, a global business unit of Sanofi S.A.Kite which became effective on April 5, 2018 (“Sanofi”)Effective Date”), and was amended and restated in September 2019, for the research, development, and commercialization of potential engineered cell therapies for cancer. The collaboration and license agreement relates to develop therapeutics for hemoglobinopathies, focused on beta thalassemiathe design of zinc finger nucleases (“ZFNs”) and sickle cell diseaseviral vectors to disrupt and insert certain genes in T-cells and natural killer cells (“SCD”NK-cells”). The agreement was originally signed with BIMA, who subsequently assigned it including the insertion of genes that encode chimeric antigen receptors (“CARs”), T-cell receptors (“TCRs”), and NK-cell receptors (“NKRs”) directed to Bioverativ Inc., which was later acquired by Sanofi.mutually agreed targets. Under the agreement, the Company is jointly conducting 2 research programs: the beta thalassemia program and the SCD program. In the beta thalassemia program, the CompanyKite is responsible for all discovery, research and development activities through the first human clinical trial. In the SCD program, both parties are responsible for research and development activities through the submission of an IND application for ZFP therapeutics intended to treat SCD.
Under both programs, Sanofi is responsible for subsequent worldwide clinical development, manufacturing and commercialization of licensed products developed under the agreement. At the end of the specified research terms for each program or under certain specified circumstances, Sanofi has the right to step in and take over any of the Company’s remaining activities. Furthermore, the Company has an option to co-promote in the U.S. any licensed products to treat beta thalassemia and SCD developed under the agreement, and Sanofi will compensate the Company for such co-promotion activities. resulting products.
Subject to the terms of thethis agreement, the Company has granted SanofiKite an exclusive, royalty-bearing, worldwide sublicensable license withunder the rightCompany’s relevant patents and know-how to grant sublicenses, to use certain ZFPdevelop, manufacture and other technology controlled by the Companycommercialize, for the purpose of researching, developing, manufacturingtreating cancer, specific cell therapy products that may result from the research program and commercializing licensed productsthat are engineered ex vivo using selected ZFNs and viral vectors developed under the agreement. The Company also granted Sanofi a non-exclusive worldwide, royalty-free fully paid license withresearch program to express CARs, TCRs or NKRs directed to candidate targets.
Following the right to grant sublicenses, under the Company’s interest in certain other intellectual property developed pursuant to the agreement. During the term of the agreement, the Company is not permitted to research, develop, manufacture or commercialize, outside of the agreement, certain gene therapy products that target genes relevant to the licensed products.
Under the agreement,Effective Date, the Company received ana $150.0 million upfront license fee of $20.0 million andpayment from Kite. In addition, Kite reimburses the Company’s direct costs to conduct the joint research program. Sangamo is also eligible to receive contingent development- and sales-based milestone payments that could total up to $115.8 million$3.0 billion if all of the specified milestones set forth in payments uponthis agreement are achieved. Of this amount, approximately $1.3 billion relates to the achievement of specified research, clinical development, regulatory and regulatoryfirst commercial sale milestones, as well as upand approximately $1.8 billion relates to $160.5 million in payments upon the achievement of specified sales-based milestones if annual worldwide net sales milestones. of licensed products reach specified levels. Each development- and sales-based milestone payment is payable (i) only once for each licensed product, regardless of the number of times that the associated milestone event is achieved by such licensed product, and (ii) only for the first ten times that the associated milestone event is achieved regardless of the number of licensed products that may achieve such milestone event. In addition, the Company is entitled to receive escalating, tiered royalty payments with a percentage in the single digits based on future annual worldwide net sales of licensed products. These royalty payments are subject to reduction due to patent expiration, entry of biosimilar products to the market and payments made under certain licenses for third-party intellectual property.
The total amount of potential regulatory, clinical development, and sales milestone payments, assuming the achievement of all specified milestonesinitial research term in the agreement is six years from the Effective Date and will expire in April 2024. Kite had an option to extend the research term for up to $276.3 million. In addition, the Company will receive royalty paymentstwo additional one-year periods for each licensed product that are a tiered double-digit percentageseparate upfront fee of annual net sales of each product. Sanofi reimburses Sangamo for agreed upon costs incurred$10.0 million per year, which was not exercised and expired in connection with research and development activities conducted by Sangamo. To date, a $6.0 million milestone has been achieved related to ST-400 for beta thalassemia and another $7.5 million milestone has been achieved related to SCD, however 0 products have been approved and therefore 0 royalty fees have been earned under the Sanofi agreement.
The agreement may be terminated by (i) the Company or Sanofi for the uncured material breach of the other party, (ii) the Company or Sanofi for the bankruptcy or other insolvency proceeding of the other party; (iii) Sanofi, upon 180 days’ advance written notice to the Company and (iv) Sanofi, for certain safety reasons upon written notice to, and after consultation with, the Company. As a result, actual future milestone payments could be lower than the amounts stated above.
October 2023. All contingent payments under the agreement, when earned, will be non-refundable and non-creditable. Through the amendment and restatement of the agreement in September 2019, the Company and Kite agreed to expand the scope of the collaboration program to incorporate the use of lentiviral or retroviral vectors provided by Kite. Kite has the right to terminate this agreement in its entirety or on a per licensed product or per candidate target basis for any reason after a specified notice period. Each party has the right to terminate this agreement on account of the other party’s bankruptcy or material, uncured breach.
The Company assessed the agreement with Kite in accordance with ASC Topic 606 and concluded that Kite is a customer. The transaction price as of December 31, 2020 of $93.3 million includes the upfront license fee of $20.0 million, two unconstrained milestones of $13.5$150.0 million and estimated researchreimbursable service costs of $59.8 million for identifiedthe research projects over the estimated performance period,period. None of the clinical or regulatory milestones have been included in
the transaction price, as none of the milestones have yet been achieved, and all unachieved milestone amounts are fully constrained. As part of its evaluation of the constraint, the Company considered numerous factors, including the fact that achievement of the milestones at this time is uncertain and contingent upon future periods when the uncertainty related to the variable consideration is resolved.
The transaction price also includes actual and estimated payments by Kite for the work by Company researchers and reimbursement of the Company’s costs incurred with third parties. The Company uses the expected value method to estimate payments related to the Company’s researchers’ work, taking into account the impact of constraint. Variable consideration is included in the transaction price only to the extent it is probable a significant reversal of cumulative revenues recognized would not occur. The Company will re-
evaluatere-evaluate the transaction price including the estimated variable consideration included in the transaction price and all constrained amounts in each reporting period and as uncertain events are resolved or other changes in circumstances occur. None
The Company has identified four performance obligations within the Kite agreement as follows: (1) a license to the technology combined with the obligation to perform research and development services to apply the Company’s technology to Kite-selected targets; (2) production of research materials; and (3-4) two material rights, each for an extension of the constrained clinicalresearch period for an additional one-year term. Such extensions contain material rights because their exercise does not require payment of a fee that is commensurate with the value of the incremental research term. The license to the Company’s intellectual property is not distinct from the related research and development activities as the licensed technology is not shared with and cannot be utilized by Kite without the research services performed by the Company.
The Company allocated variable consideration (payments by Kite for the work performed by the Company’s researchers and third-party costs, as well as any future milestones and royalties) to the specific performance obligations to which they relate, as such allocation would meet the allocation objective in ASC Topic 606. The Company allocated the fixed consideration of $150.0 million to the performance obligations based on their relative standalone selling prices. Standalone selling prices of optional research years are similar to those of the initial year, but additionally take into account the intrinsic value of the discount upon exercise and the likelihood of exercise.
Fees allocated to options with material rights are deferred until the options are exercised or regulatory milestones have beenexpire. The exercise of options is accounted for as contract continuation, with target selection fees and estimated variable consideration included in the transaction price.price at that time and allocated specifically to the respective target’s performance obligation.
Revenue for the combined license and research services performance obligations is recognized over time, as Kite consumes the benefit of such services as they are being performed by the Company. For the license combined with research and development services performance obligation, the Company recognizes revenue based on proportional performance of the ongoing research services over the period during which the Company performs the services. The estimation of progress towards the satisfaction of this performance obligation and project costs are reviewed quarterly and adjusted, as needed, to reflect the Company’s assumptions regarding the estimated volume of required activities. The production of research materials performance obligation is accounted for under the right to invoice practical expedient, as the Company has the right to invoice Kite for these services in an amount that corresponds directly with the value of the services.
As of December 31, 2023, and 2022 the Company had a receivable of $0.2 million and $0.7 million, respectively, and deferred revenue of zero and $19.4 million, respectively, related to this agreement. Changes in deferred revenue balances during the year ended December 31, 2023 relate to a reduction in the estimated future level of the Company’s research and development services under the collaboration agreement with Kite, ongoing normal progress in delivery of the performance obligations, and expiration of the option to extend the research term. The amounts of transaction price (excluding the amounts recognized as invoiced for the production of research materials performance obligation) remaining to be recognized were zero and $19.7 million as of December 31, 2023 and 2022, respectively.
Revenues recognized under the agreement were as follows (in thousands):
| | | | | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2023 | | 2022 | | 2021 | | | |
| Revenue related to Kite agreement: | | | | | | | | |
| Recognition of license fee fixed consideration | $ | 19,423 | | | $ | 37,032 | | | $ | 24,977 | | | | |
| Research services variable consideration | 1,097 | | | 1,560 | | | 476 | | | | |
| Total | $ | 20,520 | | | $ | 38,592 | | | $ | 25,453 | | | | |
During the year ended December 31, 2023, the Company recorded additional revenue related to changes in estimates in connection with the collaboration agreement with Kite. These changes in estimates were driven by reductions in the estimated future level of the Company’s research and development services in March and September 2023, and as a result, future project costs. These changes resulted in an increase in proportional cumulative performance on this collaboration and
increased revenue by $13.9 million, decreased net loss by $13.9 million, and decreased the Company’s basic and diluted net loss per share by $0.08 for the year ended December 31, 2023.
Novartis Institutes for BioMedical Research, Inc.
On July 27, 2020, the Company entered into a collaboration and license agreement with Novartis Institutes for BioMedical Research, Inc. (“Novartis”) for the research, development and commercialization of gene regulation therapies to treat three neurodevelopmental disorders. Under the agreement, which was effective upon execution, the Company granted Novartis an exclusive, royalty bearing and worldwide license, under its relevant patents and know-how, to develop, manufacture and commercialize certain of its ZF-transcriptional regulators targeted to three undisclosed genes that are associated with certain neurodevelopmental disorders, including autism spectrum disorder and intellectual disability. The Company was performing early research activities over the collaboration period for each gene target and manufacture the ZF-transcriptional regulators required for such research, costs of which are funded by Novartis. Novartis was responsible for additional research activities, studies enabling investigational new drug (“IND”) applications, clinical development, regulatory approvals, manufacturing of preclinical, clinical and approved products, and global commercialization. Subject to certain exceptions set forth in the agreement, the Company was prohibited from developing, manufacturing or commercializing any therapeutic product targeting any of the three genes that are the subject of the collaboration. Novartis also had the option to license certain of the Company’s proprietary AAVs for the sole purpose of developing, manufacturing and commercializing licensed products arising from the collaboration.
In March 2023, Novartis notified the Company of its termination for convenience, effective June 11, 2023 (the “Novartis Termination Date”), of the collaboration agreement. Novartis had indicated to the Company that the termination relates to a recent strategic review. As of the Novartis Termination Date, the collaboration agreement was terminated in its entirety and following the Novartis Termination Date the Company is not entitled to receive any further milestone payments or royalties from Novartis. As of the Novartis Termination Date, the parties have no further obligations to develop or to fund the development of any collaboration research programs under the collaboration agreement.
Upon entering the agreement, Novartis paid the Company a $75.0 million upfront license fee. Novartis was also obligated to pay the Company for the use of its resources and reimburse third-party costs incurred in the Company’s conduct of early research activities. The Company was also eligible to earn from Novartis development and commercial milestones and royalties on potential commercial sales of licensed products arising from the collaboration, none of which were triggered or earned. The agreement was going to continue, on a product-by-product and country-by-country basis, until the expiration of the applicable royalty term.
All payments received under the agreement were non-refundable and non-creditable. The transaction price of $95.1 million included the upfront license fee of $75.0 million and research costs of $20.1 million. All clinical or regulatory milestone amounts were considered fully constrained throughout the term of the agreement.
The Company assessed the agreement with SanofiNovartis in accordance with ASC Topic 606 and concluded that Sanofi isNovartis was a customer. The Company hashad identified thea single performance obligationsobligation within this arrangement as a license to the technology and ongoing research services activities.services. The Company concluded that the license iswas not discrete as it doesdid not have stand-alone value to SanofiNovartis apart from the research services to be performed pursuant to the agreement. As a result, the Company recognizesrecognized revenue from the upfront payment based on proportional performance of the ongoing research services through 2022, the estimated period the Company will perform research services.period. The estimateestimation of progress towards the satisfaction of performance obligation and project cost iswas reviewed quarterly and adjusted, as needed, to reflect the Company’s current assumptions regarding the timing of its deliverables. Revenue fromperformance obligation.
The notice of termination was accounted for as a modification of the reimbursable costscontract, as it changed both the scope of the Company’s remaining services and the consideration to which the Company was entitled. The effect of the modification was not material, as the Company was nearing the completion of its assigned early research activities, and consequently, of its sole performance obligation.
As of December 31, 2023 and 2022, the Company had a receivable of zero and $2.2 million, respectively, and deferred revenue of zero and $9.6 million, respectively, related to this agreement.
Revenues recognized under the agreement were as follows (in thousands): | | | | | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2023 | | 2022 | | 2021 | | | |
| Revenue related to Novartis agreement: | | | | | | | | |
| Recognition of upfront license fee | $ | 9,568 | | | $ | 31,344 | | | $ | 29,945 | | | | |
| Research services | 2,611 | | | 8,384 | | | 7,999 | | | | |
| Total | $ | 12,179 | | | $ | 39,728 | | | $ | 37,944 | | | | |
The Company paid $1.5 million for financial advisory fees during the year ended December 31, 2020, equal to 2% of $75.0 million received for the upfront license fee related to the integrated service deliverablecollaboration and license agreement with Novartis. The Company recognized $1.5 million as a contract asset as such amount represents a cost of obtaining the agreement. This balance was amortized and included in general and administrative expenses on a systematic basis consistent with the transfer of the services to Novartis in accordance with ASC Topic 340, Other Assets and Deferred Costs (“ASC Topic 340”). The Company amortized $0.2 million, $0.6 million and $0.6 million during the years ended December 31, 2023, 2022, and 2021 respectively.
Biogen MA, Inc.
In February 2020, the Company entered into a collaboration and license agreement with Biogen MA, Inc. (“BIMA”) and Biogen International GmbH (together with BIMA, “Biogen”) for the research, development and commercialization of gene regulation therapies for the treatment of neurological diseases. The companies planned to leverage the Company’s proprietary ZF technology delivered via AAV to modulate expression of key genes involved in neurological diseases. Concurrently with the execution of the collaboration agreement, the Company entered into a stock purchase agreement with BIMA, pursuant to which BIMA agreed to purchase 24,420,157 shares of the Company’s common stock (the “Biogen Shares”), at a price per share of $9.2137, for an aggregate purchase price of approximately $225.0 million. The collaboration agreement became effective in April 2020.
In March 2023, Biogen notified the Company of its termination for convenience, effective June 15, 2023 (the “Biogen Termination Date”), of the collaboration agreement. Biogen had indicated to the Company that the termination relates to a recent strategic review. As of the Biogen Termination Date, the collaboration agreement was terminated in its entirety and following the Biogen Termination Date the Company is not entitled to receive any further milestone payments or royalties from Biogen. As of the Biogen Termination Date, the parties have no further obligations to develop or to fund the development of any collaboration research programs under the collaboration agreement.
Under the collaboration agreement, Biogen paid the Company an upfront license fee of $125.0 million in May 2020. The Company was also eligible to receive target selection, research, development, regulatory and commercial milestone payments and royalties on potential net commercial sales of licensed products arising from the collaboration, none of which were triggered or earned.
Under the collaboration agreement, the Company granted to Biogen an exclusive, royalty bearing and worldwide license, under its relevant patents and know-how, to develop, manufacture and commercialize ZF and/or AAV-based products directed to certain neurological disease gene targets selected by Biogen. Biogen had selected four targets over the course of the collaboration and had exclusive rights to nominate up to seven additional targets. These rights expired upon the Biogen Termination Date. For each gene target selected by Biogen, the Company performed early research activities, costs of which were shared by the companies, aimed at the development of the combination of proprietary central nervous system delivery vectors and ZF-transcriptional regulators (or potential other ZF products) targeting therapeutically relevant genes.
The Company assessed the collaboration agreement with Biogen in accordance with ASC Topic 606 and concluded that Biogen is a customer. The transaction price included the upfront license fee of $125.0 million and the excess consideration from the stock purchase of $79.6 million, which represented the difference between the $225.0 million received for the purchase of the Biogen Shares and the $145.4 million estimated fair value of the equity issued. The equity issued to Biogen was valued using an option pricing model to reflect certain holding period restrictions. None of the clinical or regulatory milestones were included in the transaction price, as all such amounts were fully constrained throughout the term of the collaboration agreement. The transaction price also included actual and estimated cost-sharing payments by Biogen for the work by Company researchers and reimbursement of the Company’s costs incurred with third parties. The amounts paid and expected to be paid to Biogen for the use of Biogen’s resources and its expenses were consideration paid to a customer. Since the Company did not acquire distinct goods or services in exchange for these payments, they reduced the transaction price and were recorded as a reduction in revenue. The Company used the expected value method to estimate cost sharing payments, taking into account the impact of the constraint. Variable consideration was included in the transaction price only to the extent it was probable a significant reversal of cumulative revenues recognized would not occur. The Company re-evaluated the transaction price as uncertain events were resolved or other changes in circumstances occurred.
The Company concluded that the licenses to its intellectual property for each target were not distinct from the related research and development activities, as the licensed technology was not shared with and could not be utilized by Biogen without the research services to be performed by the Company pursuant to the agreement. On the other hand, each combination of a license to the Company’s intellectual property as applied to a specific target and the related research and development activities are performed.a discrete research project that is distinct from any other target’s project. The targets Biogen could select were options that provided Biogen with material rights, as the exercise of the options did not require payment of a fee commensurate with the value of the incremental license rights. As a result, such options also represented performance obligations.
At contract inception, the Company allocated fixed consideration of $204.6 million included in the initial transaction price to the existing targets’ license and research services performance obligations and those performance obligations for options that include material rights, based on their relative standalone selling prices. Through June 30, 2023, all such material rights have expired.
The notice of termination was accounted for as a modification of the contract, as it changed both the scope of the Company’s remaining services and the consideration to which the Company was entitled. The remaining research and development activities to be undertaken by the Company after the notice of termination were not distinct from the related activities performed prior to the modification on the same targets but were distinct from the activities on other targets. The remaining material rights were also distinct from the prior research and development activities. To account for the effects of the modification, the Company updated its estimate of the transaction price and allocated the remaining transaction consideration based on the relative standalone selling prices of the remaining distinct goods and services. Progress for each ongoing performance obligation was then remeasured using an updated estimate of the total level of effort required for each performance obligation and the total revised transaction price and a cumulative catch-up in revenue was recorded. The modification resulted in a decrease to the transaction price of $17.3 million and an increase in revenue of $127.1 million.
As of December 31, 2023 and 2022, the Company had a receivable of zero and $0.5 million, respectively, and deferred revenue of zero and $132.2 million, respectively, related to this agreement. Changes in deferred revenue balances during the year ended December 31, 2023 relate primarily to the impact of the contract modification. The amounts of transaction price remaining to be recognized were zero and $151.3 million as of December 31, 2023 and 2022, respectively.
Revenues recognized under the agreement were as follows (in thousands): | | | | | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2023 | | 2022 | | 2021 | | | |
| Revenue related to Biogen agreement: | | | | | | | | |
| Recognition of license and other fixed consideration | $ | 132,165 | | | $ | 21,820 | | | $ | 29,224 | | | | |
| Cost-sharing payments for research services, net variable consideration | 2,684 | | | 6,599 | | | 13,076 | | | | |
| Total | $ | 134,849 | | | $ | 28,419 | | | $ | 42,300 | | | | |
The Company paid $7.0 million for financial advisory fees during the year ended December 31, 2020, equal to 2% of $225.0 million received for the sale of shares and 2% of $125.0 million received for the upfront fee. The fees incurred related to both the collaboration agreement with Biogen and to the stock purchase agreement for the sale of shares. The Company believes that the allocation of fees on a relative fair value basis between the two agreements is reasonable. The Company recognized $4.1 million, which represents 2% of the initial transaction price of $204.6 million, as a contract cost asset. This balance was released into general and administrative expenses on a systematic basis consistent with the transfer of the services to Biogen in accordance with ASC Topic 340. In March, as a result of the notice of termination and resulting modification of the contract, the progress for each performance obligation was remeasured and the Company recognized $2.5 million as a cumulative catch-up to expense. The Company recognized as expense $2.6 million, $0.4 million and $0.6 million during the years ended December 31, 2023, 2022 and 2021, respectively.
Sanofi S.A.
In January 2014, the Company entered into an exclusive worldwide collaboration and license agreement (“2014 Collaboration Agreement”) to develop therapeutics for hemoglobinopathies, focused on beta thalassemia and sickle cell disease (“SCD”). The 2014 Collaboration Agreement was originally signed with BIMA, who subsequently assigned it to Bioverativ Inc., which was later acquired by Sanofi. Under the 2014 Collaboration Agreement, the Company was originally jointly conducting two research programs: a beta thalassemia program, which was discontinued in the third quarter of 2021, and the SCD program, which resulted in the development of SAR445136 (now known as BIVV003), a ZFN, gene-edited cell therapy product candidate for the treatment of SCD. In December 2021, Sanofi notified the Company of its termination for convenience, effective June 28, 2022 (the “Termination Date”), of the 2014 Collaboration Agreement. A termination and transition agreement (the “Termination and Transition Agreement”) was executed by the parties on September 6, 2022.
In the SCD program, the Company and Sanofi were jointly responsible for research and development activities prior to filing of an IND, but Sanofi was responsible for subsequent worldwide clinical development, manufacturing and commercialization of licensed products developed under the agreement. Subject to the terms of the agreement, the Company had granted Sanofi an exclusive, royalty-bearing license, with the right to grant sublicenses, to use certain ZF and other technology controlled by the Company for the purpose of researching, developing, manufacturing and commercializing licensed products developed under the agreement. The Company had also granted Sanofi a non-exclusive worldwide, royalty-free fully paid license with the right to grant sublicenses, under the Company’s interest in certain other intellectual property developed pursuant to the agreement. During the term of the agreement, the Company was not permitted to research, develop, manufacture or commercialize, outside of the agreement, certain gene therapy products that target genes relevant to the licensed products.
Under the 2014 Collaboration Agreement, the Company received an upfront license fee of $20.0 million and was eligible to receive additional payments upon the achievement of specified clinical development, regulatory milestones, and sales milestones, as well as royalty payments for each licensed product based on net sales of such product. Sanofi was also to reimburse Sangamo for agreed upon costs incurred in connection with research and development activities conducted by Sangamo. Through the Termination Date, a total of $13.5 million was received based on achievement of clinical development milestones. No products have been approved and therefore no royalty fees have been or will be earned under the 2014 Collaboration Agreement.
In its termination notice to the Company, Sanofi indicated that its termination relates to Sanofi’s change in strategic direction to focus on allogeneic universal genomic medicine approaches rather than autologous personalized cell therapies. As of the Termination Date, the 2014 Collaboration Agreement was terminated in its entirety and following the Termination Date, the Company will not be entitled to receive any further milestone payments or royalties from Sanofi. As of the Termination Date, Sanofi has no further obligations under the 2014 Collaboration Agreement to develop or to fund the development of any collaboration research programs under the 2014 Collaboration Agreement. The licenses granted to Sanofi under the 2014 Collaboration Agreement have been terminated, and the license rights have reverted to the Company.
As part of the Termination and Transition Agreement, Sanofi granted to the Company exclusive, worldwide, fully paid, royalty-free, perpetual, irrevocable licenses, with the right to grant sublicenses through multiple tiers, to certain of its intellectual property, to develop, manufacture, have manufactured, use, sell, offer to sale, import and otherwise commercialize BIVV003, the product candidate in development under the SCD program. The Company agreed to take on responsibilities for all clinical trials related to BIVV003, including completion of the ongoing clinical trial and the related long-term follow-up study. The Company also assumed all regulatory responsibilities related to BIVV003. Sanofi transferred and assigned to the Company all documentation, materials and contracts with third parties related to BIVV003, and the right to use certain Sanofi-owned or leased equipment related to BIVV003.
Sanofi has also agreed to reimburse the Company for the costs of conducting the ongoing clinical trial of BIVV003 and the costs of the long-term follow-up study through December 31, 2023, up to $7.0 million. In addition, should the Company elect not to continue the development of BIVV003 past December 31, 2023, Sanofi will become obligated to reimburse the Company for the costs of the long-term follow-up study incurred after 2023, up to $5.3 million. Sanofi’s reimbursement obligations will terminate upon certain triggering events, including the Company’s entering into a contract with a third party for collaboration, partnership, sale, licensing, or divestiture of BIVV003, or if the FDA permits early closure of the clinical trial and/or the long-term follow-up study.
The Company assessed the 2014 Collaboration Agreement in accordance with ASC Topic 606 and concluded that Sanofi was a customer, under that arrangement. The Company identified the performance obligation within this arrangement as a license to the technology combined with ongoing research services activities. The Company concluded that the license was not distinct as it did not have stand-alone value to Sanofi without the research services. As a result, the Company recognized revenue from the upfront payment and the milestones based on progress of performance of the ongoing research services. The estimation of progress towards the satisfaction of the performance obligation and project cost was reviewed quarterly and adjusted, as needed, to reflect the Company’s then current assumptions regarding the timing of its deliverables. Related costs and expenses under these arrangements have historically approximated the revenues recognized. Sanofi’s December 2021 notice of termination of the 2014 Collaboration Agreement represented a modification that reduced the expected scope of the Company’s services and the estimated transaction price and shortened the remaining performance timeline. Consistent with this change, all services provided by the Company under the 2014 Collaboration Agreement were completed by June 28, 2022, and all amounts ultimately included in the transaction price were recognized by such date. The final transaction price of $96.3 million included the upfront license fee of $20.0 million, two milestone payments in the aggregate amount of $13.5 million and reimbursement of research costs of $62.8 million. As of December 31, 20202023 and 2019,2022, the Company had deferred revenuea receivable of $1.2 millionzero and $1.7$0.6 million, respectively, related to this agreement.the 2014 Collaboration Agreement. Deferred revenue related to the 2014 Collaboration Agreement was zero and $1.1 million, respectively.
In August 2019,The Company concluded that Sanofi is not a customer under the Termination and Transition Agreement as Sanofi is not entitled to receive and cannot use the results of the ongoing clinical trial or the long-term follow-up study. This relationship
is also not a collaboration in the scope of ASC Topic 808, Collaborative Arrangements. The Company concluded that the assets acquired from Sanofi do not represent a business, as substantially all of their value is concentrated in the acquired or re-acquired licenses to intellectual property. The Company has no obligation to repay Sanofi for its ongoing funding of the clinical trial or long-term follow-up study costs. Therefore, the Company achieved a $6.0 million milestone withwill recognize Sanofi upon dosing of the third subject in the ST-400 beta thalassemia Phase 1 clinical trial. The cumulative revenue recognized in connection with this milestone was approximately $5.8 millionreimbursements as of December 31, 2020reductions to its research and included $0.1 million recognized duringdevelopment expense. During the year ended December 31, 2020.
In December 2019,2023, the Company achieved a $7.5decreased its research and development expense by $2.1 million, milestone with Sanofi upon dosing of which $0.5 million is included within prepaid expenses and other current assets on the first subject in the SCD Phase 1 clinical trial. The cumulative revenue recognized in connection with this milestone was approximately $7.2 millionCompany’s Consolidated Balance Sheet as of December 31, 2020 and included $0.1 million recognized during the year ended December 31, 2020.2023.
Revenues recognized under the agreement were as follows (in thousands): | | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2023 | | 2022 | | 2021 |
| Revenue related to Sanofi agreement: | | | | | |
| Recognition of upfront fee | $ | — | | | $ | 677 | | | $ | 34 | |
| Research services | — | | | 2,126 | | | 3,057 | |
| Milestone achievement | — | | | 457 | | | 23 | |
| Total | $ | — | | | $ | 3,260 | | | $ | 3,114 | |
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2020 | | 2019 | | 2018 |
| Revenue related to Sanofi agreement: | | | | | |
| Recognition of upfront fee | $ | 298 | | | $ | 3,494 | | | $ | 4,013 | |
| Research services | 4,823 | | | 6,367 | | | 9,503 | |
| Milestone achievement | 201 | | | 12,819 | | | 0 | |
| Total | $ | 5,322 | | | $ | 22,680 | | | $ | 13,516 | |
In March 2020,During the year ended December 31, 2021, the Company recorded an adjustmentadjustments to revenue related to a changechanges in estimateestimates in connection with the collaboration agreement with Sanofi. This adjustment wasThese changes in estimates were driven by a direct result of the decisionchange in March 2020 to increase the project scope and related project costs in September 2021 and subsequent notification of termination of the corresponding costs, both ofcollaboration agreement which resulted in a decrease inchanges to the measure of proportional cumulative performance. This adjustmentThese adjustments decreased revenue by $2.2$1.6 million, increased net loss by $2.2$1.6 million and increased the Company’s basic and diluted net loss per share by $0.02$0.01 for the year ended December 31, 2020.2021.
California Institute for Regenerative Medicine
In May 2018, the California Institute for Regenerative Medicine (“CIRM”) granted a Strategic Partnership Award for $8.0 million to fund the clinical studies of a potentially curative ZFPZF therapeutic for the treatment of beta thalassemia based on the application of Sangamo’s ZFN genome editing technology. The grant exists through December 31, 2022 and providesprovided matching funds to support the evaluate ST-400, a gene-edited cell therapy candidate for people with transfusion-dependent beta thalassemia. As of December 31, 2020, the Company had received $5.2 million under the award.
Under the terms of the CIRM grants,grant, the Company iswas obligated to pay royalties and licensing fees based on a low single digit royalty percentage on net sales of CIRM-funded product candidates or CIRM-funded technology. The Company hashad the option to decline any and all amounts awarded by CIRM and as an alternative to revenue sharing, the Company hashad the option to convert the award to a loan. Noloan, however no such election hashad been made as of December 31, 2020. The Company had received $5.2 million under the date of the issuance of these Consolidated Financial Statements. In the event that the Company terminates a CIRM-funded clinical trial, it will be obligated to repay the remaining CIRM funds on hand. Asaward as of December 31, 2020 and 2019,2020. The Company had recorded $6.4 million, and $5.7 million, respectively, including accrued interest of $1.2 million, as a loan related to this award is recorded as a loan in other non-current liabilities on the Consolidated Balance Sheets.Sheet as of December 31, 2020.
Amended Collaboration and License Agreement with Takeda
In January 2012, the Company entered into a collaboration and license agreement with Shire International GmbH, a wholly-owned subsidiary of Takeda Pharmaceutical Company Limited (“Takeda”), to research, develop and commercialize a ZFP therapeutic for treating Huntington’s disease. The Company received an upfront license fee of $13.0 million in 2012 and
recognized a $1.0 million milestone payment in 2014. Takeda does not have any milestone payment obligations, but is required to pay single digit percentage royalties to the Company, up to a specified maximum cap, on the commercial sales of therapeutic products for Huntington’s disease. The Company is required to pay single digit percentage royalties to Takeda, up to a specified maximum cap, on commercial sales of therapeutic products from programs returned under the original agreement (which include blood clotting Factors VIII and IX) that use two zinc fingers.
Pursuant to the agreement, the Company granted Takeda an exclusive, world-wide, royalty-bearing license, with the right to grant sublicenses, to use the Company’s ZFP technology for the purpose of developing and commercializing human therapeutic and diagnostic products for the huntingtin gene (“HTT gene”). During the term of the agreement, the Company is not permitted to research, develop or commercialize, outside of the agreement, certain products that target the HTT gene. The agreement may be terminated by (i) the Company or Takeda, in whole or in part, for the uncured material breach of the other party, (ii) the Company or Takeda for the bankruptcy or other insolvency proceeding of the other party and (iii) Takeda, in its entirety, effective upon at least 90 days’ advance written notice.
The Company assessed the agreement with Takeda in accordance with ASC Topic 606 and concluded that Takeda is a customer. The Company has concluded that the license is not a separate unit of accounting as it does not have stand-alone value to Takeda apart from the research services to be performed pursuant to the Takeda agreement. The Company satisfied the deliverables and research services responsibilities within the amended arrangement which were completed in 2017. As a result of the November 2021 decision to discontinue the development of ST‑400 in order to prioritize the development of other product candidates, the grant was terminated. In connection with the termination and discontinuation of the program, the Company elected not to convert the award to a loan and recognized the remaining $2.3non-refundable award amount of $5.2 million as a reduction of research and development expenses, and $1.2 million of deferred revenue fromaccrued interest on the upfront payment duringaward as interest and other income, net, on the Company’s Consolidated Statements of Operations for the year ended December 31, 2017.
The Company did 0t recognize any revenues under2021. No amounts related to this award were included on the Takeda agreement for the years endedConsolidated Balance Sheets as of December 31, 2020, 20192023 and 2018.2022.
Agreement with Sigma-Aldrich Corporation
In 2007, Sangamo entered into a license agreement with Sigma-Aldrich Corporation (“Sigma”) to provide Sigma with access to Sangamo’s proprietary ZFPZF technology and the exclusive right to use the technology to develop and commercialize research reagent products and services in the research field, excluding certain agricultural research uses that Sangamo previously licensed to Dow AgroSciences LLC (“DAS”), a wholly-owned subsidiary of Dow Chemical Company. Sangamo developed laboratory research reagents using its ZFPZF technology over a three-year research services period. Sangamo has since transferred the ZFPZF manufacturing technology to Sigma.
In October 2009, Sangamo expanded its license agreement with Sigma. In addition to the original terms of the license agreement, Sigma received exclusive rights to develop and distribute ZFP-modifiedZF-modified cell lines for commercial production of protein pharmaceuticals and certain ZFP-engineeredZF-engineered transgenic animals for commercial applications. Under the terms of the agreement, Sigma made an upfront cash payment of $20.0 million consisting of a $4.9 million purchase of 636,133 shares of Sangamo common stock, valued at $4.9 million, and a $15.1 million upfront license fee. Sangamo is also eligible to receive commercial license fees of $5.0 million based upon a percentage of net sales and sublicensing revenue and thereafter a reduced royalty rate of 10.5% of net sales and sublicensing revenue. In addition, upon the achievement of certain cumulative
commercial milestones, Sigma will make milestone payments to Sangamo up to an aggregate of $25.0$42.0 million. Sangamo does not have additional ongoing performance obligations under the agreement.
Revenues recognized under the agreement with Sigma for the years ended December 31, 2020, 20192023, 2022 and 2018,2021, were $0.5$4.7 million, $0.6$0.9 million and $0.5$1.1 million, respectively.
Agreement with DAS
In 2005, Sangamo entered into an exclusive commercial license with DAS, with an initial three-year research term. Under this agreement, Sangamo is providing DAS with access to its proprietary ZFP technology and the exclusive right to use the technology to modify the genomes or alter the nucleic acid or protein expression of plant cells, plants, or plant cell cultures. Sangamo has retained rights to use plants or plant-derived products to deliver ZFP-TFs or ZFNs into humans or animals for diagnostic, therapeutic or prophylactic purposes. In 2008, DAS exercised its option and obtained a commercial license to sell products incorporating or derived from plant cells generated using the Company’s ZFP technology. The exercise of the option triggered a one-time commercial license fee of $6.0 million, payment of the remaining $2.3 million of the previously agreed upon $4.0 million in research milestones, development and commercialization milestone payments for each product, and royalties on sales of products. Furthermore, DAS has the right to sublicense Sangamo’s ZFP technology to third parties for use in plant cells, plants, or plant cell cultures, and Sangamo will be entitled to 25% of any cash consideration received by DAS under such sublicenses. In December 2010, the Company amended its agreement with DAS to extend the period of reagent manufacturing services and research services through December 31, 2012.
The agreement with DAS also provides for minimum sublicense fees each year due to Sangamo every October, provided the agreement is not terminated by DAS. Annual fees range from $0.3 million to $3.0 million and total $25.3 million
over 11 years. The Company has identified the performance obligation within this arrangement as a license to the technology. In the event of any termination of the agreement, all rights to use the Company’s ZFP technology will revert to Sangamo, and DAS will no longer be permitted to practice Sangamo’s ZFP technology or to develop or, except in limited circumstances, commercialize any products derived from the Company’s ZFP technology.
Revenues under the agreement with DAS were $3.0 million, $3.0 million, and $3.0 million during 2020, 2019 and 2018, respectively.
NOTE 5 – ACQUISITION OF SANGAMO FRANCE
On July 20,In 2018, Sangamo entered into various agreements with the goal of eventually acquiring 100% of Sangamo France’s share capital. The Company entered into the Sangamo France SPA with certain shareholders of Sangamo France, pursuant to which it acquired 13,519,036 ordinary shares of Sangamo France (“Ordinary Shares”) as part of a block transaction that closed on October 1, 2018 (the “Acquisition Date”). Additionally, the Company and Sangamo France entered into a Tender Offer Agreement pursuant to which Sangamo agreed to acquire 11,528,635 Ordinary Shares for the same price per share as the Sangamo France SPA via a cash tender offer that closed on November 23, 2018. Following the block transaction, cash tender offer, and other open market purchases of shares, the Company owned 98.2% of the Ordinary Shares as of December 31, 2018 (or 25,047,671 Ordinary Shares). In addition to the Sangamo France SPA and the tender offer agreement, the Company also entered intocapital, including arrangements with the holders of approximately 477,000 “free shares”free shares of Sangamo France pursuant to which the Company hashad the right to purchase such shares from the holders (a call option), and such holders havehad the right to sell to the Company such shares from time to time through mid-2021 (a put option) (collectively the “Free Shares Options”). During 2019,As of December 31, 2021, the Company acquired approximately 111,000 vestedall of the 477,000 free shares, increasing itsresulting in 100% ownership of the Ordinary Shares from 98.2% to 98.7%. During 2020, the Company acquired approximately 322,000 vested free shares, pursuant to the exercise of the put options for approximately $0.7 million of cash, increasing its ownership of the Ordinary Shares to 99.8% as of December 31, 2020.
At the Acquisition Date, the fair value of the Free Shares Options was estimated to be a liability of $0.2 million. See Note 2 – Fair Value Measurement – Free Shares Asset for information regarding the valuation method. The fair value of the Free Shares Options will vary based on future changes in the Company’s stock price during the option period. The fair value of the Free Shares Options was estimated to be an asset of $0.1 million as of December 31, 2020.Sangamo France.
The acquisition of Sangamo France was accounted for as a business combination in accordance with ASC Topic 805, Business Combinations, in exchange for total consideration of approximately $45.9 million at the Acquisition Date. The operating results of Sangamo France after the Acquisition Date have been included in the Company’s Consolidated Statements of Operations. Based on the Company’s impairment assessment performed during the year ended December 31, 2023, it recognized a pre-tax goodwill impairment charge of $38.1 million and as a result the goodwill was fully impaired as of December 31, 2023, see Note 6 –
Impairment of Goodwill, Indefinite-lived Intangible Assets and Other Long-lived Assets. There was 0no goodwill impairment during the years ended December 31, 2020, 20192022 or 2018 and, as noted below, substantially all of the non-controlling interest on the Acquisition Date was subsequently acquired by the Company and, accordingly, substantially all of the goodwill is allocated to the Company as of December 31, 2020 and 2019.2021.
Non-controlling Interest
ThePrior to the acquisition of all the free shares, the fair value of the remaining non-controlling interest was determined based on the number of outstanding free shares comprising the non-controlling interest and the $2.99 acquisition price per share as of the Acquisition Date. The non-controlling interest iswas presented as a component of stockholders’ equity on the Company’s Consolidated Balance Sheets.
Non-controlling interestSheet as of December 31, 20202020. As of December 31, 2023 and 2022, after acquisition of 100% of ordinary shares of Sangamo France, the carrying amount of the non-controlling interest was recorded as follows (in thousands):additional paid‑in capital on the Company’s Consolidated Balance Sheet.
| | | | | |
| Total |
| |
| |
| |
| |
| |
| |
Balance at beginning of year | $ | 185 | |
Fair value of additional shares acquired | (927) | |
Loss attributable to non-controlling interest | (126) | |
Balance at end of year | $ | (868) | |
Three months ended March 31, 2023
During the three months ended March 31, 2023, as a result of the sustained decline in the Company’s stock price and related market capitalization, termination of the collaboration agreements with Biogen and Novartis, the Company performed an impairment assessment of goodwill, indefinite-lived intangible assets, and long-lived assets.
The Company operates as a single reporting unit based on its business and reporting structure. For goodwill, a quantitative impairment assessment was performed using a market approach, whereby the Company’s fair value of equity was compared to its carrying value. The fair value of equity was derived using both the market capitalization of the Company and an estimate of a reasonable range of values of a control premium applied to the Company’s implied business enterprise value. The control premium was estimated based upon control premiums observed in comparable market transactions. This represents a level 2 nonrecurring fair value measurement. Based on this analysis, the Company recognized a pre-tax goodwill impairment charge of $38.1 million during the three months ended March 31, 2023. As a result, the goodwill was fully impaired as of March 31, 2023.
Before completing the goodwill impairment assessment, the Company also tested its indefinite-lived intangible assets and then its long-lived assets for impairment. Based on the qualitative assessment, the Company determined it was more likely than not that its indefinite-lived intangible assets were not impaired. The Company determined all of its long-lived assets represent one asset group for purposes of long-lived asset impairment assessment. The Company concluded that the carrying value of the asset group was not recoverable as it exceeded the future undiscounted cash flows the assets are expected to generate from the use and eventual disposition. To allocate and recognize the impairment loss, the Company determined individual fair values of its long-lived assets. The Company applied a discounted cash flow method to estimate fair values of its leasehold improvements and right-of-use assets, including leasehold improvements in the process of construction and a cost replacement method to estimate the fair value of its furniture, fixtures and laboratory and manufacturing equipment. These represented level 3 nonrecurring fair value measurements. Based on this analysis, the Company recognized pre-tax long-lived asset impairment charges of $11.2 million on the right-of-use assets, $5.0 million on the related leasehold improvements, and $4.2 million on construction-in-progress, during the three months ended March 31, 2023. No impairment was recognized on the remaining long-lived assets as their carrying values were not in excess of their fair values.
Three months ended June 30, 2023
During the three months ended June 30, 2023, the Company’s stock price and the related market capitalization continued to decline. In April 2023, the Company announced a restructuring of operations and a corresponding reduction in force, see Note 11 – Restructuring Charges. The Company also initiated discussions around several actions aimed at reducing costs, preserving liquidity and improving operational performance metrics. These actions include but are not limited to deferral and reprioritization of certain research and development programs, further reduction in force, and closing or downsizing its facilities.
The Company reassessed its indefinite-lived and long-lived assets for impairment as of June 30, 2023. Given the actions contemplated above, the Company determined that it was more likely than not that its indefinite-lived intangible assets were impaired. Accordingly, the Company developed an estimate of the fair value of its indefinite-lived intangible assets using the multi-period excess earnings model (income approach) and concluded the carrying value of its indefinite-lived intangible assets were fully impaired. This represents a level 3 nonrecurring fair value measurement. As a result, an indefinite-lived intangible assets impairment charge of $51.4 million, as well as the related income tax benefit of $6.3 million due to the reversal of a deferred tax liability associated with the indefinite-lived intangible assets, were recognized during the three months ended June 30, 2023. The impairment charge was primarily driven by a higher discount rate applied to future cash flows based on market participants’ view of increased risk related to the asset.
The Company determined that there were indicators of impairment in its long-lived asset group as of June 30, 2023, based on the same factors above as well as the impairment of its indefinite-lived intangible assets. As the estimated fair value of this asset group, based on a market approach, exceeded its carrying value, no impairment loss was recognized. This represented a level 3 nonrecurring fair value measurement.
Three months ended September 30, 2023
During the three months ended September 30, 2023, the Company’s stock price and the related market capitalization continued to decline, and as such, the Company reassessed its long-lived assets for impairment as of September 30, 2023.
The Company determined all of its long-lived assets continued to represent one asset group for purposes of long-lived asset impairment assessment. The Company concluded that the carrying value of the asset group was not recoverable and the estimated fair value of this asset group was below its carrying value. The lower fair value of the asset group was mainly driven by the sustained decline in the Company’s stock price and the related market capitalization. To recognize the impairment loss, the Company determined individual fair values of its long-lived assets. The Company applied a discounted cash flow method to estimate fair values of its leasehold improvements and right-of-use assets, including leasehold improvements in the process of construction, and a market approach to estimate the fair value of its furniture, fixtures and laboratory and manufacturing equipment. These represented level 3 nonrecurring fair value measurements. Based on this analysis, the Company concluded the fair values of the long-lived assets were lower than their net book values due to declines in the market prices for leases, furniture, fixtures, and equipment. The Company recognized pre-tax long-lived asset impairment charges of $17.6 million on the right-of-use assets, $13.7 million on the related leasehold improvements and construction-in-progress, and $13.5 million on furniture, fixtures, and laboratory and manufacturing equipment during the three months ended September 30, 2023.
Three months ended December 31, 2023
During the three months ended December 31, 2023, the Company’s stock price and the related market capitalization continued to decline, and as such, the Company reassessed its long-lived assets for impairment as of December 31, 2023.
The Company determined all of its long-lived assets continued to represent one asset group for purposes of long-lived asset impairment assessment. The Company concluded that the carrying value of the asset group was not recoverable and the estimated fair value of this asset group was below its carrying value. The lower fair value of the asset group was mainly driven by the decline in the Company’s stock price and the related market capitalization. To recognize the impairment loss, the Company determined individual fair values of its long-lived assets. The Company applied a discounted cash flow method to estimate fair values of its leasehold improvements and right-of-use assets, including leasehold improvements in the process of construction, and a market approach to estimate the fair value of its furniture, fixtures and laboratory and manufacturing equipment. These represented level 3 nonrecurring fair value measurements. Based on this analysis, the Company concluded the fair values of certain lease related long-lived assets were lower than their net book values due to declines in the market prices for leases. The Company recognized pre-tax long-lived asset impairment charges of $0.1 million on the right-of-use assets and $0.2 million on the related leasehold improvements and construction-in-progress during the three months ended December 31, 2023.
The Company will continue to assess whether its long-lived assets are impaired in future periods. As the Company finalizes and implements its plans related to cost reductions and liquidity preservation, it is reasonably possible that additional impairment charges will be recognized if the Company changes how it uses various long-lived assets or elects to dispose of
them, and the cash flows associated with these assets become separately identifiable. In this case, such assets will be tested for impairment separately from the remaining long-lived assets of the Company.
NOTE 67 – OTHER BALANCE SHEET DETAILS
Property and Equipment, Net
Property and equipment, net consist of the following (in thousands):
| | | | | | | | | | | |
| December 31, |
| 2020 | | 2019 |
| Laboratory equipment | $ | 24,737 | | | $ | 17,179 | |
| Furniture and fixtures | 4,870 | | | 4,639 | |
| Leasehold improvements | 15,953 | | | 13,888 | |
| Manufacturing equipment | 1,089 | | | 0 | |
| Construction in progress | 12,091 | | | 5,901 | |
| 58,740 | | | 41,607 | |
| Less: accumulated depreciation and amortization | (17,416) | | | (11,681) | |
| Property and equipment, net | $ | 41,324 | | | $ | 29,926 | |
| | | | | | | | | | | |
| December 31, |
| 2023 | | 2022 |
| Laboratory equipment | $ | 34,630 | | | $ | 39,080 | |
| Leasehold improvements | 29,809 | | | 30,572 | |
| Furniture and fixtures | 4,610 | | | 5,731 | |
| Manufacturing equipment | 7,784 | | | 9,908 | |
| Construction in progress | 1,087 | | | 14,770 | |
| 77,920 | | | 100,061 | |
| Less: accumulated depreciation and amortization | (51,046) | | | (36,530) | |
| Property and equipment, net | $ | 26,874 | | | $ | 63,531 | |
Depreciation and amortization expense was $5.7$15.1 million in 2020, $3.92023, $12.1 million in 20192022 and $2.4$9.4 million in 2018.2021.
During the year ended December 31, 2023, the Company recorded impairment losses of $23.1 million for its leasehold improvements, $8.0 million for its laboratory equipment, $2.3 million for its manufacturing equipment, $1.6 million for its furniture and fixtures, and $1.6 million for its construction in progress. The Company did not record any impairment losses in 2022 and 2021.
Intangible Assets
The changes in intangible assets were as follows (in thousands):
| | | | | | | | | | | |
| December 31, |
| 2020 | | 2019 |
| Balance at beginning of year | $ | 53,156 | | | $ | 54,243 | |
| | | |
| Foreign currency translation adjustment | 4,972 | | | (1,087) | |
| Balance at end of year | $ | 58,128 | | | $ | 53,156 | |
| | | | | | | | | | | |
| December 31, |
| 2023 | | 2022 |
| Balance at beginning of year | $ | 50,729 | | | $ | 53,760 | |
| | | |
| Foreign currency translation adjustment | 618 | | | (3,031) | |
| | | |
| Impairment of indefinite-lived intangible asset | (51,347) | | | — | |
| Balance at end of year | $ | — | | | $ | 50,729 | |
Goodwill
The changes in goodwill were as follows (in thousands): | | | | | | | | | | | |
| December 31, |
| 2023 | | 2022 |
| Balance at beginning of year | $ | 37,552 | | | $ | 39,702 | |
| | | |
| Foreign currency translation adjustment | 586 | | | (2,150) | |
| | | |
| Impairment of goodwill | (38,138) | | | — | |
| Balance at end of year | $ | — | | | $ | 37,552 | |
| | | | | | | | | | | |
| December 31, |
| 2020 | | 2019 |
| Balance at beginning of year | $ | 39,273 | | | $ | 40,044 | |
| | | |
| Foreign currency translation adjustment | 3,525 | | | (771) | |
| Balance at end of year | $ | 42,798 | | | $ | 39,273 | |
Other Accrued Liabilities
Other accrued liabilities consist of the following (in thousands):
| | | | | | | | | | | |
| December 31, |
| 2020 | | 2019 |
| Customer advance | $ | 5,000 | | | $ | 0 | |
| Accrued research and development expenses | 4,257 | | | 4,102 | |
| Operating lease liabilities – current | 3,690 | | | 3,214 | |
| Accrued professional fees | 1,532 | | | 1,118 | |
| Other | 4,133 | | | 2,451 | |
| Total other accrued liabilities | $ | 18,612 | | | $ | 10,885 | |
| | | | | | | | | | | |
| December 31, |
| 2023 | | 2022 |
| Accrued restructuring expenses | $ | 11,733 | | | $ | — | |
Operating lease liabilities – current | 4,589 | | | 4,122 | |
| Accrued research and development expenses | 3,763 | | | 7,115 | |
| Accrued professional fees | 1,505 | | | 1,704 | |
| | | |
| Other | 1,964 | | | 3,066 | |
| Total other accrued liabilities | $ | 23,554 | | | $ | 16,007 | |
NOTE 78 – COMMITMENTS AND CONTINGENCIES
Leases
SangamoSangamo’s corporate headquarters occupies approximately 87,70059,485 square feet of research and office space, pursuant to a lease that expires in August 2031, and approximately 7,700 of office space, pursuant to a lease that expires in August 2026, in Richmond, California. Sangamo also occupies approximately 103,089 square feet of office and research and development laboratory facilities in Brisbane, California pursuant to a lease that expires in May 2029. Sangamo also occupies approximately 54,200 square feet of research and office space in Richmond, California, pursuant to leases that expire in August 2026. In addition, the Company
leases approximately 20,80028,048 square feet of office and research and development space in Valbonne, France, subjectpursuant to leases that expire beginning in June 2025 through March 2028.January 2030.
In May 2020,January 2021, the Company entered into an amendment to an existing lease to acquire approximately 8,5005,000 square feet of research and office space in Richmond, California thatCalifornia. With this amendment, the existing lease expires in August 2026. Total lease payments over the life of this amended lease are approximately $1.6$0.9 million. Variable lease payments include the Company’s allocated share of costs incurred and expenditures made by the landlord in the operation and management of the building. On February 1, 2021, the lease commencement date, the Company recorded an operating lease right-of-use asset and a corresponding lease liability of $0.7 million.
In January 2021, the Company also entered into a new lease to acquire approximately 5,800 square feet of research and office space in Valbonne, France, which expires in January 2030. Total lease payments over the life of this amended lease are approximately $0.8 million. Variable lease payments include the Company’s allocated share of costs incurred and expenditures made by the landlord in the operation and management of the building. On January 29, 2021, the lease commencement date, the Company recorded an operating lease right-of-use asset and a corresponding lease liability of $0.6 million.
In October 2021, the Company entered into an agreement to extend the lease of its research and office space in Richmond, California by five years until August 2031. The Company also leased an additional 7,997 square feet of office space at the same location from November 2021 through August 2031. The amended lease was effective October 1, 2020,2021, and the Company recorded aan adjustment to the lease liability and the corresponding ROUright-of-use asset of $1.3$9.1 million upon inception of this amended lease. Pursuant to the terms of the amended lease, the landlord agreed to reimburse the Company up to $2.6 million, related to a tenant improvement allowance.
Certain of these leases also include renewal options at the election of the Company to renew or extend the lease for an additional five to 10ten years. These optional periods have not been considered in the determination of the ROUright-of-use assets or lease liabilities associated with these leases as the Company did not consider it reasonably certain it would exercise the options.
The Company performed evaluations of its contracts and determined each of its identified leases are operating leases. For the year ended December 31, 2020, the Company incurred $10.4 millionComponents of lease costs included in operating expenses in the Consolidated Statement of Operations in relation to these operating leases. leases were as follows (in thousands):
| | | | | | | | | | | |
| December 31, |
| 2023 | | 2022 |
| Operating lease cost | $ | 9,423 | | | $ | 11,029 | |
| Variable lease cost | 3,126 | | | 3,305 | |
| Total | $ | 12,549 | | | $ | 14,334 | |
Variable lease expense was $2.3 million for the year ended December 31, 2020 and wasexpenses were not included in the measurement of the Company’s operating ROUright-of-use assets and lease liabilities. TheThis variable expense consists primarily of the Company’s proportionate share of operating expenses, property taxes and insurance and is classified as lease expense, due to the Company’s election to not separate lease and non-lease components.
Cash paid for amounts included in the measurement of operating lease liabilities for the year ended December 31, 20202023, 2022 and 2021 was $6.4$7.3 million, $10.1 million and $6.9 million, respectively and was included in net cash provided byused in operating activities in the Company’s Consolidated StatementStatements of Cash Flows.
Rent expense related to lease agreements was $10.4$9.4 million, $7.9$11.0 million and $2.3$10.8 million for 2020, 2019the years ended December 31, 2023, 2022 and 2018,2021, respectively. Future minimum payments under lease obligations at December 31, 20202023 consist of the following (in thousands):
| | Total |
| 2021 | $ | 6,191 | |
| 2022 | 6,756 | |
| 2023 | 6,851 | |
| Total | | | Total |
| 2024 | 2024 | 6,995 | |
| 2025 | 2025 | 7,058 | |
| 2026 | |
| 2027 | |
| 2028 | |
| Thereafter | Thereafter | 19,571 | |
| Total lease payments | Total lease payments | 53,422 | |
| Less: | Less: | |
| Imputed interest | Imputed interest | (11,336) | |
| Imputed interest | |
| Imputed interest | |
| Tenant improvement allowance included in contra-lease liability | |
| Total | Total | $ | 42,086 | |
| | Reported as of December 31, 2020: | |
| Operating lease liabilities - current (included in Other accrued liabilities on the Consolidated Balance Sheet) | $ | 3,690 | |
| Operating lease liabilities - long-term | 38,396 | |
| Reported as of December 31, 2023: | |
| Reported as of December 31, 2023: | |
| Reported as of December 31, 2023: | |
| Short-term portion of lease liabilities (included in other accrued liabilities on the Consolidated Balance Sheet) | |
| Short-term portion of lease liabilities (included in other accrued liabilities on the Consolidated Balance Sheet) | |
| Short-term portion of lease liabilities (included in other accrued liabilities on the Consolidated Balance Sheet) | |
| Long-term portion of lease liabilities | |
| Total | Total | $ | 42,086 | |
As of December 31, 2020,2023, the weighted-average remaining lease term is 7.66.1 years and the weighted-average incremental borrowing rate used to determine the operating lease liability was 6.1%5.6% for the Company’s operating leases.
TheDuring the year ended December 31, 2023, the Company does not have any financing leases.recorded impairment losses of $28.9 million related to its right-of-use assets. See Note 6 – Impairment of Goodwill, Indefinite-lived Intangible Assets and Other Long-lived Assets for more information.
Contractual Commitments
The following table sets forth theCompany’s non-cancelable material contractual commitments under manufacturing-related supplier arrangements as of December 31, 2020 (in thousands):
| | | | | | | | | | | | | | |
Party | | Total commitments | | Expiry date |
Brammer Bio MA - a Thermo Fisher Scientific Inc. subsidiary | | $ | 7,736 | | | December 2022 |
Lonza Netherlands, B.V. | | 13,771 | | | December 2022 |
Total contractual commitments | | $ | 21,507 | | | |
2023 related to Lonza Netherlands, B.V. amount to $5.2 million and expire in October 2024. The Company also had $1.0$0.6 million of license obligations related to its intellectual property as of December 31, 2020.2023.
Contingencies
SangamoThe Company is not party to any material pending legal proceeding. From time to time, Sangamothe Company is, and may bebecome, involved in legal proceedingslitigation and regulatory compliance matters incidental to the Company’s business, including employment and wage and hour claims, antitrust, tax, product liability, environmental, health and safety, commercial disputes, intellectual property, contracts and other matters arising out of the normal conduct of the Company’s business. Since litigation is inherently unpredictable and unfavorable resolutions can occur, assessing contingencies is highly subjective and requires judgments about future events. Sangamo regularly reviews and accrues for contingencies related to litigation and regulatory compliance matters, if it is probable that a liability has been incurred and the amount of the loss can be reasonably estimated. Based on current information, in the ordinary courseopinion of business.the Company, the ultimate resolution of these matters, individually or in aggregate, will not have a material adverse effect on the Company’s financial condition, results of operations or cash flows.
NOTE 89 – STOCKHOLDERS’ EQUITY
Preferred Stock
The Company’s Certificate of Incorporation authorizes the Company to issue up to 5,000,000 shares of preferred stock, which may be issued at the discretion of the Company’s Board of Directors. As of December 31, 2020, 02023, no shares of the Company’s preferred stock have been issued or are outstanding.
Common Stock
In June 2020, the Company’s stockholders approved an amendment to the Company’s Certificate of Incorporation to increase the total number of shares of the Company’s common stock authorized for issuance from 160,000,000 shares to 320,000,000 shares. Additionally, in June 2023, the Company’s stockholders approved an amendment to the Company’s Certificate of Incorporation to increase the total number of shares of the Company’s common stock authorized for issuance from 320,000,000 shares to 640,000,000 shares. As of December 31, 2020, 142,063,2032023, 178,133,548 shares of the Company’s common stock are outstanding.
In connection with the collaboration agreement with BIMA described in Note 4 of these Consolidated Financial Statements, the Company entered into a stock purchase agreement with BIMA, pursuant to which BIMA agreed to purchase the Biogen Shares at a price per share of $9.2137, for an aggregate purchase price of $225.0 million. The Company closed the sale of the Biogen Shares in April 2020.
In April 2019, the Company completed an underwritten public offering of its common stock, in which the Company sold an aggregate of 12.7 million shares of its common stock at a public offering price of $11.50 per share. The net proceeds to the Company from the sale of shares in this offering, after deducting underwriting discounts and commissions and other offering expenses, were approximately $136.3 million.
In April 2018, the Company completed an underwritten public offering of its common stock, in which the Company sold an aggregate of 14.2 million shares of its common stock at a public offering price of $16.25 per share. The net proceeds to the Company from the sale of shares in this offering, after deducting underwriting discounts and commissions and other offering expenses, were approximately $215.8 million.
At-the-Market Offering Agreement
In August 2020, the Company entered into an Open Market Sale Agreement℠ with Jefferies LLC (“Jefferies”) with respect to an at-the-market offering program under which the Company may offer and sell, from time to time at its sole discretion, shares of the Company’s common stock having an aggregate offering price of up to $150.0 million through Jefferies as the Company’s sales agent or principal. In December 2022, the Company entered into Amendment No. 2 to the Open Market Sale Agreement℠ which increased the aggregate offering price under the at-the-market offering program by an additional $175.0 million. Approximately $194.5 million remained available under the sales agreement as of December 31, 2023. The Company is not obligated to sell any shares under the sales agreement. As ofDuring the years ended December 31, 2020, 02023 and 2022, the Company sold 8,249,261 and 19,300,743 shares had been sold under the sales agreement.of its common stock, respectively, for net proceeds of approximately $15.1 million and $84.9 million, respectively.
2018 Equity Incentive Plan
In June 2018,May 2020, the Company’s stockholders approved an amendment and restatement of the Sangamo Therapeutics, Inc. 2018 Equity Incentive Plan (the “2018 Plan”). In connection with, to, among other things, increase the approvalaggregate number of shares of the Company’s common stock reserved for issuance under the 2018 Plan 0 additional equity awards will be granted under the previous 2013 Plan, however all outstanding equity awards under the 2013 Plan will continue to be subject to the terms and conditions as set forth in the agreements evidencing such awards and the terms of the 2013 Plan.
by 9,900,000 shares. In May 2020,2022, the Company’s stockholders approved an amendment and restatement of the 2018 Plan to, among other things, increase the aggregate number of shares of the Company’s common stock reserved for issuance under the 2018 Plan by 9,900,0007,900,000 shares. Additionally, in June 2023, the Company’s stockholders approved an amendment and restatement of the 2018 Plan to, among other things, increase the aggregate number of shares of the Company’s common stock reserved for issuance under the 2018 Plan by 10,000,000 shares.
The exercise price of a stock option granted under the 2018 Plan may not be less than 100% of the fair market value of the Company'sCompany’s common stock subject to the stock option on the date of grant, and the option term will not exceed 10ten years. If the person to whom the stock option is granted is a 10% stockholder of the Company, and the stock option granted qualifies as an incentive stock option, then the exercise price per share will not be less than 110% of the fair market value of the Company’s common stock on the date of grant, and the option term will not exceed five years. Generally, stock options granted under the 2018 Plan vest over three or four years at a rate of 25% on the one-year anniversary of the date of grant and 1/48 per month thereafter and expire 10 years after the date of grant, or earlier upon termination of employment or services to the Company.
The number of shares of common stock reserved for issuance under the 2018 Plan will be reduced: (i) on a 1-for-1 basis for each share of common stock subject to a stock option or stock appreciation right granted under the plan, (ii) by a fixed
ratio of 1.33 shares of common stock for each share of common stock issued pursuant to a full-value award granted under the plan.
Shares subject to any outstanding stock options or other awards under the 2018 Plan that expire or otherwise terminate prior to the issuance of the shares subject to those stock options or awards will be available for subsequent issuance under the 2018 Plan. Any unvested shares issued under the 2018 Plan that the Company subsequently purchases, pursuant to repurchase rights under the 2018 Plan, will be added back to the number of shares reserved for issuance under the 2018 Plan on a 1-for-1 basis or a 1.33-for-1 basis (depending on the ratio at which the share reserve was debited for the original award) and will accordingly be available for subsequent issuance in accordance with the terms of the 2018 Plan.
As of December 31, 2020,2023, there were 10,942,57613,738,867 shares of the Company’s common stock reserved for future awards under the Company’s 2018 Plan.
20102020 Employee Stock Purchase Plan
On June 2018,In May 2021, the Company’s stockholders approved an amendment and restatement of the Company’s 20102020 Employee Stock Purchase Plan (“the ESPP”). As amended, theThe ESPP provides for a total of 4.65.0 million shares of common stock reserved for issuance thereunder. Eligible employees may purchase common stock at 85% of the lesser of the fair market value of the Company’s common stock on the first day of the applicable two-year offering period or the last day of the applicable six-month purchase period. As of December 31, 2020,2023, there were 2,483,2182,858,653 shares of the Company’s common stock reserved for future issuance under the ESPP. The ESPP expired on April 30, 2020. The ongoing offering will continue through the end
Stock Option Activity
A summary of the Company’s stock option activity is as follows:
| | | | | | | | | | | | | | | | | | | | | | | |
| Number of
Shares | | Weighted-
Average
Exercise per
Share Price | | Weighted-Average
Remaining
Contractual Term | | Aggregate
Intrinsic
Value |
| | | | | (In years) | | (In thousands) |
| Options outstanding at December 31, 2019 | 9,829,287 | | | $ | 10.71 | | | | | |
| Options granted | 4,563,425 | | | $ | 8.09 | | | | | |
| Options exercised | (1,162,268) | | | $ | 8.02 | | | | | |
| Options canceled | (1,751,746) | | | $ | 10.19 | | | | | |
| Options outstanding at December 31, 2020 | 11,478,698 | | | $ | 10.02 | | | 7.80 | | $ | 69,616 | |
| Options vested and expected to vest at December 31, 2020 | 11,478,698 | | | $ | 10.02 | | | 7.80 | | $ | 69,616 | |
| Options exercisable at December 31, 2020 | 5,127,517 | | | $ | 10.71 | | | 6.48 | | $ | 29,025 | |
| | | | | | | | | | | | | | | | | | | | | | | |
| Number of
Shares | | Weighted-
Average
Exercise per
Share Price | | Weighted-Average
Remaining
Contractual Term | | Aggregate
Intrinsic
Value |
| | | | | (in years) | | (in thousands) |
| Options outstanding at December 31, 2022 | 13,174,995 | | | $ | 9.22 | | | | | |
| Options granted | 5,265,429 | | | $ | 2.38 | | | | | |
| Options exercised | — | | | $ | — | | | | | |
| Options canceled | (3,578,420) | | | $ | 8.58 | | | | | |
| Options outstanding at December 31, 2023 | 14,862,004 | | | $ | 6.95 | | | 5.57 | | $ | — | |
| | | | | | | |
| Options exercisable at December 31, 2023 | 8,618,752 | | | $ | 9.12 | | | 4.61 | | $ | — | |
The intrinsic value of options exercised was $5.4 million, $4.7 millionzero, zero and $27.0$2.8 million during 2020, 2019 and 2018, respectively.
Atthe years ended December 31, 2020, the aggregate intrinsic values of outstanding2023, 2022 and exercisable options were $69.6 million and $29.0 million, respectively. The aggregate intrinsic value of options vested and expected to vest as of December 31, 2020, 2019 and 2018 was $69.6 million, $7.5 million and $24.5 million,2021, respectively.
Restricted Stock Units
During 2020, 2019the years ended December 31, 2023, 2022 and 2018,2021, the Company awarded 2,517,101, 834,745,4,811,834, 4,349,795 and 346,0552,140,785 RSUs, respectively. The RSUs awarded in 2020, 2019the years ended December 31, 2023, 2022 and 20182021 had an average grant date fair value per award of $8.06, $9.49$2.29, $5.52 and $17.87,$11.16, respectively. These awards generally vest in a series of 3 successive equal annual installments.over three years. The aggregate fair value of RSUs vested during 2020, 2019the years ended December 31, 2023, 2022 and 20182021 was $3.7$17.8 million, $2.0$13.1 million and $0.6$9.0 million, respectively.
A summary of the Company’s RSU activity is as follows:
| | | | | | | | | | | | | | | | | |
| Number of
Shares | | Weighted-Average
Remaining
Contractual Term | | Aggregate Intrinsic
Value |
| | | (in years) | | (in thousands) |
| RSUs outstanding at December 31, 2022 | 5,243,898 | | | | | |
| RSUs awarded | 4,811,834 | | | | | |
| RSUs released | (2,532,354) | | | | | |
| RSUs forfeited | (1,470,455) | | | | | |
| RSUs outstanding at December 31, 2023 | 6,052,923 | | | 0.92 | | $ | 3,289 | |
| | | | | |
| | | | | | | | | | | | | | | | | |
| Number of
Shares | | Weighted-Average
Remaining
Contractual Term | | Aggregate Intrinsic
Value |
| | | (In years) | | (In thousands) |
| RSUs outstanding at December 31, 2019 | 862,850 | | | | | |
| RSUs awarded | 2,517,101 | | | | | |
| RSUs released | (324,305) | | | | | |
| RSUs forfeited | (384,118) | | | | | |
| RSUs outstanding at December 31, 2020 | 2,671,528 | | | 1.19 | | $ | 41,689 | |
| RSUs vested and expected to vest at December 31, 2020 | 2,671,528 | | | 1.19 | | $ | 41,689 | |
RSUs that vested in 2020, 2019the years ended December 31, 2023, 2022 and 20182021 were net-share settled such that the Company withheld shares with value equivalent to the employees’ minimum statutory obligation for the applicable income and other employment taxes and remitted the cash to the appropriate taxing authorities. The total shares withheld were approximately 90,617, 39,160,788,236, 380,917 and 20,193293,120 for 2020, 2019the years ended December 31, 2023, 2022 and 2018,2021, respectively, and were based on the value of the RSUs on their respective issuance dates as determined by the Company’s closing stock price. Total payments for the employees’ tax obligations to taxing authorities were $0.8$1.5 million, $0.4$2.1 million and $0.3$3.3 million in 2020, 2019the years ended December 31, 2023, 2022 and 2018,2021, respectively and are reflected as a financing activity within the accompanying Consolidated Statements of Cash Flows. These net-share settlements had the effect of share repurchases by the Company as they reduced and retired the number of shares that would have otherwise been issued as a result of the vesting and did not represent an expense to the Company.
NOTE 910 – STOCK-BASED COMPENSATION
The following table shows total stock-based compensation expense recognized in the accompanying Consolidated Statements of Operations (in thousands): | | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2023 | | 2022 | | 2021 |
| Research and development | $ | 15,215 | | | $ | 18,404 | | | $ | 19,534 | |
| General and administrative | 12,148 | | | 13,246 | | | 13,422 | |
| Total stock-based compensation expense | $ | 27,363 | | | $ | 31,650 | | | $ | 32,956 | |
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2020 | | 2019 | | 2018 |
| Research and development | $ | 13,523 | | | $ | 10,135 | | | $ | 8,249 | |
| General and administrative | 12,185 | | | 9,195 | | | 6,428 | |
| Total stock-based compensation expense | $ | 25,708 | | | $ | 19,330 | | | $ | 14,677 | |
As of December 31, 2020,2023, total stock-based compensation expense to be recognized in future periods related to unvested stock options was $35.5$13.3 million, which is expected to be expensed over a weighted-average period of 2.721.9 years. As of December 31, 2020,2023, total stock-based compensation expense to be recognized in future periods related to unvested RSUs was $17.1$14.4 million, which is expected to be expensed over a weighted-average period of 2.071.7 years. There was 0no capitalized stock-based employee compensation expense as of December 31, 2020, 20192023, 2022 or 2018.2021.
Valuation Assumptions
Employee stock-based compensation expense was determined using the Black-Scholes option valuation model for stock options and employee share purchases under the ESPP. Option valuation models require the input of subjective assumptions and these assumptions can vary over time. The fair value of RSUs was based on the closing price of the underlying common stock on the date of grant.
The Company bases its determination of expected volatility through its assessment of the historical volatility of its common stock. The Company relied on its historical exercise and post-vested termination activity for estimating its expected term for use in determining the fair value of these options.
The weighted-average estimated fair value per share of options granted during 2020, 2019the years ended December 31, 2023, 2022 and 20182021 was $5.25, $6.37,$1.53, $3.73 and $11.39,$7.34, respectively, based upon the assumptions used in the Black-Scholes valuation model. The assumptions used for estimating the fair value of the employee stock options were as follows:
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2020 | | 2019 | | 2018 |
| Risk-free interest rate | 0.34-0.61% | | 1.68-2.25% | | 2.53-2.96% |
| Expected term (in years) | 5.51-5.57 | | 5.50-5.62 | | 5.59-5.61 |
| Expected dividend yield of stock | 0 | | | 0 | | | 0 | |
| Expected volatility | 77.61-80.32% | | 76.46-78.39% | | 72.33-75.49% |
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2023 | | 2022 | | 2021 |
| Risk-free interest rate | 3.44-4.00% | | 2.15-3.69% | | 0.95-1.22% |
| Expected term (in years) | 5.47-5.56 | | 5.46-5.49 | | 5.46-5.52 |
| Expected dividend yield of stock | — | | | — | | | — | |
| Expected volatility | 72.18-76.05% | | 72.38-76.01% | | 77.30-79.77% |
Employees purchased 274,382, 249,3641,346,849, 576,950 and 328,710433,107 shares of common stock through the ESPP at an averagea weighted-average exercise price of $7.34, $8.53,$0.69, $3.07 and $4.51$7.78 per share during 2020, 2019the years ended December 31, 2023, 2022 and 2018,2021, respectively. The weighted-average estimated fair values of shares purchased under the Company’s ESPP during 2020, 2019the years ended December 31, 2023, 2022 and 20182021 were $8.02, $4.70$0.91, $1.85 and $7.07,$4.48, respectively, based upon the assumptions used in the Black-Scholes valuation model.
The assumptions used for estimating the fair value of the ESPP purchase rights are as follows:
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2020 | | 2019 | | 2018 |
| Risk-free interest rate | 1.53-2.80% | | 1.53-2.42% | | 2.16-2.80% |
| Expected term (in years) | 0.5-2.0 | | 0.5-2.0 | | 0.5-2.0 |
| Expected dividend yield of stock | 0 | | | 0 | | | 0 | |
| Expected volatility | 51.02-91.96% | | 51.02-91.96% | | 73.21-83.25% |
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2023 | | 2022 | | 2021 |
| Risk-free interest rate | 4.28-5.37% | | 1.62-4.61% | | 0.01-2.80% |
| Expected term (in years) | 0.5-2.0 | | 0.5-2.0 | | 0.0-2.0 |
| Expected dividend yield of stock | — | | | — | | | — | |
| Expected volatility | 65.71-100.60% | | 57.97-72.14% | | 32.54-97.88% |
NOTE 11—RESTRUCTURING CHARGES
April Restructuring
On April 26, 2023, the Company executed a restructuring of operations and a corresponding reduction in workforce (the “April Restructuring”), designed to reduce costs and increase focus on certain strategic priorities. The April Restructuring resulted in the elimination of approximately 110 roles, including 55 full-time employees and 55 contracted employees and eliminated open positions, in the United States, or approximately 23% of the total United States workforce as of April 26, 2023, and included one-time severance payments and other employee-related costs, including additional vesting of service-based stock compensation awards. As of December 31, 2023, the Company has estimated that it will incur $5.0 million in expenses related to employee severance and notice period payments, benefits and related restructuring charges for the April Restructuring, of which $3.8 million has been included in research and development expense and $1.2 million has been included in general and administrative expense in the accompanying Consolidated Statements of Operations. The Company expects the April Restructuring and the cash payments related to the April Restructuring to be substantially complete by the third quarter of 2024.
November Restructuring
On November 1, 2023, the Company executed a restructuring of operations and a corresponding reduction in workforce (the “November Restructuring”), designed to reduce costs and advance its strategic transformation into a neurology-focused genomic medicine company. The November Restructuring resulted in the elimination of approximately 162 roles, including 108 full-time employees and 54 contracted employees and eliminated open positions, in the United States, or approximately 40% of the total United States workforce, and included one-time severance payments and other employee-related costs, including additional vesting of service-based stock compensation awards. As of December 31, 2023, the Company has estimated it will incur approximately $8.7 million to $9.7 million in expenses related to employee severance and notice period payments, benefits, Brisbane facility close-out costs, and other related restructuring charges for the November Restructuring. The Company incurred $6.7 million of expenses in the year ended December 31, 2023, of which $5.0 million is included in research and development expense and $1.7 million is included in general and administrative expense in the accompanying Consolidated Statements of Operations. The Company expects to incur additional estimated costs of $2.0 million to $3.0 million through the second quarter of 2024. The Company expects the November Restructuring and the cash payments related to the November Restructuring to be substantially complete by the second quarter of 2024.
France Restructuring
In November 2023, the Company initiated an information and consultation procedure with the Works Council for its Valbonne, France workforce regarding a planned wind-down of Sangamo’s French operations and a corresponding reduction in workforce, including planned closure of the Company’s cell therapy manufacturing facility and research labs in Valbonne, France. The information and consultation procedure with the Works Council resulted in the definition of an acceptable set of termination provisions including payouts to departing employees and were a required step before the Company could eliminate positions at Sangamo France. The information and consultation procedure of the Works Council was completed in the first quarter of 2024 and the Board of Directors approved the wind-down and reduction in workforce (the “France Restructuring”) on March 1, 2024.
The France Restructuring is estimated to eliminate all 93 roles in France, or approximately 24% of the total global workforce as of March 1, 2024. The Company is expected to make severance payments as required by French law and the terms of the applicable collective bargaining agreements, and other employee-related costs. The Company concluded that payouts under the France Restructuring are a result of an ongoing post-employment benefit plan, and that payments were probable and could be estimated as of December 31, 2023. The Company estimates it will incur approximately $7.8 million to $11.5 million in expenses related to employee severance and notice period payments, benefits, contract termination costs, and other related restructuring charges for the France Restructuring. The Company incurred $4.7 million of expenses during the year ended December 31, 2023, of which $3.6 million is included in research and development expense and $1.1 million is included in general and administrative expense in the accompanying Consolidated Statements of Operations. The Company expects to incur additional estimated costs of $3.1 million to $6.8 million through the fourth quarter of 2024. The Company expects the France Restructuring and its related cash payments to be completed no later than the fourth quarter of 2024.
The following table is a summary of accrued April, November, and France Restructuring charges included within other accrued liabilities on the Company’s Consolidated Balance Sheet as of December 31, 2023 (in thousands):
| | | | | |
| Year Ended December 31, 2023 |
| Balance at December 31, 2022 | $ | — | |
| Restructuring charges | 16,364 | |
| Cash payments | (4,282) | |
| Non-cash adjustments | (349) | |
| Balance at December 31, 2023 | $ | 11,733 | |
Sangamo may also incur other cash expenses or charges not currently contemplated or estimable due to events that may occur as a result of, or associated with, the April, November and France Restructurings.
NOTE 1012 – EMPLOYEE BENEFIT PLAN
The Company sponsors a defined-contribution savings plan under Section 401(k) of the Internal Revenue Code covering all full-time employees (“Sangamo 401(k) Plan”). The Sangamo 401(k) Plan is intended to qualify under Section 401 of the Internal Revenue Code.
The Company matched employee contributions equal to 50% for the first 8%100% in 2020, 20192023, 2022, and 2018,2021, up to a limit of $5,000 in 2023 and $4,000 in 2020, 20192022 and 2018.2021. Matching funds are fully vested when contributed. Contributions to the Sangamo 401(k) Plan by
the Company were $1.2$1.8 million, $0.9$1.5 million and $0.8$1.5 million for the years ended December 31, 2020, 20192023, 2022 and 2018,2021, respectively.
NOTE 1113 – INCOME TAXES
The domestic and foreign components of loss before income taxes were as follows (in thousands): | | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2023 | | 2022 | | 2021 |
| Domestic | $ | (173,375) | | | $ | (216,573) | | | $ | (185,216) | |
| Foreign | (89,528) | | | 24,724 | | | 7,225 | |
| Loss before income taxes | $ | (262,903) | | | $ | (191,849) | | | $ | (177,991) | |
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2020 | | 2019 | | 2018 |
| Domestic | $ | (126,624) | | | $ | (77,354) | | | $ | (65,695) | |
| Foreign | 5,847 | | | (18,065) | | | (3,194) | |
| Loss before income taxes | $ | (120,777) | | | $ | (95,419) | | | $ | (68,889) | |
TheIncome tax benefit for the year 2023 and the income tax expense
for the years 2022 and 2021 consisted of the following (in thousands):
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2023 | | 2022 | | 2021 |
| Income tax expense: | | | | | |
| Current: | | | | | |
| Federal | $ | — | | | $ | — | | | $ | — | |
| State | — | | | — | | | — | |
| Foreign | 186 | | | 500 | | | 886 | |
| Subtotal | 186 | | | 500 | | | 886 | |
| Deferred: | | | | | |
| Federal | — | | | — | | | — | |
| State | — | | | — | | | — | |
| Foreign | (5,258) | | | (71) | | | (580) | |
| Subtotal | (5,258) | | | (71) | | | (580) | |
| Income tax expense | $ | (5,072) | | | $ | 429 | | | $ | 306 | |
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2020 | | 2019 | | 2018 |
| Income tax expense: | | | | | |
| Current: | | | | | |
| Federal | $ | 0 | | | $ | 0 | | | $ | 0 | |
| State | 133 | | | 0 | | | 0 | |
| Foreign | 686 | | | 0 | | | 0 | |
| Subtotal | 819 | | | 0 | | | 0 | |
| Deferred: | | | | | |
| Federal | 0 | | | 0 | | | 0 | |
| State | 0 | | | 0 | | | 0 | |
| Foreign | (474) | | | 0 | | | 0 | |
| Subtotal | (474) | | | 0 | | | 0 | |
| Income tax expense | $ | 345 | | | $ | 0 | | | $ | 0 | |
The difference between the income tax benefit for the year 2023 and the income tax expense for the years 2022 and 2021 and the amount computed by applying the federal statutory income tax rate to loss before income taxes is explained as follows (in thousands):
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2020 | | 2019 | | 2018 |
| Tax at federal statutory rate | $ | (25,363) | | | $ | (20,038) | | | $ | (14,467) | |
| State taxes, net | (3,168) | | | (9,597) | | | (2,849) | |
| Foreign rate differential | 376 | | | (665) | | | (177) | |
| Global Intangible Low-Taxed Income | 1,335 | | | 0 | | | 0 | |
| Non-deductible stock-based compensation | 4,232 | | | 2,817 | | | (2,729) | |
| Research credits | (3,657) | | | (3,429) | | | (1,005) | |
| Change in valuation allowance | 26,537 | | | 29,655 | | | 20,271 | |
| Other | 53 | | | 1,257 | | | 956 | |
| Income tax expense | $ | 345 | | | $ | 0 | | | $ | 0 | |
In March and December 2020, in response to the COVID-19 pandemic, the CARES Act and the Consolidated Appropriations Act, 2021 were passed into law and provide additional economic stimulus to address the impact of the COVID-19 pandemic. The Company does not expect any significant benefit to its income tax provision as a result of this legislation. | | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2023 | | 2022 | | 2021 |
| Tax at federal statutory rate | $ | (55,210) | | | $ | (40,288) | | | $ | (37,372) | |
| State taxes, net | (1,372) | | | (6,895) | | | (6,734) | |
| Foreign rate differential | (4,273) | | | 309 | | | 362 | |
| Global Intangible Low-taxed Income | 791 | | | 1,002 | | | 637 | |
| Non-deductible stock-based compensation | 4,770 | | | 3,545 | | | 2,770 | |
| Research credits | (7,020) | | | (6,694) | | | (5,230) | |
| Change in valuation allowance | 49,016 | | | 44,005 | | | 45,373 | |
| Transfer pricing settlement | — | | | 4,343 | | | — | |
| Goodwill impairment | 9,764 | | | — | | | — | |
| Other | (1,538) | | | 1,102 | | | 500 | |
| Income tax expense | $ | (5,072) | | | $ | 429 | | | $ | 306 | |
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of the Company’s deferred tax assets and liabilities are as follows (in thousands):
| | December 31, |
| 2020 | | 2019 |
| December 31, | | | December 31, |
| 2023 | | | 2023 | | 2022 |
| Assets: | Assets: | | | |
| Deferred tax assets: | Deferred tax assets: | |
| Deferred tax assets: | |
| Deferred tax assets: | |
| Net operating loss carryforwards | |
| Net operating loss carryforwards | |
| Net operating loss carryforwards | Net operating loss carryforwards | $ | 164,276 | | | $ | 133,765 | |
| Research and development tax credit carryforwards | Research and development tax credit carryforwards | 27,679 | | | 21,459 | |
| Stock-based compensation | Stock-based compensation | 5,321 | | | 4,194 | |
| Deferred revenue | Deferred revenue | 20,681 | | | 32,171 | |
| Capitalized research | |
| Fixed assets | Fixed assets | 10,525 | | | 11,282 | |
| Intangible assets | |
| Lease liability | Lease liability | 10,251 | | | 11,722 | |
| Accruals and reserves | Accruals and reserves | 430 | | | 675 | |
| Other | Other | 308 | | | 151 | |
| Total deferred tax asset | Total deferred tax asset | 239,471 | | | 215,419 | |
| Valuation allowance | Valuation allowance | 214,351 | | | 187,724 | |
| Deferred tax assets | Deferred tax assets | 25,120 | | | 27,695 | |
| Liabilities: | Liabilities: | | | |
| Intangible assets | Intangible assets | (14,321) | | | (13,609) | |
| Intangible assets | |
| Intangible assets | |
| Operating lease right-of-use assets | Operating lease right-of-use assets | (17,510) | | | (20,656) | |
| Deferred tax liabilities | Deferred tax liabilities | (31,831) | | | (34,265) | |
| Total net deferred tax liabilities | Total net deferred tax liabilities | $ | (6,711) | | | $ | (6,570) | |
A valuation allowance is recorded when it is more likely than not that all or some portion of the deferred income tax assets will not be realized. The Company regularly assesses the need for a valuation allowance against its deferred income tax assets by considering both positive and negative evidence related to whether it is more likely than not that the Company’s deferred income tax assets will be realized. In evaluating the Company’s ability to recover its deferred income tax assets within the jurisdiction from which they arise, the Company considers all available positive and negative evidence, including scheduled reversals of deferred income tax liabilities, projected future taxable income, tax-planning strategies, and results of recent operations. Accordingly, based upon the Company’s analysis of these factors theThe Company continues to maintain a full valuation allowance on its U.S. federal and state net deferred tax assets have been substantially offset byand on the Sangamo France net deferred tax assets, as the Company believes it is not more likely than not that these benefits will be realized. The Company recorded a full valuation allowance.allowance on the U.K. net deferred tax assets in 2023 due to the uncertainty regarding future income projections for Sangamo U.K., which outweighed positive evidence related to realization of the deferred tax assets for Sangamo U.K. The valuation allowance increased by $26.6$49.6 million, $29.6$42.0 million and $45.3$45.5 million for the years ended December 31, 2020, 20192023, 2022 and 2018,2021, respectively.
to expire in 2024 and will keep expiring through 2037, if not utilized. Federal net operating loss generated infrom 2018 will carry forward indefinitely. If not utilized, the state net operating loss carryforwards will begin to expire in 2029, respectively. The Company’s French net operating loss carryforward balance is $145.9$118.7 million, which carries over indefinitely. The Company
The Company’s policy is to reinvest the earnings of its non-U.S. subsidiaries in those operations. The Company does not provide for U.S. taxes on the earnings of foreign subsidiaries because the Company intends to reinvest such earnings offshore indefinitely. However, if these funds were repatriated, the Company would be required to accrue and pay applicable U.S. taxes and withholding taxes. Due to the cumulative losses generated in foreign countries there are no earnings to repatriate.
The Company files federal and state income tax returns with varying statutes of limitations. The tax years from 20022003 forward remain open to examination due to the carryover of net operating losses or tax credits. The Company also files United Kingdomthe U.K. and French income tax returns, and the tax years from 2008 and thereafter remain open in the United KingdomU.K., and 2016the tax years 2019 and thereafter in France are still subject to examination.
The Company’s practice is to recognize interest and/or penalties related to income tax matters in income tax expense. AsThe Company had $0.3 million and $0.2 million accrued interest and/or penalties as of December 31, 2020,2023 and 2022, respectively. Unrecognized tax benefits are not expected to change materially over the Company had 0 accrued interest and/or penalties.next 12 months. The amount of unrecognized tax benefits may change during the next year for items that, arise in the ordinary course of business. In the event that any unrecognized tax benefits areif recognized, would impact the effective tax rate will not be affected.is $1.2 million, $1.2 million and $1.2 million as of December 31, 2023, 2022 and 2021, respectively.
ITEM 9 – CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
We maintain disclosure controls and procedures that are designed to provide reasonable assurance that information required to be disclosed in our Exchange Act reports is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms and that such information is accumulated and communicated to our management, including our principal executive officer and principal financial officer, as appropriate, to allow timely decisions regarding required disclosure.
Our management is responsible for establishing and maintaining an adequate internal control over financial reporting (as such term is defined in Rules 13a-15(f) and 15d-15(f) of the Exchange Act) for our company. Our management, including our principal executive officer and acting principal financial officer, conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework set forth in the “Internal Control—Integrated Framework” issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework). Based on an evaluation under that framework, our management concluded that our internal control over financial reporting was effective at the reasonable assurance level as of December 31, 2020.2023.
The effectiveness of our internal control over financial reporting as of December 31, 20202023 has been audited by Ernst & Young LLP, an independent registered public accounting firm, as stated in their report, which is included herein.
There have been no changes in our internal control over financial reporting identified in connection with the evaluation required by Rules 13a-15(d) and 15d-15(d) of the Exchange Act that occurred during the three months ended December 31, 20202023 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
To the Stockholders and the Board of Directors of Sangamo Therapeutics, Inc.
We have audited Sangamo Therapeutics, Inc.’s internal control over financial reporting as of December 31, 2020,2023, based on criteria established in Internal Control – Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the COSO criteria). In our opinion, Sangamo Therapeutics, Inc. (the Company) maintained, in all material respects, effective internal control over financial reporting as of December 31, 2020,2023, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the 20202023 consolidated financial statements of the Company and our report dated February 24, 2021March 13, 2024 expressed an unqualified opinion thereon.
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects.
Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
None.
ITEM 12 – SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information required by this item with respect to equity compensation plans is to be included in our 20212024 Proxy Statement under the section entitled “Equity Compensation Plan Information” and the information required by this item with respect to security ownership of certain beneficial owners and management is to be included in our 20212024 Proxy Statement under the section entitled “Security Ownership of Certain Beneficial Owners and Management” and in each case is incorporated herein by reference, provided that if the 20212024 Proxy Statement is not filed within 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K, the omitted information will be included in an amendment to this Annual Report on Form 10-K filed not later than the end of such 120-day period.
ITEM 13 – CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
(a) The following documents are included as part of this Annual Report on Form 10-K:
1. Financial Statements—See Index to Consolidated Financial Statements in Item 8.
2. Financial Statement Schedules—Not Applicable.
3. Exhibits
(+) Indicates management contract or compensatory plan or arrangement.
None.
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Alexander D. Macrae and Gary Loeb,Scott Willoughby, and each of them, as his or her true and lawful attorneys-in-fact and agents, each with the full power of substitution, for him or her and in his or her name, place or stead, in any and all capacities, to sign any and all amendments (including post-effective amendments) to this Annual Report on Form 10-K, and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done in and about the premises, as fully to all intents and purposes as he or she might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents, or their, his or her substitute or substitutes, may lawfully do or cause to be done by virtue hereof.