UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
_________________________
FORM 10-K
_________________________
(Mark One)
| | | | | |
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 20212023
OR
| | | | | |
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission file number: 001-39634
_________________________
Foghorn Therapeutics Inc.
(Exact name of registrant as specified in its charter)
_________________________
| | | | | | | | |
| Delaware | | 47-5271393 |
(State or other jurisdiction of
incorporation or organization) | | (I.R.S. Employer
Identification Number) |
| |
500 Technology Square, Ste 700 Cambridge, Massachusetts | | 02139 |
| (Address of principal executive offices) | | (Zip Code) |
Registrant’s telephone number, including area code: 617-586-3100
Securities registered pursuant to Section 12(b) of the Act:
| | | | | | | | | | | | | | |
| Title of each class | | Trading
| | Name of each exchange on which registered |
| Common Stock, $0.0001 Par Value | | FHTX | | The Nasdaq Global Market |
Securities registered pursuant to Section 12(g) of the Act:
None
_________________________
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes x No ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| | | | | | | | | | | | | | |
| Large accelerated filer | ¨ | | Accelerated filer | ¨ |
| | | | |
| Non-accelerated filer | x | | Smaller reporting company | x |
| | | | |
| | | Emerging growth company | x |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ¨
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ¨
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No x
The aggregate market value of common stock held by non-affiliates of the registrant computed by reference to the price of the registrant’s common stock as of June 30, 2021,2023, the last business day of the registrant's most recently completed second fiscal quarter, was approximately (based on the last reported sale price on the NASDAQ Global Market as of such date) $208.8$199.7 million.
As of February 25, 202229, 2024 there were 41,421,96442,565,904 shares of the registrant’s common stock, par value $0.0001 per share, outstanding.
Foghorn Therapeutics Inc.
Index
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains forward-looking statements that are based on management’s beliefs and assumptions and on information currently available to management. All statements other than statements of historical facts contained in this Annual Report on Form 10-K are forward-looking statements. In some cases, you can identify forward-looking statements by terms such as “may,” “will,” “should,” “expect,” “plan,” “anticipate,” “could,” “intend,” “target,” “project,” “contemplate,” “believe,” “estimate,” “predict,” “potential” or “continue” or the negative of these terms or other similar expressions, although not all forward-looking statements contain these words. Forward-looking statements include, but are not limited to, statements concerning:
•the initiation, timing, progress, enrollment, results, and timing of results and regulatory filings of our research and development programs and our preclinical and clinical studies;studies, including potential combination trials involving FHD-286 and our collaboration with Loxo Oncology at Eli Lilly and Company (“Lilly”);
•our ability to advance any product candidates that we may develop and to successfully complete preclinical and clinical studies;
•our ability to leverage our initial programs to develop additional product candidates using our Gene Traffic Control® platform;
•the impact of the COVID-19 pandemic on our and our collaborators’ business operations, including our research and development programs and preclinical and clinical studies, as well as recent geopolitical instability and other developments that may negatively impact the ability to utilize contract development and manufacturing organizations, or CDMOs, and contract research organizations, or CROs, that are located outside of the United States;
•developments related to our competitors and our industry;
•our ability to expand the target populations of our programs and the availability of patients for clinical testing;
•our ability to obtain regulatory approval for FHD-286 FHD-609 and any future product candidates from the U.S. Food and Drug Administration or FDA,(the “FDA”) and other regulatory authorities;
•our ability to identify and enter into future license agreements and collaborations;
•our ability to continue to rely on our CDMOscontract development and CROsmanufacturing organizations (“CDMOs”) or contract research organizations (“CROs”), including those located outside the United States, such as those located in China, for our manufacturing and research needs;
•regulatory developments in the United States and foreign countries;
•general economic conditions, including recessionary conditions, interest rates, monetary fluctuations and supply chain constraints;
•geopolitical instability and armed conflict, including those in Ukraine, Gaza, and the Red Sea;
•our ability to attract and retain key scientific and management personnel; and
•the scope of protection we are able to establish, maintain and enforce for intellectual property rights covering FHD-286, FHD-609, our future productsproduct candidates, and our Gene Traffic Control® platform.
The forward-looking statements in this Annual Report on Form 10-K are only predictions and are based largely on our current expectations and projections about future events and financial trends that we believe may affect our business, financial condition and results of operations. These forward-looking statements speak only as of the date of this Annual Report on Form 10-K and are subject to a number of known and unknown risks, uncertainties and assumptions, including those described under the section entitled “Item 1A. Risk Factors” in this Annual Report on Form 10-K. Because forward-looking statements are inherently subject to risks and uncertainties, some of which cannot be predicted or quantified and some of which are beyond our control, you should not rely on these forward-looking statements as predictions of future events. Moreover, we operate in an evolving environment. New risks and uncertainties may emerge from time to time, and it is not possible for management to predict all risks and uncertainties. Except as required by applicable law, we do not plan to publicly update or revise any forward-looking statements contained herein, whether as a result of any new information, future events, changed circumstances or otherwise.
SUMMARY OF RISK FACTORS
Below is a summary of the principal factors that make an investment in our common stock speculative or risky. This summary does not address all of the risks that we face. Additional discussion of the risks summarized in this risk factor summary and other risks that we face can be found below under the heading “Item 1A. Risk Factors” and should be carefully considered, together with other information in this Annual Report on Form 10-K and our other filings with the SEC, before making an investment decision regarding our common stock.
•We have a limited operating history and have no products approved for commercial sale, which may make it difficult for you to evaluate our current business and predict our future success and viability.
•We have incurred significant losses since inception. We expect to incur losses for the foreseeable future and may never achieve or maintain profitability.
•We will need substantial additional funding. If we are unable to raise capital when needed, we could be forced to delay, reduce or eliminate our research and product development programs or future commercialization efforts.
•We are heavily dependent on the success of our product candidates, which are in preclinical and Phase 1 clinical development. We may not be successful in our efforts to identify and develop potential product candidates. If these efforts are unsuccessful, or if we experience significant delays, we may never become a commercial stage company or generate any revenues, and our business could be materially harmed.
•Our clinical trials may fail to demonstrate substantial evidence of the safety and efficacy of our product candidates, which would delay or prevent regulatory approval of the product candidates, limit their commercial potential or result in significant negative consequences following any potential marketing approval.
•We or our collaboration partner may not be able to file Investigational New Drug Applications or INDs,(“INDs”) or IND amendments to commence clinical trials of our product candidates on the timelines we expect, and even if we or they are able to, the FDA may not permit us to proceed..
•There is substantial competition inproceed. For our field, whichpartnered programs, we may result in others developing or commercializing products before we do.not be able to exert unilateral control over the development of such product candidates.
•Our lead product candidates utilizecandidate utilizes a novel mechanismsmechanism of action, which may result in greater research and development expenses, regulatory issues that could delay or prevent approval, or discovery of unknown or unanticipated adverse effects.
•There is substantial competition in our field, which may result in others developing or commercializing products before we do.
•We are highly dependent on our key personnel. If we are not successful in attracting and retaining highly qualified personnel, we may not be able to successfully implement our business strategy.
•If we are unable to adequately protect our proprietary technology and platform or obtain and maintain patent protection for our technology and products or if the scope of the patent protection obtained is not sufficiently broad, our competitors could develop and commercialize technology and products similar or identical to ours, and our ability to successfully develop and commercialize our technology and products may be impaired.
•The continuing outbreak of COVID-19 in the United StatesUnfavorable global macroeconomic conditions, geopolitical trends, and armed conflict, together with legislative and administrative actions meant to address these and other countries mayconditions, could adversely affect our business, and the market pricefinancial condition or results of our common stock.
•Our clinical trials may fail to demonstrate substantial evidence of the safety and efficacy of our product candidates, which would delay or prevent regulatory approval of the product candidates, limit their commercial potential or result in significant negative consequences following any potential marketing approval.
PART I
Unless the context otherwise requires, the terms “Foghorn,” “Foghorn Therapeutics,” the “Company,” “we,” “us” and “our” relate to Foghorn Therapeutics Inc., together with its consolidated subsidiary.
ITEM 1. BUSINESS
Overview
Foghorn is a clinical stage, precision therapeutics biotechnology company pioneering a new class of medicines that treat serious diseases by correcting abnormal gene expression through selectively targeting the chromatin regulatory system, an untapped opportunity for therapeutic intervention in oncology and with potential in a wide spectrum of other diseases including virology, autoimmune diseases and neurology.
The chromatin regulatory system orchestrates gene expression—the turning on and off of genes—which is fundamental to how all our cells function. BreakdownsThe chromatin regulatory system is implicated in this system lead to a wide rangeapproximately 50 percent of diseases impacting millions of patients. Understanding the mechanism ofall cancers, and understanding how this system works could lead to an entirely new class of therapeutics.precision medicines. To our knowledge, we are the only company with the ability to study and target the chromatin regulatory system at scale, in context, and in an integrated way.
We are a clinical stage biotechnology company pioneering a new class of medicines that modulate gene expression through selectively targeting the chromatin regulatory system an untapped opportunity for therapeutic intervention in oncology and a wide spectrum of other diseases. Our proprietary Gene Traffic Control® platform gives usprovides an integrated and mechanistic understanding of how the various components of the chromatin regulatory system interact, allowing us to identify, validate and potentially drug targets within thethis system. BreakdownsWe have developed unique capabilities that have yielded new insights and scalability in the chromatin regulatory system are associated with over 50 percent of all cancers. Addressing these breakdowns could potentially provide therapies for over 2.5 million patients with cancer. Consequently,drugging this new, previously untapped and promising area.
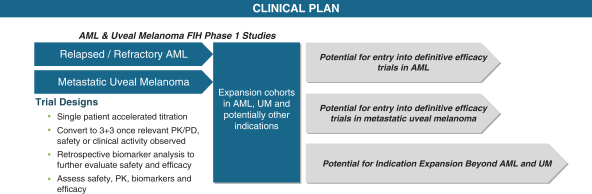
At present, we are initially focusedworking on more than 10 programs with one clinical-stage drug candidate currently in oncology.Phase 1 development and one drug candidate anticipated to begin clinical development this year. We have discovered highly selective chemical matter for some of the most challenging targets in oncology including BRM, CBP, EP300 and ARID1B, as well as other undisclosed targets. We believe our current pipeline has the potential to help more than 500,000 cancer patients. We take a small molecule modality agnostic approach to drugging targets which includes protein degraders, allosteric enzymatic inhibitors, and transcription factor disruptors. We are developinga biology first company which means we focus first on the underlying genetics and biology of a disease relevant target and then leverage the most appropriate drugging approach to impact the disease biology.
We are currently conducting a Phase 1 dose escalation study of FHD-286, a selective, allosteric ATPase inhibitor of BRM and are currently enrolling two separate Phase 1 studiesBRG1, in (i) metastatic uveal melanoma and (ii)combination with either decitabine or cytarabine in relapsed and/or refractory acute myeloid leukemia or AML,(“AML”) patients. As part of our collaboration with Loxo Oncology at Eli Lilly and myelodysplastic syndrome, or MDS. We are developing FHD-609, a targeted protein degrader, and are currently enrollingCompany (“Lilly”), we anticipate that Lilly will begin a Phase 1 dose escalation study in synovial sarcoma. with FHD-909, a selective ATPase inhibitor of BRM, later this year.
We believe Foghorn has the potential to be a major biopharmaceutical company with our current pipeline addressing more than 20 tumor types impacting more than 500,000 new patients annually. We believe that we have the potential to file six new Investigational New Drug Applications (“INDs”) over the next four years.
Our visioncurrent pipeline of product candidates and discovery programs is focused on oncology and is shown below:
Foghorn’s science and potential have been validated by strategic collaborations with world-leading pharmaceutical companies, including Lilly. In December 2021, we entered into a strategic collaboration agreement with Lilly (the “Lilly Collaboration Agreement”).Under the terms of the Lilly Collaboration Agreement, we are leveraging our platform technology to use our Gene Traffic Control platform toresearch, discover and develop drugstherapeutic molecules directed to the selective BRM target and an additional undisclosed oncology target, and up to three additional discovery programs. In February 2024, we announced that FHD-909, a selective BRM inhibitor, has been selected by Lilly to advance to the clinic, and we anticipate an IND filing by Lilly in oncologythe second quarter of 2024.
We believe this strategic collaboration confirms the rigor of our science, highlights the importance of the targets we are tackling and other therapeutic areas, including virology, autoimmune disease and neurology.underscores the relevance of the biology on which we are focused.
How the Chromatin RegulatoryRegulatory System Orchestrates Gene Expression
In order for DNA to fit in the nucleus of each human cell, DNA is densely packed into what is called chromatin, which needs to be unpacked as a necessary first step to allow for gene expression. Cells have evolved a system known as the chromatin regulatory system that can locate and unpack particular regions of chromatin, thereby enabling and orchestrating gene expression. Two of theThe major components of the chromatin regulatory system are chromatin remodeling complexes, and transcription factors, helicases and these componentsother chromatin related factors which work in concert to orchestrate gene expression. One important role for this system is to control the accessibility of chromatin which in turn determines if other factors necessary for gene expression can access the genetic material. In addition, the system controls the structure, modification, and repair of chromatin which are all necessary for proper control of gene expression. Because of the central role this system plays in orchestrating normal gene expression, aberrations in the system may result in disease. We believe our platform is uniquely suited to address these aberrations and treat these diseases.
Our Gene Traffic Control Platform
We have built ourOur proprietary Gene Traffic Control platform to givegives us an integrated and mechanistic understanding of how the various components of the chromatin regulatory system interact, allowing us to identify, validate and potentially drug targets within the system. We are initially using our Gene Traffic Control platform in oncology. In cancer, the mutations that are in or impinge on the chromatin regulatory system create genetically determined dependencies, on which the cancer cells rely for survival. These genetic dependencies result in diseased cell vulnerabilities, creating potential opportunities to selectively drug and kill diseased cells while minimizing impact to healthy cells. With ourOur platform we are ableenables us to produce components of the chromatin regulatory system at scale, thereby allowing us to identify these genetic dependencies, understand their mechanism and target their vulnerabilities. We combine our genomic and epi-genomic tools, our proprietary high throughput screening technology and our expertise in medicinal chemistry to develop enzymatic inhibitors, protein degraders and transcription factor disruptors that target the chromatin regulatory system. While initially focused in oncology, we believe our platform is broadly applicable across other disease areas.
Our Gene Traffic Control platform encompasses the following:
•Target Identification and Validation—We use genomic screens, and a suite of epi-genome sequencing and computational tools, including aspects of artificial intelligence or AI, and machine learning, to characterize, identify, and validate targets within the chromatin regulatory system. Our epi-genome sequencing tools allow us to understand
the mechanisms of how our drugs are modifying the chromatin structure. Our platform allows for the identification of genetically determined dependencies associated with the chromatin regulatory system.
•Production of Chromatin Regulatory System Components at Scale and Proprietary Assays—We have built unique capabilities to purify and synthesize chromatin remodeling complexes, transcription factors, helicases, and transcriptionother chromatin related factors. These capabilities allow us to study the chromatin regulatory system at scale and in a context that, to our knowledge, is unavailable to others, and yields unique insights that are critical to systematically drugging this system.
•Discovery and Optimization of Chemical MatterMatter——We perform proprietary high throughput screens that leverage our ability to produce the chromatin regulatory system components at scale. For example, we are able to screen for inhibitors of chromatin remodeling complexregulatory system component activity, for binders that we can turn into protein degraders, and for disruptors of transcription factor-chromatin remodeling complex interactions. Once we identify hits from our screens, we use our unique suite of assays involving the relevant component of the chromatin regulatory system to characterize, validate, and optimize our chemical matter.
•Targeted Protein DegradationDegradation——In cases where our drugging efforts are directed at targets that have no enzymatic activity, we seek to degrade the protein of interest. We have built extensive targeted protein degrader capabilities encompassing linkers and E3 ligase binders,proprietary chemistry, high-throughput cellular screening capabilities, mechanistic assays to measure proteintriage and rank compounds against multiple parameters including kinetics of degradation, and guide optimization, and ternary complex formation understanding through both biophysical structural determination and computational modeling. After completing screensWe develop both heterobifunctional degraders and finding small molecule bindersnon-cereblon based molecular glues that serve to bridge an interaction between an E3 ligase and target protein of interest. This induced proximity results in driving the target protein of interest for degradation via the ubiquitin-proteasome pathway. A demonstrated strength of our platform is leveraging degradation to enable selectivity which we usehave done now for several programs including BRM, CBP, and EP300. We have developed capabilities with long-acting formulation of our protein degradation know-howdegraders which we believe has the potential to convert binders into selective protein degraders.enable enhanced convenience and route of administration.
•Translation to Clinic and Identification of Biomarkers—Early in the drug discovery process, we use various genome and epi-genome analyses to understand the mechanism of the genetic dependency of the disease on the chromatin regulatory system. Our understanding of the mechanism of the dependency enables us to identify biomarkers for patient identification and treatment. We seek to enrich our clinical studies with the genetically relevant patient populations that are most likely to benefit from treatment.
Using our proprietary Gene Traffic Control platform, we are developing a broad pipeline of product candidates that target genetically determined dependencies within the chromatin regulatory system. Our current pipeline of product candidates and discovery programs is focused on oncology and is shown below, along with anticipated milestones.
Within the chromatin regulatory system, we have initially focused our development efforts on the BAF chromatin remodeling complex, or the BAF complex, the most mutated among a family of chromatin remodeling complexes, and its interactions with transcription factors. Our precision approach consists of designing novel small molecules to inhibit the ATPase activity of BAF complexes, to selectively degrade mutated or dependent subunits, or to disrupt the interaction between the BAF complex and associated transcription factors. We believe our platform is broadly applicable to other chromatin remodeling complexes and transcription factors.
Our first product candidate, FHD-286, is a highly potent, selective, allosteric and orally available, small-molecule, enzymatic inhibitor of BRG1 and BRM, that we are initially developing in two phase one studies for the potential treatment of (i) metastatic uveal melanoma and (ii) relapsed and/or refractory AML and MDS and. BRG1 and BRM are two highly similar proteins that are the ATPases, or the catalytic engines, across all forms of BAF. In our preclinical studies, we have observed in both AML and uveal melanoma animal xenograft models anti-tumor effects at tolerated doses. As FHD-286 progresses through clinical testing, our intention is to expand into other indications beyond uveal melanoma, AML and MDS.
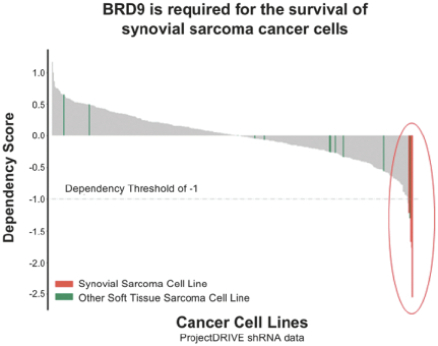
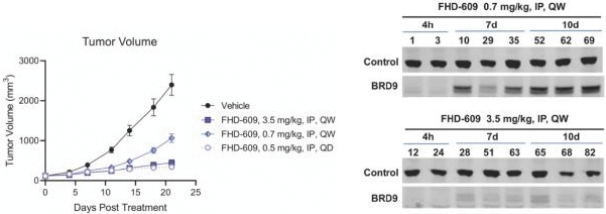
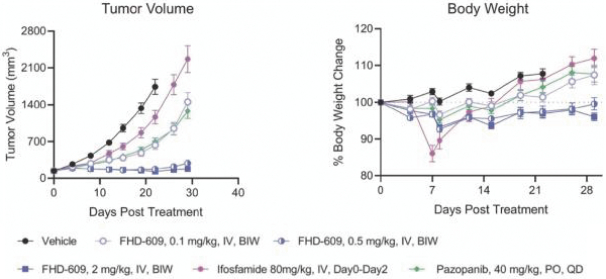
Our second product candidate, FHD-609, is a highly potent, selective and intravenous, small molecule protein degrader of BRD9, a component of a form of the BAF complex that we are developing in a Phase 1 clinical study in synovial sarcoma. Nearly all synovial sarcoma cancers contain a translocation, a type of mutation, between a BAF subunit gene, SS18, and another set of genes, SSX1, SSX2 and SSX4. These mutations render the cancer genetically dependent upon BRD9. FHD-609 has two domains: one that binds with high potency and selectivity to BRD9 and the other that binds to a receptor on the E3 ligase complex that directs proteins for destruction. In our preclinical studies in synovial sarcoma animal xenograft models, we have observed anti-tumor effects at tolerated doses. As FHD-609 progresses through clinical testing, our intention is to expand into other indications beyond synovial sarcoma.
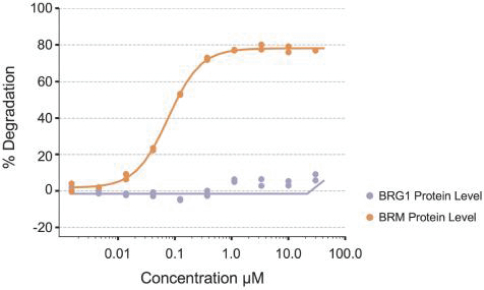
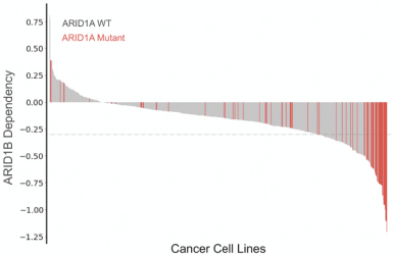
We have used our Gene Traffic Control platform to generate additional programs targeting both large and small patient populations. Examples of programs targeting large populations include selective BRM and selective ARID1B modulators, which have potential implications in over 100,000 cancer patients and 175,000 cancer patients that harbor BRG1 and ARID1A mutations respectively. We are pursuing other programs with genetically determined dependencies on other chromatin remodeling complexes beyond the BAF complex.
In December of 2021, we entered into a strategic collaboration with Loxo Oncology at Lilly, a research and development group of Eli Lilly and Company, to create novel oncology medicines by applying Foghorn’s proprietary Gene Traffic Control platform. The collaboration includes a co-development and co-commercialization agreement for the aforementioned selective BRM oncology program and an additional undisclosed oncology target. In addition, the collaboration includes three additional discovery programs using Foghorn’s proprietary Gene Traffic Control platform. Under the terms of the agreement, Foghorn received upfront consideration of $300 million in cash for the collaboration agreement and an equity investment by Lilly of $80 million in shares of Foghorn common stock.
For the BRM-selective program and the additional undisclosed target program, Foghorn will lead discovery and early research activities, while Lilly will lead development and commercialization activities with participation from Foghorn in operational activities and cost sharing. Foghorn and Lilly will share 50/50 in the U.S. economics, and Foghorn is eligible to receive royalties on Ex-U.S. sales starting in the low double-digit range and escalating into the twenties based on revenue levels.
For the additional discovery programs, Foghorn will lead discovery and early research activities. Foghorn may receive up to a total of $1.3 billion in potential development and commercialization milestones. Additionally, Foghorn will have an option to participate in a percentage of the U.S. economics and is eligible to receive tiered royalties from the mid-single digit to low-double digit range on sales outside the U.S.
In addition, we are developing compounds that disrupt the interactions between the transcription factors and BAF complexes. We believe that there are more than 100 transcription factors that could be amenable to our approach, one that disrupts the interaction of the transcription factor with the BAF complex. Preclinical activities of these early programs are underway.
Our approach to disrupting the interactions between transcription factors and the BAF complex is the basis of a collaboration signed with Merck Sharp & Dohme Corp., or Merck, in July 2020. In this collaboration, we are applying our Gene Traffic Control platform to identify disruptors of a single predetermined transcription factor. As part of the collaboration, we received an upfront payment of $15.0 million, and are also eligible to receive up to $245.0 million upon achievement of specified research, development and regulatory milestones by any product candidate generated by the collaboration, and up to $165.0 million upon achievement of specified sales-based milestones.
Our TeamLeadership
We have assembled a team with deep scientific, clinical, manufacturing, business, and leadership expertise in biotechnology, platform research, drug discovery, and development. Our management team has extensive experience discovering, developing, and commercializing drugs to treat patients with serious diseases. Adrian Gottschalk, our President and Chief Executive Officer, has more than 15 years of experience as a biopharmaceutical executive. Prior to joining Foghorn, Mr. Gottschalk served in various roles at Biogen, Inc., where he was most recently Senior Vice President and Neurodegeneration Therapeutic Area Head. In this role, he was responsible for late-stage development and commercialization of drugs to treat Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis. Our Chief Medical Officer, Samuel Agresta,Alfonso Quintas-Cardama, M.D., M.P.H. & T.M., previously served as Chief Medical Officer at Infinity PharmaceuticalsTCR2 and led the development of the marketed oncology drugs TIBSOVO® and IDHIFA® at Agios.their cell therapy platform. Our Chief Scientific Officer, Steven Bellon, PhD. was appointed to serve as the Company’s Chief Scientific Officer, effective as of January 10, 2022. Dr. Bellon joined Foghorn Therapeutics in 2016 as head of drug discovery, bringinghas more than 2025 years of drug discovery experience from multiple drug classes. Our research efforts are also guided by world-class scientistsclasses with prior roles at Vertex Pharmaceuticals, Amgen, and physicians on our Scientific Advisory Board, including David Schenkein, M.D., formerly the chief executive officer of Agios and presently a general partner and co-leader of Google Ventures life science team, Tony
Kouzarides, Ph.D., F.Med.Sci., FRS, professor of cancer biology at the University of Cambridge and deputy director of the Gurdon Institute, United Kingdom, Gerald Crabtree, M.D., founder of Ariad Pharmaceuticals, a Howard Hughes Medical Institute investigator and professor at Stanford University, and Charles Sawyers, M.D., chair of the Human Oncology and Pathogenesis Program at Memorial Sloan Kettering Cancer center, a Howard Hughes Medical Institute investigator, and past president of the American Association for Cancer Research, or AACR.Constellation Therapeutics. We have assembled an exceptional team of 119116 employees as of December 31, 2021.2023.
Our Beginnings: Foghorn Therapeutics and Flagship Pioneering
Foghorn Therapeutics was founded in 2015 by Flagship Pioneering, working together with academic co-founders Dr. Cigall Kadoch (Dana Farber Cancer Institute, Harvard University, Broad Institute and Howard Hughes Medical Institute) and Dr. Gerald Crabtree (Stanford University, Howard Hughes Medical Institute) to develop and commercialize a new category of first-in-class therapeutics to treat patients with cancer and other serious diseases. Our platform was inspired by work in the academic co-founders’ laboratories at the Dana Farber Cancer Institute and Stanford. This seminal work made it possible to understand how mutations cause disease by disrupting the machinery—the chromatin regulatory system—that orchestrates how cells turn genes on and off. Such mutations are associated with up to 50 percent of cancer and play roles in many other diseases. A Flagship Labs innovation team at Flagship Pioneering, led by Flagship Managing Partner, Dr. Douglas Cole, and, subsequently, Foghorn’s research and development team, established a fully integrated drug discovery platform based on this seminal work, which we call our Gene Traffic Control platform.
Our Strategy
Our mission is to leverage our unique insights into the chromatin regulatory system to pioneer the discovery, development and commercialization of a new class of therapies that transform the lives of patients suffering from a wide spectrum of diseases with high unmet need.
Our approach is to identify and drug genetically determined dependencies within the chromatin regulatory system. Our initial focus is in cancer with a precision oncology approach. Every program we pursuehave pursued to date is based on a genetic dependency on the chromatin regulatory system.
To achieve our mission, we are executing a strategy with the following key elements:
•Advance our lead precision oncology product candidates, FHD-286 and FHD-609,FHD-909, through clinical development in patients with select solid tumors and hematological cancers. FHD-286 and FHD-609 areis a highly selective and potent enzymatic inhibitor that targets both the BRM and protein degrader, respectively, that target two different componentsBRG1 enzymes of the BAF chromatin remodeling complex. FHD-909 is a highly selective and potent enzymatic inhibitor of just the BRM enzyme of the BAF chromatin remodeling complex. We believe our lead product candidates have the potential to address significant unmet medical needs across multiple oncology indications. We are conducting separate Phase 1 studies for (i) metastatic uveal melanoma and (ii) relapsed and/or refractory AML and MDS and expect initial clinical data for FHD-286 in the first half of 2022. We began dosing patients with FHD-609 in 2021 for the treatment of synovial sarcoma. We anticipate initial clinical data for FHD-609 as early as the first half of 2022.
•Expand our precision oncology pipeline by developing proprietary enzymatic inhibitors, degraders and disruptors that target genetically defined dependencies within the chromatin regulatory system. Based on our unique insights and understanding of the chromatin regulatory system, we continue to develop proprietary selective inhibitors, protein degraders and disruptors that modulate both chromatin remodeling complexes and transcription factors, two keyvarious components of the chromatin regulatory system. For example, using our proprietary platform, we are pursuing twohave disclosed four distinct targets,targets: BRM, ARID1B, CBP and ARID1B,EP300, that have genetically determined dependencies within the chromatin regulatory system with the combined potential impact into treat over 275,000 cancer patients.five hundred thousand patients per year. We intend to utilizeuse our platform to consistently develop novel product candidates to further deepen our precision oncology pipeline.pipeline and have the potential to file six INDs over the next four years.
•Harness our platform to develop novel product candidates to address therapeutic areas beyond oncology. As the orchestrator of gene expression, the chromatin regulatory system has implications in a large array of diseases. Based on academic literature and our research efforts, we believe our platform has significant potential across multiple therapeutic areas. We are committed to applying our Gene Traffic Control platform to additional therapeutic areas including virology, autoimmune diseases and neurology.over time. We believe our platform will allow us to continue to build a long-term pipeline of novel product candidates to address areas of high unmet medical need.
•Continue to enhance our platform to extend our leading position in developing novel therapeutics targeting the chromatin regulatory system. Our platform and unique understanding of the chromatin regulatory system is built upon the groundbreaking work of our academic co-founders and has been further developed by our
experienced team. We are committed to continuously integrating new insights, tools, technologies and capabilities to enhance our platform.
•Selectively enter into additional strategic partnerships to maximize the potential of our pipeline and our platform. Given the breadth of opportunities that are implicated by the chromatin regulatory system and the versatility of our platform, we may opportunistically enter into strategic collaborations intended to advance and accelerate our development programs, expand into new therapeutic areas and enhance the capabilities of our platform. In July 2020, we entered into a collaboration with Merck to discover and develop novel oncology therapeutics against a transcription factor target. In December 2021, we entered into a strategic collaboration with Loxo Oncology at Lilly a research and development group of Eli Lilly and Company, to create novel oncology medicines. The Loxo OncologyLilly collaboration includes a co-development and co-commercialization agreement for the selective BRM oncology program and an additional undisclosed oncology target. In addition, the collaboration includes three additional discovery programs using Foghorn’s proprietary Gene Traffic Control platform.
Chromatin Regulatory System: An Untapped Opportunity for Therapeutic Intervention
The chromatin regulatory system orchestrates gene expression. In order for DNA to fit in the nucleus of each human cell, it is densely packed into what is called chromatin. This packing of DNA occurs by winding it around a core of proteins called histones to form what is known as a nucleosome, having the appearance of thread (the DNA) wrapped around a spool (the histones). Multiple nucleosomes cluster further to form more densely packed chromatin. Before DNA can be transcribed to RNA and then translated into protein, chromatin needs to be “unpacked” to allow access for the cellular machinery responsible for DNA transcription. Cells have therefore evolved a system known as the chromatin regulatory system that can locate and unpack particular regions of the chromatin to orchestrate and allow for gene expression.
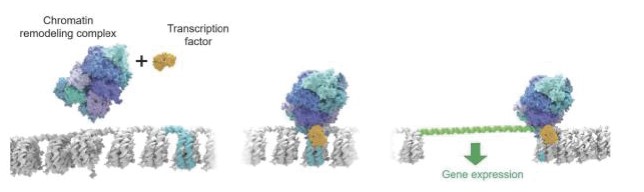
Figure 1: Chromatin Regulatory System Biology: Chromatin remodeling complexes and transcription factors work in concert to unpack chromatin to enable gene expression. The left portion of the figure shows “packed” or closed chromatin and the right portion of the figure shows “unpacked” or open chromatin with DNA highlighted in green.
Two of the major components of the chromatin regulatory system are chromatin remodeling complexes, and transcription factors. Transcription factors specify the locations of genes to be transcribed by binding to specific locations on DNA. Chromatin remodeling complexes, guided by transcription factors, unpack thehelicases and other chromatin to expose DNA for transcription. These two componentsrelated factors which work in concert to orchestrate gene expression. One important role for this system is to control the accessibility of chromatin which in both healthyturn determines if other factors necessary for gene expression can access the genetic material. In addition, the system controls the structure, modification, and diseased cells. repair of chromatin which are all necessary for proper control of gene expression. Because of the central role this system plays in orchestrating normal gene expression, aberrations in the system may result in disease. Our platform is uniquely suited to correct these aberrations and treat these diseases.
While chromatin remodeling complexes have been known in the scientific community for decades, disease relevance was not initially recognized, and consequently chromatin remodeling complexes were underappreciated as a set of relevant drug targets. Transcription factors, helicases and other chromatin related factors, on the other hand, while linked decades ago to cancer and
understood as relevant targets, have led to few approved oncology drugs, as companies seeking to drug these targets have historically lacked a systematic approach to doing so. Recently, ground-breaking work by our academic co-founders has revealed that alterations in chromatin remodeling complexes as well as their interactions with transcription factors are strongly associated with various cancers. Broad cancer sequencing initiatives have shown that mutations in the chromatin regulatory system are found in over 50 percent of all cancers, potentially impacting over 2.5 million cancer patients across the United States, Europe and Japan. Further work in the field by our founders and others has highlighted the association of this system in other therapeutic areas, including virology, autoimmune disease and neurology, implying even greater potential for therapeutic intervention.
Vulnerabilities in Cancer Created by Genetic Dependencies on the Chromatin Regulatory System
Cancer cells often contain many different mutations that lead to their abnormal growth and proliferation. Within cancer cells, these mutations give rise to genetically determined dependencies, upon which the cancer cells rely for their survival. The creation of these dependencies can be directly related to the mutation or to other cellular biology, thereby creating vulnerabilities for cancer cells and the opportunity for therapeutic intervention. In contrast, healthy cells, which lack these mutations and therefore these dependencies, are less susceptible to a therapeutic that targets these genetically determined dependencies.
There are three primary mechanisms by which geneticallyGenetically determined dependencies onmay arise from mutations in various components of the chromatin regulatory system arise. They are:
1.Mutations in(e.g., chromatin remodeling complexes,
2.Mutations helicases, transcription factors, chromatin related factors) or overexpression of transcription factors
3.Mutationsthrough mutations elsewhere in the cell that impingecreate dependencies on chromatin remodeling complexes and/or transcription factorsthe system.
Our platform enables us to identify these genetic dependencies and thereby discover the cancer cells’ vulnerability within the chromatin regulatory system. We believe these vulnerabilities create opportunities to selectively drug and kill cancer cells while minimizing impact to healthy cells. These genetically determined dependencies enable us to select specific patient populations and enrich our clinical trials using a precision approach. Every program we pursuehave pursued to date is based on a genetically determined dependency on the chromatin regulatory system.
Our Initial Focus—BAF Complexes and Associated Transcription Factors
There are 28 types of chromatin remodeling complexes. All types of chromatin remodeling complexes use ATP as an energy source for opening and closing chromatin. These remodeling complexes contain a catalytic subunit that is capable of breaking down ATP, known as the ATPase. The ATPase serves as the catalytic engine that drives the function of each chromatin remodeling complex. The breakdown or hydrolysis of each ATP molecule by the ATPase creates energy that, in turn, drives chromatin remodeling. These chromatin remodeling complexes are mutated in approximately 25 percent of cancers.
BAF, which stands for BRG1/BRM-associated factors, one type of chromatin remodeling complex, is mutated in approximately 20 percent of cancers, thus being the most mutated in the family of ATPase chromatin remodelers and among the most mutated targets in cancer. Given the breadth of mutations in cancer, the BAF complex is our initial focus among the ATPase dependent chromatin remodeling complexes.
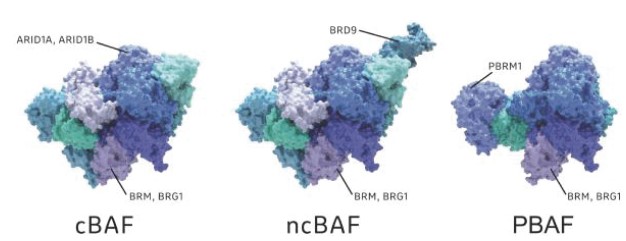
The BAF complex is a multicomponent protein structure containing twelve to fifteen protein subunits taken from a larger set of a possible 29 subunits. Three common forms of BAF are known as canonical BAF, or cBAF; non-canonical BAF, or ncBAF; and polybromo BAF, or PBAF. While the exact compositions of these forms of BAF are different, each form contains a number of common subunits, one such being the ATPase catalytic subunit. Each BAF complex contains one of two possible ATPases, either ATPase known as BRM, also known as SMARCA2, or ATPase known as BRG1, also known as SMARCA4.
Different cell types and tissues contain different forms of BAF. This cell and tissue specificity gives rise to the possibility of additional pharmacological selectivity when drugging potential targets.
Figure 2. Schematic depicting biochemical subunit compositions of mammalian BAF, ncBAF and PBAF complexes.
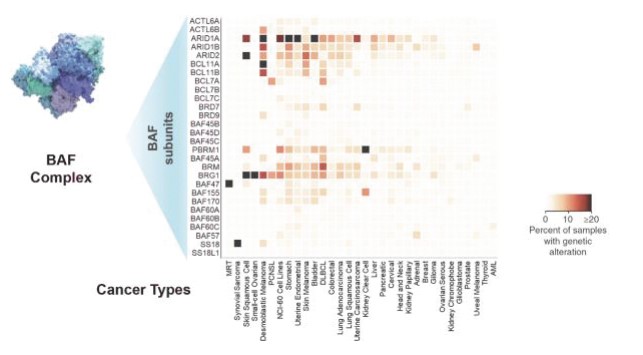
Figure 3. Genetic alterations are commonly found in subunits of the BAF complex in tumors.
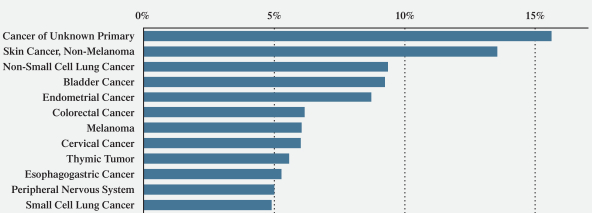
The BAF complex, when aggregating mutations across all of its subunits, is the second most mutated target to the well-known cancer target TP53. Genetic alterations of various subunits of the BAF complex have been observed in a wide range of cancers. These include but are not limited to the following:
•More than 90 percent of ovarian cancer patients;
•34 percent of uterine endometrial patients;
•34 percent of stomach cancer patients;
•29 percent of bladder cancer patients;
•28 percent of non-small cell lung cancer, or NSCLC, patients; and
•27 percent of skin cancer patients.
The following mechanistic insights provide strategies to target the BAF complex in cancer:
•Dependency exists between BAF complex subunits;
One example is:
•In some cancer cells, the gene encoding BRG1, a catalytic subunit of the BAF complex, is mutated causing a loss of function in BRG1
•Often this loss of function leads to a dependency on BRM, a similar protein to BRG1 that is the other catalytic subunit of the BAF complex
•This loss of BRG1 and subsequent dependency on BRM creates a vulnerability by rendering these cancer cells highly sensitive to targeting BRM
•Mutations elsewhere in the cell confer a dependency on the BAF complex;
One example is:
•Mutations in G-protein coupled receptors (GNAQ/GNA11) are found in 85 percent to 95 percent of uveal melanoma, a cancer of the eye
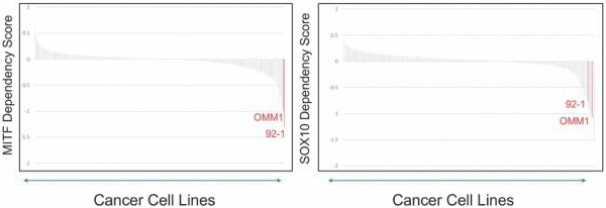
•In uveal melanoma cell lines with these mutations, we have established a dependency on two transcription factors, MITF and SOX10.
•These two transcriptions interact with the BAF complex
•Targeting the BAF complex then inhibits MITF and SOX10 mediated transcription
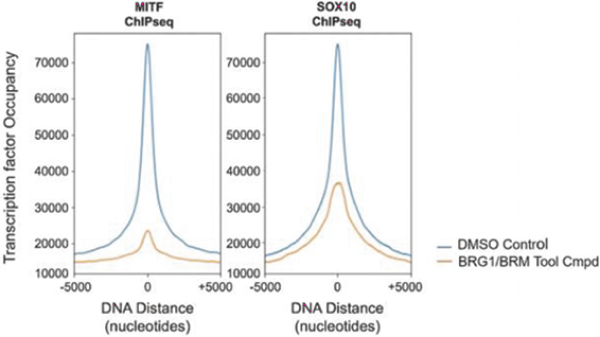
Transcription factors, the proteins that guide the chromatin remodeling complexes, help determine which genes are expressed and have long been desirable but elusive targets for drug discovery efforts. Work by our academic co-founder Cigall Kadoch, as well as others in the field, revealed that transcription factors work in concert with chromatin remodeling complexes, BAF as one example, to orchestrate gene expression. A transcription factor recognizes specific guidepost-like sequences, or locations, on DNA. The transcription factor binds to the chromatin remodeling complex and in doing so directs the remodeling complex to the appropriate location on chromatin. Once recruited to the appropriate location, the chromatin remodeling complex unpacks the chromatin, exposing the DNA and allowing transcription machinery to transcribe the corresponding gene.
Some transcription factors, such as the estrogen receptor, or ER, have long been the targets of approved and efficacious drugs for the treatment of cancers such as breast cancer. However, the majority of transcription factors have not been amenable to traditional small molecule drug inhibition. While directly blocking the DNA binding site on transcription factors would be an effective way of inhibiting their activity, it is usually not possible to find small molecules that can bind to these sites with the potency and selectivity needed to advance as therapeutics.
Different healthy cell types, such as heart, brain, or muscle cells, use different types of transcription factors. In cancer cells, mutated and/or abnormal levels of specific transcription factors are found. Because many transcription factors are cell and tissue specific, there is the possibility of additional pharmacological selectivity when drugging potential transcription factor-chromatin remodeling complex interactions. We believe that there are more than 100 transcription factors that could be amenable to our approach of disrupting the interactions of transcription factors with the BAF complex.
Our Approach to Drugging the Chromatin Regulatory System
We are focused on developing small molecule product candidates that target the chromatin regulatory system through the use of enzyme inhibitors, protein degraders and transcription factor disruptors.
•Enzyme inhibitors. These candidates have the potential to act on targets such as the ATPases BRG1 and BRM of the BAF complex. Our screening capabilities enable us to find allosteric inhibitors which afford additional selectivity over orthosteric, or direct, inhibitors.
•Protein degraders. These candidates are bifunctionaleither heterobifunctional or molecular glue degraders which serve to specifically recruit a target to an E3 ligase component, resulting in which one portion of the molecule specifically recognizes the target while the other portion is able to direct the destructionremoval of the target protein by the cell’s native protein degradation system.
•Transcription factor disruptors. These candidates will be direct small-molecule disruptors of the protein-protein interactions between transcription factors and chromatin remodeling complexes.
We leverage the appropriate mechanism based on the target in the chromatin regulatory system. In some cases, we may take multiple approaches and remain modality agnostic in order to ensure we achieve the best approach and most appropriate molecule.
The two main approaches that we are taking to drugging chromatin remodeling complexes are inhibiting its ATPase activity and degrading mutated or dependent subunits withinFor components of the chromatin remodeling complex. We are taking a different approach to modulating the activity of transcription factors than previously attempted by the field. We believe this approach can be applied across the broad set ofregulatory system that have an enzymatic function (e.g., chromatin remodeling complexes and transcription factors with which they interact, as illustrated by the BAF complex. Because transcription factors require collaboration with the BAF complex, disrupting the interaction between the two shuts down the abilityhelicases), we may leverage enzymatic inhibitors. For components of the system that are not amenable to enzymatic inhibition or where selectivity through inhibition may not be possible, we may leverage targeted protein degradation.
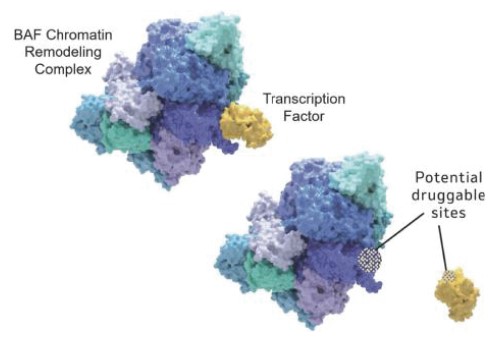
For transcription factor to drive transcription. Our approach is to findtargets, we are leveraging where appropriate protein degradation and/or small molecule disruptors that can bind either to either the transcription factor or its relevant binding partner (e.g., the BAF complex in order to break the interaction between the two. In order to understand whether it is possible to selectively drug these interactions, there are two important aspects that need to be understood. One is where specifically the transcription factor binds to the BAF complex and the second is how tightly it binds.
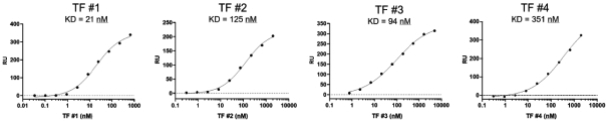
Based on our work, we have observed that individual transcription factors bind to the BAF complex at specific sites rather than all binding to a single site, implying that it should be possible to specifically interfere with the binding of one transcription factor to the BAF complex without affecting the binding of every other transcription factor. This is a critical success factor for the specificity of drug candidates binding to the BAF complex. We have also observed that the potencies of these interactions are roughly equivalent to those observed in other protein-protein interactions that have been successfully disrupted by small molecule drugs. Because many transcription factors are cell and tissue specific, there is the possibility of additional pharmacological selectivity when drugging potential transcription factor-chromatinchromatin remodeling complex interactions. We believe these findings provide the opportunity to systematically discover and develop a novel class of product candidates that are specific, selective and that will be designed to disrupt the interaction between transcription factors and the BAF complex.
Figure 4. We are disrupting transcription factor activity by blocking interactions with the BAF complex.complex).
Our Gene Traffic Control Platform
The chromatin regulatory system has remained an untapped opportunity for therapeutic intervention due to the inability to systematically characterize and study the chromatin remodeling complexes and associated transcription factors.its various components. Building upon the groundbreaking discoveries of our academic co-founders, we have developed our proprietary Gene Traffic Control platform which allows us to identify and validate targets within the chromatin regulatory system. We have unique capabilities to isolate, synthesize, characterize, and interrogate components of the BAF complexsystem at a level of scale, precision, and efficiency, that to our knowledge, no others have achieved. We have unique capabilities to understand how transcription factors interact with the BAF complex and have generated unique insights into where and how transcription factors bind. We believe our platform is broadly applicable to other chromatin remodeling complexes and transcription factors.
Our capabilities and insights have allowed forenabled the development of a suite of unique biochemical, biophysical, structural, and functional assays. We use these assays to discover and optimize novel small molecule chemical matter which include enzymatic inhibitors, protein degraders, and transcription factor disruptors to various targets within the chromatin regulatory system. To
our knowledge, we are the only company that has the ability to study the chromatin regulatory system at scale, in context, and in an integrated way.
Our Gene Traffic Control platform encompasses the following:
•Target Identification and Validation
•Production of Chromatin Regulatory System Components at Scale and Proprietary Assays
•Discovery and Optimization of Chemical Matter
•Targeted Protein Degradation
•Translation to Clinic and Identification of Biomarkers
The key features and capabilities of our platform are described below:
Target Identification and Validation
We use genomic screens and a suite of epi-genome sequencing and computational tools to characterize, identify and validate targets within the chromatin regulatory system. Our epi-genome sequencing tools allow us to understand the mechanisms of how our drugs are modifying the chromatin structure. Our platform allows for the identification of genetically determined dependencies associated with the chromatin regulatory system. Specifically, we:
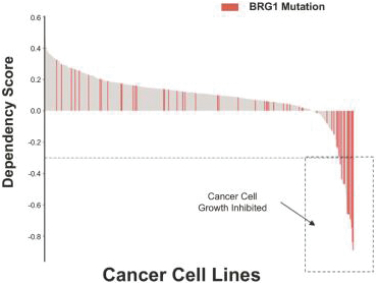
•Conduct and leverage genomic screens to identify dependencies and relationships. We utilize both broad and specific genomic screens to identify dependencies and relationships associated with the chromatin regulatory system. We use a mix of internal and external data sets that apply CRISPR and shRNA technology to understand relationships across and within a range of cancer cell lines.
•Perform broad epi-genome sequencing to validate dependencies in vitro. We apply cutting edge epi-genome sequencing tools in combination with proprietary tool compounds to further validate targets and enhance our understanding of the impact of drugging the chromatin regulatory system. These tools allow us to rapidly understand the gene expression profiles of specific cancer cell lines, the open / closed state of chromatin, and give us mechanistic understanding of how components of the system work together.
•Apply machine learning and artificial intelligence to enhance discovery efforts. We have built tools that allow us to mine and interpret external and internal datasets that aid in our discovery efforts yielding unbiased and unsupervised computer analyses to identify targets and genetic dependencies on the chromatin regulatory system and to further understand mechanism of action. Examples of external data sets include data from Thethe Cancer Genome Atlas (TCGA) and the Broad Institute. Internal data sets include data from cell lines, data from xenograft models and epi-genomic information (RNA-seq, ATAC-seq, CHiP-seq, SNAP-seq). We also use these tools in the preclinical stage to evaluate cancer cell lines & patient samples to identify biomarkers for patient stratification and patient population identification.
•Validate dependencies in vivo. Where possible, we endeavor to validate targets in various animal models with implanted cancer cells relevant to the disease we are aiming to treat. Specifically, we use mouse xenograft models with inducible CRISPR / shRNA to validate that knockdown of our target of interest results in tumor growth inhibition. We also apply epi-genome sequencing tools in the animal model setting to identify potential biomarkers.
Production of Chromatin Regulatory System Components at Scale and Proprietary Assays
We have built unique capabilities to purify and synthesize components of the BAF complex andchromatin regulatory system (chromatin remodeling complexes, transcription factors.factors helicases, chromatin related factors). These capabilities allow us to study the chromatin regulatory system at scale and in context that, to our knowledge, is unavailable to others, and yields insights that are critical to systematically drugging this system. Specifically, we:
•Purify and synthesize chromatin remodeling complexes and transcription factors at scale. Our platform has the unique ability to purify and synthesize chromatin remodeling complexes such as the BAF complex, with potential applications to other chromatin remodeling complexes. Importantly, we are able to purify disease relevant and mutated forms of BAF directly from the cancer cell lines of interest. To our knowledge, we are the only company that has developed the ability to purify and manipulate BAF, sub-complexes of BAF, as well as its hostmutant forms of subunits, in quantities that enable us both to generate structural data to identify potential binding sites for drug candidatesthese complexes. We also produce and to conduct high throughput screensscreen full length version of small molecule drug candidates against these sites.transcription factors and other chromatin regulatory system components.
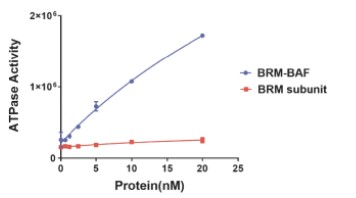
•Study chromatin remodeling complexes in context leading to relevant insights into the impact of drug intervention.Structural Biology. We have foundbelieve that the propertiesthree-dimensional structure of subunits of BAF, such as BRG1 or BRM, are different when they are incorporated into the BAF complex than when we test them in isolation. For example, the catalytic activity of these subunits using nucleosomes was increased by over 15-fold when they were incorporated into the BAF complex compared to their activities in isolation (see Figure 5 below). This is the biologically relevant activity of the complex. Additionally, the ability to screen the full complex greatly improves the potential of finding allosteric modulators which may afford additional pharmacological selectivity. These examples underscore the importance of assaying and screening the full complex.
•Utilize advanced analytical methods to develop and understand critical insights into how transcription factors interact with chromatin remodeling complexes. We integrate multiple technologies and methodologies, including high-throughput-screening, biophysics, affinity screening, and surface mapping to gain unparalleled insights into the chromatin regulatory system components provides a mechanistic understanding of the targets and how its primary two components, transcription factors andthus enables drug discovery. We have repeatably been able to determine three dimensional structures for various chromatin remodeling complexes, interact. Based on our protein-protein interaction mapping technology, we have determined precisely the binding sites of multiple transcription factors to the BAF complex, providing us with critical insights into how these factors bind to the BAF complex, and details on how these binding interactions can be disrupted using small, drug-like molecules.regulatory system targets, including x-ray
We believe that our unique capabilities as applied to the BAF complex and associated transcription factors can be applied to other chromatin remodeling complexes and transcription factors. It is our intention to further leverage our capabilities on other chromatin remodeling complex targets.
ATPase activity using nucleosome substrate
Figure 5. ATPase activitystructures of full BAFthe enzymes targets, ternary structures of protein degrader targets, and mass spectrometry mapping of transcription factor - chromatin remodeling complex underscores the importance of assaying in the appropriate biological context.interactions.
Discovery and Optimization of Chemical Matter
We perform proprietary high throughput screens that leverage our ability to produce the chromatin regulatory system components at scale. An example screen is the use of the fully assembled BAF complex which is specific to its mutated or disease relevant form (e.g., screening the BRM form of BAF which corresponds to BRG1 mutated cancer). Furthermore, we are able to screen the BAF complex when bound to a relevant transcription factor. We utilize both proprietary and publicly available chemical libraries in our screens.
Once we find hits from our screens, we use our unique suite of biophysical assays involving the relevant component of the chromatin regulatory system to characterize, validate, and optimize our chemical matter. These assays provide us with biologically relevant insights that guide our medicinal chemistry efforts.
Targeted Protein Degradation
In cases whereFor targets in the portfolio whose biology demonstrates that degradation could offer a therapeutic advantage, we develop small molecule heterobifunctional or non-cereblon based molecular glue degraders. Many of our drugging effortstargets play important scaffolding roles in chromatin remodeling complexes and/or are directed at targets that have no enzymatic activity, we seek to degrade the protein of interest through targeted protein degraders.not enzymes. Therefore, inhibition would not be effective or possible. Protein degraders are bifunctional small molecules in which one portionrecruit target proteins to specific E3 ligase complexes and by doing so, promote the removal of the molecule specifically recognizes the target while the other portion directs the destruction of the targetprotein by harnessing the cell’s ubiquitin and proteasome-based degradation system. The two chemical functionalities of the molecule are connected by a variable linker. This approach affordsresults in rapid loss and clearance from the cell of disease driving proteins and is a general method of degrading protein targets of interest.powerful complement to our inhibitor capabilities.
After completing screens, as described above,We have a broad and finding small molecule binders to the target of interest, we use our proteinhighly efficient degradation know-how to convert binders into selective protein degraders.development, screening, and triaging platform. This know-how and capabilities include:
•Proprietary library of linkers and E3 ligase binders for heterobifunctional degrader development;
•Proprietary screening strategy for novel non-cereblon based molecular glue discovery;
•Biochemical, biophysical, and cellular assays that measurecharacterize protein degradationdegrader mechanism of action and guide optimization, including protein synthesisdegradation kinetics, ubiquitination, and degradation kineticspermeability;
•Ternary complex modelingstructural determination and characterizationmolecular modeling;
•Genome wide proteomic analysis of degradationGlobal proteomics and mass spectrometry to measure selectivity in an unbiased fashion;
•Exploration of novel ligases;
•Long-acting formulation of protein degraders which enhances route of administration and frequency of delivery; and
•Degraders that may be used in conjunction with antibody technology.
Translation to Clinic and Identification of Biomarkers
We seek to enrich our clinical studies with the genetically relevant patient populations that are most likely to benefit from treatment. Early in the drug discovery process, we use various genome and epi-genome analyses to understand the genetic dependency of the cancer on the chromatin regulatory system. Our intent is to have clear genetic markers for patients whom we seek to potentially treat.
As we progress a drug candidate, we analyze tumor models and where available direct patient samples to understand biomarkers of response (e.g., change in expression level of a particular gene or set of genes, change in protein level of a component of the chromatin regulatory system). We intend to use these biomarkers in our clinical studies to understand tumor response to our drug candidates. Additionally, we will retrospectively analyze our clinical studies for any other biomarkers that will further enhance patient stratification and response.
Our Product Candidates
We are developing a broad pipeline of product candidates that target genetically determined dependencies within the chromatin regulatory system. Our programs consist of enzyme inhibitors, protein degraders and transcription factor disruptors. Our most advanced product candidates are FHD-286 and FHD-909. For FHD-286, following a monotherapy dose escalation Phase 1 in relapsed and/or refractory AML/myelodysplastic syndromes (“MDS”), we initiated two separatea Phase 1 studies for (i) metastatic uveal melanoma andcombination study with either decitabine or cytarabine in relapsed and/or refractory AML and MDS in May 2021. For our second product candidate, FHD-609, we initiated a Phase 1 study for the treatment of synovial sarcoma in August 2021. Our pipeline is as follows2023. In February 2024, we announced FHD-909 had been selected by Lilly for clinical development pursuant to the Lilly Collaboration Agreement, and we anticipate that Lilly will file an IND in the second quarter of 2024.
FHD-286
Overview
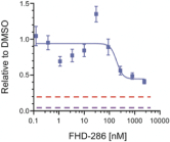
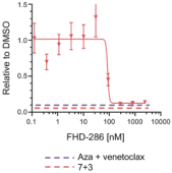
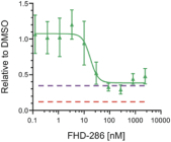
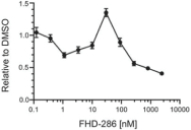
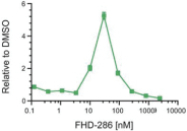
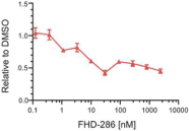
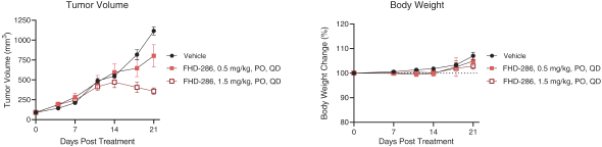
We are currently advancing our lead product candidate, FHD-286, in two separatea Phase 1 clinical studiesstudy in (i) metastatic uveal melanoma and (ii)patients with relapsed and/or refractory AML and MDS.in combination with either decitabine or cytarabine. FHD-286 is a highly potent, selective, allosteric and orally available, small-molecule, enzymatic inhibitor of BRG1 and BRM, for the potential treatment of uveal melanoma, AML and MDS. BRG1 and BRM are two highly similar proteins that serve as the ATPases, or the catalytic engines, across all forms of BAF. Our preclinical data in both AML and uveal melanoma animal xenograft models demonstrated encouraging anti-tumor activity. Additionally, the clinical data from our Phase 1 monotherapy study of FHD-286 in relapsed and/or refractory AML and MDS suggested that FHD-286 is a differentiation agent that could provide complimentary benefit if combined with other therapeutic agents. The multi-center, Phase 1 studies will study is primarily assess focused on assessing the safety and tolerability of FHD-286 in adultscombination with uveal melanoma,either decitabine or cytarabine in adult patients with relapsed and/or refractory AML and MDS.AML. Secondary endpoints are expected to include the pharmacokinetic and pharmacodynamic properties of FHD-286 as well as clinical activity. Proof of mechanism will be based on indicators of target engagement in association with FHD-286 combination treatment. As we further understand the therapeutic potential of FHD-286 in the course of these initial clinical studies,this study, we may pursue additional clinical studies in these and other indications as a single agent. We expect initial clinical data for the combination dose escalation Phase 1 study of FHD-286 in patients with relapsed and/or in combination with novel or standard of care agents. Initial data from these studies are expectedrefractory AML in the firstsecond half of 2022.2024.
AML Disease Overview
Acute myeloid leukemia, or AML is a heterogeneous group of hematologic cancers characterized by a proliferation of myeloid precursors, commonly known as blasts, with limited ability to differentiate into more mature myeloid cells. These blasts replace normal hematopoietic tissue in the bone marrow, resulting in decreased hematologica decrease in all blood cell numbers,types, or pancytopenia, and the morbidities associated with the cancer.therefrom.
AML is the second most common subtype of leukemia in adults. In major markets (United States, EU4, UK and Japan), AML has an incidence of approximately 35,000 cases annually and is generally a disease of elderly people with more than 60AML are diagnosed annually. This incidence is expected to increase approximately 17 percent over the next five years. Median age at diagnosis for people with AML is 69 and median age at death is 73, underscoring the short course of diagnosed patients being older than 60 years. life in people with this disease.
The average five-year survival rate for patients with AML is 20 percent, and there are significant differences in prognosis depending on several factors, including the age of the patient and co-morbidities at diagnosis. For patients under the age of 60, the five-year survival rate is approximately 33 percent, while for those over the age of 60 it is less than 15 percent. There are likely multiple reasons for this discrepancy, including the ability of younger patients to tolerate more aggressive therapies.
Current first-line treatments for patients with AML typically involve aggressive combination chemotherapy regimens with or without hematopoietic stem cell transplantation (HSCT)(“HSCT”). Older patients or patients who cannot tolerate HSCT, typically those with comorbidities, are often treated with cytarabine and daunorubicin induction followed by high-dose cytarabine consolidation. Patients who cannot tolerate combination chemotherapy receive low dose cytarabine, azacitidine, Venclexta ® ,decitabine, venetoclax, some combination of these therapies, and/or enroll in clinical trials. There is a single biologic, gemtuzumab ozogamicin or Mylotarg,(Mylotarg®), approved by the FDA for newly diagnosed and relapsed-refractory AML. Other, more recently approved therapeutics for AML target subsets of patients with tumors containing specific mutations such as midostaurin marketed as Rydapt® by Novartis for those with FLT3 mutations, enasidenib marketed as Idhifa® by Bristol Myers for those with mutations in IDH2, and ivosidenib, marketed as Tibsovo® by Agios for those with mutations in IDH1.
Despite these advances, the five-year disease-free survival rate among patients who do achieve remission five-year disease-free survival is only 30-40 percent because the majority of patients experience relapse. Patients in the elderly populationElderly patients with AML have a relapse rate of 80-90 percent. Younger patients have a relapse rate of between 60-80 percent. There remains a significant need for safe, durable and broadly effective AML treatments.
Uveal Melanoma Overview
Uveal melanoma is the most frequent type of ocular cancer with approximately 5,000 cases each year in the major markets (United States, EU4, UK and Japan), typically presenting upon a routine eye exam in patients without specific symptoms. Local treatment, primarily with radiation therapy, is effective in preventing local recurrence in over 95 percent of cases. Due to the asymptomatic nature of uveal melanoma, at the time of the diagnosis, a considerable portion of these patients already have metastatic disease, typically in the liver. Roughly half of all patients will eventually develop metastases. For those diagnosed with metastatic disease, the one-year survival is only 15 percent. The poor prognosis associated with metastatic disease and the lack of effective therapy highlights the need for novel therapeutic approaches that specifically target metastatic uveal melanoma.
Between 85 percent and 95 percent of uveal melanoma tumors contain mutations in one of two G-protein-coupled receptor subunits: GNAQ or GNA11. We have established through uveal melanoma cell lines with the GNAQ/GNA11 mutations that there is a dependency of these cell lines on two over expressed transcription factors, MITF and SOX10. In uveal melanoma, these two transcription factors abnormally interact with the BAF complex.
Our Solution: FHD-286
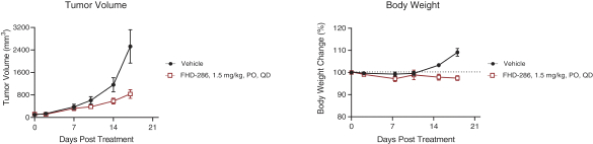
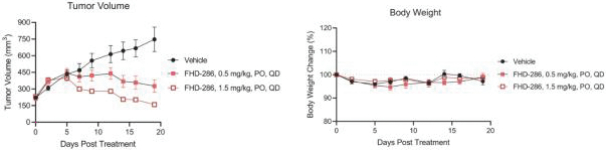
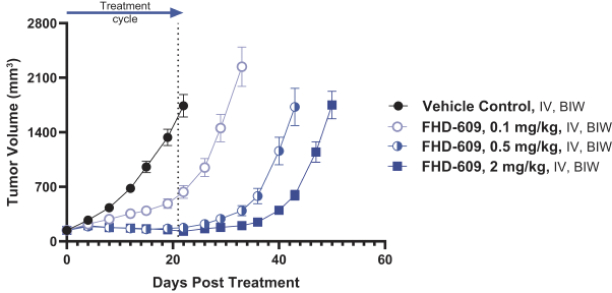
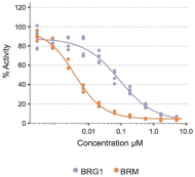
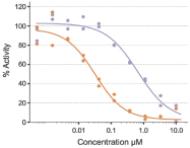
FHD-286 is a highly potent, selective, allosteric and orally available, small molecule inhibitor of the enzymatic activity of both BRG1 and BRM. In established AML cell line-derived xenograft, or CDX, models MV4-11 and OCI-AML2, we observed robust tumor growth inhibition. In established uveal melanoma CDX models, specifically MP-46 and 92-1, we observed significant tumor growth inhibition and tumor regression, respectively. We are currently conducting separate Phase 1 studies for (i) metastatic uveal melanoma and (ii) relapsed and/or refractory AML and MDS. Initial data from both Phase 1 studies is anticipated in the first half of 2022. Either BRG1 or BRM can serve as the primary ATPase, or catalytic engine, of the BAF complex. BAF complexes will contain only BRG1 or BRM, as they are mutually exclusive subunits, as shown in the figure below. BRG1 or BRM are two proteins which are 76 percent identical at the amino acid level over their entire length and over 90 percent identical in the catalytic region. We are currently advancing FHD-286 in a Phase 1 clinical study in patients with relapsed and/or refractory AML in combination with either decitabine or cytarabine. We expect initial clinical data for the combination dose escalation Phase 1 study of FHD-286 in patients with relapsed and/or refractory AML in the second half of 2024.
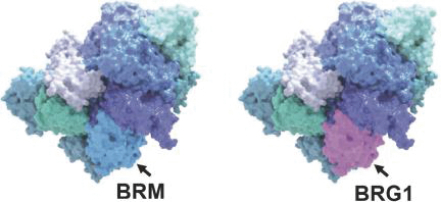
Figure 6.1. The enzymatic activity of the BAF complex is provided by the BRM or BRG1 subunits.
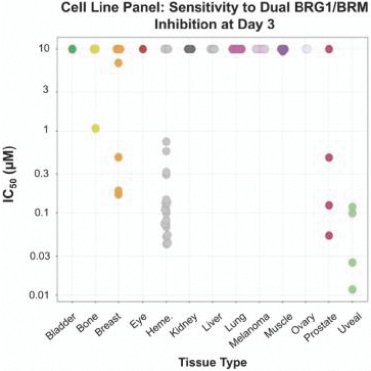
When we conducted compound screening against a panel of tumor cell lines, a number of these tumor cell lines were shown to be highly sensitive to BRG1 or BRM inhibition over a three-day period. These cell lines include nineteen of twenty-one of the
hematopoietic malignancy cell lines tested, all four of the uveal melanoma cell lines, three out of four prostate tumor cell lines, and three out of seven breast tumor cell lines. We observed additional sensitivity in other tumor cell lines tested over a seven-day period.
Figure 7. Certain cell lines, including those derived from uveal melanoma, hematological cancers, prostate cancer, and breast cancer were highly sensitive to BRG1/BRM inhibition.
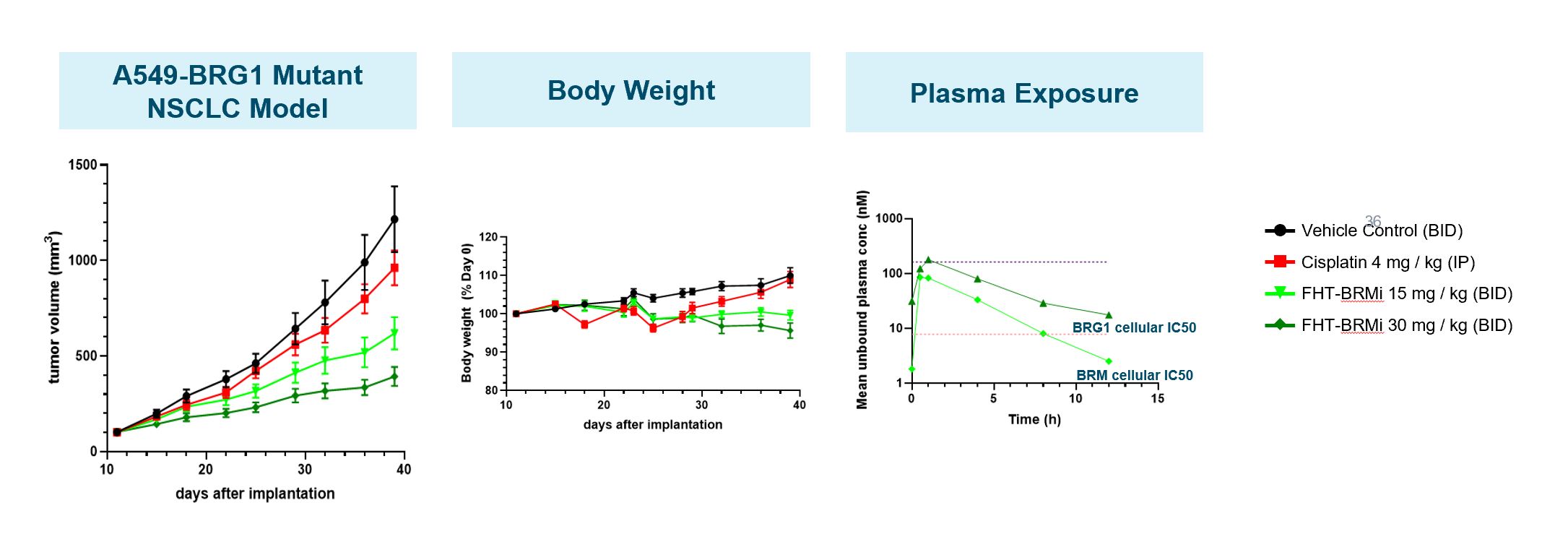
Our Preclinical Data for AML
Genetic studies have identified a critical role of BRG1 in the maintenance of the undifferentiated state of AML cells. Knockdown of the expression of BRG1 was found both to inhibit the expression of genes associated with high proliferation and to induce the expression of genes associated with mature myeloid cells. In a mouse model of AML, partial genetic inactivation of BRG1 led to a greater than two-fold increase in overall survival. These data suggest that pharmacological inhibition of BRG1 may provide a therapeutic benefit.
We have generated in vivo proof of concept data that demonstrated antitumor activityPhase 1 Monotherapy Study of FHD-286 in Relapsed and/or Refractory AML patient samples as well as multiple AML CDX models. Using tumor cells isolated from AML patients, we demonstrated that treatment with FHD-286 allowed for appropriate differentiation of AML cells. and MDS
We treated these tumor cells withconducted a single dosePhase 1 monotherapy study of FHD-286 at increasing exposures and assessed the effects on both myeloid cellular differentiation and cell death. We observed myeloid cellular differentiation at a lower nanomolar exposure relative to where we observed cell death. The data support that pharmacologic inhibition of BRG1 can release the differentiation block associated with BRG1 overexpression in AML. Ongoing research has revealed that transcription factors interacting with over-expressed BRG1 containing BAF complexes are implicated in AML. Targeted treatment that releases a differentiation block has been observed to be clinically meaningful with ATRA treatment in acute promyelocytic leukemia as well as IDH1 and IDH2 inhibition in IDH-mutated AML. The cell killing observed was comparable to the effect of standard of care combinations: cytarabine plus daunorubicin and azacytidine plus venetoclax.
| | | | | | | | |
Patient #1 Sample
BM-de novo AML | Patient #2 Sample
BM-secondary AML | Patient #3 Sample
BM-secondary AML |
| | |
Figure 8. Treatment of patient-derived AML tumor samples with FHD-286 stimulated differentiation and cell death. Dose-dependent reduction of blast counts in samples from three patients. The blast count was normalized and plotted as relative to the level in vehicle DMSO-treated samples. The level of blast count reduction achieved by standards of care (Aza + venetoclax and “7+3”) are indicated by the dashed lines. BM = Bone Marrow.
| | | | | | | | |
Total Blasts | Differentiation-like Blasts | Immature Blasts |
| | |
Figure 9: Evidence of a dose-dependent differentiation effect in patient #1 sample.
We have confirmed the sensitivity observed in our three-day cell line panel in CDX models created using OCI-AML2 and MV4-11, two AML cell lines with different underlying genetic mutations. In addition, we have observed robust dose response in further evaluation of FHD-286 in MV4-11 CDX models. We have also observed synergy of FHD-286 in combination with cytarabine.
MV4-11 AML CDX Model
FLT3 ITD, MLL-AF4
OCI-AML-2 AML CDX Model
MII-AF6, DNMTa3 mut.
Figure 10. FHD-286, dosed as monotherapy, led to tumor growth inhibition in two AML xenograft models MV4-11 and OCI-AML-2.
Our Preclinical Data for Uveal Melanoma
In uveal melanoma cell lines that contain GNAQ/GNA11 mutations, genetic studies have revealed that these cells overexpressed two transcription factors, MITF and SOX10.Our data showed that the MITF and SOX10 transcription factors abnormally over-interacted with the BAF complex in uveal melanoma cell lines. By inhibiting the ATPase activity, both BRG1 and BRM, of the BAF complex, we observed anti-tumor effects in several CDX and patient-derived xenograft, or PDX, uveal melanoma models.
We established the genetic dependency of uveal melanoma cell lines on MITF and SOX10 by analyzing data from the Project Achilles, a functional genomics screen conducted by the Broad Institute. We found that established uveal melanoma cell lines such as 92-1 and OMM1 were highly dependent on MITF or SOX10.
Figure 11. Uveal melanoma cell lines, such as 92-1 and OMM1, were highly dependent on MITF or SOX10.
We found that inhibition of BRG1 and BRM led to suppression of gene expression from several MITF and SOX10-dependent genes. A broader measure of the effect of dual inhibition of BRG1 and BRM on transcription of MITF and SOX10-dependent genes was obtained using a technique known as chromatin immunoprecipitation sequencing, or ChIP-seq. ChIP-seq allows us to find where particular proteins, in this case transcription factors, are binding to chromatin. Treatment of uveal melanoma cells with a research compound with similar properties to that of FHD-286 resulted in decreased binding of both MITF and SOX10 transcription factors to their respective chromatin binding sites. These results validate the mechanism of action of FHD-286 in uveal melanoma cells.
Figure 12. BRG1/BRM inhibitor blocked the ability of MITF and SOX10 to bind to their target sequences as determined by ChIP-seq.
We have generated in vivo proof of concept data that demonstrated antitumor activity of FHD-286 in multiple uveal melanoma CDX and PDX models. In two uveal melanoma models, 92-1 and MP-46, oral dosing of FHD-286 at 1.5 mg/kg as monotherapy
resulted in tumor growth regression and inhibition, respectively. Importantly, doses of FHD-286 of up to 1.5 mg/kg were well-tolerated in that FHD-286 at these doses did not lead to changes in body weight considered to be clinically meaningful compared to controls (e.g., changes greater than 10 percent of body weight), a commonly used measure of safety.
92-1 Uveal Melanoma Model
MP-46 Uveal Melanoma Model
Figure 13. FHD-286 led to dose-dependent tumor growth inhibition in two uveal melanoma xenograft models 92-1 and MP-46.
Clinical Plans for FHD-286 in AML and Uveal Melanoma
We are currently conducting separate Phase 1 studies with FHD-286 in (i) metastatic uveal melanoma and (ii) relapsed and/or refractory AML and MDS. Initial data from both Phase 1 studies are anticipated in the first half of 2022.
The first-in-human phase I study in AML and MDS is an accelerated titration design with two parts. Part one is the dose escalation phase that will enroll a single patient per dose (n=1) until certain criteria are met. The trial will convert to a 3+3 design once relevant pharmacokinetics/pharmacodynamics, or PK/PD, safety and or clinical activity are observed. The dose escalation portion will evaluate once daily oral, multiple ascending doses of FHD-286, with a starting dose determined by our GLP toxicology studies. Dose escalation will include patients with relapsed and/or refractory AML or MDS. The second partprimary objective of this study was to assess the safety and tolerability of multiple ascending doses of FHD-286. The secondary objectives of this study included an evaluation of preliminary clinical activity and pharmacokinetics.
In this Phase 1 monotherapy study, the adverse event profile was consistent with a late-line AML population. The most frequently observed grade 3 or greater treatment related adverse events included: increased blood bilirubin, hypocalcemia, differentiation syndrome (“DS”), stomatitis, and increased alanine aminotransferase, or ALT. The study was placed on a full clinical hold by the Food and Drug Administration (the “FDA”) in August of 2022 due to the observation of potential DS and potential linkage to grade 5 safety events. DS is an expansion phase. This phase may include multiple distinct cohorts of patientsassociated with AML informedtherapeutics that induce differentiation of blast cells into normal myeloid cells, an effect that is believed to be on target for the proposed mechanism of FHD-286. In June of 2023, the FDA lifted the full clinical hold. An expert panel was assembled to adjudicate the rate and severity of DS in this study. The adjudicated rate of DS by findings from the dose escalation phase. Initially, biomarkers, suchpanel was determined to be 15 percent (n=6 out of 40 patients) and classified one case as the associationdefinitive DS, five cases as indeterminate and with none contributing to a patient’s death.
The Phase 1 monotherapy study provided an initial evaluation of clinical activity and BRG1efficacy. In the Phase 1 dose escalation study, reductions in both peripheral and bone marrow blast counts, as well as recoveries in absolute neutrophil count, were observed in a subset of heavily pre-treated patients with relapsed and/or refractory AML or MDS, irrespective of mutational status. Across a broad range of patients, differentiation was demonstrated both morphologically as well as through the expression of specific differentiation biomarkers. Patients with evaluable paired bone marrow biopsies (i.e., at screening and during FHD-286 therapy) experienced differentiation as measured by changes in CD11b+ cells and CD34+ cells. Data shown below in Figure 2 demonstrate that in paired bone marrow biopsies across the range of dose levels will be evaluated retrospectively. tested, markers of myeloid
differentiation (CD11b+) increased while markers of leukemic stemness (CD34+) decreased across a range of different mutations.
Figure 2. Paired bone marrow biopsies from the Phase 1 monotherapy study of FHD-286 in relapsed and/or refractory AML/MDS demonstrate differentiation based on increases in CD11b and decreases in CD34.
We performed single cell RNA-Seq of matched patient bone marrow samples on a panel of genes to evaluate changes from screening to on treatment. At screening, bone marrow samples were heavily infiltrated with leukemic stem cell like blasts and the gene signatures aligned accordingly. On FHD-286 treatment, the bone marrow lost the leukemic stem cell phenotype and shifted to a more mature myeloid phenotype.
 Figure 3. Peripheral blood and bone marrow blast reductions and absolute neutrophil count recovery data at the 10 mg and 7.5 mg dose levels in the Phase 1 FHD-286 monotherapy study in patients with relapsed and/or refractory AML/MDS.
Figure 3. Peripheral blood and bone marrow blast reductions and absolute neutrophil count recovery data at the 10 mg and 7.5 mg dose levels in the Phase 1 FHD-286 monotherapy study in patients with relapsed and/or refractory AML/MDS.
 Figure 4. Peripheral blood and bone marrow blast reductions and absolute neutrophil count recovery data at the 5 mg and 2.5 mg dose levels in the Phase 1 FHD-286 monotherapy study in patients with relapsed and/or refractory AML/MDS.
Figure 4. Peripheral blood and bone marrow blast reductions and absolute neutrophil count recovery data at the 5 mg and 2.5 mg dose levels in the Phase 1 FHD-286 monotherapy study in patients with relapsed and/or refractory AML/MDS.Phase 1 Combination Study of FHD-286 with Decitabine or Cytarabine in Patients with Relapsed and/or Refractory AML
We are currently conducting a Phase 1 dose escalation study of FHD-286 in combination with either decitabine or cytarabine in patients with relapsed and/or refractory AML patients. This Phase 1 study uses a 3+3 design which enrolls three patients at each dose level and expands to include additional patients if a dose limiting toxicity is observed. The dose escalation portion is designed to evaluate multiple ascending oral doses of FHD-286 in combination with either decitabine or cytarabine.
The primary objective of this first-in-human study will be theis an evaluation of safety and tolerability, and the identification of the maximum tolerated dose and the recommended Phase 2 dose.dose for combination. The secondary objectives are expected to include an evaluation of preliminary clinical activity and pharmacokinetics. Biomarkers will be evaluated in an exploratory fashion, evaluating markers associated with response. Prospective enrollment based on biomarker findings may be included in
At present there are two arms to this combination study. The first arm investigates the expansion phase of the study.
The first-in-human Phase 1 study in uveal melanoma is an accelerated titration design with two parts. Part one is the dose escalation phase that will enroll a single patient per dose (n=1) until certain criteria are met. The trial will convert to a 3+3 design once relevant PK/PD, safety and or clinical activity are observed. The dose escalation portion will evaluate once daily oral, multiple ascending dosescombination of FHD-286 with decitabine in patients not receiving an azole antifungal classified as a strong CYP3A4 inhibitor. The second arm is testing the combination of FHD-286 with decitabine in the presence of an azole anti-fungal agent classified as a strong CYP3A4 inhibitor. The rationale for having both arms is to determine the impact of azole antifungals commonly administered to patients with AML on FHD-286 exposure given that such agents may inhibit the CYP3A4 through which FHD-286 is metabolized. The starting dose determined by our GLP toxicology studies. Dose escalationfor the weak azole arm is 2.5 mg oral once daily and the starting dose for the strong azole arm is 1.5 mg oral once daily. Both arms will include patients with metastatic uveal melanoma. The second part of the study is an expansion phase, which will be informed by findings from the dose escalation phase.escalate in parallel and are predicted to dose no higher than 7.5 mg oral daily.
The primary objective will be the evaluation of safety and tolerability and identification of the maximum tolerated dose and/or recommended Phase 2 dose. The secondary objectives are expected to include an evaluation of pharmacokinetics and
preliminary clinical activity. Biomarkers will be evaluated in an exploratory fashion, evaluating target engagement as well as markers associated with response. Prospective enrollment based on biomarker findings may be included in the expansion phase of the study.
As shown in the graphic below, these two Phase 1 studies are being conducted in parallel. We intend to explore the potential value of multiple biomarkers to further understand and accelerate drug development. Biomarkers will include assessment of various tumor mutations, as well as expression levels of BRG1 and BRM.various proteins. These biomarkers may be used for future patient selection, measurements of target engagement and biochemical and cellular measures associated with efficacy.
A recent data publication highlighted neuroendocrine prostate cancer as being dependent on BAF. We are evaluating multiple tumor types in the preclinical setting to determine our indication expansion strategy for FHD-286.
FHD-609
Overview
We are currently advancing FHD-609, a highly potent, selective and intravenous, small molecule protein degrader of BRD9, a subunit of a form of the BAF complex. Nearly all synovial sarcoma cancers harbor SS18-SSX mutations. These mutations render the cancer genetically dependent upon BRD9. FHD-609 has two domains: one that binds with high potency and selectivity to BRD9 and the other that binds to a receptor on the E3 ligase complex that directs proteins for destruction. Our preclinical data in synovial sarcoma animal xenograft models demonstrate anti-tumor effects and supported progressing FHD-609 into clinical studies. We are currently conducting a Phase 1 study with FHD-609 for the treatment of synovial sarcoma and anticipate preliminary data as early as the first half of 2022. This multi-center Phase 1 study is primarily assessing the safety and tolerability of FHD-609 in patients with synovial sarcoma. Secondary endpoints include the PK/PD properties of FHD-609 as well as clinical activity. Proof of mechanism will be based on indicators of target engagement in association with FHD-609 treatment. As we further understand the therapeutic potential of FHD-609 in the course of the initial clinical studies, we may pursue additional clinical studies in synovial sarcoma, as a single agent and/or in combination with novel or standard of care agents. In parallel, and as early as Phase 1 expansion studies, we plan to evaluate FHD-609 in other indications, including SMARCB1-deleted cancers.
Synovial Sarcoma Overview
Synovial sarcoma is a cancer of the connective tissue and most commonly originates in the arms or legs. Synovial sarcoma occurs most frequently in adolescents and young adults. There is an incidence over 1,800 new cases of synovial sarcoma in the United States, EU4, UK and Japan. Approximately 30 percent of synovial sarcomas occur in patients under 20 years of age with 84 percent of cases occurring in patients under 50 years of age.
Delay in diagnosis and treatment of synovial sarcoma is common because it is recognized simply by a lump that gradually grows over time. The primary treatment for synovial sarcoma is surgical excision of the tumor and surrounding normal tissue with the goal of sparing the limb if possible. Failure to adequately excise a sufficient area of tissue surrounding the tumor leads to recurrence rates of over 70 percent. Surgical resection is then followed by adjuvant chemotherapy or radiation therapy or both. However, there appears to be minimal benefit of these post-surgical treatments other than for palliative reasons. Radiation and chemotherapy are used in the neoadjuvant setting, or before surgery, to improve the chances of a successful limb sparing surgery.
Approximately ten percent of cases originally present as metastatic disease, and half of all cases eventually develop into metastatic disease. Eighty percent of metastases are localized in the lungs. Five-year survival rates for younger patients with
early-stage disease are approximately 76 percent; however, this decreases to approximately 20 percent in patients over age 30 with advanced disease.
There are no therapies specifically approved by the FDA for synovial sarcoma patients with metastatic disease. Pazopanib, marketed as Votrient® by Novartis has been approved by the FDA for treatment of soft tissue sarcoma in patients who had received prior chemotherapy. In a Phase 3 soft tissue sarcoma trial that included a total of 369 patients, the progression-free survival time for the subset of patients with synovial sarcoma was 4.1 months (N=25) compared to 0.9 months for those who received placebo (N=13). Other chemotherapeutic agents that may be used for palliative purposes include ifosfamide.
Synovial sarcomas are characterized by a chromosomal translocation that results in the fusion of the SS18 gene to one of three genes: SSX1, SSX2 and SSX4, creating SS18-SSX gene fusions. These gene fusions are unique in synovial sarcoma and create a protein not found in healthy patients that fuels the growth and proliferation of the cancer cells. In the scientific literature, the process has been described as the gene fusion “hijacking” the BAF complex, altering its function and causing it to unpack chromatin at wrong locations.
SS18 is a component of the BAF complex. The SS18-SSX fusion protein can also be incorporated into the BAF complex, leading to synovial sarcoma. Genomic screening in synovial sarcoma cells has identified a genetic dependency between synovial sarcoma cells containing SS18-SSX fusions and BRD9, a subunit of the ncBAF complex.
Figure 14. Synovial sarcoma cell lines were highly dependent on BRD9.
Our Solution: FHD-609
FHD-609 is a highly potent, selective and intravenous, small molecule protein degrader of BRD9. Unlike many traditional intracellular drug targets, BRD9 is not an enzyme and does not exhibit enzymatic activity. We therefore designed FHD-609 as a protein degrader, a molecule with two binding domains connected by a linker that drives the removal of targeted proteins by the cell’s protein degradation system. In cells, these protein degrader molecules bring their target into proximity of the E3 ligase which marks these target proteins for destruction by the cell’s proteasome system.
One domain of FHD-609 is a potent and selective binder of BRD9. This is chemically linked to a domain that binds to a receptor on the E3 ligase complex. FHD-609 led to the specific degradation of BRD9 in multiple synovial sarcoma tumor cell lines, with a DC50 of less than 1 (one) nM. This resulted in the elimination of detectable BRD9 protein and the concomitant inhibition of proliferation of these synovial sarcoma cell lines.
Our Preclinical Data for Synovial Sarcoma
We have generated in vivo proof of concept data that demonstrated antitumor activity of FHD-609 in synovial sarcoma CDX models. In the synovial sarcoma SYO1 CDX model containing the SS18-SSX2 mutation, dosing with FHD-609 led to potent inhibition of tumor growth. Intraperitoneal doses of FHD-609 yielded similar antitumor activity whether dosing was delivered as a once-weekly (every 7 days for three weeks) or an equivalent drug amount delivered daily over seven days for three weeks
(3.5 mg/kg delivered every week versus 0.5 mg/kg delivered daily over 7 days). This suggests that sustained tumor regression can occur with a less frequent dosing regimen, which will be explored in clinical development. In the model, tumor growth inhibition levels were associated with levels of BRD9 degradation as indicated below.
SYO-1 Synovial Sarcoma CDX Model
Figure 15. FHD-609 led to dose-dependent tumor growth inhibition of synovial sarcoma tumors equivalently at a once weekly or daily treatment schedule. On the right, the western blot shows dose-dependent BRD9 degradation correlating with the anti-tumor activity.
In the synovial sarcoma ASKA CDX model containing the SS18-SSX1 mutation, the antitumor activity of FHD-609 was comparable and superior to that observed for other systemic therapeutic agents. In this model FHD-609 was dosed intravenously twice per week, ifosfamide as a monotherapy intravenously on days one through three every three weeks, and pazopanib orally once daily. FHD-609 led to robust tumor suppression, with meaningful suppression observed through 40 days at the highest studied dose of 2 mg/kg.
ASKA Synovial Sarcoma CDX Model
Figure 16. FHD-609 resulted in tumor regression in the ASKA synovial sarcoma xenograft model. FHD-609 demonstrated significant tumor growth inhibition compared to either ifosfamide or pazopanib.
Importantly, after discontinuation of FHD-609, treatment with FHD-609 was associated with sustained tumor growth inhibition. Following discontinuance of FHD-609 treatment at 2 mg/kg, at approximately day 21 tumor regrowth was not detectable for at least another 15 days. We believe these results support the targeted degradation of BRD9 and its importance in synovial sarcoma.
ASKA Synovial Sarcoma CDX Model
Figure 17. FHD-609 treatment was associated with sustained tumor suppression after treatment withdrawal.
Clinical Plans for FHD-609 in Synovial Sarcoma
The first-in-human study in synovial sarcoma will include a standard dose escalation and expansion phase. The dose escalation portion is a Phase 1 design with a starting dose determined by the GLP toxicology studies. Dose escalation may include treatment-naïve or treatment-experienced patients with metastatic synovial sarcoma. The expansion phase may include multiple distinct cohorts of synovial sarcoma patients, informed by findings from the dose escalation phase. Initially, biomarkers such as the association of clinical activity and SS18-SSX mutational status will be evaluated retrospectively.
The primary endpoints of this first-in-human study will be safety, the identification of any dose-limiting toxicities, the maximum tolerated dose, the recommended Phase 2 dose, and the evaluation of pharmacokinetics and pharmacodynamics. The secondary endpoints are expected to include an evaluation of clinical activity: overall response rate, duration of response and additional time to event analyses. Biomarkers will be evaluated in an exploratory fashion, evaluating target engagement as well as markers associated with response and/or resistance. Prospective enrollment based on biomarker findings may be included in later studies.
Potential Areas for Expansion and Other Ongoing Exploratory Activities for FHD-286
Potential Immunomodulatory Applications
Our study of FHD-286 in metastatic uveal melanoma (“mUM”) along with data from syngeneic mouse models in several tumor types, have shown an impact of FHD-286 on specific immune cells in the expansion phasetumor microenvironment and synergism with anti-PD-1 antibodies, respectively. Specifically, in patients with mUM treated with FHD-286, we observed a log fold decrease in T-regulatory cells, a reduction of the study. As we further understand the therapeutic potentialpresence of FHD-609macrophages with an M2 phenotype (i.e., immunosuppressive tumor associated macrophages), and a reduction in PD-1 expression on CD4 and CD8 T cells in the coursetumor microenvironment. Based on these data, there may be potential applications of the initial clinical studies, we may pursue additional clinical studies in synovial sarcoma, as a single agent and/orFHD-286 in combination with novel immuno-oncology agents, such as immune checkpoint inhibitors, in certain tumors.
Potential Cancer Resistance Applications
Based on published research as well as work performed at Foghorn, we are exploring the potential of FHD-286 to combine with various tyrosine kinase inhibitors (“TKIs”) to (i) delay resistance to the TKI and/or standard(ii) overcome resistance in the setting of care agents. In parallel,prior exposure to a TKI. Pre-clinical work in both an in vitro and as early as Phase 1in vivo context is ongoing.
Other Potential Indications
We have evaluated multiple tumor types in the preclinical setting to inform our indication expansion studies, we plan to evaluate FHD-609 in other indications, including SMARCB1-deleted cancers.
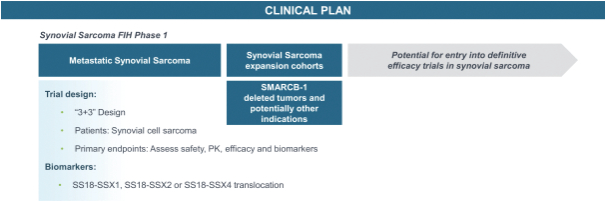
BRM-Selective Modulators
Overview
Broad cancer sequencing initiatives have shown that BRG1 is one of the most highly mutated subunits of the BAF complex. BRG1 was found to be mutated in approximately five percent of tumors sequenced as part of the Memorial Sloan Kettering Cancer Center MSK-IMPACT study, and in up to ten percent of NSCLCNon-Small Cell Lung Cancer (“NSCLC”) tumors. Beyond NSCLC, the MSK-IMPACT study highlighted BRG1 mutations in over thirty different types of tumors. In many cases, these mutations lead to a loss of enzymatic
activity in the BRG1 subunit, creating a genetically determined dependency on BRM. This loss of BRG1 and subsequent dependency on BRM leads to a drugging opportunity. We are currently developing selective modulators of BRM to target this genetic dependency in BRG1 mutated cancers. In December 2021, we entered into a strategic collaboration with Loxo Oncology at Lilly a research and development group of Eli Lilly and Company, to create novel oncology medicines. The Loxo OncologyLilly collaboration includes a co-development and co-commercialization agreement for the selective BRM oncology program.
Chromatin Regulatory System: An Untapped Opportunity for Therapeutic Intervention In February 2024, Lilly declared FHD-909, a first-in-class BRG1 inhibitor, a development candidate pursuant to the Lilly Collaboration Agreement and is targeting an IND filing in the second quarter of 2024.
12 Tumor Types with Highest Prevalence of BRG-1 Mutations
Figure 18.5. The above chart highlights the cancers with the highest prevalence of BRG1 mutations from the MSK-IMPACT study.
Non-Small Cell Lung Cancer (NSCLC) Overview
Lung cancer is the leading cause of cancer-related death, accounting for approximately 18 percent of all cancer deaths globally, or an estimated 1.8 million deaths per year. There are an estimated 228,000238,000 new cases of lung cancer diagnosed and 135,000127,000 deaths in the United States annually. NSCLC accounts for 80 to 85 percent of lung cancer cases. Genetic profiling of tumors has identified a number of genes that are altered in NSCLC. The standard of care for NSCLC has included conventional chemotherapy with or without a checkpoint inhibitor. Targeted therapies developed for the proteins encoded by some of these genes such as the epidermal growth factor receptor or EGFR,(“EGFR”) and anaplastic lymphoma kinase gene or ALK, have been approved and(“ALK”) are now part of the standard of care of patentsfor patients with NSCLC.NSCLC harboring such actionable mutations. However, less than 30 percent of NSCLC patients have alterations in these two genes. Up to two thirds of NSCLC patients who are ineligible for or resistant to treatment with EGFR or ALK targeted therapies have tumors that express PD-L1 and are candidates for checkpoint inhibitor therapies, which lead to significant improvements in progression free survival and overall survival compared to standard chemotherapy. Despite the availability of both targeted and conventional therapies, the prognosis in NSCLC remains poor, with an overall relative five-year survival for all patients diagnosed with NSCLC of 1928 percent.
An analysis of genomic data in NSCLC cancer patients, collected as part of MSK-IMPACT, revealed that gene alterations in BRG1 were found in ten percent of NSCLC samples. In a retrospective analysis conducted by MSKCC it was observed that among patients with BRG1-deficient NSCLC who received first-line platinum doublet chemotherapy or chemotherapy plus immunotherapy, median progression-free survival was 38 days and 35 days, respectively. Prognosis is poor in patients with BRG1-deficient NSCLC, highlighting the importance of developing novel therapeutics that address this unmet need.
MSK-IMPACT: BRG-1 Mutated in 10% of NSCLC
Figure 19.6. BRG1 gene alterations are found in 10 percent of NSCLC tumors and have minimal overlap with other actionable mutations present in NSCLC, such as EGFR and ALK.
Genomic screening of over 400 cancer cell lines that remove BRM via CRISPR revealed a genetic dependency of certain BRG1-mutated cancers on BRM. This finding suggests that selective inhibition or selective degradation of BRM has the potential to be therapeutically meaningful in certain cancers with BRG1 mutations.
Figure 20.7. In a screen of over 400 cancer cell lines, inactivation of the BRM gene resulted in selective inhibition of cell lines containing mutations in BRG1.
Our Solution: Selective BRM Modulators
With our collaboration partner, Loxo Oncology at Lilly, we are advancing two classes of molecules, an enzymatic inhibitor and a protein degrader, as selective modulators of BRM.
One class consists of selective, allosteric inhibitors of the ATPase activity of BRM. We are designing these inhibitors to be more selective for BRM than the very similar ATPase BRG1. Through our proprietary methods of isolating and screening BAF complexes that contain either BRG1 or BRM,gene control platform, we have identified and optimized highly selective small molecule inhibitors that are over 10 times more selective for BRM than BRG1. We have shown that this selectivity was also observed in cellular assays. Pharmacokinetic profiles of these molecules have been consistent with the ability to potently inhibit BRM while having minimal inhibitory activity against BRG1.targeting BRM.
| | | | | | | | |
Enzyme assay using BRG1 and BRM subunits | Enzyme assay using BRG1 and BRM containing full BAF complexes | Cellular assays for BRG1 and BRM |
| | |
Figure 21. This panel showed biochemical (percent of ATPase activity) and cellular selectivity of a 20X selective inhibitor of BRG1 versus BRM.
Figure 22.8. This panel showed BRM enzymatic inhibitor in vivo efficacy in a A549-BRG1 Mutant NSCLC Model with corresponding body weight and plasma exposure versus the vehicle control and Cisplatin.
Our other approach to selective BRM modulation consists of protein degrader molecules that activate the cell’s proteinubiquitin proteasome degradation system to selectively destroy BRM. One domain of the BRM degrader molecule is a potent and selective binder of BRM. This is chemically linked to a domain that binds to a receptor on the E3 ligase complex. In cells, these protein degrader molecules bring their target into proximity of the E3 ligase which marks these target proteins for destruction by the cell’s proteinubiquitin proteasome degradation system. We have shown that it is possible to identify protein degraders that lead to the destruction of BRM while leaving BRG1 untouched.
Selective Degradation of BRM
Figure 23.9. Selective BRM degrading molecules led to the degradation of over 75 percent of BRM while leaving the levels of BRG1 virtually unchanged.
ARID1B Selective ModulatorsCBP Degrader for EP300 Mutated Cancers
CREB binding protein serves as a critical co-activator for transcription factors involved in signaling pathways in a subset of cancers including bladder, colorectal, breast, gastric and Other Opportunitieslung.
Figure 10. In a screen of over 400 cancer cell lines, inactivation of the EP300 gene resulted in selective growth inhibition of cell lines containing mutations in CBP, establishing the dependency on EP300 in these cell lines.
CBP and EP300 are chromatin regulators and histone acetyltransferases and are highly homologous with similar domain structure and architecture. Functional genomics screens have shown that CBP and EP300 share a bi-directional synthetic lethal relationship. As a result, loss of function of one of these proteins leads to dependency on the other. Data suggest that there are potentially over 100,000 patients with EP300 mutations that could benefit from a therapy selectively targeting CBP.
We are developing selective CBP degraders and plan to exploit the bi-directional synthetic lethal relationship it shares with its paralog acetyltransferase, EP300, to identify and treat those patients with EP300 mutated cancers. We believe selectively targeting and degrading CBP will potentially offer a tolerability advantage compared with non-selectively degrading both targets.
We have tested in vitro a selective degrader of CBP across multiple cancers including gastric, colorectal, and bladder, which has demonstrated significant responses in cell proliferation assays as shown in the Chromatin Regulatory Systemfigure below. As demonstrated in Figure 11 below, we have developed highly selective degraders of CBP that rapidly and durably suppress the CBP target with no degradation observed for the counter target EP300.
Figure 11. A selective degrader of CBP tested across multiple cancers has demonstrated CBP-dependent cell killing in cell proliferation assays.
With more advanced degraders of CBP, we have generated data in several cell derived xenograft (“CDX”) mouse models which include gastric, colorectal, and bladder models. As seen in Figure 13 below, the degrader denoted as FHT-CBPd-9 appears well-tolerated based on the limited mouse body weight percentage changes and achieves tumor growth inhibition in the bladder model and tumor regression in the gastric model. FHT-CBPd-8, a slightly earlier version of the CBP degrader, achieves tumor growth inhibition in a colorectal model.
Potential subsets of tumor types that harbor a mutation in EP300 and therefore would be reliant on CBP for their survival include but may not be limited to bladder cancer, melanoma, endometrial, gastric, breast, NSCLC, colorectal, and pancreatic.
Figure 12. A selective degrader of CBP tested in a CDX model of gastric cancer demonstrates regression and in CDX models of colorectal and bladder cancer demonstrates tumor growth inhibition.
Historically, targeting CBP and EP300 has been attempted with dual inhibitors – therapeutics that simultaneously inhibit both the function of CBP and EP300. It has been reported in the literature that these compounds in both pre-clinical as well as the clinical setting cause thrombocytopenia, low counts of platelet cells that are important in the clotting of blood.
We have demonstrated that selective degradation of either CBP alone or EP300 alone in animal models does not cause thrombocytopenia as shown in Figure 13 below. In the figure, we show that a dual bromodomain inhibitor which inhibits both CBP and EP300 causes a meaningful drop in platelets. In contrast, our degraders of EP300 and CBP, FHT-EP300d and FHT-CBPd respectively, do not cause a drop in platelets at doses that are relevant and achieve efficacy in the animal models shown in Figure 12 and Figure 16.
Figure 13. Selective degraders of CBP and EP300 demonstrate that they do not reduce platelet counts as compared to a dual inhibitor of both CBP and EP300.
Selective EP300 Degrader for EP300 Dependent Cancers and CBP Mutated Cancers
We are developing a selective EP300 degrader targeting EP300 dependent cancers and CBP mutant cancers. The Selective EP300 program has potential in various cancers which include androgen receptor, or AR, positive prostate cancer, bladder cancer, NSCLC, various lymphomas and leukemias and could provide a new therapeutic option for potentially more than 100,000 patients a year.
Figure 14. In a screen of over 400 cancer cell lines, inactivation of the CBP gene resulted in selective growth inhibition of cell lines containing mutations in EP300, establishing the dependency on CBP in these cell lines.
We have tested in vitro a selective degrader of EP300 in a number of cell lines which has demonstrated degradation and significant responses in cell proliferation assays as shown in the figure below:
Figure 15. A selective degrader of EP300 tested in a number of cell lines has demonstrated degradation and EP300-dependent cell killing in cell proliferation assays.
With more advanced degraders of EP300, we have generated data in several CDX mouse models which include an AR positive prostate cancer and diffuse large b-cell lymphoma (“DLBCL”). As seen in Figure 16 below, the degrader denoted as FHT-EP300d appears well-tolerated based on the limited mouse body weight percentage changes and achieves tumor growth
inhibition in the prostate and DLBCL models. In the AR+ prostate model, FHD-EP300d achieves better tumor growth inhibition than enzalutamide, an androgen receptor inhibitor that is presently used to treat patients with prostate cancer.
Figure 16. A selective degrader of EP300 tested in a CDX model of AR+ Prostate Cancer and in a CDX model of DLBCL demonstrates tumor growth inhibition.
Selective ARID1B Degrader for ARID1A Mutated Cancers
The ARID1A subunit is the most mutated subunit within the BAF complex. Mutations in ARID1A confer a dependency on the ARID1B subunit of the BAF complex. ARID1A mutations are implicated in ovarian, endometrial, colorectal, bladder, and gastric cancers. Data suggest that there potentially are over 175,000 patients with ARID1A mutations that would potentiallycould benefit from a therapy selectively targeting ARID1B.
Figure 24.17. In a screen of over 400 cancer cell lines, inactivation of the ARID1B gene resulted in selective growth inhibition of cell lines containing mutations in ARID1A, establishing the dependency on ARID1B in these cell lines.
Since ARID1B is not an enzyme, our strategy is to selectively degrade ARID1B. Our platform allows us to generate full BAF complexes containing only ARID1A or ARID1B. Using our platform, we have conducted high throughput screens and have discoveredvalidated selective binderschemical matter to the ARID1B protein that we are seeking to optimize as protein degrader product candidates.
Targeting Transcription Factors: Disrupting Transcription Factor Binding to Chromatin Remodeling Complexes
Transcription factors work in concert with chromatin remodeling complexes, BAF as one example, to orchestrate gene expression. In tumor cells, genes encoding transcription factors are often amplified, deleted, rearranged via chromosomal translocation or subjected to point mutations that result in a gain or loss of function. We have developed a set of tools to visualize and study the interactions between transcription factors and chromatin remodeling complexes. To our knowledge, we are the only company with these capabilities.
We are using these capabilities to drive our drug discovery efforts across multiple transcription factor programs for a variety of cancer indications. Our strategy is to disrupt the interaction between transcription factors and chromatin remodeling complexes.
Our initial focus is on disrupting transcription factor interactions with the BAF complex. We believe that there are over 100 transcription factors in oncology that would be amenable to this new approach. Based on these insights, we are developing small molecule disruptors that block the interaction between transcription factors and the BAF complex. In addition to applications in cancer, we believe that such disruptors could be applied in other therapeutic areas.
Our approach to disrupting the interactions between transcription factors and the BAF complex is the basis of a collaboration signed with Merck in July 2020. In this collaboration, we intend to apply our Gene Traffic Control platform to identify disruptors of a single predetermined transcription factor. As part of the collaboration, we received an upfront payment of $15.0 million, and are also eligible to receive up to $245.0 million upon achievement of specified research, development and regulatory milestones by any product candidate generated by the collaboration, and up to $165.0 million upon achievement of specified sales-based milestones.
A prototypical example of a chromatin remodeling complex–transcription factor interaction is exemplified by the ERG transcription factor and BAF. In approximately half of all prostate cancers, the gene encoding ERG is fused to the TMPRRS2 promotor, resulting in the overexpression of ERG and the upregulation of a broad set of additional genes. Furthermore, genetic suppression of ERG expression in cells containing the TMPRRS2-ERG gene fusion has been shown to inhibit cell proliferation. These results support our approach that disrupting the interaction of over expressed transcription factors, such as ERG, with a chromatin remodeling complex has the potential to be therapeutically beneficial to patients.
Similar to ERG, the ability of individual transcription factors to interact with the BAF complex has previously been reported in the literature, but to our knowledge, there have not previously been systematic studies quantifying and describing these binding interactions. We used our Gene Traffic Control platform to produce and purify BAF complexes and multiple transcription factors to study the structural details as well as the biochemical and biophysical properties of their interactions.
We observed that different transcription factors bind to different sites on the surface of the BAF complex. This suggests that there is specificity in these interactions. Therefore, it may be possible to block the interaction of a specific transcription factor with the BAF complex without blocking the interactions of other transcription factors.
Figure 25.19. Illustrative locations of the binding sites of multiple transcription factors to the BAF complex.
We also observed that the binding affinities that describe how tightly transcription factors bind to the BAF complex were roughly comparable to those observed for other protein-protein interactions for which small molecule disruptors have been developed. We found that the interactions between multiple transcription factors and the BAF complex had a Kd, a measure of binding affinity, in the range of 20 to 350 nM.
Figure 26. Interactions between transcription factors and the BAF complex had Kds in the range of 20 to 350 nM. Smaller numbers reflected higher affinity binding.
Using the insights of where and how tightly transcription factors bind, we have developed as part of our Gene Traffic Control platform the ability to conduct high throughput screens on chromatin remodeling complex–transcription factor interactions. We have already validated eightnumerous BAF-transcription factor interactions for targets of interest in various cancers. We are applying our know-how to screen several of theseselect BAF-transcription factor interactions to discover and develop transcription factor disruptors. We intend to use our platform to validate and drug additional transcription factors that interact with BAF and other chromatin remodeling complexes both in oncology and other therapeutic areas.
Competition
The biotechnology and pharmaceutical industries are characterized by the rapid evolution of technologies and understanding of disease etiology, intense competition and a strong emphasis on intellectual property. We believe that our approach, strategy, scientific capabilities, know-how and experience provide us with competitive advantages, including, to our knowledge, our being the only company with the ability to study the chromatin regulatory system at scale, in context, and in an integrated way. However, we expect substantial competition from multiple sources, including major pharmaceutical, specialty pharmaceutical, and existing or emerging biotechnology companies, academic research institutions and governmental agencies and public and private research institutions worldwide. Many of our competitors, either alone or through collaborations, have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These companies may be or may become interested in the chromatin regulatory system and rapidly develop programs that may compete with ours by studying the chromatin regulatory system at scale, in context and in an integrated way. Even if they do not advance programs with the same mechanism of action as ours, these companies could develop products or product candidates that are competitive with ours or that have a superior product profile and may do so at a rapid pace. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient enrollment in clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs. As a result, our competitors may discover, develop, license or commercialize products before or more successfully than we do.
We face competition from segments of the pharmaceutical, biotechnology and other related markets that pursue the development of therapies that target broad genetic expression mechanisms, including the chromatin regulatory system. In addition, we may face competition from companies developing product candidates that utilize protein degradation approaches,
including Arvinas, Inc., Kymera Therapeutics, Inc., Nurix Therapeutics, Inc., C4 Therapeutics, Inc., and VividionC4 Therapeutics, Inc. Further, several large pharmaceutical companies have disclosed preclinical investments in this field. Our competitors will also include companies that are or will be developing other targeted therapies, including small molecule, antibody, or protein degraders for the same indications that we are targeting.targeting including Prelude Therapeutics Incorporated, Plexium, Inc., Amgen Inc., Abbvie Inc., Genentech, Inc., and SK Life Science, Inc.
We could see a reduction or elimination in our commercial opportunity if our competitors develop and commercialize drugs that are safer, more effective, have fewer or less severe side effects, are more convenient to administer, are less expensive or with more favorable labeling than our product candidates, regardless of whether they target the chromatin regulatory system as a mechanism of action. Our competitors also may obtain FDA or other regulatory approval for their drugs more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market. The key competitive factors affecting the success of all of our product candidates, if approved, are likely to be their efficacy, safety, convenience, price, the level of generic competition and the availability of reimbursement from government and other third-party payors.
Intellectual Property
We seek to protect the intellectual property and proprietary technology that we consider important to our business, including by pursuing patent applications that cover our product candidates and methods of using the same, as well as other relevant inventions and improvements that we believe to be commercially important to the development of our business. We also rely on trade secrets, know-how and continuing technological innovation to develop and maintain our proprietary and intellectual property position. Our commercial success depends, in part, on our ability to obtain, maintain, enforce and protect our intellectual property and other proprietary rights for the technology, inventions and improvements we consider important to our business, and to defend any patents we may own or in-license in the future, prevent others from infringing any patents we may own or in-license in the future, preserve the confidentiality of our trade secrets, and operate without infringing, misappropriating or otherwise violating the valid and enforceable patents and proprietary rights of third parties.
As with other biotechnology and pharmaceutical companies, our ability to maintain and solidify our proprietary and intellectual property position for our product candidates and technologies will depend on our success in obtaining effective patent claims
and enforcing those claims if granted. However, our pending provisional and PCTPatent Cooperation Treaty (“PCT”) patent applications, and any patent applications that we may in the future file or license from third parties, may not result in the issuance of patents and any issued patents we may obtain do not guarantee us the right to practice our technology or commercialize our product candidates. We also cannot predict the breadth of claims that may be allowed or enforced in any patents we may own or in-license in the future. Any issued patents that we may own or in-license in the future may be challenged, invalidated, circumvented or have the scope of their claims narrowed. In addition, because of the extensive time required for clinical development and regulatory review of a product candidate we may develop, it is possible that, before any of our product candidates can be commercialized, any related patent may expire or remain in force for only a short period following commercialization, thereby limiting the protection such patent would afford the respective product and any competitive advantage such patent may provide.
The term of individual patents depends upon the date of filing of the patent application, the date of patent issuance and the legal term of patents in the countries in which they are obtained. In most countries, including the United States, the patent term is 20 years from the earliest filing date of a non-provisional patent application. In the United States, a patent’s term may be lengthened by patent term adjustment, which compensates a patentee for administrative delays by the U.S. Patent and Trademark Office or USPTO,(the “USPTO”), in examining and granting a patent, or may be shortened if a patent is terminally disclaimed over an earlier filed patent. The term of a patent claiming a new drug product may also be eligible for a limited patent term extension when FDA approval is granted, provided statutory and regulatory requirements are met. The restoration period granted on a patent covering a product is typically one-half the time between the effective date of a clinical investigation involving human beings is begun and the submission date of an application, plus the time between the submission date of an application and the ultimate approval date. The restoration period cannot be longer than five years and the total patent term, including the restoration period, must not exceed 14 years following FDA approval. Only one patent applicable to an approved product is eligible for the extension, and only those claims covering the approved product, a method for using it, or a method for manufacturing it may be extended. Additionally, the application for the extension must be submitted prior to the expiration of the patent in question. A patent that covers multiple products for which approval is sought can only be extended in connection with one of the approvals. The USPTO reviews and approves the application for any patent term extension or restoration in consultation with the FDA. In the future, if our product candidates receive approval by the FDA, we expect to apply for patent term extensions on any issued patents covering those products, depending upon the length of the clinical studies for each product and other factors. There can be no assurance that patents will issue from our current or future pending patent applications, or that we will benefit from any patent term extension or favorable adjustments to the terms of any patents we may own or in-license in the future. In addition, the actual protection afforded by a patent varies on a product-by-product basis, from country-to-country, and depends upon many factors, including the type of patent, the scope of its coverage, the
availability of regulatory-related extensions, the availability of legal remedies in a particular country and the validity and enforceability of the patent. Patent term may be inadequate to protect our competitive position on our products for an adequate amount of time.
As of March 1, 2022,2024, we owned sixmore than 10 pending U.S. provisional patent applications, 20more than 25 pending U.S. non-provisional patent applications, 14more than 10 pending Patent Cooperation Treaty, or PCT applications, and 28more than 100 pending ex-U.S. patent applications. We currently do not own or in-license any issued patents with respect to any of our product candidates, including FHD-286, and FHD-609, or our platform technology, and our intellectual property portfolio is in its very early stages.technology.
FHD-286
As of March 1, 2022,2024, we owned two U.S. patents, seven pending U.S. provisional patent applications, fiveten pending U.S. non-provisional patent applications two pendingand PCT patent applications, and 14more than 25 pending ex-U.S. patent applications that relate to FHD-286, including its composition and various methods of use. Any U.S. or ex-U.S. patent that may issue from these patent applications would be scheduled to expire between 2039-2042,2039-2044, excluding any additional term for patent term adjustment or patent term extension, if applicable.
FHD-609
As of March 1, 2022, we owned three pending U.S. provisional patent applications, three pending U.S. non-provisional patent applications, one pending PCT patent application, and 11 pending ex-U.S. patent applications that relate to FHD-609, including its composition and various methods of use. Any U.S. or ex-U.S. patent that may issue from a non-provisional patent application claiming priority to these applications would be scheduled to expire between 2039-2042, excluding any additional term for patent term adjustment or patent term extension, if applicable.
Prosecution of most of our PCT patent applications and our provisional patent applications has not commenced and will not commence unless and until they are timely converted into U.S. non-provisional or national stage applications. Prosecution is a lengthy process, during which the scope of the claims initially submitted for examination by the USPTO or other foreign jurisdiction are often significantly narrowed by the time they issue, if they issue at all. Any of our pending PCT patent
applications are not eligible to become issued patents until, among other things, we file national stage patent applications within 30 months in the countries in which we seek patent protection. If we do not timely file any national stage patent applications, we may lose our priority date with respect to our PCT patent applications and any patent protection on the inventions disclosed in such PCT patent applications. Our provisional patent applications may never result in issued patents and are not eligible to become issued patents until, among other things, we file a non-provisional patent application within 12 months of filing the related provisional patent application. If we do not timely file non-provisional patent applications, we may lose our priority date with respect to our provisional patent applications and any patent protection on the inventions disclosed in our provisional patent applications. While we intend to timely file non-provisional and national stage patent applications relating to our provisional and PCT patent applications, we cannot predict whether any of our current or future patent applications related to FHD-286, FHD-609, or any of our other product candidates, will issue as patents. If we do not successfully obtain patent protection, or, even if we do obtain patent protection, if the scope of the patent protection we obtain with respect to FHD-286, FHD-609, or our other product candidates or technology is not sufficiently broad, we will be unable to prevent others from using our technology or from developing or commercializing technology and products similar or identical to ours or other competing products and technologies.
In addition to patent applications, we rely on unpatented trade secrets, know-how and continuing technological innovation to develop and maintain our competitive position. However, trade secrets and confidential know-how are difficult to protect. In particular, we consider various aspects of our Gene Traffic Control platform to constitute our trade secrets and know-how. We seek to protect our proprietary information, in part, by executing confidentiality agreements with our collaborators and scientific advisors and non-competition, non-solicitation, confidentiality and invention assignment agreements with our employees and consultants. We cannot guarantee that we will have executed such agreements with all applicable employees and contractors, or that these agreements will afford us adequate protection of our intellectual property and proprietary information rights. In addition, our trade secrets and/or confidential know-how may become known or be independently developed by a third party or misused by any person to whom we disclose such information. These agreements may also be breached, and we may not have an adequate remedy for any such breach. Despite any measures taken to protect our intellectual property, unauthorized parties may attempt to copy aspects of our products or to obtain or use information that we regard as proprietary. Although we take steps to protect our proprietary information, third parties may independently develop the same or similar proprietary information or may otherwise gain access to our proprietary information. As a result, we may be unable to meaningfully protect our trade secrets and proprietary information. For more information regarding the risks related to our intellectual property, please see “Risk Factors—Risks Related to our Intellectual Property.”
License Agreement with Merck
In July 2020, we entered into a Research Collaboration and Exclusive License Agreement, or the Merck Collaboration Agreement, with Merck Sharp & Dohme Corp., or Merck, to apply our Gene Traffic Control platform to discover and develop novel therapeutics based on disruptors of a specified transcription factor target.
Under the terms of the Merck Collaboration Agreement, we are responsible for certain preclinical research activities under a mutually agreed research plan, such as the use of our high throughput screening and compound optimization technology to identify and validate disruptors directed to this transcription factor target, up until our delivery to Merck of a hit package that identifies validated disruptors directed to the transcription factor target. Merck will be responsible for further preclinical research under the Merck Collaboration Agreement and for the clinical development and commercialization of therapeutics arising from the agreement. Merck will have a limited right to substitute the transcription factor target that is the subject of the collaboration for other transcription factors.
Under the terms of the Merck Collaboration Agreement, we have granted Merck an exclusive, worldwide, sublicensable license under certain patent rights and know-how to make, have made, use, import, offer to sell and sell therapeutics arising from the collaboration that disrupt the specified transcription factor target.
We have received an upfront payment of $15.0 million from Merck, and are eligible to receive up to $245.0 million upon achievement of specified research, development and regulatory milestones by any product candidate generated by the collaboration, and up to $165.0 million upon achievement of specified sales-based milestones per approved product from the collaboration, if any. We will be eligible to receive tiered royalties, calculated on a product-by-product basis, on net sales of approved products from the collaboration, if any, at royalty rates ranging from the mid-single digits to low tens, depending on whether the products are covered by patent rights we license to Merck.
The Merck Collaboration Agreement will expire upon expiration of all of Merck’s royalty obligations under the agreement. Merck may terminate the Merck Collaboration Agreement for convenience, and either party may terminate the Merck Collaboration Agreement in the event of the other party’s uncured material breach or such party’s bankruptcy or insolvency. If
Merck terminates the Merck Collaboration Agreement as a result of our breach, the licenses and other rights granted to Merck under the agreement will remain in effect and become perpetual. If the term of the agreement expires, then such licenses and other rights will become fully paid-up.
Strategic Collaboration with Lilly
On December 10, 2021, we entered into a strategic collaboration with Loxo Oncology at Lilly. Under the terms of the collaboration agreement,Lilly Collaboration Agreement, the parties will seek to leverage our platform technology to research, discover and develop therapeutic molecules directed to the selective BRM target and an additional undisclosed oncology target, and to three additional discovery programs. Lilly will pursue the clinical development, manufacture and commercialization of products derived from or containing certain compounds developed and Foghorn will have the right to participate in the development and commercialization of these products for the U.S. market.
Under the collaboration agreement,Lilly Collaboration Agreement, Lilly made an upfront payment of $300$300.0 million, and a concurrent $80$80.0 million equity investment in Foghorn. We are eligible to receive a share of U.S. profits for co-commercialized products. Lilly and Foghorn will share 50/50 in the U.S. economics for products directed to the BRM-selective program and one other undisclosed target. For the three Discovery Programs, Foghorn will have an option to participate in a percentage of the U.S. economics following the successful completion of dose-finding toxicity studies. For these programs, Foghorn is eligible to receive development and commercialization milestones of up to an aggregate of approximately $1.3 billion if Foghorn does not exercise its option to participate in the U.S. economics for any discovery program. In addition, Lilly will pay the Company tiered royalties on product sales on a country-by-country and product-by-product basis (1) at royalty rates ranging from low-double digits to the twenties on ex-U.S. sales for products directed to the BRM-selective program and one other undisclosed target and (2) at royalty rates ranging from mid-single digits to low-double digits on sales outside the U.S. for products directed to the Discovery Programs, during the applicable royalty term and subject to certain royalty step-down provisions.
Manufacturing
We do not have any manufacturing facilities or personnel. We currently rely, and expect to continue to rely, on third parties for the manufacture of our product candidates undergoing preclinical testing, as well as for clinical testing and commercial manufacture if our product candidates receive marketing approval.
All of our drug candidates are small molecules and are manufactured in synthetic processes from available starting materials. The chemistry appears amenable to scale up and does not currently require unusual equipment in the manufacturing process. We expect to continue to develop product candidates that can be produced cost-effectively at contract manufacturing facilities.
We generally expect to rely on third parties for the manufacture of companion diagnostics for our products, which are assays or tests to identify an appropriate patient population. Depending on the technology solutions we choose, we may rely on multiple third parties to manufacture and sell a single test.
Commercialization
Subject to receiving marketing approvals, we expect to commence commercialization activities by building a focused sales and marketing organization in the United States to sell our products. We believe that such an organization will be able to address the community of oncologists who are the key specialists in treating the patient populations for which our product candidates are being developed. Outside the United States, we expect to enter into distribution and other marketing arrangements with third parties for any of our product candidates that obtain marketing approval.
We also plan to build a marketing and sales management organization to create and implement marketing strategies for any products that we market through our own sales organization and to oversee and support our sales force. The responsibilities of the marketing organization would include developing educational initiatives with respect to approved products and establishing relationships with researchers and practitioners in relevant fields of medicine.
Government Regulation
The FDA and other regulatory authorities at federal, state and local levels, as well as in ex-United States countries, extensively regulate, among other things, the research, development, testing, manufacture, quality control, import, export, safety, effectiveness, labeling, packaging, storage, distribution, recordkeeping, approval, advertising, promotion, marketing, post-approval monitoring and post-approval reporting of drugs. We, along with our vendors, contract research organizations and contract manufacturers, will be required to navigate the various preclinical, clinical, manufacturing and commercial approval
requirements of the governing regulatory agencies of the countries in which we wish to conduct studies or seek approval of our product candidates. The process of obtaining regulatory approvals of drugs and ensuring subsequent compliance with appropriate federal, state, local and ex-United States statutes and regulations requires the expenditure of substantial time and financial resources.
In the United States, where we are initially focusing our drug development, the FDA regulates drug products under the Federal Food, Drug, and Cosmetic Act or FD(the “FD&C Act,Act”) as amended, its implementing regulations and other laws. If we fail to comply with applicable FDA or other requirements at any time with respect to product development, clinical testing, approval or any other legal requirements relating to product manufacture, processing, handling, storage, quality control, safety, marketing, advertising, promotion, packaging, labeling, export, import, distribution, or sale, we may become subject to administrative or judicial sanctions or other legal consequences. These sanctions or consequences could include, among other things, the FDA’s refusal to approve pending applications, issuance of clinical holds for ongoing studies, suspension or revocation of approved applications, warning or untitled letters, product withdrawals or recalls, product seizures, relabeling or repackaging, total or partial suspensions of manufacturing or distribution, injunctions, fines, civil penalties or criminal prosecution.
The process required by the FDA before our product candidates are approved as drugs for therapeutic indications and may be marketed in the United States generally involves the following:
•completion of extensive preclinical studies in accordance with applicable regulations, including studies conducted in accordance with good laboratory practice or GLP,(“GLP”) requirements;
•completion of the manufacture, under cGMP conditions, of the drug substance and drug product that the sponsor intends to use in human clinical trials along with required analytical and stability testing;
•submission to the FDA of an IND, which must become effective before clinical trials may begin;
•approval by an institutional review board or IRB,(“IRB”) or independent ethics committee at each clinical trial site before each trial may be initiated;
•performance of adequate and well-controlled clinical trials in accordance with applicable IND regulations, good clinical practice or GCP,(“GCP”) requirements and other clinical trial-related regulations to establish the safety and efficacy of the investigational product for each proposed indication;
•submission to the FDA of a New Drug Application or NDA;(“NDA”);
•a determination by the FDA within 60 days of its receipt of an NDA, to accept the filing for review;
•satisfactory completion of one or more FDA pre-approval inspections of the manufacturing facility or facilities where the drug will be produced to assess compliance with cGMP requirements to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity;
•potentially, satisfactory completion of FDA audit of the clinical trial sites that generated the data in support of the NDA;
•payment of user fees for FDA review of the NDA; and
•FDA review and approval of the NDA, including consideration of the views of any FDA advisory committee, prior to any commercial marketing or sale of the drug in the United States.
Preclinical Studies and Clinical Trials for Drugs
Before testing any drug in humans, the product candidate must undergo rigorous preclinical testing. Preclinical studies include laboratory evaluations of drug chemistry, formulation and stability, as well as in vitro and animal studies to assess safety and in some cases to establish the rationale for therapeutic use. The conduct of preclinical studies is subject to United States federal and state regulation, including GLP requirements for safety/toxicology studies. The results of the preclinical studies, together with manufacturing information and analytical data, must be submitted to the FDA as part of an IND. An IND is a request for authorization from the FDA to administer an investigational product to humans and must become effective before clinical trials may begin. Some long-term preclinical testing may continue after the IND is submitted. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, raises concerns or questions about the conduct of the clinical trial, including concerns that human research subjects will be exposed to unreasonable health risks, and imposes a full or partial clinical hold. FDA must notify the sponsor of the grounds for the hold and any identified deficiencies must be resolved before the clinical trial can begin. Submission of an IND may result in the FDA not allowing clinical trials to commence or not allowing clinical trials to commence on the terms originally specified in the IND. A clinical
hold can also be imposed once a trial has already begun, thereby halting the trial until the deficiencies articulated by FDA are corrected.
The clinical stage of development involves the administration of the product candidate to healthy volunteers or patients under the supervision of qualified investigators, who generally are physicians not employed by or under the trial sponsor’s control, in accordance with GCP requirements, which include the requirements that all research subjects provide their informed consent for their participation in any clinical trial. Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria and the parameters and criteria to be used in monitoring safety and evaluating effectiveness. Each protocol, and any subsequent amendments to the protocol, must be submitted to the FDA as part of the IND. Furthermore, each clinical trial must be reviewed and approved by an IRB for each institution at which the clinical trial will be conducted to ensure that the risks to individuals participating in the clinical trials are minimized and are reasonable compared to the anticipated benefits. The IRB also approves the informed consent form that must be provided to each clinical trial subject or his or her legal representative and must monitor the clinical trial until completed. Regulatory authorities, the IRB or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects are being exposed to an unacceptable health risk or that the trial is unlikely to meet its stated objectives. Some studies also include oversight by an independent group of qualified experts organized by the clinical study sponsor, known as a data safety monitoring board, which provides authorization for whether or not a study may move forward at designated check points based on access to certain data from the study and may halt the clinical trial if it determines that there is an unacceptable safety risk for subject or other grounds, such as a lack of observed efficacy. There also are requirements governing the reporting of ongoing clinical trials and completed clinical trials to public registries. Information about clinical trials, including results for clinical trials other than Phase 1 investigations, must be submitted within specific timeframes for publication on www.ClinicalTrials.gov, a clinical trials database maintained by the National Institutes of Health.
A sponsor who wishes to conduct a clinical trial outside of the United States may, but need not, obtain FDA authorization to conduct the clinical trial under an IND. If a foreign clinical trial is not conducted under an IND, FDA will nevertheless accept the results of the study in support of an NDA if the study was conducted in accordance with GCP requirements, and the FDA is able to validate the data through an onsite inspection if deemed necessary.
Clinical trials to evaluate therapeutic indications to support NDAs for marketing approval are typically conducted in three sequential phases, which may overlap.
•Phase 1—Phase 1 clinical trials involve initial introduction of the investigational product into healthy human volunteers or patients with the target disease or condition. These studies are typically designed to test the safety, dosage tolerance, absorption, metabolism and distribution of the investigational product in humans, excretion the side effects associated with increasing doses, and, if possible, to gain early evidence of effectiveness.
•Phase 2—Phase 2 clinical trials typically involve administration of the investigational product to a limited patient population with a specified disease or condition to evaluate the drug’s potential efficacy, to determine the optimal dosages and dosing schedule and to identify possible adverse side effects and safety risks.
•Phase 3—Phase 3 clinical trials typically involve administration of the investigational product to an expanded patient population to further evaluate dosage, to provide statistically significant evidence of clinical efficacy and to further test for safety, generally at multiple geographically dispersed clinical trial sites. These clinical trials are intended to establish the overall risk/benefit ratio of the investigational product and to provide an adequate basis for product approval and physician labeling.
Post-approval trials, sometimes referred to as Phase 4 clinical trials or post-marketing studies, may be conducted after initial marketing approval. These trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication and are commonly intended to generate additional safety data regarding use of the product in a clinical setting. In certain instances, the FDA may mandate the performance of Phase 4 clinical trials as a condition of NDA approval.
Progress reports detailing the results of the clinical trials, among other information, must be submitted at least annually to the FDA. Written IND safety reports must be submitted to the FDA and the investigators fifteen days after the trial sponsor determines the information qualifies for reporting for serious and unexpected suspected adverse events, findings from other studies or animal or in vitro testing that suggest a significant risk for human volunteers and any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigator brochure. The sponsor must also notify the FDA of any unexpected fatal or life-threatening suspected adverse reaction as soon as possible but in no case later than seven calendar days after the sponsor’s initial receipt of the information.
the Food and Drug Omnibus Reform Act of 2022 (“FDORA”) signed by President Biden on December 29, 2022 as part of the Consolidated Appropriations Act, 2023 (H.R. 2617), Congress added a requirement for sponsors to develop and submit a diversity action plan for each Phase 3 clinical trial or any other “pivotal study” of a new drug or biological product. Action plans must include the sponsor’s goals for enrollment, the underlying rationale for those goals, and an explanation of how the sponsor intends to meet them. This requirement will apply with respect to clinical investigations for which enrollment commences 180 days after the publication of a final guidance by the FDA on diversity action plans. The statute directs FDA to issue new or revised draft guidance on diversity action plans by the end of 2023, and final guidance within 9 months of closing the comment period on such draft guidance. FDA has not yet published new or revised draft guidance. During the development of a new drug, sponsors are given opportunities to meet with the FDA at certain points. These points may be prior to submission of an IND, at the end of Phase 2, and before an NDA is submitted. Meetings at other times may be requested. These meetings can provide an opportunity for the sponsor to share information about the data gathered to date, for the FDA to provide advice, and for the sponsor and the FDA to reach agreement on the next phase of development.
Concurrent with clinical trials, companies usually complete additional animal studies and must also develop additional information about the chemistry and physical characteristics of the product candidate and finalize a process for manufacturing the drug product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate and manufacturers must develop, among other things, methods for testing the identity, strength, quality and purity of the final drug product. Additionally, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life.
U.S. Marketing Approval for Drugs
Assuming successful completion of the required clinical testing, the results of the preclinical studies and clinical trials, together with detailed information relating to the product’s chemistry, manufacture, controls and proposed labeling, among other things, are submitted to the FDA as part of an NDA package requesting approval to market the product for one or more indications. An NDA is a request for approval to market a new drug for one or more specified indications and must contain proof of the drug’s safety and efficacy for the requested indications. The marketing application is required to include both negative and ambiguous results of preclinical studies and clinical trials, as well as positive findings. Data may come from company-sponsored clinical trials intended to test the safety and efficacy of a product’s use or from a number of alternative sources, including studies initiated by investigators. To support marketing approval, the data submitted must be sufficient in quality and quantity to establish the safety and efficacy of the investigational product to the satisfaction of the FDA. FDA must approve an NDA before a drug may be marketed in the United States.
The FDA reviews all submitted NDAs before it accepts them for filing and may request additional information rather than accepting the NDA for filing. The FDA must make a decision on accepting an NDA for filing within 60 days of receipt, and such decision could include a refusal to file by the FDA. Once the submission is accepted for filing, the FDA begins an in-depth substantive review of the NDA. The FDA reviews an NDA to determine, among other things, whether the drug is safe and
effective for the indications sought and whether the facility in which it is manufactured, processed, packaged or held meets standards designed to assure the product’s continued safety, quality and purity. Under the goals and polices agreed to by the FDA under the Prescription Drug User Fee Act or PDUFA,(“PDUFA”) the FDA targets ten months, from the filing date, in which to complete its initial review of a new molecular entity NDA and respond to the applicant, and six months from the filing date of a new molecular entity NDA for priority review. The FDA does not always meet its PDUFA goal dates for standard or priority NDAs, and the review process is often extended by FDA requests for additional information or clarification.
Further, under PDUFA, as amended, each NDA must be accompanied by a substantial user fee. The FDA adjusts the PDUFA user fees on an annual basis. Fee waivers or reductions are available in certain circumstances, including a waiver of the application fee for the first application filed by a small business. Additionally, no user fees are assessed on NDAs for products designated as orphan drugs, unless the product also includes a non-orphan indication.
The FDA also may require submission of a Risk Evaluation and Mitigation Strategy or REMS,(“REMS”) if it believes that a risk evaluation and mitigation strategy is necessary to ensure that the benefits of the drug outweigh its risks. A REMS can include use of risk evaluation and mitigation strategies like medication guides, physician communication plans, assessment plans, and/or elements to assure safe use, such as restricted distribution methods, patient registries, or other risk-minimization tools.
The FDA may refer an application for a novel drug to an advisory committee. An advisory committee is a panel of independent experts, including clinicians and other scientific experts, which reviews, evaluates and provides a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
Before approving an NDA, the FDA typically will inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and are adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA, the FDA may inspect one or more clinical trial sites to assure compliance with GCP and other requirements and the integrity of the clinical data submitted to the FDA.
After evaluating the NDA and all related information, including the advisory committee recommendation, if any, and inspection reports regarding the manufacturing facilities and clinical trial sites, the FDA may issue an approval letter, or, in some cases, a complete response letter. A complete response letter generally contains a statement of specific conditions that must be met in order to secure final approval of the NDA and may require additional clinical or preclinical testing in order for the FDA to reconsider the application. Even with submission of this additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval. If and when those conditions have been met to the FDA’s satisfaction, the FDA will typically issue an approval letter. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications.
Even if the FDA approves a product, depending on the specific risk(s) to be addressed it may limit the approved indications for use of the product, require that contraindications, warnings or precautions be included in the product labeling, require that post-approval studies, including Phase 4 clinical trials, be conducted to further assess a drug’s safety after approval, require testing and surveillance programs to monitor the product after commercialization, or impose other conditions, including distribution and use restrictions or other risk management mechanisms under a REMS, which can materially affect the potential market and profitability of the product. The FDA may prevent or limit further marketing of a product based on the results of post-marketing studies or surveillance programs. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes, and additional labeling claims, are subject to further testing requirements and FDA review and approval.
Orphan Drug Designation and Exclusivity
Under the Orphan Drug Act, the FDA may grant orphan designation to a drug intended to treat a rare disease or condition, which is a disease or condition that affects fewer than 200,000 individuals in the United States, or that affects more than 200,000 individuals in the United States where there is no reasonable expectation that the cost of developing and making the product available in the United States for the disease or condition will be recovered from sales of the product. Orphan designation must be requested before submitting an NDA. Orphan designation does not convey any advantage in or shorten the duration of the regulatory review and approval process, though companies developing orphan products are eligible for certain incentives, including tax credits for qualified clinical testing and waiver of application fees.
If a product that has orphan designation subsequently receives the first FDA approval for the disease or condition for which it has such designation, the product is entitled to a seven-year period of marketing exclusivity during which the FDA may not approve any other applications to market the same therapeutic agent for the same indication, except in limited circumstances, such as a subsequent product’s showing of clinical superiority over the product with orphan drug exclusivity or where the original applicant cannot produce sufficient quantities of product. Competitors, however, may receive approval of different
therapeutic agents for the indication for which the orphan product has exclusivity or obtain approval for the same therapeutic agent for a different indication than that for which the orphan product has exclusivity. Orphan drug exclusivity could block the approval of one of our products for seven years if a competitor obtains approval for the same therapeutic agent for the same indication before we do, unless we are able to demonstrate that our product is clinically superior. If an orphan designated product receives marketing approval for an indication broader than what is designated, it may not be entitled to orphan drug exclusivity. Further, orphan drug exclusive marketing rights in the United States may be lost if the FDA later determines that the request for designation was materially defective or the manufacturer of the approved product is unable to assure sufficient quantities of the product to meet the needs of patients with the rare disease or condition.
Expedited Development and Review Programs for Drugs
The FDA maintains several programs intended to facilitate and expedite development and review of new drugs to address unmet medical needs in the treatment of serious or life-threatening diseases or conditions. These programs include Fast Track designation, Breakthrough Therapy designation, Priority Review and Accelerated Approval, and the purpose of these programs is to either expedite the development or review of important new drugs to get them to patients more quickly than standard FDA review timelines typically permit.
A new drug is eligible for Fast Track designation if it is intended to treat a serious or life-threatening disease or condition and demonstrates the potential to address unmet medical needs for such disease or condition. Fast Track designation provides increased opportunities for sponsor interactions with the FDA during preclinical and clinical development, in addition to the potential for rolling review once a marketing application is filed. Rolling review means that the agency may review portions of the marketing application before the sponsor submits the complete application. In addition, a new drug may be eligible for Breakthrough Therapy designation if it is intended to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. Breakthrough
Therapy designation provides all the features of Fast Track designation in addition to intensive guidance on an efficient drug development program beginning as early as Phase 1, and FDA organizational commitment to expedited development, including involvement of senior managers and experienced review staff in a cross-disciplinary review, where appropriate.
Any product submitted to the FDA for approval, including a product with Fast Track or Breakthrough Therapy designation, may also be eligible for additional FDA programs intended to expedite the review and approval process, including Priority Review designation and Accelerated Approval. A product is eligible for Priority Review, once an NDA or BLA is submitted, if the drug that is the subject of the marketing application has the potential to provide a significant improvement in safety or effectiveness in the treatment, diagnosis or prevention of a serious disease or condition. Under priority review, the FDA’s goal date to take action on the marketing application is six months compared to ten months for a standard review. Products are eligible for Accelerated Approval if they can be shown to have an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit, or an effect on a clinical endpoint that can be measured earlier than an effect on irreversible morbidity or mortality, which is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments.
Accelerated Approval is usually contingent on a sponsor’s agreement to conduct additional post-approval studies to verify and describe the product’s clinical benefit. The FDA may withdraw approval of a drug or an indication approved under Accelerated Approval if, for example, the confirmatory trial fails to verify the predicted clinical benefit of the product. In addition, the FDA generally requires, as a condition for Accelerated Approval, that all advertising and promotional materials intended for dissemination or publication within 120 days of marketing approval be submitted to the agency for review during the pre-approval review period. After the 120-day period has passed, all advertising and promotional materials must be submitted at least 30 days prior to the intended time of initial dissemination or publication. FDORA signed by President Biden on December 29, 2022 as part of the Consolidated Appropriations Act, 2023 (H.R. 2617) includes numerous reforms to the Accelerated Approval process for drugs and biologics and enables the FDA to require, as appropriate, that a post-approval study be underway prior to granting accelerated approval. FDORA also expands the expedited withdrawal procedures already available to the FDA to allow the agency to use expedited procedures if a sponsor fails to conduct any required post-approval study of the product with due diligence including with respect to “conditions specified by the Secretary [of HHS].” FDORA also adds the failure of a sponsor of a product approved under Accelerated Approval to conduct with due diligence any required post-approval study with respect to such product or to submit timely reports with respect to such product to the list of prohibited acts in the FD&C Act.
Even if a product qualifies for one or more of these programs, the FDA may later decide that the product no longer meets the conditions for qualification or the time period for FDA review or approval may not be shortened. Furthermore, Fast Track designation, Breakthrough Therapy designation, Priority Review and Accelerated Approval do not change the scientific or medical standards for approval or the quality of evidence necessary to support approval, though they may expedite the development or review process.
Pediatric Information and Pediatric Exclusivity
The Pediatric Research Equity Act or PREA,(“PREA”) requires a sponsor to conduct pediatric clinical trials for most drugs, for a new active ingredient, new indication, new dosage form, new dosing regimen or new route of administration. Under PREA, as amended, certain NDAs and NDA supplements must contain data that can be used to assess the safety and efficacy of the drug for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may grant deferrals for submission of pediatric data or full or partial waivers. The FD&C Act requires that a sponsor who is planning to submit a marketing application for a drug that includes a new active ingredient, new indication, new dosage form, new dosing regimen or new route of administration submit an initial Pediatric Study Plan or PSP,(“PSP”) within 60 days of an end-of-Phase 2 meeting or, if there is no such meeting, as early as practicable before the initiation of the Phase 3 or Phase 2/3 study. The initial PSP must include an outline of the pediatric study or studies that the sponsor plans to conduct, including study objectives and design, age groups, relevant endpoints and statistical approach, or a justification for not including such detailed information, and any request for a deferral of pediatric assessments or a full or partial waiver of the requirement to provide data from pediatric studies along with supporting information. The FDA and the sponsor must reach an agreement on the PSP. A sponsor can submit amendments to an agreed-upon initial PSP at any time if changes to the pediatric plan need to be considered based on data collected from preclinical studies, early phase clinical trials and/or other clinical development programs.
A drug can also obtain pediatric market exclusivity in the United States. Pediatric exclusivity, if granted, adds six months to existing exclusivity periods and patent terms. This six-month exclusivity, which runs from the end of other exclusivity protection or patent term, may be granted based on the voluntary completion of a pediatric study in accordance with an FDA-issued “Written Request” for such a study.
U.S. Post-Approval Requirements for Drugs
Drugs manufactured or distributed pursuant to FDA approvals are subject to continuing regulation by the FDA, including, among other things, requirements relating to recordkeeping, periodic reporting, product sampling and distribution, reporting of adverse experiences with the product, complying with promotion and advertising requirements, which include restrictions on promoting products for unapproved uses or patient populations (known as “off-label use”) and limitations on industry-sponsored scientific and educational activities. Although physicians may prescribe legally available products for off-label uses,
manufacturers may not market or promote such uses. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability, including investigation by federal and state authorities. Prescription drug promotional materials must be submitted to the FDA in conjunction with their first use or first publication. Further, if there are any modifications to the drug, including changes in indications, labeling or manufacturing processes or facilities, the applicant may be required to submit and obtain FDA approval of a new NDA or NDA supplement, which may require the generation of additional data or the conduct of additional preclinical studies and clinical trials.
The FDA may impose a number of post-approval requirements as a condition of approval of an NDA. For example, the FDA may require post-market testing, including Phase 4 clinical trials, and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization. In addition, drug manufacturers and their subcontractors involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and certain state agencies and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with ongoing regulatory requirements, including cGMPs, which impose certain procedural and documentation requirements. Failure to comply with statutory and regulatory requirements may subject a manufacturer to legal or regulatory action, such as warning letters, suspension of manufacturing, product seizures, injunctions, civil penalties or criminal prosecution. There is also a continuing, annual prescription drug product program user fee.
Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information, requirements for post-market studies or clinical trials to assess new safety risks, or imposition of distribution or other restrictions under a REMS. Other potential consequences include, among other things:
•restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls;
•the issuance of safety alerts, Dear Healthcare Provider letters, press releases or other communications containing warnings or other safety information about the product;
•fines, warning letters or holds on post-approval clinical trials;
•refusal of the FDA to approve applications or supplements to approved applications, or suspension or revocation of product approvals;
•product seizure or detention, or refusal to permit the import or export of products;
•injunctions or the imposition of civil or criminal penalties; and
•consent decrees, corporate integrity agreements, debarment or exclusion from federal healthcare programs; or mandated modification of promotional materials and labeling and issuance of corrective information.
Companion diagnostics are designed to identify patients who are most likely to benefit from a particular therapeutic product; identify patients likely to be at increased risk for serious side effects as a result of treatment with a particular therapeutic product; or monitor response to treatment with a particular therapeutic product for the purpose of adjusting treatment to achieve improved safety or effectiveness. Companion diagnostics are regulated as medical devices by the FDA. In the United States, the FD&C Act, and its implementing regulations, and other federal and state statutes and regulations govern, among other things, medical device design and development, preclinical and clinical testing, premarket clearance or approval, registration and listing, manufacturing, labeling, storage, advertising and promotion, sales and distribution, export and import, and post-market surveillance. Unless an exemption or FDA exercise of enforcement discretion applies, diagnostic tests generally require marketing clearance or approval from the FDA prior to commercialization. The two primary types of FDA marketing authorization applicable to a medical device are clearance of a premarket notification, or 510(k), and approval of a premarket approval application or PMA.(“PMA”).
To obtain 510(k) clearance for a medical device, or for certain modifications to devices that have received 510(k) clearance, a manufacturer must submit a premarket notification demonstrating that the proposed device is substantially equivalent to a previously cleared 510(k) device or to a pre-amendment device that was in commercial distribution before May 28, 1976, or a predicate device, for which the FDA has not yet called for the submission of a PMA. In making a determination that the device is substantially equivalent to a predicate device, the FDA compares the proposed device to the predicate device and assesses whether the subject device is comparable to the predicate device with respect to intended use, technology, design and other
features which could affect safety and effectiveness. If the FDA determines that the subject device is substantially equivalent to the predicate device, the subject device may be cleared for marketing. The 510(k) premarket notification pathway generally takes from three to twelve months from the date the application is completed, but can take significantly longer.
A PMA must be supported by valid scientific evidence, which typically requires extensive data, including technical, preclinical, clinical and manufacturing data, to demonstrate to the FDA’s satisfaction the safety and effectiveness of the device. The process for developing a PMA, including the gathering of clinical and preclinical data and submission to FDA can take several years or longer. For diagnostic tests, a PMA typically includes data regarding analytical and clinical validation studies. As part of its review of the PMA, the FDA will conduct a pre-approval inspection of the manufacturing facility or facilities to ensure compliance with the quality system regulation, or QSR, which requires manufacturers to follow design, testing, control, documentation and other quality assurance procedures. The FDA’s review of an initial PMA is required by statute to take between six to ten months, although the process typically takes longer, and may require several years to complete, and PMA approval is not guaranteed. If the FDA evaluations of both the PMA and the manufacturing facilities are favorable, the FDA will either issue an approval letter or an approvable letter, which usually contains a number of conditions that must be met in order to secure the final approval of the PMA. If the FDA’s evaluation of the PMA or manufacturing facilities is not favorable, the FDA will deny the approval of the PMA or issue a not approvable letter. A not approvable letter will outline the deficiencies in the application and, where practical, will identify what is necessary to make the PMA approvable. Once granted, PMA approval may be withdrawn by the FDA if compliance with post-approval requirements, conditions of approval or other regulatory standards is not maintained or problems are identified following initial marketing.
On July 31, 2014, the FDA issued a final guidance document addressing the development and approval process for “In Vitro Companion Diagnostic Devices.” According to the guidance document, for novel therapeutic products that depend on the use of a diagnostic test and where the diagnostic device could be essential for the safe and effective use of the corresponding therapeutic product, the companion diagnostic device should be developed and approved or cleared contemporaneously with the therapeutic, although the FDA recognizes that there may be cases when contemporaneous development may not be possible. However, in cases where a drug cannot be used safely or effectively without the companion diagnostic, the FDA’s guidance indicates it will generally not approve the drug without the approval or clearance of the diagnostic device. The FDA also issued a draft guidance in July 2016 setting forth the principles for co-development of an in vitro companion diagnostic device with a therapeutic product. The draft guidance describes principles to guide the development and contemporaneous marketing authorization for the therapeutic product and its corresponding in vitro companion diagnostic.
Once cleared or approved, the companion diagnostic device must adhere to post-marketing requirements including the requirements of the FDA’s QSR, which cover the methods and documentation of the design, testing, production, processes, controls, quality assurance, labeling, packaging, and shipping of all medical devices, as well as adverse event reporting, recalls and corrections along with product marketing requirements and limitations. Medical devices, including companion diagnostics,
may be marketed only for the uses and indications for which they are cleared or approved. Device manufacturers must also establish registration and device listings with the FDA. Like drug makers, companion diagnostic makers are subject to unannounced FDA inspections at any time during which the FDA will conduct an audit of the product(s) and the company’s facilities, facility records, and manufacturing processes for compliance with its authorities.
Marketing Exclusivity
Market exclusivity provisions authorized under the FD&C Act can delay the submission or the approval of certain marketing applications. The FD&C Act provides a five-year period of non-patent marketing exclusivity within the United States to the first applicant to obtain approval of an NDA for a new chemical entity. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not approve or even accept for review an abbreviated new drug application or ANDA,(“ANDA”) or an NDA submitted under Section 505(b)(2), or 505(b)(2) NDA, submitted by another company for another drug based on the same active moiety, regardless of whether the drug is intended for the same indication as the original innovative drug or for another indication, where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement to one of the patents listed with the FDA by the innovator NDA holder.
The FD&C Act alternatively provides three years of marketing exclusivity for an NDA, or supplement to an existing NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example new indications, dosages or strengths of an existing drug. This three-year exclusivity covers only the modification for which the drug received approval on the basis of the new clinical investigations and does not prohibit the FDA from approving ANDAs or 505(b)(2) NDAs for drugs containing the active agent for the original indication or condition of use. Five-year and three-year exclusivity will not delay the submission or
approval of a full NDA. However, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to any preclinical studies and adequate and well-controlled clinical trials necessary to demonstrate safety and effectiveness.
Pediatric exclusivity is another type of marketing exclusivity available in the United States. Pediatric exclusivity provides for an additional six months of marketing exclusivity attached to another period of exclusivity if a sponsor conducts clinical trials in children in response to a written request from the FDA. The issuance of a written request does not require the sponsor to undertake the described clinical trials. In addition, orphan drug exclusivity, as described above, may offer a seven-year period of marketing exclusivity, except in certain circumstances.
Other Regulatory Matters
Manufacturing, sales, promotion and other activities of product candidates following product approval, where applicable, or commercialization are also subject to regulation by numerous regulatory authorities in the United States in addition to the FDA, which may include the Centers for Medicare & Medicaid Services or CMS,(“CMS”) other divisions of the U.S. Department of Health and Human Services, the Department of Justice, the Drug Enforcement Administration, the Consumer Product Safety Commission, the Federal Trade Commission, the Occupational Safety & Health Administration, the Environmental Protection Agency and state and local governments and governmental agencies.
Other Healthcare Laws
Healthcare providers, physicians, and third-party payors will play a primary role in the recommendation and prescription of any products for which we obtain marketing approval. Our business operations and any current or future arrangements with third-party payors, healthcare providers and physicians may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we develop, market, sell and distribute any drugs for which we obtain marketing approval. In the United States, these laws include, without limitation, federal and state fraud and federal anti-kickback, false claims, physicianabuse laws, transparency laws, and patient data privacy and security laws and regulations, including but not limited to those described below.below, some of which will not apply to us unless or until we have a marketed product.
•The federal Anti-Kickback Statute, which prohibits, among other things, persons and entities from knowingly and willfullyoffering, soliciting, offering, paying, receiving or providing any remuneration, (including any kickback, bribe, or certain rebate), directly or indirectly, overtly or covertly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase order or recommendationordering of, anya good or service for which payment may be made in whole or in part, under a federal healthcare programprograms such as Medicare and Medicaid; a person or entity need not have actual knowledge of the federal Anti-Kickback Statute or specific intent to violate it in order to have committed a violation. Violations are subject to civil and criminal fines and penalties for each violation, plus up to three times the remuneration involved, imprisonment, and exclusion from government healthcare programs. In addition, the government may assert that a claim that includes items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the civil False Claims Act.
•The federal civil and criminalFederal false claims, false statement and civil monetary penalties laws including the civil False Claims Act, or FCA, prohibit individuals or entities from,prohibiting, among other things, any person from knowingly presenting, or causing to be presented, to the federala false claim for payments of government claims for paymentfunds or approval that are false, fictitious or fraudulent; knowingly making, using, or causing to be made, or used, a false statement or record material to a false or fraudulent claim or obligation to pay or transmit money or property to the federal government; or knowingly concealing or knowingly and improperly avoiding or decreasing an obligation to pay money to the federal government. Manufacturers can be held liable under the FCA even when they do not submit claims directly to government payors if they are deemed to “cause” the submission of false or fraudulent claims. The FCA also permits a private individual acting as a “whistleblower” to bring actions on behalf of the federal government alleging violations of the FCA and to share in any monetary recovery. When an entity is determined to have violated the federal civil False Claims Act, the government may impose civil fines and penalties for each false claim, plus treble damages, and exclude the entity from participation in Medicare, Medicaid and other federal healthcare programs.
•The federal civil monetary penalties laws impose civil fines for, among other things, the offering or transfer or remuneration to a Medicare or state healthcare program beneficiary, if the person knows or should know it is likely to influence the beneficiary’s selection of a particular provider, practitioner, or supplier of services reimbursable by Medicare or a state health care program, unless an exception applies.claim;
•The Health Insurance Portability and Accountability Act of 1996 or HIPAA, imposes criminal(“HIPAA”),which, in addition to privacy protections applicable to healthcare providers and civil liability for knowingly and willfullyother entities, prohibits executing a scheme, or attempting to execute a scheme to defraud any healthcare benefit program including private payors, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense, or falsifying, concealing or covering up a material fact or making any materially false statements in connection with the delivery of or payment forrelating to healthcare benefits, items or services.matters;
•HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009,So-called federal “sunshine” law, or HITECH, and their respective implementing regulations, impose, among other things, specified requirements on covered entities and their business associates relating to the privacy and security of individually identifiable health information including mandatory contractual terms and required implementation of technical safeguards of such information. HITECH also created new tiers of civil monetary penalties, amended HIPAA to make civil and criminal penalties directly applicable to business associates in some cases, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorneys’ fees and costs associated with pursuing federal civil actions.
•The PhysicianOpen Payments, Sunshine Act, which imposes annual reporting requirements for certain manufacturers of drugs, devices, biologics,requires pharmaceutical and medical supplies for which payment is available under Medicare, Medicaid, or the Children’s Health Insurance Program, fordevice companies to report information related to certain payments and “transferstransfers of value”value provided to physicians (currently definedcertain healthcare providers to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals,CMS, as well as ownership and investment interests held by physicians and their immediate family members. Effective January 1, 2022, these reporting obligations will extend to include transfers of value made in the previous year to certain non-physician providers such as physician assistants and nurse practitioners.members;
•Federal consumer protection and unfair competition laws broadly regulate marketplace activities and activities that potentially harm consumers.
•The Federal Food, Drug, and Cosmetic Act, which among other things, strictly regulates drug product and medical device marketing, prohibits manufacturers from marketing such products prior to approval or for unapproved indications and regulates the distribution of samples;
•Federal laws, including the Medicaid Drug Rebate Program, that require pharmaceutical manufacturers to report certain calculated product prices to the government or provide certain discounts or rebates to government authorities or private entities, often as a condition of reimbursement under government healthcare programs; and
•Analogous state and foreign laws and regulations, may be broader in scope than the provisions described abovesuch as state anti-kickback, anti-bribery and false claims laws, which may apply regardless of payor. Someto healthcare items or services that are reimbursed by non-governmental third-party payors, including private insurers, as well as other state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntaryspecific compliance guidelinesstandards, restrict financial interactions between companies and relevant federal government compliance guidance;healthcare providers, require drug manufacturerscompanies to report information related to payments and other transfers of value to physicians and other healthcare providers; restrictproviders, marketing practicesexpenditures or pricing or require disclosurethe licensing or registration of marketing expenditures and pricing information. State and foreign laws may governsales representatives.
Given the privacy and security of health information in some circumstances. These data privacy and security laws may differ from each other in significant ways and often are not pre-empted by HIPAA, which may complicate compliance efforts.
The scope and enforcement of each of these laws is uncertain and subject to rapid change in the current environment of healthcare reform, especially in lightbreadth of the lacklaws and regulations, narrowness of applicable precedentexceptions, limited guidance for certain laws and regulations.regulations, and evolving government interpretations of the laws and regulations, ensuring compliance is challenging. Federal and state enforcement bodies have recently increased their scrutiny ofagencies scrutinize interactions between healthcare companies and healthcare providers, which has led to a number of investigations, prosecutions, convictions and settlements in the healthcare industry. It is possible that governmental authorities will conclude that our business practices do not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other related governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, imprisonment, disgorgement, exclusion from government funded healthcare programs, such as Medicare and Medicaid, reputational harm, additional oversight and reporting obligations if we become subject to a corporate integrity agreement or similar settlement to resolve allegations of non-compliance with these laws and the curtailment or restructuring of our operations. If any of the physicians or other healthcare providers or entities with whom we expect to do business is found not to be in compliance with applicable laws, they may be subject to similar actions, penalties and sanctions. Ensuring business arrangements comply with applicable healthcare laws, as well as responding to possible investigations by government authorities, can be time- and resource-consuming and can divert a company’s attention from its business.
Coverage and Reimbursement by Third-Party Payors
In the United States and markets in other countries, patients who are prescribed treatments for their conditions and providers performing the prescribed services generally rely on third-party payors to reimburse all or part of the associated healthcare costs. Thus, even if a product candidate isSales of an approved sales of thedrug product will depend, in part, on the extent to which third-party payors, including government health programs in the United States such as Medicare and Medicaid, commercialand private health insurers andinsurance such as managed care organizations,plans, provide coverage, and establish adequate reimbursement levels for the product. In the United
States, the Medicare and Medicaid programs are increasingly used as models for how private and other governmental payors develop their coverage and reimbursement policies for drugs. No uniform policy of coverage and reimbursement for drug products exists however, among third-party payors. Therefore, coverage and reimbursement for drug products can differ significantly from payor to payor. The process for determining whether a third-party payor will provide coverage for a product may be separate from the process for setting the price or reimbursement rate that the payor will pay for the product once coverage is approved. A third-party payor’s decision to provide coverage for a product therefore does not imply that an adequate reimbursement rate will be approved. Third-party payors are increasingly challenging the prices charged, examining the medical necessity, reviewing the cost-effectiveness of medical products and services and imposing controls to manage costs. Third-party payors may limitseek to control costs and manage utilization by, for example, excluding products from lists of approved covered products (known as “formularies”), imposing step edits that require patients to try alternative treatments before authorizing payment for products, limiting the types of diagnoses for which coverage will be provided, requiring pre-approval (known as “prior authorization”) for coverage of a prescription for each patient (to allow the payor to specificassess medical necessity) or imposing a moratorium on coverage for products on an approved list, also known aswhile the payor makes a formulary, which might not include all of the approved products for a particular indication.coverage decision.
In order to secure coverage and reimbursement for any product that might be approved for sale, a company may need to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost-effectiveness of the product, which will require additional expenditure above and beyond the costs required to obtain FDA or other comparable
regulatory approvals. Nonetheless, product candidates may not be considered medically necessary or cost effective. Additionally, companies may also need to provide discounts to purchasers, private health plans or government healthcare programs. Nonetheless, product candidates may not be considered medically necessary or cost effective. A decision by a third-party payor not to cover a product could reduce physician utilization once the product is approved and have a material adverse effect on sales, our operations and financial condition. Additionally, a third-party payor’s decision to provide coverage for a product does not imply that an adequate reimbursement rate will be approved. Further, one payor’s determination to provide coverage for a product does not assure that other payors will also provide coverage and reimbursement for the product, and the level of coverage and reimbursement can differ significantly from payor to payor.
The containment of healthcare costs has become a priority of federal, state and foreign governments, and the prices of products have been a focus in this effort. Governments have shown significant interest in implementing cost-containment programs, including price controls, restrictions on reimbursement and requirements for substitution of generic products. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit a company’s revenue generated from the sale of any approved products. Coverage policies and third-party payor reimbursement rates may change at any time. Even if favorable coverage and reimbursement status is attained for one or more products for which a company or its collaborators receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
Current and Future Healthcare Reform Legislation
In the United States and some foreign jurisdictions, there have been, and likely will continue to be, a number of legislative and regulatory changes and proposed changes regarding the healthcare system directed at broadening the availability of healthcare, improving the quality of healthcare, and containing or lowering the cost of healthcare.
For example, in March 2010, the United States Congress enacted the Patient Protection and Affordable Care Act, as amended, the Health Care and Education Reconciliation Act (the “Affordable Care Act”), which, among other things, includesexpanded health care coverage through Medicaid expansion and the implementation of the individual mandate for health insurance coverage and which included a number of changes to the coverage and payment forreimbursement of drug products under government health carehealthcare programs.
Since its enactment, there have been numerous judicial, administrative, executive, and legislative challenges to certain aspects of Beyond the Affordable Care Act, there have been ongoing health care reform efforts. Drug pricing and we expect there willpayment reform was a focus of the Trump Administration and has been a focus of the Biden Administration. For example, federal legislation enacted in 2021 eliminates a statutory cap on Medicaid drug rebate program rebates effective January 1, 2024. As another example, the Inflation Reduction Act (“IRA”) of 2022 includes a number of changes intended to address rising prescription drug prices in Medicare Parts B and D. These changes, which have varying implementation dates, include caps on Medicare Part D out-of-pocket costs, Medicare Part B and Part D drug price inflation rebates, a new Medicare Part D manufacturer discount drug program and a drug price negotiation program for certain high spend Medicare Part B and D drugs. Although the impact of the IRA remains uncertain pending ongoing implementation, the IRA is likely to have a significant effect on the healthcare industry and prescription drug pricing overall. As another example, in 2022, subsequent to the enactment of the IRA, the Biden administration released an executive order directing the HHS to report on how the Center for Medicare and Medicaid Innovation (“CMMI”) could be additional challengesleveraged to test new models for lowering drug costs for Medicare and amendmentsMedicaid beneficiaries. The report was issued in 2023 and proposed various models that CMMI is currently developing which seek to lower the cost of drugs, promote accessibility and improve quality of care, including a model addressing payment for drugs that receive accelerated approval from the FDA.
Healthcare reform efforts have been and may continue to be subject to scrutiny and legal challenge. For example, with respect to the Affordable Care Act, intax reform legislation was enacted that eliminated the future. For example, various portions of the Affordable Care Act are currently facing legal and constitutional challenges in the Fifth Circuit Court of Appeals and the United States Supreme Court. Additionally, the current administration has issued various Executive Orders which eliminated cost sharing subsidies and various provisions that would impose a fiscal burden on states or a cost, fee, tax penalty or regulatory burden onestablished for individuals healthcare providers,who do not maintain mandated health insurers, or manufacturers of pharmaceuticals or medical devices,insurance coverage beginning in 2019 and, Congress has introduced several pieces of legislation aimed at significantly revising or repealingin 2021, the Affordable Care Act. It is unclear whetherU.S. Supreme Court dismissed the Affordable Care Act will be overturned, repealed, replaced, or further amended. We cannot predict what effect further changeslatest judicial challenge to the Affordable Care Act would havebrought by several states without specifically ruling on our business.
Other legislative changes have been proposed and adopted in the United States sinceconstitutionality of the Affordable Care ActAct. As another example, revisions to regulations under the federal anti-kickback statute would remove protection for traditional Medicare Part D discounts offered by pharmaceutical manufacturers to pharmacy benefit managers and health plans. Pursuant to court order, the removal was enacted. In August 2011,delayed and recent legislation imposed a moratorium on implementation of the Budget Control Act of 2011, among other things, included aggregate reductions of Medicare paymentsrule until January 2032. As another example, the IRA drug price negotiation program has been challenged in litigation filed by various pharmaceutical manufacturers and industry groups.
There have also been efforts by federal and state government officials or legislators to providers of 2% per fiscal year, which went into effect in April 2013 and, dueimplement measures to subsequent legislative amendments to the statute, will remain in effect through 2030, with a temporary suspension from May 1, 2020 through December 31, 2020, unless additional Congressional action is taken. In addition, in January 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, further reduced Medicare payments to several providers,regulate prices or payment for pharmaceutical products, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
Moreover, payment methodologies may be subject to changes in healthcare legislation and regulatory initiatives. For example, CMS may develop new payment and delivery models, including bundled payment models. In addition, recentlyon drug importation. Recently, there has been
heightened governmental scrutiny over the manner in which manufacturers set prices for their commercial products, which has resulted in several Congressional inquiries and proposed and enacted state and federal legislation designed to, among other things, bring more transparency to product pricing, review the relationship betweenpharmaceutical pricing and manufacturer patient programs, and reform government program reimbursement methodologies for pharmaceutical products. Although a numberproposals to address the perceived high cost of these measures may require additional authorization to become effective, Congress and the current administration have each indicated that it will continue to seek new legislative and/or administrative measures to control drug costs.
On July 24, 2020, President Trump announced a number of executive orders related to prescription drug pricing that attempt to implement several of the Administration’s proposals, including a policy that would tie Medicare Part B drug prices to international drug prices; one that directs HHS to finalize the Canadian drug importation proposed rule previously issued by HHS and makes other changes allowing for personal importation of drugs from Canada; and one that directs HHS to finalize the rulemaking process on modifying the anti-kickback law safe harbors for plans, pharmacies, and pharmaceutical benefit managers after HHS confirms that the action is not projected to increase federal spending, Medicare beneficiary premiums, or patients’ total out-of-pocket costs. The probability of success of these newly announced policies and their impact on the U.S. prescription drug marketplace is unknown. There are likely to be political and legal challenges associated with implementing these reforms as they are currently envisioned.
pharmaceuticals. Individual states in the United States have also increasingly passed legislation and implemented regulations designed to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing.
General legislative cost control measures may also affect reimbursement for our product candidates. The Budget Control Act, as amended, resulted in the imposition of reductions in Medicare (but not Medicaid) payments to providers in 2013 and will remain in effect through 2032 unless additional Congressional action is taken. Any significant spending reductions affecting Medicare, Medicaid or other publicly funded or subsidized health programs that may be implemented and/or any significant taxes or fees that may be imposed on us could have an adverse impact on our results of operations.
Adoption of new legislation at the federal or state level could affect demand for, or pricing of, our current or future products if approved for sale. We cannot, however, predict the ultimate content, timing or effect of any federal and state reform efforts. There is no assurance that federal or state health care reform will not adversely affect our future business and financial results.
Outside the United States, ensuring coverage and adequate payment for a product also involves challenges. Pricing of prescription pharmaceuticals is subject to government control in many countries. Pricing negotiations with government authorities can extend well beyond the receipt of regulatory approval for a product and may require a clinical trial that compares the cost-effectiveness of a product to other available therapies. The conduct of such a clinical trial could be expensive and result in delays in commercialization. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any products, if approved in those countries.
Other U.S. Environmental, Health and Safety Laws and Regulations
We may be subject to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment and disposal of hazardous materials and wastes. From time to time and in the future, our operations may involve the use of hazardous and flammable materials, including chemicals and biological materials, and may also produce hazardous waste products. Even if we contract with third parties for the disposal of these materials and waste products, we cannot completely eliminate the risk of contamination or injury resulting from these materials. In the event of contamination or injury resulting from the use or disposal of our hazardous materials, we could be held liable for any resulting damages, and any liability could exceed our resources. We also could incur significant costs associated with civil or criminal fines and penalties for failure to comply with such laws and regulations.
We maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees, but this insurance may not provide adequate coverage against potential liabilities. However, we do not maintain insurance for environmental liability or toxic tort claims that may be asserted against us.
In addition, we may incur substantial costs in order to comply with current or future environmental, health and safety laws and regulations. Current or future environmental laws and regulations may impair our research, development or production efforts. In addition, failure to comply with these laws and regulations may result in substantial fines, penalties or other sanctions.
Government Regulation of Drugs Outside of the United States
To market any product outside of the United States, we would need to comply with numerous and varying regulatory requirements of other countries and jurisdictions regarding quality, safety and efficacy and governing, among other things, clinical trials, marketing authorization, commercial sales and distribution of drug products. Whether or not we obtain FDA approval for a product, we would need to obtain the necessary approvals by the comparable foreign regulatory authorities before we can commence clinical trials or marketing of the product in those countries or jurisdictions. The approval process ultimately varies between countries and jurisdictions and can involve additional product testing and additional administrative review periods. The time required to obtain approval in other countries and jurisdictions might differ from and be longer than that required to obtain FDA approval. Regulatory approval in one country or jurisdiction does not ensure regulatory approval in
another, but a failure or delay in obtaining regulatory approval in one country or jurisdiction may negatively impact the regulatory process in others. As in the United States, post-approval regulatory requirements, such as those regarding product manufacture, marketing, or distribution would apply to any product that is approved outside the United States.
Data Privacy Regulations
The conduct of our clinical trials may be subject to privacy restrictions based on U.S. and non-U.S. regulations. For example, we may be subject to the California Consumer Privacy Act, or CCPA. As currently written, the CCPA may impact our business activities and exemplifies the vulnerability of our business to the evolving regulatory environment related to personal data and protected health information. Additionally, the collection, use, storage, disclosure, transfer, or other processing of personal data regarding individuals in the EU and the UK, including personal health data, is subject to the EU General Data Protection Regulation or GDPR,(“GDPR”) including as it forms part of the law of England and Wales, Scotland and Northern Ireland by virtue of section 3 of the European Union (Withdrawal) Act 2018 and as amended by the Data Protection, Privacy and Electronic Communications (Amendments etc.) (EU Exit) Regulations 2019 (SI 2019/419), known as UK GDPR. Compliance with the GDPR and the UK GDPR will be a rigorous and time-intensive process that may increase our cost of doing business or require us to change our business practices, and despite those efforts, there is a risk that we may be subject to fines and penalties, litigation, and reputational harm in connection with our European activities. The UK’s data protection authority, the Information Commissioner’s Office, has indicated that following Brexit it will continue to enforce the UK GDPR in line with the enforcement of the GDPR in the EU. However, the UK government recently announced its intention to adopt a more flexible approach to the regulation of data, and as a result, there remains a risk of future divergence between the EU and UK data protection regimes. In addition, we may be subject to the California Consumer Privacy Act (“CCPA”) and other U.S. privacy laws. Although the CCPA does not apply directly to our clinical trials, it does impact our collection of information regarding investigators, business contacts, website users and other
data subjects. As currently written, the CCPA may impact our business activities and exemplifies the vulnerability of our business to the evolving regulatory environment related to personal data and protected health information.
Human Capital Resources
As of December 31, 2021,2023, we had 119116 full-time employees. 49We consider our employees to be our greatest asset and have assembled a team with deep scientific, clinical, manufacturing, business, and leadership expertise in biotechnology, platform research, drug discovery, and development. 54 of our employees have M.D. or Ph.D. degrees. Within our workforce, 9488 employees are engaged in research and development and 2528 are engaged in business development, finance, legal, and general management and administration. None of our employees are represented by labor unions or covered by collective bargaining agreements. We consider our relationship with our employees to be good.
Our Corporate Information
We were formed as a Delaware corporation in October 2015 under the name Foghorn Therapeutics Inc. Our principal executive office is located at 500 Technology Square, Suite 700, Cambridge, Massachusetts, 02139, and our phone number is 617-586-3100. Our website address is https://foghorntx.com. Our website and the information contained on, or that can be accessed through, the website will not be deemed to be incorporated by reference in, and are not considered part of, this Annual Report on Form 10-K.
We are an “emerging growth company” as defined in the Jumpstart Our Business Startups Act of 2012. We will remain an emerging growth company until the earlier of: (i) the last day of the fiscal year (a) following the fifth anniversary of the completion of the IPO,our initial public offering (“IPO”), (b) in which we have total annual gross revenue of at least $1.07$1.235 billion, or (c) in which we are deemed to be a large accelerated filer, which means the market value of our common stock that is held by non-affiliates exceeds $700.0 million as of the prior June 30th, and (ii) the date on which we have issued more than $1.0 billion in non-convertible debt during the prior three-year period.
We are also a “smaller reporting company” as defined in the Securities and Exchange Act of 1934, as amended or the Exchange Act.(the “Exchange Act”). We may continue to be a smaller reporting company even after we are no longer an emerging growth company. We may take advantage of certain of the scaled disclosures available to smaller reporting companies until the fiscal year following the determination that our voting and non-voting common stock held by non-affiliates is more than $250 million measured on the last business day of our second fiscal quarter, or our annual revenues are more than $100 million during the most recently completed fiscal year and our voting and non-voting common stock held by non-affiliates is more than $700 million measured on the last business day of our second fiscal quarter.
Available Information
Our Internet address is https://foghorntx.com. Our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, including exhibits, proxy and information statements and amendments to those reports filed or furnished pursuant to Sections 13(a) and 15(d) of the Exchange Act are available through the “Investors” portion of our website free of charge as soon as reasonably practicable after we electronically file such material with, or furnish it to, the SEC. Information on
our website is not part of this Annual Report on Form 10-K or any of our other securities filings unless specifically incorporated herein by reference. In addition, our filings with the SEC may be accessed through the SEC’s Electronic Data Gathering, Analysis and Retrieval system at http://www.sec.gov. All statements made in any of our securities filings, including all forward-looking statements or information, are made as of the date of the document in which the statement is included, and we do not assume or undertake any obligation to update any of those statements or documents unless we are required to do so by law.
We have adopted a written code of business conduct and ethics that applies to our directors, officers and employees, including our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions. A current copy of the code is posted to the “Investors” portion of our website. In addition, we intend to post on our website all disclosures that are required by law or listing rules concerning any amendments to, or waivers from, any provision of the code.
ITEM 1A. RISK FACTORS
Investing in our common stock involves a high degree of risk. You should carefully consider the risks and uncertainties described below together with all of the other information contained in this Annual Report on Form 10-K, including our consolidated financial statements and related notes, before deciding to invest in our common stock. Some of the following risks and uncertainties are, and will be, exacerbated by the COVID-19 pandemic (including any resurgences thereof) and any worsening of the global business and economic environment as a result. If any of the events or developments described below were to occur, our business, prospects, operating results and financial condition could suffer materially, the trading price of our common stock could decline and you could lose all or part of your investment. The risks and uncertainties described below are not the only ones we face. Additional risks and uncertainties not presently known to us or that we currently believe to be immaterial may also adversely affect our business.
Risks Related to Our Financial Position and Need for Additional Capital
We have a limited operating history and have no products approved for commercial sale, which may make it difficult for you to evaluate our current business and predict our future success and viability.
We are a clinical-stage biopharmaceutical company with a limited operating history. We were incorporated in October 2015, and our operations to date have been focused on building our proprietary Gene Traffic Control platform, organizing and staffing our company, business planning, raising capital, conducting discovery and research activities, conducting early stage clinical trials, protecting our trade secrets, filing patent applications, identifying potential product candidates, undertaking preclinical studies and establishing arrangements with third parties for the manufacture of initial quantities of our product candidates and component materials. We are currently in a Phase 1 clinical trialstrial for FHD-286 and FHD-609, and allanticipate an IND filing by Lilly for FHD-909 in the second quarter of our2024. Our other product candidates are in preclinical development. We have not yet demonstrated an ability to successfully complete any clinical trials, obtain marketing approvals, manufacture a commercial-scale product or arrange for a third party to do so on our behalf, or conduct sales, marketing and distribution activities necessary for successful product commercialization.
In addition, as a young business, we may encounter unforeseen expenses, difficulties, complications, delays and other known and unknown factors. We will need to transition at some point from a company with a research and development focus to a company capable of supporting commercial activities. We may not be successful in such a transition.
We expect our financial condition and results of operations to continue to fluctuate significantly from quarter to quarter and year to year due to a variety of factors, many of which are beyond our control. Accordingly, you should not rely upon the results of any quarterly or annual periods as indications of future operating performance.
We have incurred significant losses since inception. We expect to incur losses for the foreseeable future and may never achieve or maintain profitability.
Since inception, we have incurred significant operating losses. As of December 31, 2021,2023, we had an accumulated deficit of $264.3$471.6 million. We have financed our operations primarily through private placements of our preferred stock and our initial public offering, as well as through a loan with Oxford Finance LLC, or Oxford Finance, which was repaid in full during fiscal year 2021;IPO; our former collaboration agreement with Merck; and our strategic collaboration with Loxo Oncology at Lilly and Lilly's concurrent investment in our equity. For further information about our collaborations and Lilly's equity investment, see “Business—License AgreementStrategic Collaboration with Merck” and "Business—Strategic Collaboration."Lilly.” We have devoted all of our efforts to research and development. We expect to continue to incur significant expenses and increasing operating losses for the foreseeable future. The net losses we incur may fluctuate significantly from quarter to quarter. We anticipate that our expenses will increase substantially if and as we:
•advance our FHD-286 and FHD-609 product candidatescandidate and continue our preclinical and clinical development of product candidates from our current research programs;programs, including those partnered with Lilly;
•identify additional research programs and additional product candidates;
•initiate preclinical testing for any new product candidates we identify and develop;
•obtain, maintain, expand, enforce, defend and protect our trade secrets and intellectual property portfolio and provide reimbursement of third-party expenses related to our patent portfolio;
•hire additional research and development personnel;
•add operational, legal, compliance, financial and management information systems and personnel to support our research, product development and operations as a public company;
•expand the capabilities of our platform;
•acquire or in-license product candidates, intellectual property and technologies;
•operate as a public company;
•seek marketing approvals for any of our product candidates that successfully complete clinical trials; and
•ultimately establish a sales, marketing, and distribution infrastructure to commercialize any products for which we may obtain marketing approval.
We have twoone product candidatescandidate in Phase 1 clinical development and anticipate an IND filing for our partnered candidate with Lilly in the second quarter of 2024. In April 2023, we decided not to pursue a Phase 1 independent dose expansion study for FHD-609 after the study was placed on partial clinical hold. We have not initiated clinical development of anyour other product candidatecandidates and expect that it will be many years, if ever, before we have a product candidate ready for commercialization. To become and remain profitable, we must develop and, either directly or through collaborators, eventually commercialize a medicine or medicines with significant market potential. This will require us to be successful in a range of challenging
activities, including identifying product candidates, completing preclinical testing and clinical trials of product candidates, obtaining marketing approval for these product candidates, manufacturing, marketing, and selling those medicines for which we may obtain marketing approval, and satisfying any post-marketing requirements. We may never succeed in these activities and, even if we do, may never generate revenues that are significant or large enough to achieve profitability. We are unable to predict the extent of any future losses or when we will become profitable, if at all. If we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis.
We will need substantial additional funding. If we are unable to raise capital when needed, we would be forced to delay, reduce, or eliminate our research and product development programs or future commercialization efforts.
We expect our expenses to increase in connection with our ongoing activities, particularly as we identify, continue the research and development of, initiate clinical trials of, and seek marketing approval for, our product candidates. Accordingly, we will need to obtain substantial additional funding in connection with our continuing operations. If we are unable to raise capital when needed or on attractive terms, we would be forced to delay, reduce, or eliminate our research and product development programs or future commercialization efforts.
As of December 31, 2021, our cash, cash equivalents and marketable securities were $154.3 million. The net proceeds of our initial public offering were $107.9 million, after deducting underwriting discounts and commissions and other offering expenses. On November 19, 2020, we received additional net proceeds of $14.2 million in connection with the underwriters’ partial exercise of their option to purchase additional shares of common stock in our initial public offering. On December 10, 2021, we issued and sold to Lilly 4,000,000 shares of our common stock at a price of $20.00 per share for aggregate gross proceeds of $80.0 million,
Additional fundraisingcapital raising efforts, when needed, may divert our management’s attention from their day-to-day activities, which may adversely affect our ability to advance our product candidates or develop new product candidates. We cannot be certain that additional funding will be available on acceptable terms, or at all. If we are unable to raise additional capital in sufficient amounts or on terms acceptable to us, we may have to significantly delay, scale back or discontinue the development or commercialization of our product candidates or other research and development initiatives.
If we are unable to obtain funding on a reasonable and timely basis, we may be required to significantly curtail, delay or discontinue one or more of our research or development programs, clinical research, or the commercialization of any product candidate. We may be unable to expand our operations or otherwise capitalize on our business opportunities as desired. Any of the above events could significantly harm our business, prospects, financial condition and results of operations and cause the price of our common stock to decline.
We have never generated revenue from product sales and may never be profitable.
We are currently in the Phase 1 clinical development stage for our two most advanced product candidates,candidate, FHD-286 and FHD-609, andanticipate an IND filing by Lilly for FHD-909 in the second quarter of 2024. We are in the preclinical development stage for our other lead research programs. We expect that it will be many years, if ever, before we have a product candidate ready for commercialization. To become and remain profitable, we must succeed in
developing, obtaining marketing approval for and commercializing products that generate significant revenue. This will require us to be successful in a range of challenging activities, including completing preclinical testing and clinical trials of our current or future product candidates, establishing and maintaining arrangements with third parties for the manufacture of clinical supplies of our product candidates, obtaining marketing approval for our product candidates and manufacturing, marketing, selling and obtaining reimbursement for any products for which we may obtain marketing approval. We may never succeed in these activities and, even if we do, may never generate revenues that are significant enough to achieve profitability.
Unfavorable global macroeconomic conditions, geopolitical trends, and armed conflict could adversely affect our business, financial condition or results of operations.
Our results of operations could be adversely affected by general conditions in the global economy and financial markets, including inflation, rising interest rates, economic sanctions, natural disasters, pandemics, political instability, armed conflicts and wars, including the Russia-Ukraine war, the Israeli-Palestine Conflict, and attacks in the Red Sea. A severe or prolonged economic downturn, or additional global financial or political crises, could result in a variety of risks to our business, including weakened demand for our product candidates, if approved, or our ability to raise additional capital when needed on acceptable terms, if at all. A weak or declining economy could also strain our suppliers, possibly resulting in supply disruption. Any of the foregoing could harm our business and we cannot anticipate all of the ways in which the current economic climate and financial market conditions.
U.S. federal income tax reform could adversely affect our business and financial condition.
The rules dealing with U.S. federal, state, and local income taxation are constantly under review by persons involved in the legislative process and by the Internal Revenue Service and the U.S. Treasury Department. Changes to tax laws (which changes may have retroactive application) could adversely affect us or holders of our common stock. In recent years, many such changes have been made and changes are likely to continue to occur in the future. For example, on March 27, 2020, President Trump signed into law the Coronavirus Aid, Relief, and Economic Security Act, which included certain changes in tax law intended to stimulate the U.S. economy in light of the COVID-19 coronavirus outbreak, including temporary beneficial changes to the treatment of net operating losses, interest deductibility limitations and payroll tax matters. Additionally, on December 22, 2017, President Trump signed into law the Tax Cuts and Jobs Act, or the TCJA, which significantly reformed the Internal Revenue Code of 1986, as amended, or the Code. The TCJA included significant changes to corporate and individual taxation, some of which could adversely impact an investment in our common stock. Future changes in tax laws could have a material adverse effect on our business, cash flow, financial condition or results of operations. We urge investors to consult with their legal and tax advisers regarding the implications of potential changes in tax laws on an investment in our common stock.
Our future ability to utilize our net operating loss carryforwards and certain other tax attributes may be limited.
We have incurred substantial losses during our history and we may never achieve profitability. To the extent that we continue to generate taxable losses, unused losses will carry forward to offset a portion of future taxable income, if any, subject to expiration in the case of carryforwards generated prior to January 1, 2018. Additionally, we continue to generate business tax credits, including research and development tax credits, which generally may be carried forward to offset a portion of future taxable income, if any, subject to expiration of such credit carryforwards. Under Sections 382 and 383 of the Code, if a corporation undergoes an “ownership change,” generally defined as a greater than 50 percentage point change (by value) in its equity ownership over a three-year period, the corporation’s ability to use its pre-change net operating loss carryforwards, or NOLs, and other pre-change tax attributes (such as research and development tax credits) to offset its post-change income or taxes may be limited. Our prior equity offerings and other changes in our stock ownership may have resulted in such ownership changes. We may also experience ownership changes in the future or subsequent shifts in our stock ownership, some of which are outside of our control. As a result, if we earn net taxable income, our ability to use our pre-change NOLs or other pre-change tax attributes to offset U.S. federal taxable income may be subject to limitations, which could potentially result in increased future tax liability to us. Additionally, for taxable years beginning after December 31, 2021, the deductibility of such U.S. federal NOLs is limited to 80% of our taxable income in any future taxable year. There is a risk that under existing tax laws, changes thereto, regulatory changes, or other unforeseen reasons, our existing NOLs or business tax credits could expire or otherwise be unavailable to offset future income tax liabilities. At the state level, there may also be periods during which the use of NOLs or business tax credits is suspended or otherwise limited, which could accelerate or permanently increase state taxes owed. For these reasons, we may not be able to realize a tax benefit from the use of our NOLs or tax credits, even if we attain profitability.
Risks Related to Discovery and Development
We are heavily dependent on the success of our product candidates, which are in preclinical and Phase 1early clinical development. We may not be successful in our efforts to identify and develop potential product candidates. If these efforts are unsuccessful, or if we experience significant delays, we may never become a commercial stage company or generate any revenues, and our business will be materially harmed.
The success of our business depends primarily upon our ability to identify, develop, and commercialize product candidates based on our platform. All of our product development programs are still in the research or preclinical or Phase 1early clinical stage of development. Our research programs may fail to identify potential product candidates for clinical development for a number of reasons. Our research methodology may be unsuccessful in identifying potential product candidates, our potential product candidates may be shown to have harmful side effects in preclinical in vitro experiments or animal model studies, they may not show promising signals of therapeutic effect in such experiments or studies or they may have other characteristics that may make the product candidates impractical to administer or market.
If any of these events occurs, we may be forced to abandon our research or development efforts for a program or programs, which would have a material adverse effect on our business, financial condition, results of operations, and prospects. Research programs to identify new product candidates require substantial technical, financial, and human resources. We may focus our efforts and resources on potential programs or product candidates that ultimately prove to be unsuccessful, which would be costly and time-consuming.
The success of our product candidates will depend on several factors, including but not limited to the following:
•successful completion of preclinical studies;
•successful submission of INDs and initiation of clinical trials;
•establishing an acceptable safety profile of the products and maintaining such a profile following approval;
•achieving desirable therapeutic properties for our product candidates’ intended indications;
•making arrangements with third-party manufacturers, or establishing manufacturing capabilities, both for clinical and commercial supplies of our product candidates;
•receipt and related terms of marketing approvals from applicable regulatory authorities;
•obtaining and maintaining patent and trade secret protection and regulatory exclusivity of our product candidates;
•establishing sales, marketing and distribution capabilities and launching commercial sales of our products; if and when approved, whether alone or in collaboration with others; acceptance of our products, if and when approved, by patients, the medical community and third-party payors;
•obtaining and maintaining third-party coverage and adequate reimbursement;
•effectively competing with other therapies; and
•sufficiency of our financial and other resources.
If we do not successfully achieve one or more of these factors in a timely manner, or at all, we could experience significant delays or an inability to successfully commercialize our product candidates, which could materially harm our business. Moreover, if we do not receive regulatory approvals, we may not be able to continue our operations.
Our clinical trials may fail to demonstrate substantial evidence of the safety and efficacy of our product candidates, which would delay or prevent regulatory approval of the product candidates, limit the commercial potential, or result in significant negative consequences following any potential marketing approval.
To obtain the requisite regulatory approvals to market and sell any of our product candidates, we must demonstrate through extensive preclinical studies and clinical trials that such product candidates are safe and effective for use in each targeted indication. Failure can occur at any time during the clinical development process. Most product candidates that begin clinical trials are never approved by regulatory authorities for commercialization. It is impossible to predict when or if any product candidates we may develop will prove safe in humans. Our clinical trials may fail to demonstrate with substantial evidence from adequate and well-controlled trials, and to the satisfaction of the FDA or comparable foreign regulatory authorities, that such product candidates are safe and effective for their intended uses. There can be no assurance that our clinical trials will not cause undesirable side effects.
If any product candidates we develop are associated with or cause serious adverse events, undesirable side effects, or unexpected characteristics, we may need to abandon their development or limit development to certain uses or subpopulations in which the serious adverse events, undesirable side effects or other characteristics are less prevalent, less severe, or more acceptable from a risk-benefit perspective, any of which would have a material adverse effect on our business, financial condition, results of operations, and prospects. Many product candidates that initially showed promise in early stage testing for treating cancer or other diseases have later been found to cause side effects that prevented further clinical development of the product candidates. Additionally, any safety concerns observed in any one of our clinical trials in our targeted indications could limit the prospects for regulatory approval of our product candidates in those and other indications. For example, in May 2022, our Phase 1 study of FHD-286 in AML/MDS was placed on a full clinical hold by the FDA in August of 2022 due to the observation of potential DS and potential linkage to grade 5 safety events. DS is associated with AML therapeutics that induce differentiation of blast cells into normal myeloid cells, an effect that is believed to be on target for the proposed mechanism of FHD-286. The full clinical hold was lifted in June of 2023 by the FDA. An expert panel was assembled to adjudicate the rate and severity of DS in this study. The adjudicated rate of DS by the panel was determined to be 15 percent (n=6 out of 40 patients) and classified one case as definitive DS, five cases as indeterminate and none contributing to a patient’s death. Even if we are able to demonstrate that all future serious adverse events are not product-related, such occurrences could affect patient recruitment or the ability of enrolled patients to complete the trial, and the FDA could potentially impose or reimpose a clinical hold in the future on studies evaluating FHD-286.
Moreover, if our product candidates are associated with undesirable side effects in preclinical studies or clinical trials or have characteristics that are unexpected, we may elect to abandon their development or limit their development to more narrow uses or subpopulations in which the undesirable side effects or other characteristics are less prevalent, less severe or more acceptable from a risk-benefit perspective, which may limit the commercial expectations for the product candidate if approved.
Additionally, adverse developments in clinical trials of pharmaceutical and biopharmaceutical products conducted by others may cause the FDA or other regulatory oversight bodies to suspend or terminate our clinical trials or to change the requirements for approval of any of our product candidates.
Any of these events could prevent us from achieving or maintaining market acceptance of any product candidates we may identify and develop and could have a material adverse effect on our business, financial condition, results of operations, and prospects.
Even if our clinical trials are successfully completed, clinical data are often subject to varying interpretations and analyses, and we cannot guarantee that the FDA or comparable foreign regulatory authorities will interpret the results as we do. Results acceptable to support approval in one jurisdiction may be deemed inadequate by another regulatory authority to support regulatory approval in that other jurisdiction. Even if regulatory approval is secured for a product candidate, the terms of such approval may also limit its commercial potential.
We may not be able to file INDs or IND amendments to commence clinical trials of our product candidates on the timelines we or our partners expect, and even if we are able to, the FDA may not permit us to proceed.
In order to commence a clinical trial in the United States, we and our partner are required to seek FDA acceptance of an IND for each of our product candidates. We cannot be sure any IND we and our partners submit to the FDA, or any similar clinical trial application we and our partners submit in other countries, will be accepted. We may also be required to conduct additional
preclinical testing prior to filing or acceptance of an IND for any of our product candidates, and the results of any such additional preclinical testing may not be positive.
Further, we may experience manufacturing delays or other delays with IND-enabling studies. Moreover, we cannot be sure that even once clinical trials have begun, issues will not arise that suspend or terminate clinical trials. Additionally, even if the FDA agrees with the design and implementation of the clinical trials set forth in an IND, we cannot guarantee that the FDA will not change its requirements in the future. These considerations also apply to new clinical trials we may submit as amendments to existing INDs or to a new IND. Any failure to file INDs on the timelines we expect or to obtain regulatory authorizations for our trials to proceed may prevent us from completing our clinical trials or commercializing our product candidates on a timely basis, if at all.
There is substantial competition in our field, which may result in others developing or commercializing products before we do.
The biotechnology and pharmaceutical industries utilize rapidly advancing technologies and are characterized by intense competition. While we believe that our scientific knowledge and platform development expertise provide us with competitive advantages, we face potential competition from many different sources, including major pharmaceuticals, specialty pharmaceuticals and biotechnology companies, academic institutions and government agencies, and public and private research institutes that conduct research, development, manufacturing and commercialization. Many of our competitors have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, enrolling and conducting clinical trials, and seeking regulatory approvals and product marketing than we do, and have potential to advance products competitive with our product candidates or other programs addressing the chromatin regulatory system at a
rapid pace. In addition, our competitors may compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs. Our competitors may advance competing product candidates that have a more attractive product profile than our product candidates, make progress examining the chromatin regulatory system or bring a product to market before we can. Any of these developments could put us at a significant competitive disadvantage and have a material adverse effect on the prospects of our business.
Product candidates that we and our collaborators successfully develop and commercialize will compete with existing therapies and new therapies that may become available in the future. While we are not aware of other companies addressing the chromatin regulatory system at scale, in context and in an integrated way, we are aware of efforts to bring products to market that could be competitive with ours if our programs are successful. Specifically, we expect that our product candidates will compete against approved drugs for the treatment of AML, including Idhifa® by Celgene Corporation,Bristol Myers Squibb, Tibsovo® by AgiosServier Pharmaceuticals, and Rydapt® by Novartis International AG .AG. If our product candidates are approved for the indications for which we are currently planning clinical trials, they will likely compete with the competitor drugs mentioned above and with other drugs that are currently in development. Key product features that would affect our ability to effectively compete with other therapeutics include the efficacy, safety and convenience of our products. Our competitors may obtain patent protection or other intellectual property rights that limit our ability to develop or commercialize our product candidates. Our competitors may also obtain FDA or other regulatory approval for their products more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market. For additional information regarding our competition, see “Business—Competition.”
Our lead product candidates utilizecandidate utilizes a novel mechanismsmechanism of action, which may result in greater research and development expenses, regulatory issues that could delay or prevent approval, or discovery of unknown or unanticipated adverse effects.
Our lead product candidates utilizecandidate, FHD-286, utilizes a novel mechanismsmechanism of action, which may result in greater research and development expenses, regulatory issues that could delay or prevent approval, or discovery of unknown or unanticipated adverse effects. For example, FHD-609 is a protein degrader. Currently there are no approved medicines using this mechanism of action. Because FHD-609 in particular utilizes a novel mechanism of action that has not been the subject of extensive study compared to more well-known product candidates, there is also an increased risk that we may discover previously unknown or unanticipated adverse effects during our preclinical studies and clinical trials. Any such events could adversely impact our business prospects, financial condition and results of operations.
In addition, a novel mechanism of action may lengthen the regulatory review process, require us to conduct additional studies or clinical trials, increase our development costs, lead to changes in regulatory positions and interpretations, delay or prevent approval and commercialization of our product candidates or lead to significant post-approval limitations or restrictions. The novel mechanism of action also means that fewer people are trained in or experienced with product candidates of this type, which may make it more difficult to find, hire and retain personnel for research, development and manufacturing positions.
Our clinical trials may fail to demonstrate substantial evidence of the safety and efficacy of our product candidates, which would delay or prevent regulatory approval of the product candidates, limit the commercial potential, or result in significant negative consequences following any potential marketing approval.
To obtain the requisite regulatory approvals to market and sell any of our product candidates, we must demonstrate through extensive preclinical studies and clinical trials that such product candidates are safe and effective for use in each targeted indication. Failure can occur at any time during the clinical development process. Most product candidates that begin clinical trials are never approved by regulatory authorities for commercialization. We only began evaluating two product candidates in human clinical trials during 2021. Moreover, we are not aware of any other clinical trials involving products that interact with BAF complexes to affect the chromatin regulatory system in a similar manner to our products. It is impossible to predict when or if any product candidates we may develop will prove safe in humans. Our clinical trials may fail to demonstrate with substantial evidence from adequate and well-controlled trials, and to the satisfaction of the FDA or comparable foreign regulatory authorities, that such product candidates are safe and effective for their intended uses. There can be no assurance that our clinical trials will not cause undesirable side effects.
If any product candidates we develop are associated with or cause serious adverse events, undesirable side effects, or unexpected characteristics, we may need to abandon their development or limit development to certain uses or subpopulations in which the serious adverse events, undesirable side effects or other characteristics are less prevalent, less severe, or more acceptable from a risk-benefit perspective, any of which would have a material adverse effect on our business, financial condition, results of operations, and prospects. Many product candidates that initially showed promise in early stage testing for treating cancer or other diseases have later been found to cause side effects that prevented further clinical development of the
product candidates. Additionally, any safety concerns observed in any one of our clinical trials in our targeted indications could limit the prospects for regulatory approval of our product candidates in those and other indications.
Moreover, if our product candidates are associated with undesirable side effects in preclinical studies or clinical trials or have characteristics that are unexpected, we may elect to abandon their development or limit their development to more narrow uses or subpopulations in which the undesirable side effects or other characteristics are less prevalent, less severe or more acceptable from a risk-benefit perspective, which may limit the commercial expectations for the product candidate if approved.
Additionally, adverse developments in clinical trials of pharmaceutical and biopharmaceutical products conducted by others may cause the FDA or other regulatory oversight bodies to suspend or terminate our clinical trials or to change the requirements for approval of any of our product candidates.
Any of these events could prevent us from achieving or maintaining market acceptance of any product candidates we may identify and develop and could have a material adverse effect on our business, financial condition, results of operations, and prospects.
Even if our clinical trials are successfully completed, clinical data are often subject to varying interpretations and analyses, and we cannot guarantee that the FDA or comparable foreign regulatory authorities will interpret the results as we do. Results acceptable to support approval in one jurisdiction may be deemed inadequate by another regulatory authority to support regulatory approval in that other jurisdiction. Even if regulatory approval is secured for a product candidate, the terms of such approval may also limit its commercial potential.
Product development is a lengthy and expensive process with an uncertain outcome. We may incur unexpected costs or experience delays in completing, or ultimately be unable to complete, the development and commercialization of our product candidates.
We have twoone product candidatescandidate, FHD-286, in Phase 1 clinical development; all ofdevelopment and anticipate that Lilly will file an IND and begin a Phase 1 for our partnered product candidate, FHD-909, later this year; our other product candidates are in preclinical
development, and their risk of failure is high. We are unable to predict when or if any of our product candidates will prove effective or safe in humans or will receive marketing approval. Before obtaining marketing approval from regulatory authorities for the sale of any product candidate, we must conduct extensive clinical trials to demonstrate the safety and efficacy of our product candidates in humans. Before we can commence clinical trials for a product candidate, we must complete extensive preclinical testing and studies that support our planned INDs in the United States or similar applications in other jurisdictions. We cannot be certain of the timely completion or outcome of our preclinical testing and studies and cannot predict if the FDA or similar regulatory authorities outside the United States will accept our proposed clinical programs or if the outcome of our preclinical testing and studies ultimately will support the further development of our programs.
Clinical testing is expensive, difficult to design and implement, can take many years to complete and is uncertain as to the outcome. A failure of one or more clinical trials can occur at any stage of testing. We cannot guarantee that any of our ongoing and planned clinical trials will be conducted as planned or completed on schedule, if at all. Moreover, we may experience numerous unforeseen events during, or as a result of, clinical trials, that could delay or prevent our ability to receive marketing approval or commercialize our product candidates, including:
•delays in discussions with or obtaining alignment with regulators regarding trial design;
•the supply or quality of our product candidates or other materials necessary to conduct clinical trials of our product candidates may be insufficient or inadequate, including as a result of delays in the testing, validation, manufacturing and delivery of product candidates to the clinical sites by us or by third parties with whom we have contracted to perform certain of those functions;
•we may experience delays in reaching, or may fail to reach, agreement on acceptable clinical trial contracts or clinical trial protocols with prospective trial sites;
•we may experience delays in enrolling patients or may compete with other trials to enroll patients, including due to our targeted disease having small patient populations;patients;
•regulators or institutional review boards may not authorize us or our investigators to commence a clinical trial or conduct a clinical trial at a prospective trial site;
•we may experience difficulty in designing clinical trials and in selecting endpoints for diseases that have not been well-studied and for which the natural history and course of the disease is poorly understood;
•the selection of certain clinical endpoints may require prolonged periods of clinical observation or analysis of the resulting data;
•the number of patients required for clinical trials of our product candidates may be larger than we anticipate, enrollment in these clinical trials may be slower than we anticipate or participants may drop out of these clinical trials at a higher rate than we anticipate;
•we may fail to perform clinical trials in accordance with the FDA’s or any other regulatory authority’s good clinical practices or GCP,(“GCP”) requirements, or regulatory guidelines in other countries;
•our product candidates may have undesirable side effects or other unexpected characteristics, or adverse events associated with the product candidate may occur which are viewed to outweigh its potential benefits, causing us or our investigators, regulators or institutional review boards to suspend or terminate the trials;
•we may have to suspend or terminate clinical trials of our product candidates for various reasons, including a finding that the participants are being exposed to unacceptable health risks;
•our third-party contractors may fail to comply with regulatory requirements or meet their contractual obligations to us in a timely manner, or at all;
•regulators or institutional review boards may require that we or our investigators suspend or terminate clinical trials for various reasons, including noncompliance with regulatory requirements;
•clinical trials of our product candidates may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional clinical trials or abandon product development programs;
•the cost of clinical trials of our product candidates may be greater than we anticipate;
•disruptions caused by the COVID-19 pandemic may increase the likelihood that we encounter such difficulties or delays in initiating, enrolling, conducting, or completing our planned and ongoing clinical trials; and
•we could be required to conduct additional clinical trials or testing of our product candidates beyond those that we currently contemplate, which may result in a delay in our market approval, limitation of approval for patient populations, distribution limitations, or not obtaining marketing approval at all.
We could also encounter delays if a clinical trial is suspended or terminated by us, the IRBs of the institutions in which such trials are being conducted, or the FDA or comparable foreign regulatory authorities, or is recommended for suspension or termination by the data monitoring committee for such trial. A suspension or termination may be imposed due to a number of
factors, including failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols, inspection of the clinical trial operations or trial site by the FDA or comparable foreign regulatory authorities resulting in the imposition of a clinical hold, unforeseen safety issues or adverse side effects, failure to demonstrate a benefit from using a product or treatment, failure to establish or achieve clinically meaningful trial endpoints, changes in governmental regulations or administrative actions or lack of adequate funding to continue the clinical trial. Many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of our product candidates. Further, the FDA or comparable foreign regulatory authorities may disagree with our clinical trial design and our interpretation of data from clinical trials, or may change the requirements for approval even after they have reviewed and commented on the design for our clinical trials.
Our product development costs also will increase if we experience delays in preclinical studies or clinical trials or in obtaining marketing approvals. We do not know whether any of our preclinical studies or clinical trials will begin as planned, will need to be restructured or will be completed on schedule, or at all. Significant preclinical study or clinical trial delays also could shorten any periods during which we may have the exclusive right to commercialize our product candidates, or could allow our competitors to bring products to market before we do and impair our ability to successfully commercialize our product candidates, which may harm our business, results of operations, financial condition and prospects.
We may not be able to exert unilateral control over the development of product candidates when part of a collaboration.
Under our Lilly Collaboration Agreement, we influence, but do not control, the development activity of any of the product candidates covered by the Lilly Collaboration Agreement, including FHD-909. This may result in delayed and/or diminished visibility and predictability of certain aspects of development strategy, which may impact timelines and ultimate success of the product candidate.
If we experience delays or difficulties in the enrollment and dosing of patients in our clinical trials, our receipt of necessary regulatory approvals for our product candidates could be delayed or prevented.
Identifying and qualifying patients to participate in clinical trials of FHD-286 FHD-609 or any other product candidates is critical to our success. The timing of our clinical trials depends on our ability to recruit patients to participate in our studies as well as the dosing of such patients and completion of required follow-up periods. Our competitors may compete for the same limited patient populations. If we or our collaborators are unable to locate and enroll a sufficient number of eligible patients to participate in these trials as required by the FDA or other analogous regulatory authorities outside the United States, or as needed to provide appropriate statistical power for a given trial, we may not be able to initiate or continue clinical trials for our
current and future product candidates. Additionally, we may face similar challenges or delays in our other or potential future clinical trials. If patients are unwilling to participate in our studies because of negative publicity from adverse events related to the biotechnology field, competitive clinical trials for similar patient populations or for other reasons, the timeline for recruiting patients, conducting studies and obtaining regulatory approval of FHD-286 FHD-609 or any other product candidates may be delayed. These delays could result in increased costs, delays in advancing our product candidates, delays in testing the effectiveness of our product candidates or termination of the clinical trials altogether. Furthermore, our ability to enroll patients may be significantly delayed by the evolving COVID-19 pandemic and we do not know the extent and scope of such delays at this point.
Patient enrollment is also affected by other factors, including:
•severity of the disease under investigation;
•size of the patient population and process for identifying patients;
•design of the trial protocol;
•availability and efficacy of approved medications for the disease under investigation;
•convenience and ease of administration compared to approved or other investigational medications for the disease under investigation and the willingness of patients to undergo the surgical procedures necessary to administer our product candidates, such as biopsy;
•ability to obtain and maintain patient informed consent;
•risk that enrolled patients will drop out before completion of the trial;
•eligibility and exclusion criteria for the trial in question;
•perceived risks and benefits of the product candidate under trial;
•efforts to facilitate timely enrollment in clinical trials;
•patient referral practices of physicians;
•ability to monitor patients adequately during and after treatment;
•proximity and availability of clinical trial sites for prospective patients; and
•factors we may not be able to control, such as current or potential pandemics that may limit patients, principal investigators or staff or clinical site availability (for example, outbreak of COVID-19).availability.
Enrollment delays in our clinical trials may result in increased development costs for FHD-286 FHD-609 or any other product candidates, which would cause the value of our company to decline and limit our ability to obtain additional financing. If we have difficulty enrolling a sufficient number of patients to conduct our clinical trials as planned, we may need to delay, limit, or terminate clinical trials for FHD-286 FHD-609 or our other product candidates, or expand to additional jurisdictions, which could impose additional challenges on our company and expose us to risks. If we are not successful in conducting our clinical trials as planned, it would have an adverse effect on our business, financial condition, results of operations, and prospects.
Any favorable preclinical results may not be predictive of results that may be observed in clinical trials.
Data obtained from preclinical activities are subject to varying interpretations and analyses, which may delay, limit or prevent regulatory approval. Many companies that have believed their product candidates performed satisfactorily in preclinical studies have nonetheless failed to demonstrate results in clinical studies. As we generate preclinical results, such results will not ensure that later preclinical studies or clinical trials will demonstrate similar results. There is a high failure rate for drugs and biologics proceeding through clinical trials. Even product candidates that reach the clinical trial stage may fail to show the desired safety and efficacy in a later stage of clinical development. A number of companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in later-stage clinical trials even after achieving promising results in the preclinical and early stage clinical trials.
Our approach to the discovery of product candidates is unproven, and we may not be successful in our efforts to use and expand our platform to build a pipeline of product candidates with commercial value.
A key element of our strategy is to use and expand our Gene Traffic Control platform to build a pipeline of product candidates and progress these product candidates through clinical development for the treatment of various cancers and other therapeutic areas. Although our research and development efforts to date have resulted in our discovery and preclinical development of
FHD-286, FHD- 909 and FHD-609 for the treatment of cancer, FHD-286, FHD-909 and FHD-609 may not be safe or effective as cancer treatments, and we may not be able to develop any other product candidates. We may not be successful in identifying further targets in the chromatin regulatory system that are relevant in cancer, or other diseases, and which can be “basketed” into a group that is large enough to present a sufficient commercial opportunity or that is druggable with one chemical compound. Even if we are successful in building our pipeline of product candidates, the potential product candidates that we identify may not be suitable for clinical development or generate acceptable clinical data, including as a result of being shown to have unacceptable toxicity or other characteristics that indicate that they are unlikely to be products that will receive marketing approval from the FDA or other regulatory authorities or achieve market acceptance. If we do not successfully develop and commercialize product candidates, we will not be able to generate product revenue in the future, which likely would result in significant harm to our financial position and adversely affect our stock price.
We rely on third parties to perform pre-clinical experiments, to manufacture our preclinical and clinical product supplies, to produce and process clinical quantities of our product candidates and to assist with clinical trials.
We currently rely on third parties to perform certain pre-clinical experiments, manufacture preclinical and clinical product supplies and to manufacture clinical supplies of our product candidates.candidates, and certain of these third parties are located outside the United States, including in China. We need to negotiate and maintain contractual arrangements with these outside vendors for the supply of our product candidates and we may not be able to do so on favorable terms. We have not yet caused any product candidates to be manufactured on a commercial scale and may not be able to do so for any of our product candidates.
The facilities used by our contract manufacturers to manufacture our product candidates must be approved by the FDA or other foreign regulatory authorities following inspections that will be conducted after we submit an application to the FDA or other foreign regulatory authorities. We will be completely dependent on our contract manufacturing partners for compliance with cGMP and any other regulatory requirements of the FDA or other regulatory authorities for the manufacture of our product candidates. Beyond periodic audits, we have no control over the ability of our contract manufacturers to maintain adequate quality control, quality assurance and qualified personnel. If the FDA or a comparable foreign regulatory authority does not approve these facilities for the manufacture of our product candidates, if it withdraws any approval in the future, or if it otherwise identifies noncompliance with cGMPs at these facilities, we may need to find alternative manufacturing facilities, which would require the incurrence of significant additional costs and significantly impact our ability to develop, obtain regulatory approval for or market our product candidates, if approved. Similarly, if any third-party manufacturers on which we will rely fail to manufacture quantities of our product candidates at quality levels necessary to meet regulatory requirements and at a scale sufficient to meet anticipated demand at a cost that allows us to achieve profitability, our business, financial condition, results of operations, and prospects could be materially and adversely affected.
In addition, we have relied upon and plan to continue to rely upon third-party clinical investigators, contract research organizations, or CROs, and consultants. Relying on third-party clinical investigators, CROs and consultants may force us to encounter delays that are outside of our control.control, including delays and restrictions that may be imposed by legislation or executive order or other administrative action. We may be unable to identify and contract with a sufficient number of investigators, CROs and consultants on a timely basis or at all. There can be no assurance that we will be able to negotiate and enter into any additional master services agreement with other CROs, as necessary, on terms that are acceptable to us on a timely basis or at all.
Disruptions at the FDA and other government agencies caused by funding shortages or global health concerns could hinder their ability to hire, retain or deploy key leadership and other personnel, or otherwise prevent new or modified products from being developed, approved or commercialized in a timely manner or at all, which could negatively impact our business.
The ability of the FDA to review, make decisions relating to development, and approve new products can be affected by a variety of factors, including government budget and funding levels, statutory, regulatory, and policy changes, the FDA’s ability to hire and retain key personnel and accept the payment of user fees, and other events that may otherwise affect the FDA’s ability to perform routine functions. Average review times at the agency have fluctuated in recent years as a result. In addition, government funding of other government agencies that fund research and development activities is subject to the political process, which is inherently fluid and unpredictable. Disruptions at the FDA and other agencies may also slow the time necessary for new drugs or modifications to approved drugs to be reviewed and/or approved by necessary government agencies, which would adversely affect our business. For example, over the last several years, including for 35 days beginning on December 22, 2018, the U.S. government has shut down several times and certain regulatory agencies, such as the FDA, have had to furlough critical FDA employees and stop critical activities.
Separately, in response to the global COVID-19 pandemic, on March 10, 2020 the FDA announced its intention to postpone most inspections of foreign manufacturing facilities, and subsequently, on March 18, 2020, the FDA temporarily postponed routine surveillance inspections of domestic manufacturing facilities. In July 2020, the FDA resumed certain on-site inspections of domestic manufacturing facilities subject to a risk-based prioritization system. The FDA utilized this risk-based assessment
system to assist in determining when and where it was safest to conduct prioritized domestic inspections. Additionally, on April 15, 2021, the FDA issued a guidance document in which the FDA described its plans to conduct voluntary remote interactive evaluations of certain drug manufacturing facilities and clinical research sites, among other facilities. According to the guidance, the FDA may request such remote interactive evaluations where the FDA determines that remote evaluation would be appropriate based on mission needs and travel limitations. In May 2021, the FDA outlined a detailed plan to move toward a more consistent state of inspectional operations, and in July 2021, the FDA resumed standard inspectional operations of domestic facilities and was continuing to maintain this level of operation as of September 2021. More recently, the FDA has continued to monitor and implement changes to its inspectional activities to ensure the safety of its employees and those of the firms it regulates as it adapts to the evolving COVID-19 pandemic. In February 2022, the FDA determined that it would conduct both foreign and domestic mission-critical inspections and would proceed with certain foreign surveillance inspections. Regulatory authorities outside the United States may adopt similar restrictions or other policy measures in response to the COVID-19 pandemic. If a prolonged government shutdown occurs, or if global health concerns prevent the FDA or other regulatory authorities from conducting their regular inspections, reviews, or other regulatory activities, it could significantly impact the ability of the FDA or other regulatory authorities to timely review and process our regulatory submissions, which could have a material adverse effect on our business.
Risks Related to Employee Matters, Managing Growth and Information Technology
We are highly dependent on our key personnel. If we are not successful in attracting and retaining highly qualified personnel, we may not be able to successfully implement our business strategy.
We are highly dependent on Adrian Gottschalk, our Chief Executive Officer. In addition, the loss of the services of any of our executive officers, other key employees and other scientific and medical advisors, and an inability to find suitable replacements could result in delays in product development and harm our business.
Despite our efforts to retain Mr. Gottschalk and other valuable employees, members of our management, scientific and development teams may terminate their employment with us on short notice. Although we have employment agreements with our key employees, these employment agreements provide for at-will employment, which means that any of our employees could leave our employment at any time, with or without notice.
We willmay need to grow the size of our organization, and we may experience difficulties in managing this growth.growth and other issues relating to our employees.
As of December 31, 2021,2023, we had 119116 full-time employees. We intend to hire new employees to assume activities and responsibilities within the company, including conducting our research and performing development activities in the future.
Our ability to compete in the highly competitive biotechnology and pharmaceutical industries depends upon our ability to attract and retain highly qualified managerial, scientific and medical personnel. We conduct our operations at our facilities in Cambridge, Massachusetts. The Massachusetts region is headquarters to many other biopharmaceutical companies and many academic and research institutions. Competition for skilled personnel in our market is intense and may limit our ability to hire and retain highly qualified personnel on acceptable terms or at all. ChangesOn January 5, 2023, the Federal Trade Commission released a notice of proposed rulemaking that, if finalized, would ban employers from entering into or maintaining post-termination non-compete clauses with employees, which could adversely effect our business in the event key personnel leave us for employment at key competitors. Additionally, changes to U.S. immigration and work authorization laws and regulations, including those that restrain the flow of scientific and professional talent, can be significantly affected by political forces and levels of economic activity. Our business may be materially adversely affected if legislative or administrative changes to immigration or visa laws and regulations impair our hiring processes and goals or projects involving personnel who are not U.S. citizens.
Any delay or disruption in hiring such new employees could result in delays in our research and development activities and would harm our business. As our development and commercialization plans and strategies develop, and as we transition into operating as a public company, we expect to need additional managerial, operational, sales, marketing, financial and other personnel, as well as additional facilities to expand our operations.
In the future, we may hire new employees to assume activities and responsibilities within the company, including conducting our research and performing development activities. If we are not able to effectively expand our organization by hiring new employees and expanding our groups of consultants and contractors, or we are not able to effectively build out new facilities to accommodate this expansion, we may not be able to successfully implement the tasks necessary to further develop and
commercialize our product candidates and, accordingly, may not achieve our research, development and commercialization goals.
Our internal computer systems, or those used by our third-party CROs or other contractors or consultants, may fail or suffer security breaches, which could result in a material disruption of the development programs of our product candidates.
Despite the implementation of security measures, our internal computer systems and those of our current and future service providers, including our CROs and other contractors and consultants are vulnerable to damage from computer viruses, ransomware, unauthorized access, denial of service attacks, internal or external hacking, among other cyber attacks. Other events like natural disasters, and telecommunication and electrical failures.failures could also impact the availability of our or our service providers networks. While to our knowledge, we have not experienced any sucha material system failure or security breach to date, like other companies we are subject to attacks and if such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our development programs and our business operations. For example, the loss of data from completed or future preclinical studies and clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. Such an attack could also have reputational impact, result in regulatory investigations, fines, litigation, remediation costs, increased insurance premiums or impact the availability of insurance, and other costs. Likewise, we rely on third parties for the manufacture of our product candidates and to conduct clinical trials, and similar events relating to their computer systems could also have a material adverse effect on our business. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and the further development and commercialization of our product candidates could be delayed.
Business disruptions could seriously harm our future revenue and financial condition and increase our costs and expenses.
We rely on multiple CROs to mitigate potential impacts that may affect any one of our CROs. However, CDMOs and other contractors and consultants could be subject to adverse legislation or administrative restrictions, earthquakes, power shortages, telecommunications failures, water shortages, floods, hurricanes, typhoons, fires, extreme weather conditions, pandemics and other natural or man-made disasters or business interruptions, for which we are predominantly self-insured. The occurrence of any of these business disruptions could seriously harm our operations and financial condition and increase our costs and expenses. We rely on third-party manufacturers to produce our product candidates. Our ability to obtain clinical supplies of our product candidates could be disrupted if the operations of these suppliers are affected by a man-made or natural disaster or other business interruption.
The continuing outbreak of COVID-19 in the United States and other countries may adversely affect our business and the market price of our common stock.
The evolving global pandemic of COVID-19 is impacting worldwide economic activity, particularly economic activity in the United States, and poses the risk that we or our employees, contractors, suppliers or other partners may be prevented or delayed from conducting business activities for an indefinite period of time, including due to shutdowns that may be requested or mandated by governmental authorities. The continued prevalence of COVID-19 and the measures taken by the governments of countries affected could disrupt the supply chain and the manufacture or shipment of both drug substance and finished drug product for our product candidates for preclinical testing or clinical trials, cause diversion of healthcare resources away from the conduct of preclinical and clinical trial matters to focus on pandemic concerns, limit travel in a manner that interrupts key trial activities, such as trial site initiations and monitoring, delay regulatory filings with regulatory agencies in affected areas or adversely affect our ability to obtain regulatory approvals. These disruptions could also affect other facets of our business, including but not limited to:
•our ability to recruit employees from outside of the United States;
•the ability of our CROs to conduct preclinical studies and clinical trials in countries outside of the United States;
•our ability to import materials from outside of the United States; and
•our ability to export materials to our CROs and other third parties located outside of the United States.
The COVID-19 outbreak and mitigation measures also may have an adverse impact on global economic conditions, which could adversely impact our business, financial condition or results of operations. Additionally, the COVID-19 outbreak has resulted in significant financial market volatility and uncertainty. A continuation or worsening of the levels of market disruption and volatility seen in the recent past as a result of the COVID-19 outbreak could have an adverse effect on our ability to access capital and on the market price of our common stock.
Risks Related to Our Intellectual Property
If we are unable to adequately protect our proprietary technology and platform or obtain and maintain patent protection for our technology and products or if the scope of the patent protection obtained is not sufficiently broad, our competitors could develop and commercialize technology and products similar or identical to ours, and our ability to successfully develop and commercialize our technology and products may be impaired.
Our commercial success will depend in part on our ability and the ability of our licensors to obtain and maintain proprietary or intellectual property protection in the United States and other countries for our product candidates, and our core technologies, including aspects of our Gene Traffic Control platform. We rely on trade secrets, know-how and continuing technological
innovation to develop and maintain our proprietary and intellectual property position. In particular, our Gene Traffic Control platform is not the subject of patent applications.
We seek to protect our proprietary product candidates by filing patent applications in the United States and abroad related to our product candidates that are important to our business. If we or our licensors are unable to obtain or maintain patent protection with respect to our current and future product candidates, competitors and other third parties could develop and commercialize products similar or identical to ours, and our ability to successfully commercialize our product candidates and other product candidates that we may pursue may be impaired. As a result, our business, financial condition, results of operations and prospects could be materially harmed.
Currently, our patent portfolio, including our portfolio related to our product candidatescandidate FHD-286, and FHD-609, is in its earliest stages, primarily consistingconsists of provisional patent applications which do not themselves issue as patents, and patent applications filed pursuant to the Patent Cooperation Treaty or PCT.(the “PCT”), both of which do not themselves issue as patents. We have notwo issued U.S. patents related to FHD-286 or FHD-609.FHD-286. In order to continue to pursue protection based on provisional patent applications, we will need to file PCT, foreign applications and/or U.S. non-provisional patent applications prior to applicable deadlines. In order to continue to pursue protection based on PCT applications, we will need to file national phase applications in the U.S. and ex-U.S. jurisdictions prior to applicable deadlines. Even then, patents may never issue from our patent applications, or the scope of any patent may not be sufficient to provide a competitive advantage.
The degree of patent protection we require to successfully commercialize our product candidates may be unavailable or severely limited in some cases and may not adequately protect our rights or permit us to gain or keep any competitive advantage. We cannot provide any assurances that any of our pending patent applications will issue, or that any of our pending patent applications that mature into issued patents will include claims with a scope sufficient to protect FHD-286 or FHD-609 or our other current or future product candidates. In addition, the laws of foreign countries may not protect our rights to the same extent as the laws of the United States. Furthermore, patents have a limited lifespan. In the United States, the natural expiration of a patent is generally twenty years after it is filed. Various extensions may be available; however, the life of a patent, and the protection it affords, is limited. Given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. As a result, our owned and licensed patent portfolio may not provide us with adequate and continuing patent protection sufficient to exclude others from commercializing products similar or identical to our product candidates, including generic versions of such products.
Other parties have developed technologies that may be related or competitive to our own, and such parties may have filed or may file patent applications, or may have received or may receive patents, claiming inventions that may overlap or conflict with those claimed in our own patent applications, in either case that they may rely upon to dominate our patent position in the market. Publications of discoveries in the scientific literature often lag behind the actual discoveries, and patent applications in the United States and other jurisdictions are typically not published until 18 months after filing, or in some cases not at all. Therefore, we cannot know with certainty whether we were the first to make the inventions claimed in our owned or licensed pending patent applications, or that we were the first to file for patent protection of such inventions. As a result, the issuance, scope, validity, enforceability and commercial value of our patent rights cannot be predicted with any certainty.
In addition, the patent prosecution process is expensive and time-consuming, and we may not be able to file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner. Further, with respect to most of the pending patent applications covering our product candidates, prosecution has yet to commence. Patent prosecution is a lengthy process, during which the scope of the claims initially submitted for examination by the U.S. Patent and Trademark Office or USPTO,(the “USPTO”) have been significantly narrowed by the time they issue, if at all. It is also possible that we will fail to identify patentable aspects of our research and development output before it is too late to obtain patent protection.
Even if we acquire patent protection that we expect should enable us to maintain such competitive advantage, third parties may challenge the validity, enforceability or scope thereof, which may result in such patents being narrowed, invalidated or held unenforceable. The issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability, and our owned and licensed patents may be challenged in the courts or patent offices in the United States and abroad. For example, we may be subject to a third-party submission of prior art to the USPTO challenging the priority of an invention claimed within one of our patents, which submissions may also be made prior to a patent’s issuance, precluding the granting of any of our pending patent applications. We may become involved in opposition, derivation, reexamination, inter partes review, post-grant review or interference proceedings challenging our patent rights or the patent rights of others from whom we have obtained licenses to such rights. Competitors may claim that they invented the inventions claimed in our issued patents or patent applications prior to us, or may file patent applications before we do. Competitors may also claim that we are infringing on their patents and that we therefore cannot practice our technology as claimed under our patents, if issued. Competitors may also contest our patents, if
issued, by showing the patent examiner that the invention was not original, was not novel or was obvious. In litigation, a competitor could claim that our patents, if issued, are not valid for a number of reasons. If a court agrees, we would lose our rights to those challenged patents.
In addition, we may in the future be subject to claims by our former employees or consultants asserting an ownership right in our patent applications or technologies as a result of the work they performed on our behalf. Although we generally require all of our employees, consultants and advisors and any other third parties who have access to our proprietary know-how, information, or technology to assign or grant similar rights to their inventions to us, we cannot be certain that we have executed such agreements with all parties who may have contributed to our intellectual property, nor can we be certain that our agreements with such parties will be upheld in the face of a potential challenge, or that they will not be breached, for which we may not have an adequate remedy.
An adverse determination in any such submission or proceeding may result in loss of exclusivity or freedom to operate or in patent claims being narrowed, invalidated or held unenforceable, in whole or in part, which could limit our ability to stop others from using or commercializing similar or identical technology and products, without payment to us, or could limit the duration of the patent protection covering our technology and product candidates. Such challenges may also result in our inability to manufacture or commercialize our product candidates without infringing third party patent rights. In addition, if the breadth or strength of protection provided by our patents and patent applications is threatened, it could dissuade companies from collaborating with us to license, develop or commercialize current or future product candidates.
Even if our patent portfolio is unchallenged, it may not provide us with any meaningful protection or prevent competitors from designing around our patent claims to circumvent our owned or licensed patents by developing similar or alternative
technologies or products in a non-infringing manner. For example, a third party may develop a competitive product that provides benefits similar to one or more of our product candidates but that has a different composition that falls outside the scope of our patent protection. If the patent protection provided by the patents and patent applications we hold or pursue with respect to our product candidates is not sufficiently broad to impede such competition, our ability to successfully commercialize our product candidates could be negatively affected, which would harm our business.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position may be harmed.
In addition to the protection afforded by patents, we rely upon unpatented trade secret protection, unpatented know-how and continuing technological innovation to develop and maintain our competitive position. With respect to the various aspects of our Gene Traffic Control platform, including our proprietary libraries, we consider trade secrets and know-how to be our primary intellectual property. We seek to protect our proprietary technology and processes, in part, by entering into confidentiality agreements with our collaborators, scientific advisors, employees and consultants, and invention assignment agreements with our consultants and employees. We may not be able to prevent the unauthorized disclosure or use of our technical know-how or other trade secrets by the parties to these agreements, however, despite the existence generally of confidentiality agreements and other contractual restrictions. Monitoring unauthorized uses and disclosures is difficult, and we do not know whether the steps we have taken to protect our proprietary technologies will be effective. If any of the collaborators, scientific advisors, employees and consultants who are parties to these agreements breaches or violates the terms of any of these agreements, we may not have adequate remedies for any such breach or violation, and we could lose our trade secrets as a result. We also seek to preserve the integrity and confidentiality of our confidential proprietary information by maintaining physical security on our premises, and physical and electronic security of our information technology systems, but it is possible that these security measures could be breached. Enforcing a claim that a third party illegally obtained and is using our trade secrets, like patent litigation, is expensive and time-consuming, and the outcome is unpredictable. In addition, courts outside the United States are sometimes less willing to protect trade secrets.
Our trade secrets could otherwise become known or be independently discovered by our competitors. Competitors could purchase our product candidates and attempt to replicate some or all of the competitive advantages we derive from our development efforts, willfully infringe our intellectual property rights, design around our protected technology or develop their own competitive technologies that fall outside of our intellectual property rights. If any of our trade secrets were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent them, or those to whom they communicate it, from using that technology or information to compete with us. If our trade secrets are not adequately protected so as to protect our market against competitors’ products, our competitive position could be adversely affected, as could our business.
Obtaining and maintaining patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
The USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. In addition, periodic maintenance fees on issued patents often must be paid to the USPTO and foreign patent agencies over the lifetime of the patent. While an unintentional lapse can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Non-compliance events that could result in abandonment or lapse of a patent or patent application include, but are not limited to, failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. If we fail to maintain the patents and patent applications covering our products or procedures, we may not be able to stop a competitor from marketing products that are the same as or similar to our product candidates, which would have a material adverse effect on our business.
The intellectual property landscape around our technology, including our Gene Traffic Control platform, is highly dynamic, and third parties may obtain intellectual property rights that could affect our ability to use our platform or otherwise develop and commercialize product candidates.
The field of protein modeling, especially in the area of targeting transcription factors, is still in its infancy. Due to the intense research and development that is taking place by several companies, including us and our competitors, in this field, the intellectual property landscape is evolving and in flux, and it may remain uncertain for the coming years. There may be significant intellectual property related litigation and proceedings relating to our owned and in-licensed, and other third party, intellectual property and proprietary rights in the future.
Our commercial success depends upon our ability and the ability of our collaborators and licensors to develop, manufacture, market, and sell any product candidates that we may develop and use our proprietary technologies without infringing,
misappropriating, or otherwise violating the intellectual property and proprietary rights of third parties. As the biotechnology and pharmaceutical industries expand and more patents are issued, the risk increases that our Gene Traffic Control platform and related technology and product candidates may give rise to claims of infringement of the patent rights of others. Moreover, it is not always clear to industry participants, including us, which patents cover various types of therapies, products or their methods of use or manufacture. There may be third-party patents of which we are currently unaware with claims to technologies, methods of manufacture or methods for treatment related to the use or manufacture of our product candidates. Because patent applications can take many years to issue, there may be currently pending patent applications that may later result in issued patents that our product candidates may infringe. In addition, third parties may obtain patents in the future and claim that use of our technologies infringes upon these patents. We may be unable to obtain a license to such patents held by third-parties on commercially reasonable terms or at all. In the event that we are unable to obtain licenses to such patents, our ability to develop and commercialize one or more product candidates may become severely limited. Even if we were able to obtain such a license, it could be granted on non-exclusive terms, thereby providing our competitors and other third parties access to the same technologies licensed to us.
We may initiate or become involved in legal proceedings involving allegations that we are infringing a third party’s intellectual property rights, the outcome of which would be uncertain and could have a material adverse effect on the success of our business.
Our commercial success depends in part upon our ability and the ability of our collaborators to develop, manufacture and sell our product candidates and use our proprietary technologies without infringing the propriety rights and intellectual property of third parties.
The biotechnology and pharmaceutical industries are characterized by extensive and frequent litigation regarding patents and other intellectual property rights. We may in the future become party to, or threatened with, adversarial proceedings or litigation regarding intellectual property rights with respect to our product candidates and technology. Our competitors or other third parties may assert infringement claims against us, alleging that our products or technologies are covered by their patents. Given the vast number of patents in our field of technology, we cannot be certain that we do not infringe existing patents or that we will not infringe patents that may be granted in the future. If a patent holder believes our product or product candidate infringes on its patent, the patent holder may sue us even if we have received patent protection for our technology. Moreover, we may face patent infringement claims from non-practicing entities that have no relevant product revenue and against whom our own patent portfolio may thus have no deterrent effect.
If we are found to infringe a third party’s intellectual property rights, we could be required to obtain a license from such third party to continue developing and marketing our product candidates and technology. We may choose to obtain a license, even in the absence of an action or finding of infringement. In either case, we may not be able to obtain any required license on commercially reasonable terms or at all. Even if we were able to obtain such a license, it could be granted on non-exclusive terms, thereby providing our competitors and other third parties access to the same technologies licensed to us. Without such a license, we could be forced, including by court order, to cease developing and commercializing the infringing technology or product candidates. In addition, we could be found liable for monetary damages, including treble damages and attorneys’ fees if we are found to have willfully infringed such third-party patent rights. A finding of infringement could prevent us from commercializing our product candidates or force us to cease some of our business operations, which could materially harm our business. If we lose a foreign patent lawsuit, alleging our infringement of a competitor’s patents, we could be prevented from marketing our products in one or more foreign countries, which would have a materially adverse effect on our business.
We may be subject to damages resulting from claims that we or our employees have wrongfully used or disclosed alleged trade secrets of our competitors or are in breach of non-competition or non-solicitation agreements with our competitors.
We could in the future also be subject to claims that we or our employees have inadvertently or otherwise used or disclosed alleged trade secrets or other proprietary information of former employers or competitors. Although we try to ensure that our employees and consultants do not use the intellectual property, proprietary information, know-how or trade secrets of others in their work for us, we may in the future be subject to claims that we caused an employee to breach the terms of his or her non-competition or non-solicitation agreement, or that we or these individuals have, inadvertently or otherwise, used or disclosed the alleged trade secrets or other proprietary information of a former employer or competitor. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and could be a distraction to management. If our defenses to these claims fail, in addition to requiring us to pay monetary damages, a court could prohibit us from using technologies or features that are essential to our product candidates, if such technologies or features are found to incorporate or be derived from the trade secrets or other proprietary information of the former employers. An inability to incorporate such technologies or features would have a material adverse effect on our business and may prevent us from successfully commercializing our product candidates. In addition, we may lose valuable intellectual property rights or personnel as a result of such claims. Moreover, any such litigation or the threat thereof may adversely affect our ability to hire employees or contract with independent sales representatives. A loss of key personnel or
their work product could hamper or prevent our ability to commercialize our product candidates, which would have an adverse effect on our business, results of operations and financial condition.
We may become involved in lawsuits to protect or enforce our patents and other intellectual property rights, which could be expensive, time-consuming and unsuccessful.
Competitors and other third parties may infringe, misappropriate or otherwise violate our patents, if obtained, and other intellectual property rights. To counter infringement or unauthorized use, we may be required to file infringement claims. A court may disagree with our allegations, however, and may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the third-party technology in question. Further, such third parties could counterclaim that we infringe their intellectual property or that a patent we have asserted against them is invalid or unenforceable. In patent litigation in the United States, defendant counterclaims challenging the validity, enforceability or scope of asserted patents are commonplace. In addition, third parties may initiate legal proceedings against us to assert such challenges to our intellectual property rights. The outcome of any such proceeding is generally unpredictable.
An adverse result in any litigation proceeding could put one or more of our patents at risk of being invalidated or interpreted narrowly. If a defendant were to prevail on a legal assertion of invalidity or unenforceability of our patents covering one of our product candidates, we would lose at least part, and perhaps all, of the patent protection covering such product candidate. Competing products may also be sold in other countries in which our patent coverage might not exist or be as strong.
Intellectual property litigation could cause us to spend substantial resources and distract our personnel from their normal responsibilities.
Litigation or other legal proceedings relating to intellectual property claims, with or without merit, is unpredictable and generally expensive and time-consuming and is likely to divert significant resources from our core business, including distracting our technical and management personnel from their normal responsibilities. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments and if securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our common stock. Such
litigation or proceedings could substantially increase our operating losses and reduce the resources available for development activities or any future sales, marketing or distribution activities.
We may not have sufficient financial or other resources to adequately conduct such litigation or proceedings. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their greater financial resources and more mature and developed intellectual property portfolios. Accordingly, despite our efforts, we may not be able to prevent third parties from infringing upon or misappropriating or from successfully challenging our intellectual property rights, or we may be unable to successfully defend ourselves from allegations of infringement or misappropriation. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could have a material adverse effect on our ability to compete in the marketplace.
We may not be able to effectively enforce our intellectual property rights throughout the world.
Filing, prosecuting and defending patents on our product candidates in all countries throughout the world would be prohibitively expensive. The requirements for patentability may differ in certain countries, particularly in developing countries. Moreover, our ability to protect and enforce our intellectual property rights may be adversely affected by unforeseen changes in foreign intellectual property laws. Additionally, the patent laws of some foreign countries do not afford intellectual property protection to the same extent as the laws of the United States. Many companies have encountered significant problems in protecting and defending intellectual property rights in certain foreign jurisdictions. The legal systems of some countries, particularly developing countries, do not favor the enforcement of patents and other intellectual property rights. This could make it difficult for us to stop the infringement of our patents or the misappropriation of our other intellectual property rights. For example, many foreign countries have compulsory licensing laws under which a patent owner must grant licenses to third parties. Consequently, we may not be able to prevent third parties from practicing our inventions in all countries outside the United States. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and, further, may export otherwise infringing products to territories where we have patent protection, if our ability to enforce our patents to stop infringing activities is inadequate. These products may compete with our product candidates, and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
Proceedings to enforce our patent rights in foreign jurisdictions, whether or not successful, could result in substantial costs and divert our efforts and resources from other aspects of our business. Furthermore, while we intend to protect our intellectual property rights in the major markets for our product candidates, we cannot ensure that we will be able to initiate or maintain
similar efforts in all jurisdictions in which we may wish to market our product candidates. Accordingly, our efforts to protect our intellectual property rights in such countries may be inadequate.
We may be subject to claims challenging the inventorship or ownership of any intellectual property, including any patents we may own or in-license in the future.
We may be subject to claims that former employees, collaborators or other third parties have an interest in any patents we may own or in-license in the future, trade secrets, or other intellectual property as an inventor or co-inventor. We may have inventorship disputes arise from conflicting obligations of employees, consultants or others who are involved in developing our product candidates or other technologies. We generally enter into confidentiality and intellectual property assignment agreements with our employees, consultants, and contractors. These agreements generally provide that inventions conceived by the party in the course of rendering services to us will be our exclusive property. However, those agreements may not be honored and may not effectively assign intellectual property rights to us. Moreover, there may be some circumstances where we are unable to negotiate for such ownership rights. Disputes regarding ownership or inventorship of intellectual property can also arise in other contexts, such as collaborations and sponsored research. If we are subject to an inventorship dispute, such dispute may lead to litigation which could be expensive and time-consuming. If we are unsuccessful, in addition to paying monetary damages, we could lose valuable rights in intellectual property that we regard as our own, such as exclusive ownership of, or right to use, intellectual property that is important to our product candidates and our Gene Traffic Control platform. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees. Any of the foregoing could have a material adverse effect on our business, financial condition, results of operations and prospects.
If we do not obtain patent term extension and data exclusivity for any product candidates we may develop, our business may be materially harmed.
Depending upon the timing, duration and specifics of any FDA marketing approval of any product candidates we may develop, one or more of our U.S. patents, if obtained, may be eligible for limited patent term extension under the Drug Price Competition
and Patent Term Restoration Action of 1984 or Hatch-Waxman Amendments.(the “Hatch-Waxman Amendments”). The Hatch-Waxman Amendments permit a patent extension term of up to five years as compensation for patent term lost during the FDA regulatory review process. A patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval, only one patent may be extended and only those claims covering the approved drug, a method for using it or a method for manufacturing it may be extended. However, we may not be granted an extension because of, for example, failing to exercise due diligence during the testing phase or regulatory review process, failing to apply within applicable deadlines, failing to apply prior to expiration of relevant patents or otherwise failing to satisfy applicable requirements. Moreover, the applicable time period or the scope of patent protection afforded could be less than we request. If we are unable to obtain patent term extension or the term of any such extension is less than we request, our business, financial condition, results of operations and prospects could be materially harmed.
Intellectual property rights do not necessarily address all potential threats.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations and may not adequately protect our business or permit us to maintain our competitive advantage. For example:
•aspects of our Gene Traffic Control platform are protected by trade secrets, which may be inadequate to safeguard our competitive advantage, and some aspects of our platform may not be protectable by intellectual property rights at all;
•others may be able to make products that are similar to our product candidates or utilize similar technology but that are not covered by the claims of any patents that may issue to us, our licensors or our collaborator;
•we or our licensors or collaborators, might not have been the first to make the inventions covered by our pending patent applications, or any patents that may issue in the future;
•we or our licensors or collaborators, might not have been the first to file patent applications covering certain of our or their inventions;
•others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing or misappropriating our intellectual property rights;
•it is possible that our present or future pending patent applications will not lead to issued patents;
•issued patents that we hold rights to may be held invalid or unenforceable, including as a result of legal challenges by our competitors or other third parties;
•our competitors or other third parties might conduct research and development activities in countries where we do not have patent rights and then use the information learned from such activities to develop competitive products for sale in our major commercial markets;
•changes to the patent law in the United States and other jurisdictions could diminish the value of patents in general, thereby impairing our ability to protect our product candidates;
•the patents of others may harm our business; and
•we may choose not to file a patent application in order to maintain certain trade secrets or know-how, and a third party may subsequently file a patent covering such intellectual property.
Should any of these events occur, they could have a material adverse effect on our business, financial condition, results of operations and prospects.
Changes to the patent law in the United States and other jurisdictions could diminish the value of patents in general, thereby impairing our ability to protect our product candidates.
As is the case with other biopharmaceutical companies, our success is heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the biopharmaceutical industry involve both technological and legal complexity and is therefore costly, time-consuming and inherently uncertain. Recent patent reform legislation in the United States and other countries, including the Leahy-Smith America Invents Act or Leahy-Smith Act,(the “Leahy-Smith Act”) signed into law on September 16, 2011, could increase those uncertainties and costs. The Leahy-Smith Act includes a number of significant changes to U.S. patent law. These include provisions that affect the way patent applications are prosecuted, redefine prior art and provide more efficient and cost-effective avenues for competitors to challenge the validity of patents. In addition, the Leahy-Smith Act has transformed the U.S. patent system into a “first to file” system. The first-to-file provisions, however, only became effective on March 16, 2013. Accordingly, it is not yet clear what, if any, impact the Leahy-Smith Act will have on the operation of our business. However,
the Leahy-Smith Act and its implementation could make it more difficult to obtain patent protection for our inventions and increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents, all of which could harm our business, results of operations and financial condition.
The U.S. Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. Additionally, there have been recent proposals for additional changes to the patent laws of the United States and other countries that, if adopted, could impact our ability to obtain patent protection for our proprietary technology or our ability to enforce rights in our proprietary technology. Depending on future actions by the U.S. Congress, the U.S. courts, the USPTO and the relevant law-making bodies in other countries, the laws and regulations governing patents could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce any patents that we may obtain in the future.
Risks Related to Our Reliance on Third Parties
We rely, and expect to continue to rely, on third parties, including independent clinical investigators, CROs and CDMOs to conduct certain aspects of our discovery and preclinicalpre-clinical studies and development, and our clinical trials. If these third parties do not successfully carry out their contractual duties, comply with applicable regulatory requirements or meet expected deadlines, we may not be able to obtain regulatory approval for or commercialize our product candidates and our business could be substantially harmed.
We have relied upon and plan to continue to rely upon third parties, including independent clinical investigators and third-party CROs and CDMOs, as well as potential collaboration partners to conduct certain aspects of our discovery, preclinicalpre-clinical studies and development and clinical trials and to monitor and manage data for our ongoing preclinical and clinical programs. We rely on these parties for execution of our preclinical studies and planned clinical trials, and control only certain aspects of their activities. Nevertheless, we are responsible for ensuring that each of our studies and trials is conducted in accordance with the applicable protocol, legal, regulatory and scientific standards, and our reliance on these third parties does not relieve us of our regulatory responsibilities. We and our third-party contractors, CROs and CDMOs are required to comply with GCP requirements, which are regulations and guidelines enforced by the FDA and comparable foreign regulatory authorities for all of our product candidates in clinical development. Regulatory authorities enforce these GCPs through periodic inspections of trial sponsors, principal investigators and trial sites. If we or any of these third parties, our CROs or our CDMOs fail to comply with applicable GCPs, the clinical data generated in our clinical trials may be deemed unreliable and the FDA or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. Moreover, our business may be adversely affected if any of these third parties violates federal or state fraud and abuse or false claims laws and regulations or healthcare privacy and security laws.
Further, these investigators, CROs and CDMOs are not our employees and we are not able to control, other than by contract, the amount of resources, including time, which they devote to our product candidates and clinical trials. These third parties may also have relationships with other commercial entities, including our competitors, for whom they may also be conducting clinical trials or other product development activities, which could affect their performance on our behalf. If independent investigators, CROs and CDMOs fail to devote sufficient resources to the development of our product candidates, or if CROs do not successfully carry out their contractual duties or obligations or meet expected deadlines, if they need to be replaced or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols, regulatory requirements or for other reasons, our clinical trials may be extended, delayed or terminated and we may not be able to obtain regulatory approval for or successfully commercialize our product candidates. As a result, our results of operations and the commercial prospects for our product candidates would be harmed, our costs could increase and our ability to generate revenues could be delayed or precluded entirely.
If any of our relationships with these third-party CROs terminate, we may not be able to enter into arrangements with alternative CROs or to do so on commercially reasonable terms. Switching or adding additional CROs involves additional cost and requires management time and focus. In addition, there is a natural transition period when a new CRO commences work. As a result, delays occur, which can materially impact our ability to meet our desired clinical development timelines. Additionally, CROs may lack the capacity to absorb higher workloads or take on additional capacity to support our needs. Though we carefully manage our relationships with our CROs, there can be no assurance that we will not encounter similar challenges or delays in the future or that these delays or challenges will not have a material adverse impact on our business, financial condition and prospects.
In addition, certain of our CDMOs and CROs located in China may experience adverse legal and regulatory restrictions, which could adversely affect their ability to provide services to Foghorn and, thereby, harm our business.
We currently rely and expect to rely in the future on the use of manufacturing suites in third-party facilities or third parties to manufacture our product candidates. Our business could be harmed if we are unable to use third-party manufacturing
suites or if third-party manufacturers fail to provide us with sufficient quantities of our product candidates or fail to do so at acceptable quality levels or prices.
We do not currently own any facility that may be used as our clinical-scale manufacturing and processing facility and instead must currently rely on outside vendors to manufacture our product candidates in clinical quantities.
Our reliance on third parties for clinical quantities exposes us to a number of risks, including:
•our third-party manufacturers might be unable to timely manufacture our product candidates or produce the quantity and quality required to meet our clinical and commercial needs, if any;
•contract manufacturers may not be able to execute our manufacturing procedures and other logistical support requirements appropriately and incompliancein compliance with cGMP; and
•our third-party manufacturers could breach or terminate their agreements with us.
Each of these risks could delay or prevent the completion of our clinical trials or the approval of any of our product candidates by the FDA or result in higher costs. In addition, we will rely on third parties to perform certain specification tests on our product candidates prior to delivery to patients. If these tests are not appropriately done and test data are not reliable, patients could be put at risk of serious harm and the FDA could place significant restrictions on our company until deficiencies are remedied.
If our third-party manufacturers use hazardous and biological materials in a manner that causes injury or violates applicable law, we may be liable for damages.
Our research and development activities involve the controlled use of potentially hazardous substances, including chemical and biological materials, by our third-party manufacturers. Our manufacturers are subject to numerous environmental, health and safety laws and regulations, including those governing the use, manufacture, storage, handling and disposal of medical and hazardous materials. Although we believe that our manufacturers’ procedures for using, handling, storing and disposing of these materials comply with legally prescribed standards, we cannot completely eliminate the risk of contamination or injury resulting from medical or hazardous materials. As a result of any such contamination or injury, we may incur liability or local, city, state or federal authorities may curtail the use of these materials and interrupt our business operations. In the event of an accident, we could be held liable for damages or penalized with fines, and the liability could exceed our resources. We do not have any insurance for liabilities arising from medical or hazardous materials. Compliance with applicable environmental laws and regulations is expensive, and current or future environmental regulations may impair our research, development and production efforts, which could harm our business, prospects, financial condition or results of operations.
Risks Related to Regulatory and Other Legal Compliance Matters
Our clinical trials may fail to demonstrate adequately the safety and efficacy of any of our product candidates, which would delay or prevent further clinical development of those candidates.
To obtain the requisite regulatory approvals to market and sell any of our product candidates, including FHD-286 and FHD-609,FHD-909, and any other future product candidates, we must demonstrate through extensive preclinical studies and clinical trials that our products are safe and effective in humans.
Clinical trials that we conduct may not demonstrate the efficacy and safety necessary to obtain regulatory approval to market our product candidates. In some instances, there can be significant variability in safety or efficacy results between different clinical trials of the same product candidate due to numerous factors, including changes in trial procedures set forth in protocols, differences in the size and type of the patient populations, changes in and adherence to the clinical trial protocols and the rate of dropout among clinical trial participants. If the results of our ongoing or future clinical trials are inconclusive with respect to the efficacy of our product candidates, if we do not meet the clinical endpoints with statistical and clinically meaningful significance, or if there are safety concerns associated with our product candidates, we may be delayed in obtaining marketing approval, if at all.
Even if the trials are successfully completed, clinical data are often susceptible to varying interpretations and analyses, and we cannot guarantee that the FDA or other comparable foreign regulatory authorities will interpret the results as we do, and more trials could be required before we submit our product candidates for approval. We cannot guarantee that the FDA or other comparable foreign regulatory authorities will view our product candidates as having sufficient efficacy to support the indication studied in the clinical trial even if positive results are observed in early clinical trials. To the extent that the results of the trials are not satisfactory to the FDA or other comparable foreign regulatory authorities for support of a marketing
application, approval of our product candidates may be significantly delayed, or we may be required to expend significant additional resources, which may not be available to us, to conduct additional trials in support of potential approval of our product candidates. Additionally, any safety or efficacy concerns observed in any tumor-specific subgroup of our clinical trials could limit the prospects for regulatory approval of our product candidates for a tumor-agnostic indication, which could have a material adverse effect on our business, financial condition and results of operations.
We may in the future seek orphan drug status for FHD-286 and FHD-609 and some of our other future product candidates, but we may be unable to obtain such designations or to maintain the benefits associated with orphan drug status, including market exclusivity, which may cause our future revenue, if any, to be reduced.
Under the Orphan Drug Act, the FDA may grant orphan designation to a drug or biologic intended to treat a rare disease or condition, defined as a disease or condition with a patient population of fewer than 200,000 in the United States, or a patient population greater than 200,000 in the United States when there is no reasonable expectation that the cost of developing and making available the drug or biologic in the United States will be recovered from sales in the United States for that drug or biologic. Orphan drug designation must be requested before submitting an NDA. In the United States, orphan drug designation entitles a party to financial incentives such as opportunities for grant funding towards clinical trial costs, tax advantages and user-fee waivers. After the FDA grants orphan drug designation, the generic identity of the drug and its potential orphan use are disclosed publicly by the FDA. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process.
If a product that has orphan drug designation subsequently receives the first FDA approval for a particular active ingredient for the disease for which it has such designation, the product is entitled to orphan drug exclusivity, which means that the FDA may not approve any other applications, including an NDA, to market the same drug for the same indication for seven years, except in limited circumstances such as a showing of clinical superiority to the product with orphan drug exclusivity or if the FDA finds that the holder of the orphan drug exclusivity has not shown that it can assure the availability of sufficient quantities of the orphan drug to meet the needs of patients with the disease or condition for which the drug was designated. As a result, even if one of our product candidates receives orphan drug exclusivity, the FDA can still approve other drugs that have a different active ingredient for use in treating the same indication or disease. The FDA has historically taken the position that the scope of orphan exclusivity aligns with the approved indication or use of a product, rather than the disease or condition for which the product received orphan designation. However, on September 30, 2021, the U.S. Court of Appeals for the 11th Circuit issued a decision in Catalyst Pharms., Inc. v. Becerra holding that the scope of orphan drug exclusivity must align with the disease or condition for which the product received orphan designation, even if the product’s approval was for a narrower use or indication. It remains to be seen how this decision affects orphan drug exclusivity going forward. The FDA announced on January 24, 2023 that despite the Catalyst decision, it will continue to apply its longstanding regulations, which tie the scope of orphan exclusivity to the uses or indications for which the drug is approved, rather than to the designation.The FDA’s application of its orphan drug regulations post-Catalyst could be the subject of future legislation or to further challenges in court, which could impact our ability to obtain or seek to work around orphan exclusivity, and might affect our ability to retain
orphan exclusivity that the FDA previously has recognized for our products. Furthermore, the FDA can waive orphan drug exclusivity if we are unable to manufacture sufficient supply of our product.
We may seek orphan drug designation for some or all of our other future product candidates, where applicable, in addition to orphan indications in which there is a medically plausible basis for the use of these products. Even when we obtain orphan drug designation, exclusive marketing rights in the United States may be limited if we seek approval for an indication broader than the orphan designated indication and may be lost if the FDA later determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantities of the product to meet the needs of patients with the rare disease or condition. In addition, although we intend to seek orphan drug designation for other product candidates, we may never receive such designations. For example, the FDA has expressed concerns regarding the regulatory considerations for orphan drug designation as applied to tissue agnostic therapies, and the FDA may interpret the federal Food, Drug and Cosmetic Act, as amended, or the FD&C Act and regulations promulgated thereunder in a way that limits or blocks our ability to obtain orphan drug designation or orphan drug exclusivity, if our product candidates are approved, for our targeted indications.
A Breakthrough Therapy designation by the FDA, even if granted for any of our product candidates, may not lead to a faster development or regulatory review or approval process and it does not increase the likelihood that our product candidates will receive marketing approval.
We may seek Breakthrough Therapy designation from the FDA for FHD-286, and FHD-609, and for some or all of our future product candidates. A breakthrough therapy is defined as a drug or biologic that is intended, alone or in combination with one or more other drugs or biologics, to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the drug or biologic may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. For product candidates that have been designated as breakthrough therapies, interaction and communication between the FDA and the sponsor of the trial can help to identify the most efficient path for clinical development while minimizing the number of patients placed in ineffective control regimens. Drugs designated as breakthrough therapies by the FDA may also be eligible for other expedited approval programs, including accelerated approval.
Designation as a breakthrough therapy is within the discretion of the FDA. Accordingly, even if we believe one of our product candidates meets the criteria for designation as a breakthrough therapy, the FDA may disagree and instead determine not to make such designation. In any event, the receipt of a Breakthrough Therapy designation for a product candidate may not result
in a faster development process, review or approval compared to candidate products considered for approval under non-expedited FDA review procedures and does not assure ultimate approval by the FDA. In addition, even if one or more of our product candidates qualify as breakthrough therapies, the FDA may later decide that the product no longer meets the conditions for qualification. Thus, even though we intend to seek Breakthrough Therapy designation for some or all of our future product candidates for the treatment of various cancers, there can be no assurance that we will receive breakthrough therapy designation.
Our relationships with healthcare providers, physicians, and third-party payors will be subject to applicable anti-kickback, fraud and abuse, anti-bribery, physician payment transparency and other healthcare laws and regulations, which could expose us to criminal sanctions, civil penalties, contractual damages, reputational harm, and diminished profits and future earnings.
Our future arrangements with third-party payors and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we research, market, sell, and distribute our medicines for which we obtain marketing approval. Restrictions under applicable federal and state healthcare laws and regulations include the following:following, some of which will not apply unless or until we have a marketed product:
•federal Anti-Kickback Statute, which prohibits, among other things, persons from offering, soliciting, receiving or providing remuneration, directly or indirectly, to induce either the referral of an individual for, or the purchasing or ordering of, a good or service for which payment may be made under federal healthcare programs such as Medicare and Medicaid;
•federal false claims, false statements and civil monetary penalties laws prohibiting, among other things, any person from knowingly presenting, or causing to be presented, a false claim for payment of government funds or knowingly making, or causing to be made, a false statement material to a false claim;
•the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which, in addition to privacy protections applicable to healthcare providers and other entities, prohibits executing a scheme to defraud any healthcare benefit program and making false statements relating to healthcare matters;
•the Physicianso-called federal “sunshine” law, or Open Payments, Sunshine Act, which requires pharmaceutical and medical device companies to report information related to certain payments and transfers of value to certain healthcare providers
to the Center for Medicare & Medicaid Services, as well as ownership and investment interests held by physicians and their immediate family members;
•federal consumer protection and unfair competition laws broadly regulate marketplace activities and activities that potentially harm consumers;
•the Federal Food, Drug, and Cosmetic Act, which among other things, strictly regulates drug product and medical device marketing, prohibits manufacturers from marketing such products prior to approval or for unapproved indications and regulates the distribution of samples;
•federal laws, including the Medicaid Drug Rebate Program, that require pharmaceutical manufacturers to report certain calculated product prices to the government or provide certain discounts or rebates to government authorities or private entities, often as a condition of reimbursement under government healthcare programs; and
•analogous state and foreign laws and regulations, such as state anti-kickback, anti-bribery and false claims laws, which may apply to healthcare items or services that are reimbursed by non-governmental third-party payors, including private insurers, as well as other state laws that require companies to comply with specific compliance standards, restrict financial interactions between companies and healthcare providers, and require companies to report information related to payments to health care providers, marketing expenditures or marketing expenditures. pricing, or require the licensing or registration of sales representatives.
Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations will involve substantial costs. Given the breadth of the laws and regulations, limited guidance for certain laws and regulations and evolving government interpretations of the laws and regulations, governmental authorities may possibly conclude that our business practices may not comply with healthcare laws and regulations, including, without limitation, certain of our advisory board agreements with physicians who receive stock or stock options as compensation for services provided to us. If our operations are found to be in violation of any of the laws described above or any other government regulations that apply to us, we may be subject to penalties, including civil and criminal penalties, damages, fines, exclusion from participation in government health care programs, such as Medicare and Medicaid, imprisonment, and the curtailment or restructuring of our operations, any of which could adversely affect our business, financial condition, results of operations, and prospects. Further, defending against any such actions can be costly, time-consuming and may require significant personnel resources. Therefore, even if we are successful in defending against any such actions that may be brought against us, our business may be impaired.
Healthcare legislative reform measures may have a material adverse effect on our business and results of operations.
The U.S. and many foreign jurisdictions have enacted or proposed legislative and regulatory changes affecting the healthcare system that could prevent or delay marketing approval of our current or future product candidates or any future product candidates, restrict or regulate post-approval activities and affect our ability to profitably sell a product for which we obtain marketing approval. In particular, in the U.S., there have been and continue to be a number of legislative initiatives at the federal and state level to contain healthcare costs.costs, including specifically the cost of drugs. For example, in March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, of collectively, the ACA, was enacted, which substantially changed the way healthcare is financed by both government and private payors. Since its enactment, there have been judicial and Congressional challenges to certain aspectsimplementation of the ACA,IRA enacted in 2022 was intended in part to address the high cost of prescription drugs. The IRA includes caps on Medicare Part D out-of-pocket costs, Medicare Part B and we expect there will be additional challengesPart D drug price inflation rebates, a new Medicare Part D manufacturer discount drug program and amendments
the IRA remains uncertain pending ongoing implementation, the IRA it is likely to have a significant effect on the ACA in the future. It is unclear how any efforts to challenge, repeal, or replace the ACA will impact the ACA or our business.healthcare industry and prescription drug pricing overall. See “Business Section—Government Regulation—Current and Future Healthcare Reform Legislation”.
Changes in regulations, statutes or the interpretation of existing regulations could impact our business in the future by requiring, for example: (i) changes to our manufacturing arrangements, (ii) additions or modifications to product labeling, (iii) the recall or discontinuation of our products or (iv) additional record-keeping requirements. Further, healthcare reform may result in changes to payment methodologies, the implementation of pharmaceutical and biological product price controls, and reductions in Medicare and other healthcare funding. If any such changes were to be imposed, they could adversely affect the operation of our business.
Adoption of new legislation at the federal or state level could affect demand for, or pricing of, our current or future products if approved for sale. We cannot, however, predict the ultimate content, timing or effect of any federal and state reform efforts. There is no assurance that federal or state health care reform will not adversely affect our future business and financial results.
The successful commercialization of our product candidates will depend in part on the extent to which third-party payors establish coverage, adequate reimbursement levels and pricing policies.
Our ability to obtain coverage and adequate reimbursement for our product candidates by governmental healthcare programs, private health insurers, and other third-party payors will have an effect on our ability to successfully commercialize our product
candidates. We cannot be sure that coverage and reimbursement will be available for our product candidates or any future product candidate that we may develop, and any reimbursement that may become available may not be adequate or may be decreased or eliminated in the future. No uniform policy for coverage and reimbursement for products exists among third-party payors in the United States. Therefore, coverage and reimbursement for products can differ significantly from payor to payor. As a result, the coverage determination process is often a time-consuming and costly process that will require us to provide scientific and clinical support for the use of our product candidates to each payor separately, with no assurance that coverage and adequate reimbursement will be applied consistently or obtained in the first instance. If reimbursement is not available or is available only at limited levels, we may not be able to successfully commercialize our product candidates and may not be able to obtain a satisfactory financial return on our product candidates.
We are subject to U.S. and international restrictive regulations governing the use, processing and cross-border transfer of data and personal information.
The conduct of our clinical trials may be subject to privacy restrictions based on U.S. and non-U.S. regulations. For example, we may be subject to the California Consumer Privacy Act, or CCPA. As currently written, the CCPA may impactimpact our business activities and exemplifies the vulnerability of our business to the evolving regulatory environment related to personal data and protected health information. Additionally, the collection, use, storage, disclosure, transfer, or other processing of personal data regarding individuals in the EU and the UK, including personal health data, is subject to the EU General Data Protection Regulation, or GDPR including as it forms part of the law of England and Wales, Scotland and Northern Ireland by virtue of section 3 of the European Union (Withdrawal) Act 2018 and as amended by the Data Protection, Privacy and Electronic Communications (Amendments etc.) (EU Exit) Regulations 2019 (SI 2019/419), known as UK GDPR. See “Business—Government Regulation.” Compliance with the GDPR and the UK GDPR will be a rigorous and time-intensive process that may increase our cost of doing business or require us to change our business practices, and despite those efforts, there is a risk that we may be subject to fines and penalties, litigation and reputational harm in connection with our European activities. The UK’s data protection authority, the Information Commissioner’s Office, has indicated that following Brexit it will continue to enforce the UK GDPR in line with the enforcement of the GDPR in the EU. However, the UK government recently announced its intention to adopt a more flexible approach to the regulation of data, and as a result there remains a risk of future divergence between the EU and UK data protection regimes.
Compliance with U.S. and international data protection laws and regulations could require us to take on more onerous obligations in our contracts, restrict our ability to collect, use and disclose data, or in some cases, impact our ability to operate in certain jurisdictions. Failure to comply with U.S. and international data protection laws and regulations could result in government enforcement actions (which could include civil or criminal penalties), private litigation or adverse publicity and could negatively affect our operating results and business. Moreover, clinical trial subjects about whom we or our potential collaborators obtain information, as well as the providers who share this information with us, may contractually limit our ability to use and disclose the information. Claims that we have violated individuals’ privacy rights, failed to comply with data protection laws, or breached our contractual obligations, even if we are not found liable, could be expensive and time-consuming to defend and could result in adverse publicity that could harm our business.
General Risk Factors
The market price of our common stock may be volatile, which could result in substantial losses for our stockholders.
Our stock price has been and may continue to be volatile. Since our initial public offeringIPO in October 2020, the closing price of our common stock as reported on the Nasdaq Global Market has ranged from a low of $8.33$2.82 on March 10, 2021February 5, 2024 to a high of $25.88 on December 18, 2020. Some of the factors that may cause the market price of our common stock to fluctuate include:
•the success of existing or new competitive product candidates or technologies;
•the timing and results of preclinical studies and clinical trials for any product candidates that we may develop;
•the failure or discontinuation of any of our product development and research programs;
•results of preclinical studies, clinical trials, or regulatory approvals of product candidates of our competitors, or announcements about new research programs or product candidates of our competitors;
•commencement or termination of collaborations for our product development and research programs;
•regulatory or legal developments in the United States and other countries;
•developments or disputes concerning patent applications, issued patents or other proprietary rights;
•the recruitment or departure of key personnel;
•the level of expenses related to any of our research programs or product candidates that we may develop;
•the results of our efforts to develop additional product candidates or products;
•actual or anticipated changes in estimates as to financial results, development timelines or recommendations by securities analysts;
•the announcement or expectation of additional financing efforts;
•sales of our common stock by us, our insiders or other stockholders;
•expiration of market stand-off or lock-up agreements;
•the effects of geopolitical crises and the outbreak or worsening of wars or other armed conflicts;
•effects of public health crises, pandemics and epidemics, such as COVID-19;
•variations in our financial results or those of companies that are perceived to be similar to us;
•changes in estimates or recommendations by securities analysts, if any, that cover our stock;
•changes in the structure of healthcare payment systems;
•market conditions in the pharmaceutical and biotechnology sectors;
•general economic, industry and market conditions; and
•the other factors described in this “Risk Factors” section.
In recent years, the stock market in general, and the market for pharmaceutical and biotechnology companies in particular, has experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to changes in the operating performance of the companies whose stock is experiencing those price and volume fluctuations. Broad market and industry factors may seriously affect the market price of our common stock, regardless of our actual operating performance. Following periods of such volatility in the market price of a company’s securities, securities class action litigation has often been brought against that company. Because of the potential volatility of our stock price, we may become the target of securities litigation in the future.
Securities litigation could result in substantial costs and divert management’s attention and resources from our business.
If securities analysts cease to publish research or reports about our business or if they publish negative evaluations of our stock, the price of our stock could decline.
The trading market for our common stock relies in part on the research and reports that industry or financial analysts publish about us or our business. If one or more of the analysts covering our business downgrade their evaluations of our stock, the price of our stock could decline. If one or more of these analysts cease to cover our stock, we could lose visibility in the market for our stock, which in turn could cause our stock price to decline.
A significant portion of our total outstanding shares is eligible to be sold into the market, in the near future, which could cause the market price of our common stock to drop significantly, even if our business is doing well.
Sales of a substantial number of shares of our common stock in the public market, or the perception in the market that the holders of a large number of shares intend to sell shares, could reduce the market price of our common stock. Certain holders of shares of our common stock have rights, subject to specified conditions, to require us to file registration statements covering their shares or to include their shares in registration statements that we may file for ourselves or other stockholders, until such shares can otherwise be sold without restriction under Securities Act Rule 144 or until the rights terminate pursuant to the terms of the investors’ rights agreement between us and such holders. If additional shares are sold, or if it is perceived that they will be sold, in the public market, the market price of our common stock could decline.
Insiders have substantial influence over us, which could limit your ability to affect the outcome of key transactions, including a change of control.
Our directors and executive officers and their affiliates beneficially own shares representing approximately 46%39% of our outstanding common stock. As a result, these stockholders, if they act together, will be able to influence our management and affairs and all matters requiring stockholder approval, including the election of directors and approval of significant corporate transactions. This concentration of ownership may have the effect of delaying or preventing a change in control of our company and might adversely affect the market price of our common stock.
We are an “emerging growth company,” and the reduced disclosure requirements applicable to emerging growth companies may make our common stock less attractive to investors.
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012 or the JOBS Act,(the “JOBS Act”), and may remain an emerging growth company for up to five years. For so long as we remain an emerging growth company, we are permitted and plan to rely on exemptions from certain disclosure requirements that are applicable to other public companies that are not emerging growth companies. These exemptions include not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002 or SOX,(“SOX”), not being required to comply with any requirement that may be adopted by the Public Company Accounting Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements, reduced disclosure obligations regarding executive compensation, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. As a result, the information we provide stockholders will be different than the information that is available with respect to other public companies. In this annual report, we have not included all of the executive compensation related information that would be required if we were not an emerging growth company. We cannot predict whether investors will find our common stock less attractive if we rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock, and our stock price may be more volatile.
In addition, the JOBS Act provides that an emerging growth company can take advantage of an extended transition period for complying with new or revised accounting standards. This allows an emerging growth company to delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have elected not to “opt out” of such extended transition period, which means that when a standard is issued or revised and it has different application dates for public or private companies, we will adopt the new or revised standard at the time private companies adopt the new or revised standard and will do so until such time that we either (i) irrevocably elect to “opt out” of such extended transition period or (ii) no longer qualify as an emerging growth company. Therefore, the reported results of operations contained in our consolidated financial statements may not be directly comparable to those of other public companies.
We incur certain costs as a result of operating as a public company, and our management will be required to devote substantial time to compliance initiatives and corporate governance practices.
In October 2020, we completed our initial public offering.IPO. As a public company, we incur significant legal, accounting, and other expenses that we did not incur as a private company. These expenses will increase once we are no longer an "emerging“emerging growth company"company” pursuant to applicable securities rules and regulations. The Sarbanes-Oxley Act of 2002, the Dodd-Frank Wall Street Reform and Consumer Protection Act, the listing requirements of the Nasdaq Global Market, and other applicable securities rules and regulations impose various requirements on public companies, including establishment and maintenance of effective disclosure and financial controls and corporate governance practices. We expect that we will need to hire additional accounting, finance, and other personnel in connection with our becoming, and our efforts to comply with the requirements of being, a public company. Our management and other personnel will need to devote a substantial amount of time towards maintaining compliance with these requirements, which will increase our legal and financial compliance costs and will make
certain activities more time-consuming and costly. For example, we expect that the rules and regulations applicable to us as a public company may make it more difficult and more expensive for us to obtain director and officer liability insurance, which could make it more difficult for us to attract and retain qualified members of our board of directors. We are currently evaluating these rules and regulations and cannot predict or estimate the amount of additional costs we may incur or the timing of such costs. These rules and regulations are often subject to varying interpretations, in many cases due to their lack of specificity, and, as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies. This could result in continuing uncertainty regarding compliance matters and higher costs necessitated by ongoing revisions to disclosure and governance practices.
Pursuant to SOX Section 404, we are required to furnish a report by our management on our internal control over financial reporting beginning with our second filing of an Annual Report on Form 10-K with the SEC after we become a public company. However, while we remain an emerging growth company, we will not be required to include an attestation report on internal control over financial reporting issued by our independent registered public accounting firm. To achieve compliance with SOX Section 404 within the prescribed period, we will be engaged in a process to document and evaluate our internal control over financial reporting, which is both costly and challenging. In this regard, we will need to continue to dedicate internal resources, potentially engage outside consultants, adopt a detailed work plan to assess and document the adequacy of internal control over financial reporting, continue steps to improve control processes as appropriate, validate through testing that controls are functioning as documented, and implement a continuous reporting and improvement process for internal control over financial reporting. Despite our efforts, there is a risk that we will not be able to conclude, within the prescribed timeframe or at all, that our internal control over financial reporting is effective as required by SOX Section 404. If we identify one or more material weaknesses, it could result in an adverse reaction in the financial markets due to a loss of confidence in the reliability of our financial statements.
Unfavorable global economic conditions could adversely affect our business, financial condition or results
Our results of operations could be adversely affected by general conditions in the global economy and in the global financial markets. A severe or prolonged economic downturn, or additional global financial crises, could result in a variety of risks to our business, including weakened demand for our product candidates, if approved, or our ability to raise additional capital when needed on acceptable terms, if at all. For example, the global financial crisis caused extreme volatility and disruptions in the capital and credit markets. Similarly, the recent significant volatility associated with the COVID-19 outbreak has caused significant instability and disruptions in the capital and credit markets. A weak or declining economy could also strain our suppliers, possibly resulting in supply disruption. Any of the foregoing could harm our business and we cannot anticipate all of the ways in which the current economic climate and financial market conditions could adversely impact our business.
Provisions in our amended and restated certificate of incorporation, our amended and restated by-laws and Delaware law may have anti-takeover effects that could discourage an acquisition of us by others, even if an acquisition would be beneficial to our stockholders and may prevent attempts by our stockholders to replace or remove our current management.
Our amended and restated certificate of incorporation, amended and restated by-laws and Delaware law contain provisions that may have the effect of discouraging, delaying or preventing a change in control of us or changes in our management that stockholders may consider favorable, including transactions in which you might otherwise receive a premium for your shares. Our amended and restated certificate of incorporation and by-laws include provisions that:
•authorize “blank check” preferred stock, which could be issued by our board of directors without stockholder approval and may contain voting, liquidation, dividend and other rights superior to our common stock;
•create a classified board of directors whose members serve staggered three-year terms;
•specify that special meetings of our stockholders can be called only by our board of directors;
•prohibit stockholder action by written consent;
•establish an advance notice procedure for stockholder approvals to be brought before an annual meeting of our stockholders, including proposed nominations of persons for election to our board of directors;
•provide that vacancies on our board of directors may be filled only by a majority of directors then in office, even though less than a quorum;
•provide that our directors may be removed only for cause;
•specify that no stockholder is permitted to cumulate votes at any election of directors;
•expressly authorize our board of directors to modify, alter or repeal our amended and restated by-laws; and
•require supermajority votes of the holders of our common stock to amend specified provisions of our amended and restated certificate of incorporation and amended and restated by-laws.
These provisions, alone or together, could delay or prevent hostile takeovers and changes in control or changes in our management. These provisions could also limit the price that investors might be willing to pay in the future for shares of our common stock, thereby depressing the market price of our common stock.
In addition, because we are incorporated in the State of Delaware, we are governed by the provisions of Section 203 of the General Corporation Law of the State of Delaware or the DGCL,(the “DGCL”) which prohibits a person who owns in excess of 15% of our outstanding voting stock from merging or combining with us for a period of three years after the date of the transaction in which the person acquired in excess of 15% of our outstanding voting stock, unless the merger or combination is approved in a prescribed manner.
Any provision of our amended and restated certificate of incorporation, amended and restated by-laws or Delaware law that has the effect of delaying or deterring a change in control could limit the opportunity for our stockholders to receive a premium for their shares of our common stock, and could also affect the price that some investors are willing to pay for our common stock.
Our amended and restated certificate of incorporation designates the state or federal courts within the State of Delaware as the exclusive forum for certain types of actions and proceedings that may be initiated by our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers or employees.
Our amended and restated certificate of incorporation provides that, subject to limited exceptions, the state or federal courts (as appropriate) within the State of Delaware will be exclusive forums for (i) any derivative action or proceeding brought on our behalf, (ii) any action asserting a claim of breach of a fiduciary duty owed by any of our directors, officers or other employees to us or our stockholders, (iii) any action asserting a claim against us arising pursuant to any provision of the DGCL, our amended and restated certificate of incorporation or our amended and restated by-laws, (iv) action against us or any of our directors or officers involving a claim or defense arising pursuant to the Exchange Act or the Securities Act or (v) any other action asserting a claim against us that is governed by the internal affairs doctrine. Any person or entity purchasing or otherwise acquiring any interest in shares of our capital stock shall be deemed to have notice of and to have consented to the provisions of our amended and restated certificate of incorporation described above. This choice of forum provision may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, officers or other employees, which may discourage such lawsuits against us and our directors, officers and employees. Alternatively, if a court were to find these provisions of our amended and restated certificate of incorporation inapplicable to, or unenforceable in respect of, one or more of the specified types of actions or proceedings, we may incur additional costs associated with resolving such matters in other jurisdictions, which could adversely affect our business and financial condition.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
ITEM 1C. CYBERSECURITY
Risk Management and Strategy
We have established an enterprise risk management program, with cybersecurity representing a relevant component of our overall approach to risk management. The cybersecurity portion of our risk management program shares common methodologies, reporting channels and governance processes that apply across the program to other legal, compliance, strategic, operational, and financial risk areas. In general, we seek to address cybersecurity risks through a data security framework that is focused on information classification and location, access and acceptable use controls, alerts and protections, vendor risk assessment, and backup and recovery. We engage third parties to perform assessments of our cybersecurity measures, including tabletop exercises, penetration testing, and other cybersecurity audits. As part of our cybersecurity risk management procedures, we also have a third-party cyber risk management process for service providers, suppliers, and vendors.Additionally, we provide regular, mandatory training for all employees regarding cybersecurity threats.
As of the date of this Annual Report on Form 10-K, we have not experienced a cybersecurity incident or other threats that resulted in a material effect on our business strategy, results of operations, or financial condition, but we cannot provide assurance that we will not be materially affected in the future by such risks or any future material incidents. For more information on our cybersecurity related risks, see Item 1A Risk Factors of this Annual Report on Form 10-K.
Governance
Our Board of Directors and Audit Committee oversee our enterprise risk management process, including the management of risks arising from cybersecurity threats. On at least an annual basis, the Board of Directors and Audit Committee receive presentations and reports on cybersecurity risks from our Vice President of Information Technology, which may address topics including our overall assessment of cybersecurity risks, compliance with cybersecurity policies and procedures, third-party test results, the current threat environment, and technological trends and information security considerations arising with respect to our peers in the biotechnology space. The Board and Audit Committee also receive ad hoc reporting as appropriate. Our Vice President of Information Technology, who reports to our Chief Strategy and Business Operations Officer, is primarily responsible for assessing such risks. This individual has over 20 years of information technology and cybersecurity experience in the biotechnology industry.
ITEM 2. PROPERTIES
Our corporate headquarters is located at 500 Technology Square, Suite 700, Cambridge, MA 02139, where we lease and occupy approximately 81,441 square feet of office and laboratory space. The current term of our 500 Technology Square lease expires in September 2028, with an option to extend the term five additional years with 12 months’ notice at an agreed upon market rate.
We believe our existing facilities are sufficient for our needs for the foreseeable future. To meet the future needs of our business, we may lease additional or alternate space, and we believe suitable additional or alternative space will be available in the future on commercially reasonable terms.
ITEM 3. LEGAL PROCEEDINGS
From time to time, we may become involved in litigation or other legal proceedings. We are not currently a party to any litigation or legal proceedings that, in the opinion of our management, are probable to have a material adverse effect on our business. Regardless of outcome, litigation can have an adverse impact on our business, financial condition, results of operations and prospects because of defense and settlement costs, diversion of management resources and other factors.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Certain Information Regarding the Trading of Our Common Stock
Our common stock trades under the symbol “FHTX” on the Nasdaq Global Market and has been publicly traded since October 23, 2020. Prior to this time, there was no public market for our common stock.
Holders of Our Common Stock
As of February 25, 2022,29, 2024, there were approximately 3529 holders of record of shares of our common stock. This number does not include stockholders for whom shares are held in “nominee” or “street” name.
Securities Authorized for Issuance Under Equity Compensation Plans
The information required by Item 5 of Form 10-K regarding equity compensation plans is incorporated herein by reference to Item 12 of Part III of this Annual Report.
Recent Sales of Unregistered Securities
The following sets forth information as to all securities we sold in 2021 which were not registered under the Securities Act:
On December 10, 2021 the Company entered into a stock purchase agreement, or the Lilly SPA, with Eli Lilly and Company, or Lilly, pursuant to which the Company issued and sold 4,000,000 shares of our common stock at a price of $20.00 per share for aggregate gross proceeds of $80.0 million. The issuance of the shares did not involve any underwriters, underwriting discounts or commissions or a public offering. The Company issued the shares in reliance on the exemption from registration provided for under Section 4(a)(2) of the Securities Act of 1933, as amended. The Company relied on this exemption from registration for private placements based in part on the representations made by Lilly, including the representations with respect to Lilly’s investment intent. The offer and sale of the shares have not been registered under the Securities Act.
Use of Proceeds from Initial Public Offering
On October 27, 2020, we closed our initial public offering, or IPO, of our common stock pursuant to which we issued and sold 7,500,000 shares of our common stock at a price to the public of $16.00 per share for aggregate gross proceeds of $120.0 million, before deducting underwriting discounts and commissions and other offering expenses. On November 19, 2020, we sold an additional 951,837 shares of our common stock pursuant to the underwriters’ option to purchase additional shares in the IPO at the public offering price for an additional $15.2 million in gross proceeds.
All of the shares issued and sold in the IPO were registered under the Securities Act pursuant to the Registration Statement (File No. 333-249264), which was declared effective by the SEC on October 22, 2020. Goldman Sachs & Co. LLC, Morgan Stanley & Co. LLC and Cowen and Company, LLC acted as joint book-running managers and Wedbush Securities Inc. acted as lead manager of our IPO.
We received aggregate net proceeds of approximately $122.1 million after deducting underwriting discounts and commissions and other offering expenses. None of the underwriting discounts and commissions or offering expenses were incurred or paid to directors or officers of ours or their associates or to persons owning 10 percent or more of our common stock or to any of our affiliates.
We have used approximately $65.7 million of the net proceeds from the IPO as of December 31, 2021 to advance FHD-286 and FHD-609 towards the clinic, to further invest in our pipeline and platform targeting the chromatin regulatory system and for working capital and other general corporate purposes. There has been no material change in our planned use of the net proceeds from the offering as described in our Registration Statement.None.
Issuer Purchases of Equity Securities by the Issuer and Affiliated Purchasers
None.
ITEM 6. [RESERVED]
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion and analysis of our financial condition and results of operations should be read in conjunction with our consolidated financial statements and the related notes to those statements included elsewhere in this Annual Report on Form 10-K. In addition to historical financial information, the following discussion and analysis contains forward-looking statements that involve risks, uncertainties and assumptions. Some of the numbers included herein have been rounded for the convenience of presentation. Our actual results may differ materially from those anticipated in these forward-looking statements as a result of many factors, including those discussed under “Risk Factors” and elsewhere in this Annual Report on Form 10-K.
Overview
We areFoghorn is a clinical stage, precision therapeutics biotechnology company pioneering a new class of medicines that modulatetreat serious diseases by correcting abnormal gene expression through selectively targeting the chromatin regulatory system, an untapped opportunity for therapeutic intervention. intervention in oncology and with potential in a wide spectrum of other diseases including virology, autoimmune diseases and neurology.
The chromatin regulatory system orchestrates gene expression—the turning on and off of genes—which is fundamental to how all our cells function. The chromatin regulatory system is implicated in approximately 50 percent of all cancers, and understanding how this system works could lead to an entirely new class of precision medicines. To our knowledge, we are the only company with the ability to study and target the chromatin regulatory system at scale, in context, and in an integrated way.
Our proprietary Gene Traffic Control® platform gives usprovides an integrated and mechanistic understanding of how the various components of the chromatin regulatory system interact, allowing us to identify, validate and potentially drug targets within thethis system. BreakdownsWe have developed unique capabilities that have yielded new insights and scalability in the chromatin regulatory system are associated with over 50 percent of all cancers. Addressing these breakdowns could potentially provide therapies for over 2.5 million patients globally. Consequently,drugging this new, previously untapped and promising area.
At present, we are initially focusedworking on more than 10 programs with one clinical-stage drug candidate currently in oncology.Phase 1 development and one drug candidate anticipated to begin clinical development this year. We have discovered highly selective chemical matter for some of the most challenging targets in oncology including BRM, CBP, EP300 and ARID1B, as well as other undisclosed targets. We believe our current pipeline has the potential to help more than 500,000 cancer patients. We take a
small molecule modality agnostic approach to drugging targets which includes protein degraders, allosteric enzymatic inhibitors, and transcription factor disruptors. We are developinga biology first company which means we focus first on the underlying genetics and biology of a disease relevant target and then leverage the most appropriate drugging approach to impact the disease biology.
We are currently conducting a Phase 1 dose escalation study of FHD-286, a selective, allosteric ATPase inhibitor that is currently being evaluatedof BRM and BRG1, in two separate Phase 1 studiescombination with either decitabine or cytarabine in (i) metastatic uveal melanoma and (ii) relapsed and/or refractory AMLacute myeloid leukemia (“AML”) patients. As part of our collaboration with Loxo Oncology at Eli Lilly and MDS. We are developing FHD-609, a targeted protein degraderCompany (“Lilly”), we anticipate that is currently being evaluated in a Phase 1 dose escalation study will start later this year with FHD-909, a selective ATPase inhibitor of BRM.
During the third quarter of 2023, we transitioned the BRM Selective inhibitor program to Lilly which triggered the 50/50 cost share for the BRM programs. Costs related to the cost-share are included in synovial sarcoma. Our vision is to use our Gene Traffic Control platform to discoverresearch and develop drugs in oncologydevelopment expenses on the consolidated statements of operations and other therapeutic areas, including virology, autoimmune disease and neurology.comprehensive loss.
Since our inception, in October 2015, we have focused substantially all of our resources on building our Gene Traffic Control platform, organizing and staffing our company, business planning, raising capital, conducting discovery and research activities, protecting our trade secrets, filing patent applications, identifying potential product candidates, undertaking preclinical studies and clinical trial activities, establishing arrangements with third parties for the manufacture of initial quantities of our product candidates and component materials and initiating two strategic collaborations. We do not have any products approved for sale and have not generated any revenue from product sales.
On October 27, 2020, we completed our initial public offering, or IPO, pursuant to which we issued and sold 7,500,000 shares of our common stock at a public offering price of $16.00 per share, resulting in net proceeds of $107.9 million, after deducting underwriting discounts and commissions and other offering expenses. On November 19, 2020, we issued and sold an additional 951,837 shares of common stock at the IPO price of $16.00 per share pursuant to the underwriters’ partial exercise of their option to purchase additional shares of common stock, resulting in additional net proceeds of $14.2 million after deducting underwriting discounts and commissions. Prior to our IPO, we funded our operations with proceeds from sales of preferred stock, term loans and an upfront payment of $15.0 million we received in July 2020 under our collaboration agreement with Merck Sharp & Dohme Corp., or Merck.
On December 10, 2021, we entered into a collaboration agreement or the Lilly(the “Lilly Collaboration Agreement,Agreement”) with Eli Lilly and Company, or Lilly, for which we received an upfront payment of $300.0 million in January 2022 (see Note 98 to our notes to consolidated financial statements included elsewhere in this Annual Report on Form 10-K). Concurrent with the Lilly Collaboration Agreement, we also entered into the Lilly SPAa stock purchase agreement (the “Lilly SPA”) and issued and sold Lilly 4,000,000 shares of our common stock at a price of $20.00 per share, resulting in net proceeds of $80.0 million, of which $42.2$42.2 million was allocated to equity upon the issuance of our common stock.
In the Company’s common stock.third quarter of 2022, we achieved a research milestone related to a Research Collaboration and Exclusive License Agreement (the “Merck Collaboration Agreement”) with Merck Sharp & Dohme Corp. (“Merck”) and received a $5.0 million milestone payment from Merck.
We have incurred significant operating losses since our inception. For the years ended December 31, 20212023 and 2020,2022, we reported net losses of $101.3$98.4 million and $68.8$108.9 million, respectively. As of December 31, 2021,2023, we had an accumulated deficit of $264.3$471.6 million. We expect to continue to incur significant expenses and increasing operating losses for at least the next several years. Our ability to generate any product revenue or product revenue sufficient to achieve profitability will depend on the successful development and eventual commercialization of one or more product candidates we are developing and may develop.
We expect that our expenses and capital requirements will increase substantially in connection with our ongoing activities, particularly if and as we:
•advance our FHD-286 and FHD-609 product candidatescontinue preclinical and continue our preclinicalclinical development of product candidates from our current research programs;portfolio, including those partnered with Lilly;
•identify and advance additional research programs and additional product candidates;
•initiate preclinical testing for any new product candidates we identify and develop;
•obtain, maintain, expand, enforce, defend and protect our trade secrets and intellectual property portfolio and provide reimbursement of third-party expenses related to our patent portfolio;
•hire additional research and development personnel;
•add operational, legal, compliance, financial and management information systems and personnel to support our research, product development and operations;
•expand the capabilities of our platform;
•acquire or in-license product candidates, intellectual property and technologies;
•operate as a public company;
•seek marketing approvals for any product candidates that successfully complete clinical trials; and
•ultimately establish a sales, marketing and distribution infrastructure to commercialize any products for which we may obtain marketing approval.
We will not generate revenue from product sales unless and until we successfully commercialize one of our product candidates, after completing clinical development and obtaining regulatory approval. If we obtain regulatory approval for any of our product candidates, we expect to incur significant expenses related to developing our commercialization capability to support product sales, marketing, manufacturing and distribution. Further, we expect to incur additional costs associated with operating as a public company.
As a result, we will need substantial additional funding to support our continuing operations and pursue our growth strategy. Until such time as we can generate significant revenue from product sales, if ever, we expect to finance our operations through a combination of equity offerings, debt financings and collaborations or licensing arrangements and our collaboration agreements with Merck and Lilly.the Lilly Collaboration Agreement. We may be unable to raise additional funds or enter into such other agreements or arrangements when needed on favorable terms, or at all. If we fail to raise capital or enter into such agreements as, and when, needed, we may have to significantly delay, scale back our development or commercialization plans for one or more of our product candidates.
Because of the numerous risks and uncertainties associated with pharmaceutical product development, we are unable to accurately predict the timing or amount of increased expenses or when, or if, we will be able to achieve or maintain profitability. Even if we are able to generate product sales, we may not become profitable. If we fail to become profitable or are unable to sustain profitability on a continuing basis, then we may be unable to continue our operations at planned levels and be forced to reduce or terminate our operations.
COVID-19
In March 2020, COVID-19 was declared a global pandemic by the World Health Organization, and to date the COVID-19 pandemic continues to present a substantial public health and economic challenge around the world. The length of time and full extent to which the COVID-19 pandemic will directly or indirectly impact our business, results of operations and financial condition will depend on future developments that are highly uncertain, subject to change and are difficult to predict. While we continue to conduct our research and development activities, the COVID-19 pandemic may cause disruptions that affect our ability to initiate and complete preclinical studies, ongoing and future clinical trials or to procure items that are essential for our research and development activities.
We plan to continue to closely monitor the ongoing impact of the COVID-19 pandemic on our employees and our business operations. In an effort to provide a safe work environment for our employees, we have, among other things, increased the cadence of sanitization of our office and lab facilities, implemented various social distancing measures in our office and labs including replacing in-person meetings with virtual interactions, and are providing personal protective equipment for our employees present in our office and lab facilities. We expect to continue to take actions as may be required or recommended by government authorities or as we determine are in the best interests of our employees and other business partners in light of the pandemic.
Components of Our Results of Operations
Collaboration Revenue
To date, we have not generated any revenue from product sales and do not expect to generate any revenue from the sale of productsdo so in the near future. If our development efforts for our product candidates are successful and result in regulatory approval or licenses with third parties, we may generate revenue in the future from product sales, milestone payments under our existing collaboration agreements or payments from other license agreements that we may enter into with third parties.
In December of 2021, we entered into a strategic collaboration with Loxo Oncology at Lilly a research and development group of Eli Lilly and Company, to create novel oncology medicines by applying Foghorn’s proprietary Gene Traffic Control platform. The collaboration includes a co-development and co-commercialization agreement for the aforementioned selective BRM oncology program and an additional undisclosed oncology target. In addition, the collaboration includes three additional discovery programs using Foghorn’s proprietary Gene Traffic Control platform. Under the terms of the collaboration, Foghorn received upfront consideration of $300.0 million in cash pursuant to the Lilly Collaboration Agreement, together with an equity investment by Lilly of $80.0 million in shares of Foghorn common stock pursuant to the Lilly SPA.
For the BRM-selectiveBRM selective program and the additional undisclosed target program, Foghorn will lead discovery and early research activities, while Lilly will lead development and commercialization activities with participation from Foghorn in operational activities and cost sharing. Foghorn and Lilly will share 50/50 in the U.S. economics, and Foghorn is eligible to receive royalties on ex-U.S. sales starting in the low double-digit range and escalating into the twenties based on revenue levels.
For the additional discovery programs, Foghorn will lead discovery and early research activities. Foghorn may receive up to a total of $1.3 billion in potential development and commercialization milestones. Additionally, Foghorn will have an option to participate in a percentage of the U.S. economics and is eligible to receive tiered royalties from the mid-single digit to low-double digit range on sales outside the U.S. that may be exercised after the successful completion of the dose-finding toxicity studies.
We cannot provide assurances as to the timing of future milestones, royalty payments and economics associated with the strategic collaboration with Loxo Oncology at Lilly.Lilly, if any.
The CompanyIn the third quarter of 2023, we transitioned the BRM Selective inhibitor program to Lilly, for which Lilly will lead and we will participate and share in 50% of the costs until registrational trials. Costs incurred will continue to be included in research and development expenses on the consolidated statements of operations and comprehensive loss.
We recognized total deferred revenue of $337.8 million related to the Lilly Collaboration Agreement and the Lilly SPA, which included the $300.0 million upfront payment under the Lilly Collaboration Agreement as well as $37.8 million allocated to deferred revenue from the gross proceeds of the Lilly SPA to be recognized over the performance period. DuringFor the years ended December 31, 2021 ,2023 and 2022, we recognized $0.6$17.1 million and $17.4 million, respectively, of revenue under the Lilly Collaboration Agreement and, as of December 31, 2021,2023, we had $337.2$302.7 million of deferred revenue related to the above mentioned upfront payment and revenue allocation from the Lilly SPA remaining on our consolidated balance sheets. In September 2023 we re-evaluated our estimates related to the Lilly Collaboration Agreement. As a result of this re-evaluation, expected future costs increased over the course of the Lilly Collaboration Agreement, resulting in an adjustment that decreased revenue by $3.2 million for the year ended December 31, 2023.
In July 2020, we entered into a strategic research collaboration and license agreement, or the Merck Collaboration Agreement, with Merck, pursuant to which we willagreed to apply our proprietary Gene Traffic Control platform to discover and develop novel therapeutics. Under the Merck Collaboration Agreement, we granted Merck exclusive global rights to develop and commercialize drugs that target dysregulation of a single transcription factor. Under the terms of the Merck Collaboration Agreement, we received a nonrefundable upfront payment of $15.0 million from Merck, and are eligible to receive up to $245.0 million upon achievementMerck. In the third quarter of specified2022, we achieved a research development and regulatory milestones by any product candidate generated by the collaboration, and up to $165.0 million upon achievement of specified sales-based milestones per approved product from the collaboration, if any, as well as royalties on sales of any approved product from the collaboration. We cannot provide assurance as to the timing of future milestone or royalty payments or that we will receive any of these payments at all.
We record revenue over the research term as we satisfy our performance obligation under the Merck Collaboration Agreement. Accordingly, the upfront payment of $15.0 million is being recognized as revenue using the cost-to-cost method, which we believe best depicts the transfer of control to the customer over time. Under the cost-to-cost method, the extent of progress towards completion is measured based on the ratio of actual costs incurred to the total estimated costs expected upon satisfying the identified single performance obligation. We expect revenue to fluctuate as the achievement of milestones becomes probable and as our efforts to satisfy our performance obligation vary from period to period. In estimating the total costs to satisfy our performance obligation pursuant to the Merck Collaboration Agreement, we are required to make significant estimates including an estimate of the number of transcription factor substitutions and the expected time and expected costs to fulfill the performance obligation. The cumulative effect of revisions to the total estimated costs to complete our performance obligation will be recorded in the period in which the changes are identified, and amounts can be reasonably estimated. While such revisions will have no impact on our cash flows, a significant change in these assumptions and estimates could have a material impact on the timing and amount of revenue recognized in future periods. In June 2021, we re-evaluated our estimates related to the Merck Collaboration Agreement resulting inand received a reclassification$5.0 million milestone payment from Merck.
On August 9, 2023 we received notice from Merck that it was terminating the Merck Collaboration Agreement effective November 7, 2023. At the time of $1.9 million from short-term deferred revenue to long-term deferred revenue, net of current portion onnotice, no material obligations remained under the Company's consolidated balance sheets. During the years ended December 31, 2021 and 2020,Merck Collaboration Agreement. As such, we recognized $0.7 million and $0.4 million, respectively, of revenue under the Merck
Collaboration Agreement and, as of December 31, 2021, we had $13.9remaining $16.1 million of deferred revenue, related to the original $15.0 million upfront payment and $5.0 million milestone payment received from Merck, as revenue, for year ended December 31, 2023.
For the years ended December 31, 2023 and 2022, we recognized $17.0 million and $1.8 million, respectively, of revenue under the Merck Collaboration Agreement. As of December 31, 2023, we had no deferred revenue related to the upfront payment and milestone achievement remaining on our consolidated balance sheets.
Operating Expenses
Our operating expenses are comprised of research and development expenses and general and administrative expenses.
Research and Development Expenses
Research and development expenses consist primarily of costs incurred forto progress our research activities,proprietary and partnered pipeline, including our discovery efforts, and progressing our programs, which include:
•personnel-related costs, including salaries, benefits and stock-based compensation expense, for employees engaged in research and development functions;
•expenses incurred in connection with our research programs and preclinical and clinical development of our product candidates, including under agreements with third parties, such as consultants and contractors and contract research organizations, or CROs;
•the cost of manufacturing drug substance and drug product for use in our research and preclinical studies and clinical trials under agreements with third parties, such as consultants and contractors and contract development and manufacturing organizations, or CDMOs;
•laboratory supplies and research materials;
•facilities, depreciation and amortization and other expenses, which include direct and allocated expenses for rent and maintenance of facilities and insurance; and
•payments made under third-party licensing agreements.
We track our direct external research and development expenses on a program-by-program basis. These consist of costs that include fees, reimbursed materials, and other costs paid to consultants, contractors, CDMOs, and CROs in connection with our preclinical, clinical and manufacturing activities. We do not allocate employee costs, costs associated with our discovery efforts, laboratory supplies, and facilities expenses, including depreciation or other indirect costs, to specific product development programs because these costs are deployed across multiple programs and our platform and, as such, are not separately classified.
We expect that our research and development expenses willmay increase substantiallyin the future as we advance our programs into clinical development and expandcontinue our discovery, research and preclinical activities in the near term and in the future. At this time, we cannot accurately estimate or know the nature, timing and costs of the efforts that will be necessary to complete the preclinical and clinical development of any product candidates we may develop. A change in the outcome of any number of variables with respect to product candidates we may develop could significantly change the costs and timing associated with the development of that product candidate. We may never succeed in obtaining regulatory approval for any product candidates we may develop.
General and Administrative Expenses
General and administrative expenses consist primarily of personnel-related costs, including salaries, benefits and stock-based compensation, for employees engaged in executive, legal, finance and accounting and other administrative functions. General and administrative expenses also include professional fees for legal, patent, consulting, investor and public relations, human resources, and accounting and audit services as well as direct and allocated facility-related costs.
We anticipate that our general and administrative expenses willmay increase in the future as we increase our headcountcontinue to support our continued research activities and development of our programs and platform. We also anticipate that we will continue to incur increased accounting, audit, legal, regulatory, compliance, director and officer insurance costs and investor and public relations expenses associated with operating as a public company.
Other Income (Expense)
Interest Expense
Interest expense consists of interest expense associated with outstanding borrowings under our loan agreements as well as the amortization of debt discount associated with such agreements.
Interest Income and Other Income (Expense), Net
Interest Income
Interest income consists of interest earned on our invested cash balances.
Other Income, Net
Other income (expense), net consists of sublease income and miscellaneous expense unrelated to our core operations.
Provision for Income Taxes
Since our inception, we have not recorded any federal or state income tax benefits for the net losses we have incurred in any year or for theour federal or state earned research and development tax credits, earneddue to our uncertainty of realizing a benefit from those items. During the year ended December 31, 2023, we recorded a provision for income taxes related to the $300.0 million upfront payment from the Lilly Collaboration Agreement which will be recognized as taxable income in each period, as we believe, based upon the weightcurrent year, and the required capitalization of available evidence, that it is more likely than not that all of our net operating loss carryforwardsresearch and tax credit carryforwards will not be realized.
development costs pursuant to Internal Revenue Code Section 174. As of December 31, 2021, we2023, the Company had U.S. federal and state net operating loss carryforwards of $228.9 million and $206.0 million, respectively, which may be available to offset future taxable income. The federal net operating loss carryforwards include $12.5$3.0 million which expire at various dates beginning in 2035 and $216.4 million which carryforwardcan be carried forward indefinitely but in some circumstances may be limited to offset 80% of annual taxable income. The state net operating loss carryforwards expire at various dates beginning in 2036. As of December 31, 2021, we2023, the Company also had U.S. federal and state research and development tax credit carryforwards of $6.1$3.7 million and $4.1$2.6 million, respectively, which may be available to reduceoffset future tax liabilities and expire at various dates beginning in 20362041 and 2031,2037, respectively. Due to our history of cumulative net losses since inception and uncertainties surrounding our ability to generate future taxable income, we have recorded a full valuation allowance against our net deferred tax assets at each balance sheet date.
Critical Accounting Estimates
Our consolidated financial statements are prepared in accordance with generally accepted accounting principles in the United States, or GAAP.(“GAAP”). The preparation of our consolidated financial statements and related disclosures requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenue, costs and expenses and the disclosure of contingent assets and liabilities in our consolidated financial statements. We base our estimates on historical experience, known trends and events and various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. We evaluate our estimates and assumptions on an ongoing basis. Our actual results may differ from these estimates under different assumptions or conditions.
While our significant accounting policies are described in more detail in Note 2 to our consolidated financial statements appearing elsewhere in this Annual Report on Form 10-K, we believe that the following accounting policies are those most critical to the judgments and estimates used in the preparation of our consolidated financial statements.
Revenue Recognition
We received significant non-refundable upfront payments under our collaboration agreements with Merck and Lilly, from which we recognize revenue over time using the cost-to-cost method. Under the cost-to-cost method, the extent of progress towards completion is measured based on the ratio of actual costs incurred to the total estimated costs expected upon satisfying the identified single performance obligation. In estimating the total costs to satisfy our performance obligation, we are required to make significant estimates including an estimate of the expected time and expected internal and external costs to fulfill the performance obligation. In developing these estimates we consider historical experience, relevant entity-specific factors, known market trends and conditions, and a variety of other factors we believe are relevant to estimating the total cost to fulfill the performance obligation. We periodically evaluate estimates against the actual time and costs incurred as well as any anticipated changes to the timing or estimated costs. Any cumulative effect of revisions to the total estimated costs to complete our performance obligation will be recorded in the period in which the changes are identified, and amounts can be reasonably estimated. While such revisions will have no impact on our cash flows, a significant change in these assumptions and estimates could have a material impact on the timing and amount of revenue recognized in future periods and the classification of deferred revenue between short-term and long-term.
Accrued Research and Development Expenses
As part of the process of preparing our consolidated financial statements, we are required to estimate certain accrued research and development expenses. This process involves estimating the level of service performed and the associated cost incurred for the service when we have not yet been invoiced or otherwise notified of actual costs. We make estimates of our accrued expenses as of each balance sheet date in our consolidated financial statements based on facts and circumstances known to us at that time. We periodically confirm the accuracy of the estimates with the service providers and make adjustments if necessary. Examples of estimated accrued research and development expenses include those related to fees paid to:
•vendors in connection with discovery, preclinical and clinical development activities;
•CROs in connection with preclinical studies and testing and clinical trials; and
•CDMOs in connection with the process development and scale up activities and the production and manufacturing of materials.
We base the expense recorded related to contract research and manufacturing on our estimates of the services received and efforts expended pursuant to quotes and contracts with multiple CROs and CDMOs that conduct services and produce and supply materials. The financial terms of these agreements are subject to negotiation, vary from contract to contract and may result in uneven payment flows. In accruing service fees, we estimate the time period over which services will be performed and the level of effort to be expended in each period. If the actual timing of the performance of services or the level of effort varies from the estimate, we adjust the accrual accordingly. Although we do not expect our estimates to be materially different from amounts actually incurred, our understanding of the status and timing of services performed relative to the actual status and timing of services performed may vary and may result in reporting amounts that are too high or too low in any particular period. While the majority of our service providers invoice us in arrears for services performed, on a pre-determined schedule or when contractual milestones are met, some require advance payments. There may be instances in which payments made to our vendors will exceed the level of services provided and result in a prepayment of the expense. We record these as prepaid expenses on our consolidated balance sheets.
Results of Operations
Comparison of the Years Ended December 31, 20212023 and 20202022
The following table summarizes our results of operations for the years ended December 31, 20212023 and 2020:2022:
| | | | | Year Ended December 31, | | Change |
| | | | Year Ended December 31, | | Change |
| | | 2021 | | 2020 | |
| | | (in thousands) | (in thousands) |
| Collaboration revenue | Collaboration revenue | $ | 1,319 | | | $ | 430 | | | $ | 889 | |
| Operating expenses: | Operating expenses: | | | | | |
| Research and development | Research and development | 80,325 | | | 57,715 | | | 22,610 | |
| Research and development | |
| Research and development | |
| General and administrative | General and administrative | 21,728 | | | 11,246 | | | 10,482 | |
| Total operating expenses | Total operating expenses | 102,053 | | | 68,961 | | | 33,092 | |
| Loss from operations | Loss from operations | (100,734) | | | (68,531) | | | (32,203) | |
| Other income (expense): | Other income (expense): | | | | | |
| Interest expense | (1,906) | | | (979) | | | (927) | |
| Interest income and other income (expense), net | 2,553 | | | 1,001 | | | 1,552 | |
| Change in fair value of preferred stock warrant liability | — | | | (69) | | | 69 | |
| Loss on debt extinguishment | (1,233) | | | (222) | | | (1,011) | |
| Total other expense, net | (586) | | | (269) | | | (317) | |
| Interest income | |
| Interest income | |
| Interest income | |
| Other income, net | |
| | Total other income, net | |
| | Total other income, net | |
| | Total other income, net | |
| Loss before income taxes | |
| Provision for income taxes | |
| Net loss | Net loss | $ | (101,320) | | | $ | (68,800) | | | $ | (32,520) | |
Collaboration Revenue
Under our collaboration agreements, revenue is recognized based on the work performed during the period. Collaboration revenue was $1.3$34.2 million for the year ended December 31, 2021,2023, compared to $0.4$19.2 million for the year ended December 31, 2020.2022. The increase in collaboration revenue is attributed to the following:to:
•an increase in Merck collaboration revenue recognition of $0.3$15.2 million, due primarily to more work completed underdriven by the termination of the Merck Collaboration Agreement in 2021 compared to2023 and the work completed in 2020;subsequent recognition of the remaining deferred revenue; and
•an increasea decrease in Lilly collaboration revenue recognition of $0.6$0.3 million due to a change in estimated total costs to satisfy the remaining performance obligation during 2023 offset by increased work performed pursuant to theon Lilly Collaboration Agreement entered into in December 2021.partnered targets.
Research and Development Expenses
The following table summarizes our research and development expenses for the years ended December 31, 20212023 and 2020:2022:
| | | | | Year Ended December 31, | | Change |
| | | | Year Ended December 31, | | Change |
| | | 2021 | | 2020 | |
| | | (in thousands) | (in thousands) |
| Research and development program expenses: | Research and development program expenses: | |
| FHD-286 | FHD-286 | $ | 10,289 | | | $ | 5,570 | | | $ | 4,719 | |
| FHD-286 | |
| FHD-286 | |
| FHD-609 | FHD-609 | 7,604 | | | 7,137 | | | 467 | |
| Platform, research and discovery, and unallocated expenses: | Platform, research and discovery, and unallocated expenses: | |
| Platform and other early stage research external costs | Platform and other early stage research external costs | 18,865 | | | 13,707 | | | 5,158 | |
| Platform and other early stage research external costs | |
| Platform and other early stage research external costs | |
| Personnel related (including stock-based compensation) | Personnel related (including stock-based compensation) | 25,405 | | | 17,822 | | | 7,583 | |
| Facility related and other | 18,162 | | | 13,479 | | | 4,683 | |
| Facilities and IT related expenses and other | |
| Total research and development expenses | Total research and development expenses | $ | 80,325 | | | $ | 57,715 | | | $ | 22,610 | |
Research and development expenses were $80.3$109.7 million for the year ended December 31, 2021,2023, compared to $57.7$105.6 million for the year ended December 31, 2020.2022. The increase is attributed to the following:
•an increase in personnel-related costs of $7.6 million, including a $2.6 million increase in stock-based compensation expense, due primarily to increased headcount in our research and development function;
•an increase in platform and other early stage research costs of $5.2$10.2 million, including an increase of $2.4 million related to the advancement of Lilly partnered targets, which was due to continued investment and development of our platform and early research pipeline;
•an increase in personnel-related costs of $4.1 million, including a $0.4 million increase in stock-based compensation expense, due to higher average headcount in our research and development functions compared to prior year, and $1.4 million in one-time separation costs; and
•an increase in facility-related and other expenses of $4.7$1.1 million was due to the increased costs of supporting a growingour research and development organization and their research efforts, including increased rent and maintenance expense related to our new facility lease, and purchases of lab supplies, consumables, and equipment;efforts;
Partially offset by:
•an increasea decrease in our FHD-286 program costs of $4.7$7.7 million, driven by a decline in development spend in 2023 as the Company initiatedCompany’s Phase 1 study in AML/MDS was on clinical trials in Uveal Melanomahold until June 2023 and AML and MDSthe shutdown of the Phase 1 uveal melanoma study beginning in the first halfsecond quarter of 2021;2023; and
•an increasea decrease in our FHD-609 program costcosts of $0.5$3.7 million asassociated with the Company initiated enrollment in apartial clinical hold and subsequent shutdown of the Phase 1 clinical trial for FHD-609 in synovial sarcoma and SMARCAB1-loss tumors, which started in the second halfquarter of 2021 in Synovial Sarcoma.2023.
General and Administrative Expenses
The following table summarizes our general and administrative expenses for the years ended December 31, 20212023 and 2020:2022:
| | | |
| | | | Year Ended December 31, | |
| | | 2021 | | 2020 | | Change | 2023 | | 2022 | | Change |
| | | (in thousands) | | (in thousands) |
| Personnel related (including stock-based compensation) | Personnel related (including stock-based compensation) | $ | 12,062 | | | $ | 6,026 | | | $ | 6,036 | |
| Professional and consultant | 4,852 | | | 3,388 | | | 1,464 | |
| Facility related and other | 4,814 | | | 1,832 | | | 2,982 | |
| Professional and consulting | |
| Facilities and IT related expenses and other | |
| Total general and administrative expenses | Total general and administrative expenses | $ | 21,728 | | | $ | 11,246 | | | $ | 10,482 | |
General and administrative expenses were $21.7$32.4 million for the year ended December 31, 2021,2023, compared to $11.2$30.7 million for the year ended December 31, 2020.2022. The increase is attributed to the following:
•an increase in personnel-related costs of $6.0$2.3 million, including a $2.8$1.4 million increase in stock-based compensation expense, which was a result of an increase indue to higher average headcount in our general and administrative function to support our business; and
•an increasea decrease in facility related and other expense of $3.0$0.9 million, which was primarily due to including increased rent, maintenance,a decrease in insurance related expense and less overall purchases of non-capital equipment expense related to our new facility lease; andin 2023.
•an increase in professional and consulting costs of $1.5 million, which was due to increased audit, tax, and consulting services associated with being a publicly traded company.
Other Income (Expense)
Interest expenseTotal other income, net was $1.9$13.7 million for the year ended December 31, 2021,2023, compared to $1.0$8.3 million for the year ended December 31, 2020.2022. The increase in total other income, net was primarily due to an increase in interest income driven by an increase in the increased outstanding debt balance and higheraverage interest rates on our borrowingsrate partially offset by lower average cash balances throughout 2021the year ended December 31, 2023 compared to 2020.the year ended December 31, 2022.
InterestProvision for income taxes
For the year ended December 31, 2023, we recorded an income tax provision of $4.2 million. The income tax provision is primarily driven by the current federal and otherstate taxes related to the $300.0 million upfront payment for Lilly Collaboration Agreement, which will be recognized as taxable income (expense), net was $2.6 millionin the current year, and the required capitalization of research and development costs pursuant to Internal Revenue Code Section 174. We did not record a provision for income taxes for the year ended December 31, 2021 and consisted primarily of sublease income of $2.5 million related to the sublease that began in July 2020 and $0.1 million of interest income. Interest income and other income (expense), net of $1.0 million for the year ended December 31, 2020 consisted primarily of sublease income of $1.0 million related to the sublease that began in July 2020 and $0.1 million of interest income.
We recorded a loss on debt extinguishment of $1.2 million for the year ended December 31, 2021 compared to $0.2 million for the year ended December 31, 2020. We used cash on hand to repay early the balance of our loan outstanding at the time, including a final payment fee and a prepayment fee during December 31, 2021. During the year ended December 31, 2020, we repaid our then outstanding loan in November 2020, which was contractually lower than the prepayment costs incurred in 2021.2022.
Liquidity and Capital Resources
Since our inception in October 2015, we have incurred significant operating losses. We expect to incur significant expenses and operating losses for the foreseeable future as we support our continued research activities and development of our programs and platform. Through December 31, 2021,2023, we have funded our operations with proceeds from our IPOinitial public offering (“IPO”) in October 2020, sales of preferred stock, term loans, an upfront payment of $15.0 million we received in July 2020 under the Merck Collaboration Agreement, and proceeds we received in December 2021 under the Lilly SPA of $80.0 million, an upfront payment of which $42.2300.0 million was allocated to equity uponreceived in January 2022 under the issuanceLilly Collaboration Agreement and a payment of $5.0 million received from Merck under the Company’s common stockMerck Collaboration Agreement for the .achievement of a research milestone. As of December 31, 2021,2023, we had cash, cash equivalents and marketable securities of $154.3$234.1 million.
Cash Flows
The following table summarizes our sources and uses of cash for each of the periods presented:
| | | | Year Ended December 31, | | Year Ended December 31, |
| | | 2021 | | 2020 | | 2023 | | 2022 |
| | | (in thousands) | | (in thousands) |
| Net cash used in operating activities | $ | (50,250) | | | $ | (31,286) | |
| Net cash provided by (used in) operating activities | |
| Net cash provided by (used in) investing activities | Net cash provided by (used in) investing activities | 36,173 | | | (108,914) | |
| Net cash provided by financing activities | Net cash provided by financing activities | 22,418 | | | 217,473 | |
| Net increase in cash, cash equivalents and restricted cash | $ | 8,341 | | | $ | 77,273 | |
| Net increase (decrease) in cash, cash equivalents and restricted cash | |
Operating Activities
DuringFor the year ended December 31, 2021,2023, operating activities used $50.3$118.1 million of cash, resulting from our net loss of $101.3$98.4 million partially offset by net cash providedto fund our operations and by changes in our operating assets and liabilities of $33.4 million and by net non-cash charges of $17.6 million . Net cash provided by changes in our operating assets and liabilities for the year ended December 31, 2021 consisted primarily of $37.8 million of proceeds from the Lilly SPA partially offset by a $4.0 million decrease in operating lease liabilities and a $0.3 million net increase in working capital.
During the year ended December 31, 2020, operating activities used $31.3 million of cash, resulting from our net loss of $68.8$41.9 million partially offset by net non-cash charges of $8.8 million and net cash provided by changes in our operating assets and liabilities of $28.7$22.2 million. Net cash provided by changes in our operating assets and liabilities for the year ended December 31, 20202023 consisted primarily of a $15.1decrease of $34.2 million increase in operating lease liabilities resulting from our landlord incentives received, a $14.6 million increase in deferred revenue resulting from the recognition of revenue on the upfront paymentpayments received in connection with our collaboration agreements, a $7.5 million decrease in operating lease liabilities and a $0.2 million net decrease in working capital.
For the Merck Collaboration Agreementyear ended December 31, 2022, operating activities provided $193.6 million of cash, resulting from net cash provided by changes in our operating assets and liabilities of $281.5 million and by net non-cash charges of $20.9 million partially offset by a net loss of $108.9 million to fund our operations. Net cash provided by changes in our operating assets and liabilities for the year ended December 31, 2022 consisted primarily of a $302.7 million net increase in working capital. This was primarily driven by to the $300.0 million of cash received related to our collaboration receivable from Lilly and an increase of $3.7$4.3 million in accounts payable and accrued expenses and other current liabilities,deferred revenue related to the $5.0 million milestone payment from Merck, partially offset by an increasea $18.5 million decrease in deferred revenue resulting from the recognition of $4.7revenue on the upfront and milestone payments received in connection with our collaboration agreements and a $7.0 million decrease in prepaid expenses and other assets.operating lease liabilities.
Investing Activities
For the year ended December 31, 2023, net cash provided by investing activities was $144.5 million primarily due to $219.6 million of marketable securities maturing, offset by $73.9 million of purchases of marketable securities and $1.2 million in purchases of property and equipment.
DuringFor the year ended December 31, 2021, net cash provided by investing activities was $36.2 million primarily due to the maturity of marketable securities of $139.5 million partially offset by the purchases of marketable securities of $100.0 million and the acquisition of property and equipment of $3.3 million. Property and equipment purchases for the year ended December 31, 2021 were primarily related to leasehold improvements for our new facility in Cambridge, Massachusetts.
During the year ended December 31, 2020,2022, net cash used in investing activities was $108.9$244.3 million primarily due to the$409.3 million of purchases of marketable securities of $93.0and $1.2 million and the acquisitionin purchases of property and equipment partially offset by $166.2 million of $16.2 million. Property and equipment purchases formaturities of marketable securities.
Financing Activities
For the year ended December 31, 2020 were primarily related to leasehold improvements for our new facility in Cambridge, Massachusetts.
Financing Activities
During the year ended December 31, 2021,2023 and 2022, net cash provided by financing activities was $22.4$1.8 million, consisting primarily of proceeds from the Lilly SPA of $42.2 million andnet proceeds from the exercise of common stock options of $1.4 million partially offset byand the repayment of notes payable of $21.2 million.
During the year ended December 31, 2020, net cash provided by financing activities was $217.5 million, consisting primarily of proceeds from our IPO, net of underwriting discounts and commissions and other offering expenses of $122.1 million, net proceeds from the sale of our Series B preferredemployee stock of $89.9 million, net borrowings of $4.3 million and proceeds from the exercise of common stock options of $1.2 million.purchase plan.
Funding Requirements
We expect our expenses to increase substantially in connection with our ongoing activities, particularly as we advance the preclinical activities, initiate clinical trials for our product candidates in development, including those partnered with Lilly, and continue to fund on-going clinical trials.activities. As of the issuance date of the consolidated financial statements included elsewhere in this Annual Report on Form 10-K, we expect that our cash, cash equivalents and marketable securities will be sufficient to fund our operating expenses and capital expenditure requirements for at least twelve months. We have based these estimates as to how long we expect we will be able to fund our operationsthis estimate on assumptions that may prove to be inaccurate. We could use our available capital resources sooner than we currently expect, in which case we would be required to obtain additional financing sooner than planned, which may not be available to us on acceptable terms, or at all. Our failure to raise capital as and when needed would have a negative impact on our financial condition and our ability to pursue our long-term business strategy. We will be required to obtain further funding through public or private equity offerings, debt financings, collaborations and licensing arrangements or other sources.
If we are unable to raise sufficient capital as and when needed, we may be required to significantly curtail, delay or discontinue one or more of our research or development programs or the commercialization of any product candidate we may develop, or be unable to expand our operations or otherwise capitalize on our business opportunities. If we raise additional funds through collaborations or licensing arrangements with third parties, we may have to relinquish valuable rights to future revenue streams or product candidates or grant licenses on terms that may not be favorable to us.
Off-balance Sheet Arrangements
We did not have during the periods presented, and we do not currently have, any off-balance sheet arrangements, as defined in the rules and regulations of the Securities and Exchange Commission.
Recently Issued Accounting Pronouncements
A description of recently issued accounting pronouncements that may potentially impact our financial position and results of operations is disclosed in Note 2 to our consolidated financial statements appearing elsewhere in this Annual Report on Form 10-K.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
We are a smaller reporting company, as defined in Rule 12b-2 under the Securities Exchange Act of 1934, as amended, for this reporting period and are not required to provide the information required under this item.
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Stockholders and the Board of Directors of Foghorn Therapeutics Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Foghorn Therapeutics Inc. and its subsidiary (the "Company"), as of December 31, 2023 and 2022, the related consolidated statements of operations and comprehensive loss, stockholders' equity (deficit), and cash flows for the years then ended, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2023 and 2022, and the results of its operations and its cash flows for the years then ended, in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company's financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ Deloitte & Touche LLP
Boston, Massachusetts
March 7, 2024
We have served as the Company’s auditor since 2018.
ITEM 8. CONSOLIDATED FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
FOGHORN THERAPEUTICS INC.
Index to Consolidated Financial Statements
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Stockholders and the Board of Directors of Foghorn Therapeutics Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Foghorn Therapeutics Inc. and its subsidiary (the “Company”), as of December 31, 2021 and 2020, the related consolidated statements of operations and comprehensive loss, convertible preferred stock and stockholders’ equity, and cash flows for the years then ended, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2021 and 2020, and the results of its operations and its cash flows for the years then ended, in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ Deloitte & Touche LLP
Boston, Massachusetts
March 10, 2022
We have served as the Company’s auditor since 2018.
Foghorn Therapeutics Inc.
Consolidated Balance Sheets
(In thousands, except share and per share amounts)
| | December 31, |
| 2021 | | 2020 |
| December 31, | | | December 31, |
| 2023 | | | 2023 | | 2022 |
| Assets | Assets | | | |
| Current assets: | Current assets: | |
| Current assets: | |
| Current assets: | |
| Cash and cash equivalents | |
| Cash and cash equivalents | |
| Cash and cash equivalents | Cash and cash equivalents | $ | 101,136 | | | $ | 92,795 | |
| Marketable securities | Marketable securities | 53,153 | | | 92,975 | |
| Collaboration receivable | 300,000 | | | — | |
| | Prepaid expenses and other current assets | |
| Prepaid expenses and other current assets | |
| Prepaid expenses and other current assets | Prepaid expenses and other current assets | 5,273 | | | 4,917 | |
| Total current assets | Total current assets | 459,562 | | | 190,687 | |
| Property and equipment, net | Property and equipment, net | 17,563 | | | 19,528 | |
| Restricted cash | Restricted cash | 1,733 | | | 1,733 | |
| Other assets | Other assets | 2,400 | | | 842 | |
| Operating lease right-of-use assets | Operating lease right-of-use assets | 38,516 | | | 42,804 | |
| Total assets | Total assets | $ | 519,774 | | | $ | 255,594 | |
| Liabilities and Stockholders’ Equity | | | |
| Liabilities and Stockholders’ Equity (Deficit) | |
| Current liabilities: | Current liabilities: | |
| Current liabilities: | |
| Current liabilities: | |
| Accounts payable | |
| Accounts payable | |
| Accounts payable | Accounts payable | $ | 3,819 | | | $ | 3,680 | |
| Accrued expenses and other current liabilities | Accrued expenses and other current liabilities | 9,562 | | | 9,161 | |
| Operating lease liabilities | Operating lease liabilities | 6,994 | | | 3,981 | |
| Deferred revenue | Deferred revenue | 28,317 | | | 2,024 | |
| Total current liabilities | Total current liabilities | 48,692 | | | 18,846 | |
| Notes payable, net of discount | — | | | 19,654 | |
| Operating lease liabilities, net of current portion | Operating lease liabilities, net of current portion | 51,338 | | | 58,361 | |
| Deferred revenue, net of current portion | Deferred revenue, net of current portion | 322,730 | | | 12,546 | |
| Other liabilities | Other liabilities | 143 | | | — | |
| Total liabilities | Total liabilities | 422,903 | | | 109,407 | |
| Commitments and contingencies (Note 13) | 0 | | 0 |
| Stockholders’ equity: | |
| Preferred stock, $0.0001 par value; 25,000,000 shares authorized as of December 31, 2021 and 2020; no shares issued or outstanding | — | | | — | |
| Common stock, $0.0001 par value; 175,000,000 shares authorized at December 31, 2021 and 2020; 41,299,720 shares issued and outstanding at December 31, 2021 and 36,790,946 shares issued and outstanding at December 31, 2020 | 4 | | | 4 | |
| Commitments and contingencies (Note 11) | | Commitments and contingencies (Note 11) | | | |
| Stockholders’ equity (deficit): | |
| Preferred stock, $0.0001 par value; 25,000,000 shares authorized as of December 31, 2023 and 2022; no shares issued and outstanding as of December 31, 2023 and 2022 | |
| Preferred stock, $0.0001 par value; 25,000,000 shares authorized as of December 31, 2023 and 2022; no shares issued and outstanding as of December 31, 2023 and 2022 | |
| Preferred stock, $0.0001 par value; 25,000,000 shares authorized as of December 31, 2023 and 2022; no shares issued and outstanding as of December 31, 2023 and 2022 | |
| Common stock, $0.0001 par value; 175,000,000 shares authorized at December 31, 2023 and 2022; 42,282,040 shares issued and outstanding at December 31, 2023 and 41,803,436 shares issued and outstanding at December 31, 2022 | |
| Additional paid-in capital | Additional paid-in capital | 361,133 | | | 309,126 | |
| Accumulated other comprehensive loss | Accumulated other comprehensive loss | (10) | | | (7) | |
| Accumulated deficit | Accumulated deficit | (264,256) | | | (162,936) | |
| Total stockholders’ equity | 96,871 | | | 146,187 | |
| Total liabilities and stockholders’ equity | $ | 519,774 | | | $ | 255,594 | |
| Total stockholders’ equity (deficit) | |
| Total liabilities and stockholders’ equity (deficit) | |
The accompanying notes are an integral part of these consolidated financial statements.
Foghorn Therapeutics Inc.
Consolidated Statements of Operations and Comprehensive Loss
(In thousands, except share and per share amounts)
| | | | Year Ended December 31, | | Year Ended December 31, |
| | | 2021 | | 2020 | | 2023 | | 2022 |
| Collaboration revenue | Collaboration revenue | $ | 1,319 | | | $ | 430 | |
| Operating expenses: | Operating expenses: | | | |
| Research and development | Research and development | 80,325 | | | 57,715 | |
| Research and development | |
| Research and development | |
| General and administrative | General and administrative | 21,728 | | | 11,246 | |
| Total operating expenses | Total operating expenses | 102,053 | | | 68,961 | |
| Loss from operations | Loss from operations | (100,734) | | | (68,531) | |
| Other income (expense): | Other income (expense): | | | |
| Interest expense | (1,906) | | | (979) | |
| Interest income and other income (expense), net | 2,553 | | | 1,001 | |
| Change in fair value of preferred stock warrant liability | — | | | (69) | |
| Loss on debt extinguishment | (1,233) | | | (222) | |
| Total other expense, net | (586) | | | (269) | |
| Interest income | |
| Interest income | |
| Interest income | |
| Other income, net | |
| Total other income, net | |
| Loss before income taxes | |
| Provision for income taxes | |
| Net loss | Net loss | $ | (101,320) | | | $ | (68,800) | |
| Net loss per share attributable to common stockholders—basic and diluted | Net loss per share attributable to common stockholders—basic and diluted | $ | (2.73) | | | $ | (6.23) | |
| Weighted average common shares outstanding—basic and diluted | Weighted average common shares outstanding—basic and diluted | 37,171,147 | | | 11,046,802 | |
| Comprehensive loss: | Comprehensive loss: | | | |
| Net loss | Net loss | $ | (101,320) | | | $ | (68,800) | |
| Net loss | |
| Net loss | |
| Other comprehensive loss: | Other comprehensive loss: | |
| Unrealized losses on marketable securities | (3) | | | (7) | |
| Total other comprehensive loss | (3) | | | (7) | |
| Unrealized gains (losses) on marketable securities | |
| Unrealized gains (losses) on marketable securities | |
| Unrealized gains (losses) on marketable securities | |
| Total other comprehensive gain (loss) | |
| Total comprehensive loss | Total comprehensive loss | $ | (101,323) | | | $ | (68,807) | |
The accompanying notes are an integral part of these consolidated financial statements.
Foghorn Therapeutics Inc.
Consolidated Statements of Convertible Preferred Stock and Stockholders’ Equity (Deficit)
(In thousands, except share amounts)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Series A-1, A-2 and B
Convertible
Preferred Stock | | | Common Stock | | Additional
Paid-in
Capital | | Accumulated
Other
Comprehensive
Loss | | Accumulated
Deficit | | Total
Stockholders’
Equity
(Deficit) |
| | Shares | | Amount | | | Shares | | Amount | |
| Balances at December 31, 2019 | 28,615,546 | | | $ | 86,544 | | | | 4,870,851 | | | $ | — | | | $ | 6,120 | | | $ | — | | | $ | (94,136) | | | $ | (88,016) | |
| Issuance of Series B convertible preferred stock, net of issuance costs of $198 | 12,007,867 | | | 89,861 | | | | — | | | — | | | — | | | — | | | — | | | — | |
| Conversion of preferred stock to common stock | (40,623,413) | | | (176,405) | | | | 21,958,588 | | | 3 | | | 176,402 | | | — | | | — | | | 176,405 | |
| Issuance of common stock upon initial public offering net of underwriting discounts, commissions and offering costs | — | | | — | | | | 8,451,837 | | | 1 | | | 122,120 | | | — | | | — | | | 122,121 | |
| Conversion of warrant liability to equity upon closing of initial public offering | — | | | — | | | | — | | | — | | | 114 | | | — | | | — | | | 114 | |
| Issuance of common stock in connection with the cashless exercise of warrants | — | | | — | | | | 6,728 | | | — | | | — | | | — | | | — | | | — | |
| Issuance of common stock upon exercise of stock options | — | | | — | | | | 611,048 | | | — | | | 1,221 | | | — | | | — | | | 1,221 | |
| Issuance of warrants in connection with notes payable | — | | | — | | | | — | | | — | | | 188 | | | — | | | — | | | 188 | |
| Vesting of restricted stock | — | | | — | | | | 891,894 | | | — | | | — | | | — | | | — | | | — | |
| Stock-based compensation expense | — | | | — | | | | — | | | — | | | 2,961 | | | — | | | — | | | 2,961 | |
| Unrealized losses on marketable securities | — | | | — | | | | — | | | — | | | — | | | (7) | | | — | | | (7) | |
| Net loss | — | | | — | | | | — | | | — | | | — | | | — | | | (68,800) | | | (68,800) | |
| Balances at December 31, 2020 | — | | | — | | | | 36,790,946 | | | 4 | | | 309,126 | | | (7) | | | (162,936) | | | 146,187 | |
| Issuance of common stock upon exercise of stock options | — | | | — | | | | 508,774 | | | — | | | 1,418 | | | — | | | — | | | 1,418 | |
| Issuance of common stock related to stock purchase agreement | — | | | — | | | | 4,000,000 | | | — | | | 42,205 | | | — | | | — | | | 42,205 | |
| Stock-based compensation expense | — | | | — | | | | — | | | — | | | 8,384 | | | — | | | — | | | 8,384 | |
| Unrealized losses on marketable securities | — | | | — | | | | — | | | — | | | — | | | (3) | | | — | | | (3) | |
| Net loss | — | | | — | | | | — | | | — | | | — | | | — | | | (101,320) | | | (101,320) | |
| Balances at December 31, 2021 | — | | | $ | — | | | | 41,299,720 | | | $ | 4 | | | $ | 361,133 | | | $ | (10) | | | $ | (264,256) | | | $ | 96,871 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Common Stock | | Additional
Paid-in
Capital | | Accumulated
Other
Comprehensive
Loss | | Accumulated
Deficit | | Total
Stockholders’
Equity
(Deficit) |
| | Shares | | Amount | |
| Balances at December 31, 2021 | 41,299,720 | | | $ | 4 | | | $ | 361,133 | | | $ | (10) | | | $ | (264,256) | | | $ | 96,871 | |
Issuance of common stock upon exercise of stock
options and employee stock purchase plan | 503,716 | | | — | | | 1,763 | | | — | | | — | | | 1,763 | |
| Stock-based compensation expense | — | | | — | | | 14,336 | | | — | | | — | | | 14,336 | |
| Unrealized losses on marketable securities | — | | | — | | | — | | | (3,976) | | | — | | | (3,976) | |
| Net loss | — | | | — | | | — | | | — | | | (108,882) | | | (108,882) | |
| Balances at December 31, 2022 | 41,803,436 | | | 4 | | | 377,232 | | | (3,986) | | | (373,138) | | | 112 | |
Issuance of common stock upon exercise of stock
options and employee stock purchase plan | 478,604 | | | $ | — | | | 1,778 | | | — | | | — | | | 1,778 | |
| Stock-based compensation expense | — | | | — | | | 16,186 | | | — | | | — | | | 16,186 | |
| Unrealized gains on marketable securities | — | | | — | | | — | | | 3,160 | | | — | | | 3,160 | |
| Net loss | — | | | — | | | — | | | — | | | (98,426) | | | (98,426) | |
| Balances at December 31, 2023 | 42,282,040 | | | $ | 4 | | | $ | 395,196 | | | $ | (826) | | | $ | (471,564) | | | $ | (77,190) | |
The accompanying notes are an integral part of these consolidated financial statements.
Foghorn Therapeutics Inc.
Consolidated Statements of Cash Flows
(In thousands)
| | | | Year ended December 31, | | Year ended December 31, |
| | | 2021 | | 2020 | | 2023 | | 2022 |
| Cash flows from operating activities: | Cash flows from operating activities: | | | |
| Net loss | Net loss | $ | (101,320) | | | $ | (68,800) | |
| Adjustments to reconcile net loss to net cash used in operating activities: | |
| Stock-based compensation expense related to issuance of options and employee stock purchase plan | 8,384 | | | 2,961 | |
| Net loss | |
| Net loss | |
| Adjustments to reconcile net loss to net cash provided by (used in) operating activities: | |
| Stock-based compensation expense | |
| Stock-based compensation expense | |
| Stock-based compensation expense | |
| Depreciation and amortization expense | Depreciation and amortization expense | 3,227 | | | 1,327 | |
| Gain on disposal of property and equipment | — | | | (206) | |
| Loss on debt extinguishment | 1,233 | | | 222 | |
| Change in fair value of preferred stock warrant liability | — | | | 69 | |
| Loss on disposal of property and equipment | |
| Noncash lease expense | Noncash lease expense | 4,288 | | | 4,201 | |
| Noncash interest expense | 318 | | | 238 | |
| Amortization (accretion) of premium/discount on marketable securities | 179 | | | (1) | |
| Accretion of discount on marketable securities | |
| Changes in operating assets and liabilities: | Changes in operating assets and liabilities: | |
| Prepaid expenses and other current assets | |
| Prepaid expenses and other current assets | |
| Prepaid expenses and other current assets | Prepaid expenses and other current assets | (1,814) | | | (4,670) | |
| Collaboration receivable | Collaboration receivable | (300,000) | | | — | |
| Accounts payable | Accounts payable | 135 | | | 474 | |
| Accrued expenses and other liabilities | Accrued expenses and other liabilities | 2,653 | | | 3,194 | |
| Operating lease liabilities | Operating lease liabilities | (4,010) | | | 15,135 | |
| Deferred revenue | Deferred revenue | 336,477 | | | 14,570 | |
| Net cash used in operating activities | (50,250) | | | (31,286) | |
| Net cash provided by (used in) operating activities | |
| Cash flows from investing activities: | Cash flows from investing activities: | | | |
| Purchases of property and equipment | Purchases of property and equipment | (3,313) | | | (16,183) | |
| Proceeds from sale of property and equipment | — | | | 250 | |
| Purchases of property and equipment | |
| Purchases of property and equipment | |
| Purchases of marketable securities | Purchases of marketable securities | (99,984) | | | (92,981) | |
| Proceeds from maturities of marketable securities | Proceeds from maturities of marketable securities | 139,470 | | | — | |
| Net cash provided by (used in) investing activities | Net cash provided by (used in) investing activities | 36,173 | | | (108,914) | |
| Cash flows from financing activities: | Cash flows from financing activities: | | | |
| Proceeds from stock purchase agreement | 42,205 | | | — | |
| Proceeds from initial public offering, net of underwriting discounts, commissions and offering costs | — | | | 122,121 | |
| Proceeds from issuance of convertible preferred stock, net of issuance costs | — | | | 89,861 | |
| Proceeds from issuance of common stock upon exercise of stock options | 1,418 | | | 1,221 | |
| Proceeds from issuance of notes payable, net of issuance costs | — | | | 19,800 | |
| Repayments of notes payable | (21,205) | | | (15,530) | |
| Proceeds from issuance of common stock upon exercise of stock options and employee stock purchase plan | |
| Proceeds from issuance of common stock upon exercise of stock options and employee stock purchase plan | |
| Proceeds from issuance of common stock upon exercise of stock options and employee stock purchase plan | |
| Net cash provided by financing activities | Net cash provided by financing activities | 22,418 | | | 217,473 | |
| Net increase in cash, cash equivalents and restricted cash | 8,341 | | | 77,273 | |
| Net increase (decrease) in cash, cash equivalents and restricted cash | |
| Cash, cash equivalents and restricted cash at beginning of period | Cash, cash equivalents and restricted cash at beginning of period | 94,528 | | | 17,255 | |
| Cash, cash equivalents and restricted cash at end of period | Cash, cash equivalents and restricted cash at end of period | $ | 102,869 | | | $ | 94,528 | |
| Supplemental cash flow information: | Supplemental cash flow information: | | | |
| Cash paid for interest | $ | 1,716 | | | $ | 660 | |
| Cash paid for taxes | |
| Cash paid for taxes | |
| Cash paid for taxes | |
| Supplemental disclosure of noncash investing and financing information: | Supplemental disclosure of noncash investing and financing information: | |
| Purchases of property and equipment included in accounts payable, accrued expenses and other liabilities | $ | 295 | | | $ | 2,400 | |
| Conversion of redeemable convertible preferred stock to common stock | $ | — | | | $ | 176,405 | |
| Conversion of warrant liability to equity upon closing of initial public offering | $ | — | | | $ | 114 | |
| Issuance of warrants in connection with notes payable | $ | — | | | $ | 188 | |
| Purchases of property and equipment included in accounts payable and accrued expenses | |
| Purchases of property and equipment included in accounts payable and accrued expenses | |
| Purchases of property and equipment included in accounts payable and accrued expenses | |
| Reconciliation of cash, cash equivalents and restricted cash: | Reconciliation of cash, cash equivalents and restricted cash: | |
| Cash and cash equivalents | Cash and cash equivalents | $ | 101,136 | | | $ | 92,795 | |
| Restricted cash (current and non-current) | 1,733 | | | 1,733 | |
| Cash and cash equivalents | |
| Cash and cash equivalents | |
| Restricted cash (non-current) | |
| Total cash, cash equivalents and restricted cash shown in the statement of cash flows | Total cash, cash equivalents and restricted cash shown in the statement of cash flows | $ | 102,869 | | | $ | 94,528 | |
The accompanying notes are an integral part of these consolidated financial statements.
Foghorn Therapeutics Inc.
Notes to Consolidated Financial Statements
1. Nature of Business, Going Concern and Basis of Presentation
Foghorn Therapeutics Inc. (the “Company”) is a clinical-stage biopharmaceutical company discovering and developing a new class of medicines targeting genetically determined dependencies within the chromatin regulatory system. The Company uses its proprietary Gene Traffic Control platform to identify, validate and potentially drug targets within the system. The Company was founded in October 2015 as a Delaware corporation. The Company is headquartered in Cambridge, Massachusetts.
The Company is subject to risks similar to those of other early-stage companies in the biopharmaceutical industry, including dependence on key individuals, the need to develop commercially viable products, competition from other companies, many of whom are larger and better capitalized, the impact of the COVID-19 pandemic and the need to obtain adequate additional financing to fund the development of its products. There can be no assurance that the Company’s research and development will be successfully completed, that adequate protection for the Company’s intellectual property will be maintained, that any products developed will obtain required regulatory approval or that any approved products will be commercially viable. Even if the Company’s development efforts are successful, it is uncertain when, if ever, the Company will generate significant revenue from the sale of its products.
In March 2020, the World Health Organization declared the global novel coronavirus disease 2019 (“COVID-19”) outbreak a pandemic. The Company’s operations have not been significantly impacted by the COVID-19 pandemic. However, the Company cannot at this time predict the specific extent, duration, or full impact that the COVID-19 pandemic will have on its financial condition and operations, including ongoing and planned clinical trials. The impact of the COVID-19 outbreak on the Company’s financial performance will depend on future developments, including the duration and spread of the pandemic and related governmental advisories and restrictions. These developments and the impact of COVID-19 on the financial markets and the overall economy are highly uncertain and cannot be predicted. If the financial markets and/or the overall economy are impacted for an extended period, the Company’s results may be materially adversely affected.
On October 27, 2020, the Company completed its initial public offering (“IPO”) pursuant to which it issued and sold 7,500,000 shares of its common stock at a public offering price of $16.00 per share, resulting in net proceeds of $107.9 million, after deducting underwriting discounts and commissions and other offering expenses. On November 19, 2020, the Company issued and sold an additional 951,837 shares of common stock at the IPO price of $16.00 per share pursuant to the underwriters’ partial exercise of their option to purchase additional shares of common stock, resulting in additional net proceeds of $14.2 million after deducting underwriting discounts and commissions.Going concern
On December 10, 2021, the Company executed a collaboration agreement with Eli Lilly and Company (or "Lilly") (See Note 9). Concurrent with the collaboration agreement the Company completed a stock purchase agreement (the "SPA") with Lilly and issued and sold Lilly 4,000,000 shares of its common stock at a price of $20.00 per share, resulting in net proceeds of $80.0 million of which $42.2 million was allocated to equity upon the issuance of the Company’s common stock (see Note 9).
The accompanying consolidated financial statements have been prepared on the basis of continuity of operations, realization of assets and the satisfaction of liabilities and commitments in the ordinary course of business. Since inception, the Company has funded its operations primarily with proceeds from sales of preferred stock, debt financing, an upfront payment of $15.0 million the Company received in July 2020 underfrom a collaboration agreement, with Merck Sharp & Dohme Corp. (see Note 9), proceeds from the sale of commona public offering and a stock in the IPO completed in October 2020 and most recently, with sale of common stock in the SPA.purchase agreement all prior to 2022. In January 2022, the Company received an upfront payment of $300.0 million pursuant to thea collaboration agreement (the “Lilly Collaboration Agreement”) completed with Loxo Oncology at Eli Lilly (Seeand Company (“Lilly”) (see Note 9), which was included in current assets on8). In the consolidated balance sheets asthird quarter of December 31, 2021.2022, the Company achieved a research milestone related to a collaboration agreement (the “Merck Collaboration Agreement”) with Merck Sharp & Dohme Corp. (“Merck”) and received a $5.0 million milestone payment. The Company has incurred recurring losses, including net losses of $101.3$98.4 million and $68.8$108.9 million for the years ended December 31, 20212023 and 2020,2022, respectively. As of December 31, 2021,2023, the Company had an accumulated deficit of $264.3$471.6 million. The Company expects to continue to generate operating losses in the foreseeable future. As of the issuance date of these consolidated financial statements the Company expects that its cash, cash equivalents and marketable securities will be sufficient to fund its operating expenses and capital expenditure requirements for at least 12 months.
The Company will need to obtain additional funding through public or private equity offerings, debt financings, collaborations, strategic alliances and/or licensing arrangements to continue to fund its operations. The Company may not be able to obtain financing on acceptable terms, or at all, and the Company may not be able to enter into collaborative or strategic alliances or licensing arrangements. The terms of any financing may adversely affect the holdings or the rights of the Company’s stockholders. Arrangements with collaborators or others may require the Company to relinquish rights to certain of its
technologies or programs. If the Company is unable to obtain funding, the Company could be forced to delay, reduce or eliminate some or all of its research and development programs, pipeline expansion or commercialization efforts, which could adversely affect its business prospects. Although management will continue to pursue these plans, there is no assurance that the Company will be successful in obtaining sufficient funding in the future on terms acceptable to the Company to fund continuing operations when needed or at all.
Basis of presentation
The Company’s consolidated financial statements have been prepared in conformity with accounting principles generally accepted in the United States of America (“GAAP”). The accompanying consolidated financial statements include the accounts of the Company and its wholly owned subsidiary. All intercompany accounts and transactions have been eliminated in consolidation. Any reference in these notes to applicable guidance is meant to refer to the authoritative GAAP as found in the Accounting Standards Codification (“ASC”) and Accounting Standards Update (“ASU”) of the Financial Accounting Standards Board (“FASB”).
2. Summary of Significant Accounting Policies
Use of estimates
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting periods. Significant estimates and
assumptions reflected in these consolidated financial statements include, but are not limited to, revenue recognition and the accrual of research and development expenses. The Company bases its estimates on historical experience, known trends and other market-specific or other relevant factors that it believes to be reasonable under the circumstances. On an ongoing basis, management evaluates its estimates as there are changes in circumstances, facts and experience. Actual results may differ from those estimates or assumptions.
Concentrations of credit risk and of significant suppliers
Financial instruments that potentially expose the Company to concentrations of credit risk consist primarily of cash, cash equivalents and marketable securities. As of December 31, 2021, the Company maintained cash, cash equivalents and marketable securities at financial institutions in excess of federally insured limits. The Company does not believe that it is subject to unusual credit risk beyond the normal credit risk associated with commercial banking relationships.
The Company relies, and expects to continue to rely, on a small number of vendors to provide services, supplies and materials for certain activities related to its programs. These programs could be adversely affected by a significant interruption in these services or the availability of materials.
Cash equivalents
The Company considers all highly liquid investments with original maturities of three months or less at the date of purchase to be cash equivalents.
Marketable Securities
The Company’s marketable securities are classified as available-for-sale and are carried at fair value with the unrealized gains and losses reported as a component of accumulated other comprehensive loss in stockholders’ equity. Realized gains and losses and declines in value judged to be other than temporary are included as a component of interest income and other income (expense), net based on the specific identification method. The Company classifies its marketable securities with maturities beyond one year as short-term, based on their highly liquid nature and because such marketable securities are available for current operations.
The Company reviews marketable securities whenever the fair value of an investment is less than the amortized cost and evidence indicates that an investment’s carrying value is not recoverable. For available-for-sale debt securities, the credit allowance is limited to the amount that fair value is less than amortized cost. Unrealized gains (losses) are evaluated for impairment under ASC 326, Financial Instruments - Credit Losses, to determine if the impairment is credit-related or noncredit-related. Credit-related impairment is recognized as an allowance on the consolidated balance sheets with a corresponding adjustment to earnings, and noncredit-related impairment is recognized in other comprehensive loss. Evidence considered in this assessment includes reasons for the impairment, compliance with our investment policy, the severity of the impairment, collectability of the security, and any adverse conditions specifically related to the security, an industry, or geographic area.
Unrealized gains and losses are included as a component of accumulated other comprehensive loss in the consolidated balance sheets and consolidated statements of stockholders’ equity (deficit) and a component of total comprehensive loss. Realized gains and losses are included as a component of other income (expense), net based on the specific identification method.
Restricted cash
Amounts included in restricted cash represent amounts pledged as collateral for letters of credit required for security deposits on the Company’s leased facilities and credit cards.facilities. These amounts are classified as restricted cash in the Company’s consolidated balance sheets.
Property and equipment
Property and equipment are stated at cost less accumulated depreciation and amortization. Depreciation and amortization expense is recognized using the straight-line method over the estimated useful life of each asset as follows:
| | | | | |
| Estimated Useful Life |
| Laboratory equipment | 5 years |
| Furniture and fixtures | 5 years |
| Computer equipment and software | 3 years |
| Leasehold improvements | Shorter of useful life or remaining term of lease |
Costs for capital assets not yet placed into service are depreciated once placed into service. Upon retirement or sale, the cost of assets disposed of and the related accumulated depreciation and amortization are removed from the accounts and any resulting gain or loss is included in loss from operations. Expenditures for repairs and maintenance are charged to expense as incurred.
Impairment of long-lived assets
The Company evaluates its long-lived assets, which consist primarily of property and equipment and operating lease right-of-use assets, for impairment whenever events or changes in circumstances indicate that the carrying amount of such assets may not be recoverable. Recoverability of assets to be held and used is measured by a comparison of the carrying amount of an asset to the future undiscounted net cash flows expected to be generated by the asset. If such assets are considered to be impaired, the impairment to be recognized is measured by the amount by which the carrying amount of the asset exceeds the fair value of the asset. The Company did not record anyrecorded no impairment losses on long-lived assets duringfor the years ended December 31, 20212023 or 2020.2022.
Fair value measurements
Certain assets and liabilities are carried at fair value under GAAP. Fair value is defined as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. Valuation techniques used to measure fair value must maximize the use of observable inputs and minimize the use of unobservable inputs. Financial assets and liabilities carried at fair value are to be classified and disclosed in one of the following three levels of the fair value hierarchy, of which the first two are considered observable and the last is considered unobservable:
•Level 1—Quoted prices in active markets for identical assets or liabilities.
•Level 2—Observable inputs (other than Level 1 quoted prices), such as quoted prices in active markets for similar assets or liabilities, quoted prices in markets that are not active for identical or similar assets or liabilities, or other inputs that are observable or can be corroborated by observable market data.
•Level 3—Unobservable inputs that are supported by little or no market activity and that are significant to determining the fair value of the assets or liabilities, including pricing models, discounted cash flow methodologies and similar techniques.
The Company’s cash equivalents and marketable securities are carried at fair value, determined according to the fair value hierarchy described above (see Note 3). The carrying values of the Company’s accounts payable and accrued expenses approximate their fair values due to the short-term nature of these liabilities. The carrying value of the Company’s long-term debt approximates its fair value (a level 2 measurement) due to its variable interest rate.
Collaboration Agreements
The Company analyzes its collaboration arrangements to assess whether they are within the scope of ASC 808, Collaborative Arrangements (ASC 808)(“ASC 808”) to determine whether such arrangements involve joint operating activities performed by parties that are both active participants in the activities and exposed to significant risks and rewards dependent on the commercial success of such activities. This assessment is performed throughout the life of the arrangement based on changes in the responsibilities of all parties in the arrangement. When the Company has concluded that it has a customer relationship with one of its collaborators, the Company follows the guidance in ASC Topic 606, Revenue From Contracts With Customers (“ASC 606”).
Revenue recognition
The Company accounts for its 2 collaboration arrangements under ASC 606, as the Company concluded that it has a customer relationship with each of its collaborators. For additional information on the Company’s collaboration agreements, see Note 9,8, Collaboration Agreements, to these consolidated financial statements. Under ASC 606, an entity recognizes revenue when its customer obtains control of promised goods or services in an amount that reflects the consideration that the entity expects to receive in exchange for those goods or services. To determine revenue recognition for arrangements within the scope of ASC 606, the entity performs the following five steps: (i) identify the contract(s) with a customer; (ii) identify the performance obligations in the contract; (iii) determine the transaction price, including variable consideration, if any; (iv) allocate the transaction price to the performance obligations in the contract; and (v) recognize revenue when (or as) the entity satisfies a performance obligation. The Company only applies the five-step model to contracts when it is probable that the entity will collect the consideration to which it is entitled in exchange for the goods or services it transfers to the customer.
The Company assesses the goods or services promised within each contract and determines those that are performance obligations. The promised goods or services in the Company’s arrangements would likely consist of licenses, rights to the Company’s intellectual property, research and development services and related supporting activities. Arrangements that include rights to additional goods or services that are exercisable at a customer’s discretion are generally considered options. The Company assesses if these options provide a material right to the customer and if so, they are considered performance obligations.
The Company assesses whether each promised good or service is distinct for the purpose of identifying the performance obligations in the contract. This assessment involves subjective determinations and requires management to make judgments about the individual promised goods or services and whether such are separable from the other aspects of the contractual
relationship. Promised goods and services are considered distinct provided that: (i) the customer can benefit from the good or service either on its own or together with other resources that are readily available to the customer (that is, the good or service is capable of being distinct) and (ii) the entity’s promise to transfer the good or service to the customer is separately identifiable from other promises in the contract (that is, the promise to transfer the good or service is distinct within the context of the contract). In assessing whether a promised good or service is distinct, the Company considers factors such as the research, development, manufacturing and commercialization capabilities of the customer and the availability of the associated expertise in the general marketplace. The Company also considers the intended benefit of the contract in assessing whether a promised good or service is separately identifiable from other promises in the contract. If a promised good or service is not distinct, an entity is required to combine that good or service with other promised goods or services until it identifies a bundle of goods or services that is distinct.
The transaction price is then determined and allocated to the identified performance obligations in proportion to their standalone selling prices (“SSP”) on a relative SSP basis. SSP is determined at contract inception and is not updated to reflect changes between contract inception and when the performance obligations are satisfied. Determining the SSP for performance obligations requires significant judgment. In developing the SSP for a performance obligation, the Company considers applicable market conditions and relevant entity-specific factors, including factors that were contemplated in negotiating the agreement with the customer and estimated costs. The Company validates the SSP for performance obligations by evaluating whether changes in the key assumptions used to determine the SSP will have a significant effect on the allocation of arrangement consideration between multiple performance obligations.
If the consideration promised in a contract includes a variable amount, the Company estimates the amount of consideration to which it will be entitled in exchange for transferring the promised goods or services to a customer. The Company determines the amount of variable consideration by using the expected value method or the most likely amount method. The Company includes the unconstrained amount of estimated variable consideration in the transaction price. The amount included in the transaction price is constrained to the amount for which it is probable that a significant reversal of cumulative revenue recognized will not occur. At the end of each subsequent reporting period, the Company re-evaluates the estimated variable consideration included in the transaction price and any related constraint, and if necessary, adjusts its estimate of the overall transaction price. Any such adjustments are recorded on a cumulative catch-up basis in the period of adjustment.
If an arrangement includes development and regulatory milestone payments, the Company evaluates whether the milestones are considered probable of being reached and estimates the amount to be included in the transaction price using the most likely amount method. If it is probable that a significant revenue reversal would not occur, the associated milestone value is included in the transaction price. Milestone payments that are not within the Company’s control such as regulatory approvals, are generally not considered probable of being achieved until those approvals are received.
For arrangements with licenses of intellectual property that include sales-based royalties, including milestone payments based on the level of sales, and the license is deemed to be the predominant item to which the royalties relate, the Company recognizes royalty revenue and sales-based milestones at the later of (i) when the related sales occur, or (ii) when the performance obligation to which the royalty has been allocated has been satisfied.
The Company records amounts as accounts receivable when the right to consideration is deemed unconditional. When consideration is received, or such consideration is unconditionally due, from a customer prior to transferring goods or services to the customer under the terms of a contract, a contract liability is recorded for deferred revenue.
In determining the transaction price, the Company adjusts consideration for the effects of the time value of money if the timing of payments provides the Company with a significant benefit of financing. The Company does not assess whether a contract has a significant financing component if the expectation at contract inception is such that the period between the transfer of the promised goods or services to the customer and the payment by the customer will be one year or less. The Company then recognizes as revenue the amount of the transaction price that is allocated to the respective performance obligation when (or as) each performance obligation is satisfied, either at a point in time or over time, and if over time, recognition is based on the use of an output or input method.
Research and development costs
Research and development expenses consist of costs incurred in performing research and development activities, including salaries and bonuses, stock-based compensation, employee benefits, facilities costs, laboratory supplies, depreciation, and external costs of vendors engaged to conduct research, preclinical and clinical development activities as well as the cost of licensing technology.
Upfront payments and milestone payments made for the licensing of technology are expensed as research and development over the period to which they relate. Costs for research and development activities are expensed in the period in which they are incurred. Payments for such activities are based on the terms of the individual arrangements, which may differ from the pattern of costs incurred, and are reflected in the consolidated financial statements as prepaid expense or accrued research and
development expense. Determining the prepaid and accrued balances at the end of any reporting period incorporate certain judgments and estimates by management that are based on information available to the Company including information provided by vendors regarding the progress to completion of specific tasks or costs incurred.
Patent costs
All patent-related costs incurred in connection with filing and prosecuting patent applications are expensed as incurred due to the uncertainty about the recovery of the expenditure. Amounts incurred are classified as general and administrative expenses.
Leases
In accordance with ASC 842, Leases, theThe Company accounts for a contract as a lease when it has the right to control the asset for a period of time while obtaining substantially all of the asset’s economic benefits. The Company determines if an arrangement is a lease or contains an embedded lease at inception. For arrangements that meet the definition of a lease, the Company determines the initial classification and measurement of its right-of-use asset and lease liability at the lease commencement date and thereafter if modified. The lease term includes any renewal options that the Company is reasonably assured to exercise. The present value of lease payments is determined by using the interest rate implicit in the lease, if that rate is readily determinable; otherwise, the Company uses its estimated secured incremental borrowing rate for that lease term. The Company’s policy is to not record leases with an original term of twelve months or less on its consolidated balance sheets and recognizes those lease payments in the income statement on a straight-line basisconsolidated statements of operations and comprehensive loss as incurred over the lease term. The Company’s existing leases are for office and laboratory space and an equipment lease.
In addition to rent, the leases may require the Company to pay additional costs, such as utilities, maintenance and other operating costs, which are generally referred to as non-lease components. The Company has elected to not separate lease and non-lease components. Only the fixed costs for lease components and their associated non-lease components are accounted for as a single lease component and recognized as part of a right-of-use asset and liability. Rent expense for operating leases is recognized on a straight-line basis over the reasonably assured lease term based on the total lease payments and is included in operating expense in the consolidated statements of operations and comprehensive loss.
the Company’s primary office lease (see Note 10). The Company recognizes sublease income on a straight-line basis over the lease term in other income (expense), net on the consolidated statements of operations and comprehensive loss, net of any revenue share due to the Company’s lessor. Stock-based compensation
The Company measures stock options with service-based vesting or performance-based vesting granted to employees, non-employees and directors based on the fair value on the date of grant using the Black-Scholes option-pricing model. The Company measures restricted common stock awards using the difference between the purchase price per share of the award, if any, and the fair value of the Company’s common stock at the date of grant. Compensation expense for the awards is recognized over the requisite service period for employees and directors and as services are delivered for non-employees, both of which are generally the vesting period of the respective award. The Company uses the straight-line method to record the expense of awards with only service-based vesting conditions. The Company uses the graded-vesting method to record the expense of awards with both service-based and performance-based vesting conditions, commencing once achievement of the performance condition becomes probable. The Company accounts for forfeitures of share-based awards as they occur.
The Company classifies stock-based compensation expense in its consolidated statements of operations and comprehensive loss in the same manner in which the award recipient’s payroll costs are classified or in which the award recipient’s service payments are classified.
Net loss per share
Basic net income (loss) per share attributable to common stockholders is computed by dividing net income (loss) attributable to common stockholders by the weighted average number of common shares outstanding for the period. Diluted net income (loss) per share attributable to common stockholders is computed by dividing net income(loss)income (loss) attributable to common stockholders by the weighted average number of common shares outstanding for the period, adjusted for potential dilutive common shares.
The Company’s participating securities contractually entitled the holders of such shares to participate in dividends but did not contractually require the holders of such shares to participate in losses of the Company. Accordingly, in periods in which the Company reported a net loss, such losses were not allocated to such participating securities. In periods in which the Company reported a net loss attributable to common stockholders, diluted net loss per share attributable to common stockholders is the same as basic net loss per share attributable to common stockholders, since dilutive common shares are not assumed to have been issued if their effect is anti-dilutive.
The Company reported a net loss attributable to common stockholders for the years ended December 31, 20212023 and 2020.2022.
The following common stock equivalents presented based on amounts outstanding at each period end, were excluded from the calculation of diluted net loss per share because including them would have had an anti-dilutive impact:
| | December 31, |
| 2021 | | 2020 |
| December 31, | | | December 31, |
| 2023 | | | 2023 | | 2022 |
| Stock options to purchase common stock | Stock options to purchase common stock | 5,920,509 | | | 5,016,460 | |
| Warrants to purchase common stock | Warrants to purchase common stock | 18,445 | | | 18,445 | |
| 5,938,954 | | | 5,034,905 | |
| 8,970,033 | |
Segments
Operating segments are defined as components of an entity for which separate discrete financial information is made available and that is regularly evaluated by the chief operating decision maker (“CODM”) in making decisions regarding resource allocation and assessing performance. The Company’s CODM is its chief executive officer and the Company manages its operations as a single segment for the purposes of assessing performance and making operating decisions. The Company is focused on pioneering the discovery and development of a new class of medicines targeting genetically determined dependencies within the chromatin regulatory system.
Comprehensive loss
Comprehensive loss includes net loss as well as other changes in stockholders’ equity that result from transactions and economic events other than those with stockholders. For the yearyears ended December 31, 20212023 and 2020,2022, the Company’s only element of other comprehensive loss was unrealized lossesgains (losses) on available for sale debt securities.
Income taxes
The Company accounts for income taxes using the asset and liability method, which requires the recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have been recognized in the consolidated financial statements or in the Company’s tax returns. Deferred tax assets and liabilities are determined on the basis of the differences between the financial statements and tax basis of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to reverse. Changes in deferred tax assets and liabilities are recorded in the provision for income taxes. The Company assesses the likelihood that its deferred tax assets will be recovered from future taxable income and, to the extent it believes, based upon the weight of available evidence, that it is more likely than not that all or a portion of the deferred tax assets will not be realized, a valuation allowance is established through a charge to the provision for income taxes. Potential for recovery of deferred tax assets is evaluated by estimating the future taxable profits expected and considering prudent and feasible tax planning strategies.
The Company accounts for uncertainty in income taxes recognized in the consolidated financial statements by applying a two-step process to determine the amount of tax benefit to be recognized. First, the tax position must be evaluated to determine the likelihood that it will be sustained upon external examination by the taxing authorities. If the tax position is deemed more-likely-than-not to be sustained, the tax position is then assessed to determine the amount of benefit to recognize in the consolidated financial statements. The amount of the benefit that may be recognized is the largest amount that has a greater than 50% likelihood of being realized upon ultimate settlement. Any resulting unrecognized tax benefits are recorded within the provision for income taxes.
Recently issued accounting pronouncements
From time to time, new accounting pronouncements are issued by the FASB or other standard setting bodies that the Company adopts as of the specified effective date. The Company qualifies as an “emerging growth company” as defined in the Jumpstart Our Business Startups Act of 2012 and has elected not to “opt out” of the extended transition related to complying with new or revised accounting standards, which means that when a standard is issued or revised and it has different application dates for public and nonpublic companies, the Company can adopt the new or revised standard at the time nonpublic companies adopt the new or revised standard and can do so until such time that the Company either (i) irrevocably elects to “opt out” of such extended transition period or (ii) no longer qualifies as an emerging growth company.
In June 2016, the FASB issued ASU No. 2016-13, Financial Instruments – Credit Losses (Topic 326). The new standard adjusts the accounting for assets held at amortized costs basis including marketable securities accounted for as available for sale. The standard eliminatesby eliminating the probable initial recognition threshold and requires an entity to reflect its current estimate of all expected credit losses. Thelosses as an allowance for credit losses is a valuation account that is deducted from the amortized cost basis of the financial assets to present the net amount expected to be collected. For public entities, the guidance was effective for annual reporting periods beginning after December 15, 2019 and for interim periods within those fiscal years. For nonpublic entities and emerging growth companies that choose to take advantage of the extended transition period, the guidance is effective for annual reporting periods beginning after December 15, 2020. Early adoption is permitted for all entities. In November 2019, the FASB issued ASU No. 2019-10, which deferred the effective date for nonpublic entities to annual reporting periods beginning after December 15, 2022, including interim periods within those fiscal years.losses. The Company is evaluating when to adoptadopted this standardas of January 1, 2023 and the effectadoption had no material impact to the adoption will have on itsCompany’s consolidated financial statements.position, results of operation, or cash flows.
3. Marketable Securities and Fair Value Measurements
As of December 31, 2021,2023, available for sale marketable securities by security type consisted of (in thousands):
| | Amortized
Cost | | | Amortized
Cost | | Gross
Unrealized
Gains | | Gross
Unrealized
Losses | | Estimated
Fair
Value |
| | Amortized
Cost | | Gross
Unrealized
Gains | | Gross
Unrealized
Losses | | Estimated
Fair
Value |
| U.S. government agencies (due within one year) | |
| U.S. government agencies (due within one year) | |
| U.S. government agencies (due within one year) | |
| Commercial paper (due within one year) | Commercial paper (due within one year) | $ | 36,246 | | | $ | — | | | $ | (3) | | | $ | 36,243 | |
| Corporate notes and bonds (due within one year) | Corporate notes and bonds (due within one year) | 16,917 | | | — | | | (7) | | | 16,910 | |
| | Corporate notes and bonds (due after one year through two years) | |
| Corporate notes and bonds (due after one year through two years) | |
| Corporate notes and bonds (due after one year through two years) | |
| Total | Total | $ | 53,163 | | | $ | — | | | $ | (10) | | | $ | 53,153 | |
As of December 31, 2020,2022, available for sale marketable securities by security type consisted of (in thousands):
| | Amortized
Cost | | Gross
Unrealized
Gains | | Gross
Unrealized
Losses | | Estimated
Fair
Value |
| Amortized
Cost | | | Amortized
Cost | | Gross
Unrealized
Gains | | Gross
Unrealized
Losses | | Estimated
Fair
Value |
| U.S. treasury notes (due within one year) | U.S. treasury notes (due within one year) | $ | 92,982 | | | $ | — | | | $ | (7) | | | $ | 92,975 | |
| Commercial paper (due within one year) | |
| Corporate notes and bonds (due within one year) | |
| Corporate notes and bonds (due after one year through three years) | |
| Total | Total | $ | 92,982 | | | $ | — | | | $ | (7) | | | $ | 92,975 | |
The following tables present the Company’s fair value hierarchy for its assets and liabilities, which wereare measured at fair value on a recurring basis (in thousands):
| | Fair Value Measurements at
December 31, 2021 Using: |
| Level 1 | | Level 2 | | Level 3 | | Total |
| Fair Value Measurements at
December 31, 2023 Using: | | | Fair Value Measurements at
December 31, 2023 Using: |
| Level 1 | | | Level 1 | | Level 2 | | Level 3 | | Total |
| Assets: | Assets: | | | | | | | |
| Cash equivalents: | Cash equivalents: | |
| Cash equivalents: | |
| Cash equivalents: | |
| Money market funds | Money market funds | $ | 82,252 | | | $ | — | | | $ | — | | | $ | 82,252 | |
| Money market funds | |
| Money market funds | |
| U.S. treasury notes | |
| | Commercial paper | — | | | 18,496 | | | — | | | 18,496 | |
| Marketable securities: | Marketable securities: | |
| Marketable securities: | |
| Marketable securities: | |
| | U.S. government agencies | |
| | U.S. government agencies | |
| | U.S. government agencies | |
| Commercial paper | Commercial paper | — | | | 36,243 | | | — | | | 36,243 | |
| Corporate notes and bonds | Corporate notes and bonds | — | | | 16,910 | | | — | | | 16,910 | |
| Total | Total | $ | 82,252 | | | $ | 71,649 | | | $ | — | | | $ | 153,901 | |
| | | | | | | | | | | | | | | | | | | | | | | |
| Fair Value Measurements at
December 31, 2020 Using: |
| Level 1 | | Level 2 | | Level 3 | | Total |
| Assets: | | | | | | | |
| Cash equivalents: | | | | | | | |
| Money market funds | $ | 48,770 | | | $ | — | | | $ | — | | | $ | 48,770 | |
| U.S. treasury notes | — | | | 42,997 | | | — | | | 42,997 | |
| Marketable securities: | | | | | | | |
| U.S. treasury notes | — | | | 92,975 | | | — | | | 92,975 | |
| Total | $ | 48,770 | | | $ | 135,972 | | | $ | — | | | $ | 184,742 | |
During | | | | | | | | | | | | | | | | | | | | | | | |
| Fair Value Measurements at
December 31, 2022 Using: |
| Level 1 | | Level 2 | | Level 3 | | Total |
| Assets: | | | | | | | |
| Cash equivalents: | | | | | | | |
| Money market funds | $ | 28,077 | | | $ | — | | | $ | — | | | $ | 28,077 | |
| U.S. treasury notes | — | | | 1,998 | | | — | | | 1,998 | |
| Commercial paper | — | | | 22,139 | | | — | | | 22,139 | |
| Marketable securities: | | | | | | | |
| U.S. treasury notes | — | | | 7,872 | | | — | | | 7,872 | |
| Commercial paper | — | | | 56,398 | | | — | | | 56,398 | |
| Corporate notes and bonds | — | | | 229,314 | | | — | | | 229,314 | |
| Total | $ | 28,077 | | | $ | 317,721 | | | $ | — | | | $ | 345,798 | |
For the years ended December 31, 20212023 and 2020,2022, there were no transfers between Level 1, Level 2 and Level 3.
4. Property and Equipment, Net
Property and equipment, net consisted of the following (in thousands):
| | December 31, |
| 2021 | | 2020 |
| December 31, | | | December 31, |
| 2023 | | | 2023 | | 2022 |
| Laboratory equipment | Laboratory equipment | $ | 4,889 | | | $ | 3,740 | |
| Furniture and fixtures | Furniture and fixtures | 815 | | | 815 | |
| Computer equipment and software | Computer equipment and software | 100 | | | 100 | |
| Leasehold improvements | Leasehold improvements | 17,059 | | | 16,961 | |
| Assets not yet placed in service | 30 | | | 15 | |
| 22,893 | | | 21,631 | |
| 24,973 | |
| Less: Accumulated depreciation and amortization | Less: Accumulated depreciation and amortization | (5,330) | | | (2,103) | |
| $ | 17,563 | | | $ | 19,528 | |
| $ | |
Depreciation and amortization expense was $3.2$3.5 million and $1.3$3.3 million for the years ended December 31, 20212023 and 2020,2022, respectively.
5. Accrued Expenses and Other Current Liabilities
Accrued expenses consisted of the following (in thousands):
| | | | | | | | | | | |
| December 31, |
| 2021 | | 2020 |
| Accrued employee compensation and benefits | $ | 5,596 | | | $ | 3,513 | |
| Accrued tenant improvements | 13 | | | 2,385 | |
| Accrued external research and development expenses | 3,079 | | | 2,146 | |
| Accrued professional fees | 495 | | | 979 | |
| Other | 379 | | | 138 | |
| $ | 9,562 | | | $ | 9,161 | |
| | | | | | | | | | | |
| December 31, |
| 2023 | | 2022 |
| Accrued employee compensation and benefits | $ | 4,347 | | | $ | 4,743 | |
| Accrued external research and development expenses | 3,610 | | | 5,262 | |
| Accrued income taxes | 674 | | | — | |
| Accrued professional fees | 441 | | | 678 | |
| Other | 36 | | | 318 | |
| $ | 9,108 | | | $ | 11,001 | |
6. Notes Payable
Long-term debt consisted of the following (in thousands):
| | | | | | | | | | | |
| December 31, |
| 2021 | | 2020 |
| Principal amount of long-term debt | $ | — | | | $ | 20,000 | |
| Less: Current portion of long-term debt | — | | | — | |
| Long-term debt, net of current portion | — | | | 20,000 | |
| Final payment fee | — | | | 1,000 | |
| Debt discount, net of accretion | — | | | (1,346) | |
| Long-term debt, net of discount and current portion | $ | — | | | $ | 19,654 | |
Loan and Security Agreements
On November 19, 2020, the Company entered into a loan and security agreement (the "Oxford Loan Agreement"), with Oxford Finance LLC, or Oxford, for an aggregate principal amount of $20.0 million (the "Oxford Term Loan A") and up to an additional $5.0 million (the "Oxford Term Loan B"). On November 19, 2020, the Company borrowed $20.0 million under the Oxford Term Loan A. Oxford Term Loan A and Oxford Term Loan B (the "Term Loans") bear interest at a floating per annum rate equal to the greater of (i) 8.0% and (ii) the sum of (a) thirty-day LIBOR rate plus (b) 7.84%. In addition, upon loan maturity or prepayment, the Company is required to make a final payment fee equal to 5.0% of the aggregate principal amount borrowed which is being amortized to interest expense over the term of the debt using the effective interest method.
The Company is required to make monthly interest only payments under the Oxford Loan each month beginning on January 1, 2021. Beginning on December 1, 2023, the Company is required to make consecutive equal monthly payments of principal, together with applicable interest, in arrears, based upon a repayment schedule equal to 24 months, with a final maturity date of November 1, 2025 (the “Maturity Date”). At the Company's option, the Company may elect to prepay the loans subject to a prepayment fee equal to the following percentage of the principal amount being prepaid: 2% if an advance is prepaid during the first 12 months following the applicable advance date, 1% if an advance is prepaid after 12 months but prior to 24 months following the applicable advance date, and 0.5% if an advance is prepaid any time after 24 months following the applicable advance date but prior to the Maturity Date.
In addition, in connection with the Oxford Loan Agreement, the Company granted warrants to purchase 18,445 shares of the Company’s common stock at $16.26 per share. The issued warrants are exercisable for 10 years. The Company valued the warrants using the Black-Scholes option pricing model and determined the fair value of the warrants to be $0.2 million. The Company determined the warrants met the criteria for equity classification, and, as such, the fair value of the warrants were recorded as additional paid-in capital upon issuance and as a discount to the debt which is being amortized to interest expense over the term of the Oxford Loan of five years. In addition, the Company incurred debt issuance costs of $0.2 million.
In December 2021, the Company repaid early the outstanding principal balance under the Term Loans as described above plus unpaid accrued interest, the final payment fee and the prepayment fee. The Company recorded a loss on debt extinguishment of $1.2 million related to this repayment for the year ended December 31, 2021.
During the years ended December 31, 2021 and 2020, the weighted average effective interest rate on outstanding borrowings was approximately 9.5% and 6.4%, respectively.
7. Common Stock
On October 27, 2020, the Company completed its initial public offering (“IPO”) pursuant to which it issued and sold 7,500,000 shares of its common stock at a public offering price of $16.00 per share, resulting in net proceeds of $107.9 million, after deducting underwriting discounts and commissions and other offering expenses. On November 19, 2020, the Company issued and sold an additional 951,837 shares of common stock at the IPO price of $16.00 per share pursuant to the underwriters’ partial exercise of their option to purchase additional shares of common stock, resulting in additional net proceeds of $14.2 million after deducting underwriting discounts and commissions. Upon the closing of the IPO, all of the Company’s outstanding convertible preferred stock automatically converted into shares of common stock.
On December 10, 2021, the Company completed a collaboration agreement with Lilly (See Note 9). Concurrent with the collaboration agreement the Company completed a stock purchase agreement with Lilly and issued and sold Lilly 4,000,000 shares of its common stock at a price of $20.00 per share, resulting in net proceeds of $80.0 million of which $42.2 million was allocated to equity upon the issuance of the Company’s common stock (see Note 9).
Each share of common stock entitles the holder to one vote on all matters submitted to a vote of the Company's stockholders. Common stockholders are not entitled to receive dividends, unless declared by the board of directors.
8.
7. Stock-Based Compensation
2016 Stock2020 Equity Incentive Plan
The Company’s 2016 Stock Incentive Plan, (the “2016 Plan”) provided for the Company to grant incentive stock options or nonqualified stock options and other equity awards to employees, directors and consultants of the Company. Upon the effectiveness of the 2020 Equity Incentive Plan (the “2020 Plan”) in October 2020, the Company ceased granting additional awards under the 2016 Plan.
2020 Equity Incentive Plan
The 2020 Plan provides for the grant of incentive stock options, non-qualified options, stock appreciation rights, restricted stock awards, restricted stock units and other stock-based awards. The number of shares initially reserved for issuance under the 2020 Plan was (i) 2,200,000 shares (the “share pool”), plus (ii) the number of shares of common stock available for issuance under the Company’s 2016 Stock Incentive Plan (the “2016 Plan”) as of the effective date of the 2020 Plan, plus the number of shares of common stock underlying awards under the 2016 Plan that on or after the date of adoption expire or become unexercisable without delivery of shares, are forfeited to, or repurchased for cash, are settled in cash, or otherwise become available again for grant under the 2016 Plan, in each case, in accordance with its terms (up to an aggregate of 5,078,295 shares). As of December 31, 2021, 2,271,8472023, 1,638,199 shares remained available for future grant under the 2020 Plan.
The share pool will automatically increase on January 1 of each year from 2021 to 2030 by the lesser of (i) 4four percent of the number of shares of our common stock outstanding as of the close of business on the immediately preceding December 31 and (ii) the number of shares determined by the board of directors on or prior to such date for such year. The number of shares reserved for issuance under the 2020 Plan was increased by 1,651,9891,691,282 shares effective January 1, 2022.2024.
The 2020 Plan is administered by the board of directors or, at the discretion of the board of directors, by a committee of the board of directors. The exercise prices, vesting and other restrictions are determined at the discretion of the board of directors, or its committee if so delegated. Stock options granted with service-based vesting conditions generally vest over four years and expire after ten years. The exercise price for stock options granted is not less than the fair value of common stock on the date of grant. The Company bases fair value of common stock on the quoted market price. Prior to the IPO, the board of directors determined the value the Company’s common stock, taking into consideration its most recently available valuation of common stock performed by third parties as well as additional relevant factors which may have changed since the date of the most recent contemporaneous valuation through the date of grant.
2020 Employee Stock Purchase Plan
On October 21, 2020, the Company’s board of directors adopted and its stockholders approved the 2020 Employee Stock Purchase Plan (the “ESPP”), which became effective on October 21, 2020. The aggregate number of shares of common stock
available for purchase pursuant to the exercise of options under the ESPP is 360,000 shares, plus an automatic annual increase, as of January 1 of each year from 2021 to 2030, equal to the lesser of 1one percent of the number of shares of common stock outstanding as of the close of business on the immediately preceding December 31 and (ii) the number of shares determined by the board of directors on or prior to such date for such year (up to a maximum of 3,220,520 shares). As of December 31, 2023, 1,143,300 shares remained available for future grant under the ESPP. The number of shares reserved for issuance under the ESPP was increased by 367,894 shares to 780,891422,820 shares effective January 1, 2022.2024.
Eligible employees may authorize payroll deductions of up to 15% of their eligible compensation during an offering period. The purchase of shares is done at a 15% discount on the lesser of (i) the Fair Market Value of a share of Stock on the first day of the offering period and (ii) the Fair Market Value of a share of Stock on the last day of the offering period. The Company currently holds two offering periods, September 1 and March 1, respectively. The first offering period commenced under the ESPP on September 1, 2021. For the yearyears ended December 31, 2021,2023 and 2022, the Company recognized a de minimis amount$0.1 million of expense related to the ESPP.
Stock option valuation
The fair value of stock option grants is estimated using the Black-Scholes option-pricing model. The Company currently lacks company-specific historical and implied volatility information. Therefore, it estimates its expected stock volatility based on the historical volatility of a publicly traded set of peer companies. The expected term of the Company’s stock options has been determined utilizing the “simplified” method for awards that qualify as “plain-vanilla” options. The risk-free interest rate is determined by reference to the U.S. Treasury yield curve in effect at the time of grant of the award for time periods approximately equal to the expected term of the award. Expected dividend yield is based on the fact that the Company has never paid cash dividends and does not expect to pay any cash dividends in the foreseeable future.
The following table presents, on a weighted average basis, the assumptions used in the Black-Scholes option-pricing model to determine the grant-date fair value of stock options granted duringfor the years ended December 31, 20212023 and 20202022 which was as follows:
| | | | | | | | | | | |
| Year Ended December 31, |
| 2021 | | 2020 |
| Risk-free interest rate | 0.7 | % | | 0.4 | % |
| Expected volatility | 78.8 | % | | 78.5 | % |
| Expected dividend yield | — | | | — | |
| Expected term (in years) | 6.0 | | 6.1 |
| | | | | | | | | | | |
| Year Ended December 31, |
| 2023 | | 2022 |
| Risk-free interest rate | 3.7 | % | | 2.1 | % |
| Expected volatility | 87.9 | % | | 83.8 | % |
| Expected dividend yield | — | | | — | |
| Expected term (in years) | 6.0 | | 6.1 |
The following table summarizes the Company’s option activity duringfor the year ended December 31, 20212023 which was as follows:
| | | | | | | | | | | | | | | | | | | | | | | |
| Number of
Shares | | Weighted
Average
Exercise
Price | | Weighted
Average
Contractual
Term | | Aggregate
Intrinsic
Value |
| | | | | (in years) | | (in thousands) |
| Outstanding as of December 31, 2020 | 5,016,460 | | | $ | 4.93 | | | 8.5 | | $ | 76,941 | |
| Granted | 1,719,563 | | | 15.41 | | | | | |
| Exercised | (508,774) | | | 2.79 | | | | | |
| Forfeited | (306,740) | | | 10.22 | | | | | |
| Outstanding as of December 31, 2021 | 5,920,509 | | | $ | 7.88 | | | 7.9 | | $ | 88,720 | |
| Vested and expected to vest as of December 31, 2021 | 5,920,509 | | | $ | 7.88 | | | 7.9 | | $ | 88,720 | |
| Options exercisable as of December 31, 2021 | 2,435,439 | | | $ | 3.62 | | | 7.0 | | $ | 46,882 | |
| | | | | | | | | | | | | | | | | | | | | | | |
| Number of
Shares | | Weighted
Average
Exercise
Price | | Weighted
Average
Contractual
Term | | Aggregate
Intrinsic
Value |
| | | | | (in years) | | (in thousands) |
| Outstanding as of December 31, 2022 | 7,911,211 | | | $ | 10.36 | | | 7.8 | | $ | 8,648 | |
| Granted | 2,481,884 | | | 8.17 | | | | | |
| Exercised | (437,407) | | | 3.61 | | | | | |
| Forfeited | (1,004,100) | | | 11.28 | | | | | |
| Outstanding as of December 31, 2023 | 8,951,588 | | | $ | 9.97 | | | 7.4 | | $ | 7,598 | |
| Vested and expected to vest as of December 31, 2023 | 8,951,588 | | | $ | 9.97 | | | 7.4 | | $ | 7,598 | |
| Options exercisable as of December 31, 2023 | 4,879,493 | | | $ | 9.16 | | | 6.4 | | $ | 7,530 | |
The aggregate intrinsic value of stock options is calculated as the difference between the exercise price of the stock options and the fair value of the Company’s common stock for those stock options that had exercise prices lower than the fair value of the Company’s common stock. The aggregate intrinsic value of stock options exercised duringfor the years ended December 31, 20212023 and 20202022 was $5.3$1.8 million and $3.5$4.3 million, respectively.
The weighted average grant-date fair value of stock options granted duringfor the years ended December 31, 20212023 and 20202022 was $10.40$6.12 per share and $6.01$10.08 per share, respectively.
Restricted common stock
During 2015, the Company issued and sold 4,459,459 shares of restricted common stock at par value to the scientific founders of the Company. The shares were subject to vesting over a period of five years and began vesting in April 2016 and were fully vested as of December 31, 2020.
The aggregate fair value of restricted stock that vested during the years ended December 31, 2020 was $4.8 million, respectively.
Stock-based compensation
The Company recorded stock-based compensation expense related to common stock options and restricted common stock in the following expense categories of its consolidated statements of operations and comprehensive loss duringfor the years ended December 31, 20212023 and 20202022 which was as follows (in thousands):
| | Year Ended December 31, |
| 2021 | | 2020 |
| Year Ended December 31, | | | Year Ended December 31, |
| 2023 | | | 2023 | | 2022 |
| Research and development expenses | Research and development expenses | $ | 4,324 | | | $ | 1,724 | |
| General and administrative expenses | General and administrative expenses | 4,060 | | | 1,237 | |
| $ | 8,384 | | | $ | 2,961 | |
| $ | |
As of December 31, 2021,2023, total unrecognized compensation cost related to unvested options was $20.8$26.2 million, which is expected to be recognized over a weighted average period of 2.72.2 years.
9.8. Collaboration Agreements
Lilly Collaboration Agreement and Stock Purchase Agreement
In December 2021, the Company entered into a collaboration agreement (the “Lillythe Lilly Collaboration Agreement")Agreement with Lilly to create novel oncology medicines by applying Foghorn’s proprietary Gene Traffic Control platform. The collaboration includes a co-development and co-commercialization agreement for the Company’s selective BRM oncology target, consisting of two programs, and one additional undisclosed oncology target (collectively, the “Joint Programs”). The collaboration also includes 3three additional discovery targets or programs (collectively, the “Discovery Programs”) for a total of 6six programs directed at 5five targets.
With respect to the Joint Programs, the Company will lead discovery and early research activities through the successful completion of dose finding toxicity studies for the identified compound, while Lilly will lead development and commercialization activities of the identified compound with participation from the Company in development activities and
50% cost sharing until registrational clinical trials. The Company and Lilly may jointly develop and commercialize the Joint Program compound though the Company may, in its sole discretion, opt-out on a program-by-program basis of further participation in the development and commercialization efforts prior to the first registrational clinical trial. If the Company does not opt-out, Lilly and the Company will continue to share in the costs to further develop and commercialize the Joint Program compound on a worldwide basis, equally share in the U.S. profits on product sales, subject to certain adjustments, and receive royalties on sales outside of the United States (“Ex-U.S.”) at royalty rates ranging from low double digits to high twenties. If the Company opts-out of further development and commercialization efforts, it will have no further obligations to share in the development and commercialization costs, will receive royalties rather than profit share on U.S. sales and will receive royalties at a lower rate on Ex-U.S. sales.
With respect to the Discovery Programs, the Company will lead discovery and early research activities through the successful completion of dose finding toxicity studies for the identified compounds. The Company may, in its sole discretion, opt inopt-in on a program-by-program basis after the successful completion of dose finding toxicity to participate in the further development and commercialization efforts of the Discovery Program compounds. If the Company opts-in to the development and commercialization of the Discovery Program compounds, it will be eligible to receive milestone payments of up to $10.0 million per program upon specified research and development milestones and up to $180.0 million per program upon achievement of specified regulatory and commercial milestones and will also be eligible to share in the U.S. profits at pre-determined percentages on product sales. The Company would also be eligible to receive tiered Ex-U.S. royalty rates, calculated on a product-by-product and country-by-country basis, on net sales outside of the United States, if any, ranging from low single digits to low double digits, but less then teens. If the Company does not opt inopt-in to further development and commercialization efforts for the Discovery Programs, it will be eligible to receive milestone payments of up to $70.0 million per program upon specified research and development milestones and up to $360.0 million per program upon achievement of specified regulatory and commercial milestones per approved product, if any. The Company would also be eligible to receive
tiered royalties on net sales of products worldwide at royalty rates ranging from low single digits to low double digits, but less thenthan teens.
Lilly has the right to make substitutions for each of the five targets during the research term of each program, subject to certain limitations. Pursuant to the Lilly Collaboration Agreement, the Company will also participate in joint decision-making through the joint steering committee and subcommittees. Unless terminated earlier, the Lilly Collaboration Agreement will continue on a product-by-product basis until the expiration of all royalty obligations under the Lilly Collaboration Agreement and when neither the Company nor Lilly is developing, commercializing or manufacturing any product under the Lilly Collaboration Agreement. The Company or Lilly may terminate the Lilly Collaboration Agreement upon an uncured material breach by the other party. Lilly may also terminate the Lilly Collaboration Agreement in its entirety or on a target-by-target, program-by-program or product-by-product basis for any reason upon certain notice to the Company.
Under the terms of the Lilly Collaboration Agreement, Lilly agreed to make a nonrefundable upfront payment of $300.0 million to the Company within thirty (30) business days following the effective date of the agreement. Simultaneously with the execution of the Lilly Collaboration Agreement, the Company and Lilly entered into thea stock purchase agreement with Lilly SPA,(the “Lilly SPA”), pursuant to which Lilly purchased 4,000,000 shares of the Company’s common stock at $20.00 per share, for an aggregate purchase price of $80.0 million.
The Company determined that the Lilly Collaboration Agreement and the stock purchase agreement entered into with Lilly concurrently (the "Lilly SPA")SPA should be evaluated as a combined contract in accordance with ASC 606. The Company determined the fair value of the shares sold under the Lilly SPA to be $37.8 million less than the contractual purchase price stipulated in the Lilly SPA. In accordance with the applicable accounting guidance in ASC 815-40, Contracts in Entity’s Own Equity, the Company determined that the sale of stock should be recorded at fair value and therefore allocated the excess consideration received under the Lilly SPA to the Lilly Collaboration Agreement, which along with the non-refundable payment of $300.0 million will be recognized as revenue over the total estimated period of performance.
The Company accounted for the arrangement under ASC 606 as the Company concluded it has a customer relationship with Lilly. The Company determined thatthat: (1) the research activities performed by the Company for both the Joint Programs and the Discovery Programs, (2) the development activities and cost sharing for the Joint Program development efforts after dose finding toxicity until registrational clinical trials, (3) the research, development, manufacture and commercialization licenses, and (4) service on the joint steering committee and subcommittees represent a single performance obligation under the Lilly Collaboration Agreement. The Company determined that Lilly cannot benefit from the licenses separately from the research activities, the development activities until registrational clinical trials and participation on the joint steering committee and subcommittees because these services are specialized and rely on the Company’s expertise such that these activities are highly interrelated and therefore not distinct. Accordingly, the promised goods and services represent one combined performance obligation, and the entire transaction price was allocated to that single combined performance obligation. The performance obligation will be satisfied over time as the Company performs the research activities, participates and shares in the cost of the
development activities for the Joint Programs and participates in a joint steering committee and subcommittees to oversee these activities.
The Company’s options to share in further development and commercialization efforts via its opt-in/opt-out rights will be assessed and accounted for as separate units of accounting under the relevant guidance if, and when, such options are exercised by the Company.
During the third quarter of 2023, the Company has transitioned the BRM Selective inhibitor program into development activities for which Lilly will lead and the Company will participate and share in 50% of the costs until registrational trials. Costs incurred will continue to be included in research and development expenses on the consolidated statements of operations and comprehensive loss.
The transaction price of $337.8 million was initially recorded as deferred revenue and is being recognized as revenue as the performance obligation is satisfied. The Company recognizes revenue using the cost-to-cost method, which it believes best depicts the transfer of control to the customer over time. Under the cost-to-cost method, the extent of progress towards completion is measured based on the ratio of actual costs incurred to the total estimated costs expected upon satisfying the identified performance obligation. Under this method, revenue is recorded as a percentage of the estimated transaction price based on the extent of progress towards completion. As of December 31, 2021, 2023, the potential research, development and regulatory milestone payments that the Company is eligible to receive were excluded from the transaction price as they were fully constrained by uncertain events. The Company will reevaluate the transaction price at the end of each reporting period and as uncertain events are resolved or other changes in circumstances occur, and if necessary, the Company will adjust its estimate of the transaction price. Any additions to the transaction price would be reflected in the period as a cumulative revenue catch-up based on the ratio of costs incurred to the total estimated costs expected applied to the revised transaction price. Sales-based royalties and milestone payments, which predominantly relate to the license, will be recognized if and when the related sales occur.
As of December 31, 20212023, the aggregate amount of the transaction price related to the unsatisfied portion of the performance obligation was $337.2 $302.7 million, which is expected to be recognized as revenue through 2029.2029 or beyond depending on the timing of certain clinical development activities. The Company does not expect collaboration revenue to be recognized evenly over this period as it will be recognized on a percentage of completion basis (using cost-to-cost method) as the Company performs the research and development activities and participates on the joint
steering committee and subcommittees, which will likely vary from period to period. In estimating the total costs to satisfy its single performance obligation pursuant to the Lilly Collaboration Agreement, the Company is required to make significant estimates including an estimate of the number of target substitutions Lilly may elect to make and the expected time and expected costs to fulfill the performance obligation. The cumulative effect of revisions to the total estimated costs to complete the Company’s single performance obligation will be recorded in the period in which the changes are identified, and amounts can be reasonably estimated. While such revisions will have no impact on the Company’s cash flows, a significant change in these assumptions and estimates could have a material impact on the timing and amount of revenue recognized in future periods and the classification of deferred revenue between short-term and long-term. During the year ended December 31, 2023, the Company re-evaluated its estimates related to the Lilly Collaboration Agreement. As a result of this re-evaluation, expected future costs over the course of the Lilly Collaboration Agreement increased, resulting in an adjustment that decreased revenue by $3.2 million for the year ended December 31, 2023.
At inception, the Company assessed the Lilly Collaboration Agreement to determine whether a significant financing component exists and concluded that a significant financing component does not exist. For the yearyears ended December 31, 2021,2023 and 2022, the Company had recorded $0.6$17.1 million and $17.4 million, respectively, of revenue under the Lilly Collaboration Agreement, which was included in deferred revenue at contract inception.
the beginning of the respective periods. As of December 31, 2021,2023, the upfront paymentCompany had a payable to Lilly of $300.0$1.5 million was recorded as collaboration receivablein accrued expenses and other current liabilities on the Company'sCompany’s consolidated balance sheets. In JanuaryAs of December 31, 2022, the $300.0 million was collected fromCompany did not have a receivable or payable with Lilly.
Merck Collaboration Agreement
In July 2020, the Company entered into a research collaboration and license agreement (the “Merckthe Merck Collaboration Agreement”)Agreement with Merck, Sharp & Dohme Corp. (“Merck”). Thepursuant to which the Company and Merck will applyapplied Foghorn’s proprietary Gene Traffic Control platform to discover and develop novel therapeutics. Under the Merck Collaboration Agreement, the Company granted Merck exclusive global rights to develop, manufacture and commercialize drugs that target dysregulation of a single transcription factor. Under the terms of the Merck Collaboration Agreement, the Company and Merck are each responsible to perform certain research activities in accordance with a mutually agreed upon research plan. Merck may substitute the selected transcription factor during certain stages of the research program, subject to certain limitations. Following completion of the research program, Merck is responsible for the development and commercialization of the compounds developed pursuant to the research program and any product containing such compounds. Pursuant to the Merck Collaboration Agreement, the Company will also participate on a joint steering committee.
Under the terms of the agreement, Foghorn received a nonrefundable upfront payment of $15.0 million from Merck, and is eligible to receive up to $245.0 million upon achievement of specified research, development and regulatory milestones by any product candidate generated by the collaboration, and up to $165.0 million upon achievement of specified sales-based milestones per approved product from the collaboration, if any. The Company will be eligible to receive tiered royalties, calculated on a product-by-product and country-by-country basis, on net sales of approved products from the collaboration, if any, at royalty rates ranging from the mid-single digits to low tens, depending on whether the products are covered by patent rights it licenses to Merck.
Unless terminated earlier, the Merck Collaboration Agreement will continue in full force and effect until one or more products has received marketing authorization and, thereafter, until expiration of all royalty obligations under the Merck Collaboration Agreement. The Company or Merck may terminate the Merck Collaboration Agreement upon an uncured material breach by the other party or insolvency of the other party. Merck may also terminate the Merck Collaboration Agreement for any reason upon certain notice to the Company.
The Company determined that the (1) research, development, manufacture and commercialization licenses, (2) the research activities performed by the Company and (3) service on the joint committees represent a single performance obligation under the Merck Collaboration Agreement. The Company determined that Merck cannot benefit from the licenses separately from the research activities and participation on the joint steering committee because these services are specialized and rely on the Company’s expertise such that these activities are highly interrelated and therefore not distinct. Accordingly, the promised goods and services represent one combined performance obligation and the entire transaction price was allocated to that single combined performance obligation. The performance obligation will be satisfied over the research term as the Company performs the research activities and participates in a joint steering committee to oversee research activities.
The upfront payment of $15.0 million was initially recorded as deferred revenue and is being recognized as revenue as the performance obligation is satisfied. The Company recognizes revenue using the cost-to-cost method, which it believes best depicts the transfer of control to the customer over time. Under the cost-to-cost method, the extent of progress towards completion is measured based on the ratio of actual costs incurred to the total estimated costs expected upon satisfying the identified performance obligation.method. Under this method, revenue is recorded as a percentage of the estimated transaction price based on the extent of progress towards completion. As
In the third quarter of December 31, 2021, the potential research, development and regulatory milestone payments that2022, the Company is eligibleachieved a research milestone related to receive were excludedthe Merck Collaboration Agreement and received a $5.0 million milestone payment from the transaction price as they were fully constrained by uncertain events. The Company will reevaluate the transaction price at the end of each reporting period andMerck.
as uncertain events are resolved or other changes in circumstances occur, and if necessary,On August 9, 2023 the Company will adjust its estimatereceived notice from Merck that it is terminating the Merck Collaboration Agreement effective November 7, 2023. At the time of notice, no material obligations remained under the transaction price. Any additionsMerck Collaboration Agreement. As such, the Company recognized the remaining $16.1 million of deferred revenue, related to the transaction price would be reflectedoriginal $15.0 million upfront payment and the $5.0 million milestone payment received from Merck, in the periodyear ended December 31, 2023 as a cumulative revenue catch-up based on the ratio of costs incurred to the total estimated costs expected applied to the revised transaction price. Sales-based royalties and milestone payments, which predominantly relate to the license, will be recognized if and when the related sales occur.collaboration revenue.
As of December 31, 2021, the aggregate amount of the transaction price related to the unsatisfied portion of the2023, there was no remaining performance obligation is $13.9 million, which is expected to be recognized as revenue through 2028. The Company does not expect collaboration revenue to be recognized evenly over this period as it will be recognized on a percentage of completion basis (using cost-to-cost method) as the Company performs the research activities and participates on the joint steering committee, which will likely vary from period to period. In estimating the total costs to satisfy its single performance obligation pursuant tounder the Merck Collaboration Agreement, the Company is required to make significant estimates including an estimate of the number of transcription factor substitutions and the expected time and expected costs to fulfill the performance obligation. The cumulative effect of revisions to the total estimated costs to complete the Company’s single performance obligation will be recorded in the period in which the changes are identified, and amounts can be reasonably estimated. While such revisions will have no impact on the Company’s cash flows, a significant change in these assumptions and estimates could have a material impact on the timing and amount of revenue recognized in future periods and the classification of deferred revenue between short-term and long-term. In June 2021, the Company re-evaluated its estimates related to the Merck Collaboration Agreement resulting in a reclassification of $1.9 million from short-term deferred revenue to long-term deferred revenue, net of current portion on the Company's consolidated balance sheets.Agreement.
At inception, the Company assessed the Merck Collaboration Agreement to determine whether a significant financing component exists and concluded that a significant financing component does not exist. For the year ended December 31, 2021,2023 and 2022, the Company had recorded $0.7$17.0 million and $1.8 million of revenue under the Merck Collaboration Agreement. For the years ended December 31, 2023 and 2022, the Company had recorded $17.0 million and $1.1 million, respectively, of revenue under the Merck Collaboration Agreement, which was included in deferred revenue at the beginning of the periodrespective periods and the classification of deferred revenue between short-term and long-term.
10.9. Income Taxes
DuringFor the yearsyear ended December 31, 2021 and 2020,2023 the Company recorded a provision for income taxes of $4.2 million, representing an effective tax rate of 4.6%. The income tax provision and the effective tax rate is primarily driven by the current federal and state taxes related to the $300.0 million upfront payment for Lilly Collaboration Agreement, which will be recognized as taxable income in the current year, and the required capitalization of research and development costs pursuant to Internal Revenue Code Section 174. As a result, the Company expects to have taxable income in the current year, which will be reduced by utilizing available net operating loss carryforwards and research and development tax credit carryforwards.
The Company has evaluated the positive and negative evidence bearing upon its ability to realize the deferred tax assets. Management has considered the Company’s history of cumulative net losses incurred since inception and its lack of commercialization of any products that would generate revenue from product sales and has concluded that it is more likely than not that the Company will not realize the benefits of the deferred tax assets. Accordingly, a full valuation allowance has been established against the net deferred tax assets.
For the year ended December 31, 2023, the Company recorded income tax expense on pre-tax losses of $94.2 million. Historically, no income tax benefits for the net deferred tax assets comprised primarily of net operating losses incurred and research and development tax credits generated in each year, due to its uncertainty of realizing a benefit from these items.
All of the Company’s operating losses since inception have been generated in the United States.
A reconciliation of the U.S. federal statutory income tax rate to the Company’s effective income tax rate was as follows:
| | Year Ended December 31, |
| 2021 | | 2020 |
| Year Ended December 31, | | | Year Ended December 31, |
| 2023 | | | 2023 | | 2022 |
| Federal statutory income tax rate | Federal statutory income tax rate | (21.0) | % | | (21.0) | % | Federal statutory income tax rate | (21.0) | % | | (21.0) | % |
| State taxes, net of federal benefit | State taxes, net of federal benefit | (5.8) | | | (6.4) | |
| Federal and state research and development tax credits | Federal and state research and development tax credits | (4.4) | | | (3.7) | |
| Other | Other | 0.8 | | | (0.5) | |
| Change in deferred tax asset valuation allowance | Change in deferred tax asset valuation allowance | 30.4 | | | 31.6 | |
| Effective income tax rate | Effective income tax rate | 0.0 | % | | 0.0 | % | Effective income tax rate | 4.6 | % | | 0.0 | % |
Net deferred tax assets consisted of the following (in thousands):
| | December 31, |
| 2021 | | 2020 |
| December 31, | | | December 31, |
| 2023 | | | 2023 | | 2022 |
| Deferred tax assets: | Deferred tax assets: | | | |
| Net operating loss carryforwards | Net operating loss carryforwards | $ | 61,083 | | | $ | 39,289 | |
| Net operating loss carryforwards | |
| Net operating loss carryforwards | |
| Capitalized research expenditures | |
| Research and development tax credit carryforwards | Research and development tax credit carryforwards | 9,367 | | | 4,955 | |
| Capitalized start-up costs | Capitalized start-up costs | 157 | | | 174 | |
| Deferred revenue | |
| Accrued expenses | Accrued expenses | 5,300 | | | 1,010 | |
| Stock-based compensation | Stock-based compensation | 1,403 | | | 436 | |
| Operating lease liabilities | Operating lease liabilities | 15,952 | | | 17,032 | |
| Total deferred tax assets | Total deferred tax assets | 93,262 | | | 62,896 | |
| Deferred tax liabilities: | Deferred tax liabilities: | |
| Depreciation | Depreciation | (3,551) | | | (2,819) | |
| Depreciation | |
| Depreciation | |
| Operating lease right-of-use assets | Operating lease right-of-use assets | (10,566) | | | (11,694) | |
| Total deferred tax liabilities | Total deferred tax liabilities | (14,117) | | | (14,513) | |
| Valuation allowance | Valuation allowance | 79,145 | | | 48,383 | |
| Net deferred tax assets | Net deferred tax assets | $ | — | | | $ | — | |
As of December 31, 2021,2023, the Company had U.S. federal and state net operating loss carryforwards of $228.9 million and $206.0 million, respectively, which may be available to offset future taxable income. The federal net operating loss carryforwards include $12.5$3.0 million which expire at various dates beginning in 2035 and $216.4 million which carryforwardcan be carried forward indefinitely but in some circumstances may be limited to offset 80% of annual taxable income. The state net operating loss carryforwards expire at various dates beginning in 2036. As of December 31, 2021,2023, the Company also had U.S. federal and state research and development tax credit carryforwards of $6.1$3.7 million and $4.1$2.6 million, respectively, which may be available to offset future tax liabilities and expire at various dates beginning in 20362041 and 2031,2037, respectively.
UtilizationBeginning in 2022, the Tax Cuts and Jobs Act of 2017 eliminated the U.S. federal and state net operating loss carryforwards andoption to deduct research and development tax credit carryforwards may be subjectexpenditures currently and requires taxpayers to a substantial annual limitation under Sections 382amortize them, over five years for domestically incurred expenditures and 383 of theover fifteen years for foreign incurred expenditures, pursuant to Internal Revenue Code Section 174. As of 1986, and corresponding provisions of state law, due to ownership changes that have occurred previously or that could occur in the future. These ownership changes may limit the amount of carryforwards that can be utilized annually to offset future taxable income or tax liabilities. In general, an ownership change, as defined by Section 382, results from transactions increasing the ownership of certain stockholders or public groups in the stock of a corporation by more than 50% over a three-year period. The Company has not conducted a study to assess whether a change of control has occurred or whether there have been multiple changes of control since inception due to the significant complexity and cost associated with such a study. IfDecember 31, 2023, the Company has experiencedrecorded a changegross deferred tax asset of control, as defined by$170.6 million related to the Capitalized IRC Section 382, at any time since inception, utilization of the net operating loss carryforwards or research and development tax credit carryforwards would be subject to an annual limitation under Section 382. Any limitation may result in expiration of a portion of the net operating loss carryforwards or research and development tax credit carryforwards before utilization. Further, until a study is completed by the Company and any limitation is known, no amounts are being recorded as an uncertain tax position.174 expenditures.
The Company has evaluated the positive and negative evidence bearing upon its ability to realize the deferred tax assets. Management has considered the Company’s history of cumulative net losses incurred since inception and its lack of commercialization of any products that would generate revenue from product sales and has concluded that it is more likely than not that the Company will not realize the benefits of the deferred tax assets. Accordingly, a full valuation allowance has been established against the net deferred tax assets as of December 31, 20212023 and 2020.2022. Management reevaluates the positive and negative evidence at each reporting period.
The valuation allowance increased by $30.8$31.1 million and $21.8$32.8 million duringfor the years ended December 31, 20212023 and 2020,2022, respectively, primarily as a result of the increase in net operating loss carryforwards.total deferred tax assets.
As of December 31, 20212023 and 2020,2022, the Company had not recorded any amounts for unrecognized tax benefits. The Company files income tax returns in the U.S. Massachusetts, Florida and Massachusetts,Tennessee as prescribed by the tax laws of the jurisdictions in which it operates. In the normal course of business, the Company is subject to examination by federal and state jurisdictions, where applicable. There are currently no pending tax examinations. Since the Company is in a loss carryforward position, it is generally subject to
examination by the U.S. federal, state, and local income tax authorities for all tax years in which a loss carryforward is available.
11.10. Leases
In October 2019, the Company entered into a lease for 81,441 square feet of office and laboratory space in Cambridge, Massachusetts, commencing in January 2020 (the “New“Office Lease”). The initial term of the NewOffice Lease was eight years with a five-year option to extend at fair-market rent at the time of the extension. In June 2020, the Company amended the Office Lease to defer payment of a portion of the base rent and operating expenses and to extend the lease term by nine months to September 2028. The base rent payments escalate annually over the eight-year lease term and totaled approximately $60.3 million. In connection with the NewOffice Lease, the landlord agreed to fund up to $3.0 million in tenant improvements to the leased facility as well as up to an additional $16.3 million, which resulted in additional rent payments to the landlord over the lease term. DuringFor the yearyears ended December 31, 20212023 and 2020, $2.52022, $0.1 million and $15.4$0.1 million, respectively, of leasehold improvements were
reimbursed by the landlord, which resulted in an increase to operating lease liabilities. The Company is obligated to pay its portion of real estate taxes and costs related to the premises, including costs of operations and management of the leased premises. The Company is required to maintain a cash balance of $1.7 million to secure a letter of credit associated with the lease. This amount was classified as restricted cash (non-current) on the consolidated balance sheets as of December 31, 20212023 and 2020. On January 1, 2020, the lease commencement date, the Company recorded an operating lease asset of $38.6 million and corresponding lease liability of $38.3 million.
In June 2020, the Company amended the New Lease to defer payment of a portion of the base rent and operating expenses and to extend the lease term by nine months to September 2028. The amendment was accounted for as a lease modification and the right-of-use asset and lease liability were remeasured at the modification date of June 29, 2020 resulting in an increase of $7.4 million to both the right-of-use asset and lease liabilities.
The Company had a lease for office and laboratory facilities in Cambridge, Massachusetts under a noncancelable operating lease that began in August 2017 and expired in March 2025. In April 2020, this lease was assigned and assumed by a related party which became effective in October 2020.2022.
The components of lease expense waswere as follows for the years ended December 31, 20212023 and 20202022 (in thousands):
| | Year Ended December 31, |
| 2021 | | 2020 |
| Year Ended December 31, | | | Year Ended December 31, |
| 2023 | | | 2023 | | 2022 |
| Operating lease cost | Operating lease cost | $ | 7,547 | | | $ | 7,032 | |
| Short-term lease cost | — | | | 61 | |
| Variable lease cost | Variable lease cost | 2,892 | | | 1,217 | |
| $ | 10,439 | | | $ | 8,310 | |
| $ | |
Supplemental disclosure of cash flow information related to leases was as follows (in thousands):
| | Year Ended December 31, |
| 2021 | | 2020 |
| Year Ended December 31, | | | Year Ended December 31, |
| 2023 | | | 2023 | | 2022 |
| Cash paid for amounts included in the measurement of operating lease liabilities | Cash paid for amounts included in the measurement of operating lease liabilities | $ | 9,737 | | | $ | 3,086 | |
| Operating lease liabilities arising from obtaining right-of-use assets | Operating lease liabilities arising from obtaining right-of-use assets | $ | — | | | $ | 38,306 | |
| Increase in operating lease liabilities and right-of-use assets due to lease remeasurement | $ | — | | | $ | 7,384 | |
|
The weighted average remaining lease term and discount rate was as follows:
| | | | | | | | | | | |
| 2021 | | 2020 |
| Weighted-average remaining lease term—operating leases (in years) | 6.7 | | 7.7 |
| Weighted-average discount rate—operating leases | 5.35 | % | | 5.35 | % |
| | | | | | | | | | | |
| 2023 | | 2022 |
| Weighted-average remaining lease term—operating leases (in years) | 4.6 | | 5.7 |
| Weighted-average discount rate—operating leases | 5.24 | % | | 5.35 | % |
Future annual minimum lease payments under operating leases as of December 31, 20212023 was as follows (in thousands):
| | 2022 | $ | 9,954 | |
| 2023 | 10,051 | |
| 2024 | 2024 | 10,378 | |
| 2025 | 2025 | 10,626 | |
| 2026 | 2026 | 10,881 | |
| Thereafter | 19,705 | |
| 2027 | |
| 2028 | |
| Total future minimum lease payments | Total future minimum lease payments | 71,595 | |
| Less: imputed interest | Less: imputed interest | (11,802) | |
| Less: estimated lease incentives | (1,461) | |
| | Total operating lease liabilities | Total operating lease liabilities | $ | 58,332 | |
| Total operating lease liabilities | |
| Total operating lease liabilities | |
| | Year Ended December 31, |
| Year Ended December 31, | | | Year Ended December 31, |
| Included in the consolidated balance sheets (in thousands): | Included in the consolidated balance sheets (in thousands): | 2021 | | 2020 | Included in the consolidated balance sheets (in thousands): | 2023 | | 2022 |
| Current operating lease liabilities | Current operating lease liabilities | $ | 6,994 | | | $ | 3,981 | |
| Operating lease liabilities, net of current portion | Operating lease liabilities, net of current portion | 51,338 | | | 58,361 | |
| Total operating lease liabilities | Total operating lease liabilities | $ | 58,332 | | | $ | 62,342 | |
Sublease agreement
In April 2020,During the year 2022, the Company entered into a two-yearwas given notice by its sublessee (the “Former Sublessee”) that it would terminate the sublease agreementin accordance with its terms and vacate the subleased premises, prior to sublet approximately 16,843the original expiration date. At the time, the Company was subletting 19,823 square feet of office space under the NewOffice Lease, as amended, which began in July 2020. In February 2021,to the Former Sublessee.
On August 2, 2022, the Company entered into a sublease with another party (the “New Sublease”) to sublease the same premises. The New Sublease became effective during the fourth quarter of 2022 and has an amendment toinitial term of 18 months. During the sublease to sublet approximately 2,980 square feet of additional office space toyear ended December 31, 2023, the sublessee for the remaining lease term. In July 2021,notified the Company entered into an amendment to, among other things, extendthat they will exercise the sublease12-month renewal option, extending the New Sublease through November 30, 2022.April 28, 2025. As of December 31, 2021,2023, the remaining rent payments due to the Company under the amended subleaseNew Sublease were $1.9$3.4 million. DuringFor the years ended December 31, 20212023 and 2020,2022, the Company recorded other income of $2.5$2.9 million and $1.0$2.6 million, respectively, related to this sublease.its subleases.
11. Commitments and Contingencies
Leases
The Company’s commitments under its leases are described in Note 11.10.
License agreements
The Company has entered into various exclusive and non-exclusive license agreements for certain technologies. Under the terms of these license agreements, the Company could be required to reimburse the licensors for patent expenses and remit amounts in the low single-digit as sales-based royalties upon the occurrence of specific events as outlined in the corresponding license agreements. The Company is also required to make annual license maintenance fees of less than $0.1$0.3 million and pay up to $1.1 million in regulatory milestones on each licensed product upon the occurrence of specific events as outlined in one of the license agreements. None of our current product candidates utilize technologies covered by these licenses.
Indemnification agreements
In the ordinary course of business, the Company may provide indemnification of varying scope and terms to vendors, lessors, contract research organizations, business partners and other parties with respect to certain matters including, but not limited to, losses arising out of breach of such agreements or from intellectual property infringement claims made by third parties. In addition, the Company has entered into indemnification agreements with members of its board of directors and its executive officers that will require the Company, among other things, to indemnify them against certain liabilities that may arise by reason of their status or service as directors or officers. The maximum potential amount of future payments the Company could be required to make under these indemnification agreements is, in many cases, unlimited. The Company has not incurred any material costs as a result of such indemnifications and is not currently aware of any indemnification claims.
Legal Proceedings
From time to time, the Company may become involved in litigation or other legal proceedings. The Company is not currently a party to any material litigation or legal proceedings.
13.12. Defined Contribution Plan
The Company has a 401(k) defined contribution plan (the “401(k) Plan”) for its employees. Eligible employees may make pretax contributions to the 401(k) Plan up to statutory limits. ThereFor the year ended December 31, 2023, the Company recorded $0.4 million of expense related to 401(k) discretionary match. For the year ended December 31, 2022, there was no discretionary match made under the 401(k) Plan as of December 31, 2021 and 2020.Plan.
14.13. Related Parties
In October 2015, the Company entered into a five-year service agreement with Flagship Pioneering (“Flagship”), an affiliate of one of its stockholders, Flagship Venture Funds,to provide general and administrative services to the Company, including certain consulting services and the provision of employee health and dental benefit plans for the Company’s employees. The Company made cash payments to Flagship, for services received under this agreement, of $1.2 million during the year ended December 31, 2020. As of December 31, 2021 and 2020, the Company had no accounts payable to Flagship for costs related to the service agreement. This agreement expired in 2020.
In October 2015, the Company entered into a five-year consulting agreement with a scientific founder of the Company who is also a board member and a shareholder. In October 2020, thisThis agreement was amended and extended to January 1, 2022,2024, and is subject to automatic one-year renewal terms until terminated. DuringFor the years ended December 31, 20212023 and 2020,2022, the Company paid the scientific founder $0.1 million and $0.2 million, respectively.million. As of December 31, 20212023 and 2020,2022, the Company had less than $0.1 million in accounts payable due to thisthe scientific founder.
On December 10, 2021, we entered into the Lilly Collaboration Agreement with Lilly. Concurrent with the Lilly Collaboration Agreement the Company also entered into the Lilly SPA where the Company issued and sold Lilly 4,000,000 shares of our common stock at a price of $20.00 per share, making them a 5% or greater shareholder in the Company for the years ended December 31, 2023 and 2022. For the years ended December 31, 2023 and 2022, the Company had recorded $17.1 million and $17.4 million, respectively, of revenue under the Lilly Collaboration Agreement. As of December 31, 2023 and 2022, the Company had $302.7 million and $319.8 million, of deferred revenue related to the Lilly Collaboration Agreement. As of December 31, 2023, the Company had a payable of $1.5 million due to Lilly. As of December 31, 2022, the Company did not have a receivable or payable with Lilly.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
Our management, under the supervision and with the participation of our Principal Executive Officer (our Chief Executive Officer) and Principal Financial and Accounting Officer (our Chief Financial Officer), has evaluated the effectiveness of our disclosure controls and procedures as of period end. The term “disclosure controls and procedures,” as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended, or the Exchange Act, means controls and other procedures of a company that are designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is accumulated and communicated to the company’s management, including its principal executive and principal financial officers, as appropriate to allow timely decisions regarding required disclosure. Management recognizes that any disclosure controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives, and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on the evaluation of our disclosure controls and procedures as of December 31, 2021,2023, our Principal Executive Officer and Principal Financial Officer concluded that, as of such date, our disclosure controls and procedures were effective at the reasonable assurance level.
Internal Control Over Financial Reporting
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act) to provide reasonable assurance regarding the reliability of our financial reporting and the preparation of consolidated financial statements for external purposes in accordance with U.S. generally accepted accounting principles (“GAAP”). Internal control over financial reporting is a process designed by, or under the supervision of, our Chief Executive Officer and Chief Financial Officer, and effected by our Board of Directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the
preparation of financial statements for external purposes in accordance with generally accepted accounting principles and includes those policies and procedures that:
•Pertain to the maintenance of records that accurately and fairly reflect in reasonable detail the transactions and dispositions of the assets of our company;
•Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorizations of our management and directors; and
•Provide reasonable assurances regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material adverse effect on our financial statements.
Management assessed our internal control over financial reporting as of December 31, 2021,2023, the end of our fiscal year. Management based its assessment on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework). Management’s assessment included evaluation of elements such as the design and operating effectiveness of key financial reporting controls, process documentation, accounting policies, and our overall control environment.
Based on this assessment, management has concluded that our internal controls over financial reporting were effective as of December 31, 20212023 and provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external reporting purposes in accordance with GAAP. We reviewed the results of management’s assessment with the Audit Committee of our Board of Directors.
Internal control over financial reporting has inherent limitations. Internal control over financial reporting is a process that involves human diligence and compliance and is subject to lapses in judgment and breakdowns resulting from human failures. Internal control over financial reporting also can be circumvented by collusion or improper management override. Because of such limitations, there is a risk that material misstatements will not be prevented or detected on a timely basis by internal control over financial reporting. However, these inherent limitations are known features of the financial reporting process. Therefore, it is possible to design into the process safeguards to reduce, though not eliminate, this risk.
Attestation Report of the Registered Public Accounting Firm
This Annual Report does not include an attestation report of our independent registered public accounting firm regarding internal control over financial reporting. Our report was not subject to attestation by our independent registered public accounting firm pursuant to the rules of the Securities and Exchange Commission for “emerging growth companies” that permit us to provide only management’s report in this report.
Changes in Internal Control Over Financial Reporting
No change in our internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act) occurred during the three months ended December 31, 20212023 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Inherent Limitations on Effectiveness of Internal Controls
In designing and evaluating the disclosure controls and procedures, management does not expect that our internal control over financial reporting will prevent or detect all errors and all fraud. A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control systems are met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. The design of any disclosure controls and procedures also is based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions. Our management, including our Chief Executive Officer and Chief Financial Officer, believes that our disclosure controls and procedures and internal control over financial reporting are designed to provide reasonable assurance of achieving their objectives and are effective at the reasonable assurance level. However, our management does not expect that our disclosure controls and procedures or our internal control over financial reporting will prevent all errors and all fraud.
ITEM 9B. OTHER INFORMATION
None.
ITEM 9C. DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS
Not applicable.
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
OUR BOARD OF DIRECTORS
Below is a list of the names, ages as of March 10, 2022February 29, 2024 and positions of the individuals who serve on our Board of Directors.
| | | | | | | | | | | | | | |
| Name | | Age | | Position |
| Adrian Gottschalk | | 4648 | | Chief Executive Officer and Director |
| Douglas G. Cole, M.D. | | 6163 | | Chairman of the Board and Director |
| Scott Biller, Ph.D. | | 6668 | | Director |
| Balkrishan (Simba) Gill, Ph.D. | | 5759 | | Director |
Cigall Kadoch, Ph.D. | | 36 | | Director |
| Adam M. Koppel, M.D., Ph.D. | | 54 | | Director |
| Thomas J. Lynch, Jr., M.D. | | 5263 | | Director |
| Michael Mendelsohn, M.D. | | 6668 | | Director |
| B. Lynne Parshall | | 69 | | Director |
| Ian F. Smith | | 5658 | | Director |
In accordance with our amended and restated certificate of incorporation, our Board of Directors is divided into three classes of directors. At each annual meeting of stockholders, a class of directors will be elected for a three-year term to succeed the class whose terms are then expiring, to serve from the time of election and qualification until the third annual meeting following their election or until their earlier death, resignation or removal. Our directors are divided among the three classes as follows:
The Class I directors are Scott Biller, Cigall KadochThomas J. Lynch and Michael Mendelsohn, and their terms will expire at our 2024 annual meeting of stockholders.
The Class II directors are Adrian Gottschalk, Adam M. Koppel and Ian F. Smith, and their terms will expire at our 20222025 annual meeting of stockholders.
The Class III directors are Douglas G. Cole, Balkrishan (Simba) Gill and Simba Gill,B. Lynne Parshall, and their terms will expire at our 20232026 annual meeting of stockholders.
Our amended and restated certificate of incorporation provides that the authorized number of directors may be changed only by resolution of our Board of Directors. Any additional directorships resulting from an increase in the number of directors will be distributed among the three classes so that, as nearly as possible, each class will consist of one-third of the directors. The division of our Board of Directors into three classes with staggered three-year terms may delay or prevent a change of our management or a change in control.
DIRECTOR BIOGRAPHIES
Information concerning our directors is set forth below. The biographical description of each director includes the specific experience, qualifications, attributes and skills that led to the Board of Directors’ conclusion at the time of filing of this Annual Report that each person listed below should serve as a director.
DIRECTORS WITH TERMS EXPIRING IN 2022 (CLASS II DIRECTORS)
Adrian Gottschalk has served as our President, Chief Executive Officer and as a member of our Board of Directors since May 2017. Prior to joining Foghorn and since 2004, Mr. Gottschalk worked at Biogen Inc. where he was most recently a Senior Vice President and Neurodegeneration Therapeutic Area Head from November 2015 to May 2017. In this role, he was responsible for the late-stage development and commercialization of medicines for Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis, or ALS. Mr. Gottschalk holds a B.S. from Texas A&M University, an MBA from the Sloan School of Management at the Massachusetts Institute of Technology and an M.S. from the joint Harvard Medical School / Massachusetts Institute of Technology Division of Health Sciences & Technology (HST) Biomedical Enterprise Program. We believe that Mr. Gottschalk’s experience as our President and Chief Executive Officer along with over 15 years of experience in the biotechnology field provides him with the qualifications and skills necessary to serve as a member of our Board of Directors.
Adam M. Koppel, M.D., Ph.D., has served as a member of our Board of Directors since July 2017. Dr. Koppel currently serves as Managing Director of Bain Capital Life Sciences, which position he has held since June 2016. He had initially joined Bain Capital Public Equity in 2003 where he was a leader within the healthcare sector until mid-2014. During the period between mid-2014 and mid-2016, Dr. Koppel was at Biogen, where he served as Executive Vice President of Corporate Development and Chief Strategy Officer. Prior to initially joining Bain Capital in 2003, Dr. Koppel was an Associate Principal at McKinsey & Co. in New Jersey where he served a variety of healthcare companies. Dr. Koppel currently sits on the boards
of directors of Solid BioSciences, Dicerna Pharmaceuticals, Cerevel Therapeutics, Aptinyx and Viacyte. He has also previously served on the boards of Trevena and PTC Therapeutics. Dr. Koppel received an M.D. and Ph.D. in Neuroscience from the University of Pennsylvania School of Medicine. He also received an MBA from The Wharton School at the University of Pennsylvania, where he was a Palmer Scholar. He graduated magna cum laude from Harvard University with an A.B. and A.M. in History and science. We believe Dr. Koppel is qualified to serve on our Board of Directors due to his background as an executive officer, director and venture capital investor in biopharmaceutical companies, as well as his scientific and medical background.
Ian F. Smith has served as a member of our Board of Directors since April 2021. Mr. Smith currently serves as a Senior Advisor to Bain Capital Life Sciences, which position he has held since January 2021, Chair of the board of directors of Solid Biosciences since April 2020, Executive Chair of the board of directors of Viacyte, member of the board of directors of AavantiBio, and provides advisory and consulting services to these companies and other biotechnology companies. Prior to his current roles, Mr. Smith was Executive Vice President and Chief Operating Officer of Vertex Pharmaceuticals from September 2017 to January 2019 and prior to that served as Chief Financial Officer from October 2001 to September 2017. Prior to 2001, Mr. Smith was a partner in the Life Science and Technology Practice of the accounting firm Ernst & Young LLP. Mr. Smith received a B.A. with honors in accounting and finance from Manchester Metropolitan (UK). We believe Mr. Smith is qualified to serve on our Board of Directors due to his knowledge and experience across multiple roles in the biotechnology industry.
DIRECTORS WITH TERMS EXPIRING IN 2023 (CLASS III DIRECTORS)
Douglas Cole, M.D., has served as a member of our Board of Directors since October 2015. Dr. Cole joined Flagship Pioneering, which conceives, creates, resources and develops first-in-category bioplatform companies, in 2001, and is currently a Managing Partner focused on life science investments. Dr. Cole currently serves on the board of directors of Denali Therapeutics, Sigilon Therapeutics, Sana Biomedicine and a number of private companies. In the past five years, Dr. Cole served on the boards of directors of Quanterix Corporation and Editas Medicine. Dr. Cole received his M.D. from the University of Pennsylvania School of Medicine and his B.A. in English from Dartmouth College. We believe Dr. Cole is qualified to sit on our Board of Directors given his substantial experience as an investor in emerging biopharmaceutical and life sciences companies as well as his experience serving on the boards of directors of multiple public and private biopharmaceutical companies.
Balkrishan (Simba) Gill, Ph.D., has served as a member of our Board of Directors since July 2017. Dr. Gill is the President, Chief Executive Officer and a member of the board of directors of Evelo Biosciences, which positions he has held since June 2015. In addition, Dr. Gill has served as the Executive Chairman of Blackfynn Inc. since 2015. Dr. Gill has also served as a venture partner at Flagship Pioneering, an innovation enterprise that conceives, creates, resources and develops first-in-category life sciences companies, since 2015. From 2016 to 2019, Dr. Gill served on the board of directors of Realm Therapeutics PLC. Dr. Gill received his Ph.D. from King’s College, London and his MBA from INSEAD. We believe Dr. Gill is qualified to serve on our Board of Directors due to his knowledge and experience in the venture capital and pharmaceutical industries.
DIRECTORS WITH TERMS EXPIRING IN 2024 (CLASS I DIRECTORS)
Scott Biller, Ph.D., has served as a member of our Board of Directors since January 2020. Dr. Biller currently serves as a Senior Advisor for Agios Pharmaceuticals, where he previously served as the company’s Chief Scientific Officer from September 2010 to December 2019. Dr. Biller is also currently the sole proprietor of Biller Consulting, a consulting company serving the biopharmaceutical industry, and since December 2021 he has served as an Executive Venture Partner at the venture capital fund GV. From 2003 to 2010, Dr. Biller was Vice President and Head of Global Discovery Chemistry at the Novartis Institutes for Biomedical Research. Prior to that, Dr. Biller held the positions of Vice President, Pharmaceutical Candidate Optimization at the Bristol Myers Squibb, or BMS, Pharmaceutical Research Institute and Executive Director of Drug Discovery chemistry for the BMS research site in Lawrenceville, New Jersey. Since June 2020, Dr. Biller has served on the Board of Directors of Remix Therapeutics and, since February 2021, has served on the Board of Directors of Rome Therapeutics. Dr. Biller earned a S.B. degree from the Massachusetts Institute of Technology, a Ph.D. from Caltech and was an NIH Postdoctoral Fellow at Columbia University in natural product synthesis. We believe Dr. Biller is qualified to serve on our Board of Directors because of his extensive experience in drug discovery and development and his significant leadership experience in the biotechnology industry.
Thomas J. Lynch, Jr., M.D.our academic co-founder,, has served as a member of our Board of Directors since October 2022. Dr. Lynch is a world-renowned scientist, highly respected oncologist and successful leader of National Cancer Institute-Designated Comprehensive Cancer Centers. Since February 2020, Dr. Lynch has served as president and director of Fred Hutchinson Cancer Research Center, where he sets the strategic direction of the center, oversees center-wide initiatives and represents the Hutch’s interests to major partners and governmental bodies. He also directs the Fred Hutch/University of Washington Cancer Consortium and is principal investigator of its Cancer Center Support Grant. Before taking over as the Hutch’s sixth president, Dr. Lynch was most recently chief scientific officer of Bristol-Myers Squibb from March 2016. She is currently Associate Professor2017 to October 2019. Prior to that he held leadership roles as CEO of Pediatric OncologyMassachusetts General Physicians Organization from September 2015 to March 2017, director of Yale Cancer Center and physician-in-chief at the Dana-FarberYale’s Smilow Cancer Institute, which position she has held since January 2014.Hospital from April 2009 through March 2015. Prior to those positions, Dr. Kadoch also currently serves as Associate ProfessorLynch was chief of Pediatrics,hematology-oncology at Massachusetts General Hospital and professor of medicine at Harvard Medical School, which she has held since April 2014School. Dr. Lynch is a member of the American Association for Cancer Research, the American Society of Clinical Oncology, and as Institute Member and Epigenomics Program Co-Director at the Broad InstituteInternational Association for the Study of Massachusetts InstituteLung Cancer. Earlier in his career he was part of Technology and Harvard University, which she has held since April 2014. In September 2021, Dr. Kadoch was named a Howard Hughes Medical Institute Investigator. Dr. Kadoch received her B.A. from the University of California, Berkeley and her Ph.D. from Stanford University School of Medicine.first research team to discover how targeted therapies could dramatically change outcomes for lung cancer patients with mutations in the EGFR, or epidermal growth factor receptor, gene. We believe that Dr. KadochLynch is qualified to serve on our Board of Directors due to her expertise andbecause of his extensive experience as our academic co-foundera clinician and her deep understanding ofscientist, as well as his leadership experience in the role of chromatin regulation in human cancer and other serious diseases.oncology space.
Michael Mendelsohn, M.D., has served as a member of our Board of Directors since April 2017. Dr. Mendelsohn is also member of the Founder and Board of Directors of Cyclerion Pharmaceuticals, where he has served since April 2019,Chairman, as well as the Executive Chairman and PresidentChief Strategy Officer, of Cardurion Pharmaceuticals, where he has served since May 2016. He is also a Professor of Medicine and Cardiology at Tufts Medical Center and Tufts University School of Medicine. Since April 2015, Dr. Mendelsohn has also beenserved as a senior advisor and consultant to the chief medicalHead of Research and scientific officer ofDevelopment at Takeda Pharmaceutical Co. Ltd. From December 2014 to December 2018, he served as senior advisor and consultantwas Senior Advisor, Consultant and a member of the pharmaceuticals advisory committeePharmaceuticals Advisory Committee at Ironwood Pharmaceuticals for the chief scientific officer and president of research and development at Ironwood Pharmaceuticals.R&D. From May 2014 to July 2017, Dr. Mendelsohn was a venture partner for SV Health Investors. Prior to that, fromFrom June 2010 to November 2013, Dr. Mendelsohn served as Senior Vice President and Global Head of Cardiovascular Research at Merck Research Laboratories. From 1993 to 2010, Dr. Mendelsohn was a faculty memberProfessor of Medicine and Cardiology at Tufts Medical Center and Tufts University School of Medicine, where he founded and was the executive director of the Molecular Cardiology Research Institute from 1997 to 2010. From 2008 to 2010, andhe also served as Chief Scientific Officer from 2008at Tufts. From 1987 to 2010.1993, Dr. Mendelsohn was previously a member of the cardiovascular faculty of the Cardiovascular Division at Brigham and Women’s Hospital and Harvard Medical School. Dr. Mendelsohn received a B.A. in Chemistry and English from Amherst College and an M.D. from Harvard Medical School. We believe Dr. Mendelsohn is qualified to serve on our Board of Directors because of his extensive experience as a clinician and scientist, along with experience and insights as an active advisor and consultant to leadership in research and development for multinational biopharmaceutical companies.
DIRECTORS WITH TERMS EXPIRING IN 2025 (CLASS II DIRECTORS)
Adrian Gottschalk has served as our President, Chief Executive Officer and as a member of our Board of Directors since May 2017. Prior to joining Foghorn and since 2004, Mr. Gottschalk worked at Biogen Inc. where he was most recently a Senior Vice President and Neurodegeneration Therapeutic Area Head from November 2015 to May 2017. In this role, he was responsible for the late-stage development and commercialization of medicines for Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis, or ALS. Mr. Gottschalk holds a B.S. from Texas A&M University, an MBA from the Sloan School of Management at the Massachusetts Institute of Technology and an M.S. from the joint Harvard Medical School / Massachusetts Institute of Technology Division of Health Sciences & Technology (HST) Biomedical Enterprise Program. We believe that Mr. Gottschalk’s experience as our President and Chief Executive Officer along with 20 years of experience in the biotechnology field provides him with the qualifications and skills necessary to serve as a member of our Board of Directors.
Adam M. Koppel, M.D., Ph.D.,has served as a member of Foghorn’s board of directors since July 2017. Dr. Koppel currently serves as a partner of Bain Capital Life Sciences, a position he has held since June 2016. He initially joined Bain Capital Public Equity in 2003, where he was a leader within the healthcare sector until 2014. From 2014 to 2016, Dr. Koppel was executive vice president of corporate development and chief strategy officer at Biogen, Inc. Prior to joining Bain Capital Public Equity in 2003, Dr. Koppel was an associate principal at McKinsey & Co. in New Jersey where he served a variety of healthcare companies. Dr. Koppel sits on the board of directors of Solid Biosciences, Inc. and Cerevel Therapeutics Holdings, Inc. He also serves on the board of directors of Areteia Therapeutics, Inc. and Cardurion Pharmaceuticals, Inc., both private companies. Dr. Koppel previously served on the board of directors of BCLS Acquisition Corp., Trevena, Inc., Aptinyx Inc. and Dicerna Pharmaceuticals, Inc. Dr. Koppel graduated magna cum laude from Harvard University with an A.B. and A.M. in history and science. He received an M.D. and Ph.D. in neuroscience from the University of Pennsylvania School of Medicine and an M.B.A. from The Wharton School at the University of Pennsylvania, where he was a Palmer Scholar. We believe Dr. Koppel is qualified to serve on our Board of Directors due to his background as an executive officer, director and venture capital investor in biopharmaceutical companies, as well as his scientific and medical background.
Ian F. Smith has served as a member of our Board of Directors since April 2021. Mr. Smith currently serves as a Senior Advisor to Bain Capital Life Sciences, which position he has held since January 2021. He has also served as Executive Chair of the board of directors of Solid Biosciences since April 2020, Chairman of the board of directors of iVexSol Inc. since August 2023, the Chairman of the board of directors of Rivus Pharmaceuticals since November 2023, and as a board member of Alkeus Pharmaceuticals and Stoke Therapeutics since December 2022 and September 2023, respectively. Mr. Smith also provides advisory and consulting services to these companies and other biotechnology companies. Prior to his current roles, Mr. Smith was Executive Vice President and Chief Operating Officer of Vertex Pharmaceuticals from September 2017 to January 2019 and prior to that served as Chief Financial Officer from October 2001 to September 2017. Prior to 2001, Mr. Smith was a partner in the Life Science and Technology Practice of the accounting firm Ernst & Young LLP. Mr. Smith received a B.A. with honors in accounting and finance from Manchester Metropolitan (UK). We believe Mr. Smith is qualified to serve on our Board of Directors due to his knowledge and experience across multiple roles in the biotechnology industry.
DIRECTORS WITH TERMS EXPIRING IN 2026 (CLASS III DIRECTORS)
Douglas Cole, M.D.,has served as a member of our Board of Directors since October 2015. Dr. Cole joined Flagship Pioneering, which conceives, creates, resources and develops first-in-category bioplatform companies, in 2001, and is currently a Managing Partner focused on life science investments. Dr. Cole currently serves on the board of directors of Sana Biomedicine and a number of private companies. In the past five years, Dr. Cole served on the boards of directors of Denali Therapeutics, Sigilon Therapeutics, Quanterix Corporation and Editas Medicine. Dr. Cole received his M.D. from the University of Pennsylvania School of Medicine and his B.A. in English from Dartmouth College. We believe Dr. Cole is qualified to sit on our Board of Directors given his substantial experience as an investor in emerging biopharmaceutical and life sciences companies as well as his experience serving on the boards of directors of multiple public and private biopharmaceutical companies.
Balkrishan (Simba) Gill, Ph.D.,has served as a member of our Board of Directors since July 2017. Dr. Gill was the CEO of Evelo Biosciences from 2015 to 2023. During the same period Dr. Gill was a special advisor and senior partner at Flagship Pioneering, an innovation enterprise that conceives, creates, resources and develops first-in-category life sciences companies. From 2016 to 2019, Dr. Gill served on the board of directors of Realm Therapeutics PLC. Dr. Gill received his Ph.D. from King’s College, London and his MBA from INSEAD. We believe Dr. Gill is qualified to serve on our Board of Directors due to his knowledge and experience in the venture capital and pharmaceutical industries.
B. Lynne Parshall has served as a member of our Board of Directors since August 2022. She has also served as a Director of Ionis Pharmaceuticals, Inc. since September 2000 and as a Senior Strategic Advisor to Ionis from January 2018 to December 2022. Previously she served as Ionis’s Chief Operating Officer from December 2007 through January 2018 and as its Chief Financial Officer from June 1994 through December 2012. She also served as its Corporate Secretary through 2014 and has served with Ionis in various executive roles since November 1991. Prior to joining Ionis, Ms. Parshall practiced law at Cooley LLP, where she was a partner from 1986 to 1991. Ms. Parshall has also served as the CEO of Lyme Pinnacle Consulting LLC, a strategic consulting firm in the life sciences industry, since 2018. Ms. Parshall has served on the boards of directors of Cytokinetics, Inc., a public biopharmaceutical company, since February 2013; Repertoire Immune Medicines, Inc., a private company in the life sciences industry, since 2021; Ring Therapeutics, Inc., a private company in the life sciences industry, since 2022; Alltrna, Inc., a private company in the life sciences industry since 2023; and Celdara Medical, LLC, a private company in the life sciences industry, since 2023. She also served on the board of directors of Akcea Therapeutics, Inc., a public biopharmaceutical company, from January 2015 until its acquisition by Ionis in October 2020. Ms. Parshall received her NACD Directorship Certification in 2023. Ms. Parshall received an A.B. from Harvard University and her J.D. from Stanford University. We believe Ms. Parshall is qualified to serve on our Board of Directors due to her knowledge and experience in the life sciences industry as well as her experience serving as a director on other public and private company boards.
Board Membership Criteria
Our Nominating and Corporate Governance Committee is responsible for developing and recommending to our Board of Directors criteria for Board membership and, consistent with those criteria, recommending to the Board of Directors director candidates and nominees for the next annual meeting of stockholders. As reflected in our Corporate Governance Guidelines, it is the policy of the Board of Directors that all directors should possess the highest personal and professional ethics, integrity and values, and be committed to representing the long-term interests of the Company’s stockholders. The Board of Directors believes that each director should possess the requisite ability, judgment and experience to oversee the Company’s business, and should contribute to the overall diversity of the Board of Directors. The Board of Directors considers the qualifications of directors and director candidates individually and in the broader context of its overall composition and the Company’s current and anticipated future needs. Stockholders may also nominate persons to be elected as directors in accordance with our bylaws and applicable law. The Nominating and Corporate Governance Committee does not have a written policy regarding
stockholder nominations, but it has determined that it is the practice of the committee to consider candidates proposed by stockholders if made in accordance with our bylaws.
Board Meetings and Attendance
The Board of Directors held fourseven meetings during the year ended December 31, 2021.2023. Each of the directors attended at least seventy-five percent (75%) of the meetings of the Board of Directors and the committees of the Board of Directors on which he or she served during the year ended December 31, 20212023 (in each case, which were held during the period for which he or she was a director and/or a member of the applicable committee and excluding any meetings in which a director was an interested party).
The non-employee directors met in executive session during each of the regularly scheduled Board of Directors meetings during the year ended December 31, 2021.2023.
Director Attendance at Annual Meeting of Stockholders
One director attended our 20212023 annual meeting, which was held as a virtual-only meeting due to the ongoing COVID-19 pandemic.meeting.
Role of the Board in Risk Oversight
Our Board of Directors has an active role, as a whole and also at the committee level, in overseeing the management of our risks. Our Board of Directors is responsible for general oversight of risks and regular review of information regarding our risks, including liquidity risks and operational risks. The compensation committee is responsible for overseeing the management of risks relating to our executive compensation plans and arrangements. The audit committee is responsible for overseeing the management of risks relating to accounting matters and financial reporting. The nominating and governance committee is responsible for overseeing the management of risks associated with the independence of our Board of Directors and potential conflicts of interest. Although each committee is responsible for evaluating certain risks and overseeing the management of such risks, the entire Board of Directors is regularly informed through discussions from committee members about such risks. Our Board of Directors believes its administration of its risk oversight function has not negatively affected our Board of Directors’ leadership structure.
BOARD COMMITTEES
Our Board of Directors has established an audit committee, a compensation committee and a nominating and corporate governance committee, each of which operateoperates pursuant to a charter adopted by our Board of Directors. The Board of Directors may also establish other committees from time to time to assist us and the Board of Directors in their duties. The composition and functioning of all of our committees comply with all applicable requirements of the Sarbanes-Oxley Act, the Nasdaq Stock Market and the Exchange Act. Each committee’s charter is available on the corporate governance section of our website at
https://foghorntx.com. Information contained on our website is not incorporated by reference into this prospectus,Annual Report on Form 10-K, and you should not consider information contained on our website to be part of this prospectus or in deciding whether to purchase shares of our common stock.
The following table describes which directors serve on each of the Board of Directors’ committees.
| | | | | | | | | | | | | | | | | | | | |
| Name | | Audit Committee | | Compensation Committee | | Nominating and Corporate Governance Committee |
| Adrian Gottschalk | | | | | | |
| Douglas G. Cole, M.D. | | | | X | | X(1) |
| Scott Biller, Ph.D. | | | | | | |
| Balkrishan (Simba) Gill, Ph.D. | | X | | X(1) | | |
Cigall Kadoch, Ph.D. | | | | | | |
| Adam M. Koppel, M.D., Ph.D. | | X | | X(1) | | | X |
| Thomas J. Lynch, Jr., M.D. | | | | | | |
| Michael Mendelsohn, M.D. | |
| | X | | |
| B. Lynne Parshall | | XX(1) | | | | X |
| Ian F. Smith | | | | | | |
| | | | | | |
| (1) Chair of the committee | | | | | | |
Audit Committee
Our Audit Committee is composed of Adam M. Koppel, SimbaBalkrishan (Simba) Gill and Michael Mendelsohn,B. Lynne Parshall, with Dr. KoppelMs. Parshall serving as Chair of the committee. The Board of Directors has determined that each member of the Audit Committee meets the independence requirements of Rule 10A-3 under the Exchange Act and the applicable listing standards of Nasdaq. The Board of Directors has determined that each of Drs. Koppel and Gill and Ms. Parshall is an “audit committee financial expert” within the meaning of the SEC regulations and applicable listing standards of Nasdaq. The audit committee’s responsibilities include:
• appointing, approving the compensation of, and evaluating the qualifications, performance and independence of our independent registered public accounting firm;
• pre-approving all audit and permitted non-audit services to be performed by our independent registered public accounting firm;
• overseeing the work of our independent registered public accounting firm, including through the receipt and consideration of reports from such firm;
• reviewing and discussing with management and our independent registered public accounting firm our annual and quarterly financial statements and related disclosures, including earnings releases;
• reviewing and discussing with management and our independent registered public accounting firm any material issues regarding accounting principles and financial statement presentations;
• coordinating our Board of Directors’ oversight of our internal control over financial reporting, disclosure controls and procedures, code of business conduct and ethics, procedures for complaints and legal and regulatory matters;
• discussing our risk management policies with management;
• establishing policies regarding hiring employees from our independent registered public accounting firm and procedures for the receipt and retention of accounting related complaints and concerns;
• meeting independently with our independent registered public accounting firm and management;
• reviewing and approving any related person transactions;
• overseeing our guidelines and policies governing risk assessment and risk management;
• overseeing the integrity of our information technology systems, process and data;
• preparing the audit committee report required by SEC rules;
• reviewing and assessing, at least annually, the adequacy of the Audit Committee’s charter; and
• performing, at least annually, an evaluation of the performance of the Audit Committee.
During the year ended December 31, 2023, the Audit Committee met four times.
During the year ended December 31, 2021, the Audit Committee met four times.
Nominating and Governance Committee
Our Nominating and Governance Committee is composed of Douglas G. Cole, Adam M. Koppel and Adam Koppel,B. Lynne Parshall, with Dr. Cole serving as Chair of the committee. Our nominating and corporate governance committee’s responsibilities include:
• identifying individuals qualified to become members of our Board of Directors consistent with criteria approved by the board and receiving nominations for such qualified individuals;
• recommending to our Board of Directors the persons to be nominated for election as directors and to each of its committees;
• establishing a policy under which our shareholders may recommend a candidate to the nominating and corporate governance committee for consideration for nomination as a director;
• reviewing and recommending committee slates on an annual basis;
• recommending to our Board of Directors qualified candidates to fill vacancies on our Board of Directors;
• developing and recommending to our Board of Directors a set of corporate governance principals applicable to us and reviewing the principles on at least an annual basis;
• reviewing and making recommendations to our boardBoard of Directors with respect to our board leadership structure and board committee structure;
• reviewing, in concert with our Board of Directors, our policies with respect to significant issues of corporate public responsibility;
• making recommendations to our Board of Directors processes for annual evaluations of the performance of our Board of Directors, our chief executive officer and committees of our Board of Directors;
• overseeing the process for annual evaluations of our Board of Directors, chief executive officer and committees of our Board of Directors and certifying that performance of our chief executive officer and other members of executive management is being properly evaluated;
• considering and reporting to our Board of Directors any questions of possible conflicts of interest of members of our Board of Directors;
• providing new director orientation and continuing education for existing directors on a periodic basis;
• overseeing the maintenance and presentation to our Board of Directors of management’s plans for succession to senior management positions in the Company;
• reviewing and assessing, at least annually, the adequacy of the nominating and corporate governance committee’s charter; and
• performing, on an annual basis, an evaluation of the performance of the nominating and corporate governance committee.
During the year ended December 31, 2021,2023, the Nominating and Corporate Governance Committee met fivefour times.
Compensation Committee
Our Compensation Committee is composed of SimbaBalkrishan (Simba) Gill, Douglas G. Cole and Michael Mendelsohn, with Dr. Gill serving as Chair of the committee. The Board of Directors has determined that each member of the Compensation Committee is “independent” as defined under the applicable listing standards of Nasdaq and meets the independence criteria set forth in Rule 10C-1 under the Exchange Act. Our Compensation Committee’s responsibilities include:
• assisting our Board of Directors in developing and reviewing potential candidates for executive positions;
• reviewing our overall compensation strategy, including base salary, incentive compensation and equity-based grants;
• reviewing, determining and approving corporate, individual and other goals and objectives relevant to compensation of our chief executive officer and approving the compensation of the CEO;
• reviewing and approving the compensation of our other executive officers;
• reviewing and making recommendations to the Board of Directors with respect to director compensation;
• overseeing and administering our cash and equity incentive plans;
• reviewing, considering and selecting, to the extent determined to be advisable, a peer group of appropriate companies for purposingpurposes of benchmarking and analysis of compensation for our executive officers and directors;
• reviewing and approving all employment contractcontracts and other compensation, severance and change-in-control arrangements for our executive officers;
• recommending to our Board of Directors any stock ownership guidelines for our executive officers and non-employee directors;
• retaining, appointing or obtaining advice of a compensation consultant, legal counsel or other advisor, and determining the compensation and independence of such consultant or advisor;
• preparing, if required, the compensation committee report on executive compensation for inclusion in our annual proxy statement in accordance with the proxy rules;
• monitoring our compliance with the requirements of Sarbanes-Oxley relating to loans to directors and officers;
• overseeing our compliance with applicable SEC rules regarding shareholder approval of certain executive compensation matters;
• reviewing the risks associated with our compensation policies and practices;
• reviewing and assessing, at least annually, the adequacy of the compensation committee’s charter; and
• performing, on an annual basis, an evaluation of the performance of the compensation committee.
During the year ended December 31, 2021,2023, the Compensation Committee met five times.
Compensation ConsultantsConsultant
As a part of determining compensation for our executive officers and directors, the Compensation Committee engaged Pay Governance, LLC or (“Pay Governance,Governance”) as its independent compensation consultant during 2021.2023. Pay Governance provided analysis and recommendations to the Compensation Committee regarding cash and equity compensation for such officers and directors.
In determining to engage Pay Governance, the Compensation Committee considered its independence, taking into consideration relevant factors, including the absence of other services provided to the Company, the amount of fees the Company paid to Pay Governance as a percentage of its respective total revenue, Pay Governance’s policies and procedures that are designed to prevent conflicts of interest, any business or personal relationship any individual compensation advisor has with an executive officer of the Company, any business or personal relationship any individual compensation advisor has with any member of the Compensation Committee and any stock of the Company owned by Pay Governance or its individual compensation advisors. The Compensation Committee determined, based on its analysis in light of all relevant factors, including the factors listed above, that the work of Pay Governance and its individual compensation advisors as compensation consultants to the Compensation Committee has not created any conflicts of interest, and that Pay Governance is independent pursuant to the independence standards set forth in the Nasdaq listing standards promulgated pursuant to Section 10C of the Exchange Act.
Delegation of Authority and the Role of Management
The Compensation Committee may delegate to subcommittees, consisting of one or more members of the Compensation Committee, any of the responsibilities of the full committee.
Our Chief Executive Officer makes compensation-related recommendations to the Compensation Committee with respect to annual base salary, target bonus opportunities and long-term incentive award grants for the named executive officers (other than himself). No member of the management team, including our Chief Executive Officer, has a role in determining his or her own compensation.
Code of Ethics and Conduct
We have adopted a written code of business conduct and ethics that applies to our directors, officers and employees, including our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions. A current copy of the code is posted on the investor section of our website. In addition, we intend to post on our website all disclosures that are required by law or listing rules concerning any amendments to, or waivers from, any provision of the code.
Policy on Clawback and Recovery of Compensation
In 2023, we adopted a clawback policy (the “Clawback Policy”) in compliance with the requirements of the Dodd-Frank Act, final SEC rules and applicable Nasdaq listing standards, which covers our current and former executive officers. Under the Clawback Policy, if there is a restatement of our financial results due to material noncompliance with financial reporting
requirements, certain incentive-based compensation paid or awarded to covered executives will be subject to reduction and/or repayment if the amount of such compensation was calculated based upon the achievement of financial results that were the subject of the restatement and the amount of such compensation that would have been received by the covered executives had the financial results been properly reported would have been lower than the amount actually awarded.
OUR EXECUTIVE OFFICERS
Below is a list of the names, ages as of March 10, 2022,February 29, 2024, and positions, and a brief account of the business experience of the individuals who serve as our executive officers.
| | | | | | | | | | | | | | | | | |
| Name | | Age | | Position | |
| Adrian Gottschalk | | 4648 | | Chief Executive Officer and Director (Class II) | |
| Stephen DiPalma | | Allan Reine, M.D.65 | | 47 | | Interim Chief Financial Officer | |
Samuel Agresta, M.D. | | 49 | | Chief Medical Officer | |
| Steven Bellon, Ph.D. | | 58 | | 57 | | Chief Scientific Officer | |
| Fanny Cavalie | | 46 | |
Michael LaCascia | | 57 | | Chief Legal Officer | |
Fanny Cavalie | | 44 | | Chief Strategy and Business Operations Officer | |
| Carlos Costa | | 51 | | Chief People Officer | |
| Michael LaCascia | | 59 | | Chief Legal Officer | |
| Alfonso Quintás-Cardama, M.D. | | 53 | | Chief Medical Officer | |
EXECUTIVE OFFICER BIOGRAPHIES
Biographical information for Adrian Gottschalk, our Chief Executive Officer and President, is included herein under “Director Biographies—Directors with Terms Expiring in 20222025 (Class II Directors).”
Allan Reine, M.D.,Stephen DiPalma has served as our Interim Chief Financial Officer since September 2019. Prior to joining Foghorn, Dr. Reine served as Chief Financial Officer of Pieris Pharmaceuticals from August 2017 to September 2019. From August 2012 through August 2017, Dr. Reine was a portfolio manager at Lombard Odier Asset Management, where he ran a healthcare portfolio focused on biotechnology and pharmaceutical companies. Before joining Lombard Odier, from 2003 through August 2012, Dr. ReineJanuary 2024. He has served as a healthcare portfolio managermanaging director at various funds, including Citi Principal Strategies, SAC Capital, Trivium CapitalDanforth Advisors, LLC, a consultancy firm specializing in working with life sciences companies, since April 2014. Prior to and Alexandra Investment Management. Dr. Reine beganduring his career in 2001tenure at CIBC World Markets where he worked in both biotechnology investment banking and biotechnology equity research. Dr. Reine is currently a member of the board of directors of ONK Pharmaceuticals where heDanforth, Mr. DiPalma has served since March 2022. Dr. Reineas chief financial officer to a number of public and privately held companies. Mr. DiPalma received his M.D.a B.S. in finance and management information systems from the University of TorontoMassachusetts-Lowell and his B.S. in Statistical Sciencesan M.B.A. from the University of Western Ontario.Babson College.
Steven Bellon, Ph.D., has served as our Chief Scientific Officer since January 2022. He previously served in the role of Senior Vice President, Drug Discovery, since October 2019, and prior to that served in the role of Vice President, Drug Discovery since June 2016. Prior to joining Foghorn, Dr. Bellon was Senior Director and Executive Director of Constellation Pharmaceuticals, starting in September 2008. At Constellation Pharmaceuticals, Dr. Bellon built the structural biology group and the bromodomain platform. Dr. Bellon has also held positions at Amgen and Vertex Pharmaceuticals and has more than 2025 years of experience in drug discovery. Dr. Bellon received his B.S. from Haverford College and his Ph.D. from the Massachusetts Institute of Technology.
Samuel Agresta, M.D., M.P.H. & T.M.,has served as our Chief Medical Officer since September 2019. Prior to joining Foghorn, Dr. Agresta served as Chief Medical Officer of Infinity Pharmaceuticals from August 2018 to August 2019. Prior to that, Dr. Agresta was Vice President, Clinical Development of Agios Pharmaceuticals from December 2011 to August 2018. During that time, he led the development of the IDH inhibitors from drug candidate stage to FDA approval. Dr. Agresta is currently a member of the board of directors of Infinity Pharmaceuticals where he served since September 2019. Dr. Agresta received his B.S. from Georgetown University and his M.D. and M.P.H. & T.M. from Tulane University.
Michael LaCascia has served as our Chief Legal Officer since November 2020. Prior to joining Foghorn, Mr. LaCascia served as Senior Vice President and General Counsel from May 2019 to April 2020 and Chief Legal Officer from April 2020 through October 2020 of Q-State BioSciences, Inc. From January 2014 through October 2015, Mr. LaCascia was a Vice President, Corporate Legal at Vertex Pharmaceuticals Incorporated and later became Vertex’s Senior Vice President and General Counsel and Corporate Secretary from October 2015 through February 2019. Prior to his time at Vertex, Mr. LaCascia was an equity partner at WilmerHale and an adjunct lecturer at Boston University School of Law. Mr. LaCascia received his A.B. from Harvard College and his J.D. from Boston University School of Law.
Fanny Cavalie has served as our Chief Strategy and Business Operations Officer since February 2022. She previously served in the role of Senior Vice President, Business and Operations, since August 2020, and prior to that served in the role of Vice President, Business and Operations, since March 2018. Prior to joining Foghorn, Ms. Cavalie was Senior Director, Global Commercial Development Lead, Alzheimer's Disease at Biogen Inc., where she led the global marketing and new product planning team for a multiple-asset portfolio. Ms. Cavalie also worked for eleven years in the healthcare practice of McKinsey & Company. Ms. Cavalie received her M.B.A. with a concentration in International Management from Hautes Etudes Commerciales Paris.
Carlos Costa has served as our Chief People Officer since August 2022. He previously served as our SVP and VP of People & Organization. Prior to joining the company, Carlos held various roles of increasing responsibility at Biogen, in multiple areas and geographies ranging from human resources business partner for Spain, Portugal, and Italy, to HR lead for global commercial functions in the United States and the Emerging Markets region. In his most recent role at Biogen, Mr. Costa was the head of human resources for Europe, Canada, and International Partner Markets. Before joining Biogen, he spent 10 years serving as HR business partner in companies like Gillette and Roche, where he built teams, led transformations and served as strategic partner to the business. Mr. Costa holds a BS in law from UNED in Spain and received his human resources master’s degree from EADA in Barcelona. He also holds a PDD from IE Instituto de Empresa, Madrid and the Executive Leadership Program at Harvard Business School, Boston.
Michael LaCascia has served as our Chief Legal Officer since November 2020. Prior to joining Foghorn, Mr. LaCascia served as Senior Vice President and General Counsel from May 2019 to April 2020 and Chief Legal Officer from April 2020 through October 2020 of Q-State BioSciences, Inc. From January 2014 through October 2015, Mr. LaCascia was a Vice President, Corporate Legal at Vertex Pharmaceuticals Incorporated and later became Vertex’s Senior Vice President and General Counsel and Corporate Secretary from October 2015 through February 2019. Prior to his time at Vertex, Mr. LaCascia was an equity
partner at WilmerHale and an adjunct lecturer at Boston University School of Law. Mr. LaCascia received his A.B. from Harvard College and his J.D. from Boston University School of Law.
Alfonso Quintás-Cardama, M.D., has served as our Chief Medical Officer since September 2023. Dr. Quintás-Cardama previously served as Chief Medical Officer of TCR2 Therapeutics Inc., a position he held since 2017. Prior to that, he was the Clinical Development Head of the Cell & Gene Therapies Unit at GlaxoSmithKline PLC in 2017. Between 2014 and 2016, Dr. Quintás-Cardama served as Global Clinical Leader, Cell & Gene Therapy, at Novartis AG and was an Assistant Professor in the Department of Leukemia at the University of Texas, MD Anderson Cancer Center from 2009 to 2014. Dr. Quintás-Cardama received his M.D. from the Universidad de Santiago de Compostela School of Medicine in Spain. He completed an internship and residency in the Department of Medicine of the Albert Einstein College of Medicine, and a hematology and oncology fellowship and a leukemia fellowship at the University of Texas, MD Anderson Cancer Center.
ITEM 11. EXECUTIVE COMPENSATION
EXECUTIVE OFFICER AND DIRECTOR COMPENSATION
Introduction
This section provides an overview of the compensation awarded to, earned by, or paid to our principal executive officer and our next threetwo most highly compensated executive officers, listed below, in respect of their service to us for the fiscal year ended December 31, 2021.2023. We refer to these individuals as our named executive officers. Our named executive officers are:
• Adrian Gottschalk, our President and Chief Executive Officer;
• Allan Reine, M.D.Steven Bellon, Ph.D., our Chief FinancialScientific Officer; and
• Carl Decicco, Ph.D.Alfonso Quintás-Cardama, our former Chief Scientific Officer*;
• Samuel Agresta, M.D., our Chief Medical Officer.
* Dr. Decicco stepped down from his position as Chief Scientific Officer effective January 10, 2022, and served as a Special Advisor to our newly appointed Chief Scientific Officer until January 31, 2022, at which date he retired from the Company.
The Compensation Committee of our Board of Directors was responsible for determining the compensation of our executive officers during fiscal year 2021,2023, subject, in the case of our Chief Executive Officer, to the approval of our Board of Directors. Our Chief Executive Officer made recommendations to the Compensation Committee about the compensation of his direct reports in respect of fiscal years 20212023 and 2020.2022.
Summary Compensation Table
The following table sets forth the compensation awarded to, earned by, or paid to our named executive officers in respect of their service to us for the fiscal years ended December 31, 20212023 and 2020:2022, as applicable:
| | | | | | | | | | | | | | | | | | | | | | | |
Name and principal position | Year |
Salary ($)(1) |
Bonus ($) |
Option awards ($)(2) | Non-equity incentive plan compensation ($)(3) |
All other compensation ($)(4) |
Total ($) |
Adrian Gottschalk President and Chief Executive Officer | 2021 | $ | 540,000 | | — | | $ | 3,370,482 | | $ | 405,000 | | $ | — | | $ | 4,315,482 | |
| 2020 | $ | 478,950 | | — | | $ | 1,834,007 | | $ | 311,318 | | $ | — | | $ | 2,624,275 | |
Allan Reine, M.D. Chief Financial Officer | 2021 | $ | 416,000 | | — | | $ | 1,348,746 | | $ | 249,600 | | | $ | 2,014,346 | |
| | | | | | | |
Carl Decicco, Ph.D Former Chief Scientific Officer(5) | 2021 | $ | 428,500 | | — | | $ | 1,124,004 | | $ | 257,100 | | $ | — | | $ | 1,809,604 | |
| 2020 | $ | 412,000 | | — | | $ | 542,224 | | $ | 214,240 | | $ | 50,000 | | $ | 1,218,464 | |
Samuel Agresta, M.D. Chief Medical Officer | 2021 | $ | 428,500 | | — | | $ | 1,124,004 | | $ | 257,100 | | $ | — | | $ | 1,809,604 | |
| 2020 | $ | 412,000 | | — | | $ | 542,224 | | $ | 99,573 | | $ | — | | $ | 1,053,797 | |
| | | | | | | | | | | | | | | | | | | | | | | |
Name and principal position | Year |
Salary ($)(1) |
Bonus ($)(2) |
Option awards ($)(3) | Non-equity incentive plan compensation ($)(4) |
All other compensation ($)(5) |
Total ($) |
Adrian Gottschalk President and Chief Executive Officer | 2023 | $ | 579,000 | | — | | $ | 1,719,292 | | $ | 202,650 | | $ | 3,000 | | $ | 2,503,942 | |
| 2022 | $ | 575,000 | | — | | $ | 4,645,819 | | $ | 215,625 | | $ | — | | $ | 5,436,444 | |
Steven Bellon, Ph.D. Chief Scientific Officer | 2023 | $ | 445,000 | | — | | $ | 614,894 | | $ | 124,600 | | $ | 3,000 | | $ | 1,187,494 | |
| 2022 | $ | 435,000 | | — | | $ | 1,580,167 | | $ | 130,500 | | $ | — | | $ | 2,145,667 | |
Alfonso Quintás-Cardama, M.D. (6) Chief Medical Officer | 2023 | $ | 144,432 | | $ | 110,000 | | $ | 1,544,374 | | $ | 43,400 | | $ | 2,329 | | $ | 1,844,535 | |
(1) The amount reported for Mr. Gottschalk and Drs. Reine, Decicco,Bellon and AgrestaQuintás-Cardama includes employee contributions made to
our 401(k) plan.
(2) The amount reported for Dr. Quintás-Cardama reflects a sign-on bonus.
(3) The amounts reported represent the aggregate grant date fair value of options to purchase our common stock granted to
Mr. Gottschalk and Drs. Reine, DeciccoBellon and AgrestaQuintás-Cardama in fiscal year 20212023 and/or 2020,2022, as applicable, computed in accordance
with FASB ASC 718, excluding the effect of estimated forfeitures. The assumptions used to value the stock options for
this purpose are set forth in Note 87 to our consolidated financial statements included elsewhere in this Annual Report.Report on Form 10-K.
(3)(4) The amounts reported represent the annual bonus earned by each of the named executive officers for the relevant fiscal
year, to the extent applicable, based on the attainment of corporate performance goals as described below under “Annual
Bonuses.”
(4) The amount reported for Dr. Decicco reflects a travel and lodging allowance.
(5) The amounts reported represent matching contributions made by the Company under our 401(k) plan on behalf of each of our named executive officers in 2023.
(6) Dr. Decicco stepped down from his position as our Chief Scientific Officer effective January 10, 2022.Quintás-Cardama commenced employment with us on September 11, 2023.
Narrative Disclosure to Summary Compensation Table
Base Salary
The letter agreement with each named executive officer, described below, establishes aan initial base salary for the named executive officer. For 2021,2023, the base salary of Mr. Gottschalk was $579,000 (increased from $575,000 for 2022), the base salaries of Mr. GottschalkDr. Bellon was $540,000$445,000 (increased from $435,000 for 2022), and Drs. Decicco and Agresta were $428,500. Mr. Reine'sthe base salary for 2021
Dr. Quintás-Cardama was $416,000.$465,000. For 2022, Dr. Reine's2024, Mr. Gottschalk’s base salary was increased to $435,000, and$601,500, Dr. Agresta’sBellon’s base salary was increased to $445,000.$485,000 and Dr. Quintás-Cardama’s base salary was increased to $471,000.
Annual Bonuses
With respect to fiscal year 2021,2023, each of Mr. Gottschalk and Drs. Reine, DeciccoBellon and AgrestaQuintás-Cardama was eligible to receive an annual bonus, with the initial target amount of such bonus for each named executive officer set forth in his letter agreement with us, described below. For fiscal year 2021,2023, the target bonus amount, expressed as a percentage of base salary, for each of Mr. Gottschalk and Drs. Reine, DeciccoBellon and AgrestaQuintás-Cardama was as follows: up to 50%, up to 40%, up to 40%, and up to 40%, respectively.respectively, with Dr. Quintás-Cardama’s 2023 annual bonus prorated based on his September 2023 employment commencement date, as described below. Annual bonuses for fiscal year 20212023 for our named executive officers were based on the attainment of certain corporate performance goals as determined by the Compensation Committee, including those related to capital raising and financing, senior management recruitment, development of pipeline candidates, and research and development targets. For 2021,2023, the compensation committee determined that, based on the level of attainment of the applicable corporate performance goals and other factors determined relevant by the committee, each eligible executive would be eligible to earn 150%70% of his bonus target. As a result, Mr. Gottschalk earned a bonus of $405,000,$202,650, Dr. ReineBellon earned a bonus of $249,600,$124,600, and Dr. DeciccoQuintás-Cardama earned a bonus of $257,100, and Dr. Agresta earned a$43,400. For 2024, Mr. Gottschalk’s annual bonus target amount was increased to up to 55% of $257,100.his base salary.
Agreements With Our Named Executive Officers
Each named executive officer is (or was,party to a letter agreement (as amended and restated, in the case of Mr. Gottschalk and Dr. Decicco) party to an amended and restated letter agreementBellon) with us that sets forth the terms and conditions of his employment. The material terms of the agreements are described below. The terms “cause,” “good reason event”reason” and “change of control” referred to below are defined in the respective named executive officer’s agreement.
Mr. Gottschalk. In connection with our initial public offering, we entered into an amended and restated letter agreement with Mr. Gottschalk that provides for an initial base salary of $478,950 per year and an initial target annual bonus, each of 50% of his annual base salary.which has subsequently been increased, as described above. The amended and restated letter agreement provides that, as long as he is our Chief Executive Officer, we will nominate Mr. Gottschalk to serve on our Board of Directors and he will serve as a member of our Board of Directors if elected.
Mr. Gottschalk also entered into an Employee Non-Competition, Non-Solicitation, Confidentiality and Assignment of Inventions Agreement under which he has agreed not to compete with us during his employment and for 12 months following the termination of his employment, except for any termination due to layoff or without cause (as defined in this agreement), in exchange for garden leave pay during the post-employment non-competition period equal to 50% of his highest annual base salary during the two years prior to termination of his employment. We may elect to waive the post-employment non-competition period, in which case no garden leave pay would be due. Pursuant to the terms of this agreement, Mr. Gottschalk also has agreed to a perpetual obligation of confidentiality, the assignment of intellectual property, the protection and return of documents and other materials, and not to solicit our customers or business partners, or solicit or hire our employees or independent contractors, during his employment and for 12 months following termination of his employment.
Dr. ReineBellon. . In connection with our initial public offering,On January 26, 2022, we entered into an amended and restated letter agreement with Dr. ReineBellon that provided for an initial base salary, of $400,000 per yearwhich has subsequently been increased, as described above, and a target annual bonus of 40% of his annual base salary.
Dr. ReineBellon also entered into an Employee Non-Competition, Non-Solicitation, Confidentiality and Assignment of Inventions Agreement, which has terms substantially similar to Mr. Gottschalk’s Employee Non-Competition, Non-Solicitation, Confidentiality and Assignment of Inventions Agreement, as described above.
Dr. DeciccoQuintás-Cardama. . In connection with our initial public offering,On August 4, 2023, we entered into an amended and restateda letter agreement with Dr. DeciccoQuintás-Cardama that provided for an initial base salary, of $412,000 per yearwhich has subsequently been increased as described above, and a target annual bonus of 40% of his annual base salary.
Dr. Decicco also entered into an Employee Non-Competition Agreement and an Employee Non-Solicitation, Confidentiality and Assignment of Inventions Agreement, which together have terms substantially similar to Mr. Gottschalk’s Employee Non-Competition, Non-Solicitation, Confidentiality and Assignment of Inventions Agreement, as described above.
In January 2022, Dr. Decicco entered into a Transition Agreement with the Company. Pursuant With respect to the Transition Agreement,2023 fiscal year, any annual bonus earned by Dr. Decicco stepped down from his position as Chief Scientific OfficerQuintás-Cardama was to be a pro-rated annual bonus equal to one-third of the Company effective as of January 10, 2022, served as Special Advisor to the Company’s newly appointed Chief Scientific Officer through January 31, 2022, and retired frombonus he would have received had he been employed by the Company on January 31, 2022 (the “Separation Date”). Dr. Decicco continued to receive his then-current base salary throughfor the Separation Date and was eligible to receive an annual bonus based on the achievement of the pre-established performance objectives in respect of fiscal year 2021, which was earned and paid as described above. Under the Transition Agreement, any vested stock options held by Dr. Decicco as of the Separation Date will remain exercisable until July 5, 2022.In connection with entering into the Transition Agreement, Dr. Decicco signed a customary release of claims in favor of the Company.
Dr. Agresta. In connection with our initial public offering, we entered into an amended and restated letter agreement with Dr. Agresta that provides for an initial base salary of $412,000 per year and a target annual bonus of 40% of his annual base salary. The amended and restated letter agreement also provided for repayment by Dr. Agresta if we terminate his employment for cause or if he resigns without good reason, in each case, prior to September 16, 2021, of a one-time transition payment of $65,000 previously paid to him.full calendar year.
In connection with his commencement of employment, pursuant to his letter agreement, Dr. Agresta Quintás-Cardama received a one-time signing bonus in the amount of $110,000, which is subject to repayment by Dr. Quintás-Cardama if he resigns without good reason or the Company terminates his employment for cause, in each case, prior to September 11, 2024. Pursuant to his letter agreement, Dr. Quintás-Cardama was granted an option to purchase 272,000 shares of our common stock, as described in more detail below under “Equity Compensation.”
Dr. Quintás-Cardamaalso entered into an Employee Non-Competition, Non-Solicitation, Confidentiality and Assignment of Inventions Agreement, which has terms substantially similar to Mr. Gottschalk’s Employee Non-Competition, Non-Solicitation, Confidentiality and Assignment of Inventions Agreement, as described above.
Severance Upon Termination of Employment; Change in Control.
Mr. Gottschalk. If Mr. Gottschalk’s employment is terminated by us without cause or if he resigns for good reason outside of a change of control, he will be entitled to receive (i) severance in an amount equal to 12 months of his then-current base salary, payable in installments over six months; (ii) payment of the employer portion of COBRA premiums for 12 months, subject to his eligibility for, and timely election of, COBRA coverage; and (iii) any earned but unpaid bonus relating to the calendar year prior to the year of termination, payable at the same time bonuses otherwise are paid to active employees.
If Mr. Gottschalk’s employment is terminated by us without cause or if he resigns for good reason within the four months prior to or 12 months following a change of control, he will be entitled to receive (i) severance in an amount equal to 1.5 times the sum of (A) his then-current base salary plus (B) his target annual bonus for the year of termination, payable in installments over 12 months; (ii) payment of the employer portion of COBRA premiums for 18 months, subject to his eligibility for, and timely election of, COBRA coverage; (iii) any earned but unpaid bonus relating to the calendar year prior to the year of termination, payable at the same time bonuses otherwise are paid to active employees; and (iv) full acceleration of time-based stock options and other time-based equity awards.
Dr. ReineBellon. . If Dr. Reine'sBellon’s employment is terminated by us without cause or if he resigns for good reason outside of a change of control, he will be entitled to receive (i) severance in an amount equal to nine months of his then-current base salary, payable in installments over nine months; (ii) payment of the employer portion of COBRA premiums for nine months, subject to his eligibility for, and timely election of, COBRA coverage; and (iii) any earned but unpaid bonus relating to the calendar year prior to the year of termination, payable at the same time bonuses otherwise are paid to active employees.
If Dr. Reine’sBellon’s employment is terminated by us without cause or if he resigns for good reason within the three months prior to or 12 months following a change of control, he will be entitled to receive (i) severance in an amount equal to the sum of (A) his then-current base salary plus (B) his target annual bonus for the year of termination, payable in installments over 12 months; (ii) payment of the employer portion of COBRA premiums for 12 months, subject to his eligibility for, and timely election of, COBRA coverage; (iii) any earned but unpaid bonus relating to the calendar year prior to the year of termination, payable at the same time bonuses otherwise are paid to active employees; and (iv) full acceleration of time-based stock options and other time-based equity awards.
Dr. AgrestaQuintás-Cardama. If Dr. Agresta’sQuintás-Cardama’s employment is terminated by us without cause or if he resigns for good reason outside of a change of control, he will be entitled to receive (i) severance in an amount equal to nine months of his then-current base salary, payable in installments over nine months; (ii) payment of the employer portion of COBRA premiums for nine months, subject to his eligibility for, and timely election of, COBRA coverage; and (iii) any earned but unpaid bonus relating to the calendar year prior to the year of termination, payable at the same time bonuses otherwise are paid to active employees.
If Dr. Agresta’sQuintás-Cardama’s employment is terminated by us without cause or if he resigns for good reason within the three months prior to or 12 months following a change of control, he will be entitled to receive (i) severance in an amount equal to the sum of (A) his then-current base salary plus (B) his target annual bonus for the year of termination, payable in installments over 12 months; (ii) payment of the employer portion of COBRA premiums for 12 months, subject to his eligibility for, and timely election of, COBRA coverage; (iii) any earned but unpaid bonus relating to the calendar year prior to the year of termination, payable at the same time bonuses otherwise are paid to active employees; and (iv) full acceleration of time-based stock options and other time-based equity awards.
Dr. Decicco. Dr. Decicco had been entitled to receive certain severance benefits under his amended and restated employment agreement prior to his termination of employment. Under Dr. Decicco's Transition Agreement, any vested stock options held by Dr. Decicco as of the date he terminated employment with us will remain exercisable until July 5, 2022, and Dr. Decicco will remain eligible to participate in our health, dental and vision plans until December 31, 2022 as if his employment had not terminated.
Severance Subject to Compliance with Restrictive Covenant Obligations and Release of Claims. Our obligation to provide severance payments and other benefits under each of the named executive officers’officer’s letter agreements (as amended and restated, letter agreementsin the case of Mr. Gottschalk and Dr. Bellon) is conditioned on (i) the executive providing a timely and effective separation agreement containing a release of claims in favor of us; and (ii) the executive’s continued compliance with applicable restrictive covenant obligations, including any non-competition, non-solicitation and confidentiality restrictions.
Section 280G of the Code. Each named executive officer’s letter agreement (as amended and restated, in the case of Mr. Gottschalk and Dr. Bellon) provides for a Section 280G “better of provision” such that payments or benefits that each named executive officer receives in connection with a change in control will be reduced to the extent necessary to avoid the imposition of any excise tax under Sections 280G and 4999 of the Code if such reduction would result in a greater after tax payment
amount for such named executive officer than if he had been paid the full amount of such payments or benefits, with such payments and benefits subject to the excise tax.
Equity Compensation
Each of our of named executive officers received a grant of options to purchase shares of our common stock in the fiscal year 20212023 pursuant to the terms of the 2020 Equity Incentive Plan.
On January 28, 2021,26, 2023, each of Mr. Gottschalk and Drs. Reine, Decicco and Agresta wereDr. Bellon was granted optionsan option to purchase 300,000, 120,000, 100,000280,000 and 100,000 shares of our common stock, respectively, each of which option vestsvested as to 25% of the underlying shares on January 28, 2022,26, 2024, and vests as to 6.25% of the underlying shares on the first day of each calendar quarter thereafter, for the
subsequent 12 calendar quarters, generally subject to the applicable executive’s continued employment with us through the applicable vesting date.
On September 11, 2023, Dr. Quintás-Cardama was granted an option to purchase 272,000 shares of our common stock, which will vest as to 25% of the underlying shares on September 11, 2024 and as to 6.25% of the underlying shares on the first day of each calendar quarter thereafter, for the subsequent 12 calendar quarters, generally subject to Dr. Quintás-Cardama’s continued employment with us through the applicable vesting date.
Severance and Change of Control Payments and Benefits
Each of our currently employed named executive officers is entitled to severance and change of control benefits pursuant to the terms of his letter agreement (as amended and restated, letter agreementin the case of Mr. Gottschalk and Dr. Bellon) as described above under “Agreements With Our Named Executive Officers.”
Employee and Retirement Benefits
We currently provide broad-based health and welfare benefits, and certain commuter benefits, that are available to our full-time employees, including our currently employed named executive officers, including health, life, disability, vision and dental insurance. In addition, we maintain a 401(k) retirement plan for our full-time employees. The 401(k) plan permits us to make discretionary employer contributions. We did not make anyIn 2023, we made employer contributions to the 401 (k)401(k) plan in 2019.of up to $3,000 per employee, including for our named executive officers. Other than the 401(k) plan, we do not provide any qualified or non-qualified retirement or deferred compensation benefits to our employees, including our named executive officers.
Outstanding Equity Awards at Fiscal Year-end Table
The following table sets forth information about outstanding equity awards held by each of our named executive officers as of December 31, 2021:2023:
| | | | | | | | | | | | | | |
Name
|
Number of securities underlying unexercised options exercisable (#) | Number of securities underlying unexercised options unexercisable (#) |
Option exercise price ($/share) |
Option expiration date |
| Adrian Gottschalk | 458,926 | | — | | $ | 0.54 | | 5/29/2027 (1) |
| 172,354 | | 78,343 | | $ | 3.72 | | 2/20/2029 (2) |
| 97,128 | | 213,682 | | $ | 8.77 | | 8/17/2030 (3) |
| — | | 300,000 | | $ | 16.63 | | 1/27/2031 (4) |
| Allan Reine M.D. | 121,320 | | 136,193 | | $ | 3.72 | | 9/16/2029 (5) |
| 28,717 | | 63,174 | | $ | 8.77 | | 8/17/2030 (6) |
| 120,000 | | $ | 16.63 | | 1/27/2031 (7) |
| Carl Decicco, Ph.D | 69,476 | | 117,095 | | $ | 3.72 | | 2/20/2029 (8) |
| — | | 34,273 | | $ | 3.72 | | 2/20/2029 (9) |
| 28,716 | | 63,175 | | $ | 8.77 | | 8/17/2030 (10) |
| — | | 100,000 | | $ | 16.63 | | 1/27/2031 (11) |
| Samuel Agresta, M.D | 175,104 | | 136,193 | | $ | 3.72 | | 9/16/2029 (12) |
| 28,716 | | 63,175 | | $ | 8.77 | | 8/17/2030 (13) |
| — | | 100,000 | | $ | 16.63 | | 1/27/2031 (14) |
| | | | | | | | | | | | | | |
| Name | Number of securities underlying unexercised options exercisable (#) | Number of securities underlying unexercised options unexercisable (#) | Option exercise price ($/share) | Option expiration date |
| Adrian Gottschalk | 418,926 | | — | | 0.54 | 5/29/2027 (1) |
| 250,697 | | — | | 3.72 | 2/20/2029 (2) |
| 252,533 | | 58,277 | | 8.77 | 8/17/2030 (3) |
| 206,250 | | 93,750 | | 16.63 | 1/27/2031 (4) |
| 192,500 | | 247,500 | | 14.87 | 1/25/2032 (5) |
| — | | 280,000 | | 8.38 | 1/25/2033 (6) |
| Steven Bellon, Ph.D. | 3,716 | | — | | 3.72 | 2/12/2029 (7) |
| 21,622 | | 8,108 | | 8.77 | 8/18/2030 (8) |
| 52,702 | | 12,162 | | 8.77 | 8/20/2030 (9) |
| 34,375 | | 15,625 | | 16.63 | 1/27/2031 (10) |
| 65,625 | | 84,375 | | 14.87 | 1/25/2032 (11) |
| — | | 100,000 | | 8.38 | 1/25/2033 (12) |
| Alfonso Quintas-Cardama, M.D. | — | | 272,000 | | 7.57 | 9/10/2033 (13) |
(1) Represents an option to purchase 905,405 shares of our common stock, granted on May 30, 2017, which vested as to 25%
of the underlying shares on May 30, 2018, and vestsvested as to 6.25% of the underlying shares on the first day of each calendar
quarter thereafter, for the subsequent 12 calendar quarters, generally subject to Mr. Gottschalk’s continued employmentquarters.
with us through the applicable vesting date.
(2) Represents an option to purchase 250,697 shares of our common stock, granted on February 20, 2019, which vested as to
25% of the underlying shares on January 30, 2020, and vestsvested as to 6.25% of the underlying shares on the first day of each
calendar quarter thereafter, for the subsequent 12 calendar quarters, generally subject to Mr. Gottschalk’s continued
employment with us through the applicable vesting date.quarters.
(3) Represents an option to purchase 310,810 shares of our common stock, granted on August 18, 2020, which vested as to
25% of the underlying shares on August 17,18, 2021, and vests as to 6.25% of the underlying shares on the first day of each
calendar quarter thereafter, for the subsequent 12 calendar quarters, generally subject to Mr. Gottschalk’s continued
employment with us through the applicable vesting date.
(4) Represents an option to purchase 300,000 shares of our common stock, granted on January 28, 2021, which vested as to
25% of the underlying shares on January 28, 2022, and vests as to 6.25% of the underlying shares on the first day of each
calendar quarter thereafter, for the subsequent 12 calendar quarters, generally subject to Mr. Gottschalk'sGottschalk’s continued
employment with us through the applicable vesting date.
(5) Represents an option to purchase 311,297440,000 shares of our common stock, granted on September 17, 2019,January 26, 2022, which vested as
to 25% of the underlying shares on September 16, 2020,January 26, 2023, and vests as to 6.25% of the underlying shares on the first day of
each calendar quarter thereafter, for the subsequent 12 calendar quarters, generally subject to Dr. Reine’sMr. Gottschalk’s continued
employment with us through the applicable vesting date.
(6) Represents an option to purchase 91,891280,000 shares of our common stock, granted on January 26, 2023, which vested as to 25% of the underlying shares on January 26, 2024, and vests as to 6.25% of the underlying shares on the first day of each calendar quarter thereafter, for the subsequent 12 calendar quarters, generally subject to Mr. Gottschalk’s continued employment with us through the applicable vesting date.
(7) Represents an option to purchase 29,729 shares of our common stock, granted on February 13, 2019, which vested as to 25% of the underlying shares on February 13, 2020, and vested as to 6.25% of the underlying shares on the first day of each calendar quarter thereafter, for the subsequent 12 calendar quarters.
(8) Represents an option to purchase 43,243 shares of our common stock, granted on August 18, 2020, which vested as to
25% of the underlying shares on August 17, 2021, and vests as to 6.25% of the underlying shares on the first day of each
calendar quarter thereafter, for the subsequent 12 calendar quarters, generally subject to Dr. Reine’sBellon’s continued
employment with us through the applicable vesting date.
(7)(9) Represents an option to purchase 120,00064,864 shares of our common stock, granted on August 20, 2020, which vested as to 25% of the underlying shares on August 17, 2021, and vests as to 6.25% of the underlying shares on the first day of each calendar quarter thereafter, for the subsequent 12 calendar quarters, generally subject to Dr. Bellon’s continued employment with us through the applicable vesting date.
(10) Represents an option to purchase 50,000 shares of our common stock, granted on January 28, 2021, which vested as to
25% of the underlying shares on January 28, 2022, and vests as to 6.25% of the underlying shares on the first day of each
calendar quarter thereafter, for the subsequent 12 calendar quarters, generally subject to Dr. Reine'sBellon’s continued
employment with us through the applicable vesting date.
(8) Represents an option to purchase 468,378 shares of our common stock, granted on February 20, 2019, which vested as to
25% of the underlying shares on December 10, 2019, and vested as to 6.25% of the underlying shares on the first day of
each calendar quarter thereafter, for the subsequent 12 calendar quarters, generally subject to Dr. Decicco’s continued
employment with us through the applicable vesting date. The unvested portion of this option, together with the unvested
portion of other options held by Dr. Decicco, terminated in connection with his termination of employment.
(9) Represents an option to purchase 109,673 shares of our common stock, granted on February 20, 2019, which vested as to
25% of the underlying shares on January 30, 2020, and vested as to 6.25% of the underlying shares on the first day of
each calendar quarter thereafter, for the subsequent 12 calendar quarters, generally subject to Dr. Decicco’s continued
employment with us through the applicable vesting date.
(10) Represents an option to purchase 91,891 shares of our common stock, granted on August 18, 2020, which vest as to 25%
of the underlying shares on August 17, 2021, and vested as to 6.25% of the underlying shares on the first day of each
calendar quarter thereafter, for the subsequent 12 calendar quarters, generally subject to Dr. Decicco’s continued
employment with us through the applicable vesting date.
(11) Represents an option to purchase 100,000150,000 shares of our common stock, granted on January 28, 2021,26, 2022, which vested as to
25% of the underlying shares on January 28, 2022,26, 2023, and vestedvests as to 6.25% of the underlying shares on the first day of
each calendar quarter thereafter, for the subsequent 12 calendar quarters, generally subject to Dr. Decicco'sBellon’s continued
employment with us through the applicable vesting date.
(12) Represents an option to purchase 311,297100,000 shares of our common stock, granted on September 17, 2019,January 26, 2023, which vested as
to 25% of the underlying shares on September 16, 2020,January 26, 2024, and vests as to 6.25% of the underlying shares on the first day of
each calendar quarter thereafter, for the subsequent 12 calendar quarters, generally subject to Dr. Agresta’sBellon’s continued
employment with us through the applicable vesting date.
(13) Represents an option to purchase 91,891272,000 shares of our common stock, granted on August 18, 2020,September 11, 2023, which vestvests as to 25%
of the underlying shares on August 17, 2021,September 11, 2024, and vests as to 6.25% of the underlying shares on the first day of each
calendar quarter thereafter, for the subsequent 12 calendar quarters, generally subject to Dr. Agresta’sQuintás-Cardama’s continued
employment with us through the applicable vesting date.
(14) Represents an option to purchase 100,000 shares of our common stock, granted on January 28, 2021, which vested as to
25% of the underlying shares on January 28, 2022, and vests as to 6.25% of the underlying shares on the first day of each
calendar quarter thereafter, for the subsequent 12 calendar quarters, generally subject to Dr. Agresta's continued
employment with us through the applicable vesting date.
Director Compensation
The following table sets forth information concerning the compensation awarded to, earned by, or paid to our non-employee directors during the fiscal year ended December 31, 2021.2023. Mr. Gottschalk’s compensation for 20212023 is included with that of our other named executive officers above.
| | | | | | | | | | | | | | | |
Name | Fees Earned or Paid in Cash ($)(1) | | Option Awards
($)(2)(3)(4)(5) | All other compensation ($)(6)(7) |
Total ($) |
| Jose Baselga, M.D., Ph.D | $ | 11,750 | | | $ | — | | $ | 92,406 | | $ | 104,156 | |
| Douglas Cole, M.D. | $ | 99,656 | | | $ | 75,127 | | $ | — | | $ | 174,783 | |
| Simba Gill, Ph. | $ | 52,500 | | | $ | 75,127 | | $ | — | | $ | 127,627 | |
| Cigall Kadoch, Ph. | $ | 48,750 | | | $ | 75,127 | | $ | 128,640 | | $ | 252,517 | |
| Scott Biller, Ph. | $ | 42,000 | | | $ | 75,127 | | $ | — | | $ | 117,127 | |
| Adam Koppel, M.D., Ph | $ | 54,000 | | | $ | 75,127 | | $ | — | | $ | 129,127 | |
| Michael Mendelsohn, M. | $ | 48,953 | | | $ | 75,127 | | $ | — | | $ | 124,080 | |
| Ian Smith | $ | 23,722 | | | $ | 195,490 | | $ | 80,446 | | $ | 299,659 | |
| | | | | | | | | | | | | | | |
Name | Fees Earned or Paid in Cash ($)(1) | | Option Awards
($)(2)(3)(4) | All other compensation ($) |
Total ($) |
| Douglas Cole, M.D. | $ | 83,000 | | | $ | 101,573 | | $ | — | | $ | 184,573 | |
| Simba Gill, Ph.D. | $ | 57,500 | | | $ | 101,573 | | $ | — | | $ | 159,073 | |
| B. Lynne Parshall | $ | 56,079 | | | $ | 101,573 | | $ | — | | $ | 157,652 | |
| | | | | | | | | | | | | | | |
| Scott Biller, Ph.D. | $ | 44,000 | | | $ | 101,573 | | $ | — | | $ | 145,573 | |
| Adam Koppel, M.D., Ph.D. | $ | 51,500 | | | $ | 101,573 | | $ | — | | $ | 153,073 | |
| Michael Mendelsohn, M.D. | $ | 53,000 | | | $ | 101,573 | | $ | — | | $ | 154,573 | |
| Ian Smith | $ | 40,000 | | | $ | 211,572 | | $ | — | | $ | 251,572 | |
| Thomas Lynch, M.D. | $ | 44,000 | | | $ | 101,573 | | $ | — | | $ | 145,573 | |
(1) All cash fees payable in respect of Dr. Cole's board service are remitted to an affiliate of Flagship Pioneering, Inc.
(2) In 2021,2023, each of Drs. Cole, Gill, Kadoch, Biller, Koppel and Mendelsohnnon-employee director who served on our Board of Directors was granted an option to purchase 12,530 shares
of our common stock on July 13, 2021, following the 2021 annual meeting of stockholders.
(3) Mr. Smith was granted an option to purchase 25,06016,000 shares of our common stock on April 27, 2021 upon his appointmentJune 21, 2023, following the 2023 annual meeting of stockholders. Mr. Smith was also granted an option to purchase 26,977 shares of our common stock on December 8, 2023 pursuant to a consulting agreement between Mr. Smith and the Company entered into on December 8, 2023.
to the Board of Directors.
(4)(3) The amounts reported represent the aggregate grant date fair value of options to purchase our common stock granted to
our non-employee directors in fiscal year 2021,2023, computed in accordance with FASB ASC 718, excluding the effect of
estimated forfeitures. The assumptions used to value the stock options for this purpose are set forth in Note 87 to our
consolidated financial statements included elsewhere in this Annual Report.
(5)(4) Non-employee directors who served on our Board of Directors during 20212023 held the following unexercised stock options
as of December 31, 2021:2023:
| | | | | |
| Name | Number of Shares Underlying Stock Options (#) |
Jose Baselga, M.D., Ph.D | — | |
| Douglas Cole, M.D. | 12,53044,530 | |
Simba Gill, Ph.DPh.D. | 201,718151,718 | |
Cigall Kadoch, Ph.DB. Lynne Parshall | 12,53048,000 | |
Scott Biller, Ph.DPh.D. | 107,124139,124 | |
| Adam Koppel, M.D., Ph.D. | 107,124139,124 | |
| Michael Mendelsohn, M.D. | 107,124139,124 | |
| Ian Smith | 61,914120,891 | |
| Thomas Lynch, M.D. | 48,000 | |
(6) Following the passing of Dr. Baselga in March 2021, the Board of Directors authorized the Company to make a one-time
cash payment in the amount of $92,406 to Dr. Baselga's estate.
(7) The amounts reported in this column represent consulting fees earned by Dr. Kadoch and Mr. Smith in fiscal year 2021.
Consulting Agreements
Each of Dr. Kadoch and Mr. Smith is party to a consulting agreement with us that sets forth the terms and conditions of certain consulting services provided by the applicable director.
Dr. Kadoch. We entered into a consulting agreement with Dr. Kadoch, which as amended provides for an annual consulting fee of $225,000. Pursuant to the terms of the consulting agreement, Dr. Kadoch has agreed to a 10-year post-termination obligation of confidentiality, an assignment of intellectual property covenant, and not to compete with us or solicit our customers, business partners, employees or independent contractors during the term of the consulting agreement and for 12 months thereafter. In October 2020, the term of this agreement was extended to January 1, 2022, subject to automatic one-year renewal terms until terminated.
Mr. Smith. WeOn December 8, 2023, we entered into a consulting agreement with Mr. Smith, which provides for compensation in the form of an option to purchase up to 12,53026,977 shares of our common stock at an option exercise price of $10.24$5.63 per share, the closing price of a share of our common stock on the date the option was granted. Pursuant to the terms of the consulting agreement, the option vests as to one-twelfth of the underlying shares on the first day of each month starting in August 2021,January 2024, with the balance of then-unvested shares to vest on June 23, 2022. Pursuant to the terms of the consulting agreement, Mr. Smith has agreed to a perpetual obligation of confidentiality and an assignment of intellectual property covenant.December 8, 2024.
Director Compensation Policy
Our Board of Directors adopted a non-employee director compensation policy in connection with our initial public offering. Under the non-employee director compensation policy, our non-employee directors are compensated as follows:
•each non-employee director receives an annual cash fee of $35,000$40,000 ($65,00070,000 for the chair of our Board of Directors);
•each non-employee director who is a member of the audit committee receives an additional annual cash fee of $7,500 ($15,000 for the audit committee chair);
•each non-employee director who is a member of the compensation committee receives an additional annual cash fee of $5,000 ($10,000 for our compensation committee chair);
•each non-employee director who is a member of the nominating and corporate governance committee or the science committee receives an additional annual cash fee of $4,000 ($8,000 for the nominating and corporate governance or science committee chair);
•each non-employee director who is first elected or appointed to our Board of Directors after the our initial public offering is granted an option under the Foghorn Therapeutics Inc. 2020 Equity Incentive Plan, or the 2020 Plan to purchase 25,06032,000 shares of our common stock (but in no event will a non-employee director’s initial grant have a grant date fair value, determined in accordance with FASB ASC 718, that exceeds $600,000); and
•each non-employee director who has served as a member of our Board of Directors for at least a six-month period prior to the first meeting of our Board of Directors following the annual meeting of our stockholders is annually granted an option under the 2020 Plan to purchase 12,53016,000 shares of our common stock (but in no event will a non-
employeenon-employee director’s annual grant have a grant date fair value, determined in accordance with FASB ASC 718, that exceeds $300,000).
The stock options granted to our non-employee directors will have a per share exercise price at least equal to the closing price of a share of our common stock on the date of grant and will expire not later than ten years after the date of grant. The stock option granted to a non-employee director upon his or her initial election to our Board of Directors will vest as to one-third of the underlying shares on each of the first three anniversaries of the date of grant, subject to such director’s continued service on our Board of Directors. The annual stock options granted to our non-employee directors will vest in full on the first anniversary of the date of grant, subject to the director’s continued service on our Board of Directors.
Each non-employee director is entitled to reimbursement for reasonable travel and other expenses incurred in connection with attending meetings of our Board of Directors and any committee on which he or she serves.
Pursuant to the terms of the 2020 Plan, the aggregate value of all compensation granted or paid to any director with respect to any calendar year, including awards granted under the 2020 Plan and cash fees or other compensation paid by us to such director outside of the 2020 Plan for his or her services as a director during such calendar year, is subject to a limit of $750,000 in the aggregate ($1,000,000 in the aggregate with respect to a director’s first year of service on our Board of Directors).
Delinquent Section 16(a) Reports
Section 16(a) of the Securities Exchange Act of 1934 requires our executive officers, directors and persons who own more than 10% of Foghorn’s common stock to file reports of ownership and changes in ownership with the SEC. As a matter of practice, the Company assists its executive officers and directors in preparing initial reports of ownership and reports of changes in ownership and files those reports on their behalf. Based on our review of such forms, as well as information provided by the reporting persons, we believe that each of our executive officers, directors and beneficial owners of more than 10% of its common stock complied with the reporting requirements of Section 16(a) during the year ended December 31, 2023.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT
The following table sets forth certain information as of February 25, 202229, 2024 (unless otherwise specified), with respect to the beneficial ownership of our common stock by each person who is known to own beneficially more than 5% of the outstanding shares of common stock, each person currently serving as a director, each nominee for director, each named executive officer (as set forth in the Summary Compensation Table above) and all directors and executive officers as a group. The number of shares beneficially owned by each entity or person is determined in accordance with the rules of the SEC, and the information is not necessarily indicative of beneficial ownership for any other purpose. Under such rules, beneficial ownership includes any shares over which the individual has sole or shared voting power or investment power as well as any shares that the individual has the right to acquire within 60 days of February 25, 2022,29, 2024, through the exercise of any stock options, warrants or other rights. Except as otherwise indicated, and subject to applicable common property laws, the persons in the table have sole voting and investment power with respect to all shares of common stock held by that person.
Shares of common stock subject to options, warrants or other rights that are now exercisable or are exercisable within 60 days after February 25, 2022,29, 2024, are considered outstanding for purposes of computing the percentage ownership of the persons holding these options, warrants or other rights but are not to be considered outstanding for the purpose of computing the percentage ownership of any other person. As of February 25, 2022,29, 2024, there were 41,421,96442,565,904 shares of common stock outstanding. Unless otherwise indicated, the address for each beneficial owner is c/o Foghorn Therapeutics Inc., 500 Technology Square, Ste 700, Cambridge, MA 02139.
| | Name and address of beneficial owner 5% or greater stockholders: | Name and address of beneficial owner 5% or greater stockholders: | | Number of Shares | | Percentage of Shares | Name and address of beneficial owner 5% or greater stockholders: | | Number of Shares | | Percentage of Shares |
| Funds affiliated with Flagship Pioneering, Inc. (1) | Funds affiliated with Flagship Pioneering, Inc. (1) | | 12,674,120 | | | 30.6 | Funds affiliated with Flagship Pioneering, Inc. (1) | | 12,674,120 | | | 29.78 | | 29.78 |
| Klarman Family Foundation (2) | Klarman Family Foundation (2) | | 2,139,639 | | | 5.17 | Klarman Family Foundation (2) | | 2,139,639 | | | 5.03 | | 5.03 |
| Entities affiliated with Fidelity Investments (3) | Entities affiliated with Fidelity Investments (3) | | 2,484,834 | | | 6 | Entities affiliated with Fidelity Investments (3) | | 2,810,009 | | | 6.6 | | 6.6 |
| Eli Lilly and Company (4) | Eli Lilly and Company (4) | | 4,000,000 | | | 9.66 | Eli Lilly and Company (4) | | 4,000,000 | | | 9.4 | | 9.4 |
| Cigall Kadoch, Ph.D. (5) | | Cigall Kadoch, Ph.D. (5) | | 3,652,909 | | | 8.58 |
| Directors and named executive officers: | Directors and named executive officers: | |
Adrian Gottschalk (5)(6) | |
Adrian Gottschalk (5)(6) | |
Adrian Gottschalk (5)(6) | Adrian Gottschalk (5)(6) | | 1,364,049 | | | 3.22 | | 2,051,461 | | | 4.65 | | 4.65 |
Douglas G. Cole, M.D. (6)(7) | Douglas G. Cole, M.D. (6)(7) | | — | | | 0 | Douglas G. Cole, M.D. (6)(7) | | 28,530 | | | * | | * |
Scott Biller, Ph.D. (7)(8) | Scott Biller, Ph.D. (7)(8) | | 53,209 | | | * | Scott Biller, Ph.D. (7)(8) | | 123,124 | | | * | | * |
Balkrishan (Simba) Gill, Ph.D. (8)(9) | Balkrishan (Simba) Gill, Ph.D. (8)(9) | | 189,188 | | | * | Balkrishan (Simba) Gill, Ph.D. (8)(9) | | 169,779 | | | * | | * |
| Cigall Kadoch, Ph.D. (9) | | 3,624,379 | | | 8.75 |
| Adam M. Koppel, M.D., Ph.D. (10) | Adam M. Koppel, M.D., Ph.D. (10) | | 94,594 | | | * | Adam M. Koppel, M.D., Ph.D. (10) | | 123,124 | | | * | | * |
| Michael Mendelsohn, M.D. (11) | | 94,594 | | | * |
| Samuel Agresta, M.D., M.P.H. & T.M. (12) | | 285,469 | | | * |
| Allan Reine, M.D. (13) | | 291,720 | | | * |
| Carl Decicco (14) | | 527,771 | | | 1.27 |
| Ian Smith (15) | | 105,792 | | | * |
| Thomas Lynch, M.D. (11) | | Thomas Lynch, M.D. (11) | | 10,667 | | | * |
| Michael Mendelsohn, M.D. (12) | | Michael Mendelsohn, M.D. (12) | | 123,124 | | | * |
| B. Lynne Parshall (13) | | B. Lynne Parshall (13) | | 10,667 | | | * |
| Ian Smith (14) | | Ian Smith (14) | | 152,713 | | | * |
| Steven Bellon, Ph.D. (15) | | Steven Bellon, Ph.D. (15) | | 419,760 | | | * |
| Alfonso Quintas-Cardama, M.D. | |
| All executive officers and directors as a group (14 persons) (16) | All executive officers and directors as a group (14 persons) (16) | | 7,042,754 | | | 16.15 | All executive officers and directors as a group (14 persons) (16) | | 4,014,732 | | | 8.78 | | 8.78 |
* Represents beneficial ownership of less than one percent of our outstanding common stock.
(1) Consists of (i) 9,330,878 shares of common stock held by Flagship Ventures Fund V, L.P. (“Flagship Fund V”); (ii) 1,491,441 shares of common stock held by Flagship Ventures Opportunities Fund I, L.P. (“Flagship Opportunities Fund I”); and (iii) 1,851,801 shares of common stock held by Flagship Pioneering Special Opportunities Fund II, L.P, (“Flagship Opportunities Fund II,” and together with Flagship Opportunities Fund I and Flagship Fund V, the “Flagship Funds”). Flagship Ventures Fund V General Partner LLC (“Fund V GP”) is the general partner of Flagship Fund V. Flagship Ventures Opportunities Fund I General Partner LLC (“Opportunities Fund I GP”) is the general partner of Flagship Opportunities Fund I. Flagship Ventures Opportunities Fund II General Partner LLC (“Opportunities Fund II GP”) is the general partner of Flagship Opportunities Fund II. Flagship Pioneering, Inc. (“Flagship Pioneering” and together with Opportunities Fund I GP, Opportunities Fund II GP, and Fund V GP, the “Flagship General Partners”) is the manager of Opportunities Fund II GP. Noubar B. Afeyan, Ph.D. is sole director of Flagship Pioneering and may be deemed to have voting and investment control over all shares held by Opportunities Fund II. In addition, Dr. Afeyan serves as the sole manager of Fund V GP and Opportunities Fund I GP and may be deemed to possess sole voting and investment control over all the shares held by Flagship Fund V and Opportunities Fund I. Neither Dr. Afeyan or the Flagship General Partners directly own any of the shares held by the Flagship Funds and each of the Flagship General Partners and Dr. Afeyan disclaims beneficial ownership of such shares except to the extent of his or its pecuniary interest therein. The mailing address of the Flagship Funds is 55 Cambridge Parkway, Suite 800E, Cambridge, Massachusetts 02142. This information is based solely on a Schedule 13D filed by the Flagship Funds, the Flagship General Partners and Dr. Afeyan with the SEC on October 29, 2020.
(2) Consists of 2,139,639 shares of common stock held directly by the Klarman Family Foundation. Mr. Seth A. Klarman may be deemed to share beneficial ownership of such shares held by the Klarman Family Foundation, however he has no pecuniary interest therein. The mailing address for the Klarman Family Foundation is c/o KFO, LLC, P.O. Box 171627, Boston, Massachusetts 02117. This information is based solely on a Schedule 13G filed by The Klarman Family Foundation and Mr. Klarman with the SEC on February 4, 2021, which reported ownership as of December 31, 2020.
(3) Members of the Johnson family, including Abigail P. Johnson, are the predominant owners, directly or through trusts, of Series B voting common shares of FMR LLC, representing 49% of the voting power of FMR LLC. The Johnson family group and all other Series B shareholders have entered into a shareholders’ voting agreement under which all Series B voting common shares will be voted in accordance with the majority vote of Series B voting common shares. Accordingly, through their ownership of voting common shares and the execution of the shareholders’ voting agreement, members of the Johnson family may be deemed, under the Investment Company
Act of 1940, to form a controlling group with respect to FMR LLC. Neither FMR LLC nor Abigail P. Johnson has the sole power to vote or direct the voting of the shares owned directly by the various investment companies registered under the Investment Company Act of 1940 (“Fidelity Funds”) advised by Fidelity Management &
Research Company (“FMR Co”), a wholly owned subsidiary of FMR LLC, which power resides with the Fidelity Funds’ Boards of Trustees. Fidelity Management & Research Company carries out the voting of the shares under written guidelines established by the Fidelity Funds’ Boards of Trustees. The address for FMR LLC is 245 Summer Street, Boston, MA 02210. This information is based solely on Amendment No. 13 to Schedule 13G filed by FMR LLC and its affiliates with the SEC on February 9, 2022,2024, which reported ownership as of December 31, 2021.2023.
(4) Consists of 4,000,000 shares of common stock held directly by Eli Lilly and Company. This information is based solely on a Schedule 13G filed by Eli Lilly and Company with the SEC on December 13, 2021.
(5) Consists of (i) 25,2253,329,379 shares of common stock held by the Cigall Kadoch Revocable Trust of 2021, of which Dr. Kadoch is the sole trustee, (ii) 295,000 shares of common stock held directly by Dr. Kadoch, and (iii) options to purchase 28,530 shares of common stock that are exercisable within 60 days of February 29, 2024.
(6) Consists of (i) 300,000 shares of common stock held by the Adrian H. Gottschalk Living2023 Grantor Retained Annuity Trust, of which Mr. Gottschalk is the trustee, (ii) 300,000110,690 shares of common stock held by the Adrian H. Gottschalk 2021 Grantor Retained Annuity Trust, of which Mr. Gottschalk is the trustee, and (iii) 146,479101,014 shares of common stock held by the Adrian H. Gottschalk Living Trust, of which Mr. Gottschalk is the trustee, and (iv) options to purchase 892,3451,539,757 shares of common stock that are exercisable within 60 days of February 25, 202229, 2024 held directly by Mr. Gottschalk.
(6)(7) Consists of options to purchase 28,530 shares of common stock that are exercisable within 60 days of February 29, 2024. Dr. Cole is a managing partner of Flagship Pioneering but has no voting or investment power with respect to the securities described in footnote 1.
(7)(8) Consists of options to purchase 53,209123,124 shares of common stock that are exercisable by Dr. Biller within 60 days of February 29, 2024.
(9) Consists of (i) 34,061 shares of common stock held directly by Dr. Gill and (ii) options to purchase 135,718 shares of common stock that are exercisable within 60 days of February 25, 2022.29, 2024.
(8)(10) Consists of options to purchase 189,188123,124 shares of common stock that are exercisable by Dr. Koppel within 60 days of February 25, 2022.29, 2024.
(9) Consists of (i) 3,329,379 shares of common stock held by the Cigall Kadoch Revocable Trust of 2021, of which Dr. Kadoch is the sole trustee, and (ii) 295,000 shares of common stock held directly by Dr. Kadoch.
(10)(11) Consists of options to purchase 94,59410,667 shares of common stock that are exercisable by Dr. Lynch within 60 days of February 25, 2022.29, 2024.
(11)(12) Consists of options to purchase 94,594123,124 shares of common stock that are exercisable by Dr. Mendelsohn within 60 days of February 25, 2022.29, 2024.
(12)(13) Consists of options to purchase an aggregate of 285,46910,667 shares of common stock exercisable by Ms. Parshall within 60 days of February 25, 2022.
(13) Consists of (i) 53,784 shares of common stock held directly by Dr. Reine and (ii) options to purchase an aggregate of 237,936 shares of common stock that are exercisable within 60 days of February 25, 2022.29, 2024.
(14) Consists of (i) 389,599 shares of common stock held directly by Dr. Decicco and (ii) options to purchase an aggregate of 138,172 shares of common stock that are exercisable within 60 days of February 25, 2022.
(15) Consists of (i) 72,072 shares of common stock held directly by Mr. Smith and (ii) options to purchase an aggregate of 33,72080,641 shares of common stock that are exercisable within 60 days of February 25, 2022.29, 2024.
(15) Consists of (i) 171,957 shares of common stock held directly by Dr. Bellon and (ii) options to purchase an aggregate of 247,803 shares of common stock that are exercisable within 60 days of February 29, 2024.
(16) Includes options to purchase an aggregate of 2,178,5173,137,946 shares of common stock exercisable within 60 days of February 25, 2022. Includes the securities beneficially owned by Dr. Decicco as set forth in footnote 13.29, 2024.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
Except as described below, there have been no transactions since January 1, 2021, in which we were a party, the amount involved exceeded or will exceed $120,000 and in which any related person had a direct or indirect material interest.
Consulting Agreement with Cigall Kadoch, Ph.D.
In October 2015, we entered into a consulting agreement with Cigall Kadoch, Ph.D., our academic co-founder and a member of our Board of Directors, pursuant to which Dr. Kadoch provides advisory services related to the manufacturing and sale of products and services related to chromatin remodeling. Under the terms of the consulting agreement, Dr. Kadoch received a grant of 3,953,469 shares of our common stock. Additionally, we agreed to pay Dr. Kadoch a consulting fee of $150,000 per year payable in monthly installments in arrears beginning with the effective date of the consulting agreement, and we agreed to reimburse her for reasonable business expenses incurred in connection with the performance of the services under the agreement. In January 2019, we agreed to increase the consulting fee payable to Dr. Kadoch to $225,000 per year, payable in monthly installments in arrears.
In October 2020, this agreement was extended to January 1, 2022, subject to automatic one-year renewal terms until terminated. During the years ended December 31, 2021 and 2020, we paid Dr. Kadoch $128,640 and $219,500, respectively, pursuant to this agreement.
Indemnification Agreements and Directors’ and Officers’ Liability Insurance
We have entered into indemnification agreements each of our directors and executive officers against certain liabilities, costs and expenses, and have purchased directors’ and officers’ liability insurance. We also maintain a general liability insurance policy which covers certain liabilities of directors and officers arising out of claims based on acts or omissions in their capacities as directors or officers.
Related Person Transactions Policy
Our Board of Directors has adopted a written related person transaction policy setting forth the policies and procedures for the review and approval or ratification of related person transactions. This policy covers, with certain exceptions set forth in Item 404 of Regulation S-K under the Securities Act, any transaction, arrangement or relationship, or any series of similar transactions, arrangements or relationships, in which we were or are to be a participant, where the amount involved exceeds $120,000 in any fiscal year and a related person had, has or will have a direct or indirect material interest, including without limitation, purchases of goods or services by or from the related person or entities in which the related person has a material interest, indebtedness, guarantees of indebtedness and employment by us of a related person. Under the policy, a “related person” includes the company’s executive officers, directors (including director nominees) and stockholders owning more than 5% of our Common Stock. In reviewing and approving any such transactions, our audit committee is tasked with considering all relevant facts and circumstances, including, but not limited to, whether the transaction is on terms comparable to those that could be obtained in an arm’s length transaction and the extent of the related person’s interest in the transaction.
Except as described below, there have been no transactions since January 1, 2023, in which we were a party, the amount involved exceeded or will exceed $120,000 and in which any related person had a direct or indirect material interest.
Consulting Agreement with Cigall Kadoch, Ph.D.
In October 2015, we entered into a consulting agreement with Cigall Kadoch, Ph.D., our academic co-founder, former director and holder of 8.58% of our Common Stock, pursuant to which Dr. Kadoch provides advisory services related to the manufacturing and sale of products and services related to chromatin remodeling. Under the terms of the consulting agreement, Dr. Kadoch received a grant of 3,953,469 shares of our common stock. Additionally, we agreed to pay Dr. Kadoch a consulting fee of $150,000 per year payable in monthly installments in arrears beginning with the effective date of the consulting agreement, and we agreed to reimburse her for reasonable business expenses incurred in connection with the performance of the services under the agreement. In January 2019, we agreed to increase the consulting fee payable to Dr. Kadoch to $225,000 per year, payable in monthly installments in arrears. Starting in October 2022, the consulting fee payable to Dr. Kadoch was stepped down to an annualized rate of $114,000. In September 2022, the consulting fee payable to Dr. Kadoch was increased to $159,000 per year.
In September 2022, this agreement was extended to January 1, 2024, subject to automatic one-year renewal terms until terminated. During the years ended December 31, 2023 and 2022, we paid Dr. Kadoch $132,500 and $116,390, respectively, pursuant to this agreement.
Collaboration with Loxo Oncology at Eli Lilly and Company
On December 10, 2021, we entered into a strategic collaboration (the “Lilly Collaboration Agreement”) with Loxo Oncology at Eli Lilly and Company (“Lilly”). Concurrent with the Lilly Collaboration Agreement the Company also entered into a stock purchase agreement where the Company issued and sold Lilly 4,000,000 shares of our common stock at a price of $20.00 per share, making them a 5% or greater shareholder in the Company for the years ended December 31, 2023 and 2022. During the third quarter of 2023, pursuant to the terms of the Lilly Collaboration Agreement, we transitioned the BRM Selective inhibitor program into development activities for which Lilly will lead and the Company will participate and share in 50% of the costs until registrational trials. As of December 31, 2023, the Company had a payable to Lilly of $1.5 million recorded in accrued expenses and other current liabilities on the Company’s consolidated balance sheets.
Indemnification Agreements and Directors’ and Officers’ Liability Insurance
We have entered into indemnification agreements each of our directors and executive officers against certain liabilities, costs and expenses, and have purchased directors’ and officers’ liability insurance. We also maintain a general liability insurance policy which covers certain liabilities of directors and officers arising out of claims based on acts or omissions in their capacities as directors or officers.
Director Independence
Under the rules of the Nasdaq Stock Market, independent directors must comprise a majority of a listed company’s board of directors within one year of the completion of its initial public offering. In addition, the Nasdaq Stock Market rules require that, subject to specified exceptions, each member of a listed company’s audit and compensation committees be independent and that director nominees be selected or recommended for the board’s selection by independent directors constituting a majority of the independent directors or by a nominating and corporate governance committee comprised solely of independent directors. Under the Nasdaq Stock Market rules, a director will only qualify as “independent” if, in the opinion of that company’s board of directors, that person does not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director and that such person is “independent” as defined under Nasdaq Stock Market rules and the Exchange Act rules.
Audit committee members must also satisfy the independence criteria set forth in Rule 10A-3 under the Exchange Act. In order to be considered independent for purposes of Rule 10A-3, a member of an audit committee of a listed company may not, other than in his or her capacity as a member of the audit committee, the board of directors or any other board committee: (1) accept, directly or indirectly, any consulting, advisory or other compensatory fee from the listed company or any of its subsidiaries or (2) be an affiliated person of the listed company or any of its subsidiaries.
Based upon information requested from and provided by each director concerning his or her background, employment and affiliations, including family relationships, our Board of Directors has determined that each of our directors, with the exception of Dr. Kadoch and Messrs.Mr. Gottschalk, and Smith, is an “independent director” as defined under applicable rules of the Nasdaq Stock Market, including, in the case of all the members of our audit committee, with the exception of Dr. Cole, the independence criteria set forth in Rule 10A-3 under the Exchange Act, and in the case of all the members of our compensation committee, the independence criteria set forth in Rule 10C-1 under the Exchange Act and are “non-employee directors” as defined in Section 16b-3 of the Exchange Act. In making such determination, our Board of Directors considered the relationships that each such non-employee director has with our Company and all other facts and circumstances that our Board of Directors deemed relevant in determining his or her independence,
including the beneficial ownership of our capital stock by each non-employee director. Mr. Gottschalk is not an independent director under these rules because he is our President and Chief Executive Officer. Each of Dr. Kadoch and Mr. Smith is not an independent director under these rules because of the amount Foghorn has paid to them under their respective consulting agreements.
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
Principal Accountant Fees and Services
We regularly review the services and fees of Deloitte & Touche LLP, our independent registered public accounting firm. These services and fees are also reviewed by the Audit Committee on an annual basis. The aggregate fees billed for the fiscal years ended December 31, 20212023 and 20202022 for each of the following categories of services are as follows (in thousands):
| | Fee Category | Fee Category | 2021 | | 2020 | Fee Category | 2023 | | 2022 |
| Audit Fees (1) | Audit Fees (1) | $ | 671,519 | | | $ | 459,281 | |
| Audit-Related Fees (2) | — | | | 893,000 | |
| Tax Fees | — | | | — | |
| Audit Related Fees | |
| Tax Fees (2) | |
| All Other Fees (3) | All Other Fees (3) | 1,895 | | | 1,895 | |
| Total Fees | Total Fees | $ | 673,414 | | | $ | 1,354,176 | |
(1) Audit fees in 20212023 and 20202022 consist of fees billed for professional services for the audit of our annual consolidated financial statements, the review of our interim consolidated financial statements included in our quarterly reports on Form 10-Q and other professional services normally provided in connection with statutory and regulatory filings, including the issuance of comfort letters and the issuance of consents on registration statements.
(2) Audit Related FeesTax fees consist of fees billed for services provided in connection with our initial public offering.tax planning and consulting purposes.
(3) All Other Fees consist of an annual license fee for the use of accounting research software.
The Audit Committee pre-approved 100% of services performed since the pre-approval policy was adopted.
Policy on Audit Committee Pre-Approval of Audit and Permissible Non-Audit Services of Independent Registered Public Accounting Firm
The Audit Committee pre-approves all auditing services, internal control related services and permitted non-audit services (including the fees and terms thereof) to be performed by Deloitte & Touche LLP. The Audit Committee may delegate pre-approval authority to one or more members of the Audit Committee consistent with applicable law and listing standards, provided that the decisions of such Audit Committee member or members must be communicated to the Audit Committee at its next scheduled meeting.
PART IV
ITEM 15. EXHIBIT AND FINANCIAL STATEMENT SCHEDULES
1. Financial Statements
For a list of the financial statements included herein, see Index to the Consolidated Financial Statements on page 8589 of this Annual Report on Form 10-K, incorporated into this Item by reference. 2. Financial Statement Schedules
Financial statement schedules have been omitted because they are either not required or not applicable or the information is included in the consolidated financial statements or the notes thereto.
3. Exhibits
The exhibits required by Item 601 of Regulation S-K and Item 15(b) of this Annual Report on Form 10-K are listed in the Exhibit Index immediately preceding the signature page of this Annual Report on Form 10-K.
ITEM 16. FORM 10-K SUMMARY
None.
Exhibit Index | | | | | | | | |
Exhibit
number | | Description of document |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | | | | | | | |
Exhibit
number | | Description of document |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | | | | | | | |
Exhibit
number | | Description of document |
| | |
| | |
| | | | | | | | |
Exhibit
number | | Description of document |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| 101* | | Financial statements from the Annual Report on Form 10-K of the Company as of and for the period ended December 31, 2021, formatted in Extensible Business Reporting Language (XBRL): (i) Balance Sheets; (ii) Statements of Operations; (iii) Statements of Changes in Redeemable Preferred Stock and Stockholders’ Equity; (iv) Statements of Cash Flows; and (v) Notes to Financial Statements. |
* Filed herewith
** Furnished herewith
^ Indicates management contract or compensatory plan, contract or arrangement.
++ Portions of this exhibit (indicated by asterisks) have been omitted because the Registrant has determined they are not material and would likely cause competitive harm to the Registrant if publicly disclosed.
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| | | | | | | | |
| FOGHORN THERAPEUTICS INC. |
| | |
Date: March 10, 20227, 2024 | By: | /s/ Allan ReineStephen DiPalma |
| | Allan ReineStephen DiPalma |
| | Interim Chief Financial Officer
(Principal Accounting and Financial Officer) |
SIGNATURES AND POWER OF ATTORNEY
Each person whose individual signature appears below hereby authorizes and appoints Adrian Gottschalk and Allan Reine, and each of them, with full power of substitution and resubstitution and full power to act without the other, as his or her true and lawful attorney-in-fact and agent to act in his or her name, place and stead and to execute in the name and on behalf of each person, individually and in each capacity stated below, and to file any and all amendments to this Annual Report on Form 10-K and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing, ratifying and confirming all that said attorneys-in-fact and agents or any of them or their or his substitute or substitutes may lawfully do or cause to be done by virtue thereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| | | | | | | | | | | | | | |
| Signature | | Title | | Date |
| | | | |
| /s/ Adrian Gottschalk | | President, Chief Executive Officer and Director
(Principal Executive Officer) | | March 10, 20227, 2024 |
| Adrian Gottschalk | | | |
| | | | |
/s/ Allan ReineStephen DiPalma | | Interim Chief Financial Officer
(Principal Accounting and Financial Officer) | | March 10, 20227, 2024 |
Allan Reine, M.D.Stephen DiPalma. | | | |
| | | | |
| /s/ Scott Biller | | Director | | March 10, 20227, 2024 |
| Scott Biller, Ph.D. | | | | |
| | | | |
| /s/ Douglas Cole | | Director | | March 10, 20227, 2024 |
| Douglas Cole, M.D. | | | | |
| | | | |
| /s/ Simba Gill | | Director | | March 10, 20227, 2024 |
| Simba Gill, Ph.D. | | | | |
| | | | |
/s/ Cigall KadochThomas Lynch Jr. | | Director | | March 10, 20227, 2024 |
Cigall Kadoch, Ph.D.Thomas Lynch Jr., M.D. | | | | |
| | | | |
| /s/ Adam Koppel | | Director | | March 10, 20227, 2024 |
| Adam Koppel, M.D., Ph.D. | | | | |
| | | | |
| /s/ Michael Mendelsohn | | Director | | March 10, 20227, 2024 |
| Michael Mendelsohn, M.D. | | | | |
| | | | |
| /s/ B. Lynne Parshall | | Director | | March 7, 2024 |
| B. Lynne Parshall, Esq. | | | | |
| | | | |
| /s/ Ian Smith | | Director | | March 10, 20227, 2024 |
| Ian Smith | | | | |