UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
(Mark One)
☒ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the Fiscal Year Ended Ended: December 31, 2020
☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from __________________ to __________________
Commission file number: 001-37969
ENDRA Life Sciences Inc. |
(Exact Name of Registrant as Specified in Its Charter) |
Delaware | 26-0579295 | |
(State or Other Jurisdiction of Incorporation or Organization) | (I.R.S. Employer Identification No.) | |
3600 Green Court, Suite 350, Ann Arbor, MI | 48105-1570 | |
(Address of Principal Executive Offices) | (Zip Code) |
(734) 335-0468
(Registrant’s telephone number, including area code)
Title of each class | Trading Symbol | Name of each exchange on which registered | ||
Common Stock, par value $0.0001 per share | NDRA | The Nasdaq Stock Market LLC | ||
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company or an emerging growth company. See definitions of "large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer | ☐ | Accelerated filer | ☐ |
Non-accelerated | ☒ | Smaller reporting company | ☒ |
Emerging growth company | ☐ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☐
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether the registrant has filedany of those error corrections are restatements that required a report on and attestation to its management’s assessmentrecovery analysis of incentive-based compensation received by any of the effectiveness of its internal control over financial reporting under Section 404(b) ofregistrant’s executive officers during the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. relevant recovery period pursuant to § 240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act): Yes ☐ No ☒
The aggregate market value of voting and non-voting common equity held by non-affiliates of the registrant, as of June 30, 2020,2023, was approximately $23,166,096$10,256,564 based on the closing sales price of the common stock on such date as reported on the Nasdaq Capital Market.
As of March 24, 2021,2024, there were 41,599,70911,035,659 shares of the registrant’s common stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
None.
ENDRA LIFE SCIENCES INC.
TABLE OF CONTENTS
Page | ||||
4 | ||||
17 | ||||
36 | ||||
36 | ||||
36 | ||||
36 | ||||
36 | ||||
37 | ||||
37 | ||||
Management's Discussion and Analysis of Financial Condition and | 38 | |||
41 | ||||
42 | ||||
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure. | 43 | |||
43 | ||||
43 | ||||
Disclosure Regarding Foreign Jurisdictions that Prevent Inspections. | 43 | |||
44 | ||||
47 | ||||
Security Ownership of Certain Beneficial Owners and Management and Related Stockholders Matters. | 51 | |||
Certain Relationships and Related Transactions, and Director Independence. | 53 | |||
53 | ||||
54 | ||||
55 |
| 2 |
| Table of Contents |
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K (this “Annual Report”) contains “forward-looking statements” within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended, that are intended to be covered by the “safe harbor” created by those sections. Forward-looking statements, which are based on certain assumptions and describe our future plans, strategies and expectations, can generally be identified by the use of forward-looking terms such as “believe,” “expect,” “may,” “will,” “should,” “would,” “could,” “seek,” “intend,” “plan,” “goal,” “project,” “estimate,” “anticipate,” “strategy”, “future”, “likely” or other comparable terms and references to future periods. All statements other than statements of historical facts included in this Annual Report regarding our strategies, prospects, financial condition, operations, costs, plans and objectives are forward-looking statements. Examples of forward-looking statements include, among others, statements we make regarding expectations for revenues, cash flowsregarding: estimates of the timing of future events and financial performance, the anticipated results of our development efforts, andincluding the timing of submission for and receipt of required regulatory approvals and product launches.
Forward-looking statements are neither historical facts nor assurances of future performance. Instead, they are based only on our current beliefs, expectations and assumptions regarding the future of our business, future plans and strategies, projections, anticipated events and trends, the economy and other future conditions. Because forward-looking statements relate to the future, they are subject to inherent uncertainties, risks and changes in circumstances that are difficult to predict and many of which are outside of our control. Our actual results and financial condition may differ materially from those indicated in the forward-looking statements. Therefore, you should not rely on any of these forward-looking statements. Important factors that could cause our actual results and financial condition to differ materially from those indicated in the forward-looking statements include, among others, the following:
· | our limited commercial experience, limited cash and history of losses; | |
· | our ability to obtain adequate financing to fund our business operations in the future; | |
· | our ability to achieve profitability; | |
· | delays and changes in regulatory requirements, policy and guidelines, including potential delays in submitting required regulatory applications or other submissions with respect to U.S. Food and Drug Administration (“FDA”) or other regulatory agency approval; | |
· | our ability to obtain and maintain required CE mark certifications and secure required FDA and other governmental approvals for our Thermo-Acoustic Enhanced Ultrasound (“TAEUS”) applications; | |
· | our ability to develop a commercially feasible application based on our TAEUS technology; | |
· | market acceptance of our technology; | |
· | the effect of macroeconomic conditions on our business; | |
· | results of our human studies, which may be negative or inconclusive; | |
· | our ability to find and maintain development partners; | |
· | our reliance on third parties, collaborations, strategic alliances and licensing arrangements to complete our business strategy; | |
· | the amount and nature of competition in our industry; | |
· | our ability to protect our intellectual property; | |
· | potential changes in the healthcare industry or third-party reimbursement practices; | |
· | our ability to comply with regulation by various federal, state, local and foreign governmental agencies and to maintain necessary regulatory clearances or approvals; | |
· | our ability to maintain compliance with Nasdaq listing standards; | |
· | our dependence on our senior management team; and | |
· | the other risks and uncertainties described in the Risk Factors and in Management’s Discussion and Analysis of Financial Condition and Results of Operations sections of this Annual Report. |
Any forward-looking statement made by us in this report is based only on information currently available to us and speaks only as of the date on which it is made. We undertake no obligation to publicly update any forward-looking statement, whether written or oral, that may be made from time to time, whether as a result of new information, future developments or otherwise.
As used in this Annual Report, unless the context otherwise requires, the terms “ENDRA,” “we,” “us,” “our,” and the “Company” refer to ENDRA Life Sciences Inc., a Delaware corporation.
Overview
We were incorporated as a Delaware corporation in 2007. We are leveraging experience with pre-clinicaldeveloping a next-generation enhanced ultrasound devices to develop technology for increasing the capabilities of clinical diagnostic ultrasoundplatform—Thermo Acoustic Enhanced Ultrasound, or TAEUS® in order to broaden patient access to the safe diagnosis and treatment of a number of significant medical conditions in circumstances where expensive X-ray computed tomography (“CT”) and, magnetic resonance imaging (“MRI”) technology, or other diagnostic technologies such as surgical biopsy, are unavailable or impractical.
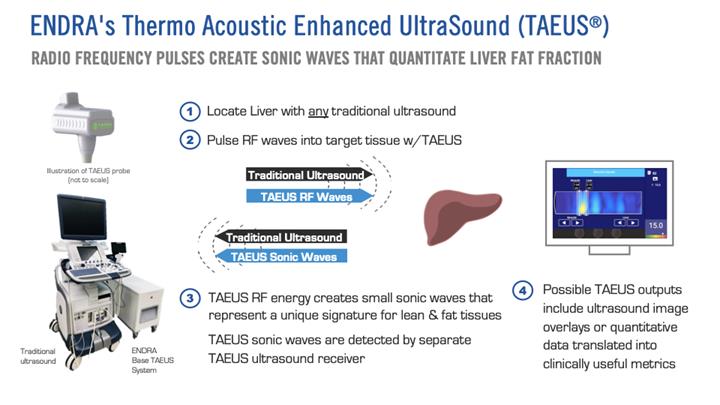
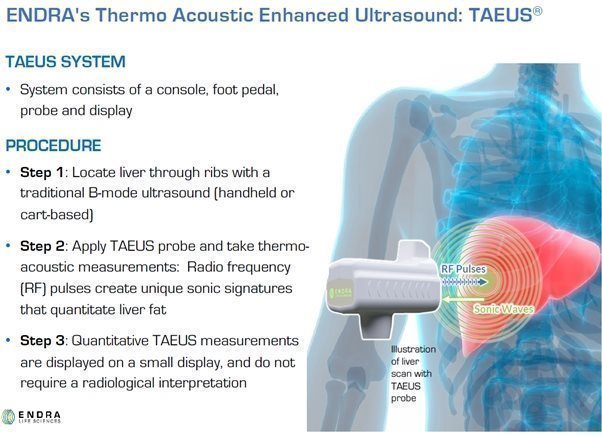
Our TAEUS technology uses radio frequency (“RF”) pulses to stimulate tissues, using a small fraction (less than 1%) of the amount of energy that would be transmitted into the body during an MRI scan. The use of RF energy allows our TAEUS technology to penetrate deep into tissue, enabling the imaging of human anatomy at depths equivalent to those of conventional ultrasound. The RF pulses are absorbed by tissue and converted into ultrasound signals, which are detected by an external ultrasound receiver and a digital acquisition system that is part of the TAEUS system. The detected ultrasound is processed into images and other forms of data using our proprietary software and algorithms and then displayed to complement conventional gray-scale ultrasound images.
We use suppliers of components, such as Blatek Industries, Inc. and Elite RF, LLC, and contract manufacturers, such as Starfish Product Engineering, Inc., to assemble and test the TAEUS liver system for commercial sale. Suppliers are vetted before engaging in work with the Company and are reviewed annually, as part of our quality management system, to assure their performance meets our needs. We have implemented internal processes to monitor designs, inventory and supply of key components needed to manufacture our TAEUS liver system. We plan production in accordance with anticipated market demand and availability and lead times of needed materials.
As described below, our first TAEUS platform application focuses on quantifying fat in the liver and stage progression of nonalcoholic fatty liver disease (“NAFLD”) which, untreated, can progress to Nonalcoholic Steatohepatitis (“NASH”), fibrosis, cirrhosis and liver cancer. In April 2016, we entered into a Collaborative Research Agreement with General Electric Company, acting through its GE Healthcare business unit and the GE Global Research Center (collectively, “GE Healthcare”), under which GE Healthcare has agreed to assist us in our efforts to commercialize this application. In November 2017, we contracted with the Centre for Imaging Technology Commercialization (“CIMTEC”) to initiate human studies, through Canada-based Robarts Research Institute, with our TAEUS device targeting NAFLD. In October 2018, we received an Investigational Testing Authorization (“ITA”) from Health Canada to commence the first human studies in healthy volunteers with our TAEUS clinical system targeting NAFLD, guiding our algorithm development, and comparing our technology to MRI. The feasibility study the first of several planned human studies, was conducted in collaboration with the widely respected Robarts Research Institute in London, Ontario, Canada. We reported the completion and top-level findings of this study in September 2019. The data collected from the study, including additional usability inputs, was included in our TAEUS liver device technical file submission for device CE mark, which we submitted in December 2019. We received CE mark approval for our NAFLD TAEUS application in March 20202020. We have registered the product in each of our primary target European markets (i.e., Germany, France, and the United Kingdom). As of December 31, 2023, we had in effect seven clinical evaluation agreements with research hospitals in North America, Europe and Asia for the conduct of clinical studies comparing our TAEUS clinical system to MRI-Proton Density Fat Fraction (“MRI-PDFF”) in the measurement of liver fat.
In June 2020, we completed thesubmitted a 510(k) Premarket Notification submissionApplication to the FDA for the application.
Diagnostic Imaging Technologies
Diagnostic imaging technologies such as CT, MRI and ultrasound allow physicians to look inside a person’s body to guide treatment or gather information about medical conditions such as broken bones, cancers, signs of heart disease or internal bleeding. The type of imaging technology a physician uses depends on a patient’s symptoms and the part of the body being examined. CT technology is well suited for viewing bone injuries, diagnosing lung and chest problems, and detecting cancers. MRI technology excels at examining soft tissue in ligament and tendon injuries, spinal cord injuries, and brain tumors. CT scans can take as little as 5 minutes, while an MRI scan can take up to 30 minutes.
| 4 |
| Table of Contents |
Unfortunately, while CT and MRI systems are versatile and create high quality images, they are also expensive and not always accessible to patients. A CT system costs approximately $1 million and an MRI system can cost up to $3 million. CT and MRI systems are large and can weigh several tons, typically requiring significant modifications to existing healthcare facilities to safely site the CT and MRI equipment. Because of their size and weight, CT and MRI systems are usually fixed-in-place at major medical facilities. As a result, they are less accessible to primary care and rural clinics, economically developing markets, and patient bedsides. As of 2018,2024, there were onlyare approximately 63,00080,000 CT systems and 50,00058,000 MRI systems in the world, approximately 50% of which wereworldwide, with a significant portion located in the U.S. and Japan.
While CT and MRI systems create high quality images, their use is not always practical. For example, the diagnosis and treatment of the estimated 1.42.5 billion people suffering from NAFLD requires ongoing surveillance of the patients’ livers to assess the progression of the disease and the efficacy of treatment. However, the use of CT and MRI systems to perform that surveillance is impractical for a number of reasons, including the high cost of the scan and the limited availability of CT and MRI systems and the required use of contrast agents, including those containing radioactive substances that can cause allergic reactions and reduced kidney functions.systems. Patient exposure to the ionizing radiation generated by a CT system must be limited for safety reasons. Similarly, because of the strong magnetic field created by an MRI machine, patients with metal joint replacements or cardiac pacemakers cannotmay be imaged withlimited for safety reasons in their use of an MRI system.
Ultrasound Technology
An ultrasound machine transmits sound waves, which bounce off tissues, organs and blood in the body. The ultrasound machine captures these echoes and uses them to create an image. Ultrasound technology excels at imaging the structure of internal organs, muscles and bone surfaces. Due to its utility, cost-effectiveness and safety profile, ultrasound imaging is frequently used in a physician’s examination room or at a patient’s bedside as a first-line diagnostic tool, which has resulted in an overall increase in the number of ultrasound scans performed.
Ultrasound systems are more broadly available to patients than either CT or MRI systems. There are an estimated one1.6 million diagnostic ultrasound systems globally in use today. Ultrasound systems are relatively inexpensive compared to CT and MRI systems, with smaller portable ultrasound systems costing as little as $10,000$5,000 and new cart-based ultrasound systems costing between $75,000 and $200,000. Ultrasound systems are also more mobile than CT and MRI systems and many are designed to be moved by an operator from room to room, or closer to patients. Ultrasound technology does not present the same safety concerns as CT and MRI technology, since ultrasound does not emit ionizing radiation and ultrasound contrast agents are generally considered to be safe.
However, ultrasound’s imaging capabilities are more limited compared to CT and MRI technology. For example, ultrasound systems cannot measure tissue temperature during thermal ablation surgery or quantify fat to diagnose early-stage liver disease -- instancesdisease-instances where CT and MRI systems are used.
Ultrasound Market
The global diagnostic ultrasound procedures globally. Additionally, an estimated 30,000device market size was valued at $7.7 billion in 2023 and is anticipated to 50,000 new and replacement systems are sold into the market each year.expand at a CAGR of 4.07% from 2022 to 2030. These numbers include both portable and cart-based ultrasound systems, and cover all types of diagnostic ultrasound procedures, including systems intended for cardiology, prenatal and abdominal use. We do not currently intend to address cart-based ultrasound systems focused on applications in prenatal care, nor certain portable ultrasound applications such as emergency room medicine, where we believe our TAEUS technology willmay not substantially impact patient care. Accordingly, we defineestimate our addressable market for one or more of our current or future TAEUS applications atto include approximately 365,000 cart-based700,000 ultrasound systems currently in use throughout the world.
We believe that demand for ultrasound systems is driven primarily by the following factors:
· | Population growth and age demographics that increase the demand for diagnostic screening for cancer, cardiology, and prenatal applications. | |
· | Economic development broadening investment in healthcare in underserved markets such as China and Latin America, where ultrasound technology has significant appeal due to its price point and flexibility at point-of-care. | |
· | Expanding ultrasound applications and improving image quality that drive demand for new ultrasound technologies, such as software enhancements, bi-axial probes, and dedicated single application systems. | |
· | Positive insurance reimbursement rate trends for ultrasound diagnostics due to the technology’s safety and cost-effectiveness. |
| 5 |
| Table of Contents |
Unmet Need
We believe that the limited availability of high-utility and cost-effective imaging technology represents a significant unmet medical need. We believe that expanding the capability of ultrasound technology to perform more of the imaging tasks presently available only on expensive CT and MRI systems will help to satisfy this unmet need.
Our Solutions
Our TAEUS technology uses a pulsed energy source – specifically, radio-frequency (“RF”) – source-specifically, RF—to generate ultrasonic waves in tissue. These waves are then detected with ultrasound equipment and used to create high-contrast images and other forms of data using our proprietary algorithms. Unlike conventional ultrasound, which creates images based on the scattering properties of tissue, thermoacoustic imaging provides tissue absorption maps of the pulsed energy, similar to those generated by CT scans. Ultrasound is only utilized to transmit the absorption signal to the imaging system outside of the body.
Our TAEUS Technology Platform for Clinical Applications
To increase the utility of our thermoacoustic technology, in 2013 we began to develop our TAEUS technology platform. Unlike the near-infrared light pulses used in our earlier photoacoustic systems, our TAEUS technology uses RF pulses to stimulate tissues, using a small fraction of the energy transmitted into the body during an MRI scan. Using RF energy enables our TAEUS technology to penetrate deep into tissue, enabling the imaging of human anatomy at depths equivalent to those of conventional ultrasound. The RF pulses are absorbed by tissue and converted into ultrasound signals, which are detected by an external ultrasound receiver and a digital acquisition system that is part of the TAEUS system. Our RF-based thermoacoustics imaging is not adversely affected by blood-filled organs, enabling our TAEUS technology to be used in clinical liver applications, among others. The detected ultrasound can then be processed into ultrasound overlays or quantitative data that may be translated into clinically useful metrics using our proprietary algorithms and displayed to complement conventional gray-scale ultrasound images. The TAEUS imaging concept is illustrated below:
After required regulatory approvals, our TAEUS technology can be added as an accessory to existing ultrasound systems, helping to improve clinical decision-making on the front lines of patient care, without requiring substantially new clinical workflows or large capital investments. We are also developingintend to offer a license for our TAEUS technology to OEMs, such as ultrasound and thermoablative capital equipment makers, for incorporation intoin their new ultrasound systems manufactured by companies such as GE Healthcare, described more fully below.
| 6 |
| Table of Contents |
We believe that our TAEUS technology has the potential to add a number of new capabilities to conventional ultrasound and other types of capital equipment, thereby enhanceenhancing the utility of both existing and new ultrasound systems and extendextending the use of ultrasound technologythese technologies to circumstances that either currently require the use of expensive CT or MRI imaging systems, where imaging is not practical using existing technology, or where other assessment tools such as surgical biopsy are required. To demonstrate the capabilities of our TAEUS platform, we have conducted various internal ex-vivo laboratory experiments and limited internal in-vivo large animal studies. In our ex-vivo and in-vivo testing, we have demonstrated that the TAEUS platform has the following capabilities and potential clinical applications:
· | Tissue Composition: Our TAEUS technology enables ultrasound to distinguish fat from lean tissue. This capability would enable the use of TAEUS-enhanced ultrasound for the early identification, staging and monitoring of NAFLD, a precursor to NASH, liver fibrosis, cirrhosis and liver cancer. | |
· | Temperature Monitoring: Our TAEUS technology enables traditional ultrasound to visualize changes in tissue temperature, in real time. This capability would enable the use of TAEUS-enhanced ultrasound to guide thermoablative therapy, which uses heat or cold to affect tissue, such as in the treatment of cardiac atrial fibrillation, or removal of cancerous liver and kidney lesions, with greater accuracy, and perform cosmetology procedures such as lipolysis of abdominal fat. | |
· | Vascular Imaging: Our TAEUS technology has the potential to enable visualization of blood vessels from any angle, using only a saline solution contrasting agent, unlike Doppler ultrasound, which requires precise viewing angles. This capability would enable the use of TAEUS-enhanced ultrasound to assist in identifying arterial plaques or malformed vessels. | |
· | Tissue Perfusion: Our TAEUS technology has the potential to image blood flow at the capillary level in a region, organ or tissue. This capability could be used to assist physicians in characterizing abnormalities in tissue perfusion symptomatic of damaged tissue, such as internal bleeding from trauma, or diseased tissue, such as certain cancers. |
Because of the large number of traditional ultrasound systems currently in global use, we are first developing our TAEUS technology for sale as an aftermarket accessory that works with existing ultrasound systems. Because our TAEUS technology is designed to enhance the utility of, not replace, conventional ultrasound, we believe healthcare providers will be able to increase the utilization of, and generate new revenue from, their existing ultrasound systems once we obtain required regulatory approval for specific applications. We further believe that clinicians will be attracted to our technology because it will enable them to perform more procedures with existing ultrasound equipment, thereby retaining more imaging patients in their clinics rather than referring patients out to a regional medical center for a CT or MRI scan.
ENDRA’s first clinical product is designed to interface with a conventional ultrasound scanner, utilizing the scanner’s B-mode imaging to guide the selected region for assessment of liver fat content. The following sub-systems will comprise ENDRA’s first generation product.
Radio Frequency (RF) Source and Computer:
The RF source consists of a low power waveform generator and an amplifier. Together, these components provide the characteristic pulses required to excite thermoacoustic signals in tissue. The computer provides processing capability to both utilize the conventional ultrasound data for navigation to the measurement site of interest, and the calculations required to convert digitized thermoacoustic signals to measurements of fat in liver tissue. The entire sub-system will reside in a single enclosure, on wheels, and sit adjacent to the ultrasound imaging system.
Specialized Transducer:
A single channel ‘receive only’“receive only” ultrasound transducer is specifically designed and optimized for thermoacoustic imaging. The transducer sub-system will detect thermoacoustic signals excited by the RF source within the liver. The transducer assembly includes electronics for signal amplification, digitization, and signal processing. The specialized transducer will attach towork in concert with the conventional ultrasound probe used for liver imaging.
RF Applicator:
The RF applicator transmits pulses of energy, provided by the RF source, into tissue. The applicator is positioned in proximity to the target region for measurement.
TAEUS platforms will provide two dimensionaltwo-dimensional imaging with a transducer composed of multiple receive elements. The RF source and applicator would be similar to those in the first generationfirst-generation product but the multi-element transducer would allow for multiple applications including: reading tissue composition, temperature, vascular flow, tissue perfusion, and other potential applications. Ultimately, we expect our technology will be incorporated into conventional ultrasound systems and our business model will transition from producing stand-alone systems to licensing our technology, IP and specialized components to ultrasound OEMs. Existing ultrasound equipment already includes power supplies, computation, high speed electronics, and ultrasound transducers, which may be leveraged by our thermoacoustic imaging applications. The RF source and applicator are the principal hardware components that will be added to OEM ultrasound systems for the OEM fully integrated form of our product.
| 7 |
| Table of Contents |
We are following a model that mirrors the approach used by companies in the past to introduce new ultrasound imaging capabilities to existing conventional ultrasound scanners. Color Doppler, elastography, 3-D imaging, and high channel count systems were all introduced by new companies (not already involved in conventional ultrasound imaging). Historically, ultrasound imaging has grown through the introduction of unique technology and capabilities that expanded the applications and use of clinical ultrasound in a form that often added separate hardware to existing ultrasound systems. Ultimately, as these new technologies gained acceptance in the marketplace they were incorporated into OEM-designed and built systems that were sold by the leading ultrasound imaging vendors.
TAEUS System for our TAEUS Technology
Our first TAEUS platform application will focusfocuses on quantifying fat in the liver and stage progression of NAFLD which, untreated, can progress to NASH, fibrosis, cirrhosis and liver cancer. In 2015,2022, over 1.42 billion people globally were estimated to be affected by NAFLD/NASH.NAFLD. The World Gastroenterology OrganisationOrganization considers NAFLD/NASH a global pandemic affecting rich and poor countries alike. Obesity, hepatitis, and diabetes are leading contributors to the development of NAFLD.
Left untreated, an estimated 30% of NAFLD cases progress to NASH, a condition in which liver fat causes inflammation and decreased liver function, possibly resulting in fatigue, weight loss, muscle pain and abdominal pain. Excess liver fat remains a root cause of and key clinical concern for both of NASH and NAFLD.
Approximately 25% of NASH cases progress to liver fibrosis, in which liver inflammation causes scar tissue which eventually prevents the liver from functioning properly. The scar tissue blocks the flow of blood through the liver and slows the processing of nutrients, hormones, drugs, and naturally produced toxins. It also slows the production of proteins and other substances made by the liver. Once a patient develops cirrhosis of the liver, the only life-saving therapy is a liver transplant. Additionally, cirrhosis patients may develop liver cancer. In 2018,January 2023, the World Health OrganizationAmerican Cancer Society estimated that liver cancer kills 782,000over 700,000 people annually. Because of the increased incidence of obesity, hepatitis and diabetes throughout the world, NAFLD has become the most common chronic liver disease and an important cause of cirrhosis and liver cancer worldwide.
Despite the increased incidence of NAFLD and its role in the development of NASH, cirrhosis and liver cancer, we believe that no low-cost, accurate and safe method exists for measuring fat in the liver. Current liver enzyme blood tests are indicative, but cannot reliably confirm early stage NAFLD or NASH, and liver enzyme levels are normal in a large percentage of patients with NAFLD. Existing ultrasound technology can only measure fat qualitatively in the liver at moderate to severe levels, typically greater than 30% liver fat, and ultrasound has low accuracy when used on obese patients. While early stage NAFLD and NASH can be confirmed by an MRI scan, an MRI scan is expensive, and MRI systems are not widely available or practical for many patients. A surgical biopsy can be used to confirm NAFLD and NASH, but is also expensive, involves a painful procedure and exposes patients to the risk of infection and bleeding. Furthermore, MRIs and surgical biopsies are impractical for repeated screening and monitoring of liver disease. We believe these limitations negatively impact the diagnosis and treatment of patients with NAFLD.
Billions of dollars are spent annually on the global diagnosis and treatment of NAFLD and related liver diseases. In the United States, annual direct medical costs for NAFLD were estimated in 2016 to be $103 billion, and in the Europe-4 countries (Germany, France, Italy, and United Kingdom), about €35 billion. Patients diagnosed with NAFLD and related liver diseases are typically treated with available therapies such as statins, insulin sensitizers and other compounds and are encouraged to adopt lifestyle changes to reduce their weight and improve their overall health.
In addition, patients receiving treatment for NAFLD-spectrum liver diseases must continue to be monitored to assess disease progression and the efficacy of treatment. Because of the high cost and limited global availability, CT and MRI technology is not typically used for this function.
We believe our TAEUS technology will enable primary care physicians, radiologists and hepatologists to diagnose NAFLD earlier and monitor patients with NAFLD-spectrum liver diseases more accurately and cost-effectively than is possible with existing technology.
Potential Licensing and Partnership Opportunities
A pipeline of 20+ pharmaceutical compounds targeting liver disease are in development by companies such as Viking Therapeutics, Inventiva, Madrigal Pharmaceuticals, Inc., Akero Therapeutics and Regeneron Pharmaceuticals. The pharmaceutical industry’s increased presence in the liver disease space represents a synergistic opportunity for ENDRA, as early detection of NAFLD could enable prescription of drug treatment at the most advantageous time for patients. The companies can also benefit from simpler, non-invasive measurements of biomarkers, such as liver fat, in the clinical stage. To this end, in March 2021, ENDRA announced a collaboration agreement with Hepion Pharmaceuticals to incorporate TAEUS as an add-on technology to support Hepion’s patient screening and biomarker measurements during its Phase 2b study of its lead drug candidate, and is working with Hepion to identify a target site at which to utilize TAEUS.
| 8 |
| Table of Contents |
In April 2016, we entered into a Collaborative Research Agreement with General Electric Company, acting through its GE Healthcare business unit and the GE Global Research Center (collectively, “GE Healthcare”).Healthcare. Under the terms of the agreement, GE Healthcare has agreed to assist us in our efforts to commercialize our TAEUS technology for use in a fatty liver application by, among other things, providing equipment and technical advice, and facilitating introductions to GE Healthcare clinical ultrasound customers. In return for this assistance, we have agreed to afford GE Healthcare certain rights of first offer with respect to manufacturing and licensing rights for the target application. More specifically, we have agreed that, prior to commercially releasing our NAFLD TAEUS application, we will offer to negotiate an exclusive ultrasound manufacturer relationship with GE Healthcare for a period of at least one year of commercial sales. The commercial sales would involve, within our sole discretion, either our commercially selling GE Healthcare ultrasound systems as the exclusive ultrasound system with our TAEUS fatty liver application embedded, or GE Healthcare being the exclusive ultrasound manufacturer to sell ultrasound systems with our TAEUS fatty liver application embedded. The agreement with GE Healthcare does not prevent us from selling our TAEUS fatty liver application technology to distributors or directly to non-manufacturer purchasers. Additionally, the agreement provides that (1) prior to offering to license any of our TAEUS fatty liver application intellectual property to a third party, we will first offer to negotiate to license our TAEUS fatty liver application intellectual property to GE Healthcare and (2) prior to selling any equity interests to a healthcare device manufacturer, we must first offer to negotiate in good faith to sell such equity interests to GE Healthcare. The agreement is subject to termination by either party upon not less than 60 days’ notice. On December 16, 2020,2022, we and GE Healthcare entered into an amendment to our agreement, extending its term to December 16, 2022.
Clinical Studies
In October 2018, we received an Investigational Testing Authorization (“ITA”) from Health Canadaauthorization to commence the first human studies in healthy volunteers with our TAEUS clinical system targeting NAFLD, guiding our algorithm development, and comparing our technology to MRI. The feasibility study was conducted in collaboration with the widely respected Robarts Research Institute in London, Canada. The data Robarts Research Institute collected with our investigational device included the following:
Commercialization
We received CE mark approval for our TAEUS FLIP (Fatty Liver Imaging Probe) system in March 2020, indicating that the TAEUS FLIP system complies with all applicable European Directives and Regulations in the European Union (“EU”) and other CE mark geographies, including the 27 EU member states. In support of our commercialization efforts in the EU, we have a full time sales representative in each of France, the United Kingdom, and Germany and expect to expand marketing efforts into other CE markets as we grow. We actively attend various trade shows and clinical conferences across the UK and EU to drive our marketing presence amongst medical professionals that constitute our target market. We have also entered into agreements with clinical evaluation sites in Switzerland, Germany, UK and France to collect clinical evidence with the aim to underscore the clinical utility of the TAEUS device for assessing NAFLD.
We are pursuing FDA premarket clearance of our TAEUS FLIP system to enable sales in the United States. See further discussion above in “Item 1. Business – Overview.”
Other Potential Clinical Applications for our TAEUS Technology
Temperature Monitoring of Thermoablative Surgery
We also intend to develop a TAEUS platform application to guide thermal ablation surgery, such as in the treatment of cardiac atrial fibrillation, chronic pain and lesions of the liver, thyroid, kidneys and other soft tissues. We plan to target clinical users of thermoablative technology, including interventional radiologists, cardiologists, gynecologists and surgical oncologists.
Thermoablation involves the use of heat or cold to remove malfunctioning or diseased tissue in surgical oncology, cardiology, neurology, gynecology, urology and urologycosmetology applications. Thermoablative technologies include RF, microwave, laser and cryogenic ablation. The worldwideglobal radiofrequency ablation devices market for RF surgical ablation procedures alonesize was estimated in 2015 to be $3.7 billion per annum, generating over 5 million annual RF ablation procedures and growingvalued at approximately 18% annually. We believe that$3.6 billion in 2021 and is expected to surpass $10.2 billion by 2030, representing a CAGR of 11% during the growth of this market is driven primarily by the aging global population requiring more cardiac and cancer procedures, as well as the relative ease-of-use and low cost of thermoablative technologies when compared to open surgery.
However, RF and other thermoablative surgery technologies pose risks, including under-treatment of diseased tissue and unintended thermal damage to areas outside the treatment area. For example, it has been reported that patients receiving RF ablation of liver tumors have experienced thermal injury to the diaphragm, gallbladder, bile ducts and gastrointestinal tract, some of which have resulted in patient deaths.
| 9 |
| Table of Contents |
Clinicians must rely on printed manufacturer guidelines to plan procedures using thermal ablation technologies or, when available, monitor tissue temperature changes in real-time with MRI imaging or surgical temperature probes. We believe these existing methods either lack real-time precision or are impractical due to cost, poor availability and other factors.
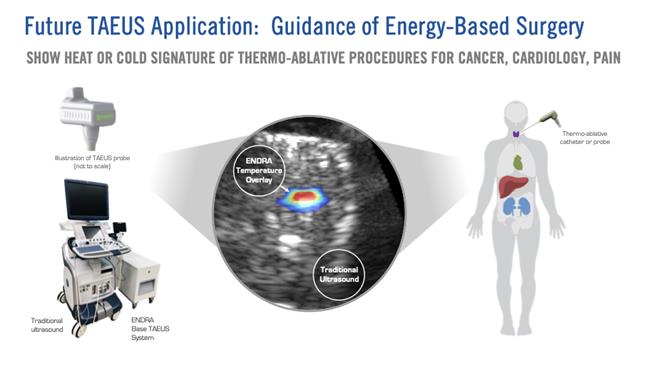
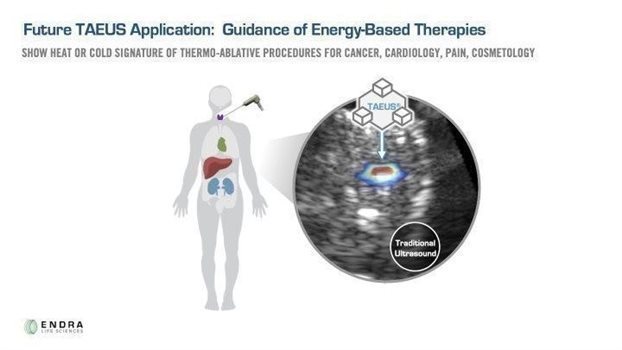
We believe that the ability to visualize changes in tissue temperature in real time could potentially enhance the effectiveness and safety of thermoablation therapies and that our TAEUS technology platform combined with traditional ultrasound has the potential to guide thermoablation surgery more cost-effectively and more accurately than existing methods.
Image below: Depiction of ex-vivo TAEUS tissue temperature analysis overlaid on traditional ultrasound image.
Vascular Imaging
We believe that our TAEUS technology can be used to image blood vessels and distinguish them from the surrounding tissue. In addition to our NAFLD and thermoablation applications, we intend to develop a cardiovascular application based on our TAEUS technology that, with the use of a standard saline contrast agent, can enable existing ultrasound systems to perform a number of cardiovascular diagnostic functions, such as identifying arterial plaque or blocked or malformed vessels, as well as safely guiding biopsies away from vital vasculature.
Conventional ultrasound imaging systems use Doppler imaging in a variety of vascular applications. Doppler ultrasound, which images the velocity of blood, is effective in larger vessels and regions where blood velocity is high. However, Doppler ultrasound is not sufficiently sensitive for use in very small vessels or in vascular imaging applications where blood velocities are very low. For these applications, contrast enhanced CT and MRI angiography is used which requires the patient to be injected with a contrast agent, iodinated compounds and gadolinium, respectively. Contrast-enhanced CT and MRI scans both require referral for examination after initial screening with ultrasound and carry risks associated with their respective contrast agents. We believe that our TAEUS platform has the potential to offer the advantages of CT and MR contrast enhanced imaging at the point of care using only a safe electrolyte solution as the contrast agent.
Tissue Perfusion or “Leakiness”
We believe that our TAEUS technology can be used to image tissue perfusion, or the absorption of fluids into an organ or tissue. We intend to develop an application for our TAEUS platform that would enable ultrasound detection of microvasculature fluid flows symptomatic of tissue compromised by trauma or disease.
| 10 |
| Table of Contents |
When a person’s body is affected by disease or trauma, blood and other fluids may leak from damaged tissues in subtle ways. Traditional ultrasound cannot effectively image these disruptions in microvascular permeability, but we believe ultrasound combined with our TAEUS technology can.
We believe that, using our TAEUS technology, physicians will be able to quickly and clearly see tissue compromised by disease, such as cancer or trauma, especially with the use of a standard saline contrast agent, when CT or MRI is not readily available.
Intellectual Property
We rely on a combination of patent, copyright, trademark and trade secret laws and other agreements with employees and third parties to establish and protect our proprietary intellectual property rights. We require our officers, employees and consultants to enter into standard agreements containing provisions requiring confidentiality of proprietary information and assignment to us of all inventions made during the course of their employment or consulting relationship. We also enter into nondisclosure agreements with our commercial counterparties and limit access to, and distribution of, our proprietary information.
We are committed to developing and protecting our intellectual property and, where appropriate, filing patent applications to protect our technology. Our issued and pending patents claims are directed at the following areas related to our technology:
· | Methods to induce and enhance thermoacoustic signal generation; | |
· | System configurations, devices and novel hardware for transmission of RF pulses into tissue and detection of acoustic signals; | |
· | Methods for integrating our devices with existing conventional ultrasound systems; and | |
· | Methods and algorithms for signal processing, image formation and analysis. |
As of the date of this Annual Report,December 31, 2023, we maintainmaintained a patent portfolio consisting of fourteen (14)forty (40) patents issued in the United States and thirteen (13)thirty-two (32) issued patents in foreign jurisdictions, twenty-one (21)four (4) patent applications pending in the United States and twenty-six (26)twenty-three (23) patent applications pending in foreign jurisdictionsinternationally relating to our technology. These patents and patent applications mostlylargely cover certain innovations relating to fat imaging, fat quantitation, and temperature monitoring in the liver and other tissues. In addition, we have three (3) licensed U.S. patents.
Each of our utility patents generally has a term of 20 years from its respective priority (earliest filing) date. Design patents have a term of 14 years from athe filing date of the respective filing date.application. Among our issued utility patents in the U.S., the first patent is set to expire in 2033 and the last patent is set to expire in 2039. Our licensed patents are set2041.
In November 2023, we engaged PatentVest, Inc. (“PatentVest”), a specialized consulting firm focused on intellectual property valuation, intellectual property portfolio management and intellectual property M&A for clients seeking to expire on June 19, 2021.
Sales and Marketing
In parallel to securing all necessary government marketing approvals, we have begun to hirehired a small internal sales and marketing team to engage and support channel partners and clinical customers. As we previously did with our Nexus 128 system, wecustomers in primary geographic markets - initially in France, the UK, and Germany, expected to be followed in the U.S. after FDA approval. We also intend to partner with several geographically-focused independent clinical ultrasoundmedical device equipment distributors to market and sell our TAEUS applications.applications in secondary markets. For instance, we have entered into a distribution agreement with a third-party covering future sales in Vietnam. We believe that these distributors have existing customer relationships, a strong knowledge of diagnostic imaging technology and the capabilities to support the installation, customer training and post-sale service of capital equipment and software.
We also intend to work with original equipment manufacturers, or OEMs, of capital medical equipment (e.g., ultrasound equipment and thermal ablation equipmentequipment) to sell our TAEUS applicationstechnology alongside their own new systems and into their existing installed base systems. We believe that these OEMs will find our applications attractive as the applications wouldcould enable them to generate additional revenue from their installed systems –- as they currently do with aftermarket accessory portfolios. We believe our relationship with GE Healthcare will facilitate this strategy.
Based on our design work and our understanding of the ultrasound accessorymedical capital equipment market, we intend to price our initial NAFLDliver TAEUS applicationsystem at a price point approximating $35,000 to $55,000,of approximately $65,000, which shouldwe believe could enable clinical purchasers to recoup their investment in less than one year by performing a relatively small number of additional ultrasoundprocedures, initially paid out-of-pocket by patients until government and private insurance reimbursement is secured for the TAEUS liver procedures.
| 11 |
| Table of Contents |
Some of our future TAEUS offerings are expected to be implemented via a hardware platform that can run multiple individual software applications that we plan to offer TAEUS users for a one-time licensing fee, enabling users to perform more procedures with their existing ultrasound equipment and retaining more patients in their clinics rather than referring them out to a regional imaging medical center for a CT or MRI scan.
We also intend to offer a license for our TAEUS technology to OEMs, such as GE Healthcare,ultrasound and thermoablative capital equipment makers, for incorporation in their new ultrasoundcapital equipment systems.
Engineering, Design and Manufacturing
Development of TAEUS Device
We contracted with StarFish Product Engineering, Inc. (“StarFish”), a medical device contract manufacturing company, to develop ENDRA’s prototype TAEUS device into a clinical product that met CE regulatory requirements required for commercial launch.regulatory clearance in the EU. We leveraged StarFish’s expertise infor the preparation and submission of our CE Technical File documentation, submitted in December 2019, which enabled us to secure the European Union CE Mark for the TAEUS liver application in March 2020. We also leveraged StarFish’s expertise infor preparation of documentation for the 510(k) submission made to the FDA in June 2020.
Regulatory Approval Pathway and Human Study
Each of our TAEUS platform applications will require regulatory approvals before we are able to sell or license the application. Based on certain factors, such as the installed base of ultrasound systems, availability of other imaging technologies, such as CT and MRI, economic strength and applicable regulatory requirements, we sought initial approval of our applications for sale in the European Union, followed by the United States and plan to seek additional approval in China.
The first TAEUS application we intend to commercialize is our NAFLD TAEUS application. Our initial target market for this application is the European Union. For commercial reasons and to support our application for CE marking, we contracted with CIMTEC, a medical imaging research group, to conduct human studies through Canada-based Robarts Research Institute to demonstrate our NAFLD TAEUS application’s ability to distinguish fat from lean tissue. In December 2018, Robarts Research Institute completed its initial healthy subject enrollment and data collection of 25 subjects and received authorization from Health Canada to expand the study to 50 subjects. In September 2019, we announced the completion and reported top-level findings of the expandedRobarts Research Institute’s initial healthy subject study and data collection of 50 subjects, which was included in our TAEUS liver device technical file submission for device CE mark. We received CE mark approval for our NAFLD TAEUS application in March 2020. We are now in the process of notifying the competent authorities that we have received the CE mark and registeringregistered the product in each of the initial target markets.
In May 2021, Regulation (EU)2017/745 on medical devices (the “Medical Device Regulation” or “MDR”) will comecame into effect. The MDR amended the prior existing regulatory framework in the EU and imposes significant additional obligations on medical device-related companies. Changes imposed by the MDR include more restrictive requirements for clinical evidence and pre-market assessment of safety and performance, revised classifications to indicate risk levels, stricter requirements for third party testing by government accredited groups for some types of medical devices, and tightened and streamlined quality management system assessment procedures.procedures, including post marketing surveillance obligations. These new rules couldalso impose additional requirements on our business, such as a requirement to conduct clinical trials to maintain our existing and obtain additional CE mark applicationsnew or renewed conformity assessment certification for existing and new products. Also, the MDR provides for additional post-market surveillance obligations, and further requirements for the traceability of products, transparency, refined responsibilities for economic operators (including manufacturer, distributors and importers) as well as a tightened and more comprehensive quality management system.
In March 2020, we received a positive certification from a government-accredited group (“Notified Body”) for our NAFLD TAEUS application, enabling us to market this application in the EU with the necessary CE Mark. The certification, which has been issued under the then applicable framework of the Medical Device Directive but taking into consideration the transitionary provisions of the MDR, should initially expire in May 2024 and re-certification under the MDR will be required in order to continue marketing of the application in the EU. However, there is currently a significant lag for recertification of medical devices under the MDR, due to the requirement to have the competent Notified Bodies be re-designated for purposes of the MDR, as there is a shortage of available Notified Bodies that have already been re-designated for all the medical devices requiring (re-)certification. In light of this development, in February 2023, the European Parliament adopted a Regulation to amend the MDR transition period and to remove the sell-off provisions in the MDR. Specifically, through the newly adopted Regulation the validity of the CE certification for Class I, Class IIa and certain Class IIb devices (which includes ENDRA’s Class IIa device) has been extended until December 31, 2028, subject to certain conditions (including, among others, continued compliance with the MDR, no significant changes to design or intended purpose, a quality management system, and engagement with a Notified Body to obtain conformity assessment). ENDRA is working with its Notified Body to ensure a timely MDR CE Mark transition, while aligning to the extended transition deadline.
| 12 |
| Table of Contents |
In June 2020, we submitted to the FDA our application under the Federal Food, Drug and Cosmetic Act (the “FD&C Act”) to sell our NAFLD TAEUS application in the U.S.United States. The application was submitted for approvalclearance under Section 510(k) of the FD&C ActAct. Following meetings with the FDA in connection with its review of our application, we determined that the 510(k) pathway was not the optimal option due to the novel nature of our TAEUS system and, we anticipate that any other TAEUS applicationsin February 2022, announced that we developwould pursue the “de novo” pathway to request the classification of our NAFLD TAEUS application as a Class II device, as described below under “FDA Approval or Clearance of Medical Devices”. In the third quarter of 2023, we submitted a de novo request to the FDA that included as support clinical data gathered from human studies comparing liver fat measurements by our TAEUS device to measurements by MRI-PDFF. In the fourth quarter of 2023, the FDA sent us an AI request related to our de novo application. Since we received the AI request, we have had several interactions with the FDA and have provided additional information. In order to fully respond to the FDA’s questions, we will similarly be submitted under Section 510(k). need to compile additional clinical data, provide additional device test data, and respond to cybersecurity related questions in a new de novo submission. We have a scheduled in-person pre-submission meeting with the FDA in the second quarter of 2024. We currently anticipate completing the necessary clinical studies by the fourth quarter of 2024 and submitting the new de novo request to the FDA in the first half of 2025.
We expect that, should we be successful in obtaining the FDA’s grant of our initial FDAde novo request, we will have clearance will allow us to sellmarket the NAFLDliver fat fraction TAEUS application in the U.S. with general imagingspecific tissue fat content claims. However, we will need to obtain additional FDA clearances to be able to make diagnostic claims for fatty tissue content determination. Accordingly, to support our commercialization efforts we expect that, following receipt of the FDA’s grant of our initial FDA clearance,de novo request, we would submit one or more additional applications to the FDA, each of which would need to include additional clinical trial data, so that following receipt of the necessary clearances we may make those diagnostic claims.
Regulation
European Union
The primary regulatory environment in Europe is the European Union, which consists of 27 member states encompassing most of the major countries in Europe.Union. In the European Union, applications incorporating our TAEUS technology are regulated as Class IIa medical devices by the European Medicines Agency (the “EMA”) and the European Union Commission.devices. As described above, our NAFLD TAEUS application has received, and we expect our future applications will need to receive, a CE mark from an appropriate Competent Authority or government-accredited group (a “Notified Body”),Notified Body, as the case may be, as a result of successful review of one or more submissions prepared by our contract engineering and manufacturer(s), so that such applications can be marketed and distributed within the European Economic Area. Each of our applications will be required to be regularly recertified for CE marking, which recertification may require an annual audit. The audit procedure, which will include on-site visits at our facility, and the contract manufacturer’s(s’) facility(ies), will require us to provide the contract manufacturer(s) with information and documentation concerning our quality management system and all applicable documents, policies, procedures, manuals, and other information.
In the European Union, the manufacturer of medical devices is subject to current Good Manufacturing Practice, or cGMP, as set forth in the relevant laws and guidelines of the European Union and its member states. Compliance with cGMP is generally assessed by a Notified Body accredited by a Competent Authority. For a Class IIa device, typically, quality system evaluation is performed by the Notified Body, which also provides the certifications necessary to fix a CE mark to the products. The Notified Body may conduct inspections of relevant facilities, and review manufacturing procedures, operating systems and personnel qualifications. In addition to obtaining approval for each application, in many cases each device manufacturing facility must be audited on a periodic basis by the Notified Body. Further inspections may occur over the life of the application.
We also must comply with data privacy regulations in the European Union and the UK. The collection and use of health data and other personal data including data collected in clinical trials is governed in the EU by the General Data Protection Regulation (“GDPR”), which imposes substantial obligations upon companies and new rights for individuals. The GDPR also forms part of the law of Great Britain (England and Wales, Scotland and Northern Ireland) by virtue of section 3 of the European Union (Withdrawal) Act 2018 and as amended by the Data Protection, Privacy and Electronic Communications (Amendments etc.) (EU Exit) Regulations 2019 (SI 2019/419) (“UK GDPR”). Failure to comply with the GDPR may result in fines of the higher of (i) €20,000,000 or (ii) 4% of the preceding fiscal year’s total annual global revenues of the noncompliant company, among other administrative penalties. Although we do not expect to obtain possession of any personal data from the operation of our products, the GDPR has increased our responsibility and potential liability in relation to personal data involved in the operation of our products, and we may be required to implement additional measures in order to comply with the GDPR and with other laws, rules, regulations and standards in the EU and UK relating to privacy and data protection. This may be onerous and if our efforts to comply with GDPR or other applicable laws, rules, regulations and standards are not successful, or are perceived to be unsuccessful, it could adversely affect our business.
| 13 |
| Table of Contents |
FDA Regulation
Each of our products must be approved or cleared by the FDA before it is marketed in the United States. Before and after approval or clearance in the United States, our applications are subject to extensive regulation by the FDA under the FD&C Act and/or the Public Health Service Act, as well as by other regulatory bodies. The FDA regulations govern, among other things, the development, testing, manufacturing, labeling, safety, storage, record-keeping, market clearance or approval, advertising and promotion, import and export, marketing and sales, and distribution of medical devices and pharmaceutical products.
FDA Approval or Clearance of Medical Devices
In the United States, medical devices are subject to varying degrees of regulatory control and are classified in one of three classes depending on the extent of controls the FDA determines are necessary to reasonably ensure their safety and efficacy:
· | Class I: general controls, such as labeling and adherence to quality system regulations; | |
· | Class II: special controls, clearance of a premarket notification, or 510(k) submission, specific controls such as performance standards, patient registries and post-market surveillance and additional controls such as labeling and adherence to quality system regulations; and | |
· | Class III: special controls and approval of a premarket approval, or PMA, application. |
We expect all of our products to be classified as, or subject to reclassification as, Class II medical devices and thus require FDA authorization prior to marketing by means of a 510(k) clearance or de novo request, rather than a PMA application.
To request marketing authorization by means of a 510(k) clearance, we must submit a notification demonstrating that the proposed device is substantially equivalent to another legally marketed medical device, has the same intended use, and is as safe and effective as a legally marketed device and does not raise different questions of safety and effectiveness than a legally marketed device. 510(k) submissions generally include, among other things, a description of the device and its manufacturing, device labeling, medical devices to which the device is substantially equivalent, safety and biocompatibility information and the results of performance testing. In some cases, a 510(k) submission must include data from human clinical studies. Marketing may commence only when the FDA issues an order finding substantial equivalence. Historically, the typical 510(k) review time has been approximately nine to twelve months from the date of the initial 510(k) submission. However, the COVID-19 pandemic has resulted in the FDA reallocating a number of its reviewers to address emergency use authorizations for COVID-19-related products, which may result in longer 510(k) review times for other devices.
In many instances, the 510(k) pathway for product marketing requires only non-clinical testing as proof of substantial equivalence to a lawfully marketed predicate device for a given indication. However, in some instances the FDA may require clinical studies to demonstrate substantial equivalence to the predicate device. Whether clinical data is provided or not, the FDA may decide to reject the substantial equivalence argument we present. If that happens, the device is automatically designated as a Class III device. The device, unless the sponsor must then fulfill more rigorous PMA requirements, or can requestrequests a risk-based classification determination for the device in accordance with the “de novo” process, which may determine that the new device is of low to moderate risk and that it can be appropriately be regulated as a Class I or II device. In the denovo process, the FDA must determine that general and special controls are sufficient to provide reasonable assurance of the safety and effectiveness of a device which has no predicate. Upon receipt of a de novo request, the FDA will conduct an acceptance review to assess the completeness of the application and whether it meets the minimum threshold of acceptability. If the de novo request is accepted for substantive review, the FDA will conduct a classification review of legally marketed device types and analyze whether an existing legally marketed device of the same type exists, which information is used to confirm the subject device is eligible for de novo classification. During the course of review, the FDA may address any issues through interactive review or send a formal request for additional information in order for the review to proceed. If a de novo request is granted, the device may be legally marketed, and a new classification is established. If the device is classified as Class II, the device may serve as a predicate for future 510(k) submissions. If the device is not approved through de novo review, then it must go through the standard PMA process for Class III devices.
After a device receives 510(k) clearance, including following classification as a Class I or II device upon an approved de novo request, any product modification that could significantly affect the safety or effectiveness of the product, or that would constitute a significant change in intended use, requires a new 510(k) clearance. If the FDA determines that the changed product does not qualify for 510(k) clearance, then a company must submit, and the FDA must approve, a PMA before marketing can begin.
Clinical Trials of Medical Devices
Depending on the nature of the device, one or more clinical trials are generally required to support a PMA application and more recently are becomingmay be necessary to support a 510(k) submission.submission and, potentially, for EU CE certification, as well as generally required for PMA applications. Clinical studies of unapproved or uncleared medical devices or devices being studied for uses for which they are not approved or cleared (investigational devices) must be conducted in compliance with FDA requirements.requirements (and/or, if conducted in another jurisdiction, the applicable laws and regulations of the jurisdiction in which the trial is conducted). If an investigational device could pose a significant risk to patients, the sponsor company must submit an investigational device exemption application to the FDA prior to initiation of the clinical study. An investigational device exemption application must be supported by appropriate data, such as animal and laboratory test results, showing that it is safe to test the device on humans and that the testing protocol is scientifically sound. The investigational device exemption will automatically become effective 30 days after receipt by the FDA unless the FDA notifies the company that the investigation may not begin. Clinical studies of investigational devices may not begin until an institutional review board has approved the study.
| 14 |
| Table of Contents |
During the study, the sponsor must comply with the FDA’s investigational device exemption requirements. These requirements include investigator selection, trial monitoring, adverse event reporting, and record keeping. The investigators must obtain patient informed consent, rigorously follow the investigational plan and study protocol, control the disposition of investigational devices, and comply with reporting and record keeping requirements. The sponsor, the FDA, or the institutional review board at each institution at which a clinical trial is being conducted may suspend a clinical trial at any time for various reasons, including a belief that the subjects are being exposed to an unacceptable risk. During the approval or clearance process, the FDA typically inspects the records relating to the conduct of one or more investigational sites participating in the study supporting the application.
Post-Approval U.S. Regulation of Medical Devices
After a device is cleared or approved for marketing, numerous and pervasive regulatory requirements continue to apply. These include:
· | the FDA’s Quality Systems Regulation (“QSR”), which governs, among other things, how manufacturers design, test, manufacture, exercise quality control over, and document manufacturing of their products; | |
· | labeling and claims regulations, which prohibit the promotion of products for unapproved or “off-label” uses and impose other restrictions on labeling; and | |
· | the Medical Device Reporting regulation, which requires reporting to the FDA of certain adverse experiences associated with use of the product. |
Post-Approval EU Regulation of Medical Devices
Notwithstanding the FDA quality systems regulation, which governs, among other things, howcertification and the CE marking on approved medical devices, economic operators such as the manufacturers, design, test, manufacture, exercise quality control over, and document manufacturingimporters or distributors of their products;
· | the manufacturer maintaining an authorized representative in the EU; | |
· | maintaining an appropriate system for obtaining, reviewing, assessing and appropriately collecting and registering reports from patients, users, distributors or healthcare professionals of suspected incidents, complaints, non-confirming products, recalls and/or withdrawals; | |
· | ensuring the traceability of all devices placed onto the market by the manufacturer, including through a unique devise identification system; | |
· | preparing and maintaining SOPs for product withdrawal, recall or other field safety corrective and preventive actions (“CAPA”) as well as maintaining a system to manage CAPA that ensures collection and evaluation of internal and external quality information, the identification of failure causes and the implementation of enduring corrective actions to eliminate failure causes and to prevent recurrence; | |
· | preparing of post-marketing surveillance reports and additional studies on the medical devices; and | |
· | regular (and, if required, ad hoc) reporting to the competent authorities in accordance with MDR. |
Good Manufacturing Practices Requirements
Manufacturers of medical devices are required to comply with the good manufacturing practices set forth in the quality system regulationQSR promulgated under Section 520 of the FD&C Act. Current good manufacturing practices regulations require,The QSR requires, among other things, quality control and quality assurance as well as the corresponding maintenance of records and documentation. The manufacturing facility for an approved product must be registered with the FDA and meet current good manufacturing practicesQSR requirements to the satisfaction of the FDA pursuant to a pre-PMA approval inspection before the facility can be used. Manufacturers, including third party contract manufacturers, are also subject to periodic inspections by the FDA and other authorities to assess compliance with applicable regulations. Failure to comply with statutory and regulatory requirements subjects a manufacturer to possible legal or regulatory action, including the seizure or recall of products, injunctions, consent decrees placing significant restrictions on or suspending manufacturing operations, and civil and criminal penalties. Adverse experiences with the product must be reported to the FDA and could result in the imposition of marketing restrictions through labeling changes or in product withdrawal. Product approvals or clearances may be withdrawn if compliance with regulatory requirements is not maintained or if problems concerning safety or efficacy of the product occur following the approval.
| 15 |
| Table of Contents |
China Regulation
China’s regulatory approval framework includes nationwide approval based on a showing that the device for which approval is sought has been previously approved in the country of origin. Alternatively, we understand it is also possible to receive approval at the provincial level or to work exclusively with hospitals that do not require such nationwide or provincial approval. We intend to explore these potential paths to regulatory compliance in China.
Other Regulations
We and our contractors must comply with numerous federal, state and local laws relating to matters such as safe working conditions, manufacturing practices, environmental protection, fire hazard control, and hazardous substance disposal. Furthermore, we are subject to various reporting requirements including those prescribed by the Affordable Care Act and the Dodd-Frank Wall Street Reform and Consumer Protection Act. We cannot be sure that we will not be required to incur significant costs to comply with these laws and regulations in the future or that these laws or regulations will not adversely affect our business, financial condition, and results of operations. Unanticipated changes in existing regulatory requirements or the adoption of new requirements could adversely affect our business, financial condition, and results of operations.
We are also become subject to regulations and product registration requirements in many foreign countries in which we have received approval may sell our products,TAEUS liver device, including in the areas of product standards, packaging requirements, labeling requirements, import and export restrictions and tariff regulations, duties and tax requirements. Additionally, third parties designing, manufacturing or conducting human studies of our devices will beare subject to local regulations, such as those of Health Canada. The time required to obtain clearance required by foreign countries may be longer or shorter than that required for EMA or FDA clearance, and requirements for licensing a product in a foreign country may differ significantly from EMA and FDA requirements.
Environmental
Our manufacturing processes involve the use, generation, and disposal of hazardous materials and wastes, including alcohol, adhesives, and cleaning materials. As such, we are subject to stringent federal, state, and local laws relating to the protection of the environment, including those governing the use, handling, and disposal of hazardous materials and wastes. Future environmental laws may require us to alter our manufacturing processes, thereby increasing our manufacturing costs. We believe that our products and manufacturing processes at our facilities comply in all material respects with applicable environmental laws. However, the risk of environmental liabilities cannot be completely eliminated.
Competition
While we believe that we are the only company developing RF-based thermoacoustic ultrasound products, we will face direct and indirect competition from a number of competitors, many of whom have greater financial, sales and marketing and other resources than we do.
Manufacturers of CT and MRI systems include multi-national corporations such as Royal Philips, Siemens AG and Hitachi, Ltd.,Fujifilm Corporation, many of whom also manufacture and sell ultrasound equipment. In the NAFLD diagnosis market we will compete with makers of surgical biopsy tools, such as Cook Medical and Sterylab S.r.l. In the thermal ablation market, we will compete with manufacturers of surgical temperature probes, such as Medtronic plc and St. Jude Medical, Inc.
Employees
As of December 31, 2020,2023, we had 1821 employees, all of whom are employed on a full-time basis. 12Twelve full-time employees were engaged in research and development activities, 2three full-time employees were engaged in sales activities, 1three full-time employee wasemployees were engaged in product assembly, and 3three full-time employees were engaged in administrative activities. Geographically, we employ 14fifteen people in the United States, 3three people in Canada, one person in France, one person in Germany and 1one person in the United Kingdom. None of our employees are covered by a collective bargaining agreement, and we believe our relationship with our employees is good.
We also employ technical advisors, on an as-needed basis, to supplement existing staff. We believe that these technical advisors provide us with necessary expertise in clinical ultrasound applications, ultrasound technology, and intellectual property.
Investing in our common stock involves a high degree of risk. You should carefully consider the following risks and all other information contained in this Annual Report, including our financial statements and the related notes, before investing in our securities. The risks and uncertainties described below are not the only ones we face but include the most significant factors currently known by us that make investing in our securities speculative or risky. Additional risks and uncertainties that we are unaware of, or that we currently believe are not material, also may become important factors that affect us. If any of the following risks materialize, our business, financial condition and results of operations could be materially harmed. In that case, the trading price of our securities could decline, and you may lose some or all of your investment.
RISK FACTOR SUMMARY
Below is a bulleted summary of our principal risk factors, however this list does not fully represent all of our known risk factors. We encourage you to carefully review the full risk factors contained in this Annual Report in their entirety for additional information regarding the material factors that make an investment in our securities speculative or risky. These risks and uncertainties include, but are not limited to, the following:
Risks Related to our Business
· | We have a history of operating losses, we may never achieve or maintain profitability, and we will need to raise significant additional capital if we are going to continue as a going concern. | |
· | Our efforts may never result in the successful development of commercial applications based on our TAEUS technology, on which our success is substantially dependent. | |
· | Our TAEUS platform applications may not achieve adequate market acceptance by the physicians, patients, third-party payors and others in the medical community. | |
· | The outbreak of pandemics, such as COVID-19, could adversely impact our business, including our pre-sales activities, clinical trials and ability to obtain regulatory approvals. | |
· | We may not remain commercially viable if there is an inadequate level of reimbursement by governmental programs and other third-party payors for our planned products or associated procedures. | |
· | We have limited resources and depend on third parties to design and manufacture, and seek regulatory approval of, our TAEUS applications. | |
· | We will need to develop marketing and distribution capabilities both internally and through our relationships with third parties in order to sell any of our TAEUS products receiving regulatory approval. | |
· | Competition in the medical imaging market is intense and we may be unable to successfully compete. | |
· | We market our TAEUS liver device in the EU and are subject to the risks of doing business outside of the United States. | |
· | We depend on our senior management team and the loss of one or more key employees or an inability to attract and retain highly skilled employees could harm our business. | |
· | Misdiagnosis, warranty and other claims, as well as product field actions and regulatory proceedings, initiated against us could increase our costs, delay or reduce our sales and damage our reputation. |
Risks Related to Intellectual Property and Other Legal Matters
· | If we are unable to protect our intellectual property, which entails significant expense and resources, then our financial condition, results of operations and the value of our technology and products could be adversely affected. | |
· | Policing unauthorized use of our proprietary rights can be difficult, expensive and time-consuming, and we might be unable to determine the extent of this unauthorized use. | |
· | Intellectual property rights may not provide adequate protection, which may permit third parties to compete against us more effectively. |
Risks Related to Government Regulation
· | If we fail to obtain and maintain necessary regulatory clearances or approvals for our TAEUS applications, or if clearances or approvals for future applications and indications are delayed or not issued, our commercial operations will be harmed. | |
· | Healthcare reform measures could hinder or prevent our planned products' commercial success. | |
· | If we fail to comply with healthcare regulations, we could face substantial penalties and our business, operations and financial condition could be adversely affected. |
| 17 |
| Table of Contents |
Risks Related to Owning Our Securities
· | Our quarterly and annual results may fluctuate significantly, may not fully reflect the underlying performance of our business and may result in volatility in the price of our securities. | |
· | Our stock price has fluctuated in the past, has recently been volatile and may be volatile in the future for reasons unrelated to our operating performance or prospects, and as a result, investors in our common stock could incur substantial losses. | |
· | We may be subject to securities litigation, which is expensive and could divert management attention. | |
· | If we are unable to implement and maintain effective internal control over financial reporting, including by remediating current material weaknesses in our internal control over financial reporting, investors may lose confidence in the accuracy and completeness of our financial reports and the market price of our securities may decrease. | |
· | Our disclosure controls and procedures may not prevent or detect all errors or acts of fraud. | |
· | Future sales and issuances of our common stock or rights to purchase common stock, including pursuant to our at-the-market offering program or equity incentive plan, could result in dilution of the percentage ownership of our stockholders and could cause the price of our securities to fall. | |
· | Our charter documents and Delaware law may inhibit a takeover that stockholders consider favorable. |
General Risk Factors
· | Our business is affected by macroeconomic conditions. | |
· | Our cash and cash equivalents could be adversely affected if the financial institutions in which we hold our cash and cash equivalents fail. | |
· | The ongoing military action in Ukraine and the Middle East could have negative impact on the global economy, which could materially adversely affect our business, operations, operating results and financial condition. | |
· | Our business could be negatively impacted by corporate social responsibility and sustainability matters. |
Risks Related to Our Business
We have a history of operating losses we may never achieve or maintain profitability, and we will need to raise significant additional capital ifto continue our business and operations. If we are goingunable to continue asraise capital or secure financing on favorable terms, or at all, to meet our capital and operating needs, we will be forced to delay or reduce our product development program and commercialization efforts, which would have a going concern.
We have limited commercial experience upon which investors may evaluate our prospects.are experiencing financial and operating challenges. We have only generated limited revenues to date and have a history of losses from operations. As of December 31, 2020,2023, we had an accumulated deficit of $57,338,489.$91.9 million. Our independent registered public accounting firm, in its report on our financial statements for the year ended December 31, 2020,2023, has raised substantial doubt about our ability to continue as a going concern.
We also expectare actively exploring additional sources of liquidity and may seek to incur costs and expenses related to consulting, laboratory development, andraise such capital through, among other means, public or private equity offerings (including sales of our common stock under our at-the-market equity offering program), debt financings, corporate collaborations and/or licensing arrangements. However, general market conditions or the hiringmarket price of scientists and other operational personnel.
| 18 |
| Table of Contents |
· | the expectation that we will continue to incur losses and generate negative cash flows from operations; | |
· | our substantially limited liquidity and capital resources to meet our obligations as they become due; | |
· | the potential that our common stock will be delisted by Nasdaq in the event we fail to maintain compliance with the minimum bid price requirement; and | |
· | risks and uncertainties that are described in more detail in the Risk Factors and Management’s Discussion and Analysis of Financial Condition and Results of Operations sections in this Annual Report on Form 10-K. |
To date, we have financed our operations primarily through the net proceeds from offerings of common and preferred stock, warrants and convertible notes, as well as sales of our discontinued Nexus 128 system. We do not know when or if our operations will generate sufficient cash to fund our ongoing operations. Therefore, we will require additional capital in order to: (i) continue to conduct research and development activities; (ii) continue to conduct clinical studies; (iii) fund the costs of seeking regulatory approval of TAEUS applications; (iv) expand our sales and marketing infrastructure; (v) acquire complementary business technology or products; and (vi) respond to business opportunities, challenges, increased regulatory obligations or unforeseen circumstances.notes. Our future funding requirements will depend on many factors, including, but not limited to:
· | the costs, timing and outcomes of regulatory reviews associated with our future products, including TAEUS applications; | |
· | the progress, timing, costs and outcomes of our clinical studies, including the ability to timely enroll patients in such clinical trials; | |
· | the costs and expenses of expanding our sales and marketing infrastructure; | |
· | the costs and timing of developing variations of our TAEUS applications and, if necessary, obtaining regulatory clearance of such variations; | |
· | the degree of success we experience in commercializing our products, particularly our TAEUS applications; | |
· | the extent to which our TAEUS applications are adopted by hospitals for use by primary care physicians, hepatologists, radiologists and oncologists for diagnosis of fatty liver disease and the thermal ablation of lesions; | |
· | the number and types of future products we develop and commercialize; | |
· | the costs of preparing, filing and prosecuting patent applications and maintaining, enforcing and defending intellectual property-related claims; | |
· | the extent and scope of our general and administrative expenses; | |
· | the outcome, timing and cost of regulatory approvals, including the potential that the FDA or comparable regulatory authorities may require that we perform more studies than those that we currently expect; | |
· | the amount of sales and other revenues from technologies and products that we may commercialize, if any, including the selling prices for such potential products and the availability of adequate third-party reimbursement; | |
· | the terms and timing of any potential future collaborations, licensing or other arrangements that we may establish; | |
· | cash requirements of any future acquisitions and/or the development of other products; | |
· | the costs of operating as a public company; | |
· | the cost and timing of completion of commercial-scale, outsourced manufacturing activities; and | |
· | the time and cost necessary to respond to technological and market developments. |
Our TAEUS platform applications may not achieve adequate market acceptance by the physicians, patients, third-party payors and others in the medical community.
Our TAEUS applications that receive regulatory approval may nonetheless fail to gain sufficient market acceptance by physicians, patients, third-party payors and others in the medical community. If our TAEUS applications do not achieve an adequate level of acceptance, we may not generate significant product revenues or any profits from sales. The degree of market acceptance of products based on our TAEUS platform will depend on a number of factors, including:
| 19 |
| Table of Contents |
· | potential or perceived advantages or disadvantages compared to alternative products; | |
· | pricing relative to competitive products and availability of third-party coverage or reimbursement; | |
· | the timing of bringing our product to market as compared to possible other new entrants to the market; | |
· | our ability to effectively raise market awareness and explain product benefits and whether we have resources sufficient to do so; | |
· | relative convenience, dependability and ease of administration; and | |
· | willingness of the target patient population to try new products and of physicians to utilize such products. |
Our revenues will be adversely affected if, due to these or other factors, the products we are able to commercialize do not gain significant market acceptance.
Public health crises, such asCOVID-19 or other similar pandemics in the novel strain of coronavirus, SARS-CoV-2, which causes COVID-19, hasfuture, can adversely impactedimpact our business, including our pre-sales activities, clinical trials and ability to obtain regulatory approvals.
Public health crises such as pandemics or similar outbreaks could adversely impact our business. In December 2019, a novel strain of coronavirus, SARS-CoV-2, which causes coronavirus disease 2019 (“COVID-19”), surfaced in Wuhan, China. Since then, COVID-19 has spread to countries around the world and has been declared a pandemic by the World Health Organization. The level and nature of the disruption caused by COVID-19 is unpredictable, may be cyclical and long-lasting and may vary from location to location. Beginning in March 2020, we undertook temporary precautionary measures to help minimize the risk of the virus to our employees, including by requiring most employees to work remotely, pausing all non-essential travel worldwide for our employees, and limiting employee attendance at industry events and in-person work-related meetings, to the extent those events and meetings are continuing. As a cash-conserving measure taken in light of the adverse economic conditions caused byFor instance, the COVID-19 pandemic in April 2020 we reduced the cash salaries of members of management by 33% for the remainder of 2020, including the salaries of our executive officers. In lieu of cash, the Company paid this portion of management salaries in the form of restricted stock units that vested over the remainder of the year. Additionally, we amended our Non-Employee Director Compensation Policy to provide that our non-employee directors’ annual retainers for the second, third and fourth fiscal quarters of 2020 were paid in in the form of restricted stock units rather than cash. We may take additional measures to mitigate the effects to our business caused by COVID-19, any of which could negatively affect our business.
We may not remain commercially viable if there is an inadequate level of reimbursement by governmental programs and other third-party payors for our planned products or associated procedures.
Medical imaging products are purchased principally by hospitals, physicians and other healthcare providers around the world that typically bill various third-party payors, including governmental programs (e.g.(e.g., Medicare and Medicaid in the United States), private insurance plans and managed care programs, for the services provided to their patients.
Third-party payors and governments may approve or deny coverage for certain technologies and associated procedures based on independently determined assessment criteria. Reimbursement decisions by payors for these services are based on a wide range of methodologies that may reflect the services’ assessed resource costs, clinical outcomes and economic value. These reimbursement methodologies and decisions confer different, and sometimes conflicting, levels of financial risk and incentives to healthcare providers and patients, and these methodologies and decisions are subject to frequent refinements. Third-party payors are also increasingly adjusting reimbursement rates, often downwards, indirectly challenging the prices charged for medical products and services. There can be no assurance that our products will be covered by third-party payors, that adequate reimbursement will be available or, even if payment is available, that third-party payors’ coverage policies will not adversely affect our ability to sell our products profitably.
We have limited data regarding the efficacy of our TAEUS platform applications. If any of our applications that receive regulatory approval do not perform in accordance with our expectations, we are unlikely to successfully commercialize our applications.
Although we have completed a number of studies with respect to our TAEUS liver device, we have limited data regarding the efficacy of other TAEUS platform applications. Since our success depends in large part on the medical and third-party payorspayor community’s acceptance of our TAEUS applications, even if we receive regulatory approval for our applications, we believe that we will need to obtain additional clinical data from users of our applications to persuade medical professions to use our applications. We may also be required to conduct post-approval clinical testing to obtain such additional data. Clinical testing is expensive, can take a significant amount of time to complete and can have uncertain outcomes. Negative results of these clinical studies could have a material, adverse impact on our business.
We cannot be certain that results from limited animal and human studies of any of our TAEUS applicationsliver device will be indicative of future studies or that any of our TAEUS applications will be successfully commercialized.
To successfully commercialize any application based on our TAEUS platform technology, we expect it will be necessary to conduct various pre-clinical and human studies to demonstrate that the product is safe and effective for human use. In October 2018For instance, we initiated certainhave conducted a number of human studies ofwith respect to our TAEUS device targeting NAFLD. In September 2019, we reported top-level findings fromliver device. These studies have initially demonstrated a clinical study conductedmeaningful correlation between the measurement of liver fat by CIMTEC relating to the feasibility of our TAEUS application for NAFLD. This data enabled us to obtain CE mark approval for our NAFLD TAEUS applicationFLIP product and make a 510(k) submission to the FDA for that application.by MRI-PDFF. However, there can be no assurance that results from these studies are indicative of results that would be achieved in future animal studies or human clinical studies of this or any future TAEUS applications, which may be required in order for our applications incorporating our technology to obtain or maintain regulatory approval. Even if clinical trials or other studies demonstrate the safety and effectiveness of any applications of our technology and the necessary regulatory approvals are obtained, the commercial success of any of such application will depend upon their acceptance by patients, the medical community, and third-party payers and on our partners’ ability to successfully manufacture and commercialize a device for such application.
| 20 |
| Table of Contents |
Our limited commercial experience makes it difficult to evaluate our business, predict our future results or forecast our financial performance and growth.
We were incorporated in 2007 and began commercializingdiscontinued our initial pre-clinical Nexus 128 product in 2010. Our NAFLD2019 and our TAEUS liver device has obtained CE mark approval but has not yet been fully commercialized. This limited commercial experience makes it difficult to evaluate our business, predict our future results or forecast our financial performance and growth. If our assumptions regarding the risks and uncertainties we face, which we use to plan our business, are incorrect or change due to circumstances in our business or our markets, or if we do not address these risks successfully, our operating and financial results could differ materially from our expectations and our business could suffer.
We have formed, and may in the future form or seek, strategic alliances and collaborations or enter into licensing arrangements, and we may not realize the benefits of such alliances, collaborations or licensing arrangements.
In April 2016, we entered into a Collaborative Research Agreement with GE Healthcare, under which GE Healthcare has agreed to support our efforts to commercialize our TAEUS technology for use in an NAFLD application by, among other things, providing equipment and technical advice, and facilitating introductions to GE Healthcare clinical ultrasound customers. This agreement does not commit GE Healthcare to a long-term relationship and it may disengage with us at any time. This agreement has a term lasting until December 16, 20222024 and is subject to termination by either party upon not less than 60 days’ notice. See the section of this Annual Report titled “Collaboration with GE Healthcare”“TAEUS System for Early Assessment and Monitoring of Nonalcoholic Fatty Liver Disease, or NAFLD” under “Item 1. Business” for further description of this agreement.
We intend in the future to form or seek additional strategic alliances, create joint ventures or collaborations or enter into licensing arrangements with third parties that we believe will complement or augment our development and commercialization efforts with respect to our technologies and applications.
Any of these relationships may require us to incur non-recurring and other charges, increase our near- and long-term expenditures, issue securities that dilute our existing stockholders, restrict our ability to collaborate with other third parties or otherwise disrupt our management and business. In addition, we face significant competition in seeking appropriate strategic partners and the negotiation process is time-consuming and complex. If we license technologies or applications, we may not be able to realize the intended benefit of such transactions. Further, strategic alliances and collaborations are subject to numerous risks, which may include the following:
· | collaborators have significant discretion in determining the efforts and resources that they will apply to a collaboration; | |
· | collaborators may not pursue development and commercialization of our technologies and applications or may elect not to continue or renew development or commercialization programs based on clinical trial results, changes in their strategic focus due to the acquisition of competitive products, availability of funding, or other external factors, such as a business combination that diverts resources or creates competing priorities; | |
· | collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with our applications and technologies; | |
· | collaborators may not properly maintain or defend our intellectual property rights or may use our intellectual property or proprietary information in a way that gives rise to actual or threatened litigation that could jeopardize or invalidate our intellectual property or proprietary information or expose us to potential liability; | |
· | disputes may arise between us and a collaborator that cause the delay or termination of the research, development or commercialization of our technologies and applications, or that result in costly litigation or arbitration that diverts management attention and resources; | |
· | collaborations may be terminated and, if terminated, may result in a need for additional capital to pursue further development or commercialization of the applicable applications or technologies; and | |
· | collaborators may own or co-own intellectual property covering our products that results from our collaborating with them, and in such cases, we would not have the exclusive right to commercialize such intellectual property. |
| 21 |
| Table of Contents |
As a result, if we enter into collaboration agreements and strategic partnerships or license our applications or technologies, we may not be able to realize the benefit of such transactions if we are unable to successfully integrate them with our existing operations and company culture, which could delay our timelines or otherwise adversely affect our business. We also cannot be certain that, following a strategic transaction or license, we will achieve the revenue or specific net income that justifies such transaction. Any delays in entering into new strategic partnership agreements related to our applications could delay the development and commercialization of our technologies and applications in certain geographies or for certain applications, which would harm our business prospects, financial condition and results of operations.
We have limited resources and depend on third parties to design and manufacture, and seek regulatory approval of, our TAEUS applications. If any third party fails to successfully design, manufacture or obtain regulatory approval of TAEUS applications, our business will be materially harmed.
We do not currently have, nor do we plan to acquire, the infrastructure or capability to design or manufacture our TAEUS applications. To support our design and manufacturing efforts, we have contracted StarFish Product Engineering, Inc., a medical device contract manufacturing company, rather than design or manufacture our TAEUS applications ourselves. We have limited control over the efforts and resources that these and any other third-party OEMs will devote to developing and manufacturing our TAEUS applications and their capabilities to serve our needs, including quality control, quality assurance and qualified personnel. In addition, for any future applications of our TAEUS technology we currently expect to depend on OEMs to acquire CE marks for the device or devices that they develop and manufacture which are necessary to permit marketing of those devices in the European Union followed by corresponding FDA approval.
An OEM may not be able to successfully design and manufacture the products it develops based on our TAEUS technology, may not devote sufficient time and resources to support these efforts or may fail in gaining the required regulatory approvals of our TAEUS applications. The failure by an OEM to perform in accordance with our expectations would substantially harm the value of our TAEUS technology, brand and business.
We will need to develop marketing and distribution capabilities both internally and through our relationships with third parties in order to sell any of our TAEUS products receiving regulatory approval. If we experience problems in developing these capabilities, our ability to sell our products could be limited.
We have limited experience selling our products and will need to develop marketing, sales and distribution capabilities in order to sell our TAEUS applications that receive the necessary regulatory approval. We have limited experience managing a sales force and customer support operations and may be unable to attract, retain and manage the collaborative manufacturing and distribution arrangements or the specialized workforce necessary to successfully commercialize our products. In addition, our sales and marketing organization must effectively explain the uses and benefits of our products as compared to alternatives in order to promote market acceptance and demand for our products. Although we have begun to hire a small internal sales and marketing team to engage and support channel partners and clinical customers, further developing these functions will be time consumingtime-consuming and expensive and our efforts may not be successful.
We intend to partner with others to assist us with some or all of these functions. However, we may be unable to find appropriate third parties with which to enter into these arrangements and any such third parties may not perform as expected.
Furthermore, third-party distributors that are in the business of selling other medical products may not devote a sufficient level of resources and support required to generate awareness of our TAEUS applications and grow or maintain product sales. If these distributors are unwilling or unable to market and sell our products, or if they do not perform to our expectations, we could experience delayed or reduced market acceptance and sales of our products. In addition, disagreements with our distributors or non-performance by these third parties could lead to costly and time-consuming litigation or arbitration and disrupt distribution channels for a period of time and require us to re-establish a distribution channel.
If we are unable to manage the growth of our business, our future revenues and operating results may be harmed.
Because of our small size, growth in accordance with our business plan, if achieved, will place a significant strain on our financial, technical, operational and management resources. As we expand our activities, there will be additional demands on these resources. The failure to continually upgrade our technical, administrative, operating and financial control systems or the occurrence of unexpected expansion difficulties, including issues relating to our research and development activities and retention of experienced scientists, managers and technicians, could have a material adverse effect on our business, financial condition and results of operations and our ability to timely execute our business plan. If we are unable to implement these actions in a timely manner, our results may be adversely affected.
| 22 |
| Table of Contents |
Competition in the medical imaging market is intense and we may be unable to successfully compete.
In general, competition in the medical imaging market is very significant and characterized by extensive research and development and rapid technological change. Competitors in this market include very large companies with significantly greater resources than we have. To successfully compete in this market we will need to develop TAEUS applications that offer significant advantages over alternative imaging products and procedures for such applications.
While we believe the technology behind our TAEUS platform is unique in the industry, developments by other medical imaging companies of new or improved products, processes or technologies may make our products or proposed products obsolete or less competitive. Alternative medical imaging devices may be more accepted or cost-effective than our products. Competition from these companies for employees with experience in the medical imaging industry could result in higher turnover of our employees. If we are unable to respond to these competitive pressures, we could experience delayed or reduced market acceptance of our products, higher expenses and lower revenue. If we are unable to compete effectively with current or new entrants to these markets, we will be unable to generate sufficient revenue to maintain our business.
Our competitors include producers of CT and MRI systems that include multi-national corporations such as Royal Philips, Siemens AG and Fujifilm Corporation, many of whom also manufacture and sell ultrasound equipment. In the NAFLD diagnosis market we will compete with makers of surgical biopsy tools, such as Cook Medical and Sterylab S.r.l. In the thermal ablation market, we will compete with manufacturers of surgical temperature probes, such as Medtronic plc and St. Jude Medical, Inc. These competitors and other potential competitors have substantially greater financial, technical and other resources, such as larger R&D staff, more robust manufacturing capabilities and more experienced marketing and manufacturing organizations. These competitors may succeed in developing, acquiring or licensing on an exclusive basis, products that are more effective or less costly than TAEUS applications that we may develop, or achieve earlier patent protection, regulatory approval, product commercialization and market penetration than us. Additionally, technologies developed by our competitors may render our potential product candidates uneconomical or obsolete, and we may not be successful in marketing our product candidates against those of our competitors.
Changes in the healthcare industry could result in a reduction in the size of the market for our products or may require us to decrease the selling price for our products, either of which could have a negative impact on our financial performance.
Trends toward managed care, healthcare cost containment, and other changes in government and private sector initiatives in Europe, the United States and China are placing increased emphasis on lowering the cost of medical services, which could adversely affect the demand for or the prices of our products. For example:
· | major third-party payors of hospital and non-hospital based healthcare services could revise their payment methodologies and impose stricter standards for reimbursement of imaging procedures charges and/or a lower or more bundled reimbursement; | |
· | there has been a consolidation among healthcare facilities and purchasers of medical devices who prefer to limit the number of suppliers from whom they purchase medical products, and these entities may decide to stop purchasing our products or demand discounts on our prices; and | |
· | there are proposed and existing laws and regulations in international and domestic markets regulating pricing and profitability of companies in the healthcare industry. |
These trends could lead to pressure to reduce prices for our products and could cause a decrease in the demand for our products in any given market that could adversely affect our revenue and profitability, which could harm our business.
We intend to market our approved TAEUS applications if approved, globally, and currently market our TAEUS liver probe in which case we will bethe EU, and are therefore subject to the risks of doing business outside of the United States.
Because we intend to market our approved TAEUS applications if approved, globally, and currently market our TAEUS liver probe in the EU, our business may beis subject to risks associated with doing business globally. Accordingly, our business and financial results in the future could be adversely affected due to a variety of factors, including:
· | changes in a specific country’s or region’s political and cultural climate or economic condition; | |
· | local outbreaks of sickness or disease; | |
· | war or terrorist attack, including cyberterrorism; | |
· | unexpected changes in laws and regulatory requirements in local jurisdictions; | |
· | difficulty of effective enforcement of contractual provisions in local jurisdictions; | |
· | inadequate intellectual property protection in certain countries; |
| 23 |
| Table of Contents |
· | trade-protection measures, import or export licensing requirements such as Export Administration Regulations promulgated by the United States Department of Commerce and fines, penalties or suspension or revocation of export privileges; | |
· | effects of applicable local tax structures and potentially adverse tax consequences; and | |
· | significant adverse changes in currency exchange rates. |
We depend on our senior management team and the loss of one or more key employees or an inability to attract and retain highly skilled employees could harm our business.
Our success largely depends upon the continued services of our executive management team and key employees. The loss of one or more of our executive officers or key employees could harm us and directly impact our financial results. Our employees may terminate their employment with us at any time. Our executive management team has significant experience and knowledge of medical devices and ultrasound systems, and the loss of any team member could impair our ability to design, identify, and develop new intellectual property and new scientific or product ideas. Additionally, if we lose the services of any of these persons, we would likely be forced to expend significant time and money in the pursuit of replacements, which may result in a delay in the implementation of our business plan and plan of operations. We can give no assurance that we could find satisfactory replacements for these individuals on terms that would not be unduly expensive or burdensome to us.
To execute our growth plan, we must attract and retain highly qualified personnel. Competition for skilled personnel is intense, especially for engineers with high levels of experience in designing and developing medical devices. In addition, we will need to identify and hire sales executives and competition for commercial and marketing talent is significant. We may experience difficulty in hiring and retaining employees with appropriate qualifications. Many of the companies with which we compete for experienced personnel have greater resources than we have. In addition, we invest significant time and expense in training our employees, which increases their value to competitors who may seek to recruit them. If we fail to attract new personnel or fail to retain and motivate our current personnel, our business and future growth prospects would be harmed.
Our employees, independent contractors, consultants, commercial partners and vendors may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements.
We are exposed to the risk of fraud, misconduct or other illegal activity by our employees, independent contractors, consultants, commercial partners and vendors. Misconduct by these parties could include intentional, reckless and negligent conduct that fails to: comply with the FD&C Act and similar laws of other countries, or the rules and regulations of the FDA and other similar foreign regulatory bodies; provide true, complete and accurate information to the FDA and other similar foreign regulatory bodies; comply with manufacturing standards we establish; comply with healthcare fraud and abuse laws in the United States and similar foreign fraudulent misconduct laws; or report financial information or data accurately or to disclose unauthorized activities to us. For any products for which we obtain regulatory approval and begin commercializing in Europe, China or the United States, respectively, our potential exposure under such laws will increase significantly, and our costs associated with compliance with such laws are also likely to increase. In particular, the promotion, sales and marketing of healthcare items and services, as well as certain business arrangements in the healthcare industry, are subject to extensive laws designed to prevent fraud, kickbacks, self-dealing and other abusive practices. TheseOur sales team in the European Union marketing our TAEUS liver probe are subject to these laws, andas well as regulations maythat restrict or prohibit a wide range of pricing, discounting, marketing and promotion, structuring and commissions, certain customer incentive programs and other business arrangements generally. It is not always possible to identify and deter misconduct by employees and other parties, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
Misdiagnosis, warranty and other claims, as well as product field actions and regulatory proceedings, initiated against us could increase our costs, delay or reduce our sales and damage our reputation, adversely affecting our financial condition.
Our business exposes us to the risk of malpractice, warranty or product liability claims inherent in the sale and support of medical device products, including those based on claims that the use or failure of one of our products resulted in a misdiagnosis or harm to a patient. Such claims may cause financial loss, damage our reputation by raising questions about our products’ safety and efficacy, adversely affect regulatory approvals and interfere with our efforts to market our products. Although to date we have not been involved in any medical malpractice or product liability litigation, we may incur significant liability if such litigation were to occur. We may also face adverse publicity resulting from product field actions or regulatory proceedings brought against us. Claims could also be asserted under state consumer protection acts. If we cannot successfully defend ourselves against product liability or related claims, we may incur substantial liabilities or be required to limit the distribution of our products. Even a successful defense would require significant financial and management resources. Regardless of the merits or eventual outcome, liability claims may result in:
| 24 |
| Table of Contents |
· | decreased demand for our products; | |
· | injury to our reputation and negative media attention; | |
· | initiation of investigations by regulators and adverse impacts to our ability to obtain regulatory approvals; | |
· | costs to defend the related litigation; | |
· | a diversion of management’s time and our resources; | |
· | substantial monetary awards to trial participants or patients; | |
· | product recalls, withdrawals or labeling, marketing or promotional restrictions; | |
· | loss of revenue; | |
· | exhaustion of any available insurance and our capital resources; | |
· | the inability to commercialize a product at all or for particular applications; and | |
· | a decline in the price of our securities. |
Although we currently maintain liability insurance in amounts that we believe are commercially reasonable, any liability we incur may exceed our insurance coverage. Our insurance policies may also have various exclusions, and we may be subject to a claim for which we have no coverage. Liability insurance is expensive and may cease to be available on acceptable terms, if at all. A malpractice, warranty, product liability or other claim or product field action not covered by our insurance or exceeding our coverage could significantly impair our financial condition. In addition, a product field action or a liability claim against us could significantly harm our reputation and make it more difficult to obtain the funding and commercial relationships necessary to maintain our business.
Our internal computer systems, or those used by third-party manufacturers or other contractors or consultants, may fail or suffer security breaches.
Despite the implementation of security measures, our internal computer systems and those of our future manufacturers and other contractors and consultants are vulnerable to damage from computer viruses and unauthorized access. Although to our knowledge we have not experienced any such material system failure or security breach to date, if such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our research and development programs and our business operations. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and the further development and commercialization of our products could be delayed.
Risks Related to Intellectual Property and Other Legal Matters
If we are unable to protect our intellectual property, which entails significant expense and resources, then our financial condition, results of operations and the value of our technology and products could be adversely affected.
Much of our value arises out offrom our proprietary technology and intellectual property for the design, manufacture and use of medical imaging systems, including development of our TAEUS applications. We rely on patent, copyright, trade secret and trademark laws to protect our proprietary technology and limit the ability of others to compete with us using the same or similar technology. Third parties may infringe or misappropriate our intellectual property, which could harm our business.
Expenses related to a patent portfolio include periodic maintenance fees, renewal fees, annuity fees, various other governmental fees on patents and/or applications due in several stages over the lifetime of patents and/or applications, as well as the cost associated with complying with numerous procedural provisions during the patent application process. We may or may not choose to pursue or maintain protection for particular inventions. In addition, there are situations in which a failure to make certain payments or noncompliance with certain requirements in the patent process can result in abandonment or lapse of a patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction.
| 25 |
| Table of Contents |
Policing unauthorized use of our proprietary rights can be difficult, expensive and time-consuming, and we might be unable to determine the extent of this unauthorized use.
Policing unauthorized use of our intellectual property is difficult, costly and time-intensive. We may fail to stop or prevent misappropriation of our technology, particularly in countries where the laws may not protect our proprietary rights to the same extent as do the laws of the United States. Proceedings to enforce our patent and other intellectual property rights in non-U.S. jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business. If we cannot prevent other companies from using our proprietary technology or if our patents are found invalid or otherwise unenforceable, we may be unable to compete effectively against other manufacturers of ultrasound systems, which could decrease our market share. In addition, the breach of a patent licensing agreement by us may result in termination of a patent license.
We may not be able to prevent the unauthorized disclosure or use of our technical knowledge or other trade secrets by consultants, vendors or former or current employees, despite the existence generally of confidentiality agreements and other contractual restrictions. Monitoring unauthorized use and disclosure of our intellectual property is difficult, and we do not know whether the steps we have taken to protect our intellectual property will be adequate.
If we are unable to protect the confidentiality of our proprietary information and know-how, the value of our technology and products could be adversely affected.
In addition to our patent activities, we rely upon, among other things, unpatented proprietary technology, processes, trade secrets and know-how. Any involuntary disclosure to or misappropriation by third parties of our confidential or proprietary information could enable competitors to duplicate or surpass our technological achievements, potentially eroding our competitive position in our market. We seek to protect confidential or proprietary information in part by confidentiality agreements with our employees, consultants and third parties. While we require all of our employees, consultants, advisors and any third parties who have access to our proprietary know-how, information and technology to enter into confidentiality agreements, we cannot be certain that this know-how, information and technology will not be disclosed or that competitors will not otherwise gain access to our trade secrets or independently develop substantially equivalent information and techniques. These agreements may be terminated or breached, and we may not have adequate remedies for any such termination or breach. Furthermore, these agreements may not provide meaningful protection for our trade secrets and know-how in the event of unauthorized use or disclosure. To the extent that any of our staff was previously employed by other pharmaceutical, medical technology or biotechnology companies, those employers may allege violations of trade secrets and other similar claims in relation to their former employee’s therapeutic development activities for us.
We may in the future be a party to intellectual property litigation or administrative proceedings that could be costly and could interfere with our ability to sell our TAEUS applications.
The medical device industry has been characterized by extensive litigation regarding patents, trademarks, trade secrets, and other intellectual property rights, and companies in the industry have used intellectual property litigation to gain a competitive advantage. It is possible that U.S. and foreign patents and pending patent applications or trademarks controlled by third parties may be alleged to cover our products, or that we may be accused of misappropriating third parties’ trade secrets. Other medical imaging market participants, many of which have substantially greater resources and have made substantial investments in patent portfolios, trade secrets, trademarks, and competing technologies, may have applied for or obtained or may in the future apply for or obtain, patents or trademarks that will prevent, limit or otherwise interfere with our ability to make, use, sell and/or export our products or to use product names. We may become a party to patent or trademark infringement or trade secret claims and litigation as a result of these and other third partythird-party intellectual property rights being asserted against us. The defense and prosecution of these matters are both costly and time consuming. Vendors from whom we purchase hardware or software may not indemnify us in the event that such hardware or software is accused of infringing a third party’s patent or trademark or of misappropriating a third party’s trade secret.
Further, if such patents, trademarks, or trade secrets are successfully asserted against us, this may harm our business and result in injunctions preventing us from selling our products, license fees, damages and the payment of attorney fees and court costs. In addition, if we are found to willfully infringe third-party patents or trademarks or to have misappropriated trade secrets, we could be required to pay treble damages in addition to other penalties. Although patent, trademark, trade secret, and other intellectual property disputes in the medical device area have often been settled through licensing or similar arrangements, costs associated with such arrangements may be substantial and could include ongoing royalties. We may be unable to obtain necessary licenses on satisfactory terms, if at all. If we do not obtain necessary licenses, we may not be able to redesign our TAEUS applications to avoid infringement.
Similarly, interference or derivation proceedings provoked by third parties or brought by the U.S. Patent and Trademark Office (“USPTO”) may be necessary to determine the priority of inventions or other matters of inventorship with respect to our patents or patent applications. We may also become involved in other proceedings, such as re-examination, inter partes review, or opposition proceedings, before the USPTO or other jurisdictional body relating to our intellectual property rights or the intellectual property rights of others. Adverse determinations in a judicial or administrative proceeding or failure to obtain necessary licenses could prevent us from manufacturing and selling our TAEUS applications or using product names, which would have a significant adverse impact on our business.
| 26 |
| Table of Contents |
Additionally, we may need to commence proceedings against others to enforce our patents or trademarks, to protect our trade secrets or know-how, or to determine the enforceability, scope and validity of the proprietary rights of others. These proceedings would result in substantial expense to us and significant diversion of effort by our technical and management personnel. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded, if any, may not be commercially meaningful. We may not be able to stop a competitor from marketing and selling products that are the same or similar to our products or from using product names that are the same or similar to our product names, and our business may be harmed as a result.
Risks Related to Government Regulation
Failure to comply with laws and regulations could harm our business.
Our business is or in the future may be subject to regulation by various federal, state, local and foreign governmental agencies, including agencies responsible for monitoring and enforcing employment and labor laws, workplace safety, environmental laws, consumer protection laws, anti-bribery laws, import/export controls, securities laws and tax laws and regulations. In certain jurisdictions, these regulatory requirements may be more stringent than those in the United States. Noncompliance with applicable regulations or requirements could subject us to investigations, sanctions, mandatory recalls, enforcement actions, adverse publicity, disgorgement of profits, fines, damages, civil and criminal penalties or injunctions and administrative actions. If any governmental sanctions, fines or penalties are imposed, or if we do not prevail in any possible civil or criminal litigation, our business, operating results and financial condition could be harmed. In addition, responding to any action will likely result in a significant diversion of management's attention and our resources and substantial costs. Enforcement actions and sanctions could further harm our business, operating results and financial condition.
If we fail to obtain and maintain necessary regulatory clearances or approvals for our TAEUS applications, or if clearances or approvals for future applications and indications are delayed or not issued, our commercial operations will be harmed.
The medical devices that we manufacture and market will be subject to regulation by numerous worldwide regulatory bodies, including the EMA, FDA and other comparable regulatory agencies. Additionally, third parties designing, manufacturing or conducting human studies of our devices will be subject to local regulations, such as those of Health Canada. These agencies and regulations require manufacturers of medical devices to comply with applicable laws and regulations governing development, testing, manufacturing, labeling, marketing and distribution of medical devices. Devices are generally subject to varying levels of regulatory control, based on the risk level of the device. Governmental regulations specific to medical devices are wide-ranging and govern, among other things:
· | product design, development and manufacture; | |
· | laboratory, pre-clinical and clinical testing, labeling, packaging storage and distribution; | |
· | premarketing clearance or approval; | |
· | record keeping; | |
· | product marketing, promotion and advertising, sales and distribution; and | |
· | post-marketing surveillance, including reporting of deaths or serious injuries and recalls and correction and removals. |
The European Union has revised its regulatory system for medical devices by implementing regulation (EU) 2017/745 on medical devices (“Medical Device Regulation” or “MDR”) and regulation (EU) 2017/746 on in vitro diagnostic medical devices. The MDR was originally meant to apply as frombecame effective on May 26, 20202021 (the “Date of Application” or “DoA”) but, in light of the COVID-19 pandemic, the Date of Application was postponed to May 26, 2021.. The changes to the regulatory system implemented by the MDR include stricter requirements for clinical evidence and pre-market assessment of safety and performance, refined classifications to indicate risk levels, requirements for third party testing by Notified Bodies, tightened and streamlined quality management system assessment procedures and additional requirements for the quality management system, additional requirements for traceability of products and transparency as well a refined responsibility of economic operators.
We are currently in a transitional period, where our existing certified products will be required to continue to comply with applicable medical device directives (including the Medical Devices Directive and the Active Implantable Medical Devices Directive) and as of the DoA, with the Medical Device Regulation and to obtain CE mark certification in order to marketcontinue or commence marketing medical devices. The CE mark is applied following approval from an independent notified bodya Notified Body or declaration of conformity. It is an international symbol of adherence to quality assurance standards and compliance with applicable European Medical Devices Directives or the MDR, as the case may be. CE mark approvals issued prior to May 26, 2021 will, subject to certain conditions (including, among others, continued compliance with the DoA willMDR, no significant changes to design or intended purpose, a quality management system, and engagement with a notified body to obtain conformity assessment), remain in place for the duration of such approvals or conformity assessments and such products may be made available for this transitional period, but no later than June 2024.valid until December 31, 2028. In March 2020, we received CE mark approval for our TAEUS FLIP (Fatty Liver Imaging Probe) System. The CE marking indicates that TAEUS FLIP System complies with all applicable European Directives and Regulationsregulations in the European Union, including the MDR,EU, and other CE mark geographies, including the 27 EU member states. We believe that future TAEUS applications will qualify for sale in the European Union as Class IIa medical devices. Although existing regulations do not require clinical trials to obtain CE marks for Class IIa medical devices, theThe MDR requires a clinical evaluation for all medical devices and clinical trials for selected medical devices.devices to be (re-)certified under the rules of the MDR. Depending on the classification of our applications, future CE mark certifications or recertification of our applications may require additional clinical evaluations or trials, as the case may be.
| 27 |
| Table of Contents |
We are also required to comply with the regulations of each other country where we commercialize products, such as the requirement that we obtain approval from the FDA and the China Food and Drug Administration before we can launch new products in the United States and China, respectively.
Our NAFLD TAEUS device is being reviewed under a “de novo” process for use in the United States, or that are banned or deviate from lawful performance standards, are subject to FDA export requirements. Exported devices are subject to the regulatory requirements of each country to whicha risk-based classification determination whether the device is exported. Frequently, regulatory approval may first be obtained in a foreign country priorof low to application in the United States due to differing regulatory requirements; however, other countries, such as China for example, require approval in the country of origin first.
We may not be able to obtain the necessary clearances or approvals or may be unduly delayed in doing so, which could harm our business. Furthermore, even
Even if we obtain regulatory approval for our TAEUS device, our product will remain subject to regulatory oversight.
Even if we are granted regulatory clearances or approvals, they may include significant limitations on the indicated uses for the product, which may limit the market for the product. Therefore, even if we believe we have successfully developed our TAEUS technology, we may not be permitted to market TAEUS applications in the United States if we do not obtain FDA regulatory clearance to market such applications. Delays in obtaining clearance or approval could increase our costs and harm our revenues and growth.
In addition, we are required to timely file various reports with the FDA, including reports required by the medical device reporting regulations that require us to report to certain regulatory authorities if our devices may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if the malfunction were to recur. If these reports are not filed timely, regulators may impose sanctions and sales of our products may suffer, and we may be subject to product liability or regulatory enforcement actions, all of which could harm our business.
If we initiate a correction or removal for one of our devices to reduce a risk to health posed by the device, we would be required to submit a publicly available Correction and Removal report to the FDA and, in many cases, similar reports to other regulatory agencies. This report could be classified by the FDA as a device recall which could lead to increased scrutiny by the FDA, other international regulatory agencies and our customers regarding the quality and safety of our devices. Furthermore, the submission of these reports has been and could be used by competitors against us in competitive situations and cause customers to delay purchase decisions or cancel orders and would harm our reputation.
The FDA and the Federal Trade Commission (the “FTC”) also regulate the advertising and promotion of our planned products to ensure that the claims we make are consistent with our regulatory clearances, that there are adequate and reasonable data to substantiate the claims and that our promotional labeling and advertising is neither false nor misleading in any respect. If the FDA or FTC determines that any of our advertising or promotional claims are misleading, not substantiated or not permissible, we may be subject to enforcement actions, including warning letters, and we may be required to revise our promotional claims and make other corrections or restitutions.
The FDA and state authorities have broad enforcement powers. Our failure to comply with applicable regulatory requirements could result in enforcement action by the FDA or state agencies, which may include any of the following sanctions:
· | adverse publicity, warning letters, fines, injunctions, consent decrees and civil penalties; | |
· | repair, replacement, refunds, recall or seizure of our products; | |
· | operating restrictions, partial suspension or total shutdown of production; | |
· | refusing our requests for 510(k) clearance or premarket approval of new products, new intended uses or modifications to existing products; | |
· | withdrawing 510(k) clearance or premarket approvals that have already been granted; and | |
· | criminal prosecution. |
| 28 |
| Table of Contents |
If any of these events were to occur, our business and financial condition would be harmed.
We have experienced and may in the future experience delays and other difficulties in enrolling a sufficient number of patients in our clinical trials which could delay or prevent the receipt of necessary regulatory approvals.
We may not be able to initiate or complete as planned any clinical trials if we are unable to identify and enroll a sufficient number of eligible patients to participate in the clinical trials required by the FDA or other regulatory authorities. We also may be unable to engage a sufficient number of clinical trial sites to conduct our trials.
We may face challenges in enrolling patients to participate in our clinical trials. Patients suffering from diseases within target indications may enroll in competing clinical trials, which could negatively affect our ability to complete enrollment of our trials. Additionally, enrollment may be delayed by unforeseen circumstances, as occurred with the COVID-19 pandemic. Enrollment challenges in clinical trials often result in increased development costs for a product candidate, significant delays and potentially the abandonment of the clinical trial.
We may have other delays in completing our clinical trials and we may not complete them at all.
Since we lack significant experience in completing clinical trials and bringing a medical device through commercialization, we have hired outside consultants with such experience. Clinical trials for our TAEUS devicemay be delayed or terminated as a result of many factors, including the following:
· | patients failing to complete clinical trials due to dissatisfaction with the procedure, side effects, or other reasons; | |
· | failure by regulators to authorize us to commence a clinical trial; | |
· | suspension or termination by regulators of clinical research for many reasons, including concerns about patient safety, the failure of study sites and/or investigators in our clinical research program to comply with GCP requirements, or our failure, or the failure of our contract manufacturers, to comply with current cGMP requirements; | |
· | delays or failure to obtain clinical supply for our products necessary to conduct clinical trials from contract manufacturers; | |
· | treatment candidates demonstrating a lack of efficacy during clinical trials; | |
· | inability to continue to fund clinical trials or to find a partner to fund the clinical trials. |
Any delay or failure to complete clinical trials could have a material adverse effect on our cost to develop and commercialize, and our ability to generate revenue from, our TAEUS device.
Our TAEUS applications may require recertification or new regulatory clearances or premarket approvals and we may be required to recall or cease marketing our TAEUS applications until such recertification or clearances are obtained.
Most countries outside of the United States require that product approvals be recertified on a regular basis, generally every five years. The recertification process requires that we evaluate any device changes and any new regulations or standards relevant to the device and, where needed, conduct appropriate testing to document continued compliance. Where recertification applications are required, they must be approved in order to continue selling our products in those countries.
In the United States, material modifications to the intended use or technological characteristics of our TAEUS applications will require new 510(k) clearances or premarket approvals or require us to recall or cease marketing the modified devices until these clearances or approvals are obtained. Based on FDA published guidelines, the FDA requires device manufacturers to initially make and document a determination of whether or not a modification requires a new approval, supplement or clearance; however, the FDA can review a manufacturer’s decision. Any modification to an FDA-cleared device that would significantly affect its safety or efficacy or that would constitute a major change in its intended use would require a new 510(k) clearance or possibly a premarket approval.
We may not be able to obtain recertification or additional 510(k) clearances or premarket approvals for our applications or for modifications to, or additional indications for, our TAEUS technology in a timely fashion, or at all. Delays in obtaining required future governmental approvals would harm our ability to introduce new or enhanced products in a timely manner, which in turn would harm our future growth. If foreign regulatory authorities or the FDA require additional approvals, we may be required to recall and to stop selling or marketing our TAEUS applications, which could harm our operating results and require us to redesign our applications. In these circumstances, we may be subject to significant enforcement actions.
| 29 |
| Table of Contents |
If any OEMs fail to comply with the FDA’s Quality System Regulations or other regulatory bodies’ equivalent regulations, manufacturing operations could be delayed or shut down and the development of our TAEUS platform could suffer.
The manufacturing processes of OEMs are required to comply with the FDA’s Quality System Regulations and other regulatory bodies’ equivalent regulations, which cover the procedures and documentation of the design, testing, production, control, quality assurance, labeling, packaging, storage and shipping of our TAEUS applications. They may also be subject to similar state requirements and licenses and engage in extensive recordkeeping and reporting and make available their manufacturing facilities and records for periodic unannounced inspections by governmental agencies, including the FDA, state authorities and comparable agencies in other countries. If any OEM fails such an inspection, our operations could be disrupted and our manufacturing interrupted. Failure to take adequate corrective action in response to an adverse inspection could result in, among other things, a shut-down of our manufacturing operations, significant fines, suspension of marketing clearances and approvals, seizures or recalls of our products, operating restrictions and criminal prosecutions, any of which would cause our business to suffer. Furthermore, these OEMs may be engaged with other companies to supply and/or manufacture materials or products for such companies, which would expose our OEMs to regulatory risks for the production of such materials and products. As a result, failure to meet the regulatory requirements for the production of those materials and products may also affect the regulatory clearance of a third-party manufacturers’ facility. If the FDA or a foreign regulatory agency does not approve these facilities for the manufacture of our products, or if it withdraws its approval in the future, we may need to find alternative manufacturing facilities, which would impede or delay our ability to develop, obtain regulatory approval for or market our products, if approved. Additionally, our key component suppliers may not currently be or may not continue to be in compliance with applicable regulatory requirements, which may result in manufacturing delays for our product and cause our results of operations to suffer.
Our TAEUS applications may in the future be subject to product recalls that could harm our reputation.
Governmental authorities in Europe, the United States and China have the authority to require the recall of commercialized products in the event of material regulatory deficiencies or defects in design or manufacture. A government-mandated or voluntary recall by us could occur as a result of component failures, manufacturing errors or design or labeling defects. Recalls of our TAEUS applications would divert managerial attention, be expensive, harm our reputation with customers and harm our financial condition and results of operations. A recall announcement would negatively affect the price of our securities.
Healthcare reform measures could hinder or prevent our planned products' commercial success.
There have been, and we expect there will continue to be, a number of legislative and regulatory changes to the healthcare system in ways that could harm our future revenues and profitability and the future revenues and profitability of our potential customers. In the EU, the Medical Devices Directive is being replaced with the more expansive Medical Devices Regulation, which may increase the costs of obtaining and maintaining required regulatory approvals for our products. We cannot predict what other healthcare initiatives, if any, will be implemented by EU member countries, or the effect any future legislation or regulation will have on us.
In the United States, federal and state lawmakers regularly propose and, at times, enact legislation that would result in significant changes to the healthcare system, some of which are intended to contain or reduce the costs of medical products and services. For example, one of the most significant healthcare reform measures in decades, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act (the “Affordable Care Act”), was enacted in 2010. The Affordable Care Act contains a number of provisions, including those governing enrollment in federal healthcare programs, reimbursement changes and fraud and abuse measures, all of which will impact existing government healthcare programs and will result in the development of new programs. The Affordable Care Act, among other things, imposes an excise tax of 2.3% on the sale of most medical devices, including ours, and any failure to pay this amount could result in the imposition of an injunction on the sale of our products, fines and penalties.
It remains unclear whether changes will be made to the Affordable Care Act, or whether it will be repealed or materially modified. For example, the Tax Cuts and Jobs Act of 2017 modified certain aspects of the Affordable Care Act and the Biden Administration and U.S. Congress may take further action regarding the Affordable Care Act. Therefore, we cannot assure you that the Affordable Care Act, as currently enacted or as may be further amended or discontinued in the future, will not harm our business and financial results and we cannot predict how future federal or state legislative or administrative changes relating to healthcare reform will affect our business.
There likely will continue to be legislative and regulatory proposals at the federal and state levels directed at containing or lowering the cost of healthcare. We cannot predict the initiatives that may be adopted in the future or their full impact. The continuing efforts of the government, insurance companies, managed care organizations and other payors of healthcare services to contain or reduce costs of healthcare may harm:
●
●
●
If we fail to comply with healthcare regulations, we could face substantial penalties and our business, operations and financial condition could be adversely affected.
Even though we do not and will not control referrals of healthcare services or bill directly to Medicare, Medicaid or other third party payors, certain federal and state healthcare laws and regulations pertaining to fraud and abuse and patients’ rights are and will be applicable to our business. We could be subject to healthcare fraud and abuse and patient privacy regulation by both the federal government and the states in which we conduct our business. Other jurisdictions such as the European Union have similar laws. The regulations that will affect how we operate include:
| 30 |
| Table of Contents |
· | the federal healthcare program Anti-Kickback Statute, which prohibits, among other things, any person from knowingly and willfully offering, soliciting, receiving or providing remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual for, or the purchase, order or recommendation of, any good or service for which payment may be made under federal healthcare programs, such as the Medicare and Medicaid programs; | |
· | the federal False Claims Act, which prohibits, among other things, individuals or entities from knowingly presenting, or causing to be presented, false claims, or knowingly using false statements, to obtain payment from the federal government; | |
· | federal criminal laws that prohibit executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters; | |
· | the federal Physician Payment Sunshine Act, created under the Affordable Care Act, and its implementing regulations, which require manufacturers of drugs, medical devices, biologicals and medical supplies for which payment is available under Medicare, Medicaid, or the Children’s Health Insurance Program to report annually to the U.S. Department of Health and Human Services, or HHS, information related to payments or other transfers of value made to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members; | |
· | the Health Insurance Portability and Accountability Act of 1996 (“HIPAA”), as amended by the Health Information Technology for Economic and Clinical Health Act, which governs the conduct of certain electronic healthcare transactions and protects the security and privacy of protected health information; and | |
· | state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payor, including commercial insurers. |
The Affordable Care Act, among other things, amends the intent requirement of the Federal Anti-Kickback Statute and criminal healthcare fraud statutes. A person or entity no longer needs to have actual knowledge of this statute or specific intent to violate it. In addition, the Affordable Care Act provides that the government may assert that a claim including items or services resulting from a violation of the Federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act.
Efforts to ensure that our business arrangements will comply with applicable healthcare laws may involve substantial costs. It is possible that governmental and enforcement authorities will conclude that our business practices do not comply with current or future statutes, regulations or case law interpreting applicable fraud and abuse or other healthcare laws and regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of civil, criminal and administrative penalties, damages, disgorgement, monetary fines, possible exclusion from participation in Medicare, Medicaid and other federal and similar foreign healthcare programs, contractual damages, reputational harm, diminished profits and future earnings, and curtailment of our operations, any of which could harm our ability to operate our business and our results of operations.
Compliance with environmental laws and regulations could be expensive. Failure to comply with environmental laws and regulations could subject us to significant liability.
Our research and development and manufacturing operations may involve the use of hazardous substances and are subject to a variety of federal, state, local and foreign environmental laws and regulations relating to the storage, use, discharge, disposal, remediation of, and human exposure to, hazardous substances and the sale, labeling, collection, recycling, treatment and disposal of products containing hazardous substances. In addition, our research and development and manufacturing operations produce biological waste materials, such as human and animal tissue, and waste solvents, such as isopropyl alcohol. These operations are permitted by regulatory authorities, and the resultant waste materials are disposed of in material compliance with environmental laws and regulations. Liability under environmental laws and regulations can be joint and several and without regard to fault or negligence. Compliance with environmental laws and regulations may be expensive and non-compliance could result in substantial liabilities, fines and penalties, personal injury and third part property damage claims and substantial investigation and remediation costs. Environmental laws and regulations could become more stringent over time, imposing greater compliance costs and increasing risks and penalties associated with violations. We cannot assure you that violations of these laws and regulations will not occur in the future or have not occurred in the past as a result of human error, accidents, equipment failure or other causes. The expense associated with environmental regulation and remediation could harm our financial condition and operating results.
Risks Related to Owning Our Securities, Our Financial Results and Our Need for Financing
Our quarterly and annual results may fluctuate significantly, may not fully reflect the underlying performance of our business and may result in volatility in the price of our securities.
| 31 |
| Table of Contents |
Our operating results will be affected by numerous factors such as:
· | variations in the level of expenses related to our proposed products; | |
· | status of our product development efforts; | |
· | execution of collaborative, licensing or other arrangements, and the timing of payments received or made under those arrangements; | |
· | intellectual property prosecution and any infringement lawsuits to which we may become a party; | |
· | regulatory developments affecting our products or those of our competitors, including the timing and success of obtaining various regulatory approvals for our products’ testing, production and marketing; | |
· | our ability to obtain and maintain FDA clearance and approval from foreign regulatory authorities for our products, which have not yet been approved for marketing; | |
· | market acceptance of our TAEUS applications; | |
· | the availability of reimbursement for our TAEUS applications; | |
· | our ability to attract new customers and grow our business with existing customers; | |
· | the timing and success of new product and feature introductions by us or our competitors or any other change in the competitive dynamics of our industry, including consolidation among competitors, customers or strategic partners; | |
· | the amount and timing of costs and expenses related to the maintenance and expansion of our business and operations; | |
· | changes in our pricing policies or those of our competitors; | |
· | general economic, industry and market conditions; | |
· | the hiring, training and retention of key employees, including our ability to expand our sales team; | |
· | litigation or other claims against us; | |
· | our ability to obtain additional financing; and | |
· | advances and trends in new technologies and industry standards. |
Any or all of these factors could adversely affect our cash position requiring us to raise additional capital which may be on unfavorable terms and result in substantial dilution. Additionally, the risks surrounding our business, as well as the limited market for our common stock, have resulted, and will likely continue to result, in volatility in the price of our common stock and warrants. From January 1, 2020 through December 31, 2020, intra-day trading prices on the Nasdaq Capital Market have fluctuated from a low of $0.60 to a high of $2.25 with respect to shares of our common stock, and from a low of $0.06 to a high of $0.60 with respect to our warrants, and may continue to fluctuate significantly in the future.
Our stock price has fluctuated in the past, has recently been volatile and may be volatile in the future for reasons unrelated to our operating performance or prospects, and as a result, investors in our common stock could incur substantial losses.
Our stock price has fluctuated in the past, has recently been volatile and may be volatile in the future. By wayFrom January 1, 2023 through December 31, 2023, intra-day trading prices of example, on December 29, 2020, the priceshares of our common stock closed at $0.71 per share while on January 26, 2021, our stock price closed at $2.85 per share with no discernible material announcements or developments relatingthe Nasdaq Capital Market fluctuated from a low of $0.87 to our operations. On January 20, 2021, the intra-day sales pricea high of our common stock fluctuated between a reported low sale price of $1.81$5.39, and a reported high sales price of $2.58. We may incur rapid and substantial decreases in our stock pricecontinue to fluctuate significantly in the foreseeable future that are unrelated to our operating performance or prospects. In addition, the COVID-19 pandemic has caused broad stock market and industry fluctuations.future. The stock market in general and the market for healthcare companies in particular have experienced extreme volatility that has often been unrelated to the operating performance of particular companies. As a result of this volatility, investors may experience losses on their investment in our common stock.
Additionally, recently, securities of certain companies have experienced significant and extreme volatility in stock price due to a sudden increase in demand for stock resulting in aggregate short positions in the stock exceeding the number of shares available for purchase, forcing investors with short exposure to pay a premium to repurchase shares for delivery to share lenders. This is known as a “short squeeze.” These short squeezes have led to the price per share of those companies to trade at a significantly inflated rate that is disconnected from the underlying value of the company. Many investors who have purchased shares in those companies at an inflated rate face the risk of losing a significant portion of their original investment as the price per share declines steadily as interest in those stocks abates. While we have no reason to believe our shares would be the target of a short squeeze, there can be no assurance that they will not be in the future, and you may lose a significant portion or all of your investment if you purchase our shares at a rate that is significantly disconnected from our underlying value.
| 32 |
| Table of Contents |
Our stock is subject to minimum requirements to remain listed on the Nasdaq Capital Market, including a minimum bid price requirement, and may be delisted if it does not maintain compliance with those requirements.
On January 5, 2022, the Company received a notification letter from the Listing Qualifications Department of Nasdaq notifying the Company that, because the closing bid price for the Company’s common stock listed on Nasdaq was below $1.00 for 30 consecutive trading days, the Company no longer met the minimum bid price requirement for continued listing on The Nasdaq Capital Market under Nasdaq Marketplace Rule 5550(a)(2), requiring a minimum bid price of $1.00 per share (the “Minimum Bid Price Requirement”).
The Company held a special meeting of the stockholders in November 2022 for the purpose of approving a reverse stock split. Following stockholder approval, the Company filed a Certificate of Amendment to the Company’s Fourth Amended and Restated Certificate of Incorporation with the Secretary of State of Delaware to effect a 1-for-20 reverse stock split of the shares of the Company’s Common Stock, effective as of December 9, 2022 (the “Reverse Stock Split”). As a result of the Reverse Stock Split, the Company regained compliance with the Nasdaq Minimum Bid Price Requirement. If we fall below the Minimum Bid Price Requirement again, we cannot be certain that our stockholders will approve a reverse stock split or, if approved, how the market would respond to such a reverse stock split.
We may be subject to securities litigation, which is expensive and could divert management attention.
In the past, companies that have experienced volatility in the market price of their securities have been subject to an increased incidence of securities class action litigation. We may be the target of this type of litigation in the future. Securities litigation against us could result in substantial costs and divert our management’s attention from other business concerns, which could seriously harm our business.
There is a limited market for our common stock.
Although our common stock is traded on the Nasdaq Capital Market, the volume of trading has historically been limited. Our average daily trading volume of our shares from January 1, 20202023 to December 31, 20202023 was approximately 301,47866,369 shares. Thinly traded stock can be more volatile than stock trading in a more active public market. While we have made efforts to increase trading in our stock, we cannot predict the extent to which an active public market for our common stock will develop or be sustained. Therefore, a holder of our common stock who wishes to sell his or her shares may not be able to do so immediately or at an acceptable price.
If securities or industry analysts do not publish research reports about our business, or if they issue an adverse opinion about our business, the price of our securities and trading volume could decline.
The trading market for our securities is influenced by the research and reports that industry or securities analysts publish about us or our business. We do not currently have andIf any of the securities or industry analysts who cover us or may never obtain research coverage bycover us in the future change their recommendation regarding our common stock adversely, or provide more favorable relative recommendations about our competitors, the price of our common stock would likely decline. If any securities andor industry analysts. If noanalyst who covers us or few analysts commence researchmay cover us in the future were to cease coverage of us or one or more of the analysts who cover us issues an adverse opinion about our company, the price of our securities would likely decline. If one or more of these analysts ceases research coverage of us or failsfail to regularly publish reports on us, we could lose visibility in the financial markets, which in turn could cause the price of our securities or trading volume of our common stock to decline.
If we are unable to implement and maintain effective internal control over financial reporting, including by remediating current material weaknesses in our internal control over financial reporting, investors may lose confidence in the accuracy and completeness of our financial reports, and the market price of our securities may decrease.
As a public company, we are required to maintain internal control over financial reporting and to report any material weaknesses in such internal controls. Section 404 of the Sarbanes-Oxley Act of 2002 (the “Sarbanes-Oxley Act”) requires that we evaluate and determine the effectiveness of our internal control over financial reporting and provide a management report on our internal control over financial reporting.
Currently, we have material weaknesses in our internal control over financial reporting and, as a result, we may not detect errors on a timely basis and our financial statements may be materially misstated. Specifically, we have insufficient personnel resources within the accounting function to segregate the duties over financial transaction processing and reporting. We are in the process of improvingintend to improve our internal control over financial reporting, whichreporting; however, the process is time-consuming, costly and complicated and could limitcomplicated. We are constrained in the improvements we are able to make due to our limited resources. Until our internal controls are improved our ability to maintain effective internal controls over financial reporting.
| 33 |
| Table of Contents |
Until such time as we are no longer an “emerging growth company” or a smaller reporting company, our auditors will not be required to attest as to our internal control over financial reporting. If we continue to identify material weaknesses in our internal control over financial reporting, if we are unable to comply with the requirements of Section 404 in a timely manner, if we are unable to assert that our internal control over financial reporting is effective or, if required, if our independent registered public accounting firm is unable to attest that our internal control over financial reporting is effective, investors may lose confidence in the accuracy and completeness of our financial reports and the market price of our common stock could decrease. We could also become subject to stockholder or other third-party litigation as well as investigations by the stock exchange on which our securities are listed, the Securities and Exchange Commission (the “SEC”) or other regulatory authorities, which could require additional financial and management resources and could result in fines, trading suspensions or other remedies.
We are subject to the periodic reporting requirements of the Exchange Act. Our disclosure controls and procedures are designed to reasonably assure that information required to be disclosed by us in reports we file or submit under the Exchange Act is accumulated and communicated to management, and recorded, processed, summarized and reported within the time periods specified by the rules and forms of the SEC. We believe that any disclosure controls and procedures or internal controls and procedures, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met.
These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur because of simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people or by an unauthorized override of the controls. Accordingly, because of the inherent limitations in our control system, misstatements due to error or fraud may occur and not be detected.
We have not paid dividends in the past and have no immediate plans to pay dividends.
We plan to reinvest all of our earnings, to the extent we have earnings, in order to further develop our technology and potential products and to cover operating costs. We do not plan to pay any cash dividends with respect to our securities in the foreseeable future. We cannot assure you that we will, at any time, generate sufficient surplus cash that would be available for distribution to the holders of our common stock as a dividend.
We incur significant costs as a result of being a public company that reports to the SEC and our management is required to devote substantial time to meet compliance obligations.
As a public company listed in the United States, we incur significant legal, accounting and other expenses relating to our compliance obligations. We are subject to reporting requirements of the Exchange Act and the Sarbanes-Oxley Act, as well as rules subsequently implemented by the SEC and Nasdaq that impose significant requirements on public companies, including requiring the establishment and maintenance of effective disclosure and financial controls and corporate governance practices. In addition, there are significant corporate governance and executive compensation-related provisions in the Dodd-Frank Act Wall Street Reform and Protection Act that contribute to our legal and financial compliance costs, make some activities more difficult, time-consuming or costly and also place undue strain on our personnel, systems and resources. Our management and other personnel need to devote a substantial amount of time to these compliance initiatives. Furthermore, these rules and regulations may make it more difficult and more expensive for us to obtain director and officer liability insurance, and we may be required to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. As a result, it may be more difficult for us to attract and retain qualified people to serve on our board of directors, our board committees or as executive officers.
Future sales and issuances of our common stock or rights to purchase common stock, including pursuant to our equity incentive plan and our at-the-market equity offering program, could result in dilution of the percentage ownership of our stockholders and could cause the price of our securities to fall.
We expect that significant capital will be needed in the future to continue our planned operations. To the extent we raise capital by issuing common stock, convertible securities or other equity securities, our stockholders may experience substantial dilution, and new investors could gain rights superior to our existing stockholders.
Our charter documents and Delaware law may inhibit a takeover that stockholders consider favorable.
Certain provisions of our Fourth Amended and Restated Certificate of Incorporation, as amended (our “Certificate of Incorporation”) and Amended and Restated Bylaws (our “Bylaws”) and applicable provisions of Delaware law may delay or discourage transactions involving an actual or potential change in control or change in our management, including transactions in which stockholders might otherwise receive a premium for their shares, or transactions that our stockholders might otherwise deem to be in their best interests. The provisions in our Certificate of Incorporation and Bylaws:
· | authorize our board of directors to issue preferred stock without stockholder approval and to designate the rights, preferences and privileges of each class; if issued, such preferred stock would increase the number of outstanding shares of our capital stock and could include terms that may deter an acquisition of us; | |
· | limit who may call stockholder meetings; |
| 34 |
| Table of Contents |
· | do not provide for cumulative voting rights; | |
· | provide that all vacancies in our board of directors may be filled by the affirmative vote of a majority of directors then in office, even if less than a quorum; | |
· | provide that stockholders must comply with advance notice procedures with respect to stockholder proposals and the nomination of candidates for director; | |
· | provide that stockholders may only amend our Certificate of Incorporation upon a supermajority vote of stockholders; and | |
· | provide that the Court of Chancery of the State of Delaware will be the exclusive forum for certain legal claims. |
In addition, section 203 of the Delaware General Corporation Law limits our ability to engage in any business combination with a person who beneficially owns 15% or more of our outstanding voting stock unless certain conditions are satisfied. This restriction lasts for a period of three years following any such person’s share acquisition. These provisions may have the effect of entrenching our management team and may deprive stockholders of the opportunity to sell their shares to potential acquirers at a premium over prevailing prices. This potential inability to obtain a control premium could reduce the price of our common stock.
General Risk Factors
Unfavorable national or global economic conditions or political developments could adversely affect our business, financial condition or results of operations.
Our results of operations could be adversely affected by general conditions in the national or global economy and financial markets. For example, governmental statements, actions or policies, political unrest and global financial crises can cause extreme volatility and disruptions in the capital and credit markets. A severe or prolonged economic downturn, political unrest or additional global financial crises, including those resulting from the COVID-19 pandemic and the ongoing Russia-Ukraine war, Israel-Hamas war and the conflict between China and Taiwan, could result in a variety of risks to our business, including weakened demand for our products, if approved, or our ability to raise additional capital when needed on acceptable terms, if at all. A weak or declining economy could also strain our suppliers, possibly resulting in supply disruption. Any of the foregoing could harm our business and we cannot anticipate all of the ways in which the current economic climate, further political developments and financial market conditions could adversely impact our business.
Our cash and cash equivalents could be adversely affected if the financial institutions in which we hold our cash and cash equivalents fail.
We regularly maintain cash balances at third-party financial institutions in excess of the Federal Deposit Insurance Corporation insurance limit. Any failure of a depository institution to return these deposits on demand, or if a depository institution is subject to other adverse conditions in the financial or credit markets, could impact access to our invested cash or cash equivalents and could adversely impact our operating liquidity and financial performance.
Our business and operations are subject to risks related to climate change.
The effects of global climate change present risks to our business. Natural disasters, extreme weather and other conditions caused by or related to climate change could adversely impact our supply chain, the courier delivery services we use, the availability and cost of raw materials and components, energy supply, transportation, or other inputs necessary for the operation of our business. Climate change and natural disasters could also result in physical damage to our facilities as well as those of our suppliers, health care providers and other business partners, which could cause disruption in our business and operations. Our facilities and our laboratory equipment would be costly to replace and could require substantial lead time to repair or replace. Although we believe we possess adequate insurance for the disruption of our business from causalities, such insurance may not be sufficient to cover all of our potential losses and may not continue to be available to us on acceptable terms, or at all.
Our business could be negatively impacted by corporate social responsibility and sustainability matters.
There has been an increased focus from investors, customers, employees and other stakeholders concerning corporate social responsibility and sustainability matters, including addressing climate change and diversity in company management, which may result in increases in our costs to operate our business or restrict certain aspects of our activities. The standards by which corporate social responsibility and sustainability efforts and related matters are measured are developing and evolving, and certain areas are subject to assumptions that could change over time and the extent and severity of climate change impacts are unknown. In addition, we could be criticized for the scope of such initiatives or goals or a lack of diversity on our board of directors or among our executive officers, or perceived as not acting responsibly in connection with these matters. Any such matters could have a material adverse impact on our future results of operations, financial position and cash flows.
| 35 |
| Table of Contents |
Item 1B. Unresolved Staff Comments
Not applicable.
Item 1C. Cybersecurity
We have established procedures for evaluating, recognizing, and managing significant risks stemming from potential unauthorized events occurring on or through our electronic information systems. These procedures comprise an important part of our overall enterprise risk management systemand are aimed at preventing, detecting, or mitigating data breaches, theft, misuse, unauthorized access, or any other security incidents or vulnerabilities affecting digitally stored data. Internally we have an Internet, Email and Computer Use Policy and all of our employees have been trained on the policy and related tools. Additionally, we employ processes to manage and identify risks arising from cybersecurity threats linked to supplier and customer relationships and our utilization of third-party technology and systems.
We adhere to a risk management framework based on applicable laws and regulations to handle cybersecurity risks across our products, services, infrastructure and corporate assets. We regularly conduct risk assessments to gauge the effectiveness of our systems, identifying areas for improvement. These processes enable us to make informed, risk-based decisions and prioritize cybersecurity measures and risk mitigation strategies. Our risk mitigation efforts encompass a range of technical and operational actions.
Our cybersecurity risks and related responses are evaluated by senior leadership, including as part of our enterprise risk assessments that are reviewed by our Board of Directors. Our management team supervises efforts to prevent, detect, mitigate and remediate cybersecurity risks and incidents. However, we cannot guarantee that our efforts will prevent any cybersecurity incident from occurring.
As of the date of this report, we have not identified any risks from cybersecurity threats, including as a result of any previous cybersecurity incidents, that we believe have, or are likely to, materially affect us, our business strategy, results of operations, or financial condition.
Item 2. Properties
Our principal office is located at 3600 Green Court, Suite 350, Ann Arbor, Michigan 48105-1570. We currently lease approximately 3,9507,200 square feet of office and light industrial/research space which will expand to approximately 7,200 square feet effective May 1, 2021, under a lease that is due to expire in December 2025. The rent is approximately $8,232 per month, increasing to $15,452$16,393 per month effective MayJanuary 1, 2021,2023, subject to moderate annual increases.
We also maintain an office in London, Ontario, Canada under a lease that is terminable by either party with 60 days’ written notice. The rent is approximately $900 per month per the agreement with the landlord, subject to moderate annual increases at the discretion of the landlord.
We believe that, with respect to both of our facilities, equivalent suitable space is available at similar rents.
We are not currently a party to any pending legal proceedings that we believe will have a material adverse effect on our business or financial conditions. We may, however, be subject to various claims and legal actions arising in the ordinary course of business from time to time.
Not applicable.
| 36 |
| Table of Contents |
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Market Information
Our common stock and our warrants issued in our initial public offering havehas been listed on the Nasdaq Capital Market under the symbolssymbol “NDRA” and “NDRAW,” respectively, since June 28, 2017 upon the separation of units sold in our initial public offering. Prior to that date, our common stock and suchtraded together with warrants traded togetherissued in our initial public offering as units beginning on May 9, 2017. Each of ourOur publicly traded warrants is exercisable for a share of our common stock at a price of $6.25 per share and expiresexpired on May 12, 2022.
As of March 25, 2021,24, 2024, there were 2523 holders of record of our common stock and one holder of record of our publicly traded warrants.
Dividend Policy
We have never paid cash dividends on our securities and we do not anticipate paying any cash dividends on our shares of common stock in the foreseeable future. We intend to retain any future earnings for reinvestment in our business. Any future determination to pay cash dividends will be at the discretion of our board of directors, and will be dependent upon our financial condition, results of operations, capital requirements and such other factors as our board of directors deems relevant.
Securities Authorized for Issuance Under Equity Compensation Plans
The information required by Item 5 of Form 10-K regarding equity compensation plans is incorporated herein by reference to Item 12 of Part III of this Annual Report on Form 10-K.
Recent Sales of Unregistered Securities
On November 30, 2023, the Company issued 202,020 shares of restricted common stock (the “Restricted Stock”) of the Company to PatentVest, Inc. (“PatentVest”) pursuant to a Restricted Stock Agreement and Consulting Services Agreement, each with PatentVest, in exchange for certain services related to the Company’s patent portfolio. The Restricted Stock is subject to a vesting schedule pursuant to the Restricted Stock Agreement and the shares may not be sold, assigned, transferred, pledged, hypothecated, disposed of or otherwise encumbered prior to becoming vested. The Restricted Stock was offered and sold in reliance upon the exemption from the registration set forth under Section 4(a)(2) of the Securities Act, and the regulations promulgated thereunder relating to sales by an issuer not involving any public offering, and in reliance on similar exemptions under applicable state laws.
Issuer Purchases of Equity Securities
There were no repurchases of our common stock during the year ended December 31, 2023.
Item 6. Selected Financial Data
| 37 |
| Table of Contents |
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
The following discussion of our financial condition and results of operations should be read in conjunction with the financial statements and the related notes thereto included elsewhere in this Annual Report. This discussion and analysis contain forward-looking statements that are based on our management’s current beliefs and assumptions, which statements are subject to substantial risks and uncertainties. Our actual results may differ materially from those expressed or implied by these forward-looking statements as a result of many factors, including those discussed in “Risk Factors” in Item 1A of this Annual Report. Please also see “Cautionary Note Regarding Forward-Looking Statements”and “Risk Factor Summary” at the beginning of this Annual Report.
Overview
We are leveraging experience with pre-clinical enhanced ultrasound devices to develop technology for increasing the capabilities of clinical diagnostic ultrasound and other types of capital equipment, to broaden patient access to the safe diagnosis and treatment of a number of significant medical conditions in circumstances where expensive X-ray computed tomography (“CT”)CT and magnetic resonance imaging (“MRI”)MRI technology, or other diagnostic technologies such as surgical biopsy, are unavailable or impractical.
The use of RF energy allows our TAEUS technology to penetrate deep into tissue, enabling the imaging of human anatomy at depths equivalent to those of conventional ultrasound. The RF pulses are absorbed by tissue and converted into ultrasound signals, which are detected by an external ultrasound receiver and a digital acquisition system that is part of the TAEUS system. The detected ultrasound is processed into images and other forms of data using our proprietary algorithms and displayed to complement conventional gray-scale ultrasound images.
Each of our TAEUS platform applications will require regulatory approvals before we are able to sell or license the application. Based on certain factors, such as the installed base of ultrasound systems, availability of other imaging technologies, such as CT and MRI, economic strength and applicable regulatory requirements, we intend to seek initial approval of our applications for sale in the European Union and the United States, followed by China.
In March 2020, we received CE mark approval for our TAEUS FLIP (Fatty(“Fatty Liver Imaging Probe) System. The CE marking indicates that TAEUS FLIPProbe”) System, complies with all applicable European Directivesenabling its marketing and Regulationssales in the European Union and other CE mark geographies, including the 27 EU member states.
In June 2020, we submitted a 510(k) Application to the FDA for our TAEUS Fatty Live Imaging Probe (“FLIP”) System. In February 2022, we announced that we would pursue FDA reclassification and clearance of our TAEUS FLIP System.
Financial Operations Overview
Revenue
No revenue has been generated by our TAEUS technology, which we have not commercially sold as of December 31, 2020.
Research and Development Expenses
Our research and development expenses primarily include wages, fees and equipment for the development of our TAEUS technology platform and the proposed applications. Additionally, we incur certain costs associated with the protection of our products and inventions through a combination of patents, licenses, applications and disclosures.
· | employee-related expenses, such as salaries, bonuses and benefits, consultant-related expenses such as consultant fees and bonuses, stock-based compensation, overhead related expenses and travel-related expenses for our research and development personnel; | |
· | expenses incurred under agreements with contract research organizations (“CROs”), contract manufacturing organizations (“CMOs”) as well as consultants that support the implementation of our clinical and non-clinical studies; | |
· | manufacturing and packaging costs in connection with conducting clinical trials; | |
· | formulation, research and development expenses related to our TAEUS technology; and | |
· | costs for sponsored research. |
38 |
| Table of Contents |
We plan to incur research and development expenses for the foreseeable future as we expect to continue the development of TAEUS and pursue FDA approval of the NAFLD TAEUS system. At this time, due to the inherently unpredictable nature of clinical development and regulatory approvals, we are unable to estimate with certainty the costs we will incur and the timelines we will require in our continued development efforts.
Sales and Marketing Expenses
Sales and marketing expenses consist primarily of headcount and consulting costs, and marketing and tradeshow expenses. Currently, our marketing efforts are through our website and attendance of key industry meetings and conferences. In connection with the commercialization of our TAEUS applications, we are building a small sales and marketing team to train and support global ultrasound distributors and expect to execute traditional marketing activities such as promotional materials, electronic media and participation in industry events and conferences. In September 2020,As of December 31, 2023, we hiredhad a full-time sales representative in each of the United Kingdom.
General and Administrative Expenses
General and administrative expenses consist primarily of salaries and related expenses for our management and personnel, and professional fees, such as for accounting, consulting and legal services.
Critical Accounting Policies and Estimates
Use of Estimates
The preparation of the financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, and disclosure of contingent liabilities at the date of the financial statements and the reported amounts of expenses during the reporting period. Actual results could differ from those estimates.
Management makes estimates that affect certain accounts including inventory reserve, deferred income tax assets, accrued expenses, fair value of equity instruments and reserves for any other commitments or contingencies. Any adjustments applied to estimates are recognized in the period in which such adjustments are determined.
Share-based Compensation
Our 2016 Omnibus Incentive Plan (the “Omnibus Plan”) permits the grant of stock options and other stock awards to our employees, consultants and non-employee members of our board of directors. Each January 1 the pool of shares available for issuance under the Omnibus Plan automatically increases by an amount equal to the lesser of (i) the number of shares necessary such that the aggregate number of shares available under the Omnibus Plan equals 25% of the number of fully-diluted outstanding shares on the increase date (assuming the conversion of all outstanding shares of preferred stock and other outstanding convertible securities and exercise of all outstanding options and warrants to purchase shares) and (ii) if the board of directors takes action to set a lower amount, the amount determined by the board. On January 1, 2021,2024, the pool of shares issuable under the Omnibus Plan automatically increased by 1,599,5701,717,783 shares from 5,861,6581,322,169 shares to 7,461,228.3,039,952 shares. As of December 31, 2020, prior to the 1,599,570 share increase,2023, there were 3,891,521663,633 shares of common stock remaining available for issuance under the Omnibus Plan.
We record share-based compensation in accordance with the provisions of the Share-based Compensation Topic of the FASB Codification. The guidance requires the use of option-pricing models that require the input of highly subjective assumptions, including the option’s expected life and the price volatility of the underlying stock. The fair value of each option grant is estimated on the date of grant using the Black-Scholes option valuation model which uses certain assumptions related to risk-free interest rates, expected volatility, expected life of the common stock options, and future dividends, and the resulting charge is expensed using the straight-line attribution method over the vesting period.
| 39 |
| Table of Contents |
Stock compensation expense recognized during the period is based on the value of share-based awards that were expected to vest during the period adjusted for estimated forfeitures. The estimated fair value of grants of stock options and warrants to non-employees is charged to expense, if applicable, in the financial statements.
Recent Accounting Pronouncements
See Note 2 of the accompanying financial statements for a discussion of recently issued accounting standards.
Results of Operations
Years ended December 31, 20202023 and 2019
Revenue
We had no revenue during the years ended December 31, 20202023 and 2019.
Cost of Goods Sold
We had no cost of goods sold during the years ended December 31, 20202023 and 2019.
Research and Development
Research and development expenses were $5,917,944$5,003,695 for the year ended December 31, 2020,2023, as compared to $6,574,999$6,554,194 for the year ended December 31, 2019,2022, a decrease of $657,055,$1,550,499 or 10%24%. The costs include primarily wages, fees and equipment for the development of our TAEUS product line. Research and development expenses decreased from the same period for the prior year as we completed development of our initial TAEUS product and began focusing our spending on commercialization of the product that has been developed.
Sales and Marketing
Sales and marketing expenses were $581,893$820,554 for the year ended December 31, 2020,2023, as compared to $412,434$1,429,150 for the year ended December 31, 2019, an increase2022, a decrease of $169,459,$608,596, or 41%43%. The increase wascosts include primarily due to additional headcount and pre-selling activities for our TAEUS product line. Sales and marketing expenses decreased largely due to the departure of our Chief Commercial Officer. Currently, our marketing efforts are through our website and attendance of key industry meetings. Subsequent to the period ending December 31, 2020 we began hiring and training additional staff to support our sales efforts.
General and Administrative
Our general and administrative expenses for the year ended December 31, 20202023 were $5,002,080,$4,696,486, compared to $3,856,159$5,174,215 for the year ended December 31, 2019, an increase2022, a decrease of $1,145,921,$477,729, or 30%9%. Our wage and related expenses for the year ended December 31, 20202023 were $2,070,747,$1,554,670, compared to $1,878,265$2,123,291 for the year ended December 31, 2019.2022. Wage and related expenses in the year ended December 31, 20202023 included $215,988 for bonuses and $811,871$339,696 of stock compensation expense related to the issuance and vesting of options, and RSU’s, compared to $147,915 for bonuses, $720,030$416,508 of stock compensation expense related to the issuance and vesting of options, for the year ended December 31, 2019.2022. Our professional fees, which include legal, audit, and investor relations, for the year ended December 31, 20202023 were $2,512,878,$1,980,464, compared to $1,455,346$2,047,964 for the year ended December 31, 2019. The increase was due to additional costs2022.
Other Income
Other income for investor relations related activity, and legal fees associated with our financing transactions.
Net Loss
As a result of the foregoing, for the year ended December 31, 2020,2023, we recorded a net loss of $11,725,501,$10,060,250, compared to a net loss of $13,305,964$13,179,092 for the year ended December 31, 2019.
Near-Term Liquidity and Capital Resources
Since inception, we have incurred losses and expect to continue to incur losses for the foreseeable future. As of December 31, 2023, we had an accumulated deficit of $91,930,152 and had $2,833,907 in cash. To date we have funded our operations through private and public sales of our securities. As of December 31, 2020,securities and will need to raise additional funds in order to execute on our business plan, fully commercialize our TAEUS technology, and generate revenues. If we had $7,227,316are unable to obtain adequate financing or financings in cash. We completed the following financing events from January 2019 through December 2020:
| 40 |
| Table of Contents |
We issued an aggregate of 1,421,858 shares of our common stock in return for gross proceeds of $1,372,437 from the at-the-market facility.
The consolidated financial statements included in this Form 10-K have been prepared assuming we will continue as a going concern, which contemplates the realization of assets and the settlement of liabilities and commitments in the normal course of business. As reflected in the accompanying consolidated financial statements, during the year ended December 31, 2020,2023, we incurred net losses of $11,725,501$10,060,250 and used cash in operations of $10,746,595. These$9,548,775. In light of our cash balance as of December 31, 2023, we will need to raise additional capital in order to fund operations through the next twelve months, and other factors raise substantial doubt about ourprior to any ability to continue as a going concern for one yearfund operations from revenue generated from the issuancesale of the accompanying financial statements.our products. The financial statements do not include any adjustments that might be necessary should we be unable to continue as a going concern.
Operating Activities
During the year ended December 31, 2020,2023, we used $10,746,595$9,548,775 of cash in operating activities primarily as a result of our net loss of $11,725,501,$10,060,250, offset by share-based compensation of $2,102,352,$996,430, amortization of debt discountright of $232,426,use assets of $151,725, inventory reserve of $138,045, depreciation expense of $99,342, amortization$123,726, fixed assets write-off of Right of Use assets of $65,907,$24,868, and net changes in operating assets and liabilities of $(1,521,121)$(923,319).
During the year ended December 31, 2019,2022, we used $8,588,851$12,769,371 of cash in operating activities primarily as a result of our net loss of $13,305,964$13,179,092, offset by share-based compensation of $1,399,547, depreciation and amortization expenses of $80,577,$1,199,838, amortization of debt discountright of $2,355,469,use assets of $137,597, depreciation expense of $96,661, fixed assets write-off of $1,391, and net changes in operating assets and liabilities of $597,830.
Investing Activities
During the year ended December 31, 2020, the Company2023, we used $75,333$33,844 in investing activities related to purchases of equipment.
During the year ended December 31, 2019, the Company2022, we used $43,595$202,577 in investing activities related to purchasepurchases of equipment.
Financing Activities
During the year ended December 31, 2020,2023, our financing activities provided $11,875,037, including $6,780,942$6,483,393 in proceeds from issuanceissuances of common stock $4,757,011$20,053 in proceeds from issuances of common stock warrants, and $1,014,859 in proceeds from warrant exercises, and $337,084 in proceeds from loans.
During the year ended December 31, 2019,2022, our financing activities provided $8,335,278, including $5,344,257 from the issuance of Series A Preferred Stock and warrants, $375,520 from the issuance of Series B Preferred Stock and warrants, $2,490,501$8,399,512 in proceeds from the placementissuances of senior secured convertible promissory notes and warrants, and $125,000 from the issuance common stock and warrants.
Long-Term Liquidity
We have not completed the commercialization of any of our TAEUS technology platform applications. We expect to continue to incur significant expenses for the foreseeable future. We anticipate that our expenses will increase substantially as we:
· | advance the engineering design and development of our TAEUS technology; | |
· | acquire parts and build finished goods inventory of the TAEUS FLIP system; | |
· | complete regulatory filings required for marketing approval of our NAFLD TAEUS application in the United States, including clinical studies to advance our de novo application with the FDA; | |
· | seek to hire a small internal marketing team to engage and support channel partners and clinical customers for our NAFLD TAEUS application; | |
· | expand marketing of our NAFLD TAEUS application; | |
· | advance development of our other TAEUS applications; and | |
· | add operational, financial and management information systems and personnel, including personnel to support our product development, planned commercialization efforts and our operation as a public company. |
It is possible that we will not achieve the progress that we expect because the actual costs and timing of completing the development and regulatory approvals for a new medical device are difficult to predict and are subject to substantial risks and delays. We have no committed external sources of funds.funds except for the February 2024 ATM Agreement, the use of which may be limited due to registration statement rules relating to public float. We do not expect that our existing cash will be sufficient for us to complete the commercialization of our NAFLD TAEUS application or to complete the development of any other TAEUS application and we will need to raise substantial additional capital for those purposes. As a result, we will need to finance our future cash needs through public or private equity offerings, debt financings, corporate collaboration and licensing arrangements or other financing alternatives. Our forecast of the period of time through which our financial resources will be adequate to support our operations is a forward-looking statement and involves risks and uncertainties, and actual results could vary as a result of a number of factors, including the factors discussed in the Risk Factors section of this Annual Report on Form 10-K entitled “Risk Factors”.10-K. We have based this estimate on assumptions that may prove to be wrong, and we could utilize our available capital resources sooner than we currently expect.
Until we can generate a sufficient amount of revenue from our TAEUS platform applications, if ever, we expect to finance future cash needs through public or private equity offerings, debt financings or corporate collaborations and licensing arrangements. Additional funds may not be available when we need them on terms that are acceptable to us, or at all. As described below, the COVID-19 pandemic has impacted our business operations to some extent and is expected to continue to do so and, in light of the effect of such pandemic on financial markets, these impacts may include reduced access to capital. If adequate funds are not available, we may be required to delay, reduce the scope of or eliminate one or more of our research or development programs or our commercialization efforts or perhaps even cease the operation of our business. To the extent that we raise additional funds by issuing equity securities, our stockholders may experience additional dilution, and debt financing, if available, may involve restrictive covenants. To the extent that we raise additional funds through collaborations and licensing arrangements, it may be necessary to relinquish some rights to our technologies or applications or grant licenses on terms that may not be favorable to us. We may seek to access the public or private capital markets whenever conditions are favorable, even if we do not have an immediate need for additional capital at that time.
Off-Balance Sheet Transactions
At December 31, 2020,2023, the Company did not have any transactions, obligations or relationships that could be considered off-balance sheet arrangements.
Not applicable.
| 41 |
| Table of Contents |
Item 8. Financial Statements and Supplementary Data.
Index to Financial Statements
ENDRA Life Sciences Inc.
December 31, 2020
Page | |||
F-1 | |||
F-2 | |||
F-3 | |||
F-4 | |||
F-5 | |||
F-6 |
| 42 |
| Table of Contents |
| New York Office: 805 Third Avenue New York, NY 10022 212.838-5100 www.rbsmllp.com |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of
ENDRA Life Sciences Inc. and Subsidiaries
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of ENDRA Life Sciences Inc. and Subsidiaries (collectively, the “Company”) as of December 31, 20202023 and 2019,2022, the related consolidated statements of operations, shareholders’ equity and cash flows for each of the two years in the period ended December 31, 2020,2023, and the related notes and schedules (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 20202023 and 2019,2022, and the results of its operations and its cash flows for each of the two years in the period ended December 31, 2020,2023 and 2022 in conformity with accounting principles generally accepted in the United States of America.
The Company's Ability to Continue as a Going Concern
The accompanying consolidated financial statements have been prepared assuming the Company will continue as a going concern. As discussed in Note 2 to the accompanying consolidated financial statements, the Company has suffered recurring losses from operations, generated negative cash flows from operating activities, has an accumulated deficit and has stated that substantial doubt exists about Company’s ability to continue as a going concern. Management's evaluation of the events and conditions and management’s plans in regarding these matters are also described in Note 2. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matters
The critical audit matters communicated below are matters arising from the current period audit of the financial statements that were communicated or required to be communicated to the audit committee and that: (1) relate to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective or complex judgments.
We determined that there are no critical audit matters.
/s/ RBSM LLP
We have served as the Company’s auditor since 2015.
New York, NY
March 25, 2021
New York, NY Washington DC Mumbai & Pune, India Boca Raton, FL
San Francisco, CA Las Vegas, NV Beijing, China Athens, Greece
Member: ANTEA International with affiliated offices worldwide
| F-1 |
| Table of Contents |
ENDRA Life Sciences Inc.
Consolidated BalanceBalance Sheets
December 31, | December 31, | |
| Assets | 2020 | 2019 |
| Current Assets | ||
| Cash | $7,227,316 | $6,174,207 |
| Prepaid expenses | 390,800 | 116,749 |
| Inventory | 589,620 | 113,442 |
| Other current assets | 5,986 | 130,701 |
| Total Current Assets | 8,213,722 | 6,535,099 |
| Non-Current Assets | ||
| Fixed assets, net | 212,242 | 236,251 |
| Right of use assets | 339,012 | 404,919 |
| Total Assets | $8,764,976 | $7,176,269 |
| Liabilities and Stockholders’ Equity | ||
| Current Liabilities | ||
| Accounts payable and accrued liabilities | $910,183 | $1,708,525 |
| Convertible notes payable, net of discount | - | 298,069 |
| Lease liabilities, current portion | 76,480 | 66,193 |
| Total Current Liabilities | 986,663 | 2,072,787 |
| Long Term Debt | ||
| Loans | 337,084 | - |
| Lease liabilities | 271,908 | 342,812 |
| Total Long Term Debt | 608,992 | 342,812 |
| Total Liabilities | 1,595,655 | 2,415,599 |
| Stockholders’ Equity | ||
| Series A Convertible Preferred Stock, $0.0001 par value; 10,000 shares authorized; 196.794 and 6,338.490 shares issued and outstanding, respectively | 1 | 1 |
| Series B Convertible Preferred Stock, $0.0001 par value; 1,000 shares authorized; no shares and 351.711 shares issued and outstanding, respectively | - | - |
| Common stock, $0.0001 par value; 80,000,000 shares authorized; 34,049,704 and 8,421,401 shares issued and outstanding, respectively | 3,404 | 842 |
| Additional paid in capital | 64,493,611 | 49,933,736 |
| Stock payable | 10,794 | 43,528 |
| Accumulated deficit | (57,338,489) | (45,217,437) |
| Total Stockholders’ Equity | 7,169,321 | 4,760,670 |
| Total Liabilities and Stockholders’ Equity | $8,764,976 | $7,176,269 |
|
| December 31, |
|
| December 31, |
| ||
Assets |
| 2023 |
|
| 2022 |
| ||
Current Assets |
|
|
|
|
|
| ||
Cash and cash equivalents |
| $ | 2,833,907 |
|
| $ | 4,889,098 |
|
Prepaid expenses |
|
| 198,905 |
|
|
| 490,299 |
|
Total Current Assets |
|
| 3,032,812 |
|
|
| 5,379,397 |
|
Non-Current Assets |
|
|
|
|
|
|
|
|
Inventory |
|
| 2,622,865 |
|
|
| 2,644,717 |
|
Fixed assets, net |
|
| 111,782 |
|
|
| 235,655 |
|
Right of use assets |
|
| 354,091 |
|
|
| 505,816 |
|
Prepaid expenses, long term |
|
| 626,610 |
|
|
| 502,576 |
|
Other assets |
|
| 5,986 |
|
|
| 5,986 |
|
Total Assets |
| $ | 6,754,146 |
|
| $ | 9,274,147 |
|
|
|
|
|
|
|
|
|
|
Liabilities and Stockholders’ Equity |
|
|
|
|
|
|
|
|
Current Liabilities |
|
|
|
|
|
|
|
|
Accounts payable and accrued liabilities |
| $ | 700,754 |
|
| $ | 1,523,012 |
|
Lease liabilities, current portion |
|
| 173,857 |
|
|
| 152,228 |
|
Loans |
|
| 28,484 |
|
|
| 28,484 |
|
Total Current Liabilities |
|
| 903,095 |
|
|
| 1,703,724 |
|
|
|
|
|
|
|
|
|
|
Long Term Debt |
|
|
|
|
|
|
|
|
Loans, long term |
|
| - |
|
|
| - |
|
Lease liabilities |
|
| 192,062 |
|
|
| 365,919 |
|
Total Long Term Debt |
|
| 192,062 |
|
|
| 365,919 |
|
|
|
|
|
|
|
|
|
|
Total Liabilities |
|
| 1,095,157 |
|
|
| 2,069,643 |
|
|
|
|
|
|
|
|
|
|
Stockholders’ Equity |
|
|
|
|
|
|
|
|
Series A Convertible Preferred Stock, $0.0001 par value; 10,000 shares authorized; 141.397 shares issued and outstanding |
|
| 1 |
|
|
| 1 |
|
Series B Convertible Preferred Stock, $0.0001 par value; 1,000 shares authorized; no shares issued and outstanding |
|
| - |
|
|
| - |
|
Series C Convertible Preferred Stock, $0.0001 par value; 100,000 shares authorized; no shares issued and outstanding |
|
| - |
|
|
| - |
|
Common stock, $0.0001 par value; 80,000,000 shares authorized; 10,390,150 and 3,169,103 shares issued and outstanding, respectively |
|
| 1,039 |
|
|
| 317 |
|
Additional paid in capital |
|
| 97,582,868 |
|
|
| 89,068,015 |
|
Stock payable |
|
| 5,233 |
|
|
| 6,073 |
|
Accumulated deficit |
|
| (91,930,152 | ) |
|
| (81,869,902 | ) |
Total Stockholders’ Equity |
|
| 5,658,989 |
|
|
| 7,204,504 |
|
Total Liabilities and Stockholders’ Equity |
| $ | 6,754,146 |
|
| $ | 9,274,147 |
|
The accompanying notes are an integral part of these consolidated financial statements.
| F-2 |
| Table of Contents |
ENDRA Life Sciences Inc.
Consolidated Statements of Operations
|
| Year Ended |
|
| Year Ended |
| ||
|
| December 31, |
|
| December 31, |
| ||
|
| 2023 |
|
| 2022 |
| ||
Operating Expenses |
|
|
|
|
|
| ||
Research and development |
| $ | 5,003,695 |
|
| $ | 6,554,194 |
|
Sales and marketing |
|
| 820,554 |
|
|
| 1,429,150 |
|
General and administrative |
|
| 4,696,486 |
|
|
| 5,174,215 |
|
Total operating expenses |
|
| 10,520,735 |
|
|
| 13,157,559 |
|
|
|
|
|
|
|
|
|
|
Operating loss |
|
| (10,520,735 | ) |
|
| (13,157,559 | ) |
|
|
|
|
|
|
|
|
|
Other Income (Expenses) |
|
|
|
|
|
|
|
|
Other income (expenses) |
|
| 460,485 |
|
|
| (21,533 | ) |
Total other income (expenses) |
|
| 460,485 |
|
|
| (21,533 | ) |
|
|
|
|
|
|
|
|
|
Loss from operations before income taxes |
|
| (10,060,250 | ) |
|
| (13,179,092 | ) |
|
|
|
|
|
|
|
|
|
Provision for income taxes |
|
| - |
|
|
| - |
|
|
|
|
|
|
|
|
|
|
Net Loss |
| $ | (10,060,250 | ) |
| $ | (13,179,092 | ) |
|
|
|
|
|
|
|
|
|
Net loss per share – basic and diluted |
| $ | (1.58 | ) |
| $ | (4.56 | ) |
|
|
|
|
|
|
|
|
|
Weighted average common shares – basic and diluted |
|
| 6,363,759 |
|
|
| 2,891,292 |
|
The accompanying notes are an integral part of these consolidated financial statements.
| F-3 |
| Table of Contents |
ENDRA Life Sciences Inc.
Consolidated Statements of Stockholders’ Equity
Year Ended December 31, 2022 |
| Series A Convertible Preferred Stock |
|
| Series B Convertible Preferred Stock |
|
| Common stock |
|
| Additional Paid in |
|
| Stock |
|
| Accumulated |
|
| Total Stockholders' |
| |||||||||||||||||||
|
| Shares |
|
| Amount |
|
| Shares |
|
| Amount |
|
| Shares |
|
| Amount |
|
| Capital |
|
| Payable |
|
| Deficit |
|
| Equity |
| ||||||||||
Balance as of December 31, 2021 |
|
| 141.397 |
|
| $ | 1 |
|
|
| - |
|
|
| - |
|
|
| 2,127,726 |
|
| $ | 212 |
|
| $ | 79,460,980 |
|
| $ | 13,863 |
|
| $ | (68,690,810 | ) |
| $ | 10,784,246 |
|
Common stock issued for cash, net of funding costs |
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| 1,041,377 |
|
|
| 105 |
|
|
| 8,399,407 |
|
|
| - |
|
|
| - |
|
|
| 8,399,512 |
|
Fair value of vested stock options |
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| 1,199,838 |
|
|
| - |
|
|
| - |
|
|
| 1,199,838 |
|
Stock payable towards preference dividend |
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| 7,790 |
|
|
| (7,790 | ) |
|
| - |
|
|
| - |
|
Net loss |
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| (13,179,092 | ) |
|
| (13,179,092 | ) |
Balance as of December 31, 2022 |
|
| 141.397 |
|
| $ | 1 |
|
|
| - |
|
|
| - |
|
|
| 3,169,103 |
|
| $ | 317 |
|
| $ | 89,068,015 |
|
| $ | 6,073 |
|
| $ | (81,869,902 | ) |
| $ | 7,204,504 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||||||||||||||||
Year Ended December 31, 2023 |
| Series A Convertible Preferred Stock |
|
| Series B Convertible Preferred Stock |
|
| Common stock |
|
| Additional Paid in |
|
| Stock |
|
| Accumulated |
|
| Total Stockholders' |
| |||||||||||||||||||
|
| Shares |
|
| Amount |
|
| Shares |
|
| Amount |
|
| Shares |
|
| Amount |
|
| Capital |
|
| Payable |
|
| Deficit |
|
| Equity |
| ||||||||||
Balance as of December 31, 2022 |
|
| 141.397 |
|
| $ | 1 |
|
|
| - |
|
|
| - |
|
|
| 3,169,103 |
|
| $ | 317 |
|
| $ | 89,068,015 |
|
| $ | 6,073 |
|
| $ | (81,869,902 | ) |
| $ | 7,204,504 |
|
Common stock issued for cash, net of funding costs |
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| 5,637,547 |
|
|
| 564 |
|
|
| 6,482,829 |
|
|
| - |
|
|
| - |
|
|
| 6,483,393 |
|
Common stock issued for warrant exercise |
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| 1,583,500 |
|
|
| 158 |
|
|
| 1,014,701 |
|
|
| - |
|
|
| - |
|
|
| 1,014,859 |
|
Warrants issued for cash, net of funding costs |
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| 20,053 |
|
|
| - |
|
|
| - |
|
|
| 20,053 |
|
Fair value of vested stock options |
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| 996,430 |
|
|
| - |
|
|
| - |
|
|
| 996,430 |
|
Stock payable towards preference dividend |
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| 840 |
|
|
| (840 | ) |
|
| - |
|
|
| - |
|
Net loss |
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| (10,060,250 | ) |
|
| (10,060,250 | ) |
Balance as of December 31, 2023 |
|
| 141.397 |
|
| $ | 1 |
|
|
| - |
|
|
| - |
|
|
| 10,390,150 |
|
| $ | 1,039 |
|
| $ | 97,582,868 |
|
| $ | 5,233 |
|
| $ | (91,930,152 | ) |
| $ | 5,658,989 |
|
The accompanying notes are an integral part of these consolidated financial statements.
| F-4 |
| Table of Contents |
ENDRA Life Sciences Inc.
Consolidated Statements of Operations
Year Ended | Year Ended | |
December 31, | December 31, | |
2020 | 2019 | |
| Operating Expenses | ||
| Research and development | $5,917,944 | $6,574,999 |
| Sales and marketing | 581,893 | 412,434 |
| General and administrative | 5,002,080 | 3,856,159 |
| Total operating expenses | 11,501,917 | 10,843,592 |
| Operating loss | (11,501,917) | (10,843,592) |
| Other Expenses | ||
| Amortization of debt discount | (232,426) | (2,355,469) |
| Other income (expense) | 8,842 | (106,903) |
| Total other expenses | (223,584) | (2,462,372) |
| Loss from operations before income taxes | (11,725,501) | (13,305,964) |
| Provision for income taxes | - | - |
| Net Loss | $(11,725,501) | $(13,305,964) |
| Deemed dividend | (395,551) | (4,219,777) |
| Net Loss attributable to common stockholders | $(12,121,052) | $(17,525,741) |
| Net loss per share – basic and diluted | $(0.63) | $(2.34) |
| Weighted average common shares – basic and diluted | 19,192,226 | 7,499,984 |
|
| Year Ended |
|
| Year Ended |
| ||
|
| December 31, |
|
| December 31, |
| ||
|
| 2023 |
|
| 2022 |
| ||
Cash Flows from Operating Activities |
|
|
|
|
|
| ||
Net loss |
| $ | (10,060,250 | ) |
| $ | (13,179,092 | ) |
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
|
|
|
|
Depreciation and amortization |
|
| 123,726 |
|
|
| 96,661 |
|
Fixed assets write off |
|
| 24,868 |
|
|
| 1,391 |
|
Inventory reserve |
|
| 138,045 |
|
|
| - |
|
Stock compensation expense including common stock issued for RSUs |
|
| 996,430 |
|
|
| 1,199,838 |
|
Amortization of right of use assets |
|
| 151,725 |
|
|
| 137,597 |
|
Changes in operating assets and liabilities: |
|
|
|
|
|
|
|
|
Decrease in prepaid expenses |
|
| 167,360 |
|
|
| 355,128 |
|
Increase in inventory |
|
| (116,193 | ) |
|
| (1,360,139 | ) |
Decrease in accounts payable and accrued liabilities |
|
| (822,258 | ) |
|
| 111,575 |
|
Decrease in lease liability |
|
| (152,228 | ) |
|
| (132,330 | ) |
Net cash used in operating activities |
|
| (9,548,775 | ) |
|
| (12,769,371 | ) |
|
|
|
|
|
|
|
|
|
Cash Flows from Investing Activities |
|
|
|
|
|
|
|
|
Purchases of fixed assets |
|
| (33,884 | ) |
|
| (202,577 | ) |
Proceeds from sale of fixed assets |
|
| 9,163 |
|
|
| - |
|
Net cash used in investing activities |
|
| (24,721 | ) |
|
| (202,577 | ) |
|
|
|
|
|
|
|
|
|
Cash Flows from Financing Activities |
|
|
|
|
|
|
|
|
Proceeds from issuance of common stock |
|
| 6,483,393 |
|
|
| 8,399,512 |
|
Proceeds from issuance of warrants |
|
| 20,053 |
|
|
| - |
|
Proceeds from warrant exercise |
|
| 1,014,859 |
|
|
| - |
|
Net cash provided by financing activities |
|
| 7,518,305 |
|
|
| 8,399,512 |
|
|
|
|
|
|
|
|
|
|
Net decrease in cash |
|
| (2,055,191 | ) |
|
| (4,572,436 | ) |
|
|
|
|
|
|
|
|
|
Cash, beginning of year |
|
| 4,889,098 |
|
|
| 9,461,534 |
|
|
|
|
|
|
|
|
|
|
Cash, end of year |
| $ | 2,833,907 |
|
| $ | 4,889,098 |
|
|
|
|
|
|
|
|
|
|
Supplemental disclosures of cash items |
|
|
|
|
|
|
|
|
Interest paid |
| $ | 44,985 |
|
| $ | 59,113 |
|
Income tax paid |
| $ | - |
|
| $ | - |
|
|
|
|
|
|
|
|
|
|
Supplemental disclosures of non-cash items |
|
|
|
|
|
|
|
|
Stock dividend payable |
| $ | 840 |
|
| $ | 7,790 |
|
Right of use asset |
| $ | 354,091 |
|
| $ | 505,816 |
|
The accompanying notes are an integral part of these consolidated financial statements.
| F-5 |
| Table of Contents |
ENDRA Life Sciences Inc.
| Year Ended December 31, 2019 | Series A Convertible | Series B Convertible | Total | |||||||
Preferred Stock | Preferred Stock | Common stock | Additional | Accumulated | Stockholders' | |||||
Shares | Amount | Shares | Amount | Shares | Amount | Paid in Capital | Stock Payable | Deficit | Equity | |
| Balance as of December 31, 2018 | - | $- | - | $- | 7,422,642 | $742 | $33,939,162 | - | (27,691,696) | 6,248,208 |
| Series A Convertible Preferred Stock issued | 6,338.490 | 1 | - | - | 904,526 | 90 | 7,412,361 | - | - | 7,412,452 |
| Series B Convertible Preferred Stock issued | - | - | 351.711 | - | - | - | 375,520 | - | - | 375,520 |
| Common stock issued for note conversions | - | - | - | - | 94,233 | 10 | 140,396 | - | - | 140,406 |
| Fair value of vested stock options | - | - | - | - | - | - | 1,399,547 | - | - | 1,399,547 |
| Debt discount | - | - | - | - | - | - | 2,490,501 | - | - | 2,490,501 |
| Deemed dividend on preferred stock | - | - | - | - | - | 4,219,777 | - | (4,219,777) | - | |
| Stock to be issued | - | - | - | - | - | - | (43,528) | 43,528 | - | - |
| Net loss | - | - | - | - | - | - | - | - | (13,305,964) | (13,305,964) |
| Balance as of December 31, 2019 | 6,338.490 | $1 | 351.711 | $- | 8,421,401 | $842 | $49,933,736 | 43,528 | (45,217,437) | 4,760,670 |
| Year Ended December 31, 2020 | Series A Convertible | Series B Convertible | Total | |||||||
Preferred Stock | Preferred Stock | Common stock | Additional | Accumulated | Stockholders' | |||||
Shares | Amount | Shares | Amount | Shares | Amount | Paid in Capital | Stock Payable | Deficit | Equity | |
| Balance as of December 31, 2019 | 6,338.490 | $1 | 351.711 | $- | 8,421,401 | $842 | $49,933,736 | 43,528 | (45,217,437) | 4,760,670 |
| Series A Convertible Preferred Stock converted to common stock | (6,141.696) | - | - | - | 7,178,400 | 717 | 79,997 | (80,714) | - | - |
| Series B Convertible Preferred Stock converted to common stock | - | - | (351.711) | - | 360,279 | 36 | 1,633 | (1,669) | - | - |
| Common stock issued for cash | - | - | - | - | 9,862,777 | 986 | 6,779,956 | - | - | 6,780,942 |
| Common stock issued for note conversions | - | - | - | - | 331,441 | 33 | 493,814 | - | - | 493,847 |
| Common stock issued for warrant exercise | - | - | - | - | 7,098,108 | 710 | 4,756,301 | - | - | 4,757,101 |
| Common stock issued for services | - | - | - | - | 149,025 | 15 | 125,486 | - | - | 125,501 |
| Common stock issued for vested RSUs | - | - | - | - | 648,273 | 65 | 453,725 | - | - | 453,790 |
| Fair value of vested stock options | - | - | - | - | - | - | 1,523,061 | - | - | 1,523,061 |
| Fair value adjustment related to warrants repricing | - | - | - | - | - | - | 395,551 | - | (395,551) | - |
| Stock payable towards preference dividend | - | - | - | - | - | - | (49,649) | 49,649 | - | - |
| Net loss | - | - | - | - | - | - | - | - | (11,725,501) | (11, 725,501) |
| Balance as of December 31, 2020 | 196.794 | $1 | - | $- | 34,049,704 | $3,404 | $64,493,611 | 10,794 | (57,338,489) | 7,169,322 |
Year Ended | Year Ended | |
December 31, | December 31, | |
2020 | 2019 | |
| Cash Flows from Operating Activities | ||
| Net loss | $(11, 725,501) | $(13,305,964) |
| Adjustments to reconcile net loss to net cash used in operating activities: | ||
| Depreciation and amortization | 99,342 | 80,577 |
| Common stock, options and warrants issued for services | 2,102,352 | 1,399,547 |
| Amortization of debt discount | 232,426 | 2,355,469 |
| Impairment of other assets | - | 249,256 |
| Amortization of right of use assets | 65,907 | 34,434 |
| Changes in operating assets and liabilities: | ||
| Increase in prepaid expenses | (274,051) | 28,675 |
| Decrease in lease liability | (60,617) | (30,348) |
| Increase in inventory | (476,178) | (53,998) |
| Decrease in Other Current Assets | 124,715 | (106,642) |
| Decrease in accounts payable and accrued liabilities | (834,990) | 760,143 |
| Net cash used in operating activities | (10,746,595) | (8,588,851) |
| Cash Flows from Investing Activities | ||
| Purchases of fixed assets | (75,333) | (43,595) |
| Net cash used in investing activities | (75,333) | (43,595) |
| Cash Flows from Financing Activities | ||
| Proceeds from senior secured convertible promissory notes, net of fees | - | 2,490,501 |
| Proceeds from issuance of Series A Convertible Preferred Stock | - | 5,344,257 |
| Proceeds from issuance of Series B Convertible Preferred Stock | - | 375,520 |
| Proceeds from warrant exercise | 4,757,011 | - |
| Proceeds from loans | 337,084 | - |
| Proceeds from issuance of common stock | 6,780,942 | 125,000 |
| Net cash provided by financing activities | 11,875,037 | 8,335,278 |
| Net decrease in cash | 1,053,109 | (297,168) |
| Cash, beginning of period | 6,174,207 | 6,471,375 |
| Cash, end of period | $7,227,316 | $6,174,207 |
| Supplemental disclosures of cash items | ||
| Interest paid | $1,920 | $- |
| Income tax paid | $- | $- |
| Supplemental disclosures of non-cash items | ||
| Discount on convertible notes | $- | $2,490,501 |
| Conversion of convertible notes and accrued interest | $493,814 | $140,406 |
| Exchange of balance in convertible notes and accrued interest for Series A Convertible Preferred Stock | $- | $1,943,195 |
| Deemed dividend | $395,551 | $4,219,777 |
| Conversion of Series A Convertible Preferred Stock | $(717) | $- |
| Conversion of Series B Convertible Preferred Stock | $(36) | $- |
| Shares issued for financing cost | 27,300 | - |
| Stock dividend payable | $(49,649) | $- |
| Right of use asset | $339,012 | $404,919 |
| Lease liability | $348,388 | $409,005 |
For the years ended December 31, 20202023 and 2019
Note 1 –- Nature of the Business
ENDRA Life Sciences Inc. (“ENDRA” or the “Company”) has developed and is continuing to develop technology for increasingcharacterizing tissue non-invasively, at the capabilitiespoint of clinical diagnostic ultrasoundpatient care, to broaden patient access to the safe diagnosis and treatment of a number of significant medical conditions in circumstances where expensive X-ray computed tomography (“CT”) and, magnetic resonance imaging (“MRI”) technology isor other technologies are unavailable or impractical.
ENDRA was incorporated on July 18, 2007 as a Delaware corporation.
Certain reclassifications have been made to the laws of Ontario, Canada on July 6, 2017, and is wholly owned by2022 consolidated financial statements in order to conform to the Company.
Note 2 –- Summary of Significant Accounting Policies
Use of Estimates
The preparation of the financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, and disclosure of contingent liabilities at the date of the financial statements and the reported amounts of expenses during the reporting period. Actual results could differ from those estimates.
Management makes estimates that affect certain accounts including inventory reserve, deferred income tax assets, accrued expenses, fair value of equity instruments and reserves for any other commitments or contingencies. Any adjustments applied to estimates are recognized in the period in which such adjustments are determined.
Principles of Consolidation
The Company’s consolidated financial statements include all accounts of the Company and its consolidated subsidiarysubsidiaries and/or entities as of reporting period ending date(s) and for the reporting period(s) then ended. All inter-company balances and transactions have been eliminated.
Basis of Presentation
The financial statements and related disclosures have been prepared pursuant to the rules and regulations of the Securities and Exchange Commission (“SEC”). These financial statements have been prepared using the accrual basis of accounting in accordance with Generally Accepted Accounting Principles (“GAAP”) of the United States.
Cash and Cash Equivalents
The Company considers all cash on hand and in banks, including accounts in book overdraft positions, certificates of deposit, and other highly-liquidhighly liquid investments with maturities of one year or less, when purchased, to be cash. As of December 31, 2020Cash equivalents include investments in an institutional money market fund, which invests in U.S. Treasury bills, notes and December 31, 2019, the Company had no cash equivalents.bonds, and/or repurchase agreements, backed by such obligations. Carrying value approximates fair value. The Company maintains its cash in bank deposit accounts which, at times, may exceed federally insured limits. The Company has not experienced any losses in such accounts and periodically evaluates the credit worthinesscreditworthiness of the financial institutions and has determined the credit exposure to be negligible.
Inventory
The Company’s inventory is stated at the lower of cost or estimated net realizable value, with cost primarily determined on a weighted-average cost basis on the first-in, first-out method. The Company periodically determines whether a reserve should be taken for devaluation or obsolescence of inventory.
| F-6 |
| Table of Contents |
Capitalization of Fixed Assets
The Company capitalizes expenditures related to property and equipment, subject to a minimum rule, that have a useful life greater than one year for: (1) assets purchased; (2) existing assets that are replaced, improved or the useful lives have been extended; or (3) all land, regardless of cost. Acquisitions of new assets, additions, replacements and improvements (other than land) costing less than the minimum rule in addition to maintenance and repair costs, including any planned major maintenance activities, are expensed as incurred.
Leases
Accounting Standards Update (“ASU”) No. 2016-02, “Leases.” ASU 2016-02 requires a lessee to record a right of use asset and a corresponding lease liability on the balance sheet for all leases with terms longer than 12 months. ASU 2016-02 is effective for all interim and annual reporting periods beginning after December 15, 2018. Early adoption is permitted. A modified retrospective transition approach is required for lessees for capital and operating leases existing at, or entered into after, the beginning of the earliest period presented in the financial statements. At December 31, 20202023 and 2022 the Company recorded a right of use asset of $354,091 and $505,816, respectively. At December 31, 20192023 and 2022 the Company recorded a lease liability of $348,388$365,919 and $409,005,$518,147, respectively.
Revenue Recognition
ASU No. 2014-09, “Revenue from Contracts with Customers” (“ASC Topic 606”). This standard provides a single set of guidelines for revenue recognition to be used across all industries and requires additional disclosures. The updated guidance introduces a five-step model to achieve its core principal of the entity recognizing revenue to depict the transfer of goods or services to customers at an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. The Company adopted the updated guidance effective January 1, 2018 using the full retrospective method. The new standard did not have a material impact on its financial position and results of operations, as it did not change the manner or timing of recognizing revenue.
Under ASC Topic 606, in order to recognize revenue, the Company is required to identify an approved contract with commitments to perform respective obligations, identify rights of each party in the transaction regarding goods to be transferred, identify the payment terms for the goods transferred, verify that the contract has commercial substance and verify that collection of substantially all consideration is probable. The adoption of ASC Topic 606 did not have an impact on the Company’s operations or cash flows.
Research and Development Costs
The Company follows FASB Accounting Standards Codification (“ASC”) Subtopic 730-10, “Research and Development”. Research and development costs are charged to the statement of operations as incurred. During the years ended December 31, 20202023 and 2019,2022, the Company incurred $5,917,944$5,003,695 and $6,574,999$6,554,194 of expenses related to research and development costs, respectively.
Net Earnings (Loss) Per Common Share
The Company computes earnings per share under ASC Subtopic 260-10, “Earnings Per Share”. Basic earnings (loss) per share is computed by dividing the net income (loss) attributable to the common stockholders (the numerator) by the weighted average number of shares of common stock outstanding (the denominator) during the reporting periods. Diluted loss per share is computed by increasing the denominator by the weighted average number of additional shares that could have been outstanding from securities convertible into common stock (using the “treasury stock” method), unless their effect on net loss per share is anti-dilutive. There were 10,047,0101,514,715 and 24
December 31, 2020 | December 31, 2019 | |
| Options to purchase common stock | 3,569,707 | 3,449,319 |
| Warrants to purchase common stock | 6,251,103 | 13,496,924 |
| Shares issuable upon conversion of notes | - | 362,568 |
| Shares issuable upon conversion of Series A Convertible Preferred Stock | 226,200 | 7,285,651 |
| Shares issuable upon conversion of Series B Convertible Preferred Stock | - | 355,263 |
| Potential equivalent shares excluded | 10,047,010 | 24,949,725 |
|
| December 31, 2023 |
|
| December 31, 2022 |
| ||
Options to purchase common stock |
|
| 624,240 |
|
|
| 391,902 |
|
Warrants to purchase common stock |
|
| 882,349 |
|
|
| 10,330 |
|
Shares issuable upon conversion of Series A Convertible Preferred Stock |
|
| 8,126 |
|
|
| 8,126 |
|
Potential equivalent shares excluded |
|
| 1,514,715 |
|
|
| 410,358 |
|
Fair Value Measurements
Disclosures about fair value of financial instruments require disclosure of the fair value information, whether or not recognized in the balance sheet, where it is practicable to estimate that value.
In accordance with ASC Topic 820, “Fair Value Measurements and Disclosures,” the Company measures certain financial instruments at fair value on a recurring basis. ASC Topic 820 defines fair value, established a framework for measuring fair value in accordance with accounting principles generally accepted in the United States, and expands disclosures about fair value measurements.
| F-7 |
| Table of Contents |
Fair value is defined as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. ASC Topic 820 established a three-tier fair value hierarchy, which prioritizes the inputs used in measuring fair value. The hierarchy gives the highest priority to unadjusted quoted prices in active markets for identical assets or liabilities (Level 1 measurements) and the lowest priority to unobservable inputs (Level 3 measurements). These tiers include:
· | Level 1, defined as observable inputs such as quoted prices for identical instruments in active markets; |
· | Level 2, defined as inputs other than quoted prices in active markets that are either directly or indirectly observable such as quoted prices for similar instruments in active markets or quoted prices for identical or similar instruments in markets that are not active; and |
· | Level 3, defined as unobservable inputs in which little or no market data exists, therefore requiring an entity to develop its own assumptions, such as valuations derived from valuation techniques in which one or more significant inputs or significant value drivers are unobservable. |
Financial assets are considered Level 3 when their fair values are determined using pricing models, discounted cash flow methodologies or similar techniques and at least one significant model assumption or input is unobservable.
The carrying amounts of the Company’s financial assets and liabilities, including cash, accounts receivable, prepaid expenses, accounts payable, accrued expenses, and other current liabilities, approximate their fair values because of the short maturity of these instruments. The fair value of notes payable and convertible notes approximates their fair values since the current interest rates and terms on these obligations are the same as prevailing market rates.
Share-based Compensation
The Company’s 2016 Omnibus Incentive Plan (the “Omnibus Plan”) permits the grant of stock options and other share-based awards to its employees, consultants and non-employee members of the board of directors. Each January 1 the pool of shares available for issuance under the Omnibus Plan automatically increases by an amount equal to the lesser of (i) the number of shares necessary such that the aggregate number of shares available under the Omnibus Plan equals 25% of the number of fully-diluted outstanding shares on the increase date (assuming the conversion of all outstanding shares of preferred stock and other outstanding convertible securities and exercise of all outstanding options and warrants to purchase shares) and (ii) if the board of directors takes action to set a lower amount, the amount determined by the board. OnEffective January 1, 2021,2024, the pool of shares available for issuanceissuable under the Omnibus Plan automatically increased by 1,599,5701,717,783 shares from 5,861,6581,322,169 shares to 7,461,228.
The Company records share-based compensation in accordance with the provisions of the Share-based Compensation Topic of the FASB Codification. The guidance requires the use of option-pricing models that require the input of highly subjective assumptions, including the option’s expected life and the price volatility of the underlying stock. The fair value of each option grant is estimated on the date of grant using the Black-Scholes option valuation model, and the resulting charge is expensed using the straight-line attribution method over the vesting period.
Stock compensation expense recognized during the period is based on the value of share-based awards that were expected to vest during the period adjusted for estimated forfeitures. The estimated fair value of grants of stock options and warrants to non-employees of the Company is charged to expense, if applicable, in the financial statements. These options vest in the same manner as the employee options granted under the stock incentive plan as described above.
Going Concern
The Company’s financial statements are prepared using accounting principles generally accepted in the United States (“U.S. GAAP”) applicable to a going concern, which contemplates the realization of assets and liquidation of liabilities in the normal course of business. The Company has limited commercial experience and had a cumulative net loss from inception to December 31, 20202023 of $57,338,489.$91,930,152. The Company had working capital of $7,227,059$2,129,717 as of December 31, 2020.2023. The Company has not established an ongoing source of revenue sufficient to cover its operating costs and to allow it to continue as a going concern. The accompanying financial statements for the period ended December 31, 2020 have been prepared assuming the Company will continue as a going concern. The Company’s cash resources will likely be insufficient to meet its anticipated needs during the next twelve months. The Companyconcern and will require additional financing to fund its future planned operations, including research and development and commercialization of its products.
| F-8 |
| Table of Contents |
Recent Accounting Pronouncements
The Company considered
recent accounting pronouncements issued by the FASB, including its Emerging Issues Task Force, the American Institute of Certified Public Accountants, and the SEC, did not or in management’s opinion will not have a material impact on the Company’s present or future consolidated financial statements.Note 3 –- Inventory
As of December 31, 20202023 and December 31, 2019,2022, inventory consisted of raw materials, and subassemblies to be used in the assembly of a TAEUS system.systems, and finished goods. As of December 31, 2020,2023, the Company had no orders pending for the sale of a TAEUS system.
As of December 31, 2020 and December 31, 2019,2023, the Company had
As of December 31, 20202023 and 2022, the Company had inventory valued at $2,622,865 and $2,644,717, respectively.
Note 4 - Fixed Assets
As of December 31, 2019,2023 and 2022, fixed assets consisted of the following:
December 31, 2020 | December 31, 2019 | |
| Property, leasehold and capitalized software | $718,902 | $679,179 |
| TAEUS development and testing | 79,207 | 43,596 |
| Accumulated depreciation | (585,867) | (486,524) |
| Fixed assets, net | $212,242 | $236,251 |
|
| December 31, 2023 |
|
| December 31, 2022 |
| ||
Property, leasehold and capitalized software |
| $ | 587,030 |
|
| $ | 738,720 |
|
TAEUS development and testing |
|
| 125,151 |
|
|
| 140,617 |
|
Accumulated depreciation |
|
| (600,399 | ) |
|
| (643,682 | ) |
Fixed assets, net |
| $ | 111,782 |
|
| $ | 235,655 |
|
Depreciation expense for the yearsyear ended December 31, 20202023 and 20192022 was $99,342$123,726 and $80,577, respectively.
Note 5 –- Accounts Payable and Accrued Liabilities
As of December 31, 20202023 and December 31, 2019,2022, current liabilities consisted of the following:
December 31, 2020 | December 31, 2019 | |
| Accounts payable | $402,910 | $1,278,431 |
| Accrued payroll | 48,260 | 94,862 |
| Accrued bonuses | 369,393 | 295,794 |
| Accrued employee benefits | 5,750 | 5,750 |
| Accrued interest | - | 9,738 |
| Insurance premium financing | 83,870 | 23,950 |
| Total | $910,183 | $1,708,525 |
|
| December 31, 2023 |
|
| December 31, 2022 |
| ||
Accounts payable |
| $ | 360,401 |
|
| $ | 613,961 |
|
Accrued payroll |
|
| 150,293 |
|
|
| 60,638 |
|
Accrued bonuses |
|
| 35,518 |
|
|
| 683,738 |
|
Accrued employee benefits |
|
| 5,750 |
|
|
| 5,750 |
|
Insurance premium financing |
|
| 148,792 |
|
|
| 158,925 |
|
Total |
| $ | 700,754 |
|
| $ | 1,523,012 |
|
Note 6 –- Bank Loans
Toronto-Dominion Bank Loan
On April 27, 2020, the Company entered into a commitment loan with TD Bank under the Canadian Emergency Business Account, in the principal aggregate amount of CAD 40,000, which is due and payable upon the expiration of the initial term on December 31, 2022.2022, which was later extended to December 31, 2023. This note bears interest on the unpaid balance at the rate of zero percent (0%) per annum during the initial term. Under this note no interest payments are due until January 1, 2023.2024. Under the conditions of the loan, twenty-five percent (25%) of the loan will be forgiven if seventy-five percent (75%) is repaid prior to the initial term date.
| F-9 |
| Table of Contents |
Note 7 –- Capital Stock
Reverse Stock Split
On June 17, 2020,December 7, 2022, the Company filed a Certificate of Amendment to its Fourth Amended and Restated Certificate of Incorporation with the Secretary of State of the State of Delaware effecting ana certificate of amendment (the “Certificate of Amendment”) to increase the numberits certificate of authorized sharesincorporation, which Certificate of Amendment effectuated as of December 19, 2022 at 12:01 a.m. Eastern Time (the “Effective Time”) a reverse split of the Company’s common stock from 50,000,000by a ratio of one-for-20 (the “Reverse Split”). All per share amounts and number of shares in the consolidated financial statements and related notes have been retroactively restated to 80,000,000 shares.reflect the Reverse Split. No fractional shares were, or shall be, issued in connection with the Reverse Split. A stockholder who would otherwise be entitled to receive a fractional share of common stock is entitled to receive the fractional share rounded up to the next whole share. The CertificateReverse Split did not change the number of Amendment was approved byshares of common or preferred stock that the Company is authorized to issue, or the par value of the Company’s stockholders atcommon or preferred stock.
The Reverse Split resulted in a proportionate adjustment to the per share conversion or exercise price and the number of shares of common stock issuable upon the conversion or exercise of outstanding preferred stock, stock options and warrants, as well as the number of shares of common stock eligible for issuance under the Company’s annual meeting of stockholders on June 16, 2020.
Capital Stock
At December 31, 2020,2023, the authorized capital of the Company consisted of 90,000,000 shares of capital stock, comprised of 80,000,000 shares of common stock with a par value of $0.0001 per share, and 10,000,000 shares of preferred stock with a par value of $0.0001 per share. The Company has designated 10,000 shares of its preferred stock as Series A Convertible Preferred Stock (“Series A Preferred Stock”) and, 1,000 shares of its preferred stock as Series B Convertible Preferred Stock (“Series B Preferred Stock”), 100,000 shares of its preferred stock as Series C Preferred Stock, and the remainder of 9,989,000the 9,889,000 preferred shares remain authorized but undesignated.
As of December 31, 2020,2023, there were 34,049,70410,390,150 shares of common stock, 196.794(which exclude 202,020 unvested shares of restricted stock described in Note 8 below) 141.397 shares of Series A Preferred Stock, and no shares of Series B Preferred Stock or Series C Preferred Stock issued and outstanding, and a stock payable balance of $10,794.
During the year ended December 31, 2020,2023, the Company issued a total of 25,628,3037,221,047 shares of its common stock, as follows:
- 4,312,500 shares of its Series A Preferred Stock;
- 1,325,047 shares of its at-the-market equity offering programs;
- 1,583,500 upon warrant exercises for an aggregate net proceeds of $1,014,859.
During the year ended December 31, 2022, the Company issued a total of 1,041,377 shares uponof its common stock in return for aggregate net proceeds of $8,399,512 under the conversion of $493,847 principal and accrued interest on convertible promissory notes issued in July 2019;
At-the-Market Equity Offering of Common Stock
On December 15, 2020,June 21, 2021, the Company entered into an underwriting agreementthe At-The-Market Issuance Sales Agreement with Ascendiant (the “Underwriting Agreement”) with ThinkEquity, a division of Fordham Financial Management, Inc. (the “Underwriter”), relating to an underwritten public offering for the issuance and sale of 7,143,000 shares of the Company’s common stock, par value $0.0001 per share (the “Common Stock”). In addition, under the terms of the Underwriting Agreement, the Company granted the Underwriter a 45-day option to purchase up to an additional 714,286 shares of its Common Stock to cover over-allotments, if any. The Underwriter exercised in full its option to purchase the additional 714,286 shares on December 16, 2020.
| F-10 |
| Table of Contents |
Note 8 –- Common Stock Options and Restricted Stock Units (RSU’s)
Common Stock Options
Stock options are awarded to the Company’s employees, consultants and non-employee members of the board of directors under the 2016 Omnibus Incentive Plan (the “Omnibus Plan”) and are generally granted with an exercise price equal to the market price of the Company’s common stock at the date of grant. The aggregate fair value of these stock options granted by the Company during the year ended December 31, 20202023 was determined to be $378,422$1,017,534 using the Black-Scholes-Merton option-pricing model based on the following assumptions: (i) volatility rate of 94%105% to 119%107%, (ii) discount rate of 0%, (iii) zero expected dividend yield, (iv) risk free rate of 3.68% to 3.86%, (v) price of $1.30 to $4.16, and (iv)(vi) expected life of 10 years. A summary of option activity under the Company’s Omnibus Plan as of December 31, 2020,2023, and changes during the year then ended, is presented below:
Number of Options | Weighted Average Exercise Price | Weighted Average Remaining Contractual Term (Years) | |
| Balance outstanding at December 31, 2019 | 3,449,319 | $2.32 | 8.26 |
| Granted | 327,918 | 1.26 | 9.26 |
| Exercised | - | - | - |
| Forfeited | - | - | - |
| Cancelled or expired | (207,530) | - | - |
| Balance outstanding at December 31, 2020 | 3,569,707 | $2.13 | 7.50 |
| Exercisable at December 31, 2020 | 1,458,069 | $2.83 | 6.60 |
|
| Number of Options |
|
| Weighted Average Exercise Price |
|
| Weighted Average Remaining Contractual Term (Years) |
| |||
Balance outstanding at December 31, 2022 |
|
| 391,902 |
|
| $ | 31.47 |
|
|
| 7.41 |
|
Granted |
|
| 274,128 |
|
|
| 4.02 |
|
|
| 8.84 |
|
Exercised |
|
| - |
|
|
| - |
|
|
| - |
|
Forfeited |
|
| - |
|
|
| - |
|
|
| - |
|
Cancelled or expired |
|
| (41,790 | ) |
|
| 33.89 |
|
|
| - |
|
Balance outstanding at December 31, 2023 |
|
| 624,240 |
|
| $ | 19.25 |
|
|
| 7.26 |
|
Exercisable at December 31, 2023 |
|
| 269,255 |
|
| $ | 31.41 |
|
|
| 5.34 |
|
Restricted Common Stock
On November 30, 2023, the Company issued 202,020 shares of restricted common stock (the “Restricted Stock”) of the Company to PatentVest, Inc. (“PatentVest”) pursuant to a Restricted Stock Units
Note 9 - Common Stock Warrants
Warrant Conversions
On May 2, 2023, the Company hadconducted the Offering in which the Company issued 2,156,250 warrants to purchase shares of common stock for an exercise price per share equal to $1.40. The warrants expire May 2, 2028. In December 2023, the Board approved the reduction of the exercise price per share from $1.40 to $0.70. The Company also issued to the placement agent and vested the following RSU’s:
Restricted Stock Units Outstanding | Weighted Average Grant Date Fair Value | |
| Balance Outstanding at December 31, 2019 | - | $- |
| Granted | 674,019 | 0.70 |
| Vested / Released | (674,019) | - |
| Forfeited | - | - |
| Cancelled or expired | - | - |
| Balance outstanding at December 31, 2020 | - | $- |
The following table summarizes all stock warrant activity of the Company for the year ended December 31, 2020:
Number of Warrants | Weighted Average Exercise Price | Weighted Average Contractual Term (Years) | |
| Balance outstanding at December 31, 2019 | 13,496,924 | $2.02 | 4.07 |
| Granted | - | - | - |
| Exercised | (7,098,108) | 0.70 | 3.39 |
| Forfeited | - | - | - |
| Expired | (147,713) | - | - |
| Balance outstanding at December 31, 2020 | 6,251,103 | $2.79 | 2.79 |
| Exercisable at December 31, 2020 | 6,251,103 | $2.79 | 2.79 |
|
| Number of Warrants |
|
| Weighted Average Exercise Price |
|
| Weighted Average Contractual Term (Years) |
| |||
Balance outstanding at December 31, 2022 |
|
| 10,330 |
|
| $ | 25.01 |
|
|
| 1.78 |
|
Granted |
|
| 2,458,125 |
|
|
| 1.41 |
|
|
| 4.16 |
|
Exercised |
|
| (1,583,500 | ) |
|
| 0.70 |
|
|
| 4.34 |
|
Forfeited |
|
| - |
|
|
| - |
|
|
| - |
|
Expired |
|
| (2,606 | ) |
|
| 46.95 |
|
|
| - |
|
Balance outstanding at December 31, 2023 |
|
| 882,349 |
|
| $ | 1.58 |
|
|
| 3.79 |
|
Exercisable at December 31, 2023 |
|
| 882,349 |
|
| $ | 1.58 |
|
|
| 3.79 |
|
Note 10 –- Related Party Transactions
On May 2, 2023, the Company conducted the Offering in which the Company sold 83,333 shares of its common stock and 41,667 warrants to the Company’s director, Anthony DiGiandomenico, for cash at the public offering price, which was less than 5% of beneficial ownership in the Company.
On October 17, 2023, the Company entered into a consulting agreement with one of its directors, Alex Tokman, pursuant to which Mr. Tokman provides commercialization services. Under the terms of the agreement, Mr. Tokman is compensated at a rate of $150 per hour for his services.
| F-11 |
| Table of Contents |
On November 30, 2023, the Company entered into a Restricted Stock Agreement and Consulting Services Agreement, each with PatentVest, in exchange for certain services related to the Company’s patent portfolio. PatentVest is a wholly-owned subsidiary of MDB Capital Holdings, LLC (“MDB”). Anthony DiGiandomenico, a member of the Company’s board of directors, is the Chief of Transactions and a director of MDB. Lou Basenese, a member of our board of directors, is President and Chief Market Strategist at Public Ventures LLC, a wholly-owned subsidiary of MDB.
Note 11 - Commitments &and Contingencies
Office Lease
Effective January 1, 2015, the Company entered into an office lease agreement with Green Court, LLC, a Michigan limited liability company, for approximately 3,657 rentable square feet of space, for the initial monthly rent of $5,986, which commenced on January 1, 2015 for an initial term of 60 months. On October 10, 2017 this lease was amended increasing the rentable square feet of space to 3,950 and the monthly rent to $7,798.
On July 16, 2019, the Company exercised its option to extend the lease for an additional 5 years past the initial term originally expiring on December 31, 2019.
The Company records the lease asset and lease liability at the present value of lease payments over the lease term. The lease typically does not provide an implicit rate; therefore, the Company uses its estimated incremental borrowing rate at the time of lease commencement to discount the present value of lease payments. The Company’s discount rate for operating leases at December 31, 20202023 was 10%. Lease expense is recognized on a straight-line basis over the lease term to the extent that collection is considered probable. As a result, the Company has been recognizing rents as they become payable based on the adoption of ASC Topic 842. The weighted-average remaining lease term is 4.672.0 years.
As of December 31, 2020,2023, the maturities of operating lease liabilities are as follows:
Operating Lease | |
| 2021 | 101,752 |
| 2022 | 104,793 |
| 2023 | 107,954 |
| 2024 and beyond | 111,192 |
| Total | $425,691 |
| Less: amount representing interest | (77,303) |
| Present value of future minimum lease payments | 348,388 |
| Less: current obligations under leases | 76,480 |
| Long-term lease obligations | $271,908 |
|
| Operating Lease |
| |
2024 |
|
| 202,624 |
|
2025 and beyond |
|
| 202,624 |
|
Total |
| $ | 405,247 |
|
Less: amount representing interest |
|
| (39,328 | ) |
Present value of future minimum lease payments |
|
| 365,919 |
|
Less: current obligations under leases |
|
| (173,857 | ) |
Long-term lease obligations |
| $ | 192,062 |
|
For the yearsyear ended December 31, 20202023 and 2019,2022, the Company incurred rent expenses of $120,275$218,815 and $105,514,$213,912, respectively.
Employment and Consulting Agreements
Francois Michelon
If Mr. Michelon’s employment is terminated by the Company without cause or Mr. Michelon terminates his employment for good reason, Mr. Michelon will be entitled to receive 12 months’ continuation of his current base salary and a lump sum payment equal to 12 months of continued healthcare coverage (or 24 months’ continuation of his current base salary and a lump sum payment equal to 24 months of continued healthcare coverage if such termination occurs within one year following a change in control).
Under his employment agreement, Mr. Michelon is eligible to receive benefits that are substantially similar to those of the Company’s other senior executive officers.
| F-12 |
| Table of Contents |
Michael Thornton - The Company granted Mr. Michelon 123,064 restricted stock units in lieu of $86,145 of his cash salary. The restricted stock units vested on a daily basis through December 31, 2020.
If Mr. Thornton’s employment is terminated by the Company without cause or Mr. Thornton terminates his employment for good reason, Mr. Thornton will be entitled to receive 12 months’ continuation of his current base salary and a lump sum payment equal to 12 months of continued healthcare coverage (or 24 months’ continuation of his current base salary and a lump sum payment equal to 24 months of continued healthcare coverage if such termination occurs within one year following a change in control).
Under his employment agreement, Mr. Thornton is eligible to receive benefits that are substantially similar to those of the Company’s other senior executive officers.
Litigation
From time to time the Company may become a party to litigation in the normal course of business. As of December 31, 2020,2023, there were no legal matters that management believes would have a material effect on the Company’s financial position or results of operations.
Note 11 –12 - Income Taxes
The components of earnings before income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of the Company’s deferred tax assets as of December 31, 2020 and 2019 are summarized below.
2020 | 2019 | |
| Net operating loss carryforward | $(11,827,295) | $(8,106,070) |
| Stock based compensation | 150,595 | -- |
| Fair value of options | 395,996 | 289,528 |
| Total deferred tax assets | (11,280,704) | (7,816,542) |
| Valuation allowance | $11,280,704 | $7,816,542 |
| Net deferred tax asset | $- | $- |
|
| For the Years Ended December 31, |
| |||||
Income (loss) before income taxes |
| 2023 |
|
| 2022 |
| ||
|
|
|
|
|
|
| ||
Domestic |
|
| (8,403,400 | ) |
|
| (11,548,400 | ) |
Foreign |
|
| (1,593,300 | ) |
|
| (1,630,700 | ) |
Total income (loss) before income taxes |
| $ | (9,996,700 | ) |
| $ | (13,179,100 | ) |
Income tax provision (benefit) consists of the losses incurred during such periods. Reconciled below is the difference between the income tax rate computed by applying the U.S. federal statutory rate and the effective tax ratefollowing for the years ended December 31, 20202023 and 2019.
2020 | 2019 | |
| U.S. federal statutory income tax | -21.00% | -21.00% |
| State tax, net of federal tax benefit | -5.80% | -5.80% |
| Stock based compensation | 0.00% | 0.00% |
| Change in valuation allowance | 26.80% | 26.80% |
| Effective tax rate | 0.00% | 0.00% |
Income tax provision (benefit): | For the Years Ended December 31, | |||||||
Current | 2023 | 2022 | ||||||
Federal | - | - | ||||||
State | - | - | ||||||
Foreign | - | - | ||||||
Total Current | - | - | ||||||
Deferred | ||||||||
Federal | - | - | ||||||
State | - | - | ||||||
Foreign | - | - | ||||||
Total Deferred | - | - | ||||||
Total income tax provision (benefit) | $ | - | $ | - | ||||
A reconciliation of the Company has availableincome tax provision (benefit) by applying the statutory United States federal income tax rate to income (loss) before income taxes is as follows:
Rate Reconciliation |
| For the Years Ended December 31, |
|
|
|
| ||||||||||
|
| 2023 |
|
|
|
| 2022 |
|
|
| ||||||
Expected tax at statutory rates |
| $ | (2,099,400 | ) |
|
| 21 | % |
| $ | (2,767,700 | ) |
|
| 21 | % |
Permanent Differences |
| $ | (83,000 | ) |
|
| 1 | % |
|
| 3,000 |
|
|
| 0 | % |
State Income Tax, Net of Federal benefit |
| $ | (448,100 | ) |
|
| 4 | % |
|
| (589,200 | ) |
|
| 4 | % |
State Rate Change-Federal Impact |
| $ | - |
|
|
| 0 | % |
|
| 53,200 |
|
|
| 0 | % |
State Rate Change Adjustment |
| $ | - |
|
|
| 0 | % |
|
| (253,200 | ) |
|
| 2 | % |
Foreign taxes at rate different than US Taxes |
| $ | (33,800 | ) |
|
| 0 | % |
|
| (35,800 | ) |
|
| 0 | % |
Current Year Change in Valuation Allowance |
| $ | 2,630,700 |
|
|
| -26 | % |
|
| 5,134,200 |
|
|
| -39 | % |
Prior Year True-Ups |
| $ | 33,600 |
|
|
| 0 | % |
|
| (1,544,500 | ) |
|
| 12 | % |
Income tax provision (benefit) |
| $ | - |
|
|
| 0 | % |
| $ | - |
|
|
| 0 | % |
| F-13 |
| Table of Contents |
Deferred tax assets and liabilities are provided for significant income and expense items recognized in different years for tax and financial reporting purposes. Temporary differences, which give rise to a net deferred tax asset is as follows:
Deferred Tax Assets/(Liabilities) Detail |
| For the Years Ended December 31, |
| |||||
|
| 2023 |
|
| 2022 |
| ||
Deferred Tax Assets (Liabilities): |
|
|
|
|
|
| ||
Stock Based Compensation |
| $ | 1,406,400 |
|
|
| 1,145,700 |
|
Accrued Bonus |
| $ | 63,300 |
|
|
| 13,100 |
|
Depreciation |
| $ | (7,800 | ) |
|
| (12,500 | ) |
ROU (Asset) |
| $ | (92,600 | ) |
|
| (132,300 | ) |
ROU Liability |
| $ | 95,700 |
|
|
| 135,500 |
|
Capitalized R&D |
| $ | 1,960,100 |
|
|
| 1,314,202 |
|
R&D Credit |
| $ | 29,800 |
|
|
| 29,800 |
|
Net Operating Losses (US) |
| $ | 16,665,000 |
|
|
| 15,364,200 |
|
Net Operating Losses (Foreign) |
| $ | 1,042,600 |
|
|
| 674,100 |
|
Net deferred tax assets (liabilities) |
|
| 21,162,500 |
|
|
| 18,531,802 |
|
Valuation allowance |
|
| (21,162,500 | ) |
|
| (18,531,802 | ) |
Net deferred tax assets (liabilities) |
| $ | - |
|
| $ | - |
|
The domestic U.S. net operating loss carryforwards for federalcarryforward increased from $57,008,606 at December 31, 2022 to $62,033,535 at December 31, 2023. After consideration of all the evidence, both positive and statenegative, management has recorded a full valuation allowance at December 31, 2023 and 2022, due to the uncertainty of realizing the deferred income tax purposesassets. Out of approximately $43.4 million, which $27.4 million has an indefinite life and $16 millionthe $62,033,535 net operating losses carry forward, $16,012,698 will begin to expire in 2027 through 2037.
The Internal Revenue Code includes a provision, referred to as Global Intangible Low-Taxed Income (“GILTI”), which provides for a 10.5% tax on certain income of controlled foreign corporations. We have elected to account for GILTI as a period cost if and when occurred, rather than recognizing deferred taxes for basis differences expected to reverse.
The Company is subject to taxation in the U.S. and various states and foreign jurisdictions. U.S. federal income tax returns for 2020 and 2019after remain open to examination. We and thereforeour subsidiaries are also subject to income tax in multiple states and foreign jurisdictions. Generally, foreign income tax returns after 2020 remain open to examination. No income tax returns are currently under examination. As of December 31, 2023 and 2022, the entity had no significant impact onCompany does not have any unrecognized tax benefits, and continues to monitor its current and prior tax positions for any changes. The Company recognizes penalties and interest related to unrecognized tax benefits as income tax expense. For the year-end 2020 and 2019 tax provision. Generally, all expenses relating to research & development that are incurred in Canada are the responsibility and owned by the United States parent company, since it is the owner of all of the Company’s intangibles.
Note 13 - Subsequent Events
Subsequent to the year ended December 31, 2020,2023, the Company issued an aggregatea total of 3,567,899118,904 shares of its common stock upon Private Warrantwarrant exercises for grossan aggregate net proceeds of $2,918,472.
Subsequent to the year ended December 31, 2023, the Company received written notice from the Listing Qualifications Staffissued a total of The Nasdaq Stock Market LLC (“Nasdaq”) notifying the Company that for at least ten consecutive business days, from January 15, 2021 to February 1, 2021, the closing bid price for the Company’s316,963 shares of its common stock was $1.00 per share or greater. Accordingly, the written notice stated that the Company had regained compliance with the minimum bid price listing requirement set forth under Nasdaq Listing Rule 5550(a)(2).
Subsequent to the 2021 Ascendiant ATM Agreement, Ascendiant may sell the shares in sales deemed to be “at-the-market” equity offerings as defined in Rule 415 under the Securities Act, including sales made directly on or through the Nasdaq Capital Market. If agreed to in a separate terms agreement, the Company may sell shares to Ascendiant as principal at a purchase price agreed upon by Ascendiant and the Company. Ascendiant may also sell shares in negotiated transactions with our prior approval.
| F-14 |
| Table of Contents |
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure.
None.
Evaluation of Disclosure Controls and Procedures
As of the end of the period covered by this report, management performed, with the participation of our principal executive and principal financial officers,officer, an evaluation of the effectiveness of our disclosure controls and procedures as defined in Rules 13a-15(e) and 15d-15(e) of the Exchange Act. Our disclosure controls and procedures are designed to ensure that information required to be disclosed in the reports we file or submit under the Exchange Act is recorded, processed, summarized, and reported within the time periods specified in the SEC’s forms, and that such information is accumulated and communicated to our management, including our principal executive officer and principal financial officer, to allow timely decisions regarding required disclosures. Based on the evaluation, our principal executive and principal financial officersofficer concluded that, as of December 31, 2020,2023, our disclosure controls and procedures were not effective due to a material weakness in internal control over financial reporting, as described below.
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining effective internal control over financial reporting as defined in Rules 13a-15(f) and 15d-15(f) of the Exchange Act. Internal control over financial reporting is a process designed to provide reasonable assurance to the Company’s management and board of directors regarding the reliability of our financial reporting for external purposes in accordance with accounting principles generally accepted in the United States of America.
Because of its inherent limitations, internal control over financial reporting is not intended to provide absolute assurance that a misstatement of our consolidated financial statements would be prevented or detected. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate. Therefore, even those systems determined to be effective can only provide reasonable assurance with respect to financial statement preparation and presentation.
Management conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework in Internal Control –- Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission.
A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of our annual or interim financial statements will not be prevented or detected on a timely basis. Management identified the following material weakness as of December 31, 2020:2023: insufficient personnel resources within the accounting function to segregate the duties over financial transaction processing and reporting. Because of this material weakness, management concluded that the Company’s internal control over financial reporting was not effective as of December 31, 2020.
Continuing Remediation Efforts
To remediate its internal control weakness, management intends to implement the following measures, during 2021:
· | Add additional accounting personnel or outside consultants, such as a new controller, to properly segregate duties and to effect timely, accurate preparation of the financial statements; and | |
· | Complete the development of and maintain adequate written accounting policies and procedures. |
As we are not an “accelerated filer” under SEC rules, we are not required to properly segregate duties and to effect timely, accurate preparationprovide an auditor’s attestation of the financial statements; and
Changes in Internal Control of Financial Reporting
During the quarter ended December 31, 2020,2023, except as described above under “Continuing Remediation Efforts,” there were no changes that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Not applicable.
Item 9C. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections
Not applicable.
| 43 |
| Table of Contents |
PART III
Item 10. Directors, Executive Officers and Corporate Governance.
The following table sets forth the names and ages of all of our executive officers and directors. Our officers are appointed by, and serve at the pleasure of, the board of directors.
Name | Age | Position | ||
Francois Michelon | 58 | Chief Executive Officer and Chairman | ||
Michael Thornton | 55 | Chief Technology Officer | ||
Irina Pestrikova | 38 | Senior Director, Finance | ||
Louis J. Basenese | 46 | Director | ||
Anthony DiGiandomenico | 57 | Director | ||
Michael Harsh | 69 | Director | ||
Alexander Tokman | 62 | Director |
Biographical information required by this Item is incorporated by referencewith respect to our Proxy Statementexecutive officers and directors is provided below. There are no family relationships between any of our executive officers or directors.
Francois Michelon joined ENDRA as Chief Executive Officer and Chairman of our board of directors in 2015. He has over 20 years of healthcare technology experience in general management, operations, strategy and marketing across the diagnostic imaging, surgical instrument and dental sectors.
From 2012 to 2014, Mr. Michelon served as Vice President of Global Marketing for the 3i division of Biomet, Inc. (now Zimmer Biomet Holdings, Inc.), a provider of oral reconstruction technologies, where he was responsible for the upstream and downstream development of the division’s global portfolio. From 2004 to 2011, Mr. Michelon served as Group Director of Global Services and Visualization for Smith & Nephew plc’s Advanced Surgical Devices division, where he led in the B2B service and capital equipment sectors and had responsibility over the financial performance of these as well. From 1997 to 2004, Mr. Michelon worked at GE Healthcare in a variety of global upstream and downstream marketing roles.
Mr. Michelon received an MBA from Carnegie-Mellon University and a BA in Economics from the University of Chicago. He has also earned his Six Sigma Black Belt certification.
Mr. Michelon’s extensive industry and executive experience and his intimate understanding of our business as our Chief Executive Officer position him well to serve as a member of our Board of Directors.
Michael Thornton joined ENDRA as Chief Operating Officer in 2007 and became our Chief Technology Officer in 2008 and has served in that role since. Prior to that, Mr. Thornton was a founder and President of Enhanced Vision Systems Corp., or EVS, a developer and supplier of medical imaging equipment to the pharmaceutical, biotech, and academic sectors.
In 2002, EVS was acquired by General Electric Company and was integrated into the Functional and Molecular Imaging business unit of GE Medical Systems (now GE Healthcare, a subsidiary of General Electric Company). Following the acquisition of EVS by GE Medical Systems, Mr. Thornton held a number of positions at GE Healthcare, including Sales Manager, Global Product Manager, and Site Leader. He was a member of the leadership team that expanded the pre-clinical imaging business to include: computed tomography, optical, and positron emission tomography imaging technologies, with global market reach. He is also a founder of Volumetrics Medical Corp., a developer and manufacturer of quality assurance devices for diagnostic imaging.
Prior to founding EVS, Mr. Thornton developed medical imaging related technologies at the Robarts Research Institute (London, Ontario, Canada) for which he obtained an MSc in Electrical Engineering from the University of Western Ontario. Mr. Thornton also holds a BASc in Electrical Engineering from the University of Toronto and is a member of the American Association of Physicists in Medicine.
Irina Pestrikova joined ENDRA as Senior Director, Finance in June 2021. From 2014 to 2021, Ms. Pestrikova was the Director of Operations of Wells Compliance Group. Wells Compliance Group is a technology-based services firm supporting the financial reporting needs of publicly traded companies and privately held firms whose investor or shareholder base requires timely GAAP-compliant financial reporting. In her role as Director of Operations, Ms. Pestrikova provided accounting and bookkeeping services to a number of public companies, including ENDRA. Ms. Pestrikova has also been the Director of Finance & Operations of Atlas Bookkeeping, Inc., a provider of financial reporting, modeling and analysis, since 2020. She holds an MBA in Finance from Pepperdine University.
| 44 |
| Table of Contents |
Louis J. Basenese, joined our Board of Directors in April 2020. Mr. Basenese is the President, Chief Market Strategist at Public Ventures, LLC, a registered broker-dealer, Member FINRA/SIPC. Previously, he was Founder and Chief Analyst of Disruptive Tech Research, LLC, an independent equity research and advisory firm focused exclusively on Schedule 14A relatingdisruptive technology companies that has served the investment management community from June 2014 through September 2022. Since 2005, Mr. Basenese has also managed The Basenese Group, LLC, a consulting business focused on communications and business development for private and public small and microcap businesses.
Mr. Basenese holds an M.B.A. in Finance from the Crummer Graduate School of Business at Rollins College and a Bachelor of Arts from the University of Florida. He is also a former Series 7 and Series 66 license holder.
Mr. Basenese’s experience with investor relations and business development of technology-focused companies, as well as financing and strategic planning, provides him with the qualifications and skills necessary to serve as a member of our Board of Directors.
Anthony DiGiandomenico joined our Board of Directors in 2013. A co-founder of MDB Capital Group LLC, Mr. DiGiandomenico focuses on corporate finance and capital formation for growth-oriented companies. He has participated in all areas of corporate finance including private capital, public offerings, PIPEs, business consulting and strategic planning, and mergers and acquisitions.
Mr. DiGiandomenico has also worked on a wide range of transactions for growth-oriented companies in biotechnology, nutritional supplements, manufacturing and entertainment industries. Prior to forming MDB Capital Group LLC in 1997, Mr. DiGiandomenico served as President and CEO of the Digian Company, a real estate development company. Mr. DiGiandomenico has also served on the board of directors of Cue Biopharma, Inc., an immunotherapy company, and on the board of directors of Provention Bio, Inc., a clinical-stage biopharmaceutical company.
Mr. DiGiandomenico holds an MBA from the Haas School of Business at the University of California, Berkeley and a BS in Finance from the University of Colorado.
Mr. DiGiandomenico’s financial expertise, general business acumen and significant executive leadership experience position him well to make valuable contributions to our 2021Board of Directors.
Michael Harsh joined our Board of Directors in 2015. He is a Portfolio Executive for the National Institutes of Health (NIH) Rapid Acceleration of Diagnostics (RADx) COVID-19 Response Program and a co-founder and Chief Product Officer of Terapede Systems, a digital Xray startup that focuses on developing an ultra-high resolution medical flat panel X-ray detector. He co-founded Terapede in 2015. Prior to Terapede, Mr. Harsh had a 36-year career with General Electric (“GE”). He held numerous positions within GE and served as Vice President and Chief Technology Officer of GE Healthcare, a multi-billion dollar division of GE, where he led its global science and technology organization and research and development teams in diagnostics, healthcare IT and life sciences. In 2004, Mr. Harsh was named Global Technology Leader - Imaging Technologies at the GE Global Research Center, where he led the research for imaging technologies across the company as well as the research associated with computer visualization and superconducting systems.
Additionally, Mr. Harsh is a member of the boards of directors of Magnetic Insights, Imagion Biosystems (ASX: IBX.AX), and EmOpti, Inc., as well as a member of the Radiological Society of North America (RSNA), Research & Education Foundation Board of Trustees. He had previously served as a director for Compute Health Acquisition Corp. until its merger with Allurion Technologies and as a director for FloDesign Sonics until its acquisition by MilliporeSigma, a division of the Merck Group. He is also a McKinsey Senior Advisor and a consultant in the medical device industry.
Mr. Harsh is a graduate of Marquette University, where he earned a bachelor’s degree in Electrical Engineering. He holds numerous U.S. patents in the field of medical imaging and instrumentation. In 2008, Mr. Harsh was elected to the American Institute for Medical and Biological Engineering College of Fellows for his significant contributions to the medical and biological engineering field.
Mr. Harsh’s extensive industry, executive and board experience position him well to serve on our Board of Directors.
Alexander Tokman joined our Board of Directors in 2008. He currently serves as a President of iUNU, a privately held AI/Computer Vision SaaS company and recently was a CEO-in-Residence at the Allen Institute for Artificial Intelligence (AI2), from 2019 to 2020. Mr. Tokman also serves as an independent board director for Izotropic Corporation (CSE: IZO), a company commercializing a dedicated breast CT imaging platform, and on the board of the American Academy of Thermography, a non-profit organization focused on bringing novel infrared imaging applications for disease diagnosis.
| 45 |
| Table of Contents |
Mr. Tokman served as President, Chief Executive Officer, and a director of Microvision, Inc., a publicly traded laser beam scanning display and imaging company, from January 2006 to December 2017.
Previously, Mr. Tokman completed a 10+ year tenure as an executive with GE Healthcare, where he led several global businesses, the latest being its Global Molecular Imaging and Radiopharmacy multi-technology business unit, of which he was General Manager from 2003 to 2005.
Between 1995 and 2003, Mr. Tokman served in various leadership roles at GE Healthcare, where he led the definition and successful commercialization of several product segments, including PET/CT.
Mr. Tokman is a certified Six Sigma and Design for Six Sigma (DFSS) Black Belt and Master Black Belt and as one of General Electric Company’s Six Sigma pioneers, he drove the quality culture change across GE Healthcare in the late 1990s. From 1989 to 1995, Mr. Tokman served as development programs lead and a head of Industry and Regional Development at Tracor Applied Sciences (BAE Systems). Mr. Tokman has both an MS and BS in Electrical Engineering from the University of Massachusetts, Dartmouth.
Mr. Tokman’s industry expertise and significant executive leadership and director experience position him well to make valuable contributions to our Board of Directors.
Board Independence
The Board of Directors has determined that each of Mr. Basenese, Mr. DiGiandomenico, Mr. Harsh and Mr. Tokman is an independent director within the meaning of the director independence standards of The Nasdaq Stock Market (“Nasdaq”). Furthermore, the Board has determined that all of the members of the Audit Committee, Compensation Committee and Corporate Governance and Nominating Committee are independent within the meaning of the director independence standards of Nasdaq and the rules of the SEC applicable to each such committee.
Committees
Audit Committee. Our Audit Committee consists of Mr. Basenese, Mr. DiGiandomenico, and Mr. Harsh. The Board of Directors has determined that each member of the Audit Committee is independent within the meaning of the Nasdaq director independence standards and applicable rules of the SEC for audit committee members. The Board of Directors has elected Mr. DiGiandomenico as Chairperson of the Audit Committee and has determined that he qualifies as an “audit committee financial expert” under the rules of the SEC. The Audit Committee is responsible for assisting the Board of Directors in fulfilling its oversight responsibilities with respect to financial reports and other financial information. The Audit Committee (1) reviews, monitors and reports to the Board of Directors on the adequacy of the Company’s financial reporting process and system of internal controls over financial reporting, (2) has the ultimate authority to select, evaluate and replace the independent auditor and is the ultimate authority to which the independent auditors are accountable, (3) in consultation with management, periodically reviews the adequacy of the Company’s disclosure controls and procedures and approves any significant changes thereto, (4) provides the audit committee report for inclusion in our proxy statement for our annual meeting of stockholders and (5) recommends, establishes and monitors procedures for the receipt, retention and treatment of complaints relating to be filed pursuantaccounting, internal accounting controls or auditing matters and the receipt of confidential, anonymous submissions by employees of concerns regarding questionable accounting or auditing matters. The Audit Committee met twice in 2023 as well as acted by written consent.
Compensation Committee. Our Compensation Committee presently consists of Mr. Basenese, Mr. DiGiandomenico, Mr. Harsh and Mr. Tokman, each of whom is a non-employee director as defined in Rule 16b-3 of the Exchange Act. The Board has also determined that each member of the Compensation Committee is also an independent director within the meaning of Nasdaq’s director independence standards. Mr. Tokman serves as Chairperson of the Compensation Committee. The Compensation Committee (1) discharges the responsibilities of the Board of Directors relating to Regulation 14Athe compensation of our directors and executive officers, (2) oversees the Company’s procedures for consideration and determination of executive and director compensation, and reviews and approves all executive compensation, and (3) administers and implements the Company’s incentive compensation plans and equity-based plans. The Compensation Committee [did not meet separately from the board of directors in 2023] but acted by unanimous written consent.
Corporate Governance and Nominating Committee. Our Corporate Governance and Nominating Committee consists of Mr. Harsh and Mr. Tokman. The Board of Directors has determined that each member of the Corporate Governance and Nominating Committee is an independent director within the meaning of the Nasdaq director independence standards and applicable rules of the SEC. Mr. Harsh serves as Chairperson of the Corporate Governance and Nominating Committee. The Corporate Governance and Nominating Committee (1) recommends to the Board of Directors persons to serve as members of the Board of Directors and as members of and chairpersons for the committees of the Board of Directors, (2) considers the recommendation of candidates to serve as directors submitted from the stockholders of the Company, (3) assists the Board of Directors in evaluating the performance of the Board of Directors and the Board committees, (4) advises the Board of Directors regarding the appropriate board leadership structure for the Company, (5) reviews and makes recommendations to the Board of Directors on corporate governance and (6) reviews the size and composition of the Board of Directors and recommends to the Board of Directors any changes it deems advisable. The Corporate Governance and Nominating Committee did not meet separately from the Board of Directors in 2023 but acted by written consent.
| 46 |
| Table of Contents |
Delinquent Section 16(a) Reports
Section 16(a) of the Exchange Act within 120 days afterrequires our directors, executive officers and persons who own more than ten percent of a registered class of our equity securities to file reports of ownership and changes in ownership with the endSEC. Such persons are required by SEC regulations to furnish us with copies of all such filings. Based solely on our review of the copies of the reports that we received and written representations that no other reports were required, we believe that our executive officers, directors and greater than 10% stockholders complied with all applicable filing requirements on a timely basis during 2023.
Code of Business Conduct and Ethics
We have in place a Code of Business Conduct and Ethics (the “Code of Ethics”) that applies to all of our directors, officers and employees. The Code of Ethics is designed to deter wrongdoing and to promote:
· | honest and ethical conduct, including the ethical handling of actual or apparent conflicts of interest between personal and professional relationships; | |
· | full, fair, accurate, timely and understandable disclosure in reports and documents that we file with, or submit to, the SEC and in other public communications that we make; | |
· | compliance with applicable governmental laws, rules and regulations; | |
· | the prompt internal reporting of violations of the Code of Ethics to an appropriate person identified in the Code of Ethics; and | |
· | accountability for adherence to the Code of Ethics. |
A current copy of the Code of Ethics is available at www.endrainc.com. A copy may also be obtained, free of charge, from us upon a request directed to ENDRA Life Sciences, Inc., 3600 Green Court, Suite 350, Ann Arbor, Michigan 48105, attention: Investor Relations. We intend to disclose any amendments to or waivers of a provision of the Code of Ethics required to be disclosed by applicable SEC rules by posting such information on our website available at www.endrainc.com and/or in our public filings with the SEC.
Nasdaq Rule 5608 Clawback Policy
The Company has adopted an incentive-based compensation recovery policy as required by the rules of the Nasdaq Stock Market, which is filed as Exhibit 97 to this report.
Item 11. Executive Compensation
Our compensation philosophy is to offer our executive officers compensation and benefits that are competitive and meet our goals of attracting, retaining and motivating highly skilled management, which is necessary to achieve our financial and strategic objectives and create long-term value for our stockholders. We believe the levels of compensation we provide should be competitive, reasonable and appropriate for our business needs and circumstances. Our board of directors uses benchmark compensation studies in determining compensation elements and levels. The principal elements of our executive compensation program have to date included base salary, annual bonus opportunity and long-term equity compensation in the form of stock options. We believe successful long-term Company performance is more critical to enhancing stockholder value than short-term results. For this reason and to conserve cash and better align the interests of management and our stockholders, we emphasize long-term performance-based equity compensation over base annual salaries.
The following table sets forth information concerning the compensation earned by the individual that served as our principal executive officer during 2023 and our two most highly compensated executive officers other than the individual who served as our principal executive officer during 2023 (collectively, the “named executive officers”):
| 47 |
| Table of Contents |
2023 Summary Compensation Table
Name and Principal Position |
| Year |
| Salary ($) |
|
| Option Awards ($) (1) |
|
| Non-equity Incentive Plan Compensation ($) |
|
| All Other Compensation ($)(2) |
|
| Total ($) |
| |||||
Francois Michelon(3) |
| 2023 |
|
| 387,859 |
|
|
| 258,341 |
|
|
| - |
|
|
| 587 |
|
|
| 646,787 |
|
Chief Executive Officer |
| 2022 |
|
| 423,000 |
|
|
| 212,609 |
|
|
| - |
|
|
| 587 |
|
|
| 636,196 |
|
Michael Thornton(3) |
| 2023 |
|
| 278,851 |
|
|
| 252,734 |
|
|
| - |
|
|
| 392 |
|
|
| 531,977 |
|
Chief Technology Officer |
| 2022 |
|
| 301,278 |
|
|
| 208,128 |
|
|
| - |
|
|
| 392 |
|
|
| 509,798 |
|
Irina Pestrikova |
| 2023 |
|
| 185,000 |
|
|
| 29,724 |
|
|
| - |
|
|
| - |
|
|
| 214,724 |
|
Senior Director, Finance |
| 2022 |
|
| 174,952 |
|
|
| 12,373 |
|
|
| - |
|
|
| - |
|
|
| 187,325 |
|
______________
(1) | The amounts shown in this column indicate the grant date fair value of option awards granted in the subject year computed in accordance with FASB ASC Topic 718. For additional information regarding the assumptions made in calculating these amounts, see notes 2 and 8 to the financial statements included in Part II, Item 8 of this Annual Report. The shares underlying these option awards vest and become exercisable in three equal annual installments beginning on the first anniversary of their respective grant dates. |
(2) | Represents insurance premiums paid by the Company with respect to life insurance for the benefit of the named executive officer. |
(3) | In consideration of the Company’s limited resources, in September 2023, Mr. Michelon and Mr. Thornton each agreed to a 30% reduction in each of their base salaries received for the remainder of 2023 in order to preserve cash for the Company’s operations. |
Employment Agreements and Change of Control Arrangements
The following is a summary of the employment arrangements with our named executive officers.
Francois Michelon. Effective May 12, 2017, the Company entered into an amended and restated employment agreement with Francois Michelon, our Chief Executive Officer and Chairman of the Board of Directors, which agreement was amended on December 27, 2019. Mr. Michelon’s employment with the Company is “at will” and may be terminated by him or the Company at any time and for any reason. Pursuant to the employment agreement, Mr. Michelon receives an annual base salary that is subject to adjustment at the Board of Directors’ discretion. Effective January 1, 2022, the Compensation Committee increased Mr. Michelon’s annual salary to $423,000. In September 2023, Mr. Michelon agreed to a 30% reduction of his base salary received for the remainder of 2023 in order to preserve cash for the Company’s operations. Mr. Michelon is also eligible for an annual cash bonus based upon the achievement of performance-based objectives established by the Board of Directors.
If Mr. Michelon’s employment is terminated by the Company without cause (as defined in the 2016 Plan) or if Mr. Michelon resigns for good reason (as defined in the employment agreement), Mr. Michelon will be entitled to receive, subject to his execution of a standard release agreement, 12 months’ continuation of his current base salary and a lump sum payment equal to 12 months of continued healthcare coverage (or 24 months’ continuation of his current base salary and a lump sum payment equal to 24 months of continued healthcare coverage if such termination occurs within one year following a change in control).
Under his employment agreement, Mr. Michelon is eligible to receive benefits that are substantially similar to those of the Company’s other senior executive officers.
Michael Thornton. Effective May 12, 2017, the Company entered into an amended and restated employment agreement with Michael Thornton, our Chief Technology Officer, which agreement was amended on December 27, 2019. The employment agreement provides that Mr. Thornton’s employment with the Company is “at will” and may be terminated by him or the Company at any time and for any reason. Pursuant to the employment agreement, Mr. Thornton receives an annual base salary that is subject to adjustment at the Board of Directors’ discretion. Effective January 1, 2022, the Compensation Committee increased Mr. Thornton’s annual salary to $324,000. In September 2023, Mr. Thornton agreed to a 30% reduction of his base salary received for the remainder of 2023 in order to preserve cash for the Company’s operations.
If Mr. Thornton’s employment is terminated by the Company without cause (as defined in the 2016 Plan) or if Mr. Thornton resigns for good reason (as defined in the employment agreement), Mr. Thornton will be entitled to receive, subject to his execution of a standard release agreement, 12 months’ continuation of his current base salary and a lump sum payment equal to 12 months of continued healthcare coverage (or 24 months’ continuation of his current base salary and a lump sum payment equal to 24 months of continued healthcare coverage if such termination occurs within one year following a change in control).
| 48 |
| Table of Contents |
Under his employment agreement, Mr. Thornton is eligible to receive benefits that are substantially similar to those of the Company’s other senior executive officers.
Irina Pestrikova. Ms. Pestrikova is employed by the Company pursuant to an Offer Letter by and between the Company and Ms. Pestrikova, dated as of June 9, 2021. Ms. Pestrikova’s employment is “at will” and may be terminated by the Company at any time and for any reason. Effective as of December 31, 2023, Ms. Pestrikova’s annual salary was set by the Board at $185,000. Per the terms of her offer letter, Ms. Pestrikova is eligible to receive employee benefits plans including medical, dental, vision, and 401(k) plans.
Additionally, our named executive officers are eligible to participate in our health and welfare programs and 401(k) plan, and other benefit programs on the same basis as other employees.
Outstanding Equity Awards at 2023 Fiscal Year End
The following table provides information regarding equity awards held by the named executive officers as of December 31, 2023.
|
| Option Awards | |||||||||||||
|
| Number of Securities Underlying Unexercised Options (#) Exercisable |
|
| Number of Securities Underlying Unexercised Options (#) Unexercisable |
|
| Option Exercise Price ($) |
|
| Option Expiration Date | ||||
Francois Michelon |
|
| 15,366 |
|
|
| - |
|
|
| 100.00 |
|
| 5/12/25 | |
Chief Executive Officer |
|
| 1,598 |
|
|
| - |
|
|
| 91.00 |
|
| 5/12/25 | |
|
|
| 6,250 |
|
|
| - |
|
|
| 45.00 |
|
| 12/13/26 | |
|
|
| 30,600 |
|
|
| - |
|
|
| 18.00 |
|
| 12/11/29 | |
|
|
| 1,667 |
|
|
| 833 | (1) |
|
| 52.80 |
|
| 4/5/31 | |
|
|
| - |
|
|
| 15,000 | (2) |
|
| 52.80 |
|
| 4/5/31 | |
|
|
| 11,886 |
|
|
| 23,771 | (3) |
|
| 7.60 |
|
| 3/28/32 | |
|
|
| - |
|
|
| 69,531 | (4) |
|
| 4.02 |
|
| 1/30/33 | |
Michael Thornton |
|
| 1,598 |
|
|
| - |
|
|
| 91.00 |
|
| 5/12/25 | |
Chief Technology Officer |
|
| 15,667 |
|
|
| - |
|
|
| 100.00 |
|
| 5/12/25 | |
|
|
| 6,250 |
|
|
| - |
|
|
| 45.00 |
|
| 12/13/26 | |
|
|
| 28,796 |
|
|
| - |
|
|
| 18.00 |
|
| 12/11/29 | |
|
|
| 1,667 |
|
|
| 833 | (1) |
|
| 52.80 |
|
| 4/5/31 | |
|
|
| - |
|
|
| 15,000 | (2) |
|
| 52.80 |
|
| 4/5/31 | |
|
|
| 11,635 |
|
|
| 23,271 | (3) |
|
| 7.60 |
|
| 3/28/32 | |
|
|
| - |
|
|
| 68,022 | (4) |
|
| 4.02 |
|
| 1/30/33 | |
Irina Pestrikova |
|
| 133 |
|
|
| 67 | (5) |
|
| 44.60 |
|
| 2/5/31 | |
Senior Director, Finance |
|
| 2,500 |
|
|
| 1,250 | (6) |
|
| 42.20 |
|
| 6/18/31 | |
|
|
| 200 |
|
| - |
|
|
| 14.00 |
|
| 10/27/30 | ||
|
|
| 692 |
|
|
| 1,383 | (3) |
|
| 7.60 |
|
| 3/28/32 | |
|
|
| - |
|
|
| 8,000 | (4) |
|
| 4.02 |
|
| 1/30/33 | |
(1) | Represents unvested portion of stock option award which vests in three equal annual installments beginning on April 5, 2022. |
(2) | Represents unvested portion of stock option award which vests as follows: (i) 25% vests upon the Company’s earning $5 million or more of revenue with a gross margin of 10% or greater, (ii) 25% vests upon the Company’s earning $10 million or more of revenue with a gross margin of 35% or greater, (iii) 25% vests upon the Company’s earning $15 million or more of revenue with a gross margin of 40% or greater, and (iv) 25% vests upon the Company’s earning $20 million or more of revenue with a gross margin of 50% or greater. |
(3) | Represents unvested portion of stock option award which vests in three equal annual installments beginning on March 28, 2023. |
(4) | Represents unvested portion of stock option award which vests in three equal annual installments beginning on January 30, 2024. |
(5) | Represents unvested portion of stock option award which vests in three equal annual installments beginning on February 5, 2022. |
(6) | Represents unvested portion of stock option award which vests in three equal annual installments beginning on June 18, 2022. |
| 49 |
| Table of Contents |
Equity Compensation Plan Table
The following table presents information on the Company’s equity compensation plans as of December 31, 2023. All outstanding awards relate to our common stock.
Plan Category |
| Number of Securities to Be Issued upon Exercise of Outstanding Options, Warrants and Rights (a) |
|
| Weighted-Average Exercise Price of Outstanding Options, Warrants and Rights (b) |
|
| Number of Securities Remaining Available for Future Issuance under Equity Compensation Plans (Excluding Securities Reflected in Column (a)) (c) |
| |||
Equity compensation plans approved by security holders |
|
| 624,240 | (1) |
| $ | 19.25 |
|
|
| 663,633 | (2) |
Equity compensation plans not approved by security holders |
|
| - |
|
|
| - |
|
|
| - |
|
Total |
|
| 624,240 |
|
| $ | 19.25 |
|
|
| 663,633 |
|
_________________
(1) | Consists of outstanding stock options exercisable for shares of common stock issued under the 2016 Plan. |
(2) | As of January 1, 2024, as a result of an automatic increase to the pool of shares available for issuance under the 2016 Plan on such date, the number of shares available for future issuance under the 2016 Plan was 2,381,416 shares. |
Director Compensation
Effective January 30, 2023, the Company adopted a non-employee director compensation policy (the “Compensation Policy”) pursuant to which each of our non-employee directors receives, upon his or her initial election to the Board of Directors, a stock option exercisable for 2,500 shares of common stock with a per share exercise price equal to the closing price of the common stock on the Nasdaq on the grant date. All such stock options vest in three equal annual installments beginning on the one-year anniversary of the grant date. Under the Compensation Policy, on the first trading day of each calendar year, each non-employee director is awarded a stock option exercisable for 600 shares of common stock, with a per share exercise price equal to the closing price of the common stock on the Nasdaq on the grant date, which becomes exercisable in three equal annual installments beginning on the first anniversary of the grant date. Additionally, pursuant to the Compensation Policy, each non-employee director is paid an annual cash retainer of $40,000, prorated for partial years of service and paid quarterly in arrears.
The following table sets forth information with respect to compensation earned by or awarded to each of our non-employee directors who served on the Board of Directors during the fiscal year ended December 31, 2020.
Name |
| Fees Earned or Paid in Cash ($) |
|
| Option Awards ($) (1) (2) |
|
| All Other Compensation ($) |
|
| Total ($) |
| ||||
Anthony DiGiandomenico |
|
| 40,000 |
|
|
| 35,747 |
|
|
|
|
|
| 75,747 |
| |
Michael Harsh |
|
| 40,000 |
|
|
| 33,566 |
|
|
|
|
|
| 73,566 |
| |
Alexander Tokman |
|
| 40,000 |
|
|
| 35,747 |
|
|
| 27,600 |
|
|
| 103,347 |
|
Louis Basenese |
|
| 40,000 |
|
|
| 25,065 |
|
|
|
|
|
|
| 65,065 |
|
(1) | The amounts shown in this column indicate the grant date fair value of option awards granted in the subject year computed in accordance with FASB ASC Topic 718. For additional information regarding the assumptions made in calculating these amounts, see Notes 2 and 8 to the audited financial statements included in our Annual Report on Form 10-K for the fiscal year ended December 31, 2023. The following table shows the number of shares subject to outstanding option awards held by each non-employee director as of December 31, 2023: |
| 50 |
| Table of Contents |
Name | Shares Subject to Outstanding Option Awards (#) | |||
Louis Basenese | 17,921 | |||
Anthony DiGiandomenico | 26,022 | |||
Michael Harsh | 25,460 | |||
Alexander Tokman | 26,022 | |||
(2) | In addition to annual awards granted pursuant to the Compensation Policy, in 2023 the Company awarded each director additional stock options in order to realign the incentive nature of the Company’s equity compensation with the price of the Company’s common stock. |
(3) | Represent fees paid for consulting services pursuant to that certain Consulting Agreement, dated October 17, 2023, between the Company and Mr. Tokman. |
Item is incorporated by reference to our Proxy Statement on Schedule 14A relating to our 2021 annual meeting of stockholders to be filed pursuant to Regulation 14A of the Exchange Act within 120 days after the end of the Company’s fiscal year ended December 31, 2020.
The following tables set forth certain information regarding beneficial ownership of our voting stock as of March 24, 2024 by:
· | each person or group of affiliated persons known by us to be the beneficial owner of more than 5% of any class of our voting stock; | |
· | each named executive officer included in the Summary Compensation Table above; | |
· | each of our directors; | |
· | each person nominated to become director; and | |
· | all executive officers, directors and nominees as a group. |
Unless otherwise noted below, the address of each person listed in the tables is c/o ENDRA Life Sciences Inc. at 3600 Green Court, Suite 350, Ann Arbor, Michigan 48105. To our knowledge, each person listed below has sole voting and investment power over the shares shown as beneficially owned except to the extent jointly owned with spouses or otherwise noted below.
Beneficial ownership is determined in accordance with the rules of the SEC. The information requireddoes not necessarily indicate ownership for any other purpose. Under these rules, shares of stock which a person has the right to acquire (i.e., by this Item is incorporated by reference to our Proxy Statement on Schedule 14A relating to our 2021 annual meetingthe exercise of stockholdersany option or warrant) within 60 days after March 24, 2024 are deemed to be filed pursuantbeneficially owned and outstanding for purposes of calculating the number of shares and the percentage beneficially owned by that person. However, these shares are not deemed to Regulation 14Abe beneficially owned and outstanding for purposes of computing the Exchange Act within 120 days after the endpercentage beneficially owned by any other person. The applicable percentages of the Company’s fiscal year ended December 31, 2020.
| 51 |
| Table of Contents |
Beneficial Ownership
Name of Beneficial Owner |
| Shares of Common Stock Beneficially Owned |
|
|
| Percentage of Common Stock Beneficially Owned |
|
| Shares of Series A Preferred Stock Beneficially Owned |
|
| Percentage of Series A Preferred Stock Beneficially Owned |
| ||||
Francois Michelon |
|
| 108,229 | (1) |
|
| * |
|
|
| - |
|
|
| - |
|
|
Michael Thornton |
|
| 136,122 | (2) |
|
| 1.2 | % |
|
| - |
|
|
| - |
|
|
Irina Pestrikova |
|
| 6,950 | (3) |
|
| * |
|
|
|
|
|
|
|
|
|
|
Louis Basenese |
|
| 14,636 | (4) |
|
| * |
|
|
| - |
|
|
| - |
|
|
Anthony DiGiandomenico |
|
| 161,887 | (5) |
|
| 1.5 | % |
|
| - |
|
|
| - |
|
|
Michael Harsh |
|
| 21,570 | (6) |
|
| * |
|
|
| - |
|
|
| - |
|
|
Alexander Tokman |
|
| 25,929 | (7) |
|
| * |
|
|
| - |
|
|
| - |
|
|
All directors and executive officers as a group (7 persons) |
|
| 475,323 |
|
|
| 4.2 | % |
|
| - |
|
|
| - |
|
|
5% Stockholders |
|
|
|
|
|
|
|
|
|
| - |
|
|
| - |
|
|
Mark R. Busch(8) |
|
|
|
|
|
|
|
|
|
| 17.488 |
|
|
| 50.0 | % |
|
Juan R. Rivero(9) |
|
|
|
|
|
|
|
|
|
| 17.488 |
|
|
| 50.0 | % |
|
* Less than one percent.
(1) | Consists of 4,860 shares of common stock, 103,262 shares of common stock issuable upon the exercise of options that are presently exercisable or becoming exercisable within 60 days of March 24, 2024 and 107 shares of common stock issuable upon the exercise of restricted warrants. |
(2) | Consists of 34,139 shares of common stock, 100,770 shares of common stock issuable upon the exercise of options that are presently exercisable or becoming exercisable within 60 days of March 24, 2024 and 1,213 shares of common stock issuable upon the exercise of restricted warrants. |
(3) | Consists of 6,950 shares of common stock issuable upon the exercise of options that are presently exercisable or becoming exercisable within 60 days of March 24, 2024. |
(4) | Consists of 2,454 shares of common stock and 12,182 shares of common stock issuable upon the exercise of options that are presently exercisable or becoming exercisable within 60 days of March 24, 2024. |
(5) | Consists of 102,434 shares of common stock, 17,786 shares of common stock issuable upon the exercise of options that are presently exercisable or becoming exercisable within 60 days of March 24, 2024 and 41,667 shares of common stock issuable upon the exercise of restricted warrants. |
(6) | Consists of 3,457 shares of common stock, 17,810 shares of common stock issuable upon the exercise of options that are presently exercisable or becoming exercisable within 60 days of March 24, 2024 and 303 shares of common stock issuable upon the exercise of restricted warrants. |
(7) | Consists of 8,143 shares of common stock and 17,786 shares of common stock issuable upon the exercise of options that are presently exercisable or becoming exercisable within 60 days of March 24, 2024. |
(8) | Shares jointly owned with spouse. Mr. Busch’s address is 300 S. Tryon St., Suite 1000, Charlotte, NC 28202. |
(9) | Mr. Rivero’s address is 14521 Jockey Circle, N. Davie, FL 33330. |
| 52 |
| Table of Contents |
Item 13. Certain Relationships and Related Transactions, and Director Independence
Policy for Review of Related Person Transactions
The informationBoard of Directors has adopted a written policy with regard to related person transactions, which sets forth our procedures and standards for the review, approval or ratification of any transaction required by this Item is incorporated by reference to our Proxy Statement on Schedule 14A relating to our 2021 annual meeting of stockholders to be filed pursuant to Regulation 14Areported in our filings with the SEC or in which one of our executive officers or directors has a direct or indirect material financial interest, with limited exceptions. Our policy is that the Corporate Governance and Nominating Committee shall review the material facts of all related person transactions (as defined in the related person transaction approval policy) and either approve or disapprove of the Exchange Act within 120 days afterentry into any related person transaction. In the endevent that obtaining the advance approval of the Corporate Governance and Nominating Committee is not feasible, the Corporate Governance and Nominating Committee shall consider the related person transaction and, if the Corporate Governance and Nominating Committee determines it to be appropriate, may ratify the related person transaction. In determining whether to approve or ratify a related person transaction, the Corporate Governance and Nominating Committee will take into account, among other factors it deems appropriate, whether the related person transaction is on terms comparable to those available from an unaffiliated third-party under the same or similar circumstances and the extent of the related person's interest in the transaction.
Related Person Transactions
SEC regulations define the related person transactions that require disclosure to include any transaction, arrangement or relationship in which the amount involved exceeds the lesser of (a) $120,000 or (b) one percent of the average of the Company’s total assets at year-end for the last two completed fiscal year ended December 31, 2020.
Other than as set forth below, since January 1, 2022, the Company has not participated in any such related party transaction.
On May 2, 2023, the Company conducted a public offering in which Anthony DiGiandomenico, a director of the Company, purchased 83,333 shares of the Company’s common stock and 41,667 warrants at the public offering price, for an aggregate purchase price of approximately $100,000.
Item 14. Principal Accountant Fees and Services
RBSM LLP (“RBSM”) audited our Proxy Statement on Schedule 14A relating to our 2021 annual meeting of stockholders to be filed pursuant to Regulation 14A offinancial statements for the Exchange Act within 120 days after the end of the Company’s fiscal year ended December 31, 2020.
Fee Category |
| 2023 |
|
| 2022 |
| ||
Audit Fees (1) |
| $ | 161,448 |
|
| $ | 146,000 |
|
Audit-Related Fees |
| ‒ |
|
| ‒ |
| ||
Tax Fees (2) |
| $ | 60,000 |
|
| $ | 30,000 |
|
Total |
| $ | 221,448 |
|
| $ | 176,000 |
|
_________________
(1) | Audit fees include fees for professional services rendered for the audit of our annual statements, quarterly reviews, consents and assistance with and review of documents filed with the SEC. |
(2) | Tax fees include fees (or, for 2023, estimated fees) for professional services rendered for tax compliance, tax advice and tax planning. |
| 53 |
| Table of Contents |
PART IV
(a) List of documents filed as part of this report:
1. Financial Statements (see “Financial Statements and Supplementary Data” at Item 8 and incorporated herein by reference)
2. Financial Statement Schedules (Schedules to the Financial Statements have been omitted because the information required to be set forth therein is not applicable or is shown in the accompanying Financial Statements or notes thereto)
3. Exhibits
The following is a list of exhibits filed as part of this Annual Report:
| Incorporated by Reference | |||||
| Exhibit Number | Exhibit Description | Filed Herewith | Form | Exhibit | Filing Date |
| Fourth Amended and Restated Certificate of Incorporation of the Company | 8-K | 3.2 | 05/12/17 | ||
| Amended and Restated Bylaws of the Company | S-1 | 3.4 | 12/06/16 | ||
| Specimen Certificate representing shares of common stock of the Company | S-1 | 4.1 | 11/21/16 | ||
| Form of Warrant Agreement and Warrant comprising a part of the Company’s units issued in its 2017 initial public offering | S-1 | 4.2 | 11/21/16 | ||
| Form of Underwriters’ Warrant issued to certain designees of the underwriters in the Company’s 2017 initial public offering | S-1 | 4.3 | 11/21/16 | ||
| Form of Warrant issued in June 2018 Private Placement | 8-K | 4.2 | 07/02/18 | ||
| Form of Underwriters’ Warrant issued to certain designees of the underwriters in the Company’s October 2018 offering | 10-Q | 4.6 | 11/05/18 | ||
| Form of Convertible Promissory Note issued in July 2019 Private Placement | 8-K | 4.1 | 07/29/19 | ||
| Form of Warrant issued in July 2019 Private Placement | 8-K | 4.2 | 07/29/19 | ||
| Certificate of Designations of Series A Convertible Preferred Stock | 8-K | 4.1 | 12/11/19 | ||
| Form of Warrant issued in December 2019 Series A Convertible Preferred Stock Offering | 8-K | 4.2 | 12/11/19 | ||
| Certificate of Designations of Series B Convertible Preferred Stock | 8-K | 4.1 | 12/26/19 | ||
| Form of Warrant issued in December 2019 Series B Convertible Preferred Stock Offering | 8-K | 4.2 | 12/26/19 | ||
| Description of Securities | X | ||||
| ENDRA Life Sciences Inc. 2016 Omnibus Incentive Plan * | S-1 | 10.4 | 12/06/16 | ||
| First Amendment to ENDRA Life Sciences Inc. 2016 Omnibus Incentive Plan* | DEF 14A | Appx. A | 05/10/18 | ||
| Form of Stock Option Award under 2016 Omnibus Incentive Plan* | S-1 | 10.5 | 12/06/16 | ||
| Form of Restricted Stock Unit Award under 2016 Omnibus Incentive Plan* | S-1 | 10.6 | 12/06/16 | ||
| Non-Employee Director Compensation Policy* | 10-Q | 10.2 | 08/14/2020 | ||
| Form of Indemnification Agreement by and between the Company and each of its directors and executive officers* | S-1 | 10.8 | 11/21/16 | ||
| Amended and Restated Employment Agreement, dated May 12, 2017, by and between the Company and Francois Michelon* | 8-K | 10.1 | 05/12/17 | ||
| First Amendment to Employment Agreement, dated December 27, 2019, by and between the Company and Francois Michelon* | 8-K | 10.1 | 12/27/19 | ||
| Amended and Restated Employment Agreement, dated May 12, 2017, by and between the Company and Michael Thornton* | 8-K | 10.2 | 05/12/17 | ||
| First Amendment to Employment Agreement, dated December 27, 2019, by and between the Company and Michael Thornton* | 8-K | 10.2 | 12/27/19 | ||
| Collaborative Research Agreement, dated April 22, 2016, by and between the Company and General Electric Company | S-1 | 10.17 | 11/21/16 | ||
| Amendment to Collaborative Research Agreement, dated April 21, 2017, by and between the Company and General Electric Company | S-1 | 10.21 | 05/03/17 | ||
| Amendment 2 to Collaborative Research Agreement, dated January 30, 2018, by and between the Company and General Electric Company | 8-K | 10.1 | 01/30/18 | ||
| Amendment 3 to Collaborative Research Agreement, dated January 13, 2020, by and between the Company and General Electric Company | 8-K | 10.1 | 01/15/20 | ||
| 10.15 | Amendment 4 to Collaborative Research Agreement, dated December 16, 2020, by and between the Company and General Electric Company | 8-K | 10.1 | 12/21/20 | |
| 10.16 | Gross Lease, dated January 1, 2015, between the Company and Green Court LLC | S-1 | 10.18 | 11/21/16 | |
| 10.17 | Amendment to Gross Lease, dated October 10, 2017, by and between the Company and Green Court LLC | 10-Q | 10.2 | 05/15/18 | |
| Second Amendment to Lease, dated March 15, 2021, by and between the Company and Green Court LLC | X | ||||
| 10.19 | Sublicense Agreement, dated August 2, 2007, by and between the Company and Optosonics, Inc. | S-1 | 10.19 | 11/21/16 | |
| 10.20 | Amendment to Sublicense Agreement, dated January 18, 2011, by and between the Company and Optosonics, Inc. | S-1 | 10.20 | 11/21/16 | |
| 10.21 | Master Services Agreement, dated October 24, 2017, by and between the Company and CriTech Research, Inc. | 10-K | 10.15 | 03/20/18 | |
| 10.22 | Consulting Agreement, dated October 31, 2017, by and between the Company and StarFish Product Engineering, Inc. | 10-K | 10.16 | 03/20/18 | |
| 10.23 | Employment Agreement, dated May 13, 2019, by and between the Company and David Wells* | 10-Q | 10.2 | 05/14/19 | |
| 10.24 | Employment Agreement, dated April 20, 2019, by and between the Company and Renaud Maloberti* | 10-Q | 10.2 | 08/08/19 | |
| 10.25 | U.S. Small Business Administration Paycheck Protection Program Note, issued by the Company to First Republic Bank | 10-Q | 10.2 | 05/14/2020 | |
| Subsidiaries of the Company | X | ||||
| Consent of RBSM LLP, Independent Registered Public Accounting Firm (with respect to Form S-3) | X | ||||
| Consent of RBSM LLP, Independent Registered Public Accounting Firm (with respect to Form S-8) | X | ||||
| 24.1 | Power of Attorney (included on signature page) | X | |||
| Certification Pursuant to Rule 13a-14(a) or Rule 15d-14(a) of the Securities Exchange Act of 1934 | X | ||||
| Certification Pursuant to Rule 13a-14(a) or Rule 15d-14(a) of the Securities Exchange Act of 1934 | X | ||||
| Certification Pursuant to 18 U.S.C Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | X | ||||
| 101.INS | XBRL Instance Document | X | |||
| 101.SCH | XBRL Taxonomy Schema | X | |||
| 101.CAL | XBRL Taxonomy Extension Calculation Linkbase | X | |||
| 101.DEF | XBRL Taxonomy Extension Definition Linkbase | X | |||
| 101.LAB | XBRL Taxonomy Extension Label Linkbase | X | |||
| 101.PRE | XBRL Taxonomy Extension Presentation Linkbase | X |
| 54 |
| Table of Contents |
________________
*
Indicates management compensatory plan, contract or arrangement.None.
| 55 |
| Table of Contents |
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
ENDRA Life Sciences Inc. | |||
Dated: March | By: | /s/ Francois Michelon | |
Francois Michelon | |||
Chief Executive Officer and Director (Principal Executive Officer) | |||
POWER OF ATTORNEY AND SIGNATURES
We, the undersigned officers and directors of ENDRA Life Sciences Inc., hereby severally constitute and appoint Francois Michelon our true and lawful attorney, with full power to him to sign for us and in our names in the capacities indicated below, any amendments to this Annual Report on Form 10-K, and generally to do all things in our names and on our behalf in such capacities to enable ENDRA Life Sciences Inc. to comply with the provisions of the Securities Exchange Act of 1934, as amended, and all the requirements of the Securities Exchange Commission.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Signatures | Title | Date | ||
/s/ Francois Michelon | Chief Executive Officer and Director (Principal Executive Officer) | March | ||
Francois Michelon | ||||
/s/ | Senior Director, Finance (Principal Financial and Accounting Officer) | March | ||
Irina Pestrikova | ||||
/s/ Louis J. Basenese | Director | March | ||
Louis J. Basenese | ||||
/s/ Anthony DiGiandomenico | Director | March 28, 2024 | ||
Anthony DiGiandomenico | ||||
/s/ Michael Harsh | Director | March 28, 2024 | ||
Michael Harsh | ||||
/s/ Alexander Tokman | Director | March 28, 2024 | ||
Alexander Tokman |
| 56 | ||||
