Unless otherwise indicated, “Sanara MedTech,” “we,“Sanara,” the “Company,” “our,” “us,” “our,” and “the Company,or “we,” refer to Sanara MedTech Inc. and its consolidated subsidiaries.
| 2 |
CAUTIONARY STATEMENT REGARDING FORWARD-LOOKING STATEMENTS
This report contains forward-looking statements within the meaning of the federal securities laws. Forward-looking statements generally relate to future events or our future financial or operating performance. In some cases, you can identify forward-looking statements because they contain words such as “aims,” “anticipates,” “believes,” “continue,“contemplates,” “contemplates,“continue,” “could,” “estimates,” “expects,” “forecast,” “guidance,” “intends,” “may,” “plans,” “possible,” “potential,” “predicts,” “preliminary,” “projects,” “seeks,” “should,” “target,” “will” or “would” or the negative of these words, variations of these words or other similar terms or expressions that concern our expectations, strategy, plans, or intentions. Such forward-looking statements are subject to certain risks, uncertainties and assumptions relating to factors that could cause actual results to differ materially from those anticipated in such statements, including, without limitation, the following:
| ● | shortfalls in forecasted revenue growth; |
| ● | our ability to implement our comprehensive wound and skincare strategy through acquisitions and investments and our ability to realize the anticipated benefits of such acquisitions and investments; |
| ● | our ability to meet our future capital requirements; |
| ● | our ability to retain and recruit key personnel; |
| ● | the intense competition in the markets in which we operate and our ability to compete within our markets; |
| ● | the failure of our products to obtain market acceptance; |
| ● | the effect of security breaches and other disruptions; |
| ● | our ability to maintain effective internal controls over financial reporting; |
| ● | our ability to develop and commercialize new products and products under development, including the manufacturing, distribution, marketing and sale of such products; |
| ● | our ability to maintain and further grow clinical acceptance and adoption of our products; |
| ● | the impact of competitors inventing products that are superior to ours; |
| ● | disruptions of, or changes in, our distribution model, consumer base or the supply of our products; |
| ● | our ability to manage product inventory in an effective and efficient manner; |
| ● | the failure of third-party assessments to demonstrate desired outcomes in proposed endpoints; |
| ● | our ability to successfully expand into wound and skincare virtual consult and other services; |
| ● | our ability and the ability of our research and development partners to protect the proprietary rights to technologies used in certain of our products and the impact of any claim that we have infringed on intellectual property rights of others; |
| ● | our dependence on technologies and products that we license from third parties; |
| ● | the effects of current and future laws, rules, regulations and reimbursement policies relating to the labeling, marketing and sale of our products and our planned expansion into wound and skincare virtual consult and other services and our ability to comply with the various laws, rules and regulations applicable to our business; and |
| ● | the effect of defects, failures or quality issues associated with our products. |
All forward-looking statements speak only as of the date on which they are made. For a more detailed discussion of these and other factors that may affect our business, see the discussion in “Item 1A. Risk Factors” and “Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations” in this report. We caution that the foregoing list of factors is not exclusive, and new factors may emerge, or changes to the foregoing factors may occur, that could impact our business. We do not undertake any obligation to update any forward-looking statement, whether written or oral, relating to the matters discussed in this report, except to the extent required by applicable securities laws.
| 3 |
PART I
Overview
We are a medical technology company focused on developing and commercializing transformative technologies to improve clinical outcomes and reduce healthcare expenditures in the surgical, chronic and surgical wound and skin careskincare markets. Our portfolio of products and services will allow us to deliver comprehensive wound and skin care solutions for patients in all care settings, including acute (hospitals and long-term acute care hospitals (“LTACHs”)) and post-acute (wound care clinics, physician offices, skilled nursing facilities (“SNFs”), home health, hospice, and retail). Each of our products, services and technologies contributeare designed to achieve our overall goal of achievingproviding better clinical outcomes at a lower overall cost for patients regardless of where they receive care. We strive to be one of the most innovative and comprehensive providers of effective surgical, wound and skin care products and technologiesskincare solutions and are continually seeking to expand our offerings for patients requiring wound and skin care treatments across the entire continuum of care in the United States.
We currently market sevenseveral products across chronicsurgical and surgicalchronic wound care applications and have multiple products in our pipeline. We licenseOn August 1, 2023, we acquired, among other things, the underlying intellectual property of, as well as the rights to manufacture and sell, CellerateRX Surgical Activated Collagen (“CellerateRX Surgical”), our productsprimary product, and HYCOL Hydrolyzed Collagen (“HYCOL”) from research and development partners Applied Nutritionals, LLC (“AN”Applied”) (throughfor human wound care use (such transaction, the “Applied Asset Purchase”). Prior to such time, we had licensed the rights to these products through a sublicense agreement (the “Sublicense Agreement”) with CGI Cellerate RX, LLC (“CGI Cellerate RX”), an affiliate of The Catalyst Group, Inc. (“Catalyst”)) and, both of which are related parties. In connection with the asset purchase, Applied assigned its license agreement with CGI Cellerate RX to a wholly owned subsidiary of the Company. We also license certain products from Rochal Industries, LLC (“Rochal”) and have the right to exclusively distribute certain products under development by Cook Biotech Inc. (“Cook Biotech”).
In 2021,April 2022, we intendentered into a merger agreement through which Precision Healing Inc. (“Precision Healing”) became a wholly owned subsidiary of the Company. Precision Healing is developing a diagnostic imager and lateral flow assay (“LFA”) for assessing a patient’s wound and skin conditions. This comprehensive wound and skin assessment technology is designed to begin marketing three biologic productsquantify biochemical markers to determine the trajectory of a wound’s condition to enable better diagnosis and treatment protocol. In December 2023, we received 510(k) clearance from the U.S. Food and Drug Administration (“FDA”) for surgicalthe Precision Healing diagnostic imager. We are evaluating regulatory pathways for the Precision Healing LFA.
In July 2022, we entered into a membership interest purchase agreement with Scendia Biologics, LLC (“Scendia”) and Ryan Phillips (“Phillips”) pursuant to which we acquired 100% of the issued and outstanding membership interests in Scendia from Phillips. Since our acquisition of Scendia, we have been selling a full line of regenerative and orthobiologic technologies including (i) TEXAGEN Amniotic Membrane Allograft (“TEXAGEN”), (ii) BiFORM Bioactive Moldable Matrix (“BiFORM”), (iii) ACTIGEN Verified Inductive Bone Matrix (“ACTIGEN”) and (iv) ALLOCYTE Advanced Cellular Bone Matrix (“ALLOCYTE”).
In November 2022, we established a partnership with InfuSystem Holdings, Inc. (“InfuSystem”) focused on delivering a complete wound care applications pursuantsolution targeted at improving patient outcomes, lowering the cost of care, and increasing patient and provider satisfaction. The partnership enables InfuSystem to offer innovative products including negative pressure wound therapy (“NPWT”) devices and supplies and our marketingadvanced wound care product line and distribution agreement with Cook Biotech.
In November 2023, we launched BIASURGE Advanced Surgical Solution (“BIASURGE”). BIASURGE is a no-rinse, advanced surgical solution used for wound irrigation. It contains an antimicrobial preservative effective against a broad spectrum of pathogenic microorganisms. BIASURGE is indicated for use in the mechanical cleansing and removal of debris, including microorganisms, from surgical wounds.
Comprehensive Value-Based Care Strategy
In June 2020, we formed a subsidiary, United Wound and Skin Solutions, LLC (“UWSS”(formerly known as “WounDerm”), to hold certain investments and operations in wound and skin careskincare virtual consult services. In 2023, WounDerm was renamed and is now doing business as “Tissue Health Plus” (“THP”). THP is continuing its mission to simplify skin health, starting with wound care through a refined business plan. Through THP, we plan to offer a first of its kind value-based wound care program to payers and risk-bearing entities such as accountable care organizations and value-based care (“VBC”) primary care companies, with Medicare Advantage payers as the initial target segment for this program.
THP’s programs are expected to enable payers to divest wound care spend risk, reduce wound related hospitalizations and improve patient quality of life. THP plans to coordinate delivery of community and home-based wound care for its managed patients. Community based care spans a variety of settings including physician offices, skilled nursing homes, assisted living facilities and senior living facilities. THP programs are intended to integrate science and evidence-based medicine protocols to standardize wound prevention and treatment.
We anticipate that these various service offeringsTHP’s customer contracts will allow clinicians/physicians utilizinghave three-to-five-year terms. These contracts are expected to incorporate a mix of value-based pricing methodologies including episodic, “per member per month”, and “fee for value” pricing. We believe this approach is aligned with the financial goals of the payers and will help deliver outstanding clinical outcomes for the patients.
| 4 |
Our vision for our technologiescomprehensive approach consists of three key sets of planned capabilities:
| (a) | Care Hub – This virtual patient monitoring, care coordination and navigation center is expected to help doctors and nurses support their patients throughout their wound care journey, from prevention to treatment. We expect to have Care Hub staffed by wound care certified nurse practitioners (“NPs”) and registered nurses (“RNs”), incorporating care delivery best practices from partnerships with Direct Dermatology Inc. (“DirectDerm”) and certain physician-led multispecialty wound care groups. With NPs leading the care hub, RNs would assume the role of wound specialists, providing patients with expert review and support of the overarching plan of care on each patient’s journey through the process. In addition, care navigators are expected to serve as a primary point of contact for patients and their providers, coordinating care, managing appointments and ensuring seamless communication among all team members. |
| (b) | Managed Services Organization (“MSO”) Network – With respect to patient-side wound care, our plan is that THP’s programs would be performed by a network of third-party providers who will be contracted through managed services agreements. These providers would include podiatrists, wound care provider groups, primary care physicians, and home health agencies. The providers in the THP network are expected to leverage THP’s standard of care, patient education and tools to deliver optimal patient outcomes with high predictability and efficiency. |
| (c) | Technology Platform – THP’s technology platform will focus on scaling workflows of THP’s Care Hub and MSO Network through automation and integration. We expect the technology platform to enable enhanced patient empowerment and self-healthcare. We anticipate that our technology platform will leverage our technology investments and partnerships with Precision Healing, Pixalere Healthcare, Inc. (“Pixalere”) and others, by leveraging modern technology including artificial intelligence and machine learning. Our platform technology is expected to manage program economics, standards of care, patient monitoring, wound assessments, network performance monitoring, and revenue cycle management. We expect that each of these components will work in concert with each other, constantly improving economics and care delivery. |
We are seeking a partner to collectfacilitate commercialization of Tissue Health Plus and analyze large amounts of data on patient conditions and outcomes that will improve treatment protocols and ultimately lead to more evidence-based formulary to improve patient outcomes. We intend to launch our initial virtual consult service offerings in 2021. Through a combination of our UWSS services and our Sanara products, we believe we will be able to offer patient care solutions at every stepshare in the continuumcost of wound and skin care from diagnosis through healing.
Market Scale
A study by a physician at the Department of Surgery for the Indiana University Health Comprehensive Wound Center found that approximately 8.2 million patients suffer from surgical and chronic wounds each year in the United States. Furthermore, according to an article published by the American College of Surgeons and Surgical Infection Society,, in the United States, the annual treatment cost projections for all wounds is approximately $28 billion with the estimated annual cost of surgical site infections ranging from $3.5 billion to $10 billion. Looking at the target markets for our specific products, according to SmartTRAK Advanced Wound Care, the U.S. advanced dressings market is estimated to be $1.3 billion and the U.S. wound biologics market is estimated to be $2 billion. The U.S. teledermatology market alone is estimated to grow from $5 billion in 2019 to $45 billion by 2027 according to a research report by Fortune Business Insights. In addition to our surgical wound and chronic wound divisions, the Company is planning to launch virtual consult services through UWSS for both virtual wound and virtual skin (dermatology) consultations.
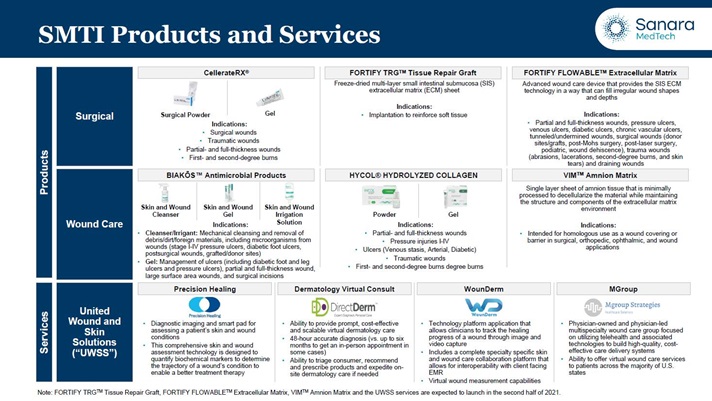
Summary of ourOur Product, & Service and Technology Offerings and Development Programs
We are committed to developing and commercializing innovative products that address the challenges physicians face in diagnosing and treating wound and skin careskincare ailments. The following table sets forth our product and service portfolio:
In 2021, we launched FORTIFY TRG and UWSS technology-based services will become meaningful drivers of revenueFORTIFY FLOWABLE, which we license from Cook Biotech. FORTIFY TRG and FORTIFY FLOWABLE, are currently 510(k) cleared for use in the future.surgical segment. We believe that both products are a complementary offering to CellerateRX Surgical and we are working to increase both the number of contract opportunities and facility approvals to drive further sales growth.
In July 2022, through the acquisition of Scendia, we expanded our surgical product offerings to include regenerative and orthobiologic technologies. We chose to focus our sales and marketing on four key products: TEXAGEN, BiFORM, ACTIGEN and ALLOCYTE. As part of the integration of Scendia into Sanara, we are continuing to work to add facility approvals for these products.
During the third quarter of 2022, we began to experience supply issues with the ALLOCYTE product line. From the fourth quarter of 2022 through September of 2023, we were unable to fill certain orders for this product, which negatively impacted our sales growth. The supply constraint was caused by significant supplier limits on qualifying eligible donor tissue and supplier necessity to subcontract all processing to secondary suppliers. We have since expanded the ALLOCYTE product line with the release of ALLOCYTE Plus, which is processed by an alternative supplier with in-house processing capabilities. Our first sales of ALLOCYTE Plus occurred in October 2023.
| 5 |
In November 2023, we launched BIASURGE. BIASURGE is a no-rinse, advanced surgical solution used for wound irrigation. It contains an antimicrobial preservative that is effective against a broad spectrum of pathogenic microorganisms. BIASURGE is indicated for use in mechanical cleansing and removal of debris, including microorganisms, from surgical wounds.
In December 2023, we entered into an exclusive license agreement with Tufts University (“Tufts”) to develop and commercialize certain new bioactive collagen peptide-based applications using patented technology licensed from Tufts. We believe the licensed technology, supported by the expertise of our research and development team, will help us expand our product offering of collagen products.
Our chronicpost-acute wound care products, HYCOL, Hydrolyzed Collagen (Powder and Gel) (collectively, “HYCOL”), BIAKŌS Antimicrobial Skin and Wound Cleanser (“BIAKŌBIAKŌS AWC”) and BIAKŌBIAKŌS Skin andAntimicrobial Wound Gel (“BIAKŌS AWG”), are usedtargeted for use across the post-acute continuum of care, including home health, hospice, physician offices, podiatrists, retail, SNFs,skilled nursing facilities (“SNFs”), and wound care centers.clinics. Our chronicpost-acute wound care products can be usedare indicated for use on stage I-IV pressure ulcers, diabetic foot ulcers (“DFUs”), venous stasis, arterial, post-surgical wounds, first- and second-degree burns and donor sites. BIAKŌBIAKŌS AWC is also available in an irrigation bottle (BIAKŌ(BIAKŌS Antimicrobial Skin and Wound Irrigation Solution) that can be used in conjunction with negative pressure wound therapy installationNPWT instillation and dwell (“NPWTi-d”) andas well as other wound irrigation needs.
Further, we have a robust pipeline of products under development for bothto further expand our position in the chronicsurgical and surgical wound care and virtual consult markets. We believe our pipeline effortsinitiatives will deepen our comprehensive portfolio of offerings as well asand allow us to address additional clinical applications. Wound care products in our pipeline include an over-the-counter handbuild product and skin cleanser, an antimicrobial skin protectant, a debrider product that leverages the body’s own enzymes and moisture, a newtechnology platforms related to hydrolyzed collagen, wound bed preparation, device for use with BIAKŌS AWC, next generation CellerateRX and HYCOL, a novel dressing that delivers oxygen to the wound bed, and a sterile BIAKŌS product for use in surgical settings. Additionally, Sanara expects to commercialize three products with Cook Biotech in the second half of 2021. The first two, FORTIFY TRG Tissue Repair Graft and FORTIFY FLOWABLE Extracellular Matrix, are currently 510(k) cleared for useantimicrobials in the surgical and wound care segment, and VIM Amnion Matrix is categorized by the U.S. Food and Drug Administration (“FDA”) as an HCT/P, subject to regulation under Section 361 of the Public Health Service Act (“PHSA”) (for which no premarket approval or clearance is required).

Strategy
●
●
●
● Seek and establish partnerships with Medicare Advantage, at-risk payors and other types of healthcare at-risk models. We believe we have assembled the products, services and technologies to offer a comprehensive strategy to help improve outcomes and lower wound care costs across the continuum of care. Looking ahead, we plan to leverage these capabilities to partner with value-based care models to aid in the treatment of their wound care patients who currently are a significant cost for the healthcare system and challenging population to heal.
● Capture patients throughout the entire continuum of care.
| 6 |
Competitive Strengths
●
●
● Wound care products for all care settings. Our wound care product portfolio allows clinicians to personalize solutions to meet the needs of individual wound care patients in all care settings including acute (hospitals and long-term acute care hospitals) and post-acute (wound care clinics, physician offices, SNFs, home health, hospice, podiatrists and retail).
● Innovative pipeline and proven clinical performance. We have a robust pipeline of surgical, wound and skincare products that we expect to market in the near term. We believe the efficacy of our offerings will be proven via statistically significant collected and analyzed clinical and health economic outcomes data, resulting in expanded adoption of our products at a lower overall cost to payors.
● Proven executive leadership team with a long-term track record of value creation.
Market Opportunities for ourOur Products, Services and Technology-Based Services
According to a study published by the Value in Health journal, roughly 15% of the Medicare beneficiary population has chronic nonhealing wounds. Chronic wounds do not advance through the phases of healing in a normal progression and do not show significant progress toward healing in 30 days. Factors contributing to the chronicity of the wound may include pressure / pressure/friction, trauma;trauma, insufficient blood flow and oxygenation in locations such as the lower extremities;extremities, increased bacterial load;load, excessive proteases;proteases, degraded growth factors;factors, matrix metalloproteinases, (“MMPs”); senescent / senescent/aberrant cells;cells or inappropriate treatment. Examples of chronic wounds include DFUs, venous leg ulcers (“VLUs”), arterial ulcers, pressure ulcers and hard-to-heal surgical/traumatic wounds. In each of the various wound types, the presence of biofilms is a frequent cause for chronificationchronicity of wounds and the removal of biofilms is a crucial step to commence healing. Biofilms need to be eradicated to prevent further deterioration of the wound that may result in additional negative patient outcomes. If not effectively treated, these wounds can lead to potentially severe complications, including further infection, osteomyelitis, fasciitis, amputation and increased mortality. Chronic wounds are primarily seen in the elderly population. For example, a 2019 study published in Advances in Wound Care reported that in the United States, 3% of the population over the age of 65 had open wounds. According to the same study, in 2020, the U.S. government estimated that the elderly population totaled 55 million people, suggesting that chronic wounds will continue to be an increasingly persistent problem in this population. Four common chronic and other hard-to-heal wounds are:
●
● Diabetic Foot Ulcers. Diabetes can lead to a reduction in blood flow, which can cause patients to lose sensation in their feet and may prevent them from noticing injuries, sometimes leading to the development of DFUs, which are open sores or ulcers on the feet that may take several weeks to heal, if ever. According to the 2020 National Diabetes Statistics Report by the Center for Disease Control and Prevention, in the United States alone, over 34 million people, or approximately 10% of the population, suffer from diabetes, a chronic, life-threatening disease.
● Venous Leg Ulcers. VLUs develop as a result of vascular insufficiency, or the inability for the vasculature of the leg to return blood back toward the heart properly and, according to a 2013 report published by the International Journal of Tissue Repair and Regeneration, VLUs affect approximately 600,000 people per year in the United States alone. These ulcers usually form on the sides of the lower leg, above the ankle and below the calf, and are slow to heal and often recur if preventative steps are not taken. The risk of VLUs can be increased as a result of a blood clot forming in the deep veins of the legs, obesity, smoking, lack of physical activity or work that requires many hours of standing.
● Pressure Injury/Ulcers. Pressure injury/ulcers are injuries to the skin and underlying tissue resulting from prolonged pressure, or pressure in combination with shear or friction. Constant pressure on an area of skin reduces blood supply to the area and over time can cause the skin to break down and form an open ulcer. These often occur in patients who are hospitalized or confined to a chair or bed and most often form on the skin over bony areas, where there is little cushion between the bone and the skin, such as heels, ankles, hips and the tailbone. Annually, 2.5 million pressure injury/ulcers are treated in the United States in acute care facilities alone, according to a 2006 study published in the Journal of the American Medical Association.
| 7 |
Sanara Products
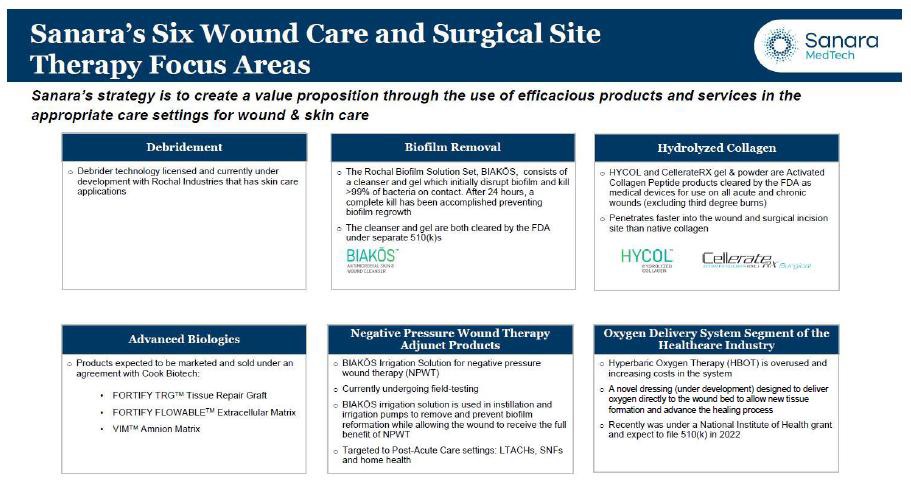
We market and distribute surgical, wound and skin careskincare products and services to physicians, hospitals, clinics, and post-acute care settings. Our products are primarily sold in the U.S. surgical tissue repair and advanced wound care and surgical tissue repair markets. We are actively seekingbelieve we have the ability to expand withindrive our six focus areas of wound and skin care for the acute, post-acute, and surgical markets: (1) debridement, (2) biofilm removal, (3) hydrolyzed collagen, (4) advanced biologics, (5) negative pressure wound therapy adjunct products, and (6) the oxygen delivery system segment of the wound and skin care market. The table below summarizes how we believe our current products and product pipeline address our sixfrom concept to preclinical and clinical development while meeting quality and regulatory requirements. We are constantly seeking long-term strategic partnerships with a focus areas:
CellerateRX Surgical is a medical hydrolysate of Type I bovine collagen indicated for the management of surgical, traumatic, and partial- and full-thickness wounds as well as first- and second-degree burns. It is manufactured in what we believe to bewith a trade secret process and theproprietary process. CellerateRX Surgical powder is further processedsterilized, packaged and designed specifically for use in athe operating room or other sterile surgical environment. The gel is typically applied post-operatively. CellerateRX Surgical products are primarily purchased by hospitals and ambulatory surgical centers for use by surgeons on surgical wounds. The predominance of CellerateRX Surgical isproducts are used in foot and ankle, neuro/spinal, orthopedic/hip and knee replacement, orthofor a variety of surgical wounds, including those associated with orthopedic, spine, trauma and ortho oncology surgeries.oncologic procedures. Additional specialties benefitingsurgical wounds that may benefit from the use of CellerateRX Surgical include cardiothoracic, colorectal, general, general trauma,cardiovascular, gynecologic, oncology, hand, head and neck, Mohs, obstetrics and gynecology (including caesarean deliveries), plastic/reconstructive, urologic and vascular.
CellerateRX Surgical is used in operative cases where patients might have trouble healing normally due to underlying health complications. There is always a risk of complication with surgical incisions.wounds. This is especially true in patients with certain comorbidities, including obesity, diabetes and hypertension. These complications can include surgical sitewound infections, dehiscence (where an incision opens after primary closure) and necrosis. Surgeons use CellerateRX Surgical to complement the body’s normal healing process. By helpingsupporting the body to heal normally without complications, improved patient outcomes are achieved, thereby reducing downstream costs related to complications (such as re-operation, longer hospitalization, re-admittance, extended rehabilitative care and other additional treatments). Wound infectionsSurgical wound complications have become increasingly problematic due to the high rates of surgical patient comorbidities and the financial strain on insurance carrierspayors as well as hospitals who suffer exorbitant costs for readmission of these patients within 30 days of surgery.
HYCOL Hydrolyzed Collagen products are a medical hydrolysate of Type I bovine collagen intended for the management of full and partial thickness wounds including pressure ulcers, venous and arterial leg ulcers and DFUs. HYCOL is primarily used in or targeted to post-acute care settings such as home health, SNFs, wound care centersclinics and physician offices and is currently approvedeligible for reimbursement under Medicare Part B. HYCOL provides the benefit of hydrolyzed collagen fragments directly in the wound bed. Therefore, unlike with the body’s own native collagen or native collagen products, the body does not have to break HYCOL down before use, which is extremely beneficial when treating elderly and otherwise compromised patients with comorbidities such as diabetes and cardiovascularperipheral vascular disease.
BIAKŌS AWC is an FDAa 510(k) cleared, patented product that laboratory tests show effectively disrupts extracellular polymeric substancescontains synergistic ingredients that have been shown to eradicateimpact mature biofilm microbes. BIAKŌS AWC is indicated for the mechanical removal of debris, dirt, foreign materials, and microorganisms from wounds including stage I-IV pressure ulcers, DFUs, post-surgical wounds, firstfirst- and second-degree burns, as well as grafted and donor sites. BIAKŌS AWC is effective in killing free-floatingplanktonic microbes and immature and mature bacterial biofilms and fungal biofilms.biofilms within the wound. In addition, safety studies demonstrated that BIAKŌS AWC is biocompatible and supports the wound healing process. Initial sales of BIAKŌS AWC occurred in July 2019.
BIAKŌS Antimicrobial Wound GelAWG is an antimicrobial hydrogel wound dressing that can be used alone or in combination with BIAKŌS AWC. In February 2020, we received notification of FDAThe BIAKŌS AWG is also 510(k) clearance for BIAKŌS Antimicrobial Wound Gelcleared and launched the product launched in November 2020 to complement BIAKŌS AWC.
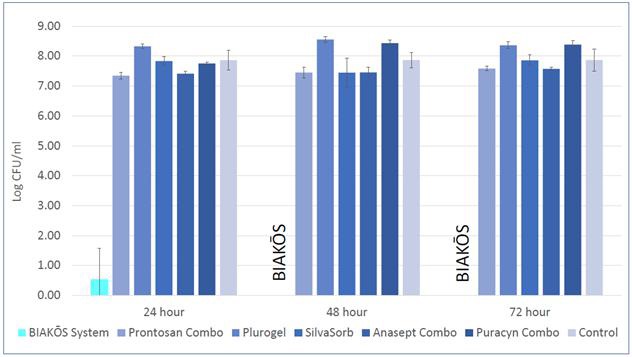
BIAKŌS AWC and BIAKŌS Antimicrobial Wound GelAWG are effective against planktonic microbes as well as immature and mature biofilms.biofilms within the wound. When used together, the cleanser can be used initially to clean a wound and disrupt biofilms (removing 99% in 10 minutes). The gel can then be applied and remains in the wound for up to 72 hours, helping to continue disrupting biofilm microbes. In a study conducted in 2020, BIAKŌS Antimicrobial Wound Gel,AWG, in combination with BIAKŌS AWC, was compared to a number of commercially available wound cleansers to treat chronic wounds such as pressure, diabetic and venous ulcersulcers. The BIAKŌS combination of cleansing and gel dressing demonstrated a reduction in the inflammatory phase of wound healing. The BIAKŌS system reduced the biofilm burdenmicrobes by 7.5 logs (>99.99% reduction) by the 24-hour time point and eradicated it by the 48-hour time point while the remaining commercial controls reduced the Methicillin-resistant Staphylococcus aureus (“MRSA”) biofilms by less than 1 log. Below.
BIASURGE is a graphic summarizingno-rinse advanced surgical solution used for wound irrigation. It contains an antimicrobial preservative effective against a broad spectrum of pathogenic microorganisms. BIASURGE is indicated for use in the efficacymechanical cleansing and removal of the usedebris, including microorganisms, from surgical wounds. First sales of BIAKŌS Antimicrobial Wound GelBIASURGE occurred in combination with BIAKŌS AWC when reducing the MRSA mature biofilm.
FORTIFY TRG Tissue Repair Graft;
| 8 |
FORTIFY FLOWABLE Extracellular Matrix is an advanced wound care device that presents the SIS ECMSmall Intestine Submucosa Extracellular Matrix technology in a way that can fill irregular wound shapes and depths. FORTIFY FLOWABLE Extracellular Matrix is indicated for the management of wounds including: partial and full-thickness wounds, pressure ulcers, venous leg ulcers, diabetic foot ulcers, chronic vascular ulcers, tunneled/undermined wounds, surgical wounds (donor sites/grafts, post-Mohs surgery, post-laser surgery, podiatric, wound dehiscence)dehiscence sites), traumatraumatic wounds (abrasions, lacerations, second-degree burns, and skin tears) and draining wounds. FORTIFY FLOWABLE Extracellular Matrix is provided sterile and is intended for one-time use. It is a 510(k) cleared product.
TEXAGEN is a single layer sheetmulti-layer amniotic membrane allograft used as an anatomical barrier with robust handling that can be sutured for securement if needed. BiFORM is an osteoconductive, bioactive, porous implant that allows for bony ingrowth across the graft site. It can be hydrated and used as a strip or molded into a putty to fill a bone defect. ACTIGEN is a verified inductive allograft putty with conformable handling properties. ALLOCYTE and ALLOCYTE Plus are human allograft cellular bone matrices containing bone-derived progenitor cells and conformable bone fibers. These viable cellular allografts are ready to use upon thawing and have fibrous handling properties.
Recent Published Studies on CellerateRX Surgical
CellerateRX Surgical is a medical hydrolysate of amnion tissueType I bovine collagen indicated for the management of surgical, traumatic, and partial- and full-thickness wounds as well as first- and second-degree burns. CellerateRX Surgical is sterilized, packaged and designed, specifically for use in the operating room (or sterile field), including additional sterility validation. It can be applied in the operating room to surgical wounds that is minimally processedmay result from a wide variety of surgical procedures to decellularizesupport the material while maintainingwound healing environment.
An animal study model by the structureIndiana University Center for Regenerative Medicine and componentsEngineering and the McGowan Institute for Regenerative Medicine was published in Advances in Wound Care in September of 2023. The study, titled “Hydrolyzed Collagen Powder Dressing Improves Wound Inflammation, Perfusion, and Breaking Strength of Repaired Tissue,” demonstrated the effects of CellerateRX Surgical powder on resolution of wound inflammation, perfusion, closure, and breaking strength of the extracellular matrix environment. All tissues are collected from consenting donors, testedrepaired skin. Moreover, the study provided translational research validating published clinical case series and further highlighting mechanistic effects of hydrolyzed collagen. Future empirical and clinical research revealing the unique support hydrolyzed collagen provides the wound environment is currently ongoing.
Several research findings involving CellerateRX Surgical powder have been noted in scientific literature. For example, in November 2021, Dr. William Hotchkiss published a retrospective study of 154 patients in JSM Neurosurgery and Spine, in which patients underwent spinal surgery and CellerateRX Surgical was utilized in the surgical wound. The study found a lower wound dehiscence rate in a high-risk patient population, when compared to previously published wound complication rates in the literature. Another retrospective case study regarding the use of CellerateRX Surgical was recently published by Dr. Alex Gitelman in November 2022. This study of 54 patients undergoing spinal surgery demonstrated no incidence of surgical wound complication.
In a retrospective study published in the Journal of Surgery in October 2023, the impact of CellerateRX Surgical collagen on surgical site infection rates in elective multispecialty surgical procedures was case matched 1:3 for infectious diseases,a total of 5,335 patients and determined eligible for transplantation by a licensed Medical Director. It is provided in multiple sizes and terminally sterilized. The VIM Amnion Matrix is intended for homologous use as a wound covering or barrierdemonstrated an overall reduction of 59% in surgical orthopedic, ophthalmic, and wound applications. It is air-dried for off-the-shelf room temperature storage with no product preparation. The graft is supplied sterile and is intended for one-time use in a single patient.
Intellectual Property
Since the acquisitions of Rochal and Precision Healing and the Applied Asset Purchase, our quality standards, we intend to relaunchresearch and development activities have included internally developing additional proprietary products, services and technologies for the product, which we expect to occur, if at all, in late 2021.
In July 2021, we expectacquired certain assets from Rochal, including intellectual property. With respect to report its operationsthe assets we acquired from Rochal and products developed following the Rochal acquisition, our patent portfolio includes, among others, eight issued U.S. patents, including U.S. Patent No. 8,829,053 entitled “Biocidal Compositions and Methods of Using the Same” (expiring December 7, 2031) relating to BIAKŌS AWC, BIAKŌS AWG and BIASURGE Surgical Wash, as a separate business divisionwell as over 100 issued patents in 2021. UWSS currently owns WounDermforeign jurisdictions. Following our acquisition by merger of Precision Healing in April 2022, described in further detail below, our patent portfolio now includes, among others, five pending U.S. patent applications as well as one pending international patent application. Following the Applied Asset Purchase in August 2023, described in further detail below, our portfolio also now includes, among others, nine additional U.S. patent applications, five trademarks, four 510(k) clearances and has investment interests in, or has exclusive affiliations with, three companies, which include DirectDerm, MGroup,various domain names.
In addition to the above patents, our pending patent applications and Precision Healing.new filings are representative of our ongoing efforts to broaden our portfolio as we continue developing new products for the surgical and chronic wound and skincare markets. We intend to begin launching UWSS’s service offerings in 2021.
| 9 |
Sales and Marketing
As of December 31, 2023, we employed 39 field sales managers (“RSMs”) and five wound care division RSMs.representatives covering 42 states. Our RSMsfield sales representatives are recruited based on their previous industry experience and professional performance and are required to have a minimum of three years of experience successfully selling into similar markets.performance. We constantly evaluate new markets and sales opportunities to add to our sales teams as warranted.
Field sales representatives are initially trained through an internal learning management system, SanaraU,“SanaraU”, which gives them further product and surgical specialty training including wound etiology, operating room etiquette and credentialing requirements. After completing their internal training, new hire RSMsfield sales representatives participate in field training with experienced RSM field trainers to get insights into best practice as well as real world training. The initial training period lasts approximately fiveeight weeks. RSMsField sales representatives are supported by regular updated training modules on product information and best practices.
A key component of our sales and marketing efforts involves working with physicians and clinicians to champion our products in their facilities. Our surgical division worksWe work closely with surgeons and health system stakeholders to demonstrate the efficacy and beneficial impact of our surgical products and successfully navigate the hospital value analysis committee (more commonly known as the “VAC”), approval process, allowing our products to be sold in those facilities. Similarly, our wound care division workswe work with clinicians to demonstrate the efficacy of our wound care products in their respective care settings. If our sales and marketing efforts are successful, the clinicians then advocate for the use of our products when medically necessary and push for their suppliers to carry our products.
Manufacturing, Supply and Production
We do not own or operate and do not intend to establish our own manufacturing facilities. We rely on, and plan to continue relying on contract manufacturing for our products. Our contract manufacturing strategy is intended to drive cost leverage andthrough scale and avoid the high capital outlays and fixed costs associated with constructing and operating manufacturing facilities. Our manufacturing partners have internal compliance processes to maintain the high quality and reliability of our products. They utilize annual internal audits, combined with external audits by regulatory agencies and commercial partners to monitor their quality control practices. We believe our contract manufacturers are well-positioned to support future expansion of our product sales. We do source some packaging and marketing materials separate from our licensing partners.
During the third quarter of 2022, we began to experience supply issues with the ALLOCYTE product line. The supply constraint was caused by significant supplier limits on qualifying eligible donor tissue and supplier necessity to subcontract all processing to secondary suppliers. From the fourth quarter of 2022 through September 2023, we were unable to fill certain orders for this product, which negatively impacted our sales. We have since expanded the ALLOCYTE product line with the release of ALLOCYTE Plus, which is processed by an alternative supplier with in-house processing capabilities. Our first sales of ALLOCYTE Plus occurred in October 2023. We have a sufficient supply of ALLOCYTE Plus to meet currently expected demand and believe we have measures in place to regularly stock the product in the future.
Reimbursement, Clinical Validation and Clinical Utility
We do not promote our products based on their reimbursement status, however, we are mindful of the benefits of a favorable reimbursement coverage status to increase patient access and support our research and development efforts to supply the highest efficacy solutions.
Three of our chronic wound care products (BIAKŌS Antimicrobial Skin and Wound Gel,AWG, HYCOL Hydrolyzed Collagen Powder and HYCOL Hydrolyzed Collagen Gel) have HCPCSHealthcare Common Procedure Coding System A codes and are eligible for reimbursement through Medicare Part B. There is currently no reimbursement for BIAKŌS AWC orand BIAKŌS Antimicrobial Skin and Wound Irrigation Solution. CellerateRX Surgical is currently captured as part of the cost of the surgical procedure.
We anticipate that our UWSS services,THP strategy, once launched, will provide a wealthsignificant amount of patient data to help us measure our products’ effectiveness on improving patient outcomes while simultaneously reducing healthcare costs. We believe our reimbursement strategy, including establishing the clinical validation, clinical utility and health economics of our products, will allow us to drive improved reimbursement coverage for our products and technologies.
Competition
The wound care market is served by a number ofseveral large, multi-product line companies as well as a number of small companies. Our products compete with primary dressings, advanced wound care products, collagen matrices and other biopharmaceutical products. Manufacturers and distributors of competitive products include Smith & Nephew plc, Medline Industries, Inc., ConvaTec Group plc, Mölnlycke Health Care AB, 3M Company, Integra LifeSciences Holdings Corporation (which acquired ACell Inc. on January 20, 2021) and numerous others. Many of our competitors are significantly larger than we are and have greater financial and personnel resources. We believe, however, that
With respect to our products outperform our competitors’ currently available equivalent products for the specific application in which they are intended by providing improved efficacy, better outcomes, and reduced cost of patient care.
| 10 |
Licensing Agreements
CellerateRX Surgical Activated Collagen
In August 27, 2018, we entered into an exclusive, world-wide sublicense agreementSublicense Agreement with CGI Cellerate RX to distribute CellerateRX Surgical and HYCOL products into the surgical and wound care and surgical markets. We payPursuant to the Sublicense Agreement, we paid royalties of 3-5% of annual collected net sales of CellerateRX Surgical and HYCOL. As amended,In August 2023, we acquired the termunderlying intellectual property of, as well as the sublicense extends through May 2050,rights to manufacture and sell, CellerateRX Surgical and HYCOL products from Applied. In connection with automatic year-to-year renewal terms thereafter so long as our Net Sales (as defined in the sublicense agreement) each year are equal to or in excess of $1,000,000. If our Net Sales fall below $1,000,000 for any year after the initial expiration date,this acquisition, Applied assigned its license agreement with CGI Cellerate RX will have the right to terminate the sublicense agreement upon written notice. Minimum royalties of $400,000 per year are payable for the first five yearsa wholly owned subsidiary of the sublicense agreement.
BIAKŌS Antimicrobial Wound Gel and BIAKŌS Antimicrobial Skin and Wound Cleanser
In July 7, 2019, we executed a license agreement with Rochal wherebypursuant to which we acquired an exclusive world-wide license to market, sell and further develop antimicrobial products for the prevention and treatment of microbes on the human body utilizing certain Rochal patents and pending patent applications (the “BIAKŌS License Agreement”). Currently, the products covered by the BIAKŌS License Agreement are BIAKŌS Antimicrobial Wound GelAWG and BIAKŌS Antimicrobial Skin and Wound Cleanser.AWC. Both products are 510(k) approved.cleared. Our Executive Chairman is a director of Rochal, and indirectly a significant shareholder of Rochal, and through the potential exercise of warrants, a majority shareholder of Rochal. Another one of our directors is also a director and significant shareholder of Rochal.
Future commitments under the terms of the BIAKŌS License Agreement include:
| ● | We pay Rochal a royalty of 2-4% of net sales. The minimum annual royalty due to Rochal was $130,000 in 2023 and will increase by $10,000 each subsequent calendar year up to a maximum amount of $150,000. |
| ● | We could pay an additional annual royalty based on specific net profit targets from sales of the licensed products, subject to a maximum of $1,000,000 during any calendar year. |
Unless previously terminated by the parties, the BIAKŌS License Agreement will expireexpires with the related patents in December 2031.
CuraShield Antimicrobial Barrier Film and No Sting Skin Protectant
In October 1, 2019, we executed a license agreement with Rochal wherebypursuant to which we acquired an exclusive world-wide license to market, sell and further develop certain antimicrobial barrier film and skin protectant products for use in the human health care market utilizing certain Rochal patents and pending patent applications (the “ABF License Agreement”). Currently, the products covered by the ABF License Agreement are CuraShield Antimicrobial Barrier Film and a no sting skin protectant product.
Future commitments under the terms of the ABF License Agreement include:
| ● | The Company will pay Rochal a royalty of 2-4% of net sales. The minimum annual royalty due to Rochal will be $50,000 beginning with the first full calendar year following the year in which first commercial sales of the products occur. The annual minimum royalty will increase by $5,000 each subsequent calendar year up to a maximum amount of $75,000. |
| ● | We could pay additional royalties annually based on specific net profit targets from sales of the licensed products, subject to a maximum of $500,000 during any calendar year. |
Unless previously terminated or extended by the parties, the ABF License Agreement will terminate upon expiration of the last U.S. patent in October 2033. No commercial sales or royalty payments hadroyalties have been maderecognized under ABF License Agreementthis agreement as of December 31, 2020.2023.
| 11 |
Debrider License Agreement
In May 4, 2020, we executed a product license agreement with Rochal, wherebypursuant to which we acquired an exclusive world-wide license to market, sell and further develop a debrider for human medical use to enhance skin condition or treat or relieve skin disorders, excluding uses primarily for beauty, cosmetic, or toiletry purposes (the “Debrider License Agreement”).
Future commitments under the terms of the Debrider License Agreement include:
| ● | Upon FDA clearance of the licensed products, we will pay Rochal $500,000 in cash and an additional $1,000,000, which, at our option, may be paid in any combination of cash and our common stock. |
| ● | We will pay Rochal a royalty of 2-4% of net sales. The minimum annual royalty due to Rochal will be $100,000 beginning with the first full calendar year following the year in which first commercial sales of the licensed products occur and increase by $10,000 each subsequent calendar year up to a maximum amount of $150,000. |
| ● | We could pay additional royalties annually based on specific net profit targets from sales of the licensed products, subject to a maximum of $1,000,000 during any calendar year. |
Unless previously terminated or extended by the parties, the Debrider License Agreement will expire in October 2034. No commercial sales or royalties hadhave been recognized under the Debrider License Agreementthis agreement as of December 31, 2020.
Cook Biotech Marketing and Distribution Agreement
In December 17, 2020, we entered into a marketing and distribution agreement with Cook Biotech whereby we were appointed as the exclusive distributor in the United States of threecertain Cook Biotech advanced biologic products. The first two products, FORTIFY TRG Tissue Repair Graft and FORTIFY FLOWABLE Extracellular Matrix, are for use in the surgical wound care segment, and VIM Amnion Matrix is for use in the chronic wound care and surgical wound care segments. We expect to commercialize these products in the second half of 2021.
Under the terms of the agreement, we will purchase the products from Cook Biotech at initial transfer prices stipulated in the agreement. Cook Biotech may update the transfer prices annually based on changes in the US Producer’s Price Index. Minimum annual order quantities will beare agreed upon by both parties after the firsteach year of the contract term. The agreement will terminate onWhile the third anniversaryterm of the date on whichagreement ends in June 2024, the first commercial sale to us from Cook Biotech is made, withagreement provides for automatic two-year renewal terms unlessif notice of non-renewalnonrenewal is not given by one party at least one year prior to the end of the initial term or renewal term that is then in effect.
Tufts University License Agreement
In December 2023, we signed an exclusive license agreement with Tufts to develop and delivery system for orthopedic bone void fillers. This patent is notcommercialize patented technology covering 18 unique collagen peptides. As part of our long-term strategic focus. We subsequentlythis agreement, we formed a new subsidiary, Sanara Collagen Peptides, LLC (“SCP”) and have issued 10% of SCP’s outstanding units to Tufts. SCP has exclusive rights to develop and commercialize new products based on the licensed patents and patents pending. SCP will pay royalties to Tufts based on net sales of licensed products and technologies. Pursuant to the patent toexclusive license agreement, royalties will be calculated at a third party to marketrate of 1.5% or 3%, depending on the type of product or technology developed. SCP will pay Tufts a bone void filler product for which we receiveminimum annual royalty of $50,000 on January 1 of the year following the first anniversary of the first commercial sale of the licensed products or technologies. SCP will pay Tufts a 3%$100,000 minimum annual royalty on product sales overJanuary 1 of each subsequent year during the life ofroyalty term specified in the patent, which expires in 2023, with annual minimum royalties of $201,000. We pay two unrelated third parties a combined royalty equal to eight percent (8%) of our net revenues and royalties generated from products that utilize the acquired patented bone hemostat and delivery system. To date, royalties received by us related to this licensing agreement have not exceeded the annual minimum of $201,000 ($50,250 per quarter). Therefore, our annual royalty obligation under the terms of theexclusive license agreement has been $16,080 ($4,020 per quarter).
Government Regulation
Our operations are subject to comprehensive federal, state and local laws and regulations in the jurisdictions in which we or our research and development partners or affiliates do business. The laws and regulations governing our business and interpretations of those laws and regulations and are subject to frequent change. Our ability to operate profitably will depend in part upon our ability, and that of our research and development partners and affiliates, to operate in compliance with applicable laws and regulations. The laws and regulations relating to medical products and healthcare services that apply to our business and that of our partners and affiliates continue to evolve, and we must, therefore, devote significant resources to monitoring developments in legislation, enforcement, and regulation in such areas. As the applicable laws and regulations change, we are likely to make conforming modifications in our business processes from time to time. We cannot provide assurance that a review of our business by courts or regulatory authorities will not result in determinations that could adversely affect our operations or that the regulatory environment will not change in a way that restricts our operations.
FDA Regulation
Our medical products and operations are regulated by the FDA and other federal and state agencies. TheMost of the products we currently market are regulated as medical devices in the United States under the Federal Food, Drug, and Cosmetic Act (“FDCA”), as implemented and enforced by the FDA. The FDA regulates the development, testing, manufacturing, labeling, packaging, storage, installation, servicing, advertising, promotion, marketing, distribution, import, export and market surveillance of our medical devices.
In addition, we have entered into an agreement to market and distribute VIM Amnion Matrixcertain products for use in the chronic wound care and surgical wound care segments. VIM Amnion Matrix is a tissue-based product regulated by the FDA under Section 361 of the PHSAPublic Health Service Act (“PHSA”) (42 U.S.C. § 264) and 21 C.F.R. Part 1271.
| 12 |
Device Premarket Regulatory Requirements
Before being introduced into the U.S. market, each medical device must obtain marketing clearance or approval from the FDA through the 510(k) premarket notification process, the de novo classification process (summarized below under De Novo Classification Process)below), or the premarket approval application (“PMA”) process, unless they are determined to be Class I devices or to otherwise qualify for an exemption from one of these available forms of premarket review and authorization by the FDA. Under the FDCA, medical devices are classified into one of three classes—Class I, Class II or Class III—depending on the degree of risk associated with each medical device and the extent of control needed to provide reasonable assurance of safety and effectiveness. Classification of a device is important because the class to which a device is assigned determines, among other things, the necessity and type of FDA review required prior to marketing the device. Class I devices are those for which reasonable assurance of safety and effectiveness can be assured by adherence to general controls that include compliance with the applicable portions of the FDA’s Quality System Regulation (“QSR”), as well as regulations requiring facility registration and product listing, reporting of adverse medical events, and appropriate, truthful and non-misleading labeling, advertising and promotional materials. The Class I designation also applies to devices for which there is insufficient information to determine that general controls are sufficient to provide reasonable assurance of the safety and effectiveness of the device or to establish special controls to provide such assurance, but that are not life-supporting or life-sustaining or for a use which is of substantial importance in preventing impairment of human health, and that do not present a potential unreasonable risk of illness ofor injury.
Class II devices are those for which general controls alone are insufficient to provide reasonable assurance of safety and effectiveness and there is sufficient information to establish “special controls.” These special controls can include performance standards, post-market surveillance requirements, patient registries and FDA guidance documents describing device-specific special controls. While most Class I devices are exempt from the 510(k) premarket notification requirement, most Class II devices require a 510(k) premarket notification prior to commercialization in the United States; however, the FDA has the authority to exempt Class II devices from the 510(k) premarket notification requirement under certain circumstances. As a result, manufacturers of most Class II devices must submit 510(k) premarket notifications to the FDA under Section 510(k) of the FDCA (21 U.S.C. § 360(k)) in order to obtain the necessary clearance to market or commercially distribute such devices. To obtain 510(k) clearance, manufacturers must submit to the FDA adequate information demonstrating that the proposed device is “substantially equivalent” to a predicate device already on the market. A predicate device is a legally marketed device that is not subject to PMA, meaning, (i) a device that was legally marketed prior to May 28, 1976 (“preamendments device”) and for which a PMA is not required, (ii) a device that has been reclassified from Class III to Class II or I, or (iii) a device that was found substantially equivalent through the 510(k) process. If the FDA agrees that the device is substantially equivalent to a predicate device currently on the market, it will grant 510(k) clearance to commercially market the device. If there is no adequate predicate to which the manufacturer can compare its proposed device, the proposed device is automatically classified as a Class III device. In such cases, the device manufacturer must then fulfill the more rigorous PMA requirements or can request a risk-based classification determination for the device in accordance with the de novo classification process.
The de novo classification process allows a manufacturer whose novel device is automatically classified into Class III to request down-classification of its device to Class I or Class II on the basis that the device presents low or moderate risk, rather than requiring the submission and approval of a PMA application.PMA. Under the Food and Drug Administration Safety and Innovation Act of 2012 (“FDASIA”), the FDA is required to classify a device within 120 days following receipt of the de novo classification request. If the manufacturer seeks reclassification into Class II, the classification request must include a draft proposal for special controls that are necessary to provide a reasonable assurance of the safety and effectiveness of the medical device. The FDA may reject the classification request if it identifies a legally marketed predicate device that would be appropriate for a 510(k) or determines that the device is not low to moderate risk or that general controls would be inadequate to control the risks and special controls cannot be developed.
Devices that are intended to be life sustaining or life supporting, devices that are implantable, devices that present a potential unreasonable risk of harm or are of substantial importance in preventing impairment of health and devices that are not substantially equivalent to a predicate device are placed in Class III and generally require FDA approval through the PMA process, unless the device is a preamendments device not yet subject to a regulation requiring premarket approval. The PMA process is more demanding than the 510(k) premarket notification process. For a PMA, the manufacturer must demonstrate through extensive data, including data from preclinical studies and clinical trials, that the device is safe and effective. The PMA must also contain a full description of the device and its components, a full description of the methods, facilities and controls used for manufacturing, and proposed labeling. Following receipt of a PMA, the FDA determines whether the application is sufficiently complete to permit a substantive review. If the FDA accepts the application for review, it has 180 days under the FDCA to complete its review of a PMA, although in practice, the FDA’s review often takes significantly longer, and can take up to several years. Before approving a PMA, the FDA generally also performs an on-site inspection of manufacturing facilities for the product to ensure compliance with the QSR.
Thus far, all of the medical devices that we currently market and distribute have been cleared through 510(k) premarket notifications filed by our third-party research and development partners, who are the manufacturers of such devices.notifications. We also are continuing to work through the development process for a number of products in our pipeline. We are currently working on final formulation and the development of a retail marketing strategy for an over-the-counter hand and skin cleanser. Our autolytic debrider product as well a novel dressing that delivers oxygen to the wound bed and a sterile BIAKŌSantimicrobial surgical wash product are currently under development at Rochal, and wedevelopment. We are in discussions concerning the best path for seeking clearance and approval for these products. In December 2023, we received 510(k) clearance from the FDA for the Precision Healing diagnostic imager. We are also exploring new indications of usecurrently evaluating commercial and improved formulasregulatory pathways for a next generation CellerateRX and a next generation HYCOL.the Precision Healing LFA.
| 13 |
Clinical trials are almost always required to support PMAs and are sometimes required to support 510(k) submissions. All clinical investigations of devices to determine safety and effectiveness must be conducted in accordance with the FDA’s investigational device exemption (“IDE”), regulations that govern investigational device labeling, prohibit promotion of the investigational device and specify recordkeeping, reporting and monitoring responsibilities of study sponsors and study investigators. If the device presents a “significant risk,” as defined by the FDA, the agency requires the device sponsor to submit an IDE application to the FDA, which must become effective prior to commencing human clinical trials. The IDE will automatically become effective 30 days after receipt by the FDA, unless the FDA denies the application or notifies the company that the investigation is on hold and may not begin until the sponsor provides supplemental information about the investigation that satisfies FDA’s concerns. If the FDA determines that there are deficiencies or other concerns with an IDE that require modification of the study, the FDA may permit a clinical trial to proceed under a conditional approval. In addition, the study must be approved by, and conducted under the oversight of, an institutional review board (“IRB”), for each clinical site. If the device presents a non-significant risk to the patient according to criteria established by the FDA as part of the IDE regulations, a sponsor may begin the clinical trial after obtaining approval for the trial by one or more IRBs without separate authorization from the FDA, but must still comply with abbreviated IDE requirements, such as monitoring the investigation, ensuring that the investigators obtain informed consent, and labeling and record-keeping requirements.
Device Post-marketPostmarket Regulatory Requirements
After a device is cleared or approved for commercialization, and prior to marketing, numerous regulatory requirements apply to the various entities responsible for preparing a device for distribution, including the manufacturer (including specification developer), contract manufacturers, relabelers/repackagers, sterilizers and initial importer, as applicable. These include:
| ● | establishment registration and device listing; |
| ● | development of a quality management system, including establishing and implementing procedures to design and manufacture devices in compliance with the QSR (unless a device category is exempt from this requirement by the FDA, such as in the case of many Class I devices); |
| ● | labeling regulations that prohibit the promotion of products for uncleared or unapproved uses (known as off-label uses), as well as requirements to provide accurate and non-misleading information and adequate information on both risks and benefits of the device; |
| ● | FDA’s unique device identification requirements that call for a unique device identifier on device labels, packages, and in some cases, on the device itself, and submission of data to the FDA’s Global Unique Device Identification Database; |
| ● | medical device reporting regulations that require manufacturers to report to the FDA if a device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if it were to recur; |
| ● | corrections and removal reporting regulations that require manufacturers report to the FDA field corrections and product recalls or removals if undertaken to reduce a risk to health posed by the device or to remedy a violation of the FDCA that may present a risk to health; and |
| ● | postmarket surveillance regulations, which apply to Class II or Class III devices if the FDA has issued a postmarket surveillance order and the failure of the device would be reasonably likely to have serious adverse health consequences, the device is expected to have significant use in the pediatric population, the device is intended to be implanted in the human body for more than one year, or the device is intended to be used to support or sustain life and to be used outside a user facility. |
Our research and development partners and their contract manufacturers may be subject to periodic scheduled or unscheduled inspections by the FDA. If we are required to register with the FDA, by becoming the manufacturer or specification developer of any medical device for instance, then we also may be subject to such inspections by the FDA. If the FDA believes we or any of our research and development partners or their contract manufacturers are not in compliance with the QSR, or other post-marketpostmarket requirements, it has broad authority to take significant enforcement actions to compel compliance. Specifically, if the FDA determines that we or our research and development partners or their contract manufacturers failed to comply with applicable regulatory requirements, the agency can take a variety of compliance or enforcement actions, which may result in any of the following sanctions:
| ● | untitled letters, warning letters, fines, injunctions, consent decrees and civil penalties; |
| ● | customer notifications or repair, replacement or refunds; |
| ● | mandatory recalls, withdrawals, or administrative detention or seizure of our products; |
| ● | operating restrictions or partial suspension or total shutdown of production; |
| ● | refusing or delaying requests for 510(k) marketing clearance or approval of premarket approval applications relating to new products or modified products; |
| ● | reclassifying a 510(k)-cleared device or withdrawing PMA approval; |
| ● | refusal to grant export approvals for our products; or |
| ● | pursuing criminal prosecution. |
Any such enforcement action by the FDA would have a material adverse effect on our business. In addition, these regulatory controls, as well as any changes in FDA policies, can affect the time and cost associated with the development, introduction, and continued availability of new products.
| 14 |
HCT/P Regulatory Requirements
Some of the products we currently market are regulated as biologics, more specifically as human cells, tissues, and cellular and tissue-based products (“HCT/Ps”). They include (i) TEXAGEN, (ii) BiFORM, (iii) ACTIGEN, (iv) ALLOCYTE and (v) ALLOCYTE Plus. HCT/Ps are regulated by the FDA’s Center for Biologics Evaluation and Research (“CBER”) or Center for Devices and Radiological Health (“CDRH”) depending on the type of product, how it is manufactured and its intended uses. HCT/Ps that meet all of the criteria described in 21 C.F.R. § 1271.10(a) are regulated by the CBER under Section 361 of the PHSA (42 U.S.C. § 264) and 21 C.F.R. Part 1271 only (“361 products”). Although 361 products do not require premarket review by the FDA prior to commercialization, manufacturers of 361 products must register with the FDA, submit a list of HCT/Ps manufactured, and comply with current good tissue practices (“cGTP”), among other things.
Federal Trade Commission Regulatory Oversight
Our advertising for our products and services is subject to federal truth-in-advertising laws enforced by the Federal Trade Commission or FTC,(the “FTC”), as well as comparable state consumer protection laws. Under the Federal Trade Commission Act (“FTC Act”), the FTC is empowered, among other things, to (a) prevent unfair methods of competition and unfair or deceptive acts or practices in or affecting commerce; (b) seek monetary redress and other relief for conduct injurious to consumers; and (c) gather and compile information and conduct investigations relating to the organization, business, practices, and management of entities engaged in commerce. The FTC has very broad enforcement authority, and failure to abide by the substantive requirements of the FTC Act and other consumer protection laws can result in administrative or judicial penalties, including civil penalties, injunctions affecting the manner in which we would be able to market services or products in the future, or criminal prosecution.
Fraud and Abuse and Transparency Laws and Regulations
Our business activities (and the business activities of our research and development partners and affiliates), including, but not limited to, research, sales, promotion, distribution and medical education, are subject to regulation by numerous federal and state regulatory and law enforcement authorities in the United States, including the Department of Justice, the Department of Health and Human Services and its various divisions, CMS, the Health Resources and Services Administration, the Department of Veterans Affairs, the Department of Defense, and state and local governments. Our business activities must comply with numerous healthcare laws, including, but not limited to, anti-kickback and false claims laws and regulations as well as data privacy and security laws and regulations, which are described below.
The federal Anti-Kickback Statute prohibits, among other things, any person or entity, from knowingly and willfully offering, paying, soliciting, or receiving any remuneration, directly or indirectly, overtly or covertly, in cash or in kind, to induce or in return for purchasing, leasing, ordering, or arranging for or recommending the purchase, lease, furnishing, or order of any item or service reimbursable under Medicare, Medicaid, or other federal healthcare programs, in whole or in part. The term “remuneration” has been interpreted broadly to include anything of value. The Anti-Kickback Statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on one hand and prescribers, purchasers, formulary managers, and beneficiaries on the other. There are certain statutory exceptions and regulatory safe harbors protecting some common activities from prosecution. The exceptions and safe harbors are drawn narrowly, and practices that involve remuneration that may be alleged to be intended to induce prescribing, purchases, or recommendations may be subject to scrutiny if they do not qualify for an exception or safe harbor. Failure to meet all of the requirements of a particular applicable statutory exception or regulatory safe harbor does not make the conduct per se illegal under the Anti-Kickback Statute. Instead, the legality of the arrangement will be evaluated on a case-by-case basis based on a cumulative review of all of its facts and circumstances. Several courts have interpreted the statute’s intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of federal healthcare covered business, the statute has been violated. The Patient Protection and Affordable Care Act, of 2010, as amended (the “ACA”), modified the intent requirement under the Anti-Kickback Statute to a stricter standard, such that a person or entity no longer needs to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. In addition, the ACA also provided that a violation of the federal Anti-Kickback Statute is grounds for the government or a whistleblower to assert that a claim for payment of items or services resulting from such violation constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act (the “FCA”). The ACA further created new federal requirements for reporting, by applicable manufacturers of covered drugs, payments and other transfers of value to physicians and teaching hospitals, and ownership and investment interests held by physicians and other healthcare providers and their immediate family members.
| 15 |
The federal civil FCA, prohibits, among other things, any person or entity from knowingly presenting, or causing to be presented, a false or fraudulent claim for payment to, or approval by, the federal government, knowingly making, using, or causing to be made or used a false record or statement material to a false or fraudulent claim to the federal government, or avoiding, decreasing, or concealing an obligation to pay money to the federal government. A claim includes “any request or demand” for money or property presented to the U.S. government. The civil FCA has been used to assert liability on the basis of kickbacks and other improper referrals, improperly reported government pricing metrics such as Best Price or Average Manufacturer Price, or submission of inaccurate information required by government contracts, improper use of Medicare provider or supplier numbers when detailing a provider of services, improper promotion of off-label uses not expressly approved by the FDA in a drug’s label, and allegations as to misrepresentations with respect to the products supplied or services rendered. Several pharmaceutical and other healthcare companies have further been sued under these laws for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. Intent to deceive is not required to establish liability under the civil FCA; however, a change in Department of Justice policy now prohibits enforcement actions for knowing violations of law based on non-compliancenoncompliance with agency subregulatory guidance. Civil FCA actions may be brought by the government or may be brought by private individuals on behalf of the government, called “qui tam” actions. If the government decides to intervene in a qui tam action and prevails in the lawsuit, the individual will share in the proceeds from any fines or settlement funds. If the government declines to intervene, the individual may pursue the case alone. Since 2004, these FCA lawsuits against pharmaceutical companies have increased significantly in volume and breadth, leading to several substantial civil and criminal settlements, as much as $3.0 billion, regarding certain sales practices and promoting off labeloff-label drug uses. Civil FCA liability may be imposed for Medicare or Medicaid overpayments, for example, overpayments caused by understated rebate amounts, that are not refunded within 60 days of discovering the overpayment, even if the overpayment was not caused by a false or fraudulent act.
The government may further prosecute conduct constituting a false claim under the criminal FCA. The criminal FCA prohibits the making or presenting of a claim to the government knowing such claim to be false, fictitious, or fraudulent and, unlike the civil FCA, requires proof of intent to submit a false claim. The civil monetary penalties statute is another potential statute under which drug and device companies may be subject to enforcement. Among other things, the civil monetary penalties statute imposes fines against any person who is determined to have presented, or caused to be presented, claims to a federal healthcare program that the person knows, or should know, is for an item or service that was not provided as claimed or is false or fraudulent.
HIPAA also created federal criminal statutes that prohibit knowingly and willfully executing, or attempting to execute, a scheme to defraud or to obtain, by means of false or fraudulent pretenses, representations, or promises, any of the money or property owned by, or under the custody or control of, a healthcare benefit program, regardless of whether the payor is public or private, knowingly and willfully embezzling or stealing from a health care benefit program, willfully obstructing a criminal investigation of a health care offense, and knowingly and willfully falsifying, concealing, or covering up by any trick or device a material fact or making any materially false statements in connection with the delivery of, or payment for, healthcare benefits, items, or services relating to healthcare matters. The ACA, as amended, modified the intent requirement under the certain portions of these federal criminal statutes such that a person or entity no longer needs to have actual knowledge of the statute or specific intent to violate it.
Many states have also adopted laws similar to each of the above federal laws, which may be broader in scope and apply to items or services reimbursed by any third-party payor, including commercial insurers, and some have transparency laws that require reporting price increases and related information. Certain state laws also regulate manufacturers’ use of prescriber-identifiable data. Certain states also require implementation of commercial compliance programs and compliance with the pharmaceutical industry’s voluntary compliance guidelines and the applicable compliance guidance promulgated by the federal government, or otherwise restrict payments or the provision of other items of value that may be made to healthcare providers and other potential referral sources; impose restrictions on marketing practices; or require drug manufacturers to track and report information related to payments, gifts, and other items of value to physicians and other healthcare providers. These laws may affect our future sales, marketing, and other promotional activities by imposing administrative and compliance burdens.
If our operations are found to be in violation of any of the laws or regulations described above or any other laws that apply to us, we may be subject to penalties or other enforcement actions, including criminal and significant civil monetary penalties, damages, fines, disgorgement, imprisonment, exclusion from participation in government healthcare programs, corporate integrity agreements, debarment from receiving government contracts or refusal of new orders under existing contracts, reputational harm, diminished profits and future earnings, and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations.
Telemedicine Standards, and Related Laws and Guidelines
We have entered into a management agreement with Mgroup, which is a physician-owned multispecialty wound care group with active medical licenses in over 40 states. Through this partnership, we expect to make available coordinated telemedicine services on our platform. In connection with this arrangement, we expect to administer non-clinical services to support the delivery of telemedicine services, including billing, scheduling and a wide range of other administrative and support services, and will be paid a pre-determined, fair market value amount for those services. We have also entered into a professional services agreement with and made a minority investment in DirectDerm, a dermatology telemedicine company based in California, which has an exclusive network of dermatologists licensed across 23 states.
The delivery of telemedicine services directly or through contractual relationships is subject to various federal, state, and local laws, regulations and approvals, relating to, among other things, the health provider licensure, adequacy and continuity of medical care, medical practice standards (including specific requirements when providing healthcare utilizing telemedicine technologies and consulting services among providers), medical records maintenance, personnel supervision, and prerequisites for the prescription of medication. The application of some of these laws to telemedicine is unclear and subject to differing interpretation. Further, laws and regulations specific to delivering medical services utilizing telemedicine technologies continues to evolve with some states incorporating modality and consent requirements for certain telemedicine encounters.
| 16 |
Telemedicine services also implicate state corporate practice of medicine and fee-splitting laws which vary from state to state and are not always consistent among states. In addition, these requirements are subject to broad powers of interpretation, enforcement discretion by state regulators, and, in some cases, dated (yet still valid) case law. Some of these requirements may apply to us or our partners, even if we do not have a physical presence in the state, based solely on the engagement of a provider licensed in the state or the provision of telemedicine to a resident of the state. However, regulatory authorities or other parties, including providers in our affiliated provider network, may assert that, despite these arrangements, we are engaged in the corporate practice of medicine or that our contractual arrangements with affiliated physician groups constitute unlawful fee-splitting. In this event, failure to comply could lead to adverse judicial or administrative action against us and/or our providers, civil or criminal penalties, receipt of cease-and-desist orders from state regulators, loss of provider licenses, or the need to make changes to the arrangements with our affiliated provider network; each of which could interfere with our business or prompt other materially adverse consequences.
U.S. Federal and State Health Information Privacy and Security Laws
There are numerous U.S. federal and state laws and regulations related to the privacy and security of personally identifiable information (“PII”), including health information. In particular, HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, and its respective implementing regulations, establishes privacy and security standards that limit the use and disclosure of PHI,protected health information (“PHI”), and require the implementation of administrative, physical, and technical safeguards to ensure the confidentiality, integrity and availability of individually identifiable health information in electronic form. Our affiliated network providers and our hospital, health system and other provider clients are all regulated as covered entities under HIPAA. Since the effective date of the HIPAA Omnibus Final Rule on September 23, 2013, HIPAA’s requirements are also directly applicable to the independent contractors, agents and other “business associates” of covered entities that create, receive, maintain or transmit protected health information (“PHI”)PHI in connection with providing services to covered entities. We are a business associate under HIPAA when we are working on behalf of our affiliated providers.
Violations of HIPAA may result in civil and criminal penalties. The civil penalties range from $119 to $59,522 per violation, with a cap of $1.8 million per year for violations of the same standard during the same calendar year. However, aA single breach incident can result in violations of multiple standards. We must also comply with HIPAA’s breach notification rule. Under the breach notification rule, covered entities must notify affected individuals without unreasonable delay in the case of a breach of unsecured PHI, which may compromise the privacy, security or integrity of the PHI. In addition, notification must be provided to Health and Human Services (“HHS”) and the local media in cases where a breach affects more than 500 individuals. Breaches affecting fewer than 500 individuals must be reported to HHS on an annual basis. The regulations also require business associates of covered entities to notify the covered entity of breaches by the business associate.
State attorneys general also have the right to prosecute HIPAA violations committed against residents of their states. While HIPAA does not create a private right of action that would allow individuals to sue in civil court for a HIPAA violation, its standards have been used as the basis for the duty of care in state civil suits, such as those for negligence or recklessness in misusing personal information. In addition, HIPAA mandates that HHS conduct periodic compliance audits of HIPAA covered entities and their business associates for compliance. It also tasks HHS with establishing a methodology whereby harmed individuals who were the victims of breaches of unsecured PHI may receive a percentage of the Civil Monetary Penalty fine paid by the violator. In light of the HIPAA Omnibus Final Rule, recent enforcement activity, and statements from HHS, we expect increased federal and state HIPAA privacy and security enforcement efforts.
Many states in which we or our research and development partners may operate also have laws that protect the privacy and security of sensitive and personal information, including health information. These laws may be similar to or even more protective than HIPAA and other federal privacy laws. For example, the laws of the State of California are more restrictive than HIPAA. Where state laws are more protective than HIPAA, we must comply with the state laws to which we are subject, in addition to HIPAA. In certain cases, it may be necessary to modify our planned operations and procedures to comply with these more stringent state laws. Not only may some of these state laws impose fines and penalties upon violators, but also some, unlike HIPAA, may afford private rights of action to individuals who believe their personal information has been misused. In addition, state laws are changing rapidly, and there is discussion of a new federal privacy law or federal breach notification law, to which we may be subject.
In addition to HIPAA and state health information privacy laws, we may be subject to other state and federal privacy laws, including laws that prohibit unfair privacy and security practices and deceptive statements about privacy and security and laws that place specific requirements on certain types of activities, such as data security and texting.
In recent years, there have been a number of well-publicized data breaches involving the improper use and disclosure of PII and PHI. Many states have responded to these incidents by enacting laws requiring holders of personal information to maintain safeguards and to take certain actions in response to a data breach, such as providing prompt notification of the breach to affected individuals and state officials. In addition, under HIPAA and pursuant to the related contracts that we enter into with our business associates, we must report breaches of unsecured PHI to our contractual partners following discovery of the breach. Notification must also be made in certain circumstances to affected individuals, federal authorities and others.
| 17 |
Employees
As of March 30, 2021,December 31, 2023, we had a staff of 41, consisting of 40107 full-time employees and 1 part-time employee.
Corporate Information
We were incorporated in Texas on December 14, 2001. On March 15, 2019, we entered into a Share Exchange Agreement with CGI Cellerate RX, an affiliate of Catalyst, pursuant to which we acquired Catalyst’s 50% equity interest in Cellerate, LLC (“Cellerate”) in exchange for 1,136,815 shares of our newly created Series F Convertible Preferred Stock (the “Cellerate Acquisition”). Prior to the consummation of the Cellerate Acquisition, we and Catalyst each owned a 50% equity interest in Cellerate. The Cellerate Acquisition was accounted for as a reverse merger, and Cellerate was deemed to be the accounting acquirer. In May 2019, we changed our name to Sanara MedTech Inc.
Available Information
The Company electronically files reports with the Securities and Exchange Commission (the “SEC”). The SEC maintains an Internet site (www.sec.gov) that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC. Copies of the Company’s Annual Report on Form 10-K, Quarterly Reports on Form 10-Q and Current Reports on Form 8-K and amendments to those reports filed or furnished to the SEC are also available free of charge through the Company’s investor relations website (http://www.sanaramedtech.com/investor-relations/), as soon as reasonably practicable after electronically filing with or otherwise furnishing such information to the SEC, and are available in print to any shareholder who requests it.
Item 1A. RISKRISK FACTORS
The risks below are those that we believe are the material risks that we currently face but are not the only risks facing us and our business. If any of these risks actually occur, our business, financial condition and results of operations could be materially adversely affected. Below is a summary of our risk factors with a more detailed discussion following.
| ● | We have had a history of losses, which may continue as we expand our selling efforts. |
| ● | Our revenue growth for a particular period is difficult to predict, and a shortfall in forecasted revenues may harm our operating results. |
| ● | Our current comprehensive wound and skincare strategy involves growth through acquisitions and investments, which requires us to incur substantial costs and potential liabilities for which we may never realize the anticipated benefits. |
| ● | Our indebtedness could adversely affect our financial condition and prevent us from fulfilling our obligations. |
| ● | Our outstanding indebtedness is subject to certain operating and financial covenants that restrict our business and financing activities and may adversely affect our cash flow and our ability to operate our business. |
| ● | Failure to manage our growth strategy could harm our business. |
| ● | We may fail to realize all the anticipated benefits of the Applied Asset Purchase, or such benefits may take longer to realize than expected. |
| ● | If we are unable to compete within our markets or our products, services and technologies do not gain market acceptance, our financial condition and operating results could suffer. |
| ● | Security breaches and other disruptions could compromise our information and expose us to liability, which would cause our business and reputation to suffer. |
| ● | If we fail to maintain an effective system of internal controls over financial reporting, we may not be able to accurately report our financial results or prevent fraud and our business may be harmed and our stock price may be adversely impacted. |
| ● | We rely heavily on our research and development partners to design, manufacture and supply the products we have licensed for marketing. If we or one of our partners fails to perform adequately, fulfill our needs, or comply with regulations, we may be required to incur significant costs or even be subject to enforcement actions. We also may face significant delays in our product introductions and commercialization. |
| ● | Our revenue generated from the sale of Scendia products is heavily dependent on license agreements with certain manufacturers, and the termination of any of these license agreements could harm our business. |
| ● | Certain of our product candidates are still under development and we may not be able to successfully commercialize any of these product candidates. |
| ● | Our future success will largely depend on our ability to maintain and further grow clinical acceptance and adoption of our products, and we may be unable to adequately educate healthcare practitioners on the use and benefits of our products. |
| 18 |
| ● | Disruption of, or changes in, our distribution model or customer base could harm our sales and margins. |
| ● | Interruptions in the supply of our products or inventory loss may adversely affect our business, financial condition and results of operations. |
| ● | If we are unable to manage product inventory in an effective manner, our profitability could be impaired. |
| ● | Failure of any third-party assessments to demonstrate desired outcomes in proposed endpoints could have a negative impact on our business performance. |
| ● | Increased prices for, or unavailability of, raw materials used in our products could adversely affect our business, financial condition and results of operations. |
| ● | Our planned expansion into wound and skincare virtual consult and other services could have a material adverse effect on our business, financial condition and results of operations. |
| ● | Our planned expansion into wound and skincare virtual consult and other services will require entrance into several markets in which we have little or no experience, which may not be successful and could be costly. |
| ● | Our planned expansion into the telehealth business is dependent on our relationships with affiliated professional entities to provide physician services, and our business would be adversely affected if those relationships were disrupted. |
| ● | Recent and frequent state legislative and regulatory changes specific to telemedicine may present us with additional requirements and state compliance costs, with potential operational impacts in certain jurisdictions. |
| ● | If we are unable to adequately protect our intellectual property rights, we may not be able to compete effectively. |
| ● | CellerateRX Surgical is not currently protected by any pending patent application or any unexpired patent. Currently the substantial majority of our net revenue is derived from the sale of CellerateRX Surgical. CellerateRX Surgical may be subject to competition from the sale of substantially equivalent products that could adversely affect our business and operations. |
| ● | We are heavily dependent on technologies and products we have licensed from third parties, and we may need to license technologies and products in the future, and if we fail to obtain licenses we need, or fail to comply with our payment and other obligations in the agreements under which we in-license intellectual property and other rights from third parties, we could lose our ability to develop and commercialize our products. |
| ● | We may be found to infringe on or violate intellectual property rights of others. |
| ● | Our business is affected by numerous regulations relating to the development, manufacture, distribution, labeling, marketing and sale of our products. |
| ● | We are subject to various governmental regulations relating to the labeling, marketing and sale of our products. |
| ● | If we fail to obtain or experience significant delays in obtaining regulatory clearances or approvals to market future medical device products, we will be unable to commercialize these products until such clearance or approval is obtained. |
| ● | Delays in or changes to the FDA clearance and approval processes or ongoing regulatory requirements could make it more difficult for us to obtain FDA clearance or approval of new products or comply with ongoing requirements. |
| ● | Failure to obtain or maintain adequate reimbursement or insurance coverage for drugs, if any, could limit our ability to market those drugs and decrease our ability to generate revenue. Changes in reimbursement policies and regulations by governmental or other third-party payors may have an adverse impact on the use of our products. |
| ● | We rely heavily on our research and development partners to comply with applicable laws and regulations relating to product classification and when and what types of FDA marketing authorization are needed to lawfully commercialize a new or updated medical product in the United States. |
| ● | We and our employees and contractors are subject, directly or indirectly, to federal, state and foreign healthcare fraud and abuse laws, including false claims laws. If we are unable to comply, or have not fully complied, with such laws, we could face substantial penalties. |
| ● | We and our or our research and development partners’ use and disclosure of PII, including health information, is subject to federal and state privacy and security regulations, and our failure to comply with those regulations or to adequately secure the information we hold could have a material adverse effect on our client base, business, financial condition and results of operations. |
| ● | Our officers, employees, independent contractors, principal investigators, consultants and commercial partners may engage in misconduct or activities that are improper under other laws and regulations, which would create liability for us. |
| ● | We could be adversely affected if healthcare reform measures substantially change the market for medical care or healthcare coverage in the United States. |
| ● | Defects, failures or quality issues associated with our products could lead to product recalls or safety alerts, adverse regulatory actions, product liability lawsuits and other litigation and negative publicity that could materially adversely affect our reputation, business, results of operations and financial condition. |
| 19 |
Risks Related to negatively impact our business, financial condition and results of operations.
We have had a history of losses, which may continue as we expand our selling efforts.
We have incurred net losses in most years since we began our current operations in 2004. We plan to continue making significant investments in our sales force and clinical programs, which substantially increase our operating expenses. Consequently, we will need to continue our revenue growth to become profitable in future periods. We cannot offer any assurance that we will be able to generate future sales growth. If we fail to achieve profitability, our stock price may decline, and you may lose part or all of your investment.
Our revenue growth for a particular period is difficult to predict, and a shortfall in forecastforecasted revenues may harm our operating results.
Because we are a relatively small company, our revenue growth and, consequently, results of operations are difficult to predict. We plan our operating expense levels based primarily on forecasted revenue levels. A shortfall in revenue could lead to operating results being below expectations as we may not be able to quickly reduce our fixed expenses in response to short-term revenue shortfalls. We have experienced fluctuations in revenue and operating results from quarter to quarter and anticipate that these fluctuations will continue until we achieve a critical mass with our product and service sales. These fluctuations can result from a variety of factors, including:
| ● | economic conditions worldwide, including increases in inflation, as well as economic conditions specific to the healthcare industry, which could affect the ability of surgical and post-acute facilities to purchase our products and could result in a reduction in elective operative procedures; |
| ● | governmental regulations, including those adopted in response to the COVID-19 pandemic or other potential outbreaks; |
| ● | the uncertainty surrounding our ability to attract new customers and retain existing customers; |
| ● | changes in reimbursement rates for our products by government and private insurers; |
| ● | the length and variability of our sales cycle, especially gaining approvals for the use of our products in additional hospitals and surgery centers, which makes it difficult to forecast the quarter in which our sales will occur; |
| ● | issues including delays in the sourcing of our products; |
| ● | the timing of regulatory approvals; |
| ● | the timing of operating expense relating to the expansion of our business and operations; |
| ● | changes in the pricing of our products and those of our competitors; |
| ● | the development of new wound care products or product enhancements by our competitors; and |
| ● | actual events, circumstances, outcomes and amounts differing from assumptions and estimates used in preparing our operating plan and how well we execute our strategy and operating plans. |
As a consequence, operating results for a particular future period are difficult to predict and prior results are not necessarily indicative of future results. Any of the foregoing factors, or any other factors discussed elsewhere herein, could have a material adverse effect on our business.
| 20 |
Our current comprehensive wound and skin careskincare strategy involves growth through acquisitions and investments, which requires us to incur substantial costs and potential liabilities for which we may never realize the anticipated benefits.
We may be unable to continue implementing our growth strategy, and our strategy ultimately may be unsuccessful. We engage in evaluations of potential acquisitions and investments and are in various stages of discussion regarding possible acquisitions, certain of which, if consummated, could be significant to us. Any new acquisition or investment could result in material transaction expenses, increased interest and amortization expense, increased depreciation expense and increased operating expense, any of which could have a material adverse effect on our operating results. In addition, if we are unable to integrate businesses and operations that we have acquired or will acquire in the future, our profitability could suffer. These acquisitions and investments also involve other risks, including diversion of management resources otherwise available for the running of our business and the development of our business, as well as risks associated with entering markets in which our marketing teams and sales force has limited experience or where experienced distribution alliances are not available. We may not be able to identify suitable acquisition or investment candidates in the future, obtain acceptable financing or consummate any future acquisitions or investments. In addition, certain potential acquisitions may be subject to antitrust and competition laws, which could impact our ability to pursue strategic acquisitions and could result in mandated divestitures. If we are unsuccessful in our current strategy to expand intocomprehensive wound and skin care virtual consult and other services,skincare strategy, we may be unable to meet our financial targets and our financial performance could be materially and adversely affected.
Failure to manage our growth strategy could harm our business.
Our ability to successfully implement our business plan and develop, market and sell our surgical, wound and skin careskincare products, services and servicestechnologies requires an effective plan for managing our future growth. We plan to increase the scope of our operations at a rapid rate. Future expansion efforts will be expensive and may strain our internal operating resources. To manage future growth effectively, we must maintain and enhance our financial and accounting systems and controls, integrate new personnel and manage expanded operations. If we do not manage growth properly, it could harm our operating results and financial condition.
We operatemay fail to realize all the anticipated benefits of the Applied Asset Purchase, or such benefits may take longer to realize than expected.
We may fail to realize all of the anticipated benefits of the Applied Asset Purchase, including expected operational, research and development and cost synergies, or such benefits may not be achieved within the anticipated timeframe. In addition, in highly competitive marketsconnection with the Applied Asset Purchase, we acquired control of the manufacturing of our CellerateRX Surgical and face competition from large, well-established medical device manufacturersHYCOL products, which was previously controlled by the Sellers. If we are unable to maintain or replace the Sellers’ manufacturing process, our sales of CellerateRX Surgical and telehealth providers as well as new market entrants,HYCOL products could be jeopardized.
Furthermore, we have incurred significant transaction costs in connection with the Applied Asset Purchase, including payment of certain fees and expenses incurred in connection with the Applied Asset Purchase and the financing of the Applied Asset Purchase, and our future financial results could be impacted if the intangible assets we acquired in the Applied Asset Purchase become impaired. The failure to realize the anticipated benefits of the Applied Asset Purchase or any of our other recent acquisitions could cause an interruption of, or a loss of momentum in, our operations and could result in a material adverse effect on our financial condition, results of operations or cash flows.
If we are unable to compete within our markets or our products, services and servicestechnologies do not gain market acceptance, our operating results and financial condition could suffer.
Competition from other medical device companies is significant and we could be significantly affected by new product introductions and other activities of market participants. We compete with other companies in acquiring rights to products or technologies from third-party developers. In addition, many specialized products companies have formed collaborations with large, established companies to support research, development and commercialization of wound care products which may be competitive with ours. Academic institutions, government agencies and other public and private research organizations are also conducting research activities and may commercialize wound care products on their own or through joint ventures. Although our products have performed well in customer evaluations, we are a relatively unknown brand in a market dominated by companies with extensive product lines and large customer bases. We may not, even with more efficacious products, be able to secure contracts and achieve significant growth with large national accounts.
In addition, if wethe anticipated full launch of our wound and skin careskincare virtual consult and other service offerings is successful, we will face competition from other telehealth providers. The public health emergency caused by the COVID-19 pandemic has led to the widespread adoption of telemedicine for most health care clinical specialties, including wound care and dermatology. As such, any clinical wound care or dermatology physician and/or provider group that has incorporated telemedicine into their practice could be considered competitive. If we are unable to compete with other telehealth providers, our operating results and financial condition may suffer.
Several factors may limit the market acceptance of our products, services and services,technologies, including the timing of regulatory approvals and market entry relative to competitive products, services and services,technologies, the availability of alternative products, services and services,technologies, the price of our products, services and servicestechnologies relative to alternative products, services and services,technologies, the availability of third-party reimbursement and the extent of marketing efforts by third-party distributors or agents that we retain. There can be no assurance that ourOur products, services or services willtechnologies may not receive market acceptance in a commercially viable period of time, if at all. Furthermore, there can be no assurance that we canour competitors may develop products, and services or technologies that are more effective or achieve greater market acceptance than competitive products and services, or that our competitors will not succeed in developing or acquiring products and technologies that are more effective than those being developed by us, thatwhich would render our products, services and technologies less competitive or obsolete.
| 21 |
Our competitors enjoy several competitive advantages over us, including but not limited to:
| ● | large and established distribution networks in the United States and/or in international markets; |
| ● | greater financial, managerial and other resources for products research and development, sales and marketing efforts and protecting and enforcing intellectual property rights; |
| ● | greater name recognition; |
| ● | larger consumer bases; |
| ● | more expansive portfolios of products and intellectual property rights; and |
| ● | greater experience in obtaining and maintaining regulatory approvals and/or clearances from the FDA and other regulatory agencies. |
The presence of competition in our market may lead to pricing pressure which would make it more difficult to sell our products, services and servicestechnologies at a profitable price or may prevent us from selling our products at all. Our failure to compete effectively would have a material adverse effect on our business.
Security breaches and other disruptions could compromise our information and expose us to liability, which would cause our business and reputation to suffer.
In the ordinary course of our business, we use networks to collect and store sensitive data, including intellectual property, proprietary business information and important information of our customers, suppliers and business partners, as well as personally identifiable information of our customers and employees. The secure processing, maintenance and transmission of this information is critical to our operations. Despite our security measures, our information technology and infrastructure may be vulnerable to attacks by hackers or breached due to employee error, malfeasance or other disruptions. Any such breach could compromise our networks and the information stored there could be accessed, publicly disclosed, lost or stolen. Any such access, disclosure or other loss of information could result in the loss of existing customers, difficulty in attracting new customers, backlash from negative public relations, legal claims or proceedings, liability under laws that protect the privacy of personal information, and regulatory penalties. Further, such access, disclosure or loss may cause disruption of our operations and the services we provide to customers, damage to our reputation, and cause a loss of confidence in our products and services, which could adversely affect our business.
We have programs, processes and technologies in place to prevent, detect, contain, respond to and mitigate security related threats and potential incidents. We undertake considerable ongoing improvements to our systems, connected devices and information-sharing products in order to minimize vulnerabilities, in accordance with industry and regulatory standards. Because the techniques used to obtain unauthorized access change frequently and can be difficult to detect, anticipating, identifying or preventing these intrusions or mitigating them if and when they occur, may be challenging.
If we fail to maintain an effective system of internal controls over financial reporting, we may not be able to accurately report our financial results or prevent fraud and our business may be harmed and our stock price may be adversely impacted.
Effective internal controls over financial reporting are necessary for us to provide reliable financial reports and to effectively prevent fraud. Any inability to provide reliable financial reports or to prevent fraud could harm our business. The Sarbanes-Oxley Act of 2002 (the “Sarbanes-Oxley Act”) requires management to evaluate and assess the effectiveness of our internal control over financial reporting. In order to comply with the requirements of the Sarbanes-Oxley Act, we are required to continuously evaluate and, where appropriate, enhance our policies, procedures and internal controls. If we fail to maintain the adequacy of our internal controls over financial reporting, we could be subject to litigation or regulatory scrutiny and investors could lose confidence in the accuracy and completeness of our financial reports. We cannot provide any assurance that in the future we will be able to fully comply with the requirements of the Sarbanes-Oxley Act or that management will conclude that our internal control over financial reporting is effective. If we fail to fully comply with the requirements of the Sarbanes-Oxley Act or our management concludes that our internal controls over financial reporting are not effective, our business may be harmed and our stock price may decline. In addition, because of its inherent limitations, internal controlcontrols over financial reporting may not prevent or detect misstatements.
| 22 |
Risks Related to Our Indebtedness
Our indebtedness could adversely affect our assessment, testingfinancial condition and evaluation of the design and operating effectivenessprevent us from fulfilling our obligations.
A significant portion of our internal control over financial reporting resulted infuture cash flow is required to pay interest and principal on our conclusion that as of December 31, 2019, our internal control over financial reporting was not effective, dueoutstanding indebtedness, and we may be unable to our small size and limited segregation of duties. As a result of such determination, we implemented additional controls, including the hiring of an additional full-time accounting professional in January 2020, which enabledgenerate sufficient cash flow from operations, or have future borrowings available, to enable us to properly segregate duties, and concluded that the material weakness was remediated as of June 30, 2020. While we have concluded that the material weakness has been remediated, projections of any evaluation of the effectiveness of internal control over financial reporting for future periods arerepay our indebtedness or to fund other liquidity needs. Among other consequences, this indebtedness could:
| ● | require us to use a significant percentage of our cash flow from operations for debt service and the satisfaction of repayment obligations, and not for other purposes, such as funding working capital and capital expenditures or making future acquisitions; | |
| ● | cause our interest expense to increase if there is a general increase in interest rates, because our indebtedness bears interest at floating rates; | |
| ● | limit our flexibility in planning for or reacting to changes in our business and limit our ability to exploit future business opportunities; and | |
| ● | cause us to be more highly leveraged than some of our competitors, which may place us at a competitive disadvantage. |
Our outstanding indebtedness is subject to the riskcertain operating and financial covenants that controlsrestrict our business and financing activities and may become inadequate because of changes in conditions or if the degree of compliance with the policies or procedures deteriorate over time.
The Loan Agreement governing our revolving linerequires us as guarantor and Sanara MedTech Applied Technologies, LLC, a wholly owned subsidiary of credit includes restrictive terms, and our failure to comply with any of these terms could result in a default, which would have an adverse effect on our business and prospects.
| ● | create or permit the existence of additional liens on our assets; | |
| ● | enter into any pledge agreements or arrangements; | |
| ● | with respect to SMAT, make investments in or provide capital contributions to other entities; | |
| ● | enter into a merger or consolidation or acquire any assets outside of the ordinary course of business; | |
| ● | dispose of a material part of our assets or property or enter into any sale-leaseback transactions; | |
| ● | with respect to SMAT, pay dividends or distributions of our capital stock, without the prior written consent of the Bank; | |
| ● | make or permit to remain outstanding any loans; | |
| ● | change the nature of our business; | |
| ● | enter into transactions with affiliates other than in the ordinary course of business on an arm’s-length basis; or | |
| ● | amend, modify or obtain or grant a waiver of any provision of any document or instrument evidencing or securing any of our subordinated indebtedness. |
In addition, starting with the quarter ending September 30, 2023, SMAT is required to maintain a minimum liquidityDebt Service Coverage Ratio of $1.0 million1.2 to 1.0, which ratio is calculated as of December 31, 2020 and March 31, 2021,the last day of the applicable fiscal quarter. SMAT is also required to maintain as of the end of each fiscal quarter a minimum Tangible Net Worth (as defined inmaximum Cash Flow Leverage Ratio of not more than (a) 4.5 to 1.0 as of the Loan Agreement)last day of $1.0 million and, beginning with the fiscal quarter ending on September 30, 2023, (b) 4.0 to 1.0 as of the last day of each fiscal quarter ending on December 31, 2023, and March 31, 2024, (c) 3.5 to 1.0 as of the last day of each fiscal quarter ending on June 30, 2021,2024, and September 30, 2024, and (d) 3.0 to 1.0 as of the last day of each fiscal quarter thereafter.
Further, in the event that SMAT fails to comply with its financial covenants, we are required to contribute cash to SMAT in an amount equal to the amount required to satisfy the financial covenants. During the three months ended September 30, 2023 and December 31, 2023, we contributed an immaterial amount of cash to SMAT to ensure that SMAT was in compliance with the financial covenants under the Loan Agreement as of September 30, 2023 and December 31, 2023. If we are required to make additional cash contributions to SMAT in future periods, we may not have sufficient funds available to fulfill such obligation.
A breach of any of the covenants under our loan agreements, subject to certain cure periods, will result in an event of default, which could cause all of our outstanding indebtedness under the Loan Agreement to become immediately due and payable, and a minimum Interest Coverage Ratio (as defineddefault interest rate of an additional 5.0% per annum may be applied to the outstanding loan balance. If our indebtedness is accelerated, we cannot be certain that we will have sufficient funds available to pay the accelerated indebtedness or that we will have the ability to refinance the accelerated indebtedness on terms favorable to us or at all.
As a result of the negative covenants contained in the Loan Agreement)Agreement and the need for cash to pay interest and principal amortization payments required under the Loan Agreement, the Company may not have access to cash it otherwise would have for working capital, capital expenditures, acquisitions, general corporate purposes or other purposes.
Pursuant to the Loan Agreement, SMAT is subject to restrictions on making certain payments, including dividends, other distributions and loans, to the Company. As a result, although SMAT may use its cash to repay the Term Loan, the Company may not have access to cash it otherwise would have for working capital, capital expenditures, acquisitions, general corporate purposes or other purposes.
| 23 |
If our Executive Chairman ceases to serve as the Chairman of 1.5 to 1.0. A breachthe Board of these covenantsDirectors of the Company or SMAT, such event could result in aan event of default under the Loan Agreement. If amounts owedAgreement.
The Loan Agreement provides that if Ron Nixon, our Executive Chairman, ceases to serve as Chairman of the Board of Directors of the Company or SMAT, an event of default will occur under the Loan Agreement, are accelerated becauseunless a successor, acceptable to the Bank, is elected or appointed within 10 business days of a defaultsuch event. If such an event were to occur and we are unable to pay such amounts, Cadence may havefind a replacement suitable to the right to assume control of substantiallyBank, we would be in default and all of our assets.
Risks Related to Our Products
We rely heavily on our research and development partners to design, manufacture and supply the products we have licensed for marketing. If we or one of our partners fails to perform adequately, or fulfill our needs, or comply with regulations, we may be required to incur significant costs.costs or even be subject to enforcement actions. We may also face significant delays in our product introductions and commercialization.
While we expect to have the capability to develop certain of our pipeline in-house, we do not currently own any facility that may be used as a manufacturing and processing facility, and wetherefore rely heavily on our research and development partners, from whom we license allmost of the products we currently commercialize, to design, manufacture and supply such products.
We and our research and development partners responsible for manufacturing our products and their contract manufacturers are obliged to operate in accordance with FDA’s current good manufacturing practices (“cGMP”), current good tissue practices (“cGTP”), and the Quality System Regulation (“QSR”),QSR, as applicable, as well as other regulations applicable to medical product manufacturers. The manufacture of regulated medical products in compliance with cGMP, cGTP, and the QSR, as applicable, requires significant expertise and capital investment, including the development of advanced manufacturing techniques and process controls. Manufacturers of medical products often encounter difficulties in production, including difficulties with production costs and yields, quality control, including product stability and quality assurance testing, shortages of qualified personnel, as well as compliance with strictly enforced regulatory requirements, other federal and state regulatory requirements and foreign regulations. If we or our research and development partners or their contract manufacturers were to encounter any of these difficulties or otherwise fail to comply with their obligations to us or under applicable regulations, our ability to commercialize our products would be jeopardized. Any delay or interruption in
We and the supplymanufacturers of products could have a material adverse effect on our business.
Our revenue generated from the sale of Scendia products is heavily dependent on license agreements with certain manufacturers, and the termination of any of these license agreements could harm our business.
In July 2022, we entered into a membership interest purchase agreement with Scendia and the Seller, pursuant to which we acquired 100% of the issued and outstanding membership interests in Scendia from the Seller. Scendia provides clinicians and surgeons with a full line of regenerative and orthobiologic technologies including (i) TEXAGEN, (ii) BiFORM, (iii) ACTIGEN and (iv) ALLOCYTE. We rely on license agreements with certain manufacturers in order to sell Scendia products. These license agreements are nonexclusive and generally have a term between one and five years. The license agreements are subject to renewal; however, the manufacturers may determine not to renew the agreements or to terminate the contracts pursuant to their terms. We cannot be certain that these license agreements will continue to be available to us or will be available to us on reasonable terms. If any of these agreements are terminated, we may be unable to reacquire the necessary license on satisfactory terms or at all. The loss of, or inability to maintain, any of these license agreements could negatively impact our ability to sell Scendia products, which could have a material adverse effect on our business, financial condition and results of operations.
| 24 |
Certain of our product candidates are still under development, and we may not be able to successfully commercialize any of these product candidates.
Our pipeline contains products and product candidates for mitigation of opportunistic pathogens and biofilm, wound re-epithelialization and closure, necrotic tissue debridement, and cell compatible substrates. We may also decide to develop other product candidates. Certain of our research and development programs are in developmental stages. One or more of our product candidates may fail to meet safety and efficacy standards in human testing, even if those product candidates are found to be effective in animal studies. To develop and commercialize product candidates, we must provide the FDA and foreign regulatory authorities with human clinical and nonclinical animal data that demonstrate adequate safety and effectiveness. To generate this data, we will have to subject our product candidates to significant additional research and development efforts, including extensive nonclinical studies and clinical testing. Our approach to product discovery may not be effective or may not result in the development of any product. It can take several years for a product to be approved and we may not be successful in bringing any therapeutic candidates to the market. A new product candidate may appear promising at an early stage of development or after clinical trials and never reach the market, or it may reach the market and not sell, for a variety of reasons. For example, the product may:
| ● | be shown to be ineffective or to cause harmful side effects during preclinical testing or clinical trials; | |
| ● | fail to receive regulatory approval on a timely basis or at all; | |
| ● | be difficult to manufacture on a large scale; | |
| ● | not be economically viable; | |
| ● | not be prescribed by doctors or accepted by patients; | |
| ● | fail to receive a sufficient level of reimbursement from government, insurers or other third-party payors; or | |
| ● | infringe on intellectual property rights of any other party. |
If our delivery platform technologies or product development efforts fail to generate product candidates that lead to the successful development and commercialization of products, or if the product candidates we have (or may in the future) acquired are not approved or cleared for commercialization in the United States or, otherwise experience adverse regulatory action, our business and financial condition will be materially adversely affected.
Our future success will largely depend on our ability to maintain and further grow clinical acceptance and adoption of our products, and we may be unable to adequately educate healthcare practitioners on the use and benefits of our products.
Healthcare practitioners play a significant role in determining the course of a patient’s treatment and, ultimately, the type of products, if any, that will be used to treat the patient. As a result, our commercial success is heavily dependent on our ability to educate practitioners on the use of our products in both surgical and post-acute care settings. Acceptance and adoption of our products in our markets depends on educating healthcare practitioners as to the distinctive characteristics, benefits, safety, clinical efficacy and cost-effectiveness of our products, including potential comparisons to our competitors’ products, and on training healthcare practitioners in the proper application of our products. If we are not successful in convincing healthcare practitioners of the merits and advantages of our products compared to our competitors’ products, they may not use our products and we will be unable to increase our sales and sustain growth or profitability.
Convincing healthcare practitioners to dedicate the time and energy necessary to properly train to use new products and techniques is challenging as healthcare practitioners may be hesitant to change their medical practices, and we may not be successful in these efforts. In particular, as healthcare resources are strained due to the ongoing COVID-19 pandemic, it may be more difficult to convince healthcare practitioners to commit their time and resources to learning to use a new product. If healthcare practitioners are not properly trained, they may use our products ineffectively, resulting in unsatisfactory patient outcomes, negative publicity or lawsuits against us. Accordingly, even if our products show superior benefits, safety or efficacy, based on head-to-head clinical trials, in comparison to alternative treatments, our success will depend on our ability to gain and maintain market acceptance for our products. If we fail to do so, our sales will not grow and our business, financial condition and results of operations will be adversely affected. We may not have adequate resources to effectively educate the medical community and our efforts may not be successful due to physician resistance or negative perceptions regarding our products.
Disruption of, or changes in, our distribution model or customer base could harm our sales and margins.
If we fail to manage the distribution of our products properly, or if the financial condition or operations of our reseller channels weaken, there may be a material adverse effect on our business. Furthermore, a change in the mix of our customers between service provider and enterprise, or a change in the mix of direct and indirect sales, could adversely affect our business.
| 25 |
Interruptions in the supply of our products or inventory loss may adversely affect our business, financial condition and results of operations and financial condition.
Our products are manufactured using technically complex processes requiring specialized facilities, highly specific raw materials and other production constraints. The complexity of these processes, as well as strict company and government standards for the manufacture and storage of our products, subjects us to production risks. In addition to ongoing production risks, process deviations or unanticipated effects of approved process changes may result in non-compliancenoncompliance with regulatory requirements including stability requirements or specifications. Most of our products must be stored and transported within a specified temperature range. For example, if environmental conditions deviate from that range, our products’ remaining shelf-lives could be impaired or their safety and efficacy could be adversely affected, making them unsuitable for use. These deviations may go undetected. Severe weather conditions and natural disasters may make compliance with these processes and maintenance of these standards more difficult, and climate change threatens more extreme weather events, which could increase our production risks. The occurrence of actual or suspected production and distribution problems can lead to lost inventories, and in some cases recalls, with consequential reputational damage and the risk of product liability. The investigation and remediation of any identified problems can cause production delays and result in substantial additional expenses. Any unforeseen failure in the storage of our products or loss in supply could result in a loss of our market share and negatively affect our revenues and operations.
In addition, Scendia operates a state-licensed and FDA registered tissue bank for the storage and distribution of allograft tissue-based products to hospitals, surgery centers and clinicians across the United States. During the third quarter of 2022, we began to experience supply issues with the ALLOCYTE product line. From the fourth quarter of 2022 through September or 2023, we were unable to fill certain orders for this product, which negatively impacted our sales growth. The supply constraint was caused by significant supplier limits on qualifying eligible donor tissue and supplier necessity to subcontract all processing to secondary suppliers. While we have since expanded the ALLOCYTE product line with the release of ALLOCYTE Plus, which is processed by an alternative supplier with in-house processing capabilities, any prolonged disruptions to the supply of our products or inventory could materially affect our business, financial condition and results of operations.
If we are unable to manage product inventory in an effective and efficient manner, our profitability could be impaired.
Many factors affect the efficient use and planning of product inventory, such as effectiveness of predicting demand, effectiveness of preparing manufacturing to meet demand, efficiently meeting product mix and product demand requirements and product expiration. Our products have a shelf life of between 18 months and 5 years. If we are unable to manage our product inventory efficiently or within expected budget goals or keep sufficient finished and in-process product on hand to meet demand, our operating margins and long-term growth prospects could be impaired.
We place orders with our suppliers based on forecasts of demand and, in some instances, may acquire additional inventory to accommodate anticipated demand. Our forecasts are based on management’s judgment and assumptions, each of which may introduce error into our estimates. If we overestimate customer demand, our excess or obsolete inventory may increase significantly, which would reduce our gross margin and adversely affect our financial results. Conversely, if we underestimate customer demand or if insufficient manufacturing capacity is available, we would miss revenue opportunities and potentially lose market share and damage our customer relationships.
Failure of any third-party assessments to demonstrate desired outcomes in proposed endpoints could have a negative impact on our business performance.
Our collaborators regularly conduct clinical studies designed to test a variety of endpoints associated with product performance and use across a number of applications. If a clinical study conducted by us or our collaborators fails to demonstrate statistically significant results supporting performance, use benefits or compelling health economic outcomes from using our products, physicians may elect not to use our products as a treatment for conditions that may benefit from them. Furthermore, in the event of an adverse clinical study outcome, our products may not achieve “standard-of-care” designations, where they exist, for the conditions in question, which could deter the adoption of our products. Also, if serious adverse events are reported during the conduct of a study, it could affect continuation of the study, product marketing authorization by regulatory authorities and product adoption by healthcare professionals or could cause regulatory authorities to impose other restrictions on the product or require additional warning or precaution statements to appear on the product labeling. If we or our collaborators are unable to develop a body of statistically significant evidence from our clinical studies, whether due to adverse results or the inability to complete properly designed studies, public and private payors could refuse to cover our products, limit the manner in which they cover our products, or reduce the price they are willing to pay or reimburse for our products.
Increased prices for, or unavailability of, raw materials used in our products could adversely affect our business, financial condition and results of operations and financial condition.
Our profitability is affected by the prices of the raw materials used in the manufacture of our products. These prices may fluctuate based on a number of factors beyond our control, including changes in supply and demand, general economic conditions, labor costs, fuel related delivery costs, competition, import duties, excises and other indirect taxes, currency exchange rates, and government regulation. Due to the highly competitive nature of the healthcare industry and the cost containment efforts of our customers and third-party payors, we may be unable to pass along cost increases for key components or raw materials through higher prices to our customers. If the cost of key components or raw materials increases, and we are unable fully to recover these increased costs through price increases or offset these increases through other cost reductions, we could experience lower margins and profitability. Significant increases in the prices of raw materials that cannot be recovered through productivity gains, price increases or other methods could adversely affect our business, results of operations and financial condition.
| 26 |
Risks Related to Our Planned Expansion into Wound and Skin CareSkincare Virtual Consult and Other Services
Our planned expansion into wound and skin careskincare virtual consult and other services, including THP programs, could have a material adverse effect on our business, financial condition orand results of operations.
Our planned expansion into wound and skin careskincare virtual consult and other services, including our planned THP programs, subjects us to risks associated with the use of new and novel technologies, operational, financial, regulatory, legal and reputational risks, as well as the risk that we may be unable to timely or successfully launch our service offerings. The success of UWSS’sthese operations depends upon our ability to commercialize UWSS’sour service offerings, and our failure to do so could negatively affect our ability to generate revenue from these activities.
Our planned expansion into wound and skin careskincare virtual consult and other services will require entrance into several markets in which we have little or no experience, which may not be successful and could be costly.
As part of our planned expansion into wound and skin careskincare virtual consult services, we will be required to enter into other markets in which we have little to no experience, including EMR, telehealth and healthcare diagnostics. While we intend to expand our staff with individuals with more experience in the EMR, telehealth and diagnostic markets and will closely scrutinize individuals we engage, we cannot provide assurance that we willmay not be able to retain or continue to hire well-qualified and experienced individuals or that our assessment of individuals we retain willmay not be accurate. As we enter new markets, we will face new technological and operational risks and challenges with which we are unfamiliar and may incur significant costs. Entering new markets requires substantial management efforts and skills to mitigate these risks and challenges. Our lack of experience with certain of these new markets may result in unsuccessful new market entries. If we do not manage our entry into new markets properly, these costs and risks could harm our business, financial condition or results of operations.
Our planned expansion into the telehealth business, is dependent on our relationships with affiliated professional entities to provide physician services, and our business would be adversely affected if those relationships were disrupted.
There is a risk that U.S. state authorities in some jurisdictions may find that any future contractual relationships we enter into with our affiliated professional entities who provide telehealth services violate laws prohibiting the corporate practice of medicine and professional fee-splitting laws. These laws generally prohibit the practice of medicine by lay persons or entities or sharing of professional fees with lay persons and are intended to prevent unlicensed persons or entities from interfering with or inappropriately influencing a physician’s professional judgment. The corporate practice of medicine prohibition exists in some form, by statute, regulation, board of medicine or attorney general guidance, or case law, in most states and is subject to change and to evolving interpretations by state boards of medicine, state attorneys general and state courts. As such, we will be required to continually monitor our compliance with laws in every jurisdiction in which we plan to operate, and we cannot guarantee that subsequent interpretation of the corporate practice of medicine laws will not circumscribe our future business operations. State corporate practice of medicine doctrines could also subject physicians to penalties for aiding the corporate practice of medicine, which could discourage physicians from participating in our network of providers.
Due to the prevalence of the corporate practice of medicine doctrine, including in the states where we plan to conduct our telehealth business, we expect to continue contracting with provider-entities through management services agreements. Although we expect that these relationships will continue, we cannot guarantee that they will. A material change in our relationships with these provider entities, whether resulting from a dispute among the parties, a change in government regulation, or the loss of these affiliations, could impair our ability to provide services to our future clients and could have a material adverse effect on our business, financial condition and results of operations. Any scrutiny, investigation or litigation with regard to our future arrangements with these professional entities could have a material adverse effect on our business, financial condition, and results of operations.
Recent and frequent state legislative and regulatory changes specific to telemedicine may present us with additional requirements and state compliance costs, with potential operational impacts in certain jurisdictions.
The state laws and regulations specific to telemedicine vary from state to state and are continually evolving. In some cases, these laws and regulations target “direct to consumer” telehealth service offerings rather than specialty consultative services, such as our planned acute telemedicine service offerings, and incorporate informed consent, modality, medical records and follow up care and other requirements. Thus, where new legislation and regulations apply to our planned expansion into telemedicine services, we may incur costs to monitor, evaluate, and modify operational processes for compliance. All such activities will increase our costs and could, in certain circumstances, impact our ability to make telemedicine services available in a particular state.
| 27 |
Risks Related to Intellectual Property
If we are unable to adequately protect our intellectual property rights, we may not be able to compete effectively.
Part of our success depends on our and our research development partners’ ability to protect proprietary rights to technologies used in certain of our products. We and our research development partners rely on patents, copyrights, trademarks and trade secret laws to establish and maintain proprietary rights in our technology and products. However, these legal means afford only limited protection and may not adequately protect our or our research development partners’ rights or permit us to gain or keep a competitive advantage. Patents and patent applications for the products we have may not be sufficient or broad enough to prevent competitors from introducing similar products into the market. Our or our research development partners’ patents or attempts to enforce them may not be upheld by the courts.courts and the damages or other remedies awarded if we were to prevail in upholding such patents may not be commercially meaningful. Efforts to enforce any of our or our research development partners’ proprietary rights could be time-consuming and expensive, which could adversely affect our business and prospects and divert management’s attention. There can be no assurance that our or our research and development partners’ proprietary rights will not be challenged, invalidated or circumvented or that such rights will in fact provide competitive advantages to us.
Furthermore, the issuance of a patent, while presumed valid and enforceable, is not conclusive as to its validity or its enforceability and it may not provide us with adequate proprietary protection or competitive advantages against competitors with similar products. Competitors may also be able to design around our patents. Other parties may develop and obtain patent protection for more effective technologies, designs or methods. In addition, we may not be able to prevent the unauthorized disclosure or use of our technical knowledge or trade secrets by consultants, vendors, former employees and current employees.
Patent rights are territorial, and patent protection extends only to those countries where we have issued patents. Filing, prosecuting and defending patents on our products and product candidates in all countries and jurisdictions throughout the world would be prohibitively expensive, and our intellectual property rights in some countries outside the United States could be less or more extensive than those in the United States, and their litigation processes differ. Competitors may successfully challenge or avoid our patents or manufacture products in countries where we have not applied for patent protection. Changes in the patent laws in the United States or other countries may diminish the value of our patent rights. As a result of these and other factors, the scope, validity, enforceability, and commercial value of our and our research development partners’ patent rights are uncertain and unpredictable.
The patent positions of life sciences companies, including our and our research development partners’ patent positions, involve complex legal and factual questions, and, therefore, the issuance, scope, validity and enforceability of any patent claims that we and our research development partners may obtain cannot be predicted with certainty. Patents, if issued, may be challenged, deemed unenforceable, invalidated, or circumvented. A third-party may submit prior patents, or we may become involved in opposition, derivation, reexamination, inter partes review, post-grant review, supplemental examination, or interference proceedings challenging our patent rights or the patent rights of our licensors or development partners. The costs of defending or enforcing our proprietary rights in these proceedings can be substantial, and the outcome can be uncertain. An adverse determination in any such submission or proceeding could reduce the scope of, or invalidate, our patent rights, allow third parties to commercialize our technology or products and compete directly with us, or reduce our ability to manufacture or commercialize products. Furthermore, if the scope or strength of protection provided by our patents and patent applications is threatened, it could discourage companies from collaborating with us to license, develop or commercialize current or future products. The ownership of our proprietary rights could also be challenged.
Our and our research development partners’ ability to enforce our respective patent rights depends on the ability to detect infringement. It is difficult to detect infringers who do not advertise the components that are used in their products. Moreover, it may be difficult or impossible to obtain evidence of infringement in a competitor’s or potential competitor’s product, particularly in litigation in countries other than the United States that do not provide an extensive discovery procedure.
CellerateRX Surgical is not currently protected by any pending patent application noror any unexpired patent. Accordingly,Currently the substantial majority of our net revenue is derived from the sale of CellerateRX Surgical. CellerateRX Surgical may be subject to competition from the sale of substantially equivalent products that could adversely affect our business and operations.
Our CellerateRX Surgical hasproducts, from which we derive a substantial majority of our net revenue, have no patent protection, and therefore, in order to continue to obtain commercial benefits from CellerateRX Surgical, we will rely on product manufacturing trade secrets, know-how and related non-patentnonpatent intellectual property.property, such as potential regulatory rights that would require various resources to separately obtain. The effect of CellerateRX Surgical’s lack of patent protection depends, among other things, upon the nature of the market and the position of our products in the market from time to time, the size of the market, the complexities and economics of manufacturing a competitive product and applicable regulatory approval requirements. In the event that competition develops substantially equivalent products, this competition could have a material adverse effect on our business, financial condition and operating results. Trade secret protection is effective only against wrongful acquisition, use or disclosure of confidential information. A competitor can avoid a claim of trade secret misappropriation by showing independent development without use of a trade secret owner’s information, however, this typically requires some time, effort and financial resources to develop independently. The entrance into the market of a product substantially equivalent to CellerateRX Surgical may erode our product’s market share, which may have a material adverse effect on our business, financial condition and results of operations.
| 28 |
We are heavily dependent on technologies and products we have licensed from third parties, and we may need to license technologies and products in the future, and if we fail to obtain licenses we need, or fail to comply with our payment and other obligations in the agreements under which we in-license intellectual property and other rights from third parties, we could lose our ability to develop and commercialize our products.
We are heavily dependent on licenses from our research and development partners for allmost of our technologies and products and are party to a sublicense agreement with CGI Cellerate RX, license agreements with Rochal and a marketing and distribution agreement with Cook Biotech. Our sublicense agreement and license agreements require that we comply with various obligations and provisions, and that we pay royalties to the sublicensor or licensor, as applicable, based on our net sales of the sublicensed and licensed products.
No assurance can be given that our existing sublicense agreement, license agreements or marketing and distribution agreement will continue to be extended on reasonable terms or at all. In addition, we expect we will need to license intellectual property, technology and products from third parties in the future and that these licenses will be material to our business. No assurance can be given that we will meet our minimum performance obligations or generate sufficient revenue or raise additional financing to meet our payment obligations in our agreements with CGI Cellerate RX, Rochal and Cook Biotech or other license or marketing and distribution agreements we enter into with third parties in the future. Any failure to meet our minimum performance obligations or make the payments required by our agreements may permit the licensor or supplier to terminate the agreement. If we were to lose or otherwise be unable to maintain these licenses or marketing and distribution agreements for any reason, it would halt our ability to commercialize one or more of our pipeline products. Furthermore, such loss of these licenses or marketing and distribution agreements may enable development of new products that may compete with our pipeline products, and our competitors may gain proprietary position. Any of the foregoing could result in a material adverse effect on our business or results of operations.
In certain cases, we may rely on our licensors to conduct prosecution, maintenance and/or defense of patents or trademarks on our behalf. Our ability to ensure that these patents and trademarks are properly prepared, prosecuted, maintained, enforce or defended is therefore limited, which may adversely affect our licensed intellectual property rights. Any failure by our licensors to properly prepare, prosecute, maintain, enforce, and defend patents or trademarks or other licensed rights could materially harm our ability to protect our products, thereby materially reducing our potential profits.
We may be found to infringe on or violate the intellectual property rights of others.
We may not have identified all patents, published applications or published literature that affect our business either by blocking our ability to commercialize our products or R&D candidates, by preventing the patentability of one or more aspects of our products or R&D candidates to us or our licensors, or by covering the same or similar technologies that may affect our ability to market our products and R&D candidates. For example, we (or the licensor of a product or R&D candidate to us) may not have conducted a patent clearance search sufficient to identify potentially obstructing third party patent rights. Moreover, patent applications in the United States are maintained in confidence for up to 18 months after their filing. In some cases, however, patent applications remain confidential in the USPTO, for the entire time prior to issuance as a U.S. patent. Patent applications filed in countries outside of the United States are not typically published until at least 18 months from their first filing date. Similarly, publication of discoveries in the scientific or patent literature often lags behind actual discoveries. We cannot be certain that we or our licensors were the first to invent, or the first to file, patent applications covering our products and candidates. We also may not know if our competitors filed patent applications for technology covered by our pending applications or if we were the first to invent the technology that is the subject of our patent applications. Competitors may have filed patent applications or received patents and may obtain additional patents and proprietary rights that block or compete with our patents.
Such third parties, including customers, may in the future assert claims or initiate litigation related to exclusive patent, copyright, trademark and other intellectual property rights to technologies and related standards that are relevant to us.us, our operations and our products. These assertions may emerge over time as a result of our growth and the general increase in the pace of patent claim assertions, particularly in the U.S.United States. Because of the existence of a large number of patents in the healthcare field, the secrecy of some pending patent applications and the rapid rate of issuance of new patents, we believe that it is not economically practical or even possible to determine in advance whether a product or any of its components infringes or will infringeis completely free of infringement of the patent rights of others.others even when we take reasonably objective steps to determine hat relevant patent rights might exist and, if so, to evaluate such patent rights relative to our proposed and actual products and methods with patent counsel. The asserted claims or initiated litigation can include claims against us or our manufacturers, suppliers or customers alleging infringement of their proprietary rights with respect to our existing or future products or components of those products. We may not have sufficient resources to bring these actions to a successful conclusion. In addition, intellectual property litigation or claims could force us to cease developing, selling or otherwise commercializing one or more of our products; to pay substantial damages for past use of the asserted intellectual property; to obtain a license from the holder of the asserted intellectual property, which may not be available on reasonable terms, if at all; and redesign, or rename in the case of trademark claims, our product(s) to avoid such third party rights, which may not be possible or which could be costly and time-consuming. Any of these risks coming to fruition could have a material adverse effect on our business, results of operations, financial condition and prospects. Regardless of the merit of these claims, they can be time-consuming, result in costly litigation and diversion of technical and management personnel, or require us to develop a non-infringingnoninfringing technology or enter into license agreements. Where claims are made by customers, resistance even to unmeritorious claims could damage customer relationships. There can be no assurance that licenses will be available on acceptable terms and conditions, if at all, or that our indemnification by our suppliers will be adequate to cover our costs if a claim were brought directly against us or our customers. Furthermore, because of the potential for high court awards that are not necessarily predictable and the resources required to engage in a full defense of such allegations, it is not unusual to find even arguably unmeritorious claims settled for significant amounts. If any infringement or other intellectual property claim made against us by any third party is successful, or if we fail to develop non-infringingnoninfringing technology or license the proprietary rights on commercially reasonable terms and conditions, our business could be materially and adversely affected.
| 29 |
Risks Related to Regulations
Our business is affected by numerous regulations relating to the development, manufacture, distribution, labeling, marketing and sale of our products.
Government regulation by the FDA and similar agencies in other countries is a significant factor in the development, manufacturing and marketing of our products and in the acquisition or licensing of new products. Complying with government regulations is often time consuming and expensive and may involve delays or actions adversely impacting the marketing and sale of our current or future products.
Following initial regulatory approval or clearance of any products that we or our research and development partners may develop, we and/or our research and development partners will be subject to continuing regulatory review, including, but not limited to:
| ● | appropriate establishment registration and product listing requirements; |
| ● | FDA’s cGMP, cGTP and QSR regulations, which govern the methods used in, and the facilities and controls used for, the design, manufacture, packaging, labeling, storage, installation, and servicing of finished devices, drugs and/or biologics, as applicable; |
| ● | FDA labeling requirements, which mandate the inclusion of certain content in medical product labels and labeling, and which also prohibit the promotion of products for uncleared or unapproved, i.e., “off-label,” indications; |
| ● | adverse event reporting regulations, which, generally, require applicable establishments (such as manufacturers and importers, among others) report to the FDA any adverse reactions, events, or experiences that meet the FDA’s reporting thresholds for the given product type (e.g., under FDA’s adverse-event reporting regulations under its device framework, adverse events must be reported if they may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if it were to recur); and |
| ● | the Reports of Corrections and Removals regulation, which requires that manufacturers and importers report to the FDA corrective actions and product removals (both of which are defined under applicable regulations) that meet the definition of a “recall” if undertaken to reduce a risk to health posed by the product or to remedy a violation of the FDCA that may present a risk to health and that manufacturers and importers keep records of recalls that they determine to be not reportable. |
Failure to comply with applicable regulatory requirements can result in, among other things, the FDA or other governmental authorities:
| ● | imposing fines and penalties on us; |
| ● | preventing us from manufacturing or selling our products; |
| ● | delaying or denying pending applications for approval or clearance of our products or of new uses or modifications to our existing products, or withdrawing or suspending current approvals or clearances; |
| ● | ordering or requesting a recall of our products; |
| ● | issuing warning letters, untitled letters, or “It has Come to Our Attention” letters; |
| ● | imposing operating restrictions, including a partial or total shutdown of production or investigation of any or all of our products; |
| ● | refusing to permit to import or export of our products; |
| ● | detaining or seizing our products; |
| ● | obtaining injunctions preventing us from manufacturing or distributing any or all of our products; |
| ● | commencing criminal prosecutions or seeking civil penalties; and |
| ● | requiring changes in our advertising and promotion practices. |
| 30 |
In addition, private consumer and penalties on us;
The manufacturing facilities we or our research and development partners use (and may use) to make any of our FDA-regulated products are or may become subject to periodic review and inspection by the FDA. If a previously unknown problem with a product or a manufacturing or laboratory facility used or contracted by us or one of our research and development partners is discovered, the FDA may impose restrictions on that product or on the manufacturing facility, including requiring us and/or our research and development partner to withdraw the product from the market. Any changes to an approved or cleared product, including the way it is manufactured or promoted, often requires FDA review and separate approval or clearance before the product, as modified, may be marketed. In addition, for products we develop in the future, we and our contract manufacturers may be subject to ongoing FDA requirements for submission of safety and other post-market approval information. If we violate regulatory requirements at any stage, whether before or after marketing approval or clearance is obtained, we may be fined, be forced to remove a product from the market or experience other adverse consequences, which would materially harm our financial results. Additionally, due to limitations imposed on us by the scope of the cleared or approved indications or intended use of our products and by FDA and Federal Trade Commission (“FTC”) regulations relating to promotional claims, we may not be able to obtain the labeling claims necessary or desirable for product promotion.
Distribution of our products outside the United States is subject to extensive government regulation. These regulations, including the requirements for marketing authorizations or product licenses necessary to bring a medical product to market, the time required for regulatory review and the sanctions imposed for violations, vary from country to country. We do not know whether we will obtain the marketing authorizations or product licenses necessary to market our products in such countries or that we will not be required to incur significant costs in obtaining or maintaining these regulatory approvals.
We are subject to various governmental regulations relating to the labeling, marketing and sale of our products.
Both before and after a product is commercially released, we have ongoing responsibilities under regulations promulgated by the FDA, the FTC, and similar U.S. and foreign regulations governing product labeling and advertising, distribution, sale and marketing of our products. Medical devices and biological products may only be marketed or promoted for the uses and indications set forth in the approved or cleared product labeling. A number of enforcement actions have been taken against companies that promoted products for “off-label” uses (i.e., uses that are not described in the approved or cleared labeling), including actions alleging that claims submitted to government healthcare programs for reimbursement of products that were promoted for “off-label” uses are fraudulent in violation of the Federal False Claims Act or other federal and state statutes and that the submission of those claims was caused by off-label promotion. The failure to comply with prohibitions on off-label promotion can result in significant monetary penalties, revocation or suspension of a company’s business license, suspension of sales of certain products, product recalls, civil or criminal sanctions, exclusion from participating in federal healthcare programs, or other enforcement actions. In the United States, allegations of such wrongful conduct could also result in a corporate integrity agreement with the U.S. government that imposes significant administrative obligations and costs.
If we fail to obtain or experience significant delays in obtaining regulatory clearances or approvals to market future medical device products, we will be unable to commercialize these products until such clearance or approval is obtained.
The developing, testing, manufacturing, marketing and selling of medical devices is subject to extensive regulation by governmental authorities in the U.S.United States and other countries. The process of obtaining regulatory clearance and approval of certain medical technology products is costly and time consuming. Inherent in the development of new medical products is the potential for delay because product testing, including clinical evaluation, is typically required, especially for drugs, biologics and high-risk devices, before such products can be approved for human use. With respect to medical devices, such as those that we currently market, before a new medical device, or a new indicated use of, or claim for, an existing product can be marketed (unless it is a Class I device), it must first receive either premarket clearance under Section 510(k) of the Federal Food, Drug and Cosmetic ActFDCA or approval of a PMA from the FDA, or be reclassified and receive marketing authorization through the de novo classification process, unless an exemption applies.
In the 510(k)-clearance process, the FDA must determine that the proposed device is “substantially equivalent” to a Class I or II device legally on the market, known as a “predicate” device, with respect to intended use, technology, safety and effectiveness to clear the proposed device for marketing. Clinical data is sometimes required to support substantial equivalence for certain device types. The PMA pathway requires an applicant to demonstrate the safety and effectiveness of the device for its intended use based, in part, on extensive data including, but not limited to, technical, preclinical, clinical trial, manufacturing and labeling data. The PMA process is typically required for devices that are deemed to pose the greatest risk, such as life-sustaining, life-supporting or implantable devices. If a device is novel and there is no appropriate predicate to which the applicant can demonstrate substantial equivalence, the device will be automatically classified as a Class III device and require approval through the PMA process prior to commercialization, unless the applicant submits a de novo classification request demonstrating that the novel device should be reclassified into Class I or II. Demonstrating that a novel device should be reclassified to Class I or II from Class III typically requires extensive information and data on the benefits and risks of the device, including performance data and frequently data from one or more clinical studies. The 510(k), PMA and de novo classification approval processes can be expensive and lengthy.
| 31 |
Failure to comply with applicable regulatory requirements can result in, among other things, suspension or withdrawal of clearances or approvals, seizure or recall of products, injunctions against the manufacture, holding, distribution, marketing and sale of a product and civil and criminal sanctions. Furthermore, changes in existing regulations or the adoption of new regulations could prevent us from obtaining, or affect the timing of, future regulatory clearances or approvals. Meeting regulatory requirements and evolving government standards may delay marketing of any new products developed by us for a considerable period of time, impose costly procedures upon our activities and result in a competitive advantage to larger companies that compete against us.
The FDA or other regulatory agencies willmay not clear or approve any products developed by us on a timely basis, ifor at all, or, if granted, that clearance or approval will notmay entail limiting the indicated uses for which we may market the product, which could limit the potential market for any of these products.
Delays in or changes to the FDA clearance and approval processes or ongoing regulatory requirements could make it more difficult for us to obtain FDA clearance or approval of new products or comply with ongoing requirements.
New government regulations may be enacted and changes in FDA policies and regulations and, their interpretation and enforcement, could prevent or delay regulatory clearance or approval of new products. We cannot predict the likelihood, nature or extent of adverse government regulation that may arise from future legislation or administrative action, either in the U.S.United States or abroad. Therefore, we do not know whether we or our research and development partners will be able to continue to comply with such regulations or whether the costs of such compliance will have a material adverse effect on our business. Changes could, among other things, require different labeling, monitoring of patients, interaction with physicians, education programs for patients or physicians, curtailment of necessary supplies, or limitations on product distribution. These changes, or others required by the FDA could have an adverse effect on our business, and specifically, on the sales of affected products. The evolving and complex nature of regulatory science and regulatory requirements, the broad authority and discretion of the FDA and the generally high level of regulatory oversight results in a continuing possibility that from time to time, we will be adversely affected by regulatory actions despite ongoing efforts and commitment to achieve and maintain full compliance with all regulatory requirements. If we or our research and development partners are not able to maintain regulatory compliance, we may not be permitted to market our products and our business would suffer.
Failure to obtain or maintain adequate reimbursement or insurance coverage for drugs, if any, could limit our ability to market those drugs and decrease our ability to generate revenue. Changes in reimbursement policies and regulations by governmental or other third-party payors may have an adverse impact on the use of our products.
The pricing, coverage, and reimbursement of our products, if any, must be sufficient to support our commercial efforts and other development programs, and the availability and adequacy of coverage and reimbursement by third-party payors, including governmental and private insurers, are essential for most patients to be able to afford medical treatments. Sales of our products depend substantially, both domestically and abroad, on the extent to which the costs of our products, if any, will be paid for or reimbursed by health maintenance, managed care, and similar healthcare management organizations, or government payers and private payors. If coverage and reimbursement are not available, or are available only in limited amounts, we may have to subsidize or provide medical products for free or we may not be able to successfully commercialize our products.
A significant portion of our wound care products are purchased principally for the Medicare and Medicaid eligible population by hospital outpatient clinics, wound care clinics, durable medical equipment (“DME”) suppliers and SNFs, which typically bill various third-party payors, primarily state and federal healthcare programs (e.g., Medicare and Medicaid), and managed care plans, for the products and services provided to their patients. Although the majority of our wound care products are currently eligible for reimbursement under Medicare Part B, adjustments to our reimbursement amounts or a change in CMS’s reimbursement policies could have an adverse effect on our market opportunities in this area. The ability of our customers to obtain appropriate reimbursement for products and services from third-party payors is critical to the success of our business because reimbursement status affects which products our customers purchase. In addition, our ability to obtain reimbursement approval in foreign jurisdictions may affect our ability to expand our product offerings internationally.
Third-party payors have adopted, and are continuing to adopt, a number of policies intended to curb rising healthcare costs. These policies include the imposition of conditions of payment by foreign, state and federal healthcare programs as well as private insurance plans, and the reduction in reimbursement amounts applicable to specific products and services.
Changes in healthcare systems in the U.S.United States or internationally in a manner that significantly reduces reimbursement for procedures using our products or denies coverage for these procedures would also have an adverse impact on the acceptance of our products and the prices which our customers are willing to pay for them.
Moreover, increasing efforts by governmental and private payors in the United States and abroad to limit or reduce healthcare costs may result in restrictions on coverage and the level of reimbursement for new medical products and, as a result, they may not cover or provide adequate payment for our products. We expect to experience pricing pressures in connection with our products due to the increasing trend toward managed healthcare, including the increasing influence of health maintenance organizations and additional legislative changes. The downward pressure on healthcare costs in general, and prescription drugs or biologics in particular, has and is expected to continue to increase in the future. As a result, profitability of our current or future products may be more difficult to achieve.
| 32 |
We rely heavily on our research and development partners to comply with applicable laws and regulations relating to product classification and when and what types of FDA marketing authorization.
We rely heavily on our research and development partners, from whom we license allmost of the products we currently commercialize, to determine the appropriate classification for each such product and to comply with applicable regulations related to obtaining the proper marketing authorization. With respect to each medical device product we license, our respective research and development partner designs the product and determines whether the device should be classified as a Class I, II, or III device and the appropriate FDA marketing authorization pathway to pursue (i.e., 510(k), PMA or de novo classification). In addition, we rely on our research and development partners to determine whether specific legal or regulatory definitions or exemptions apply to particular medical products, which individually may be subject to FDA oversight as a device, drug, biologic or human cellular or tissue-based product. The FDA has broad regulatory authority to interpret and enforce the laws and regulations that govern medical products in commercial distribution, and any adverse determination by the FDA relating to one of our licensed products could require significant cost and effort to comply.
For example, our research and development partner, Cook Biotech, from whom we have the right to exclusively distribute three biologic products for surgical and wound care applications, has determined that one such product, VIM Amnion Matrix, is intended for homologous use as a wound covering or barrier. It is possible that the FDA, after evaluating the product, itspromotional claims and other information pertinent to the FDA’s determination of the product’s intended use, and any promotional claims, may not agree with Cook Biotech’s conclusion that the VIM Amnion Matrix product is intended for homologous use, which would change the legal framework under which the product is regulated and may require Cook Biotech and us to incur substantial costs and expend significant effort to bring the product into compliance with applicable regulations. Such action by the FDA may also require us to cease marketing operations relating to the VIM Amnion Matrix product until the appropriate corrections are complete.
Similarly, some of the devices that we market under a license (or that we have acquired or have, otherwise, obtained commercialization rights in the United States) have been updated or modified since their initial 510(k) clearance. Depending on the nature of the updates or modifications made to a 510(k)-cleared device, the FDA may require the submission (and clearance) of a new 510(k). More specifically, any modification that could significantly affect the cleared device’s safety or effectiveness, or that would constitute a significant change in its intended use, will require a new 510(k) clearance. The FDA requires device manufacturers to make the initial determination as to whether a proposed modification to a cleared device requires a new 510(k) submission, but the FDA can review any such decision not to submit a new 510(k) (if it becomes aware of the modifications during an inspection or otherwise) and may disagree with the manufacturer’s determination that the given modification(s) did not require new clearance. If the FDA finds that a manufacturer has improperly marketed a modified device (for which the FDA has determined that a new 510(k) is required) under the original device’s 510(k), the FDA may mandate that the manufacturer cease marketing and/or recall the modified device until the requisite clearance is obtained, in addition to one or more other enforcement actions. FDA may disagree with our partners’ decisions not to submit new 510(k) notifications for those of our 510(k)-cleared devices that have been updated or modified since their initial clearance, in which case, we may be subject to a wide range of FDA enforcement actions, including, but not limited to, warning letters, fines, and other penalties, and our business will be adversely affected, as we would likely be required to cease commercialization (and, possibly, conduct a recall) of the modified product(s) at-issue and may incur additional expenses in connection with the preparation and submission of a new 510(k).
We and our employees and contractors are subject, directly or indirectly, to federal, state and foreign healthcare fraud and abuse laws, including false claims laws. If we are unable to comply, or have not fully complied, with such laws, we could face substantial penalties.
Our operations are subject to various federal, state and foreign fraud and abuse laws. These laws may affect our operations, including the financial arrangements and relationships through which we market, sell and distribute our products. U.S. federal and state laws that affect our ability to operate include, but are not limited to:
| ● | the federal Anti-Kickback Statute, which prohibits, among other things, persons or entities from knowingly and willfully soliciting, receiving, offering or paying any remuneration (including any kickback, bribe, or rebate), directly or indirectly, overtly or covertly, in cash or in kind in return for, the purchase, recommendation, leasing or furnishing of an item or service reimbursable under a federal healthcare program, such as the Medicare and Medicaid programs; |
| ● | federal civil and criminal false claims laws and civil monetary penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment or approval from Medicare, Medicaid, or other government payors that are false or fraudulent; |
| ● | Section 242 of HIPAA codified at 18 U.S.C. § 1347, which created new federal criminal statutes that prohibit a person from knowingly and willfully executing a scheme or from making false or fraudulent statements to defraud any healthcare benefit program (i.e., public or private); |
| ● | federal transparency laws, including the so-called federal “sunshine” law, which requires the tracking and disclosure to the federal government by pharmaceutical and medical device manufacturers of payments and other transfers of value to physicians and teaching hospitals as well as ownership and investment interests that are held by physicians and their immediate family members; and |
| ● | state law equivalents of each of these federal laws, such as anti-kickback and false claims laws that may apply to items or services reimbursed by any third-party payer, including commercial insurers, state laws that require pharmaceutical and medical device companies to comply with their industry’s voluntary compliance guidelines and the applicable compliance guidance promulgated by the federal government or otherwise restrict certain payments that may be made to healthcare providers and other potential referral sources, state laws that require drug and medical device manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures, state laws that prohibit giving gifts to licensed healthcare professionals and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts in certain circumstances, such as specific disease states. |
| 33 |
In particular, activities and arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, waste and other abusive practices. These laws and regulations may restrict or prohibit a wide range of activities or other arrangements related to the development, marketing or promotion of products, including pricing and discounting of products, provision of customer incentives, provision of reimbursement support, other customer support services, provision of sales commissions or other incentives to employees and independent contractors and other interactions with healthcare practitioners, other healthcare providers and patients.
Because of the breadth of these laws and the narrow scope of the statutory or regulatory exceptions and safe harbors available, our business activities could be challenged under one or more of these laws. Relationships between medical product manufacturers and health care providers are an area of heightened scrutiny by the government. We engage in various activities, including the conduct of speaker programs to educate physicians, the provision of reimbursement advice and support to customers, and the provision of customer and patient support services, that have been the subject of government scrutiny and enforcement action within the medical device industry.
Government expectations and industry best practices for compliance continue to evolve and past activities may not always be consistent with current industry best practices. Further, there is a lack of government guidance as to whether various industry practices comply with these laws, and government interpretations of these laws continue to evolve, all of which create compliance uncertainties. Any non-compliancenoncompliance could result in regulatory sanctions, criminal or civil liability and serious harm to our reputation. Although we have a comprehensive compliance program designed to ensure that our employees’ and commercial partners’ activities and interactions with healthcare professionals and patients are appropriate, ethical, and consistent with all applicable laws, regulations, guidelines, policies and standards, it is not always possible to identify and deter misconduct, and the precautions we take to detect and prevent this activity may not be effective in preventing such conduct, mitigating risks, or reducing the chance of governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations.
If a government entity opens an investigation into possible violations of any of these laws (which may include the issuance of subpoenas), we would have to expend significant resources to defend ourselves against the allegations. Allegations that we, our officers, or our employees violated any one of these laws can be made by individuals called “whistleblowers” who may be our employees, customers, competitors or other parties. Government policy is to encourage individuals to become whistleblowers and file a complaint in federal court alleging wrongful conduct. The government is required to investigate all of these complaints and decide whether to intervene. If the government intervenes and we are required to pay damages, which in such cases are typically set at three times the actual monetary damages, to the government, the whistleblower, as a reward, is awarded a percentage of such damages or any settlement amount. If the government declines to intervene, the whistleblower may proceed on hertheir own and, if she isthey are successful, shethey will receive a percentage of any judgment or settlement amount the company is required to pay. The government may also initiate an investigation on its own. If any such actions are instituted against us, those actions could have a significant impact on our business, including the imposition of significant fines, and other sanctions that may materially impair our ability to run a profitable business. In particular, if our operations are found to be in violation of any of the laws described above or if we agree to settle with the government without admitting to any wrongful conduct or if we are found to be in violation of any other governmental regulations that apply to us, we, our officers and employees may be subject to sanctions, including civil and criminal penalties, damages, fines, exclusion from participation in government health care programs, such as Medicare and Medicaid, imprisonment, the curtailment or restructuring of our operations and the imposition of a corporate integrity agreement, any of which could adversely affect our business, results of operations and financial condition.
We and our research and development partners’ use and disclosure of personally identifiable information,PII, including health information, is subject to federal and state privacy and security regulations, and our failure to comply with those regulations or to adequately secure the information we hold could result in significant liability or reputational harm and, in turn,have a material adverse effect on our client base, business, financial condition and results of operations.
Numerous state and federal laws and regulations, including HIPAA, govern the collection, dissemination, use, privacy, confidentiality, security, availability and integrity of PII, including protected health information. HIPAA establishes a set of basic national privacy and security standards for the protection of PHI by health plans, healthcare clearinghouses and certain healthcare providers, referred to as covered entities, and the business associates with whom such covered entities contract for services, which likely includes us. HIPAA requires healthcare providers and business associates to develop and maintain policies and procedures with respect to PHI that is used or disclosed, including the adoption of administrative, physical, and technical safeguards to protect such information. HIPAA also implemented the use of standard transaction code sets and standard identifiers that covered entities must use when submitting or receiving certain electronic healthcare transactions, including activities associated with the billing and collection of healthcare claims. HIPAA imposes mandatory penalties for certain violations. HIPAA also authorizes state attorneys general to file suit on behalf of their residents. Courts will be able to award damages, costs and attorneys’ fees related to violations of HIPAA in such cases. While HIPAA does not create a private right of action allowing individuals to sue us in civil court for violations of HIPAA, its standards have been used as the basis for duty of care in state civil suits such as those for negligence or recklessness in the misuse or breach of PHI.
| 34 |
In addition, HIPAA mandates that the Secretary of HHS conduct periodic compliance audits of HIPAA covered entities or business associates for compliance with the HIPAA Privacy and Security Standards. HIPAA further requires that patients be notified of any unauthorized acquisition, access, use or disclosure of their unsecured PHI that compromises the privacy or security of such information, with certain exceptions related to unintentional or inadvertent use or disclosure by employees or authorized individuals. HIPAA specifies that such notifications must be made without unreasonable delay and in no case later than 60 calendar days after discovery of the breach. If a breach affects 500 patients or more, it must be reported to HHS without unreasonable delay, and HHS will post the name of the breaching entity on its public web site. Breaches affecting 500 patients or more in the same state or jurisdiction must also be reported to the local media. If a breach involves fewer than 500 people, the covered entity must record it in a log and notify HHS at least annually.
Numerous other federal and state laws protect the confidentiality, privacy, availability, integrity and security of PII, including PHI. These laws in many cases are more restrictive than, and may not be preempted by, the HIPAA rules and may be subject to varying interpretations by courts and government agencies, creating complex compliance issues for us and our clients and potentially exposing us to additional expense, adverse publicity and liability. In addition, new health information standards, whether implemented pursuant to HIPAA, congressional action or otherwise, could have a significant effect on the manner in which we must handle healthcare related data, and the cost of complying with standards could be significant. If we do not comply with existing or new laws and regulations related to PHI, we could be subject to criminal or civil sanctions.
Because of the extreme sensitivity of the PII we or our partners may store and transmit, the security features of our technology platforms are very important. If our security measures, some of which may be managed by third parties, are breached or fail, unauthorized persons may be able to obtain access to sensitive client and patient data, including HIPAA-regulated PHI. As a result, our reputation could be severely damaged, adversely affecting client or investor confidence. Clients may curtail their use of or stop using our products and services, which would cause our business to suffer. In addition, we could face litigation, damages for contract breach, penalties and regulatory actions for violation of HIPAA and other applicable laws or regulations and significant costs for remediation, notification to individuals and for measures to prevent future occurrences. Any potential security breach could also result in increased costs associated with liability for stolen assets or information, repairing system damage that may have been caused by such breaches, incentives offered to client or other business partners in an effort to maintain our business relationships after a breach and implementing measures to prevent future occurrences, including organizational changes, deploying additional personnel and protection technologies, training employees and engaging third-party experts and consultants. While we maintain insurance covering certain security and privacy damages and claim expenses, our coverage may not be sufficient to compensate for all liability.
Our officers, employees, independent contractors, principal investigators, consultants and commercial partners may engage in misconduct or activities that are improper under other laws and regulations, which would create liability for us.
We are exposed to the risk that our officers, employees, independent contractors (including contract research organizations CROs)(“CROs”)), principal investigators, consultants and commercial partners may engage in fraudulent conduct or other illegal activity and/or may fail to disclose unauthorized activities to us. Misconduct by these parties could include, but is not limited to, intentional, reckless and/or negligent failures to comply with:
| ● | the laws and regulations of the FDA and its foreign counterparts requiring, among other things, compliance with good manufacturing practice and/or quality system requirements, post-market vigilance reporting, product marketing authorization requirements, facility registration requirements, the reporting of true, complete and accurate information to such regulatory bodies, including but not limited to safety problems associated with the use of our products; |
| ● | laws and regulations of the FDA and its foreign counterparts concerning the conduct of clinical trials and the protection of human research subjects, including but not limited to good clinical practices; |
| ● | other laws and regulations of the FDA and its foreign counterparts relating to the manufacture, processing, packing, holding, investigating or distributing in commerce of medical devices, biological products and/or HCT/Ps; |
| ● | manufacturing standards we have established; or |
| ● | healthcare fraud and abuse laws, including but not limited to, the Anti-Kickback Statute, the Stark Law, the FCA, and state law equivalents. |
| 35 |
In particular, companies involved in the manufacture of medical products are subject to laws and regulations intended to ensure that medical products that will be used in patients are safe and effective, and, specifically, that they are not adulterated or misbranded, that they are properly labeled, and have the identity, strength, quality and purity that which they are represented to possess. Further, companies involved in the research and development of medical products are subject to extensive laws and regulations intended to protect research subjects and ensure the integrity of data generated from clinical trials and of the regulatory review process. Any misconduct in any of these areas, whether by our own employees or by contractors, vendors, business associates, consultants, or other entities acting as our agents, could result in regulatory sanctions, criminal or civil liability and serious harm to our reputation. Although we have a comprehensive compliance program designed to ensure that our employees’, CRO partners’, principal investigators’, consultants’, and commercial partners’ activities and interactions with healthcare professionals and patients are appropriate, ethical, and consistent with all applicable laws, regulations, guidelines, policies and standards, it is not always possible to identify and deter misconduct, and the precautions we take to detect and prevent this activity may not be effective in preventing such conduct, mitigating risks, or reducing the chance of governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. If any such actions are instituted against us, or our CRO partners, principal investigators, consultants, or commercial partners, those actions could have a significant impact on our business, including the imposition of significant fines, and other sanctions that may materially impair our ability to run a profitable business.
We could be adversely affected if healthcare reform measures substantially change the market for medical care or healthcare coverage in the United States.
Third party payors, governmental authorities, and other applicable stakeholders have developed, and are continuing to develop, increasingly sophisticated methods of controlling healthcare costs. In both the Patient ProtectionUnited States and certain foreign jurisdictions, there have been numerous legislative and regulatory changes to the healthcare system that could impact our ability to sell our products profitably. In particular, the Affordable Care Act (P.L. 111-148) and on March 30, 2010, the signed the Health Care and Education Reconciliation Act (P.L. 111-152), collectively commonly referred to as the “Healthcare Reform Law.” The Healthcare Reform Law included a number of new rules regarding health insurance, the provision of healthcare, conditions to reimbursement for healthcare services provided to Medicare and Medicaid patients, and other healthcare policy reforms. Through the law-making process, substantial changes have been and continue to be made to the system for paying for healthcarewas enacted in the United States including changes made to extend medical benefits to certain Americans who lacked insurance coveragein 2010, and to containvarious analogous or reduce healthcare costs (such as by reducing or conditioning reimbursement amounts for healthcare services and drugs, and imposing additional taxes, fees, and rebate obligations on pharmaceutical and medical device companies). This legislation was one of the most comprehensive and significant reforms ever experienced by the healthcare industry in the United States and has significantly changed the way healthcare is financed by both governmental and private insurers. This legislation has impacted the scope of healthcare insurance and incentives for consumers and insurance companies, among others. Additionally, the Healthcare Reform Law’s provisions were designed to encourage providers to find cost savings in their clinical operations. This environment has caused changes in the purchasing habits of consumers and providers and resulted in specific attention to the pricing negotiation, product selection and utilization review surrounding pharmaceuticals and medical devices. This attention may result in our current commercial products, products we may commercialize or promote in the future, and our therapeutic candidates, being chosen less frequently or the pricing being substantially lowered. At this stage, it is difficult to estimate the full extent of the direct or indirect impact of the Healthcare Reform Law on us.
There have been, and likely will continue to be, legislative and regulatory proposals at the foreign, federal and state levels directed at containing or results of operations.
Defects, failures or quality issues associated with our products could lead to product recalls or safety alerts, adverse regulatory actions, product liability lawsuits and other litigation and negative publicity that could materially adversely affect our reputation, business, results of operations and financial condition.
Quality is extremely important to us and our customers due to the serious and costly consequences of product failure. Quality and safety issues may occur with respect to any of our products, and our future operating results will depend on our ability to maintain an effective quality control system and effectively train and manage our workforce with respect to our quality system. The development, manufacture and control of medical devicesproducts are subject to extensive and rigorous regulation by numerous government agencies, including the FDA and similar foreign agencies. Compliance with these regulatory requirements including but not limited to the FDA’s QSR, good manufacturing practices and adverse events/recall reporting requirements in the United States and other applicable regulations worldwide, is subject to continual review and is monitored rigorously through periodic inspections by the FDA and foreign regulatory authorities. The FDA and foreign regulatory authorities may also require post-market testing and surveillance to monitor the performance of products cleared or approved for use in their jurisdictions. Our manufacturing facilities and those of our suppliers and independent sales agencies are also subject to periodic regulatory inspections. If the FDA or other regulatory authority were to conclude that we or our suppliers have failed to comply with any of these requirements, it could institute a wide variety of enforcement actions, ranging from a public warning letter to more severe sanctions, such as product recalls or seizures, withdrawals, monetary penalties, consent decrees, injunctive actions to halt the manufacture or distribution of products, import detentions of products made outside the United States, export restrictions, restrictions on operations or other civil or criminal sanctions. Civil or criminal sanctions could be assessed against our officers, employees, or us. Any adverse regulatory action, depending on its magnitude, may restrict us from effectively manufacturing, marketing, and selling our products.
| 36 |
Relatedly, although we have contractual indemnity from the manufacturers of our current products for certain liability claims related to their production, we could face product liability lawsuits or other similar proceedings relating to actual or alleged injuries, defects, deficiencies, failures, and/or representations relating to our products that could fall outside of the scope of the contractual indemnities. We do not have, and do not anticipate obtaining, contractual indemnification from parties supplying raw materials or parties marketing the products we sell. In any event, indemnification from the manufacturers of our products or from any other party is limited by the terms of the indemnity and by the creditworthiness of the indemnifying party. A successful product liability or other applicable claim or series of claims brought against us could result in judgments, fines, damages and liabilities that could have a material adverse effect on our business. We may incur significant expense investigating and defending these claims, even if they do not result in liability. Moreover, even if no judgments, fines, damages or liabilities are imposed on us, our reputation could suffer as result of any such claim, which could have a material adverse effect on our business.
Product liability insurance for the healthcare industry may become prohibitively expensive, to the extent it is available at all. We may not be able to maintain such insurance on acceptable terms or be able to secure increased coverage as commercialization of our products progresses, nor can we be sure that existing or future claims against us will be covered by our product liability insurance. In the event that we do not have adequate insurance or contractual indemnification, product liability claims relating to defective products could have a material adverse effect on our business.
In addition, we cannot predict the results of future legislative activity or future court decisions, any of which could increase regulatory requirements, subject us to government investigations or expose us to unexpected litigation. Any regulatory action or litigation, regardless of the merits, may result in substantial costs, divert management’s attention from other business concerns and place additional restrictions on our sales or the use of our products. In addition, negative publicity, including regarding a quality or safety issue, could damage our reputation, reduce market acceptance of our products, cause us to lose customers and decrease demand for our products. Any actual or perceived quality issues may also result in issuances of physician’s advisories against our products or cause us to conduct voluntary recalls. Any product defects or problems, regulatory action, litigation, negative publicity or recalls could disrupt our business and have a material adverse effect on our business, results of operations and financial condition.
Risks Related to Ownership of Our Common Stock
The issuance of shares upon the exercise of derivative securities or as earnout payments may cause immediate and substantial dilution to our existing shareholders.
As of March 30, 2021,December 31, 2023, we had 11,500approximately 110,617 shares of common stock that were issuable upon the exercise of vested outstanding stock options.options and warrants. The issuance of shares upon the exercise of these stock options and warrants may result in dilution to the equity interest and voting power of holders of our common stock.
In the future, we may also issue additional shares of common stock or other securities convertible into or exchangeable for shares of common stock. For instance, certain of the product license agreements we have entered into with third parties require us to make payments to such third parties upon the occurrence of certain events. Pursuant to these product license agreements, we may choose to make such payments in shares of our common stock. In addition, the Precision Healing merger agreement and the Scendia membership interest purchase agreement require us to pay up to an aggregate of $20.0 million upon the achievement of certain performance thresholds. In each case and subject to certain limitations, the earnout consideration is payable in cash or, at our election, in shares of our common stock.
The issuance of additional shares of our common stock may substantially dilute the ownership interests of our existing shareholders. Furthermore, sales of a substantial amount of our common stock in the public market, or the perception that these sales may occur, could reduce the market price of our common stock. This could also impair our ability to raise additional capital through the sale of our securities.
It is possible that we will require additional capital to meet our financial obligations and support business growth.
We intend to continue to make significant investments to support our business growth and expect to require additional funds to respond to business challenges. Accordingly, we may need to engage in equity or debt financings to secure additional funds. If we raise additional funds through future issuances of equity or convertible debt securities, our existing shareholders could suffer significant dilution, and any new equity securities we issue could have rights, preferences and privileges superior to those of holders of our common stock. Any debt financing that we secure in the future could involve restrictive covenants relating to our capital raising activities and other financial and operational matters, which may make it more difficult for us to obtain additional capital and to pursue business opportunities, including potential acquisitions. We may not be able to obtain additional financing on terms favorable to us, if at all. If we are unable to obtain adequate financing or financing on terms satisfactory to us when and if we require it, our ability to continue to support our business growth and to respond to business challenges could be significantly impaired, and our business may be harmed.
| 37 |
The trading price of the shares of our common stock is highly volatile, and purchasers of our common stock could incur substantial losses.
The market price of our common stock has been and is likely to continue to be highly volatile and could fluctuate widely in response to various factors, many of which are beyond our control, including the following:
| ● | technological innovations or new products and services by us or by our competitors, including announcements by us or our competitors of significant contracts, acquisitions, strategic partnerships or capital commitments; |
| ● | additions or departures of key personnel; |
| ● | changes in expectations as to our future financial performance; |
| ● | sales of our common stock; |
| ● | our ability to execute our business plan; |
| ● | loss of any strategic relationship; |
| ● | industry developments; |
| ● | changes in financial estimates by any securities analysts who follow our common stock, our failure to meet these estimates or failure of those analysts to initiate or maintain coverage of our common stock; |
| ● | general market conditions, including market volatility and inflation; |
| ● | fluctuations in stock market prices and trading volumes of similar companies; |
| ● | economic, political and other external factors; |
| ● | period-to-period fluctuations in our financial results; |
| ● | applicable regulatory developments in the United States and foreign countries, both generally or specific to us and our products; and |
| ● | intellectual property, product liability or other litigation against us. |
Although publicly traded securities are subject to price and volume fluctuations, it is likely that our common stock will experience these fluctuations to a greater degree than the securities of more established and better capitalized organizations.
Our common stock does not have a vigorous trading market, and you may not be able to sell your securities at or near ask prices, or at all.
Although there is a public market for our common stock, trading volume has been historically low, which could impact our stock price and your ability to sell shares of our common stock at or near ask prices, or at all. We can give no assurance that a more active and liquid public market for the shares of our common stock will develop in the future.
The potential sale of large amounts of common stock may have a negative effect upon the market value of our shares.
Sales of a significant number of shares of our common stock in the public market or the perception that these sales might occur could harm the market price of our common stock and make it more difficult for us to raise funds through future offerings of common stock. As additional shares of our common stock become available for resale in the public market, the supply of our common stock will increase, which could decrease the price of our common stock.
We have not paid, and we are unlikely to pay, cash dividends on our securities in the near future. Because we have no current plans to pay cash dividends on our common stock for the foreseeable future, you may not receive any return on investment unless you sell your common stock for a price greater than that which you paid for it.
We have not paid and do not currently intend to pay dividends on our common stock, which may limit the current return available on an investment in our stock. Future dividends on our stock, if any, will depend on our future earnings, capital requirements, financial condition and such other factors as our management may consider relevant. Currently, we intend to retain earnings, if any, to increase our net worth and reserves. Consequently, shareholders will only realize an economic gain on their investment in our common stock if the price appreciates. Shareholders should not purchase our common stock expecting to receive cash dividends. Because we currently do not pay dividends, and there may be limited trading in our common stock, shareholders may not have any manner to liquidate or receive any payment on their common stock. Therefore, our failure to pay dividends may cause shareholders to not see any return on their common stock even if we are successful in our business operations. In addition, because we do not pay dividends, we may have trouble raising additional funds, which could affect our ability to expand our business operations.
A few of our existing shareholders own a large percentage of our voting stock and have control over matters requiring shareholder approval and may delay or prevent a change in control or otherwise lead to actual or potential conflicts of interest.
As of March 30, 2021,22, 2024, our directors beneficially owned, andincluding through their affiliates, controlled, more than 50%approximately 53% of our outstanding common stock. As a result, our directors and their affiliates could have the ability to exert substantial influence over all matters requiring approval by our shareholders, including (i) the election and removal of directors, (ii) any proposed merger, consolidation or sale of all or substantially all of our assets as well as other corporate transactions and (iii) any amendment to our Certificate of Formation, as amended (the “Certificate of Formation”). This concentration of control could be disadvantageous to other shareholders having different interests. This significant concentration of share ownership may adversely affect the trading price for our common stock because investors sometimes perceive disadvantages in owning stock in companies with controlling shareholders.
In addition, our Certificate of Formation contains a provision which under the Texas Business Organizations Code (the “TBOC”) could allow the shareholders who own a majority of our common stock to approve certain major transactions without the approval of other shareholders that otherwise would be required under Texas corporation law.
| 38 |
Our Certificate of Formation includes provisions limiting the personal liability of our directors for breaches of fiduciary duties under Texas law.
Our Certificate of Formation contains a provision eliminating a director’s personal liability to the fullest extent permitted under Texas law. Pursuant to the TBOC, a corporation has the power to indemnify its directors and officers against judgments and certain expenses other than judgments that are actually and reasonably incurred in connection with a proceeding, provided that there is a determination that the individual acted in good faith and in a manner reasonably believed to be in or not opposed to the best interests of the corporation and, with respect to any criminal proceeding, had no reasonable cause to believe the individual’s conduct was unlawful. However, no indemnification may be made in respect of any proceeding in which such individual is liable to the corporation or improperly received a personal benefit and is found liable for willful misconduct, breach of the duty of loyalty ownedowed to the corporation, or an act or omission deemed not to be committed in good faith.
The principal effect of the limitation on liability provision is that a shareholder will be unable to prosecute an action for monetary damages against a director unless the shareholder can demonstrate a basis for liability for which indemnification is not available under the TBOC. This provision, however, should not limit or eliminate our rights or any shareholder’s rights to seek non-monetarynonmonetary relief, such as an injunction or rescission, in the event of a breach of a director’s fiduciary duty. This provision will not alter a director’s liability under federal securities laws. The inclusion of this provision in our Certificate of Formation may discourage or deter shareholders or management from bringing a lawsuit against directors for a breach of their fiduciary duties, even though such an action, if successful, might otherwise have benefited us and our shareholders.
Texas law, and our Certificate of Formation and bylawsour Amended and Restated Bylaws contain anti-takeover provisions that could delay or discourage takeover attempts that shareholders may consider favorable.
Under our Certificate of Formation, our board of directors can authorize the issuance of preferred stock, which could diminish the rights of holders of our common stock and make a change of control of the Company more difficult even if it might benefit our shareholders. The board of directors is authorized to issue shares of preferred stock in one or more series and to fix the voting powers, preferences and other rights and limitations of the preferred stock. Accordingly, we may issue shares of preferred stock with a preference over our common stock with respect to dividends or distributions on liquidation or dissolution, or that may otherwise adversely affect the voting or other rights of the holders of common stock. Issuances of preferred stock, depending upon the rights, preferences and designations of the preferred stock, may have the effect of delaying, deterring or preventing a change of control, even if that change of control might benefit our shareholders.
In addition, provisions of our Certificate of Formation and bylawsour Amended and Restated Bylaws (“Bylaws”) may delay or discourage transactions involving an actual or potential change in our control or change in our management, including transactions in which shareholders might otherwise receive a premium for their shares, or transactions that our shareholders might otherwise deem to be in their best interests. For example, our Certificate of Formation and bylawsBylaws (i) do not provide for cumulative voting rights (therefore allowing the holders of a majority of the shares of common stock entitled to vote in any election of directors to elect all of the directors standing for election, if they should so choose), (ii) require that special meetings of the shareholders be called by the Chairman of the board of directors, the President or the board of directors, or by the holders of not less than tentwenty-five percent (10%(25%) of all the shares issued, outstanding and entitled to vote, (iii) permit our board of directors to alter, amend or repeal our bylawsBylaws or to adopt new bylaws, and (iv) enable our board of directors to increase the number of persons serving as directors and to fill vacancies created as a result of the increase by a majority vote of the directors present at a meeting of directors.
While we are subject to the provisions of Title 2, Chapter 21, Subchapter M of the TBOC, which provides that a Texas corporation that qualifies as an “issuing public corporation” (as defined in the TBOC) may not engage in specified types of business combinations, including mergers, consolidations and asset sales, with a person, or an affiliate or associate of that person, who is an “affiliated shareholder,” the restrictions in Title 2, Chapter 21, Subchapter M of the TBOC do not apply to us because we have elected, in the manner provided under the TBOC, not to be subject to such provisions.
Our failure to meet the continued listing requirements of The Nasdaq Capital Market could result in a delisting of our common stock.
Our shares of common stock are currently listed for trading on The Nasdaq Capital Market under the symbol “SMTI.” If we fail to satisfy the continued listing requirements of The Nasdaq Stock Market, LLC (“Nasdaq”), such as the corporate governance requirements, the shareholder’s equity requirement or the minimum closing bid price requirement, Nasdaq may take steps to delist our common stock. Such a delisting or even notification of failure to comply with such requirements would likely have a negative effect on the price of our common stock and would impair your ability to sell or purchase our common stock when you wish to do so. In the event of a delisting, we expect that we would take actions to restore our compliance with Nasdaq’s listing requirements, but we can provide no assurance that any such action taken by us would allow our common stock to become listed again, stabilize the market price or improve the liquidity of our common stock, prevent our common stock from dropping below the Nasdaq minimum bid price requirement or prevent future non-compliancenoncompliance with Nasdaq’s listing requirements.
| 39 |
ITEM 1B. UNRESOLVEDUNRESOLVED STAFF COMMENTS
None.
ITEM 1C. CYBERSECURITY
We recognize the critical importance of cybersecurity in safeguarding sensitive information and maintaining operational resilience. We have processes for assessing, identifying and managing cybersecurity risks, which are built into our information technology function and are designed to help protect our information assets and operations from internal and external cyber threats, protect employee and customer information from unauthorized access or attack, as well as secure our networks and systems.
The Audit Committee of the Board of Directors (the “Audit Committee”) is responsible for overseeing cybersecurity risk and periodically updates our Board of Directors on such matters. The Audit Committee receives periodic updates from management regarding cybersecurity matters and is notified between such updates regarding any significant new cybersecurity threats or incidents. We do not believe that there are currently any known risks from cybersecurity threats that are reasonably likely to materially affect us or our business strategy, results of operations or financial condition.
As a company, we leverage the National Institute of Standards and Technology (NIST) Cybersecurity Framework to align with industry best practices and to develop policies and procedures, which include a Cybersecurity Response Plan focusing on detection, validation, mitigation, recovery, and refinement. In addition to the Cybersecurity Response Plan, we provide cybersecurity awareness training to our employees, including training on social engineering, phishing, password protection, confidential data protection, mobile security, and incident reporting, to help prevent and reduce the impact of potential cybersecurity events.
We have strategically invested resources and tools to combat the ever-changing cyber threat landscape. These investments include growing our internal technology team headcount and bolstering our partnerships with entities with external expertise, resulting in increased protection, monitoring, and response capabilities. We have also made investments to carry cybersecurity insurance that provides additional protection and resources to reduce a cybersecurity event’s impact and potential losses.
Our technology and management teams meet regularly with key internal and external stakeholders to review cybersecurity concerns and areas for continued focus and improvement.
We face risks from cybersecurity threats that could have a material adverse effect on our business, financial condition, results of operations, cash flows or reputation. To date, we have not experienced any cyber incidents that have had a material adverse effect on our business, financial condition, results of operations, or cash flows. See “Risk Factors – Risks Related to Our Business – Security breaches and other disruptions could compromise our information and expose us to liability, which would cause our business and reputation to suffer.”
ITEM 2. PROPERTIES
We do not own any buildings or other real property. We have onethree material operating lease which consists of anleases for office space lease with a remaining lease term of 42 months as of December 31, 2020.space. Our leased office space in Fort Worth, Texas consists of 5,877approximately 11,414 square feet of rentable space located in Summit Office Park, a twin-building, mid-rise, 242,000 square foot office park located on the perimeter of the Fort Worth Texas central business district. The monthly base rental payments forIn March and September of 2023, we amended our primary office lease areto obtain additional space, as follows:
Our leased office space in San Antonio, Texas consists of approximately 7,289 square feet of rentable space located in a discount rate14,500 square foot office park in an industrial district in San Antonio, Texas. This lease has a remaining lease term of 6.25%.
Our leased office space in Orlando, Florida represents a lease we assumed in connection with our acquisition of Scendia. The lease consists of approximately 7,684 square feet in a 22,947 square foot office building. However, we are co-tenants in the lease with an unaffiliated company with our share of the lease being agreed to as 40% or approximately 3,074 square feet. The lease has a remaining term of 23 months as of December 31, 2023.
We periodically enter into operating lease contracts for office space and equipment. Arrangements are evaluated at inception to determine whether such arrangements constitute a lease. In accordance with the transition guidance of Accounting Standards Codification (“ASC”) Topic No. 842, such arrangements are included in our balance sheet as of January 1, 2019.
See Note 78 to the consolidated financial statements for additional information on our operating leases.
ITEM 3. LEGALLEGAL PROCEEDINGS
From time to time, we may be involved in claims and legal actions that arise in the ordinary course of business. To our knowledge, there are no material pending legal proceedings to which we are a party or of which any of our property is the subject.
This item is not applicable.
| 40 |
PART II
ITEM 5. MARKETMARKET FOR REGISTRANT’S COMMON EQUITY, RELATED SHAREHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Our common stock is traded on The Nasdaq Capital Market under the trading symbol “SMTI.” The closing price of our common stock as reported by Nasdaq on March 29, 202122, 2024, was $28.75.
Record Holders
As of March 30, 2021,22, 2024, there were 220283 shareholders of record and there were 7,617,1228,622,805 shares of common stock issued and outstanding. The number of shareholders of record does not reflect the number of persons or entities who held stock in nominee or street name through various brokerage firms.
Dividends
We have never declared or paid any cash dividends on our common stock and we do not intend to pay cash dividends in the foreseeable future. We currently expect to retain any future earnings to fund our operations and the expansion of our business.
Recent Sales of Unregistered Securities
There were no sales of unregistered securities during the year ended December 31, 2023 that were not previously reported on a Quarterly Report on Form 10-Q or a Current Report on Form 8-K.
Issuer Purchases of Equity Securities
We did not repurchase any of our equity securities during the fourth quarter of the fiscal year ended December 31, 2023.
ITEM 6. SELECTED FINANCIAL DATA
ITEM 7. MANAGEMENT’SMANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion and analysis contains forward-looking statements about future revenues, operating results, plans and expectations. Forward-looking statements are based on a number of assumptions and estimates that are inherently subject to significant risks and uncertainties and our results could differ materially from the results anticipated by our forward-looking statements as a result of many known or unknown factors, including, but not limited to, those factors discussed in Part I, Item 1A. Risk Factors. Also, please read the “Cautionary Statement Regarding Forward-Looking Statements” set forth at the beginning of this Annual Report on Form 10-K.
In addition, the following discussion should be read in conjunction with Part I of this Annual Report on Form 10-K as well as our consolidated financial statementsConsolidated Financial Statements and the related Notes to Consolidated Financial Statements contained elsewhere in this Annual Report on Form 10-K.
Overview
We are a medical technology company focused on developing and commercializing transformative technologies to improve clinical outcomes and reduce healthcare expenditures in the surgical, and chronic wound and skin careskincare markets. Our portfolio of products and services will allow us to deliver comprehensive wound and skin care solutions for patients in all care settings, including acute (hospitals and long-term acute care hospitals (“LTACHs”)) and post-acute (wound care clinics, physician offices, skilled nursing facilities (“SNFs”), home health, hospice, and retail). Each of our products, services and technologies contributesare designed to achieve our overall goal of achievingproviding better clinical outcomes at a lower overall cost for patients regardless of where they receive care. We strive to be one of the most innovative and comprehensive providers of effective surgical, wound and skin care products and technologiesskincare solutions and are continually seeking to expand our offerings for patients requiring wound and skin care treatments across the entire continuum of care in the United States.
We currently market sevenseveral products across chronicsurgical and surgicalchronic wound care applications and have multiple products in our pipeline. We licenseOn August 1, 2023, we acquired, among other things, the underlying intellectual property of, as well as the rights to manufacture and sell, CellerateRX Surgical Activated Collagen (“CellerateRX Surgical”), our productsprimary product, and HYCOL Hydrolyzed Collagen (“HYCOL”) from research and development partners Applied Nutritionals, LLC (“AN”Applied”) (throughfor human wound care use (for more information regarding this acquisition, see the “Recent Acquisitions” section below). Prior to such time, we had licensed the rights to these products through a sublicense agreement (the “Sublicense Agreement”) with CGI Cellerate RX, LLC (“CGI Cellerate RX”), an affiliate of The Catalyst Group, Inc. (“Catalyst”)) and, both of which are related parties. In connection with the asset purchase, Applied assigned its license agreement with CGI Cellerate RX to a wholly owned subsidiary of the Company. We also license certain products from Rochal Industries, LLC (“Rochal”) and have the right to exclusively distribute certain products under development by Cook Biotech Inc. (“Cook Biotech”).
In 2021,April 2022, we intendentered into a merger agreement through which Precision Healing Inc. (“Precision Healing”) became a wholly owned subsidiary of the Company. Precision Healing is developing a diagnostic imager and lateral flow assay (“LFA”) for assessing a patient’s wound and skin conditions. This comprehensive wound and skin assessment technology is designed to begin marketing three biologic productsquantify biochemical markers to determine the trajectory of a wound’s condition to enable better diagnosis and treatment protocol. In December 2023, we received 510(k) clearance from the U.S. Food and Drug Administration (“FDA”) for surgicalthe Precision Healing diagnostic imager. We are currently evaluating regulatory pathways for the Precision Healing LFA.
In July 2022, we entered into a membership interest purchase agreement with Scendia Biologics, LLC (“Scendia”) and Ryan Phillips (“Phillips”) pursuant to which we acquired 100% of the issued and outstanding membership interests in Scendia from Phillips. Since our acquisition of Scendia, we have been selling a full line of regenerative and orthobiologic technologies including (i) TEXAGEN Amniotic Membrane Allograft (“TEXAGEN”), (ii) BiFORM Bioactive Moldable Matrix (“BiFORM”), (iii) ACTIGEN Verified Inductive Bone Matrix (“ACTIGEN”) and (iv) ALLOCYTE Advanced Cellular Bone Matrix (“ALLOCYTE”).
In November 2022, we established a partnership with InfuSystem Holdings, Inc. (“InfuSystem”) focused on delivering a complete wound care applications pursuantsolution targeted at improving patient outcomes, lowering the cost of care, and increasing patient and provider satisfaction. The partnership is expected to enable InfuSystem to offer innovative products, including Cork Medical, LLC’s negative pressure wound therapy devices and supplies, and our marketingadvanced wound care product line and distribution agreement with Cook Biotech.associated services to new customers.
In November 2023, we launched BIASURGE Advanced Surgical Solution (“BIASURGE”). BIASURGE is a no-rinse, advanced surgical solution used for wound irrigation. It contains an antimicrobial preservative effective against a broad spectrum of pathogenic microorganisms. BIASURGE is indicated for use in the mechanical cleansing and removal of debris, including microorganisms, from surgical wounds.
| 41 |
Comprehensive Value-Based Care Strategy
In June 2020, we formed a subsidiary, United Wound and Skin Solutions, LLC (“UWSS”(formerly known as “WounDerm”), to hold certain investments and operations in wound and skin careskincare virtual consult services. In 2023, WounDerm was renamed and is now doing business as “Tissue Health Plus” (“THP”). THP is continuing its current mission to simplify skin health, starting with wound care through a refined business plan. Through THP, we plan to offer a first of its kind value-based wound care program to payers and risk-bearing entities such as accountable care organizations and value-based care (“VBC”) primary care companies, with Medicare Advantage payers as the initial target segment for this program.
THP’s programs are expected to enable payers to divest wound care spend risk, reduce wound related hospitalizations and improve patient quality of life. THP plans to coordinate delivery of community and home-based wound care for its managed patients. Community based care spans a variety of settings including physician offices, skilled nursing homes, assisted living facilities and senior living facilities. THP programs are intended to integrate science and evidence-based medicine protocols to standardize wound prevention and treatment.
We anticipate that THP’s customer contracts will have three-to-five-year terms. These contracts are expected to incorporate a mix of value-based pricing methodologies including episodic, “per member per month”, and “fee for value” pricing. We believe this approach is aligned with the financial goals of the payers and will help deliver outstanding clinical outcomes for the patients.
Our vision for our various service offerings will allow clinicians/physicians utilizing our technologiescomprehensive approach consists of three key sets of planned capabilities:
| (a) | Care Hub – This virtual patient monitoring, care coordination and navigation center is expected to help doctors and nurses support their patients throughout their wound care journey, from prevention to treatment. We expect to have Care Hub staffed by wound care certified nurse practitioners (“NPs”) and registered nurses (“RNs”), incorporating care delivery best practices from partnerships with Direct Dermatology Inc. (“DirectDerm”) and certain physician-led multispecialty wound care groups. With NPs leading the care hub, RNs are expected to be the wound specialists, providing patients with expert review and support of the overarching plan of care on each patient’s journey through the process. In addition, care navigators are expected to serve as a primary point of contact for patients and their providers, coordinating care, managing appointments and ensuring seamless communication among all team members. |
| (b) | Managed Services Organization (“MSO”) Network – With respect to patient-side wound care, our plan is that THP’s programs would be performed by a network of third-party providers who will be contracted through managed services agreements. These providers would include podiatrists, wound care provider groups, primary care physicians and home health agencies. The providers in the THP network are expected to leverage THP’s standard of care, patient education and tools to deliver optimal patient outcomes with high predictability and efficiency. |
| (c) | Technology Platform – THP’s technology platform will focus on scaling workflows of THP’s Care Hub and MSO Network through automation and integration. We expect the THP technology platform to enable enhanced patient empowerment and self-healthcare. We anticipate that our platform will leverage our technology investments and partnerships with Precision Healing, Pixalere Healthcare, Inc. (“Pixalere”) and others, by leveraging modern technology including artificial intelligence and machine learning. Our platform technology is expected to manage program economics, standards of care, patient monitoring, wound assessments, network performance monitoring, and revenue cycle management. We expect that each of these components will work in concert with each other, constantly improving economics and care delivery. |
We are seeking a partner to collectfacilitate commercialization of Tissue Health Plus and analyze large amounts of data on patient conditions and outcomes that will improve treatment protocols and ultimately lead to more evidence-based formulary to improve patient outcomes. We intend to launch our initial virtual consult service offerings in 2021. Through a combination of our UWSS services and our Sanara products, we believe we will be able to offer patient care solutions at every stepshare in the continuumcost of development of the program. Excluding noncash items, our full year operating expenses for THP in 2023 were approximately $5.2 million.
Recent Acquisitions
Precision Healing
In April 2022, we closed a merger transaction with Precision Healing, pursuant to which Precision Healing became a wholly owned subsidiary of the Company. Precision Healing is developing a diagnostic imager and LFA for assessing a patient’s wound and skin care fromconditions. This comprehensive wound and skin assessment technology is designed to quantify biochemical markers to determine the trajectory of a wound’s condition to enable better diagnosis through healing.
Pursuant to the merger agreement, among other things, we agreed to (i) pay the holders of Precision Healing common stock and preferred stock closing consideration consisting of 165,738 shares of our common stock, which was issued to accredited investors, and $125,966 in cash, which was paid to stockholders who were not accredited investors (ii) pay approximately $0.6 million of transaction expenses on behalf of the COVID-19 Pandemic
Scendia
In July 2022, we entered into a membership interest purchase agreement by and among the Company, Scendia and Seller pursuant to which we acquired 100% of the issued and outstanding membership interests in Scendia from the Seller. Scendia provides clinicians and surgeons with a full line of regenerative and orthobiologic technologies. Beginning in March 2020, many states issued orders suspending elective surgeries in order to free-up hospital resources to treat COVID-19 patients. This resulted in a reduction in demand for our surgicalearly 2022, we began co-promoting certain products beginning in the second half of March 2020. Additionally, most states limited access to SNFs to only resident caregivers, which impeded our ability to provide educationwith Scendia, including: (i) TEXAGEN, (ii) BiFORM, (iii) ACTIGEN, and product training(iv) ALLOCYTE. Prior to the clinicians who use our productsacquisition, Scendia owned 50% of the issued and outstanding membership interests in these facilities. These restrictions resulted in an overall decline in sales forSanara Biologics, LLC (“Sanara Biologics”), and we owned the second quarterremaining 50% of 2020. During the third and fourth quarters of 2020, we saw a strong rebound in product sales as restrictions on elective surgeries eased in our primary markets in Texas, Florida, and the southeastern United States.
Pursuant to be performed with the exception of certain geographic COVID-19 hotspots. We will continue to closely monitorpurchase agreement, the COVID-19 pandemic in order to ensureaggregate consideration at closing for the safety of our people and our ability to serve our customers and patients.
In addition to the Cellerate Acquisition was treated as a recapitalization of Sanara MedTech. As part ofcash and stock consideration, the reverse merger and recapitalization, the net liabilities existingpurchase agreement provides that Phillips is entitled to receive two potential earnout payments, payable on an annual basis, not to exceed $10.0 million in the Company as of the date of the Cellerate Acquisition totaling approximately $1,666,537, which included $508,973 ofaggregate. The earnout consideration is payable to Phillips in cash were convertedor, at our election, in up to equity as part of the Cellerate Acquisition. No step-up in basis or intangible assets or goodwill was recorded in this transaction.
| 42 |
Applied Asset Purchase
On August 1, 2023, we entered into an Asset Purchase Agreement (the “Applied Purchase Agreement”) by and among the Company, as guarantor, Sanara MedTech Applied Technologies, LLC, a wholly owned subsidiary of the Company (“SMAT”), Applied, The Hymed Group Corporation (“Hymed” and together with Applied, the “Sellers”), and Dr. George D. Petito (the “Owner”), pursuant to which SMAT acquired certain assets of the Sellers and the Owner including the Sellers’ and Owner’s inventory, intellectual property, manufacturing and related equipment, goodwill, rights and claims, other than certain excluded assets, all as more specifically set forth in the Applied Purchase Agreement (collectively, the “Applied Purchased Assets”), and assumed certain Assumed Liabilities (as defined in the Applied Purchase Agreement), upon the terms and subject to the conditions set forth in the Applied Purchase Agreement (such transaction, the “Applied Asset Purchase”). The Applied Purchased Assets include the underlying intellectual property of, as well as the rights to manufacture and sell, CellerateRX Surgical and HYCOL products for human wound care use.
The Applied Purchased Assets were purchased for an initial aggregate purchase price of $15.25 million, consisting of (i) $9.75 million in cash (the “Cash Closing Consideration”), (ii) 73,809 shares of our common stock (the “Stock Closing Consideration”) with an agreed upon value of $3.0 million and (iii) $2.5 million in cash (the “Installment Payments”), to be paid in four equal installments on each of the next four anniversaries of the closing of the Applied Asset Purchase (the “Closing”).
In addition to the Cash Closing Consideration, Stock Closing Consideration and Installment Payments, the Applied Purchase Agreement provides that the Sellers are entitled to receive up to an additional $10.0 million (the “Applied Earnout”), which is payable to the Sellers in cash, upon the achievement of certain performance thresholds relating to SMAT’s collections from net sales of a collagen-based product currently under development. Upon expiration of the seventh anniversary of the Closing, to the extent the Sellers have not earned the entirety of the Applied Earnout, SMAT shall pay the Sellers a pro-rata amount of the Applied Earnout based on collections from net sales of the product, with such amount to be due credited against any Applied Earnout payments already made by SMAT (the “True-Up Payment”). The Applied Earnout, minus the True-Up Payment and any Applied Earnout payments already made by SMAT, may be earned at any point in the future, including after the True-Up Payment is made.
In connection with the Applied Asset Purchase and pursuant to the Applied Purchase Agreement, effective August 1, 2023, we entered into a professional services agreement (the “Petito Services Agreement”) with the Owner, pursuant to which the Owner, as an independent contractor, agreed to provide certain services to us, including, among other things, assisting with the development of products already in development and assisting with research, development, formulation, invention and manufacturing of any future products (the “Petito Services”). As consideration for the Petito Services, the Owner is entitled to receive: (i) a base salary of $12,000 per month during the term of the Petito Services Agreement, (ii) a royalty payment equal to three percent (3%) of the actual collections from net sales of certain products the Owner develops or co-develops that reach commercialization, (iii) a royalty payment equal to five percent (5%) for the first $50.0 million in aggregate collections from net sales of certain future products and a royalty payment of two and one-half percent (2.5%) on aggregate collections from net sales of certain future products on any amounts exceeding $50.0 million but up to $100.0 million, (iv) $500,000 in cash in the event that 510(k) clearance is issued for any future product accepted by the Company and (v) $1.0 million in cash in the event that a U.S. patent is issued for a certain product; provided that with respect to the incentive payments described in (iv) and (v) of the foregoing, the Owner shall not earn more than $2.5 million. The Petito Services Agreement has an initial term of three years and is subject to automatic successive one-month renewals unless earlier terminated in accordance with its terms. The Petito Services Agreement may be terminated upon the Owner’s death or disability or by us or the Owner “For Cause” (as defined in the Petito Services Agreement); provided, however, that the base salary described in (i) of the foregoing paragraph shall survive termination through the three-year initial term and the royalty payments and incentive payments described in (ii)-(v) of the foregoing paragraph shall survive termination of the Petito Services Agreement.
Recent Developments
Loan Agreement
In connection with the entry into the Applied Purchase Agreement, on August 1, 2023, we, as guarantor, and SMAT, as borrower, entered into a loan agreement (the “Loan Agreement”) with Cadence Bank (the “Bank”) providing for, among other things, an advancing term loan in the aggregate principal amount of $12.0 million (the “Term Loan”). Pursuant to the Loan Agreement, the Bank agreed to make, at any time and from time to time prior to February 1, 2024, one or more advances to SMAT. On August 1, 2023, the Bank made an advance under the Term Loan for $9.75 million, the proceeds of which were used to fund the Cash Closing Consideration for the Applied Asset Purchase. For more information regarding the Loan Agreement, see the “Liquidity and Capital Resources” section below.
Tufts University License Agreement
In December 2023, we signed an exclusive license agreement with Tufts University (“Tufts”) to develop and commercialize patented technology covering 18 unique collagen peptides. As part of this agreement, we formed a new subsidiary, Sanara Collagen Peptides, LLC (“SCP”) and have issued 10% of SCP’s outstanding units to Tufts. SCP has exclusive rights to develop and commercialize new products based on the licensed patents and patents pending. SCP will pay royalties to Tufts based on net sales of licensed products and technologies. Pursuant to the exclusive license agreement, royalties will be calculated at a ratiorate of one share for every 100 shares. All share and per share information1.5% or 3%, depending on the type of product or technology developed. SCP will pay Tufts a minimum annual royalty of $50,000 on January 1 of the year following the first anniversary of the first commercial sale of the licensed products or technologies. SCP will pay Tufts a $100,000 minimum annual royalty on January 1 of each subsequent year during the royalty term specified in this discussion and analysis has been adjusted to reflect the reverse stock split.exclusive license agreement.
| 43 |
Components of Results of Operations
Sources of Revenues
Our revenue is derived primarily from sales of our surgical wound caresoft tissue repair and bone fusion products to hospitals and other acute care facilities, andfacilities. In particular, the substantial majority of our product sales revenue is derived from sales of our chronic wound care products to customers across the post-acute continuum of care.CellerateRX Surgical. Our revenue is driven by direct orders shipped by us to our customers, and to a lesser extent, direct sales to customers through delivery at the time of procedure by one of our sales representatives. We generally recognize revenue when a purchase order is received from the customer and our product is received by the customer.
Revenue streams from product sales and royalties are summarized below for the years ended December 31, 20202023 and 2019. All revenue was generated in the United States.
Year Ended December 31, | ||
2020 | 2019 | |
| Surgical | $14,463,182 | $10,597,234 |
| Wound Care | 922,794 | 1,010,404 |
| Royalty revenue | 201,000 | 159,125 |
| Total Revenue | $15,586,976 | $11,766,763 |
| For the Year Ended | ||||||||
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| Soft tissue repair products | $ | 54,836,410 | $ | 41,653,954 | ||||
| Bone fusion products | 9,952,432 | 3,987,891 | ||||||
| Royalty revenue | 201,000 | 201,000 | ||||||
| Total Net Revenue | $ | 64,989,842 | $ | 45,842,845 | ||||
We recognize royalty revenue from a development and licensing agreement with BioStructures, LLC. We record revenue each calendar quarter as earned per the terms of the agreement which stipulates that we will receive quarterly royalty payments of at least $50,250. Under the terms of the development and license agreement, royalties of 2.0% are recognized on sales of products containing our patented resorbable bone hemostasis. The minimum annual royalty due to our Companyus is $201,000 per year throughoutthrough the lifeend of the patent which expires in 2023. These royalties are payable in quarterly installments of $50,250. To date, royalties related to this development and licensing agreement have not exceeded the annual minimum of $201,000 ($50,250 per quarter).
Cost of Goods Sold
Cost of goods sold consists primarily of the acquisition costs from the manufacturers of our licensed products, raw material costs for certain components sourced directly by our Company,us, and all related royalties related due as a result of the sale of our products. Our gross profit represents total net revenue less the cost of goods sold, and gross margin isrepresents gross profit expressed as a percentage of total revenue.
Operating Expenses
Selling, general and administrative expenses (“SG&A”) expenses consist primarily of salaries, sales commissions, benefits, bonuses and stock-based compensation. SG&A also includes outside legal counsel fees, audit fees, insurance premiums, rent and other corporate expenses. We expense all SG&A expenses as incurred.
Research and development (“R&D”) expenses include costs related to enhancements to our currently available products and additional investments in our product, services and technologies development pipeline. This includes personnel-related expenses, including salaries and benefits for all personnel directly engaged in R&D activities, contracted services, materials, prototype expenses and allocated overhead, which is comprised of lease expense and other facilities related costs. We expense R&D costs as incurred. We generally expect our SG&Athat R&D expenses towill increase in absolute dollars and decrease as a percent of revenue as we growcontinue to support product enhancements and to bring new products to market.
Depreciation and amortization expenses include depreciation of fixed assets and amortization of intangible assets that have a finite life, such as product licenses, patents and intellectual property, customer relationships and assembled workforces.
Change in fair value of earnout liabilities represents our commercial organization.measurement of the change in fair value at the balance sheet date of our earnout liabilities that were established at the time of our Precision Healing and Scendia acquisitions.
| 44 |
Other Income (Expense)
Other income (expense) is primarily comprised of interest income,losses on equity method investments, interest expense, and other non-operatingnonoperating activities. Interest expense during 2020 consisted primarily of interest expense associated with our unsecured promissory note under the Paycheck Protection Program (the “PPP”) pursuant to the Coronavirus Aid, Relief and Economic Security Act, and for 2019, consisted primarily of interest on amounts due on our revolving line of credit that matured in June 2020. Interest income consists of interest earned on our cash and cash equivalents.
Results of Operations
Net Revenues.
For the year ended December 31,During the third and fourth quartersquarter of 2020,2022, we saw a strong rebound inbegan to experience supply issues with the ALLOCYTE product sales as restrictions on elective surgeries eased in our primary markets in Texas, Florida, andline. The amount of qualifying eligible donor tissue was significantly reduced industry wide due to the southeastern United States. Fourth quarter revenues of $4,789,138 were up 43% compared tostringent screening required. During the fourth quarter of 2019,2022 and representedthe nine months ended September 30, 2023, we were unable to fill certain orders for this product, which negatively impacted our sales growth. The supply constraint was caused by significant supplier limits on qualifying eligible donor tissue and supplier necessity to subcontract all processing to secondary suppliers. We have since expanded the ALLOCYTE product line with the release of ALLOCYTE Plus, which is processed by an alternative supplier with in-house processing capabilities. Our first sales of ALLOCYTE Plus occurred in October 2023. We have a record high sales quarter forsufficient supply of ALLOCYTE Plus to meet currently expected demand and believe we have measures in place to regularly stock the Company.
Cost of goods sold
. Cost of goods sold for the year ended December 31,Selling, general
andadministrativeexpensesResearch and development expenses.
Depreciation and amortization expense. Depreciation and amortization expense for the year ended December 31, 2023, was $3.7 million compared to $2.4 million for the year ended December 31, 2022. The increase in depreciation and amortization expense during 2023 was primarily due to declinesthe amortization of intangible assets acquired as part of the Precision Healing, Scendia and Applied transactions.
Change in draws onfair value of earnout liabilities. Change in fair value of earnout liabilities was a benefit of $3.4 million for the year ended December 31, 2023 compared to expense of $0.3 million for the year ended December 31, 2022. The current year benefit is as a result of a decrease in the estimated fair value of the earnout liabilities established at the time of our revolving linePrecision Healing and Scendia acquisitions. The decrease in the estimated fair value was due to a change in the discount factor utilized in the valuation models, a decrease in the projected undiscounted amounts to be paid, as well as adjustments to the projected timing of creditthe payments to be made, partially offset by accretion. The prior year period expenses were due to the accretion of the earnout liabilities.
Other income (expense). Other income (expense) for the year ended December 31, 2023 was $0.2 million compared to $1.4 million for the year ended December 31, 2022. Other income (expense) for 2023 included interest expense, and amortization of debt issuance costs related to the Term Loan entered into in conjunction with the Applied Asset Purchase. The higher other income (expense) in 2022 was primarily due to a $1.0 million loss recognized due to the dissolution of our subsidiary, Sanara Pulsar, LLC (“Sanara Pulsar”). Sanara Pulsar had minimal sales since its inception and was dissolved effective December 2022. The higher other income (expense) was also due to the recognition of $0.4 million of loss from our equity method investment in Precision Healing prior to our acquisition of the remaining interest in April 2022.
| 45 |
Loss before income taxes. We had a loss before income taxes of $4.4 million for the year ended December 31, 2023, compared to a loss before income taxes of $13.9 million for the year ended December 31, 2022. The lower loss in 2023 was due to increased gross profits and changes in fair value of earnout liabilities, partially offset by higher SG&A costs, higher R&D expenses, and higher amortization of our acquired intangible assets as discussed above.
Income tax benefit. We recognized net deferred tax liabilities associated with the Precision Healing and Scendia transactions. As of December 31, 2022, prior to consideration of these deferred tax liabilities, we had net deferred tax assets in excess of the deferred tax liabilities being recognized, however, a 100% valuation allowance had previously been provided against our net deferred tax assets. As a result of the recording of the net deferred tax liabilities related to the Precision Healing merger and Scendia acquisition, we have reviewed the valuation allowance and determined that maturedit should be reduced by the amount of the net deferred tax liabilities that were recognized. This resulted in June 2020 and the conversionrecognition of an interest-bearing promissory note to common stock in early 2020.
Net income / loss
Liquidity and Capital Resources
Cash on hand at December 31, 20202023 was $455,366,$5.1 million, compared to $6,611,928$9.0 million at December 31, 2019.2022. Historically, we have financed our operations primarily from the sale of equity securities. During 2019 and 2020, our principal sources of liquidity have been cash generated from operations, utilization of our bank line of credit that matured in June 2020, cash provided by an unsecured promissory note in the principal amount of $583,000In February 2023, we entered into a Controlled Equity Offering SM Sales Agreement (the “Sales Agreement”) with Cantor Fitzgerald & Co., as sales agent (“the PPP Loan”Cantor”) to Cadence Bank, N.A. (“Cadence”), pursuant to the PPP,which we could offer and $10,000,000 in proceeds receivedsell from a private placement of our common stock in October 2019.
Sales of the shares, pursuant to the Sales Agreement, were made in sales deemed to be an “at the market offering” as defined in Rule 415(a)(4) promulgated under the Securities Act of 1933, as amended. Upon delivery of a placement notice and subject to the terms and conditions of the Sales Agreement, Cantor agreed to use commercially reasonable efforts consistent with its normal trading and sales practices, applicable state and federal law, rules and regulations and the rules of The Nasdaq Capital Market to sell the shares from time to time based upon our instructions, including any price, time period or size limits specified by us. We had no obligation to sell any of the shares under the Sales Agreement and could suspend or terminate the offering of our common stock pursuant to the Sales Agreement upon notice to Cantor and subject to other conditions. Pursuant to the Sales Agreement, we paid Cantor a commission of 3.0% of the aggregate gross proceeds from each sale of the shares.
In 2023, we sold an aggregate of 26,143 shares of common stock for gross proceeds of $31,625,000, before deducting underwriting discountsapproximately $1.1 million and commissionsnet proceeds of approximately $1.0 million pursuant to the Sales Agreement. We paused the offering at the end of the first quarter of 2023 and estimateddid not reactivate it during the remainder of 2023. The Registration Statement on Form S-3 relating to this offering expenses. expired in January 2024.
On August 1, 2023, we, as guarantor, and SMAT, as borrower, entered into the Loan Agreement with the Bank providing for, among other things, a Term Loan in the aggregate principal amount of up to $12.0 million, which was evidenced by an advancing promissory note. Pursuant to the Loan Agreement, the Bank agreed to make, at any time and from time to time prior to February 1, 2024, one or more advances to SMAT. On August 1, 2023, the Bank made an advance under the Term Loan for $9.75 million, the proceeds of which were used to fund the Cash Closing Consideration for the Applied Asset Purchase. For more information regarding the Loan Agreement, see the “Loan Agreement” section below.
We expect our future needs for cash to use the net proceeds from the offering to expandinclude funding potential acquisitions, further developing our salesforce and for further development of its products, services and technologies pipeline and clinical studies, expanding our sales force, repayment of debt as it becomes due and for general corporate purposes, including working capital (see Note 13purposes. If we seek to consummate acquisitions in the consolidated financial statements contained elsewhere in this Annual Report on Form 10-K for more information on this offering).future, we expect to finance such acquisitions with the proceeds from equity or debt issuances. Based on our current plan of operations, including acquisitions, we believe our cash on hand, when combined with expected cash flows from operations, and amounts available under our revolving credit facility, will be sufficient to fund our growth strategy and to meet our anticipated operating expenses and capital expenditures for at least the next twelve months.
Precision Healing Merger
In November 2020, we were informed that the full amount of the PPP Loan was forgiven. The forgiveness of the loan was recognized as Other income at that time. If appropriate, we may pursue additional financing including issuing additional stock and incurring additional debt to support our strategic initiatives. If we are unable to obtain additional funding for operations at any time in the future, we may not be able to continue expanding the business as currently planned which would require us to modify various aspects of our operations.
In April 2022, we closed a senior liquidity preference relative to the common shareholders. As stipulated in the agreementsmerger transaction with Precision Healing UWSS invested an additional $600,000 in February 2021 for 150,000 additional shares of Series A Stock. The additional shares of Series A Stock will convert into shares of common stock ofpursuant to which Precision Healing at a ratio based on the date Precision Healing delivers a development milestone relatedbecame our wholly owned subsidiary. Pursuant to a wound diagnostic tool currently under development.
| 46 |
Upon the closing of the merger, the outstanding Precision Healing options previously granted under the Precision Healing Inc. 2020 Stock Option and Grant Plan (the “Precision Healing Plan”) converted, pursuant to their terms, into options to acquire an aggregate of 144,191 shares of our common stock with a weighted exercise price of $10.71 per share. These options expire between August 2030 and April 2031. In addition, outstanding and unexercised Precision Healing warrants converted into rights to receive warrants to purchase (i) 4,424 shares of our common stock with an initial exercise price of $7.32 per share and an expiration date of April 22, 2031, and (ii) 12,301 shares of our common stock with an initial exercise price of $12.05 per share and an expiration date of August 10, 2030. Concurrent with the assumption of the Precision Healing Plan, we terminated the ability to offer future awards under the Precision Healing Plan.
Pursuant to the merger agreement, upon the achievement of certain performance thresholds, the securityholders of Precision Healing, including the holders of options and warrants to purchase Precision Healing common stock and certain persons promised options to purchase Precision Healing common stock, are also entitled to receive payments of up to $10.0 million, which was accounted for as fullcontingent consideration pursuant to Accounting Standards Codification Topic 805, Business Combinations. The earnout consideration is payable in cash or, at our election, is payable to accredited investors in shares of our common stock at a price per share equal to the greater of (i) $27.13 or (ii) the average closing price of our common stock for the 20 trading days prior to the date such earnout consideration is due and payable. Pursuant to the merger agreement, a minimum percentage of the earnout consideration may be required to be issued to accredited investors in shares of our common stock for tax purposes. The amount and composition of the portion of earnout consideration payable is subject to adjustment and offsets as set forth in the merger agreement. We do not anticipate making an earnout consideration payment prior to January 2025.
Scendia Acquisition
In July 2022, we entered into a membership interest purchase agreement by and among the Company, Scendia and Phillips pursuant to which, and in accordance with the terms and conditions set forth therein, we acquired 100% of the issued and outstanding membership interests in Scendia from Phillips.
Pursuant to the purchase agreement, Phillips was entitled to receive closing consideration consisting of (i) approximately $1.6 million of cash, subject to certain adjustments, and (ii) 291,686 shares of our common stock. Pursuant to the purchase agreement, at closing, we withheld 94,798 shares of common stock with an agreed upon value of $1.95 million (the “Indemnity Holdback Shares”), which such Indemnity Holdback Shares were withheld to the extent provided in the purchase agreement to satisfy Phillips’ indemnification obligations and subsequently issued and released to Phillips in July 2023.
In addition to the cash consideration and the stock consideration, the purchase agreement provides that Phillips is entitled to receive two potential earnout payments, payable on an annual basis, not to exceed $10.0 million in the aggregate. The earnout consideration is payable to Phillips in cash or, at our election, in up to 486,145 shares of our common stock upon the achievement of certain performance thresholds relating to net revenue attributable to sales of Scendia products during the two-year period following the closing. We made the first earnout payment of approximately $693,000 in cash in August 2023. We expect the $750,000final earnout payment to be made in the third quarter of 2024.
Applied Asset Purchase
On August 1, 2023, we entered into the Applied Purchase Agreement by and among the Company, SMAT, Hymed, Applied and the Owner, pursuant to which became dueSMAT acquired the Applied Purchased Assets and assumed certain Assumed Liabilities upon the Company’s completionterms and subject to the conditions set forth in the Applied Purchase Agreement. The transaction closed on August 1, 2023. The Applied Purchased Assets were purchased for an initial aggregate purchase price of $15.25 million, consisting of (i) $9.75 million in cash, (ii) 73,809 shares of our common stock, with an agreed upon value of $3.0 million and (iii) $2.5 million in cash, to be paid in four equal installments on each of the next four anniversaries of the Closing.
In addition to the Cash Closing Consideration, Stock Closing Consideration and Installment Payments, the Applied Purchase Agreement provides that the Sellers are entitled to receive up to an additional $10.0 million, which is payable to the Sellers in cash, upon the achievement of certain performance thresholds relating to SMAT’s collections from net sales of a collagen-based product currently under development. Upon expiration of the seventh anniversary of the Closing, to the extent the Sellers have not earned the entirety of the Applied Earnout, SMAT shall pay the Sellers the True-Up Payment. The Applied Earnout, minus the True-Up Payment and any Applied Earnout payments already made by SMAT, may be earned at any point in the future, including after the True-Up Payment is made.
Since the closing of the Applied Asset Purchase, we make intercompany royalty payments to SMAT at the same rate as set forth in the Sublicense Agreement. SMAT intends to use the royalties received to repay borrowings under the Term Loan. As described under “Loan Agreement” below, SMAT is required to maintain compliance with certain maintenance covenants and is limited in its ability to distribute or lend cash to the Company without consent of the Bank.
| 47 |
Loan Agreement
In connection with the entry into the Applied Purchase Agreement, on August 1, 2023, we, as guarantor, and SMAT, as borrower, entered into the Loan Agreement with the Bank providing for, among other things, a Term Loan in the aggregate principal amount of $12.0 million, which was evidenced by an advancing promissory note. Pursuant to the Loan Agreement, the Bank agreed to make, at any time and from time to time prior to February 1, 2024, one or more advances to SMAT.
The proceeds of the advances under the Loan Agreement were used for working capital raiseand for purposes of financing up to one hundred percent (100%) of the Cash Closing Consideration and Installment Payments for the Applied Asset Purchase and related fees and expenses, including any subsequent payments that may be due to the Sellers after the Closing. On August 1, 2023, the Bank, at the request of SMAT, made an advance for $9.75 million. The proceeds from the advance were used to fund the Cash Closing Consideration for the Applied Asset Purchase.
Advances under the Term Loan will begin amortizing in February 2021.monthly installments commencing on August 5, 2024. All remaining unpaid balances under the Term Loan are due and payable in full on August 1, 2028 (the “Maturity Date”). SMAT may prepay amounts due under the Term Loan. All accrued but unpaid interest on the unpaid principal balance of outstanding advances is due and payable monthly, beginning on September 5, 2023 and continuing monthly on the fifth day of each month thereafter until the Maturity Date. The unpaid principal balance of outstanding advances bears interest, subject to certain conditions, at the lesser of the Maximum Rate (as defined in the Loan Agreement) or the Base Rate, which is for any day, a rate per annum equal to the term secured overnight financing rate (Term SOFR) (as administered by the Federal Reserve Bank of New York) for a one-month tenor in effect on such day plus three percent (3.0%).
The obligations of SMAT under the Loan Agreement and the other loan documents delivered in connection therewith are guaranteed by us and are secured by a first priority security interest in substantially all of the existing and future assets of SMAT.
The Loan Agreement contains customary representations and warranties and certain covenants that limit (subject to certain exceptions) the ability of SMAT and us to, among other things, (i) create, assume or guarantee certain liabilities, (ii) create, assume or suffer liens securing indebtedness, (iii) make or permit loans and advances, (iv) acquire any assets outside the ordinary course of business, (v) consolidate, merge or sell all or a material part of its assets, (vi) pay dividends or other distributions on, or redeem or repurchase, interest in an obligor, including us as guarantor, (vii) cease, suspend or materially curtail business operations or (viii) engage in certain affiliate transactions. In addition, the Loan Agreement contains financial covenants that require SMAT to maintain (i) a minimum Debt Services Coverage Ratio and (ii) a maximum Cash Flow Leverage Ratio, in each case, as defined and calculated according to the procedures set forth in the Loan Agreement. Pursuant to the Loan Agreement, in the event that SMAT fails to comply with the financial covenants described above, we are required to contribute cash to SMAT in an amount equal to the amount required to satisfy the financial covenants. SMAT is limited in its ability to distribute or lend cash to the Company without consent of the Bank.
Pursuant to the Loan Agreement, starting with the three months ended September 30, 2023 and for each quarter thereafter, SMAT will be required to maintain a minimum Debt Service Coverage Ratio (as defined below) of 1.2 to 1.0, which ratio is calculated as of the last day of the applicable fiscal quarter. The “Debt Service Coverage Ratio” is the ratio of (a) the sum of the following during the preceding twelve (12) month period, subject to annualization in certain circumstances: (i) earnings before interest, taxes, depreciation, amortization, stock compensation expense and gains or losses on sales of assets outside the ordinary course of business (“EBITDA”) minus (ii) capital expenditures, minus (iii) cash taxes, minus (iv) dividends and distributions, to (b) the sum of (i) the current portion of long-term debt, (ii) Installment Payments made during the preceding twelve (12) month period and (iii) interest expense during the preceding twelve (12) month period, subject to annualization in certain circumstances. As of December 31, 2023, following an immaterial cash contribution from Sanara, SMAT’s Debt Service Coverage Ratio was 1.3 to 1.0.
The Loan Agreement also requires SMAT to, subject to certain conditions, maintain a maximum Cash Flow Leverage Ratio (as defined below) of not more than (a) 4.5 to 1.0 as of the last day of the fiscal quarter ending on September 30, 2023, (b) 4.0 to 1.0 as of the last day of each fiscal quarter ending on December 31, 2023, and March 31, 2024, (c) 3.5 to 1.0 as of the last day of each fiscal quarter ending on June 30, 2024, and September 30, 2024, and (d) 3.0 to 1.0 as of the last day of each fiscal quarter thereafter. The “Cash Flow Leverage Ratio” is the ratio of all Funded Debt (as defined in the Loan Agreement) to certain multiples of EBITDA during the preceding twelve (12) month period, subject to annualization in certain circumstances. SMAT was in compliance with all financial covenants under the Loan Agreement as of December 31, 2023. As of December 31, 2023, following an immaterial cash contribution from Sanara, SMAT’s Cash Flow Leverage Ratio was 4.0 to 1.0.
The Loan Agreement also contains customary events of default. If such an event of default occurs, the Bank would be entitled to take various actions, including the acceleration of amounts due under the Loan Agreement and actions permitted to be taken by a secured creditor.
| 48 |
Cash Flow Analysis
For the year ended December 31, 2020,2023, net cash used in operating activities was $4,034,518$3.2 million compared to $2,167,401$5.6 million used in operating activities for the year ended December 31, 2019.2022. The higherlower use of cash in 20202023 was primarily due to net revenue growth outpacing the growth of our investment in sales force expansion, corporate infrastructure,cash operating expenses and start-up costs related to telehealth services including the developmenttiming of electronic imagerycash expenditures for certain accrued payables and data sharing technology to support virtual consultation and diagnostics.
For the year ended December 31, 2020,2023, net cash used in investing activities was $2,744,374$10.2 million compared to $1,197,097$3.5 million used in investing activities during the year ended December 31, 2019.2022. The higher use of cash used in investing activities during 20202023 was primarily due to our entry into a product license agreement with Rochal in May 2020, which included an initial cash payment of $600,000, along with a $500,000 milestone payment madepaid pursuant to Rochal during the first quarter of 2020 as a result of FDA clearance of BIAKŌS Antimicrobial Wound Gel. During the third quarter of 2020, a $500,000 long-term investment was made to purchase a minority interest in DirectDerm. During the fourth quarter of 2020, a $600,000 long-term investment was made in Precision Healing.
For the year ended December 31, 2020,2023, net cash provided by financing activities was $622,330$9.6 million as compared to $9,800,005 provided by$0.6 million used in financing activities for the year ended December 31, 2019.2022. The cash provided by financing activities in 2020 was primarily related to funds received from the PPP Loan. Cash provided by financing activities for the year ended December 31, 2019during 2023 was due to net loan proceeds of $9.7 million utilized for the Applied Asset Purchase and net proceeds received from a $10,000,000 private placementpursuant to sales of our common stock in October 2019.
Material Transactions with Related Parties
CellerateRX Surgical Sublicense Agreement
We have an exclusive, world-wide sublicense to distribute CellerateRX Surgical and HYCOL products into the surgical and wound care and surgical markets from an affiliate of Catalyst, CGI Cellerate RX, which, licensesprior to the Applied Asset Purchase, licensed the rights to CellerateRX from AN.Applied. Sales of CellerateRX haveSurgical comprised the substantial majority of our sales during 2018, 2019the twelve months ended December 31, 2023 and 2020. On January 26, 2021,2022. Prior to the Applied Asset Purchase discussed above, we amended the term of the sublicense agreement to extend the term to May 17, 2050, with automatic one-year renewals so long as annual net sales of CellerateRX exceed $1,000,000. We paypaid royalties based on ourthe annual net salesNet Sales of CellerateRXlicensed products (as defined in the Sublicense Agreement) consisting of 3% of all collected net salesNet Sales each year up to $12,000,000,$12.0 million, 4% of all collected net salesNet Sales each year that exceed $12,000,000$12.0 million up to $20,000,000,$20.0 million, and 5% of all collected net salesNet Sales each year that exceed $20,000,000. Minimum royalties of $400,000 per year are payable for the first five years of the sublicense agreement, which was entered on August 27, 2018.$20.0 million. For the yearsyear ended December 31, 20202023 and 2019, royalties due2022, royalty expense was $1.0 million and $1.8 million, respectively, under the terms of this agreement totaled $479,809 and $400,000, respectively.
In August 2023, we acquired the underlying intellectual property of, as well as the rights to manufacture and Catalyst, collectivelysell, CellerateRX Surgical and HYCOL products from Applied. In connection with their affiliates, includingthis acquisition, Applied assigned its license agreement with CGI Cellerate RX beneficiallyto a wholly owned 3,481,406 sharessubsidiary of our common stockthe Company and no further royalties will be due to Applied thereunder. Since the Closing of the Applied Asset Purchase, we indirectly make intercompany royalty payments to SMAT at the same rate as set forth in the Sublicense Agreement. These intercompany royalty payments and the offsetting cost of goods sold were eliminated in consolidation effective as of December 31, 2020.
Consulting Agreement
In connectionJuly 2021, we entered into an asset purchase agreement with Rochal, a related party. Concurrent with the Cellerate Acquisition,Rochal asset purchase, we issuedentered into a 30-month convertible promissory note to CGI Cellerate RX, an affiliate of Catalyst, in the principal amount of $1,500,000, bearing interest at a 5% annual interest rate, compounded quarterly. Interest on the promissory note was payable quarterly but could have been deferred at our election to the maturity of the promissory note. Outstanding principal and interest were convertible at CGI Cellerate RX’s option into shares of our common stock at a conversion price of $9.00 per share.
Catalyst Transaction Advisory Services Agreement
In March 2023, we entered into a Transaction Advisory Services Agreement (the “Catalyst Services Agreement”) effective March 1, 2023 with Catalyst, a related party. Pursuant to the Catalyst Services Agreement, Catalyst, by and through its directors, officers, employees and affiliates that are not simultaneously serving as directors, officers or employees of the Company (collectively, the “Covered Persons”), agreed to perform certain transaction advisory, business and organizational strategy, finance, marketing, operational and strategic planning, relationship access and corporate development services for us in connection with any merger, acquisition, recapitalization, divestiture, financing, refinancing, or other similar transaction in which we may be, or may consider becoming, involved, and any such additional services as mutually agreed upon in writing by and between Catalyst and us (the “Catalyst Services”).
Pursuant to the Catalyst Services Agreement, we agreed to reimburse Catalyst for (i) compensation actually paid by Catalyst to any of the Covered Persons at a rate no more than a rate consistent with industry practice for the performance of services similar to the Catalyst Services, as documented in reasonably sufficient detail, and (ii) all reasonable out-of-pocket costs and expenses payable to unaffiliated third parties, as documented in customary expense reports, as each of (i) and (ii) is incurred in connection with the Catalyst Services rendered under the Catalyst Services Agreement, with all reimbursements being contingent upon the prior approval of the Audit Committee of our Board of Rochal.Directors. We incurred $174,486 of costs pursuant to the Catalyst Services Agreement during 2023.
| 49 |
Receivables and Payables
We had outstanding related party receivables totaling $8,400 at December 31, 2023, and $98,548 at December 31, 2022. We had outstanding related party payables totaling $77,805 at December 31, 2023, and $34,036 at December 31, 2022.
Impact of Inflation and Changing Prices
Inflation and changing prices have not had a material impact on our historical results of operations. We do not currently anticipate that inflation and changing prices will have a material impact on our future results of operations.
Critical Accounting Policies
Our discussion and analysis of our financial condition and results of operations are based on our consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the U.S.United States. The preparation of these consolidated financial statements requires us to make estimates and judgmentsassumptions that affect the reported amounts of assets and liabilities, disclosure of contingent assets and expenses.liabilities at the date of the consolidated financial statements, and the reported revenue and expenses during the reporting period. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances. The results of these assumptions form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Under different assumptions or conditions, actual results may differ from these estimates.
We have identified certain significant accounting policiesestimates which involve a higher degree of judgment and complexity in making certain estimates and assumptions that affect amounts reported in our consolidated financial statements, as summarized below.
Revenue Recognition
We recognize revenue in accordance with Accounting Standards Codification (“ASC”) Topic 606, Revenue from Contracts with Customers, which we adopted on January 1, 2018 using the modified retrospective method.Customers. Revenues are recognized when a purchase order is received from the customer and control of the promised goods or services is transferred to the customer in an amount that reflects the consideration we expect to be entitled to receive in exchange for transferring those goods or services. Revenue is recognized based on the following five stepfive-step model:
| - Identification of the contract with a customer | |
| - Identification of the performance obligations in the contract | |
| - Determination of the transaction price | |
| - Allocation of the transaction price to the performance obligations in the contract | |
| - Recognition of revenue when, or as, we satisfy a performance obligation |
Inventories
Inventories are stated at the lower of cost or net realizable value, with cost computed on a first-in, first-out basis. Inventories consist primarily of finished goods, and also include an immaterial amount of raw materials and related packaging components. We recorded inventory obsolescence expense of $406,812 in 2023 and $540,090 in 2022. The allowance for obsolete and slow-moving inventory had a balance of $446,917 at December 31, 2023, and $523,832 at December 31, 2022.
Goodwill
The excess of purchase price over the fair value of identifiable net assets acquired in business combinations is recorded as goodwill. As of December 31, 2023 and 2022, all of our goodwill relates to the acquisition of Scendia. Goodwill has an indefinite useful life and is not amortized. Goodwill is tested annually as of December 31 for impairment, or more frequently if circumstances indicate impairment may have occurred. We may first perform a qualitative assessment to determine if it is more likely than not that the fair value of the contract withreporting unit is less than the respective carrying value. If it is determined that it is more likely than not that a customer
| 50 |
Impairment of Long-Lived Assets
Long-lived assets, including certain identifiable intangibles held and to be used by our Company,us, are reviewed for impairment whenever events or changes in circumstances including the COVID-19 pandemic, indicate that the carrying amount of such assets may not be recoverable. We continuously evaluate the recoverability of our long-lived assets based on estimated future cash flows and the estimated liquidation value of such long-lived assets and provide for impairment if such undiscounted cash flows are insufficient to recover the carrying amount of the long-lived assets. If impairment exists, an adjustment is made to write the asset down to its fair value, and a loss is recorded as the difference between the carrying value and fair value. Fair values are determined based on quoted market values, undiscounted cash flows or internal and external appraisals, as applicable. Assets to be disposed of are carried at the lower of carrying value or estimated net realizable value.fair value less cost to sell. No impairment was recorded during the years ended December 31, 20202023 and 2019.
Investments in Equity Securities
Our equity investments consist of non-marketablenonmarketable equity securities in privately held companies without readily determinable fair values, andvalues. Unless accounted for under the equity method of accounting, the investments are reported at cost minus impairment, if any, plus or minus changes resulting from observable price changes in orderly transactions for the identical or similar investment of the same issuer.
We apply the equity method of accounting for investments when we have significant influence, but not controlling interest, in the investee. Judgment regarding the level of influence over each equity method investment includes considering key factors such as ownership interest, representation on the board of directors, participation in policy-making decisions and material intercompany transactions. Our proportionate share of the net income (loss) resulting from these investments is reported under the line item captioned “Share of losses from equity method investment” in our Consolidated Statements of Operations. Our equity method investment is adjusted each period for our share of the investee’s income or loss and dividend paid, if any. We classify distributions received from our equity method investment using the cumulative earnings approach on the Consolidated Statements of Cash Flows. As a result of the Precision Healing merger in April 2022, we did not have any investments which are recorded applying the equity method of accounting as of December 31, 2023 or 2022.
We reviewed the carrying value of our investments and have determined there was no impairment or observable price changes as of December 31, 2020.
Income Taxes
We account for income taxes in accordance with ASC Topic No. 740, “IncomeIncome Taxes.” This standard requires us to provide a net deferred tax asset or liability equal to the expected future tax benefit or expense of temporary reporting differences between book and tax accounting and any available operating loss or tax credit carry forwards.
2020 | 2019 | |
| Net operating loss carry forwards (21% as of December 31, 2019) | $2,827,835 | $1,876,114 |
| Valuation allowance | (2,827,835) | (1,876,114) |
| Net non-current deferred tax asset | $- | $- |
We recognized net deferred tax liabilities associated with the Precision Healing merger and the Scendia acquisition. As of the dates of these acquisitions, prior to consideration of these acquired deferred tax liabilities, we had net deferred tax assets in excess of the deferred tax liabilities being recognized, however, a 100% valuation allowance had previously been provided against our net deferred tax assets. As a result of the recording of the net deferred tax liabilities related to the Precision Healing merger and Scendia acquisition, we reviewed the valuation allowance and determined that it should be reduced by the amount of the deferred tax liabilities that were recognized. This resulted in a 2022 income tax benefit of $5.8 million.
A 100% valuation allowance has been provided for allthe remaining net deferred tax assets as theof December 31, 2023 and 2022, as our ability of the Company to generate sufficient taxable income in the future is uncertain.
Off-Balance Sheet Arrangements
None.
ITEM 7A. QUANTITATIVEQUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
As a smaller reporting company, we are not required to provide this information.
| 51 |
ITEM 8. FINANCIALFINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
SANARA MEDTECH INC. AND SUBSIDIARIES
Index to Consolidated Financial Statements
| (Weaver and Tidwell, L.L.P. PCAOB No. 410) | |
F-5 | |
| F-1 |
Report of Independent Registered Public Accounting Firm
Board of Directors of
Sanara MedTech Inc.
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheets of Sanara MedTech Inc. and its subsidiaries (collectively, the “Company”)Company) as of December 31, 20202023 and 2019,2022, and the related consolidated statements of operations, changes in stockholders’shareholders’ equity and cash flows for each of the two years thenin the period ended December 31, 2023, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 20202023 and 2019,2022, and the results of theirits operations and theirits cash flows for each of the two years thenin the period ended December 31, 2023 in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These financial statements are the responsibility of the Company’sentity’s management. Our responsibility is to express an opinion on the Company’sentity’s financial statements based on our audits.audit. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) ("PCAOB"(“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the auditaudits to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company'sentity’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our auditsaudit also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provideaudit provides a reasonable basis for our opinion.
Critical Audit Matters
The critical audit matters are mattersmatter communicated below is a matter arising from the current period audit of the financial statements that werewas communicated or required to be communicated to the audit committee and that: (1) relaterelates to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective, or complex judgments. We determined that there are noThe communication of a critical audit matters.
Applied Asset Purchase (Refer to Note 5 to the financial statements)
Critical Audit Matter Description
The Company entered into an asset purchase agreement to acquire certain assets from the Sellers (as defined in Note 5). Management has recorded the acquisition as an asset acquisition due to its determination that substantially all of the fair value of the assets acquired was concentrated in a group of similar identifiable assets.
We identified the determination of the fair values of the acquired intangible assets as a critical audit matter because of the subjective process when developing the estimated fair values. This in turn led to significant auditor judgment and subjectivity in performing procedures relating to the valuation of acquired intangible assets and consideration paid, and significant audit effort was necessary in evaluating the significant assumptions relating to the estimates, including the timing and amounts of future cash flows. In addition, the audit effort involved the use of professionals with specialized skill and knowledge.
How the Critical Audit Matter Was Addressed in the Audit
Our audit procedures related to the Company’s estimated fair values included the following, among others:
● We obtained an understanding of the design and implementation of management’s controls over the acquisition, including the fair value of the acquired intangible assets.
● We obtained and evaluated the relevant asset purchase agreement to evaluate the completeness of the identified assets.
● With the assistance of our internal fair value specialists, we evaluated the appropriateness of the methodologies applied in determining the fair values of the acquired intangible assets, and the reasonableness of the significant inputs and assumptions used by management, and the skills, knowledge and experience of the fair value specialists engaged by management to assist in determining the fair value of the acquired intangible assets. This testing included inquiries with management, evaluation of the methods applied by management’s specialists, consideration of positive and negative evidence impacting management’s forecasts and assumptions, and evaluation of relevant market and industry factors.
● We evaluated management’s determination that substantially all of the value of the acquired assets was concentrated in a single asset or group of similar assets, and management’s conclusion that the transaction should therefore be accounted for as an asset acquisition.
We have served as the Company'sCompany’s auditor since 2014.2021.
/s/ Weaver and Tidwell, L.L.P.
Austin, Texas
March 25, 2024
| F-2 |
SANARA MEDTECH INC. AND SUBSIDIARIES
CONSOLIDATED BALANCEBALANCE SHEETS
December 31, | December 31, | |
| Assets | 2020 | 2019 |
| Current assets | ||
| Cash | $455,366 | $6,611,928 |
| Accounts receivable, net of allowances of $100,189 and $73,162 | 2,217,533 | 1,285,165 |
| Royalty receivable | 49,344 | 50,250 |
| Inventory, net of allowance for obsolescence of $276,603 and $43,650 | 1,148,253 | 746,519 |
| Prepaid and other assets | 611,817 | 161,902 |
| Total current assets | 4,482,313 | 8,855,764 |
| Long-term assets | ||
| Property, plant and equipment, net of accumulated depreciation of $124,691 and $60,694 | 678,589 | 204,953 |
| Right of use assets – operating leases | 467,653 | 585,251 |
| Intangible assets, net of accumulated amortization of $827,108 and $603,580 | 3,097,666 | 1,471,194 |
| Investment in equity securities | 1,100,000 | - |
| Total long-term assets | 5,343,908 | 2,261,398 |
| Total assets | $9,826,221 | $11,117,162 |
| Liabilities and shareholders' equity | ||
| Current liabilities | ||
| Accounts payable | $271,251 | $337,504 |
| Accounts payable – related parties | 223,589 | 68,668 |
| Accrued royalties and expenses | 502,191 | 528,060 |
| Accrued bonus and commissions | 2,417,277 | 1,588,056 |
| Operating lease liability - current | 125,587 | 117,533 |
| Total current liabilities | 3,539,895 | 2,639,821 |
| Long-term liabilities | ||
| Operating lease liability – long term | 355,797 | 481,384 |
| Convertible notes payable – related party | - | 1,500,000 |
| Accrued interest - related party | - | 103,557 |
| Other long-term liabilities | 90,293 | - |
| Total long-term liabilities | 446,090 | 2,084,941 |
| Total liabilities | 3,985,985 | 4,724,762 |
| Shareholders' equity | ||
| Series F Convertible Preferred Stock: $10 par value, 1,200,000 shares authorized; none issued and outstanding as of December 31, 2020 and 1,136,815 issued and outstanding as of December 31, 2019 | - | 11,368,150 |
| Common Stock: $0.001 par value, 20,000,000 shares authorized; 6,297,008 issued and outstanding as of December 31, 2020 and 3,571,001 issued and outstanding as of December 31, 2019 | 6,297 | 3,571 |
| Additional paid-in capital | 13,176,576 | (2,081,829) |
| Accumulated deficit | (7,032,242) | (2,675,802) |
| Total Sanara MedTech shareholders' equity | 6,150,631 | 6,614,090 |
| Equity attributable to noncontrolling interest | (310,395) | (221,690) |
| Total shareholders' equity | 5,840,236 | 6,392,400 |
| Total liabilities and shareholders' equity | $9,826,221 | $11,117,162 |
| December 31, | December 31, | |||||||
| 2023 | 2022 | |||||||
| Assets | ||||||||
| Current assets | ||||||||
| Cash | $ | 5,147,216 | $ | 8,958,995 | ||||
| Accounts receivable, net | 8,474,965 | 6,805,761 | ||||||
| Accounts receivable – related parties | 8,400 | 98,548 | ||||||
| Accounts receivable | 8,400 | 98,548 | ||||||
| Royalty receivable | 49,344 | 99,594 | ||||||
| Inventory, net | 4,717,533 | 3,549,000 | ||||||
| Prepaid and other assets | 608,411 | 1,104,611 | ||||||
| Total current assets | 19,005,869 | 20,616,509 | ||||||
| Long-term assets | ||||||||
| Intangible assets, net | 44,926,061 | 31,509,980 | ||||||
| Goodwill | 3,601,781 | 3,601,781 | ||||||
| Investment in equity securities | 3,084,278 | 3,084,278 | ||||||
| Right of use assets – operating leases | 1,995,204 | 806,402 | ||||||
| Property and equipment, net | 1,257,956 | 1,416,436 | ||||||
| Total long-term assets | 54,865,280 | 40,418,877 | ||||||
| Total assets | $ | 73,871,149 | $ | 61,035,386 | ||||
| Liabilities and shareholders’ equity | ||||||||
| Current liabilities | ||||||||
| Accounts payable | $ | 1,924,082 | $ | 1,392,701 | ||||
| Accounts payable – related parties | 77,805 | 34,036 | ||||||
| Accounts payable | 77,805 | 34,036 | ||||||
| Accrued bonuses and commissions | 7,676,770 | 7,758,284 | ||||||
| Accrued royalties and expenses | 2,047,678 | 2,144,475 | ||||||
| Earnout liabilities – current | 1,100,000 | 1,162,880 | ||||||
| Current portion of debt | 580,357 | - | ||||||
| Operating lease liabilities – current | 361,185 | 313,933 | ||||||
| Total current liabilities | 13,767,877 | 12,806,309 | ||||||
| Long-term liabilities | ||||||||
| Long-term debt, net of current portion | 9,113,123 | - | ||||||
| Earnout liabilities – long-term | 2,723,001 | 6,003,811 | ||||||
| Operating lease liabilities – long-term | 1,737,445 | 505,291 | ||||||
| Other long-term liabilities | 1,941,686 | - | ||||||
| Total long-term liabilities | 15,515,255 | 6,509,102 | ||||||
| Total liabilities | 29,283,132 | 19,315,411 | ||||||
| Commitments and contingencies (Note 10) | - | |||||||
| Shareholders’ equity | ||||||||
| Common Stock: $ par value, shares authorized; issued and outstanding as of December 31, 2023 and issued and outstanding as of December 31, 2022 | 8,535 | 8,300 | ||||||
| Additional paid-in capital | 72,860,556 | 65,213,987 | ||||||
| Accumulated deficit | (28,036,814 | ) | (23,394,757 | ) | ||||
| Total Sanara MedTech shareholders’ equity | 44,832,277 | 41,827,530 | ||||||
| Equity attributable to noncontrolling interest | (244,260 | ) | (107,555 | ) | ||||
| Total shareholders’ equity | 44,588,017 | 41,719,975 | ||||||
| Total liabilities and shareholders’ equity | $ | 73,871,149 | $ | 61,035,386 | ||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-3 |
SANARA MEDTECH INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF OPERATIONS
Year Ended | ||
December 31, | ||
2020 | 2019 | |
| Net Revenue | $15,586,976 | $11,766,763 |
| Cost of goods sold | 1,616,625 | 1,209,300 |
| Gross profit | 13,970,351 | 10,557,463 |
| Operating expenses | ||
| Selling, general and administrative expenses | 18,683,594 | 13,067,569 |
| Depreciation and amortization | 291,370 | 119,951 |
| Bad debt expense | 30,000 | 110,000 |
| Total operating expenses | 19,004,964 | 13,297,520 |
| Operating loss | (5,034,613) | (2,740,057) |
| Other income / (expense) | ||
| Other income | 14,822 | 10,198 |
| Interest expense | (11,528) | (105,919) |
| Debt forgiveness | 586,174 | - |
| Total other income / (expense) | 589,468 | (95,721) |
| Net loss | (4,445,145) | (2,835,778) |
| Less: Net loss attributable to noncontrolling interest | (88,705) | (21,690) |
| Net loss attributable to Sanara MedTech common shareholders | $(4,356,440) | $(2,814,088) |
| Basic loss per share of Common stock | $(0.76) | $(1.32) |
| Diluted loss per share of Common stock | $(0.76) | $(1.32) |
| Weighted average number of common shares outstanding basic | 5,734,537 | 2,132,745 |
| Weighted average number of common shares outstanding diluted | 5,734,537 | 2,132,745 |
| Year Ended | ||||||||
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| Net Revenue | $ | 64,989,842 | $ | 45,842,845 | ||||
| Cost of goods sold | 7,852,686 | 6,360,851 | ||||||
| Gross profit | 57,137,156 | 39,481,994 | ||||||
| Operating expenses | ||||||||
| Selling, general and administrative expenses | 56,994,753 | 45,976,328 | ||||||
| Research and development | 4,132,425 | 3,367,032 | ||||||
| Depreciation and amortization | 3,675,026 | 2,371,068 | ||||||
| Change in fair value of earnout liabilities | (3,449,895 | ) | 284,746 | |||||
| Total operating expenses | 61,352,309 | 51,999,174 | ||||||
| Operating loss | (4,215,153 | ) | (12,517,180 | ) | ||||
| Other income (expense) | ||||||||
| Interest expense and other | (475,783 | ) | - | |||||
| Share of losses from equity method investment | - | (379,633 | ) | |||||
| Gain (loss) on disposal of investment | 251,034 | (1,040,311 | ) | |||||
| Total other income (expense) | (224,749 | ) | (1,419,944 | ) | ||||
| Loss before income taxes | (4,439,902 | ) | (13,937,124 | ) | ||||
| Income tax benefit | - | 5,844,796 | ||||||
| Net loss | (4,439,902 | ) | (8,092,328 | ) | ||||
| Less: Net loss attributable to noncontrolling interest | (136,705 | ) | (154,831 | ) | ||||
| Net loss attributable to Sanara MedTech shareholders | $ | (4,303,197 | ) | $ | (7,937,497 | ) | ||
| Net loss per share of common stock, basic and diluted | $ | ) | $ | ) | ||||
| Weighted average number of common shares outstanding, basic and diluted | ||||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-4 |
SANARA MEDTECH INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS’ EQUITY
Preferred Stock Series F | Common Stock | Additional | Total | |||||||
$10 par value | $0.001 par value | Paid-In | Treasury Stock | Accumulated | Noncontrolling | Shareholders' | ||||
Shares | Amount | Shares | Amount | Capital | Shares | Amount | Income/(Deficit) | Interest | Equity | |
| Balance at December 31, 2018 | 1,136,815 | $11,368,150 | - | $- | $(10,919,639) | - | $- | $138,286 | $- | $586,797 |
| Reverse recapitalization | - | - | 2,366,465 | 2,366 | (1,159,929) | (41) | - | - | - | (1,157,563) |
| Treasury stock retirement | - | - | (41) | - | - | 41 | - | - | - | - |
| Repurchase and cancellation of fractional shares | - | - | (243) | - | (1,061) | - | - | - | - | (1,061) |
| Private placement stock issue | - | - | 1,204,820 | 1,205 | 9,998,800 | - | - | - | - | 10,000,005 |
| Advance on future noncontrolling interest distribution | - | - | - | - | - | - | - | - | (200,000) | (200,000) |
| Net loss | - | - | - | - | - | - | - | (2,814,088) | (21,690) | (2,835,778) |
| Balance at December 31, 2019 | 1,136,815 | $11,368,150 | 3,571,001 | $3,571 | $(2,081,829) | - | $- | $(2,675,802) | $(221,690) | $6,392,400 |
| Conversion of Preferred Shares to Common Stock | (1,136,815) | (11,368,150) | 2,273,630 | 2,274 | 11,365,876 | - | - | - | - | - |
| Conversion of Promissory Note to Common Stock | - | - | 179,101 | 179 | 1,611,732 | - | - | - | - | 1,611,911 |
| Issuance of Common Stock for intangible asset | - | - | 60,000 | 60 | 749,940 | - | - | - | - | 750,000 |
| Employee stock purchase program | - | - | 3,735 | 4 | 39,326 | - | - | - | - | 39,330 |
| Share-based compensation | - | - | 209,541 | 209 | 1,491,531 | - | - | - | - | 1,491,740 |
| Net loss | - | - | - | - | - | - | - | (4,356,440) | (88,705) | (4,445,145) |
| Balance at December 31, 2020 | - | $- | 6,297,008 | $6,297 | $13,176,576 | - | $- | $(7,032,242) | $(310,395) | $5,840,236 |
| Shares | Amount | Capital | Deficit | Interest | Equity | |||||||||||||||||||
| Common Stock | Additional | Total | ||||||||||||||||||||||
| $0.001 par value | Paid-In | Accumulated | Noncontrolling | Shareholders’ | ||||||||||||||||||||
| Shares | Amount | Capital | Deficit | Interest | Equity | |||||||||||||||||||
| Balance at December 31, 2021 | 7,676,662 | $ | 7,677 | $ | 45,867,768 | $ | (15,235,044 | ) | $ | (488,397 | ) | $ | 30,152,004 | |||||||||||
| Issuance of common stock for acquisitions | 457,424 | 457 | 11,128,053 | - | (2,638 | ) | 11,125,872 | |||||||||||||||||
| Issuance of common stock options and warrants for acquisitions | - | - | 4,612,645 | - | - | 4,612,645 | ||||||||||||||||||
| Share-based compensation | 173,959 | 174 | 3,790,643 | - | - | 3,790,817 | ||||||||||||||||||
| Net settlement and retirement of equity-based awards | (8,088 | ) | (8 | ) | (185,122 | ) | (222,216 | ) | - | (407,346 | ) | |||||||||||||
| Distribution to noncontrolling interest member | - | - | - | - | (220,000 | ) | (220,000 | ) | ||||||||||||||||
| Dissolution of investment | - | - | - | - | 758,311 | 758,311 | ||||||||||||||||||
| Net loss | - | - | - | (7,937,497 | ) | (154,831 | ) | (8,092,328 | ) | |||||||||||||||
| Balance at December 31, 2022 | 8,299,957 | $ | 8,300 | $ | 65,213,987 | $ | (23,394,757 | ) | $ | (107,555 | ) | $ | 41,719,975 | |||||||||||
| Balance | 8,299,957 | $ | 8,300 | $ | 65,213,987 | $ | (23,394,757 | ) | $ | (107,555 | ) | $ | 41,719,975 | |||||||||||
| Share-based compensation | 100,829 | 100 | 3,442,622 | - | - | 3,442,722 | ||||||||||||||||||
| Net settlement and retirement of equity-based awards | 34,501 | 35 | 203,031 | (338,860 | ) | - | (135,794 | ) | ||||||||||||||||
| Issuance of common stock for acquisitions | 73,809 | 74 | 3,089,571 | - | - | 3,089,645 | ||||||||||||||||||
| Issuance of common stock in equity offering | 26,143 | 26 | 911,345 | - | - | 911,371 | ||||||||||||||||||
| Net loss | - | - | - | (4,303,197 | ) | (136,705 | ) | (4,439,902 | ) | |||||||||||||||
| Balance at December 31, 2023 | 8,535,239 | $ | 8,535 | $ | 72,860,556 | $ | (28,036,814 | ) | $ | (244,260 | ) | $ | 44,588,017 | |||||||||||
| Balance | 8,535,239 | $ | 8,535 | $ | 72,860,556 | $ | (28,036,814 | ) | $ | (244,260 | ) | $ | 44,588,017 | |||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-5 |
SANARA MEDTECH INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CASHCASH FLOWS
Year Ended | ||
December 31, | ||
2020 | 2019 | |
| Cash flows from operating activities: | ||
| Net loss | $(4,445,145) | $(2,835,778) |
| Adjustments to reconcile net loss to net cash used in operating activities | ||
| Depreciation and amortization | 291,370 | 119,951 |
| Interest expense on convertible debt | 8,354 | 61,934 |
| Interest expense on PPP loan | 3,174 | - |
| Loss on disposal of asset | 2,897 | 15,944 |
| Bad debt expense | 30,000 | 110,000 |
| Inventory obsolescence | 318,076 | 120,442 |
| Share-based compensation | 1,402,897 | - |
| Noncash lease expense | 117,598 | 99,009 |
| Debt forgiveness, including interest | (586,174) | - |
| Changes in operating assets and liabilities: | ||
| Accounts receivable | (961,462) | (324,368) |
| Inventory | (719,810) | (401,647) |
| Prepaid and other assets | (449,915) | (554,969) |
| Accounts payable | (66,253) | (65,037) |
| Accounts payable - related parties | 154,921 | (19,599) |
| Accrued royalties and expenses | (25,870) | 282,004 |
| Accrued liabilities | 890,824 | 1,224,713 |
| Net cash used in operating activities | (4,034,518) | (2,167,401) |
| Cash flows from investing activities: | ||
| Purchase of property and equipment | (544,374) | (182,825) |
| Cash received in reverse acquisition | - | 508,973 |
| Repurchase and cancellation of fractional shares | - | (1,061) |
| Proceeds from disposal of assets | - | 301 |
| Purchase of intangible assets | (1,100,000) | (1,522,485) |
| Investment in equity securities | (1,100,000) | - |
| Net cash used in investing activities | (2,744,374) | (1,197,097) |
| Cash flows from financing activities: | ||
| Draw on line of credit | - | 2,200,000 |
| Pay off line of credit | - | (2,200,000) |
| Proceeds from PPP Loan | 583,000 | - |
| Private placement stock issue | - | 10,000,005 |
| Advance on future noncontrolling interest distribution | - | (200,000) |
| Common stock issued for Employee Stock Purchase Plan | 39,330 | - |
| Net cash provided by financing activities | 622,330 | 9,800,005 |
| Net increase (decrease) in cash | (6,156,562) | 6,435,507 |
| Cash, beginning of period | 6,611,928 | 176,421 |
| Cash, end of period | $455,366 | $6,611,928 |
| Cash paid during the period for: | ||
| Interest | $- | $43,985 |
| Income taxes | - | - |
| Supplemental noncash investing and financing activities: | ||
| Common stock issued for conversion of Series F Preferred Stock | 11,368,150 | - |
| Common stock issued for conversion of related party debt and interest | 1,611,911 | - |
| Common stock issued for purchase of intangible asset | 750,000 | - |
| Common stock issued in reverse capitalization; less cash received of $508,973 | - | 1,666,537 |
| 2023 | 2022 | |||||||
| Year Ended | ||||||||
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| Cash flows from operating activities: | ||||||||
| Net loss | $ | (4,439,902 | ) | $ | (8,092,328 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Depreciation and amortization | 3,675,026 | 2,371,068 | ||||||
| Loss on disposal of property and equipment | - | 2,634 | ||||||
| Bad debt expense | 202,941 | 280,000 | ||||||
| Inventory obsolescence | 406,812 | 540,090 | ||||||
| Share-based compensation | 3,442,722 | 2,702,633 | ||||||
| Noncash lease expense | 342,972 | 263,518 | ||||||
| Loss on equity method investment | - | 379,633 | ||||||
| (Gain) Loss on disposal of investment | (251,034 | ) | 1,040,311 | |||||
| Benefit from deferred income taxes | - | (5,844,796 | ) | |||||
| Accretion of finance liabilities | 98,926 | - | ||||||
| Amortization of debt issuance costs | 5,138 | - | ||||||
| Change in fair value of earnout liabilities | (3,449,895 | ) | 284,746 | |||||
| Changes in operating assets and liabilities: | ||||||||
| Accounts receivable, net | (1,821,895 | ) | (2,250,223 | ) | ||||
| Accounts receivable – related parties | 90,148 | (18,761 | ) | |||||
| Inventory, net | (1,545,339 | ) | (517,271 | ) | ||||
| Prepaid and other assets | 496,200 | (159,592 | ) | |||||
| Accounts payable | 531,380 | (301,966 | ) | |||||
| Accounts payable – related parties | 43,768 | (121,781 | ) | |||||
| Accrued royalties and expenses | (739,645 | ) | 1,156,073 | |||||
| Accrued bonuses and commissions | (81,513 | ) | 2,994,512 | |||||
| Operating lease liabilities | (252,366 | ) | (263,370 | ) | ||||
| Net cash used in operating activities | (3,245,556 | ) | (5,554,870 | ) | ||||
| Cash flows from investing activities: | ||||||||
| Purchases of property and equipment | (265,246 | ) | (147,015 | ) | ||||
| Proceeds from disposal of property and equipment | 650 | 1,549 | ||||||
| Purchases of intangible assets | - | (600,000 | ) | |||||
| Investment in equity securities | - | (250,000 | ) | |||||
| Acquisitions, net of cash acquired | (9,942,750 | ) | (2,516,164 | ) | ||||
| Net cash used in investing activities | (10,207,346 | ) | (3,511,630 | ) | ||||
| Cash flows from financing activities: | ||||||||
| Loan proceeds, net | 9,688,341 | - | ||||||
| Equity offering net proceeds | 911,371 | - | ||||||
| Net settlement of equity-based awards | (135,794 | ) | (407,346 | ) | ||||
| Cash payment of finance and earnout liabilities | (822,795 | ) | - | |||||
| Distribution to noncontrolling interest member | - | (220,000 | ) | |||||
| Net cash provided by (used in) financing activities | 9,641,123 | (627,346 | ) | |||||
| Net decrease in cash | (3,811,779 | ) | (9,693,846 | ) | ||||
| Cash, beginning of period | 8,958,995 | 18,652,841 | ||||||
| Cash, end of period | $ | 5,147,216 | $ | 8,958,995 | ||||
| Cash paid during the period for: | ||||||||
| Interest | $ | 283,948 | $ | 206 | ||||
| Supplemental noncash investing and financing activities: | ||||||||
| Right of use assets obtained in exchange for lease obligations | 1,531,773 | - | ||||||
| Equity issued for acquisitions | 3,089,645 | 15,738,518 | ||||||
| Earnout and other liabilities generated by acquisitions | 3,759,642 | 6,882,151 | ||||||
| Investment in equity securities converted in asset acquisition | - | 1,803,440 | ||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-6 |
SANARA MEDTECH INC. AND SUBSIDIARIES
NOTE 1 – NATURE OF BUSINESS AND BACKGROUND
Sanara MedTech Inc. (“we”, “our”,(together with its wholly owned and majority owned subsidiaries on a consolidated basis, the “Company”) is a medical technology company focused on developing and commercializing transformative technologies to improve clinical outcomes and reduce healthcare expenditures in the surgical, and chronic wound and skin careskincare markets. Our portfolio of products and services will allow us to deliver comprehensive wound and skin care solutions for patients in all care settings, including acute (hospitals and long-term acute care hospitals) and post-acute (wound care clinics, physician offices, skilled nursing facilities (“SNFs”), home health, hospice, and retail). Each of ourthe Company’s products, services and technologies contributesare designed to our overallachieve the Company’s goal of achievingproviding better clinical outcomes at a lower overall cost for patients regardless of where they receive care. We striveThe Company strives to be one of the most innovative and comprehensive providers of effective surgical, wound and skin care productsskincare solutions and technologies and areis continually seeking to expand ourits offerings for patients requiring wound and skin care treatments across the entire continuum of care in the United States.
NOTE 2 -- — SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Principles of Consolidation
The accompanying consolidated financial statements include the accounts of Sanara MedTech Inc. and its wholly owned subsidiaries. The consolidatedand majority-owned subsidiaries, as well as other entities in which the Company has a controlling financial statements also include the accounts of Sanara Pulsar, which is owned 60% by the Company’s wholly owned subsidiary Cellerate, LLC, and 40% owned by Wound Care Solutions, Limited, an unaffiliated company registered in the United Kingdom (“WCS”).interest. All significant intercompany profits, losses, transactions and balances have been eliminated in consolidation.
Use of Estimates
The preparation of financial statements in conformity with generally accepted accounting principles requires management to make estimates and assumptions that affect the reported amounts reported inof assets and liabilities, disclosure of contingent assets and liabilities at the date of the consolidated financial statements, and accompanying notes. The extent to which the COVID-19 pandemic may directly or indirectly impact our business, financial condition,reported revenue and results of operations is highly uncertain and subject to change. We consideredexpenses during the potential impact of the COVID-19 pandemic on our estimates and assumptions and determined there was not a material impact on our estimates and assumptions used in preparing our consolidated financial statements as of and for the
Cash and Cash Equivalents
The Company considers all highly liquid investments purchased with an original maturity of three months or less to be cash equivalents.
The Company computes incomeincome/loss per share in accordance with Accounting Standards Codification (“ASC”) Topic 260, Earnings per Share, which requires the Company to present basic and dilutivediluted income per share when the effect is dilutive. Basic income per share is computed by dividing income available to common shareholders by the weighted average number of shares of common stock outstanding. Diluted income per share is computed similarsimilarly to basic income per share, except that the denominator is increased to include the number of additional shares of common stock that would have been outstanding if the potential shares of common stock had been issued and if the additional shares of common stock were dilutive. All convertible instrumentscommon stock equivalents were excluded from the current and prior period calculations as their inclusion would have been anti-dilutive during the years ended December 31, 2020 2023 and 2019 2022 due to the Company'sCompany’s net loss.
December 31, | ||
2020 | 2019 | |
| Numerator for basic and diluted net loss per share: | ||
| Net loss attributable to Sanara MedTech common shareholders | $(4,356,440) | $(2,814,088) |
| Denominator for basic and diluted net loss per share: | ||
| Weighted average shares used to compute diluted net loss per share | 5,734,537 | 2,132,745 |
| Basic and diluted net loss per share attributable to common shareholders | $(0.76) | $(1.32) |
SCHEDULE OF COMPUTATION OF DILUTED NET LOSS PER SHARE
| As of December 31, | ||||||||
| 2023 | 2022 | |||||||
| Stock options (a) | 93,892 | 146,191 | ||||||
| Warrants (b) | 16,725 | 16,725 | ||||||
| Unvested restricted stock | 144,211 | 181,102 | ||||||
| Anti-dilutive securities | 144,211 | 181,102 | ||||||
| (a) | Shares underlying stock options assumed pursuant to the merger agreement with Precision Healing, Inc. (“Precision Healing”) in April 2022. See Note 3 for more information regarding the Precision Healing merger. | |
| (b) | Shares underlying warrants assumed pursuant to the merger agreement with Precision Healing in April 2022. See Note 3 for more information regarding the Precision Healing merger. |
| F-7 |
As of December 31, | ||
2020 | 2019 | |
| Stock options | 11,500 | 11,500 |
| Convertible debt | - | 178,173 |
| Preferred shares | - | 2,273,630 |
| Unvested restricted stock | 170,178 | - |
Revenue Recognition
The Company recognizes revenue in accordance with ASC Topic 606, Revenue from Contracts with Customers (“ASC 606”), which the Company adopted on January 1, 2018 using the modified retrospective method.. Revenues are recognized when a purchase order is received from the customer and control of the promised goods or services is transferred to the customer in an amount that reflects the consideration the Company expects to be entitled to receive in exchange for transferring those goods or services. Revenue is recognized based on the following five stepfive-step model:
- Identification of the contract with a customer
- Identification of the performance obligations in the contract
- Determination of the transaction price
- Allocation of the transaction price to the performance obligations in the contract
- Recognition of revenue when, or as, the Company satisfies a performance obligation
Details of this five-step process are as follows:
Identification of the contract with a customer
Customer purchase orders are generally considered to be contracts under ASC 606. Purchase orders typically identify the specific terms of products to be delivered, create the enforceable rights and obligations of both parties and result in commercial substance. No other forms of contract revenue recognition, such as the completed contract or percentage of completion methods, were utilized by the Company in either
Performance obligations
The Company’s performance obligation is generally limited to delivery of the requested items to its customers at the agreed upon quantities and prices.
Determination and allocation of the transaction price
The Company has established prices for its products. These prices are effectively agreed to when customers place purchase orders with the Company. Rebates and discounts, if any, are recognized in full at the time of sale as a reduction of net revenue. Allocation of transaction prices is not necessary where only one performance obligation exists.
Recognition of revenue as performance obligations are satisfied
Product revenues are recognized when a purchase order is received from the customer, the products are delivered and control of the goods and services passes to the customer.
Disaggregation of Revenue
Revenue streams from product sales and royalties are summarized below for the
Year Ended | ||
December 31, | ||
2020 | 2019 | |
| Product sales revenue | $15,385,976 | $11,607,638 |
| Royalty revenue | 201,000 | 159,125 |
| Total Revenue | $15,586,976 | $11,766,763 |
SCHEDULE OF REVENUE FROM PRODUCT SALES AND ROYALTIES
| 2023 | 2022 | |||||||
| For the Year Ended | ||||||||
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| Soft tissue repair products | $ | 54,836,410 | $ | 41,653,954 | ||||
| Bone fusion products | 9,952,432 | 3,987,891 | ||||||
| Royalty revenue | 201,000 | 201,000 | ||||||
| Total Net Revenue | $ | 64,989,842 | $ | 45,842,845 | ||||
The Company recognizes royalty revenue from a development and licensinglicense agreement betweenwith BioStructures, LLC and the Company.LLC. The Company records revenue each calendar quarter as earned per the terms of the agreement, which stipulates the Company will receive quarterly royalty payments of at least $50,250.$50,250. Under the terms of the development and license agreement, royalties of 2.0%2.0% are recognized on sales of products containing the Company’s patented resorbable bone hemostasis. The minimum annual royalty due to the Company is $201,000$201,000 per year throughoutthrough the lifeend of the patent which expires in 2023. These royalties are payable in quarterly installments of $50,250.$50,250. To date, royalties related to this development and licensinglicense agreement have not exceeded the annual minimum amount of $201,000$201,000 ($50,250 per quarter).
| F-8 |
Accounts Receivable Allowances
Accounts receivable are typically due within 30 days of invoicing. The Company establishes an allowance for doubtful accounts to ensureprovide for an estimate of accounts receivable which are not overstated dueexpected to uncollectible accounts.be collectible. The Company bases the allowance on an assessment of customer creditworthiness, historical payment experience, the age of outstanding receivables and other information as applicable and will record its allowance based on the estimated credit losses. The Company recorded bad debt expense of
Inventories
Inventories are stated at the lower of cost or net realizable value, with cost computed on a first-in, first-out basis. Inventories consist primarily of finished goods, and also include an immaterial amount of raw materials and related packaging components. The Company recorded inventory obsolescence expense of $318,076$406,812 in 20202023 and $120,442$540,090 in 2019.2022. The allowance for obsolete and slow-moving inventory had a balance of $276,603$446,917 at December 31, 2020,2023, and $43,650$523,832 at December 31, 2019. The Company considered the impact of COVID-19 on its recorded value of inventory and determined no adjustment was necessary as of December 31, 2020.
Property and Equipment
Property and equipment are stated at cost, less accumulated depreciation. Depreciation is recorded using the straight-line method over the estimated useful lives of the related assets, ranging from threetwo to ten years. Below is a summary of property and equipment for the periods presented:
December 31, | December 31, | |
2020 | 2019 | |
| Computers | $87,252 | $87,310 |
| Office equipment | 22,597 | 22,312 |
| Furniture and fixtures | 205,871 | 153,995 |
| Leasehold improvements | 2,030 | 2,030 |
| Capitalized software development costs | 485,530 | - |
803,280 | 265,647 | |
| Less accumulated depreciation | (124,691) | (60,694) |
| Property and equipment, net | $678,589 | $204,953 |
SCHEDULE OF PROPERTY AND EQUIPMENT
| Useful | December 31, | December 31, | ||||||||
| Life | 2023 | 2022 | ||||||||
| Computers | 3-5 years | $ | 194,788 | $ | 172,154 | |||||
| Office equipment | 3-7 years | 201,785 | 87,225 | |||||||
| Furniture and fixtures | 5-10 years | 304,338 | 258,414 | |||||||
| Leasehold improvements | 2-5 years | 134,170 | 19,631 | |||||||
| Internal use software | 5 years | 1,618,999 | 1,618,998 | |||||||
| Property and equipment, gross | 2,454,080 | 2,156,422 | ||||||||
| Less accumulated depreciation | (1,196,124 | ) | (739,986 | ) | ||||||
| Property and equipment, net | $ | 1,257,956 | $ | 1,416,436 | ||||||
Depreciation expense related to property and equipment was $67,842$456,138 and $407,769 for the twelve monthsyear ended December 31, 2020,2023 and $29,940 for the twelve months ended December 31, 2019.2022, respectively.
| F-9 |
Internal Use Software
The Company accounts for costs incurred to develop or acquire computer software for internal use in accordance with ASC 350-40.Topic 350-40, Intangibles – Goodwill and Other. The Company capitalizes the costs incurred during the application development stage, which generally includes third-party developer fees to design the software configuration and interfaces, coding, installation and testing.
The Company begins capitalization of qualifying costs when both the preliminary project stage is completed and management has authorized further funding for the completion of the project. Costs incurred during the preliminary project stage along with post implementation stages of internal-use computer software are expensed as incurred. The Company also capitalizes costs related to specific upgrades and enhancements when it is probable the expenditures will result in additional functionality. Capitalized development costs are classified as property“Property and equipment, netnet” in the consolidated balance sheetsConsolidated Balance Sheets and are amortizeddepreciated over the estimated useful life of the software, which is generally five years.
Goodwill
The excess of purchase price over the fair value of identifiable net assets acquired in business combinations is recorded as goodwill. As of December 31, 2023 and December 31, 2022, all of the Company’s goodwill relates to seven years.
Intangible Assets
Intangible Assetsassets are stated at cost of acquisition less accumulated amortization and impairment loss, if any. Cost of acquisition includes the purchase price and any cost directly attributable to bringing the asset to its working condition for the intended use. The Company amortizes its finite-lived intangible assets on a straight-line basis over the estimated useful life of the respective assets which is generally the life of the related patents (if applicable).
Impairment of Long-Lived Assets
Long-lived assets, including certain identifiable intangibles held and to be used by the Company, are reviewed for impairment whenever events or changes in circumstances including the COVID-19 pandemic, indicate that the carrying amount of such assets may not be recoverable. The Company continuously evaluates the recoverability of its long-lived assets based on estimated future cash flows and the estimated liquidation value of such long-lived assets and provides for impairment if such undiscounted cash flows are insufficient to recover the carrying amount of the long-lived assets. If impairment exists, an adjustment is made to write the asset down to its fair value, and a loss is recorded as the difference between the carrying value and fair value. Fair values are determined based on quoted market values, undiscounted cash flows or internal and external appraisals, as applicable. Assets to be disposed of are carried at the lower of carrying value or estimated net realizable value.fair value less cost to sell. No impairment was recorded
Investments in Equity Securities
The Company’s equity investments consist of non-marketablenonmarketable equity securities in privately held companies without readily determinable fair values, andvalues. Unless accounted for under the equity method of accounting, the investments are reported at cost minus impairment, if any, plus or minus changes resulting from observable price changes in orderly transactions for the identical or similar investment of the same issuer.
The Company applies the equity method of accounting to investments when it has significant influence, but not controlling interest, in the investee. Judgment regarding the level of influence over each equity method investment includes considering key factors such as ownership interest, representation on the board of directors, participation in policy-making decisions and material intercompany transactions. The Company’s proportionate share of the net income (loss) resulting from these investments is reported under the line item captioned “Share of losses from equity method investment” in the Company’s Consolidated Statements of Operations. The Company’s equity method investment is adjusted each period for the Company’s share of the investee’s income or loss and dividend paid, if any. The Company classifies distributions received from its equity method investment using the cumulative earnings approach in the Company’s Consolidated Statements of Cash Flows. As a result of the Precision Healing merger in April 2022 (see Note 3), as of December 31, 2022, the Company does not have any investments which are recorded applying the equity method of accounting.
The Company has reviewed the carrying value of its investments and has determined there was no impairment or observable price changes as of December 31, 2020.2023 and 2022.
| F-10 |
Fair Value Measurement
As defined in ASC Topic 820, Fair Value Measurement (“ASC 820”), fair value is the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date (exit price). The Company utilizes market data or assumptions that market participants would use in pricing the asset or liability, including assumptions about risk and the risks inherent in the inputs to the valuation technique. These inputs can be readily observable, market corroborated, or generally unobservable. ASC 820 establishes a fair value hierarchy that prioritizes the inputs used to measure fair value. The hierarchy gives the highest priority to unadjusted quoted prices in active markets for identical assets or liabilities (level(Level 1 measurement) and the lowest priority to unobservable inputs (level(Level 3 measurement). This fair value measurement framework applies at both initial and subsequent measurement.
The three levels of the fair value hierarchy defined by ASC 820 are as follows:
Level 1 – Quoted prices are available in active markets for identical assets or liabilities as of the reporting date. Active markets are those in which transactions for the asset or liability occur in sufficient frequency and volume to provide pricing information on an ongoing basis. Level 1 primarily consists of financial instruments such as exchange-traded derivatives, marketable securities and listed equities.
Level 2 – Pricing inputs are other than quoted prices in active markets included in Level 1, which are either directly or indirectly observable as of the reported date. Level 2 includes those financial instruments that are valued using models or other valuation methodologies. These models are primarily industry-standard models that consider various assumptions, including quoted forward prices for commodities, time value, volatility factors, and current market and contractual prices for the underlying instruments, as well as other relevant economic measures. Substantially all of these assumptions are observable in the marketplace throughout the full term of the instrument, can be derived from observable data or are supported by observable levels at which transactions are executed in the marketplace. Instruments in this category generally include non-exchange-tradednonexchange-traded derivatives such as commodity swaps, interest rate swaps, options and collars.
Level 3 – Pricing inputs include significant inputs that are generally less observable from objective sources. These inputs may be used with internally developed methodologies that result in management’s best estimate of fair value.
The carrying amounts of cash, accounts receivable, accounts payable and accrued expenses, other than acquisition-related expenses, approximate fair value because of the short-term nature of these instruments. The Company does not have any assets or liabilities thatfair value of acquisition-related accrued expenses is categorized as Level 2 of the fair value hierarchy. The value of these instruments has been estimated using discounted cash flow analysis based on the Company’s incremental borrowing rate. The carrying value of the Company’s debt, which has variable interest rates determined each month, approximates fair value based on instruments with similar terms (Level 2 inputs). The fair value of the contingent earnout consideration and the acquisition date fair value of goodwill and intangibles related to the acquisitions discussed in Notes 3, 4 and 5 are required to bebased on Level 3 inputs.
Liabilities for contingent consideration for the Company’s Precision Healing, Scendia and Applied acquisitions are measured and recorded at fair value oneach reporting period, with the acquisition-date fair value included as part of the consideration transferred. Subsequent changes in fair value for the Precision Healing and Scendia acquisitions are reported under the line item captioned “Change in fair value of earnout liabilities” in the Company’s Consolidated Statements of Operations. Due to the Applied Asset Purchase being accounted for as an asset acquisition and given that the transaction did not include contingent shares, subsequent revaluations of contingent consideration for the Applied acquisition results in an adjustment to the contingent consideration liability and the intellectual property intangible asset with a recurring basis.
cumulative catch-up amortization adjustment. The current year changes in fair value of earnout liabilities below are as a result of a net decrease in the estimated fair value of the earnout liabilities established at the time of the Company’s Precision Healing and Scendia acquisitions. The current year revaluation of earnout liability is a result of a decrease in the estimated earnout liability established at the time of the Company’s Applied acquisition. The following table sets forth a summary of the changes in fair value for the Level 3 contingent earnout considerations.
SCHEDULE OF CHANGES IN FAIR VALUE FOR CONTINGENT EARNOUT CONSIDERATION
| Balance at December 31, 2022 | $ | 7,166,691 | ||
| Additions | 893,000 | |||
| Changes in fair value of earnout liabilities | (3,449,895 | ) | ||
| Revaluation of earnout liability | (94,000 | ) | ||
| Settlements | (692,795 | ) | ||
| Balance at December 31, 2023 | $ | 3,823,001 |
| F-11 |
Income Taxes
Income taxes are accounted for under the asset and liability method, whereby deferred income taxes are recorded for temporary differences between financial statement carrying amounts and the tax basis of assets and liabilities. Deferred tax assets and liabilities reflect the tax rates expected to be in effect for the years in which the differences are expected to reverse. A valuation allowance is provided if it is more likely than not that some or all of the deferred tax asset will not be realized.
The Company accounts for stock-based compensation to employees and nonemployees in accordance with Accounting Standards UpdateASC Topic 718, Compensation – Stock Compensation (“ASU”ASC 718”) 2018-07 Topic 718.. Stock-based compensation is measured at the grant date, based on the fair value of the award, and is recognized as expense over the stipulated vesting period, (if any).if any. The Company estimates the fair value of stock-based payments using the Black-Scholes option-pricing model for common stock options and warrants, and the closing price of the Company’s common stock for grants of common stock, issuances.
Research and Development Costs
Research and development (“R&D”) expenses consist of personnel-related expenses, including salaries and benefits for all personnel directly engaged in R&D activities, contracted services, materials, prototype expenses and allocated overhead which is comprised of lease expense and other facilities-related costs. R&D expenses include costs for contracted services related to improvements to manufacturing processes, enhancements to the Company’s currently available products and additional investments in the product and platform development pipeline. The Company expenses research and developmentR&D costs as incurred.
Recently Adopted Accounting Pronouncements
In 2019,June 2016, the Financial Accounting Standards Board (“FASB”) issued ASU 2016-13, Financial Instruments - Credit Losses (Topic 326). This update amends the impairment model by requiring entities to use a forward-looking approach based on expected losses to estimate credit losses for most financial assets and certain other instruments that are not measured at fair value through net income. The Company changed its methodadopted the new guidance effective January 1, 2023. The adoption did not have a material impact on the Company’s consolidated financial position, results of accounting for leases due to the adoption of ASU No. 2016-02, Leases; as modified by ASUs 2018-01, 2018-10, 2018-11, and 2018-20.
Recent Accounting Pronouncements
In December 2019,November 2023, the FASB issued ASU 2019-12, Simplifications2023-07, Segment Reporting (Topic 280): Improvements to Accounting for Income Taxes,Reportable Segment Disclosures (“ASU 2023-07”), which removes certain exceptions to the general principlesrequires disclosure of Topic 740incremental segment information on an annual and adds guidance to reduce complexity in accounting for income taxes. Theinterim basis. ASU 2023-07 is effective for annualfiscal years beginning after December 15, 2023, and interim periods inwithin fiscal years beginning after December 15, 2020.2024 on a retrospective basis. The Company is currently evaluating the impact of the adoptioneffect of this standardpronouncement on its disclosures.
In December 2023, the Company's consolidated financial statements.
NOTE 3 – NOTES PAYABLEPRECISION HEALING MERGER
In April 2022, the Company entered into a merger agreement by and among the Company, United Wound and Skin Solutions, LLC, a Delaware limited liability company and wholly owned subsidiary of the Company, Precision Healing, PH Merger Sub I, Inc., a Delaware corporation, PH Merger Sub II, LLC, a Delaware limited liability company, and Furneaux Capital Holdco, LLC (d/b/a BlueIO), solely in its capacity as the representative of the securityholders of Precision Healing. On April 4, 2022 (the “Closing Date”), the merger parties closed the transactions contemplated by the merger agreement and Precision Healing became a wholly owned subsidiary of the Company.
Precision Healing is developing a diagnostic imager and lateral flow assay for assessing a patient’s wound and skin conditions. This comprehensive skin and wound assessment technology is designed to quantify biochemical markers to determine the trajectory of a wound’s condition to enable better diagnosis and treatment protocol. To date, Precision Healing has not generated revenues.
Pursuant to the terms of the merger agreement, holders of Precision Healing common stock and preferred stock, other than the Company, were entitled to receive closing consideration, consisting of $125,966 in cash, which was paid to stockholders who were not accredited investors, shares of the Company’s common stock, which was paid only to accredited investors, and the payment in cash of approximately $0.6 million of transaction expenses of Precision Healing. The Company recorded the issuance of the shares to accredited investors and cash payments to nonaccredited investors based on the closing price per share of the Company’s common stock on the Closing Date, which was $.
On the Closing Date, the outstanding Precision Healing options previously granted under the Precision Healing Inc. 2020 Stock Option and Grant Plan (the “Precision Healing Plan”) converted, pursuant to their terms, into options to acquire an aggregate of shares of Company common stock with a weighted exercise price of $10.71 per share. These options expire between August 2030 and April 2031. In addition, outstanding and unexercised Precision Healing warrants converted into rights to receive warrants to purchase (i) 4,424 shares of Company common stock with an initial exercise price of $7.32 per share and an expiration date of April 22, 2031, and (ii) 12,301 shares of the Company’s common stock with an initial exercise price of $12.05 per share and an expiration date of August 10, 2030.
Pursuant to the merger agreement, the Company assumed sponsorship of the Precision Healing Plan, effective as of the Closing Date, as well as the outstanding awards granted thereunder, the award agreements evidencing the grants of such awards and the remaining shares available under the Precision Healing Plan, in each case adjusted in the manner set forth in the merger agreement to such awards. Concurrent with the assumption of the Precision Healing Plan, the Company terminated the ability to offer future awards under the Precision Healing Plan.
| F-12 |
Pursuant to the merger agreement, upon the achievement of certain performance thresholds, the securityholders of Precision Healing, including the holders of options and warrants to purchase Precision Healing common stock and certain persons promised options to purchase Precision Healing common stock, are also entitled to receive payments of up to $10.0 million, which was accounted for as contingent consideration pursuant to ASC 805. The earnout consideration is payable in cash or, at the Company’s election, is payable to accredited investors in shares of Company common stock at a price per share equal to the greater of (i) $27.13 or (ii) the average closing price of Company common stock for the 20 trading days prior to the date such earnout consideration is due and payable. Pursuant to the merger agreement, a minimum percentage of the earnout consideration may be required to be issued to accredited investors in shares of Company common stock for tax purposes. The amount and composition of the portion of earnout consideration payable is subject to adjustment and offsets as set forth in the merger agreement.
As the contingent earnout payments are not subject to any specific individual performance by the shareholders, the contingent shares are not subject to ASC 718. Further, as the contingent consideration was negotiated as part of the Cellerate Acquisition,transfer of assets, the obligation was measured at fair value and included in the total purchase consideration transferred. Additionally, the contingent earnout payments meet the criteria under ASC Topic 480, Distinguishing Liabilities from Equity (“ASC 480”) as the monetary value of the shares to be issued is predominantly based on the exercise contingency (i.e., revenue targets). Accordingly, the consideration is classified as a liability and will be remeasured at its estimated fair value at each reporting period with the subsequent change in fair value recognized as a gain or loss in accordance with ASC 480.
The total purchase consideration as determined by the Company was as follows:
SCHEDULE OF PURCHASE CONSIDERATIONS
| Consideration | Equity Shares | Dollar Value | ||||||
| Fair value of Sanara common shares issued | $ | 5,096,444 | ||||||
| Fair value of assumed options | 4,109,750 | |||||||
| Fair value of assumed warrants | 502,895 | |||||||
| Cash paid to nonaccredited investors | 125,370 | |||||||
| Cash paid for fractional shares | ||||||||
| Carrying value of equity method investment in Precision Healing | 1,803,440 | |||||||
| Fair value of contingent earnout consideration | 3,882,151 | |||||||
| Direct transaction costs | 1,061,137 | |||||||
| Total purchase consideration | $ | 16,581,783 | ||||||
Based on guidance provided by ASC 805, the Company recorded the Precision Healing merger as an asset acquisition due to the determination that substantially all the fair value of the assets acquired was concentrated in a group of similar identifiable assets. The Company believes the “substantially all” criterion was met with respect to the acquired intellectual property based on the Company’s valuation models. These models assigned value to the acquired intellectual property based on estimated future cash flows. Accordingly, the Company accounted for the merger as an asset acquisition.
The purchase consideration, plus transaction costs, was allocated to the individual assets according to their fair values as a percentage of the total fair value of the assets purchased, with no goodwill recognized. Based on the estimated fair value of the gross assets acquired, the total fair value of the net assets acquired was primarily attributable to, and classified as, finite-lived intellectual property and assembled workforce in the second quarter of 2022. The total purchase consideration was allocated based on the relative estimated fair value of such assets as follows:
SCHEDULE OF PURCHASE CONSIDERATION ON FAIR VALUE OF ASSETS ACQUIRED
| Description | Amount | |||
| Cash | $ | 32,202 | ||
| Net working capital (excluding cash) | (308,049 | ) | ||
| Fixed assets, net | 9,228 | |||
| Deferred tax assets | 278,661 | |||
| Intellectual property | 20,325,469 | |||
| Assembled workforce | 664,839 | |||
| Deferred tax liabilities | (4,420,567 | ) | ||
| Net assets acquired | $ | 16,581,783 | ||
| F-13 |
NOTE 4 – SCENDIA PURCHASE AGREEMENT
In July 2022, the Company entered into a membership interest purchase agreement by and among the Company, Scendia, a Delaware limited liability company, and Ryan Phillips (“Phillips”) pursuant to which, and in accordance with the terms and conditions set forth therein, the Company acquired 100% of the issued and outstanding membership interests in Scendia from Phillips.
Scendia provides clinicians and surgeons with a 30-month convertible promissory notefull line of regenerative and orthobiologic technologies for their patients through certain customer accounts. Beginning in early 2022, the Company began co-promoting certain products with Scendia, including: (i) TEXAGEN Amniotic Membrane Allograft, (ii) BiFORM Bioactive Moldable Matrix, (iii) ACTIGEN Verified Inductive Bone Matrix and (iv) ALLOCYTE Advanced Cellular Bone Matrix. Prior to
Pursuant to the purchase agreement, Phillips was entitled to receive closing consideration consisting of (i) approximately $1.6 million of cash, subject to certain adjustments, and (ii) shares of common stock of the Company. Pursuant to the purchase agreement, at closing, the Company withheld shares of common stock with an agreed upon value of $1.95 million (the “Indemnity Holdback Shares”), which such Indemnity Holdback Shares were withheld to the extent provided in the purchase agreement to satisfy Phillips’ indemnification obligations and subsequently issued and released to Phillips in July 2023.
In addition to the cash consideration and the stock consideration, the purchase agreement provides that Phillips is entitled to receive two potential earnout payments, payable on an annual basis, not to exceed $10.0 million in the aggregate, which was accounted for as contingent consideration pursuant to ASC 805. The earnout consideration is payable to Phillips in cash or, at the Company’s election, in up to shares of the Company’s common stock upon the achievement of certain performance thresholds relating to net revenue attributable to sales of Scendia products during the two-year period following the closing. The Company made the first earnout payment of approximately $693,000 in cash in August 2023.
As the contingent earnout payments are not subject to any specific individual performance by Phillips, the contingent shares are not subject to ASC 718. Further, as the contingent consideration was negotiated as part of the transfer of assets, the obligation was measured at fair value and included in the total purchase consideration transferred. Additionally, the contingent earnout payments meet the criteria under ASC 480, as the monetary value of the shares to be issued is predominantly based on the exercise contingency (i.e., revenue targets). Accordingly, the contingent consideration is classified as a liability at its estimated fair value at each reporting period with the subsequent change in fair value recognized as a gain or loss in accordance with ASC 480.
The total purchase consideration, subject to typical post-closing adjustments, as determined by the Company was as follows:
SCHEDULE OF PURCHASE CONSIDERATIONS
| Consideration | Equity Shares | Dollar Value | ||||||
| Fair value of Sanara common shares issued | $ | 6,032,066 | ||||||
| Cash consideration | 1,562,668 | |||||||
| Fair value of contingent earnout consideration | 3,000,000 | |||||||
| Total purchase consideration | $ | 10,594,734 | ||||||
| F-14 |
Based on guidance provided by ASC 805, the Company recorded the Scendia acquisition as a business combination. The purchase consideration was allocated to the individual assets according to their fair values as a percentage of the total fair value of the net assets purchased. The excess of the purchase consideration over the net assets purchased was recorded as goodwill. The total purchase consideration was allocated as follows:
SCHEDULE OF PURCHASE CONSIDERATION ON FAIR VALUE OF ASSETS ACQUIRED
| Description | Amount | |||
| Cash | $ | 201,406 | ||
| Net working capital (excluding cash) | 1,294,499 | |||
| Fixed assets, net | 42,300 | |||
| Noncontrolling interest in Sanara Biologics, LLC | 2,638 | |||
| Customer relationships | 7,155,000 | |||
| Deferred tax liabilities | (1,702,890 | ) | ||
| Goodwill | 3,601,781 | |||
| Net assets acquired | $ | 10,594,734 | ||
The goodwill acquired consists of expected synergies from the acquisition to the Company’s overall corporate strategy. The Company does not expect any of the goodwill to be deductible for income tax purposes. The Company incurred acquisition costs of approximately $187,000 in 2022, which is included in “Selling, general and administrative expenses” in the accompanying Consolidated Statements of Operations.
NOTE 5 – APPLIED ASSET PURCHASE
On August 1, 2023, the Company entered into an Asset Purchase Agreement (the “Applied Purchase Agreement”) by and among the Company, as guarantor, Sanara MedTech Applied Technologies, LLC, a Texas limited liability company and wholly owned subsidiary of the Company (“SMAT”), The Hymed Group Corporation, a Delaware corporation (“Hymed”), Applied Nutritionals, LLC, a Delaware limited liability company (“Applied”, and together with Hymed, the “Sellers”), and Dr. George D. Petito (the “Owner”), pursuant to which SMAT acquired certain assets of the Sellers and the Owner, including, among others, the Sellers’ and Owner’s inventory, intellectual property, manufacturing and related equipment, goodwill, rights and claims, other than certain excluded assets, all as more specifically set forth in the Applied Purchase Agreement (collectively, the “Applied Purchased Assets”), and assumed certain Assumed Liabilities (as defined in the Applied Purchase Agreement), upon the terms and subject to the conditions set forth in the Applied Purchase Agreement (such transaction, the “Applied Asset Purchase”). The Applied Purchased Assets include the underlying intellectual property of, as well as the rights to manufacture and sell, CellerateRX Surgical Activated Collagen (“CellerateRX Surgical”) and HYCOL Hydrolyzed Collagen (“HYCOL”) products for human wound care use.
The Applied Purchased Assets were purchased for an initial aggregate purchase price of $15.25 million, consisting of (i) $9.75 million in cash (the “Cash Closing Consideration”), (ii) shares of the Company’s common stock (the “Stock Closing Consideration”) with an agreed upon value of $3.0 million and (iii) $2.5 million in cash (the “Installment Payments”), to be paid in four equal installments on each of the next four anniversaries of the closing of the Applied Asset Purchase (the “Closing”).
Prior to the Closing, the Company licensed certain of its products from Applied through a sublicense agreement (the “Sublicense Agreement”) with CGI Cellerate RX, LLC (“CGI Cellerate RX”), an affiliate of The Catalyst Group, Inc. (“Catalyst”), in the principal amount of $1,500,000, bearing interest at a 5% annual interest rate, compounded quarterly. Interest on the promissory note was payable quarterly but could have been deferred at the Company’s electionrelated party (see Note 14 for additional information regarding transactions with related parties). Pursuant to the maturity ofSublicense Agreement, the promissory note. Outstanding principalCompany has an exclusive, world-wide sublicense to distribute CellerateRX Surgical and interest were convertible atHYCOL products into the surgical and wound care markets. In connection with the Applied Asset Purchase, Applied assigned its license agreement with CGI Cellerate RX’s option to SMAT. Since the Closing of the Applied Asset Purchase, Sanara indirectly makes intercompany royalty payments to SMAT at the same rate as set forth in the Sublicense Agreement. Effective August 1, 2023, these intercompany royalty payments and the offsetting cost of goods sold are eliminated on a consolidated basis.
In addition to the Cash Closing Consideration, Stock Closing Consideration and Installment Payments, the Applied Purchase Agreement provides that the Sellers are entitled to receive up to an additional $10.0 million (the “Applied Earnout”), which is payable to the Sellers in cash, upon the achievement of certain performance thresholds relating to SMAT’s collections from net sales of a collagen-based product currently under development. Upon expiration of the seventh anniversary of the Closing, to the extent the Sellers have not earned the entirety of the Applied Earnout, SMAT shall pay the Sellers a pro-rata amount of the Applied Earnout based on collections from net sales of the product, with such amount to be due credited against any Applied Earnout payments already made by SMAT (the “True-Up Payment”). The Applied Earnout, minus the True-Up Payment and any Applied Earnout payments already made by SMAT, may be earned at any point in the future, including after the True-Up Payment is made.
In connection with the Applied Asset Purchase and pursuant to the Applied Purchase Agreement, effective August 1, 2023, the Company entered into a professional services agreement (the “Petito Services Agreement”) with the Owner, pursuant to which the Owner, as an independent contractor, agreed to provide certain services to the Company, including, among other things, assisting with the development of products already in development and assisting with research, development, formulation, invention and manufacturing of any future products (the “Petito Services”). As consideration for the Petito Services, the Owner is entitled to receive: (i) a base salary of $12,000 per month during the term of the Petito Services Agreement, (ii) a royalty payment equal to three percent (3%) of the actual collections from net sales of certain products the Owner develops or co-develops that reach commercialization, (iii) a royalty payment equal to five percent (5%) for the first $50.0 million in aggregate collections from net sales of certain future products and a royalty payment of two and one-half percent (2.5%) on aggregate collections from net sales of certain future products on any amounts exceeding $50.0 million but up to $100.0 million, (iv) $500,000 in cash in the event that 510(k) clearance is issued for any future product accepted by the Company and (v) $1.0 million in cash in the event that a U.S. patent is issued for a certain product; provided that with respect to the incentive payments described in (iv) and (v) of the foregoing, the Owner shall not earn more than $2.5 million.
| F-15 |
The Petito Services Agreement has an initial term of three years and is subject to automatic successive one-month renewals unless earlier terminated in accordance with its terms. The Petito Services Agreement may be terminated upon the Owner’s death or disability or by the Company or the Owner “For Cause” (as defined in the Petito Services Agreement); provided, however, that the base salary described in (i) of the foregoing paragraph shall survive termination through the three-year initial term and the royalty payments and incentive payments described in (ii)-(v) of the foregoing paragraph shall survive termination of the Petito Services Agreement.
As the contingent consideration was negotiated as part of the transfer of assets, the contingent obligation was measured at fair value and included in the total purchase consideration transferred. Accordingly, since the Applied Asset Purchase was accounted for as an asset acquisition and did not include contingent shares, the contingent consideration is classified as a liability at its estimated fair value at each reporting period with subsequent revaluations recognized as an adjustment to the intellectual property intangible asset and the earnout liability with a cumulative catch up amortization adjustment.
The total purchase consideration for the Applied Asset Purchase as determined by the Company was as follows:
SCHEDULE OF ASSET PURCHASE CONSIDERATIONS
| Consideration | Equity Shares | Dollar Value | ||||||
| Cash Closing Consideration | $ | 9,750,000 | ||||||
| Fair value of Stock Closing Consideration | 3,089,645 | |||||||
| Fair value of Installment Payments | 2,040,808 | |||||||
| Cash paid for inventory | 30,007 | |||||||
| Fair value of Petito Services Agreement defined payments | 825,834 | |||||||
| Fair value of Petito Services Agreement contingent consideration | 893,000 | |||||||
| Direct transaction costs | 162,743 | |||||||
| Total purchase consideration | $ | 16,792,037 | ||||||
Based on guidance provided by ASC 805, the Company recorded the Applied Asset Purchase as an asset acquisition due to the determination that substantially all the fair value of the assets acquired was concentrated in a group of similar identifiable assets. The Company believes the “substantially all” criterion was met with respect to the acquired intellectual property being the only significant asset acquired. Accordingly, the Company accounted for the transaction as an asset acquisition.
The purchase consideration, plus transaction costs, was allocated to the individual assets according to their fair values as a percentage of the total fair value of the assets purchased, with no goodwill recognized. Based on the estimated fair value of the gross assets acquired, the total fair value of the net assets acquired was primarily attributable to, and classified as, finite-lived intellectual property in the third quarter of 2023. The total purchase consideration was allocated based on the relative estimated fair value of such assets as follows:
SCHEDULE OF PURCHASE CONSIDERATION ON FAIR VALUE OF ASSETS ACQUIRED
| Description | Amount | |||
| Inventory | $ | 30,007 | ||
| Equipment | 33,062 | |||
| Intellectual property | 16,728,968 | |||
| Net assets acquired | $ | 16,792,037 | ||
| F-16 |
NOTE 6 – GOODWILL AND INTANGIBLES, NET
The changes in the carrying amount of the Company’s common stock at a conversion price of $9.00 per share.
Principal Amount | Accrued Interest | ||||
| Note Payable | Terms of the agreement | 2020 | 2019 | 2020 | 2019 |
| August 27, 2018 Promissory Note | A $1,500,000 note payable (i) interest accrues at 5% per annum and compounds quarterly (ii) original maturity date of March 1, 2021 | $- | $1,500,000 | $- | $103,557 |
| Total | $- | $1,500,000 | $- | $103,557 | |
SCHEDULE OF CHANGES IN THE CARRYING AMOUNT OF THE GOODWILL
| Total | ||||
| Balance as of December 31, 2021 | $ | - | ||
| Scendia Acquisition | 3,601,781 | |||
| Balance as of December 31, 2022 | 3,601,781 | |||
| Acquisitions | - | |||
| Balance as of December 31, 2023 | $ | 3,601,781 | ||
The carrying values of the Company’s finite-lived intangible assets were as follows:
December 31, 2020 | December 31, 2019 | |||||
Accumulated | Accumulated | |||||
Cost | Amortization | Net | Cost | Amortization | Net | |
| Product Licenses | $3,350,000 | $(264,909) | $3,085,091 | $1,500,000 | $(48,876) | $1,451,124 |
| Patent | 510,310 | (510,310) | - | 510,310 | (510,310) | - |
| Software and Other | 64,464 | (51,889) | 12,575 | 64,464 | (44,394) | 20,070 |
| Total | $3,924,774 | $(827,108) | $3,097,666 | $2,074,774 | $(603,580) | $1,471,194 |
SCHEDULE OF FINITE LIVED INTANGIBLE ASSETS
| December 31, 2023 | December 31, 2022 | |||||||||||||||||||||||
| Accumulated | Accumulated | |||||||||||||||||||||||
| Cost | Amortization | Net | Cost | Amortization | Net | |||||||||||||||||||
| Amortizable Intangible Assets: | ||||||||||||||||||||||||
| Product Licenses | $ | 4,793,879 | $ | (1,342,626 | ) | $ | 3,451,253 | $ | 4,793,879 | $ | (980,583 | ) | $ | 3,813,296 | ||||||||||
| Patents and Other IP | 38,570,549 | (3,181,186 | ) | 35,389,363 | 21,935,580 | (1,492,057 | ) | 20,443,523 | ||||||||||||||||
| Customer relationships and other | 7,947,332 | (1,861,887 | ) | 6,085,445 | 7,947,332 | (694,171 | ) | 7,253,161 | ||||||||||||||||
| Total | $ | 51,311,760 | $ | (6,385,699 | ) | $ | 44,926,061 | $ | 34,676,791 | $ | (3,166,811 | ) | $ | 31,509,980 | ||||||||||
As of December 31, 2020,2023, the weighted-average amortization period for allfinite-lived intangible assets was 12.814.5 years. Amortization expense related to intangible assets was $223,528$3,218,888 and $1,963,299 for the twelve monthsyear ended December 31, 20202023 and $90,011 for the twelve months ended December 31, 2019.2022, respectively. The estimated remaining amortization expense as of December 31, 20202023 for finite-lived intangible assets is as follows:
| 2021 | $258,059 |
| 2022 | 255,645 |
| 2023 | 250,564 |
| 2024 | 250,564 |
| 2025 | 250,564 |
| Thereafter | 1,832,270 |
| Total | $3,097,666 |
SCHEDULE OF FUTURE AMORTIZATION EXPENSE
| 2024 | $ | 3,889,804 | ||
| 2025 | 3,889,804 | |||
| 2026 | 3,872,548 | |||
| 2027 | 3,758,696 | |||
| 2028 | 3,725,454 | |||
| Thereafter | 25,789,755 | |||
| Total | $ | 44,926,061 |
The Company has reviewed the carrying value of intangible assets dueand has determined there was no impairment during either of the years ended December 31, 2023 or 2022.
NOTE 7 – INVESTMENTS IN EQUITY SECURITIES
The Company’s equity investments consist of nonmarketable equity securities in privately held companies without readily determinable fair values. Unless accounted for under the equity method of accounting, the investments are reported at cost minus impairment, if any, plus or minus changes resulting from observable price changes in orderly transactions for the identical or similar investment of the same issuer.
In July 2020, the Company made a $ long-term investment to purchase certain nonmarketable securities consisting of Series B-2 Preferred Shares of Direct Dermatology Inc. (“DirectDerm”), representing approximately % ownership of DirectDerm at that time. Through this investment, the eventsCompany received exclusive rights to utilize DirectDerm’s technology in all acute and circumstances surroundingpost-acute care settings such as skilled nursing facilities, home health and wound clinics. In 2021, the COVID-19 pandemic.
| F-17 |
In November 2020, the Company entered into agreements to purchase certain nonmarketable securities consisting of COVID-19 has created shares of Series A Convertible Preferred Stock (the “Series A Stock”) of Precision Healing for an impairment loss onaggregate purchase price of $600,000. The Series A Stock was convertible into shares of common stock of Precision Healing and had a senior liquidation preference relative to the common shareholders. This initial investment represented approximately 12.6% ownership of Precision Healing’s outstanding voting securities. In February 2021, the Company invested $600,000 to purchase additional shares of Series A Stock which was convertible into shares of common stock of Precision Healing. This resulted in ownership of approximately 22.4% of Precision Healing’s outstanding voting securities. With this level of significant influence, the Company transitioned to the equity method of accounting for this investment. In June 2021, the Company invested $500,000 for additional shares of Series A Stock, which increased the Company’s intangible assets asownership of Precision Healing’s outstanding voting securities to approximately 29.0%. In October and in December 31of 2021, and more shares of Series A Stock were purchased for $500,000 and $600,000, 2020. Accordingly, there was no impairment loss recognized onrespectively.
As discussed above, in April 2022, the Company closed a merger transaction with Precision Healing pursuant to which Precision Healing became a wholly owned subsidiary of the Company (see Note 3 for more information). As a result of the merger, the Company’s intangible assets during the
In June 2021, the Company invested $ to purchase Class A Preferred Shares (the “Shares”) of Canada based Pixalere Healthcare Inc. (“Pixalere”). The Company had one customer which accounted forShares are convertible into approximately 10% of Company’s sales in 2019, and one customer that accounted for 10%% of the outstanding accounts receivable at the endequity of 2019.
The Company has reviewed the characteristics of the Pixalere Shares in accordance with ASC Topic 323, Investments – Equity Method and Joint Ventures. Due to the substantive liquidation preferences of the Shares over Pixalere’s common stock, the Pixalere Shares are not “in-substance” common stock, and therefore, the Company will not utilize the equity method of accounting for this investment. In accordance with ASC Topic 321, Investments - Equity Securities, this investment was reported at cost as of December 31, 2023.
The following summarizes the Company’s investments for the periods presented:
SCHEDULE OF INVESTMENTS
| December 31, 2023 | December 31, 2022 | |||||||||||||||
| Carrying Amount | Economic Interest | Carrying Amount | Economic Interest | |||||||||||||
| Equity Method Investment | ||||||||||||||||
| Precision Healing Inc. | $ | - | - | % | $ | - | - | % | ||||||||
| Cost Method Investments | ||||||||||||||||
| Direct Dermatology, Inc. | 1,000,000 | 1,000,000 | ||||||||||||||
| Pixalere Healthcare Inc. | 2,084,278 | 2,084,278 | ||||||||||||||
| Total Cost Method Investments | 3,084,278 | 3,084,278 | ||||||||||||||
| Total Investments | $ | 3,084,278 | $ | 3,084,278 | ||||||||||||
| F-18 |
The following summarizes the loss from the equity method investment reflected in the Consolidated Statements of Operations:
SCHEDULE OF LOSS FROM EQUITY METHOD INVESTMENT
| 2023 | 2022 | |||||||
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| Investment | ||||||||
| Precision Healing Inc. | $ | - | $ | (379,633 | ) | |||
| Total | $ | - | $ | (379,633 | ) | |||
| Loss from equity method investment | $ | - | $ | (379,633 | ) | |||
NOTE 6 - COMMITMENTS AND CONTINGENCIES
The Company periodically enters into operating lease contracts for office space and equipment. Arrangements are evaluated at inception to determine whether such arrangements constitute a lease. In accordance with the transition guidance of ASC 842, such arrangements are included on the consolidated balance sheets as of January 1, 2019.
The Company has twothree material operating leases: anleases for office space. In March and September of 2023, the Company amended its primary office lease to obtain additional space, lease with aas well as extend the term. The leases have remaining lease termterms of
In accordance with ASC Topic 842, Leases, the Company has recorded leaseROU assets of $467,653$1,995,204 and a related lease liability of $481,384$2,098,630 as of December 31, 2020. 2023. The Company recorded amortizationlease expense of $117,598 in 2020$434,066 for the year ended December 31, 2023 for its leased assets.assets and $297,136 for the year ended December 31, 2022. Cash paid in 2020 for amounts included in the measurement of operating lease liabilities was $380,576 for the year ended December 31, 2023 and $296,988 for the year ended December 31, 2022. The present value of the Company’s operating lease liabilities as of December 31, 2020 was $150,886. The present value of our operating lease liabilities2023 is shown below.
Maturity of Operating Lease Liabilities
December 31, 2020 | |
| 2021 | $151,317 |
| 2022 | 151,333 |
| 2023 | 154,271 |
| 2024 | 77,870 |
| 2025 | - |
| Thereafter | - |
| Total lease payments | 534,791 |
| Less imputed interest | (53,407) |
| Present Value of Lease Liabilities | $481,384 |
| Operating lease liability - current | 125,587 |
| Operating lease liability – long term | 355,797 |
SCHEDULE OF OPERATING LEASE LIABILITY
| Year | Total | |||
| 2024 | $ | 505,017 | ||
| 2025 | 532,053 | |||
| 2026 | 379,529 | |||
| 2027 | 297,947 | |||
| 2028 | 295,689 | |||
| Thereafter | 604,050 | |||
| Total lease payments | $ | 2,614,285 | ||
| Less imputed interest | (515,655 | ) | ||
| Present Value of Lease Liabilities | $ | 2,098,630 | ||
| Operating lease liabilities – current | $ | 361,185 | ||
| Operating lease liabilities – long-term | $ | 1,737,445 | ||
As of December 31, 2020, our2023, the Company’s operating leases have a weighted average remaining lease term of 3.55.9 years and a weighted average discount rate of 6.25%7.6%.
| F-19 |
NOTE 89 – STOCKHOLDERS’ EQUITY
Term Loan
In connection with the reverse stock split,entry into the Applied Purchase Agreement, on August 1, 2023, SMAT, as borrower, and the Company, also madeas guarantor, entered into a corresponding adjustmentloan agreement (the “Loan Agreement”) with Cadence Bank (the “Bank”) providing for, among other things, an advancing term loan in the aggregate principal amount of $12.0 million (the “Term Loan”), which was evidenced by an advancing promissory note. Pursuant to the Company’s authorizedLoan Agreement, the Bank agreed to make, at any time and from time to time prior to February 1, 2024, one or more advances to SMAT.
The proceeds of the advances under the Loan Agreement will be used for working capital stockand for purposes of financing up to reduceone hundred percent (100%) of the authorized common stockCash Closing Consideration and Installment Payments for the Applied Asset Purchase and related fees and expenses, including any subsequent payments that may be due to 20,000,000 sharesthe Sellers after the Closing. On August 1, 2023, the Bank, at the request of SMAT, made an advance for $9.75 million. The proceeds from the advance were used to fund the Cash Closing Consideration for the Applied Asset Purchase.
Advances under the Term Loan will begin amortizing in monthly installments commencing on August 5, 2024. All remaining unpaid balances under the Term Loan are due and payable in full on August 1, 2028 (the “Maturity Date”). SMAT may prepay amounts due under the Term Loan. All accrued but unpaid interest on the unpaid principal balance of outstanding advances is due and payable monthly, beginning on September 5, 2023 and continuing monthly on the fifth day of each month thereafter until the Maturity Date. The unpaid principal balance of outstanding advances bears interest, subject to certain conditions, at the lesser of the Maximum Rate (as defined in the Loan Agreement) or the Base Rate, which is for any day, a rate per annum equal to the term secured overnight financing rate (Term SOFR) (as administered by the Federal Reserve Bank of New York) for a one-month tenor in effect on such day plus three percent (3.0%).
The obligations of SMAT under the Loan Agreement and the authorized preferred stock to 2,000,000 shares, effective May 10, 2019.
The purchasersLoan Agreement contains customary representations and warranties and certain covenants that limit (subject to certain exceptions) the ability of SMAT and the Company to, among other things, (i) create, assume or guarantee certain liabilities, (ii) create, assume or suffer liens securing indebtedness, (iii) make or permit loans and advances, (iv) acquire any assets outside the ordinary course of business, (v) consolidate, merge or sell all or a material part of its assets, (vi) pay dividends or other distributions on, or redeem or repurchase, interest in an obligor, including the Company, as guarantor (vii) cease, suspend or materially curtail business operations or (viii) engage in certain affiliate transactions. In addition, the Loan Agreement contains financial covenants that require SMAT to maintain (i) a minimum Debt Services Coverage Ratio of 1.2 to 1.0 as of the last day of each applicable fiscal quarter and (ii) a maximum Cash Flow Leverage Ratio of not more than (a) 4.5 to 1.0 as of the last day of the fiscal quarter ending on September 30, 2023, (b) 4.0 to 1.0 as of the last day of each fiscal quarter ending on December 31, 2023 and March 31, 2024, (c) 3.5 to 1.0 as of the last day of each fiscal quarter ending June 30, 2024 and September 30, 2024 and (d) 3.0 to 1.0 as of the last day of each fiscal quarter thereafter. Pursuant to the Loan Agreement, in the offering were related party entitiesevent that SMAT fails to three members ofcomply with the Company’s board of directors.
The Loan Agreement contains customary events of default. If such an event of default occurs, the Bank would be entitled to certain vesting provisionstake various actions, including the acceleration of amounts due under the Loan Agreement and other terms and conditions set forth in each recipient’s restricted stock agreement.
The Company granted and issued 209,541 shares, nettable below presents the components of forfeitures,outstanding debt for the periods presented:
As of restricted common stock to Company employees, directors, and certain consultants ofDecember 31, 2023, the Company. The fair value of these awards is basedinterest rate on the closing priceadvance under the Term Loan was 8.3%.
SCHEDULE OF LONG-TERM DEBT
December 31, 2023 | December 31, 2022 | |||||||
| Term Loan | $ | 9,750,000 | $ | - | ||||
| Total debt | 9,750,000 | - | ||||||
| Less: debt issuance costs, net of accumulated amortization of $5,138 and zero | (56,520 | ) | - | |||||
| Long-term debt | 9,693,480 | - | ||||||
| Less: Current portion of long-term debt | 580,357 | - | ||||||
| Long-term debt | $ | 9,113,123 | $ | - | ||||
| F-20 |
The table below presents the aggregate maturities of the Company’s common stockoutstanding debt as of December 31, 2023:
SCHEDULE OF MATURITIES OUTSTANDING DEBT
| Year | Total | |||
| 2024 | $ | 580,357 | ||
| 2025 | 1,625,000 | |||
| 2026 | 1,950,000 | |||
| 2027 | 1,950,000 | |||
| 2028 | 3,644,643 | |||
| Thereafter | - | |||
| Total debt | $ | 9,750,000 | ||
In connection with the Term Loan, the Company incurred $61,658 in debt issuance costs during year ended December 31, 2023. Debt issuance costs are amortized to “Interest expense and other” on the respective grant dates; then, is recognized as compensation expense on a straight-line basisConsolidated Statement of Operations over the vesting periodlife of the award.
Revolving Line of Credit
In January 2021, the twelve months ended December 31, 2019.
For the Year Ended | ||
December 31, 2020 | ||
Shares | Weighted Average Grant Date Fair Value | |
| Non-vested at beginning of period | - | $- |
| Granted | 214,894 | 14.05 |
| Vested | (43,098) | 13.58 |
| Forfeited | (1,618) | 11.15 |
| Non-vested at December 31, 2020 | 170,178 | $14.20 |
For the Year Ended | |||
December 31, 2020 | |||
Weighted Average | Weighted Average Remaining | ||
Options | Exercise Price | Contract Life | |
| Outstanding at beginning of period | 11,500 | $6.00 | |
| Granted | - | - | |
| Exercised | - | - | |
| Forfeited | - | $- | |
| Expired | - | - | |
| Outstanding at December 31, 2020 | 11,500 | $6.00 | 2.0 |
| Exercisable at December 31, 2020 | 11,500 | $6.00 | 2.0 |
2020 | 2019 | |
| Net operating loss carry forwards | $2,827,835 | $1,876,114 |
| Valuation allowance | (2,827,835) | (1,876,114) |
| Net non-current deferred tax asset | $- | $- |
2020 | 2019 | |
| Expected federal income tax benefit | $914,852 | $605,767 |
| Goodwill amortization | - | 65,957 |
| Change in valuation allowance | (1,020,111) | (294,050) |
| NOL carryover adjusted for expiration | 111,345 | (302,134) |
| Pass through entity income allocation | - | (94,151) |
| Meals and entertainment | (24,859) | (29,741) |
| Stock-based compensation | (103,657) | 48,352 |
| PPP Loan Forgiveness | 122,430 | - |
| Income tax expense (benefit) | $- | $- |
NOTE 10 – DEBT- COMMITMENTS AND CREDIT FACILITIES
License Agreements and Royalties
CellerateRX Surgical
In August 2018, the Company entered an exclusive, world-wide sublicense agreement with CGI Cellerate RX, LLC (“CGI Cellerate RX”) to distribute CellerateRX Surgical and HYCOL products into the surgical and wound care markets. Pursuant to the sublicense agreement, the Company pays royalties of Credit
Under this agreement, royalty expense, which is recorded in “Cost of credit was usedgoods sold” in the accompanying Consolidated Statements of Operations, totaled $991,099 and $1,771,219, respectively for the years ended December 31, 2023 and 2022. Sales of CellerateRX Surgical comprised the substantial majority of the Company’s sales during 2023 and 2022.
As discussed further in Note 5, on August 1, 2023, the Company purchased certain assets from Applied, including the rights to supportmanufacture and sell CellerateRX Surgical and HYCOL products. In connection with the short-term working capital requirementsApplied Asset Purchase, Applied assigned its license agreement with CGI Cellerate RX to SMAT. Since the Closing, Sanara indirectly makes intercompany royalty payments to SMAT at the same rate as set forth in the Sublicense Agreement. Effective August 1, 2023, these intercompany royalty payments and the offsetting cost of Cellerate, LLC. On June 21,goods sold are eliminated on a consolidated basis.
| F-21 |
BIAKŌS Antimicrobial Wound Gel and BIAKŌS Antimicrobial Skin and Wound Cleanser
In July 2019, the Company modifiedexecuted a license agreement with Rochal Industries, LLC (“Rochal”), a related party, pursuant to which the revolving lineCompany acquired an exclusive world-wide license to market, sell and further develop antimicrobial products for the prevention and treatment of creditmicrobes on the human body utilizing certain Rochal patents and pending patent applications (the “BIAKŌS License Agreement”). Currently, the products covered by the BIAKŌS License Agreement are BIAKŌS Antimicrobial Wound Gel and BIAKŌS Antimicrobial Skin and Wound Cleanser. Both products are 510(k) cleared.
Future commitments under the terms of the BIAKŌS License Agreement include:
| ● | The Company pays Rochal a royalty of 2-4% of net sales. The minimum annual royalty due to Rochal was $120,000 for 2022 and will increase by $10,000 each subsequent calendar year up to a maximum amount of $150,000. |
| ● | The Company pays additional royalty annually based on specific net profit targets from sales of the licensed products, subject to a maximum of $1,000,000 during any calendar year. |
Unless previously terminated by the parties, the BIAKŌS License Agreement expires with Cadence to increase the maximum principal amount from $1,000,000 to $2,500,000. Onrelated patents in December 2031.
Under this agreement, royalty expense, which is recorded in “Cost of goods sold” in the accompanying Consolidated Statements of Operations, was $130,000 and $120,000 for the years ended December 31, 2023 and 2022, respectively. The Company’s Executive Chairman is a director of Rochal, and indirectly a significant shareholder of Rochal, and through the potential exercise of warrants, a majority shareholder of Rochal. Another one of the Company’s directors is also a director and significant shareholder of Rochal.
CuraShield Antimicrobial Barrier Film and No Sting Skin Protectant
In October 16, 2019, the Company paid downexecuted a license agreement with Rochal pursuant to which the entire $2,200,000 balanceCompany acquired an exclusive world-wide license to market, sell and further develop certain antimicrobial barrier film and skin protectant products for use in the human health care market utilizing certain Rochal patents and pending patent applications (the “ABF License Agreement”). Currently, the products covered by the ABF License Agreement are CuraShield Antimicrobial Barrier Film and a no sting skin protectant product.
Future commitments under the terms of the revolving lineABF License Agreement include:
| ● | The Company will pay Rochal a royalty of 2-4% of net sales. The minimum annual royalty due to Rochal will be $50,000 beginning with the first full calendar year following the year in which first commercial sales of the products occur. The annual minimum royalty will increase by 10% each subsequent calendar year up to a maximum amount of $75,000. |
| ● | The Company will pay additional royalties annually based on specific net profit targets from sales of the licensed products, subject to a maximum of $500,000 during any calendar year. |
Unless previously terminated or extended by the parties, the ABF License Agreement will terminate upon expiration of creditthe last U.S. patent in October 2033. No commercial sales or royalties have been recognized under this agreement as of December 31, 2023.
Debrider License Agreement
In May 2020, the Company executed a product license agreement with Rochal, pursuant to which the Company acquired an exclusive world-wide license to market, sell and further develop a debrider for human medical use to enhance skin condition or treat or relieve skin disorders, excluding uses primarily for beauty, cosmetic, or toiletry purposes (the “Debrider License Agreement”).
Future commitments under the terms of the Debrider License Agreement include:
| ● | Upon FDA clearance of the licensed products, the Company will pay Rochal $500,000 in cash and an additional $1,000,000, which at the Company’s option may be paid in any combination of cash and its common stock. |
| ● | The Company will pay Rochal a royalty of 2-4% of net sales. The minimum annual royalty due to Rochal will be $100,000 beginning with the first full calendar year following the year in which first commercial sales of the licensed products occur and increase by 10% each subsequent calendar year up to a maximum amount of $150,000. |
| ● | The Company will pay additional royalty annually based on specific net profit targets from sales of the licensed products, subject to a maximum of $1,000,000 during any calendar year. |
Unless previously terminated or extended by the parties, the Debrider License Agreement will expire in October 2034. No commercial sales or royalties have been recognized under this agreement as of December 31, 2023.
| F-22 |
Rochal Asset Acquisition
The Company entered into an asset purchase agreement with Rochal effective July 1, 2021, pursuant to which the Company purchased certain assets of Rochal. Pursuant to the asset purchase agreement, for the three-year period after the effective date, Rochal is entitled to receive consideration for any new product relating to the business that is directly and primarily based on an invention conceived and reduced to practice by a member or members of Rochal’s science team. For the three-year period after the effective date, Rochal is also entitled to receive an amount in cash equal to twenty-five percent of the proceeds received for any Grant (as defined in the asset purchase agreement) by either the Company or Rochal. In addition, the Company agreed to use commercially reasonable efforts to perform Minimum Development Efforts (as defined in the asset purchase agreement) with respect to certain products under development, which if obtained, will entitle the Company to intellectual property rights from Rochal in respect of such products.
Precision Healing Merger Agreement
In April 2022, the Company closed a private placementmerger transaction with Precision Healing pursuant to which Precision Healing became a wholly owned subsidiary of the Company. Pursuant to the terms of the merger agreement, holders of Precision Healing common stock and preferred stock, other than the Company, were entitled to receive closing consideration, consisting of $125,966 in cash consideration, which was paid to stockholders who were not accredited investors, shares of the Company’s common stock. This revolving linestock, which was paid only to accredited investors, and the payment in cash of credit maturedapproximately $0.6 million of transaction expenses of Precision Healing. The Company recorded the issuance of the shares to accredited investors and cash payments to nonaccredited investors based on June 19, 2020.
Upon the closing of the merger, the Precision Healing outstanding options previously granted under the Precision Healing Plan converted, pursuant to their terms, into options to acquire an aggregate of shares of Company common stock with a new credit facility established in early 2021.
Pursuant to purchasethe merger agreement, upon the achievement of certain non-marketable securities consisting of 150,000 shares of Series A Convertible Preferred Stock (the “Series A Stock”)performance thresholds, the securityholders of Precision Healing, Inc. (“including the holders of options and warrants to purchase Precision Healing”)Healing common stock and certain persons promised options to purchase Precision Healing common stock, are also entitled to receive payments of up to $10.0 million, which was accounted for an aggregate purchaseas contingent consideration pursuant to ASC 805. The earnout consideration is payable in cash or, at the Company’s election, is payable to accredited investors in shares of Company common stock at a price per share equal to the greater of (i) $ or (ii) the average closing price of $600,000.Company common stock for the 20 trading days prior to the date such earnout consideration is due and payable. Pursuant to the merger agreement, a minimum percentage of the earnout consideration may be required to be issued to accredited investors in shares of Company common stock for tax purposes. The Series A Stockamount and composition of the portion of earnout consideration payable is convertible into 150,000subject to adjustment and offsets as set forth in the merger agreement. See Note 3 for more information regarding the merger with Precision Healing.
Scendia Purchase Agreement
In July 2022, the Company closed the Scendia acquisition pursuant to which Scendia became a wholly owned subsidiary of the Company. Pursuant to the purchase agreement, the aggregate consideration for the acquisition at closing was approximately $7.6 million, subject to customary post-closing adjustments. The consideration consisted of (i) approximately $1.6 million of cash, subject to certain adjustments, and (ii) shares of common stock of the Company. Pursuant to the purchase agreement, at closing, the Company withheld Indemnity Holdback Shares, which such Indemnity Holdback Shares were withheld to the extent provided in the purchase agreement to satisfy Phillips’ indemnification obligations and subsequently issued and released to Phillips in July 2023.
In addition to the cash consideration and the stock consideration, the purchase agreement provides that Phillips is entitled to receive two potential earnout payments, payable on an annual basis, not to exceed $10.0 million in the aggregate, which was accounted for as contingent consideration pursuant to ASC 805. The earnout consideration is payable to Phillips in cash or, at the Company’s election, in up to shares of the Company’s common stock upon the achievement of certain performance thresholds relating to net revenue attributable to sales of Scendia products during the two-year period following the closing. The Company made the first earnout payment of approximately $693,000 in cash in August 2023. The Company expects the final earnout payment to be made in the third quarter of 2024. See Note 4 for more information regarding the acquisition of Scendia.
| F-23 |
Applied Asset Purchase
On August 1, 2023, the Company closed the Applied Asset Purchase. The Applied Purchased Assets were purchased for an initial aggregate purchase price of $15.25 million, consisting of (i) the Cash Closing Consideration, (ii) the Stock Closing Consideration and (iii) the Installment Payments.
In addition to the Cash Closing Consideration, Stock Closing Consideration and Installment Payments, the Applied Purchase Agreement provides that the Sellers are entitled to receive the Applied Earnout, which is payable to the Sellers in cash, upon the achievement of certain performance thresholds relating to SMAT’s collections from net sales of a collagen-based product currently under development. Upon expiration of the seventh anniversary of the Closing, to the extent the Sellers have not earned the entirety of the Applied Earnout, SMAT shall pay the Sellers the True-Up Payment. The Applied Earnout, minus the True-Up Payment and any Applied Earnout payments already made by SMAT, may be earned at any point in the future, including after the True-Up Payment is made.
In connection with the Applied Asset Purchase and pursuant to the Applied Purchase Agreement, effective August 1, 2023, the Company entered into the Petito Services Agreement with the Owner, pursuant to which the Owner, as an independent contractor, agreed to provide the Petito Services. As consideration for the Petito Services, the Owner is entitled to receive: (i) a base salary of $12,000 per month during the term of the Petito Services Agreement, (ii) a royalty payment equal to three percent (3%) of the actual collections from net sales of certain products the Owner develops or codevelops that reach commercialization, (iii) a royalty payment equal to five percent (5%) for the first $50.0 million in aggregate collections from net sales of certain future products and a royalty payment of two and one-half percent (2.5%) on aggregate collections from net sales of certain future products on any amounts exceeding $50.0 million but up to $100.0 million, (iv) $500,000 in cash in the event that 510(k) clearance is issued for any future product accepted by the Company and (v) $1.0 million in cash in the event that a U.S. patent is issued for a certain product; provided that with respect to the incentive payments described in (iv) and (v) of the foregoing, the Owner shall not earn more than $2.5 million.
The Petito Services Agreement has an initial term of three years and is subject to automatic successive one-month renewals unless earlier terminated in accordance with its terms. The Petito Services Agreement may be terminated upon the Owner’s death or disability or by the Company or the Owner “For Cause” (as defined in the Petito Services Agreement); provided, however, that the base salary described in (i) of the foregoing paragraph shall survive termination through the three-year initial term and the royalty payments and incentive payments described in (ii)-(v) of the foregoing paragraph shall survive termination of the Petito Services Agreement.
Other Commitments
On December 20, 2023, the Company signed an exclusive license agreement with Tufts University (“Tufts”) to develop and commercialize patented technology covering 18 unique collagen peptides. As part of this agreement, the Company formed a new subsidiary, Sanara Collagen Peptides, LLC (“SCP”) and has issued 10% of SCP’s outstanding units to Tufts. SCP has exclusive rights to develop and commercialize new products based on the licensed patents and patents pending. SCP will pay royalties to Tufts based on net sales of licensed products and technologies. Under the exclusive license agreement, royalties will be calculated at a rate of 1.5% or 3%, depending on the type of product or technology developed. SCP will pay Tufts a minimum annual royalty of $50,000 on January 1 of the year following the first anniversary of the first commercial sale of the licensed products or technologies. SCP will pay Tufts a $100,000 minimum annual royalty on January 1 of each subsequent year during the royalty term specified in the exclusive license agreement. There have been no material accounting impacts related to this arrangement as of December 31, 2023.
NOTE 11 – SHAREHOLDERS’ EQUITY
Common Stock
At the Company’s Annual Meeting of Shareholders held in July 2020, the Company approved the Restated 2014 Omnibus Long Term Incentive Plan (the “LTIP Plan”) in which the Company’s directors, officers, employees and consultants are eligible to participate. A total of shares had been issued under the LTIP Plan and were available for issuance as of December 31, 2023.
In April 2022, the Company closed a merger transaction with Precision Healing and haspursuant to which Precision Healing became a senior liquidity preference relativewholly owned subsidiary of the Company. Pursuant to the terms of the merger agreement, holders of Precision Healing common shareholders.stock and preferred stock, other than the Company, were entitled to receive closing consideration, consisting of $125,966 in cash consideration, which was paid to stockholders who were not accredited investors, shares of the Company’s common stock, which was paid only to accredited investors, and the payment in cash of approximately $0.6 million of transaction expenses of Precision Healing. The Company investedrecorded the issuance of the shares to accredited investors and cash payments to nonaccredited investors based on the closing price per share of the Company’s common stock on April 4, 2022, which was $.
Upon the closing of the merger, the Precision Healing outstanding options previously granted under the Precision Healing Plan converted, pursuant to their terms, into options to acquire an additional $600,000 in February 2021 for 150,000 additionalaggregate of shares of Series A Stock. The additionalCompany common stock with a weighted exercise price of $ per share. These options expire between August 2030 and April 2031. In addition, outstanding and unexercised Precision Healing warrants converted into rights to receive warrants to purchase (i) 4,424 shares of Series A Stock will convert intothe Company’s common stock with an initial exercise price of $7.32 per share and an expiration date of April 22, 2031, and (ii) 12,301 shares of the Company’s common stock with an initial exercise price of $12.05 per share and an expiration date of August 10, 2030. Concurrent with the assumption of the Precision Healing Plan, the Company terminated the ability to offer future awards under the Precision Healing Plan.
| F-24 |
Pursuant to the merger agreement, upon the achievement of certain performance thresholds, the securityholders of Precision Healing, including the holders of options and warrants to purchase Precision Healing common stock and certain persons promised options to purchase Precision Healing common stock, are also entitled to receive payments of up to $10.0 million, which was accounted for as contingent consideration pursuant to ASC 805. The earnout consideration is payable in cash or, at the Company’s election, is payable to accredited investors in shares of Company common stock at a price per share equal to the greater of (i) $ or (ii) the average closing price of Company common stock for the 20 trading days prior to the date such earnout consideration is due and payable. Pursuant to the merger agreement, a minimum percentage of the earnout consideration may be required to be issued to accredited investors in shares of Company common stock for tax purposes. The amount and composition of the portion of earnout consideration payable is subject to adjustment and offsets as set forth in the merger agreement. See Note 3 for more information regarding the merger with Precision Healing.
In July 2022, the Company closed the Scendia acquisition pursuant to which Scendia became a wholly owned subsidiary of the Company. Pursuant to the purchase agreement, the aggregate consideration at closing for the acquisition was approximately $7.6 million, subject to customary post-closing adjustments. The consideration consisted of (i) approximately $1.6 million of cash, subject to certain adjustments, and (ii) shares of common stock of Precision Healingthe Company. Pursuant to the purchase agreement, at a ratio basedclosing, the Company withheld Indemnity Holdback Shares, which such Indemnity Holdback Shares were withheld to the extent provided in the purchase agreement to satisfy Phillips’ indemnification obligations and subsequently issued and released to Phillips in July 2023.
In addition to the cash consideration and the stock consideration, the purchase agreement provides that Phillips is entitled to receive two potential earnout payments, payable on an annual basis, not to exceed $10.0 million in the date Precision Healing delivers a development milestone relatedaggregate, which was accounted for as contingent consideration pursuant to a wound diagnostic tool currently under development.
In accordance with applicable accounting standards for the disclosure of events that occur after the balance sheet date but before the financial statements are issued, all significant events or transactions that occurred after December 31, 2020, are outlined below.
Sales of the shares ofwere made in sales deemed to be an “at the Company’s common stock was exempt from registrationmarket offering” as defined in Rule 415(a)(4) promulgated under the Securities Act of 1933, as amended (the “Securities Act”),amended. Upon delivery of a placement notice and subject to the terms and conditions of the Sales Agreement, Cantor agreed to use commercially reasonable efforts consistent with its normal trading and sales practices, applicable state and federal law, rules and regulations and the rules of The Nasdaq Capital Market to sell the shares from time to time based upon the Company’s instructions, including any price, time period or size limits specified by the Company. The Company had no obligation to sell any of the shares under the Sales Agreement and could suspend or terminate the offering of its common stock pursuant to the exemption provided in Section 4(a)(2)Sales Agreement upon notice to Cantor and subject to other conditions. Cantor’s obligations to sell the shares under the Sales Agreement were subject to satisfaction of certain conditions, including customary closing conditions. Pursuant to the Sales Agreement, the Company paid Cantor a commission of 3.0% of the Securities Actaggregate gross proceeds from each sale of the shares.
In 2023, the Company sold an aggregate of shares of common stock for gross proceeds of approximately $1.1 million and Rule 506(b) promulgated thereundernet proceeds of approximately $0.9 million pursuant to the Sales Agreement. The Company paused the offering at the end of the first quarter of 2023 and did not reactivate it during the remainder of 2023. The Form S-3 registration statement for this offering expired at the end of 2023.
On August 1, 2023, the Company closed the Applied Asset Purchase. Included in the purchase price was shares of the Company’s common stock. See Note 5 for more information regarding the acquisition of Applied.
Restricted Stock Awards
The Company issues restricted stock awards under the LTIP Plan which are subject to certain vesting provisions and other terms and conditions set forth in each recipient’s respective restricted stock agreement. The Company granted and issued shares, net of forfeitures, of restricted common stock to employees, directors, and certain advisors of the Company under the LTIP Plan during the year ended December 31, 2023. The fair value of these awards was $4,045,377 based on the closing price of the Company’s common stock on the respective grant dates, which will be recognized as compensation expense on a straight-line basis over the vesting period of the awards.
Share-based compensation expense of $ and $ was recognized in “Selling, general and administrative expenses” in the accompanying Consolidated Statements of Operations during the years ended December 31, 2023 and 2022, respectively. Equity awards totaling $1,038,183, which were accrued as a saleliability as of December 31, 2021, were reclassed to accredited investorsequity in 2022 upon settlement of these awards.
| F-25 |
At December 31, 2023, there was $ of total unrecognized share-based compensation expense related to unvested share-based equity awards. Unrecognized share-based compensation expense is expected to be recognized over a weighted-average period of years.
SUMMARY OF RESTRICTED STOCK ACTIVITY
| For the Year Ended | ||||||||
| December 31, 2023 | ||||||||
| Shares | Weighted Average Grant Date Fair Value | |||||||
| Nonvested at beginning of period | 181,102 | $ | 22.89 | |||||
| Granted | 118,912 | 39.14 | ||||||
| Vested | (137,720 | ) | 23.75 | |||||
| Forfeited | (18,083 | ) | 33.66 | |||||
| Nonvested at December 31, 2023 | 144,211 | $ | 34.07 | |||||
Stock Options
SCHEDULE OF STOCK OPTION ACTIVITY
| For the Year Ended | ||||||||||||
| December 31, 2023 | ||||||||||||
| Weighted Average | Weighted Average | |||||||||||
| Options | Exercise Price | Remaining Contract Life | ||||||||||
| Outstanding at beginning of period | 146,191 | $ | 10.65 | |||||||||
| Granted or assumed | - | - | ||||||||||
| Exercised | (52,299 | ) | 11.41 | |||||||||
| Forfeited | - | - | ||||||||||
| Expired | - | - | ||||||||||
| Outstanding at December 31, 2023 | 93,892 | $ | 10.22 | |||||||||
| Exercisable at December 31, 2023 | 93,892 | $ | 10.22 | |||||||||
| F-26 |
Warrants
A summary of the status of outstanding warrants to purchase common stock at December 31, 2023 and changes during the year then ended is presented below:
SCHEDULE OF WARRANTS TO PURCHASE COMMON STOCK
| For the Year Ended | ||||||||||||
| December 31, 2023 | ||||||||||||
| Weighted Average | Weighted Average | |||||||||||
| Exercise | Remaining | |||||||||||
| Warrants | Price | Contract Life | ||||||||||
| Outstanding at beginning of period | 16,725 | $ | 10.80 | |||||||||
| Granted or assumed | - | - | ||||||||||
| Exercised | - | - | ||||||||||
| Forfeited | - | - | ||||||||||
| Expired | - | - | ||||||||||
| Outstanding at December 31, 2023 | 16,725 | $ | 10.80 | |||||||||
| Exercisable at December 31, 2023 | 16,725 | $ | 10.80 | |||||||||
NOTE 12 – CUSTOMERS AND SUPPLIERS
The Company had no customers in 2023 that accounted for at least 10% of the Company’s annual sales or whose accounts receivable balance exceeded 10% of the year-end balance. The Company had no customers in 2022 that accounted for at least 10% of Company’s annual sales and whose accounts receivable exceeded 10% of the year-end balance.
The Company’s principal revenue producing products are purchased from one manufacturer. If this supplier became unable to provide finished goods inventory in a timely manner, the Company’s business, operating results, and financial condition could be materially adversely affected.
NOTE 13 – INCOME TAXES
The Company accounts for income taxes in accordance with whomASC Topic No. 740, Income Taxes. This standard requires the Company to provide a net deferred tax asset or liability equal to the expected future tax benefit or expense of temporary reporting differences between book and tax accounting and any available operating loss or tax credit carry forwards.
As discussed in Notes 3 and 4, the Company recognized net deferred tax liabilities associated with the Precision Healing merger and the Scendia acquisition. As of the dates of these acquisitions, prior to consideration of these acquired deferred tax liabilities, the Company had net deferred tax assets in excess of the deferred tax liabilities being recognized, however, a pre-existing relationship.100% valuation allowance had previously been provided against the Company’s net deferred tax assets. As a result of the recording of the net deferred tax liabilities related to the Precision Healing merger and Scendia acquisition, the Company reviewed the valuation allowance and determined that it should be reduced by the amount of the deferred tax liabilities that were recognized. This resulted in a year-to-date income tax benefit of $5.8 million for the year ending December 31, 2022.
After applying the provisions of Section 382 of the Internal Revenue Code, the unexpired net operating loss (“NOL”) carryforward at December 31, 2023 was approximately $55.7 million, of which, approximately $27.0 million, generated in 2017 and prior, will expire between 2024 and 2037. Under the Tax Cuts and Jobs Act, the NOL generated from 2018 through 2023, of approximately $28.7 million, will have an indefinite carryforward period but can generally only be used to offset 80% of taxable income in any particular year. The Company may be subject to certain limitations in its annual utilization of NOL carry forwards to off-set future taxable income pursuant to Section 382 of the Internal Revenue Code, which could result in NOLs expiring unused.
The components of the deferred income tax assets and liabilities consisted of the following:
SCHEDULE OF DEFERRED TAX ASSETS
| 2023 | 2022 | |||||||
| As of December 31, | ||||||||
| 2023 | 2022 | |||||||
| Deferred tax assets | ||||||||
| Net operating loss carry forwards | $ | 12,467,570 | $ | 6,818,547 | ||||
| Research and development costs | 2,119,193 | 707,076 | ||||||
| Stock compensation expense | 711,598 | 232,250 | ||||||
| Accrued expenses | 528,148 | - | ||||||
| Lease liability | 512,668 | - | ||||||
| Contingent liability | 221,028 | - | ||||||
| Acquisition liability | 176,629 | - | ||||||
| Bad debt and other reserves | 129,924 | 92,286 | ||||||
| Inventory reserves | 109,176 | 43,678 | ||||||
| Other temporary differences | 203,340 | 26,102 | ||||||
| Total deferred tax assets | 17,179,274 | 7,919,939 | ||||||
| Deferred tax liabilities | ||||||||
| Depreciation and amortization | (6,180,688 | ) | (5,919,626 | ) | ||||
| Right of Use assets | (487,402 | ) | - | |||||
| Accrued expenses | (424,423 | ) | (26,903 | ) | ||||
| Contingent liability | (130,802 | ) | - | |||||
| Other temporary differences | (166,712 | ) | - | |||||
| Valuation allowance | (9,789,247 | ) | (1,973,410 | ) | ||||
| Net deferred tax asset | $ | - | $ | - | ||||
A 100% valuation allowance has been provided for all deferred tax assets, as the ability of the Company to generate sufficient taxable income in the future is uncertain.
| F-27 |
Reconciliations of the expected federal income tax benefit based on the statutory income tax rate of 21% to the actual benefit for the years ended December 31, 2023 and 2022 are listed below.
SCHEDULE OF INCOME TAX EXPENSE (BENEFIT)
| 2023 | 2022 | |||||||
For the Year Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| Expected federal income tax (expense) benefit | $ | (903,220 | ) | $ | 2,851,758 | |||
| State and local taxes, net of federal (expense) benefit | (226,714 | ) | - | |||||
| Fair value adjustments | (819,270 | ) | - | |||||
| Share-based compensation | (557,168 | ) | (249,516 | ) | ||||
| Other permanent differences | 118,765 | - | ||||||
| NOL carryover adjusted for expiration | - | 46,936 | ||||||
| Equity method investment loss | - | (79,723 | ) | |||||
| Meals and entertainment | - | (4,843 | ) | |||||
| Research and development costs | - | (707,076 | ) | |||||
| Other temporary differences | - | 329,829 | ||||||
| NOL carryover adjustments | (5,148,128 | ) | - | |||||
| Intangibles | (720,407 | ) | - | |||||
| Other true ups | (230,474 | ) | - | |||||
| Changes in tax rates | 673,449 | - | ||||||
| Change in valuation allowance | 7,813,167 | 3,657,431 | ||||||
| Income tax (expense) benefit | $ | - | $ | 5,844,796 | ||||
All tax years starting with 2021 are open for examination.
NOTE 14 – RELATED PARTIES
CellerateRX Sublicense Agreement
The Company has an exclusive, world-wide sublicense to distribute CellerateRX Surgical and HYCOL products into the surgical and wound care markets from an affiliate of The Catalyst Group, Inc. (“Catalyst”), CGI Cellerate RX, which licenses the rights to CellerateRX Surgical and HYCOL from Applied. Sales of CellerateRX have comprised the substantial majority of the Company’s sales during 2023 and 2022. In January 2021, the Company amended the term of the sublicense agreement to extend the term to May 17, 2050, with automatic successive one-year renewals so long as annual net sales of the licensed products exceed $1,000,000. The Company pays royalties based on the annual Net Sales of licensed products (as defined in the sublicense agreement) consisting of 3% of all collected Net Sales each year up to $12,000,000, 4% of all collected Net Sales each year that exceed $12,000,000 up to $20,000,000, and 5% of all collected Net Sales each year that exceed $20,000,000.
As discussed further in Note 5, on August 1, 2023, the Company purchased certain assets from Applied, including the underlying intellectual property of, as well as the rights to manufacture and sell, CellerateRX Surgical and HYCOL products. In connection with the Applied Asset Purchase, Applied assigned its license agreement with CGI Cellerate RX to SMAT. Since the Closing, Sanara indirectly makes intercompany royalty payments to SMAT at the same rate as set forth in the Sublicense Agreement. Ronald T. Nixon, the Company’s Executive Chairman, is the founder and managing partner of Catalyst.
| F-28 |
Product License Agreements
In July 2019, the Company executed a license agreement with Rochal, a related party, whereby the Company acquired an exclusive world-wide license to market, sell and further develop antimicrobial products for the prevention and treatment of microbes on the human body utilizing certain Rochal patents and pending patent applications. Currently, the products covered by the BIAKŌS License Agreement are BIAKŌS Antimicrobial Wound Gel and BIAKŌS Antimicrobial Skin and Wound Cleanser. Both products are 510(k) cleared. Mr. Nixon is a director of Rochal, and indirectly a significant shareholder of Rochal, and through the potential exercise of warrants, a majority shareholder of Rochal. Another one of the Company’s directors is also a director and significant shareholder of Rochal.
In October 2019, the Company executed the ABF License Agreement with Rochal whereby the Company acquired an exclusive world-wide license to market, sell and further develop certain antimicrobial barrier film and skin protectant products for use in the human health care market utilizing certain Rochal patents and pending patent applications. Currently, the products covered by the ABF License Agreement are CuraShield Antimicrobial Barrier Film and a no sting skin protectant product.
In May 2020, the Company executed a product license agreement with Rochal, whereby the Company acquired an exclusive world-wide license to market, sell and further develop a debrider for human medical use to enhance skin condition or treat or relieve skin disorders, excluding uses primarily for beauty, cosmetic, or toiletry purposes.
See Note 10 for more information on these product license agreements.
Consulting Agreement
Concurrent with the Rochal asset purchase, in July 2021, the Company entered into a consulting agreement with Ann Beal Salamone pursuant to which Ms. Salamone agreed to provide the Company with consulting services with respect to, among other things, writing new patents, conducting patent intelligence, and participating in certain grant and contract reporting. In consideration for the consulting services to be provided to the Company, Ms. Salamone is entitled to receive an annual consulting fee of $177,697, with payments to be paid once per month. The consulting agreement has an initial term of three years, unless earlier terminated by the Company, and is subject to renewal. Ms. Salamone is a director of the Company and is a significant shareholder and the current Chair of the board of directors of Rochal.
Catalyst Transaction Advisory Services Agreement
In March 2023, the Company entered into a Transaction Advisory Services Agreement (the “Catalyst Services Agreement”) effective March 1, 2023 with Catalyst, a related party. Pursuant to the Catalyst Services Agreement, Catalyst, by and through its directors, officers, employees and affiliates that are not simultaneously serving as directors, officers or employees of the Company (collectively, the “Covered Persons”), agreed to perform certain transaction advisory, business and organizational strategy, finance, marketing, operational and strategic planning, relationship access and corporate development services for the Company in connection with any merger, acquisition, recapitalization, divestiture, financing, refinancing, or other similar transaction in which the Company may be, or may consider becoming, involved, and any such additional services as mutually agreed upon in writing by and between Catalyst and the Company (the “Catalyst Services”).
Pursuant to the Catalyst Services Agreement, the Company agreed to reimburse Catalyst for (i) compensation actually paid by Catalyst to any of the Covered Persons at a rate no more than a rate consistent with industry practice for the performance of services similar to the Catalyst Services, as documented in reasonably sufficient detail, and (ii) all reasonable out-of-pocket costs and expenses payable to unaffiliated third parties, as documented in customary expense reports, as each of (i) and (ii) is incurred in connection with the Catalyst Services rendered under the Catalyst Services Agreement, with all reimbursements being contingent upon the prior approval of the Audit Committee of the Company’s Board of Directors. The Company incurred $174,486 of costs pursuant to the Catalyst Services Agreement in the year ended December 31, 2023.
| F-29 |
ITEM 9. CHANGESCHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLSCONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
We maintain disclosure controls and procedures that are designed to ensure that information required to be disclosed by us in the reports that we file or submit to the SECSecurities and Exchange Commission (the “SEC”) under the Securities Exchange Act of 1934, as amended (the “Exchange Act”), is recorded, processed, summarized and reported within the time periods specified by the SEC’s rules and forms, and that information is accumulated and communicated to our management, including our principal executive and principal financial officers (whom we refer to in this periodic report as our Certifying Officers), as appropriate to allow timely decisions regarding required disclosure. Our management evaluated, with the participation of our Certifying Officers, the effectiveness of our disclosure controls and procedures as of
Management’s Report on Internal Control over Financial Reporting
Management is responsible for establishing and maintaining adequate internal control over financial reporting, as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act. Our internal control over financial reporting is designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of the consolidated financial statements for external reporting purposes in accordance with generally accepted accounting principles.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness of internal control over financial reporting to future periods are subject to the risk that controls may become inadequate because of changes in conditions or that the degree of compliance with the policies or procedures may deteriorate over time.
Management believes that our policies and procedures provide reasonable assurance that our operations are conducted with a high standard of business ethics. In management'smanagement’s opinion, our financial statements present fairly, in all material respects, our financial position, results of operations, and cash flows. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives. Management applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures.
The Company’s management, specifically its Certifying Officers, has assessed the effectiveness of the Company’s internal control over financial reporting as of
December 31,Changes in Internal Control over Financial Reporting
There were no changes in our internal control over financial reporting that occurred during the quarter ended December 31, 20202023 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting. We will continue to evaluate the effectiveness of internal controls and procedures on an on-going basis.
No Attestation Report of Registered Public Accounting Firm
This Annual Report on Form 10-K does not include an attestation report of our registered public accounting firm regarding internal control over financial reporting. Management’s report was not subject to attestation by our registered public accounting firm pursuant to the rules of the SEC that permit us to provide only management’s report in this Annual Report on Form 10-K.
ITEM 9B. OTHEROTHER INFORMATION
None.
ITEM 9C. DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS
Not applicable.
| 52 |
PART III
ITEM 10. DIRECTORS,DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The following table sets forthinformation required in response to this Item 10 is incorporated herein by reference to our Definitive Proxy Statement on Schedule 14A to be filed with the names, ages and positionsSEC no later than 120 days after the end of the current directorsfiscal year covered by this Annual Report on Form 10-K.
ITEM 11. EXECUTIVE COMPENSATION
The information required in response to this Item 11 is incorporated herein by reference to our Definitive Proxy Statement on Schedule 14A to be filed with the SEC no later than 120 days after the end of the Company.
| NAME | AGE | POSITION | YEAR FIRST ELECTED |
| Ronald T. Nixon | 65 | Executive Chairman | 2019 |
| J. Michael Carmena | 65 | Vice Chairman | 2019 |
| James W. Stuckert | 83 | Director | 2015 |
| Ann Beal Salamone | 70 | Director | 2019 |
| Kenneth E. Thorpe | 64 | Director | 2019 |
| Robert A. DeSutter | 52 | Director | 2020 |
| Sara N. Ortwein | 62 | Director | 2020 |
Salary | Bonus | Stock awards | All other compensation | Total | |||
Name and Principal Position | Year | ($)(1) | ($)(2) | ($)(3) | ($)(4) | ($) | |
| J. Michael Carmena, Principal Executive Officer | 2020 | 185,000 | 75,000 | 232,226 | 8,000 | 500,226 | |
| 2019 | 200,000 | 75,000 | - | 9,600 | 284,600 | ||
| Michael D. McNeil, Chief Financial Officer | 2020 | 161,875 | 87,500 | 153,519 | 6,837 | 409,731 | |
| 2019 | 168,000 | 70,500 | - | 5,085 | 243,585 | ||
| Zachary B. Fleming, Co-Chief Operating Officer and President, Surgical | 2020 | 208,125 | 112,500 | 246,755 | 24,785 | 592,165 | |
| 2019 | 204,167 | 115,000 | - | 11,746 | 330,913 | ||
| Shawn M. Bowman, Co-Chief Operating Officer and President, Wound Care | 2020 | 208,125 | 90,000 | 225,159 | 28,305 | 551,589 | |
| 2019 | 204,167 | 105,000 | - | 11,746 | 320,913 | ||
Base Salaries | |||
| Name | 2019(1) | 2020(2) | % Change |
| J. Michael Carmena | $200,000 | $180,000 | -10% |
| Michael D. McNeil | $175,000 | $157,500 | -10% |
| Zachary B. Fleming | $225,000 | $202,500 | -10% |
| Shawn M. Bowman | $225,000 | $202,500 | -10% |
OPTION AWARDS | STOCK AWARDS | |||||
| Name | Number of securities underlying unexercised options(Exercisable) (#) | Number of securities underlying unexercised options(Unexercisable) (#) | Option exercise price ($) | Option expiration date | Number of shares or units of stock that have not vested (#) (1) | Market value of shares of units of stock that have not vested ($) |
| Zachary B. Fleming | 2,000 | - | 6 | 12/31/2022 | 13,674 | 682,333 |
| Shawn M. Bowman | - | - | - | - | 12,314 | 614,469 |
| J. Michael Carmena | 5,000 | - | 6 | 12/31/2022 | 16,941 | 845,356 |
| Michael D. McNeil | 1,000 | - | 6 | 4/13/2023 | 11,038 | 550,796 |
| Name | Fees earned or paid in cash | Stock awards (2) | All other compensation | Total |
($) | ($) | ($) | ($) | |
| Ronald T. Nixon | - | 80,603 | - | 80,603 |
| James W. Stuckert | - | 80,603 | - | 80,603 |
| Ann Beal Salamone | - | 26,872 | - | 26,872 |
| Kenneth E. Thorpe | - | 26,872 | - | 26,872 |
| Robert A. DeSutter | - | - | - | - |
| Sara N. Ortwein | - | - | - | - |
| S. Oden “Denny” Howell Jr. (1) | - | 80,603 | - | 80,603 |
ITEM 12. SECURITYSECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The following table provides information as of December 31, 2020, as adjusted for the 1-for-100 reverse stock split which became effective May 10, 2019, regarding shares of common stock that may be issued under the Company's existing equity compensation plans:
| Plan Category | Number of shares to be issued upon the exercise of outstanding options | Weighted average exercise price of outstanding options | Number of shares remaining available for future issuance under equity compensation plans (excluding shares reflected in the 2nd column) |
| Equity compensation plans approved by shareholders | 11,500 | $6.00 | 1,751,724 |
| Equity compensation plans not approved by shareholders | — | — | — |
Common Stock | ||
| NAME | Number of shares beneficially owned | Beneficial ownership percentage |
| Directors and Named Executive Officers | ||
| Ronald T. Nixon (1) | 3,502,240 | 46.0% |
| James W. Stuckert (2) | 946,403 | 12.4% |
| J. Michael Carmena (3) | 14,014 | * |
| Zachery Fleming (4) | 10,353 | * |
| Shawn Bowman (5) | 6,992 | * |
| Michael McNeil (6) | 6,016 | * |
| Ann Beal Salamone (7) | 1,606 | * |
| Kenneth E. Thorpe (8) | 1,606 | * |
| All directors and executive officers as a group (9 persons) | 4,502,948 | 59.1% |
| Certain Beneficial Owners | ||
| S. Oden “Denny” Howell Jr. (9) | 490,394 | 6.4% |
| CGI Cellerate RX, LLC (10) | 2,452,731 | 32.2% |
| FA Sanara, LLC (11) | 963,856 | 12.7% |
ITEM 13. CERTAINCERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The Company was a participantinformation required in the following transactions with related parties since January 1, 2019.
ITEM 14. PRINCIPALPRINCIPAL ACCOUNTING FEES AND SERVICES
The information required by this Item 14 is incorporated herein by reference to conduct annual audits and reviewour Definitive Proxy Statement on Schedule 14A to be filed with the SEC no later than 120 days after the end of quarterly financial statements for the
| 53 |
PART IV
ITEM 15. EXHIBITSEXHIBITS AND FINANCIAL STATEMENT SCHEDULES
| (a) | Financial Statements |
Refer to Index to Financial Statements appearing on page F-1.
| (b) | Financial Statement Schedules |
No financial statement schedules are provided because the information called for is not required or is shown in the financial statements or the notes thereto.
| (c) | Exhibits |
The exhibits listed below are filed or incorporated by reference as a part of this report.
| 54 |
| Certification of Principal Financial Officer in accordance with 18 U.S.C. Section 1350, as adopted by Section 906 of the Sarbanes-Oxley Act of 2002. | ||
| Sanara MedTech Inc. Compensation Recovery Policy. | ||
| 101.INS | Inline XBRL Instance Document. | |
| 101.SCH | Inline XBRL Taxonomy Extension Schema Document. | |
| 101.CAL | Inline XBRL Taxonomy Extension Calculation Linkbase Document. | |
| 101.DEF | Inline XBRL Taxonomy Extension Definition Linkbase Document. | |
| 101.LAB | Inline XBRL Taxonomy Extension Label Linkbase Document. | |
| 101.PRE | Inline XBRL Taxonomy Extension Presentation Linkbase Document. | |
| 104 | Cover Page Interactive Data |
* Filed herewith
# Certain schedules and exhibits to this agreement have been omitted pursuant to Item 601(a)(5) of Regulation S-K. A copy of any omitted schedule or exhibit will be furnished supplementally to the Securities and Exchange Commission or its staff upon request. If indicated on the first page of such agreement, certain confidential information has been excluded pursuant to Item 601(b)(2)(ii) of Regulation S-K. Such excluded information is not material and is the type that the Company treats as private or confidential.
+ Certain schedules and exhibits to this agreement have been omitted pursuant to Item 601(a)(5) of Regulation S-K. A copy of any omitted schedule or exhibit will be furnished supplementally to the Securities and Exchange Commission or its staff upon request. Certain confidential information has been excluded pursuant to Item 601(b)(10)(iv) of Regulation S-K. Such excluded information is not material and is the type that the Company treats as private or confidential.
** The certifications attached as Exhibit 32.1 and Exhibit 32.2 are not deemed “filed” with the Securities and Exchange Commission and are not to be incorporated by reference into any filing of Sanara MedTech Inc. under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended, whether made before or after the date of this Annual Report on Form 10-K, irrespective of any general incorporation language contained in such filing.
† Identifies a management contract or compensatory plan
ITEM 16. FORMFORM 10-K SUMMARY
None.
| 55 |
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| SANARA MEDTECH INC. | |||
| March | By: | /s/ Michael D. McNeil | |
| Michael D. McNeil | |||
Chief Financial Officer | |||
| (Principal Financial Officer and duly authorized officer) | |||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signature | Title | Date | ||
/s/ Zachary B. Fleming | March | |||
/s/ Michael D. McNeil | Chief Financial Officer (Principal Financial and Accounting Officer) | March | ||
| Michael D. McNeil | ||||
/s/ Ronald T. Nixon | Chairman | March 25, 2024 | ||
| Ronald T. Nixon | ||||
| /s/ Robert DeSutter | Director | March 25, 2024 | ||
| Robert DeSutter | ||||
| /s/ Roszell Mack III | Director | March 25, 2024 | ||
| Roszell Mack III | ||||
| /s/ Eric D. Major | Director | March 25, 2024 | ||
| Eric D. Major | ||||
| /s/ Sara Ortwein | Director | March 25, 2024 | ||
| Sara Ortwein | ||||
| /s/ Ann Beal Salamone | Director | March 25, 2024 | ||
| Ann Beal Salamone | ||||
| /s/ James W. Stuckert | Director | March | ||
| James W. Stuckert | ||||
/s/ Eric D. Tanzberger | March | |||
| 56 |