Denali Therapeutics Inc.
The aggregate market value of the common stock held by non-affiliates of the registrant as of June 30, 20202023 (the last business day of the registrant’s most recently completed second fiscal quarter) was approximately $850.6 million,$2.0 billion, based on the closing price of the registrant’s common stock, as reported by the NASDAQNasdaq Global Select Market on June 30, 20202023 of $24.18$29.51 per share. Shares of the registrant’s common stock held by each executive officer, director and holder of 5% or more of the outstanding common stock have been excluded in that such persons may be deemed to be affiliates. This calculation does not reflect a determination that certain persons are affiliates of the registrant for any other purpose.
Denali Therapeutics Inc.
This Annual Report on Form 10-K contains forward-looking statements. All statements other than statements of historical facts contained in this report, including statements regarding our future results of operations and financial position, business strategy, product candidates, planned preclinical studies and clinical trials, research and development costs, regulatory approvals, timing and likelihood of success, as well as plans and objectives of management for future operations, are forward-looking statements. These statements involve known and unknown risks, uncertainties and other important factors that are in some cases beyond our control and may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by the forward-looking statements.
In some cases, you can identify forward-looking statements by terms such as “may,” “will,” “should,” “would,” “expect,” “plan,” “anticipate,” “could,” “intend,” “target,” “project,” “contemplate,” “believe,” “estimate,” “predict,” “potential” or “continue” or the negative of these terms or other similar expressions. Forward-looking statements contained in this report include, but are not limited to, statements about:
We have based these forward-looking statements largely on our current expectations and projections about our business, the industry in which we operate, and financial trends that we believe may affect our business, financial condition, results of operations and prospects, and these forward-looking statements are not guarantees of future performance or development. These forward-looking statements speak only as of the date of this report and are subject to a number of risks, uncertainties and assumptions described in the section titled “Risk Factors” and elsewhere in this report. Because forward-looking statements are inherently subject to risks and uncertainties, some of which cannot be predicted or quantified, you should not rely on these forward-looking statements as predictions of future events. The events and circumstances reflected in our forward-looking statements may not be achieved or occur and actual results could differ materially from those projected in the forward-looking statements. Except as required by applicable law, we undertake no obligation to update publicly any forward-looking statements for any reason after the date of this Annual Report on Form 10-K to conform these statements to actual results or to changes in expectations.
In addition, statements that “we believe” and similar statements reflect our beliefs and opinions on the relevant subject. These statements are based upon information available to us as of the date of this report, and while we believe such information forms a reasonable basis for such statements, such information may be limited or incomplete, and our statements should not be read to indicate that we have conducted an exhaustive inquiry into, or review of, all potentially available relevant information. These statements are inherently uncertain and you are cautioned not to unduly rely upon these statements.
This report contains estimates, projections and other information concerning our industry, our business and the markets for our product candidates. We obtained the industry, market and similar data set forth in this report from our own internal estimates and research and from academic and industry research, publications, surveys and studies conducted by third parties, including governmental agencies. Information that is based on estimates, forecasts, projections, market research or similar methodologies is inherently subject to uncertainties and actual events or circumstances may differ materially from events and circumstances that are assumed in this information. While we believe that the data we use from third parties are reliable, we have not separately verified these data. Further, while we believe our internal research is reliable, such research has not been verified by any third party. You are cautioned not to give undue weight to any such information, projections and estimates.
ITEM 1. BUSINESS
Our goal is to discover, develop and deliver therapeutics to defeat degeneration.
As part of our strategy, we identify and validate biomarkers which are relevant for both animal models and human trials and are critical for selecting patients, predicting and measuring target engagement, supporting dose selection and enabling decisions on progression of product candidates to the next phase of development. When practicable, we are developing patient selection biomarkers for our programs to enable identification of patients with the relevant disease biology and stage of disease likely to benefit from targeted therapy in order to increase the likelihood of success of clinical trials. Ultimately, by reducing the number of patients that are likely to experience a low treatment response, we expect to positively impact market acceptance of these targeted therapies, driven by high and meaningful response rates within the targeted population as defined by the patient selection biomarkers. In certain indications, regulatory approval may limit the market of a product candidate to target patient populations when patient selection biomarkers are used. In these indications, regulatory authorities may require us to run additional clinical trials prior to expanding the label for approval that includes a broader patient population.
Our Programs
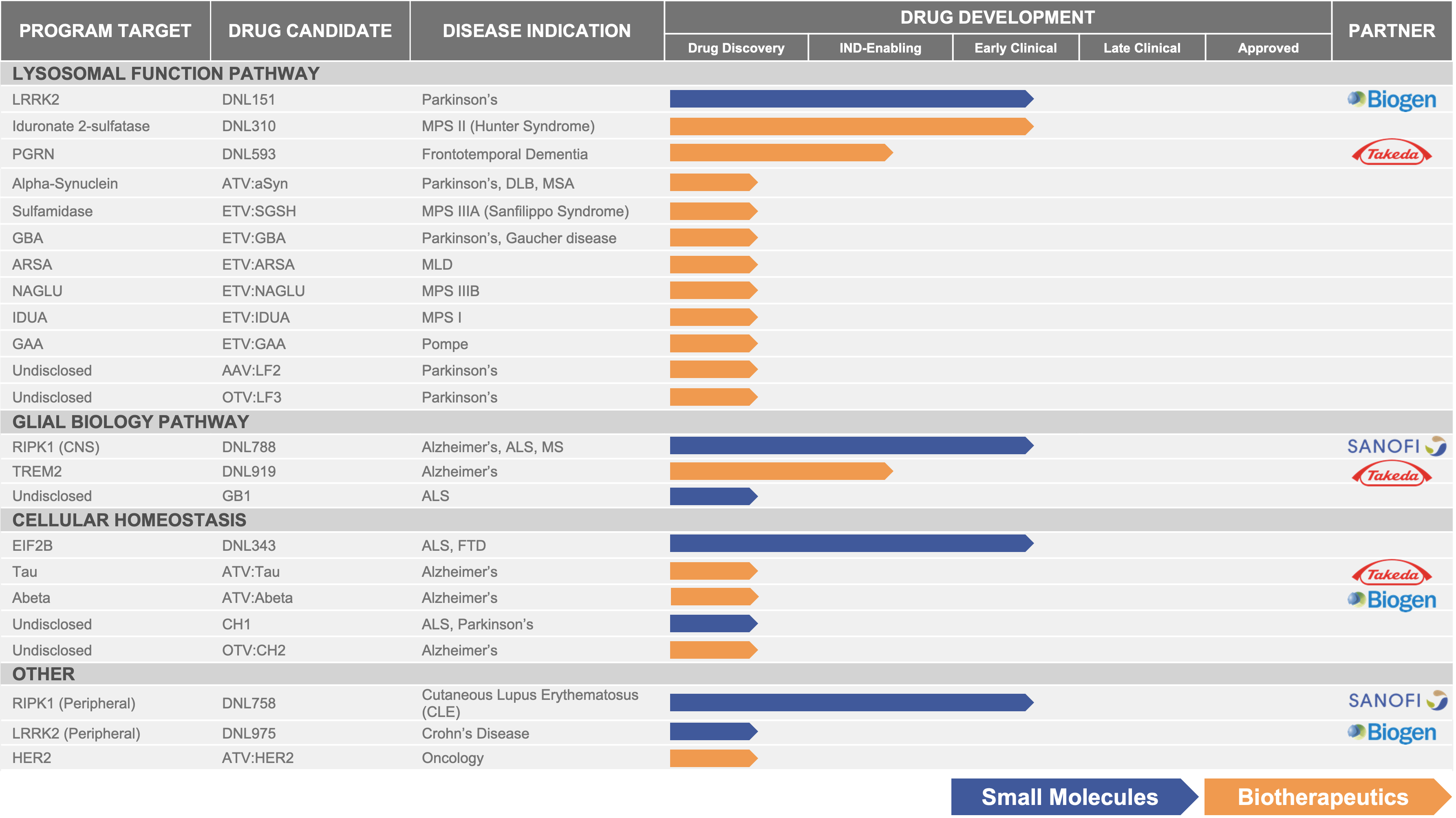
The following table summarizes key information about our clinical programs and pipeline:
Figure 4:5: Denali’s current pipelinedevelopment pipeline.
We are developing a broad portfolio of targeted therapeutic candidates for neurodegenerative diseases.diseases and LSDs. Our programs are at different stages of clinical and preclinical development. In addition to our current clinical development portfolio, we expect to advance at least three new molecule entities (“NMEs”) into clinical development in the 2024 to 2025 timeframe. We discuss our most advanced programs in further detail below.
Late-Stage And Mid-Stage Clinical Programs
Tividenofusp alfa(DNL310, ETV:IDS) Enzyme Replacement Therapy Program for MPS II (Hunter Syndrome)
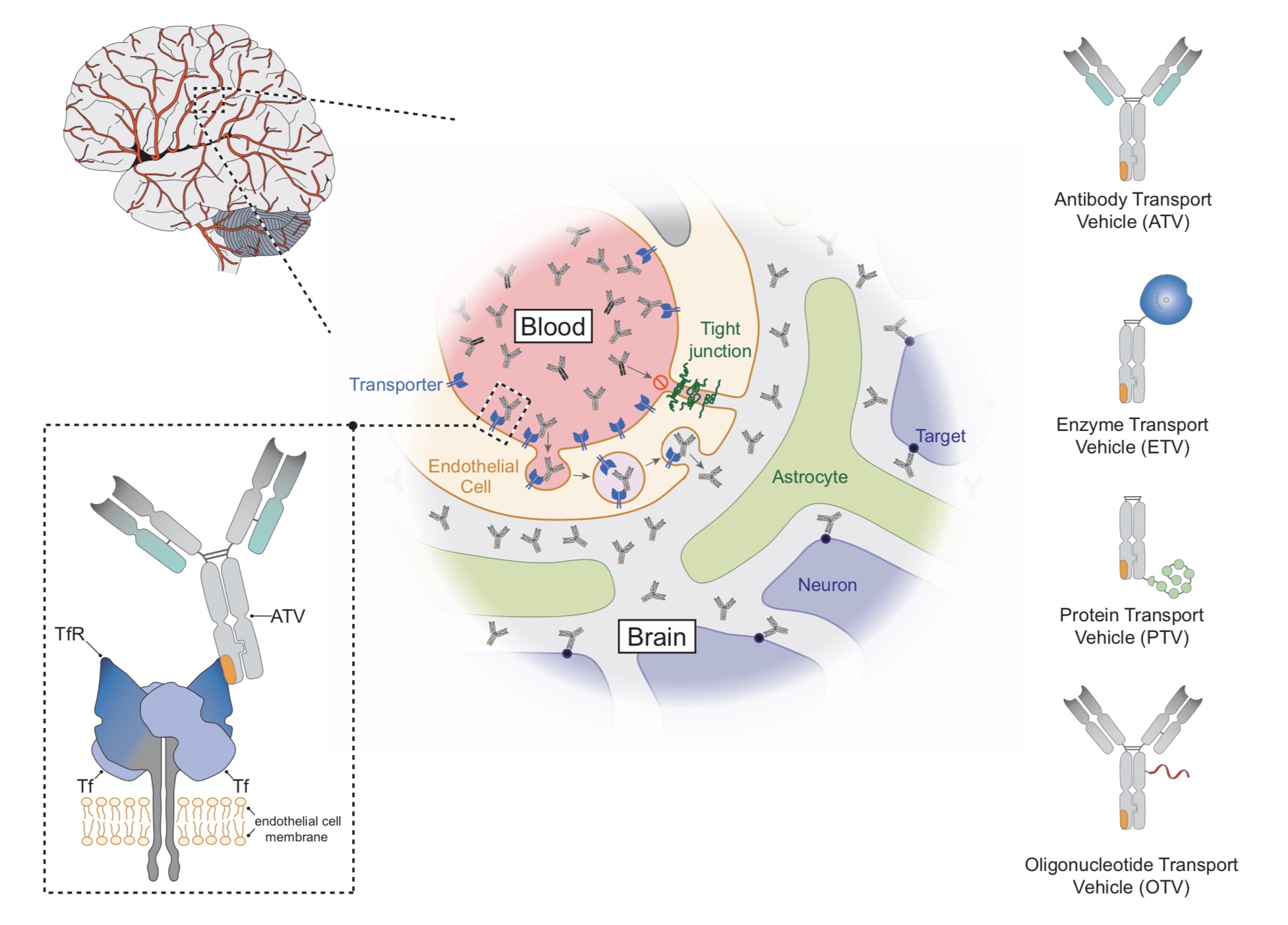
MPS II, also called Hunter syndrome, is a rare genetic disease that affects over2,000 individuals, primarily males, world-wide, and leads to behavioral, cognitive, and physical symptoms ultimately resulting in shortened lifespan. MPS II is caused by mutations in the iduronate-2-sulfatase (IDS) gene, which leads to a deficiency of the IDS enzyme responsible for the breakdown of the glycosaminoglycans (GAGs) heparan and dermatan sulfate in lysosomes. Symptoms often begin emerging around age two and include physical complications, including organ dysfunction, joint stiffness, hearing loss and impaired growth leading to short stature, and neurocognitive symptoms with impaired development. The disease is characterized by a buildup of GAGs in lysosomes — the part of the cell that breaks down materials including GAGs. The current standard of care enzyme replacement therapy ("ERT") partially treats the physical symptoms but does not cross the blood-brain barrier, and as a result, cognitive and behavioral symptoms experienced by the majority of patients with MPS II are not addressed. Therapies that address behavioral, cognitive, and somatic manifestations of the disease are one of the greatest unmet needs for this community.
DNL310 is a fusion protein composed of IDS fused to Denali’s proprietary ETV, which is engineered to cross the BBB via receptor-mediated transcytosis into the brain and to enable broad delivery of IDS into cells and tissues throughout the body with the goal of addressing the behavioral, cognitive, and physical manifestations of MPS II. In March 2021, the U.S. Food and Drug Administration granted Fast Track designation to DNL310 for the treatment of patients with MPS II. In May 2022, the European Medicines Agency ("EMA") granted DNL310 Priority Medicines designation. DNL310 is an investigational product candidate and has not been approved by any Health Authority.
Based on supportive clinical and preclinical data to date, we are enrolling the Phase 2/3 COMPASS study in North America, South America, and Europe. The Phase 2/3 COMPASS study is expected to enroll 54 participants with MPS II, with and without neuronopathic disease. The participants are randomized 2:1 to receive either DNL310 or idursulfase, respectively. Cohort A includes children ages ≥2 to <6 years with neuronopathic disease; cohort B includes children ages ≥6 to <17 without neuronopathic disease. Upon completion of the ongoing Phase 1/2 study, and together with data from the global COMPASS study, this combined data package is intended to support registration.
In parallel with the Phase 2/3 COMPASS study, we are also conducting a Phase 1/2 trial for DNL310 in patients with MPS II as a multicenter, multiregional, open-label, single-arm trial to assess the safety, pharmacokinetics, and pharmacodynamics of DNL310 administered once weekly by intravenous infusion. We have previously reported interim analyses from the Phase 1/2 study and most recently at the 2024 WORLDSymposiumTM in February 2024. Key results are summarized below.
The additional data reported at the 2024 WORLDSymposiumTM demonstrated that DNL310 enabled rapid and sustained normalization of heparan sulfate in cerebrospinal fluid ("CSF") with mean reductions from baseline of approximately 90% at weeks 24, 49, and 104. Normalizationof CSF heparan sulfate was observed even in participants with high levels of preexisting serum anti-iduronate-2-sulfatase antibodies (Figure 6). In urine, a high-magnitude and sustained reduction of heparan sulfate and dermatan sulfate after switching from standard-of-care ERT to DNL310 suggested added peripheral activity.
Figure 6: Normal levels of CSF HS were achieved and sustained over time, including in those with pre-existing high serum ADA.
Following treatment with DNL310, mean reductions of 64%, 60%, and 71%, representing near normalization or complete normalization, were observed in levels of gangliosides GM2 and GM3 and glucosylsphingosine lipids, respectively, at week 104 in participants receiving DNL310, which is consistent with improved lysosomal function.
Robust and sustained reduction in serum neurofilament light ("NfL"), a marker of neuronal damage, was observed, reaching statistical significance after 61 weeks and a reduction of 64% after two years of treatment with DNL310 (Figure 7). Changes in serum and CSF NfL from baseline to week 104 were correlated; at week 104, there was a statistically significant 54% mean reduction from baseline in CSF NfL.
Figure 7: Robust reduction of serum NfL, a marker of neuronal damage, was observed with long-term dosing.
Exploratory clinical outcomes data from Vineland Adaptive Behavior Scales (VABS)-II and Bayley Scales of Infant and Toddler Development (BSID)-III assessments out to 104 weeks of treatment with DNL310 were reported for the first time at the 2024 WORLDSymposiumTM. Continued improvement and stabilization of adaptive behavior and cognitive scores suggest DNL310 has positive effects (Figure 8).
Figure 8: Continued improvement and stabilization in sum of VABS-II scores and cognitive scores suggest DNL310 has positive effects.
Hearing, as assessed by auditory brainstem response ("ABR") testing, numerically improved over time after initiation of DNL310 across all frequencies (Figure 9). At week 104, ABR thresholds showed statistically significant improvements across three of the four frequencies, with a trend toward greater improvement at higher frequencies.
Figure 9: Changes of ABR thresholds with DNL310 treatment suggest improved hearing.
Generally positive somatic activity on multiple endpoints was observed in participants receiving DNL310. Liver volume in standard-of-care ERT-naïve participants decreased by a mean of 25% from baseline at weeks 24 and 49. In standard-of-care ERT-experienced participants liver volume decreased by a mean of 5% from baseline at week 49. Spleen volume was normal in all participants at weeks 24 and 49 of DNL310 treatment. At baseline, spleen volume was normal in all participants except 1 standard-of-care ERT-naïve participant, who had normal spleen volumes at weeks 24 and 49 of DNL310 treatment.
DNL310 continues to be generally well tolerated (median treatment duration 100 weeks and maximum treatment duration 161 weeks). The most frequent treatment-emergent adverse events were infusion related reactions, which decreased in frequency and severity with continued dosing. An independent data monitoring committee met in December 2023 and recommended that the study may continue without modifications.
2024 expected progress and milestones:
•Presentation of additional interim Phase 1/2 data at the Society for the Study of Inborn Errors of Metabolism (SSIEM) Annual Symposium.
•Complete enrollment of global Phase 2/3 COMPASS study in MPS II.
DNL343 eIF2B Activator Program for ALS
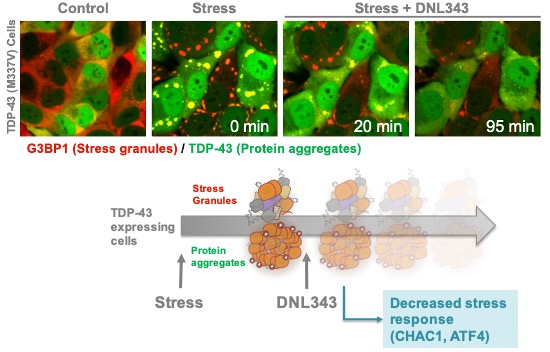
We are developing DNL343 as a novel eIF2B activator with first-in-class potential for the treatment of ALS and other indications. eIF2B is an intracellular protein complex that regulates protein synthesis and is required for neuronal health and function. When neurons experience stress, activation of the integrated stress response ("ISR") pathway leads to suppression of eIF2B activity, resulting in impaired protein synthesis and formation of stress granules. Stress granules are thought to be a precursor of TDP-43 aggregation, which is a hallmark pathology in ALS. DNL343 is designed to activate eIF2B and thereby restore protein synthesis, disperse TDP-43 aggregates, and improve neuronal survival.
In May 2023, the first participant with ALS was dosed with DNL343 (Regimen G) in the Phase 2/3 HEALEY ALS Platform Trial led by the Sean M. Healey & AMG Center for ALS ("Healey Center") at Massachusetts General Hospital ("MGH") in collaboration with the Northeast ALS Consortium ("NEALS") clinical trial network. The HEALEY ALS Platform Trial is a large-scale collaborative effort made possible by contributions from patients and families, clinical trial sites, industry partners and research collaborators to evaluate multiple investigational therapies simultaneously with the goal of accelerating the development of potential new treatments for ALS. Therapeutic candidates that enter the platform trial are chosen by a group of expert ALS scientists and members of the Healey & AMG Center. Recruitment is ongoing in Regimen G (DNL343) of the Phase 2/3 HEALEY ALS Platform Trial.
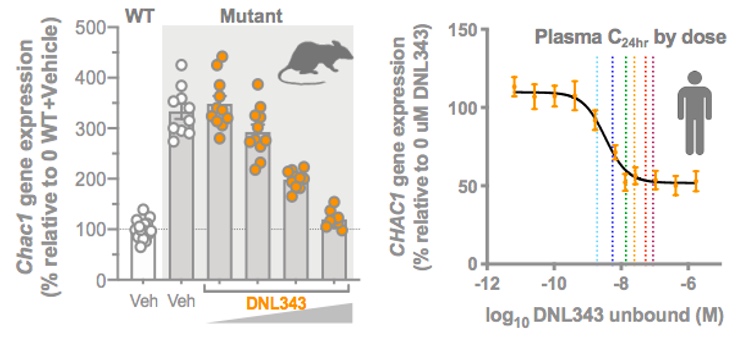
Late-stage development of DNL343 is supported by results of Phase 1/1b studies in 95 healthy volunteers and 28 participants with ALS. In April 2023, we presented final data from the 28-day treatment period of the Phase 1b study of DNL343 in participants with ALS at the 75th Annual Meeting of the American Academy of Neurology ("AAN"). The Phase 1b study is a multicenter, randomized, placebo-controlled, double-blind, 28-day trial followed by an 18-month open-label extension, designed to evaluate the safety, pharmacokinetics and pharmacodynamics of DNL343 participants with ALS. Study results have demonstrated that once-daily oral dosing with DNL343 for 28 days was generally well tolerated and demonstrated extensive CSF penetration. In addition, robust inhibition of biomarkers associated with the ISR pathway was observed in blood samples from study participants (Figure 10).

Figure 10: Phase 1b results demonstrated 28-day dosing with DNL343 reduced ISR biomarkers (e.g., ATF4 protein and CHAC1 mRNA) in blood samples from ALS patients.
2024 expected progress and milestones:
•Complete enrollment of participants in Regimen G (DNL343) in Phase 2/3 HEALEY ALS Platform Trial.
SAR443820/DNL788 RIPK1 Inhibitor Program for CNS Disease: ALS, MS
As described in more detail in “Business - Licenses and Collaborations” below, we are collaborating with Sanofi on the development of small molecules that inhibit RIPK1, a critical signaling protein in the tumor necrosis factor (“TNF”) receptor pathway and a regulator of inflammation and cell death. Increased RIPK1 activity in the brain drives neuroinflammation and cell necroptosis and contributes to neurodegeneration. RIPK1 inhibition has been shown to have beneficial effects in preclinical models of ALS, MS, Alzheimer's disease, and other diseases.
SAR443820/DNL788 is our lead CNS-penetrant RIPK1 inhibitor in clinical development. Sanofi completed a Phase 1 trial of SAR443820/DNL788 in healthy volunteers, which demonstrated high brain penetrance and robust target engagement at doses that were generally well tolerated.
In February 2024, Sanofi informed us that the Phase 2 HIMALAYA study evaluating SAR443820/DNL788 in participants with amyotrophic lateral sclerosis (ALS) did not meet the primary endpoint of change in ALS Functional Rating Scale-Revised (ALSFRS-R). Sanofi intends to present the detailed efficacy and safety results of the ALS Phase 2 HIMALAYA study at a future scientific forum.
Sanofi is evaluating SAR443820/DNL788 in another Phase 2 clinical trial in participants with multiple sclerosis (MS), and the outcome of HIMALAYA study has no impact on the ongoing MS study.
2024 expected progress and milestones:
•Continue Phase 2 K2 study in MS
BIIB122/DNL151 LRRK2 Inhibitor Program for Parkinson's disease
Parkinson's disease is one of the most common brain diseases, affecting approximately 610 million people world-wide.worldwide. It is commonly thought of asconsidered to be a movement disorder because patients can experience tremors, slowness of movement, stiffness and difficulty with walking and balance. In addition, Parkinson's patients can have other non-motor type problems such as constipation, depression and memory loss. The disease results fromParkinson symptoms are a result of the loss of dopamine-producing cells in the brain, andwhich is likelycurrently thought to be caused by a combination of genetic and environmental risk factors.
Mutations in the LRRK2 isgene are one of the most common genetic risk factorfactors for Parkinson's disease, anddisease. LRRK2 is involved in maintaining a healthy cellular environment by regulating lysosomal function through modification of Rab proteins. Increased levels of LRRK2 kinase activity lead to lysosomal dysfunction, which contributesis believed to neurodegeneration and the formation of Lewy bodies, a central pathology of Parkinson's disease.contribute to neurodegeneration. Inhibition of LRRK2 activity mayhas the potential to slow the progression of Parkinson’s disease in patients, with and without known genetic risks based on restoration of lysosomal function.
We have advanced two chemically distinct CNS-penetrant small molecules that inhibit LRRK2 into clinical development, DNL201 and DNL151. Based on the totality of preclinical and clinical data to date, both DNL201 and DNL151 have met our requirements to proceed into further late-stage clinical studies. In August 2020, we, in collaboration with Biogen, announced the selection of DNL151 to advance into two late-stage studies in Parkinson’s disease: one in Parkinson's patients who carry LRRK2 mutations and the other in Parkinson's patients independent of mutation status.
DNL151 was selected for its pharmacokinetic properties that provide additional dosing regimen flexibility. DNL201 remains a backup molecule for this program. As described in more detail in “Business - Licenses and Collaborations” below, we are collaborating with Biogen onto co-develop and co-commercialize our small molecule inhibitors of LRRK2 for Parkinson's disease. BIIB122/DNL151 is the most clinically advanced small molecule inhibitor of LRRK2 programcurrently in clinical testing for Parkinson's disease. Biogen is conducting the global Phase 2b LUMA study, which commenced in May 2022 and intendis evaluating the efficacy and safety of BIIB122/DNL151 as compared to initiate late-stage developmentplacebo in the second half of 2021.
Biomarker-Driven Developmentapproximately 640 participants with early-stage Parkinson's disease.
We have developed assays that measureResults from Phase 1 and Phase 1b trials of BIIB122/DNL151 in healthy volunteers and patients with Parkinson's disease, respectively, showed robust target and pathway engagement as measured by pS935 LRRK2 and pRab10 phosphorylation as markers ofpT73 Rab10 (“pRab10”), respectively. Furthermore, reduction in total LRRK2 kinase activity to demonstrate target engagement in humans. Phosphorylation of LRRK2 at serine 935 ("pS935") is a well-established biomarker of LRRK2 kinase activity that has been demonstrated to respond to pharmacological inhibition. Rab10 is a member of the Rab GTPase family involved in endolysosomal function and is a direct substrate of LRRK2 kinase.
To provide greater insight into the effects of LRRK2 inhibitors on lysosomal biology in humans, in addition to pRab10 phosphorylation, we have measured di-22:6-bis (monoacylglycerol) phosphate ("BMP di22:6") in the urine of subjects treated with LRRK2 inhibitors. BMP di22:6 is a lysosomal lipid biomarker that indicates changes in lysosome function associated with LRRK2 inhibition and is elevated in individuals that carry LRRK2 disease-associated mutations.
Preliminary Results of DNL151 (BIIB122) Phase 1 Clinical Trial
DNL151 (BIIB122) has completed dosing of 162 healthy volunteers in an ongoing Phase 1 trial in healthy volunteer subjects and met safety,CSF demonstrated central target engagement, and a dose-dependent reduction in urine of the lysosomal lipid 22:6-bis[monoacylglycerol] phosphate (“BMP”), a biomarker goals. In January 2020, we announced that preliminary results from the ongoing Phase 1 trial for of lysosomal function, suggested improvement of lysosomal function. BIIB122/DNL151 (BIIB122) in healthy volunteers demonstrated an encouraging safety and pharmacokinetic/pharmacodynamic profile (Figure 5A). DNL151 (BIIB122) was generally well tolerated at allacross a broad range of doses tested, and the majority of subjects experienced either no or mild AEs. Target and pathway engagement of greater than 50 percent and a dose-dependent reduction of BMP in urine offor up to 50 percent were observed at clinically relevant doses. We are currently completing further dose escalation cohorts28 days, the longest treatment duration in the Phase 1 trial to define the full therapeutic window of DNL151 (BIIB122).both studies.
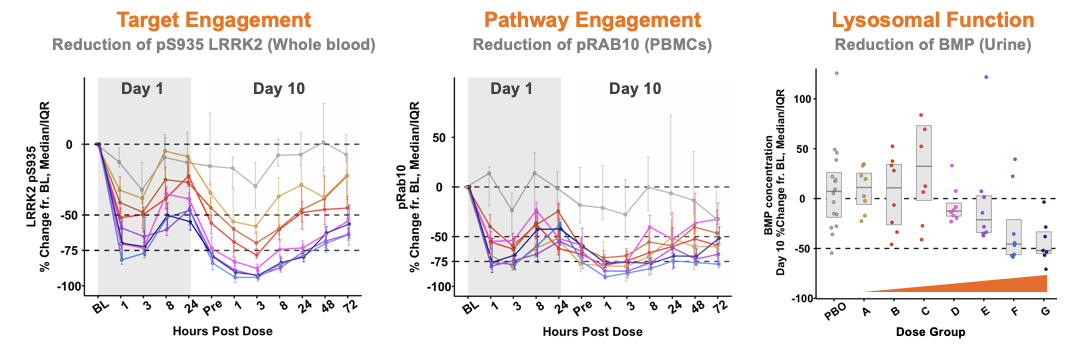
Figure 5A: Left: DNL151 (BIIB122) demonstrated: pS935 LRRK2 inhibition, a biomarkerIn June 2023, Denali in conjunction with Biogen, and based on review of portfolio timelines and resource prioritization, announced plans to revise the clinical development program for BIIB122/DNL151. Prior to the planned revisions, the BIIB122 clinical development program encompassed two global late-stage clinical trials: the Phase 2b LUMA study in participants with early-stage Parkinson’s disease noted above; and the Phase 3 LIGHTHOUSE study in participants with Parkinson’s disease related to pathogenic variants of LRRK2, kinase activity and target engagement. Middle: pRab10 inhibition, a biomarker of LRRK2 kinase pathway engagement linked to lysosomal regulation. Right: reductionwhich commenced in urinary BMP.September 2022.
DNL151 (BIIB122) Phase 1b trialIn consideration of the LIGHTHOUSE study’s complexity, including the long timeline with anticipated study completion in 2031, we and Biogen agreed to refocus efforts to enable a timely readout on efficacy in idiopathic early-stage Parkinson’s disease patients
The DNL151 (BIIB122) Phase 1b trial (NCT04056689)while gaining further clinical data in Parkinson's disease patients was a 28-day, multicenter, randomized, placebo controlled, double-blind clinical trial in patients with mild-to-moderate Parkinson’s disease conducted in the United States and Europe. Its purpose was to evaluate safety, tolerability, pharmacokinetics, pharmacodynamics, including target and pathway engagement biomarkers as well as certain exploratory clinical endpoints, after multiple oral doses of DNL151 (BIIB122). A total of 36 Parkinson's patients were enrolled. In January 2021, we announced completion of the Phase 1b trial and that target engagement and pathway engagement goals (the same as for DNL201) were met. We plan to present data from the Phase 1b trial at upcoming medical congresses.
DNL151 (BIIB122) Future Development Plan
We and Biogen expect to initiate late-stage clinical development of DNL151 (BIIB122) in Parkinson's patients by year-end 2021. Two clinical trials are planned: one in Parkinson's patients who carry LRRK2 mutations and the other in Parkinson's patients independent of mutation status.
Results of DNL201 Phase 1 Clinical Trial
The DNL201 Phase 1 clinical trial in healthy volunteers was completed in July 2018. This was a randomized, double-blind, placebo-controlled, single-center clinical trial in 105 healthy young and 17 healthy elderly subjects to investigate the safety and tolerability of single and multiple oral doses of DNL201 and characterize the pharmacokinetics and pharmacodynamics of DNL201 in plasma and CSF. In this trial, DNL201 met all objectives including cerebrospinal fluid ("CSF") exposure levels and LRRK2 inhibition, as well as evidence of pathway engagement, at doses that were well tolerated. The data from this clinical trial supported our decision to initiate the Phase 1b clinical trial in Parkinson’s disease patients.
Results of DNL201 Phase 1b Clinical Trial
The DNL201 Phase 1b clinical trial in Parkinson’s disease patients was completed in December 2019, and safety and biomarker data from this trial, including biomarkers of lysosomal function, support advancement to the next stage of clinical development (Figure 5B below).
This trial (NCT03710707) was a 28-day, randomized, placebo-controlled Phase 1b clinical trial in patients with mild to moderate Parkinson’s disease with and without geneticpathogenic variants in LRRK2. The planned revisions to the BIIB122 clinical development program were not based on any safety or efficacy data from studies of BIIB122. We have since modified the LUMA study’s enrollment criteria to allow for inclusion of eligible participants with Parkinson’s disease and a confirmed pathogenic variant of LRRK2, mutations. Its purpose wasin addition to continuing to enroll eligible participants with idiopathic early-stage Parkinson’s disease.
In February 2024, we announced the execution of a Collaboration and Development Funding Agreement in January 2024 with a third party related to a global Phase 2a study of BIIB122/DNL151, which Denali plans to solely operationalize to evaluate safety tolerability, pharmacokinetics, pharmacodynamics, and target and pathway engagement biomarkers associated with BIIB122 in multiple oral doses of DNL201. Exploratory endpoints included certain clinical endpoints. The 28 patients dosed in the trial were randomized to receive either a low dose of DNL201, a high dose of DNL201, or placebo.
Safety
DNL201 was generally well tolerated at the low dose and the majority of subjects experienced either no or mild adverse events (“AEs”). There was one serious adverse event considered unrelated to drug. At the high dose, the majority of subjects experienced either mild or moderate AEs and there was one severe AE (headache) leading to dose reduction and one study withdrawal (headache and nausea). All treatment-related AEs were manageable and reversible.
Target and Pathway Engagement
The Phase 1b results with DNL201 in patientsparticipants with Parkinson’s disease met all biomarker goals by demonstrating greater than 50 percent inhibitionand confirmed pathogenic variants of pS935LRRK2. This agreement includes committed funding of $75.0 million, of which $12.5 million was received in January 2024, and the remainder will be triggered based on time and operational milestones in the study. Biogen will continue to conduct the ongoing global Phase 2b LUMA study in early-stage Parkinson’s disease. Denali and Biogen will co-commercialize BIIB122/DNL151 assuming regulatory approval. The third party will be eligible to receive low single-digit royalties from Denali on annual worldwide net sales of LRRK2 and pRAB10 in bloodinhibitors for both doses tested and improvementthe treatment of Parkinson’s disease, with royalty amounts varying based on the scope of the lysosomal biomarker BMP by 20 percent and 60 percent in urine at the low and high dose, respectively.

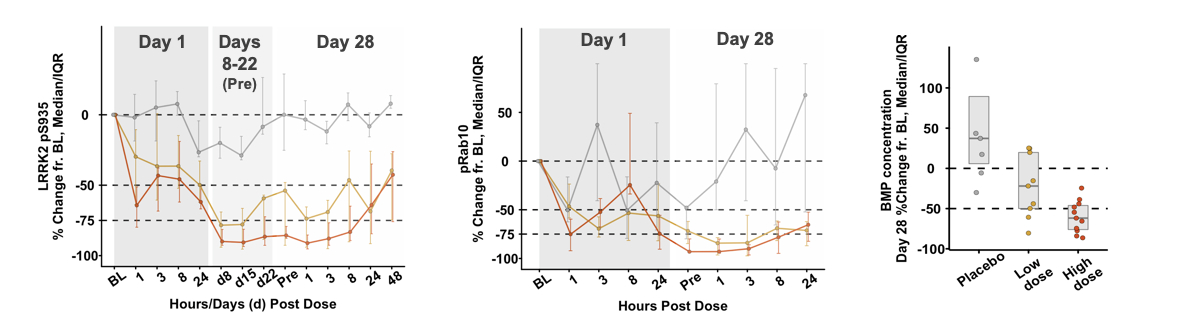

Figure 5B: Left:label DNL201 demonstrated: pS935 LRRK2 inhibition, a biomarker of LRRK2 kinase activity and target engagement. Middle: pRab10 inhibition, a biomarker of LRRK2 kinase pathway engagement linked to lysosomal regulation. Right: reduction in urinary BMP, an established biomarker of lysosomal dysfunction in biofluids that is elevated in LRRK2 mutation carriers..
•Continue Phase 2b LUMA study in early-stage PD.
•Initiate Phase 2a study in PD related to pathogenic variants of LRRK2.
Other LRRK2 Compounds
Genetic and functional studies have linked LRRK2 and other proteins that modulate lysosomal function to Crohn's disease. Excessive LRRK2 activity leads to a reduction in lysosomal function, which contributes to the inflammation and intestinal dyshomeostasis that are characteristic of this disorder. We have discovered potent and selective small molecule inhibitors of LRRK2 and have selected a lead clinical candidate (“DNL975”)(DNL975) for treatment of Crohn's disease. As described in more detail in “Business - Licenses and Collaborations” below, we are collaborating with Biogen on the Peripheral LRRK2 program.
ETV:IDS Enzyme Replacement TherapyEclitasertib (SAR443122/DNL758) RIPK1 Inhibitor Program for Hunter Syndrome
Hunter syndrome is an X-linked, monogenic LSD, which affects approximately 2,000 boys world-wide. Hunter syndrome is caused by a mutation in the gene that encodes for the enzyme iduronate-2-sulfatase (IDS). IDS is an enzyme responsible for breaking down glycosaminoglycans ("GAGs") in the lysosome thereby maintaining lysosomal homeostasis. In Hunter syndrome, the reduction or loss of IDS enzyme activity leads to accumulation of GAGs, which causes lysosomal dysfunction and neurodegeneration as well as progressive damage to multiple organs including bone, cartilage, heart, lung and brain. Approximately two-thirds of Hunter syndrome patients suffer from central manifestations of the disease, which is characterized by intellectual disability and a progressive cognitive decline that emerges between three and five years of age. Hunter syndrome is currently treated with IV infusions of recombinant IDS protein. These treatments do not efficiently distribute to the brain and, therefore, cannot address the neurological symptoms of the disease. There is a demonstrated need for enzyme replacement therapies ("ERTs") that effectively cross the BBB so as to treat both central and peripheral manifestations of Hunter syndrome and other LSDs.
DNL310 ("ETV:IDS") is an intravenously, centrally administered ERT biotherapeutic enabled by our enzyme transport vehicle ("ETV"), designed to address both central and peripheral manifestations of the disease by delivering IDS and reducing GAGs, both peripherally and in the brain, in patients with Hunter syndrome. DNL310 is currently being evaluated in a Phase 1/2 trial in patients with Hunter syndrome.
Biomarker-Driven Development
We have developed and validated assays to quantify GAGs in CSF and urine of patients receiving DNL310. These assays are designed to measure changes in primary substrate accumulation that is a result of enzyme deficiency in these patients. In patients with Hunter syndrome receiving standard ERT, a reduction in urine GAG levels is associated with improvements in peripheral manifestations of the disease whereas there is no effect on CSF GAG levels or neurocognitive manifestations.Based on our preclinical models with ETV:IDS, we anticipate that reductions in both urine and CSF GAG levels may result in improvements in both peripheral and neurocognitive manifestations of the disease. In addition, we have identified downstream pathway and disease biomarkers that are dysregulated in animal models of Hunter syndrome, and we are exploring these biomarkers in clinical studies.
Phase 1/2 Trial in Patients with Hunter Syndrome
The investigational new drug application ("IND") for DNL310 was accepted in January 2020. We initiated a Phase 1/2 trial for DNL310 in Hunter syndrome patients in August 2020 as a multicenter, multiregional, open-label trial to assess the safety, pharmacokinetics, and pharmacodynamics of increasing dose levels of DNL310 administered once weekly by intravenous infusion.The initial trial design included two staggered cohorts: the first (Cohort A) enrolled a total of five neuronopathic patients aged five to ten years; the second (Cohort B) is enrolling either neuronopathic or non-neuronopathic patients aged two to eighteen years. A third cohort (Cohort C) is planned to enroll neuronopathic patients aged two to under four years.
In November 2020, we announced achieving first-in-human biomarker proof of concept for our TV technology based on safety and biomarker results after patients in Cohort A had received four weekly IV doses of DNL310. In February 2021, we presented an interim analysis of additional safety and biomarker data from the five patients enrolled in Cohort A, who all received three months of weekly IV doses of DNL310 after switching from idursulfase ERT on Day 1 of the trial. Key results included:
•Normal levels of heparan sulfate, a GAG, in CSF that were seen after four weeks of dosing in four of the five patients were sustained after three months of dosing (mean 85% reduction; p<0.001); heparan sulfate levels were further significantly reduced and approached normal levels in the fifth patient (from 25% to 73% reduction from one to three months, respectively). (Figure 6)
•Reductions in downstream exploratory CSF biomarkers, GM3 and BMP (lysosomal lipids), of 39% and 15%, respectively, were observed after eight weeks of dosing with DNL310, consistent with improvement in lysosomal function.
•Reductions in urine heparan sulfate (Figure 6) and dermatan sulfate following a switch from idursulfase, of 76% and 82%, respectively, were observed after eight weeks of dosing of DNL310, supporting potential for improved peripheral effects relative to standard of care.
•DNL310 was generally well tolerated with no dose reductions and all five patients continue in the trial. The most frequently observed adverse events were mild or moderate infusion-related reactions in three of the five patients, which is consistent with other ERTs.
•Based on three-month clinical data, doses of DNL310 from 3 mg/kg to 30 mg/kg are generally well tolerated and provide flexibility for dose selection in clinical trials.
We believe the Phase 1/2 data to date continue to support the overall safety profile and biomarker effects of DNL310 as an investigational treatment in Hunter syndrome. Importantly, at dose levels resulting in robust and durable biomarker response, DN310 appears generally well tolerated with a safety profile consistent with standard of care ERT. In addition, early biomarker effects initially seen after four weeks of treatment with DNL310 were sustained after three months of dosing, and the reductions observed in CSF lipid biomarkers indicate, for the first time, an improvement in lysosomal function. The urine GAG results also support the potential for systemic administration of TV-enabled therapeutics to address peripheral disease. Overall, the magnitude and durability of biomarker response and tolerability seen with DNL310 in this Phase 1/2 trial provide strong support for the potential application of our TV technology to deliver enzymes and other therapeutic modalities to the brain.
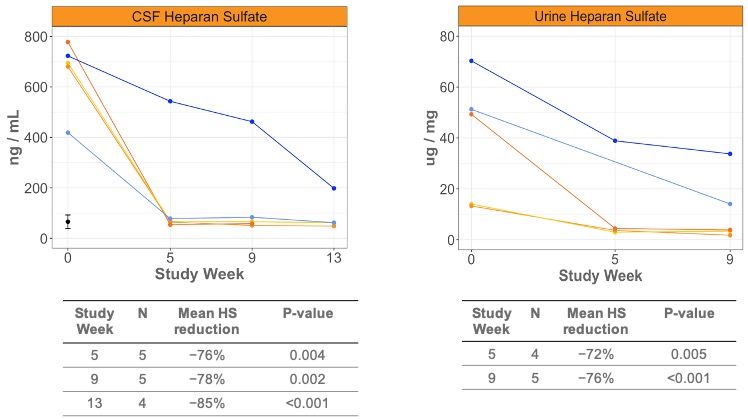
Figure 6: Heparan sulfate levels as measured in the CSF (left) and urine (right) from patients with Hunter syndrome enrolled into Cohort A of the ongoing Phase 1/2 trial of DNL310. Normal range of CSF heparan sulfate (black circle on left).
Preclinical Studies in a Mouse Model of Hunter Syndrome
In a mouse model of Hunter syndrome, we have shown that ETV:IDS treatment reduced CSF GAGs and that these reductions were correlated with GAG reductions in the brain (Figure 7). Furthermore, reduction in CSF GAG levels was associated with subsequent improvements in lysosomal function, neurodegeneration biomarkers, neurobehavioral outcomes and correction of skeletal disease manifestations in the mouse model.
Figure 7: ETV:IDS reduced peripheral and CNS GAGs in an MPS II mouse model.
Future Development Plan
In January 2021, based on strong proof-of-concept data, we announced the expansion of our DNL310 development program with the addition of a third cohort (Cohort C, approximately 12 patients) to the ongoing Phase 1/2 trial. Cohort C will enable further exploration of clinical endpoints related to neuronopathic manifestations in patients at early disease stages. We plan to initiate a pivotal Phase 2/3 trial with DNL310 in the first half of 2022.
EIF2B Program
ALS and FTD are rare neurodegenerative diseases collectively affecting over 200,000people world-wide. ALS, often called Lou Gehrig's disease, refers to a group of progressive neurodegenerative diseases that affect nerve cells in the brain and spinal cord, leading to loss of voluntary muscle control and movement. FTD refers to a rare group of brain disorders that are characterized by atrophy of the frontal and temporal lobes of the brain and are associated with changes to personality and behavior as well as difficulties with language.
Mutations in genes associated with ALS and FTD alter RNA homeostasis, which contributes to the aggregation of TDP-43 or other RNA binding proteins. TDP-43 inclusions are found in over 95% of ALS patients and approximately 50% of FTD patients. Activators of EIF2B have demonstrated benefits in resolving TDP-43 aggregation, restoring protein translation and attenuating neurodegeneration via inhibition of the cellular integrated stress response in numerous in vitro and in vivo models. Our most advanced EIF2B activator, DNL343, is a brain-penetrant small molecule designed to rescue EIF2B function and restore normal RNA metabolism.
Biomarker-Driven Development
We have developed assays to measure ATF4 protein and other ISR-dependent changes in gene expression as proximal pathway markers of EIF2B activity in humans. ATF4 is a transcription factor that modulates ISR gene expression and is upregulated when cellular stress modifies EIF2B activity. These biomarkers have been characterized in in vitro assays and in human peripheral blood mononuclear cells ("PBMCs").
Clinical Development Plan
We initiated a Phase 1 clinical trial of DNL343 in healthy volunteers in February 2020. The Phase 1 trial is a randomized, double-blind, placebo-controlled, single-center clinical trial in 88 healthy subjects to investigate the safety and tolerability of single and multiple ascending oral doses of DNL343 and characterize the PK and PD of DNL343 in plasma and CSF. Initial safety and biomarker results from this trial are expected in the first half of 2021. We plan to initiate a Phase 1b trial in ALS patients in the second half of 2021.
Figure 8A: DNL343 treatment dissolved existing TDP-43 aggregates in a cell based model and was associated with decreased stress response gene expression.
Figure 8B: Left: DNL343 treatment resulted in a dose dependent reduction in the expression of the ISR-dependent gene Chac1 and other ISR-dependent genes in the brain of a mouse neurodegeneration model. Right: A similar reduction in CHAC1 expression is observed in ex vivo DNL343 studies using PBMCs from healthy volunteers; overlay of single ascending dose (SAD) pharmacokinetics data from healthy volunteers in Phase 1 treated with DNL343 demonstrated clinical exposures achieved in SAD cohorts. A dose-dependent reduction in gene expression of CHAC1 and ATF4 protein levels consistent with ex vivo studies was observed (data not shown).
RIPK1 Inhibitor Program
RIPK1 is a critical signaling protein in the tumor necrosis factor receptor pathway and is a regulator of inflammation and cell death. Increased RIPK1 activity in the brain drives neuroinflammation and cell necroptosis and contributes to neurodegeneration. RIPK1 inhibition has been shown to have beneficial effects in preclinical models of Alzheimer’s disease, ALS and other diseases.
As described in more detail in “Business - Licenses and Collaborations” below, we are collaborating with Sanofi on the RIPK1 inhibitor program in neurological diseases, including Alzheimer’s disease, ALS and multiple sclerosis ("MS").
In June 2020, we announced the joint decision with Sanofi to pause clinical studies with DNL747, previously the program's lead brain-penetrant RIPK1 inhibitor molecule, and focus our efforts on accelerating development of DNL788 (SAR443820), which we believe has superior drug properties and a more rapid path toward proof-of-concept clinical studies.
Biomarker-Driven Development
Target engagement has been characterized using a marker of RIPK1 activity, autophosphorylation of RIPK1 at Serine166 ("pS166"). This biomarker has been characterized in in vitro assays in human and monkey PBMCs and has been demonstrated to be robustly reduced by RIPK1 inhibitors.
DNL747 Phase 1b Results and Decision to Advance DNL788 (SAR443820)
In June 2020, we announced data from 31 patients in two 29-day Phase 1b trials in Alzheimer’s disease and ALS, and additional data from 6 ALS patients in an open label extension trial, showing that DNL747 was safe and well tolerated at the dose tested with no significant treatment related adverse events. Target engagement of approximately 80% median inhibition of pRIPK1 in blood at trough drug concentration was achieved.
In parallel to the clinical trials, chronic toxicity studies with DNL747 in cynomolgus monkeys showed dose- and duration-dependent adverse nonclinical findings at exposures higher than those tested in the clinic. These findings, which are considered off-target and molecule-specific, impact the ability to increase the dose of DNL747 and achieve higher levels of target inhibition without time consuming additional clinical safety studies in patients to evaluate the long-term safety and tolerability.
Due to the emerging evidence that higher levels of target inhibition may be required for maximizing efficacy, and challenges to achieving higher doses imposed by molecule-specific toxicity findings with DNL747, we made the decision with Sanofi to pause additional studies with DNL747. We are encouraged by the emerging pathway biomarker data in Alzheimer’s disease and ALS patients, and our experience and learnings with DNL747 should allow us to progress quickly with clinical studies for DNL788 (SAR443820). DNL788 (SAR443820) appears to have a superior preclinical therapeutic window compared to DNL747, facilitating development in multiple indications, including Alzheimer’s disease, ALS and MS.
Future Development Plan
Sanofi submitted an IND application for DNL788 (SAR443820) in October 2020. First-in-human dosing in a Phase 1 healthy volunteer trial commenced in December 2020; data are expected to be available in the second half of 2021.
Other RIPK1 CompoundsPeripheral Inflammatory Diseases: UC
As part of our parallel development strategy, we have also developed a number of other structurally diverse CNS-penetrant and peripherally-restricted RIPK1 inhibitor molecules, which are included as part of the collaboration agreement with Sanofi. Sanofi, described in more detail in “Business - Licenses and Collaborations” below. Sanofi is solely responsible for the development and commercialization of peripherally restricted RIPK1 inhibitors.
In July 2020, weOctober 2023, Sanofi announced that Sanofi commenced athe development of eclitasertib in cutaneous lupus erythematosus (CLE) was being discontinued because the Phase 1b trial of DNL758 (SAR443122), a Peripheral Product (as defined below),2 proof-of-concept study did not meet its primary endpoint (percent change from baseline in hospitalized adult patients with severe COVID-19 lung disease. The Phase 1b trial was completed Cutaneous Lupus Erythematosus Disease Area
and DNL758 (SAR443122)Severity Index-A (CLASI-A) at week 12). Eclitasertib was found to be generally well tolerated and resulted in changes in disease relevant biomarkers and clinical outcome trends consistent with the proof of mechanism. Based on the rapidly evolving landscape of treatment and prevention options for COVID-19, Sanofi has made a sponsor decision to hold further development in COVID-19 at this time. Sanofi continues to develop DNL758 (SAR443122) as a potential treatment for cutaneous lupus erythematosus ("CLE"). CLE is a type of interface dermatitis characterized by immune cell infiltration. Biopsy-derived T-cells from CLE patients are dominated by IFN-γ and TNF-α positive cells. Furthermore, primary pro-inflammatory cytokines involved in the pathophysiology of CLE are strongly linked to RIPK1 activation and downstream signaling. Therefore,inhibition of RIPK1 activity downstream of TNF and IFN receptor signaling is considered an important target to modulate the pathophysiology of CLE.Sanofi plans to initiate a Phase 2 clinical trial of DNL758 (SAR443122) in CLE patients in the first half of 2021. Sanofi leads all clinical development activities with DNL758 (SAR443122) for systemic inflammatory diseases.
PTV:PGRN Program for FTDwell-tolerated.
PGRNSanofi is conducting a soluble lysosomal protein that has critical functionsPhase 2 study of eclitasertib in lysosomes and an innate immunity in the brain. The GRN gene encodespatients with ulcerative colitis. Recruitment for the PGRN protein. Loss of function genetic mutations in the trial is ongoing.GRN genecauses FTD by decreasing PGRN levels in the brain.
2024 expected progress and milestones:
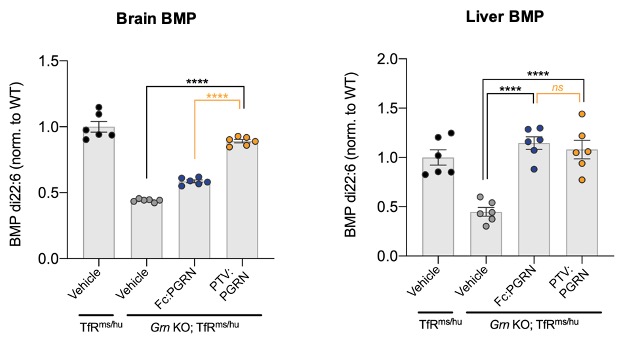
DNL593 (“PTV:PGRN”) is an intravenously, centrally administered recombinant PGRN biotherapeutic enabled by our protein transport vehicle ("PTV"). DNL593 is designed to restore normal levels of PGRN in multiple cell types in the brain without interfering with normal PGRN processing. DNL593 restored lysosomal function and reduced microglial activation in the brain of granulin knock-out mice. BMP has been identified as a translatable biomarker for FTD in humans that reflects lysosomal function. DNL593 has demonstrated the ability to rescue BMP levels both peripherally and in the brain and rescue brain BMP levels in granulin knock-out mice (Figure 9).•Continue Phase 2 UC study.
EARLY-STAGE CLINICAL AND PRECLINICAL PROGRAMS
TAK-594/DNL593 (PTV:PGRN) Program for FTD-GRN
Figure 9: PTV:PGRN but not Fc:PGRN rescues brain BMP levels (left) and both rescue Liver BMP levels (right) in granulin knock-out mice.
In December 2020, we initiated IND-enabling studies for DNL593 and we plan to file an IND application or a clinical trial application ("CTA") in late 2021. As described in more detail in “Business - Licenses and Collaborations” below, we are collaborating with Takeda to co-develop and co-commercialize TAK-594/DNL593 (PTV:PGRN), an investigational, brain-penetrant PGRN replacement therapy enabled by Denali's PTV platform and designed to restore PGRN levels in the brain without interfering with normal PGRN transport and processing. Preclinical proof of concept demonstrates that PTV enhances uptake of recombinant PGRN by multiple cell types in the brain, including neurons and microglia, as compared to non-PTV PGRN. In addition, TAK-594/DNL593 rescued both neurodegeneration and microglial dysfunction in PGRN-deficient mice. Our improved mechanistic understanding of the role of PGRN in lysosomal function indicates that intravenous delivery of TAK-594/DNL593 followed by PTV-enhanced transport to the brain may be an effective therapeutic approach to increase PGRN levels in lysosomes.
Together with Takeda, we initiated a Phase 1/2 clinical trial of TAK-594/DNL593 for FTD-GRN in 2022. Results from Part A of this study evaluating TAK-594/DNL593 in healthy subjects were presented at the Alzheimer's Association International Conference in July 2023. Single doses of TAK-594/DNL593 were generally well-tolerated and resulted in substantial increases in CSF PGRN levels, suggesting that brain delivery of TAK-594/DNL593 was achieved and that TAK-594/DNL593 has the rightpotential to optaddress PGRN deficiency (Figure 11).
Figure 11: Dose-dependent increase in to our PTV:CSF PGRN program. The initiation of IND-enabling studies triggered a milestone payment of $8.0 million from Takeda which we received in January 2021.
ATV:TREM2 Program for Alzheimer's disease
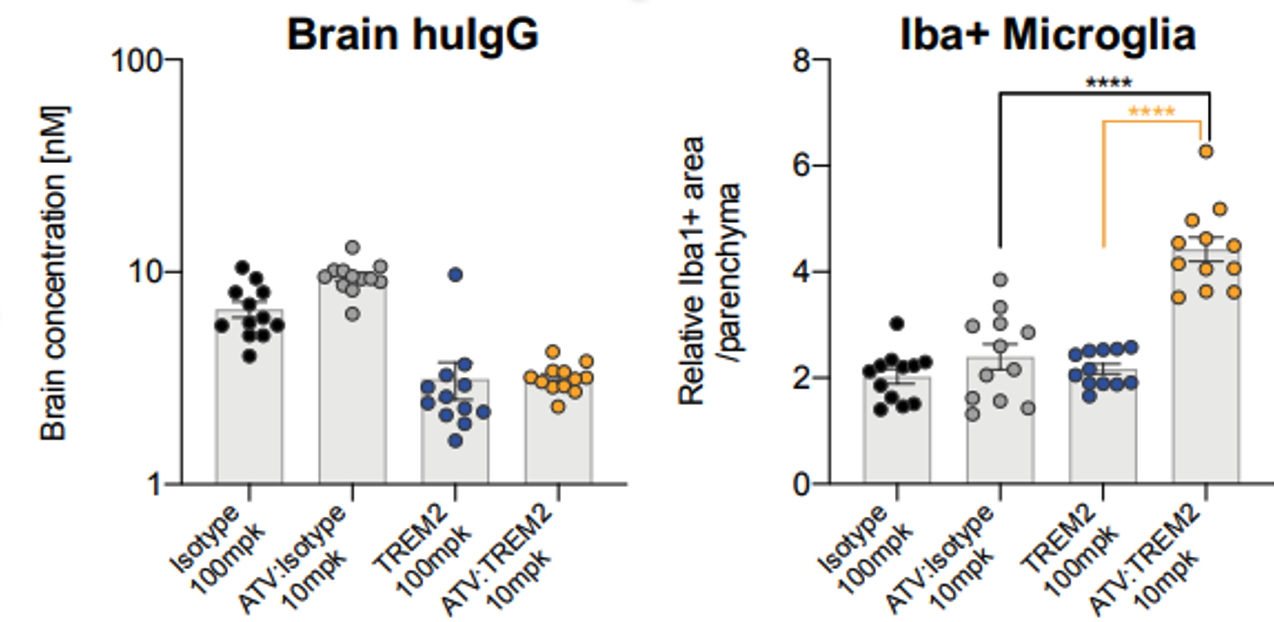
Dementias affect approximately 50 million people world-wide. Alzheimer's disease is the most common cause of dementia accounting for 60-70% of cases. TREM2 is a protein expressed in microglia, the resident immune cells of the brain. Mutations in the TREM2 gene are strongly associatedhealthy volunteers with an increased risk for Alzheimer's disease. DNL919 (“ATV:TREM2”) is a TV-enabled antibody designed to modulate TREM2 signaling and thereby normalize microglial function. We have demonstrated significantly higher brain uptake of ATV:TREM2 compared to standard TREM2 antibodies as well as modulation of microglial gene expression in mouse models (Figure 10).
intravenous
2024 expected progress and milestones:
•Resume Part B of the Phase 1/2 study in FTD-GRN.
DNL126 (ETV:SGSH) Program for MPS IIIA (Sanfilippo Syndrome A)
DNL126 (ETV:SGSH) is our second most advanced ETV program following DNL310 (ETV:IDS). DNL126 is an investigational therapy in development for the potential treatment of MPS IIIA (Sanfilippo syndrome A), a rare LSD that causes fatal brain damage. MPS IIIA is caused by genetic defects that result in a reduction in the activity of SGSH, an enzyme responsible for degrading heparan sulfates in the lysosome. There are no approved treatments for MPS IIIA.
In January 2024, we commenced dosing in a Phase 1/2 study of DNL126 in MPS IIIA. This is a small, multicenter, open-label, Phase 1/2 study to assess the safety, tolerability, pharmacokinetics, pharmacodynamics, and exploratory clinical efficacy of DNL126 in participants with Sanfilippo syndrome Type A (MPS IIIA). The core study period is 25 weeks (approximately 6 months) and is followed by a 72-week (approximately 18 month) open-label extension.
In February 2024, we presented supportive preclinical data at the 2024 WORLDSymposiumTM demonstratingthat DNL126 improved lysosomal and microglial morphology, neurodegeneration, and cognitive function in adult MPS IIIA mice. As shown in (Figure 10: 12),Single dose 10 mg/kg ATV:TREM2 induces robust pharmacodynamic response peripheral treatment with ETV:SGSH lowered substrate accumulation (heparan sulfate) in brain. the brain and in CSF, which was correlated with improved cognitive behavioral performance in adult MPS IIIA mice.
Left
Figure 12: Similar (A) In MPS IIIA mice, treatment with ETV:SGSH (low and high doses) lowered accumulation of heparan sulfate (HS) in the brain concentrationsand in CSF compared to vehicle-treated mice. Bar graphs display mean ± SEM and p values: one-way ANOVA with Dunnett’s multiple comparison test; **p < 0.01, ****p < 0.0001. (B) Lower HS levels were correlated with improved cognitive performance of adult MPS IIIA mice on a spatial learning and memory assay, the active place avoidance (APA) task. Correlations were calculated using nonparametric Spearman’s correlation coefficient with two-tailed 95% confidence intervals. Correlation graphs depict linear regression line.
2024 expected progress and milestones:
•Biomarker proof of concept and safety data from the Phase 1/2 study in late 2024.
DNL622 (ETV:IDUA) for MPS I (Hurler syndrome)
Alpha-L-iduronidase (“IDUA”) is an enzyme responsible for degrading heparan and dermatan sulfate in the lysosome. Genetic defects in IDUA result in a reduction or absence of lysosomal IDUA activity and cause Hurler syndrome (or MPS I), which is characterized by alterations in the skeleton, heart, respiratory system, and brain. DNL622 (ETV:IDUA) is a recombinant IDUA enzyme engineered to cross the BBB, to replace IDUA, and to treat the cognitive, behavioral and physical manifestations of the disease. DNL622 is currently in the IND-enabling stage of preclinical development.
Antibody Transport Vehicle Amyloid beta (ATV:Abeta) program
Targeting Abeta plaques in the brain has been investigated by the field as a therapeutic approach to treat AD, and two Abeta-targeted antibodies have received accelerated approval from the U.S. FDA. These accelerated approvals were based on clinical data demonstrating that treatment was associated with a reduction in the accumulation of Abeta plaque in the brain, a defining feature of AD. Subsequently, the U.S. FDA converted one of the antibodies to traditional approval following a determination that a confirmatory trial verified clinical benefit.
Our ATV:Abeta program utilizes the TfR TV platform to enable increased brain exposure and target engagement of anti-Abeta. In preclinical studies in mice, superior amyloid plaque binding (Figure 13A) and reduction (Figure 13B) with ATV:Abeta was demonstrated compared to a conventional Abeta antibody, which may enable a wider therapeutic window in treating AD as compared to conventional anti-Abeta therapy. In addition, Biogen has shown in a mouse model of AD the potential for ATV:Abeta to reduce the risk of amyloid-related imaging abnormalities (ARIA) associated with the treatment of Alzheimer’s disease (Figure 13C).
Figure 13: Preclinical data showing superior amyloid plaque binding (A), superior plaque reduction (B), and fewer to no ARIA events (C).
In April 2023, Biogen exercised its exclusive option to license ATV:Abeta using TVTfR and is responsible for its development and commercialization.
2024 expected progress and milestones:
•IND-enabling studies.
Oligonucleotide Transport Vehicle ("OTV") Platform
Oligonucleotides, such as ASOs, are a novel class of biotherapeutics with the potential to address the root cause of many diseases through modulation of gene expression. This class, however, has been limited in its potential for treatment of neurodegenerative diseases, primarily due to the challenge of delivering effective amounts of drug to relevant brain regions. Direct injection into the CSF (e.g., intrathecal injection) or certain brain regions has not achieved with 10 mg/kg ATV:TREM2the robust biodistribution into deep brain tissue, which may be necessary for effective therapeutic activity.
Nonhuman primate data demonstrated that intravenous delivery of an ASO enabled by our OTV technology resulted in broad brain biodistribution of the ASO (Figure 14), which was superior to intrathecal administration of the ASO. These data support the potential of our OTV platform to enable peripheral administration of oligonucleotide therapeutics and 100 mg/kg standard TREM2 antibody. to address a wide range of neurodegenerative and other neurological diseases.
Figure 14:Right: ATV:TREM2 has greater impact on microglia than standard TREM2 antibody using similar doses and exposure in mice. Broad brain biodistribution of an intravenously administered ASO enabled by our OTV technology (right) as compared to limited brain biodistribution of ASO delivered intrathecally (left).
In January, 2021, we initiatedannounced that OTV:MAPT targeting tau for Alzheimer’s disease and OTV:SNCA targeting αSyn for Parkinson’s disease are the first programs in the IND-enabling studies for DNL919, and we plan to file an IND application or a stage of development. (CTAFigure 15). As shown in late 2021 or early 2022. As described in more detail in “Business - Licenses and Collaborations” below, Takeda has the right to opt into our ATV:TREM2 program. The initiation of IND-enabling studies triggered a milestone payment of $8.0 million from Takeda which we expect to receive in early 2021.(
Our portfolio includes additional preclinical programs, including programs targeting alpha-Synuclein ("ATV:aSyn"), Tau ("ATV:Tau"), Abeta (“ATV:Abeta”), SGSH ("ETV:SGSH"), GBA (“ETV:GBA”), GAA (“ETV:GAA”), IDUA (“ETV:IDUA”), NAGLU (“ETV:NAGLU”), and ARSA (“ETV:ARSA”) and oligonucleotide delivery (“OTV”) which are enabled by our TV platform technology. In additionwe have optimized lead ASOs to ATV:TREM2 and PTV:PGRN, our ATV:Tau program is subject to our collaboration agreement with Takeda and our ATV:Abeta program is subject to our collaboration agreement with Biogen, as describedachieve robust knock-down of human MAPT expression in more detail in “Business - Licenses and Collaborations” below.
Licenses and Collaborations
Takeda Option and Collaboration Agreement
Overview
In January 2018, we entered into the Collaboration Agreement with Takeda ("Takeda Collaboration Agreement"), pursuant to which we granted Takeda an option with respect to our ATV:BACE1/Tau, ATV:TREM2 and PTV:PGRN programs. The Takeda Collaboration Agreement provided that Takeda pay a $40.0 million upfront payment related to the collaboration, as well as $110.0 million under a share purchase agreement, both of which were received in February 2018. The Takeda Collaboration Agreement became effective in February 2018 when the requirements of the Hart-Scott-Rodino Antitrust Improvements Act of 1976 were satisfied. In February 2019, we amended the agreement to replace the ATV:BACE1/Tau program with the ATV:Tau program.hTau x TfRmu/hu mouse brain.
Research
Figure 15: OTV is designed to open a large potential indication space in neurodegeneration and beyond by enabling superior biodistribution of ASOs across brain regions, providing superior knockdown of target gene expression across cell types, and enabling intravenous dosing of oligonucleotide therapeutics for CNS delivery. OTV:MAPT and OTV:SNCA are lead OTV programs in the IND-enabling stage of development.
Figure 16: OTV:MAPT in an unmodified format achieves approximately 20% knockdown. After optimizing the OTV molecule through protein, linker, and/or oligonucleotide engineering, the five new OTV:MAPT Leads (L1-L5) provided >50% human MAPT knockdown in hTau x TfRmu/hu KI mice, with L4 providing 75% human MAPT knockdown.
2024 expected progress and milestones:
•IND-enabling studies with OTV:MAPT and OTV:SNCA.
Other TV-Enabled Discovery Programs
Denali continues to use deep scientific expertise in neurodegeneration biology and the BBB to discover and develop medicines and platforms with the focus on programs enabled by the TV and targeting common neurodegenerative disease, including Alzheimer’s and Parkinson’s, and LSDs.
Our portfolio includes additional preclinical programs, including programs targeting TREM2 (“ATV:TREM2”) and HER2 (“ATV:HER2”).
In August 2023 we announced that, in agreement with Takeda, the companies would discontinue clinical development of TAK-920/DNL919 (ATV:TREM2) in Alzheimer’s disease. This was a strategic decision based on the totality of clinical data emerging from the single ascending dose Phase 1 study of TAK-920/DNL919 in healthy volunteers and Takeda’sin consideration of the rapidly evolving treatment landscape for Alzheimer's disease whereby an understanding of drug combinations with newly approved therapies will be important. A preliminary analysis of Phase 1 data indicated robust target engagement and effects on microglial biomarkers (e.g., CSF1R, SPP1, IL1RA, IP10, MIP1b, MCP-1), which were consistent with preclinical studies that demonstrated that ATV:TREM2 induced robust changes to a responsive microglial cell state (van Lengerich B, et al. Nat Neurosci. 2023). In the Phase 1 study, TAK-920/DNL919 was clinically well tolerated at doses with demonstrated changes in CSF biomarkers and there were no serious adverse events or severe treatment emergent adverse events; however, safety signals of moderate, reversible hematologic effects were observed at the highest dose tested, suggesting a narrow therapeutic window for the Alzheimer’s disease patient population. The Phase 1 safety findings are believed to be specific to properties of TAK-920/DNL919 and TREM2 biology. Denali and Takeda will continue to focus research efforts on molecules in preclinical development, including exploration of potential combination therapy given recent new drug approvals in Alzheimer's disease.
Human epidermal growth factor receptor 2 ("HER2") is a growth factor receptor that is over-expressed in multiple cancers, including breast, colorectal, and gastric cancer. Up to half of patients diagnosed with metastatic HER2-positive breast cancer have brain metastases for which limited treatment options exist. Using ATV, we have engineered mono- and bispecific formats of HER2 antibodies. In preclinical mouse studies, we have demonstrated improved anti-tumor activity of ATV-enabled HER2 antibodies in a HER2-positive peripheral tumor model. Our bispecific ATV:HER2 antibody demonstrated improved peripheral anti-tumor activity as compared to non-ATV HER2 antibodies as well as enhanced brain uptake as compared to a non-ATV HER2 antibody. The data support the potential for ATV:HER2 to treat HER2-positive peripheral tumors and brain metastases and to further validate the potential for TV applications in oncology.
Licenses and Collaborations
Biogen License and Collaboration Agreement and Right of First Negotiation, Option and License Agreement
Overview
In October 2020, we entered into a Definitive LRRK2 Collaboration and License Agreement (“LRRK2 Agreement”) pursuant to which we granted Biogen a license to co-develop and co-commercialize our small molecule LRRK2 inhibitor program (the "LRRK2 Program"), and a Right of First Negotiation, Option and License Agreement (the "ROFN and Option Agreement"), pursuant to which we granted an option and right of first negotiation to certain of our programs utilizing our TV technology platform, including our amyloid beta program (collectively the "Biogen Collaboration Agreement"), with Biogen Inc.’s subsidiaries, Biogen MA Inc. (“BIMA”) and Biogen International GmbH (“BIG”) (BIMA and BIG, collectively, “Biogen”). In August 2023, we executed an Amendment to the Definitive LRRK2 Collaboration and License Agreement and Waiver of and Amendment to Right of First Negotiation, Option, and License Agreement (the "Biogen Amendment").
LRRK2 Agreement
The LRRK2 Agreement includes our small molecule LRRK2 inhibitors (“LRRK2 Products”) that penetrate the BBB, including DNL201 and BIIB122/DNL151, as well as those that do not penetrate the BBB. Based on the totality of preclinical and clinical data to date, both DNL201 and BIIB122/DNL151 (two chemically distinct LRRK2 inhibitors) have met our requirements to proceed into further late-stage clinical testing, however, BIIB122/DNL151 has been selected to proceed due to pharmacokinetic properties that provide additional dosing regimen flexibility.
Payments
Under the terms of the LRRK2 Agreement, Biogen paid us a $400.0 million upfront payment in October 2020. With respect to the LRRK2 Program, Biogen is required to make milestone payments up to approximately $1.125 billion upon achievement of certain development and sales milestone events. Such milestone payments include $375.0 million in development, $375.0 million upon first commercial sale, and $375.0 million in net sales-based milestones. The Biogen Amendment changed certain milestone criteria while the total amount of development, regulatory, and commercial milestones across all indications remained the same. We will share profits and losses equally with Biogen for LRRK2 Products in the United States and will share profits and losses in China with Biogen sharing 60% of such profits and losses and us sharing 40% of such profits and losses. We will be entitled to receive royalties in the high teens to low twenties percentages on net sales for LRRK2 Products outside of the United States and China. Information on cost sharing reimbursements between us and Biogen is included in this Annual Report on Form 10-K in our financial statements and the related notes and the section titled “Management’s Discussion and Analysis of Financial Condition and Results of Operations.”
License Grant to LRRK2 Program
Under the Takeda CollaborationLRRK2 Agreement, and unless we otherwise agreed jointly with Takeda, we will be responsible, at our cost, for conducting activities relating to pre-IND development of biologic products directed to the three identified targets and enabled by our BBB delivery technology targeting transferrin receptor during the applicable option period. The option period continues for each target until the first biologic product candidate directed to the relevant target is IND-ready or about five years after selection of the target, whichever is earlier.
Takeda is obligated to pay us up to an aggregate of $25.0 million with respect to each of the three programs under the Takeda Collaboration Agreement directed to a target and based upon the achievement of certain preclinical milestone events, up to $75.0 million in total, of which $5.0 million was received when the Takeda Collaboration Agreement became effective. Additional payments totaling $26.0 million have become due based on the achievement of certain preclinical milestone events, of which $10.0 million was received by December 31, 2020, and an additional $8.0 million was received in January 2021.
Collaboration Activities Following Takeda’s Option Exercise
If Takeda exercises its option with respect to a particular target and collaboration program (i.e., the biologic products directed to the target for which Takeda has exercised its option), then Takeda will have the right to develop and commercialize, jointly with us, a specified number of biologic products enabled by our BBB technology that were developed during the option period and which are directed to the relevant target, and we will grant to Takedagranted Biogen a co-exclusive, worldwide license under the intellectual property that we control related to those biologic products.
Takeda is obligatedour LRRK2 inhibitors, including certain intellectual property licensed to pay us by a $5.0 million option fee for each target for which Takeda exercises its option, up to $15.0 million in total.
In addition, if Takeda exercises its option for all three collaboration programs, Takeda may be obligated to pay us up to an aggregate of $407.5 million upon achievement of certain clinical milestone events and up to an aggregate of $300.0 million in regulatory milestone events relating to receipt of regulatory approval in the United States, certain European countries and Japan. Further, Takeda may also be obligated to pay us up to $75.0 million per biologic product upon achievement of a certain sales-based milestone, or an aggregate of $225.0 million if one biologic product from each program achieves the milestone.
Once Takeda has exercised its optionDevelopment and Commercialization of LRRK2 Program
We and Biogen are jointly developing LRRK2 Products pursuant to a clinical development plan set forth within the LRRK2 Agreement. We and Biogen share responsibility and costs for global development of LRRK2 Products pursuant to a particular target, wemutually agreed development plan and Takeda will share equallybudget, with Biogen funding 60% of such costs and us funding 40% of such costs. We have the ability to opt out of the development and commercialization costs, and, if applicable, the profits, for each collaboration program. However, for each collaboration program, we may elect not to continue sharing development and commercialization costs, or Takeda may elect to terminate our cost-profit sharing rights and obligations if, following notice from Takeda and a cure period, we fail to satisfy our cost sharing obligationsarrangement, as further described below.
Biogen will lead commercialization activities globally for LRRK2 Products. We will co-commercialize the LRRK2 Products with respectBiogen in the United States and China, provided that the profit-sharing arrangement for the LRRK2 Products is still in effect, as further described below.
We may opt out of development cost sharing worldwide and upon such election, from any further profit-sharing from the LRRK2 Program. We also have the right to opt out of the relevant collaboration program.profit-sharing arrangement for the LRRK2 Program or for only those LRRK2 Products that do not penetrate the BBB (“Peripheral LRRK2 Products”), in each of the United States and China. After such an election by us or termination by Takeda becomes effective,opt out, we will no longer be obligated to share in the development and commercialization costs for, the relevant collaboration program, and we will notor be entitled to share in any profitsthe applicable revenues from, that collaboration program. Insteadsuch LRRK2 Program (or from the Peripheral LRRK2 Products) for such country, as applicable. If we choose to exercise our opt out rights, we will be entitled to receive tiered royalties.royalties on net sales of the applicable LRRK2 Program in the relevant country (or countries). The royalty rates for the applicable LRRK2 Program will be a percentage in the low-high teens to mid-teen percentages on net sales, or low-low twenties, but may increase to high-teen percentages on net salesthe low twenties to mid-twenties if we have met a certain co-funding thresholdthresholds or there has been a first commercial sale at the time of our electionelection.
LRRK2 Program Manufacturing
Biogen will be responsible for delivering all supplies for clinical trials and commercial production for LRRK2 Products, except that we will deliver such supplies until the point of transition which will be mutually agreed by us and Biogen, but in no event later than commencement of activities to opt out of co-development or Takeda’s termination of our cost-profit sharing rights and obligations,support commercial launch, and in each case, these royalty rates will be subjectany event we retain manufacturing rights for certain independent clinical activities.
LRRK2 Program Royalty Term
For any LRRK2 Product for which Biogen is required to certain reductions specified in the Takeda Collaboration Agreement. Takedapay royalties, Biogen will pay theseus royalties to us for each biologic product included in the relevant collaboration program, on a country-by-country basis and product-by-product basis until the latest of (i) the expiration of certain patents covering the relevant biologic product, (ii) the expiration of all regulatory exclusivity for that biologic product in the applicable country, and (iii) an agreed period of time after the first commercial sale of that biologic product in the applicable country. If, in a particular country, unless biosimilara LRRK2 Product for which Biogen is required to pay royalties is not covered by specified patent rights in that country or where generic competition in excess of a significant level specified in the Takeda Collaboration Agreement occurs earlier, in which case Takeda’sexists, Biogen’s royalty obligations in the applicable country would terminate.
For each collaboration program for which we are sharing costs and profits with Takeda, we will lead the conduct of clinical activities for each indication up to the first trial with a clinical outcomes-based efficacy endpoint, and Takeda will lead the conduct of all subsequent clinical activities for that indication. For each collaboration program for which we are sharing costs and profits with Takeda, we and Takeda will jointly commercialize biologic products included in the relevant collaboration program in the United States and China. Unless we have opted out of cost-sharing for two collaboration programs, we have the right to lead commercialization activities in the United States for one collaboration program and Takeda will lead commercialization activities in the United States for all collaboration programs for which we do not lead commercialization activities. Further, Takeda will lead commercialization activities in China and will solely conduct commercialization activities in all other countries.
We have the right to lead all manufacturing activities for all collaboration programs for which the parties are sharing costs and profits.be reduced.
Exclusivity of LRRK2 Program
During the option period for a particular target and, ifterm of the applicable option is exercised by Takeda (unless the Takeda CollaborationLRRK2 Agreement, is terminated earlier), until expiration of an agreed period of time after the first regulatory approval in the United States or Europe of a biologic product within the applicable collaboration program, neither partywe nor Biogen may conduct preclinical, clinical or commercial activities involving antibodies or protein-based therapeutic products directed to the same target (orany small molecule that targets LRRK2 as its primary mechanism of action anywhere in the case of a bi-specific program,world, unless such molecule is included under the same combination of targets) that have an intended therapeutic effect in diseasescollaboration and conditions of the CNS (including lysosomal storage diseases), exceptonly to the extent such activity is permitted under the Takeda Collaboration Agreement.LRRK2 Agreement or, with respect to Biogen, the molecule is an ASO product that is the subject of a collaboration between Biogen and a particular third party.
ROFN and Option Agreement
Option & ROFN Programs
In addition to the LRRK2 Program, Biogen also received an exclusive option to license two preclinical programs enabled by our TV technology platform, including our ATV:Abeta program ("Option Programs"). In April 2023, Biogen exercised its option to develop and commercialize our ATV:Abeta program. As Biogen exercised its option with respect to the ATV:Abeta Program, we granted Biogen an exclusive, worldwide license under certain intellectual property to develop, manufacture, and commercialize products that are the subject of the ATV:Abeta Program. In August 2023, Biogen agreed to waive the remaining option upon execution of the Biogen Amendment.
In August 2023, upon execution of the Biogen Amendment, Biogen also agreed to waive its right of first negotiation on two additional TV-enabled therapeutics, which we initially granted to Biogen under the ROFN and Option Agreement.
Payments
Under the ROFN and Option Agreement, Biogen paid us a $160.0 million upfront payment in October 2020 and a $5.0 million option exercise payment in April 2023 for the ATV:Abeta program. With respect to the ATV:Abeta license granted by us to Biogen, Biogen is obligated to pay to us an aggregate of up to $142.5 million in development milestone payments, an aggregate of up to $180.0 million upon first commercial sale, and up to $190.0 million of net sales-based milestone payments. Furthermore, Biogen is obligated to pay us royalties in the mid-single digit percentages, upon the achievement of certain sales thresholds.
Exclusivity of ROFN and Option Agreement
Until termination or expiration of the ROFN and Option Agreement, neither party may conduct preclinical, clinical or commercial activities involving certain therapeutic products directed to ATVTfR:Abeta, unless the therapeutic is included under the collaboration and only to the extent such activity is permitted under the ROFN and Option Agreement or the therapeutic is a product that includes AAVs, oligonucleotides or small molecules.
Termination
Each party may terminate the TakedaBiogen Collaboration Agreement in its entirety, or with respect to a particular collaboration program, as applicable, if the other party remains in material breach of the TakedaBiogen Collaboration Agreement following a cure period to remedy the material breach. Takeda may terminate the Takeda Collaboration Agreement in its entirety or with respect to any particular collaboration program, for convenience and afterAfter giving a specified amount of prior notice to us, but TakedaBiogen may not do soterminate the Biogen Collaboration Agreement for a certain period of time afterconvenience in its entirety or, in the effective datecase of the Takeda Collaboration Agreement. Takeda may also terminateLRRK2 Agreement, with respect to one or more specified regions of the Takedaworld, and in the case of the Biogen Collaboration Agreement, with respect to any collaboration program if the joint steering committee established under the Takeda Collaboration Agreement unanimously agrees that a material safety event has occurred with respect to the applicable collaboration program.Option Program for one or more specified regions or in its entirety. We may terminate the Takeda CollaborationLRRK2 Agreement with respect to a particular collaboration program if TakedaBiogen fails to conduct materialmeaningful activities to advance the development and commercial activitiesor commercialization of any LRRK2 Products for a specified period of time, with respect to a collaboration program, unless TakedaBiogen cures such failure within a certain period of time. We may terminate the Biogen Collaboration Agreement if Biogen challenges any patents licensed to it under the Biogen Collaboration Agreement. We and TakedaBiogen may each terminate the TakedaBiogen Collaboration Agreement in its entirety if the other party is declared insolvent or in similar financial distress or if, subject to a specified cure period, the other party challenges any patents licensed to it under the Takeda Collaboration Agreement.distress.
Following any termination
Common Stock Purchase Agreement
Pursuant to the terms of the Takeda Collaboration Agreement,In August 2020, in connection with our collaboration with Biogen, we entered into a common stock purchase agreement (the “Purchase Agreement”) with Takeda in January 2018,BIMA, pursuant to which we agreed to issue and sell, and Takeda agreed to purchase, 4,214,559sold 13,310,243 shares of our common stock (the “Shares”)to BIMA for an aggregate purchase price of $110.0$465.0 million pursuant to the terms and conditions thereof. We closedin September 2020. In connection with the sale of the Shares to Takeda in February 2018.
We and Takeda alsoshares, we entered into a standstill and stock restriction agreement (the “Standstill Agreement”"Biogen Standstill Agreement"). Pursuant with Biogen, pursuant to the terms of the Standstill Agreement, Takedawhich Biogen agreed to certain transfer and standstill restrictions, including a restriction on acquiring more than 10%which have now expired, with the exception of our capital stock, for a specified period of time following the closing of the sale of the Shares to Takeda, or earlier upon our change of control or, with respect to the transfer restrictions, termination of the Takeda Collaboration Agreement. In addition, Takeda iscertain volume limitations. Biogen remains entitled to certain demand registration rights with respect tofor the Shares following terminationshares received in the transaction under the terms of the transfer restrictions if the Shares cannot be resold without restriction pursuant to Rule 144 promulgated under the Securities Act of 1933, as amended.Biogen Standstill Agreement.
Sanofi Collaboration and License Agreement
Overview
In October 2018, we entered into the Collaboration Agreement with Genzyme Corporation, a wholly owned subsidiary of Sanofi S.A. ("Sanofi") pursuant to which certain small molecule compounds that bind to and inhibit RIPK1 (“RIPK1 Inhibitors”) contributed by Sanofi and by us will be developed and commercialized. The Sanofi Collaboration Agreement became effective in November 2018 when the requirements of the Hart-Scott-Rodino Antitrust Improvements Act of 1976 were satisfied.
We and Sanofi plan toare jointly developdeveloping products containing RIPK1 Inhibitors for neurological indications, such as Alzheimer’s disease ALS and MS, and Sanofi plans to develop and commercializeis developing products containing RIPK1 Inhibitors for systemic inflammatory indications, such as cutaneous lupus, rheumatoid arthritis and psoriasis.UC.
The Sanofi Collaboration Agreement includes our and Sanofi’s RIPK1 Inhibitors that measurably penetrate the BBB ("CNS Products"), and our and Sanofi’s RIPK1 Inhibitors that do not measurably penetrate the BBB ("Peripheral Products"). The two most advanced RIPK1 Inhibitors in the collaboration are SAR443820/DNL788, (SAR443820),and eclitasertib (SAR443122/DNL758). SAR443820/DNL788 is a CNS Product that was discovered by us and is currently in alicensed to Sanofi who has completed Phase 1 trialtrials in healthy volunteers, and DNL758 (SAR443122), a Peripheral Product discovered by us, which has completed a Phase 1b trial in COVID-19 patients and in which Sanofi plans to initiateis leading a Phase 2 clinical trial in patients with cutaneous lupus in the first half of 2021. In June 2020, together withMS. Eclitasertib is a Peripheral Product discovered by us, and licensed to Sanofi we decided to pausewho is leading a Phase 2 clinical studies with DNL747 and focus our efforts on accelerating development of DNL788 (SAR443820), which we believe has superior drug properties and a more rapid path toward proof-of-concept clinical studiestrial in patients in multiple neurological indications.with UC.
License Grant
Under the Sanofi Collaboration Agreement, we granted Sanofi an exclusive, worldwide license under intellectual property that we control related to our RIPK1 Inhibitors, including certain intellectual property licensed to us by an academic institution.
Payments
When the Sanofi Collaboration Agreement became effective in November 2018, Sanofi paid us $125.0 million upfront, and in August 2019, Sanofi paid us $10.0 million for the milestone triggered by the commencement of a DNL758 (SAR443122) Phase 1 clinical trial in healthy volunteers.upfront. Under the Sanofi Collaboration Agreement, Sanofi is required to make milestone payments up to approximately $1.1 billion upon achievement of certain clinical, regulatory and sales milestone events. Such milestone payments include $215.0 million in clinical milestone payments and $385.0 million in regulatory milestone payments for CNS Products, as defined, that are developed and approved in the United States, by the European Medicines Agency ("EMA")Europe and in Japan for three indications, including Alzheimer's disease. TheseSanofi has made payments of $65.0 million for CNS clinical milestones alsothrough December 31, 2023. The cumulative earned and potential milestones include $120.0 million in clinical milestone payments, $175.0 million in regulatory milestone payments and $200.0 million in commercial milestone payments for Peripheral Products, as defined, that are developed and approved in the United States, by the EMAEurope and in Japan for three indications. Sanofi has made payments of $35.0 million for peripheral clinical milestones through December 31, 2023.
We will share profits and losses equally with Sanofi for CNS Products sold in the United States and China, and receive variable royalties on net sales for CNS Products sold outside of the United States and China and for Peripheral Products sold worldwide, each as further described below.
RIPK1 Inhibitors contributed by Sanofi and developed and commercialized under the Sanofi Collaboration Agreement will be subject to lower milestone and royalty payments to us compared to RIPK1 Inhibitors contributed by us. We will also retain responsibility for certain payment obligations under our agreement with an academic institution which licensed certain intellectual property to us that we are sublicensing to Sanofi under the Sanofi Collaboration Agreement.
Program for Development and Commercialization of CNS Products
We and Sanofi will jointly develop CNS Products pursuant to a global development plan ("CNS Development Plan"). We will be responsible, at our cost, for conducting Phase 1 and Phase 2 trials for CNS Products for Alzheimer’s disease and any activities required to support such clinical trials and specific for Alzheimer’s disease. Sanofi will beis responsible, at its cost, for all other Phase 1 and Phase 2 trials for CNS Products, including for ALS and multiple sclerosis.MS. Sanofi will lead the conduct of all Phase 3 and later stage development trials for CNS Products, with Sanofi and us funding 70% and 30% of such costs, respectively. The Sanofi Collaboration Agreement contains certain protections for us with respect to Phase 3 development costs not included in the initial budget for the CNS Development Plan agreed to by the parties, including a deferral mechanism for costs incurred above the budgeted amounts for such trials and for costs incurred in respect of Phase 3 and other clinical trials not contemplated in the initial CNS Development Plan. In addition, we have the ability to opt out of the cost-profit sharing provisions of the Sanofi Collaboration Agreement, as further described below.
Sanofi will lead commercialization activities globally for CNS Products. We may elect to conduct certain co-commercialization activities outside of MS with respect to each CNS Product in the United States and/or China, provided that the cost-profit sharing provisions of the Sanofi Collaboration Agreement for the relevant CNS Product are still in effect, as further described below.
We may opt out of the cost-profit sharing provisions of the Sanofi Collaboration Agreement for CNS Products in the United States and China on a CNS product-by-CNS Product and country-by-country basis. Sanofi may also terminate our cost-profit sharing provisions of the Sanofi Collaboration Agreement in its entirety if, following notice from Sanofi and a cure period, we fail to satisfy our cost-sharing obligations. After such an opt out by us or termination by Sanofi, we will no longer be obligated to share in the development and commercialization costs for the applicable CNS Products and we will not share in the applicable profits from such CNS Products. Instead, we will be entitled to receive tiered royalties on net sales of the applicable CNS Products in the relevant country (or countries). The royalty rates will be a percentage in the low double digits to mid-teens, but may increase to the mid-teens to low-twenties percentages for all countries in which Sanofi is paying royalties on the applicable CNS Products, if we have met certain co-funding thresholds at the time of our election or Sanofi’s termination of our cost-profit sharing rights and obligations.
Program for Development and Commercialization of Peripheral Products
Sanofi iswill be responsible, at its cost, for conducting activities relating to the development and commercialization of all Peripheral Products.
Sanofi will lead commercialization activities globally for Peripheral Products. We will be entitled to receive tiered royalties in the low- to mid- teen percentages on net sales of Peripheral Products.
Manufacturing
Sanofi will be responsible for delivering all supplies for current and future clinical trials and commercial production for CNS Products and Peripheral Products. However, we retain manufacturing rights for certain independent clinical activities.
Royalty Term
For any CNS Product with respect to any country for which Sanofi is required to pay royalties on net sales and for each Peripheral Product, Sanofi will pay royalties to us on a country-by-country basis until the latest of (i) the expiration of certain patents covering the relevant product, (ii) the expiration of all regulatory exclusivity for that product in the applicable country, and (iii) an agreed period of time after the first commercial sale of that product in the applicable country. If, in a particular country, a CNS Product for which Sanofi is required to pay royalties or a Peripheral Product is not covered by specified patent rights in that country or net sales in that country decrease below specified thresholds as a result of generic competition, Sanofi’s royalty obligations in the applicable country would be reduced or would terminate as specified in the Sanofi Collaboration Agreement.
Exclusivity
During the term of the Sanofi Collaboration Agreement, neither we nor Sanofi may conduct IND-enabling, clinical or commercial activities involving any RIPK1 Inhibitor, anywhere in the world, unless the RIPK1 Inhibitor is included by us or Sanofi, as the case may be, under the collaboration and only to the extent such activity is permitted under the Sanofi Collaboration Agreement.
Termination
Each party may terminate the Sanofi Collaboration Agreement in its entirety, or with respect to a particular program (i.e., the CNS Products program or Peripheral Products program), as applicable, if the other party remains in material breach of the Sanofi Collaboration Agreement following a cure period to remedy the material breach. After giving a specified amount of prior notice to us, Sanofi may terminate the Sanofi Collaboration Agreement for convenience in its entirety, with respect to any particular program, or with respect to one or more specified regions of the world. Sanofi may also terminate the Sanofi Collaboration Agreement with respect to any program or a particular RIPK1 Inhibitor if a material safety event has occurred and cessation of all development and commercialization of all RIPK1 Inhibitors in the affected program or the affected RIPK1 Inhibitor is recommended. We and Sanofi may each terminate the Sanofi Collaboration Agreement in its entirety if the other party is declared insolvent or in similar financial distress or if, subject to a specified cure period, the other party challenges any patents licensed to it under the Sanofi Collaboration Agreement.
Following any termination of the Sanofi Collaboration Agreement with respect to a particular program or a particular region (or regions) of the world or termination of the Sanofi Collaboration Agreement in its entirety, our rights to each of our RIPK1 Inhibitors that were licensed to Sanofi will revert to us. Sanofi will conduct certain development, manufacturing and commercialization activities on a transitional basis following termination of the Sanofi Collaboration Agreement, as outlined in the Sanofi Collaboration Agreement or agreed by Sanofi, depending upon the basis for the applicable termination.
If the Sanofi Collaboration Agreement is terminated for any reason other than by Sanofi for our material uncured breach, our insolvency or our challenge to any of the patents licensed to us by Sanofi, Sanofi will grant us an exclusive license to certain intellectual property controlled by Sanofi with respect to such RIPK1 Inhibitors (which could be subject to low single digit royalties payable to Sanofi).
Biogen LicenseTakeda Option and Collaboration Agreement and Right of First Negotiation, Option and License Agreement
Overview
In August 2020,January 2018, we entered into a binding Provisional Collaboration and License Agreement (“Provisional Biogen("Takeda Collaboration Agreement”Agreement") with Biogen Inc.’s subsidiaries, Biogen MA Inc. (“BIMA”Takeda Pharmaceutical Company Limited ("Takeda") and Biogen International GmbH (“BIG”) (BIMA and BIG, collectively, “Biogen”), pursuant to which we granted Biogen a license to co-develop and co-commercialize our small molecule LRRK2 inhibitor program (the “LRRK2 Program”),Takeda an option to our amyloid beta program utilizing our TV technology platform to cross the BBB as well as one other unnamed program also utilizing our TV technology platform (the “Option Programs”), and a right of first negotiation with respect to two additional unnamed programs for indications within Alzheimer’s disease, Parkinson’s disease, ALSour ATV:BACE1/Tau, ATV:TREM2 and multiple sclerosis utilizing our TV technology platform (the “ROFN Programs”) should we decide to seek a collaboration with a third party for suchPTV:PGRN programs. In October 2020, we entered into a Definitive LRRK2 Collaboration and License Agreement (“LRRK2 Agreement”) and a Right of First Negotiation, Option and License Agreement (the “ROFN and Option Agreement”) with Biogen (collectively the "Biogen Collaboration Agreement"). The material terms of the LRRK2 Agreement and the ROFN and Option Agreement were consistent with, and superseded, the Provisional Biogen Collaboration Agreement.
LRRK2 Agreement
The LRRK2 Agreement includes our small molecule LRRK2 inhibitors (“LRRK2 Products”) that penetrate the BBB, including DNL201 and DNL151 (BIIB122), as well as those that do not penetrate the BBB. Based on the totality of preclinical and clinical data to date, both DNL201 and DNL151 (BIIB122) (two chemically distinct LRRK2 inhibitors) have met our requirements to proceed into further late-stage clinical testing, however, DNL151 (BIIB122) has been selected to proceed due to pharmacokinetic properties that provide additional dosing regimen flexibility.
Payments
Under the terms of the LRRK2 Agreement, BiogenTakeda paid us a $400.0$40.0 million upfront payment in October 2020. With respectrelated to the LRRK2 Program, Biogen collaboration, and an additional $110.0 millionis required to make milestone payments up to approximately under a share purchase agreement in February 2018. $1.125 billion upon achievementThe Takeda Collaboration Agreement became effective in February 2018, following satisfaction of certain development and sales milestone events. Such milestone payments include $375.0 million in development, $375.0 million upon first commercial sale, and $375.0 million in net sales-based milestones. We will share profits and losses equally with Biogen for LRRK2 Products in the United States and will share profits and losses in China with Biogen sharing 60% of such profits and losses and us sharing 40% of such profits and losses. We will be entitled to receive royalties in the high teens to low twenties percentages on net sales for LRRK2 Products outsiderequirements of the United StatesHart-Scott-Rodino Antitrust Improvements Act of 1976. In February 2019, we amended the agreement to replace the ATV:BACE1/Tau program with the ATV:Tau program and China.in March 2022, the parties mutually agreed to terminate activity on the ATV:Tau program over which Takeda had its option to develop and commercialize jointly with the Company. Takeda exercised the options for the PTV:PGRN and ATV:TREM2 programs in November 2021 and December 2021, respectively.
License Grant to LRRK2 ProgramResearch Phase and Takeda’s Option
Under the LRRK2Takeda Collaboration Agreement we granted Biogen a co-exclusive, worldwide license under intellectual property that we control relatedwere responsible, at our cost, for conducting activities relating to pre-IND development of biologic products directed to the identified targets and enabled by our LRRK2 inhibitors, including certain intellectual property licensedBBB delivery technology targeting TfR during the applicable option period. The option period continued for each target until the first biologic product candidate directed to us by a third party.the relevant target was IND-ready or about five years after selection of the target, whichever was earlier.
DevelopmentTakeda was obligated to pay us up to an aggregate of $25.0 million with respect to each of the programs under the Takeda Collaboration Agreement directed to a target and Commercializationbased upon the achievement of LRRK2 Programcertain preclinical milestone events, up to $55.0 million in total after the ATV:Tau program termination, all of which was earned and received as of September 30, 2022.
Collaboration Activities Following Takeda’s Option Exercise
We and Biogen will jointly develop LRRK2 Products pursuantSubsequent to a clinical development plan set forth in the LRRK2 Agreement. We and Biogen will share responsibility and costs for global development of LRRK2 Products, with Biogen funding 60% of such costs and us funding 40% of such costs. We have the ability to opt out of the development cost sharing arrangement, as further described below.
Biogen will lead commercialization activities globally for LRRK2 Products. We will co-commercialize the LRRK2 Products with Biogen in the United States and China, provided that the profit-sharing arrangement for the LRRK2 Products is still in effect, as further described below.
We may opt out of development cost sharing worldwide and upon such election, of all further profit sharing from the LRRK2 Program. We also have the right to opt out of the profit-sharing arrangement for the LRRK2 Program or for only those LRRK2 Products that do not penetrate the BBB (“Peripheral LRRK2 Products”), in each of the United States and China. After such an opt out, we will no longer be obligated to share in the development and commercialization costs for, and we will not share in the applicable revenues from, such LRRK2 Program (or from the Peripheral LRRK2 Products), in such country, as applicable. In such cases, we will be entitled to receive tiered royalties on net sales of the applicable LRRK2 Program in the relevant country (or countries). The royalty rates for the applicable LRRK2 Program will be a percentage in the high teens to low twenties, but may increase to the low twenties to mid-twenties, if we have met certain co-funding thresholds or there has been a first commercial sale at the time of our election.
LRRK2 Program Manufacturing
Biogen will be responsible for delivering all supplies for clinical trials and commercial production for LRRK2 Products, except that we will deliver such supplies until the point of transition which will be mutually agreed by us and Biogen, but in no event later than commencement of activities to support commercial launch, and in any event we retain manufacturing rights for certain independent clinical activities.
LRRK2 Program Royalty Term
For any LRRK2 Product for which Biogen is required to pay royalties, Biogen will pay us royalties on a country-by-country basis and product-by-product basis until the latest of (i) the expiration of certain patents covering the relevant product, (ii) the expiration of all regulatory exclusivity for that product in the applicable country, and (iii) an agreed period of time after the first commercial sale of that product in the applicable country. If, in a particular country, a LRRK2 Product for which Biogen is required to pay royalties is not covered by specified patent rights in that country or where generic competition exists, Biogen’s royalty obligations in the applicable country would be reduced.
Exclusivity of LRRK2 Program
During the term of the LRRK2 Agreement, neither we nor Biogen may conduct preclinical, clinical or commercial activities involving any small molecule that targets LRRK2 asTakeda exercising its primary mechanism of action anywhere in the world, unless such molecule is included under the collaboration and only to the extent such activity is permitted under the LRRK2 Agreement or, with respect to Biogen, the molecule is an antisense oligonucleotide product that is the subject of a collaboration between Biogen and a particular third party.
Termination
Each party may terminate the LRRK2 Agreement in its entirety, if the other party remains in material breach of the LRRK2 Agreement following a cure period to remedy the material breach. After giving a specified amount of prior notice to us, Biogen may terminate the LRRK2 Agreement for convenience in its entirety, or with respect to one or more specified regions of the world, but Biogen may not do so for a certain period of time after the effective date of the LRRK2 Agreement. We may terminate the LRRK2 Agreement if Biogen fails to conduct meaningful activities to advance the development or commercialization of any LRRK2 Products for a specified period of time, unless Biogen cures such failure within a certain period of time or if Biogen challenges any patents licensed to it under the LRRK2 Agreement. We and Biogen may each terminate the LRRK2 Agreement in its entirety if the other party is declared insolvent or in similar financial distress.
Following any termination of the LRRK2 Agreementoption with respect to a particular region (or regions)target and collaboration program (i.e., the biologic products directed to the target for which Takeda has exercised its option), Takeda has the right to develop and commercialize, jointly with us, a specified number of the world or termination of the LRRK2 Agreement in its entirety,biologic products enabled by our rights to each of our LRRK2 inhibitorsBBB technology that were licenseddeveloped during the option period and which are directed to Biogen will revertthe relevant target, and we are obligated to us. Biogen will conduct certain development, manufacturing and commercialization activities ongrant to Takeda a transitional basis following termination ofco-exclusive license under the LRRK2 Agreement, as outlined in the LRRK2 Agreement.
If the LRRK2 Agreement is terminated, Biogen will grant us an exclusive license to certain intellectual property controlled by Biogen with respectwe control related to such LRRK2 inhibitors.those biologic products.
ROFN and Option Agreement
Option Programs
In addition to the LRRK2 Program, Biogen also received an exclusive option to license two preclinical programs that utilize our TV technology platform, including our ATV:Abeta program and a second program for an unnamed target, excluding small molecules, AAVs and oligonucleotides (each, an "Option Program"). Biogen’s option may be triggered up to initiation of IND-enabling studies for each program and continues for each program until a specified period of time after delivery of an option data package or 30 business days after the 5th anniversary of the effective date of the Provisional Biogen Collaboration Agreement, whichever is earlier.
ROFN Programs
Further, Biogen will have the right of first negotiation on two additional TV-enabled therapeutics in the fields of Alzheimer’s disease, Parkinson’s disease, ALS or multiple sclerosis should we decide to seek a collaboration with a third party for such programs, but this does not include any of our small molecule, AAVs and oligonucleotide programs. The ROFN period continues until seven years after the effective date of the Provisional Biogen Collaboration Agreement or the date on which we have offered Biogen two ROFN Programs and for which Biogen has agreed to trigger a ROFN for such program, whichever is earlier. However, if we do not execute an agreement with a third party with respect to a particular ROFN Program offered to Biogen within a specified amount of time, then Biogen will have one additional right to exercise the ROFN again with respect to such ROFN Program.
Payments
Under the ROFN and Option Agreement, Biogen paid us a $160.0 million upfront payment in October 2020. With respect to the options granted by us to Biogen, Biogen is obligated to pay to us an aggregate of up to $270.0 million in option exercise and development milestone payments and an aggregate of up to $615.0 million in commercial milestone payments, following the achievement of certain prespecified milestone events and if Biogen exercises both of its options. Furthermore, Biogen isTakeda was obligated to pay us royalties in the mid-single digit to mid-teens percentages, depending on the programa $5.0 million option fee for each target for which Biogen exercisesTakeda exercised its option, and we received fees totaling $10.0 million from Takeda in 2021 for option exercise payments for the PTV:PGRN and ATV:TREM2 programs.
Since Takeda exercised its option for both collaboration programs, Takeda may be obligated to pay us up to an aggregate of $280.0 million upon the achievement of certain sales thresholds.
In addition, if Biogen exercises its ROFN with respectclinical milestone events and up to an eligible Denali program,aggregate of $200.0 million in regulatory milestone events relating to receipt of regulatory approval in the parties areUnited States, certain European countries and Japan. Further, Takeda may also be obligated to negotiate in good faith forpay us up to $75.0 million per biologic product upon achievement of a specified periodcertain sales-based milestone, or an aggregate of time regarding$150.0 million if one biologic product from each program achieves the financial and other terms of an agreement pursuant to which Biogen would obtain rights to such program.
License Grant to Option Programs
If Biogen exercises its option with respect to an Option Program, then we will grant Biogen an exclusive, worldwide license under certain intellectual property that we control that is specific to that Option Program, and a non-exclusive license under certain intellectual property that we control that pertains to our TV platform, in each case to develop, manufacture and commercialize products that are the subject of such Option Program.milestone.
ExclusivityFurther, we and Takeda share equally the development and commercialization costs, and, if applicable, the profits, for each collaboration program. However, for each collaboration program, we may elect not to continue sharing development and commercialization costs, or Takeda may elect to terminate our cost-profit sharing rights and obligations if, following notice from Takeda and a cure period, we fail to satisfy our cost sharing obligations with respect to the relevant collaboration program. After such an election by us or termination by Takeda becomes effective, we will no longer be obligated to share in the development and commercialization costs for the relevant collaboration program, and we will not share in any profits from that collaboration program. Instead, we will be entitled to receive tiered royalties. The royalty rates will be in the low- to mid-teen percentages on net sales, or low- to high-teen percentages on net sales if we have met a certain co-funding threshold at the time of ROFNour election to opt out of co-development or Takeda’s termination of our cost-profit sharing rights and Optionobligations, and, in each case, these royalty rates will be subject to certain reductions specified in the Takeda Collaboration Agreement. Takeda will pay these royalties to us for each biologic product included in the relevant collaboration program, on a country-by-country basis, until the latest of (i) the expiration of certain patents covering the relevant biologic product, (ii) the expiration of all regulatory exclusivity for that biologic product, and (iii) an agreed period of time after the first commercial sale of that biologic product in the applicable country, unless biosimilar competition in excess of a significant level specified in the Takeda Collaboration Agreement occurs earlier, in which case Takeda’s royalty obligations in the applicable country would terminate.
IfFor each collaboration program for which we are sharing costs and profits with Takeda, we will lead the conduct of clinical activities for each indication up to the first trial with a clinical outcomes-based efficacy endpoint, and Takeda will lead the conduct of all subsequent clinical activities for that indication. For each collaboration program for which we are sharing costs and profits with Takeda, we and Takeda will jointly commercialize biologic products included in the relevant collaboration program in the United States and China. Unless we have opted out of cost-sharing for both collaboration programs, we have the right to lead commercialization activities in the United States for one collaboration program and Takeda will lead commercialization activities in the United States for all collaboration programs for which we do not lead commercialization activities. Further, Takeda will lead commercialization activities in China and will solely conduct commercialization activities in all other countries.
We have the right to lead all manufacturing activities for all collaboration programs for which the parties are sharing costs and profits.
Information on cost sharing reimbursements between us and Takeda is included in this Annual Report on Form 10-K in our financial statements and the related notes and the section titled “Management’s Discussion and Analysis of Financial Condition and Results of Operations.”
Exclusivity
Unless the Takeda Collaboration Agreement is terminated earlier, until expiration of an agreed period of time after the first regulatory approval in the United States or Europe of a biologic product within the applicable option is exercised by Biogen, then until termination or expiration of the ROFN and Option Agreement,collaboration program, neither party may conduct preclinical, clinical or commercial activities involving certainantibodies or protein-based therapeutic products directed to the same target unless(or in the case of a bi-specific program, the same combination of targets) that have an intended therapeutic is included undereffect in diseases and conditions of the collaboration and onlyCNS (including LSDs), except to the extent such activity is permitted under the ROFN and Option Agreement or the therapeutic is a product that includes AAVs, oligonucleotides or small molecules.Takeda Collaboration Agreement.
Termination
Each party may terminate the ROFN and OptionTakeda Collaboration Agreement in its entirety, or with respect to a particular collaboration program, as applicable, if the other party remains in material breach of the ROFN and OptionTakeda Collaboration Agreement following a cure period to remedy the material breach. AfterTakeda may terminate the Takeda Collaboration Agreement in its entirety or with respect to any particular collaboration program, for convenience and after giving a specified amount of prior notice to us, Biogenus. Takeda may also terminate the ROFN and OptionTakeda Collaboration Agreement for convenience in its entirety or with respect to any Option Program for one or more specified regions or in its entirety.collaboration program if the joint steering committee established under the Takeda Collaboration Agreement unanimously agrees that a material safety event has occurred with respect to the applicable collaboration program. We may terminate the ROFNTakeda Collaboration Agreement with respect to a particular collaboration program if Takeda fails to conduct material development and Option Agreement if Biogen challenges any patents licensedcommercial activities for a specified period of time with respect to it under the ROFN and Option Agreement.a collaboration program, unless Takeda cures such failure within a certain period of time. We and BiogenTakeda may each terminate the ROFN and OptionTakeda Collaboration Agreement in its entirety if the other party is declared insolvent or in similar financial distress.distress or if, subject to a specified cure period, the other party challenges any patents licensed to it under the Takeda Collaboration Agreement.
Following any termination of the ROFN and OptionTakeda Collaboration Agreement with respect to a particular collaboration program or the Takeda Collaboration Agreement in its entirety, orour rights to each terminated collaboration program will revert to us, Takeda will grant us a license to intellectual property owned by Takeda with respect to an Option Program (including with respectsuch collaboration program (which could be subject to certain royalty payments that would be negotiated at the time of such a region),termination) and, unless the licenses that we granted to Biogen with respect to each such terminated Option Program will be terminated. Biogentermination was by Takeda on the basis of a material safety event, Takeda will conduct certain development, manufacturing and commercialization activities for certain of such products on a transitional basis following termination of the ROFN and Option Agreement, as outlined in the ROFN and Option Agreement.
If the ROFN and Option Agreement is terminated following exercise of the applicable option with respect to an Option Program, Biogen will grant us an exclusive license to certain intellectual property controlled by Biogen with respect to certain of such products that were the subject of such Option Program.
Common Stock Purchase Agreement
In August 2020, in connection withPursuant to the Provisional Biogenterms of the Takeda Collaboration Agreement, we entered into a common stock purchase agreement (the “Stock Purchase Agreement”) with BIMA,Takeda in January 2018, pursuant to which we agreed to issue and sell, and BIMA agreed to purchase, 13,310,243sold 4,214,559 shares of our common stock (the “Shares”) to Takeda for an aggregate purchase price of $465.0 million pursuant$110.0 million. We closed the sale to the terms and conditions thereof. The sale of the Shares closed, and payment was received,Takeda in September 2020.February 2018.
Pursuant to the terms ofAt closing, we also entered into a standstill and stock restriction agreement (the “Standstill“Takeda Standstill Agreement”) entered into between Biogen and us atwith Takeda. Pursuant to the closingterms of the sale of the Shares, Biogen hasTakeda Standstill Agreement, Takeda agreed to certain transfer and standstill restrictions including a restriction on acquiring more than 10% of our capital stock, and to vote the Shares in the same manner and proportion as the votes cast by the other holders of our common stock on certain matters, in each case for a specified period of eighteen monthstime following the closing of the sale, of the Shares, or earlier upon a change of control of us. In addition, Biogen will bewhich have now expired. Takeda remains entitled to certain demand registration rights with respect to the Shares following termination of the transfer restrictions.restrictions if the Shares cannot be resold without restriction pursuant to Rule 144 promulgated under the Securities Act of 1933, as amended (the "Securities Act").
F-star License and Collaboration Agreement
Overview
In August 2016, we entered into a licenseLicense and collaboration agreementCollaboration Agreement (“F-star Collaboration Agreement”) with F-star Gamma Limited (“F-star Gamma”), F-star Biotechnologische Forschungs-und Entwicklungsges m.b.H ("F-star GmbH") and F-star Biotechnology Limited ("F-star Ltd") (collectively "F-star"). The goal of the collaboration was the development of certain constant Fc domains of an antibody with non-native antigen binding activity, or Fcabs, to enhance delivery of therapeutics across the BBB into the brain. The collaboration was designed to leverage F-star’s modular antibody technology and our expertise in the development of therapies for neurodegenerative diseases. In connection with the entry into the F-star collaboration agreement, we also purchased an option for an upfront option fee of $0.5 million, which we refer to as the buy-out-option, to acquire all of the outstanding shares of F-star Gamma pursuant to a pre-negotiated buy-out option agreement (the “Option Agreement”).
In May 2018, we exercised such buy-out option and entered into a Share Purchase Agreement (the “Purchase Agreement”) with the shareholders of F-star Gamma and Shareholder Representative Services LLC, pursuant to which we acquired all of the outstanding shares of F-star Gamma (the “Acquisition”).
As a result of the Acquisition, F-star Gamma became a wholly-owned subsidiary of the Company and we changed the entity’s name to Denali BBB Holding Limited. In addition, we became a direct licensee of certain intellectual property of F-star Ltd (by way of the Company’s assumption of F-star Gamma’s license agreement with F-star Ltd, dated August 24, 2016, (the “F-star Gamma License”)). We made initial exercise payments under the Purchase Agreement and the F-star Gamma License of $18.0 million in the aggregate, less the net liabilities of F-star Gamma, which were approximately $0.2 million. In addition, we are required to make future contingent payments, to F-star Ltd and the former shareholders of F-star Gamma, up to a maximum amount following completion of the research phase of the F-star collaboration of $447.0243.0 million in the aggregate upon the achievement of certain defined preclinical, clinical, regulatory and commercial milestones. These include up to $27.03.0 million in preclinical contingent payments, $50.030.0 million in clinical contingent payments, $120.060.0 million in regulatory contingent payments and $250.0150.0 million in commercial contingent payments. The amount We have made payments of the contingent payments will vary based on whether F-star delivers an Fcab (constant Fc-domains with antigen-binding activity) that meets pre-defined criteria and whether the Fcab has been identified solely by us or solely by F-star or jointly by us and F-star.$49.8 million through In June 2019, we made a payment of $1.5 million to F-star Ltd upon the achievement of a specified preclinical milestoneDecember 31, 2023 in the aggregate consisting of up-front, preclinical and clinical contingent consideration. This contingent consideration payment fully satisfies the Company's ETV:IDS program.clinical contingent consideration obligations under the Purchase Agreement.
Under the terms of the original F-star Collaboration Agreement, we could nominate up to three Fcab targets (“Accepted Fcab Targets”) within the first three years of the date of the F-star Collaboration Agreement. Upon entering into the F-star Collaboration Agreement, we had selected transferrin receptor (“TfR”)TfR as the first Accepted Fcab Target and paid F-star Gamma an upfront fee of $5.5 million, which included selection of the first Accepted Fcab Target. In May 2018, we exercised our right to nominate two additional Fcab Targets and identified aCD98 as the second Accepted Fcab Target. We made a one-time payment for the two additional Accepted Fcab Targets of, in the aggregate, $6.0 million and extended the time period for our selection of the third Accepted Fcab Target until approximately the fourth anniversary of the date of the original F-star Collaboration Agreement. We havedid not identifiedidentify a third Fcab Target.
We arewere also responsible for certain research costs incurred by F-star Ltd in conducting activities under an agreed development plan for each Fcab, for up to 24 months after the target Fcab is accepted.
Genentech Exclusive License Agreement
In June 2016, we entered into an exclusive license agreement with Genentech, Inc. (“Genentech”). The agreement gives us access to Genentech’s LRRK2 inhibitor small molecule program for Parkinson’s disease. Under the agreement, Genentech granted us (i) an exclusive, worldwide, sublicensable license under Genentech’s rights to certain patents and patent applications directed to small molecule compounds which bind to and inhibit LRRK2 and (ii) a non-exclusive, worldwide, sublicensable license to certain related know-how, in each case, to develop and commercialize certain compounds and licensed products incorporating any such compound. We are obligated to use commercially reasonable efforts during the first three years of the agreement to research, develop and commercialize at least one licensed product.
Our financial obligations underupon entering the agreement with Genentech included an upfront payment of $8.5 million and a technology transfer fee of $1.5 million. In addition, we may owe Genentech milestone payments upon the achievement of certain development, regulatory and commercial milestones, up to a maximum of $315.0 million in the aggregate.aggregate, which are subject to equal cost sharing with Biogen since execution of the Biogen Collaboration Agreement. These milestones include up to $37.5 million in clinical milestone payments, $102.5 million in regulatory milestone payments and $175.0 million in commercial milestone payments. We have made milestone payments to Genentech of $15.0 million through December 31, 2023.
In addition, we are obligated to pay royalties on net sales of licensed products ranging from low to high single-digit percentages, with the exact royalty rate dependent on various factors, including (i) whether the compound incorporated in the relevant licensed product is a Genentech-provided compound or a compound acquired or developed by us, (ii) the date a compound was first discovered, derived or optimized by us, (iii) the existence of patent rights covering the relevant licensed product in the relevant country, (iv) the existence of orphan drug exclusivity covering a licensed product that is a Genentech-provided compound and (v) the level of annual net sales of the relevant licensed product. We also have the right to credit a certain amount of third-party royalty and milestone payments against royalty and milestone payments owed to Genentech, but such credit cannot reduce our royalty obligation to Genentech by more than fifty percent. Our royalty payment obligations will expire on a country-by-country and licensed product-by-licensed product basis upon the later of (a) ten years after the first commercial sale of such licensed product in such country or (b) the expiration of the last valid claim of a licensed patent covering such licensed product in such country. If one of our licensed products incorporates a compound provided to us by Genentech, has orphan drug exclusivity, and is not covered by a valid claim of a licensed patent, we must pay royalties on net sales of such licensed products on a country-by-country and licensed product-by-licensed product basis until such orphan drug exclusivity in such country expires, but our obligation to pay these royalties may be eliminated or reduced if there is a clinically superior product marketed in such country. Under the terms of our LRRK2 Agreement with Biogen, Biogen is responsible for 50% of any payment obligation to Genentech under this agreement accruing after October 2020.
Unless earlier terminated, our agreement with Genentech will continue in effect until all of our royalty and milestone payment obligations to Genentech expire. Following expiration of the agreement, we will retain our licenses under the intellectual property Genentech licensed to us on a non-exclusive, royalty-free basis. Genentech may terminate the agreement if we challenge any of the patent rights licensed to us by Genentech, or if we materially breach the agreement, subject to specified notice and cure provisions, or enter into bankruptcy or insolvency proceedings. If Genentech terminates the agreement for our material breach, bankruptcy or insolvency after we have made a milestone payment to Genentech, then we are obligated to grant to Genentech an exclusive right of first negotiation with respect to certain of our patents, know-how and regulatory filings directed to Genentech-provided compounds. We do not have the right to terminate the agreement without cause, but may terminate the agreement for Genentech’s material breach, subject to specified notice and cure provisions.
Manufacturing
We believe it is important to our business success to have a reliable, high-quality preclinical and clinical drug supply chain. As we mature as a company and approach commercial stage operations, securing reliable high-quality commercial drug supply will be critical.
We do not currently own or operate facilities for product manufacturing, storage, distribution or testing. We are actively working towards building our preclinical and clinical manufacturing capabilities as part of our goal to become a fully integrated global organization.
We currently rely on third-party contract development and manufacturing organizations ("CDMOs"), to manufacture and supply our preclinical and clinical materials used during the development of our product candidates. We have established relationships with several CDMOs, including Lonza Sales AG ("Lonza"), and WuXi Biologics Limited and Thermo Fisher Scientific.("Wuxi"). Effective September 2017, we entered into a development and manufacturing services agreement with Lonza, which we have subsequently amended to add scope of work. We refer to this agreement, as amended, as the DMSA or the Lonza agreement. Pursuant to the Lonza agreement, Lonza agreed to provide clinical development and manufacturing services with respect to certain of our biologic products on a fee-for-service basis.
We do not currently operate facilities for product manufacturing, storage, distribution or testing. In April 2023 we entered into, and subsequently amended, a lease for a clinical manufacturing site in Salt Lake City, Utah after terminating our previous SLC lease in March 2023. We plan to use the facility to expand our clinical manufacturing capabilities for biologic therapeutics including the manufacture of materials for toxicology studies and drug substance for early human clinical studies, with the goal of increasing flexibility and speed in advancing new investigational therapies into clinical trials. The build-out of the Utah site is in process, and we currently expect to begin operations and manufacturing at the facility in 2024.
We currently do not need commercial manufacturing capacity. When and if this becomes relevant, we intend to evaluate both third-party manufacturers as well as building out internal capabilities and capacity.
Commercialization Plan
We do not currently have any approved drugs and we do not expect to have any approved drugs in the near term.2024. Therefore, we have no sales, marketing or commercial product distribution capabilities at the current time.
Our vision, however, is to become a fully integrated, independent global leader in neurodegeneration with capabilities spanning discovery, development, manufacturing and commercial in order to optimize speed, quality and level of patient access to our medicines. We look to grow strategically both in terms of therapeutic areas of high unmet need, starting with lysosomal storage diseasesLSDs with CNS pathology and expanding into large neurodegenerative disorders, as well as from a geographic perspective, with an initial focus on establishing a commercial presence in the United States with a subsequent expansion worldwide (includingand the European Union ("EU") and, with subsequent global expansion (including China).
For programs covered by collaboration agreements (including those with Takeda, Sanofi and Biogen), we expect to commercialize only in certain geographies, as defined by the terms of the agreements with the counterpart, and rely on our partners to provide commercialization infrastructure for the rest of the world.
Competition
The biotechnology and pharmaceutical industries, including in the neurodegenerative disease field, are characterized by rapidly advancing technologies, strong competition and an emphasis on intellectual property. We face substantial competition from many different sources, including large and specialty pharmaceutical and biotechnology companies, academic research institutions, governmental agencies and public and private research institutions. We believe that the key competitive factors affecting the success of any of our product candidates will include efficacy, safety profile, method of administration, cost, level of promotional activity and intellectual property protection.
Our product candidates will compete with current therapies approved for the treatment of neurodegenerative diseases, which to date have been primarily targeted at treating the symptoms of such diseases rather than halting or slowing the progression of the disease. However, in addition to such currently approved therapies, we believe that our product candidates, if approved, may also compete with other potential therapies intended to halt or slow the progression of neurodegenerative disease that are being developed by a number of companies and institutions, including but not limited to:
•Alzheimer’s Disease: PotentiallyTreatments currently approved in some geographies for Alzheimer’s Disease include amyloid beta-directed antibody therapies from Biogen and Eisai. Additionally, potentially disease modifying therapeutics are being developed by several large and specialty pharmaceutical and biotechnology companies, including Biogen, Eli Lilly Eisai,(including Prevail Therapeutics, its wholly owned subsidiary), Roche (including Genentech, its wholly owned subsidiary), Alector, and AbbVie in various stages of clinical trials.development.
•Parkinson’s Disease: Potentially disease modifying therapeutics are being developed by several large and specialty pharmaceutical and biotechnology companies, including Prothena, Prothena/Roche (including Genentech, its wholly owned subsidiary), Sanofi, Biogen/Novartis/UCB, Biogen, Ionis, Prevail/Eli Lilly (including Prevail Therapeutics, its wholly owned subsidiary), AstraZeneca, Takeda, Oncodesign/Servier, and TakedaNeuron 23 in various stages of clinical trials.development.
•ALS: PotentiallyTreatments currently approved in some geographies for ALS include Relyvrio/Albrioza (Amylyx Pharmaceuticals), Radicava (Mitsubishi Tanabe Pharma) and Qalsody (Biogen). Additionally, potentially disease modifying therapeutics are being developed by several large and specialty pharmaceutical and biotechnology companies, and academic institutions, including Biogen/Ionis, Amylyx,Clene Nanomedicine, AB Science, Novartis, Bristol Myers Squibb, and NovartisCalico/AbbVie in various stages of clinical trials.development.
•Lysosomal Storage Diseases: FTD-GRN:The currently approved treatments for LSDs are enzyme-based therapies. Potentially disease modifying therapeutics are being developed by several large and specialty pharmaceutical and biotechnology companies, including BioMarin,Alector/GSK, Eli Lilly (including Prevail Therapeutics, its wholly owned subsidiary), Passage Bio, AviadoBio, Vesperbio, Arkuda Therapeutics, Biomarin, and Orchard Therapeutics in various stages of development.
•LSDs: The currently approved treatments for LSDs are enzyme-based therapies. Various BBB-penetrant and direct to CNS delivered ERTs and gene therapies are being developed by several large and specialty pharmaceutical and biotechnology companies, including JCR Pharmaceuticals, RegenxBio, Sangamo, Kyowa Kirin/Orchard Therapeutics, Takeda and Ultragenyx in various stages of clinical trials.development.
In addition, there are companies that are developing technologies that would compete directly with our technologies, including:
•Blood-Brain Barrier Technology: There are several large and specialty pharmaceutical and biotechnology companies developing BBB delivery technologies that utilize RMT, including AbbVie, biOasis Technologies, ABL Bio, BioArtic, JCR Pharmaceuticals, and Roche (including Genentech, its wholly owned subsidiary), Alector, Bioarctic, Bicycle Therapeutics among others.
Intellectual Property
Our intellectual property is critical to our business and we strive to protect it, including by obtaining and maintaining patent protection in the United States and internationally for our product candidates, novel biological discoveries and BBB platform technology, including new targets and applications, and other inventions that are important to our business. We also rely on trademarks, trade secrets, know-how, continuing technological innovation and licensing opportunities to develop and maintain our proprietary position.
As of December 31, 2020,2023, our owned and licensed patent portfolio includes over 9001,700 patents and patent applications, including over 30 licensed U.S. issued patents and 1430 owned U.S. issued patents, covering certain aspects of our proprietary technology, our product candidates, and related inventions and improvements. The patent portfolio also includes over 500 licensed patents issued in jurisdictions outside of the United States, and over 400800 owned patent applications pending in jurisdictions outside of the United States that, in many cases, are counterparts to the foregoing U.S. patents and patent applications. For our product candidates and our BBB platform technology, we generally pursue or in-license patent protection covering compositions of matter, methods of use, and manufacture. For example, we
BBB Platform
We own 11 patent applications in the United States and internationally that are directedfamilies related to the composition of matter of certain antibodies and small molecule product candidates that we intend to develop or are developing, as well as the Fc domain portion of our BBB platform technology.
For our BBB platform technology we own pending patent applications These include a family directed to the composition and sequences of our TfR-binding TVs, the earliest of which are expected to expire in 2038, not including any patent term adjustments and twoany patent term extensions. We also have 3 issued U.S. patents, which are also expected to expire in 2038, not including any patent term adjustments and any patent term extensions, as well as pending patent applications, to other BBB platform technology. Furthermore,Other families related to BBB platform technology, if issued, are expected to expire in 2038 or later, all not including any patent term adjustments and any patent term extensions. In addition, we license multiple patent families from F-star, the earliest issued patents of which are expected to expire in 2026, not including any patent term adjustments and any patent term extensions.
ETV Platform, ETV:IDS, and ETV:SGSH Programs
We own 10 patent applicationsfamilies directed to our ETV platform and related products, including ETV:IDS, ATV:TREM2,ETV: SGSH, and PTV:PGRN programs. In particular, we own anETV:IDUA. This includes 1 issued U.S. patent, which is expected to expire in 2038, not including any patent term adjustments and any patent term extensions, directed to the composition of matter of our ETV:IDS molecules, including DNL310. In
addition, we license multipleWe also own 6 additional patent families from F-star, the earliestdirected to various aspects of our DNL310 program, which if issued, patents of which are expected to expire in 2026,2039 or later, all not including any patent term adjustments and any patent term extensions. Of the 10 patent families, 2 families relate to the composition of matter of our ETV:SGSH structures, including DNL126. Any patents issuing from these families are expected to expire in 2039 and 2041, respectively, not including any patent term adjustments and any patent term extensions.
ForPTV:PGRN Program
We own 4 patent families directed to our PTV:PGRN program. This includes an issued U.S. patent, which is expected to expire in 2040, not including any patent term adjustments and any patent term extensions, directed to the composition of matter of our PTV:PGRN molecules, including TAK-594/DNL593. We also own an additional patent family directed to the composition of matter of our PTV:PGRN structures, including TAK-594/DNL593, the earliest of which are expected to expire in 2039, not including any patent term adjustments and any patent term extensions. We also own additional patent families directed to various aspects of our TAK-594/DNL593 program, which, if issued, are expected to expire in 2039 or later, all not including any patent term adjustments and any patent term extensions. Our PTV:PGRN program is subject to our Takeda collaboration.
ATV:TREM2 Program
We own 6 patent families related to our ATV:TREM2 program. These families include a granted U.S. patent, which is expected to expire in 2041, not including any patent term adjustments and any patent term extensions, directed to the composition of matter of our ATV:TREM2 molecules. The other 5patent families, if issued, are expected to expire between 2038 and 2043, all not including any patent term adjustments and any patent term extensions. Our ATV:TREM2 program is subject to our Takeda collaboration.
Oligonucleotide Transport Vehicle Platform
We own3 patent families related to our OTV platform. These families are directed to compositions and methods of use of our OTVs, and if issued, are expected to expire between 2042 and 2043, not including any patent term adjustments and any patent term extensions.
LRRK2 Inhibitor Program
Our LRRK2 program which is subject to our collaboration agreement with Biogen,Biogen. For this program, we license multiple patent families from Genentech directed to, among other things, DNL201, BIIB122/DNL151 (BIIB122) and other related compounds, which are expected to expire in 2031, not including any patent term adjustments and any patent term extensions. Furthermore, we own additional patent families that have projected expiration dates in 2038 or later, not accounting for any patent term adjustments and any patent term extensions.extensions, related to the LRRK2 program. We also own two a patent family that includes 3issued U.S. patents, which are expected to expire in 2037, not including any patent term adjustments and any patent term extensions, directed to the composition of matter of BIIB122/DNL151 (BIIB122) and methods of treatment using BIIB122/DNL151, (BIIB122), respectively, as well as pending patent applications and granted patents in jurisdictions outside the U.S.
RIPK1 Inhibitor Program
Our RIPK1 program is subject to our collaboration agreement with Sanofi. We own 7 patent families directed to our RIPK1 program, weinhibitor program. These include a family with one issued U.S. patent, directed to the composition of matter of our current RIPK1 lead, SAR443820/DNL788, which is expected to expire in 2038, not including any patent term adjustments and any patent term extensions. We also own foura patent family that includes 2 issued U.S. patents, which are expected to expire in 2037, not including any patent term adjustments and any patent term extensions, and pending patent applications, which areinclude those directed to the composition of matter of our prior lead DNL747eclitasertib (SAR443122/DNL758), as well as other RIPK1 inhibitor compounds.
eIF2b Activator Program
We additionally have pending applications both in and outside the U.S.own 9 patent families directed to the composition of matter of our current RIPK1 lead, DNL788 (SAR443820), as well as other RIPK1 inhibitor compounds, which, if issued, wouldeIF2B activator program, including 4 patent families directed to DNL343 that are expected to expire in 2038 or later, with the remaining families directed to other eIF2B compounds expiring between 2038 and 2040, not including any patent term adjustments and any patent term extensions.
We cannot guarantee that our owned and licensed pending patent applications, or any patent applications that we may in the future file or license from third parties, will result in the issuance of patents. We also cannot predict the scope of claims that may be allowed or enforced in our patents. In addition, the coverage claimed in a patent application can be significantly reduced before the patent is issued, and its scope can be reinterpreted after issuance. Consequently, we may not obtain or maintain adequate patent protection for any of our programs and product candidates. For more information regarding the risks related to our intellectual property, see "Risk Factors - Risks Related to Our Intellectual Property."
The term of individual patents depends upon the legal term of the patents in the countries in which they are obtained. In most countries in which we file, the patent term is 20 years from the earliest date of filing a non-provisional patent application. In the United States, the patent term of a patent that covers an FDA-approved drug may also be eligible for patent term extension, which permits patent term restoration as compensation for the patent term lost during the FDA regulatory review process. The Hatch-Waxman Act permits a patent term extension of up to five years beyond the expiration of the patent. The length of the patent term extension is related to the length of time the drug is under regulatory review. Patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval, only one patent applicable to an approved drug may be extended and only those claims covering the approved drug, a method for using it, or a method for manufacturing it may be extended. Similar provisions are available in Europe and other foreign jurisdictions to extend the term of a patent that covers an approved drug. In the future, if and when our products receive FDA approval, we expect to apply for patent term extensions on patents covering those products. We plan to seek patent term extensions to any of our issued patents in any jurisdiction where these are available, however there is no guarantee that the applicable authorities, including the FDA in the United States, will agree with our assessment of whether such extensions should be granted, and if granted, the length of such extensions. For more information regarding the risks related to our intellectual property, see "Risk Factors - Risks Related to Our Intellectual Property."
In addition to patent protection, we also rely on trademark registration, trade secrets, know how, other proprietary information and continuing technological innovation to develop and maintain our competitive position. We seek to protect and maintain the confidentiality of proprietary information to protect aspects of our business that are not amenable to, or that we do not consider appropriate for, patent protection. Although we take steps to protect our proprietary information and trade secrets, including through contractual means with our employees and consultants, third parties may independently develop substantially equivalent proprietary information and techniques or otherwise gain access to our trade secrets or disclose our technology. Thus, we may not be able to meaningfully protect our trade secrets. It is our policy to require our employees, consultants, outside scientific collaborators, sponsored researchers and other advisors to execute confidentiality agreements upon the commencement of employment or consulting relationships with us. These agreements provide that all confidential information concerning our business or financial affairs developed or made known to the individual during the course of the individual’s relationship with us is to be kept confidential and not disclosed to third parties except in specific circumstances. Our agreements with employees also provide that all inventions conceived by the employee in the course of employment with us or from the employee’s use of our confidential information are our exclusive property. However, such confidentiality agreements and invention assignment agreements can be breached and we may not have adequate remedies for any such breach. For more information regarding the risks related to our intellectual property, see "Risk Factors - Risks Related to Our Intellectual Property."
The patent positions of biotechnology companies like ours are generally uncertain and involve complex legal, scientific and factual questions. Our commercial success will also depend in part on not infringing upon the proprietary rights of third parties. It is uncertain whether the issuance of any third-party patent would require us to alter our development or commercial strategies, or our drugs or processes, obtain licenses or cease certain activities. Our breach of any license agreements or our failure to obtain a license to proprietary rights required to develop or commercialize our future products may have a material adverse impact on us. If third parties prepare and file patent applications in the United States that also claim technology to which we have rights, we may have to participate in interference or derivation proceedings in the United States Patent and Trademark Office (the "USPTO") to determine priority of invention. For more information, see "Risk Factors - Risks Related to Our Intellectual Property."
Government Regulation
Government authorities in the United States at the federal, state and local level and in other countries regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, post-approval monitoring and reporting, marketing and export and import of drug and biological products. Generally, before a new drug or biologic can be marketed, considerable data demonstrating its quality, safety and efficacy must be obtained, organized into a format specific for each regulatory authority, submitted for review and approved by the regulatory authority.
U.S. Drug Development
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act ("FDCA"), and its implementing regulations, and biologics under the FDCA, the Public Health Service Act ("PHSA"), and their implementing regulations. Both drugs and biologics also are subject to other federal, state and local statutes and regulations. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations requires the expenditure of resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or post-market may subject an applicant to administrative or judicial sanctions. These sanctions could include, among other actions, the FDA’s refusal to approve pending applications, withdrawal of an approval, a clinical hold, untitled or warning letters, product recalls or market withdrawals, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement and civil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect on us.
Any future product candidates must be approved by the FDA through either a new drug application ("NDA"), or a biologics license application ("BLA"), process before they may be legally marketed in the United States. The process generally involves the following:
•Completion of extensive preclinical studies in accordance with applicable regulations, including studies conducted in accordance with good laboratory practice ("GLP"), requirements;
•Submission of an IND to the FDA, or a CTA to the EMA or to an appropriate National Competent Authority, one of which must become effective before human interventional clinical trials may begin;
•Approval by an independent institutional review board ("IRB"), or ECs at each clinical trial site before each trial may be initiated;
•Performance of adequate and well-controlled human clinical trials in accordance with applicable IND or CTA regulations, good clinical practice ("GCP"), requirements and other clinical trial-related regulations to establish the safety and efficacy of the investigational product for each proposed indication;
•Submission to the FDA of an NDA or BLA;
•A determination by the FDA within 60 days of its receipt of an NDA or BLA to accept the filing for review;
•Satisfactory completion of a FDA pre-approval inspection of the manufacturing facility or facilities where the drug or biologic will be produced to assess compliance with current good manufacturing practices ("cGMP"), requirements to assure that the facilities, methods and controls are adequate to preserve the drug or biologic’s identity, strength, quality and purity;
•Potential FDA auditinspection of the preclinical and/or clinical trial sites that generated the data in support of the NDA or BLA;
•FDA review and approval of the NDA or BLA, including consideration of the views of any FDA advisory committee, prior to any commercial marketing or sale of the drug or biologic in the United States; and
•Compliance with any post-approval requirements, including the potential requirement to implement a Risk Evaluation and Mitigation Strategy ("REMS"), and the potential requirement to conduct post-approval studies.
The data required to support an NDA or BLA are generated in two distinct developmental stages: preclinical and clinical. The preclinical and clinical testing and approval process requires substantial time, effort and financial resources, and we cannot be certain that any approvals for any future product candidates will be granted on a timely basis, or at all.
Preclinical Studies and IND
The preclinical developmental stage generally involves laboratory evaluations of drug chemistry, formulation and stability, as well as studies to evaluate pharmacology, pharmacokinetics and toxicity in animals, which support subsequent clinical testing. The sponsor must submit the results of the preclinical studies, together with manufacturing information, analytical data, any available clinical data or literature, and a proposed clinical protocol, to the FDA as part of the IND. An IND is a request for authorization from the FDA to administer an investigational product to humans, and must become effective before human clinical trials may begin.
Preclinical studies include laboratory evaluation of product chemistry and formulation, as well as in vitro and animal studies to assess the pharmacology, pharmacokinetics, and potential for adverse events and in some cases to establish a rationale for therapeutic use. The conduct of preclinical studies is subject to federal regulations and requirements, including GLP regulations for safety/toxicology studies. An IND sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and plans for clinical studies, among other things, to the FDA as part of an IND. Some long-term preclinical testing, such as animal tests of reproductive adverse events, chronic toxicity and carcinogenicity, may continue after the IND is submitted. An IND automatically becomes effective 30 days after receipt by the FDA, unless before that time, the FDA raises concerns or questions related to one or more proposed clinical trials that precludes study initiation and places the trial on clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. As a result, submission of an IND may not result in the FDA allowing clinical trials to commence.
Clinical Trials
The clinical stage of development involves the administration of the investigational product to healthy volunteers or patients under the supervision of qualified investigators, generally physicians not employed by or under the trial sponsor’s control, in accordance with GCP requirements, which include the requirement that all research subjects provide their informed consent for their participation in any clinical trial. ClinicalInterventional clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria and the parameters to be used to monitor subject safety and assess efficacy. Each protocol, and any subsequent amendments to the protocol, must be submitted to the FDA as part of the IND. Furthermore, each clinical trial must be reviewed and approved by an IRB for each institution at which the clinical trial will be conducted to ensure that the risks to individuals participating in the clinical trials are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the informed consent form that must be provided to each clinical trial subject or his or her legal representative, and must monitor the clinical trial until completed. There also are requirements governing the reporting of ongoing clinical trials and completed clinical trial results to public registries.
A sponsor who wishes to conduct a clinical trial outside of the United States may, but need not, obtain FDA authorization to conduct the clinical trial under an IND. If a foreign clinical trial is not conducted under an IND, the sponsor may submit data from the clinical trial to the FDA in support of an NDA or BLA. The FDA will accept a well-designed and well-conducted foreign clinical trial not conducted under an IND if the trial was conducted in accordance with GCP requirements and the FDA is able to validate the data through an onsite inspection if deemed necessary.
Clinical trials in the United States generally are conducted in three sequential phases, known as Phase 1, Phase 2 and Phase 3, andwhich may overlap.overlap or be combined:
•Phase I clinical trials generally involve a small number of healthy volunteers or disease-affected patients who are initially exposed to a single dose and then multiple doses of the product candidate. The primary purpose of these clinical trials is to assess the metabolism, pharmacologic action, and initial side effect tolerability and safety of the drug.
•Phase II clinical trials involve studies in a limited number of disease-affected patients to determineevaluate the dose requiredpreliminary efficacy, optimal dosages, and dosing schedule and to produce the desired benefits. At the same time, safety and further PK and PD information is collected,identify possible adverse side effects and safety risks are identified, and a preliminary evaluation of efficacy is conducted.risks.
•Phase III clinical trials generally involve a large number of patients at multiple sites and are designed to provide the data necessary to demonstrate the effectiveness of the product for its intended use, its safety in use and to establish the overall benefit/risk relationship of the product and provide an adequate basis for product approval. These trials may include comparisons with placebo and/or other comparator treatments. The duration of treatment is often extended to mimic the actual use of a product during marketing.
When these phases overlap or are combined, the trials may be referred to as Phase 1/2 or Phase 2/3. Post-approval trials, sometimes referred to as Phase 4 clinical trials, may be conducted after initial marketing approval. These trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication. In certain instances, the FDA may mandate the performance of Phase 4 clinical trials as a condition of approval of an NDA or BLA.
Progress reports detailing the results of the clinical trials, among other information, must be submitted at least annually to the FDA. Expedited written IND safety reports must be submitted to the FDA, IRBs, ECs and the investigators for serious and unexpected suspected adverse events, findings from other studies suggesting a significant risk to humans exposed to the drug, findings from animal or in vitro testing that suggest a significant risk for human subjects and any clinically important increase in the rate or severity of a serious suspected adverse reaction over that listed in the protocol or investigator brochure.
Further, as a result of the COVID-19 pandemic, we may be required to develop and implement additional clinical trial policies and procedures designed to help protect subjects from the COVID-19 virus. For example, the FDA has issued guidance on conducting clinical trials during the pandemic, which describes a number of considerations for sponsors of clinical trials impacted by the pandemic, including certain reporting requirements, and additional guidance on the good manufacturing practice considerations for responding to COVID-19 infection and other topics. We may be required to make further adjustments to our clinical trials or business operations based on current or future guidance and regulatory requirements as a result of the COVID-19 pandemic.
Phase 1, Phase 2 and Phase 3 clinical trials may not be completed successfully within any specified period, if at all. The FDA or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug or biologic has been associated with unexpected serious harm to patients. Additionally, some clinical trials are overseen by an independent group of qualified experts organized by the clinical trial sponsor, known as a data safety monitoring board or committee. This group provides authorization for whether a trial may move forward at designated check points based on access to certain data from the trial. Concurrent with clinical trials, companies usually complete additional animal studies and also must develop additional information about the chemistry and physical characteristics of the drug or biologic as well as finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product and, among other things, companies must develop methods for testing the identity, strength, quality and purity of the final product. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that our product candidates do not undergo unacceptable deterioration over their shelf life.
We may be required to develop and implement additional clinical trial policies and procedures designed to help protect subjects from public health concerns, such as the COVID-19 pandemic. For example, the FDA has issued various guidance documents on conducting clinical trials during the pandemic, including certain reporting requirements and additional guidance on good manufacturing practice considerations for responding to COVID-19 infection.NDA/BLA Review Process
Following completion of the clinical trials, data are analyzed to assess whether the investigational product is safe and effective for the proposed indicated use or uses. The results of preclinical studies and clinical trials are then submitted to the FDA as part of an NDA or BLA, along with the proposed labeling, and information relating to the product's chemistry, manufacturing, and manufacturingcontrols, among other information, to ensure consistent product quality, safety, and other relevant data.efficacy. In short, the NDA or BLA is a request for approval to market the drug or biologic for one or morethe specified indicationsindication(s) and must contain proof of safety and efficacy for a drug or safety, purity and potency for a biologic. The application may include both negative and ambiguous results of preclinical studies and clinical trials, as well as positive findings. Data may come from company-sponsored clinical trials intended to test the safety and efficacy of a product’s use and/or from a number of alternative sources, including studies initiated by investigators or cooperative clinical groups. To support marketing approval, the data submitted must be sufficient in quality and quantity to establish the safety and efficacy of the investigational product to the satisfaction of FDA. FDA approval of an NDA or BLA must be obtained before a drug or biologic may be marketed in the United States.
Under the Prescription Drug User Fee Act ("PDUFA"), as amended, each NDA or BLA must be accompanied by a user fee. FDA adjusts the PDUFA user fees on an annual basis. According to the FDA’s FY 20212024 user fee schedule, effective through September 31, 2021,30, 2024, the user fee for an application requiring clinical data, such as an NDA or BLA, is $2,875,842.$4,048,695. PDUFA also imposes an annual program fee for each marketed human drug or biologic of $336,432. $416,734.Fee waivers or reductions are available in certain circumstances, including a waiver of the application fee for the first application filed by a small business.limited circumstances. Additionally, no user fees are assessed on NDAs or BLAs for products designated as orphan drugs for an orphan indication submission.
The FDA reviews all submitted NDAs and BLAs before it accepts them for filing, and may request additional information rather than accepting the NDA or BLA for filing.filing, such as the issuance of a Refuse to File (RTF) letter. The FDA must make a decision on accepting an NDA or BLA for filing within 60 days of receipt. Once the submission is accepted for filing, the FDA begins an in-depth review of the NDA or BLA. Under the goals and policies agreed to by the FDA under PDUFA, the FDA has ten months, from the filing date, in which to complete its initial review of a new molecular-entity NDA or original BLA and respond to the applicant, and six months from the filing date of a new molecular-entity NDA or original BLA designated for priority review. The FDA does not always meet its PDUFA goal dates for standard and priority NDAs or BLAs, and the review process may be extended by FDA requests for additional information or clarification.
Before approving an NDA or BLA, the FDA will conduct a pre-approval inspection of the manufacturing facilities for the new product to determine whether they comply with cGMP requirements. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. The FDA also may audit data from clinical trials to ensure compliance with GCP requirements. Additionally, the FDA may refer applications for novel drug products or drug products which present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and under what conditions, if any. The FDA is not bound by recommendations of an advisory committee, but it considers such recommendations when making decisions on approval. The FDA likely will reanalyze the clinical trial data, which could result in extensive discussions between the FDA and the applicant during the review process. After the FDA evaluates an NDA or BLA, it will issue an approval letter or a Complete Response Letter. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications. A Complete Response Letter indicates that the review cycle of the application is complete and the application will not be approved in its present form. A Complete Response Letter usually describes all of the specific deficiencies in the NDA or BLA identified by the FDA. The Complete Response Letter may require additional clinical data, additional pivotal Phase 3 clinical trial(s) and/or other significant and time-consuming requirements related to clinical trials, preclinical studies or manufacturing. If a Complete Response Letter is issued, the applicant may either resubmit the NDA or BLA, addressing all of the deficiencies identified in the letter, or withdraw the application. Even if such data and information are submitted, the FDA may decide that the NDA or BLA does not satisfy the criteria for approval. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than we interpret the same data.
If regulatory approval of a product is granted, such approval will be granted for particular indications and may entail limitations on the indicated uses for which such product may be marketed. For example, the FDA may approve the BLA with a Risk Evaluation and Mitigation Strategy, or REMS, to ensure the benefits of the product outweigh its risks. A REMS is a safety strategy to manage a known or potential serious risk associated with a product and to enable patients to have continued access to such medicines by managing their safe use, and could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. The FDA also may condition approval on, among other things, changes to proposed labeling or the development of adequate controls and specifications. Once approved, the FDA may withdraw the product approval if compliance with pre- and post-marketing requirements is not maintained or if problems occur after the product reaches the marketplace. The FDA may require one or more Phase 4 post-market studies and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization, and may limit further marketing of the product based on the results of these post-marketing studies.
Orphan Drugs
Under the Orphan Drug Act, the FDA may grant orphan designation to a drug or biological product intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making the product available in the United States for this type of disease or condition will be recovered from sales of the product.
Orphan drug designation must be requested before submitting an NDA or BLA. After the FDA grants orphan drug designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Orphan drug designation does not convey any advantage in or shorten the duration of the regulatory review and approval process.
If a product that has orphan designation subsequently receives the first FDA approval for the disease or condition for which it has such designation, the product is entitled to orphan drug exclusivity, which means that the FDA may not approve any other applications to market the same drug for the same indication for seven years from the date of such approval, except in limited circumstances, such as a showing of clinical superiority to the product with orphan exclusivity by means of greater effectiveness, greater safety or providing a major contribution to patient care or in instances of drug supply issues. Competitors, however, may receive approval of either a different product for the same indication or the same product for a different indication but that could be used off-label in the orphan indication. Orphan drug exclusivity also could block the approval of one of our products for seven years if a competitor obtains approval before we do for the same product, as defined by the FDA, for the same indication we are seeking approval, or if a product candidate is determined to be contained within the scope of the competitor’s product for the same indication or disease. If one of our products designated as an orphan drug receives marketing approval for an indication broader than that which is designated, it may not be entitled to orphan drug exclusivity. Orphan drug status in the EU has similar, but not identical, requirements and benefits.
In view of the court decision in Catalyst Pharms., Inc. v. Becerra, 14 F.4th 1299 (11th Cir. 2021), in January 2023, the FDA published a notice in the Federal Register to clarify that while the agency complies with the court’s order in Catalyst, the FDA intends to continue to apply its longstanding interpretation of the regulations to matters outside of the scope of the Catalyst order – that is, the agency will continue tying the scope of orphan-drug exclusivity to the uses or indications for which a drug is approved, which permits other sponsors to obtain approval of a drug for new uses or indications within the same orphan designated disease or condition that have not yet been approved. It is unclear how future litigation, legislation, agency decisions, and administrative actions will impact the scope of the orphan drug exclusivity.
Expedited Development and Review Programs
The FDA has a fast track program that is intended to expedite or facilitate the process for reviewing new drugs and biologics that meet certain criteria. Specifically, new drugs and biologics are eligible for fast track designation if they are intended to treat a serious or life-threatening condition and preclinical or clinical data demonstrate the potential to address unmet medical needs for the condition. Fast track designation applies to the combination of the product and the specific indication for which it is being studied. The sponsor can request the FDA to designate the product for fast track status any time before receiving NDA or BLA approval, but ideally no later than the pre-NDA or pre-BLA meeting.
Additionally, a drug or biologic may be eligible for designation as a breakthrough therapy if the product is intended, alone or in combination with one or more other drugs or biologics, to treat a serious or life-threatening condition and preliminary clinical evidence indicates that the product may demonstrate substantial improvement over currently approved therapies on one or more clinically significant endpoints. The benefits of breakthrough therapy designation include the same benefits as fast track designation, plus intensive guidance from the FDA to facilitate an efficient drug development program.
Any product submitted to the FDA for marketing, including under a fast track or breakthrough therapy designation program, may be eligible for other types of FDA programs intended to expedite development and review, such as priority review and accelerated approval.
Any product is eligible for priority review if it treats a serious or life-threatening condition and, if approved, would provide a significant improvement in safety and effectiveness compared to available therapies. Priority review reduces the review time for an initial or supplemental marketing application by four months.
A product may be eligible for accelerated approval, if it treats a serious or life-threatening condition and generally provides a meaningful advantage over available therapies based on an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality ("IMM"), that is reasonably likely to predict an effect on IMM or other clinical benefit. As a condition of accelerated approval, the FDA requires that a sponsor of a drug or biologic receiving accelerated approval subsequently provide additional data confirming the anticipated clinical benefit, for example by performing adequate and well-controlled post-marketing clinical trials. If clinical benefit is not confirmed, accelerated approval may be revoked. If the FDA concludes that a drug or biologic shown to be effective can be safely used only if distribution or use is restricted, it may require such post-marketing restrictions, as it deems necessary to assure safe use of the product. Further, the Food and Drug Omnibus Reform Act made several changes to the FDA’s authorities and its regulatory framework, including, among other changes, requiring the FDA to specify conditions for post-approval study requirements and setting forth procedures for the FDA to withdraw a product on an expedited basis for non-compliance with post-approval requirements.
Fast track designation, breakthrough therapy designation, priority review, and accelerated approval do not change the standards for approval, but may expedite the development or approval process.
Abbreviated Licensure Pathway of Biological Products as Biosimilar or Interchangeable
The Patient Protection and Affordable Care Act ("PPACA"), Affordable Care Act ("ACA"), signed into law in 2010, includes a subtitle called the Biologics Price Competition and Innovation Act of 2009 ("BPCIA"), created an abbreviated approval pathway for biological products shown to be highly similar to an FDA-licensed reference biological product. The BPCIA attempts to minimize duplicative testing, and thereby lower development costs and increase patient access to affordable treatments. An application for licensure of a biosimilar product must include information demonstrating biosimilarity based upon the following, unless the FDA determines otherwise:
•analytical studies demonstrating that the proposed biosimilar product is highly similar to the approved product notwithstanding minor differences in clinically inactive components; and
•animal studies (including the assessment of toxicity); and.
•a clinical trial or trials (including the assessment of immunogenicity and PK or PD) sufficient to demonstrate safety, purity and potency in one or more conditions for which the reference product is licensed and intended to be used.
In addition, an application must include information demonstrating that:
•the proposed biosimilar product and reference product utilize the same mechanism of action for the condition(s) of use prescribed, recommended, or suggested in the proposed labeling, but only to the extent the mechanism(s) of action are known for the reference product;
•the condition or conditions of use prescribed, recommended, or suggested in the labeling for the proposed biosimilar product have been previously approved for the reference product;
•the route of administration, the dosage form, and the strength of the proposed biosimilar product are the same as those for the reference product; and
•the facility in which the biological product is manufactured, processed, packed or held meets standards designed to assure that the biological product continues to be safe, pure, and potent.
Biosimilarity means that the biological product is highly similar to the reference product notwithstanding minor differences in clinically inactive components; and that there are no clinically meaningful differences between the biological product and the reference product in terms of the safety, purity, and potency of the product. In addition, the law provides for a designation of “interchangeability” between the reference and biosimilar products, whereby the biosimilar may be substituted for the reference product without the intervention of the health care provider who prescribed the reference product. The higher standard of interchangeability must be demonstrated by information sufficient to show that:
•the proposed product is biosimilar to the reference product;
•the proposed product is expected to produce the same clinical result as the reference product in any given patient; and
•for a product that is administered more than once to an individual, the risk to the patient in terms of safety or diminished efficacy of alternating or switching between the biosimilar and the reference product is no greater than the risk of using the reference product without such alternation or switch.
FDA approval is required before a biosimilar may be marketed in the United States. However, complexities associated with the large and intricate structures of biological products and the process by which such products are manufactured pose significant hurdles to the FDA’s implementation of the law that are still being worked out by the FDA. For example, the FDA has discretion over the kind and amount of scientific evidence—laboratory, preclinical and/or clinical—required to demonstrate biosimilarity to a licensed biological product.
The FDA intends to consider the totality of the evidence, provided by a sponsor to support a demonstration of biosimilarity, and recommends that sponsors use a stepwise approach in the development of their biosimilar products. Biosimilar product applications thus may not be required to duplicate the entirety of preclinical and clinical testing used to establish the underlying safety and effectiveness of the reference product. However, the FDA may refuse to approve a biosimilar application if there is insufficient information to show that the active ingredients are the same or to demonstrate that any impurities or differences in active ingredients do not affect the safety, purity or potency of the biosimilar product. In addition, as with BLAs, biosimilar product applications will not be approved unless the product is manufactured in facilities designed to assure and preserve the biological product’s safety, purity and potency.
The submission of a biosimilar application does not guarantee that the FDA will accept the application for filing and review, as the FDA may refuse to accept applications that it finds are insufficiently complete. The FDA will treat a biosimilar application or supplement as incomplete if, among other reasons, any applicable user fees assessed under the Biosimilar User Fee Act of 2012 have not been paid. In addition, the FDA may accept an application for filing but deny approval on the basis that the sponsor has not demonstrated biosimilarity, in which case the sponsor may choose to conduct further analytical, preclinical or clinical studies and submit a BLA for licensure as a new biological product.
The timing of final FDA approval of a biosimilar for commercial distribution depends on a variety of factors, including whether the manufacturer of the branded product is entitled to one or more statutory exclusivity periods, during which time the FDA is prohibited from approving any products that are biosimilar to the branded product. The FDA cannot approve a biosimilar application for twelve years from the date of first licensure of the reference product. Additionally, a biosimilar product sponsor may not submit an application for four years from the date of first licensure of the reference product. A reference product may also be entitled to exclusivity under other statutory provisions. For example, a reference product designated for a rare disease or condition (an “orphan drug”) may be entitled to seven years of exclusivity, in which case no product that is biosimilar to the reference product may be approved until either the end of the twelve-year period provided under the biosimilarity statute or the end of the seven-year orphan drug exclusivity period, whichever occurs later. In certain circumstances, a regulatory exclusivity period can extend beyond the life of a patent, and thus block biosimilarity applications from being approved on or after the patent expiration date. In addition, the FDA may under certain circumstances extend the exclusivity period for the reference product by an additional six months if the FDA requests, and the manufacturer undertakes, studies on the effect of its product in children, a so-called pediatric extension.
The first biological product determined to be interchangeable with a branded product for any condition of use is also entitled to a period of exclusivity, during which time the FDA may not determine that another product is interchangeable with the reference product for any condition of use. This exclusivity period extends until the earlier of: (1) one year after the first commercial marketing of the first interchangeable product; (2) 18 months after resolution of a patent infringement against the applicant that submitted the application for the first interchangeable product, based on a final court decision regarding all of the patents in the litigation or dismissal of the litigation with or without prejudice; (3) 42 months after approval of the first interchangeable product, if a patent infringement suit against the applicant that submitted the application for the first interchangeable product is still ongoing; or (4) 18 months after approval of the first interchangeable product if the applicant that submitted the application for the first interchangeable product has not been sued.
Post-Approval Requirements
Following approval of a new product, the manufacturer and the approved product are subject to continuing regulation by the FDA, including, among other things, monitoring and record-keeping requirements, requirements to report adverse experiences, and complying with promotion and advertising requirements, which include restrictions on promoting drugs for unapproved uses or patient populations (known as “off-label use”) and limitations on industry-sponsored scientific and educational activities. Although physicians in the United States may prescribe legally available drugs for off-label uses, manufacturers may not market or promote such uses. Prescription drug promotional materials must be submitted to the FDA in conjunction with their first use. Further, if there are any modifications to the drug or biologic, including changes in indications, labeling or manufacturing processes or facilities, thean applicant maywould be required to submit and obtain FDA approval of a new NDA/BLA or NDA/BLAa supplement before any material modifications can be implemented for a drug or biologic, including changes in labeling or manufacturing processes or facilities, which may require the development of additional data or preclinicalnonclinical studies and clinical trials.
The FDA may also place other conditions on approvals including the requirement for a Risk Evaluation and Mitigation Strategy ("REMS"),REMS, to assure the safe use of the product. A REMS could include medication guides, physician communication plans or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. Any of these limitations on approval or marketing could restrict the commercial promotion, distribution, prescription or dispensing of products. Product approvals may be withdrawn for non-compliance with regulatory standards or if problems occur following initial marketing.
The FDA may withdraw approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical studies to assess new safety risks; or imposition of distribution restrictions or other restrictions under a REMS program. Other potential consequences include, among other things:
•restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls;
•fines, warning letters or holds on post-approval clinical studies;
•refusal of the FDA to approve pending applications or supplements to approved applications;
•applications, or suspension or revocation of product license approvals;
•product seizure or detention, or refusal to permit the import or export of products; or
•injunctions or the imposition of civil or criminal penalties.
The FDA strictly regulates marketing, labeling, advertising, and promotion of products that are placed on the market. Drugs and biologics may be promoted only for the approved indications and in accordance with the provisions of the approved label. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability.
Other U.S. Regulatory Matters
Manufacturing, sales, promotion and other activities following product approval are also subject to regulation by numerous regulatory authorities in the United States in addition to the FDA, including the Centers for Medicare & Medicaid Services, other divisions of the Department of Health and Human Services, the Department of Justice, the Drug Enforcement Administration, the Consumer Product Safety Commission, the Federal Trade Commission, the Occupational Safety & Health Administration, the Environmental Protection Agency and state and local governments.
For example, in the United States, sales, marketing and scientific and educational programs also must comply with state and federal fraud and abuse laws. These laws include the federal Anti-Kickback Statute, which makes it illegal for any person, including a prescription drug manufacturer (or a party acting on its behalf), to knowingly and willfully solicit, receive, offer or pay any remuneration that is intended to induce or reward referrals, including the purchase, recommendation, order or prescription of a particular drug, for which payment may be made under a federal healthcare program, such as Medicare or Medicaid. Violations of this law are punishable by up to five years in prison, criminal fines, administrative civil money penalties and exclusion from participation in federal healthcare programs. Moreover, the ACA provides that the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act.
Pricing and rebate programs must comply with the Medicaid rebate requirements of the U.S. Omnibus Budget Reconciliation Act of 1990 and more recent requirements in the ACA. If products are made available to authorized users of the Federal Supply Schedule of the General Services Administration, additional laws and requirements apply. Products must meet applicable child-resistant packaging requirements under the U.S. Poison Prevention Packaging Act. Manufacturing, sales, promotion and other activities also are potentially subject to federal and state consumer protection and unfair competition laws.
The distribution of biologic and pharmaceutical products is subject to additional requirements and regulations, including extensive record-keeping, licensing, storage and security requirements intended to prevent the unauthorized sale of pharmaceutical products.
The failure to comply with any of these laws or regulatory requirements subjects firms to possible legal or regulatory action. Depending on the circumstances, failure to meet applicable regulatory requirements can result in criminal prosecution, fines or other penalties, injunctions, requests for recall, seizure of products, total or partial suspension of production, denial or withdrawal of product approvals or refusal to allow a firm to enter into supply contracts, including government contracts. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business. Prohibitions or restrictions on sales or withdrawal of future products marketed by us could materially affect our business in an adverse way.
Changes in regulations, statutes or the interpretation of existing regulations could impact our business in the future by requiring, for example: (i) changes to our manufacturing arrangements; (ii) additions or modifications to product labeling; (iii) the recall or discontinuation of our products; or (iv) additional record-keeping requirements. If any such changes were to be imposed, they could adversely affect the operation of our business.
U.S. Patent-Term Restoration and Marketing Exclusivity
Depending upon the timing, duration and specifics of FDA approval of any future product candidates, some of our U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, commonly referred to as the Hatch-Waxman Act. The Hatch-Waxman Act permits restoration of the patent term of up to five years as compensation for patent term lost during product development and FDA regulatory review process. Patent-term restoration, however, cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent-term restoration period is generally one-half the time between the effective date of an IND and the submission date of an NDA or BLA plus the time between the submission date of an NDA or BLA and the approval of that application, except that the review period is reduced by any time during which the applicant failed to exercise due diligence. Only one patent applicable to an approved drug is eligible for the extension and the application for the extension must be submitted prior to the expiration of the patent. The USPTO, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. In the future, we may apply for restoration of patent term for our currently owned or licensed patents to add patent life beyond its current expiration date, depending on the expected length of the clinical trials and other factors involved in the filing of the relevant NDA or BLA.
Market exclusivity provisions under the FDCA also can delay the submission or the approval of certain applications. The FDCA provides a five-year period of non-patent marketing exclusivity within the United States to the first applicant to gain approval of a NDA for a new chemical entity. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept for review an abbreviated new drug application ("ANDA"), or a 505(b)(2) NDA submitted by another company for another version of such drug where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement. The FDCA also provides three years of marketing exclusivity for a NDA, 505(b)(2) NDA or supplement to an existing NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example, new indications, dosages or strengths of an existing drug. This three-year exclusivity covers only the conditions of use associated with the new clinical investigations and does not prohibit the FDA from approving ANDAs for drugs containing the original active agent. Five-year and three-year exclusivity will not delay the submission or approval of a full NDA. However, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the preclinical studies and adequate and well-controlled clinical trials necessary to demonstrate safety and effectiveness.
A reference biological product is granted twelve years of data exclusivity from the time of first licensure of the product, and the FDA will not accept an application for a biosimilar or interchangeable product based on the reference biological product until four years after the date of first licensure of the reference product. “First licensure” typically means the initial date the particular product at issue was licensed in the United States. Date of first licensure does not include the date of licensure of (and a new period of exclusivity is not available for) a biological product if the licensure is for a supplement for the biological product or for a subsequent application by the same sponsor or manufacturer of the biological product (or licensor, predecessor in interest, or other related entity) for a change (not including a modification to the structure of the biological product) that results in a new indication, route of administration, dosing schedule, dosage form, delivery system, delivery device or strength, or for a modification to the structure of the biological product that does not result in a change in safety, purity, or potency. Therefore, one must determine whether a new product includes a modification to the structure of a previously licensed product that results in a change in safety, purity, or potency to assess whether the licensure of the new product is a first licensure that triggers its own period of exclusivity. Whether a subsequent application, if approved, warrants exclusivity as the “first licensure” of a biological product is determined on a case-by-case basis with data submitted by the sponsor.
European Union Drug Development
As in the United States, medicinal products can be marketed only if a marketing authorization from the competent regulatory agencies has been obtained.
Similar to the United States, the various phases of preclinical and clinical research in the EU are subject to significant regulatory controls. AlthoughIn the EU, a clinical trial application, or CTA, must be submitted to each country’s national health authority and an independent ethics committee, respectively. Once the CTA is approved in accordance with a country’s requirements, clinical study development may proceed. The clinical studies must be conducted in accordance with GCP and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki. The Clinical Trials Regulation EU No 536/2014, which replaced the Clinical Trials Directive, 2001/20/EC has sought to harmonizeentered into application on January 31, 2022. The Clinical Trials Regulation harmonizes the EU clinical trials regulatory framework, setting out common rulesprocesses for the controlassessment and authorizationsupervision of clinical trials inthroughout the EU, the EU Member States have transposed and applied the provisions of the Directive differently. This has led to significant variations in the member state regimes.EU. Under the current regime, beforeRegulation, sponsors can submit one online application via a single online platform known as the Clinical Trials Information System (CTIS) for approval to run a clinical trial can be initiatedin several European countries, making it must be approved in each of the EU countries where the trial ismore efficient to be conducted by two distinct bodies: the National Competent Authority ("NCA"), and one or more Ethics Committees ("ECs"). Under the current regime all suspected unexpected serious adverse reactionscarry out such multinational trials. A transition period applies to the investigated drug that occur during the clinical trial havesubmissions under the Regulation. For example, from 31 January 2023 onwards, clinical trial sponsors need to be reportedapply via the Clinical Trials Information System to start a clinical trial. From 31 January 2025, any trials approved under the NCAClinical Trials Directive that continue running will need to comply with the Clinical Trials Regulation and ECs oftheir sponsors must enter information on the Member State where they occurred.trials in the CTIS.
The EU clinical trials legislation currently is undergoing a transition process mainly aimed at harmonizing and streamlining clinical-trial authorization, simplifying adverse-event reporting procedures, improving the supervision of clinical trials and increasing their transparency. Clinical Trials Regulation EU No 536/2014, which is expected to be enacted, will ensure that the rules for conducting clinical trials in the EU will be identical. In the meantime, Clinical Trials Directive 2001/20/EC continues to govern all clinical trials performed in the EU.
European Union Drug Review and Approval
In the European Economic Area ("EEA"), which is comprised of the 27 Member States of the EU, plus Norway, Iceland and Liechtenstein, medicinal products can only be commercialized after obtaining a Marketing Authorization ("MA"). There are two types of marketing authorizations.
•The Community MA is issued by the European Commission through the Centralized Procedure, based on the opinion of the Committee for Medicinal Products for Human Use ("CHMP"), of the EMA, and is valid throughout the entire territory of the EEA. The Centralized Procedure is mandatory for certain types of products, such as biotechnology medicinal products, orphan medicinal products, advanced-therapy medicines such as gene-therapy, somatic cell-therapy or tissue-engineered medicines and medicinal products containing a new active substance indicated for the treatment of HIV, AIDS, cancer, neurodegenerative disorders, diabetes, auto-immune and other immune dysfunctions and viral diseases. The Centralized Procedure may also apply for products containing a new active substance not yet authorized in the EEA, or for products that constitute a significant therapeutic, scientific or technical innovation or which are in the interest of public health in the EU.
•National MAs, which are issued by the competent authorities of the Member States of the EEA and only cover their respective territory, are available for products not falling within the mandatory scope of the Centralized Procedure. Where a product has already been authorized for marketing in a Member State of the EEA, this National MA can be recognized in another Member States through the Mutual Recognition Procedure. If the product has not received a National MA in any Member State at the time of application, it can be approved simultaneously in various Member States through the Decentralized Procedure. Under the Decentralized Procedure an identical dossier is submitted to the competent authorities of each of the Member States in which the MA is sought, one of which is selected by the applicant as the Reference Member State ("RMS"). The competent authority of the RMS prepares a draft assessment report, a draft summary of the product characteristics ("SPC"), and a draft of the labeling and package leaflet, which are sent to the other Member States (referred to as the Member States Concerned) for their approval. If the Member States Concerned raise no objections, based on a potential serious risk to public health, to the assessment, SPC, labeling, or packaging proposed by the RMS, the product is subsequently granted a national MA in all the Member States (i.e., in the RMS and the Member States Concerned).
Under the procedures described above, before granting the MA, the EMA or the competent authorities of the Member States of the EEA assess the risk-benefit balance of the product on the basis of scientific criteria concerning its quality, safety and efficacy. Starting in January 2021, the Medicines and Healthcare products Regulatory Agency (MHRA) will take onMHRA assumed additional regulatory responsibilities for medical products marketed in the UK, as pan-EU regulatory procedures before the EMA will no longer apply in the UK. MHRA and the National Institute for Biological Standards and Control (NIBSC)("NIBSC") recently issued new guidance documents to the industry regarding regulation under the UK system. Proposals set forth in the new MHRA guidance will take effect through legislative changes that are subject to parliamentary approval, which may increase the amount of resources and time needed for obtaining regulatory approval in the UK and delay our clinical development and commercialization. The full impactOn January 1, 2024, MHRA launched a new streamlined international recognition framework replacing the current European Commission Decision Reliance Procedure (ECDRP) and allow the MHRA to rely on or give regard to a European decision or decision of Brexit on our business remains unclear.
Coverage and Reimbursement
Sales of our products will depend, in part, on the extent to which our products will be covered by third-party payors, such as government health programs, commercial insurance and managed healthcare organizations. In the United States no uniform policy of coverage and reimbursement for drug or biological products exists. Accordingly, decisions regarding the extent of coverage and amount of reimbursement to be provided for any of our products will be made on a payor-by-payor basis. As a result, the coverage determination process is often a time-consuming and costly process that will require us to provide scientific and clinical support for the use of our products to each payor separately, with no assurance that coverage and adequate reimbursement will be obtained.
The United States government, state legislatures and foreign governments have shown significant interest in implementing cost containment programs to limit the growth of government-paid health care costs, including price-controls, restrictions on reimbursement and requirements for substitution of generic products for branded prescription drugs. For example, the ACA contains provisions that may reduce the profitability of drug products through increased rebates for drugs reimbursed by Medicaid programs, extension of Medicaid rebates to Medicaid managed care plans, mandatory discounts for certain Medicare Part D beneficiaries and annual fees based on pharmaceutical companies’ share of sales to federal health care programs. Adoption of general controls and measures, coupled with the tightening of restrictive policies in jurisdictions with existing controls and measures, could limit payments for pharmaceutical drugs.
The Medicaid Drug Rebate Program requires pharmaceutical manufacturers to enter into and have in effect a national rebate agreement with the Secretary of the Department of Health and Human Services as a condition for states to receive federal matching funds for the manufacturer’s outpatient drugs furnished to Medicaid patients. The ACA made several changes to the Medicaid Drug Rebate Program, including increasing pharmaceutical manufacturers’ rebate liability by raising the minimum basic Medicaid rebate on most branded prescription drugs from 15.1% of average manufacturer price ("AMP"), to 23.1% of AMP and adding a new rebate calculation for “line extensions” (e.g., new formulations, such as extended release formulations) of solid oral dosage forms of branded products, as well as potentially impacting their rebate liability by modifying the statutory definition of AMP. The ACA also expanded the universe of Medicaid utilization subject to drug rebates by requiring pharmaceutical manufacturers to pay rebates on Medicaid managed care utilization and by enlarging the population potentially eligible for Medicaid drug benefits. The Centers for Medicare & Medicaid Services ("CMS"), have proposed to expand Medicaid rebate liability to the territories of the United States as well.
The Medicare Prescription Drug, Improvement, and Modernization Act of 2003 ("MMA"), established the Medicare Part D program to provide a voluntary prescription drug benefit to Medicare beneficiaries. Under Part D, Medicare beneficiaries may enroll in prescription drug plans offered by private entities that provide coverage of outpatient prescription drugs. Unlike Medicare Part A and B, Part D coverage is not standardized. While all Medicare drug plans must give at least a standard level of coverage set by Medicare, Part D prescription drug plan sponsors are not required to pay for all covered Part D drugs, and each drug plan can develop its own drug formulary that identifies which drugs it will cover and at what tier or level. However, Part D prescription drug formularies must include drugs within each therapeutic category and class of covered Part D drugs, though not necessarily all the drugs in each category or class. Any formulary used by a Part D prescription drug plan must be developed and reviewed by a pharmacy and therapeutic committee. Government payment for some of the costs of prescription drugs may increase demand for products for which we receive marketing approval. However, any negotiated prices for our products covered by a Part D prescription drug plan likely will be lower than the prices we might otherwise obtain. Moreover, while the MMA applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own payment rates. Any reduction in payment that results from the MMA may result in a similar reduction in payments from non-governmental payors.
For a drug product to receive federal reimbursement under the Medicaid or Medicare Part B programs or to be sold directly to U.S. government agencies, the manufacturer must extend discounts to entities eligible to participate in the 340B drug pricing program. The required 340B discount on a given product is calculated based on the AMP and Medicaid rebate amounts reported by the manufacturer.
There have been several recent U.S. Congressional inquiries and proposed federal and state legislation designed to, among other things, bring more transparency to drug pricing, reduce the cost of prescription drugs under Medicare, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drugs. At
In August 2022, Congress passed the Inflation Reduction Act of 2022, which includes prescription drug provisions that have significant implications for the pharmaceutical industry and Medicare beneficiaries. These include allowing the federal level, in 2020, the Trump administration announced several executive orders relatedgovernment to prescription drug pricing that seek to implement several of the administration's proposals. The FDA also releasednegotiate a final rule in September 2020 providing guidance for states to build and submit importation plans for drugs from Canada. Further, in November 2020, the U.S. Secretary of the Department of Health and Human Services ("HHS") finalized a regulation removing safe harbor protection formaximum price reductions from pharmaceutical manufacturers to plan sponsors under Part D, either directly or through pharmacy benefit managers, unless the price reduction is requiredpaid by law. The rule also creates a new safe harbor for price reductions reflected at the point-of-sale, as well as a safe harborMedicare for certain fixed fee arrangements between pharmacy benefit managerssingle source drugs responsible for significant Medicare spending, imposing penalties and manufacturers. The CMS also issued an interim final rule implementing President Trump’s Most Favored Nation executive order, which would tieexcise tax for manufacturers that fail to comply with the drug price negotiation requirements, requiring inflation rebates for all Medicare Part B paymentsand Part D drugs, with limited exceptions, if their drug prices increase faster than inflation, and redesigning Medicare Part D to reduce out-of-pocket prescription drug costs for beneficiaries, among other changes. Various industry stakeholders, including certain physician-administered drugs topharmaceutical companies and the lowestPharmaceutical Research and Manufacturers of America, have initiated lawsuits against the federal government asserting that the price paid in other economically advanced countries, effective January 1, 2021. On December 28, 2020, the United States District Court in Northern California issued a nationwide preliminary injunction against implementationnegotiation provisions of the interim final rule. In December 2020, CMS issued a final rule implementing significant manufacturer price reporting changes under the Medicaid Drug Rebate Program, including regulations that affect manufacturer-sponsored patient assistance programs subject to pharmacy benefit manager accumulator programsIRA are unconstitutional. The impact of these judicial challenges, legislative, executive, and Best Price reporting related to certain value-based purchasing arrangements. It is unclear to what extent these new regulationsadministrative actions, and any future regulationshealthcare measures and legislationagency rules implemented by the Biden administration will have on our business, including our abilityus and the pharmaceutical industry as a whole is unclear. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, and achieve profitability.attain profitability, or commercialize our product candidates if approved.
At the state level, legislatures have also increasingly passed legislation and implemented regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. For example, a number of states are considering or have recently enacted state drug price transparency and reporting laws that could substantially increase our compliance burdens and expose us to greater liability under such state laws once we begin commercialization after obtaining regulatory approval for any of our products. We are unable to predict the future course of federal or state healthcare legislation in the United States directed at broadening the availability of healthcare and containing or lowering the cost of healthcare. These and any further changes in the law or regulatory framework that reduce our revenue or increase our costs could also have a material and adverse effect on our business, financial condition and results of operations.
As noted above, the marketability of any products for which we receive regulatory approval for commercial sale may suffer if the government and third-party payors fail to provide adequate coverage and reimbursement. An increasing emphasis on cost containment measures in the United States has increased and we expect will continue to increase theindicates continued pressure on pharmaceutical pricing. Coverage policies and third-party reimbursement rates may change at any time. Even if favorable coverage and reimbursement status is attained for one or more products for which we receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
In addition, in most foreign countries, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing and reimbursement vary widely from country to country. For example, the EU provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. A member state may approve a specific price for the medicinal product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of our products. Historically, products launched in the EU do not follow price structures of the United States and generally prices tend to be significantly lower.
Financial Information about Segments
We manage our operations as a single reportable segment for the purposes of assessing performance and making operating decisions. See "Note 1 - Significant Accounting Policies" in the notes to the consolidated financial statements included elsewhere in this Annual Report on Form 10-K.
Employees and Human Capital Resources
As of December 31, 2020,2023, we had 291 employees, all of whom wereapproximately 445 full-time and approximately two-thirds of which hold Ph.D. or M.D. degrees. Of these full-time employees, 234 were engaged in research and development activities and 57 were engaged in finance, legal, business development, human resources, information technology, facilities and other general administrative functions. Substantially allemployees. A large majority of our employees are locatedwork out of our headquarters location in South San Francisco, California. NoneCA, with the remainder working out of our employees is represented by a labor union or covered by a collective bargaining agreement. We consider our relations with our employees to be good.locations in Salt Lake City, Utah and Zurich, Switzerland.
Our human capital resources objectives include,strategy aims to attract new talent and retain and incentivize existing employees by investing in their professional development, as applicable, identifying, recruiting, retaining, incentivizingwell as providing them with challenging and integratingrewarding opportunities for personal growth. Our working environment is guided by the core Denali values of trust, growth, grit and unity, and reinforced by developing quality leadership, fostering diversity and inclusion, emphasizing continuous growth, creating opportunities for engagement, and embracing our existing and new employees, advisors and consultants. The principal purposes ofgoal to defeat degeneration. Our values-driven culture is complemented by our equity and cash incentive plans, arewhich serve to (1) attract and retain and reward personnelemployees through the granting of stock-based and cash-based compensation awards, in order to increase stockholder valueawards; and the success of our company by motivating such individuals(2) motivate employees to perform to the best of their abilities and achieve our objectives.objectives, thereby increasing the value and success of our company.
Key areas of focus for Denali include:
Health and Safety. Our health and safety programs are designed around global standards with specifications addressing regulations, specific hazards, and the unique working environment of our operations. We mandate employee health and safety training and ergonomic assessments, and require specialized training for all lab-based employees. We conduct regular internal safety audits to ensure that proper safety policies and program procedures are in place. In addition, we engage both internal and third-party compliance assessments and audit selected operations for adherence to health and safety standards. Denali’s safety programs have been highly effective: since we commenced operations in 2015, we have had zero reportable regulatory safety incidents.
Diversity and Inclusion. Denali embraces differences and acknowledges the valuable perspectives that a diverse workforce brings to problem-solving. Our Unity in Diversity team spearheads action-oriented diversity programs, such as social responsibility through community leadership and volunteerism, investment in STEM-focused outreach, and creating a safe place for expression and ideas. As part of our plan to create a more inclusive workplace, we have adopted a zero tolerance policy towards harassment and discrimination and created safe avenues for employees to submit complaints, including an anonymous hotline and a formal complaint system that allows for direct access to our human resources department.
We have implemented several measures to ensure that we are accountable for making progress on our diversity and inclusion initiatives. Diversity and inclusion objectives are embedded in our annual performance goals. We also ensure pipeline diversity by partnering with each division in their workforce planning forecasts to develop initiatives and goals to recruit diverse talent across all leadership and skill areas. As of December 31, 2023, approximately 54% of our workforce and 48% of managers were female. As of December 31, 2023, ethnic or racial minorities represented approximately 54% of our workforce and 46% of our managers.
Training and Development. We believe training and development are an important part of creating a safe, productive, fair, and equal environment. We encourage continuous feedback, improvement, and growth for our employees. We provide technical, leadership and compliance training to all employees in several formats, including through live seminars, online trainings and professional organizations. Managers are given annual training to hone their supervisory skills and better support their employees’ development; they are, in turn, accountable for guiding the development of a personal and professional growth plan for each employee. In addition, Denali has designed employee development programs to help employees develop essential skills that are aligned to promote growth and Denali’s values.
Flexible Work Options. The global pandemic has accelerated our capabilities and culture with respect to flexible work. Denali values workplace flexibility and hybrid ways of working, and has introduced a policy which we believe balances more workplace flexibility with time together to collaborate and connect in person. We use enhanced tools and technology designed to help us optimize productivity and collaboration while facilitating a hybrid work environment for our diverse workforce.
Corporate Information
We were incorporated in Delaware in 2013. Our principal executive offices are located at 161 Oyster Point Blvd., South San Francisco, California 94080. Our telephone number is (650) 866-8548. Our website address is www.denalitherapeutics.com. We also use our website as a channel of distribution of important company information. Important information, including news or announcements regarding our financial performance, investor events and press releases, is posted on and accessible from our website.releases. We intend to use Denali’sour website as a means of disclosing material non-public information and for complying with our disclosure obligations under Regulation FD.
We file electronically with the SECSecurities and Exchange Commission ("SEC") our annual reports on Form 10-K, quarterly reports on Form 10-Q, and current reports on Form 8-K pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended or the ("Exchange Act.Act"). We make available on our website at www.denalitherapeutics.com, free of charge, copies of these reports, including amendments to such reports, as soon as reasonably practicable after we electronically file such material with, or furnish it to, the SEC. The public may read or copy any materials we file with the SEC at the SEC's Public Reference Room at 100 F Street NE, Washington, D.C. 20549. The public may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. The SEC maintains a website that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC. The address of that website is www.sec.gov. The information in or accessible through the SEC and our website or social media sites does not constitute part of this Annual Report on Form 10-K or any other report or document we file with the SEC, and any references to our website and social media sites are intended to be inactive textual references only.
We use Denali®, the Denali Therapeutics logo, and other marks as trademarks in the United States and other countries. This Annual Report on Form 10-K contains references to our trademarks and service marks and to those belonging to other entities. Solely for convenience, trademarks and trade names referred to in this Annual Report on Form 10-K, including logos, artwork and other visual displays, may appear without the ® or ™ symbols, but such references are not intended to indicate in any way that we will not assert, to the fullest extent under applicable law, our rights or the rights of the applicable licensor to these trademarks and trade names. We do not intend our use or display of other entities’ trade names, trademarks or service marks to imply a relationship with, or endorsement or sponsorship of us by any other entity.
ITEM 1A. RISK FACTORS
Investing in our common stock involves a high degree of risk. You should carefully consider the risks described below, as well as the other information in this Annual Report on Form 10-K, including our financial statements and the related notes and the section titled “Management’s Discussion and Analysis of Financial Condition and Results of Operations.” The occurrence of any of the events or developments described below could harm our business, financial condition, results of operations and growth prospects. In such an event, the market price of our common stock could decline and you may lose all or part of your investment. Additional risks and uncertainties not presently known to us or that we currently deem immaterial also may impair our business operations and the market price of our common stock.
Risk Factor Summary
TheThis summary of risks below provides an overview of the principal risks we are exposed to. These risks are described more fully in the section entitled “Risk Factors” in this Form 10-K.
Risks Related to Our Business, Financial Condition and Capital Requirements
•We are in the earlyclinical stages of clinical drug development and have a very limited operating history and no products approved for commercial sale.sale, which may make it difficult to evaluate our current business and predict our future success and viability.
•We have incurred significant net losses since our inception and anticipate that we will continue to incur net losses for the foreseeable future.
•A pandemic, epidemic or outbreak of an infectious disease, such as COVID-19, or the perception of its effects, may materially and adversely affect our business, operations and financial condition.
•Drug development is a highly uncertain undertaking. We have never generated any revenue from product sales, and may never do so.
•Due to the significant resources required for the development of our programs, and depending on our ability to access capital, we must prioritize development of certain product candidates.
Risks Related to the Discovery, Development and Commercialization of Our Product Candidates
•We are heavily dependent on the successful development of our BBB technology and the programs currently in our pipeline, which are in the early stages of development. There’s no assurance that any of our product candidates will receive regulatory approval.preclinical and clinical development stages.
•We may not be successful in our efforts to continue to create a pipeline of product candidates or to develop commercially successful products.
•We have concentrated a substantial portion of our efforts on the treatment of neurodegenerative diseases, a fieldand LSDs, fields that hashave seen limited success in drug development.
•We may encounter substantial delays in our clinical trials, or may not be able to conduct or complete our clinical trials on the timelines we expect, if at all.
•We may encounter difficulties enrolling and/or retaining patients in our clinical trials, and our clinical development activities could thereby be delayed or otherwise adversely affected.
•Our clinical trials may reveal significant adverse events, toxicities, or other side effects and may fail to demonstrate substantial evidence of the safety and efficacy or potency of our product candidates, which would prevent, delay, or limit the scope of regulatory approval and commercialization.
•We face significant competition and there is a possibility that our competitorsoperating results may achieve regulatory approval before us or develop therapies that are safer or more effective than ours.suffer if we fail to compete effectively.
•The manufacture of our product candidates, particularly those that utilize our BBB platform technology, is complex and we may encounter difficulties in production.
•If we are unable to establish sales and marketing capabilities or enter into agreements with third parties, we may not be successful in commercializing product candidates if and when they are approved.
•If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to limit commercialization of our product candidates.
Risks Related to Regulatory Approval and Other Legal Compliance Matters
•The regulatory approval processes of the FDA, EMA and comparable foreign regulatory authorities are lengthy, time consuming, and inherently unpredictable. If we are ultimately unable to obtain regulatory approval for our product candidates, we will be unable to generate product revenue.
•We may in the futurecurrently conduct clinical trials outside the United States, and the FDA, EMA and applicable foreign regulatory authorities may not accept data from such trials.
•Healthcare legislative measures aimed at reducing healthcare costs may have a material adverse effect on our business and results of operations.
•Our business is subject to complex and evolving U.S. and foreign laws and regulations, information security policies, and contractual obligations relating to privacy and data protection.
Risks Related to Our Reliance on Third Parties
•We expect to depend on collaborations with third parties for the research, development, and commercialization of certain product candidates. If any such collaborations are not successful, we may not be able to realize the market potential of those product candidates.
•We expect to rely on third parties to conduct our clinical trials and some aspects of our research and preclinical testing, and those third parties may not perform satisfactorily.
•We contract withOur reliance on third parties for the manufacture of the significant majority of the materials for our research programs, preclinical studies, and clinical trials. This reliance on third partiestrials may increase the risk that we will not have sufficient quantities of such materials or product candidates.
•We depend on third-party suppliers for key raw materials used in our manufacturing, and the loss of these suppliers or their inability to supply us with adequate raw materials could harm our business.
Risks Related to Our Intellectual Property
•If we are unable to obtain and maintain patent protection for our product candidates or our BBB technology, our competitors could develop and commercialize products or technology similar or identical to ours, and adversely affect our ability to commercialize any product candidates.
•If any of our owned or in-licensed patent applications do not issue as patents in any jurisdiction, we may not be able to compete effectively.
•Our rights to develop and commercialize our BBB technology and product candidates are subject, in part, to the terms of licenses granted to us by others or licenses granted by us to others.
•We may not be able to protect our intellectual property and proprietary rights throughout the world.
•Our patent protection could be reduced or eliminated if we are unable to comply with requirements imposed by government patent agencies.
•Changes in U.S. patent law could impair our ability to protect our products.
•Issued patents covering our BBB technology, product candidates and other technologies could be found invalid or unenforceable if challenged.
•Patent terms may be inadequate to protect our competitive position on our product candidates for an adequate amount of time.
•We may be subject to claims challenging the inventorship of our intellectual property.
•If we are unable to protect the confidentiality of our trade secrets, our business would be harmed.
•We may not be successful in obtaining, through acquisitions, in-licenses or otherwise, necessary rights to our BBB platform technology, product candidates or other technologies.
•We may be subject to claims that our employees, consultants, or advisors have wrongfully used or disclosed alleged trade secrets of their current or former employers.
•Third party intellectual property claims against us, our licensors or our collaborators may prevent or delay the development of our BBB platform technology, product candidates and other technologies.
Risks Related to Our Operations
•If we are not successful in attracting, motivating and retaining highly qualified personnel, we may not be able to successfully implement our business strategy.
•We have engaged in and may in the future engage in acquisitions or strategic partnerships, which may increase our capital requirements, dilute our stockholders, or cause us to incur debt or assume contingent liabilities.
•The proposed spin out of our preclinical small molecule portfolio is subject to various risks and uncertainties or may not result in the expected benefits.
•Our internal computer systems, or those used by our collaborators, CROs or other contractors, may fail or suffer security breaches or incidents that could compromise the confidentiality, integrity, and availability of such systems and data, and expose us to liability.liability, and affect our reputation.
•Our business is subject to risks associated with international operations.
Risks Related to Ownership of Our Common Stock
•The market price of our common stock has been and may continue to be volatile, which could result in substantial losses for investors.
•If securities analysts publish negative evaluations of our stock, or if they do not publish research or reports about our business or if they publish negative evaluations of our stock,business; the price of our stock and trading volume could decline.
•Raising additional capital may cause dilution to our existing stockholders, restrict our operations or require us to relinquish rights to our technologies or product candidates.
•Delaware law and provisions in our charter documents might prevent a change in control of our company or changes in our management, depressing the trading price of our common stock.
•Our amended and restated certificate of incorporation provides exclusive forums for disputes between us and our stockholders, limiting their ability to obtain a favorable judicial forum.
Risks Related to Our Business, Financial Condition and Capital Requirements
We are in the earlyclinical stages of clinical drug development and have a very limited operating history and no products approved for commercial sale, which may make it difficult to evaluate our current business and predict our future success and viability.
We are an earlya clinical-stage biopharmaceutical company with a very limited operating history, focused on developing therapeutics for neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease and ALS.ALS, and LSDs, including Hunter syndrome and Sanfilippo syndrome. We commenced operations in May 2015, have no products approved for commercial sale and have not generated any revenue from product sales. Drug development is a highly uncertain undertaking and involves a substantial degree of risk. WeOur clinical-stage programs are in various phases ranging from Phase 1 1b or 1/2 clinical trials for our LRRK2, EIF2B, ETV:IDS and RIPK1 programs and have not initiated clinical trials for any of our other current product candidates.through Phase 3. To date, we have not initiated or completed a pivotal clinical trial, obtained marketing approval for any product candidates, manufactured a commercial scale product or arranged for a third party to do so on our behalf, or conducted sales and marketing activities necessary for successful product commercialization. Our shortlimited operating history as a company makes any assessment of our future success and viability subject to significant uncertainty. We will encounter risks and difficulties frequently experienced by early-stageclinical-stage biopharmaceutical companies, in rapidly evolving fields, and we have not yet demonstrated an ability to successfully overcome such risks and difficulties. If we do not address these risks and difficulties successfully, our business will suffer.
We have incurred significant net losses since our inception and anticipate that we will continue to incur net losses for the foreseeable future.
We have incurred significant net losses in every year since our inception until the year ended December 31, 2020, includinginception. Our net losses $197.6were $145.2 million, $326.0 million, and $36.2$290.6 million for the years ended December 31, 20192023, 2022, and 2018,2021, respectively. We had net income of $71.1 million for the year ended December 31, 2020, as a result of revenue recognized associated with our collaboration arrangement with Biogen. As of December 31, 2020,2023, we had an accumulated deficit of $354.4 million.$1.12 billion.
We have invested significant financial resources in research and development activities, including for our preclinical and clinical product candidates and our TV platform. We do not expect to generate revenue from product sales for several years, if at all. The amount of our future net losses will depend, in part, on the level of our future expenditures and revenue. Moreover, our net losses may fluctuate significantly from quarter to quarter and year to year, such that a period-to-period comparison of our results of operations may not be a good indication of our future performance.
We expect to continue to incur significant expenses and increasingly higher operating losses for the foreseeable future. We anticipate that our expenses will increase substantially if and as we:
•continue our research and discovery activities;
•progress our current and any future product candidates through preclinical and clinical development;
•initiate and conduct additional preclinical, clinical, or other studies for our product candidates;
•work with our contract manufacturers to scale up the manufacturing processes for our product candidates or, in the future, establish and operate a manufacturing facility;
•change or add additional contract manufacturers or suppliers;
•seek regulatory approvals and marketing authorizations for our product candidates;
•establish sales, marketing and distribution infrastructure to commercialize any products for which we obtain approval;
•acquire or in-license product candidates, intellectual property, and technologies;
•make milestone, royalty, or other payments due under any license or collaboration agreements;
•obtain, maintain, protect, and enforce our intellectual property portfolio, including intellectual property obtained through license agreements;
•attract, hire, and retain qualified personnel;personnel and incur increased stock-based compensation, especially in light of a competitive compensation environment;
•provide additional internal infrastructure to support our continued research and development operations and any planned commercialization efforts in the future;
•implement additional internal systems and infrastructure related to cybersecurity;
•experience any delays or encounter other issues related to our operations;
•meet the requirements and demands of being a public company; and
•defend against any product liability claims or other lawsuits related to our products.products; and
•build clinical manufacturing capabilities and capacity.
Our prior losses and expected future losses have had and will continue to have an adverse effect on our stockholders’ equity and working capital. In any particular quarter or quarters, our operating results could be below the expectations of securities analysts or investors, which could cause our stock price to decline.
A pandemic, epidemic or outbreak of an infectious disease, such as COVID-19, or the perception of its effects, may materially and adversely affect our business, operations and financial condition.
Public health outbreaks, such as epidemics or pandemics involving infectious or contagious diseases, such as COVID-19, may significantly disrupt our business. Such outbreaks pose the risk that we or our employees, contractors, suppliers, and other partners may be prevented from conducting business activities for an indefinite period of time due to the spread of the disease, due to shutdowns that may be requested or mandated by federal, state and local governmental authorities or certain employers, or due to the economic consequences associated with the pandemic. Business disruptions could include disruptions or restrictions on our ability to travel, as well as temporary closures of our facilities and the facilities of our partners, clinical trial sites, service providers, suppliers or contract manufacturers. While it is not possible at this time to estimate the overall impact that the COVID-19 pandemic could have on our business, the continued rapid spread of COVID-19, both across the United States and throughout much of the world, and the measures taken by the governments of countries and local authorities affected has disrupted and could delay the initiation of new clinical trials, the progress of our ongoing clinical trials, and could disrupt and delay our preclinical activities, and potentially the manufacture or shipment of both drug substance and finished drug product of our product candidates for preclinical testing and clinical trials and adversely impact our business, financial condition or operating results.
For example, as a result of the COVID-19 pandemic the state of California, where our corporate offices are located, has issued orders limiting activities to varying levels, including at the most restrictive level, an order for all residents to remain at home, except for the performance of essential activities, which include biomedical research. We have implemented policies that enable some of our employees to work remotely, and such policies may continue for an indefinite period. We have also implemented various safety protocols for all on-site personnel, including regular, mandatory COVID-19 testing procedures and compliance measures for social distancing and use of personal protective equipment. Our priority is to protect the health and safety of our employees, community, partners and clinical trial participants, while working to ensure the sustainability of our business operations as this unprecedented situation continues to evolve.We continue to evaluate the impact COVID-19 may have on our ability to effectively conduct our business operations as planned, and work with healthcare providers supporting our clinical studies to mitigate risk to patients while taking into account regulatory, institutional, and government guidance and policies, but there can be no assurance that we will be able to avoid part or all of any impact from the spread of COVID-19 or its consequences.
As the COVID-19 pandemic continues to spread around the globe, we have experienced and may continue to experience disruptions that could severely impact our business, preclinical programs and clinical trials, including:
•delays or difficulties in enrolling patients in our clinical trials, particularly elderly subjects, who are at a higher risk of severe illness or death from COVID-19, which can be further complicated by the presence of comorbidities that are often present in subjects with neurodegenerative diseases;
•difficulties interpreting data from our clinical trials due to the possible effects of COVID-19 on subjects enrolled in our clinical trials that contract COVID-19;
•delays or difficulties in clinical site initiation, including difficulties in recruiting clinical site investigators and clinical site staff;
•diversion of healthcare resources away from the conduct of clinical trials, including the diversion of hospitals serving as our clinical trial sites and hospital staff supporting the conduct of clinical trials;
•interruption of key clinical trial activities, such as clinical trial site monitoring, due to limitations on travel imposed or recommended by federal or state governments, employers and others; limitations in resources that would otherwise be focused on the conduct of our business or our clinical trials, including because of sickness or the desire to avoid contact with large groups of people or as a result of government-imposed “shelter in place” or similar working restrictions;
•delays in receiving approval from local regulatory authorities to initiate our planned clinical trials;
•interruption of, or delays in receiving, supplies of our product candidates from our contract manufacturing organizations due to staffing shortages, production slowdowns or stoppages and disruptions in delivery systems, as well as delays in clinical sites receiving the supplies and materials needed to conduct our clinical trials;
•delays or difficulties in furthering our preclinical and clinical programs, due to interruptions or limitations in our third party service providers’ business operations;
•interruption in global shipping that may affect the transport of clinical trial materials, such as investigational drug product used in our clinical trials;
•changes in clinical trial site procedures and requirements as well as regulatory requirements for conducting clinical trials during the COVID-19 pandemic which may require us to change the ways in which our clinical trials are conducted, implement additional procedures and policies, or to discontinue the clinical trials altogether, or which may result in unexpected costs and delays;
•delays or interruptions in the operations of or necessary interactions with the U.S. Food and Drug Administration ("FDA") or other regulators, ECs and other important agencies and contractors due to limitations in employee resources or forced furlough of government or contractor personnel, which may impact review and approval timelines and result in unexpected costs and delays;
•refusal of the FDA to accept data from clinical trials in affected geographies outside the United States;
•limitations on employee resources that would otherwise be focused on the conduct of our nonclinical studies and clinical trials, either because of sickness of employees and their families or the desire of employees to avoid contact with large groups of people; and
•negative impact on our ability to raise capital.
We have clinical trial sites for our clinical studies in the United States and Europe, certain of which have been affected by the COVID-19 pandemic due to prioritization of hospital resources toward the COVID-19 outbreak, travel or quarantine restrictions imposed by federal, state or local governments, and the inability to access sites for initiation and patient monitoring and enrollment. Healthcare facilities and offices may be required to focus limited resources on non-clinical trial matters, including treatment of COVID-19 patients, and may not be available, in whole or in part, for clinical trial services related to our clinical trials. As a result, patient screening, new patient enrollment, monitoring and data collection may be affected. We experienced a pause in enrollment in our DNL151 (BIIB122) Phase 1 and Phase 1b trials, our DNL343 Phase 1 trial, and our ETV:IDS program observational biomarker study. In all cases, recruitment has resumed. However, further pauses or delays could occur depending on the progression of the pandemic. The completion of such trials could be delayed as a result.
We may be required to develop and implement additional clinical trial policies and procedures designed to help protect subjects from the COVID-19 virus. For example, in March 2020, the FDA issued a guidance, which the FDA subsequently updated, on conducting clinical trials during the pandemic, which describes a number of considerations for sponsors of clinical trials impacted by the pandemic, including the requirement to include in the clinical trial report contingency measures implemented to manage the clinical trial, and any disruption of the clinical trial as a result of the COVID-19 pandemic; a list of all subjects affected by the COVID-19-pandemic related study disruption by unique subject identifier and by investigational site and a description of how the individual’s participation was altered; and analyses and corresponding discussions that address the impact of implemented contingency measures (e.g., participant discontinuation from investigational product and/or study, alternative procedures used to collect critical safety and/or efficacy data) on the safety and efficacy results reported for the clinical trial. In June 2020, FDA also issued a guidance on good manufacturing practice considerations for responding to COVID-19 infection in employees in drug and biologic products manufacturing, including recommendations for manufacturing controls to prevent contamination of the products.
Some third-party manufacturers that supply us materials for product candidates or other materials necessary to manufacture product to conduct preclinical tests and clinical trials are located in countries affected by COVID-19, and we may experience delays in advancing these tests and trials, if our third-party manufacturers experience disruptions such as temporary closures or suspension of services. The spread of an infectious disease, including COVID-19, may also result in the inability of our suppliers to deliver components or raw materials on a timely basis or at all. Currently, we do not expect delays to our clinical trials due to manufacturing or supply-chain issues, and we believe we have sufficient drug supplies to complete ongoing trials as well as additional drug substance supplies expected to be sufficient to support planned clinical trials well into 2021. Furthermore, the spread of the virus may affect the operations of key governmental agencies, such as the FDA, which may delay the development of our product candidates. Such events may result in a period of business disruption, and in reduced operations, or doctors and medical providers may be unwilling to participate in our clinical trials, any of which could materially affect our business, operations and financial condition.
The COVID-19 pandemic continues to rapidly evolve. The extent to which COVID-19 impacts our business will depend on future developments, which are highly uncertain and cannot be predicted, such as the ultimate geographic spread of the disease, the duration of the pandemic, travel restrictions and social distancing in the United States and other countries, business closures or business disruptions and the effectiveness of actions taken in the United States and other countries to contain and treat the disease and to address its impact, including on financial markets or otherwise. While the extent of the impact of the COVID-19 pandemic on our business and financial results is uncertain, a continued and prolonged public health crisis could have a material negative impact on our business, operations and financial condition.
To the extent the COVID-19 pandemic adversely affects our business, operations and financial condition, it may also have the effect of heightening many of the risks described in this “Risk Factors” section.
Drug development is a highly uncertain undertaking and involves a substantial degree of risk. We have never generated any revenue from product sales, and we may never generate product revenue or be profitable.
We have no products approved for commercial sale and have not generated any revenue from product sales. To obtain revenue from the sales of our product candidates that are significant or large enough to achieve profitability, we must succeed, either alone or with third parties, in developing, obtaining regulatory approval for, manufacturing and marketing therapies with significant commercial success.
Our ability to generate revenue and achieve profitability depends significantly on many factors, including:
•successfully prioritizing and completing research and preclinical and clinical development of our product candidates;
•obtaining regulatory approvals and marketing authorizations for product candidates for which we successfully complete clinical development and clinical trials;
•developing a sustainable and scalable manufacturing process for our product candidates, including those that utilize our TV platform, as well as establishing and maintaining commercially viable supply relationships with third parties that can provide adequate products and services to support clinical activities and commercial demand of our product candidates;
•identifying, assessing, acquiring, and/or developing new product candidates;
•negotiating favorable terms in any collaboration, licensing, or other arrangements into which we may enter;
•launching and successfully commercializing product candidates for which we obtain regulatory and marketing approval, either by collaborating with a partner or, if launched independently, by establishing a sales, marketing, and distribution infrastructure;
•obtaining and maintaining an adequate price for our product candidates, both in the United States and in foreign countries where our products are commercialized;
•obtaining adequate reimbursement for our product candidates from payors;
•obtaining market acceptance of our product candidates as viable treatment options;
•addressing any competing technological and market developments;
•receiving milestone and other payments under our current and any future collaboration arrangements;
•maintaining, protecting, expanding, and enforcing our portfolio of intellectual property rights, including patents, trade secrets and know-how; andrights;
•attracting, hiring, and retaining qualified personnel.personnel;
•general economic conditions, including conditions resulting from rising inflation and interest rates, recent bank failures and instability in the financial services sector, geopolitical uncertainty and instability or war; and
•addressing any delays in our clinical trials or other impacts from a pandemic or other global health emergency.
Because of the numerous risks and uncertainties associated with drug development, we are unable to predict the timing or amount of our expenses, or when we will be able to generate any meaningful revenue or achieve or maintain profitability, if ever. In addition, our expenses could increase beyond our current expectations if we are required by the FDA, or foreign regulatory agencies, to perform studies in addition to those that we currently anticipate, or if there are any delays in any of our current or our future collaborators’ clinical trials or the development of any of our product candidates. Even if one or more of our product candidates is approved for commercial sale, we anticipate incurring significant costs associated with commercializing any approved product candidate and ongoing compliance efforts.
Even if we are able to generate revenue from the sale of any approved products, we may not become profitable and may need to obtain additional funding to continue operations. Revenue from the sale of any product candidate for which regulatory approval is obtained will be dependent, in part, upon the size of the markets in the territories for which we gain regulatory approval, the accepted price for the product, the ability to get reimbursement at any price, and whether we own the commercial rights for that territory. If the number of addressable patients is not as significant as we anticipate, the indication approved by regulatory authorities is narrower than we expect, or the reasonably accepted population for treatment is narrowed by competition, physician choice, or treatment guidelines, we may not generate significant revenue from sales of such products, even if approved. Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis.
Our failure to become and remain profitable would decrease the value of our company and could impair our ability to raise capital, expand our business, maintain our research and development efforts, diversify our pipeline of product candidates, or continue our operations and cause a decline in the value of our common stock, all or any of which may adversely affect our viability.
If we fail to obtain additional financing, we may be unable to complete the development and, if approved, commercialization of our product candidates.
Our operations have required substantial amounts of cash since inception. We currently fund our operations primarily with the proceeds from our initial public offering ("IPO"), our follow-on offeringofferings completed in January 2020 and October 2022, and payments received from our Takeda Collaboration Agreement,collaboration agreements with Biogen, Sanofi, Collaboration Agreement and Biogen Collaboration Agreement.Takeda. We are currently advancing three product candidates, DNL151 (BIIB122), DNL343, have a diversified portfolio with numerous programs at various stages of research, discovery, preclinical and DNL310 through clinical development, and have several other product candidates in preclinical development, as well as early-stage research projects.development. Developing our product candidates is expensive, and we expect to continue to spend substantial amounts as we fund our early-stage research projects, and continue to advance our programs through preclinical and clinical development. Even if we are successful in developing our product candidates, obtaining regulatory approvals and launching and commercializing any product candidate will require substantial additional funding.
As of December 31, 2020,2023, we had $1.5$1.03 billion in cash, cash equivalents, and marketable securities. We believe that our existing cash, cash equivalents, and marketable securities will be sufficient to fund our projected operations through at least the next 12twelve months. Our estimate as to how long we expect our existing cash, cash equivalents, and marketable securities to be available to fund our operations is based on assumptions that may be provedproven inaccurate, and we could use our available capital resources sooner than we currently expect. In addition, changingChanging circumstances, some of which may be beyond our control, such as recent bank failures, geopolitical uncertainty, rising inflation or interest rates, or a perceived or actual economic downturn, may cause us to increase our spending significantly faster than we currently anticipate, and we may need to spend more moneyseek additional funds sooner than currently expected because of circumstances beyond our control.planned. We may also need to raise additional funds sooner than we anticipate if we choose to expand more rapidly than we presently anticipate.
We will requirecannot be certain that additional capital for the further development and, if approved, commercialization of our product candidates. Additional capital may notfunding will be available when we need it, on terms acceptable to us or at all. WeOther than the PIPE financing that was announced on February 27, 2024, we have no committed source of additional capital. If adequate capital is not available to us on a timely basis, we may be required to significantly delay, scale back, or discontinue our research and development programs or the commercialization of any product candidates, if approved, or be unable to continue or expand our operations or otherwise capitalize on our business opportunities, as desired, which could materially affect our business, financial condition, results of operations, and growth prospects and cause the price of our common stock to decline.
Due to the significant resources required for the development of our programs, and depending on our ability to access capital, we must prioritize development of certain product candidates. Moreover, we may expend our limited resources on programs that do not yield a successful product candidate and fail to capitalize on product candidates or indications that may be more profitable or for which there is a greater likelihood of success.
We have a diversified portfolio with more than fifteen programs. Thesenumerous programs require significant capital investment. Our programs are at various stages of research, discovery, preclinical and early clinical development. These programs require significant capital investment. We seek to maintain a process of prioritization and resource allocation to maintain an optimal balance between aggressively advancing lead programs and ensuring replenishment ofreplenishing our portfolio. We regularly review the programs in our portfolio, and terminate those programs which do not meet our development criteria, which we have done a number of times in the past.
Due to the significant resources required for the development of our programs, we must focus our programs on specific diseases and disease pathways and decide which product candidates to pursue and advance and the amount of resources to allocate to each. Our decisions concerning the allocation of research, development, collaboration, management, and financial resources toward particular product candidates or therapeutic areas may not lead to the development of any viable commercial product and may divert resources away from better opportunities. Similarly, our potential decisions to delay, terminate, spin out, or collaborate with third parties in respect of certain programs may subsequently also prove to be suboptimal and could cause us to miss valuable opportunities. If we make incorrect determinations regarding the viability or market potential of any of our programs or product candidates or misread trends in the biopharmaceutical industry, in particular for neurodegenerative diseases,and LSDs, our business, financial condition, results of operations and growth prospects could be materially adversely affected. As a result, we may fail to capitalize on viable commercial products or profitable market opportunities, be required to foregoforgo or delay pursuit of opportunities with other product candidates or other diseases and disease pathways that may later prove to have greater commercial potential than those we choose to pursue, or relinquish valuable rights to such product candidates through collaboration, licensing or other royalty arrangements in cases in which it would have been advantageous for us to invest additional resources to retain sole development and commercialization rights.
A pandemic, epidemic or outbreak of an infectious disease, such as COVID-19, or the perception of its effects, may materially and adversely affect our business, operations and financial condition.
Public health outbreaks, such as epidemics or pandemics may significantly disrupt our business. Such outbreaks pose the risk that we or our employees, contractors, suppliers, and other partners may be prevented from conducting business activities for an indefinite period of time due to the spread of the disease, due to shutdowns that may be requested or mandated by federal, state, and local governmental authorities or certain employers, or due to the economic consequences associated with the pandemic. Business disruptions could include disruptions or restrictions on our ability to travel, as well as temporary closures of our facilities and the facilities of our partners, clinical trial sites, service providers, suppliers, or contract manufacturers. For example, the COVID-19 pandemic caused a temporary disruption in our ability to recruit participants for our clinical trials in the calendar year 2020 and the first quarter of 2021. While it is not possible to predict whether another pandemic, epidemic, or infectious disease outbreak similar to COVID-19 will materialize, any measures taken by governments and local authorities in response to such future health crises have the potential to disrupt and delay the initiation of new clinical trials, the progress of our ongoing clinical trials and our preclinical activities, and potentially the manufacture or shipment of both drug substance and finished drug product of our product candidates for preclinical testing and clinical trials, as well as adversely impact our business, financial condition, or operating results.
The continued impact of the COVID-19 pandemic may materially and adversely affect our business, operations and financial condition.
On May 11, 2023, the federal government ended the COVID-19 public health emergency, which ended a number of temporary changes made to federally funded programs, while some remain in effect. The full impact of the termination of the public health emergency on the FDA and other regulatory policies and operations remains unclear. In response to the COVID-19 pandemic, we implemented policies that enabled some of our employees to work remotely, which policies may continue for an indefinite period. Due to telecommuting patterns, modified work schedules, and enhanced safety protocols, our laboratory operations have at times and may again operate with decreased efficiency. Furthermore, our clinical trial sites for our clinical studies were impacted by the COVID-19 pandemic: in 2020, we experienced a pause in enrollment in our BIIB122/DNL151 Phase 1 and Phase 1b trials, our DNL343 Phase 1 and Phase 2/3 trials, and our ETV:IDS program observational biomarker study, and we have subsequently experienced certain delays in patient enrollment.
The FDA issued a number of COVID-19 related guidance documents for manufacturers and clinical trial sponsors in 2020 and 2021, many of which have expired or were withdrawn with the expiration of the COVID-19 public health emergency in May 2023, although some COVID-19 related guidance documents remain in effect. Should the FDA issue additional guidance that mandates material changes to our clinical trials in response to a pandemic or other public health outbreaks, the costs of such clinical trials may increase. To the extent we experience any ongoing pandemic disruptions or other public health emergencies, including a resurgence of COVID-19 cases, potential impacts to our business may include delays or difficulties in enrolling patients, difficulties interpreting data impacted by trial disruptions, supply chain issues, staffing shortages, and disruptions to the operations of our service providers, any of which could have a material adverse effect on our business and clinical development plans.
To the extent another pandemic or other public health outbreak adversely affects our business, operations and financial condition in the future, it may also have the effect of heightening many of the risks described in this “Risk Factors” section.
Risks Related to the Discovery, Development, and Commercialization of Our Product Candidates
Research and development of biopharmaceutical products is inherently risky. We are heavily dependent on the successful development of our BBB platform technology and the programs currently in our pipeline, which are in the early stages of preclinical and clinical development.development stages. We cannot give any assurance that any of our product candidates will receive regulatory, including marketing approval, which is necessary before they can be commercialized.
We are at an early stage of development of many of the product candidates currently in our programs and are further developing our BBB platform technology. To date, we have invested substantially all of our efforts and financial resources to identify, acquire intellectual property for, and develop our BBB platform technology and our programs, including conducting preclinical studies and early-stage clinical trials, and providing general and administrative support for these operations. Our future success is dependent on our ability to successfully develop, obtain regulatory approval for, and then successfully commercialize our product candidates, and we may fail to do so for many reasons, including the following:
•our product candidates may not successfully complete preclinical studies or clinical trials;
•our drug delivery platform technology designed to deliver large molecule therapeutics across the BBB may not be clinically viable;
•a product candidate may on further study be shown to have harmful side effects or other characteristics that indicate it is unlikely to be effective or otherwise does not meet applicable regulatory criteria;
•our competitors may develop therapeutics that render our product candidates obsolete or less attractive;
•our competitors may develop platform technologies to deliver large molecule therapeutics across the BBB that render our platform technology obsolete or less attractive;
•the product candidates and BBB platform technology that we develop may not be sufficiently covered by intellectual property for which we hold exclusive rights;
•the product candidates and BBB platform technology that we develop may be covered by third parties’ patents or other intellectual property or exclusive rights;
•the market for a product candidate may change so that the continued development of that product candidate is no longer reasonable or commercially attractive;
•a product candidate may not be capable of being produced in commercial quantities at an acceptable cost, or at all;
•if a product candidate obtains regulatory approval, we may be unable to establish sales and marketing capabilities, or successfully market such approved product candidate, to gain market acceptance;candidate; and
•a product candidate may not be accepted as safe and effective by patients, the medical community, or third-party payors, if applicable.
If any of these events occur, we may be forced to abandon our development efforts for a program or programs, which wouldcould have a material adverse effect on our business and could potentially cause us to cease operations.business.
We may not be successful in our efforts to further develop our BBB platform technology and current product candidates. We are not permitted to market or promote any of our product candidates before we receive regulatory approval from the FDA or comparable foreign regulatory authorities, and we may never receive such regulatory approval for any of our product candidates. Each of ourOur product candidates isare in the early stages of development and will require significant additional clinical development, management of preclinical, clinical, and manufacturing activities, regulatory approval, adequate manufacturing supply, a commercial organization, and significant marketing efforts before we generate any revenue from product sales, if at all.
We have never completed a clinical development program. We have previously discontinued the development of certain molecules prior to completion of preclinical development because we did not believe they met our criteria for potential clinical success. None of our product candidates have advanced into late-stage development or a pivotal clinical trial and it may be years before any such trial is initiated, if at all. Further, we cannotcannot be certain that any of our product candidates will be successful in clinical trials. For instance, in 2016, we initiated a Phase 1 clinical trial in a former RIPK1 inhibitor product candidate, DNL104, which we subsequently discontinued based on liver test abnormalities in some clinical trial healthy volunteer participants. Further, in June 2020,August 2023, together with our collaboration partner Sanofi,Takeda, we paused clinical activities with DNL747 to acceleratediscontinued development of DNL788 (SAR443820),TAK-920/DNL919 (ATV:TREM2) in part dueAlzheimer’s disease, based on data from the Phase 1 study and the rapidly evolving treatment landscape and shifted our efforts to DNL747 preclinical chronic toxicity studies.exploring back-up molecules. We may in the future advance product candidates into clinical trials and terminate such trials prior to their completion.
If any of our product candidates successfully complete clinical trials, we generally plan to seek regulatory approval to market our product candidates in the United States, the EU, and in additional foreign countries where we believe there is a viable commercial opportunity. We have never commenced, compiled, or submitted an application seeking regulatory approval to market any product candidate. Wecandidate, and may never receive such regulatory approval to market any product candidates even if sucha product candidatescandidate successfully completecompletes clinical trials, which would adversely affect our viability. To obtain regulatory approval in countries outside the United States, we must comply with numerous and varying regulatory requirements of such other countries regarding safety, efficacy or potency, purity, chemistry, manufacturing and controls, clinical trials, commercial sales, pricing, and distribution of our product candidates. We may also rely on our collaborators or partners to conduct the required activities to support an application for regulatory approval, and to seek approval, for one or more of our product candidates. We cannot be sure that our collaborators or partners will conduct these activities or do so within the time frame we desire. Even if we (or our collaborators or partners) are successful in obtaining approval in one jurisdiction, we cannot ensure that we will obtain approval in any other jurisdictions. If we are unable to obtain approval for our product candidates in multiple jurisdictions, our revenue, business, financial condition, results of operations and growth prospects could be negatively affected.
Even if we receive regulatory approval to market any of our product candidates, whether for the treatment of neurodegenerative diseasesand LSDs or other diseases, we cannot assure you that any such product candidate will be successfully commercialized, widely accepted in the marketplace, or more effective than other commercially available alternatives.
Investment in biopharmaceutical product development involves significant risk that any product candidate will fail to demonstrate adequate efficacy or potency, or an acceptable safety profile, gain regulatory approval, and become commercially viable. We cannot provide any assurance that we will be able to successfully advance any of our product candidates through the development process or, if approved, successfully commercialize any of our product candidates.
We may not be successful in our efforts to continue to create a pipeline of product candidates or to develop commercially successful products. If we fail to successfully identify and develop additional product candidates, our commercial opportunity may be limited.
One of our strategies is to identify and pursue clinical development of additional product candidates. We currently have several programs in the research, discovery, and preclinical stages of development. Identifying, developing, obtaining regulatory approval for, and commercializing additional product candidates for the treatment of neurodegenerative diseasesand LSDs will require substantial additional funding and is prone to the risks of failure inherent in drug development. We cannot provide you any assurance that we will be able to successfully identify or acquire additional product candidates, advance any of these additional product candidates through the development process, successfully commercialize any such additional product candidates, if approved, or assemble sufficient resources to identify, acquire, develop or, if approved, commercialize additional product candidates. If we are unable to successfully identify, acquire, develop, and commercialize additional product candidates, our commercial opportunity may be limited.
We have concentrated a substantial portion of our research and development efforts on the treatment of neurodegenerative diseases, a fieldand LSDs, fields that hashave seen limited success in drug development. Further, our product candidates are based on new approaches and novel technology, which makes it difficult to predict the time and cost of product candidate development and subsequently obtaining regulatory approval.
We have focused our research and development efforts on addressing neurodegenerative diseases.and LSDs. Collectively, efforts by biopharmaceutical companies in the fieldfields of neurodegenerative diseasesand LSDs have seen limited success in drug development. There are few effective therapeutic options available for patients with neurodegenerative diseases, such as Alzheimer’s disease, Parkinson’s disease, and ALS, and other neurodegenerative diseases.LSDs, such as Hunter syndrome and Sanfilippo syndrome. Our future success is highly dependent on the successful development of our BBB platform technology and our product candidates for treating neurodegenerative diseases.and LSDs. Developing and, if approved, commercializing our product candidates for treatment of neurodegenerative diseasesand LSDs subjects us to a number of challenges, including engineering product candidates to cross the BBB to enable optimal concentration of the therapeutic in the brain and obtaining regulatory approval from the FDA and other regulatory authorities who have only a limited set of precedents to rely on.
Our approach to the treatment of neurodegenerative diseasesand LSDs aims to identify and select targets with a genetic link to neurodegenerative diseases,and LSDs, as applicable, identify and develop molecules that engage the intended target, identify and develop biomarkers, which are biological molecules found in blood, other bodily fluids or tissues that are signs of a normal or abnormal process or of a condition or disease, to select the right patient population and demonstrate target engagement, pathway engagement and impact on disease progression of our molecules, and engineer our molecules to cross the BBB and act directly in the brain. This strategy may not prove to be successful. We may not be able to discover, develop, and utilize biomarkers to demonstrate target engagement, pathway engagement, and the impact on disease progression of our molecules. We cannot be sure that our approach will yield satisfactory therapeutic products that are safe and effective, scalable, or profitable. Moreover, public perception of drug safety issues, including adoption of new therapeutics or novel approaches to treatment, may adversely influence the willingness of subjects to participate in clinical trials, or if approved, of physicians to subscribe to novel treatments.
We may encounter substantial delays in our clinical trials, or may not be able to conduct or complete our clinical trials on the timelines we expect, if at all.
Clinical testing is expensive, time consuming, and subject to uncertainty. We cannot guarantee that any clinical trials will be conducted as planned or completed on schedule, if at all. We cannot be sure that submission of an IND, or a CTA,clinical trial application ("CTA"), will result in the FDA or EMA, as applicable, allowing clinical trials to begin in a timely manner, if at all. Moreover, even if these trials begin, issues may arise that could suspend or terminate such clinical trials. A failure of one or more clinical trials can occur at any stage of testing, and our future clinical trials may not be successful. Events that may prevent successful or timely initiation or completion of clinical trials include:
•inability to generate sufficient preclinical, toxicology, or other in vivo or in vitro data to support the initiation or continuation of clinical trials;
•delays in confirming target engagement, patient selection, or other relevant biomarkers to be utilized in preclinical and clinical product candidate development;
•delays in reaching a consensus with regulatory agencies on trial design;
•delays in reaching agreement on acceptable terms with prospective CROs and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and clinical trial sites;
•delays in identifying, recruiting and training suitable clinical investigators;
•delays in obtaining required IRB approval at each clinical trial site;
•imposition of a temporary or permanent clinical hold by regulatory agencies for a number of reasons, including after review of an IND or amendment, CTA or amendment, or equivalent application or amendment; as a result of a new safety finding that presents unreasonable risk to clinical trial participants; a negative finding from an inspection of our clinical trial operations or trial sites; developments on trials conducted by competitors for related technology that raises FDA or EMA concerns about risk to patients of the technology broadly; or if the FDA or EMA finds that the investigational protocol or plan is clearly deficient to meet its stated objectives;
•delays in identifying, recruiting, and enrolling suitable patients to participate in our clinical trials, and delays caused by patients withdrawing from clinical trials or failing to return for post-treatment follow-up;
•difficulty collaborating with patient groups and investigators;
•failure by our CROs, other third parties, or us to adhere to clinical trial requirements;
•failure to perform in accordance with the FDA’s or any other regulatory authority’s current good clinical practices ("cGCPs") requirements, or applicable EMA or other regulatory guidelines in other countries;
•occurrence of adverse events associated with the product candidate that are viewed to outweigh its potential benefits;
•changes in regulatory requirements and guidance that require amending or submitting new clinical protocols;
•changes in the approval policies or regulations of the FDA or other regulatory authorities;
•changes in the standard of care on which a clinical development plan was based, which may require new or additional trials;
•the cost of clinical trials of our product candidates being greater than we anticipate;
•clinical trials of our product candidates producing negative or inconclusive results, which may result in us or our collaborators deciding, or regulators requiring us, to conduct additional clinical trials or abandon product development programs;
•transfer of manufacturing processes from our academic collaborators to larger-scale facilities operated by a CDMO or by us, and delays or failure by our CDMOs or us to make any necessary changes to such manufacturing process;
•delays in manufacturing, testing, releasing, validating, or importing/exporting sufficient stable quantities of our product candidates for use in clinical trials or the inability to do any of the foregoing; and
•delays associated with the COVID-19 global pandemic.a pandemic or other public health emergency.
Any inability to successfully initiate or complete clinical trials could result in additional costs to us or impair our ability to generate revenue. In addition, if we make manufacturing or formulation changes to our product candidates, we or our collaborators may be required to or we may elect to conduct additional studies to bridge our modified product candidates to earlier versions. Clinical trial delays could also shorten any periods during which our products have patent protection and may allow our competitors to bring products to market before we do, which could impair our ability to successfully commercialize our product candidates and may harm our business and results of operations.
We could also encounter delays if a clinical trial is suspended or terminated by us or our collaborators, by the data safety monitoring board for such trial, or by the FDA, EMA or any other regulatory authority, or if the IRBs of the institutions in which such trials are being conducted suspend or terminate the participation of their clinical investigators and sites subject to their review. Such authorities may suspend or terminate a clinical trial due to a number of factors, including failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols, inspection of the clinical trial operations or trial site by the FDA, EMA, or other regulatory authorities resulting in the imposition of a clinical hold, unforeseen safety issues or adverse side effects, failure to demonstrate a benefit from using a product candidate, changes in governmental regulations or administrative actions, or lack of adequate funding to continue the clinical trial.
DNL201, a former LRRK2 inhibitor product candidate, completed a Phase 1b clinical trialFor example, in Parkinson's disease patients with and withoutJanuary 2022, we announced that the genetic LRRK2 mutation. This program was previously subject to a partialTAK-920/DNL919 (ATV:TREM2) IND application had been placed on clinical hold due to preclinical toxicity data. The partial clinical hold was removed in December 2017 based on additional clinical and preclinical data provided toby the FDA. In our completed Phase 1bAugust 2023 we announced that, in agreement with Takeda, we would discontinue clinical trialdevelopment of DNL201TAK-920/DNL919 in patients with Parkinson’s disease, there was one SAE considered unrelated to drug, and at the high dose, there was one severe AE (headache) leading to dose reduction and one study withdrawal (headache and nausea). Our clinical-stage product candidates are DNL151 (BIIB122) for Parkinson's disease, DNL343 for ALS and FTD, and DNL310 for Hunter syndrome. In the nonclinical safety studies for these product candidates, toxicities were observed at high doses in rat and/or cynomolgus monkey above doses and exposures that will be tested in the clinic.Alzheimer’s disease. We cannot assure you that DNL151 (BIIB122), DNL343 and DNL310, orwe will ever resume the clinical program for TAK-920/DNL919, nor can we assure you that our other product candidates will not be subject to new, partial, or full clinical holds in the future.future, which may impact development plans.
We or our collaborators may in the future advance product candidates into clinical trials and terminate such trials prior to their completion, such as we did for DNL104, which could adversely affect our business. Further, after the commencement of clinical trials, we or our collaborators may discontinue advancement of lead molecules, such as the TAK-920/DNL919 program, or pause the advancement of lead molecules in favor of a backup molecule with a superior safety or efficacy profile, such as we did in our RIPK1 program, switching our focus from DNL747 to DNL788 (SAR443820).SAR443820/DNL788.
Delays in the completion of any clinical trial of our product candidates will increase our costs, slow down our product candidate development and approval process and delay or potentially jeopardize our ability to commence product sales and generate revenue. In addition, many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of our product candidates.
We may encounter difficulties enrolling and/or retaining patients in our clinical trials, and our clinical development activities could thereby be delayed or otherwise adversely affected.
The timely completion of clinical trials in accordance with their protocols depends, among other things, on our ability to enroll a sufficient number of patients who remain in the trial until its conclusion. We may experience difficulties in patient enrollment and retention in our clinical trials for a variety of reasons, including:
•inability or delay in enrollment of patients due to a variety of reasons, including outbreaks and public health crises, such as the COVID-19 global pandemic;
•the size and nature of the patient population;
•the patient eligibility criteria defined in the protocol, including biomarker-driven identification and/or certain highly-specific criteria related to stage of disease progression, which may limit the patient populations eligible for our clinical trials to a greater extent than competing clinical trials for the same indication that do not have biomarker-driven patient eligibility criteria;
•the size of the study population required for analysis of the trial’s primary endpoints;
•the proximity of patients to a trial site;
•the design of the trial;
•our ability to recruit clinical trial investigators with the appropriate competencies and experience;
•competing clinical trials for similar therapies or targeting patient populations meeting our patient eligibility criteria;
•clinicians’ and patients’ perceptions as to the potential advantages and side effects of the product candidate being studied in relation to other available therapies and product candidates;
•our ability to obtain and maintain patient consents; and
•the risk that patients enrolled in clinical trials will not complete such trials, for any reason, including the risk of higher drop-out rates if participants become infected with the COVID-19 virus or other infectious diseases that impact their participation in our trials.
Our inability to enroll and retain a sufficient number of patients for our clinical trials would result in significant delays or may require us to abandon one or more clinical trials altogether. Enrollment delays in our clinical trials may result in increased development costs for our product candidates and jeopardize our ability to obtain marketing approval for the sale of our product candidates. Furthermore, even if we are able to enroll a sufficient number of patients for our clinical trials, we may have difficulty maintaining participation in our clinical trials through the treatment and any follow-up periods, which could delay or negatively impact the anticipated readouts from our clinical trials, delay our regulatory submissions, and increase the costs of the clinical trials.
Our clinical trials may reveal significant adverse events, toxicities, or other side effects and may fail to demonstrate substantial evidence of the safety and efficacy or potency of our product candidates, which would prevent, delay, or limit the scope of regulatory approval and commercialization.
Before obtaining regulatory approvals for the commercial sale of any of our product candidates, we must demonstrate through lengthy, complex, and expensive preclinical studies and clinical trials that our product candidates are both safe and effective for use in each target indication. For those product candidates that are subject to regulation as biological drug products, we will need to demonstrate that they are safe, pure, and potent for use in their target indications. Each product candidate must demonstrate an adequate risk versus benefit profile in its intended patient population and for its intended use.
Clinical testing is expensive and can take many years to complete, and its outcome is inherently uncertain. Failure can occur at any time during the clinical trial process. The results of preclinical studies of our product candidates may not be predictive of the results of early-stage or later-stage clinical trials, and results of early clinical trials of our product candidates may not be predictive of the results of later-stage clinical trials. The results of clinical trials in one set of patients or disease indications may not be predictive of those obtained in another. In some instances, there can be significant variability in safety or efficacy or potency results between different clinical trials of the same product candidate due to numerous factors, including changes in trial procedures set forth in protocols, differences in the size and type of the patient populations, changes in and adherence to the dosing regimen and other clinical trial protocols and the rate of dropout among clinical trial participants. Open-label extension studies may also extend the timing and increase the cost of clinical development substantially. Product candidates in later stages of clinical trials may fail to show the desired safety and efficacy or potency profile despite having progressed through preclinical studies and initial clinical trials. A number of companies in the biopharmaceutical industry have suffered significant setbacks in advanced clinical trials due to lack of efficacy or potency or unacceptable safety issues, notwithstanding promising results in earlier trials. This is particularly true in neurodegenerative diseases,and LSDs, where failure rates historically have been higher than in many other disease areas. Most product candidates that begin clinical trials are never approved by regulatory authorities for commercialization.
We have limited experience in designing clinical trials and may be unable to design and execute a clinical trial to support marketing approval. We cannot be certain that our current clinical trials or any other future clinical trials will be successful. Additionally, any safety concerns observed in any one of our clinical trials in our targeted indications could limit the prospects for regulatory approval of our product candidates in those and other indications, which could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
In addition, evenEven if such clinical trials are successfully completed, we cannot guarantee that the FDA or foreign regulatory authorities will interpret the results as we do, or thatapprove the product candidates will be approved for the currently proposed indications, and more trials could be required before we submit our product candidates for approval. To the extent that the results of the trials are not satisfactory to the FDA or foreign regulatory authorities for support of a marketing application, we may be required to expend significant resources, which may not be available to us, or to conduct additional trials in support of potential approval of our product candidates. Even if regulatory approval is secured for any of our product candidates, the terms of such approval, such as requiring us to narrow our indications to a smaller subset, of patient population, may limit the scope and use of our product candidate, which may also limit its commercial potential.
Interim, topline, and preliminary data from our clinical trials that we announce or publish from time to time may change as more patient data become available, and are subject to audit and verification procedures that could result in material changes in the final data.
From time to time, we may publicly disclose preliminary, interim, or topline data from our nonclinical studies and clinical trials. These interim updatestrials, which are based on a preliminary analysis of then-available data, and the results and related findings and conclusions are subject to change following a more comprehensive review of the data related to the particular study or trial. We also make assumptions, estimations, calculations, and conclusions as part of our analyses of data, and we may not have received or had the opportunity to fully and carefully evaluate all data. As a result, the topline results that we report may differ from future results of the same studies, or different conclusions or considerations may qualify such results, once additional data have been received and fully evaluated. Topline data also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary data we previously published. As a result, preliminary, interim, or topline data should be viewed with caution until the final data are available. In addition, we may report interim analyses of only certain endpoints rather than all endpoints. Interim data from clinical trials that we may complete are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues and more patient data become available. Adverse changes between interim data and final data could significantly harm our business and prospects. Further, additional disclosure of interim data by us or by our competitors in the future could result in volatility in the price of our common stock.
Further, others, including our collaborators or regulatory agencies, may not accept or agree with our assumptions, estimates, calculations, conclusions, or analyses or may interpret or weigh the importance of data differently, which could impact the value of the particular program, the approvabilityapproval or commercialization of the particular product candidate and our company in general. In addition, the information we choose to publicly disclose regarding a particular study or clinical trial is typically selected from a more extensive amount of available information. You or others may not agree with what we determine is the material or otherwise appropriate information to include in our disclosure, and any information we determine not to disclose may ultimately be deemed significant with respect to future decisions, conclusions, views, activities or otherwise regarding a particular product candidate or our business. If the preliminary or topline data that we report differ from late, final, or actual results, or if others, including our collaborators or regulatory authorities, disagree with the conclusions reached, our ability to obtain approval for, and commercialize our product candidates may be harmed, which could harm our business, financial condition, results of operations and prospects.harmed.
We face significant competition in an environment of rapid technological and scientific change, and there is a possibility that our competitorsoperating results may achieve regulatory approval before us or develop therapies that are safer, more advanced or more effective than ours, which may negatively impact our abilitysuffer if we fail to successfully market or commercialize any product candidates we may develop and ultimately harm our financial condition.compete effectively.
The development and commercialization of new drug products is highly competitive. Moreover, the neurodegenerative field isand lysosomal storage fields are characterized by strong and increasing competition, and a strong emphasis on intellectual property. We may face competition with respect to any product candidates that we seek to develop or commercialize in the future from majorcompetition. Our potential competitors include pharmaceutical companies, specialty pharmaceutical companies, and biotechnology companies, worldwide. Potential competitors also include academic institutions, government agencies, and other public and private research organizations that conduct research, seekresearch. Our competitors, either alone or with collaborative partners, may succeed in developing, acquiring, or licensing on an exclusive basis drug or biologic products that are more effective, safer, more easily commercialized, or less costly than our product candidates or may develop proprietary technologies or secure patent protection that we may need for the development of our technologies and establish collaborative arrangements for research, development, manufacturing, and commercialization.products.
There are a
A number of large pharmaceutical and biotechnology companies that are currently pursuing the development ofdeveloping products for the treatment of the neurodegenerative diseaseand LSD indications for which we have research programs, including Alzheimer’s disease, Parkinson’s disease, Hunter syndrome, and ALS. Companies that we are aware are developing therapeutics in the neurodegenerative disease areaand LSD areas include companies with significant financial resources, such as AbbVie, Alector, AstraZeneca, Biogen, Bristol-Myers Squibb, Eli Lilly, E-Scape Bio, GlaxoSmithKline, Ionis, JCR Pharmaceuticals, Johnson & Johnson, Novartis, Prevail Therapeutics, Roche, Sanofi and Takeda.resources. In addition to competition from other companies targeting neurodegenerative indications, any products we may develop may also face competition from other types of therapies, such as gene-editing therapies.
Many of our current or potential competitors either alone or with their strategic partners, have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals, and marketing approved products than we do. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. TheseOur competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs. Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient, or are less expensive than any products that we may develop. Furthermore, currently approved products could be discovered to have application for treatment of neurodegenerative diseaseor LSD indications, which could give such products significant regulatory and market timing advantages over any of our product candidates. Our competitors also may obtain FDA, EMA or other regulatory approval for their products more rapidly than we may obtain approval for oursdo, and may obtain orphan product exclusivity from the FDA for indications our product candidates are targeting, which could result in our competitors establishing a strong market position before we are able to enter the market. Additionally, products or technologies developed by our competitors may render our potential product candidates uneconomical or obsolete, and we may not be successful in marketing any product candidates we may develop against competitors.
In addition, we could face litigation or other proceedings with respect to the scope, ownership, validity and/or enforceability of our patents relating to our competitors’ products and our competitors may allege that our products infringe, misappropriate or otherwise violate their intellectual property. The availability of our competitors’ products could limit the demand, and the price we are able to charge, for any products that we may develop and commercialize. See “Risks Related to Our Intellectual Property.”
The manufacture of our product candidates, particularly those that utilize our BBB platform technology, is complex and we may encounter difficulties in production. If we or any of our third-party manufacturers encounter such difficulties, orWe may fail to meet rigorously enforcedsuccessfully manufacture our product candidates, operate our own manufacturing facility, or obtain regulatory standards,approval to utilize or commercialize from our ability to provide supplymanufacturing facility, which could adversely affect our clinical trials and the commercial viability of our product candidates for clinical trials or our products for patients, if approved, could be delayed or stopped, or we may be unable to maintain a commercially viable cost structure..
The processes involved in manufacturing our drug and biological product candidates, particularly those that utilize our BBB platform technology, are complex, expensive, highly regulated and subject to multiple risks. Additionally, the manufacture of biologics involves complex processes, including developing cells or cell systems to produce the biologic, growing large quantities of such cells, and harvesting and purifying the biologic produced by them. As a result, the cost to manufacture a biologic is generally far higher than traditional small molecule chemical compounds, and the biologics manufacturing process is less reliable and is difficult to reproduce. Manufacturing biologics is highly susceptible to product loss due to contamination, equipment failure, improper installation or operation of equipment, vendor or operator error, inconsistency in yields, variability in product characteristics, and difficulties in scaling the production process. Even minor deviations from normal manufacturing processes could result in reduced production yields, product defects, and other supply disruptions. Further, as product candidates are developed through preclinical studies to late-stage clinical trials towards approval and commercialization, it is common that various aspects of the development program, such as manufacturing methods, are altered along the way in an effort to optimize processes and results. Such changes carry the risk that they will not achieve these intended objectives, and any of these changes could cause our product candidates to perform differently and affect the results of planned clinical trials or other future clinical trials.
In order to conduct clinical trials of our product candidates, or supply commercial products, if approved, we will need to manufacture them in small and large quantities. Our manufacturing partners may be unable to successfully increase the manufacturing capacity for any of our product candidates in a timely or cost-effective manner, or at all. In addition, quality issues may arise during scale-up activities. If our manufacturing partners are unable to successfully scale up the manufacture of our product candidates in sufficient quality and quantity, the development, testing, and clinical trials of that product candidate may be delayed or become infeasible, and regulatory approval or commercial launch of any resulting product may be delayed or not obtained, which could significantly harm our business. The same risks would apply to our internal manufacturing facilities and capabilities, which we are actively working towards building.building in Salt Lake City, Utah. Under an operating lease for approximately 60,000 rentable square feet of laboratory, office, and warehouse premises, we have initiated the build-out of our Utah site to expand our clinical manufacturing capabilities for biologic therapeutics including the manufacture of materials for toxicology studies and drug substance for early human clinical studies. In addition, building internal manufacturing capacity carries significant risks in terms of being able to plan, design, and execute on a complex project to build manufacturing facilities in a timely and cost-efficient manner,manner. To date, we have experienced delays with the manufacturing site build-out, and there can be no assurance that our current and future efforts to scale our internal manufacturing capabilities may notwill succeed.
In addition, the manufacturing process, including any material modifications in the manufacturing process for any products that we may develop, is subject to FDA, EMA and foreign regulatory authority approval processes and continuous oversight, and we will need to contract with manufacturers who can meet all applicable FDA, EMA and foreign regulatory authority requirements, including complying with current good manufacturing practices ("cGMPs"), on an ongoing basis. If we or our third-party manufacturers are unable to reliably produce products to specifications acceptable to the FDA, EMA or other regulatory authorities, we may not obtain or maintain the approvals we need to commercialize such products. Even if we obtain regulatory approval for any of our product candidates, there is no assurance that either we or our CDMOs will be able to manufacture the approved product to specifications acceptable to the FDA, EMA or other regulatory authorities, to produce it in sufficient quantities to meet the requirements for the potential launch of the product, or to meet potential future demand. Any of these challenges could delay completion of clinical trials, require bridging clinical trials or the repetition of one or more clinical trials, increase clinical trial costs, delay approval of our product candidate, impair commercialization efforts, increase our cost of goods, and have an adverse effect on our business, financial condition, results of operations and growth prospects.
If, in the future, we are unable to establish sales and marketing capabilities or enter into agreements with third parties to sell and market any product candidates we may develop, we may not be successful in commercializing those product candidates if and when they are approved.
We do not have a sales or marketing infrastructure and have no experience in the sale, marketing, or distribution of pharmaceutical products. To achieve commercial success for any approved product for which we retain sales and marketing responsibilities, we must either develop a sales and marketing organization or outsource these functions to third parties. In the future, we may choose to build a focused sales, marketing, and commercial support infrastructure to sell, or participate in sales activities with our collaborators for, some of our product candidates if and when they are approved.
There are risks involved with both establishing our own commercial capabilities and entering into arrangements with third parties to perform these services. For example, recruiting and training a sales force or reimbursement specialists is expensive and time consuming and could delay any product launch. If the commercial launch of a product candidate for which we recruit a sales force and establish marketing and other commercialization capabilities is delayed or does not occur for any reason, we would have prematurely or unnecessarily incurred these commercialization expenses. This may be costly, and our investment would be lost if we cannot retain or reposition our commercialization personnel.
Factors that may inhibit our efforts to commercialize any approved product on our own include:
•our inability to recruit and retain adequate numbers of effective sales, marketing, reimbursement, customer service, medical affairs, and other support personnel;
•the inability of sales personnel to obtain access to physicians or persuade adequate numbers of physicians to prescribe any future approved products;
•the inability of reimbursement professionals to negotiate arrangements for formulary access, reimbursement, and other acceptance by payors;
•the inability to price our products at a sufficient price point to ensure an adequate and attractive level of profitability;
•restricted or closed distribution channels that make it difficult to distribute our products to segments of the patient population;
•the lack of complementary products to be offered by sales personnel, which may put us at a competitive disadvantage relative to companies with more extensive product lines; and
•unforeseen costs and expenses associated with creating an independent commercialization organization.
If we enter into arrangements with third parties to perform sales, marketing, commercial support, and distribution services, our product revenue or the profitability of product revenue may be lower than if we were to market and sell any products we may develop ourselves. In addition, we may not be successful in entering into arrangements with third parties to commercialize our product candidates or may be unable to do so on terms that are favorable to us. We may have little control over such third parties, and any of them may fail to devote the necessary resources and attention to sell and market our products effectively. If we do not establish commercialization capabilities successfully, either on our own or in collaboration with third parties, we will not be successful in commercializing our product candidates if approved.
Even if any product candidates we develop receive marketing approval, they may fail to achieve the degree of market acceptance by physicians, patients, healthcare payors, and others in the medical community necessary for commercial success.
The commercial success of any of our product candidates will depend upon its degree of market acceptance by physicians, patients, third-party payors, and others in the medical community. Even if any product candidates we may develop receive marketing approval, they may nonetheless fail to gain sufficient market acceptance by physicians, patients, healthcare payors, and others in the medical community. The degree of market acceptance of any product candidates we may develop, if approved for commercial sale, will depend on a number of factors, including:
•the efficacy or potency and safety of such product candidates as demonstrated in pivotal clinical trials and published in peer-reviewed journals;
•the potential and perceived advantages compared to alternative treatments;
•the ability to offer our products for sale at competitive prices;
•the ability to offer appropriate patient access programs, such as co-pay assistance;
•the extent to which physicians recommend our products to their patients;
•convenience and ease of dosing and administration compared to alternative treatments;
•the clinical indications for which the product candidate is approved by FDA, EMA or other regulatory agencies;
•product labeling or product insert requirements of the FDA, EMA or other comparable foreign regulatory authorities, including any limitations, contraindications or warnings contained in a product’s approved labeling;
•restrictions on how the product is distributed;
•the timing of market introduction of competitive products;
•publicity concerning our products or competing products and treatments;
•the strength of marketing and distribution support;
•sufficient third-party coverage or reimbursement; and
•the prevalence and severity of any side effects.
If any product candidates we develop do not achieve an adequate level of acceptance, we may not generate significant product revenue, and we may not become profitable.
Even if we are able to commercialize any product candidates, such products may become subject to unfavorable pricing regulations, third-party reimbursement practices, or healthcare reform initiatives, which would harm our business.
The regulations that govern marketing approvals, pricing, and reimbursement for new drugs vary widely from country to country. In the United States, recently enacted legislation may significantly change the approval requirements in ways that could involve additional costs and cause delays in obtaining approvals. Some countries require approval of the sale price of a drug before it can be marketed. In many countries, the pricing review period begins after marketing or product licensing approval is granted. In some foreign markets, prescription pharmaceutical pricing remains subject to continuing governmental control even after initial approval is granted. As a result, we might obtain marketing approval for a product in a particular country, but then be subject to price regulations that delay our commercial launch of the product, possibly for lengthy time periods, and negatively impact the revenue we are able to generate from the sale of the product in that country. Adverse pricing limitations may hinder our ability to recoup our investment in one or more product candidates, even if any product candidates we may develop obtain marketing approval.
Our ability to successfully commercialize any products that we may develop also will depend in part on the extent to which reimbursement for these products and related treatments will be available from government health administration authorities, private health insurers, and other organizations. Government authorities and third-party payors, such as private health insurers and health maintenance organizations, decide which medications they will pay for and establish reimbursement levels. A primary trend in the U.S. healthcare industry and elsewhere is cost containment. Government authorities and third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medications. Government authorities currently impose mandatory discounts for certain patient groups, such as Medicare, Medicaid and Veterans Affairs ("VA"), hospitals, and may seek to increase such discounts at any time. Future regulation may negatively impact the price of our products, if approved. Increasingly, third-party payors are requiring that drug companies provide them with predetermined discounts from list prices and are challenging the prices charged for medical products. We cannot be sure that reimbursement will be available for any product candidate that we commercialize and, if reimbursement is available, the level of reimbursement. Reimbursement may impact the demand for, or the price of, any product candidate for which we obtain marketing approval. In order to get reimbursement, physicians may need to show that patients have superior treatment outcomes with our products compared to standard of care drugs, including lower-priced generic versions of standard of care drugs. If reimbursement is not available or is available only to limited levels, we may not be able to successfully commercialize any product candidate for which we obtain marketing approval. In the United States, no uniform policy of coverage and reimbursement for products exists among third-party payors and coverage and reimbursement levels for products can differ significantly from payor to payor. As a result, the coverage determination process is often a time consuming and costly process that may require us to provide scientific and clinical support for the use of our products to each payor separately, with no assurance that coverage and adequate reimbursement will be applied consistently or obtained in the first instance.
There may be significant delays in obtaining reimbursement for newly approved drugs, and coverage may be more limited than the purposes for which the medicine is approved by the FDA, EMA or other comparable foreign regulatory authorities. Moreover, eligibility for reimbursement does not imply that any drug will be paid for in all cases or at a rate that covers our costs, including research, development, manufacture, sale, and distribution. Interim reimbursement levels for new drugs, if applicable, may also not be sufficient to cover our costs and may not be made permanent. Reimbursement rates may vary according to the use of the drug and the clinical setting in which it is used, may be based on reimbursement levels already set for lower cost drugs and may be incorporated into existing payments for other services. Net prices for drugs may be reduced by mandatory discounts or rebates required by government healthcare programs or private payors and by any future relaxation of laws that presently restrict imports of drugs from countries where they may be sold at lower prices than in the United States. Third-party payors often rely upon Medicare coverage policy and payment limitations in setting their own reimbursement policies. Our inability to promptly obtain coverage and profitable payment rates from both government-funded and private payors for any approved products we may develop could have a material adverse effect on our operating results, our ability to raise capital needed to commercialize product candidates, and our overall financial condition.
If any of our small molecule product candidates that are small molecules obtain regulatory approval, additional competitors could enter the market with generic versions of such drugs, which may result in a material decline in sales of affected products.
Under the Drug Price Competition and Patent Term Restoration Act of 1984 (the "Hatch-Waxman Act"), a pharmaceutical manufacturer may file an abbreviated new drug application ("ANDA") seeking approval of a generic copy of an approved, small molecule innovator product. Under the Hatch-Waxman Act, a manufacturer may also submit a new drug application ("NDA") under section 505(b)(2) that references the FDA’s prior approval of the small molecule innovator product. A 505(b)(2) NDA product may be for a new or improved version of the original innovator product. The Hatch-Waxman Act also provides for certain periods of regulatory exclusivity, which preclude FDA approval (or in some circumstances, FDA filing and reviewing) of an ANDA or 505(b)(2) NDA. These include, subject to certain exceptions, the period during which an FDA-approved drug is subject to orphan drug exclusivity. In addition to the benefits of regulatory exclusivity, an innovator NDA holder may have patents claiming the active ingredient, product formulation or an approved use of the drug, which would be listed with the product in the FDA publication, “Approved Drug Products with Therapeutic Equivalence Evaluations,” known as the “Orange Book.” If there are patents listed in the Orange Book, a generic or 505(b)(2) applicant that seeks to market its product before expiration of the patents must include in the ANDA a “Paragraph IV certification,” challenging the validity or enforceability of, or claiming non-infringement of, the listed patent or patents. Notice of the certification must be given to the innovator, too, and if within 45 days of receiving notice the innovator sues to protect its patents, approval of the ANDA is stayed for 30 months, or as lengthened or shortened by the court.
Accordingly, if any of our small molecule product candidates are approved, competitors could file ANDAs for generic versions of our small molecule drug products or 505(b)(2) NDAs that reference our small molecule drug products, respectively. If there are patents listed for our small molecule drug products in the Orange Book, those ANDAs and 505(b)(2) NDAs would be required to include a certification as to each listed patent indicating whether the ANDA applicant does or does not intend to challenge the patent. We cannot predict which, if any, patents in our current portfolio or patents we may obtain in the future will be eligible for listing in the Orange Book, how any generic competitor would address such patents, whether we would sue on any such patents, or the outcome of any such suit.
We may not be successful in securing or maintaining proprietary patent protection for products and technologies we develop or license. Moreover, if any of our owned or in-licensed patents that are listed in the Orange Book are successfully challenged by way of a Paragraph IV certification and subsequent litigation, the affected product could immediately face generic competition and its sales would likely decline rapidly and materially. Should sales decline, we may have to write off a portion or all of the intangible assets associated with the affected product and our results of operations and cash flows could be materially and adversely affected. See “Risks Related to Our Intellectual Property.”
Our biologic, or large molecule, product candidates for which we intend to seek approval may face competition sooner than anticipated.
Even if we are successful in achieving regulatory approval to commercialize a product candidate faster than our competitors, our large molecule product candidates may face competition from biosimilar products. In the United States, our large molecule product candidates are regulated by the FDA as biologic products and we intend to seek approval for these product candidates pursuant to the biologics license application ("BLA"), pathway. The Biologics Price Competition and Innovation Act of 2009 (the "BPCIA"), created an abbreviated pathway for the approval of biosimilar and interchangeable biologic products. The abbreviated regulatory pathway establishes legal authority for the FDA to review and approve biosimilar biologics, including the possible designation of a biosimilar as “interchangeable” based on its similarity to an existing brand product. Under the BPCIA, an application for a biosimilar product cannot be approved by the FDA until 12 years after the original branded product was approved under a BLA. The law is complex and is still being interpreted and implemented by the FDA. As a result, its ultimate impact, implementation, and meaning are subject to uncertainty. While it is uncertain when such processes intended to implement BPCIA may be fully adopted by the FDA, any such processes could have a material adverse effect on the future commercial prospects for our large molecule product candidates.
We believe that any of our large molecule product candidates approved as a biologic product under a BLA should qualify for the 12-year period of exclusivity. However, there is a risk that this exclusivity could be shortened due to congressional action or otherwise, or that the FDA will not consider our product candidates to be reference products for competing products, potentially creating the opportunity for generic competition sooner than anticipated. Moreover, the extent to which a biosimilar product, once approved, will be substituted for any one of our reference products in a way that is similar to traditional generic substitution for non-biologic products is not yet clear, and will depend on a number of marketplace and regulatory factors that are still developing. In addition, a competitor could decide to forego the biosimilar approval path and submit a full BLA after completing its own preclinical studies and clinical trials. In such cases, any exclusivity to which we may be eligible under the BPCIA would not prevent the competitor from marketing its product as soon as it is approved.
In Europe, the European Commission has granted marketing authorizations for several biosimilar products pursuant to a set of general and product class-specific guidelines for biosimilar approvals issued over the past few years. In Europe, a competitor may reference data supporting approval of an innovative biological product, but will not be able to get it on the market until 10 years after the time of approval of the innovative product. This 10-year marketing exclusivity period will be extended to 11 years if during the first eight of those 10 years, the marketing authorization holder obtains an approval for one or more new therapeutic indications that bring significant clinical benefits compared with existing therapies. In addition, companies may be developing biosimilar products in other countries that could compete with our products, if approved.
If competitors are able to obtain marketing approval for biosimilars referencing our large molecule product candidates, if approved, such products may become subject to competition from such biosimilars, with the attendant competitive pressure and potential adverse consequences. Such competitive products may be able to immediately compete with us in each indication for which our product candidates may have received approval.
If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to limit commercialization of our product candidates.
We face an inherent risk of product liability as a result of the clinical testing of our product candidates and will face an even greater risk when and if we commercialize any products. For example, we may be sued if our product candidates cause or are perceived to cause injury or are found to be otherwise unsuitable during clinical testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability or a breach of warranties. Claims could also be asserted under state consumer protection acts. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit testing and commercialization of our product candidates. Even successful defense would require significant financialcosts to defend litigation and managementa diversion of management's time and resources. Regardless of the merits or eventual outcome, liability claims may result in:
•in a decreased or interrupted demand for our products;
•products, injury to our reputation;
•reputation, withdrawal of clinical trial participants and inability to continue clinical trials;
•trials, and initiation of investigationsinvestigation by regulators;
•costs to defend the related litigation;
•a diversion of management’s time and our resources;
•regulators. Any successful liability claims could result in substantial monetary awards to trial participants or patients;
•product recalls, withdrawals, or labeling, marketing or promotional restrictions;
•loss of revenue;
•exhaustion of any available insurance and our capital resources;
•the inability to commercialize any product candidate; and
•a decline in our share price.
Our inability to obtain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization of products we develop, alone or with collaborators. Our insurance policies may have various exclusions, and we may be subject to a product liability claim for which we have no coverage. We may have to pay any amounts awarded by a court or negotiated in a settlement that exceed our coverage limitations or that are not covered by our insurance, and we may not have, or be able to obtain, sufficient capital to pay such amounts. Even if our agreements with any future corporate collaborators entitle us to indemnification against losses, such indemnification may not be available or adequate should any claim arise.
Risks Related to Regulatory Approval and Other Legal Compliance Matters
The regulatory approval processes of the FDA, EMA, and comparable foreign regulatory authorities are lengthy, time consuming, and inherently unpredictable. If we are ultimately unable to obtain regulatory approval for our product candidates, we will be unable to generate product revenue and our business will be substantially harmed.
The time required to obtain approval by the FDA, EMA and comparable foreign regulatory authorities is unpredictable, typically takes many years following the commencement of clinical trials, and depends upon numerous factors, including the type, complexity and novelty of the product candidates involved. In addition, approval policies, regulations, or the type and amount of clinical data necessary to gain approval may change during the course of a product candidate’s clinical development and may vary among jurisdictions, which may cause delays in the approval or the decision not to approve an application. For example, if the Supreme Court reverses or curtails the Chevron doctrine, which gives deference to regulatory agencies in litigation against the FDA and other agencies, more companies may bring lawsuits against the FDA to challenge longstanding decisions and policies of the FDA, which could undermine the FDA's authority, lead to uncertainties in the industry, and disrupt the FDA's normal operations, which could delay the FDA's review of our marketing applications.
Regulatory authorities have substantial discretion in the approval process and may refuse to accept any application or may decide that our data are insufficient for approval and require additional preclinical, clinical or other studies. Moreover, the FDA, EMA or other regulatory authorities may fail to approve companion diagnostics that we contemplate using with our therapeutic product candidates. We have not submitted for, or obtained regulatory approval for any product candidate, and it is possible that none of our existing product candidates or any product candidates we may seek to develop in the future will ever obtain regulatory approval.
Applications for our product candidates could fail to receive regulatory approval in an initial or subsequent indication for many reasons, including but not limited to the following:
•the FDA, EMA or comparable foreign regulatory authorities may disagree with the design, implementation, or results of our clinical trials;
•the FDA, EMA or comparable foreign regulatory authorities may determine that our product candidates are not safe and effective, only moderately effective or have undesirable or unintended side effects, toxicities, or other characteristics that preclude our obtaining marketing approval or prevent or limit commercial use;
•the population studied in the clinical program may not be sufficiently broad or representative to assure efficacy or potency and safety in the full population for which we seek approval;
•we may be unable to demonstrate to the FDA or comparable foreign regulatory authorities that a product candidate’s risk-benefit ratio when compared to the standard of care is acceptable;
•the FDA, EMA or comparable foreign regulatory authorities may disagree with our interpretation of data from preclinical studies or clinical trials;
•the data collected from clinical trials of our product candidates may not be sufficient to support the submission of an NDA, BLA, or other submission or to obtain regulatory approval in the United States or elsewhere;
•we may be unable to demonstrate to the FDA, EMA or comparable foreign regulatory authorities that a product candidate’s risk-benefit ratio for its proposed indication is acceptable;
•the FDA, EMA or comparable foreign regulatory authorities may fail to approve the manufacturing processes, test procedures and specifications, or facilities of third-party manufacturers with which we contract for clinical and commercial supplies; and
•the approval policies or regulations of the FDA, EMA or comparable foreign regulatory authorities may significantly change in a manner rendering our clinical data insufficient for approval.
This lengthy approval process, as well as the unpredictability of the results of clinical trials, may result in our failing to obtain regulatory approval to market any of our product candidates, which would significantly harm our business, results of operations, and prospects.
Our product candidates may cause undesirable side effects or have other properties that could halt their clinical development, prevent their regulatory approval, limit their commercial potential, or result in significant negative consequences.
Adverse events or other undesirable side effects caused by our product candidates could cause us, our collaborators, or regulatory authorities to interrupt, delay, or halt clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA, EMA, or other comparable foreign regulatory authorities.
Our most advanced product candidates, BIIB122/DNL151, (BIIB122)DNL310, SAR443820/DNL788, eclitasertib (SAR443122/DNL758), DNL343, and DNL310TAK-594/DNL593 are currently our only clinical stage product candidates. In our completed Phase 1b clinical trial of former product candidate DNL201 in patients with Parkinson’s disease, there was one SAE considered unrelated to drug, and at the high dose, there was one severe AE (headache) leading to dose reduction and one study withdrawal (headache and nausea). Adverse events and other side effects may result from higher dosing, repeated dosing, and/or longer-term exposure to DNL151 (BIIB122), DNL343 and/or DNL310our product candidates and could lead to delays and/or termination of the development of these product candidates.
In 2016,
For example, in August 2023, together with our collaboration partner Takeda,we initiatedmade the strategic decision to discontinue clinical development of TAK-920/DNL919 in Alzheimer's disease following a Phase 1 clinical trial in a former RIPK1 inhibitor product candidate, DNL104, which we subsequently discontinued based on liver function test abnormalities in some clinical trial healthy volunteer participants. hold by the FDA.
In 2020, we paused clinical studies with DNL747 in our RIPK1 program. Chronic toxicity studies with DNL747 in cynomolgus monkeys showed dose- and duration-dependent adverse preclinical findings at exposures higher than those tested in the clinic. These findings, which are considered off-target and molecule-specific, may impact the ability to increase the dose of DNL747 and achieve higher levels of target inhibition without time consuming additional clinical safety studies in patients to evaluate the long-term safety and tolerability.
Drug-related side effects could affect patient recruitment, the ability of enrolled patients to complete the trial, and/or result in potential product liability claims. We are required to maintain product liability insurance pursuant to certain of our license agreements. We may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses due to liability. A successful product liability claim or series of claims brought against us could cause our stock price to decline and, if judgments exceed our insurance coverage, could adversely affect our results of operations and business. In addition, regardless of merit or eventual outcome, product liability claims may result in impairment of our business reputation, withdrawal of clinical trial participants, costs due to related litigation, distraction of management’s attention from our primary business, initiation of investigations by regulators, substantial monetary awards to patients or other claimants, the inability to commercialize our product candidates, and decreased demand for our product candidates, if approved for commercial sale.
Additionally, if one or more of our product candidates receives marketing approval, and we or others, including our collaborators, later identify undesirable side effects or adverse events caused by such products, a number of potentially significant negative consequences could result, including but not limited to:
•regulatory authorities may withdraw approvals of such product and cause us to recall our product;
•regulatory authorities may require additional warnings on the label;
•we may be required to change the way the product is administered or conduct additional clinical trials or post-approval studies;
•we may be required to create a Risk Evaluation and Mitigation Strategy plan which could include a medication guide outlining the risks of such side effects for distribution to patients, a communication plan for healthcare providers, and/or other elements, such as boxed warning on the packaging, to assure safe use;
•we could be sued and held liable for harm caused to patients; and
•our reputation may suffer.
Any of these events could prevent us from achieving or maintaining market acceptance of the particular product candidate, if approved, and could significantly harm our business, financial condition, results of operations, and growth prospects. We cannot predict whether our product candidates will cause toxicities in humans that would preclude or lead to the revocation of regulatory approval based on nonclinical studies or early-stage clinical trials.
We currently and may in the future conduct clinical trials for our product candidates outside the United States, and the FDA, EMA, and applicable foreign regulatory authorities may not accept data from such trials.
We may in the future choose tocurrently conduct one or more of our clinical trials outside the United States, including in Europe.Europe, and may continue to do so in the future. The acceptance of data from clinical trials conducted outside the United States or another jurisdiction by the FDA, EMA, or applicable foreign regulatory authority may be subject to certain conditions. In cases where data from foreign clinical trials are intended to serve as the basis for marketing approval in the United States, the FDA will generally not approve the application on the basis of foreign data alone unless (i) the data are applicable to the United States population and United States medical practice; and (ii) the trials were performed by clinical investigators of recognized competence and pursuant to cGCP regulations. Additionally, the FDA’s clinical trial requirements, including sufficient size of patient populations and statistical powering, must be met. Many foreign regulatory bodies have similar approval requirements. In addition, such foreign trials would be subject to the applicable local laws of the foreign jurisdictions where the trials are conducted. There can be no assurance that the FDA, EMA, or any applicable foreign regulatory authority will accept data from trials conducted outside of the United States or the applicable jurisdiction. If the FDA, EMA, or any applicable foreign regulatory authority does not accept such data, it would result in the need for additional trials, which would be costly and time-consuming and delay aspects of our business plan, and which may result in our product candidates not receiving approval or clearance for commercialization in the applicable jurisdiction.
Obtaining and maintaining regulatory approval of our product candidates in one jurisdiction does not mean that we will be successful in obtaining regulatory approval of our product candidates in other jurisdictions.
Obtaining and maintaining regulatory approval of our product candidates in one jurisdiction does not guarantee that we will be able to obtain or maintain regulatory approval in any other jurisdiction, butand a failure or delay in obtaining regulatory approval in one jurisdiction may have a negative effect on the regulatory approval process in others. For example, even if the FDA or EMA grants marketing approval of a product candidate, comparable regulatory authorities in foreign jurisdictions must also approve the manufacturing, marketing and promotion of the product candidate in those countries. Approval procedures vary among jurisdictions and can involve requirements and administrative review periods different from those in the United States, including additional preclinical studies or clinical trials as clinical trials conducted in one jurisdiction may not be accepted by regulatory authorities in other jurisdictions. In many jurisdictions outside the United States, a product candidate must be approved for reimbursement before it can be approved for sale in that jurisdiction. In some cases, the price that we intend to charge for our products is also subject to approval.
Obtaining foreign regulatory approvals and compliance with foreign regulatory requirements could result in significant delays, difficulties, and costs for us and could delay or prevent the introduction of our products in certain countries. If we or any partner we work with fail to comply with the regulatory requirements in international markets or fail to receive applicable marketing approvals, our target market will be reduced and our ability to realize the full market potential of our product candidates will be harmed.
Even if we obtain regulatory approval for a product candidate, our products will remain subject to extensive regulatory scrutiny.
If any of our product candidates are approved, they will be subject to ongoing regulatory requirements, for manufacturing, labeling, packaging, storage, advertising, promotion, sampling, record-keeping, conduct of post-marketing studies, and submission of safety, efficacy or potency, and other post-market information, including both federal and state requirements in the United States and requirements of comparable foreign regulatory authorities.
While healthcare professionals are free to use and prescribe drug products for off-label uses, the FDA strictly regulates manufacturers’ promotional claims of drug products. In particular, a product may not be promoted for uses that are not approved by the FDA as reflected in the FDA-approved labeling. The FDA, the Department of Justice, the Inspector General of the Department of Health and Human Services, among other government agencies, actively enforce the laws and regulations prohibiting the promotion of off-label uses, and aA company that is found to have improperly promoted off-label uses may be subject to significant liability, including large civil and criminal fines, penalties, and enforcement actions. If we cannot successfully manage the promotion of our approved product candidates, we could become subject to significant liability, which wouldcould materially adversely affect our business and financial condition.
Manufacturers and manufacturers’ facilities are required to comply with extensive requirements imposed by the FDA, EMA and comparable foreign regulatory authorities, including ensuring that quality control and manufacturing procedures conform to cGMP regulations. As such, we and our contract manufacturers will be subject to continual review and inspections to assess compliance with cGMP and adherence to commitments made in any NDA, BLA or marketing authorization application ("MAA"). Accordingly, we and others with whom we work must continue to expend time, money, and effort in all areas of regulatory compliance, including manufacturing, production and quality control.
Any regulatory approvals that we receive for our product candidates will be subject to limitations on the approved indicated uses for which the product may be marketed and promoted or to the conditions of approval (including the requirement to implement a Risk Evaluation and Mitigation Strategy) or contain requirements for potentially costly post-marketing testing. We will be required to report certain adverse reactions and production problems, if any, to the FDA, EMA, and comparable foreign regulatory authorities. Any new legislation addressing drug safety issues could result in delays in product development or commercialization, or increased costs to assure compliance. The FDA and other agencies, including the Department of Justice, closely regulate and monitor the post-approval marketing and promotion of products to ensure that they are manufactured, marketed and distributed only for the approved indications and in accordance with the provisions of the approved labeling. We will have to comply with requirements concerning advertising and promotion for our products. Promotional communications with respect to prescription drugs are subject to a variety of legal and regulatory restrictions and must be consistent with the information in the product’s approved label. As such, we may not promote our products for indications or uses for which they do not have approval. The holder of an approved NDA, BLA, or MAA must submit new or supplemental applications and obtain approval for certain changes to the approved product, product labeling, or manufacturing process. We could also be asked to conduct post-marketing clinical trials to verify the safety and efficacy of our non-biologic products or safety, purity, and potency for our biologic products, in general or in specific patient subsets. If original marketing approval was obtained via the accelerated approval pathway, we could be required to conduct a successful post-marketing clinical trial to confirm clinical benefit for our products. The Food and Drug Omnibus Reform Act reformed the accelerated approval pathway, such as requiring the FDA to specify conditions for post-approval study requirements and setting forth procedures for the FDA to withdraw a product on an expedited basis for non-compliance with post-approval requirements. An unsuccessful post-marketing study or failure to complete such a study could result in the withdrawal of marketing approval.
Manufacturers and manufacturers’ facilities are required to comply with extensive requirements imposed by the FDA, EMA, and comparable foreign regulatory authorities, including ensuring that quality control and manufacturing procedures conform to cGMP regulations. If a regulatory agency discovers previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, or disagrees with the promotion, marketing, or labeling of a product, such regulatory agency may impose restrictions on that product or us, including requiring withdrawal of the product from the market. If we fail to comply with applicable regulatory requirements, a regulatory agency or enforcement authority may, among other things:
•things, issue warning letters, that would result in adverse publicity;impose penalties, suspend regulatory approvals, or require a product recall. Any of these actions by a regulatory agency could require us to expend significant time and resources, generate negative publicity, and adversely affect the value of our company.
•To the extent wimpose civil or criminal penalties;
•suspend or withdraw regulatory approvals;
•suspend any of our ongoing clinical trials;
•refuse to approve pending applications or supplements to approved applications submitted by us;
•impose restrictions on our operations, including closing our contract manufacturers’ facilities;
•seize or detain products; and/or
•require a product recall.
Any government investigation of alleged violations of law could require us to expend significant time and resources in response, and could generate negative publicity. Any failure to comply with ongoing regulatory requirements may significantly and adversely affect our ability to commercialize and generate revenue from our products. If regulatory sanctions are applied or if regulatory approval is withdrawn, the value of our company and our operating results will be adversely affected.
We plan toe seek orphan drug designation for someany of our product candidates, but we may be unable to obtain such designations or to maintain the benefits associated with orphanorphan drug status, including market exclusivity, which may cause our revenue, if any, to be reduced.
Under the Orphan Drug Act, the FDA may grant orphan designation to a drug or biologic intended to treat a rare disease or condition defined as a disease or condition with a patient population of fewer than 200,000 in the United States, or a patient population greater than 200,000 in the United States whenwhere there is no reasonable expectation that the cost of developing and making available the drug or biologic in the United States will be recovered from sales in the United States for that drug or biologic. Orphan drug designation must be requested before submitting an NDA or BLA. In the United States,Once granted, orphan drug designation entitles a party to financial incentives such as opportunities for grant funding towards clinical trial costs, tax advantages, and user-fee waivers. After the FDA grants orphan drug designation, the generic identity of the drug and its potential orphan use are disclosed publicly by the FDA. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process.certain exclusivity protections. In February 2019, the FDA granted orphan drug designation for our DNL310 program in Hunter syndrome. We plan to seek orphan drug designations for some other product candidates and may be unable to obtain such designations.
If a product that has orphan drug designation subsequently receives the first FDA approval for a particular active ingredient for the disease for which it has such designation, the product is entitled to orphan product exclusivity, which means that the FDA may not approve any other NDA or BLA applications to market the same drug or biologic for the same indication for seven years, except in limited circumstances such as a showing of clinical superiority to the product with orphan exclusivity or if FDA finds that the holder of the orphan exclusivity has not shown that it can assure the availability of sufficient quantities of the orphan product to meet the needs of patients with the disease or condition for which the drug was designated. As a result, even though DNL310 has been granted orphan drug designation and even if one of our product candidates receives orphan exclusivity,However, the FDA can still approve other drugs that have a different active ingredient for use in treating the same indication or disease. Furthermore, the FDAdisease, and can waive orphan exclusivity if we are unable to manufacture sufficient supply of our product. We plan to seek orphan drug designations for some other product candidates, but we may be unable to obtain such designations.
Further, in response to Catalyst Pharms., Inc. v. Becerra, 14 F.4th 1299 (11th Cir. 2021), the FDA clarified in a January 2023 notice that the FDA intends to continue to apply its longstanding interpretation of the regulations to matters outside of the scope of the Catalyst order – that is, the agency will continue tying the scope of orphan-drug exclusivity to the uses or indications for which a drug is approved, which permits other sponsors to obtain approval of a drug for new uses or indications within the same orphan designated disease or condition that have not yet been approved. It is unclear how future litigation, legislation, agency decisions, and administrative actions will impact the scope of the orphan drug exclusivity.
We have received Fast Track designation from the FDA for SAR443820/DNL788, and may seek Fast Track designation from the FDA for additional product candidates. Even if one or more of our product candidates receives Fast Track designation, we may be unable to obtain or maintain the benefits associated with the Fast Track designation.
The FDA has granted Fast Track designation to SAR443820/DNL788. Fast Track designation is designed to facilitate the development and expedite the review of therapies to treat serious conditions and fill an unmet medical need. However, if we do not continue to meet the criteria of the Fast Track designation, or if our clinical trials are delayed, suspended or terminated, or put on clinical hold due to unexpected adverse events or issues with clinical supply, we will not receive the benefits associated with the Fast Track program. Furthermore, Fast Track designation does not change the standards for approval. Fast Track designation alone does not guarantee qualification for the FDA’s priority review procedures. Fast Track designation also does not guarantee our product candidate will be approved in a timely manner, if at all.
Healthcare legislative measures aimed at reducing healthcare costs may have a material adverse effect on our business and results of operations.
We may face difficulties from changes to current regulations and future legislation. Current and future legislation may increase the difficulty and cost for us to commercialize our drugs, if approved, and affect the prices we may obtain, including changes in coverage and reimbursement policies in certain market segments for our product candidates, which could make it difficult for us to sell our product candidates, if approved, profitably. Third-party payors, whether domestic or foreign, or governmental or commercial, are developing increasingly sophisticated methods of controlling healthcare costs.
In both the United States and certain foreign jurisdictions, there have been a number of legislative and regulatory changes to the health care system that could impact our ability to sell our products profitably. In particular, in 2010,These include the enactment of the Affordable Care Act ("ACA"of 2010 (“ACA”), was enacted,the American Rescue Plan Act of 2021, which among other things, subjected biologic products to potential competition by lower-cost biosimilars, addressedwill eliminate a new methodology by which rebates owed by manufacturers under thestatutory cap on Medicaid Drug Rebate Program are calculatedrebates that manufacturers pay to state Medicaid programs, and the July 2021 executive order, "Promoting Competition in the American Economy," with multiple provisions aimed at increasing competition for drugsprescription drugs. In August 2022, Congress passed the Inflation Reduction Act of 2022 ("IRA"), which includes prescription drug provisions that are inhaled, infused, instilled, implanted or injected, increasedhave significant implications for the minimum Medicaid rebates owed by most manufacturers underpharmaceutical industry and Medicare beneficiaries, including allowing the Medicaid Drug Rebate Program, extended the Medicaid Drug Rebate Programfederal government to utilization of prescriptions of individuals enrolled in Medicaid managed care organizations, subjected manufacturers to new annual fees and taxesnegotiate a maximum fair price for certain brandedhigh-priced single source Medicare drugs, imposing penalties and excise tax for manufacturers that fail to comply with the drug price negotiation requirements, requiring inflation rebates for all Medicare Part B and Part D drugs, with limited exceptions, if their drug prices increase faster than inflation, and redesigning Medicare Part D to reduce out-of-pocket prescription drugs,drug costs for beneficiaries, among other changes.Various industry stakeholders, including pharmaceutical companies, the U.S. Chamber of Commerce, and provided incentives to programs that increasethe Pharmaceutical Research and Manufacturers of America, have initiated lawsuits against the federal government’s comparative effectiveness research. Recent changes ingovernment asserting that the U.S. administration could lead to repeal of or changes in some or allprice negotiation provisions of the ACA,Inflation Reduction Act are unconstitutional. The impact of these judicial changes, legislative, executive, and complyingadministrative actions and any future healthcare measures and agency rules implemented by the government on us and the pharmaceutical industry as a whole is unclear. At the state level, a number of states are considering or have recently enacted state drug price transparency and reporting laws that could substantially increase our compliance burdens and expose us to greater liability under such state laws once we begin commercialization after obtaining regulatory approval for any of our products.
Since its enactment, there have been executive, judicial, and congressional challenges to certain aspects of the ACA. It is unclear how future litigation or healthcare measures promulgated by the Biden administration will impact our business, financial condition, and results of operations. Complying with any new legislation or reversing changes implemented under the ACAin healthcare regulation could be time-intensive and expensive, resulting in a material adverse effect on our business. Until the ACA is fully implemented or there is more certainty concerning the future of the ACA, it will be difficult to predict its full impact and influence on our business.
In addition, other legislative changes have been proposed and adopted in the United States since the ACA was enacted. In August 2011, the Budget Control Act of 2011, among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions of Medicare payments to providers of 2% per fiscal year, which went into effect in 2013, and will remain in effect through 2030, with the exception of a temporary suspension implemented under various COVID-19 relief legislation from May 1, 2020 through March 31, 2021, unless additional Congressional action is taken. The American Taxpayer Relief Act of 2012 further reduced Medicare payments to several providers, including hospitals and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
There have been, and likely will continue to be, legislative and regulatory proposals at the foreign, federal and state levels directed at containing or lowering the cost of healthcare. We cannot predict the initiatives that may be adopted in the future. The continuing efforts of the government, insurance companies, managed care organizations and other payors of healthcare services to contain or reduce costs of healthcare and/or impose price controls may adversely affect:
•the demand for our product candidates, if we obtain regulatory approval;
•our ability to receive or set a price that we believe is fair for our products;
•our ability to generate revenue and achieve or maintain profitability;
•the level of taxes that we are required to pay; and
•the availability of capital.
We expect that the ACA and IRA, as well as other healthcare reform measures that may be adopted in the future, may result in additional reductions in Medicare and other healthcare funding, more rigorous coverage criteria, lower reimbursement, and new payment methodologies. This could lower the price that we receive for any approved product. Any denial in coverage or reduction in reimbursement from Medicare or other government-funded programs may result in a similar denial or reduction in payments from private payors, which may prevent us from being able to generate sufficient revenue, attain profitability or commercialize our product candidates, if approved. In addition, increased scrutiny by the U.S. Congress of the FDA’s approval process may significantly delay or prevent marketing approval, as well as subject us to more stringent product labeling and post-marketing testing and other requirements.
There have been, and likely will continue to be, legislative and regulatory proposals at the foreign, federal and state levels directed at containing or lowering the cost of healthcare. We cannot predict the initiatives that may be adopted in the future. The continuing efforts of the government, insurance companies, managed care organizations, and other payors of healthcare services to contain or reduce costs of healthcare and/or impose price controls may adversely affect the demand for any product candidates that are approved, our ability to receive or set a price we believe is fair for our products, our ability to attract investment, our ability to generate revenue or achieve profitability, the level of taxes we are required to pay, and the availability of capital.
Our employees, independent contractors, consultants, commercial partners, and vendors may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements.
We are exposed to the risk of fraud, misconduct, or other illegal activity by our employees, independent contractors, consultants, commercial partners, and vendors. Misconduct by these parties could include intentional, recklessreckless. and negligent conduct that fails to: comply with the laws of the FDA, EMA, and other comparable foreign regulatory authorities; provide true, completecomplete. and accurate information to the FDA, EMA and other comparable foreign regulatory authorities; comply with manufacturing standards we have established; comply with healthcare fraud and abuse laws in the United States and similar foreign fraudulent misconduct laws; or report financial information or data accurately or to disclose unauthorized activities to us. If we obtain FDA approval of any of our product candidates and begin commercializing those products in the United States, our potential exposure under such laws will increase significantly, and our costs associated with compliance with such laws are also likely to increase. In particular, research, sales, marketing, education and other business arrangements in the healthcare industry are subject to extensive laws designed to prevent fraud, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, educating, marketing and promotion, sales and commission, certain customer incentive programs and other business arrangements generally. Activities subject to these laws also involve the improper use of information obtained in the course of patient recruitment for clinical trials, which could result in regulatory sanctions and cause serious harm to our reputation. We have adopted a code of business conduct and ethics, but it is not always possible to identify and deter misconduct by employees and third parties, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
If we fail to comply with healthcare laws, we could face substantial penalties and our business, operations and financial conditions could be adversely affected.
If we obtain FDA approval for any of our product candidates and begin commercializing those products in the United States, our operations will be subject to various federal, state, local, and stateforeign healthcare fraud and abuse laws. The laws that may impact our operations include:
•include the federal Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, receiving, offering or paying any remuneration (including any kickback, bribe, or rebate), directly or indirectly, overtly or covertly, in cash or in kind, to induce, or in return for, either the referral of an individual, or the purchase, lease, order or recommendation of any good, facility, item or service for which payment may be made, in whole or in part, under a federal healthcare program, such as the Medicare and Medicaid programs. The intent standard under the federal Anti-Kickback Statute was amended by the ACA to eliminate the need to prove specific intent and actual knowledge to establish an Anti-Kickback Statute violation. In addition, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act;
•federal civil and criminal false claims laws, including the False Claims Act, which can be enforced through civil “qui tam” or “whistleblower” actions, and civil monetary penalty laws generally prohibit individuals or entities, among other things, from knowingly presenting, or causing to be presented, claims for payment or approval from Medicare, Medicaid, or other third-party payors that are false or fraudulent or knowingly making a false statement to improperly avoid, decrease or conceal an obligation to pay money to the federal government. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of these statutes or specific intent to violate them in order to have committed a violation;
•the federal Health Insurance Portability and Accountability Act of 1996 ("HIPAA"), which created new federal criminal statutes that prohibit knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program or obtain, by means of false or fraudulent pretenses, representations, or promises, any of the money or property owned by, or under the custody or control of, any healthcare benefit program, regardless of the payor (e.g., public or private) and knowingly and willfully falsifying, concealing or covering up by any trick or device a material fact or making any materially false statements in connection with the delivery of, or payment for, healthcare benefits, items or services relating to healthcare matters;
•HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009 ("HITECH"), and their respective implementing regulations, which impose requirements on certain covered healthcare providers, health plans, and healthcare clearinghouses as well as their respective business associates that perform services for them that involve the use, or disclosure of, individually identifiable health information, relating to the privacy, security and transmission of individually identifiable health information without appropriate authorization;
•the federal Physician Payment Sunshine Act, created under the ACA, and its implementing regulations, which require manufacturers of drugs, devices, biologicals and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program to report annually to the U.S. Department of Health and Human Services under the Open Payments Program, information related to payments or other transfers of value made to physicians, as defined by law, and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. Effective January 1, 2022, these reporting and transparency requirements will extend to include payments and transfers of value made during the previous year to certain non-physician covered recipients, including physician assistants, nurse practitioners and other mid-level practitioners;
•federal consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers; and
•analogous state and foreign laws and regulations, suchregulations. These laws may impact, among other things, our clinical research program, as statewell as our proposed and foreign anti-kickback, false claims, consumer protectionfuture sales, marketing, and unfair competition laws which may apply to pharmaceutical business practices, including but not limited to, research, distribution,education programs. In particular, the promotion, sales, and marketing arrangements as well as submitting claims involvingof healthcare items orand services reimbursed by any third-party payer, including commercial insurers; stateis subject to extensive laws that require pharmaceutical companiesand regulations designed to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government that otherwise restricts payments that may be made to healthcare providersprevent fraud, kickbacks, self-dealing, and other potential referral sources; stateabusive practices. These laws that require drug manufacturers to file reports with states regardingand regulations may restrict or prohibit a wide range of pricing, discounting, marketing and marketing information, such as the tracking and reporting of gifts, compensationspromotion, sales commission, customer incentive, and other remuneration and items of value provided to healthcare professionals and entities; and state and foreign laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts.business arrangements.
Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available, it is possible that some of our business activities could, despite our efforts to comply, be subject to challenge under one or more of such laws. Efforts to ensure that our business arrangements will comply with applicable healthcare laws may involve substantial costs. It is possible that governmental and enforcement authorities will conclude that our business practices may not comply with current or future statutes, regulations, or case law interpreting applicable fraud and abuse or other healthcare laws and regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of civil, criminal, and administrative penalties, damages, disgorgement, monetary fines, possible exclusion from participation in Medicare, Medicaid, and other federal healthcare programs, contractual damages, reputational harm, diminished profits and future earnings, and curtailment of our operations, any of which could adversely affect our ability to operate our business and our results of operations. In addition, the approval and commercialization of any of our product candidates outside the United States will also likely subject us to foreign equivalents of the healthcare laws mentioned above, among other foreign laws.
Our business is subject to complex and evolving U.S. and foreign laws and regulations, information security policies, and contractual obligations relating to privacy and data protection, including the use, processing, and cross-border transfer of personal information. These laws and regulations are subject to change and uncertain interpretation, and could result in claims, changes to our business practices, or monetary penalties, and otherwise may harm our business.
We receive, generate, and store significant and increasing volumes of sensitive information and business-critical information, including employee and personal data (including protected health information), research and development information, commercial information, and business and financial information. We heavily rely on external security and infrastructure vendors to manage our information technology systems and data centers. We face a number of risks relative to protecting this critical information, including the loss of access, risk, inappropriate use or disclosure, inappropriate modification, and the risk of our being unable to adequately monitor, audit, and modify our controls over our critical information. This risk extends to the third-party vendors and subcontractors we use to manage this sensitive data.
A wide variety of provincial, state, national, and international laws and regulations apply to the collection, use, retention, protection, disclosure, transfer, and other processing of personal data. These data protection and privacy-related laws and regulations are evolving and may result in ever-increasing regulatory and public scrutiny and escalating levels of enforcement and sanctions. For example, the collection and use of personal data in the EU are governed by the EU General Data Protection Regulation ("GDPR"), which became fully effective on May 25, 2018. The GDPR imposes stringent data protection requirements, including, for example, more robust disclosures to individuals and a strengthened individual data rights regime, shortened timelines for data breach notifications, limitations on retention of information, increased requirements pertaining to special categories of data, such as health data, and additional obligations when we contract with third-party processors in connection with the processing of the personal data. The GDPR also imposes strict rules on the transfer of personal data out of the EU to the United States and other third countries, and in the context of clinical trials we currently rely on patient informed consent as the legal basis for such transfers. In addition, the GDPR provides that EU member states may make their own further laws and regulations limiting the processing of personal data, including genetic, biometric or health data. The GDPR provides for penalties for noncompliance of up to the greater of €20 million or four percent of worldwide annual revenues. The GDPR applies extraterritorially, and we may be subject to the GDPR because of our data processing activities that involve the personal data of individuals located in the EU, such as in connection with any EU clinical trials. Additionally, the UK has implemented legislation that substantially implements the GDPR (the "UK GDPR"), with penalties for noncompliance of up to the greater of £17.5 million or four percent of worldwide revenues. Aspects of UK data protection laws and regulations remain unclear. On June 28, 2021, the European Commission announced a decision of "adequacy" concluding that the UK ensures an equivalent level of data protection to the GDPR, which provides some relief regarding the legality of continued personal data flows from the European Economic Area ("EEA") to the UK. Some uncertainty remains, however, as this adequacy determination must be renewed after four years and may be modified or revoked in the interim. We cannot fully predict how the UK GDPR and data protection laws or regulations may impose additional responsibility and liabilitydevelop in relation to the personal data that we process and we may be required to put in place additional mechanisms to ensure compliance with the new data protection rules. Beginning in 2021, the UK will be a “third country” under the GDPR. medium-to-long term.
We may however, incur liabilities, expenses, costs, and other operational losses under the GDPR and UK GDPR as well as applicable EU Member Statesprivacy and data protection laws of Switzerland, the United Kingdom, privacy laws.and applicable EU member states. We may find it necessary or appropriate to make additional changes to the ways we or our service providers collect, disclose, transfer, and otherwise process data within the EEA, Switzerland, and the UK, and to our related policies and practices. This may be onerous and may interrupt or delay our development activities, and adversely affect our business, financial condition, results of operations and prospects.
Further, various states, such as California, Massachusetts, and Massachusetts,Washington have implemented similar privacy laws and regulations that impose restrictive requirements regulating the use and disclosure of health information and other personally identifiablepersonal information. These laws and regulations are not necessarily preempted by HIPAA, particularly if a state affords greater protection to individuals than HIPAA. Where state laws are more protective than HIPAA, we have tomust comply with the stricter provisions. In addition to fines and penalties imposed upon violators, some of these state laws also afford private rights of action to individuals who believe their personal information has been misused. For example, California recentlyhas enacted legislation, the California Consumer Privacy Act ("CCPA"), that, will, among other things, requirerequires covered companies to provide new disclosures to California consumers, and affordaffords such consumers new abilities to opt-out of certain sales of personal information, thatinformation. The CCPA became effective on January 1, 2020. The CCPA was amended several times throughout 2018 and 2019, and it is unclear whether further modifications will be made to this legislation or how it will be interpreted. In addition, the CCPA, as amended and expanded by the California Privacy Rights Act ("CPRA"), requires covered companies to provide new disclosures to individuals and consumers in California, and afford such individuals and consumers new data protection rights, including the ability to opt-out of certain sales of personal information. Numerous other states in the United States have proposed or enacted similar legislation. Further, some states have enacted more specific legislation, such as Washington's enactment of the My Health, My Data Act, which includes a private right of action. The U.S. federal government is also contemplating federal privacy legislation. The GDPR, UK GDPR, CCPA, CPRA, and many other federal, state, and foreign laws and regulations relating to privacy and data protection are still being tested in courts, and they are subject to new and differing interpretations by courts and regulatory officials. Additionally, the interplay of federal and state laws may be subject to varying interpretations by courts and government agencies, creating complex compliance issues for us and data we receive, use and share, potentially exposing us to additional expense, adverse publicity and liability. We are working to comply with the GDPR, UK GDPR, CCPA, CPRA and other privacy and data protection laws and regulations that apply to us, and we anticipate needing to devote significant additional resources to complying with these laws and regulations. These and future laws and regulations may increase our compliance costs and potential liability.
It is possible that the GDPR, UK GDPR, CCPA, CPRA, or other laws and regulations relating to privacy and data protection may be interpreted and applied in a manner that is inconsistent from jurisdiction to jurisdiction or inconsistent with our current policies and practices and compliance with such laws and regulations could require us to change our business practices and compliance procedures in a manner adverse to our business.practices. We cannot guarantee that we are in compliance with all such applicable data protection laws and regulations and we cannot be sure how these regulations will be interpreted, enforced or applied to our operations. Furthermore, other jurisdictions outside the EU are similarly introducing or enhancing privacy and data security laws, rules, and regulations, which could increase our compliance costs and the risks associated with noncompliance. It is possible that these laws may be interpreted and applied in a manner that is inconsistent with our practices and our efforts to comply with the evolving data protection rules may be unsuccessful. We cannot guarantee that we or our vendors may be in compliance with all applicable international laws and regulations as they are enforced now or as they evolve. For example, our privacy policies may be insufficient to protect any personal information we collect, or may not comply with applicable laws. Our non-compliance could result in government-imposed fines or orders requiring that we change our practices, which could adversely affect our business. In addition to the risks associated with enforcement activities and potential contractual liabilities, our ongoing efforts to comply with evolving laws and regulations at the federal and state level may be costly and require ongoing modifications to our policies, procedures and systems. In addition, if we are unable to properly protect the privacy and security of protected health information, we could be alleged or found to have breached our contracts.
Our actual or perceived failure to adequately comply with applicable laws and regulations or other actual or asserted obligations relating to privacy and data protection, or to protect personal data and other data we process or maintain, could result in regulatory enforcement actions against us, including fines, imprisonment of company officials and public censure, claims for damages by affected individuals, other lawsuits or reputational and damage, all of which could materially affect our business, financial condition, results of operations and growth prospects.
If we or any contract manufacturers and suppliers we engage fail to comply with environmental, health, and safety laws and regulations, we could become subject to fines or penalties or incur costs that could have a material adverse effect on the success of our business.
We and any contract manufacturers and suppliers we engage are subject to numerous federal, state, and local environmental, health, and safety laws, regulations, and permitting requirements, including those governing laboratory procedures; the generation, handling, use, storage, treatment, and disposal of hazardous and regulated materials and wastes; the emission and discharge of hazardous materials into the ground, air, and water; and employee health and safety. Our operations involve the use of hazardous and flammable materials, including chemicals and biological and radioactive materials. Our operations also produce hazardous waste. We generally contract with third parties for the disposal of these materials and wastes. We cannot eliminate the risk of contamination or injury from these materials. In the event of contamination or injury resulting from our use of hazardous materials, we could be held liable for any resulting damages, and any liability could exceed our resources. Under certain environmental laws, we could be held responsible for costs relating to any contamination at our current or past facilities and at third-party facilities. We also could incur significant costs associated with civil or criminal fines and penalties.
Compliance with applicable environmental laws and regulations may be expensive, and current or future environmental laws and regulations may impair our research, product development, and manufacturing efforts. In addition, we cannot entirely eliminate the risk of accidental injury or contamination from these materials or wastes. Although we maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees resulting from the use of hazardous materials, this insurance may not provide adequate coverage against potential liabilities. We do not carry specific biological or hazardous waste insurance coverage, and our property, casualty, and general liability insurance policies specifically exclude coverage for damages and fines arising from biological or hazardous waste exposure or contamination. Accordingly, in the event of contamination or injury, we could be held liable for damages or be penalized with fines in an amount exceeding our resources, and our clinical trials or regulatory approvals could be suspended, which could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
Our business activities may be subject to the Foreign Corrupt Practices Act and similar anti-bribery and anti-corruption laws, as well as U.S. and certain foreign export controls, trade sanctions, and import laws and regulations.
Our business activities may be subject to the Foreign Corrupt Practices Act of 1977, as amended (the "FCPA"), and similar anti-bribery or anti-corruption laws, regulations or rules of other countries in which we operate, including the U.K. Bribery Act. The FCPA generally prohibits offering, promising, giving, or authorizing others to give anything of value, either directly or indirectly, to a non-U.S. government official in order to influence official action, or otherwise obtain or retain business. The FCPA also requires public companies to make and keep books and records that accurately and fairly reflect thecertain transactions of the corporation and to devise and maintain an adequate system of internal accounting controls. Our business is heavily regulated and therefore involves significant interaction with public officials, including officials of non-U.S. governments. Additionally, in many other countries, the health care providers who prescribe pharmaceuticals are employed by their government, and the purchasers of pharmaceuticals are government entities; therefore, our dealings with these prescribers and purchasers are subject to regulation under the FCPA. Recently the Securities and Exchange Commission (the "SEC"), and Department of Justice have increased their FCPA enforcement activities with respect to biotechnology and pharmaceutical companies. There is no certainty that all of our employees, agents, contractors, or collaborators, or those of our affiliates, will comply with all applicable laws and regulations, particularly given the high level of complexity of these laws. Violations of these laws and regulations could result in fines, criminal sanctions against us, our officers, or our employees, the closing down of our facilities, requirements to obtain export licenses, cessation of business activities in sanctioned countries, implementation of compliance programs, and prohibitions on the conduct of our business. Any such violations could include prohibitions on our ability to offer our products in one or more countries and could materially damage our reputation, our brand, our international expansion efforts, our ability to attract and retain employees, and our business, prospects, operating results, and financial condition.
In addition, in the future once we enter a commercialization phase, our products may be subject to U.S. and foreign export controls, trade sanctions, and import laws and regulations. Governmental regulation of the import or export of our products, or our failure to obtain any required import or export authorization for our products, when applicable, could harm our international sales and adversely affect our revenue. Compliance with applicable regulatory requirements regarding the export of our products may create delays in the introduction of our products in international markets or, in some cases, prevent the export of our products to some countries altogether. Furthermore, U.S. export control laws and economic sanctions prohibit the shipment of certain products and services to countries, governments, and persons targeted by U.S. sanctions. If we fail to comply with export and import regulations and such economic sanctions, we may be fined or other penalties could be imposed, including a denial of certain export privileges. Moreover, any new export or import restrictions, new legislation or shifting approaches in the enforcement or scope of existing regulations, or in the countries, persons, or technologies targeted by such regulations, could result in decreased use of our products by, or in our decreased ability to export our products to existing or potential customers with international operations. Any limitation on our ability to export or sell access to our products would likely adversely affect our business.
Inadequate funding for the FDA, the SEC and other government agencies could hinder their ability to hire and retain key leadership and other personnel, prevent new products and services from being developed or commercialized in a timely manner, or otherwise prevent those agencies from performing normal business functions on which the operation of our business may rely, which could negatively impact our business.
The ability of the FDA to review and approve new products can be affected by a variety of factors, including government budget and funding levels, ability to hire and retain key personnel and accept the payment of user fees, and statutory, regulatory, and policy changes. Average review times at the agencyFDA have fluctuated in recent years as a result. In addition, government funding of the SEC and other government agencies on which our operations may rely, including those that fund research and development activities is subject to the political process, which is inherently fluid and unpredictable.
Disruptions at the FDA and other government agencies may also slow the time necessary for new drugs to be reviewed and/or approved by necessary government agencies, which would adversely affect our business. For example, over the last several years including from December 22, 2018 until January 25, 2019, the U.S. government has shut down several times and certain regulatory agencies, such as the FDA and the SEC, have had to furlough critical FDA, SEC and other government employees and stop critical activities. If a prolonged government shutdown or other disruption occurs, it could significantly impact the ability of the FDA or other regulatory authorities to timely review and process our regulatory submissions, or to provide feedback on our clinical development plans, which could have a material adverse effect on our business. Further, in our operations as a public company, future government shutdowns or other disruptions to normal operations could impact our ability to access the public markets and obtain the funding necessary capital in order to properly capitalize and continue our operations.
Risks Related to Our Reliance on Third Parties
We expect to depend on collaborations with third parties for the research, development, and commercialization of certain of the product candidates we may develop.candidates. If any such collaborations are not successful, we may not be able to realize the market potential of those product candidates.
We anticipate seeking third-party collaborators for the research, development, and commercialization of certain of the product candidates we may develop. For example, we have collaborations with F-star, Takeda, Sanofi, Biogen, and others to further our development of product candidates and to enhance our research efforts directed to better understanding neurodegenerative diseases.and LSDs. Our likely collaborators for any other collaboration arrangements include large and mid-size pharmaceutical companies, regional and national pharmaceutical companies, biotechnology companies, and academic institutions. If we enter into any such arrangements with any third parties, we will likely have shared or limited control over the amount and timing of resources that our collaborators dedicate to the development or potential commercialization of any product candidates we may seek to develop with them. Our ability to generate revenue from these arrangements with commercial entities will depend on our collaborators’ abilities to successfully perform the functions assigned to them in these arrangements. We cannot predict the success of any collaboration that we enter into.
Collaborations involving our research programs, or any product candidates we may develop, pose the following risks to us:
•collaborators generally have significant discretion in determining the efforts and resources that they will apply to these collaborations;
•collaborators may not properly obtain, maintain, enforce, or defend intellectual property or proprietary rights relating to our product candidates or research programs or may use our proprietary information in such a way as to expose us to potential litigation or other intellectual property related proceedings, including proceedings challenging the scope, ownership, validity and enforceability of our intellectual property;
•collaborators may own or co-own intellectual property covering our product candidates or research programs that results from our collaboration with them, and in such cases, we may not have the exclusive right to commercialize such intellectual property or such product candidates or research programs;
•we may need the cooperation of our collaborators to enforce or defend any intellectual property we contribute to or that arises out of our collaborations, which may not be provided to us;
•collaborators may control certain interactions with regulatory authorities, which may impact on our ability to obtain and maintain regulatory approval of our products candidates;
•disputes may arise between the collaborators and us that result in the delay or termination of the research, development, or commercialization of our product candidates or research programs or that result in costly litigation or arbitration that diverts management attention and resources;
•collaborators may decide to not pursue development and commercialization of any product candidates we develop or may elect not to continue or renew development or commercialization programs based on clinical trial results, changes in the collaborator’s strategic focus or available funding or external factors such as an acquisition that diverts resources or creates competing priorities;
•collaborators may delay clinical trials, provide insufficient funding for a clinical trial program, stop a clinical trial or abandon a product candidate, repeat or conduct new clinical trials, or require a new formulation of a product candidate for clinical testing;
•collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with our product candidates or research programs if the collaborators believe that competitive products are more likely to be successfully developed or can be commercialized under terms that are more economically attractive than ours;
•collaborators may restrict us from researching, developing, or commercializing certain products or technologies without their involvement;
•collaborators with marketing and distribution rights to one or more product candidates may not commit sufficient resources to the marketing and distribution of such product candidates;
•we may lose certain valuable rights under circumstances identified in our collaborations,agreements with our collaborators, including if we undergo a change of control;
•collaborators may grant sublicenses to our technology or product candidates or undergo a change of control and the sublicensees or new owners may decide to take the collaboration in a direction which is not in our best interest;
•collaborators may become bankrupt, which may significantly delay our research or development programs, or may cause us to lose access to valuable technology, know-how, or intellectual property of the collaborator relating to our products, product candidates, or research programs;
•key personnel at our collaborators may leave, which could negatively impact our ability to productively work with our collaborators;
•collaborations may require us to incur short and long-term expenditures, issue securities that dilute our stockholders, or disrupt our management and business;
•if our collaborators do not satisfy their obligations under our agreements with them, or if they terminate our collaborations with them, we may not be able to develop or commercialize product candidates as planned;
•collaborations may require us to share in development and commercialization costs pursuant to budgets that we do not fully control and our failure to share in such costs could have a detrimental impact on the collaboration or our ability to share in revenue generated under the collaboration;
•collaborations may be terminated in their entirety or with respect to certain product candidates or technologies and, if so terminated, may result in a need for additional capital to pursue further development or commercialization of the applicable product candidates or technologies, including our BBB platform technology; and
•collaboration agreements may not lead to development or commercialization of product candidates in the most efficient manner or at all. If a present or future collaborator of ours were to be involved in a business combination, the continued pursuit and emphasis on our development or commercialization program under such collaboration could be delayed, diminished, or terminated.
We may face significant competition in seeking appropriate collaborations. Recent business combinations among biotechnology and pharmaceutical companies have resulted in a reduced number of potential collaborators. In addition, the negotiation process is time-consuming and complex, and we may not be able to negotiate collaborations on a timely basis, on acceptable terms, or at all. If we are unable to do so, we may have to curtail the development of the product candidate for which we are seeking to collaborate, reduce or delay its development program or one or more of our other development programs, delay its potential commercialization or reduce the scope of any sales or marketing activities, or increase our expenditures and undertake development or commercialization activities at our own expense. If we elect to increase our expenditures to fund development or commercialization activities on our own, we may need to obtain additional capital, which may not be available to us on acceptable terms or at all. If we do not have sufficient funds, we may not be able to further develop product candidates or bring them to market and generate product revenue.
If we enter into collaborations to develop and potentially commercialize any product candidates, we may not be able to realize the benefit of such transactions if we or our collaborator elects not to exercise the rights granted under the agreement or if we or our collaborator are unable to successfully integrate a product candidate into existing operations and company culture. The failure to develop and commercialize a product candidate pursuant to our agreements with our current or future collaborators could prevent us from receiving future payments under such agreements, which could negatively impact our revenues. In addition, if our agreement with any of our collaborators terminates, our access to technology and intellectual property licensed to us by that collaborator may be restricted or terminate entirely, which may delay our continued development of our product candidates utilizing the collaborator’s technology or intellectual property or require us to stop development of those product candidates completely. We may also find it more difficult to find a suitable replacement collaborator or attract new collaborators, and our development programs may be delayed or the perception of us in the business and financial communities could be adversely affected. Many of the risks relating to product development, regulatory approval, and commercialization described in this “Risk Factors” section also apply to the activities of our collaborators and any negative impact on our collaborators may adversely affect us.
We expect to rely on third parties to conduct our clinical trials and some aspects of our research and preclinical testing, and those third parties may not perform satisfactorily, including failing to meet deadlines for the completion of such trials, research, or testing.
We currently rely and expect to continue to rely on third parties, such as CROs, clinical data management organizations, medical institutions, and clinical investigators, to conduct some aspects of our research and preclinical testing and our clinical trials. Any of these third parties may terminate their engagements with us or be unable to fulfill their contractual obligations. If we need to enter into alternative arrangements, it would delay our product development activities.
Our reliance on these third parties for research and development activities reduces our control over these activities but does not relieve us of our responsibilities. For example, we remain responsible for ensuring that each of our clinical trials is conducted in accordance with the general investigational plan and protocols for the trial. Moreover, the FDA requires us to comply with cGCPs for conducting, recording, and reporting the results of clinical trials to assure that data and reported results are credible, reproducible, and accurate and that the rights, integrity, and confidentiality of trial participants are protected. We also are required to register ongoing clinical trials and post the results of completed clinical trials on a government-sponsored database within certain time frames. Failure to do so can result in fines, adverse publicity, and civil and criminal sanctions.
Our third-party service providers are not our employees, and we are therefore unable to directly monitor whether or not they devote sufficient time and resources to our clinical and nonclinical programs. These third-party service providers may also have relationships with other commercial entities, including our competitors, for whom they may also be conducting clinical trials or other drug development activities that could harm our competitive position. If these third parties do not successfully carry out their contractual duties, meet expected deadlines, or conduct our clinical trials in accordance with regulatory requirements or our stated protocols, we will not be able to obtain, or may be delayed in obtaining, marketing approvals for any product candidates we may develop and will not be able to, or may be delayed in our efforts to, successfully commercialize our medicines.
We also expect to rely on other third parties to store and distribute drug supplies for our clinical trials. Any performance failure on the part of our distributors, including with the shipment of any drug supplies, could delay clinical development or marketing approval of any product candidates we may develop or commercialization of our medicines, producing additional losses and depriving us of potential product revenue.
We currently contract withOur reliance on third parties for the manufacture of the significant majority of the materials for our research programs, preclinical studies, and clinical trials and expect to continue to do so for commercialization of some or all product candidates that we may develop. This reliance on third parties may increase the risk that we will not have sufficient quantities of such materials, product candidates, or any medicines that we may develop and commercialize, or that such supply will not be available to us at an acceptable cost, which could delay, prevent, or impair our development or commercialization efforts.
WeAlthough we have initiated the build-out of our Utah site to expand our clinical manufacturing capabilities for biologic therapeutics, we do not have any operational manufacturing facilities. We currently rely on third-party manufacturers for the manufacture of our materials for preclinical studies and clinical trials and expect to continue to do so for some or all of our materials for preclinical studies, clinical trials and for commercial supply of any product candidates that we may develop.
We may be unable to establish any further agreements with third-party manufacturers or to do so on acceptable terms. Even if we are able to establish agreements with third-party manufacturers, reliance on third-party manufacturers entails additional risks, including:
•including the possible breach, of the manufacturing agreement by the third party;
•the possible termination, or nonrenewalnon-renewal of the agreement by the third party, at a time that iswhich may be costly or inconvenient, for us;
•relianceand the inability of the third party to produce the required volume in a timely manner. We may also be exposed to the risks of relying on the third party for regulatory compliance, quality assurance, safety, and pharmacovigilance and related reporting; andreporting.
•the inability to produce required volume in a timely manner and to quality standards.
Third-party manufacturers may not be able to comply with U.S. export control regulations, cGMP regulations, or similar regulatory requirements outside the United States. Our failure, or the failure of our third-party manufacturers, to comply with applicable regulations could result in a need to replace current third-party manufacturers including the possibility of supply delays, clinical holds on our trials, sanctions being imposed on us, including fines, injunctions, civil penalties, delays, suspension or withdrawal of approvals, license revocations, seizures or recalls of product candidates or medicines, operating restrictions, and criminal prosecutions, any of which could significantly and adversely affect supplies of our medicines and harm our business, financial condition, results of operations, and growth prospects.
Any medicines that we may develop may compete with other product candidates and products for access to manufacturing facilities. There are a limited number of manufacturers that operate under cGMP regulations and that might be capable of manufacturing for us.
Any performance failure on the part of our existing or future manufacturers could delay clinical development or marketing approval. We do not currently have arrangements in place for redundant supply for anymany components of our product candidates. If any one of our current contract manufacturers cannot perform as agreed, we may be required to replace that manufacturer and may incur added costs and delays in identifying and qualifying any such replacement. Furthermore, securing and reserving production capacity with contract manufacturers may result in significant costs.
Our current and anticipated future dependence upon othersthird parties for the manufacture of any product candidates we may develop or medicines may adversely affect our future profit margins and our ability to commercialize any medicines that receive marketing approval on a timely and competitive basis.
We depend on third-party suppliers for key raw materials used in our manufacturing processes, and the loss of these third-party suppliers or their inability to supply us with adequate raw materials could harm our business.
We rely on third-party suppliers for the raw materials required for the production of our product candidates. Our dependence on these third-party suppliers and the challenges we may face in obtaining adequate supplies of raw materials involve several risks, including limited control over pricing, availability, quality and delivery schedules. As a small company, our negotiation leverage is limited and we are likely to get lower priority than our competitors who are larger than we are.competitors. We cannot be certain that our suppliers will continue to provide us with the quantities of these raw materials that we require or satisfy our anticipated specifications and quality requirements. Any supply interruption
Further, we have in limited or sole sourcedthe past and may in the future experience delayed shipments of raw materials could materially harm our abilitydue to manufacture our product candidates until a new source of supply, if any, could be identified and qualified.interruptions relating to the aforementioned events. We may be unable to find a sufficient alternative supply channel in a reasonable time or on commercially reasonable terms. Any performance failure on the part of our suppliers could delay the development and potential commercialization of our product candidates, including limiting supplies necessary for clinical trials and regulatory approvals, which would have a material adverse effect on our business.
Risks Related to Our Intellectual Property
If we are unable to obtain and maintain patent protection for any product candidates we develop or for our BBB platform technology, our competitors could develop and commercialize products or technology similar or identical to ours, and our ability to successfully commercialize any product candidates we may develop, and our technology may be adversely affected.
Our success depends in large part on our ability to obtain and maintain patent protection in the United States and other countries with respect to our BBB platform technology and any proprietary product candidates and other technologies we may develop. We seek to protect our proprietary position by in-licensing intellectual property and filing patent applications in the United States and abroad relating to our BBB platform technology, programs and product candidates, as well as other technologies that are important to our business. Given that the development of our technology and product candidates is at an early stage, our intellectual property portfolio with respect to certain aspects of our technology and product candidates is also at an early stage. In addition, we cannot be certain that any patents we own or in-license in the United States adequately cover the Fc domain portion of our BBB platform technology that binds to transferrin receptor, or adequately cover the antibodies, enzymes or proteins being developed in our ATV:TREM2, ETV:IDS, ETV:SGSH, ETV:IDUA, PTV:PGRN, ATV:Abeta, OTV, or PTV:PGRNother TV-enabled programs. We have filed or intend to file patent applications on these aspects of our technology and product candidates; however, there can be no assurance that any such patent applications will issue as granted patents. Furthermore, in some cases, we have only filed provisional patent applications on certain aspects of our technology and product candidates and each of these provisional patent applications is not eligible to become an issued patent until, among other things, we file a non-provisional patent application within 12twelve months of the filing date of the applicable provisional patent application. Any failure to file a non-provisional patent application within this timeline could cause us to lose the ability to obtain patent protection for the inventions disclosed in the associated provisional patent applications. Furthermore, in some cases, we may not be able to obtain issued claims covering compositions relating to our BBB platform technology, programs and product candidates, as well as other technologies that are important to our business, and instead may need to rely on filing patent applications with claims covering a method of use and/or method of manufacture for protection of such BBB platform technology, programs, product candidates, and other technologies. There can be no assurance that any such patent applications will issue as granted patents, and even if they do issue, such patent claims may be insufficient to prevent third parties, such as our competitors, from utilizing our technology. Any failure to obtain or maintain patent protection with respect to our BBB platform technology, programs and product candidates could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
If any of our owned or in-licensed patent applications do not issue as patents in any jurisdiction, we may not be able to compete effectively.
Changes in either the patent laws or their interpretation in the United States and other countries may diminish our ability to protect our inventions, obtain, maintain, and enforce our intellectual property rights and, more generally, could affect the value of our intellectual property or narrow the scope of our owned and licensed patents. With respect to both in-licensed and owned intellectual property, we cannot predict whether the patent applications we and our licensors are currently pursuing will issue as patents in any particular jurisdiction or whether the claims of any issued patents will provide sufficient protection from competitors or other third parties.
The patent prosecution process is expensive, time-consuming, and complex, and we may not be able to file, prosecute, maintain, enforce, or license all necessary or desirable patent applications at a reasonable cost or in a timely manner.manner, including delays as a result of the COVID-19 pandemic impacting our or our licensors' operations. It is also possible that we will fail to identify patentable aspects of our research and development output in time to obtain patent protection. Although we enter into nondisclosure and confidentiality agreements with parties who have access to confidential or patentable aspects of our research and development output, such as our employees, corporate collaborators, outside scientific collaborators, CROs, contract manufacturers, consultants, advisors, and other third parties, any of these parties may breach the agreements and disclose such output before a patent application is filed, thereby jeopardizing our ability to seek patent protection. In addition, our ability to obtain and maintain valid and enforceable patents depends on whether the differences between our inventions and the prior art allow our inventions to be patentable over the prior art. Furthermore, publications of discoveries in the scientific literature often lag behind the actual discoveries, and patent applications in the United States and other jurisdictions are typically not published until 18 months after filing, or in some cases not at all. Therefore, we cannot be certain that we or our licensors were the first to make the inventions claimed in any of our owned or licensed patents or pending patent applications, or that we or our licensors were the first to file for patent protection of such inventions.
If the scope of any patent protection we obtain is not sufficiently broad, or if we lose any of our patent protection, our ability to prevent our competitors from commercializing similar or identical technology and product candidates would be adversely affected.
The patent position of biotechnology and pharmaceutical companies generally is highly uncertain, involves complex legal and factual questions, and has been the subject of much litigation in recent years. As a result, the issuance, scope, validity, enforceability, and commercial value of our patent rights are highly uncertain. Our owned or in-licensed pending and future patent applications may not result in patents being issued which protect our BBB platform technology, product candidates or other technologies or which effectively prevent others from commercializing competitive technologies and product candidates.
Moreover, the coverage claimed in a patent application can be significantly reduced before the patent is issued, and its scope can be reinterpreted after issuance. Even if patent applications we license or own currently or in the future issue as patents, they may not issue in a form that will provide us with any meaningful protection, prevent competitors or other third parties from competing with us, or otherwise provide us with any competitive advantage. Any patents that we own or in-license may be challenged, narrowed, circumvented, or invalidated by third parties. Consequently, we do not know whether our BBB platform technology, product candidates or other technologies will be protectable or remain protected by valid and enforceable patents. Our competitors or other third parties may be able to circumvent our patents by developing similar or alternative technologies or products in a non-infringing manner which could materially adversely affect our business, financial condition, results of operations and growth prospects.
The issuance of a patent is not conclusive as to its inventorship, scope, validity, or enforceability, and our patents may be challenged in the courts or patent offices in the United States and abroad. We or our licensors may be subject to a third-party preissuance submission of prior art to the USPTO, or become involved in opposition, derivation, revocation, reexamination, post-grant and inter partes review, or interference proceedings or other similar proceedings challenging our owned or licensed patent rights. An adverse determination in any such submission, proceeding or litigation could reduce the scope of, or invalidate or render unenforceable, our owned or in-licensed patent rights, allow third parties to commercialize our BBB platform technology, product candidates or other technologies and compete directly with us, without payment to us, or result in our inability to manufacture or commercialize products without infringing third-party patent rights. Moreover, we, or one of our licensors, may have to participate in interference proceedings declared by the USPTO to determine priority of invention or in post-grant challenge proceedings, such as oppositions in a foreign patent office, that challenge our or our licensor’s priority of invention or other features of patentability with respect to our owned or in-licensed patents and patent applications. Such challenges may result in loss of patent rights, loss of exclusivity, or in patent claims being narrowed, invalidated, or held unenforceable, which could limit our ability to stop others from using or commercializing similar or identical technology and products, or limit the duration of the patent protection of our BBB platform technology, product candidates and other technologies. Such proceedings also may result in substantial cost and require significant time from our scientists and management, even if the eventual outcome is favorable to us.If we or our collaborators are unsuccessful in any such proceeding or other priority or inventorship dispute, we may be required to obtain and maintain licenses from third parties, including parties involved in any such interference proceedings or other priority or inventorship disputes. Such licenses may not be available on commercially reasonable terms or at all, or may be non-exclusive. If we are unable to obtain and maintain such licenses, we may need to cease the development, manufacture, and commercialization of one or more of the product candidates we may develop. The loss of exclusivity or the narrowing of our owned and licensed patent claims could limit our ability to stop others from using or commercializing similar or identical technology and products.
In addition, given the amount of time required for the development, testing, and regulatory review of new product candidates, patents protecting such product candidates might expire before or shortly after such product candidates are commercialized. As a result, our intellectual property may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours.
Some of our owned and in-licensed patents and patent applications are, and may in the future be, co-owned with third parties. For example, we may currently, and may in the future, co-own certain patents and patent applications relating to our BBB platform technology with F-star. In addition, certain of our licensors co-own the patents and patent applications we in-license with other third parties with whom we do not have a direct relationship. Our exclusive rights to certain of these patents and patent applications are dependent, in part, on inter-institutional or other operating agreements between the joint owners of such patents and patent applications, who are not parties to our license agreements. If our licensors do not have exclusive control of the grant of licenses under any such third-party co-owners’ interest in such patents or patent applications or we are otherwise unable to secure such exclusive rights, such co-owners may be able to license their rights to other third parties, including our competitors, and our competitors could market competing products and technology. In addition, we may need the cooperation of any such co-owners of our patents in order to enforce such patents against third parties, and such cooperation may not be provided to us. Any of the foregoing could have a material adverse effect on our competitive position, business, financial conditions, results of operations, and growth prospects.
Our rights to develop and commercialize our BBB platform technology and product candidates are subject, in part, to the terms and conditions of licenses granted to us by others or licenses granted by us to others.
We are heavily reliant upon licenses to certain patent rights and proprietary technology from third parties that are important or necessary to the development of our BBB platform technology and product candidates. For example, in June 2016, we entered into a license agreement with Genentech pursuant to which we received an exclusive license to certain of Genentech’s intellectual property relating to our LRRK2 program, including our BIIB122/DNL151 (BIIB122) product candidate.
Our agreements with F-star and other license agreements may not provide exclusive rights to use certain licensed intellectual property and technology in all relevant fields of use and in all territories in which we may wish to develop or commercialize our technology and products in the future. For example, F-star retains the right to use itself, and to license to others, its modular antibody technology for any purpose other than the targets which we have agreed with F-star would or may be exclusively available to us. As a result, we may not be able to prevent competitors or other third parties from developing and commercializing competitive products that also utilizes technology that we have in-licensed.
In addition, subject to the terms of any such license agreements, we do not have the right to control the preparation, filing, prosecution and maintenance, and we may not have the right to control the enforcement, and defense of patents and patent applications covering the technology that we license from third parties. For example, under our agreements with F-star and Genentech, the licensors control prosecution and, in the case of F-star and in specified circumstances, enforcement of certain of the patents and patent applications licensed to us. Also, under our agreements with Takeda, Sanofi and Biogen, they control prosecution, and in specified circumstances, enforcement of certain of the patents and patent applications licensed to them. We cannot be certain that our in-licensed or out-licensed patents and patent applications that are controlled by our licensors or licensees will be prepared, filed, prosecuted, maintained, enforced, and defended in a manner consistent with the best interests of our business. If our licensors or licensees fail to prosecute, maintain, enforce, and defend such patents, or lose rights to those patents or patent applications, the rights we have licensed may be reduced or eliminated, our right to develop and commercialize our BBB platform technology and any of our product candidates that are subject of such licensed rights could be adversely affected, and we may not be able to prevent competitors from making, using and selling competing products. In addition, even where we have the right to control patent prosecution of patents and patent applications we have licensed to and from third parties, we may still be adversely affected or prejudiced by actions or inactions of our licensees, our licensors and their counsel that took place prior to the date upon which we assumed control over patent prosecution.
Furthermore, our owned and in-licensed patents may be subject to a reservation of rights by one or more third parties. For example, our license to certain intellectual property owned by Genentech is subject to certain research rights Genentech granted to third parties prior to our license agreement. In addition, certain of our in-licensed intellectual property relating to RIPK1 was funded in part by the U.S. government. As a result, the U.S. government may have certain rights to such intellectual property. When new technologies are developed with U.S. government funding, the U.S. government generally obtains certain rights in any resulting patents, including a non-exclusive license authorizing the U.S. government to use the invention or to have others use the invention on its behalf. The U.S. government’s rights may also permit it to disclose the funded inventions and technology to third parties and to exercise march-in rights to use or allow third parties to use the technology we have licensed that was developed using U.S. government funding. The U.S. government may exercise its march-in rights if it determines that action is necessary because we fail to achieve practical application of the government-funded technology, or because action is necessary to alleviate health or safety needs, to meet requirements of federal regulations, or to give preference to U.S. industry. In addition, our rights in such inventions may be subject to certain requirements to manufacture products embodying such inventions in the United States in certain circumstances and if this requirement is not waived. Any exercise by the U.S. government of such rights or by any third party of its reserved rights could have a material adverse effect on our competitive position, business, financial condition, results of operations and growth prospects.
If we fail to comply with our obligations in the agreements under which we license intellectual property rights from third parties or otherwise experience disruptions to our business relationships with our licensors, we could lose license rights that are important to our business.
We have entered into license agreements with third parties and may need to obtain additional licenses from others to advance our research or allow commercialization of product candidates we may develop or our BBB platform technology. It is possible that we may be unable to obtain additional licenses at a reasonable cost or on reasonable terms, if at all. In that event, we may be required to expend significant time and resources to redesign our technology, product candidates, or the methods for manufacturing them or to develop or license replacement technology, all of which may not be feasible on a technical or commercial basis. If we are unable to do so, we may be unable to develop or commercialize the affected product candidates or continue to utilize our existing BBB platform technology, which could harm our business, financial condition, results of operations, and growth prospects significantly. We cannot provide any assurances that third-party patents do not exist which might be enforced against our current technology, including our BBB platform technology, manufacturing methods, product candidates, or future methods or products resulting in either an injunction prohibiting our manufacture or future sales, or, with respect to our future sales, an obligation on our part to pay royalties and/or other forms of compensation to third parties, which could be significant.
In addition, each of our current license agreements, and we expect our future agreements, will impose various development, diligence, commercialization, and other obligations on us. Certain of our license agreements also require us to meet development timelines, or to exercise commercially reasonable efforts to develop and commercialize licensed products, in order to maintain the licenses. In spite of our efforts, our licensors might conclude that we have materially breached our obligations under such license agreements and might therefore terminate the license agreements, thereby removing or limiting our ability to develop and commercialize products and technology covered by these license agreements. If these in-licenses are terminated, or if the underlying patents fail to provide the intended exclusivity, competitors or other third parties would have the freedom to seek regulatory approval of, and to market, products identical to ours and we may be required to cease our development and commercialization of certain of our product candidates or of our current BBB platform technology. Any of the foregoing could have a material adverse effect on our competitive position, business, financial conditions, results of operations, and growth prospects.
Moreover, disputes may arise regarding intellectual property subject to a licensing agreement, including:
•the scope of rights granted under the license agreement and other interpretation-related issues;
•the extent to which our technology and processes infringe on intellectual property of the licensor that is not subject to the licensing agreement;
•the sublicensing of patent and other rights under our collaborative development relationships;
•our diligence obligations under the license agreement and what activities satisfy those diligence obligations;
•the inventorship and ownership of inventions and know-how resulting from the joint creation or use of intellectual property by our licensors and us and our partners; and
•the priority of invention of patented technology.
In addition, the agreements under which we currently license intellectual property or technology from third parties are complex, and certain provisions in such agreements may be susceptible to multiple interpretations. The resolution of any contract interpretation disagreement that may arise could narrow what we believe to be the scope of our rights to the relevant intellectual property or technology, or increase what we believe to be our financial or other obligations under the relevant agreement, either of which could have a material adverse effect on our business, financial condition, results of operations and growth prospects. Moreover, if disputes over intellectual property that we have licensed prevent or impair our ability to maintain our current licensing arrangements on commercially acceptable terms, we may be unable to successfully develop and commercialize the affected product candidates, which could have a material adverse effect on our business, financial conditions, results of operations and growth prospects.
In addition, the United States federal government retains certain rights in inventions produced with its financial assistance under the Patent and Trademark Law Amendments Act, or the Bayh-Dole Act. The federal government retains a “nonexclusive, nontransferable, irrevocable, paid-up license” for its own benefit. The Bayh-Dole Act also provides federal agencies with “march-in rights.” March-in rights allow the government, in specified circumstances, to require the contractor or successors in title to the patent to grant a “nonexclusive, partially exclusive, or exclusive license” to a “responsible applicant or applicants.” If the patent owner refuses to do so, the government may grant the license itself. If, in the future, we co-own or license in technology which is critical to our business that is developed in whole or in part with federal funds subject to the Bayh-Dole Act, our ability to enforce or otherwise exploit patents covering such technology may be adversely affected
We may not be able to protect our intellectual property and proprietary rights throughout the world.
Filing, prosecuting, and defending patents on our BBB platform technology, product candidates and other technologies in all countries throughout the world would be prohibitively expensive, and the laws of foreign countries may not protect our rights to the same extent as the laws of the United States. Further, our ability to pursue patents throughout the world may be delayed or affected due to the COVID-19 global pandemic. Consequently, we may not be able to prevent third parties from practicing our inventions in all countries outside the United States, or from selling or importing products made using our inventions in and into the United States or other jurisdictions. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and, further, may export otherwise infringing products to territories where we have patent protection but enforcement is not as strong as that in the United States. These products may compete with our products, and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents, trade secrets, and other intellectual property protection, particularly those relating to biotechnology products, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our intellectual property and proprietary rights generally. European applications will soon have the option, upon grant of a patent, of becoming a Unitary Patent which will be subject to the jurisdiction of the Unitary Patent Court ("UPC"). This will be a significant change in European patent practice. As the UPC is a new court system, there is no precedent for the court, increasing the uncertainty. Proceedings to enforce our intellectual property and proprietary rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly, could put our patent applications at risk of not issuing, and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate, and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property and proprietary rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
TableGeopolitical actions in the United States and in foreign countries could increase the uncertainties and costs surrounding the prosecution or maintenance of Contentsour patent applications or those of any current or future licensors and the maintenance, enforcement or defense of our issued patents or those of any current or future licensors. For example, the United States and foreign government actions related to Russia's invasion of Ukraine may limit or prevent filing, prosecution, and maintenance of patent applications in Russia. Government actions may also prevent maintenance of issued patents in Russia. These actions could result in abandonment or lapse of our patents or patent applications, resulting in partial or complete loss of patent rights in Russia. If such an event were to occur, it could have a material adverse effect on our business. In addition, a decree was adopted by the Russian government in March 2022, allowing Russian companies and individuals to exploit inventions owned by patentees that have citizenship or nationality in, are registered in, or predominately have primary place of business or profit-making activities in the United States and other countries that Russia has deemed unfriendly without consent or compensation. Consequently, we may be unable to prevent third parties from practicing our inventions in Russia or from selling or importing products made using our inventions in and into Russia. Accordingly, our competitive position may be impaired, and our business, financial condition, results of operations and prospects may be adversely affected.
Many countries have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. In addition, many countries limit the enforceability of patents against government agencies or government contractors. In these countries, the patent owner may have limited remedies, which could materially diminish the value of such patent. If we or any of our licensors is forced to grant a license to third parties with respect to any patents relevant to our business, our competitive position may be impaired, and our business, financial condition, results of operations, and growth prospects may be adversely affected.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment, and other requirements imposed by government patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees, renewal fees, annuity fees, and various other government fees on patents and applications will be due to be paid to the USPTO and various government patent agencies outside of the United States over the lifetime of our owned or licensed patents and applications. In certain circumstances, we rely on our licensing partners to pay these fees due to U.S. and non-U.S. patent agencies. The USPTO and various non-U.S. government agencies require compliance with several procedural, documentary, fee payment, and other similar provisions during the patent application process. We are also dependent on our licensors to take the necessary action to comply with these requirements with respect to our licensed intellectual property. In some cases, an inadvertent lapse can be cured by payment of a late fee or by other means in accordance with the applicable rules. There are situations, however, in which non-compliance can result in abandonment or lapse of the patent or patent application, resulting in a partial or complete loss of patent rights in the relevant jurisdiction. In such an event, potential competitors might be able to enter the market with similar or identical products or technology, which could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
Geopolitical actions in the United States and in foreign countries could prevent us from continuing to make these periodic payments in certain locations. For example, the United States and foreign government actions related to Russia's invasion of Ukraine may limit our ability to make or prevent us from making these payments in Russia. These actions could result in abandonment or lapse of our patents or patent applications, resulting in partial or complete loss of patent rights in Russia, which could adversely affect our business.
Changes in U.S. patent law could diminish the value of patents in general, thereby impairing our ability to protect our products.
Changes in either the patent laws or interpretation of the patent laws in the United States could increase the uncertainties and costs surrounding the prosecution of patent applications and the enforcement or defense of issued patents. Assuming that other requirements for patentability are met, prior to March 2013, in the United States, the first to invent the claimed invention was entitled to the patent, while outside the United States, the first to file a patent application was entitled to the patent. After March 2013, under the Leahy-Smith America Invents Act (the "America Invents Act"), enacted in September 2011, the United States transitioned to a first inventor to file system in which, assuming that other requirements for patentability are met, the first inventor to file a patent application will be entitled to the patent on an invention regardless of whether a third party was the first to invent the claimed invention. A third party that files a patent application in the USPTO after March 2013, but before us could therefore be awarded a patent covering an invention of ours even if we had made the invention before it was made by such third party. This will require us to be cognizant going forward of the time from invention to filing of a patent application. Since patent applications in the United States and most other countries are confidential for a period of time after filing or until issuance, we cannot be certain that we or our licensors were the first to either (i) file any patent application related to our BBB platform technology, product candidates or other technologies or (ii) invent any of the inventions claimed in our or our licensor’s patents or patent applications.
The America Invents Act also includes a number of significant changes that affect the way patent applications will be prosecuted and also may affect patent litigation. These include allowing third-party submission of prior art to the USPTO during patent prosecution and additional procedures to attack the validity of a patent by USPTO administered post-grant proceedings, including post-grant review, inter partes review, and derivation proceedings. Because of a lower evidentiary standard in USPTO proceedings compared to the evidentiary standard in United States federal courts necessary to invalidate a patent claim, a third party could potentially provide evidence in a USPTO proceeding sufficient for the USPTO to hold a claim invalid even though the same evidence would be insufficient to invalidate the claim if first presented in a district court action. Accordingly, a third party may attempt to use the USPTO procedures to invalidate our patent claims that would not have been invalidated if first challenged by the third party as a defendant in a district court action. Therefore, the America Invents Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our owned or in-licensed patent applications and the enforcement or defense of our owned or in-licensed issued patents, all of which could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
In addition, the patent positions of companies in the development and commercialization of biologics and pharmaceuticals are particularly uncertain. Recent U.S. Supreme Court rulings have narrowed the scope of patent protection available in certain circumstances and weakened the rights of patent owners in certain situations. For example, the Supreme Court of the United States held in Amgenv. Sanofi (2023) that a functionally claimed genus was invalid for failing to comply with the enablement requirement of the Patent Act. In addition, the Federal circuit recently issued a decision involving the interaction of patent term adjustment (PTA), terminal disclaimers, and obvious-type double patenting. This combination of events has created uncertainty with respect to the validity and enforceability of patents, once obtained. Depending on future actions by the U.S. Congress, the federal courts, and the USPTO, the laws and regulations governing patents could change in unpredictable ways that could have a material adverse effect on our existing patent portfolio and our ability to protect and enforce our intellectual property in the future. For example, in Assoc. for Molecular Pathology v. Myriad Genetics, Inc. (2013), the U.S. Supreme Court held that certain claims to DNA molecules are not patentable. While we do not believe that any of the patents owned or licensed by us will be found invalid based on this decision, we cannot predict how future decisions by the courts, the U.S. Congress or the USPTO may impact the value of our patents.
Issued patents covering our BBB platform technology, product candidates and other technologies could be found invalid or unenforceable if challenged in court or before administrative bodies in the United States or abroad.
If we or one of our licensors initiated legal proceedings against a third party to enforce a patent covering our BBB platform technology, product candidates or other technologies, the defendant could counterclaim that such patent is invalid or unenforceable or raise a defense to infringement. In patent litigation in the United States, defendant counterclaims alleging invalidity or unenforceability are commonplace. Grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements, including lack of subject matter eligibility for patenting, novelty, obviousness, or non-enablement. Grounds for an unenforceability assertion could be an allegation that someone connected with prosecution of the patent withheld relevant information from the USPTO, or made a misleading statement, during prosecution. Grounds for defenses to infringement include statutory exemptions to patent infringement for uses related to submitting information to regulatory authorities to seek certain regulatory approvals. Third parties may raise claims challenging the validity or enforceability of our owned or in-licensed patents before administrative bodies in the United States or abroad, even outside the context of litigation. Such mechanisms include re-examination, post-grant review, inter partes review, interference proceedings, derivation proceedings, and equivalent proceedings in foreign jurisdictions (e.g., opposition proceedings). Such proceedings could result in the revocation of, cancellation of, or amendment to our patents in such a way that they no longer cover our BBB platform technology, product candidates or other technologies. The outcome following legal assertions of invalidity and unenforceability is unpredictable. With respect to the validity question, for example, a judge or jury could find that our patent claims laws of nature or are otherwise ineligible for patenting, and we cannot be certain that there is no invalidating prior art, of which we or our licensing partners and the patent examiner were unaware during prosecution. If a third party were to prevail on a legal assertion of invalidity or unenforceability, we would lose at least part, and perhaps all, of the patent protection on our BBB platform technology, product candidates or other technologies. Such a loss of patent protection would have a material adverse impact on our business, financial condition, results of operations and growth prospects.
If we do not obtain patent term extension and data exclusivity for any product candidates we may develop, our business may be materially harmed.
Depending upon the timing, duration and specifics of any FDA marketing approval of anyPatent terms may be inadequate to protect our competitive position on our product candidates for an adequate amount of time.
Patent rights are of limited duration. In the United States, if all maintenance fees are paid timely, the natural expiration of a patent is generally 20 years after its first effective filing date. Given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such product candidates are commercialized. Even if patents covering our product candidates are obtained, once the patent life has expired for a product, we may develop, onebe open to competition from biosimilar or moregeneric products. As a result, our patent portfolio may not provide us with sufficient rights to exclude others from commercializing product candidates similar or identical to ours. Upon issuance in the United States, the term of our owned or in-licensed U.S. patents maya patent can be eligible for limitedincreased by patent term extension underadjustment, which is based on certain delays caused by the Hatch-Waxman Act.USPTO, but this increase can be reduced or eliminated based on certain delays caused by the patent applicant during patent prosecution. The Hatch-Waxman Act permitterm of a United States patent term extension of up to five years as compensation formay also be shortened if the patent term lost during the FDA regulatory review process.is terminally disclaimed over an earlier-filed patent. A patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval, only one patent(PTE) based on regulatory delay may be extended and only those claims covering the approved drug, a method for using it, or a method for manufacturing it may be extended. Similar extensions as compensation for patent term lost during regulatory review processes are also available in certainthe United States. However, only a single patent can be extended for each marketing approval, and any patent can be extended only once, for a single product. Moreover, the scope of protection during the period of the PTE does not extend to the full scope of the claim, but instead only to the scope of the product as approved. Laws governing analogous PTEs in foreign countries and territories, suchjurisdictions vary widely, as in Europe underdo laws governing the ability to obtain multiple patents from a Supplementary Patent Certificate. However,single patent family. Additionally, we may not be grantedreceive an extension in the United States and/or foreign countries and territories because of, for example, failingif we fail to exercise due diligence during the testing phase or regulatory review process, failing to apply within applicable deadlines, failingfail to apply prior to expiration of relevant patents or otherwise failingfail to satisfy applicable requirements. Moreover, the applicable time period or the scope of patent protection afforded could be less than we request. If we are unable to obtain patent term extensionPTE or restoration, or the term of any such extension is shorterless than what we request, the period during which we will have the right to exclusively market our product will be shortened and our competitors may obtain approval of competing products following our patent expiration and may take advantage of our business, financial condition, results of operationsinvestment in development and growth prospectsclinical trials by referencing our clinical and nonclinical data to launch their product earlier than might otherwise be the case, and our revenue could be materially harmed.reduced, possibly materially.
We may be subject to claims challenging the inventorship of our patents and other intellectual property.
We or our licensors may be subject to claims that former employees, collaborators, or other third parties have an interest in our owned or in-licensed patents, trade secrets, or other intellectual property as an inventor or co-inventor. For example, we or our licensors may have inventorship disputes arise from conflicting obligations of employees, consultants or others who are involved in developing our BBB platform technology, product candidates, or other technologies. Litigation may be necessary to defend against these and other claims challenging inventorship or our or our licensors’ ownership of our owned or in-licensed patents, trade secrets, or other intellectual property. If we or our licensors fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights, such as exclusive ownership of, or right to use, intellectual property that is important to our BBB platform technology, product candidates and other technologies. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees. Any of the foregoing could have a material adverse effect on our business, financial condition, results of operations, and growth prospects.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed.
In addition to seeking patents for our BBB platform technology, product candidates, and other technologies, we also rely on trade secrets and confidentiality agreements to protect our unpatented know-how, technology, and other proprietary information and to maintain our competitive position. Trade secrets and know-how can be difficult to protect. We expect our trade secrets and know-how to over time be disseminated within the industry through independent development, the publication of journal articles describing the methodology, and the movement of personnel from academic to industry scientific positions.
We seek to protect these trade secrets and other proprietary technology, in part, by entering into nondisclosure and confidentiality agreements with parties who have access to them, such as our employees, corporate collaborators, outside scientific collaborators, CROs, contract manufacturers, consultants, advisors, and other third parties. We also enter into confidentiality and invention or patent assignment agreements with our employees and consultants as well as train our employees not to bring or use proprietary information or technology from former employers to us or in their work, and remind former employees when they leave their employment of their confidentiality obligations. We cannot guarantee that we have entered into such agreements with each party that may have or have had access to our trade secrets or proprietary technology and processes. Despite our efforts, any of these parties may breach the agreements and disclose our proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for such breaches. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive, and time-consuming, and the outcome is unpredictable. In addition, some courts inside and outside the United States are less willing or unwilling to protect trade secrets. If any of our trade secrets were to be lawfully obtained or independently developed by a competitor or other third party, we would have no right to prevent them from using that technology or information to compete with us. If any of our trade secrets were to be disclosed to or independently developed by a competitor or other third party, our competitive position would be materially and adversely harmed.
We may not be successful in obtaining, through acquisitions, in-licenses, or otherwise, necessary rights to our BBB platform technology, product candidates or other technologies.
We currently have rights to intellectual property, through licenses from third parties, to identify and develop our BBB platform technology and product candidates. Many pharmaceutical companies, biotechnology companies, and academic institutions are competing with us in the fieldfields of neurodegenerationneurodegenerative and LSDs and BBB technology andor may have patents and have filed and are likely filingplan to file patent applications potentially relevant to our business. In order to avoid infringing these third-party patents, we may find it necessary or prudent to obtain licenses to such patents from such third-party intellectual property holders. We may also require licenses from third parties for certain BBB technologies that we are evaluating for use with our current or future product candidates. In addition, with respect to any patents we co-own with third parties, we may require licenses to such co-owners’ interest to such patents. However, we may be unable to secure such licenses or otherwise acquire or in-license any compositions, methods of use, processes, or other intellectual property rights from third parties that we identify as necessary for our current or future product candidates and our BBB platform technology. The licensing or acquisition of third-party intellectual property rights is a competitive area, and several more established companies may pursue strategies to license or acquire third-party intellectual property rights that we may consider attractive or necessary. These established companies may have a competitive advantage over us due to their size, capital resources, and greater clinical development and commercialization capabilities. In addition, companies that perceive us to be a competitor may be unwilling to assign or license rights to us. We also may be unable to license or acquire third-party intellectual property rights on terms that would allow us to make an appropriate return on our investment or at all. If we are unable to successfully obtain rights to required third-party intellectual property rights or maintain the existing intellectual property rights we have, we may have to abandon development of the relevant program or product candidate, which could have a material adverse effect on our business, financial condition, results of operations, and growth prospects.
We may be subject to claims that our employees, consultants, or advisors have wrongfully used or disclosed alleged trade secrets of their current or former employers or claims asserting ownership of what we regard as our own intellectual property.
Many of our employees, consultants, and advisors are currently or were previously employed at universities or other biotechnology or pharmaceutical companies, including our licensors, competitors, and potential competitors. Although we try to ensure that our employees, consultants, and advisors do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or these individuals have used or disclosed intellectual property, including trade secrets or other proprietary information, of any such individual’s current or former employer. Litigation may be necessary to defend against these claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management.
In addition, while it is our policy to require our employees and contractors who may be involved in the conception or development of intellectual property to execute agreements assigning such intellectual property to us, we may be unsuccessful in executing such an agreement with each party who, in fact, conceives or develops intellectual property that we regard as our own. The assignment of intellectual property rights may not be self-executing or the assignment agreements may be breached, and we may be forced to bring claims against third parties or defend claims that they may bring against us to determine the ownership of what we regard as our intellectual property. Such claims could have a material adverse effect on our business, financial condition, results of operations, and growth prospects.
Third partyThird-party claims of intellectual property infringement, misappropriation, or other violation against us, our licensors, or our collaborators may prevent or delay the development and commercialization of our BBB platform technology, product candidates, and other technologies.
The fieldfields of discovering treatments for neurodegenerative diseases,and LSDs, especially using BBB technology, is highly competitive and dynamic. Due to the focused research and development that is taking place by several companies, including us and our competitors, in this field,these fields, the intellectual property landscape is in flux, and it may remain uncertain in the future. As such, there may be significant intellectual property litigation and proceedings relating to our owned, and in-licensed, and other third-party intellectual property and proprietary rights in the future.
Our commercial success depends in part on our, our licensors’ and our collaborators’ ability to avoid infringing, misappropriating, and otherwise violating the patents and other intellectual property rights of third parties. There is a substantial amount of complex litigation involving patents and other intellectual property rights in the biotechnology and pharmaceutical industries, as well as administrative proceedings for challenging patents, including interference, derivation, and reexamination proceedings before the USPTO or oppositions and other comparable proceedings in foreign jurisdictions. As discussed above, recently, due to changes in U.S. law referred to as patent reform, new procedures including inter partes review and post-grant review have been implemented. As stated above, this reform adds uncertainty to the possibility of challenge to our patents in the future.
Numerous U.S. and foreign issued patents and pending patent applications owned by third parties exist relating to BBB technology and in the fields in which we are developing our product candidates. As the biotechnology and pharmaceutical industries expand and more patents are issued, the risk increases that our BBB platform technology, product candidates, and other technologies may give rise to claims of infringement of the patent rights of others. We cannot assure you that our BBB platform technology, product candidates, and other technologies that we have developed, are developing or may develop in the future will not infringe existing or future patents owned by third parties. We may not be aware of patents that have already been issued and that a third party, for example, a competitor in the fields in which we are developing our BBB platform technology, product candidates, and other technologies might assert are infringed by our current or future BBB platform technology, product candidates or other technologies, including claims to compositions, formulations, methods of manufacture or methods of use or treatment that cover our BBB platform technology, product candidates, or other technologies. It is also possible that patents owned by third parties of which we are aware, but which we do not believe are relevant to our BBB platform technology, product candidates, or other technologies, could be found to be infringed by our BBB platform technology, product candidates, or other technologies. In addition, because patent applications can take many years to issue, there may be currently pending patent applications that may later result in issued patents that our BBB platform technology, product candidates, or other technologies may infringe.
Third parties may have patents or obtain patents in the future and claim that the manufacture, use, or sale of our BBB platform technology, product candidates, or other technologies infringes upon these patents. In the event that any third-party claims that we infringe their patents or that we are otherwise employing their proprietary technology without authorization and initiates litigation against us, even if we believe such claims are without merit, a court of competent jurisdiction could hold that such patents are valid, enforceable, and infringed by our BBB platform technology, product candidates, or other technologies. In this case, the holders of such patents may be able to block our ability to commercialize the applicable product candidate or technology unless we obtain a license under the applicable patents, or until such patents expire or are finally determined to be held invalid or unenforceable. Such a license may not be available on commercially reasonable terms or at all. Even if we are able to obtain a license, the license would likely obligate us to pay license fees or royalties or both, and the rights granted to us might be nonexclusive, which could result in our competitors gaining access to the same intellectual property. If we are unable to obtain a necessary license to a third-party patent on commercially reasonable terms, we may be unable to commercialize our BBB platform technology, product candidates, or other technologies, or such commercialization efforts may be significantly delayed, which could in turn significantly harm our business.
Defense of infringement claims, regardless of their merit, would involve substantial litigation expense and would be a substantial diversion of management and other employee resources from our business, and may impact our reputation. In the event of a successful claim of infringement against us, we may be enjoined from further developing or commercializing our infringing BBB platform technology, product candidates, or other technologies. In addition, we may have to pay substantial damages, including treble damages and attorneys’ fees for willful infringement, obtain one or more licenses from third parties, pay royalties, and/or redesign our infringing product candidates or technologies, which may be impossible or require substantial time and monetary expenditure. In that event, we would be unable to further develop and commercialize our BBB platform technology, product candidates, or other technologies, which could harm our business significantly.
Engaging in litigation to defend against third parties alleging that we have infringed, misappropriated, or otherwise violated their patents or other intellectual property rights is very expensive, particularly for a company of our size, and time-consuming. Some of our competitors may be able to sustain the costs of litigation or administrative proceedings more effectively than we can because of greater financial resources. Patent litigation and other proceedings may also absorb significant management time. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings against us could impair our ability to compete in the marketplace. The occurrence of any of the foregoing could have a material adverse effect on our business, financial condition, or results of operations or growth prospects.
We may become involved in lawsuits to protect or enforce our patents and other intellectual property rights, which could be expensive, time consuming, and unsuccessful.
Competitors may infringe our patents or the patents of our licensing partners, or we may be required to defend against claims of infringement. In addition, our patents or the patents of our licensing partners also may become involved in inventorship, priority, or validity disputes. To counter or defend against such claims can be expensive and time consuming. In an infringement proceeding, a court may decide that a patent in which we have an interest is invalid or unenforceable, the other party’s use of our patented technology falls under the safe harbor to patent infringement under 35 U.S.C. §271(e)(1), or may refuse to stop the other party from using the technology at issue on the grounds that our owned and in-licensed patents do not cover the technology in question. An adverse result in any litigation proceeding could put one or more of our owned or in-licensed patents at risk of being invalidated or interpreted narrowly. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation.
Even if resolved in our favor, litigation or other legal proceedings relating to intellectual property claims may cause us to incur significant expenses and could distract our personnel from their normal responsibilities. In addition, there could be public announcements of the results of hearings, motions, or other interim proceedings or developments, and if securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our common stock. Such litigation or proceedings could substantially increase our operating losses and reduce the resources available for development activities or any future sales, marketing, or distribution activities. We may not have sufficient financial or other resources to conduct such litigation or proceedings adequately. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their greater financial resources and more mature and developed intellectual property portfolios. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could have a material adverse effect on our ability to compete in the marketplace.
If our trademarks and trade names are not adequately protected, then we may not be able to build name recognition in our markets of interest and our business may be adversely affected.
Our registered or unregistered trademarks or trade names may be challenged, infringed, circumvented, or declared generic, or determined to be infringing on other marks. We may not be able to protect our rights to these trademarks and trade names, which we need to build name recognition among potential partners or customers in our markets of interest. At times, competitors or other third parties may adopt trade names or trademarks similar to ours, thereby impeding our ability to build brand identity and possibly leading to market confusion. In addition, there could be potential trade name or trademark infringement claims brought by owners of other registered trademarks or trademarks that incorporate variations of our registered or unregistered trademarks or trade names. Over the long term, if we are unable to establish name recognition based on our trademarks and trade names, then we may not be able to compete effectively and our business may be adversely affected. Our efforts to enforce or protect our proprietary rights related to trademarks, trade secrets, domain names, copyrights, or other intellectual property may be ineffective and could result in substantial costs and diversion of resources and could adversely affect our business, financial condition, results of operations, and growth prospects.
Intellectual property rights do not necessarily address all potential threats.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations and may not adequately protect our business or permit us to maintain our competitive advantage. For example:
•others may be able to make products that are similar to our product candidates or utilize similar technology but that are not covered by the claims of the patents that we license or may own;
•we, or our current or future licensors or collaborators, might not have been the first to make the inventions covered by the issued patent or pending patent application that we license or own now or in the future;
•we, or our current or future licensors or collaborators, might not have been the first to file patent applications covering certain of our or their inventions;
•others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our owned or licensed intellectual property rights;
•it is possible that our current or future pending owned or licensed patent applications will not lead to issued patents;
•issued patents that we hold rights to may be held invalid or unenforceable, including as a result of legal challenges by our competitors or other third parties;
•our competitors or other third parties might conduct research and development activities in countries where we do not have patent rights and then use the information learned from such activities to develop competitive products for sale in our major commercial markets;
•we may not develop additional proprietary technologies that are patentable;
•the patents of others may harm our business; and
•we may choose not to file a patent in order to maintain certain trade secrets or know-how, and a third party may subsequently file a patent covering such intellectual property.
Should any of these events occur, they could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
Risks Related to Our Operations
We are highly dependent on our key personnel, and if we are not successful in attracting, motivating and retaining highly qualified personnel, we may not be able to successfully implement our business strategy.
Our ability to compete in the highly competitive biotechnology and pharmaceutical industries depends upon our ability to attract, motivate and retain highly qualified managerial, scientific, and medical personnel. We are highly dependent on our management, particularly our Chief Executive Officer, Dr. Ryan Watts, and our scientific and medical personnel. The loss of the services provided by any of our executive officers, other key employees, and other scientific and medical advisors, and our inability to find suitable replacements, could result in delays in the development of our product candidates and harm our business.
We primarily conduct our operations at our facility in South San Francisco, California, in a region that is headquarters to many other biopharmaceutical companies and many academic and research institutions. Competition for skilled personnel is intense and the turnover rate can be high, which may limit our ability to hire and retain highly qualified personnel on acceptable terms or at all. We expect that we may need to recruit talent from outside of our region, and doing so may be costly and difficult.
To induce valuable employees to remain at our company, in addition to salary and cash incentives, we have provided restricted stock and stock option grants that vest over time. The value to employees of these equity grants that vest over time may be significantly affected by movements in our stock price that are beyond our control, and may at any time be insufficient to counteract more lucrative offers from other companies. Although we have employment agreements with our key employees, these employment agreements provide for at-will employment, which means that any of our employees could leave our employment at any time, with or without notice. We do not maintain “key man” insurance policies on the lives of all of these individuals or the lives of any of our other employees. If we are unable to attract and incentivize quality personnel on acceptable terms, or at all, it may cause our business and operating results to suffer.
We will need to grow the size and capabilities of our organization, and we may experience difficulties in managing this growth.
As of December 31, 2020,2023, we had 291approximately 445 employees, all of whom were full-time. As our development plans and strategies develop, we must add a significant number of additional managerial, operational, financial, and other personnel. Future growth will impose significant added responsibilities on members of management, including:
•identifying,including recruiting, integrating, retaining, and motivatingretaining additional employees;
•managing our internal development efforts effectively, including the clinicalefforts; and FDA review process for our current and future product candidates, while complying with our contractual obligations to contractors and other third parties;
•expanding our operational, financial and management controls, reporting systems, and procedures; and
•managing increasing operational and managerial complexity. procedures.
Our future financial performance and our ability to continue to develop and, if approved, commercialize our product candidates will depend, in part, on our ability to effectively manage any future growth. Our management may also have to divert a disproportionate amount of its attention away from day-to-day activities in order to manage these growth activities. Our ability to successfully manage our expected growth is uncertain given the fact that all of our executive officers have joined us since February 2015. This lack of long-term experience working together as a company may adversely impact our senior management team’s ability to effectively manage our business and growth.
We currently rely, and for the foreseeable future will continue to rely, in substantial part on certain independent organizations, advisors, and consultants to provide certain services. There can be no assurance that the services of these independent organizations, advisors, and consultants will continue to be available to us on a timely basis when needed, or that we can find qualified replacements. In addition, if we are unable to effectively manage our outsourced activities or if the quality or accuracy of the services provided by consultants is compromised for any reason, our clinical trials may be extended, delayed, or terminated, and we may not be able to obtain regulatory approval of our product candidates or otherwise advance our business. There can be no assurance that we will be able to manage our existing consultants or find other competent outside contractors and consultants on economically reasonable terms, if at all.
If we are not able to effectively expandmanage our organization by hiring new employees and expanding our groups of consultants and contractors,growth, we may not be able to successfully implement the tasks necessary to further develop our product candidates and, accordingly, may not achieve our research, development, and commercialization goals.
We have engaged in and may in the future engage in acquisitions or strategic partnerships, which may increase our capital requirements, dilute our stockholders, cause us to incur debt or assume contingent liabilities, and subject us to other risks.
We have in the past engaged in acquisitions and strategic partnerships, and we may engage in various acquisitions and strategic partnerships, or spin outs of businesses in the future, including licensing or acquiring complementary products, intellectual property rights, technologies, or businesses. For instance, in January 2018 we entered into the Takeda Collaboration Agreement,businesses as amended in February 2019, and in connection therewith we issued and sold to Takeda 4,214,559 sharespart of our commonbusiness strategy. For example, we have collaboration agreements with Takeda, Sanofi and Biogen, and issued stock for an aggregate purchase price of $110.0 million in February 2018. On May 30, 2018, we exercised our buy-out option in connection with the F-star Collaboration Agreemententering into certain of those agreements in 2018 and entered into a Purchase Agreement pursuant to which we acquired all of the outstanding shares of F-star Gamma. Further, on October 29, 2018, we entered into the Sanofi Collaboration Agreement. In August 2020, we entered into the Provisional Biogen Collaboration Agreement, and in connection therewith we sold 13,310,243 shares of our common stock to Biogen in September 2020 for an aggregate purchase price of $465.0 million.2020. Any acquisition or strategic partnershipsuch transaction may entail numerous risks, including:
•increased operating expenses and cash requirements;
•the assumption of indebtedness or contingent liabilities;
•the issuance of our equity securities which would result in dilution to our stockholders;
•assimilation of operations, intellectual property, products and product candidates of an acquired company, including difficulties associated with integrating new personnel;
•the diversion of our management’s attention from our existing product programs and initiatives in pursuing such an acquisition or strategic partnership;
•retention of key employees, the loss of key personnel,employees, and uncertainties in our ability to maintain key business relationships;
•risks and uncertainties associated with the other party to such a transaction, including the prospects of that party and their existing products or product candidates and regulatory approvals; and
•our inability to generate revenue from acquired intellectual property, technology and/or products sufficient to meet our objectives or even to offset the associated transaction and maintenance costs.
In addition, if we undertake such a transaction, we may issue dilutive securities, assume or incur debt obligations, incur large one-time expenses and acquire intangible assets that could result in significant future amortization expense.
The proposed spin out of our preclinical small molecule portfolio is subject to various risks and uncertainties and may not be completed on the terms or timeline currently contemplated, or at all, or may not result in the expected benefits, which might harm our business or operations.
On January 8, 2024, we announced our intention to spin out our preclinical small molecule portfolio into a new, independently-funded company ("NewCo"). Unanticipated developments could delay, prevent, or otherwise adversely affect the proposed spin out, including but not limited to disruptions in general or financial market conditions, litigation or other legal challenges to the spin out, or potential problems or delays in licensing our intellectual property to NewCo. We cannot assure you that we will be able to complete the spin out on the terms or the timeline that we announced, if at all. Furthermore, the anticipated benefits of the spin out are based on a number of assumptions; if some of these prove incorrect, the Company may not be able to achieve the full expected strategic and financial benefits of the transaction.
The price of the Company's common stock may be more volatile around the time of the spin out, and the Company cannot predict the effect of the spin out on the trading price of shares of its common stock. Moreover, following the spin out, the price of shares of the Company's common stock may fluctuate.
Our internal computer systems, or those used by our third-party research institution collaborators, CROs, or other contractors or consultants, may fail or suffer other breakdowns, cyberattacks, or information security breaches or incidents that could compromise the confidentiality, integrity, and availability of such systems and data, expose us to liability, and affect our reputation.
We are increasingly dependent upon information technology systems, infrastructure, and data to operate our business. We also rely on third-party vendors and their information technology systems. Despite the implementation of security measures, our internal computer systems and those of our collaborators, CROs, and other contractors and consultants may be vulnerable to damage, outages and interruptions resulting from computer viruses and other malicious code or unauthorized access, or breached, compromised, or otherwise subject to security incidents due to operator error, malfeasance, or other system disruptions. Geopolitical events, such as war and armed conflicts, may increase the risks of cyber-attacks, disruptions, and security breaches and incidents that we and these third parties face. As the cyber-threat landscape evolves, these attacks are growing in frequency, sophistication, and intensity, and are becoming increasingly difficult to detect. TheseSecurity threats can come from a variety of sources, ranging in sophistication from an individual hacker to a state-sponsored attack. Cyber threats may be broad-based or otherwise generic in nature, or they may be custom-crafted against our information systems. systems or those of our collaborators, CROs, or other contractors or consultants.
Over the past few years, cyber-attacks have become more prevalent, intense, sophisticated, and much harder to detect and defend against. Such attacks could include the use of key loggers or other harmful and virulent malware, including ransomware or other denials of service, and can be deployed through malicious websites, the use of social engineering and/or other means. We and our third-party vendorscollaborators, CROs, or other contractors and consultants may not be able to anticipate all types of security threats, and we may not be able to implement preventive measures effective against all such security threats. The techniques used by cyber criminals change frequently, may not be recognized until launched, and can originate from a wide variety of sources. Although to our knowledge we and our vendors have not experienced any such material system failure or security breach or incident to date, if a breakdown, cyberattack or other information security breach or incident were to occur and cause interruptions in our operations, it could result in a material disruption of our development programs and our business operations, whether due to a loss or misappropriation of trade secrets or loss of, or unauthorized modification, unavailability, disclosure, or other unauthorized processing of other proprietary information or other similar disruption and we could incur liability and reputational damage. For example, theany corruption, loss, or other unavailability of clinical trial data from completed, ongoing or future clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. Likewise, we rely on our third-party research institution collaborators for research and development of our product candidates and other third parties for the manufacture of our product candidates and to conduct clinical trials, and similar events relating to their computer systems could also have a material adverse effect on our business.
Cyber-attacks, breaches, interruptions, or other data security incidents could result in legal claims or proceedings by private parties or governmental authorities, liability under federal or state laws that protect the privacy of personal information, regulatory penal-ties,penalties, significant remediation costs, disrupt key business operations, and divert attention of management and key information technology resources. In the United States, notice of breaches must be made to affected individuals, the U.S. Secretary of the Department of Health and Human Services ("HHS"), and for extensive breaches, notice may need to be made to the media or U.S. state attorneys general. Such a notice could harm our reputation and our ability to compete. The HHS has the discretion to impose penalties without attempting to resolve violations through informal means. In addition, U.S. state attorneys general are authorized to bring civil actions seeking either injunctions or damages in response to violations that threaten the privacy of state residents. There can be no assurance that we, our collaborators, CROs, vendors,contractors, consultants, and any other business counterparties will be successful in efforts to detect, prevent, protect against, or fully recover systems or data from all break-downs, service interruptions, attacks, or security breaches of systems. In addition,or incidents. Although we do not maintain standalone cyber-securitycybersecurity insurance, and have limited insurance coverage in the event of any breach or disruption of our or our collaborators’, CROs’, or vendors’ systems, including any unauthorized access or loss of any personal data that we may collect, store or otherwise process. The costs related to significant security breaches, incidents, or disruptions could be material and exceed the limits of any insurance coverage we have, and may have. Toresult in increases in our insurance costs. Relevant insurance may in the extent that anyfuture become unavailable to us on commercially reasonable terms or at all. Any disruption or security breach wereor incident that results in or is perceived to resulthave resulted in a loss of, or damage to, our data or systems, or inappropriate disclosure, use, acquisition, transfer, modification, unavailability, or other processing of confidential or proprietary information, including data related to our personnel, we could result in the loss, unauthorized modification, use, unavailability, disclosure or other unauthorized processing of critical or sensitive data, and could cause us to incur liability andliability. Further, in any such event, the further development and commercialization of our product candidates could be delayed and our business and operations could be adversely affected and/or could result inaffected. Any of the loss or disclosure of critical or sensitive data, whichforegoing could result in financial, legal, business, or reputational harm to us.
Business disruptions could seriously harm our future revenue and financial condition and increase our costs and expenses.
Our operations, and those of our third-party research institution collaborators, CROs, CDMOs, suppliers, and other contractors and consultants, could be subject to earthquakes, power shortages, telecommunications failures, water shortages, floods, hurricanes, typhoons, fires, extreme weather conditions, medicalhealth epidemics such as COVID-19, and other natural or man-made disasters or business interruptions, for which we are partly uninsured. In addition, we rely on our third-party research institution collaborators for conducting research and development of our product candidates, and they may be affected by bank failures or instability in the financial services sector, government shutdowns, or withdrawn funding. The occurrence of any of these business disruptions could seriously harm our operations and financial condition and increase our costs and expenses. We rely on third-party manufacturers to produce and process our product candidates. Our ability to obtain clinical supplies of our product candidates could be disrupted if the operations of these suppliers are affected by a man-made or natural disaster or other business interruption.
AllThe majority of our operations including our corporate headquarters are located in a single facility in South San Francisco, California. Damage or extended periods of interruption to our corporate, development or research facilities due to fire, extreme weather conditions or natural disaster, power loss, communications failure, unauthorized entry, or other events could cause us to cease or delay development of some or all of our product candidates. Although we maintain property damage and business interruption insurance coverage on these facilities, our insurance might not cover all losses under such circumstances and our business may be seriously harmed by such delays and interruption.
Our business is subject to economic, political, regulatory, and other risks associated with international operations.
Our business is subject to risks associated with conducting business internationally. Some of our suppliers and collaborative relationships are located outside the United States. Accordingly, our future results could be harmed by a variety of factors, including:
•economic weakness, including inflation, rising interest rates or political instability in particularcertain non-U.S. economies and markets;
•differing and changing regulatory requirements in non-U.S. countries;
•challenges enforcing our contractual and intellectual property rights, especially in those foreignnon-U.S. countries that do not respect and protectoffer the same level of intellectual property rights to the same extentprotection as the United States;
•difficulties in compliance with non-U.S. laws and regulations;
•changes in non-U.S. regulations and customs, tariffs, and trade barriers;
•changes in non-U.S. currency exchange rates and currency controls;
•changes in a specific country’s or region’s political or economic environment;
•trade protection measures, import or export licensing requirements, or other restrictive actions by U.S. or non-U.S. governments;government actions;
•negative consequences from changes in tax laws;
•compliance with tax, employment, immigration, and labor laws for employees living or traveling abroad;
•workforce uncertainty in countries where labor unrest is more common than in the United States;
•difficulties associated with staffing and managing international operations, including differing labor relations;
•potential liability under the FCPA, UK Bribery Act, or comparable foreign laws;
•business interruptions resulting from geo-politicalgeopolitical actions, including war and armed conflict, terrorism, natural disasters including earthquakes, typhoons, floods, and fires, or health epidemics such as COVID-19; and
•cyberattacks, which are growing in frequency, sophistication and intensity, and are becoming increasingly difficult to detect.
These and other risks associated with our planned international operations may materially adversely affect our ability to attain profitable operations.
Our ability to use our net operating loss carryforwards and certain other tax attributes may be limited.
As of December 31, 2020,2023, we had federal net operating loss carryforwards of approximately $212.7$290.6 million, federal research and development tax credit carryforwards of approximately $20.4$53.1 million, and orphan tax credit carryforwards of approximately $3.3$37.4 million, some of which will begin to expire in 2035.2034. Under Sections 382 and 383 of the United States Internal Revenue Code of 1986, as amended, (the "Code"), if a corporation undergoes an “ownership change” (generally defined as a greater than 50-percentage-point cumulative change (by value) in the equity ownership of certain stockholders over a rolling three-year period), the corporation’s ability to use its pre-change net operating loss carryforwards and other pre-change tax attributes to offset its post-change taxable income or taxes may be limited. As a result of our IPO in December 2017, our follow-on offering in January 2020, and private placements and other transactions that have occurred since our incorporation, we mayWe have experienced such an ownership change. Wechanges in the past, and we may also experience ownership changes in the future as a result of subsequent shifts in our stock ownership, including in connection with our October 2022 offering, some of which are outside our control. As a result, our ability to use our pre-change net operating loss carryforwards and other pre-change tax attributes to offset post-change taxable income or taxes may be subject to limitation.
We may be subject to adverse legislative or regulatory tax changes that could negatively impact our financial condition.
The rules dealing with U.S. federal, state, and local income taxation are constantly under review by persons involved in the legislative processlegislators and by the IRS and the U.S. Treasury Department. Changes to tax laws (which changes may have retroactive application) could adversely affect our stockholders or us. In recent years, many such changes have been madeoccurred and changes are likely to continue to occur in the future.future, which could adversely affect our shareholders. For example, in December 2017, Congress passedAugust 2022, the Tax Cuts and JobsUnited States enacted the Inflation Reduction Act, which made broadimplemented a 15% minimum tax on book income for certain companies and complexintroduced a 1% excise tax on stock buybacks. In addition, the current tax administration has proposed changes to the orphan drug tax laws. We cannot predict whether, when,credit. Changes in what form, or with what effective dates, tax laws, regulations and rulings may be enacted, promulgatedregulation, or decided, whichenforcement could result in an increase inadversely affect our or our stockholders’, tax liabilitystockholders or require us to implement changes in the manner in which we operate in order to minimize increases in our tax liability.
Risks Related to Ownership of Our Common Stock
The market price of our common stock has been and may continue to be volatile, which could result in substantial losses for investors.
The trading price of our common stock has been and may continue to be highly volatile and could be subject to wide fluctuations in response to various factors, some of which are beyond our control. In addition to the factors discussed in this "Risk Factors" section and elsewhere in this report, these factors include:
•the success of existing or new competitive products or technologies;
•the timing and results of clinical trials for our current product candidates and any future product candidates that we may develop;
•commencement or termination of collaborations for our product development and research programs;
•failure to achieve development, regulatory, or commercialization milestones under our collaborations;
•failure or discontinuation of any of our product development and research programs;
•failure to develop our BBB platform technology;
•results of preclinical studies, clinical trials, or regulatory approvals of product candidates of our competitors, or announcements about new research programs or product candidates of our competitors;
•regulatory or legal developments in the United States and other countries;
•developments or disputes concerning patent applications, issued patents, or other proprietary rights;
•the recruitment or departure of key personnel;
•the level of expenses related to any of our research programs, clinical development programs, or product candidates that we may develop;
•the results of our efforts to develop additional product candidates or products;
•actual or anticipated changes in estimates as to financial results, development timelines, or recommendations by securities analysts;
•announcement or expectation of additional financing efforts;
•sales of our common stock by us, our insiders, or other stockholders;
•variations in our financial results or those of companies that are perceived to be similar to us;
•changes in estimates or recommendations by securities analysts, if any, that cover our stock;
•changes in the structure of healthcare payment systems;
•adoption of new accounting standardssystems or changes in accounting standards;
•ineffectiveness of our internal controls;
•significant lawsuits, including patent or stockholder litigation;
•market conditions in the pharmaceutical and biotechnology sectors; and
•other events or factors affecting general economic, industry, and market conditions, including those caused bybank failures or instability in the COVID-19 pandemic.
In recent years, the stock market in general, and the market for pharmaceutical and biotechnology companies in particular, has experienced significant price and volume fluctuations that have often been unrelated or disproportionate to changes in the operating performance of the companies whose stock is experiencing those price and volume fluctuations. Broad market and industry factors may seriously affect the market price of our common stock, regardless of our actual operating performance. In the past, when the market price of a stock has been volatile, holders of that stock have instituted securities class action litigation against the company that issued the stock. If any of our stockholders were to bring a lawsuit against us, the defense and disposition of any such lawsuits could be costly and divert the time and attention of our management and harm our operating results, regardless of the merits of such a claim.
If securities analysts publish negative evaluations of our stock, or if they do not publish research or reports about our business, or if they publish negative evaluations of our stock, the price of our stock and trading volume could decline.
The trading market for our common stock relies in part on the research and reports that industry or financial analysts publish about us or our business. If one or more of the analysts covering our business downgrade their evaluations of our stock, or if we fail to meet the expectations of analysts, the price of our stock could decline. If one or more of these analysts cease to cover our stock, we could lose visibility in the market for our stock, which in turn could cause our stock price or trading volume to decline.
Sales of substantial amounts of our common stock in the public markets, or the perception that such sales might occur, could cause the market price of our common stock to decline significantly, even if our business is doing well.
Sales of a substantial number of shares of our common stock in the public market, or the perception that these sales might occur, could depress the market price of our common stock and could impair our ability to raise capital through the sale of additional equity securities. We are unable to predict the effect that sales may have on the prevailing market price of our common stock.
Sales of our common stock by current stockholders may make it more difficult for us to sell equity or equity-related securities in the future at a time and price that we deem reasonable or appropriate, and make it more difficult for you to sell shares of our common stock.
Certain holders of shares of our common stock have rights, subject to conditions, to require us to file registration statements covering their shares or to include their shares in registration statements that we may file for ourselves or other stockholders. RegistrationFor example, on February 27, 2024, we entered into a securities purchase agreement (the “Purchase Agreement”) with certain existing accredited investors in connection with the recently announced PIPE financing, pursuant to which the investors were granted certain registration rights. Furthermore, in connection with the PIPE financing, we have agreed to enter into a registration rights agreement with an investor following such time that investor may be deemed an affiliate of these shares under the Securities Act would result inCompany, pursuant to which the shares becoming freely tradeable in the public market, subjectinvestor is entitled to the restrictions of Rule 144 in the case of our affiliates.certain resale registration rights. Any sales of securities by these stockholders, or the perception that sales will be made in the public market, could have a material adverse effect on the market price for our common stock.
We have registered on Form S-8 all shares of common stock that are issuable under our 2017 Equity Incentive Plan and 2017 Employee Stock Purchase Plan. As a consequence, these shares can be freely sold in the public market upon issuance, subject to volume limitations applicable to affiliates.
Raising additional capital may cause dilution to our existing stockholders, restrict our operations, or require us to relinquish rights to our technologies or product candidates.
We may seek additional capital through a combination of public and private equity offerings, debt financings, strategic partnerships and alliances, and licensing arrangements. For example, in August 2020, we entered into the Provisional Biogen Collaboration Agreement, and in connection therewith issued and sold 13,310,243 shares of our common stock to Biogen in September 2020 for an aggregate purchase price of $465$465.0 million. We, and indirectly, our stockholders, will bear the cost of issuing and servicing all such securities. Additionally, collaborations we enter into with third parties may provide capital in the near term but limit our potential cash flow and revenue in the future. If we raise additional funds through strategic partnerships and alliances and licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies or product candidates, or grant licenses on terms unfavorable to us.
TableIn January 2020, we sold 9.0 million shares of Contentscommon stock in an underwritten follow-on offering pursuant to a shelf registration statement filed in March 2019 and, in October 2022, we sold 11.9 million shares of common stock in an underwritten public offering pursuant to a second shelf registration statement filed in February 2022. Also in February 2022, we entered into an equity distribution agreement with Goldman Sachs & Co. LLC, SVB Securities LLC, and Cantor Fitzgerald & Co., as sales agents, to establish an at-the-market facility pursuant to which we may offer and sell from time to time up to $400.0 million in shares of our common stock. On February 27, 2024, we announced a PIPE financing in which we have agreed to sell 3,244,689 shares of our common stock and pre-funded warrants to purchase 26,046,065 shares of our common stock and which is expected to result in gross proceeds of approximately $499.7 million. We have also granted a certain investor certain director nomination and additional registration rights, subject to certain exceptions, conditions, and limitations.
Because ourOur decision to issue debt or equity securities in any future offering will depend on market conditions and other factors beyond our control, and therefore we cannot predict or estimate the amount, timing, or nature of any future offerings. To the extent that we raise additional capital through the sale of equity or debt securities, your ownership interest will be diluted, and the terms may include liquidation or other preferences that adversely affect your rights as a stockholder. The incurrence of indebtedness would result in increased fixed payment obligations and could involve restrictive covenants, such as limitations on our ability to incur additional debt, limitations on our ability to acquire, sell, or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business.
On March 12, 2019, we filed a In addition, any sales of our common stock or other securities under our shelf registration statement could put downward pressure on Form S-3 (File No. 333-230232) with the Securities and Exchange Commission, which became effective upon filing. In January 2020, we sold 9.0 million shares of commonour stock in a follow-on offering pursuant to this registration statement. The shelf registration continues to allow us to sell, from time to time, an unspecified number of shares of common stock; shares of preferred stock; debt securities; warrants to purchase shares of common stock, preferred stock, or other securities; purchase contracts; and units representing two or more of the foregoing securities.price. Additionally, collaborations we enter into with third parties may provide capital in the near term but limit our potential cash flow and revenue in the future. If we raise additional funds through strategic partnerships and alliances and licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies or product candidates, or grant licenses on terms unfavorable to us.
Our principal stockholders and management own a significant percentage of our stock and will be able to exercise significant influence over matters subject to stockholder approval.
As of December 31, 2020, ourOur directors, executive officers, holders of more than 5% of our outstanding stock, and their respective affiliates beneficially own shares representing more than 50.0%a significant percentage of our outstanding common stock. As a result, these stockholders, if they act together, may significantly influence all matters requiring stockholder approval, including the election of directors and approval of significant corporate transactions. This concentration of ownership may have the effect of delaying or preventing a change in control of our company that our other stockholders may believe is in their best interests. This in turn could have a material adverse effect on our stock price and may prevent attempts by our stockholders to replace or remove the board of directors or management.
If we are unable to maintain effective internal controls, our business, financial position and results of operations and growth prospects could be adversely affected.
As a public company, we are subject to reporting and other obligations under the Securities Exchange Act of 1934, as amended, (the "Exchange("Exchange Act"), including the requirements of Section 404 of the Sarbanes-Oxley Act, which require annual management assessments of the effectiveness of our internal control over financial reporting.
The rules governing the standards that must be met for management and our auditors to assess our internal control over financial reporting are complex and require significant documentation, testing, and possible remediation to meet the detailed standards under the rules. During the course of its testing, our management or auditors may identify material weaknesses or deficiencies which may not be remedied in time to meet the deadline imposed by the Sarbanes-Oxley Act. These reporting and other obligations place significant demands on our management and administrative and operational resources, including accounting resources.
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Our internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with accounting principles generally accepted in the United States ("U.S. GAAP"). Any failure to maintain effective internal controls could have an adverse effect on our business, financial position, results of operations, and growth prospects.
Our disclosure controls and procedures may not prevent or detect all errors or acts of fraud.
We are subject to the periodic reporting requirements of the Exchange Act. We designed our disclosure controls and procedures to reasonably assure that information we must disclose in reports we file or submit under the Exchange Act is accumulated and communicated to management, and recorded, processed, summarized and reported within the time periods specified in the rules and forms of the SEC. We believe that any disclosure controls and procedures or internal controls and procedures, no matter how well-conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met.
These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur because of simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people or by an unauthorized override of the controls. Accordingly, because of the inherent limitations in our control system, misstatements due to error or fraud may occur and not be detected.
We have not paid and do not expect to pay any dividends for the foreseeable future. Any return on investment may be limited to the value of our common stock. Investors may never obtain a return on their investment.
We have never paid cash dividends on our common stock and do not anticipate that we will pay any dividends in the foreseeable future. We currently intend to retain our future earnings, if any, to maintain and expand our existing operations. If we do not pay dividends, our common stock may be less valuable because a return on your investment will only occur if our stock price appreciates, which may never occur.
Delaware law and provisions in our charter documents might discourage, delay, or prevent a change in control of our company or changes in our management and, therefore, depress the trading price of our common stock.
Provisions in our amended and restated certificate of incorporation and amended and restated bylaws may discourage, delay, or prevent a merger, acquisition, or other change in control that stockholders may consider favorable, including transactions in which you might otherwise receive a premium for your shares of our common stock. These provisions may also prevent or frustrate attempts by our stockholders to replace or remove our management. Therefore, these provisions could adversely affect the price of our common stock. Among other things, our charter documents:
•establish that our board of directors is divided into three classes, Class I, Class II, and Class III, with each class serving staggered three-year terms;
•provide that vacancies on our board of directors may be filled only by a majority of directors then in office, even though less than a quorum;
•provide that our directors may only be removed for cause;
•eliminate cumulative voting in the election of directors;
•authorize our board of directors to issues shares of preferred stock and determine the price and other terms of those shares, including preferences and voting rights, without stockholder approval;
•provide our board of directors with the exclusive right to elect a director to fill a vacancy or newly created directorship;
•permit stockholders to only take actions at a duly called annual or special meeting and not by written consent;
•prohibit stockholders from calling a special meeting of stockholders;
•require that stockholders give advance notice to nominate directors or submit proposals for consideration at stockholder meetings;
•authorize our board of directors, by a majority vote, to amend the bylaws; and
•require the affirmative vote of at least 66 2/3% or more of the outstanding shares of common stock to amend many of the provisions described above.
In addition, Section 203 of the General Corporation Law of the State of Delaware, (the "DGCL"), prohibits a publicly-held Delaware corporation from engaging in a business combination with an interested stockholder, generally a person which together with its affiliates owns, or within the last three years has owned, 15.0% of our voting stock, for a period of three years after the date of the transaction in which the person became an interested stockholder, unless the business combination is approved in a prescribed manner.
Any provision of our amended and restated certificate of incorporation, amended and restated bylaws, or Delaware law that has the effect of delaying or preventing a change in control could limit the opportunity for our stockholders to receive a premium for their shares of our capitalcommon stock and could also affect the price that some investors are willing to pay for our common stock.
Our amended and restated certificate of incorporation provides that the Court of Chancery of the State of Delaware and the federal district courts of the United States of America will be the exclusive forums for substantially all disputes between us and our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers, or employees.
Our amended and restated certificate of incorporation provides that the Court of Chancery of the State of Delaware is the exclusive forum for:
•any derivative action or proceeding brought on our behalf;
•any action asserting a claim of breach of fiduciary duty;
•any action asserting a claim against us arising under the DGCL, our amended and restated certificate of incorporation, or our amended and restated bylaws; and
•any action asserting a claim against us that is governed by the internal-affairs doctrine.
Our amended and restated certificate of incorporation further provides that the federal district courts of the United States of America will be the exclusive forum for resolving any complaint asserting a cause of action arising under the Securities Act.
These exclusive-forum provisions may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, officers, or other employees, which may discourage lawsuits against us and our directors, officers, and other employees. If a court were to find either exclusive-forum provision in our amended and restated certificate of incorporation to be inapplicable or unenforceable in an action or we do not enforce such provision, we may incur additional costs associated with resolving the dispute in other jurisdictions, which could seriously harm our business.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
ITEM 1C. CYBERSECURITY
Denali's approach to cybersecurity seeks to defend the confidentiality, integrity and availability of our systems and information for our people and our patients.
Risk Management and Strategy
We have established policies and processes for assessing, identifying, and managing material risk from cybersecurity threats, and have integrated these processes into our overall risk management systems and processes. We routinely assess material risks from cybersecurity threats, including any potential unauthorized occurrence on or conducted through our information systems that may result in adverse effects on the confidentiality, integrity, or availability of our information systems or any information residing therein.
We conduct periodic risk assessments to identify cybersecurity threats, as well as assessments in the event of a material change in our business practices that may affect information systems that are vulnerable to such cybersecurity threats. These risk assessments include identification of reasonably foreseeable internal and external risks, the likelihood and potential damage that could result from such risks, and the sufficiency of existing policies, procedures, systems, and safeguards in place to manage such risks.
Following these risk assessments, we re-design, implement, and maintain reasonable safeguards to minimize identified risks; reasonably address any identified gaps in existing safeguards; and regularly monitor the effectiveness of our safeguards. We devote significant resources and designate high-level personnel, including our Head of IT and Cybersecurity, to manage the risk assessment and mitigation process.
As part of our overall risk management system, we conduct annual security awareness training for personnel at all levels and functions, issue periodic simulated social engineering tests, and have established protocols to escalate cybersecurity incidents from identification through remediation. These activities are undertaken in collaboration with human resources, IT, and management.
We also engage consultants to assist us in monitoring and testing our safeguards. Performance of our cybersecurity controls for certain systems is periodically reviewed by our internal quality functions. We further require third-party service providers who will be handling our company’s sensitive information to certify that they implement appropriate security measures, consistent with all applicable laws, to maintain reasonable security measures in connection with their work with us, and to promptly report any suspected breach of its security measures that may affect our company.
Additional information regarding whether any risks from cybersecurity threats, including as a result of any previous cybersecurity incidents, have materially affected or are reasonably likely to materially affect our company, including our business strategy, results of operations, or financial condition, are included in this Annual Report on Form 10-K in Item 1A, “Risk Factors”.
Governance
One of the key functions of our board of directors is informed oversight of our risk management process, including risks from cybersecurity threats. Our board of directors is responsible for monitoring and assessing strategic risk exposure, and our executive officers are responsible for the day-to-day management of the material risks we face. Our board of directors administers its cybersecurity risk oversight function directly as a whole, as well as through the audit committee.
Our Head of IT is primarily responsible to assess and manage our material risks from cybersecurity threats and has prior work experience in cybersecurity, holds relevant degrees, and current industry recognized cybersecurity certifications.
Our Head of IT oversees our cybersecurity policies and processes, including those described in “Risk Management and Strategy” above. The processes by which our Head of Information Technology is informed about and monitors the prevention, detection, mitigation, and remediation of cybersecurity incidents includes the following the identification and assessment of assets and assessing potential associated risks, implementation of protective measures, continuous monitoring and detection of unusual or suspicious activities, incident response and recovery management, regular security awareness and training, review and alignment of cybersecurity practices with industry recognized cybersecurity practices, compliance and regulation concerns
Our Head of IT provides quarterly briefings to the audit committee regarding our company’s cybersecurity risks and activities, including any recent cybersecurity incidents and related responses, cybersecurity systems testing, activities of third parties, and the like. Our audit committee provides regular updates to the board of directors on such reports; the Head of IT may be called upon to brief the board as well.
ITEM 2. PROPERTIES
Our corporate headquarters are located in South San Francisco, California, comprising 148,020 square feet of office, research and development, engineering and laboratory space pursuant to a lease agreement which commenced on April 12, 2019 and expires on April 30, 2029, with an option to extend for a period of ten years. This facility houses allthe majority of our personnel.
Utah
The build-out of approximately 60,000 rentable square feet of office, lab and clinical manufacturing premises in Salt Lake City, Utah ("SLC Facility") is in process. The lease ("SLC Lease") will commence when the space is available for use, which is anticipated to be during 2024, and is expected to terminate in 2039. We have two five year renewal period options to extend the lease for a further ten years at the end of the lease term.
Switzerland
Our European headquarters encompasses office space in a shared facility located in Zürich, Switzerland. The current lease agreement is through December 2024, with the option to renew. The lease terms provide flexibility to increase our space allocation on short term notice.
We believe that our existing premises are adequate for our current needs.
ITEM 3. LEGAL PROCEEDINGS
From time to time, we may become involved in litigation or other legal proceedings. We are not currently a party to any litigation or legal proceedings that, in the opinion of our management, are likely to have a material adverse effect on our business. Regardless of outcome, litigation can have an adverse impact on us because of defense and settlement costs, diversion of management resources and other factors.
On September 10, 2020, we and all directors were named in a shareholder derivative action filed in the Delaware Court of Chancery challenging the compensation we paid to our directors since the IPO in December 2017.
On January 13, 2021, the parties to the derivative action entered into a settlement agreement, subject to Court approval and certain other closing conditions, the terms of which were disclosed in a Form 8-K filed on February 5, 2021. Amounts owed by us pursuant to the settlement agreement are not material to us.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Our common stock has been listed on The NASDAQNasdaq Global Select Market under the symbol “DNLI” since December 8, 2017. Prior to that date, there was no public trading market for our common stock.
Holders of Common Stock
As of February 17, 2021,20, 2024, there were 145approximately 144 holders of record of our common stock. The actual number of stockholders is greater than this number of record holders and includes stockholders who are beneficial owners but whose shares are held in street name by brokers and other nominees. This number of holders of record also does not include stockholders whose shares may be held in trust or by other entities.
Dividend Policy
We have never declared or paid any cash dividends on our common stock or any other securities. We anticipate that we will retain all available funds and any future earnings, if any, for use in the operation of our business and do not anticipate paying cash dividends in the foreseeable future. In addition, future debt instruments may materially restrict our ability to pay dividends on our common stock. Payment of future cash dividends, if any, will be at the discretion of the board of directors after taking into account various factors, including our financial condition, operating results, current and anticipated cash needs, the requirements of current or then-existing debt instruments and other factors the board of directors deems relevant.
Performance Graph
This graph is not “soliciting material” or deemed “filed” with the SEC for purposes of Section 18 of the Exchange Act, or otherwise subject to liabilities under that Section, and shall not be deemed incorporated by reference into any filing of Denali Therapeutics Inc. under the Securities Act of 1933, as amended (the "Securities Act"), whether made before or after the date hereof and irrespective of any general incorporation language in any such filing.
The following graph compares the cumulative total return to stockholder return on our common stock relative to the cumulative total returns of the NASDAQNasdaq Composite Index and the NASDAQNasdaq Biotechnology Index. An investment of $100 is assumed to have been made in our common stock and each index on December 8, 2017 (the first day of trading of our common stock)31, 2018 and its relative performance is tracked through December 31, 2020.2023. Pursuant to applicable Securities and Exchange Commission rules, all values assume reinvestment of the full amount of all dividends, however no dividends have been declared on our common stock to date. The stockholder returns shown on the graph below are based on historical results and are not necessarily indicative of future performance, and we do not make or endorse any predictions as to future stockholder returns.
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 12/8/17 | | 12/29/17 | | 3/31/18 | | 6/30/18 | | 9/30/18 | | 12/31/18 | | 3/31/19 | | 6/30/19 | | 9/30/19 | | 12/31/19 | | 3/31/20 | | 6/30/20 | | 9/30/20 | | 12/31/20 |
| Denali Therapeutics Inc. | $ | 100.00 | | | $ | 87.03 | | | $ | 109.57 | | | $ | 84.86 | | | $ | 120.98 | | | $ | 114.97 | | | $ | 129.22 | | | $ | 115.53 | | | $ | 85.25 | | | $ | 96.94 | | | $ | 97.44 | | | $ | 134.56 | | | $ | 199.39 | | | $ | 466.11 | |
| NASDAQ Composite Index | 100.00 | | | 101.37 | | | 103.99 | | | 110.87 | | | 119.09 | | | 98.49 | | | 115.04 | | | 119.50 | | | 119.71 | | | 134.63 | | | 115.84 | | | 151.69 | | | 168.74 | | | 195.11 | |
| NASDAQ Biotechnology Index | 100.00 | | | 102.71 | | | 102.77 | | | 105.93 | | | 117.79 | | | 93.61 | | | 108.16 | | | 105.72 | | | 96.61 | | | 117.11 | | | 105.08 | | | 133.35 | | | 132.25 | | | 148.06 | |

Recent Sales of Unregistered Securities
On September 22, 2020,February 27, 2024, we issued and sold 13,310,243entered into a securities purchase agreement (the “Purchase Agreement”) with certain existing accredited investors for the private placement of (i) 3,244,689 shares of our common stock at a price of $17.07 per share and (ii) pre-funded warrants to Biogen forpurchase an aggregate of 26,046,065 shares of our common stock (the “Pre-Funded Warrants”) at a purchase price of $465.0 million pursuant$17.06 per Pre-Funded Warrant, resulting in anticipated gross proceeds of $499.7 million. The Pre-Funded Warrants are exercisable at an exercise price of $0.01 and will be exercisable until exercised in full. The holders of Pre-Funded Warrants may not exercise a Pre-Funded Warrant if the holder, together with its affiliates, would beneficially own more than 4.99% of the number of shares of common stock outstanding immediately after giving effect to such exercise. The holders of Pre-Funded Warrants may increase or decrease such percentage not in excess of 19.99%, in the case of an increase, by providing at least 61 days’ prior notice to the termsCompany. The private placement is expected to close on February 29, 2024, subject to customary closing conditions. We intend to use the net proceeds from the private placement to support our ongoing research and development activities, the acceleration and expansion of its proprietary BBB-crossing TV technology, as well as general corporate purposes and working capital. We have agreed to file a registration statement for purposes of registering the shares of common stock purchase agreement with BIMA and in connection with the Biogen Collaboration Agreement. No underwriters were involvedsold in the saleprivate placement (including the shares of common stock underlying the Pre-Funded Warrants). We have also granted a certain investor certain director nomination and additional registration rights, subject to certain exceptions, conditions, and limitations.
We are relying on the book entry position representing the securities sold and issued contain legends restricting transferexemptions from registration available under Section 4(a)(2) and/or Rule 506(b) of the securities without registrationRegulation D promulgated under the Securities Act or an applicable exemption from registration.
The offer, sale and issuance of the securities described above was exempt from registration under the Securities Act under Section 4(a)(2) of the Securities Act as a transactionwith respect to transactions by an issuer not involving any public offering, and we expect to file a public offering. The recipient of securities in this transaction acquired the securities for investment only and notForm D with a view to or for sale in connection with any distribution thereof and appropriate legends were affixedrespect to the securities issued in this transaction. The recipient of securities in this transaction was an accredited person and had adequate access, through employment, business or other relationships, to information about the registrant.
Use of Proceeds from Registered Securities
On December 7, 2017, our Registration Statement on Form S-1 (File No. 333-221522) was declared effective by the SEC for our initial public offering of common stock. We started trading on The NASDAQ Global Select Market on December 8, 2017, and the transaction formally closed on December 12, 2017. In connection with the initial public offering,October 2022, we sold an aggregate of 15,972,221 shares of common stock, including 2,083,333 shares sold pursuant to the underwriters' full exercise of their option to purchase additional shares, at a price to the public of $18.00 per share. The aggregate offering price for shares sold in the offering was $287.5 million. The joint book-running managers for the initial public offering were Goldman, Sachs & Co. LLC, Morgan Stanley & Co. LLC, and J.P. Morgan Securities LLC. After deducting underwriting discounts, commissions and offering expenses paid or payable by us of approximately $23.2$11.9 million the net proceeds from the offering were approximately $264.3 million. No offering expenses were paid or are payable, directly or indirectly, to our directors or officers, to persons owning 10.0% or more of any class of our equity securities or to any of our affiliates. There has been no material change in the planned use of proceeds from our initial public offering as described in our final prospectus filed with the SEC on December 8, 2017 pursuant to Rule 424(b)(4). We invested the funds received in short-term, interest-bearing investment-grade securities and government securities.
In January 2020, we sold 9.0 million shares of common stock (inclusive of shares sold pursuant to an overallotment option granted to the underwriters in connection with the offering) through an underwritten public offering at a price of $23.00$26.50 per share for aggregate net proceeds of approximately $296.2 million.
There hashave been no material changechanges in the planned use of the net proceeds from the follow-on public offering as described in the Registration Statement. We have invested or plan to invest the funds received in short-term, interest-bearing investment-grade securities and government securities.
Issuer Purchases of Equity Securities
Not applicable.
ITEM 6. [RESERVED]
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion and analysis of our financial condition and results of operations should be read together with our consolidated financial statements and the related notes to those statements included elsewhere in this report. This discussion and analysis and other parts of this report contain forward-looking statements based upon current beliefs, plans and expectations related to future events and our future financial performance that involve risks, uncertainties and assumptions, such as statements regarding our intentions, plans, objectives, expectations, forecasts and projections. Our actual results and the timing of selected events could differ materially from those anticipated in these forward-looking statements as a result of several factors, including those set forth under the section titled “Risk Factors” and elsewhere in this report.
Overview
Our goal is to discover, develop and developdeliver therapeutics to defeat degeneration.
Our discovery and development strategy is guided by three overarching principles that we believe will significantly increase the probability of success and will accelerate the timing to bring effective therapeutics to patientspeople living with neurodegenerative diseases:
•Genetic Pathway Potentialdiseases and LSDs: We select our therapeutic targets and disease pathways based on genes that, when mutated, cause, or are major risk factors for, neurodegenerative diseases. We refer to these genes as degenogenes;
•Engineering Degenogenes: Genetic Pathway Realization – each of our programs addresses a molecular target or biological pathway that is genetically validated to cause or increase the risk for neurodegenerative diseases.
•Brain DeliveryDelivery: Validation and Optionality: We – we engineer our product candidates to cross the blood-brain barrier ("BBB")BBB and act directly in the brain;brain. Our proprietary TV platform technology is designed to effectively deliver large therapeutic molecules, such as enzymes, proteins, antibodies, and oligonucleotides, across the BBB after intravenous administration.
•Biomarker-Driven Development and Approval – we discover, develop and use biomarkers to inform dose selection, assess clinical activity, and identify patients most likely to respond to our therapies. We are actively engaged in discussions with health authorities regarding the potential use of biomarkers as primary clinical endpoints to support faster paths to approval.
Our clinical-stage large molecule programs are as follows:
•Tividenofusp alfa (DNL310, ETV:IDS), our lead ERT program enabled by our ETV, ETV:IDS is designed to cross the BBB and restore IDS and reduce GAGs, both peripherally and in the brain, in patients with MPS II (Hunter syndrome);
•TAK-594/DNL593 (PTV:PGRN), our recombinant PGRN biotherapeutic enabled by our PTV, being developed in collaboration with Takeda, to address certain types of FTD, specifically FTD-GRN caused by PGRN deficiency;
•DNL126 (ETV:SGSH), our second most advanced ETV enabled program, which is designed to restore lysosomal activity of SGSH, an enzyme responsible for degrading heparan sulfates in the lysosome, in patients with MPS IIIA (Sanfilippo syndrome Type A).
Our clinical-stage small molecule programs are as follows:
•BIIB122/DNL151 (LRRK2), our LRRK2 inhibitor program, being developed in collaboration with Biogen, to address Parkinson’s disease;
•DNL343 (eIF2B), our eIF2B activator program to address diseases such as ALS and FTD;
•Biomarker-Driven DevelopmentSAR443820/DNL788 (CNS-penetrant RIPK1 inhibitor): We discover, develop and utilize biomarkers to select the right patient population and demonstrate target engagement, pathway engagement and impact on disease progression of, our product candidates.
We are developing a broad portfolio of targeted therapeutic candidates for neurodegenerative diseases. Our programs are at different stages of clinical and preclinical development, including five programs in clinical studies.
We have also developed a proprietary BBB platform technology, our transport vehicle ("TV"), which enables multiple modality-based platforms to deliver a wide range of large-molecule therapeutics across the BBB, including enzymes, antibodies, proteins and oligonucleotides. This technology is designed to engage specific BBB transport receptors, which are ubiquitously expressed in brain capillaries and facilitate transport of proteins into the brain. We are currently optimizing and broadening this platform technology.
Our five clinical-stage programs are:
•our leucine-rich repeat kinase 2 ("LRRK2") inhibitor program, in collaboration with Biogen, to address Parkinson’s disease;
•our Eukaryotic initiation factor 2 B ("EIF2B”) activator program to address diseases such as amyotrophic lateral sclerosis ("ALS") and Frontotemporal Dementia ("FTD”);
•our ETV:IDS program, our most advanced program enabled by our TV technology, which is designed to restore iduronate 2-sulfatase ("IDS"), and reduce glycosaminoglycans, both peripherally and in the brain, in patients with mucopolysaccharidosis II ("MPS II", or "Hunter syndrome");
•our receptor interacting serine/threonine protein kinase 1 ("RIPK1")CNS-penetrant RIPK1 inhibitor program, partnered with Sanofi, to address neurological diseases such as Alzheimer’s diseaseMS and ALS as well as multiple sclerosis ("MS”);Alzheimer's disease; and
•Eclitasertib (SAR443122/DNL758, peripheral RIPK1 inhibitor), a second non-CNS penetrant RIPK1 inhibitor, partnered with Sanofi, to address peripheral inflammatory diseases such as cutaneous lupus.UC.
The following table summarizes key information about our clinical stage programs:
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| Program | | Product Candidate(s)Candidate | | Clinical PhaseStudy(ies) | | Indication(s)Indication | | Operational Control |
LRRK2 | | DNL151 (BIIB122) | | Ph 1 and Ph 1b | | Parkinson's disease | | Joint with Biogen |
| ETV:IDS | | Tividenofusp alfa, or DNL310 | | Ph 1/2 | | Hunter syndrome (MPS II) | | Denali |
| | Ph 2/3 | | |
| PTV:PGRN | | TAK-594/DNL593 | | Ph 1/2 (paused)1 | | FTD-GRN | | Joint with Takeda |
| ETV:SGSH | | DNL126 | | Ph 1/2 | | Sanfilippo syndrome Type A (MPS IIIA) | | Denali |
EIF2BLRRK2 | | BIIB122/DNL151 | | Ph 2a (planned) | | Parkinson's disease | | Denali |
| | Ph 2b | | | Joint with Biogen |
| eIF2B | | DNL343 | | Ph 11b | | ALS and FTD | | Denali |
| | Ph 2/3 | | ALS | | Joint with Healey Center |
| RIPK1 (CNS-penetrant) | | SAR443820/DNL788 | | Ph 2 (closing)2 | | ALS | | Sanofi |
| | Ph 2 | | MS | | Sanofi |
| | | | | | | | |
| RIPK1 (Peripheral) | | Eclitasertib, or SAR443122/DNL758 (SAR443122) | | Ph 1b2 | | UC | | Systemic inflammatory diseases | Sanofi |
RIPK1 (CNS) | | DNL788 (SAR443820) | | Ph 1 | | Neurological diseases | | Sanofi |
To complement our internal capabilities, we have entered into arrangements with biopharmaceutical companies, patient-focused data companies, numerous leading academic institutions and foundations to gain access to new product candidates, enable and accelerate the development of our existing programs and deepen our scientific understanding of certain areas of biology. We currently rely on third-party contract manufacturers to manufacture and supply our preclinical and clinical materials to(2)Study will be used during the development of our product candidates. We currently doclosing as it did not need commercial manufacturing capacity.meet its primary endpoint.
Since we commenced operations, we have devoted substantially all of our resources to discovering, acquiring and developing product candidates, building our BBB platform technology and assembling our core capabilities in understanding key neurodegenerative disease pathways.
Key operational and financing milestones for the year ended December 31, 20202023 and in 20212024 to date include:
•LRRK2Tividenofusp alfa DNL310 (ETV:IDS)
◦In February 2023, at the WORLDSymposiumTM, we reported additional interim data from the open-label, single-arm Phase 1/2 study of DNL310. Over 49 weeks of DNL310 treatment in the Phase 1/2 study, positive changes across measures of exploratory clinical outcomes including VABS-II (adaptive behavior) and BSID-III (cognitive capabilities) scores and global impression scales were observed. The data also suggested that DNL310 improved hearing, as assessed by auditory brainstem response testing. Additional biomarker data out to 49 weeks continued to demonstrate that DNL310 enabled rapid and sustained normalization of cerebrospinal fluid ("CSF") heparan sulfate to normal healthy levels and improvement in lysosomal function biomarkers. Reduction in urine heparan sulfate and dermatan sulfate after switch from standard of care to DNL310 suggested additional sustained peripheral activity of DNL310. The DNL310 safety profile, with up to two years of treatment, remained consistent with standard of care;
◦In June 2023, we announced a robust reduction in Neurofilament Light ("NfL") with DNL310 treatment in MPS II (Hunter syndrome). Interim results demonstrated average reduction of 64% (p <0.001) from baseline in serum NfL after two years of dosing with DNL310 in the Phase 1/2 study. The FDA has recommended assessment of NfL, a marker of neuroaxonal damage, as a possible biomarker in MPS II;
◦In August 2023, we presented new interim data from the Phase 1/2 study of DNL310 in MPS II in an oral presentation at the Society for the Study of Inborn Errors of Metabolism ("SSIEM") Annual Symposium 2023. The interim clinical outcomes data, safety profile, and biomarker effects, including normalization of CSF heparan sulfate and reduction in NfL, continue to support development of DNL310 in MPS II;
◦In January 2020,2024, we announced positive results from our LRRK2 program.that enrollment continues in the global Phase 1b results2/3 COMPASS study and is expected to be completed in 2024. We continue to engage with DNL201 in patients with Parkinson’s disease demonstrated high levels of targetthe U.S. Food and pathway engagementDrug Administration on a faster path to approval; and improvement of lysosomal biomarkers. Interim Phase 1 results with DNL151 (BIIB122) in more than 150 healthy volunteers also met all safety and biomarker goals. Both clinical trials showed dose-dependent target engagement, improvement in biomarkers of lysosomal function and demonstrated safety profiles supporting progression to further development;
◦In August 2020,February 2024, we entered into a binding Provisional Collaborationpresented additional interim data from the open-label, single-arm Phase 1/2 study of DNL310 at the 2024 WORLDSymposiumTM. Data out to 104 weeks showed additional improvement and License Agreement with Biogen Inc.’s subsidiaries, Biogen MA Inc. (“BIMA”stabilization in multiple measures of clinical outcomes, including those of adaptive behavior, cognition, hearing, and growth trajectory. In addition, robust and sustained responses in biomarkers of neuronal health (e.g., CSF heparan sulfate, neurofilament light (NfL)) and Biogen International GmbH (“BIG”) (BIMAperipheral activity (e.g. urine heparan sulfate and BIG, collectively, “Biogen”)dermatan sulfate) were observed. DNL310 continued to co-develop and co-commercialize our small molecule inhibitors of LRRK2 for Parkinson’s disease. Under the Provisional Collaboration and License Agreement, Biogen also received rights to opt into two programs and a right of first negotiation for two additional programs, in each case for neurodegenerative diseases leveraging our TV technology platform to cross the BBB and excluding small molecules, AAVs and oligonucleotides. In October 2020, the Provisional Collaboration and License Agreement expired upon the execution of the Definitive LRRK2 Collaboration and License Agreement and Right of First Negotiation, Option and License Agreement with Biogen (collectively the "Biogen Collaboration Agreement"). In connection with the Biogen Collaboration Agreement, we received an equity investment of $465.0 million in September 2020 and an aggregate of $560.0 million in upfront payments in October 2020. We may be eligible to receive up to $1.1 billion in potential milestone payments plus profit sharing and royalties for the LRRK2 program;generally well tolerated.
•TAK-594/DNL593 (PTV:PGRN)
◦In August 2020, we announced, together with our collaboration partner Biogen, the selectionMarch 2023, a $10.0 million milestone payment from Takeda was triggered upon achievement of DNL151 (BIIB122) to progress into late-stage development. We expect to initiate late-stagea specified clinical development of DNL151 (BIIB122) in Parkinson's patientsmilestone in the second halfPhase 1/2 study of 2021. Two clinical studies are planned: oneTAK-594/DNL593 in patients who carry LRRK2 mutationswith FTD-GRN, which we received in May 2023;
◦In July 2023, we presented healthy volunteer data from Part A of the Phase 1/2 study of TAK-594/DNL593 at the Alzheimer's Association International Conference, which continued to demonstrate that single doses of TAK-594/DNL593 resulted in substantial increases in CSF PGRN levels and the other in Parkinson's patients independent of mutation status;were generally well tolerated; and
◦In January 2021,2024, we announced completion ofthat Part B in the DNL151 (BIIB122)TAK-594/DNL593 Phase 1b trial1/2 study in Parkinson’s patients. Target engagementparticipants with FTD-GRN has been voluntarily paused to implement protocol modifications and pathway engagement goals were met. We are currently completing further dose escalation cohorts in an expanded Phase 1 trialis expected to define the full therapeutic window of DNL151 (BIIB122);resume this year.
•ETV:IDS:DNL126 (ETV:SGSH)
◦In August 2020,February 2024, we commencedannounced that dosing had been initiated in athe Phase 1/2 clinical trialstudy of DNL310DNL126 in Hunter syndrome patients. In November 2020, we announcedMPS IIIA; biomarker proof of concept was achieved inand safety data are expected by the Phase 1/2 trial after four weekly dosesend of DNL310, and2024. Further, in February 2021,2024, we announced additional positive interim results after three months of dosing. Based on strong proof-of-conceptpresented preclinical data we are expanding our DNL310 development programat WORLDSymposiumTM demonstrating that DNL126 improves lysosomal and microglial morphology, degeneration, and cognitive behavior in the ongoing Phase 1/2 trial, which will enable further exploration of clinical endpoints related to neuronopathic manifestations in patients. We plan to initiate a Phase 2/3 trial in the first half of 2022;MPS IIIA mice.
•EIF2B: In February 2020, we initiated a Phase 1 clinical trial for DNL343 in healthy volunteers. Results from this trial are expected to be available in the first half of 2021. We plan to initiate a Phase 1b trial of DNL343 in patients with ALS in the second half of 2021;
•RIPK1BIIB122/DNL151 (LRRK2)
◦In December 2020,June 2023, in conjunction with Biogen, and based on review of portfolio timelines and resource prioritization, we announced plans to revise the clinical development program for BIIB122/DNL151. Prior to the planned revisions, the BIIB122 clinical development program encompassed two global late-stage clinical trials: the Phase 2b LUMA study in participants with early-stage Parkinson’s disease, which commenced in May 2022; and the Phase 3 LIGHTHOUSE study in participants with Parkinson’s disease related to LRRK2 mutations, which commenced in September 2022. In consideration of the LIGHTHOUSE study’s complexity, including the long timeline with anticipated study completion in 2031, Biogen and we plan to refocus our efforts to enable a timely readout on efficacy in idiopathic early-stage Parkinson’s disease while gaining further clinical data in Parkinson’s disease with and without a LRRK2 mutation. The planned revisions to the BIIB122 clinical development program are not based on any safety or efficacy data from studies of BIIB122. Biogen and we plan to modify the LUMA study’s enrollment criteria to allow for inclusion of eligible participants with Parkinson’s disease and a confirmed pathogenic variant of LRRK2, in addition to continuing to enroll eligible participants with idiopathic early-stage Parkinson’s disease. Collectively, data from the LUMA study will inform next steps for the development of BIIB122 in Parkinson’s disease;
◦In August 2023, we executed an amendment to the Definitive LRRK2 Collaboration and License Agreement and Waiver of and Amendment to Right of First Negotiation (ROFN), Option, and License Agreement. As part of the amendment, certain milestone criteria were changed while the total amount of development, regulatory, and commercial milestones across all indications remains the same. In addition, Biogen agreed to waive the remaining option and rights of first negotiation under the ROFN and Option Agreement; and
◦In February 2024, we announced the execution of a Collaboration and Development Funding Agreement in January 2024 with a third party related to a global Phase 2a study of BIIB122/DNL151, which we plan to solely operationalize to evaluate safety and biomarkers associated with BIIB122 in participants with Parkinson’s disease and confirmed pathogenic variants of LRRK2. This agreement includes committed funding of $75.0 million, of which $12.5 million was received in January 2024, and the remainder will be triggered based on time and operational milestones in the study. Biogen will continue to conduct the ongoing global Phase 2b LUMA study in early-stage Parkinson’s disease. Denali and Biogen will co-commercialize BIIB122/DNL151 assuming regulatory approval. The third party will be eligible to receive low single-digit royalties from Denali on annual worldwide net sales of LRRK2 inhibitors for the treatment of Parkinson’s disease, with royalty amounts varying based on the scope of the label.
•SAR443820/DNL788 and Eclitasertib (SAR443122/DNL758) (RIPK1)
◦In January 2023, our collaboration partner Sanofi commenced dosing in the Phase 2 study of SAR443820/DNL788 (SAR443820), a potent, selective brain-penetrant small molecule inhibitor of RIPK1, in a Phase 1 clinical trial in healthy volunteers. Earlier in 2020, we decided, together with our collaboration partner Sanofi, to pause clinical studies with DNL747 and focus our efforts on accelerating development of DNL788 (SAR443820), the backup molecule, which we believe has superior drug properties and a more rapid path toward proof-of-concept clinical studies in patients with MS, triggering a $25.0 million milestone payment, which was received in multiple neurological indications;January 2023. In November 2023 we announced that Sanofi has completed enrollment in this study;
◦Separately,In August 2023, we announced that Sanofi had completed enrollment in July 2020,the global Phase 2 HIMALAYA study of SAR443820/DNL788in ALS in July;
◦In August 2023, we announced that Sanofi commenced dosing of DNL758 (SAR443122), a peripherally-restricted small molecule inhibitor of RIPK1, incompleted a Phase 1b clinical trial2 study of eclitasertib, also known as SAR443122/DNL758, in hospitalized adult patients with severe COVID-19 lung disease. TheCLE in June. In October 2023, Sanofi announced that the development of eclitasertib in CLE is being discontinued because the Phase 1b trial was completed and DNL758 (SAR443122)2 proof-of-concept study did not meet its primary endpoint (percent change from baseline in CLASI-A at week 12). Eclitasertib was found to be generally well toleratedwell-tolerated. Sanofi continues to recruit the Phase 2 study of eclitasertib in patients with UC; and resulted
◦In February 2024, we announced that the Phase 2 HIMALAYA study evaluating SAR443820/DNL788 in changesparticipants with ALS did not meet the primary endpoint of change in disease relevant biomarkersALS Functional Rating Scale-Revised (ALSFRS-R). Sanofi intends to present the detailed efficacy and clinical outcome trends consistent withsafety results of the proof of mechanism. Based on the rapidly evolving landscape of treatment and prevention options for COVID-19,ALS Phase 2 HIMALAYA study at a future scientific forum. Sanofi has made a sponsor decision to hold further developmentis evaluating SAR443820/DNL788 in COVID-19 at this time. Sanofi plans to initiate aanother Phase 2 clinical trial in participants with MS, and the outcome of DNL758 (SAR443122)HIMALAYA study has no impact on the ongoing MS study.
•DNL343 (eIF2B)
◦In April 2023, we presented final data from the 28-day treatment period of the Phase 1b study of DNL343 in patientsparticipants with cutaneous lupusALS at the 75th Annual Meeting of the American Academy of Neurology ("AAN"). The results continued to demonstrate that once-daily oral dosing with DNL343 for 28 days was generally well tolerated and demonstrated extensive CSF penetration. In addition, robust inhibition of biomarkers associated with the ISR pathway was observed in blood samples from study participants; and
◦In May 2023, the first patient was dosed with DNL343 in the first halfPhase 2/3 HEALEY ALS Platform Trial. Recruitment of 2021;participants continues for this trial.
•Other
◦In January 2020, we sold 9.0 million shares of common stock (inclusive of shares sold pursuant to an overallotment option granted to the underwriters in connection with the offering) through an underwritten public offering at a price of $23.00 per share for aggregate net proceeds of $193.9 million;
◦In December 2020March 2023, a contingent consideration payment of $30.0 million associated with our acquisition of F-star Gamma was triggered upon the achievement of a specified clinical milestone in the ETV:IDS program. This payment fully satisfies our clinical contingent consideration obligations under the Purchase Agreement;
◦In April 2023, we entered into a new operating lease in Salt Lake City, Utah for a 59,336 square foot laboratory (the "Utah site"), office and January 2021, GLPwarehouse premises, after terminating our previous SLC lease in March 2023, decreasing future lease payments by $6.1 million while increasing the lease term by approximately five and a half years. The Utah site will expand our clinical manufacturing capabilities for biologic therapeutics (large molecules) as we plan to use the premises for the manufacture of materials for toxicology studies and drug substance for early human clinical studies with the goal of increasing flexibility and speed in advancing new investigational therapies into clinical trials;
◦In April 2023, our collaboration partner Biogen exercised its option to develop and commercialize our ATV program targeting amyloid-beta, triggering an option exercise payment of $5.0 million, which we received in May 2023;
◦In August 2023 we announced that, in agreement with Takeda, the companies will discontinue clinical development of TAK-920/DNL919 in Alzheimer’s disease. This is a strategic decision based on the totality of clinical data emerging from the single ascending dose Phase 1 study of TAK-920/DNL919 in healthy volunteers and in consideration of the rapidly evolving treatment landscape for Alzheimer's disease whereby an understanding of drug combinations with newly approved therapies will be important. A preliminary analysis of Phase 1 data indicates robust target engagement and effects on microglial biomarkers (e.g., CSF1R, SPP1, IL1RA, IP10, MIP1b, MCP-1), which were initiated for PTV:PGRN andconsistent with preclinical studies that demonstrate that ATV:TREM2 respectively, triggeringinduces robust changes to a responsive microglial cell state (van Lengerich B, et al. Nat Neurosci. 2023). In the secondPhase 1 study, TAK-920/DNL919 was clinically well tolerated at doses with demonstrated changes in CSF biomarkers and there were no serious adverse events or severe treatment emergent adverse events; however, safety signals of moderate, reversible hematologic effects were observed at the highest dose tested, suggesting a narrow therapeutic window for the Alzheimer’s disease patient population. The Phase 1 safety findings are believed to be specific to properties of TAK-920/DNL919 and TREM2 biology. Denali and Takeda will continue to focus research efforts on back-up molecules in preclinical milestone payments under the Takeda Collaboration Agreementdevelopment, including exploration of $8.0 million for each program. The milestone payment for PTV:PGRN was receivedpotential combination therapy given recent new drug approvals in January 2021;Alzheimer's disease;
◦In January 2021, following achievement of human biomarker proof of concept with DNL310 for Hunter syndrome,2024, we announced the additionour intention to spin out our preclinical small molecule portfolio. We will maintain ownership of, five new brain-penetrant enzyme replacement therapy programs in its ETVand continue to advance, our current portfolio including: (1) ETV:GBAof clinical stage small molecule programs. The decision was made based on clinical validation and prioritization of our TV-enabled platforms for Gaucher diseasebrain delivery of large molecules; and Parkinson’s disease; (2) ETV:GAA for Pompe disease; (3) ETV:IDUA for MPS I; (4) ETV:NAGLU for MPS IIIB; and (5) ETV:ARSA for Metachromatic Leukodystrophy ("MLD"); and
◦To address risks posed byIn February 2024, we announced that on February 27, 2024 we entered into a securities purchase agreement with certain existing accredited investors for the COVID-19 pandemic, we have implemented policies that enable someprivate placement of 3,244,689 shares of our employeescommon stock at a price of $17.07 per share and pre-funded warrants to work remotely. For all on-site personnel, wepurchase an aggregate of 26,046,065 shares of our common stock at a purchase price of $17.06 per pre-funded warrant, resulting in anticipated gross proceeds of approximately $499.7 million. The pre-funded warrants will have implemented several safety protocols, including regular, mandatory COVID-19 testing proceduresan exercise price of $0.01 per share of Common Stock, be immediately exercisable and compliance measures for social distancing and use of personal protective equipment. After initial COVID-19 pandemic shutdown restrictions were put into placeremain exercisable until exercised in March 2020, we experienced a pause in patient recruitment in several clinical trials. Recruitment has since resumed for all affected clinical trials.full. The private placement is expected to close on February 29, 2024, subject to customary closing conditions.
We do not have any products approved for sale and have not generated any product revenue since our inception. We have funded our operations primarily from the issuance and sale of convertible preferred stock, and the proceeds from our IPO, follow-on offering,sale of common stock in public offerings, and payments received from our collaboration agreements with Takeda, Sanofi and Biogen.
We have incurred significant operating losses to date and expect to continue to incur operating losses for the foreseeable future. Our ability to generate product revenue will depend on the successful development and eventual commercialization of one or more of our product candidates. Due to revenue recognized from our collaboration arrangement with Biogen, weWe had net incomelosses of $71.1$145.2 million, for the year ended December 31, 2020. Our net losses were $197.6$326.0 million, and $88.2$290.6 million for the years ended December 31, 20192023, 2022, and 2018,2021, respectively. As of December 31, 2020,2023, we had an accumulated deficit of $354.4 million.$1.12 billion. We expect to continue to incur significant expenses and operating losses as we advance our current clinical stage programs through healthy volunteer and patient trials; broaden and improve our BBB platform technology; acquire, discover, validate and develop additional product candidates; obtain, maintain, protect and enforce our intellectual property portfolio; and hire additional personnel.
LicenseWe currently rely on third-party contract manufacturers to manufacture and Collaboration Agreements
Takeda
In January 2018, we entered intosupply our preclinical and clinical materials to be used during the Collaboration Agreement with Takeda (“Takeda Collaboration Agreement”) pursuant to which we granted Takeda an option with respect to threedevelopment of our programs to develop and commercialize, jointly with us, certain biologic products that are enabled by our BBB delivery technology and intended for the treatment of neurodegenerative disorders. The three programs were our ATV:BACE1/Tau, ATV:TREM2 and PTV:PGRN programs. The Takeda Collaboration Agreement became effective in February 2018 when the requirements of the Hart-Scott-Rodino Antitrust Improvements Act of 1976 ("HSR") were satisfied. In February 2019, we amended the Takeda Collaboration Agreement to replace the ATV:BACE1/Tau program with the ATV:Tau program.
Under the terms of the Takeda Collaboration Agreement, Takeda paid us a $40.0 million upfront payment, and is obligated to pay us up to an aggregate of $25.0 million with respect to each program under the Takeda Collaboration Agreement directed to a target and based upon the achievement of certain preclinical milestone events, up to $75.0 million in total, of which we have earned and received $15.0 million to date. Takeda is also obligated to pay us a $5.0 million option fee for each target for which Takeda exercises its option, up to $15.0 million in total.
Pursuant to the terms of the Takeda Collaboration Agreement, we entered into the Purchase Agreement with Takeda in January 2018, pursuant to which we agreed to issue and sell, and Takeda agreed to purchase, 4,214,559 shares of our common stock for an aggregate purchase price of $110.0 million.product candidates. We closed the sale of the 4,214,559 shares of our common stock to Takeda on February 23, 2018.
After Takeda exercises its option for a particular target, we and Takeda will share equally the development and commercialization costs, and, if applicable, the profits, for each collaboration program. However, for each collaboration program, we may electcurrently do not to continue sharing development and commercialization costs, or Takeda may elect to terminate our cost-profit sharing rights and obligations if, following notice from Takeda and a cure period, we fail to satisfy our cost sharing obligations with respect to the relevant collaboration program. After such an election by us or termination by Takeda becomes effective, we will no longer be obligated to share in the development and commercialization costs for the relevant collaboration program, and we will not share in any profits from that collaboration program. Instead we will be entitled to receive tiered royalties. The royalty rates will be in the low- to mid-teen percentages on net sales, or low- to high-teen percentages on net sales if we have met a certain co-funding threshold at the time of our election to opt out of co-development or Takeda’s termination of our cost-profit sharing rights and obligations, and, in each case, these royalty rates will be subject to certain reductions specified in the Takeda Collaboration Agreement. Takeda will pay these royalties to us for each biologic product included in the relevant collaboration program, on a country-by-country basis, until the latest of (i) the expiration of certain patents covering the relevant biologic product, (ii) the expiration of all regulatory exclusivity for that biologic product, and (iii) an agreed period of time after the firstneed commercial sale of that biologic product in the applicable country, unless biosimilar competition in excess of a significant level specified in the Takeda Collaboration Agreement occurs earlier, in which case Takeda’s royalty obligations in the applicable country would terminate.manufacturing capacity.
License and Collaboration Agreements
Collaborations and partnering are central components of our strategy to build, develop and commercialize our portfolio of product candidates. We have numerous arrangements with biopharmaceutical companies, technology companies, academic institutions, foundations, and patient-focused data companies.
Biogen
In October 2020, we entered into the LRRK2 Agreement with Biogen pursuant to which we granted Biogen a license to co-develop and co-commercialize our small molecule LRRK2 Program, in addition to the ROFN and Option Agreement pursuant to which we granted an option and a right of first negotiation to certain of our programs utilizing our TV technology platform; collectively the "Biogen Collaboration Agreement." In connection with our collaboration with Biogen, we also entered into a common stock purchase agreement pursuant to which we sold 13,310,243 shares of our common stock to BIMA for an aggregate purchase price of $465.0 million. Under the terms of the Biogen Collaboration Agreement, we received $560.0 million in upfront payments in October 2020.
In April 2023, Biogen exercised its option to license our ATV:Abeta program and we received additional consideration of $5.0 million for an option exercise fee. In August 2023, we executed an Amendment (the “Biogen Amendment”) to the LRRK2 Agreement and ROFN and Option Agreement with Biogen. Pursuant to the Biogen Amendment, the schedule of potential LRRK2 Agreement milestones was amended, while maintaining the same total value of milestones that Denali is eligible to receive. In addition, if Takeda exercisesBiogen waived its option right to the second option program and waived its rights of first negotiation for all three collaborationtwo other TV-enabled programs Takeda may be obligated to payunder the ROFN and Option Agreement. Further details regarding the terms of the agreements between us up to an aggregate of $407.5 million upon achievement of certain clinical milestone events and up to an aggregate of $300.0 millionBiogen are included in regulatory milestone events relating to receipt of regulatory approvalthis Annual Report on Form 10-K in the United States, certain European countriessection titled "Business - Licenses and Japan. Takeda may also be obligatedCollaborations."
In relation to pay us up to $75.0 million per biologic product upon achievement of a certain sales-based milestone, or an aggregate of $225.0 million if one biologic product from each program achieves this milestone.
Wethe Biogen Collaboration Agreement, we have recognized related party collaboration revenue of $27.2$295.5 million, $5.9$3.1 million, and $5.7$3.7 million associated with the Takeda Collaboration Agreement in the years ended December 31, 2020, 20192023, 2022 and 2018, respectively.2021, respectively, related party research and development expense of $17.7 million and $8.2 million for cost sharing payments to Biogen in the year ended December 31, 2023 and 2022, respectively, and a related party offset to research and development expense as a result of cost sharing reimbursements from Biogen of $6.5 million in the year ended December 31, 2021. We have recorded a receivablecost sharing payables of $8.0$3.2 million from Takedaand $4.4 million on the Consolidated Balance Sheet as of December 31, 2020, payment for which was received in January 2021,2023 and no receivable as of December 31, 2019.2022, respectively. Through December 31, 2020,2023, we have received $15.0had earned $5.0 million in preclinical milestoneoption fee payments from Takeda and havebut had not recorded any milestone revenue or product sales under the TakedaBiogen Collaboration Agreement.
Sanofi
In October 2018, we entered into the Sanofi Collaboration Agreement with Sanofi pursuant to which certain small molecule CNS and peripheral RIPK1 inhibitors contributed by Sanofi and by us are being, or will be developed and commercialized. The Sanofi Collaboration Agreement became effective in November 2018 when the HSR requirements were satisfied. UnderFurther details regarding the terms of the Collaboration Agreement,agreement between us and Sanofi, paid us a $125.0 million upfront payment. Underand historic payments between the Sanofi Collaboration Agreement, Sanofi is required to make milestone payments up to approximately $1.1 billion upon achievement of certain clinical, regulatory and sales milestone events. Such milestone payments include $215.0 millionparties under the agreement, are included in clinical milestone payments and $385.0 million in regulatory milestone payments for CNS Products, as defined, that are developed and approvedthis Annual Report on Form 10-K in the United States, by the European Medicines Agency ("EMA")section titled "Business - Licenses and in Japan for three indications, including Alzheimer's disease. These milestones also include $120.0 million in clinical milestone payments, $175.0 million in regulatory milestone payments and $200.0 million in commercial milestone payments for Peripheral Products, as defined, that are developed and approved in the United States, by the EMA and in Japan for three indications. Through December 31, 2020, we have received milestone payments of Collaborations."$10.0 million under the Sanofi Collaboration Agreement.
We will share profits and losses equally with Sanofi for CNS Products sold in the United States and China, and receive royalties on net sales for CNS Products outside of the United States and China and for Peripheral Products sold worldwide, each as further described below. RIPK1 Inhibitors contributed by Sanofi and developed and commercialized under the Sanofi Collaboration Agreement will be subject to lower milestone and royalty payments to us compared to RIPK1 Inhibitors contributed by us. We will also retain responsibility for certain payment obligations under our agreement with an academic institution which licensed certain intellectual property to us that we are sublicensing to Sanofi under the Sanofi Collaboration Agreement.
We and Sanofi will jointly develop CNS Products pursuant to a global development plan. We will be responsible, at our cost, for the conduct of Phase 1 and Phase 2 trials for CNS Products for Alzheimer’s disease and any activities required to support such clinical trials and specific for Alzheimer’s disease. Sanofi will be responsible, at its cost, for all other Phase 1 and Phase 2 trials for CNS Products, including for ALS and multiple sclerosis. Sanofi will lead the conduct of all Phase 3 and later stage development trials for CNS Products, with Sanofi funding 70% of such costs and us funding 30% of such costs. We have the ability to opt out of the cost-profit sharing provisions of the Sanofi Collaboration Agreement, as further described below.
Sanofi will lead commercialization activities globally for CNS Products. We may elect to conduct certain co-commercialization activities outside of MS with respect to each CNS Product in the United States and/or China, provided that the cost-profit sharing provisions of the Sanofi Collaboration Agreement for the relevant CNS Product are still in effect, as further described below.
We may opt out of the cost-profit sharing provisions of the Sanofi Collaboration Agreement for CNS Products in the United States and China on a CNS Product-by-CNS Product and country-by-country basis. Sanofi may also terminate our cost-profit sharing provisions of the Sanofi Collaboration Agreement in its entirety if, following notice from Sanofi and a cure period, we fail to satisfy our cost-sharing obligations. After such an opt out by us or termination by Sanofi, we will no longer be obligated to share in the development and commercialization costs for the applicable CNS Products and we will not share in the applicable profits from such CNS Products. Instead, we will be entitled to receive tiered royalties on net sales of the applicable CNS Products in the relevant country (or countries). The royalty rates will be a percentage in the low double digits to mid-teens, but may increase to the mid-teens to low-twenties percentages for all countries in which Sanofi is paying royalties on the applicable CNS products, if we have met certain co-funding thresholds at the time of our election or Sanofi’s termination of our cost-profit sharing rights and obligations.
Sanofi will be responsible, at its cost, for conducting activities relating to the development and commercialization of all Peripheral Products. Sanofi will lead commercialization activities globally for Peripheral Products. We will be entitled to receive tiered royalties in the low- to mid- teen percentages on net sales of Peripheral Products.
We have recognized collaboration revenue of $1.1$25.0 million, $20.8$53.4 million and $123.5$15.0 million associated with the Sanofi Collaboration Agreement in the years ended December 31, 2020, 20192023, 2022 and 2018,2021, respectively, and recorded ano receivable from Sanofi of $44,303 and $1.2 million on the Consolidated Balance Sheets as of December 31, 20202023 and 2019, respectively.2022. Through December 31, 2020,2023, we havehad received milestone payments of $10.0$100.0 million and have we had not recorded any product sales under the Sanofi Collaboration Agreement.
Biogen
In August 2020, we entered into the Provisional Biogen Collaboration Agreement with Biogen pursuant to which we granted Biogen a license to co-develop and co-commercialize our LRRK2 Program, an option in respect of two Option Programs, and a right of first negotiation with respect to the ROFN Programs should we decide to seek a collaboration with a third party for such programs. In October 2020, we entered into the LRRK2 Agreement and the ROFN and Option Agreement with Biogen. The material terms of the LRRK2 Agreement and the ROFN and Option Agreement were consistent with, and superseded, the Provisional Biogen Collaboration Agreement.
The LRRK2 Agreement
The LRRK2 Agreement includes our LRRK2 Products that penetrate the BBB, including DNL201 and DNL151 (BIIB122), as well as those that do not penetrate the BBB. Based on the totality of preclinical and clinical data to date, both DNL201 and DNL151 (BIIB122) (two chemically distinct LRRK2 inhibitors) have met our requirements to proceed into further late-stage clinical testing, however, DNL151 (BIIB122) has been selected to proceed due to pharmacokinetic properties that provide additional dosing regimen flexibility.
Takeda
Under
In January 2018, we entered into the Takeda Collaboration Agreement pursuant to which we granted Takeda an option to develop and commercialize, jointly with us, our ATV:TREM2, PTV:PGRN and ATV:BACE1/Tau programs, the latter of which was later replaced with our ATV:Tau program. Pursuant to the terms of the LRRK2 Agreement, Biogen paid us a $400 million upfront payment in October 2020. With respect to the LRRK2 Program, Biogen is required to make milestone payments up to approximately $1.125 billion upon achievement of certain development and sales milestone events. Such milestone payments include $375.0 million in development, $375.0 million upon first commercial sale, and $375.0 million in net sales-based milestones. We will share profits and losses equally with Biogen for LRRK2 Products in the United States and will share profits and losses in China with Biogen sharing 60% of such profits and losses and us sharing 40% of such profits and losses. We will be entitled to receive royalties in the high teens to low twenties percentages on net sales for LRRK2 Products outside of the United States and China.
We and Biogen will jointly develop LRRK2 Products pursuant to a clinical development plan set forth in the LRRK2 Agreement. We and Biogen will share responsibility and costs for global development of LRRK2 Products, with Biogen funding 60% of such costs and us funding 40% of such costs. We have the ability to opt out of the development cost sharing arrangement, as further described below. Biogen will lead commercialization activities globally for LRRK2 Products. We will co-commercialize the LRRK2 Products with Biogen in the United States and China, provided that the profit-sharing arrangement for the LRRK2 Products is still in effect, as further described below.
We may opt out of development cost sharing worldwide and upon such election, of all further profit sharing from the LRRK2 Program. We also have the right to opt-out of the profit-sharing arrangement for the LRRK2 Program or for only those LRRK2 Products that do not penetrate the BBB (“Peripheral LRRK2 Products”), in each of the United States and China. After such an opt out, we will no longer be obligated to share in the development and commercialization costs for, and we will not share in the applicable revenues from, such LRRK2 Program (or from the Peripheral LRRK2 Products) in such country, as applicable. In such cases, we will be entitled to receive tiered royalties on net sales of the applicable LRRK2 Program in the relevant country (or countries). The royalty rates for the applicable LRRK2 Program will be a percentage in the high teens to low twenties, but may increase to the low twenties to mid-twenties, if we have met certain co-funding thresholds or there has been a first commercial sale at the time of our election.
The ROFN and Option Agreement
In addition to the LRRK2 Program, Biogen also received an exclusive option to license two preclinical programs from our TV technology platform, including our ATV:Abeta program and a second program utilizing our TV technology for an unnamed target, excluding small molecules, AAVs and oligonucleotides. Biogen’s option may be triggered up to initiation of IND-enabling studies for each program and continues for each program until a specified period of time after delivery of an option data package or 30 business days after the 5th anniversary of the effective date of the Provisional Biogen Collaboration Agreement, whichever is earlier.
Further, Biogen will have the right of first negotiation on two additional TV-enabled therapeutics in the field of Alzheimer’s disease, Parkinson’s disease, ALS or multiple sclerosis should we decide to seek a collaboration with a third party for such programs, but this does not include any of our small molecule, AAVs and oligonucleotide programs. The ROFN period continues until seven years after the effective date of the Provisional Biogen Collaboration Agreement or the date on which we have offered Biogen two ROFN Programs and for which Biogen has agreed to trigger a ROFN for such program, whichever is earlier. However, if we do not execute an agreement with a third party with respect to a particular ROFN Program offered to Biogen within a specified amount of time, then Biogen will have one additional right to exercise the ROFN again with respect to such ROFN Program.
Under the ROFN and Option Agreement, Biogen paid us a $160 million upfront payment in October 2020. With respect to the options granted by us to Biogen, Biogen is obligated to pay to us an aggregate of up to $270 million in option exercise and development milestone payments and an aggregate of up to $615 million in commercial milestone payments, following the achievement of certain prespecified milestone events and if Biogen exercises both of its options. Furthermore, Biogen is obligated to pay us royalties in the mid-single digit to mid-teens percentages, depending on the program for which Biogen exercises its option and upon the achievement of certain sales thresholds.
In addition, if Biogen exercises its ROFN with respect to an eligible Denali program, the parties are obligated to negotiate in good faith for a specified period of time regarding the financial and other terms of an agreement pursuant to which Biogen would obtain rights to such program.
Common Stock Purchase Agreement
In August 2020, in connection with the Provisional BiogenTakeda Collaboration Agreement, we also entered into a common stock purchase agreement (the “Stockthe Purchase Agreement”)Agreement with BIMA,Takeda in January 2018, pursuant to which we agreed to issue and sell, and BIMA agreed to purchase, 13,310,243sold 4,214,559 shares of our common stock (the “Shares”)to Takeda for an aggregate purchase price of $465.0 million pursuant to the terms and conditions thereof. The sale of the Shares closed, and payment was received, in September 2020.$110.0 million.
In November 2021 and December 2021, Takeda exercised its options for the PTV:PGRN and ATV:TREM2 programs, respectively, subsequent to which we have shared equally in the development costs for the programs. In March 2022, we mutually agreed under the Takeda Collaboration Agreement to terminate activity on the ATV:Tau program. Further details regarding the terms of the agreement between us and Takeda, and historic payments between the parties under the agreements, are included in this Annual Report on Form 10-K in the section titled "Business - Licenses and Collaborations."
We have recognized related party collaboration revenue of $307.4of $10.0 million, $51.9 million and a related party offset$29.9 million associated with the Takeda Collaboration Agreement in the years ended December 31, 2023, 2022 and 2021, respectively, and offsets to research and development expense for cost sharing reimbursements of $9.3$12.2 million, $18.2 million and $13.7 million in the yearyears ended December 31, 2020 associated with the Biogen Collaboration Agreement.2023, 2022, and 2021, respectively. We have recorded cost sharing reimbursements duereceivables of $2.7 million and $8.9 million from Biogen of $5.7 million on the consolidated balance sheetTakeda as of December 31, 2020.2023 and 2022, respectively. Through December 31, 2020,2023, we have received $65.0 million in milestone payments and $10.0 million of option exercise fees from Takeda, and we have not received any milestone payments from Biogen and have not recorded any product sales under the BiogenTakeda Collaboration Agreement.
F-star
In August 2016, we entered into a License andthe F-star Collaboration Agreement ("F-star Collaboration Agreement") with the F-star Gamma Limited (“F-star Gamma”), F-star Biotechnologische Forschungs-und Entwicklungsges m.b.H ("F-star GmbH") and F-star Biotechnology Limited ("F-star Ltd") (collectively, “F-star”)entities. . The goal of the collaboration iswas the development of Fcabs to enhance delivery of therapeutics across the BBB into the brain. The collaboration leverages F-star’s modular antibody technology and our expertise in the development of therapies for neurodegenerative diseases. In connection with the entry into the F-star Collaboration Agreement, we also purchased an option for an upfront option fee of $0.5 million, or the buy-out option to acquire all of the outstanding shares of F-star Gamma pursuant to a pre-negotiated buy-out option agreement ("Option Agreement").agreement.
In May 2018, we exercised such buy-out option and entered into a Sharethe F-star Purchase Agreement (the “Purchase Agreement”) with the shareholders of F-star Gamma and Shareholder Representative Services LLC, pursuant to which we acquired all of the outstanding shares of F-star Gamma (the “Acquisition”).
Gamma. As a result of the Acquisition, F-star Gamma became our wholly-owned subsidiary and the entity's name was changed to Denali BBB Holding Limited. In addition, we became a direct licensee of certain intellectual property of F-star Ltd by way of our assumption of F-star Gamma’s license agreement with F-star Ltd, dated August 24, 2016, (the “F-star Gamma License”). We made initial exercise payments2016. Further details regarding the terms of $18.0 million in aggregate under the Purchase Agreementarrangements between us and the F-star Gamma License, lessentities, and historic payments between the net liabilities of F-star Gamma, which were approximately $0.2 million. In addition, weparties under the agreements, are required to make future contingent payments, to F-star Ltd and the former shareholders of F-star Gamma, up to a maximum amount of $447.0 millionincluded in this Annual Report on Form 10-K in the aggregate uponsection titled "Business - Licenses and Collaborations."
Through December 31, 2023 we have recognized consideration paid under the achievementF-star Purchase Agreement of certain defined$49.8 million as research and development expenses consisting of up-front, preclinical clinical, regulatory and commercial milestones. These include up to $27.0 million in preclinical contingent payments, $50.0 million in clinical contingent consideration payments, $120.0of which $30.0 million of research and development expense was recognized in regulatorythe year ended December 31, 2023 for a contingent payments and $250.0 millionconsideration payment triggered in commercial contingent payments. The amount of the contingent payments will vary based on whether F-star delivers an Fcab (constant Fc-domains with antigen-binding activity) that meets pre-defined criteria and whether the Fcab has been identified solely by us or solely by F-star Ltd. or jointly by us and F-star Ltd. In June 2019, we made a payment of $1.5 million to F-star LtdMarch 2023 upon the achievement of a specified preclinicalclinical milestone in ourthe ETV:IDS program.
Under the terms of the original F-star Collaboration Agreement, we could nominate up to three Fcab targets (“Accepted Fcab Targets”) within the first three years of the date of the F-star Collaboration Agreement. Upon entering into the F-star Collaboration Agreement, we selected transferrin receptor (“TfR”) as the first Accepted Fcab Target and paid F-star Gamma an upfront fee of $5.5 million, which included selection of the first Accepted Fcab Target. In May 2018, we exercised our right to nominate two additional Fcab Targets and identified a second Accepted Fcab Target. We made a one-time payment for the two additional Accepted Fcab Targets of $6.0 million in the aggregate and extended the time period for our selection of the third Accepted Fcab Target until approximately the fourth anniversary of the date of the original F-star Collaboration Agreement, or August 2020. We have not identified a third Fcab Target.
We did not recognize any There was no contingent consideration on the acquisition date. We recognized $1.5 million and $18.3 million of consideration as research and development expense recognized in the years ended December 31, 2019 and 2018, respectively. No research and development expense was recognized for consideration under the F-star Purchase Agreement in the year ended December 31, 2020. $6.0 million for the nomination of two additional Accepted Fcab Targets under the F-star Collaboration Agreement was reco2022 or 2021. gnized as research and development expense in the year ended December 31, 2018, along with the upfront option fee of $0.5 million previously included within other non-current assets. We recognized an additional $1.2 million, $1.1 million and $0.80.1 million of research and development expense related to the funding of F-star research costs for the year ended December 31, 2021, but none for the years ended December 31, 2020, 2019 and 20182023 or , respectively. 2022.
Genentech
In June 2016, we entered into an Exclusive License Agreementexclusive license agreement with Genentech. The agreement gives us access to Genentech’s LRRK2 inhibitor program. Our collaboration partner in the LRRK2 program, Biogen, is responsible for 50% of any payment obligation to Genentech under the Biogen Collaboration Agreement.
As consideration to date, we have paid Genentech $12.5$25.0 million in the aggregate, including an upfront fee, a technology transfer fee and athree clinical milestone payment, allpayments, with $18.8 million of which wasthis recorded as research and development expense as incurred. No amounts were recordedincurred, net of cost sharing reimbursements from Biogen. Included within this amount, in the year ended December 31, 2022, we paid Genentech a $7.5 million clinical milestone payment triggered upon the commencement of dosing in the global Phase 2b LUMA study to evaluate the efficacy and safety of BIIB122/DNL151 by Biogen, and a further $5.0 million clinical milestone payment triggered upon the commencement of dosing in the global Phase 3 LIGHTHOUSE study to evaluate the efficacy and safety profile of BIIB122/DNL151 by Biogen. Of these milestones, we recognized $6.3 million as research and development expense as incurred after cost sharing reimbursements from Biogen inthe year ended December 31, 2022. We recognized no expenses under this agreement in the years ended December 31, 2020 or 2019. The first clinical milestone payment of $12.5 million was recorded as research2023 and development expense during the year ended December 31, 2017.
We may owe Genentech milestone payments upon the achievement of certain development, regulatory, and commercial milestones, up to a maximum of $315.0 million in the aggregate. These milestones include up to $37.5 million in clinical milestone payments, $102.5 million in regulatory milestone payments and $175.0 million in commercial milestone payments. In addition, we may owe royalties on net sales of licensed products ranging from low to high single-digit percentages, with the exact royalty rate dependent on various factors, including (i) whether the compound incorporated in the relevant licensed product is a Genentech-provided compound or a compound acquired or developed by us, (ii) the date a compound was first discovered, derived or optimized by us, (iii) the existence of patent rights covering the relevant licensed product in the relevant country, (iv) the existence of orphan drug exclusivity covering a licensed product that is a Genentech-provided compound and (v) the level of annual net sales of the relevant licensed product. We also have the right to credit a certain amount of third-party royalty and milestone payments against royalty and milestone payments owed to Genentech, up to a maximum reduction of fifty percent. Under the terms of our LRRK2 Agreement with Biogen, Biogen is responsible for 50% of any payment obligation to Genentech under this agreement accruing after October 2020.
Unless earlier terminated, the agreement with Genentech will continue in effect until all of our royalty and milestone payment obligations to Genentech expire. Following expiration of the agreement, we will retain the licenses under the intellectual property Genentech licensed to us on a non-exclusive, royalty-free basis.
Components of Operating Results
Collaboration Revenue
To date, we have not generated any revenue from product sales and do not expect to generate any revenue from product sales for the foreseeable future. All revenue recognized to date has been collaboration and license revenue from our collaboration agreements with Takeda, Sanofi and Biogen.
In the future, we will continue to recognizeFuture revenue may be recognized from the Takeda Collaboration Agreement, Sanofi Collaboration Agreement, and Biogen Collaboration Agreement, and may generate revenuebe generated from product sales or milestones,milestone payments, royalties and costprofit sharing reimbursement from other collaboration agreements, strategic alliances and licensing arrangements. We expect that our revenue will fluctuate from quarter-to-quarter and year-to-year as a result of the timing and amount of license fees, milestones,option exercise fees, milestone payments, profit sharing reimbursement, of costs incurred and other payments and product sales, to the extent any are successfully commercialized. If we fail to complete the development of our product candidates in a timely manner or obtain regulatory approval for them, our ability to generate future revenue, and our results of operations and financial position, would be materially adversely affected.
Operating Expenses
Research and Development
Research and development activities account for a significant portion of our operating expenses. We record research and development expenses as incurred. Research and development expenses incurred by us for the discovery and development of our product candidates and BBB platform technology include:
• external research and development expenses, including:
–expenses incurred under arrangements with third parties, such as contract research organizations ("CROs"), preclinical testing organizations, contract development and manufacturing organizations ("CDMOs"), academic and non-profit institutions and consultants;
–expenses to acquire technologies to be used in research and development that have not reached technological feasibility and have no alternative future use;
–fees related to our license and collaboration agreements;
•personnel related expenses, including salaries, benefits and stock-based compensation expense; and
•other expenses, which include direct and allocated expenses for laboratory, facilities and other costs.
A portion of our research and development expenses are direct external expenses, which we track on a program-specific basis once a program has commenced late-stage IND-enabling studies.
Program expenses include expenses associated with our most advanced product candidates and the discovery and development of backup or next-generation molecules. We also track external expenses associated with our TV platform. These expenses include thoseexternal expenses incurred by us relating to our Takeda Collaboration Agreement, Sanofi Collaboration Agreement and Biogen Collaboration Agreement. All external costs associated with earlier stage programs, or that benefit the entire portfolio, are tracked as a group. We do not trackalso incur personnel orand other operating expenses incurred for our research and development programs on a program-specific basis.which are presented in aggregate. These expenses primarily relate to salaries and benefits, stock-based compensation, facility expenses including rent and depreciation, and lab consumables.
Where we share costs with our collaboration partners, such as in our Biogen Collaboration Agreement and Takeda Collaboration Agreement, research and development expenses may include cost sharing reimbursements from, or payments to, our partner, respectively.collaboration partners.
It is challenging to predict the nature, timing and estimated long-range costs of the efforts that will be necessary to complete the development of, and obtain regulatory approval for, any of our product candidates. This is made more challenging by events outside of our control, such as the recent COVID-19 pandemic.pandemic and increased geopolitical uncertainty. We are also unable to predict when, if ever, material net cash inflows will commence from sales or licensing of our product candidates. This is due to the numerous risks and uncertainties associated with drug development, including the uncertainty of:
•our ability to add and retain key research and development personnel;
•our ability to establish an appropriate safety profile with IND-enabling toxicology studies;
•our ability to successfully develop, obtain regulatory approval for, and then successfully commercialize, our product candidates;
•our successful enrollment in and completion of clinical trials;
•the costs associated with the development of any additional product candidates we identify in-house or acquire through collaborations;
•our ability to discover, develop and utilize biomarkers to demonstrate target engagement, pathway engagement and the impact on disease progression of our molecules;
•our ability to establish agreements with third-party manufacturers for clinical supply for our clinical trials and commercial manufacturing, if our product candidates are approved;
•the terms and timing of any collaboration, license or other arrangement, including the terms and timing of any milestone payments thereunder;
•our ability to obtain and maintain patent, trade secret and other intellectual property protection and regulatory exclusivity for our product candidates if and when approved;
•our receipt of marketing approvals from applicable regulatory authorities;
•our ability to commercialize products, if and when approved, whether alone or in collaboration with others; and
•the continued acceptable safety profiles of the product candidates following approval.
A change in any of these variables with respect to the development of any of our product candidates would significantly change the costs, timing and viability associated with the development of that product candidate. We expect our research and development expenses to increase at least over the next several years as we continue to implement our business strategy, advance our current programs, expand our research and development efforts, seek regulatory approvals for any product candidates that successfully complete clinical trials, access and develop additional product candidates and incur expenses associated with hiring additional personnel to support our research and development efforts. In addition, product candidates in later stages of clinical development generally incur higher development costs than those in earlier stages of clinical development, primarily due to the increased size and duration of later-stage clinical trials.
General and Administrative
General and administrative expenses include personnel-relatedpersonnel related expenses, such as salaries, benefits, travel and stock-based compensation expense, expenses for outside professional services and allocated expenses. Outside professional services consist of legal, accounting and audit services and other consulting fees. Allocated expenses consist of rent, depreciation and other expenses related to our office and research and development facility not otherwise included in research and development expenses.
We expect to continue to incur certain expenses as a result of operating as a public company, including expenses related to compliance with the rules and regulations of the SEC and those of any national securities exchange on which our securities are traded, insurance expenses, investor relations activities and other administrative and professional services. We expect to increase our administrative headcount as we advance our product candidates through clinical development, which will increase our general and administrative expenses.
Interest and Other Income, Net
Interest and other income, net, consists primarily of interest income and investment income earned on our cash, cash equivalents, and marketable securities, gains and losses on foreign currency hedges, and sublease income.
Results of Operations
Comparison of the years ended December 31, 20202023 and 20192022
The following table sets forth the significant components of our results of operations (in thousands):
| | Year Ended December 31, | | Change | |
| 2020 | | 2019 | | $ | | % | |
| Year Ended December 31, | |
| 2023 | |
| 2023 | |
| 2023 | |
| Collaboration revenue: | |
| Collaboration revenue: | |
| Collaboration revenue: | Collaboration revenue: | | | | | | | |
| Collaboration revenue from customers | Collaboration revenue from customers | 335,561 | | | 26,320 | | | 309,241 | | | * | |
| Collaboration revenue from customers | |
| Collaboration revenue from customers | | 330,531 | | | 105,065 | | | 225,466 | | | * | % |
| Other collaboration revenue | Other collaboration revenue | 98 | | | 358 | | | (260) | | | (73) | | |
| Total Collaboration revenue | 335,659 | | | 26,678 | | | 308,981 | | | * | |
| Total collaboration revenue | |
| Total collaboration revenue | |
| Total collaboration revenue | |
| Operating expenses: | |
| Operating expenses: | |
| Operating expenses: | Operating expenses: | |
| Research and development | Research and development | 212,615 | | | 193,382 | | | 19,233 | | | 10 | | |
| Research and development | |
| Research and development | |
| General and administrative | |
| General and administrative | |
| General and administrative | General and administrative | 60,326 | | | 46,480 | | | 13,846 | | | 30 | | |
| Total operating expenses | Total operating expenses | 272,941 | | | 239,862 | | | 33,079 | | | 14 | | |
| Income (loss) from operations | 62,718 | | | (213,184) | | | 275,902 | | | * | |
| Total operating expenses | |
| Total operating expenses | |
| Loss from operations | |
| Loss from operations | |
| Loss from operations | |
| Interest and other income, net | Interest and other income, net | 9,241 | | | 15,219 | | | (5,978) | | | (39) | | |
| Income (loss) before income taxes | 71,959 | | | (197,965) | | | 269,924 | | | * | |
| Income tax (expense) benefit | (823) | | | 351 | | | (1,174) | | | * | |
| Net income (loss) | $ | 71,136 | | | $ | (197,614) | | | $ | 268,750 | | | * | % |
| Interest and other income, net | |
| Interest and other income, net | |
| Loss before income taxes | |
| Loss before income taxes | |
| Loss before income taxes | |
| Income tax expense | |
| Income tax expense | |
| Income tax expense | |
| Net loss | |
| Net loss | |
| Net loss | | $ | (145,224) | | | $ | (325,991) | | | $ | 180,767 | | | (55) | | % |
Refer to “Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations—Results of Operations” in our 20192022 Annual Report on Form 10-K for a discussion of the results of operations for the year ended December 31, 20192022 compared to the year ended December 31, 2018.2021.
To date, we have not generated any product revenue. We do not expect to generate any product revenue unless and until we obtain regulatory approval of and commercialize any of our product candidates, and we do not know when, or if, either will occur.
We expect to continue to incur significant losses for the foreseeable future, and we expect the losses to increase as we expand our research and development activities and continue the development of, and seek regulatory approvals for, our product candidates, and begin to commercialize any approved products. Further, we expect general and administrative expenses to increase as we continue to incur additional costs associated with supporting our growing operations. We are subject to all of the risks typically related to the development of new product candidates, and we may encounter unforeseen expenses, difficulties, complications, delays and other unknown factors that may adversely affect our business. We anticipate that we will need substantial additional funding in connection with our continuing operations.
Until we can generate a sufficient amount of revenue from the commercialization of our product candidates or from our existing collaboration agreements, or future agreements with other third parties, if ever, we expect to finance our future cash needs through public or private equity or debt financings. Additional capital may not be available on reasonable terms, if at all. If we are unable to raise additional capital in sufficient amounts or on terms acceptable to us, we may have to significantly delay, scale back or discontinue the development or commercialization of one or more of our product candidates. If we raise additional funds through the issuance of additional debt or equity securities, it could result in dilution to our existing stockholders, increased fixed payment obligations and the existence of securities with rights that may be senior to those of our common stock. If we incur indebtedness, we could become subject to covenants that would restrict our operations and potentially impair our competitiveness, such as limitations on our ability to incur additional debt, limitations on our ability to acquire, sell or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business. Additionally, any future collaborations we enter into with third parties may provide capital in the near term but limit our potential cash flow and revenue in the future. Any of the foregoing could significantly harm our business, financial condition and prospects.
A change in the outcome of any of these or other variables with respect to the development of any of our product candidates could significantly change the costs and timing associated with the development of that product candidate. Furthermore, our operating plans may change in the future, and we may need additional funds to meet operational needs and capital requirements associated with such operating plans.
The following table sets forth a summary of the primary sources and uses of cash for each of the periods presented below (in thousands):
Refer to “Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations—Liquidity and Capital Resources” in our 20192022 Annual Report on Form 10-K for a discussion of the cash flows for the years ended December 31, 20192022 and 2018.
This discussion and analysis of our financial condition and results of operations is based on our consolidated financial statements, which have been prepared in accordance with U.S. GAAP. The preparation of these consolidated financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the consolidated financial statements, as well as the reported revenues recognized and expenses incurred during the reporting periods. Our estimates are based on our historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions. While ourOur significant accounting policies are described in more detail in the notes to our consolidated financial statements included elsewhere in this report, wereport. We believe that the following accounting policies are critical to understandingestimates involve a significant level of estimation uncertainty which could have a material impact on our historical and future performance, as these policies relate to the more significant areas involving management’s judgments and estimates.financial condition or results of operations.
See Note 1 to our consolidated financial statements included in Part II, Item 8, “Financial Statements and Supplementary Data,” of this Annual Report on Form 10-K for a description of recent accounting pronouncements applicable to our business.
We are exposed to market risks in the ordinary course of our business, primarily related to interest rate and foreign currency sensitivities.
The primary objective of our investment activities is to preserve capital to fund our operations. We also seek to maximize income from our investments without assuming significant risk. To achieve our objectives, we maintain a portfolio of investments in a variety of securities of high credit quality and short-term duration, according to our board-approved investment policy. Our investments are subject to interest rate risk and could fall in value if market interest rates increase. A hypothetical 10% relative change in interest rates during any of the periods presented would not have had a material impact on our consolidated financial statements.
The majority of our transactions occur in U.S. dollars. However, we do have certain transactions that are denominated in currencies other than the U.S. dollar, primarily the Euro and British Pound, and we therefore are subject to foreign exchange risk. The fluctuation in the value of the U.S. dollar against other currencies affects the reported amounts of expenses, assets and liabilities primarily associated with a limited number of preclinical, clinical and manufacturing activities.
To the Stockholders and the Board of Directors of Denali Therapeutics Inc.
We have audited the accompanying consolidated balance sheets of Denali Therapeutics Inc. (the Company) as of December 31, 20202023 and 2019,2022, the related consolidated statements of operations and comprehensive income (loss),loss, stockholders' equity and cash flows for each of the three years in the period ended December 31, 2020,2023, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 20202023 and 2019,2022, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2020,2023, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company's internal control over financial reporting as of December 31, 2020,2023, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework), and our report dated February 26, 202127, 2024 expressed an unqualified opinion thereon.
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
We have served as the Company’s auditor since 2015.
Denali Therapeutics Inc.
See accompanying notes to consolidated financial statements.
Denali Therapeutics Inc.
See accompanying notes to consolidated financial statements.
Denali Therapeutics Inc.
See accompanying notes to consolidated financial statements.
Denali Therapeutics Inc.
See accompanying notes to consolidated financial statements.
Denali Therapeutics Inc.
Denali Therapeutics Inc. ("Denali" or the “Company”) is a biopharmaceutical company, incorporated in Delaware, that discovers and develops therapeutics to defeat neurodegenerative diseases. The Company is headquartered in South San Francisco, California.
These consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America (“U.S. GAAP”) and pursuant to the rules and regulations of the Securities and Exchange Commission (“SEC”).
These consolidated financial statements include the accounts of the Company and its wholly-owned subsidiary.subsidiaries. All intercompany balances and transactions have been eliminated in consolidation. For the Company and its subsidiary,subsidiaries, the functional currency has been determined to be U.S. dollars. Monetary assets and liabilities denominated in foreign currency are remeasured at period-end exchange rates. Non-monetaryrates, non-monetary assets and liabilities denominated in foreign currencies are remeasured at historical rates, and transactions in foreign currencies are remeasured at average exchange rates. Foreign currency transaction gains and losses resulting from remeasurement are recognized in interest and other income, net in the Consolidated Statements of Operations and Comprehensive Income (Loss).Loss.
The preparation of financial statements in conformity with U.S. GAAP requires the Company to make certain estimates, judgments and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements, as well as the reported amounts of expenses during the reporting period. Actual results could differ from those estimates, and such differences could be material to the Consolidated Balance Sheets and Consolidated Statements of Operations and Comprehensive Income (Loss).Loss.
Financial instruments that potentially subject the Company to significant concentrations of credit risk consist primarily of cash, cash equivalents, and marketable securities and forward foreign currency exchange contracts.securities. Substantially all of the Company’s cash and cash equivalents are deposited in accounts with financial institutions that management believes are of high credit quality. Such deposits have and will continue to exceed federally insured limits. The Company maintains its cash with accredited financial institutions and accordingly, such funds are subject to minimal credit risk.
Accounting Standards Codification ("ASC") Topic 820, Fair Value Measurement (“ASC 820”), establishes a fair value hierarchy for instruments measured at fair value that distinguishes between assumptions based on market data (observable inputs) and the Company’s own assumptions (unobservable inputs). Observable inputs are inputs that market participants would use in pricing the asset or liability based on market data obtained from sources independent of the Company. Unobservable inputs are inputs that reflect the Company’s assumptions about the inputs that market participants would use in pricing the asset or liability, and are developed based on the best information available in the circumstances.
ASC 820 identifies fair value as the exchange price, or exit price, representing the amount that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants. As a basis for considering market participant assumptions in fair value measurements, ASC 820 establishes a three-tier fair value hierarchy that distinguishes between the following:
To the extent that the valuation is based on models or inputs that are less observable or unobservable in the market, the determination of fair value requires more judgment. Accordingly, the degree of judgment exercised by the Company in determining fair value is greatest for instruments categorized in Level 3. A financial instrument’s level within the fair value hierarchy is based on the lowest level of any input that is significant to the fair value measurement.
The carrying amounts reflected in the Consolidated Balance Sheets for cash and cash equivalents, prepaid expenses and other current assets, accounts payable, and accrued liabilities approximate their fair values due to their short-term nature.
The Company considers all highly liquid investments with original maturities of 90 days or less at the date of purchase to be cash and cash equivalents. Cash equivalents are reported at fair value.
Cash, cash equivalents, and restricted cash reported within the Consolidated Statements of Cash Flows is composed of Cashcash and Cashcash equivalents reported in the Consolidated Balance Sheets and $1.6 million and $1.5 million of restricted cash for the letter of credit for the Company’s headquarters building lease which is included within other non-current assets in the Consolidated Balance Sheets.Sheets as of December 31, 2023 and 2022, respectively.
The Company generally invests its excess cash in money market funds and investment grade short to intermediate-term fixed income securities. Such investments are included in cash and cash equivalents, short-term marketable securities, or long-termshort-term marketable securities on the Consolidated Balance Sheets, are considered available-for-sale, and are reported at fair value with net unrealized gains and losses included as a component of stockholders’ equity.
The Company classifies investments in securities with remaining maturities of less than one year, or where its intent is to use the investments to fund current operations or to make them available for current operations, as short-term investments. The Company classifies investments in securities with remaining maturities of over one year as long-term investments.investments, unless intended to fund current operations. The amortized cost of debt securities is adjusted for amortization of premiums and accretion of discounts to maturity, which is included in interest and other income, net in the Consolidated Statements of Operations and Comprehensive Income (Loss).Loss. Realized gains and losses and declines in value determined to be due to credit losses on marketable securities, if any, are included in interest and other income, net.
The Company periodically evaluates the need for an allowance for credit losses. This evaluation includes consideration of several qualitative and quantitative factors, including whether it has plans to sell the security, whether it is more likely than not it will be required to sell any marketable securities before recovery of its amortized cost basis, and if the entity has the ability and intent to hold the security to maturity, and the portion of any unrealized loss that is the result of a credit loss. Factors considered in making these evaluations include quoted market prices, recent financial results and operating trends, implied values from any recent transactions or offers of investee securities, credit quality of debt instrument issuers, expected cash flows from securities, other publicly available information that may affect the value of the marketable security, duration and severity of the decline in value, and the Company's strategy and intentions for holding the marketable security.
Accounts receivable are included within prepaid expenses and other current assets on the Consolidated Balance Sheets. The accounts receivable balance represents amounts receivable from the Company's collaboration partners, excluding related parties, net of an allowance for credit losses, if required.
Property and equipment are stated at cost, net of accumulated depreciation. Depreciation is computed using the straight-line method over the related estimated useful lives as presented in the table below. Significant additions and improvements are capitalized, while repairs and maintenance are charged to expense as incurred.
The Company periodically evaluates property and equipment for impairment whenever events or changes in circumstances indicate that a potential impairment may have occurred. If such events or changes in circumstances arise, the Company compares the carrying amount of the long-lived assets to the estimated future undiscounted cash flows expected to be generated by the long-lived assets. If the estimated aggregate undiscounted cash flows are less than the carrying amount of the long-lived assets, an impairment charge, calculated as the amount by which the carrying amount of the assets exceeds the fair value of the assets, is recorded. The fair value of the long-lived assets is determined based on the estimated discounted cash flows expected to be generated from the long-lived assets. The Company has not recorded any suchmaterial impairment charges during the years presented.
ROU assets represent the Company’s right to use the underlying asset for the lease term and lease liabilities represent the Company’s obligation to make lease payments arising from the lease. Operating lease ROU assets and liabilities are recognized at the lease commencement date based on the present value of lease payments due over the lease term, with the ROU assets adjusted for lease incentives received. When determining the present value of lease payments, the Company uses its incremental borrowing rate on the date of lease commencement, or the rate implicit in the lease, if known. The Company does not assume renewals in its determination of the lease term unless the renewals are deemed by management to be reasonably certain at lease inception.
The Company analyzes its collaboration arrangements to assess whether they are within the scope of ASC 808, Collaborative Arrangements (“ASC 808”) to determine whether such arrangements involve joint operating activities performed by parties that are both active participants in the activities and exposed to significant risks and rewards dependent on the commercial success of such activities. This assessment is performed throughout the life of the arrangement based on changes in the responsibilities of all parties in the arrangement. For collaboration arrangements within the scope of ASC 808 that contain multiple elements, the Company first determines which elements of the collaboration are deemed to be within the scope of ASC 808 and those that are more reflective of a vendor-customer relationship and, therefore, within the scope of Topic 606. For elements of collaboration arrangements that are accounted for pursuant to ASC 808, an appropriate recognition method is determined and applied consistently, generally by analogy to Topic 606. The accounting treatment pursuant to Topic 606 is outlined below.
In determining the appropriate amount of revenue to be recognized as the Company fulfills its obligations under each of its agreements, the Company performs the following steps: (i) identification of the promised goods or services in the contract; (ii) determination of whether the promised goods or services are performance obligations including whether they are distinct in the context of the contract; (iii) measurement of the transaction price, including the constraint on variable consideration; (iv) allocation of the transaction price to the performance obligations based on estimated selling prices; and (v) recognition of revenue when (or as) the Company satisfies each performance obligation.
Amounts received prior to satisfying the revenue recognition criteria are recorded as contract liabilities in the Company’s Consolidated Balance Sheets. If the related performance obligation is expected to be satisfied within the next twelve months this will be classified in current liabilities. Amounts recognized as revenue prior to the Company having an unconditional right (other than a right that is conditioned only on the passage of time) to receipt are recorded as contract assets in the Company's Consolidated Balance Sheets. If the Company expects to have an unconditional right to receive the consideration in the next twelve months, this will be classified in current assets. A net contract asset or liability is presented for each contract with a customer.
At contract inception, the Company assesses the goods or services promised in a contract with a customer and identifies those distinct goods and services that represent a performance obligation. A promised good or service may not be identified as a performance obligation if it is immaterial in the context of the contract with the customer, if it is not separately identifiable from other promises in the contract (either because it is not capable of being separated or because it is not separable in the context of the contract), or if the promised good or service does not provide the customer with a material right.
The Company considers the terms of the contract to determine the transaction price. The transaction price is the amount of consideration to which the Company expects to be entitled in exchange for transferring promised goods or services to a customer. The consideration promised in a contract with a customer may include fixed amounts, variable amounts, or both. Variable consideration will only be included in the transaction price when it is not considered constrained, which is when it is probable that a significant reversal in the amount of cumulative revenue recognized will not occur.
If it is determined that multiple performance obligations exist, the transaction price is allocated at the inception of the agreement to all identified performance obligations based on the relative standalone selling prices ("SSP"). The relative SSP for each deliverable is estimated using external sourced evidence if it is available. If external sourced evidence is not available, the Company uses its best estimate of the SSP for the deliverable.
Revenue is recognized when, or as, the Company satisfies a performance obligation by transferring a promised good or service to a customer. An asset is transferred when, or as, the customer obtains control of that asset, which for a service is considered to be as the services are received and used. The Company recognizes revenue over time by measuring the progress toward complete satisfaction of the relevant performance obligation using an appropriate input or output method based on the nature of the service promised to the customer.
After contract inception, the transaction price is reassessed at every period end and updated for changes such as resolution of uncertain events. Any change in the transaction price is allocated to the performance obligations on the same basis as at contract inception, or to a single performance obligation as applicable. The Company accounts for the exercise of a material right as either a contract modification or as a continuation of the existing contract, as is most appropriate based on the facts and circumstances.
Management may be required to exercise considerable judgment in estimating revenue to be recognized. Judgment is required in identifying performance obligations, estimating the transaction price, estimating the SSP of identified performance obligations, which may include forecasted revenue, development timelines, reimbursement rates for personnel costs, discount rates and probabilities of technical and regulatory success, and estimating the progress towards satisfaction of performance obligations.
The Company has acquired and may continue to acquire the rights to develop and commercialize new product candidates from third parties. The upfront payments to acquire license, product or rights, as well as any future milestone payments, are immediately recognized as research and development expense provided that there is no alternative future use of the rights in other research and development projects.
The Company’s stock-based compensation programs grant awards that have included stock options, restricted stock units, restricted stock awards, and shares issued under its employee stock purchase plan. Grants are awarded to employees, including directors, and non-employees.non-employee service providers.
Income taxes are accounted for using the liability method, under which deferred tax assets and liabilities are determined based on the temporary differences between the financial reporting and tax bases of assets and liabilities and consideration is given to net operating losses and tax credit carryforwards. Deferred tax assets and liabilities are measured using the enacted tax rates that are expected to be in effect when the differences are expected to reverse.
The Company assesses the likelihood that deferred tax assets will be recovered from future taxable income, and a valuation allowance is established when necessary to reduce deferred tax assets to the amounts more likely than not expected to be realized.
The Company’s policy is to recognize interest and penalties accrued on any unrecognized tax benefits as a component of income tax expense.
Assets and liabilities measured at fair value at each balance sheet date are as follows (in thousands):
The Company’s Level 2 securities are valued using third-party pricing sources. The pricing services utilize industry standard valuation models, including both income and market-based approaches, for which all significant inputs are observable, either directly or indirectly.
4. Derivative Financial Instruments
In August 2016, the Company entered into a License and Collaboration Agreement (“F-star Collaboration Agreement”) with F-star Gamma Limited (“F-star Gamma”), F-star Biotechnologische Forschungs-und Entwicklungsges M.B.H ("F-star GmbH") and F-star Biotechnology Limited ("F-star Ltd") (collectively, “F-star”) to leverage F-star’s modular antibody technology and the Company’s expertise in the development of therapies for neurodegenerative diseases. Under the F-star Collaboration Agreement, the Company made payments to F-star totaling $11.5 million. In connection with the entry into the F-star Collaboration Agreement, the Company also purchased an option for an upfront option fee of $0.5 million (the “buy-out-option”), to acquire all of the outstanding shares of F-star Gamma pursuant to a pre-negotiated buy-out option agreement (the “Option Agreement”).
In May 2018, the Company exercised the Option Agreement and entered into a Share Purchase Agreement (the “Purchase Agreement”) with the shareholders of F-star Gamma and Shareholder Representative Services LLC, pursuant to which the Company acquired all of the outstanding shares of F-star Gamma (the “Acquisition”).
The Company may opt out of development cost sharing worldwide and upon such election, from any further profit-sharing from the LRRK2 Program. The Company also has the right to opt-out of the profit sharing arrangement for the LRRK2 Program or for only those LRRK2 Products that do not penetrate the BBB (“Peripheral LRRK2 Products”), in each of the United States and China. After such an opt out, the Company will no longer be obligated to share in the development and commercialization costs for, or be entitled to share in the applicable revenues from, such LRRK2 Program (or from the Peripheral LRRK2 Products) for such country, as applicable. If the Company chooses to exercise its opt out rights, the Company will be entitled to receive tiered royalties on net sales of the applicable LRRK2 Program in the relevant country (or countries). The royalty rates for the applicable LRRK2 Program will be a percentage in the high teens to low twenties, but may increase to the mid-twenties if the Company has met certain co-funding thresholds or there has been a first commercial sale at the time of the Company's election.
In connection with the Provisional Biogen Collaboration Agreement, the Company entered into a common stock purchase agreement (the "Stock Purchase Agreement") with BIMA on August 5, 2020, pursuant to which the Company agreed to issue and sell, and BIMA agreed to purchase,sold 13,310,243 shares of the Company’s common stock (the “Shares”) to BIMA for an aggregate purchase price of $465.0 million pursuant to the terms and conditions thereof. Since the shares of common stock owned by Biogen as of December 31, 2020 represent more than 10% of the voting interest of the Company, Biogen is considered a related party as defined in ASC 850.million. Management determined that it was appropriate to account for the Provisional Biogen Collaboration Agreement and the SPA as one arrangement because they were entered into at the same time with interrelated financial terms.
The Company identified the following distinct performance obligations associated with the Biogen Collaboration Agreement that had not yet been delivered under the original contract: the LRRK2 Program license, the research services for the ATV:Abeta and TV programs (“Option Research Services”) which include option joint steering committee ("JSC") participation, and a material right for an option under the ROFN and Option Agreement. Further, the LRRK2 Development Activities which includes LRRK2 JSC and joint development committee (“JDC”) participation was identified as a unit of account under ASC 808. The LRRK2 Development Activities, JSC and JDC participation are considered to be a single unit of account since the development activities are highly interrelated with the JSC and JDC involvement and these are not distinct in the context of the contract. Further, the same was considered to be true for the option research services and option JSC participation performance obligation.
The Company believes that the Biogen Collaboration Agreement is a collaboration arrangement as defined in ASC 808, Collaborative Agreements.Arrangements. The Company also believes that Biogen meets the definition of a customer as defined in ASC 606, Revenue From Contracts With Customers for all of the performance obligations identified at inception except for the LRRK2 Development Activities. Since ASC 808 does not address recognition and measurement, the Company looked to other accounting literature for guidance where the performance obligation does not fall under ASC 606, and determined that for the interim LRRK2 development activities subject to cost sharing provisions, the guidance in ASC 730, Research and Development should be applied.
The respective standalone value for each of the performance obligations was determined at inception by applying the SSP method and the transaction price of $604.9 million at inception was allocated based on the relative SSP method with revenue recognition timing to be determined either by delivery, resolution of an option, or the provision of services.
In assessing the Biogen Collaboration Agreement, management exercised considerable judgment in estimating revenue to be recognized, specifically related to estimating the discount for lack of marketability associated with the stock issuance, determining the separate performance obligations under the Biogen Collaboration Agreement, and estimating the standalone selling price of those performance obligations.
In October 2018, the Company entered into a Collaboration and License Agreement ("Sanofi Collaboration Agreement") with Genzyme Corporation, a wholly owned subsidiary of Sanofi S.A. ("Sanofi") pursuant to which certain small molecule CNS and peripheral receptor interacting serine/threonine protein kinase 1 ("RIPK1") inhibitors contributed by Sanofi and by the Company will be developed and commercialized. The Sanofi Collaboration Agreement became effective in November 2018 at which time Sanofi paid the Company an upfront payment of $125.0 million.million which was included in the transaction price at inception. Under the Sanofi Collaboration Agreement, the Company is eligible to receive milestone payments from Sanofi up to approximately $1.1 billion upon achievement of certain clinical, regulatory and sales milestone events. Such milestone payments include $215.0 million in clinical milestone payments and $385.0 million in regulatory milestone payments for CNS Products, as defined, that are developed and approved in the United States, by the European Medicines Agency ("EMA") and in Japan for 3three indications, including Alzheimer's disease. These milestones also include $120.0 million in clinical milestone payments, $175.0 million in regulatory milestone payments and $200.0 million in commercial milestone payments for Peripheral Products, as defined, that are developed and approved in the United States, by the EMAEurope and Japan for 3three indications.
The Company will share profits and losses equally with Sanofi for CNS Products sold in the United States and China, and receive variable royalties on net sales for CNS Products sold outside of the United States and China and for Peripheral Products sold worldwide.
Sanofi will be responsible, at its cost, for conducting activities relating to the development and commercialization of all Peripheral Products. Denali will be entitled to receive tiered royalties in the low- to mid- teen percentages on net sales of Peripheral Products.
The Company identified the following distinct performance obligations associated with the Sanofi Collaboration Agreement upon inception: the CNS program license, the Peripheral program license, the Phase 1 and Phase 2 trials for CNS Products for Alzheimer’s disease ("Alzheimer's Disease Services"), and the Phase 1b trial for DNL747 for ALS and associated activities ("Retained Activities").
The Company believes that the Sanofi Collaboration Agreement is a collaboration arrangement as defined in ASC 808, Collaborative Agreements.Arrangements. The Company also believes that Sanofi meets the definition of a customer as defined in ASC 606, Revenue From Contracts With Customers for three of the performance obligations identified at inception, but does not meet the definition of a customer for the Alzheimer's Disease Services. Further, Sanofi does not meet the definition of a customer for all Phase 3 and later stage development trials for CNS Products led by Sanofi for which the Company will fund 30% of total costs. Since ASC 808 does not address recognition and measurement, the Company looked to other accounting literature for guidance where the performance obligation does not fall under ASC 606, and determined that for the Alzheimer's Disease Services, the guidance in ASC 606 should be analogized for the recognition, measurement and reporting of this performance obligation, and for the cost sharing provisions, the Company determined that the guidance in ASC 730, Research and Development should be applied.
The Company used an adjusted market assessment approach to estimate the selling price for the program licenses, and an expected cost plus margin approach for estimating the Alzheimer’s Disease Services and the Retained Activities. The program licenses and existing know-how were delivered on the effective date of the Sanofi Collaboration Agreement.Agreement, and as such upfront revenue allocated to these performance obligations were recognized at this time. Further, clinical milestones are recognized as earned since these relate to the underlying program licenses previously delivered. The Alzheimer’s Disease Services and the Retained Activities were expected to be delivered over time as the services are performed. For the Alzheimer's Disease Services, revenue will bewas recognized over time using the input method, based on costs incurred to perform the services, since the level of costs incurred over time iswas thought to best reflect the transfer of services to Sanofi. For the Retained Activities, revenue will bewas recognized over time using the output method, based on amounts invoiced to Sanofi, since this is believed to directly correlate to the value of the services performed.
Under the Takeda Collaboration Agreement and unless otherwise agreed jointly between both parties, the Company will bewas responsible, at its cost, for conducting activities relating to pre-IND development of biologic products directed to the 3three identified targets and enabled by its BBB delivery technology targeting TfR during the applicable research period. The period through which the option can be exercised continues for each target until the first biologic product directed
The Company identified performance obligations during the research period consisting of the license, the development options, and joint steering committee ("JSC") participation together with the research services for each collaboration program. The license rights, JSC involvement, option and research services arewere considered to be a single performance obligation for each program since the research services arewere highly interrelated with the option and JSC involvement and will significantly modify the license. The performance obligations under each of the 3three programs are separate since the activities and risks under the programs are distinct.
In June 2016, the Company entered into an Exclusive License Agreement with Genentech, Inc. (“Genentech”). TheThis agreement gives the Company access to Genentech’s LRRK2 inhibitor small molecule program for Parkinson's disease. Under the agreement, Genentech granted the Company (i) an exclusive, worldwide, sublicensable license under Genentech’s rights to certain patents and patent applications directed to small molecule compounds which bind to and inhibit LRRK2 and (ii) a non-exclusive, worldwide, sublicensable license to certain related know-how, in each case, to develop and commercialize certain compounds and licensed products incorporating any such compound.
Accrued expenses and other current liabilities consists of the following (in thousands):
Management exercised judgment in applying the requirements of ASC 842, including the determination as to whether certain contracts contain a lease, the lease consideration, and the commencement date of the lease, and for leases identified under the Headquarters Lease Amendment,standard, the discount rate used to determine the measurement of the lease liability. As the implicit rateThe discount rates of our operating leases are an approximation of the Headquarters Lease Amendment was not known,Company's incremental borrowing rate and are dependent upon the Company estimated a 9.0% discount rate, which was management’s estimateterm and economics of the Company’s incremental borrowing rate.agreement. To estimate the incremental borrowing rate, management consideredconsiders observable debt yields of comparable market instruments, as well as benchmarks within the Headquarters Lease Amendmentlease agreement that may be indicative of the rate implicit in the lease.
Effective September 2017, the Company entered into a Development and Manufacturing Services Agreement as amended (“DMSA”) with Lonza Sales AG (“Lonza”) for the development and manufacture of biologic products. Under the DMSA, the Company will execute purchase orders based on project plans authorizing Lonza to provide development and manufacturing services with respect to certain of the Company's antibody and enzyme products, and will pay for the services provided and batches delivered in accordance with the DMSA and project plan. Unless earlier terminated, the DMSA will expire on September 6, 2022.
From time to time, the Company may be involved in lawsuits, arbitration, claims, investigations and proceedings consisting of intellectual property, employment and other matters which arise in the ordinary course of business. The Company records accruals for loss contingencies to the extent that the Company concludes that it is probable that a liability has been incurred and the amount of the related loss can be reasonably estimated.
Aggregate intrinsic value represents the difference between the fair value of the Company's common stock and the exercise price of outstanding options. The total intrinsic value of options exercised was $43.8$12.1 million, $13.1$12.6 million, and $9.8$47.9 million for the years endedas of December 31, 2020, 20192023, 2022 and 2018,2021, respectively. During the years ended December 31, 2020, 2019,2023, 2022, and 20182021 the weighted-average grant-date fair value of the options vested was $11.51, $10.09,$24.30, $23.96, and $3.85$11.83 per share, respectively. The weighted-average grant date fair value of all options granted during the years ended December 31, 2020, 20192023, 2022 and 20182021 was $15.67, $11.86,$17.95, $26.00, and $14.79$41.30 per share, respectively.
The reconciliation of federal statutory income tax rate to our effective income tax rate is as follows:
The components of the Company’s net deferred tax assets are as follows (in thousands):
A reconciliation of the beginning and ending amounts of unrecognized tax benefits is as follows (in thousands):
Since the Company is in a loss carryforward position, the Company is generally subject to examination by the U.S. federal, state and local income tax authorities for all tax years in which a loss carryforward is available.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
Any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objective and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on this evaluation, our Chief Executive Officer and Chief Financial Officer concluded that, as of December 31, 2020,2023, the design and operation of our disclosure controls and procedures were effective at a reasonable assurance level.
There was no change in our internal control over financial reporting identified in connection with the evaluation required by Rule 13a-15(d) and 15d-15(d) of the Exchange Act that occurred during the quarter ended December 31, 20202023 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Our management is responsible for establishing and maintaining adequate internal control over financial reporting (as defined in Rule 13a-15(f) under the Exchange Act). Under the supervision of and with the participation of our principal executive officer and principal financial officer, our management assessed the effectiveness of our internal control over financial reporting as of December 31, 20202023 based on the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission in “Internal Control—Integrated Framework” (2013). Based on this assessment, management concluded that our internal control over financial reporting was effective as of December 31, 2020.2023.
The effectiveness of our internal control over financial reporting as of December 31, 20202023 has been audited by Ernst & Young LLP, an independent registered public accounting firm, as stated in their report which is included herein.
To the Stockholders and the Board of Directors of Denali Therapeutics Inc.
We have audited Denali Therapeutics Inc.’s internal control over financial reporting as of December 31, 2020,2023, based on criteria established in Internal Control - IntegratedControl-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the COSO criteria). In our opinion, Denali Therapeutics Inc. (the Company) maintained, in all material respects, effective internal control over financial reporting as of December 31, 2020,2023, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the consolidated balance sheets of the Company as of December 31, 20202023 and 2019,2022, the related consolidated statements of operations and comprehensive income (loss),loss, stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2020,2023, and the related notes and our report dated February 26, 202127, 2024 expressed an unqualified opinion thereon.
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Annual Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects.
Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Information required by this item will be contained in our definitive proxy statement to be filed with the SEC on Schedule 14A in connection with our 20212024 Annual Meeting of Stockholders (the “Proxy Statement”), which is expected to be filed not later than 120 days after December 31, 2020,2023, and is incorporated herein by reference.
Information required by this item will be contained in the Proxy Statement and is incorporated herein by reference.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
Information required by this item will be contained in the Proxy Statement and is incorporated herein by reference.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS AND DIRECTOR INDEPENDENCE
Information required by this item will be contained in the Proxy Statement and is incorporated herein by reference.
Information required by this item will be contained in the Proxy Statement and is incorporated herein by reference.
1. Financial Statements
See Index to Financial Statements in Part II Item 8 of this Annual Report on Form 10-K.
2. Financial Statement Schedules
All schedules are omitted because they are not applicable or the required information is shown in the financial statements or notes thereto.
3. Exhibits
The documents listed in the Exhibit Index are incorporated by reference or are filed with this report, in each case as indicated therein (numbered in accordance with Item 601 of Regulation S-K).
None.
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this Annual Report on Form 10-K to be signed on its behalf by the undersigned, thereunto duly authorized.
Each person whose signature appears below constitutes and appoints Ryan J. Watts, Ph.D. and Steve E. Krognes,Alexander O. Schuth, M.D., and each of them acting individually, as his or her true and lawful attorney-in-fact and agent, with full power of substitution and resubstitution, for him or her and in his or her name, place and stead, in any and all capacities, to sign any and all amendments to this Annual Report on Form 10-K, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he or she might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents, or any of them, or their or his substitutes, may lawfully do or cause to be done by virtue thereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated: