| | | | | | | | |
UNITED STATES SECURITIES AND EXCHANGE COMMISSION |
| WASHINGTON, D.C. 20549 |
| | |
| FORM 10-K | |
(Mark one)
☒ ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended December 31, 20202023
ORor
☐ TRANSITION REPORT PURSUANT TO SECTION 13 OR 15 (d)15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the transition period from to
Commission File Number: 001-39452
| | | | | | | | |
| | |
| INHIBRX, INC. |
|
| (Exact name of registrant as specified in its charter) |
| | |
| Delaware | | 82-4257312 |
(State or other jurisdiction of incorporation or organization) | | (I.R.S. Employer Identification Number)No.) |
| | |
| 11025 N. Torrey Pines Road, Suite 200 | | |
| La Jolla, California | | 92037 |
| (Address of principal executive offices) | | (Zip Code) |
| | |
| (858) 795-4220 | |
| (Registrant’s telephone number, including area code) |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | |
| Securities registered pursuant to Section 12(b) of the Act |
| | | | | | |
| Title of each class | | Trading symbol(s) | | Name of each exchange on which registered |
| Common Stock, par value $0.0001 | | INBX | | The Nasdaq Global Market |
| | | | | | |
| Securities registered pursuant to Section 12(g) of the Act |
| | | None | | | |
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐☒ No ☒☐
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant:registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports);, and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| | | | | | | | | | | | | | |
| Large Accelerated Filer | ☐☒ | | Accelerated filer | ☐ |
| Non-accelerated filer | ☒☐ | | Smaller reporting company | ☒ |
| | | Emerging growth company | ☒☐ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 7(a)(2)(B)13(a) of the SecuritiesExchange Act. ☒☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. Yes ☐☒ No ☒☐
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to § 240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
As of August 31, 2020,June 30, 2023, the aggregate market value of the registrant’s common stock held by non-affiliates of the registrant was approximately $261.4$892.9 million, based on the closing price of the registrant’s common stock on the Nasdaq Global Market of $17.49$25.96 per share. The registrant has elected to use August 31, 2020 as the calculation date, as on June 30, 2020 (the last business day of the registrant’s most recently completed second fiscal quarter) the registrant was a privately-held concern.
As of February 28, 2021,21, 2024, the registrant had 37,747,66447,392,447 shares of common stock outstanding.
Documents Incorporated By Reference
The following documents (or parts thereof) are incorporated by reference into the following parts of this Form 10-K: Certain information required in Part III of this Annual Report on Form 10-K is incorporated from the Registrant’s Proxy Statement for the 2021 Annual Meeting of Stockholders to be filed with the Securities and Exchange Commission.None.
INHIBRX, INC.
ANNUAL REPORT ON FORM 10-K
For the Year Ended December 31, 20202023
| | | | | | | | |
| TABLE OF CONTENTS | |
| | PAGEPage |
| |
| |
| | |
| | |
| Item 1B. | Unresolved Staff Comments | |
| Item 1C. | | |
| | |
| | |
| | |
| | |
| |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| Item 9C. | Disclosure Regarding Foreign Jurisdictions that Prevent Inspections | |
| | |
| |
| | |
| | |
| | |
| | |
| | |
| | |
| |
| | |
| | |
| | |
| |
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K, or this Annual Report of Inhibrx, Inc., or Inhibrx, or the Company (also referred to as “we,” “us,” and “our”) contains forward-looking statements that involve risks and uncertainties. Except as otherwise indicated by the context, references in this Annual Report to “we,” “us” and “our” are to the consolidated business of the Company. All statements other than statements of historical facts contained in this Annual Report are forward-looking statements. In some cases, you can identify forward-looking statements by words such as “anticipate,” “believe,” “contemplate,” “continue,” “could,” “estimate,” “expect,” “intend,” “may,” “plan,” “possible,” “potential,” “predict,” “project,” “design,” “seek,” “should,” “target,” “will,” “would,”“would” or the negative of these words or other comparable terminology. These forward-looking statements include, but are not limited to, statements about:
•our ability to complete the Merger (as defined below) and related spin-out transaction (as described below);
•the initiation, timing, progress and results of our research and development programs as well as our preclinical studies and clinical trials;
•our ability to advance therapeutic candidates into, and successfully complete, clinical trials;
•our interpretation of initial, interim or preliminary data from our clinical trials, including interpretations regarding disease control and disease response;
•the timing or likelihood of regulatory filings and approvals;
•the commercialization of our therapeutic candidates, if approved;
•the pricing, coverage and reimbursement of our therapeutic candidates, if approved;
•our ability to utilize our technology platform to generate and advance additional therapeutic candidates;
•the implementation of our business model and strategic plans for our business and therapeutic candidates;
•our ability to successfully manufacture our therapeutic candidates for clinical trials and commercial use, if approved;
•our ability to contract with third-party suppliers and manufacturers and their ability to perform adequately;
•the scope of protection we are able to establish and maintain for intellectual property rights covering our therapeutic candidates;
•our ability to enter into strategic partnerships and the potential benefits of such partnerships;
•our estimates regarding expenses, capital requirements and needs for additional financing;
•our ability to raise funds needed to satisfy our capital requirements, which may depend on financial, performance;
•our expectations regarding the impact of the COVID-19 pandemic on our business;economic and market conditions and other factors, over which we may have no or limited control;
•our and our third party partnersthird-party partners’ and service providers’ ability to continue operations and advance our therapeutic candidates through clinical trials, andas well as the ability of our third party manufacturers to provide the required raw materials, antibodies and other biologics for our preclinical research and clinical trials, in light of the COVID-19 pandemic and the recent political developments in Hong Kong;current market conditions or any pandemics, regional conflicts, sanctions, labor conditions, geopolitical events, natural disasters or extreme weather events;
•our ability to retain the continued service of our key professionals and to identify, hire and retain additional qualified professionals; and
•developments relating to our competitors and our industry.
These forward-looking statements are subject to a number of risks, uncertainties and assumptions, including those described in the section titled “Risk Factors” elsewhere in this Annual Report. Moreover, we operate in a very competitive and rapidly changing environment, and new risks emerge from time to time. It is not possible for our management to predict all risks, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements we may make. In light of these risks, uncertainties and assumptions, the forward-looking events and circumstances discussed in this Annual Report may not occur and actual results could differ materially and adversely from those anticipated or implied in the forward-looking statements. In addition, statements that “we believe” and similar statements reflect our beliefs and opinions on the relevant subject. These statements are based upon information available to us as of the date of this Annual Report, and while we believe such information forms a reasonable basis for such statements, such information may be limited or incomplete, and our statements should not be read to indicate that we have conducted an exhaustive inquiry into, or review of, all potentially available relevant
information. These statements are inherently uncertain and investors are cautioned not to unduly rely upon these statements.
You should not rely upon forward-looking statements as predictions of future events. Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee that the future results, levels of activity, performance or events and circumstances reflected in the forward-looking statements will be achieved or occur. We undertake no obligation to update publicly any forward-looking statements for any reason after the date of this Annual Report to conform these statements to new information, actual results or to changes in our expectations, except as required by law.
You should read this Annual Report and the documents that we file with the Securities and Exchange Commission, or the SEC, with the understanding that our actual future results, levels of activity, performance, and events and circumstances may be materially different from what we expect.
This annual reportAnnual Report includes trademarks, tradenames and service marks that are the property of other organizations. Solely for convenience, trademarks and tradenames referred to in this annual reportAnnual Report appear without the ® and ™ symbols, but those references are not intended to indicate, in any way, that we will not assert, to the fullest extent under applicable law, our rights, or that the applicable owner will not assert its rights, to these trademarks and tradenames.
Part I.
Item 1. Business.
Overview
We are a clinical-stage biotechnologybiopharmaceutical company with a pipeline of novel biologic therapeutic candidates, developed using our proprietary modular protein engineering expertiseplatforms. We leverage our innovative protein engineering technologies and proprietary single domain antibody, or sdAb, platform. Our sdAb platform allows usdeep understanding of target biology to pursuecreate therapeutic candidates with attributes and mechanisms superior to current approaches and applicable to a range of challenging, validated targets with clinical promise, but where other antibodyhigh potential.
Recent Developments
Sale of INBRX-101 to Sanofi
In January 2024, we announced that we entered into a definitive agreement, or the Merger Agreement, with Aventis Inc., or Aventis, a wholly owned indirect subsidiary of Sanofi, whereby Sanofi will indirectly acquire, through Aventis, all the assets and biologic based approaches have failed. Highly modular, our sdAbs can be combined with precise valencies and multiple specificities, creating therapeutic candidates designedliabilities primarily related to be capable of enhanced cell signaling, conditional activationINBRX-101, or combined synergistic functions.
Wethe Merger, an optimized, recombinant alpha-1 antitrypsin, or AAT, augmentation therapy currently have four programs in ongoing clinical trials, with the most advanced expected to initiate a registration-enablingregistrational trial in mid-2021. Three of these programs are for the treatment of various cancers, and one for the treatment of Alpha-1 Antitrypsin Deficiency,patients with alpha-1 antitrypsin deficiency, or AATD. Our most advanced therapeutic candidate, INBRX-109, is a tetravalent death receptor 5, or DR5, agonist currently being evaluated in patients diagnosed with difficult-to-treat cancers, such as chondrosarcoma, synovial sarcoma, mesothelioma and pancreatic adenocarcinoma. We plan to submit an amended Investigational New Drug Application, or IND,Immediately prior to the U.S. Foodclosing of the Merger, all assets and Drug Administration,liabilities not primarily related to INBRX-101 will be spun out into a new publicly traded company, Inhibrx Biosciences, Inc., or New Inhibrx.
Under the FDA, for INBRX-109 during March 2021. We expect to initiateterms of the definitive agreements, Aventis will acquire all of our outstanding shares through a registration-enabling trial evaluating INBRX-109 in patients diagnosed with conventional chondrosarcoma around mid-2021. INBRX-106 is a hexavalent OX40 agonist, currently being investigated as a single agentmerger, and in combinationturn, each of our shareholders will receive: (i) $30.00 per share in cash, (ii) one contingent value right per share, representing the right to receive a contingent payment of $5.00 in cash upon the achievement of a regulatory milestone and (iii) one SEC-registered, publicly listed, share of New Inhibrx for every four shares of Inhibrx common stock held. In addition, in connection with Keytruda,the transaction, Aventis will (1) assume and retire our outstanding third-party debt with Oxford Finance, LLC, or Oxford, (2) cause New Inhibrx to be funded with $200 million in patients with locally advanced or metastatic solid tumors. Both INBRX-109cash, and INBRX-106 programs are designed(3) retain an equity interest in New Inhibrx of approximately 8%. Subject to achieve target agonism through precise controlthe satisfaction of therapeutic valency. INBRX‑105 is a conditional 4-1BB agonist,customary closing conditions, including the receipt of regulatory approvals, we currently being investigated in patients with locally advanced or metastatic solid tumors, as a single agent and initiating in combination with Keytrudaexpect the transaction to close during the second quarter of 2021. 2024.
Discontinuation of INBRX-105
We have decided to terminate our INBRX-105 program, a tetravalent programmed death-ligand 1, or PD-L1, targeted 4-1BB agonist. During the length of our Phase 1/2 trial, we dosed approximately 150 patients. We initially observed single agent complete and partial responses in non-small cell lung cancer, or NSCLC, and head and neck squamous cell carcinoma, or HNSCC. We also observed partial responses with INBRX-105 in combination with Keytruda® (pembrolizumab). However, after evaluation of the totality of the data from the expansion cohorts, the initial signal was not sufficiently validated to support the continuation of this program. We are in the process of winding down the clinical trial and expect it to be complete within the first half of 2024. Consequently and in conjunction with this decision, Elpiscience Biopharmaceuticals, Inc, or Elpiscience, terminated its rights to commercialize INBRX-105 in greater China.
Current Clinical Pipeline
Our fourth program, current clinical pipeline includes therapeutic candidates in the following categories:
•INBRX-101, is an optimized, recombinant alpha 1which seeks to maintain the natural function of Alpha-1 antitrypsin, or AAT, augmentation therapyin a recombinant format, optimized for AATD. We anticipate additional data releases from all fourless frequent dosing and greater potential therapeutic activity as compared to plasma-derived AAT, or pdAAT; and
•INBRX-109 and INBRX-106, both of which utilize our clinical programsmultivalent formats where the precise valency can be optimized in 2021.
Our Pipeline
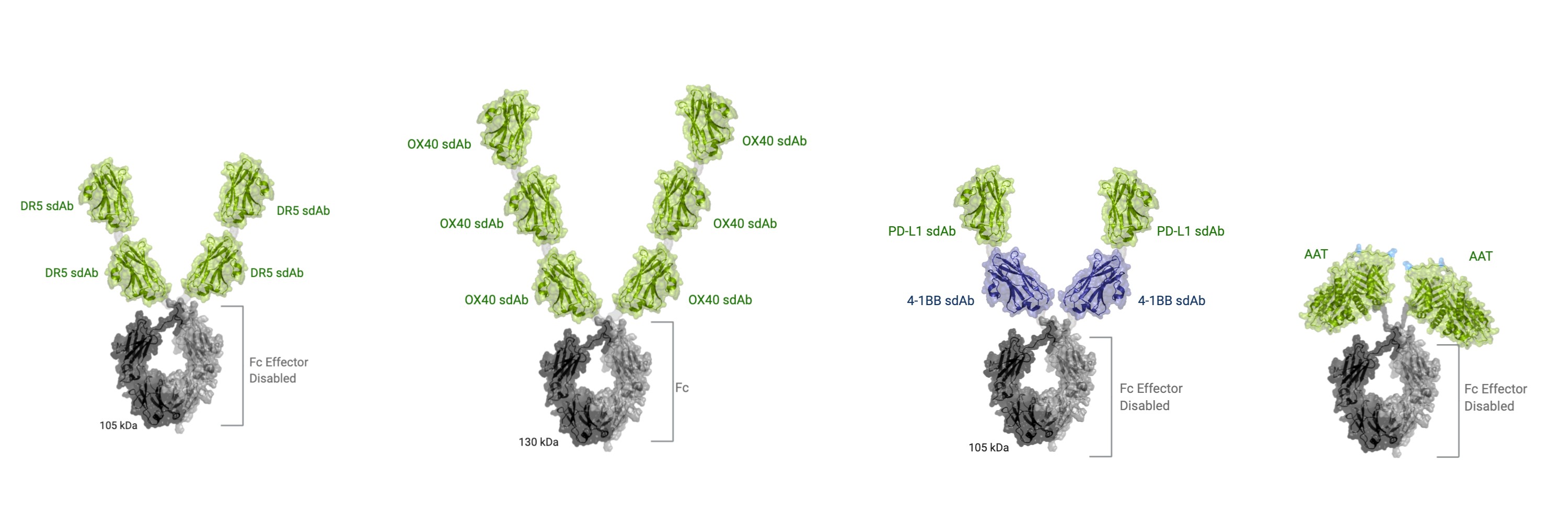
We have developed a diverse pipeline of therapeutic candidates that are specifically designedtarget-centric way to leveragemediate what we believe to be the power of our core sdAb platform and protein engineering expertise, as shown below:
| | | | | | | | | | | | | | | | | | | | |
|
INBRX-109most appropriate agonist function. | | INBRX-106 | | INBRX-105 | | INBRX-101 |
Tetravalent DR5 agonist | | Hexavalent OX40 agonist | | PD-L1x4-1BB tetravalent conditional agonist | | AAT-Fc fusion protein |
| | | | | | | | | | | | | | | | |
| | | | | | |
| INBRX-101 | | INBRX-109 | | INBRX-106 | | |
AAT-Fc fusion
protein | | Tetravalent DR5
agonist | | Hexavalent OX40
agonist | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Program | Therapeutic Area | Target(s)/Format | STAGE OF DEVELOPMENT | Anticipated Next Milestones |
| Preclinical | Phase 1 | Phase 2 | Phase 3 |
| INBRX-101* | Orphan/Respiratory | Neutrophil Elastase Inhibitor
AAT-Fusion Protein | |
| | | |
|
| INBRX-109** | Oncology | DR5
Tetravalent Agonist | | • Initiation of Trials-
◦Registration-enabling Phase 2 trial in conventional chondrosarcoma –Mid-2021^
◦Ewing combination cohort – Mid-2021
• Initial Data-
◦Mesothelioma and pancreatic adenocarcinoma chemotherapy combination cohorts – Q4 2021
◦Synovial sarcoma cohort (single agent) – 2H 2021
◦Ewing combination cohort –1H 2022
|
| |
|
| INBRX-106*** | Oncology | OX40
Hexavalent Agonist | | • Initial Data-
◦ Escalation with Keytruda – Q3 2021
◦ Expansion with Keytruda cohorts – Mid-2022
|
| | | | | |
|
| INBRX-105** | Oncology | PD-L1 x 4-1BB
Tetravalent Conditional Agonist | • Initial Data-
◦ Escalation with Keytruda cohorts – Q4 2021
◦ Expansion with Keytruda cohorts – Q3 2022
|
| |
|
| INBRX-101*** | Orphan/Respiratory | Neutrophil Elastase
AAT-Fusion Protein | • Initial multiple ascending dose data – Q4 2021 |
| |
|
__________________
* Subject to potential acquisition by Aventis as described above.
** Third party partnership with Chinese biotechnology company, Transcenta Holding, Ltd. (formerly Hangzhou Just Biotherapeutics Co., Ltd.), or Transcenta, currently in place for development and commercialization in China, Hong Kong, Macau and/or Taiwan.
*** Third party partnership with Chinese biotechnology company, Elpiscience Biopharmaceuticals, Inc., or Elpiscience, currently in place for development and commercialization in China, Hong Kong, Macau and/or Taiwan.
*** Commercialization and development rights outside of the United States and Canada, subject to an option agreement with Chiesi Farmaceutici S.p.A., or Chiesi.
^ The FDA granted fast track designation to INBRX-109 for the treatment of patients with unresectable or metastatic conventional chondrosarcoma in January 2021. Advancing INBRX-109 into registration-enabling randomized Phase 2 trials is contingent upon the FDA’s approval of the Company’s IND amendment, which is planned to be submitted in March 2021.
INBRX-109
INBRX-109 is a precisely engineered tetravalent sdAb-based therapeutic candidate that agonizes DR5 to induce tumor selective programmed cell death. The IND for INBRX-109 became effective in August 2018, and we initiated a three-part, Phase 1 clinical trial in the United States in November 2018. Part 1, the traditional single agent 3+3 dose escalation portion of this trial, was completed in August 2019, with enrollment of 20 patients. INBRX-109 was well-tolerated, with no significant toxicities observed at doses up to and including the maximum administered dose of 30 mg/kg. No maximum tolerated dose, or MTD, was reached.
In September 2019, we commenced Part 2 of this trial with single agent dose expansion cohorts, enrollment of which is still ongoing. During the fourth quarter of 2020, we initiated Part 3, chemotherapy combination cohorts, in malignant pleural mesothelioma and pancreatic adenocarcinoma. Initial data from the combination dose escalation cohorts are expected in the fourth quarter of 2021. As of February 2, 2021, we have enrolled over 104 patients in Part 1 and in the following cancer type specific cohorts: colorectal, gastric and pancreatic adenocarcinomas, malignant pleural mesothelioma, chondrosarcoma and synovial sarcoma.
In January 2021, the FDA granted fast track designation to INBRX-109 for the treatment of patients with unresectable or metastatic conventional chondrosarcoma, which is a serious, life-threatening medical condition with no currently approved effective systemic therapy options. We plan to initiate a registration-enabling Phase 2 trial with progression free survival as the primary endpoint in mid-2021.
We plan to investigate the therapeutic utility of INBRX-109 through a broad clinical development plan across difficult-to-treat cancers. Future clinical development of INBRX-109 may include combinations with additional chemotherapies and potentially synergistic agents in multiple solid tumors and hematologic malignancies. We are currently conducting preclinical studies with INBRX-109 and various rational combination agents, including targeted therapeutics and apoptotic pathway modulators, aimed at guiding our future clinical plans.
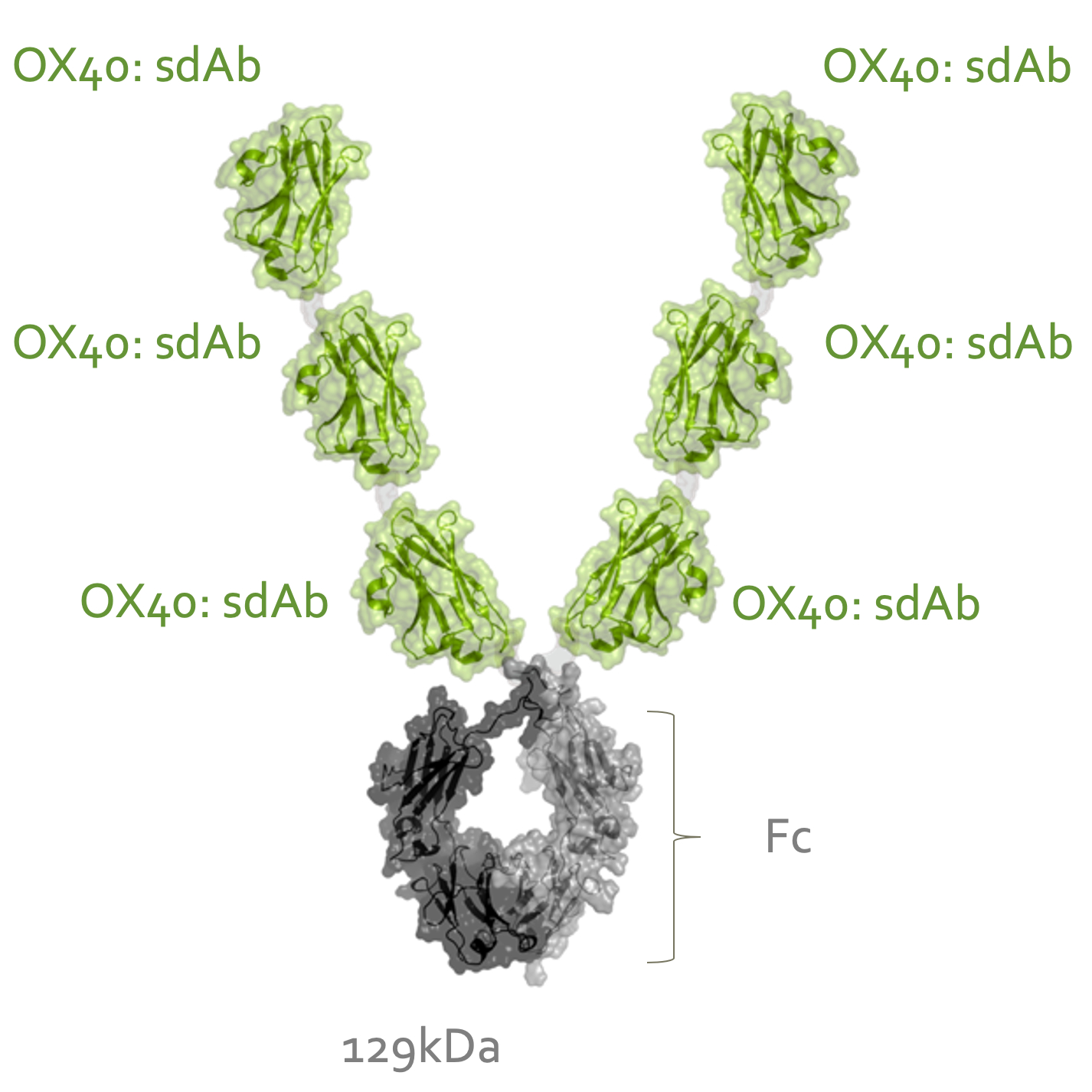
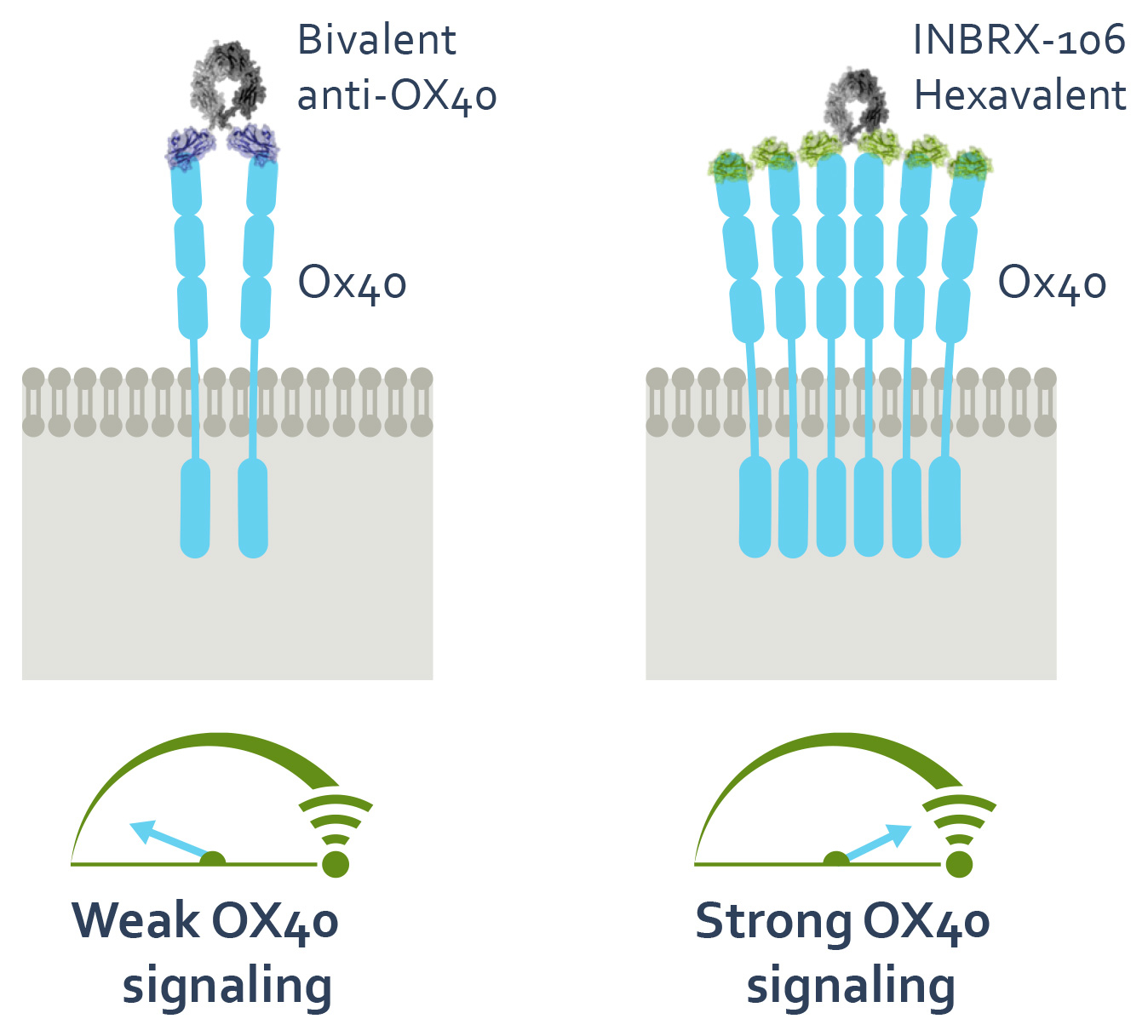
INBRX-106
INBRX-106 is a precisely engineered hexavalent sdAb-based therapeutic candidate targeting OX40 that has the potential to treat numerous cancer indications, both as a single agent and in combination. OX40, also known as TNFRSF4, is a member of the tumor necrosis factor receptor superfamily, or TNFRSF, and is predominately expressed on activated T-cells. Signaling through OX40 provides co-stimulation that promotes T-cell expansion, enhanced effector function and memory cell formation, and prevents activation induced cell death. Based on the capacity for OX40 signaling to enhance anti-tumor immunity in preclinical studies, there have been many efforts to therapeutically exploit this pathway for cancer immunotherapy.
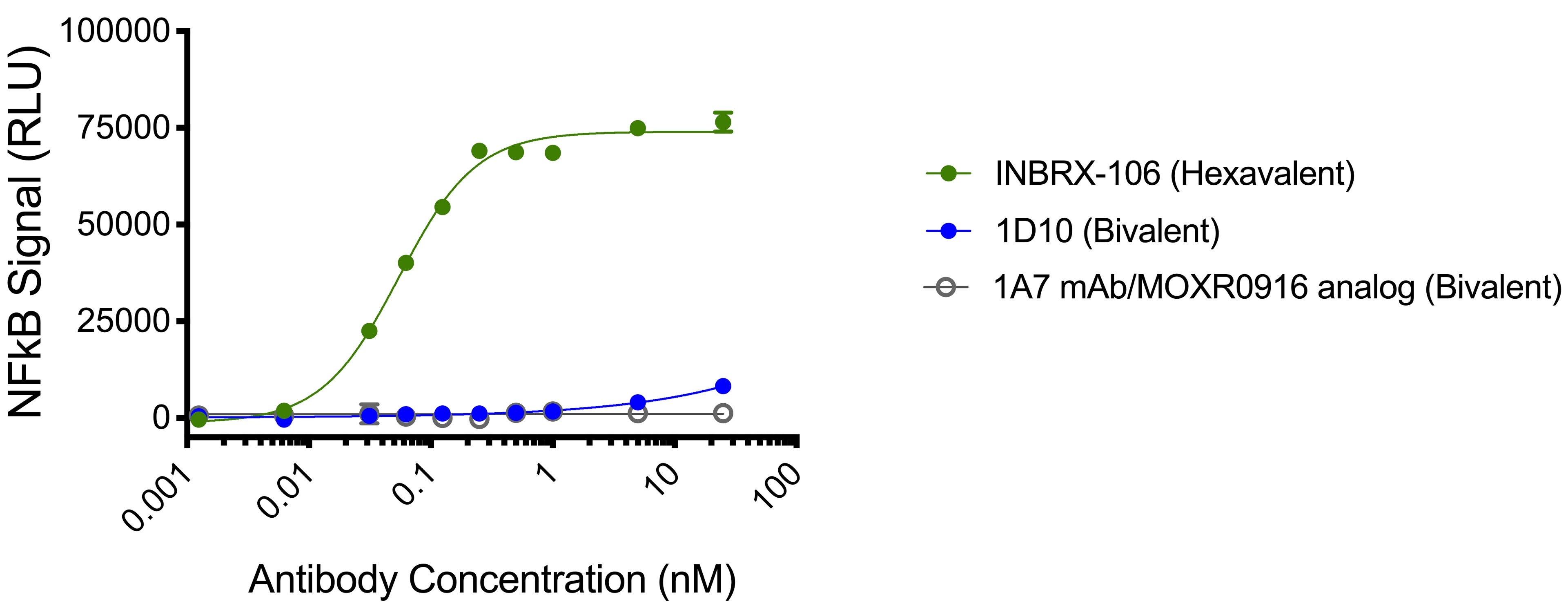
To date, OX40 agonism has mainly been clinically explored with bivalent antibodies. We believe the lack of success with many of these prior attempts resulted from insufficient valency to achieve receptor clustering. We believe INBRX-106, a hexavalent therapeutic candidate with the ability to bind six OX40 molecules per molecule of drug, has the potential to achieve improved receptor clustering and downstream signaling. In preclinical studies, we have observed that INBRX-106 mediated T-cell co-stimulation and also reduced the suppressive activity of regulatory T-cells, with superior activity to bivalent comparators.
The IND for INBRX-106 became effective in June 2019, and we initiated a four-part Phase 1 clinical trial in the United States in December 2019. Part 1 of this trial, single agent dose escalation, was completed in January 2021 with enrollment of 20 patients. INBRX-106 was well-tolerated, without reaching an MTD up to the maximum administered dose of 3 mg/kg.
Part 2 of this trial, single-agent dose expansion, and Part 3 of this trial, dose escalation in combination with Keytruda, were both initiated in February 2021. Initial data from the combination dose escalation cohort of this trial is expected to be announced during the third quarter of 2021. We expect to initiate Part 4 of this trial, the combination expansion cohorts, upon the completion of Part 3 of this trial and to announce initial data from those combination expansion cohorts during the middle of 2022.
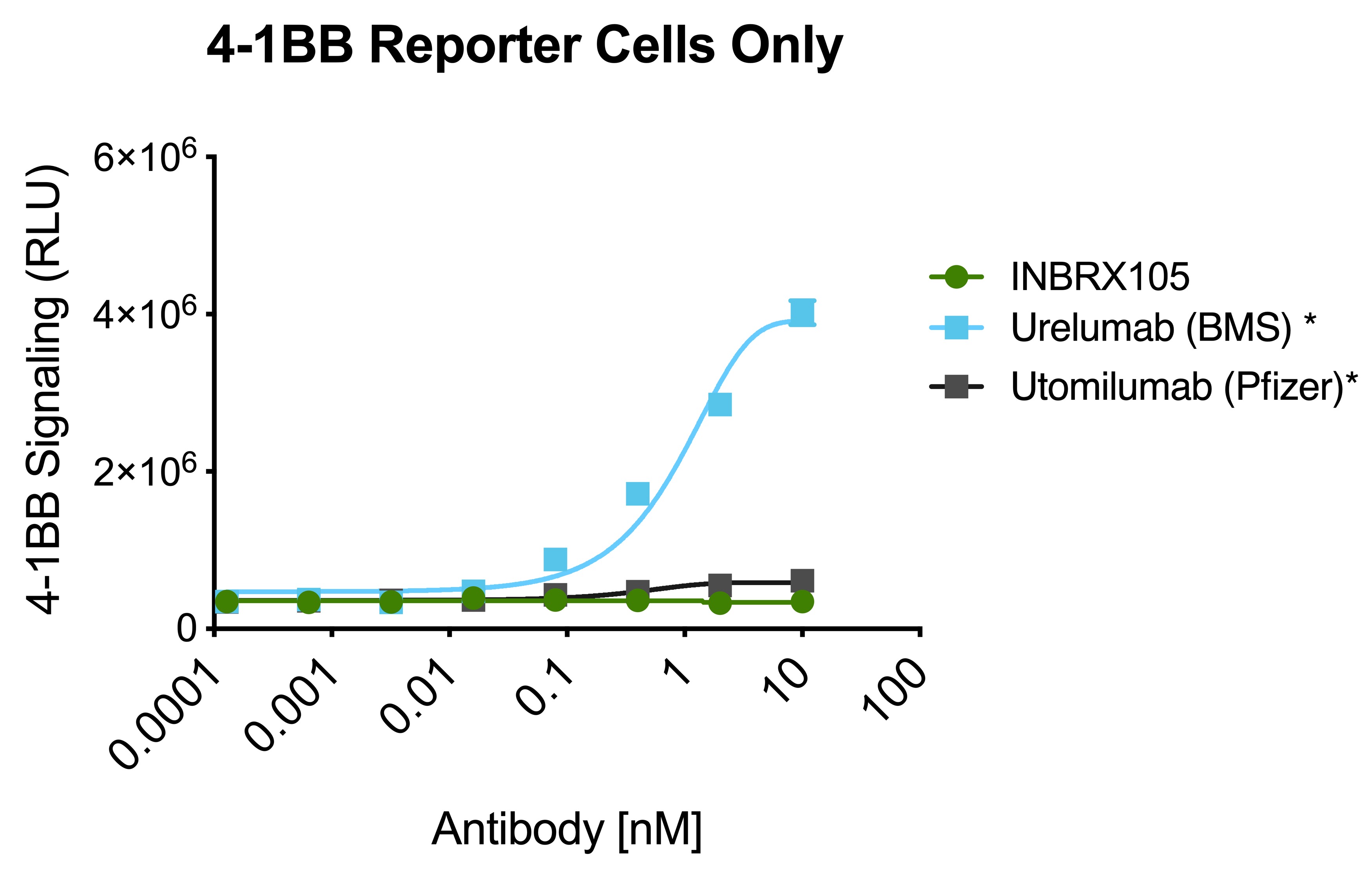
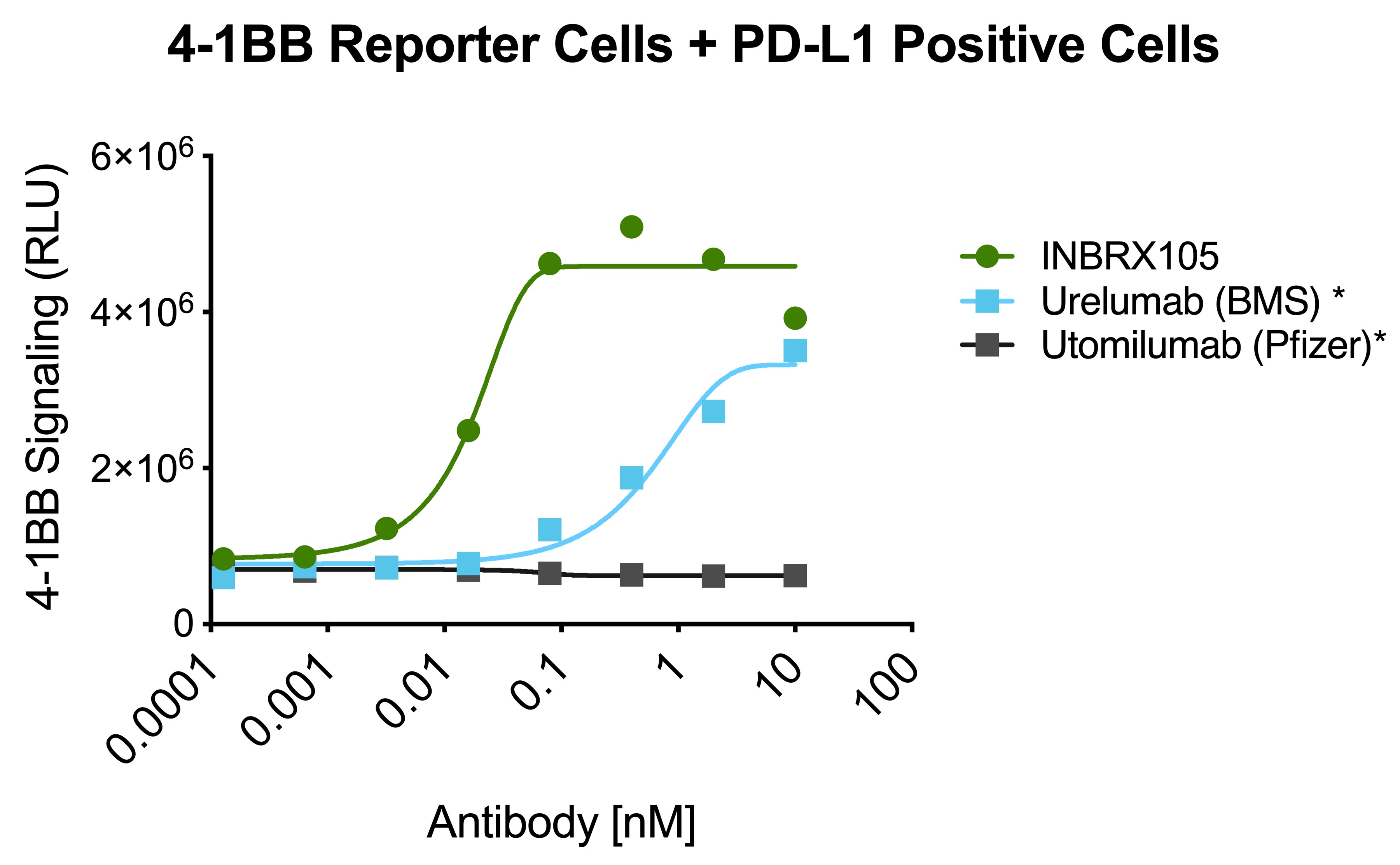
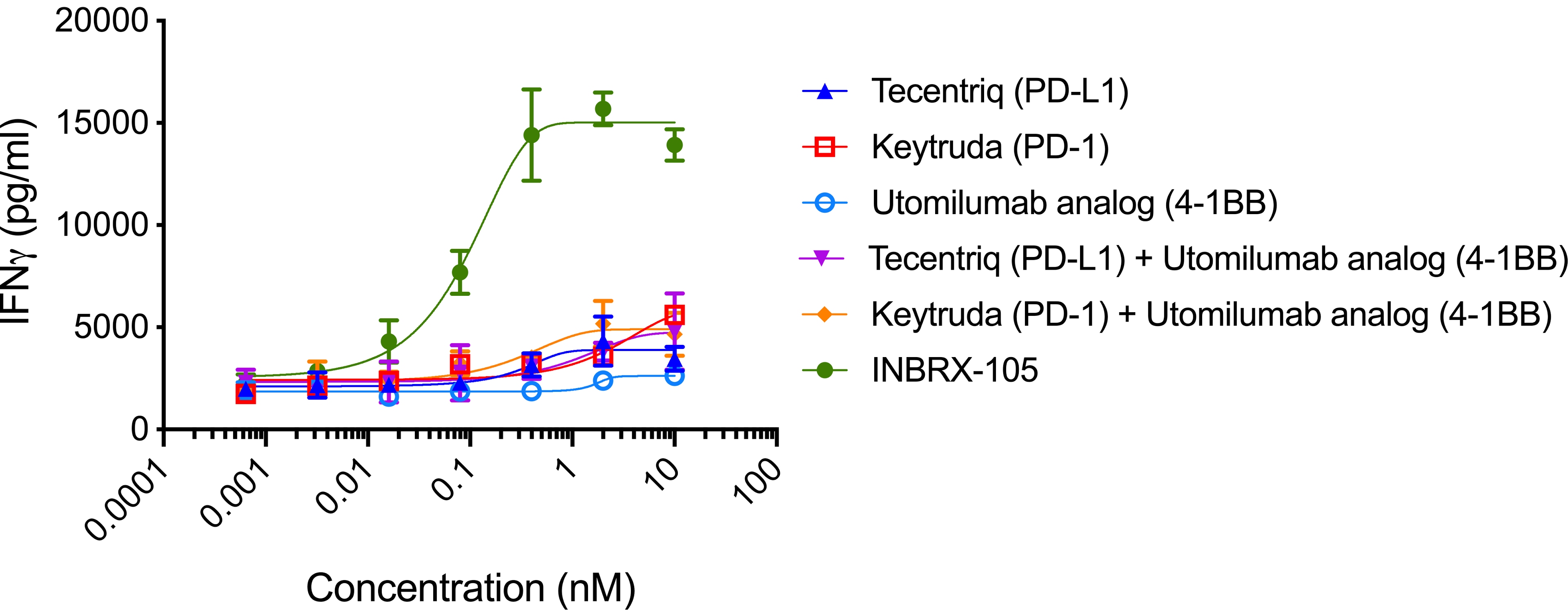
INBRX-105
INBRX-105 is a precisely engineered multi-specific sdAb-based therapeutic candidate that is designed to agonize 4-1BB selectively in the presence of PD-L1, which is typically found in the tumor microenvironment and associated lymphoid tissues. We believe that the unique format of INBRX-105, comprised of two PD-L1 targeting domains, each arranged adjacent to a 4-1BB targeting domain, will result in a selective PD-L1 targeted 4-1BB agonist. Based on the enrichment of PD-L1 expression in the tumor microenvironment, we believe INBRX-105 has the potential to selectively activate 4-1BB + T-cells in the tumor microenvironment, thus potentially overcoming the toxicity issues that have limited the success of prior 4-1BB agonists. PD-L1 is a validated biomarker for tumors with the potential for anti-tumor immune activation and thus we believe represents an ideal anchor antigen to drive 4-1BB agonism.
We initiated a four-part Phase 1 clinical trial in February 2019. Part 1, single-agent dose escalation, was completed with enrollment of 32 patients. Part 2, single-agent expansion, was initiated during the first quarter of 2021 and we expect to initiate Part 3 of this trial, escalation in combination with Keytruda, during the second quarter of 2021. We expect to announce initial data from Part 3 of this trial during the fourth quarter of 2021 and Part 4 of this trial, Keytruda combination expansion cohorts, during the third quarter of 2022.
INBRX-101
INBRX-101 is a precisely engineered recombinant human AAT-Fc fusion protein therapeutic candidate, in development for the treatment of AATD. The current standard of care for patients with AATD has been unchanged for decades and relies on weekly infusions of plasma derived AAT, or pdAAT, therapeutics. In spite of the frequent dosing, this therapy is incapable of maintaining serum AAT levels in the normal range for individuals affected by the disease.
Combining proprietary protein engineering with robust process development in INBRX-101, we are seeking to overcome the previous challenge in this field of maintaining the function of recombinant AAT, while manufacturing at commercial scale. We believe INBRX-101 has the potential to be dosed every three weeks, while maintaining patients in the normal range of AAT exposure. This would be a significant improvement for patients, currently receiving weekly infusions and sub-optimal augmentation.
The IND for INBRX-101 became effective in November 2018, and we initiated a Phase 1 dose escalation clinical trial in July 2019. As a result of the COVID-19 pandemic, we temporarily suspended enrollment for this trial but resumed recruitment during the fourth quarter of 2020. This temporary suspension was not a result of an observation of adverse events and was solely related to circumstances relating to the COVID-19 pandemic. Assuming enrollment continues as we currently anticipate, we expect to complete the multi-dose cohorts during the fourth quarter of 2021.
In May 2019, we entered into an option agreement, as amended in August 2019, or the Chiesi Option Agreement, with Chiesi, pursuant to which we granted Chiesi an exclusive option to obtain an exclusive license to develop and commercialize INBRX-101 outside of the United States and Canada following completion of the Phase 1 trial.
Our Leadership Team
We have assembled a team with deep scientific, manufacturing, and clinical experience in discovering and developing protein therapeutics.therapeutics, as well as an accomplished commercial team with the expertise to successfully bring our therapeutic candidates, if approved, to market. Our in-house capabilities span the disciplines of discovery, protein engineering, cell biology, translational research, chemistry, manufacturing and controls, or CMC, clinical development, and clinical development.commercialization. Members of our team bring experience from multiple organizations including Genentech, Inc., Gilead Sciences, Inc., Merck & Co., Novartis AG, Pfizer Inc., and Novartis AG.Roche. Our board of directors is comprised of individuals with proven business and scientific accomplishments and significant operating knowledge of our company.
Our Strategy
Our mission is to discover and develop effective biologic treatments for people with life-threatening conditions and to evolve Inhibrx into a commercial-stage biotechnologybiopharmaceutical company with a differentiated and sustainable product portfolio by focusing on the following:
Rapidly advance and optimize the clinical development of our lead programs.
We are focused on rapidly advancingEach of our four therapeutic candidates, each withclinical programs has key clinical data readoutsor milestone events expected in 2021 and 2022. To augment our U.S.-centric clinical strategy for our oncology therapeutic candidates,2024/2025. Since entering the clinic, we have formed collaborations in China designed to provide access to patient populations for clinical trials not readily available in the United States, including treatment-naïve patients,made great strides and to facilitate rapid patient enrollment with the goal of generating more robust early clinical data. We believe this harmonized clinical strategy may allow us to accelerate our development timelines. In addition, for each of our therapeutic candidates, should single agent efficacy be demonstrated, we plan to maximizehave the potential benefit to patients by broadly exploring combinationsreach the marketplace as soon as 2026 with standard of care agents and other rational combination agents.our first commercial therapeutic.
Apply our sdAb platform and other protein technologiesengineering platforms to create differentiated, next-generation therapeutics in focused disease areas with particular emphasis on oncology.
We have developed an sdAb platform and other protein technologies that we believe can be applied to meet the specific challenges of complex target biology. Our current pipeline is focused on oncology and orphan diseases. We plancontinue to focus our internal clinical development where we believe we can create effective and flexible solutions to address the challenges of both validated and novel targets and generate differentiated therapeutics that can be advancedin areas with a high unmet medical need. Our modular protein engineering platforms enable us to efficiently through clinical development.identify optional therapeutic formats customized to the target biology.
Maintain our culture of innovation, execution and efficiency.
Over the last 11 years weWe have successfully built an innovative culture that encourages scientific risk-taking within the bounds of our data-driven philosophy. This enables our research and development team to discover numerous promising preclinical candidates cost effectively, from which we select what we believe are highly differentiated programs for clinical development.
Maximize the potential of our therapeutic pipeline by retaining rights as well as selectively entering into additional strategic partnerships depending on the therapeutic candidate, indication and geography.pipeline.
We have a disciplined strategy to maximize the potential of our therapeutic pipeline by retaining developmentin order to bring the greatest value to our shareholders and commercialization rightsthe most significant impact to those therapeutic candidates, indicationspatients. We are continuously looking to streamline operations to increase efficiency and geographies thatto ensure maximum value is achieved with the capital we believeraise. Additionally, such as with Sanofi, we can develop and eventually commercialize successfully and efficiently on our own if they are approved, while also seeking towill enter into additional strategic partnerships and transactions in instances where we believe partnering will accelerate our development timelines and/or maximize the worldwide commercial potential of any approved therapeutic candidates. Forcandidate.
Our Pipeline
INBRX-101
INBRX-101 is a precisely engineered recombinant human AAT-Fc fusion protein therapeutic candidate that we are developing for the treatment of patients with AATD. We believe INBRX-101 for AATD has the potential to be dosed monthly, while maintaining patients in the normal range of AAT exposure, which would be a significant improvement for patients currently receiving weekly plasma-derived AAT infusions that do not maintain normal AAT exposure. Through our proprietary engineering capabilities, we were able to overcome the challenges of producing AAT recombinantly and manufacturing at commercial scale. Additionally, we believe there is an opportunity for INBRX-101 to be used as a treatment for Acute graft-versus-host disease. It has the potential for a more sustainable dosing schedule and could provide a superior safety benefit over existing approved therapies coupled with greater efficacy.
In January 2024, we announced that we entered into a definitive agreement with Aventis, a wholly owned indirect subsidiary of Sanofi, whereby Sanofi will, indirectly through Aventis, acquire the INBRX-101 program. Subject to the satisfaction of customary closing conditions, including the receipt of regulatory approvals, we expect the transaction to close during the second quarter of 2024.
Overview of AATD
AATD is an inherited disease that causes an increased risk of developing pulmonary disease defined by progressive loss of lung tissue and function and is associated with decreased life expectancy. The pulmonary manifestations of AATD include the entire spectrum of emphysema and disorders associated with chronic obstructive pulmonary disease. Patients with AATD harbor mutations in the AAT-encoding gene Serpin family A member 1, which causes AAT protein misfolding, loss of activity, and retention in the liver. AAT is a protease inhibitor that primarily targets human neutrophil elastase, or NE, an enzyme that is released by white blood cells in response to infections and has the capacity to degrade normal tissues, especially in the lung, if not tightly controlled by AAT.
Unmet Medical Need
This disease affects roughly 100,000 people in the United States, with a similar number of patients in Europe. Since AATD must be diagnosed using laboratory testing and cannot be diagnosed by symptoms or by a medical examination alone, it is believed that many individuals with AATD are likely undiagnosed or misdiagnosed. In April 2017, the United States Food and Drug Administration, or FDA, allowed for the marketing of direct-to-consumer tests that provide genetic risk information for certain conditions, including AATD. We believe such tests will lead to earlier diagnosis and an increase in the number of patients identified with AATD.
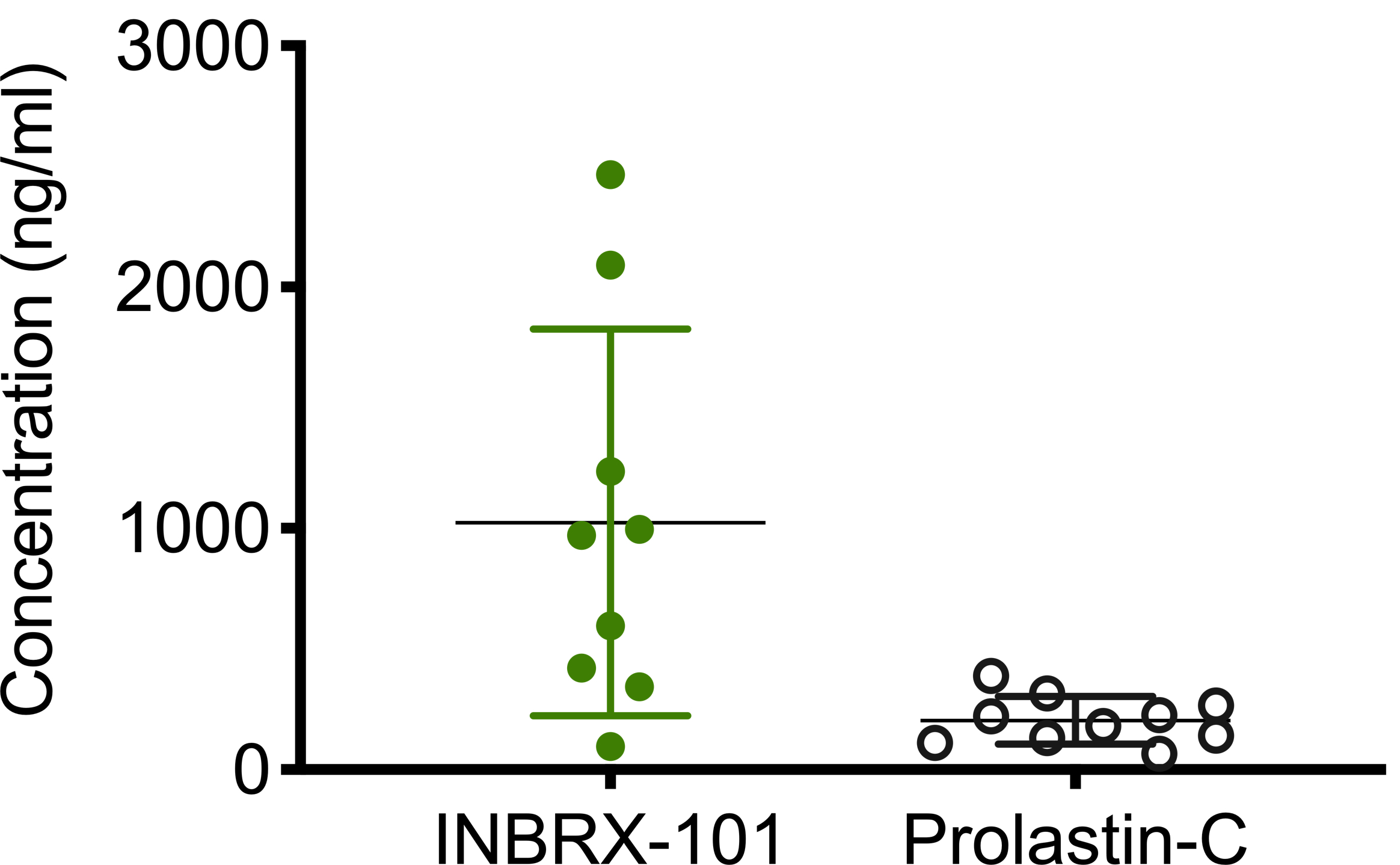
We performed a study where functional AAT levels were measured in plasma samples from 65 normal MM genotype individuals. Consistent with other published data, this analysis revealed the 5th and 95th percentiles of functional AAT levels in the normal MM genotype individuals were 21 and 54 µM, respectively, with a median of 36 µM.
Functional AAT levels in 65 healthy volunteers and 30 Phase 1 AAT study participants at baseline prior to dosing of INBRX-101
–Box plots show the minimum, lower quartile, median, upper quartile and maximum.
–The shaded region represents the 5th-95th percentiles of the normal range of functional AAT in healthy MM genotype adults.
–AAT variant determination was conducted by the Mayo Clinic Laboratories using an LC-MS/MS method (A1ALC).
–The Ph 1 baseline data represents the functional AAT levels measured in patients at the beginning of the study prior to dosing of INBRX-101.
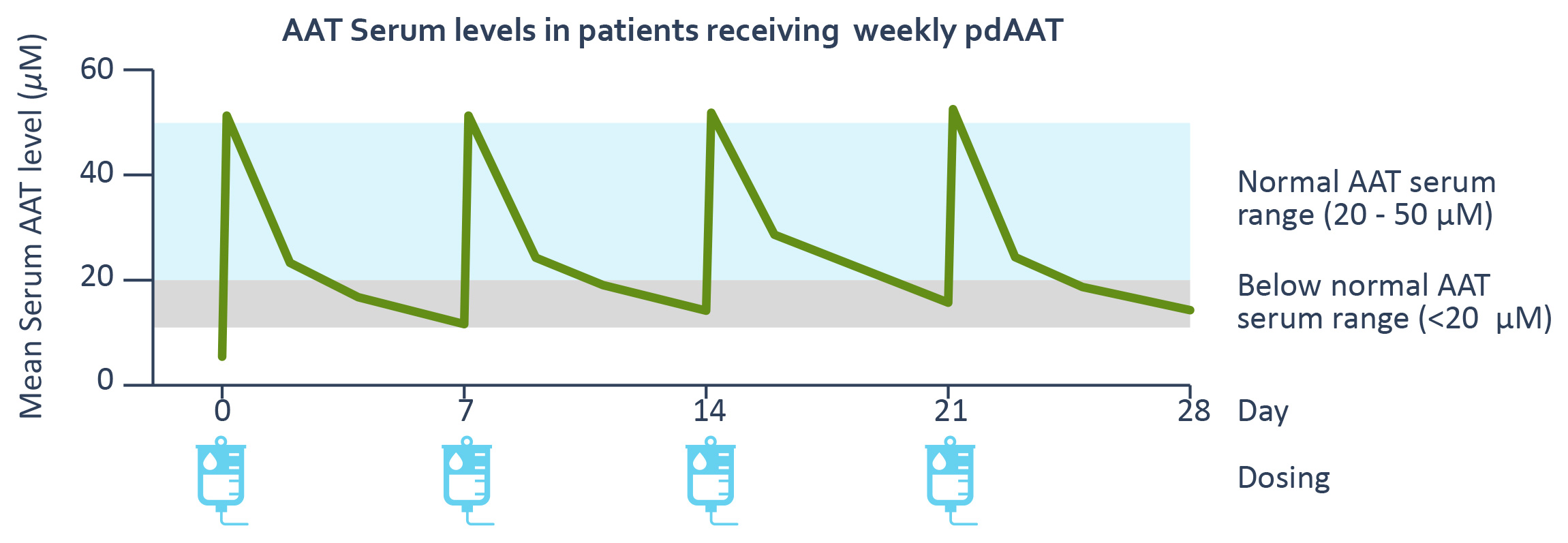
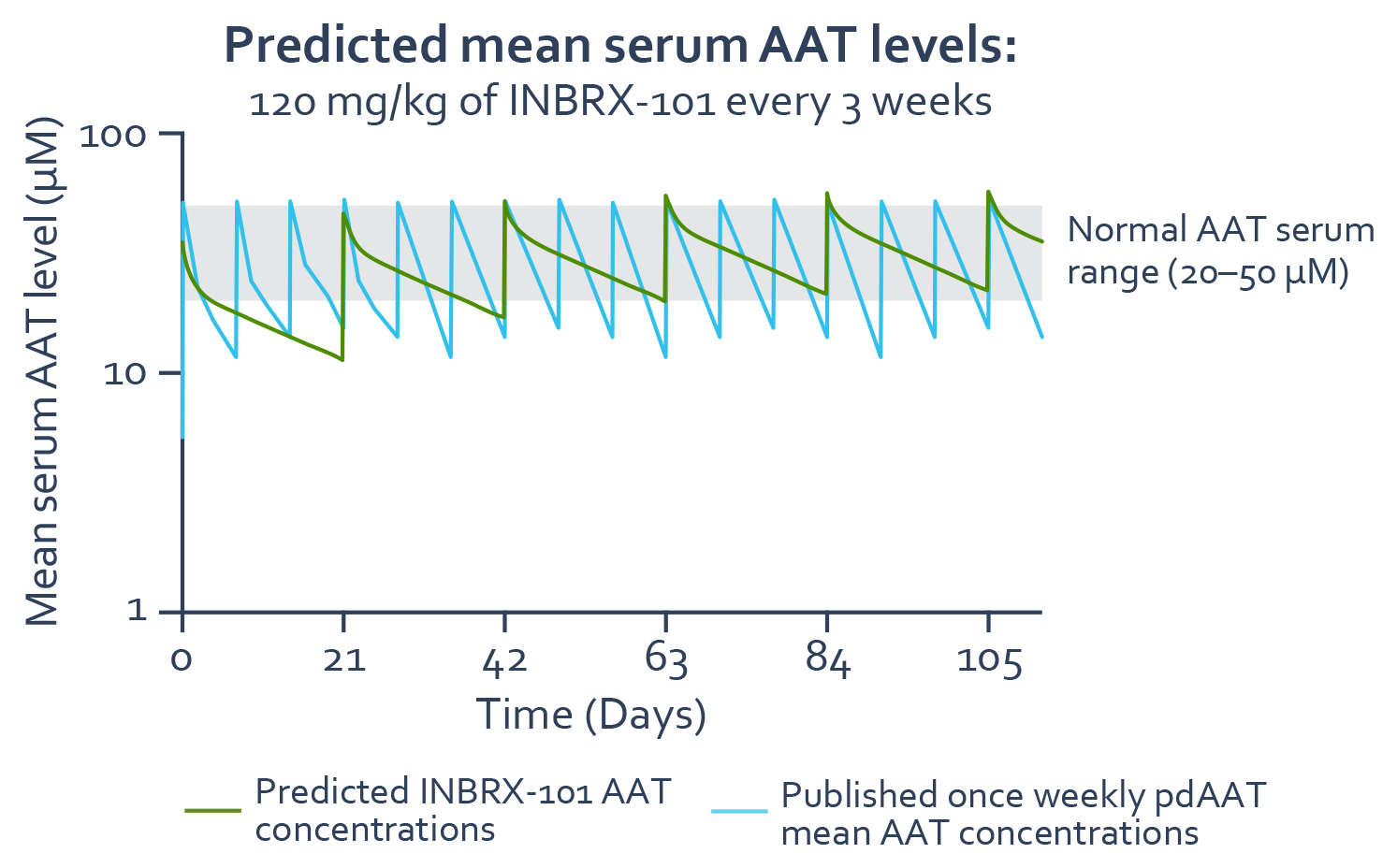
example, we have granted Chiesi an exclusive optionThe approved standard of care for AATD has been augmentation therapy using pdAAT and has not been substantially improved since 1987. Plasma-derived treatments are highly dependent on human donor blood supply, which is costly and can be limited. Due to obtain an exclusive licensethe short half-life of pdAAT therapeutics, patients require weekly infusions to developachieve and commercialize INBRX-101 outsidemaintain serum concentration above the protective threshold, presumed to be 11 µM. Even with frequent treatments, the AAT serum concentration following plasma-derived treatment remains considerably below the normal range of 21 to 54 µM as seen in the United States and Canada following completionadaptation graph below that depicts the pharmacokinetic profile of the Phase 1 trial. In addition, wepdAAT based on published data. There is clinical evidence suggesting that maintenance of higher AAT serum trough levels may better protect against lung function decline.

Additionally, there are currently party to license agreements for the development and, if approved, commercialization of INBRX-105, INBRX-106 and INBRX-109 solely in China, Hong Kong, Macau and Taiwan. We otherwise possess the worldwide rights to these candidates. Where appropriate, we plan to seek to enter into additional strategic partnershipsother significant barriers with the goalcurrent standard of maximizing the potentialcare for patients with AATD, including under-diagnosis, high cost of our therapeutic pipeline.
Selectively enter into additional strategic partnerships to maximize the potential of our protein engineering technologies in non-core areas or indications ancillary to oncology.
We believe that the broad potential of our protein engineering technologies, coupled with our strong intellectual property portfolio, can lead to the generation of multiple additional novel product candidates addressing a wide range of therapeutic areas. For example, we licensed our sdAb technologies for specific binders to bluebird bio, Inc., or bluebird, for use in cell therapy. Moreover, we have several preclinical programs focused on non-oncology indications that we are actively seeking to license. Where appropriate, we plan to continue to seek to opportunistically enter into additional strategic partnerships in areas that are considered non-core or indications ancillary to oncology to maximize the potential of our technology platform.
Our Approach
With the goal of addressing unmet medical needs, we leverage our deep understanding of target biology and innovative protein engineering technologies to create therapeutic candidates with attributes and mechanisms of action that are superior to current approaches and applicable to a range of challenging, validated targets withchronic augmentation therapy, high potential.
Our proprietary sdAb platform allows us to address complex target biology where other biologic approaches have failed or are sub-optimal. Our precision engineering enables the generation of therapeutic candidates with defined valencies and specificities, which we believe can result in optimal mechanisms of action. Initially, we have focused on applying these technologies to targets with clinically validated mechanistic rationales but where prior approaches have lacked sufficient therapeutic activity or safety.
Our clinical pipeline is enabled by our protein and process engineering capabilities, and includes therapeutic candidates in the following categories:
•INBRX-109 and INBRX-106 utilize our multivalent formats, for which the precise valency was optimized in a target-centric way to mediate what we believe to be the most appropriate agonist function;
•INBRX-105’s mechanism of action is enabled by a multi-specific format that we believe enables PD-L1 targeted 4-1BB agonism; and
•INBRX-101 seeks to maintain the natural function of AAT in a recombinant format, optimized for less frequent dosing and greater potential therapeutic activity as compared to pdAAT.
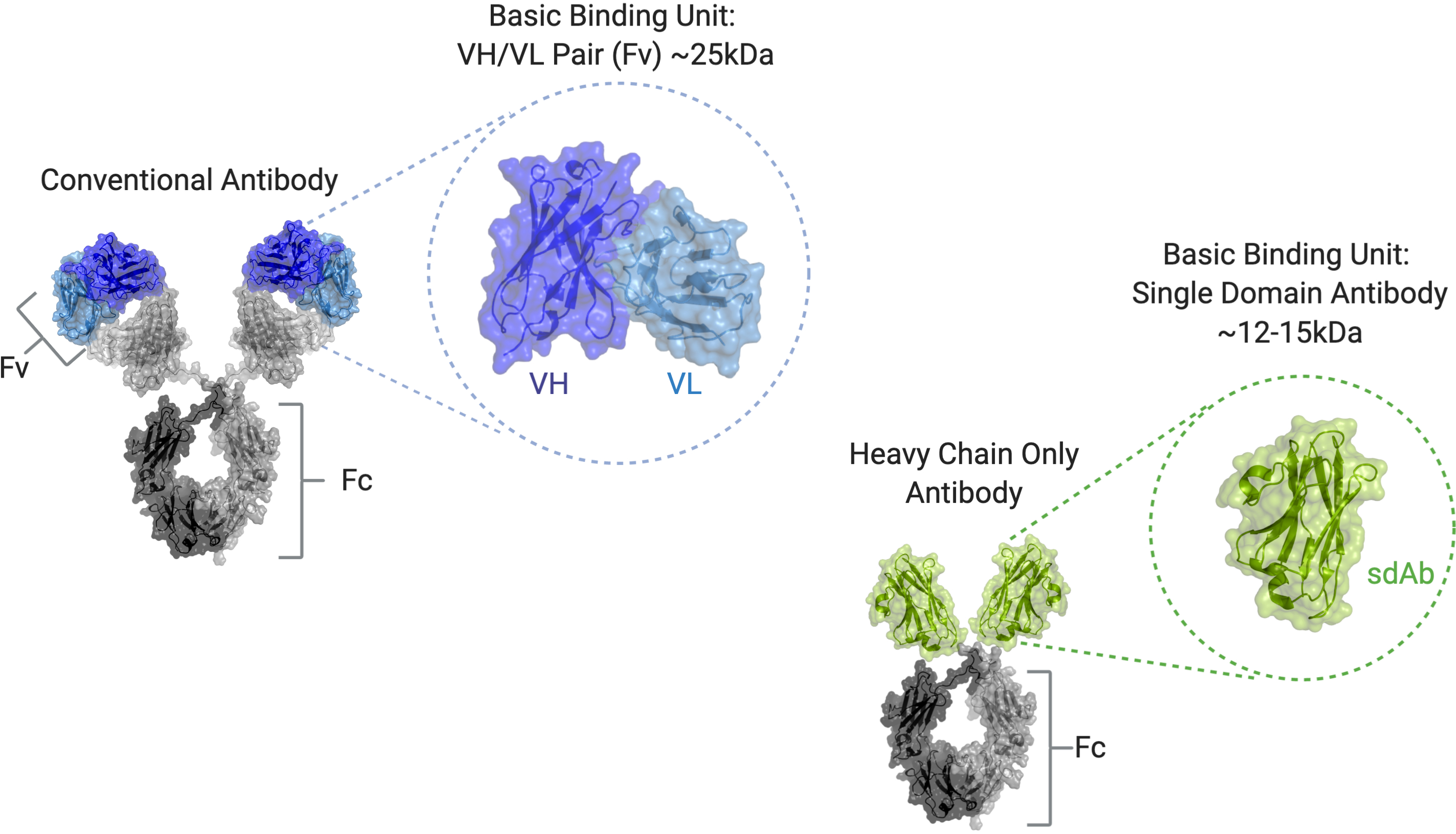
Conventional Antibodies and Their Limitations
Antibodies are multifunctional, Y-shaped proteins, made up of a pair of interacting heavy and light chains to form a symmetric, dimeric molecule with 4 total protein chains. Each arm of the antibody contains an antigen binding region (also referred to as the variable fragment, or Fv), which consists of both a heavy and light variable domain, or VH and VL, respectively, held together through interaction of constant domains. The Fc portion of the heavy-chain constant domain extends the half-life of the antibody in circulation and has the potential to modulate the function of immune cells through interaction with various Fc receptors found on the surface of cells of the immune system.
We believe that antibody Fv domains are not optimal building blocks for multispecific and multivalent therapeuticspatient burden due to frequent infusions, and the extensive protein engineering required to ensure the correct pairinginherent risks of the appropriate VH and VL domains to construct functional Fvs.
One common approach used to achieve correct pairing is the production of single chain variable fragments, or scFvs, which are generated by combining the VH and VL domain antigen binding domains using a flexible linker. Despite the addition of this linker, VH or VL sequences within an scFv have the propensity to errantly pairreceiving human blood products with a neighboring scFv, and can lead to disruption of proper Fv formation or aggregation. This issue canplasma collection practices that may not be exacerbated insustainable.
constructsOur Solution - INBRX-101
INBRX-101 is a recombinant AAT protein that contain multiple scFvs, with each entity requiring significant up-front optimization. These formatting challenges may restrict broad applicabilityis comprised of this technology.
To address these limitations, we have developed our sdAb platform to enable the streamlined production of protein formats with multiple antigen specificities. sdAbs are conceptually similartwo human AAT molecules covalently linked to the VH domainFc region of human immunoglobulin G4. AAT has proven difficult to develop recombinantly, often displaying loss of activity and experiencing accelerated degradation. Fusion of AAT to the Fc region allows for production using a conventionalstandard antibody but do not contain nor require a paired VL domain, as shownexpression system, which, when combined with our proprietary AAT-function preserving purification process, generates substantial and scalable yields of active recombinant AAT protein.
Clinical Data
In July 2019, we initiated an open-label, dose escalating Phase 1 trial which was conducted in approximately 10 clinical sites in the image below. DenotedUnited States, the United Kingdom and New Zealand and enrolled a total of 31 patients. The trial was designed as heavy-chain only antibodies, sdAbs are half the molecular weightsingle ascending dose, or SAD, administrations of an ScFv, but maintain specific target bindingINBRX-101, with an affinity range that is comparabledoses of 10 mg/kg, 40 mg/kg, 80 mg/kg or 120 mg/kg, followed by multiple ascending dose, or MAD, administrations of INBRX-101, with doses of 40 mg/kg, 80 mg/kg or 120 mg/kg. These doses were administered every three weeks intravenously to those achieved by conventional antibodies. sdAbs are derived from heavy-chain only antibodies that are naturally produced by animals in the camelid family, which includes camels, alpacas and llamas. Unlike the antibody systems of other mammals, a subsetpatients diagnosed with AATD who were either treatment naïve or previously treated with pdAAT therapeutics. The primary objective of the camelid antibody repertoire is composedtrial was safety and tolerability. Secondary objectives included AAT serum exposure, immunogenicity, as measured by frequency of heavy-chain onlyanti-drug antibodies, that can be miniaturized into sdAbs. Modularand NE activity. In addition, we measured pharmacokinetic and pharmacodynamic biomarkers in nature, they can be linked tobronchoalveolar lavage fluid, or BALF, for those patients who received MAD administrations of INBRX-101 at doses of 80 mg/kg or 120 mg/kg.
In March 2022, the FDA granted orphan drug designation for INBRX-101 for the treatment of AATD.
In May 2022, we announced topline results from the Phase 1 trial. Data from this Phase 1 trial included patients with AATD with the ZZ genotype, SZ genotype, and the MZ genotype of the SERPINA1 gene. There were no drug-related severe or serious adverse events, or AEs. Drug-related AEs were predominantly mild with a constant domain to capture half-life extendingfew moderate events, and immune cell modulating function. We believe that the small sizeall were transient and reversible. No signs of sdAbs, along with their stable nature and simple structure, make them ideal building blocks to construct novel biologics with multiple specificities and functions.
Comparison of Conventional Antibody and Single Domain Antibody Structure
Overview of Our sdAb Platform Technology
sdAbs provide a small, simple, modular target binding domain that can be combined in a variety of ways to meet the unique needs of each biological target. We have deep domain expertise surrounding discovery, humanization and optimization of sdAbs and their incorporation into unique configurations enabling the generation of therapeutic candidates with favorable drug-like properties and differentiated mechanisms of action. We have created various multivalent and multispecific therapeutic formats that are each designed to achieve unique functions specifically tailored to the requirements of a given biological target, including the ability to:
•effectively cluster receptors with precisely defined valency;
•simultaneously engage multiple antigens or epitopes;
•combine synergistic functions in a single molecule; and
•restrict therapeutic activity to specific micro environments in the body.neutralizing anti-drug antibodies were seen. Dose-related
Whileincreases in maximal and total INBRX-101 exposure occurred across the benefits of sdAb proteins are well documented, the presence of pre-existing anti-drug antibodies in human serum may limit the broad use of these agents as therapeutics. We have developed a series of patented modifications to our humanized sdAb scaffold that are designed to eliminate recognition by these pre-existing anti-drug antibodies, without compromising affinity, specificity or stabilityentirety of the sdAb. We believe this improvement, combined with our extensive knowledgesingle and multiple ascending dose ranges.
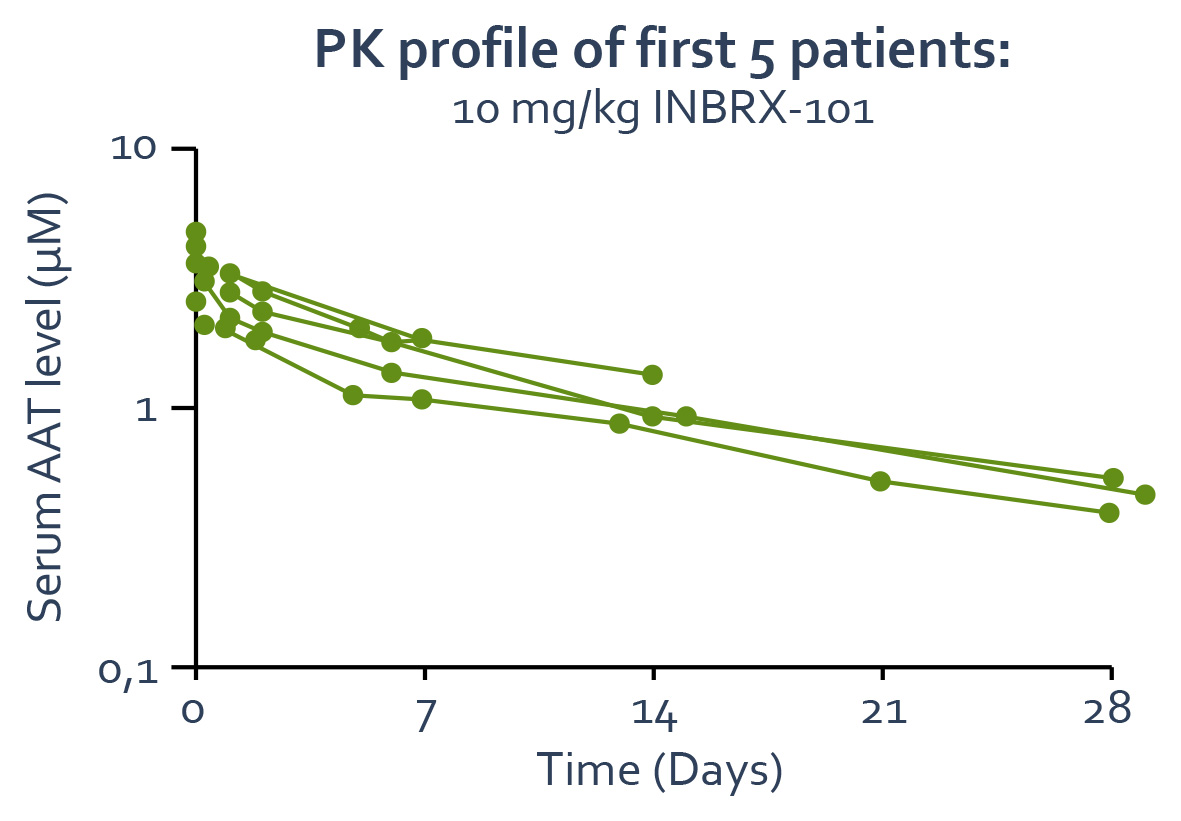
The topline data from the multiple ascending dose cohorts of INBRX-101 at 40, 80 and 120 mg/kg IV every three weeks showed the expected accumulation of functional AAT levels. Based on and assuming the accuracy of the structural nuancesPK modeling, accumulation is expected to continue following subsequent doses and to reach a steady state after a total of sdAbs, will enableapproximately five to six consecutive doses, administered every three weeks.
Functional AAT levels over time in AATD patients administered
40, 80 or 120 mg/kg INBRX-101 every three weeks
–Arrows indicate INBRX-101 IV dosing.
–The shaded region represents the streamlined developmentnormal range of therapeutic candidates with favorable biophysical properties,functional AAT in healthy adults.
–Functional AAT samples were not collected immediately following the second dose (Day 21).
–Results shown are from preliminary, unaudited data. Data shown from the 120 mg/kg multiple dose cohort are from the first four evaluable patients; the full dataset is still being collected and analyzed and will greatly extend the utility and applicability of our sdAb platform. Through our experience, we have found that our sdAb-based therapeutic candidates can be readily manufacturedpresented at high yields with established processes used to produce therapeutic proteins.
Overview of Our Multivalent Agonists
Cell surface receptors often require clustering to induce activation and effect downstream signaling. Receptors that require clustering are inherently difficult targets for conventional bivalent therapeutic antibodies, which are limited to interaction with no more than two receptors per molecule. In preclinical studies, we and third parties have observed that increased valency enhances receptor clustering and achieves the desired agonism of specific targets. Our sdAb platform enables us to build therapeutic candidates with a defined number of binding domains, customized to the valency requirements of a given target. We believe this precise protein engineering will increase the potential to achieve optimal clinical benefit by appropriately engaging these clustering-dependent receptors. Using our sdAb platform, we have developed higher order, multivalent antibodies that target TNFRSF agonists, as described below, that contain four (tetravalent) or six (hexavalent) antigen binding domains. Our multivalent therapeutic candidates are generally smaller than conventional antibodies and other multivalent formats that utilize fusions of natural ligands or immunoglobulin M-based, or IgM-based, constant regions, which we believe may confer advantages in the clinic.later date.
The current standard of care, plasma-derived AAT, dosed once weekly at 60 mg/kg, achieves an average concentration, or Cavg, of functional AAT of 17.8 µM over the weekly dosing interval as calculated from steady-state area under the curve, or AUC. INBRX-101 achieved a mean Cavg of functional AAT of 40.4 µM over the 21-day dosing interval following the third 80 mg/kg dose.
In October 2022, we reported BALF data where we observed the post-dose presence of INBRX-101 in every patient and in all three lobes of the lung collected from each of these patients. This data showed emerging evidence of a dose dependent increase of INBRX-101 lung exposure. These BALF samples were from eleven AATD patients in the 80mg/kg and 120mg/kg multiple ascending dose cohorts of the Phase 1 trial and were analyzed using a proprietary and validated mass spectrometry assay developed by Inhibrx to detect INBRX-101 specifically.
In April 2023, we initiated ElevAATe, a registration-enabling trial for INBRX-101 for the treatment of patients with emphysema due to AATD. The primary endpoint of the trial is the mean change in the average functional AAT, or fAAT, concentration as measured by anti-neutrophil elastase capacity from baseline to average serum trough fAAT concentration at steady state (Ctrough,ss). The initial read-out from the ElevAATe trial is expected to occur in mid-2025.
INBRX-109
INBRX-109 is a precisely engineered tetravalent sdAb-based therapeutic candidate targeting DR5.death-receptor 5, or DR5, a TNFRSF member, also known as tumor necrosis factor-related apoptosis-inducing ligand, or TRAIL, receptor 2. DR5 activation induces cancer-specific programmed cell death. The valency of INBRX-109 was selected to maximize the therapeutic index. In patients treated with INBRX-109 to date in our Phase 1 clinical trial, we have observed signs of single agent activity in serious,
life-threatening medical conditions that havewith limited treatment options. We observed signs of activity with INBRX-109 in combination with standard chemotherapies, the trial of which is still ongoing. In particular, there are no FDA-approved systemic therapiesJune 2021, based on the initial Phase 1 data results observed, we initiated a registration- enabling Phase 2 trial for the treatment of unresectable or metastatic conventional chondrosarcoma. In addition toData from the developmentregistration-enabling trial in unresectable or metastatic conventional chondrosarcoma is expected during the first half of INBRX-109 as a single agent, we have initiated cohorts with INBRX-109 in combination with chemotherapies in two solid tumor indications. We continue to conduct preclinical studies with INBRX-109 in combination with various rational combination agents, including targeted therapeutics and apoptotic pathway modulators, aimed at guiding these future clinical plans.2025.
Background on DR5
Apoptosis is a critical process for maintaining healthy tissue homeostasis, but this process is frequently altered in cancer patients leading to the accumulation of malignant cells. Apoptotic signaling pathways are tightly regulated by the balance of pro- and anti-apoptotic factors, and their therapeutic modulation has the potential to be exploited for the treatment of cancer. Targeting the anti-apoptotic proteins has been a clinically successful strategy. For example, Venetoclax, an inhibitor of B-cell lymphoma 2, or Bcl-2, was approved by the FDA for the treatment of chronic lymphocytic leukemia in 2016.
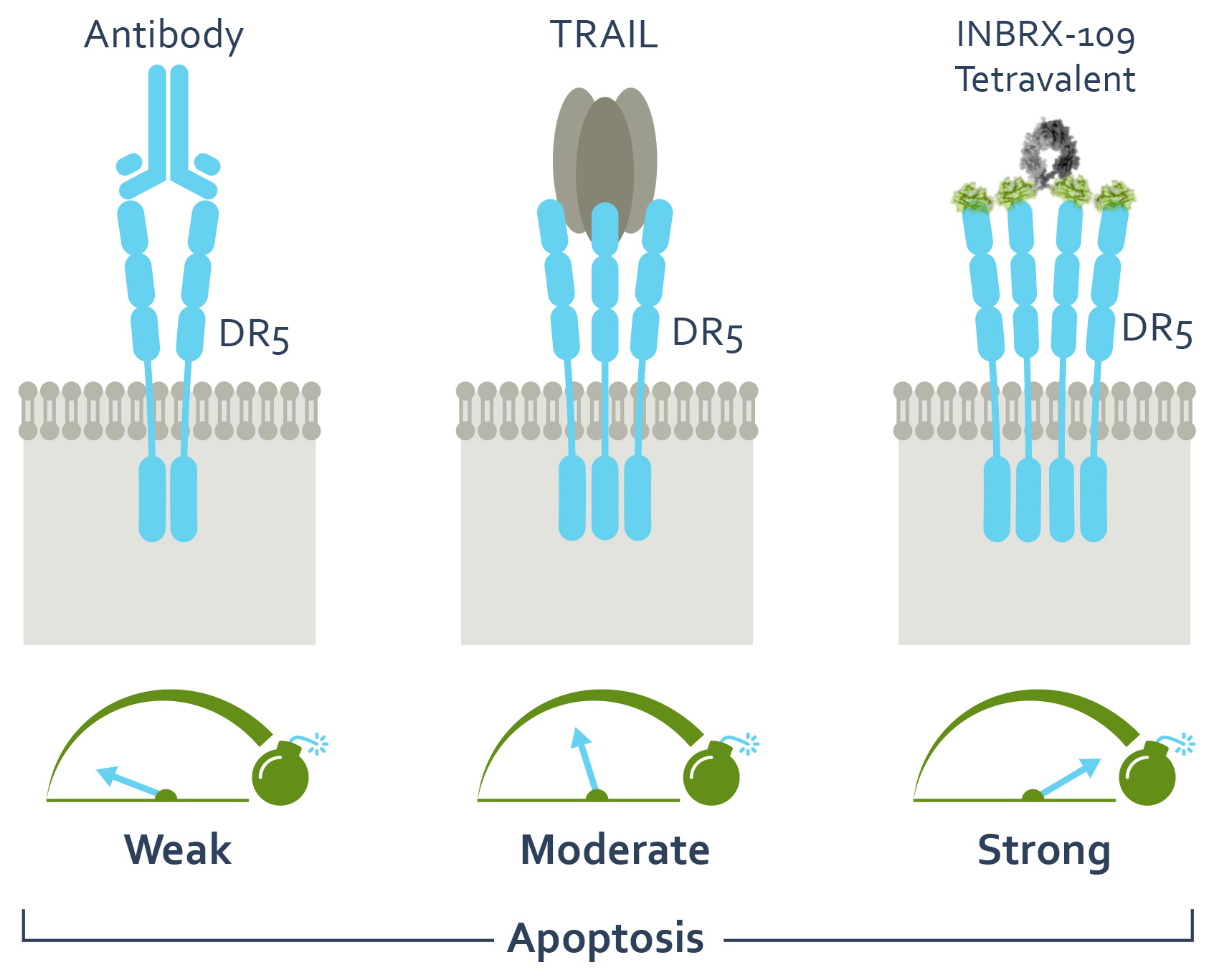
Alternatively, we believe therapeutically targeting pro-apoptotic proteins such as DR5 a TNFRSF member, also known as tumor necrosis factor-related apoptosis-inducing ligand, or TRAIL, receptor 2, is a promising oncology treatment strategy. DR5 signaling is induced by clustering of multiple receptors, which initiates an apoptotic signaling pathway resulting in cell death. The strength of apoptotic signaling is proportional to the degree of DR5 clustering. Importantly, although DR5 is expressed throughout the body, cancer cells have been shown to be more sensitive to DR5 signaling compared to healthy cells of normal tissues.
The promise of inducing cancer-specific cell death has led to extensive efforts by pharmaceutical and biotechnology companies to therapeutically exploit the DR5 pathway for the treatment of cancer. These initial efforts centered around developing recombinant versions of the DR5 ligand, TRAIL, and agonistic bivalent DR5 antibodies. Despite demonstrated clinical safety as single agents and in combination with chemotherapies, these first generation DR5
agonists failed to meet clinical efficacy endpoints. We believe these failures were caused by insufficient clustering of DR5, which is necessary for activation of this pathway.
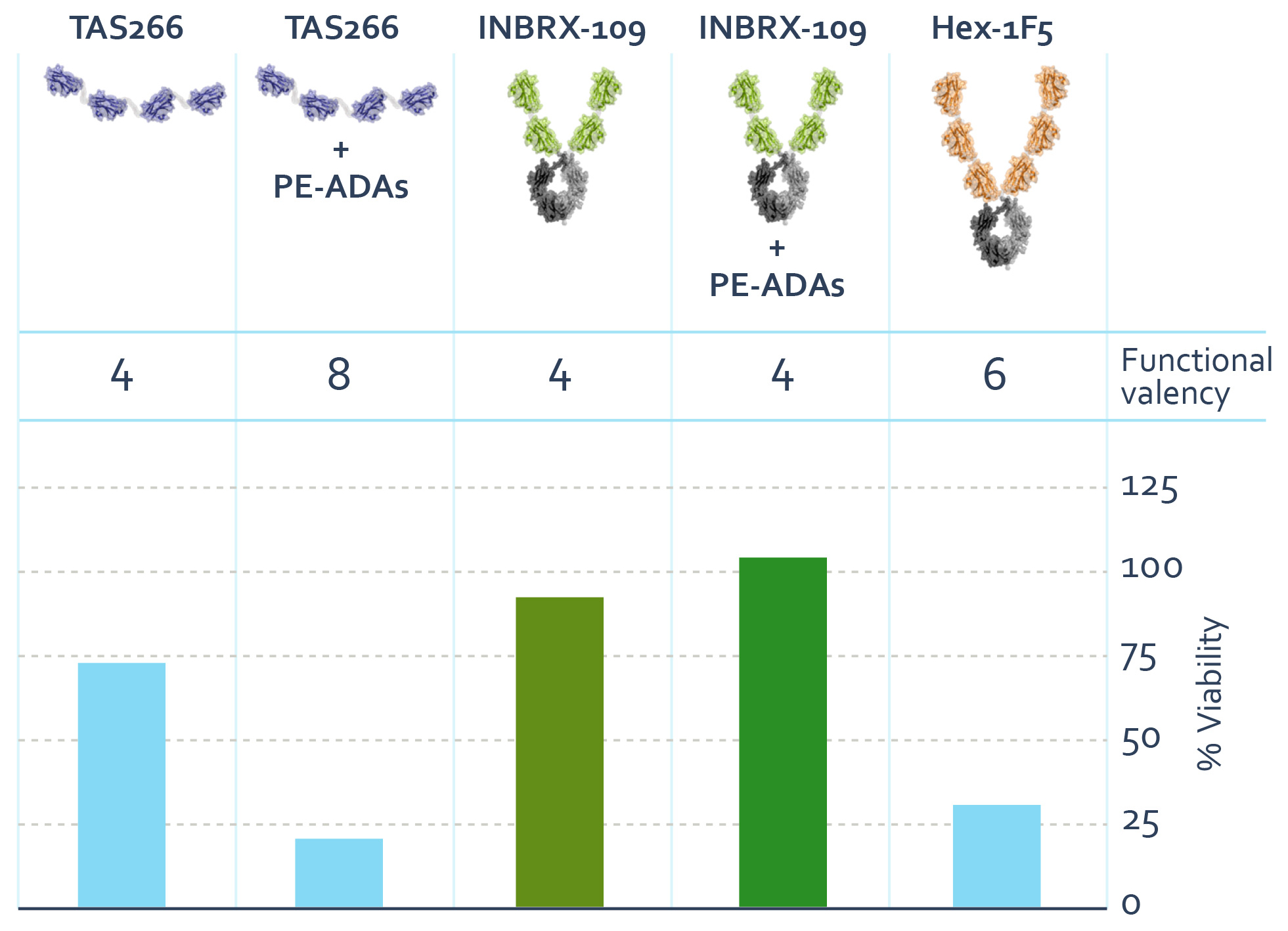
The prospect of improved efficacy through enhanced DR5 clustering led to the development by a third party of TAS266, a tetravalent DR5 agonist comprised of four DR5 binding sdAbs. This therapeutic candidate showed significantly more potency as compared to the previous generations of DR5 agonists. TAS266 was evaluated preclinicallyUnmet Medical Need
We are currently investigating INBRX-109 in over 600 human cancer cell lineschondrosarcoma, Ewing sarcoma and demonstrated the greatest activity for mesothelioma, gastric cancer, colorectal cancer, pancreatic cancer and non-small cell lung cancer. Despite these preclinical data, an initial clinical trial indicated that TAS266 caused dose-limiting liver toxicity, or hepatotoxicity. Subsequent analysis suggested pre-existing anti-sdAb antibodies, or PE-ADAs, to TAS266 led to the hyper-clustering of TAS266, adversely increasing the effective valencycertain other solid tumor types. These are some of the drug candidatemost aggressive diseases, some of which are also orphan oncology indications that have shown signs of activity in preclinical studies. These indications and causing apoptosismany of liver cells. These findings highlight the importance of tightly controlling valency when developingthese cancer subtypes do not respond well to currently approved therapies and represent a drug candidate against this high potential oncology target.significant unmet need.
Our Solution - INBRX-109
INBRX-109 is a tetravalent agonist of DR5 that we designed with our proprietary single domain antibody, or sdAb, platform to drive cancer-selective programmed cell death and to maximize potency while minimizing on-target liver toxicity arising from hepatocyte apoptosis. We believe INBRX-109 has the potential to overcome the limitations of previous DR5 agonists. As shown in the diagram below, INBRX-109 is comprised of four DR5 targeted sdAbs fused to an Fc region that has been modified to prevent Fc receptor interactions. In preclinical studies, we have observed that INBRX-109 has the ability to potently agonize DR5 through efficient receptor clustering, causing cancer cell death. Based upon the observation that TAS266 induced hepatotoxicity in a Phase 1 clinical trial,experience with earlier generation DR5 agonists, hepatocytes appear to be a non-cancerous cell type particularly sensitive to DR5 agonism. We have engineered INBRX-109 with our proprietary sdAb modifications to reduce recognition by pre-existing anti-drug antibodies in humans, which can lessen the potential for hyper-clustering.hyper-clustering and thereby reduce potential hepatotoxicity.
Primary objectives of the Phase 1 trial are safety, tolerability, and determination of the maximum tolerated dose, or MTD, and recommended Phase 2 dose. Secondary objectives are serum exposure and immunogenicity, as measured by frequency of anti-drug antibodies. Exploratory objectives include clinical anti-tumor efficacy, based on response rate, duration of response, disease control rate, progression-free survival and overall survival, as well as evaluation of potential predictive diagnostic and pharmacodynamic biomarkers.
INBRX-109: Tetravalent DR5 Agonistic Antibody
Clinical Data
We measuredinitiated a Phase 1 clinical trial in the ability of INBRX-109 to kill cancer cells at various antibody concentrations, and compared INBRX-109 to other DR5 binding test articles including a bivalent DR5 antibody and a trivalent TRAIL protein. The figure below shows the viabilityUnited States in November 2018. This Phase 1 clinical trial is designed as an open-label, three-part trial in patients with locally advanced or metastatic solid tumors.
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | |
| Phase 1 INBRX-109 Trial Design and Status | |
| First in human Phase 1 trial started November 2018 | |
| Part 1: Single-Agent Dose Escalation |
⇨
| Part 2: Single-Agent Dose Expansion |
⇨
| Part 3: Dose Expansion with Chemotherapy | |
| Completed | Completed | Ongoing | |
| Dose Range: 0.3 to 30 mg/kg
N= 20
Result: Maximum tolerated dose not reached | N=20 | Colorectal adenocarcinoma | N=10 | Mesothelioma with carboplatin or cisplatin | |
| |
| N=10 | Gastric adenocarcinoma | N=10 | Mesothelioma with carboplatin and pemetrexed or cisplatin and pemetrexed | |
| |
| N=20 | Malignant pleural mesothelioma | |
| N=20 | Pancreatic adenocarcinoma 2L with fluorouracil and irinotecan (mFOLFIRI) | |
| N=20 | Chondrosarcoma | |
| |
| N=10 | Synovial sarcoma | N=20 | Colorectal adenocarcinoma with FOLFIRI | |
| |
| N=12 | IDH1/2-mutant conventional chondrosarcoma | N=20-50 | Ewing sarcoma 2-4L with irinotecan and temozolomide | |
| |
| N=12 | Nonconventional chondrosarcoma | N=20 | SDH-def solid tumors or GIST with temozolomide | |
| |
| N=12 | Solid tumors, BMI>30 | | | |
| | | |
| | | | | | | | |
Part 1
Part 1 of the human chondrosarcoma cell line, H-EMC-SS, ontrial utilized a traditional 3+3 dose escalation design escalating INBRX-109 as a single agent from 0.3 mg/kg to 30 mg/kg. Twenty patients were enrolled in this portion of the vertical Y-axis, and
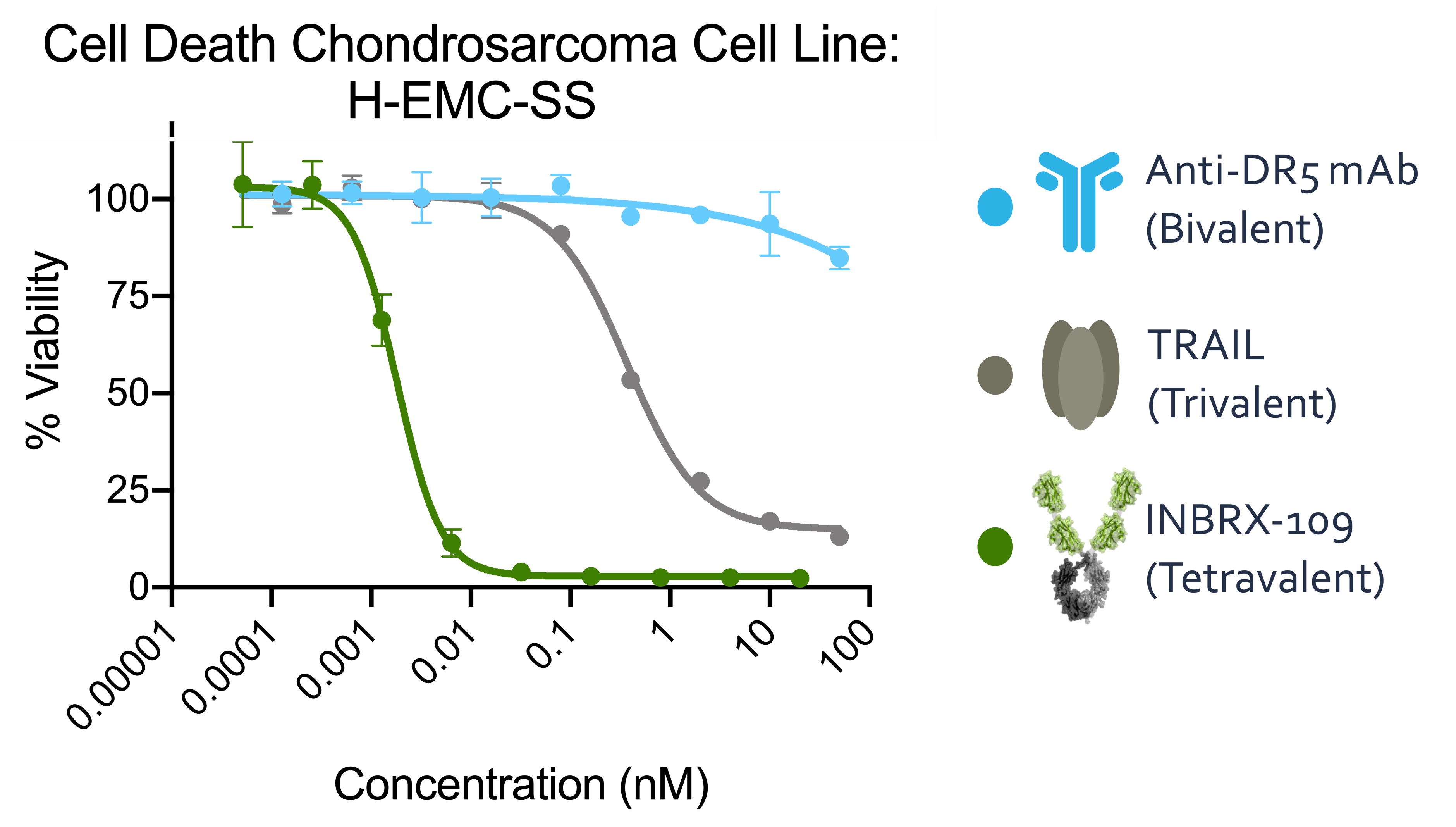
a range of test article concentration on the horizontal X-axistrial, which was completed in nanomolar, or nM, units. This assay was used to measure whether these test articles kill cancer cells by reducing the viability from approximately 100% down toward 0%, and also to measure the minimal concentration of test article needed to kill these cancer cells. We observed that only test articles that bound three or more DR5 molecules killed this cancer cell line, while the bivalent conventional antibody did not have any effect. Additionally, we found that the tetravalent DR5 agonist, INBRX‑109, killed H-EMC-SS cells at concentrations over 100 times lower than trivalent TRAIL. Our preclinical data and early clinical data indicate INBRX‑109 may be substantially more potent than recombinant TRAIL or conventional DR5 antibodies previously studied in clinical trials.
Impact of Valency on DR5 Induced Tumor Cell Death
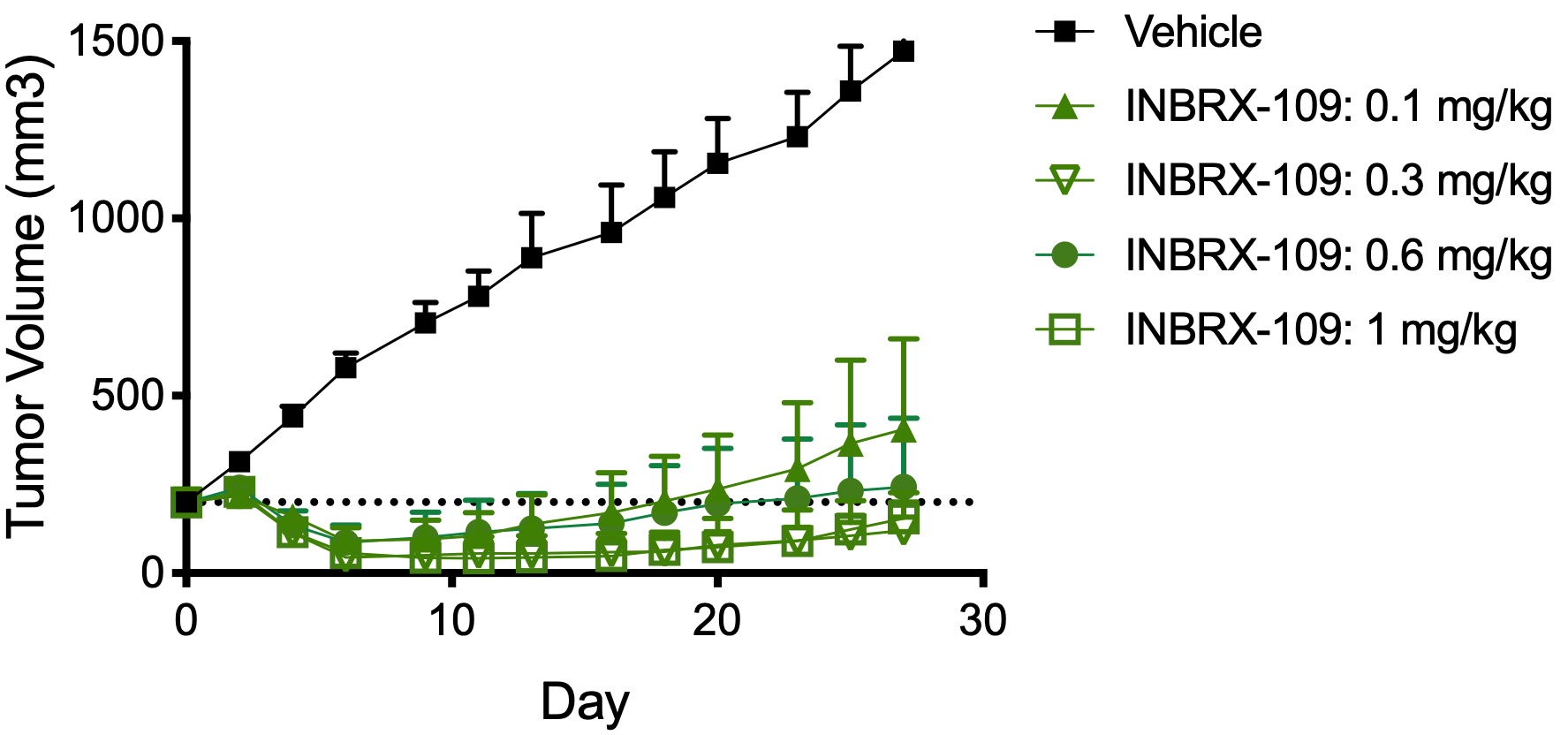
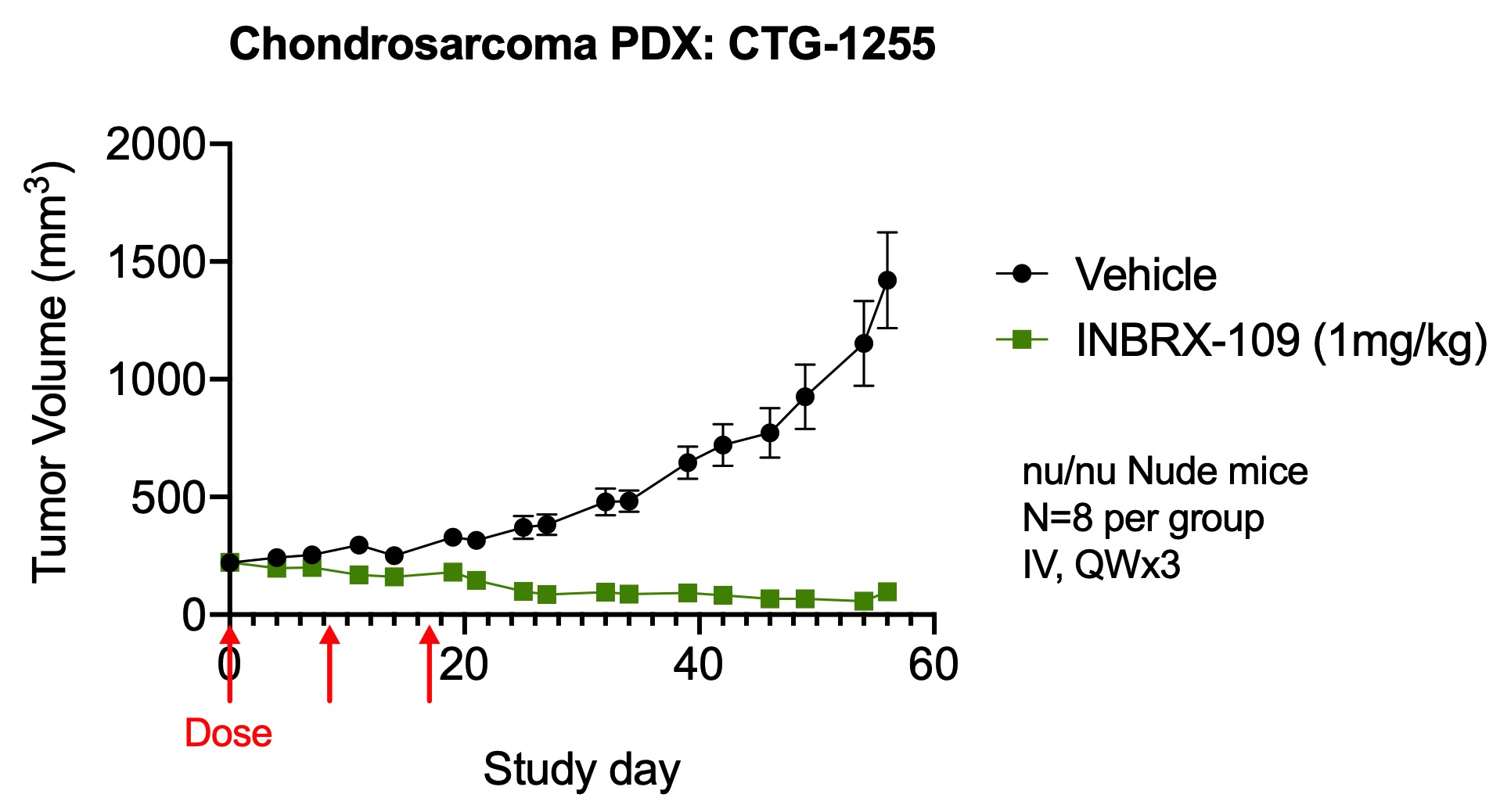
The anti-tumor activity of INBRX-109 was further assessed in multiple in vivo xenograft models using primary patient-derived, or PDX, tumors or cancer cell lines. In these models, immunodeficient mice were engrafted with either human primary tumor cells or the tumor cell lines. For example, mice bearing tumors of Colo-205 were treated with different doses of INBRX-109 when their tumors reached an average size of about 300 mm3. As shown in the image below, we observed that tumor volume was reduced in mice treated with a single dose of INBRX-109 as low as 0.1mg/kg, administered intravenously, as compared to vehicle-treated mice. Additionally, mice bearing tumors of chondrosarcoma PDX were treated intravenously with 1 mg/kg of INBRX-109 when their tumors reached an average size of about 220 mm3. As shown in the second image below, webe well-tolerated without significant toxicities observed that tumor volume was reduced in mice treated with three doses of INBRX-109.
Colo-205 Dose Response
The observed clinical hepatotoxicity of hyper-clustered TAS266 highlights the importance of valency toward the therapeutic index of DR5 agonism. We developed an in vitro human hepatocyte assay, that enables us to compare constructs of different valencies and the impact of PE-ADA recognition on hepatocyte viability. Shown below as percentage viability, in this assay, a hexavalent construct, Hex-1F5, composed of six DR5 binding domains, was found to mediate significant hepatocyte death. Additionally, an analog of TAS266 caused the death of human hepatocytes in the presence of pooled immunoglobulins known to contain PE‑ADAs, that recognize single domain antibodies, while meaningful hepatocyte death with INBRX-109 was not observed. We believe that the hepatocyte death mediated by TAS266 observed in this assay was due to hyper-clustering of TAS266 by the PE-ADAs and resulted in an increase in the functional valency. Furthermore, the finding of hepatocyte cell death with the hexavalent DR5 agonist represents a collapsing of the therapeutic index as compared to a tetravalent agonist. We
believe our assay provides meaningful evidence of an upper bound of the optimal valency and that a tetravalent agonist with reduced recognition by PE-ADAs is the preferred therapeutic strategy for exploiting the DR5 pathway.
Human Hepatocyte Toxicity Assay
Additionally, we observed that INBRX-109 did not induce toxicities in cynomolgus monkeys after up to five weekly INBRX-109 administrations ofat doses up to and including the maximum administered dose of 30 mg/kg. No MTD was reached.
Part 2
In September 2019, we commenced single agent dose expansion cohorts, Part 2 is now complete and enrolled 116 patients in single agent dose cohorts in the following tumor types: colorectal and gastric adenocarcinomas, malignant pleural mesothelioma, chondrosarcoma, synovial sarcoma, and solid tumors with BMI>30.
Part 3
In February 2021, we initiated chemotherapy combination cohorts in Part 3 of this trial and will enroll up to 100 mg/kg.
Unmet Medical Need
patients. We are currently investigating INBRX-109 in chondrosarcoma, synovialEwing sarcoma, malignant pleural mesothelioma,colorectal cancer cohorts, and gastricSDH-deficient solid tumors, or GIST.
Initial Results from Phase 1 INBRX-109 Ewing
We announced preliminary efficacy and pancreatic adenocarcinomas. These are somesafety data from the Phase 1 trial of INBRX-109 in combination with Irinotecan, or IRI, and Temozolomide, or TMZ, for the treatment of advanced or metastatic, unresectable Ewing sarcoma. Among the 13 patients evaluable as of the most aggressive diseases, certaindata cut of which are also orphan oncology indications, that showed signsSeptember 8, 2023, the observed disease control rate was 76.9%, or 10 out of activity13 patients as measured by RECISTv1.1, with seven patients achieving partial responses (53.8%) and three patients achieving stable disease (23.1%). Overall, INBRX-109 in preclinical studies. These indications and manycombination with IRI/TMZ was well- tolerated from a safety perspective. We have expanded recruitment of this cohort as a result of these cancer subtypes do not respond well to currently approved immunotherapies and represent a significant unmet need.preliminary findings.
Initial Results from Phase 1 INBRX-109 Chondrosarcoma
Chondrosarcoma is a rare malignant bone tumor composed of cartilage matrix-producing cells. It is reported to be the second most common primary bone sarcoma with an incidence of 1 in 200,000 per year globally. The incidence in the United States is reported to be about 1,400 cases per year. There is currently no approved systemic treatment for patients with unresectable or metastatic disease. The placebo arm of a placebo-controlled trial, which tested the hedgehog pathway inhibitor IPI-926 in this indication had a progression free survival of approximately three months. In this trial, IPI-926 did not result in any partial responses and only a small subset of patients had minor reductions in their tumor size.
Malignant Pleural Mesothelioma
Mesothelioma is a malignant tumor most often caused by inhaled asbestos fibers which accumulate in the lining of the lungs, abdomen or heart. The epithelioid subtype is the most common histology of malignant pleural mesothelioma, reportedly comprising about 70% of all cases. It is estimated that as many as 43,000 people worldwide die from mesothelioma each year. There are currently limited approved therapeutic treatments. The placebo arm of a recent large double-blind, placebo-controlled trial testing tremelimumab, an anti-CTLA-4 antibody
(DETERMINE trial), in malignant pleural or peritoneal mesothelioma with second and third-line patients had a progression free survival rate of 2.7 months and an overall survival rate of 7.3 months. In this trial, tremelimumab did not significantly prolong progression free survival or overall survival compared with placebo in patients with previously treated malignant mesothelioma.
Synovial Sarcoma
Synovial sarcoma is a cancer that can arise from different types of soft tissue, such as muscle or ligaments. It is often found in the arm, leg, or foot and near joints such as the wrist or ankle. It can also form in soft tissues in the lung or abdomen. It is estimated that one to three people per 1,000,000 people are diagnosed with synovial sarcoma each year. Current therapeutic treatments available for unresectable or metastatic synovial sarcoma are doxorubicin or ifosfamide. Pazopanib and trabectedin have also shown activity.
Pancreatic Adenocarcinoma
Pancreatic cancer is a malignant tumor that originates in the pancreas and quickly metastasizes. The most common type of pancreatic cancer, comprising over 90% of all cases, begins in the cells that line the ducts that carry digestive enzymes out of the pancreas, or pancreatic adenocarcinoma. It is estimated that pancreatic cancer accounts for about 3% of all cancers in the U.S. and about 7% of all cancer deaths. Unresectable or metastatic pancreatic adenocarcinoma is often treated with fluorouracil, or 5FU, irinotecan and oxaliplatin, or gemcitabine and albumin-bound paclitaxel chemotherapies, with generally short-lived benefit.
Gastric Adenocarcinoma
Gastric cancer is the second most common cancer worldwide. Greater than 50% of patients present with unresectable locally advanced or metastatic gastric adenocarcinoma. Standard protocols include fluorouracil or capecitabine and oxaliplatin with or without nivolumab, fluorouracil or capecitabine and cisplatin, or other combinations containing irinotecan, paclitaxel, carboplatin, docetaxel, and trastuzumab for HER2 positive gastric adenocarcinoma.
Clinical Data
The IND for INBRX-109 became effective in August 2018, and we initiated a Phase 1 clinical trial in the United States in November 2018. This Phase 1 clinical trial is designed as an open-label, three-part trial in patients with locally advanced or metastatic solid tumors.
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | |
| | Phase 1 INBRX-109 Trial Design and Status | | |
| | First in human Phase 1 trial started November 2018 | | |
| | Part 1:
Dose Escalation |
⇨
⇨ | Part 2:
Dose Expansion Cohorts |
⇨
⇨ | Part 3:
Combination Studies | | |
| | Completed August 2019 | Ongoing | Initiated | | |
| | Dose Range:
0.3 to 30 mg/kg
N= 20
Result:
Maximum tolerated
dose not reached
| N=32
N=10
N=20
N=20
N=10 | Chondrosarcoma
Synovial sarcoma
Malignant pleural mesothelioma
Colorectal adenocarcinoma
Gastric adenocarcinoma | N=20
N=20
| Pancreatic adenocarcinoma 2nd line with FOLFIRI
Mesothelioma with Cisplatin/Carboplatin
or
Cisplatin/Carboplatin & Pemetrexed | | |
| | | | | | | | | | |
Part 1
Part 1 of the trial utilized a traditional 3+3 dose escalation design escalating INBRX-109 as a single agent from 0.3 mg/kg to 30 mg/kg. Twenty patients were enrolled in this portion of the trial, which was completed in August 2019. INBRX-109 was observed to be well-tolerated without significant toxicities observed at doses up to and including the maximum administered dose of 30 mg/kg. No MTD was reached.
Part 2
In September 2019, we commenced Part 2 of this trial with single agent dose expansion cohorts, enrollment of which is still ongoing. As of February 2, 2021, we have enrolled over 104 patients in single agent dose cohorts in the following tumor types: colorectal and gastric adenocarcinomas, malignant pleural mesothelioma, chondrosarcoma and synovial sarcoma. We expect to announce initial data from the synovial sarcoma cohort during the second half of 2021.
Part 3
During the fourth quarter of 2020, we initiated chemotherapy combination cohorts in malignant pleural mesothelioma and pancreatic adenocarcinoma. We expect to announce initial data from these combination cohorts during the fourth quarter of 2021.
Primary objectives of the Phase 1 trial are safety, tolerability, and determination of the MTD and recommended Phase 2 dose. Secondary objectives are serum exposure and immunogenicity, as measured by frequency of anti-drug antibodies. Exploratory objectives include clinical anti-tumor efficacy, based on response rate, duration of response, disease control rate, progression-free survival and overall survival, as well as evaluation of potential predictive diagnostic and pharmacodynamic biomarkers.
INBRX-109 Chondrosarcoma
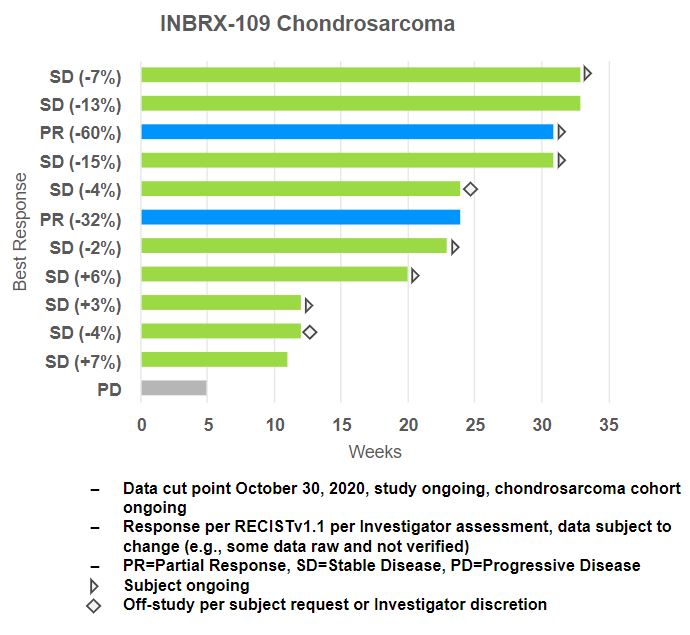
Based on initial data from the chondrosarcoma cohort of our Phase 1 trial, stable disease or partial responses were observed in 9 out of 12 patients with chondrosarcoma, with six of these patients still on treatment as of October 30, 2020. Per investigator requests, an additional 10 patient enrollment slots were added to the chondrosarcoma cohort of the ongoing trial, which were filled as of January 2021. We plan to provide a more mature data set from the chondrosarcoma cohort during the second half of 2021, which will include data on conventional and non-conventional chondrosarcoma patients.
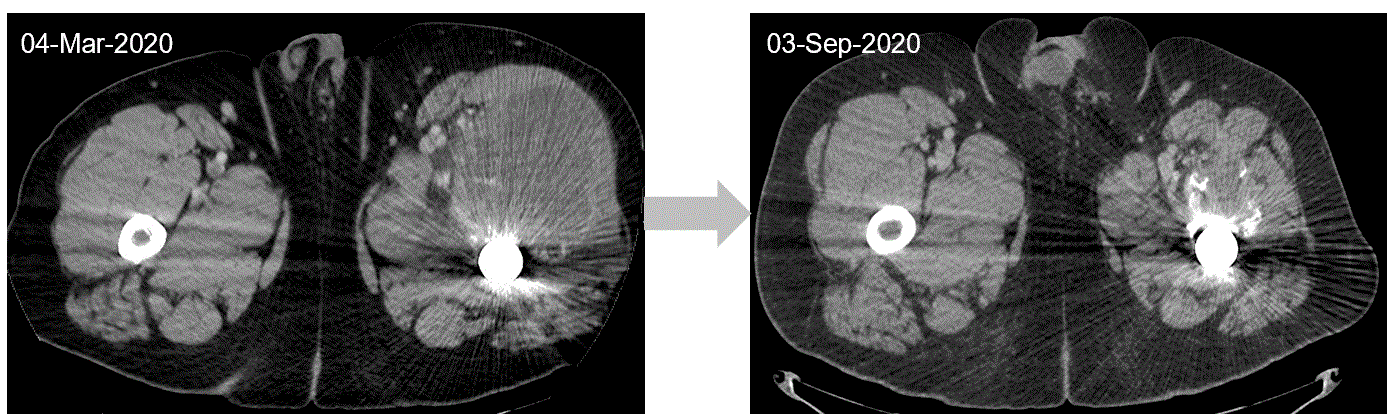
The CT tumor imaging scan of a chondrosarcoma patient below shows a target lesion, at baseline and at tumor assessment after eight cycles of INBRX-109. This patient achieved a best response of partial response with 60% shrinkage in the target lesions per RECIST (version 1.1).
In January 2021, the FDA granted fast trackFast Track designation to INBRX-109 for the treatment of patients with unresectable or metastatic conventional chondrosarcoma. We plan to submit an IND amendmentIn November 2021, the FDA granted orphan drug designation for INBRX-109 duringfor the first quartertreatment of 2021. We plan to initiateconventional chondrosarcoma. In August 2022, the EMA granted orphan drug designation for INBRX-109 for the treatment of conventional chondrosarcoma.
In November 2022, we announced efficacy and safety data from the ongoing Phase 1 INBRX-109 expansion cohorts for the treatment of chondrosarcoma. Among the 33 patients evaluable as of November 8, 2022, the observed disease control rate was 87.9%, or 29 out of 33 patients as measured by RECISTv1.1, with two patients achieving partial responses (6.1%) and 27 patients achieving stable disease (81.8%). Disease control was observed in patients with and without IDH1/IDH2 mutations. Of those achieving stable disease, 55.6% had decreases from baseline in tumor size. Clinical benefit was durable, 14 of 33 patients (42.4%) who achieved disease control had a clinical benefit lasting greater than 6 months, and the longest duration of stable disease observed was 20 months. At that time, the median progression-free survival, or PFS, was 7.6 months, and five patients remained on study.
Treatment-related AE, were reported in less than 5% of the patients with the most common being increased alanine aminotransferase, or ALT, increased aspartate aminotransferase, or AST, and increased blood bilirubin and fatigue. There were no grade 4 or 5 events reported among patients with treatment-related AEs.
Phase 2 INBRX-109 Chondrosarcoma
In June 2021, we initiated a registration-enabling Phase 2 trial in this patient population, which will enroll approximately 200 patients in total at 50 different sites worldwide, with progression free survival, or mPFS, as the endpoint, in mid-2021.primary endpoint.
Primary objectives of the ongoing Phase 2 trial are to evaluate the efficacy of INBRX-109 as measured by mPFS, assessed by central real-time independent radiology review. Secondary objectives are to evaluate the overall survival, mPFS by investigator assessment, quality of life, objective response rate, duration of response, disease control rate, safety and tolerability, pharmacokinetics and anti-drug antibodies to INBRX-109.
In early 2023, the Phase 2 trial was placed on partial clinical hold by the FDA, and we paused patient enrollment in the trial, following the occurrence of a grade 5 hepatotoxicity event and pre-defined stopping rules built into the protocol. The additional insight gained on patients at risk of significant hepatotoxicity led us to believe that elderly patients with fatty liver disease are the at-risk population. We implemented the Hepatic Steatosis Index, or HSI, into the protocol and excluded elderly patients with an HSI score of 36 or higher, which has helped to address the patients at risk of significant hepatotoxicity. The FDA lifted the hold in April 2023 after we amended the trial protocol. We expect to announce data from this trial during the first half of 2025.
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | |
| | Planned Phase 2 Trial Design | | |
| | Randomized, Blinded, Placebo-controlled, Phase 2 Study of INBRX-109 in Conventional Chondrosarcoma, Grades 2 and 3, Unresectable or Metastatic Conventional Chondrosarcoma | | |
| | Randomization 2:1
Key eligibility criteria: • Conventional chondrosarcoma • Grade 2 and 3 • Unresectable or metastatic
Stratification by histologic grade and line of therapytherapy. Grade and IDH1/2 mutation status | | | | ⇨
⇨Until progressive disease (PD) or toxicity with cross-over
Including interim futility analysis
• Primary Endpoint: progression free survival (PFS)
• Secondary Endpoints: overall survival, (OS),quality of life, overall response rate, (ORR)/duration of response, (DOR), disease control rate, (DCR), quality of life (QoL)safety, etc. | | |
| ⇨ | INBRX-109 N= 134
3 mg/kg every three weeks
(Ongoing) | ⇨ | | INBRX-109
(2) | |
| | | < | | |
| ⇨ | Placebo N= 67 (allows for crossover)
3 mg/kg every three weeks
(Ongoing) | ⇨ | | Placebo
(1) | |
| | | | | |
| | | | | | | | |
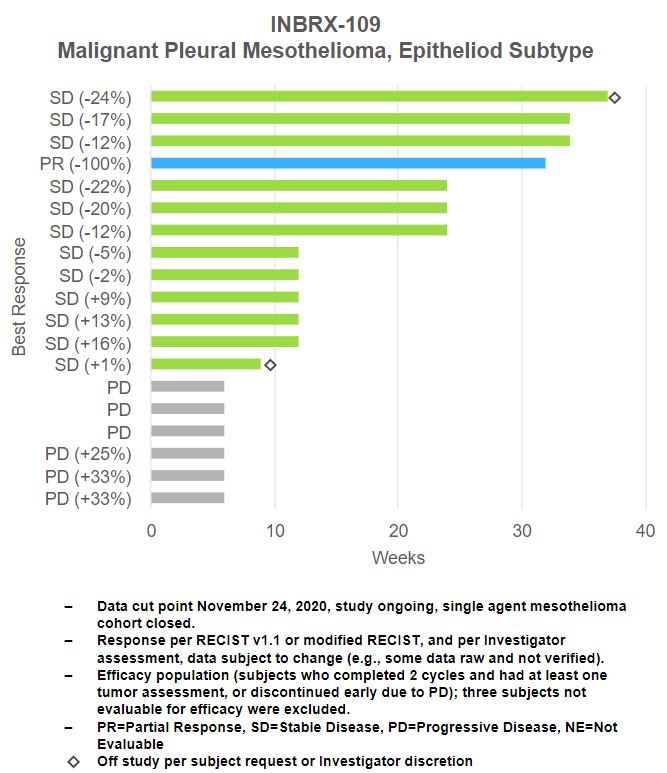
MesotheliomaINBRX-106
In initial data fromINBRX-106 is a hexavalent OX40 agonist, currently being investigated as a single agent and in combination with Keytruda, a PD-1 blocking checkpoint inhibitor, in patients with locally advanced or metastatic solid tumors. We continue to enroll and/or have active patients in Part 2, of this trial, we have observed signs of single agent activity in malignant pleural mesothelioma, epithelioid subtype. As of November 24, 2020, time on treatment with INBRX-109 was equal to or greater than 24 weeks in seven of the 19 patients evaluable for response (~37%).
Phase 1 Safety Data
As of February 2, 2021,dose expansion, and based on preliminary Phase 1 data, INBRX-109 has been generally well tolerated in the majority of patients. The chart below is based on 92 patients with available adverse event data and details the adverse events, or AEs, related to INBRX-109 per Investigator assessments. There has been a low incidence of Grade 1, 2, and 3 AEs. Few serious adverse events, or SAEs (including one Grade 5 liver failure and death), and one dose-limiting toxicity, or DLT, were observed.
While the majority of the liver-related AE (e.g., AST increased and/or ALT increased) have been Grade 1 & 2, four patients developed ≥ Grade 3 AE (n=4/92, 4.3%) related to INBRX-109. The first patient, who had an alanine transaminase, or ALT, increase of Grade 2 at baseline and liver metastases from gastric cancer, experienced Grade 3 aspartate transaminase, or AST, and ALT increases during the first cycle of INBRX-109, which were related to
INBRX-109 per Investigator assessment. The AST and ALT increases were observed to be partially reversible, with a trend towards baseline before the patient enrolled to hospice for progressive gastric cancer. The second patient, who had pleural mesothelioma, was admitted for acute respiratory failure and sepsis due to pneumonia and was found to have Grade 3 AST and ALT increases, which were related to INBRX-109 per Investigator assessment. The AST and ALT increases were observed to be partially reversible, with a trend towards baseline before the patient passed away from acute respiratory failure due to pneumonia. The third patient, who had pleural mesothelioma, was found to have Grade 2 AST and Grade 3 ALT increases during the first cycle of INBRX-109. Per Investigator assessment, these increases were related to INBRX-109 but were transient and fully reversible before the patient began the second cycle of INBRX-109.
The single event of Grade 5 acute liver failure, in a pleural mesothelioma patient, was possibly attributed to INBRX-109 per Investigator assessment. Other possible contributing causes for the Grade 5 acute liver failure considered for this patient were hypoxic liver injury, accidental or intentional overdose of acetaminophen-containing pain medications, and treatment with several other potential hepatotoxic medications and their cumulative toxicity. Notably, this patient screened negative for pre-existing anti-drug antibodies against INBRX-109.
| | | | | | | | | | | |
| AE Term | AE related to INBRX-109 (very likely, probable, possible) |
| Grade per CTCAEv5.0 | Grade 1 & 2
(N) | Grade 3
(N) | All Grade
(N) |
| Fatigue | 16 | 1 | 17 |
| AST increased | 9 | 2 | 11 |
| ALT increased | 8 | 3 | 11 |
| Nausea | 7 | 1 | 8 |
| Diarrhea | 7 | 0 | 7 |
| Pyrexia | 5 | 0 | 5 |
| Abdominal pain | 3 | 1 | 4 |
| Rash | 3 | 0 | 3 |
| Decreased appetite | 3 | 0 | 3 |
| Headache | 3 | 0 | 3 |
One death possibly attributed to study drug, acute hepatic failure in patient with mesothelioma (N=1/92,~1%)
9 out of 92 subjects with INBRX-109-related liver-related AE (~ 10%)
Data cut point February 2, 2021
Disclaimers:
-Total number of subjects: 92 with reported adverse event data, study ongoing
- AE reported ≥ 3 times, causality to INBRX-109 per Investigator assessment
- Some data preliminary and subject to change (raw, unvalidated, and verification pending)
- Excluded data: uncoded AE; missing AE terms; missing causality; missing severity |
Future Clinical Development Plans
We plan to investigate the therapeutic utility of INBRX-109 through a broad clinical development plan across difficult-to-treat cancers. Ewing sarcoma is a rare type of cancer that occurs in bones or in the soft tissue around the bones, more commonly found in children. There are an estimated 1,000 cases of Ewing sarcoma in the United States each year. We plan to initiate enrollment in Ewing sarcoma chemotherapyPart 4, combination cohort during the middle of 2021 andexpansion cohorts. We expect to announce initial data from this combination cohortthese cohorts during the firstsecond half of 2022.2024.
Other futureBackground on Immunotherapy
A notable recent success in cancer treatment is the approval of checkpoint inhibitor immunotherapies as therapeutic agents. Immune checkpoints are key mechanisms that fine-tune and control the body’s immune response. In the cancer setting, tumors have developed strategies for hijacking these checkpoints, preventing an immune response to the cancer and allowing the tumor cells to proliferate unchecked. Checkpoint inhibitor immunotherapies were developed to overcome this phenomenon by relieving immune cell inhibition, resulting in a potentially long-lasting amplification of the anti-tumor immune response. Therapies against checkpoint proteins, such as PD-1 and PD-L1, produced impressive results in clinical development, resulting in marketing approvals in a number of INBRX-109 may include combinations with additional chemotherapies and potentially synergistic agents in several solid tumors, and hematologic malignancies. We are currently conducting preclinical studies with INBRX-109 and various rational combination agents, including targeted therapeutics and apoptotic pathway modulators, aimed at guiding our future clinical plans.
INBRX-106
INBRX-106 is a precisely engineered hexavalent sdAb-based therapeutic candidate targeting OX40, designed to be an optimized agonist of this co-stimulatory receptor.
Background on OX40
OX40, also known as TNFRSF4, is a member of the TNFRSF; OX40TNFRSF, is predominately expressed and is a key co-stimulatory receptor on activated T-cells. Signaling through OX40 provides co-stimulation that promotes T-cell expansion, enhanced effector function and memory cell formation, and prevents activation-induced cell death. The natural mechanism of OX40 activation is via the interaction with its trimeric ligand, OX40L, which serves to effectively cluster multiple OX40 molecules and facilitate downstream signaling leading to nuclear factor kappa-light-chain-enhancer of activated B cells, or NFkB, activation. Based on the capacity for OX40 signaling to enhance anti-tumor immunity in preclinical studies, there have been many efforts to therapeutically exploit this pathway for cancer immunotherapy. Most previously developed agents were bivalent OX40 agonists;agonists, the configuration of which we believe the bivalent configuration isto be poorly suited for efficient receptor clustering, which is evidenced by the disappointinglack of clinical responses observed for such molecules.
Our Solution - INBRX-106
As shown in the diagram below, INBRX-106 is composed of six OX40 targeting sdAbs and a functional Fc domain. INBRX-106 is designed to bind six OX40 molecules on the cell surface to mediate efficient receptor clustering and downstream signaling. Additionally, INBRX-106 is able to exploit IgG-mediated effector function via the Fc domain. In preclinical studies, we observed that INBRX-106 can mediate T-cell co-stimulation and reduce the suppressive activity of regulatory T-cells.
INBRX-106: Hexavalent OX40 Agonist
Through a series of preclinical studies, we compared the potential agonist activity of INBRX-106 to bivalent OX40 agonist antibodies. In these studies, INBRX-106 elicited superior OX40 agonism when compared to the bivalent antibodies, 1A7 (analog of MOXR-0916) or 1D10 using an OX40 expressing NFkB reporter cell line, wherein clustering of OX40 receptor mediated signaling culminated in luciferase expression as shown below in relative luminescence units, or RLU, on the Y-axis.
Valency Drives OX40 Agonism
OX40 Agonism: NFKB Reporter Cell Line
To assess the ability of INBRX-106 to provide co-stimulation and modulate T-cell receptor, or TCR, driven T-cell responses, we conducted various in vitro assays using primary human immune cells. Following anti-CD3 mediated TCR activation of T-cells, INBRX-106 mediated co-stimulation in these assays was superior to a bivalent OX40 antibody at enhancing T-cell proliferation and activation. Representative results are shown below wherein CD4 T-cell proliferation (top left) was monitored by flow cytometric measurement of cell division as shown on the Y-axis as % proliferated Interferon gamma, or IFNγ, production (top right) was assessed by ELISA as shown on the Y-axis in pg/ml. CD4 T-cell activation as determined by flow cytometric measurement of CD25 (bottom left) and CD71
(bottom right) as shown on the Y‑axis as mean fluorescence intensity, or MFI. INBRX-106 was found to mediate similar co-stimulation of CD4 and CD8 T-cells.
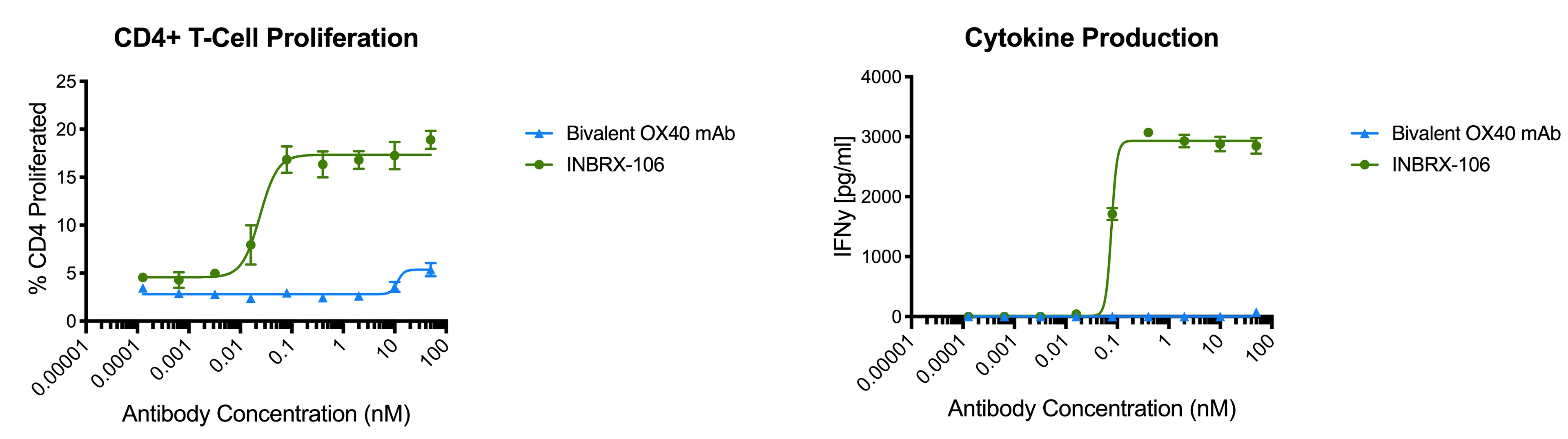
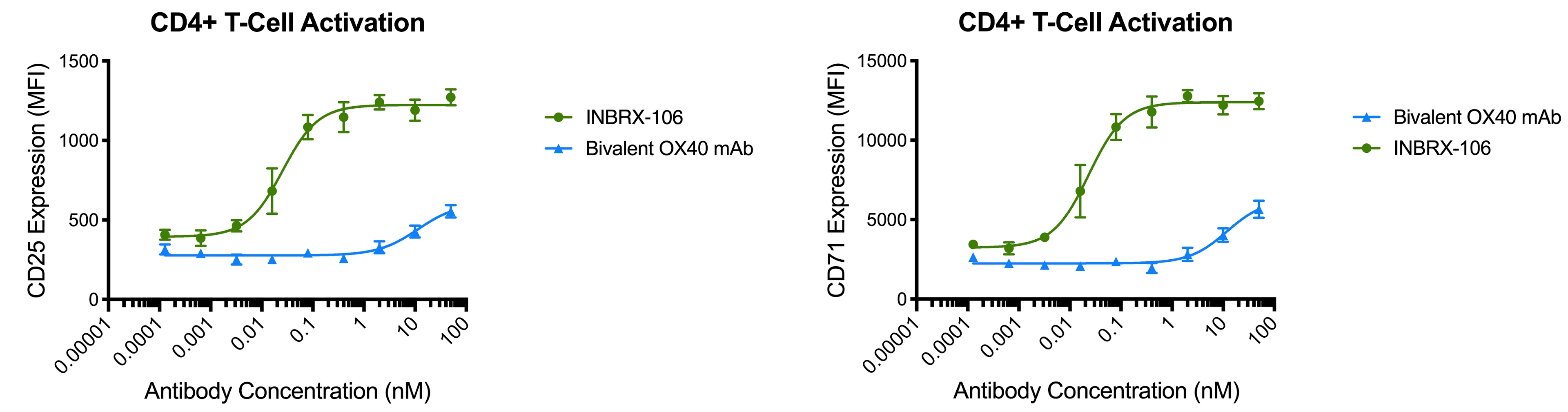
Syngeneic tumor models are a commonly used tool to investigate the anti-tumor activity of immunomodulatory agents. INBRX-106, however, does not interact with mouse OX40. Therefore, in order to investigate the potential of an INBRX-106-like mechanism to mediate anti-tumor immunity in a preclinical model, we developed mu106, a molecule that has a matched hexavalent, sdAb-based therapeutic format and binds mouse OX40 with a similar affinity to that of INBRX-106 on human OX40. We tested mu106 in numerous syngeneic tumor models and observed robust single agent anti-tumor activity, as well as enhanced anti-tumor activity in combination with a PD‑1 blocking antibody. As shown below, presented as the individual tumor growth curve of each mouse in a given treatment group (n=10 mice/group), mu106 (Hexavalent OX40 mAb) elicited superior anti-tumor activity compared to a bivalent OX40 antibody or a PD-1 blocking antibody, or a combination thereof. Furthermore, the anti-tumor
activity of mu106 was enhanced in combination with a PD-1 blocking antibody. It is generally accepted that PD-1/PD-L1 blocking agents display minimal anti-tumor activity in the B16-F10 syngeneic tumor model.
Unmet Medical Need
The approval of immune checkpoint inhibitors that block CTLA-4 or PD-1, or their ligands such as PD-L1 respectively, has caused a paradigm shift in oncology treatment due to their substantial response rates and overall survival benefit across numerous cancers. However, the checkpoint inhibitors achieve lasting benefit or cure only in a subset of patients and therefore, additional therapeutics are required to further stimulate an anti-cancer immune response. We plan to clinically evaluate INBRX-106 as a single agent to reach into tumors not responsive to current checkpoint inhibitor therapy ("cold" tumors) and to combine INBRX-106 synergistically with PD-1 or other checkpoint inhibitors, vaccines, and other immune stimulatory approaches.
Clinical Data
We initiated a Phase 1 clinical trial in December 2019 for INBRX-106. This Phase 1 trial is designed as an open-label, four-part trial in patients with locally advanced or metastatic solid tumors.
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | Phase 1 INBRX-106 Trial Design and Status | | |
| | First in human Phase 1 trial started December 2019 | | |
| | Part 1:
Single Agent
Dose Escalation |
⇨
⇨ | Part 2:
Single Agent
Dose Expansion |
⇨
⇨ | Part 3:
Combination
Dose Escalation |
⇨
⇨ | Part 4:
Combination
Dose Expansion | | |
| | Completed January 2021 | Started February 2021 | Started February 2021 | Planned | | |
| | Dose Range:
0.0003 to 3 mg/kg
N= 20
Result:
Maximum tolerated
dose not reached
| INBRX-106:
RP2D
Two
Dosing
Schedules
N=24
Basket |
INBRX-106 + Keytruda
Dose Range:
0.01 to 0.3 mg/kg
Minimum N=12
| INBRX-106 + Keytruda
(post PD-1/PD-L1
Checkpoint Inhibitor)
N= 60
PD-L1+ NSCLC (1st cohort)
PD-L1+ NSCLC (2nd cohort)
PD-L1+ basket
| | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
Part 1 of the trial utilized a traditional 3+3 algorithm for single agent dose escalation from 0.0003 mg/kg to 3 mg/kg. Twenty patients were enrolled in this portion of the trial, which was completed in January 2021. INBRX-106 was observed to be generally well tolerated in humans, with only three serious adverse events, one Grade 3 AE and one DLT. The most common AEs reported for INBRX-106 were Grade 1 and 2 rashes or dermatitis, cytokine release syndrome, fatigue, and infusion-related reactions. Notably, cutaneous toxicities are common immune-related AEs associated with immune checkpoint inhibitors. These mostly mild or moderate and likely immune-related toxicities were in line with the mechanism of action of this candidate therapeutic. The maximum administered dose was 3 mg/kg and the MTD level was not reached.
Signs of clinical benefit to date were observed in patients with a range of cancer types, including tumors generally considered to be “hot” (a tumor that has been infiltrated by T-cells) and “cold” (a tumor that has not been infiltrated by T-cells), as well as in patients that were relapsed or refractory to checkpoint inhibitors. Activity was observed at dose levels in a range from 0.0003 to 0.3 mg/kg and peripheral biomarker sampling confirmed agonist activity across these low doses. As of March 3, 2021, preliminary single agent data from the first 20 patients treated in the dose escalation portion of this trial, and with available tumor assessment data, demonstrated stable disease for eight patients as shown below. These stable diseases occurred at low dose levels. As of March 3, 2021, the longest duration of stable disease was 50 weeks, or approximately eleven and a half months, and the greatest reduction in tumor volume per RECIST was 23%.
| | | | | | | | | | | | | | | | | | | | | | | |
Phase 1 INBRX-106 Dose Escalation Status
Part 1 as of March 3, 2021
|
| | | | | | | |
| Dose Level
mg/kg | Demographics | Tumor Type | Notable Prior Therapies and Best Response | INBRX-106 Best Response | Notable Time on INBRX-106 | |
| 0.0003 - 0.003 | 59 M W | Small intestine cancer (GIST) | S/p 8 lines, no CPI | SD (+2%) | 32 weeks | |
| 0.001 - 0.01 | 85 F W | Melanoma (cutaneous) | S/p Pembrolizumab SD ~3.5y | SD (-23%) | 40 weeks | |
| 0.003 - 0.01 | 69 M W | Bladder CA | S/p 2 lines, Nivolumab PD 5mo | PD | | |
| 0.01 - 0.1 | 56 F O | NSCLC, AD (PD-L1 40%) | S/p Carboplatin/Pemetrexed/Pembrolizumab PD 2mo | SD (Non-measurable) | 50 weeks, ongoing | |
| 0.03 - 0.1 | 54 M W | Esophageal AD | | PD | | |
| 0.1 | 55 M W | Esophageal AD | S/p 4 lines, Pembrolizumab PD 3mo | SD (+12%) | 12 weeks | |
| 0.1 | 78 M W | Prostate AD (CRPC) | S/p 6 lines, no CPI | SD (+6%; PSA -52%) | 18 weeks | |
| 0.1 | 58 F W | RCC (sarcomatoid features) | | PD | | |
| 0.3 | 70 F W | Chondrosarcoma | S/p 1 line, Spartalizumab/IL-15/IL-2 12mo SD | SD (+7%) | 12 weeks | |
| 0.3 | 71 M W | HNSCC | | NE (DLT) | | |
| 0.3 | 47 M W | Colon AD (KRAS mutation) | | PD | | |
| 0.3 | 60 M W | NSCLC, AD (PD-L1 0%) | S/p 6 lines, Nivolumab SD ~3.5y, Atezolizumab/Bevacizumab ~SD 1.5y | SD (-2%) | 30 weeks, ongoing | |
| 0.3 | 74 M O | RCC (clear cell) | S/p 2 lines, Nivolumab UK ~3y | SD (+6%) | 9 weeks | |
| 0.3 | 80 F W | Breast AD (IDC) | | PD | | |
| 1.0 | 72 F W | Atypical carcinoid pulmonary | | PD | | |
| 1.0 | 46 M W | Rectal AD | | PD | | |
| 1.0 | 71 M W | Pancreatic AD | | PD | | |
| 3.0 | 55 F W | RCC | | PD | | |
| 3.0 | 50 F W | Colon AD | | iuPD | | |
| 3.0 | 65 M W | Pancreatic AD | | NE | | |
| *Stable disease (SD); Progressive disease (PD); Immune unconfirmed progressive disease (iuPD);Not evaluable (NE); Unknown (UK); Adenocarcinoma (AD); Renal cell carcinoma (RCC); Castration-resistant Prostate Cancer (CRPC); Prostate-specific antigen (PSA); Head and Neck Squamous Cell Carcinoma (HNSCC); Non-small Cell Lung Cancer (NSCLC); Invasive Duct Carcinoma (IDC); Small Cell Lung Cancer (SCLC); Checkpoint Inhibitor (CPI) Male (M); Female (F); White (W); Other (O) | |
Future Clinical Development Plan
With the conclusion of Part 1of the trial, single agent dose escalation, Part 2 of the trial, single agent dose expansion, was initiated in February 2021. Utilization of extensive biomarker data guided the determination of the optimal dose for Part 2 of the trial, which we believe to be 0.03 mg/kg. Key attributes leading to the choice of this dose level included OX40 occupancy data, evidence of peripheral memory T-cell activation and proliferation, and sufficient drug clearance to allow target recovery prior to the next dose administration. Part 2 treatment cohorts of this trial are in the following tumor types: non-small cell lung cancer, melanoma, head and neck squamous cell carcinoma, gastric or gastro-esophageal junction adenocarcinoma, renal cell carcinoma, and urothelial (transitional) cell carcinoma.
In Parts 3 and 4 of this trial, INBRX-106 will be evaluated in combination with Keytruda, a PD-1 blocking checkpoint inhibitor. In Part 3 of the trial, which initiated in February 2021, INBRX-106 will be escalated in combination with Keytruda. Patients in the INBRX-106 combination expansion cohorts in Part 4 will have non-small cell lung cancer or melanoma, head and neck squamous cell carcinoma, gastric or gastro-esophageal junction adenocarcinoma, renal cell carcinoma, and urothelial (transitional) cell carcinoma, which are positive for PD-L1 expression, as determined by immunohistochemistry and possessing adequate hematologic and organ function, to qualify for enrollment.
We expect the entire trial will be conducted at approximately 10-12 clinical sites in the United States and enroll approximately 120 patients.
Primary objectives of the trial are safety and tolerability, and the determination of the MTD and recommended Phase 2 dose of INBRX-106 as a single agent, and INBRX-106 in combination with Keytruda. Secondary objectives will be serum exposure, immunogenicity, as measured by frequency of anti-drug antibodies, and clinical anti-tumor efficacy per RECIST (version 1.1) and immune RECIST based on response rate, duration of response, disease control rate, progression-free survival and overall survival. Exploratory objectives will include evaluation of potential predictive diagnostic and pharmacodynamic biomarkers.
We expect to announce initial data from dose escalation in combination with Keytruda during the third quarter of 2021 and dose expansion in combination with Keytruda during the third quarter of 2022.
Overview of Our Conditional Agonists
Our pipeline includes conditional agonist therapeutic candidates designed to restrict functional activity to the specific site where a given antigen is expressed, for example, restricting the co-stimulation of T-cells to the tumor microenvironment. By agonizing receptors that function to promote an anti-tumor immune response in a manner that is dependent on a tumor-biased target, conditional agonists have the potential to induce anti-tumor activity without generating systemic toxicities. Our sdAb platform allows for careful selection of the affinity and valency of targeting domains, which we believe may provide optimal biologic activity and systemic exposure, potentially achieving a superior therapeutic index.
INBRX-105
Background on Immunotherapy and Unmet Medical Need
A notable recent success in cancer treatment is the approval of checkpoint inhibitor immunotherapies as therapeutic agents. Immune checkpoints are key mechanisms that fine-tune and control the body’s immune response. In the cancer setting, tumors have developed strategies for hijacking these checkpoints, preventing an immune response to the cancer and allowing the tumor cells to proliferate unchecked. Checkpoint inhibitor immunotherapies were developed to overcome this phenomenon by relieving immune cell inhibition, resulting in a potentially long-lasting amplification of the anti-tumor immune response. Therapies against checkpoint proteins, such as PD-1 and PD-L1, produced impressive results in clinical development, resulting in regulatory approvals in a number of malignancies.
Despite unprecedented clinical response rates, the majority of patients fail to respond to therapies targeting PD-1 and PD-L1. We believe this is in part because T-cells require co-stimulation for full functionality. Thus, checkpoint inhibition alone is likely insufficient to fully enable the immune system to attack a tumor, and we believe further benefit could be derived by the addition of immune co-stimulatory agents.
We believe a promising co-stimulatory receptor is 4-1BB, also known as TNFRSF9 or cluster of differentiation 137, which has been identified on T-cells isolated from primary tumors. 4-1BB is a member of the TNFRSF and is expressed on recently activated T-cells but is largely absent from circulating T-cells under normal conditions. Agonistic 4-1BB antibodies have exhibited anti-tumor activity in mouse models of cancer, especially in combination with other immunomodulatory or tumor targeted therapeutics. These observations motivated the clinical advancement of antibodies designed to agonize 4-1BB and co-stimulate tumor reactive T-cells. Most recently, urelumab, a 4-1BB agonist, showed initial signs of single agent therapeutic efficacy, but clinical investigation was halted due to dose-limiting liver toxicities. The resulting data suggest there is potential for 4-1BB agonists to exert anti-cancer function, but also suggest a potentially significant safety advantage for therapeutics able to enrich for 4-1BB activation in the tumor microenvironment.
Our Solution - INBRX-105
INBRX-105INBRX-106 is a precisely engineered multi-specifichexavalent sdAb-based therapeutic candidate that istargeting OX40, designed to agonize 4-1BB selectively in the presence of PD-L1, which is typically found in the tumor microenvironment and associated lymphoid tissues. We believe that the unique format of INBRX-105, comprised of two PD-L1 targeting domains, each arranged adjacent to a 4-1BB targeting domain, should result in a best-in-class PD-L1 targeted 4-1BB agonist. Based on the enrichment of PD-L1 expression in the tumor microenvironment, we believe INBRX-105 has the potential to selectively activate 4-1BB + T-cells in the tumor microenvironment, thus overcoming the toxicity issues that have limited the success of prior 4-1BB agonists. PD-L1 is a validated biomarker for tumors with the potential
for anti-tumor immune activation and thus we believe representsbe an ideal anchor antigen to drive 4-1BB agonism. The image below illustrates the unique structure of INBRX-105.
INBRX-105: A PD-L1 Targeted 4-1BB Agonist
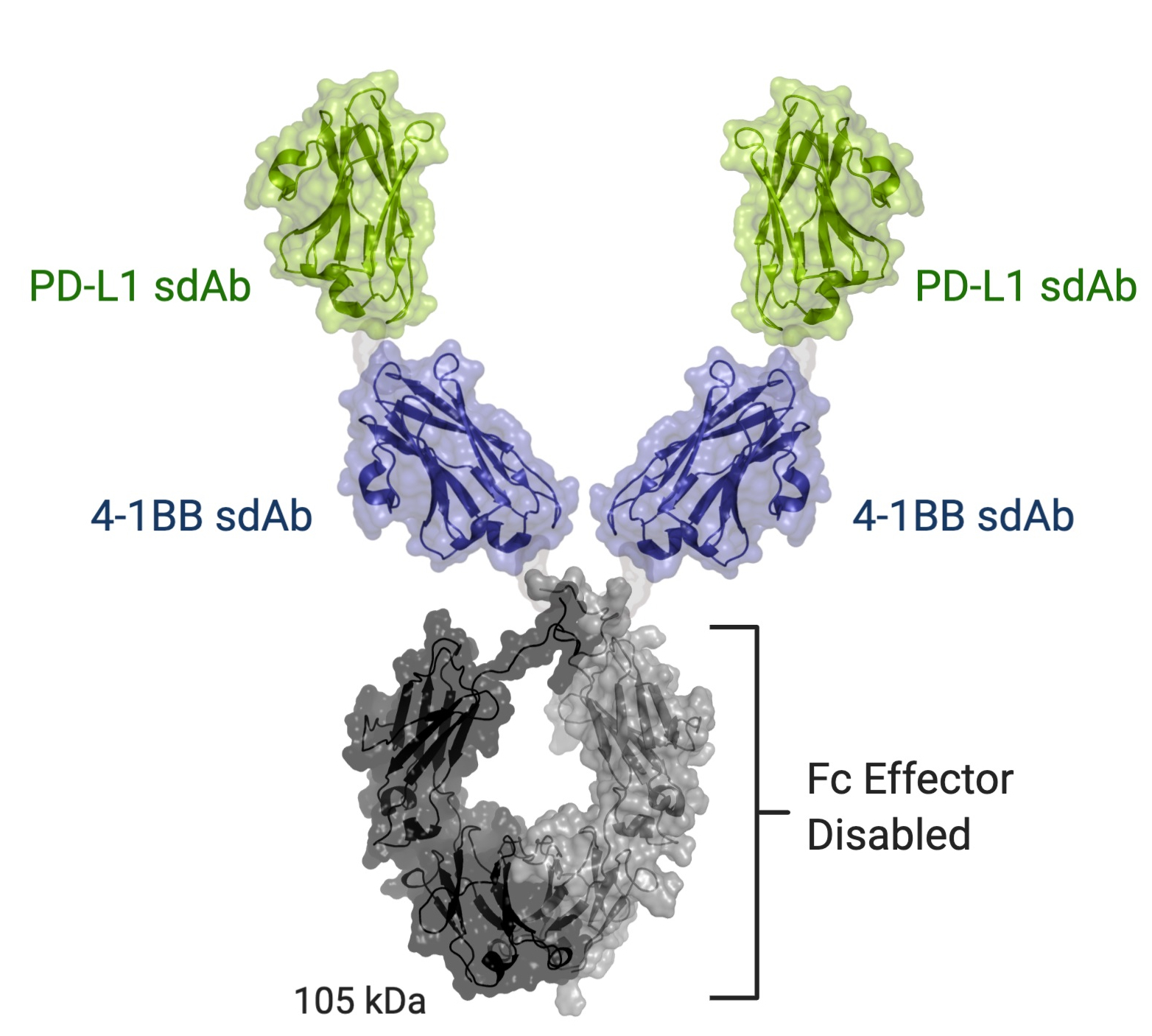
We tested INBRX-105 in a variety of in vitro assays to evaluate its ability to mediate PD-L1 dependent 4-1BB agonism. In some cases, we compared the 4-1BB agonistic activity to analogs of conventional anti-4-1BB antibodies with published mechanisms and clinical experience, urelumab or utomilumab. The figure below shows the results of a study in which reporter cells expressing 4-1BB on the surface were mixed with a second cell population that either did or did not express PD-L1 on the surface. Activation of 4-1BB receptors through clustering drives signaling and subsequent luciferase expression in the reporter cell line, read as RLUs, and shown on the vertical axis. We observed that INBRX-105 only activated 4-1BB signaling in the presence of PD-L1 expressing cells and had no effect on 4-1BB signaling in the presence of cells lacking PD-L1. Furthermore, these assays revealed that the analog of urelumab was a constitutiveoptimized agonist of 4-1BB signaling, whereas the analog of utomilumab did not display agonistic activity, demonstrating distinct mechanisms of action among the three therapeutic candidates.
INBRX-105 PD-L1-Dependent 4-1BB Agonism
To assess the potential ability of INBRX-105 to modulate the activity of T-cells activated through TCR engagement, we conducted various in vitro assays using primary human immune cells. One such assay, an allogeneic mixed lymphocyte reaction, or MLR, is commonly used to evaluate the propensity of immunomodulatory agents to provide T-cell co-stimulation. Therein, T-cells are induced to secrete the cytokine IFNγ, as a result of TCR clustering and activation. This cytokine secretion can be greatly enhanced through concurrent activation ofthis co-stimulatory receptors, such as 4-1BB. In an MLR, we observed superior IFNγ production by INBRX-105, shown on the vertical axis in the figure below, as compared to Keytruda, Tecentriq or the combination of these therapeutics and an analog of the 4‑1BB antibody, utomilumab. We have also observed similar effects in human T-cells stimulated ex vivo with a specific viral antigen to achieve TCR activation.
Mixed Lymphocyte Reaction: IFNγ Production
Syngeneic tumor models are a commonly used tool to investigate the anti-tumor activity of immunomodulatory agents. However, INBRX-105 does not interact with either mouse PD-L1 or 4-1BB. Therefore, in order to investigate the potential to mediate anti-tumor immunity preclinically, we developed a surrogate version of INBRX-105, mu105, with analogous format and function on mouse PD-L1 and 4-1BB. To test the tumor centric activation of 4-1BB, we evaluated the activity of mu105 in the MC38 colorectal carcinoma syngeneic model, as this model is known to express PD-L1 on both tumor and infiltrating immune cells.receptor. As shown in the figurediagram below, INBRX-106 is composed of six OX40 targeting sdAbs and a 2 mg/kg dosefunctional Fc domain.

As a hexavalent therapeutic candidate, INBRX-106 is designed to bind six OX40 molecules on the cell surface to mediate efficient receptor clustering and downstream signaling. In preclinical studies, we observed that INBRX-106 elicited superior OX40 agonism when compared to the bivalent antibodies, 1A7 (analog of mu105 activated 4-1BB, as shown byMOXR-0916) or 1D10 using an increaseOX40 expressing NFkB reporter cell line, wherein clustering of OX40 receptor mediated signaling culminated in luciferase expression. We also observed that INBRX-106 can mediate T-cell co-stimulation and reduce the suppressive activity of regulatory T-cells. Additionally, INBRX-106 is able to exploit IgG-mediated effector memory T-cells, or Tem cells, as a percentage of total CD8 T-cells. This dose level provided a short duration of 4-1BB agonism, but maintainedfunction via the Fc domain.
ability to activate anti-tumor immune cells, as evidenced by the robust tumor control in combination with an anti-PD-1 blocking antibody. We believe that this data demonstrates the potential for 4-1BB agonism through an INBRX-105 like mechanism to meaningfully alter the T-cell population, enrich for cells with anti-tumor capacity, and provide co-stimulation with potential for synergistic activity with an anti-PD-1 check point inhibitor.
Clinical Data
We initiated a Phase 11/2 clinical trial in the United States in February 2019.December 2019 for INBRX-106. This Phase 1 trial is designed as an open-label,open- label, four-part trial in patients with locally advanced or metastatic solid tumors and is being conducted at approximately 10-12 clinical sites in the United States.
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | |
| | Phase 1 INBRX-105 Trial Design and Status | |
| | First in human Phase 1 trial started February 2019 | |
| | Part 1:
Single Agent
Dose Escalation |
⇨
⇨ | Part 2:
Single Agent
Dose Expansion |
⇨
⇨ | Part 3:
Combination
Dose Escalation |
⇨
⇨ | Part 4:
Combination
Dose Expansion | |
| | Completed | Started Q1 2021 | Planned | Planned | |
| | INBRX-105
N=32
Dose range:
0.001 to 3 mg/kg | INBRX-105
N=32
Dose range: RP2D
PD-L1+ NSCLC
PD-L1+ Melanoma
PD-L1+ Head & Neck Cancer
PD-L1+ Basket
Indications based on known sensitivity to 4-1BB and/or checkpoint blockade | INBRX-105 + Keytruda
Minimum N=9
Dose range: RP2D
Locally advanced or
metastatic solid tumors | INBRX-105 + Keytruda
N= 80
Post PD-1/PD-L1
Checkpoint Inhibitor:
PD-L1+ NSCLC
PD-L1+ Melanoma
PD-L1+ Basket
CPI naive: PD-L1+ NSCLC | |
| | | | | | | | | | | | |
| | | | | | | | | | | | |
Single agent dose escalation was completed with a total of 32 patients enrolled. We observed dose-limiting toxicities, which were consistent with immune related adverse events, such as hepatitis, arthritis and myalgia/hyperthyroidism, at several dose levels and determined the 1 mg/kg dose level as the MTD of INBRX-105. Patients in this part of the study were not pre-screened for PD-L1 expression. Eight out of 18 evaluable patients (44%), who were dosed at the 0.1 mg/kg dose level or above, achieved stable disease, with the greatest reduction in tumor volume observed to be 20% by RECISTv1.1. The longest duration on treatment with INBRX-105 was 41 weeks, or approximately nine and a half months. Notably, seven patients with stable disease tested positive for PD-L1 expression, with a minimum of 1% positivity as determined by immunohistochemistry (Range 1 to 95%), and the
results of one patient are pending. Four of these eight patients were refractory to or progressed on prior PD-1 checkpoint inhibitors.
Single agent dose expansion began enrolling during the first quarter of 2021. Part 2 of this trial will consist of 32 patients in expansion cohorts in each of non-small cell lung cancer, melanoma, head and neck squamous cell carcinoma, as well as a basket cohort of gastric or gastro-esophageal junction adenocarcinoma, renal cell carcinoma, and urothelial (transitional) cell carcinoma. Patients in the expansion cohorts for Part 2 of this trial will have to be positive for PD-L1 expression, as determined by immunohistochemistry, to qualify for enrollment.
Future Clinical Development Plan
We expect to achieve the maximal therapeutic benefit of INBRX-105 in combination with PD-1 checkpoint blockade. Our preclinical studies demonstrate that acute exposure to PD-L1 dependent 4-1BB agonism is sufficient to derive maximal anti-tumor activity when co-dosed with another PD-1 blocking agent at a target saturating dose. As such, a dose escalation cohort of INBRX-105 in combination with Keytruda is targeted to initiate enrollment during the second quarter of 2021 and we expect to announce initial data during the fourth quarter of 2021.
Part 4 of this trial will consist of combination expansion cohorts in each of non-small cell lung cancer, melanoma, head and neck squamous cell carcinoma, as well as a basket cohort of gastric or gastro-esophageal junction adenocarcinoma, renal cell carcinoma, and urothelial (transitional) cell carcinoma, which are positive for PD-L1 expression and are refractory to or progressed on prior PD-1 checkpoint inhibitors. Additionally, we are planning to evaluate INBRX-105 in combination with Keytruda in checkpoint inhibitor naive non-small cell lung cancer patients that are positive for PD-L1 expression. We expect to announce initial data from the Part 4 combination expansion cohorts during the third quarter of 2022.
tumors. Primary objectives of the Phase 1 trial are safety and tolerability, and the determination of the MTD and recommended Phase 2 dose of INBRX-105INBRX-106 as a single agent and INBRX-105 in combination with Keytruda.
Secondary objectives are serum exposure, immunogenicity, as measured by the frequency of anti-drug antibodies. Exploratory objectives includeantibodies, and clinical anti-tumoranti- tumor efficacy per RECIST (version 1.1) and immune RECIST based on response rate, duration of response, disease control rate, progression-free survival and overall survival, as well assurvival. Exploratory objectives will include evaluation of potential predictive diagnostic and pharmacodynamic biomarkers.
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | |
| Phase 1/2 INBRX-106 Study Design | |
| Phase 1/2 Study of Single Agent INBRX-106 and INBRX-106 in Combination with Keytruda in Patients with Locally Advanced or Metastatic Solid Tumors | |
| | |
| Part 1: INBRX-106 Single-Agent Dose Escalation |
⇨
⇨ | Part 2: INBRX-106 Single-Agent Dose Expansion |
⇨
⇨ | Part 3: Dose Escalation with Keytruda |
⇨
⇨ | Part 4: Dose Expansion with Keytruda | |
| Complete | Ongoing | Complete | Ongoing | |
|
N=20 All-comers |
N=52-64
CPI r/r: Basket Dose Regimen #1
Basket Dose Regimen #2
Basket Dose Regimen #3
|
N=21
|
N=136
CPI r/r: Basket PDL1+
NSCLC PDL1+
Basket, MMR-def/MSI-high
Uveal melanoma
| |
| | | NSCLC PD-L1+ (TPS≥50%) or TMB high (Ongoing) CPI r/r | | | | NSCLC TMB or PDL1 high (Ongoing) CPI r/r -Dose Regimen #1 a -Dose Regimen #2 b
Basket HNSCC, PDL1 + (CPS≥1) (ongoing) CPI naive | |
| a | | Alternating treatment: INBRX-106 0.3 mg/kg followed by Keytruda 400 mg/kg 3 weeks later; Alternates every three weeks | |
| b | | Priming: INBRX-106 0.3 mg/kg ⇨ 0.1 mg/kg Q3W + Keytruda | |
| | | | | | | | | | | |
OverviewPart 1 of Our Therapeutic Candidate Utilizing Unique Protein Engineering
INBRX-101
INBRX-101 isthe trial utilized a precisely engineered recombinant human AAT-Fc fusion protein therapeutic candidate that we are developingtraditional 3+3 algorithm for the treatment of patients with AATD. We believe INBRX-101 has the potentialsingle agent dose escalation from 0.0003 mg/kg to 3 mg/kg in twenty patients. INBRX-106 was observed to be dosed every three weeks, while maintaining patientsgenerally well tolerated in the normal range of AAT exposure,humans. The most common AEs reported for INBRX-106 were Grade 1 and 2 and notably, cutaneous toxicities, which would be a significant improvement for patients currently receiving weekly infusions. Through our proprietary engineering capabilities, we were able to overcome the challenges of producing AAT recombinantly and manufacturing at commercial scale.
Overview of AATD and Unmet Medical Need
AATD is an inherited disease that causes an increased risk of developing pulmonary disease defined by progressive loss of lung tissue and function and isare common immune-related AEs associated with decreased life expectancy. The pulmonary manifestations of AATD include the entire spectrum of emphysemaimmune checkpoint inhibitors. These mostly mild or moderate and disorders associated with chronic obstructive pulmonary disease. Patients with AATD harbor mutationslikely immune-related toxicities were in the AAT-encoding gene Serpin family A member 1 which causes AAT protein misfolding, loss of activity and retention in the liver. AAT is a protease inhibitor that primarily targets human neutrophil elastase, or NE, an enzyme that is released by white blood cells in response to infections and has the capacity to degrade normal tissues, especially in the lung, if not tightly controlled by AAT.
According to the Alpha-1 Foundation, the disease affects roughly 100,000 people in the United States, with a similar number of patients in Europe. Since AATD must be diagnosed using laboratory testing and cannot be diagnosed by symptoms or by a medical examination alone, it is believed that many individuals with AATD are likely undiagnosed or misdiagnosed. In April 2017, the FDA allowed for the marketing of direct-to-consumer tests that
provide genetic risk information for certain conditions, including AATD. We believe such tests will lead to earlier diagnosis and an increase in the number of patients identified with AATD.
The approved standard of care for AATD has been augmentation therapy using pdAAT and has not been substantially improved since 1987. Plasma-derived treatments are highly dependent on human donor blood supply, which is costly and can be limited. Due to the short half-life of pdAAT therapeutics, patients require weekly infusions to achieve and maintain serum concentration above the protective threshold, presumed to be 11 µM. However, even with frequent treatments, the AAT serum concentration following treatment remains considerably below the normal range of 20 to 50 µM. There is clinical evidence suggesting that maintenance of higher AAT serum trough levels may better protect against lung function decline. The graph below is an adaptation based on published data that depicts the pharmacokinetic profile of pdAAT. The published results from the RAPID trial revealed the correlation of serum AAT through levels and lung density decline in AATD patients. Additionally, there are other significant barriers to treatment for patients with AATD, including under-diagnosis, high cost of chronic augmentation therapy, and difficulties associated with administering intravenous infusions on a weekly basis. It has been reported that the global AATD augmentation therapy market size was valued at $1.1 billion in 2018 and is projected to reach $1.9 billion by the end of 2026.
Our Solution - INBRX-101
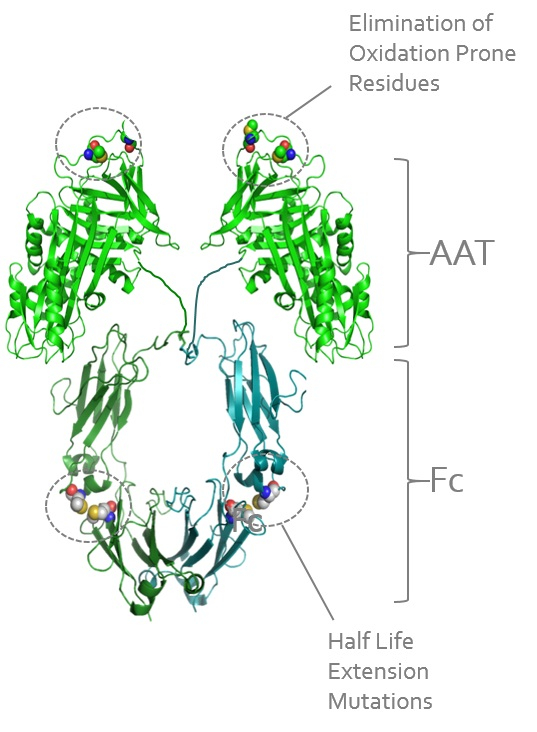
INBRX-101 is a recombinant AAT protein that is comprised of two human AAT molecules covalently linked to the Fc region of human immunoglobulin G4. AAT has proven difficult to develop recombinantly, often displaying loss of activity and experiencing accelerated degradation. We believe our novel approach has the potential to overcome these challenges in our clinical trials. Fusion of AAT to the Fc region allows for production using a standard antibody expression system, which, when combined with our proprietary AAT-function preserving purification process, generates substantial and potentially scalable yields of active recombinant AAT protein. In preclinical studies, we have observed the Fc region to significantly improve pharmacokinetic properties for INBRX-101 as compared to pdAAT therapeutics. Our clinical trials will seek to demonstrate that this extended exposure supports the potential for dosing every three weeks while maintaining patients in the normal range of AAT exposure, which in turn would reduce the frequency of annual infusions and could significantly improve patient quality of life.
In our preclinical studies, we compared INBRX-101 to pdAAT therapeutics across several parameters. INBRX-101 is intended to maximize the functional activity of AAT, particularly in the lung.
It is known that AAT is inactivated by oxidation, which can occur from the oxidative burst of neutrophils or inhalation of pollutants. Therefore, we designed INBRX-101 with amino acid changes in the AAT sequence to mitigate oxidation-induced inactivation,line with the goal of preventing loss of activity at the desired site of action.
The primary mechanism of action of INBRX-101 isthis candidate therapeutic. The maximum administered dose was 3 mg/kg and the MTD level was not reached.
Part 2 of the trial, single agent dose expansion, administered INBRX-106 in different dosing schedules to inhibit NE. In a preclinical assay, we measured the abilitypatients with tumor types responsive to checkpoint inhibitors. Part 2 treatment cohorts of INBRX-101 to inhibit NE by comparing INBRX-101 to the FDA-approved pdAAT, Prolastin-C,this trial were in the presencefollowing tumor types: NSCLC, melanoma, HNSCC, gastric or gastroesophageal junction adenocarcinoma, renal cell carcinoma, and urothelial (transitional) cell carcinoma. We expanded the enrollment for Part 2 in order to increase the dataset in the single agent cohorts and to enroll additional NSCLC patients. We expect to announce additional data from Part 2 in 2025.
In Parts 3 and 4 of NE and a substrate that becomes florescent when it is cleaved by NE. The vertical Y-axis shows the activity of NE as the rate of relative fluorescence units over time, orRFU/sec, while the X-axis shows the concentration of either INBRX-101 or Prolastin-C. The figure below shows that as the concentration of INBRX-101 or Prolastin-C increases, the RFU/sec decreases indicating that NEthis trial, INBRX-106 is being inhibited by both test articlesevaluated in combination with equivalent potency.
INBRX-101 vs. Prolastin-C: Neutrophil Elastase Inhibition
The primary pathologyKeytruda, a PD-1 blocking checkpoint inhibitor. KEYTRUDA® is a registered trademark of AATD is lung density decline due to NE degradationMerck Sharp & Dohme LLC, a subsidiary of elastin in the lung. Therefore, it is critical that INBRX-101 penetrates the physiological site where NE is degrading elastin, which is inside the lung. We performed a lung drug distribution study to measure the concentration of INBRX-101 and Prolastin-C in mice expressing human FcRn treated with either test article. The figure below shows the concentration of both INBRX-101 and Prolastin-C in the broncheoalveolar lavage fluid, or BALF (fluid in the lungs), of hFcRN transgenic mice 48 hours after intravenous treatment, with each circle representing an individually dosed animal.
INBRX-101 vs. Prolastin-C: MerckLung Exposure huFcRN Tg Mice
Clinical Data
We initiated a Phase 1 clinical trial in July 2019. The Phase 1 trial is an open-label, dose escalating trial and is designed as single ascending dose administrations followed by multiple ascending dose administrations of INBRX-101 with doses of 10 mg/kg, 40 mg/kg, 80 mg/kg or 120 mg/kg, administered to patients diagnosed with
AATD who are either treatment naïve& Co., Inc., Rahway, NJ, USA. In the all-comer Part 3 of the trial, INBRX-106 was escalated in combination with Keytruda and enrolled patients with locally advanced or previously treated with pdAAT therapeutics. As of July 24, 2020, and based on preliminary Phase 1 safety data in 5 patients, INBRX-101 has been well tolerated in humans, without any significant possibly- or probably-related AEs, and without any SAEs or DLTs.
The graph on the left below depicts the preliminary pharmacokinetic, or PK, profiles for the first five patients dosed with INBRX-101 at a dose of 10 mg/kg, demonstrating an estimated half-life of approximately 13 days. The PK profile of INBRX-101 in humansmetastatic solid tumors. It was observed to be consistentwell tolerated, with preclinical findings in cynomolgus monkeyspredominantly mild or moderate immune-related toxicities noted. We observed durable responses across multiple tumor types.
In Part 4, INBRX-106 combination expansion cohorts, we are enrolling patients who are positive for PD-L1 expression, as determined by immunohistochemistry, and rats. The graph on the right below depicts the resultspossess adequate hematologic and organ function, to qualify for enrollment. Most of a PK model based on preclinical data, to estimate the exposure of INBRX-101 over time when dosed at 120 mg/kg dosed every three weeks (green). This modeling data is overlaid on the published PK profile of pdAAT dosed weekly at the approved 60 mg/kg (blue). Based on this model and the observed consistency between the human PK data collected to date and preclinical studies, we expect that INBRX-101, dosed significantly less frequently than pdAAT,these patients will be ablecheckpoint refractory with NSCLC (both TPS>1% and >50%), and a basket of other indications as well as uveal melanoma. The most common AEs observed in this study related to maintain AAT serumINBRX-106 were infusion-related reaction, fatigue, rash, nausea, pruritus, and diarrhea. Most patients with related treatment emergent adverse events had events that were mild or moderate (Grade 1 or 2) in severity.
We continue to enroll patients with NSCLC and HNSCC in Part 4, both in combination with Keytruda. The initial data observed from 24 NSCLC patients who all had previous checkpoint inhibitor exposure was tumor reduction or stabilization of target lesions in more than half the patients. Of those patients, one complete response and four partial responses were observed. The initial data observed from 14 HNSCC patients, seven of which were checkpoint failures and seven of which were checkpoint naive, was tumor reduction of target lesions in half the patients. Of those patients, two complete responses and five partial responses were observed. We are in the normal range.
process of expanding these cohorts and expect to initiate at least one additional cohort by mid 2024. We expect the trial to be conducted in approximately 12 clinical sites in the United States, the United Kingdom and New Zealand and will enroll approximately 36 patients. Based on preclinical pharmacokinetic data, we have designed the dose escalation trial to administer INBRX-101 by intravenous infusion every three weeks with the intent to select a dosage necessary to achieve sustained, normal, serum levels of AAT within this dosing interval.
Primary endpoints of the trial are safety and tolerability. Secondary endpoints are serum exposure and immunogenicity, as measured by frequency of anti-drug antibodies. Exploratory endpoints include NE activity and its biomarkers. In addition, we will measure pharmacokinetic and pharmacodynamic biomarkers in BALF.
In May 2019, we entered into the Chiesi Option Agreement with Chiesi, pursuant to which we granted Chiesi an option to obtain an exclusive license to develop and commercialize INBRX-101 outside of the United States and Canada following completion of the Phase 1 trial.
As a result of the COVID-19 pandemic, including patient travel restrictions, we temporarily suspended enrollment in this trial but resumed enrollmentmore mature dataset during the fourththird quarter of 2020. The temporary suspension of enrollment was not related to any observed adverse events from our Phase 1 trial or required by any third party or regulatory authority. Rather, this suspension was made at our discretion2025 and was directly related to circumstances involving the COVID-19 pandemic. Assuming enrollment continues as we anticipate it will, we expect to announce initial Phase 1 data from the multi-dose cohorts during the fourth quarter of 2021. Assuming positive data from this trial, we believe we could initiate potential registration-enabling Phase 2 trials in late 2022.
Other sdAb Development Programs
We believe that the breadth of our technological platform and our strong intellectual property portfolio will enable us to generate multiple future clinical candidates representing significant future revenue opportunities. We plan to continue to advance our sdAb platform and other protein technologies in areasprovide an update at that are considered non-core or indications ancillary to oncology via out-licensing agreements and other corporate partnerships.time.
License and Collaboration Agreements
We have entered into various license and collaboration agreements. The license agreements discussed below pertain to intellectual property that we own and license to the parties identified below. We retain ownership of the intellectual property subject to the license agreements.
Elpiscience Agreements
We have entered into two different license agreements with Elpiscience Biopharmaceuticals, Inc., or Elpiscience. Each agreement is a standalone license to Elpiscience of a distinct and differentiated protein therapeutic candidate with a separate biological target.
On February 28, 2018, we entered into a license agreement, or the PD-L1 and 4-1BB License Agreement, with Elpiscience, pursuant to which we granted Elpiscience an exclusive license to our bi-specific therapeutic candidate designed to target PD-L1 and 4-1BB, also referred to as INBRX-105. On April 30, 2018, we entered into a license agreement, or the OX40 License Agreement, with Elpiscience, pursuant to which we granted Elpiscience an exclusive license to our multivalent protein therapeutic directed to the biological target OX40, or INBRX-106. The PD-L1 and 4-1BB License Agreement was terminated in February 2024 in conjunction with our decision to terminate the INBRX-105 program.
Each of the PD-L1 and 4-1BB License Agreement and OX40 License Agreement providesprovided Elpiscience with the right to further advance the respective therapeutic candidatescandidate through clinical trials, as well as to manufacture and commercialize these therapeutic candidates in China, Hong Kong, Macau and Taiwan. The PD-L1 and 4-1BB License Agreement and OX40 License Agreement also requirerequired us to provide Elpiscience with know-how and materials specific to INBRX-105 and INBRX-106, respectively, including process development and manufacturing data and information necessary to develop INBRX-105 and INBRX-106. Additionally, pursuant to each of the license agreements, Elpiscience granted to us a royalty-free, worldwide, non-exclusive research license to intellectual property created by Elpiscience that incorporates intellectual property we licensed to Elpiscience. In the PD-L1 and 4-1BB License Agreement and OX40 License Agreement, we also agreed to negotiate an agreement to supply Elpiscience with INBRX-105 and INBRX-106, respectively, for their development in China, Hong Kong, Macau and Taiwan.
As payment for the license granted in the PD-L1 and 4-1BB License Agreement, we received a non-refundable upfront payment of $2.5 million and reimbursement of $3.0 million for certain costs we incurred. In December 2018, we received the first milestone payment of $2.0 million under the PD-L1 and 4-1BB License Agreement. Additionally, we received reimbursement for specific supply-related costs in the amounts of $1.7 million under the
PD-L1 and 4-1BB License Agreement. Following its termination, there are no further payments under this agreement.
As payment for the license granted in the OX40 License Agreement, we received a non-refundable fee of $2.5 million, payable in two installments, and reimbursement of $3.4 million for certain toxicology study costs and chemistry, manufacturing and controls costs. In December 2018,Additionally, we received reimbursement for specific supply-related costs in the first milestone paymentamounts of $2.0$1.5 million under the PD-L1 and 4-1BBOX40 License Agreement. We are also eligible to receive specified developmental and commercial milestone payments of up to an aggregate of $100.0 million under each of the PD-L1 and 4-1BB License Agreement and the OX40 License Agreement. Additionally, we received reimbursement for specific supply-related costs in the amounts of $1.7 million and $1.5 million related to the PD-L1 and 4-1BB License Agreement and the OX40 License Agreement respectively. Further, each license agreement provides us within addition to the right to receive percentage tiered royalties on future product sales with rates in the high single-digits.
Each of the PD-L1 and 4-1BB License Agreement and the OX40 License Agreement containscontained representations and warranties, insurance, indemnification and confidentiality provisions customary for similar agreements and arrangements. The terms of the PD-L1 and 4-1BB License Agreement and the OX40 License Agreement commenced on February 28, 2018 and April 30, 2018, respectively, and expire on a country-by-country basis upon the full expiration of Elpiscience’s milestone- and royalty-based payment obligations under each license agreement. The royalty obligations expire upon the later of (i) the expiration of the last valid patent claim for the applicable product in the relevant jurisdiction, and (ii) 12 years following that date of the first commercial sale of the applicable product in the relevant jurisdiction. Each license agreement may be terminated by (i) either party for the uncured material breach of the other party, (ii) either party for bankruptcy or other insolvency proceedings of the other party, and (iii) Elpiscience at any time upon 90 days’ written notice to us. The PD-L1 and 4-1BB License Agreement was terminated in February 2024 in conjunction with our decision to terminate the INBRX-105 program. As of December 31, 20202023, and based on current plans, the last to expire issued patent (assuming the issuance of a patent based on our pending patent application) previously licensed under the PD-L1 and 4-1BB License Agreement is projected to expire in January 2037 and the last to expire issued patent (assuming the issuance of a patent based on our pending patent application) licensed under the OX40 License Agreement is projected to expire in August 2039.2039, in each case absent any extension of term.
Chiesi Option Agreement
In May 2019, we entered into the Chiesian Option Agreement with Chiesi Farmaceutici S.p.A., or Chiesi, which was later amended by the First Amendment to Option Agreement, dated August 19, 2019, or the Chiesi Option Agreement, pursuant to which we granted Chiesi an exclusive option to obtain an exclusive license to develop and commercialize INBRX-101 outside of the United States and Canada. UnderAdditionally, the Chiesi Option Agreement provided Chiesi with a right of negotiation for INBRX-101 development and commercialization rights in the United States and Canada in the event we engaged in discussions with any third parties for such rights during the term of the Chiesi Option Agreement or, as applicable, the term of a definitive exclusive license agreement between Chiesi and the Company. In August 2019, under the terms of the Chiesi Option Agreement, we received a one-time, non-refundable option initiation payment of $10.0 million in August 2019. If Chiesi exercises its option undermillion. Pursuant to the Chiesi Option Agreement, thenresearch and development services would be performed for Chiesi must pay us a one-time, non-refundable fee of $12.5 million uponduring the effective date of the definitive agreement granting Chiesi the exclusive license.
The option period, under the Chiesi Option Agreement began on August 19, 2019. The option period under the Chiesi Option Agreement expireswhich would continue until 60 days following the last-to-occur of (a) our(i) the Company’s delivery to Chiesi of the trial phase data for the first Phase I Clinical Trial for INBRX-101 (including the complete clinical study report); (b) our, (ii) the Company’s delivery to Chiesi of the finalized minutes from the definitive FDA scientific advice meeting conducted following completion of such Phase I Clinical Trial with the use of the full clinical study report if required by the FDA;FDA, and (c) our(iii) the Company’s delivery to Chiesi of the finalized minuteswritten scientific advice from the definitive parallel EMA-HTA scientific advice meeting conductedEMA following completion of such Phase I Clinical Trial with full use of the clinical study report if required by the EMA. The
On July 24, 2023, we received written scientific advice from the EMA which confirmed CT lung densitometry as the established primary regulatory endpoint to support a marketing authorization application in the European Union, or EU, for the treatment of emphysema secondary to AATD. We provided a copy of the EMA scientific advice to Chiesi upon receipt which, as described above, fulfilled the necessary deliverables to Chiesi and triggered the start of its 60-day option period window. On September 18, 2023, Chiesi declined to exercise its option for the ex-North American rights, resulting in the expiration of the Chiesi Option Agreement may be terminated by (i) Chiesi upon 30 days’ written notice to us, (ii) either party for(upon the uncured material breachlapse of the other party,option period), and (iii) either party for bankruptcy or other insolvency proceedings of the other party. The Chiesi Option Agreement contains representations, warrantieswith us retaining global rights to develop and indemnification provisions customary for similar agreements and arrangements.commercialize INBRX-101.
Transcenta License Agreement and Technical Services Agreements
On June 21, 2017, we entered into a license agreement, or the Transcenta License Agreement, with Transcenta Holding, Ltd. (formerly Hangzhou Just Biotherapeutics Co., Ltd.), or Transcenta, pursuant to which we granted to Transcenta exclusive, non-transferable rights to develop, manufacture, and sell its products containing our mono-specific protein therapeutic candidate, designed to target DR5, or INBRX-109, or a derivative thereof, within China, Hong Kong, Macau and Taiwan. We also agreed to provide Transcenta with the know-how and materials, including assays and cell lines, necessary to develop the therapeutic candidate. Additionally, pursuant to the Transcenta License Agreement, Transcenta granted to us a royalty-free, worldwide, non-exclusive research license to intellectual property created by Transcenta that incorporates intellectual property we licensed to Transcenta. As payment for the license we granted in the Transcenta License Agreement, we received a non-refundable upfront payment of $2.5 million. In August 2018, the Company achieved the first milestone under the contract for $0.5 million. In June 2019, the Company achieved another milestone under the contract for $1.0 million. Additionally, we are eligible to receive certain developmental and commercial milestone payments of up to an aggregate of approximately $100.0 million, as well as percentage tiered royalties on future product sales with rates ranging between the high single-digits to the low teens.
The Transcenta License Agreement contains representations and warranties, insurance, indemnification and confidentiality provisions customary for similar agreements and arrangements. The term of the Transcenta License Agreement commenced on June 21, 2017, and expires on a country-by-country basis upon the full expiration of Transcenta’s milestone- and royalty-based payment obligations under the Transcenta License Agreement. The royalty payment obligations expire on a product-by-product and country-by-country basis at such time that there is (i) no valid patent claim for the applicable product in the relevant jurisdiction, and (ii) no Generic Product (as defined in this Transcenta License Agreement) of an applicable product available in the relevant jurisdiction. As of December 31, 20202023, and based on current plans, the last to expire issued patent (assuming the issuance of a patent based on our pending patent application) licensed under the Transcenta License Agreement is projected to expire in July 2036.2036, absent any extension of term. The Transcenta License Agreement may be terminated by (i) either party for the uncured material breach of the other party, (ii) either party for bankruptcy or other insolvency proceedings of the other party, and (iii) Transcenta at any time upon 90 days’ written notice to us. In May 2020, we notified Transcenta of potential disputes or disagreements regarding its performance under the Transcenta License Agreement. We are currently in discussions with Transcenta regarding these matters.
On March 19, 2018, we also entered into a technical services agreement, or the March TSA, with Transcenta pursuant to which we provided reasonable assistance with the drug substance transfer between our contract manufacturer and Transcenta. We were reimbursed $0.7 million in April 2018 for the March TSA. The March TSA
had a six monthsix-month term and expired in September 2018. On October 6, 2018, we entered into a second technical services agreement, or the October TSA, to provide reasonable assistance pertaining to the transfer of the drug substance between our contract manufacturer and Transcenta. We were reimbursed $0.4 million in October 2018 for the October TSA. The October TSA also had a six monthsix-month term and expired in April 2019.
Celgene Agreement
On July 1, 2013, we entered into a license agreement with Celgene, a Bristol Meyers Squibb Company, or Celgene, as amended on November 23, 2018, or the Celgene Agreement, pursuant to which we granted Celgene an exclusive, global license for the development, manufacture and commercialization of our proprietary CD47 binding domain, or the Celgene Licensed Intellectual Property. Per the terms of the Celgene Agreement, Celgene is operationally and financially responsible for the development, manufacturing and commercialization activities of Celgene Licensed Intellectual Property and any additional related antibodies covered by the Celgene Agreement.
As payment for the license granted in the Celgene Agreement, we may be eligible to receive development and regulatory milestones of up to an aggregate of $934.1 million, assuming the achievement of all potential milestones in the Celgene Agreement, as well as percentage tiered royalties based on future worldwide sales, with rates ranging from between the high single-digits to the low teens, subject to potential reduction when and if comparable third-party products attain certain levels of competitive market share (on a country-by-country basis) and, subject to certain limitations, payments to third parties for third-party intellectual property rights. Celgene’s royalty obligations expire (on a country-by-country basis) upon the later of (i) the expiration of the last valid patent claim for the applicable Celgene Licensed Intellectual Property or related product in a country, and (ii) 12 years following that date of the first commercial sale of the applicable Celgene Licensed Intellectual Property or related product in a country. We
are obligated to pay 2% of future amounts received under the Celgene Agreement to advisors who assisted us with the negotiations and other matters in connection with the Celgene Agreement.
The Celgene Agreement contains representations and warranties, insurance, indemnification and confidentiality provisions customary for similar agreements and arrangements. In addition, Celgene has the sole right, but not the obligation, to enforce the intellectual property included in the Celgene Agreement.
The term of the Celgene Agreement commenced on July 1, 2013 and expires on a licensed product-by-licensed product and country-by-country basis on the date of expiration of Celgene’s applicable royalty obligations. As of December 31, 20202023 and based on our current plans, the last to expire issued patent licensed under the Celgene Agreement is projected to expire in 2033, absent any extension of term. The Celgene Agreement may be terminated by (i) either party for the uncured material breach of the other party, (ii) either party for bankruptcy or other insolvency proceedings of the other party, (iii) Celgene at any time upon 30 days’ written notice to us, and (iv) us, upon Celgene’s, or any of its affiliates’, legal challenge to the validity or enforceability of a licensed patent.
bluebird bio2seventy Agreements
The 2018 bluebird2seventy Agreement
On December 20, 2018, we entered into aan exclusive license agreement or the 2018 bluebird Agreement, with bluebird bio, Inc., or bluebird, pursuant to which we granted to bluebird an exclusive license to use our proprietary sdAb platform to research, develop and commercialize chimeric antigen receptor, or CAR, T-cell therapies. In November 2021, bluebird assigned the agreement, which we refer to as the 2018 2seventy Agreement, to its affiliate, 2seventy bio, Inc., or 2seventy, in connection with an internal restructuring and subsequent spin-out of 2seventy. Under the terms of the 2018 bluebird2seventy Agreement, we provided bluebird2seventy the exclusive worldwide rights to develop, manufacture and commercialize certain cell therapy products containing sdAbs directed to various cancer targets. As payment for the license granted in the 2018 bluebird2seventy Agreement, we received a non-refundable upfront payment of $7.0 million.million in January 2019. We are also entitled to receive certain developmental milestone payments of up to an aggregate of $51.5 million per therapeutic, as well as percentage tiered royalties on future product sales with rates in the mid-single-digits. In August 2021, we received a $2.0 million milestone payment under this agreement.
The 2018 bluebird2seventy Agreement contains representations and warranties, indemnification and confidentiality provisions customary for similar agreements and arrangements. The term of the 2018 bluebird2seventy Agreement commenced on December 20, 2018, and expires on a product-by-product and country-by-country basis upon the full expiration of bluebird’s2seventy’s milestone- and royalty-based payment obligations under the 2018 bluebird2seventy Agreement. The royalty obligations under the 2018 bluebird2seventy Agreement expire on a product-by-product and country-by-country basis on the later of (i) the expiration of the last valid patent claim for the applicable product in the relevant jurisdiction; (ii) the date on which any applicable regulatory, pediatric, orphan drug or data exclusivity, which provides bluebird2seventy with the exclusive right to market the applicable product in the relevant jurisdiction, expires; and (iii) 12 years following that date of the first commercial sale of the applicable product in a country. As of December 31, 20202023 and based on current plans, the last to expire issued patent (assuming the issuance of one or more patents based on our pending patent applications) licensed under the 2018 bluebird2seventy Agreement is projected to expire in May 2040.2040, absent any extension of term. The 2018 bluebird2seventy Agreement may be terminated by (i) either party for the uncured material breach of the other party,
(ii) either party for bankruptcy or other insolvency proceedings of the other party, and (iii) bluebird2seventy at any time upon 30 days’ written notice to us.
The 2020 bluebird2seventy Agreement
On June 9, 2020, we entered into an option and license agreement or the 2020 bluebird Agreement, with bluebird, pursuant to which we granted to bluebird exclusive worldwide rights to develop binders and cell therapy products containing sdAbs directed to specified targets, consisting of two initial programs and up to an additional 8 programs. Inhibrx retains all rights to the specific sdAbs outside of the cell therapy field. WeThis agreement, which we refer to as the 2020 2seventy Agreement, was also assigned to 2seventy in November 2021 in connection with bluebird’s internal restructuring and subsequent spin-out of 2seventy. In June 2020, we received a non-refundable upfront option fee of $0.2 million in connection with each of the two initial programs, or $0.4 million in aggregate, and we are entitled to an upfront option fee for each additional program. WeIn June 2022, 2seventy selected a third program and paid a non-refundable upfront option fee of $0.2 million in exchange for a development license. Under each of the three programs, we also
granted to bluebird an option toin which 2seventy may acquire an exclusive license with respect to all binders and cell therapy products developed under the 2020 bluebird2seventy Agreement, which entitles us to additional fees upon exercise of the option.
bluebird 2seventy may exercise its option on a program-by-program basis during the option term. Additionally, bluebird2seventy may extend the option term for up to six months in the event that there are additional bona fide development activities that bluebird2seventy desires to undertake. We are also entitled to receive certain developmental milestone payments of up to an aggregate of $51.5 million per therapeutic, as well as percentage tiered royalties on future product sales with rates in the mid-single-digits.mid-single digits.
The 2020 bluebird2seventy Agreement contains representations and warranties, indemnification and confidentiality provisions customary for similar agreements and arrangements. The term of the 2020 bluebird2seventy Agreement commenced on June 9, 2020, and expires on a product-by-product and country-by-country basis upon the full expiration of bluebird’s2seventy’s milestone- and royalty-based payment obligations under the 2020 bluebird2seventy Agreement. The royalty obligations under the 2020 bluebird2seventy Agreement expire on a product-by-product and country-by-country basis on the later of (i) the expiration of the last valid patent claim for the applicable product in the relevant country; (ii) the date on which any applicable regulatory, pediatric, orphan drug or data exclusivity, which provides bluebird2seventy with the exclusive right to market the applicable product in the relevant country, expires; and (iii) 12 years following that date of the first commercial sale of the applicable product in such country. As of December 31, 2020 and based on current plans, the last to expire issued patent (assuming the issuance of one or more patents based on our pending patent applications) licensed under the 2020 bluebird Agreement is projected to expire in May 2040. The 2020 bluebird2seventy Agreement may be terminated by (i) either party for the uncured material breach of the other party, (ii) either party for bankruptcy or other insolvency proceedings of the other party, and (iii) bluebird2seventy at any time upon prior written notice within a specified number of days before the termination.
WuXi Agreement
On August 28, 2018, we entered into an Amended and Restated Master Services Agreement, as amended by Amendment No. 1 to Amended and Restated Master Services Agreement dated December 11, 2019, or the WuXi Agreement, with WuXi Biologics (Hong Kong) Limited, or WuXi,In May 2021, pursuant to which we agreed,the option extension terms in the 2020 2seventy Agreement, bluebird requested to extend the option term for three years and subjectone of the initial programs by an additional six months in exchange for an option extension fee of $0.1 million. In August 2021, pursuant to certain conditions,the option exercise terms in the agreement, bluebird exercised its option to exclusively use WuXi to manufacture our therapeutic candidates for which we plan to first initiate clinical studies outside of China. Under the WuXi Agreement, WuXi and certain of its affiliates will provide biologics development and manufacturing services on a project-by-project basis.
Per the termslicense one of the WuXi Agreement, we will own all intellectual property created, developed or reducedinitial programs in exchange for an option exercise fee of $2.1 million, the payment of which was received in October 2021. In May 2023, 2seventy declined to practice by WuXi in the course of providing services to us; provided that, certain intellectual property created or developed by WuXi may be owned exclusively by WuXi if this intellectual property (i) relates to generally applicable experimental methods, (ii) relates to generally applicable manufacturing processes, or (iii) is derivative of WuXi’s pre-existing intellectual property.
Per the termsexercise its option for one of the WuXi Agreement, fees will be mutually agreed upon on a project-by-project basis. We also may be eligible to receive discounts in the low- to mid-single digits for certain projects and services per the terms of the WuXi Agreement.
The WuXi Agreement contains representations and warranties, insurance, indemnification and confidentiality provisions customary for similar agreements and arrangements.
The term of the WuXi Agreement commenced on August 28, 2018 and ends on the date that either party elects to terminate it by providing 30-day written notice to the other party, provided that the WuXi Agreement may only be terminated in this manner if all services requested by pending work orders under the WuXi Agreement have been completed. Work orders commence on the date indicated in each such work order and will terminate upon completion of the services requested therein. Work orders may be terminated by (i) us at any time upon three months’ written notice to WuXi, and (ii) either party for the uncured material breach of the other party, provided that the material breach was not caused by the party terminating the work order.two initial programs.
Intellectual Property
We strive to protect the proprietary technology and information commercially or strategically important to our business. We seek to obtain and maintain, patent rights intended to cover the technologies incorporated into, or used to produce, our therapeutic candidates, the compositions of matter of our therapeutic candidates and their methods of use and manufacture, as well as other inventions that are important to our business. We also seek to obtain strategic or commercially valuable patent rights in the United States and other jurisdictions.
To cover our proprietary technologies and our current pipeline of proprietary products and related methods, such as methods of use, we have filed patent applications representing 3942 patent families. As of December 31, 2020,2023, our patent estate which is solely owned, included 1625 issued United States patents, 1828 United States pending non-provisional patent applications, 146 United States pending provisional patent applications, 13 pending Patent Cooperation Treaty, or PCT, applications, 164233 issued foreign patents and 213367 foreign patent applications currently pending in various foreign jurisdictions.
Specifically, we own more than sixseven patent families with claims directed to various sdAb and/or multivalent therapeutic antibodies including, for example, our INBRX-105, INBRX-106 and INBRX-109 therapeutic candidates, and related methods of using the same to treat diseases, e.g., cancer, inflammatory disease, or infectious disease. Patent applications in these families are pending in multiple jurisdictions, including, for example, the United States, Australia, European Patent Organization, Canada, China, Japan, Korea, and Russia; as well as PCT applications and several U.S. provisional applications. Patents in these patent families, if granted, are expected to expire between 2036 and 2039,2043, depending upon their respective filing dates and absent any patent term adjustments or extensions.
We also own twothree patent families directed to protease inhibitor fusion proteins including, for example, our INBRX-101 therapeutic candidate. SevenNine United States and 5367 foreign patents (Australia, Canada, Mexico, Europe (validated in 3937 countries), Hong Kong, India, Israel, Japan, South Korea, Mexico, New Zealand, Russia, and Russia)Ukraine) were granted in these families. These patents are expected to expire in 2032 or 2035, absent any patent
term extension. Additional patents in thisthese patent family,families, if granted, are expected to expire in 2032, 2035, or 2035,2043, depending upon their respective filing dates and absent any patent term adjustments or extensions.
Additionally, we own two patent families relating to the Celgene Licensed Intellectual Property that are licensed to Celgene. Two United States and 6390 foreign patents (Australia, Chile, Colombia, China, Europe (validated in up to 39 countries), Eurasia (validated in Russia), Hong Kong, Indonesia, Israel, Japan, South Korea, Mexico, Malaysia, Macao, New Zealand, Peru, Philippines, Russia, Singapore, South Africa, Ukraine, and Ukraine)Vietnam) have been granted in these families and include claims directed to anti-CD47 monoclonal antibodies and/or methods of using the same to treat cancer. These patents are expected to expire in 2033, absent any patent term extension. Patent applications in these families are pending in multiple jurisdictions, including, for example, the United States, Australia, European Patent Organization, Canada, China, Eurasia, Japan, and South Korea, and Russia.Korea. Additional patents in these patent families, if granted, are expected to expire in 2033, depending upon their respective filing dates and absent any patent term adjustments or extensions.
The following patents (including expected 20 year20-year expiration dates) relate to our fusion protein technologies.
| | | | | | | | | | | | | | |
| Jurisdiction | Patent Number | Expected Expiration Date | Type | Title |
| US | 8,986,688 | 6/28/2032 | Composition and method | WAP DOMAIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| US | 9,914,765 | 6/28/2032 | Composition | WAP DOMAIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| Mexico | 356433 | 6/28/2032 | Composition and use | WAP DOMAIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| US | 8,980,266 | 6/28/2032 | Composition and method | SERPIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| US | 9,920,109 | 4/6/2034 | Method | SERPIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| US | 10,730,929 | 6/28/2032 | Composition | SERPIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| US | 10,723,785 | 6/28/2032 | Composition | SERPIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| US | 10,400,029 | 6/28/2032 | Composition | SERPIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| US | 11,046,752 | 6/28/2032 | Method | SERPIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| US | 11,827,691 | 6/28/2032 | Method | SERPIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| Australia | 2012275287 | 6/28/2032 | Composition, method and use | SERPIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| Australia | 2017279724 | 6/28/2032 | Method | SERPIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| Australia | 2015339507 | 10/27/2035 | Composition, method | SERPIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| Australia | 2019202904 | 6/28/2032 | Composition, method and use | SERPIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| Australia | 2021202131 | 6/28/2032 | Composition, method and use | SERPIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| Canada | 2839619 | 6/28/2032 | Composition, method and use | SERPIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| | | | | | | | | | | | | | |
| Europe (validated in AL, AT, BA, BE, BG, CH, CY, CZ, DE, DK, EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LI, LT, LU, LV, MC, ME, MK, MT, NL, NO, PL, PT, RO, RS, SE, SK, SM, TR) | 2726092 | 6/28/2032 | Use | SERPIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| Hong Kong | 1197544 | 6/28/2032 | Use | SERPIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| India | 373030 | 6/28/2032 | Composition | SERPIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| India | 366213 | 10/27/2035 | Composition | SERPIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| Israel | 230209 | 6/28/2032 | Use | SERPIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| Japan | 6674604 | 6/28/2032 | Method | SERPIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| Japan | 6737781 | 6/28/203210/27/2035 | MethodUse | SERPIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| Korea | 10-2084944 | 6/28/2032 | Composition | SERPIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| Korea | 10-2231139 | 6/28/2032 | Composition | SERPIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| Mexico | 356517 | 6/28/2032 | Composition and use | SERPIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| Mexico | 381183 | 6/28/2032 | Method | SERPIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| New Zealand | 619023 | 6/28/2032 | Composition and use | SERPIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| New Zealand | 727846 | 6/28/2032 | Method | SERPIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| New Zealand | 744257 | 6/28/2032 | Method | SERPIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| Russia | 2642310 | 6/28/2032 | Composition and use | SERPIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| Russia | 2698655 | 6/28/2032 | Method | SERPIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| Russia | 2728861 | 6/28/2032 | Composition and use | SERPIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| Russia | 2727452 | 6/28/2032 | Composition and use, method | SERPIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| Russia | 2746550 | 10/27/2035 | Use | SERPIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| Ukraine | 124083 | 6/28/2032 | Composition and use | SERPIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
| Ukraine | 127305 | 10/27/2035 | Composition and use | SERPIN FUSION POLYPEPTIDES AND METHODS OF USE THEREOF |
The following patentpatents (including expected 20 year20-year expiration date) relatesrelate to our single domain antibody technologies.
| | | | | | | | | | | | | | |
| Jurisdiction | Patent Number | Expected Expiration Date | Type | Title |
| US | 10,526,397 | 2/24/2036 | Composition | NON-IMMUNOGENIC SINGLE DOMAIN ANTIBODIES |
| AU | 2016209247 | 2/7/2036 | Composition | NON-IMMUNOGENIC SINGLE DOMAIN ANTIBODIES |
| JP | 7001474 | 2/7/2036 | Composition | NON-IMMUNOGENIC SINGLE DOMAIN ANTIBODIES |
The following patents (including expected 20 year20-year expiration dates) relate to our multivalent therapeutic antibody technologies.
| | | | | | | | | | | | | | |
| Jurisdiction | Patent Number | Expected Expiration Date | Type | Title |
| US | 10,308,720 | 7/18/2036 | Composition | MULTIVALENT AND MULTISPECIFIC DR5-BINDING FUSION PROTEINS |
| US | 11,117,973 | 11/22/2036 | Method | MULTIVALENT AND MULTISPECIFIC DR5-BINDING FUSION PROTEINS AND METHODS OF TREATING NEOPLASMS |
| AU | 2,016,291,701 | 7/18/2036 | Composition and use | MULTIVALENT AND MULTISPECIFIC DR5-BINDING FUSION PROTEINS |
| CN | ZL201680041274.2 | 7/18/2036 | Composition and use | MULTIVALENT AND MULTISPECIFIC DR5-BINDING FUSION PROTEINS |
EP (Validated in: AL, AT, BA, BE, BG, CH, CY, CZ, DE, DK, EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LI, LT, LU, LV, MC, ME, MK, MT, NL, NO, PL, PT, RO, RS, SE, SI, SK, SM, TR) | 3322734 | 7/18/2036 | Composition and use | MULTIVALENT AND MULTISPECIFIC DR5-BINDING FUSION PROTEINS |
| HK | 1254433 | 7/18/2036 | Composition and use | MULTIVALENT AND MULTISPECIFIC DR5-BINDING FUSION PROTEINS |
| ID | P000075513 | 7/18/2036 | Composition and use | MULTIVALENT AND MULTISPECIFIC DR5-BINDING FUSION PROTEINS |
| IL | 256772 | 7/18/2036 | Composition and use | MULTIVALENT AND MULTISPECIFIC DR5-BINDING FUSION PROTEINS |
| IN | 401979 | 7/18/2036 | Composition and use | MULTIVALENT AND MULTISPECIFIC DR5-BINDING FUSION PROTEINS |
| JP | 6807606 | 7/18/2036 | Composition and method | MULTIVALENT AND MULTISPECIFIC DR5-BINDING FUSION PROTEINS |
| JP | 7244938 | 7/18/2036 | Composition and method | MULTIVALENT AND MULTISPECIFIC DR5-BINDING FUSION PROTEINS |
| MX | 400419 | 7/18/2036 | Composition and use | MULTIVALENT AND MULTISPECIFIC DR5-BINDING FUSION PROTEINS |
| RU | 2748620 | 7/18/2036 | Composition and use | MULTIVALENT AND MULTISPECIFIC DR5-BINDING FUSION PROTEINS |
| SG | 11201800223W | 7/18/2036 | Composition and use | MULTIVALENT AND MULTISPECIFIC DR5-BINDING FUSION PROTEINS |
| ZA | 2018/00238 | 7/18/2036 | | MULTIVALENT AND MULTISPECIFIC DR5-BINDING FUSION PROTEINS |
| | | | | | | | | | | | | | |
| US | 10,093,742 | 7/22/2036 | Composition and method | MULTIVALENT AND MULTISPECIFIC GITR-BINDING FUSION PROTEINS |
| US | 10,844,129 | 7/22/2036 | Composition and method | MULTIVALENT AND MULTISPECIFIC GITR-BINDING FUSION PROTEINS |
| AU | 2,016,297,249 | 7/22/2036 | Composition, method and use | MULTIVALENT AND MULTISPECIFIC GITR-BINDING FUSION PROTEINS |
| NZ | 739130 | 7/22/2036 | Composition, method and use | MULTIVALENT AND MULTISPECIFIC GITR-BINDING FUSION PROTEINS |
| | | | |
| | | | |
| | | | |
| | | | |
| | | | |
| US | 11,117,972 | 5/4/2037 | Composition | MULTIVALENT AND MULTISPECIFIC OX40- BINDING FUSION PROTEINS |
| ID | P000085528 | 1/11/2037 | Composition | MULTIVALENT AND MULTISPECIFIC OX40-BINDING FUSION PROTEINS |
| IL | 260,531 | 1/11/2037 | Composition and use | MULTIVALENT AND MULTISPECIFIC OX40-BINDING FUSION PROTEINS |
| JP | 7,328,761 | 1/11/2037 | Composition and use | MULTIVALENT AND MULTISPECIFIC OX40-BINDING FUSION PROTEINS |
| RU | 2,773,052 | 7/22/2036 | Composition and use | MULTIVALENT AND MULTISPECIFIC OX40- BINDING FUSION PROTEINS |
| US | 11,447,556 | 2/8/2040 | Composition | MULTIVALENT AND MULTISPECIFIC OX40- BINDING FUSION PROTEINS |
| RU | 2,802,070 | 8/12/2039 | Composition, method and use | OX40-BINDING POLYPEPTIDES AND USES THEREOF |
| US | 10,501,551 | 8/14/2037 | Composition and method | MULTIVALENT AND MULTISPECIFIC 41BB-BINDING FUSION PROTEINS |
| US | 11,566,078 | 2/23/2038 | Composition | PDL1-BINDING PROTEINS (AS AMENDED) |
| AU | 2,017,207,742 | 1/11/2037 | Composition and method | MULTIVALENT AND MULTISPECIFIC 41BB-BINDING FUSION PROTEINS |
| ID | P000089844 | 1/11/2037 | Composition, method and use | MULTIVALENT AND MULTISPECIFIC 41BB-BINDING FUSION PROTEINS |
| IL | 260,530 | 1/11/2037 | Composition and use | MULTIVALENT AND MULTISPECIFIC 41BB-BINDING FUSION PROTEINS |
| JP | 7,022,993 | 1/11/2037 | Composition and method | MULTIVALENT AND MULTISPECIFIC 41BB-BINDING FUSION PROTEINS |
| RU | 2,789,648 | 1/11/2037 | Composition, method and use | MULTIVALENT AND MULTISPECIFIC 41BB-BINDING FUSION PROTEINS |
| US | 11,434,297 | 12/24/2040 | Composition, method and use | CD123-BINDING POLYPEPTIDES AND USES THEREOF |
| US | 11,560,428 | 12/4/2040 | Composition and use | CD33-BINDING POLYPEPTIDES AND USES THEREOF |
| ID | P000090304 | 12/4/2040 | Composition and use | CD33-BINDING POLYPEPTIDES AND USES THEREOF |
| JP | 7,164,544 | 4/11/2038 | Composition, method and use | MULTISPECIFIC POLYPEPTIDE CONSTRUCTS HAVING CONSTRAINED CD3 BINDING AND METHODS OF USING THE SAME |
| NZ | 758,264 | 4/11/2038 | Composition, method and use | MULTISPECIFIC POLYPEPTIDE CONSTRUCTS HAVING CONSTRAINED CD3 BINDING AND METHODS OF USING THE SAME |
The following patents (including expected 20 year20-year expiration dates) relate to our CD47 antibody technologies.
| | | | | | | | | | | | | | |
| Jurisdiction | Patent Number | Expected Expiration Date | Type | Title |
| US | 9,045,541 | 2/6/2033 | Composition | CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| US | 9,663,575 | 2/6/2033 | Method | CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| | | | | | | | | | | | | | |
| Australia | 2013217114 | 2/6/2033 | Composition and method | CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| Chile | 60943 | 2/6/2033 | | CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| | | | |
| China | ZL201380017900.0 | 2/6/2033 | Composition and use | CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| Colombia | 14171778 | 2/6/2033 | | CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| Costa Rica | 4109 | 2/6/2033 | | CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| Eurasia (validated in Russia) | 34778 | 2/6/2033 | | CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| | | | | | | | | | | | | | |
| Europe (validated in AL, AT, BA, BE, BG, CH, CY, CZ, DE, DK, EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LI, LU, LV, MC, ME, MK, MT, NL, NO, PL, PT, RO, RS, SE, SI, SK, SM, TR) | 2812443 | 2/6/2033 | Composition and use | CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| Hong Kong | 1205195 | 2/6/2033 | | CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| Indonesia | IDP00005873 | 2/6/2033 | | CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| Israel | 233934 | 2/6/2033 | Composition and use | CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| India | 395453 | 2/6/2033 | | CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| Japan | 6273212 | 2/6/2033 | Composition and use | CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| Korea | 10-2100388 | 2/6/2033 | Composition and use | CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| Korea | 10-2338833 | 2/6/2033 | | CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| Mexico | 360772 | 2/6/2033 | Composition and use | CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| Malaysia | MY-169341-A | 2/6/2033 | | CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| New Zealand | 628314 | 2/6/2033 | Composition and use | CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| Philippines | 1/2014/501758 | 2/6/2033 | | CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| Singapore | 11201404638S | 2/6/2033 | | CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| Ukraine | 116772 | 2/6/2033 | | CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| South Africa | 201405864 | 2/6/2033 | | CD47 ANTIBODIES AND METHODS OF USE THEREOF |
Hong KongVietnam | HK120519510028772 | 2/6/2033 | | CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| Australia | 2013377886 | 8/6/2033 | Composition, method, and use | NON-PLATELET DEPLETING AND NON-RED BLOOD CELL DEPLETING CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| | | | | | | | | | | | | | |
ChinaAustralia | ZL201380075409.32019200942 | 8/6/2033 | Composition, method, and use | NON-PLATELET DEPLETING AND NON-RED BLOOD CELL DEPLETING CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| Chile | CL63005B | 2/6/2033 | | NON-PLATELET DEPLETING AND NON-RED BLOOD CELL DEPLETING CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| China (also registered in Macao) | ZL201380075409.3 | 8/6/2033 | Composition and use | NON-PLATELET DEPLETING AND NON-RED BLOOD CELL DEPLETING CD47 ANTIBODIES AND METHODS OF USE THEREOF |
Eurasia
(validated in Russia) | 036963 | 8/6/2033 | | NON-PLATELET DEPLETING AND NON-RED BLOOD CELL DEPLETING CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| Europe (validated in DE, ES, FR, GB, IT) | 2953643 | 8/6/2033 | | NON-PLATELET DEPLETING AND NON-RED BLOOD CELL DEPLETING CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| Hong Kong | 1218863 | 8/6/2033 | | NON-PLATELET DEPLETING AND NON-RED BLOOD CELL DEPLETING CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| Indonesia | IDP000058855 | 8/6/2033 | | NON-PLATELET DEPLETING AND NON-RED BLOOD CELL DEPLETING CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| Israel | 240390 | 8/6/2033 | | NON-PLATELET DEPLETING AND NON-RED BLOOD CELL DEPLETING CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| Israel | 274566 | 8/6/2033 | | NON-PLATELET DEPLETING AND NON-RED BLOOD CELL DEPLETING CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| Japan | 6726238 | 8/6/2033 | | NON-PLATELET DEPLETING AND NON-RED BLOOD CELL DEPLETING CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| Korea | 10-2170196 | 8/6/2033 | | NON-PLATELET DEPLETING AND NON-RED BLOOD CELL DEPLETING CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| Korea | 10-2276974 | 8/6/2033 | | NON-PLATELET DEPLETING AND NON-RED BLOOD CELL DEPLETING CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| Macao | J/003573 | 8/6/2033 | | NON-PLATELET DEPLETING AND NON-RED BLOOD CELL DEPLETING CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| Mexico | MX393557 | 8/6/2033 | | | | | | | | | | | | NON-PLATELET DEPLETING AND NON-RED BLOOD CELL DEPLETING CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| New Zealand | 710695 | 8/6/2033 | | NON-PLATELET DEPLETING AND NON-RED BLOOD CELL DEPLETING CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| Peru | 9897 | 8/6/2033 | | NON-PLATELET DEPLETING AND NON-RED BLOOD CELL DEPLETING CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| | | | | | | | | | | | | | |
| Philippines | 12015501729 | 8/6/2033 | | NON-PLATELET DEPLETING AND NON-RED BLOOD CELL DEPLETING CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| Singapore | 10201706383X | 8/6/2033 | | NON-PLATELET DEPLETING AND NON-RED BLOOD CELL DEPLETING CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| South Africa | 2015/05745 | 8/6/2033 | | NON-PLATELET DEPLETING AND NON-RED BLOOD CELL DEPLETING CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| South Africa | 201905292 | 8/6/2033 | | NON-PLATELET DEPLETING AND NON-RED BLOOD CELL DEPLETING CD47 ANTIBODIES AND METHODS OF USE THEREOF |
| Vietnam | 10029478 | 8/6/2033 | | NON-PLATELET DEPLETING AND NON-RED BLOOD CELL DEPLETING CD47 ANTIBODIES AND METHODS OF USE THEREOF |
We continually assess and refine our intellectual property strategy as we develop new technologies and therapeutic candidates. As our business evolves, we may, among other activities, file additional patent applications in pursuit of our intellectual property strategy, to adapt to competition or to seize potential opportunities.
The term of individual patents depends upon the laws of the countries in which they are obtained. In most countries in which we file, the patent term is 20 years from the earliest date of filing of a non-provisional patent application. However, the term of United States patents may be extended for delays incurred due to compliance with the FDA requirements or by delays encountered during prosecution that are caused by the United States Patent and Trademark Office, or USPTO. For example, the Drug Price Competition and Patent Term Restoration Act of 1984, or the Hatch-Waxman Act, permits a patent term extension for FDA-approved drugs of up to five years beyond the expiration of the patent. The length of the patent term extension is related to the length of time the drug is under regulatory review. Patent extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval, and only one patent applicable to an approved drug may be extended. Similar provisions are available in Europe and other jurisdictions to extend the term of a patent that covers an approved drug. In the future, if and when our therapeutic candidates receive FDA approval, we expect to apply for patent term extensions on patents covering those therapeutic candidates. We intend to seek patent term extensions in any jurisdiction where these are available and where we also have a patent that may be eligible; however, there is no guarantee that the applicable authorities, including the USPTO and FDA, will agree with our assessment of whether such extensions should be granted, and even if granted, the length of such extensions.
We also rely on trade secrets to protect aspects of our technology and business not amenable to, or that we do not consider appropriate for, patent protection. We seek to protect this intellectual property, in part, by requiring our employees, consultants, outside scientific collaborators, sponsored researchers and other service providers and advisors to execute confidentiality agreements upon the commencement of employment or other relationship with us. In general, these agreements provide that confidential information concerning our business or financial affairs developed or made known to the individual during the course of the individual’s relationship with us is to be kept confidential and not disclosed to third parties except in specific circumstances. In the case of employees, the agreements further provide that inventions and discoveries conceived or reduced to practice by the individual that are related to our business, or actual, or demonstrably anticipated, research or development, or made during normal working hours, on our premises or using our equipment, supplies, or proprietary information, are our exclusive property. In many cases our agreements with consultants, outside scientific collaborators, sponsored researchers and other service providers and advisors require them to assign, or grant us licenses to, inventions resulting from the work or services they render under such agreements or grant us an option to negotiate a license to use such inventions.
Further, we expect to rely on data exclusivity, market exclusivity, patent term adjustment and patent term extensions when available.
We seek trademark protection in the United States and in certain other jurisdictions where available and when we deem appropriate. We currently have a registration for “Inhibrx” in the United States. We intend to file applications for trademark registrations in connection with our therapeutic candidates in various jurisdictions, including the United States.
Competition
The biotechnologybiopharmaceutical industry is characterized by rapid evolution of technologies, fierce competition and strong defense of intellectual property. While we believe that our platforms, technology, knowledge, experience, and scientific resources provide us with competitive advantages, we face competition from major pharmaceutical and biotechnology companies, academic institutions, governmental agencies and public and private research institutions, among others.
Any therapeutic candidates that we successfully develop and commercialize will compete with currently approved therapies and new therapies that may become available in the future. Key product features that would affect our ability to effectively compete with other therapeutics include the efficacy, safety and convenience of our therapeutics, the ease of use and effectiveness of any complementary diagnostics and/or companion diagnostics, and price and levels of reimbursement. Our primary competitors fall into the following groups:
•Companies developing novel therapeutics based on sdAb or alternative scaffold product candidates, including Alligator Bioscience AB, Camel-IDS N.V., Crescendo Biologics Ltd., GlaxoSmithKline plc, IGM Biosciences, Inc., Lava Therapeutics N.V., Molecular Partners AG, Pieris Pharmaceuticals, Inc., Precirix, and Sanofi S.A. and VHsquared Ltd.;
•Antibody drug discovery companies that may compete with us in the search for novel therapeutic antibody targets, including Regeneron Pharmaceuticals, Inc., Adimab LLC, Genmab A/S, Macrogenics, Inc., Merus N.V., MorphoSys AG, Numab Therapeutics AG, TeneoBio,Amgen, Inc., Xencor Inc., and Zymeworks Inc.; and
•Companies developing therapeutics designed to treat AATD, including CSL Limited, Grifols, S.A., Kamada Ltd., Takeda Pharmaceutical Company Limited, and Vertex Pharmaceuticals, Inc.Mereo BioPharma Group plc.
Our competitors also include other large pharmaceutical and biotechnology companies who may be developing therapeutic candidates with mechanisms similar to or targeting the same indications as our therapeutic candidates.
The availability of reimbursement from government and other third-party payors will also significantly affect the pricing and competitiveness of our therapeutic candidates. Our competitors also may obtain FDA or other regulatorymarketing approval for their products more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market.
Many of the companies against which we may compete have significantly greater financial resources and expertise in research and development, manufacturing, preclinicalnonclinical testing, conducting clinical trials, obtaining regulatorymarketing approvals and marketing approved products than we do. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These early stage and more established competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
Government Regulation
Governmental authorities in the United States, at the federal, state and local level, and other countries extensively regulate, among other things, the research, development, testing, manufacture, labeling, packaging, promotion, storage, advertising, distribution, marketing and export and import of products such as those we are developing. Our therapeutic candidates must be approved by the FDA through the Biologics License Application, or BLA process before they may be legally marketed in the United States and will be subject to similar requirements in other countries prior to marketing in those countries. The process of obtaining regulatorymarketing approvals in the U.S. and in foreign countries and jurisdictions, and the subsequent compliance with applicable federal, state, local and foreign statutes and regulations, require the expenditure of substantial time and financial resources.
United States Government Regulation
In the United States, the FDA regulates biopharmaceutical products under the Federal Food, Drug, and Cosmetic Act and the Public Health Services Act, or PHSA, and implementing regulations. Failure to comply with the applicable U.S. requirements at any time during the product development or approval process, or after approval, may subject an applicant to administrative or judicial sanctions brought by the FDA and the Department of Justice, or
DOJ, or other governmental entities, any of which could have a material adverse effect on us. These sanctions could include:
•refusal to approve pending applications;
•withdrawal of an approval;
•imposition of a clinical hold;
•warning or untitled letters;
•seizures or administrative detention of product;
•total or partial suspension of production or distribution; or
•injunctions, fines, disgorgement, or civil or criminal penalties.
BLA Approval Processes
The process required by the FDA before a therapeutic biologic may be marketed in the United States generally involves the following:
•completion of nonclinical laboratory tests, animal studies and formulation studies conducted according to good laboratory practices and other applicable regulations;regulations and guidance;
•submission to the FDA of an Investigational New Drug Application, or IND, which must become effective before human clinical trials may begin;
•performance of adequate and well-controlled human clinical trials according to good clinical practices, or GCPs, to establish the safety and efficacy of the therapeutic candidate for its intended use;
•submission to the FDA of a BLA;
•satisfactory completion of an FDA inspectioninspections of the manufacturing facility or facilities at which the therapeutic candidate is produced to assess readiness for commercial manufacturing and conformance to the manufacturing-related elements of the application, to conduct a data integrity audit, and to assess compliance with current Good Manufacturing Practices, or cGMPs, to assure that the facilities, methods and controls are adequate to preserve the therapeutic candidate’s identity, strength, quality and purity; and
•FDA review and approval of the BLA.
Preclinical Studies and IND Application
Once a biopharmaceutical candidate is identified for development, it enters the preclinical or nonclinical testing stage. Nonclinical tests include laboratory evaluations of product chemistry, toxicity and formulation, as well as potential animal studies. The Consolidated Appropriations Act for 2023, signed into law on December 29, 2022 (P.L. 117-328), amended both the FDCA and the Public Health Service Act, or PHSA, to specify that nonclinical testing for drugs and biologics, respectively, may, but is not required to, include in vivo animal testing. According to the amended language, a sponsor may fulfill nonclinical testing requirements by completing various in vitro assays (e.g., cell-based assays, organ chips, or microphysiological systems), in silico studies (i.e., computer modeling), other human or non-human biology based tests (e.g., bioprinting), or in vivo animal tests. An IND sponsor must submit the results of the nonclinical tests, together with manufacturing information and analytical data, to the FDA as part of the IND. SomeAdditional nonclinical testing may continuecontinues even after the IND is submitted. In addition to including the results of the nonclinical studies, the IND will also include a study protocol detailing, among other things, the objectives of the clinical trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated if the first phase lends itself to an efficacy determination. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, places the IND on clinical hold. In this case, the IND sponsor and the FDA must resolve any outstanding concerns before clinical trials can begin. A clinical hold may occur at any time during the life of an IND and may affect one or more specific studies or all studies conducted under the IND.
Human Clinical Trials in Support of a BLA
All clinical trials must be conducted under the supervision of one or more qualified investigators in accordance with GCPs. They must be conducted under protocols detailing the objectives of the trial, dosing procedures, research subject selection and exclusion criteria and the safety and effectiveness criteria to be evaluated. Each protocol, and any subsequent material amendment to the protocol, must be submitted to the FDA as part of the IND, and progress reports detailing the status of the clinical trials must be submitted to the FDA annually. Sponsors also must report to the FDA serious and unexpected adverse reactions in a timely manner, such as any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigationinvestigator’s brochure or any findings from other studies or animal or in vitro testing that suggest a significant risk in humans exposed to the product or therapeutic candidate. An institutional review board, or IRB, at each institution participating in the clinical trial must review and approve the protocol before a clinical trial commences at that institution and must also approve the information regarding the trial and the consent form that must be provided to each research subject or the subject’s legal representative, monitor the trial until completed and otherwise comply with IRB regulations.
There are also requirements governing the reporting of ongoing clinical trials and completed clinical trial results to public registries.
Human clinical trials are typically conducted in three sequential phases that may overlap or be combined.
Phase 1-The product or therapeutic candidate is initially introduced into healthy human subjects and tested for safety, dosage tolerance, absorption, metabolism, distribution and elimination. In the case of some therapeutic candidates for severe or life-threatening diseases, such as cancer, especially when the product or therapeutic candidate may be inherently too toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in patients.
Phase 2-Clinical trials are performed on a limited patient population intended to identify possible adverse effectsAEs and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage.
Phase 3-Clinical trials are undertaken to further evaluate dosage, clinical efficacy and safety in an expanded patient population at geographically dispersed clinical trial sites. These trials are intended to establish the overall risk-benefit ratio of the product and provide an adequate basis for product labeling.
A pivotal trial is a clinical trial that adequately meets regulatory agency requirements for the evaluation of a product or therapeutic candidate’s efficacy and safety such that it can be used to justify the approval of the product. Generally, pivotal trials are also Phase 3 trials but may be Phase 2 trials if the trial design provides a reliable assessment of clinical benefit, particularly in situations where there is an unmet medical need. Human clinical trials are inherently uncertain and Phase 1, Phase 2 and Phase 3 testing may not be successfully completed. The FDA or the sponsor may suspend a clinical trial at any time for a variety of reasons, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the product or therapeutic candidate has been associated with unexpected serious harm to patients.
Congress also recently amended the FDCA, as part of the Consolidated Appropriations Act for 2023, to require sponsors of a Phase 3 clinical trial, or other “pivotal study” of a new drug to support marketing authorization, to design and submit a diversity action plan for such clinical trial. The action plan must include the sponsor’s diversity goals for enrollment, as well as a rationale for the goals and a description of how the sponsor will meet them. Sponsors must submit a diversity action plan to the FDA by the time the sponsor submits the relevant clinical trial protocol to the agency for review. The FDA may grant a waiver for some or all of the requirements for a diversity action plan. It is unknown at this time how the diversity action plan may affect Phase 3 trial planning and timing or what specific information FDA will expect in such plans, but if the FDA objects to a sponsor’s diversity action plan or otherwise requires significant changes to be made, it could delay initiation of the relevant clinical trial.
During the development of a new product or therapeutic candidate, sponsors are given opportunities to meet with the FDA at certain points; specifically, prior to the submission of an IND, at the end of Phase 2 and before a BLA is submitted. Meetings at other times may be requested. These meetings can provide an opportunity for the sponsor to share information about the data gathered to date and for the FDA to provide advice on the next phase of
development. Sponsors typically use the meeting at the end of Phase 2 to discuss their Phase 2 clinical results and present their plans for the pivotal Phase 3 clinical trialtrials that they believe will support the approval of the new therapeutic.
Post-approval trials, sometimes referred to as “Phase 4” clinical trials, may be conducted after initial marketing approval. These trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication. In certain instances, FDA may mandate the performance of “Phase 4” clinical trials.
Concurrent with clinical trials, sponsors usually complete additional animal safety studies, develop additional information about the chemistry and physical characteristics of the product or therapeutic candidate and finalize a process for manufacturing commercial quantities of the product or therapeutic candidate in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product or therapeutic candidate and the manufacturer must develop methods for testing the quality, purity and potency of the product or therapeutic candidate. To help reduce the risk of the introduction of adventitious agents with use of biological products, the PHSA emphasizes the importance of manufacturing control for products whose attributes cannot be precisely defined. The manufacturing process must be capable of consistently producing quality batches of the product or therapeutic candidate and, among other criteria, the sponsor must develop methods for testing the identity, strength, quality, potency and purity of the final biological product. Additionally, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate that the biological product or therapeutic candidate does not undergo unacceptable deterioration over its shelf life.
Sponsors of clinical trials of certain FDA-regulated products, including prescription drugs and biologics, are required to register and disclose certain clinical trial information on a public registry maintained by the U.S. National Institutes of Health, or NIH. In particular, information related to the product, patient population, phase of investigation, study sites and investigators and other aspects of the clinical trial is made public as part of the registration of the clinical trial. Competitors may use this publicly available information to gain knowledge regarding the progress of development programs. Although sponsors are also obligated to disclose the results of their clinical trials after completion, disclosure of the results can be delayed in some cases for up to two years after the date of completion of the trial. Failure to timely register a covered clinical study or to submit study results as provided for in the law can give rise to civil monetary penalties and also prevent the non-compliant party from receiving future grant funds from the federal government. The NIH’s Final Rule on ClinicalTrials.gov registration and reporting requirements became effective in 2017, and the government has brought enforcement actions against clinical trial sponsors that fail to comply with such requirements.
Marketing Application Submission and FDA Review
Assuming successful completion of the required clinical testing, the results of the preclinicalnonclinical studies and clinical trials, along with detailed descriptions of the product’s chemistry, manufacturing, and controls,CMC, proposed labeling and other relevant information are submitted to the FDA as part of a BLA requesting approval to market the product.
Under the Prescription Drug User Fee Act, or PDUFA, as amended, each BLA must be accompanied by a significant user fee. The FDA adjusts the PDUFA user fees on an annual basis. PDUFA also imposes an annual program fee forthat is assessed on sponsors of approved prescription biological or drug products. Fee waivers or reductions are available in certain circumstances, such as where a waiver is necessary to protect the public health, where the fee would present a significant barrier to innovation, or where the applicant is a small business submitting its first human therapeutic application for review.
Under the goals and policies agreed to by the FDA under PDUFA, for original BLAs, the FDA has ten months from the filing date in which to complete its initial review of a standard application and respond to the applicant, and six months from the filing date for an application with priority review. The FDA does not always meet its PDUFA goal dates, and the review process is often significantly extended by FDA requests for additional information or clarification and athe sponsor’s process to respond to such inquiries. This FDA review typically takes twelve months from the date the BLA is submitted to the FDA (for a standard review) and eight months from the date the BLA is submitted (for a “priority review”) because the FDA has approximately two months, or 60 days, after BLA submission to make a “filing” decision.
The FDA conducts a preliminary review of a submitted BLA within 60 days of receipt and informs the sponsor by the 74th day after the FDA’s receipt of the submission whether the application is substantially complete to permit a substantive review. The FDA may refuse to file any BLA that it deems incomplete or not properly reviewable at the time of submission, and may request additional information. In this event, the BLA must be resubmitted with the additional information. The resubmitted application also is subject to review before the FDA accepts it for filing.
Once the submission is accepted for filing, the FDA begins an in-depth substantive review of the BLA. The FDA reviews the BLA to determine, among other things, whether the proposed product is safe, potent, and/or effective for its intended use, whether it has an acceptable purity profile and whether the product is being manufactured in accordance with cGMP. The FDA may refer applications for novel products or products that present difficult questions of safety or efficacy to an advisory committee. Typically, an advisory committee is a panel of independent experts, including clinicians and other scientific experts, that reviews, evaluates and provides a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers these recommendations carefully when making decisions.
The FDA likely will re-analyze the clinical trial data, which could result in extensive discussions between the FDA and the applicant during the review process. The FDA also may require the submissiondevelopment of a risk evaluation and mitigation strategy, or REMS, plan if it determines that a REMS is necessary to ensure that the benefits of the drug outweigh its risks and to assure the safe use of the biological product. The REMS plan could include medication guides, physician communication plans, assessment plans and/or elements to assure safe use, such as restricted distribution methods, patient registries or other risk minimization tools. The FDA determines the requirement for a REMS, as well as the specific REMS provisions, on a case-by-case basis. If the FDA concludes a REMS plan is needed, the sponsor of the BLA must submit a proposed REMS plan. The FDA will not approve a BLA without a REMS plan, if required.
Before approving a BLA, the FDA will typically inspect the facilities at which the product is manufactured. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving a BLA, the FDA will typically inspect one or more clinical sites to assure that the clinical trials were conducted in compliance with IND trial requirements and GCP requirements and to assure the integrity of the clinical data submitted to the FDA. To ensure cGMP and GCP compliance by its employees and third-party contractors, an applicant must incur significant expenditure of time, money and effort in the areas of training, record keeping, production and quality control.
The approval process is lengthy and often difficult, and notwithstanding the submission of relevant data and information, the FDA may ultimately decide that the BLA does not satisfy its regulatory criteria for approval and deny approval or may require additional clinical or other data and information. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than we interpret the same data. If the agency decides not to approve a BLA, the FDA will issue a complete response letter, or CRL, that describes all of the specific deficiencies in the BLA identified by the FDA. A CRL indicates that the review cycle of the application is complete and the application will not be approved in its present form. The deficiencies identified may be minor, for example, requiring labeling changes, or major, for example, requiring additional clinical trials. Additionally, the complete response letterCRL may include recommended actions that the applicant might take to place the application in a condition for approval. If a complete response letterCRL is issued, the applicant may either resubmit the BLA, addressing all of the deficiencies identified in the letter, or withdraw the application. If and when those deficiencies have been addressed to the FDA’s satisfaction in a resubmission of the BLA, the FDA will issue an approval letter to the applicant. The FDA has committed to reviewing such resubmissions in response to an issued CRL in either two or six months depending on the type of information included. Even with the submission of this additional information, however, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
Even if a product receives regulatorymarketing approval, the approval may be limited to specific indications and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling. The FDA may impose restrictions and conditions on product distribution, prescribing, or dispensing in the form of a
REMS plan, or otherwise limit the scope of any approval. In addition, the FDA may require post marketing clinical trials, sometimes referred to as “Phase 4” clinical trials, designed to further assess a biological product’s safety and effectiveness, and/or testing and surveillance programs to monitor the safety of approved products that have been commercialized. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further testing requirements and FDA review and approval.
Companion Diagnostics
The FDA issued a final guidance document in July 2014 addressing agency policy in relation to in vitro companion diagnostic tests. The guidance explains that for some drugs and therapeutic biologics, the use of a companion diagnostic test is essential for the safe and effective use of the product, such as when the use of a product is limited to a specific patient subpopulation that can be identified by using the test. According to the guidance, the FDA generally will not approve such a product if the companion diagnostic is not also approved or cleared for the appropriate indication, and accordingly the therapeutic product and the companion diagnostic should be developed and approved or cleared contemporaneously. However, the FDA may decide that it is appropriate to approve such a product without an approved or cleared in vitro companion diagnostic device when the drug or therapeutic biologic is intended to treat a serious or life-threatening condition for which no satisfactory alternative treatment exists and the FDA determines that the benefits from the use of a product with an unapproved or uncleared in vitro companion diagnostic device are so pronounced as to outweigh the risks from the lack of an approved or cleared in vitro companion diagnostic device. The FDA encourages sponsors considering developing a therapeutic product that requires a companion diagnostic to request a meeting with both relevant device and therapeutic product review divisions to ensure that the product development plan will produce sufficient data to establish the safety and effectiveness of both the therapeutic product and the companion diagnostic. Because the FDA’s policy on companion diagnostics is set forth only in guidance, this policy is subject to change and is not legally binding.
Fast Track, Breakthrough Therapy, RTOR, and Priority Review Designations
The FDA is authorized to designate certain products for expedited development or review if they are intended to address an unmet medical need in the treatment of a serious or life-threatening disease or condition. These programs include fast trackFast Track designation, breakthrough therapy designation and priority review designation. In January 2021, INBRX-109 was granted Fast Track designation for patients with unresectable or metastatic conventional chondrosarcoma.
To be eligible for a fast trackFast Track designation, the FDA must determine, based on the request of a sponsor, that a product is intended to treat a serious or life-threatening disease or condition and demonstrates the potential to address an unmet medical need by providing a therapy where none exists or a therapy that may be potentially superior to existing therapy based on efficacy or safety factors. Fast trackTrack designation provides opportunities for more frequent
interactions with the FDA review team to expedite development and review of the product. The FDA may also review sections of the BLA for a fast trackFast Track product on a rolling basis before the complete application is submitted, if the sponsor and the FDA agree on a schedule for the submission of the application sections, and the sponsor pays any required user fees upon submission of the first section of the BLA. In addition, fast trackFast Track designation may be withdrawn by the sponsor or rescinded by the FDA if the designation is no longer supported by data emerging in the clinical trial process.
In addition, with the enactment of the FDA Safety and Innovation Act, or FDASIA, in 2012, Congress created a new regulatory program in 2012 for product and therapeutic candidates designated by the FDA as “breakthrough therapies” upon a request made by the IND sponsors. A breakthrough therapy is defined as a drug or biologic that is intended, alone or in combination with one or more other drugs or biologics, to treat a serious or life-threatening disease or condition, and preliminary clinical evidence indicates that the drug or biologic may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. Drugs or biologics designated as breakthrough therapies aremay also be eligible for accelerated approval of their respective marketing applications. The FDA must take certain actions with respect to breakthrough therapies, such as holding timely meetings with and providing advice to the product sponsor, intended to expedite the development and review of an application for approval of a breakthrough therapy.
Specific to oncology drug applications, FDA’s Oncology Center of Excellence has developed a program called Real-Time Oncology Review, or RTOR. RTOR facilitates earlier submission of topline results (i.e., efficacy and safety results from clinical studies before the study report is completed) and datasets, after database lock, to support an earlier start to the agency’s review of a marketing application review. The intent of RTOR is to provide FDA reviewers earlier access to data, to identify data quality and potential review issues, and to potentially enable early feedback to the applicant, which can allow for a more streamlined and efficient review process for the product’s BLA. Applicants can apply for review under RTOR when the database for a pivotal trial has been locked and the oncology product is eligible under FDA’s criteria for the program. Eligibility requires (a) clinical evidence indicating that the drug may demonstrate substantial improvement on one or more clinically relevant endpoints over available therapies; (b) the use of straightforward study designs and easily interpreted clinical trial endpoints (e.g., overall survival, response rates); and (c) that no aspect of the BLA is likely to require a longer review time (e.g., requirement for new REMS or input from an advisory committee). In November 2023, the agency finalized guidance for industry on RTOR.
Finally, the FDA may designate a product for priority review if it is a drug or biologic that treats a serious condition and, if approved, would provide a significant improvement in safety or effectiveness. The FDA determines at the time that the marketing application is submitted, on a case-by-case basis, whether the proposed drug represents a significant improvement in treatment, prevention or diagnosis of disease when compared with other available therapies. Significant improvement may be illustrated by evidence of increased effectiveness in the treatment of a condition, elimination or substantial reduction of a treatment-limiting drug reaction, documented enhancement of patient compliance that may lead to improvement in serious outcomes, or evidence of safety and effectiveness in a new subpopulation. A priority review designation is intended to direct overall attention and resources to the evaluation of such applications, and to shorten the FDA’s goal for taking action on a marketing application from ten months to six months for an original BLA from the date of filing.
Even if a product qualifies for one or more of these programs, the FDA may later decide that the product no longer meets the conditions for qualification or decide that the time period for FDA review or approval will not be shortened. Furthermore, fast trackFast Track designation, breakthrough therapy designation, RTOR, and priority review do not change the standards for approval and may not ultimately expedite the development or approval process.
Accelerated Approval Pathway
In addition, products studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit over existing treatments may receive accelerated approval from the FDA and may be approved on the basis of adequate and well-controlled clinical trials establishing that the drug product has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit. The FDA may also grant accelerated approval for such a drug or biologic when the product has an effect on an intermediate clinical endpoint that can be measured earlier than an effect on irreversible morbidity or mortality, or IMM, and that is reasonably likely to predict an effect on IMM or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. As a condition of approval, the FDA may require that a sponsor of a drug receiving accelerated approval perform post-marketing clinical trials to verify and describe the predicted effect on IMM or other clinical endpoint, and the product may be subject to expedited withdrawal procedures. Drugs and biologics granted accelerated approval must meet the same statutory standards for safety and effectiveness as those granted traditional approval. Further, all promotional materials for product and therapeutic candidates being considered and approved under the Accelerated Approval Program are subject to prior review by the FDA.
For the purposes of accelerated approval, a surrogate endpoint is a marker, such as a laboratory measurement, radiographic image, physical sign, or other measure that is thought to predict clinical benefit, but is not itself a measure of clinical benefit. Surrogate endpoints can often be measured more easily or more rapidly than clinical endpoints. An intermediate clinical endpoint is a measurement of a therapeutic effect that is considered reasonably likely to predict the clinical benefit of a drug, such as an effect on IMM. The FDA has limited experience with accelerated approvals based on intermediate clinical endpoints, but has indicated that such endpoints generally may support accelerated approval when the therapeutic effect measured by the endpoint is not itself a clinical benefit and
basis for traditional approval, if there is a basis for concluding that the therapeutic effect is reasonably likely to predict the ultimate long-term clinical benefit of a drug.
The accelerated approval pathway is most often used in settings in which the course of a disease is long and an extended period of time is required to measure the intended clinical benefit of a drug, even if the effect on the surrogate or intermediate clinical endpoint occurs rapidly. For example, accelerated approval has been used extensively in the development and approval of drugs for treatment of a variety of cancers in which the goal of therapy is generally to improve survival or decrease morbidity and the duration of the typical disease course requires lengthy and sometimes large clinical trials to demonstrate a clinical or survival benefit.
The accelerated approval pathway is usually contingent on a sponsor’s agreement to conduct, in a diligent manner, additional post-approval confirmatory studies to verify and describe the drug’s clinical benefit. As a result, a product or therapeutic candidate approved on this basis is subject to rigorous post-marketing compliance requirements, including the completion of Phase 4 or post-approval clinical trials to confirm the effect on the clinical endpoint. In addition, as part of the Consolidated Appropriations Act for 2023, Congress provided FDA additional statutory authority to mitigate potential risks to patients from continued marketing of ineffective drugs previously granted accelerated approval. Under these recent amendments to the FDCA, the agency may require a sponsor of a product granted accelerated approval to have a confirmatory trial underway prior to approval. The sponsor must also submit progress reports on a confirmatory trial every six months until the trial is complete, and such reports will be published on the FDA’s website. Failure to conduct required post-approval studies, or to confirm the predicted clinical benefit of the product during post-marketing studies, would allowallows the FDA to withdraw approval of the drug. All promotional materialsCongress also recently amended the law to give FDA the option of using expedited procedures to withdraw product approval if the sponsor’s confirmatory trial fails to verify the claimed clinical benefits of the product. Prior to the recent statutory amendments enacted by Congress, several oncology sponsors voluntarily withdrew specific indications for producttheir drug products that were being marketed pursuant to accelerated approval, and therapeutic candidates being considered and approved underthe FDA’s Oncology Center of Excellence launched an initiative called Project Confirm, aimed at promoting transparency in the area of accelerated approvals for oncology indications. Scrutiny of the accelerated approval program are subjectpathway is likely to prior review bycontinue in the FDA.coming years and may lead to further legislative and/or administrative changes in the future.
Patent Term Restoration and Reference Product Marketing Exclusivity for Biological Products
Depending upon the timing, duration and specifics of FDA approval of the use of our therapeutic candidates, some of our United States patents may be eligible for limited patent term extension under the Hatch-Waxman Act. The Hatch-Waxman Act permits a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product or therapeutic candidate’s approval date. The patent term restoration period is generally one half of the time between the effective date of an IND and the submission date of a BLA, plus the time between the submission date of a BLA and the approval of that application, except that the review period is reduced by any time during which the applicant failed to exercise due diligence. Only one patent applicable to an approved product or therapeutic candidate is eligible for the extension and the application for extension must be made prior to expiration of the patent. The USPTO, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. In the future, we intend to apply for restorations of patent term for some of our currently owned or licensed patents to add patent life beyond their current expiration date, depending on the expected length of clinical trials and other factors involved in the submission of the relevant BLA.
The Biologics Price Competition and Innovation Act, or the BPCIA, enacted in 2010, amended the PHSA to authorize the FDA to approve similar versions of innovative biologics such as ours, which are also known as “reference biological products.” The new pathway authorized under the BPCIA allows FDA to approve, under an abbreviated application, a biological product that has been shown to be highly similar to a reference product notwithstanding minor differences in clinically inactive components; there also can be no clinically meaningful differences between the follow-on product and the reference product in terms of safety, purity and potency. Follow-on biological products approved for marketing using this pathway are commonly known as “biosimilars,” and companies seeking approval of biosimilar products must submit in their abbreviated BLAs data from analytical studies, animal studiesan assessment of toxicity, and a clinical trial or trials sufficient to meet the statutory standard in order to
obtain FDA approval. In addition, an interchangeable product is a biosimilar product that can be expected to produce the same clinical results as the reference product in any given patient and, for products administered multiple times to an individual, that the product and the reference product may be alternated or switched after one has been previously administered without increasing safety risks or risks of diminished efficacy relative to exclusive use of the reference biological product without such alternation or switch. Upon licensure by the FDA, an interchangeable biosimilar may be substituted for the reference product without the intervention of the health care provider who prescribed the reference product. FDA approved the first interchangeable biosimilars, including an interchangeable monoclonal antibody biosimilar, in 2021.
The BPCIA bars the FDA from approving biosimilar applications for 12 years after a reference biological product receives initial marketing approval. FDA also cannot accept an application for a biosimilar product that is based on the reference biological product until four years after the date of first licensure of the reference product in question. “First licensure” typically means the initial date the particular product at issue was licensed in the United States. Date of first licensure does not include the date of licensure of a supplement for the reference product for a subsequent application filed by the same sponsor or manufacturer of the reference product for a change (not including a modification to the structure of the biological product) that results in a new indication, route of administration, dosing schedule, dosage form, delivery system, delivery device or strength or for a modification to the structure of the biological product that does not result in a change in safety, purity or potency. Therefore, one must determine whether a new product includes a modification to the structure of a previously licensed product that
results in a change in safety, purity or potency to assess whether the licensure of the new product is a first licensure that triggers its own period of exclusivity. Whether a subsequent application, if approved, warrants exclusivity as the “first licensure” of a biological product is determined on a case-by-case basis with data submitted by the sponsor.
The BPCIA is complex and only beginning to beis still being interpreted and implemented by the FDA.FDA and by federal judges. In addition, recent government proposals have sought to reduce the 12-year reference product exclusivity period. Other aspects of the BPCIA, some of which may impact the BPCIA exclusivity provisions, have also been the subject of recent litigation and legislative amendments. As a result, the ultimate impact, implementation and meaning of the BPCIA iscontinues to be subject to significant uncertainty.
Orphan Drug Designation and Orphan Product Exclusivity
Under the Orphan Drug Act, the FDA may grant Orphan Drug Designation to a therapeutic candidate intended to treat a rare disease or condition, which is generally a disease or condition that affects either (i) fewer than 200,000 individuals in the United States, or (ii) more than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making available in the United States a product or therapeutic candidate for this type of disease or condition will be recovered from sales in the United States for that product or therapeutic candidate. Orphan Drug Designation must be requested before submitting a BLA. After the FDA grants Orphan Drug Designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Orphan Drug Designation does not convey any advantage in or shorten the duration of the regulatory review and approval process. In November 2021, INBRX-109 was granted designation as an orphan product by the FDA for the treatment of chondrosarcoma, and in March 2022, INBRX-101 was granted designation as an orphan product by the FDA for the treatment of AATD.
If a product or therapeutic candidate that has Orphan Drug Designation subsequently receives the first FDA approval for the disease for which it has such designation, the approved product is entitled to orphan product exclusivity, which means that the FDA may not approve any other marketing applications for the same drug for the same indication, except under limited circumstances (such as showing of clinical superiority to the product with orphan exclusivity by means of greater effectiveness, greater safety or providing a major contribution to patient care or in instances of drug supply issues), for seven years. Competitors, however, may still receive approval of either a different product for the same indication for which the orphan product has exclusivity or for the same product but for a different indication (which could then be used off-label in the orphan indication). Orphan product exclusivity, however, could also block the approval of one of our therapeutic candidates for seven years if a competitor obtains approval of the same drug as defined by the FDA, or if our therapeutic candidate is determined to be contained within a competitor’s approved drug for the same indication or disease. Further, if a drug designated as an orphan product ultimately receives marketing approval for an indication broader than what was designated in its orphan
product application, it may not be entitled to exclusivity. Recent court cases have challenged FDA’s approach to determining the scope of orphan drug exclusivity; however, at this time the agency continues to apply its long-standing interpretation of the governing regulations and has stated that it does not plan to change any orphan drug implementing regulations.
In addition, an orphan drug credit is available for qualifying costs incurred between the date the FDA designates a drug as an orphan drug and the date the FDA approves the drug.
Pediatric Exclusivity and Pediatric Use
Pediatric exclusivity is another type of non-patent marketing exclusivity available in the United States and, if granted, it provides for the attachment of an additional six months of marketing protection to the term of any existing regulatory exclusivity or listed patents. Under the Best Pharmaceuticals for Children Act, or BPCA, certain therapeutic candidates may obtain an additional six months of exclusivity if the sponsor submits information requested in writing by the FDA, referred to as a Written Request, relating to the use of the active moiety of the product or therapeutic candidate in children. The data do not need to show the product to be effective in the pediatric population studied; rather, the additional protection is granted if the pediatric clinical trial is deemed to have fairly responded to the FDA’s Written Request. Although the FDA may issue a Written Request for studies on either approved or unapproved indications, it may only do so where it determines that information relating to that use of a product or therapeutic candidate in a pediatric population, or part of the pediatric population, may produce health benefits in that population. The issuance of a Written Request does not require the sponsor to undertake the described trials. This is not a patent term extension, but it effectively extends the regulatory period during which the FDA cannot approve another application.
In addition, the Pediatric Research Equity Act, or PREA, requires a sponsor to conduct pediatric trials for most therapeutic drugs and biologics, for a new active ingredient, new indication, new dosage form, new dosing regimen or new route of administration. Under PREA, original new drug applications, BLAs and supplements thereto must contain a pediatric assessment unless the sponsor has received a deferral or waiver, and with enactment of FDASIA in 2012, sponsorswaiver. Sponsors must also submit Pediatric Study Plans, or PSPs, to the agency for review within sixty days of an end-of-Phase 2 meeting or, if there is no such meeting, as early as practicable before the initiation of the Phase 3 or Phase 2/3 clinical trial. The initial PSP must include an outline of the pediatric trial or trials that the sponsor plans to conduct, including trial objectives and design, age groups, relevant endpoints and statistical approach, or a
justification for not including such detailed information, and any request for a deferral of pediatric assessments or a full or partial waiver of the requirement to provide data from pediatric trials along with supporting information. The FDA and the sponsor must reach an agreement on the PSP. A sponsor can submit amendments to an agreed upon initial PSP at any time if changes to the pediatric plan need to be considered based on data collected from preclinical studies, early phase clinical trials or other clinical development programs.
The required assessment must assess the safety and effectiveness of the product or therapeutic candidate for the claimed indications in all relevant pediatric subpopulations and support dosing and administration for each pediatric subpopulation for which the product and therapeutic candidate is safe and effective. The sponsor or FDA may request a deferral of pediatric trials for some or all of the pediatric subpopulations. A deferral may be granted for several reasons, including a finding that the product or therapeutic candidate is ready for approval for use in adults before pediatric trials are complete or that additional safety or effectiveness data needs to be collected before the pediatric trials begin. The law now requires the FDA is required to send a PREA Non-Compliance letter to sponsors who have failed to submit their pediatric assessments required under PREA, have failed to seek or obtain a deferral or deferral extension or have failed to request approval for a required pediatric formulation. It further requiresformulation, and the FDA to publicly post theposts such PREA Non-Compliance letterletters and sponsor’s response. Unless otherwise required by regulation, the pediatric data requirements do not apply to products with orphan designation, although FDA has recently taken steps to limit what it considers abuse of this statutory exemption in PREA by announcing that it does not intend to grant any additional orphan drug designations for rare pediatric subpopulations of what is otherwise a common disease.
As partCongress periodically considers enacting new incentives or mandates applicable to pediatric drug development, and the regulatory requirements applicable to pediatric drug developers may change in the future. For example, bipartisan legislation introduced in 2023 in the House of the FDASIA, the United States Congress made several changesRepresentatives would increase funding for pediatric trials;
mandate that drugs for rare diseases be studied in children; and grant FDA authority to the BPCA and PREA, and permanently reauthorized both laws.assess penalties against companies that do not complete required pediatric studies.
Post-Approval Requirements
Following approval of a new product, the manufacturer and the approved biological product are subject to pervasive and continuing regulation by the FDA, including, among other things, monitoring and recordkeeping activities, reporting of adverse experiences with the product, product sampling and distribution restrictions, complying with promotion and advertising requirements, which include restrictions on promoting drugs for unapproved uses or patient populations (i.e., “off-label use”) and limitations on industry-sponsored scientific and educational activities. Although physicians may prescribe legally available products for off-label uses, manufacturers may not market or promote such uses. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability. If there are any modifications to the product, including changes in indications, labeling or manufacturing processes or facilities, the applicant may be required to submit and obtain FDA approval of a new BLA or a BLA supplement, which may require the applicant to develop additional data or conduct additional preclinicalnonclinical studies and clinical trials.
Once an approval is granted, the FDA may withdraw the approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product or therapeutic reaches the market. Later discovery of previously unknown problems with a product or therapeutic candidate, including adverse eventsAEs of unanticipated severity or frequency, may result in in mandatory revisions to the approved labeling to add new safety information; imposition of post-market or clinical trials to assess new safety risks; or imposition of distribution or other restrictions under a REMS program. Other potential consequences of non-compliance include, among other things:
•restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls;
•mandated modification of promotional materials or labeling and the issuance of corrective information;
•fines, warning letters or other enforcement-related letters or clinical holds on post-approval clinical trials;
•refusal of the FDA to approve pending BLAs or supplements to approved BLAs, or suspension or revocation of product approvals;
•product seizure or detention, or refusal to permit the import or export of products;
•injunctions or the imposition of civil or criminal penalties; and
•consent decrees, corporate integrity agreements, debarment, or exclusion from federal health care programs; or mandated modification of promotional materials and labeling and the issuance of corrective information.programs.
Accordingly, aAny therapeutic candidate manufactured or distributed by us pursuant to FDA approvals arewill be subject to continuing regulation by the FDA, including, among other things:
•record-keeping requirements;
•reporting of adverse experiences with the therapeutic candidate;
•providing the FDA with updated safety and efficacy information;
•therapeutic sampling and distribution requirements;
•notifying the FDA and gaining its approval of specified manufacturing or labeling changes; and
•complying with FDA promotion and advertising requirements, which include, among other things, standards for direct-to-consumer advertising, restrictions on promoting products for uses or in-patient populations that are not described in the product’s approved labeling, limitations on industry-sponsored scientific and educational activities and requirements for promotional activities involving the internet.
FDA regulations require that products be manufactured in specific approved facilities and in accordance with cGMPs. The cGMP regulations include requirements relating to organization of personnel, buildings and facilities, equipment, control of components and drug product containers and closures, production and process controls, packaging and labeling controls, holding and distribution, laboratory controls, records and reports and returned or salvaged products. Therapeutic manufacturers and other entities involved in the manufacture and distribution of approved therapeutic products are required to register their establishments with the FDA and certain state agencies and are subject to periodic unannounced inspections by the FDA and some state agencies for compliance with
cGMPs and other laws. The FDA periodically inspects manufacturing facilities to assess compliance with cGMP requirements. In addition, changes to the manufacturing process are strictly regulated, and, depending on the significance of the change, may require FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP and impose reporting and documentation requirements upon the BLA applicant and any third-party manufacturers involved in producing the approved biological product. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of quality control and quality assurance.
In addition, the distribution of prescription pharmaceutical products is subject to the Prescription Drug Marketing Act, or the PDMA, which regulates the distribution of drugs and drug samples at the federal level, and sets minimum standards for the registration and regulation of drug distributors by the states. Both the PDMA and state laws limit the distribution of prescription pharmaceutical product samples and impose requirements to ensure accountability in distribution. MostMore recently, the Drug Supply Chain Security Act, or the DSCSA, was enacted with the aim of building an electronic system to identify and trace certain prescription drugs distributed in the United States, including most biological products. The DSCSA mandates phased-in and resource-intensive obligations for pharmaceutical manufacturers, wholesale distributors, and dispensers over a 10‑year period that is expected to culminateculminated in November 2023. However, FDA announced a one-year stabilization period, until November 2024, to give entities subject to the DSCSA additional time to finalize interoperable tracking systems and to ensure supply chain continuity. From time to time, new legislation and regulations may be implemented that could significantly change the statutory provisions governing the approval, manufacturing and marketing of products regulated by the FDA. For example, the FDA released proposed regulations in February 2022 to amend the national standards for licensing of wholesale drug distributors by the states; establish new minimum standards for state licensing third-party logistics providers; and create a federal system for licensure for use in the absence of a State program, each of which is mandated by the DSCSA. It is impossible to predict whether further legislative or regulatory changes will be enacted, or FDA regulations, guidance or interpretations changed or what the impact of such changes, if any, may be.
Regulation Outside of the United States
In addition to regulations in the United States, we will be subject to regulations of other jurisdictions governing any clinical trials and commercial sales and distribution of our therapeutic candidates. Whether or not we obtain FDA approval for a product, we must obtain approval by the comparable regulatory authorities of countries outside of the United States before we can commence clinical trials in such countries and approval of the regulators of such countries or economic areas, such as the European Union,EU, before we may market products in those countries or areas. The approval process and requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from place to place, and the time may be longer or shorter than that required for FDA approval.
Under European UnionEU regulatory systems, a company can consider applying for marketing authorization in several European UnionEU member states by submitting its marketing authorization application(s) under a centralized, decentralized or mutual recognition procedure. The centralized procedure provides for the grant of a single marketing authorization that is valid for all European UnionEU member states. The centralized procedure is compulsory
for medicines derived from biotechnology, orphan medicinal products, or those medicines with an active substance not authorized in the European UnionEU on or before May 20, 2004 intended to treat acquired immune deficiency syndrome, cancer, neurodegenerative disorders or diabetes and optional for those medicines containing a new active substance not authorized in the European UnionEU on or before May 20, 2004, medicines which are highly innovative, or medicines to which the granting of a marketing authorization under the centralized procedure would be in the interest of patients at the European Union-level.EU-level. The decentralized procedure provides for recognition by European UnionEU national authorities of a first assessment performed by one of the member states. Under this procedure, an identical application for marketing authorization is submitted simultaneously to the national authorities of several European UnionEU member states, one of them being chosen as the “Reference Member State,” and the remaining being the “Concerned Member States.” The Reference Member State must prepare and send drafts of an assessment report, summary of product characteristics and the labelling and package leaflet within 120 days after receipt of a valid marketing authorization application to the Concerned Member States, which must decide within 90 days whether to recognize approval. If any Concerned Member State does not recognize the marketing authorization on the grounds of potential serious risk to public health, the disputed points are eventually referred to the European
Commission, whose decision is binding on all member states. The mutual recognition procedure is similar to the decentralized procedure except that a medicine must have already received a marketing authorization in at least one of the member states, and that member state acts as the Reference Member State.
As in the United States, we may apply for designation of a therapeutic candidate as an orphan drug for the treatment of a specific indication in the European UnionEU before the application for marketing authorization is made.
Orphan drugs in the European UnionEU enjoy economic and marketing benefits, including up to ten years of market exclusivity for the approved indication unless another applicant can show that its product is safer, more effective or otherwise clinically superior to the orphan-designated product, the marketing authorization holder is unable to supply sufficient quantity of the medicinal product or the marketing authorization holder has given its consent.
It is not yet clear howOn January 31, 2020, the United Kingdom’s recent withdrawalKingdom formally withdrew from the EU, also known as Brexit. The United Kingdom and the EU entered into a trade agreement known as the Trade and Cooperation Agreement, which went into effect on January 1, 2021. As a result of Brexit, United Kingdom licensing decisions were transferred from European UnionMedicines Agency, or EMA, to the Medicines and Healthcare Products Regulatory Agency, or MHRA, the United Kingdom Regulatory Body. For a period of three years following January 1, 2021, the United Kingdom will affectcontinue to adopt decisions taken by the European Commission on the approval of medicinalnew marketing authorizations. However, companies will be required to submit an identical application to the MHRA upon the Medicinal Products for Human Use, or CHMP, positive opinion of the application. The MHRA will then wait for the European Commission decision on approval. More recently, in March 2023, the United Kingdom government and the European Commission reached agreement on a regulatory framework to replace the Northern Ireland Protocol, referred to as the Windsor Framework. The Windsor Framework is expected to apply as of January 1, 2025 and will change the existing system under the Northern Ireland Protocol, including the regulation of pharmaceutical products in the United Kingdom. Specifically, the MHRA will be responsible for approving all medicines intended to be marketed in the United Kingdom (i.e., Great Britain and Northern Ireland), while the EMA will no longer be involved in approving medicines intended for sale in Northern Ireland.
Since the regulatory framework in the United Kingdom covering the quality, safety and efficacy of pharmaceutical products, clinical trials, marketing authorization, commercial sales and distribution of pharmaceutical products is derived from European UnionEU directives and regulations, Brexit could materially impact the future regulatory regime theas it applies to medicinal products and the approval of product candidates in the United Kingdom. The United Kingdom and the European Union entered into a trade agreement known as the Trade and Cooperation Agreement, which is provisionally applicable as of January 1, 2021 but has not yet been ratified by the European Parliament. In addition, the UK Medicines and Healthcare products Regulatory Agency, or MHRA, recently issued guidance concerning the UK’s recognition, for purposes of granting UK marketing authorizations, of marketing authorizations issued through the European Union’s decentralised and mutual recognition procedures. We are currently evaluating the potential impacts on our business of the new Trade and Cooperation Agreement and the MHRA’s new guidance on approval pathways for medicinal products.
Coverage, Pricing and Reimbursement
Sales of our products will depend, in part, on the extent to which our products will be covered by third-party payors, such as government health programs, commercial insurance and managed healthcare organizations. There may be significant delays in obtaining coverage and reimbursement for approved products, and coverage may be more limited than the purposes for which the product is approved by the FDA or regulatory authorities in other countries. It is time consuming and expensive to seek reimbursement from third-party payors. Moreover, eligibility for reimbursement does not imply that any product will be paid for in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution. Interim payments for new products, if applicable, may also not be sufficient to cover our costs and may not be made permanent. Payment rates may vary according to the use of the product and the clinical setting in which it is used, may be based on payments allowed for lower cost products that are already reimbursed and may be incorporated into existing payments for other services. Net prices for products may be reduced by mandatory discounts or rebates required by third-party payors and by any future relaxation of laws that presently restrict imports of products from countries where they may be sold at lower prices than in the United States.U.S. In the U.S., third-party payors often rely upon Medicare coverage policy and payment limitations in setting their own reimbursement policies, but they also have their own methods and approval
process apart from Medicare coverage and reimbursement determinations. Accordingly, one third-party payor’s determination to provide coverage for a product does not assure that other payors will also provide coverage for the product. In August 2022, President Biden signed into the law the Inflation Reduction Act of 2022, or the IRA. Among other things, the IRA has multiple provisions that may impact the prices of drug products that are both sold into the Medicare program and throughout the United States. A manufacturer of drugs or biological products covered by Medicare Parts B or D must pay a rebate to the federal government if their drug product’s price increases faster than the rate of inflation. This calculation is made on a drug product by drug product basis and the amount of
the rebate owed to the federal government is directly dependent on the volume of a drug product that is paid for by Medicare Parts B or D. Additionally, starting for payment year 2026, the Centers for Medicare & Medicaid Services, or CMS, will negotiate drug prices annually for a select number of single source Part D drugs without generic or biosimilar competition. CMS will also negotiate drug prices for a select number of Part B drugs starting for payment year 2028. If a drug product is selected by CMS for negotiation, it is expected that the revenue generated from such drug will decrease. CMS has begun to implement these new authorities and entered into the first set of agreements with pharmaceutical manufacturers to conduct price negotiations in October 2023. However, the IRA’s impact on the pharmaceutical industry in the United States remains uncertain, in part because multiple large pharmaceutical companies and other stakeholders (e.g., the U.S. Chamber of Commerce) have initiated federal lawsuits against CMS arguing the program is unconstitutional for a variety of reasons, among other complaints. Those lawsuits are currently ongoing.
Additionally, the containment of healthcare costs has become a priority of federal and state governments and the prices of therapeutics have been a focus in this effort. The United States government, state legislatures and foreign governments have shown significant interest in implementing cost-containment programs, including price controls, restrictions on reimbursement and requirements for substitution of generic and biosimilar products. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit our net revenue and results. If these third-party payors do not consider our products to be cost-effective compared to other therapies, they may not cover our products after approval as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow us to sell our products on a profitable basis. In addition, companion diagnostic tests require coverage and reimbursement separate and apart from the coverage and reimbursement for their companion pharmaceutical or biological products. Similar challenges to obtaining coverage and reimbursement for the pharmaceutical or biological products apply to companion diagnostics.
Moreover, in some foreign countries, the proposed pricing for a product and therapeutic candidate must be approved before it may be lawfully marketed. The requirements governing therapeutic pricing vary widely from country to country. For example, the European UnionEU provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. A member state may approve a specific price for the medicinal product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of our therapeutic candidates. Historically, therapeutic candidates launched in the European UnionEU do not follow price structures of the United States and generally tend to be significantly lower.
Healthcare Reform
In the United States and some foreign jurisdictions, there have been, and continue to be, several legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing approval of product and therapeutic candidates, restrict or regulate post-approval activities, and affect the ability to profitably sell product and therapeutic candidates that obtain marketing approval. The FDA’s and other regulatory authorities’ policies may change and additional government regulations may be enacted that could prevent, limit or delay regulatorymarketing approval of our product and therapeutic candidates. For example, in December 2016, the 21st Century Cures Act, or Cures Act, was signed into law. The Cures Act, among other things, is intended to modernize the regulation of drugs and devices and to spur innovation, but its ultimate implementation is uncertain. In addition, in August 2017, the FDA Reauthorization Act was signed into law, which reauthorized the FDA’s user fee programs and included additional drug and device provisions that build on the Cures Act. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we otherwise may have obtained and we may not achieve or sustain profitability, which would adversely affect our business, prospects, financial condition and results of operations. Moreover, among policy makers and payors in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality and/or expanding access.
For example, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or collectively the ACA, was enacted in March 2010 and has had a significant impact on the health care industry in the U.S. The ACA expanded coverage for the uninsured while at the same time containing overall healthcare costs. It also included the BPCIA, which created an abbreviated approval pathway for biological
products that are biosimilar to or interchangeable with an FDA-licensed reference biological product. With regard to biopharmaceutical products, the ACA, among other things, addressed a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected, increased the minimum Medicaid rebates owed by manufacturers under the Medicaid Drug
Rebate Program and extended the rebate program to individuals enrolled in Medicaid managed care organizations, established annual fees on manufacturers of certain branded prescription drugs, and created a new Medicare Part D coverage gap discount program. As another example, the 2021 Consolidated Appropriations Act signed into law on December 27, 2020 incorporated extensive healthcare provisions and amendments to existing laws, including a requirement that all manufacturers of drugs and biological products covered under Medicare Part B report the product’s average sales price, or ASP, to the United States Department of Health and Human Services, or HHS, beginning on January 1, 2022, subject to enforcement via civil money penalties.
Since its enactment, there have been executive, judicial and Congressional challenges The American Rescue Plan Act of 2021 also included a provision that eliminated the statutory cap on rebates that drug manufacturers pay to certain aspectsMedicaid. Beginning in January 2024, Medicaid rebates are no longer capped at 100 percent of the ACA and wequarterly average manufacturer price, or AMP.
We expect there will be additional challenges and amendmentsthat further legislative changes or additions to the ACA, the Medicare and Medicaid programs, and changes stemming from other healthcare reform measures, especially with regard to healthcare access, financing or other legislation in the future. Members of the US Congressindividual states, could have indicated that they may continue to seek to modify, repeal or otherwise invalidate all, or certain provisions of, the ACA. For example, the Tax Cuts and Jobs Act, or TCJA, was enacted in 2017 and, among other things, removed penalties, starting January 1, 2019, for not complying with the ACA’s individual mandate to carry health insurance, commonly referred to as the “individual mandate.” In December 2018, a U.S. District Court Judge in the Northern District of Texas ruled that the individual mandate was a critical and inseverable feature of the ACA, and therefore, because it was repealed as part of the TCJA, the remaining provisions of the ACA were invalid and the law in its entirety was unconstitutional. On December 18, 2019, the U.S. Court of Appeals for the Fifth Circuit upheld the District Court ruling that the individual mandate was unconstitutional but remanded the case back to the District Court to determine whether other reforms enacted as part of the ACA but not specifically related to the individual mandate or health insurance, including the provisions comprising the BPCIA, could be severed from the rest of the ACA so as not to be declared invalid as well. On March 2, 2020, the United States Supreme Court granted the petitions for writs of certiorari to review this case and allocated one hour for oral arguments, which occurred on November 10, 2020. A decision from the Supreme Court is expected to be issued in spring 2021. It is unclear how this litigation and other efforts to repeal and replace the ACA will impact the implementation of the ACA, the pharmaceutical industry more generally, and our business. Complying with any new legislation or reversing changes implemented under the ACA could be time-intensive and expensive, resulting in a material adverse effect on our business.
In addition, other legislative changes have been proposed and adoptedthe health care industry in the United States since the ACA that affect health care expenditures. These changes include aggregate reductions to Medicare payments to providers of up to 2% per fiscal year pursuant to the Budget Control Act of 2011, which began in 2013 and will remain in effect through 2030 unless additional Congressional action is taken. The Coronavirus Aid, Relief, and Economic Security Act, or the CARES Act, which was signed into law on March 27, 2020 and was designed to provide financial support and resources to individuals and businesses affected by the COVID-19 pandemic, suspended the 2% Medicare sequester from May 1, 2020 through December 31, 2020, and extended the sequester by one year, through 2030, in order to offset the added expense of the 2020 cancellation. The 2021 Consolidated Appropriations Act was subsequently signed into law on December 27, 2020 and extends the CARES Act suspension period to March 31, 2021.
Additionally, on December 20, 2019, President Trump signed the Further Consolidated Appropriations Act for 2020 into law, or P.L. 116-94, that includes a piece of bipartisan legislation called the Creating and Restoring Equal Access to Equivalent Samples Act of 2019, or the CREATES Act. The CREATES Act aims to address the concern articulated by both the FDA and others in the industry that some brand manufacturers have improperly restricted the distribution of their products, including by invoking the existence of a REMS for certain products, to deny generic and biosimilar product developers access to samples of brand products. Because generic and biosimilar product developers need samples to conduct certain comparative testing required by the FDA, some have attributed the inability to timely obtain samples as a cause of delay in the entry of generic and biosimilar products. To remedy this concern, the CREATES Act establishes a private cause of action that permits a generic or biosimilar product developer to sue the brand manufacturer to compel it to furnish the necessary samples on “commercially reasonable, market-based terms.” Whether and how generic and biosimilar product developments will use this new pathway, as well as the likely outcome of any legal challenges to provisions of the CREATES Act, remain highly uncertain and its potential effects on our future commercial products are unknown.States.
Moreover, there has been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products, which has resulted in several Congressional inquiries and proposed and enacted federal and
state legislation designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drug products. The United States Department of Health and Human Services, or HHS, has solicited feedback on some of various measures intended to lower drug prices and reduce the out of pocket costs of drugs and implemented others under its existing authority. For example, in May 2019, HHS issued a final rule to allow Medicare Advantage plans the option to use step therapy for Part B drugs beginning January 1, 2020. This final rule codified an HHS policy change that was effective January 1, 2019. As part of the Trump administration’s so-called “Blueprint” to lower drug prices, HHS and FDA also released on July 31, 2019 their Safe Importation Action Plan proposing two different pathways for the importation of foreign drug products. One pathway focuses on the importation of certain drugs from Canada, which required the agencies to go through notice-and-comment rulemaking, while the second pathway allows manufacturers to distribute their drugs manufactured abroad and was released as agency policy in an FDA guidance document first issued in December 2019. FDA’s notice of proposed rulemaking to implement a system whereby state governmental entities could lawfully import and distribute prescription drugs sourced from Canada was published at the end of December 2019 and in September 2020, the rulemaking was finalized by the FDA. Those new regulations became effective on November 30, 2020, although the impact of such future programs is uncertain, in part because lawsuits have been filed challenging the government’s authority to promulgate them. The final resolutions may also be vulnerable to being overturned by a joint resolution of disapproval from Congress under the procedures set forth in the Congressional Review Act, which could be applied to regulatory actions taken by the Trump administration on or after August 21, 2020 (i.e., in the last 60 days of legislative session of the 116th Congress. Congress and the executive branch have each indicated that it will continue to seek new legislative and/or administrative measures to control drug costs making this area subject to ongoing uncertainty.
As another example, in July 2020, President Trump announced four executive orders related to prescription drug pricing that attempted to implement several of his Administration’s proposals, including a policy that would tie Medicare Part B drug prices to international drug prices (which was subsequently implemented by HHS and then challenged in federal court by industry groups in December 2020); on that directed HHS to finalize the Canadian drug importation proposed rule previously issued by HHS (which has since been finalized, as noted above) and makes other changes allowing for personal importation of drugs from Canada; one that directed HHS to finalize the rulemaking process on modifying the anti-kickback law safe harbors for plans, pharmacies, and pharmaceutical benefit managers after HHS confirms that the action is not projected to increase federal spending, Medicare beneficiary premiums, or patients’ total out-of-pocket costs (which HHS finalized in November 2020, also making those rules subject to potentially being overturned under the Congressional Review Act); and one that reduces costs of insulin and epipens to patients of federally qualified health centers. The probability of success of these newly enacted policies and their impact on the U.S. prescription drug marketplace is unknown. There are likely to be political and legal challenges associated with implementing these reforms as they are currently envisioned, and the January 20, 2021 transition to a new Democrat-led presidential administration created further uncertainty. Following his inauguration, President Biden took immediate steps to order a regulatory freeze on all pending substantive executive actions in order to permit incoming department and agency heads to review whether questions of fact, policy, and law may be implicated and to determine how to proceed.
Individual states in the United States have also increasingly passed legislation and implemented regulations designed to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. In December 2020, the U.S. Supreme Court held unanimously that federal law does not preempt the states’ ability to regulate pharmaceuticalpharmacy benefit managers, or PBMs, and other members of the health care and pharmaceutical supply chain, an important decision that may leadhas led to further and more aggressive efforts by states in this area. The Federal Trade Commission in mid-2022 also launched sweeping investigations into the practices of the PBM industry that could lead to additional federal and state legislative or regulatory proposals targeting such entities’ operations, pharmacy networks, or financial arrangements. During the current congressional session, numerous PBM reforms are being considered in both the Senate and the House of Representatives; they include diverse legislative proposals such as eliminating rebates; divorcing service fees from the price of a drug, discount, or rebate; prohibiting spread pricing; limiting administrative fees; requiring PBMs to report formulary placement rationale; promoting transparency. Significant efforts to change the PBM industry as it currently exists in the U.S. may affect the entire pharmaceutical supply chain and the business of other stakeholders, including therapeutic biological product developers like us.
We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative or executive action, either in the United States or abroad. We expect that additional state and federal health care reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for health care products and services.
Other Healthcare Laws
Our current and future business operations are subject to healthcare regulation and enforcement by the federal government and the states and foreign governments where we research, and, if approved, market, sell and distribute our therapeutic candidates. These laws include, without limitation, state and federal anti-kickback, fraud and abuse, false claims, privacy and security, physician sunshine and drug pricing transparency laws and regulations such as:
•The federal Anti-Kickback Statute prohibits, among other things, any person from knowingly and willfully offering, soliciting, receiving or providingpaying remuneration, directly or indirectly, to induce either the referral of
an individual, for an item or service or the purchasing or ordering of a good or service, for which payment may be made under federal healthcare programs such as the Medicare and Medicaid programs. The federal Anti-Kickback Statute is subject to evolving interpretations.broadly interpreted and aggressively enforced with the result that beneficial commercial arrangements can be penalized in the healthcare industry. In the past, the government has enforced the federal Anti-Kickback Statute to reach large settlements with healthcare companies based on a variety of arrangements, including sham consulting and other financial arrangements with physicians. Penalties for violating the federal Anti-Kickback Statute include imprisonment, fines and possible exclusion from federal healthcare programs such as Medicare and Medicaid. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. In addition, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the civil False Claims Act;
•The federal civil and criminal false claims laws, including the civil False Claims Act, and civil monetary penalty laws, prohibit, among other things, knowingly presenting or causing the presentation of a false, fictitious or fraudulent claim for payment to the U.S. government, knowingly making, using, or causing to be made or used a false record or statement material to a false or fraudulent claim to the U.S. government, or from knowingly making a false statement to avoid, decrease or conceal an obligation to pay money to the U.S. government. Actions under these laws may be brought by the Attorney General or as a qui tam action by a private individual in the name of the government. The federal government uses these laws, and the accompanying threat of significant liability, in its investigation and prosecution of pharmaceutical and biotechnology companies throughout the U.S., for example, in connection with the promotion of products for unapproved uses and other allegedly unlawful sales and marketing practices;practices. If an entity is found to have violated the False Claims Act, it must pay three times the actual damages sustained by the government, plus mandatory and substantial civil penalties;
•The U.S. federal Health Insurance Portability and Accountability Act of 1996 and its accompanying regulations, or HIPAA, created new federal, civil and criminal statutes that prohibit among other actions, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payors, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense, and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation;
•The Physician Payments Sunshine Act, enacted as part of the ACA, among other things, imposes reporting requirements on manufacturers of FDA-approved drugs, devices, biologics and medical supplies covered by Medicare or Medicaid to report, on an annual basis, to HHSCMS information related to payments and other transfers of value to physicians (defined to include doctors, dentists, optometrists, podiatrists, chiropractors and beginning in 2022 for paymentschiropractors), teaching hospitals and other transfers of value provided in the previous year, certain advanced non-physician health care practitioners), teaching hospitals,healthcare practitioners, as well as ownership and investment interests held by physicians and their immediate family members;
•HIPAA, as amended bymembers. The law provides for the Health Information Technology for Economicimposition of civil monetary penalties, and Clinical Health Act, or HITECH,payments reported also have the potential to draw scrutiny on payments and their respective implementing regulations impose specified requirements relating torelationships with physicians, which may have implications under the privacy, securityAnti-Kickback Statute and transmission of individually identifiable health information. Among other things, HITECH makes HIPAA’s privacy and security standards directly applicable to “business associates,” defined as independent contractors or agents of covered entities, which include certain healthcare providers, health plans, and healthcare clearinghouses, that create, receive, maintain or transmit protected health information in connection with providing a service for or on behalf of a covered entity. HITECH also increased the civil and criminal penalties that may be imposed against covered entities, business associates and possibly other persons, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce HIPAA and seek attorney’s fees and costs associated with pursuing federal civil actions; andlaws;
•Analogous state laws and regulations, such as state anti-kickback and false claims laws which may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers; state laws which require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government in addition to requiring drug and therapeutic biologics manufacturers to report information related to payments to physicians and other healthcare providers or marketing expenditures and pricing information; state and local laws which require the registration of pharmaceutical sales representatives; and state laws and non-United States laws and regulations (particularly European UnionEU laws regarding personal data relating to individuals based in Europe) that govern the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways, thus complicating compliance efforts.
Moreover, in November 2020, HHS finalized significant changes to the regulations implementing the Anti-Kickback Statute, as well as the Physician Self-Referral Law, or Stark Law, and the civil monetary penalty rules regarding beneficiary inducements, with the goal of offering the healthcare industry more flexibility and reducing the regulatory burden associated with those fraud and abuse laws, particularly with respect to value-based arrangements among industry participants. As noted above in the section called “Healthcare Reform,” however, those final rules may be potentially overturned under the Congressional Review Act following the change in control of the legislative and executive branches in January 2021.
Ensuring that our current and future business arrangements with third parties comply with applicable healthcare laws and regulations could involve substantial costs. It is possible that governmental authorities will conclude that our business practices do not comply with current or future statutes, regulations, agency guidance or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any such requirements, we may be subject to significant civil, criminal and administrative penalties, including monetary damages, fines, disgorgement, imprisonment, loss of eligibility to obtain approvals from the FDA, exclusion from participation in government contracting, healthcare reimbursement or other government programs, including Medicare and Medicaid, reputational harm, diminished profits and future earnings, additional reporting requirements if we become subject to a corporate integrity agreement or other agreement to resolve allegations of non-compliance with any of these laws, and the curtailment or restructuring of our operations.
U.S. and European Data Security and Data Privacy Laws
HIPAA, as well as a number of other federal and state privacy-related laws, extensively regulate the use and disclosure of individually identifiable health information, known as “protected health information” or “PHI”.
HIPAA applies to health plans, healthcare providers who engage in certain standard healthcare transactions electronically, such as electronic billing, and healthcare clearinghouses, all of which are referred to as “covered entities” under HIPAA. State imposed health information privacy and security laws typically apply based on licensure, for example, licensed providers or licensed entities are limited in their ability to use and share health information. State and federal consumer protection laws, including the Federal Trade Commission Act, govern the collection, use disclosure and protection of health and other personal information and could apply to our operations and the operations of our collaborators.
Additionally, all states have enacted legislation protecting the privacy and security of “personal information” such as identifiable financial or health information, social security number and credit card information. These laws overlap and apply simultaneously with federal privacy and security requirements and regulated entities must comply with all of them. The California Consumer Privacy Act, or CCPA, that went into effect January 1, 2020, is one of the most restrictive state privacy laws, protecting a wide variety of personal information and granting significant rights to California residents with respect to their personal information. Regulations under CCPA have been modified several times. While there is currently an exception under CCPA for protected health information that is subject to HIPAA and clinical trial regulations, CCPA still applies to the personal information of employees and may otherwise impact our business. Additionally, an amendment to the CCPA, the California Privacy Rights Act, or CPRA, was approved by California voters in the election of November 3, 2020 and took full effect on January 1, 2023. CPRA modifies CCPA significantly, potentially resulting in further uncertainty, additional costs and expenses stemming from efforts to comply, and additional potential for harm and liability for failure to comply. Among other things, CPRA established a new regulatory authority, the California Privacy Protection Agency, or CPPA, with expanded enforcement authority. The CPPA is currently promulgating additional regulations under the CPRA, which could lead to added uncertainty and operational risks. Other states in the U.S. are considering privacy laws similar to CCPA increasing the regulatory and enforcement risk. In addition to California, several states have similar omnibus privacy laws which took effect in 2023, including Virginia, Colorado, Connecticut, and Utah, that may have operational impacts on our business. In dealing with health information for the development of our technology or for commercial purposes, we will be indirectly affected by HIPAA and state-imposed health information privacy and security laws because these laws regulate the ability of our potential customers and research collaborators to share health information with us. Additionally, we must identify and comply with all applicable state laws for the protection of personal information with respect to employee information or other personal information that the company collects. See Part I, Item 1A “Risk Factors-We face regulation and potential liability related to privacy, data protection and information security which may require significant resources and may adversely affect our business, operations and financial performance.”
In the EU, increasingly stringent data protection and privacy rules that have and will continue to have substantial impact on the use of personal and patient data across the healthcare industry became stronger in May 2018. The EU General Data Protection Regulation, or GDPR, applies across the EU and includes, among other things, a requirement for prompt notice of data breaches to data subjects and supervisory authorities in certain circumstances and significant fines for non-compliance. The GDPR fine framework can be up to 20 million euros, or up to 4% of
the company’s total global turnover of the preceding fiscal year, whichever is higher. The GDPR sets out a number of requirements that must be complied with when processing the personal data of such EU based data subjects including: providing expanded disclosures about how their personal data will be used; higher standards for organizations to demonstrate that they have obtained valid consent or have another legal basis in place to justify their data processing activities; the obligation to appoint data protection officers in certain circumstances; new rights for individuals to be “forgotten” and rights to data portability, as well as enhanced current rights (e.g., access requests); the principal of accountability and demonstrating compliance through policies, procedures, training and audit; and the new mandatory data breach regime. In particular, medical or health data, genetic data and biometric data where the latter is used to uniquely identify an individual are all classified as “special category” data under the GDPR and are afforded greater protection and require additional compliance obligations. Noncompliance could result in the imposition of fines, penalties, or orders to stop noncompliant activities. We may be subject to GDPR if we undertake operations in the EU, offer products or services to individuals in the EU or monitor the behavior of individuals within the EU.
We could also be subject to evolving EU laws on data export, for transfers of data outside the EU to us, group companies or third parties. The GDPR only permits exports of data outside the EU to jurisdictions that ensure an adequate level of data protection. The United States has not been deemed to offer an adequate level of protection, so in order for us to transfer personal data from the EU to the United States, we must identify a legal basis for data transfer (e.g., the EU Commission approved Standard Contractual Clauses or certification under the recently-adopted EU-US Data Privacy Framework). On July 16, 2020, the Court of Justice of the EU or the CJEU, issued a landmark opinion in the case Maximilian Schrems vs. Facebook (Case C-311/18), called Schrems II. This decision (i) calls into question commonly relied upon data transfer mechanisms as between the EU member states and the United States (such as the Standard Contractual Clauses) and (ii) invalidates the EU-U.S. Privacy Shield on which many companies had relied as an acceptable mechanism for transferring such data from the EU to the United States. The CJEU is the highest court in Europe and the Schrems II decision heightens the burden on data importers to assess U.S. national security laws on their business and future actions of EU data protection authorities are difficult to predict. While the recently-adopted EU-US Data Privacy Framework was meant to address the concerns raised by the CJEU in Schrems II and provide an approved method for cross-border data transfer from the EU to the US, it will likely be subject to future legal challenges and we have not certified to participate in the EU-US Data Privacy Framework.
Where we rely on third parties to carry out services for us, including processing personal data on our behalf, we are required under GDPR to enter into contractual arrangements to help ensure that these third parties only process such data according to our instructions and have sufficient security measures in place. Any security breach or non-compliance with our contractual terms or breach of applicable law by such third parties could result in enforcement actions, litigation, fines and penalties or adverse publicity and could cause our customers to lose trust in us, which could have an adverse impact on our reputation and business. Any contractual arrangements requiring the transfer of personal data from the EU to us in the United States will require greater scrutiny and assessments as required under Schrems II and may have an adverse impact on cross-border transfers of personal data, or increase costs of compliance.
Other Laws and Regulations
Our present business is, and our future business may be, subject to regulation under the Clean Air Act, the Clean Water Act, the Comprehensive Environmental Response, Compensation and Liability Act, the National Environmental Policy Act, the Toxic Substances Control Act, the Resource Conservation and Recovery Act, national restrictions, and other current and potential future local, state, federal, and foreign regulations. See Part I, Item 1A “Risk Factors- If we do not comply with laws regulating the protection of the environment and health and human safety, our business could be adversely affected.”
Manufacturing
We do not own or operate manufacturing facilities for the production of any of our therapeutic candidates, nor do we have plans to develop our own manufacturing operations in the foreseeable future. We presently have relationships with suppliers for the manufacture of supplies for all of our required raw materials, antibodies and other biologics
for our preclinical research, clinical trials, and if and when applicable, commercialization. We currently employ internal resources to manage our manufacturing relationships with these third parties.relationships.
FacilitiesHuman Capital Resources
Our headquartersemployees are locateda key factor in La Jolla, California where we currently lease approximately 34,000 square feet of laboratoryour ability to achieve our mission to discover and office space under a lease that expires in 2025. We have an option to extend the lease an additional five years. In May 2019, we executed an amendment to our lease agreement to expand our facilities by approximately 9,000 square feet and occupancydevelop effective biologic therapeutics for that space commenced in January 2020.people with life-threatening conditions. We believe that this space is sufficientour future success depends on our continued ability to meetidentify, recruit, retain, and incentivize our needs for the foreseeable futuremanagement team and that any additional space we may require will be availableour clinical, scientific, and other employees. Our ability to do so depends on commercially reasonable terms.factors including our Company culture, compensation and benefits, growth and development opportunities, and prioritization of employee safety.
EmployeesEmployee Profile
As of December 31, 2020,2023, we had 91172 employees, 84166 of whom were full-time, and 79137 of whom were engaged in research and development activity.activity, and 78 of which hold advanced degrees, including but not limited to Ph.D., M.D., PharmD, J.D., MBA, and other master’s degrees. None of our employees are represented by a labor union and we believe we maintain good relations with our employees.
Diversity, Equity and Inclusion
Our employees represent a broad range of backgrounds and bring a wide array of perspectives and experiences. A key component of our culture is our commitment to diversity, equity, and inclusion, or DEI. We believe this commitment allows us to better drive innovation and achieve our mission. Our DEI principles are reflected in our efforts in building a better workplace where employees can be themselves and succeed, and use their voice and influence to create a better world. We are consciously expanding the diversity of our workforce, creating growth and development opportunities for our employees, embracing different perspectives and fostering an inclusive work environment.
Employee Conduct & Ethics
Inhibrx adopted and regularly reviews corporate policies, including a Code of Conduct and Ethics and Whistleblower Policy, which apply to all of our employees. All employees complete a mandatory public company training session and are required to abide by, review and confirm compliance to the Company’s Corporate Code of Conduct and Ethics and Whistleblower Policy, as well as our Insider Trading Policy governing trading by Company personnel in the Company’s securities. We have established a Whistleblower reporting hotline to enable our employees to anonymously report any suspected violations of these policies. In addition, the Company requires employees to complete Anti-Harassment Training, with employees who work in a management capacity required to complete additional trainings in Harassment Prevention and Diversity, Equity and Inclusion.
Employee Compensation and Benefits
Our compensation programs are designed to reward and support our employees in order to continue to attract and retain top talent. Our compensation includes:
•Employee base salaries that are competitive and consistent with employee positions, skill levels, experience, and knowledge;
•Stock-based compensation awards which help to align the interests of our stockholders with those of our employees;
•Bonus award plans for all full-time employees;
•Retirement savings options and matching contributions;
•Fully covered healthcare benefits for all full-time employees and their dependents;
•Unlimited vacation benefit for all full-time employees; and
•Parental leave and other leave options available to all employees.
Employee Growth and Development
Inhibrx is committed to fostering and growing talent within the biopharmaceutical and life sciences space. We provide internship opportunities for students interested in biotechnology and science within our research and development departments. Many of our interns have continued on to join the Company in a full-time position after graduation. Our hiring process is transparent and we are an equal opportunity employer. Many of our employees
hold advanced degrees, as well as professional licenses and certifications; however, the Company equally commits resources to advancing all of our employees with a range of educational backgrounds. We offer tuition reimbursement aimed at growth and career development, as well as the opportunity for employees to attend relevant conferences and symposiums. In addition, we offer in-house coaching opportunities to refine or develop professional skills as our employees become managers and plan their career growth.
Employee Wellness, Health, and Safety
We are strongly committed to the health and safety of our employees and strive to maintain the highest possible level of safety in our workplace. We require annual workplace safety training to reinforce workplace safety procedures that may be useful in the event of emergency situations and to assist our employees in helping to prevent workplace accidents. Our Environmental Health and Safety Committee, which is comprised of numerous cross-departmental members, meets regularly to review workplace safety and adherence to safety policies.
Corporate Information
We were incorporated under the laws of the State of Delaware on November 17, 2017 under the name Tenium Therapeutics, Inc. In April 2018, we changed our name to Inhibrx, Inc.
Available Information
Our internet address is www.inhibrx.com, to which we regularly post copies of our press releases as well as additional information about us. Our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and all amendments to those reports, are available to you free of charge through the Investors section on our website as soon as reasonably practicable after such materials have been electronically filed with, or furnished to, the SEC. The SEC maintains an internet site (http://www.sec.gov) that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC. We include our web site address in this Annual Report on Form 10-K only as an inactive textual reference. Information contained in our website does not constitute a part of this report or our other filings with the SEC.
Item 1A. Risk Factors.
Investing in our common stock involves a high degree of risk. You should carefully consider the risks and uncertainties described below, together with all of the other information in this Annual Report, including our consolidated financial statements and related notes, before investing in our common stock. The risks and uncertainties described below are not the only ones we face. Additional risks and uncertainties that we are unaware of, or that we currently believe are not material, may also become important factors that affect us. If any of the following risks occur, our business, operating results, financial condition, and prospects could be materially harmed. In that event, the price of our common stock could decline, and you could lose part or all of your investment.
Summary Risk Factors
Our business is subject to a number of risks of which you should be aware before making an investment decision. These risks are discussed more fully in the section of this Annual Report titled “Risk Factors”. These risks include, among others, the following:
Risks Related to the Proposed Merger and Spin-Off
•The consummation of the Merger is subject to a number of conditions, many of which are largely outside of the parties’ control, and, if these conditions are not satisfied or waived on a timely basis, the Merger Agreement may be terminated and the Merger may not be completed.
•Failure to complete the Merger could adversely affect the stock price and future business and financial results of the Company.
•While the Merger is pending, the Company will be subject to business uncertainties and certain contractual restrictions that could adversely affect the business and operations of the Company.
•Litigation against the Company, Aventis, or the members of their respective boards, could prevent or delay the completion of the Merger or result in the payment of damages following completion of the Merger.
Risks Related to Our Financial Condition and Need for Additional Capital
•We have a limited operating history, have incurred significant losses since our inception and anticipate that we will continue to incur significant losses for the foreseeable future. We may never generate product revenue or become profitable, or if we achieve profitability, we may not be able to sustain it.
•Biotechnology product development is a highly speculative undertaking and involves a substantial degree of uncertainty. We have never generated any revenue from product sales and may never be profitable.
•We expect we will need to raise substantial additional funds to advance development of our therapeutic candidates, and we cannot guarantee this additional funding will be available on acceptable terms or at all. Failure to obtain this funding when needed may force us to delay, limit or terminate our development efforts and, if any of our therapeutic candidates are approved, our commercialization efforts.
•We may be adversely affected by the effects of inflation.
Risks Related to the Development, Clinical Testing and Commercialization of Our Therapeutic Candidates
•Our therapeutic candidates are in earlyvarious stages of development and may fail or suffer delays that materially and adversely affect their commercial viability, and the results of any preclinical studies or clinical trials may not be predictive of the results of later clinical trials.viability. If we, or our collaboration partners, are unable to advance our therapeutic candidates through clinical development, obtain regulatorymarketing approval and ultimately commercialize our therapeutic candidates, or experience significant delays in doing so, our business will be materially harmed.
•The regulatory approval processes of the FDA and comparable foreign authorities are lengthy, time consuming and inherently unpredictable, and if we are ultimately unable to obtain regulatory approval for our therapeutic candidates, our business will be substantially harmed.
•We will need substantial additional funding in order to complete the development and commercialization of our therapeutic candidates.
•We rely on third parties to conduct alla portion of our clinical trials and certain of our preclinical studies and for the manufacture of our therapeutic candidates and we intend to continue to do so.nonclinical studies. If these third parties do not perform as contractually required, fail to satisfy regulatory or legal requirements or miss expected deadlines, our development programs could be delayed with material and adverse effects on our business, financial condition, results of operations and prospects.
•If we are not ableRisks Related to obtainOur Organization and enforce patent protection for our technologies or therapeutic candidates, development and commercialization of our therapeutic candidates may be adversely affected.Operations
•We face significant competition and if our competitors develop and market products that are more effective, safer or less expensive than the therapeutic candidates we develop, our commercial opportunities will be negatively impacted.
•Our business, the conduct of our clinical trials, regulatory approvalresults of operations, financial condition, and prospects access to capital, performance of third parties and our business may be adversely affected by circumstancesthe widespread outbreak of an illness or any other communicable disease, or any other public health crisis, including the coronavirus disease.
Risks Related to Intellectual Property
•If we are not able to obtain and uncertainties relatedenforce patent protection for our technologies or therapeutic candidates, development and commercialization of our therapeutic candidates may be adversely affected.
Risks Related to Government Regulation
•We or our collaborators may be unable to obtain marketing approval for any product that we or a collaborator may develop and the marketing approval processes of the FDA and other comparable regulatory authorities outside the United States are lengthy, time consuming and inherently unpredictable.
Risks Related to Ownership of Our Common Stock
•We do not know whether an active, liquid and orderly trading market will continue for our common stock and as a result it may be difficult for you to sell your shares of our common stock.
•We expect that our stock price may fluctuate significantly.
•Our executive officers, directors and holders of more than 5% of our capital stock own a significant percentage of our stock and will be able to exercise significant control over matters subject to stockholder approval.
•Raising additional capital may cause dilution to our existing stockholders, restrict our operations or require us to relinquish rights to our technologies or product candidates.
Risks Related to the COVID-19 pandemic.Proposed Merger and Spin-Off
The consummation of the Merger is subject to a number of conditions, many of which are largely outside of the parties’ control, and, if these conditions are not satisfied or waived on a timely basis, the Merger Agreement may be terminated and the Merger may not be completed.
The Merger is subject to certain customary closing conditions, including: (i) adoption of the Merger Agreement by holders of a majority of the outstanding shares of Company common stock entitled to vote on such matters at the Company’s shareholders meeting and who are present at the shareholders meeting, in person or by proxy; (ii) the expiration, termination or receipt of any approval or clearances applicable to the consummation of the Merger under applicable antitrust laws, including the Hart-Scott-Rodino Antitrust Improvements Act of 1976 and the receipt of certain additional clearances or approvals of certain other governmental bodies applicable to the Merger; (iii) the absence of any law or order prohibiting or making illegal the consummation of the Merger; (iv) effectiveness of the registration statement to be filed with respect to registration of the common stock of New Inhibrx that will be distributed in the spin-off; (v) completion of the spin-off; (vi) subject to certain qualifications, the accuracy of representations and warranties of the Company, Aventis and Art Acquisition Sub, Inc., or Merger Sub, as applicable, under the Merger Agreement and the performance in all material respects by the Company, Aventis and Merger Sub, as applicable, of their obligations under the Merger Agreement; and (vii) the absence of any Company Material Adverse Effect (as defined in the Merger Agreement).
The failure to satisfy all of the required conditions could delay the completion of the Merger by a significant period of time or prevent it from occurring. Any delay in completing the Merger could cause the parties to not realize some or all of the benefits that are expected to be achieved if the Merger is successfully completed within the expected timeframe. There can be no assurance that the conditions to closing of the Merger will be satisfied or waived or that the Merger will be completed within the expected timeframe or at all.
Failure to complete the Merger could adversely affect the stock price and future business and financial results of the Company.
There can be no assurance that the conditions to the closing of the Merger will be satisfied or waived or that the Merger will be completed. If the Merger is not completed within the expected timeframe or at all, the ongoing business of the Company could be adversely affected and the Company will be subject to a variety of risks and possible consequences associated with the failure to complete the Merger, including the following: (i) upon termination of the Merger Agreement under specified circumstances, the Company is required to pay Aventis a termination fee of $54,500,000; (ii) the Company will incur certain transaction costs, including legal, accounting,
financial advisor, filing, printing and mailing fees, regardless of whether the Merger closes; (iii) under the Merger Agreement, the Company is subject to certain restrictions on the conduct of its business prior to the closing of the Merger, which may adversely affect its ability to execute certain of its business strategies; and (iv) the proposed Merger, whether or not it closes, will divert the attention of certain management and other key employees of the Company from ongoing business activities, including the pursuit of other opportunities that could be beneficial to the Company as an independent company.
If the Merger is not completed, these risks could materially affect the business and financial results of the Company and its stock price, including to the extent that the current market price of the Company’s common stock is positively affected by a market assumption that the Merger will be completed.
While the Merger is pending, the Company will be subject to business uncertainties and certain contractual restrictions that could adversely affect the business and operations of the Company.
In connection with the pending Merger, some customers, vendors or other third parties of the Company may react unfavorably, including by delaying or deferring decisions concerning their business relationships or transactions with the Company, which could adversely affect the revenues, earnings, funds from operations, cash flows and expenses of the Company, regardless of whether the Merger is completed. In addition, due to certain restrictions in the Merger Agreement on the conduct of business prior to completing the Merger, the Company may be unable (without the other party’s prior written consent), during the pendency of the Merger, to pursue strategic transactions, undertake significant capital projects, undertake certain significant financing transactions and otherwise pursue other actions, even if such actions would prove beneficial and may cause the Company to forego certain opportunities it might otherwise pursue. In addition, the pendency of the Merger may make it more difficult for the Company to effectively retain and incentivize key personnel and may cause distractions from the Company’s strategy and day-to-day operations for its current employees and management.
The Company will incur substantial transaction fees and Merger-related costs in connection with the Merger that could adversely affect the business and operations of the Company if the Merger is not completed.
The Company expects to incur non-recurring transaction fees, which include legal and advisory fees and substantial Merger-related costs associated with completing the Merger, and which could adversely affect the business operations of the Company if the Merger is not completed.
The termination fee and restrictions on solicitation contained in the Merger Agreement may discourage other companies from trying to acquire the Company.
The Merger Agreement prohibits the Company from initiating, soliciting, proposing or knowingly encouraging or knowingly facilitating any competing acquisition proposals, subject to certain limited exceptions. The Merger Agreement also contains certain termination rights, including, but not limited to, the right of the Company to terminate the Merger Agreement to accept a Superior Proposal (as defined in the Merger Agreement), subject to and in accordance with the terms and conditions of the Merger Agreement, and provides that, upon termination of the Merger Agreement by the Company to enter into an alternative acquisition agreement with respect to a Superior Proposal, the Company will be required to pay Aventis a termination fee of $54,500,000 in cash. The termination fees and restrictions could discourage other companies from trying to acquire the Company even though those other companies might be willing to offer greater value to the Company’s stockholders than Aventis has offered in the Merger.
Litigation against the Company, Aventis, or the members of their respective boards, could prevent or delay the completion of the Merger or result in the payment of damages following completion of the Merger.
It is a condition to the Merger that no injunction or other order preventing the consummation of the Merger shall have been issued by any court of competent jurisdiction or other governmental authority of competent jurisdiction and remain in effect. It is possible that lawsuits may be filed by the Company’s stockholders challenging the Merger. The outcome of such lawsuits cannot be assured, including the amount of costs associated with defending these claims or any other liabilities that may be incurred in connection with the litigation of these claims. If plaintiffs are successful in obtaining an injunction prohibiting the parties from completing the Merger on the agreed-upon terms, such an injunction may delay the consummation of the Merger in the expected timeframe, or may prevent the Merger from being consummated at all. Whether or not any plaintiff’s claim is successful, this type of litigation can
Uncertainty about the Merger may adversely affect the relationships between the Company and its customers, vendors and employees, whether or not the Merger is completed.
TableIn response to the announcement of Contentsthe Merger, existing or prospective customers, vendors and other third party relationships of the Company may delay, defer or cease providing goods or services, delay or defer other decisions concerning the Company, refuse to extend credit to the Company, or otherwise seek to change the terms on which they do business with the Company. Any such delays or changes to terms could materially harm the Company’s business.In addition, as a result of the Merger, current and prospective employees could experience uncertainty about their future with the Company. These uncertainties may impair the Company’s ability to retain, recruit or motivate key management and technical, manufacturing, and other personnel.
If the Merger is not consummated by September 22, 2024, unless extended in accordance with the Merger Agreement, either the Company or Aventis may terminate the Merger Agreement, subject to certain exceptions.
Either the Company or Aventis may terminate the Merger Agreement if the Merger has not been consummated by September 22, 2024, subject to certain extensions in the Merger Agreement. However, this termination right will not be available to a party if that party failed to fulfill its obligations under the Merger Agreement and that failure was the proximate cause of the failure to consummate the Merger on time. In the event the Merger Agreement is terminated by either party due to the failure of the Merger to close by September 22, 2024, unless extended, the Company will have incurred significant costs and will have diverted significant management focus and resources from other strategic opportunities and ongoing business activities without realizing the anticipated benefits of the Merger.
Risks Related to Our Financial Condition and Need for Additional Capital
We have a limited operating history, have incurred significant losses since our inception and anticipate that we will continue to incur significant losses for the foreseeable future. We may never generate product revenue or become profitable, or if we achieve profitability, we may not be able to sustain it.
We are a clinical-stage biotechnologybiopharmaceutical company. To date, we have financed our operations through equity and debt financings, license and milestone revenue and grants. We have incurred significant recurring operating losses since our inception. Our net loss for the yearsyear ended December 31, 2020 and 20192023 was $76.1 million and $51.4 million, respectively.$241.4 million. As of December 31, 2020,2023, we had an accumulated deficit of $145.4$613.7 million. We expect to incur additional losses in future years as we execute our plan to continue our discovery, research and development activities, including the manufacturing of and ongoing and planned preclinical and clinical development and commercialization of our therapeutic candidates, and as we incur the additional costs of operating as a public company.candidates. We are unable to predict the extent of any future losses or when we will become profitable, if ever. Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our failure to become and remain profitable would decrease the value of our company and could impair our ability to raise capital, maintain our research and development efforts, expand our business or continue our operations. A decline in the value of our company could also cause you to lose all or part of your investment.
Biotechnology product development is a highly speculative undertaking and involves a substantial degree of uncertainty. We have never generated any revenue from product sales and may never be profitable.
We have devoted substantially all of our financial resources and efforts to developing our therapeutic candidates, identifying potential therapeutic candidates and conducting preclinicalnonclinical studies and clinical trials. We are still in the early stagesdevelopment stage for all of developing our therapeutic candidates, and while we have not yet demonstrated an ability to successfully conduct orand complete anycertain of our clinical trials, including large-scale,we have yet to demonstrate an ability to conduct pivotal clinical trials, obtain marketing approval, manufacture a commercial scale product or arrange for a third party to do so on our behalf or conduct sales and marketing activities necessary for successful product commercialization. Consequently, we have no meaningful operations upon which to evaluate our business and predictions about our future success or viability may not be as accurate as they could be if we had more experience developing therapeutic candidates. We previously entered into the Chiesi Option Agreement and a license agreement, or the Celgene Agreement, with Celgene Corporation, a Bristol-Myers Squibb Company, or Celgene.certain collaboration agreements. Our ability to generate revenue and achieve
profitability including any revenue we may receive pursuant to the Chiesi Option Agreement or the Celgene Agreement, depends in large part on our ability, alone or with license partners, to achieve milestones and to successfully complete the development of, obtain the necessary regulatorymarketing approvals for, and commercialize, our therapeutic candidates. For example, while we believe it is possible Celgene may incorporate our proprietary CD47 binding domain, or the Celgene Licensed Intellectual Property, into a multispecific format with another antibody for future development pursuant to the Celgene Agreement, this may not occur. We believe it is unlikely we will receive additional milestone payments pursuant to the Celgene Agreement unless Celgene develops a therapeutic candidate under the Celgene Agreement and advances that therapeutic candidate beyond Phase 1 clinical trials. Even if we achieve development or commercial milestones, generate product royalties or generate product sales, including any milestones and royalties we may be eligible to receive pursuant to the Celgene Agreement or the Chiesi Option Agreement, we may never achieve or sustain profitability on a quarterly or annual basis. We do not anticipate generating revenue from sales of products for the foreseeable future. Our ability to generate future revenue from product sales depends heavily on our and our collaborators’ success in:
•completing clinical trials through all phases of clinical development of our current therapeutic candidates, including INBRX-101, INBRX-109, INBRX-106, INBRX-105, and INBRX-101;INBRX-106;
•advancing our preclinical therapeutic candidates into clinical development;
•seeking and obtaining marketing approvals for our therapeutic candidates that successfully complete clinical trials;
•obtaining satisfactory acceptance, formulary placement coverage and adequate reimbursement for our approved products from third-party payors, including private health insurers, managed care providers and governmental payor programs, including Medicare and Medicaid;
•launching and commercializing products for which we obtain marketing approval, with a collaborator or, if launched independently, successfully establishing a sales force, marketing and distribution infrastructure;
•establishing and maintaining supply and manufacturing relationships with third parties;
•obtaining market acceptance of any approved products by physicians, patients, third-party payors and the medical community;
•maintaining, protecting, expanding and enforcing our intellectual property portfolio;
•implementing additional internal systems and infrastructure, as needed; and
•attracting, hiring and retaining qualified personnel.
Because of the numerous risks and uncertainties associated with biological product development, we are unable to accurately predict the timing or amount of increased expenses or when, or if, we will be able to achieve profitability. If we are required by the FDA, the European Medicines Agency, or EMA, or other comparable foreign authorities to perform preclinicalnonclinical studies or clinical trials in addition to those we currently anticipate, or if there are any delays in completing our clinical trials or the development of any of our therapeutic candidates, our expenses could increase and revenue could be further delayed.
We expect we will need to raise substantial additional funds to advance development of our therapeutic candidates, and we cannot guarantee this additional funding will be available on acceptable terms or at all. Failure to obtain this funding when needed may force us to delay, limit or terminate our development efforts and, if any of our therapeutic candidates are approved, our commercialization efforts.
As of December 31, 2020,2023, we had $128.7$277.9 million in cash.cash and cash equivalents. We expect our expenses to increase in future years as we execute our plan to continue our discovery, research and development activities, including the ongoing and planned preclinical and clinical development and commercialization of our therapeutic candidates. Identifying potential therapeutic candidates and conducting preclinical testing and clinical trials are time consuming, expensive and uncertain processes that take years to complete, and we, or our collaborators, may never generate the necessary data or results required to obtain regulatorymarketing approval and achieve product sales. In addition, our therapeutic candidates, if approved, may not achieve commercial success.
We believe that our existing cash and cash equivalents will be sufficient to fund our planned operations through at least the 12 month period following the date of this Annual Report. However, changing circumstances or inaccurate estimates by us may cause us to use capital significantly faster than we currently anticipate, and we may need to spend more money than currently expected because of circumstances beyond our control. For example, our current and our planned preclinical studies and clinical trials for our current therapeutic candidates or other therapeutic candidates we may seek to develop may encounter technical, enrollment or other issues that could cause our development costs to increase more than we expect, or we could expand our clinical trials to additional indications which could increase clinical trial expenses. Because successful development of our therapeutic candidates is uncertain, we are unable to estimate the actual funds we will require to complete research and development and commercialize our therapeutic candidates. Our ability to raise additional funds will depend on financial, economic and market conditions and other factors, over which we may have no or limited control. In addition, our ability to
obtain future funding when needed through equity financings, debt financings or strategic collaborations may be particularly challenging in light of the uncertaintiesrecent market and circumstances regarding the COVID-19 pandemic (as defined below).macroeconomic conditions, which have been particularly challenging for research and development life science companies.
If adequate funds are not available on commercially acceptable terms when needed, we may be forced to delay, reduce or terminate the development or commercialization of all or part of our research programs or clinical therapeutic candidates, or we may be unable to take advantage of future business opportunities. In addition, any additional capital raising efforts may divert our management from their day-to-day activities, which may adversely affect our ability to develop and commercialize our current and future therapeutic candidates.
Raising additional capital by issuing equity or debt securities may cause dilution to existing stockholders, and raising funds through lending and licensing or collaboration agreements may restrict our operations or require us to relinquish proprietary rights.
Until such time as we can generate substantial revenue from product sales, if ever, we expect to finance our cash needs through a combination of equity and debt financings, strategic collaborations and license and development agreements. We do not have any committed external source of funds.funds and pursuant to the terms of the Merger Agreement, we cannot incur additional indebtedness or issue additional equity prior to the closing of, or other termination of, the Merger. To the extent that we raise additional capital by issuing equity securities, our existing stockholders’ ownership may experience substantial dilution, and the terms
of these securities may include liquidation or other preferences that adversely affect your rights as a stockholder. Equity and debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as redeeming our shares, making investments, incurring additional debt, making capital expenditures or declaring dividends.
The incurrence of indebtedness could result in increased fixed payment obligations and we may be required to agree to certain restrictive covenants therein, such as limitations on our ability to incur additional debt, limitations on our ability to acquire, sell or license intellectual property rights and other operating restrictions that could adversely affect our ability to conduct our business. Additionally, in Julyduring 2020, 2021, and November 2020,2022, we borrowed a total of $200.0 million in seven separate tranches from Oxford (as defined below) $10.0 millionunder our loan and $20.0 million, respectively undersecurity agreement and its amendments, which we refer to collectively as the Amended 2020 Loan Agreement, (as defined below), which, as further described below, also has covenants restricting our ability to declare dividends or incur additional indebtedness.
If we raise additional capital through collaborations, strategic alliances or third-party licensing agreements, we may have to relinquish valuable rights to our intellectual property, future revenue streams, research programs or therapeutics candidates, or grant licenses on terms that may not be favorable to us. If we are unable to raise additional capital through equity or debt financings when needed, (including if we are unable to do so as a result of the COVID-19 pandemic), we may be required to delay, limit, reduce or terminate our product development or future commercialization efforts, or grant rights to develop and market therapeutic candidates that we would otherwise develop and market ourselves.
We may be adversely affected by the effects of inflation.
Inflation has the potential to adversely affect our liquidity, business, financial condition and results of operations by increasing our overall cost structure. The existence of inflation in the economy has resulted in, and may continue to result in, higher interest rates and capital costs, shipping costs, supply shortages, increased costs of labor, weakening exchange rates and other similar effects. Recently, inflation has increased throughout the U.S. economy. Inflation can adversely affect us by increasing the costs of clinical trials and research, the development of our therapeutic candidates, administration and other costs of doing business. We may experience increases in the prices of labor and other costs of doing business. In an inflationary environment, cost increases may outpace our expectations, causing us to use our cash and other liquid assets faster than forecasted. If this happens, we may need to raise additional capital to fund our operations, which may not be available in sufficient amounts or on reasonable terms, if at all, sooner than expected.
We maintain our cash and cash equivalents at financial institutions. The failure of financial institutions could adversely affect our ability to pay our operational expenses or make other payments.
Recent and potential future disruptions in access to bank deposits or lending commitments due to bank failure have contributed to increased volatility and could materially and adversely affect our liquidity, our business and financial condition. The closures of Silicon Valley Bank and Signature Bank and their placement into receivership with the Federal Deposit Insurance Corporation, or FDIC, created bank-specific and broader financial institution liquidity risk and concerns. Although the Department of the Treasury, the Federal Reserve, and the FDIC jointly released a statement that depositors at Silicon Valley Bank and Signature Bank would have access to their funds, even those in excess of the standard FDIC insurance limits, future adverse developments with respect to specific financial institutions or the broader financial services industry may lead to market-wide liquidity shortages. The failure of any bank in which we deposit our funds could reduce the amount of cash we have available for our operations or delay our ability to access such funds. Any such failure may increase the possibility of a sustained deterioration of financial market liquidity, or illiquidity at clearing, cash management and/or custodial financial institutions. In the event we have a commercial relationship with a bank that has failed or is otherwise distressed, we may experience delays or other issues in meeting our financial obligations. If other banks and financial institutions enter receivership or become insolvent in the future in response to financial conditions affecting the banking system and financial markets, our ability to access our cash and cash equivalents and investments may be threatened and could have a material adverse effect on our business and financial condition.
Risks Related to the Development, Clinical Testing and Commercialization of Our Therapeutic Candidates
Our therapeutic candidates are in earlyvarious stages of development and may fail or suffer delays that materially and adversely affect their commercial viability. If we, or our collaborators, are unable to advance our therapeutic candidates through clinical development, obtain regulatorymarketing approval and ultimately commercialize our therapeutic candidates, or experience significant delays in doing so, our business will be materially harmed.
We are early in our development efforts, with only fourthree therapeutic candidates currently in clinical trials (INBRX-109, INBRX-106, INBRX-105(INBRX-101, INBRX-109, and INBRX-101)INBRX-106). We have no products on the market and our ability to achieve and sustain profitability depends on obtaining regulatorymarketing approvals for and successfully commercializing our therapeutic candidates, either alone or with our collaborators. Before obtaining regulatorymarketing approval for the commercial distribution of our therapeutic candidates, we, or our collaborators, must conduct extensive preclinicalnonclinical tests and clinical trials to demonstrate sufficient safety and efficacy of our therapeutic candidates in patients. Failure can occur at any time during the clinical trial process and our future clinical trial results may not be successful. AsWe have previously experienced delays in our clinical trials due to enrollment delays and clinical holds. If we experience additional delays or fail to develop or again terminate development of a result,therapeutic candidate in our pipeline, we may not have the financial resources to continue development of, or to modify existing or to enter into new license or collaboration for, a therapeutic candidate if we experience anycandidate. Other issues that may again delay or potentially prevent regulatorymarketing approval of, or our ability to commercialize, our therapeutic candidates, including:include:
•negative or inconclusive results from our clinical trials, the clinical trials of our collaborators or the clinical trials of others for therapeutic candidates similar to ours, leading to a decision or requirement to conduct additional preclinicalnonclinical testing or clinical trials or abandon a program;
•therapeutic-related side effects experienced by participants in our clinical trials, the clinical trials of our collaborators or by individuals using drugs or therapeutic biologics similar to our therapeutic candidates;
•delays in submitting INDs or comparable foreign applications or delays or failure in obtaining the necessary approvals from regulators to commence a clinical trial, or a suspension, partial suspension or termination of a clinical trial once commenced;
•conditions imposed by the FDA or comparable foreign authorities regarding the scope or design of our clinical trials;
•delays in enrolling research subjects or high drop-out rates of research subjects enrolled in clinical trials;
•delays or difficulties in our clinical trials due to quarantines or other restrictions resulting from the COVID-19 pandemic, including further delays or suspensions in enrollment in our Phase 1 clinical trial for INBRX-101;
•unfavorable FDA or other regulatory agency inspection and review of a clinical trial site or the manufacturing location(s) for a therapeutic candidate;
•inadequate supply or quality of therapeutic candidate clinical material or other raw materials or supplies necessary for the conduct of our clinical trials or the clinical trials of our collaborators;
•delay in the development or approval of companion diagnostic tests for our therapeutic candidates;
•failure of our third-party contractors or investigators to comply with regulatory requirements or otherwise meet their contractual obligations in a timely manner, or at all;
•delays and changes in regulatory requirements, policy and guidelines, including the imposition of additional regulatory oversight for human clinical testing generally or with respect to our technology in particular; or
•varying interpretations of data by the FDA and similar foreign regulatory agencies.
The therapeutic candidates we or our collaborators pursue may not demonstrate the necessary safety or efficacy requirements for regulatorymarketing approval. For instance, we independently decided to cease development of INBRX-105 as we determined, after evaluation of the totality of the data from the expansion cohorts, that the initial signal was not sufficiently validated to support the continuation of the program. Further, a clinical trial may be suspended, partially suspended or terminated by us, our collaborators, the Institutional Review Boards of the institutions in whichIRBs overseeing such trials, are being conducted, the Data Safety Monitoring Board for such trial or by the FDA or other regulatory authorities due to a number of factors, including failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols, inspection of the clinical trial operations or trial site by the FDA or other regulatory authorities resulting in the imposition of a clinical hold or partial clinical hold, unforeseen safety issues or adverse side effects, failure to demonstrate a benefit from using a drug or therapeutic biologic, changes in governmental regulations, administrative actions or lack of adequate funding to continue the clinical trial. Clinical holds may be placed prior to a clinical trial even beginning, in order to address potential safety and risk concerns of regulatory authorities, and partial or complete clinical holds can be imposed at any time during a trial. For example, in early 2023, the Phase 2 trial of INBRX-109 was placed on partial clinical hold by the FDA, and we paused patient enrollment in the trial, following the occurrence of a grade 5 hepatotoxicity event and pre-defined stopping rules built into the protocol. The FDA lifted the hold in April 2023 after we amended the trial protocol to include additional screening criteria and to make other changes to address patients who may be at risk of significant hepatotoxicity. Furthermore, while we perform certain similar functions internally, we expect to rely on contract research organizations, or CROs, and clinical trial sites to ensure proper and timely conduct of our clinical trials and while we expect to enter into and have entered into agreements governing those CROs’ committed activities, we have limited influence over their actual performance.
If we or our collaborators experience delays in the completion of, or termination of, any clinical trial of our therapeutic candidates, the commercial prospects of our therapeutic candidates willmay be harmed, and our ability to generate product revenue or receive royalties from any of these therapeutic candidates willmay be delayed. In addition, anyAny delays in completing our clinical trials willmay increase our costs, slow down our product development and approval process and jeopardize our ability to commence product sales and generate revenue. If we were to cancel the development of any of our therapeutic candidates, we may still be required to pay certain non-cancellable commitments to our CROs under the terms of our various CRO contracts. Our approach to protein engineering is novel and unproven, and as such, the cost, time needed to develop, and likelihood of success of our therapeutic candidates may be more uncertain than if we employed more established drug development approaches. Any of these occurrences may materially and adversely affect our business, financial condition, results of operations and prospects. In addition, many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatorymarketing approval of our therapeutic candidates.
The results of preclinical studies and early stage clinical trials of our therapeutic candidates may not be predictive of the results of later stage clinical trials. Initial results or observations in our ongoing clinical trials may not be indicative of results obtained when these trials are completed or in later stage trials.
Success in preclinical studies and early clinical trials does not ensure that later and pivotal clinical trials will generate the same results, or otherwise provide adequate data to demonstrate the safety and efficacy of a therapeutic candidate. Frequently, therapeutic candidates that have shown promising results in preclinical studies or early clinical trials have subsequently suffered significant setbacks in later or pivotal clinical trials. The initiation of registration-enabling trials for INBRX-109,INBRX-106 or for any of our other therapeutic candidates, is and would be predicated on positive Phase 1 trial results and acceptance of our IND amendment by the FDA. Our therapeutic candidates in clinical trials, including INBRX-101, INBRX-109, INBRX-106, INBRX-105 and INBRX-101,INBRX-106, may ultimately fail to show the desired safety and efficacy in clinical trials despite having progressed through preclinical studies and despite any initial observations of single agent activity, stable disease or partial responses. As noted above, we ceased development of INBRX-105 after determining that the initial signal was not sufficiently validated to support the continuation of the
program even though we observed certain partial and complete responses. There can be no assurance that any of our clinical trials will ultimately be successful or support further clinical development, including development in registration-enabling trials, of any of our therapeutic candidates. A number of companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in clinical development even
after achieving promising results in earlier studies or trials, and any of these setbacks in our clinical development could have a material adverse effect on our business and operating results.
Our therapeutic candidates may cause undesirable side effects that could delay or prevent their regulatorymarketing approval, limit the commercial profile of an approved label, or result in significant negative consequences following marketing approval, if any.
Undesirable side effects caused by our therapeutic candidates have caused and could again cause us, our collaborators or regulatory authorities, to interrupt, delay or halt clinical trials andtrials. These circumstances could result in a more restrictive label or the delay or denial of regulatorymarketing approval by the FDA or other regulatory authorities. Results of our clinical trials or the clinical trials of our collaborators could reveal a high and unacceptable severity of adverse side effects and it is possible that patients enrolled in these clinical trials could respond in unexpected ways. For instance, INBRX-109 and INBRX-106 and INBRX-105 are all therapeutic candidates targeting oncology indications that are clinically evaluated in very sick populations,populations. Certain trial participants, including participants evaluated in our trials for INBRX-105 and INBRX-109, have in the past and others may in the future, experience side effects or serious AEs that could be related to one of our therapeutic candidates. INBRX-101 is a therapeutic candidate focused on AATD, a rare disease with a relatively small patient population. It may be difficult to establish safety and efficacy in these types of patient populations. Further, we intend to develop certain of our therapeutic candidates in combination with one or more cancer therapies. This combination may have additional side effects that were not present in preclinical studies or clinical trials of our therapeutic candidates conducted as a monotherapy or in combination with other cancer therapies. The uncertainty resulting from the use of our therapeutic candidates in combination with other cancer therapies may make it difficult to accurately predict side effects in future clinical trials.
If our clinical trials or the clinical trials of our collaborators reveal adverse side effects, our trials or the clinical trials of our collaborators could be suspended or terminated and the FDA or comparable foreign regulatory authorities could impose a clinical hold, order us to cease further development of or deny approval of our therapeutic candidates for any or all targeted indications. SuchThese side effects could also affect patient recruitment or the ability of enrolled patients to complete the trial or result in potential product liability claims. Further, clinical trials by their nature utilize a sample of the potential patient population. Rare and severe side effects of our therapeutic candidates may only be uncovered with a significantly larger number of patients exposed to our therapeutic candidates.
In the event that any of our therapeutic candidates receives regulatorymarketing approval and we, our collaborators or others identify undesirable side effects caused by aour product or any(or potentially other therapeutics with similar therapeutics,mechanisms of action), any of the following adverse eventsAEs could occur:
•regulatory authorities may withdraw their approval of the product;
•additional restrictions may be imposed on the marketing of the particular product or the manufacturing processes for the product or any component of the product;
•we may be subject to fines, injunctions or the imposition of civil or criminal penalties;
•regulatory authorities may require the addition of safety-related labeling statements, such as a “black box” warning or a contraindication;
•we may be required to create a Medication Guide outlining the risks of such side effects for distribution to patients or to implement other aspects of a REMS such as a restricted distribution program or educational programs for prescribers;
•we could be sued and held liable for harm caused to patients;
•the product may become less competitive; and
•our reputation may suffer.
In addition, adverse side effects caused by any therapeutics that may be similar in nature to our therapeutic candidates could delay or prevent regulatorymarketing approval of our therapeutic candidates, limit the commercial profile of an approved label for our therapeutic candidates, or result in significant negative consequences for our therapeutic candidates following marketing approval.
We believe that anyAny of the above described events could prevent us from achieving or maintaining market acceptance of our therapeutic candidates, if approved, and could delay, impede and/or substantially increase the costs of commercializing our therapeutic candidates thus significantly impacting our ability to successfully commercialize
our therapeutic candidates and generate revenue. Any of the above described occurrences may materially and adversely affect our business, financial condition, results of operations and prospects.
We expect to develop certain of our therapeutic candidates in combination with other therapies, and safety or supply issues with combination use products may delay or prevent development and approval of our therapeutic candidates.
We intend to develop certain of our therapeutic candidates in combination with one or more approved or investigational cancer therapies. Even if any therapeutic candidate we develop were to receive marketing approval or be commercialized for use in combination with other existing therapies, we would continue to be subject to the risks that the FDA or similar regulatory authorities outside of the United States could revoke approval of the therapy used in combination with our product or that safety, efficacy, manufacturing or supply issues could arise with any of those existing therapies. If the therapies we use in combination with our therapeutic candidates are replaced as the standard of care for the indications we choose for any of our therapeutic candidates, the FDA or similar regulatory authorities outside of the United States may require us to conduct additional clinical trials. The occurrence of any of these risks could result in our own products, if approved, being removed from the market or being less successful commercially.
We also may evaluate our therapeutic candidates in combination with one or more cancer therapies that have not yet been approved for marketing by the FDA or similar regulatory authorities outside of the United States. We will not be able to market and sell any therapeutic candidate we develop in combination with an unapproved cancer therapy if that unapproved cancer therapy does not ultimately obtain marketing approval. In addition, unapproved cancer therapies face the same risks described with respect to our therapeutic candidates currently in development and clinical trials, including the potential for serious adverse effects,AEs, delay in their clinical trials and lack of FDA approval.
If the FDA or similar regulatory authorities outside of the United States do not approve these other drugs or revoke their approval of, or if safety, efficacy, manufacturing, or supply issues arise with, the drugs we choose to evaluate in combination our therapeutic candidates, we may be unable to obtain approval of or market any such therapeutic candidate.
If we experience delays or difficulties in the enrollment of patients in clinical trials, our receipt of necessary regulatorymarketing approvals could be delayed or prevented.
We may not be able to initiate or continue clinical trials for our therapeutic candidates if we are unable to locate and enroll a sufficient number of eligible patients to participate in these trials as required by the FDA or similar regulatory authorities outside the United States. In particular, we may have particular difficulty enrolling patients for our clinical trials for INBRX-101 since patients with AATD are particularly susceptible to COVID-19 and the physicians acting as clinical investigators for our clinical sites may be otherwise engaged with treating COVID-19 patients. For instance, during the second and third quarters of 2020 as a result of the COVID-19 pandemic, we temporarily suspended enrollment for our trial for INBRX-101. This trial resumed enrollment during the fourth quarter of 2020 but there is no guarantee that we will not have to suspend enrollment for this trial again in the future. Should any competitors have ongoing clinical trials for therapeutic candidates treating the same indications as our therapeutic candidates, patients who would otherwise be eligible for our clinical trials may instead enroll in clinical trials of our competitors’ therapeutic candidates.
Patient enrollment is affected by other factors including:
•the severity of the disease under investigation;
•the patient eligibility criteria for the study in question;
•the perceived risks and benefits of the therapeutic candidate under study;
•our payments for conducting clinical trials;
•the patient referral practices of physicians; and
•the proximity and availability of clinical trial sites for prospective patients.
Our inability to enroll a sufficient number of patients for any of our clinical trials could result in significant delays and could require us to abandon one or more clinical trials altogether. Enrollment delays in our clinical trials may result in increased development costs for our therapeutic candidates and in delays to commercially launching our therapeutic candidates, if approved, which would materially harm our business.
If we do not achieve our projected development and commercialization goals in the timeframes we announce and expect, the commercialization of any of our therapeutic candidates may be delayed, and our business willmay be harmed.
We have provided, and will continue to provide, a number of timing estimates regarding the initiation of clinical trials and clinical development milestones, and the expected availability of data resulting from these trials for certain of our therapeutic candidates. We expect to continue to estimate the timing of these types of development milestones and our expected timing for the accomplishment of various other scientific, clinical, regulatory and other product development objectives. From time to time, we may publicly announce the expected timing of some of these events. However, theevents and we have had to adjust our previously announced timing for certain of our therapeutic candidates. The achievement of many of these milestones and events may be outside of our control. All of these timing estimations are based on a variety of assumptions we make, which may cause the actual timing of these events to differ from the timing we expect, includingincluding:
•our available capital resources and our ability to obtain additional funding as needed;
•the rate of progress, costs and results of our clinical trials and research and development activities;
•our ability to identify and enroll patients who meet clinical trial eligibility criteria;
•our receipt of clinical trial clearances or approvals by the FDA, EMA and other regulatory authorities and the timing of these approvals;
•our ability to access sufficient, reliable and affordable supplies of materials used in the manufacture of our therapeutic candidates;
•the efforts of our collaborators and licensees including Celgene, with respect to the commercialization of our therapeutics;
•the securing of, costs related to, and timing issues associated with, manufacturing our therapeutic candidates and, if any of our therapeutic candidates are approved, associated with sales and marketing activities and the commercial manufacture of our therapeutic candidates; and
•circumstances arising from or relating to the COVID-19 pandemic,pandemics, regional conflicts, sanctions, geopolitical events, natural disasters or extreme weather events, including potential effects on the global supply chain, our manufacturers and the availability of raw materials needed for the research and development of our therapeutic candidates.
If we fail to achieve announced milestones in the timeframes we expect, the future marketing approval and commercialization of any of our therapeutic candidates may be delayed, and our business, financial condition, results of operations, and prospects may be harmed and our stock price may decline.
Initial, interim, topline and preliminary data from our clinical trials that we may announce, observe or publish from time to time may change as more patient data become available and are subject to audit and verification procedures that could result in material changes in the final data.
We expect fromFrom time to time, towe publish initial, interim, topline or preliminary data from our clinical trials. In particular, we previously reported observed signsCertain of partial responses and stable disease in initial data from our Phase 1 clinical trial for INRBRX-109 and noted observed signs of partial responses, disease control and tumor regression in initial and interim data from our Phase 1 clinical trial for INBRX-106. Our clinical trials are also conducted as “open-label” trials. An open-label trial is one where both the patientstudy participant and investigator know whether the patientparticipant is receiving the therapeutic candidate and where we (as the sponsor) have access to trial data on an ongoing basis during the trial. These initial and interim data from INBRX-109 and INBRX-106 and other initial, interim, topline and preliminary data from our clinical trials that we may publish from time to time or that we may observe on an ongoing basis in our open-label trials may change as more patient data become available and, accordingly, they are not necessarily predictive of final results. Preliminary and interim data are subject to the risk that one or more of the clinical outcomes may materially change as patientparticipant enrollment continues, more patientparticipant data becomebecomes available and we issue our final clinical trial report. Initial, interim, topline and preliminary data also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary data we previously published. As a result, initial, interim, topline and preliminary data should be viewed with caution until the final data are available. Material adverse changes in the final data compared to the interim data could significantly harm our business prospects.
The market opportunities for any current or future therapeutic candidate we develop, if and when approved, may be limited to those patients who are ineligible for established therapies or for whom prior therapies have failed, and may be small.
Cancer therapies are sometimes characterized as first-, second-, or third-line, and the FDA often approves new therapies initially only for third-line use. When cancer is detected early enough, first-line therapy, usually
chemotherapy, hormone therapy, surgery, radiation therapy or a combination of these, is sometimes adequate to cure the cancer or prolong life without a cure. Second- and third-line therapies are administered to patients when prior therapy is not effective. We expect to initially seek approval of certain of our therapeutic candidates as a therapy for patients who have received one or more prior treatments. Subsequently, for those products that prove to be sufficiently beneficial, if any, we would expect to seek approval potentially as a first-line therapy, but there is no guarantee that therapeutic candidates we develop, even if approved, would be approved for first-line therapy, and, prior to any such approvals, we may have to conduct additional clinical trials.
The number of patients who have the cancers we are targeting may turn out to be lower than expected. Additionally, the potentially addressable patient population for our current programs or future therapeutic candidates may be limited, if and when approved. Even if we obtain significant market share for any therapeutic candidate, if and when approved, if the potential target populations are small, we may never achieve profitability without obtaining marketing approval for additional indications, including to be used as first- or second-line therapy.
We rely on third parties to conduct allportions of our clinical trials and certain of our preclinical studies and intend to continue to do so.nonclinical studies. If these third parties do not perform as contractually required, fail to satisfy regulatory or legal requirements or miss expected deadlines, our development programs could be delayed with material and adverse effects on our business, financial condition, results of operations and prospects.
While we expect to continue our current clinical trials and expect to initiate clinical trials in the near term for many of ourother therapeutic candidates, we do not have the ability to independently conduct clinical trials. As such, while we perform certain functions internally, we currently rely and intend to continue to rely on third-party CROs, clinical data management organizations and consultants to help us design, conduct, supervise and monitor clinical trials of our therapeutic candidates. As a result, we will have less control over the timing, quality and other aspects of our clinical trials than we would have had we conducted them on our own. There is a limited number of third party service providers that specialize or have the expertise required to achieve our business objectives. If any of our relationships with these third-party CROs or clinical investigators terminate, we may not be able to enter into arrangements with alternative CROs or investigators or to do so on commercially reasonable terms. Further, these investigators, CROs and consultants are not our employees and we have limited control over the amount of time and resources that they dedicate to our programs. These third parties may have contractual relationships with other entities, some of which may be our competitors, which may draw time and resources from our programs. The third parties with which we contract might not be diligent, careful or timely in conducting our preclinicalnonclinical studies or clinical trials. These third parties may also be susceptible to disruption as a result of health crises such as the COVID-19 pandemic or periods of societal unrest. For example, we currently retain CROs located in Hong Kong. These CROs may be negatively affected by, or subject to disruption arising from, the societal unrest and regulatory regime change related to China’s recent passage of a national security law exerting additional control by mainland China over activities in Hong Kong. If we cannot contract with acceptable third parties on commercially reasonable terms, or at all, or if these third parties do not carry out their contractual duties, satisfy the legal and regulatory requirements for the conduct of preclinicalnonclinical studies or clinical trials or meet expected deadlines for any reason, our clinical development programs could be delayed and otherwise adversely affected.
In all events, we will be responsible for ensuring that each of our preclinicalnonclinical studies and clinical trials is conducted in accordance with the general investigational plan and protocols for the relevant study or trial. The FDA requires preclinicalnonclinical studies to be conducted in accordance with good laboratory practices and clinical trials to be conducted in accordance with GCPs including practices and requirements for designing, conducting, recording and reporting the results of preclinicalnonclinical studies and clinical trials to assure that data and reported results are credible and accurate and that the rights, integrity and confidentiality of clinical trial participants are protected. Our reliance on third parties we do not control will not relieve us of these responsibilities and requirements. Any adverse development or delay in our clinical trials could have a material and adverse effect on our business, financial condition, results of operations and prospects.
We are dependent on Celgene for the successful development, manufacture and commercialization of the Celgene Licensed Intellectual Property. If Celgene does not devote sufficient resources to the development, manufacture and commercialization of the Celgene Licensed Intellectual Property, is unsuccessful in its efforts, or chooses to terminate the Celgene Agreement with us, we may not receive any further proceeds from the Celgene Agreement and our business could be materially harmed.
Pursuant to the terms of the Celgene Agreement, we have exclusively licensed (even as to us) to Celgene the right to develop, manufacture and commercialize the Celgene Licensed Intellectual Property. Celgene is obligated to use commercially reasonable efforts to clinically develop and commercialize the Celgene Licensed Intellectual Property. However, Celgene may ultimately determine it is not commercially reasonable to continue development of the Celgene Licensed Intellectual Property. This outcome could occur for many reasons, including internal business reasons or because of unfavorable regulatory feedback. Further, on review of any safety and efficacy data then available for potential candidates relating to the Celgene Licensed Intellectual Property, the FDA may impose requirements on a clinical trial program that would render the program commercially nonviable. In the event of any such decision, we would be unable to advance such program ourselves.
Per the terms of the Celgene Agreement, Celgene has full control and authority over the development and commercialization of the Celgene Licensed Intellectual Property. We may disagree with Celgene about the development strategy it employs, but we will have limited rights to impose our preferred development strategy on Celgene. Celgene may elect to discontinue the development of the Celgene Licensed Intellectual Property.
The Celgene Licensed Intellectual Property may not be scientifically or commercially successful for us due to a number of important factors, including the following:
•Celgene has wide discretion in determining the efforts and resources that it will apply to its development, manufacture and commercialization of the Celgene Licensed Intellectual Property. The timing and amount of any development milestones, regulatory milestones and royalties that we may receive under the Celgene Agreement will depend on, among other things, Celgene’s efforts, allocation of resources and successful development and commercialization of the Celgene Licensed Intellectual Property and the other antibodies that are the subject of the Celgene Agreement;
•Celgene may terminate the Celgene Agreement without cause and for circumstances outside of our control, which could make it difficult for us to attract new strategic partners or adversely affect how we are perceived in scientific and financial communities;
•Celgene may develop or commercialize the Celgene Licensed Intellectual Property in such a way as to elicit litigation that could jeopardize or invalidate our intellectual property rights or expose us to potential liability; and
•Celgene may not comply with all applicable regulatory requirements, or may fail to report safety data in accordance with all applicable regulatory requirements.
If Celgene were to breach the Celgene Agreement, we may need to enforce our right to terminate the Celgene Agreement in legal proceedings, which could be costly and cause delay in our ability to receive our rights back to develop and commercialize the Celgene Licensed Intellectual Property. If we were to terminate the Celgene Agreement due to Celgene’s breach or if Celgene terminated the Celgene Agreement without cause upon 30 days’ written notice, the development and commercialization of the Celgene Licensed Intellectual Property could be delayed, curtailed or terminated. Additionally, even if the Celgene Agreement is terminated, we may not be able to dispose of the Celgene Licensed Intellectual Property in a strategic transaction without Celgene’s consent.
Celgene became a Bristol-Myers Squibb Company in November 2019. Bristol-Myers Squibb Company may again enter into one or more transactions with third parties, including other mergers, consolidations, reorganizations, sale of substantial assets, sales of substantial stock or other changes in control involving Celgene. These types of transactions could divert the attention of Celgene’s management and adversely affect Celgene’s ability to retain and motivate key personnel who are important to the continued development of the Celgene Licensed Intellectual Property. In addition, the third party to any such transaction could determine to reprioritize Celgene’s development programs such that Celgene ceases to diligently pursue the development of the Celgene Licensed Intellectual Property, and/or cause the Celgene Agreement to terminate. If Celgene does not devote sufficient resources to the
development, manufacture and commercialization of the Celgene Licensed Intellectual Property, is unsuccessful in its efforts, or chooses to terminate the Celgene Agreement with us, we may not receive any further proceeds from the Celgene Agreement and our business could be materially harmed.
We are currently party to license agreements with Elpiscience and Transcenta for the development and, if approved, commercialization of INBRX-105 and INBRX-106 and INBRX-109, respectively. We may in the future enter into additional collaborations with third parties to develop our therapeutic candidates, including collaborations with Chiesi if Chiesi exercises its option pursuant to the Chiesi Option Agreement.candidates. If these collaborations are not successful, our business could be harmed.
We are currently party to two differenta license agreements, or the Elpiscience Agreements,agreement with Elpiscience, pursuant to which we granted Elpiscience exclusive licenses for the development and, if approved, commercialization of INBRX-105 and INBRX-106 in China, Hong Kong, Macau and
Taiwan. We are also party to the Transcenta License Agreement, with Transcenta, pursuant to which we granted Transcenta an exclusive license for the development and, if approved, commercialization of INBRX-109 in China, Hong Kong, Macau and Taiwan. Ultimately, Chiesi may not exercise its option pursuant to the Chiesi Agreement. If Chiesi does not exercise its option under the Chiesi Agreement, we could incur additional expenses for the development, and if approved, the commercialization of INBRX-101. We may enter into additional collaborations with third parties in the future, including collaborations with Chiesi if Chiesi exercises its option pursuant to the Chiesi Option Agreement.future. Any collaborations that we are party to may pose several risks, including the following:
•collaborators have significant discretion in determining the efforts and resources that they will apply to these collaborations;
•collaborators may not perform their obligations as expected;
•as occurred with our trials for INBRX-105, the clinical trials conducted as part of these collaborations may not be successful;
•collaborators may not pursue development and commercialization of any therapeutic candidates that achieve regulatorymarketing approval or may elect not to continue or renew development or commercialization programs based on clinical trial results;
•changes in the collaborators’ strategic focus or available funding or external factors, such as an acquisition, that divert resources or create competing priorities;
•collaborators may delay clinical trials, provide insufficient funding for clinical trials, stop a clinical trial or abandon a therapeutic candidate, repeat or conduct new clinical trials or require a new formulation of a therapeutic candidate for clinical testing;
•we may not have access to, or may be restricted from disclosing, certain information regarding therapeutic candidates being developed or commercialized under a collaboration and, consequently, may have limited ability to inform our stockholders about the status of such therapeutic candidates;
•collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with our therapeutic candidates if the collaborators believe that competitive products are more likely to be successfully developed or can be commercialized under terms that are more economically attractive than ours;
•therapeutic candidates developed in collaboration with us may be viewed by our collaborators as competitive with their own therapeutic candidates or products, which may cause collaborators to cease to devote, or limit, resources to the commercialization of our therapeutic candidates;
•a collaborator with marketing and distribution rights to one or more of our therapeutic candidates that achieve regulatorymarketing approval may not commit sufficient resources to the marketing and distribution of any such therapeutic candidate;
•collaborators may not properly maintain or defend our intellectual property rights or may use our proprietary information in such a way as to invite litigation that could jeopardize or invalidate our intellectual property or proprietary information or expose us to potential litigation;
•disputes may arise with respect to the ownership of intellectual property developed pursuant to our collaborations;
•collaborators may infringe the intellectual property rights of third parties, which may expose us to litigation and potential liability; and
•collaborations may be terminated for the convenience of the collaborator and, if terminated, we could be required to raise additional capital to pursue further development or commercialization of the applicable therapeutic candidates.
In addition, we may have disagreements with these collaborators, including disagreements over proprietary rights, collaborator performance, contract interpretation or the preferred course of development of any therapeutic candidates. Any disagreement we may have with our collaborators, may cause delays or termination of the research, development or commercialization of our therapeutic candidates pursuant to the applicable agreement, may lead to additional responsibilities for us with respect to our therapeutic candidates or may result in litigation or arbitration, any of which would be time-consuming and expensive and which would likely divert the attention of our management from our core research and development activities. These types of disputes could materially harm our financial condition and our business.
The manufacture of biotechnology products is complex, and manufacturers often encounter difficulties in production. If we or any of our third party manufacturers encounter such difficulties, or otherwise fail to comply
with their contractual obligations, the development or commercialization of our therapeutic candidates could be delayed or stopped.
While we have found that our sdAb-based therapeutic candidates can be readily manufactured at high yields with established processes used to produce therapeutic proteins, the manufacture of biotechnology products is generally complex and requires significant expertise and capital investment. We and our contract manufacturers must comply with cGMP regulations and guidelines for clinical trial product manufacture and for commercial product manufacture. Manufacturers of biotechnology products often encounter difficulties in production, particularly in scaling up, addressing product quality, product comparability, validating production processes and mitigating potential sources of contamination. These problems include difficulties with raw material procurement, production costs and yields, quality control, product quality, including stability of the product, quality assurance testing, operator error, shortages of qualified personnel, as well as compliance with strictly enforced federal, state and foreign regulations. Furthermore, if microbial, viral or other contaminations are discovered in our therapeutics or in the manufacturing facilities in which our therapeutics are made, such manufacturing facilities may need to be closed for an extended period of time to investigate and remedy the contamination.
We cannot assure you that manufacturing problems, including supply chain disruptions of any of our therapeutic candidates or products will not occur in the future. Any delay or interruption in the supply of preclinical or clinical trial supplies including any delays arising from circumstances related to the COVID-19 pandemic,or supply chain disruptions could delay the completion of these trials, increase the costs associated with maintaining these trial programs and, depending upon the period of delay, require us to commence new trials at additional expense or terminate trials completely.
If we were to experience an unexpected loss of supply of, or if any supplier were unable to meet our demand for any of our therapeutic candidates or future approved products we seek to commercialize, if any, we could experience delays in our research or planned clinical studies or be forced to stop our development or commercialization efforts. We could be unable to find alternative suppliers of acceptable quality, in the appropriate volumes and at an acceptable cost. Moreover, our suppliers are often subject to strict manufacturing requirements and rigorous testing requirements, which could limit or delay production. The long transition periods necessaryneeded to switch manufacturers and suppliers, if necessary, would significantly delay our clinical studies and the commercialization of our therapeutics, if approved, which would materially adversely affect our business, prospects, financial condition and results of operation.
We rely on third parties to supply and manufacture our therapeutic candidates, and we expect to continue to rely on third parties to manufacture and supply our therapeutics, if approved. The development of therapeutic candidates and the commercialization of any therapeutic candidates, if approved, could be stopped, delayed or made less profitable if any of these third parties fail to provide us with sufficient quantities of therapeutic candidates or therapeutics, fail to do so at acceptable quality levels or prices, or fail to maintain or achieve satisfactory regulatory compliance.
We do not currently have, nor do we plan to acquire, the infrastructure or capability internally to develop and manufacture our therapeutic candidates for use in the conduct of our trials or for commercial supply, if our
therapeutics are approved. Instead, we rely on, and expect to continue to rely on third-party providers to manufacture the supplies for our preclinical studies and clinical trials. We currently rely on a limited number of third-party contract manufacturers for all of our required raw materials, antibodies, and other biologics for our preclinical research and clinical trials, as well as for the manufacture of supplies for our therapeutic candidates. To the extent any of our manufacturing partners isare unable to fulfill these obligations in a timely manner, including as a result of circumstances relating to the COVID-19 pandemic or circumstances related to China’s implementation of its recently adopted security law to exert additional control over activities in Hong Kong, our clinical trials may be delayed, and our business may be adversely affected. In general, reliance on third party providers may expose us to more risk than if we were to manufacture our therapeutic candidates ourselves. We do not control the operational processes of the contract manufacturing organizations with whom we contract and are dependent on these third parties for the production of our therapeutic candidates in accordance with relevant regulations (such as cGMP), which include, among other things, quality control and the maintenance of records and documentation.
Our third-party manufacturers may be unable to successfully scale up manufacturing of our therapeutic candidates in sufficient quality and quantity, which would delay or prevent us from developing our therapeutic candidates and commercializing any approved therapeutic candidates.
Our manufacturing partners may be unable to successfully increase the manufacturing capacity for our therapeutic candidates in a timely or cost-effective manner, or at all, as needed for our development efforts or, if our therapeutic candidates are approved, our commercialization efforts. Quality issues may also arise during scale-up activities. If we, or any manufacturing partners, are unable to successfully scale up the manufacture of our therapeutic candidates in sufficient quality and quantity, the development, testing, and clinical trials of our therapeutic candidates may be delayed or infeasible, and regulatorymarketing approval or future commercial launch of any resulting therapeutic may be delayed or not obtained, which could significantly harm our business.
Failure to successfully identify, develop and commercialize additional therapeutics or therapeutic candidates could impair our ability to grow.
Although a substantial amount of our efforts will focus on the continued preclinical and clinical testing and potential approval of our therapeutic candidates in our current pipeline, we expect to continue to innovate and potentiallyexpect to expand our portfolio. Because we have limited financial and managerial resources, research programs to identify therapeutic candidates may require substantial additional technical, financial and human resources, whether or not any new potential therapeutic candidates are ultimately identified. Our success may depend in part upon our ability to identify, select and develop promising therapeutic candidates and therapeutics. We may expend resources and ultimately fail to discover and generate additional therapeutic candidates suitable for further development. All therapeutic candidates are prone to risks of failure typical of biotechnology product development, including the possibility that a therapeutic candidate may not be suitable for clinical development as a result of its harmful side effects, limited efficacy or other characteristics indicating that it is unlikely to receive approval by the FDA, the EMA and other comparable foreign regulatory authorities or to achieve market acceptance. If we do not successfully develop and commercialize new therapeutic candidates we have identified and explored, our business, prospects, financial condition and results of operations could be adversely affected.
Our business entails a significant risk of product liability and our ability to obtain sufficient insurance coverage could have a material and adverse effect on our business, financial condition, results of operations and prospects.
We are exposed to significant product liability risks inherent in the development, testing, manufacturing and marketing of biotechnology treatments of any therapeutic candidates for which we or our collaborators may conduct clinical trials. Product liability claims could delay or prevent completion of our development programs. If we succeed in marketing any approved products, these claims could result in an FDA investigation of the safety and effectiveness of our future commercial products, our manufacturing processes and facilities (or the manufacturing processes and facilities of our third-party manufacturers) or our marketing programs, a recall of our products or more serious enforcement action, limitations on the approved indications for which the product may be used or suspension or withdrawal of approvals. Regardless of the merits or eventual outcome, liability claims may also result in decreased demand for our products, injury to our reputation, costs to defend the related litigation, a diversion of management’s time and our resources, substantial monetary awards to clinical trial participants or patients and a decline in our stock price. Any insurance we have or may obtain may not provide sufficient coverage against
potential liabilities. Furthermore, clinical trial and product liability insurance is becoming increasingly expensive. As a result, we may be unable to obtain sufficient insurance at a reasonable cost to protect us against losses caused by potential product liability claims that could have a material and adverse effect on our business, financial condition, results of operations and prospects.
If our therapeutic candidates are approved for marketing and commercialization and we are unable to develop sales, marketing and distribution capabilities on our own or enter into agreements with third parties to perform these functions on acceptable terms, we will be unable to commercialize successfully any such therapeutic candidates.
We currently have no sales, marketing or distribution capabilities. While we have marketing and commercialization agreements in place for certain territories with our collaborators, we will still need to developexpand our own internal sales, marketing and distribution capabilities to commercialize our approved therapeutic candidates, if any, in the United States and other remaining worldwide territories, or will need to enter into collaborations with third parties to
perform these services. Any internal effort would be expensive and time-consuming, and we would need to commit significant financial and managerial resources to develop an internal marketing and sales force with technical expertise and the related supporting distribution, administration and compliance capabilities. If we were to rely on additional third parties with these capabilities to market our future therapeutics or were to decide to co-promote products with any of our current or future collaborators, we would need to establish and maintain or revise existing marketing and distribution arrangements with these partners, and there can be no assurance that we will be able to enter into such arrangements on acceptable terms or at all. Any revenue we receive in connection with third-party license, marketing or distribution arrangements, including the Celgene Agreement, the Chiesi Option Agreement and license agreements with our licensing partners in China, will depend upon the efforts of these third parties, and there can be no assurance these third parties will establish adequate sales and distribution capabilities or be successful in gaining market acceptance of any approved product. If we are not successful in commercializing any product approved in the future, either on our own or through third parties, our business, financial condition, results of operations and prospects could be materially and adversely affected.
The future commercial success of our therapeutic candidates will depend on the degree of market acceptance of our therapeutic candidates among physicians, patients, healthcare payors and the medical community.
Our therapeutic candidates are still in early stages ofclinical development and many of our therapeutic candidates areemerging pipeline is still in preclinical development; we may never have an approved product that is commercially successful. Due to the inherent risk in the development of biotechnologybiopharmaceutical products, it is probable that not all or none of the therapeutic candidates in our pipeline, including any that are or may be licensed to third parties, will successfully complete development and be commercialized. Furthermore, even when available on the market, our products may not achieve an adequate level of acceptance by physicians, patients and the medical community, and we may not become profitable. In addition, efforts to educate the medical community and third-party payors on the benefits of our products may require significant resources and may never be successful, which would prevent us from generating significant revenue or becoming profitable. Market acceptance of any approved products by physicians, patients and healthcare payors will depend on a number of factors, many of which are beyond our control, including, but not limited to:
•changes in the standard of care for the targeted indications for any approved product;
•wording in the FDA- or EMA-approved prescribing information;
•sales, marketing and distribution support;
•potential product liability claims;
•acceptance by physicians, patients and healthcare payors of each product as safe, effective and cost-effective;
•relative convenience, ease of use, ease of administration and other perceived advantages over alternative products;
•prevalence and severity of adverse eventsAEs or publicity;
•limitations, precautions or warnings listed in the summary of product characteristics, patient information leaflet, package labeling or instructions for use;
•the cost of treatment with our therapeutics in relation to alternative treatments;
•the extent to which products are approved for inclusion and adequately reimbursed on formularies of hospitals and third-party payors, including managed care organizations; and
•whether our products are designated in the label, under physician treatment guidelines or under reimbursement guidelines as a first, second, third or last line therapy.
Risks Related to Our Organization and Operations
We face significant competition and if our competitors develop and market products that are more effective, safer or less expensive than the therapeutic candidates we develop, our commercial opportunities will be negatively impacted.
The life sciences industry is highly competitive. We are currently developing therapeutic candidates that will compete, if approved, with other products and therapies that currently exist or are being developed. Our primary competitors fall into the following groups:
•Companies developing novel therapeutics based on sdAb or alternative scaffold product candidates, including Alligator Bioscience AB, Camel-IDS N.V., Crescendo Biologics Ltd., GlaxoSmithKline plc, IGM Biosciences,
Inc., Lava Therapeutics N.V., Molecular Partners AG, Pieris Pharmaceuticals, Inc., Precirix, and Sanofi S.A. and VHsquared Ltd.;
•Antibody drug discovery companies that may compete with us in the search for novel therapeutic antibody targets, including Regeneron Pharmaceuticals, Inc., Adimab LLC, Genmab A/S, Macrogenics, Inc., Merus N.V., MorphoSys AG, Numab Therapeutics AG, TeneoBio,Amgen, Inc., Xencor Inc., and Zymeworks Inc.; and
•Companies developing therapeutics designed to treat AATD, including CSL Limited, Grifols, S.A., Kamada Ltd., Takeda Pharmaceutical Company Limited, and Vertex Pharmaceuticals, Inc.Mereo BioPharma Group plc.
Our competitors also include other large pharmaceutical and biotechnology companies who may be developing therapeutic candidates with mechanisms similar to or targeting the same indications as our therapeutic candidates.
Products we may develop in the future are also likely to face competition from other products and therapies, some of which we may not currently be aware. We have competitors both in the United States and internationally, including major multinational pharmaceutical companies, established biotechnology companies, specialty pharmaceutical companies, universities and other research institutions. Many of our competitors have significantly greater financial, manufacturing, marketing, product development, technical and human resources than we do. Large pharmaceutical companies, in particular, have extensive experience in clinical testing, obtaining marketing approvals, recruiting patients and manufacturing pharmaceutical products. These companies also have significantly greater research and marketing capabilities than we do and may also have products that have been approved or are in late stages of development, and collaborative arrangements in our target markets with leading companies and research institutions. Established pharmaceutical companies may also invest heavily to accelerate discovery and development of novel compounds or to in-license novel compounds that could make the therapeutic candidates that we develop obsolete. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated among a smaller number of our competitors. As a result of all of these factors, our competitors may succeed in obtaining patent protection and/or marketing approval or discovering, developing and commercializing products in our field before we do.
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe effects, are more convenient, have a broader label, are marketed more effectively, are reimbursed or are less expensive than any products that we may develop. Our competitors also may obtain FDA, EMA or other marketing approval for their products more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market. Even if the therapeutic candidates we develop achieve marketing approval, they may be priced at a significant premium over competitive products if any have been approved by then, resulting in reduced competitiveness.
Smaller and other early stage companies may also prove to be significant competitors. These third parties compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our
programs. In addition, the biopharmaceutical industry is characterized by rapid technological change. If we fail to stay at the forefront of technological change, we may be unable to compete effectively. Technological advances or products developed by our competitors may render our therapeutic candidates obsolete, less competitive or not economical.
Our business, the conduct of our clinical trials, results of operations, financial condition, and prospects may be adversely affected by the widespread outbreak of an illness or any other communicable disease, or any other public health crisis, including the coronavirus disease (COVID-19) pandemic.
Our business could be adversely affected by widespread outbreak of illness or other communicable diseases, health epidemics, or any other public health crisis. On January 30, 2020, the World Health Organization, or the WHO, announced a global health emergency because of a novel strain of coronavirus named SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2), or novel coronavirus, which causes coronavirus disease 2019, or COVID-19, originating in Wuhan, China. In March 2020, the WHO classified the COVID-19 outbreak as a pandemic, based on the rapid increase in exposure globally.
During 2020, COVID-19 spread to most regions of the world, including California, where our primary office and laboratory space is located.
As a result of the COVID-19 pandemic, we may experience disruptions that could severely impact our business, preclinical studies and clinical trials, including:
•We are, with our partners, conducting clinical trials and other research in geographies that are affected by the COVID-19 pandemic. Potential impacts of the COVID-19 pandemic on our various clinical trials may include delays related to patient enrollment, dosing and study monitoring due to changes in policies at various clinical sites, federal, state, local or foreign laws, rules and regulations, including quarantines or other travel restrictions. They may also be impacted by the prioritization of healthcare resources toward pandemic efforts, including diminished attention of physicians serving as our clinical trial investigators and reduced availability of site staff supporting the conduct of our clinical trials, interruption or delays in the operations of the FDA, or other reasons related to the pandemic. For example, we temporarily suspended enrollment for our INBRX-101 Phase 1 clinical trial as a direct result of the COVID-19 pandemic during the second and third quarters of 2020. While our oncology-related clinical trials have not been materially impacted to date, it is possible that these programs may be negatively impacted in the future. If the COVID-19 pandemic continues, other aspects of our clinical trials may be adversely affected, delayed or interrupted, including, for example, site initiation, patient recruitment, further or additional delays in enrollment, availability of clinical trial materials, and data analysis. Some patients and clinical investigators may not be able to comply with clinical trial protocols and patients may choose to withdraw from our trials. It is unknown how long these suspension, pauses or disruptions could continue. FDA has issued a guidance for industry, “FDA Guidance on Conduct of Clinical Trials of Medical Products during COVID-19 Pandemic Guidance for Industry, Investigators, and Institutional Review Boards,” which is being periodically updated to continue clarifying for affected stakeholders to ensure the safety of clinical trial participants, maintain compliance with GCPs, and minimize risks to trial and clinical data integrity. We intend to follow FDA’s recommendations in order to address unavoidable protocol deviations due to COVID-19 illness or COVID-19 control measures, as appropriate.
•We currently rely on third parties to, among other things, manufacture raw materials, manufacture our therapeutic candidates, ship investigational drugs and clinical samples, perform quality testing and supply other goods and services to run our business. If any third party in our supply chain for materials are adversely impacted by restrictions resulting from the COVID-19 pandemic, including as a result of staffing shortages, production slowdowns and disruptions in delivery systems, our supply chain may be disrupted, limiting our ability to conduct our research and development operations.
•We have requested that our employees, including all of our administrative employees, work remotely and have restricted on-site staff to only those personnel who must perform essential on-site activities such as activities in our research and development laboratories. Our increased reliance on employees working from home may negatively impact productivity, or disrupt, delay, or otherwise adversely impact our business. In addition, this could increase our cybersecurity risk, create data accessibility concerns, and make us more
susceptible to communication disruptions, any of which could adversely impact our business operations or delay necessary interactions with local and federal regulators, ethics committees, manufacturing sites, research or clinical trial sites and other important agencies and contractors.
•Our employees conducting research and development activities may be more limited in their ability to access our laboratories for an extended period of time as a result of the rigorous workplace policies and guidelines implemented as a result of the COVID-19 pandemic. Additionally, it is possible that governmental authorities will further modify current restrictions. As a result, this could delay timely completion of our research and development efforts.
•Health regulatory agencies globally may experience disruptions in their operations as a result of the COVID-19 pandemic. The FDA and comparable foreign regulatory agencies may have slower response times or be under-resourced to continue to monitor our clinical trials and, as a result, review, inspection, and other timelines may be materially delayed. It is unknown how long these disruptions could continue, were they to occur. Any elongation or de-prioritization of our clinical trials or delay in regulatory review resulting from such disruptions could materially affect the development and study of our therapeutic candidates. For example, regulatory authorities may require that we not distribute a therapeutic candidate lot until the relevant agency authorizes its release. Such release authorization may be delayed as a result of the COVID-19 pandemic and could result in delays to our clinical trials.
•The trading prices for biopharmaceutical companies have been highly volatile as a result of the COVID-19 pandemic. As a result, we may face difficulties raising capital through sales of our common shares in future offerings or such sales may be on unfavorable terms. In addition, a recession, depression or other sustained adverse market event resulting from the spread of COVID-19 could materially and adversely affect our business and the value of our common shares.
The COVID-19 pandemic continues to rapidly evolve. The ultimate impact of the pandemic on our business operations is highly uncertain and subject to change and will depend on future developments, which cannot be accurately predicted, including the duration of the pandemic, the ultimate geographic spread of the disease, additional or modified government actions, new information that will emerge concerning the severity and impact of COVID-19 and the actions taken to contain COVID-19 or address its impact in the short and long term, among others. We do not yet know the full extent of potential delays or impacts on our business, our clinical trials, our research programs, healthcare systems or the global economy.
Any inability to attract and retain qualified key management and technical personnel would impair our ability to implement our business plan.
Our success largely depends on the continued service of key management, advisors and other specialized personnel, including Mark P. Lappe, our Chief Executive Officer, Brendan P. Eckelman, Ph.D., our Chief Scientific Officer, Klaus W. Wagner, M.D., Ph.D., our Chief Medical Officer, and Kelly D. Deck, our Chief Financial Officer, who are all employed at will and for whom we do not have “key man” insurance coverage. The loss of one or more members of our management team or other key employees or advisors could delay our research and development programs and have a material and adverse effect on our business, financial condition, results of operations and prospects. We are dependent on the continued service of our technical personnel because of the highly technical nature of our therapeutic candidates and technologies and the specialized
nature of the regulatorymarketing approval process. Our future success will depend in large part on our continued ability to attract and retain other highly qualified scientific, technical and management personnel, as well as personnel with expertise in clinical testing, manufacturing, governmental regulation and commercialization. We face competition for personnel from other companies, universities, public and private research institutions, government entities and other organizations (many of whom have substantially greater financial resources than us), and we might not be able to attract or retain these key employees on conditions that are economically acceptable. Our inability to attract and retain these key employees could prevent us from achieving our objectives and implementing our business strategy, which could have a material adverse effect on our business and prospects.
We have a significant amount of debt which may affect our ability to operate our business and secure additional financing in the future.
In July 2020, If we borrowed $10.0 million underfail to comply with the loan and security agreement, or the 2020 Loan Agreement, with Oxford Finance LLC, or Oxford, and in November 2020, we borrowed an additional $20.0 million under an
amendment to the 2020 Loan Agreement, or the Amended 2020 Loan Agreement, our business, prospects and results of operations could be materially and adversely affected.
During 2020, 2021, and 2022, we borrowed a total of $200.0 million in seven separate tranches from Oxford, under the Amended 2020 Loan Agreement.
Our obligations under the Amended 2020 Loan Agreement are secured by substantially all of our assets, other than our intellectual property.assets. The Amended 2020 Loan Agreement requires us, and any debt arrangements or instruments we may enter into in the future may require us, to comply with various covenants that limit our ability to, among other things:
•dispose of assets;
•complete mergers or acquisitions;
•incur or guarantee indebtedness;
•sell or encumber certain assets;
•pay dividends or make other distributions to holders of our capital stock, including by way of certain stock buybacks;
•make specified investments;
•engage in different lines of business;
•change certain key management personnel; and
•engage in certain transactions with our affiliates.
These covenants may make it difficult to operate our business. A failure by us to comply with the covenants could result in an event of default, which could adversely affect our ability to respond to changes in our business and manage our operations. Upon the occurrence of an event of default, including the occurrence of a material adverse change, the lender could elect to declare all amounts outstanding to be due and payable and exercise other remedies. If the indebtedness were to be accelerated, our future financial condition could be materially adversely affected.
We may incur additional indebtedness in the future. The instruments governing such indebtedness could contain provisions that are as, or more, restrictive than our existing debt instruments. Our obligations pursuant to the Amended 2020 Loan Agreement are secured by substantially all of our assets, including our intellectual property. If we are unable to repay, refinance or restructure our indebtedness when payment is due, the lenders could proceed against anythis collateral granted to them to secure such indebtedness or force us into bankruptcy or liquidation. Further, if our business is subject to liquidation, the right to repayment of Oxford and any other holders of indebtedness would be senior to the rights of the holders of our common stock to receive any proceeds from the liquidation.
Our employees, and independent contractors, principal investigators, contract research organizations, consultants or vendors may engage in misconduct or other improper activities, including non-compliance with governmental and regulatory standards and requirements.bodies.
We are exposed to the risk of fraud or other misconduct by our employees, independent contractors, principal investigators, contract research organizations, consultants or independent contractors.vendors. Misconduct by these parties could include intentional failures to comply with state and federal securities laws, FDA regulations, provide accurate information to the FDA, comply with manufacturing standards we may establish for our therapeutic candidates, comply with federal and state data privacy, security, fraud and abuse, and other healthcare laws and regulations, report financial information or data accurately or disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud,
kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Activities subject to these laws could also involve the improper use or misrepresentation of information obtained in the course of clinical trials, which could result in regulatory sanctions and cause serious harm to our reputation. It is not always possible to identify and deter misconduct by employees and third parties, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. Additionally, we are subject to the risk that a person or government could allege such fraud or other misconduct, even if none occurred. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a material and adverse effect on our business, financial condition, results of operations and prospects, including the imposition of significant civil, criminal and administrative penalties, monetary damages, fines, disgorgement, imprisonment, loss of eligibility to obtain marketing approvals from the FDA, exclusion from participation in government contracting, healthcare reimbursement or other government programs, including Medicare and Medicaid, reputational harm, diminished profits and future earnings, additional reporting requirements if subject to a corporate integrity agreement or other agreement to resolve allegations of non-compliance with any of these laws, and the curtailment or restructuring of our operations.
We may be subject to claims that our employees, consultants or independent contractors have wrongfully used or disclosed confidential information of their former employers or other third parties.
Certain of our employees, consultants or advisors are currently, or were previously, employed at universities or other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although we try to ensure that our employees, consultants and advisors do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that these individuals or we have used or disclosed intellectual property, including trade secrets or other proprietary information, of any such individual’s current or former employer. Litigation may be necessary to defend against these claims. As noted in Item 3 of this Annual Report, we and Dr. Eckelman are party to litigation asserting claims against us for misappropriation of trade secrets related to Dr. Eckelman’s service as an expert witness. If we fail in defending against these and any other such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management.
Our insurance may not provide adequate levels of coverage against claims which may adversely affect our financial condition.
We expectmaintain insurance that we believe is adequate for businesses of our size and type. However, there are types of losses that we believe are not economically reasonable to insure or that cannot be insured against. For instance, because directors and officers, or D&O, liability insurance has become cost prohibitive with high retentions providing minimal coverage, we have not renewed our D&O policy.
It is possible that we may be subject to securities litigation in the future, including potential class action or stockholder derivative actions. Our indemnification agreements with our directors and certain officers, as well as Delaware General Corporation Law, may require us, among other things, to indemnify them against certain liabilities that may arise by reason of their status or service as directors or officers. Without D&O insurance, the amounts we would pay to defend any such litigation or indemnify our officers and directors should they be subject to legal action based on their service to us could have a material adverse effect on our financial condition, results of operations and liquidity.
As we expand our development and regulatory capabilities, and as a result, we may encounter difficulties in managing our growth, which could disrupt our operations.
We expect tomay experience significant growth in the number of our employees and the scope of our operations, particularly in the areas of drug development and regulatory affairs, as well as sales and marketing to the extent any of our therapeutic candidates approach receipt of marketing authorization. To manage our anticipated future growth, we must continue to implement and improve our managerial, operational and financial systems, expand our facilities and continue to recruit and train additional qualified personnel. Due to our limited financial resources, we may not be able to
effectively manage the expansion of our operations or recruit and train additional qualified personnel. The expansion of our operations may lead to significant costs and may divert our management and business development resources. Any inability to manage growth could delay the execution of our business plans or disrupt our operations.
We may not be able to integrate efficiently or achieve the expected benefits of any acquisitions of complementary businesses, therapeutic candidates or technologies.
Should we in the future acquire any complementary business, therapeutic candidates or technologies, our ability to integrate and manage acquired businesses, therapeutic candidates or technologies effectively will depend upon a number of factors including the size of the acquired business, the complexity of any therapeutic candidate or technology and the resulting difficulty of integrating the acquired business’s operations, if any. Our relationship with current employees or employees of any acquired business may become impaired. We may also be subject to unexpected claims and liabilities arising from such acquisitions. These claims and liabilities could be costly to defend, could be material to our financial condition and might exceed either the limitations of any applicable indemnification provisions or the financial resources of the indemnifying parties. There can also be no assurance that we will be able to assess ongoing profitability and identify all actual or potential liabilities of a business, therapeutic candidate or technology prior to its acquisition. If we acquire businesses, therapeutic candidates or technologies that result in assuming unforeseen liabilities in respect of which it has not obtained contractual protections or for which protection is not available, this could materially adversely affect our business, prospects, financial condition and results of operations.
Our business may be adversely affected as a result of major computer system failures.
Any of the internal computer systems belonging to us or our third-party service providers are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war, and telecommunication and electrical failure. Any system failure, accident or security breach that causes interruptions in our own or in third-party service vendors’ operations could result in a material disruption of our development programs. For example, the loss of clinical trial data from completed or future clinical trials could result in delays in our or our partners’ or collaborators’ regulatorymarketing approval efforts and significantly increase our costs in order to recover or reproduce the lost data. To the extent that any disruption or security breach results in a loss or damage to our data or applications, or inappropriate disclosure of confidential or proprietary information, we may incur liability, our development programs, reputation and competitive position may be adversely affected and the further development of our therapeutic candidates may be delayed. Furthermore, we may incur additional costs to remedy the damage caused by these disruptions or security breaches.
Cybersecurity breaches could expose us to material liability, damage our reputation, compromise our confidential information or otherwise adversely affect our business.
We maintain sensitive company data on our computer networks and third-party cloud services, including our intellectual property and proprietary business information. We face a number of threats to our networks from unauthorized access, accidental acts or omissions that expose vulnerabilities, security breaches and other system disruptions. As noted above, certainOur third-party partners, including CROs and providers of data hosting or cloud services, as well as suppliers, distributors, alliances, and other third-party service providers, face similar risks, which could affect us directly or indirectly.
We are increasingly dependent upon our technology systems to operate our business and our ability to effectively manage our business depends on the security, reliability and adequacy of our technology systems and data, which includes use of cloud technologies. A breakdown, invasion, corruption, destruction or breach of our technology systems, including the cloud technologies that we utilize, and/or unauthorized access to our data and information could subject us to liability or negatively impact the operation of our business. Our technology systems, including the cloud technologies that we utilize, continue to increase in multitude and complexity, making them potentially vulnerable to breakdown, malicious intrusion and random attack. Likewise, data privacy or security breaches by individuals authorized to access our technology systems, including the cloud technologies that we utilize, may pose a risk that sensitive data, including intellectual property, trade secrets or personal information belonging to us, our patients, customers or other business partners, may be exposed to unauthorized persons or to the public.
Cyber-attacks are increasing in their frequency, sophistication and intensity, and are becoming increasingly difficult to detect. They are often carried out by motivated, well-resourced, skilled and persistent actors, including nation states, organized crime groups, “hacktivists” and employees are presentlyor contractors acting with malicious intent. Cyber-attacks could include the deployment of harmful malware and may inkey loggers, ransomware, a denial-of-service attack, a malicious website, the future be working remotely as a resultuse of social engineering and other means to affect the COVID-19 pandemic. We mayconfidentiality, integrity and availability of our technology systems and data. Our key business partners face additional cybersecuritysimilar risks as a resultand any security breach of these circumstances and despitetheir systems could adversely affect our security measures,posture. In addition, our infrastructure may be vulnerableincreased use of cloud technologies could heighten these and other operational risks, and any failure by cloud technology service providers to attacks by hackersadequately safeguard their systems and prevent cyber-attacks could disrupt our operations and result in misappropriation, corruption, or vulnerable to other disruptive problems.loss of confidential or propriety information.
The United States federal and variousall state and foreign governments have adopted or proposed requirements regarding the collection, distribution, use, security, and storage of personally identifiable information and other data relating to individuals, and federal and state consumer protection laws are being applied to enforce regulations related to the collection, use, and dissemination of data. Some of these federal, state and foreign government requirements include obligations of companies to notify individuals and others of security breaches involving particularcertain personally identifiable information, which could result from breaches experienced by us or by our vendors, contractors, or organizations with which we have formed strategic relationships. Even though we may have contractual protections with such vendors, contractors, or other organizations, notifications and follow-up actions related to a security breach could impact our reputation, prompt regulatory scrutiny and enforcement, cause us to incur significant costs, including legal expenses, harm customer confidence, hurt our expansion into new markets,or cause us to incur remediation costs or cause us to lose existing customers.that could, under such circumstances, materially harm our business.
Any such security breach may materially compromise information stored on our networks and may result in significant data losses or theft of our intellectual property or proprietary business information, it may also subject us to significant fines, penalties or liabilities for any noncompliance with certain privacy and security laws. A cybersecurityWe maintain cyber liability insurance; however, this insurance may not be sufficient to cover the financial, legal, business, or reputational losses that may result from an interruption of breach of our systems.
While we continue to build and improve our systems and infrastructure, including our business continuity plans, there can be no assurance that our efforts will prevent breakdowns or breaches in our systems that could adversely affect our reputationbusiness and operations and/or result in the loss of critical or sensitive information, which could result in financial, legal, business, operational or reputational harm to us, loss of competitive advantage or loss of consumer confidence. In addition, our liability insurance may not be sufficient in type or amount to cover us against claims related to security breaches, cyber-attacks and other related breaches. Further, a data breach could result in regulatory investigations and negative consequences, including disruptionpublicity which could damage our reputation and have an adverse effect on our business, financial condition or results of our internal operations, increased cybersecurity protection costs, lost revenue or litigation.operations.
Our current operations are concentrated in one location, and we or the third parties upon whom we depend on may be adversely affected by earthquakes, medical epidemics or pandemics, or other natural disasters.
Our current operations are located in our facilities in La Jolla, California. Any unplanned event, such as flood, fire, explosion, earthquake, extreme weather condition, medical epidemics or pandemics, power shortage, telecommunication failure or other natural or manmademan-made accidents or incidents including the COVID-19 pandemic, that results in us being unable to fully utilize our facilities, or the manufacturing facilities of our third-party contract manufacturers, may have a material and adverse effect on our ability to operate our business, particularly on a daily basis, and have significant negative consequences on our financial and operating conditions. Loss of access to these facilities may result in increased costs, delays in the development of our therapeutic candidates or interruption of our business operations. Earthquakes, medical epidemics or pandemics including the COVID-19 pandemic, or other natural disasters could further disrupt our operations, and have a material and adverse effect on our business, financial condition, results of operations and prospects. Certain of these natural disasters, including fires and severe weather events may be exacerbated by the effects of climate change. If a natural disaster, pandemic, including the COVID-19 pandemic power outage or other event occurred that prevented us from using all or a significant portion of our headquarters, that damaged critical infrastructure, such as our research facilities or if similar events occurred elsewhere effecting the manufacturing facilities of our third-party contract manufacturers, or that otherwise disrupted operations, it may be difficult or, in certain cases, impossible, for us to continue our
business for a substantial period of time. We may incur substantial expenses as a result of the limited nature of our disaster recovery and business continuity plans, which, could have a material and adverse effect on our business. As part of our risk management policy, we maintain insurance coverage at levels that we believe are appropriate for our business. However, in the event of an accident or incident at these facilities, we cannot assure you that the amounts of insurance will be sufficient to satisfy any damages and losses. If our facilities, or the manufacturing facilities of our third-party contract manufacturers, are unable to operate because of an accident or incident or for any other reason, even for a short period of time, any or all of our research and development programs may be harmed. Any business interruption may have a material and adverse effect on our business, financial condition, results of operations and prospects.
Risks Related to Intellectual Property
If we are not able to obtain and enforce patent protection for our technologies or therapeutic candidates, development and commercialization of our therapeutic candidates may be adversely affected.
Our success depends in part on our ability to obtain and maintain patents and other forms of intellectual property rights, including in-licenses of intellectual property rights of others, patents and patent applications protecting, or seeking to protect, our therapeutic candidates and methods for treating patients using our therapeutic candidates, as well as our ability to preserve our trade secrets, to prevent third parties from infringing upon our proprietary rights and to operate without infringing upon the proprietary rights of others. Our patent estate includes various issued United States patents, United States pending non-provisional patent applications, United States pending provisional applications, pending PCT applications, issued foreign patents, and foreign patent applications currently pending in various foreign jurisdictions.
While we will endeavor to protect our therapeutic candidates with intellectual property rights such as patents, as appropriate, the process of obtaining, maintaining, and enforcing patents is time-consuming, expensive and sometimes unpredictable, and we may not be able to file and prosecute all necessary or desirable patent applications, or maintain, enforce and license any patents that may issue from such patent applications, at a reasonable cost or in a timely manner. It is also possible that we will fail to identify patentable aspects of our research and development output before it is too late to obtain patent protection. We may not have the right to control the preparation, filing and prosecution of patent applications, or to maintain the rights to any patents we may license to or from third parties. Therefore, such patents and applications may not be prosecuted and enforced in a manner consistent with the best interests of our business.
Our existing issued and granted patents and any future patents we obtain may not be sufficiently broad to prevent others from using our technology or from developing competing products and technology. There is no guarantee that any of our pending patent applications will result in issued or granted patents, that any of our issued or granted patents will not later be found to be invalid or unenforceable or that any issued or granted patents will include claims that are sufficiently broad to provide meaningful protection from any competitors. Our competitors may be able to circumvent our patents by developing similar or alternative therapeutic candidates in a non-infringing manner. We will be able to protect our proprietary rights from unauthorized use by third parties only to the extent that our current and future proprietary technology and therapeutic candidates are covered by valid and enforceable patents or are effectively maintained as trade secrets. If third parties disclose or misappropriate our proprietary rights, it may materially and adversely affect our business.
The USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other provisions during the patent process. There are situations in which non-compliance can result in abandonment or lapse of a patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, competitors might be able to enter the market earlier than would otherwise have been the case. The standards applied by the USPTO and foreign patent offices in granting patents are not always applied uniformly or predictably. For example, there is no uniform worldwide policy regarding patentable subject matter or the scope of claims allowable in biotechnology patents. As such, we do not know the degree of future protection that we will have on our proprietary therapeutics and technology.
The patent position of biotechnology and pharmaceutical companies generally is highly uncertain, involves complex legal, technological and factual questions and has in recent years been the subject of much litigation. Once granted,
patents may remain open to opposition, interference, re-examination, post-grant review, inter partes review, nullification or derivation action in court or before patent offices or similar proceedings for a given period after allowance or grant, during which time third parties can raise objections against such initial grant. In the course of such proceedings, which may continue for a protracted period of time, the patent owner may be compelled to limit the scope of the allowed or granted claims thus attacked, or may lose the allowed or granted claims altogether, e.g., due to a determination that the claims are invalid or unenforceable. In addition, there can be no assurance that:
•others will not or will not be able to legally make, use or sell products or therapeutic candidates that are the same as or similar to our therapeutic candidates despite the claims of the patents that we own or license;
•we or our licensors, or our collaborators are the first to make the inventions covered by each of our issued patents and pending patent applications that we own or license;
•we or our licensors, or our collaborators are the first to file patent applications covering certain aspects of our inventions;
•others will not independently develop similar or alternative technologies or duplicate any of our technologies without infringing our intellectual property rights;
•any issued patents that we own or have licensed will provide us with any competitive advantage; or
•the patents of others will not have a material or adverse effect on our business, financial condition, results of operations and prospects.
As a result, the issuance, scope, validity, enforceability and commercial value of our patent rights are highly uncertain.
We may not be able to protect our intellectual property rights throughout the world.
Obtaining valid and enforceable issued or granted patents covering our therapeutic candidates in the United States and worldwide can be extremely costly. In jurisdictions where we have not obtained patent protection, competitors or third parties may use our technology to develop their own products and further, may export otherwise infringing products to territories where we have patent protection, but where it is more difficult to enforce a patent as compared to the United States. Third-party or competitor products may compete with our future products in jurisdictions where we do not have issued or granted patents or where our issued or granted patent claims or other intellectual property rights are not sufficient to prevent competitor activities in these jurisdictions. The legal systems of certain countries, particularly certain developing countries, make it difficult to enforce patents and such countries may not recognize other types of intellectual property protection, particularly that relating to biotechnology. This could make it difficult for us to prevent the infringement of our patents or marketing of competing products in violation of our proprietary rights generally in certain jurisdictions. Proceedings to enforce our patent rights in foreign jurisdictions could put our patents at risk of being invalidated or interpreted narrowly and our patent applications at risk of not issuing, could provoke third parties to assert claims against us, and, whether or not successful, could result in substantial cost and divert our efforts and attention from other aspects of our business.
We generally file a provisional patent application first (a priority filing) at the USPTO. An international application under the PCT is usually filed within 12 months after the priority filing. Based on the PCT filing, national and regional patent applications may be filed in the United States, Europe, Japan, Australia and Canada and, depending on the individual case, also in one, several or all of Brazil, China, India, Israel, Mexico, New Zealand, Russia or Eurasian Patent Organization, Singapore, South Africa, South Korea and other jurisdictions. We have so far not filed for patent protection in all national and regional jurisdictions where such protection may be available. In addition, we may decide to abandon national and regional patent applications before grant. Finally, the grant proceeding of each national or regional patent is an independent proceeding which may lead to situations in which applications might in some jurisdictions be refused by the relevant registration authorities, while granted by others. It is also quite common that, depending on the country, various scopes of patent protection may be granted on the same therapeutic candidate or technology.
When a patent is granted by a regional patent office (e.g., Europe or Eurasia), the patent must be validated in individual countries in order to be in effect in those countries. We may decide not to validate regional patents in every available country or at all in any country in the region. In addition, we may decide to abandon national and regional patent applications before or after grant.
The laws of some jurisdictions do not protect intellectual property rights to the same extent as the laws in the United States, and many companies have encountered significant difficulties in protecting and defending such rights in such jurisdictions. If we or any licensors encounter difficulties in protecting, or are otherwise precluded from effectively protecting, the intellectual property rights important for our business in such jurisdictions, the value of these rights may be diminished and we may face additional competition from others in those jurisdictions. Many countries have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. In addition, many countries limit the enforceability of patents against government agencies or government contractors. In these countries, the patent owner may have limited remedies, which could materially diminish the value of such
patent. If we or any licensors are forced to grant a license to third parties with respect to any patents relevant to our business, our competitive position in the relevant jurisdiction may be impaired and our business and results of operations may be adversely affected.
Changes in patent laws could diminish the value of patents in general, thereby impairing our ability to protect our products.
Changes in either the patent laws or interpretation of the patent laws in the United States and other jurisdictions in which we file patent applications could increase the uncertainties and costs surrounding the prosecution of patent applications and the enforcement or defense of issued patents. For example, under the Leahy-Smith America Invents Act, or the America Invents Act, enacted in September 2011, the United States transitioned to a first inventor to file system in which, assuming that other requirements for patentability are met, the first inventor to file a patent application is entitled to the patent on an invention regardless of whether a third party was the first to invent the claimed invention. In contrast, prior to March 2013, in the United States, the first to invent the claimed invention was entitled to the patent, assuming that other requirements for patentability were met. Furthermore, United States patent law under the America Invents Act allows for post issuance challenges to United States patents, including ex parte reexaminations, inter parte reviews and post grant oppositions. If our United States patents are challenged using such procedures, we may not prevail, possibly resulting in altered or diminished claim scope or loss of patent rights altogether. Similarly, some countries, notably members of the European Union,EU, also have post grant opposition proceedings that can result in changes in scope and/or cancellation of patent claims.
The United States Supreme Court has also ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. In addition to increasing uncertainty with regard to our ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on decisions by the United States Congress, the federal courts and the USPTO, the laws and regulations governing patents could change in unpredictable ways that could weaken our ability to obtain new patents or to enforce our existing patents and patents that we might obtain in the future.
As another example, the complexity and uncertainty of European patent laws have increased in recent years. In Europe, a new unitary patent system was launched on June 1, 2023, which significantly impacted European patents, including those granted before the introduction of such a system. Under the unitary patent system, European applications now have the option, upon grant of a patent, of becoming a Unitary Patent which are subject to the jurisdiction of the Unitary Patent Court (UPC). As the UPC is a new court system, there is no precedent for the court, increasing the uncertainty of any litigation. Patents granted before the implementation of the UPC have the option of opting out of the jurisdiction of the UPC and remaining as national patents in the UPC countries. Patents that remain under the jurisdiction of the UPC will be potentially vulnerable to a single UPC-based revocation challenge that, if successful, could invalidate the patent in all countries who are signatories to the UPC. We cannot predict with certainty the long-term effects of any potential changes.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non compliancenon-compliance with these requirements.
The USPTO, the European Patent Office and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. For example, periodic maintenance and annuity fees on any issued patent are due to be paid to the USPTO, the European Patent Office and foreign patent agencies in several stages over the lifetime of the patent. Some
jurisdictions also require payment of annuity fees during pendency of a patent application. While an inadvertent lapse can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which non-compliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Non complianceNon-compliance events that could result in abandonment or lapse of a patent or patent application include failure to respond to official actions within prescribed time limits, non paymentnon-payment of fees and failure to properly legalize and submit formal documents. If we or our licensors or collaboration partners fail to maintain the patents and patent applications covering our therapeutic candidates, our competitors might better be able to enter the market, which would have an adverse effect on our business.
We may be required to reduce the scope of our intellectual property due to intellectual property claims included in the patents or patent applications of others.
Third parties may have filed, and may in the future file, patent applications covering technology similar to ours. It is also possible that we have failed to identify relevant third-party patents or applications. For example, United States applications filed before November 29, 2000 and certain United States applications filed after that date that will not be filed outside the United States remain confidential until patents issue. Patent applications in the United States and elsewhere are published approximately 18 months after the earliest filing for which priority is claimed, with this earliest filing date being commonly referred to as the priority date. Therefore, patent applications covering our therapeutic candidates could have been filed by others without our knowledge. Additionally, pending patent
applications that have been published can, subject to certain limitations, be later amended in a manner that could cover any future approved products or our therapeutic candidates. Any such patent application may have priority over our patent applications, which could further require us to obtain rights to issued patents covering such technologies, if possible, or block us from practicing certain aspects of our technology if we are unable to successfully pursue litigation to nullify or invalidate the third-party intellectual property right concerned.
If another party has filed a United States patent application on inventions similar to ours that claims priority to an application filed prior to March 16, 2013, we may have to participate in an interference proceeding declared by the USPTO to determine priority of invention in the United States. Similarly, if another party has filed a United States patent application on inventions similar to ours that claims priority to an application filed after March 16, 2013, we may have to participate in a derivation proceeding to determine whether that party derived the claimed invention from an inventor listed on our application and then filed the third-party application without authorization. The costs of these proceedings could be substantial, and it is possible that such efforts would be unsuccessful if, unbeknownst to us, the other party had independently arrived at the same or similar invention prior to our own invention, resulting in a loss of our United States patent position with respect to such inventions. In addition, an unfavorable outcome could require us to cease using the related technology or to attempt to license rights to it from the prevailing party. Our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms or at all, or if a non-exclusive license is offered and our competitors gain access to the same technology. Further, changes enacted on March 15, 2013 to the United States patent laws under the America Invents Act resulted in the United States changing from a “first to invent” country to a “first to file” country. As a result, we may lose the ability to obtain a patent if a third party files with the USPTO first and could become involved in proceedings before the USPTO to resolve disputes related to inventorship. We may also become involved in similar proceedings in other jurisdictions.
We or our licensors, licensees or any future strategic partners may become subject to third-party claims or litigation alleging infringement of patents or other proprietary rights or seeking to invalidate patents or other proprietary rights, and we may need to resort to litigation to protect or enforce our patents or other proprietary rights, all of which could be costly, time consuming, delay or prevent the development and commercialization of our therapeutic candidates, or put our patents and other proprietary rights at risk.
Our commercial success depends in part on our avoiding infringement of the patents and proprietary rights of third parties. We or our licensors, licensees or any future strategic partners may be subject to third-party claims for infringement or misappropriation of patent or other proprietary rights. There is a substantial amount of litigation, both within and outside the United States, involving patent and other intellectual property rights in the biotechnology and pharmaceutical industries including patent infringement lawsuits, interferences, derivations, oppositions and inter partes review proceedings before the USPTO, and corresponding foreign patent offices.
Numerous United States and foreign issued patents and pending patent applications, which are owned by third parties, exist in the fields in which we are pursuing development candidates. As the biotechnology and pharmaceutical industries expand and more patents are issued, the risk increases that we may be subject to claims of infringement of the patent rights of third parties. Because patent applications can take many years to issue, there may be currently pending patent applications which may later result in issued patents that our current or future therapeutic candidates may infringe. In addition, third parties may obtain patents in the future and claim that use of our technologies infringes upon these patents. If we, our licensees or our licensors, or any future strategic partners are found to infringe a third-party patent or other intellectual property rights, we could be required to pay damages, potentially including treble damages, if we are found to have willfully infringed. In addition, we, our licensees or our licensors, or any future strategic partners may choose to seek, or be required to seek, a license from a third party, which may not be available on acceptable terms, if at all. Even if a license can be obtained on acceptable terms, the rights may be non-exclusive, which could give any competitors access to the same technology or intellectual property rights licensed to us. If we fail to obtain a required license, the holders of any such patents may be able to block us, our licensees or our collaborators from marketing therapeutic candidates based on our technology until such patents expire, which could limit our ability to generate revenue or achieve profitability and possibly prevent us from generating revenue sufficient to sustain our operations.
In addition, we may find it necessary to pursue claims or initiate lawsuits to protect or enforce our patent or other intellectual property rights. The cost to us in defending or initiating any litigation or other proceeding relating to
patent or other proprietary rights, even if resolved in our favor, could be substantial, and litigation would divert our management’s attention. Competitors may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could delay our research and development efforts and limit our ability to continue our operations.
If we were to initiate legal proceedings against a third party to enforce a patent covering one of our products or our technology, the defendant could counterclaim that our patent is invalid or unenforceable. In patent litigation in the United States, defendant counterclaims alleging invalidity or unenforceability are commonplace. Grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements, for example, lack of novelty, obviousness or non-enablement. Grounds for an unenforceability assertion could be an allegation that someone connected with prosecution of the patent withheld relevant information from the USPTO, or made a misleading statement, during prosecution. The outcome following legal assertions of invalidity and unenforceability during patent litigation is unpredictable. With respect to the validity question, for example, we cannot be certain that there is no invalidating prior art, of which we and the patent examiner were unaware during prosecution. Publications of discoveries in the scientific literature often lag behind the actual discoveries, and patent applications in the United States and other jurisdictions are typically not published until 18 months after filing or, in some cases, not at all. If a defendant were to prevail on a legal assertion of invalidity or unenforceability, we would lose at least part, and perhaps all, of the patent protection on one or more of our products or certain aspects of our technology. This loss of patent protection could have a material and adverse effect on our business, financial condition, results of operations and prospects. Patents and other intellectual property rights also will not protect our technology if competitors design around our protected technology without legally infringing our patents or other intellectual property rights.
If we fail to comply with our obligations under the agreements pursuant to which we license intellectual property rights from third parties, or otherwise experience disruptions to our business relationships with our licensors, we could lose the rights to intellectual property licensed to us.
We are a party to license agreements under which we are granted rights to third-party intellectual property, and we expect that we may need to enter into additional license agreements in the future. License agreements may impose various development obligations, payment of royalties and fees based on achieving certain milestones, as well as other obligations. If we fail to comply with our obligations under these agreements, the licensor may have the right to terminate the license. The termination of any license agreements or failure to adequately protect such license agreements could prevent us from commercializing therapeutic candidates covered by the licensed intellectual property or otherwise adversely affect our business. Our license agreements may involve sublicenses from third parties which are not the original licensor of the intellectual property at issue. Under these agreements, we would
rely on our licensor to comply with its obligations under the primary license agreements, where we may have no relationship with the original licensor of such rights. If the licensors fail to comply with their obligations under these upstream license agreements, the original third-party licensor may have the right to terminate the original license, which may terminate the sublicense. If this were to occur, we would no longer have rights to the applicable intellectual property and, in the case of a sublicense, if we were not able to secure our own direct license with the owner of the relevant rights, which we may not be able to do at a reasonable cost or on reasonable terms, it may adversely affect our ability to continue to develop and commercialize any of our therapeutic candidates incorporating the relevant intellectual property.
Disputes may arise regarding intellectual property subject to a licensing agreement, including:
•the scope of rights granted under the license agreement and other interpretation related issues;
•the extent to which our technology and processes infringe on intellectual property of the licensor that is not subject to the licensing agreement;
•the sublicensing of patent and other rights under any collaboration relationships we might enter into in the future;
•our diligence obligations under the license agreement and what activities satisfy those diligence obligations;
•the ownership of inventions and know-how resulting from the joint creation or use of intellectual property by our licensors and us and our collaborator; and
•the priority of invention of patented technology.
If disputes over intellectual property that we have licensed prevent or impair our ability to maintain any licensing arrangements on acceptable terms, we may be unable to successfully develop and commercialize the affected therapeutic candidates.
Our intellectual property agreements with our licensors, licensees, collaborators and third parties may be subject to disagreements over contract interpretation, which could narrow the scope of, or result in termination of, our rights to the relevant intellectual property or technology or increase our financial or other obligations to such third parties, or reduce the financial or other obligations our licensees have to us.
Certain provisions in our intellectual property agreements may be susceptible to multiple interpretations. For example, we may disagree with our licensors, licensees or collaborators regarding whether, when and to what extent various obligations under these agreements apply to certain of our/their therapeutic candidates and products, including various payment, development, commercialization, funding, diligence, sublicensing, insurance, patent prosecution and enforcement and/or other obligations. The resolution of any contract interpretation disagreement that may arise could affect the scope of our rights to the relevant intellectual property or technology, or affect financial or other obligations under the relevant agreement. In either case, such disagreement could have a material adverse effect on our business, financial condition, results of operations and prospects.
In addition, while it is our policy to require our employees and contractors who may be involved in the conception or development of intellectual property to execute agreements assigning such intellectual property to us, we may be unsuccessful in executing such an agreement with each party who in fact conceives or develops intellectual property that we regard as our own. Our assignment agreements may not be self-executing or may be breached, and we may be forced to bring claims against third parties, or defend claims they may bring against us, to determine the ownership of what we regard as our intellectual property.
Intellectual property litigation could cause us to spend substantial resources and distract our personnel from their normal responsibilities.
Litigation or other legal proceedings relating to intellectual property claims, with or without merit, are unpredictable, generally expensive, time consuming and are likely to divert significant resources from our core business, including distracting our technical and management personnel from their normal responsibilities. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments and if securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on
the price of our common stock. Such litigation or proceedings could substantially increase our operating losses and reduce the resources available for development activities or any future sales, marketing or distribution activities.
We may not have sufficient financial or other resources to adequately conduct such litigation or proceedings. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their greater financial resources and more mature and developed intellectual property portfolios. Accordingly, despite our efforts, we may not be able to prevent third parties from infringing upon or misappropriating or from successfully challenging our intellectual property rights. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could have a material and adverse effect on our ability to compete in the marketplace.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed.
In addition to seeking patent protection for certain aspects of our therapeutic candidates, we also consider trade secrets, including confidential and unpatented know-how important to our business. We may rely on trade secrets or confidential know-how to protect our technology, especially where patent protection is believed to be of limited value. Trade secrets and confidential know-how are difficult to maintain as confidential. We seek to protect trade secrets and confidential and unpatented know-how, in part, by entering into non-disclosure and confidentiality agreements with parties who have access to such knowledge, such as our employees, partners, outside scientific
collaborators, CROs, contract manufacturers, consultants, advisors and other third parties. In addition, while it is our policy to require our employees and contractors who may be involved in the conception or development of intellectual property to execute agreements assigning such intellectual property to us, we may be unsuccessful in executing such an agreement with each party who in fact conceives or develops intellectual property that we regard as our own. Our assignment agreements may not be self-executing or may be breached, and we may be forced to bring claims against third parties, or defend claims they may bring against us, to determine the ownership of what we regard as our intellectual property.
Moreover, even if relevant agreements are entered into, despite these efforts, any of these parties may breach the agreements and unintentionally or willfully disclose our proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for such breaches. Confidentiality agreements may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive and time-consuming, and the outcome is unpredictable. In addition, some courts in the United States and certain foreign jurisdictions are less willing or unwilling to protect trade secrets. If any of our trade secrets were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent them from using that technology or information to compete with us. Moreover, a competitor who independently develops substantially equivalent proprietary information may even apply for patent protection in respect of the same. If successful in obtaining such patent protection, our competitors could limit our use of our trade secrets or confidential know-how. Under certain circumstances, we may also decide to publish some know-how to attempt to prevent others from obtaining patent rights covering such know-how. If any of our trade secrets were to be disclosed to or independently developed by a competitor, our competitive position would be harmed. Additionally, if the steps taken to maintain our trade secrets are deemed inadequate, we may have insufficient recourse against third parties for misappropriating the trade secret.
We may be subject to claims that we or our employees or consultants have wrongfully used or disclosed alleged trade secrets of our employees’ or consultants’ former employers or their clients. These claims may be costly to defend and if we do not successfully do so, we may be required to pay monetary damages and may lose valuable intellectual property rights or personnel.
Many of our employees were previously employed at universities or biotechnology companies, including potential competitors. Although no claims against us are currently pending, we may be subject to claims that these employees or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. If we fail in defending such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. A loss of key research personnel or their work product could hamper our ability to commercialize, or prevent us from
commercializing, our therapeutic candidates, which could severely harm our business. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
If our trademarks and trade names are not adequately protected, then we may not be able to build name recognition in our markets of interest and our business may be adversely affected.
Our trademarks or trade names may be challenged, infringed, circumvented or declared generic or determined to be infringing on other marks. Our trademarks may not be approved by one or more governmental trademark offices or may not be approved for use on our products by regulatory agencies, such as the FDA. We may not be able to protect our rights to these trademarks and trade names or may be forced to stop using these names, which we need for name recognition by potential partners or customers in our markets of interest. If we are unable to establish name recognition based on our trademarks and trade names, we may not be able to compete effectively and our business may be adversely affected.
If our patent terms expire before or soon after our therapeutic candidates are approved, or if manufacturers of biosimilar drugs successfully challenge our patents, our business may be materially harmed.
Patents have a limited duration. In the United States, if all maintenance fees are timely paid, the natural expiration of a patent is generally 20 years from its earliest United States non provisional filing date. Various extensions may be available, but the life of a patent, and the protection it affords, is limited. Even if patents covering our therapeutic candidates, their manufacture, or use are obtained, once the patent life has expired, we may be open to competition from competitive medications, including biosimilar medications.
Depending upon the timing, duration and conditions of FDA marketing approval of our therapeutic candidates, one or more of our United States patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, referred to asor the Hatch-Waxman Act, and similar legislation in the European Union.EU. The Hatch-Waxman Act permits a patent term extension of up to five years for a patent covering an approved product as compensation for effective patent term lost during product development and the FDA regulatory review process. The patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval, and only one patent applicable to an approved drug may be extended. However, we may not receive an extension if we fail to apply within applicable deadlines, fail to apply prior to expiration of relevant patents or otherwise fail to satisfy applicable requirements. Moreover, the length of the extension could be less than we request. If we are unable to obtain patent term extension or the term of any such extension is less than we request, the period during which we can enforce our patent rights for that product will be shortened and our competitors may obtain approval to market competing products sooner than we expect. Also, the scope of our right to exclude during any patent term extension period may be limited or may not cover a competitor’s product or product use. As a result, our revenue from applicable therapeutic candidates, if approved, could be reduced, possibly materially.
Given the amount of time required for the development, testing and regulatory review of new drug candidates, patents protecting such drug candidates might expire before or shortly after such drug candidates are commercialized. As a result, our patents and patent applications may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours. If we are unable to obtain an exclusive license to any such third-party co-owners’ interest in such patents or patent applications, such co-owners may be able to license their rights to other third parties, including our competitors, and our competitors could market competing products and technology. In addition, we may need the cooperation of any such co-owners of our patents in order to enforce such patents against third parties, and such cooperation may not be provided to us. Any of the foregoing could have a material adverse effect on our competitive position, business, financial conditions, results of operations and prospects.
Manufacturers of biosimilar drugs may challenge the scope, validity, or enforceability of our patents in court or before a patent office, and we may not be successful in enforcing or defending those intellectual property rights and, as a result, may not be able to develop or market the relevant product exclusively, which would have a material adverse effect on any potential sales of that product. Upon the expiration of our issued patents or patents that may issue from our pending patent applications, we will not be able to assert such patent rights against potential competitors and our business, financial condition, results of operations, and prospects may be adversely affected.
Intellectual property rights do not necessarily address all potential threats to our competitive advantage.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations, and may not adequately protect our business nor permit us to maintain our competitive advantage. The following examples are illustrative:
•Others may be able to make therapeutic candidates that are the same as or similar to our therapeutic candidates but that are not covered by the claims of the patents that we own or may have exclusively licensed.
•Others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our intellectual property rights.
•Third parties might conduct research and development activities in countries where we do not have patent rights and then use the information learned from such activities to develop competitive products for sale in our major commercial markets.
•We may not develop additional technologies that are patentable.
Risks Related to Government Regulation
We or our collaborators may be unable to obtain regulatorymarketing approval for any product that we or a collaborator may develop and the regulatorymarketing approval processes of the FDA and other comparable regulatory authorities outside the United States are lengthy, time-consuming and inherently unpredictable.
Any product that we or our collaborators may attempt to develop, manufacture or market in the United States will be subject to extensive regulation by the FDA, including regulations relating to development, preclinicalnonclinical testing, performance of clinical trials, manufacturing and post-approval commercialization. PreclinicalNonclinical testing, clinical trials and manufacturing, among other activities, will be subjected to an extensive review process before a new therapeutic product may be sold in the United States. Satisfaction of these and other regulatory requirements is costly, time consuming, uncertain and subject to unanticipated delays, including delays arising from the COVID-19 pandemic.delays. The time required to obtain FDA approval, and any other required approvals for biological products is unpredictable but typically requires several years and may never be obtained.
Any product that we or our collaborators may wish to develop, manufacture or market in countries other than the United States will also be subject to numerous foreign regulatory requirements governing the conduct of clinical trials, manufacturing and marketing, pricing and third-party reimbursement among other things in such countries. The foreign regulatorymarketing approval process includes all of the risks and uncertainties associated with FDA approval described above as well as risks attributable to the satisfaction of local regulations in such foreign jurisdictions.
In particular, obtaining marketing approval for biological products requires the submission of extensive preclinicalnonclinical and clinical data and supporting information to regulatory authorities for each therapeutic indication to establish the product candidate’s safety and efficacy. Securing marketing approval also requires the submission of information about the product manufacturing process, and in many cases the inspection of manufacturing, processing, and packaging facilities by the regulatory authorities. Our product candidates may not be effective, may be only moderately effective or may prove to have undesirable or unintended side effects, toxicities or other characteristics that may preclude our obtaining marketing approval or prevent or limit commercial use, or there may be deficiencies in cGMP compliance by us or by our contract development and manufacturing organizations, or CDMOs, that could result in the candidate not being approved. Moreover, we have not obtained regulatorymarketing approval for any therapeutic candidate in any jurisdiction and it is possible that none of our existing therapeutic candidates or any therapeutic candidates we may seek to develop in the future will ever obtain regulatorymarketing approval.
Our therapeutic candidates could fail to receive, or could be materially delayed in receiving, regulatorymarketing approval for many reasons, including any one or more of the following:
•the FDA, EMA or comparable foreign regulatory authorities may disagree with the design or implementation of our clinical trials;
•we may be unable to demonstrate to the satisfaction of the FDA, EMA or comparable foreign regulatory authorities that a product candidate is safe and effective for its proposed indication;
•the results of clinical trials may not meet the level of statistical significance required by the FDA, EMA or comparable foreign regulatory authorities for marketing approval;
•we may be unable to demonstrate that a product candidate’s clinical and other benefits outweigh its safety risks;
•the FDA, EMA or comparable foreign regulatory authorities may disagree with our interpretation of data from preclinicalnonclinical studies or clinical trials;
•the data collected from clinical trials of our product candidates may not be sufficient to support the submission of a BLA or other submission or to obtain regulatorymarketing approval in the United States or elsewhere;
•upon review of our clinical trial sites and data, the FDA or comparable foreign regulatory authorities may find our record keeping or the record keeping of our clinical trial sites to be inadequate or may identify other GCP deficiencies related to the trials;
•the manufacturing processes or facilities of third-party manufacturers with which we contract for clinical and commercial supplies may fail to meet the requirements of the FDA, EMA or comparable foreign regulatory authorities; or
•the medical standard of care or the approval policies or regulations of the FDA, EMA or comparable foreign regulatory authorities may significantly change in a manner that renders our clinical data insufficient for approval.
It is possible that none of the therapeutic candidates we or our collaborators may develop will obtain the marketing approvals necessary for us or our collaborators to sell the products either in the United States or any other country. Furthermore, approval by the FDA of a therapeutic product does not assure approval by regulatory authorities outside the United States or vice versa. Even if approval for a therapeutic product is obtained, such approval may be subject to limitations on the indicated uses or appropriate patient population that could result in a significantly reduced potential market size for the product.
If we or our collaborators fail to obtain the appropriate regulatorymarketing approvals necessary for us or our collaborators to sell our therapeutic candidates, or if the approvals are more limited than those that we intend to seek, our business, financial condition and results of operations would be materially harmed.
We will be subject to stringent domestic and foreign therapeutic and drug regulation with respect to any potential products. Even if we receive regulatorymarketing approval for any of our therapeutic candidates, we will still be subject to ongoing regulatory obligations and continued review, which may result in significant additional expense. If we fail to comply with United States and foreign regulatory requirements, regulatory authorities could limit or withdraw any marketing or commercialization approvals we may receive and subject us to other penalties. Any unfavorable regulatory action may materially and adversely affect our future financial condition and business operations.
Even if we receive marketing and commercialization approval for a therapeutic candidate, we will be subject to continuing post-marketing regulatory requirements. Our potential products, further development activities and manufacturing and distribution of a future product, once developed and determined, will be subject to extensive and rigorous regulation by numerous government agencies, including the FDA and comparable foreign agencies. To varying degrees, each of these agencies monitors and enforces our compliance with laws and regulations governing the development, testing, manufacturing, labeling, marketing, distribution, and the safety and effectiveness of our therapeutic candidates and, if approved, our future products. The process of obtaining marketing approval or clearance from the FDA and comparable foreign bodies for new products, or for enhancements, expansion of the indications or modifications to existing products, could:
•take a significant, indeterminate amount of time;
•require the expenditure of substantial resources;
•involve rigorous preclinicalnonclinical and clinical testing, and possibly post-market surveillance;
•require design changes of our potential products; or
•result in our never being granted the regulatorymarketing approval we seek.
Any of these occurrences may cause our operations or potential for success to suffer, harm our competitive standing and result in further losses that adversely affect our financial condition.
The FDA, as well as its foreign regulatory counterparts, also have significant post-market authority, including the authority to require labeling changes based on new safety information and to require post-market studies or clinical trials to evaluate safety risks related to the use of a product or to require withdrawal of the product from the market. Additionally, the FDA regulates the promotional claims that may be made about prescription products, such as our products, if approved. In particular, a product may not be promoted for uses that are not approved by the FDA as reflected in the product’s approved labeling. However, we may share truthful and not misleading information with healthcare providers and payors that is otherwise consistent with the product’s FDA approved labeling.
We will have ongoing responsibilities under these and other FDA and international regulations, both before and after a product is approved and commercially released. Compliance with applicable regulatory requirements is subject to continual review and is monitored rigorously through periodic inspections by the FDA and foreign regulatory agencies. If we or our collaborators, manufacturers or service providers fail to comply with applicable continuing regulatory requirements in the United States or foreign jurisdictions in which we seek to market our products, we or they may be subject to, among other things, fines, warning letters, adverse regulatory inspection finding, holds on
clinical trials, delay of approval or refusal by the FDA or applicable authorities to approve pending applications or supplements to approved applications, suspension or withdrawal of regulatorymarketing approval, product recalls and seizures, administrative detention of products, refusal to permit the import or export of products, operating restrictions, exclusion of eligibility from government contracts, injunctions, civil penalties or criminal prosecution. Any adverse regulatory action, depending on its magnitude, may restrict us from effectively commercializing our potential products and harm our business. In addition, negative publicity and product liability claims resulting from any adverse regulatory action could have a material adverse effect on our business, financial condition, results of operations, and prospects.
The FDA, EMA and other comparable foreign regulatory authorities may not accept data from trials conducted outside of their respective jurisdictions. While we currently have partnerships in China designed to provide access to patient populations outside of the United States and in the future may conduct clinical trials in other foreign jurisdictions, there can be no assurance these data will be accepted by the FDA or EMA or other comparable foreign regulatory authorities as a basis for a product’s regulatorymarketing approval.
To augment our U.S.-centric clinical strategy, we have formed partnerships in China designed to provide access patient populations for clinical trials not readily available in the United States and to facilitate rapid patient enrollment with the goal of generating more robust early clinical data from patients in China. We may in the future pursue partnerships to conduct other clinical trials outside of the United States. The acceptance of study data from clinical trials conducted outside the United States or another jurisdiction by the FDA, EMA or applicable foreign regulatory authority may be subject to certain conditions. In cases where data from foreign clinical trials are intended to serve as the basis for marketing approval in the United States, the FDA will generally not approve the application on the basis of foreign data alone unless (i) the data are applicable to the U.S. population and U.S. medical practice and (ii) the trials were performed by clinical investigators of recognized competence and pursuant to GCP regulations. For example, in February 2022, the FDA publicly rebuked an oncology product sponsor for submitting a marketing application with Phase III clinical data solely from China and since that time, it has declined to approve other applications that contained primarily China-generated clinical data. Additionally, the FDA’s clinical trial requirements, including sufficient size of patient populations and statistical powering, must be met. Many foreign regulatory bodies have similar approval requirements. In addition, any foreign trials would be subject to the applicable local laws of the foreign jurisdictions where the trials are conducted. There can be no assurance that the FDA, EMA or any applicable foreign regulatory authority will accept data from trials conducted outside of the United States or the applicable jurisdiction, including any trials conducted in China.
Our partnerships in China subject us to risks and uncertainties relating to the laws and regulations of China and the changes in relations between the United States and China.
Under its current leadership, the government of China has been pursuing economic reform policies, including by encouraging foreign trade and investment. However, there is no assurance that the Chinese government will continue to pursue such policies, that such policies will be successfully implemented, that such policies will not be
significantly altered, or that such policies will be beneficial to our partnerships in China. China’s system of laws can be unpredictable, especially with respect to foreign investment and foreign trade. The United States government has called for substantial changes to foreign trade policy with China and has raised, and has proposed to further raise in the future, tariffs on several Chinese goods. China has retaliated with increased tariffs on United States goods. Moreover, China’s legislature has recently adopted a national security law to substantially change the way Hong Kong has been governed since the territory was handed over by the United Kingdom to China in 1997. This law increases the power of the central government in Beijing over Hong Kong, limit the civil liberties of residents of Hong Kong and could restrict the ability of businesses in Hong Kong to continue to conduct business or to continue to with business as previously conducted. The U.S. State Department has indicated that the United States no longer considers Hong Kong to have significant autonomy from China. The U.S. State Department has recentlypreviously enacted sanctions related to China’s governing of Hong Kong, and the United States may impose the same tariffs and other trade restrictions on exports from Hong Kong that it places on goods from mainland China. Any further changes in United States trade policy could trigger retaliatory actions by affected countries, including China, resulting in trade wars. For example, the Uyghur Forced Labor Prevention Act bans imports from China’s Xinjiang Uyghur Autonomous Region unless it can be shown that the goods were not produced using forced labor and this legislation may have an adverse effect on global supply chains which could adversely impact our business and results of operations. Additionally, the biopharmaceutical industry in particular in China is strictly regulated by the Chinese government. Changes to Chinese regulations affecting biopharmaceutical companies are also unpredictable. Any regulatory changes and changes in United States and China relations may have a material adverse effect on our partnerships in China which could materially harm our business and financial condition.
Unfavorable pricing regulations, third-party reimbursement practices or healthcare reform initiatives could harm our business in the future.
There is increasing pressure on biotechnology companies to reduce healthcare costs. In the United States, these pressures come from a variety of sources, such as managed care groups and institutional and government purchasers. Increased purchasing power of entities that negotiate on behalf of federal healthcare programs and private sector beneficiaries could increase pricing pressures in the future. Such pressures may also increase the risk of litigation or investigation by the government regarding pricing calculations. The biotechnology industry will likely face greater regulation and political and legal actions in the future.
Adverse pricing limitations may hinder our ability to recoup our investment in one or more future therapeutic candidates, even if our future therapeutic candidates obtain regulatorymarketing approval. Adverse pricing limitations prior to approval will also adversely affect us by reducing our commercial potential. Our ability to commercialize any potential products successfully also will depend in part on the extent to which coverage and reimbursement for these products and related treatments becomes available from third-party payors, including government health administration authorities, private health insurers and other organizations. Third-party payors decide which medications they will pay for and establish reimbursement levels. In addition, companion diagnostic tests require coverage and reimbursement separate and apart from the coverage and reimbursement for their companion pharmaceutical or biological products. Similar challenges to obtaining coverage and reimbursement, applicable to pharmaceutical or biological products, will apply to companion diagnostics.
A significant trend in the U.S. healthcare industry and elsewhere is cost containment. Third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medications. Increasingly, third-party payors are requiring that companies provide them with predetermined discounts from list prices and are challenging the prices charged for medical products. Additionally, the IRA authorized CMS to negotiate drug prices annually for a select number of single source Part D drugs without generic or biosimilar competition starting in payment year 2026, and to negotiate drug prices for a select number of Part B drugs starting for payment year 2028. If a drug product is selected by CMS for negotiation, it is expected that the revenue generated from such drug will decrease. CMS has begun to implement these new authorities and entered into the first set of agreements with pharmaceutical manufacturers to conduct price negotiations in October 2023. However, the IRA’s impact on the biopharmaceutical industry in the United States remains uncertain, in part because multiple large pharmaceutical companies and other stakeholders (e.g., the U.S. Chamber of Commerce) have initiated federal lawsuits against CMS arguing the program is unconstitutional for a variety of reasons, among other complaints. Those lawsuits are currently ongoing.
We cannot be sure that coverage and reimbursement will be available for any product that we commercialize in the future and, if reimbursement is available, what the level of reimbursement will be. Reimbursement may impact the demand for, or the price of, any product for which we obtain marketing approval in the future. If reimbursement is not available or is available only to limited levels, we may not be able to successfully commercialize any therapeutic candidate that we successfully develop.
There may be significant delays in obtaining reimbursement for approved products, and coverage may be more limited than the purposes for which the product is approved by the FDA or regulatory authorities in other countries. Moreover, eligibility for reimbursement does not imply that any product will be paid for in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution. Interim payments for new products, if applicable, may also not be sufficient to cover our costs and may not be made permanent. Payment rates may vary according to the use of the product and the clinical setting in which it is used, may be based on payments allowed for lower cost products that are already reimbursed and may be incorporated into existing payments for other services. Net prices for products may be reduced by mandatory discounts or rebates required by third-party payors and by any future relaxation of laws that presently restrict imports of products from countries where they may be sold at lower prices than in the United States. Third-party payors often rely upon Medicare coverage policy and payment limitations in setting their own reimbursement policies, but also have their own methods and approval process apart from Medicare coverage and reimbursement determinations. Accordingly, one third-party payor’s determination to provide coverage for a product does not assure that other payors will also provide coverage for the product. Our inability to promptly obtain coverage and adequate reimbursement from third-party payors for approved products could have a material adverse effect on our operating results, our ability to raise capital needed to commercialize potential products and our overall financial condition.
Healthcare legislative reform measures may have a material and adverse effect on our business, financial condition, results of operations, and prospects.
Third-party payors, whether domestic or foreign, or governmental or commercial, are developing increasingly sophisticated methods of controlling healthcare costs. In both the United States and certain foreign jurisdictions, there have been, and likely will continue to be, legislative and regulatory proposals at the foreign, federal, and state levels directed at containing or lowering the cost of healthcare. We cannot predict the initiatives that may be adopted in the future. The continuing efforts of the government, insurance companies, managed care organizations, and other
payors of healthcare services to contain or reduce costs of healthcare and/or impose price controls may adversely affect:
•the demand for our therapeutic candidates, if we obtain regulatorymarketing approval;
•our ability to receive or set a price that we believe is fair for our products;
•our ability to generate revenue and achieve or maintain profitability;
•the level of taxes that we are required to pay; and
•the availability of capital.
In March 2010, the ACA was enacted, which includes measures that have significantly changed the way healthcare is financed by both governmental and private insurers in the United States. It also included the BPCIA, which created an abbreviated approval pathway for biological products that are biosimilar to or interchangeable with an FDA-licensed reference biological product. The ACA continues to significantly impact the United States’ pharmaceutical industry.
Moreover, there has been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products, which has resulted in several Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drug products. On March 10, 2020, the Trump administration sent “principles” for drug pricing to Congress, calling for legislation that would, among other things, cap Medicare Part D beneficiary out-of-pocket pharmacy expenses, provide an option to cap Medicare Part D beneficiary monthly out-of-pocket expenses, and place limits on pharmaceutical price increases. In addition, the Trump administration previously released a “Blueprint” to lower drug prices and reduce out of pocket costs of drugs that contained proposals to increase manufacturer competition, increase the negotiating power of certain federal healthcare programs, incentivize manufacturers to lower the list price of their products and reduce the out-of-pocket costs of drug products paid by consumers. The HHS has solicited feedback on some of these measures and has implemented others under its existing authority. For example, in September 2020, the FDA finalized a rulemaking to establish a system whereby state governmental entities could lawfully import and distribute prescription drugs sourced from Canada. Those new regulations became effective on November 30, 2020, although the impact of such future programs is uncertain. While some of these and other measures may require additional authorization to become effective, Congress and the Trump administration have each indicated that they will continue to seek new legislative and/or administrative measures to control drug costs. Notably, on July 24, 2020, President Trump announced four executive orders related to prescription drug pricing that attempt to implement several of the Administration’s proposals, including a policy that would tie Medicare Part B drug prices to international drug prices; one that directs HHS to finalize the Canadian drug importation proposed rule previously issued by HHS (which has since been finalized, as noted above) and makes other changes allowing for personal importation of drugs from Canada; one that directs HHS to finalize the rulemaking process on modifying the anti-kickback law safe harbors for plans, pharmacies, and pharmaceutical benefit managers after HHS confirms that the action is not projected to increase federal spending, Medicare beneficiary premiums, or patients’ total out-of-pocket costs; and one that reduces costs of insulin epinephrine auto-injectors to patients of Federally Qualified Health Centers. President Trump issued another executive order on September 13, 2020 that directs HHS to undertake rulemaking in order to test an international reference pricing model for prescription drug products. The probability of success of these newly announced policies and their impact on the U.S. prescription drug marketplace is unknown. There are likely to be political and legal challenges associated with implementing these reforms as they are currently envisioned.
At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. In December 2020, the U.S. Supreme
Court held unanimously that federal law does not preempt the states’ ability to regulate PBMs and other members of the health care and pharmaceutical supply chain, an important decision that has led to more aggressive efforts by states in this area. The Federal Trade Commission in mid-2022 also launched sweeping investigations into the practices of the PBM industry that could lead to additional federal and state legislative or regulatory proposals targeting such entities’ operations, pharmacy networks, or financial arrangements. During the current congressional session, numerous PBM reforms are being considered in both the Senate and the House of Representatives; they include diverse legislative proposals such as eliminating rebates; divorcing service fees from the price of a drug, discount, or rebate; prohibiting spread pricing; limiting administrative fees; requiring PBMs to report formulary placement rationale; promoting transparency. Significant efforts to change the PBM industry as it currently exists in the U.S. may affect the entire biopharmaceutical supply chain and the business of other stakeholders, including therapeutic biological product developers like us.
We expect that the ACA, as well as other healthcare reform measures that may be adopted in the future may result in additional reductions in Medicare and other healthcare funding, more rigorous coverage criteria, lower reimbursement, and new payment methodologies. This could lower the price that we receive for any approved product. Any denial in coverage or reduction in reimbursement from Medicare or other government-funded
programs may result in a similar denial or reduction in payments from private payors, which may prevent us from being able to generate sufficient revenue, attain profitability, or commercialize our therapeutic candidates, if approved.
In the European UnionEU, similar political, economic and regulatory developments may affect our ability to profitably commercialize our current or any future products. In addition to continuing pressure on prices and cost containment measures, legislative developments at the European UnionEU or member state level may result in significant additional requirements or obstacles that may increase our operating costs. In international markets, reimbursement and healthcare payment systems vary significantly by country, and many countries have instituted price ceilings on specific products and therapies. Our future products, if any, might not be considered medically reasonable and necessary for a specific indication or cost-effective by third-party payors, an adequate level of reimbursement might not be available for such products and third-party payors’ reimbursement policies might adversely affect our ability to sell any future products profitably.
Legislative and regulatory proposals have also been made to expand post-approval requirements and restrict sales and promotional activities for biologic therapeutics.therapeutics, and FDA’s statutory authorities are periodically amended by Congress. For example, on December 20, 2019, President Trump signed P.L. 116-94 that includes a piece of bipartisan legislation called the CREATES Act. The CREATES Act aims to address the concern articulated by both the FDA and others in the industry that some brand manufacturers have improperly restricted the distribution of their products to deny generic and biosimilar product developers access to samples of brand products. Whether and how generic and biosimilar product developers will use this new pathway, as well as the likely outcome of any legal challenges to provisionspart of the CREATESConsolidated Appropriations Act remain highly uncertainfor 2023, Congress provided FDA additional authorities related to the accelerated approval pathway for human drugs and its potential effects onbiologics. Under these amendments to the U.S. biopharmaceutical industry are unknown.FDCA, the agency may require a sponsor of a product granted accelerated approval to have a confirmatory trial underway prior to approval. The amendments also give FDA the option of using expedited procedures to withdraw product approval if the sponsor’s confirmatory trial fails to verify the claimed clinical benefits of the product. We cannot be sure whether additional legislative changes will be enacted, or whether the FDA regulations, guidance or interpretations will be changed, or what the impact of such changes on the marketing approvals of our therapeutic candidates, if any, may be. In addition, increasedIncreased scrutiny by the U.S. Congress of the FDA’s approval process may significantly delay or prevent marketing approval, as well as subject us to more stringent product labeling and post-approval testing and other requirements.
We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, our therapeutic candidates may lose any marketing approval that may have been obtained and we may not achieve or sustain profitability, which would adversely affect our business.
If we or our partners, manufacturers or service providers fail to comply with healthcare laws and regulations, we or they could be subject to enforcement actions, which could affect our ability to develop, market and sell our products and may harm our reputation.
Healthcare providers, physicians and third-party payors play a primary role in the recommendation and prescription of any therapeutic candidates for which we may obtain marketing approval. Our current and future arrangements
with healthcare providers, third-party payors and customers expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we market, sell and distribute our therapeutic candidates for which we obtain marketing approval. In addition, we may be subject to patient data privacy and security regulation by both the U.S. federal government and the states in which we conduct our business. Restrictions under applicable federal and state healthcare laws and regulations, include the following:
•the U.S. federal Anti-Kickback Statute, which prohibits, among other things, persons or entities from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind to induce or reward either the referral of an individual for, or the purchase, or order or recommendation of, any good or service, for which payment may be made under federal and state healthcare programs such as Medicare and Medicaid. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation;
•the federal civil and criminal false claims laws, including the U.S. federal False Claims Act, which can be enforced through civil whistleblower or qui tam actions, and the civil monetary penalties laws, which
prohibit individuals or entities from knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false or fraudulent, knowingly making, using or causing to be made or used, a false record or statement material to a false or fraudulent claim, or from knowingly making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government. In addition, the government may assert that a claim including items and services resulting from a violation of the U.S. federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the U.S. federal False Claims Act;
•HIPAA, which imposes criminal and civil liability for, among other things, knowingly and willfully executing, or attempting to execute a scheme to defraud any healthcare benefit program, or knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statement in connection with the delivery of or payment for healthcare benefits, items or services; similar to the U.S. federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation;
•HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act and their implementing regulations, which imposes obligations on certain healthcare providers, health plans, and healthcare clearinghouse, known as covered entities, as well as their business associates that perform certain services involving the use or disclosure of individually identifiable health information, including mandatory contractual terms, with respect to safeguarding the privacy, security, and transmission of individually identifiable health information, and require notification to affected individuals and regulatory authorities of certain breaches of individually identifiable health information;
•the U.S. federal legislation commonly referred to as the Physician Payments Sunshine Act, enacted as part of the ACA, and its implementing regulations, which requires certain manufacturers of drugs, devices, biologics and medical supplies that are reimbursable under Medicare, Medicaid, or the Children’s Health Insurance Program to report annually to HHS information related to certain payments and other transfers of value to physicians (defined to include doctors, dentists, optometrists, podiatrists, chiropractors and beginning in 2022 for payments and other transfers of value provided in the previous year,chiropractors), certain advanced non-physician healthcare practitioners)practitioners and teaching hospitals, as well as ownership and investment interests held by the physicians described above and their immediate family members; and
•analogous state laws and regulations, such as state anti-kickback and false claims laws that may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers; state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government in addition to requiring drug and therapeutic biologics manufacturers to report information related to payments to physicians and other healthcare providers or marketing expenditures and pricing information; state and local laws that require the registration of pharmaceutical sales representatives; and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts.
Ensuring that our future business arrangements with third parties comply with applicable healthcare laws and regulations could involve substantial costs. It is possible that governmental authorities will conclude that our business practices do not comply with current or future statutes, regulations, agency guidance or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any such requirements, we may be subject to significant civil, criminal and administrative penalties, including monetary damages, fines, disgorgements, imprisonment, loss of eligibility to obtain approvals from the FDA, exclusion from participation in government contracting, healthcare reimbursement or other government programs, including Medicare and Medicaid, reputational harm, diminished profits and future earnings, additional reporting
requirements if we become subject to a corporate integrity agreement or other agreement to resolve allegations of non-compliance with any of these laws, and the curtailment or restructuring of our operations, any of which could adversely our financial results. Although effective compliance programs can mitigate the risk of investigation and prosecution for violations of these laws, these risks cannot be entirely eliminated. Any action against us for an alleged or suspected violation could cause us to incur significant legal expenses and could divert our management’s attention from the operation of our business, even if our defense is successful. In addition, achieving and sustaining compliance with applicable laws and regulations may be costly to us in terms of money, time and resources.
patients with metastatic or unresectable chondrosarcoma and for INBRX-101 for the treatment of patients with AATD. We may seek orphan drug status for one or more of ouradditional therapeutic candidates, but even if it is granted, we may be unable to maintain any benefits associated with orphan drug status, including market exclusivity.
Under the Orphan Drug Act, the FDA may grant orphan designation to a drug or biologic intended to treat a rare disease or condition or for which there is no reasonable expectation that the cost of developing and making available in the United States a drug or biologic for a disease or condition will be recovered from sales in the United States for that drug or biologic. If a product that has orphan drug designation subsequently receives the first FDA approval for the disease for which it has such designation, the product is entitled to orphan product exclusivity, which means that the FDA may not approve any other applications, including a full BLA to market the same drug or biologic for the same indication for seven years, except in limited circumstances, such as a showing of clinical superiority to the product with orphan drug exclusivity.
We received orphan drug status for INBRX-109 for the treatment of patients with metastatic or unresectable chondrosarcoma and for INBRX-101 for the treatment of patients with AATD. We may seek orphan drug status for one or more of ouradditional therapeutic candidates, but the FDA may not approvegrant any such request. Even if the FDA grantswith orphan drug status, to one or more of our therapeutic candidates, exclusive marketing rights in the United States may be limited if we seek FDA marketing approval for an indication broader than the therapeutic candidate’s orphan designated indication. Additionally, any therapeutic candidate that initially receives orphan drug status designation, may lose such designation if the FDA later determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantities of the product to meet the needs of patients with the rare disease or condition. In addition, others may obtain orphan drug status and then achieve marketing approval and exclusivity before us for products addressing the same diseases or conditions as products we are developing, thus limiting our ability to compete in the markets addressing such diseases or conditions for a significant period of time.
We received fast-track designation for INBRX-109 for the treatment of patients with metastatic or unresectable chondrosarcoma. We may seek fast-track designation for other of our therapeutic candidates. Even if received, fast-track designation may not actually lead to a faster review process.
We aim to benefit from the FDA’s fast trackFast Track and priority review processes. However, our therapeutic candidates may not receive an FDAWe received fast-track designation for INBRX-109 for the treatment of patients with metastatic or priority review. Without fast-track designation, submitting a BLA and getting through the regulatory process to gain marketing approval is a lengthier process.unresectable chondrosarcoma. Under fast-track designation, the FDA may initiate a rolling review of sections of a fast-track drug’s BLA before the application is complete. However, the FDA’s time period goal for reviewing an application does not begin until the last section of the BLA is submitted. Additionally, the fast-track designation may be withdrawn by the FDA if the FDA believes that the designation is no longer supported by data emerging in the clinical trial process. Under the FDA policies, a drug candidate is eligible for priority review, or review within a six-month time frame from the time a complete BLA is accepted for filing, if the drug candidate provides a significant improvement compared to marketed drugs in the treatment, diagnosis or prevention of a disease. A fast-track designated drug candidate would ordinarily meet the FDA’s criteria for priority review.
The fast-track designation for ourINBRX-109, or for other future therapeutic candidates, if obtained, may not actually lead to a faster review process and a delay in the review process or in the approval of our potential products will delay revenue from their potential sales and will increase the capital necessary to fund these product development programs. Without fast-track designation, submitting a BLA and getting through the regulatory process to gain marketing approval is a lengthier process.
We are working towards submitting INBRX-101 for marketing authorization through the use of the accelerated approval pathway and may pursue this accelerated approval pathway for future therapeutic candidates. There is no assurance that, upon receipt of our future marketing application for INBRX-101, the FDA will agree to file it
and conduct a substantive review of the data or that FDA will agree that we have met the substantial evidence of effectiveness standard necessary to support marketing approval. If unable to obtain approval under the accelerated approval pathway, we may be required to conduct additional nonclinical studies or clinical trials beyond those that we currently contemplate, which could increase the expense of obtaining, and delay the receipt of, necessary marketing approvals. Even if we receive accelerated approval from the FDA for INBRX-101, or other future therapeutic candidates for which the accelerated approval pathway may be appropriate, if our confirmatory trials do not verify clinical benefit, or if we do not comply with rigorous post-marketing requirements, the FDA may seek to withdraw accelerated approval.
In October 2022, we announced that, based on discussions with the FDA, there is potential to pursue an accelerated approval for INBRX-101 in patients with emphysema due to AATD using AAT serum levels as the surrogate endpoint.
Under the accelerated approval provisions in the FDCA and the FDA’s implementing regulations, the FDA may grant accelerated approval to a product or therapeutic designed to treat a serious or life-threatening condition that demonstrates an effect on a surrogate endpoint, or intermediate clinical endpoint, that is reasonably likely to predict clinical benefit or on a clinical endpoint that can be measured earlier than IMM, or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. For the purposes of accelerated approval, a surrogate endpoint is a marker, such as a laboratory measurement, radiographic image, physical sign, or other measure that is thought to predict clinical benefit but is not itself a measure of clinical benefit. An intermediate clinical endpoint is a clinical endpoint that can be measured earlier than an effect on IMM that is reasonably likely to predict an effect on irreversible morbidity, mortality, or other clinical benefit.
The FDA may not accept our application for accelerated approval for INBRX-101, may not grant this approval on a timely basis, or may not grant approval at all. The FDA or foreign regulatory authorities could require us to conduct further studies prior to considering our application or granting approval of any type. We might not be able to fulfill the FDA’s requirements in a timely manner, which would cause delays, or approval might not be granted because our submission is deemed incomplete by the FDA. A failure to obtain accelerated approval or any other form of expedited development, review, or approval for INBRX-101 would result in a longer time period to commercialization of INBRX-101, would increase the cost of development of INBRX-101, and could harm our competitive position in the marketplace. Following high-profile voluntary withdrawals of accelerated approval indications by several oncology sponsors as a result of post-approval trials failing to verify their drug products’ clinical benefit for those indications, which resulted in December 2022 amendments by Congress to the FDA’s authorities related to accelerated approval, public scrutiny of the accelerated approval pathway is likely to continue and may lead to further legislative and/or administrative changes in the future.
Moreover, even if we receive accelerated approval from the FDA, we will be subject to rigorous post-marketing requirements, including the completion of one or more confirmatory post-approval clinical trials to verify the clinical benefit of INBRX-101, and submission to the FDA of all promotional materials 30-120 days prior to their dissemination. The FDA could seek to withdraw accelerated approval for multiple reasons, including if we fail to conduct any required post-approval study, a post-approval study does not confirm the predicted clinical benefit, other evidence shows that the product is not safe or effective under the conditions of use, or we disseminate promotional materials that are found by the FDA to be false and misleading. Products that receive accelerated approval may be subject to expedited withdrawal procedures if post-approval studies fail to verify the predicted clinical benefit. In addition, as part of the Consolidated Appropriations Act for 2023, Congress provided FDA new statutory authorities to mitigate potential risks to patients from continued marketing of ineffective drugs previously granted accelerated approval. Under these recent amendments to the FDCA, the agency may require a sponsor of a product seeking accelerated approval to have a confirmatory trial underway prior to such approval being granted. At this time it has not been fully determined how a future confirmatory post-approval clinical trial for INBRX-101 would be designed or implemented, whether more than one trial will become necessary, or what the FDA would expect with respect to the timing of initiating such a confirmatory clinical trial for INBRX-101, should it be granted accelerated approval. If we fail to receive accelerated approval for INBRX-101 or fail to comply with the post-marketing requirements, our business, results of operations, prospects and the price of our common stock may be materially and adversely affected.
Our therapeutic candidates for which we intend to seek approval may face competition sooner than anticipated.
Even if we are successful in achieving regulatorymarketing approval to commercialize a therapeutic candidate ahead of our competitors, our future therapeutic candidates may face direct competition from biosimilar products. In the United States, our therapeutic candidates are regulated by the FDA as biological products, and we intend to seek approval for these therapeutic candidates pursuant to the BLA pathway. The BPCIA created an abbreviated pathway for the FDA approval of biosimilar biological products based on a previously licensed innovator, or reference, biological product. Under the BPCIA, an application for a biosimilar biological product cannot be approved by the FDA until 12 years after the original reference biological product was approved under a BLA. The law is complex and is still being interpreted and implemented by the FDA. As a result, its ultimate impact, implementation, and meaning are subject to uncertainty. While it is uncertain when such processes intended to implement BPCIA may be fully adopted by the FDA, any such processes could have a material adverse effect on the future commercial prospects for our therapeutic candidates.
We believe that any of our therapeutic candidates approved as a biological product under a BLA should qualify for the 12-year period of exclusivity available to reference biological products. However, there is a risk that this
exclusivity could be shortened due to Congressional action or otherwise, or that the FDA will not consider our therapeutic candidates to be reference biological products pursuant to its interpretation of the exclusivity provisions of the BPCIA, potentially creating the opportunity for follow-on biosimilar competition sooner than anticipated. Moreover, the extent to which a biosimilar product, once approved, will be substituted for any one of our reference products in a way that is similar to traditional generic substitution for non-biological products is not yet clear, and will depend on a number of marketplace and regulatory factors that are still developing including whether a future competitor seeks an interchangeability designation for a biosimilar of one of our products. Under the BPCIA as well as state pharmacy laws, only so-called “interchangeable” biosimilar products are considered substitutable for the reference biological product without the intervention of the healthcare provider who prescribed the original biological product. However, as with all prescribing decisions made in the context of a patient-provider relationship and a patient’s specific medical needs, healthcare providers are not restricted from prescribing biosimilar products in an off-label manner. In addition, a competitor could decide to forego the abbreviated approval pathway available for biosimilar products and to submit a full BLA for product licensure after completing its own preclinicalnonclinical studies and clinical trials. In such a situation, any exclusivity to which we may be eligible under the BPCIA would not prevent the competitor from marketing its biological product as soon as it is approved.
In Europe, the European Commission has granted marketing authorizations for severalmany biosimilar products pursuant to a set of general and product class-specific guidelines for biosimilar approvals issued over the past few years. In addition, companies may be developing biosimilar products in other countries that could compete with our products, if approved.
If competitors are able to obtain marketing approval for biosimilars referencing our therapeutic candidates, if approved, our future products may become subject to competition from such biosimilars, whether or not they are designated as interchangeable, with the attendant competitive pressure and potential adverse consequences. Such competitive products may be able to immediately compete with us in each indication for which our therapeutic candidates may have received approval.
We face regulation and potential liability related to the privacy, data protection and information security which may require significant resources and may adversely affect our business, operations and financial performance.
The regulatory environment surrounding information security, data collection and privacy is increasingly demanding. We are subject to numerous U.S. federal and state laws and non-U.S. regulations, including in Europe and China, governing the protection of personal and confidential information of our clinical subjects, clinical investigators, employees and vendors/business contacts, including in relation to medical records, credit card data and financial information. ForIn the United States, numerous federal and state laws and regulations, including federal health information privacy laws, state data breach notification laws, state health information privacy laws, and federal and state consumer protection laws (e.g., Section 5 of the Federal Trade Commission Act and CCPA), that govern the collection, use, disclosure and protection of health-related and other personal information could apply to our operations or the operations of our collaborators, and the privacy regulatory area is in constant flux. The state of California, for example, has adopted the CCPA, which went into effect beginning in January 2020. The CCPA establishes a new privacy framework for covered businesses by creating an expanded definition of personal information, establishing new data privacy rights for residents of the State of California, imposing special rules on the collection of personal information from minors, and creating a new and potentially severe statutory damages framework for violations of the CCPA and for businesses that fail to implement reasonable security procedures and
practices to prevent data breaches. While there is currently an exception under CCPA for protected health information that is subject to HIPAA and clinical trial regulations, CCPA still applies to the personal information of employees and may otherwise impact our business. In November of 2020, California voters approved the CPRA, which took full effect on January 1, 2023. The CPRA amends the CCPA significantly, potentially resulting in further uncertainty, additional costs and expenses in an effort to comply, and additional harm and liability for failure to comply. Among other things, the CPRA established a new regulatory authority, the California Privacy Protection Agency, or CPPA, which has expanded enforcement authority. Further, the CPPA has introduced significant draft regulations under the CPRA which could have operational effects on our business and further uncertainty with respect to enforcement. Comprehensive privacy laws similar to the CCPA in Virginia, Colorado, Connecticut, and Utah all took effect in 2023, and laws in Montana, Oregon and Texas will take effect in 2024, increasing the regulatory compliance risk. Like the GDPR, many of these state laws categorize medical or health data, genetic data, and biometric data that can be used to identify a natural person as “sensitive data” and the processing or collection of such will require additional compliance obligations.
In the EU, the GDPR, which came into effect on May 25, 2018, the European General Data Protection Regulation,could increase our burden of regulatory compliance. The GDPR implements more stringent operational requirements for processors and controllers of personal data, including, for example, requiring expanded disclosures about how personal information is to be used, limitations on retention of information, mandatory data breach notification requirements, and higher standards for data controllers to demonstrate that they have obtained either valid consent or have another legal basis in place to justify their data processing activities. The GDPR further provides that EU member states may make their own additional laws and regulations in relation to certain data processing activities, which could further limit our ability to use and share personal data and could require localized changes to our operating model. Under the GDPR, went into effect, significantly increasing fining levelsfines of up to €20 million or up to 4% of the total worldwide annual turnover or up to €20 million (whicheverof the preceding financial year, whichever is higher)higher, may be assessed for non-compliance with its requirements. noncompliance, which significantly increases our potential financial exposure for non-compliance.
We will be subject to the GDPR where we have a European UnionEU presence or “establishment” (e.g. European UnionEU based subsidiary or operations), when conducting clinical trials with European UnionEU based data subjects (whether the trials are conducted directly by us or through a clinical vendor or partner) or offering approved products or services (if relevant) to European UnionEU based data subjects (regardless of whether involving our European UnionEU based subsidiary or operations).
The GDPR sets out a number of requirements that must be complied with when handlingprocessing the personal data of such European UnionEU based data subjects including: providing expanded disclosures about how their personal data will be used; higher standards for organizations to demonstrate that they have obtained valid consent or have another legal basis in place to justify their data processing activities; the obligation to appoint data protection officers in certain circumstances; new rights for individuals to be “forgotten” and rights to data portability, as well as enhanced current rights (e.g. access requests); the principal of accountability and demonstrating compliance through policies, procedures, training and audit; the new mandatory data breach regime. In particular, medical or health data, genetic data and biometric data where the latter is used to uniquely identify an individual are all classified as “special category” data under the GDPR and afford greater protection and require additional compliance obligations. Further, European UnionEU member states have a broad right to impose additional conditions—including restrictions—on these data categories. This is because the GDPR allows European UnionEU member states to derogate from the requirements of the GDPR mainly in regard to specific processing situations (including special category data and processing for scientific or statistical purposes). As the European UnionEU states continue to reframe their national legislation to
harmonize with the GDPR, we will need to monitor compliance with all relevant European UnionEU member states’ laws and regulations, including where permitted derogations from the GDPR are introduced.
We will also be subject to evolving European UnionEU laws on data export, where we transfer data outside the European UnionEU to group companies or third parties. The GDPR only permits exports of data outside the European UnionEU where there is a suitable data transfer solution in place to safeguard personal data (e.g. the European UnionEU Commission approved Standard Contractual Clauses)Clauses or the recently-adopted EU-US Data Privacy Framework). On July 16, 2020, the Court of Justice of the European UnionEU or the CJEU, issued a landmark opinion in the case Maximilian Schrems vs. Facebook (Case C-311/18), called Schrems II. This decision (a)(i) calls into question certain data transfer mechanisms as between the European UnionEU member states and the U.S. (such as the Standard Contractual Clauses) and (b)(ii) invalidates the EU-U.S. Privacy Shield on which many companies had relied as an acceptable mechanism for transferring such data from the EU to the U.S. The CJEU is the highest court in Europe and the Schrems II decision heightens the burden on data importers to assess the
impact of U.S. national security laws on their business and future actions of European UnionEU data protection authorities are difficult to predict atpredict. While the early date. Consequently,recently-adopted EU-US Data Privacy Framework was meant to address the concerns raised by the CJEU in Schrems II, it will likely be subject to future legal challenges. Further, the EU-US Data Privacy Framework is only available to companies who have completed a self-certification and application. We have not self-certified to the EU-US Data Privacy Framework, and consequently, there is some risk of any of our data transfers from the European UnionEU being halted.halted by one or more EU member states.
Where we rely on third parties to carry out a number of services for us, including processing personal data on our behalf, we are required under GDPR and the U.S. state privacy laws to enter into contractual arrangements to help ensure that these third parties only process such data according to our instructions and have sufficient security measures in place. Any security breach or non-compliance with our contractual terms or breach of applicable law by such third parties could result in enforcement actions, litigation, fines and penalties or adverse publicity and could cause our customers to lose trust in us, which could have an adverse impact on our reputation and business. Any contractual arrangements requiring the transfer of personal data from the European UnionEU to us in the United States will requirerequires greater scrutiny and assessments as required under Schrems II and may have an adverse impact on cross-border transfers of personal data, or increase costs of compliance.
Further,Our partnerships in China and our CRO relationships in Hong Kong may expose us to new and stringent Chinese data security laws. The Data Security Law of the United Kingdom’s decisionPeople’s Republic of China, or the PRC Data Security Law, took effect on September 1, 2021. The PRC Data Security Law requires data processing, which includes the collection, storage, use, processing, transmission, provision and publication of data, to leavebe conducted in a legitimate and proper manner. Moreover, the European Union, often referred to as Brexit, has created uncertaintyPRC Data Security Law provides a national security review procedure for those data processing activities which affect or may affect national security and imposes export restrictions on certain data and information. In addition, the PRC Data Security Law also provides that any organization or individual within the territory of the PRC shall not provide any foreign judicial body and law enforcement body with regard toany data protection regulationstored in the United Kingdom. In particular, whileterritory of the DataPRC without the approval of the competent PRC governmental authorities. Also in China, the Personal Information Protection Act of 2018, that “implements”Law, which took effect on November 1, 2021, introduced stringent protection requirements for processing personal information. We may be required to make further significant adjustments to our business practices to comply with data security and complements the GDPR achieved Royal Assent on May 23, 2018 and is now effective in the United Kingdom, it is still unclear whether transfer of data from the European Economic Area to the United Kingdom will remain lawful under GDPR. During the period of “transition” (i.e., until December 31, 2020), European Union law will continue to apply in the United Kingdom, including the GDPR, after which the GDPR will be converted into United Kingdom law. Beginning in 2021, the United Kingdom will be a “third country” under the GDPR.
In the United States, numerous federal and state laws and regulations, including federal healthpersonal information privacy laws, state data breach notification laws, state health information privacy laws, and federal and state consumer protection laws (e.g., Section 5 of the Federal Trade Commission Act and California Consumer Privacy Act of 2018, or CCPA), that govern the collection, use, disclosure and protection of health-related and other personal information could apply to our operations or the operations of our collaborators, and the privacy regulatory area is in constant flux. The state of California, for example, has adopted the CCPA, which went into effect beginning in January 2020. The CCPA has been characterized as the first “GDPR-like” privacy statute to be enacted in the United States because it mirrors a number of the key provisions of the GDPR. The CCPA establishes a new privacy framework for covered businesses by creating an expanded definition of personal information, establishing new data privacy rights for residents of the State of California, imposing special rules on the collection of personal information from minors, and creating a new and potentially severe statutory damages framework for violations of the CCPA and for businesses that fail to implement reasonable security procedures and practices to prevent data breaches. An initiative called the California Privacy Rights Act, or CCPA 2.0, will be voted on in California on November 2, 2020 and its future application could impact our operations or that of our collaborators. Other states have been considering legislation similar to the CCPA, and several federal privacy proposals are under consideration in the current session of Congress.China.
In recent years, U.S. and European lawmakers and regulators have expressed concern over electronic marketing. In the European Union,EU, marketing is defined broadly to include any promotional material and the rules specifically on e-marketing are currently set out in the ePrivacy Directive which will be replaced by a new ePrivacy Regulation.
While the ePrivacy Regulation was originally intended to be adopted on May 25, 2018 (alongside the GDPR), it is still going through the European legislative process and commentators now expect it toprocess. Once the regulation is adopted, there will be adopted in 2021.a two-year implementation period. The current draft of the ePrivacy Regulation imposes strict opt-in e-marketing rules with limited exceptions to business to business communications and significantly increases fining powers to the same levels as GDPR (see above). In the U.S., the state privacy laws in California, Virginia, Colorado, Connecticut, and Utah include provisions regulating certain types of behavioral marketing and require companies to analyze online marketing and, in some cases, require opt-out mechanisms, increasing operational expenses and risk. The Federal Trade Commission has also issued a notice of proposed rulemaking with respect to online marketing and data collection.
We may find it necessary or desirable to join self-regulatory bodies or other privacy-related organizations, particularly relating to biopharmacy and/or scientific research that may require compliance with their rules pertaining to privacy and data security.
The introduction of the GDPR, and any resultant changes in European UnionEU member states’ national laws and regulations and the ePrivacy Regulation, will increase our compliance obligations and will necessitate the review and implementation of policies and processes relating to our collection and use of data. This increase in compliance obligations could also lead to an increase in compliance costs which may have an adverse impact on our business, financial condition or results of operations.
If any person, including any of our employees, clinical vendors or partners or those with whom we share such information, negligently disregards or intentionally breaches our established controls with respect to our clinical subject, clinical investigator or employee data, or otherwise mismanages or misappropriates that data, we could be
subject to significant monetary damages, regulatory enforcement actions, fines and/or criminal prosecution in one or more jurisdictions. As above, under the GDPR there are significant new punishments for non-compliance which could result in a penalty of up to 4% of a firm’s global annual revenue.
Applicable laws may conflict with each other, and by complying with the laws or regulations of one jurisdiction, we may find that we are violating the laws or regulations of another jurisdiction. Despite our efforts, we may not have fully complied in the past and may not in the future. If we become liable under laws or regulations applicable to us, we could be required to pay significant fines and penalties, our reputation may be harmed and we may be forced to change the way we operate. That could require us to incur significant expenses or to discontinue certain services, which could negatively affect our business.
We are increasingly dependent upon technology systems and data to operate our business. In particular, the COVID-19 pandemic has caused us to modify our business practices, including in certain cases allowing office-based employees in the United States to work from home. As a result, we are increasingly dependent upon our technology systems to operate our business and our ability to effectively manage our business depends on the security, reliability and adequacy of our technology systems and data, which includes use of cloud technologies. A breakdown, invasion, corruption, destruction or breach of our technology systems, including the cloud technologies that we utilize, and/or unauthorized access to our data and information could subject us to liability or negatively impact the operation of our business. Our technology systems, including the cloud technologies that we utilize, continue to increase in multitude and complexity, making them potentially vulnerable to breakdown, malicious intrusion and random attack. Likewise, data privacy or security breaches by individuals authorized to access our technology systems, including the cloud technologies that we utilize, may pose a risk that sensitive data, including intellectual property, trade secrets or personal information belonging to us, our patients, customers or other business partners, may be exposed to unauthorized persons or to the public.
Cyber-attacks are increasing in their frequency, sophistication and intensity, and are becoming increasingly difficult to detect. They are often carried out by motivated, well-resourced, skilled and persistent actors, including nation states, organized crime groups, “hacktivists” and employees or contractors acting with malicious intent. Cyber-attacks could include the deployment of harmful malware and key loggers, ransomware, a denial-of-service attack, a malicious website, the use of social engineering and other means to affect the confidentiality, integrity and availability of our technology systems and data. Our key business partners face similar risks and any security breach of their systems could adversely affect our security posture. In addition, our increased use of cloud technologies could heighten these and other operational risks, and any failure by cloud technology service providers to adequately safeguard their systems and prevent cyber-attacks could disrupt our operations and result in misappropriation, corruption or loss of confidential or propriety information.
While we continue to build and improve our systems and infrastructure, including our business continuity plans, there can be no assurance that our efforts will prevent breakdowns or breaches in our systems that could adversely affect our business and operations and/or result in the loss of critical or sensitive information, which could result in financial, legal, business, operational or reputational harm to us, loss of competitive advantage or loss of consumer confidence. In addition, our liability insurance may not be sufficient in type or amount to cover us against claims related to security breaches, cyber-attacks and other related breaches. Further, a data breach could result in negative publicity which could damage our reputation and have an adverse effect on our business, financial condition or results of operations.
We are subject to U.S. and certain foreign export and import controls, sanctions, embargoes, anti-corruption laws, and anti-money laundering laws and regulations. Compliance with these legal standards could impair our ability to compete in domestic and international markets. We can face criminal liability and other serious consequences for violations, which can harm our business.
We are subject to export control and import laws and regulations, including the U.S. Export Administration Regulations, U.S. Customs regulations, various economic and trade sanctions regulations administered by the U.S. Treasury Department’s Office of Foreign Assets Controls, the U.S. Foreign Corrupt Practices Act of 1977, as amended, the U.S. domestic bribery statute contained in 18 U.S.C. § 201, the U.S. Travel Act, the USA PATRIOT Act, and other state and national anti-bribery and anti-money laundering laws in the countries in which we conduct activities. Anti-corruption laws are interpreted broadly and prohibit companies and their employees, agents, contractors, and other collaborators from authorizing, promising, offering, or providing, directly or indirectly, improper payments or anything else of value to recipients in the public or private sector. We may engage third parties to sell our products sell our products outside the United States, to conduct clinical trials, and/or to obtain necessary permits, licenses, patent registrations, and other regulatorymarketing approvals. We have direct or indirect interactions with officials and employees of government agencies or government-affiliated hospitals, universities, and other organizations. We can be held liable for the corrupt or other illegal activities of our employees, agents, contractors, and other collaborators, even if we do not explicitly authorize or have actual knowledge of such activities. Any violations of the laws and regulations described above may result in substantial civil and criminal fines and penalties, imprisonment, the loss of export or import privileges, debarment, tax reassessments, breach of contract and fraud litigation, reputational harm, and other consequences.
Use of net operating loss carryforwards may be limited and U.S. federal income tax reform could adversely affect us.
Our ability to utilize our net operating loss, or NOL, carryforwards and other tax attributes to offset future taxable income or tax liabilities may be limited as a result of ownership changes. Under Sections 382 and 383 of the Internal Revenue Code, as amended, or IRC, a corporation that undergoes an “ownership change” may be subject to limitations on its ability to utilize its pre-change NOLs and other tax attributes otherwise available to offset future taxable income and/or tax liability. An ownership change is defined as a cumulative change of 50% or more in the ownership positions of certain stockholders during a rolling three-year period. We are in the process of completing a
formal study to assess whether an ownership change for purposes of Section 382 or 383 has occurred and, pending finalization, have not identified any ownership changes to date through December 31, 2023. However, we may experience ownership changes in the future as a result of shifts in our stock ownership (some of which shifts are outside our control), including as a result of the Merger. Corresponding rules may apply under state tax laws. We have not yet determined the amount of the cumulative change in our ownership resulting from the initial public offering of our common stock, or our IPO, or other transactions, or any resulting limitations, if any, on our ability to utilize our NOL carryforwards and other tax attributes. Even if there is no limitation on utilization of our NOL carryforwards as the result of an ownership change, the utilization of NOL carryforwards originating from a loss incurred in a year after 2017 is limited and may reduce taxable income in any post-2020 year by no more than 80% of the pre-NOL taxable income in such year. If we earn taxable income in a future year, such limitations on utilization of NOL carryforwards could result in increased future tax liability to us and our future cash flows could be adversely affected.
New legislation or regulation which could affect our tax burden could be enacted by any governmental authority. We cannot predict the timing or extent of such tax-related developments which could have a negative impact on our financial results. United States federal legislation affecting the tax laws was enacted in December 2017 (the TCJA), March 2020 (the Families First Coronavirus Response Act), and again in March 2020 (the CARES Act). We cannot estimate how the changes in tax law from this legislation will affect our tax liability in future years, but we have recorded a full valuation allowance related to our NOLs and other deferred tax assets due to the uncertainty of the ultimate realization of the future benefits from those assets.
Additionally, we use our best judgment in attempting to quantify and reserve for these tax obligations. However, a challenge by a taxing authority, our ability to utilize tax benefits such as carryforwards or tax credits, or a deviation from other tax-related assumptions may cause actual financial results to deviate from previous estimates.
If we do not comply with laws regulating the protection of the environment and health and human safety, our business could be adversely affected.
We maintain quantities of various flammable and toxic chemicals in our facilities in La Jolla, California required for our research and development activities. We are subject to federal, state and local laws and regulations governing the use, manufacture, storage, handling and disposal of these hazardous materials. We believe our procedures for storing, handling and disposing these hazardous materials in our La Jolla facilities comply with the relevant guidelines of La Jolla, the state of California and the Occupational Safety and Health Administration of the U.S. Department of Labor. Although we believe that our safety procedures for handling and disposing of these materials comply with the standards mandated by applicable regulations, the risk of accidental contamination or injury from these materials cannot be eliminated. If an accident occurs, we could be held liable for resulting damages, which could be substantial. We are also subject to numerous environmental, health and workplace safety laws and regulations, including those governing laboratory procedures, exposure to blood-borne pathogens and the handling of animals and biohazardous materials. Any insurance coverage we have may not be sufficient to cover these liabilities. Additional federal, state and local laws and regulations affecting our operations may be adopted in the future. We may incur substantial costs to comply with, and substantial fines or penalties if we violate, any of these laws or regulations which would adversely affect our business.
Our future growth may depend, in part, on our ability to operate in foreign markets, where we would be subject to additional regulatory burdens and other risks and uncertainties.
Our future growth may depend, in part, on our ability to develop and commercialize our therapeutic candidates, if approved, in foreign markets for which we may rely on collaboration with third parties. We are not permitted to market or promote any of our therapeutic candidates before we receive regulatorymarketing approval from the applicable regulatory authority in that foreign market, and we may never receive such regulatorymarketing approval for any of our therapeutic candidates. To obtain separate regulatorymarketing approval in many other countries we must comply with numerous and varying regulatory requirements of such countries regarding safety and efficacy and governing, among other things, clinical trials and commercial sales, pricing and distribution of our therapeutic candidates, and we cannot predict success in these jurisdictions. If we obtain approval of our therapeutic candidates and ultimately commercialize our therapeutic candidates in foreign markets, we would be subject to the risks and uncertainties, including the burden of complying with complex and changing foreign regulatory, tax, accounting and legal requirements and the reduced protection of intellectual property rights in some foreign countries. We may need to rely on third parties to market, distribute and sell our products in foreign markets.
Risks Related to Ownership of Our Common Stock
We do not know whether an active, liquid and orderly trading market will continue for our common stock and as a result it may be difficult for you to sell your shares of our common stock.
Prior to our IPO in August 2020, there was no public market for shares of our common stock. Shares of our common stock only recently began trading on the Nasdaq Global Market and we can provide no assurance that we will be able to sustain an active trading market for our shares. The lack of an active market may impair your ability to sell your shares at the time you wish to sell them or at a price that you consider reasonable. The lack of an active market
may also reduce the fair market value of your shares. Furthermore, an inactive market may also impair our ability to raise capital by selling shares of our common stock and may impair our ability to enter into strategic collaborations or acquire companies, technologies or other assets by using our shares of common stock as consideration.
We expect that our stock price may fluctuate significantly.
The trading price of shares of our common stock may be highly volatile and could be subject to wide fluctuations in response to many risk factors listed in this section, and others beyond our control, including:
•results of our clinical trials and preclinical studies or those of our competitors;
•the success of competitive products or technologies;
•regulatory or legal developments in the United States and other countries;
•geopolitical events, such as global conflicts or hostilities;
•the level of expenses related to our therapeutic candidates or development programs;
•changes in the structure of healthcare payment systems; actual or anticipated fluctuations in our financial condition and operating results;
•announcements by us, our partners or our competitors of new therapeutics or therapeutic candidates, significant contracts, strategic partnerships, joint ventures, collaborations, commercial relationships or capital commitments;
•failure to meet or exceed financial estimates and projections of the investment community or that we provide to the public;
•issuance of new or updated research or reports by securities analysts or recommendations for our stock;
•disputes or other developments related to proprietary rights, including patents, litigation matters, and our ability to obtain patent protection for our technologies;
•commencement of, or our involvement in, litigation;
•fluctuations in the valuation of companies perceived by investors to be comparable to us;
•manufacturing disputes or delays;
•any future sales of our common stock, including the exercise of warrants, or other securities;
•any change to the composition of the board of directors or key personnel;
•expiration of contractual lock-up agreements with our executive officers, directors and security holders;
•general economic conditions and slow or negative growth of our markets;
•share price and volume fluctuations attributable to inconsistent trading volume levels of our shares;
•announcement or expectation of additional debt or equity financing efforts; and
•circumstances and market conditions relating to the COVID-19 pandemic.pandemics and natural disasters.
These and other market and industry factors may cause the market price and demand for our common stock to fluctuate substantially, regardless of our actual operating performance. In addition, the stock market in general, and life science companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies. In the past, when the market price of a stock has been volatile, holders of that stock have on occasion instituted securities class action litigation against the company that issued the stock. If any of our stockholders were to bring a lawsuit against us, the defense and disposition of the lawsuit could be costly and divert the time and attention of our management and harm our operating results.
Our executive officers, directors and holders of more than 5% of our capital stock own a significant percentage of our stock and will be able to exercise significant control over matters subject to stockholder approval.
As of December 31, 2020,2023, our executive officers, directors and holders of more than 5% of our capital stock beneficially owned approximately 60.6%61% of our shares of common stock outstanding. Accordingly, this group of stockholders will continue to have significant control over our operations. This concentration of ownership could have the effect of delaying or preventing a change in our control or otherwise discouraging a potential acquirer from attempting to obtain control of us, which in turn could have a material adverse effect on our stock price and may prevent attempts by our stockholders to replace or remove the board of directors or management.
Future sales of our common stock in the public market could cause our stock price to fall.
Our stock price could decline as a result of sales of a large number of shares of our common stock or the perception that these sales could occur. These sales, or the possibility that these sales may occur, also might make it more difficult for us to sell equity securities in the future at a time and at a price that we deem appropriate.
As of December 31, 2020,2023, we had 37,712,39047,369,511 shares of common stock outstanding. Of these shares, 8,050,000 shares sold in our IPO are freely tradable without restriction or further registration under the Securities Act of 1933, as amended, or the Securities Act, unless held by our “affiliates”, as that term is defined in Rule 144 under the Securities Act, or Rule 144. The holders of the remaining 29,662,390In order to raise additional capital, we may sell shares of common stock under our Open Market Sale Agreement, or 78.7% of our outstandingthe Sales Agreement, with Jefferies LLC, or the Sales Agent, under which we may, from time to time, sell shares of common stock were prohibited or otherwise restricted as a resulthaving an aggregate offering price of securities law provisions, market standoff agreements entered into byup to $200.0 million through the Sales Agent. As of December 31, 2023, shares of common stock with an aggregate offering price of $171.4 million have been sold through our stockholders with us or lock-up agreements entered into by our stockholders withSales Agent pursuant to the underwriters. The lock-up agreements expired on February 14, 2021. We cannot predict what effect, if any, salesSales Agreement. Sales of our shares incommon stock made pursuant to the public market orSales Agreement will be made under our $400.0 million Shelf Registration on Form S-3ASR, which became automatically effective upon filing on September 3, 2021. As of the availabilitydate of this Annual Report, our employees and members of our board of directors hold approximately 3.5 million vested and in-the-money options to acquire shares for sale will have on the market price of our common stock. However, futureThese shares are registered pursuant to our previously filed registration statements on Form S-8 and are generally freely-tradeable. Future sales of substantial amounts of our common stock in the public market, including sales by our directors, employees, or other significant holders, shares issued upon exercise of then outstanding options or warrants, or the perception that such sales may occur, could adversely affect the market price of our common stock.
stock and cause significant dilution to our existing stockholders. Raising additional capital may cause dilution to our existing stockholders, restrict our operations or require us to relinquish rights to our technologies or product candidates.
We may seek additional capital through a combination of public and private equity offerings, debt financings, strategic partnerships and alliances, and licensing arrangements. We, and indirectly, our stockholders, will bear the cost of issuing and servicing such securities. Because our decision to issue debt or equity securities in any future offering will depend on market conditions and other factors beyond our control, we cannot predict or estimate the amount, timing, or nature of any future offerings. To the extent that we raise additional capital through the sale of equity or debt securities, your ownership interest will be diluted, and the terms may include liquidation or other preferences that adversely affect your rights as a stockholder. The incurrence of indebtedness would result in increased fixed payment obligations and could involve restrictive covenants, such as limitations on our ability to incur additional debt, limitations on our ability to acquire, sell, or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business. Additionally, any future collaborations we enter into with third parties may provide capital in the near term but limit our potential cash flow and revenue in the future. If we raise additional funds through strategic partnerships and alliances and licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies or product candidates, or grant licenses on terms unfavorable to us.
If securities or industry analysts do not publish research reports about our business, or if they issue an adverse opinion about our business, our stock price and trading volume could decline.
The trading market for our common stock will be influenced by the research and reports that industry or securities analysts publish about us or our business. If one or more of the analysts who cover us issues an adverse opinion about our company, our stock price would likelycould decline. If one or more of these analysts ceases coverage of us or fails to regularly publish reports on us, we could lose visibility in the public markets, which could cause our stock price or trading volume to decline.
The Nasdaq Stock Market may delist our securities from its exchange, which could limit investors’ ability to make transactions in our securities and subject us to additional trading restrictions.
Our common stock is listed on the Nasdaq Global Market. We cannot assure you that, in the future, our securities will meet the continued listing requirements to be listed on the Nasdaq Global Market. If the Nasdaq Stock Market delists our common stock, we could face significant material adverse consequences, including:
•a limited availability of market quotations for our securities;
•a determination that our common stock is a “penny stock” which will require brokers trading in our common stock to adhere to more stringent rules and possibly resulting in a reduced level of trading activity in the secondary trading market for our common stock;
•a limited amount of news and analyst coverage for our company; and
•a decreased ability to issue additional securities or obtain additional financing in the future.
WeExcept as may occur in connection with the Merger and spin-out transaction, we do not intend to pay dividends on our common stock so any returns will be limited to the value of our stock.
WeExcept as may occur in connection with the Merger and spin-out transaction, we currently anticipate that we will retain future earnings for the development, operation and expansion of our business and do not anticipate declaring or paying any cash dividends for the foreseeable future. The Amended 2020 Loan Agreement requires, and future debt financing agreements may require us to have the lender’s permission before declaring dividends on our common stock. Any return to stockholders will therefore be limited to the appreciation of their stock.
If we fail to maintain an effective system of internal control over financial reporting, we may not be able to accurately report our financial results or prevent fraud. As a result, stockholders could lose confidence in our financial statements and other public reporting, which would harm our business and the trading price of our common stock.
Effective internal controls over financial reporting are necessary for us to provide reliable financial reports and, together with adequate disclosure controls and procedures, are designed to prevent fraud. Any failure to implement required new or improved controls, or difficulties encountered in their implementation could cause us to fail to meet our reporting obligations. Pursuant to Section 404 of the Sarbanes-Oxley Act of 2002, or Section 404, our
management will be required to report upon the effectiveness of our internal control over financial reporting beginning in the year following the first required annual report.reporting.
The rules governing the standards that must be met for management to assess our internal control over financial reporting are complex and require significant documentation, testing and possible remediation. To comply with the requirements of being a reporting company under the Securities and Exchange Act of 1934, as amended, or the Exchange Act, we will need to implement additional financial and management controls, reporting systems and procedures.
We cannot assure you that there will not be material weaknesses or significant deficiencies identified in our internal control over financial reporting in the future. Any failure to maintain internal control over financial reporting could severely inhibit our ability to accurately report our financial condition, results of operations or cash flows. In the future, if we are unable to conclude that our internal control over financial reporting is effective, or if our independent registered public accounting firm determines we have a material weakness or significant deficiency in our internal control over financial reporting, investors may lose confidence in the accuracy and completeness of our financial reports, the market price of our common stock could decline, and we could be subject to sanctions or investigations by The Nasdaq Stock Market, the SEC, or other regulatory authorities. Failure to remedy any material weakness in our internal control over financial reporting, or to implement or maintain other effective control systems required of public companies, could also restrict our future access to the capital markets.
WhenEffective December 31, 2023, we lose our status as an “emerging growth company,became a “large accelerated filer,” as defined in the Jumpstart Our Business Startups Act of 2012, as amended, or the JOBS Act, and once we become an “accelerated filer” as defined in the Exchange ActAct. As a result, our independent registered public accounting firm will beis now required to attest to the effectiveness of our internal controls over financial reporting pursuant to Section 404. We could be an “emerging growth company” for up to five years from the closing of our initial public offering. An independent assessment of the effectiveness of our internal controls could detect problems that our management’s assessment might not. Undetected material weaknesses in our internal controls could lead to financial statement restatements and require us to incur the expense of remediation.
We will continue to incur significantly increased costs as a result of operating as a public company, and our management is required to devote substantial time to new compliance initiatives.
As a public company, we incur significant legal, accounting and other expenses that we did not incur as a private company. We are subject to the reporting requirements of the Exchange Act, which require, among other things, that we file with the SEC annual, quarterly and current reports with respect to our business and financial condition. In addition, the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, as well as rules subsequently adopted by the SEC and Nasdaq Stock Market to implement provisions of the Sarbanes-Oxley impose significant requirements on
public companies, including requiring establishment and maintenance of effective disclosure and financial controls and changes in corporate governance practices. Further, in July 2010, the Dodd-Frank Wall Street Reform and Consumer Protection Act, or the Dodd-Frank Act, was enacted. There are significant corporate governance and executive compensation-related provisions in the Dodd-Frank Act that require the SEC to adopt additional rules and regulations in these areas such as “say on pay” and proxy access. Legislation permits emerging growth companies to implement many of these requirements over a longer period and up to five years from the pricing of our IPO. We intend to take advantage of this legislation, but cannot guarantee that we will not be required to implement these requirements sooner than budgeted or planned and thereby incur unexpected expenses. Stockholder activism, the current political environment and the current high level of government intervention and regulatory reform may lead to substantial new regulations and disclosure obligations, which may lead to additional compliance costs and impact the manner in which we operate our business in ways we cannot currently anticipate.
We expect the rules and regulations applicable to public companies to substantially increase our legal and financial compliance costs and to make some activities more time-consuming and costly. If these requirements divert the attention of our management and personnel from other business concerns, they could have a material adverse effect on our business, financial condition and results of operations. The increased costs will decrease our net income or increase our net loss, and may require us to reduce costs in other areas of our business or increase the prices of our products or services. For example, we expect these rules and regulations to make it more difficult and more expensive for us to obtain director and officer liability insurance as compared to when we operated as a privately
held company. We cannot predict or estimate the amount or timing of additional costs we may incur to respond to these requirements. The impact of these requirements could also make it more difficult for us to attract and retain qualified persons to serve on our board of directors, our board committees or as executive officers.
We are an “emerging growth company” and a “smaller reporting company”, and will be able to avail ourselves of reduced disclosure requirements applicable to emerging growth companies and smaller reporting companies, which could make our common stock less attractive to investors.
We are an “emerging growth company,” as defined in the JOBS Act, and a “smaller reporting company” under SEC regulations. For so long as we remain an emerging growth company or smaller reporting company, we intend to take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not “emerging growth companies” or “smaller reporting companies.” These exemptions include reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. As long as we remain an emerging growth company (and thereafter for so long as we remain a non-accelerated filer), these exemptions also include an exemption from the auditor attestation requirements of Section 404. In addition, Section 107 of the JOBS Act also provides that an emerging growth company can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act for complying with new or revised accounting standards. In other words, an emerging growth company can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have irrevocably elected not to take advantage of the extended transition period afforded by the JOBS Act for the implementation of new or revised accounting standards and, as a result, will comply with new or revised accounting standards on the relevant dates on which adoption of such standards is required for non-emerging growth companies. As a result of our emerging growth company election and smaller reporting company status, investors may view our financial statements as not comparable to some other public companies. We cannot predict if investors will find our common stock less attractive because we may rely on certain of these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile. We may take advantage of these reporting exemptions until we are no longer an emerging growth company or a smaller reporting company. We will remain an emerging growth company until the earliest of (i) the last day of the fiscal year in which we have total annual gross revenue of $1.07 billion or more; (ii) the last day of our fiscal year following the fifth anniversary of the date of the completion of our IPO (December 31, 2025); (iii) the date on which we have issued more than $1.0 billion in nonconvertible debt during the previous three years; or (iv) the date on which we are deemed to be a large accelerated filer under the rules of the SEC. In addition, we will continue to be a smaller reporting company as long as we (a) have less than $250 million in public float (based on our common equity) measured as of the last business day of our most recently completed second fiscal quarter, or (b) have annual revenues of less than $100 million during the most recently completed fiscal year for which audited financial statements are available and have no public float or public float of less than $700 million (based on our common equity).
Anti-takeover provisions contained in our restated certificate of incorporation and restated bylaws, as well as provisions of Delaware law, could impair a takeover attempt.
Our restated certificate of incorporation, restated bylaws and Delaware law contain provisions which could have the effect of rendering more difficult, delaying or preventing an acquisition deemed undesirable by our board of directors. Our corporate governance documents include provisions:
•authorizing our board of directors to issue up to 15,000,000 shares of preferred stock without stockholder approval upon the terms and conditions and with the rights, privileges and preferences as our board of directors may determine;
•specifying that special meetings of our stockholders can be called only by our board of directors, the chairman of our board of directors or our Chief Executive Officer and that our stockholders may not act by written consent;
•establishing an advance notice procedure for stockholder proposals to be brought before an annual meeting of our stockholders, including proposed nominations of persons for election to our board of directors;
•providing that our board of directors may create new directorships and that vacancies on our board of directors may be filled only by a majority of directors then in office, even though less than a quorum;
•establishing that our board of directors is divided into three classes—Class I, Class II, and Class III—with each class serving staggered three-year terms;
•providing that our board of directors may amend our restated bylaws without stockholder approval; and
•requiring a super-majority of votes to amend certain of the above-mentioned provisions.
These provisions, alone or together, could delay or prevent hostile takeovers and changes in control or changes in our management.
As a Delaware corporation, we are also subject to provisions of Delaware law, including Section 203 of the Delaware General Corporation law, which prevents some stockholders holding more than 15% of our outstanding common stock from engaging in certain business combinations without approval of the holders of substantially all of our outstanding common stock.
Any provision of our amended and restated certificate of incorporation, amended and restated bylaws or Delaware law that has the effect of delaying or deterring a change in control could limit the opportunity for our stockholders to receive a premium for their shares of our common stock, and could also affect the price that some investors are willing to pay for our common stock.
Our amended and restated certificate of incorporation designates the Court of Chancery of the State of Delaware, or the Chancery Court, or the federal district court for the District of Delaware, or the District Court of Delaware, or the other federal district courts of the United States as the exclusive forum for certain types of actions and
proceedings that may be initiated by our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers or employees.
Our amended and restated certificate of incorporation requires, unless we otherwise consent, that the Chancery Court will, to the fullest extent permitted by law, be the sole and exclusive forum for the following types of actions or proceedings under Delaware statutory or common law (subject to the Chancery Court having personal jurisdiction over the indispensable parties named as defendants): (i) any derivative action or proceeding brought on our behalf, (ii) any action or proceeding asserting a claim of breach of a fiduciary duty owed by any of our current or former directors, officers and employees to us or our stockholders, (iii) any action or proceeding asserting a claim against us or any of our current or former directors, officers or employees of the Company, , arising out of or pursuant to any provision of the Delaware General Corporation Law, our amended and restated certificate of incorporation or our amended and restated bylaws, (iv) any action or proceeding to interpret, apply, enforce or determine the validity of our amended and restated certificate of incorporation or our amended and restated bylaws, (v) any action or proceeding as to which the Delaware General Corporation Law confers jurisdiction to the Chancery Court, or (vi) any action or proceeding asserting a claim against us, or our directors, officers or employees, governed by the internal affairs doctrine. If the Chancery Court does not have jurisdiction for these actions or proceedings, then the actions or proceedings must be brought in a state court located in the State of Delaware. If these state courts also do not have jurisdiction, these actions or proceedings must be brought in the District Court of Delaware. These limitations in our amended and restated certificate of incorporation will not apply to actions brought to enforce a duty or liability created by the Securities Act, the Exchange Act or to any claim for which the federal courts have exclusive jurisdiction. However, our amended and restated certificate of incorporation also provides that, unless we otherwise consent in writing, the federal district courts of the United States shall, to the fullest extent permitted by law, be the sole and exclusive forum for the resolution of any complaint asserting a cause of action arising under the Securities Act. In addition, any person holding, owning or otherwise acquiring any interest in any shares of our capital stock shall be deemed to have notice of and to have consented to this provision of our amended and restated certificate of incorporation. These Delaware and federal court choice of forum provisions may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, officers or employees, which may discourage such lawsuits against us and our directors, officers and employees even though an action, if successful, might benefit our stockholders. Stockholders who do bring a claim in the Chancery Court or the District Court of Delaware could face additional litigation costs in pursuing any such claim, particularly if they do not reside in or near the jurisdiction. The Chancery Court or the District Court of Delaware may also reach different judgments or results than would other courts, including courts where a stockholder considering an action may be located or would otherwise choose to bring the action, and such judgments or results may be more favorable to us than to our stockholders. Alternatively, if a court were to find these provisions of our amended and restated
certificate of incorporation inapplicable to, or unenforceable in respect of, one or more of the specified types of actions or proceedings, we may incur additional costs, which could have a material adverse effect on our business, financial condition or results of operations.
On March 18, 2020, the Delaware Supreme Court ruled that provisions of a Delaware corporation’s certificate of incorporation that designate a federal forum for securities claims brought pursuant to the Securities Act, or federal forum provisions, are valid and enforceable under Delaware law, or the March 2020 Ruling. Various U.S. Supreme Court cases offer support for the argument that federal forum provisions do not violate federal policy. However, the March 2020 Ruling applies only to claims brought in Delaware state courts, and it is not binding on any other state court or the federal courts. Therefore, we are unable to predict whether a state court in any other state or a federal court would enforce a federal forum provision such as the one set forth in our amended and restated certificate of incorporation.
We may be subject to securities litigation that materially diverts the attention of our management or pursuant to which is expensive and could divert management attention.we incur substantial costs.
The market price of our common stock may be volatile and, in the past, companies that have experienced volatility in the market price of their stock have been subject to securities class action litigation. We may be the target of this type of litigation in the future. Securities litigation against us could result in substantial costs and materially divert our management’s attention from other business concerns, which could seriously harm our business.
Item 1B. Unresolved Staff Comments.
None.
Item 1C. Cybersecurity.
Cybersecurity
We recognize the critical importance of Contentsmaintaining the trust and confidence of our patients, business partners and employees toward our business and are committed to protecting the confidentiality, integrity and availability of our business operations and systems. Our board of directors is actively involved in oversight of our risk management activities, and cybersecurity represents an important element of our overall approach to risk management. Our cybersecurity policies, standards, processes and practices are based on recognized frameworks established by the National Institute of Standards and Technology, or NIST, the International Organization for Standardization and other applicable industry standards. In general, we seek to address cybersecurity risks through a comprehensive, cross-functional approach that is focused on preserving the confidentiality, security and availability of the information that we collect and store by identifying, preventing and mitigating cybersecurity threats and effectively responding to cybersecurity incidents when they occur.
Cybersecurity Risk Management and Strategy; Effect of Risk
We face risks related to cybersecurity such as unauthorized access, cybersecurity attacks and other security incidents, including as perpetrated by hackers and unintentional damage or disruption to hardware and software systems, loss of data, and misappropriation of confidential information. To identify and assess material risks from cybersecurity threats, we maintain a comprehensive cybersecurity program to ensure our systems are effective and prepared for information security risks, including regular oversight of our programs for security monitoring for internal and external threats to ensure the confidentiality and integrity of our information assets. We consider risks from cybersecurity threats alongside other company risks as part of our overall risk assessment process. We employ a range of tools and services, including regular network and endpoint monitoring, audits, vulnerability assessments, penetration testing, threat modeling and tabletop exercises to inform our risk identification and assessment. As discussed in more detail under “Cybersecurity Governance” below, our audit committee provides oversight of our cybersecurity risk management and strategy processes, which are led by our Chief Financial Officer, General Counsel, and Director of Information Technology.
We also identify our cybersecurity threat risks by comparing our processes to standards set by NIST and the International Organization for Standardization, as well as by engaging experts to attempt to infiltrate our information systems. To provide for the availability of critical data and systems, maintain regulatory compliance, manage our material risks from cybersecurity threats, and protect against and respond to cybersecurity incidents, we undertake the following activities:
a.monitor emerging data protection laws and implement changes to our processes that are designed to comply with such laws;
b.through our policies, practices and contracts (as applicable), require employees, as well as third parties that provide services on our behalf, to treat confidential information and data with care;
c.employ technical safeguards that are designed to protect our information systems from cybersecurity threats, including firewalls, intrusion prevention and detection systems, anti-malware functionality and access controls, which are evaluated and improved through vulnerability assessments and cybersecurity threat intelligence;
d.provide regular, mandatory training for our employees and contractors regarding cybersecurity threats as a means to equip them with effective tools to address cybersecurity threats, and to communicate our evolving information security policies, standards, processes and practices;
e.conduct regular phishing email simulations for all employees and contractors with access to our email systems to enhance awareness and responsiveness to possible threats;
f.conduct annual cybersecurity management and incident training for employees involved in our systems and processes that handle sensitive data;
g.run tabletop exercises to simulate a response to a cybersecurity incident and use the findings to improve our processes and technologies; and
h.carry information security risk insurance that provides protection against the potential losses arising from a cybersecurity incident.
Our incident response plan coordinates the activities we take to prepare for, detect, respond to and recover from cybersecurity incidents, which include processes to triage, assess severity for, escalate, contain, investigate and remediate the incident, as well as to comply with potentially applicable legal obligations and mitigate damage to our business and reputation.
As part of the above processes, we regularly engage with consultants, auditors and other third parties, including annually having an independent third-party review our cybersecurity program to help identify areas for continued focus, improvement and compliance.
Our processes also address cybersecurity threat risks associated with our use of third-party service providers, including our suppliers and manufacturers or who have access to patient and employee data or our systems. In addition, cybersecurity considerations affect the selection and oversight of our third-party service providers. We perform diligence on third parties that have access to our systems, data or facilities that house such systems or data, and continually monitor cybersecurity threat risks identified through such diligence. Additionally, we generally require those third parties that could introduce significant cybersecurity risk to us to agree by contract to manage their cybersecurity risks in specified ways, and to agree to be subject to cybersecurity audits, which we conduct as appropriate.
We describe whether and how risks from identified cybersecurity threats, including as a result of any previous cybersecurity incidents, have materially affected or are reasonably likely to materially affect us, including our business strategy, results of operations, or financial condition, under the heading “Cybersecurity breaches could expose us to material liability, damage our reputation, compromise our confidential information or otherwise adversely affect our business,” which disclosures are incorporated by reference herein.
In the last two fiscal years, we have not experienced any material cybersecurity incidents and the expenses we have incurred from cybersecurity incidents were immaterial. This includes penalties and settlements, of which there were none.
Cybersecurity Governance; Management
Cybersecurity is an important part of our risk management processes and an area of focus for our board of directors and management. The audit committee of our board of directors is responsible for the oversight of risks from cybersecurity threats.
At least annually, our audit committee receives an update from management of our cybersecurity threat risk management and strategy processes covering topics such as data security posture, results from third-party assessments, progress towards pre-determined risk-mitigation-related goals, our incident response plan, and material cybersecurity threat risks or incidents and developments, as well as the steps management has taken to respond to such risks. In such sessions, our audit committee generally receives materials discussing current and emerging material cybersecurity threat risks, and describing our ability to mitigate those risks, as well as recent developments, evolving standards, technological developments and information security considerations arising with respect to our peers and third parties, and discusses such matters with our Director of Information Technology. Our audit committee also receives prompt and timely information regarding any cybersecurity incident that meets establishing reporting thresholds, as well as ongoing updates regarding any such incident until it has been addressed.
Our cybersecurity risk management and strategy processes, which are discussed in greater detail above, are led by our Chief Financial Officer, General Counsel, and Director of Information Technology. Such individuals have collectively several years of prior work experience in various roles involving managing information security, developing cybersecurity strategy, implementing effective information and cybersecurity programs, as well as relevant degrees and certifications. These management team members are informed about and monitor the prevention, mitigation, detection, and remediation of cybersecurity incidents through their management of, and participation in, the cybersecurity risk management and strategy processes described above, including the operation of our incident response plan. As discussed above, these management team members report to the audit committee
of our board of directors about cybersecurity threat risks, among other cybersecurity related matters, at least annually.
Item 2. Properties.
Our headquarters are located in La Jolla, California where we currently lease approximately 43,000 square feet of laboratory and office space under a lease that expires in 2025. We have an option to extend the lease an additional five years. We believe that this space is sufficient to meet our needs for the foreseeable future and that any additional space we may require will be available on commercially reasonable terms.
Item 3. Legal Proceedings.
WeExcept as disclosed below, we are not currently a party to any material legal proceedings. From time to time, we may become involved in legal proceedings arising in the ordinary course of our business. Regardless of outcome, litigation can have an adverse impact on us due to defense and settlement costs, diversion of management resources, negative publicity, reputational harm and other factors.
On March 1, 2022, I-Mab Biopharma, or I-Mab, filed a lawsuit against the Company and Brendan Eckelman, the Company’s co-founder and Chief Scientific Officer, in the United States District Court for the District of Delaware, C.A. No. 22-00276-CJB, asserting claims for misappropriation of trade secrets related to Dr. Eckelman’s service as an expert witness for Tracon Pharmaceuticals, Inc., or Tracon, in Tracon’s arbitration against I-Mab. The case is currently in the discovery process.
Item 4. Mine Safety Disclosures.
Not applicable.
Part II.
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Market Information
Our common stock began tradingis traded on the Nasdaq Global Market in August 2020 under the ticker symbol “INBX.”
Holders of Common Stock
As of February 28, 2021,21, 2024, we had 37,747,66447,392,447 outstanding shares of common stock and approximately 11414 holders of record of our common stock. The approximate number of holders is based upon the actual number of holders registered in our records at such date and excludes holders in “street name” or persons, partnerships, associations, corporations, or other entities identified in security positions listings maintained by depository trust companies.
Dividend Policy
We have never declared or paid cash dividends on our capital stock. We currently intend to retain all available funds and future earnings, if any, for use in the operation of our business and do not anticipate paying any cash dividends on our common stock in the foreseeable future. Any future determination to declare and pay dividends will be made at the discretion of our board of directors and will depend on various factors, including applicable laws, our results of operations, our financial condition, our capital requirements, general business conditions, our future prospects and other factors that our board of directors may deem relevant. In addition, the terms of the Amended 2020 Loan Agreement restrict our ability to pay dividends without the prior written consent of Oxford. Investors should not purchase our common stock with the expectation of receiving cash dividends.
Securities Authorized for Issuance Under Equity Compensation Plans
See Item 12 of Part III of this Annual Report on Form 10-K for information about our equity compensation plans which is incorporated by reference herein.
Recent Sales of Unregistered Securities
None.
Use of Proceeds
On August 18, 2020, the SEC declared effective our registration statement on Form S-1 (File No. 333-240135), as amended, filed in connection with our IPO. Our IPO closed on August 21, 2020, and we issued and sold 8,050,000 shares of our common stock at a price to the public of $17.00 per share, which included the exercise in full of the underwriters’ option to purchase additional shares. We received gross proceeds from our IPO of $136.9 million, before deducting underwriting discounts, commissions and offering costs of $11.0 million, for net proceeds of approximately $125.9 million. No offering expenses were paid directly or indirectly to any of our directors or officers (or their associates) or persons owning ten percent or more of any class of our equity securities or to any other affiliates.
The joint book-running managing underwriters of the offering were Jefferies LLC, Evercore Group L.L.C. and Credit Suisse Securities (USA) LLC, with LifeSci Capital as Co-Manager.
Upon receipt, the net proceeds from our IPO were held in cash and cash equivalents, primarily bank deposits and money market funds. Through December 31, 2020,2023, we have used a portion of the net proceeds from our IPO for research and development of our programs and for general corporate purposes. There has been no material change in the planned use of proceeds from our IPO from those disclosed in the Prospectus.
Purchases of Equity Securities by the Issuer and Affiliated Purchasers
None.
Item 6. Selected Consolidated Financial Data.
We are a smaller reporting company as defined by Rule 12b-2 of the Exchange Act and are not required to provide the information required under this item.[Reserved]
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
You should read the following discussion and analysis of our financial condition and results of operations together in conjunction with our consolidated financial statements and related notes appearing elsewhere in this Annual Report on Form 10-K, or Annual Report. Some of the information contained in this discussion and analysis or set forth elsewhere in this Annual Report contain forward-looking statements that involve risk and uncertainties, such as statements of our plans, objectives, expectations and intentions. As a result of many factors, including those factors set forth in the section of this Annual Report titled “Risk Factors,” our actual results could differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis.
Overview
We are a clinical-stage biotechnologybiopharmaceutical company with a pipeline of novel biologic therapeutic candidates, developed using our proprietary modular protein engineering expertiseplatforms. We leverage our innovative protein engineering technologies and proprietary single domain antibody, or sdAb, platform. Our sdAb platform allows usdeep understanding of target biology to pursuecreate therapeutic candidates with attributes and mechanisms superior to current approaches and applicable to a range of challenging, validated targets with clinical promise, but where other antibodyhigh potential.
Recent Developments
Sale of INBRX-101 to Sanofi
In January 2024, we announced that we entered into a definitive agreement with Aventis, a wholly owned indirect subsidiary of Sanofi, whereby Sanofi will indirectly acquire, through Aventis, all the assets and biologic based approaches have failed. Highly modular, our sdAbs can be combined with precise valencies and multiple specificities, creating therapeutic candidates designedliabilities primarily related to be capable of enhanced cell signaling, conditional activationINBRX-101, an optimized, recombinant alpha-1 antitrypsin, or combined synergistic functions.
WeAAT, augmentation therapy currently have four programs in ongoing clinical trials, with the most advanced expected to initiate a registration-enablingregistrational trial in mid-2021. Three of these programs are for the treatment of various cancers,patients with alpha-1 antitrypsin deficiency, or AATD. Immediately prior to the closing of the Merger, all assets and liabilities not primarily related to INBRX-101 will be spun out into a new publicly traded company, New Inhibrx.
Under the terms of the definitive agreement, Aventis will acquire all of our outstanding shares through a merger, and in turn, each of our shareholders will receive: (i) $30.00 per share in cash, (ii) one contingent value right per share, representing the right to receive a contingent payment of $5.00 in cash upon the achievement of a regulatory milestone and (iii) one SEC-registered, publicly listed, share of New Inhibrx for every four shares of Inhibrx common stock held. In addition, in connection with the transaction, Aventis will (1) assume and retire our outstanding third-party debt, including our debt arrangements with Oxford, (2) cause New Inhibrx to be funded with $200 million in cash, and (3) retain an equity interest in New Inhibrx of approximately 8%. Subject to the satisfaction of customary closing conditions, including the receipt of regulatory approvals, we currently expect the transaction to close during the second quarter of 2024.
Discontinuation of INBRX-105
We have decided to terminate our INBRX-105 program, a tetravalent programmed death-ligand 1, or PD-L1, targeted 4-1BB agonist. During the length of our Phase 1/2 trial, we dosed approximately 150 patients. We initially observed single agent complete responses in non-small cell lung cancer, or NSCLC, and head and neck squamous cell carcinoma, or HNSCC, as well as multiple partial responses. We also observed numerous partial responses with INBRX-105 in combination with Keytruda. However, after evaluation of the totality of the data from the expansion cohorts, we concluded the initial signal was not sufficiently validated to support the continuation of this program. We are in the process of winding down the clinical trial and expect it to be complete within the first half of 2024. Consequently, in February 2024, Elpiscience Biopharmaceuticals, Inc, or Elpiscience, terminated its rights to commercialize INBRX-105 in greater China.
Current Clinical Pipeline
Our current clinical pipeline includes therapeutic candidates in the following categories:
•INBRX-101, which seeks to maintain the natural function of Alpha-1 antitrypsin, or AAT, in a recombinant format, optimized for less frequent dosing and greater potential therapeutic activity as compared to plasma-derived AAT, or pdAAT; and
•INBRX-109 and INBRX-106, both of which utilize our multivalent formats where the precise valency can be optimized in a target-centric way to mediate what we believe to be the most appropriate agonist function.
| | | | | | | | | | | | | | | | |
| | | | | | |
| INBRX-101 | | INBRX-109 | | INBRX-106 | | |
AAT-Fc fusion
protein | | Tetravalent DR5
agonist | | Hexavalent OX40
agonist | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Program | Therapeutic Area | Target(s)/Format | STAGE OF DEVELOPMENT |
| Preclinical | Phase 1 | Phase 2 | Phase 3 |
| INBRX-101* | Orphan/Respiratory | Neutrophil Elastase
AAT-Fusion Protein | |
| | | |
|
| INBRX-109** | Oncology | DR5
Tetravalent Agonist | |
| |
|
| INBRX-106*** | Oncology | OX40
Hexavalent Agonist | |
| | | | |
|
| | | |
| | | | |
|
| | | |
| |
|
| | | |
| |
|
__________________* Subject to potential acquisition by Aventis as described above.
** Third party partnership with Chinese biotechnology company, Transcenta, currently in place for development and commercialization in China, Hong Kong, Macau and/or Taiwan.
*** Third party partnership with Chinese biotechnology company, Elpiscience, currently in place for development and commercialization in China, Hong Kong, Macau and/or Taiwan.
INBRX-101 is an optimized, recombinant alpha-1 antitrypsin, or AAT, augmentation therapy candidate for AATD. In March 2022, the United States Food and Drug Administration, or FDA, granted orphan drug designation for INBRX-101 for the treatment of Alpha-1 Antitrypsin Deficiency,AATD. In May 2022, we announced topline results from the INBRX-101 Phase 1 clinical trial. We believe the data revealed the potential to achieve normal AAT levels with less frequent dosing than the current standard of care and showed the treatment was well tolerated with no drug-related severe or serious AEs at doses up to and including 120 mg/kg in single and multi-dose administered intravenously. In May 2023, the FDA granted Fast Track designation to INBRX-101 for the treatment of patients with emphysema due to AATD.
In April 2023, we initiated ElevAATe, a registration-enabling trial for INBRX-101 for the treatment of patients with emphysema due to AATD. The primary endpoint of the trial is the mean change in the average functional AAT, or fAAT, concentration as measured by anti-neutrophil elastase capacity from baseline to average serum trough fAAT concentration at steady state (Ctrough,ss). The initial read-out from the ElevAATe trial is expected to occur in
mid-2025 and we intend to submit for regulatory approval once completed. In our end of Phase 1 meeting, the FDA requested additional information to support the correlation between serum AAT levels and the clinical benefit in AATD to further support serum AAT levels as a surrogate endpoint reasonably likely to predict clinical benefit. We are in the process of compiling this data through existing registry, health records and published data and plan to submit those analyses to the FDA as part of the BLA submission.
Our most advanced therapeutic candidate, INBRX-109, is a tetravalent death receptor 5, or DR5, agonist currently being evaluated in patients diagnosed with difficult-to-treat cancers, such as chondrosarcoma, synovialmesothelioma, colorectal cancer, Ewing sarcoma mesothelioma and pancreatic adenocarcinoma. We plan to submit an amended Investigational New Drug Application,In June 2021, based on the initial Phase 1 data results, we initiated a registration-enabling Phase 2 trial for the treatment of unresectable or IND, to the U.S. Food and Drug Administration, ormetastatic conventional chondrosarcoma for which the FDA and the European Medicine Agency, or EMA, granted orphan drug designation in November 2021 and August 2022, respectively. In November 2022, we announced updated efficacy and safety data from the ongoing Phase 1 INBRX-109 expansion cohorts for INBRX-109 during March 2021. We expect to initiate athe treatment of chondrosarcoma, which showed disease control was observed in patients with and without isocitrate dehydrogenase, or IDH, mutations. Data from the registration-enabling trial evaluatingin unresectable or metastatic conventional chondrosarcoma is expected during the first half of 2025.
On November 2, 2023, we announced preliminary efficacy and safety data from the Phase 1 trial of INBRX-109 in combination with Irinotecan, or IRI, and Temozolomide, or TMZ, for the treatment of advanced or metastatic, unresectable Ewing sarcoma. Among the 13 patients diagnosedevaluable as of the data cut of September 8, 2023, the observed disease control rate was 76.9%, or 10 out of 13 patients as measured by RECISTv1.1, with conventional chondrosarcoma around mid-2021. 7 patients achieving partial responses (53.8%) and 3 patients achieving stable disease (23.1%). Overall, INBRX-109 in combination with IRI/TMZ was well tolerated from a safety perspective. We have expanded recruitment of this cohort as a result of these preliminary findings.
INBRX-106 is a precisely engineered hexavalent sdAb-based therapeutic candidate targeting OX40, designed to be an optimized agonist of this co-stimulatory receptor. It is currently being investigated as a single agent and in combination with Keytruda in patients with locally advanced or metastatic solid tumors. Both INBRX-109Parts 1 and INBRX-106 programs are designed to achieve target agonism through precise control of therapeutic valency. INBRX‑105 is a conditional 4-1BB agonist, currently being investigated in patients with locally advanced or metastatic solid tumors,3, dose escalation as a single agent and initiating in combination with Keytruda, during the second quarter of 2021. Our fourth program, INBRX-101, is an optimized, recombinant alpha 1 antitrypsin, or AAT, augmentation therapy for AATD. We anticipate additional data releases from all four of our clinical programs in 2021.
We have developed a diverse pipeline of therapeutic candidates that are specifically designed to leverage the power of our core sdAb platform and protein engineering expertise, as shown below:
| | | | | | | | | | | | | | | | | | | | |
|
INBRX-109 | | INBRX-106 | | INBRX-105 | | INBRX-101 |
Tetravalent DR5 agonist | | Hexavalent OX40 agonist | | PD-L1x4-1BB tetravalent conditional agonist | | AAT-Fc fusion protein |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Program | Therapeutic Area | Target(s)/Format | STAGE OF DEVELOPMENT | Anticipated Next Milestones |
Preclinical | Phase 1 | Phase 2 | Phase 3 |
INBRX-109* | Oncology | DR5
Tetravalent Agonist | | • Initiation of Trials-
◦Registration-enabling Phase 2 trial in conventional chondrosarcoma –Mid-2021^
◦Ewing combination cohort – Mid-2021
• Initial Data-
◦Mesothelioma and pancreatic adenocarcinoma chemotherapy combination cohorts – Q4 2021
◦Synovial sarcoma cohort (single agent) – 2H 2021
◦Ewing combination cohort –1H 2022
|
| |
|
INBRX-106** | Oncology | OX40
Hexavalent Agonist | | • Initial Data-
◦ Escalation with Keytruda – Q3 2021
◦ Expansion with Keytruda cohorts – Mid-2022
|
| |
|
INBRX-105** | Oncology | PD-L1 x 4-1BB
Tetravalent Conditional Agonist | | • Initial Data-
◦ Escalation with Keytruda cohorts – Q4 2021
◦ Expansion with Keytruda cohorts – Q3 2022
|
| |
|
INBRX-101*** | Orphan/Respiratory | Neutrophil Elastase
AAT-Fusion Protein | | • Initial multiple ascending dose data – Q4 2021 |
| |
|
__________________
* Third party partnership with Chinese biotechnology company, Transcenta Holding, Ltd. (formerly Hangzhou Just Biotherapeutics Co., Ltd.), or Transcenta, currently in place for development and commercialization in China, Hong Kong, Macau and/or Taiwan.
** Third party partnership with Chinese biotechnology company, Elpiscience Biopharmaceuticals, Inc., or Elpiscience, currently in place for development and commercialization in China, Hong Kong, Macau and/or Taiwan.
*** Commercialization and development rights outside of the United States and Canada, subject to an option agreement with Chiesi Farmaceutici S.p.A., or Chiesi.
^ The FDA granted fast track designation to INBRX-109 for the treatment of patients with unresectable or metastatic conventional chondrosarcoma in January 2021. Advancing INBRX-109 into registration-enabling randomized Phase 2 trials is contingent upon the FDA’s approval of the Company’s IND amendment, which is plannedbeen completed. It was observed to be submittedwell tolerated, with predominantly mild or moderate immune-related toxicities notes. We observed durable responses across multiple tumor types. We expanded the enrollment for Part 2, single agent dose expansion, to increase the dataset in March 2021.
Since our inception, we have had significant operating losses. Our net loss for the years ended December 31, 2020 and 2019 was $76.1 million and $51.4 million, respectively. During August 2020, we completed an initial public offering of our common stock, or our IPO, in which we sold 8,050,000 shares of common stock at an offering price of $17.00 per share. Proceeds from the IPO, net of underwriting discounts, commissions and offering costs, were $125.9 million. As of December 31, 2020, we had an accumulated deficit of $145.4 million and cash and cash equivalents of $128.7 million. Since our inception, we have devoted substantially all of our efforts to therapeutic drug discovery and development, conducting preclinical studies and clinical trials, enabling manufacturing activities in support of our therapeutic candidates, hiring to support our research and development activities and financial reporting capabilities, establishing our intellectual property portfolio, and raising capital to support and expand these activities. Our primary use of cash has been to fund operating expenses, which consist primarily of research and development expenditures,single agent cohorts and to a lesser extent, general and administrative, or G&A, expenditures. Cash used to fund operating expenses is impacted by the timing of when we pay these expenses, as reflected in the change in our outstanding accounts payable and accrued expenses.enroll additional NSCLC patients. We expect to announce additional data from Part 2 in 2025. We continue to incur net losses forenroll patients with NSCLC and HNSCC in Part 4, combination expansion cohorts. We are in the foreseeable future,process of expanding these cohorts and we expect our research and development expenses, G&A expenses and capital expenditures will continue to increase. In particular, we expect our expenses and losses to increase as we continue our development of, and seek regulatory approvals for, our therapeutic candidates (especially as we move more candidates from preclinical to clinical development as well as study candidates in later stages of clinical development), and begin to commercialize any approved products, if ever. We also expect our expenses and losses to increase as we hire additional personnel and incur increased accounting, audit, legal, regulatory, compliance, and director and officer insurance costs and as we incur increased costs from investor and public relations expenses associated with operating as a public company. Our net losses may fluctuate significantly from quarter-to-quarter and year-to-year, depending on the timing of our clinical trials and our expenditures on other research and development activities.
Based upon our current business plan, we believe that our existing cash and cash equivalents will enable us to fund our operating expenses and capital expenditure requirements throughinitiate at least the next 12 months from the issuance date of this report. To date, we have not had any therapeutics approved for sale and have not generated any revenue
from the commercial sale of approved therapeutic products.one additional cohort by mid 2024. We do not expect to generate any revenue from therapeutic product sales unlesshave a more mature dataset during the third quarter of 2025 and until we, or our collaboration partners, successfully complete development and obtain regulatory approval for one or more of our therapeutic candidates, which we expect will take a number of years. If we obtain regulatory approval for any of our therapeutic candidates, we expect to incur significant commercialization expenses related to product sales, marketing, manufacturing and distribution. As a result, until such time, if ever, as we can generate substantial product revenue, we expect to finance our cash needs through equity offerings, debt financings or other capital sources, including strategic licensing and collaboration or other similar agreements. However, there can be no assurance as to the availability or terms upon which such finances or capital might be available in the future. If we are unable to secure adequate additional funding, we will need to reevaluate our operating plan and may be forced to make reductions in spending, extend payment terms with suppliers, liquidate assets where possible, delay, scale back or eliminate some or all of our development programs, or relinquish rights to our intellectual property on less favorable terms than we would otherwise choose. These actions could materially impact our business, results of operations, financial condition, and prospects.
We do not own or operate manufacturing facilities for the production of any of our therapeutic candidates, nor do we have plans to develop our own manufacturing operations in the foreseeable future. We currently rely on a limited number of third-party contract manufacturers for all of our required raw materials, antibodies, and other biologics for our preclinical research, clinical trials, and if and when applicable, commercial product, and employ internal resources to manage our manufacturing relationships with these third parties.
License and Collaboration Agreements
Celgene Agreement
On July 1, 2013, we entered into a license agreement with Celgene, as amended on November 23, 2018, or the Celgene Agreement, pursuant to which we granted Celgene an exclusive, global license for the development, manufacture and commercialization of our proprietary CD47 binding domain, or the Celgene Licensed Intellectual Property. Per the terms of the Celgene Agreement, Celgene is operationally and financially responsible for the development, manufacturing and commercialization activities of Celgene Licensed Intellectual Property and any additional related antibodies covered by the Celgene Agreement.
As payment for the license granted in the Celgene Agreement, we may be eligible to receive development and regulatory milestones of an aggregate of $934.1 million, assuming the achievement of all potential milestones in the Celgene Agreement, as well as percentage tiered royalties based on future worldwide sales, with rates ranging between the high single-digits and low teens, subject to potential reduction when and if comparable third party products attain certain levels of competitive market share (on a country-by-country basis) and, subject to certain limitations, payments to third parties for third-party intellectual property rights. We are obligated to pay 2% of future amounts received under the Celgene Agreement to advisors who assisted us with the negotiations and other matters in connection with the Celgene Agreement.
WuXi Agreement
Pursuant to the WuXi Agreement, we agreed, for three years and subject to certain conditions, to exclusively use WuXi to manufacture our therapeutic candidates for which we plan to first initiate clinical studies outside of China. Under the WuXi Agreement, WuXi and certain of its affiliates will provide biologics development and manufacturing services on a project-by-project basis.
Per the terms of the WuXi Agreement, we will own all intellectual property created, developed or reduced to practice by WuXi in the course of providing services to us; providedan update at that certain intellectual property created or developed by WuXi may be owned exclusively by WuXi if this intellectual property (i) relates to generally applicable experimental methods, (ii) relates to generally applicable manufacturing processes, developed solely at WuXi’s expense, or (iii) is derivative of WuXi’s pre-existing intellectual property.
Per the terms of the WuXi Agreement, fees will be mutually agreed upon on a project-by-project basis. We also may be eligible to receive discounts in the low- to mid-single digits for certain projects and services per the terms of the WuXi Agreement.
bluebird bio Agreements
On December 20, 2018, we entered into an exclusive license agreement with bluebird to research, develop and commercialize CAR T-cell therapies using our proprietary sdAb platform. Under the terms of this license agreement we will provide bluebird the exclusive worldwide rights to develop, manufacture and commercialize certain cell therapy products containing sdAbs directed to various cancer targets. In January 2019, we received a $7.0 million payment and pursuant to the license agreement, we are entitled to receive developmental milestone payments of up to an aggregate of $51.5 million per therapeutic, as well as percentage tiered royalties on future product sales with rates in the mid-single digits.
On June 9, 2020, we entered into the 2020 bluebird Agreement with bluebird, pursuant to which we granted to bluebird exclusive worldwide rights to develop binders and cell therapy products containing sdAbs directed to specified targets, consisting of two initial programs and up to an additional 8 programs. Inhibrx retains all rights to the specific sdAbs outside of the cell therapy field. We received a non-refundable upfront option fee of $0.2 million in connection with each of the two initial programs, or $0.4 million in aggregate, and we are entitled to an upfront option fee for each additional program. We also granted to bluebird an option to acquire an exclusive license with respect to all binders and cell therapy products developed under the 2020 bluebird Agreement, which entitles us to additional fees upon exercise of the option. We are also entitled to receive certain developmental milestone payments of up to an aggregate of $51.5 million per therapeutic, as well as percentage tiered royalties on future product sales with rates in the mid-single-digits.
Other Collaboration Agreements
In addition to these contracts, we have entered into strategic collaborations with other third parties, including Chiesi, Phylaxis (as defined below), Transcenta, and Elpiscience. For more information regarding these agreements, refer to Note 8 to the consolidated financial statements.time.
Components of Results of Operations
Revenue
To date, all of our revenue has been derived from licenses with collaboration partners and grant awards. We have not generated any revenue from the commercial sale of approved therapeutic products, and until, if ever, they are approved for commercial sale, we expect our revenue for the next several years will be derived primarily from payments under our current license agreements and any potential future grant awards and agreements with our collaboration partners.collaborations or strategic transactions.
Operating Expenses
Research and Development
To date, our research and development expenses have related primarily to research activities, including our discovery efforts, and preclinical and clinical development and the manufacturing of our therapeutic candidates. Research and development expenses are recognized as incurred and payments made prior to the receipt of goods or services to be used in research and development are capitalized until the goods or services are received. We do not track our internal research and development expenses on a program-by-program basis as they primarily relate to personnel, early research and consumable costs, which are deployed across multiple projects under development.
Research and development expenses consist primarily of:
•External expenses, consisting of:
•◦expenses incurred in connection with the preclinical development of our programs;
•◦clinical trials of our therapeutic candidates, including under agreements with third parties, such as consultants and contract research organizations, or CROs;
•◦expenses associated with the manufacturing of our contract development and manufacturing therapeutic candidates including under agreements with contract development and manufacturing organizations, or CDMOs;
◦expenses associated with regulatory requirements, including fees and other expenses related to our Scientific Advisory Board; and
•◦other external expenses, such as laboratory services related to our discovery and development programs and other shared services, and
•Internal expenses, consisting of:
•◦salaries, benefits and other related costs, including non-cash stock-based compensation, for personnel engaged in research and development functions;
•◦facilities, depreciation and other expenses, which include direct and allocated expenses for depreciation and amortization, rent and maintenance of facilities; and
•◦other internal expenses, such as laboratory supplies and other shared research and development costs.
We expect that research and development expense will continue to increase over the next several years as we continue development of our therapeutic candidates currently in clinical stage development, support our preclinical programs, and continue to discover new therapeutic candidates, as well as increase our headcount. In particular, clinical development of our therapeutic candidates, as opposed to preclinical development, generally has higher development costs, primarily due to the increased size and duration of later-stage clinical trials. Moreover, the costs associated with our CDMOs to manufacture our therapeutic candidates and future commercial products is also much more costly as compared to early stage preclinical development. We cannot determine with certainty the timing of initiation, the duration or the completion costs of current or future preclinical studies and clinical trials of our therapeutic candidates due to the inherently unpredictable nature of preclinical and clinical development. Preclinical and clinical development timelines, the probability of success and development costs can differ materially from expectations. We anticipate that we will make determinations as to which therapeutic candidates to pursue and how much funding to direct to each therapeutic candidate on an ongoing basis in response to the results of ongoing and future preclinical studies and clinical trials, regulatory developments and our ongoing assessments as to each therapeutic candidate’s commercial potential. We will need substantial additional capital in the future to support these efforts. In addition, we cannot forecast which therapeutic candidates may be subject to future collaborations, when such arrangements will be secured, if at all, and to what degree such arrangements would affect our development plans and capital requirements.
Our clinical development costs may vary significantly based on factors such as:
•the per patient trial costs;
•the number of trials required for approval;
•the number of sites included in the trials;
•the countries in which the trials are conductedconducted;
•the length of time required to enroll eligible patients;
•the number of patients that participate in the trials;
•the number of doses that patients receive;
•the drop-out or discontinuation rates of patients;
•the potential additional safety monitoring requested by regulatory agencies;
•the duration of patient participation in the trials and follow-up;
•the cost, timing, and timing ofsuccessful manufacturing of our therapeutic candidates;
•the phase and development of our therapeutic candidates;
•the efficacy and safety profile of our therapeutic candidates; and
•the uncertainties related to the COVID-19 pandemic.potential economic downturn, geopolitical events and widespread health events on capital and financial markets.
General and Administrative
General and administrative, or G&A, expenses consist primarily of:
•salaries, benefits and other related costs, including non-cash stock-based compensation, for personnel engaged in G&A functions;
•expenses incurred in connection with accounting and audit services, legal services, business developmentincluding costs associated with obtaining and maintaining our patent portfolio, investor relations as well asand consulting expenseexpenses under agreements with third parties, such as consultants and contractors;
•expenses incurred in connection with commercialization and business development activity; and
•facilities, depreciation and other expenses, which include direct and allocated expenses for depreciation and amortization, rent and maintenance of facilities, insurance and supplies.
We expect our G&A expenses will continue to increase overin the next several yearsfuture to support our continued research and development activities. We expect increased costs related to pre-commercialization and business development activities, including the hiring of additional personnel as we incur increasedcontinue to build our commercial team in preparation for our future product launches. Additionally, we expect other professional service fees to increase, including but not limited to, patent-related costs for filing, prosecution and maintenance of our product candidates, and compliance costs, accounting, audit, legal, regulatory, compliance, director and officer insurance costs as well as investor and public relations expenses associated with operating as a public company and as we increase our headcount.additional personnel.
Other Income (Expense)
Other income (expense)Interest expense. Interest expense consists primarily of interest expense on our loans with Oxford Finance LLC, or Oxford.
Interest income. Interest income consists of interest earned on cash and cash equivalents, which include investments held during the period in highly liquid debt securities with original maturities of less than three months from Oxfordour date of acquisition.
Loss on Equity Method Investment
Our equity interest in Phylaxis BioScience, LLC, or Phylaxis, is accounted for as an equity method investment and prior to conversion, interest expense related to the amortizationCompany’s proportionate share of the debt discount related tonet income or loss of Phylaxis is included as loss in equity method investment in the 2019 Note and the 2020 Notes. Other income (expense) also consistsconsolidated statement of gains or losses on the change in fair value of derivative liabilities, and gains or losses on the change in fair value of warrant liabilities.operations.
Results of Operations
Comparison of Years Ended December 31, 20202023 and 2019December 31, 2022
The following table summarizes our consolidated results of operations for each of the periods indicated (in thousands, except percentages):
| | YEAR ENDED DECEMBER 31, | | CHANGE |
| 2020 | | 2019 | | ($) | | (%) |
| YEAR ENDED DECEMBER 31, | | | YEAR ENDED DECEMBER 31, | | CHANGE |
| 2023 | | | 2023 | | 2022 | | ($) | | (%) |
| Revenue: | Revenue: | | | | | | | |
| License fee revenue | |
| License fee revenue | |
| License fee revenue | License fee revenue | $ | 12,808 | | | $ | 9,093 | | | $ | 3,715 | | | 41 | % | $ | 1,800 | | | $ | | $ | 2,178 | | | $ | | $ | (378) | | | (17) | | (17) | % |
| Grant revenue | Grant revenue | 80 | | | 4,118 | | | (4,038) | | | (98) | % | Grant revenue | — | | | 14 | | 14 | | | (14) | | (14) | | | (100) | | (100) | % |
| Total revenue | Total revenue | 12,888 | | | 13,211 | | | (323) | | | (2) | % | Total revenue | 1,800 | | | 2,192 | | 2,192 | | | (392) | | (392) | | | (18) | | (18) | % |
| Operating expenses: | Operating expenses: | | | | | | |
| Research and development | Research and development | 73,495 | | | 47,907 | | | 25,588 | | | 53 | % |
| Research and development | |
| Research and development | | 191,640 | | | 110,186 | | | 81,454 | | | 74 | % |
| General and administrative | General and administrative | 6,836 | | | 6,257 | | | 579 | | | 9 | % | General and administrative | 29,381 | | | 21,123 | | 21,123 | | | 8,258 | | 8,258 | | | 39 | | 39 | % |
| Abandoned offering costs | — | | | 2,761 | | | (2,761) | | | (100) | % |
| Total operating expenses | Total operating expenses | 80,331 | | | 56,925 | | | 23,406 | | | 41 | % | Total operating expenses | 221,021 | | | 131,309 | | 131,309 | | | 89,712 | | 89,712 | | | 68 | | 68 | % |
| Loss from operations | Loss from operations | (67,443) | | | (43,714) | | | (23,729) | | | 54 | % | Loss from operations | (219,221) | | | (129,117) | | (129,117) | | | (90,104) | | (90,104) | | | 70 | | 70 | % |
| Other income (expense): | Other income (expense): | | |
| Interest expense, net | (10,816) | | | (5,719) | | | (5,097) | | | 89 | % |
| Interest expense | |
| Interest expense | |
| Interest expense | | (31,840) | | | (18,181) | | | (13,659) | | | 75 | % |
| Interest income | | Interest income | 11,917 | | | 2,074 | | | 9,843 | | | 475 | % |
| Other income (expense), net | Other income (expense), net | (2) | | | (53) | | | 51 | | | (96) | % | Other income (expense), net | (580) | | | 1 | | 1 | | | (581) | | (581) | | | (58,100) | | (58,100) | % |
| Change in fair value of warrant liabilities | (24) | | | — | | | (24) | | | 100 | % |
| Change in fair value of derivative liabilities | 2,651 | | | (1,016) | | | 3,667 | | | (361) | % |
| | Total other expense | |
| | Total other expense | |
| | Total other expense | Total other expense | (8,191) | | | (6,788) | | | (1,403) | | | 21 | % | (20,503) | | | (16,106) | | (16,106) | | | (4,397) | | (4,397) | | | 27 | | 27 | % |
| Provision for income taxes | Provision for income taxes | 3 | | | 898 | | | (895) | | | (100) | % | Provision for income taxes | 3 | | | 3 | | 3 | | | — | | — | | | — | | — | % |
| Loss on equity method investment | Loss on equity method investment | 487 | | | — | | | 487 | | | 100 | % | Loss on equity method investment | 1,634 | | | — | | — | | | 1,634 | | 1,634 | | | 100 | | 100 | % |
| Net loss | Net loss | $ | (76,124) | | | $ | (51,400) | | | $ | (24,724) | | | 48 | % | Net loss | $ | (241,361) | | | $ | | $ | (145,226) | | | $ | | $ | (96,135) | | | 66 | | 66 | % |
License Fee Revenue
License fee revenue increaseddecreased by $3.7$0.4 million to $12.8from $2.2 million during the year ended December 31, 2020 from $9.12022 to $1.8 million during the year ended December 31, 2019. The $12.82023. During the years ended December 31, 2023 and December 31, 2022, we recognized $0.1 million and $0.9 million, respectively, of license fee revenue related to the Chiesi Option Agreement. Upon Chiesi’s declination of its option, all remaining deferred revenue under this agreement was recognized during the period. Additionally, during the years ended December 31, 2023 and December 31, 2022, we recognized license fee revenue of $1.6 million and $1.1 million, respectively, related to our agreements with Phylaxis. During the year ended December 31, 2022, we recognized the remaining $1.1 million of license fee revenue recordedunder the first phase of our agreements with Phylaxis as we completed our obligation to transfer the exclusive licenses for, and complete development services, to modify the two compounds. During the year ended December 31, 2023, we transferred a second-generation compound to Phylaxis, upon which we received an additional 5% equity interest in Phylaxis, which we recognized as revenue at its fair value of $1.6 million. During the year ended December 31, 2022, we also recognized $0.2 million of revenue under the 2020 2seventy Agreement following the grant of an exclusive option and development license upon initiation of a third program. No revenue was recognized under this agreement with 2seventy during the year ended December 31, 2020 primarily consisted of $7.4 million related to our option agreement with Chiesi, $2.0 million related to a milestone earned under our contract with Elpiscience, $2.0 million related to our agreements with Phylaxis (as defined below), and $0.4 million related to our option and license agreement with bluebird. The $9.1 million of license fee revenue recorded during the year ended December 31, 2019 consisted primarily of $7.0 million recorded as revenue from bluebird following the transfer of an exclusive license, $1.1 million recognized as revenue related to our option agreement with Chiesi, and $1.0 million upon completion of a milestone event from Transcenta.
Grant Revenue
Grant revenue decreased by $4.0 million to $0.1 million during the year ended December 31, 2020 from $4.1 million during the year ended December 31, 2019. GrantWe recognized approximately $14,000 of grant revenue during the year ended December 31, 20202022, all of which consisted of $0.1 millionrevenue earned under our grant with the Department of Defense, or DoD, which was awarded in February 2021 and completed in September 2020. Grant revenueFebruary 2022. The grant was completed during the year ended December 31, 2019 consisted of $3.1 million from CARB-X, $0.9 million from2022, and no revenue was earned during the NIH, and $0.2 million from the DoD. During 2019, we completed all work under our CARB-X and NIH grants.year ended December 31, 2022.
Research and Development Expense
The following table sets forth the primary external and internal research and development expenses (in thousands, except percentages):
| | YEAR ENDED DECEMBER 31, | | CHANGE |
| 2020 | | 2019 | | ($) | | (%) |
| YEAR ENDED DECEMBER 31, | | | YEAR ENDED DECEMBER 31, | | CHANGE |
| 2023 | | | 2023 | | 2022 | | ($) | | (%) |
| External expenses: | External expenses: | | | | | | | |
| Contract manufacturing | Contract manufacturing | $ | 33,214 | | | $ | 2,645 | | | $ | 30,569 | | | 1,156 | % |
| Contract manufacturing | |
| Contract manufacturing | | $ | 77,248 | | | $ | 27,367 | | | $ | 49,881 | | | 182 | % |
| Clinical trials | Clinical trials | 9,015 | | | 7,222 | | | 1,793 | | | 25 | % | Clinical trials | 42,960 | | | 28,988 | | 28,988 | | | 13,972 | | 13,972 | | | 48 | | 48 | % |
| Preclinical studies | 2,802 | | | 10,649 | | | (7,847) | | | (74) | % |
| | Other external research and development | |
| Other external research and development | |
| Other external research and development | Other external research and development | 3,145 | | | 3,277 | | | (132) | | | (4) | % | 10,480 | | | 6,760 | | 6,760 | | | 3,720 | | 3,720 | | | 55 | | 55 | % |
| Internal expenses: | Internal expenses: | |
| Personnel | |
| Personnel | |
| Personnel | Personnel | 16,655 | | | 14,926 | | | 1,729 | | | 12 | % | 47,818 | | | 35,937 | | 35,937 | | | 11,881 | | 11,881 | | | 33 | | 33 | % |
| Equipment, depreciation, and facility | Equipment, depreciation, and facility | 4,905 | | | 4,011 | | | 894 | | | 22 | % | Equipment, depreciation, and facility | 7,361 | | | 6,151 | | 6,151 | | | 1,210 | | 1,210 | | | 20 | | 20 | % |
| Other internal research and development | Other internal research and development | 3,759 | | | 5,177 | | | (1,418) | | | (27) | % | Other internal research and development | 5,773 | | | 4,983 | | 4,983 | | | 790 | | 790 | | | 16 | | 16 | % |
| Total research and development expenses | Total research and development expenses | $ | 73,495 | | | $ | 47,907 | | | $ | 25,588 | | | 53 | % | Total research and development expenses | $ | 191,640 | | | $ | | $ | 110,186 | | | $ | | $ | 81,454 | | | 74 | | 74 | % |
Research and development expense increased by $25.681.4 million to $73.5from $110.2 million during the year ended December 31, 2020 from $47.92022 to $191.6 million during the year ended December 31, 2019.2023. The overall increase in research and development expense was primarily due to the following factors:
•contract manufacturing expense increased by $30.6$49.9 million which wasdue to the nature of the development and manufacturing activities performed during the current period at our CDMO and CRO partners supporting our clinical and preclinical therapeutic candidates, primarily attributabledue to worklarge scale drug substance manufacturing services, including the utilization of raw materials, performed by one of our CDMO partners for INBRX-101, in addition to other activities performed with our CDMO partners which reflect the formulation and manufacturingstage-specific needs of each of our INBRX-101programs, including early and INBRX-109 therapeutic candidates;late stage drug substance clinical manufacturing, analytical development, QC testing, and stability studies, as well as drug product development, scale-up, robustness studies, and selected BLA-enabling activities;
•clinical trial expense increased by $1.8$14.0 million due to costs incurred upon the initiation of the registration-enabling Phase 2 trial for INBRX-101 for the treatment of emphysema due to AATD, which was primarily attributable to increased CRO costs fromwe initiated during the current year, as well as the progression of our INBRX-109 registration-enabling Phase 1 Trials;
•preclinical expense decreased by $7.8 million, which was primarily attributable to2 trial for the completiontreatment of preclinical developmentunresectable or metastatic conventional chondrosarcoma. We also incurred increased costs associated with the utilization of Keytruda used in combination with INBRX-105 in our fourPhase 1/2 clinical programs as they progress through the pipeline;trial, while costs associated with our Phase 1/2 clinical trial for INBRX-106 remained consistent in each period;
•personnel-related expense increased by $1.7$11.9 million, primarily related to an increase in headcount as a result of a significant expansion of our clinical team, as well as the issuance of additional stock options and the expansion of the bonus eligibility pool during the current year;
•facility and equipment-related expense increased by $1.2 million, which was primarily attributable to an overall increase in our headcount, certain salary adjustments,software subscriptions to support research and additional stock-based compensation expense from existingdevelopment activities, in addition to increased repairs and new option grants, as well as an increase in R&D executive bonuses during the year ended December 31, 2020;
•facilities expense increased by $0.9 million, which was primarily attributable to additional rent payments under an amended lease which commenced in January 2020;maintenance services; and
•other research and development expense decreasedincreased by $1.6$4.5 million, which was primarily attributable to a decreasean increase in travel expenses to clinical and vendor sites, clinical-related consulting expenses, the purchase of lab supplies, as we progressed our four main programs into clinical trials.and preclinical studies.
G&A Expense
G&A expense increased by $0.5$8.3 million to $6.8from $21.1 million during the year ended December 31, 2020 from $6.32022 to $29.4 million during the year ended December 31, 2019. Personnel-related2023. The overall increase was primarily due to the following factors:
•personnel-related expenses increased by $0.4$5.7 million, primarily attributable to an increase in headcount as we continue to build out our commercial strategy and medical affairs team, as well as increased expense
related to additional stock option grants to employees and the expansion of the bonus eligibility pool in the current year;
•pre-commercialization expenses increased by $1.0 million, primarily related to increases in consulting services to support our commercial operations business intelligence strategies and market research expenses related to INBRX-101 and INBRX-109; and
•professional service fees increased by $0.6 million, which was primarily attributable to increases in accounting services performed during the period as a result of our filing status in the current year, consulting services performed related to the proposed Merger, and an increase in legal expenses, including those related to intellectual property and other general corporate matters; and
•facility and equipment-related expense increased by $0.6 million, which was primarily attributable to an increase in software subscriptions and IT services related to software implementations.
Other Income (Expense)
Interest expense. Interest expense increased by $13.6 million from $18.2 million during the year ended December 31, 2020 as compared2022 to the year ended December 31, 2019, due to increased headcount during the period, as well as certain salary adjustments, G&A executive bonus increases, and additional stock-based compensation expense from existing and new option grants during the year ended December 31, 2020. Facilities expense increased by $0.1$31.8 million during the year ended December 31, 2020 as compared2023, all of which relates to the year ended
December 31, 2019 due to increased rent expense under our amended building lease which commenced in January 2020.
Abandoned Offering Costs
In August 2020, we successfully completed an IPO,interest incurred and offset all costs against the proceeds earned. Therefore, no abandoned IPO costs were incurred during the year ended December 31, 2020. During the year ended December 31, 2019, we abandoned our contemplated IPO and wrote off all related IPO costs for a total of $2.8 million.
Other Income (Expense)
Interest expense, net. Interest expense, net, increased by $5.1 million to $10.8 million for the year ended December 31, 2020 from $5.7 million for the year ended December 31, 2019. During the year ended December 31, 2020, $7.8 million of the total interest expense was related to accrual of interest and amortization of debt discounts on the 2019 Note and the 2020 Notes through their conversion date, in addition to $2.2 million of debt discounts on the 2019 Note and the 2020 Notes accelerated upon the conversion in August 2020. During the year ended December 31, 2019, $5.2 million of the total interest expense was related to the 2019 Note, which was issued in May 2019. During the year ended December 31, 2020, $0.9 million of the total interest expense was related to the Amended 2020 Loan Agreement. The increase in interest expense is the result of an increase in the average outstanding principal balance during the year ended December 31, 2022 to the year ended December 31, 2022, as well as an increase in our variable interest rate driven by overall market conditions during the year ended December 31, 2023. For more information regarding the Amended 2020 Loan Agreement, refer to Note 3 to the consolidated financial statements.
Interest income.During the year ended December 31, 2019, $0.72023, we earned $11.9 million of the total interest expense wasincome, of which $7.5 million related to interest earned on our sweep and money market account balances and $4.4 million related to the loan and security agreement, as amended, oraccretion of discounts on investments in debt securities during the 2015 Loan Agreement with Oxford, which was paid in full in March 2020.
Other income, net. Other income, net, was comparable forperiod. During the year ended December 31, 2020 as compared2022, we earned $2.1 million of interest income, of which $0.8 million related to interest earned on our sweep and money market account balances and $1.3 million related to the prior year.
Changeaccretion of discount son investments in fair value of warrant liabilities. Duringdebt securities during the third quarter of 2020, we issued warrants to Oxford, which were classified as liabilities at their fair value. Following our IPO, the warrants were remeasured and reclassified into equity.period. The changeincrease in fair value of approximately $24,000 upon the remeasurement was reflected as the change in fair value of warrant liabilitiesinterest income during the year ended December 31, 2020.
Change in fair value2023 is the result of derivative liabilities. The change in fair value of derivative liabilities of $2.7 million during the year ended December 31, 2020 related to the decrease in fair value of the derivative liabilities on the 2019 Notehigher cash and the 2020 Note to zero. The change in fair value of the derivative liabilities was assessed to be zero during the second quarter of 2020 due to the expected conversion of the notes via an embedded conversion feature which did not meet the definition of a derivative. The 2019 Note and the 2020 Notes later converted during the third quarter of 2020 and the derivative liabilities were extinguished. The change in fair value of the derivative liability of $1.0 million during the year ended December 31, 2019 related to the increase in fair value of the derivative liability on the 2019 Note after its establishment in May 2019.cash equivalent balances, coupled with rising interest rates, generating higher returns.
Income Taxes
Income tax expense decreased by $0.9 million towas approximately $3,000 forduring each of the yearyears ended December 31, 2020 from $0.9 million for the year ended2023 and December 31, 2019. This decrease was due to foreign income tax expense incurred during 2019 related to payments received from Chiesi and Transcenta.2022, respectively. For the years ended December 31, 20202023 and 2019,December 31, 2022, we have applied a 100% valuation allowance against our federal deferred tax assets since it is more likely than not that the deferred tax assets will not be realized.
Loss on Equity Method Investment
During the third quarter of 2020,year ended December 31, 2023, we entered into a joint venturereceived an entity affiliated with ArrowMark Partners, Phylaxis BioScience, LLC, or Phylaxis, and received a 10%additional 5% equity interest in Phylaxis following the company.achievement of a milestone under our agreements. Upon receipt of the equity interest, we established an equity method investment at its fair value.value of $1.6 million. The loss on equity method investment during the year ended December 31, 20202023 of $0.5$1.6 million consists of our share of losses from our investment in Phylaxis, which reduced our equity investment to zero. During the year ended December 31, 2022, we did not record a loss on our equity method investment in Phylaxis which had been valued at zero prior to this period.
Liquidity and Capital Resources and Financial Condition
Sources of Liquidity
In August 2020, we completed our IPO of 8,050,000 shares of our common stock at a price to the public of $17.00 per share, which included the exercise in full by the underwriters of their option to purchase 1,050,000 additional shares of our common stock. Our aggregate net proceeds from the offering were $125.9 million, net of underwriting discounts, commissions and offering costs.
Prior to the IPO, otherTo date, sources of capital raised to fund our operations werehave been comprised of the sale of equity securities, borrowings under the loan and security agreements for gross proceeds of $200.0 million, payments received from commercial partners for licensing rights to our therapeutic candidates under development, and grants, borrowings under loan and security agreements with Oxford including $10.0 million of gross proceeds borrowed under the 2020 Loan Agreement in July 2020, and proceeds from the sale and issuance of convertible promissory notes.
Through December 31, 2023, proceeds from the 2019 Notesale of equity securities as a public company consisted of (i) $136.9 million in gross proceeds from our initial public offering, (ii) $171.4 million in gross proceeds under our Open Market Sale Agreement, or the Sales Agreement, with Jefferies LLC, or the Sales Agent, and (iii) $200.0 million in gross proceeds from the 2020 Notes.private placement transaction, or the Private Placement, with certain institutional and other accredited investors, or Purchasers, in which we sold and issued shares of our common stock and, with respect to
certain Purchasers, pre-funded warrants to purchase our common stock pursuant to a Securities Purchase Agreement, as amended, or the Purchase Agreement. The Purchasers have certain registration rights pursuant to the Purchase Agreement which have been waived until after the close of the Merger. Sales of our common stock made pursuant to the Sales Agreement have been made under our $400.0 million Shelf Registration on Form S-3ASR, which became automatically effective upon filing on September 3, 2021. As of December 31, 2020,2023, we have used a total of $171.4 million of the $400.0 million under our Shelf Registration, with $228.6 million remaining and available for use.
Future Funding Requirements
Since our inception, we have devoted substantially all of our efforts to therapeutic drug discovery and development, conducting preclinical studies and clinical trials, enabling manufacturing activities in support of our therapeutic candidates, establishing our intellectual property portfolio, developing our commercialization strategy, hiring to support these departments and activities, and raising capital to support and expand these activities. Cash used to fund operating expenses is impacted by the timing of when we pay these expenses, as reflected in the change in our outstanding accounts payable and accrued expenses. Our net losses may fluctuate significantly from quarter-to-quarter and year-to-year, depending on the timing of our clinical trials and our expenditures on other research and development activities. Our net loss for the years ended December 31, 2023 and December 31, 2022 was $241.4 million and $145.2 million, respectively. As of December 31, 2023, we had an accumulated deficit of $613.7 million and cash and cash equivalents of $128.7$277.9 million.
In January 2024, we announced the proposed Merger with Aventis, which, subject to the satisfaction of customary closing conditions, including the receipt of regulatory approvals, we currently expect to close during the second quarter of 2024. In connection with the Merger Agreement, we have agreed to various covenants, including, among others, agreement to conduct our business in the ordinary course during the period between the execution of the Merger Agreement and the effective time of the Merger. Outside of certain limited exceptions, we may not take, authorize, commit, resolve, or agree to do certain actions without Aventis’s consent, including, but not limited to:
•acquiring businesses and disposing of significant assets;
•incurring expenditures above specified thresholds;
•issuing equity;
•issuing debt facilities; and
•repurchasing shares of our outstanding common stock.
We do not believe these restrictions will prevent us from meeting our ongoing costs of operations, working capital needs, or capital expenditure requirements.
Based upon our current operating plans, we believe that our existing cash and cash equivalents will be sufficient to fund our operations for at least the next 12 months from the issuance date of this report.these consolidated financial statements are issued. Our forecast of the period of time through which our financial resources will be adequate to support our operations is a forward-looking statement that involves risks and uncertainties, and actual results could vary materially. We have based this estimate on assumptions that may prove to be wrong, and we could deplete our capital resources sooner than we expect. Additionally, the
The process of conducting preclinical studies and testing product candidates in clinical trials is costly, and the timing of progress and expenses in these studies and trials is uncertain. We expect to continue to incur net losses for the foreseeable future until, if ever, we have an approved product and can successfully commercialize it. We expect our research and development expenses to increase as we continue our development of, and seek marketing approvals for, our therapeutic candidates (especially as we move more candidates into later stages of clinical development), and begin to commercialize any approved products, if ever. At this time, we are preparing to proceed with the commercialization of certain of our product candidates, if ever approved. As a result, we will incur significant pre-commercialization expenses in preparation for launch, the outcome of which is uncertain. Additionally, if approved, we will incur significant commercialization expenses related to product sales, marketing, manufacturing, and distribution. We also expect additional general and administrative expenses as we hire additional personnel and incur increased accounting, audit, legal, regulatory and compliance, investor and public relations expense to support the Company as we continue to expand.
Until such time we, if ever, can generate substantial product revenue, we expect to finance our cash needs through equity offerings, debt financings or other capital sources, including strategic licensing and collaborations, strategic transactions, or other similar arrangements and transactions, and from time to time, we engage in discussions with potential acquirers regarding the disposition of one or more of our product candidates. However, there can be no assurance as to the availability or terms upon which such finances or capital might be available in the future. If we are unable to secure adequate additional funding, we will need to reevaluate our operating plan and may be forced to make reductions in spending, extend payment terms with suppliers, liquidate assets where possible, delay, scale back or eliminate some or all of our development programs, or relinquish rights to our intellectual property on less favorable terms than we would otherwise choose. These actions could materially impact our business, results of operations, financial condition, and prospects.
Our future liquidity and capital funding requirements will depend on numerous factors, including:
•the outcome, costs and timing of preclinical studies and clinical trials for our current or future therapeutic candidates;
•whether and when we are able to obtain regulatorymarketing approval to market any of our therapeutic candidates;candidates and the outcome of meetings with applicable regulatory agencies, including the FDA;
•our ability to successfully commercialize any therapeutic candidates that receive regulatorymarketing approval;
•the emergence and effect of competing or complementary therapeutics or therapeutic candidates;
•our ability to maintain, expand and defend the scope of our intellectual property portfolio, including the amount and timing of any payments we may be required to make, or that we may receive, in connection with the licensing, filing, prosecution, defense and enforcement of any patents or other intellectual property rights;
•our ability to retain our current employees and the need and ability to hire additional management and scientific and medical personnel;
•the terms and timing of any strategic licensing, collaboration or other similar agreement that we have established or may establish;
•our ability to repay, refinance or restructure our indebtedness when payment is due, including in the event such indebtedness is accelerated;
•the valuation of our capital stock; and
•the continuing or future effects of the COVID-19 pandemica potential economic downturn, geopolitical events, and related uncertaintieswidespread health events on capital and financial markets.
We will continue to require additional funding in order to completedo not own or operate manufacturing and testing facilities for the developmentproduction of any of our therapeutic candidates, and commercializenor do we have plans to develop our therapies, if approved. Until such time, if ever,own manufacturing operations in which we can generate substantial product revenue, we expect to continue to fund our operations and capital funding needs through potential future equity offerings, debt financings or other capital sources, including strategic licensing, collaboration, or other similar arrangement. However, we may be unable to raise additional funds or enter into such other arrangements when needed,the foreseeable future. We currently rely on favorable terms or at all. If we are unable to secure adequate additional funding, we will need to reevaluate our operating plan and may be forced to make reductions in spending, extend payment terms with suppliers, liquidate assets where possible, delay, scale back or eliminate some ora limited number of third-party contract manufacturers for all of our development programs, or relinquish rightsrequired raw materials, antibodies and other biologics for our preclinical research, clinical trials, and if and when applicable, commercial product, and employ internal resources to manage our manufacturing relationships with these third parties.
Commitments
Our material cash requirements from known contractual and other obligations primarily relate to our intellectual property on less favorable terms thanlease obligations, debt, and services provided by our third party CROs and CDMOs.
We have two leases for our laboratory and office space, which expire in 2025, with an option to extend the leases for an additional five years. As of December 31, 2023, we have future minimum rental obligations under these leases of $3.4 million, of which $2.3 million and $1.1 million are current and non-current, respectively. For more information regarding these lease agreements, refer to Note 8 to the consolidated financial statements.
Under the Amended 2020 Loan Agreement, and assuming the proposed Merger is not consummated, we are required to make interest only payments through February 2025, with all principal payments and final fee payments beginning in March 2025 and continuing through the maturity date of January 2027. The interest-only period may be extended an additional twelve months if the Company raises at least $100.0 million in upfront licensing or partnership proceeds by February 2025, upon which principal payments would otherwise choose. These actions could materially impact our business, resultsbegin in March 2026. As of operations, financial condition, and prospects.December 31, 2023, we have a minimum obligation of $257.7 million of long-term debt, including minimum
interest payments, of Contentswhich $18.7 million and $239.0 million are current and non-current, respectively. For more information regarding the Amended 2020 Loan Agreement, refer to Note 3 to the consolidated financial statements.
We enter into contracts in the normal course of business with CROs related to our ongoing preclinical studies and clinical trials and with CDMOs for clinical supplies and manufacturing scale-up activities. These contracts are generally cancellable, with notice, at our option. We have recorded accrued expenses of approximately $33.2 million in our consolidated balance sheets for expenditures incurred by CROs and CDMOs as of December 31, 2023.
While these contracts are generally cancellable, some may contain specific activities that involve one or more noncancellable commitments, including minimum purchase commitments, binding annual forecasts and capital equipment investments. Additionally, depending on the timing and reasoning of the exit, certain termination penalties may apply and can range from the cost of work performed to date up to twelve months of future committed manufacturing costs. As of December 31, 2023, the noncancellable portion of these contracts total in aggregate, excluding amounts recorded in accounts payable and accrued expenses as of this date is approximately $62.8 million. The noncancellable purchase commitments relate to the purchase of raw materials and future contract manufacturing of drug supply for INBRX-101.
Cash Flow Summary
The following table sets forth a summary of the net cash flow activity for each of the periods indicated (in thousands):
| | YEAR ENDED DECEMBER 31, | | | YEAR ENDED DECEMBER 31, |
| 2023 | | | 2023 | | 2022 |
| | YEAR ENDED DECEMBER 31, |
| 2020 | | 2019 |
| | Net cash used in operating activities | |
| Net cash used in operating activities | |
| Net cash used in operating activities | Net cash used in operating activities | $ | (47,968) | | | $ | (32,079) | |
| Net cash used in investing activities | Net cash used in investing activities | (1,364) | | | (1,810) | |
| Net cash provided by financing activities | Net cash provided by financing activities | 166,456 | | | 43,326 | |
| Net increase in cash | Net increase in cash | $ | 117,124 | | | $ | 9,437 | |
Operating Activities
Net cash used in operating activities was $48.0$193.3 million during the year ended December 31, 20202023 and consisted primarily of a net loss of $76.1$241.4 million, adjusted for non-cash items including stock-based compensation expense of $24.8 million, accretion on our debt discount and the non-cash portion of interest expense related to our debt of $10.2 million, stock-based compensation expense of $5.0$4.9 million, depreciation and amortization of $1.0$1.2 million, and non-cash lease expense of $1.4$1.8 million. Non-cash revenue of $1.6 million a changeearned in fair value of derivative liabilities of $2.7 million, and arelation to the equity interest in Phylaxis received during the period was offset by the loss on equity method investmentsinvestment of $1.6 million reflecting our share of losses in Phylaxis. Changes in operating assets and liabilities also contributed to the cash used in operating activities, including an increase in prepaid expenses of $10.3 million, primarily due to the prepayment for clinical drug substance manufacturing services at our CDMOs during the year. Additionally, the operating lease liability decreased by $1.9 million as a result of lease payments made throughout the year. Accounts receivable increased by $0.5 million, primarily as a result of dividends earned and not received as of December 31, 2023. Deferred revenue decreased by $0.2 million following the recognition of $0.2 million of previously deferred revenue related to our option agreement with Chiesi. These uses of cash were offset by an increase in accrued expenses of $26.1 million and an increase in accounts payable of $2.1 million, primarily due to the timing of clinical drug substance manufacturing services incurred at our CDMOs and clinical activities incurred at our CROs, in addition to increases in compensation-related accruals as related to employee bonuses.
Net cash used in operating activities was $115.3 million during the year ended December 31, 2022 and consisted primarily of a net loss of $145.2 million, adjusted for non-cash items including stock-based compensation expense of $20.5 million, accretion on our debt discount and the non-cash portion of interest expense related to our debt of $3.4 million, depreciation and amortization of $1.2 million, and non-cash lease expense of $1.6 million. Changes in operating assets and liabilities also contributed to the cash used in operating activities, primarily related to increases in accounts payable of $10.4 million and to accrued expenses and other current liabilities of $8.5 million due to an overall higher volume of clinical and development activities, and a decrease in prepaid expenses of $0.7 million. These were offset in part by a decrease in operating lease liability of $1.3 million andincluding a decrease in deferred revenue of $5.6$2.0 million as a result offollowing the recognition of $7.4$0.9 million of previously deferred revenue related to
our option agreement with Chiesi and $1.1 million of previously deferred revenue related to our option agreement with Chiesi and the addition of $2.3 million of deferred revenue related to our agreements with Phylaxis, adjusted for $0.5Phylaxis. Additionally, the operating lease liability decreased by $1.7 million as a result of non-cash deferred revenue relatedlease payments made throughout the year. Other non-current assets increased by $1.3 million relating to the equity interestdeposits paid to one of our CRO partners, while our accounts payable balance decreased by $0.7 million. These uses of cash were offset in Phylaxis.
Net cash used in operating activities was $32.1 million during the year ended December 31, 2019 and consisted primarily of a net loss of $51.4 million, adjusted for non-cash items such as accretion on our debt discount and non-cash interest of $5.5 million, stock-based compensation expense of $4.0 million, write off of expenses related to offerings contemplated in early 2019 of $2.8 million, non-cash lease expense of $1.4 million, depreciation and amortization of $1.2 million, and change in fair value of derivative liabilities of $1.0 million. Changes in operating assets and liabilities also contributed to the cash used in operating activities, primarily related to an increase in deferred revenue of $8.9 million related to a new agreement andpart by an increase in accrued expenses and other current liabilities of $1.1$7.6 million due to an overall higher volume of preclinical and clinical activity, offset in part by a decrease in accounts payable of $3.5 million, a decrease in operating lease liability of $1.0 million, and an increase in prepaid expenses of $0.7$0.6 million, which are due to the timing of clinical and development activities which were incurred during the year. Additionally, accounts receivable and receivables from related toparties decreased by $0.6 million, primarily as a result of the receipt of payments made during 2019.associated with our cost sharing agreements with Elpiscience.
Investing Activities
Net cash used in investing activities was $1.4$4.6 million and $1.8$0.7 million during the years ended December 31, 20202023 and 2019,December 31, 2022, respectively, and was related to capital purchases includingof laboratory equipment and leasehold improvements for our facility in La Jolla, California.office equipment. During the year ended December 31, 2023, the increased cash outflows related to the expansion of software and office space.
Financing Activities
Net cash provided by financing activities was $166.5$202.0 million during the year ended December 31, 20202023 and consisted primarily of the gross proceeds of $136.9$200.0 million from our IPO,the issuance of common stock and pre-funded warrants in a private placement transaction, offset in part by $11.0 millionissuance costs of costs associated with the offering, gross$0.4 million. In addition, we received proceeds of $15.0$2.3 million from the sale and issuanceexercise of the 2020 Notes, and net proceeds of $29.9 million from the Amended 2020 Loan Agreement, consisting of $30.0 million in principal, net of $0.1 million in lender fees, further offset in part by $0.8 million of costs associated with the debt issuance. These proceeds were offset in part by the repayment of $3.6 million of principal and final payments on our 2015 Loan Agreement. During 2020, we also received $1.9 million in borrowings from the Paycheck Protection Program, or PPP Loan, which was subsequently repaid in full with principal payments of $1.9 million.
Net cash provided by financing activities was $43.3$258.6 million forduring the year ended December 31, 20192022 and consisted primarily of the grossnet proceeds of $40.0$128.9 million from the saleAmended 2020 Loan Agreement and issuance of the 2019 Note and gross proceeds of $12.0$127.4 million from the issuance of convertible preferred shares,our ATM offering, offset in part by the payment of costs associated with the sale and issuanceATM offering of $0.4 million. In addition, we received proceeds of $2.8 million from the 2019 Noteexercise of $2.5 million. Additionally, net cash used in financing also included the repayment of debt principal of $6.2 million.
Off-Balance Sheet Arrangements
We did not have during the periods presented, and we do not currently have, any off-balance sheet arrangements, as defined in the rules and regulations of the SEC.stock options.
Critical Accounting Estimates and Policies
Our consolidated financial statements and accompanying notes are prepared in accordance with United States generally accepted accounting principles, or GAAP, which requires management to make estimates and assumptions that affect the amounts reported. Management bases its estimates on historical experience, market and other conditions, and various other assumptions it believes to be reasonable. Although these estimates are based on management’s best knowledge of current events and actions that may impact us in the future, the estimation process is, by its nature, uncertain given that estimates depend on events over which we may not have control. If market and other conditions change from those that we anticipate, our consolidated financial statements may be materially affected. In addition, if our assumptions change, we may need to revise our estimates, or take other corrective actions, either of which may also have a material effect in our consolidated financial statements. We review our estimates, judgments, and assumptions used in our accounting practices periodically and reflect the effects of revisions in the period in which they are deemed to be necessary. We believe that these estimates are reasonable; however, our actual results may differ from these estimates.
While our significant accounting policies are described in more detail in Note 1 to our consolidated financial statements included elsewhere in this Annual Report, we believe that the following critical accounting policies and estimates have a higher degree of inherent uncertainty and require our most significant judgments:
Revenue Recognition
To date, we have generated revenue from our collaboration and licensing agreements with partners, as well as from grants from the NIH, the CDMRP funded through the DoD, and from private not-for-profit organizations including CARB-X. Billings to customers or payments received from customers are included in deferred revenue on the balance sheet until all revenue recognition criteria are met.
We recognize revenue when, or as, the promised goods or services are transferred to customers in an amount that reflects the consideration to which we expect to be entitled in exchange for those services. To determine revenue recognition for arrangements we conclude are within the scope of Accounting Standards Codification, or ASC, Topic 606, Revenue from Contracts with Customers, or ASC Topic 606, we perform the following five steps: (i) identify the contract(s) with a customer; (ii) identify the performance obligation(s) in the contract; (iii) determine the transaction price; (iv) allocate the transaction price to the performance obligation(s) in the contract; and (v) recognize revenue when (or as) the performance obligation(s) are satisfied. At contract inception, we assess the goods or services promised within each contract, assess whether each promised good or service is distinct and identifies those that are performance obligations. We recognize as revenue the amount of the transaction price that is allocated to the respective performance obligation when, or as, the performance obligation is satisfied.
Collaboration and License Agreements
We enter into collaborative agreements with partners that typically include one or more of the following: (i) license fees; (ii) nonrefundable up-front fees; (iii) payments for reimbursement of research costs; (iv) payments associated with achieving specific development, regulatory, or commercial milestones; and (v) royalties based on specified percentages of net product sales, if any. At the initiation of an agreement, we analyze each unit of account within the contract to determine if the counterparty is a customer in the context of the unit of account, in which case we apply the recognition, measurement, presentation, and disclosure requirements of ASC Topic 606. If a unit of account does not represent a transaction with a customer, it represents an arrangement with a collaborator subject to guidance under ASC Topic 808, Collaborative Arrangements, or ASC Topic 808. We early adopted Accounting Standardsaccount.
Update, or ASU, 2018-18, Collaborative Arrangements (Topic 808): Clarifying the Interaction between Topic 808 and Topic 606 upon implementation of ASC Topic 606. The standard did not have a material effect on our consolidated financial statements.
In applying the principles of ASC Topic 606, weWe consider a variety of factors which may require significant judgment in determining the appropriate estimates and assumptions under these arrangements, such as whether the elements are distinct performance obligations, whether there are observable stand-alonestandalone prices, and whether any licenses are functional or symbolic. We evaluate
each performance obligation to determine if it can be satisfied and recognized as revenue at a point in time or over time. Typically, license fees and non-refundable upfront fees are considered fixed, while milestone payments are identified as variable consideration which must be evaluated to determine if it is constrained and, therefore, excluded from the transaction price.
As of December 31, 2020, all collaboration and license revenue was within the scope of ASC Topic 808 and/or ASC Topic 606 and recognized accordingly.
See Note 86 to our consolidated financial statements included elsewhere in this Annual Report for additional information on our collaboration and license agreements.
Accrued Research and Development and Clinical Trial AccrualsCosts
Research and development costs are expensed as incurred and include the cost of compensation and related expenses, as well as expensesWe enter into contracts for third parties who conduct research and development onactivities, including with CDMOs for clinical supplies and manufacturing scale-up activities related to our behalf, pursuant to developmenttherapeutic candidates and consultingwith CROs for our preclinical studies and clinical trials. The financial terms of these agreements vary and may result in place. Clinical trial costs, including costs associated with third-party contractors,payment flows that do not match the periods over which materials or services are provided, resulting in either an accrual or a significant componentprepaid expense.
These accruals of research and development expense. We expense research and development costs based on work performed. In determining the amountexpenses require us to expense, management relies onestimate expenses incurred, including estimates of total costs based onthe time period over which services will be performed, completion of contract components, completed, the enrollment of subjects, and the completionstatus of trialsour clinical trials. Such estimates are dependent upon the timeliness and other events.accuracy of data provided by the CROs and CDMOs regarding the status and cost of the studies. If applicable, coststhe actual timing of the performance of services varies from our estimates, we adjust the accrual or prepaid expense accordingly.
Although we do not expect our estimates to be materially different from amounts actually incurred, related toif our estimates of the purchasestatus and timing of in-process researchservices performed differ from the actual status and development for early-stage products or productstiming of services performed, it could result in us reporting amounts that are not commercially viable and ready for use,too high or too low in any particular period. To date, there have been no alternative future use, are charged to expense in the period incurred. Costs incurred related to the licensingmaterial differences between our estimates of products that have not yet received regulatory approval to be marketed, or that are not commercially viable and ready for use, or have no alternative future use, are charged to expense in the period incurred.
Derivative Liabilities
Prior to their conversion upon the consummation of our IPO, the 2019 Notesuch expenses and the 2020 Notes contained embedded features that provided multiple settlement alternatives. Certain of these settlement features provided the noteholders the right to a fixed number of our shares upon conversion of the note. Other settlement features provide the noteholders the right or the obligation to receive cash or a variable number of shares upon our completion of a capital raising transaction, change of control, qualified IPO or default. We evaluated each settlement alternative within the 2019 Note and the 2020 Notes under ASC 815-15, Embedded Derivatives and ASC 815-40, Contracts in Entity’s Own Equity and determined to account for certain settlement features as redemption features meeting the requirements for separate accounting as a single, compound derivative instrument.
These embedded derivatives were accounted for as liabilities at their estimated fair value as of the date of issuance, and then subsequently remeasured to fair value as of each balance sheet date, with the related remeasurement adjustment being recognized as a component of change in fair value of derivative liabilities in the consolidated statements of operations. The aggregate outstanding principal amount plus accrued interest to date under the 2019 Note and the 2020 Notes automatically converted and settled into shares of our common stock immediately prior to the completion of our IPO at a fixed conversion price based on a specified valuation.amounts actually incurred.
Fair Value of Stock-Based Awards and Stock-Based Compensation Expense
We recognize compensation costs related to stock-based awards, including stock options, under our 2017 Employee, Director and Consultant Equity Incentive Plan to certain of our employees and certain members of our board of directors. The grant date fair value of the stock-based awards is generally recognized on a straight-line basis over the requisite service period, which is generally the vesting period of the respective awards.
We estimate the grant date fair value, and the resulting stock-based compensation, using the Black-Scholes option pricing model. The Black-Scholes option pricing model requires the use of subjective assumptions to determine the fair value of stock-based awards. These assumptions include:
•Fair Value of Common Stock—Our board of directors determines the fair value of each share of underlying common stock based on the closing price of our common stock as reported by the applicable trading market on the date of the grant. Our board of directors intended all options granted to be exercisable at a price per share not less than the per share fair value of our common stock underlying those options on the grant date.
•Expected Term—We estimate the expected term of our stock options granted to employees and non-employee directors using the simplified method, whereby, the expected term equals the average of the vesting term and the original contractual term of the option. We utilize this method as we do not have sufficient historical exercise data to provide a reasonable basis upon which to estimate the expected term.
•Expected Volatility—Due to the lack of company specific historical and implied volatility data indicative of the expected future volatility, we based our estimate of expected volatility on the estimate and expected volatilities of a guideline group of publicly traded companies. For these analyses, we select companies with comparable characteristics to ours including enterprise value, risk profiles, and with historical share price information sufficient to meet the expected life of the stock-based awards. We compute the historical volatility data using the daily closing prices for the selected companies’ shares during the equivalent period of the calculated expected term of our stock-based awards. We will continue to apply this process until a sufficient amount of historical information regarding the volatility of our own stock price becomes available.available, or we believe the volatility of our own market-traded shares best represents expected volatility.
•Risk-Free Interest Rate—For the determination of the risk-free interest rates we utilize the U.S. Treasury yield curve for instruments in effect at the time of measurement with a term commensurate with the expected term assumption.
•Expected Dividend—The expected dividend yield is assumed to be zero as we have never paid dividends and do not have current plans to pay any dividends on our common stock.
Changes in these assumptions can materially affect the fair value and ultimately how much stock-based compensation expense is recognized. These inputs are subjective and generally require significant analysis and judgment to develop. See Note 75 to our consolidated financial statements included elsewhere in this Annual Report for information regarding certain of the specific assumptions we used in applying the Black-Scholes option pricing model to determine the estimated fair value of our stock options granted in the periods discussed.
Income Taxes
Income taxes are accounted for under the asset and liability method. Deferred income taxes are recorded for temporary differences between financial statement carrying amounts and the tax basis of assets and liabilities. Deferred tax assets and liabilities reflect the tax rates expected to be in effect for the years in which the differences are expected to reverse. A valuation allowance is provided if it is more likely than not that some or all of the deferred tax assets will not be realized.
We follow the provisions of accounting for uncertainty in income taxes which prescribes a model for the recognition and measurement of a tax position taken or expected to be taken in a tax return, and provides guidance on derecognition, classification, interest and penalties, disclosure and transition.
We have not completed a formal study to determine if any ownership changes within the meaning of IRC Section 382 and 383 have occurred. It is possible that we have already incurred ownership changes and may incur additional ownership changes in the future. If an ownership change has occurred, our ability to use our NOL or tax credit carryforwards may be restricted, which could require us to pay federal or state income taxes earlier than would be required if such limitations were not in effect.
JOBS Act Accounting Election
We are an “emerging growth company,” as defined in the JOBS Act. Under the JOBS Act, emerging growth companies can delay adopting new or revised accounting standards issued subsequent to the enactment of the JOBS Act until such time as those standards apply to private companies. We have irrevocably elected not to take advantage
of this extended transition period and, as a result, will comply with new or revised accounting standards on the relevant dates on which adoption of such standards is required for non-emerging growth companies.
Recent Accounting Pronouncements
For information with respect to recently issued accounting standards and the impact of these standards, if any, on our consolidated financial statements, refer to Note 1 in our consolidated financial statements in Part II, Item 8 of this Annual Report.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk.
We are a smaller reporting company as defined by Rule 12b-2 of the Exchange Act and are not required to provide the information required under this item.Not applicable.
Item 8. Financial Statements and Supplementary Data.
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
| | | | | |
| PAGEPage |
(BDO USA, P.C.; San Diego, California; PCAOB ID No. 243) | |
| |
| |
| |
| |
| |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
Board of Directors and Stockholders
Inhibrx, Inc.
La Jolla, California
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheets of Inhibrx, Inc. (the “Company”) as of December 31, 20202023 and 2019,2022, the related consolidated statements of operations, convertible preferred stock and stockholders’ equity, (deficit), and cash flows for each of the years then ended, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 20202023 and 2019,2022, and the results of its operations and its cash flows for each of the years then ended, in conformity with accounting principles generally accepted in the United States of America.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (“PCAOB”), the Company's internal control over financial reporting as of December 31, 2023, based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”) and our report dated February 28, 2024 expressed an unqualified opinion thereon.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”)PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matter
The critical audit matter communicated below is a matter arising from the current period audit of the consolidated financial statements that was communicated or required to be communicated to the audit committee and that: (1) relates to accounts or disclosures that are material to the consolidated financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of the critical audit matter does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to which it relates.
Clinical Trial Accruals
As described in Note 1 and Note 2 to the consolidated financial statements, clinical trial activities are accrued and expensed based on estimates of the period in which services and efforts are expended by contract research organizations (“CROs”) and other third parties. Estimates are determined by reviewing cost information provided by CROs, other third-party vendors and internal clinical personnel, and contractual arrangements with CROs and the
scope of work to be performed. As of December 31, 2023, the Company recorded $9.2 million in clinical trial accruals, which was included in accrued expenses on the balance sheet.
We identified the estimation of clinical trial accruals as a critical audit matter. Management’s judgment was required in estimating the period in which services and efforts were expended used to determine the clinical trial accruals. Auditing clinical trial accruals involved especially challenging auditor judgment due to the nature and extent of audit effort required to address these matters.
The primary procedures we performed to address this critical audit matter included:
•Testing the appropriate measurement of clinical trial accruals by obtaining and inspecting certain agreements and amendments, and confirming total clinical costs incurred and total amounts billed with third-party vendors.
•Testing the completeness of the Company’s clinical trial accruals by evaluating internal materials and publicly available information (such as press releases and public databases that track clinical trials) and inquiring of clinical personnel to gain an understanding of the status of certain on-going clinical trials, and testing invoices received after year-end for certain third-party vendors.
/s/ BDO USA, LLPP.C.
We have served as the Company’s auditor since 2018.
San Diego, California
March 12, 2021February 28, 2024
Inhibrx, Inc.
Consolidated Balance Sheets
(In thousands, except share data and par value)
| | AS OF DECEMBER 31, |
| 2020 | | 2019 |
| AS OF DECEMBER 31, | | | AS OF DECEMBER 31, |
| 2023 | | | 2023 | | 2022 |
| Assets | Assets | | | |
| Current assets: | Current assets: | |
| Cash | $ | 128,664 | | | $ | 11,540 | |
| Current assets: | |
| Current assets: | |
| Cash and cash equivalents | |
| Cash and cash equivalents | |
| Cash and cash equivalents | |
| Accounts receivable | Accounts receivable | 82 | | | 199 | |
| Receivables from related parties | Receivables from related parties | 533 | | | 239 | |
| Prepaid expenses and other current assets | Prepaid expenses and other current assets | 2,893 | | | 3,583 | |
| Total current assets | Total current assets | 132,172 | | | 15,561 | |
| Property and equipment, net | Property and equipment, net | 3,492 | | | 3,230 | |
| Right-of-use asset | 7,831 | | | 7,453 | |
| Operating right-of-use asset | |
| Other non-current assets | Other non-current assets | 245 | | | 245 | |
| Total assets | Total assets | $ | 143,740 | | | $ | 26,489 | |
| Liabilities, convertible preferred stock, and stockholders’ equity (deficit) | | | |
| Liabilities and stockholders’ equity | |
| Current liabilities: | |
| Current liabilities: | |
| Current liabilities: | Current liabilities: | |
| Accounts payable | Accounts payable | $ | 13,458 | | | $ | 3,115 | |
| Accounts payable | |
| Accounts payable | |
| Accrued expenses | Accrued expenses | 13,357 | | | 4,832 | |
| Current portion of deferred revenue | 3,081 | | | 7,939 | |
| Current portion of long-term debt, net of discount | 0 | | | 3,563 | |
| Deferred revenue | |
| | Current portion of lease liability | Current portion of lease liability | 1,503 | | | 1,105 | |
| Other current liabilities | 0 | | | 16 | |
| Current portion of lease liability | |
| Current portion of lease liability | |
| | Total current liabilities | Total current liabilities | 31,399 | | | 20,570 | |
| Convertible notes, net of discount | 0 | | | 30,367 | |
| Derivative liabilities | 0 | | | 1,916 | |
| Long-term debt, net of current portion and including final payment fee | 29,244 | | | 0 | |
| Total current liabilities | |
| Total current liabilities | |
| | Long-term debt, including final payment fee | |
| | Long-term debt, including final payment fee | |
| | Long-term debt, including final payment fee | |
| | Non-current portion of lease liability | Non-current portion of lease liability | 6,707 | | | 6,698 | |
| Non-current portion of deferred revenue | 737 | | | 1,000 | |
| Other non-current liabilities | 180 | | | 0 | |
| Non-current portion of lease liability | |
| Non-current portion of lease liability | |
| | Total liabilities | Total liabilities | 68,267 | | | 60,551 | |
| Commitments and contingencies (Note 12) | 0 | | 0 |
| Convertible preferred stock, $0.0001 par value; 0 shares authorized or outstanding at December 31, 2020; 25,765,000 shares authorized at December 31, 2019; 12,534,331 shares issued and outstanding at December 31, 2019; liquidation preferences of $87.5 million at December 31, 2019. | 0 | | | 59,507 | |
| Stockholders’ equity (deficit) | |
| Preferred stock, $0.0001 par value, 15,000,000 shares authorized and 0 shares outstanding at December 31, 2020; 0 shares authorized or outstanding at December 31, 2019 | 0 | | | 0 | |
| Common stock, $0.0001 par value, 120,000,000 shares authorized at December 31, 2020 and 65,000,000 shares authorized at December 31, 2019; 37,712,390 shares issued and outstanding at December 31, 2020 and 18,154,119 issued and outstanding at December 31, 2019 | 4 | | | 2 | |
| | Total liabilities | |
| | Total liabilities | |
| Commitments and contingencies (Note 9) | | Commitments and contingencies (Note 9) | | | |
| | Stockholders’ equity: | |
| Stockholders’ equity: | |
| Stockholders’ equity: | |
| Preferred stock, $0.0001 par value, 15,000,000 shares authorized and no shares outstanding as of December 31, 2023 and December 31, 2022 | |
| Preferred stock, $0.0001 par value, 15,000,000 shares authorized and no shares outstanding as of December 31, 2023 and December 31, 2022 | |
| Preferred stock, $0.0001 par value, 15,000,000 shares authorized and no shares outstanding as of December 31, 2023 and December 31, 2022 | |
| Common stock, $0.0001 par value, 120,000,000 shares authorized as of December 31, 2023 and December 31, 2022; 47,369,511 and 43,564,283 shares issued and outstanding as of December 31, 2023 and December 31, 2022, respectively | |
| Additional paid-in-capital | Additional paid-in-capital | 220,848 | | | (24,316) | |
| Accumulated deficit | Accumulated deficit | (145,379) | | | (69,255) | |
| Total stockholders’ equity (deficit) | 75,473 | | | (93,569) | |
| Total liabilities, convertible preferred stock and stockholders’ equity (deficit) | $ | 143,740 | | | $ | 26,489 | |
| Total stockholders’ equity | |
| Total liabilities and stockholders’ equity | |
The accompanying notes are an integral part of these consolidated financial statements.
Inhibrx, Inc.
Consolidated Statements of Operations
(In thousands, except per share data)
| | YEAR ENDED DECEMBER 31, |
| 2020 | | 2019 |
| YEAR ENDED DECEMBER 31, | | | YEAR ENDED DECEMBER 31, |
| 2023 | | | 2023 | | 2022 |
| | Revenue: | Revenue: | |
| Revenue: | |
| Revenue: | |
| License fee revenue | |
| License fee revenue | |
| License fee revenue | License fee revenue | $ | 12,808 | | | $ | 9,093 | |
| Grant revenue | Grant revenue | 80 | | | 4,118 | |
| Total revenue | Total revenue | 12,888 | | | 13,211 | |
| Operating expenses: | Operating expenses: | | | |
| Research and development | Research and development | 73,495 | | | 47,907 | |
| Research and development | |
| Research and development | |
| General and administrative | General and administrative | 6,836 | | | 6,257 | |
| Abandoned offering costs | 0 | | | 2,761 | |
| | Total operating expenses | |
| Total operating expenses | |
| Total operating expenses | Total operating expenses | 80,331 | | | 56,925 | |
| Loss from operations | Loss from operations | (67,443) | | | (43,714) | |
| Other income (expense): | Other income (expense): | |
| Interest expense, net | (10,816) | | | (5,719) | |
| Other expense, net | (2) | | | (53) | |
| Change in fair value of warrant liabilities | (24) | | | 0 | |
| Change in fair value of derivative liabilities | 2,651 | | | (1,016) | |
| Interest expense | |
| Interest expense | |
| Interest expense | |
| Interest income | |
| Other income (expense), net | |
| | Total other expense | |
| | Total other expense | |
| | Total other expense | Total other expense | (8,191) | | | (6,788) | |
| Loss before provision for income taxes | Loss before provision for income taxes | (75,634) | | | (50,502) | |
| Provision for income taxes | Provision for income taxes | 3 | | | 898 | |
| Loss on equity method investment | Loss on equity method investment | 487 | | | 0 | |
| Net loss | Net loss | $ | (76,124) | | | $ | (51,400) | |
| Net loss per share, basic and diluted | Net loss per share, basic and diluted | $ | (3.01) | | | $ | (2.83) | |
| Weighted-average shares of common stock outstanding, basic and diluted | Weighted-average shares of common stock outstanding, basic and diluted | 25,261 | | | 18,154 | |
The accompanying notes are an integral part of these consolidated financial statements.
Inhibrx, Inc.
Consolidated Statements of Convertible Preferred Stock and Stockholders’ Equity (Deficit)
(In thousands)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| CONVERTIBLE PREFERRED STOCK (SHARES) | | CONVERTIBLE PREFERRED STOCK (AMOUNT) | | | | | COMMON
STOCK
(SHARES) | | COMMON
STOCK
(AMOUNT) | | ADDITIONAL
PAID-IN
CAPITAL | | ACCUMULATED
DEFICIT | | | | | | TOTAL STOCKHOLDERS’ EQUITY (DEFICIT) |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| Balance as of December 31, 2018 | 10,815 | | | $ | 47,519 | | | | | | 18,154 | | | $ | 2 | | | $ | (39,790) | | | $ | (17,855) | | | | | | | $ | (57,643) | |
| Stock-based compensation expense | — | | | — | | | | | | — | | | — | | | 3,974 | | | — | | | | | | | 3,974 | |
| Issuance of Mezzanine Preferred Stock | 1,719 | | | 11,988 | | | | | | — | | | — | | | — | | | — | | | | | | | — | |
| 2019 Note - beneficial conversion feature | — | | | — | | | | | | — | | | — | | | 11,500 | | | — | | | | | | | 11,500 | |
| Net loss | — | | | — | | | | | | — | | | — | | | — | | | (51,400) | | | | | | | (51,400) | |
| Balance as of December 31, 2019 | 12,534 | | | $ | 59,507 | | | | | | 18,154 | | | $ | 2 | | | $ | (24,316) | | | $ | (69,255) | | | | | | | $ | (93,569) | |
| Stock-based compensation expense | — | | | — | | | | | | — | | | — | | | 5,023 | | | — | | | | | | | 5,023 | |
| 2020 Notes - beneficial conversion feature | — | | | — | | | | | | — | | | — | | | 2,656 | | | — | | | | | | | 2,656 | |
| Issuance of shares upon exercise of stock options | — | | | — | | | | | | 2 | | | — | | | 27 | | | — | | | | | | | 27 | |
| Issuance of shares in IPO, net of issuance costs | — | | | — | | | | | | 8,050 | | | 1 | | | 125,859 | | | — | | | | | | | 125,860 | |
| Issuance of shares in conversion of preferred shares | (12,534) | | | (59,507) | | | | | | 7,211 | | | 1 | | | 59,506 | | | — | | | | | | | 59,507 | |
| Issuance of shares in conversion of the 2019 Note and the 2020 Notes | — | | | — | | | | | | 4,295 | | | — | | | 59,393 | | | — | | | | | | | 59,393 | |
| Reversal of unamortized BCF on the 2019 Note | — | | | — | | | | | | — | | | — | | | (5,332) | | | — | | | | | | | (5,332) | |
| Reversal of unamortized BCF on the 2020 Notes | — | | | — | | | | | | — | | | — | | | (2,107) | | | — | | | | | | | (2,107) | |
| Reclassification of warrant liabilities into equity | — | | | — | | | | | | — | | | — | | | 139 | | | — | | | | | | | 139 | |
| Net loss | — | | | — | | | | | | — | | | — | | | — | | | (76,124) | | | | | | | (76,124) | |
| Balance as of December 31, 2020 | 0 | | | $ | 0 | | | | | | 37,712 | | | $ | 4 | | | $ | 220,848 | | | $ | (145,379) | | | | | | | $ | 75,473 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | Common Stock
(Shares) | | Common Stock (Amount) | | Additional Paid-In Capital | | Accumulated Deficit | | | | | | Total Stockholders’ Equity |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| Balance as of December 31, 2021 | | | | | | | | 38,991 | | | $ | 4 | | | $ | 279,526 | | | $ | (227,147) | | | | | | | $ | 52,383 | |
| Stock-based compensation expense | | | | | | | | — | | | — | | | 20,450 | | | — | | | | | | | 20,450 | |
| Issuance of shares upon exercise of stock options | | | | | | | | 241 | | | — | | | 2,757 | | | — | | | | | | | 2,757 | |
| Issuance of shares, net of issuance costs | | | | | | | | 4,332 | | | — | | | 126,981 | | | — | | | | | | | 126,981 | |
| Issuance of warrants | | | | | | | | — | | | — | | | 712 | | | — | | | | | | | 712 | |
| Net loss | | | | | | | | — | | | — | | | — | | | (145,226) | | | | | | | (145,226) | |
| Balance as of December 31, 2022 | | | | | | | | 43,564 | | | $ | 4 | | | $ | 430,426 | | | $ | (372,373) | | | | | | | $ | 58,057 | |
| Stock-based compensation expense | | | | | | | | — | | | — | | | 24,846 | | | — | | | | | | | 24,846 | |
| Issuance of shares upon exercise of stock options | | | | | | | | 184 | | | — | | | 2,316 | | | — | | | | | | | 2,316 | |
| Issuance of common stock and pre-funded warrants in private placement, net of issuance costs | | | | | | | | 3,621 | | | 1 | | | 199,644 | | | — | | | | | | | 199,645 | |
| Net loss | | | | | | | | — | | | — | | | — | | | (241,361) | | | | | | | (241,361) | |
| Balance as of December 31, 2023 | | | | | | | | 47,369 | | | $ | 5 | | | $ | 657,232 | | | $ | (613,734) | | | | | | | $ | 43,503 | |
The accompanying notes are an integral part of these consolidated financial statements.
Inhibrx, Inc.
Consolidated Statements of Cash Flows
(In thousands)
| | YEAR ENDED DECEMBER 31, |
| 2020 | | 2019 |
| YEAR ENDED DECEMBER 31, | | | YEAR ENDED DECEMBER 31, |
| 2023 | | | 2023 | | 2022 |
| | Cash flows from operating activities | Cash flows from operating activities | |
| Cash flows from operating activities | |
| Cash flows from operating activities | |
| Net loss | |
| Net loss | |
| Net loss | Net loss | $ | (76,124) | | | $ | (51,400) | |
| Adjustments to reconcile net loss to net cash used in operating activities: | Adjustments to reconcile net loss to net cash used in operating activities: | |
| Depreciation and amortization | |
| Depreciation and amortization | |
| Depreciation and amortization | Depreciation and amortization | 1,019 | | | 1,163 | |
| Accretion of debt discount and non-cash interest expense | Accretion of debt discount and non-cash interest expense | 10,189 | | | 5,498 | |
| Stock-based compensation expense | Stock-based compensation expense | 5,023 | | | 3,974 | |
| Non-cash lease expense | Non-cash lease expense | 1,374 | | | 1,355 | |
| Change in fair value of derivative liabilities | (2,651) | | | 1,016 | |
| Change in fair value of warrant liabilities | 24 | | | 0 | |
| Loss from equity method investment | Loss from equity method investment | 487 | | | 0 | |
| Non-cash license revenue | |
| Loss on disposal of fixed assets | Loss on disposal of fixed assets | 0 | | | 69 | |
| Abandoned offering costs | 0 | | | 2,761 | |
| | Changes in operating assets and liabilities: | Changes in operating assets and liabilities: | |
| | Changes in operating assets and liabilities: | |
| | Changes in operating assets and liabilities: | |
| Accounts receivable | |
| Accounts receivable | |
| Accounts receivable | Accounts receivable | 117 | | | 491 | |
| Receivables from related parties | Receivables from related parties | (294) | | | (239) | |
| Prepaid expenses and other current assets | Prepaid expenses and other current assets | 690 | | | (719) | |
| Other non-current assets | Other non-current assets | 0 | | | (1,272) | |
| Accounts payable | Accounts payable | 10,426 | | | (3,474) | |
| Accrued expenses and other current liabilities | 8,525 | | | 1,127 | |
| Accrued expenses | |
| Operating lease liability | Operating lease liability | (1,345) | | | (1,005) | |
| Deferred revenue, current portion | Deferred revenue, current portion | (5,345) | | | 7,939 | |
| Deferred revenue, non-current portion | Deferred revenue, non-current portion | (263) | | | 1,000 | |
| Other non-current liabilities | 180 | | | (363) | |
| | Net cash used in operating activities | |
| Net cash used in operating activities | |
| Net cash used in operating activities | Net cash used in operating activities | (47,968) | | | (32,079) | |
| Cash flows from investing activities | Cash flows from investing activities | | | |
| Purchase of fixed assets | Purchase of fixed assets | (1,364) | | | (1,810) | |
| Purchase of fixed assets | |
| Purchase of fixed assets | |
| | Net cash used in investing activities | |
| Net cash used in investing activities | |
| Net cash used in investing activities | Net cash used in investing activities | (1,364) | | | (1,810) | |
| Cash flows from financing activities | Cash flows from financing activities | | | |
| Proceeds from initial public offering, gross | 136,850 | | | 0 | |
| Costs associated with initial public offering | (10,990) | | | 0 | |
| Proceeds from the issuance of convertible notes | 15,000 | | | 40,000 | |
| Costs associated with the issuance of convertible notes | 0 | | | (2,470) | |
| Proceeds from issuance of common stock and pre-funded warrants in private placement | |
| Proceeds from issuance of common stock and pre-funded warrants in private placement | |
| Proceeds from issuance of common stock and pre-funded warrants in private placement | |
| Issuance costs associated with issuance of common stock and pre-funded warrants in private placement | |
| Proceeds from ATM offering | |
| Costs associated with ATM offering | |
| | Proceeds from the issuance of debt | |
| Proceeds from the issuance of debt | |
| Proceeds from the issuance of debt | Proceeds from the issuance of debt | 29,938 | | | 0 | |
| Payment of fees associated with debt | Payment of fees associated with debt | (786) | | | 0 | |
| Repayment of principal on debt | (2,183) | | | (6,192) | |
| Final payment on debt | (1,400) | | | 0 | |
| Proceeds from the Paycheck Protection Program loan | 1,875 | | | 0 | |
| Repayment of principal on the Paycheck Protection Program loan | (1,875) | | | 0 | |
| Proceeds from exercise of stock options | Proceeds from exercise of stock options | 27 | | | 0 | |
| Proceeds from the issuance of preferred stock | 0 | | | 12,000 | |
| | Net cash provided by financing activities | |
| | Net cash provided by financing activities | |
| | Net cash provided by financing activities | |
| Net increase in cash | |
| Cash and cash equivalents at beginning of period | |
| Cash and cash equivalents at end of period | |
|
| | | | | | | | | | | |
| Costs associated with the issuance of preferred stock | 0 | | | (12) | |
| Net cash provided by financing activities | 166,456 | | | 43,326 | |
| Net increase in cash | 117,124 | | | 9,437 | |
| Cash and cash equivalents at beginning of period | 11,540 | | | 2,103 | |
| Cash and cash equivalents at end of period | $ | 128,664 | | | $ | 11,540 | |
| | | |
| Supplemental disclosure of cash flow information | | | |
| Cash paid for interest | $ | 504 | | | $ | 476 | |
| Cash paid for income taxes | $ | 3 | | | $ | 976 | |
| | | |
| Supplemental schedule of non-cash investing and financing activities | | | |
| Conversion of preferred stock to common stock | $ | 59,507 | | | $ | 0 | |
| Conversion of the 2019 Note and the 2020 Notes to common stock | $ | 59,393 | | | $ | 0 | |
| Debt discount arising from convertible note beneficial conversion features and reversal of beneficial conversion features | $ | 4,783 | | | $ | (11,500) | |
| Operating lease liabilities arising from obtaining right-of-use assets | $ | 1,752 | | | $ | 8,808 | |
| Derivative liabilities arising from the issuance of the 2019 Note and the 2020 Notes | $ | 735 | | | $ | 900 | |
| | | |
| Non-cash equity method investment | $ | 487 | | | $ | 0 | |
| Reclassification of warrant liabilities to equity | $ | 139 | | | $ | 0 | |
| Warrant liabilities issued to lender in conjunction with 2020 Loan Agreement | $ | 115 | | | $ | 0 | |
| | | |
| | | |
| Payable for purchase of fixed assets | $ | 83 | | | $ | (59) | |
| | | |
| | | |
| | | | | | | | | | | |
| Supplemental disclosure of cash flow information | | | |
| Cash paid for interest | $ | 26,717 | | | $ | 13,090 | |
| Cash paid for income taxes | $ | 3 | | | $ | 4 | |
| | | |
| Supplemental schedule of non-cash investing and financing activities | | | |
| Payable for purchase of fixed assets | $ | 519 | | | $ | — | |
| Fair value of warrants issued to lender in conjunction with February 2022 Amendment (as defined in Note 3) | $ | — | | | $ | 712 | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
The accompanying notes are an integral part of these consolidated financial statements.
Inhibrx, Inc.
Notes to Consolidated Financial Statements
1. ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Organization
Inhibrx, Inc., or the Company, or Inhibrx, is a clinical-stage biotechnologybiopharmaceutical company focused on developing a broad pipeline of novel biologic therapeutic candidates. The Company combines a deep understanding of target biology with innovative protein engineering, proprietary discovery technologies, and an integrative approach to research and development to design highly differentiated therapeutic candidates. The Company’s current pipeline is focused on oncology and orphan diseases.
The Company is subject to risks and uncertainties common to early-stage companies in the biotechnologybiopharmaceutical industry, including, but not limited to, risks associated with preclinical studies, clinical trials and regulatory applications, development by competitors of new technological innovations, dependence on key personnel, protection of proprietary technology, compliance with government regulations and the ability to secure additional capital to fund operations. The Company’s therapeutic candidates currently under development will require significant additional research and development efforts, including clinical and preclinical testing and regulatorymarketing approval prior to commercialization. These efforts require significant amounts of capital, adequate personnel and infrastructure and extensive compliance-reporting capabilities. Even if the Company’s development efforts are successful, it is uncertain when, if ever, the Company will realize significant revenue from product sales.
Initial Public Offering
On August 21, 2020, the Company completed its initial public offering, or IPO, in which it sold 8,050,000 shares of common stock at an offering price of $17.00 per share. Net proceeds from the IPO were $125.9 million, net of underwriting discounts, commissions, and offering costs.
In addition, each of the following occurred in connection with the completion of the IPO:
•the conversion of the 2019 Note and the 2020 Notes (each as defined below) into 4,294,603 shares of the Company’s common stock;
•the conversion of all outstanding convertible preferred stock into 7,211,086 shares of the Company’s common stock;
•the adjustment of an outstanding warrant to purchase convertible preferred stock into a warrant to purchase 7,354 shares of the Company’s common stock; and
•the amendment and restatement of the Company’s certificate of incorporation, authorizing 120.0 million shares of common stock and 15.0 million shares of preferred stock.
Reverse Stock Split
On August 11, 2020 the Company effected a one-for-1.7382 reverse stock split of the Company’s common stock, or the Reverse Stock Split. The par value and the authorized shares of the common stock were not adjusted as a result of the Reverse Stock Split. All issued and outstanding common stock and the conversion prices of the convertible preferred stock and convertible notes have been retroactively adjusted to reflect this Reverse Stock Split for all periods presented.
Basis of Presentation
The accompanying consolidated financial statements have been prepared in conformity with accounting principles generally accepted in the United States of America, or GAAP, and applicable rules and regulations of the Securities and Exchange Commission, or the SEC, related to an annual report on the Form 10-K.
Principles of Consolidation
The consolidated financial statements include the accounts of the Company and its wholly-owned subsidiary and have been prepared in conformity with GAAP. All intercompany accounts and transactions have been eliminated in consolidation.
Liquidity
As of December 31, 2020,2023, the Company had an accumulated deficit of $145.4$613.7 million and cash and cash equivalents of $128.7$277.9 million. From its inception and through December 31, 2020,2023, the Company has devoted substantially all of its efforts to therapeutic drug discovery and development, conducting preclinical studies and clinical trials, enabling manufacturing activities in support of its therapeutic candidates, pre-commercialization activities, organizing and staffing the Company, establishing its intellectual property portfolio and raising capital to support and expand these activities.
WhileIn August 2023, the Company received gross proceeds of $200.0 million before deducting $0.4 million of offering expenses payable by the Company in a private placement transaction, or the Private Placement, with certain institutional and other accredited investors, or Purchasers, in which the Company sold and issued 3,621,314 shares of the Company’s common stock and, with respect to certain Purchasers, pre-funded warrants to purchase 6,714,636 shares of the Company’s common stock. See Note 4 for further discussion of this equity offering.
The Company believes that its existing cash and cash equivalents will be sufficient to fund the Company’s operations for at least 12 months from the date these consolidated financial statements are issued, theissued. The Company plans to finance its future cash needs through equity offerings, debt financings or other capital sources, including potential collaborations, licenses, strategic transactions and other similar arrangements.
If the Company does raise additional capital through public or private equity or convertible debt offerings, the ownership interests of its existing stockholders will be diluted, and the terms of those securities may include liquidation or other preferences that adversely affect its stockholders’ rights. If the Company raises capital through additional debt financings, it may be subject to covenants limiting or restricting its ability to take specific actions, such as incurring additional debt or making certain capital expenditures. To the extent that the Company raises
additional capital through strategic licensing, collaboration or other similar agreement, it may have to relinquish valuable rights to its therapeutic candidates, future revenue streams or research programs at an earlier stage of development or on less favorable terms than it would otherwise choose, or to grant licenses on terms that may not be favorable to the Company. There can be no assurance as to the availability or terms upon which such financing and capital might be available in the future. If the Company is unable to secure adequate additional funding, it will need to reevaluate its operating plan and may be forced to make reductions in spending, extend payment terms with suppliers, liquidate assets where possible, delay, scale back or eliminate some or all of its development programs, or relinquish rights to its technology on less favorable terms than it would otherwise choose. These actions could materially impact its business, financial condition, results of operations and prospects.
The rules and regulations of the SEC or any other regulatory agencies may restrict the Company’s ability to conduct certain types of financing activities, or may affect the timing of and amounts it can raise by undertaking such activities.
Use of Estimates
The preparation of these consolidated financial statements in conformity with GAAP requires the Company to make estimates and judgments that affect the reported amounts of assets, liabilities, revenue and expense and the disclosure of contingent assets and liabilities in the Company’s financial statements and accompanying notes. The Company’s most significant estimates relate to evaluation of embedded derivative instruments and beneficial conversion features within convertible notes, whether revenue recognition criteria have been met, accounting for development work and preclinical studies and clinical trials, determining the assumptions used in measuring stock-based compensation, the incremental borrowing rate estimated in relation to the Company’s operating lease, and valuation allowances for the Company’s deferred tax assets. The Company bases its estimates on historical experience, known trends and other market-specific or other relevant factors that it believes to be reasonable under the circumstances. Estimates are periodically reviewed in light of changes in circumstances, facts and experience. Changes in estimates are recorded in the period in which they become known. The Company’s actual results may differ from these estimates under different assumptions or conditions.
Fair Value of Financial Instruments
The Company’s financial instruments consist principally of accounts receivable, investments in debt securities, accounts payable, accrued expense, its long-term debt, convertible notes, warrants, and derivative instruments as applicable.warrants. The carrying amounts of financial instruments such as accounts receivable, accounts payable, accrued expense, and convertible notesinvestments in debt securities classified as cash equivalents approximate their related fair values due to the short-term nature of these instruments. The carrying value of the Company’s debt approximates fair value due to its interest being reflective of current market rates for debt with similar terms and conditions. The Company’s derivative instruments and warrants are equity-classified and carried at the instruments’ fair value based on unobservable market inputs.upon classification into equity.
Cash and Cash Equivalents
Cash and cash equivalents are comprised of cash held in financial institutions including readily available checking, overnight sweep and money market accounts.accounts, and highly liquid investments in debt securities with an original maturity of three months or less. The Company’s investments in debt securities have consisted of U.S. Treasury Bills, which were recorded at their amortized cost, reflective of the amortization or accretion of premiums or discounts.
December 31, 2023, the Company held no investments in debt securities. As of December 31, 2022, these investments were recorded at their amortized cost of $196.3 million, which was adjusted for the amortization or accretion of premiums or discounts. Concentrations of Credit Risk
Financial instruments that subject the Company to significant concentrations of credit risk consist primarily of cash and cash equivalents. The Company maintains deposits in federally insured financial institutions in excess of federally insured limits.limits by the Federal Deposit Insurance Corporation, or FDIC, of up to $250,000. The Company’s cash management and investment policy limits investment instruments to investment-grade securities with the objective to preserve capital and to maintain liquidity until the funds can be used in operations. The Company has
not experienced any losses in such accounts and believes it is not exposed to significant risk on its cash balances due to the financial condition of the depository institutioninstitutions in which those deposits are held.
The Company continually evaluates its accounts receivable for all outstanding third-party balances to determine the potential exposure to a concentration of credit risk. The Company’s major third-party contracting parties, some of which account for significant balances in both accounts receivable and revenue, are generally large, credit-worthy biotechnology companies and government bodies. The Company assesses the collectability of accounts receivable through a review of its current aging, as well as an analysis of its historical collection rate, general economic conditions, and credit status of these third parties. As of December 31, 20202023 and 2019,December 31, 2022, all outstanding accounts receivable were deemed to be fully collectible, and therefore, no allowance for doubtful accounts was recorded.
The Company currently depends on third-party suppliers for key materials and services used in its research and development, as well as manufacturing processes, and is subject to certain risks related to the loss of these third-party suppliers or their inability to supply the Company with adequate materials and services. In August 2018, the Company entered into a strategic alliance with WuXi Biologics (Hong Kong), Limited, or WuXi Biologics, pursuant to which the Company agreed, for three years and subject to certain conditions, to exclusively use WuXi Biologics to manufacture its therapeutic candidates on a project-by-project basis. The Company does not control the manufacturing processes of the contract development and manufacturing organizations, or CDMOs, with whom it contracts, including WuXi Biologics, and is dependent on these third parties for the production of its therapeutic candidates in accordance with relevant regulations (such as current Good Manufacturing Practices, or cGMP, which includes, among other things, quality control, quality assurance and the maintenance of records and documentation.
Dividends
As of December 31, 2020,2023, the Company has never declared or paid any dividends on its common stock.
Additionally, the Amended 2020 Loan Agreement limits, among other things, the Company’s ability to pay dividends and make certain other payments. Any future determination to pay dividends on the Company’s common stock will be at the discretion of the Company’s board of directors and will depend upon, among other factors, the results of operations, financial condition, capital requirements, contract restrictions, business prospects and other factors the Company’s board of directors may deem relevant.
Property and Equipment, Net
Property and equipment are stated at cost less accumulated depreciation. Depreciation is provided on a straight-line basis over the estimated useful lives of the assets. The Company estimates useful lives as follows:
•machinerylaboratory and office equipment: three to five years;
•furniture, fixtures and fixtures: three toother: five years; and
•computer software: four to fivethree years.
Amortization of leasehold improvements is provided on a straight-line basis over the shorter of their estimated useful lives or the lease term. The costs of additions and betterments are capitalized, and repairs and maintenance costs are expensed in the periods incurred.
Impairment of Long-Lived Assets
The Company reviews long-lived assets for impairment whenever events or circumstances indicate that the carrying amount of an asset may not be recoverable. An impairment loss is recognized when estimated undiscounted future cash flows expected to result from the use of the asset and its eventual disposition are less than its carrying amount. If such assets are considered impaired, the amount of the impairment loss recognized is measured as the amount by which the carrying value of the asset exceeds the fair value of the asset, fair value being determined based upon future cash flows or appraised values, depending on the nature of the asset. The Company recognized no impairment losses during any of the periods presented within its consolidated financial statements.
Leases
The Company has 2two existing leases for its corporate headquarters in La Jolla, California. Its first lease commenced in June 2018. In May 2019, the Company signed an amendment to its lease agreement to expand the Company’s facilities, which commenced in January 2020 and is accounted for as a separate lease. The Company’s 2two existing leases are classified as operating leases.
For the long-term operating lease of its corporate headquarters, the Company recognized aan operating right-of-use asset and lease liability on its consolidated balance sheet. The lease liability is determined as the present value of future lease payments using an estimated incremental borrowing rate that the Company would have to pay to borrow equivalent funds on a collateralized basis at the lease commencement date. The operating right-of-use asset is based on the liability adjusted for any prepaid or deferred rent. The lease term at the commencement date is determined by considering whether renewal options and termination options are reasonably assured of exercise. Agreements are
reviewed at inception to determine if they contain a lease. Leases are reviewed and classified as operating or financing leases at commencement.
The Company has elected to exclude from its consolidated balance sheets recognition of leases having a term of 12 months or less (short-term leases) and has elected not to separate lease components and non-lease components for its long-term operating leases.
Rent expense for the operating leases are recognized on a straight-line basis over the lease term and is included in operating expense in the consolidated statements of operations for all periods presented.
Equity Method Investment in Phylaxis
The Company uses the equity method of accounting for equity investments in companies if the investment provides the ability to exercise significant influence, but not control, over operating and financial policies of the investee. As discussed in Note 8,6, the Company received an equity investment in the form of a 10% equity interest as consideration in a series of agreements with Phylaxis (as defined below), which was later increased to 15% in the fourth quarter of 2023 following the achievement of a milestone. This equity interest is accounted for as an equity method investment. The Company'sinvestment and the Company’s proportionate share of the net income or loss of these companiesPhylaxis is included as loss in equity method investment in the consolidated statement of operations. Judgment regarding the level of influence over each equity method investment includes considering key factors such as the Company’s ownership interest, representation on the board of directors, legal form of the investee (e.g. limited liability corporation), participation in policy-making decisions, and material purchase and sale transactions. During
The investment had been reduced to zero prior to the third quarterbeginning of 2020, the Company’s equity method investment was written down to 02021 as a result of the allocation of the Company’s sharesshare of prior losses of the investee. AsFollowing the Company’s increase in equity interest of 5% in the fourth quarter of 2023, the Company established an additional equity method investment and subsequently recorded its proportionate loss as loss in equity method investment in the consolidated statement of operations. Accordingly, the Company’s investment in Phylaxis as of December 31, 2020, the equity method investment remains at 0.
Derivative Liabilities
As discussed in Note 5, the convertible promissory note, or the 2019 Note, issued in May 2019 to DRAGSA 50 LLC, an entity affiliated with Viking Global Investors LP, or Viking,2023 and the convertible promissory notes, or the 2020 Notes, issued in April 2020 to Viking and certain other investors contained embedded features that provided multiple settlement alternatives. Certain of these settlement features provided the noteholders with a right to a fixed number of the Company’s shares upon conversion of the note. Other settlement features provided the noteholders the right or the obligation to receive cash or a variable number of shares upon the completion of a capital raising transaction, change of control, qualified IPO (as defined in the 2019 Note and the 2020 Notes), or default of the Company. The Company evaluated each settlement alternative within the 2019 Note and the 2020 Notes under the Financial Accounting Standards Board, or FASB, Accounting Standards Codification, or ASC, Topic 815-15, Embedded Derivatives and ASC Topic 815-40, Contracts in Entity’s Own Equity and determined certain of the settlement features would be accounted for as redemption features meeting the requirements for separate accounting as a single, compound derivative instrument.
This single, compound derivative instrument was accounted for as a liability at its estimated fair value as of the date of issuance, and then subsequently remeasured to fair value as of each balance sheet date, with the related remeasurement adjustment being recognized as a component of change in fair value of derivative liabilities in the consolidated statements of operations. During August 2020, the Company consummated its IPO, resulting in the
conversion of the 2019 Note and the 2020 Notes into shares of the Company’s common stock. As of December 31, 2020 the derivative liabilities no longer exist.
Warrant Liabilities
As discussed in Note 3, in conjunction with the 2020 Loan Agreement with Oxford, the Company issued warrants for the purchase of: 1) its preferred stock if the Company remained privately-held or 2) common stock upon the successful completion of an IPO prior to December 31, 2020. Upon the issuance of the warrants in July 2020, pursuant to ASC Topic 480, the warrants were accounted for as liabilities and measured at their estimated fair value as of the date of issuance, based on the potential conversion into preferred stock. During August 2020, upon consummation of the Company’s IPO, the warrants were deemed to be issued for the purchase of common stock and were no longer accounted for as liabilities. The change in fair value was recognized as a component of change in fair value of warrant liabilities in the consolidated statements of operations during the year ended December 31, 2020.2022 is zero.
Fair Value Measurements
The Company determines the fair value measurements of applicable assets and liabilities based on a three-tier fair value hierarchy established by accounting guidance and prioritizes the inputs used in measuring fair value. These tiers include:
•Level 1 - Quoted prices in active markets for identical assets or liabilities.
•Level 2 - Inputs other than Level 1 that are observable, either directly or indirectly, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities.
•Level 3 - Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities.
In certain cases, the inputs used to measure fair value may fall into different levels of the fair value hierarchy. In such cases, the level in the fair value hierarchy within which the fair value measurement in its entirety falls has been determined based on the lowest level input that is significant to the fair value measurement in its entirety. The Company’s assessment of the significance of a particular input to the fair value measurement in its entirety requires judgment and considers factors specific to the asset or liability.
The 2019 NoteDuring the years ended December 31, 2023 and December 31, 2022, the Company’s investments in debt securities consisted of U.S. Treasury Bills, which are classified as Level 1 in the fair value hierarchy. Due to the short-term nature of these securities which are classified as cash equivalents, the amortized value approximated fair value and the 2020 Notes contained embedded derivative liabilities (see Note 5). The 2020 Loan Agreement contained warrant liabilities (see Note 3). Prior to the completion of the IPO in August 2020,Company did not remeasure these were classified within Level 3 of the hierarchy, since they were valued by using inputs that were unobservable in the market. Upon the consummation of the IPO in August 2020 and asinstruments at fair value. As of December 31, 2020,2023, the derivative liabilities and warrant liabilitiesCompany held no longer existed.
investments in debt securities. The following table presentsCompany’s outstanding debt is classified as Level 2 in the Company’s financial liability that was measured at fair value ashierarchy. As of December 31, 2019 (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | |
| LEVEL 1 | | LEVEL 2 | | LEVEL 3 | | TOTAL |
| | | | | | | |
| Derivative liability | $ | 0 | | | $ | 0 | | | $ | 1,916 | | | $ | 1,916 | |
| | | | | | | |
| | | | | | | |
Prior to2023 and December 31, 2022, the completion of the IPO in August 2020, the Company’s liabilities that wereCompany had no financial instruments measured at fair value on a recurring basis using significant unobservable inputs (Level 3) included the derivative liabilities related to the 2019 Note and the 2020 Notes and the warrants issued with the 2020 Loan Agreement. The following table provides abasis.
reconciliation of financial instruments measured at fair value using Level 3 unobservable inputs for the year ended December 31, 2020 (in thousands):
| | | | | |
| |
| |
| |
Balance as of December 31, 2019 | $ | 1,916 | |
Establishment of derivative liability on the 2020 Notes | 735 | |
Establishment of warrant liabilities on the 2020 Loan Agreement | 115 | |
Change in fair value of derivative liabilities on the 2019 and 2020 Notes | (2,651) | |
Change in fair value of warrant liabilities | 24 | |
Reclassification of warrant liabilities into equity | (139) | |
Balance as of December 31, 2020 | $ | 0 | |
The fair value of the Company’s derivative liabilities categorized in Level 3 of the fair value hierarchy as of December 31, 2019 was determined, with the assistance of an independent third-party valuation specialist, using a lattice model. The valuation utilized unobservable inputs, including the discount rate, management’s assessment of the probability of various underlying events triggering the conversion of the Company’s 2019 Note and the 2020 Notes (as discussed in Note 5), such as an IPO, financing, or extraordinary event, and the current valuation of the Company determined with the assistance of an independent third-party valuation specialist. The warrant liabilities were valued using a Black-Scholes model upon issuance. As of December 31, 2020, the derivative liabilities no longer exist as a result of the conversion of the 2019 Note and the 2020 Notes upon the IPO. Additionally, as of December 31, 2020 the warrant liabilities have been reclassified into equity after the Oxford warrants became exercisable for common stock subsequent to the Company’s IPO.
Deferred Financing Costs and Other Debt-Related Costs
Deferred financing costs are capitalized, recorded as an offset to the Company’s debt balances and amortized as interest expense over the term of the associated debt instrument using the effective interest method, pursuant to ASC Topic 835-30, Imputation of Interest. If the maturity of the debt is accelerated as a result of default or early debt repayment, the amortization would then be accelerated. Amounts paid related to debt financing activities are presented on the consolidated balance sheet as a direct deduction from the debt liability.
Deferred Offering Costs
The Company capitalizes costs that are directly associated with equity financings until such financings are consummated at which time such costs are recorded against the gross proceeds of the offering. Should an in-process equity financing be abandoned, the deferred offering costs will be expensed immediately as a charge to operating expenses in the statement of operations.
Legal, accounting, and filing fees related directly to the Company’s IPO wereShelf Registration are capitalized as deferred IPOoffering costs. Deferred offering costs associated with the Shelf Registration are reclassified to be offset against proceeds uponadditional paid-in capital when the consummation ofCompany completes offerings under the IPO within stockholders’ equity (deficit).Shelf Registration. In August 2020,October 2022, the Company completed its IPOan offering under the Shelf Registration and offset $11.0$3.0 million of IPOoffering costs against the proceeds. AsIn August 2023, the Company completed a private placement offering, under which the Company offset $0.4 million of offering costs against the proceeds. There were no deferred offering costs as of December 31, 20202023 or December 31, 2022.
Financial Instruments with Characteristics of Both Liabilities and 2019, no deferred IPO costs were capitalizedEquity
The Company accounts for issued warrants either as a liability or equity in accordance with ASC 480-10, Accounting for Certain Financial Instruments with Characteristics of both Liabilities and Equity, or ASC 480-10, and ASC 815-40, Accounting for Derivative Financial Instruments Indexed to, and Potentially Settled in, a Company’s Own Stock, or ASC 815-40. Under ASC 480-10, warrants are considered a liability if they are mandatorily redeemable and they require settlement in cash, other assets, or a variable number of shares. If warrants do not meet liability classification under ASC 480-10, the Company considers the requirements of ASC 815-40 to determine whether the warrants should be classified as a liability or as equity. Under ASC 815-40, contracts that may require settlement for cash are liabilities, regardless of the probability of the occurrence of the triggering event. Liability-classified warrants are measured at fair value on the issuance date and at the end of each reporting period. Any change in the fair value of the warrants after the issuance date is recorded in other expense, net in the consolidated statements of operations as a gain or loss. If warrants do not require liability classification under ASC 815-40, in order to conclude warrants should be classified as equity, the Company assesses whether the warrants are indexed to its common stock and whether the warrants are classified as equity under ASC 815-40 or under another applicable GAAP standard. Equity-classified warrants are accounted for at fair value on the issuance date with no changes in fair value recognized after the issuance date. The Company’s consolidated balance sheets. Duringoutstanding warrants do not meet the yearrequirements for liability classification under ASC-480-10 or ASC-815-40. Therefore, the Company’s outstanding warrants are classified as equity as of and for the years ended December 31, 2019, the Company recorded $2.8 million of abandoned offering costs on its consolidated statement of operations.2023 and December 31, 2022.
Revenue Recognition
To date, theThe Company has generated revenue from its license and collaboration agreements with partners, as well as from grants from government agencies and private not-for-profit organizations. Payments received from customers are included in deferred revenue, allocated between current and non-current on the consolidated balance sheet until all revenue recognition criteria are met. In accordance with ASC Topic 606, Revenue from Contracts with Customers, or ASC Topic 606, the Company recognizes revenue when, or as, the promised goods or services are transferred to customers in an amount that reflects the consideration to which it expects to be entitled in exchange for those services. To determine revenue recognition for arrangements the Company concludes are within the scope of ASC Topic 606, the Company performs the following five steps: (1) identify the contract(s) with a customer; (2) identify the performance obligation(s) in the contract; (3) determine the transaction price; (4) allocate the transaction price to the performance obligation(s) in the contract; and (5) recognize revenue when (or as) the
performance obligation(s) are satisfied. At contract inception, the Company assesses the goods or services promised within each contract, assesses whether each promised good or service is distinct and identifies those that are performance obligations. The Company recognizes as revenue the amount of the transaction price that is allocated to the respective performance obligation when, or as, the performance obligation is satisfied.
Collaborative Research, Development, and License Agreements
The Company enters into collaborative agreements with partners that typicallywhich may include the transfer of licenses, options to license, and the performance of research and development activities. The terms associated with these agreements may include one or more of the following:following (1) license fees; (2) nonrefundable up-front fees; (3) payments for reimbursement of research costs; (4) payments associated with achieving specific development, regulatory, or commercial milestones; and (5) royalties based on specified percentages of net product sales, if any. AtPayments received from customers are included in deferred revenue, allocated between current and non-current on the initiationconsolidated balance sheet, until all revenue recognition criteria are met.
Typically, license fees, non-refundable upfront fees, and funding of an agreement,research activities are considered fixed, while milestone payments, including option exercise fees, are identified as variable consideration, which is constrained
and excluded from the Company analyzes whether each unit of account results in a contract with a customer under ASC Topic 606 or in an arrangement with a collaborator subject to guidance under ASC Topic 808, Collaborative Arrangements, or ASC Topic 808. The Company’s licensing arrangements are typically for functional intellectual property as it exists at a point in time, being the time that the license agreement is executed.transaction price. The Company typically does not have an ongoing performance obligation to support or maintain the licensed intellectual property.will recognize revenue for sales-based royalty if and when a subsequent sale occurs.
The Company considers a variety of factors in determining the appropriateapplies significant judgment when making estimates and assumptions under these arrangements, such asagreements, including evaluating whether the elements arecontractual obligations represent distinct performance obligations, including the assessment of whether options represent material rights, determining whether there are observable stand-alonestandalone prices and allocating transaction price to performance obligations within a contract, assessing whether any licenses are functional or symbolic.symbolic, determining when performance obligations have been met, and assessing the recognition of variable consideration. The Company evaluates each performance obligation to determine if it can be satisfied and recognized as revenue at a point in time or over time. Typically, performance obligations consisting of a transfer of a license fees, non-refundable upfront fees, and fundingor the achievement of milestones are recognized at a point in time upon the transfer, while performance obligations consisting of research activities are considered fixed, while milestone payments are identified as variable considerationrecognized over time using an input method which must be evaluated to determine if it is constrained and, therefore, excluded from the transaction price. The Company estimates the amount of variable consideration using the most likely amount, as milestone payments typically only have two possible outcomes. The Company recognizes revenue for sales-based royalty promised in exchange for the license of intellectual property only when the subsequent sale occurs. In the case of an agreement which provides the partner with an option to license a therapeutic or therapeutic candidate in the future, the Company evaluates whether this option represents a material right at the inceptionrepresentative of the agreement. If determinedCompany’s efforts to be a material right,fulfill the Company will consider the option a separate performance obligation. The Company has historically concluded that the option to grant a license in the future are not material rights as they are contingent upon future events which may not occur. When an option is exercised, the Company will identify any separate performance obligations.
The Company may allocate transaction price using a number of methods including estimating standalone selling price of performance obligations and using the residual approach when the standalone selling price of the license is highly variableobligation, based on costs incurred with third-parties or uncertain, and observable standalone selling prices exist for the other goods or services promised in the contract.internal labor hours performed.
Grant Revenue
Since inception, theThe Company has been awarded multiple grants from the National Institutes of Health, or NIH, onea grant from the Congressionally Directed Medical Research Program, or CDMRP, funded through the DepartmentDoD, under which the Company’s internal costs specifically covered by the grant for a specified project are subject to reimbursement when incurred. Since there is no transfer of Defense,control of goods or DoD,services to the granting agency, revenue is recognized as well as one grant from Combating Antibiotic Resistant Bacteria Biopharmaceutical Accelerator, or CARB-X. With respect to revenue derived from reimbursement of direct, out-of-pocket expenses for research and development costs associated with these grants, where the Company acts as a principal with discretionincurs expenses related to choose suppliers, bears credit risk, and performs part of the services requiredgrant in the transaction, the Company records revenue for the gross amount of the reimbursement. Thereimbursement and costs associated with these reimbursements are reflected as a component of research and development expense in the consolidated statements of operations.
Since there is no transfer of control of goods or services to the granting entities, the granting entities do not meet the definition of a “customer” as defined by ASC Topic 606 and therefore, these government grants and private not-for-profit institution grants are not within the scope of ASC Topic 606. Since the Company has concluded that these grants do meet the definition of a contribution and are indeed non-reciprocal transactions, the adoption of ASC Topic 606 had no impact on the Company’s accounting for grant revenue from governmental agencies or private institutions not considered customers.
Revenue from these grants are based upon internal costs incurred that are specifically covered by the grant, plus an additional rate that provides funding for overhead expenses. Revenue is recognized as the Company incurs expenses related to the grant which is consistent with the concept of transfer of control of a service over time under ASC Topic 606.
Accrued Research and Development and Clinical Trial AccrualsCosts
Research and development costs are expensed as incurred based on estimates of the period in which services and efforts are expended, and include the cost of compensation and related expenses, as well as expenses for third parties who conduct research and development on the Company’s behalf, pursuant to development and consulting agreements in place. The Company’s preclinical studies and clinical trials are performed internally, by third party contract research organizations, or CROs, and/or clinical investigators. The Company also engages with contract development and manufacturing organizations, or CDMOs, for clinical supplies and manufacturing scale-up activities related to its therapeutic candidates. Invoicing from these third parties may be monthly based upon services performed or based upon milestones achieved. The Company accrues these expenses based upon its assessment of the status of each clinical trial and the work completed, and uponestimates determined by reviewing cost information obtained from the CROs and CDMOs. The Company’s estimates are dependent upon the timeliness and accuracy of data provided by the CROs and CDMOs, regardingother third-party vendors and internal clinical personnel, and contractual arrangements with CROs and CDMOs and the status and costscope of the studies.work to be performed. Costs incurred related to the Company’s purchases of in-process research and development for early-stage products or products that are not commercially viable and ready for use, or have no alternative future use, are charged to expense in the period incurred. Costs incurred related to the licensing of products that have not yet received regulatorymarketing approval to be marketed, or that are not commercially viable and ready for use, or have no alternative future use, are charged to expense in the period incurred.
Income Taxes
Income taxes are accounted for under the asset and liability method. Deferred income taxes are recorded for temporary differences between financial statement carrying amounts and the tax basis of assets and liabilities. Deferred tax assets and liabilities reflect the tax rates expected to be in effect for the years in which the differences are expected to reverse. A valuation allowance is provided if it is more likely than not that some or all of the deferred tax assets will not be realized.
The Company also follows the provisions of accounting for uncertainty in income taxes which prescribes a model for the recognition and measurement of a tax position taken or expected to be taken in a tax return, and provides guidance on derecognition, classification, interest and penalties, disclosure and transition.
The utilization of unused federal and state net operating losses, or NOLs, and research tax credit carryforwards to offset future taxable income may be subject to an annual limitation as a result of ownership changes that have occurred previously or may occur in the future. Under Sections 382 and 383 of the Internal Revenue Code, as
amended, or IRC, a corporation that undergoes an “ownership change” may be subject to limitations on its ability to utilize its pre-change NOLs and other tax attributes otherwise available to offset future taxable income and/or tax liability. An ownership change is defined as a cumulative change of 50% or more in the ownership positions of certain stockholders during a rolling three-year period. The Company has not completedis in the process of completing a formal study to determine if any ownership changes within the meaning of IRC Section 382 and 383 have occurred.had occurred and, pending finalization, has not identified any ownership changes as of December 31, 2023. It is possible that the Company has already incurred ownership changes and may incur additional ownership changes in the future. If an ownership change has occurred,occurs, the Company’s ability to use its NOL or tax credit carryforwards may be restricted, which could require the Company to pay federal or state income taxes earlier than would be required if such limitations were not in effect.
Net Loss Per Share
Basic net loss per share is computed by dividing net loss by the weighted average number of common sharesstock outstanding during the same period. Diluted net loss per share is computed by dividing net loss by the weighted average number of common and common equivalent sharesstock equivalents outstanding during the same period. The Company excludes common stock equivalents from the calculation of diluted net loss per share when the effect is anti-dilutive.
The weighted average number of common stock used in the basic and diluted net loss per common stock calculations includes the weighted-average pre-funded warrants outstanding during the period as they are exercisable at any time for nominal cash consideration.
For purposes of the diluted net loss per share calculation, the convertible preferred stock, convertible notes,other than pre-funded warrants as discussed above, warrants for purchase of common stock and stock options are considered to be potentially dilutive securities. Basic and diluted net loss attributable to common stockholders per share is presented in conformity with the two class method
required for participating securities as the convertible preferred stock is considered a participating security. The Company’s participating securities do not have a contractual obligation to share in the Company’s losses. As such, the net loss was attributed entirely to common stockholders. Accordingly, for the years ended December 31, 20202023 and 2019,December 31, 2022, there is no difference in the number of shares used to calculate basic and diluted shares outstanding.
The followingPotentially dilutive securities that could potentially decreasenot included in the calculation of diluted net loss per share, were not included inbecause to do so would be anti-dilutive, as weighted based on the determination of diluted loss per shareperiod outstanding during each year, are as their effect is anti-dilutivefollows (in thousands):
| | | | | | | | | | | | | | | |
| YEAR ENDED DECEMBER 31, | | |
| 2020 | | 2019 | | | | |
| | | |
| Shares issuable upon conversion of convertible preferred stock | 0 | | | 7,211 | | | | | |
| Outstanding stock options | 2,525 | | | 2,012 | | | | | |
Convertible note (1) | 0 | | | 2,787 | | | | | |
| Warrant to purchase common stock | 7 | | | 0 | | | | | |
| 2,532 | | | 12,010 | | | | | |
(1)The conversion of the convertible notes into common stock as of December 31, 2019 assumes a conversion price of $14.35 per share for the 2019 Note and $12.56 per share for the 2020 Notes, and includes the conversion of the principal balance and all accrued interest as of December 31, 2019 (see Note 5). | | | | | | | | | | | | | | | |
| YEAR ENDED DECEMBER 31, | | |
| 2023 | | 2022 | | | | |
| | | |
| | | | | | | |
| Outstanding stock options | 6,336 | | | 5,018 | | | | | |
| | | | | | | |
| Warrant to purchase common stock | 47 | | | 45 | | | | | |
| 6,383 | | | 5,063 | | | | | |
Fair Value of Stock-Based Awards
The Company recognizes compensation costs related to stock options based on the estimated fair value of the awards on the date of grant. The Company estimates the grant date fair value, and the resulting stock-based compensation expense, using the Black-Scholes option pricing model. The grant date fair value of the stock-based awards is generally recognized on a straight-line basis over the requisite service period, which is generally the vesting period of the respective awards. The Company recognizes forfeitures as they occur.
Other Comprehensive Income
The Company has no material components of other comprehensive loss and accordingly, net loss is equal to comprehensive loss in all periods presented.
Segment Information
The Company operates under 1one segment which develops biologic therapeutic candidates. Operating segments are identified as components of an enterprise about which separate discrete financial information is available for evaluation by the chief operating decision-maker in making decisions regarding resource allocation and assessing performance.
Recent Accounting Pronouncements
From time to time, new accounting pronouncements are issued by the Financial Accounting Standards Board, or FASB, or other standard setting bodies. Unless otherwise stated, theThe Company believes that the impact of the recently issued accounting
pronouncements that are not yet effective will not have a material impact on its consolidated financial condition or results of operations upon adoption. The Company has irrevocably elected not to take advantage of the extended transition period afforded by the Jumpstart Our Business Startups Act of 2012, as amended, or JOBS Act, for the implementation of new or revised accounting standards and, as a result, will comply with new or revised accounting standards on the relevant dates on which adoption of such standards is required for non-emerging growth companies.
Adoption of New Accounting Pronouncements
In August 2018, the FASB issued ASU 2018-13, Fair Value Measurements (Topic 820): Disclosure Framework-Changes to the Disclosure Requirements for Fair Value Measurements, which includes amendments intended to improve the effectiveness of disclosure requirements for recurring and nonrecurring fair value measurements. The standard removes, modifies, and adds certain disclosure requirements and affects companies that are required to include fair value measurement disclosures. The amendments in ASU 2018-13 are effective for fiscal years beginning after December 15, 2019 with early adoption permitted. The Company adopted this guidance as of
January 1, 2020, which did not result in a material impact on its consolidated financial statements and related disclosures.
Accounting Standards Not Yet Adopted
In June 2016, the FASB issued Accounting Standards Update, or ASU, 2016-13,Financial Instruments - Credit Losses: Losses: Measurement of Credit Losses on Financial StatementsInstruments (Topic 362)326), which intends to improve financial reporting by requiring earlier recognition of credit losses on certain financial assets, such as available-for-saleheld-to-maturity debt securities. Subsequent to the issuance of ASU 2016-13, the FASB issued several additional ASUs to clarify implementation guidance, provide narrow-scope improvements and provide additional disclosure guidance. In November 2019, the FASB issued an amendment making thisThe Company adopted ASU effective for fiscal years beginning after December 15, 2022 for smaller reporting companies. The new standard will be effective for the Company on2016-13 as of January 1, 2023, or at such time where it is no longer a smaller reporting company. The Company is currently evaluating the potential impact that the adoption of ASU 2016-13 may have on its consolidated financial statements and related disclosures.
In December 2019, the FASB issued ASU 2019-12, Income Taxes: Simplifying the Accounting for Income Taxes (Topic 740), which simplifies the accounting for income taxes by removing certain exceptions to the general principlesdid not result in Topic 740. ASU 2019-12 also improves consistent application and simplification of other areas of Topic 740 by clarifying and amending existing guidance. ASU 2019-12 is effective for fiscal years beginning after December 15, 2020. The Company is currently evaluating the potential impact that the adoption of ASU 2019-12 may have, and does not currently expect this to have a material impact on its consolidated financial statements and related disclosures.
Recently Issued but Not Yet Adopted Accounting Pronouncements
In January 2020,December 2023, the FASB issued ASU 2020-01,2023-09, Investments-Equity Securities (Topic 321)Improvements to Income Tax Disclosures, Investments-Equity Method and Joint Ventures (Topic 323), and Derivatives and Hedging (Topic 815) - Clarifying the Interactions between Topic 321, Topic 323, and Topic 815, whichaddresses the accounting for the transition into and out of the equity method and provides clarification of the interaction of rules for equity securities, the equity method of accounting, and forward contracts and purchase options requires more detailed income tax disclosures. The guidance requires entities to disclose disaggregated information about their effective tax rate reconciliation as well as expanded information on certain types of securities.income taxes paid by jurisdiction. The amendments clarify that: (a) an entity should consider observable transactions that require it to either apply or discontinue the equity method of accounting for the purposes of applying the measurement alternative in accordance with Topic 321 immediately before applying or upon discontinuing the equity method; and (b) an entity should not consider whether, upon the settlement of the forward contract or exercise of the purchased option, individually or with existing investments, the underlying securities woulddisclosure requirements will be accounted for under the equity method in Topic 323 or the fair value option in accordanceapplied on a prospective basis, with the financial instruments guidance in Topic 825.option to apply them retrospectively. The provisions of this guidance are to be applied prospectively upon their effective date. ASU 2020-01standard is effective for fiscal years beginning after December 15, 2020, and interim periods within those years. Early2024, with early adoption is permitted, but requires simultaneous adoption of all provisions of this guidance.permitted. The Company is currently evaluating the potential impact thatdisclosure requirements related to the adoption of ASU 2020-01 may have, and does not currently expect this to have a material impact on its consolidated financial statements and related disclosures.new standard.
In August 2020,November 2023, the FASB issued ASU 2020-06,2023-07, Debt—Debt with Conversion and Other Options (Subtopic 470-20) and Derivatives and Hedging—Contracts in Entity’s Own EquitySegment Reporting (Topic 280): Improvements to Reportable Segment Disclosures, (Subtopic 815-40): Accountingwhich is intended to improve reportable segment disclosure requirements, primarily through additional disclosures about significant segment expenses, including for Convertible Instruments and Contracts in an Entity’s Own Equity, which simplifies the accounting for certain financial instruments with characteristics of liabilities and equity, including convertible instruments and contracts on an entity’s own equity. Specifically, ASU 2020-06 simplifies accounting for the issuance of convertible instruments by removing major separation models required under current GAAP. In addition, the ASU removes certain settlement conditions that are required for equity contracts to qualify for the derivative scope exception and simplifies the diluted earnings per share (EPS) calculation in certain areas. ASU 2020-06 will besingle reportable segment entities. The standard is effective for fiscal years beginning after December 15, 2023, includingand interim periods within those fiscal years. Early adoption is permitted, beginning in fiscal years which beginbeginning after December 15, 2020, including interim2024, with early adoption permitted. The amendments should be applied retrospectively to all prior periods within those fiscal years. The FASB has specified that an entity should adoptpresented in the guidance as of the beginning of its annual fiscal year.financial statements. The Company is currently evaluating the potential impact thatdisclosure requirements related to the adoption of ASU 2020-06 may have on its consolidated financial statements and related disclosures.new standard.
2. OTHER FINANCIAL INFORMATION
Prepaid Expense and Other Current Assets
Prepaid expense and other current assets were comprised of the following (in thousands):
| | AS OF DECEMBER 31, | |
| AS OF DECEMBER 31, | |
| AS OF DECEMBER 31, | |
| 2023 | |
| 2023 | |
| 2023 | |
Clinical drug substance and product manufacturing (1) | |
Clinical drug substance and product manufacturing (1) | |
Clinical drug substance and product manufacturing (1) | |
Clinical trials (2) | |
Clinical trials (2) | |
Clinical trials (2) | |
| Licenses | |
| Licenses | |
| Licenses | |
Outside research and development services (3) | |
Outside research and development services (3) | |
Outside research and development services (3) | |
| | | AS OF DECEMBER 31, | |
| Other | |
| | 2020 | | 2019 | |
| | Outside research and development services | $ | 2,204 | | | $ | 3,207 | | |
| Other | |
| | | Other | Other | 689 | | | 376 | | |
| Prepaid expense and other current assets | Prepaid expense and other current assets | $ | 2,893 | | | $ | 3,583 | | |
| Prepaid expense and other current assets | |
| Prepaid expense and other current assets | |
(1) Relates primarily to the Company’s usage of third-party CDMOs for clinical and development efforts. See “Accrued Research and Development Clinical Trial Costs” in Note 1 for further discussion of the components of research and development.
(2) Relates primarily to the Company’s prepayments to third-party CROs for management of clinical trials and prepayments for drug supply to be used in combination with the Company’s therapeutics. See “Accrued Research and Development Clinical Trial Costs” in Note 1 for further discussion of the components of research and development.
(3) Relates to the Company’s usage of third parties for other research and development efforts. See “Accrued Research and Development Clinical Trial Costs” in Note 1 for further discussion of the components of research and development.
Property and Equipment, Net
Property and equipment, net were comprised of the following (in thousands):
| | AS OF DECEMBER 31, | |
| 2020 | | 2019 | |
| | AS OF DECEMBER 31, | |
| AS OF DECEMBER 31, | |
| AS OF DECEMBER 31, | |
| 2023 | |
| 2023 | |
| 2023 | |
| Machinery and equipment | Machinery and equipment | $ | 5,652 | | | $ | 4,723 | | |
| Machinery and equipment | |
| Machinery and equipment | |
| Furniture, fixtures and other | |
| Furniture, fixtures and other | |
| Furniture, fixtures and other | |
| Leasehold improvements | Leasehold improvements | 441 | | | 344 | | |
| Furniture and fixtures | 507 | | | 317 | | |
| Construction in process | 257 | | | 229 | | |
| Leasehold improvements | |
| Leasehold improvements | |
| Computer software | |
| Computer software | |
| Computer software | |
Construction in process (1) | |
Construction in process (1) | |
Construction in process (1) | |
| Total property and equipment | |
| Total property and equipment | |
| Total property and equipment | Total property and equipment | 6,857 | | | 5,613 | | |
| Less: accumulated depreciation and amortization | Less: accumulated depreciation and amortization | (3,365) | | | (2,383) | | |
| Less: accumulated depreciation and amortization | |
| Less: accumulated depreciation and amortization | |
| Property and equipment, net | Property and equipment, net | $ | 3,492 | | | $ | 3,230 | | |
| Property and equipment, net | |
| Property and equipment, net | |
(1) As of December 31, 2023, consists of renovations to the Company’s office space and software not yet placed in service.
Depreciation and amortization expense totaled $1.0 million and $1.2 million for each of the years ended December 31, 20202023 and 2019, respectively.December 31, 2022, and consisted of the following (in thousands): | | | | | | | | | | | | | | | |
| | YEAR ENDED DECEMBER 31, | | |
| 2023 | | 2022 | | | | |
| | | | | |
| Research and development | $ | 992 | | | $ | 1,003 | | | | | |
| General and administrative | 199 | | | 222 | | | | | |
| Total depreciation and amortization expense | $ | 1,191 | | | $ | 1,225 | | | | | |
Accrued Expenses
Accrued expenses were comprised of the following (in thousands): | | | | | | | | | | | | | |
| AS OF DECEMBER 31, | | |
| 2023 | | 2022 | | |
| | | | | |
Clinical drug substance and product manufacturing (1) | $ | 22,805 | | | $ | 5,381 | | | |
Clinical trials (2) | 9,224 | | | 4,527 | | | |
| Compensation expense | 6,506 | | | 3,374 | | | |
| Interest expense | 2,348 | | | 2,124 | | | |
Other outside research and development (3) | 1,129 | | | 1,164 | | | |
| Professional fees | 780 | | | 428 | | | |
| Other | 503 | | | 226 | | | |
| Accrued expenses | $ | 43,295 | | | $ | 17,224 | | | |
| | | | | | | | | | | | | |
| AS OF DECEMBER 31, | | |
| 2020 | | 2019 | | |
| | | | | |
| Accrued research and development | $ | 11,529 | | | $ | 3,414 | | | |
| Accrued professional fees | 282 | | | 313 | | | |
| | | | | |
| Accrued compensation expense | 1,165 | | | 813 | | | |
| Other | 381 | | | 292 | | | |
| Accrued expenses | $ | 13,357 | | | $ | 4,832 | | | |
The amount accrued for research and development expense relates(1) Relates primarily to the Company’s increased usage of third-party CROs and CDMOs for clinical and development efforts as its therapeutic candidates progress.efforts. See Note 1 for further discussion of the components of research and development.
(2) Relates primarily to the Company’s usage of third-party CROs for management of clinical trials. See Note 1 for further discussion of the components of research and development.
(3) Relates to the Company’s usage of third parties for other research and development efforts. See “Accrued Research and Development Clinical Trial Costs” in Note 1 for further discussion of the components of research and development.
3. DEBT
The Company’s debt balance under the 2015 Loan Agreement and the Amended 2020 Loan Agreement consisted of the following as of December 31, 2020 and 2019:
2015 Loan Agreement
As of December 31, 2020, the Company had no outstanding balances under the 2015 Loan Agreement. As of March 31, 2020, the Company had repaid its term loans from Oxford under the 2015 Loan Agreement in full, including all principal balances, accrued interest, and the $1.4 million final payment. As of December 31, 2019, the Company’s outstanding debt balance under the 2015 Loan Agreement consisted of the following (in thousands).
| | | | | | | | | |
| | | AS OF DECEMBER 31, | | |
| | | 2019 | | |
Term A | | | $ | 831 | | | |
Term B | | | 893 | | | |
Term C | | | 930 | | | |
Term D | | | 930 | | | |
Add: debt discount | | | (21) | | | |
Total debt | | | 3,563 | | | |
Less: Current portion, including debt discount | | | (3,563) | | | |
Long-term debt, including debt discount | | | $ | 0 | | | |
On March 15, 2015, the Company entered into the 2015 Loan Agreement, pursuant to which Oxford agreed, subject to certain conditions, to make term loans to the Company in 4 $5.0 million installments for an aggregate of $20.0 million. The term loans were made on the following dates: (1) Term A: March 31, 2015; (2) Term B: September 9, 2016; (3) Term C: December 22, 2016; and (4) Term D: December 22, 2016. The proceeds from these loans were designated for working capital and general business purposes.
Pursuant to the terms of the 2015 Loan Agreement, Oxford had a senior-secured lien on all of the Company’s current and future assets, other than its intellectual property. Oxford also had the right to declare the term loan immediately due and payable in an event of default under the 2015 Loan Agreement, which includes, among other things, a material adverse change in the Company’s business, operations, or financial condition or a material impairment in the prospect of repayment of the term loan. Throughout the life of the 2015 Loan Agreement, the Company was in compliance with all covenants under the 2015 Loan Agreement.
Each term loan bore interest at the following annual rate: (1) Term A: 8.0%; (2) Term B: 8.6%; (3) Term C: 8.7%; and (4) Term D: 8.7%. The repayment schedule provided for interest-only payments in arrears until May 2016, followed by consecutive equal monthly payments of principal and interest in arrears through the maturity date, which was March 31, 2020, or the 2015 Loan Maturity Date, for all four term loans. The Company had the option to prepay the outstanding balance of the term loans in full prior to the 2015 Loan Maturity Date, subject to a prepayment fee of up to 3.0%. Upon repayment of each term loan, the Company was also required to make a final payment to Oxford equal to 7.0% of the original principal amount of each term loan. This final payment of $1.4 million was accreted over the life of the 2015 Loan Agreement using the effective interest method.
The Company’s interest expense related to the 2015 Loan Agreement for the years ended December 31, 2020 and 2019, was $0.04 million and $0.7 million, respectively. Interest expense is calculated using the effective interest method and is inclusive of non-cash amortization of the debt discount and accretion of the final payment.
2020 Loan Agreement
OnIn July 15, 2020, the Company entered into a loan and security agreement, or the 2020 Loan Agreement, with Oxford Finance LLC, or Oxford, pursuant to which it received $10.0 million in gross proceeds, net of $0.1 million of debt issuance costs. On November 12,or Term A. The 2020 the Company entered into the Amended 2020 Loan Agreement and received $20.0 million in additional gross proceeds, net of $0.02 million of debt issuance costs and $0.7 million of a one-time first amendment fee. The Amended 2020 Loan Agreement provides for 2 additional tranches of term loans in an aggregate principal amount of up to $40.0 million, as
follows: (a)Agreement was subsequently amended in November 2020, or the November 2020 Amendment, upon which a second tranche in an aggregate principal amount of $20.0 million was funded, upon the closing of the 2020 Loan Amendment, or Term B, and (b)in June 2021, or the June 2021 Amendment, upon which a third tranche in an aggregate principal amount of $20.0$40.0 million was funded, or Term C.
In February 2022, the Company entered into an additional amendment, or the February 2022 Amendment, to fund onthe 2020 Loan Agreement, collectively, the Amended 2020 Loan Agreement, upon which the Company received gross proceeds of $40.0 million, or before September 30, 2021, subjectTerm D. The February 2022 Amendment also provided for an increase in the interest rate and for three future tranches of debt to be funded upon the achievement of certain milestones. In June 2022, the Company received additional gross proceeds of $30.0 million, or Term E, following the initiation of part 4 of the Phase 1 clinical trial of INBRX-105, as well as an additional $30.0 million in gross proceeds, or Term F, following the receipt of positive topline data from the Phase 1 clinical trial of INBRX-101.
In October 2022, the Company entered into an amendment to the Amended 2020 Loan Agreement, or the October 2022 Amendment. The October 2022 Amendment amended the milestone terms of the last remaining tranche, Term G, under the Amended 2020 Loan Agreement to provide for the funding of $30.0 million upon the announcement of the regulatory path for INBRX-101 rather than upon the initiation of a potentially registration-enabling clinical trial of INBRX-101. In October 2022, the Company met this milestone and drew the final tranche for additional gross proceeds of $30.0 million.
The Company determined the Company’s therapeutic candidate, INBRX-109, in unresectable or metastatic conventional chondrosarcoma, pursuant to the termsNovember 2020, June 2021, February 2022, and October 2022 Amendments should be treated as modifications of the 2020 Loan Amendment, or Term C. Term B and Term C are in addition to the first tranche of term loans, or Term A, in an aggregate principal amount of $10.0 million, which remains outstanding following the amendment. The terms of Term A under theoriginal 2020 Loan Agreement since the terms and resulting cash flows were modifiednot substantially changed upon each of the amendments. The Company will continue to align with Term Bamortize the existing debt discounts prior to modification through the Amended Maturity Date (as defined below).
As of July 1, 2023, a LIBOR Transition Event, as defined in the Amended 2020 Loan Agreement, occurred, and pursuant to the Amended 2020 Loan Agreement.Agreement, Oxford selected a LIBOR Replacement Rate, as defined in the Amended 2020 Loan Agreement, replacing the 30 day U.S. Dollar London InterBank Offered Rate with the 1-Month Chicago Mercantile Exchange term secured overnight financing rate, or 1-Month CME Term SOFR, causing an amendment to the interest rate of the Company’s outstanding loans. No other terms were changed. The Company has elected the optional expedient under ASC Topic 848-20 and therefore deemed the modification to not be substantial.
Collectively, asAs of December 31, 20202023, the Company has $30.0$200.0 million in gross principal outstanding in term loans under the Amended 2020 Loan Agreement. The outstanding term loans will mature on NovemberJanuary 1, 2025,2027, or the Amended Maturity Date, and bear interest at a floating per annum rate equal to the greater of (1) 7.96% and8.30% or (2) the sum of (i) the 30 day U.S. Dollar LIBOR rate reported in The Wall Street Journal1 Month CME Term SOFR on the last business day of the month that immediately precedes the month in which the interest will accrue, plus (ii) 7.80%0.10%, and (iii) 8.19%. TheUnder the Amended 2020 Loan Agreement, the repayment schedule provides for interest-only payments until Januarythrough February 1, 2024. Upon funding of Term C, the interest-only payments may be extended by 12 months to January 1, 2025. The interest-only period is2025, followed by 23 months of equal payments of principal and interest payments. In the event of a qualifying financing event in which the Company raises at least $100.0 million in upfront licensing or ifpartnership proceeds by February 2025, the interest-only period ismay be extended an additional 12 months through February 1, 2026, which would then be followed by 11 months of equal payments of principal and interest.plus interest, beginning on March 1, 2026. Upon the Amended Maturity Date, a final payment of 9.0% of the original principal amount will be due to Oxford. This final payment of $2.7$18.0 million will beis being accreted over the life of the Amended 2020 Loan Agreement using the effective interest method. The Company has the option to prepay the outstanding balance of the term loans in full prior to the Amended Maturity Date, subject to a prepayment fee ranging from 1.0% to 3.0%, depending onupon the timing of the prepayment. All other terms of the original 2020 Loan Agreement remain outstanding.
As of December 31, 2020, the
The Company’s outstanding debt balance under the Amended 2020 Loan Agreement consisted of the following as of December 31, 2023 and December 31, 2022 (in thousands). | | | | | | | | | | | |
| AS OF DECEMBER 31, |
| 2023 | | 2022 |
| Term A | $ | 10,900 | | | $ | 10,900 | |
| Term B | 21,800 | | | 21,800 | |
| Term C | 43,600 | | | 43,600 | |
| Term D | 43,600 | | | 43,600 | |
| Term E | 32,700 | | | 32,700 | |
| Term F | 32,700 | | | 32,700 | |
| Term G | 32,700 | | | 32,700 | |
| Less: debt discount | (11,032) | | | (15,931) | |
| Long-term debt, including debt discount and final payment fee | $ | 206,968 | | | $ | 202,069 | |
| | | | | |
| AS OF DECEMBERAs of December 31, |
| 2020 |
Term A | $ | 10,900 | |
Term B | 21,800 | |
Add: debt discount | (3,456) | |
Total debt | 29,244 | |
Less: Current portion, including debt discount | 0 | |
Long-term debt, including debt discount | $ | 29,244 | |
Through 2023, and unless extended in accordance with the Company is only required to make interestterms described above, the Company’s interest-only period will continue through February 2025, with principal payments on the outstanding principal balance, with futurebeginning in March 2025. Future principal payments and final fee payments beginning in 2024,will be made as follows (in thousands):
| | | | | |
| AS OF DECEMBER 31, |
| 2020 |
| |
| |
| |
| 2024 | $ | 15,652 | |
| 2025 | 17,048 | |
| Total future minimum payments | 32,700 | |
| Less: unamortized debt discount | (3,456) | |
| Total debt | $ | 29,244 | |
In November 2020, the Company evaluated the amendment and determined it should be treated as a modification of the original 2020 Loan Agreement since the terms and resulting cash flow were not substantially changed in the Amended 2020 Loan Agreement. The Company will continue to amortize the existing debt discounts under the original 2020 Loan Agreement through the Amended Maturity Date.
| | | | | |
| AS OF DECEMBER 31, 2023 |
| 2025 | $ | 86,956 | |
| 2026 | 104,348 | |
| 2027 | 26,696 | |
| Total future minimum payments | 218,000 | |
| Less: unamortized debt discount | (11,032) | |
| Total debt | $ | 206,968 | |
The Company’s obligations under the Amended 2020 Loan Agreement are secured by a first priority security interest of substantially all of the Company’s assets other than itswith a positive lien on intellectual property. The Amended 2020 Loan Agreement includes customary events of default, including instances of a material adverse change in the Company’s operations, that may require prepayment of the outstanding term loans. Additionally, following the June 2021 Amendment, the Amended 2020 Loan Agreement requires the Company to maintain a minimum cash balance of $20.0 million. As of December 31, 2020,2023, the Company is in compliance with all covenants under the Amended 2020 Loan Agreement and has not received any notification or indication from Oxford of an intent to declare the loan due prior to maturity.
Concurrently with the debt issuance in July 2020,February 2022 Amendment, the Company issued 40,000 warrants to Oxford warrants to purchase shares of the Company’s capitalcommon stock equal to 1.25%at an exercise price of the funded amount, or $125,000.$45.00. Upon issuance, the warrants were exercisable for preferred stock and were classified as liabilities pursuant to Topic ASC 480equity and recorded at their fair value of $0.12$0.7 million. Upon the consummationSee Note 4 for further discussion of the Company’s IPO in August 2020, the Company remeasured the warrants and determined a fair value of approximately $0.14 million. The change in fair value was recorded as a change in fair value of warrant liabilities within other income in the Company’s consolidated statements of operations. As of December 31, 2020, the warrants are equity-classified and are exercisable for 7,354 shares of common stock of the Company at $17.00 per share.these warrants.
The Company’s interest expense related to the Amended 2020 Loan Agreement with Oxford for the year ended December 31, 2020 was $0.9 million. Interest Expense
Interest expense is calculated using the effective interest method and is inclusive of non-cash amortization of the debt discount and accretion of the final payment.
4. PPP LOAN
On March 27, 2020, the U.S. federal government enacted the Coronavirus Aid, Relief, and Economic Security Act, or CARES Act. The CARES Act includes a provision for a Paycheck Protection Program, or PPP, administered by the U.S. Small Business Administration, or SBA, and further amended by the Paycheck Protection Program Flexibility Act of 2020, or PPP Flexibility Act, which was enacted on June 5, 2020. See Note 13 for further details regarding other provisions of the CARES Act.
Under the CARES Act, the Company was approved for a loan pursuant to the PPP, or the PPP Loan, in the amount of $1.9 million and received the funds on May 11, 2020. The application for these funds required the Company to certify in good faith that current economic uncertainty made the loan request necessary to support the ongoing operations of the Company. The Company was also required to certify that the loan funds would be used to retain workers and maintain payroll or make mortgage payments, lease payments, and utility payments. The PPP Loan had a two-year term and bore interest at a rate of 1.0% per annum.
Under the terms of the CARES Act, the Company was eligible to apply for and be granted forgiveness for all or a portion of the PPP Loan. Such forgiveness, if any, would have been determined, subject to limitations, based on the use of loan proceeds for payroll costs, rent and utility costs and provided that only a portion of the use of proceeds are for non-payroll costs. The unforgiven portion of the PPP Loan was eligible be repaid by the Company at any time prior to maturity with no prepayment penalty.
Subsequent to its IPO in August 2020 with net proceeds of $125.9 million, the Company determined that it would not seek forgiveness of the PPP Loan, and on October 2, 2020 repaid the loan in full plus all interest accrued during the period outstanding. Interest expense related to the PPP Loan for the year ended December 31, 2020 was approximately $7,000. Interest expense was calculated using the effective interest method.
5. CONVERTIBLE PROMISSORY NOTES
In May 2019, the Company issued the 2019 Note in the aggregate principal amount of $40.0 million to Viking in a private placement transaction. The 2019 Note began to accrue interest in February 2020 at a rate of 1.5% per month. In April 2020, the Company issued the 2020 Notes in the aggregate principal amount of $15.0 million to Viking and
certain other investors in a private placement transaction. Upon issuance, the 2020 Notes accrued interest at a rate of 1.0% per month.
The 2019 Note and the 2020 Notes were to mature on the earlier of (1) March 31, 2022; (2) upon the consummation of certain potential settlement events (each as defined in the 2019 Note and the 2020 Notes); or (3) the time at which the balance of the 2019 Note and the 2020 Notes is due and payable upon an Event of Default.
Conversion Upon Qualified IPO
On August 21, 2020, the Company completed a qualified IPO, which was defined as the completion of an IPO with a Company valuation above $400.0 million for the 2019 Note and $350.0 million for the 2020 Notes, or Qualified IPO. As defined, a Qualified IPO triggered the automatic conversion and settlement of the aggregate outstanding principal amount plus accrued interest to date under the 2019 Note and the 2020 Notes into shares of the Company’s common stock at the lesser of (i) a conversion price equal to 90% of the IPO price per share and (ii) a fixed price based on a specified valuation.
As of the conversion date, accrued interest expense for the 2019 Note and the 2020 Notes was $3.7 million and $0.7 million, respectively for an aggregate total balance of $43.7 million and $15.7 million, respectively. Due to the Company’s valuation upon IPO being greater than $400.0 million, the 2019 Note converted at the fixed price of $14.35 per share, resulting in the issuance of 3,046,467 shares of common stock to the holder. Due to the Company’s valuation upon IPO being greater than $350.0 million, the 2020 Notes converted at a fixed price of $12.56 per share, resulting in the issuance of 1,248,136 shares of common stock to the holders.
The conversion of the 2019 Note and the 2020 Notes were evaluated for contingent beneficial conversion features, or BCF, under ASC Topic 470-20, Debt With Conversion and Other Options. See discussion of debt discount below for evaluation upon conversion.
Derivative Liabilities
The terms of the 2019 Note and the 2020 Notes contained alternative conversion options to the Qualified IPO upon the consummation of certain events, each of which had its own conversion prices and terms. For instance, upon a maturity date conversion, upon the election of the holder, the note would have converted and settled into shares of the Company’s then-most senior equity securities at the time of such maturity date conversion at the lesser of (i) a conversion price equal to 90% of the price per share to be received by the then-most senior equity securities and (ii) a fixed price based on a specified valuation.
Upon issuance of each of the 2019 Note and the 2020 Notes and prior to conversion upon completion of the Company’s IPO, the Company evaluated each settlement alternative within the 2019 Note and the 2020 Notes under ASC Topic 815-15, Embedded Derivatives and ASC Topic 815-40, Contracts in Entity’s Own Equity, in order to determine if it required bifurcation as a derivative instrument. The Company determined certain settlement features qualified as conversion features not meeting the requirements for separate accounting as embedded derivatives, while others qualified as redemption features meeting the requirements for separate accounting which were accounted for as derivative instruments.
Upon issuance of the 2019 Note on May 20, 2019, the estimated fair value of the bifurcated compound liability was approximately $0.9 million, which was recorded as a derivative liability with a corresponding debt discount on the consolidated balance sheets. The 2019 Note was amended during April 2020 to align the maturity date and certain other provisions with the 2020 Notes. During the second quarter of 2020, the Company assessed the amendment and determined it should be treated as a modification. Since there was no change in the fair value of the embedded conversion options as of the modification date, no change in the carrying amount of the convertible debt instrument was recognized.
Upon issuance of the 2020 Notes on April 6, 2020, the estimated fair value of the bifurcated compound liability was approximately $0.7 million, which was recorded as a derivative liability with a corresponding debt discount on the consolidated balance sheets.
The Company engaged a third-party valuation specialist in order to assess the fair value of the derivative liability as of December 31, 2019, which was determined based upon significant inputs not observable in the market, including the discount rate, management’s assessment of the probability of various underlying events triggering the conversion of the Company’s notes, such as an IPO, financing or extraordinary event, as well as the current valuation of the Company as determined by an independent third-party’s valuation report.
The fair value of the derivative liability at December 31, 2019 was $1.9 million. Changes in the probability of each embedded redemption feature drove changes in the fair value of the derivative liabilities and had a corresponding impact on the Company’s net loss. During the second quarter of 2020, the fair value of the derivative was determined to be 0 based upon the expected conversion of the notes via an embedded conversion feature which did not meet the definition of a derivative. Upon conversion of the 2019 Note and the 2020 Notes in August 2020 upon completion of the Company’s IPO, the derivative liabilities were extinguished. No additional expense was recorded at extinguishment since the derivative liabilities had already been assessed to have zero value. During the year ended December 31, 2020, the Company recorded other income2023, interest expense was $31.8 million, $4.9 million of $2.7 millionwhich related to changes in fair value of derivative liabilities in the consolidated statements of operations.
Debt Discount
Prior to conversion, the Company also evaluated the 2019 Note and the 2020 Notes for BCFs under ASC Topic 470-20, Debt With Conversion and Other Options. The Company determined that the maturity date conversion feature met the definition of a BCF since the embedded conversion option was in-the-money at inception and was fixed and determinable upon execution. Upon issuance of the 2019 Note during the second quarter of 2019, the Company recorded $11.5 million as a debt discount with a corresponding entry to additional paid-in-capital on its consolidated balance sheet to record the BCF on the 2019 Note. Additionally, at issuance in May 2019, the Company recorded a debt discount which consisted of $0.9 million related to the establishment of the derivative liability and $2.5 million related to debt issuance costs on the 2019 Note.
Upon issuance of the 2020 Notes during the second quarter of 2020, the Company recorded $2.7 million as a debt discount with a corresponding entry to additional paid-in-capital on its consolidated balance sheets to record the BCF on the 2020 Notes. Additionally, the Company recorded a debt discount of $0.7 million related to the establishment of the derivative liability on the 2020 Notes.
Prior to conversion, the debt discounts were amortized over the life of the notes. Amortizationnon-cash amortization of the debt discount from inceptionand accretion of the 2019 Note until conversionfinal payment. During the year ended December 31, 2022, interest expense was $18.2 million, $3.4 million of which related to non-cash amortization of the 2019 Notedebt discount and the 2020 Notes upon IPO was $8.7 million. At the time of conversion, the 2019 Note and the 2020 Notes had unamortized discounts of $6.9 million and $2.7 million, respectively.
Upon conversionaccretion of the 2019 Note and the 2020 Notes,final payment.
4. STOCKHOLDERS’ EQUITY
Open Market Sale Agreement
In September 2021, the Company remeasuredentered into the intrinsic valueOpen Market Sale Agreement, or the Sales Agreement, with Jefferies LLC, or the Sales Agent, under which it may, from time to time, sell shares of its common stock having an
aggregate offering price of up to $200.0 million through the BCFs which resulted in a reversal ofSales Agent, or the remaining unamortized discounts associated withATM Offering. Pursuant to the BCFs, resulting in a $7.4 million impact to additional paid-in-capital. Additionally,Sales Agreement, the Company acceleratedpaid the unamortized debt discounts related toSales Agent a commission for its services in acting as an agent in the debt issuance costs and the derivative liabilities, resulting in a $2.2 million charge to interest expense.
As of December 31, 2019, the Company’s 2019 Note and the 2020 Notes balance, net of discount, consisted of the following (in thousands):
| | | | | |
| AS OF DECEMBER 31,2019 |
| |
Principal amount | $ | 40,000 | |
Unamortized debt discounts | (9,633) | |
Convertible note, net | $ | 30,367 | |
6. STOCKHOLDERS’ EQUITY (DEFICIT)
Amended and Restated Certificate of Incorporation
On August 21, 2020 upon completion of the IPO, the Company’s certificate of incorporation was amended and restated to authorize 120,000,000 sharessale of common stock and 15,000,000 shares of preferred stock, each with a par value of $0.0001 per share.
Convertible Preferred Stock
Priorin an amount equal to its conversion to common stock in connection with the Company’s IPO, the convertible preferred stock was classified as temporary, or mezzanine, equity on the accompanying consolidated balance sheets since the shares contained certain redemption features that were not solely within the control3% of the Company. The Company had not previously accreted the convertible preferred stockgross sales price per share sold, which was amended to its redemption value since the shares were not currently redeemable and redemption was not deemed to be probable.
Convertible preferred stock authorized and issued as of December 31, 2019 consisted2% of the following (in thousands, except share, issue period, andtotal sales price per share amounts):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| AS OF DECEMBER 31,2019 |
| Shares Authorized | | Shares Issued and Outstanding | | Issue Period | | Price Per Share | | Aggregate Liquidation Preference |
| Series Mezzanine 1 | 4,299,319 | | 4,299,319 | | 2017-2018 | | $ | 6.98 | | | $ | 30,009 | |
| Series Mezzanine 2 | 21,465,681 | | 8,235,012 | | 2018-2019 | | $ | 6.98 | | | $ | 57,480 | |
| Total | 25,765,000 | | | 12,534,331 | | | | | | | $ | 87,489 | |
During the year ended December 31, 2019,2022, the Company issued 1.7 millionsold 4,332,354 shares of Series Mezzanine 2 Preferred Stock at a purchase price of $6.98 per share, resulting inpursuant to the Sales Agreement for net proceeds of approximately $12.0$127.4 million, after deducting offering expenses. No sharescommissions of Series Mezzanine 1 Preferred Stock and Series Mezzanine 2 Preferred Stock were issued$2.6 million. In connection with the ATM Offerings during the year ended December 31, 2020.2022, the Company incurred an additional $0.4 million of expenses which were offset against the proceeds of the offering. During the year ended December 31, 2023, the Company did not issue any shares under the Sales Agreement.
Securities Purchase Agreement
In connectionAugust 2023, the Company entered into a Securities Purchase Agreement, as amended, or the Purchase Agreement, with the completion ofPurchasers, pursuant to which the Company’s IPO in August 2020, all of the outstanding shares of convertible preferred stock were converted into 7,211,086Company sold and issued 3,621,314 shares of the Company’s common stock.
Common Stock Warrants
As discussed in Note 3, the Company issuedstock for $19.35 per share and, with respect to certain Purchasers, pre-funded warrants to Oxford concurrently with the 2020 Loan Agreement. Upon the consummationpurchase 6,714,636 shares of the Company’s IPOcommon stock in August 2020, the Private Placement. The purchase price of the pre-funded warrants was $19.3499 per pre-funded warrant, with an exercise price of $0.0001 per share. The Company received gross proceeds of $200.0 million from the Private Placement, before deducting $0.4 million of offering expenses payable by the Company. The pre-funded warrants are equity-classified and carried at the instruments’ fair value upon issuance. The pre-funded warrants are exercisable for 7,354 shares of common stock ofupon issuance pursuant to certain beneficial ownership limitations as defined in the Company at $17.00 per share.Purchase Agreement and will expire when exercised in full. As of December 31, 2023, all pre-funded warrants are still outstanding.
Warrants Issued in Connection with Amended 2020 Loan Agreement
As of December 31, 2023, the following equity-classified warrants were outstanding, in addition to the pre-funded warrants discussed above:
| | | | | | | | | | | | | | |
| Expiration Date | | Shares of Common Stock Issuable Upon Exercise of Warrants | | Exercise Price per Share |
| July 15, 2030 | | 7,354 | | $ | 17.00 | |
| February 18, 2032 | | 40,000 | | $ | 45.00 | |
The Company’s warrants are equity-classified and reflected in additional paid-in-capitalcarried at $0.14 million. Thethe instruments’ fair value upon classification into equity, with no subsequent remeasurements.
Common Stock Reserved for Future Issuance
Common stock reserved for future issuance as of December 31, 2023 and December 31, 2022 consist of the warrants was determined using the Black-Scholes model on the date of reclassification. No subsequent remeasurement is required for equity-classified warrants.following (in thousands):
| | | | | | | | | | | |
| AS OF DECEMBER 31, |
| 2023 | | 2022 |
| Options to purchase common stock issued and outstanding | 6,494 | | | 5,305 | |
| Shares available for future equity grants | 533 | | | 162 | |
| Pre-funded warrants issued and outstanding | 6,715 | | | — | |
| Warrants issued and outstanding | 47 | | | 47 | |
| Total common stock reserved for future issuance | 13,789 | | | 5,514 | |
7.5. EQUITY COMPENSATION PLANSPLAN
Stock Incentive Plan
The Company has 1Company’s share-based compensation plan, the Amended and Restated 2017 Employee, Director and Consultant Equity Incentive Plan, or the 2017 Plan, which provides for the issuance of incentive stock options, restricted
and unrestricted stock awards, and other stock-based awards. As of December 31, 2020,2023, an aggregate of 3.07.8 million shares of common stock were reservedauthorized for issuance under the 2017 Plan, of which 0.5 million were available.remain available for issuance.
Stock Option Activity
The Company recognizes compensation costs related to stock-based awards, including stock options, based on the estimated fair value of the awards on the date of grant. The Company grants options with an exercise price equal to the fair market value of the Company’s stock on the date of the option grant. The options are subject to four-year vesting with a one-year cliff and have a contractual term of 10 years.
A summary of the Company’s stock option activity under its 2017 Plan for the year ended December 31, 20202023 is as follows (in thousands, except for per share data and years):
| | NUMBER OF SHARES | | WEIGHTED AVERAGE EXERCISE PRICE | | WEIGHTED AVERAGE REMAINING CONTRACTUAL TERM (IN YEARS) | | AGGREGATE INTRINSIC VALUE |
| | Outstanding as of December 31, 2019 | 2,013 | | $ | 10.92 | | | 9.0 | | $ | 1,261 | |
| | Number of Shares | | | Number of Shares | | Weighted Average Exercise Price | | Weighted Average Remaining Contractual Term (In Years) | | Aggregate Intrinsic Value |
| Outstanding as of December 31, 2022 | |
| Granted | |
| Granted | |
| Granted | Granted | 747 | | | $ | 13.97 | | | 9.4 | | $ | 14,217 | |
| Forfeited | Forfeited | (232) | | | $ | 11.15 | | |
| Forfeited | |
| Forfeited | |
| Exercised | Exercised | (3) | | | $ | 10.52 | | | $ | 19 | |
| Outstanding as of December 31, 2020 | 2,525 | | | $ | 11.80 | | | 8.2 | | $ | 53,481 | |
| Vested and exercisable as of December 31, 2020 | 988 | | | $ | 10.84 | | | 7.6 | | $ | 21,849 | |
| Exercised | |
| Exercised | |
| Outstanding as of December 31, 2023 | |
| Outstanding as of December 31, 2023 | |
| Outstanding as of December 31, 2023 | |
| Vested and exercisable as of December 31, 2023 | |
The aggregate intrinsic value of stock options exercised during the years ended December 31, 2023 and December 31, 2022 was $2.1 million and $3.7 million, respectively. Aggregate intrinsic value of stock options outstanding at December 31, 2020exercised and December 31, 2019outstanding is calculated using the fair value of common stock at each respective date.on the date of exercise and as of December 31, 2023, respectively. The total fair value of stock options vested during the years ended December 31, 2023 and December 31, 2022 was $24.9 million and $25.0 million, respectively.
Stock-Based Compensation Expense
Stock options are valued using the Black-Scholes Merton option pricing model on the date of grant. This option pricing model involves a number of estimates, including the expected lives of the stock options, the Company’s anticipated stock volatility, and interest rates. Stock-based compensation expense is recognized using the straight-line method over the vesting period. The Company recognizes forfeitures as they occur.
The weighted-average assumptions used by the Company to estimate the fair value of stock option grants using the Black-Scholes option pricing model, as well as the resulting weighted-average fair value for the years ended December 31, 20202023 and 2019December 31, 2022 were as follows.
| | | | YEARS ENDED DECEMBER 31, | | YEAR ENDED DECEMBER 31, |
| | | 2020 | | | 2019 | | 2023 | | 2022 |
| | Risk-free interest rate | Risk-free interest rate | 0.42 | % | | | 2.07 | % |
| Risk-free interest rate | |
| Risk-free interest rate | | 3.77 | % | | 2.63 | % |
| Expected volatility | Expected volatility | 97.72 | % | | | 87.43 | % | Expected volatility | 84.31 | % | | 85.26 | % |
| Expected dividend yield | Expected dividend yield | 0 | % | | | 0 | % | Expected dividend yield | — | % | | — | % |
| Expected term | Expected term | 6.08 | | | 6.08 | Expected term | 6.08 | | 6.08 |
| | Weighted average fair value | Weighted average fair value | $ | 10.82 | | | $ | 9.82 | Weighted average fair value | $ | 16.88 | | $ | 17.72 |
The Company determines the assumptions used in the option pricing model in the following manner:
Risk-Free Interest Rate—For the determination of the risk-free interest rates, the Company utilizes the U.S. Treasury yield curve for instruments in effect at the time of measurement with a term commensurate with the expected term assumption.
Expected Volatility—Due to the Company’s limited historical stock price volatility data indicative of the expected future volatility, the Company based its estimate of expected volatility on the estimated and expected volatilities of a guideline group of publicly traded companies. For these analyses, the Company selected companies with comparable characteristics including enterprise value, risk profiles, and with historical share price information sufficient to meet the expected life of the stock-based awards. The Company computes the historical volatility data using the daily closing prices for the selected companies’ shares during the equivalent period of the calculated expected term of its stock-based awards. The Company will continue to apply this process until a sufficient amount of historical information regarding the volatility of its own stock price becomes available.available, or until the volatility of the Company’s market-traded shares best represents its expected volatility.
Expected Dividend—The expected dividend yield is assumed to be zero since the Company has never paid dividends and does not have current plans to pay any dividends on its common stock.
Expected Term—The Company estimates the expected term of its stock options granted to employees and non-employee directors using the simplified method, whereby, the expected term equals the average of the vesting term and the original contractual term of the option. The Company utilizes this method since it does not have sufficient historical exercise data to provide a reasonable basis upon which to estimate the expected term.
Prior to the Company’s IPO, given the absence of a public trading market for the Company’s common stock, its board of directors exercised their judgment and considered a number of objective and subjective factors to determine the best estimate of the fair value of the Company’s common stock underlying the stock options, such as: contemporaneous valuations performed by independent third-party specialists, its stage of development, including the status of its research and development efforts of its therapeutic candidates and the material risks related to its business and industry, its results of operations and financial condition, including its levels of capital resources, the prices at which its sold shares of its convertible preferred stock, the rights, preferences and privileges of its convertible preferred stock relative to those of its common stock, the conditions in the biotechnology industry and the economy in general, the stock price performance and volatility of comparable life sciences public companies, as well as recently completed mergers and acquisitions of peer companies, the likelihood of achieving a liquidity event for the holders of its common stock or convertible preferred stock, such as an IPO or a sale of the Company given prevailing market conditions, trends and developments in its industry, external market conditions affecting the life sciences and biotechnology sectors, and the lack of liquidity of its common stock, among other factors.
Subsequent to the Company’s IPO, theFair Value—The fair value of the Company’s common stock is determined based on its closing market price on the date of the grant. The Company’s board of directors intended all options granted to be exercisable at a price per share not less than the per share fair value of its common stock underlying those options on the grant date.
Stock-based compensation expense for stock options consisted of the following (in thousands):
| | | | YEAR ENDED DECEMBER 31, | |
| | 2020 | | 2019 | |
| | 2023 | |
| 2023 | |
| 2023 | |
| | Research and development | |
| | Research and development | |
| | Research and development | Research and development | $ | 4,300 | | | $ | 3,405 | | |
| General and administrative | General and administrative | 723 | | | 569 | | |
| General and administrative | |
| General and administrative | |
| Total stock-based compensation expense | Total stock-based compensation expense | $ | 5,023 | | | $ | 3,974 | | |
| Total stock-based compensation expense | |
| Total stock-based compensation expense | |
As of December 31, 2020,2023, the Company had $13.6$51.8 million of total unrecognized stock-based compensation expense related to its stock options, which itis expected to recognizebe recognized over a weighted-average period of 2.32.5 years.
8.6. LICENSE AND GRANT REVENUES
The following table summarizes the total revenue recorded in the Company’s consolidated statements of operations (in thousands):
| | | | | | | | | | | | | | | |
| YEAR ENDED DECEMBER 31, | | |
| 2020 | | 2019 | | | | |
| | | | | |
| License fee revenue from affiliates | | | | | | | |
| Elpiscience Biopharmaceuticals, Inc. | $ | 2,000 | | | $ | 0 | | | | | |
| Transcenta Holding, Ltd. | 0 | | | 1,000 | | | | | |
| Total license fee revenue from affiliates | 2,000 | | | 1,000 | | | | | |
| License fee revenue from non-affiliates | | | | | | | |
| Chiesi Farmaceutici S.p.A. | 7,365 | | | 1,061 | | | | | |
| Phylaxis BioScience, LLC | 1,993 | | | 0 | | | | | |
| bluebird bio, Inc. | 400 | | | 7,000 | | | | | |
| Other license revenues from non-affiliates | 1,050 | | | 32 | | | | | |
| Total license fee revenue from non-affiliates | 10,808 | | | 8,093 | | | | | |
| Grant revenue | | | | | | | |
| National Institutes of Health | 0 | | | 880 | | | | | |
| Combating Antibiotic Resistant Bacteria Accelerator | 0 | | | 3,081 | | | | | |
| Congressionally Directed Medical Research Program | 80 | | | 157 | | | | | |
| Total grant revenue | 80 | | | 4,118 | | | | | |
| Total revenue | $ | 12,888 | | | $ | 13,211 | | | | | |
| | | | | | | | | | | | | | | |
| YEAR ENDED DECEMBER 31, | | |
| 2023 | | 2022 | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| License fee revenue | | | | | | | |
| Phylaxis BioScience, LLC | $ | 1,634 | | | $ | 1,101 | | | | | |
| Chiesi Farmaceutici S.p.A. | 166 | | | 877 | | | | | |
| 2seventy bio, Inc. | — | | | 200 | | | | | |
| | | | | | | |
| Total license fee revenue | 1,800 | | | 2,178 | | | | | |
| Grant revenue | | | | | | | |
| | | | | | | |
| | | | | | | |
| Congressionally Directed Medical Research Program | — | | | 14 | | | | | |
| Total grant revenue | — | | | 14 | | | | | |
| Total revenue | $ | 1,800 | | | $ | 2,192 | | | | | |
License and Collaboration Agreements
Phylaxis Agreements
In July 2020, the Company entered into a joint venture with an entity affiliated with ArrowMark Partners, Phylaxis BioScience, LLC, or Phylaxis. In connection with the joint venture, the Company entered into the following agreements: Contribution Agreement, License Agreement, Limited Liability Company Agreement, and Master Services Agreement, or collectively the Phylaxis Agreements, pursuant to which the Company licensed certain intellectual property and know-how to Phylaxis and agreed to provide services to develop certain compounds. Upon closing, theThe Company received a $2.5$5.0 million in nonrefundable, upfront paymentpayments from Phylaxis under the Master Services Agreement, or the MSA. The Company is also entitled to an additional $2.5 million payable within 180 days from closing under the MSA, $1.25 million of which was received in October 2020. UponIn addition, upon closing, the Company received a 10% equity interest in Phylaxis as consideration for the contribution of the license of the Company’s intellectual property and know-how and is entitledknow-how. Pursuant to receivethe terms of the Phylaxis Agreements, during the fourth quarter of 2023, the Company received an additional 5% based onequity interest in Phylaxis following the achievement of certain milestones.a milestone event. Under the License Agreement, the Company is also entitled to specified development and commercialization milestone payments of up to an aggregate of $225.0 million and $175.0 million, respectively. The Company is also entitled to share in a percentage of the profits of Phylaxis under the Limited Liability Company Agreement.
The Company recognized the initial transaction price of $5.5 million, comprised of the $5.0 million in upfront payments and the fair value of the 10% equity interest of $0.5 million, over the period of performance of development services on the two compounds. During the third quarter of 2022, the Company fulfilled this performance obligation in full and recognized all remaining deferred revenue under this contract.
During the fourth quarter of 2023, the Company transferred a second generation compound to Phylaxis, which was a milestone event under the agreements. Upon transfer, the Company received an additional 5% equity interest in Phylaxis, which has a fair value of $1.6 million. The Company recognized the fair value of the 5% equity interest as revenue and accounted for the investment under the equity method. In order to determine the fair value of the equity interest in Phylaxis upon this milestone, the Company engagedutilized the investee’s independent valuations based on recent financings from third party investors at fair market value.
During the years ended December 31, 2023 and December 31, 2022, the Company recognized $1.6 million and $1.1 million of revenue under the Phylaxis agreements. As of December 31, 2023 and December 31, 2022, the Company had no deferred revenue related to this agreement.
Chiesi
In May 2019, the Company entered into an Option Agreement, as amended by the First Amendment to Option Agreement, dated August 19, 2019, or the Chiesi Option Agreement, with Chiesi Farmaceutici S.p.A., or Chiesi, pursuant to which the Company granted to Chiesi an exclusive option to obtain an exclusive license to develop and
commercialize INBRX-101 outside of the United States and Canada. Additionally, the Chiesi Option Agreement provided Chiesi with a third-party valuation specialist. The valuation report utilized a market approach to establish the total equity valueright of Phylaxis using inputs not observablenegotiation for INBRX-101 development and commercialization rights in the market, includingUnited States and Canada in the discount rate. The fair valueevent that the Company engages in discussions with any third parties for such rights during the term of the equity interest was $0.5 million, which has been accounted for underChiesi Option Agreement. Under the equity method. The fair valueterms of the equity interest uponChiesi Option Agreement, the executionCompany received a one-time, non-refundable option initiation payment of the agreements has been included$10.0 million in the transaction price, along with the $5.0 million of payments due pursuant to the MSA.August 2019.
The Company identified the transferone performance obligation as of the exclusive licenses for,effective date of the Chiesi Option Agreement, which is to perform research and performance of, development services for Chiesi during the option period, which would continue (unless the Chiesi Option Agreement is terminated earlier by Chiesi or the Company) until 60 days following the last to modifyoccur of (i) the Company’s delivery to Chiesi of the trial phase data for the first Phase I Clinical Trial, (ii) the Company’s delivery to Chiesi of the finalized minutes from the definitive U.S. Food and second compounds as 1 performance obligationDrug Administration, or FDA, scientific advice meeting conducted following completion of such Phase I Clinical Trial, and allocated(iii) the transaction price evenlyCompany’s delivery to Chiesi of the finalized minutes from the definitive parallel European Medicines Agency health technology assessment, or EMA-HTA, scientific advice meeting conducted following completion of such Phase I Clinical Trial. The Company determined that the option to grant a license in the future is not a material right.
the European Medicines Agency, or EMA, scientific advice to Chiesi upon receipt, which fulfilled the necessary deliverables to Chiesi and triggered the start of its 60-day option period window. On September 18, 2023, the Company was notified that Chiesi declined to exercise its option, resulting in the expiration of the Chiesi Option Agreement (upon the lapse of the option period), and with the Company retaining global rights to develop and commercialize INBRX-101. between the 2 compounds. Revenue relatedThe $10.0 million upfront payment was allocated to the single performance obligation will beobligation. Revenue is recognized over time as services are performed during the option period, based on the Company’s progresseffort to satisfy the performance obligation.
obligation relative to the total expense estimated to be incurred during the option period. During the yearyears ended December 31, 2020,2023 and December 31, 2022, the Company recognized $2.0 million of revenue related to this performance obligation. As of December 31, 2020, the Company has $1.6$0.1 million and $0.6$0.9 million of current and non-current deferredin revenue related to this agreement, respectively. TheAs of December 31, 2023, the Company expects to complete all workhas no deferred revenue related to this contract and recognize revenue in its entirety by the first halfagreement. As of 2022.
bluebird
2018 License Agreement
On December 20, 2018,31, 2022, the Company entered into a License Agreement with bluebirdhad $0.1 million current deferred revenue related to this agreement.
2seventy bio, Inc., or bluebird, whereby the Company granted bluebird the exclusive, worldwide rights to develop, manufacture, and commercialize certain cell therapy products containing binders. In January 2019, the Company received a non-refundable upfront fee of $7.0 million from bluebird for this exclusive license. As part of the Company’s performance obligations to bluebird, it transferred certain know-how to bluebird, the completion of which occurred in March 2019. As a result, the Company recognized the $7.0 million upfront payment as license fee revenue during March 2019 upon completion of all performance obligations.
The Company is also entitled to receive specified development milestone payments of up to an aggregate of $51.5 million per therapeutic, as well as percentage tiered royalties on future product sales with rates in the mid-single-digits. Due to the uncertainty in the achievement of the developmental milestones, the variable consideration associated with the future milestone payments has been fully constrained (excluded) from the transaction price until such time that the Company concludes that it is probable that a significant reversal of previously recognized revenue will not occur. These estimates will be re-assessed at each reporting period.
2020 Option and License Agreement
In June 2020, the Company entered into a separatean Option and License Agreement with bluebird, pursuant to which the Company granted to bluebird exclusive worldwide development licensesrights to develop binders and cell therapy products containing single domain antibodies, or sdAbs, directed to specified targets, consisting of 2two initial programs and up to an additional 8 programs. The Company retains all rights to the specific sdAbs outside of the cell therapy field. In November 2021, this agreement, or the 2020 2seventy Agreement, was assigned to 2seventy bio, Inc., or 2seventy, in connection with bluebird’s internal restructuring and subsequent spin-out of 2seventy.
In June 2020, the Company received a non-refundable upfront option fee of $0.2 million in connection with each of the 2two initial programs, or $0.4 million in aggregate, and is entitled to an upfront option fee for each additional program, on a program-by-program basis. TheIn June 2022, 2seventy selected a third program and paid a non-refundable upfront option fee of $0.2 million in exchange for a development license. Under each of the three programs, the Company alsohas granted to bluebird an option toin which 2seventy may acquire an exclusive license with respect to all binders and cell therapy products developed under this agreement, which entitles the Company to additional fees upon exercise of the option. In connection with each program for which bluebird2seventy exercises its option, bluebird2seventy will be required to pay the Company a one-time, non-refundable, non-creditable fee in the low-single-digit millions for each option bluebird chooses to exercise.millions. The Company is also entitled to receive certain developmental milestone payments of up to an aggregate of $51.5 million per therapeutic, as well as percentage tiered royalties on future product sales with rates in the mid-single-digits. Due to the uncertainty in the achievement of the developmental milestones and future sales, the variable consideration associated with the future milestone payments has been fully constrained (excluded) from the transaction price until such time that the Company concludes that it is probable that a significant reversal of previously recognized revenue will not occur. These estimates will be re-assessed at each reporting period. As of December 31, 2023, 2seventy has exercised one of its options and declined one of its options as related to the two initial programs, while the third program option remains outstanding.
As of the effective date of the agreement,2020 2seventy Agreement, the Company identified 1one performance obligation, which was the transfer of the exclusive development license to bluebird for the 2two initial programs. The Company determined that the option granted for an exclusive license in the future was not a material right. For the 8eight programs not identified upon execution of the contract, the Company evaluated the customer option for additional purchases and determined that those options for additional programs did not constitute material rights nor variable consideration. As additional programs are identified, the Company will re-assessre-assesses its performance obligations and transaction price accordingly.
TheIn June 2022, pursuant to the terms regarding the addition of new programs in the 2020 2seventy Agreement, the Company determinedreceived a $0.2 million upfront option fee related to the total transaction priceselection of a third program and transferred the contract at execution was $0.4 million for the transfer of the 2 exclusiverelated know-how and development licenses.license. The Company recognized $0.4the $0.2 million of revenue related to this agreement
at the point in time in which the exclusive development licenses were transferred to bluebird, which occurred upon execution ofprogram was added and the agreement in June 2020.program term began.
Chiesi
In May 2019, the Company entered into an Option Agreement, as amended by the First Amendment to Option Agreement, dated August 19, 2019, the Chiesi Option Agreement, with Chiesi Farmaceutici S.p.A., or Chiesi, pursuant to which the Company granted to Chiesi an exclusive option to obtain an exclusive license to develop and commercialize INBRX-101 outside of the United States and Canada. Under the terms of the Chiesi Option Agreement, the Company received a one-time, non-refundable option initiation payment of $10.0 million in August 2019. If Chiesi chooses to exercise its option under the Chiesi Option Agreement, then Chiesi must pay the Company a one-time, non-refundable fee of $12.5 million upon the effective date of the definitive agreement granting Chiesi the exclusive license. If the option is exercised, under the license agreement, the Company may be entitled to receive specified milestone payments of up to $122.5 million, as well as royalties on future product sales.
The Company has identified 1 performance obligation as of the effective date of the Chiesi Option Agreement, which is to perform research and development services for Chiesi during the option period, which will continue (unless the Chiesi Option Agreement is terminated earlier by the Chiesi or the Company) until 60 days following the last to occur of (i) the Company’s delivery to Chiesi of the trial phase data for the first Phase I Clinical Trial, (ii) the Company’s delivery to Chiesi of the finalized minutes from the definitive U.S. Food and Drug Administration, or FDA, scientific advice meeting conducted following completion of such Phase I Clinical Trial, and (iii) the Company’s delivery to Chiesi of the finalized minutes from the definitive parallel EMA-HTA scientific advice meeting conducted following completion of such Phase I Clinical Trial. The Company has determined that the option to grant a license in the future is not a material right.
The $10.0 million upfront payment has been allocated to the single performance obligation. Revenue is recognized over time as services are performed during the option period, based on the Company’s effort to satisfy the performance obligation relative to the total expense estimated to be incurred during the option period. During the yearsyear ended December 31, 2020 and 2019,2022, the Company recognized $7.4$0.2 million and $1.1 million in revenue related to this agreement, respectively. As of December 31, 2020, the Company has $1.4 million and $0.1 million of current and non-current deferred revenue related to this agreement, respectively. As of December 31, 2019, the Company had $7.9 million and $1.0 million of current and non-current deferred revenue related to this agreement. The Company expects to complete all work related to this contract anddid not recognize any revenue in its entirety by the first half of 2022.
Elpiscience
As of December 31, 2020, the Company has entered into 2 different License Agreements with Elpiscience Biopharmaceuticals, Inc., or Elpiscience. Each agreement is a standalone license of a distinct and differentiated protein therapeutic candidate with a separate biological target, providing a unique benefit. Neither the license nor the asset is reliant upon the other.
PD-L1 and 4-1BB Program
In February 2018, the Company entered into a License Agreement, or the PD-L1 and 4-1BB License Agreement, with Elpiscience, whereby the Company granted Elpiscience an exclusive license to the Company’s bi-specific therapeutic candidate designed to target PD-L1 and 4-1BB, or INBRX-105. This license provides Elpiscience with the right to further advance the therapeutic candidate through clinical trials, as well as to manufacture and commercialize it. It also requires the Company to provide Elpiscience with know-how and materials specific to INBRX-105, including process development and manufacturing data and information necessary to develop INBRX-105. In the PD-L1 and 4-1BB License Agreement, the Company also agreed to negotiate an agreement to supply Elpiscience with INBRX-105 for its development in China, Hong Kong, Macau and Taiwan. The Company is eligible to receive specified developmental and commercial milestone payments of up to an aggregate of $100.0 million, as well as percentage tiered royalties on future product sales with rates in the high single-digits. Due to the uncertainty in the achievement of the developmental milestones, the variable consideration associated with the future milestone payments has been fully constrained (excluded) from the transaction price until such time that the
Company concludes that it is probable that a significant reversal of previously recognized revenue will not occur. This will be re-assessed at each reporting period.
Under the PD-L1 and 4-1BB License Agreement, the Company is entitled to reimbursement for certain toxicology study costs and chemistry, manufacturing and controls, or CMC costs. Elpiscience paid the Company $3.0 million in March 2018 for certain costs in relation to the development of INBRX-105. The payment received in advance of the work to be completed was recorded to other current liabilities upon receipt and, since the Company was acting as an agent on behalf of Elpiscience, the Company recorded it as a contra-expense against research and development as the relevant work was completed. During the year ended December 31, 2019, the Company completed all relevant work on this program and derecognized the remaining approximately $0.7 million of other current liabilities related to the reimbursement under the PD-L1 and 4-1BB License Agreement. As of December 31, 2019, there were 0 liabilities related to this reimbursement remaining.
OX40 Program
In April 2018, the Company entered into a separate License Agreement, or the OX40 License Agreement, with Elpiscience, whereby the Company granted Elpiscience an exclusive license to the Company’s multivalent protein therapeutic directed to the biological target OX40, or INBRX-106. This license provides Elpiscience with the right to further advance the therapeutic candidate through clinical trials, as well as manufacture and commercialize it. It also requires the Company to provide Elpiscience with know-how and materials specific to INBRX-106, including process development and manufacturing data and information necessary to develop INBRX-106. In the OX40 License Agreement, the Company also agreed to negotiate an agreement to supply Elpiscience with INBRX-106 for its development in Mainland China, Hong Kong Macau and Taiwan. The Company is eligible to receive certain developmental and commercial milestones of up to an aggregate of $100.0 million, as well as percentage tiered royalties on future product sales with rates in the high single-digits. Due to the uncertainty in the achievement of the developmental and commercial milestones, the variable consideration associated with the future milestone payments has been fully constrained (excluded) from the transaction price until such time that the Company concludes that it is probable that a significant reversal of previously recognized revenue will not occur. This will be re-assessed at each reporting period.
In September 2020, the Company achieved a milestone under the contract for $2.0 million, which was received during September 2020. The Company recognized revenue of $2.0 million during the year ended December 31, 2020. NaN revenue was recognized related to this agreement during the year ended December 31, 2019.
Under the OX40 License Agreement, the Company is entitled to reimbursement for certain toxicology study costs and CMC costs. Elpiscience paid the Company $3.4 million in August 2018 for certain costs incurred in relation to the development and production of INBRX-106. The payment received in advance of the work to be completed was recorded to other current liabilities upon receipt and since the Company was acting as an agent on behalf of Elpiscience, the Company recorded it as a contra-expense against research and development as the relevant work was completed. As of December 31, 2020, the Company has $0.2 million recorded as receivables from related parties from Elpiscience for expenses incurred which have not yet been reimbursed. During the years ended December 31, 2020 and 2019, the Company derecognized approximately $0.2 million and $1.7 million, respectively, of current liabilities related to these reimbursements under the OX40 License Agreement. During the year ended December 31, 2020, the Company received $0.2 million in reimbursements for expenses already incurred. As of December 31, 2020, the Company had 0 liabilities outstanding related to this agreement. Additional reimbursements of $0.1 million related to multi-year stability studies are expected to be recognized and paid upon completion.
During 2020, the Company entered into additional cost sharing arrangements with Elpiscience related to formulation development and material generation. During the year ended December 31, 2020, the Company derecognized approximately $0.3 million of current liabilities related to these agreements, all of which is recorded as receivables from related parties as of December 31, 2020.
Transcenta
In June 2017, the Company entered into a License Agreement, or the Transcenta License Agreement, with Transcenta Holding, Ltd. (formerly Hangzhou Just Biotherapeutics Co., Ltd.), or Transcenta, whereby the Company
granted Transcenta exclusive, non-transferable rights to develop, manufacture, and sell its products containing the Company’s protein therapeutic candidate, designed to target DR5, or INBRX-109, or a derivative thereof, within mainland China, Hong Kong, Macau and Taiwan and agreed to provide Transcenta with the intellectual property and materials, including assays and cell lines, necessary to develop the therapeutic candidate. In addition, pursuant to the Transcenta License Agreement, the Transcenta License Agreement granted the Company a royalty-free, worldwide, non-exclusive research license to intellectual property created by Transcenta that incorporates intellectual property licensed to Transcenta, or the Transcenta IP. In June 2019, the Company achieved a milestone under the contract for $1.0 million, which was received during August 2019, net of $0.1 million in foreign tax withholding.
Under the Transcenta License Agreement, the Company is also eligible to receive certain developmental and commercial milestone payments of up to an aggregate of approximately $100.0 million, as well as percentage tiered royalties on future product sales with rates ranging between the high single-digits to the low teens. Due to the uncertainty in the achievement of the developmental and commercial milestones, the variable consideration associated with the future milestone payments has been fully constrained (excluded) from the transaction price until such time that the Company concludes that it is probable that a significant reversal of previously recognized revenue will not occur. These estimates will be re-assessed at each reporting period.
Celgene
In June 2012, the Company entered into a Development and Option Agreement with Celgene whereby the Company granted Celgene an option to exclusively license certain intellectual property rights relating to the Company’s CD47 checkpoint inhibitor. Upon grant of the Development and Option Agreement, Celgene made a non-refundable upfront option payment for the exclusive license. In June 2013, Celgene opted to exercise its right to license the intellectual property rights associated with the Company’s CD47 therapeutic. The Company is also eligible to receive certain developmental and commercial milestones of an aggregate of $934.1 million, assuming the achievement of all potential milestones in the agreement, as well as percentage tiered royalties based on future worldwide sales, with rates ranging between the high single-digits and low teens. The Company is obligated to pay 2% of future amounts received under the Development and Option Agreement to advisors who assisted the Company with the negotiation and other matters in connection with the Development and Option Agreement.2023.
Government Grants
Grant revenue was recognized in accordance with the Company’s stated methodology discussed in Note 1.
NIHCDMRP funded through the DoD
During August 2017,In February 2021, the Company was awarded a grant byfrom the NIH in the amount of $1.0 million for the continued development of multi-specific antibodies for the treatment of Pseudomonas aeruginosa infection, all of which has been recognized as revenue upon completion in June 2019.
Revenue from this NIH grant was based upon internal and subcontractor costs incurred by the Company that are specifically covered by the grants, and where applicable, an additional facilities and administrative rate that provided funding for overhead expenses. Revenue was recognized when the Company incurred these covered expenses related to the grant’s continued progression. The Company submitted invoices to the NIH for only those expenses which had been incurred by the Company in each period, resulting in reimbursement in arrears.
CARB-X Subaward Agreement
During July 2017, the Company was awarded a grant by Boston University in the amount of approximately $4.6 million. The program, Combating Antibiotic Resistant Bacteria Accelerator, or CARB-X, is a non-profit public-private partnership dedicated to accelerating antibacterial research to tackle the global rising threat of drug-resistant bacteria. The Company received this award in relation to its program aimed at the development of novel antimicrobial therapeutics using multiple mechanisms of action to circumvent the development of resistance, and in the case of this grant, specifically for pseudomonas. The payment structure of this grant was a cost share, where 70% of costs were reimbursed by the sponsor, or Boston University, and the remaining 30% of costs are covered by the Company. The work under this grant began in October 2017 and was discontinued during the third quarter of 2019 based on the uncertainty within the applicable regulatory environment.
Revenue from this grant was based upon internal costs incurred by the Company that were specifically covered by the grant as well as certain specified overhead expenses. Revenue was recognized when the Company incurred these covered expenses related to the grant’s continued progression. The Company submitted invoices to Boston University for only those expenses which had been incurred by the Company in each period, resulting in reimbursement in arrears. Prior to discontinuation of the work on this grant, $4.0 million in revenue was recognized, all of which has been received in cash from Boston University.
CDMRP funded through the DoD
During September 2018, the Company was awarded a grant by the CDMRP funded through the DoD by the U.S. Army Medical Research Acquisition Activity in the amount of approximately $0.3 million for the continued development of multi-specific antibodiessdAb based therapeutics targeting SARS-CoV-2 Spike protein for the treatment against Acinetobacter baumannii infection by circumventing development of bacterial resistance. The workCOVID-19, or the 2021 DoD grant. Work under thisthe grant began in December 2018February 2021 and was completed in September 2020.continued through February 2022.
Revenue from this grant is based upon internal and subcontractor costs incurred by the Company that are specifically covered by the grants, and where applicable, an additional facilities and administrative rate that provided funding for overhead expenses. Revenue is recognized when the Company incurs these covered expenses related to the grant’s continued progression. The Company submits requests for reimbursement to the DoD for only those expenses which have been incurred by the Company in each period, resulting in reimbursement in arrears. As of December 31, 2020, $0.3 million in revenue has been recognized, all of which has been received in cash from the DoD.
9. RELATED PARTY TRANSACTIONS
From time to time, the Company will enter into an agreement with a related party in the ordinary course of its business. These agreements are ratified by the Company’s Board of Directors or a committee thereof pursuant to policy.
LAV Summit Limited
LAV Summit Limited, or LAV SL, a limited company, is a principal shareholder owning more than 5% of the Company’s outstanding equity interest as of December 31, 2020. Due to this equity ownership, LAV SL is considered a related party. Moreover, Judith Li, a former member of the Company’s board of directors, is a Partner at LAV SL’s General Partner, Lilly Asia Ventures, or LAV, a biomedical venture capital firm.
In June 2017, the Company entered into the Transcenta License Agreement. LAV, through one of its funds, holds a significant equity ownership position in Transcenta. During the year ended December 31, 2020,2022, $14,000 was recognized as revenue under the Company recorded 0 revenue related to this agreement. Duringgrant. All work was completed during the year ended December 31, 2019,2022. No revenue was recognized under the Company recorded revenue of $1.0 million related to this agreement. See Note 8 for further discussion of this agreement.
In February 2018, the Company entered into the PD-L1 and 4-1BB License Agreement with Elpiscience. LAV, through one of its funds, holds a significant equity ownership position in Elpiscience and the Venture Partner of LAV is the CEO, Founder, and Director at Elpiscience. Accordingly, the Company identifies Elpiscience as a related party. Under this agreement, the Company is entitled to reimbursement for certain CMC and toxicology expenses incurred for which Elpiscience has paid the Company $4.7 million to date as of December 31, 2020. During the years ended December 31, 2020 and 2019, the Company recorded 0 revenue related to this agreement. See Note 8 for further discussion of this agreement.
In April 2018, the Company entered into the OX40 License Agreement with Elpiscience. Under this agreement, the Company is entitled to reimbursement for certain CMC and toxicology expenses incurred for which Elpiscience has paid the Company approximately $5.2 million to date as of December 31, 2020. Duringgrant during the year ended December 31, 2020, the Company recorded revenue of $2.0 million related to this agreement. During the year ended December 31, 2019, the Company recorded 0 revenue related to this agreement. See Note 8 for further discussion of this agreement.
In July 2020, the Company entered into a cost sharing agreement with Elpiscience related to OX40. Under this agreement, the Company is entitled to reimbursement for certain formulation development costs. Through
December 31, 2020, the Company has 0t received any reimbursements related to this agreement. See Note 8 for further discussion of this agreement.
WuXi Biologics Healthcare Venture
WuXi Biologics Healthcare Venture, or WuXi Bio HV, is a corporate venture arm of WuXi Biologics, with whom the Company has an existing Exclusivity Agreement. In January 2019, WuXi Bio HV purchased 1.7 million shares of the Company’s Series Mezzanine 2 Preferred Stock at a purchase price of $6.98 per share, for a purchase price of $12.0 million. These shares converted to common stock upon the consummation of the Company’s IPO in August 2020.
See Note 6 for further discussion of Preferred Stock transactions.2023.
10.7. INCOME TAXES
The components of income tax expense (benefit) were as follows for the years ended December 31, 20202023 and 2019,December 31, 2022, respectively (in thousands):
| | YEAR ENDED
DECEMBER 31, |
| 2020 | | 2019 |
| YEAR ENDED DECEMBER 31, | | | YEAR ENDED DECEMBER 31, |
| 2023 | | | 2023 | | 2022 |
| Current expense: | Current expense: | | | |
| State | State | $ | 3 | | | $ | (2) | |
| Foreign | 0 | | | 900 | |
| State | |
| State | |
| | Total current expense | Total current expense | $ | 3 | | | $ | 898 | |
| Total current expense | |
| Total current expense | |
The provision for the yearyears ended December 31, 20202023 and December 31, 2022 was related to California state income tax. The provision for the year ended December 31, 2019 was related to payments received in connection with the Transcenta License Agreement and Chiesi Option Agreement.taxes.
A reconciliation of income tax expense to the amount computed by applying the statutory federal income tax rate to the loss from operations is summarized for the years ended December 31, 20202023 and 2019December 31, 2022 as follows (in thousands):
| | YEAR ENDED
DECEMBER 31, |
| 2020 | | 2019 |
| YEAR ENDED DECEMBER 31, | | | YEAR ENDED DECEMBER 31, |
| 2023 | | | 2023 | | 2022 |
| Expected income tax benefit at federal statutory rate | Expected income tax benefit at federal statutory rate | $ | (15,985) | | | $ | (10,605) | |
| State income tax expense (benefit), net of federal benefit | State income tax expense (benefit), net of federal benefit | 4,548 | | | (4,807) | |
| Permanent items | Permanent items | 2,291 | | | 1,937 | |
| R&D credits | R&D credits | (2,878) | | | (2,910) | |
| Unrecognized tax benefits (FIN 48) | Unrecognized tax benefits (FIN 48) | 707 | | | 1,232 | |
| Foreign withholding tax | 0 | | | 711 | |
| | Return to provision true-ups | |
| Return to provision true-ups | |
| Return to provision true-ups | Return to provision true-ups | 164 | | | 0 | |
| Valuation allowance | Valuation allowance | 11,156 | | | 15,340 | |
| Income tax expense | Income tax expense | $ | 3 | | | $ | 898 | |
Income taxes are accounted for under the asset and liability method. Deferred income taxes are recorded for temporary differences between financial statement carrying amounts and the tax basis of assets and liabilities. Deferred tax assets and liabilities reflect the tax rates expected to be in effect for the years in which the differences are expected to reverse. A valuation allowance is provided if it is more likely than not that some or all of the deferred tax assets will not be realized.
The components of net deferred income taxes were as follows (in thousands):
| | AS OF DECEMBER 31, |
| AS OF DECEMBER 31, | | | AS OF DECEMBER 31, |
| | | 2020 | | 2019 | | 2023 | | 2022 |
| Deferred income tax assets | Deferred income tax assets | | | |
| Net operating loss carryforward | |
| Net operating loss carryforward | |
| Net operating loss carryforward | Net operating loss carryforward | $ | 27,055 | | | $ | 16,284 | |
| Research and development credits | Research and development credits | 6,465 | | | 4,346 | |
| Intangibles | Intangibles | 4,176 | | | 6,168 | |
| Accruals | Accruals | 233 | | | 302 | |
| Stock compensation | Stock compensation | 409 | | | 49 | |
| Change in fair value | 0 | | | 284 | |
| Capitalized research and development costs | |
| Deferred revenue | Deferred revenue | 806 | | | 280 | |
| Operating lease liabilities | Operating lease liabilities | 1,733 | | | 2,183 | |
| Fixed assets | 0 | | | 42 | |
| Other | Other | 3 | | | 78 | |
| Gross deferred tax assets | Gross deferred tax assets | 40,880 | | | 30,016 | |
| Less: Valuation allowance | Less: Valuation allowance | (39,087) | | | (27,931) | |
| Total deferred tax assets | Total deferred tax assets | 1,793 | | | 2,085 | |
| Deferred income tax liabilities | Deferred income tax liabilities | | |
| Fixed assets | Fixed assets | (140) | | | 0 | |
| Fixed assets | |
| Fixed assets | |
| Operating lease right-of-use assets | Operating lease right-of-use assets | (1,653) | | | (2,085) | |
| Other | |
| Total deferred tax liabilities | Total deferred tax liabilities | (1,793) | | | (2,085) | |
| Net deferred tax assets (liabilities) | Net deferred tax assets (liabilities) | $ | 0 | | | $ | 0 | |
As of December 31, 2020,2023, the Company had unused federal and state NOL carryforwards of approximately $122.4$305.1 million and $18.6$65.3 million, respectively. The federal NOL carryforwards generated in years beginning after 2017 may be carried forward indefinitely but federal NOL carryforwards generated in tax years beginning after December 31, 2017 will, in loss utilization years beginning after December 31, 2020,are only be available to offset up to 80% of pre-NOL taxable income each year. The state NOL carryforwards will begin to
expire in 2038.2038, if not utilized. As of December 31, 2020,2023, the Company also had federal and state research tax credit carryforwards of $6.4$12.4 million and $2.8$6.2 million, respectively. The federal research tax credit carryforwards begin to expire in 2038, while the state carryforwards have no expiration.
The utilization of NOL and research tax credit carryforwards to offset future taxable income may be subject to an annual limitation as a result of ownership changes that have occurred previously or may occur in the future. Under Sections 382 and 383 of the IRC, a corporation that undergoes an “ownership change” may be subject to limitations on its ability to utilize its pre-change NOLs and other tax attributes otherwise available to offset future taxable income and/or tax liability. An ownership change is defined as a cumulative change of 50% or more in the ownership positions of certain stockholders during a rolling three-year period. The Company has not completedis in the process of completing a formal study to determine if any ownership changes within the meaning of IRC Section 382 and 383 have occurred.had occurred and, pending the finalization of the study, has not identified any ownership changes as of December 31, 2023. It is possible that the Company has already incurred ownership changes and may incur additional ownership changes in the future. If an ownership change has occurred, the Company’s ability to use its NOL or tax credit carryforwards may be restricted, which could require the Company to pay federal or state income taxes earlier than would be required if such limitations were not in effect.
The Company has established a full valuation allowance against its deferred tax assets due to uncertainties that preclude it from determining that it is more likely than not that the Company will be able to generate sufficient taxable income to realize such assets. Management assesses the available positive and negative evidence to estimate if sufficient future taxable income will be generated to utilize the existing deferred tax assets. A significant piece of objective negative evidence evaluated was the cumulative loss incurred over the three-year period ended December 31, 2020.2023. Such objective evidence limits the ability to consider other subjective evidence such as the
Company’s projections for future growth. Based on this evaluation, as of December 31, 2020,2023, a valuation allowance of $39.1$146.0 million has been recorded in order to measure only the portion of the deferred tax asset that more likely than not will be realized. The amount of the deferred tax asset considered realizable, however, could be adjusted if objective negative evidence in the form of cumulative losses is no longer present and additional weight may be given to subjective evidence, such as estimates of future taxable income during carryforward periods and the Company’s projections for growth.
The Company recognizes liabilities for uncertain tax positions based on a two-step process. The first step is to evaluate the tax position for recognition by determining if the weight of available evidence indicates that it is more likely than not that the position will be sustained on audit, including resolution of related appeals or litigation processes, if any. The second step is to measure the tax benefit as the largest amount that is more than 50% likely of being realized upon settlement. While the Company believes that it has appropriate support for the positions taken on its tax returns, the Company regularly assesses the potential outcome of examinations by tax authorities in determining the adequacy of its provision for income taxes.
The following table summarizes the activity related to the Company’s unrecognized tax benefits (in thousands):
| | AS OF DECEMBER 31, |
| AS OF DECEMBER 31, | | | AS OF DECEMBER 31, |
| | | 2020 | | 2019 | | 2023 | | 2022 |
| Beginning balance | Beginning balance | $ | 1,499 | | | $ | 230 | |
| Increases related to current year tax positions | Increases related to current year tax positions | 656 | | | 1,269 | |
| Ending balance | Ending balance | $ | 2,155 | | | $ | 1,499 | |
As of December 31, 2020,2023, the Company had gross unrecognized tax benefits of $2.2$4.7 million, none of which would affect the effective tax rate due to the existence of a full valuation allowance. The Company does not expect any significant increases or decreases to its unrecognized tax benefits within the next 12 months. The Company’s policy is to recognize the interest expense and/or penalties related to income tax matters as a component of income tax expense. The Company had 0no accrual for interest or penalties on its consolidated balance sheets as of December 31, 20202023 or December 31, 2019,2022, and has 0tnot recognized interest and/or penalties in its consolidated statements of operations for the years ended December 31, 20202023 and 2019.December 31, 2022 as the unrecognized tax benefits relate to tax positions for which no cash tax liability has been reduced.
The Company is subject to income taxes in the United States and California. Due to the carryforward of unutilized NOLs and R&D credits, thevarious state jurisdictions. The Company’s tax years from 20142019 and forward are subject to examination by the United States and Californiastate tax authorities. The Company has not been, nor is it currently, under examination by the U.S. federal or any state tax authority.
11.8. LEASES
Operating Leases
In September 2017, the Company entered into a seven-year lease agreement for approximately 34,000 square feet as its sole location in La Jolla, California. The lease expires in June 2025 with an option to extend the lease an additional five years.years, which is not included in the right-of-use asset and lease liabilities. The lease contained an initial base rent of approximately $0.1 million per month with 2% annual escalations, plus a percentage of taxes and operating expenses incurred by the lessor in connection with the ownership and management of the property, the latter of which to be determined annually.
In May 2019, the Company executed an amendment to its lease agreement to expand its facilities by approximately 9,000 square feet and began occupying this space in January 2020. The amended lease terminates coterminously with the initial lease agreement and contains an initial base rent of approximately $30,000 per month with 2% annual escalations, plus a percentage of taxes and operating expenses incurred by the lessor in connection with the ownership and management of the property, the latter of which is to be determined annually.
The operating right-of-use asset and operating lease liability as of December 31, 20202023 and 2019December 31, 2022 are as follows:follows (in thousands):
| | AS OF DECEMBER 31, | | AS OF DECEMBER 31, |
| 2020 | | 2019 |
| Right-of-use asset | $ | 7,831 | | | $ | 7,453 | |
| AS OF DECEMBER 31, | | | AS OF DECEMBER 31, |
| 2023 | | | 2023 | | 2022 |
| Operating right-of-use asset | |
| | Operating lease liability | Operating lease liability | |
| Operating lease liability | |
| Operating lease liability | |
| Current | |
| Current | |
| Current | Current | 1,503 | | | 1,105 | |
| Non-current | Non-current | 6,707 | | | 6,698 | |
| Total operating lease liability | Total operating lease liability | $ | 8,210 | | | $ | 7,803 | |
During the years ended December 31, 20202023 and 2019,December 31, 2022, the Company recognized operating lease expense of $3.1$3.7 million and $2.4$3.4 million, respectively. During each of the years ended December 31, 2023 and December 31, 2022, the Company paid $2.2 million for amounts included in the measurement of the operating lease liability in each period.
As of December 31, 20202023 and 2019,December 31, 2022, the Company’s operating lease had a remaining term of 4.51.5 and 5.52.5 years, respectively.
The Company discounts its lease payments using its incremental borrowing rate as of the commencement of the lease. The Company has determined a weighted-average discount rate of 8.2% and 8.0% as of December 31, 20202023 and 2019, respectively.December 31, 2022.
Future minimum rental payments undercommitments for the Company’s operating leases reconciled to the operating lease liability are as follows (in thousands):
| | | | | | | | |
| | | | AS OF DECEMBER 31, 2020 |
| | | | |
| | | | |
| 2021 | | | | $ | 2,119 | |
| 2022 | | | | 2,161 | |
| 2023 | | | | 2,203 | |
| 2024 | | | | 2,247 | |
| 2025 | | | | 1,137 | |
| | | | |
| Total future minimum lease payments | | | | $ | 9,867 | |
| Less: imputed interest | | | | (1,657) | |
| Present value of operating lease liability | | | | 8,210 | |
| Less: current portion of operating lease liability | | | | (1,503) | |
| Non-current portion of operating lease liability | | | | $ | 6,707 | |
| | | | | |
| AS OF DECEMBER 31, |
| 2023 |
| |
| |
| |
| |
| 2024 | $ | 2,247 | |
| 2025 | 1,137 | |
| |
| Total future minimum lease payments | $ | 3,384 | |
| Less: imputed interest | (211) | |
| Present value of operating lease liability | 3,173 | |
| Less: current portion of operating lease liability | (2,063) | |
| Non-current portion of operating lease liability | $ | 1,110 | |
12.9. COMMITMENTS AND CONTINGENCIES
Litigation
TheOther than as described below, the Company is not party to any material legal proceedings. From time to time, it may be involved in legal proceedings or subject to claims incident to the ordinary course of business. Regardless of the outcome, such proceedings or claims can have an adverse impact on the Company because of defense and settlement costs, diversion of resources and other factors, and there can be no assurances that favorable outcomes will be obtained.
I-Mab Litigation
On March 1, 2022, I-Mab Biopharma, or I-Mab, filed a lawsuit against the Company and Brendan Eckelman, the Company’s co-founder and Chief Scientific Officer, in the United States District Court for the District of Delaware, C.A. No. 22-00276-CJB, asserting claims for misappropriation of trade secrets related to Dr. Eckelman’s service as an expert witness for Tracon Pharmaceuticals, Inc., or Tracon, in Tracon’s arbitration against I-Mab. The case is currently in the discovery process.
Inhibrx is unable to reasonably estimate possible damages or a range of possible damages in this matter given the uncertainty and therefore has not recorded a liability on its books as of December 31, 2023.
Indemnification
In the ordinary course of business, the Company enters into agreements that may include indemnification provisions. Pursuant to such agreements, the Company may indemnify, hold harmless and defend an indemnified party for losses suffered or incurred by the indemnified party. In some cases, the indemnification obligation will continue after the termination of the agreement. The maximum potential amount of future payments the Company could be required to make under these provisions is not determinable. In addition, the Company has entered into indemnification agreements with its directors and certain officers that may require the Company, among other things, to indemnify them against certain liabilities that may arise by reason of their status or service as directors or officers. To date, no demands have been made upon the Company to provide indemnification under these agreements, and thus, there are no indemnification claims that the Company is aware of that could have a material effect on the Company’s consolidated balance sheets, consolidated statements of operations, or consolidated statements of cash flows.
Purchase Commitments
The Company has several ongoing contracts with CROs for preclinical studies and clinical trials and with CDMOs for clinical supplies and manufacturing scale-up activities. These contracts are generally cancellable, with notice, at the Company’s option. The Company recorded accrued expenses of approximately $11.5 million and $3.4 million in its consolidated balance sheet for expenditures incurred by CROs and CDMOs as of December 31, 2020 and December 31, 2019, respectively.
While these contracts are generally cancellable, some may contain specific activities that involve one or more non-cancellablenoncancellable commitments, including minimum purchase commitments, binding annual forecasts, and capital equipment investments. Additionally, depending on the timing and reasoning of the exit, certain termination penalties may apply and can range from cost of work performed to date and up to twelve months of future committed manufacturing costs. As of December 31, 20202023 and December 31, 2019,2022, the non-cancellablenoncancellable portion of these contracts total in aggregate, excluding amounts recorded in accounts payable and accrued expensespaid or incurred at each
respective date, approximately $7.0$62.8 million and $31.5$74.8 million, respectively. The noncancellable purchase commitments as of December 31, 2023 relate to the purchase of raw materials and future contract manufacturing of drug supply for INBRX-101. During the years ended December 31, 20202023 and 2019,December 31, 2022, the Company incurred $30.2$44.5 million and $1.3$0.3 million of expenses related to theseits non-cancellable purchase agreements, respectively.
Other Commitments
Additionally, as of December 31, 2023 and December 31, 2022, Mark P. Lappe, the Company’s Chief Executive Officer, Brendan P. Eckelman, the Company’s Chief Scientific Officer, Klaus W. Wagner, the Company’s Chief Medical Officer,and Kelly D. Deck, the Company’s Chief Financial Officer, and certain other members of management have agreements that provide for severance compensation in the event of termination or a change in control.
Contingencies
13. COVID-19
On January 30, 2020,In connection with its proposed Merger (as defined below), the World Health Organization, or WHO, announcedCompany engaged legal counsel under a global health emergency becausecontingent fee arrangement. Under this agreement, the Company is obligated to pay a fee of a new strain of coronavirus originating in Wuhan, China, or$20.0 million, contingent upon the COVID-19 outbreak, and the risks to the international community as the virus spreads globally beyond its point of origin. In March 2020, the WHO classified the COVID-19 outbreak as a pandemic, based on the rapid increase in exposure globally.
The continued spreadconsummation of the COVID-19 pandemicproposed transaction. In the event the Merger is affectingnot completed, the United States and global economies and may affect the Company’s operations and those of third parties on which the Company relies, including by causing disruptions in the supply of the Company’s therapeutic candidates and the conduct of future clinical trials. In addition, the COVID-19 pandemic may affect the operations of the FDA which could result in delays in meetings, reviews and approvals, including with respect to any of the Company’s therapeutic candidates. For example, as a
result of the COVID-19 pandemic patient travel restrictions, the Company temporarily suspended patient enrollment for its trial for INBRX-101 during the second and third quarters of 2020. Additionally, while the potential economic impact brought by, and the duration of, the COVID-19 pandemic is difficult to assess or predict, the impact of the COVID-19 pandemic on the global financial markets may reduce the Company’s ability to access capital, which could negatively impact the Company’s short-term and long-term liquidity. The ultimate impact of the COVID-19 pandemic is highly uncertain and subject to change. The Company does not yet know the full extent of potential delays or impacts onowe any fees to its business, financing or future clinical trial activities or on healthcare systems or the global economy as a whole. However, these effects could have a material impact on the Company’s business, financial condition, results of operations, and prospects, and those of the third parties relied upon by the Company.
CARES Act
On March 27, 2020, the U.S. federal government enacted the CARES Act, which includes provisions relating to refundable payroll tax credits, deferment of employer side social security payments, net operating loss carryback periods, alternative minimum tax credit refunds, modifications to the net interest deduction limitations and technical corrections to tax depreciation methods for qualified improvement property. The CARES Act also includes provisions for PPP loans administered by the SBA. The Company applied for and received PPP funds in May 2020 and subsequently repaid the loan in full in October 2020. See Note 4 for further discussion of the Company’s PPP loan.
The CARES Act also permits employers to defer the payment of the employer share of social security taxes due for the period beginning March 27, 2020 and ending December 31, 2020. Of the amounts deferred, 50% is required to be paid by December 31, 2021 and the remaining 50% is required to be paid by December 31, 2022. The Company began deferring payment of the employer share of social security taxes in April 2020. As of December 31, 2020, the Company had deferred payment of $0.4 million of such taxes, $0.2 million of which are classified as accrued expenses and $0.2 million of which are classified as other non-current liabilities in the Company’s consolidated balance sheets.
legal counsel. The Company will continue to examineincur these costs in full upon the consummation of the Merger, if and evaluatewhen it is completed, and has not incurred any impacts the CARES Act and associated subsequent legislation may have on its business and taxes.
14. SUBSEQUENT EVENTS
For the audited consolidated financial statementsportion of this fee as of December 31, 2020, the2023.
10. SUBSEQUENT EVENTS
The Company evaluated subsequent events to assess the need for potential recognition or disclosure in this report. Based upon this evaluation, it was determined that no additional subsequent events required recognition or disclosure in these consolidated financial statements, other than disclosures related to those outlined below.
DuringMerger and Spin-Off
On January 22, 2024, the firstCompany, Aventis Inc., or Parent, a wholly owned indirect subsidiary of Sanofi, and Art Acquisition Sub, Inc., or the Merger Sub, a wholly owned subsidiary of Parent, entered into an Agreement and Plan of Merger, or the Merger Agreement. Pursuant to the terms of the Merger Agreement, Parent will acquire all outstanding shares of the Company via the merger of Merger Sub with and into the Company, or the Merger, with the Company surviving the Merger as a wholly owned subsidiary of Parent, and in turn each shareholder will receive (i) $30.00 per share in cash, (ii) one contingent value right per share, representing the right to receive a contingent payment of $5.00 in cash upon the achievement of a regulatory milestone, and (iii) one SEC-registered, publicly listed, share of Inhibrx Biosciences, Inc., or New Inhibrx, for every four shares of Inhibrx common stock held. In addition, in connection with the transaction, Parent will (1) assume and retire the Company’s outstanding third-party debt, (2) cause New Inhibrx to be funded with $200.0 million in cash, and (3) retain an equity interest in New Inhibrx of approximately 8%.
In connection with and as a condition to the Merger, the Company and New Inhibrx entered into a Separation and Distribution Agreement, dated as of January 22, 2024, or the Separation and Distribution Agreement, pursuant to which, immediately prior to the effective time of the Merger: (i) the Company will effect a pre-closing reorganization, which will result in (x) the Company owning, assuming or retaining all assets and liabilities primarily related to INBRX-101, or the 101 Business, and (y) New Inhibrx owning, assuming or retaining all other assets and liabilities of the Company and its subsidiaries; and (ii) thereafter, the Company will distribute to its stockholders as of the record date on a pro rata basis, 92% of the issued and outstanding shares of New Inhibrx common stock, at a ratio of one share of New Inhibrx common stock for every four shares of the Company’s issued and outstanding common stock held on the record date. Following the spin-off, New Inhibrx will be a separate public company and the Company will retain 8% of the issued and outstanding shares of New Inhibrx common stock as of the effective time of the spin-off.
The boards of directors of both the Company and Sanofi have unanimously approved the spin-off and the Merger. Parent will pay transaction consideration totaling approximately $2.2 billion in aggregate value. Parent will also make payments at the closing of the Merger to settle the Company’s third-party debt. Following the closing of the Merger, New Inhibrx will continue to operate under the Inhibrx name. Parent’s acquisition of the Company is subject to the completion of the New Inhibrx spin-off transaction and other customary closing conditions, including
receipt of regulatory approvals and approval by the Company’s shareholders. The companies currently expect the transaction to close in the second quarter of 2021,2024.
The Merger Agreement contains certain termination rights for each of the Company granted stock optionsand Parent. Upon termination of the Merger Agreement in accordance with its terms, under certain circumstances, the Company will be required to purchasepay Parent a termination fee in an aggregate of 1.7 million common shares at a weighted average exercise price of $33.58 per share to employees under the 2017 Equity Incentive Plan. The Company granted these options with an exercise priceamount equal to $54.5 million, including if the fair market valueMerger Agreement is terminated due to (i) the Company accepting a Superior Proposal (as defined in the Merger Agreement) or (ii) the Board changing its recommendation that stockholders vote to approve the Merger Agreement. This termination fee will also be payable by the Company if the Merger Agreement is terminated under certain circumstances and prior to such termination, a proposal to acquire the 101 Business or more than 50% of the Company’s stock onor assets is made or publicly announced and not publicly withdrawn and the dateCompany enters into a definitive agreement for, or completes, any transaction involving the acquisition of the option grant.101 Business or more than 50% of its stock or assets within twelve months of such termination. The options are subjectMerger Agreement also provides that Parent will be required to four-year vesting with a one-year cliff and have a contractual term of 10 years.
In February 2021,pay the Company was awarded a grant fromreverse termination fee of $92.1 million if the DepartmentMerger is not consummated due to the failure of Defense in the amountcertain conditions to be satisfied as a result of approximately $0.3 million for the development of sdAb based therapeutics targeting SARS-CoV-2 Spike protein for the treatment of COVID-19. Work under the grant began in February 2021 and is expectedfailure to continue through February 2022. No cash reimbursements have been received under this grant to date.
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure.
None.
Item 9A. Controls and Procedures.
Evaluation of Disclosure Controls and Procedures
We maintain disclosure controls and procedures that are designed to ensure that information required to be disclosed in our reports filed pursuant to the Exchange Act is recorded, processed, summarized, and reported within the time periods specified in the SEC’s rules and forms, and that such information is accumulated and communicated to our management, including our principal executive officer and principal financial and accounting officer, as appropriate, to allow timely decisions regarding required disclosure.
Our management, with the participation and supervision of our Chief Executive Officer and our Chief Financial Officer, evaluated the effectiveness of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act) as of the end of the period covered by this Annual Report on Form 10-K.Report. Based on that evaluation, our Chief Executive Officer and our Chief Financial Officer have concluded that, as of the end of the period covered by this Annual Report, on Form 10-K, our disclosure controls and procedures were designed and operating effectively at the reasonable assurance level.
Management’s Annual Report on Internal Control over Financial Reporting
This annual report does not include a report of management's assessment regarding internal control over financial reporting or an attestation report of the company's registered public accounting firm due to a transition period established by rules of the SEC for newly public companies.
Changes in Internal Control over Financial Reporting
There were no changes in our internal control over financial reporting that occurred during the quarter ended December 31, 2020 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Inherent Limitations on Effectiveness of Controls
Our management, including our Chief Executive Officer and our Chief Financial Officer, does not expect that our disclosure controls or our internal control over financial reporting will prevent or detect all error and all fraud. A control system, no matter how well designed and operated, can provide only reasonable and not absolute assurance of achieving the desired control objectives. In reaching a reasonable level of assurance, management necessarily was required to apply its judgment in evaluating the benefits of possible controls and procedures relative to their costs. Controls can also be circumvented by the individual acts of some persons, by collusion of two or more people, or by management override of the controls. In addition, the design of any system of controls is based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions. Over time, controls may become inadequate because of changes in conditions or deterioration in the degree of compliance with policies or procedures.
Changes in Internal Control over Financial Reporting
There were no changes in our internal control over financial reporting that occurred during the quarter ended December 31, 2023 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Management’s Annual Report on Internal Control over Financial Reporting
Internal control over financial reporting is defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act, as amended, as a process designed by, or under the supervision of, the company’s principal executive and principal financial officers and effected by the Company’s board of directors, management and other personnel to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with GAAP. The Company’s internal control over financial reporting includes those policies and procedures that:
•pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the Company;
•provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with GAAP, and that receipts and expenditures of the Company are being made only in accordance with authorizations of management and directors of the company; and
•provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the Company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become
inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
The Company’s management assessed the effectiveness of the Company’s internal control over financial reporting as of December 31, 2023. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission, or COSO, in Internal Control-Integrated Framework.
Based on our assessment, management believes that, as of December 31, 2023, the Company’s internal control over financial reporting is effective based on those criteria.
The Company’s independent registered public accounting firm has issued an audit report on our assessment of the Company’s internal control over financial reporting. This report appears further below in this Item 9A.
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
Board of ContentsDirectors and Stockholders
Inhibrx, Inc.
La Jolla, California
Opinion on Internal Control over Financial Reporting
We have audited Inhibrx, Inc.’s (the “Company’s”) internal control over financial reporting as of December 31, 2023, based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (the “COSO criteria”). In our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2023, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (“PCAOB”), the consolidated balance sheets of the Company as of December 31, 2023 and 2022, the related consolidated statements of operations, stockholders’ equity, and cash flows for each of the years then ended, and the related notes and our report dated February 28, 2024 expressed an unqualified opinion thereon.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying “Item 9A, Management’s Annual Report on Internal Control over Financial Reporting”. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit of internal control over financial reporting in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audit also included performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ BDO USA, P.C.
San Diego, California
February 28, 2024
Item 9B. Other Information.
None.During the fiscal quarter ended December 31, 2023, none of our directors or officers (as defined in Section 16 of the Securities Exchange Act of 1934, as amended) adopted or terminated any contract, instruction or written plan for the purchase or sale of our securities that was intended to satisfy the affirmative defense conditions of Rule 10b5-1(c) or any “non-Rule 10b5-1 trading arrangement,” as defined in Item 408(a) of Regulation S-K.
On February 26, 2024, we terminated the PD-L1 and 4-1BB License Agreement with Elpiscience, pursuant to which we granted Elpiscience an exclusive license to our bi-specific therapeutic candidate designed to target PD-L1 and 4-1BB, also referred to as INBRX-105, including all ancillary agreements related to the PD-L1 and 4-1BB License Agreement. After evaluation of the totality of the data from the expansion cohorts for INBRX-105, the initial signal was not sufficiently validated to support the continuation of the INBRX-105 program. Immediately prior to the termination of the PD-L1 and 4-1BB License Agreement, no amounts were outstanding under the agreement.
Item 9C. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections.
Not applicable.
Part III.
Item 10. Directors, Executive Officers and Corporate Governance.
The response to this item is incorporated by reference from the discussion responsive thereto under the captions “Management
Management and Corporate Governance”Governance
The Company is committed to maintaining strong corporate governance practices that benefit the long-term interests of our stockholders by providing for effective oversight and “Codemanagement of the Company. The board of directors and management believe that the Company has a sound governance structure in place for both management and the board of directors. The Company’s corporate governance documents and policies can be accessed on the Investors page of our website at www.inhibrx.com. The board of directors and its committees regularly review the Company’s corporate governance documents and policies to ensure that they take into account developments at the Company, changes in regulations and listing requirements, and the continuing evolution of best practices in the area of corporate governance.
Corporate Code of Conduct and Ethics”Ethics
Inhibrx has adopted a Corporate Code of Conduct and Ethics and Whistleblower Policy that applies to all of our officers, directors, and employees. The Corporate Code of Conduct and Ethics and Whistleblower Policy is available on the Governance Documents section of the Investors page on our website at www.inhibrx.com. We intend to disclose any amendments to the Corporate Code of Conduct and Ethics and Whistleblower Policy, or any waivers of our requirements, on our website to the extent required by applicable laws.
Board Leadership and Risk Oversight
We do not have a policy regarding the separation of the roles of Chief Executive Officer and Chairman of the board of directors, as our Board believes it is in the best interests of the Company to make that determination based on the position and direction of the Company and the membership of the board of directors. Mark P. Lappe is our proxy statementcurrent Chief Executive Officer and our current Chairman of the board of directors. Our Board has determined that having an employee director serve as Chairman is in the best interests of our stockholders at this time because of the efficiencies achieved in having the role of Chief Executive Officer and Chairman of the Board combined, and because the detailed knowledge of our day-to-day operations and business that the Chief Executive Officer possesses greatly enhances the decision-making processes of our Board as a whole.
We do not have a lead independent director. The Chairman of the board of directors and the other members of the Board work in concert to provide oversight of our management and affairs. Our Board encourages communication among its members and between management and the Board to facilitate productive working relationships. Working with the other members of the board of directors, our Chairman also strives to ensure that there is an appropriate balance and focus among key responsibilities such as strategic development, review of operations, and risk oversight.
Risk is inherent with every business, and how well a business manages risk can ultimately determine its success. We face a number of risks, including, but not limited to, risks relating to financial condition, therapeutic candidate development, dependence on collaborative partners, uncertainty regarding patents and proprietary rights, comprehensive government regulations, cybersecurity, and dependence on key personnel. Management is responsible for the 2021day-to-day management of the risks we face, while our Board as a whole, and through its committees, is responsible for the oversight of risk management. In its risk oversight role, our Board has the responsibility to satisfy itself that the risk management processes designed and implemented by management are adequate and functioning as designed. Our Board satisfies this responsibility, in part, through reports by each committee chairperson regarding the committee’s considerations and actions. The Board also has the responsibility of ensuring compliance with the risk management processes designed and implemented by management, which it satisfies through reports directly from the officers responsible for oversight of particular risks within our Company. The Board believes that full and open communication between management and the Board is essential for effective risk management and oversight.
Director Independence
Our Board annually reviews the independence of all of our directors and affirmatively makes a determination as to the independence of each director based on whether such director satisfies the definition of “independent director” as set forth in the applicable rules of the Nasdaq Stock Market and the SEC. Our Board has determined that all of our directors are independent directors other than Mark P. Lappe, by virtue of his position as Chief Executive Officer of the Company. There are no family relationships among any of our directors or executive officers.
Stockholder Communications with the Board of Directors
The Company desires that the views of stockholders be heard by the board of directors, and that appropriate responses be provided to stockholders on a timely basis. Stockholders wishing to formally communicate with the Board or any individual director may send communications directly to the Company by mail at Inhibrx, Inc., 11025 N. Torrey Pines Rd., Suite 200, La Jolla, California 92037, Attention: Corporate Secretary, or electronically using the “Send us a Message” option on the Contact Us page on our website at www.inhibrx.com. Communications will be distributed to our board of directors, or to any individual director or directors as appropriate, depending on the facts and circumstances outlined in the communications.
Our Board of Directors
Our amended and restated certificate of incorporation provides that our business is to be managed by or under the direction of our board of directors. Our Board is divided into three classes for purposes of election. One class is elected at each annual meeting of stockholders to serve for a three-year term.
The table below sets forth the names of the Board’s five directors and their ages, their positions in the Company, if any, and their length of service. Also provided below is information regarding each person’s principal occupations or employment for at least the past five years, additional public company directorships held during the past five years, and each person’s specific experience, qualifications, attributes, or skills that led to our board of directors’ conclusion at the time of filing of this Annual Report that each person should serve as a director.
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| Name | | Age | | Position(s) | | Director Since | | Expiration of Term |
| Mark P. Lappe | | 57 | | Chairman of the Board of Directors, President, and Chief Executive Officer | | 2010 | | 2026 |
| Douglas G. Forsyth | | 55 | | Director | | 2018 | | 2025 |
| Jon Faiz Kayyem, Ph.D. | | 60 | | Director | | 2018 | | 2024 |
| Kimberly Manhard | | 64 | | Director | | 2020 | | 2025 |
| Kristiina Vuori, M.D., Ph.D. | | 56 | | Director | | 2021 | | 2024 |
Mark P. Lappe
Mr. Lappe co-founded Inhibrx in 2010 and has served as our Chief Executive Officer since our inception and served as our Chief Financial Officer until April 2020. Mr. Lappe also serves as the Chairman of our board of directors. Mr. Lappe has extensive expertise in the biotechnology industry with over thirty years of experience in executive management, investment management and executive recruiting, having built the executive teams of over forty start-up biotechnology and medical device companies. Prior to founding Inhibrx, Mr. Lappe was the founder and Managing Partner of Efficacy Biotech Fund, a fund focused on strategic investment in public biotechnology companies. We believe Mr. Lappe’s service as our co-founder and Chief Executive Officer and his extensive experience in the biotechnology and biotherapeutic industries qualifies him to serve on our board of directors.
Douglas G. Forsyth
Mr. Forsyth has served as a member of our Board since April 2018. From 1994 until February 2022, Mr. Forsyth served as a portfolio manager, a managing director and Chief Investment Officer of U.S. Income & Growth Strategies with Allianz Global Investors GmbH, or Allianz. Mr. Forsyth was also the head of the Allianz Income and Growth Strategies team, during which time he had portfolio management, trading and research responsibilities, and oversaw all aspects of the Income and Growth platform’s business, including product development and
implementation. Mr. Forsyth was the lead portfolio manager for the Allianz U.S. High Yield Bond strategy since its inception in August 1994 and assumed lead portfolio management responsibility for the firm’s U.S. Convertible strategy in 1998. In addition to management responsibility for institutional clients worldwide, Mr. Forsyth supervised multiple open-end and closed-end mutual funds. Mr. Forsyth has 30 years of investment-industry experience. Mr. Forsyth currently serves, or has served, on a variety of non-public company boards across a variety of industries as well as several charitable boards. Mr. Forsyth received a Bachelor of Business Administration from the University of Iowa. We believe that Mr. Forsyth is qualified to serve as a member of our board of directors due to his extensive experience as an investment fund portfolio manager and his extensive business strategy experience.
Jon Faiz Kayyem, Ph.D.
Dr. Kayyem has served as a member of our Board since April 2018. He is the founder of GenMark Diagnostics Inc., or GenMark, and from May 2010 until February 2018, Dr. Kayyem served in various leadership positions at GenMark including Senior Vice President of Research and Development, Chief Scientific Officer, President, and Chief Executive Officer as the company evolved from Osmetech plc. In 2018, Dr. Kayyem became a consultant to GenMark's board of directors and executive team. In March 2021, GenMark agreed to merge with Roche, which subsequently acquired GenMark through a tender offer. In 2006, Dr. Kayyem founded and served as a member of the board of directors of privately-held Calimmune, Inc. until its acquisition by CSL Behring in 2017. In 1995, he founded Clinical Micro Sensors, Inc. to commercialize technical innovations he developed while serving as a Senior Research Fellow at the California Institute of Technology, or Caltech. In 2000, Clinical Micro Sensors, Inc. was sold to Motorola, Inc. and was subsequently purchased by Osmetech plc in 2005. From June 2000 until December 2004, Dr. Kayyem served as Vice President of Life Sciences at Motorola. In October 2004, he co-founded the biotechnology fund management company, Efficacy Capital Limited, and, until September 2009, served as a managing partner. Dr. Kayyem currently serves as a member of the board of directors of Biodesix, Inc. He also currently serves, or has served, as a director or advisor on various non-public company boards, including non-profit and educational institutions. Dr. Kayyem received a combined B.S. and M.S. in Molecular Biophysics and Biochemistry from Yale University and a Ph.D. in Molecular Biology from Caltech. We believe that Dr. Kayyem’s extensive experience as an executive and serving on other boards of directors in the biotechnology and biotherapeutic industry qualifies him to serve as a member of our board of directors.
Kimberly Manhard
Ms. Manhard has served as a member of our Board since June 2020. Ms. Manhard has served as the Senior Vice President of Global Strategic Planning and Execution for Acadia Pharmaceuticals Inc., or Acadia, since February 2024. Prior to Acadia, Ms. Manhard served as the Executive Vice President, Drug Development of Heron Therapeutics, Inc., or Heron, from January 2016 to June 2023. She also served as a director of Heron from 2014 to 2016 and from 2019 to February 2023. From 2006 to 2016, she served as the Senior Vice President of Regulatory Affairs and Development Operations at Ardea Biosciences, Inc., or Ardea. In her role at Ardea, Ms. Manhard was instrumental in the development and 2015 regulatory approval of lesinurad (Zurampic). Prior to joining Ardea in 2006, she was President of her own consultancy firm, Vice President of Regulatory Affairs for Exelixis, Inc., and held multiple regulatory positions at Agouron Pharmaceuticals, Inc., a division of the Warner-Lambert Company, supporting development and commercialization of anticancer and antiviral products, including nelfinavir (Viracept). Ms. Manhard was also previously with Bristol-Myers Squibb Company in regulatory affairs, responsible for oncology compounds, including paclitaxel (Taxol) and infectious disease compounds, including didanosine (Videx) and stavudine (Zerit). Ms. Manhard began her career in clinical research with Eli Lilly and Company and G.H. Besselaar Associates (Covance Inc.). Ms. Manhard is a member of the board of trustees for the Fleet Science Center. She received a B.S. degree in zoology and a B.A. degree in French from the University of Florida. We believe that Ms. Manhard is qualified to serve on our board of directors due to her experience in executive management and as a director with other biotechnology and pharmaceutical companies, as well as her experience in drug development, regulatory affairs, and pharmaceutical operations.
Kristiina Vuori, M.D., Ph.D.
Dr. Vuori has served as a member of our Board since October 2021. Since January 2023, she has served as President and CEO and a member of the board of directors of Sanford Laboratories for Innovative Medicines, or Sanford Labs, an independent, not-for-profit biomedical research institution with a mission to discover and deliver the next-generation of molecular medicines. Since January 1995, Dr. Vuori has also served as Professor, and since January
2010 as Pauline and Stanley Foster Distinguished Chair, at the National Cancer Institute-designated Cancer Center, or Cancer Center, of Sanford Burnham Prebys Medical Discovery Institute, or SBP, a non-profit research organization with major research programs in cancer, neurodegeneration, diabetes, and infectious, inflammatory, and childhood diseases. In addition, Dr. Vuori served as President and a member of the board of directors of SBP from January 2010 to June 2022, and as SBP’s interim Chief Executive Officer from January 2013 to September 2014, and from September 2017 to June 2020. She also served as SBP’s Executive Vice President for Scientific Affairs in 2008-2010, as Director of the Cancer Center in 2005-2013, and as Deputy Director of the Cancer Center in 2003-2005. Dr. Vuori has previously served or is currently serving on the board of directors of Bionano Genomics, Inc., Sio Gene Therapies, Inc., Forian Inc., WebMD Health Corp, Inc., the American Association for Cancer Research, the California Institute for Regenerative Medicine and the California Breast Cancer Research Program. Dr. Vuori received her M.D. and Ph.D. from the University of Oulu, Finland. We believe that Dr. Vuori’s experience as a physician-scientist in biomedical research and drug discovery and as an educator of research scientists, her experience managing a large non-profit biomedical research organization, and her various leadership roles in non-profit, for-profit, and public boards qualify her to serve as a member of our board of directors.
Board Meetings and Attendance
During the fiscal year ended December 31, 2023, our board of directors met eight times and acted by written consent zero times. No director attended fewer than 75% of the total number of meetings of our board of directors and of Board committees on which he or she served during the fiscal year ended December 31, 2023. Our board of directors has adopted a policy requiring that each member of the Board make every effort to attend each annual meeting of our stockholders. All the individuals who were directors of the Company at the time of the 2023 annual meeting of stockholders attended that meeting.
Committees of our Board of Directors
Our Board has established three standing committees: the Audit Committee, the Compensation Committee, and the Nominating Committee, each of which is described more fully below.
Audit Committee
Our Audit Committee is comprised of Douglas G. Forsyth, Jon Faiz Kayyem, Ph.D., and Kristiina Vuori, M.D., Ph.D., with Mr. Forsyth serving as the Chairperson. All members of the Audit Committee satisfy the current independence standards promulgated by the SEC and by Nasdaq, as such standards apply specifically to members of audit committees. Our Board has determined that Mr. Forsyth is an “audit committee financial expert,” as the SEC has defined that term in Item 407 of Regulation S-K. The Audit Committee met four times during 2023.
Our Audit Committee’s roles and responsibilities are set forth in the Audit Committee’s written charter and include the authority to retain and terminate the services of our independent registered public accounting firm. In addition, the Audit Committee reviews annual financial statements, considers matters relating to accounting policy and internal controls, and reviews the scope of annual audits.
A copy of the Audit Committee’s written charter is publicly available on the Governance Documents section of the Investors page on our website at www.inhibrx.com.
Compensation Committee
Our Compensation Committee is comprised of Jon Faiz Kayyem, Ph.D., Douglas G. Forsyth, and Kimberly Manhard, with Dr. Kayyem serving as the Chairperson. Our Board has determined that all members of the Compensation Committee qualify as independent under the definition promulgated by Nasdaq. During 2023, the Compensation Committee met two times and acted by written consent two times.
Our Compensation Committee’s roles and responsibilities are set forth in the Compensation Committee’s written charter and include reviewing, approving, and making recommendations regarding our compensation policies, practices, and procedures to ensure that legal and fiduciary responsibilities of our board of directors are carried out and that such policies, practices, and procedures contribute to our success. Our Compensation Committee also administers our equity compensation plans. The Compensation Committee may delegate authority for day-to-day administration and interpretation of the Company’s various compensation plans, including the selection of
participants, the determination of award levels, and the approval of award documents to our non-executive officer employees. However, the Compensation Committee may not delegate any authority under those plans for matters affecting the compensation and benefits of the Company’s executive officers. The Chief Executive Officer is not present for any discussions relating to his compensation.
A copy of the Compensation Committee’s written charter is publicly available on the Governance Documents section of the Investors page on our website at www.inhibrx.com.
Nominating Committee
Our Nominating Committee is comprised of Douglas G. Forsyth, Kimberly Manhard, and Kristiina Vuori, M.D., Ph.D., with Mr. Forsyth serving as the Chairperson. Our Board has determined that all members of the Nominating Committee qualify as independent under the definition promulgated by Nasdaq. During 2023, the Nominating Committee met two times.
The Nominating Committee’s responsibilities are set forth in the Nominating Committee’s written charter and include, among other matters:
•evaluating and making recommendations to the full Board as to the composition, organization, and governance of the Board and its committees,
•evaluating, nominating, and recommending individuals for membership on our board of directors,
•periodically reviewing and evaluating director performance,
•overseeing the process for CEO and other executive officer succession planning, and
•developing and recommending governance policies and guidelines for the Company.
A copy of the Nominating Committee’s written charter is publicly available on the Governance Documents section of the Investors page on our website at www.inhibrx.com.
Our Executive Officers
As of the date of this Annual Report, our current executive officers and their respective ages and positions are set forth in the following table:
| | | | | | | | | | | | | | |
| Name | | Age | | Position |
Mark P. Lappe (1) | | 57 | | Chief Executive Officer and President |
| Kelly D. Deck, C.P.A. | | 44 | | Chief Financial Officer |
| Brendan P. Eckelman, Ph.D. | | 45 | | Chief Scientific Officer |
________________(1) Mr. Lappe is also a director of the Company and his biographical information appears on page 153. Kelly D. Deck, C.P.A.
Ms. Deck currently serves as our Chief Financial Officer and leads our accounting, finance, information technology, and investor relations functions. She joined Inhibrx in October 2018, bringing decades of experience in the life science industry. Prior to joining our team, Ms. Deck held various accounting and finance positions of increasing responsibility with Apricus Biosciences, Inc., Hologic, Inc., Gen-Probe, Inc., and Cytori Therapeutics, Inc. Ms. Deck is currently a member of the board of trustees for the Fleet Science Center. Ms. Deck holds a B.S. and M.S. in Accounting and is an active licensed Certified Public Accountant in the state of California.
Brendan P. Eckelman, Ph.D.
Dr. Eckelman co-founded Inhibrx in April 2010 and currently serves as our Chief Scientific Officer. Until October 2021, he also served as our Executive Vice President of Corporate Strategy. He served as a member of our board of directors from April 2018 until October 2021. From August 2015 to November 2018, he served as our Chief Operating Officer and Vice President of Biotherapeutics. From 2010 until August 2015, Dr. Eckelman served as our Vice President of Scientific Operations. Dr. Eckelman is the head of our research team, overseeing several key functional areas spanning discovery and protein engineering to therapeutic development. Prior to co-founding Inhibrx, Dr. Eckelman was a Research Investigator in the biotherapeutics group at the Genomics Institute of the Novartis Research Foundation. He conducted his graduate research at the Sanford-Burnham-Prebys Medical Discovery Institute and received a Ph.D. in Molecular Pathology from the University of California, San Diego, or UCSD, School of Medicine. Dr. Eckelman received his B.S. in Molecular Biology and his M.S. in Biology from UCSD.
Item 11. Executive Compensation.
Non-Employee Director Compensation Table
The response to this item is incorporatedfollowing table sets forth a summary of the compensation earned by reference fromour non-employee directors for their service on our Board during the discussion responsive thereto underyear ended December 31, 2023:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Name | | Fees Earned or Paid in Cash ($) | | Stock Awards
($) | | Option Awards ($)(1) | | All Other Compensation ($)(2) | | Total
($) |
| Douglas G. Forsyth | | $ | 63,000 | | | $ | — | | | $ | 254,030 | | | $ | — | | | $ | 317,030 | |
| Jon Faiz Kayyem, Ph.D. | | $ | 52,500 | | | $ | — | | | $ | 254,030 | | | $ | — | | | $ | 306,530 | |
| Kimberly Manhard | | $ | 44,000 | | | $ | — | | | $ | 254,030 | | | $ | — | | | $ | 298,030 | |
| | | | | | | | | | |
| Kristiina Vuori, M.D., Ph.D. | | $ | 46,500 | | | $ | — | | | $ | 254,030 | | | $ | — | | | $ | 300,530 | |
________________
(1)These amounts represent the caption “Executive Officer and Director Compensation”aggregate grant date fair value of time-based stock option awards granted by the Company during the periods presented, determined in accordance with FASB ASC Topic 718. All awards are amortized over the vesting life of the award. For the assumptions used in our proxy statementvaluations, see “Note 5 – Equity Compensation Plans” to our consolidated financial statements included elsewhere in this Annual Report.
(2)Excludes amounts reimbursed for out-of-pocket expenses incurred in connection with service as a director.
Non-Employee Director Compensation - Equity
As of December 31, 2023, non-employee directors held options to purchase shares of our common stock pursuant to the 2021terms of our Amended and Restated 2017 Employee, Director and Consultant Equity Incentive Plan, or 2017 Plan, as follows:
| | | | | | | | | | | | | | |
| Name | | Stock Awards Outstanding at December 31, 2023 (#) | | Option Awards Outstanding at December 31, 2023 (#) |
| Douglas G. Forsyth. | | — | | | 62,259 | |
| Jon Faiz Kayyem, Ph.D. | | — | | | 62,259 | |
| Kimberly Manhard | | — | | | 65,759 | |
| Kristiina Vuori, M.D., Ph.D. | | — | | | 60,000 | |
| | | | |
Non-Employee Director Compensation Policy
We have adopted a Non-Employee Director Compensation Policy that is designed to enable us to attract and retain, on a long-term basis, highly qualified non-employee directors. Under the Non-Employee Director Compensation Policy, each non-employee director is eligible to receive the following amounts for their service on our board of directors:
•Initial Stock Option Grant. For new non-employee directors, a stock option to purchase 30,000 shares of the Company’s common stock.
•Annual Stock Option Grants. A stock option to purchase 15,000 shares of the Company’s common stock, on the date of the first Board meeting held following the Company’s annual meeting of stockholders.stockholders each year.
•Annual Fees. Annual retainers in the following amounts, payable quarterly in arrears:
| | | | | | | | |
| Position | | Retainer |
| Board Member | | $ | 35,000 | |
| Board Chairperson | | $ | 35,000 | |
| Audit Committee Chair | | $ | 15,000 | |
| Compensation Committee Chair | | $ | 10,000 | |
| Nominating Committee Chair | | $ | 8,000 | |
| Audit Committee Member | | $ | 7,500 | |
| Compensation Committee Member | | $ | 5,000 | |
| Nominating Committee Member | | $ | 4,000 | |
All options granted under our Non-Employee Director Compensation Policy will have an exercise price equal to the fair market value of the Company’s common stock on the date of grant and terminate on the tenth anniversary of the date of grant. Subject to the continued service of each non-employee director, each annual stock option grant vests on the first anniversary of the date of grant and each initial stock option grant vests in equal monthly installments until the third anniversary of the date of grant.
Directors may be reimbursed for reasonable out-of-pocket expenses incurred in connection with their service as directors. Directors are also entitled to the protection provided by their indemnification agreements and the indemnification provisions in our amended and restated certificate of incorporation and amended and restated bylaws.
Summary Compensation Table
The following table sets forth a summary of cash and non-cash compensation awarded, earned, or paid for services rendered to us during the fiscal years ended December 31, 2023 and December 31, 2022 by our named executive officers, consisting of (i) each individual serving as principal executive officer during the fiscal year ended December 31, 2023 and (ii) our other two most highly compensated officers serving during the fiscal year ended December 31, 2023.
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Name and Principal Position | | Year | | Salary
($) | | Bonus ($) | | Option Awards ($)(1) | | Non-Equity Incentive Plan Compensation ($)(2) | | All Other Compensation ($)(3) | | Total
($) |
| Mark P. Lappe | | 2023 | | 675,000 | | | — | | | 1,706,771 | | | 405,000 | | | 15,250 | | | 2,802,021 | |
| Chief Executive Officer and President | | 2022 | | 650,000 | | | — | | | — | | | 357,500 | | | 15,250 | | | 1,022,750 | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| Brendan P. Eckelman, Ph.D. | | 2023 | | 520,000 | | | — | | | 682,708 | | | 234,000 | | | 15,250 | | | 1,451,958 | |
| Chief Scientific Officer | | 2022 | | 475,000 | | | — | | | — | | | 213,750 | | | 15,250 | | | 704,000 | |
| Kelly D. Deck, C.P.A. | | 2023 | | 430,000 | | | — | | | 512,031 | | | 193,500 | | | 13,368 | | | 1,148,899 | |
| Chief Financial Officer | | 2022 | | 380,000 | | | — | | | 491,327 | | | 133,000 | | | 18,168 | | | 1,022,495 | |
________________(1)These amounts represent the aggregate grant date fair value of time-based stock option awards granted by the Company during the periods presented, determined in accordance with FASB ASC Topic 718. All awards are amortized over the vesting life of the award. For the assumptions used in our valuations, see “Note 5 – Equity Compensation Plans” to our consolidated financial statements included elsewhere in this Annual Report. Awards issued during the year ended December 31, 2023 are reflected in the “Outstanding Equity Awards as of December 31, 2023” table below.
(2)These amounts represent performance-based cash bonuses earned during the year ended December 31, 2023, which were paid out during the first quarter of 2024. The annual performance-based cash bonus for each
named executive officer is defined within their respective employment agreement and is measured based upon the achievement of Company objectives for the periods presented.
(3)These amounts represent matching employer contributions under the Company’s 401(k) retirement plan and employer contributions to health savings accounts. The formula for determining the matching contributions is the same for named executive officers as it is for all salaried employees (and is subject to the same statutory maximum).
Narrative Disclosure to Summary Compensation Table
Employment Arrangements with our Named Executive Officers
We have entered into executive employment agreements with each of our named executive officers in connection with their employment with us, the material terms of which are described below. Except as noted below, these executive employment agreements provide for “at will” employment. In addition, each named executive officer has entered into a Proprietary Information and Inventions Assignment Agreement obligating the officer to refrain from disclosing any of our proprietary information received during the course of employment and to assign to us any inventions conceived or developed during the course of employment.
Employment Agreement with Mark P. Lappe
On January 1, 2019, we entered into an employment agreement with Mark P. Lappe, setting forth the terms and conditions of his employment with the Company. Mr. Lappe was entitled to an annual base salary of $450,000 and eligible to receive an annual bonus of up to 50% of his then-current base salary based on achievement of certain corporate goals. In addition, in consideration of the payments and benefits provided under his employment agreement, Mr. Lappe agreed to certain restrictive covenants, including, among other things, non-competition and non-solicitation provisions that apply during the term of Mr. Lappe’s employment and for 12 months thereafter.
Mr. Lappe is also entitled to certain severance and change in control benefits pursuant to his employment agreement, the terms of which are described below under “Potential Payments upon Termination or Change of Control.”
On August 7, 2020, Mr. Lappe entered into an amended and restated employment agreement, pursuant to which his annual base salary increased to $650,000 per year. All other terms remained the same as the prior agreement.
Effective January 1, 2022, Mr. Lappe entered into an amended and restated employment agreement, pursuant to which he is eligible to receive an annual bonus of up to 55% of his then-current base salary based on achievement of certain corporate goals. All other terms remained the same as the prior agreement.
Effective January 1, 2023, Mr. Lappe entered into an amended and restated employment agreement, pursuant to which his annual base salary increased to $675,000 per year and the annual bonus that he is eligible to receive increased to up to 60% of his then-current base salary based on achievement of certain corporate goals. All other terms remained the same as the prior agreement.
Effective January 1, 2024, Mr. Lappe’s annual base salary increased to $695,250 per year. No other terms of his employment agreement changed and thus his employment agreement was not amended and restated.
Employment Agreement with Brendan P. Eckelman, Ph.D.
On January 1, 2019, we entered into an employment agreement with Brendan P. Eckelman, Ph.D., setting forth the terms and conditions of his employment with the Company. Dr. Eckelman was entitled to an annual base salary of $350,000 and eligible to receive an annual bonus of up to 40% of his then-current base salary based on achievement of certain corporate goals. In addition, in consideration of the payments and benefits provided under his employment agreement, Dr. Eckelman agreed to certain restrictive covenants, including, among other things, non-competition and non-solicitation provisions that apply during the term of Dr. Eckelman’s employment and for 12 months thereafter.
Dr. Eckelman is also entitled to certain severance and change in control benefits pursuant to his employment agreement, the terms of which are described below under “Potential Payments upon Termination or Change of Control.”
On August 7, 2020, Dr. Eckelman entered into amended and restated employment agreement, pursuant to which his annual base salary increased to $450,000 per year. All other terms remained the same as the prior agreement, other than payments upon termination or change of control, which are described below.
Effective January 1, 2022, Dr. Eckelman entered into an amended and restated employment agreement, pursuant to which his annual base salary increased to $475,000 per year and he is eligible to receive an annual bonus of up to 45% of his then-current base salary based on achievement of certain corporate goals. All other terms remained the same as the prior agreement.
Effective January 1, 2023, Dr. Eckelman's annual base salary increased to $520,000 per year. No other terms of his employment agreement changed and thus his employment agreement was not amended and restated.
Effective January 1, 2024, Dr. Eckelman's annual base salary increased to $535,600 per year. No other terms of his employment agreement changed and thus his employment agreement was not amended and restated.
Employment Agreement with Kelly D. Deck, C.P.A.
On August 7, 2020, we entered into an employment agreement with Kelly D. Deck, C.P.A., setting forth the terms and conditions of her employment with the Company. Ms. Deck was entitled to an annual base salary of $330,000 and eligible to receive a performance bonus of up to 25% of her then-current base salary based on achievement of certain corporate goals. In addition, in consideration of the payments and benefits provided under her employment agreement, Ms. Deck agreed to certain restrictive covenants, including, among other things, non-competition and non-solicitation provisions that apply during the term of Ms. Deck’s employment and for 12 months thereafter.
Ms. Deck is also entitled to certain severance and change in control benefits pursuant to her employment agreement, the terms of which are described below under “Potential Payments upon Termination or Change of Control.”
Effective January 1, 2022, Ms. Deck entered into an amended and restated employment agreement, pursuant to which her annual base salary increased to $380,000 per year and she is eligible to receive an annual bonus of up to 35% of her then-current base salary based on achievement of certain corporate goals. All other terms remained the same as the prior agreement.
Effective January 1, 2023, Ms. Deck entered into an amended and restated employment agreement, pursuant to which her annual base salary increased to $430,000 per year and the annual bonus that she is eligible to receive increased to up to 45% of her then-current base salary based on achievement of certain corporate goals. All other terms remained the same as the prior agreement.
Effective January 1, 2024, Ms. Deck’s annual base salary increased to $460,100 per year. No other terms of her employment agreement changed and thus her employment agreement was not amended and restated.
Potential Payments Upon Termination or Change In Control
The employment agreements of Mr. Lappe, Dr. Eckelman, and Ms. Deck as of December 31, 2023 provided for the following severance payments upon termination by the Company without Cause (as defined below), or by the executive for Good Reason (as defined below): (i) payment of the executive’s then-current base salary for a period of 12 months following termination; (ii) acceleration of unvested equity awards that would have vested during the 12 months following the date of termination; and (iii) continued coverage under our group health insurance plan with the cost of such coverage shared in the same relative proportion by the Company and the executive as in effect on their last day of employment until the earlier of 12 months from termination or the date the executive becomes eligible for medical benefits with another employer.
Further, the agreements provided that upon termination by the Company without Cause or by the executive for Good Reason within a period of one year following a Change of Control (as defined below), or 90 days preceding the earlier to occur of a Change of Control or the execution of a definitive agreement the consummation of which would result in a Change of Control, the executive would be entitled to receive: (i) a lump sum payment equal to 18 months of the executive’s then-current base salary; (ii) a lump sum payment equal to 1.5 times the target bonus for the year of termination; (iii) acceleration of all unvested equity awards as of the date of termination; and (iv) continued
coverage under our group health insurance plan with the cost of such coverage shared in the same relative proportion by the Company and the executive as in effect on their last day of employment until the earlier of 18 months from termination or the date the executive becomes eligible for medical benefits with another employer. Payment in each case would be subject to the executive’s execution of a release satisfactory to us following such termination. In addition, if the executive’s employment terminated as a result of disability or death, they would be entitled to receive a pro-rated target bonus for the period during which the executive was employed in the year of termination.
The following definitions apply to the severance terms under each executive’s amended and restated employment agreement:
“Cause” means (i) the executive’s conviction of (A) a felony or (B) any misdemeanor involving moral turpitude, deceit, dishonesty or fraud; (ii) the executive’s willful failure or refusal to comply with lawful directions of such executive’s supervisor, which failure or refusal continues for more than five business days after written notice is given to the executive by such executive’s supervisor, which notice sets forth in reasonable detail the nature of such failure or refusal; (iii) willful and material breach by the executive of a material written Company policy applicable to the executive or the executive’s covenants and/or obligations under his or her employment agreement, provided that the breach is not cured within 5 business days; and/or (iv) misconduct by the executive that materially damages us or any of our affiliates.
“Good Reason” means (i) relocation of the executive’s principal business location to a location more than 30 miles from the executive’s then-current business location; (ii) a material diminution in the executive’s duties, authority or responsibilities; (iii) a material reduction in the executive’s base salary; or (iv) willful and material breach by us or our covenants and/or obligations under the executive’s employment agreement.
“Change of Control” means the occurrence of any of the following events: (i) any person (as such term is used in Sections 13(d) and 14(d) of the Exchange Act) becomes the beneficial owner, directly or indirectly, of securities of the Company representing fifty percent (50%) or more of the total voting power represented by the Company’s then outstanding voting securities (excluding for this purpose any such voting securities held by the Company, or any affiliate, parent or subsidiary of the Company, or by any employee benefit plan of the Company) pursuant to a transaction or a series of related transactions; (ii) a merger or consolidation of the Company other than a merger or consolidation which would result in the voting securities of the Company outstanding immediately prior thereto continuing to represent (either by remaining outstanding or by being converted into voting securities of the surviving entity or the parent of such corporation) at least fifty percent (50%) of the total voting power represented by the voting securities of the Company or such surviving entity or parent of such corporation, as the case may be, outstanding immediately after such merger or consolidation; (iii) our stockholders approve an agreement for the sale or disposition by the Company of all or substantially all of our assets; or (iv) a change in the composition of our board of directors, as a result of which fewer than a majority of the directors are incumbent directors.
The following table sets forth the amounts payable to each of our current named executive officers based on an assumed termination as of December 31, 2023 based upon certain designated events.
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| Name | | Cash Severance | | Health and Other Insurance Benefits | | Stock Options (Unvested and Accelerated) | | Total |
| Mark P. Lappe | | | | | | | | |
| Termination for reasons other than Cause, death or Disability, or for Good Reason | | $ | 675,000 | | | $ | 28,271 | | | $ | 1,739,781 | | | $ | 2,443,052 | |
| Termination in connection with a Change of Control | | $ | 1,620,000 | | | $ | 42,406 | | | $ | 2,705,552 | | | $ | 4,367,958 | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| Brendan P. Eckelman, Ph.D. | | | | | | | | |
| Termination for reasons other than Cause, death or Disability, or for Good Reason | | $ | 520,000 | | | $ | 28,425 | | | $ | 581,467 | | | $ | 1,129,892 | |
| Termination in connection with a Change of Control | | $ | 1,131,000 | | | $ | 42,638 | | | $ | 958,225 | | | $ | 2,131,863 | |
| Kelly D. Deck, C.P.A. | | | | | | | | |
| Termination for reasons other than Cause, death or Disability, or for Good Reason | | $ | 430,000 | | | $ | 28,271 | | | $ | 836,837 | | | $ | 1,295,108 | |
| Termination in connection with a Change of Control | | $ | 935,250 | | | $ | 42,406 | | | $ | 1,273,677 | | | $ | 2,251,333 | |
Elements of Compensation
Base Salary
The Compensation Committee reviews the base salaries of our executive officers, including our named executive officers, from time to time and makes adjustments as it determines to be reasonable and necessary to reflect the scope of an executive officer’s performance, role, responsibilities, skills, and experience.
In 2023, the annual base salaries for Mr. Lappe, Dr. Eckelman, and Ms. Deck were increased to $675,000, $520,000, and $430,000, respectively.
Annual Performance Bonus
The Company also provides executive officers with annual performance-based cash bonuses, which are specifically designed to reward executives for overall Company performance in a given year. Corporate goals are established by the Compensation Committee with input from senior management. The target annual cash bonus amounts relative to base salary vary depending on each executive’s accountability, scope of responsibilities, and potential impact on the Company’s performance.
The Compensation Committee considers the Company’s overall performance for the preceding fiscal year in deciding whether to award a bonus and, if one is to be awarded, the amount of the bonus. The annual cash bonus for each executive officer is based 100% on overall Company performance. The Compensation Committee retains the ability to apply discretion in making adjustments to the final bonus payouts.
The evaluation of Company performance for 2023 bonus purposes was based on the achievement, or failure to achieve, a set of weighted performance goals. The Company’s 2023 performance goals generally fell into the categories of executing on the progression of our clinical programs, obtaining sufficient funding through various methods, expanding existing and new business development relationships, and increasing the visibility of the Company to outside investors and the public. For fiscal year 2023, the Compensation Committee determined that the Company achieved 100% of the performance goals and thus the executive officers should be paid their bonuses at 100% of the targeted levels. Accordingly, the following table sets forth the target bonus for each of the named executive officers for fiscal year 2023 and the resulting incentive payout, based on the level of achievement of the 2023 corporate goals:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Name | | Title | | Fiscal Year 2023 Incentive Bonus Rate at Target | | 2023 Evaluation of Company Performance | | Final Ratio Incentive Bonus as a Percentage of Base Salary | | Fiscal Year 2023 Incentive Bonus Award ($) |
| Mark P. Lappe | | Chief Executive Officer and President | | 60% | | 100% | | 60% | | $ | 405,000 | |
| | | | | | | | | | |
| Brendan P. Eckelman, Ph.D. | | Chief Scientific Officer | | 45% | | 100% | | 45% | | $ | 234,000 | |
| Kelly D. Deck, C.P.A. | | Chief Financial Officer | | 45% | | 100% | | 45% | | $ | 193,500 | |
Equity-Based Compensation
Although we do not have a formal policy with respect to the grant of equity incentive awards to our executive officers, or any formal equity ownership guidelines applicable to them, we believe that equity grants provide our executive officers with a strong link to our long-term performance, create an ownership opportunity, and help to align the interests of our executive officers and our stockholders. In addition, we believe that equity grants with a time-based vesting feature promote executive retention. Accordingly, our Compensation Committee periodically reviews the equity incentive compensation of our executives and other employees, including our named executive officers, and from time to time may grant equity incentive awards to them in the form of stock options.
Outstanding equity awards of our named executive officers as of December 31, 2023 are reflected in the “Outstanding Equity Awards as of December 31, 2023” table below.
Rule 10b5-1 Trading Plans
Certain of our employees, including directors and executive officers, have adopted, or may in the future adopt, written plans, known as Rule 10b5-1 Plans, in which they will contract with a broker to buy or sell shares of our common stock on a periodic basis. Under a Rule 10b5-1 Plan, a broker executes trades pursuant to parameters established by the individual when entering into the plan, without further direction from the individual. The individual may amend or terminate the 10b5-1 Plan in limited circumstances.
401(k) Retirement Plan
We maintain a tax-qualified retirement plan that provides eligible employees, including our named executive officers, with an opportunity to save for retirement on a tax-advantaged basis. All participants’ interests in their contributions are 100% vested when contributed. Contributions are allocated to each participant’s individual account and are then invested in selected investment alternatives according to the participants’ directions. Our retirement plan is intended to qualify under Section 401(a) of the Code. Our plan authorizes employer safe harbor matching contributions up to 5% of an employee’s annual compensation, which were suspended beginning in the second quarter of 2020 and reinstated at the beginning of 2021.
Health and Welfare Benefits
Our named executive officers are eligible to participate in our employee benefit plans and programs, which include medical, dental, and vision benefits, life and disability insurance, and wellness and tuition reimbursement, to the same extent as our other full-time employees generally, subject to the terms and eligibility requirements of those plans and programs.
Hedging
The Company maintains an insider trading policy that generally prohibits executives and directors from engaging in any transaction in which they may profit from short-term speculative swings in the value of the Company’s securities. This includes “short sales” (selling borrowed securities that the seller hopes can be purchased at a lower price in the future) or “short sales against the box” (selling owned, but not delivered securities), and “put” and “call” options. To date, no directors or executives have engaged in these activities.
Compensation Consultant
The Compensation Committee also retained an independent compensation consultant in 2023 to assist in evaluating the Company’s executive compensation program. Market pay data from both the peer group and industry data sources are collectively a primary factor for the Compensation Committee in setting target levels of base salary, annual incentive awards, and long-term incentive awards, and are taken into account along with performance in determining base pay increases. In 2023, Aon Consulting, Inc., or Aon, an independent consulting firm, was retained to conduct a competitive review and analysis of the Company’s executive compensation program. Aon, in coordination with Company management, identified a peer group of similarly situated companies across business segment and size metrics (e.g., business sector, stage of development, market cap, and revenue) and presented an assessment of the compensation of the Company’s executive officers relative to market/peer group pay data for various elements of executive compensation, including base salary, target bonus opportunities, target total cash compensation, and equity ownership. The Compensation Committee analyzed this data in consultation with Company management and Aon, and thereafter established the 2023 compensation packages for the Company’s executive officers.
Outstanding Equity Awards as of December 31, 2023
The following table shows certain information regarding outstanding equity awards as of December 31, 2023 for our named executive officers:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Option Awards |
| Name | | Number of Securities Underlying Unexercised Options
(#)
Exercisable | | Number of Securities Underlying Unexercised Options
(#)
Unexercisable | | Equity Incentive Plan Awards: Number of Securities Underlying Unexercised Unearned Options (#) | | Option Exercise Price ($) | | Option Expiration Date | |
| Mark P. Lappe | | 105,729 | | | 39,271 | | | — | | | $ | 33.63 | | | 1/15/2031 | (1) |
| | — | | | 100,000 | | | — | | | $ | 23.30 | | | 1/3/2033 | (2) |
| Brendan P. Eckelman, Ph.D. | | 29,167 | | | 10,833 | | | — | | | $ | 33.63 | | | 1/15/2031 | (1) |
| | — | | | 40,000 | | | — | | | $ | 23.30 | | | 1/3/2033 | (2) |
| Kelly D. Deck, C.P.A. | | 80,543 | | | — | | | — | | | $ | 10.52 | | | 11/26/2028 | (3) |
| | 31,641 | | | 2,877 | | | — | | | $ | 11.25 | | | 4/21/2030 | (4) |
| | 51,042 | | | 18,958 | | | — | | | $ | 33.63 | | | 1/15/2031 | (1) |
| | 9,583 | | | 10,417 | | | — | | | $ | 34.16 | | | 1/11/2032 | (5) |
| | — | | | 30,000 | | | — | | | $ | 23.30 | | | 1/3/2033 | (2) |
________________(1)These stock option grants were made pursuant to our 2017 Plan with the following vesting schedule: 25% of the shares vested on the first anniversary of the vesting commencement date, or January 15, 2022, and 1/48th of the shares vest monthly thereafter.
(2)These stock option grants were made pursuant to our 2017 Plan with the following vesting schedule: 25% of the shares vested on the first anniversary of the vesting commencement date, or January 3, 2024, and 1/48th of the shares vest monthly thereafter.
(3)This stock option grant was made pursuant to our 2017 Plan with the following vesting schedule: 25% of the shares vested on the first anniversary of the vesting commencement date, or November 26, 2019, and 1/48th of the shares vest monthly thereafter.
(4)This stock option grant was made pursuant to our 2017 Plan with the following vesting schedule: 25% of the shares vested on the first anniversary of the vesting commencement date, or April 21, 2021, and 1/48th of the shares vest monthly thereafter.
(5)This stock option grant was made pursuant to our 2017 Plan with the following vesting schedule: 25% of the shares vested on the first anniversary of the vesting commencement date, or January 11, 2023, and 1/48th of the shares vest monthly thereafter.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
Equity Compensation Plan Information
The responsefollowing table provides certain aggregate information with respect to this item is incorporatedall of the Company’s equity compensation plans in effect as of December 31, 2023:
| | | | | | | | | | | | | | | | | | | | | | | |
| Plan Category | | Number of Securities to be Issued upon Exercise of Outstanding Options, Warrants and Rights (a) | | Weighted Average Exercise Price of Outstanding Options, Warrants and Rights (b) | | Number of Securities Remaining Available for Future Issuance under Equity Compensation Plan (Excluding Securities Reflected in Column (a)) (c) | |
Equity compensation plans approved by security holders (1) | | 6,493,526 | | | $ | 23.22 | | | 532,550 | | |
| Equity compensation plans not approved by security holders | | — | | | — | | | — | | |
| Total | | 6,493,526 | | | $ | 23.22 | | | 532,550 | | (2) |
________________(1)This consists of our 2017 Plan.
(2)The reserve for shares available under our 2017 Plan will be increased on the first fiscal day of each year of the Company during the period beginning in fiscal year 2021 and ending on the second day of fiscal year 2030, in an amount equal to the lesser of (i) 4% of the number of outstanding shares of common stock on such date and (ii) an amount determined by reference from the discussion responsive thereto2017 Plan administrator. Notwithstanding the foregoing, the maximum number of shares that may be issued as incentive stock options under the captions “Security2017 Plan is 60 million.
Security Ownership of Certain Beneficial Owners
The following table sets forth certain information with respect to the beneficial ownership of our common stock as of February 21, 2024 by (i) each person who, to our knowledge, beneficially owns more than 5% of our common stock; (ii) each of our directors, director nominees, and Management”named executive officers (as disclosed in this Annual Report); and “Equity Compensation Plan Information”(iii) all of our executive officers, directors, and director nominee as a group. Unless otherwise indicated in our proxy statementthe table or the footnotes to the following table, each person named in the table has sole voting and investment power and such person’s address is c/o Inhibrx, Inc., 11025 N. Torrey Pines Rd., Suite 200, La Jolla, California 92037.
We determined the number of shares of common stock beneficially owned by each person under rules promulgated by the SEC, based on information obtained from Company records and filings with the SEC on or before February 21, 2024. In cases of holders who are not directors, director nominee, and named executive officers, Schedules 13G or 13D filed with the SEC (and, consequently, ownership reflected here) often reflect holdings as of a date prior to February 21, 2024. The information is not necessarily indicative of beneficial ownership for any other purpose. Under these rules, beneficial ownership includes any shares as to which the individual or entity has sole or shared voting power or investment power and also any shares which the individual or entity had the right to acquire within sixty (60) days of February 21, 2024. These shares, however, are not deemed outstanding for the 2021 annual meetingpurpose of stockholders.computing the percentage ownership of any other individual.
Applicable percentages are based on 47,392,447 shares of common stock outstanding as of February 21, 2024, as adjusted as required by the rules promulgated by the SEC.
| | | | | | | | | | | |
| Beneficial Ownership (1) |
| Name and Address of Beneficial Owner | Number of Common Stock | | Percent of Common Stock |
| 5% or Greater Stockholders | | | |
Entities affiliated with Viking Global Opportunities Illiquid Investments Sub-Master LP (2) | 7,150,826 | | | 15.09 | % |
Entities affiliated with RA Capital Healthcare Fund, L.P. (3) | 4,735,648 | | | 9.99 | % |
Entities affiliated with Perceptive Life Sciences Master Fund, Ltd. (4) | 4,787,602 | | | 9.99 | % |
| | | |
Entities affiliated with BlackRock, Inc. (5) | 2,715,672 | | | 5.73 | % |
| | | |
| Named Executive Officers and Directors | | | |
Mark P. Lappe (6) | 3,194,717 | | | 6.72 | % |
Brendan P. Eckelman, Ph.D. (7) | 2,400,553 | | | 5.06 | % |
Kelly D. Deck, C.P.A. (8) | 192,561 | | | * |
Jon Faiz Kayyem, Ph.D. (9) | 3,309,060 | | | 6.98 | % |
Douglas G. Forsyth (10) | 633,016 | | | 1.33 | % |
Kimberly Manhard (11) | 36,743 | | | * |
Kristiina Vuori, M.D., Ph.D. (12) | 24,375 | | | * |
| All current executive officers and directors as a group (7 persons) | 9,791,025 | | | 20.62 | % |
________________* Indicates beneficial ownership of less than 1%.
(1)Beneficial ownership of shares and percentage ownership are determined in accordance with the rules of the SEC. Unless otherwise indicated and subject to community property laws where applicable, the individuals and entities named in the table above have sole voting and investment power with respect to all shares of our common stock shown as beneficially owned by them.
(2)Consists of (i) 6,305,866 shares of common stock held by Viking Global Opportunities Illiquid Investments Sub-Master LP, or VGOP, (ii) 333,333 shares of common stock held by KAVRA 104 LLC, or KAVRA 104, and (iii) 511,627 shares of common stock held by Viking Global Opportunities Drawdown (Aggregator) LP, or VGOD. Excludes pre-funded warrants to purchase 1,038,765 shares of common stock held by VGOP. The pre-funded warrants do not allow for an exercise that would result in the holder of such pre-funded warrants (together with its affiliates, any “group” or any other persons whose beneficial ownership could be aggregated with the holder) would beneficially own more than 9.99% of the number of shares of common stock outstanding immediately following exercise. Viking Global Investors LP, or VGI, provides managerial services to VGOP, KAVRA 104, and VGOD. VGI has the authority to dispose of and vote the shares of common stock directly held by VGOP, KAVRA 104 and VGOD. Viking Global Opportunities Parent GP LLC, or Opportunities Parent, is the sole member of Viking Global Opportunities GP LLC, or Opportunities GP, which has the authority to dispose of and vote the shares of common stock controlled by Viking Global Opportunities Portfolio GP LLC (which consists of the shares of common stock directly held by VGOP), and KAVRA 104, and is the sole member of Viking Global Opportunities Drawdown GP LLC, which has the authority to dispose of and vote the shares of common stock controlled by Viking Global Opportunities Drawdown Portfolio GP LLC (which consists of the shares of common stock directly held by VGOD). Mr. O. Andreas Halvorsen, Mr. David C. Ott and Ms. Rose S. Shabet, as Executive Committee Members of Viking Global Partners LLC (general partner of VGI) and Opportunities GP have shared authority to dispose of and vote the shares of common stock beneficially owned by VGI and Opportunities Parent. None of Mr. Halvorsen, Mr. Ott, and Ms. Shabet directly owns any shares of common stock. The address for these entities is 600 Washington Boulevard, Floor 11, Stamford, Connecticut, 06901.
(3)Consists of 4,724,207 shares of common stock held by RA Capital Healthcare Fund, L.P., or the Fund, and 11,441 shares of common stock the Fund beneficially owns based on the right to acquire upon exercise of pre-funded warrants, subject to the beneficial ownership blocker of the pre-funded
warrants. Excludes pre-funded warrants to purchase 2,735,804 shares of common stock held by the Fund. The pre-funded warrants do not allow for an exercise that would result in the holder of such pre-funded warrants (together with its affiliates, any “group” or any other persons whose beneficial ownership could be aggregated with the holder) would beneficially own more than 9.99% of the number of shares of common stock outstanding immediately following exercise. RA Capital Healthcare Fund GP, LLC is the general partner of the Fund. The general partner of RA Capital Management, L.P., or RA Capital, is RA Capital Management GP, LLC, of which Dr. Peter Kolchinsky and Mr. Rajeev Shah are the controlling persons. RA Capital serves as investment adviser for the Fund. RA Capital, Dr. Kolchinsky and Mr. Shah may be deemed to have voting and investment power over the shares held by the Fund. RA Capital, Dr. Kolchinsky and Mr. Shah disclaim beneficial ownership of the securities listed above except to the extent of any pecuniary interest therein. The address for the individuals and entities listed above is c/o RA Capital Management, L.P., 200 Berkeley Street, 18th Floor, Boston, Massachusetts 02116.
(4)Consists of 4,256,104 shares of common stock held by Perceptive Life Sciences Master Fund, Ltd, or the Master Fund, and 531,498 shares of common stock the Master Fund beneficially owns based on the right to acquire upon exercise of pre-funded warrants, subject to the beneficial ownership blocker of the pre-funded warrants. Excludes pre-funded warrants to purchase 2,397,128 shares of common stock held by the Fund. The pre-funded warrants do not allow for an exercise that would result in the holder of such pre-funded warrants (together with its affiliates, any “group” or any other persons whose beneficial ownership could be aggregated with the holder) would beneficially own more than 9.99% of the number of shares of common stock outstanding immediately following exercise. Perceptive Advisors LLC serves as the investment manager to the Master Fund and Mr. Joseph Edelman is the managing member of Perceptive Advisors LLC. The address for the individual and entities listed above is 51 Astor Place, 10th Floor, New York, New York 10003.
(5)Consists of 2,715,672 shares of common stock held by BlackRock, Inc. and its affiliates. The address for this entity is BlackRock, Inc., 50 Hudson Yards, New York, New York 10001.
(6)Consists of (i) 2,486,192 shares of common stock held by The Lappe Family Trust, (ii) 59,462 shares of common stock held by The Mark Paul Lappe Roth IRA, (iii) 500,000 shares of common stock held by The Lappe Descendants’ Trust-SD DTD 12-21-20, and (iv) 149,063 options to purchase shares of common stock that are exercisable as of February 21, 2024 or that will become exercisable within 60 days after such date pursuant to the terms of the 2017 Plan.
(7)Consists of (i) 2,035,553 shares of common stock held by the Eckelman Living Trust dated February 5, 2014, of which Dr. Eckelman is the trustee, (ii) 160,000 shares of common stock held by a trust for the benefit of Dr. Eckelman’s immediate family, (iii) 160,000 shares of common stock held by a trust for the benefit of Dr. Eckelman’s immediate family; and (iv) 45,000 options to purchase shares of common stock that are exercisable as of February 21, 2024 or that will become exercisable within 60 days after such date pursuant to the terms of the 2017 Plan.
(8)Consists of 192,561 options to purchase shares of common stock that are exercisable as of February 21, 2024 or that will become exercisable within 60 days after such date pursuant to the terms of the 2017 Plan.
(9)Consists of (i) 3,224,301 shares of common stock held by The Jon F. Kayyem and Paige Gates-Kayyem Family Trust, of which Dr. Kayyem is the trustee, (ii) 25,000 shares of common stock held in a custodial account managed by Dr. Kayyem for the benefit of Dr. Kayyem's immediate family, and (iii) 25,000 shares of common stock held in a custodial account managed by Dr. Kayyem for the benefit of Dr. Kayyem's immediate family. Also includes 34,759 options to purchase shares of common stock that are exercisable as of February 21, 2024 or that will become exercisable within 60 days after such date pursuant to the terms of the 2017 Plan.
(10)Consists of 598,257 shares of common stock held by the Douglas G. Forsyth and Rosanna Forsyth as Co-Trustees of the Forsyth Family Trust, Dated July 20, 2001. Also includes 34,759 options to purchase shares of common stock that are exercisable as of February 21, 2024 or that will become exercisable within 60 days after such date pursuant to the terms of the 2017 Plan.
(11)Consists of 36,743 options to purchase shares of common stock that are exercisable as of February 21, 2024 or that will become exercisable within 60 days after such date pursuant to the terms of the 2017 Plan.
(12)Consists of 24,375 options to purchase shares of common stock that are exercisable as of February 21, 2024 or that will become exercisable within 60 days after such date pursuant to the terms of the 2017 Plan.
Item 13. Certain Relationships and Related Transactions, and Director Independence
The response
Director Independence
Our Board annually reviews the independence of all of our directors and affirmatively makes a determination as to the independence of each director based on whether such director satisfies the definition of “independent director” as set forth in the applicable rules of the Nasdaq Stock Market and the SEC. Our Board has determined that all of our directors are independent directors other than Mark P. Lappe, by virtue of his position as Chief Executive Officer of the Company. There are no family relationships among any of our directors or executive officers.
Policies and Procedures on Related Party Transactions
We have adopted a policy that requires all future transactions between us and any director, executive officer, holder of 5% or more of any class of our capital stock, or any member of the immediate family of, or entities affiliated with, any of them, or any other related persons, as defined in Item 404 of Regulation S-K, or their affiliates, in which the amount involved is equal to or greater than $120,000, be approved in advance by our Audit Committee. Any request for such a transaction must first be presented to our Audit Committee for review, consideration, and approval. In approving or rejecting any such proposal, our Audit Committee is to consider all available information deemed relevant by the Audit Committee, including, but not limited to, the extent of the related person’s interest in the transaction, and whether the transaction is on terms no less favorable to us than terms we could have generally obtained from an unaffiliated third party under the same or similar circumstances.
Related Party Transactions
LAV Summit Limited, or LAV SL, a limited company, is a principal shareholder which owned more than 5% of the Company’s outstanding equity interest during the year ended December 31, 2021. Due to this itemequity ownership, LAV SL was considered a related party. LAV SL’s General Partner, Lilly Asia Ventures, or LAV, through one of its funds, holds a significant equity ownership position in Chinese biotechnology company, Elpiscience, and the Venture Partner of LAV is incorporatedthe CEO, Founder, and Director at Elpiscience. Accordingly, the Company identified Elpiscience as a related party and all agreements entered into between the Company and Elpiscience through December 31, 2021 are deemed related party agreements. As of December 31, 2021, LAV SL no longer holds more than 5% of the Company’s outstanding equity interest. Accordingly, any future contracts entered into with LAV SL affiliates will not be considered related party transactions.
In April 2018, the Company entered into a license agreement, or the OX40 License Agreement, with Elpiscience, whereby the Company granted Elpiscience an exclusive license to the Company’s multivalent protein therapeutic directed to the biological target OX40, or INBRX-106. Under this agreement, the Company is entitled to reimbursement for certain CMC and toxicology expenses incurred. The Company received $0.2 million in reimbursements under this agreement during the year ended December 31, 2022, and has received $5.4 million to date as of December 31, 2023 and December 31, 2022. During the year ended December 31, 2022, the Company derecognized $32,000 as contra-expenses, all of which was received in cash as of December 31, 2022. The Company did not receive any reimbursements or derecognize any contra-expense during the year ended December 31, 2023. No further reimbursements remain under this contract.
In July 2020, the Company entered into an additional cost sharing agreement with Elpiscience related to the OX40 License Agreement. Under this agreement, the Company is entitled to reimbursement for certain formulation development costs. Reimbursements under this contract are recognized as contra-expense as incurred. The Company received $0.3 million in reimbursements under this agreement during the year ended December 31, 2022 and has received $0.5 million of reimbursements to date as of December 31, 2023 and December 31, 2022. The Company did not receive any reimbursements or derecognize any contra-expense during the year ended December 31, 2023. No further reimbursements remain under this contract.
In August 2023, the Company entered into the Purchase Agreement with certain beneficial owners of more than 5% of the Company’s common stock, pursuant to which the Company sold and issued 3,621,314 shares of the Company’s common stock for $19.35 per share and pre-funded warrants to purchase 6,714,636 shares of the Company’s common stock in the Private Placement. The purchase price of the pre-funded warrants was $19.3499
per pre-funded warrant, with an exercise price of $0.0001 per share. The holders of more than 5% of the Company’s outstanding common stock shown in the table below purchased securities in the Private Placement.
| | | | | | | | | | | | | | | | | | | | |
| Purchaser | | Shares of Common Stock | | Warrant Shares Underlying Pre-Funded Warrant | | Aggregate Purchase Price |
| Viking Global Opportunities Illiquid Investments Sub-Master LP | | — | | | 1,038,765 | | | $ | 20,099,998.87 | |
| Viking Global Opportunities Drawdown (Aggregator) LP | | 511,627 | | | — | | | $ | 9,899,982.45 | |
| RA Capital Healthcare Fund, L.P. | | 870,340 | | | 2,747,245 | | | $ | 69,999,995.03 | |
| Perceptive Life Sciences Master Fund, Ltd. | | 688,960 | | | 2,928,626 | | | $ | 69,999,996.24 | |
In connection with the execution of the Merger Agreement, the Company entered into an Agreement relating to the Pre-Funded Warrant to Purchase Common Stock and Securities Purchase Agreement, dated as of January 22, 2024, by reference fromand between the discussion responsive thereto underCompany and each holder of the captions “Certain Relationshipspre-funded warrants, or the Private Placement Warrant Amendment, clarifying the treatment of the pre-funded warrants in connection with the Merger and the spin-off such that the holders of the pre-funded warrants will receive the Merger Consideration (as defined in the Merger Agreement) and, pursuant to the terms of the pre-funded warrants, receive warrants to purchase up to an aggregate of 1,678,661 shares of New Inhibrx common stock with terms, rights and obligations equivalent to the pre-funded warrants, in each case, subject to conditions specified in such amendment.
During the years ended December 31, 2023 and December 31, 2022, there were no other transactions or series of similar transactions to which we were or are a party in which the amount involved exceeded or exceeds $120,000 and in which any of our directors or executive officers, any holder of more than 5% of any class of our voting securities or any member of the immediate family of any of the foregoing persons had or will have a direct or indirect material interest, other than the compensation arrangements with our executive officers and non-employee directors described in “Executive Compensation” and “Director Compensation,” respectively.
Related Person Transactions”Party Transaction related to the Merger and “ManagementSpin Out
In January 2024, we entered into the Merger Agreement with Aventis, a wholly owned indirect subsidiary of Sanofi, whereby Sanofi will indirectly acquire, through Aventis, all the assets and Corporate Governance”liabilities primarily related to INBRX-101. Immediately prior to the closing of the Merger, all assets and liabilities not primarily related to INBRX-101 will be spun out into New Inhibrx.
Under the terms of the Merger Agreement, Aventis will acquire all of our outstanding shares through a merger, and in turn, each of our proxy statementshareholders will receive: (i) $30.00 per share in cash, (ii) one contingent value right per share, representing the right to receive a contingent payment of $5.00 in cash upon the achievement of a regulatory milestone and (iii) one SEC-registered, publicly listed, share of New Inhibrx for every four shares of Inhibrx common stock held. In addition, in connection with the 2021 annual meetingtransaction, Aventis will (1) assume and retire our outstanding third-party debt, including our debt arrangements with Oxford, (2) cause New Inhibrx to be funded with $200 million in cash, and (3) retain an equity interest in New Inhibrx of stockholders.approximately 8%. Subject to the satisfaction of customary closing conditions, including the receipt of regulatory approvals, we currently expect the transaction to close during the second quarter of 2024.
In connection with the execution of the Merger Agreement, the Company entered into the Private Placement Warrant Amendment clarifying the treatment of the pre-funded warrants in connection with the Merger and the spin-off such that the holders of the pre-funded warrants will receive the Merger Consideration (as defined in the Merger Agreement) and, pursuant to the terms of the pre-funded warrants, receive warrants to purchase up to an aggregate of 1,678,661 shares of New Inhibrx common stock with terms, rights and obligations equivalent to the pre-funded warrants, in each case, subject to conditions specified in such amendment.
As of February 21, 2024, our directors and executive officers owned, in the aggregate, approximately 20.6% of the outstanding voting shares of our common stock. See “Security Ownership of Certain Beneficial Owners” above.
Item 14. Principal Accountant Fees and Services
The response to this item is incorporated by reference from the discussion responsive thereto under the caption “Independent
Fees for Independent Registered Public Accounting Firm”Firm
The following table presents the fees billed to the Company by BDO USA, P.C., or BDO, for professional services rendered for the years ended December 31, 2023 and December 31, 2022, respectively:
| | | | | | | | | | | | | | |
| | 2023 | | 2022 |
| Audit Fees | | $ | 690,153 | | | $ | 532,697 | |
| Audit-Related Fees | | — | | | — | |
| Tax Fees | | 93,456 | | | 44,400 | |
| All Other Fees | | — | | | — | |
| Total | | $ | 783,609 | | | $ | 577,097 | |
Audit Fees: For the years ended December 31, 2023 and December 31, 2022, the aggregate audit fees billed by our independent registered public accounting firm were for professional services performed by BDO for the audit of our annual financial statements included in our proxy statementForm 10-K filing and review of financial statements included in our quarterly Form 10-Q filings, reviews of registration statements and issuances of consents, comfort letters, and services that are normally provided in connection with statutory and regulatory filings or engagements.
Audit-Related Fees: For the years ended December 31, 2023 and December 31, 2022, there were no fees billed by our independent registered accounting firm for audit-related fees.
Tax Fees: For the years ended December 31, 2023 and December 31, 2022, the tax-related fees billed by an affiliate of our independent registered accounting firm pertained to services related to tax return preparation, tax planning services, and Section 382 analysis related to the Company’s NOLs.
All Other Fees: For the years ended December 31, 2023 and December 31, 2022, there were no fees billed by our independent registered accounting firm for other services, other than the fees described above.
Pre-Approval Policies and Procedures
All audit and non-audit services provided by BDO must be pre-approved by the Audit Committee. BDO will provide the Audit Committee with an engagement letter during the first half of the fiscal year, outlining the scope of the proposed services and estimated fees for the 2021 annual meetingfiscal year. Pre-approval may be given for a category of stockholders.services, provided that (i) the category is reasonably narrow and detailed and (ii) the Audit Committee establishes a fee limit for such category. The Audit Committee may delegate to any member of the Audit Committee the authority to grant pre-approval of permitted non-audit services to be provided by BDO between Audit Committee meetings; provided, however, that any such pre-approval shall be presented to the full Audit Committee at its next scheduled meeting. The Audit Committee pre-approved all audit and permitted non-audit services provided by BDO for the years ended December 31, 2023 and December 31, 2022.
Part IV.
Item 15. Exhibits and Financial Statement Schedules
Exhibits
The exhibits listed in the accompanying “Index to Exhibits” below are filed or incorporated by reference as part of this Annual Report on Form 10-K.Report.
Financial Statement Schedules
See “Index to Consolidated Financial Statements” at Item 8 to this Annual Report on Form 10-K.Report. Other financial statement schedules have not been included because they are not applicable or the information is included in the financial statements or notes thereto.
Index to Exhibits
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Exhibit No. | | Description of Exhibit | | Filed Herewith | | Form | | Incorporated By Reference File No. | | Date Filed | | | | | | | | | | | | | | | | | | | | | | |
| 2.1 | | EXHIBIT
| | | | DESCRIPTION OF EXHIBIT8-K | | 001-39452 | | 1/23/2024 | | | | | | | | | | | | | | | | | | | | | | |
| 2.2 | | FILED
| | | | FORM8-K | | INCORPORATED BY REFERENCE FILE NO.001-39452 | | DATE FILED1/23/2024 | | | | | | | | | | | | | | | | | | | | | | |
| 3.1 | | | | | | 8-K | | 001-39452 | | 8/21/2020 | | | | | | | | | | | | | | | | | | | | | | |
| 3.2 | | | | | | 8-K | | 001-39452 | | 8/21/2020 | | | | | | | | | | | | | | | | | | | | | | |
| 4.1 | | | | | | S-1 | | 333-231907 | | 6/3/2019 | | | | | | | | | | | | | | | | | | | | | | |
| 4.2 | | | | | | S-1 | | 333-240135 | | 7/28/2020 | | | | | | | | | | | | | | | | | | | | | | |
4.3^4.3^ | | | | | | S-1 | | 333-240135 | | 7/28/2020 | | | | | | | | | | | | | | | | | | | | | | |
| 4.4 | | | | | | 8-K | | 001-39452 | | 1/23/2024 | | | | | | | | | | | | | | | | | | | | | | |
| 4.5 | | | | | | X10-K | | 001-39542 | | 3/12/2021 | | | | | | | | | | | | | | | | | | | | | | |
| 4.6 | | | | | | 8-K | | 001-39452 | | 8/29/2023 | | | | | | | | | | | | | | | | | | | | | | |
| 4.7 | | | | | | 8-K | | 001-39452 | | 1/23/2024 | | | | | | | | | | | | | | | | | | | | | | |
| 10.1Δ | | | | | | S-1 | | 333-240135 | | 7/28/2020 | | | | | | | | | | | | | | | | | | | | | | |
| 10.2Δ | | | | | | S-1/A10-Q | | 333-240135001-39452 | | 8/12/202011/07/2022 | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 10.3Δ | | | | | | S-1/A | | 333-240135 | | 8/12/2020 | | | | | | | | | | | | | | | | | | | | | | |
10.4Δ | | | | | | S-1/A10-Q | | 333-240135001-39452 | | 8/12/202011/09/2021 | | | | | | | | | | | | | | | | | | | | | | |
10.5Δ10.4Δ | | | | | | S-1/A10-Q | | 333-240135001-39452 | | 8/12/202011/07/2022 | | | | | | | | | | | | | | | | | | | | | | |
10.6Δ10.5Δ | | | | | | S-1/A | | 333-240135 | | 8/12/2020 | | | | | | | | | | | | | | | | | | | | | | |
10.7Δ10.6Δ | | | | | | S-1 | | 333-231907 | | 6/3/2019 | | | | | | | | | | | | | | | | | | | | | | |
10.8Δ10.7Δ | | | | | | S-1 | | 333-231907 | | 6/3/2019 | | | | | | | | | | | | | | | | | | | | | | |
10.9Δ10.8Δ | | | | | | S-1 | | 333-240135 | | 7/28/2020 | | | | | | | | | | | | | | | | | | | | | | |
10.10†10.9† | | | | | | S-1 | | 333-231907 | | 6/3/2019 | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
10.1110.10 | | | | | | S-1 | | 333-231907 | | 6/3/2019 | | | | | | | | | | | | | | | | | | | | | | |
10.12†10.11† | | | | | | S-1 | | 333-231907 | | 6/3/2019 | | | | | | | | | | | | | | | | | | | | | | |
10.1310.12 | | | | | | S-1 | | 333-231907 | | 6/3/2019 | | | | | | | | | | | | | | | | | | | | | | |
10.1410.13 | | | | | | S-1 | | 333-231907 | | 6/3/2019 | | | | | | | | | | | | | | | | | | | | | | |
10.15†10.14† | | | | | | S-1 | | 333-231907 | | 6/3/2019 | | | | | | | | | | | | | | | | | | | | | | |
10.16†10.15 | | | | X | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 10.16† | | | | | | S-1 | | 333-231907 | | 6/3/2019 | | | | | | | | | | | | | | | | | | | | | | |
| 10.17† | | | | | | S-1 | | 333-231907 | | 6/3/2019 | | | | | | | | | | | | | | | | | | | | | | |
| 10.18 | | | | | | S-1 | | 333-231907 | | 6/3/2019 | | | | | | | | | | | | | | | | | | | | | | |
| 10.19 | | | | | | S-1 | | 333-231907 | | 6/3/2019 | | | | | | | | | | | | | | | | | | | | | | |
| 10.20† | | | | | | S-1 | | 333-231907 | | 6/3/2019 | | | | | | | | | | | | | | | | | | | | | |
| 10.21 | | | | | | S-1/A | | 333-231907 | | 8/20/2019 | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
10.22†10.20† | | | | | | S-1 | | 333-240135 | | 7/28/2020 | | | | | | | | | | | | | | | | | | | | | | |
10.2310.21 | | | | | | S-1 | | 333-240135 | | 7/28/2020 | | | | | | | | | | | | | | | | | | | | | | |
10.2410.22 | | | | | | 10-Q | | 001-39452 | | 11/13/2020 | | | | | | | | | | | | | | | | | | | | | | |
| 10.23 | | | | | | 8-K | | 001-39452 | | 6/21/2021 | | | | | | | | | | | | | | | | | | | | | | |
| 10.24 | | | | | | 8-K | | 001-39452 | | 2/22/2022 | | | | | | | | | | | | | | | | | | | | | | |
| 10.25 | | | | | | 8-K | | 001-39452 | | 6/16/2022 | | | | | | | | | | | | | | | | | | | | | | |
| 10.26 | | | | | | 8-K | | 001-39452 | | 10/4/2022 | | | | | | | | | | | | | | | | | | | | | | |
| 10.27† | | | | | | S-1 | | 333-231907 | | 6/3/2019 | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
10.26†10.28† | | | | | | S-1 | | 333-240135 | | 7/28/2020 | | | | | | | | | | | | | | | | | | | | | | |
| 10.29 | | | | | | S-3ASR | | 333-259313 | | 9/3/2021 | | | | | | | | | | | | | | | | | | | | | | |
| 10.30^ | | | | | | 8-K | | 001-39452 | | 8/29/2023 | | | | | | | | | | | | | | | | | | | | | | |
| 21.1 | | | | | | S-1 | | 333-231907 | | 6/3/2019 | | | | | | | | | | | | | | | | | | | | | | |
| 23.1 | | | | X | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 31.1 | | | | X | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 31.2 | | | | X | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 32.1* | | | | X | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 32.2* | | | | X | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 97.1Δ | | | | | | 10-Q | | 001-39452 | | 11/9/2023 | | | | | | | | | | | | | | | | | | | | | | |
| 101.INS | | Inline XBRL Instance Document - the instance document does not appear in the Interactive Data File because XBRL tags are embedded within the Inline XBRL document | | X | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 101.SCH | | Inline XBRL Taxonomy Extension Schema Document | | X | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 101.CAL | | Inline XBRL Taxonomy Extension Calculation Linkbase Document | | X | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 101.DEF | | Inline XBRL Taxonomy Extension Definition Linkbase Document | | X | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 101.LAB | | Inline XBRL Taxonomy Extension Label Linkbase Document | | X | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 101.PRE | | Inline XBRL Taxonomy Extension Presentation Linkbase Document | | X | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 104 | | Cover Page Interactive Data File - the cover page XBRL tags are embedded within the Inline XBRL document contained in Exhibit 101 | | X | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
^ Certain exhibits and schedules have been omitted pursuant to Item 601(a)(5) of Regulation S-K. The Company hereby undertakes to furnish supplementally a copy of any omitted exhibit or schedule upon request by the SEC.
† Pursuant to Item 601(b)(10) of Regulation S-K, certain confidential portions of this exhibit have been omitted by means of marking such portions with asterisks as the identified confidential portions (i) are not material and (ii) would be competitively harmful if publicly disclosed.
Δ Management Compensation Plan or arrangement.
* This certification is deemed not filed for purpose of section 18 of the Exchange Act or otherwise subject to the liability of that section, nor shall it be deemed incorporated by reference into any filing under the Securities Act or the Exchange Act.
Item 16. Form 10-K Summary
None.
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, as amended, the Registrant has duly caused this Report to be signed on its behalf by the undersigned, thereunto duly authorized.
| | |
| INHIBRX, INC. |
|
| /s/ Mark P. Lappe |
| Name: Mark P. Lappe |
| Title: Chief Executive Officer and Chairman |
|
Date: March 12, 2021February 28, 2024 |
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, as amended, this Report has been signed below by the following persons on behalf of the Registrant in the capacities and on the dates indicated.
| | | | | | | | | | | | | | |
| SIGNATURE | | TITLE | | DATE |
| | | | |
| /s/ Mark P. Lappe | | Chief Executive Officer and Chairman (principal executive officer) | | March 12, 2021February 28, 2024 |
| Mark P. Lappe | | |
| | | | |
| /s/ Kelly D. Deck, C.P.A. | | Chief Financial Officer (principal financial officer and principal accounting officer) | | March 12, 2021February 28, 2024 |
| Kelly D. Deck, C.P.A. | | |
| | | | |
| /s/ Brendan P. Eckelman, Ph.D. | | Director | | March 12, 2021
| Brendan P. Eckelman, Ph.D. | |
| | | | |
| /s/ Jon Faiz Kayyem, Ph.D. | | Director | | March 12, 2021February 28, 2024 |
| Jon Faiz Kayyem, Ph.D. | | |
| | | | |
| /s/ Douglas G. Forsyth | | Director | | March 12, 2021February 28, 2024 |
| Douglas G. Forsyth | | |
| | | | |
| /s/ Kimberly Manhard | | Director | | March 12, 2021February 28, 2024 |
| Kimberly Manhard | | |
| | | | |
| /s/ Kristiina Vuori, M.D., Ph.D. | | Director | | February 28, 2024 |
| Kristiina Vuori, M.D., Ph.D. | | |