UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| | | | | |
| ☒ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 20202023
OR
| | | | | |
| ☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from _______ to _______
Commission File Number 012-34567001-39614
TARSUS PHARMACEUTICALS, INC.
(Exact name of Registrant as specified in its charter)
| | | | | | | | |
| Delaware | | 81-4717861 |
(State or other jurisdiction of
incorporation or organization) | | (I.R.S. Employer
Identification Number) |
| | | | | | | | |
| 15440 Laguna Canyon Road, Suite 160 | | |
| Irvine, California | | 92618 |
| (Address of principal executive offices) | | (Zip Code) |
(949) 409-9820418-1801
(Registrant's telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| | | | | | | | |
| Title of each class | Trading Symbol | Name of each exchange on which registered |
| Common Stock, $0.0001 par value per share | TARS | The Nasdaq Global Market LLC (Nasdaq Global Select Market) |
Securities registered pursuant to Section 12(g) of the Act: None.
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒ No ☐
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and "emerging growth company" in Rule 12b-2 of the Exchange Act.
| | | | | | | | | | | | | | |
| Large accelerated filer | ☐ | | Accelerated filer | ☐ |
| Non-accelerated filer | ☒ | | Smaller reporting company | ☒ |
| | | Emerging growth company | ☒ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☒
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☐
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by an of the registrant's executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ☐ No ☒
The aggregate market value of the registrant’s voting and non-voting common equity held by non-affiliates of the registrant, based on the closing price of the registrant’s common stock as reported by the Nasdaq Global Select Market on October 16, 2020,June 30, 2023, was approximately $312$460.0 million. The registrant has elected to use October 16, 2020 as the calculation date, which was the initial trading date of the registrant’s common stock on the Nasdaq Global Select Market, because on June 30, 2020 (the last business day of the registrant’s second fiscal quarter), the registrant was a privately-held company. Shares of common stock held by each executive officer, director, and holder of 5%10% or more of the outstanding common stock have been excluded in that such persons may be deemed to be affiliates. This determination of affiliate status is not necessarily a conclusive determination for other purposes.
As of March 19, 2021,February 21, 2024, the number of outstanding shares of the registrant’s common stock, par value $0.0001 per share, was 20,503,096.34,218,886.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the information called for by Part III of this Annual Report on Form 10-K is hereby incorporated by reference to portions of the registrant’s definitive proxy statement for its 20212024 annual meeting of stockholders, which will be filed with the Securities and Exchange Commission not later than 120 days after the registrant’s fiscal year ended December 31, 2020.2023.
TABLE OF CONTENTS
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains “forward-looking statements”forward-looking statements within the meaning of Section 21E of the Securities Exchange Act of 1934, as amended ("Exchange Act"). All statements other than statements of historical facts contained in this Annual Report on Form 10-K, including statements regarding our future results of operations and financial position, business strategy, product candidates, planned preclinical studies and clinical trials, results of clinical trials, research and development costs, regulatory approvals, timing and likelihood of success, as well as plans and objectives of management for future operations, are forward-looking statements. These statements involve known and unknown risks, uncertainties and other important factors that are in some cases beyond our control and may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by the forward-looking statements. Factors that may cause actual results to differ from expected results include, among others:
•our ability to successfully commercialize XDEMVY®, formerly known as TP-03, for the treatment of Demodex blepharitis;
•the prevalence of Demodex blepharitis and the size of the market opportunity for XDEMVY;
•our plans relating to commercializing XDEMVY and our product candidates, if approved, including commercialization timelines and sales strategy;
•any statements regarding our ability to achieve distribution and patient access for our products including XDEMVY and timing and breadth of payer coverage; our expectations of the potential market size, pricing, gross-to-net yields, eye care provider and patient acceptance of our product and product candidates, opportunity and patient populations for our product and product candidates, including XDEMVY;
•the rate and degree of market acceptance and clinical utility of XDEMVY and our product candidates;
•the likelihood of our clinical trials demonstrating safety and efficacy of our product candidates, and other positive results;
•the timing and progress of our current clinical trials and timing of initiation of our future clinical trials, and the reporting of data from our current and future trials;
•the timing or likelihood of regulatory filings and approval for our product candidates and our ability to meet existing or future regulatory standards or comply with post-approval requirements;
•our plans relating to the clinical development of our current and future product candidates, including the size, number and disease areas to be evaluated;
•the prevalenceimpact of Demodex blepharitishealth epidemics on our business and the size of the market opportunity for our product candidates;
•the rate and degree of market acceptance and clinical utility of our product candidates;
•our plans relating to commercializing our product candidates, if approved, including sales strategy;operations;
•the impact of COVID-19unfavorable global and geopolitical economic conditions on our business and operations;
•the success of competing therapies that are or may become available;
•our estimates of the number of patients in the United States ("U.S.") or globally, as applicable, who suffer from Demodex blepharitis, Meibomian Gland Disease ("MGD"), rosacea, Lyme disease and malaria and the number of patients that will enroll in our clinical trials;
•the beneficial characteristics, safety, efficacy, therapeutic effects and potential advantages of our product candidates;
•the timing or likelihood of regulatory filings and approval for our product candidates;
•our ability to obtain and maintain regulatory approval of our product candidates and our product candidates to meet existing or future regulatory standards;
•our plans relating to the further development and manufacturing of our product and product candidates, including additional indications for which we may pursue;
•our ability to identify additional products, product candidates or technologies with significant commercial potential that are consistent with our commercial objectives;
•the expected potential benefits of strategic collaborations with third parties (including, for example, the receipt of payments, achievement and timing of milestones under license agreements, and the ability of our third party collaborators to commercialize our product candidates in the territories under license) and our ability to attract collaborators with development, regulatory and commercialization expertise;
•existing regulations and regulatory developments in the United StatesU.S. and other jurisdictions;
•our plans and ability to obtain, maintain, and/or protect intellectual property rights, including extensions of existing patent terms where available;
•our continued reliance on third parties to conduct additional clinical trials of our product candidates, and for the manufacture of our product candidates for preclinical studies and clinical trials;
•the need to hire additional personnel, in particular sales personnel and our ability to attract and retain such personnel;
•the accuracy of our estimates regarding expenses, future revenue, capital requirements and needs for additional financing;
•our financial performance;
•the sufficiency of our existing capital resources to fund our future operating expenses and capital expenditure requirements;
•our competitive position;
•our expectations regarding the period during which we will qualify as an emerging growth company under the JOBS Act; and
•our anticipated use of our existing resources and the proceeds from our Initial Public Offering.
We have based these forward-looking statements largely on our current expectations and projections about our business, the industry in which we operate and financial trends that we believe may affect our business, financial condition, results of operations and growth prospects, and these forward-looking statements are not guarantees of future performance or development. These forward-looking statements speak only as of the date of this Annual Report on Form 10-K and are subject to a number of risks, uncertainties and assumptions, including those described in the section titled “Risk Factors” and elsewhere in this Annual Report on Form 10-K. BecauseMoreover, we operate in a very competitive and rapidly changing environment. New risks emerge from time to time. It is not possible for our management to predict all risks, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements are inherently subject towe may make. In light of these risks, uncertainties and uncertainties, some of which cannot be predicted or quantified, you should not rely on theseassumptions, the forward-looking statements as predictions of future events. The events and circumstances reflecteddiscussed in our forward-looking statementsthis report may not be achieved or occur and actual results could differ materially and adversely from those projectedanticipated or implied in the forward-looking statements. Except as required by applicable law, we do not plan to publicly update or revise any forward-looking statements after the date of this Annual Report on Form 10-K, whether as a result of any new information, future events or otherwise.
You should not rely upon forward-looking statements as predictions of future events. Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee that the future results, advancements, discoveries, levels of activity, performance or events and circumstances reflected in the forward-looking statements will be achieved or occur. Moreover, except as required by law, neither we nor any other person assumes responsibility for the accuracy and completeness of the forward-looking statements. We undertake no obligation to update
In addition,publicly any forward-looking statements that “we believe” and similar statements reflect our beliefs and opinions on the relevant subject. These statements are based upon information available to us as offor any reason after the date of this Annual Report on Form 10-K,report to conform these statements to actual results or to changes in our expectations.
You should read this report and whilethe documents that we believe such information forms a reasonable basis for such statements, such informationreference in this report and have filed with the SEC as exhibits to this report with the understanding that our actual future results, levels of activity, performance and events and circumstances may be limited or incomplete, and our statements should not be read to indicate thatmaterially different from what we have conducted an exhaustive inquiry into, or review of, all potentially available relevant information. These statements are inherently uncertain and you are cautioned not to unduly rely upon these statements.
Unless the context otherwise requires, all references in this Annual Report on Form 10-K to the “Company”, “we,” “us,” “our,” “Tarsus”“Tarsus,” and “Tarsus Pharmaceuticals” refer to Tarsus Pharmaceuticals, Inc. We primarily conduct our business activities as Tarsus Pharmaceuticals.
***
Tarsus Pharmaceuticals, Tarsus, and Tarsus Pharmaceuticals, Inc., our logo, XDEMVY, and other registered or common law trade names, trademarks or service marketsmarks of Tarsus appearing in this report are the property of the Company. This report contains additional trade names, trademarks and service marks of other companies that are the property of their respective owners. We do not intend our use or display of other companies’ trade names, trademarks or service marks to imply a relationship with, or endorsement or sponsorship of us by, these other companies. Solely for convenience, our trade names, trademarks and service marks referred to in this report appear without the ®, ®, ™ or SM symbols, but such references are not intended to indicate, in any way, that we will not assert, to the fullest extent under applicable law, our rights or the right of the applicable licensor to these trade names, trademarks and service marks.
SUMMARY OF RISKS ASSOCIATED WITH OUR BUSINESS
We face risks and uncertainties associated with our business, many of which are beyond our control. Some of the more significant risks associated with our business include the following:
•We are a late clinical-stagecommercial-stage biopharmaceutical company with a limited operating history.history and a single product approved for commercial sale. We have incurred significant losses and negative cash flows from operations since our inception and anticipate that we will continue to incur significant expenses and losses for the foreseeable future.
•Due to the recently initiated commercialization of XDEMVY and our continued development of our pipeline of product candidates through clinical trials and other indications, our capital requirements are difficult to predict and may change. We may need to obtain substantial additional funding to complete the developmentachieve our goals and any commercialization of our product candidates, if approved. If we are unablea failure to raiseobtain this necessary capital when needed we would be forcedon acceptable terms, or at all, could force us to delay, reduce or eliminate our product development programs, commercialization efforts or other operations.
•We are heavily dependent on the successsuccessful commercialization of XDEMVY and the development, regulatory approval, and commercialization of our leadcurrent and future product candidate, TP-03 for the treatment of Demodex blepharitis.
•The COVID-19 pandemic, which began in late 2019 and has spread worldwide, may continue to affect our ability to initiate and complete preclinical studies and clinical trials, disrupt regulatory activities, disrupt our manufacturing and supply chain or have other adverse effects on our business and operations.
•Even if TP-03 or any other product candidate that we develop receives marketing approval, weWe may not be successful in educating eye care physicianproviders ("ECPs"), and the market about the need for treatments specifically for Demodex blepharitis and or other diseases or conditions targeted by XDEMVY or our product candidates, and TP-03candidates. XDEMVY or other product candidates that we may develop may fail to achieve market acceptance by ECPs, other healthcare providers and patients, or adequate formulary coverage, pricing or reimbursement by third-party payorspayers and others in the medical community, and the market opportunity for these products may be smaller than we estimate.
•The development XDEMVY and commercialization of our products, including our leadany product candidate, TP-03candidates for the potential treatment of Demodex blepharitis and Meibomian Gland Disease ("MGD"), TP-04 for the potential treatment of rosacea and TP-05 for potential Lyme prophylaxis and community malaria reduction, is dependent on intellectual propertywhich we license from Elanco Tiergesundheit AG ("Elanco"). If we breach our agreements with Elancoobtain marketing approval may become subject to unfavorable pricing regulations, third-party coverage or the agreements are terminated, we could lose license rights that are important to our business. Furthermore, some of our revenue is dependent on receipt of payments and achievement and timing of milestones under the terms of our out-license agreement with LianBio Ophthalmology Limited ("LianBio"), granting exclusive commercial rights of TP-03 for the treatment of Demodex blepharitis and MGD within the People's Republic of China, Macau, Hong Kong, and Taiwan (the "Territory"). Adverse effects on our business could occur if LianBio is not able to make payments, achieve milestones, and/reimbursement practices or successfully commercialize TP-03 in the Territory.
•We will need to develop and expand our company and we may encounter difficulties in managing our growth,healthcare reform initiatives, which could disruptharm our operations.
•The sizes of the market opportunity for our product candidates, particularlyXDEMVY for the treatment of Demodex blepharitis and TP-03 for the treatment of Demodex blepharitis and MGD, as well as our other product or product candidates, have not been established with precision and may be smaller than we estimate, possibly materially. If our estimates of the sizes overestimate these markets, our sales growth may be adversely affected. We may also not be able to grow the markets for our product candidates as intended or at all.
•The development and commercialization of our products, including XDEMVY, for the treatment of Demodex blepharitis, TP-03 for the potential treatment of MGD, TP-04 for the potential treatment of rosacea and TP-05 for potential Lyme disease prophylaxis and community malaria reduction, is dependent on intellectual property we license from Elanco Tiergesundheit AG ("Elanco").
•We expect to expand our development, regulatory and operational capabilities, and, as a result, we may encounter difficulties in managing our growth, which could disrupt our operations.
•We contract with third parties for the commercial manufacture of XDEMVY and for the manufacture of our product candidates for preclinical studies, clinical trials and for eventual commercialization. This reliance on third parties increases the risk that we will not have sufficient quantities of XDEMVY or our product candidates or compounds or that such supply will not be available to us at an acceptable cost, which could delay, prevent or impair our commercialization or development efforts.
•Clinical drug development involvesis a lengthy, expensive and expensiverisky process with uncertain timelines and uncertain outcomes, and results of earlier studies and trials may not be predictive of future results.
•Any termination or suspension of, or delays in the commencement or completion of, our planned clinical trials could result in increased costs to us, delay or limit our ability to generate revenue and adversely affect our commercial prospects.
•We rely on third parties to conduct our clinical trials and perform some of our research and preclinical studies.
•If we are unable to obtain and maintain sufficient intellectual property protection for XDEMVY or our product candidates, or if the scope of the intellectual property protection is not sufficiently broad, our competitors could develop and
commercialize products similar or identical to ours, and our ability to successfully commercialize our products may be adversely affected.
•Patent terms may be inadequate to protect our competitive position on our product candidates and preclinical programs for an adequate amount of time.
•The concentration of our stock ownership will likely limit your ability to influence corporate matters, including the ability to influence the outcome of director elections and other matters requiring stockholder approval.
PART I
Item 1. Business
Overview
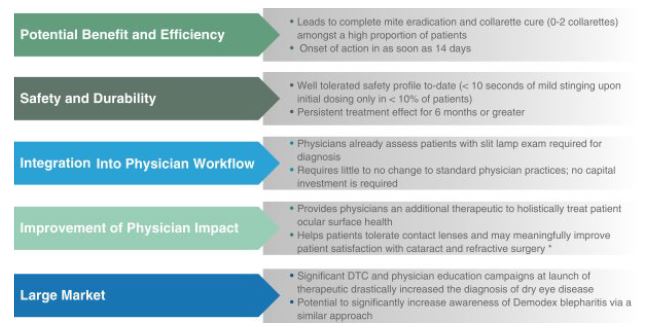
We are a late clinical-stagecommercial-stage biopharmaceutical company focused on the development and commercialization of therapeutic candidates to address large market opportunities, initially in ophthalmic conditions, where there are limited treatment alternatives.therapeutics, starting with eye care. Our lead commercial product, candidate,XDEMVY (lotilaner ophthalmic solution) 0.25%, formerly known as TP-03, is a novel therapeutic in Phase 2b/3 that is being developedwas approved by the U.S. Food and Drug Administration ("FDA") on July 24, 2023 for the treatment of Demodexblepharitis, caused by the infestation of Demodex mites, which is referred to as Demodex blepharitis. Blepharitis (“Blephar” is a reference toan eyelid and “itis” is a reference to inflammation) is a conditionmargin disease characterized by inflammation, of the eyelid margin, redness and ocular irritation, including a specific type of eyelash dandruff called collarettes in Demodex blepharitis. The healthy interaction of the eyelid and the surface of the eyeball is crucial to ocular health. Poorly controlled and progressive blepharitis can lead to worsening of corneal damage over time and, in extreme cases, blindness.
According to published studies, thereirritation. There are an estimated 2025 million patientspeople in the United StatesU.S. who suffer from blepharitis, with approximately 45% or nine million of cases caused by Demodex infestation. Further, blepharitis. XDEMVY is the possible number of Demodex blepharitis cases may be as high as approximately 25 million, based on our internal research indicating approximately 58% of patients presenting to eye care clinics have collarettesfirst and a published study estimating that at least 45 million people annually visit an eye care clinic.
We believe that TP-03 has the potential to be the firstonly FDA-approved therapeutic for the treatment of Demodex blepharitis and becomewe believe it is the definitive standard of care.
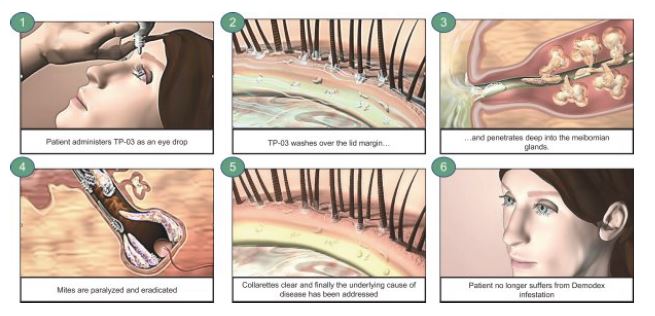
XDEMVY targets and eradicates the root cause of Demodex blepharitis – Demodex mite infestation. The active pharmaceutical ingredient ("API") of TP-03,XDEMVY, lotilaner, is designed to paralyzeparalyzes and eradicateeradicates mites and other parasites through the inhibition of parasite-specific gamma-aminobutyric acid-gated chloride ("GABA-Cl") channels.
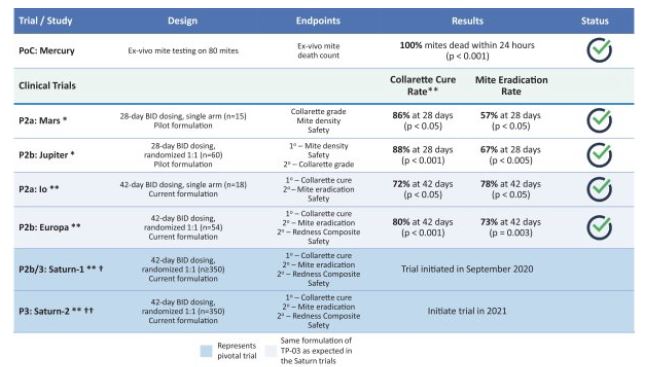
To date, we have completed seven clinical trials that include a Phase 3 trial (the "Saturn-2 trial"), a Phase 2b/3 trial (the "Saturn-1 trial"), four Phase 2 trials, and a Phase 1 trial (the "Hyperion trial") for TP-03XDEMVY in Demodex blepharitis, all of which met their primary, secondary and/or certain exploratory endpoints, as applicable, and during which TP-03 was well tolerated. Our Phase 2b/3 trial, Saturn-1, commenced in September 2020. We expect to begin our Phase 3 trial, Saturn-2, in the second quarter of 2021, both with primary and secondary endpoints consistent with those of our Europa and Io Phase 2 trials. If successful, we expect the Saturn-1 and Saturn-2 trials to support our submission of a new drug application ("NDA") with the United States Food and Drug Administration ("FDA")drug well tolerated throughout each trial. We have also completed, and/or have ongoing clinical trials for TP-03 for the potential treatment of Demodex blepharitis.
In December 2020 we had a Type C meeting with the FDA for TP-03. This meeting confirmed our planned NDA pathway with respect to the data and information required in our forthcoming NDA filing. Furthermore, we intend to pursue marketing authorizations in jurisdictions outside the United States, including Europe and Japan.
On March 26, 2021, we executed an out-license agreement with LianBio Ophthalmology Limited ("LianBio"), granting exclusive commercial rights of TP-03MGD, TP-04 for the potential treatment of Demodex blepharitisrosacea, and MGD within the People’s Republic of China, Macau, Hong Kong,TP-05 for potential Lyme disease prophylaxis and Taiwan (the "Territory"). We are contractually entitled to receive (i) an aggregate $25 million by June 30, 2021, (ii) regulatory and sales milestone receipts totaling $75 million and $100 million, respectively, (iii) tiered royalties in the low double-digits on the net sales of TP-03 within the Territory, and (iv) a minority interest in LianBio that vests upon the achievement of certain clinical and regulatory milestones.
We intend to further advance our pipeline with the lotilaner API to address several diseases across therapeutic categories in human medicine, including eye care, dermatology, and other diseases. Theseinfectious disease prevention. We are investigating the development of our product candidates to address targeted diseases with high unmet medical needneeds, which currently include TP-03 for the potential treatment of MGD, TP-04, a novel gel formulation of lotilaner for the potential treatment of rosacea, and TP-05, a novel investigative oral formulation of lotilaner, for potential Lyme disease prophylaxis and malaria.community malaria reduction.
TP-03 for the Potential Treatment of Meibomian Gland Disease (MGD)
TP-04 for the Potential Treatment of Rosacea
We are exploring the therapeutic potential of TP-04 as a novel topical gel formulation for the treatment of rosacea, a chronic skin disease characterized by facial redness, inflammatory lesions, burning and stinging, which can flare up in response to certain triggers such as sun exposure or emotional stress. There may be several factors that contribute to the cause of rosacea, including genetics, environmental factors, an overactive immune system, and Demodex mites. According to the U.S. National Rosacea Society, approximately 16 million people in the U.S. are affected by rosacea and rosacea prevalence can represent up to 5% of the global population.
inflammatory lesions and Investigator's Global Assessment (IGA) score (change in baseline and success rate) compared to vehicle at Week 12. We plan to discuss and determine the potential regulatory path with the FDA.
TP-05 for the Potential Prevention of Lyme Disease
Lyme disease can potentially cause severe, often debilitating symptoms with permanent and irreversible damage. The disease can result in inflammation, nerve, joint and muscle pain or swelling, numbness, shortness of breath and, in severe cases, neurological complications such as facial palsy, vision issues and meningitis, including severe headaches and neck stiffness. Lyme disease can often go undetected and untreated because the ticks are not always noticed before they transmit the disease. People who are in high-risk areas and/or spend extended amounts of time outdoors in wooded, grassy areas are at higher risk of contracting the infection. Data from the Centers for Disease Control and Prevention ("CDC") show that the risk of Lyme disease is spreading to new geographic areas, resulting in a significant need for prophylactic solutions.
Currently, there are no FDA-approved pharmacological prophylactic options for Lyme disease. We believe TP-05 is currently the only non-vaccine, drug-based, preventive therapeutic in clinical development that targets the ticks, and potentially prevents Lyme disease transmission. It is designed to rapidly provide systemic blood levels of lotilaner, a well-characterized anti-parasitic agent that paralyzes and kills infected ticks attached to the human body through selective targeting of parasite-specific GABA-CI channels, before they can transmit the Borrelia burgdorferi infection that causes Lyme disease.
In December 2022, we announced positive topline results from the completed Phase 1 Callisto trial (the "Callisto trial") and enrollment of the first patient in the Phase 2a clinical trial (the "Carpo trial"). The Carpo trial is designed to evaluate TP-05, a novel investigative oral, non-vaccine prophylactic for the potential prevention of Lyme disease in humans. The Carpo trial is a randomized, double-blind, placebo-controlled trial that evaluated the efficacy of TP-05 in killing lab grown, non-disease carrying ticks after they have attached to the skin of healthy volunteers, as well as confirm the safety, tolerability, and blood concentration of TP-05. Sterile, non-pathogenic nymphal ticks were placed on the skin of healthy human volunteers at two separate instances (one day prior to dosing and 30 days after dosing). Tick mortality was evaluated within 24 hours of attachment after each placement. In most cases, ticks must be attached for 36-48 hours or more before Lyme disease can be transmitted, so killing ticks within 24 hours of attachment can greatly increase the probability of disease prevention.
On February 22, 2024, we announced positive topline results from the Carpo trial which demonstrated a statistically significant benefit in killing ticks compared to (p < 0.0001). Specifically, after the Day 1 tick challenge, mean tick mortality was 97.0% (± 1.4 standard error, SE) and 92.0% (± 6.3 SE) for the high and low doses of TP-05, respectively, compared to only 5.0% (± 2.5 SE) for placebo. Similarly, at the 30-day challenge, mean tick mortality at 24 hours after placement was 89.0% (± 11.1 SE) and 91.0% (± 6.1 SE) for the high and low doses of TP-05, respectively, compared to only 9.0% (± 8.0 SE) for placebo (p<0.001). No statistically significant differences in tick mortality were observed between the two TP-05 treatment arms, and TP-05 was generally well tolerated. We plan to discuss and determine the potential regulatory path with the FDA.
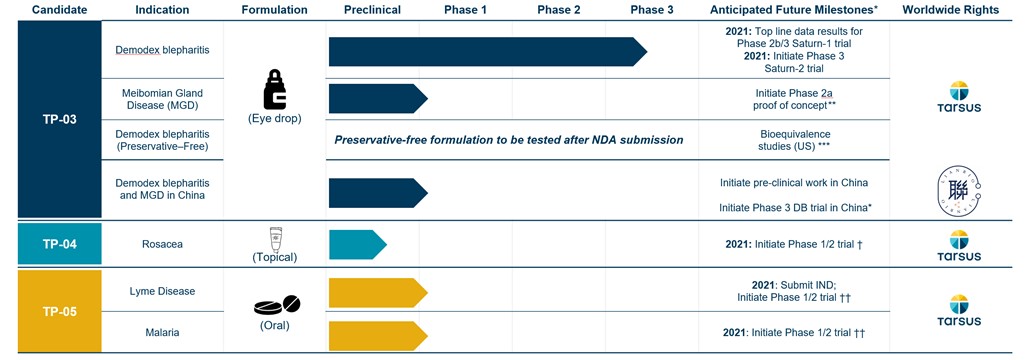
The following pipeline chart presents our wholly-ownedwholly owned product candidates and clinical development status:
Figure 1: Pipeline Chart
| | | | | | | | |
| WW | | | | | Worldwide |
*EU | | Anticipated milestones are subject to the impact of the ongoing COVID-19 pandemic on our business and those of our partners.European Union |
**OUS | | Outside the U.S. |
Our Strategy
Our goal is to transform the treatment of Demodex blepharitis with XDEMVY, the first ever FDA-approved pharmaceutical therapeutic for the treatment of Demodex blepharitis, and to develop our pipeline of innovative therapies that target certain parasite-mediated diseases with large market opportunities. We intend to achieve these goals by pursuing the following key strategic objectives:
| | | | | | | | | | | |
| • | | Continue to obtain payer coverage and grow sales of XDEMVY for the treatment of Demodex blepharitis. We intendlaunched XDEMVY for the treatment of Demodex blepharitis in August 2023 after receiving FDA approval in July 2023. |
| | | | | | | | | | | |
| • | | Continue to rely on preclinical studieseducate eye care providers ("ECPs"). Active disease education is continuing to drive awareness and clinical safety assessmentsencourage ECPs to proactively diagnose Demodex blepharitis through a standard eye examination. |
| | | | | | | | | | | |
| • | | Expand the eye care applications of TP-03 for other indications, including MGD. Similar to blepharitis, MGD may also be caused by Demodex infestation, and we are exploring TP-03 in this indication. In December 2023, we announced positive topline results from the Demodex blepharitis program. We have not conductedErsa Phase 2a clinical trial and do not intendplan to conduct any preclinical studiesdiscuss and determine the regulatory path with the FDA. |
| | | | | | | | | | | |
| • | | Continue to advance and expand our pipeline, bringing novel products utilizing lotilaner to unmet needs across human medicine, including MGD, rosacea and Lyme disease prophylaxis. The mechanism of lotilaner coupled with our insights into disease where it can demonstrate clinical benefit, provides an opportunity to expand into new indications for treatment or prevention. In December 2023, we announced positive topline results from the Ersa trial, designed to evaluate TP-03 for the treatment of MGDMGD. TP-03 demonstrated statistically significant and clinically meaningful improvements compared to baseline in order to advance to Phase 2a. |
*** | We intend to leverage all preclinical, Phase 2two objective measures of the disease – the presence and Phase 3 dataquality of liquid secretion as measured by the Meibomian Gland Secretion Score ("MGSS") and the number of glands secreting normal (clear) liquid and was well tolerated. In February 2024, we announced positive topline results from the TP-03 Demodex blepharitis program. We intendGalatea and Carpo trials and plan to conduct discuss and determine the potential regulatory paths with the FDA.
|
| | | | | | | | | | | |
| • | | in vitroEvaluate and selectively enter collaborations to maximize the potential of our pipeline and the scope of our eye care product offerings. or in vivo bioequivalence studiesApart from the development and license agreement (the "China Out-License") with our preservative-free formulation to compare it to the current preserved formulationLianBio Ophthalmology Limited ("LianBio") of TP-03 in for the treatment of Demodex blepharitis after NDA submission and file a supplement.MGD within the China Territory, as described below within
|
† | We intend License Agreements: LianBio Agreement, we have retained our rights globally to leverage systemic preclinical data fromall of our TP-03 program and augment with additional dermal preclinical studies to select formulation in order to advance to Phase 1/2, which we intend to conduct outside the United States. We may need to address this approach with the FDA if we were to conduct a clinical trial in the United States. We have not conducted any preclinical studies in rosacea with TP-04 to date. |
†† | In relation to Lyme disease and malaria, we intend to leverage oral systemic preclinical data from our TP-03 program as well as third-party oral systemic preclinical studies for Lyme disease or community malaria reduction, respectively (and will not conduct our own preclinical studies for Lyme disease and malaria). The formulations used in preclinical studies use the common approach of a gavage that is scaled as appropriateindications for use in animals. However, human administration, while continuinghumans, including for XDEMVY for the treatment of Demodex blepharitis, TP-03 for the potential treatment of MGD, TP-04 for the potential treatment of rosacea and TP-05 for potential Lyme disease prophylaxis and community malaria reduction. Given the potential to be oral, will take the form of a tablet or capsule. In relation to lyme, we had a successful pre-IND meeting with the FDA in February 2021 and gained agreement on our proposed Phase1 study design. We plan to file an IND in the US in the second quarter of 2021 and, subject to FDA approval of the IND, we intend to initiate our Phase 1 trials to evaluate safety and pharmacokinetics of TP-05 from single ascending dose (SAD) and multiple ascending dose (MAD) studies in normal healthy volunteers. In relation to malaria,treat patients worldwide we may conduct our Phase 1/2 trial outside the United States.opportunistically enter into additional strategic collaborations around certain product candidates, disease and/or geographic regions. |
In August 2023 we launched XDEMVY in the U.S. with a specialty sales force, social and digital media, and ECP education campaigns and targeted prescribing ophthalmologists and optometrists. In our work with key opinion leaders and various associations to increase Demodex blepharitis awareness and education, we have highlighted prevalence, impact, and simplicity of diagnosis of Demodex blepharitis. Our goal is to continue to educate ECPs about the prevalence of Demodex blepharitis, simplicity and efficient diagnosis, and the positive profile of our product. In addition to educating ECPs, we believe that patient awareness and identification is important and plan to continue to increase awareness through education and marketing efforts directed toward patients with Demodex blepharitis.
Consistent with our goal to educate ECPs, we established a field medical team that is actively communicating with ECPs across the country. We also launched a physician-facing disease education campaign geared at increased disease awareness and encouraging ECPs to more proactively diagnose Demodex blepharitis by incorporating eyelid screening as part of their routine exams.
Blepharitis: Market Overview
Blepharitis is a common, chronic ophthalmic conditionlid margin disease characterized by inflammation of the eyelid margin, redness and ocular irritation. It is also a progressive disease that often manifests with more severe symptoms if left untreated, such as blurring of vision, missing eyelashes, corneal damage and potentially, in extreme cases, blindness. According to published studies, an estimated 20 million patients suffer fromDemodex blepharitis in the United States, and there is growing recognition within the ophthalmic community about Demodex mitesmay affect as an underlying cause of blepharitis. Demodex mites are the most common ectoparasite found on humans. Demodex mites potentially cause approximately 45%, or approximately nine million, of blepharitis cases in the United States and we believe that the number of Demodex blepharitis patients in the United States may bemany as high as approximately 25 million Americans based on our internal researchan extrapolation from the Titan study indicating approximately 58% of patients presenting to U.S. eye care clinics have collarettes, and a published study estimatingpathognomonic sign of Demodex infestation, and that at least 45 million people annually visit an eye care clinic. In addition, there is growing awareness among ECPs of the pathognomonic sign of Demodex infestation called collarettes or cylindrical dandruff; which is a specific type of debris found at the base of the eyelashes, known as collarettes, or cylindrical dandruff, which are highly correlated with Demodex infestation.eyelashes. Collarettes are composed of partially digested epithelial cells, mite waste products and eggs among other things and can be easily diagnosed by ECPs withas part of a standard eye examination. The prevalence of Demodex blepharitis increases progressively with aging, which is theone main risk factor for the condition.disease, though other frequently presenting patients can also suffer from dry eye, contact lens intolerance, and cataracts. These aging patients commonly present to the offices of ECPs for other ophthalmic conditionsdiseases besides blepharitis, such as cataract surgery evaluation and contact lens discomfort. Accordingly, we believe that there is significant opportunity to increase the diagnosis rate of Demodex blepharitis through ECP and patient education that encourages examination of the conditiondisease in standard practice.
Despite the high prevalence of patients with Demodex blepharitis and growing awareness of the condition amongstdisease among ECPs, there arewere no FDA-approved therapeutics for the treatment of blepharitis, or for let alone Demodex blepharitis. blepharitis, until XDEMVY was approved in July 2023. Although we believe blepharitis and Demodex blepharitis are significantly underdiagnosed conditions,under-diagnosed diseases with limited treatment alternatives, there are already an estimated 2.1approximately 1.5 million annual Demodexblepharitis
diagnoses in the United States withU.S. based on findings from the Titan study and data that show blepharitis classified per the International Classification of Diseases, Tenth Revision, Clinical Modification ("ICD-10-CM"). Demodex blepharitis is
currently treated with a variety of over the counter remedies such as tea tree oil, lid wipes and artificial tears, as well as off-label prescription products, which often show sub-optimal efficacy, are poorly tolerated and lead to significant irritation and dissatisfaction for patients, and do not eradicate the Demodex mites.
We have conducted epidemiology and market research on the prevalence of blepharitis and potential adoption of TP-03.XDEMVY. Our research indicates approximately 58% of patients presenting to ECP offices have collarettes and, based on the Gao (2005),study, all patients with collarettes were also found to have Demodex. This further validates the accessible opportunity to increase diagnosis rates among patients.Demodex mites. In addition, our market research suggests the potential for a high level of adoption of TP-03, if approved.XDEMVY. In our surveys completed during 2023, we interviewed 50approximately 250 ECPs, 96%over 90% of whom indicated they would prescribe TP-03an FDA approved prescription therapy as a first-line treatment for Demodex blepharitis after exposure to the TP-03 target product profile. See the section titled “Market Opportunity in Blepharitis—Our Market Research Studies and Surveys” for more information regarding our surveys. Further, patients continue to have underlying risk of Demodex infestation, so there could be a recurrence based on the presence of Demodex mites in the skin even after eradication of Demodex mites from the eyelid. Our Phase 2 data from the Mars and Jupiter clinical trials followed patients to one year after treatment and showed meaningful recurrence of Demodex blepharitis within six to nine months, which increased considerably one year after treatment. We believe this data suggests TP-03, if approved, may be used on a chronic, intermittent basis in Demodex blepharitis.
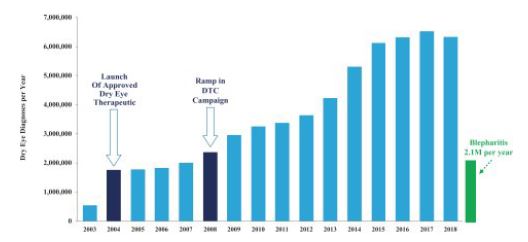
We believe the blepharitis market has the potential to be analogous to other ophthalmic markets that grew significantly once there was a product to address the large, latent demand for an effective therapy, such as dry eye. For example, another ocular surface disease, dry eye, had no approved therapeutic for the condition until 2003. With the approval of a therapeutic to treat dry eye in 2003 and concurrent ECP and patient education, the diagnosis rate increased by approximately 12 times, growing from 500,000 annual diagnoses in 2003 to over six million annual diagnoses in 2015. Annual diagnoses rates have been maintained at similar levels since 2015. Blepharitis already has 2.1 million diagnoses per year with blepharitis classified per the ICD-10-CM, despite no approved therapies to help with market awareness, but we believe there is potential for significant market expansion. The markets may be analogous because both Demodex blepharitis and dry eye are diseases that affect the front of the eye, are treated by ECPs, can cause an inflamed ocular surface or eyelids and have similar patient demographics. The potential market for Demodex blepharitis, however, may not be analogous to the market for dry eye due to differences in symptoms, regulatory approval and market dynamics and certain other factors. See “Risk Factors – Risk Related to Development and Commercialization of Our Product Candidates—The market for blepharitis and Demodex blepharitis may be not be similar to the market for dry eye.” for additional information and risks related to the comparison of the Demodex blepharitis market to that of dry eye.
We believe there is a significant opportunity to increase the diagnosis rate of Demodex blepharitis and build a significant new market with the approval ofXDEMVY, a safe and effective therapeutic alternativetherapy that addresses the underlying root cause of the condition.disease.
Our Approach: TP-03
We are developing TP-03, formulated infestation, as an eye drop, which we believe, if approved, has the potential to become the standard of care for Demodex blepharitis. TP-03 isthere could be a novel therapeuticrecurrence or reinfestation based on the drug, lotilaner, which is designed to paralyze and eradicatepresence of Demodex mites and other parasites throughin the inhibitionfacial pores even after eradication of parasite-specific GABA-Cl channels.
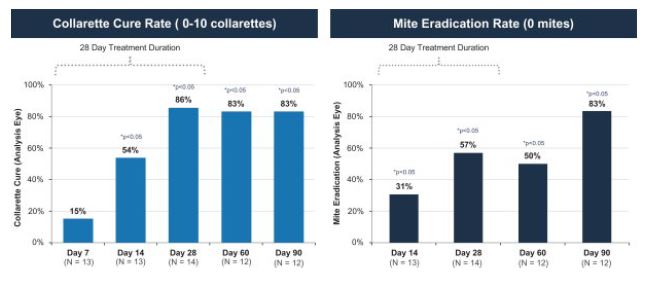
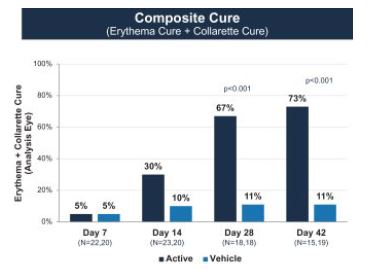
We have completed four Phase 2 clinical trials to date, along with one additional ex vivoDemodex study. Key efficacy endpoints for our Mars and Jupiter clinical trials included collarette grade and mite density and key efficacy endpoints for our Io and Europa clinical trials included collarette cure rate based on collarette grade, which we refer to herein as collarette cure rate, and mite eradication rate. TP-03 met its primary, secondary and/or exploratory endpoints, as applicable, in such trials, and showed statistically significant cure and eradication rates in our two most recent trials, Io and Europa. TP-03 was generally well tolerated throughout these trials. The Phase 2a Mars trial was a smaller single arm trial evaluating the safety and efficacy of TP-03 with a 28-day twice per day, or BID, dosing regimen, with exploratory endpoints including collarette grade and mite density. We utilized the datamites from the exploratory endpoints to determine collarette cure rate, defined as 10 or fewereyelid, that may potentially necessitate retreatment. The Saturn-2 trial enrolled 412 adults having, among other things, more than ten collarettes per lid and mite eradication rate, defined as zero mites per lash. Both collarette cure and mite eradication rates were assessed at 28 days, which were 86% and 57%, respectively.
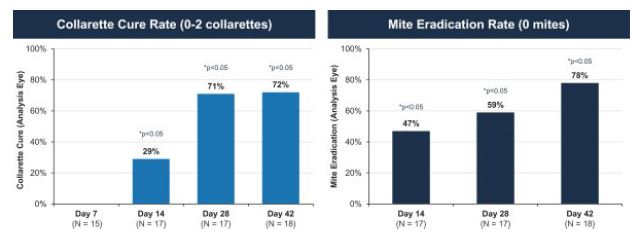
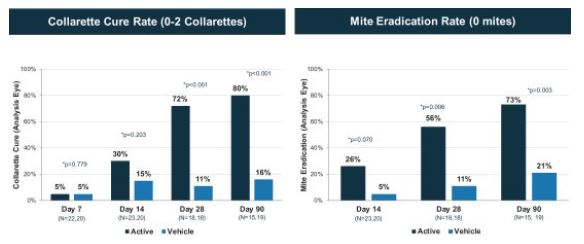
The Phase 2b Jupiter trial was a larger randomized, controlled double-blind trial with mite density and collarette grade asleast mild lid erythema. All pre-specified primary and secondary endpoints respectively. Similarlywere met, XDEMVY was well tolerated, and improvement in lids (reduction of collarettes to the Marsno more than 2 collarettes per upper lid) was demonstrated in 55% of patients treated with XDEMVY. The Saturn-1 trial we utilized the collarette grade and mite density data to determine collarette cure and mite eradication rates, which were both defined consistently with those used in the Mars trial. The efficacy observed in the Jupiter trial appeared consistent with the Mars trial, with a collarette cure rate of 88%, and mite eradication rate of 66%, which were statistically significant compared to the vehicle control. We subsequently conducted the Io Phase 2a and Europa Phase 2b trials to evaluate the safety and efficacy of TP-03 formulation for the treatment of Demodex blepharitis. The design of the Io trial took into account feedback from the FDA, and accordingly we defined collarette cure rate as aenrolled 421 adult patients having more stringent standard of two or fewerthan ten collarettes on the eyelid, with a treatment duration of 42 days.
In the Io trial, we utilized for the first time in clinical trials the formulation of TP-03 that is expected to support our NDA submission for Demodex blepharitis. The change in formulation consisted of replacing LAK with sorbate as the preservative as well as adding a chelating agent. The key differences between the Ioupper lid and Europa trials, were that Io was a smaller single-arm trial, while Europa was a slightly larger randomized vehicle-controlled trial. The endpoints achieved in Io and Europa were consistent across both trials, with a collarette cure rate of 72% and 80%, respectively, and a mite eradication rate of 78% and 73%, respectively. The achievement of primary, secondary and/or exploratory endpoints, as applicable, in such trials, and safety results across our comprehensive Phase 2 program provided us with the basis to design and initiate our pivotal Phase 2b/3 and Phase 3 clinical trials for TP-03 for the treatment of Demodex blepharitis.
We plan to evaluate TP-03 in two pivotal registration trials, referred to as Saturn-1 and Saturn-2. Saturn-1 is a Phase 2b/3, randomized, controlled, double-blind trial to evaluate the safety and efficacy of TP-03 that is expected to enroll at least 350 Demodex blepharitis patients in multiple centers in the United States. Saturn-1 commenced in September 2020 and top-line data is expected in 2021, subject to the impactmild erythema of the ongoing COVID-19 pandemic. Saturn-2, our confirmatory Phase 3 randomized, controlled, double-blindupper eyelid margin. The Saturn-1 trial has a highly comparable designresults showed the pre-specified primary and secondary endpoints were met, and improvement in lids (reduction of collarettes to that of Saturn-1 and is also expected to enroll approximately 350 Demodex blepharitis patients. Enrollment of Saturn-2 is expected to begin in 2021, subject to the impact of the ongoing COVID-19 pandemic.
In connection with our IND application, we have received a “no-objection” letter from the FDA regarding the trial design for Saturn-1. The trial design for Saturn-2 is highly comparable to that of Saturn-1 and we expect to update the IND protocol prior to commencing Saturn-2. We expect these trials to support the potential submission of an NDA for TP-03 for the treatment of Demodex blepharitis. In December 2020 we had a Type C meeting with the FDA; this meeting confirmed our planned NDA pathway with respect to the data and information required in our forthcoming NDA filing.
We also intend to explore the therapeutic potential of TP-03 for a second ophthalmic condition, MGD, commonly characterized in part by a widespread clogging of the meibomian glands that can result in tear film deficiency, and is one of the leading causes of dry eye disease. There are no FDA-approved therapeutics for MGD. In the United States, MGD prevalence has been found to be approximately two-thirds of the estimated 34 million dry eye patient population. One species of Demodex mite, Demodex brevis, is known to infest the meibomian gland, and clinical signs of MGD have been shown to be correlated with infestation of Demodex brevis. While dry eye is a multi-factorial disease, TP-03 is designed to relieve some of the key elements of MGD by virtue of causing the death of the Demodex brevis mites.
Our Approach: TP-04 and TP-05
We are also developing additional clinical-stage product candidates with lotilaner. These include TP-04 for the potential treatment of rosacea as well as TP-05 for potential Lyme prophylaxis and community malaria reduction.
Rosacea is a chronic skin disease characterized by facial redness, inflammatory lesions, burning and stinging, which can flare up in response to certain triggers such as sun exposure or emotional stress. According to the U.S. National Rosacea Society, approximately 16 million people in the United States are affected by rosacea and a study estimates rosacea prevalence can represent up to 5.4% of the global population. We intend to develop TP-04 as a topical formulation, and we plan to initiate a Phase 1/2 trial of TP-04 outside the United States, for the treatment of rosacea in 2021. We initiated preclinical studies to select Phase 1/2 formulation for TP-04.
Lyme disease is the most common vector-borne disease in the United States, caused by infection of Borrelia bacteria following bite by a tick vector. Estimates of annual cases of Lyme disease in the United States range from approximately 300,000 to 400,000. Malaria is one of the world’s highest unmet public health needs, with approximately 228 million cases and more than 400,000 deaths caused2 collarettes per upper lid) was demonstrated in 44% of patients treated with XDEMVY by malaria worldwide. We are developing TP-05 as an oral formulation that is designed as a prophylactic drug against Lyme disease to eradicate the tick before it can transmit the Borrelia bacteria. Further, we believe TP-05 also has the potential to significantly reduce malaria transmission through reducing the lifespan of mosquitos that transmit malaria. This may in turn provide herd protection against the spread of malaria. TP-05 is not intended to treat the disease, but to limit its transmission. We have obtained FDA feedback in a pre-IND meeting and plan to submit an IND and initiate a Phase 1/2 trial of TP-05 for Lyme disease in 2021. For malaria, we may conduct the Phase 1/2 trial outside of the United States.
Our Strategy
Our goal is to transform the treatment of Demodex blepharitis with a first ever FDA approved pharmaceutical therapeutic, and to develop our pipeline of innovative therapies that target certain parasite-mediated diseases with large market opportunities. We intend to achieve these goals by pursuing the following key strategic objectives:
| | | | | | | | | | | |
| • | | Advance TP-03 through clinical development and eventual approval for the treatment of Demodex blepharitis. We have observed in multiple Phase 2 trials across 147 patients that TP-03 results in the achievement of clinical endpoints, which are generally the same clinical endpoints that will be utilized in our pivotal Phase 2b/3 and Phase 3 trials. We have commenced our first pivotal trial, Saturn-1, a Phase 2b/3 trial, in September 2020, and we expect top-line data in 2021, subject to the impact of the ongoing COVID-19 pandemic. Enrollment of our second pivotal trial, Saturn-2, which will be a Phase 3 trial, is expected to begin in 2021, subject to the impact of the ongoing COVID-19 pandemic.
|
| | | | | | | | | | | |
| • | | Educate ECPs and establish our own specialty sales organization to commercialize TP-03 in the United States. If approved by the FDA for Demodex blepharitis, we intend to commercialize TP-03 by developing our own sales organization targeting a subset of the approximately 25,000 prescribing ECPs in the United States. Throughout our commercialization efforts, we intend to educate ECPs on Demodex blepharitis and how to diagnose it with a standard eye examination.
|
| | | | | | | | | | | |
| • | | Expand the label of TP-03 for other indications, including MGD. Like blepharitis, MGD may also be caused by Demodex infestation, and we intend to explore the clinical potential for TP-03 in the indication.
|
| | | | | | | | | | | |
| • | | Develop our pipeline, bringing novel products utilizing lotilaner to unmet needs across human medicine, including rosacea, Lyme disease and malaria. We plan to expand our pipeline of novel, differentiated product candidates that target parasites to treat or prevent important diseases. The mechanism of lotilaner coupled with our insights into disease where it can demonstrate clinical benefit, provides an opportunity to expand into new indications for treatment or prevention. By utilizing new formulations of lotilaner, we intend to develop a topical formulation designed to treat rosacea, and oral formulations for the prophylaxis of Lyme and community malaria reduction. We intend to expand into Phase 1/2 clinical trials for these indications in 2021.
|
| | | | | | | | | | | |
| • | | Evaluate and selectively enter into strategic collaborations to maximize the potential of our pipeline and the scope of our eye care product offerings. Other than the recent out-license to LianBio of TP-03 for the treatment of Demodex blepharitis and MGD within the Territory as noted above, we have retained our rights globally to all of our indications for use in humans, including our lead product candidate, TP-03, for the potential treatment of Demodex blepharitis and MGD, TP-04 for the potential treatment of rosacea and TP-05 for potential prophylaxis of Lyme and community malaria reduction. We are dependent on licenses from Elanco for the development and commercialization of these products. Given the potential to treat patients worldwide we may opportunistically enter into strategic collaborations around certain product candidates, disease or geographic regions.
|
Blepharitis Overview
Blepharitis
Ocular surface disease represents a broad category of disease that affects at least 35 million people in the United States. The ocular surface comprises the cornea, conjunctiva, eyelids and lacrimal glands and as such any diseases in these structures can be classified as ocular surface disease. Common ocular surface diseases include dry eye disease, ocular allergy, blepharitis and MGD. Almost all of the ocular surface diseases are associated with eye redness and ocular surface inflammation and in some cases conjunctival and/or lid edema. Patients often present with multiple ocular surface diseases and the symptoms have significant overlap, leading to frequent misdiagnosis of the various conditions.
Blepharitis is a common, chronic ophthalmic conditionlid margin disease, which may lead to or exacerbate ocular surface disease. Blepharitis is characterized by inflammation of the eyelid margin, redness and ocular irritation, and is primarily diagnosed and treated by ECPs, including ophthalmologists and optometrists. Based on published studies, an estimated 20 million patients suffer from blepharitis in the United States. Typical signs and symptoms of blepharitis include debris on the eyelashes, redness of the eye and eyelid, missing or misdirected eyelashes, blurring of vision, irritation, lid itchiness and ocular discomfort. It isBlepharitis can be challenging to manage, recurs frequently, and its progression can lead to scarring of the eyelid, loss of proper eyelid and tear-film function, eyelid and lash abnormalities, inflammation of the conjunctiva and surrounding skin, suboptimal surgical outcomes, corneal damage, and potentially in extreme cases, blindness. Further, approximately 67% of cataract patients have Demodex infestation, which can increase the risk for both infection after cataract and refractive surgery. Therefore, treating Demodex blepharitis may improve patient satisfaction with cataract and refractive surgery. Additionally, the primary reason people stop wearing contact lenses is discomfort and blepharitis has been shown to cause contact lens intolerance. Therefore, treating Demodex blepharitis may reduce contact lens intolerance. We believe these benefits may lead to better vision and an improved quality of life for patients.
Multiple factors can cause blepharitis, including infestation by Demodex mites, bacterial infection, clogging of the meibomian glands and seborrheic dermatitis.
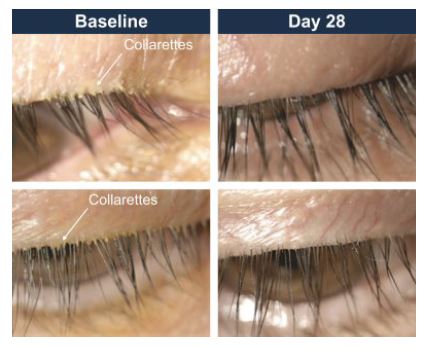
The following image showsimages illustrate representative eyelids with blepharitis:Demodex blepharitis demonstrating the characteristic sign of collarettes:
Figure 2: Eyelids With Demodex Blepharitis
Demodex Blepharitis
Demodex infestation is a major cause of blepharitis, implicated in approximately 45% of blepharitis cases, or approximately nine million patients. Weand we estimate that the number of Demodex blepharitis patients in the United StatesU.S. may be as high as approximately 25 million. Although we believe blepharitis and Demodex blepharitis are significantly under-diagnosed diseases, with limited treatment options, there are already an estimated 1.5 million annual Demodex blepharitis diagnoses in the U.S. based on our internal research indicating approximately 58% of patients presenting to eye care clinics have collarettesthe Titan study and a published study estimating that at least 45 million people annually visit an eye care clinic. Thesecoding for blepharitis cases are referred to classified per the ICD-10-CM. Demodex blepharitis. Demodex mites are the most common ectoparasite found on humans and are more likely to cause infestation and disease with aging. Demodex blepharitis typically presents bilaterally in patients with the condition.disease. There are two species of Demodex, folliculorum and brevis, that live on the skin of the face and eyelids. Demodex folliculorum, which is commonly found in the follicle, is the more common sub-species of mite that causes Demodex blepharitis.
The key clinicalpathognomonic sign of Demodex blepharitis is a specific type of eyelid debris known as the collarette, which is also sometimes referred to as cylindrical dandruff, sleeves, or waxy scurf. Collarettes are composed of partially digested epithelial cells, mite waste products and eggs among other things and can be easily diagnosed by ECPs with a standard eye examination known as the slit lamp examination. Other bothersome signs and symptoms of Demodex blepharitis that can lead to further disease progression include missing or misdirected eyelashes, crusting, redness of the lid margin, inflammation of the lid margin, inflammation of the conjunctiva and/or inflammation of the cornea, also known as blepharoconjunctivitis and blepharokeratitis. Demodex blepharitis is a progressive disease that often manifests with more severe signs and symptoms if left untreated, such as blurring of vision, missing eyelashes, corneal damage and potentially, in extreme cases, blindness. Furthermore, Demodex blepharitis can negatively impact quality of lifedaily activities and create an emotional burden for individuals with the condition.
disease. According to studies we have conducted, approximately 56% of cataract patients have
The following figures demonstrateillustrate how Demodex folliculorum mites enter and reside in the eyelash follicles:
Figure 3: Demodex folliculorum Mites Entering and Residing in Eyelash Follicles
Demodex infestation can lead to Demodex blepharitis in three main ways:
| | | | | | | | |
| 1) | Mechanical: Overcrowded mites scrape the epithelial cell lining of the eyelash follicles with their claws and lay eggs, causing follicular distention, misdirected lashes, eyelash loss and irritation. Dead mites and collarettes also obstruct the hair follicle opening, leading to inflammation. |
| | | | | | | | |
| 2) | Chemical: Mites excrete digestive enzymes as they feed and exude digestive waste when they die, resulting in inflammation, redness, irritation and epithelial hyperplasia. |
| | | | | | | | |
| 3) | Bacterial: Bacteria living on the mite surface or in its gut may cause inflammation of the surrounding ocular tissues. |
As mites scratch and feed on the skin, the partially digested epithelial cells, keratin, mite waste and eggs combine to form collarettes. These collarettes are typically found at the base of the lash but can migrate away as the hair shaft grows.
The following figure showsillustrates collarettes at the base of an eye lash:eyelash:
Figure 4: Collarettes Are the Pathognomonic Sign of Demodex Blepharitis

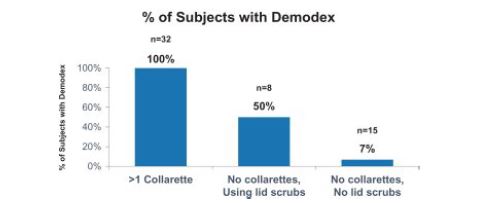
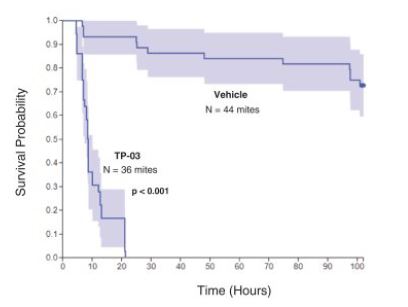
AThe Gao study conducted by Gao et al. (Gao et al. (2005): High Prevalence of Demodex in Eyelashes with Cylindrical Dandruff. Invst Ophth and Vis Sci, September 2005, Vol. 46, No. 3089-3094.) confirmed the pathognomonic relationship of the collarette to Demodex infestation. The study included 55 patients seen at the Ocular Surface Center in Miami, Florida to determine the prevalence of Demodex in eyelashes with collarettes. All patients underwent a routine, complete eye examination and external photography. Patients were divided into three main groups: those with collarettes; those without collarettes and those who had been using daily lid scrubs for a full year; and as well as those without collarettes who were not performing theusing daily lid scrubs. As illustratedOf the thirty-two patients in the figure below,study, 100% (n=32) of patients with at least one collarette had Demodex present. Those patients without collarettes were divided into two groups; patients who were using lid scrubs for a full year and those who were not. Only 7% (n=15) of patients without collarettes and who were not performing the daily lid scrubs had Demodex, while 50% (n=8) of those subjects without collarettes, but who were using daily lid scrubs with shampoo for a full year, had Demodex infestation, implying that hygiene alone did not eradicate the mites. All 55 patients were seen at one location and may not be representative of the United StatesU.S. population. However, subsequent studies including the two TP-03 pivotal Saturn studies that had >800 patients, consistently demonstrated a correlation between the presence of collarettes and Demodex mites.
The following figure shows that 100% of patients with at least one collarette had Demodex infestation:
Figure 5: 100% of Patients With at Least One Collarette Had Demodex Infestation
Demodex blepharitis can be easily diagnosed by ECPs with the standard eye examination, thea slit lamp examination, by confirming the presence of collarettes. The slit lamp examination is routinely performed by ECPs as part of standard practice during a customary eye examination, so diagnosing Demodex blepharitis via presence of collarettes would not involve any additional equipment, training or workflow alterations on the part of the ECP.
Treatment Options Before Approval of XDEMVY and Their Limitations
Market Opportunity in Blepharitis
According to published studies, there are an estimated 20 million patients inOther than the United States who suffer from blepharitis, with approximately 45%, or approximately nine million,use of cases caused by Demodex infestation. Further, we estimate thatXDEMVY, the number of Demodex blepharitis cases in the United States may be as high as approximately 25 million based on our internal research indicating approximately 58% of patients presenting to eye care clinics have collarettes and a published study estimating that at least 45 million people annually visit an eye care clinic. Despite the high prevalence of patients with Demodex blepharitis and growing awareness of the condition amongst ECPs, there are nofirst ever FDA-approved therapeuticspharmaceutical therapy for the treatment of Demodex blepharitis, or for Demodex blepharitis. Although we believe blepharitis and Demodex blepharitis are significantly underdiagnosed conditions, with limited treatment alternatives, there are already an estimated 2.1 million annual blepharitis diagnoses in the United States with blepharitis classified per the ICD-10-CM.
Our Market Research Studies and Surveys
Our market research studies include an ECP survey of 50 ECPs, or the ECP Survey, that we commissioned and which was conducted in 2019 and 2020 tohas been generally determine market awareness and current management of Demodex blepharitis and introduce a hypothetical TP-03 product profile. Twenty ECPs were interviewed in 2019 and 30 ECPs were interviewed in 2020. ECPs were chosen based on a random sample of ophthalmologists and optometrists nationwide that had sufficient exposure to blepharitis patients to provide a representative sample of ECPs who see and manage blepharitis patients, and would potentially be in a position to prescribe TP-03 if it were approved and available. The ECP Survey generally asked ECPs questions regarding their patient base and also introduced the TP-03 product profile to them. The ECP Survey showed that 44% of all diagnosed patients were over the age of 65, and 57% of diagnosed patients were female. Distribution of diagnoses across race and income metrics was approximately proportional to that of the United States population, implying little correlation of the condition with socioeconomic status. The ECP Survey also showed that in April through May of 2020, the number of blepharitis patients in the eye care clinic, which represented 60% of the total patients, was approaching the level of dry eye patients, which represented approximately 65% of the total patients. We believe this demonstrates that the market size for blepharitis may be similar to the market size of dry eye and that both are common in the eye care clinics, which informs us on the potential to increase diagnoses through ECP education. As the population of the United States continues to age, we believe this disease will become more prevalent. While we believe the ECP survey provides insight to the potential market size of Demodex patients, since it was a random sampling of ECPs nationwide that had sufficient exposure to blepharitis patients, the sample size of ECPs and corresponding potential patient population was relatively small and the number of blepharitis patients may not be comparable to the number of dry eye patients. The patients may have had overlapping diagnoses but the ECP Survey did not measure any such potential overlap.
The following figures show that according to the ECP Survey, the number of blepharitis diagnoses approached the number of dry eye diagnoses. This trend is also supported by independent epidemiology studies:
Figures 6 and 7: Blepharitis Diagnoses Approaching Number of Dry Eye Diagnoses
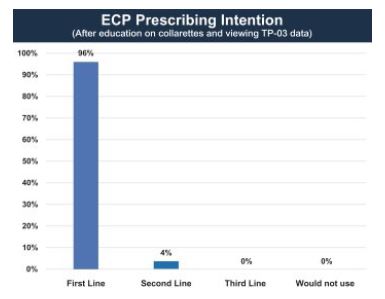
Additionally, the ECP Survey obtained feedback on a hypothetical TP-03 product profile, based on the Mars and Jupiter clinical trial data. In the ECP Survey, a total of 25 ophthalmologists and 25 optometrists were educated on the pathognomonic relationship of collarettes to Demodex blepharitis and exposed to the Mars and Jupiter clinical trial data. After exposure, ECPs were asked their intent to prescribe TP-03 if it were available for their patients presenting with Demodex blepharitis as indicated by collarettes. In these studies, 48 of 50 (96%) ECPs indicated that they would prescribe TP-03 as first-line treatment, and 2 of 50 (4%) indicated that they would prescribe it as second-line treatment. No ECPs indicated that they would prescribe TP-03 as third line or not prescribe TP-03.
The following figure shows that 96% of the surveyed ECPs indicated they would prescribe TP-03 as a first-line treatment:
Figure 8: 96% of Surveyed ECPs Indicated Prescribing Intention TP-03 as First-Line Treatment
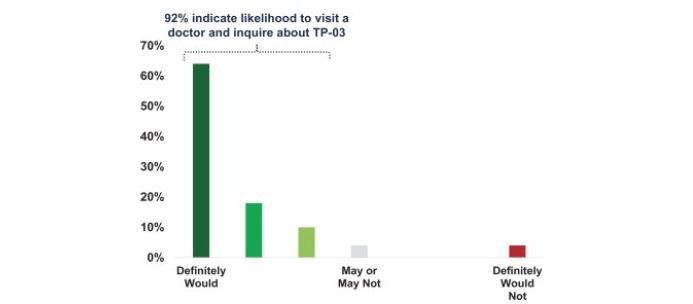
We also sponsored a separate market research survey of 50 patients with blepharitis symptoms, where the presence of “eye crust” on eye lashes was noted as a primary symptom. These patients were randomly selected nationwide and not chosen in connection with the identity of his or her ECP. The individuals with blepharitis symptoms were educated on collarettes and that Demodex mites cause collarettes, and provided with a description of a hypothetical product with the target profile of TP-03, which can potentially eradicate Demodex mites in the eyelid and eliminate collarettes in a majority of patients. After considering the target product profile, the individuals with blepharitis symptoms were asked how likely they were to visit a doctor and ask about whether a product such as TP-03 would be right for them (on a scale of 1-7). Forty-six of 50 (92%) of the individuals with blepharitis symptoms indicated a likelihood to visit a doctor and ask about a product with a target profile of TP-03, and 32 of 50 (64%) indicated a response of “definitely would.” Two of 50 (4%) of the individuals with blepharitis symptoms indicated that they “may or may not,” and two of 50 (4%) of the individuals with blepharitis symptoms indicated that they “definitely would not.”
The following figure shows the results of this survey:
Figure 9: 92% of Individuals with Blepharitis Symptoms Indicated Likelihood To Inquire About a TP-03-Type Product
We believe that blepharitis is underdiagnosed due to the lack of an approved treatment and associated patient and physician education, and the lack of awareness of the role of Demodex mites in the disease process. The ECP Survey showed that only 10-15% of ECPs were aware that collarettes are highly correlated with Demodex infestation. Despite the current lack of awareness by ECPs, we believe that the ocular surface disease marketplace is highly responsive to education of both the ECP and the patient. For example, as a result of ECP and patient education, the dry eye market grew significantly once there was a product to address the large, latent demand for an effective therapy. Dry eye had no approved therapeutic until 2003. Once a therapeutic was approved, the diagnosis rate increased by approximately 12 times, growing from 500,000 annual diagnoses in 2003 to over six million annual diagnoses by 2015. Annual diagnoses rates have been maintained at similar levels since 2015. We believe the Demodex blepharitis and dry eye markets may be analogous because both Demodex blepharitis and dry eye are diseases that affect the front of the eye, are treated by ECPs, can cause an inflamed ocular surface or eyelids and have similar patient demographics. The potential market for Demodex blepharitis, however, may not be analogous to the market for dry eye
due to differences in symptoms, regulatory approval and market dynamics and other factors. See “Risk Factors—Risk Related to Development and Commercialization of Our Product Candidates—The market for blepharitis and Demodex blepharitis may be not be similar to the market for dry eye” for additional information and risks related to the comparison of the Demodex blepharitis market to that of dry eye.
The following figure shows the annual diagnoses of dry eye disease prior to and after and the launch of an approved therapy for dry eye, with the 2.1 million diagnoses for blepharitis in 2019 shown on the plot for reference:
Figure 10: Dry Eye Diagnoses Per Year
Demodex mites have been implicated in approximately 45% of all blepharitis cases in a meta-analysis of 11 studies consisting of an aggregate of 4,741 patients with blepharitis. The incidence of Demodex blepharitis increases progressively with aging, which is the main risk factor for the condition, with the majority of people over the age of 60 having been shown to have the disease.
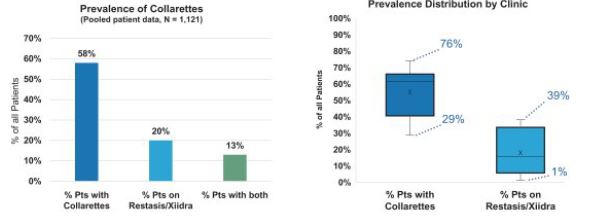
Our market research studies also included an epidemiological evaluation we conducted of 1,121 consecutive patients in eight ophthalmology and optometry practices conducted in 2020 (the "Patient Epidemiology Survey"), which showed that 58% of all patients entering the practices had collarettes and all patients entering practices with collarettes were also found to have Demodex blepharitis. By comparison, 20% of those same 1,121 patients were currently on a prescription therapeutic for dry eye disease (Restasis or Xiidra). Additionally, 13% of the 1,121 patients presented with collarettes and were also on a prescription therapeutic for dry eye at the same time. The clinics in the Patient Epidemiology Survey were geographically diverse and represented both high and low dry eye prescription rates. Each practice was required to enroll between 100 and 180 consecutive patients. The clinic with the lowest prevalence demonstrated 29% of consecutive patients had collarettes, and the clinic with the highest prevalence demonstrated 76% of consecutive patients had collarettes.
The following figure shows the prevalence of collarettes and prevalence distribution by clinic, from the Patient Epidemiology Survey:
Figures 11 and 12: Prevalence of Collarettes
We believe the data has demonstrated that there is a significant population for blepharitis, and for Demodex blepharitis more specifically, since the number of patients with collarettes exceeded by almost three times the number of patients on a prescription therapeutic for dry eye disease. While there is overlap between the number of patients with collarettes and the number of patients with dry eye and the number of patients with dry eye is most likely higher than the number of patients currently on a prescription for dry eye, and we believe the data shows a potential significant population for Demodex blepharitis. We also believe the data illustrates a significant opportunity to increase the diagnosis rate of Demodex blepharitis by educating ECPs on the high correlation of the presence of collarette to Demodex blepharitis in order for ECPs to incorporate the examination of the condition into standard practice.
Over the past several years, an increasing number of clinical studies on Demodex have been published in the context of ocular surface disease, with more than half of these studies published in the last four years. We believe this shows an increased understanding of the importance of Demodex in the scientific ophthalmic community and represents an important step in educating ECPs about Demodex blepharitis.
Current Treatment Options and Their Limitations
Despite the high prevalence of patients with Demodex blepharitis and growing awareness of the condition amongst ECPs, there are no FDA-approved therapeutics for the treatment of blepharitis or for Demodex blepharitis. Demodex blepharitis is currently treatedmanaged with a variety of over the counterover-the-counter remedies such as tea tree oil, lid wipes and artificial tears, as well as off-label prescription products for dry eye.
These approaches have significant limitations, including:
| | | | | | | | | | | |
| | | |
| • | | limited efficacy of over-the-counter and off-label treatments as well as device-based treatments administered in clinic by ECPs; |
| | | | | | | | | | | |
| • | | insufficient definitive knowledge of diagnostic criteria to guide treatment decisions; |
| | | | | | | | | | | |
| • | | prohibitive side effects (significant burning and stinging) from treatments that target Demodex mites (e.g. tea tree oil); |
| | | | | | | | | | | |
| • | | current treatments do not typically eradicate the Demodex mites, leading to a chronic and progressive disease; and |
| | | | | | | | | | | |
| • | | some treatments may be harmful (e.g. to meibomian gland epithelial cells). |
limited efficacyAccording to the Titan real-world prevalence study, 75% of over-the-counter and off-label treatments as well as device-based treatments administered in clinic by ECPs;
insufficient definitive knowledge of diagnostic criteria to guide treatment decisions;
prohibitive side effects (significant burning and stinging) from treatments that target Demodex mites (e.g.patients using tea tree oil);oils and
57% of those using lid wipes were found to have a high prevalence of collarettes, indicating that current treatments do not eradicatemanagement tools for this disease are ineffective. The Titan study was an institutional review board ("IRB") -approved, retrospective chart review of 1,032 patients across six U.S.-based ophthalmology and optometry practices conducted by seven investigators. The study was designed to better understand the prevalence of Demodex mites, leading to a chronic and progressive condition.
blepharitis via collarettes in U.S. eye care clinics.
Our Approach: Treating Demodex Mites, a Root Cause of Disease
To address these limitations and high unmet need for effectively treating Demodex blepharitis, we developed and are developing in the initial stages of commercializing XDEMVY, formerly known as TP-03, formulated as an eye drop, which we believe, if approved, hasis the potential to become thedefinitive standard of care for the treatment of Demodex blepharitis. TP-03XDEMVY is a novel therapeutic based on the drug lotilaner, which is designed to paralyze and eradicate mites and other parasites through the inhibition of parasite-specific GABA-Cl channels. To date, TP-03 has successfullyXDEMVY met the primary, secondary and/or exploratory,all endpoints as applicable, in each of the four completed Phase 2 trials. TP-03its clinical trials and was generally well tolerated throughout each of these trials. As a result, XDEMVY was approved by the FDA in July 2023 for the treatment of Demodex blepharitis and we believe that TP-03 has the potential to bebegan commercializing XDEMVY in August 2023. XDEMVY is the first ever approvedFDA-approved therapeutic for Demodex blepharitis.
The following figure summarizes the key advantages of TP-03:
Figure 13: Summary of TP-03 Key Advantages
| | | | | |
* | Blepharitis can increase the risk for both infection after cataract and refractive surgery and discomfort and can cause intolerance to contact lenses so treating Demodex blepharitis may improve tolerance to contact lenses and improve patient satisfaction with cataract and refractive surgery |
Commercial Strategy for Demodex Blepharitis Launch
If approved, we plan to commercialize TP-03 in the United States with a focused, specialty sales force, direct-to-consumer, or DTC, campaign, and ECP education campaigns with key opinion leaders, and professional associations. In our work with key opinion leaders and various associations to increase Demodex blepharitis awareness and education, we plan to highlight prevalence, impact, and simplicity of diagnosis of Demodex blepharitis. Our goal is to educate ECPs about the prevalence of Demodex blepharitis and the safety and efficacy of our products. In addition to educating ECPs, we believe that patient awareness and identification is important and plan to increase awareness through education and marketing efforts directed toward patients.
TP-03XDEMVY Eye Drops – Mechanism of Action
| | | | | | | | |
| | |
The active ingredient in TP-03 eye dropsXDEMVY is lotilaner, a member of a new class of anti-parasitic molecules called isoxazolines. It is a potent non-competitive antagonist of insect and arachnid GABA-Cl channels. Lotilaner is designed to eradicate Demodex mites by selectively inhibiting GABA-Cl channels, causing mite paralysis and eventual death. It has a low affinitydemonstrated no binding to mammalianhuman GABA-Cl and other ion channels (e.g. hERG) and thus likely has no impact on the human nervous system. Lotilaner is a lipophilic molecule, which may promote its uptake in the oily sebum of the hair follicle, where the mites reside. In clinical trials, TP-03 isXDEMVY was topically applied to the eye twice dailyBID to ensure delivery of the drug to the eyelid margin. Following mite eradication, collarettes eventually clear from the eyelid since they are composed of mite-related waste. | | |
The following figures showillustrate the intended paralysis of mites in the hair follicle by TP-03XDEMVY administration:
Figure 14:5: Progression of TP-03XDEMVY Application, Mite Paralysis and Eradication
Clinical Development Program
WeTo date we have completed one Phase 3 trial, one Phase 2b/3 trial, four Phase 2 clinical trials, to date, along withand one additionalPhase 1 trial for XDEMVY in ex vivoDemodex study. blepharitis, all of which met primary, secondary and/or certain exploratory endpoints, while demonstrating XDEMVY was well tolerated. These pivotal trial results (Phase 2b/3 and Phase 3) supported the FDA approval of XDEMVY in July 2023.
The table below summarizes completed and planned clinical investigations evaluating TP-03 for Demodex blepharitis. We plan to evaluate TP-03 in two pivotal registration trials, referred to as Saturn-1 and Saturn-2. Saturn-1 isSaturn-2 Trial
In May 2021, we initiated the Saturn-2 trial. It was a Phase 2b/3, randomized, controlled, double-blindmulticenter, double-masked trial to evaluatestudying the safety and efficacy of TP-03 is expected to enroll at least 350 Demodex blepharitis patients in multiple centers in the United States. We commenced Saturn-1 in September 2020 and top-line data is expected in 2021, subject to the impact of the ongoing COVID-19 pandemic. Saturn-2, our confirmatory Phase 3 randomized, controlled, double-blind trial, has a highly comparable design to that of Saturn-1 and is also expected to enroll approximately 350 Demodex blepharitis patients. Enrollment of Saturn-2 is expected to begin in 2021, subject to the impact of the ongoing COVID-19 pandemic. In connection with our IND application, we have received a “no-objection” letter from the FDA regarding the trial design for Saturn-1. The trial design for Saturn-2 is highly comparable to that of Saturn-1 and we expect to update the IND protocol prior to commencing Saturn-2. We expect these trial results to support the potential submission of an NDA for TP-03XDEMVY for the treatment of Demodex blepharitis.
The following figure shows our completedSaturn-2 trial was similar in design and planned investigations evaluating TP-03 for Demodex blepharitis:
Figure 15: Completedsize to Saturn-1, which met the primary and Planned Investigations Evaluating TP-03 for Demodex Blepharitis
* The Mars and Jupiter trials used collarette grade as an endpoint, which has been translated into apatients achieving collarette cure, (defined as <10 collarettes). This is different from the collarette cure (defined as <2 collarettes) endpoint used in Io, Europa, Saturn-1 and the planned Saturn-2 trials. The Mars and Jupiter trials also used mite density as an endpoint, which is different from mite eradication. Mite density is translated into mite eradication, which is defined as zero to two collarettes per lid. Secondary endpoints included the eradication of Demodexmites per lash consistently throughout trials.
** Primary endpoint in Io, Europa, Saturn-1 and intended in Saturn-2 is collarettethe proportion of patients achieving a cure based on a composite of collarette grade.
† In connection with our IND application, a no-objection letter was received from the FDA regarding the trial design of the Saturn-1 trial.
†† Saturn-2 design is highly comparable to that of Saturn-1 with respect to which the FDA raised “no-objection”cure and we expect to update the IND protocol prior to commencing Saturn-2.
TP-03 is a presentation of lotilaner, the API, formulated into a topical eye drop formulation. Only one dose strength of TP-03, 0.25% lotilaner solution was used across all clinical trials and studies.
erythema cure (eyelid redness). A statistically significant outcome for primary efficacy endpoints is typically one of the requirements for FDA approval of a product. A statistically significant outcome indicates that the probability of the outcome occurring at random is less than the pre-established allowed error level, frequently set at 0.05 (or 1 in 20).
Mercury Study
The Mercury preclinical study was an ex vivo study of the efficacy of TP-03 in causing the paralysis and death of Demodex mites performed at a single center in the United States in 2019 through 2020. Mites were collected following lash plucking at a single center in the United States. Mites were placed in small wells and submersed in a drop of either TP-03 (pilot formulation) or a vehicle control (formulation with no active ingredient). The ex vivo efficacyIn May 2022, we announced positive topline results of the Mercury study show that 100% of mites subjected to the pilot formulation of TP-03 were dead within 24 hours andSaturn-2 trial, our second XDEMVY pivotal trial. The Saturn-2 trial enrolled 412 adults having, among other things, more than 70% of mites subjected to the formulation with no active ingredient were still viable after four days of observation.
The following figure presents the efficacy results on Demodex mite survival probability over time, with the shaded regions representing the 95% confidence interval for each of the two groups:
Figure 16: Mercury StudyEx VivoEfficacy Results
Mars Phase 2a Single-Arm Clinical Trial
Mars was a Phase 2a single-arm clinical trial which evaluated the safetyten collarettes per lid and efficacy of TP-03 for the treatment of Demodex blepharitis completed in 2019 in Mexico City, Mexico at a well-established site for ocular therapy trials. Subjects were treated with TP-03 (pilot formulation) BID for 28 daysleast mild lid erythema. All pre-specified primary and followed to 90 days (62 days post-treatment). A total of 18 subjects were enrolled in the trial, with 15 patients receiving treatment beyond Day 1. Fourteen of the 15 subjects who received treatment completed the trial through Day 28. Twelve of the 15 subjects continued through 90 days (62 days post-treatment) and three subjects did not continue the trial after Day 28. In data described below, one subject was lost to follow-up after Day 2, one subject missed the Day 7 visit and one subject missed the Day 14 visit.
Exploratory endpoints included collarette grading score and mite density, as assessed by microscopic evaluation of plucked lashes. As this was an exploratory Phase 2a trial, no primary or secondary endpoints were pre-specified. We utilized the data from the exploratory endpointsmet, XDEMVY was well tolerated and improvement in lids (reduction of collarettes to determineno more than 2 collarettes per upper lid) was demonstrated in 55% of patients treated with XDEMVY.
Primary Endpoint:
•55% of patients on XDEMVY achieved complete collarette cure, and mite eradication rates. We defined collarette cure as collarette count of 10 or fewer, consistent with prior studies in the field. We defined mite eradication as zero mites per lash.
The following figure illustrates representative photos of two Mars subjects at baseline and Day 28 who exhibited a collarette cure:
Figure 17: Two Mars Trial Subjects at Baseline and Day 28 Who Exhibited a Collarette Cure
The proportion of subjects who exhibited a collarette cure was statistically significant compared to baseline by Day 14 with 54% of subjects (7 of 13 subjects, p < 0.05) exhibiting a cure, at Day 28 with 86% of subjects (12 of 14 subjects, p < 0.05) exhibiting a cure, and post-treatment at Day 60 with 83% of subjects (10 of 12 subjects, p < 0.05) exhibiting a cure and at Day 90 with 83% of subjects (10 of 12 subjects, p < 0.05) exhibiting a cure.
The proportion of subjects who exhibited mite eradication was statistically significant compared to baseline at Day 14 with 31% of subjects (4 of 13 subjects, p < 0.05) exhibiting eradication, at Day 28 with 57% of subjects (8 of 14 subjects, p < 0.05) exhibiting eradication, and post-treatment at Day 60 with 50% of subjects (6 of 12 subjects, p < 0.05) exhibiting eradication and Day 90 with 83% of subjects (10 of 12 subjects, p < 0.05) exhibiting eradication.
The following figure shows the data for each visit day:
Figures 18 and 19: Mars Phase 2a Trial Collarette Cure and Mite Eradication Rates
There were no adverse events reported and TP-03 was well tolerated. No subjects discontinued the trial due to adverse events.
Jupiter Phase 2b Trial
Jupiter was a Phase 2b randomized, controlled, double-blind trial which evaluated the safety and efficacy of TP-03 for the treatment of Demodex blepharitis completed in 2019 in Mexico City, Mexico at a well-established site for ocular therapy trials. Subjects were treated with either TP-03 (pilot formulation) or with a vehicle control BID for 28 days. Subjects
were followed to 90 days (62 days post-treatment) to evaluate post-treatment duration of effect. The primary endpoint was mite density for the treatment group as compared to the mite density for the vehicle control group at Day 28 and the secondary endpoint was collarette grade as compared to the collarette grade for the vehicle control group at Day 28. We utilized the collarette grading and mite density data to determine collarette cure and mite eradication rates. The definitions of collarette cure and mite eradication in the Jupiter trial were consistent with those used in the Mars trial. A total of 60 subjects were enrolled in the trial, with 51 subjects receiving treatment through Day 28 and 49 subjects completing the trial through Day 90.
The proportion of subjects who exhibited a collarette cure was statistically significant for TP-03 compared to vehicle control at Day 14 (78% vs. 44% subjects, p = 0.020), Day 28 (88% vs. 22%, p < 0.001) and post-treatment at Day 60 (95% vs. 26%, p < 0.001) and Day 90 (96% vs. 15%, p < 0.001).
The proportion of subjects who exhibited mite eradication was statistically significant for TP-03 compared to vehicle control at Day 14 (35% vs. 4% subjects, p = 0.009), Day 28 (67% vs. 26%, p = 0.005) and post-treatment at Day 60 (85% vs. 7%, p < 0.001) and Day 90 (68% vs. 19%, p = 0.001).
The following figures show the collarette cure and mite eradication rates for each visit day:
Figures 20 and 21: Jupiter Trial Collarette Cure and Mite Eradication Rates
There were no serious adverse events reported and TP-03 was well tolerated. Any adverse events were not related to treatment and no subjects discontinued the trial due to adverse events.
Trials With Updated Protocol and Formulation Designed to Support Pivotal Trials
Io Phase 2a Trial
Io was a Phase 2a single-arm trial which evaluated the safety and efficacy of TP-03 for the treatment of Demodex blepharitis completed in 2020 in Mexico City, Mexico at a well-established site for ocular therapy trials. The design of the Io trial took into account feedback from the FDA, and accordingly we subsequently defined collarette cure rate as a more stringent standard of less than or equal to two collarettes per lid at day 43, compared to 12% on the eyelid. The definitionvehicle (p<0.0001).
Secondary Endpoints:
•50% of patients on XDEMVY achieved mite eradication defined as zero mites per lash remained consistent with that of the Mars and Jupiter trials. In the Io trial we also utilized for the first time in clinical trials the formulation of TP-03 which is expectedat day 43, compared to support our NDA submission for Demodex blepharitis. The Io trial design, as well as the formulation of TP-03 utilized, are consistent with the design and formulation to be utilized in our Saturn-1 Phase 2b/3 trial. The primary efficacy endpoint was the proportion14% on vehicle (p<0.0001).
•30% of patients experiencingon XDEMVY compared to 9% of patients on vehicle (p<0.0001) achieved complete lid erythema cure at day 43.
•19% of patients on XDEMVY achieved a complete composite cure, based on collarette grade, or collarette cure, at Day 42. The secondary endpoint was the mite eradication rate at Day 42. A total of 18 subjects were enrolled in the trial, all of whom were crossed over from the vehicle control group of the Jupiter trial. In data described below, three subjects missed the Day 7 visit, one subject missed the Day 14 visit and one subject missed the Day 28 visit.
The proportion of subjects who exhibited a collarette cure was statistically significant for TP-03 compared to baseline by Day 14 with 29% of subjects (5 of 17 subjects, p < 0.05), at Day 28 with 71% of subjects (12 of 17 subjects, p < 0.05) and at Day 42 with 72% of subjects (13 of 18 subjects, p < 0.05).
The proportion of subjects who exhibited mite eradication was statistically significant for TP-03 compared to baseline by Day 14 with 47% of subjects (8 of 17 subjects, p < 0.05), at Day 28 with 59% of subjects (10 of 17 subjects, p < 0.05) and at Day 42 with 78% of subjects (14 of 18 subjects, p < 0.05).
The following figures present theachieving both complete collarette cure and mite eradication rates for each visit day:complete lid erythema cure, compared to 4% on vehicle (p<0.0001) at day 43.
Safety Profile:
Figures 22 and 23: Io Trial Collarette Cure and Mite Eradication Rates▪91% of XDEMVY patients reported that the drop comfort was neutral to very comfortable.
All 18 subjects completed the trial through Day 42 of dosing. There were no serious treatment-related adverse events reported and TP-03 was well tolerated. No subjects discontinued the trial duenor any treatment-related adverse events leading to adverse events. The most common adverse event included mild transient burning after administration for less than 10 seconds.treatment discontinuation.
Additional Analysis:
The following table provides a summary of treatment emergent adverse events for the trial:
Figure 24: Summary of Treatment Emergent Adverse Events
Europa Phase 2bSaturn-1 Trial
EuropaThe Saturn-1 trial was a Phase 2b randomized, controlled, double-blindmulticenter, double-masked Phase 2b/3 trial whichthat evaluated the safety and efficacy of TP-03 forXDEMVY in adults with Demodex blepharitis. In September 2020, we commenced the treatmentSaturn-1 trial and completed enrollment of Demodex blepharitis completed in 2020 in Mexico City, Mexico at a well-established site for ocular therapy trials. Like the Io trial, the Europa trial was designed with the same protocols and formulation as will be utilized in our Saturn-1 Phase 2b/3 trial. Subjects were treated with either TP-03 or with a vehicle control BID for 42 days. A total of 54 subjects were enrolled in the trial with 34 subjects completing the trial through Day 42.
The primary efficacy endpoint was the proportion of421 adult patients experiencing a cure based on collarette grade of two or fewerhaving more than ten collarettes on the upper lid and at least mild erythema of the upper eyelid margin. Each patient had at least 1.5 mites per lash on the upper and lower eyelids combined. One drop of XDEMVY was self-administered twice per day in each eye for six weeks. Enrolled patients received no treatment for blepharitis symptoms (i.e. lid hygiene) during the trial or 14 days prior to enrollment. The primary endpoint was complete collarette cure (grade zero defined as - zero to two collarettes per lid) and the secondary endpoints included complete mite
eradication (mite density of zero mites per lash) and composite cure (the presence of zero to two collarettes on the upper eyelid and the absence of erythema (redness).
In June 2021, we announced positive results of the Saturn-1 trial. The pre-specified primary and secondary endpoints were met, and improvement in lids (reduction of collarettes to no more than 2 collarettes per upper lid) was demonstrated in 44% of patients treated with XDEMVY.
44% of patients on XDEMVY achieved the primary endpoint of complete collarette reduction at day 43 compared to the7% on vehicle control,(p<0.0001). 81% of patients achieved a significant, clinically meaningful collarette count reduction defined as zero to ten collarettes per lid at Day 42. Secondary efficacy endpoints included (1)day 43 compared to 23% of those on vehicle (p<0.0001). Additionally, a significant, clinically meaningful collarette reduction was seen in 23% of patients on XDEMVY compared to 11% on vehicle as early as day 8 (p=0.03).
The secondary endpoint of complete mite eradication achieved statistically significant results by day 15, and 68% of patients on XDEMVY achieved mite eradication compared to 17% on vehicle control (defined as zero mites per lash)(p<0.0001) at Day 42, and (2)day 43.
For composite cure, 13.4% of patients on TP-03 achieved a complete cure based on a composite cure rateendpoint of collarette cure and zero lid erythema (redness)cure compared to 1.0% on vehicle control. Subjects(p<0.0001) at day 43. Results for complete erythema cure (19% of patients on XDEMVY compared to 7% of patients on vehicle, p<0.0001) and one grade or more erythema improvement (45% of patients on XDEMVY compared to 28% of patients on vehicle, p=0.0002) were also asked to rate the comfortstatistically significant. Additionally, 92% of the trial eye drop at Day 42.
The proportion of subjects who exhibited a collarette cure was statistically significant for TP-03 compared to vehicle control at Day 28 (72% vs 11% subjects, p < 0.001) and Day 42 (80% vs. 16%, p < 0.001).
The proportion of subjects who exhibited a mite eradication was statistically significant for TP-03 compared to vehicle control at Day 28 (56% vs 11% subjects, p = 0.006) and Day 42 (73% vs. 21%, p = 0.003).
The following figures show the collarette cure and mite eradication rates for each visit day:
Figures 25 and 26: Europa Trial Collarette Cure and Mite Eradication Rates
As provided in the figures below, the proportion of subjects who exhibited composite cure rate of zero lid erythema combined with collarette cure was statistically significant for TP-03 compared to vehicle control at Day 28 (67% vs 11% subjects, p < 0.001) and Day 42 (73% vs. 11%, p < 0.001).
The following figure shows data for each visit day:
Figure 27: Composite Cure Rate was Statistically Significant for TP-03 Compared to Vehicle Control
The figure below shows how subjects rated drop comfort at each visit in each of the active and vehicle control groups. The bars shows the percentage ofXDEMVY patients who rated the drop as either “very comfortable,” “slightly comfortable” or “neither comfortable or uncomfortable” in both the active and vehicle control groups for each visit day. There was only one subject, in the vehicle group,reported that rated the drop as “very uncomfortable” and only at Days 14 and 28. All other subjects that did not rate the drop as “very comfortable”, “slightly comfortable” or “neither comfortable or uncomfortable” rated the drop as “slightly uncomfortable.” At Day 7, the drop was rated as “slightly uncomfortable” by 6/22 (27.3%) in the active group, 2/20 (10.0%) in the vehicle control group and 8/42 (19.0%) overall.
The following figure shows the drop comfort ratings at each visit day:
Figure 28: Comfort Ratings
No subjects discontinued the trial duewas neutral to an adverse event and TP-03 was well tolerated. Allvery comfortable. There were no serious treatment-related adverse events were mild in severity.
The following table provides a summary of treatment related or possibly related adverse events for the trial:
Figure 29: No Subjects Discontinued Europa Trial Due to Adverse Event
TP-03 Safety Profile From Phase 2 Clinical Trials
Our Phase 2 trial data indicates that TP-03 was well tolerated with no significantnor any treatment-related adverse events leading to treatment discontinuation acrossdiscontinuation.
In July 2021, we presented additional data from the four trials. A totalSaturn-1 Trial at the American Society of 82 subjects have received one or more dosesCataract and Refractive Surgery 2021 Annual Meeting demonstrating high treatment response rates, and reinforcing the potential of TP-03 and have hadXDEMVY to be the standard of care forDemodex blepharitis patients.
•95% of XDEMVY patients showed a significant improvement in mite count, achieving ≤0.5 mites per lash
•93% of XDEMVY patients improved by at least one follow-up visit; while 52 subjects received the vehicle control. Only nine subjects treated with TP-03collarette grade
We also announced results from an additional Saturn-1 safety analysis, which reinforced XDEMVY’s positive profile, revealing that XDEMVY had treatment-relatedno clinically significant adverse events, versus one subject treated with the vehicle control. The most common treatment-related adverse events, or TRAEs, reported were burning after administration, which was reported in seven subjects (8.5%) receiving TP-03 versus one subject (1.9%) receiving the vehicle control. Other TRAEs reported for subjects treated with TP-03 were blurred vision in two subjects (2.4%effect on multiple safety measures including Corrected Distance Visual Acuity ("CDVA"), dysgeusia in one subject (1.2%corneal staining, and intraocular pressure ("IOP"), and instillation site erythema in one subject (1.2%); with no corresponding TRAEs reported in subjects treated with vehicle.
Adverse events deemed unrelated to treatment occurred in five subjects treated with TP-03, with diarrhea reported in two subjects (2.4%), hypertension reported in one subject (1.2%), cataract operation reported in one subject (1.2%), and pharyngotonsillitis reported in one subject (1.2%).
For subjects treated with the vehicle control, the six subjects reported adverse events unrelated to treatment, which consisted of diarrhea in three subjects (5.7%) and viral upper respiratory tract infection in three subjects (5.7%).
Furthermore, there was littlesignificant findings from slit lamp biomicroscopy or no change in mean corrected distance visual acuity across all four trials, and only one subject treated with TP-03 showed a worsening of 0.22 logMAR from baseline to Day 28fundus exam in the left eye. This subject did not returnstudy. In addition, no impact to endothelial cell density ("ECD") was demonstrated in a subset of 21 patients. ECD was further evaluated as part of the Saturn-2 trial plan and also demonstrated no impact.
Phase 2 Clinical Trials
We completed four Phase 2 clinical trials for their follow-up visit on Day 42. Intraocular pressure was recorded in theXDEMVY, along with one additional ex vivo study, which included our Mars, Jupiter, Io, and Europa trials, and there were only minor reported changes observed in both the active and the vehicle control. The most common clinically significant slit lamp examination finding was an increase from baseline in corneal staining. The increases in staining were typically transient, occurring at only a single visit, and, in the controlled trials, occurred more often in subjects treated with the vehicle control versus subjects treated with TP-03. The only other slit lamp examination finding was conjunctival hyperemia in one subject receiving TP-03 at Day 7 and one subject receiving the vehicle control at Day 14.
Disease Recurrence Following End of Treatment Course
Subjects from theclinical trials. Key efficacy endpoints for our Mars and active arm of the Jupiter clinical trials were followed for one year after enrollment. Combined interim data for recurrence of clinicalincluded collarette grade Demodex blepharitis was defined as a collarette score of two or more, and mite density of one or more per lash. The data show a persistent treatment effect of TP-03and key efficacy endpoints for six months or greater for collarette score, with meaningful recurrence of collarettes occurring after six months with the recurrence increasing considerably one year after treatment. Mites recurred more quickly than collarettes, consistent with collarettes being composed of mite byproducts. Subsequent to initial treatment, we believe that TP-03 may be utilized by patients and ECPs for intermittent re-treatment based on the disease recurrence observed.
Ongoing and Planned Pivotal Trials
Saturn-1 and Saturn-2 Trials
Saturn-1 is a randomized, controlled, double-blind Phase 2b/3 trial. In September 2020, commenced the trial to evaluate the safety and efficacy of TP-03 for the treatment of Demodex blepharitis in multiple centers in the United States. Target enrollment for the trial is at least 350 subjects, enrolled 1:1 between a BID dose of TP-03 and a vehicle control. Saturn-1 is designed for subjects to receive treatment for 42 days and be subsequently followed for safety under a separate protocol for one year. The primary efficacy endpoint is proportion of subjects with cure based on a collarette grade of zero (< 2) at Day 43. The two secondary endpoints are (1) proportion of subjects with Demodex eradication (zero mites per lash) at Day 43 and (2) proportion of subjects with a composite of collarette and erythema cure at Day 43 (collarette grade of zero combined with erythema score of zero). Top-line data for Saturn-1 is anticipated in 2021, subject to the impact of the ongoing COVID-19 pandemic.
Saturn-2 is designed to be a confirmatory Phase 3 trial that we plan to initiate promptly after completing enrollment in Saturn-1. It is a randomized, controlled, double-blind trial to evaluate the safety and efficacy of TP-03 for the treatment of Demodex blepharitis in multiple centers in the Unites States. Target enrollment for the trial is approximately 350 subjects, enrolled 1:1 between a BID dose TP-03 arm and a vehicle control. Saturn-2 is designed for subjects to be followed for 43 days and receive treatment for 42 days. The primary efficacy endpoint is proportion of subjects with cure based on a collarette grade of zero at Day 43. The two secondary endpoints are (1) proportion of subjects with Demodex eradication (zero mites per lash) at Day 43 and (2) proportion of subjects with a composite of collarette and erythema cure at Day 43 (collarette grade of zero combined with erythema score of zero). Enrollment of Saturn-2 is expected to begin in 2021, subject to the impact of the ongoing COVID-19 pandemic.
The protocol, including the subject inclusion and exclusion criteria and endpoints, and formulation are designed to be consistent with those of our Io and Europa Phase 2clinical trials included collarette cure rate based on collarette grade, which we refer to herein as collarette cure rate, and mite eradication rate. The primary, secondary and/or certain exploratory endpoints were met, as applicable, in such trials, and showed statistically significant cure and eradication rates in Io and Europa. XDEMVY was generally well tolerated throughout these trials. We have received a “no-objection” letter from the FDA regarding the trial design for Saturn-1 and Saturn-2 has a highly comparable design to that of Saturn-1. We expect that the data form the Saturn-1 and Saturn-2 trials will support our NDA filing for TP-03 in Demodex blepharitis. Prior to submitting an NDA, we plan to perform a clinical pharmacokinetic study for TP-03 to support our NDA submission for Demodex blepharitis, and the FDA is recommending we perform carcinogenicity testing as well as embryofetal development studies in a second species. While we have factored these recommendations into our timeline and expenses, any adverse results, or any additional or similar requirements or recommendations from the FDA could cause delay in obtaining regulatory approval and increased costs.
Development of TP-03 for Meibomian Gland Disease
Beyond Demodex blepharitis, we intend to exploreare exploring the clinical potential for the use of TP-03 in the treatment of MGD, a chronic abnormality of the meibomian glands.glands, which are glands on the inner part of the eyelid that secrete lipids and other molecules that are critical to maintaining a healthy tear film on the surface of the eye. MGD is commonly characterized in part by a widespread clogging or destruction of the meibomian glands that can result in tear film deficiency and is one of the leading causes of dry eye disease. In the United States,U.S., MGD prevalence has been found to be approximately two-thirds of the estimated 3430-40 million dry eye patient population.patients who are impacted by the disease. One species of Demodex mite, Demodex brevis, is known to infest the meibomian gland, and clinical signs of MGD have been shown to be correlated with Demodex brevis infestation. While dry eye is a multi-factorial disease, TP-03 is designed to relieve some of the key elements associated withthat may be contributing to MGD by virtue of causing the death oferadicating the Demodex brevis mites.mites as well as decreasing the inflammation of the eyelid caused by Demodex blepharitis. In December 2023, we reported positive topline results from the Ersa trial which demonstrated a statistically significant improvement in two objective measures
of the disease following treatment for 12-weeks with TP-03 as measured by the MGSS and the number of glands secreting normal (clear) liquid as measured in the central 15 glands of the lower eyelid. We plan to discuss and determine the potential regulatory path forward with the FDA.
There areis no FDA-approved therapeuticsmedicine for MGD. MGD, which is currently treated with a variety of over-the-counter remedies such as tea tree oil, lid wipes and artificial tears, as well as off-label prescription products for dry eye. MGD is also treated with in-office device procedures for gland expression (de-clogging) by an ECP. Despite these existing treatments, there is still a significant and unmet need for an effective prescription therapeutic in MGD.
The following figures showillustrate a representation of Demodex mites infesting the meibomian gland:
Figure 30: 6: Demodex Mites Infesting the Meibomian Gland
Our Additional Product Candidates
TP-04 Topical Formulation for the Treatment of Rosacea
Rosacea
Rosacea is a chronic skin disease characterized by facial redness, inflammatory lesions, burning and stinging, which can flare up in response to certain triggers such as sun exposure or emotional stress. According to the U.S. National Rosacea Society, approximately 16 million people in the United StatesU.S. are affected by rosacea and a study estimates rosacea prevalence can represent up to 5.4% of the global population. There are two phenotypes for rosacea: patients who present with papulopustular rosacea or PPR,("PPR"), and patients who do not present with PPR (non-PPR), with approximately 55% of patients presenting with the PPR phenotype and 45% presenting with the non-PPR phenotype. According to primary research we conducted ofwith dermatologists, of patients who are being treated with topicals for rosacea, an estimated 18% of overall rosacea patients are seeking treatments, with PPR patients generally more likely to seek treatment given the noticeable symptoms.
The cause of rosacea remains unclearmultifactorial but there is increasing evidence that Demodex mites play a role in the disease. Studies have found a correlation between Demodex infestation and rosacea, with a higher density of Demodex mites found in the skin of rosacea patients than in controls.patients. A proposed theory suggests that the Bbacillusacillus oleronius bacteria has a pathogenic role, contributing to skin inflammation and the signs and symptoms of rosacea; these bacteria are also known to be sensitive to the antibiotics typically prescribed to treat rosacea. Meanwhile, Demodex mites have been shown to carry Bbacillusacillus oleronius in their digestive tracts, suggesting that Demodex may contribute to rosacea by being a transporter for the bacteria that causes the disease. Furthering that point, there is evidence to suggest that Bbacillusacillus oleronius forms a symbiotic relationship with Demodex, and that both species must be present in order to cause the signs and symptoms of rosacea. Other bacteria such as Sstaphylococcustaphylococcus epidermidis, also potentially carried by Demodex mites, may play a role in the disease as well. In fact, Soolantra, the current standard of care for rosacea, has an anti-parasitic and Demodex targeting mechanism, suggesting that the clearance of Demodex plays a role in resolving the signs of symptoms of rosacea.
Current Treatment Options and Their Limitations
Rosacea is currently treated through topical anti-parasitic creams like ivermectin and other topicals including azelaic acid, and antibiotics like metronidazole, as well as other mechanismstherapies such as alpha agonists. An estimated 50% of rosacea patients are on at least one prescription topical treatment. TheA current standard of care, Soolantra,standard-of-care, is a brandedan anti-parasitic drug composed of 1% ivermectin cream which targets Demodex mites but is only modestly effective, takes 8 to 12 weeks to showthat has demonstrated modest efficacy and is expensive at around approximately $500$700 wholesale acquisition cost or WAC, for a 30-day supply.
Other treatments in developmentavailable products include Sol-Gel’sGalderma's Epsolay, a topical cream containing 5% encapsulated benzoyl peroxide and Foamix’s FMX103,Journey Medical Corporation's Zilxi, a topical foam containing 1.5% minocycline, both of which are being commercialized for the treatment of Subtype II rosacea,
characterized by small, dome-shaped erythematous papules that resemble acne but are associated with burning and stinging sensations.
While current treatments can address the symptoms of the disease by reducing redness and decreasing papules and pustules, for the majority of patients, complete clearance of these features is not currently achievable and there still exists an unmet medical need.
Our Approach,Approach: TP-04 Topical Formulation for Rosacea
To address this unmet need in the rosacea market, we are developing lotilaner is being developed as a topical dermatology product, TP-04, initially for the treatment of PPR. ItTP-04 is designed to be active after topical administration in skin with no systemic activity. Lotilaner’s mechanism of targeting and killing Demodex mites has been established through our preclinical study and clinical trials evaluating TP-03XDEMVY in Demodex blepharitis, which is why we believe it may be effective in another Demodex driven condition.disease. We believe we can improve upon existing treatments in the market, like Soolantra, with an API that is potentially more effective (longer half-life, more lipophilic, greater therapeutic window). We believe a longer half-life is expected to leadleads to a more durable and long-lasting treatment and that more lipophilicity is expected to provide better bioavailability in the sebum in the follicle and sebaceous glands where mites reside, thus increasing the opportunity to target and eradicate mites and a greater therapeutic window.
We further believe there is potentialhave completed the initial preclinical studies and the Galatea Phase 1 trial for TP-04 to be evaluated in combination withand have selected a topical anti-inflammatory agents or vasoconstrictors.
We have initiated the preclinical studiesformulation for TP-04.early clinical studies. We intend to leverage systemic preclinical data from our TP-03XDEMVY program (suchsuch as embryofetal development studies, genotoxicity studies and safety pharmacology studies, and augment with additional dermal preclinical studies such as in vitro skin permeation and penetration studies,the dermal toxicology and dermal pharmacodynamics testing we intend to conduct) to identify a lead formulation that maximizes dermal levels of TP-04 and which we believe can compare favorably to Soolantra.studies. We may need to address this approach with the FDA if we were to conduct a clinical trial in the United States and if the FDA rejected our use of data from these preclinical studies, it could cause delay in any trial we conduct in the United States for TP-04. We plan to evaluateare evaluating whether TP-04 is safe and effective at treating the symptoms of PPR in the Galatea trial, a Phase 1/22a trial outside the United States,being conducted in Canada, which is expected to begin enrollmentwe initiated in 2021.March 2023 and in February 2024 announced positive topline results. Prior to commencing clinical trials in the United StatesU.S. for TP-04, we will need to obtain an INDinvestigational new drug application ("IND") from the FDA.
TP-05 Oral Formulation for the Prophylactic Protection against Lyme Disease
Lyme Disease
Lyme disease is the most common vector-borne disease in the United States,U.S., caused by infection of Borrelia bacteria following a bite by a tick vector, predominantly ticks of the Ixodes genus (namely Ixodes scapularis in the United States)U.S.). Estimates of annual cases of Lyme disease in the United States range fromThere are approximately 300,000 to 400,000, with many cases being misdiagnosed or diagnosed late. According to 2018 data from the U.S. Centers for Disease Control, or CDC, in 2018, over 3080 million people in the United States areU.S. at risk of contracting Lyme disease exposure with over 20more than 30 million of those people residingwhich are moderate to high risk, according to a report commissioned by the Company. Further, there are approximately 400,000 reported cases in statesthe U.S. each year, but it is believed that havethe actual number of cases could be more. We estimate a “high incidence”greater than $1.3 billion impact to the U.S. healthcare system as a result of Lyme disease per the CDC; and the geographic footprint of Lyme disease carrying ticks is expanding.disease. Lyme disease occurs most commonly in geographical areas where the Ixodes scapularis tick is prevalent, namely in the Northeast and Mid-Atlantic regions of the United States.U.S., but also in other regions of the U.S. Lyme disease also occurs in certain parts of Europe, typically resulting from a different Ixodes species vector.
The mechanism of Lyme disease infection is well understood. Borrelia bacteria colonizes the salivary glands of the ticks, and the infected saliva is transmitted to the human host when a tick attaches to a person for feeding. The transfer usually occurs at the conclusion of the feeding and therefore, the probability of Borrelia transmission, and thereforethus the risk of Lyme disease, increases with the duration of the tick’s attachment. Borrelia is rarely transferred during the first or even second day of feeding but transfers quite efficiently during and after the third day of feeding.feeding (greater than 48 hours). This window from the time of bite to the time of transmission offers an opportunity for intervention to prevent Lyme disease.disease if the tick can be killed prior to the transfer of the Borrelia bacteria.
Lyme disease can be a serious condition that may affect multiple bodilyorgan systems and produce a broad range of symptoms. Early symptoms include a localized rash, fever and fatigue. More severe, sometimes chronic, symptoms may evolve as the infection spreads, including fever, muscle and joint pain, peripheral and central neurological deficits and lymphocytic meningitis. Lyme disease can be successfully treated with oral antibiotics when diagnosed sufficiently early, but chronic symptoms can commonly persist beyond antibiotic treatment.
Current Lyme Disease Prophylaxis Options and Their Limitations
Lyme disease is currently prevented through behavior modification – avoiding areas where ticks are prevalent, wearing clothing which minimizes tick exposure, using insect repellants, and physically removing ticks that have attached. With the exception of removing attached ticks, none of these approaches prevents the transmission of Borrelia post-bite.
Moreover, there are currently no FDA-approved therapeuticssmall molecules or biologics for the prevention of Lyme disease. A vaccine for Lyme disease, LYMERix, was developed and launched by SmithKline Beecham in 1999. Approximately 1.5 million doses of the vaccine were sold in 1999, but the product was quickly pulledremoved from the market following negative press and class actiona class-action litigation claiming a dangerous side effect profile. We are aware of a second vaccinevaccines currently under development including a multivalent recombinant protein vaccine, VLA-15, being developed by Valneva in partnership with Pfizer for Lyme disease, VLA-15.
There is significant unmet need fordisease; mRNA-based vaccine mRNA-1982/1975, being developed by Moderna and elicits high levels of anti-OspA antibodies; and a safe, effective and easy to use prophylactic for at-risk individuals in Lyme disease.
Our Approach,Approach: TP-05 Oral Formulation for the Prophylactic Protection against Lyme Disease
Since Borrelia is usually transferred during the second or third day following a tick bite, our approach is to eradicate the tick before it can transmit the bacteria. To do this, we are developing TP-05 as an oral tablet formulation of lotilaner. We are targeting potentially at least 30 days of prophylactic protection against Lyme disease with a singlesimple oral doseregimen of TP-05. Given that lotilaner was developed specifically, in part, to eradicate ticks with systemic administration to companiondomesticated animals such as dogs or cats, the pharmacology of lotilaner infor Lyme disease prophylaxis is well understood. Similar to its mechanism against Demodex mites, lotilaner is a potent non-competitive antagonist of tick GABA-Cl channels. Antagonism of these channels in ticks induces paralysis and eventual death. While lotilaner results in the paralysis and eventual death of the I. scapularis vector, it does also result in the death of B. burgdorferi, a non-free-living bacterium whose entire survival is conditional upon its living host. The high preferenceselectivity for insect and arachnid GABA-Cl channels over mammalianhuman channels promoteswhere there has been no demonstrated binding, is a highly advantageous part of the safety profile of the molecule. Extensive preclinical systemic toxicology and safety pharmacology studies have been performed by third parties to date and support advancing TP-05 into clinical development. Lotilaner has a long, approximateapproximately 30-day systemic half-life in dogs, which we believe could provide for a convenient monthlyoral tablet administration.
We have identifiedIn December 2022, we announced positive topline results from the Phase 1 Callisto trial for TP-05, a lead formulationnovel, oral, non-vaccine therapeutic for the potential prevention of Lyme disease. The Callisto trial was a randomized, double-blind, single and multiple-ascending dose trial that evaluated the safety, tolerability, and PK of TP-05 for Phase 1/2 trials. Forin healthy subjects. Results from the trial showed that TP-05 and its use in Lyme prophylaxis, we intend to leverage oral systemic preclinicalwas well tolerated with no dose-related or drug-related serious adverse events. Pharmacokinetic data from our TP-03 program (embryofetal development studiesthe trial demonstrated rapid absorption and safety pharmacology studies)an extended half-life of TP-05 that potentially supports a convenient oral regiment supporting its potential as a rapid onset, prophylactic therapy for Lyme disease. Additionally, exploratory ex-vivo tick kill modeling that utilized serum from TP-05 treated subjects demonstrated potent, rapid killing of adult and nymph ticks. In December 2022, we also announced the initiation of the Phase 2a Carpo trial, evaluating TP-05 for the potential prevention of Lyme disease in humans. The Carpo trial is a randomized, double-blind, placebo-controlled trial that evaluated the efficacy of TP-05 in killing lab grown, non-disease carrying ticks after they have attached to the skin of healthy volunteers, as well as third-party oral systemic preclinical studies, such as 28-dayconfirm the safety, tolerability, and 13-week rat toxicology studies, eight-month dog toxicology studies,blood concentration of TP-05. In February 2024, we announced positive topline results from the Carpo trial and systemic genetoxicity studies. These preclinical studies useplan to discuss and determine the same route of administration (oral) and coverpotential regulatory path with the dose range intended for clinical studies, but whereas the preclinical studies used a gavage administration, we intend to administer TP-05 in humans via a tablet or capsule.FDA.
We believe TP-05 is a presentationcurrently the only non-vaccine, drug-based prophylaxis in development designed to target ticks, and potentially prevent Lyme disease transmission. It is designed to rapidly and durably provide systemic blood levels of lotilaner which ispotentially sufficient to kill infected ticks attached to the same ashuman body before they can transmit the Elanco API but formulated into a tablet. We obtained rights to these studies through the All Human Uses Elanco Agreement and we are not currently planning to conduct any additional preclinical studies to support our Phase 1/2 clinical trial. We had a successful pre-IND meeting with the FDA in February 2021 and gained agreement on our proposed Phase1 study design. We plan to file an IND in the US in the second quarter of 2021 and, subject to FDA approval of the IND, we intend to initiate our Phase 1 trials to evaluate safety and pharmacokinetics of TP-05 from single ascending dose (SAD) and multiple ascending dose (MAD) studies in normal healthy volunteers.
Malaria
Malaria is one of the world’s highest unmet public health needs, with approximately 228 million cases and more than 400,000 deaths caused by malaria worldwide in 2018 alone. The disease is caused by several species of plasmodium, a parasite transmitted to humans by a mosquito vector. Symptoms and outcomes include fever, vomiting, fatigue, seizure, coma and death. The disease is widespread in tropical and subtropical regions near the equator, with sub-Saharan Africa, parts of Asia and parts of Latin America impacted the most. The majority of deaths occur in Africa.
There has been a massive years-long effort to combat malaria using a variety of techniques, including administration of multiple types of prophylactic and therapeutic drugs, insecticide spraying and mosquito nets. While worldwide rates of malaria have dropped over the last two decades, meaningful reduction in malaria rates has stalled since approximately 2015. It is widely hoped that new vector control and prophylactic modalities will play an important role in further reducing the burden of the disease.
Our Approach, TP-05 Oral Formulation, Long-acting Endectocide for Community Malaria Reduction
The malaria community, including, the World Health Organization, Medicines for Malaria Venture, Bill and Melinda Gates Foundation and others, has called for the development of a long-acting endectocide, or antiparasitic drug active against endoparasites such as plasmodium, for the control and reduction of malaria in endemic regions. An endectocide for malaria is a drug taken by a person bacteria that causes the death or incapacity of a mosquito before it can infect another person. Field studies and modeling have shown that reducing the lifespan of the mosquitos may significantly reduce malaria transmission. With mass drug administration, which is a common approach for other anti-malarial medications, a community could attainLyme disease.
herd protection against spread of malaria using an endectocide. For instance, a modeling study on mosquito populations published in PNAS in 2018 (the "PNAS Study"), showed that mass administration of an isoxazoline endectocide could reduce community malaria rates by as much as 65% with administration to just 30% of a community’s population.
The pharmacology of isoxazoline endectocides in malaria control is well understood. Similar to its mechanism against Demodex and ticks, the isoxazoline endectocides are potent non-competitive antagonists of GABA-Cl channels in mosquitos. Furthermore, according to the PNAS Study, the isoxazoline endectocides showed significant ability to cause the death of various disease carrying mosquito vectors that were fed drug-supplemented human blood in a membrane feeding assay. Additional transmission modeling in the PNAS Study of these isoxazolines on malaria incidence, which assumed a 90-day efficacy period, translated to survival of less than 1% of mosquitos. We believe our treatment concept of the oral administration of TP-05 to a human population may likewise lead to the death of blood-fed insect vectors and a decline in disease transmission. This is based on the relatively long half-life of lotilaner, an isoxazoline endectocide, which has been demonstrated in preclinical studies. However, modeling studies may not be accurate representations of malaria transmission in human populations and the efficacy of lotilaner or TP-05 to cause the death of mosquitos and reduced transmission rates has not been tested in human populations.
For TP-05 and its use in malaria community reduction, we intend to leverage oral systemic preclinical data from our TP-03 program (embryofetal development studies and safety pharmacology studies), as well as third-party oral systemic preclinical studies, such as 28-day and 13-week rat toxicology studies, eight-month dog toxicology studies, and systemic genetoxicity studies. These preclinical studies use the same route of administration (oral) and cover the dose range intended for clinical studies, but whereas the preclinical studies used a gavage administration, we intend to administer TP-05 in humans via a tablet or capsule. We obtained rights to these studies through the All Human Uses Elanco Agreement and we will not conduct any additional preclinical studies to support our Phase 1/2 clinical trial. We intend to evaluate whether TP-05 is safe and effective at eradicating mosquitos in a planned Phase 1/2 trial, which may be conducted outside the United States.
Chemistry, Manufacturing and Controls
We do not currently own or operate and currently have no plans to establish facilities for manufacturing, storing, distributing or testing our product or product candidates. We rely and expect to continue to rely for the foreseeable future on CMOscontract manufacturing organizations ("CMOs") to manufacture and supply our preclinical and clinical materials to be used during the development of our product candidates. We have assembled a team of employees and consultants to oversee our technical quality and CMOs.
TheOur lead product, XDEMVY, and our product candidate, TP-03, is a presentation of lotilaner, the API, formulated into a topical eye drop formulation. Only one dose strength of TP-03 is currently being developed, 0.25% lotilaner solution. Since TP-03 is an anti-infective, only one strength, at the maximum level that be formulated (0.25%), is required to be evaluated clinically.
We believe that our supply of TP-03 is sufficient to complete Saturn-1, and that our supply of cGMP lotilaner is sufficient to complete Saturn-2. We believe that the existing capacity of our current API supplier will beis sufficient to support commercial scale-up, validation and commercial launch activities if TP-03 is approved.activities. Our current supplier currently manufactures cGMPcurrent good manufacturing practice ("cGMP") lotilaner at multiple geographically distinct facilities.
Although we have relied on a single supplier for both non-clinical and clinical supply for lotilaner under cGMP protocols and a single CMO to manufacture TP-03XDEMVY and to perform analytical testing services, it is our intent to identifywe have identified and qualifyare in the process of qualifying an additional manufacturersmanufacturer to provide lotilaner and drug product manufacturing and analytical testing services, if possible, prior to submission of our NDA for TP-03.services. The drug product manufacturing is a compounding and aseptic filling operation that we believe could be transferred to additional CMOs as necessary. We are evaluating a supplierhave suppliers for TP-04 topical formulation for rosacea and have identified a supplier for TP-05 oral formulation for our Phase 1/2 trials for Lyme disease and malaria.
Our third-party service providers, our third-party supply chain providers, their facilities and the TP-03XDEMVY used in our clinical trials or for commercial sale are required to be in compliance with the requirements of cGMP. The cGMP regulations govern manufacturing processes and procedures, including requirements relating to organization of personnel, buildings and facilities, equipment, control of components and packaging containers and closures, production and process controls, packaging and labeling controls, holding and distribution, laboratory controls, records and reports, and returned or salvaged products. Product candidates used in late-stage clinical trials must be manufactured in accordance with cGMP requirements and manufacturing specifications and processes must satisfy FDA or other authorities’ requirements before any product is approved and before we can manufacture commercial products. Our third-party manufacturers are also subject to periodic inspections of facilities by the FDA and other authorities, including procedures and operations used in the testing and manufacture of TP-03XDEMVY to assess compliance with applicable regulations. Our failure, or the failure of our third-party providers and supply chain providers, to comply with such statutory and regulatory requirements could subject us to possible legal or regulatory action, including clinical holds, fines, injunctions, civil penalties, delays, suspension or withdrawal of approvals, license revocation, suspension of production, warning letters, the seizure or recall of products, operating restrictions and criminal prosecutions.
Any of these actions could have a material impact on commercial supplies of XDEMVY, clinical supplies of TP-03 or our other product candidates. Contract manufacturers at times encounter difficulties involving production yields, quality control and quality assurance, as well as shortages of qualified personnel.
Competition
The biotechnology and pharmaceutical industries are characterized by rapid technological advancement, significant competition and an emphasis on intellectual property. We face potential competition from many different sources, including major and specialty pharmaceutical and biotechnology companies, academic research institutions, governmental agencies and public and private research institutions. Any products or product candidates that we successfully develop and commercialize will compete with existing approaches and new therapies that may become available in the future. We believe that the key competitive factors affecting the success of any of our products or product candidates will include efficacy, combinability, safety profile, convenience, cost, level of promotional activity devoted to them and intellectual property protection.
WhileOther than XDEMVY, there are currently no currently availableother on-label prescription pharmaceutical treatments available for the treatment of blepharitis or Demodex blepharitis specifically, a number of other treatments are currently available for the treatment of blepharitis in the United States. CurrentU.S. Other than XDEMVY, current treatments for blepharitis in the United StatesU.S. include over the counter and off-label remedies such as tea tree oil, lid wipes and artificial tears. We are aware of other companies developing potential prescription therapies for blepharitis, including Azura Ophthalmics, Aperta Biosciences, LLC, Eyevance Pharmaceuticals, Formosa Pharmaceuticals, Inc., Glaukos Corp., Hovione Scientia, Nicox, SA, Novaliq GmbH, Premark Pharma, Quorum Innovations and Quorum Innovations.Viatris. To our knowledge, Azura Ophthalmics, Aperta Biosciences, LLC isand Glaukos Corp. are the only companycompanies currently focused on Demodex blepharitis and Nicox SAare in pre-clinical stage, and Premark Pharma, Azura Ophthalmics and Nicox are the only companies with blepharitis programs that have completed Phase 2 trials.
Many of the companies against which we may compete have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory
approvals and marketing approved products than we do. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
Sales and Marketing
In light of our stage of development, we have not yet established a commercial organization or distribution capabilities. If TP-03 receives marketing approval, we plan to commercialize TP-03(lotilaner ophthalmic solution) 0.25% in the United States by developing our ownU.S. in August 2023, and hired an approximately 100 person sales organizationforce targeting a subsetapproximately 15,000 ECPs that we believe represent approximately 80% of the approximately 25,000 prescribing ECPs in the United States.potential prescribers. Throughout our commercialization efforts, we intendhave continued to drive awareness and further educate ECPs on Demodex blepharitis and how to properly diagnose it withby having patients look down during a simplestandard slit lamp examination.eye examination and identifying collarettes, the pathognomonic sign of the disease. Given the importance of increasing awareness and educating patients with blepharitis, we also anticipate deployinghave deployed focused DTCsocial and digital marketing campaigns for TP-03. We expect to conduct most of the buildout of this organization following NDA submission and approval of TP-03, if obtained.XDEMVY.
There are an estimated 25 million people in the U.S. who suffer from Demodex blepharitis, however, we are initially targeting the approximately 7 million people who are proactively seeking treatment for Demodex blepharitis or seeking treatment for complementary eye conditions or treatments.

Outside the United States,U.S., we intend to further develop commercialization strategies for TP-03, which may include collaborations with other companies. OnIn March 26, 2021, we executed an out-license agreemententered into the China Out-License with LianBio, Ophthalmology Limited, granting exclusive commercial rights of TP-03 for the treatment of Demodex blepharitis and MGD within the China Territory. The People’s Republicterms of China, Macau, Hong Kong, and Taiwan. Wethis agreement are contractually entitled to receive (i) an aggregate $25 million by June 30, 2021, (ii) regulatory and sales milestone receipts totaling $75 million and $100 million, respectively, (iii) tiered royalties in the low double-digits on the net sales of TP-03further described below within this greater China territory, and (iv) a minority interest inLicense Agreements: LianBio that vests upon the achievement of certain clinical and regulatory milestones.
Agreement
Intellectual Property
We protect our intellectual property rights and proprietary technology with a combination of patent rights that we own or license in certain fields of use, trademark rights, confidentiality procedures and contractual provisions. We seek not only to protect our intellectual property rights and proprietary technology in select key global markets, but also to supplement our intellectual property portfolio with new filings and applications to enhance such protection and support commercialization of current and future product candidates. To that end, we continue to seek protection for our technological innovations and branding efforts by filing new patent and trademark applications when and where appropriate. In the normal course of business, we intend to pursue, when possible, composition, method of use, dosing and formulation patent protection, as well as manufacturing and drug development processes and technology.
The term of individual patents varies depending upon the date of filing of the patent application, the date of patent issuance, and the legal term of patents in the countries in which they are obtained. Generally, patents issued for applications
filed in the United States are effective for 20 years from the earliest effective filing date. In addition, in certain instances, a patent term can be extended to restore a portion of the term effectively lost as a result of the FDA regulatory review period. The restoration period cannot be longer than five years and the total patent term, including the restoration period, must not exceed 14 years following FDA approval. The duration of patents outside of the United States varies in accordance with provisions of applicable local law, but typically is also 20 years from the earliest effective filing date.
Our patent portfolio includes a combination of issued patents and pending patent applications licensed from third parties, as well as those assigned solely to us based on our ongoing development activities. The patents and applications in our portfolio can be categorized as related to XDEMVY, TP-03, TP-04, TP-05 or future pipeline product candidates and alternative technologies. Some of our issued patents and patent applications are exclusively licensed to us in therapeutic fields of use from Elanco. TheAs of December 31, 2023, the patents and patent applications owned by or licensed to us worldwide include approximately 3745 issued patents and approximately 2572 pending patent applications.
We in-license certain of such patents and patent applications from Elanco. These patents and patent applications relate to lotilaner and are issued or pending in, for example, the United States,U.S., Argentina, Australia, Brazil, Canada, Chile, China,
Columbia, several European territories, India, Japan, South Korea, Mexico, New Zealand, the Russian Federation, South Africa and Taiwan. The licensed-in portfolio includes approximately 3638 issued patents and approximately 417 pending patent applications; the issued patents and at least some of the pending patent applications include composition of matter claims. The estimated natural expiration date of the issued in-licensed patents is approximately 2029 to 2030 with a potential extension until 2032.
Approximately 1844 of our owned patents and pending patent applications include treatment and composition of matter claims which relate to our TP-03 product candidateXDEMVY with respect to our lead indication (e.g., isoxazoline parasiticides for the treatment of Demodex blepharitis), as well as other conditions. These pending patent applications include applications in the United States,U.S., Australia, Brazil, Canada, China, Europe, Hong Kong, Israel, India, Japan, South Korea, Mexico, New Zealand, the Russian Federation, and South Africa, and the Patent Cooperation Treaty (PCT).Africa. We received an issuance on onehave a total of our owned pending patent applications with treatment claims, U.S. Pat. No. 10,835,517, on November 17, 2020.7 granted patents. The estimated natural expiration datedates of thisthese issued patent ispatents are 2038, and if additional patents issue on thesethe pending applications of ours, the estimated natural expiration dates are between approximately 2038 and 2040.
While estimated expiration dates and descriptions of patents and patent applications are listed above, the actual protection afforded by a patent varies on a product-by-product basis, from country-to-country, and depends upon many factors, including the type of patent, the scope of its coverage, patent term adjustments, the availability of regulatory-related extensions, disclaimers, the availability of legal remedies in a particular country, and the validity and enforceability of the patent.
Our continuing research and development activities, technical expertise and contractual arrangements supplement our existing intellectual property protection and help us maintain our competitive position, and we rely on trade secrets to protect our proprietary information and technologies, especially where we do not believe patent protection is appropriate or obtainable, or where such patents would be difficult to enforce. In order to maintain such trade secrets and other proprietary information, we rely in part on confidentiality agreements with our employees, consultants, contractors, outside scientific collaborators and other advisors.
We also protect our brand through trademark rights. We areAs of December 31, 2023, we own i) 1 trademark registration in the registered owner of six U.S., ii) 9 pending trademark applications.applications in the U.S., iii) 18 trademark registrations in foreign countries, and iv) 3 pending trademark applications in foreign countries. In order to supplement the protection of our brand, we also own at least one6 registered internet domain name.names.
We continue to make substantial investments to augment the capabilities of our people, processes, and technologies in order to address our cybersecurity risks. Our cybersecurity risks, and the controls designed to mitigate those risks, are integrated into our overall risk management governance and are reviewed quarterly by our Board of Directors.
As of December 31, 2023, we've implemented a set of comprehensive cybersecurity and data protection policies and procedures. Our employees and contractors receive regular cybersecurity awareness trainings, including specific topics related to social engineering and email fraud. We have capable employees and consultants with significant expertise and certifications in cybersecurity related to our industry, as well as access to additional resources and other third parties as needed. We invest in advanced technologies for continuous cybersecurity monitoring across our information technology environment which are designed to prevent, detect, and minimize cybersecurity attacks, as well as alert management of such attacks.
Our Information Technology General Controls ("ITGCs") are firmly established based on recognized industry standards and cover areas such as risk management, data backup, and disaster recovery. We have implemented processes to monitor security threats and vulnerabilities and respond to all cybersecurity incidents affecting us, including prompt escalation and communication of major security incidents to senior business leadership and our Board of Directors. We conduct cybersecurity penetration testing annually to identify and remediate cybersecurity gaps. We also perform cybersecurity assessments of all our third-party providers who have access to our information technology systems and data.
License Agreements
Elanco In-License Agreement for Skin and Eye and Derm Elanco Agreement
In January 2019, we entered into the Eye and Derman agreement with Elanco Agreement granting us an exclusive, worldwide, sublicensable license rights to certain intellectual property for the development, marketing, and commercialization of lotilaner for the treatment, palliation, prevention or cure of any eye or skin disease or condition in humans.humans (as amended and restated in June 2022, the "Eye and Derm Elanco Agreement"). We are obligated to use commercially reasonable efforts to develop and commercialize products comprising lotilaner and must achieve certain developmental milestones within specified achievement deadlines. If we fail to meet these obligations, Elanco has the right to terminate the Eye and Derm Elanco Agreement. We utilize the intellectual property licensed under the Eye and Derm Elanco Agreement in our TP-03 and TP-04 product candidates. We are permitted to have certain third parties manufacture lotilaner for us and, upon ElancoElanco's consent, additional third parties.
WeUnder the Eye and Derm Elanco Agreement, we have made ancash payments to Elanco totaling $9.0 million for clinical milestone achievements, including: a $1.0 million upfront payment at execution in 2019 and a total of $1.0$4.0 million for three specified clinical milestone achievements in September 2020, April 2021, and March 2023, which were recorded as research and development expense in the accompanying Statement of Operations and Comprehensive Loss in the respective periods incurred. Additionally, during the third quarter of 2023, a milestone of $4.0 million was achieved and paid to Elanco at contract execution,upon the first commercial sale of XDEMVY in the U.S, which wewas recorded within “research and development” expense within our statements of operations foras an intangible asset in the year ended December 31, 2019. accompanying Balance Sheets.
In accordance with the terms of the Eye and Derm Elanco Agreement, we are obligated to make further cash payments to Elanco upon our achievement of various clinical milestones up to an aggregate maximum of $6.0$2.0 million and various sales milestones up to an aggregate
maximum of $79.0$75.0 million. If we receive certain types of payments from sublicensees, we are obligated to pay Elanco a variable percentage beginning in the low double-digitsmid to high single digits of such proceeds and decreasing after certain milestones are met, except for sublicense revenue generated after achieving regulatory approval for the use of lotilaner to treat or cure any eye or skin disease or condition in humans. We owe Elanco tiered royalties during the royalty term in the mid-to-high single digits on our future net sales and those of our sublicensees. The royalty term for any licensed product in a given country commences on the date of first commercial sale of such licensed product and ends on the latest of (i) expiration of the last-to-expire of the licensed patents which has at least one valid claim, (ii) the expiration of regulatory exclusivity and (iii) ten years after the first commercial sale of such licensed product in such country. As a result of the commercialization of XDEMVY in August 2023, we began accruing royalties payable, which were recorded to cost of sales in the accompanying Statement of Operations and Comprehensive Loss for the year ended December 31, 2023.
The Eye and Derm Elanco Agreement shall expire on a licensed product-by-licensed product and country-by-country basis upon the expiration of the applicable royalty term with respect to such licensed product in such country. The achievement deadlines for eye-related diligence milestones range between 18 months after contract execution to six years after contract execution. The achievement deadlines for dermatological diligence milestones range between 24 months after contract execution to nine years after contract execution.
Either party may terminate the Eye and Derm Elanco Agreement upon a material breach by the other party, solely in the country pertaining to such breach, that is not cured within 60 days after receiving written notice thereof. If we fail to comply with our development obligations under the Eye and Derm Elanco Agreement, and fail to remedy such failure or cure such breach within 60 days, Elanco will have the right to terminate the Eye and Derm Elanco Agreement. If we fail to meet any diligence milestones by the achievement deadlines set forth in the Eye and Derm Elanco Agreement for any reason other than those outside of our reasonable control, and such milestones remain unmet for 120 days after Elanco notifies us thereof, Elanco may terminate the Eye and Derm Elanco Agreement. If we fail to meet certain dermatological milestones by the achievement deadlines set forth in the Eye and Derm Elanco Agreement for any reasons other than those outside of our reasonable control, and such milestones remain unmet for 120 days after Elanco notifies us thereof, Elanco may limit our field of use under the Eye and Derm Elanco Agreement to the treatment, palliation, prevention or cure of eye diseases or conditions in humans only. If Elanco terminates the Eye and Derm Elanco Agreement for our failure to achieve a development milestone by the specified achievement deadline, then we must grant Elanco a non-exclusive, sublicensable, royalty free license to our patents and know-how relating to lotilaner to develop, manufacture and commercialize lotilaner and any licensed products for the treatment, palliation, prevention or cure of any eye or skin disease or condition in humans. Elanco may also terminate the Eye and Derm Elanco Agreement if we, our affiliates or sublicensees initiate proceedings to oppose Elanco’s licensed patents and such proceeding is not withdrawn within 30 days of Elanco providing notice to us; provided that Elanco may not terminate the Eye and Derm Elanco Agreement for a challenge by a sublicensee if we terminate the sublicense with such sublicensee within such 30 day period.
Under the terms of the Eye and Derm Elanco Agreement, we grant togranted Elanco a worldwide, sublicensable, royalty-free, perpetual license to our patents related to lotilaner and the licensed products and to our know-how to research, develop, make and commercialize lotilaner and the licensed products for all applications in non-human animals, agricultural application, seed treatment applications and urban pest applications related to structural, turf, lawns and gardens. We also grantgranted Elanco an exclusive royalty-free, perpetual license to any intellectual property we conceive from our use of lotilaner applications in non-human animals, agricultural applications, seed treatment applications and urban pest applications related to structural, turf, lawns and gardens.
Elanco retains the sole responsibility to prosecute the patents they license to us and has the first right to enforce the licensed intellectual property against third parties in the licensed field of use but cannot settle or dispose of any such action without our written consent.
Elanco In-License Agreement for All Human Uses Elanco Agreement
In September 2020, we entered into the All Human Uses Elanco Agreement with Elanco granting us an exclusive, worldwide, sublicensable license to certain intellectual property for the development, marketing, and commercialization of lotilaner for all applications in humans other than the treatment, palliation, prevention or cure of any eye or skin disease or condition. We are obligated to use commercially reasonable efforts to develop and commercialize products comprising lotilaner and must achieve certain developmental milestones within specified achievement deadlines. If we fail to meet these obligations, Elanco has the right to terminate the All Human Uses Elanco Agreement. We utilize the intellectual property licensed under the All Human Uses Elanco Agreement in our TP-05 product candidates. We are permitted to have certain third parties manufacture lotilaner for us and, upon ElancoElanco's consent, additional third parties.
Pursuant toUnder the terms of the All Human Uses Elanco Agreement, we issued 1,652,346222,460 shares of our common stock to Elanco at the execution of the agreement in September 2020, with an estimated fair value of $3.1 million.
In March 2021, we issued 187,500 shares of our common stock to Elanco, which we recorded within “research and development” expense within our statementswith an estimated fair value of operations for the year ended December 31, 2020. We are also obligated to issue Elanco an additional 187,500 shares of our common stock in the second quarter of 2021$5.5 million, to maintain the All Human Uses Elanco Agreement. In accordance withDecember 2022, we achieved a clinical milestone of $0.5 million related to the termsinitiation of this agreement,our Phase 2a Carpo trial for the potential treatment of Lyme disease.
Under the All Human Uses Elanco Agreement, we have made cash payments to Elanco totaling $0.5 million for clinical milestone achievements. We are obligatedrequired to make furtheradditional cash payments to Elanco upon ourthe achievement of various clinical milestones up tofor an aggregate maximum of $4.5$4.0 million and various commercial and sales threshold milestones up tofor an aggregate maximum of $77.0 million. We may owe further
sales milestone paymentsIn addition, we are obligated to pay contractual royalties to Elanco for sales in certain countries. If we receive payments from sublicensees, we are obligated to pay Elanco a variable percentage beginning in the low to mid double-digits of such proceeds and decreasing after certain milestones are met, except for sublicense revenue generated after achieving regulatory approval for the use of lotilaner to applications in humans other than to treat or cure any eye or skin disease or condition. We owe Elanco tiered royalties during the royalty term in the mid-to-high single digits on our future net sales and those of our sublicensees. The royalty term for any licensed product in a given country commences on the date of first commercial sale of such licensed product and ends on the latest of (a) expiration of the last-to-expire of the licensed patents which has at least one valid claim, (b) the expiration of regulatory exclusivity, and (c) ten years after the first commercial sale of such licensed product in such country. The All Human Uses Elanco Agreement shall expire on a licensed product-by-licensed product and country-by-country basis upon the expiration of the applicable royalty term with respect to such licensed product in such country. The achievement deadlines for diligence milestones range between 24 months after contract execution to six years after contract execution.
Either party may terminate the All Human Uses Elanco Agreement upon a material breach by the other party, solely in the country pertaining to such breach, that is not cured within 60 days after receiving written notice from the other party. If we fail to comply with our development obligations under the All Human Uses Elanco Agreement, and fail to remedy such failure or cure such breach within 60 days, Elanco will have the right to terminate the All Human Uses Elanco Agreement. If we fail to meet any diligence milestones by the achievement deadlines set forth in the All Human Uses Elanco Agreement for any reason other than those outside of our reasonable control, and such milestones remain unmet for 120 days after Elanco notifies us of the failure to meet such diligence milestone, Elanco may terminate the All Human Uses Elanco Agreement. If Elanco terminates the All Human Uses Elanco Agreement for our failure to achieve a development milestone by the specified achievement deadline, then we must grant Elanco a non-exclusive, sublicensable, royalty free license to our patents and know-how relating to lotilaner to develop, manufacture and commercialize lotilaner and any licensed products for all applications in humans other than the treatment, palliation, prevention or cure of any eye or skin disease or condition. Elanco may also terminate the All Human Uses Elanco Agreement if we, our affiliates or sublicensees initiate proceedings to oppose Elanco’s licensed patents and such proceeding is not withdrawn within 30 days of Elanco providing notice to us; provided that Elanco may not terminate the All Human Uses Elanco Agreement for a challenge by a sublicensee if we terminate the sublicense with such sublicensee within such 30 day period.
Under the terms of the All Human Uses Elanco Agreement, we grantgranted to Elanco a non-exclusive worldwide, sublicensable, royalty-free, perpetual license to our patents related to lotilaner and the licensed products and to our know-how to research, develop, make and commercialize lotilaner and the licensed products for all applications in non-human animals and other non-human-use applications, agricultural applications, seed treatment applications and urban pest applications related to structural, turf, lawns and gardens. We also grant to Elanco an exclusive, royalty-free, perpetual license to any intellectual property we conceive from our use of lotilaner for all applications in non-human animals agricultural applications, seed treatment applications and urban pest applications related to structural, turf, lawns and gardens.other non-human applications.
Elanco retains the sole responsibility to prosecute the patents they license to us and has the first right to enforce the licensed intellectual property against third parties in the licensed field of use but cannot settle or dispose of any such action without our written consent.
LianBio Agreement
On March 26, 2021, we entered into athe China Out-License with LianBio for its exclusive development and license agreement (the “LianBio Agreement”) with LianBio, pursuant to which, among other things, we licensed the productcommercialization rights for the development and commercialization of TP-03 (lotilaner ophthalmic solution, 0.25%) in the People’sThe People's Republic of China, Macau, Hong Kong, Macau, and Taiwan (the “Territory”"China Territory") for the treatment of Demodex blepharitis and Meibomian Gland Disease.
Under the termsThrough December 31, 2023, we received aggregate payments from LianBio totaling $82.5 million comprised of the LianBio Agreement, we are entitled to an upfront paymentinitial consideration of $15.0 million within fifteen (15) daysand $67.5 million for the achievement of signing with an additional unconditional paymentspecified milestone events.
As of $10.0 million within 45 days of signing. WeDecember 31, 2023, we will be eligible to receive developmentfurther consideration from LianBio upon the achievement of additional TP-03 events, including: (i) additional regulatory milestones and commercializationone-time payments of up to an aggregate of $22.5 million, (ii) China-based TP-03 sales threshold milestone payments of up to $75.0 million andan aggregate of $100.0 million, respectively, as well as(iii) tiered mid-to-high-teenlow-to-high-teen royalties onfor the saleChina Territory TP-03 product sales, and (iv) vesting of TP-03 in the Territory. We also received a LianBio equity warrant to purchase ordinary shares of LianBio, which will vest upon the achievement of certain development and commercializationregulatory milestones. The warrant will be exercisable at the fair market value at the time of issuance. The term of the LianBio AgreementChina Out-License will expire upon the expiration of the royalty term in the China Territory, as defined in the agreement, unless earlier terminated. LianBio may also terminate the LianBio AgreementChina Out-License for any reason upon ninety (90) days’ prior notice to us. The foregoing summary
On February 13, 2024, LianBio announced its completion of a comprehensive strategic review and determined to initiate the wind down of its operations, including the sale of remaining pipeline assets, the delisting of its American Depositary Shares, deregistration under Section 12(b) of the LianBio Agreement doesExchange Act and workforce reductions. As of the date of this filing, it is uncertain if and when we will receive any royalties or future milestone consideration under the China Out-License, including but not purport to be complete and is qualified in its entirety by referencelimited to the full textmilestone achievement of the LianBio Agreement, which will be filed as an exhibit to our Quarterly Report on form 10-Q for the quarter ended March 31, 2021.
Government Regulation
Government authorities in the United StatesU.S. at the federal, state and local level and in other countries extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, post-approval monitoring and reporting, marketing and export and import of drug products such as those we are developing. Generally, before a new drug can be marketed, considerable data demonstrating its quality, safety and efficacy must be obtained, organized into a format specific for each regulatory authority, submitted for review and approved by the regulatory authority. We will be required to navigate the various preclinical, clinical and commercial approval requirements of the governing regulatory agencies of the countries in which we wish to conduct studies or trials or seek approval of our product candidates. The processes for obtaining regulatory approvals in the United StatesU.S. and other countries, as appropriate, along with subsequent compliance with appropriate federal, state, local and foreign statutes and regulations, require the expenditure of substantial time and resources.
United StatesU.S. Drug Regulation
In the United States,U.S., we are subject to extensive regulation by the FDA, which regulates drugs under the Federal Food, Drug, and Cosmetic Act (the "FDCA"), and its implementing regulations. FDA approval is required before any new unapproved drug or dosage form, including a new use of a previously approved drug, can be marketed in the United States.U.S. Drugs also are subject to other federal, state and local statutes and regulations. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations requires the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. or foreign requirements at any time during the product development process, approval process or post-marketing may subject an applicant to a variety of administrative or judicial sanctions, such as the FDA’FDA’s refusal to approve pending NDAs, withdrawal of an approval, imposition of a clinical hold, issuance of warning or untitled letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement or civil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect on our business, market acceptance of our products, and our reputation.
Our product candidates are considered small molecule drugs and must be approved by the FDA through the NDA process before they may be legally marketed in the United States.U.S. The process generally involves the following:
completion of extensive preclinical studies in accordance with applicable regulations, including studies conducted in accordance with good laboratory practice, or GLP, requirements;
submission to the FDA of an IND, which must become effective before human clinical trials may begin in the United States and must be updated annually or when significant changes are made;
approval by an independent IRB or independent ethics committee at each clinical trial site before each trial may be initiated;
performance of adequate and well-controlled human clinical trials in accordance with applicable IND regulations, GCP, requirements and other clinical trial-related regulations to establish substantial evidence of the safety and efficacy of the investigational product for each proposed indication;
submission to the FDA of an NDA;
a determination by the FDA within 60 days of its receipt of an NDA to accept the submission for review;
satisfactory completion of an FDA pre-approval inspection of the manufacturing facility or facilities where the drug will be produced to assess compliance with cGMP requirements, and of selected clinical investigational sites to assess compliance with GCD;
potential FDA audit of the preclinical study and/or clinical trial sites that generated the data in support of the NDA filing;
payment of user fees for FDA review of the NDA;
FDA review and approval of the NDA, including consideration of the views of any FDA advisory committee, prior to any commercial marketing or sale of the drug in the United States; and
compliance with any post-approval requirements, including the potential requirement to implement a REMS and the potential requirement to conduct post-approval studies.
| | | | | | | | | | | |
| | | |
| • | | completion of extensive preclinical studies in accordance with applicable regulations, including studies conducted in accordance with good laboratory practice ("GLP") requirements; |
| | | | | | | | | | | |
| • | | submission to the FDA of an IND, which must become effective before human clinical trials may begin in the U.S. and must be updated annually or when significant changes are made; |
| | | | | | | | | | | |
| • | | approval by an independent IRB or independent ethics committee at each clinical trial site before each trial may be initiated; |
| | | | | | | | | | | |
| • | | performance of adequate and well-controlled human clinical trials in accordance with applicable IND regulations, GCP, requirements and other clinical trial-related regulations to establish substantial evidence of the safety and efficacy of the investigational product for each proposed indication; |
| | | | | | | | | | | |
| • | | submission to the FDA of an NDA; |
| | | | | | | | | | | |
| • | | a determination by the FDA within 60 days of its receipt of an NDA to accept the submission for review; |
| | | | | | | | | | | |
| • | | satisfactory completion of an FDA pre-approval inspection of the manufacturing facility or facilities where the drug will be produced to assess compliance with cGMP requirements, and of selected clinical investigational sites to assess compliance with GCP; |
| | | | | | | | | | | |
| • | | potential FDA audit of the preclinical study and/or clinical trial sites that generated the data in support of the NDA filing; |
| | | | | | | | | | | |
| • | | payment of user fees for FDA review of the NDA as well as annual fees after NDA approval; |
| | | | | | | | | | | |
| • | | FDA review and approval of the NDA, including consideration of the views of any FDA advisory committee, prior to any commercial marketing or sale of the drug in the U.S.; and |
| | | | | | | | | | | |
| • | | compliance with any post-approval requirements, including the potential requirement to implement a Risk Evaluation and Migration Strategy ("REMS") and the potential requirement to conduct post-approval studies. |
The data required to support an NDA are generated in two distinct developmental stages: preclinical and clinical. The preclinical and clinical testing and approval process can take many years and the actual time required to obtain approval, if any, may vary substantially based upon the type, complexity and novelty of the product or condition being treated.
Preclinical Studies and IND Submission
Before testing any drug product candidate in humans, the product candidate must undergo rigorous preclinical testing. The preclinical developmental stage generally involves laboratory evaluations of drug chemistry, formulation and
stability, as well as in vitro and animal studies to assess the potential for adverse events and in some cases to establish a rationale for therapeutic use. The conduct of preclinical studies is subject to federal regulations and requirements, including GLP regulations for certain safety/toxicology studies. An IND is a request for authorization from the FDA to administer an investigational product to humans, and must become effective before human clinical trials may begin in the United States.
An IND sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and plans for clinical studies, among other things, to the FDA as part of an IND. Some long-term preclinical testing, such as animal tests of reproductive adverse events and carcinogenicity, may continue after the IND is submitted. An IND automatically becomes effective 30 days after receipt by the FDA and clinical trials may proceed under such IND at such time, unless before that time the FDA raises concerns or questions related to one or more proposed clinical trials and places the trial on clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. As a result, submission of an IND may not result in the FDA allowing clinical trials to commence. A separate submission to an existing IND must also be made for each successive clinical trial conducted during product development along with any subsequent changes to the investigational plan.
Clinical Trials
The clinical stage of development involves the administration of the investigational product to healthy volunteers or patients under the supervision of qualified investigators, generally physicians not employed by or under the trial sponsor’s control, in accordance with GCP requirements, which include the requirement that all research subjects provide their informed
consent for their participation in any clinical trial. Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria and the parameters to be used to monitor subject safety and assess efficacy. Each protocol, and any subsequent amendments to the protocol, must be submitted to the FDA as part of the IND. Furthermore, each clinical trial must be reviewed and approved by an IRB for each institution at which the clinical trial will be conducted to ensure that the risks to individuals participating in the clinical trials are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the informed consent form that must be provided to each clinical trial subject or his or her legal representative, and must monitor the clinical trial until completed. There also are requirements governing the reporting of ongoing clinical trials and completed clinical trial results to public registries, including the website maintained by the U.S. National Institutes of Health, ClinicalTrials.gov.
A sponsor who wishes to conduct a clinical trial outside of the United StatesU.S. may, but need not, obtain FDA authorization to conduct the clinical trial under an IND. If a foreign clinical trial is not conducted under an IND, the sponsor may still submit data from the clinical trial to the FDA in support of an NDA. The FDA may agree to accept a well-designed and well-conducted foreign clinical trial not conducted under an IND if the trial was conducted in accordance with GCP requirements and the FDA is able to validate the data through an onsite inspection, if deemed necessary, and the practice of medicine in the foreign country is consistent with the United States.
Clinical trials in the United StatesU.S. generally are conducted in three sequential phases, known as Phase 1, Phase 2 and Phase 3. Although the phases are usually conducted sequentially, they may overlap or be combined.
Phase 1 clinical trials generally involve a small number of healthy volunteers or disease-affected patients who are initially exposed to a single dose and then multiple doses of the product candidate. The primary purpose of these clinical trials is to assess the metabolism, pharmacologic action, tolerability and safety of the drug in humans, the side effects associated with increasing doses, and if possible, to gain early evidence on effectiveness.
Phase 2 clinical trials generally involve studies in disease-affected patients to determine the dose and dosing schedule required to produce the desired benefits. At the same time, safety and further pharmacokinetic and pharmacodynamic information is collected, possible adverse effects and safety risks are identified and a preliminary evaluation of efficacy is conducted.
Phase 3 clinical trials generally involve a large number of patients at multiple sites and are designed to provide the data necessary to demonstrate the safety and effectiveness of the product for its intended use and to establish the overall benefit/risk relationship of the product to provide an adequate basis for product approval. These trials may include comparisons with placebo and/or other comparator treatments. The duration of treatment is often extended to mimic the actual use of a product during marketing.
A Phase 1/2 clinical trial has elements of a Phase 1 trial and a Phase 2 trial. We have designated our TP-04 and TP-05 trials as Phase 1/2 trials since we intend to go beyond the typical safety and tolerability assessments of a Phase 1 trial and intend to have these trials include additional efficacy assessments as well.
A Phase 2b/3 clinical trial has elements of a late Phase 2 trial and a Phase 3 trial. We have designated Saturn-1 as a Phase 2b/3 trial as it is both our first multi-center trial based in the United States, and also a pivotal trial for the United States.
| | | | | | | | | | | |
| • | | Phase 1 clinical trials generally involve a small number of healthy volunteers or disease-affected patients who are initially exposed to a single dose and then multiple doses of the product candidate. The primary purpose of these clinical trials is to assess the metabolism, pharmacologic action, tolerability and safety of the drug in humans, the side effects associated with increasing doses, and if possible, to gain early evidence on effectiveness. |
| | | | | | | | | | | |
| • | | Phase 2 clinical trials generally involve studies in disease-affected patients to determine the dose and dosing schedule required to produce the desired benefits. At the same time, safety and further pharmacokinetic and pharmacodynamic information is collected, possible adverse effects and safety risks are identified and a preliminary evaluation of efficacy is conducted. |
| | | | | | | | | | | |
| • | | Phase 3 clinical trials generally involve a large number of patients at multiple sites and are designed to provide the data necessary to demonstrate the safety and effectiveness of the product for its intended use and to establish the overall benefit/risk relationship of the product to provide an adequate basis for product approval. These trials may include comparisons with placebo and/or other comparator treatments. The duration of treatment is often extended to mimic the actual use of a product during marketing. |
| | | | | | | | | | | |
| • | | A Phase 1/2 clinical trial has elements of a Phase 1 trial and a Phase 2 trial. We have designated our TP-04 and TP-05 trials as Phase 1/2 trials since we intend to go beyond the typical safety and tolerability assessments of a Phase 1 trial and intend to have these trials include additional efficacy assessments as well. |
| | | | | | | | | | | |
| • | | A Phase 2b/3 clinical trial has elements of a late Phase 2 trial and a Phase 3 trial. We have designated Saturn-1 as a Phase 2b/3 trial as it is both our first multi-center trial based in the U.S., and also a pivotal trial for the U.S. |
Post-approval trials, sometimes referred to as Phase 4 clinical trials, may be conducted after initial marketing approval. These trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication. In certain instances, the FDA may mandate the performance of Phase 4 clinical trials as a condition of approval of an NDA.
Progress reports detailing the results of the clinical trials, among other information, must be submitted at least annually to the FDA and written IND safety reports must be submitted to the FDA and the investigators for serious and unexpected suspected adverse events, findings from other studies suggesting a significant risk to humans exposed to the drug, findings from animal or in vitro testing that suggest a significant risk for human subjects and any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol.
Phase 1, Phase 2 and Phase 3 clinical trials may not be completed successfully within any specified period, if at all. The FDA or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients.
Equivalent, and similarly detailed, obligations will apply to the conduct of clinical trials in third countries including the European Union (“EU”).
NDA Review and Marketing Approval
Following completion of clinical trials, data are analyzed to assess whether the investigational product is safe and effective for the proposed indicated use or uses. The results of preclinical studies and clinical trials are then submitted to the FDA as part of an NDA, along with proposed labeling, chemistry and manufacturing information, and other information in a request for approval to market the drug for one or more specified indications. The application must include both negative and ambiguous results of preclinical studies and clinical trials, as well as positive findings. Data may come from company-sponsored clinical trials intended to test the safety and efficacy of a product’s use or from a number of alternative sources, including studies initiated by investigators. To support marketing approval, the data submitted must be sufficient in quality and quantity to establish the safety and efficacy of the investigational product to the satisfaction of FDA. FDA approval of an NDA must be obtained before a drug may be marketed in the United States.
Under the Prescription Drug User Fee Act, or "PDUFA",PDUFA, as amended, each NDA must be accompanied by an application user fee. FDA adjusts the PDUFA user fees on an annual basis. PDUFA also imposes an annual program fee for each marketed human drug. Fee waivers or reductions are available in certain circumstances, including a waiver of the application fee for the first application filed by a qualifying small business. Additionally, no user fees are assessed on NDAs for products designated as orphan drugs, unless the product NDA also includes a non-orphan indication.
The FDA reviewsconducts a preliminary review of all submitted NDAs before it accepts them for filing to determine if they are sufficiently complete to permit a substantive review, and the FDA may request additional information rather than accepting the NDA for filing. In this event, the application must be resubmitted with the additional information. The resubmitted application is also subject to review before the FDA accepts it for filing. The FDA must make a decision on accepting an NDA for filing within 60 days of receipt. Once the submission is accepted for filing, the FDA begins an in-depth review of the NDA. NDA to determine, among other things, whether the drug is safe and effective and whether the facility in which it is manufactured, processed, packaged or held meets standards designed to assure the product’s continued safety, quality and purity.
Under PDUFA, the FDA has agreed to certain performance goals in the review of NDAs through a two-tiered classification system, standard review and priority review. According to PDUFA performance goals, the FDA endeavors to review applications subject to standard review within ten months, whereas the FDA’s goal is to review priority review applications within six months, depending on whether the drug is a new molecular entity. The FDA does not always meet its PDUFA goal dates for standard and priority NDAs, and the review process is often extended by FDA requests for additional information or clarification.
The FDA conducts a preliminary review of all NDAs within the first 60 days after submission, before accepting them for filing, to determine whether they are sufficiently complete to permit substantive review. The FDA may request additional information rather than accept an NDA for filing. In this event, the application must be resubmitted with the additional information. The resubmitted application is also subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review. The FDA reviews an NDA to determine, among other things, whether the drug is safe and effective and whether the facility in which it is manufactured, processed, packaged or held meets standards designed to assure the product’s continued safety, quality and purity.
In addition, under the Pediatric Research Equity Act of 2003 as amended and reauthorized, certain NDAs or supplements to an NDA must contain data that are adequate to assess the safety and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations, and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may, on its own initiative or at the request of the applicant, grant deferrals for submission of some or all pediatric data until after approval of the product for use in adults, or full or partial waivers from the pediatric data requirements.
Before approving an NDA, the FDA will conduct a pre-approval inspection of the manufacturing facilities for the new product to determine whether they comply with cGMP requirements. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. The FDA may also may audit data from clinical trials to ensure compliance with GCP requirements.
The FDA generally accepts data from foreign clinical trials in support of an NDA if the trials were conducted under an IND. If a foreign clinical trial is not conducted under an IND, the FDA nevertheless may accept the data in support of an NDA if the study was conducted in accordance with GCP requirements and the FDA is able to validate the data through an on-site inspection, if deemed necessary. Although the FDA generally requests that marketing applications be supported by some data from domestic clinical studies, the FDA may accept foreign data as the sole basis for marketing approval if (1) the foreign data are applicable to the U.S. population and U.S. medical practice, (2) the studies were performed by clinical investigators with recognized competence, and (3) the data may be considered valid without the need for an on-site inspection or, if the FDA considers the inspection to be necessary, the FDA is able to validate the data through an on-site inspection or other appropriate means.
Additionally, the FDA may refer applications for novel drug products or drug products which present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and under what conditions, if any. The FDA is not bound by recommendations of an advisory committee, but it considers such recommendations when making decisions on approval. The FDA also closely analyzes the clinical trial data, which could result in extensive discussions between the FDA and the applicant during the review process.
After the FDA evaluates an NDA, it will issue an approval letter or a Complete Response Letter. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications and potentially subject to other requirements. A Complete Response Letter indicates that the review cycle of the application is complete and the application will not be approved in its present form. A Complete Response Letter usually describes all of the specific deficiencies in the NDA identified by the FDA. The Complete Response Letter may require additional clinical data, including the potential requirement to conduct additional pivotal Phase 3 clinical trial(s) and/or other significant and time-consuming requirements related to clinical trials, or to conduct additional preclinical studies or manufacturing changes. If a Complete Response Letter is issued, the applicant may either resubmit the NDA, addressing all of the deficiencies identified in the letter, or withdraw the application. Even if such data and information are submitted, the FDA may decide that the NDA does not satisfy the criteria for approval. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than we interpret the same data.
Post-Approval Requirements
Following approval of a new product, the product is subject to continuing regulation by the FDA, including, among other things, requirements relating to facility registration and drug listing monitoring and record keeping, periodic reporting, product sampling and distribution, advertising and promotion, and reporting of adverse experiences with the product. After approval, most changes to the approved product, such as adding new indications or other labeling claims, are subject to prior FDA review and approval. There also are continuing, annual user fee requirements for any marketed products and the establishments at which such products are manufactured, as well as new application fees for supplemental applications with clinical data. The FDA strictly regulates marketing, labeling, advertising and promotion of drugs, including after they are placed on the market. Drugs may be promoted only for the approved indications and in accordance with the provisions of the approved label.
Although physicians may prescribe legally available drugs for off-label uses, manufacturers may not market or promote such uses. Prescription drug promotional materials must be submitted to the FDA in conjunction with their first use or first publication.
The FDA may also place other conditions on approvals including the requirement for REMS,a Risk Evaluation and Mitigation Strategy ("REMS"), to assure the safe use of the product. If the FDA concludes that a REMS is needed, the NDA sponsor must submit a proposed REMS. The FDA will not approve the FDA without an approved REMS, if required. Any of these limitations on approval or marketing could restrict the commercial promotion, distribution, prescription or dispensing of products. Product approvals may be withdrawn for non-compliance with regulatory standards or if problems occur following initial marketing.
FDA regulations require that products be manufactured in specific approved facilities and in accordance with cGMP regulations. We rely, and expect to continue to rely, on third parties for the production of clinical and commercial quantities of our products in accordance with cGMP regulations. These manufacturers must comply with cGMP regulations that require, among other things, quality control and quality assurance, the maintenance of records and documentation, and the
obligation to investigate and correct any deviations from cGMP. Manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP requirements and other laws. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain cGMP compliance. The discovery of violative conditions, including failure to conform to cGMP regulations, could result in enforcement actions, and the discovery of problems with a product after approval may result in restrictions on a product, its manufacturer or the NDA holder, including recalls.
The FDA may withdraw approval of a product if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Corrective action could delay drug distribution and require significant time and financial expenditures. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information; imposition of post-market studies
or clinical studies to assess new safety risks or imposition of distribution restrictions or other restrictions under a REMS program. Other potential consequences include, among other things:
restrictions on the marketing or manufacturing of the product, suspension of the approval, complete withdrawal of the product from the market, or product recalls;
fines, warning letters, or holds on post-approval clinical trials;
refusal of the FDA to approve pending applications or supplements to approved applications;
suspension or revocation of product approvals;
product seizure or detention, or refusal to permit the import or export of products; or
injunctions or the imposition of civil or criminal penalties.
| | | | | | | | | | | |
| • | | restrictions on the marketing or manufacturing of the product, suspension of the approval, complete withdrawal of the product from the market, or product recalls; |
| | | | | | | | | | | |
| • | | fines, warning letters, or holds on post-approval clinical trials; |
| | | | | | | | | | | |
| • | | refusal of the FDA to approve pending applications or supplements to approved applications; |
| | | | | | | | | | | |
| • | | suspension or revocation of product approvals; |
| | | | | | | | | | | |
| • | | product seizure or detention, or refusal to permit the import or export of products; or |
| | | | | | | | | | | |
| • | | injunctions or the imposition of civil or criminal penalties. |
Other Regulatory Matters
Pharmaceutical companies are subject to additional healthcare regulation and enforcement by the federal government and by authorities in the states and foreign jurisdictions in which they conduct their business. Manufacturing, sales, promotion and other activities following product approval are subject to regulation by numerous regulatory authorities in the United States in addition to the FDA, including CMS, other divisions of the Department of Health and Human Services, the Department of Justice, the Drug Enforcement Administration, the Consumer Product Safety Commission, the Federal Trade Commission, the Occupational Safety & Health Administration, the Environmental Protection Agency, and state and local governments. For example, in the United States,U.S., sales, marketing and scientific and educational programs also must comply with state and federal fraud and abuse laws, false claims laws, transparency laws, government price reporting, and health information privacy and security laws. These laws include the following:
| | | | | | | | | | | |
| | | |
| • | | the federal Anti-Kickback Statute (“AKS”) which prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration (i.e., anything of value), directly or indirectly, in cash or in kind, to induce or in return either for the referral of an individual for, or for purchasing, leasing, ordering or arranging for the purchase, lease or order of any healthcare item or service reimbursable under Medicare, Medicaid or other federally financed healthcare programs. This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on one hand and prescribers, purchasers, and formulary managers on the other. Although there are a number of statutory exceptions and regulatory safe harbors protecting certain common business arrangements and activities from prosecution or regulatory sanctions, the exceptions and safe harbors are drawn narrowly and practices that involve remuneration intended to induce prescribing, purchasing or recommending may be subject to scrutiny if they do not fit squarely within an exception or safe harbor. Our practices may not in all cases meet all of the criteria for safe harbor protection from AKS liability. Moreover, there are no safe harbors for many common practices, such as educational and research grants, charitable donations, product support and patient assistance programs. The regulatory safe harbors also are subject to regulatory revision and interpretation by a number of government agencies. Liability under the AKS may be established without proving actual knowledge of the statute or specific intent to violate it. In addition, the government may assert that a claim including items or services resulting from a violation of the AKS constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act (discussed below). Violations of the AKS are punishable by imprisonment, criminal fines, damages, civil monetary penalties, and exclusion from participation in federal healthcare programs; |
| | | | | | | | | | | |
| • | | the federal civil False Claims Act (“FCA”) which prohibits any person from, among other things, knowingly presenting, or causing to be presented, a false or fraudulent claim for payment of government funds, or knowingly making, using, or causing to be made or used a false record or statement material to an obligation to pay money to the government, or knowingly concealing or knowingly and improperly avoiding, decreasing, or concealing an obligation to pay money to the federal government. Actions under the FCA may be brought by the Attorney General or as a qui tam action by a private individual in the name of the government. Such private individuals may share in amounts paid by the entity to the government in recovery or settlement. Recently, several pharmaceutical and other healthcare companies have been prosecuted under these laws for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. Other companies have been prosecuted for causing false claims to be submitted because of the companies’ marketing of the product for unapproved, and thus non-reimbursable, uses. FCA liability is potentially significant in the healthcare industry because the statute provides for treble damages and significant mandatory penalties per false or fraudulent claim or statement for violations, as well as exclusion from participation in federal healthcare programs. Pharmaceutical and other healthcare companies also are subject to other federal false claims laws, including, among others, federal criminal healthcare fraud and false statement statutes that extend to non-government health benefit programs; |
| | | | | | | | | | | |
| • | | the federal Health Insurance Portability and Accountability Act ("HIPAA"), which imposes criminal liability for, among other things, knowingly and willfully executing or attempting to execute a scheme to defraud any healthcare benefit program, knowingly and willfully embezzling or stealing from a health care benefit program, willfully obstructing a criminal investigation of a health care offense, or knowingly and willfully making false statements relating to healthcare matters; |
| | | | | | | | | | | |
| • | | HIPAA and its implementing regulations, also imposes obligations, on certain covered entity health care providers, health plans and health care clearinghouses as well as their business associates that perform certain services involving the use or disclosure of individually identifiable health information, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information; |
| | | | | | | | | | | |
| • | | federal consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers; |
| | | | | | | | | | | |
| • | | the FDCA, which prohibits, among other things, the adulteration or misbranding of drugs, biologics and medical devices, including off-label or pre-approval promotion; |
| | | | | | | | | | | |
| • | | the federal Physician Payments Sunshine Act, which requires applicable manufacturers of covered drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program, with specific exceptions, to annually report to Centers for Medicare & Medicaid Services ("CMS") information regarding direct or indirect payments and other transfers of value to physicians and teaching hospitals (and certain other practitioners as of 2022), as well as information regarding ownership and investment interests held by physicians and their immediate family members; and |
| | | | | | | | | | | |
| • | | analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws which may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non- governmental third-party payers, including private insurers, state laws that require pharmaceutical manufacturers to comply with the industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government and may require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures, state laws that require pharmaceutical manufacturers to report information on the pricing of certain drug products, state and local laws that require the licensure and registration of pharmaceutical sales representatives, and state and foreign laws that govern the privacy and security of health information in some circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts. |
Furthermore, states are constantly adopting new laws or amending existing laws, requiring attention to frequently changing regulatory requirements. For example, California has enacted the federal Anti-Kickback Statute, which makes it illegal for any person, including a prescription drug manufacturer (or a party acting on its behalf), to knowingly and willfully solicit, receive, offer or pay any remuneration, directly or indirectly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, lease, order or recommendation of an item or service for which payment may be made, in whole or in part, under a federal healthcare program, such as Medicare or Medicaid. Moreover, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the civil False Claims Act;
the federal False ClaimsCalifornia Consumer Privacy Act which imposes civil penalties, sometimes pursued through civil whistleblower or qui tam actions, against individuals or entities for, among other things, knowingly presenting, or causing to be presented, claims for payment of government funds that are false or fraudulent or making a false statement material to an obligation to pay money to the government or knowingly concealing or knowingly and improperly avoiding, decreasing, or concealing an obligation to pay money to the federal government;
the Federal HIPAA, which imposes criminal liability for, among other things, knowingly and willfully executing or attempting to execute a scheme to defraud any healthcare benefit program, knowingly and willfully embezzling or stealing from a health care benefit program, willfully obstructing a criminal investigation of a health care offense, or knowingly and willfully making false statements relating to healthcare matters;
HIPAA,("CCPA"), as amended by the Health Information Technology for Economic and Clinical HealthCalifornia Privacy Rights Act and their implementing regulations, also imposes obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information;
federal consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers;
the FDCA, which prohibits, among other things, the adulteration or misbranding of drugs, biologics and medical devices, including off-label or pre-approval promotion;
the federal Physician Payments Sunshine Act, which requires applicable manufacturers of covered drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program, with specific exceptions, to annually report to CMS information regarding direct or indirect payments and other transfers of value to physicians and teaching hospitals (and certain other practitioners beginning in 2022), as well as information regarding ownership and investment interests held by physicians and their immediate family members; and
analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws which may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non- governmental third-party payors, including private insurers, state laws that require pharmaceutical manufacturers to comply with the industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government and may require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures, state laws that require pharmaceutical manufacturers to report information on the pricing of certain drug products, and state and foreign laws that govern the privacy and security of health information in some circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts.
Additionally,("CPRA"). The CCPA created new transparency requirements, granted California recently enacted legislation that has been dubbed the first “GDPR-like” law in the United States. CCPA creates new individual privacy rights for consumers (as that word is broadly defined in the law) several new rights with regard their personal information, and places increased privacy and security obligations on entities handling personal data of consumers or households. The CCPA went into effect on January 1, 2020 and requires covered companies to provide new disclosures to California consumers, provide such consumers new ways to opt-out of certain sales of personal information, and allow for a new cause of action for data breaches. In addition, the CPRA introduced significant amendments to the CCPA and established and funded a dedicated California privacy regulator, the California Privacy Protection Agency (“CPPA”).The amendments introduced by the CPRA went into effect on January 1, 2023, and implementing regulations continue to be introduced by the CPPA. In addition, California residents have the right to bring a private right of action in connection with certain types of incidents. These claims may result in significant liability and potential damages. Other states, including Virginia, Colorado, Utah, Indiana, Iowa, Tennessee, Montana, Texas, and Connecticut, have enacted privacy laws similar to the CCPA that impose new obligations or limitations in areas affecting our business and we continue to assess the impact of these state legislations on our business as additional information and guidance becomes available. Similarly, there are a number of legislative proposals in the United States, at both the federal and state level, that could impose new obligations or limitations in areas affecting our business. Failure to comply with the CCPA and other state law may result in, among other things, significant civil penalties and injunctive relief, or potential statutory or actual damages. These laws could impact our business activities depending on how it isthey are interpreted and exemplifiesexemplify the vulnerability of our business to not only cyber threats but also the evolving regulatory environment related to personal data and protected health information.
Products must meet applicable child-resistant packaging requirements under the U.S. Poison Prevention Packaging Act. Manufacturing, sales, promotion and other activities also are potentially subject to federal and state consumer protection and unfair competition laws.
The distribution of pharmaceutical products is subject to additional requirements and regulations, including extensive record-keeping, licensing, storage and security requirements intended to prevent the unauthorized sale of pharmaceutical products.
We may also be subject to state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, as well as state laws that require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers, marketing expenditures or drug pricing. In addition, we may be subject to state and local laws that require the registration of pharmaceutical sales representatives.
The failure to comply with any of these laws or regulatory requirements subjects firms to possible legal or regulatory action. Depending on the circumstances, failure to meet applicable regulatory requirements can result in significant civil, criminal and administrative penalties, including damages, fines, disgorgement, individual imprisonment, exclusion from participation in government funded healthcare programs, such as Medicare and Medicaid, compliance oversight and reporting obligations, contractual damages, reputational harm, diminished profits and future earnings, injunctions, requests for recall, seizure of products, total or partial suspension of production, denial or withdrawal of product approvals or refusal to allow a firm to enter into supply contracts, including government contracts.
Equivalent, and similarly detailed, obligations will apply to the conduct of clinical trials in third countries including the EU.
United StatesU.S. Patent-Term Restoration and Marketing Exclusivity
Depending upon the timing, duration and specifics of FDA approval of any future product candidates, some of our U.S. patents may be eligible for limited patent term extension under the Hatch-Waxman Act. The Hatch-Waxman Act permits restoration of the patent term of up to five years as compensation for patent term lost during product development and FDA regulatory review process. Patent-term restoration, however, cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent-term restoration period is generally one-half the time between the
effective date of an IND or the issue date of the patent, whichever is later, and the submission date of an NDA plus the time between the submission date of an NDA or the issue date of the patent, whichever is later, and the approval of that application, except that the review period is reduced by any time during which the applicant failed to exercise due diligence. Only one patent applicable to an approved drug is eligible for the extension and the application for the extension must be submitted prior to the expiration of the patent. The USPTO,U.S. Patent and Trademark Office ("USPTO"), in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. In the future, we may apply for restoration of patent term for our currently owned or licensed patents to add patent life beyond its current expiration date, depending on the expected length of the clinical trials and other factors involved in the filing of the relevant NDA.
Market exclusivity provisions under the FDCA also can delay the submission or the approval of certain applications. The FDCA provides a five-year period of non-patent marketing exclusivity within the United StatesU.S. to the first applicant to gain approval of an NDA for a new chemical entity. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept for review an abbreviated new drug application or ANDA,("ANDA") or a 505(b)(2) NDA submitted by another company for another version of such drug where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement. The FDCA also provides three years of marketing exclusivity for an NDA, 505(b)(2) NDA or supplement to an existing NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example, new indications, dosages or strengths of an existing drug. This three-year exclusivity covers only the conditions of use associated with the new clinical investigations and does not prohibit the FDA from approving ANDAs for drugs containing the original active agent. Five-year and three-year exclusivity will not delay the submission or approval of a full 505(b)(1) NDA. However, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the preclinical studies and adequate and well-controlled clinical trials necessary to demonstrate safety and effectiveness.
European Union Drug Development
Similar to the United States,U.S., the various phases of preclinical and clinical research in the European Union are subject to significant regulatory controls. Although the European Union Clinical Trials Directive 2001/20/EC has sought to harmonize the EU clinical trials regulatory framework, setting out common rules for the control and authorization of clinical trials in the EU, the EU Member States have transposed and applied the provisions of the Directive differently. This has led to significant variations in the member state regimes. Under the current regime of the Clinical Trials Directive, before a clinical trial can be initiated it must be approved in each of the EU countries where the trial is to be conducted by two distinct bodies: the National Competent Authority or NCA,("NCA"), and one or more ethics committees. Under the current regime of the Clinical Trials Directive all suspected unexpected serious adverse reactions to the investigated drug that occur during the clinical trial have to be reported to the NCA and ECs of the Member State where they occurred.
The EU clinical trials legislation currently is undergoing a transition process mainly aimed at harmonizing and streamlining clinical-trial authorization, simplifying adverse-event reporting procedures, improvingIn order to streamline the supervisionregulation of clinical trials and increasing their transparency. Recently enactedacross the EU, the EU legislator has adopted Regulation (EU) No 536/2014 (the "EU Clinical Trials Regulation"). The new EU Clinical Trials Regulation, which has repealed and replaced the EU No 536/2014 ensuresClinical Trials Directive, introduced a complete overhaul of the former regulation of clinical trials for
medicinal products in the EU. The main characteristics of the regulation include: a streamlined application procedure through a single entry point, the “EU portal”; a single set of documents to be prepared and submitted for the application as well as simplified reporting procedures for clinical trial sponsors; and a harmonized procedure for the assessment of applications for clinical trials, which is divided in two parts. The EU Clinical Trials Regulation is applicable as of January 31, 2022 and is applicable directly in all countries of the European Economic Area ("EEA") (which is comprised of 27 Member States of the EU plus Norway, Iceland and Liechtenstein). Clinical trials that were authorized under the rulesClinical Trials Directive before January 31, 2023 can continue to be conducted under the Clinical Trials Directive until January 31, 2025.An application to transition ongoing trials from the current Clinical Trials Directive to the new Clinical Trials Regulation will need to be submitted and authorized in time before the end of the transitional period. From January 31, 2023 onwards, the application for conductingnew clinical trials must be done in accordance with the new Clinical Trials Regulation. The Clinical Trials Regulation is intended to simplify and streamline the approval of clinical trials in the EU will be identical. In the meantime, Clinical Trials Directive 2001/20/EC continues to govern all clinical trials performed in the EU.
European Union Drug Review and Approval
In order to market any product outside of the United States,U.S., a company must also comply with numerous and varying regulatory requirements of other countries and jurisdictions regarding quality, safety, and efficacy, and governing, among other things, clinical trials, marketing authorization, commercial sales, and distribution of products. Whether or not it obtains FDA approval for a product, an applicant will need to obtain the necessary approvals by the comparable foreign regulatory authorities before it can commence clinical trial or marketing of the product in those countries or jurisdictions.
Clinical Trial Approval
Pursuant to the currently applicable Clinical Trials Directive 2001/20/EC and the Directive 2005/28/EC on GCP, in order to undertake a clinical trial in a European Union member state, an applicant must obtain approval from the competent national authority of the European Union member state in which the clinical trial is to be conducted, or in multiple member states if the clinical trial is to be conducted in a number of member states. The EEA is comprised of the 27 Member States of the European Union and the three members of the EFTA pillar of the EEA, Iceland, Liechtenstein and Norway. Furthermore, the applicant may only start a clinical trial at a specific site after the competent ethics committee has issued a favorable opinion. The clinical trial application must be accompanied by an IMP dossier with supporting information prescribed by Directive
2001/20/EC and Directive 2005/28/EC and corresponding national laws of the member states and further detailed in applicable guidance documents.
In April 2014, the European Union adopted a new Clinical Trials Regulation (EU) No 536/2014, but it is not yet applicable in the European Union. The new legislation, which will be directly applicable in all European Union member states, aims to streamline the approval of clinical trials in the European Union by applying consistent rules and harmonizing the approvals process throughout the European Union. For instance, the new Clinical Trials Regulation provides for a streamlined application procedure via a single-entry point facilitating a harmonized assessment across multiple member states. The timing of implementation of the new Clinical Trials Regulation will be dependent on the development and launch of a fully functional clinical trials portal and database, which would be confirmed by an independent audit. The new legislation will come into effect six months after the European Commission publishes a confirmation of full functionality of the clinical trials information system. The website indicated that the audit was expected to commence in December 2020.
Parties conducting certain clinical trials in the European Union must, as in the United States, post clinical trial information at the EudraCT website: https://eudract.ema.europa.eu.
Marketing Authorization
In the EEA, medicinal products can only be commercialized after obtaining a Marketing Authorization or MA.("MA"). There are a number of types of marketing authorizations.
The Community MA is adopted by the European Commission in the form of a decision through the Centralized Procedure. The decision, which is based on the opinion of the Committee for Medicinal Products for Human Use, or CHMP, of the EMA, and is valid throughout the entire territory of the EEA. The Centralized Procedure is mandatory for certain types of products, such as biotechnology medicinal products, orphan medicinal products, advanced-therapy medicines such as gene-therapy, somatic cell-therapy or tissue-engineered medicines and medicinal products containing a new active substance indicated for the treatment of HIV, AIDS, cancer, neurodegenerative disorders, diabetes, auto-immune and other immune dysfunctions and viral diseases. The Centralized Procedure is optional, on approval by the EMA for products containing a new active substance not yet authorized in the EEA, or for products that constitute a significant therapeutic, scientific or technical innovation or which are in the interest of public health in the European Union.
| | | | | | | | | | | |
| | | |
| • | | The Community MA is adopted by the European Commission in the form of a decision through the Centralized Procedure. The decision, which is based on the opinion of the Committee for Medicinal Products for Human Use ("CHMP") of the EMA, is valid throughout the entire territory of the EEA. The Centralized Procedure is mandatory for certain types of products, such as biotechnology medicinal products, orphan medicinal products, advanced-therapy medicines such as gene-therapy, somatic cell-therapy or tissue-engineered medicines and medicinal products containing a new active substance indicated for the treatment of HIV, AIDS, cancer, neurodegenerative disorders, diabetes, auto-immune and other immune dysfunctions and viral diseases. The Centralized Procedure is optional, on approval by the EMA for products containing a new active substance not yet authorized in the EEA, or for products that constitute a significant therapeutic, scientific or technical innovation or which are in the interest of public health in the European Union. |
Under the Centralized Procedure, the CHMP established at the EMA is responsible for conducting an initial scientific assessment of a product. The maximum timeframe for the evaluation of an MA under the Centralized Procedure is 210 days, excluding clock stops when additional information or written or oral explanation is to be provided by the applicant in response to questions of the CHMP.
Accelerated evaluation may be granted by the CHMP in exceptional cases, when a medicinal product is of major interest from the point of view of public health and, in particular, from the viewpoint of therapeutic innovation. If the CHMP accepts such a request, the time limit of 210 days will be reduced to 150 days, but it is possible that the CHMP may revert to the standard time limit for the Centralized Procedure if it determines that it is no longer appropriate to conduct an accelerated assessment.
| | | | | | | | | | | |
| • | | MAs based on the Mutual Recognition Procedure or the Decentralized Procedure are available for products not falling within the mandatory scope of the Centralized Procedure. Where a product is first authorized by a Reference Member State this may be recognized by other Concerned Member States through the Mutual Recognition Procedure. Alternatively, a product can be approved simultaneously in various EU Member States through the Decentralized Procedure. Under the Decentralized Procedure an identical dossier is submitted to the competent authorities of each of the EU Member States in which the MA is sought, one of which is selected by the applicant as the Reference Member State. The competent authority of the Reference Member State prepares a draft assessment report, a draft Summary of Product Characteristics ("SmPC") and a draft of the labeling and package leaflet, which are sent to the other Member States (referred to as the "Concerned Member States") for their approval. If the Concerned Member States raise no objections, based on a potential serious risk to public health, to the assessment, SmPC, labeling or packaging proposed by the Reference Member State, the product is subsequently granted a national MA in all the Member States (i.e., in the Reference Member State and the Concerned Member States). |
Regulatory data protection and market exclusivity in the EU
In the EU, new medicinal products are granted a protection period of 8 years of data exclusivity and an additional 2 years of market exclusivity. As such, for a period of 8 years, generics cannot use the data of the innovator to obtain a marketing authorization. Only after 8 years have lapsed, other parties that apply for a marketing authorization (generics or biosimilars) may make reference to the dossier of the originator product. Only after another 2 years (i.e. a total of 10 years) may such generic or biosimilar medicinal product be placed on the Mutual Recognition Procedure ormarket. In April 2023, the Decentralised Procedure are availableEuropean Commission published a proposal to reform this system. In this proposal, the current standard period of regulatory data protection will be reduced from eight years to six years. The legislative process for products not falling withinthis reform is expected to take several years. It is currently uncertain if the mandatory scope ofproposal will be adopted in its current form and it is uncertain if and when the Centralized Procedure. Where a product is first authorized by a Reference Member State this may be recognized by other Concerned Member States through the Mutual Recognition Procedure. Alternatively, a product can be approved simultaneously in various EU Member States through the Decentralized Procedure. Under the Decentralized Procedure an identical dossier is submitted to the competent authorities of each of the EU Member States in which the MA is sought, one of which is selected by the applicant as the Reference Member State. The competent authority of the Reference Member State prepares a draft assessment report, a draft Summary of Product Characteristics, or SmPC, and a draft of the labeling and package leaflet, which are sent to the other Member States (referred to as the Concerned Member States) for their approval. If the Concerned Member States raise no objections, based on a potential serious risk to public health, to the assessment, SPC, labeling or packaging proposed by the Reference Member State, the product is subsequently granted a national MA in all the Member States (i.e., in the Reference Member State and the Concerned Member States).
Coverage and Reimbursement
Sales of our products will depend, in part, on the extent to which our products will be covered by third-party payors,payers, such as government health programs, commercial insurance, and managed healthcare organizations. There is significant
uncertainty related to third-party payor coverage and reimbursement of newly approved products. In the United States,U.S., for example, principal decisions about reimbursement for new products are typically made by CMS, or by Medicare’sthe agency that administers the Medicare program through regional contractors, state Medicaid programs, third-party payers, and druginsurance plans. These entities decidesdecide whether and to what extent a new product will be covered and reimbursed under Medicare,based on clinical needs and private third-party payors often follow their decisions regarding coverage and reimbursement to a substantial degree. However,economic impact. To date, no uniform policy of coverage and reimbursement for drug products exists. Accordingly, decisions regarding the extent of coverage and amount of reimbursement to be provided for any of our products will be made on a payor-by-payorpayer-by-payer basis.
Increasingly, third-party payorspayers are requiring that drug companies provide them with predetermined discounts usually in the form of rebates from list prices and are challenging the prices charged for medical products. Further, such payorspayers are increasingly challenging the price, examining the medical necessity and reviewing the cost effectiveness of medical product candidates.newly launched drugs. There may be especially significant delays in obtaining coverage and reimbursement for newly approved drugs.drugs as several large payers have implemented new to market blocks that can last anywhere between six to twelve months. Third-party payorspayers may limit coverage to specific product candidates on an approved list, known as a formulary, which might not include all FDA-approved drugs for a particular indication. We may need to conduct additional expensive pharmaco-economic Phase 4 real-world studies to demonstrate the medical necessity and cost effectiveness of our products. As a result, the coverage determination process is often a time-consuming and costly process that will require us to provide scientific and clinical support for the use of our products, to each payor separately, with no assurance that coverage and adequate reimbursement will be obtained.
If we successfully commercialize any of our product candidates, we mayWe participate in the Medicaid Drug Rebate Program and Medicare Part D Coverage Gap Discounts Program. Participation is required for federal funds to be available for our covered outpatient drugs under Medicaid and if applicable, Medicare Part B.B, and under Medicare Part D, respectively. Under the Medicaid Drug Rebate Program, we would beare required to pay a mandatory rebate to each state Medicaid program for our covered outpatient drugs that are dispensed to Medicaid beneficiaries and paid for by a state Medicaid program as a condition of having federal funds being made available to the states for our drugs under MedicaidMedicaid. Those rebates are based on pricing data reported by the manufacturer on a monthly and if applicable,quarterly basis to CMS. These data include the average manufacturer price and, in the case of innovator products, the best price for each drug, which, in general, represents the lowest price available from the manufacturer to any wholesaler, retailer, provider, health maintenance organization, nonprofit entity, or governmental entity in the U.S. in any pricing structure, calculated to include all sales and associated rebates, discounts, and other price concessions. Under the Medicare Part BD Coverage Gap Discount Program,
manufacturers, including us, are currently required to provide to CMS a 70% discount on brand name prescription drugs utilized by Medicare Part D beneficiaries when those beneficiaries are in the coverage gap phase of the MedicarePart D benefit design. The IRA sunsets the coverage gap discount program starting in 2025 and replaces it with a new manufacturer discount program.
Federal law requires that any company that participates in the Medicaid Drug Rebate Program also participate in the Public Health Service’s 340B drug pricing program in order for federal funds to be available for the manufacturer’s drugs under Medicaid and Medicare Part B. The 340B drug pricing program requires participating manufacturers to agree to charge statutorily-definedstatutorily defined covered entities no more than the 340B “ceiling price”ceiling price for the manufacturer’s covered outpatient drugs. These 340B covered entities include a variety of community health clinics and other entities that receive health services grants from the Public Health Service, as well as hospitals that serve a disproportionate share of low-income patients. The 340B ceiling price is calculated using a statutory formula, which is based on the average manufacturer price and rebate amount for the covered outpatient drug as calculated under the Medicaid Drug Rebate Program.
In addition, in order to be eligible to have its products paid for with federal funds under the Medicaid and Medicare Part B programs and purchased by certain federal agencies and grantees, a manufacturer also must participate in the VA FSSU.S. Department of Veterans Affairs ("VA") Federal Supply Schedule ("FSS") pricing program. Under this program, the manufacturer is obligated to make its innovator and single source products available for procurement on an FSS contract and charge a price to four federal agencies, the VA, U.S. Department of Veterans Affairs, U.S. DoD,Defense ("DoD"), Public Health Service and U.S. Coast Guard - that is no higher than the statutory Federal Ceiling Price.Price ("FCP"). Manufacturers also are obligated to calculate and submit to the VA on a quarterly and annual basis, their Non-Federal Average Manufacturer Price (“Non-FAMP”), which the VA uses to calculate the FCP. Moreover, pursuant to regulations issued by the DoD Defense Health Agency to implement Section 703 of the National Defense Authorization Act for Fiscal Year 2008, manufacturers are required to provide rebates on utilization of their innovator and single source products that are dispensed to TRICARE beneficiaries by TRICARE network retail pharmacies.
The requirements under the Medicaid, 340B, FSS, and TRICARE programs could reduce the revenue we may generate from any products that are commercialized in the future and could adversely affect our business and operating results. If we fail to comply with any applicable obligations under governmental pricing programs that we participate in, we could be subject to additional reimbursement requirements, significant civil monetary penalties, sanctions and fines, and those could negatively impact our business, financial condition, results of operations and growth prospects.
In addition, in most foreign countries, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing and reimbursement vary widely from country to country. For example, the European Union provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. A member state may approve a specific price for the medicinal product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. Pharmaceutical products may face competition from lower-priced products in foreign countries that have placed price controls on pharmaceutical products and may also compete with imported foreign products. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing.
Healthcare Reform
The United StatesU.S. government, state legislatures, and foreign governments have shown significant interest in implementing cost containment programs to limit the growth of government-paid healthcare costs, including price-controls,
restrictions on reimbursement, and requirements for substitution of generic products and/or lower cost over the counter alternatives for branded prescription drugs. For example, in March 2010, the Affordable Care Act was passed, which substantially changed the way healthcare is
financed by both the government and private insurers, and significantly impacts the U.S. pharmaceutical industry. The Affordable Care Act contains provisions that may reduce the profitability of drug products through increased rebates for drugs reimbursed by Medicaid programs, extension of Medicaid rebates to Medicaid managed care plans, mandatory discounts for certain Medicare Part D beneficiaries and annual fees based on pharmaceutical companies’ share of sales to federal health care programs. The Affordable Care Act made several changes to the Medicaid Drug Rebate Program, including increasing pharmaceutical manufacturers’ rebate liability by raising the minimum basic Medicaid rebate. The Affordable Care Act also expanded the universe of Medicaid utilization subject to drug rebates by requiring pharmaceutical manufacturers to pay rebates on Medicaid managed care utilization and by enlarging the population potentially eligible for Medicaid drug benefits. Effective April 1, 2022, Medicaid rebate liability will be expanded to include the territories of the United States as well.
There have been judicial challenges to certain aspects of the Affordable Care Act, as well as efforts by Congress to repeal or replace,modify, and by the Trump administrationagencies to alter the implementation of, certain aspects of the Affordable Care Act. For example, on December 22, 2017, President Trump signed into law new federalCongress eliminated the tax legislation commonly referred to as the Tax Cuts and Jobs Act (the Tax Act) which includes a provision repealing, effective January 1, 2019, the tax-based shared responsibility payment imposed bypenalty for not complying with the Affordable Care Act on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate.” On December 14, 2018, a federal district court in Texas ruled theAct’s individual mandate in the absence of the tax penalty is unconstitutional and, because it is a critical and inseverable feature of the Affordable Care Act, the remaining provisions of the Affordable Care Act are invalid as well. On December 18, 2019, the U.S. Court of Appeals for the Fifth Circuit affirmed the District Court’s ruling, but remanded the case back to the District Court as to the question of severability. On March 2, 2020, the United States Supreme Court granted certiorari to review this case, which is expected to be decided in mid-2021. Additionally, the Further Consolidated Appropriations Act of 2020, Pub. L. No. 116-94, permanently eliminated, effective January 1, 2020, the Affordable Care Act-mandated “Cadillac” tax on high-cost employer-sponsoredcarry health coverage and the medical device excise tax on non-exempt medical devices and, effective January 1, 2021, also eliminates the annual fee imposed on certain health insurance providers based on market share.insurance. Further, the Bipartisan Budget Act of 2018, among other things, amended the Affordable Care Act effective January 1, 2019, to increase from 50 percent to 70 percent the point-of-sale discount that is owed by pharmaceutical manufacturers who participate in Medicare Part D and to close the coverage gap in most Medicare drug plans, commonly referred to as the “donut hole.”"donut hole"(the Inflation Reduction Act of 2022 sunsets the coverage gap discount program and replaces it with a new manufacturer discount, beginning in 2025).
It is possible that the ACA, as currently enacted or may be amended in the future, as well as other healthcare reform measures including those that may be adopted in the future, may result in more rigorous coverage criteria, and less favorable payment methodologies, or other downward pressure on coverage and payment and the price that we receive for any approved product. Any reduction in reimbursement or restriction on coverage under Medicare or other government programs may result in a similar reduction or restriction by private payers.
Other legislative changes have been proposed and adopted in the United StatesU.S. since the Affordable Care Act was enacted. These changes included aggregate reductions to Medicare payments to providers of 2% per fiscal year effective April 1, 2013, which, due to subsequent legislative amendments, will stay in effect through 2030 unless additional Congressional action is taken, with the exception of a temporary suspension of the 2% cut in Medicare payments from May 1, 2020 through December 31, 2020, pursuant to the CARES Act signed into law in March 2020 and designed to provide financial support and resources to individuals and businesses affectedrequired by the COVID-19 pandemic. In January 2013,Budget Control Act of 2021, as amended by the American Taxpayer Relief Act of 2012 was signed into law, which,(“ATRA”).Subsequent legislation extended the 2% reduction, generally to 2031. Sequestration is currently set at 2% and will increase to 2.25% for the first half of fiscal year 2030, to 3% for the second half of fiscal year 2030, and to 4% for the remainder of the sequestration period that lasts through the first six months of fiscal year 2031. ATRA, among other things, also reduced Medicare payments to several types of providers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. Other new laws may result in additional reductions in Medicare and other healthcare funding, which could have a material adverse effect on customers for our drugs, if approved, and accordingly, our financial operations.
Additionally, there has been heightened governmental scrutiny over the manner in which drug manufacturers set prices for their marketed products, which has resulted in several Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drug products. For example, at the federal level, the Trump administration’s budget for fiscal year 2021 includes a $135 billion allowance to support legislative proposals seeking to reduce drug prices, increase competition, lower out-of-pocket drug costs for patients, and increase patient access to lower-cost generic and biosimilar drugs. On March 10, 2020, the Trump administration sent “principles” for drug pricing to Congress, calling for legislation that would, among other things, cap Medicare Part D beneficiary out-of-pocket pharmacy expenses, provide an option to cap Medicare Part D beneficiary monthly out-of-pocket expenses, and place limits on pharmaceutical price increases. On July 24, 2020, President Trump signed several executive orders directed toward lowering drug prices. On October 9, 2019, HHS, OIG and CMS issued two proposed rules that set forth modifications to the Federal Anti-Kickback Statute, Civil Monetary Penalties Law and Physician Self-Referral Law (or the Stark Law) regulations to promote value-based and coordinated care arrangements. At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing.
Moreover, the Medicare Prescription Drug, Improvement, and Modernization Act of 2003 or MMA,("MMA") established the Medicare Part D program to provide a voluntary prescription drug benefit to Medicare beneficiaries. Under Part D, Medicare beneficiaries may enroll in prescription drug plans offered by private entities that provide coverage of outpatient prescription drugs. Unlike Medicare Part A and B, Part D coverage is not standardized. While all Medicare drug plans must give at least a standard level of coverage set by Medicare, Part D prescription drug plan sponsors are not required to pay for all covered Part D drugs, and each drug plan can develop its own drug formulary that identifies which drugs it will cover and at what tier or level. However, Part D prescription drug formularies must include drugs within each therapeutic category and class of covered Part D drugs, though not necessarily all the drugs in each category or class. Any formulary used by a Part D prescription drug plan must be developed and reviewed by a pharmacy and therapeutic committee. Government payment for some of the costs of prescription drugs may increase demand for products for which we receive marketing approval. However, any negotiated prices for our products covered by a Part D prescription drug plan likely will be lower than the prices we might otherwise obtain. Moreover, while the MMA applies only to drug benefits for Medicare beneficiaries, private third-party payorspayers often follow Medicare coverage policy and payment limitations in setting their own payment rates.rates and in establishing their formulary placement.
Further, the Inflation Reduction Act of 2022 ("IRA") introduces several changes to the Medicare Part D benefit, including a limit on annual out-of-pocket costs and a change in manufacturer liability under the program which could negatively affect the profitability of our product candidates. The IRA sunsets the current Part D coverage gap discount program starting in 2025 and replaces it with a new manufacturer discount program. Failure to pay a discount under this new program will be subject to a civil monetary penalty. In addition, the IRA established a Medicare Part B inflation rebate scheme effective January 2023 and a Medicare Part D inflation rebate scheme effective October 2022, under which, generally speaking, manufacturers will owe rebates if the price of a Part B or Part D drug increases faster than the pace of inflation. Failure to timely pay a Part B or D inflation rebate is subject to a civil monetary penalty. The IRA also creates a drug price negotiation program under which the prices for Medicare units of certain high Medicare spend drugs and biologicals without generic or biosimilar competition will be capped by reference to, among other things, a specified non-federal average manufacturer price
starting in 2026. Failure to comply with requirements under the drug price negotiation program is subject to an excise tax and/or a civil monetary penalty. Congress continues to examine various policy proposals that may result in pressure on the prices of prescription drugs with respect to the government health benefit programs and otherwise. The IRA or other legislative changes could impact the market conditions for our product candidates.
Additionally, there has been heightened governmental scrutiny over the manner in which drug manufacturers set prices for their marketed products, which has resulted in several Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drug products. At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. Finally, some states have established Prescription Drug Affordability Boards (or similar entities) to review high-cost drugs and, in some cases, set upper payment limits.
Human Capital Resources
Human Capital Resources
As of December 31, 2020,2023, we had 20244 employees, all of whomwhich were full-time employees, and twelve of whom were engaged in research and development activities.employees. None of our employees are represented by a labor union or covered under a collective bargaining agreement.
Our human capital resources objectives include, as applicable, identifying, recruiting, retaining, incentivizing and integrating our existing and new employees, advisors and consultants. The principal purposes of our equity and cash incentive plans are to attract, retain and reward personnel through the granting of stock-based and cash-based compensation awards, in order to increase stockholder value and the success of our company by motivating such individuals to perform to the best of their abilities and achieve our objectives.
Employee Development and Training
Our values-based culture and our employees are a critical component of our success. We strive to create a supportive and professional environment for our employees. We expend considerable management time and attention, and financial resources, to attracting, retaining, and motivating exceptional individuals at our company.
Diversity, Equity, and Inclusion
We are committed to creating and maintaining a workplace free from discrimination or harassment on the basis of race, color, citizenship, religion, creed, national origin, ancestry, gender, sexual orientation, age, marital status, veteran status, disability, medical condition, or any other status protected by applicable law. Our employment policies and compliance trainings prohibit such discrimination and harassment. Our management team and employees are also expected to exhibit and promote honest, ethical, and respectful conduct in the workplace. All of our employees must adhere to a code of business conduct and ethics that sets standards for appropriate behavior and are required to attend annual training on the code of business conduct and ethics.
Corporate Information
We were incorporated under the laws of the State of Delaware in November 2016. Our principal executive offices are located at 15440 Laguna Canyon Road, Suite 160, Irvine, California 92618. Our telephone number is (949) 409-9820.418-1801. Our website address is www.tarsusrx.com. Information contained on the website is not incorporated by reference into this Annual Report on Form 10-K. We have included our website address in this Annual Report on Form 10-K solely as an inactive textual reference.
Facilities
We currently lease approximately 20,00032,145 square feet of office and laboratory space in Irvine, California under certain leases that last expire in January 2024.2027, with a renewal option for a term of three years. We believe that this space will be sufficient to meet our needs for the foreseeable future and that any additional space we may require will be available on commercially reasonable terms.
Legal Proceedings
We are not currently a party to any material legal proceedings. From time to time, we may become involved in legal proceedings arising in the ordinary course of our business. Regardless of outcome, litigation can have an adverse impact on us due to defense and settlement costs, diversion of management resources, negative publicity, reputational harm and other factors.
Item 1A. Risk Factors
Investing in our common stock is speculative and involves a high degree of risk. Before investing in our common stock, you should consider carefully the risks described below, together with the other information contained in this Annual Report on Form 10-K, including our financial statements and the related notes appearing at the end of this Annual Report on Form 10-K. If any of the following risks occur, our business, financial condition, results of operations and future growth prospects could be materially and adversely affected. In these circumstances, the market price of our common stock could decline, and you may lose all or part of your investment. The risks described below are not the only ones we face. Additional risks that we are unaware of, or that we currently believe are not material, may also become important factors that affect us. This Annual Report on Form 10-K also contains forward-looking statements that involve risks and uncertainties. Our actual results could differ materially from those anticipated in the forward-looking statements as a result of a number of factors, including the risks described below. See “Special Note Regarding Forward-Looking Statements.”
Risks Related to our Financial PositionBusiness and NeedOperations
We are an early commercial-stage biopharmaceutical company with a limited operating history and a single product approved for Additional Capital
commercial sale. We have incurred significant losses and negative cash flows from operations since our inception and anticipate that we will continue to incur significant expenses and losses for the foreseeable future.
We do not have any products approvedone product, XDEMVY, which recently obtained FDA approval for sale, we have not generated any revenue andthe treatment of Demodex blepharitis in the U.S. in July 2023. We have incurred net losses in each reporting periodyear since our company’s formation in 2016. We have funded our operations primarily from the sale and issuance of redeemable convertible preferred stock, convertible promissory notes and the sale of our common stock in our IPO.IPO, subsequent Follow-On Public Offerings, and under our 2023 ATM Prospectus, as well as proceeds from product sales, net, our China Out-License and draws from our Credit Facility. For the years ended December 31, 20192023 and 2018,2022, our net losses were $4.7$135.9 million and $1.3$62.1 million, respectively. For the twelve months ended December 31, 2020, our net losses were $26.8 million. As of December 31, 20202023 and December 31, 2019,2022, we had an accumulated deficit of $32.8$244.7 million and $6.0$108.8 million, respectively. Additionally, the net losses we incur may fluctuate significantly from quarter to quarter such that a period-to-period comparison of our results of operations may not be a good indicator of our future performance. The size of our future net losses will depend, in part, on the rate of future growth of our expenses and our ability to generate revenue. We expect that it will be a few years, if ever, before we have a product candidate ready for commercialization.recently initiated sales and marketing activities to commercialize XDEMVY in August 2023. We expect to incur increasing levels of operating losses over the next several years and for the foreseeable future asuntil our revenue from product sales from XDEMVY and any other approved products exceeds expenses, which may never occur. We may never achieve profitability and, even if we advance and commercialize, if approved,do, we may not be able to sustain or increase our product candidates.profitability. Our prior losses, combined with expected future losses, have had and will continue to have an adverse effect on our accumulated deficit and working capital.
We expect to continue incurring significant expenses and increasing operating losses for the foreseeable future. We expect that our expenses will increase substantially if and as we:
•continue to commercialize XDEMVY and any other products for which we may obtain marketing approval;
•enhance our product development and planned future commercialization efforts of our product candidates, including through hiring additional clinical, regulatory, quality control and scientific personnel;
•seek marketing approvals and reimbursement for our product candidates;
•prepare for and initiate additional preclinical, clinical and other studies for our product candidates;
•change or add additional manufacturers or suppliers, some of which may require additional permits or other governmental approvals;
•create additional infrastructure to support our operations as a public company, including adding operational, financial and management information systems and personnel;
•enhance our product development and planned future commercialization efforts, including through hiring additional clinical, regulatory, quality control and scientific personnel;
•seek marketing approvals and reimbursement for our product candidates;
•establish a sales, marketing, and distribution infrastructure to commercialize any products for which we may obtain marketing approval;
•seek to identify, assess, acquire or develop additional product candidates;
•acquire or in-license other product candidates and technologies;
•make milestone or other payments in connection with the development or approval of our product candidates;
•maintain, protect, enforce and expand our intellectual property portfolio; and
•experience any delays or encounter issues with any of the above. Because of the numerous risks and uncertainties associated with biopharmaceutical product development, we are unable to accurately predict the timing or amount of increased expenses or when, or if, we will be able to achieve profitability. Our expenses could increase beyond our expectations if, among other things:
•we are required by regulatory authorities to perform trials or studies in addition to, or different than, those that we currently expect;
•there are any delays in establishing appropriate manufacturing arrangements for or completing the development of any of our product candidates; or
we are required by regulatory authorities to perform clinical trials or studies in addition to, or different than, those that we currently expect; or •there are any third-party challenges to our intellectual property or we need to defend against any intellectual property-related claim.
Even ifWe expect to continue to expend substantial resources in connection with our commercialization efforts. If we obtain regulatory approval for and are successful in commercializing one or more of our product candidates, we expect to incur substantial additional research and development and other expenditures to develop and market additional product candidates or to expand the approved indications of any marketed product. We may encounter unforeseen expenses, difficulties, complications, delays and other unknown factors that may adversely affect our business.
Our limited operating history may make it difficult for youWe expect to evaluate the success ofexpand our business to date and to assess our future viability.
We commenced activities in 2016. Our limited operating history may make it difficult to evaluate the success of our business to date and to assess our future viability. Our operations to date have been limited to organizing our company, raising capital, identifying and developing product candidates, establishing licensing arrangements and/or acquiring necessary technology, undertaking research, preclinical studies and clinical trials of our product candidates, establishing arrangements for the manufacture of product candidates and longer-term planning for potential commercialization. Our prospects must be considered in light of the uncertainties, risks, expenses and difficulties frequently encountered by companies in their early stages of operations. We have limited experience in and have not yet demonstrated our ability to obtain marketing approvals, manufacture a commercial scale product or arrange for a third party to do so on our behalf, or conductdevelopment, regulatory, operational, sales, marketing, and distribution activities necessary for successful product commercialization. Consequently, any predictions you make about our future success or viability may not becapabilities and, as accurate as they could be if we had a longer operating history or a history of successfully developing, obtaining marketing approval for and commercializing products. In addition, as our business grows,result, we may encounter unforeseen expenses, difficulties complications, delays and other known and unknown obstacles. We will need to transition at some point from a company with ain managing our growth, which could disrupt our operations.
As we advance our research and development focusprograms and commercialization efforts, we expect to a company capableexperience continued growth in the number of supporting commercial activities. We may not be successfulour employees and the scope of our operations, particularly in such a transition.the areas of clinical development, quality, regulatory affairs, manufacturing, quality control, sales, marketing, and distribution. To manage our anticipated future growth, we must:
•identify, recruit, integrate, maintain and motivate additional qualified personnel;
•manage our development efforts effectively, including the initiation and execution of clinical trials for our product candidates; and
•improve our operational, financial and management controls, reporting systems and procedures.
We are heavily dependentOur future financial performance and our ability to develop, manufacture and commercialize our product candidates will depend, in part, on our ability to effectively manage any future growth, and our management may also have to divert financial and other resources, and a disproportionate amount of its attention away from day-to-day activities in order to devote a substantial amount of time, to managing these growth activities. If we do not effectively manage the successexpansion of our operations, we could experience weaknesses in our infrastructure, operational mistakes, loss of business opportunities, loss of employees and reduced productivity among remaining employees. The expansion of our operations also could lead product candidate, TP-03, forto significant costs and may divert our management and business development resources. Any inability to manage growth could delay the treatmentexecution of Demodex blepharitis. If we are unable to successfully complete the clinical development program, obtain regulatory approval for, or commercialize, TP-03, or experience significant delays in doing so, our business will be materially harmed.plans or disrupt our operations.
We currently have no products that are approvedrely, and for commercial salethe foreseeable future will continue to rely, in substantial part on certain third-party contract organizations, advisors and we have never had any products approvedconsultants to provide certain services, including assuming substantial responsibilities for sale or commercialized. To date, we have invested a substantial majoritythe conduct of our business efforts and financial resources to the preclinical and clinical development of TP-03 for the treatment of Demodex blepharitis. Our future success is dependent on our ability to successfully develop, obtain regulatory approval for, and commercialize TP-03 for the treatment of Demodex blepharitis, and we cannot accurately predict when or if TP-03 will be proven to be effective or safe in humans or whether it will receive regulatory approval. Before we can generate any revenue from sales of TP-03, we will be required to conduct additional clinical development, seek and obtain regulatory approval, secure adequate manufacturing supply to support commercial sales and build a commercial organization. We have not yet demonstrated our ability to complete pivotal clinical trials. Further, the commercial success of TP-03 will also depend on patent protection, successfully educating “ECPs” about Demodex blepharitis and related diagnosis, acceptance of TP-03 by patients, the medical community and third-party payors, TP-03’s ability to compete with other therapies, secure adequate healthcare coverage and reimbursement, and maintenance of an acceptable safety profile following approval, among other factors. If we do not achieve one or more of these factors in a timely manner or at all, we could experience significant delays or an inability to successfully commercialize TP-03, which would materially harm our business. If we were required to discontinue development of TP-03, or if TP-03 does not receive regulatory approval, fails to achieve significant market acceptance, or fails to receive adequate reimbursement, we would be delayed by many years in our ability to achieve profitability, if ever, and may not be able to generate sufficient revenue to continue our business.
We may need to obtain additional funding to complete the development and any commercialization of our product candidates, if approved. If we are unable to raise this necessary capital when needed, we would be forced to delay, reduce or eliminate our product development programs, commercialization efforts or other operations.
Since our inception, we have funded our operations through private placements of preferred stock, convertible promissory notestrials and the sale of our common stock in our IPO. Over the next few years, we expect our expenses to increase substantially and we will require a larger amount of capital to fund the developmentmanufacture of our product candidates. As our product candidates enterWe cannot assure you that the services of such third-party contract organizations, advisors and advance through preclinical studies and clinical trials, weconsultants will need substantial additional fundscontinue to expand our clinical, regulatory, quality and manufacturing capabilities. In addition, if we obtain marketing approval for any of our product candidates, we expect to incur significant commercialization expenses related to marketing, sales, manufacturing and distribution. Furthermore, as a public company, we incur significant legal, accounting and other costs associated with operating as a public company.
As of December 31, 2020, we had $168.1 million in cash and cash equivalents. Based on our current business plans, in addition to the completed IPO in October 2020, we believe that our existing cash and cash equivalents will be sufficient to fund our anticipated level of operations through at least the next 12 months. We believe that our cash and cash equivalents will be sufficient to fund our operating expenses and capital expenditure requirements into the fourth quarter of 2022. Accordingly, our existing cash and cash equivalents will be insufficient for us to concurrently fund our product candidates through regulatory approval and commercialization. We will need to raise substantial additional capital to complete the development and commercialization of our product candidates through equity offerings, debt financings, marketing and distribution arrangements and other collaborations, strategic alliances and licensing arrangements or other sources. We may also need to raise additional funds earlier than currently anticipated if we choose to pursue additional indications for our product candidates, acquire new product candidates or otherwise expand our business more rapidly than we presently planned.
We have based these estimates on assumptions that may prove to be incorrect or require adjustment because of our ongoing business decisions, and we could utilize our available capital resources sooner than we currently expect. Our future capital requirements will depend on many factors, including:
•the scope, rate of progress, costs and results of our drug discovery, preclinical development activities, laboratory testing and clinical trials for our product candidates;
•the number and scope of clinical programs we decide to pursue;
•the scope and costs of manufacturing development and commercial manufacturing activities and our ability to scale them up;
•the extent to which we acquire or in-license other product candidates and technologies;
•the cost, timing and outcome of regulatory review of our product candidates, including the potential for regulatory authorities to require that we conduct more studies and trials than those that we currently expect to conduct and the costs of post-marketing studies or risk evaluation and mitigation strategies that could be required by regulatory authorities;
•potential changes in the regulatory environment and enforcement rules;
•the cost and timing of establishing sales and marketing capabilities, if any of our product candidates receive marketing approval;
•the costs of preparing, filing and prosecuting patent applications, maintaining and enforcing our intellectual property rights and defending intellectual property-related claims;
•our ability to establish and maintain collaborations on favorable terms, if at all;
•our efforts to enhance operational systems and our ability to attract, hire and retain qualified personnel, including personnel to support the development of our product candidates;
•potential changes in pharmaceutical pricing and reimbursement infrastructure;
•the costs associated with being a public company; and
•the cost associated with commercializing our product candidates, if they receive marketing approval.
Identifying potential product candidates and conducting preclinical studies and clinical trials is a time consuming, expensive and uncertain process that takes years to complete, and we may never generate the necessary data or results required to obtain marketing approval. In addition, our product candidates, if approved, may not achieve adequate product sales or commercial success. We do not expect to have any products commercially available for sale for many years, if at all. Accordingly, we will need to obtain substantial additional funding in connection with our continuing operations. Adequate additional financing may not be available to us on acceptable terms,a timely basis when needed, or at all, and may be impacted by the economic climate and market conditions. For example, market volatility resulting from the COVID-19 pandemic or any other future infectious diseases, epidemics or pandemics could also adversely impact our ability to access capital as and when needed. Ifthat we can find qualified replacements. In addition, if we are unable to raise capital when neededeffectively manage our outsourced activities or on attractive terms, we would be forced to delay, limit, reduceif the quality or eliminateaccuracy of the services provided by our research and
development programsvendors or future commercialization efforts or grant rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves. In addition, attempting to secure additional financing may divert the time and attention of management from day-to-day activities and distract fromconsultants is compromised for any reason, our research and development efforts. Alternatively, we may seek additional capital due to favorable market conditions or strategic considerations, even if we believe we have sufficient funds for our current or future operating plans.
We currently generate no revenues from sales of any products and may never generate revenue or be profitable.
We have no products approved for commercial sale and do not anticipate generating any product revenue, unless and until either TP-03 or another product candidate receives the regulatory approvals necessary for commercialization in one or more jurisdictions. Our ability to generate revenue and achieve profitability depends significantly on our ability, or any future collaborator’s ability, to achieve a number of challenging objectives, including:
•successful and timely completion of preclinical and clinical development of our product candidates, including the clinical development of TP-03 for the treatment of Demodex blepharitis or other indications and any other future product candidates;
•establishing and maintaining relationships with contract research organizations, or "CROs", and clinical sites for the clinical development, both in the United States and internationally, of our product candidates;
•timely receipt of regulatory approvals from applicable regulatory authorities for TP-03 or any other product candidates for which we successfully complete clinical development;
•making any required post-marketing approval commitments to applicable regulatory authorities;
•establishing and maintaining commercially viable supply and manufacturing relationships with third parties that can provide adequate, in both amount and quality, products and services to support clinical development and meet the market demand for product candidates that we develop, if approved;
•obtaining an investigational new drug application, or "IND", prior to commencing clinical trials in the United States for a particular indication, such as TP-04 for the potential treatment of rosacea (although for TP-04 we first intend to conduct a Phase 1/2 trial outside the United States and thus do not plan to submit an "IND" prior to this trial) and TP-05 for potential Lyme prophylaxis and community malaria reduction;
•successful commercial launch following any regulatory approval, including the development of a commercial infrastructure, whether in-house or with one or more collaborators;
•a continued acceptable safety and efficacy profile both prior to and following any marketing approval of our product candidates;
•successfully educating ECPs about Demodex blepharitis and related diagnosis;
•commercial acceptance of TP-03 and any of our other product candidates by patients, the medical community and third-party payors;
•identifying, assessing and developing new product candidates;
•obtaining, maintaining and expanding patent protection, trade secret protection and regulatory exclusivity, both in the United States and internationally;
•protecting our rights in our intellectual property portfolio;
•defending against third-party interference or infringement claims, if any;
•obtaining favorable terms in any collaboration, licensing or other arrangements that may be necessaryextended, delayed or desirable to develop, manufacture or commercialize our existing or acquired product candidates;
•obtaining coverageterminated, and adequate reimbursement for customers and patients from government and third-party payors for product candidates that we develop;
•addressing any competing therapies and technological and market developments; and
•attracting, hiring and retaining qualified personnel.
We may never be successful in achieving our objectives and, even if we do, may never generate revenue that is significant or large enough to achieve profitability. If we do achieve profitability, we may not be able to sustainsuccessfully commercialize XDEMVY, obtain marketing approval of our product candidates or increase profitability on a quarterly or annual basis andotherwise advance our business. We cannot assure you that we will continuebe able to incur substantial researchproperly
manage our existing vendors or consultants or find other competent outside vendors and development and other expenditures to develop and market additional product candidates. In addition, as a young business, we may encounter unforeseen expenses, difficulties, complications, delays and other known and unknown challenges. Our failure to become and remain profitable would decrease the value of our company and could impair our ability to maintainconsultants on economically reasonable terms, or further our research and development efforts, raise additional necessary capital, grow our business, retain key employees and continue our operations.at all.
The COVID-19 pandemic,Many of the biotechnology and pharmaceutical companies that we compete against for qualified personnel and consultants have greater financial and other resources, different risk profiles and a longer history in the industry than we do. If we are unable to continue to attract and retain high quality personnel and consultants, the rate and success at which began in late 2019we can discover and has spread worldwide, may affect our ability to initiatedevelop product candidates and complete preclinical studies and clinical trials, disrupt regulatory activities, disrupt our manufacturing and supply chain or have other adverse effects onoperate our business and operations. In addition, this pandemic has caused substantial disruption in the financial markets and may adversely impact economies worldwide, both of which could result in adverse effects onwill be limited. If we are not able to effectively expand our business and operations.
The COVID-19 pandemic, which began in December 2019 and has spread worldwide, caused many governments to implement measures to slow the spread of the outbreak through quarantines, travel restrictions, heightened border scrutiny and other measures. The outbreak and government measures taken in response have also had a significant impact, both directly and indirectly, on businesses and commerce, as worker shortages have occurred, supply chains have been disrupted, facilities and production have been suspended, and demand for certain goods and services, such as medical services and supplies, has spiked, while demand for other goods and services, such as travel, has fallen. The future progression of the outbreak and its effects on our business and operations are uncertain.
Our business, operations and clinical development timelines and plans have been and could continue to be adversely affectedorganization by COVID-19, and could be adversely impacted by other health epidemics in regions where we have concentrations of clinical trial sites or other business operations, and could cause significant disruption in the operations of CROs upon whom we rely. The COVID-19 pandemic has affected multiple countries worldwide, including those where we have planned and ongoing preclinical studies and clinical trials. In addition, in response to the COVID-19 pandemic, many state, local and foreign governments have put in place quarantines, executive orders, shelter-in-place orders and similar government orders and restrictions in order to control the spread of the disease. Such orders or restrictions, and the perception that such orders or restrictions could continue or, after being lifted, be reinstated for a period of time, have resulted in business closures, work stoppages, slowdowns and delays, work-from-home policies, travel restrictions and cancellation of events, among other effects that could negatively impact productivity and disrupt our business and operations. We have implemented a work-from-home policy for ourhiring new employees and have also implemented enhanced travel-safe policies forexpanding our employees’ travel to our clinical sites. We may take further actions that alter our operations as may be required by federal, state or local authorities, or whichgroups of consultants and contractors, we determine are in the best interests of our employees.
Moreover, our clinical development timelines and plans could be affected by the COVID-19 pandemic as we and the third-party manufacturers and clinical research organizations that we engage may face disruptions. Site initiation and patient enrollment could be delayed or suspended due to prioritization of hospital resources toward the COVID-19 pandemic or patients not having a desire to enroll in clinical trials due to concerns regarding COVID-19. While we have not experienced any material delays in enrollment, we cannot be certain that we will not experience future delays in enrollment. In addition, some patients may not be able to comply with clinical trial protocolssuccessfully implement the tasks necessary to further develop and the ability to conduct follow up visits with treated patients may be limited if patients do not want to participate in follow up visits due to concerns regarding COVID-19 or if quarantines impede patient movement or interrupt healthcare services. There may be shortages in the raw materials used in the manufacturing ofcommercialize our product candidates or laboratory supplies for our preclinical studies and, clinical trials, in each case, because of ongoing efforts to address the outbreak.
We cannot assure that the inability to collect such clinical data would not have an adverse impact on our clinical trial results. Similarly, our ability to recruit and retain patients and principal investigators and site staff who, as healthcare providers, may have heightened exposure to COVID-19 could be adversely impacted.
We may experience disruptions that could severely impact our business, preclinical studies, and clinical trials, including:
•delays in receiving approval from local regulatory authorities to initiate our planned clinical trials, including receiving any required "IND";
•delays or difficulties in enrolling and retaining patients in our clinical trials;
•delays or difficulties in clinical site initiation, including difficulties in recruiting clinical site investigators and clinical site staff;
•manufacturing disruptions;
•delays in clinical sites receiving the supplies and materials needed to conduct our clinical trials;
•delays in the transport of clinical trial materials;
•changes in local regulations as part of a response to the COVID-19 coronavirus pandemic which may require us to change the ways in which our clinical trials are conducted, which may result in unexpected costs, or to discontinue the clinical trials altogether;
•diversion of healthcare resources away from the conduct of clinical trials, including the diversion of hospitals serving as our clinical trial sites and hospital staff supporting the conduct of our clinical trials;
•difficulties recruiting or retaining patients for our planned clinical trials if patients are affected by the virus or are fearful of visiting or traveling to clinical trial sites because of the outbreak;
•interruption of or changes in key clinical trial activities, such as clinical trial site monitoring, implementation of virtual monitoring, use of local testing labs, or home delivery of study drugs, due to limitations on travel imposed or recommended by federal or state governments, employers and others, use of new digital technologies for subject visits or interruption of clinical trial subject visits and study procedures, the occurrence of which could affect the integrity of clinical trial data;
•risk that participants enrolled in our clinical trials will acquire COVID-19 while the clinical trial is ongoing, which could impact the results of the clinical trial, including by increasing the number of observed adverse events;
•delays in necessary interactions with local regulators, ethics committees and other important agencies and contractors due to limitations in employee resources or forced furlough of government employees;
•limitations in employee resources that would otherwise be focused on the conduct of our clinical trials, including because of sickness of employees or their families or the desire of employees to avoid contact with large groups of people;
•interruption or delays in the operations of the FDA which may impact review and approval timelines;
•delays in regulatory approvals for our product candidates due to the FDA focusing on clinical trials related to therapies and vaccines targeting COVID-19;
•refusal of the FDA to accept data, including from clinical trials in affected geographies or failure to comply with updated FDA guidance and expectations related to the conduct of clinical trials during the COVID-19 pandemic; and
•interruption or delays to our sourced discovery and clinical activities.
The response to the COVID-19 pandemic may redirect resources with respect to regulatory matters in a way that would adversely impact our ability to pursue marketing approvals. In addition, we may face impediments to regulatory meetings and potential approvals due to measures intended to limit in-person interactions. Furthermore, third parties, including manufacturers, medical institutions, clinical investigators, CROs and consultants with whom we conduct business, are similarly adjusting their operations and assessing their capacity in light of the COVID-19 pandemic. If these third parties continue to experience shutdowns or business disruptions, our ability to conduct our business in the manner and on the timelines presently planned could be materially and negatively impacted.
The extent to which the COVID-19 pandemic impacts our business, clinical trials, results of operations and financial condition will depend on future developments, which are highly uncertain and cannot be predicted, including, but not
limited to, the duration of the pandemic, its severity, the actions to contain the virus or address its impact, and how quickly and to what extent normal economic and operating activities can resume. Further, while the potential economic impact brought by and the duration of COVID-19 may be difficult to assess or predict, the COVID-19 pandemic has resulted in significant disruptions of global financial markets, which could reduce our ability to access capital, which could in the future negatively affect our liquidity. To the extent the COVID-19 pandemic adversely affects our business, clinical trials, results of operations and financial condition, it may also have the effect of heightening many of the other risks described in this “Risk Factors” section.
The ultimate impact of the COVID-19 pandemic or a similar health epidemic is highly uncertain and subject to change.
Raising additional capital may cause dilution to our stockholders, restrict our operations or require us to relinquish rights to our technologies or product candidates.
Until such time, if ever, as we can generate substantial product revenue, we expect to finance our cash needs through possible combinations of equity offerings, debt financings, collaborations, strategic alliances and licensing arrangements. To the extent that we raise additional capital through the sale of equity or convertible debt securities, your ownership interest will be diluted and the terms of these securities may include liquidation or other preferences that adversely affect your rights as a common stockholder. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt or liens, making acquisitions, selling or licensing our assets, redeeming our stock, making certain investments, making capital expenditures or declaring dividends.
If we raise funds through collaborations, strategic alliances or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates or grant licenses on terms thataccordingly, may not be favorable to us. If we raise funds through research grants, we may be subject to certain requirements, which may limit our ability to use the funds or require us to share information fromachieve our research, development and development. Raising additional capital through any of these or other means could adversely affect our business and the holdings or rights of our stockholders, and may cause the market price of our shares to decline. If we are unable to raise additional funds through equity or debt financings when needed, we may be required to delay, limit, reduce or terminate our product development or future commercialization efforts or grant rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves.goals.
Our future success depends on our ability to retain key employees, consultants and advisors and to attract, retain and motivate qualified personnel.
We are highly dependent on the expertise of our executive officers, as well as the other members of our scientific and clinical teams and certain advisors to develop and soundly execute our business strategy. Although we have employment offer letters with each of our executive officers, each of them may terminate their employment with us at any time. We do not maintain “key person”key person insurance for any of our executives or employees.
Recruiting and retaining qualified scientific, and clinical, personnel and if we are successful in obtaining marketing approval for TP-03 or other product candidates, sales and marketing personnel, are critical to our success. The loss of the services of our executive officers or other key employees could impede the achievement of our research, development and commercialization objectives and seriously harm our ability to successfully implement our business strategy. Furthermore, replacing executive officers and key employees may be difficult and may take an extended period of time because of the limited number of individuals in our industry with the breadth of skills and experience required to successfully develop, gain regulatory approval for and commercialize our product candidates. Competition to hire qualified personnel in our industry is intense, and we may be unable to hire, train, retain or motivate these key personnel on acceptable terms given the competition among numerous pharmaceutical and biotechnology companies for similar personnel.
Furthermore, to the extent we hire personnel from competitors, we may be subject to allegations that they have been improperly solicited or that they have divulged proprietary or other confidential information, or that their former employers own their research output. We also experience competition for the hiring of scientific and clinical personnel from universities and research institutions. In addition, we rely on consultants and advisors, including scientific and clinical advisors, to assist us in formulating our research and development and commercialization strategy. Our consultants and advisors may be employed by employers other than us and may have commitments under consulting or advisory contracts with other entities that may limit their availability to us. If we are unable to continue to attract and retain high quality personnel, our ability to pursue our growth strategy will be limited, and our business, prospects, financial condition and results of operations may be adversely affected.
Many of our employees have become or will soon become vested in a substantial amount of our common stock or a number of common stock options. Our employees may be more likely to leave us if the shares they own have significantly appreciated in value relative to the original purchase prices of the shares, or if the exercise prices of the options that they hold are significantly below the market price of our common stock, particularly after the expiration of the lock-up agreements described herein.stock. Our future success also depends on our ability to continue to attract and retain additional executive officers and other key employees.
Our information technology systems, or those of our third-party CROs or other contractors or consultants, may fail or suffer security breaches, loss or leakage of data, and other disruptions, which could result in a material disruption of XDEMVY and our product candidates’ development programs, compromise sensitive information related to our business or prevent us from accessing critical information, potentially exposing us to liability or otherwise adversely affecting our business.
We are increasingly dependent upon information technology systems, infrastructure and data to operate our business. In the ordinary course of business, we collect, store and transmit confidential information (including but not limited to intellectual property, proprietary business information and personal information). It is critical that we do so in a secure manner to maintain the confidentiality, availability and integrity of such confidential information. We also have outsourced elements of our operations to third parties, and as a result we manage a number of third-party contractors who have access to our confidential information.
Despite the implementation of security measures, given their size and complexity and the increasing amounts of confidential information that they maintain, our internal information technology systems and those of our third-party CROs, CMO, and other contractors and consultants are potentially vulnerable to breakdown or other damage or interruption from service interruptions, system malfunction, natural disasters, interruptions or cyber incidents resulting from the conflict between
Russia and Ukraine, terrorism, war and telecommunication and electrical failures, as well as security breaches from inadvertent or intentional actions by our employees, contractors, consultants, business partners, and/or other third parties, or from cyber-attacks by malicious third parties (including the deployment of harmful malware, ransomware, denial-of-service attacks, social engineering and other means to affect service reliability and threaten the confidentiality, integrity and availability of information), which may compromise our system infrastructure or lead to data leakage. Further, due to the political uncertainty involving Russia and Ukraine, there is an increased likelihood that escalation of tensions could result in cyber attacks that could either directly or indirectly impact our operations. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and reputational damage and the commercial operations of XDEMVY and further development of our product candidates could be delayed.
While we have not experienced any such system failure, accident or security breach to date, we cannot assure you that our data protection efforts and our investment in information technology and cybersecurity will prevent significant breakdowns, data leakages, breaches in our systems or other cyber incidents that could have a material adverse effect upon our reputation, business, operations or financial condition, whether due to a loss of our trade secrets or other proprietary information or other similar disruptions. Our inability to use or access our information systems at critical points in time could adversely affect the timely and efficient operation of our business. Any delayed sales, significant costs or lost customers resulting from these technology failures could adversely affect our business, operations, and financial results. For example, if such an event were to occur and cause interruptions in our operations, it could result in a material disruption to our commercial operations of XDEMVY and further development of our product candidates could be delayed. In addition, the loss of clinical trial data for our product candidates could result in delays in our marketing approval efforts and significantly increase our costs to recover or reproduce the data. Furthermore, significant disruptions of our information technology systems or security breaches could result in the loss, misappropriation, and/or unauthorized access, use, or disclosure of, or the prevention of access to, confidential information (including trade secrets or other intellectual property, proprietary business information, and personal information), which could result in financial, legal, business, and reputational harm to us. For example, any such event that leads to unauthorized access, use, or disclosure of personal information, including personal information regarding our clinical trial subjects or employees, could harm our reputation directly, compel us to comply with federal and/or state breach notification laws and foreign law equivalents, subject us to mandatory corrective action, and otherwise subject us to liability under laws and regulations that protect the privacy and security of personal information, including private lawsuits or class actions under the California Consumer Privacy Act, which could result in significant legal and financial exposure and reputational damages that could potentially have an adverse effect on our business.
We maintain specific coverage to mitigate losses associated with certain cybersecurity incidents that impact our or our third parties’ systems, networks, and technologies.
Product liability lawsuits against us could cause us to incur substantial liabilities, could divert our resources and could limit or delay our commercialization of XDEMVY or any product candidates that we may develop.
We face an inherent risk of product liability exposure related to the commercialization of XDEMVY and the testing of our product candidates in human clinical trials and will continue to face risk if we commercially sell any future products we may develop. The sale of XDEMVY and any approved products in the future as well as the use of product candidates by us in clinical trials may expose us to liability claims. These claims might be made by patients that use the product, healthcare providers, pharmaceutical companies or others selling such products. On occasion, large judgments have been awarded in class action lawsuits based on products that had unanticipated adverse effects. If we cannot successfully defend ourselves against claims that XDEMVY or our product candidates caused injuries, we could incur substantial liabilities. Regardless of merit or eventual outcome, product liability claims may result in:
•the inability or delay of our efforts to commercialize XDEMVY or any products that we may develop;
•decreased demand for XDEMVY or any product candidates or products that we may develop;
•withdrawal of regulatory approval, recall, restriction on the approval or a black box warning or contraindication for XDEMVY or any future product candidates, if approved;
•delay, variation or termination of clinical trials;
•injury to our reputation and significant negative media attention;
•withdrawal of clinical trial subjects or challenges with clinical trial enrollment;
•initiation of investigations by regulators;
•significant costs to defend the related litigation and diversion of management’s time and our resources;
•substantial monetary awards to study subjects or patients;
•product recalls, withdrawals or new labeling requirements, marketing or promotional restrictions; and
•loss of revenue.
Although we maintain product liability insurance coverage, it may not be adequate to cover all liabilities that we may incur. We anticipate that we will need to develop and expand our company, and we may encounter difficulties in managing this development and expansion, which could disrupt our operations.
In connection with becoming a public company, we expect to increase our number of employees and the scope ofinsurance coverage as our operations. To manage our anticipated development and expansion, we must continue to implement and improve our managerial, operational and financial systems, expand our facilities and continue to recruit and train additional qualified personnel. Also, our management may need to divert a disproportionate amount of its attention away from its day-to-day activities and devote a substantial amount of time to managing these development activities. This may result in weaknesses in our infrastructure, give rise to operational mistakes, loss of business opportunities, loss of employees and reduced productivity among remaining employees. If our managementproduct candidates advance through clinical trials. Insurance coverage is unable to effectively manage our expected development and expansion, our expenses may increase more than expected, our ability to generate revenue could be reduced andincreasingly expensive, thus we may not be able to implementmaintain insurance coverage at a reasonable cost or in an amount adequate to satisfy any liability that may arise. If a successful product or clinical trial liability claim or series of claims is brought against us for uninsured liabilities or in excess of insured liabilities, our assets may not be sufficient to cover such claims and our business strategy. Our future financial performance and our ability to commercialize our product candidates, if approved, and compete effectively will depend, in part, on our ability to effectively manage the future development and expansion of our company.operations could be impaired.
WeOur employees, independent contractors, including our CROs and CMOs, commercial partners, consultants, suppliers, service providers, and other vendors may not realize the benefits of any acquisitions, in-licenseengage in misconduct or other collaborations or strategic alliances that we enter into.
We may form or seek strategic alliances, create joint ventures or collaborations or enter into additional licensing arrangementsimproper activities, including noncompliance with third parties that we believe will complement or augmentregulatory standards and requirements, which could have an adverse effect on our development and commercialization efforts with respect to our product candidates and any future product candidates that we may develop. Any of these relationships may require us to incur non-recurring and other charges, increase our near and long-term expenditures, issue securities that dilute our existing stockholders or disrupt our management and business. In addition, we face significant competition in seeking appropriate strategic partners and the negotiation process is time-consuming and complex. Moreover, we may not be successful in our efforts to establish a strategic partnership or other alternative arrangements for our product candidates because we are unable to agree on commercial terms, or because third parties may not view our product candidates as having the requisite potential to demonstrate safety and efficacy. If we license products or businesses, we may not be able to realize the benefit of such transactions if we are unable to successfully integrate them with our existing operations and company culture. We cannot be certain that, following a strategic transaction or license, we will achieve the revenue or specific net income that justifies such transaction. Any delays in entering into new strategic partnership agreements related to our product candidates could delay the development and commercialization of our product candidates in certain geographies for certain indications, which would harm our business prospects, financial condition and results of operations.
We expectare exposed to expandthe risk that our development,employees, independent contractors, including our CROs and CMOs, commercial partners, consultants, suppliers, service providers, and other vendors may engage in misconduct or other illegal activity. Misconduct by these parties could include intentional, reckless and/or negligent conduct or other unauthorized activities that violate the laws and regulations of the FDA and other similar foreign regulatory authorities, including those laws that require the reporting of true, complete, and operational capabilitiesaccurate information to such foreign regulatory authorities; manufacturing standards; U.S. federal and potentially implement sales, marketingstate healthcare fraud and distribution capabilitiesabuse, data privacy laws and as a result, we may encounter difficultiesother similar non-U.S. laws; or laws that require the true, complete, and accurate reporting of financial information or data. Activities subject to these laws also involve the improper use or misrepresentation of information obtained in managingthe course of clinical trials, the creation of fraudulent data in our growth,nonclinical studies or clinical trials, or illegal misappropriation of product, which could disruptresult in regulatory sanctions and cause serious harm to our reputation. It is not always possible to identify and deter misconduct by employees and other third parties, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. In addition, we are subject to the risk that a person or government could allege such fraud or other misconduct, even if none occurred. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business and financial results, including, without limitation, the imposition of significant civil, criminal and administrative penalties, damages, monetary fines, disgorgements, possible exclusion from participation in Medicare, Medicaid and other U.S. healthcare programs, imprisonment, other sanctions, contractual damages, reputational harm, future earnings and curtailment of our operations, any of which could adversely affect our ability to operate our business and our results of operations.
As of December 31, 2020, we had 20 full-time employees. As we advanceHealth epidemics may affect our researchability to initiate and development programs, we expect to experience significant growthcomplete preclinical studies and clinical trials, disrupt regulatory activities, disrupt our manufacturing and supply chain or have other adverse effects on our business and operations. In addition, health epidemics could cause substantial disruption in the numberfinancial markets and may adversely impact economies worldwide, both of which could result in adverse effects on our employeesbusiness and operations.
Our business, operations and clinical development timelines could be adversely affected by health epidemics in regions where we have concentrations of clinical trial sites or other business operations, and could cause significant disruption in the operations of CROs upon whom we rely. Moreover, our clinical development timelines and plans could be affected by health epidemics as we and the scopethird-party manufacturers and clinical research organizations that we engage may face disruptions. Site initiation and patient enrollment could be delayed or suspended due to prioritization of our operations, particularlyhospital resources toward the health epidemics or patients not having a desire to enroll in clinical trials due to concerns. In addition, some patients may not be able to comply with clinical trial protocols and the ability to conduct follow up visits with treated patients may be limited if patients do not want to participate in follow up visits due to concerns regarding health epidemics or if quarantines impede patient movement or interrupt healthcare services. There may be shortages in the areas of clinical development, quality, regulatory affairs,raw materials used in the manufacturing and quality control and, if any of our product candidates receives marketing approval, sales, marketingor laboratory supplies for our preclinical studies and distribution. To manageclinical trials, in each case, because of ongoing efforts to address the outbreak.
We cannot assure that the inability to collect such clinical data would not have an adverse impact on our anticipated future growth, we must:clinical trial results. Similarly, our ability to recruit and retain patients and principal investigators and site staff who, as healthcare providers, may have heightened exposure to health epidemics could be adversely impacted.
We may experience disruptions that could severely impact our business, preclinical studies, and clinical trials, including:
•identify, recruit, integrate, maintain and motivate additional qualified personnel;delays in receiving approval from local regulatory authorities to initiate our planned clinical trials, including receiving any required IND;
•managedelays or difficulties in enrolling and retaining patients in our development efforts effectively,clinical trials;
•delays or difficulties in clinical site initiation, including difficulties in recruiting clinical site investigators and clinical site staff;
•manufacturing disruptions;
•delays in clinical sites receiving the initiationsupplies and materials needed to conduct our clinical trials;
•delays in the transport of clinical trial materials;
•changes in local regulations as part of a response to a health epidemic which may require us to change the ways in which our clinical trials are conducted, which may result in unexpected costs, or to discontinue the clinical trials altogether;
•diversion of healthcare resources away from the conduct of clinical trials, forincluding the diversion of hospitals serving as our product candidates;clinical trial sites and hospital staff supporting the conduct of our clinical trials;
•improvedifficulties recruiting or retaining patients for our operational, financial and management controls, reporting systems and procedures.planned clinical trials if patients are affected by the virus or are fearful of visiting or traveling to clinical trial sites because of the outbreak;
Our future financial performance and•risk that participants enrolled in our abilityclinical trials will acquire a particular disease related to develop, manufacture and commercialize our product candidates will depend,a health epidemic while the clinical trial is ongoing, which could impact the results of the clinical trial, including by increasing the number of observed adverse events;
•delays in part, on our ability to effectively manage any future growth, and our management may also have to
divert financialnecessary interactions with local regulators, ethics committees and other important agencies and contractors due to limitations in employee resources and a disproportionate amountor forced furlough of its attention away from day-to-day activities in order to devote a substantial amount of time, to managing these growth activities. If we do not effectively manage the expansion of our operations, we could experience weaknesses in our infrastructure, operational mistakes, loss of business opportunities, loss of employees and reduced productivity among remaining employees. The expansion of our operations also could lead to significant costs and may divert our management and business development resources. Any inability to manage growth could delay the execution of our business plans or disrupt our operations.government employees;
We currently rely, and for the foreseeable future will continue to rely,limitations in substantial partemployee resources that would otherwise be focused on certain third-party contract organizations, advisors and consultants to provide certain services, including assuming substantial responsibilities for the conduct of our clinical trials, andincluding because of sickness of employees or their families or the manufacturedesire of our product candidates. We cannot assure you thatemployees to avoid contact with large groups of people;
•interruption or delays in the services of such third-party contract organizations, advisors and consultants will continue to be available to us on a timely basis when needed, or that we can find qualified replacements. In addition, if we are unable to effectively manage our outsourced activities or if the quality or accuracyoperations of the services provided by our vendors or consultants is compromisedFDA which may impact review and approval timelines;
•delays in regulatory approvals for any reason, our clinical trials may be extended, delayed or terminated, and we may not be able to obtain marketing approval of our product candidates due to the FDA focusing on clinical trials related to therapies and vaccines targeting health epidemics;
•refusal of the FDA to accept data, including from clinical trials in affected geographies or otherwise advancefailure to comply with updated FDA guidance and expectations related to the conduct of clinical trials during a health epidemic; and
•interruption or delays to our business. We cannot assure you that we will be able to properly manage our existing vendors or consultants or find other competent outside vendorssourced discovery and consultants on economically reasonable terms, or at all.clinical activities.
Many of the biotechnology and pharmaceutical companies that we compete against for qualified personnel and consultants have greater financial and other resources, different risk profiles and a longer history in the industry than we do. If we are unable to continue to attract and retain high quality personnel and consultants, the rate and success at which we can discover and develop product candidates and operate our business will be limited. If we are not able to effectively expand our organization by hiring new employees and expanding our groups of consultants and contractors, we may not be able to successfully implement the tasks necessary to further develop and commercialize our product candidates and, accordingly, may not achieve our research, development and commercialization goals.
WeThe response to a health epidemic may engageredirect resources with respect to regulatory matters in acquisitions or strategic partnershipsa way that could disruptwould adversely impact our business, cause dilutionability to our stockholders, reduce our financial resources, cause or to incur debt or assume contingent liabilities, and subject us to other risks.
In the future, we may enter into transactions to acquire other businesses, products or technologies or enter into strategic partnerships, including licensing. If we do identify suitable acquisition or partnership candidates, we may not be able to make such acquisitions or partnerships on favorable terms, or at all. Any acquisitions or partnerships we make may not strengthen our competitive position, and these transactions may be viewed negatively by customers or investors. We may decide to incur debt in connection with an acquisition or issue our common stock or other equity securities to the stockholders of the acquired company, which would reduce the percentage ownership of our existing stockholders. For example, pursuant to the terms of the license agreement with Elanco Tiergesundheit AG (“Elanco”), granting us a worldwide, sublicensable license for the development andpursue marketing of lotilaner for all applications in humans outside the treatment or cure of any eye or skin condition in humans (the “All Human Uses Elanco Agreement”), if it is not terminated, or if we have not provided notice to terminate the All Human Uses Elanco Agreement, within 18 months of its effective date, we will be required to issue Elanco additional shares of our common stock equating to $3.0 million of aggregate value.
We could incur losses resulting from undiscovered liabilities of the acquired business or partnership that are not covered by the indemnification we may obtain from the seller or our partner.approvals. In addition, we may not be ableface impediments to successfully integrate any acquired personnel, technologiesregulatory meetings and potential approvals due to measures intended to limit in-person interactions. Furthermore, third parties, including manufacturers, medical institutions, clinical investigators, CROs and consultants with whom we conduct business, are similarly adjusting their operations intoand assessing their capacity in light of a health epidemic. If these third parties continue to experience shutdowns or business disruptions, our existingability to conduct our business in an effective, timelythe manner and non-disruptive manner. Acquisitionson the timelines presently planned could be materially and negatively impacted.
The extent to which the health epidemic impacts our business, clinical trials, results of operations and financial condition will depend on future developments, which are highly uncertain and cannot be predicted, including, but not limited to, the duration of the pandemic, its severity, the actions to contain the virus or partnershipsaddress its impact, and how quickly and to what extent government orders and mandates are lifted and normal economic and operating activities can resume. Further, while the potential economic impact of any health epidemic may be difficult to assess or predict, it could result in significant disruptions of global financial markets, which could reduce our ability to access capital, which could in the future negatively affect our liquidity. To the extent a health epidemic adversely affects our business, clinical trials, results of operations and financial condition, it may also divert management attention from day-to-day responsibilities, lead to a loss of key personnel, increase our expenses and reduce our cash available for operations and other uses. We cannot predict the number, timing or size of future acquisitions or partnerships orhave the effect that any such transactions might have on our operating results.
of heightening many of the other risks described in this “Risk Factors” section. The ultimate impact of a health epidemic is highly uncertain and subject to change.
We or the third parties upon whom we depend on may be adversely affected by earthquakes, fires or other natural disasters, or geopolitical events and our business continuity and disaster recovery plans may not adequately protect us from a serious disaster.
EarthquakesAny unplanned event, such as earthquakes, fires, flood, explosion, extreme weather, health epidemics, pandemics, power outages, telecommunication failures, war or other military conflict, terrorist activities or other natural disastersor manmade accidents or incidents could severely disrupt our operations, and have a material adverse effect on our business, results of operations, financial condition and prospects. If a natural disaster, power outage or other event occurred that prevented us from using all or a significant portion of our headquarters, that damaged critical infrastructure, such as the manufacturing facilities of our third-party contract manufacturers, or that otherwise disrupted operations, it may be difficult or, in certain cases, impossible for us to continue our business for a substantial period of time. The disaster recovery and business continuity plans we have in place currently are limited and are unlikely to prove adequate in the event of a serious disaster or similar event. We may incur substantial expenses as a result of the limited nature of our disaster recovery and business continuity plans, which, particularly when taken together with our lack of earthquake insurance, could have a material adverse effect on our business.
Unfavorable global and geopolitical economic conditions could adversely affect our business, financial condition or results of operations.
Our results of operations could be adversely affected by general conditions in the global and geopolitical economy and in the global financial markets. Financial pressures may cause government or other third-party payers to more aggressively seek cost containment measures in healthcare and other settings. As a result of global economic conditions, some third-party payers may delay or be unable to satisfy their reimbursement obligations. Job losses or other economic hardships (including inflation) may also affect patients’ ability to afford healthcare as a result of increased co-pay or deductible obligations, greater cost sensitivity to existing co-pay or deductible obligations, lost healthcare insurance coverage or for other reasons. We believe such conditions have led and could continue to lead to reduced demand for our products, which could have a material adverse effect on our product sales, net, business and results of operations. The recent global financial crisis caused extreme volatilitycurrent inflationary environment related to increased aggregate demand, supply chain constraints and disruptionsthe effects from the armed conflict in Ukraine (including the capital and credit markets. Additionally,effects of the COVID-19 pandemic has caused significant disruptionssanctions that were implemented in response to the U.S.conflict and the resulting impacts on the commodity market and supply chains) and Israel have also increased our operating expenses and may continue to affect our operating expenses. Our operational costs, including the cost of energy, materials, labor, distribution and our other operational and facilities costs are subject to market conditions and are being adversely affected by inflationary pressures. Global and geopolitical economic conditions may also adversely affect the ability of our distributors, customers and suppliers to obtain the liquidity required to buy inventory or raw materials and to perform their obligations under agreements with us, which could disrupt our operations. Although we monitor our distributors’, regionalcustomers’ and global economiessuppliers’ financial condition and has contributedtheir liquidity to significant volatility and negative pressure in financial markets. A severe or prolonged economic downturn, or any prolonged economic downturn caused by the COVID-19 pandemic, could result in a variety of risks tomitigate our business including reduced ability to raise additional capital when neededrisks, some of our distributors, customers and suppliers may become insolvent, which could have a material adverse effect on acceptable terms, if at all. A weak or declining economy could also strain our suppliers, possibly resulting in supply disruption. Any of the foregoing could harm ourproduct sales, business and we cannot anticipate allresults of the ways in which the currentoperations. A significant worsening of global and geopolitical economic climate and financial market conditions could adversely impact our business.
Our ability to use our net operating loss carryforwards and certainprecipitate or materially amplify the other tax attributes may be limited.risks described herein.
We have incurred substantial losses duringmaintain a significant portfolio of investments disclosed as cash equivalents and marketable securities on our history which we expect to continue, do not expect to become profitableaccompanying Balance Sheets. The value of our investments may be adversely affected by interest rate fluctuations, inflation, downgrades in credit ratings, illiquidity in the near future, and we may never achieve profitability. Under Sections 382 and 383 of the Internal Revenue Code of 1986, as amended (the “Code”), if a corporation undergoes an “ownership change,” generally defined as a greater than 50 percentage point change (by value) in its equity ownership by certain stockholders over a three-year period, the corporation’s ability to use its pre-change net operating loss carryforwards (“NOLs”),capital markets, health epidemics and other pre-change tax attributes (such as research tax credits) to offset its post-change income or taxesfactors that may be limited. We have not yet completed an ownership change analysis. If a requisite ownership change occurs,result in other-than-temporary declines in the amount of remaining tax attribute carryforwards available to offset taxable income and reduce income tax expense in future years may be restricted or eliminated. Similar provisions of state tax law may also apply to limit our use of accumulated state tax attributes. In addition, at the state level, there may be periods during which the use of NOLs is suspended or otherwise limited, which could accelerate or permanently increase state taxes owed. As a result, even if we attain profitability, we may be unable to use a material portionvalue of our NOLs and other tax attributes, whichinvestments. Any of those events could adversely affectcause us to record impairment charges with respect to our future cash flows.investment portfolio or to realize losses on sales of investments.
U.S. federal income tax reform and the implementation of such reforms could adversely affect us.
On December 22, 2017, the United States enacted the “Tax Cuts and Jobs Act” (the “ TCJA”) that significantly reformed the Code. The TCJA, among other things, includes contained significant changes to corporate taxation, including a reduction of the corporate tax rate from a top marginal rate of 35% to a flat rate of 21%, the limitation of the tax deduction for net interest expense to 30% of adjusted earnings (except for certain small businesses), the limitation of the deduction for NOLs arising in taxable years beginning after December 31, 2017 to 80% of current year taxable income and elimination of NOL carrybacks for losses arising in taxable years ending after December 31, 2017 (though any such NOLs may be carried forward indefinitely), the imposition of a one-time taxation of offshore earnings at reduced rates regardless of whether they are repatriated, the elimination of U.S. tax on foreign earnings (subject to certain important exceptions), the allowance of immediate deductions for certain new investments instead of deductions for depreciation expense over time, and the modification or repeal of many business deductions and credits. The financial statements contained herein reflect the effects of the TCJA based on current guidance. However, there remain uncertainties and ambiguities in the application of certain provisions of the TCJA, and, as a result, we made certain judgments and assumptions in the interpretation thereof.
As part of Congress’s response to the COVID-19 pandemic, the Families First Coronavirus Response Act (“FFCR Act”) was enacted on March 18, 2020, and the Coronavirus Aid, Relief, and Economic Security Act (“CARES Act”) was enacted on March 27, 2020. Both contain numerous tax provisions. In particular, the CARES Act retroactively and temporarily (for taxable years beginning before January 1, 2021) suspends application of the 80%-of-income limitation on the use of NOLs, which was enacted as part of the TCJA. It also provides that NOLs arising in any taxable year beginning after December 31, 2017 and before January 1, 2021 are generally eligible to be carried back up to five years. The CARES Act also temporarily (for taxable years beginning in 2019 or 2020) relaxes the limitation of the tax deductibility for net interest expense by increasing the limitation from 30% to 50% of adjusted taxable income.
Regulatory guidance under the TCJA, the FFCR Act and the CARES Act is and continues to be forthcoming, and such guidance could ultimately increase or lessen impact of these laws on our business and financial condition. It is also likely that Congress will enact additional tax legislation in connection with the COVID-19 pandemic, some of which could have an impact on our company. In addition, it is uncertain if and to what extent various states will conform to the TCJA, the FFCR Act or the CARES Act.
Risks Related to Development and Commercialization
We have only recently obtained regulatory approval for XDEMVY in the U.S. and commenced the commercial launch of XDEMVY. We have limited experience as a commercial company and generating revenue from product sales. If the commercial launch of XDEMVY is unsuccessful or any future approved products are unsuccessful, we may never be profitable.
We recently received approval by the FDA for XDEMVY for the treatment of Demodex blepharitis in the U.S. and began generating revenue from product sales during the third quarter of 2023. Our Product Candidatesability to become and remain profitable is heavily dependent on our ability to generate revenue from XDEMVY. The success of our commercialization will depend on a number of factors, including, among others, the continued development of our commercial organization, including our internal sales and marketing team and distribution capabilities, our ability to navigate the significant expenses and risks involved with the development and management of such capabilities, satisfying any post-marketing regulatory requirements, our ability to secure adequate healthcare coverage and the acceptance of XDEMVY by patients, ECPs and third-party payers. Further, our commercial success is dependent on our ability to educate ECPs, patients and others in the medical community about Demodex blepharitis. If XDEMVY, or any other future approved product, does not achieve an adequate level of acceptance, coverage, pricing or reimbursement, we may not generate significant revenue from product sales and we may not be profitable. Even if we successfully commercialize XDEMVY in the U.S., we may be unable to achieve or maintain profitability, unless XDEMVY is approved in other jurisdictions or for additional indications. Because of the uncertainties and risks associated with these activities, we are unable to accurately and precisely predict the timing and amount of revenues from product sales of XDEMVY, or any future approved products, or if or when we might achieve profitability.
If we are unsuccessful in accomplishing our objectives, or if our commercialization efforts do not develop as planned, we may not be able to successfully commercialize XDEMVY or any future approved products, we may require significant additional capital and financial resources, we may not become profitable, and we may not be able to compete against more established companies in our industry. Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis.
We are heavily dependent on the successful commercialization of XDEMVY and the development, regulatory approval, and commercialization of our current and future product candidates.
We currently have one product approved for commercial sale, XDEMVY (lotilaner ophthalmic solution) 0.25%, which was approved by the FDA in July 2023 for the treatment of Demodex blepharitis in the U.S. The success of our business, including our ability to generate revenue from product sales in the future, will primarily depend on the successful commercialization of XDEMVY and the successful development, regulatory approvals and commercialization of our product candidates in one or more jurisdictions. Our ability to generate revenue and achieve profitability depends significantly on our ability, or any future collaborator’s ability, to achieve a number of challenging objectives, including:
•timely receipt of regulatory approvals from applicable regulatory authorities for our product candidates for which we successfully complete clinical development;
•successful and timely completion of preclinical and clinical development of our product candidates;
•successfully educating ECPs about Demodex blepharitis and related diagnosis;
•successful commercial launch following any regulatory approval, including leveraging our commercial infrastructure in-house or with one or more collaborators;
•commercial acceptance of XDEMVY and any of our other product candidates by patients, the medical community and third-party payers;
•establishing and maintaining relationships with contract research organizations ("CROs") and clinical sites for the clinical development, both in the U.S. and internationally, of our product candidates;
•making any required post-marketing approval commitments to applicable regulatory authorities;
•establishing and maintaining commercially viable supply and manufacturing relationships with third parties that can provide adequate, in both amount and quality, products and services to support clinical development and meet the market demand for product candidates that we develop, if approved;
•obtaining an IND prior to commencing clinical trials in the U.S. for drug for a particular indication, such as TP-04 for the potential treatment of rosacea and TP-05 for potential Lyme disease prophylaxis and community malaria reduction;
•a continued acceptable safety and efficacy profile both prior to and following any marketing approval of our product candidates;
•identifying, assessing and developing new product candidates;
•obtaining, maintaining and expanding patent protection, trade secret protection and regulatory exclusivity, both in the U.S. and internationally;
•protecting our rights in our intellectual property portfolio;
•defending against third-party interference or infringement claims, if any;
•obtaining favorable terms in any collaboration, licensing or other arrangements that may be necessary or desirable to develop, manufacture or commercialize our existing or acquired product candidates;
•obtaining coverage and adequate reimbursement for customers and patients from government and third-party payers for XDEMVY and other potential product candidates that we develop;
•addressing any competing therapies and technological and market developments; and
•attracting, hiring and retaining qualified personnel.
We may never be successful in achieving our objectives and, even if we do, may never generate significant revenue that is large enough to achieve profitability. If we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis and we will continue to incur substantial research and development and other expenditures to develop and market additional product candidates. In addition, as a young business, we may encounter unforeseen expenses, difficulties, complications, delays and other known and unknown challenges. Our failure to become and remain profitable would decrease the value of our company and could impair our ability to maintain or further our research and development efforts, raise additional necessary capital, grow our business, retain key employees and continue our operations.
We may not be successful in educating ECPs and the market about the need for treatments specifically for Demodex blepharitis and other diseases or conditions targeted by XDEMVY or our product candidates. XDEMVY or other product candidates that we may develop may fail to achieve market acceptance by ECPs, other healthcare providers and patients, or adequate formulary coverage, pricing or reimbursement by third-party payers and others in the medical community, and the market opportunity for these products may be smaller than we estimate.
XDEMVY, or any current or future product candidate that receives marketing approval, may fail to gain sufficient market acceptance by ECPs or other healthcare providers, patients, third-party payers and others in the medical community. Before the approval of XDEMVY, there was no approved prescription therapeutic for Demodex blepharitis and the only other current treatments include over-the-counter and off-label remedies such as tea tree oil, lid wipes and artificial tears, as well as off-label prescription products. Efforts to educate the medical community, patients and third-party payers on the benefits of XDEMVY and our other product candidates may require significant resources and may not be successful.
Although XDEMVY is approved for the treatment of Demodex blepharitis, ECPs and potential patients may not have sufficient information about, or recognize the need for a treatment specifically targeting Demodex blepharitis. It is possible that ECPs may continue to rely on other treatments for treating symptoms consistent with Demodex blepharitis. A key tenet of our continued commercialization strategy is to educate ECPs on Demodex blepharitis and how to diagnose it with a simple slit lamp examination as well as raise patient awareness of Demodex blepharitis. However, our efforts may prove to be unsuccessful, and we may not be able to develop this new market for XDEMVY. We may still not achieve success in promotional efforts for XDEMVY, and ECPs may continue to use existing treatments rather than XDEMVY or any other product candidate and potential patients may not inquire as to XDEMVY. It is also possible that ECPs and patients may not be willing to adopt XDEMVY for the treatment of Demodex blepharitis because of the possibility that the disease will recur despite mite eradication and the potential for periodic use of XDEMVY.
In addition, if generic versions of any products that compete with XDEMVY or any of our product candidates are approved for marketing by the FDA or comparable foreign regulatory authorities, they could be offered at a substantially lower price than we expect to offer for XDEMVY or our other product candidates, if approved. As a result, ECPs, patients and third-party payers may choose to rely on such products rather than XDEMVY or our product candidates, if approved.
If XDEMVY or any other product candidate that we develop does not achieve an adequate level of acceptance, formulary coverage, pricing or reimbursement we may not generate significant product revenues and we may not become profitable. The degree of market acceptance of XDEMVY or any other product candidate that we develop, if approved for commercial sale, will depend on a number of factors, including:
•the efficacy, safety and potential advantages of XDEMVY, or our product candidates, if approved, compared to alternative treatments, including the existing standard-of-care, and the perceptions by members of the healthcare community of the same;
•our ability to offer our products for sale at competitive prices, particularly in light of the lower cost of alternative treatments;
•the clinical indications for which the product is approved;
•the convenience and ease of administration compared to alternative treatments;
•the willingness of the target patient population to try new therapies and of ECPs to prescribe these therapies;
•the strength and effectiveness of our marketing and distribution support, which may be adversely impacted by health epidemics;
•publicity concerning our products or competing products and treatments;
•the timing of market introduction of competitive products;
•the perception by patients or physicians that the diseases we are targeting, including Demodex blepharitis, are not burdensome;
•the potential for our competitors to limit our access to the market through anti-competitive contracts or other arrangements;
•the availability of third-party formulary coverage and adequate reimbursement;
•product labeling or product insert requirements of the FDA or other regulatory authorities;
•the prevalence and severity of any side effects; and
•any restrictions on the use of our products, if approved, together with other medications.
The sales, marketing, and distribution of XDEMVY or any future approved products may be unsuccessful or less successful than anticipated. If we are unable to establish sales and marketing capabilities or enter into agreements with third parties to sell and market XDEMVY or any future approved products on acceptable terms, we may be unable to successfully commercialize XDEMVY or any future approved products.
We recently began commercializing our first product, XDEMVY, in the United States. The success of our commercialization efforts for XDEMVY and any future approved products is subject to the effective execution of our business plan, including, among others, the continued development of our internal sales, marketing and distribution capabilities. For example, we have established an internal infrastructure as well as an ECP-focused sales and distribution infrastructure to market XDEMVY and our product candidates in the U.S., and have completed hiring in areas to support commercialization, including sales management, sales representatives, marketing, access and reimbursement, sales support and distribution. There are significant risks involved with establishing our own sales, marketing, and distribution capabilities, including our ability to hire, retain and appropriately incentivize qualified individuals, provide adequate training to sales and marketing personnel, and effectively manage geographically dispersed sales and marketing teams to generate sufficient demand. Any failure or delay in the development of these capabilities could or negatively affect the success of our commercialization efforts and business. For
example, the commercialization of XDEMVY may not develop as planned or anticipated, which may require us to, among other items, adjust or amend our business plan and incur significant expenses.
Further, given our lack of experience commercializing products, we do not have a track record of successfully executing on the commercialization of an approved product. If we are unsuccessful in accomplishing our objectives and executing on our business plan, or if the commercialization of XDEMVY or any future approved products does not develop as planned, we may require significant additional capital and financial resources, we may not become profitable, and we may not be able to compete against more established companies in our industry.
Further, if we choose to collaborate, either globally or on a territory-by-territory basis, with third parties that have direct sales forces and established distribution systems, either to augment our own sales force and distribution systems or in lieu of our own sales force and distribution systems, we will be required to negotiate and enter into arrangements with such third parties relating to the proposed collaboration. If we are unable to enter into such arrangements when needed, on acceptable terms, or at all, we may not be able to successfully commercialize any of our product candidates that receive regulatory approval or any such commercialization may experience delays or limitations. If we are unable to successfully commercialize our approved product candidates, either on our own or through collaborations with one or more third parties, our future product revenue will suffer and we may incur significant additional losses.
Further, in order to continue to commercialize XDEMVY or commercialize any product candidates, if approved, we must continue to build marketing, sales, distribution, managerial and other non-technical capabilities or make arrangements with third parties to perform these services for each of the territories in which we may have approval to sell and market our product candidates. We may not be successful in accomplishing these required tasks.
The sizes of the market opportunities for our product or product candidates, particularly XDEMVY for the treatment of Demodex blepharitis and TP-03 for the treatment of MGD, have not been established with precision and may be smaller than we estimate, possibly materially. If our estimates of the sizes overestimate these markets, our sales growth may be adversely affected. We may also not be able to grow the markets for our product candidates as intended or at all.
Our assessment of the potential market opportunity for XDEMVY and other product candidates that we develop is based on industry and market data that we obtained from industry publications and research, surveys and studies conducted by third parties and our own internal epidemiology and market research studies. Industry publications and third-party research, surveys and studies generally indicate that their information has been obtained from sources believed to be reliable, although they do not guarantee the accuracy or completeness of such information. While we believe these industry publications and third-party research, surveys and studies are reliable, we have not independently verified such data. Similarly, although the studies we have conducted are based on information that we believe to be complete and reliable, we cannot guarantee that such information is accurate or complete. The potential market opportunities for the treatment of Demodex blepharitis and for the treatment of MGD is difficult to precisely estimate, because patients often have multiple ocular surface diseases and the symptoms have significant overlap, leading to frequent misdiagnosis of the various conditions. Therefore, our estimates of the potential market opportunities for our product candidates include several key assumptions based on our industry knowledge, industry publications, third-party research and our own epidemiology studies and market research, which may be based on a small sample size and fail to accurately reflect market opportunities. While we believe that our internal assumptions and the bases of the studies and research we have conducted are reasonable, no independent source has verified such assumptions or bases. If any of our assumptions or estimates, or these publications, research, surveys or studies prove to be inaccurate, then the actual market for XDEMVY or any of our other product candidates may be smaller than we expect, and as a result our revenue from product sales may be limited and it may be more difficult for us to achieve or maintain profitability.
Due to the patients presenting at ECP clinics with multiple ocular surface diseases, there is overlap in market size estimates for blepharitis and MGD. Therefore, if XDEMVY receives regulatory approval for the treatment of both Demodex blepharitis and MGD, our opportunity could be less than our forecasts because the actual market for XDEMVY might be significantly smaller than our estimates.
Even though we obtained regulatory approval with respect to XDEMVY for Demodex blepharitis, we may not be able to obtain regulatory approval for additional indications, such as MGD, or we may be required to conduct additional trials, which would limit our ability to realize the full market potential of XDEMVY or increase the costs of developing TP-03 for MGD.
We are exploring the therapeutic potential for TP-03 in MGD as an additional indication. If we are successful, the indication for use of TP-03 could potentially be broadened beyond the treatment of Demodex blepharitis to include MGD as an additional indication. However, there can be no assurance that we will obtain approval for any other indication, including MGD or for any broadened indication beyond the treatment of Demodex blepharitis. If we fail to maintain required approvals for
these additional or broadened indications, or if regulatory approvals are delayed, we will not realize the full market potential of TP-03. Additionally, the FDA or other comparable foreign regulatory authority may require us to conduct additional clinical trials before seeking regulatory approval.
We face significant competition, and if our competitors develop and market technologies or products more rapidly than we do or that are more effective, safer or less expensive than the product candidates we develop, our commercial opportunities will be negatively impacted. XDEMVY and our product candidates, if approved, will also compete with existing branded, generic and off-label products.
The development and commercialization of new drug products is highly competitive. We face competition with respect to XDEMVY and our product candidates that we may seek to develop or commercialize in the future, from many different sources, including major pharmaceutical companies, specialty pharmaceutical companies and biotechnology companies worldwide and existing treatments. Potential competitors also include academic institutions, government agencies and other public and private research organizations that conduct research, seek patent protection and establish collaborative arrangements for research, development, manufacturing and commercialization.
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient or are less expensive than our products. Our competitors also may obtain FDA approval or other regulatory authority approval for their products more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market.
In addition, our ability to compete may be affected in many cases by insurers or other third-party payers, particularly Medicare and the competent authorities of the individual EU Member States, seeking to encourage the use of generic products. Generic products are currently being used for certain of the indications that we are pursuing, and additional products are expected to become available on a generic basis over the coming years.
Many of the companies against which we are competing or against which we may compete in the future have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller and early stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These third parties compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
Additionally, while XDEMVY is approved for the treatment of blepharitis or Demodex blepharitis specifically, a number of other treatments are currently available for blepharitis in the U.S. Current treatments for blepharitis in the U.S. include over the counter remedies such as tea tree oil, lid wipes and artificial tears, as well as off-label prescription products. If ECPs were to continue to prescribe these other existing treatments instead of XDEMVY, our business would be adversely affected.
Although we obtained FDA approval of XDEMVY, and even if we obtain FDA approval of any of our product candidates, we may never obtain approval or authorization for such product candidates, including XDEMVY, in any other jurisdiction or commercialize such product candidates in the U.S. or in any other jurisdiction, which would limit our ability to realize their full market potential.
In order to market any products, including XDEMVY, outside of the U.S., we will need to comply with additional onerous but varying regulatory requirements of other countries regarding safety and efficacy on a country-by-country basis. Approval by the FDA in the United States does not ensure approval by comparable regulatory authorities in other countries or jurisdictions nor does it ensure that we will be able to successfully commercialize XDEMVY or any other approved products in the U.S. or in other jurisdictions. In addition, clinical trials conducted in one country may not be accepted by regulatory authorities in other countries, and regulatory approval in one country does not mean that regulatory approval will be obtained in any other country. Further, successful commercialization in the United States does not guarantee successful commercialization in other jurisdictions. Approval procedures vary among countries and can involve additional product testing and validation and additional administrative review periods. Seeking foreign regulatory approvals could result in significant delays, difficulties and costs for us and may require additional preclinical studies or clinical trials which would be costly and time consuming. Regulatory requirements can vary widely from country to country and could delay or prevent the introduction of our products in those countries. Satisfying these and other regulatory requirements is costly, time consuming, uncertain and subject to unanticipated delays. In addition, our failure to obtain regulatory approval in any country may delay or have negative effects on
the process for regulatory approval in other countries. We do not have any product candidates approved for sale in international markets, and we do not have experience in obtaining regulatory approval in international markets. If we, or our collaboration partners, fail to comply with regulatory requirements in international markets or to obtain and maintain required approvals, or if regulatory approvals in international markets are delayed, our ability to realize the full market potential of our products will be harmed.
Our future product candidates may cause significant adverse events, toxicities or other undesirable side effects which may delay or prevent marketing approval or cause us to abandon or limit further clinical development or commercialization of those product candidates. In addition, significant adverse events, toxicities or other undesirable side effects may be identified during post-marketing surveillance for XDEMVY, or future approved products, which could result in regulatory action or negatively affect our ability to market the product.
Adverse events or other undesirable side effects caused by our product candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA, the European Commission or other comparable foreign regulatory authorities.
During the conduct of clinical trials, subjects report changes in their health, including illnesses, injuries, and discomforts, to their study doctor. Often, it is not possible to determine whether or not the product candidate being studied caused these conditions. It is possible that as we test our product candidates in larger, longer and more extensive clinical trials, or as use of these product candidates becomes more widespread if they receive regulatory approval, illnesses, injuries, discomforts and other adverse events that were not observed in earlier trials, as well as conditions that did not occur or went undetected in previous trials, will be reported by subjects. Many times, side effects are only detectable after investigational products are tested in large-scale, Phase 3 clinical trials or, in some cases, after they are made available to patients on a commercial scale after approval.
Our understanding of the relationship between our product candidates and these adverse events may change as we gather more information, and additional unexpected adverse events or an increase in adverse event rates may occur. If additional clinical experience indicates that any of our product candidates have side effects or causes serious or life-threatening side effects, participant recruitment for trials and the ability of enrolled subjects to complete trials could be negatively impacted, and the development of the product candidate may fail or be delayed, which would severely harm our business, prospects, operating results and financial condition.
Additionally, if we or others later identify undesirable side effects or adverse events caused by XDEMVY or one of our product candidates that receives marketing approval, a number of potentially significant negative consequences could result, including, but not limited to:
•regulatory authorities may withdraw approvals of such product or require additional warnings on the label such as a black box warning, a contraindication or other limitations on the product's approved use, or issue safety alerts, Dear Healthcare Provider letters, press releases or other communications containing warnings or other safety information about the product;
•the product may be seized by regulatory authorities;
•there may be a recall of the product;
•we may be required to change the way the product is administered or conduct additional clinical trials or post-approval studies;
•we may be required to create and implement a REMS plan, which could include a medication guide outlining the risks of such side effects for distribution to patients, a communication plan for healthcare providers, including ECPs, and/or other elements to assure safe use;
•the product may become less competitive;
•we could be sued and held liable for harm caused to patients; and
•our reputation may suffer and there may be resulting harm to physician or patient acceptance of our product.
Any of these events could prevent us from achieving or maintaining market acceptance of the particular product candidate, if approved, and could significantly harm our business, results of operations, and prospects.
As we participate in the Medicaid Drug Rebate Program and other governmental pricing programs, failure to comply with obligations under these programs could result in additional reimbursement requirements, penalties, sanctions and fines, which could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
Underthe Medicaid Drug Rebate program, a participating manufacturer is required to pay a rebate to each state Medicaid program for its covered outpatient drugs that are dispensed to Medicaid beneficiaries and paid for by the state Medicaid program as a condition of having federal funds being made available for drugs under Medicaid and Medicare Part B. Those rebates are based on pricing data reported by the manufacturer on a monthly and quarterly basis to CMS. These data include the average manufacturer price and, in the case of innovator products, the best price for each drug, which, in general, represents the lowest price available from the manufacturer to any wholesaler, retailer, provider, health maintenance organization, nonprofit entity, or governmental entity in the U.S. in any pricing structure, calculated to include all sales and associated rebates, discounts, and other price concessions. If we fail to pay the required rebate amount or report pricing data on a timely basis, we may be subject to civil monetary penalties and/or termination from the Medicaid Drug Rebate program. Additionally, civil monetary penalties can be applied if we are found to have knowingly submitted any false price or product information to the government, if we fail to submit the required price data on a timely basis, or if we misclassify or misreport product information.CMS could also decide to terminate our Medicaid drug rebate agreement, in which case federal payments may not be available under Medicaid or Medicare Part B for our covered outpatient drugs.
The ACA (addressed further above in the section on “Business – Government Regulatory – Coverage and Reimbursement”) made significant changes to the Medicaid Drug Rebate Program, and CMS issued a final regulation to implement the changes to the Medicaid Drug Rebate Program under the ACA. CMS also issued a final regulation that modified prior Medicaid Drug Rebate Program regulations to permit reporting multiple best price figures with regard to value based purchasing arrangements; and provide definitions for “line extension,” “new formulation,” and related terms, with the practical effect of expanding the scope of drugs considered to be line extensions that are subject to an alternative rebate formula. While the regulatory provisions that purported to affect the applicability of the best price and average manufacturer price exclusions of manufacturer-sponsored patient benefit programs, in the context of PBM “accumulator” programs were invalidated by a court, such programs may continue to negatively affect us in other ways. Our failure to comply with these price reporting and rebate payment options, as well as pharmaceutical benefit manager “accumulator” programs, could negatively impact our financial results.
Federal law requires that a manufacturer also participate in the 340B Drug Pricing program in order for federal funds to be available for the manufacturer’s drugs under Medicaid and Medicare Part B. The 340B program requires participating manufacturers to agree to charge no more than the 340B “ceiling price” for the manufacturer’s covered outpatient drugs to a specified “covered entities,” including community health centers and other entities that receive certain federal grants, as well as hospitals that serve a disproportionate share of low-income patients. The 340B ceiling price is calculated using a statutory formula, which is based on the average manufacturer price and rebate amount for the covered outpatient drug as calculated under the Medicaid Drug Rebate Program. If we are found to have knowingly and intentionally charged 340B covered entities more than the statutorily mandated ceiling price, we could be subject to significant civil monetary penalties and/or such failure also could be grounds for HRSA to terminate our agreement to participate in the 340B program, in which case our covered outpatient drugs would no longer be eligible for federal payment under the Medicaid or Medicare Part B program.
Further, the IRA establishes a Medicare Part D inflation rebate scheme (the first rebate period is in fourth quarter 2022 through third quarter 2023) and a drug price negotiation program, with the first negotiated prices to take effect in 2026. It also makes several changes to the Medicare Part D benefit, including the creation of a new manufacturer discount program in place of the current coverage gap discount program (beginning in 2025). Manufacturers may be subject to civil monetary penalties for certain violations of the negotiation and inflation rebate provisions and an excise tax during a noncompliance period under the negotiation program. Drug manufacturers may also be subject to civil monetary penalties with respect to their compliance with the new Part D manufacturer drug discount program.
Pricing and rebate calculations are complex, vary across products and programs, and are often subject to interpretation by the manufacturer, governmental agencies, and courts. A manufacturer that becomes aware that its Medicaid reporting for a prior quarter was incorrect, or has changed as a result of recalculation of the pricing data, is obligated to resubmit corrected data up to three years after those data originally were due. Restatements and recalculations increase the costs for complying with the laws and policies governing the Medicaid Drug Rebate program and could result in an overage or
underage in our rebate liability for past quarters. They also may affect the 340B ceiling price and therefore liability under the 340B program.
Finally, in order to be eligible to have its products paid for with federal funds under the Medicaid and Medicare programs and purchased by the Department of Veterans Affairs (“VA”), Department of Defense (“DoD”), Public Health Service, and Coast Guard (the “Big Four agencies”) and certain federal grantees, a manufacturer is required to participate in the VA Federal Supply Schedule (“FSS”) pricing program, established under Section 603 of the Veterans Health Care Act of 1992. Under this program, the manufacturer is obligated to make its covered drugs available for procurement on an FSS contract and charge a price to the Big Four agencies that is no higher than the Federal Ceiling Price (“FCP”), which is a price calculated pursuant to a statutory formula. The FCP is derived from a calculated price point called the “non-federal average manufacturer price” (“Non FAMP”), which the manufacturer calculates and reports to the VA on a quarterly and annual basis. Pursuant to applicable law, knowing provision of false information in connection with a Non FAMP filing can subject a manufacturer to significant penalties for each item of false information. The FSS contract also contains extensive disclosure and certification requirements. If we overcharge the government in connection with the FSS contract or Tricare Retail Pharmacy Rebate Program, whether due to a misstated FCP or otherwise, we will be required to refund the difference to the government. Failure to make necessary disclosures and/or to identify contract overcharges can result in allegations against us under the False Claims Act and other laws and regulations. Unexpected refunds to the government, and any response to government investigation or enforcement action, would be expensive and time-consuming, and could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
Under Section 703 of the National Defense Authorization Act for FY 2008, the manufacturer is required to pay quarterly rebates to DoD on utilization of its innovator products that are dispensed through DoD’s Tricare network pharmacies to Tricare beneficiaries. The rebates are calculated as the difference between the annual Non FAMP and FCP for the calendar year that the product was dispensed. A manufacturer that overcharges the government in connection with the FSS contract or Tricare Retail Pharmacy Rebate Program, whether due to a misstated FCP or otherwise, is required to refund the difference to the government. Failure to make necessary disclosures and/or to identify contract overcharges can result in allegations against us under the False Claims Act and other laws and regulations
We may expend our limited resources on the commercialization of XDEMVY for the treatment of Demodex blepharitis and fail to capitalize on product candidates or indications that may be more profitable or for which there is a greater likelihood of success.
Because we have limited financial and managerial resources, we must prioritize our research programs and will need to focus our product candidates on the potential treatment of certain indications. We are currently focused on the commercialization, of XDEMVY for the treatment of Demodex blepharitis. As a result, we may forego or delay pursuit of opportunities with other product candidates or for other indications that later prove to have greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on the most viable commercial products or profitable market opportunities. Our spending on current and future research and development programs for TP-03 and other product candidates may not yield any commercially viable products. If we do not accurately evaluate the commercial potential or target market for XDEMVY, TP-03 for other indications and other product candidates, we may also relinquish valuable rights to that product candidate through collaboration, licensing or other royalty arrangements in cases in which it would have been more advantageous for us to retain sole development and commercialization rights to such product candidate.
The terms of approvals and ongoing regulation of XDEMVY and any other current product candidates or product candidates we develop could require substantial expenditure of resources and may limit how we manufacture and market our products, which could materially impair our ability to generate revenue from product sales.
XDEMVY, and any other product candidate for which we obtain regulatory approval, along with the manufacturing processes, post-approval clinical data, labeling, advertising, and promotional activities for such product, will be subject to continual requirements of and review by the FDA, the EMA and other regulatory authorities. These requirements include submissions of safety and other post-marketing information and reports, registration and listing requirements, cGMP requirements relating to quality control, quality assurance and corresponding maintenance of records and documents, and requirements regarding the distribution of samples to physicians and recordkeeping. Even if regulatory approval of a product candidate is granted, the approval may be subject to limitations on the indicated uses for which the product may be marketed or to the conditions of approval, or contain requirements for costly post-marketing testing and surveillance to monitor the safety or efficacy of the product.
Accordingly, we and our contract manufacturers will continue to expend time, money, and effort in all areas of regulatory compliance, including manufacturing, production, product surveillance, and quality control for XDEMVY and any
other approved products. If we are not able to comply with post-approval regulatory requirements, we could have the regulatory approvals for our products, including XDEMVY, withdrawn by regulatory authorities and our ability to market XDEMVY or any future products could be limited, which could adversely affect our ability to achieve or sustain profitability. Further, the cost of compliance with post-approval regulations may have a negative effect on our business, operating results, financial condition, and prospects.
If XDEMVYor any of our product candidates that are approved for marketing are found to have been improperly promoted for off-label uses by us, or if ECPs misuse our products or use our products off-label, we may become subject to prohibitions on the sale or marketing of our products, product liability claims and significant fines, penalties and sanctions, and our brand and reputation could be harmed.
The FDA and other foreign regulatory authorities strictly regulate the marketing of and promotional claims that are made about drug products. In particular, a product may not be promoted for uses or indications that are not approved by the FDA or such other foreign regulatory authorities as reflected in the product’s approved labeling. Any regulatory approval that the FDA or a foreign regulatory authority grants is limited to those specific diseases and indications for which a product is deemed to be safe and effective. For example, the FDA-approved label for XDEMVY is limited to the treatment of Demodex blepharitis, and we are not permitted to promote XDEMVY for any other uses, unless and until such uses are approved.
In addition, although we believe XDEMVY and our product candidates may exhibit a lower risk of side effects or more favorable tolerability profile or better symptomatic improvement than other products for the indications we are studying, without head-to-head data, we will be unable to make comparative claims for XDEMVY or our product candidates, if approved. If we receive regulatory approval for any of our products and are found to have promoted XDEMVY or any of our products or product candidates, if approved, for off-label uses, we may become subject to significant liability, which would materially harm our business. Both federal and state governments have levied large civil and criminal fines against companies for alleged improper promotion and have enjoined several companies from engaging in off-label promotion. If we become the target of such an investigation or prosecution based on our marketing and promotional practices, we could face similar sanctions, which would materially harm our business. In addition, our management’s attention could be diverted from our business operations, significant legal expenses could be incurred, and our brand and reputation could be damaged. The FDA has also previously requested that companies enter into consent decrees or permanent injunctions under which specified promotional conduct is changed or curtailed. If we are deemed by the FDA to have engaged in the promotion of our products for off-label use, we could be subject to FDA regulatory or enforcement actions, including the issuance of an untitled letter, a warning letter, injunction, seizure, civil fine, or criminal penalties. It is also possible that other federal, state, or foreign enforcement authorities might take action if they determine our business activities constitute promotion of an off-label use, which could result in significant penalties, including criminal, civil or administrative penalties, damages, fines, disgorgement, exclusion from participation in government healthcare programs and the curtailment or restructuring of our operations. We cannot, however, prevent an ECP from using XDEMVY or our product candidates in ways that fall outside the scope of the approved indications, as he or she may deem appropriate in his or her medical judgment. ECPs may also misuse XDEMVY or our product candidates, if approved, or use improper techniques, which may lead to adverse results, side effects or injury and, potentially, subsequent product liability claims. Furthermore, the use of XDEMVY or our product candidates, if approved, for indications other than those approved by the FDA and/or other regulatory authorities may not effectively treat such conditions, which could harm our brand and reputation among ECPs and patients.
Clinical drug development is a lengthy, expensive and risky process with uncertain timelines and uncertain outcomes, and results of earlier studies and trials may not be predictive of future results. If clinical trials of our product candidates particularly TP-03 for the treatment of Demodex blepharitis, do not meet safety or efficacy endpoints or are prolonged or delayed, we may be unable to obtain required regulatory approvals, and therefore be unable to commercialize our product candidates on a timely basis or at all.
Before obtaining marketing approval from regulatory authorities for the sale of our product candidates, we must conduct extensive clinical trials to demonstrate the safety and efficacy of the product candidates in humans. The research and development of drugs is an extremely risky industry. Only a small percentage of product candidates that enter the development process ever receive marketing approval. Failure or delay can occur at any time during the clinical trial process. To date, we have focused substantially all of our efforts and financial resources on identifying, acquiring, and developing our product candidates, including conducting preclinical studies and clinical trials. Clinical testing is expensive and can take many years to complete, and we cannot be certain that any clinical trials will be conducted as planned or completed on schedule, if at all. Furthermore, product candidates are subject to continued preclinical safety studies, which may be conducted concurrently with our clinical testing. The outcomes of these safety studies may delay the launch of or enrollment in future clinical trials and could impact our ability to continue to conduct our clinical trials. Our inability to successfully complete preclinical and clinical development could result in additional costs to us and negatively impact our ability to generate revenue. Our future success is dependent on our ability to successfully develop, obtain regulatory approval for, and then successfully commercialize product
candidates. We currently generate no revenuesrevenue from product sales of any products,for one product, and we may never be able to develop or commercialize aadditional marketable product.products.
We have not yet completed any Phase 2b/3 or Phase 3 trials for any product candidate. The results of preclinical and early clinical trials of our product candidates and other products with the same mechanism of action may not be predictive of the results of later-stage clinical trials. For example, we may not be able to replicate the safety and efficacy results of our Phase 22b/3 clinical trials for Demodex blepharitis in our Phase 2b/3 trial, Saturn-1, or our Phase 3 trial, Saturn-2.clinical trials for other indications in the future. Clinical trial failure may result from a multitude of factors including flaws in trial design, dose selection, placebo effect, patient enrollment criteria and other challenges with enrolling and maintaining trial subjects, relatively smaller sample size in earlier trials, and failure to demonstrate favorable safety or efficacy traits. As such, failure in clinical trials can occur at any stage of testing. A number of companies in the biopharmaceutical industry have suffered setbacks in the advancement of clinical trials due to lack of efficacy or adverse safety profiles, notwithstanding promising results in earlier trials, and we cannot be certain that we will not face similar setbacks. Based upon negative or inconclusive results, we may decide, or regulators may require us, to conduct additional clinical trials or preclinical studies. In addition, data obtained from clinical trials are susceptible to varying interpretations, and regulators may not interpret our data as favorably as we do, which may further delay, limit or prevent marketing approval. Furthermore, as more product candidates within a particular class of drugs proceed through clinical development to regulatory review and approval, the amount and type of clinical data that may be required by regulatory authorities may increase or change. The outcome of preclinical testing and early clinical trials may not be predictive of the success of later clinical trials, and preliminary or interim results of a clinical trial do not necessarily predict final results. For example, our product candidates may fail to show the desired safety and efficacy in clinical development despite positive results in preclinical studies or having successfully advanced through initial clinical trials. The failure of any of our product candidates to demonstrate safety and efficacy in any clinical trial could negatively impact the perception of our other product candidates or cause regulatory authorities to require additional testing before approving any of our product candidates.
We currently have one product candidate, TP-03, in clinical development and its risk of failure is high. For example, use of TP-03 requires the patient to follow a prescribed technique to administer the eye drops. Failure to properly administer the eye drops by the patient or inappropriate technique demonstration by the eye care practitioners, may adversely affect the outcome of TP-03 in demonstrating efficacy in one or more clinical trials. We are unable to predict if this product candidate or any of our future product candidates that advance into clinical trials will prove safe or effective in humans or will obtain marketing approval. If we are unable to complete preclinical or clinical trials of current or future product candidates, due to safety concerns, or if the results of these trials are not satisfactory to convince regulatory authorities of their safety or efficacy, we will not be able to obtain marketing approval for commercialization. Even if we are able to obtain marketing approvals for any of our product candidates, those approvals may be for indications that are not as broad as desired or may contain other limitations that would adversely affect our ability to generate revenue from sales of those products. Moreover, if we are not able to differentiate our product against other approved products within the same class of drugs, or if any of the other circumstances described above occur, our business would be materially harmed and our ability to generate revenue from that class of drugs would be severely impaired.
Each of our product candidates will require additional clinical development, management of clinical, preclinical (for some of our product candidates) andand/or manufacturing activities, regulatory approval in multiple jurisdictions, achieving and maintaining commercial-scale supply, building of a commercial organization, substantial investment and significant marketing
efforts before we generate any revenues from product sales. We are not permitted to market or promote any of our product candidates before we receive regulatory approval from the FDA or comparable foreign regulatory authorities, and we may never receive such regulatory approval for any of our product candidates. We may experience delays in our ongoing clinical trials, and we do not know whether planned clinical trials will begin on time, need to be redesigned, enroll patients on time or be completed on schedule, if at all. For example, the FDA has recommended that for TP-03 we conduct carcinogenicity testing as well as embryofetal development studies in a second species. Any further recommendations by the FDA regarding our applications or clinical trials could cause delay of any regulatory approval by the FDA and cause our expenses to increase. We may experience numerous unforeseen events during, or as a result of, clinical trials that could delay or prevent our ability to receive marketing approval or commercialize TP-03, our other product candidates, or any other product candidates that we may develop, including:
•we may experience delays in or failure to reach agreement on acceptable terms with prospective CROs, vendors and clinical sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs, vendors and trial sites;
•we may fail to obtain sufficient enrollment in our clinical trials, our enrollment needs may grow larger than we anticipate, or participants may fail to complete our clinical trials at a higher rate than we anticipate;
•clinical trials of our product candidates may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional clinical trials or abandon product development programs;
•we may decide, or regulators or institutional review boardsIRBs or ethics committees may require us, to suspend or terminate clinical research for various reasons, including noncompliance with regulatory requirements or a finding that the participants are being exposed to unacceptable health risks;
•regulators or institutional review boardsIRBs or ethics committees may not authorize us or our investigators to commence a clinical trial at a prospective clinical trial site or at all or may require us to perform additional or unanticipated clinical trials to obtain approval or we may be subject to additional post-marketing testing requirements to maintain regulatory approval;
•regulators may revise the requirements for approving our product candidates, or such requirements may not be as we anticipate;
•the cost of clinical trials of our product candidates may be greater than we anticipate, and we may need to delay or suspend one or more trials until we complete additional financing transactions or otherwise receive adequate funding;
•the supply or quality of our product candidates or other materials necessary to conduct clinical trials of our product candidates may be insufficient or inadequate or may be delayed;
•our product candidates may have undesirable side effects or other unexpected characteristics, causing us or our investigators, regulators or institutional review boardsIRBs or ethics committees to suspend or terminate trials;
•regulatory authorities may determine that the planned design of our clinical trials is flawed or inadequate;
•regulatory authorities may suspend or withdraw their approval of a product or impose restrictions on its distribution;
•we may not be able to timely or at all obtain INDs for a product candidate;
•we may modify a preclinical study or clinical trial protocol;
•third-party contractors may fail to comply with regulatory requirements or meet their contractual obligations to us in a timely manner, or at all;
•we may be unable to establish clinical endpoints that applicable regulatory authorities consider clinically meaningful, or, if we seek accelerated approval, biomarker efficacy endpoints that applicable regulatory authorities consider likely to predict clinical benefit;
•we may experience delays due to the ongoing COVID-19 pandemic,outbreak of health epidemics, including with respect to the conduct of ongoing clinical trials, receipt of product candidates or other materials, submission of New Drug Applications ("NDAs"),NDAs, filing of INDs, and starting any clinical trials for other indications or programs; and
•we may experience manufacturing delays due to the recent COVID-19 pandemichealth epidemics in our supply chain caused by a shortage of raw materials, a lack of employees on site at our suppliers due to illness, or a lack of productivity at our suppliers due to local or national government quarantine restrictions on coming to the workplace.
If we are required to conduct additional clinical trials or other testing of our product candidates beyond those that we currently contemplate, if we are unable to successfully complete clinical trials of our product candidates or other testing, if the results of these trials or tests are not positive or are only modestly positive, if there are safety concerns or if we determine that the observed safety or efficacy profile would not be competitive in the marketplace, we may:
•incur unplanned costs;
•be delayed in obtaining marketing approval for our product candidates;
•not obtain marketing approval at all;
•obtain marketing approval in some countries and not in others;
•obtain approval for indications or patient populations that are not as broad as intended or desired;
•obtain approval with labeling that includes significant use or distribution restrictions or safety warnings;
•be subject to additional post-marketing testing requirements; or
•have the product removed from the market after obtaining marketing approval.
We cannot be certain whether any of our planned clinical trials will begin on schedule or any preclinical studies we plan to initiate will begin on our intended schedule, or whether any such studies or clinical trials will need to be restructured or will be completed on schedule, or at all. If we experience delays in the completion of, or termination of, any clinical trial of our product candidates, or are unable to achieve clinical endpoints due to unforeseen events, such as the COVID-19 pandemic,health epidemics, the commercial prospects of our product candidates will be harmed, and our ability to generate product revenues from any of these product candidates will be delayed. In addition, any delays in completing our clinical trials will increase our costs, slow down our product candidate development and approval process and jeopardize our ability to commencegenerate additional revenue from product sales and generate revenues.sales. Significant clinical trial delays could also allow our competitors to bring products to market before we do or shorten any periods during which we have the exclusive right to commercialize our product candidates and impair our ability to commercialize our product candidates and may harm our business and results of operations.
Even if TP-03 or any other product candidate that we develop receives marketing approval, we may not be successful in educating ECPs and the market about the need for treatments specifically for Demodex blepharitis and other diseases or conditions targeted by our product candidates, and TP-03 or other product candidates that we may develop may fail to achieve market acceptance by ECPs, other healthcare providers and patients, or adequate formulary coverage, pricing or reimbursement by third-party payors and others in the medical community, and the market opportunity for these products may be smaller than we estimate.
If TP-03 or any other product candidate that we are developing or develop receives marketing approval, it may nonetheless fail to gain sufficient market acceptance by ECPs or other healthcare providers, patients, third-party payors and others in the medical community. There is no approved prescription therapeutic for Demodex blepharitis and current treatments include over-the-counter and off-label remedies such as tea tree oil, lid wipes and artificial tears, as well as off-label prescription products. Efforts to educate the medical community, patients and third-party payors on the benefits of our product candidates may require significant resources and may not be successful.
Further, even if TP-03 demonstrates promising or superior clinical results and receives FDA or other regulatory marketing approval, including the treatment of both signs and symptoms of Demodex blepharitis, ECPs and potential patients may not have sufficient information about, or recognize the need for a treatment specifically targeting Demodex blepharitis, it is possible that ECPs may continue to rely on other treatments for treating symptoms consistent with Demodex blepharitis. A key tenant of our commercialization strategy is to educate ECPs on Demodex blepharitis and how to diagnose it with a simple slit
lamp examination as well as raise patient awareness of Demodex blepharitis. However, our efforts may prove to be unsuccessful, and we may not be able to develop this new market for which there is no approved treatment. If TP-03 receives FDA or other regulatory marketing approval, we may still not achieve success in promotional efforts for TP-03, and ECPs may continue to use existing treatments rather than TP-03 or any other product candidate and potential patients may not inquire as to TP-03. It is also possible that ECPs and patients may not be willing to adopt TP-03 for the treatment of Demodex blepharitis because of the likelihood that the disease will recur despite mite eradication and the necessity for periodic use of TP-03.
In addition, if generic versions of any products that compete with any of our product candidates are approved for marketing by the FDA or comparable foreign regulatory authorities, they would likely be offered at a substantially lower price than we expect to offer for our product candidates, if approved. As a result, ECPs, patients and third-party payors may choose to rely on such products rather than our product candidates.
If TP-03 or any other product candidate that we develop does not achieve an adequate level of acceptance, formulary coverage, pricing or reimbursement we may not generate significant product revenues and we may not become profitable. The degree of market acceptance of TP-03 or any other product candidate that we develop, if approved for commercial sale, will depend on a number of factors, including:
•the efficacy, safety and potential advantages of our product candidates compared to alternative treatments, including the existing standard of care, and the perceptions by members of the healthcare community of the same;
•our ability to offer our products for sale at competitive prices, particularly in light of the lower cost of alternative treatments;
•the clinical indications for which the product is approved;
•the convenience and ease of administration compared to alternative treatments;
•the willingness of the target patient population to try new therapies and of ECPs to prescribe these therapies;
•the strength and effectiveness of our marketing and distribution support, which may be adversely impacted by the COVID-19 pandemic;
•publicity concerning our products or competing products and treatments;
•the timing of market introduction of competitive products;
•the perception by patients or physicians that the diseases we are targeting, including Demodex blepharitis, are not burdensome;
•the potential for our competitors to limit our access to the market through anti-competitive contracts or other arrangements;
•the availability of third-party formulary coverage and adequate reimbursement;
•product labeling or product insert requirements of the FDA or other regulatory authorities;
•the prevalence and severity of any side effects; and
•any restrictions on the use of our products, if approved, together with other medications.
Our product candidates still require significant testing. We only recently began clinical trials to test TP-03TP-04 and TP-05 in humans and, as a company, we have limited experience in this area.
Most of our operations to date have been limited to preclinical studies and clinical trials. We have completed Phase 2 clinical trials for TP-03 for the treatment of Demodex blepharitis and commenced our Phase 2b/3 clinical trial for the same indication, Saturn-1, for TP-03 in September 2020 and expect to initiate our Phase 3 clinical trial, Saturn-2, for TP-03 in 2021 and initiate clinical trials for our other product candidate in the future. As a result, we will need to expand our clinical operations, quality and regulatory capabilities to support these activities.
Additionally, we are early in our development efforts for certain of our product candidates and indications, including TP-03 for the treatment of MGD, TP-04 for the treatment of rosacea and TP-05 for potential Lyme disease prophylaxis and community malaria reduction. The risk of failure for product candidates in early development is high. Extensive clinical trials are necessary to demonstrate the safety and efficacy of such product candidates in humans. Clinical trials may fail to demonstrate that such product candidates are safe for humans and effective for indicated uses. Further, we intend to leverage data from the TP-03 preclinical studies and clinical safety assessments for the treatment of Demodex blepharitis to satisfy the preclinical study requirements for TP-03 for the treatment of MGD, and TP-04 and TP-05 and other indications. For MGD, we intend to rely on preclinical studiesannounced the enrollment of our first patient in the Phase 2a Ersa trial studying TP-03 for the potential treatment of MGD and clinical safety assessments fromin December 2023 reported positive topline results. For rosacea, we conducted the Demodex blepharitis program. We have not conductedPhase 1 Galatea trial with TP-04 and do not intend to conduct any preclinical studies with TP-03initiated the Phase 2a Galatea trial, for the treatment of MGDrosacea in orderMarch 2023. In February 2024, we announced positive topline results and plan to advance to Phase 2a. For our preservative-free formulation of Demodex blepharitis, we intend to leverage all preclinical, Phase 2discuss and Phase 3 data from TP-03 Demodex blepharitis program. We intend to conduct in vitro or in vivo bioequivalence studiesdetermine the potential regulatory path with our preservative-free formulation to compare it to the current preserved formulation of TP-03 in Demodex blepharitis after NDA submission and file a supplement. For rosacea, we intend to leverage data from TP-03 preclinical studies and augment with additional preclinical studies to select formulation in order to advance to Phase 1/2. We have not conducted any preclinical studies in rosacea with TP-04 to date. In relationFDA. With respect to Lyme disease, in December 2022 we announced positive topline results from the completed Callisto trial and malaria, we intendenrollment of the first patient in the Carpo trial. The Carpo trial, evaluating TP-05, a novel investigative oral, non-vaccine pharmacological prophylactic for the potential prevention of Lyme disease in humans is a randomized, double-blind, placebo-controlled trial that evaluated the efficacy of TP-05 in killing lab grown, non-disease carrying ticks after they have attached to leverage data from our TP-03 preclinical studies for Demodex blepharitisthe skin of healthy volunteers, as well as third-party preclinical studies for Lyme disease or malaria, respectively (and will not conduct our own preclinical studies for Lyme diseaseconfirm the safety, tolerability, and malaria). Subjectblood concentration of TP-05. In February 2024, we announced positive topline results from the Carpo trial and plan to FDA feedback, we intend to conduct Phase 1/2 trials in rosacea, Lyme diseasediscuss and malaria based on these preclinical studies. In relation to malaria, we may conduct our Phase 1/2 trial outsidedetermine the United States.potential regulatory path with the FDA. The FDA may reject our use of data from TP-03 preclinical studies for the treatment of Demodex blepharitis for other indications or require additional studies to augment the data to advance for clinical development. The FDA may also reject our use of data from preclinical studies conducted by third parties for Lyme disease and malaria and require us to conduct additional preclinical studies before advancing to additional clinical trials. In addition, data from preclinical studies conducted by third parties may not be as reliable as data from studies conducted by us and since we did not conduct the studies, there may be weaknesses in the studies design or results that we may not be aware of.
In part because of our limited infrastructure, experience conducting clinical trials as a company and regulatory interactions, we cannot be certain that our clinical trials will be completed on time, that our planned clinical trials will be initiated on time, if at all, that our planned development programs would be acceptable to
the FDA or other comparable foreign regulatory authorities, or that, if approval is obtained, such product candidates can be successfully commercialized.
Our clinical trialsWe have and may continue to date have been small, and we may not be able to replicate our results from completed trials in Phase 3 trials.
Our clinical trials conducted to date, including for TP-03 for the treatment of Demodex blepharitis, have been small, each with fewer than 60 persons, and have advanced through Phase 1 and Phase 2. For example, the clinical trial which had the highest patient population to date was our Phase 2b Europa clinical trial, which enrolled 54 patients. Our Phase 2b/3 and Phase 3 clinical trials, Saturn-1 and Saturn-2, will be conducted with larger patient populations to evaluate TP-03 for the treatment of Demodex blepharitis, in which we expect to enroll at least 350 patients each. In these later trials, additional risks, including previously unidentified low incidence safety risks or lack of efficacy may materialize. Adverse or inconclusive results in these later clinical trials may, despite initially promising results, result in such product candidate not receiving requisite approvals for marketing and sale, and there is a risk that additional clinical trials will be required to obtain such approvals or that our clinical development program will be required to be altered, which would result in increased costs, significant delays to filing with regulatory authorities, filing for a narrower indication than previously anticipated or the abandonment of efforts to commercialization such product candidate.
If we encounter difficulties or delays enrolling patients in our clinical trials, which could cause delays in or adverse effects of our clinical development activities could be delayed or otherwise adversely affected.activities.
We have and may continue to experience difficulties in patient enrollment in our clinical trials for a variety of reasons. The timely completion of clinical trials in accordance with their protocols depends, among other things, on our ability to enroll a sufficient number of patients who remain in the study until its conclusion. We have and may continue to experience difficulties in patient enrollment in our clinical trials for a variety of reasons. For example, we recently experienced delays related to our Carpo trial with topline results pushed out to February 2024 as a result of patient enrollment delays. The enrollment of patients depends on many factors, including:
•the patient eligibility criteria defined in the protocol;
•size of the patient population required for analysis of the trial’s primary endpoints;
•the proximity of patients to study sites;
•the design of the trial;
•our ability to recruit clinical trial investigators with the appropriate competencies and experience;
•clinicians’ and patients’ perceptions as to the potential advantages of the product candidate being studied in relation to other available therapies, including any new drugs that may be approved for the indications we are investigating;
•our ability to obtain and maintain patient consents;
•costs to, or lack of adequate compensation for, prospective patients;
•difficulties of enrolling patients or patients continuing to participate in follow-up visits due to the ongoing COVID-19 pandemic;or new health epidemics; and
•the risk that patients enrolled in clinical trials will drop out of the trials before completion.
In addition, our clinical trials may compete with other clinical trials for product candidates that are in the same therapeutic areas as our product candidates, and this competition would reduce the number and types of
patients available to us, because some patients who might have opted to enroll in our trials may instead opt to enroll in a trial being conducted by one of our competitors. Since the number of qualified clinical investigators is limited, we expect to conduct some of our clinical trials at the same clinical trial sites that some of our competitors use, which will reduce the number of patients who are available for our clinical trials in such clinical trial site. Moreover, potential patients and their doctors may be inclined to use existing therapies rather than enroll patients in any future clinical trial.
Delays in patient enrollment may result in increased costs or may affect the timing or outcome of the planned clinical trials, which could prevent completion of these trials and adversely affect our ability to advance the development of our product candidates.
Our current or future product candidates may cause significant adverse events, toxicities or other undesirable side effects which may delay or prevent marketing approval or cause us to abandon or limit further clinical development of those product candidates. In addition, if we obtain approval for any of our product candidates, significant adverse events, toxicities or other undesirable side effects may be identified during post-marketing surveillance, which could result in regulatory action or negatively affect our ability to market the product.
Adverse events or other undesirable side effects caused by our product candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA, the European Commission or other comparable foreign regulatory authorities.
During the conduct of clinical trials, subjects report changes in their health, including illnesses, injuries, and discomforts, to their study doctor. Often, it is not possible to determine whether or not the product candidate being studied caused these conditions. It is possible that as we test our product candidates in larger, longer and more extensive clinical trials, or as use of these product candidates becomes more widespread if they receive regulatory approval, illnesses, injuries, discomforts and other adverse events that were not observed in earlier trials, as well as conditions that did not occur or went undetected in previous trials, will be reported by subjects. Many times, side effects are only detectable after investigational products are tested in large-scale, Phase 3 clinical trials or, in some cases, after they are made available to subjects on a commercial scale after approval.
The most commonly reported adverse events in TP-03 to date were mild transient ocular burning after administration for less than 10 seconds. One patient in the Europa clinical trial also reported mild burning and blurriness after administration that lasted the entire treatment period. Our understanding of the relationship between our product candidates and these adverse events may change as we gather more information, and additional unexpected adverse events or an increase in adverse event rates may occur. If additional clinical experience indicates that TP-03 or any other product candidate has side effects or causes serious or life-threatening side effects, participant recruitment for trials and the ability of enrolled subjects to complete trials could be negatively impacted, and the development of the product candidate may fail or be delayed, which would severely harm our business, prospects, operating results and financial condition.
Additionally, if one or more of our product candidates receives marketing approval, and we or others later identify undesirable side effects or adverse events caused by such products, a number of potentially significant negative consequences could result, including, but not limited to:
•regulatory authorities may withdraw approvals of such product or require additional warnings on the label such as a “black box” warning or a contraindication, or issue safety alerts, Dear Healthcare Provider letters, press releases or other communications containing warnings or other safety information about the product;
•the product may be seized by regulatory authorities;
•there may be a recall of the product;
•we may be required to change the way the product is administered or conduct additional clinical trials or post-approval studies;
•we may be required to create and implement a Risk Evaluation and Mitigation Strategy ("REMS") plan, which could include a medication guide outlining the risks of such side effects for distribution to patients, a communication plan for healthcare providers, including ECPs, and/or other elements to assure safe use;
•the product may become less competitive;
•we could be sued and held liable for harm caused to patients; and
•our reputation may suffer and there may be resulting harm to physician or patient acceptance of our product.
Any of these events could prevent us from achieving or maintaining market acceptance of the particular product candidate, if approved, and could significantly harm our business, results of operations, and prospects.
Any termination or suspension of, or delays in the commencement or completion of, our planned clinical trials could result in increased costs to us, delay or limit our ability to generate revenue from product sales and adversely affect our commercial prospects.
Before we can initiate clinical trials in the United StatesU.S. for our product candidates, we must submit the results of preclinical testing and any previous clinical studies to the FDA along with other information, including information about product candidate chemistry, manufacturing and controls (“CMC”) and our proposed clinical trial protocol, as part of an IND. The initiation of clinical trials in the European Union (“EU”)EU Member States will be subject to similar requirements concerning approval by competent national authorities and the receipt of a positive opinion from the relevant ethics committees. We do not know whether our planned trials will begin on time or be completed on schedule, if at all. The commencement and completion of clinical trials can be delayed for a number of reasons, including delays related to:
•the FDA or comparable foreign regulatory authorities placing the clinical trial on hold;
•subjects failing to enroll or remain in our trial at the rate we expect;
•subjects choosing an alternative treatment or other product candidates, or participating in competing clinical trials;
•lack of adequate funding to continue the clinical trial;
•subjects experiencing severe or unexpected drug-related adverse effects;
•failure to demonstrate efficacy of the product;
•any interruptions or delays in the supply of our product candidates for our clinical trials;
•a facility manufacturing any of our product candidates or any of their components being ordered by the FDA or comparable foreign regulatory authorities to temporarily or permanently shut down due to violations of good manufacturing practice ("cGMP")cGMP regulations or other applicable requirements, or infections or cross-contaminations of product candidates in the manufacturing process;
•any changes to our manufacturing process that may be necessary or desired;
•any failure or delay in reaching an agreement with CROs, vendors and clinical trial sites;
•third-party clinical investigators losing the licenses or permits necessary to perform our clinical trials, not performing our clinical trials on our anticipated schedule or consistent with the clinical trial protocol, good clinical practices ("GCP") or regulatory requirements or other third parties not performing data collection or analysis in a timely or accurate manner;
•third-party contractors becoming debarred, disqualified or suspended or otherwise penalized by the FDA or other comparable foreign regulatory authorities for violations of applicable regulatory requirements, in which case we may need to find a substitute contractor, and we may not be able to use some or all of the data produced by such contractors in support of our marketing applications;
•one or more Institutional Review Boards ("IRBs"), other ethics committees refusing to approve, suspending or terminating the trial at an investigational site, precluding enrollment of additional subjects, or withdrawing its approval of the trial; or
•changes in regulatory requirements and policies, which may require us to amend clinical trial protocols to comply with these changes and resubmit our clinical trial protocols to IRBs or ethics committees for reexamination.
For example, while we have received, in connection with our IND application, a “no-objection” letter from the FDA regarding the trial design for Saturn-1, the FDA may still require additional studies be conducted prior to submitting an NDA for TP-03. For instance, we will also need to perform a pharmacokinetic study for TP-03 to support our NDA submission for Demodex blepharitis and the FDA is recommending carcinogenicity testing for TP-03 as well as embryofetal development studies in a second species, any result of which, or any additional requests by the FDA, could cause delays in regulatory approval by the FDA. Any delays in completing our clinical trials will increase our costs, slow down our product candidate development and approval process and jeopardize the commercial prospects of our product candidates and our ability to commencegenerate revenue from product sales and generate revenue.sales.
In addition, many of the factors that cause, or lead to, termination or suspension of, or a delay in the commencement or completion of, clinical trials may also ultimately lead to the denial of regulatory approval of a product candidate. For example, if we make manufacturing or formulation changes to our product candidates, we may need to conduct additional studies to bridge our modified product candidates to earlier versions. Further, if one or more clinical trials are delayed, our competitors may be able to bring products to market before we do, and the commercial viability of our product candidates could be significantly reduced. Any of these occurrences may harm our business, financial condition and prospects significantly. Any termination of any clinical trial of our product candidates will harm our commercial prospects and our ability to generate revenue.
If we are unable to establish sales and marketing capabilities or enter into agreements with third parties to sell and market ourrevenue from product candidates on acceptable terms, we may be unable to successfully commercialize our product candidates that obtain regulatory approval.
We currently have a very limited marketing team and no sales team. In order to commercialize any product candidates, if approved, we must build marketing, sales, distribution, managerial and other non-technical capabilities or make arrangements with third parties to perform these services for each of the territories in which we may have approval to sell and market our product candidates. We may not be successful in accomplishing these required tasks.
Establishing an internal sales and marketing team with technical expertise and supporting distribution capabilities to commercialize our product candidates will be expensive and time-consuming, and will require significant attention of our executive officers to manage. Any failure or delay in the development of our internalsales, marketing and distribution capabilities could adversely impact the commercialization of any of our product candidates that we obtain approval to market, if we do not have arrangements in place with third parties to provide such services on our behalf. Alternatively, if we choose to collaborate, either globally or on a territory-by-territory basis, with third parties that have direct sales forces and established distribution systems, either to augment our own sales force and distribution systems or in lieu of our own sales force and distribution systems, we will be required to negotiate and enter into arrangements with such third parties relating to the proposed collaboration. If we are unable to enter into such arrangements when needed, on acceptable terms, or at all, we may not be able to successfully commercialize any of our product candidates that receive regulatory approval or any such commercialization may experience delays or limitations. If we are unable to successfully commercialize our approved product
candidates, either on our own or through collaborations with one or more third parties, our future product revenue will suffer and we may incur significant additional losses.
The sizes of the market opportunities for our product candidates, particularly TP-03 for the treatment of Demodex blepharitis and for the treatment of MGD, have not been established with precision and may be smaller than we estimate, possibly materially. If our estimates of the sizes overestimate these markets, our sales growth may be adversely affected. We may also not be able to grow the markets for our product candidates as intended or at all.
Our assessment of the potential market opportunity for TP-03 and other product candidates that we develop is based on industry and market data that we obtained from industry publications and research, surveys and studies conducted by third parties and our own internal epidemiology and market research studies. Industry publications and third-party research, surveys and studies generally indicate that their information has been obtained from sources believed to be reliable, although they do not guarantee the accuracy or completeness of such information. While we believe these industry publications and third-party research, surveys and studies are reliable, we have not independently verified such data. Similarly, although the studies we have conducted are based on information that we believe to be complete and reliable, we cannot guarantee that such information is accurate or complete. The potential market opportunities for the treatment of Demodex blepharitis and for the treatment of MGD is difficult to precisely estimate, because patients often have multiple ocular surface diseases and the symptoms have significant overlap, leading to frequent misdiagnosis of the various conditions. Therefore, our estimates of the potential market opportunities for our product candidates include several key assumptions based on our industry knowledge, industry publications, third-party research and our own epidemiology studies and market research, which may be based on a small sample size and fail to accurately reflect market opportunities. While we believe that our internal assumptions and the bases of the studies and research we have conducted are reasonable, no independent source has verified such assumptions or bases. If any of our assumptions or estimates, or these publications, research, surveys or studies prove to be inaccurate, then the actual market for TP-03 or any of our other product candidates may be smaller than we expect, and as a result our product revenue may be limited and it may be more difficult for us to achieve or maintain profitability.
Due to the patients presenting at ECP clinics with multiple ocular surface diseases, there is overlap in market size estimates for blepharitis and MGD. Therefore, if TP-03 receives regulatory approval for the treatment of Demodex blepharitis and MGD, our opportunity could be less than our forecasts because the actual market for TP-03 might be significantly smaller than our estimates.
The market for blepharitis and Demodex blepharitis may be not be similar to the market for dry eye.
The markets for blepharitis and Demodex blepharitis may prove to be materially different from the dry eye market that grew significantly once there was an approved product. Even if we obtain approval for TP-03 for the treatment of Demodex blepharitis and commercialize TP-03 in this indication, we may not be able to expand the market for Demodex blepharitis materially or at all or increase awareness of Demodex blepharitis to an extent similar to the dry eye market expansion induced by the commercial dry eye product or at all. Our inability to grow the market in a similar way to the dry eye market may occur due to differences in the underlying diseases, different ECP or patient attitudes towards the diseases, symptoms or treatment, changes over time in attitudes towards direct to patient marketing, different reimbursement and coverage, differences in company strategy, marketing or operations and differences in key assumptions which we have not taken into account in our analysis. Additionally, there may be differences in symptoms, regulatory approval and market dynamics. Dry eye had numerous over-the-counter options that reinforced the disease and promoted disease management. Further, patient awareness for dry eye may have been higher due to the various over-the-counter options for dry eye, which do not exist to such a degree for Demodex blepharitis.
We may not be able to demonstrate the safety and efficacy of TP-03, TP-04 and TP-05 in the indications we are pursuing even though the API underlying such product candidates is safe and effective in animals.
TP-03, TP-04 and TP-05 are presentations of lotilaner, the API, formulated into an eye drop, topical cream and oral formulation, respectively. Lotilaner, which is designed to paralyze and eradicate mites and other parasites through the inhibition of parasite-specific GABA-Cl channels, has been found to be safe and effective in animals. However, despite being safe and effective in animals, we may not be able to demonstrate that TP-03, TP-04 and TP-05 are safe and effective for human use in the indications we are pursuing for these product candidates. This may be in part because the requirements and regulations applicable to approval of a product candidate for human use are significantly more stringent than for animals and that there may be other ocular or other relevant differences between animals and humans.
We face significant competition, and if our competitors develop and market technologies or products more rapidly than we do or that are more effective, safer or less expensive than the product candidates we develop, our commercial opportunities
will be negatively impacted. Our product candidates will, if approved, also compete with existing branded, generic and off-label products.
The development and commercialization of new drug products is highly competitive. We face competition with respect to our product candidates that we may seek to develop or commercialize in the future, from many different sources, including major pharmaceutical companies, specialty pharmaceutical companies and biotechnology companies worldwide and existing treatments. Potential competitors also include academic institutions, government agencies and other public and private research organizations that conduct research, seek patent protection and establish collaborative arrangements for research, development, manufacturing and commercialization.
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient or are less expensive than our products. Our competitors also may obtain FDA approval or other regulatory authority approval for their products more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market.
In addition, our ability to compete may be affected in many cases by insurers or other third-party payors, particularly Medicare and the competent authorities of the individual EU Member States, seeking to encourage the use of generic products. Generic products are currently being used for certain of the indications that we are pursuing, and additional products are expected to become available on a generic basis over the coming years.
Many of the companies against which we are competing or against which we may compete in the future have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller and early stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These third parties compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
Additionally, while there are no currently available on-label prescription pharmaceutical treatments available for the treatment of blepharitis or Demodex blepharitis specifically, a number of other treatments are currently available for the treatment of blepharitis in the United States. Current treatments for blepharitis in the United States include over the counter remedies such as tea tree oil, lid wipes and artificial tears, as well as off-label prescription products. If ECPs were to continue to prescribe these other existing treatments instead of TP-03, our business would be adversely affected.
Product liability lawsuits against us could cause us to incur substantial liabilities, could divert our resources and could limit or delay our commercialization of any product candidates that we may develop.
We face an inherent risk of product liability exposure related to the testing of our product candidates in human clinical trials and will face an even greater risk if we commercially sell any products that we may develop. While we currently have no products that have been approved for commercial sale, the use of product candidates by us in clinical trials, and the sale of any approved products in the future, may expose us to liability claims. These claims might be made by patients that use the product, healthcare providers, pharmaceutical companies or others selling such products. On occasion, large judgments have been awarded in class action lawsuits based on products that had unanticipated adverse effects. If we cannot successfully defend ourselves against claims that our product candidates or products caused injuries, we could incur substantial liabilities. Regardless of merit or eventual outcome, product liability claims may result in:
•delay, variation or termination of clinical trials;
•decreased demand for any product candidates or products that we may develop;
•withdrawal of regulatory approval, recall, restriction on the approval or a “black box” warning or contraindication for an approved drug;
•injury to our reputation and significant negative media attention;
•withdrawal of clinical trial subjects;
•initiation of investigations by regulators;
•significant costs to defend the related litigation and diversion of management’s time and our resources;
•substantial monetary awards to study subjects or patients;
•product recalls, withdrawals or labeling, marketing or promotional restrictions;
•loss of revenue; and
•the inability or delay of our efforts to commercialize any products that we may develop.
Although we maintain product liability insurance coverage, it may not be adequate to cover all liabilities that we may incur. We anticipate that we will need to increase our insurance coverage as our product candidates advance through clinical trials and if we successfully commercialize any products. Insurance coverage is increasingly expensive, thus we may not be able to maintain insurance coverage at a reasonable cost or in an amount adequate to satisfy any liability that may arise. If a successful clinical trial or product liability claim or series of claims is brought against us for uninsured liabilities or in excess of insured liabilities, our assets may not be sufficient to cover such claims and our business operations could be impaired.
Even if we obtain FDA approval of any of our product candidates, we may never obtain approval or authorization or commercialize such products outside of the United States, which would limit our ability to realize their full market potential.
In order to market any products outside of the United States, we will need to comply with additional onerous but varying regulatory requirements of other countries regarding safety and efficacy. Clinical trials conducted in one country may not be accepted by regulatory authorities in other countries, and regulatory approval in one country does not mean that regulatory approval will be obtained in any other country. Approval procedures vary among countries and can involve additional product testing and validation and additional administrative review periods. Seeking foreign regulatory approvals could result in significant delays, difficulties and costs for us and may require additional preclinical studies or clinical trials which would be costly and time consuming. Regulatory requirements can vary widely from country to country and could delay or prevent the introduction of our products in those countries. Satisfying these and other regulatory requirements is costly, time consuming, uncertain and subject to unanticipated delays. In addition, our failure to obtain regulatory approval in any country may delay or have negative effects on the process for regulatory approval in other countries. We do not have any product candidates approved for sale in any jurisdiction, including international markets, and we do not have experience in obtaining regulatory approval in international markets. If we, or our collaboration partners, fail to comply with regulatory requirements in international markets or to obtain and maintain required approvals, our ability to realize the full market potential of our products will be harmed.sales.
Our future growth may depend, in part, on our ability to penetrate foreign markets, where we would be subject to additional regulatory burdens and other risks and uncertainties.
Our future profitability may depend, in part, on our ability to commercialize our product candidates in foreign markets for which we may rely on collaboration with third parties.parties, such as our China Out-License with LianBio. We are evaluating the opportunities for the development and commercialization of our product candidates in other foreign markets. We are not permitted to market or promote any of our product candidates before we receive regulatory approval from the applicable regulatory authority in that foreign market, and we may never receive such regulatory approval for any of our product candidates. To obtain separate regulatory approvals in other countries we may be required to comply with numerous and varying regulatory requirements of such countries regarding the safety and efficacy of our product candidates and governing, among other things, clinical trials and commercial sales, pricing and distribution of our product candidates, and we cannot predict success in these jurisdictions. If we obtain approval of our product candidates and ultimately commercialize our product candidates in foreign markets, we would be subject to additional risks and uncertainties, including:
•our customers’ ability to obtain reimbursement for our product candidates in foreign markets;
•our inability to directly control commercial activities if we are relying on third parties;
•the burden of complying with complex and changing foreign regulatory, tax, accounting and legal requirements;
•different medical practices and customs in foreign countries affecting acceptance in the marketplace;
•import or export licensing requirements;
•longer accounts receivable collection times;
•longer lead times for shipping;
•language barriers for technical training and the need for language translations;
•reduced protection of intellectual property rights in some foreign countries;
•the existence of additional potentially relevant third-party intellectual property rights;
•foreign currency exchange rate fluctuations; and
•the interpretation of contractual provisions governed by foreign laws in the event of a contract dispute.
For example, the pharmaceutical industry in the China Territory is subject to comprehensive government regulation and supervision, encompassing the approval, registration, manufacturing, packaging, licensing and marketing of new drugs. In recent years, the regulatory framework in the China Territory regarding the pharmaceutical industry has undergone significant changes, and we expect that it will continue to undergo significant changes. Any such changes or amendments may result in increased compliance costs on our business or cause delays in or prevent the successful development of TP-03 by LianBio under the China Out-License and reduce the current benefits we believe are available to us. The China Territory authorities have become increasingly vigilant in enforcing laws in the pharmaceutical industry and any failure by LianBio or our other partners to maintain compliance with applicable laws and regulations or obtain and maintain required licenses and permits may result in the suspension or termination of our partner’s business activities in the China Territory. Additionally, to the extent that we enter into collaborations with third parties for development and/or commercialization of our products or product candidates in foreign markets, we will be unable to directly control development and commercial activities or whether such third parties continue to develop or commercialize such products or product candidates. For example, on February 13, 2024, LianBio announced its completion of a comprehensive strategic review and determined to initiate the wind down of its operations, including the sale of remaining pipeline assets, the delisting of its American Depositary Shares, deregistration under Section 12(b) of the Exchange Act, and workforce reductions. As of the date of this filing, it is uncertain if and when we will receive any future milestone consideration under the China Out-License, including but not limited to the milestone achievement of an additional drug supply agreement execution.
Another example of the changing regulatory requirements is that in the EU, the European Commission has presented a proposal to reform the current EU pharmaceutical legislation. The proposal intends to reduce the regulatory data protection period and orphan market exclusivity period for new medicinal products. It is currently uncertain if the proposal will be adopted in its current form and it is uncertain if and when the revised legislation would enter into force.
Foreign sales of our product candidates could also be adversely affected by the imposition of governmental controls, political and economic instability, trade restrictions and changes in tariffs. In some countries, particularly the countries in Europe, the pricing of prescription pharmaceuticals is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take considerable time after the receipt of marketing approval for a drug. To obtain reimbursement or pricing approval in some countries, we may be required to conduct a clinical trial that compares the cost-effectiveness of our product candidate to other available therapies. If reimbursement of our products is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, our business could be harmed, possibly materially.
We have conducted alla number of our completed clinical trials for our product candidates at sites outside the United States,U.S., and the FDA may not accept data from trials conducted in such locations.
We have conducted all of our clinical trials outside the United States, including our Phase 2 clinical trials for the treatment of Demodex blepharitis which were conducted in Mexico City, Mexico, at a well-established site for ocular therapy trials. We may in the future choose to conduct other clinical trials outside the United States. Since we have yet to complete any clinical trials in the United States, it is possible that we may not be able to replicate the efficacy and safety results of our completed Phase 2 trials in the United States. We expect that the FDA will primarily consider the efficacy results of our Saturn-1 and Saturn-2 trials in addition to safety data from all human trials, our preclinical studies data as well as preclinical data for lotilaner in support of our potential NDA submission for TP-03 for the treatment of Demodex blepharitis.
Although the FDA may accept data from clinical trials conducted outside the United States,U.S., acceptance of this data is subject to conditions imposed by the FDA. For example, the clinical trial must be well designed and conducted and be performed by qualified investigators in accordance with certain ethical principles. Theand policy principles, including GCP standards. Among other requirements, the trial population must also adequately represent the U.S. population, and the data must be applicable to the U.S. population and U.S. medical practice in ways that the FDA deems clinically meaningful. In addition, while these clinical trials are subject to the applicable local laws, FDA acceptance of the data will depend on its determination that the trials also complied with all applicablecertain U.S. laws and regulations. There can be no assurance the FDA will accept data from clinical trials conducted outside of the United States.U.S. There can also be no assurance that the comparable foreign regulatory authority in any jurisdiction in which we seek regulatory approval for our product candidates will accept data
from clinical trials conducted outside such jurisdiction. If the FDA or any such foreign regulatory authority does not accept the data from any trial that we have conducted outside the United States,U.S., it would likely result in the need for additional trials, which would be costly and time-consuming and could delay or permanently halt our development of the applicable product candidates.
In addition, there are risks inherent in conducting clinical trials in multiple jurisdictions, inside and outside of the United StatesU.S. and if return towe conduct trials outside of the United States,U.S., we may face risks, such as:
•regulatory and administrative requirements of the jurisdiction where the trial is conducted that could burden or limit our ability to conduct our clinical trials;
•foreign exchange rate fluctuations;
•manufacturing, customs, shipment and storage requirements;
•cultural or legal differences in the standards for medical practice and clinical research;
•diminished protection of intellectual property in some countries;
•different cultural attitudes to self-reported adverse events (such as burning, stinging, blurry vision) leading to a different safety profile; and
•the risk that the patient populations in such trials are not considered representative as compared to the patient population in the target markets where approval is being sought.
Even if we obtain regulatory approval with respect to TP-03 for Demodex blepharitis, we may not be able to obtain regulatory approval for additional indications, such as MGD, or we may be required to conduct additional trials, which would limit our ability to realize the full market potential of TP-03 or increase the costs of developing TP-03 for MGD.
If we obtain regulatory approval with respect to TP-03 for the treatment of Demodex blepharitis, we intend explore the therapeutic potential for TP-03 in MGD as an additional indication. If we are successful, the indication for use of TP-03 could potentially be broadened beyond the treatment of Demodex blepharitis to include MGD as an additional indication. However, there can be no assurance that, even if we obtain approval for Demodex blepharitis, we will obtain approval for any other indication, including MGD or for any broadened indication beyond the treatment of Demodex blepharitis. If we fail to obtain and maintain required approvals for these additional or broadened indications, or if regulatory approvals are delayed, we will not realize the full market potential of TP-03. Additionally, the FDA or other comparable foreign regulatory authority may require us to conduct additional clinical trials before seeking regulatory approval. For example, we intend to rely on preclinical studies we have conducted with TP-03 for Demodex blepharitis instead of conducting preclinical studies for MGD. The FDA may not approve of this approach and may require us to conduct preclinical studies with TP-03 in MGD, which may delay our expected timelines for approval and increase costs. If we were required to conduct additional clinical trials, our costs for developing TP-03 for treating MGD would be substantially higher and the timing of any regulatory approval, if any, would be substantially extended, which could adverse effect our results of operations.
Changes in methods of product candidate manufacturing or formulation may result in additional costs or delay.
As product candidates proceed through pre-clinical studies to late-stage clinical trials towards potential approval and commercialization, it is common that various aspects of the development program, such as manufacturing methods and formulation, are altered along the way in an effort to optimize processes and results. Such changes carry the risk that they will not achieve these intended objectives. Any of these changes could cause our product candidates to perform differently and affect the results of planned clinical trials or other future clinical trials conducted with the altered materials. Such changes may also require additional testing, FDA notification or FDA approval. This could delay completion of clinical trials, require the conduct of bridging clinical trials or the repetition of one or more clinical trials, increase clinical trial costs, delay approval of our product candidates and jeopardize our ability to commence sales and generate revenue. For example, we intend to develop a preservative-free formulation for TP-03, which we expect to be completed after the submission of the NDA for Demodex blepharitis and used for commercialization. We intend to initiate preclinical equivalence studies with this preservative-free formulation after the NDA submission of the current formulation of TP-03 for Demodex blepharitis. However, there can be no assurance that the FDA will not require us to conduct studies or trials in addition to these preclinical equivalence studies, which would mean additional costs and potentially delays in our approval of TP-03 for the treatment of Demodex blepharitis.
Managing our obligations under our in-license and out-license agreements and other strategic agreements may divert management time and attention, causing delays or disruptions to our business.
We have entered into two license agreements with Elanco: (i) an agreement granting us a worldwide, sublicensable license for the development and marketing of lotilaner for the treatment or cure of any eye or skin condition in humans,as amended(“Eye and Derm Elanco Agreement”)Agreement and (ii) the All Human Uses Elanco Agreement, and have also entered into the LianBio AgreementChina Out-License as discussed elsewhere herein. We also may in the future enter into in-license or out-license agreements with multiple licensors and strategic agreements, which, subject us to various obligations, including diligence obligations, reporting and notification obligations, payment obligations for achievement of certain milestone as well as other material obligations. We may need to devote substantial time and attention to ensuring that we successfully integrate these transactions into our existing operations and are compliant with our obligations under these agreements, which may divert management’s time and attention away from our research and development programs or other day-to-day activities.
Our in-license, out-license, and strategic agreements are also complex and certain provisions in those agreements may be susceptible to multiple interpretations. In the event of any disagreement about the interpretation of these provisions, our management may need to devote a disproportionate amount of its attention to resolving these disagreements. Such disruptions may cause delays in our research and development programs and other business objectives.
Our operating activities may be restricted by certain covenants in our license and other strategic agreements, which could limit our development and commercial opportunities.
In connection with our in-license, out-license, or other collaborations or strategic alliances, we may agree to and be bound by negative covenants which may limit our development and commercial opportunities. For example, pursuant to the Eye and Derm Elanco Agreement and the All Human Uses Elanco Agreement, we made certain covenants to only engage with third party suppliers previously approved by Elanco, and only under certain circumstances. These provisions may inhibit our development efforts, prevent us from forming strategic collaborations to develop and potentially commercialize any other product candidates and may materially harm our business, financial condition, results of operations and prospects.
We may expend our limited resources to pursue TP-03 for the treatment of Demodex blepharitis and fail to capitalize on product candidates or indications that may be more profitable or for which there is a greater likelihood of success.
Because we have limited financial and managerial resources, we must prioritize our research programs and will need to focus our product candidates on the potential treatment of certain indications. We are currently focused on the development and commercialization, if approved, of TP-03 for the treatment of Demodex blepharitis. As a result, we may forego or delay pursuit of opportunities with other product candidates or for other indications that later prove to have greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on the most viable commercial products or profitable market opportunities. Our spending on current and future research and development programs for TP-03 for the treatment of Demodex blepharitis may not yield any commercially viable products. If we do not accurately evaluate the commercial potential or target market for TP-03, we may also relinquish valuable rights to that product candidate through collaboration, licensing or other royalty arrangements in cases in which it would have been more advantageous for us to retain sole development and commercialization rights to such product candidate.
Even if we obtain regulatory approvals for any product candidates we develop, the terms of approvals and ongoing regulation of our products could require substantial expenditure of resources and may limit how we manufacture and market our products, which could materially impair our ability to generate revenue.
Any product candidate for which we obtain regulatory approval, along with the manufacturing processes, post-approval clinical data, labeling, advertising, and promotional activities for such product, will be subject to continual requirements of and review by the FDA, the EMA and other regulatory authorities. These requirements include submissions of safety and other post-marketing information and reports, registration and listing requirements, cGMP requirements relating to quality control, quality assurance and corresponding maintenance of records and documents, and requirements regarding the distribution of samples to physicians and recordkeeping. Even if regulatory approval of a product candidate is granted, the approval may be subject to limitations on the indicated uses for which the product may be marketed or to the conditions of approval, or contain requirements for costly post-marketing testing and surveillance to monitor the safety or efficacy of the product.
Accordingly, assuming we receive regulatory approval for one or more product candidates we develop, we and our contract manufacturers will continue to expend time, money, and effort in all areas of regulatory compliance, including manufacturing, production, product surveillance, and quality control. If we are not able to comply with post-approval regulatory requirements, we could have the regulatory approvals for our products withdrawn by regulatory authorities and our ability to market any future products could be limited, which could adversely affect our ability to achieve or sustain profitability. Further, the cost of compliance with post-approval regulations may have a negative effect on our business, operating results, financial condition, and prospects.
Interim top-line and preliminary results from our clinical trials that we announce or publish from time to time may change as more participant data become available and are subject to audit and verification procedures, which could result in material changes in the final data.
From time to time, we may publish interim top-line or preliminary results from our clinical trials. Interim results from clinical trials that we may complete are subject to the risk that one or more of the clinical outcomes may materially change as participant enrollment continues and more participant data become available. We also make assumptions, estimations,
calculations, and conclusions as part of our analyses of data, and we may not have received or had the opportunity to fully evaluate all data. Preliminary or top-line results also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary data we previously published. As a result, interim and preliminary data should be viewed with caution until the final data are available. Adverse differences between preliminary or interim data
and final data could be material and could significantly harm our reputation and business prospects and may cause the trading price of our common stock to fluctuate significantly.
Risks Related to our Financial Position and Need for Additional Capital
Our limited operating history may make it difficult for you to evaluate the success of our business to date and to assess our future viability.
We commenced activities in 2016. Our limited operating history may make it difficult to evaluate the success of our business to date and to assess our future viability. Our operations to date have been limited to organizing our company, raising capital, identifying and developing product candidates, establishing licensing arrangements and/or acquiring necessary technology, undertaking research, preclinical studies and clinical trials of our product candidates, establishing arrangements for the manufacture of XDEMVY and other product candidates and longer-term planning for commercialization efforts of XDEMVY and our other potential product candidates. Our prospects must be considered in light of the uncertainties, risks, expenses and difficulties frequently encountered by companies in their early stages of operations. We have limited experience in obtaining marketing approvals, manufacturing commercial scale product or arranging for a third party to do so on our behalf, or conducting sales, marketing and distribution activities necessary for successful product commercialization. Consequently, any predictions you make about our future success or viability may not be as accurate as they could be if we had a longer operating history or a history of successfully developing, obtaining marketing approval for and commercializing products. In addition, as our business grows, we may encounter unforeseen expenses, difficulties, complications, delays and other known and unknown obstacles. We may not be successful as we transition from a company with a research and development focus to a company capable of supporting commercial activities.
Due to the recently initiated commercialization of XDEMVY and our continued development of our pipeline of product candidates through clinical trials and other indications, our capital requirements are difficult to predict and may change. We may need to obtain substantial additional funding to achieve our goals and a failure to obtain this necessary capital when needed on acceptable terms, or at all, could force us to delay, reduce or eliminate our product development programs, commercialization efforts or other operations.
Since our inception, we have funded our operations through private placements of preferred stock, convertible promissory notes, the sale of our common stock in our IPO and the Follow-On Public Offerings, and the 2023 ATM Prospectus, as well as proceeds from product sales, net, our China Out-License, and draws on our Credit Facility. We expect our expenses to increase substantially and we will require a larger amount of capital to fund our commercialization efforts, the development of our product candidates and the maintenance and expansion of our operations and capabilities. These expenditures will include costs associated with marketing and selling any products approved for sale, including XDEMVY, conducting non-clinical studies and clinical trials, obtaining regulatory approvals, securing manufacturing and supply of product candidates, costs associated with in-licensing assets consistent with our core strategy and other unanticipated costs. Further, as a public company, we incur significant legal, accounting and other costs associated with operating as a public company.
We believe that our cash, cash equivalents and marketable securities of $227.4 million as of December 31, 2023 and expected sales of XDEMVY is sufficient to fund our current and planned operations for at least the next twelve months from the date of filing this Annual Report on Form 10-K.
We will need to raise substantial additional capital to complete the development and commercialization of XDEMVY and our other product candidates through one or more of: equity offerings, draws from our Credit Facility, marketing and distribution arrangements and other collaborations, strategic alliances and licensing arrangements or other sources.
Due to the complexities of our transition to a commercial-stage company, it is challenging to estimate the actual amounts necessary to successfully commercialize any products approved for sale. We may need to raise additional funds earlier than currently anticipated if we choose to pursue additional indications for our product candidates, acquire new product candidates or otherwise expand our business more rapidly than we presently planned. We have based these estimates on assumptions that may prove to be incorrect or require adjustment because of our ongoing business decisions, and we could utilize our available capital resources sooner than we currently expect. Our future capital requirements will depend on many factors, including:
•the cost and timing, receipt and amount of sales and marketing capabilities of any current and future products, including the success of our commercialization efforts involving XDEMVY;
•market acceptance of our current and future products, including XDEMVY, and the impact of any competing products;
•the ability of patients or healthcare providers to obtain coverage of or sufficient reimbursement for any current or future products;
•the scope and costs of manufacturing development and commercial manufacturing activities and our ability to scale them up;
•the scope, rate of progress, costs and results of our drug discovery, preclinical development activities, laboratory testing and clinical trials for our product candidates;
•the number and scope of clinical programs we decide to pursue;
•the extent to which we acquire or in-license other product candidates and technologies;
•the cost, timing and outcome of regulatory review of our product candidates, including the potential for regulatory authorities to require that we conduct more studies and trials than those that we currently expect to conduct and the costs of post-marketing studies or risk evaluation and mitigation strategies that could be required by regulatory authorities;
•suspensions or delays in enrollment of our ongoing and future clinical trials, issues with data collection, or changes to the number of subjects we decide to enroll in clinical trials, including as a result of health pandemics, competing trials, or otherwise;
•the costs of commercialization activities for any current or future products that are approved for sale, including marketing, sales, and distribution costs, and any discounts or rebates to obtain access;
•potential changes in the regulatory environment and enforcement rules;
•the costs of preparing, filing and prosecuting patent applications, maintaining and enforcing our intellectual property rights and defending intellectual property-related claims;
•our ability to establish and maintain collaborations on favorable terms, if at all;
•our ability to satisfy our outstanding debt obligations;
•our efforts to enhance operational systems and our ability to attract, hire and retain qualified personnel, including personnel to support the sales and marketing activities associated with the commercialization of our products, including XDEMVY, and the development of our product candidates;
•potential changes in pharmaceutical pricing and reimbursement infrastructure;
•the costs related to any future collaboration or licensing partners upon the achievement of negotiated milestones;
•the costs associated with any product liability or other lawsuits related to our products;
•the expense needed to attract and retain skilled personnel; and
•the costs associated with being a public company.
Commercialization efforts of any current or future products, including our commercialization efforts involving XDEMVY, identifying potential product candidates and conducting preclinical studies and clinical trials is a time consuming, expensive and uncertain process that takes years to complete, and we may never generate the necessary data or results required to obtain marketing approval for our product candidates. In addition, our product candidates, if approved, may not achieve
adequate product sales or commercial success. Although we initiated commercialization of XDEMVY for the treatment of Demodex blepharitis in August 2023, we will need to continue to sustain our existing capital resources to fund our future operating expenses and capital expenditure requirements. Adequate additional financing may not be available to us on acceptable terms, or at all, and may be impacted by the economic climate and market conditions. If we are unable to raise capital when needed or on attractive terms, we would be forced to delay, limit, reduce or eliminate our research and development programs or future commercialization efforts or grant rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves. In addition, attempting to secure additional financing may divert the time and attention of management from day-to-day activities and distract from our research and development efforts. Alternatively, we may seek additional capital due to favorable market conditions or strategic considerations, even if we believe we have sufficient funds for our current or future operating plans.
In February 2022, we executed the Credit Facility, as amended in January 2023 and August 2023 (see Note 10) with Hercules Capital, Inc. (“Hercules”) and Silicon Valley Bank, a division of First-Citizens Bank & Trust Company (“SVB”). Concurrent with the execution of the Credit Facility, the Company made a $20.0 million draw. As of December 31, 2023, the Credit Facility provides for a remaining aggregate principal amount of up to $125.0 million with tranched availability as follows: $15.0 million related to the NDA submission for TP-03, $35.0 million which became available in July 2023 upon FDA approval of XDEMVY, $50.0 million upon achievement of certain quarterly revenue thresholds, and $25.0 million upon lender approval. On March 15, 2023 and September 15, 2023, respectively, we made separate draws of $5.0 million (including SVB's commitment of $1.25 million) from the $25.0 million tranche associated with the NDA submission of TP-03. Each of the tranches may be drawn down in $5.0 million increments at our election. As reported elsewhere, on March 10, 2023, SVB was closed by the California Department of Financial Protection and Innovation, which appointed the Federal Deposit Insurance Corporation ("FDIC"), as receiver, and SVB's deposits and substantially all of SVB's assets were transferred into a new entity, Silicon Valley Bridge Bank, N.A ("SVBB"). On March 12, 2023, the Department of the Treasury, the Federal Reserve, and the FDIC jointly released a statement that depositors at SVB would have access to their funds, even those in excess of the standard FDIC insurance limits, under a systemic risk exception. Such parties also announced, among other items, that SVBB had assumed the obligations and commitments of former SVB and commitments to advance under existing credit agreements with former SVB will be honored by SVBB pursuant to the terms of such credit agreements. On March 27, 2023, First Citizens Bank assumed all of SVBB’s obligations and commitments, and SVBB began operating as Silicon Valley Bank, a division of First Citizens Bank. Unless otherwise noted herein, all references to SVB or Silicon Valley Bank shall refer to Silicon Valley Bank, a division of First Citizens Bank. In light of the foregoing, the Company does not believe it has exposure to loss as a result of SVB’s receivership. There can be no assurances that the closure of SVB, or any other financial institution, or any related impacts across the financial services industry will not adversely affect our ability to access the additional availability under our Credit Facility.
Raising additional capital may cause dilution to our stockholders, restrict our operations or require us to relinquish rights to our technologies or product candidates.
Until such time we can generate substantial revenue from product sales, including from XDEMVY, our only approved product, we expect to finance our cash needs through possible combinations of equity offerings, debt financings, collaborations, strategic alliances and licensing arrangements. To the extent that we raise additional capital through the sale of equity or convertible debt securities, your ownership interest will be diluted and the terms of these securities may include liquidation or other preferences that adversely affect your rights as a common stockholder. For example, in May 2022 and August 2023, we completed the Follow-On Public Offerings, in which we received total net proceeds of $74.2 million and $99.3 million, respectively (after deducting underwriting discounts, commissions and other estimated offering-related expenses) through the issuance of 5,889,832 and 6,069,449 shares of our common stock, respectively. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions. For example, the Credit Facility restricts our ability to pursue certain transactions that we may believe to be in our best interests without the prior written consent of Hercules and SVB, including but not limited to, incurring additional indebtedness (subject to certain exceptions with respect to issuing convertible indebtedness), incurring additional liens (including a negative pledge on intellectual property), engaging in mergers, acquisitions and consolidations; conducting asset sales or exclusively licensing our assets in a transaction that constitutes legal transfer to such licensee, making investments and loans, engaging in certain corporate changes, transacting with affiliates, declaring dividends or making other distributions, and making payments on certain other indebtedness.
If we raise funds through collaborations, strategic alliances or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates or grant licenses on terms that may not be favorable to us. If we raise funds through research grants, we may be subject to certain requirements, which may limit our ability to use the funds or require us to share information from our research and development. Raising additional capital through any of these or other means could adversely affect our business and the holdings or rights of our stockholders, and may cause the market price of our shares to decline. If we are unable to raise additional funds through equity or debt financings when needed, we may be required to delay, limit, reduce or terminate our
product development or continued and future commercialization efforts or grant rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves.
Adverse developments affecting the financial services industry, such as actual events or concerns involving liquidity, defaults or non-performance by financial institutions or transactional counterparties, could adversely affect our business and our financial condition and results of operations.
Actual events involving limited liquidity, defaults, non-performance or other adverse developments that affect financial institutions, transactional counterparties or other companies in the financial services industry or the financial services industry generally, or concerns or rumors about any events of these kinds or other similar risks, have in the past and may in the future lead to market-wide liquidity problems. For example, on March 10, 2023, SVB was closed by the California Department of Financial Protection and Innovation, which appointed the FDIC, as receiver, and SVB was subsequently transferred into a new entity, SVBB. On March 12, 2023, the Department of the Treasury, the Federal Reserve, and the FDIC jointly released a statement that depositors at SVB would have access to their funds, even those in excess of the standard FDIC insurance limits, under a systemic risk exception. Such parties also announced, among other items, that Silicon Valley Bridge Bank has assumed the obligations and commitments of former SVB and commitments to advance under existing credit agreements with former SVB will be honored by SVBB in accordance with and pursuant to the terms of such credit agreements. On March 27, 2023, First Citizens Bank assumed all of SVBB’s obligations and commitments, and SVBB began operating as Silicon Valley Bank, a division of First Citizens Bank. Unless otherwise noted herein, all references to SVB or Silicon Valley Bank shall refer to Silicon Valley Bank, a division of First Citizens Bank. In light of the foregoing, the Company does not believe it has exposure to loss as a result of SVB’s receivership.
We currently maintain cash held on deposit at financial institutions in the U.S., including at SVB. These deposits are insured by the FDIC in an amount up to $250,000 for any depositor. To the extent we hold cash deposits in amounts that exceed the FDIC insurance limitation, we may incur a loss in the event of a failure of any of the financial institutions where we maintain deposits, to the extent such loss exceeds the FDIC insurance limitation, and such a failure could have a material adverse effect upon our liquidity, operations and our results of operations.
Additionally, we and other parties with whom we conduct business may be unable to access funds in such deposit account or other accounts, including money market funds, held with a financial institution or lending arrangements with such a financial institution. Our ability and any of our counter-party’s ability to pay their obligations to us or to enter into new commercial arrangements requiring additional payments to us could be adversely affected. In this regard, counterparties to SVB credit agreements and arrangements, and third parties such as beneficiaries of letters of credit (among others), may experience direct impacts from financial institutions in the future and uncertainty remains over liquidity concerns in the broader financial services industry.
Inflation and rapid increases in interest rates have led to a decline in the trading value of previously issued government securities with interest rates below current market interest rates. Although the U.S. Department of Treasury, FDIC and Federal Reserve Board have announced a program to provide up to $25 billion of loans to financial institutions secured by certain of such government securities held by financial institutions to mitigate the risk of potential losses on the sale of such instruments, widespread demands for customer withdrawals or other liquidity needs of financial institutions for immediately liquidity may exceed the capacity of such program. There is no guarantee that the U.S. Department of Treasury, FDIC and Federal Reserve Board will provide access to uninsured funds in the future in the event of the closure of other banks or financial institutions, or that they would do so in a timely fashion.
Our existing indebtedness may limit our flexibility in financing and operating our business and adversely affect our business, financial condition and results of operations.
In February 2022 we entered into the Credit Facility with Hercules and SVB, as amended in January 2023 and August 2023. As of December 31, 2023, the Credit Facility provides for a remaining aggregate principal amount of up to $125.0 million with tranched availability. In addition to these amounts, we may borrow substantial funds in the future to provide a portion of the capital needed in our business and may secure the repayment of such borrowings by placing additional liens or other encumbrances on our assets. Our Credit Facility contains customary conditions to borrowing, events of default and affirmative and negative covenants, including covenants that restrict our ability to incur additional indebtedness, incur additional liens, conduct asset sales or exclusively license our assets in a transaction that constitutes legal transfer to such licensee, make investments and loans, engage in certain corporate changes, transact with affiliates, or declare dividends or make other distributions to holders of our stock. Such restrictions could limit our ability to take certain actions and could reduce our flexibility to run and manage our business which could have an adverse effect on our results of operations. The obligations
under the Credit Facility are secured by a first priority lien on substantially all of our assets, excluding our intellectual property on which there is a negative pledge, subject to customary exceptions. If we were unable to repay amounts due under the Credit Facility, Hercules and SVB could proceed against such assets. Any declaration by Hercules or SVB of an event of default could significantly harm our business and prospects and could cause the price of our common stock to decline.
We may engage in acquisitions or strategic partnerships that could disrupt our business, cause dilution to our stockholders, reduce our financial resources, cause or to incur debt or assume contingent liabilities, and subject us to other risks.
In the future, we may enter into transactions to acquire other businesses, products or technologies or enter into strategic partnerships, including licensing. If we do identify suitable acquisition or partnership candidates, we may not be able to make such acquisitions or partnerships on favorable terms, or at all. Any acquisitions or partnerships we make may not strengthen our competitive position, and these transactions may be viewed negatively by customers or investors. We may decide to incur debt in connection with an acquisition or issue our common stock or other equity securities to the stockholders of the acquired company, which would reduce the percentage ownership of our existing stockholders. For example, our Credit Facility may restrict our ability to pursue certain mergers, acquisitions or consolidations without obtaining the prior consent of Hercules and SVB or repaying our outstanding loan amounts.
We could incur losses resulting from undiscovered liabilities of the acquired business or partnership that are not covered by the indemnification we may obtain from the seller or our partner. In addition, we may not be able to successfully integrate any acquired personnel, technologies and operations into our existing business in an effective, timely and non-disruptive manner. Acquisitions or partnerships may also divert management attention from day-to-day responsibilities, lead to a loss of key personnel, increase our expenses and reduce our cash available for operations and other uses. We cannot predict the number, timing or size of future acquisitions or partnerships or the effect that any such transactions might have on our operating results.
Our ability to use our net operating loss carryforwards and certain other tax attributes may be limited.
We have incurred substantial losses during our history which we expect to continue, we do not expect to become profitable in the near future, and we may never achieve profitability. Under Sections 382 and 383 of the Internal Revenue Code of 1986, as amended (the “Code”), if a corporation undergoes an “ownership change,” generally defined as a greater than 50 percentage point change (by value) in its equity ownership by certain stockholders over a three-year period, the corporation’s ability to use its pre-change net operating loss carryforwards (“NOLs”), and other pre-change tax attributes (such as research tax credits) to offset its post-change income or taxes may be limited. We have not yet completed an ownership change analysis. If a requisite ownership change occurs, the amount of remaining tax attribute carryforwards available to offset taxable income and reduce income tax expense in future years may be restricted or eliminated. Similar provisions of state tax law may also apply to limit our use of accumulated state tax attributes. In addition, at the state level, there may be periods during which the use of NOLs is suspended or otherwise limited, which could accelerate or permanently increase state taxes owed. As a result, even if we attain profitability, we may be unable to use a material portion of our NOLs and other tax attributes, which could adversely affect our future cash flows.
We may be subject to adverse legislative or regulatory tax changes that could negatively impact our financial condition.
The rules dealing with U.S. federal, state, and local income taxation are complex and are constantly under review by legislators, the U.S. Treasury Department, and the Internal Revenue Service. Changes to tax laws (which may have retroactive application) have occurred and are likely to continue to occur in the future, which could adversely affect our shareholders.
Risks Related to Reliance on Third Parties
We rely on third parties to conduct our clinical trials and perform some of our research and preclinical studies. If these third parties do not satisfactorily carry out their contractual duties or fail to meet expected deadlines, our development programs may be delayed or subject to increased costs, each of which may have an adverse effect on our business and prospects.
We do not have the ability to independently conduct our clinical trials. We currently rely on third parties, such as CROs, clinical data management organizations, medical institutions and clinical investigators, to conduct our current and planned clinical trials of TP-03, TP-04 and TP-05 and other product candidates, and we expect to continue to rely upon third parties to conduct additional clinical trials of potential future product candidates. Third parties have a significant role in the conduct of our clinical trials and the subsequent collection and analysis of data. These third parties are not our employees, and except for remedies available to us under our agreements with such third party, we have limited ability to control the amount or timing of resources that any such third party will devote to our clinical trials. Some of these third parties may terminate their
engagements with us at any time. If we need to enter into alternative arrangements with a third party, it would delay our development activities.
Our reliance on these third parties for such development activities will reduce our control over these activities but will not relieve us of our regulatory responsibilities. For example, we will remain responsible for ensuring that each of our clinical trials is conducted in accordance with the general investigational plan and protocols for the trial. Moreover, the FDA requires us to comply with GCP standards, regulations for conducting, recording and reporting the results of clinical trials to assure that data and reported results are credible and accurate and that the rights, integrity and confidentiality of trial participants are protected. The EC also requires us to comply with similar standards. Regulatory authorities enforce these GCP requirements through periodic inspections of trial sponsors, principal investigators and trial sites. If we or any of our CROs fail to comply with applicable GCP requirements, the clinical data generated in our clinical trials may be deemed unreliable and the FDA, EC or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. We cannot assure you that upon inspection by a given regulatory authority, such regulatory authority will determine that any of our clinical trials comply with GCP or other applicable regulations. In addition, our clinical trials must be conducted with product produced under current applicable cGMP regulations. Our failure to comply with these regulations may require us to repeat clinical trials, which would delay the marketing approval process. We also are required to register certain ongoing clinical trials and post the results of certain completed clinical trials on a government-sponsored database, ClinicalTrials.gov, within certain timeframes. Failure to do so can result in fines, adverse publicity and civil and criminal sanctions.
The third parties we rely on for these services may also have relationships with other entities, some of which may be our competitors. In addition, the operations of our CROs and other third-party service providers may be constrained or disrupted by the ongoing COVID-19 pandemic.health epidemics. If these third parties do not successfully carry out their contractual duties, meet expected deadlines or conduct our clinical trials in accordance with regulatory requirements or our stated protocols, we will not be able to obtain, or may be delayed in obtaining, marketing approvals for our product candidates and will not be able to, or may be delayed in our efforts to, successfully commercialize our product candidates.
If any of our relationships with these third parties terminate, we may not be able to enter into arrangements with alternative third parties or do so on commercially reasonable terms. Switching or adding additional CROs, investigators and other third parties involves additional cost and requires management time and focus. In addition, there is a natural transition period when a new CRO commences work. As a result, delays can occur, which could materially impact our ability to meet our desired clinical development timelines. The COVID-19 pandemic and government measures taken in response have also had a significant impact on many CROs. Although we plan to carefully manage our relationships with our CROs, investigators and other third parties, we may nonetheless encounter challenges or delays in the future, which could have a material and adverse impact on our business, financial condition and prospects.
In addition, principal investigators for our clinical trials may serve as scientific advisors or consultants to us from time to time and receive compensation in connection with such services. Under certain circumstances, we may be required to report some of these relationships to the FDA. The FDA may conclude that a financial relationship between us and a principal investigator has created a conflict of interest or otherwise affected interpretation of the trial. The FDA may therefore question the integrity of the data generated at the applicable clinical trial site and the utility of the clinical trial itself may be jeopardized. This could result in a delay in approval, or rejection, of our marketing applications by the FDA and may ultimately lead to the denial of marketing approval of any product candidates.
We contract with third parties for the commercial manufacture of XDEMVY and for the manufacture of our product candidates for preclinical studies, clinical trials and for eventual commercialization. This reliance on third parties increases the risk that we will not have sufficient quantities of XDEMVY or our product candidates or compounds or that such supply will not be available to us at an acceptable cost, which could delay, prevent or impair our developmentcommercialization or commercializationdevelopment efforts.
We do not have any, and have no plans to acquire any, manufacturing facilities. We produce in our laboratory relatively small quantities of compounds for evaluation in our research programs. We rely, and expect to continue to rely, on third parties for the commercial manufacture of XDEMVY and the manufacture of our product candidates for preclinical and clinical testing, as well as for commercial manufacture if any of our product candidates, areif approved. We currently have limited manufacturing arrangements and expect that XDEMVY and each of our product candidates will only be covered by single source suppliers for the foreseeable future. For example, we purchase our API for XDEMVY, lotilaner, from Elanco, who sources through a single source supplier. This reliance increases the risk that we will not have sufficient quantities of XDEMVY or our product candidates or any future approved products, if approved, or such quantities at an acceptable cost or quality, which could delay, prevent or impair our developmentcommercialization or commercializationdevelopment efforts.
Furthermore, all entities involved in the preparation of XDEMVY for commercial sale or other therapeutics for clinical trials or commercial sale, including our existing contract manufacturers for XDEMVY and our product candidates, are subject to extensive regulation. Components of a finished therapeutic product approved for commercial sale or used in clinical trials must be manufactured in accordance with cGMP requirements. These regulations govern manufacturing processes and procedures, including record keeping, and the implementation and operation of quality systems to control and assure the quality of XDEMVY, investigational products and future products approved for sale. Poor control of production processes can lead to the introduction of contaminants, or to inadvertent changes in the properties or stability of XDEMVY or our product candidates that may not be detectable in final product testing. We or our contract manufacturers must supply all necessary documentation in support of an NDA on a timely basis and must adhere to the FDA’s Good Laboratory Practice regulations and cGMP regulations enforced by the FDA through its facilities inspection program. Foreign regulatory authorities, including the European Commission and the competent authorities of the EU Member States, may require compliance with similar requirements. The facilities and quality systems of our third-party contractor manufacturers must pass a pre-approval inspection for compliance with the applicable regulations as a condition of marketing approval of our product candidates. We do not control the manufacturing process of, and are completely dependent on, our contract manufacturing partners for compliance with cGMP regulations. We have little or no control over the production processes of third-party manufacturers, CMOs or other suppliers. The third-party manufacturing facilities used in the production of API and our drug products are located outside of the U.S. and require FDA approval, which our third-party manufacturers may have limited experience with obtaining. Our CMOs and other suppliers are subject to inspection by the FDA and may receive observations that they may not be able to resolve in a timely or effective manner, which could impact whether our products can be approved on a timely basis, if at all.
In the event that any of our manufacturers fails to comply with such requirements or to perform its obligations to us in relation to quality, timing or otherwise, or if our supply of XDEMVY, components or other materials becomes limited or interrupted for other reasons, we may be forced to manufacture theXDEMVY or other materials ourselves, for which we currently do not have the capabilities or resources, or enter into an agreement with another third party, which we may not be able to do on commercially reasonable terms, if at all. In particular, any replacement of our manufacturers could require significant effort and expertise because there may be a limited number of qualified replacements. In some cases, the technical skills or technology required to manufacture XDEMVY or our product candidates may be unique or proprietary to the original manufacturer and we may have difficulty transferring such skills or technology to another third party and a feasible alternative may not exist. These factors would increase our reliance on such manufacturer or require us to obtain a license from such manufacturer in order to have another third party manufacture XDEMVY or our product candidates. If we elect to or are required to change manufacturers for any reason, we will be required to verify that the new manufacturer maintains facilities and procedures that comply with quality standards and with all applicable regulations and guidelines. If any of our current contract manufacturers cannot perform as agreed, we may be required to replace such manufacturers. Although we believe that there are several potential alternative manufacturers who could manufacture XDEMVY or our product candidates, we may incur added costs and delays in identifying and qualifying any such replacement or be unable to reach agreement with an alternative manufacturer. In addition, the COVID-19 pandemic may impact our ability to procure sufficient supplies for the development of our product candidates. The extent of this impact will depend on the severity and duration of the spread of the virus, and the actions undertaken to contain COVID-19 or treat its effects. The delays associated with the verification of a new manufacturer could negatively affect our ability to develop product candidates in a timely manner or within budget.
Our or a third party’s failure to execute on our manufacturing requirements, to do so on commercially reasonable terms and comply with cGMP could adversely affect our business in a number of ways, including:
•an inability to meet commercial demands for XDEMVY or any other future product that is approved;
•requirements to cease development or to recall batches of XDEMVY or our product candidates;
•an inability to initiate or continue clinical trials of our product candidates under development;
•delay in submitting regulatory applications, or receiving marketing approvals, for our product candidates;
•loss of the cooperation of an existing or future collaborator, including by Elanco under the license agreements with Elanco; and
•subjecting third-party manufacturing facilities or our manufacturing facilities to additional inspections by regulatory authorities;
•requirements to cease development or to recall batches of our product candidates; and
•in the event of approval to market and commercialize our product candidates, an inability to meet commercial demands for our product or any other future product candidates.authorities.
OurXDEMVY, our product candidates and any future products that we may develop may compete with other products and product candidates and products for access to manufacturing facilities. As a result, we may not obtain access to these facilities on a priority basis or at all. There are a limited number of manufacturers that operate under cGMP regulations and that might be capable of manufacturing for us. Any performance failure on the part of our existing or future manufacturers could prevent or delay commercialization efforts of XDEMVY or any future products, if approved, clinical development of product candidates or marketing approval.approval of current or future product candidates.
We or our third-party manufacturers may encounter shortages in the raw materials or active pharmaceutical ingredients ("APIs") necessary to produce XDEMVY or our product candidates in the quantities needed for our clinical trials or, if our product candidates are approved, in sufficient quantities for our commercialization or to meet an increase in demand, or for our clinical trials, as a result of capacity constraints or delays or disruptions in the market for the raw materials or active pharmaceutical ingredients,APIs, including shortages caused by the purchase of such raw materials or active pharmaceutical ingredientsAPIs by our competitors or others. The failure of us or our third-party manufacturers to obtain the raw materials or active pharmaceutical ingredientsAPIs necessary to manufacture sufficient quantities of XDEMVY or our product candidates, may have a material adverse effect on our business.
If any of our product candidates are approved for marketing and commercialization and we are unable to establish sales, marketing and distribution capabilities or enter into agreements with third parties to sell, market and distribute our product candidates, we will be unable to successfully commercialize our product candidates if and when they are approved.
We have no sales, marketing or distribution capabilities or experience. To achieve commercial success for any approved product for which we retain sales and marketing responsibilities, we must either develop a sales and marketing organization, which would be expensive and time consuming, or outsource these functions to other third parties, or use a hybrid model incorporating both of these approaches.
There are risks involved with both establishing our own sales and marketing capabilities and entering into arrangements with third parties to perform these services. For example, recruiting and training a sales force is expensive and time consuming and could delay any product launch. If the commercial launch of a product candidate for which we recruit a sales force and establish marketing capabilities is delayed or does not occur for any reason, we would have prematurely or unnecessarily incurred these commercialization expenses. This may be costly, and our investment would be lost if we cannot retain or reposition our sales and marketing personnel.
Factors that may inhibit our efforts to commercialize our medicines on our own include:
•our inability to recruit, train and retain adequate numbers of effective sales, marketing, coverage or reimbursement, customer service, medical affairs and other support personnel;
•our inability to equip sales personnel with effective materials, including medical and sales literature to help them educate ECPs regarding the indications we are targeting and our products, if approved;
•the inability of sales personnel to obtain access to ECPs or persuade adequate numbers of ECPs to prescribe any future medicines;
•the inability of reimbursement professionals to negotiate arrangements for formulary access, reimbursement and other acceptance by payors;
•the lack of complementary medicines to be offered by sales personnel, which may put us at a competitive disadvantage relative to companies with more extensive product lines;
•the inability to price our products at a sufficient price point to ensure an adequate and attractive level of profitability; and
•unforeseen costs and expenses associated with creating an independent sales and marketing organization.
If we enter into arrangements with third parties to perform sales, marketing and distribution services, our product revenue or the profitability of these product revenue to us are likely to be lower than if we were to market and sell any medicines that we develop ourselves. In addition, we may not be successful in entering into arrangements with third parties to
sell and market our product candidates or may be unable to do so on terms that are favorable to us. In entering into third-party marketing or distribution arrangements, any revenue we receive will depend upon the efforts of the third parties and we cannot assure you that such third parties will establish adequate sales and distribution capabilities or devote the necessary resources and attention to sell and market our medicines effectively. If we do not establish sales and marketing capabilities successfully, either on our own or in collaboration with third parties, we will not be successful in commercializing our product candidates.
We, or our third-party manufacturers, may be unable to successfully scale-up manufacturing of XDEMVY or our product candidates in sufficient quality and quantity, which would delay or prevent us from commercializing, conducting clinical trials and developing our product candidates.
In order to successfully commercialize XDEMVY and to conduct clinical trials of our product candidates, we will need to manufacture themXDEMVY and our product candidates in large quantities. We, or our manufacturing partners, may be unable to maintain or successfully increase the manufacturing capacity for XDEMVY or any of our product candidates in a timely or cost-effective manner, or at all. In addition, quality issues may arise during scale-up activities. If we, or our manufacturing partners, are unable to successfully scale up the manufacture of XDEMVY or our product candidates in sufficient quality and quantity, the commercialization of XDEMVY or the development, testing and clinical trials of that product candidate may be delayed or become infeasible, and commercialization of XDEMVY or marketing approval or commercial launch of any resulting product may be delayed or not obtained, which could significantly harm our business.
Changes in methods of product candidate manufacturing or formulation may result in additional costs or delay.
As product candidates progress through preclinical to late stage clinical trials to marketing approval and commercialization, it is common that various aspects of the development program, such as manufacturing methods and formulation, are altered along the way in an effort to optimize yield, manufacturing batch size, minimize costs and achieve consistent quality and results. Such changes carry the risk that they will not achieve these intended objectives. Any of these changes could cause our product candidates to perform differently and affect the results of planned clinical trials or other future clinical trials conducted with the altered materials. This could delay completion of clinical trials, require the conduct of bridging clinical trials or the repetition of one or more clinical trials, increase clinical trial costs, delay approval of our product candidates and jeopardize our ability to commercialize our product candidates and generate revenue.
Risks Related to Intellectual Property
Changes in patent law in the U.S. and other jurisdictions could diminish the value of patents in general, thereby impairing our ability to protect XDEMVY or our product candidates.
As is the case with other biopharmaceutical companies, our success is heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the biopharmaceutical industry involves both technological and legal complexity and is therefore costly, time-consuming and inherently uncertain. Changes in either the patent laws or interpretation of the patent laws in the U.S. could increase the uncertainties and costs. Recent patent reform legislation in the U.S. and other countries, including the Leahy-Smith America Invents Act (the “Leahy-Smith Act”), signed into law on September 16, 2011, could increase those uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents. The Leahy-Smith Act includes a number of significant changes to U.S. patent law. These include provisions that affect the way patent applications are prosecuted, redefine prior art and provide more efficient and cost-effective avenues for competitors to challenge the validity of patents. These include allowing third-party submission of prior art to the U.S. Patent and Trademark Office (“USPTO”) during patent prosecution and additional procedures to attack the validity of a patent by USPTO administered post-grant proceedings, including post-grant review, inter partes review, and derivation proceedings. After March 2013, under the Leahy-Smith Act, the U.S. transitioned to a first inventor to file system in which, assuming that the other statutory requirements are met, the first inventor to file a patent application will be entitled to the patent on an invention regardless of whether a third party was the first to invent the claimed invention. However, the Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents, all of which could have a material adverse effect on our business, financial condition, results of operations and prospects.
The U.S. Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. Depending on future actions by the U.S. Congress, the U.S. courts, the USPTO and the relevant law-making bodies in other countries, the laws and regulations governing patents could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce our existing patents and patents that we might obtain in the future.
The U.S. federal government retains certain rights in inventions produced with its financial assistance under the Bayh-Dole Act. The federal government retains a nonexclusive, nontransferable, irrevocable, paid-up license for its own benefit. The Bayh-Dole Act also provides federal agencies with “march-in rights”. March-in rights allow the government, in specified circumstances, to require the contractor or successors in title to the patent to grant a nonexclusive, partially exclusive, or exclusive license to a responsible applicant or applicants. If the patent owner refuses to do so, the government may grant the license itself. If, in the future, we co-own or license in technology that is critical to our business that is developed in whole or in part with federal funds subject to the Bayh-Dole Act, our ability to enforce or otherwise exploit patents covering such technology may be adversely affected.
Additionally, the new unitary patent system that came into effect in Europe in June 2023 has increased the complexity and uncertainty of European patent laws and would significantly impact European patents, including those granted before the introduction of such a system. Under the unitary patent system, European applications will have the option, upon grant of a patent, of becoming a Unitary Patent which will be subject to the jurisdiction of the Unitary Patent Court (“UPC”). As the UPC is a new court system, there is no precedent for the court, increasing the uncertainty of any litigation. Patents granted before the implementation of the UPC will have the option of opting out of the jurisdiction of the UPC and remaining as national patents in the UPC countries. Patents that remain under the jurisdiction of the UPC will be potentially vulnerable to a single UPC-based revocation challenge that, if successful, could invalidate the patent in all countries who are signatories to the UPC. We cannot predict with certainty the long-term effects of any potential changes.
The development and commercialization of our products, including our lead product candidate,XDEMVY, for the treatment of Demodex blepharitis, TP-03 for the potential treatment of Demodex blepharitis and MGD, TP-04 for the potential treatment of rosacea and TP-05 for potential Lyme disease prophylaxis and community malaria reduction, is dependent on intellectual property we license from Elanco. If we breach our agreements with Elanco or the agreements are terminated, we could lose license rights that are important to our business.
Pursuant to the Eye and Derm Elanco Agreement and the All Human Uses Elanco Agreement each,(each an Elanco Agreement,"Elanco Agreement" and together the "Elanco Agreements") we acquired exclusive, worldwide, sublicensable licenses to certain intellectual property of Elanco for the development, marketing and commercialization of lotilaner for (i) the treatment, prevention, palliation or cure of any eye or skin disease or condition in humans and (b) all other applications in humans, respectively. The Eye and Derm Elanco Agreement and the All Human Uses Elanco AgreementAgreements impose various development, regulatory, commercial diligence, financial and other obligations on us. If we fail to comply with our obligations under the Elanco Agreements, or otherwise materially breach either Elanco Agreement, and fail to remedy such failure or cure such breach within 60 days, Elanco will have the right to terminate the applicable Elanco Agreement. If we fail to meet any milestones by the achievement deadlines set forth in either Elanco Agreement for any reason other than those outside of our reasonable control, and such milestones remain unmet for 120 days after Elanco notifies us thereof, Elanco may terminate the applicable Elanco Agreement.
If we fail to meet certain dermatological milestones by the achievement deadlines set forth in the Eye and Derm Elanco Agreement for any reasons other than those outside of our reasonable control, and such milestones remain unmet for 120 days after Elanco notifies us thereof, Elanco may limit our field of use under the Eye and Derm Elanco Agreement to the treatment, palliation, prevention or cure of eye diseases or conditions in humans only. If either Elanco Agreement is terminated or if our field of use in the Eye and Derm Elanco Agreement is reduced to eye and skin conditions only by Elanco, we would lose our applicable license in the country where such license was terminated and all rights therein to the licensed intellectual property would revert to Elanco. The loss of the license from Elanco would prevent us from developing and commercializing TP-03, TP-04 and TP-05 in any country where the license is terminated and could subject us to claims of breach of contract and patent infringement by Elanco if any continued research, development, manufacture or commercialization of TP-03, TP-04 or TP-05 is covered by the affected patents. If Elanco terminates the Eye and Derm Elanco Agreement for our failure to achieve a development milestone by the specified achievement deadline, then we must grant Elanco a non-exclusive, sublicensable, royalty freeroyalty-free license to our patents and know-how relating to lotilaner to develop, manufacture and commercialize lotilaner and
any licensed products for the treatment, palliation, prevention or cure of any eye or skin disease or condition in humans. If Elanco terminates the All Human Uses Elanco Agreement for our failure to achieve a development milestone by the specified achievement deadline, then we must grant Elanco a non-exclusive, sublicensable, royalty freeroyalty-free license to our patents and know-how relating to lotilaner to develop, manufacture and commercialize lotilaner and any licensed products for all applications in humans other than the treatment, palliation, prevention or cure of any eye or skin disease or condition. Accordingly, the loss of our license or the termination of our license for skin diseases and conditions or of our license for other use in humans with Elanco would materially harm our business.
If we are unable to obtain and maintain sufficient intellectual property protection for XDEMVY or our product candidates, or if the scope of the intellectual property protection is not sufficiently broad, our competitors could develop and commercialize products similar or identical to ours, and our ability to successfully commercialize our products may be adversely affected.
We rely upon a combination of patents, trademarks, trade secret protection, and confidentiality agreements to protect the intellectual property related to XDEMVY, our development programs and product candidates. Our success depends
in large part on our ability to obtain and maintain patent protection in the United StatesU.S. and other countries with respect to XDEMVY, our product candidates and research programs. We seek to protect our proprietary position by filing patent applications in the United StatesU.S. and abroad related to our novel discoveries and technologies that are important to our business. Our pending and future patent applications may not result in patents being issued whichthat protect XDEMVY or our product candidates or their intended uses or whichthat effectively prevent others from commercializing competitive technologies, products or product candidates.
Obtaining and enforcing patents is expensive and time-consuming, and we may not be able to file and prosecute all necessary or desirable patent applications, or maintain and/or enforce patents that may issue based on our patent applications, at a reasonable cost or in a timely manner. Moreover, in some circumstances, we do not have the right to control the preparation, filing and prosecution of patent applications, or to maintain, enforce and defend the patents, covering technology that we license from third parties. It is also possible that we will fail to identify patentable aspects of our research and development results before it is too late to obtain patent protection. Although we enter into non-disclosure and confidentiality agreements with parties who have access to patentable aspects of our research and development output, such as our employees, corporate collaborators, outside scientific collaborators, contract research organizations, contract manufacturers, consultants, advisors and other third parties, any of these parties may breach these agreements and disclose such results before a patent application is filed, thereby jeopardizing our ability to seek patent protection.
The patent position of biotechnology and pharmaceutical companies generally is highly uncertain, involves complex legal and factual questions and has in recent years been the subject of much litigation, resulting in court decisions, including Supreme Court decisions, which have increased uncertainties as to the ability to enforce patent rights in the future. In addition, the scope of patent protection outside of the United StatesU.S. is uncertain and laws of foreign countries may not protect our rights to the same extent as the laws of the United States,U.S., or vice versa. For example, European patent law restricts the patentability of methods of treatment of the human body more than United StatesU.S. law does. With respect to both owned and in-licensed patent rights, we cannot predict whether the patent applications we and our licensors are currently pursuing or will pursue will issue as patents in any particular jurisdiction or whether the claims of any issued patents will provide sufficient protection from competitors.
Further, we may not be aware of all third-party intellectual property rights potentially relating to XDEMVY or our product candidates or their intended uses, and as a result the impact of such third-party intellectual property rights upon the patentability of our own patents and patent applications, as well as the impact of such third-party intellectual property upon our ability to commercialize our products, is highly uncertain. Because we have not yet conducted a formal freedompatent landscape analysis related to operate analysis for patents related toXDEMVY or our product candidates, we may not be aware of issued patents that a third party might assert are infringed by XDEMVY or one of our current or future product candidates, which could materially impair our ability to commercialize XDEMVY or our product candidates. Even if we diligently search third-party patents for potential infringement by our products or product candidates, including XDEMVY, TP-03, TP-04 or TP-05, we may not successfully find patents that our products or product candidates, including XDEMVY, TP-03, TP-04 or TP-05, may infringe. If we are unable to secure and maintain freedom to operate,confirm that our products do not infringe third-party patents, others could preclude us from commercializing XDEMVY or our product candidates. In addition, publications of discoveries in the scientific literature often lag behind the actual discoveries and patent applications in the United StatesU.S. and other jurisdictions are typically not published until 18 months after filing or, in some cases, not published at all. Therefore, we cannot know with certainty whether we were the first to make the inventions claimed in our patents or pending patent applications, or that we were the first to file for patent protection of such inventions. As a result, the issuance, scope, validity, enforceability and commercial value of our patent rights are highly uncertain. Our patents or pending patent applications may be challenged in the courts or patent offices in the United StatesU.S. and abroad. For example, we may be subject to a third party pre-issuance submission of prior art to the U.S. Patent and Trademark Office, or become involved in post-grant review or interference procedures, oppositions, derivations, revocations, reexaminations, or inter partes review proceedings, in the United StatesU.S. or elsewhere, challenging our patent rights or the patent rights of others. An adverse determination in any such submission, proceeding or litigation could reduce the scope of, or
invalidate, our patent rights, allow third parties to commercialize our technology or product candidates and compete directly with us, without payment to us, or result in our inability to manufacture or commercialize drugs without infringing third-party patent rights. If the breadth or strength of protection provided by our patents and patent applications is threatened, regardless of the outcome, it could dissuade companies from collaborating with us to license, develop or commercialize XDEMVY or our current or future product candidates.
Our owned and licensed patent estate includes patent applications, many of which are at an early stage of prosecution. The coverage claimed in a patent application can be significantly reduced before the patent is issued, and its scope can be reinterpreted after issuance. Even if our owned and in-licensed patent applications issue as patents, they may not issue in a form that will provide us with any meaningful protection, prevent competitors from competing with us or otherwise provide us with any competitive advantage. The issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability, and our owned and in-licensed patents may be challenged in the courts or patent offices in the United StatesU.S. and abroad. Such challenges may result in loss of exclusivity or freedomability to operatesell our products without infringing third-party patents or in patent claims being narrowed, invalidated, or held unenforceable, in whole or in part, which could limit our ability to stop others from using or commercializing similar or identical technology and products, or limit the duration of the patent protection of our technology and products. In addition, given the amount of time required for the development, testing and regulatory review of
new product candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. Furthermore, our competitors may be able to circumvent our patents by developing similar or alternative technologies or products in a non-infringing manner. As a result, our owned and in-licensed patent portfolio may not provide us with sufficient rights to exclude others from commercializing technology and products similar or identical to any of our technology and product candidates.
Furthermore, while we seek to protect the trademarks we use in the United StatesU.S. and in other countries, we may be unsuccessful in obtaining registrations and/or otherwise protecting these trademarks. If that were to happen, we may be prevented from using our names, brands and trademarks unless we enter into appropriate royalty, license or coexistence agreements, which may not be available or may not be available on commercially reasonable terms. Over the long term, if we are unable to establish name recognition based on our trademarks, trade names, service marks and domain names, then we may not be able to compete effectively, resulting in a material adverse effect on our business. Our registered or unregistered trademarks or trade names may be challenged, infringed, diluted or declared generic, or determined to be infringing on other marks. We rely on both registration and common law protection for our trademarks. We may not be able to protect our rights to these trademarks and trade names or may be forced to stop using these names, which we need to build name recognition among potential partners or customers in our markets of interest. At times, competitors may adopt trademarks and trade names similar to ours, thereby impeding our ability to build brand identity and possibly leading to market confusion. During trademark registration proceedings, we may receive rejections. Although we would be given an opportunity to respond to those rejections, we may be unable to overcome such rejections. In addition, in the United States Patent and Trademark Office (“USPTO”)USPTO and in comparable agencies in many foreign jurisdictions, third parties are given an opportunity to oppose pending trademark applications and to seek to cancel registered trademarks. Opposition or cancellation proceedings may be filed against our trademarks, and our trademarks may not survive such proceedings. Effective trademark protection may not be available or may not be sought in every country in which our products are made available. Any name we propose to use for our products in the United StatesU.S. must be approved by the FDA, regardless of whether we have registered it, or applied to register it, as a trademark. The FDA typically conducts a review of proposed product names, including an evaluation of potential for confusion with other product names. If the FDA objects to any of our proposed product names, we may be required to expend significant additional resources in an effort to identify a usable substitute name that would qualify under applicable trademark laws, that does not infringe the existing rights of third parties and that is acceptable to the FDA. If we are unable to establish name recognition based on our trademarks and trade names, we may not be able to compete effectively and our business may be adversely affected.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees, renewal fees, annuities fees and various other governmental fees on patents and/or patent applications are due to be paid to the USPTO and foreign patent agencies in several stages over the lifetime of the patent and/or patent application. The USPTO and various foreign governmental patent agencies also require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. In certain circumstances, we rely on our licensing partners to pay these fees to, or comply with the procedural and documentary rules of, the relevant patent agency While an inadvertent lapse can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Non-compliance events that could result in abandonment or lapse of a patent or patent application include, but are not limited to, failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal
documents. In such an event, potential competitors might be able to enter the market with similar or identical products or technology. If we or our licensors fail to maintain the patents and patent applications relating to XDEMVY or our product candidates, our competitive position, business, financial condition, results of operations and prospects would be adversely affected.
We may not identify relevant third-party patents or may incorrectly interpret the relevance, scope or expiration of a third-party patent which might adversely affect our ability to develop and market our products.
We cannot guarantee that any of our patent searches or analyses, including the identification of relevant patents, the scope of patent claims or the expiration of relevant patents, are complete or thorough, nor can we be certain that we have identified each and every third party patent and pending application in the United StatesU.S. and abroad that is relevant to or necessary for the commercialization of XDEMVY or our product candidates in any jurisdiction. Because we have not yet conducted a formal freedompatent landscape analysis related to operate analysis for patents related toXDEMVY or our product candidates, we may not be aware of issued patents that a third party might assert are infringed by one of XDEMVY or our current or future product candidates, which could materially impair our ability to commercialize XDEMVY or our product candidates. Even if we diligently search third-party patents for potential infringement by our products, including XDEMVY, or product candidates, we may not successfully find patents that our
products or product candidates may infringe. If we are unable to secure and maintain freedom to operate,confirm that our products, including XDEMVY, do not infringe third-party patents, others could preclude us from commercializing XDEMVY or our product candidates.
The scope of a patent claim is determined by an interpretation of the law, the written disclosure in a patent and the patent’s prosecution history. Our interpretation of the relevance or the scope of a patent or a pending application may be incorrect, which may negatively impact our ability to market our products. We may incorrectly determine that our products are not covered by a third-party patent or may incorrectly predict whether a third party’s pending application will issue with claims of relevant scope. Our determination of the expiration date of any patent in the United StatesU.S. or abroad that we consider relevant may be incorrect, which may negatively impact our ability to develop and market XDEMVY or our product candidates. Our failure to identify and correctly interpret relevant patents may negatively impact our ability to develop and market our products.products, including XDEMVY.
We may wish to acquire rights to future assets through in-licensing or may attempt to form collaborations in the future with respect to XDEMVY or our product candidates, but may not be able to do so, which may cause us to alter or delay our commercialization or development and commercialization plans.
The commercialization of XDEMVY and the development and potential commercialization of our product candidates will require substantial additional capital to fund expenses. We haveIn 2019 and 2020, we entered into the Eye and Derm Elanco Agreement and the All Human Uses Elanco Agreement.Agreement, respectively. We plan to utilize these license rights in developing and marketing XDEMVY, and our TP-03,TP-04 and TP-05 product candidates. We may, in the future, decide to collaborate with other biopharmaceutical companies for the development and potential commercialization of thoseXDEMVY in other jurisdictions or our product candidates. We will face significant competition in seeking appropriate collaborators. We may not be successful in our efforts to establish a strategic partnership or other alternative arrangements for our product candidates because they may be deemed to be at too early of a stage of development for collaborative effort and third parties may not view our product candidates as having the requisite potential to demonstrate safety and efficacy. If and when we collaborate with a third party for the commercialization of XDEMVY in other jurisdictions or the development and commercialization of a product candidate, we can expect to relinquish some or all of the control over the future success of XDEMVY or that product candidate to the third party. Our ability to reach a definitive agreement for a collaboration will depend, among other things, upon our assessment of the collaborator’s resources and expertise, the terms and conditions of the proposed collaboration and the proposed collaborator’s evaluation of a number of factors. Those factors may include the following:
•the design or results of clinical trials;
•the likelihood of approval by the FDA or comparable foreign regulatory authorities;
•the potential market for the product candidate;
•the costs and complexities of manufacturing and delivering such product candidate to patients;
•the design or results of clinical trials;
•the likelihood of approval by the FDA or comparable foreign regulatory authorities;
•the potential of competing products;
•the existence of uncertainty with respect to our ownership of technology or other rights, which can exist if there is a challenge to such ownership without regard to the merits of the challenge; and
•industry and market conditions generally.
The collaborator may also consider alternative product candidates or technologies for similar indications that may be available to collaborate on and whether such a collaboration could be more attractive than the one with us for XDEMVY or our product candidate. We may also be restricted under any license agreements from entering into agreements on certain terms or at all with potential collaborators. Collaborations are complex and time-consuming to negotiate and document. In addition, there have been a significant number of recent business combinations among large pharmaceutical companies that have resulted in a reduced number of potential future collaborators and changes to the strategies of the combined company. As a result, we may not be able to negotiate collaborations on a timely basis, on acceptable terms, or at all. If we are unable to do so, we may have to curtail the development of such product candidate, reduce or delay one or more of our other development programs, delay the potential commercialization or reduce the scope of any planned sales or marketing activities for such product candidate, or increase our expenditures and undertake development, manufacturing or commercialization activities at our own expense. If we elect to increase our expenditures to fund development, manufacturing or commercialization activities on our own, we may need to obtain additional capital, which may not be available to us on acceptable terms or at all. If we do not have sufficient funds, we may not be able to further develop our product candidates or bring them to market and generate product revenue.
Collaborations that we have entered into and may enter in the future may not be successful, and any success will depend heavily on the efforts and activities of such collaborators. Collaborations pose a number of risks, including the following:
•collaborators have significant discretion in determining the amount and timing of efforts and resources that they will apply to these collaborations;
•collaborators may not perform their obligations as expected;
•collaborators may not pursue development of our product candidates or may elect not to continue or renew development programs based on results of clinical trials or other studies, changes in the collaborators’ strategic focus or available funding, or external factors, such as an acquisition or business combination, that divert resources or create competing priorities;
•collaborators may not pursue commercialization of any product or product candidates that achieve marketing approval or may elect not to continue or renew commercialization programs based on results of clinical trials or other studies, changes in the collaborators’ strategic focus or available funding, or external factors, such as an acquisition or business combination, that may divert resources or create competing priorities;
•collaborators may delay clinical trials, provide insufficient funding for a clinical trial program, stop a clinical trial or abandon a product candidate, repeat or conduct new clinical trials or require a new formulation of a product candidate for clinical testing;
•we may not have access to, or may be restricted from disclosing, certain information regarding product candidates being developed or commercialized under a collaboration and, consequently, may have limited ability to inform our stockholders about the status of such product candidates on a discretionary basis;
•collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with XDEMVY or our product candidates and products if the collaborators believe that the competitive products are more likely to be successfully developed or can be commercialized under terms that are more economically attractive than ours;
•product candidates discovered in collaboration with us may be viewed by our collaborators as competitive with their own product candidates or products, which may cause collaborators to cease to devote resources to the commercialization of our product candidates;
•a collaborator may fail to comply with applicable regulatory requirements regarding the development, manufacture, distribution or marketing of a product candidate or product;
•a collaborator may seek to renegotiate or terminate their relationship with us due to unsatisfactory clinical results, manufacturing issues, a change in business strategy, a change of control or other reasons;
•a collaborator with marketing and distribution rights to one or more of our product candidates that achieve marketing approval may not commit sufficient resources to the marketing and distribution of such product or products;
•disagreements with collaborators, including disagreements over intellectual property or proprietary rights, contract interpretation or the preferred course of development, might cause delays or terminations of the research, development or commercialization of product candidates, might lead to additional responsibilities for us with respect to product candidates, or might result in litigation or arbitration, any of which would be time-consuming and expensive;
•collaborators may not properly obtain, maintain, enforce, defend or protect our intellectual property or proprietary rights or may use our proprietary information in such a way as to potentially lead to disputes or legal proceedings that could jeopardize or invalidate our intellectual property or proprietary information or expose us to potential litigation;
•disputes may arise with respect to the ownership of intellectual property developed pursuant to our collaborations;
•collaborators may infringe, misappropriate or otherwise violate the intellectual property or proprietary rights of third parties, which may expose us to litigation and potential liability; and
•collaborations may be terminated for the convenience of the collaborator, and, if terminated, we could be required to raise additional capital to pursue further development or commercialization of the applicable product candidates.
Collaboration agreements may not lead to development or commercialization of our products or product candidates in the most efficient manner, or at all. If any collaborations that we enter into do not result in the successful development and commercialization of products or if one of our collaborators terminates its agreement with us, we may not receive any future research funding or milestone or royalty payments under the collaboration. If we do not receive the funding we expect under these agreements, our development of our product candidates could be delayed and we may need additional resources to develop our product candidates. All of the risks relating to product development, regulatory approval and commercialization described in this report also apply to the activities of our collaborators.
In the future, we may need to obtain additional licenses of third-party technology that may not be available to us or are available only on commercially unreasonable terms or we may fail to comply with our obligations under such agreements and our business could be harmed.
In addition to the Eye and Derm Elanco Agreement and the All Human Uses Elanco Agreement, from time to time we may be required to license technology from additional third parties to further develop or commercialize our product candidates. Should we be required to obtain licenses to any third-party technology, including any such patents required to manufacture, use or sell our product candidates, such licenses may not be available to us on commercially reasonable terms, or at all.
If we are unable to license such technology, or if we are forced to license such technology on unfavorable terms, our business could be materially harmed. If we are unable to obtain a necessary license, we may be unable to develop or commercialize the affected product candidates, which could materially harm our business and the third parties owning such intellectual property rights could seek either an injunction prohibiting our sales or an obligation on our part to pay royalties and/or other forms of compensation. Even if we are able to obtain a license, it may be non-exclusive, thereby giving our competitors access to the same technologies licensed to us.
If we are unable to obtain rights to required third-party intellectual property rights or maintain the existing intellectual property rights we have, we may be required to expend significant time and resources to redesign our technology, product candidates, or the methods for manufacturing them or to develop or license replacement technology, all of which may not be feasible on a technical or commercial basis. If we are unable to do so, we may be unable to develop or commercialize the affected technology and product candidates, which could harm our business, financial condition, results of operations and prospects significantly.
Additionally, if we fail to comply with our obligations under any license agreements, our counterparties may have the right to terminate these agreements, in which event we might not be able to develop, manufacture or market, or may be forced to cease developing, manufacturing or marketing, any product that is covered by these agreements or may face other penalties under such agreements. Such an occurrence could materially adversely affect the value of the product candidate being developed under any such agreement. Termination of these agreements or reduction or elimination of our rights under these agreements, or restrictions on our ability to freely assign or sublicense our rights under such agreements when it is in the interest of our business to do so, may result in our having to negotiate new or reinstated agreements with less favorable terms,
cause us to lose our rights under these agreements, including our rights to important intellectual property or technology or impede, or delay or prohibit the further development or commercialization of one or more product candidates that rely on such agreements.
If we enter into in-bound intellectual property license agreements, we may not be able to fully protect the licensed intellectual property rights or maintain those licenses. In each of the Eye and Derm Elanco Agreement and the All Human Uses Elanco Agreement, Elanco retains, and future licensors could retain, the right to prosecute and defend the intellectual property rights licensed to us, in which case we would depend on the ability of our licensors to obtain, maintain and enforce such licensed intellectual property. These licensors may determine not to pursue litigation against other companies or may pursue such litigation less aggressively than we would. If our licensors do not adequately protect such licensed intellectual property, competitors may be able to use such intellectual property and erode or negate any competitive advantage we may have, which could materially harm our business, negatively affect our position in the marketplace, limit our ability to commercialize our products and product candidates and delay or render impossible our achievement of profitability. Further, entering into such
license agreements could impose various diligence, commercialization, royalty or other obligations on us. Future licensors may allege that we have breached our license agreement with them, and accordingly seek to terminate our license, which could adversely affect our competitive business position and harm our business prospects.
In addition to the above risks, intellectual property rights that we license in the future may include sublicenses under intellectual property owned by third parties, in some cases through multiple tiers. The actions of our licensors may therefore affect our rights to use our sublicensed intellectual property, even if we are in compliance with all of the obligations under our license agreements. Should our licensors or any of the upstream licensors fail to comply with their obligations under the agreements pursuant to which they obtain the rights that are sublicensed to us, or should such agreements be terminated or amended, our ability to develop and commercialize our product candidates may be materially harmed.
Disputes may arise regarding intellectual property subject to a licensing agreement, including:
•the scope of rights granted under the license agreement and other interpretation related issues;
•the extent to which our technology and processes infringe on intellectual property of the licensor that is not subject to the licensing agreement;
•the sublicensing of patent and other rights;
•our diligence obligations under the license agreement and what activities satisfy those diligence obligations;
•the inventorship and ownership of inventions and know-how resulting from the joint creation or use of intellectual property by our licensors and us and our partners; and
•the priority of invention of patented technology.
In addition, the agreements under which we currently license intellectual property or technology from third parties are complex, and certain provisions in such agreements may be susceptible to multiple interpretations. The resolution of any contract interpretation disagreement that may arise could narrow what we believe to be the scope of our rights to the relevant intellectual property or technology, or increase what we believe to be our financial or other obligations under the relevant agreement, either of which could have a material adverse effect on our business, financial condition, results of operations and prospects. Moreover, if disputes over intellectual property that we have licensed prevent or impair our ability to maintain our current licensing arrangements on commercially acceptable terms, we may be unable to successfully develop and commercialize the affected technology and product candidates, which could have a material adverse effect on our business, financial conditions, results of operations and prospects.
We cannot ensure that patent rights relating to inventions described and claimed in our pending patent applications will issue or that our patents or patents based on our patent applications will not be challenged and rendered invalid and/or unenforceable.
WeAlthough we have pending U.S. and foreign patent applications in our portfolio, however, we cannot predict:
•if and when patents may issue based on our patent applications;
•the scope of protection of any patent issuing based on our patent applications;
•whether the claims of any patent issuing based on our patent applications will provide protection against competitors;
•whether or not third parties will find ways to invalidate or circumvent our patent rights;
•whether or not others will obtain patents claiming aspects similar to those covered byclaimed in our patents and patent applications;
•whether we will need to initiate litigation or administrative proceedings to enforce and/or defend our patent rights which will be costly whether we win or lose; and/or
•whether the patent applications that we own or in-license will result in issued patents with claims that cover our products or product candidates or uses thereof in the United StatesU.S. or in other foreign countries.
We cannot be certain that the claims in our pending patent applications directed to our products or product candidates and/or technologies will be considered patentable by the USPTO or by patent offices in foreign countries. One aspect of the determination of patentability of our inventions depends on the scope and content of the “prior art,” information that was or is deemed available to a person of skill in the relevant art prior to the priority date of the claimed invention. There may be prior art of which we are not aware that may affect the patentability of our patent claims or, if issued, affect the validity or enforceability of a patent claim. Even if the patents do issue based on our patent applications, third parties may challenge the validity, enforceability or scope thereof, which may result in such patents being narrowed, invalidated or held unenforceable. Furthermore, even if they are unchallenged, patents in our portfolio may not adequately exclude third parties from practicing relevant technology or prevent others from designing around our claims. If the breadth or strength of our intellectual property position with respect to our product candidates is threatened, it could dissuade companies from collaborating with us to develop and threaten our ability to commercialize our product candidates. In the event of litigation or administrative proceedings, we cannot be certain that the claims in any of our issued patents will be considered valid by courts in the United StatesU.S. or foreign countries.
If we are sued for infringing, misappropriating or otherwise violating intellectual property rights of third parties, such litigation could be costly and time consuming and could prevent or delay us from developing or commercializing our product candidates.
Our commercial success depends, in part, on our ability to develop, manufacture, market and sell our product candidates without infringing, misappropriating or otherwise violating the intellectual property and other proprietary rights of third parties. Third parties may allege that we have infringed or misappropriated their intellectual property. Litigation or other legal proceedings relating to intellectual property claims, with or without merit, are unpredictable and generally expensive and time consuming and, even if resolved in our favor, are likely to divert significant resources from our core business, including distracting our technical and management personnel from their normal responsibilities. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments and if securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the market price of our common stock.
Such litigation or proceedings could substantially increase our operating losses and reduce the resources available for development activities or any future sales, marketing or distribution activities. We may not have sufficient financial or other resources to adequately conduct such litigation or proceedings. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their greater financial resources and more mature and developed intellectual property portfolios. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could have a material adverse effect on our ability to compete in the marketplace.
There is a substantial amount of intellectual property litigation in the biotechnology and biopharmaceutical industries, and we may become party to, or threatened with, litigation or other adversarial proceedings regarding intellectual property rights with respect to our product candidates. Third parties may assert infringement claims against us based on existing or future intellectual property rights, regardless of merit. The pharmaceutical and biotechnology industries have produced a significant number of patents, and it may not always be clear to industry participants, including us, which patents are directed to various types of products or methods of use. As the pharmaceutical and biotechnology industries expand and more patents are issued, the risk increases that our technologies or product candidates that we may identify may be subject to claims of infringement of the patent rights of third parties. The scope of patents is subject to interpretation by the courts, and the interpretation is not always uniform. The legal threshold for initiating litigation or contested proceedings is low, so even lawsuits or proceedings with a low probability of success might be initiated and require significant resources to defend. If we were sued for patent infringement, we would need to demonstrate that our product candidates, products or methods either do
not infringe the patent claims of the relevant patent or that the patent claims are invalid or unenforceable, and we may not be able to do this. Proving invalidity may be difficult. For example, in the United States,U.S., proving invalidity in court requires a showing of clear and convincing evidence to overcome the presumption of validity enjoyed by issued patents. Even if we are successful in these proceedings, we may incur substantial costs and the time and attention of our management and scientific personnel could be diverted in pursuing these proceedings, which could have a material adverse effect on our business and operations. In addition, we may not have sufficient resources to bring these actions to a successful conclusion.
If we are found to infringe a third party’s intellectual property rights, we could be forced, including by court order, to cease developing, manufacturing or commercializing the infringing product candidate or product. Alternatively, we may be required to obtain a license from such third party in order to use the infringing technology and continue developing, manufacturing or marketing the infringing product candidate. However, we may not be able to obtain any required license on commercially reasonable terms or at all. Even if we were able to obtain a license, it could be non-exclusive, thereby giving our
competitors access to the same technologies licensed to us and could require us to make substantial licensing and royalty payments. We could be forced, including by court order, to cease developing, manufacturing and commercializing the infringing technology or product. In addition, we could be found liable for monetary damages, including treble damages and attorneys’ fees if we are found to have willfully infringed a patent. A finding of infringement could prevent us from commercializing our product candidates or force us to cease some of our business operations, which could materially harm our business. Claims that we have misappropriated the confidential information or trade secrets of third parties could have a similar negative impact on our business, financial condition, results of operations and prospects.
If we do not obtain patent term extension for any product candidates we may develop, our business may be materially harmed.
In the United States,U.S., the term of a patent that covers an FDA-approved drug may be eligible for limited patent term extension, which permits patent term restoration as compensation for the patent term lost during the FDA regulatory review process. The Drug Price Competition and Patent Term Restoration Act of 1984, also known as the Hatch-Waxman Act, permits a patent term extension of up to five years beyond the expiration of the patent. The length of the patent term extension is related to the length of time the drug is under regulatory review. Patent extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval, and only one patent applicable to an approved drug may be extended and only those claims covering the approved drug, a method for using it, or a method for manufacturing it may be extended. Similar provisions are available in Europe and certain other non-United Statesnon-U.S. jurisdictions to extend the term of a patent that covers an approved drug. While in the future, if and when our product candidates receive FDA approval, we expect tomay apply for patent term extensions on patents covering thoseXDEMVY and other product candidates that may receive FDA approval, there is no guarantee that the applicable authorities will agree with our assessment of whether such extensions should be granted, and even if granted, the length of such extensions. We may not be granted patent term extension either in the United StatesU.S. or in any foreign country because of, for example, failing to exercise due diligence during the testing phase or regulatory review process, failing to apply within applicable deadlines, failing to apply prior to expiration of relevant patents or otherwise failing to satisfy applicable requirements. Moreover, the term of extension, as well as the scope of patent protection during any such extension, afforded by the governmental authority could be less than we request. If we are unable to obtain any patent term extension or the term of any such extension is less than we request, our competitors may obtain approval of competing products following the expiration of our patent rights, and our business, financial condition, results of operations and prospects could be materially harmed.
Our internal information technology systems, or those of our third-party CROs or other contractors or consultants, may fail or suffer security breaches, loss or leakage of data, and other disruptions, which could result in a material disruption of our product candidates’ development programs, compromise sensitive information related to our business or prevent us from accessing critical information, potentially exposing us to liability or otherwise adversely affecting our business.
We are increasingly dependent upon information technology systems, infrastructure and data to operate our business. In the ordinary course of business, we collect, store and transmit confidential information (including but not limited to intellectual property, proprietary business information and personal information). It is critical that we do so in a secure manner to maintain the confidentiality and integrity of such confidential information. We also have outsourced elements of our operations to third parties, and as a result we manage a number of third-party contractors who have access to our confidential information.
Despite the implementation of security measures, given their size and complexity and the increasing amounts of confidential information that they maintain, our internal information technology systems and those of our third-party CROs and other contractors and consultants are potentially vulnerable to breakdown or other damage or interruption from service interruptions, system malfunction, natural disasters, terrorism, war and telecommunication and electrical failures, as well as security breaches from inadvertent or intentional actions by our employees, contractors, consultants, business partners, and/or other third parties, or from cyber-attacks by malicious third parties (including the deployment of harmful malware, ransomware,
denial-of-service attacks, social engineering and other means to affect service reliability and threaten the confidentiality, integrity and availability of information), which may compromise our system infrastructure or lead to data leakage. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and reputational damage and the further development and commercialization of our drug candidates could be delayed.
While we have not experienced any such system failure, accident or security breach to date, we cannot assure you that our data protection efforts and our investment in information technology will prevent significant breakdowns, data leakages, breaches in our systems or other cyber incidents that could have a material adverse effect upon our reputation, business, operations or financial condition, whether due to a loss of our trade secrets or other proprietary information or other similar disruptions. For example, if such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our programs and the development of our product candidates could be delayed. In addition, the loss of clinical trial data for our product candidates could result in delays in our marketing approval efforts and significantly increase our costs to recover or reproduce the data. Furthermore, significant disruptions of our internal information technology systems or security breaches could result in the loss, misappropriation, and/or unauthorized access, use, or disclosure of, or the prevention of access to, confidential information (including trade secrets or other intellectual property, proprietary business information, and personal information), which could result in financial, legal, business, and reputational harm to us. For example, any such event that leads to unauthorized access, use, or disclosure of personal information, including personal information regarding our clinical trial subjects or employees, could harm our reputation directly, compel us to comply with federal and/or state breach notification laws and foreign law equivalents, subject us to mandatory corrective action, and otherwise subject us to liability under laws and regulations that protect the privacy and security of personal information, including private lawsuits or class actions under the California Consumer Privacy Act, which could result in significant legal and financial exposure and reputational damages that could potentially have an adverse effect on our business.
We may become involved in lawsuits to protect or enforce our patents and other intellectual property rights, which could be expensive, time consumingtime-consuming and unsuccessful.
Competitors may infringe, misappropriate or otherwise violate our patents, trademarks, copyrights or other intellectual property. It may be difficult to detect infringers who do not advertise the components that are used in their products. Moreover, it may be difficult or impossible to obtain evidence of infringement in a competitor’s or potential competitor’s product. To counter infringement or unauthorized use, we may be required to file infringement or other intellectual property-related claims, which can be expensive and time consuming and divert the time and attention of our management and scientific personnel. There can be no assurance that we will have sufficient financial or other resources to file and pursue such infringement claims, which typically last for years before they are concluded. Any claims we assert against perceived infringers could provoke these parties to assert counterclaims against us alleging that we infringe their patents, in addition to counterclaims asserting that our patents are invalid or unenforceable, or both. In any patent infringement proceeding, there is a risk that a court will decide that a patent of ours is invalid or unenforceable, in whole or in part, and that we do not have the right to stop the other party from making, using, or selling the invention at issue. In patent litigation in the United States,U.S., defendant counterclaims alleging invalidity or unenforceability are commonplace. Grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements, including lack of novelty, obviousness, or non-enablement. Grounds for an unenforceability assertion could be an allegation that someone connected with prosecution of the patent withheld relevantmaterial information from the USPTO, or made a misleading statement, during prosecution. Third parties may institute such claims before administrative bodies in the United StatesU.S. or abroad, even outside the context of litigation. Such mechanisms include re-examination, post-grant review, inter partes review, interference proceedings, derivation proceedings, and equivalent proceedings in foreign jurisdictions (e.g., opposition proceedings). The outcome following legal assertions of invalidity and unenforceability is unpredictable. There is also a risk that, even if the validity of such patents is upheld, the court will construe the patent’s claims narrowly or decide that we do not have the right to stop the other party from making, using or selling the invention at issue on the grounds that our patent claims do not cover the invention. An adverse outcome in a litigation or proceeding involving our patents could limit our ability to assert our patents against those parties or other competitors, and may curtail or preclude our ability to exclude third parties from making, using and selling similar or competitive products. Any of these occurrences could adversely affect our competitive business position, business prospects and financial condition. Similarly, if we assert trademark infringement claims, a court may determine that the marks we have asserted are invalid or unenforceable, or that the party against whom we have asserted trademark infringement has superior rights to the marks in
question. In this case, we could ultimately be forced to cease use of such trademarks, which could materially harm our business and negatively affect our position in the marketplace.
Even if we establish infringement, the court may decide not to grant an injunction against further infringing activity and instead award only monetary damages, which may or may not be an adequate remedy. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during litigation. There also could be public announcements of
the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a material adverse effect on the price of shares of our common stock. Moreover, we cannot assure you that we will have sufficient financial or other resources to file and pursue such infringement claims, which typically last for years before they are concluded. Even if we ultimately prevail in such claims, the monetary cost of such litigation and the diversion of the attention of our management and scientific personnel could outweigh any benefit we receive as a result of the proceedings.
Because of the expense and uncertainty of litigation, we may not be in a position to enforce our intellectual property rights against third parties.
Because of the expense and uncertainty of litigation, we may conclude that even if a third party is infringing our issued patent, any patents that may be issued as a result of our pending or future patent applications or other intellectual property rights, the risk-adjusted cost of bringing and enforcing such a claim or action may be too high or not in the best interest of our company or our stockholders. In such cases, we may decide that the more prudent course of action is to simply monitor the situation or initiate or seek some other non-litigious action or solution.
Changes in patent law in the United StatesU.S. and other jurisdictions could diminish the value of patents in general, thereby impairing our ability to protect our product candidates.
As is the case with other biopharmaceutical companies, our success is heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the biopharmaceutical industry involves both technological and legal complexity and is therefore costly, time consumingtime-consuming and inherently uncertain. Changes in either the patent laws or interpretation of the patent laws in the United StatesU.S. could increase the uncertainties and costs. Recent patent reform legislation in the United StatesU.S. and other countries, including the Leahy-Smith America Invents Act (“Leahy-Smith Act”) signed into law on September 16, 2011, could increase those uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents. The Leahy-Smith Act includes a number of significant changes to U.S. patent law. These include provisions that affect the way patent applications are prosecuted, redefine prior art and provide more efficient and cost-effective avenues for competitors to challenge the validity of patents. These include allowing third-party submission of prior art to the USPTO during patent prosecution and additional procedures to attack the validity of a patent by USPTO administered post-grant proceedings, including post-grant review, inter partes review, and derivation proceedings. After March 2013, under the Leahy-Smith Act, the United StatesU.S. transitioned to a first inventor to file system in which, assuming that the other statutory requirements are met, the first inventor to file a patent application will be entitled to the patent on an invention regardless of whether a third party was the first to invent the claimed invention. However, the Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents, all of which could have a material adverse effect on our business, financial condition, results of operations and prospects.
The U.S. Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. Depending on future actions by the U.S. Congress, the U.S. courts, the USPTO and the relevant law-making bodies in other countries, the laws and regulations governing patents could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce our existing patents and patents that we might obtain in the future.
The United StatesU.S. federal government retains certain rights in inventions produced with its financial assistance under the Bayh-Dole Act. The federal government retains a “nonexclusive,nonexclusive, nontransferable, irrevocable, paid-up license”license for its own benefit. The Bayh-Dole Act also provides federal agencies with “march-in rights”. March-in rights allow the government, in specified circumstances, to require the contractor or successors in title to the patent to grant a “nonexclusive,nonexclusive, partially exclusive, or exclusive license”license to a “responsibleresponsible applicant or applicants.” If the patent owner refuses to do so, the government may grant the license itself. If, in the future, we co-own or license in technology whichthat is critical to our business that is developed in whole or in part with federal funds subject to the Bayh-Dole Act, our ability to enforce or otherwise exploit patents covering such technology may be adversely affected.
We may not be able to protect our intellectual property rights throughout the world.
Patents are of national or regional effect, and filing, prosecuting and defending patents on all of our product candidates throughout the world would be prohibitively expensive, and the laws of foreign countries may not protect our rights to the same extent as the laws of the United States.U.S. As such, we may not be able to prevent third parties from practicing our inventions in all countries outside the United States,U.S., or from selling or importing products made using our inventions in and into the United StatesU.S. or other jurisdictions. Further, the legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents, trade secrets and other intellectual property protection, particularly those
relating to pharmaceuticals or biologics, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. In addition, certain jurisdictions do not protect to the same extent or at all inventions that constitute new methods of treatment. As such, we may not be able to prevent third parties from practicing our inventions in all countries outside the United States,U.S., or from selling or importing products made using our inventions in and into the United StatesU.S. or other jurisdictions. Furthermore, certain foreign and developing countries, including China and India, have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. In those countries, we and our licensors may have limited remedies if patents are infringed or if we or our licensors are compelled to grant a license to a third party, which could materially diminish the value of those patents. This could limit our potential revenue opportunities. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
We may rely on trade secret and proprietary know how which can be difficult to trace and enforce, and if we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed.
In addition to seeking patents for some of our technology and product candidates, we may also rely on trade secrets, including unpatented know-how, technology and other proprietary information, to maintain our competitive position. Elements of our product candidate, including processes for their preparation and manufacture, may involve proprietary know-how, information, or technology that is not covered by patents, and thus for these aspects we may consider trade secrets and know-how to be our primary intellectual property. Any disclosure, either intentional or unintentional, by our employees, the employees of third parties with whom we share our facilities or third party consultants and vendors that we engage to perform research, clinical trials or manufacturing activities, or misappropriation by third parties (such as through a cybersecurity breach) of our trade secrets or proprietary information could enable competitors to duplicate or surpass our technological achievements, thus eroding our competitive position in our market.
Trade secrets and know-how can be difficult to protect. We require our employees to enter into written employment agreements containing provisions of confidentiality and obligations to assign to us any inventions generated in the course of their employment. We further seek to protect our potential trade secrets, proprietary know-how, and information in part, by entering into non-disclosure and confidentiality agreements with parties who are given access to them, such as our corporate collaborators, outside scientific collaborators, contract research organizations, contract manufacturers, consultants, advisors and other third parties. With our consultants, contractors, and outside scientific collaborators, these agreements typically include invention assignment obligations. While it is our policy to require our employees and contractors who may be involved in the conception or development of intellectual property to execute agreements assigning such intellectual property to us, we may be unsuccessful in executing an enforceable agreement with each party who in fact conceives or develops intellectual property that we regard as our own. Despite these efforts, our assignment agreements may not be self-executing and any of these parties may breach the agreements and disclose our proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for such breaches. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive and time-consuming, and the outcome is unpredictable. If we fail in bringing or defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights. Such an outcome could materially, and adversely affect our business, financial condition, results of operations, and growth prospects. Even if we are successful in defending against such claims, litigation could result in substantial costs and distraction to management and other employees. The assignment risks of this paragraph could also pertain to any intellectual property licensed-in to us. In addition, some courts inside and outside the United StatesU.S. are less willing or unwilling to protect trade secrets. If any of our trade secrets were to be lawfully obtained or independently developed by a competitor or other third party, we would have no right to prevent them from using that technology or information to compete with us. If any of our trade secrets were to be disclosed to or independently developed by a competitor or other third party, our competitive position would be harmed.
We may be subject to claims that our employees, consultants or independent contractors have wrongfully used or disclosed confidential information of third parties.
We employ individuals who were previously employed at other biotechnology or biopharmaceutical companies, or at research institutions. Although we seek to protect our ownership of intellectual property rights by ensuring that our agreements with our employees, collaborators, and other third parties with whom we do business include provisions requiring
such parties to assign rights in inventions to us, we may be subject to claims that we or our employees, consultants or independent contractors have inadvertently or otherwise used or disclosed confidential information of our employees’ former employers or other third parties. We or our licensors may also be subject to claims that former employers or other third parties have an ownership interest in our patents. Litigation may be necessary to defend against these claims. There is no guarantee of success in defending these claims, and if we or our licensors fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights, such as exclusive ownership of, or right to use, valuable intellectual property. Even if we are successful, litigation could result in substantial cost and be a distraction to our management and other employees.
Intellectual property rights do not necessarily address all potential threats to our competitive advantage.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations, and may not adequately protect our business or permit us to maintain our competitive advantage. For example:
•others may be able to make product candidates that are similar to ours but that are not covered by the claims of the patents that we own or have exclusively licensed;
•we or our licensors or future collaborators might not have been the first to make the inventions covered by the issued patent or pending patent application that we own or have exclusively licensed;
•we or our licensors or future collaborators might not have been the first to file patent applications covering certain of our inventions;
•others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our intellectual property rights;
•it is possible that our pending patent applications will not lead to issued patents;
•issued patents that we own or have exclusively licensed may be held invalid or unenforceable, as a result of legal challenges by our competitors;
•our competitors might conduct research and development activities in countries where we do not have patent rights and then use the information learned from such activities to develop competitive products for sale in our major commercial markets;
•we cannot ensure that any of our patents, or any of our pending patent applications, if issued, or those of our licensors, will include claims having a scope sufficient to protect our product candidates;
•we cannot ensure that any patents issued to us or our licensors will provide a basis for an exclusive market for our commercially viable product candidates or will provide us with any competitive advantages;
•the Supreme Court of the United States,U.S., other U.S. federal courts, Congress, the USPTO or similar foreign authorities may change the standards of patentability and any such changes could narrow or invalidate, or change the scope of, our or our licensors’ patents;
•patent terms may be inadequate to protect our competitive position on our product candidates for an adequate amount of time;
•we cannot ensure that our commercial activities or product candidates will not infringe upon the patents of others;
•we cannot ensure that we will be able to successfully commercialize our product candidates on a substantial scale, if approved, before the relevant patents that we own or license expire;
•we may choose not to file a patent in order to maintain certain trade secrets or know-how, and a third party may subsequently file a patent covering such intellectual property;
•we may not develop additional proprietary technologies that are patentable; and
•the patents of others may have an adverse effect on our business.
Should any of these events occur, they could significantly harm our business, results of operations and prospects.
Patent terms may be inadequate to protect our competitive position on our product candidates and preclinical programs for an adequate amount of time.
Patent rights are of limited duration. In the United States,U.S., if all maintenance fees are timely paid, the natural expiration of a patent is generally 20 years from its earliest U.S. non-provisional filing date. Various extensions may be available, but the life of a patent, and the protection it affords, is limited. A patent term extension based on regulatory delay may be available in the United States.U.S. However, only a single patent can be extended for each marketing approval, and any patent can be extended only once, for a single product. Moreover, the scope of protection during the period of the patent term extension does not extend to the full scope of the claim, but instead only to the scope of the product as approved. Laws governing analogous patent term extensions in foreign jurisdictions vary widely, as do laws governing the ability to obtain multiple patents from a single patent family. Given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such product candidates are commercialized. Even if patents covering our product candidates are obtained, once the patent life has expired for a product, we may be open to competition from biosimilar or generic products. Additionally, we may not receive an extension if we fail to apply within applicable deadlines, fail to apply prior to expiration of relevant patents or otherwise fail to satisfy applicable requirements. If we are unable to obtain patent term extension or restoration, or the term of any such extension is less than we request, the period during which we will have the right to exclusively market our product will be shortened and our competitors may obtain approval of competing products following our patent expiration, and our revenue could be reduced, possibly materially.
Risks Related to Government Regulation
Our industry is highly regulated by the FDA and comparable foreign regulatory agencies.authorities. We must comply with extensive, strictly enforced regulatory requirements to develop, obtain, and maintain marketing approval for XDEMVY or any of our product candidates.candidates, if approved.
AnyXDEMVY and any product candidates we develop and the activities associated with their development and commercialization, including their design, testing, manufacture,manufacturing, safety, efficacy, recordkeeping, labeling, storage, approval, advertising, promotion, sale, and distribution are very heavily regulated. We have not received approval to market any product candidates from regulatory authorities in any jurisdiction. We have only limited experience in filing and supporting the applications necessary to gain marketing approvals and have relied and expect to continue to rely on third-party contract research organizations to assist us in this process. Securing FDA or comparable foreign regulatory approval such as a marketing authorization from the European Commission or the competent authorities of the individual EU Member States requires the submission of extensive preclinical and clinical data and supporting information for each therapeutic indication to establish the product candidate’s safety and efficacy for its intended use. Securing regulatory approval also requires the submission of information about the product manufacturing process to, and inspection of manufacturing facilities by, the relevant regulatory authority. It takes years to complete the testing of a new drug and development delays and/or failure can occur at any stage of testing. Any of our present and future clinical trials may be delayed, halted, not authorized, or approval of any of our products may be delayed or may not be obtained due to any of the following:
•any preclinical study or clinical trial may fail to produce safety and efficacy results satisfactory to the FDA or comparable foreign regulatory authorities;
•preclinical and clinical data can be interpreted in different ways, which could delay, limit or prevent marketing approval;
•negative or inconclusive results from a preclinical study or clinical trial or adverse events during a clinical trial could cause a preclinical study or clinical trial to be repeated or a development program to be terminated, even if other studies or trials relating to the development program are ongoing or have been completed and were successful;
•the FDA or comparable foreign regulatory authorities can place a clinical hold on a trial if, among other reasons, it finds that subjects enrolled in the trial are or would be exposed to an unreasonable and significant risk of illness or injury;
•the facilities that we utilize, or the processes or facilities of third-party vendors, including without limitation the contract manufacturers who are or will be manufacturing drug substance and drug product for us or any potential collaborators, may not satisfactorily complete inspections by the FDA or comparable foreign regulatory authorities; and
•we may encounter delays or rejections based on changes in FDA regulations, standards or policies or the regulations, standards or policies of comparable foreign regulatory authorities during the period in which we
develop a product candidate or the period required for review of any final marketing approval before we are able to market any product candidate.
In addition, information generated during the clinical trial process is susceptible to varying interpretations that could delay, limit, or prevent marketing approval at any stage of the approval process.
Moreover, early positive preclinical or clinical trial results may not be replicated in later clinical trials. As more product candidates within a particular class of drugs proceed through clinical development to regulatory review and approval, the amount and type of clinical data that may be required by regulatory authorities may increase or change. Failure to demonstrate adequately the quality, safety and efficacy of any of our product candidates would delay or prevent marketing approval of the applicable product candidate. We cannot assure you that if clinical trials are completed, either we or our potential collaborators will submit applications for required authorizations to manufacture or market potential products or that any such application will be reviewed and approved by appropriate regulatory authorities in a timely manner, if at all. Changes in marketing approval policies during the development period, changes in or the enactment of additional statutes or regulations, or changes in regulatory review for each submitted product application, may cause delays in the approval or rejection of an application.
Changes in healthcare law and implementing regulations, as well as changes in healthcare policy, may impact our business in ways that we cannot currently predict, and may have a significant adverse effect on our business and results of operations.
In the United StatesU.S. and some foreign jurisdictions, there have been, and continue to be, several legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing approval of product candidates, restrict or regulate post-approval activities, impact pricing and reimbursement and affect our ability to profitably sell XDEMVY or any other product candidates for which we obtain marketing approval.approval and prevent or delay marketing approval of product candidates. Among policy makers and payorspayers both federally and on the state level in the United StatesU.S. and elsewhere, including in the European Union, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality and/or expanding access. In the United States,U.S., the pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by major legislative initiatives.
The Affordable Care Act of 2010 substantially changed the way healthcare is financed by both the government and private insurers, and significantly impacts the U.S. pharmaceutical industry. The Affordable Care Act, among other things: (i) introduced a new average manufacturer price definition for drugs and biologics that are inhaled, infused, instilled, implanted or injected and not generally dispensed through retail community pharmacies; (ii) increased the minimum Medicaid rebates owed by manufacturers under the Medicaid Drug Rebate Program and expanded rebate liability from fee-for-service Medicaid utilization to include the utilization of Medicaid managed care organizations as well; (iii) established a branded prescription drug fee that pharmaceutical manufacturers of branded prescription drugs must pay to the federal government; (iv) expanded the list of covered entities eligible to participate in the 340B drug pricing program; (v) established a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 70% (increased from 50% in 2019) point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D;D (which, under the IRA, will be replaced by a new manufacturer discount program starting in 2025); (vi) expanded eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to additional individuals and by adding new mandatory eligibility categories for individuals with income at or below 133% of the federal poverty level, thereby potentially increasing manufacturers’ Medicaid rebate liability; (vii) created a licensure framework for follow on biologic products; and (viii) established a Center for Medicare & Medicaid Innovation CMMI,("CMMI"), at the CentersCenter for Medicare & Medicaid Services (“CMS”("CMS") to test innovative payment and service delivery models to lower Medicare and Medicaid spending, potentially including prescription drug spending.
Since its enactment, there have been judicial challenges to certain aspects of the Affordable Care Act, as well as efforts by Congress to repeal or replace,modify, and the Trump administrationagencies to alter the implementation of, certain aspects of the Affordable Care Act. For example, Congress eliminated the TCJA includes a provision repealing, effective January 1, 2019, the tax-based shared responsibility payment imposed bytax penalty for not complying with the Affordable Care Act on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate.” On December 14, 2018, a federal district court in Texas ruled theAct’s individual mandate in the absence of the tax penalty is unconstitutional and, because it is a critical and inseverable feature of the Affordable Care Act, the remaining provisions of the Affordable Care Act are invalid as well. On December 18, 2019, the U.S. Court of Appeals for the Fifth Circuit affirmed the District Court’s ruling but remanded the case back to the District Court as to the question of severability. On March 2, 2020, the United States Supreme Court granted certiorari to review this case, which is expected to be decided by mid-2021. Additionally, the Further Consolidated Appropriations Act of 2020, Pub. L. No. 116-94 permanently eliminated, effective January 1, 2020, the Affordable Care Act -mandated “Cadillac” tax on high-cost employer-sponsoredcarry health coverage and the medical device excise tax on non-exempt
medical devices and, effective January 1, 2021, also eliminates the annual fee imposed on certain health insurance providers based on market share.insurance. Further, the Bipartisan Budget Act of 2018, among other things, amended the Affordable Care Act, effective
January 1, 2019, to increase from 50 percent to 70 percent the point-of-sale discount that is owed by pharmaceutical manufacturers who participate in Medicare Part D and to close the coverage gap in most Medicare drug plans, commonly referred to as the “donut hole.”"donut hole" (which, under the IRA, will be replaced by a new manufacturer discount program starting in 2025). In the future, Congress may consider other legislation to repeal or replacemodify elements of the Affordable Care Act or other health care reform measures, agencies may further alter their implementation of elements of the Affordable Care Act or other such measures, and other judicial challenges to elements of the Affordable Care Act or other such measures may be brought. The extent to which any such changes may impact our business or financial condition is uncertain.
It is possible that the Affordable Care Act, as currently enacted or may be amended in the future, as well as other healthcare reform measures including those that may be adopted in the future, may result in more rigorous coverage criteria, and less favorable payment methodologies, or other downward pressure on coverage and payment and the price that we receive for any approved product. Any reduction in reimbursement or restriction on coverage under Medicare or other government programs may result in a similar reduction or restriction by private payers.
Other legislative changes have been proposed and adopted since the Affordable Care Act was enacted. These changes include aggregate reductions to Medicare payments to providers of up to 2% per fiscal year pursuant to the Budget Control Act of 2011 and subsequent laws, which went into effect on April 1, 2013laws. Subsequent legislation extended the 2% reduction, generally to 2031.Sequestration is currently set at 2% and will remain in effect through 2020 unless additional Congressional action is taken, , withincrease to 2.25% for the exceptionfirst half of a temporary suspensionfiscal year 2030, to 3% for the second half of fiscal year 2030, and to 4% for the remainder of the 2% cut in Medicare payments from May 1, 2020,sequestration period that lasts through December 31, 2020, pursuant to the CARES Act signed into law in March 2020 and designed to provide financial support and resources to individuals and businesses affected by the COVID-19 pandemic.first six months of fiscal year 2031. The American Taxpayer Relief Act of 2012 (“ATRA”("ATRA") among other things, also reduced Medicare payments to several types of providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. New laws may result in additional reductions in Medicare and other healthcare funding, which may materially adversely affect customer demand and affordability for our products and related services and, accordingly, the results of our financial operations. Additional changes that may affect our business include the expansion of new programs such as Medicare payment for performance initiatives for physicians under the Medicare Access and CHIP Reauthorization Act of 2015 (“MACRA”) which first affected physician payment in 2019. At this time, itIt is unclear how the introduction of the Medicare quality payment program will impact overall physician reimbursement.
Also, thereThe Inflation Reduction Act of 2022 ("IRA") introduces several changes to the Medicare Part D benefit, including a limit on annual out-of-pocket costs and a change in manufacturer liability under the program which could negatively affect the profitability of our product candidates. The IRA sunsets the current Part D coverage gap discount program starting in 2025 and replaces it with a new manufacturer discount program. Failure to pay a discount under this new program will be subject to a civil monetary penalty. In addition, the IRA establishes a Medicare Part B inflation rebate scheme and a Medicare Part D inflation rebate scheme, under which, generally speaking, manufacturers will owe rebates if the price of a Part B or Part D drug increases faster than the pace of inflation. Failure to timely pay a Part B or D inflation rebate is subject to a civil monetary penalty. The IRA also creates a drug price negotiation program under which the prices for Medicare units of certain high Medicare spend drugs and biologics without generic or biosimilar competition will be capped by reference to, among other things, a specified non-federal average manufacturer price starting in 2026. Failure to comply with requirements under the drug price negotiation program is subject to an excise tax and/or a civil monetary penalty. This or any other legislative change could impact the market conditions for our products.
In the EU, the European Commission has published a proposal that intends to reduce the regulatory data protection period and orphan market exclusivity period for new medicinal products. Although it is currently uncertain if the proposal will be adopted in its current form and it is uncertain if and when the revised legislation would enter into force, this reform can impact our product candidates in the EU.
There has been heightened governmental scrutiny over the manner in which drug manufacturers set prices for their marketed products, which have resulted in several Congressional inquiries and proposed bills and initiatives, as well as state efforts, designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drug products. For example, at the federal level, the Trump administration’s budget for fiscal year 2021 includes a $135 billion allowance to support legislative proposals seeking to reduce drug prices, increase competition, lower out-of-pocket drug costs for patients, and increase patient access to lower-cost generic and biosimilar drugs. On March 10, 2020, the Trump administration sent “principles” for drug pricing to Congress, calling for legislation that would, among other things, cap Medicare Part D beneficiary out-of-pocket pharmacy expenses, provide an option to cap Medicare Part D beneficiary monthly out-of-pocket expenses, and place limits on pharmaceutical price increases. On July 24, 2020, President Trump signed several executive orders directed toward lowering drug prices. Individual states in the United StatesU.S. have become increasingly active in passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures. Additionally, states have established Prescription Drug Affordability Boards (or similar entities) to review high-cost drugs and, in some cases, set upper payment limits.
We expect that these and other healthcare reform measures in the future, may result in more rigorous coverage criteria and lower reimbursement, and in additional downward pressure on the price that we receive for any approved product. Any reduction in reimbursement from Medicare or other government-funded programs may result in a similar reduction in
payments from private payors.payers. The implementation of cost containment measures or other healthcare reforms may hinder us in generating revenue, attaining profitability or commercializing our drugs, once marketing approval is obtained.
In the EU, the European Commission has published a proposal that intends to reduce the regulatory data protection period for new medicinal products, which would allow generic competitors to obtain marketing authorization for generic products relying on our data earlier than under the current laws and we may be faced with earlier generic competition and lower prices for our product on the EU market. The legislative process for this reform is expected to take several years. Although it is currently uncertain if the proposal will be adopted in its current form and it is uncertain if and when the revised legislation would enter into force, this reform could impact our product candidates in the EU.
In the European Union, coverage and reimbursement status of any product candidates for which we obtain regulatory approval are provided for by the national laws of EU Member States. The requirements may differ across the EU Member States. In markets outside of the United StatesU.S. and the European Union, reimbursement and healthcare payment systems vary significantly by country, and many countries have instituted price ceilings on specific products and therapies. Also, at the national level, actions have been taken to enact transparency laws regarding payments between pharmaceutical companies and health care professionals.
We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action in the United States,U.S., the European Union or any other jurisdiction. If we or any third parties we may engage with are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we or such third parties are not able to maintain regulatory compliance, our product candidates may lose any regulatory approval that may have been obtained and we may not achieve or sustain profitability.
Our employees, independent contractors, clinical trial investigators, CROs,contract research organizations, consultants, vendors and any potential commercial partners may engage in misconduct or other improper activities, including non-compliance with regulatory standards and requirements and insider trading.
We are exposed to the risk of fraud or other misconduct by our employees, clinical trial investigators, CROs, consultants, vendors and any potential commercial partners. Misconduct by these parties could include intentional, reckless and/or negligent conduct or disclosure of unauthorized activities to us that violates: (i) FDA laws and regulations or those of comparable foreign regulatory authorities, including those laws that require the reporting of true, complete and accurate information, (ii) manufacturing standards, (iii) federal and state health and data privacy, security, fraud and abuse, government price reporting, transparency reporting requirements, and other healthcare laws and regulations in the United StatesU.S. and abroad or (iv) laws that require the true, complete and accurate reporting of financial information or data. Specifically, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Activities subject to these laws could also involve the improper use or misrepresentation of information obtained in the course of clinical trials, creation of fraudulent data in preclinical studies or clinical trials or illegal misappropriation of drug product, which could result in regulatory sanctions and cause serious harm to our reputation. It is not always possible to identify and deter misconduct by employees and other third parties, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with such laws or regulations. We adopted a code of conduct applicable to all of our employees immediately following the completion of our IPO, as well as a disclosure program and other applicable policies and procedures, but it is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. Additionally, we are subject to the risk that a person or government could allege such fraud or other misconduct, even if none occurred. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant civil, criminal and administrative penalties, damages, monetary fines, disgorgements, possible exclusion from participation in Medicare, Medicaid, other U.S. federal healthcare programs or healthcare programs in other jurisdictions, integrity oversight and reporting obligations to resolve allegations of non-compliance, individual imprisonment, other sanctions, contractual damages, reputational harm, diminished profits and future earnings, and curtailment of our operations.
If we fail to comply with environmental, health and safety laws and regulations, we could become subject to fines or penalties or incur costs that could have a material adverse effect on the success of our business.
We, and the third parties with whom we share our facilities, are subject to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment and
disposal of hazardous materials and wastes. Each of our operations involve the use of hazardous and flammable materials, including chemicals and biological and radioactive materials. Each of our operations also produce hazardous waste products. We generally contract with third parties for the disposal of these materials and wastes. We cannot eliminate the risk of contamination or injury from these materials. We could be held liable for any resulting damages in the event of contamination or injury resulting from the use of hazardous materials by us or the third parties with whom we share our facilities, and any liability could exceed our resources. We also could incur significant costs associated with civil or criminal fines and penalties.
Although we maintain general liability insurance as well as workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees resulting from the use of hazardous materials, this insurance may not provide adequate coverage against potential liabilities. We do not maintain insurance for environmental liability or toxic tort claims that may be asserted against us in connection with our storage or disposal of biological, hazardous or radioactive materials.
In addition, we may incur substantial costs in order to comply with current or future environmental, health and safety laws and regulations. These current or future laws and regulations may impair our research and development. Failure to comply with these laws and regulations also may result in substantial fines, penalties or other sanctions.
Further, with respect to the operations of our current and any future third-party contract manufacturers, it is possible that if they fail to operate in compliance with applicable environmental, health and safety laws and regulations or properly dispose of wastes associated with our products, we could be held liable for any resulting damages, suffer reputational harm or experience a disruption in the manufacture and supply of our product candidates or products. In addition, our supply chain may be adversely impacted if any of our third-party contract manufacturers become subject to injunctions or other sanctions as a result of their non-compliance with environmental, health and safety laws and regulations. For example, we source our API for XDEMVY, lotilaner, from Elanco, who sources through a single source supplier. If such manufacturers become subject to such injunctions or sanctions due to non-compliance, it could delay, prevent or impair our commercialization efforts, which could have an adverse effect on our business.
a marketing authorization.
We willmay be subject to federal, state and foreign healthcare and abuse laws and false claims laws, as well as information privacy and security laws and regulations. If we are unable to comply, or have not fully complied, with such laws, we could face substantial penalties, criminal sanctions, contractual damages, reputational harm, and diminished profits and future earnings.
ECPs and third-party payorspayers will play a primary role in the recommendation and prescription of XDEMVY and any future product candidates we may develop and any product candidates for which we obtain marketing approval. Our arrangements with ECPs, patients, third-party payorspayers and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may affect our business or financial arrangements and relationships through which we would market, sell and distribute our products. As a biopharmaceutical company, federal and state healthcare laws and regulations pertaining to fraud and abuse are applicable to our business and may affect our ability to operate. These laws include, but are not limited to:
•the federal Anti-Kickback Statute, which prohibits any person or entity from, among other things, knowingly and willfully soliciting, receiving, offering or paying any remuneration, directly or indirectly, overtly or covertly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, order or recommendation of an item or service reimbursable, in whole or in part, under a federal healthcare program, such as the Medicare and Medicaid programs. The term “remuneration” has been broadly interpreted to include anything of value. The Anti-Kickback Statute has also been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand and prescribers, purchasers, and formulary managers on the other the other hand. Liability under the Anti-Kickback Statute may be established without proving actual knowledge of the statute or specific intent to violate it. In addition, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act. Although there are a number of statutory exceptions and regulatory safe harbors protecting some common activities from prosecution or regulatory sanctions, the exemptions and safe harbors are drawn narrowly, and practices that do not fit squarely within an exemption or safe harbor, or for which no exception or safe harbor is available, may be subject to scrutiny;
•federal civil laws, such as the False Claims Act ("FCA") which prohibits individuals or entities from, among other things, knowingly presenting, or causing to be presented, false or fraudulent claims for payment of government funds, and knowingly making, using or causing to be made or used a false record or statement material to an obligation to pay money to the government or knowingly concealing or knowingly and improperly avoiding, decreasing or concealing an obligation to pay money to the federal government. Actions under the FCA may be brought by the U.S. Attorney General or as a qui tam action by a private individual (a whistleblower)further described in the name of the government and the individual, and the whistleblower may share in any monetary recovery. Many pharmaceutical companies have been investigated and have reached substantial settlements under the federal civil FCA in connection with their alleged off-label promotion of drugs, purportedly concealing price concessions in the pricing information submitted to the government for government price reporting purposes, and allegedly providing free product to customers with the expectation that the customers would bill federal health care programs for the product. In addition, a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the FCA. In addition, manufacturers can be held liable under the FCA even when they do not submit claims directly to government payors if they are deemed to “cause” the submission of false or fraudulent claims. FCA liability is potentially significant in the healthcare industry because the statute provides for treble damages and significant mandatory penalties per false or fraudulent claim or statement for violations. Because of the potential for large monetary exposure, healthcare and pharmaceutical companies often resolve allegations without admissions of liability for significant and material amounts to avoid the uncertainty of treble damages and per claim penalties that may be awarded in litigation proceedings. Settlements may require companies to enter into corporate integrity agreements with the government, which may impose substantial costs on companies to ensure compliance. Pharmaceutical and other healthcare companies also are subject to other federal false claims laws, including, among others, federal criminal healthcare fraud and false statement statutes that extend to non-government health benefit programs;
•the federal Health Insurance Portability and Accountability Act of 1996 ("HIPAA") among other things, imposes criminal liability for knowingly or willfully executing or attempting to execute a scheme to defraud any healthcare benefit program, including private third-party payors, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense, and creates federal criminal laws that prohibit knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement or representation, or making or using any false writing or document knowing the same to contain any materially false, fictitious or fraudulent
statement or entry in connection with the delivery of or payment for healthcare benefits, items or services. Similar to the federal healthcare Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it to have committed a violation;
•HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009 ("HITECH") and its implementing regulations, which imposes privacy, security and breach reporting obligations with respect to individually identifiable health information upon entities subject to the law, such as health plans, healthcare clearinghouses and healthcare providers and their respective business associates that perform services for them that involve individually identifiable health information. HITECH also created new tiers of civil monetary penalties, amended HIPAA to make civil and criminal penalties directly applicable to business associates, and gave state attorneys general new authority to file civil actions for damages or injunctions in U.S. federal courts to enforce the federal HIPAA laws and seek attorneys’ fees and costs associated with pursuing federal civil actions;
•federal and state consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers;
•the Federal Food, Drug and Cosmetic Act, which prohibits, among other things, the adulteration or misbranding of drugs, biologics and medical devices;
•the federal Physician Payments Sunshine Act, implemented as the Open Payments Program, requires, among other things, certain manufacturers of drugs, devices, biologics and medical supplies for which payment I available under Medicare, Medicaid, or the Children’s Health Insurance Program (with certain exceptions) to report annually to the Department of Health and Human Services, Centers for Medicare and Medicaid Services, information related to payments and other transfers of value provided to physicians (defined to include doctors, dentists, optometrists, podiatrist and chiropractors) and teaching hospitals, as well as physician ownership and investment interests, including such ownership and investment interests held by a physician’s immediate family members. Beginning in 2022, applicable manufacturers also will be required to report information regarding payments and transfers of value provided to physician assistants, nurse practitioners, clinical nurse specialists, certified nurse anesthetists, and certified nurse-midwives;
•state and foreign law equivalents of each of the above federal laws, such as anti-kickback laws, false claims laws, transparency laws and misleading advertising laws, that may impose similar or more prohibitive restrictions, and may apply to items or services reimbursed by any non-governmental third-party payors, including private insurers; and
•state and foreign laws that require pharmaceutical companies to implement compliance programs, comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, or to track and report gifts, compensation and other remuneration provided to physicians and other health care providers, and other federal, state and foreign laws that govern the privacy and security of health information or personally identifiable information in certain circumstances, including state health information privacy and data breach notification laws which govern the collection, use, disclosure, and protection of health-related and other personal information, many of which differ from each other in significant ways and often are not pre-empted by HIPAA, thus requiring additional compliance efforts.section titled “Business - Government Regulation - Other Regulatory Matters.”
We have entered into consulting and scientific advisory board arrangements with physicians and other ECPs, including some who could influence the use of XDEMVY or our product candidates, if approved. Because of the complex and far-reaching nature of these laws, regulatory agencies may view these transactions as prohibited arrangements that must be restructured, or discontinued, or for which we could be subject to other significant penalties. We could be adversely affected if regulatory agencies interpret our financial relationships with providers who may influence the ordering of and use of XDEMVY or our product candidates, if approved, to be in violation of applicable laws.
The scope and enforcement of each of these laws is uncertain and subject to rapid change in the current environment of healthcare reform. Various state and federal regulatory and enforcement agencies continue actively to investigate violations of health care laws and regulations, and the United StatesU.S. Congress continues to strengthen the arsenal of enforcement tools. The BBA of 2018 increased the criminal and civil penalties that can be imposed for violating certain federal health care laws, including the Anti-Kickback Statute. Responding to investigations can be time-andtime- and resource-consuming and can divert management’s attention from the business. Any such investigation or settlement could increase our costs or otherwise have an adverse effect on our business.
Efforts to ensure that our collaborations or business arrangements with third parties, and our business generally, comply with applicable healthcare laws and regulations will likely be costly. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other current or future governmental laws and regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, disgorgement,disgorgements, individual imprisonment, exclusion from government funded healthcare programs, such as Medicare and Medicaid, contractual damages, reputational harm, diminished profits and future earnings, additional reporting obligations and oversight if we become subject to a corporate integrity agreement or other agreement to resolve allegations of non-compliance with these laws, and the curtailment or restructuring of our operations, any of which could substantially disrupt our operations.
Inadequate funding for the FDA and other government agencies could hinder their ability to hire and retain key leadership and other personnel, prevent new products and services from being developed or commercialized in a timely manner or otherwise prevent those agencies from performing normal business functions on which the operation of our business may rely, which could negatively impact our business.
The ability of the FDA to review and approve new products can be affected by a variety of factors, including government budget and funding levels, ability to hire and retain key personnel and accept the payment of user fees, and statutory, regulatory, and policy changes. Average review times at the agency have fluctuated in recent years as a result. In addition, government funding of the SEC and other government agencies on which our operations may rely, including those that fund research and development activities, is subject to the political process, which is inherently fluid and unpredictable.
Disruptions at the FDA and other agencies may also slow the time necessary for new products to be reviewed and/or approved by necessary government agencies, which would adversely affect our business. If a prolonged government shutdown occurs, it could significantly impact the ability of the FDA to timely review and process our regulatory submissions, which could have a material adverse effect on our business. Further, in our operations as a public company, future government shutdowns could impact our ability to access the public markets and obtain necessary capital in order to properly capitalize and continue our operations.
We are subject to certain U.S. and foreign anti-corruption, anti-money laundering, export control, sanctions, and other trade laws and regulations. We can face serious consequences for violations.
U.S. and foreign anti-corruption, anti-money laundering, export control, sanctions, and other trade laws and regulations, which we collectively refer to as Trade Laws, prohibit, among other things, companies and their employees, agents, clinical research organizations, legal counsel, accountants, consultants, contractors, and other partners from authorizing, promising, offering, providing, soliciting, or receiving directly or indirectly, corrupt or improper payments or anything else of value to or from recipients in the public or private sector. Violations of Trade Laws can result in substantial criminal fines and civil penalties, imprisonment, the loss of trade privileges, debarment, tax reassessments, breach of contract and fraud litigation, reputational harm, and other consequences. We have direct or indirect interactions with officials and employees of government agencies or government-affiliated hospitals, universities, and other organizations. We also expect our non-U.S. activities to increase over time. We expect to rely on third parties for research, preclinical studies, and clinical trials and/or to obtain necessary permits, licenses, patent registrations, and other marketing approvals. We can be held liable for the corrupt or other illegal activities of our personnel, agents, or partners, even if we do not explicitly authorize or have prior knowledge of such activities.
EvenFor XDEMVY, or if we receive marketing approval for aanother product candidate, we are and will becontinue being subject to ongoing regulatory obligations and continued regulatory review, which may result in significant additional expense and subject us to restrictions, withdrawal from the market, or penalties if we fail to comply with applicable regulatory requirements or if we experience unanticipated problems with our product candidates, when and if approved.
Obtaining coverage and reimbursement approval for a product from a government or other third-party payorpayer is a time-consuming and costly process that could require us to provide supporting scientific, clinical and cost effectiveness data for the use of our products to the payor.payer. There may be significant delays in obtaining such coverage and reimbursement for newly approved products, and coverage may be more limited than the purposes for which the product is approved by the FDA or comparable foreign regulatory authorities. Moreover, eligibility for coverage and reimbursement does not imply that a product will be paid for in all cases or at a rate that covers our costs, including research, development, intellectual property, manufacture, sale and distribution expenses. Interim reimbursement levels for new products, if applicable, may also not be sufficient to cover our costs and may not be made permanent. Reimbursement rates may vary according to the use of the product and the clinical setting in which it is used, may be based on reimbursement levels already set for lower cost products
and may be incorporated into existing payments for other services. Net prices for products may be reduced by mandatory
discounts or rebates required by government healthcare programs or private payors,payers, by any future laws limiting drug prices and by any future relaxation of laws that presently restrict imports of product from countries where they may be sold at lower prices than in the United States.U.S.
There is significant uncertainty related to the insurance coverage and reimbursement of newly approved products. Third-party payorspayers often rely upon Medicare coverage policy and payment limitations in setting reimbursement policies, but also have their own methods and approval process apart from Medicare coverage and reimbursement determinations.
Coverage and reimbursement by a third-party payorpayer may depend upon a number of factors, including the third-party payor’spayer’s determination that use of a product is:
•a covered benefit under its health plan;
•safe, effective and medically necessary;
•appropriate for the specific patient;
•cost-effective; and
•neither experimental nor investigational.
We cannot be sure that reimbursement will be available for XDEMVY or any other product that we commercialize and, if coverage and reimbursement are available, what the level of reimbursement will be. Obtaining reimbursement for our products may be particularly difficult because of the higher prices often associated with branded therapeutics and therapeutics administered under the supervision of a physician. Our inability to promptly obtain coverage and adequate reimbursement rates from both government-funded and private payorspayers for any approved products that we develop could have a material adverse effect on our operating results, our ability to raise capital needed to commercialize products and our overall financial condition.
Reimbursement may impact the demand for, and the price of, XDEMVY or any other product for which we obtain marketing approval. Assuming we obtain coverage for aXDEMVY or another given product by a third-party payor,payer, the resulting reimbursement payment rates may not be adequate or may require co-payments that patients find unacceptably high. Patients who are prescribed medications for the treatment of their conditions, and their prescribing physicians, generally rely on third-party payorspayers to reimburse all or part of the costs associated with those medications. Patients are unlikely to use our products unless coverage is provided and reimbursement is adequate to cover all or a significant portion of the cost of our products. Therefore, coverage and adequate reimbursement is critical to new product acceptance. Coverage decisions may depend upon clinical and economic standards that disfavor new products when more established or lower cost therapeutic alternatives are already available or subsequently become available.
We expect to experience pricing pressures in connection with the sale of XDEMVY or any of our other product candidates due to the trend toward managed healthcare, the increasing influence of health maintenance organizations, and additional legislative changes. The downward pressure on healthcare costs in general, particularly prescription medicines, medical devices and surgical procedures and other treatments, has become very intense. As a result, increasingly high barriers are being erected to the successful commercialization of new products. Further, the adoption and implementation of any future governmental cost containment or other health reform initiative may result in additional downward pressure on the price that we may receive for any approved product.
Outside of the United States,U.S., many countries require approval of the sale price of a product before it can be marketed and the pricing review period only begins after marketing or product licensing approval is granted. To obtain reimbursement or pricing approval in some of these countries, we may be required to conduct a clinical trial that compares the cost-effectiveness of our product candidate to other available therapies. In some foreign markets, prescription pharmaceutical pricing remains subject to continuing governmental control even after initial approval is granted. As a result, we might obtain marketing approval for a product candidate in a particular country, but then be subject to price regulations that delay our commercial launch of the product, possibly for lengthy time periods, and negatively impact the revenue, if any, we are able to generate from the sale of the product in that country. Adverse pricing limitations may hinder our ability to recoup our investment in one or more product candidates, even if such product candidates obtain marketing approval.
Failure to comply with health and data protection laws and regulations could lead to government enforcement actions (which could include civil or criminal penalties), significant fines, private litigation, and/or adverse publicity and could negatively affect our financial condition, operating results and business.
We and any potential collaborators may be subject to federal, state, and foreign data protection laws and regulations (i.e., laws and regulations that address privacy and data security). In the United States,U.S., numerous federal and state laws and regulations, including federal health information privacy laws, state data breach notification laws, state health information privacy laws, and federal and state consumer protection laws (e.g., Section 5 of the FTC Act)FTC), that govern the collection, use, disclosure, and protection of health-related and other personal information could apply to our operations or the operations of our collaborators. In addition, we may obtain health information from third parties (including research institutions from which we obtain clinical trial data) that are subject to privacy and security requirements under the Health Insurance Portability and Accountability Act of 1996 ("HIPAA"). Though we are not directly subject to HIPAA as amended by HITECH. Dependinginformation privacy and security provisions – other than with respect to providing certain employee benefits, depending on the facts and circumstances, we could be subject to criminal penalties if we knowingly obtain, use, or disclosereceive individually identifiable health information maintained by a HIPAA-covered entity in a manner that is not authorized or permitted by HIPAA.
Furthermore, states are constantly adopting new laws or amending existing laws, requiring attention to frequently changing regulatory requirements. For example, in California, has enacted the California Consumer Privacy Act (“CCPA”), which came into effect on January 1, 2020. The CCPA, gives California residents expanded rights to access and delete their personal information, opt out of certain personal information sharing and receive detailed information about how their personal information is used,as amended by requiring covered companies to provide new disclosuresthe CPRA, creates transparency requirements, grants to California consumers (as that term is broadly defined) several rights with regard to their personal information, and places increased privacy and security obligations on entities handling personal data of consumers or households. The CCPA requires covered companies to provide disclosures to California consumers, and provides such consumers newwith ways to opt-out of certain sales of personal information. The California Attorney GeneralCPRA introduced significant amendments to the CCPA and established and funded the CPPA.The amendments introduced by the CPRA went into effect on January 1, 2023, and implementing regulations continue to be introduced by the CPPA. Failure to comply with the CCPA may seek substantial monetaryresult in, among other things, significant civil penalties and injunctive relief, inor potential statutory or actual damages. In addition, California residents have the event of our non-compliance with the CCPA. The CCPA also providesright to bring a private right of action (within connection with certain types of incidents. These claims may result in significant liability and potential damages. Other states including Virginia, Colorado, Utah, Indiana, Iowa, Tennessee, Montana, Texas, and Connecticut, have enacted privacy laws similar to the potential for class actions) for certainCCPA that impose new obligations or limitations in areas affecting our business and we continue to assess the impact of these state legislations on our business as additional information and guidance becomes available. Similarly, there are a number of legislative proposals in the United States, at both the federal and state level, that could impose new obligations or limitations in areas affecting our business. The CCPA and other state laws could impact our business activities depending on how they are interpreted and exemplify the vulnerability of our business to not only cyber threats but also the evolving regulatory environment related to personal data breaches that is expected to increase data breach litigation.and protected health information. The CCPA may increase our compliance costs and potential liability, and similar laws have been proposed at the federal level and have been proposed and enacted in other states.
The FTC also sets expectations for failing to take appropriate steps to keep consumers’ personal information secure, or failing to provide a level of security commensurate to promises made to individual about the security of their personal information (such as in a privacy notice) may constitute unfair or deceptive acts or practices in violation of Section 5(a) of the FTC Act. The FTC expects a company’s data security measures to be reasonable and appropriate in light of the sensitivity and volume of consumer information it holds, the size and complexity of its business, and the cost of available tools to improve security and reduce vulnerabilities. Individually identifiable health information is considered sensitive data that merits stronger safeguards. With respect to privacy, the FTC also sets expectations that companies honor the privacy promises made to individuals about how the company handles consumers’ personal information; any failure to honor promises, such as the statements made in a privacy policy or on a website, may also constitute unfair or deceptive acts or practices in violation of the FTC Act. While we do not intend to engage in unfair or deceptive acts or practices, the FTC has the power to enforce promises as it interprets them, and events that we cannot fully control, such as data breaches, may result in FTC enforcement. Enforcement by the FTC under the FTC Act can result in civil penalties or enforcement actions.
International
Activities outside of the U.S. require adherence to local and national data protection standards, impose additional compliance requirements and generate additional risks of enforcement for non-compliance. EU Member States and the United Kingdom (“UK”), as well as other jurisdictions where we may in the future operate, have adopted data protection laws includingand regulations, which impose significant compliance obligations. For example, the EU General Data Protection Regulation (“GDPR”) may also applyimposes certain obligations and restrictions on the ability to collect, analyze, use, store, disclose, transfer, or otherwise process personal data, including health-related and other personal information obtained outside of the United States.from clinical trial subjects. The GDPR extendsimposes a broad range of obligations and restrictions relating to the geographical scopeprocessing and protection of EUpersonal data, protection lawincluding obligatoins to non-EU entities under certain conditions, tightens existing EUhaving a legal basis for processing personal data protection principles, creates new obligations for companies and new rights for individuals. Failure to comply with the GDPR(which may result in substantial finessome instances in obtaining the consent of the individuals to whom the personal data relates), providing detailed information about the processing activities disclosed to the individuals, dealing with restrictions on sharing of personal data with third parties, and other administrative penalties.the transferring of personal data out of the EU, having contractual arrangements in place where required (such as with clinical trial sites and vendors), reporting in certain instances personal data breaches to data protection authorities and/or affected individuals, appointing data protection officers, conducting data protection impact assessments, responding to privacy rights requests, and keeping records of processing activities. The GDPR may increase our responsibility and liability in relation to personal data that we process and we may be required to put in place additional mechanismsexpend significant capital and other resources to ensure ongoing compliance with the GDPR.applicable privacy and data security laws, to
protect against security breaches and hackers, or to alleviate problems caused by such breaches. This may be onerous and if our efforts to comply with the GDPR or other applicable EU laws and regulations are not successful, it could adversely affect our business. The GDPR prohibitsRecent scrutiny and reevaluation of legal mechanisms to allow for the transfer of personal data from the EEA, Switzerland, or UK to countries outside of the European Economic Area (“EEA”) such as the United States, which are not considered by the European CommissionU.S. may impact our ability to provide an adequate level oftransfer personal data protection. Switzerland has adopted similar restrictions.or otherwise may cause us to incur significant costs to do so legally. Although there are legal mechanisms to allow for the transfer of personal data from the EEA, Switzerland, and Switzerlandthe UK to the United States,U.S., uncertainty about compliance with EU data protection laws remains and data protection authorities from the different EU Member States may interpret the GDPR differently, and guidance on implementation and compliance practices are often updated or otherwise revised, which adds to the complexity of processing personal data in the European Union. Enforcement by EU and UK regulators is generally active, and failure to comply with the GDPR or applicable Member State/UK local law may result in substantial fines, amongst other things (such as notices requiring compliance within a certain timeframe). The GDPR provides for fines and other administrative penalties in the event of any non-compliance, including fines of up to 10,000,000 Euros or up to 2% of our total worldwide annual turnover for certain comparatively minor offenses, or up to 20,000,000 Euros or up to 4% of our total worldwide annual turnover for more serious offenses. The GDPR also confers a private right of action on data subjects and consumer associations to lodge complaints with data protection authorities, seek judicial remedies, and obtain compensation for damages resulting from violations of the GDPR. Further, the UK Government may amend/update UK data protection laws, which may result in changes to our business operations and potentially incur commercial cost.
Additionally, European/UK data protection laws, including the GDPR, generally restrict the transfer of personal data from the EEA (including the EU), UK, and Switzerland, to the U.S. and most other countries (except those deemed to be adequate by the European Commission/UK Secretary of State as applicable) unless the parties to the transfer have implemented specific safeguards to protect the transferred personal data. This may cause us to incur significant compliance costs for implementing lawful transfer mechanisms, conducting data transfer impact assessments, and implementing additional measures where necessary to ensure that personal data transferred are adequately protected in a manner essentially equivalent to the EU. The GDPR provides different transfer mechanisms we can use to lawfully transfer personal data from the EU to countries outside the EU. An example is relying on adequacy decisions of the European Commission, such as the EU-U.S. Data Privacy Framework which was adopted by the European Commission in 2023. The adequacy decision concludes that the U.S. ensures an adequate level of protection (compared to that of the EU) for personal data transferred from the EU to U.S. companies participating in the EU-U.S. Data Privacy Framework. The adequacy decisions of the European Commission are subject to periodic reviews and may be amended or withdrawn. Another example of a lawful transfer mechanism under the GDPR is using the EU Standard Contractual Clauses (“EU SCCs”) as approved by the European Commission in 2021. In particular, on July 16, 2020,order to use the EU Standard Contractual Clauses mechanism, the exporter and the importer must ensure that the importer may guarantee a level of personal data protection in the importing country’s level of protection must be adequate that is essentially equivalent to that of the EEA. It follows from case law of the Court of Justice of the European Union (“CJEU”) and the European Data Protection Board that compliance with EU data transfer obligations involves conducting transfer impact assessments, which includes documenting detailed analyses of data access and protection laws in the case ofcountries in which data importers are located, which can be costly and time-consuming. Data Protection Commissioner v. Facebook Ireland Limited, Maximillian Schrems (Case C-311/18) (“Schrems II”), invalidatedimporters must also expend resources in analyzing their ability to comply with transfer obligations, including implementing new safeguards and controls to further protect personal data In the EU-U.S. Privacy Shield ProgramUK, international transfer mechanisms have been approved, including: the International Data Transfer Agreement and the International Data Transfer Addendum to the EU SCCs. The UK Information Commissioner’s Office has issued and maintains guidance on how to approach undertaking risk assessments for transfers of personalUK data from the EU to the United States, and added further uncertainty and complexity to the use of the Standard Contractual Clauses as a compliance mechanism for transfers of personal datanon-adequate countries outside the EU.UK.
In addition, the United Kingdom leaving the EU could also leadA lack of valid transfer mechanisms for data subject to further legislative and regulatory changes. It remains unclear how the United KingdomEU/UK data protection laws could increase exposure to enforcement actions as described above, and may affect our business operations and require commercial cost (including potentially limiting our ability to collaborate/work with certain third parties and/or regulations will developrequiring an increase in our data processing capabilities in the medium to longer term and howEU/UK). Further, the European/UK data protection laws (including laws on international data transfers from the EEA to the United Kingdom willas set out above) may also be regulated, especially following the United Kingdom’s departure from the EUupdated/revised, accompanied by new guidance and/or judicial/regulatory interpretations, which could entail further impacts on January 31, 2020 without a deal. However, the United Kingdom has transposed the GDPR into domestic law with the Data Protection Act 2018, which remains in force following the United Kingdom’s departure from the EU. During the period of “transition” (i.e., until December 31, 2020), EU law will continue to apply in the UK, including the GDPR, after which the GDPR will be converted into UK law. Beginning in 2021, the UK will be a “third country” under the GDPR. We may incur liabilities, expenses, costs,our compliance efforts and other operational losses under GDPR and applicable EU Member States and the United Kingdom privacy laws in connection with any measures we take to comply with them.
Failure to comply with U.S. and international data protection laws and regulations could result in government enforcement actions (which could include civil or criminal penalties), significant fines, private litigation, and/or adverse publicity and could negatively affect our financial condition, operating results and business. Moreover, clinical trial subjects about whom we or our potential collaborators obtain information,personal data, as well as the providers who share this informationpersonal data with us, may contractually limit our ability to use and disclose the information.personal data. Claims that we have violated individuals’ privacy rights,
failed to comply with data protection laws, or breached our contractual obligations, even if we are not found liable, could be expensive and time-consuming to defend and could result in adverse publicity that could harm our business. We cannot assure you that our third-party service providers with access to our or our customers’, suppliers’, trial patients’ and employees’ personally identifiable and other sensitive or confidential information in relation to which we are responsible will not breach contractual obligations imposed by us, or that they will not experience data security breaches or attempts thereof, which could have a corresponding effect on our business, including putting us in breach of our obligations under privacy and data protection laws and regulations and/or which could in turn adversely affect our business, results of operations and financial condition. We
cannot assure you that our contractual measures and our own privacy and security- related safeguards will protect us from the risks associated with the third-party processing, storage and transmission of such information. Furthermore, the laws are not consistent, and compliance in the event of a widespread data breach is costly.
If we are able to successfully commercialize any of our products and if we participate in the Medicaid Drug Rebate Program or other governmental pricing programs, failure to comply with obligations under these programs could result in additional reimbursement requirements, penalties, sanctions and fines which could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
If we successfully commercialize any of our product candidates, we may participate in the Medicaid Drug Rebate Program. Participation is required for federal funds to be available for our covered outpatient drugs under Medicaid and, if applicable, Medicare Part B. Under the Medicaid Drug Rebate Program, we would be required to pay a rebate to each state Medicaid program for our covered outpatient drugs that are dispensed to Medicaid beneficiaries and paid for by a state Medicaid program as a condition of having federal funds being made available to the states for our drugs under Medicaid and, if applicable, Part B of the Medicare program.
Federal law requires that any company that participates in the Medicaid Drug Rebate Program also participate in the Public Health Service’s 340B drug pricing program in order for federal funds to be available for the manufacturer’s drugs under Medicaid and Medicare Part B. The 340B drug pricing program requires participating manufacturers to agree to charge statutorily-defined covered entities no more than the 340B “ceiling price” for the manufacturer’s covered outpatient drugs. These 340B covered entities include a variety of community health clinics and other entities that receive health services grants from the Public Health Service, as well as hospitals that serve a disproportionate share of low-income patients.
In addition, in order to be eligible to have its products paid for with federal funds under the Medicaid and Medicare Part B programs and purchased by certain federal agencies and grantees, a manufacturer also must participate in the U.S. Department of Veterans Affairs (“VA”), Federal Supply Schedule (“FSS”) pricing program. Under this program, the manufacturer is obligated to make its innovator and single source products available for procurement on an FSS contract and charge a price to four federal agencies, U.S. Department of Veterans Affairs, U.S. Department of Defense (the "DoD"), Public Health Service and U.S. Coast Guard, that is no higher than the statutory Federal Ceiling Price. Moreover, pursuant to regulations issued by the DoD Defense Health Agency to implement Section 703 of the National Defense Authorization Act for Fiscal Year 2008, manufacturers are required to provide rebates on utilization of their innovator and single source products that are dispensed to TRICARE beneficiaries by TRICARE network retail pharmacies.
The requirements under the Medicaid, 340B, FSS, and TRICARE programs could reduce the revenue we may generate from any products that are commercialized in the future and could adversely affect our business and operating results. If we fail to comply with any applicable obligations under governmental pricing programs that we participate in, we could be subject to additional reimbursement requirements, significant civil monetary penalties, sanctions and fines, and those could negatively impact our business, financial condition, results of operations and growth prospects.
Risks Related to Ownership of our Common Stock
The stock price of our common stock may be volatile or may decline regardless of our operating performance and you could lose all or part of your investment.
The market price of our common stock may fluctuate significantly in response to numerous factors, many of which are beyond our control, including:
•our failure to achieve product development or commercialization goals or regulatory approval milestones in the timeframe we announce;
•overall performance of the equity markets;
•our operating performance and the performance of other similar companies;
•results from our ongoing clinical trials and future clinical trials with our current and future product candidates or of our competitors;
•delays in the commencement, enrollment and the ultimate completion of clinical trials;
•changes in our projected operating results that we provide to the public, our failure to meet these projections or changes in recommendations by securities analysts that elect to follow our common stock;
•regulatory actions with respect to our product or product candidates;
•regulatory or legal developments in the United StatesU.S. and other countries;
•the level of expenses related to future product candidates or clinical development programs;
•our failure to achieve product development or commercialization goals or regulatory approval milestones in the timeframe we announce;
•changes in hospital or ECP practices;
•announcements of acquisitions, strategic alliances or significant agreements by us or by our competitors;
•developments or disputes concerning patent applications, issued patents or other intellectual property or proprietary rights;
•recruitment or departure of key personnel;
•the economy as a whole and market conditions in our industry, including conditions resulting from COVID-19;industry;
•variations in our financial results or the financial results of companies that are perceived to be similar to us;
•financing or other corporate transactions, or inability to obtain additional funding;
•trading activity by a limited number of stockholders who together beneficially own a majority of our outstanding common stock;
•the expiration of market standoff or contractual lock-up agreements;
•the size of our market float; and
•any other factors discussed in this report.
In addition, the stock markets have experienced extreme price and volume fluctuations that have affected and continue to affect the market prices of equity securities of many biopharmaceutical companies. Stock prices of many biopharmaceutical companies have fluctuated in a manner unrelated or disproportionate to the operating performance of those companies. In the past, stockholders have filed securities class action litigation following periods of market volatility. If we were to become involved in securities litigation, it could subject us to substantial costs, divert resources and the attention of management from our business and adversely affect our business.
If securities or industry analysts do not publish research or publish inaccurate or unfavorable research about our business, our stock price and trading volume could decline.
The trading market for our common stock will depend in part on the research and reports that securities or industry analysts publish about us or our business. Securities and industry analysts do not currently, and may never, publish research on our company. If nosecurities or only very few securities analysts commence coverage of us, or if industry analysts cease coverage of us, the trading price for our common stock would be negatively affected. If one or more of the analysts who cover us downgrade our common stock or publish inaccurate or unfavorable research about our business, our common stock price would likely decline. If one or more of these analysts cease coverage of us or fail to publish reports on us regularly, demand for our common stock could decrease, which might cause our common stock price and trading volume to decline.
We are an “emerging"emerging growth company”company" and "smaller reporting company" and we cannot be certain if the reduced reporting requirements applicable to emerging growth companies will make our common stock less attractive to investors.
We are an “emergingemerging growth company”company as defined in the Jumpstart our Business Startups Act of 2012, as amended (the “JOBS Act”) and we intend to take advantage of some of the exemptions from reporting requirements that are applicable to other public companies that are not emerging growth companies, including:
•the option to present only two years of audited financial statements, in addition to any required unaudited interim financial statements, with correspondingly reduced “Management’s Discussion and Analysis of Financial Condition and Results of Operations” disclosure;
•not being required to comply with the auditor attestation requirements of Section 404(b) of the Sarbanes OxleySarbanes-Oxley Act of 2002, as amended (the "Sarbanes Oxley"Sarbanes-Oxley Act");
•not being required to comply with any requirement that may be adopted by the Public Company Accounting Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements;
•not being required to disclose certain executive compensation-related items such as the correlation between executive compensation and performance and comparisons of the chief executive officer’s compensation to median employee compensation; and
•not being required to submit certain executive compensation matters to stockholder advisory votes, such as “say-on-pay,” “say-on-frequency,”"say-on-pay," "say-on-frequency," and “say-on-golden"say-on-golden parachutes.”"
The JOBS Act permits an “emergingemerging growth company”company such as us, to take advantage of an extended transition period to comply with new or revised accounting standards applicable to public companies. We have irrevocably elected to opt out of this provision and, as a result, we will comply with new or revised accounting standards as required when they are adopted.
We cannot predict if investors will find our common stock less attractive because we will rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile. We may take advantage of these reporting exemptions until we are no longer an emerging growth company. We will remain an emerging growth company until the earlier of (1) December 31, 2025, the last day of the fiscal year following the fifth anniversary of the completion of our initial public offering, (b) the last day of the fiscal year (a) in which we have total annual gross revenue of at least $1.07$1.235 billion or (b) in which we are deemed to be a large accelerated filer, which means the market value of our common stock that is held by non-affiliates exceeds $700 million as of the prior June 30th and (2) the date on which we have issued more than $1.0 billion in non-convertible debt during the prior three-year period. We are also a “smallersmaller reporting company”company as defined in the Exchange Act.Act of 1934. We may continue to be a smaller reporting company even after we are no longer an emerging growth company. We may take advantage of certain of the scaled disclosures available to smaller reporting companies and will be able to take advantage of these scaled disclosures for so long as our voting and non-voting common stock held by non-affiliates is less than $250.0 million measured on the last business day of our second fiscal quarter, or our annual revenue is less than $100.0 million during the most recently completed
fiscal year and our voting and non-voting common stock held by non-affiliates is less than $700.0 million measured on the last business day of our second fiscal quarter.
Investors may find our common stock less attractive to the extent we rely on the exemptions and relief granted by the JOBS Act. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may decline or become more volatile.
Sales of a substantial number of shares of our common stock in the public market could cause the price of our common stock to fall.
Sales of a substantial number of shares of our common stock in the public market could occur at any time. Thecause our stock price to decline. Sales of our common stock could decline if there area substantial sales of our common stock, particularly sales by our directors, executive officers and significant stockholders, or if there is a large number of shares of our common stock available for sale andin the public market could occur at any time. These sales, or the perception in the market perceives that sales will occur.the holders of a large number of shares intend to sell shares, could reduce the market price of our common stock. As of March 19, 2021,February 21, 2024, we had 20,503,09634,218,886 shares of common stock outstanding. Of these shares, approximately 6,325,000 shares sold in our IPO may be resold in the public market, unless purchased by our affiliates or existing stockholders. Of the remaining shares, a significant percentage of these shares of common stock are
currently restricted as a result of securities laws or lock-up agreements entered into in connection with the IPO. These shares will become available to be sold in the public market as early as 180 days following the date of our final prospectus filed with the SEC on October 16, 2020 pursuant to Rule 424(b) under the Securities Act of 1933, as amended (“Securities Act”). Shares held by directors, executive officers and other affiliates will be subject to volume limitations under Rule 144 under the Securities Act of 1933 and various vesting agreements. The underwriters of the IPO may, in their discretion, permit our stockholders
We have registered and intend to sell shares priorcontinue to the expiration of the restrictive provisions contained in those lock-up agreements.
Moreover, holders of an aggregate of 11,107,018 shares of our common stock have rights under our registration rights agreement, subject to specified conditions, to require us to file registration statements covering their shares and to include their shares in registration statements that we may file for ourselves or for other stockholders, subject to lockup agreements. In addition, on October 20, 2020, we filed a registration statement on Form S-8 registering 13,732,980register all shares of common stock that we have issued and may issue under our employee equity incentivecompensation plans. As a result,Once we register these shares, registered under this registration statement on Form S-8they can be freely sold in the public market upon issuance, subject to the satisfaction of vesting arrangements and the exercise of such options, volume limitations applicable to affiliatesaffiliates. We cannot predict what effect, if any, sales of our shares in the public market or lock-up agreements.
Thethe availability of shares for sale will have on the market price of our common stock. However, future sales of substantial amounts of our common stock in the public market, including shares issued upon exercise of our outstanding warrant or options, or the perception that such sales may occur, could adversely affect the market price of our common stock. We also expect that significant additional capital may be needed in the future to continue our planned operations. To raise capital, we may sell common stock, including pursuant to our ATM program, convertible securities or other equity securities in one or more transactions at prices and in a manner we determine from time to time. For example, in August 2023 we completed a follow-on public offering of 5.7 million shares of our common stock could decline asat a resultpublic offering price of $17.50 per share for net proceeds of approximately $99.3 million (after deducting underwriting discounts, commissions and other estimated offering-related expenses) and in the fourth quarter of 2023 we raised approximately $19.2 million, after deducting broker commissions and fees, through sales under our ATM program. To the extent that additional capital is raised through the sale and issuance of a substantial number ofshares or other securities convertible into shares, our shares of common stock in the public marketstockholders will be diluted. These sales, or the perception in the market that the holders of a large number of shares intend to sell their shares.shares, could reduce the market price of our common stock.
The concentration of our stock ownership will likely limit your ability to influence corporate matters, including the ability to influence the outcome of director elections and other matters requiring stockholder approval.
As of December 31, 2020,2023, our officers, directors and the holders of more than 5% of our outstanding stock collectively beneficially own approximately 80%67% of our common stock. As a result, these stockholders, acting together, will have significant influence over all matters that require approval by our stockholders, including the election of directors and approval of significant corporate transactions. Corporate actions might be taken even if other stockholders oppose them. This concentration of ownership might also have the effect of delaying or preventing a change of control of our company that other stockholders may view as beneficial.
Requirements associated with being a public company will increase our costs significantly, as well as divert significant company resources and management attention.
As a public company, we are subject to the reporting requirements of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), or the other rules and regulations of the Securities and Exchange Commission (the “SEC”), or any securities exchange relating to public companies. Compliance with the various reporting and other requirements applicable to public companies requires considerable time and attention of management and we will incur significant legal, accounting and other expenses. We cannot assure you that we will satisfy our obligations as a public company on a timely basis.
In addition, as a public company, it may be more difficult or more costly for us to obtain certain types of insurance, including directors’ and officers’ liability insurance, and we may be forced to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. The impact of these events could also make it more difficult for us to attract and retain qualified personnel to serve on our board of directors, our board committees or as executive officers.
If we fail to maintain proper and effective internal controls, our ability to produce accurate and timely financial statements could be impaired, which could result in sanctions or other penalties that would harm our business.
We are subject to the reporting requirements of the Exchange Act of 1934, the Sarbanes-Oxley Act and the rules and regulations of the Nasdaq Stock Market (“Nasdaq”).Nasdaq. The Sarbanes-Oxley Act requires, among other things, that we maintain effective disclosure controls and procedures and internal controls over financial reporting. Commencing in the fiscal year ending December 31, 2021, weWe must perform system and process design evaluation and testing of the effectiveness of our internal controls over financial reporting to allow management to report on the effectiveness of our internal controls over financial reporting, in our Form 10-K filing for that year, as required by Section 404 of the Sarbanes-Oxley Act. To achieve compliance with Section 404 within the prescribed period, we will be engaged in a process to document and evaluate our internal control over financial reporting, which is both costly and challenging. In this regard,Therefore, we will need to continue to dedicate internal resources, including through hiring additional financial and accounting personnel, potentially engageengaging outside consultants, and adopt a detailed work plancontinue to assess and document the adequacy of internal control over financial reporting, continue steps to improve control processes as appropriate, validate through testing that controls are functioning as documented and implement a continuous reporting and improvement process for internal control over financial reporting. This will require
that werequires us to incur substantial additional professional fees and internal costs to expand our accounting and finance functions and that we expend significant management efforts. Prior to our IPO, we have never been required to test our internal controls within a specified period and, as a result, we may experience difficulty in meeting these reporting requirements in a timely manner.maintain compliance.
We may discover weaknesses in our system of internal financial and accounting controls and procedures that could result in a material misstatement of our financial statements. Our internal control over financial reporting will not prevent or detect all errors and all fraud. A control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the control system’s objectives will be met. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that misstatements due to error or fraud will not occur or that all control issues and instances of fraud will be detected.
If we are not able to comply with the requirements of Section 404 of the Sarbanes-Oxley Act in a timely manner, or if we are unable to maintain proper and effective internal controls over financial reporting, we may not be able to produce timely and accurate financial statements. If that were to happen, our investors could lose confidence in our reported financial information, the market price of our stock could decline and we could be subject to sanctions or investigations by the SEC or other regulatory authorities including equivalent foreign authorities.
We do not intend to pay dividends for the foreseeable future.
We have never declared nor paid cash dividends on our capital stock. We currently intend to retain any future earnings to finance the operation and expansion of our business, and we do not expect to declare or pay any dividends in the foreseeable future. Our Credit Facility also contains a negative covenant that prohibits us from paying dividends subject to limited exceptions. Consequently, stockholders must rely on sales of their common stock after price appreciation, which may never occur, as the only way to realize any future gains on their investment.
We could be subject to securities class action litigation.
In the past, securities class action litigation has often been brought against a company following a decline in the market price of its securities. This risk is especially relevant for us because biopharmaceutical companies have experienced significant stock price volatility in recent years. If we face such litigation, it could result in substantial costs and a diversion of management’s attention and resources, which could harm our business.
Our operating results may fluctuate significantly, which makes our future operating results difficult to predict and could cause our operating results to fall below expectations or our guidance.
Our quarterly and annual operating results may fluctuate significantly in the future, which makes it difficult for us to predict our future operating results. Our operating results may fluctuate due to a variety of factors, many of which are outside of our control and may be difficult to predict, including the following:
•the cost of manufacturing XDEMVY or our other product candidates, which may vary depending on the quantity of production and the terms of our agreements with manufacturers;
•the level of demand for XDEMVY or our product candidates should they receive approval, which may vary significantly;
•the risk/benefit profile, cost and reimbursement policies with respect to XDEMVY or our product candidates, if approved, and existing and potential future drugs that compete with our product candidates;
•the gross-to-net yields for XDEMVY or our other product candidates, if approved;
•the timing and success or failure of clinical trials for our product candidates or competing product candidates, or any other change in the competitive landscape of our industry, including consolidation among our competitors or partners;
•our ability to successfully recruit patients for preclinical studies and clinical trials, and any delays caused by difficulties in such recruitment efforts;
•our ability to obtain regulatory approval for our product candidates, and the timing and scope of any such approvals we may receive;
•the timing and cost of, and level of investment in, research and development activities relating to our product candidates, which may change from time to time;
•the cost of manufacturing our product candidates, which may vary depending on the quantity of production and the terms of our agreements with manufacturers;
•our ability to attract, hire and retain qualified personnel;
•expenditures that we will or may incur to develop additional product candidates;
•the level of demand for our product candidates should they receive approval, which may vary significantly;
•the risk/benefit profile, cost and reimbursement policies with respect to our product candidates, if approved, and existing and potential future drugs that compete with our product candidates;
•the changing and volatile U.S., European and global economic environments, including the impact of COVID-19;current or future health pandemics; and
•future accounting pronouncements or changes in our accounting policies.
The cumulative effects of these factors could result in large fluctuations and unpredictability in our quarterly and annual operating results. As a result, comparing our operating results on a period-to-period basis may not be meaningful. This variability and unpredictability could also result in our failing to meet the expectations of industry or financial analysts or investors for any period. If our revenue or operating results fall below the expectations of analysts or investors or below any forecasts we may provide to the market, or if the forecasts we provide to the market are below the expectations of analysts or investors, the price of our common stock could decline substantially. Such a stock price decline could occur even when we have met any previously publicly stated guidance we may provide.
Delaware law and provisions in our amended and restated certificate of incorporation and amended and restated bylaws could make a merger, tender offer or proxy contest difficult, thereby depressing the trading price of our common stock.
Our status as a Delaware corporation and the anti-takeover provisions of the Delaware General Corporation Law may discourage, delay or prevent a change in control by prohibiting us from engaging in a business combination with an interested stockholder for a period of three years after the person becomes an interested stockholder, even if a change of control would be beneficial to our existing stockholders. In addition, our amended and restated certificate of incorporation and amended and restated bylaws contain provisions that may make the acquisition of our company more difficult, including the following:
•a classified board of directors with three-year staggered terms, which could delay the ability of stockholders to change the membership of a majority of our board of directors;
•the ability of our board of directors to issue shares of preferred stock and to determine the price and other terms of those shares, including preferences and voting rights, without stockholder approval, which could be used to significantly dilute the ownership of a hostile acquiror;
•the exclusive right of our board of directors to elect a director to fill a vacancy created by the expansion of our board of directors or the resignation, death or removal of a director, which prevents stockholders from being able to fill vacancies on our board of directors;
•a prohibition on stockholder action by written consent, which forces stockholder action to be taken at an annual or special meeting of our stockholders;
•the requirement that a special meeting of stockholders may be called only by a majority vote of our entire board of directors, the chairman of our board of directors or our chief executive officer, which could delay the ability of our stockholders to force consideration of a proposal or to take action, including the removal of directors;
•the requirement for the affirmative vote of holders of at least 66 2/3% of the voting power of all of the then-outstanding shares of the voting stock, voting together as a single class, to amend the provisions of our amended and restated certificate of incorporation or our amended and restated bylaws, which may inhibit the ability of an acquiror to effect such amendments to facilitate an unsolicited takeover attempt; and
•advance notice procedures with which stockholders must comply to nominate candidates to our board of directors or to propose matters to be acted upon at a stockholders’ meeting, which may discourage or deter a potential acquiror from conducting a solicitation of proxies to elect the acquiror’s own slate of directors or otherwise attempting to obtain control of us.
In addition, as a Delaware corporation, we are subject to Section 203 of the Delaware General Corporation Law. These provisions may prohibit large stockholders, in particular those owning 15% or more of our outstanding voting stock, from merging or combining with us for a certain period of time. A Delaware corporation may opt out of this provision by express provision in its original certificate of incorporation or by amendment to its certificate of incorporation or bylaws approved by its stockholders. However, we have not opted out of this provision.
These and other provisions in our amended and restated certificate of incorporation, amended and restated bylaws and Delaware law could make it more difficult for stockholders or potential acquirors to obtain control of our board of directors or initiate actions that are opposed by our then-current board of directors, including delay or impede a merger, tender offer or proxy contest involving our company. The existence of these provisions could negatively affect the price of our common stock and limit opportunities for you to realize value in a corporate transaction.
Our amended and restated certificate of incorporation provides that the Court of Chancery of the State of Delaware and the U.S. federal district courts are the exclusive forums for substantially all disputes between us and our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers or employees.
Our amended and restated certificate of incorporation provides that the Court of Chancery of the State of Delaware is the exclusive forum for any derivative action or proceeding brought on our behalf, any action asserting a breach of fiduciary duty, any action asserting a claim against us arising pursuant to the Delaware General Corporation Law, our certificate of incorporation or our bylaws or any action asserting a claim against us that is governed by the internal affairs doctrine.
This provision would not apply to suits brought to enforce a duty or liability created by the Exchange Act.Act of 1934. Furthermore, Section 22 of the Securities Act of 1933 creates concurrent jurisdiction for federal and state courts over all such Securities Act actions. Accordingly, both state and federal courts have jurisdiction to entertain such claims. To prevent having to litigate claims in multiple jurisdictions and the threat of inconsistent or contrary rulings by different courts, among other considerations, our certificate of incorporation will further provide that the U.S. federal district courts will be the exclusive forum for resolving any complaint asserting a cause of action arising under the Securities Act. While the Delaware courts have determined that such choice of forum provisions are facially valid, a stockholder may nevertheless seek to bring a claim in a venue other than those designated in the exclusive forum provisions. In such instance, we would expect to vigorously assert the validity and enforceability of the exclusive forum provisions of our certificate of incorporation. This may require significant additional costs associated with resolving such action in other jurisdictions and there can be no assurance that the provisions will be enforced by a court in those other jurisdictions.
This choice of forum provision may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, officers or other employees and may discourage these types of lawsuits. Alternatively, if a court were to find the choice of forum provision contained in our amended and restated certificate of incorporation to be inapplicable or unenforceable in an action, we may incur additional costs associated with resolving such action in other jurisdictions.
Item 1B. Unresolved Staff Comments.
None.
Item 1C. Cybersecurity.
We continue to make substantial investments to augment the capabilities of our people, processes, and technologies in order to address our cybersecurity risks. Our cybersecurity risks, and the controls designed to mitigate those risks, are integrated into our overall risk management governance and are reviewed quarterly by our Board of Directors.
Risk Management and Strategy
As of December 31, 2023, we've implemented a set of comprehensive cybersecurity and data protection policies and procedures. Our employees and contractors receive regular cybersecurity awareness trainings, including specific topics related to social engineering and email frauds. We have capable employees and consultants with significant expertise and certifications in cybersecurity related to our industry. We invest in advanced technologies for continuous cybersecurity monitoring across our information technology environment which are designed to prevent, detect, and minimize cybersecurity attacks, as well as alert management of such attacks.
Our ITGCs are firmly established based on recognized industry standards and cover areas such as risk management, data backup, and disaster recovery. We have implemented processes to monitor security threats and vulnerabilities and respond to all cybersecurity incidents affecting us, including prompt escalation and communication of major security incidents to senior business leadership and our Board of Directors. We conduct cybersecurity penetration testing annually to identify and remediate cybersecurity gaps. We also perform cybersecurity assessments of all our third-party providers who have access to our information technology systems and data.
Primary responsibility for assessing, monitoring and managing our cybersecurity risks rests with the Head of IT who reports to our Chief Financial Officer, to manage the risk assessment and mitigation process. We have a dedicated IT resource with expertise in cybersecurity and risk management who is dedicated to working with our internal IT team on cybersecurity risk management.
We also engage other consultants, and other third parties in connection with our risk assessment and mitigation processes. These service providers assist with the design and implementation of our cybersecurity policies and procedures, as well as monitor and test our safeguards. We require each third-party service provider to certify that it has the ability to implement and maintain appropriate security measures, consistent with all applicable laws, to implement and maintain reasonable security measures in connection with their work with us, and to promptly report any suspected breach of its security measures that may affect our company.
Governance
Our Board of Directors and Audit Committee are responsible for overseeing our cyber security risk management and strategy.
Our Head of IT provides periodic briefings to the Audit Committee including our cybersecurity risks and activities, any potential cybersecurity incidents and related responses, cybersecurity systems testing and, activities of third parties. Our Audit Committee regularly meets with our Chief Financial Officer and Head of IT about the Company’s ongoing compliance and risk management and reports to the Board regularly.
Cybersecurity Threat Disclosure
To date, we are not aware of any cybersecurity threats that have materially affected or are reasonably likely to materially affect the Company.
For further discussion of cybersecurity risks, please see Item 1A, “Risk Factors”.
Item 2. Properties.
We currently lease approximately 20,00032,145 square feet of office and laboratory space in Irvine, California under certain leases that last expire in January 2024.2027, with a renewal option for a term of three years. We believe that this space will be sufficient to meet our needs for the foreseeable future and that any additional space we may require will be available on commercially reasonable terms.
Item 3. Legal Proceedings.
We are not currently a party to any material legal proceedings. From time to time, we may become involved in legal proceedings arising in the ordinary course of our business. Regardless of outcome, litigation can have an adverse impact on us due to defense and settlement costs, diversion of management resources, negative publicity, reputational harm and other factors.
Item 4. Mine Safety Disclosures.
None.
PART II
Item 5. Market for Registrant's Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Our common stock has been publicly traded on the Nasdaq Global Select Market under the symbol “TARS” since our IPO on October 15, 2020. Prior to this date, there was no public market for our common stock.
Holders of Common Stock
As of March 19, 2021,February 21, 2024, the closing price of our common stock on the Nasdaq Global Select Market was $32.06$30.13 per share, and there were approximately 14332 holders of record of our common stock. The actual number of stockholders is greater than this number of record holders, and includes stockholders who are beneficial owners, but whose shares are held in street name by brokers and other nominees. This number of holders of record also does not include stockholders whose shares may be held in trust by other entities.
Dividend Policy
We have never declared or paid any cash dividends on our common stock and do not anticipate paying cash dividends in the foreseeable future.
Securities Authorized for Issuance under Equity Compensation Plans
The information required by this item will be contained in our definitive proxy statement to be filed with the Proxy StatementSEC in connection with our Annual Meeting of Stockholders within 120 days after December 31, 2023 and is incorporated in this Annual Report on Form 10-K by reference.
Recent Sales of Unregistered Securities
The following sets forth information regarding all unregistered securities sold by us during the year ended December 31, 2020. No underwriters were involved in the sales and the certificates representing the securities sold and issued contain legends restricting transfer of the securities without registration under the Securities Act or an applicable exemption from registration.
(a) In July 2020 we granted certain of our employees stock options to purchase an aggregate of 320,829 shares of common stock upon the exercise of options under our 2016 Plan at an exercise price per share of $2.0055, for an aggregate exercise price of approximately $0.6 million.
(b) Pursuant to the terms of that certain License Agreement between the Company and Elanco Tiergesundheit AG ("Elanco"), dated September 3, 2020 (the "All Human Uses Elanco Agreement"), in September 2020 we issued 222,460 shares of our common stock to Elanco as consideration for licenses included in the All Human Uses Elanco Agreement.
(c) In September 2020, we issued and sold to 11 accredited investors an aggregate of 2,857,079 shares of our Series C preferred stock at a purchase price of $14.0003 per share, for aggregate consideration of approximately $40.0 million.
(d) In September 2020 we granted certain of our employees stock options to purchase an aggregate of 416,688 shares of common stock upon the exercise of options under our 2016 Plan at an exercise price per share of $10.99, for an aggregate exercise price of approximately $4.6 million.
The offers, sales and issuances of the securities described in Items (a) and (d) above were exempt from registration under the Securities Act under either (1) Rule 701 in that the transactions were under compensatory benefit plans and contracts relating to compensation as provided under Rule 701 or (2) Section 4(a)(2) of the Securities Act or Regulation D promulgated thereunder as transactions by an issuer not involving any public offering. The recipients of such securities were the registrant’s directors, officers, employees, consultants or other service providers and received the securities under our 2016 Plan. The recipients of securities in each of these transactions represented their intention to acquire the securities for investment only and not with view to or for sale in connection with any distribution thereof and appropriate legends were affixed to the securities issued in these transactions.
The offers, sales and issuances of the securities described in Items (b) and (c) above were exempt from registration under the Securities Act under Section 4(a)(2) of the Securities Act or Regulation D promulgated thereunder as transactions by an issuer not involving a public offering. The recipients of securities in each of these transactions acquired the securities for investment only and not with a view to or for sale in connection with any distribution thereof and appropriate legends were affixed to the securities issued in these transactions. Each of the recipients of securities in these transactions was an accredited person and had adequate access, through employment, business or other relationships, to information about the registrant.
None.
Use of Proceeds from Initial Public Offering
On October 16, 2020, our Registration Statement on Form S-1 (File No. 333-249076) (the "Registration Statement") relating to the initial public offering of our common stock was declared effective by the SEC. Pursuant to such Registration Statement, we sold an aggregate of 6,325,000 shares of our common stock, which includesincluded 825,000 shares sold pursuant to the underwriters’ full exercise of their option to purchase additional shares, at a price of $16.00 per share. The aggregate offering price for shares sold in the offering was $101.2 million. On October 20, 2020, we closed the sale of such shares, resulting in aggregate cash proceeds to us of $91.7 million, net of underwriting discounts, commissions and offering expenses paid or payable by us. No offering expenses were paid or are payable, directly or indirectly, to our directors or officers, to persons owning 10% or more of any class of our equity securities or to any of our affiliates. BofA Securities, Inc., Jefferies LLC and Raymond James & Associates, Inc., LifeSci Capital LLC and Ledenburg Thalman & Co. Inc., acted as the joint book-running managers of the offering.
There has been no material change in the planned use of proceeds from our initial public offering as described in the final prospectus, dated October 15, 2020, filed with the SEC on October 16, 2020, pursuant to Rule 424(b) of the Securities Act.
Purchases of Equity Securities by the Issuer and Affiliated Purchases
None.
Other Information
Item 6. Selected Financial Data
The following selected consolidated financial data has been derived fromAs disclosed in our audited Consolidated Financial Statements. The audited Consolidated Financial Statements for the fiscal years ended December 31, 2020 and 2019 are included elsewhere in this AnnualCurrent Report on Form 10-K.
The information set forth below should8-K filed with the SEC on January 18, 2024, we announced that Jose Trevejo, M.D., Ph.D. would be read in conjunctionleaving his role as the Company’s Chief Medical Officer (the “Transition”), effective as of January 18, 2024 (the “Transition Date”) and leaving his employment with Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations and the Consolidated Financial Statements and Notes thereto in Item 8. Financial Statements and Supplementary Data. The information set forth below is not necessarily indicative of our future financial condition or future results of operations.
| | | | | | | | | | | |
| | Year Ended December 31, |
| | 2020 | | 2019 |
| (in thousands, except per share data) |
| Statement of Operations and Comprehensive Loss Data: | | | |
| Research and development | $ | 18,826 | | | $ | 3,162 | |
| General and administrative | 8,172 | | | 1,136 | |
| Total operating expenses | 26,998 | | | 4,298 | |
| Loss from operations | (26,998) | | | (4,298) | |
| Other income (expense): | | | |
| Interest income (expense), net | 188 | | | (40) | |
| Loss on extinguishment of convertible notes | — | | | (255) | |
| Change in fair value of derivative liabilities | — | | | (76) | |
| Total other income (expense) | 188 | | | (371) | |
| Provision for income taxes | (1) | | | (1) | |
| Net loss and comprehensive loss | $ | (26,811) | | | $ | (4,670) | |
| Net loss per share attributable to common stockholders, basic and diluted | $ | (4.32) | | | $ | (1.98) | |
| | | | | | | | | | | |
| | December 31, |
| | 2020 | | 2019 |
| (in thousands) |
| Selected Balance Sheet Data: | | | |
| Cash and cash equivalents | $ | 168,129 | | | $ | 57,952 | |
Working capital (1) | 165,268 | | | 57,211 | |
| Total assets | 171,972 | | | 58,316 | |
| Total liabilities | 5,992 | | | 919 | |
| Preferred stock | — | | | 63,402 | |
| Accumulated deficit | (32,845) | | | (6,034) | |
| Total stockholders’ equity (deficit) | 165,980 | | (6,005) | |
(1) We define working capital as current assets less current liabilities.In connection with the Transition and Dr. Trevejo’s termination of employment, on February 21, 2024, we entered into a separation and severance agreement with Dr. Trevejo, which provides for the following benefits effective upon and after the Separation Date: severance payments equal to twelve months of base salary and twelve months of company-paid continued benefits coverage, a lump sum bonus payment payable in 2024 equal to his 2023 annual target bonus adjusted based in part on the 2023 Company performance score and Company discretion, accelerated vesting of options for 13,789 shares of our common stock, and an option exercise period extension for certain options, in exchange for a release and waiver of claims and continued compliance with his confidentiality obligations.The foregoing summary of the terms of the Separation Agreement is qualified in its entirety by reference to the complete text of the Separation Agreement, a copy of which will be filed with our Quarterly Report on 10-Q for the quarter ending March 30, 2024.
Item 6. Reserved
Item 7. Management's Discussion and Analysis of Financial Condition and Results of Operations
The following discussion and analysis of our financial condition and results of operations should be read in conjunction with our “Selected Financial Data” and our financial statements and the related notes to those statements included elsewhere in this Annual Report on Form 10-K. In addition to historical financial information, the following discussion and analysis contains forward-looking statements that involve risks, uncertainties and assumptions. Our actual results and timing of selected events may differ materially from those anticipated in these forward-looking statements as a result of many factors, including, but not limited to, those discussed under the section titled “Risk Factors” and elsewhere in this Annual Report on 10-K. See the section titled “Special Note Regarding Forward-Looking Statements” elsewhere in this Annual Report on Form 10-K.
Overview
Our Business
We are a late clinical-stagecommercial stage biopharmaceutical company focused on the development and commercialization of therapeutic candidates to address large market opportunities initially in ophthalmic conditions where there are limited treatment alternatives.therapeutics, starting with eye care. Our lead product, candidate, TP-03, is a novel therapeutic in Phase 2b/3 that is being developedXDEMVY® was approved by the U.S. Food and Drug Administration ("FDA") on July 24, 2023 for the treatment of blepharitis caused by the infestation of Demodex mites, which is referred to as Demodex blepharitis. Blepharitis (“Blephar” is a reference to eyelid and “itis” is a reference to inflammation) is a conditionan ophthalmic lid margin disease characterized by inflammation of the eyelid margin, redness and ocular irritation, including a specific type of eyelash dandruff called collarettes. The healthy interaction of the eyelid and the surface of the eyeball is crucial to ocular health.collarettes, which are pathognomonic for Demodex blepharitis. Poorly controlled and progressive blepharitis can lead to worsening of corneal damage over time and, in extreme cases, blindness.
According to published studies, there are an estimated 20 million patients in the United States who suffer from blepharitis, with approximately 45% or nine million of cases caused by Demodex infestation. Further, the possible number of Demodex blepharitis cases There may be as highmany as approximately 25 million based on our internal research indicating approximately 58%people in the U.S. who suffer from Demodex blepharitis. XDEMVY is the first and only therapeutic approved by the FDA and we believe is the definitive standard of patients presenting to eye care clinics have collarettes and a published study estimating that at least 45 million people annually visit an eye care clinic.for the treatment of Demodex blepharitis.
We believe that TP-03 hasXDEMVY targets and eradicates the potential to be the first FDA-approved therapeutic for the treatmentroot cause of Demodex blepharitis and become the standard of care.– Demodex mite infestation. The active pharmaceutical ingredient ("API") of TP-03,XDEMVY, lotilaner, is designed to paralyzeparalyzes and eradicateeradicates mites and other parasites through the inhibition of parasite-specific gamma-aminobutyric acid-gated chloride ("GABA-Cl") channels).channels.
To date, we have completed seven clinical trials that include a Phase 3 Saturn-2 trial, a Phase 2b/3 Saturn-1 trial, four Phase 2 trials, and a Phase 1 trial for TP-03XDEMVY in Demodex blepharitis, all of which met their primary, secondary and/or certain exploratory endpoints, as applicable, and during which TP-03 was well tolerated. Our Phase 2b/3 trial, Saturn-1, commenced in September 2020 and was fully enrolled by the first quarter of 2021. We expect to begin our second pivotal trial, Saturn-2, in the second quarter of 2021. Saturn-1 and Saturn-2 have primary and secondary endpoints consistent with those of our Europa and Io Phase 2 trials. We expect these TP-03 pivotal trials to support our submission of a new drug application ("NDA") with the United States Food and Drug Administration ("FDA")drug well tolerated throughout each trial. We have also completed, and/or have ongoing clinical trials for TP-03 for the potential treatment of Demodex blepharitis.MGD, TP-04 for the potential treatment of rosacea and TP-05 for potential Lyme disease prophylaxis.
We intend to further advance our pipeline with the lotilaner API to address several diseases across therapeutic categories in human medicine, including eye care, dermatology, and other diseases. Theseinfectious disease prevention. We are investigating the development of our product candidates to address targeted diseases with high unmet medical needneeds, which currently include Meibomian Gland DiseaseTP-03 for the potential treatment of meibomian gland disease ("MGD"), TP-04, a novel gel formulation of lotilaner for the potential treatment of rosacea, and TP-05, a novel investigative oral formulation of lotilaner, for potential Lyme disease prophylaxis and malaria.community malaria reduction.
Recent Business and Clinical Highlights
TP-03 Type C MeetingXDEMVY: In December 2020, we had a Type C meeting withXDEMVY was approved by the FDA in July 2023, as the first and only approved therapeutic for TP-03Demodex blepharitis, a highly prevalent eyelid disease that impacts approximately 25 million eye care patients in the treatmentU.S. XDEMVY targets the root cause of Demodex blepharitis. This meeting confirmed blepharitis and in pivotal trials demonstrated significant improvement in eyelids (reduction of collarettes, the pathognomonic sign of the disease, to no more than two collarettes per upper lid), mite eradication (mite density of zero mites per lash) and erythema cure (grade zero).
•Generated strong prescription and sales growth of XDEMVY in 2023 enabled by the execution of key commercial initiatives, including the deployment of our planned NDA pathwaysales force targeting 15,000 eye care providers ("ECPs"), representing more than 80% of all eye care prescriptions and high-impact education leading to ECP adoption. We recognized $14.7 million in product sales, net of XEMVY during the year ended December 31, 2023.
◦Delivered approximately 17,400 bottles of XDEMVY to patients
◦Approximately 6,000 ECPs have started patients on XDEMVY with respectmore than 50% of ECPs prescribing XDEMVY to multiple patients as of February 23, 2024
•We are actively engaged in contracting discussions with all the key commercial and Medicare accounts and we are on track to potentially secure broad commercial coverage sequentially throughout 2024 and broad Medicare coverage in 2025
•During 2023, six manuscripts were published in peer-reviewed journals including:
◦Saturn-1, one year extension data highlighting the safety and information requireddurable response of XDEMVY
◦Two independent meta-analyses validating efficacy, safety and impact of our study results
•We presented additional Saturn-2 pivotal data at the American Academy of Optometry and the American Academy of Ophthalmology conferences further demonstrating XDEMVY as the standard of care for Demodex blepharitis
•The American Academy of Ophthalmology added XDEMVY as the first and only FDA-approved therapeutic for Demodex blepharitis in the NDA filing.their Preferred Practice Patterns (PPP) guidelines
TP-03 Pivotal TrialsMeibomian Gland Disease, Ersa Trial: Saturn-1In August 2022, we announced the enrollment was completedof our first patient in the first quarter of 2021. We expect to initiate our second pivotalCompany's Phase 2a Ersa clinical trial Saturn-2, in the second quarter 2021.
TP-03 Out-License: On March 26, 2021, we executed the LianBio Agreement with LianBio, granting exclusive commercial rights ofstudying TP-03 for the treatment of MGD. On December 11, 2023, we announced positive topline results of the Ersa Phase 2a clinical trial evaluating TP-03 (lotilaner ophthalmic solution, 0.25%) administered twice daily or three times a day for 12 weeks for the treatment of MGD in patients with Demodex mites. TP-03 demonstrated statistically significant and clinically meaningful improvements compared to baseline in two objective measures of the disease – the presence and quality of liquid secretion as measured by the Meibomian Gland Secretion Score ("MGSS") and the number of glands secreting normal (clear) liquid and was well tolerated. We plan to discuss and determine the potential regulatory path with the FDA.
TP-04 Rosacea, Galatea Trial: In March 2023, we initiated the Galatea trial, a Phase 2a trial evaluating TP-04, a novel gel formulation of lotilaner, for the treatment of rosacea. On February 27, 2024, we announced positive topline results from the Galatea trial evaluating TP-04 for the treatment of rosacea which demonstrated statistically significant improvements (p<0.05) in inflammatory lesions and Investigator's Global Assessment (IGA) score (change in baseline and success rate) were observed compared to vehicle at week 12. TP-04 was generally well tolerated. We plan to discuss and determine the potential regulatory path with the FDA.
TP-05 Lyme Disease, Callisto and Carpo Trials: In December 2022, we announced positive topline results from the completed Phase 1 Callisto trial and enrollment of the first patient in the Phase 2a Carpo trial. The Carpo trial is designed to evaluate TP-05, a novel investigative oral, non-vaccine pharmacological prophylactic for the potential prevention of Lyme disease in humans. The Carpo trial evaluated the efficacy of TP-05 in killing lab grown, non-disease carrying ticks after they have attached to the skin of healthy volunteers, as well as confirm the safety, tolerability, and blood concentration of TP-05. On February 22, 2024, we announced positive topline results from the Carpo trial, which demonstrated statistical significance in the mortality of ticks compared to vehicle (p<.001), regardless of treatment arm, and was well tolerated. We plan to discuss and determine the potential regulatory path with the FDA.
We believe TP-05 is currently the only non-vaccine, drug-based prophylaxis in development that targets ticks, and potentially prevents Lyme disease transmission. It is designed to rapidly and durably provide systemic blood levels of lotilaner potentially sufficient to kill infected ticks attached to the human body before they can transmit the Borrelia bacteria that causes Lyme disease.
TP-03 China Territory Out-License: In March 2021, we executed an out-license agreement (the "China Out-License") with LianBio Ophthalmology Limited ("LianBio"), granting exclusive commercial rights to TP-03 for the treatment of Demodex blepharitis and MGD within The People’s Republic of China, Macau, Hong Kong, and Taiwan (the "Territory""China Territory"). We are contractually entitled to receive (i)
In February 2023, a specified milestone was triggered based upon the signing of an agreement for which the Company has no ongoing obligations, resulting in $2.5 million recognized as license fees and collaboration revenue in the Statements of Operations for the year ended December 31, 2023. As of the date of this filing, we have received aggregate $25contractual cash proceeds from LianBio of $82.5 million, by June 30, 2021, (ii) regulatory and sales milestone receipts totaling $75representing initial consideration of $15.0 million and $100$67.5 million respectively, (iii) tiered royaltiesfor the achievement of specified milestone events. We also received equity in LianBio as part of this China Out-License, a portion of which remains subject to a China-based regulatory vesting provision.
On February 13, 2024, LianBio announced its completion of a comprehensive strategic review and determined to initiate the low double-digits onwind down of its operations, including the net salessale of TP-03 withinremaining pipeline assets, the Territory, and (iv) a minority interest in LianBio. We expect to receive $70 million from these contractual milestones during the next 12 months.delisting of its American Depositary
Corporate and Financial Overview
We were incorporated as a Delaware corporation in November 2016, and our headquarters isare located in Irvine, California. Since our inception, in November 2016, we have devoted substantially all of our resources to organizing and staffing our company, acquiring intellectual property, clinical development of our product candidates, building our research and development capabilities, raising capital, and enhancing our corporate infrastructure.
To date we have financed our operations through private placements of preferred stock, and convertible promissory notes. From inception through September 2020, we have raised net proceeds of approximately $101.0 million through private placements of preferred stock.
On October 20, 2020, we completed our initial public offering ("IPO") through an underwritten sale of 5,500,000 shares of our common stock at a price of $16.00 per share. The aggregatenotes, net proceeds from issuance of common stock in our Initial Public Offering ("IPO"), our subsequent follow-on public offerings in May 2022 (the "May 2022 Public Offering") and August 2023 (the "August 2023 Public Offering", collectively the offering (after deducting underwriting discounts"Follow-On Public Offerings"), and commissionsour sales agreement prospectus (the "2023 ATM Prospectus"), as well as proceeds from product sales, net, our China Out-License, and other offering expenses), inclusive of an additional 825,000 common shares sold upondrawdowns from the full exercise of the underwriters’ purchase option, were $91.7 million. Concurrent with the IPO, all then-outstanding shares of our convertible preferred stock outstanding (see Note 4) automatically converted into an aggregate of 11,107,018 issued common shares.Credit Facility.
In advance of the IPO, on October 8, 2020, our board of directors approved a 1-for-7.4276 reverse stock split of our capital stock. All share and per share information included in the accompanying financial statements has been adjusted to reflect this reverse stock split.
We have incurred significant net operating losses in every year since our inception and expect to continue to incur significant operating expenses as we commercialize XDEMVY for Demodex blepharitis, and, increasing operating losses for the foreseeable future.as we advance our other product candidates through clinical trials, regulatory submissions, and potential commercialization. Our net losses were $26.8loss was $135.9 million and $4.7$62.1 million for the years ended December 31, 20202023 and 2019,2022, respectively. Our net losses may fluctuate significantly from quarter to quarter and year to year and could be substantial. As of December 31, 2020 and 2019, we had an accumulated deficit of $32.8 million and $6.0 million, respectively, from research and development and general and administrative activities since our inception. We anticipate that our operating expenses will increase significantly as we:
•conduct additional clinical trials of our lead product candidate, TP-03, for the treatment of Demodex blepharitis including our Phase 2b/3 trial, Saturn-1,commercialize XDEMVY and our Phase 3 trial, Saturn-2;other products for which we obtain regulatory approvals;
•maintain regulatory approval for XDEMVY and seek regulatory approval for our other product candidates that successfully complete clinical development, if any;
•advance the clinical development of TP-03 for the potential treatment of MGD, TP-04 for the potential treatment of rosacea and TP-05 for the potential Lyme prophylaxis and community malaria reduction;
•seek regulatory and marketing approvals for product candidates that successfully complete clinical development, if any;
•establish our own sales force in the United States to commercialize our products for which we obtain regulatory approval;disease prophylaxis;
•engage with contract manufacturers to ensure a sufficient supply chain capacity to provide commercial quantities of XDEMVY and any other products for which we may obtain marketing approval;
•maintain, expand and protect our intellectual property portfolio;
•hire additional staff, including clinical, scientific, technical, regulatory, marketing, operations, financial, and other support personnel, to execute our business plan; and
•add information systems and personnel to support our product development and potential future commercialization efforts, and to enable us to operate as a public company.
We do not have any products approved for salebegan generating product sales during the year ended December 31, 2023 following the FDA approval of XDEMVY in July 2023 and we have not yet generated anyour subsequent commercial launch in August 2023. Our reported revenue within license fees and collaboration revenue is from product sales. We do notour China Out-License and clinical supply agreement; we expect to generate revenues from product sales unless and until we successfully complete clinical development and obtain regulatory approval for a product candidate and commercially launch such product. report additional revenue under this caption in future periods.
Until such time as we can generate significant revenue from product sales and achieve profitability, if ever, we expect to finance our operations through private or public equity or debt
financings, or collaborations, strategic alliances, or licensing arrangements with third parties. Adequate funding may not be available to us when needed on acceptable terms, or at all. If we raise additional funds through collaborations, strategic alliances, or licensing arrangements with third parties, we may have to relinquish valuable rights to our intellectual property, future revenue streams, research programs or product candidates or grant licenses on terms that may not be favorable to us. If we are unable to raise additional capital or enter into such agreements as and when needed, we could be forced to significantly delay, scale back, or discontinue our product development and/or commercialization plans, which would negatively and adversely affect our financial condition.
Because of the numerous risks and uncertainties associated with drug product development and commercialization, we are unable to accurately predict the timing or amount of increased expenses or when or if we will be able to achieve or maintain profitability. Even if we are able to generate significant revenue from product sales, net we may not become profitable. If we fail to become profitable or are unable to sustain profitability on a continuing basis, then we may be unable to continue our operations at planned levels.
As of December 31, 2020,2023, our aggregate cash, and cash equivalents and marketable securities was $168.1$227.4 million – see “Liquiditythe section below titled "Management's Discussion and Analysis of Financial Condition and Results of operations —
Liquidity and Capital Resources.”"
Impact of the COVID-19 Pandemic on our OperationsMacroeconomic Environment
EffortsRecently, the economy has experienced downward pressure, and together with high rates of inflation and energy supply issues experienced in certain regions, war and geopolitical conflicts, have led to containregional and/or global macroeconomic challenges, the spreadeffects of COVID-19 in the United States (including in California where our corporate headquarters and laboratory facility are located) and other countries have included quarantines, shelter-in-place orders, and various other government restrictions in order to control the spread of this virus.
We have been carefully monitoring the COVID-19 pandemic as it continues to progress and its potential impact on our business. We have taken important steps to ensure the workplace safety of our employees when working within our laboratory and administrative offices, or when traveling to our clinical trial sites. We have also implemented an interim work-from-home policy and we may take further actions aswhich may be required by federal, state or local authorities.of an extended duration.
To date,In addition, we have been ablemay be exposed to continuecredit risk on deposits at financial institutions to the extent our key business activities and advance our clinical programs. However,account balances exceed the amount insured by the Federal Deposit Insurance Corporation (“FDIC”). We maintain cash held in deposit at financial institutions in the future, it is possibleU.S., including SVB, a division of First Citizens Bank. While these deposits are insured by the FDIC in an amount up to $250,000 for any depositor, to the extent we hold cash deposits in amounts that our clinical development timelines and business plans could be adversely affected. We maintain regular communication with our vendors and clinical sites to appropriately plan for, and mitigate,exceed the impactFDIC insurance limitation, we may incur a loss in the event of a failure of any of the COVID-19 pandemic onfinancial institutions where we maintain deposits. We invest our operations. Specifically, for our Phase 2b/3 Saturn-1 trial, we have instituted various protocols for our sites,excess cash in highly liquid investments, including increasing health screening of individuals and providing enhanced communication and trainingmoney market fund accounts, that are readily convertible into cash without penalty. We believe the Company is not exposed to staff regarding COVID-19. We have also over-enrolled trial participants and identified additional clinical sites in case there are site closuressignificant credit risk due to COVID-19. However, the ultimate effect from this pandemic on our development timelines for TP-03financial position of the depository institution and ourthe types of accounts we hold, but we will continue to monitor regularly and adjust, if needed, to mitigate risk, including any ongoing or new events involving limited liquidity, defaults, non-performance or other product candidates is inherently uncertain.adverse developments that affect financial institutions.
See the section titled “Risk Factors” in this reportRisk Factors for a further discussion of the potential adverse impact of COVID-19unfavorable global and geopolitical economic conditions on our business, results of operations and financial condition.
Components of our Results of Operations
Operating ExpensesProduct Sales, Net
We recognize product sales, net of XDEMVY when a customer obtains control of promised goods or services, which occurs at a point in time, typically upon delivery of the Company's product to the customer. We record the amount of revenue that reflects the consideration we expect to receive in exchange for those goods or services. We apply the following five-step model in order to determine this amount: (i) identification of the promised goods in the contract; (ii) determination of whether the promised goods are performance obligations, including whether they are capable of being distinct; (iii) measurement of the transaction price, including the constraint on variable consideration; (iv) allocation of the transaction price to the performance obligations; and (v) recognition of revenue as each performance obligation is satisfied.
We sell XDEMVY to customers in the U.S., which became available for commercial sale during the third quarter of 2023. We sell XDEMVY to a limited number of specialty pharmacies and distributors (i.e., its customers) who in turn sell it directly to clinics, hospitals, pharmacies and federal healthcare programs. Revenue from product sales is primarily recognized upon physical delivery of the product (when the customer obtains control of the product), in return for agreed-upon consideration. Shipping and handling activities are considered to be fulfillment activities rather than a separate performance obligation and are recorded within selling, general and administrative expenses in the Statements of Operations and Comprehensive Loss.
Revenues from product sales are recorded at the net sales price, or the transaction price, which may include fixed or variable consideration for (i) invoice discounts for prompt payment and distribution service fees, (ii) government and private payer rebates, chargebacks, discounts and fees, (iii) product returns and (iv) costs of co-pay assistance programs for patients, as well as other incentives. Estimates of variable consideration are calculated based on the actual product sales each reporting period and the nature of the variable consideration related to those sales. Where appropriate, we utilize the expected value method to determine the appropriate amount for estimates of variable consideration based on factors such as the current contractual and statutory requirements, specific known market events and trends, industry data and forecasted customer buying and payment patterns. The amount of variable consideration that is included in the transaction price may be constrained and is included in product sales, net only to the extent that it is probable that a significant reversal in the amount of the cumulative
revenue recognized will not occur when the uncertainty associated with the variable consideration is subsequently resolved. These estimates reflect our best estimate of the amount of consideration to which we expect to be entitled based on the terms of the contract. Actual amounts of consideration ultimately received may differ materially from estimates. If actual results in the future vary from estimates, we will adjust these estimates, which would affect product sales, net and earnings in the period such variances are adjusted. We categorize product sales deduction estimates as follows:
Distribution Service Fees: We engage with wholesalers and specialty pharmacies to distribute our products to end customers. We pay the wholesalers and certain specialty pharmacies a fee for services such as: inventory management, chargeback administration, and service level commitments. We estimate the amount of distribution services fees to be paid to the customers and adjust the transaction price with the amount of such estimate at the time of sale to the customer. An accrued liability is recorded for unpaid distribution service fees.
Prompt Pay Discounts: We provide our customers with a percentage discount on their invoice if the customers pay within the agreed upon timeframe. We expect that our customers will earn prompt pay discounts. We estimate the probability of customers paying promptly based on the percentage of discount outlined in the purchase agreement between the two parties, and deducts the full amount of these discounts from gross product sales and accounts receivable at the time revenue is recognized.
Product Returns: Our operating expensescustomers are contractually permitted to return the product within the contractual allowable time before and after the applicable expiration date. In the initial sales period, we estimate a provision for returns based on industry data and adjust the transaction price at the time of the product sale to the customer. Once sufficient history has been collected for product returns, we will utilize that history to inform its returns estimate. Once the product is returned, it is destroyed since inceptionit cannot be resold.
Chargebacks: A chargeback is the difference between our invoice price to the wholesaler and the wholesaler’s customer's contract price. The wholesaler tracks these sales and charges us back for the difference between the negotiated prices paid between the wholesaler's customers and wholesaler's acquisition cost. We estimate the percentage of goods sold that are eligible for chargeback and adjust the transaction price and accounts receivable at the time of sale of the product to the customer.
Co-payment Assistance: Patients who meet certain eligibility requirements may receive co-payment assistance. We record contra-revenue for co-payment assistance based on actual program participation and estimates of program redemption using data provided by third-party administrators. An accrued liability is recorded on unredeemed co-payment assistance related to products for which control has been transferred to the customer.
Rebates and Discounts: We accrue rebates for contractually agreed-upon discounts with commercial insurance companies and mandated discounts under government programs such as the Medicaid Drug Rebate Program, Medicare Part D Prescription Drug Program, and other government health care programs in the U.S. We estimate for expected utilization of commercial insurance rebates based on data received from our customers. The estimates for rebates under government programs are based on statutory discount rates and expected utilization as well as historical data we have consisted solelyaccumulated since product launch. Our rebate calculations may require estimates, including estimates of customer mix, to determine which product sales will be subject to rebates and the amount of such rebates. We update our estimates and assumptions on a quarterly basis and record any necessary adjustments to revenue in the period identified. Rebates are generally invoiced and paid in arrears so that the accrual balance consists of an estimate of the amount expected to be incurred for the current quarter’s activity, plus an accrual balance for known prior quarters’ unpaid rebates. If actual rebates vary from estimates, we may need to adjust accruals, which would affect product sales, net in the period of adjustment. An accrued liability is recorded for unpaid rebates related to product for which control has transferred to the customer.
License Fees and Collaboration Revenue
License fees and collaboration revenue is primarily attributable to contractual milestones under the China Out-License. These amounts represent the contractual milestones achieved or allocated under the China Out-License that had been fully or partially completed by period end. These allocated amounts represented the satisfaction of the transfer of license rights to LianBio and the completion of related performance obligations. License fees and collaboration revenue also includes the satisfaction of performance obligations under an existing clinical supply agreement.
We will recognize additional license fees and collaboration revenue under the China Out-License to the extent other events occur, specifically related to (i) milestone achievement of an additional drug supply agreement execution, (ii) milestone achievement of certain regulatory events in the China Territory, and (iii) royalties and milestones from our licensee's
product sales of TP-03 in the China Territory. As of the date of this filing, it is uncertain if and when we will receive any royalties or future milestone consideration under the China Out-License, including but not limited to the milestone achievement of an additional drug supply agreement execution.
Cost of Sales
Cost of sales consists of direct and indirect costs related to the manufacturing and distribution of XDEMVY, including raw materials, third-party manufacturing costs, packaging services, and freight-in, as well as third-party royalties payable on our product sales, net and amortization of capitalized intangible assets associated with XDEMVY. Cost of sales may also include period costs related to certain inventory warehouse and distribution operations and inventory adjustment charges. Prior to FDA approval of XDEMVY, manufacturing and other inventory costs were recorded to research and development expensesexpenses. Therefore, cost of sales of XDEMVY will reflect a lower average per unit cost until the related inventory is sold, which we expect to sell during 2024.
Cost of License Fees and generalCollaboration Revenue
Cost of license fees and administrative expenses.collaboration revenue includes the proportion of expense recognized under the terms of the China Out-License payable under the terms of our in-license agreement for lotilaner.
Research and Development Expenses
Our researchResearch and development expenses consist of expenses incurred in connection with the development of our product candidates, including:
•fees paid to third parties to conduct certain research and development activities on our behalf, including under agreements with CROs;
•payments under licensing agreements, such as our upfront in-license fee for lotilaner;
•consulting costs and certain allocated payroll, employee benefit and other employee-related expensescosts (including stock-based compensation, and salaries)salaries, payroll taxes) for personnel engaged in research and development functions;
•costs related to compliance with clinical regulatory requirements;
•costs of procuring drug products for use in our preclinical studies and clinical trials; and
•facilities expenses, which include direct and allocated expenses for rent of our laboratory.
We expense both internal and external research and development expenses as incurred or as certain upfront or milestone payments become contractually due to licensors upon achievement of clinical or regulatory events. We recognize external research and development costs based on an evaluation of the progress-to-completion of (i) specific tasks performed or deliverables provided by CROs and contract manufacturing organizations ("CMOs")CMOs and (ii) patient visits for dosing or other follow-up. To estimate period expense for recognition, we use information provided to us by our service providers and we then apply the corresponding fee schedule.
We track our external research and development expenses on a program-by-program basis, such as fees paid to CROs, CMOs, and research laboratories in connection with our pre-clinical development, process development, manufacturing and clinical development activities. However, we do not currently track employee time on a program-by-program basis. ForPrior to commercialization of XDEMVY, for the years ended December 31, 20202023 and 2019, substantially all2022, the vast majority of our external and internal research and development expenses arewere attributable to our TP-03 program for Demodex blepharitis.the development of XDEMVY.
We expect our research and development expenses to increase substantially in the future, as we seek to initiate and progress additional clinical trials for our product candidates, including TP-03 for the potential treatment of MGD, TP-04 for the potential treatment of rosacea, and TP-05 for potential Lyme prophylaxis and community malaria reduction. We expect to complete our clinical programs for these product candidates, and as appropriate, pursue regulatory approval and prepare for the possible commercialization for each.
Selling, General and Administrative Expenses
OurSelling, general and administrative expenses consist of personnel-related costs including payroll,salaries, benefits, and stock-based compensation and other personnel-related expenses for our executive, finance, sales and marketing, and other administrative functions. Other selling, general and administrative expenses include sales and marketing costs to support our commercial launch, consulting fees, legal services, rent and other facilities costs, patient assistance donations, and other general operating expenses, not otherwise classified as research and development expenses.
We expect that our selling, general and administrative expenses will increase substantially in the future as a result of expanding our operations, including hiring personal,personnel, continued commercialization of XDEMVY, preparing for potential commercialization of our other product candidates, and additional facility occupancy costs, as well as various incremental costs associated with being a public company, (includingincluding: increased legal and accounting fees, regulatory costs associated with maintaining compliance with the rules of the Nasdaq Stock Market and SEC regulations, investor relations activities, directors and officers liability insurance premiums, and other accompanying compliance and governance costs).costs.
Other Income, (Expense), Net
Other income, (expense), net primarily consists primarily of (i) interest income earned on our cash, and cash equivalents, and marketable securities, (ii) interest expense on convertible promissory notes (converted tothe Credit Facility, and (iii) the change in estimated fair value of the LianBio equity instruments in December 2019; comprisedwarrants and LianBio common stock we received as part of coupon interest, amortization of debt issuance costs, and non-cash accretion of its estimated discount at issuance).the China Out-License.
Benefit from Income Tax ProvisionTaxes
Since our inception, we have not recorded any U.S. federal or state income tax benefits for the net operating losses we have incurred in each year, or for our earned research and development tax credits, due to our uncertainty of realizing a benefit from either. As a result of the Tax Cuts and Jobs Act of 2017, net operating losses (for U.S. income tax purposes) generated prior to December 31, 20182017 can be carried forward for up to 20 years, while net operating losses generated after December 31, 2017 can be carried forward indefinitely, but are limited to 80% utilization against taxable income. Our California net operating losses will begin to expire in 2037. The federal research and development tax credits begin to expire in 20372040 unless previously utilized, and the California credit carryforwards are available indefinitely.
Results of Operations
Comparison of the Years Ended December 31, 20202023 and 20192022
The following table summarizes our results of operations for the periods indicated:
| | | | | | | | | | | | | | | | | |
| | Year Ended
December 31, | | |
| | 2023 | | 2022 | | Change |
| | (in thousands) | | |
| Revenues: | | | | | |
| Product sales, net | $ | 14,729 | | | $ | — | | | $ | 14,729 | |
| License fees and collaboration revenue | 2,718 | | | 25,816 | | | (23,098) | |
| Total revenues | 17,447 | | | 25,816 | | | (8,369) | |
| Operating expenses: | | | | | |
| Cost of sales | 1,593 | | | — | | | 1,593 | |
| Cost of license fees and collaboration revenue | — | | | 955 | | | (955) | |
| Research and development | 50,312 | | | 42,624 | | | 7,688 | |
| Selling, general and administrative | 108,700 | | | 44,949 | | | 63,751 | |
| Total operating expenses | 160,605 | | | 88,528 | | | 72,077 | |
| Loss from operations | (143,158) | | | (62,712) | | | (80,446) | |
| Other income (expense): | | | | | |
| Interest income | 10,337 | | | 3,499 | | | 6,838 | |
| Interest expense | (3,346) | | | (2,199) | | | (1,147) | |
| Other (expense) income, net | (102) | | | 86 | | | (188) | |
| Unrealized gain (loss) on equity investments | 259 | | | (268) | | | 527 | |
| Change in fair value of equity warrants issued by licensee | 117 | | | (501) | | | 618 | |
| Total other income, net | 7,265 | | | 617 | | | 6,648 | |
| Loss before income taxes | (135,893) | | | (62,095) | | | (73,798) | |
| Benefit from income taxes | — | | | 4 | | | (4) | |
| Net loss | $ | (135,893) | | | $ | (62,091) | | | $ | (73,802) | |
Product Sales, Net
During the year ended December 31, 2023, in conjunction with the launch of XDEMVY, we recognized revenue of $14.7 million from product sales, net of rebates, chargebacks, discounts, and other adjustments driven by approximately 17,400 prescriptions of XDEMVY to patients. During the year ended December 31, 2022, there were no product sales, net recognized.
License Fees and Collaboration Revenue
For the years ended December 31, 20202023 and 2019 (in thousands):2022, we recognized $2.5 million and $25.8 million, respectively, of license fees and collaboration revenue primarily attributable to contractual milestones under the China Out-License. These amounts represent the contractual milestones achieved or allocated under the China Out-License that have been fully or partially completed by the period ends. These allocated amounts represented the satisfaction of the transfer of license rights to LianBio and the completion of related performance obligations.
| | | | | | | | | | | | | | | | | |
| | Year Ended
December 31, | | |
| | 2020 | | 2019 | | Change |
| | (in thousands) | | |
| Operating expenses: | | | | | |
| Research and development | $ | 18,826 | | | $ | 3,162 | | | $ | 15,664 | |
| General and administrative | 8,172 | | | 1,136 | | | 7,036 | |
| Total operating expenses | 26,998 | | | 4,298 | | | 22,700 | |
| Loss from operations before other income (expense) and income taxes | (26,998) | | | (4,298) | | | (22,700) | |
| Other income (expense): | | | | | |
| Interest income (expense), net | 188 | | | (40) | | | 228 | |
| Loss on extinguishment of convertible notes | — | | | (255) | | | 255 | |
| Change in fair value of derivative liabilities | — | | | (76) | | | 76 | |
| Total income (expense), net | 188 | | | (371) | | | 559 | |
| Provision for income taxes | (1) | | | (1) | | | — | |
| Net loss | $ | (26,811) | | | $ | (4,670) | | | $ | (22,141) | |
We will recognize additional license fees and collaboration revenue under the China Out-License to the extent other events occur, specifically related to (i) milestone achievement of an additional drug supply agreement execution, (ii) milestone achievement of regulatory events in the China Territory, and (iii) royalties and milestones from our licensee's product sales of TP-03 in the China Territory. As of the date of this filing, it is uncertain if and when we will receive any future royalties or milestone consideration under the China Out-License, including but not limited to the milestone achievement of an additional drug supply agreement execution.
Cost of Sales
For the year ended December 31, 2023, we recognized $1.6 million in cost of sales for XDEMVY. Cost of sales consists of direct and indirect costs related to the manufacturing and distribution of XDEMVY, including raw materials, third-party manufacturing costs, packaging services, and freight-in, as well as third-party royalties payable on our product sales, net and amortization of capitalized intangible assets associated with XDEMVY. During the year ended December 31, 2022, there were no cost of sales recognized.
Cost of License Fees and Collaboration Revenue
For the year ended December 31, 2023, we did not recognize any cost of license fees and collaboration revenue. For the year ended December 31, 2022, cost of license fees and collaboration revenue was $1.0 million. This amount relates to our contractual payment obligations to Elanco, in proportion to our recognized license fee and collaboration revenue under the China Out-License arrangement in the same period.
Research and Development Expenses
| | | | | | | | | | | | | | | | | |
| December 31, | | Change |
| 2023 | | 2022 | |
| Direct external expenses: | | | | | |
| TP-03 program | $ | 14,311 | | | $ | 20,405 | | | $ | (6,094) | |
| TP-04 program | 2,583 | | | 3,638 | | | (1,055) | |
| TP-05 program | 5,261 | | | 3,205 | | | 2,056 | |
| Other early-stage programs | 619 | | | — | | | 619 | |
| Indirect expenses: | | | | | |
| Compensation and personnel-related | 24,129 | | | 13,533 | | | 10,596 | |
| Other | 2,409 | | | 1,843 | | | 566 | |
| Elanco milestone expenses | 1,000 | | | — | | | 1,000 | |
| Total research and development expenses | $ | 50,312 | | | $ | 42,624 | | | $ | 7,688 | |
Research and development expenses increased by $15.7$7.7 million for the year ended December 31, 2020.2023, as compared to the year ended December 31, 2022. The increase was primarily due to (i) an upfront payment to our licensor (in the form$10.6 million of our common stock) then valued at $3.1 million as consideration for the September 2020 expansion of our in-license for lotilaner to cure or treat all diseases and conditions in humans (beyond that of the eye or skin for which we already held exclusive rights to treat with lotilaner), (ii) TP-03 clinical milestone achievement in September 2020, requiring a $1.0 million payment to our licensor, (iii) increased manufacturing, development, and research activities of $10.7 million to conduct our clinical studies for TP-03, and (iv) increased payroll and personnel-related expensespersonnel-
related costs (including increased stock-based compensation)compensation expense of $1.7 million due to additional clinical and formulation development employees$2.1 million), for 25 employee additions year-over-year to drive our product development initiatives.initiatives, (ii) $0.6 million of increased other indirect expenses, (iii) $1.0 million of milestone expense related to our in-license agreement with Elanco, (iv) $2.1 million of increased TP-05 program expenses primarily related to the Carpo trial initiated in December 2022 and the new food effect study initiated during the first quarter of 2023, and (v) $0.6 million of increased spend related to other early-stage programs. These increases were partially offset by decreases in direct external expenses for the TP-03 and TP-04 programs of $6.1 million and $1.1 million, respectively. The decrease in our TP-03 program expenses was primarily due to significantly reduced clinical trial costs of $6.9 million given the completion of our Saturn-2 trial in the first half of 2022 and $1.0 million of reduced preclinical study expenses, partially offset by $0.9 million of increased API manufacturing and $0.6 million of increased clinical trial expenses related to the Ersa trial. The decrease in our TP-04 program expenses was primarily due to $1.4 million of decreased preclinical expenses and $0.9 million of decreased clinical trial expenses from the Phase 1 Galatea trial, partially offset by $1.4 million of increased clinical trial expenses related to the Phase 2a Galatea trial that began in March 2023.
Selling, General and Administrative Expenses
GeneralSelling, general and administrative expenses increased by 7.0$63.8 million for the year ended December 31, 2020.2023, as compared to the year ended December 31, 2022. The increase was primarily due to (i) $2.5$27.7 million increase inof increased payroll and personnel-related expensescosts (including increased stock-based compensation)compensation expense of $4.1 million) for 157 employee additions year-over-year to support our business growth and commercial leadership hires for our recent commercial launch of XDEMVY, (ii) $2.2$0.9 million increaseof severance costs related to our former Chief Financial Officer's separation from the Company in legal serviceJune 2023, (iii) $22.3 million of increased commercial costs as we continued our commercial expansion and market research activities, and (iii) $2.3prepared for the recent commercial launch of XDEMVY, (iv) $6.2 million increase in consulting, insurance,of increased IT applications, legal and other professional expenses to support the continued growth and expansion of our corporate infrastructure and (v) $6.5 million of increased facilities and office and administrative expenses.
Interest Income (Expense), Net
The change in interest income (expense), net was primarily Sales and marketing headcount and associated vendor spend increased significantly during 2023 due to convertible promissory notes issued to our co-founderscommercial launch and certain other related parties, aggregating $2.0activities for XDEMVY.
Other Income, Net
Other income, net increased by $6.6 million in principal value (see below). During the year ended December 31, 2019, interest expense recognized on these notes was $0.1 million and was partially offset by interest income from our bank deposits and money market fund investments. These convertible promissory notes were all converted in December 2019 and we therefore had no interest expense for the year ended December 31, 2020.
Loss on Extinguishment of Convertible Notes
2023. The loss on extinguishment of convertible notes wasincrease is primarily due to (i) $6.8 million of increased interest income earned on our cash, cash equivalents and marketable securities, (ii) $0.6 million change in estimated fair value of the extinguishmentLianBio equity warrants we received as part of convertible promissory notes issued to our co-foundersChina Out-License in March 2021, and certain other related parties, aggregating $2.0(iii) $0.5 million in principal value (see section below). On December 13, 2019 we completed the issuance of Series B preferred stock upon which the then-outstanding notes, along with accrued interest, converted into 2.0 million shares of Series B preferred stock. Since these notes were converted in December 2019, no change in fair value of derivative liabilities was recorded for the year endedLianBio common stock. These increases to other income were partially offset by increased interest expense of $1.1 million on the Credit Facility.
Benefit from Income Taxes
We maintain a valuation allowance against our net deferred tax assets as of December 31, 2020.
Change in Fair Value of Derivative Liabilities
During May, August, and October 2019, we issued convertible promissory notes to our co-founders and certain other related parties, aggregating $2.0 million in principal value. These notes contained stock-settled redemption features that were required to be separately accounted for as derivative liabilitiesdeferred tax assets on the balance sheet until December 13, 2019 when we completed a qualified equity financing.
These then-outstanding notes converted at the option of the holder into 2.0 million shares of Series B preferred stock. Change in fair value of derivative liabilities consists of non-cash changes in the fair value of these stock-settled redemption features. We classified the rights as a derivative liability on our balance sheet that was initially recorded at fair value and that we remeasured to fair value at December 13, 2019, and we recognized changes in the fair value of the derivative associated with the rights as a component of other income (expense) in our statement of operations. Since these notes were converted in December 2019, no change in fair value of derivative liabilities was recorded for the year ended December 31, 2020.least an annual basis.
Liquidity and Capital Resources
Sources of Liquidity
Overview
Since our inception, in 2016 and through December 31, 2020,we have financed our operations have been substantially financed by cash proceeds of $61.0 million fromthrough private placements of Series Apreferred stock, net proceeds from the issuance of common stock through our IPO, Follow-on Public Offerings, and Series B preferred stock. the 2023 ATM Prospectus, as well as proceeds from product sales, net, the China Out-License, and drawdowns from the Credit Facility. As of December 31, 2023, we had cash, cash equivalents and marketable securities of $227.4 million.
IPO, Follow-On Public Offerings and ATM Prospectus
In Septemberconnection with our October 2020 IPO, we issued 2,857,079sold 6,325,000 shares of our Series C preferred stock for cash proceeds of $39.8 million. On October 20, 2020, we completed our IPO through an underwritten sale of 5,500,000 shares of its common stock at a price(inclusive of $16.00 per share. The aggregate proceeds from the IPO, inclusive of an additional 825,000 common shares sold upon the full exercise of the underwriters' option to purchase 825,000 shares of common stock). After deducting underwriting discounts, commissions and other related expenses, our IPO proceeds were $91.7 million.
In May 2022, we completed the May 2022 Public Offering in which 5,600,000 shares of our common stock were sold at a public offering price of $13.50 per share. We also granted the underwriters a 30-day option to purchase up to 840,000 additional shares of common stock at the public offering price, less underwriting discounts and commissions. In June 2022, the
underwriters partially exercised their option to purchase an additional 289,832 shares of common stock at the offering price of $13.50 per share, before underwriting discounts and commissions. After giving effect to the exercise of the underwriters’ purchase option, we sold 5,889,832 shares for total net proceeds received of $74.2 million, after underwriting discounts, commissions and other offering-related expenses.
In August 2023, we completed the August 2023 Public Offering in which 5,714,285 shares of our common stock were sold at a public offering price of $17.50 per share for aggregate net proceeds received of approximately $93.5 million, after deducting underwriting discounts, and commissions, and other IPO expenses, were $91.7 million.offering-related expenses. We also granted the underwriters a 30-day option to purchase up to 857,142 additional shares of our common stock at the public offering price. In September 2023, the underwriters partially exercised this option and the sale of an additional 355,164 shares of common stock at the public offering price of $17.50 per share. After giving effect to the exercise of the underwriters' option, we sold a total of 6,069,449 shares and received aggregate net proceeds of $99.3 million, after deducting underwriting discounts, commissions, and other offering-related expenses.
We will continueIn December 2023, we sold 1,000,000 shares of our common stock for $20.00 per share under a sales agreement prospectus filed in November 2023, pursuant to be dependent upon equity, debt financing, and/or other formsthe 2023 Shelf Registration Statement (defined below) covering the sale of capital raises at least untilup to $100.0 million of our common stock pursuant to an Open Market Sale AgreementTM (the "2023 ATM Prospectus") with Jefferies LLC ("Jefferies"). This resulted in net proceeds of $19.2 million, after deducting broker commissions and offering related expenses.
China Out-License
As of the date of this filing, we are ablehave received $82.5 million of total proceeds in connection with our China Out-License. In February 2024, LianBio announced its plan to generate significant positive cash flows from ourwind down its operations. As of December 31, 2020,the date of this filing, it is uncertain if and when we had cashwill receive any royalties or future milestone consideration under the China Out-License, including the specified milestone of $2.5 million and cash equivalentsthe remaining $120.0 million of $168.1potential future regulatory and sales achievement milestones.
Credit Facility
In February 2022, we executed the Credit Facility with Hercules and SVB. Capital draws are at our election and are in $5.0 million increments. Concurrent with the execution of the Credit Facility we drew $20.0 million. This Credit Facility was amended in January 2023 and August 2023. The Credit Facility, as amended, set a maximum interest rate, updated the terms of prepayment under the Credit facility and includes an extended period to drawdown the tranche associated with the NDA submission, from March 15, 2023 to March 15, 2024 provided at least $5.0 million was drawn on or before March 15, 2023 and at least an additional $5.0 million was drawn on or before September 15, 2023. On March 15, 2023 and September 15, 2023, respectively, we made draws of $5.0 million (including SVB's commitment of $1.25 million) from the $25.0 million tranche associated with the NDA submission of TP-03. The Credit Facility includes a four-year period of interest-only payments and is extendable for a fifth year to February 2027 maturity, upon our expected achievement of required conditions. We currently have no ongoing materialother financing commitments, such as lines of credit or guarantees, that are expectedguarantees.
As of the date of this filing, we have $125.0 million of remaining tranched availability as follows:
•$15.0 million, which became available in September 2022 upon our NDA submission of TP-03 to affect our liquidity over the next five years.FDA;
•$35.0 million, which became available in July 2023 upon FDA approval of XDEMVY;
•$50.0 million upon achievement of certain quarterly revenue thresholds; and
•$25.0 million available with lender approval.
Funding Requirements
Liquidity
Our primary use of cash is to fund operating expenditures. Theseexpenditures currently consist of cost of sales, research and development expensescosts (including activities within our pre-clinical,preclinical, clinical, regulatory, and drug manufacturing initiatives) and selling, general and administrative expenses.costs. Our use of cash is impacted by the timing and extent of the required payments for each of these activities.activities and other business requirements. We have incurred significant losses and negative cash flows from operations since our inception and had an accumulated deficit of $244.7 million as of December 31, 2023.
We believe that our cash, and cash equivalents and marketable securities of $168.1$227.4 million as of December 31, 2020, and our expected $70 million of initial proceeds from our March 2021 out-license of TP-03 in the China territory (see above and Note 11), will enable us2023 is sufficient to fund our current and planned operations for at least the next twelve months from the date of filing this Annual Report on Form 10-K. We also anticipate having at least $50.0 million of available capital from our Credit Facility through March 2024 and an additional $75.0 million of additional tranched availability through December 2024. The Credit Facility requires interest-only debt service payments that are expected to remain through its maturity in February 2027 and its remaining tranches are subject to undrawn expiry in either March 2024 or December 2024. Our cash runway estimate is predicated on current assumptions for future revenue, operating expenses, and capital expenditure requirements into the first half of 2023. We have based this cash runway estimate on our current assumptions. These assumptionsdebt availability and may require future adjustments as part of our ongoing business decisions within pipeline development and other corporate initiatives.adjustments. Accordingly, we may requirebe required to raise additional capital resources earlier than we currently expect.expect based on our cash requirements and market dynamics.
To date,Shelf Registration Statements
On November 9, 2023, we have not generatedfiled a shelf registration statement on Form S-3 that was declared effective by the SEC on November 21, 2023, (the "2023 Shelf Registration Statement"), which replaced the November 2021 Shelf Registration Statement, as defined below, and permits us to offer up to $300.0 million of common stock, preferred stock, debt securities and warrants in one or more offerings and in any product revenue. We do not expectcombination, including in units from time to generate any product revenue unlesstime.
As part of the 2023 Shelf Registration Statement, we concurrently filed the 2023 ATM Prospectus with Jefferies. Under the terms of the 2023 ATM Prospectus, Jefferies will act as the Company's sales agent and until we (1) complete developmentis entitled to compensation for its services equal to 3% of the gross proceeds of any shares of common stock sold. In December 2023, we sold 1,000,000 shares of our product candidates; (2) obtain applicable regulatory approvals;common stock under the 2023 ATM Prospectus for $20.00 per share and (3) successfully commercializereceived net proceeds of $19.2 million, after deducting broker commission and offering-related expenses.
On November 1, 2021, we filed a shelf registration statement on Form S-3 that was declared effective by the SEC on November 5, 2021 (the “2021 Shelf Registration Statement”), which permitted us to offer up to $300.0 million of common stock, preferred stock, debt securities and warrants in one or enter into other collaborative agreements formore offerings and in any combination, including in units from time to time. The 2023 Shelf Registration Statement replaced the 2021 Shelf Registration Statement.
Also, as part of the 2021 Shelf Registration Statement, we concurrently filed a sales agreement prospectus covering the sale of up to $100.0 million of our product candidates. common stock pursuant to an Open Market Sale AgreementTM (the “2021 ATM Prospectus”) with Jefferies LLC.We dodid not knowsell any shares of our common stock under the 2021 ATM Prospectus. On July 31, 2023, in connection with certainty when, or if, any of these items will ultimately occur. However,the August 2023 Public Offering, we expectterminated the sales agreement prospectus relating to recognize "license fee revenue" in the first quarter of 2021 for our TP-03 out-license in the China territory.ATM Prospectus.
Other Liquidity Risks
We expect to incur significant operating losses for the foreseeable future, and for these losses to further increase, as we ramp upexpand our clinical development programs for our other product candidates and begin activities forgiven the recent commercial launch readiness.of XDEMVY. We may also encounter unforeseen expenses, difficulties, complications, delays and other currently unknown factors that could adversely affect our business.
We will likelymay require additional capital to fully develop our product candidates and to execute our business strategy. Our requirements of a future capital raise will depend on many factors, including:
110•the amount of revenue received from commercial sales of XDEMVY or our product candidates, should any of our product candidates receive marketing approval;
the cost and timing associated with commercializing XDEMVY or our product candidates, if they receive marketing approval; •the scope, timing, rate of progress and costs of our drug discovery efforts, preclinical development activities, laboratory testing and clinical trials for our product candidates;
•the number and scope of clinical programs we decide to pursue;
•the cost, timing and outcome of preparing for and undergoing regulatory review of our product candidates;
•the scope and costs of development and commercial manufacturing activities;
•the cost and timing associated with commercializing our product candidates, if they receive marketing approval;Table of Content•the amount of revenue, if any, received from commercial sales of our product candidates, should any of our product candidates receive marketing approval;
•the achievement of milestones or occurrence of other developments that trigger payments under any collaboration agreements we might have at such time;time and availability of our Credit Facility;
•the extent to which we acquire or in-license other product candidates and technologies;
•the costs of preparing, filing and prosecuting patent applications, maintaining and enforcing our intellectual property rights and defending intellectual property-related claims;
•our ability to establish and maintain collaborations on favorable terms, if at all;
•our efforts to enhance operational systems and our ability to attract, hire and retain qualified personnel, including personnel to support the development of our product candidates and, ultimately, the sale of our products, following FDA approval;
•our implementation of various computerized information systems;
•impact of COVID-19health epidemics on our clinical development or operations; and
•the costs associated with being a public company.
A change in the outcome of any of these or other variables with respect to the development of any of our product candidates could significantly change the costs and timing associated with the development of that product candidate. Furthermore, our operating plans may change in the future, and we will continue to require additional capital to meet operational needs and capital requirements associated with such operating plans. If we raise additional funds by issuing equity securities, our stockholders may experience dilution. Any future debt financing into which we enter may impose upon us additional covenants that restrict our operations, including limitations on our ability to incur liens or additional debt, pay dividends, repurchase our common stock, make certain investments or engage in certain merger, consolidation or asset sale transactions. Any debt financing or additional equity that we raise may contain terms that are not favorable to us or our stockholders.
Adequate funding may not be available to us on acceptable terms or at all. Our potential inability to raise capital when needed could have a negative impact on our financial condition and our ability to pursue our business strategies. If we are unable to raise additional funds as required, we may need to delay, reduce, or terminate some or all development programs and clinical trials. We may also be required to sell or license our rights to product candidates in certain territories or indications that we would otherwise prefer to develop and commercialize ourselves. If we are required to enter into collaborations and other arrangements to address our liquidity needs, we may have to give up certain rights that limit our ability to develop and commercialize our product candidates or may have other terms that are not favorable to us or our stockholders, which could materially and adversely affect our business and financial prospects. See the section of this Annual Report on Form 10-K titled “Risk Factors” for additional risks associated with our substantial capital requirements.
Convertible NotesContractual Obligations and Commitments
From May 2019 through October 2019,We have entered into arrangements that contractually obligate us to make payments that will affect our liquidity and cash flows in future periods. Such arrangements include those related to the contractual obligations described below:
Lease Commitments
Our operating lease commitments reflect payments due for our active lease agreements in Irvine, California, for adjacent office and laboratory suites which expire on January 31, 2027. As of December 31, 2023, our contractual commitments for our leases were $2.4 million, which will be paid over the remaining lease term of 3.1 years.
Purchase Obligations
As of December 31, 2023, we issued convertible promissory notes with an aggregate principal amounthave entered into manufacturing supply agreements for the commercial supply of $2.0 million.XDEMVY. These notes wereamounts do not represent all converted into an aggregate of 268,056 sharesour anticipated purchases, but instead represent the contractually obligated minimum purchases or firm commitments of Series B preferred stock in December 2019 (see Note 4).non-cancelable minimum amounts, as follows:
| | | | | | | | |
| | December 31, 2023 |
| 2024 | | $ | 2,962 | |
| 2025 | | 2,829 | |
| 2026 | | 3,379 | |
| 2027 | | 3,798 | |
| 2028 | | 4,500 | |
| Thereafter | | 4,590 | |
| Total | | $ | 22,058 | |
Milestone Obligations
The terms of our Eye and Derm Elanco Agreement and All Human Uses Elanco Agreement require us to make future development milestone payments aggregating up to $6.0 million and future commercial and sales-based milestone payments aggregating up to $152.0 million upon our achievement of the specified milestones. The amount and timing of such obligations are unknown or uncertain as of December 31, 2023.
Summary Statement of Cash Flows
The following table sets forth the primary sources and uses of cash and cash equivalents and restricted cash for each of the periods presented below:
| | | | Year Ended
December 31, | | Year Ended
December 31, |
| | | 2020 | | 2019 | | 2023 | | 2022 |
| | | (in thousands) | | (in thousands) |
| Net cash (used in) provided by: | |
| Net cash provided by (used in): | |
| Operating activities | |
| Operating activities | |
| Operating activities | Operating activities | $ | (21,138) | | | $ | (3,673) | |
| Investing activities | Investing activities | (456) | | | (175) | |
| Financing activities | Financing activities | 131,771 | | | 59,445 | |
| Net increase in cash, cash equivalents and restricted cash | $ | 110,177 | | | $ | 55,597 | |
| Net increase (decrease) in cash and cash equivalents | |
Net Cash Used in Operating Activities
Net cash used in operating activities was $21.1 million for the year ended December 31, 2020, which was primarily attributable to our net loss of $26.8 million, partially offset by non-cash items (i.e., accounting for stock-issuance costs at IPO and stock-based compensation) totaling $4.2 million, and a net increase in liabilities of $1.5 million associated with accounts payable and accrued bonuses.
Net cash used in operating activities was $3.7$117.5 million for the year ended December 31, 2019,2023, which primarily attributable to ourconsisted of a net loss of $4.7$135.9 million and a change in net operating assets and liabilities of $0.3 million, partially offset by net non-cash items totaling $0.5and other charges of $18.1 million. The change in net operating assets and liabilities was primarily due to an increase in accounts receivable of $16.6 million and a netan increase in liabilitiesprepaid expenses of $0.5$2.9 million, associated withpartially offset by an increase in accounts payable and other accrued bonuses.
Net Cash Usedliabilities of $13.2 million and an increase in Investing Activitiesaccrued payroll and benefits of $7.7 million. The net non-cash and other charges were primarily related to stock-based compensation expense of $19.8 million.
Net cash used in investingoperating activities was $0.5$49.0 million for the year ended December 31, 2020,2022, which primarily consisted of propertyour net loss of $62.1 million and a change in net operating assets and liabilities of $1.0 million, partially offset by net non-cash and other charges of $14.0 million. The change in net operating assets and liabilities was primarily due to an increase in other receivables of $3.5 million partially offset by an increase in accrued payroll and benefits of $2.7 million. The net non-cash and other charges were primarily related to stock-based compensation expense of $13.5 million.
Net Cash Provided by (Used in) Investing Activities
Net cash provided by investing activities was $140.6 million for the year ended December 31, 2023, and relates to $174.8 million of proceeds from maturities of investments. This cash provided by investing activities was offset by (i) $28.7 million of purchased investments, (ii) $4.0 million of intangible asset additions, and (iii) $1.5 million of purchased furniture, fixtures, office equipment purchases and tenantleasehold improvements tofor our leased laboratory and administrative offices.
Net cash used in investing activities was $0.2$144.6 million for the year ended December 31, 2019,2022, which consisted and relates to $149.4 million of propertypurchased investments and $0.5 million of purchased office equipment purchases and tenantleasehold improvements tofor our leased laboratory and administrative offices. This cash used in investing activities was offset by $5.3 million of proceeds received from maturities of investments.
Net Cash Provided by Financing Activities
Net cash provided by financing activities was $131.8$130.2 million for the year ended December 31, 2020 due to our issuance2023 which consisted of Series C preferred stock for net proceeds(i) $99.4 million of $39.8 million and net proceeds from the IPOissuance of $91.7 million.common stock from our August 2023 Public Offering, (ii) $19.2 million of net proceeds from common stock sold under the 2023 ATM Prospectus, (iii) $10.0 million of proceeds from our Credit Facility, (iv) $1.0 million of proceeds from our employee stock purchase plan, and (v) $0.6 million of proceeds from the exercise of vested employee stock options.
Net cash provided by financing activities was $59.4$94.0 million for the year ended December 31, 2019 due to our2022 which consisted of (i) $74.4 million of net proceeds from the issuance of Series B preferredcommon stock for netfrom our May 2022 Public Offering, (ii) $20.0 million of proceeds from our Credit Facility, partially offset by $0.9 million of $57.5issuance costs, (iii) $0.5 million of proceeds from our employee stock purchase plan, and issuance(iv) $0.1 million of convertible promissory notes for proceeds of $2.0 million.
Contractual Obligations and Commitments
The following table summarizes our contractual obligations as of December 31, 2020:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | Payments Due by Period |
| | Total | | Less
than
1 year | | 1 to 3
years | | 3 to 5
years | | More
than
5 years |
| | | | (in thousands) |
Operating lease obligations (1) | $ | 953 | | | $ | 349 | | | $ | 579 | | | $ | 25 | | | $ | — | |
Contingent milestone obligations (2) | 165,500 | | | 2,000 | | | 8,500 | | | 8,000 | | | 147,000 | |
| Purchase Obligations (3) | 1,189 | | | 1,189 | | | — | | | — | | | — | |
| Total | $ | 167,642 | | | $ | 3,538 | | | $ | 9,079 | | | $ | 8,025 | | | $ | 147,000 | |
(1)The operating lease obligations are related to facility leases for our corporate headquarters and research and development facilities in Irvine, California, expiring April 30, 2022 and January 31, 2024.
(2)Milestone obligations are contingent upon our achievement of specified development, regulatory and sales milestones. Given the unpredictability of the drug development process, and the impossibility of predicting the success of current and future clinical trials and the timing of achievement (if at all) of sales milestones, these values assume that all development, regulatory, and sales milestones under our in-license agreement with Elanco, as amended, are successfully met. These values in each column represent the composite best estimates for each achievement date. If any or all milestones are met, we believe that the corresponding increase in value from the related drug program will exceed the amountexercise of our milestone obligation.
(3)Purchase obligations represent the amount of open purchase orders and contractual commitments to vendors for products and services that have not been delivered or rendered, as of December 31, 2020. We enter contracts in the normal course of business with clinical research organizations and clinical sites and with contract manufacturers for pre-clinical and clinical drug supply, as well as with various other vendors in operating our business. These contracts generally provide for termination provisions with notice. The values in each column represent the obligations that are non-cancelable as of December 31, 2020.
We enter into contracts in the normal course of business with (i) clinical research organizations and clinical sites, (ii) contract manufacturers for pre-clinical and clinical drug supply, (iii) regulatory consultants and (iv) various other vendors in operating our business. These contracts generally provide for termination provisions with notice, and therefore we believe that our non-cancelable obligations under these agreements were not material as of December 31, 2020.vested employee stock options.
Critical Accounting Policies, Significant Judgments and Use of Estimates
Our management’s discussion and analysis of financial condition and results of operations is based on our financial statements, which have been prepared in accordance with U.S. generally accepted accounting principles ("GAAP"). The preparation of these financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements, as well as the reported revenue earned and expenses incurred during the reporting periods. Our estimates are based on our historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ materially from these estimates under different assumptions or conditions. Historically, revisions to our estimates have not resulted in a material change to the financial statements.
While our significant accounting policies are described in the notes to our financial statements also included in this Annual Report on Form 10-K, we believe these critical accounting policies are the most important to understanding and evaluating our reported financial results.
Research and Development ExpensesRevenue Recognition
Research and development costs are expensed as incurred or as certain upfront or milestone payments become contractually due to licensors upon the achievement of clinical or regulatory events. These expenses also include internal costs directly attributable to in-development programs, including cost of certain salaries, payroll taxes, employee benefits, and stock-based compensation expense, as well as laboratory and clinical supplies, pre-clinical and clinical trial related expenses, and the cost of services provided by outside contractors.(i) Product Sales, Net
We have entered,recognize product sales of XDEMVY net of reserves established for applicable discounts and may continueallowances that are offered within contracts with our customers. Such reserves include estimates for rebates and discounts, product returns, and chargebacks. The liability for such rebates consists of invoices received for claims from prior quarters that remain unpaid, or for which an invoice has not been received, and estimated rebates for the current applicable reporting period. Such estimates require us to enter into, license agreementsproject the magnitude of our product sales that will be subject to accesssuch rebates and utilize certain technology. In each case, we evaluate if the assets acquired in a transaction represent the acquisition of an asset or a business, as defined under applicable GAAP. Our only executed in-license agreement was evaluated and determined to represent an asset acquisition. Because this asset had not yet received regulatory approval and has no alternative future use, its fair value was immediately recognized as research and development expense.
We make research and development expense accrual estimates as of each balance sheet date,are based on facts known to us at that time. This process involves reviewing open contracts and purchase orders, communicating withactual historical rebates, as supplemented by our personnel to identify services that have been performed on our behalf, and estimating the level of service performed and the associated cost incurred for the service when we have not yet been invoiced or otherwise notified of the actual cost.judgement. We periodically confirm the accuracy of our estimates with the service providers, including CROs, and record adjustments, if necessary. To date, there have been no material differences between our estimates of such expenses and the amounts actually incurred.
Stock-Based Compensation
We measure and recognize compensation expense for all stock optionsestimate chargebacks based on the estimated fair valuepercentage of goods sold that are eligible for chargeback and adjust the award ontransaction price, accounts receivable, and in-channel inventory at the grant date. We use the Black-Scholes option-pricing model to estimate the fair valuetime of awarded stock options. The fair value is recognized as expense on a straight-line or ratable basis over the requisite or implicit service period.
The determination of the grant date fair value of our issued stock options is affected principally by our model assumptions, including (a) the expected term of the stock option until its exercise by the recipient, (b) our assumed stock price volatility through a designated peer group of publicly-traded companies for a period equalsale to the expected option term, (c) the prevailing risk-free interest rate over the expected term, and (d) any expected dividend payments over the expected term. If any of these assumptions change, our stock-based compensation expense for future grants could materially differ.
Stock-based compensation expense for equity awards granted to our employees and members of our Board of Directorscustomer. There is recognized on a straight-line basis over each award’s vesting period. We account for forfeitures as they occur. We use the Black-Scholes option pricing model to determine the fair value of stock options (as of the date of grant).
The recognition of stock-based compensation expensetime-lag between us receiving rebate notices and the initialactual calculation of stock option fair value requires uncertain assumptions, including (a) the expected term that the stock option will remain outstanding, (b) our stock price volatility over the expected term, and (c) the prevailing risk-free interest ratechargebacks for the period matching the expected term.
Expected Term – The expected term is calculated using the simplified method which is used when there is insufficient historical data about exercise patterns and post-vesting employment termination behavior. The simplified method is based on the vesting period and the contractual term for each grant, or for each vesting-tranche for awards with graded vesting. The mid-point between the vestingin-channel inventory. To date, and the maximum contractual expiration date is used as the expected term under this method. For awards with multiple vesting-tranches, the times from grant until the mid-points for each of the tranches may be averaged to provide an overall expected term.
Expected Volatility –We estimate the volatility of our common stock on the date of grant based on our designated peer group of publicly-traded companies for a period equal to the expected option term.
Expected Dividend Rate – Weactual amounts have not paid and do not anticipate paying any dividends in the near future. Accordingly, we estimate the dividend yield to be zero.
Risk-Free Interest Rate – We estimate the risk-free interest rate based upon the U.S. Department of the Treasury yields in effect at award grant for a period equaling the expected term of the stock option.
Common Stock Valuations prior todiffered materially from our IPO
Prior to the IPO our valuations of our common stock were determined in accordance with the guidelines outlined in the American Institute of Certified Public Accountants Practice Aid, Valuation of Privately-Held-Company Equity Securities Issued as Compensation, or the Practice Aid. The assumptions underlying these valuations represented management’s best estimate, which involved inherent uncertainties and the application of management’s judgment. As a result, if we had used different assumptions or estimates, the fair value of our common stock and our stock-based compensation expense could have been materially different.
Common Stock Valuations following our IPO
Subsequent to the IPO, the fair value of our common stock is based on the closing quoted market price of our common stock as reported by the Nasdaq Global Select Market on the date of grant.estimates.
Recent Accounting Pronouncements
A description of recent accounting pronouncements that may potentially impact our financial position, results of operations or cash flows is disclosed in the notes to which they relate within our financial statements.
Off-Balance Sheet Arrangements
Since our inception, we have not engaged in any off-balance sheet arrangements, as defined in the rules and regulations of the SEC.
Indemnification Agreements
As permitted under Delaware law and in accordance with our bylaws, we indemnify our officers and directors for certain events or occurrences while the officer or director is or was serving in such capacity. We are also party to indemnification agreements with our officers and directors. We believe the fair value of the indemnification rights and
agreements is minimal. Accordingly, we have not recorded any liabilities for these indemnification rights and agreements as of December 31, 2020.2023.
JOBS Act Accounting Election
The JOBSJumpstart Our Business Startups Act of 2012 permits an “emergingemerging growth company”company such as us to take advantage of an extended transition period to comply with new or revised accounting standards applicable to public companies. We have irrevocably elected to opt out of this provision and, as a result, we will comply with new or revised accounting standards as required when they are adopted.
We will remain an emerging growth company until the earliest of (1) December 31, 2025, which is the last day of our first fiscal year (a) following the fifth anniversary of the completion of our IPO, (b)(2) the last day of our first fiscal year (a) in which we have total annual gross revenues of at least $1.07$1.235 billion or (c)(b) in which we are deemed to be a large accelerated filer, which means the market value of our common stock that is held by non-affiliates exceeds $700 million, as of the prior June 30th and (2)(3) the date on which we have issued more than $1.0 billion in non-convertible debt securities during the prior three-year period.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
We are a smaller reporting company as defined by Rule 12b-2 of the Exchange Act and are not required to provide the information required under this item.
Item 8. Financial Statements and Supplementary Data
TARSUS PHARMACEUTICALS, INC.
INDEX TO THE FINANCIAL STATEMENTS
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRMReport of Independent Registered Public Accounting Firm
To the Stockholders and the Board of Directors of Tarsus Pharmaceuticals, Inc.
Opinion on the Financial Statements
We have audited the accompanying balance sheets of Tarsus Pharmaceuticals, Inc. (the Company) as of December 31, 20202023 and 2019,2022, the related statements of operations and comprehensive loss, preferred and common stock and stockholders’ equity (deficit) and cash flows for each of the two years thenin the period ended December 31, 2023, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company at December 31, 20202023 and 2019,2022, and the results of its operations and its cash flows for each of the two years thenin the period ended December 31, 2023, in conformity with U.S. generally accepted accounting principles.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ Ernst & Young LLP
We have served as the Company’s auditor since 2020.
Irvine, California
March 31, 2021February 27, 2024
TARSUS PHARMACEUTICALS, INC.
BALANCE SHEETS
(In thousands, except share and par value amounts)
| | | | December 31, | | December 31, |
| | | 2020 | | 2019 | | 2023 | | 2022 |
| ASSETS | ASSETS | | | |
| Current assets: | Current assets: | |
| Current assets: | |
| Current assets: | |
| Cash and cash equivalents | Cash and cash equivalents | $ | 168,129 | | | $ | 57,952 | |
| Restricted cash | 20 | | | 20 | |
| Cash and cash equivalents | |
| Cash and cash equivalents | |
| Marketable securities | |
| Accounts receivable, net | |
| Inventory | |
| Other receivables | Other receivables | 20 | | | 36 | |
| Prepaid expenses | Prepaid expenses | 2,486 | | | 22 | |
| Total current assets | Total current assets | 170,655 | | | 58,030 | |
| Property and equipment, net | Property and equipment, net | 548 | | | 154 | |
| Intangible assets, net | |
| Operating lease right-of-use assets | Operating lease right-of-use assets | 688 | | | 126 | |
| Long-term investments | |
| Other assets | Other assets | 81 | | | 6 | |
| Total assets | Total assets | $ | 171,972 | | | $ | 58,316 | |
| LIABILITIES, PREFERRED STOCK AND STOCKHOLDERS’ EQUITY (DEFICIT) | | | |
| LIABILITIES AND STOCKHOLDERS' EQUITY | |
| Current liabilities: | Current liabilities: | |
| Current liabilities: | |
| Current liabilities: | |
| Accounts payable and other accrued liabilities | |
| Accounts payable and other accrued liabilities | |
| Accounts payable and other accrued liabilities | Accounts payable and other accrued liabilities | $ | 4,347 | | | $ | 520 | |
| Accrued payroll and benefits | Accrued payroll and benefits | 1,040 | | | 299 | |
| Total current liabilities | Total current liabilities | 5,387 | | | 819 | |
| Term loan, net | |
| Other long-term liabilities | Other long-term liabilities | 605 | | | 100 | |
| Total liabilities | Total liabilities | 5,992 | | | 919 | |
Commitments and contingencies (Note 10) | 0 | | 0 |
| Series A Preferred Stock, $0.0001 par value; 0 shares authorized, issued and outstanding at December 31, 2020; 1,575,030 shares authorized, issued and outstanding at December 31, 2019; liquidation preference of $3,650 at December 31, 2019 | 0 | | | 3,564 | |
| Series B Preferred Stock, $0.0001 par value; 0 shares authorized, issued and outstanding at December 31, 2020; 6,731,649 shares authorized and 6,674,909 shares issued and outstanding at December 31, 2019; liquidation preference of $60,010 at December 31, 2019 | 0 | | | 59,838 | |
| Stockholders’ equity (deficit): | |
| Preferred stock, $0.0001 par value; 10,000,000 and 0 shares authorized at December 31, 2020 and December 31, 2019, respectively; 0 shares issued and outstanding at December 31, 2020 and December 31, 2019 | 0 | | | 0 | |
| Common stock, $0.0001 par value; 200,000,000 shares authorized; 20,502,576 shares issued and 20,323,201 outstanding, which excludes 179,375 shares subject to repurchase at December 31, 2020; 2,650,919 shares issued and 2,646,619 outstanding, which excludes 4,300 shares subject to repurchase at December 31, 2019 | 4 | | | 2 | |
Commitments and contingencies (Note 8) | | Commitments and contingencies (Note 8) | | | |
| Stockholders’ equity: | |
| Preferred stock, $0.0001 par value; 10,000,000 shares authorized; no shares issued and outstanding | |
| Preferred stock, $0.0001 par value; 10,000,000 shares authorized; no shares issued and outstanding | |
| Preferred stock, $0.0001 par value; 10,000,000 shares authorized; no shares issued and outstanding | |
| Common stock, $0.0001 par value; 200,000,000 shares authorized; 34,211,190 shares issued and outstanding at December 31, 2023; 26,727,458 shares issued and outstanding at December 31, 2022 | |
| Additional paid-in capital | Additional paid-in capital | 198,821 | | | 27 | |
| Accumulated other comprehensive loss | |
| Accumulated deficit | Accumulated deficit | (32,845) | | | (6,034) | |
| Total stockholders’ equity (deficit) | 165,980 | | | (6,005) | |
| Total liabilities, preferred stock and stockholders’ equity (deficit) | $ | 171,972 | | | $ | 58,316 | |
| Total stockholders’ equity | |
| Total liabilities and stockholders’ equity | |
See accompanying notes to these financial statements.
TARSUS PHARMACEUTICALS, INC.
STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
(In thousands, except share and per share amounts)
| | | | | | | | | | | |
| | Year Ended December 31, |
| | 2020 | | 2019 |
| Operating expenses: | | | |
| Research and development | $ | 18,826 | | | $ | 3,162 | |
| General and administrative | 8,172 | | | 1,136 | |
| Total operating expenses | 26,998 | | | 4,298 | |
| Loss from operations before other income (expense) and income taxes | (26,998) | | | (4,298) | |
| Other income (expense): | | | |
| Interest income (expense), net | 188 | | | (40) | |
| Loss on extinguishment of convertible notes | 0 | | | (255) | |
| Change in fair value of derivative liabilities | 0 | | | (76) | |
| Total other income (expense) | 188 | | | (371) | |
| Provision for income taxes | (1) | | | (1) | |
| Net loss and comprehensive loss | $ | (26,811) | | | $ | (4,670) | |
| Net loss per share attributable to common stockholders, basic and diluted | $ | (4.32) | | | $ | (1.98) | |
| Weighted-average shares outstanding used in computing net loss per share attributable to common stockholders, basic and diluted | 6,207,367 | | | 2,362,768 | |
| | | | | | | | | | | |
| | Year Ended December 31, |
| | 2023 | | 2022 |
| Revenues: | | | |
| Product sales, net | $ | 14,729 | | | $ | — | |
| License fees and collaboration revenue | 2,718 | | | 25,816 | |
| Total revenues | 17,447 | | | 25,816 | |
| Operating expenses: | | | |
| Cost of sales | 1,593 | | | — | |
| Cost of license fees and collaboration revenue | — | | | 955 | |
| Research and development | 50,312 | | | 42,624 | |
| Selling, general and administrative | 108,700 | | | 44,949 | |
| Total operating expenses | 160,605 | | | 88,528 | |
| Loss from operations | (143,158) | | | (62,712) | |
| Other income (expense): | | | |
| Interest income | 10,337 | | | 3,499 | |
| Interest expense | (3,346) | | | (2,199) | |
| Other (expense) income, net | (102) | | | 86 | |
| Unrealized gain (loss) on equity investments | 259 | | | (268) | |
| Change in fair value of equity warrants issued by licensee | 117 | | | (501) | |
| Total other income, net | 7,265 | | | 617 | |
| Loss before income taxes | (135,893) | | | (62,095) | |
| Benefit from income taxes | — | | | 4 | |
| Net loss | $ | (135,893) | | | $ | (62,091) | |
| | | |
| Other comprehensive loss: | | | |
| Unrealized gain (loss) on marketable securities and cash equivalents | 72 | | | (74) | |
| Comprehensive loss | $ | (135,821) | | | $ | (62,165) | |
| | | |
| Net loss per share, basic and diluted | $ | (4.62) | | | $ | (2.52) | |
| Weighted-average shares outstanding, basic and diluted | 29,383,276 | | | 24,619,700 | |
See accompanying notes to these financial statements.
TARSUS PHARMACEUTICALS, INC.
STATEMENTS OF PREFERRED AND COMMON STOCK AND STOCKHOLDERS’ EQUITY (DEFICIT)
(In thousands, except share data)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Preferred Stock | | | Common Stock | | Additional Paid-In Capital | | Accumulated
Deficit | | Total
Stockholders’
Equity (Deficit) |
| | Shares | | Amount | | | Shares | | Amount | | |
| Balance as of December 31, 2017 | 0 | | | $ | 0 | | | | 1,434,027 | | | $ | 1 | | | $ | 0 | | | $ | (45) | | | $ | (44) | |
| Net loss | — | | | | | | | | | | | | (1,319) | | | (1,319) | |
| Recognition of stock-based compensation expense | — | | | — | | | | — | | | — | | | 9 | | | — | | | 9 | |
| Issuance of common stock under stock purchase agreements | — | | | — | | | | 5,385 | | | — | | | — | | | — | | | — | |
| Vesting of founder shares subject to repurchase | — | | | — | | | | 538,532 | | | — | | | — | | | — | | | — | |
| Lapse of repurchase rights related to common stock issued pursuant to early exercises | — | | | — | | | | 100,974 | | | — | | | — | | | — | | | — | |
| Issuance of Series A Preferred Stock for cash, net of issuance costs of $86 | 1,575,030 | | | 3,564 | | | | — | | | — | | | — | | | — | | | — | |
| Balance as of December 31, 2018 | 1,575,030 | | | $ | 3,564 | | | | 2,078,918 | | | $ | 1 | | | $ | 9 | | | $ | (1,364) | | | $ | (1,354) | |
| Net loss | — | | | — | | | | — | | | — | | | — | | | (4,670) | | | (4,670) | |
| Recognition of stock-based compensation expense | — | | | — | | | | — | | | — | | | 18 | | | — | | | 18 | |
| Vesting of founder shares subject to repurchase | | | | | | 493,654 | | | 1 | | | — | | | — | | | 1 | |
| Lapse of repurchase rights related to common stock issued pursuant to early exercises | — | | | — | | | | 74,047 | | | — | | | — | | | — | | | — | |
| Issuance of Series B Preferred Stock upon conversion of Convertible Notes | 268,056 | | | 2,410 | | | | — | | | — | | | — | | | — | | | — | |
| Issuance of Series B Preferred Stock for cash, net of issuance costs of $172 | 6,406,853 | | | 57,428 | | | | — | | | — | | | — | | | — | | | — | |
| Balance as of December 31, 2019 | 8,249,939 | | | $ | 63,402 | | | | 2,646,619�� | | | $ | 2 | | | $ | 27 | | | $ | (6,034) | | | $ | (6,005) | |
| Net loss | — | | | — | | | | — | | | — | | | — | | | (26,811) | | | (26,811) | |
| Recognition of stock-based compensation expense | — | | | — | | | | — | | | — | | | 839 | | | — | | | 839 | |
| Lapse of repurchase rights related to common stock issued pursuant to early exercises | — | | | — | | | | 4,300 | | | — | | | — | | | — | | | — | |
| Issuance of common stock upon exercise of vested stock options | — | | | — | | | | 17,804 | | | — | | | 10 | | | — | | | 10 | |
| Shares issued as consideration for in-license rights (Note 9 (b)) | — | | | $ | — | | | | 222,460 | | | — | | | 3,115 | | | — | | | 3,115 | |
| Issuance of Series C Preferred Stock in September 2020 at $14.0003 per share, net of issuance costs of $243 | 2,857,079 | | | $ | 39,756 | | | | — | | | — | | | — | | | — | | | — | |
| Issuance of common stock upon initial public offering, net of issuance costs of $2,442 | — | | | $ | — | | | | 6,325,000 | | | 1 | | | 91,673 | | | — | | | 91,674 | |
| Conversion of preferred stock into common stock upon initial public offering | (11,107,018) | | | $ | (103,158) | | | | 11,107,018 | | | 1 | | | 103,157 | | | — | | | 103,158 | |
| Balance as of December 31, 2020 | 0 | | | $ | 0 | | | | 20,323,201 | | | $ | 4 | | | $ | 198,821 | | | $ | (32,845) | | | $ | 165,980 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | Common Stock | | Additional Paid-In Capital | | Accumulated Other Comprehensive Loss | | Accumulated
Deficit | | Total
Stockholders’
Equity |
| | | | | Shares | | Amount | | | |
| Balance as of December 31, 2021 | | | | | 20,698,737 | | | $ | 4 | | | $ | 213,398 | | | $ | — | | | $ | (46,672) | | | $ | 166,730 | |
| Net loss | | | | | — | | | — | | | — | | | — | | | (62,091) | | | (62,091) | |
| Recognition of stock-based compensation expense | | | | | — | | | — | | | 13,460 | | | — | | | — | | | 13,460 | |
| Issuance of common stock upon follow-on public offering, net of issuance costs of $5.3 million | | | | | 5,889,832 | | | 1 | | | 74,233 | | | — | | | — | | | 74,234 | |
| Lapse of repurchase obligation for stock option exercises, prior to vesting | | | | | 27,840 | | | — | | | 56 | | | — | | | — | | | 56 | |
| Exercise of vested stock options | | | | | 40,979 | | | — | | | 123 | | | — | | | — | | | 123 | |
| Issuance of common stock upon the vesting of restricted stock units | | | | | 32,914 | | | — | | | — | | | — | | | — | | | — | |
| Shares issued in connection with the employee stock purchase plan | | | | | 37,156 | | | — | | | 462 | | | — | | | — | | | 462 | |
| Other comprehensive loss | | | | | — | | | — | | | — | | | (74) | | | — | | | (74) | |
| Balance as of December 31, 2022 | | | | | 26,727,458 | | | $ | 5 | | | $ | 301,732 | | | $ | (74) | | | $ | (108,763) | | | $ | 192,900 | |
| Net loss | | | | | — | | | — | | | — | | | — | | | (135,893) | | | (135,893) | |
| Recognition of stock-based compensation expense | | | | | — | | | — | | | 19,830 | | | — | | | — | | | 19,830 | |
| Issuance of common stock upon follow-on public offering, net of issuance costs of $6.9 million | | | | | 6,069,449 | | | — | | | 99,303 | | | — | | | — | | | 99,303 | |
| Issuance of common stock under an at-the-market sale agreement, net of issuance costs of $0.8 million | | | | | 1,000,000 | | | — | | | 19,199 | | | — | | | — | | | 19,199 | |
| Exercise of vested stock options | | | | | 136,310 | | | — | | | 592 | | | — | | | — | | | 592 | |
| Issuance of common stock upon the vesting of restricted stock units | | | | | 206,813 | | | — | | | — | | | — | | | — | | | — | |
| Shares issued in connection with the employee stock purchase plan | | | | | 71,160 | | | — | | | 985 | | | — | | | — | | | 985 | |
| Other comprehensive income | | | | | — | | | — | | | — | | | 72 | | | — | | | 72 | |
| Balance as of December 31, 2023 | | | | | 34,211,190 | | | $ | 5 | | | $ | 441,641 | | | $ | (2) | | | $ | (244,656) | | | $ | 196,988 | |
See accompanying notes to these financial statements.
TARSUS PHARMACEUTICALS, INC.
STATEMENTS OF CASH FLOWS
(In thousands)
| | | | | | | | | | | |
| | Year ended
December 31, |
| | 2020 | | 2019 |
| Cash Flows From Operating Activities: | | | |
| Net loss | $ | (26,811) | | | $ | (4,670) | |
| Adjustments to reconcile net loss to net cash used in operating activities: | | | |
| Depreciation and amortization | 96 | | | 37 | |
Stock-based compensation (Note 5) | 839 | | | 18 | |
Amortization of operating lease right-of-use asset (Note 10(a)) | 150 | | | 37 | |
| Impairment of operating lease right-of-use asset | 15 | | | 0 | |
Change in fair value of derivative liabilities (Note 9) | 0 | | | 76 | |
| Stock issued for in-license agreement (Note 9(b)) | 3,115 | | | 0 | |
| Non-cash related party interest expense | 0 | | | 89 | |
Loss on extinguishment of convertible notes (Note 9) | 0 | | | 255 | |
| Changes in operating assets and liabilities: | | | |
| Other receivables | 16 | | | (32) | |
| Prepaid expenses and other current assets | (2,464) | | | (18) | |
| Other non-current assets | (75) | | | (6) | |
| Accounts payable and other accrued liabilities | 3,240 | | | 245 | |
| Accrued payroll and benefits | 741 | | | 296 | |
| Net cash used in operating activities | (21,138) | | | (3,673) | |
| Cash Flows From Investing Activities: | | | |
| Purchases of property and equipment | (456) | | | (175) | |
| Cash used in investing activities | (456) | | | (175) | |
| Cash Flows From Financing Activities: | | | |
Proceeds from issuance of Series B Preferred Stock, net of issuance costs (Note 4) | (28) | | | 57,456 | |
| Proceeds from issuance of Series C Preferred Stock, net of issuance costs (Note 4) | 39,756 | | | 0 | |
Proceeds from issuance of convertible notes, net of issuance costs (Note 9) | 0 | | | 1,989 | |
| Proceeds from issuance of common stock upon initial public offering, net of issuance costs | 91,673 | | | 0 | |
| Proceeds from early exercise of stock options | 360 | | | 0 | |
| Proceeds from issuance of common stock upon exercise of vested stock options | 10 | | | 0 | |
| Net cash provided by financing activities | 131,771 | | | 59,445 | |
| Net increase in cash, cash equivalents and restricted cash | 110,177 | | | 55,597 | |
| Cash, cash equivalents, and restricted cash — beginning of year | 57,972 | | | 2,375 | |
| Cash, cash equivalents, and restricted cash — end of year | $ | 168,149 | | | $ | 57,972 | |
| Reconciliation of cash, cash equivalents and restricted cash | | | |
| Cash and cash equivalents | $ | 168,129 | | | $ | 57,952 | |
| Restricted cash | 20 | | | 20 | |
| Cash, cash equivalents and restricted cash | $ | 168,149 | | | $ | 57,972 | |
| Supplemental Disclosures Noncash Investing and Financing Activities: | | | |
| Conversion of Preferred Stock to common stock upon initial public offering | $ | 103,158 | | | $ | 0 | |
| Stock issued for in-license agreement | $ | 3,115 | | | $ | 0 | |
| Operating lease right-of-use asset obtained in exchange for operating lease liability | $ | 726 | | | $ | 163 | |
| | | |
Settlement of derivative liabilities upon conversion of convertible notes (Note 8) | $ | 0 | | | $ | 363 | |
Additions of property and equipment in accounts payable and other accrued liabilities (Note 3(b)) | $ | 34 | | | $ | 10 | |
| Series B Preferred Stock issuance costs in accounts payable and other accrued liabilities | $ | 0 | | | $ | 28 | |
Conversion of convertible notes to Series B Preferred Stock (Note 8) | $ | 0 | | | $ | 2,410 | |
| | | | | | | | | | | |
| | Year ended
December 31, |
| | 2023 | | 2022 |
| Cash Flows From Operating Activities: | | | |
| Net loss | $ | (135,893) | | | $ | (62,091) | |
| Adjustments to reconcile net loss to net cash used in operating activities: | | | |
| Depreciation | 744 | | | 325 | |
| Amortization of intangible assets | 133 | | | — | |
| Stock-based compensation | 19,830 | | | 13,460 | |
| Accretion of term loan-related costs | 385 | | | 309 | |
| Non-cash lease expense | 541 | | | 464 | |
| | | |
| | | |
| Amortization of discount on available-for-sale debt securities | — | | | (1,316) | |
| Net amortization/accretion on marketable securities | (3,163) | | | — | |
| Unrealized (gain) loss on equity investment | (259) | | | 268 | |
| Change in fair value of equity warrants issued by licensee | (117) | | | 501 | |
| Unrealized gain from transactions denominated in a foreign currency | — | | | 1 | |
| | | |
| Changes in operating assets and liabilities: | | | |
| Accounts receivable, net | (16,621) | | | — | |
| Inventory | (3,107) | | | — | |
| Other receivables | 2,490 | | | (3,490) | |
| Prepaid expenses | (2,889) | | | (721) | |
| Other non-current assets | (718) | | | (215) | |
| Accounts payable and other accrued liabilities | 13,240 | | | 821 | |
| Accrued payroll and benefits | 7,726 | | | 2,721 | |
| Other long-term liabilities | 185 | | | (67) | |
| Net cash used in by operating activities | (117,493) | | | (49,030) | |
| Cash Flows From Investing Activities: | | | |
| Proceeds from sales of marketable securities | 174,770 | | | 5,315 | |
| Purchases of marketable securities | (28,664) | | | (149,438) | |
| Intangible asset additions | (4,000) | | | — | |
| Purchases of property and equipment | (1,502) | | | (506) | |
| Net cash provided by (used in) investing activities | 140,604 | | | (144,629) | |
| Cash Flows From Financing Activities: | | | |
| Proceeds from issuance of common stock upon follow-on public offering, net of paid issuance costs | 99,355 | | | 74,352 | |
| Proceeds from issuance of common stock under an at-the-market sales agreement, net of paid issuance costs | 19,244 | | | — | |
| Proceeds from term loan | 10,000 | | | 20,000 | |
| Proceeds from sale of common stock under employee stock purchase plan | 985 | | | 462 | |
| Proceeds from exercise of vested stock options | 592 | | | 123 | |
| Payment of deferred offering costs | — | | | (75) | |
| Payment of term loan issuance costs | — | | | (875) | |
| Net cash provided by financing activities | 130,176 | | | 93,987 | |
| Net increase (decrease) in cash and cash equivalents | 153,287 | | | (99,672) | |
| Cash and cash equivalents — beginning of year | 71,660 | | | 171,332 | |
| Cash and cash equivalents — end of year | $ | 224,947 | | | $ | 71,660 | |
| Supplemental Disclosures Noncash Investing and Financing Activities: | | | |
| Operating lease right-of-use asset obtained in exchange for operating lease liability | $ | 1,846 | | | $ | — | |
| Interest expense paid in cash | $ | 2,880 | | | $ | 1,675 | |
| Additions of property and equipment included within accounts payable and other accrued liabilities | $ | 134 | | | $ | 21 | |
| | | |
| | | |
See accompanying notes to these financial statements.
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
1. DESCRIPTION OF BUSINESS AND PRESENTATION OF FINANCIAL STATEMENTS
(a) Description of Business
Tarsus Pharmaceuticals, Inc. (“Tarsus” or the “Company”) is a late clinical-stagecommercial stage biopharmaceutical company focused on the development and commercialization of novel therapeutic candidates to address large market opportunities initiallytherapeutics, starting with eye care. The Company launched XDEMVY® (lotilaner ophthalmic solution) 0.25%, formerly known as TP-03, for the treatment of Demodex blepharitis, in ophthalmic conditions where there are limited treatment alternatives.August 2023, after receiving United States ("U.S") Food and Drug Administration ("FDA") approval in July 2023.
(b) InitialFollow-On Public OfferingOfferings and Reverse Stock SplitATM Prospectus
On October 20, 2020,In August 2023, the Company completed its initiala follow-on public offering "IPO" through an underwritten saleunder its shelf registration statement on Form S-3 that was declared effective by the SEC on November 5, 2021 (the "2021 Shelf Registration Statement") of 5,500,0005,714,285 shares of common stock at a public offering price of $17.50 per share (the "August 2023 Public Offering"). The Company also granted the underwriters a 30-day option to purchase up to 857,142 additional shares of its common stock at athe public offering price. In September 2023, the underwriters partially exercised this option resulting in the Company's issuance of an additional 355,164 shares of common stock at the public offering price of $16.00$17.50 per share. The aggregate net proceeds received by the Company from the IPO, inclusive of an additional 825,000 common shares sold upon the full exercise of the underwriters’ purchase option totaled $91.7were approximately $99.3 million, after deducting underwriting discounts, and commissions, and other offeringoffering-related expenses.
ConcurrentIn 2021 we filed the 2021 Shelf Registration Statement, issuable pursuant to the terms of an Open Market Sale Agreement™ (the "2021 ATM Prospectus") dated November 1, 2021 by and between the Company and Jefferies LLC, covering the sale of up to $100.0 million of our common stock. In connection with the closingAugust 2023 Public Offering, the Company terminated the 2021 ATM Prospectus. The Company did not have any sales of common stock pursuant to the 2021 ATM Prospectus.
In November 2023, the Company filed a shelf registration statement on Form S-3 that was declared effective by the SEC on November 21, 2023, (the "2023 Shelf Registration Statement"), which replaced the 2021 Shelf Registration Statement and permits the Company to offer up to $300.0 million of common stock, preferred stock, debt securities and warrants in one or more offerings and in any combination, including in units from time to time.
As part of the 2023 Shelf Registration Statement, the Company concurrently filed a sales agreement prospectus covering the sale of up to $100.0 million of common stock pursuant to an Open Market Sale Agreement (the "2023 ATM Prospectus") with Jefferies LLC ("Jefferies"). Under the terms of the 2023 ATM Prospectus, Jefferies will act as the Company's IPO, all then-outstandingsales agent and is entitled to compensation for its services equal to 3% of the gross proceeds of any shares of common stock sold. In December 2023, the Company sold 1,000,000 shares of common stock under the 2023 ATM Prospectus for net proceeds of $19.2 million, after deducting broker commissions and offering-related expenses.
In May 2022, the Company completed a follow-on public offering under its Shelf Registration Statement for an initial underwritten sale of 5,600,000 shares of its convertible preferredcommon stock (see Note 4) were automatically converted into an aggregateat the public offering price of 11,107,018$13.50 per share. The Company also granted the underwriters a 30-day option to purchase up to 840,000 additional shares of its common shares.
On October 8, 2020,stock at the public offering price. In June 2022, the underwriters partially exercised this option and the Company's Boardsale of Directors approved a 1-for-7.4276 reverse stock split and a certificatean additional 289,832 shares at the public offering price of amendment was filed to restate the Company's certificate of incorporation to effect this reverse stock split. The par value was not adjusted as a result of the reverse stock split. All share and$13.50 per share information included inwas concurrently completed. Total aggregate net proceeds received by the accompanying financial statements give retroactive effect to the reverse stock split for all periods presented.Company were approximately $74.2 million, after deducting underwriting discounts, commissions, and other offering-related expenses.
(c) Liquidity Risks
The Company has no revenuea limited operating history, limited history of product sales and incurredhas accumulated losses and negative cash flows from operations since inception, resulting in an accumulated deficit of $32.8 million as of December 31, 2020,inception. The Company has funded its inception-to-date operations through the IPO, Follow-on Public Offerings and the income potential of2023 ATM Prospectus, as well as from proceeds from product sales, net, the Company's businessChina Out-License, and market are unproven. The Company’s cash and cash equivalents was $168.1 million and $58.0 million as of December 31, 2020 and December 31, 2019, respectively. draws on the credit facility.The Company expects to continue to incur operating losses and negative cash flows and has historically financedestimates that its operations primarily through equity capital raises.
The Company believes that existing capital resources including the net proceeds from the IPO in October 2020, will be sufficient to meet projected operating expense requirements for at least 12 months from the issuance date of issuance of the accompanying financial statements.
The financial statementsFinancial Statements that have been prepared on a going concern basis, which contemplates the realization of assets and satisfaction of liabilities in the normal course of business. The financial statements do not include any adjustments relating to the recoverability and classification of recorded asset amounts or the amounts and classification of liabilities that might result from the outcome of this uncertainty. Management expects to continue to incur additional substantial losses in the foreseeable future as a result of the Company’s research and development activities.
The Company’s operations have consisted primarily of its organization, securing financing, in-licensing intellectual property, and conducting preclinical and clinical studies. The Company faces risks associated with early-stage biotechnology companies whose product candidates are in development that require significant additional research and development efforts, including extensive preclinical and clinical testing and regulatory approval prior to commercialization. These efforts require the Company to expend large amounts of additional capital to complete research and development, achieve research and development objectives, defend intellectual property rights, and recruit and retain skilled personnel, including key members of management.
The Company will be required to raise additional capital to fund future operations, however, no assurance can be given as to whether additional needed financing will be available on terms acceptable to the Company, if at all.going-concern basis.
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
IfThe Company plans to fund its operations, capital funding and other liquidity needs using existing cash and investments and, to the extent available, cash generated from commercial operations. Management expects the Company raises additional funds by issuing equity securities, its stockholders may experience dilution. Any future debt financing into which the Company enters may impose additional covenants that restrict operations, including limitations on its abilityto continue to incur liens oroperating losses for the foreseeable future and may be required to raise additional debt, pay dividends, repurchase common stock, make certain investments or engage in certain merger, consolidation or asset sale transactions. Any debtcapital to fund its ongoing operations. However, no assurance can be given as to whether financing or additional equity raise may containwill be available on terms that are not favorableacceptable to the Company, or its stockholders. Further, adequate funding may not be available on acceptable terms, or at all. The Company’s potential inability to raise capital when needed could have a negative impact on its financial condition and ability to pursue planned business strategies. If the Company is unable to raise additional funds as required, it may need to delay, reduce, or terminate some or all of its development programs and clinical trials. The Company may also be required to sell or license its rights to product candidates in certain territories or indications that it would otherwise prefer to develop and commercialize on its own. If the Company is required toown and/or enter into collaborations and other arrangements to address its liquidity needs, it may have to give up certain rights that limit its ability to develop and commercialize product candidates or may have other terms that are not favorable to the Company or its stockholders, which could materially and adversely affect its business and financial prospects. These factors may adversely impact the Company's ability to achieve its business objectives and would likely have an adverse effect on its future business prospects, or even its ability to remain a going concern.
(d) Operating Segment
The Company operates one reportable operating segment focused on the development and commercialization of therapeutics. To date, the Company has operated, managed and managedorganized its business and financial information on an aggregate basis for the purposespurpose of evaluating financial performance and the allocation of capital and personnel resources. Accordingly,The Company’s chief operating decision-maker (CODM), its Chief Executive Officer, reviews its operating results for the Company’s management determined that it operates 1 reportable operating segment that is focused exclusively on developing pharmaceutical products for commercialization.purpose of allocating resources and evaluating financial performance.
(e) Emerging Growth Company Status
The Company is an emerging growth company, as defined in the Jumpstart Our Business Startups Act of 2012 (the "JOBS Act"). Under the JOBS Act, emerging growth companies can delay adopting new or revised accounting standards issued subsequent to the enactment of the JOBS Act until such time as those standards apply to private companies. The Company has irrevocably elected to not take this exemption and, asexemption. As a result, it will adopt new or revised accounting standards on the relevant effective dates on which adoption of such standards is required for other public companies that are not emerging growth companies.
2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES AND USE OF ESTIMATES
(i) Basis of Presentation
The preparation of financial statementsaccompanying Financial Statements have been prepared in conformityaccordance with U.S. generally accepted accounting principles (“GAAP”) in the U.S. and with the rules and regulations of the Securities and Exchange Commission (“SEC”). The preparation of financial statements in conformity with GAAP and with the rules and regulations of the SEC requires management to make informed estimates and assumptions that impactaffect the amounts reported in these financial statementsaccompanying Financial Statements and accompanying notes.Notes. These amountsestimates and assumptions are based upon historical experience, knowledge of current events and various other factors believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities and the recording of expenses that are not readily apparent from other sources and involve judgments with respect to numerous factors that are difficult to predict and may materially differ from the amounts ultimately realized and reported due to the inherent uncertainty of any estimate or assumption. On an on-going basis, management evaluates its most criticalActual results could differ materially from those estimates and assumptions used in the preparation of the accompanying Financial Statements under different assumptions and conditions.
The Company’s Financial Statements as of and for the year ended December 31, 2023 reflect the Company’s estimates of the impact of the macroeconomic and geopolitical environment, including those relatedthe impact of inflation, higher interest rates, and foreign exchange rate fluctuations. The duration and the scope of these conditions cannot be predicted; therefore, the extent to which these conditions will directly or indirectly impact the (i) fairCompany’s business, results of operations and financial condition, is uncertain. The Company is not aware of any specific event or circumstance that would require an update to its estimates, judgments and assumptions or a revision of the carrying value of stock awards and periodic expense recognitionthe Company’s assets or liabilities as of stock-based compensation, (ii) the realizationissuance date of income tax assets and estimates of tax liabilities, (iii) expense accruals related to research and development activities, including clinical trials, and, for periods prior to the IPO, (iv) valuation of convertible notes, derivative instruments, and preferred stock.accompanying Financial Statements.
AccountingThe accounting policies and estimates that most significantly impact the presented amounts within these financial statementsaccompanying Financial Statements are further described below:
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
Cash and Cash Equivalents
Cash and cash equivalents consist of bank deposits and highly liquid investments, including money market fund accounts, that are readily convertible into cash without penalty, with original maturities of three months or less from the purchase date. The carrying amounts reported in the accompanying Balance Sheets for cash and cash equivalents are valued at cost, which approximate their fair value.
Marketable Securities and Long-Term Investments
Marketable securities consist primarily of short-term fixed income investments carried at estimated fair value as determined based upon quoted market prices or pricing models for similar securities (see Note 3). Management determines the appropriate classification of its investments in fixed income securities at the time of purchase. Available-for-sale securities with original maturities beyond three months at the date of purchase, including those that have maturity dates beyond one year from the balance sheet date, are classified as current assets on the accompanying Balance Sheets due to their highly liquid nature and availability for use in current operations.
Marketable securities are recorded at fair value with unrealized gains and losses reported as a component of accumulated other comprehensive loss within the accompanying Statements of Stockholders' Equity until realized. The Company periodically evaluates whether declines in fair values of its available-for-sale securities below their book value are other-than-temporary. This evaluation consists of several qualitative and quantitative factors regarding the severity and duration of the unrealized loss as well as the Company’s ability and intent to hold the available-for-sale security until a forecasted recovery occurs. The cost of debt securities is adjusted for amortization of premiums and accretion of discounts to maturity. Such amortization and accretion, as well as interest and dividends, are included in interest income. Realized gains and losses as well as credit losses, if any, on marketable securities identified on a specific identification basis are included in other income (expense) on the accompanying Statements of Operations and Comprehensive Loss. The Company evaluated the underlying credit quality and credit ratings of the issuers during the period. To date, the Company has not identified any other-than-temporary declines in fair value of its investments and no credit losses associated with credit risk have occurred or have been recorded. Interest earned on marketable securities is included in interest income within the accompanying Statements of Operations and Comprehensive Loss.
Long-term investments consist of holdings of common stock in the publicly-traded parent company of LianBio Ophthalmology Limited ("LianBio"), reflecting the intent to hold these shares for at least one year from the balance sheet date. These equity securities are designated as available-for-sale with associated unrealized gains or losses reported in other income (expense) within the Statements of Operations and Comprehensive Loss for each reported period.
Accounts Receivable, Net
Accounts receivable generally consists of amounts due from its customers, which includes pharmaceutical wholesalers and specialty pharmacy providers related to product sales of XDEMVY in the U.S. Payment terms are typically 30-60 days following delivery to customers. Accounts receivable are recorded net of discounts, chargebacks, allowances and other adjustments. The Company monitors the financial performance and creditworthiness of its customers so it can properly assess and respond to changes in their credit profile. The Company estimates the allowance for credit losses based on existing contractual payment terms, actual payment patterns of customers and individual customer circumstances. Amounts determined to be uncollectible are written off against the reserve when it is probable that the receivable will not be collected. The Company did not record a reserve for estimated credit losses during the year ended December 31, 2023.
Inventory
Inventory is valued at the lower-of-cost or net realizable value, with cost determined on a first-in, first-out (FIFO) basis. The Company performs an assessment of the recoverability of capitalized inventory during each reporting period, and adjusts the value for any excess and obsolete inventory to net realizable value in the period in which the impairment is first identified and such charges are recorded as a component of cost of sales in the Statements of Operations and Comprehensive Loss. The determination of whether inventory costs will be realizable requires estimates by management. If actual market conditions are less favorable than projected by management, additional write-downs of inventory may be required. The
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
sheets for cash and cash equivalents are valued at cost, which approximate their fair value. Cash equivalents may consist of money market funds.
(iii) Restricted Cash
Restricted cash represents cash held as collateral for the Company’s corporate credit card program. Any cash that is legally or contractually restricted from immediate use is classified as restricted cash.
(iv) Concentration of Credit Risk and Other Risks and Uncertainties
Financial instruments that potentially subject the Company to significant concentrations of credit risk consist primarily of cash and cash equivalents in deposits at financial institutions that exceed federally insured limits.
In March 2020, the World Health Organization declared a pandemic related to the global novel coronavirus disease 2019 (“COVID-19”) outbreak. The Company’s operations have not been significantly impacted by the COVID-19 pandemic. The Company has been carefully monitoring the potential impact COVID-19 may have on the Company’s business. However, the Company cannot at this time predict the specific extent, duration, or full impact that the COVID-19 outbreak will have on its financial condition and operations, including ongoing and planned clinical trials.
The Company’s results of operations involve numerous risks and uncertainties. Factors that could adversely impact the Company’s operating results and business objectives include, but are not limited to, (1) uncertainty of results of clinical trials, (2) uncertainty ofcapitalizes inventory costs associated with products following regulatory approval of the Company’s potential product candidates, including TP-03 for ophthalmic conditions, TP-04 for treatment of skin conditionswhen future commercialization is considered probable and TP-05 for prophylaxis of Lyme and community malaria reduction, (3) uncertainty of market acceptance of its product candidates, (4) competition from substitute products and larger companies, (5) securing and protecting proprietary technology and strategic relationships, and (6) and dependence on key individuals and sole source suppliers.
The Company’s product candidates require approvals from the U.S. Food and Drug Administration (“FDA”) and comparable foreign regulatory agencies prior to commercial sales in their respective jurisdictions. There can be no assurance that any product candidates will receive the necessary approvals. If the Company is denied approval, approval is delayed or the Company is unable to maintain approval for any product candidate, it could have a materially adverse impact on the business.
(v) Property and Equipment
Property and equipment is stated at historical cost and is depreciated on a straight-line basis over an estimated useful life that corresponds with its designated asset category. Leasehold improvements are amortized on a straight-line basis over the shorter of the remaining lease term or the estimated useful lives of related improvements. The Company evaluates the recoverability of “long-lived assets” (which includes property and equipment) whenever events or changes in circumstances in the business indicate that the asset’s carrying amount may not be recoverable. Recoverability of these assets is measured by a comparison of the carrying amounts to the sum of the future undiscounted cash flows the assets areeconomic benefit is expected to generate overbe realized. Product that may be used in clinical development programs are excluded from inventory and the remaining useful lives of the assets. If a long-lived asset fails a recoverability test, the Company measures the amount by which the carrying value of the asset exceeds its fair value. Other than the right-of-use ("ROU") asset impairment discussed in Note 9, there were no events or changes in business circumstances during the years ended December 31, 2020costs are charged to research and 2019 that indicated the carrying amounts of any long-lived assets were not fully recoverable.
(vi) Derivative Instruments
The convertible notes issued in May 2019, August 2019, and October 2019 contained embedded derivative instruments, representing “contingent redemption options”. The contingent redemption options met the requirements for separate accounting and were accounted for as a single derivative instrument for each tranche of the convertible notes. The derivative instruments were recorded at fair value at inception and were subject to remeasurement to fair value, with any changes in estimated fair value recognized as a component of “other (expense) income”development expense in the Statements of Operations and Comprehensive Loss (see Note 7).
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
As the Notes were convertedXDEMVY in December 2019, no expenseJuly 2023, costs related to changes in fair valuethe production of derivative liabilities wasinventory were recorded for the twelve months ended December 31, 2020.
(vii) Research and Development Costs
Research and development costs are expensed as incurred or as certain upfront or milestone payments become contractually due to licensors upon the achievement of clinical or regulatory events. These expenses also include internal costs directly attributable to in-development programs, including cost of certain salaries, payroll taxes, employee benefits, and stock-based compensation expense, as well as laboratory and clinical supplies, pre-clinical and clinical trial related expenses, and the cost of services provided by outside contractors. The Company recognizes expense for pre-clinical studies and clinical trial activities performed by these third parties. This is typically based upon estimates of the proportion of work completed over the term of the individual study or trial, as well as patient enrollment and dosing events in accordance with agreements established with clinical research organizations ("CROs") and clinical trial or pre-clinical study sites.
The Company has entered, and may continue to enter into, license agreements to access and utilize intellectual property for drug development. In each case, the Company evaluates if the assets acquired in a transaction represent the acquisition of an asset or a business, as defined under applicable GAAP. The Company’s only executed in-license agreement (see Note 9(b)) was evaluated and determined to represent an asset acquisition. Because this asset had not yet received regulatory approval and has no alternative future use, its purchase price was immediately recognized as research and development expense. In addition, any future milestone payments made before product regulatory approval (that do not meet the definition of a derivative) will also be immediately recognized as research and development expense when paid or become payable, provided there is no alternative future use of the rights in other research and development projects.
(viii) Deferred Offering Costs
Costs directly related to the Company’s IPO were deferred for expense recognition and instead capitalized and recorded on the accompanying balance sheets. These costs consisted of legal fees, accounting fees, and other applicable professional services. These deferred offering costs were reclassified to “additional paid in capital” and offset against IPO proceeds, upon closing of the IPO in October 2020. There were 0 deferred offering costs capitalized as of December 31, 2020 and 2019.
(ix) Stock-Based Compensation
Stock-based compensation expense is recognized for all equity awards granted to employees, consultants, and members of the Company’s Board of Directors and is recognized at fair value. For stock-based awards that vest subject to the satisfaction of a service requirement, the fair value measurement date is the date of grant and the related expense is recognized on a straight-line basis over each award’s actual or implied vesting period. For stock-based awards that vest subject to a performance condition, the Company recognizes compensation cost if and when it concludes that it is probable that the performance condition will be achieved and the related expense is recognized on an accelerated attribution method. As applicable, the Company reverses previously recognized expense for forfeitures of unvested awards in the period of occurrence. The Company uses the Black-Scholes option pricing model to estimate the fair value of stock-based awards as of the date of grant.
The measurement of the fair value of stock-based awards and recognition of stock-based compensation expense requires assumptions to be estimated by management that involve inherent uncertainties and the application of management’s judgment, including (a) the fair value of the Company’s common stock on the date of the option grant, (b) the expected term of the stock option until its exercise by the recipient, (c) stock price volatility over the expected term, (d) the prevailing risk-free interest rate over the expected term, and (e) expected dividend payments over the expected term.
Management estimates the expected term of awarded stock options utilizing the “simplified method” for awards as the Company does not yet have sufficient exercise history since its November 2016 formation. Further, prior to the IPO, the Company was privately-held and therefore lacked company-specific historical and implied volatility information of its stock. Accordingly, management estimated this expected volatility based on a designated peer-group of publicly-traded companies for a look-back period, as of the date of grant, that corresponded with the expected term of the awarded stock option.
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
The Company estimates the risk-free interest rate based upon the U.S. Department of the Treasury yield curve in effect at award grant for time periods that correspond with the expected term of the awarded stock option. The Company’s expected dividend yield is 0 because it has never paid cash dividends and does not expect to for the foreseeable future.
Prior to the IPO, given the absence of a public trading market, the Company’s Board of Directors, with input from management, considered numerous objective and subjective factors to determine the fair value of its common stock. The factors included: (i) third-party valuations of the Company’s common stock; (ii) the Company’s stage of development; (iii) the status of research and development efforts; (iv) the rights, preferences and privileges of the Company’s preferred stock relative to common stock; (v) the Company’s operating results and financial condition, including the Company’s levels of available capital resources; (vi) equity market conditions affecting comparable public companies; (vii) general U.S. market conditions; and (viii) the lack of current marketability of the Company’s common stock. Subsequent to the IPO, the fair value of the Company’s common stock is based on the closing quoted market price of its common stock as reported by the NASDAQ Global Select Market on the date of grant.
All stock-based compensation costs are recorded in the Statements of Operations and Comprehensive Loss based upon the underlying employee's role within the Company.
(x) Income Taxes
Income taxes are accounted for using the asset and liability method. Deferred tax assets and liabilities are recorded based on the estimated future tax effects of temporary differences between the tax basis of assets and liabilities and amounts reported in the financial statements, as well as operating losses and tax credit carry forwards using enacted tax rates and laws that are expected to be in effect when the differences are expected to reverse. Deferred tax assets and liabilities are measured using the enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period of enactment. Realization of deferred tax assets is dependent upon future earnings, the timing and amount of which are uncertain due to the Company’s historical operating performance and recorded cumulative net losses in prior fiscal periods.
A valuation allowance is recorded to reduce deferred tax assets, because based upon a weighting of positive and negative factors, it is more likely than not that these deferred tax assets will not be realized. If/when the Company were to determine that deferred tax assets are realizable, an adjustment to the corresponding valuation allowance would increase the net income in the period that such determination was made.
In the event that the Company is assessed interest and/or penalties from taxing authorities that have not been previously accrued, such amounts would be included as a component of “income tax expense” within the Statements of Operations and Comprehensive Loss in the period the notice was received. To date there have been 0 interest or penalties charged.incurred.
(xi) Preferred StockIntangible Assets, Net
Note 8). Amortization expense for the year ended December 31, 2023 was $0.1 million. The Company classified preferred stock outsidehad no intangible assets as of stockholders’ equity (deficit) on the accompanying balance sheets. The requirements of a deemed liquidation event, as defined within its amended and restated certificate of incorporation filed in September 2020 (the “2020 Amended and Restated Certificate of Incorporation”) were not entirely within the Company’s control. In the event of such a deemed liquidation event, the proceeds from the event are distributed in accordance with the liquidation preferences, provided that the holders of preferred stock have not converted their shares into common stock. The Company recorded the issuance of preferred stock at the issuance price less related issuance costs. December 31, 2022.
As of December 31, 2019,2023, the Company did not adjustexpected future amortization expense for the Company's intangible assets is as follows:
| | | | | | | | |
| | Amounts |
| 2024 | | $ | 400 | |
| 2025 | | 400 | |
| 2026 | | 400 | |
| 2027 | | 400 | |
| 2028 | | 400 | |
| Thereafter | | 1,867 | |
| Total future amortization | | $ | 3,867 | |
Long-lived assets, including intangibles, are evaluated for impairment whenever events or changes in circumstance indicate that the carrying value of outstanding preferred stock to its liquidation preference because a deemed liquidation event wasan asset might not probable of occurring asbe fully recoverable. To do so, the Company compares the carrying value of the endintangible asset to the undiscounted net cash flows over its remaining useful life, and if not recoverable, will estimate the fair value of the reporting period. Allasset. If the fair value is less than the carrying amount, an impairment loss is recognized in the Statements of the outstanding sharesOperations and Comprehensive Loss. There have been no impairments of preferred stock were automatically converted to common stock shares upon the closing of the IPO in October 2020.
(xii) Net Loss per Share Attributable to Common Stockholders
Basic net loss per share attributable to common stockholders is calculated by dividing the net loss by the weighted-average number of shares of common stock outstandingintangible assets for the period, without consideration for potential dilutive shares ofyear ended December 31, 2023.
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
common stock. Diluted net loss per share is computed by dividing the net loss by the weighted-average number of common share equivalents outstanding for the period determined using the treasury-stock method and if-converted method, as applicable. Basic and diluted net loss per share attributable to common stockholders is presented in conformity with the two-class method required for participating securities. The Company’s participating securities include preferred stock, unvested common stock to founders, and unvested common stock awards issued upon early exercise of certain stock options. The Company’s participating securities do not have a contractual obligation to share in the Company’s losses. As such, the net loss was attributed entirely to common stockholders. Shares of common stock subject to repurchase by the Company are excluded from the weighted-average shares. Due to net losses in all periods presented, all otherwise potentially dilutive securities are antidilutive. Accordingly, basic net loss per share equals diluted net loss per share for all period presented in the accompanying financial statements.
(xiii) Fair Value Measurements
Assets and liabilities recorded at fair value on a recurring basis in the balance sheets are categorized based upon the level of judgment associated with the inputs used to measure their fair values. Fair value is defined as the exchange price that would be received for an asset or an exit price that would be paid to transfer a liability in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. Valuation techniques used to measure fair value must maximize the use of observable inputs and minimize the use of unobservable inputs. The authoritative guidance on fair value measurements establishes a three-tier fair value hierarchy for disclosure of fair value measurements as follows:
•Level 1: Quoted prices (unadjusted) in active markets for identical assets or liabilities that are publicly accessible at the measurement date.
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
•Level 2: Observable prices that are based on inputs not quoted on active markets, but that are corroborated by market data. These inputs may include quoted prices for similar assets or liabilities or quoted market prices in markets that are not active to the general public.
•Level 3: Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities.
The carrying amounts for financial instruments consisting of cash, and cash equivalents, accounts receivable, net, accounts payable and accrued liabilities approximate fair value due to theirthe short maturities. Derivative instrumentsmaturities for each. The Company's equity warrant holdings disclosed as other assets are carried at fair value based on unobservable market inputs.inputs (see Note 3).
(xiv) Assets and liabilities are classified based on the lowest level of input that is significant to the fair value measurements. The Company reviews the fair value hierarchy classification on a quarterly basis. Changes in the ability to observe valuation inputs may result in a reclassification of levels for certain assets or liabilities within the fair value hierarchy. The Company did not have any transfers of assets and liabilities between the levels of the fair value hierarchy during the years presented.
Property and Equipment, Net
Property and equipment, net are stated at historical cost less accumulated depreciation. Depreciation is calculated using the straight-line method over the estimated useful lives of the assets that range from three to five years. Leasehold improvements are amortized on a straight-line basis over the shorter of the remaining lease term or the estimated useful lives of related improvements. The Company evaluates the recoverability of its property and equipment, net whenever events or changes in circumstances of the business indicate that the asset’s carrying amount may not be recoverable. Recoverability of these assets is measured by a comparison of the carrying amounts to the sum of the future undiscounted cash flows the assets are expected to generate over the remaining useful lives of the assets. If a long-lived asset fails a recoverability test, the Company measures the amount by which the carrying value of the asset exceeds its fair value. There were no impairments recognized during the years ended December 31, 2023 and 2022.
Leases
The Company determines if an arrangement is or contains a lease at inception. Right-of-Use assets (“ROU assets”) represent the Company’s right to control an underlying asset for the lease term and lease liabilities represent the Company’s obligation to make lease payments arising from the lease. ROU assets and liabilities are recognized at the commencement date based on the present value of lease payments over the initial non-cancelable lease term, unless there is a renewal option that is reasonably certain to be exercised. The Company uses its incremental borrowing rate at the lease commencement date in determining the discount rate utilized to present value the future minimum lease payments since an implicit interest rate in each at-market lease agreement was not determinable. The Company has lease agreements with both lease and non-lease components, which are accounted for as a single component for all asset classes. Lease expense for the Company's operating leases are recognized on a straight-line basis over the lease term.
The Company's variable lease costs, consisting primarily of real estate taxes, insurance costs, and common area maintenance, are expensed as incurred and excluded from the reported ROU assets and lease liabilities amounts presented in the accompanying Balance Sheets. The noncurrent portion of the operating lease liability is included in other long-term liabilities in the accompanying Balance Sheets. Rent expense is allocated to research and development and selling, general and administrative expenses in the accompanying Statements of Operations and Comprehensive Loss.
Concentration Risk
Credit Risk
Financial instruments that potentially subject the Company to significant concentrations of credit risk consist primarily of cash, cash equivalents, marketable securities and accounts receivable. The Company maintains cash held on deposit at financial institutions in the U.S., including Silicon Valley Bank ("SVB"), a division of First Citizens Bank. These
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
deposits are insured by the Federal Deposit Insurance Corporation ("FDIC") in an amount up to $250,000 for any depositor. To the extent the Company holds cash deposits in amounts that exceed the FDIC insurance limitation, it may incur a loss in the event of a failure of any of the financial institutions where it maintains deposits. The Company invests its excess cash in highly liquid investments, including money market fund accounts, that are readily convertible into cash without penalty.
Management believes the Company is not exposed to significant credit risk due to the financial position of the depository institution, but will continue to monitor regularly and adjust, if needed, to mitigate risk, including any ongoing or new events involving limited liquidity, defaults, non-performance or other adverse developments that affect financial institutions. The Company has established guidelines regarding diversification of its investments and their maturities, which are designed to maintain principal and maximize liquidity. To date, the Company has not experienced any losses associated with this credit risk and continues to assess that this exposure is not significant.
Major Customers
The Company entered into agreements with certain limited specialty pharmacies and specialty distributors for the sale of XDEMVY in the U.S. The following table summarizes the percentage of the Company's product sales from each of its largest customers:
| | | | | | | | |
| | Year Ended December 31, 2023 |
| Customer A | | 37 | % |
| Customer B | | 24 | % |
| Customer C | | 14 | % |
| Customer D | | 13 | % |
| Customer E | | 11 | % |
As of December 31, 2023, amounts due from these five customers each exceeded 10% of gross accounts receivable and accounted for approximately 100% of the accounts receivable balance on a combined basis.
Major Suppliers
The Company does not currently own manufacturing facilities and depends on an outsourced manufacturing strategy for the production of XDEMVY for commercial use and for the production of its other product candidates for clinical trials. The Company entered into agreements with third-party manufacturers that are approved for the commercial production of XDEMVY and third-party suppliers that are approved for XDEMVY's active pharmaceutical ingredient. Although there are potential sources of supply other than the Company's existing manufacturers and suppliers, any new supplier would be required to qualify under applicable regulatory requirements. The loss of certain manufacturers and third-party suppliers could result in a temporary disruption of the Company’s commercialization efforts.
Revenue Recognition
(i) Product Sales, Net
The Company recognizes product sales, net of XDEMVY when a customer obtains control of promised goods or services, which occurs at a point in time, typically upon delivery of the Company's product to the customer. The Company records the amount of revenue that reflects the consideration that it expects to receive in exchange for those goods or services. The Company applies the following five-step model in order to determine this amount: (i) identification of the promised goods in the contract; (ii) determination of whether the promised goods are performance obligations, including whether they are capable of being distinct; (iii) measurement of the transaction price, including the constraint on variable consideration; (iv) allocation of the transaction price to the performance obligations; and (v) recognition of revenue as each performance obligation is satisfied.
The Company sells XDEMVY to customers in the U.S., which became available for commercial sale during the third quarter of 2023. The Company sells XDEMVY to a limited number of specialty pharmacies and distributors (i.e., its
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
customers) who in turn sell it directly to clinics, hospitals, pharmacies and federal healthcare programs. Revenue from product sales is primarily recognized upon physical delivery of the product (when the customer obtains control of the product), in return for agreed-upon consideration. Shipping and handling activities are considered to be fulfillment activities rather than a separate performance obligation and are recorded within selling, general and administrative expenses in the accompanying Statements of Operations and Comprehensive Loss.
Revenues from product sales are recorded at the net sales price, or the transaction price, which may include fixed or variable consideration for (i) invoice discounts for prompt payment and distribution service fees, (ii) government and private payer rebates, chargebacks, discounts and fees, (iii) product returns and (iv) costs of co-pay assistance programs for patients, as well as other incentives. Estimates of variable consideration are calculated based on the actual product sales each reporting period and the nature of the variable consideration related to those sales. Where appropriate, the Company utilizes the expected value method to determine the appropriate amount for estimates of variable consideration based on factors such as the current contractual and statutory requirements, specific known market events and trends, industry data and forecasted customer buying and payment patterns. The amount of variable consideration that is included in the transaction price may be constrained and is included in product sales, net only to the extent that it is probable that a significant reversal in the amount of the cumulative revenue recognized will not occur when the uncertainty associated with the variable consideration is subsequently resolved. These estimates reflect the Company’s best estimate of the amount of consideration to which the Company expects to be entitled based on the terms of the contract. Actual amounts of consideration ultimately received may differ materially from estimates. If actual results in the future vary from estimates, the Company will adjust these estimates, which would affect product sales, net and earnings in the period such variances are adjusted. The Company categorizes product sales deduction estimates as follows:
Distribution Service Fees: The Company engages with wholesalers and specialty pharmacies to distribute its products to end customers. The Company pays the wholesalers and certain specialty pharmacies a fee for services such as: inventory management, chargeback administration, and service level commitments. The Company estimates the amount of distribution services fees to be paid to the customers and adjusts the transaction price with the amount of such estimate at the time of sale to the customer. An accrued liability is recorded for unpaid distribution service fees.
Prompt Pay Discounts: The Company provides its customers with a percentage discount on their invoice if the customers pay within the agreed upon timeframe. The Company expects that its customers will earn prompt pay discounts. The Company estimates the probability of customers paying promptly based on the percentage of discount outlined in the purchase agreement between the two parties, and deducts the full amount of these discounts from gross product sales and accounts receivable at the time revenue is recognized.
Product Returns: The Company's customers are contractually permitted to return the product within the contractual allowable time before and after the applicable expiration date. In the initial sales period, the Company estimates its provision for returns based on industry data and adjusts the transaction price at the time of the product sale to the customer. Once sufficient history has been collected for product returns, the Company will utilize that history to inform its returns estimate. Once the product is returned, it is destroyed since it cannot be resold.
Chargebacks: A chargeback is the difference between the Company's invoice price to the wholesaler and the wholesaler’s customer's contract price. The wholesaler tracks these sales and charges back the Company for the difference between the negotiated prices paid between the wholesaler's customers and wholesaler's acquisition cost. The Company estimates the percentage of goods sold that are eligible for chargeback and adjusts the transaction price and accounts receivable at the time of sale of the product to the customer.
Co-payment Assistance: Patients who meet certain eligibility requirements may receive co-payment assistance. The Company records contra-revenue for co-payment assistance based on actual program participation and estimates of program redemption using data provided by third-party administrators. An accrued liability is recorded on unredeemed co-payment assistance related to products for which control has been transferred to the customer.
Rebates and Discounts: The Company accrues rebates for contractually agreed-upon discounts with commercial insurance companies and mandated discounts under government programs such as the Medicaid Drug Rebate Program, Medicare Part D Prescription Drug Program, and other government health care programs in the U.S. The Company's estimates
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
for expected utilization of commercial insurance rebates are based on data received from its customers. The Company's estimates for rebates under government programs are based on statutory discount rates and expected utilization as well as historical data it has accumulated since product launch. The Company’s rebate calculations may require estimates, including estimates of customer mix, to determine which product sales will be subject to rebates and the amount of such rebates. The Company updates its estimates and assumptions on a quarterly basis and records any necessary adjustments to revenue in the period identified. Rebates are generally invoiced and paid in arrears so that the accrual balance consists of an estimate of the amount expected to be incurred for the current quarter’s activity, plus an accrual balance for known prior quarters’ unpaid rebates. If actual rebates vary from estimates, the Company may need to adjust accruals, which would affect product sales, net in the period of adjustment. An accrued liability is recorded for unpaid rebates related to product for which control has transferred to the customer.
(ii) License Fees and Collaboration Revenue
China Out-License
License fees and collaboration revenue in the accompanying Statements of Operations and Comprehensive Loss has historically primarily related to one out-license agreement (the "China Out-License") that allows the third-party licensee to market the Company's TP-03 product candidate (representing functional intellectual property) in the People's Republic of China, Hong Kong, Macau, and Taiwan (the "China territory")— see Note 9. The accounting and reporting of revenue for out-license arrangements requires significant judgment for: (a) identification of the number of performance obligations within the contract; (b) the contract’s transaction price for allocation (including variable consideration); (c) the stand-alone selling price for each identified performance obligation; and (d) the timing and amount of revenue recognition in each period.
The China Out-License was analyzed under GAAP to determine whether the promised goods or services are distinct or must be accounted for as part of a combined performance obligation. In making these assessments, the Company considers factors such as the stage of development of the underlying intellectual property and the capabilities of the customer to develop the intellectual property on their own, and/or whether the required expertise is readily available. If the license is not distinct, the license is combined with other promised goods or services as a combined performance obligation for revenue recognition.
The China Out-License arrangement included the following forms of consideration: (i) non-refundable upfront license payment; (ii) equity-based consideration; (iii) sales-based royalties; (iv) sales-based threshold milestones; (v) one-time payments for executing drug supply agreements; (vi) development milestone payments; and (vii) regulatory milestone payments. Revenue is recognized in proportion to the allocated transaction price when (or as) the respective performance obligation is satisfied. The Company evaluates the progress related to each milestone at each reporting period and, if necessary, adjusts the probability of achievement and related revenue recognition. The measure of progress, and thereby periods over which revenue is recognized, is subject to estimates by management and may change over the course of the agreement.
Contractual Terms for Receipt of Payments
A performance obligation is a promise in a contract to transfer a distinct good or service and is the unit of accounting. A contract’s transaction price is allocated among each distinct performance obligation based on relative standalone selling price and recognized when, or as, the applicable performance obligation is satisfied.
The contractual terms that establish the Company’s right to collect specified amounts from its customers and that require contemporaneous evaluation and documentation under GAAP for the corresponding timing and amount of revenue recognition, are as follows:
Upfront License Fees: The Company determines whether non-refundable license fee consideration is recognized at the time of contract execution (i.e., when the license is transferred to the customer and the customer is able to use and benefit from the license) or over the actual (or implied) contractual period of the China Out-License.The Company also evaluates whether it has any other requirements to provide substantive services that are inseparable from the performance obligation of the license transfer to determine whether any combined performance obligation is satisfied over time or at a point in time.
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
Upfront payments may require deferral of revenue recognition to a future period until the Company performs obligations under these arrangements.
Development Milestones:The Company utilizes the most likely amount method to estimate the amount of consideration to which it will be entitled for achievement of development milestones as these represent variable consideration. For those payments based on development milestones (e.g., patient dosing in a clinical study or the achievement of statistically significant clinical results), the Company assesses the probability that the milestone will be achieved, including its ability to control the timing or likelihood of achievement, and any associated revenue constraint. Given the high degree of uncertainty around the occurrence of these events, the Company determines the milestone and other contingent amounts to be constrained until the uncertainty associated with these payments is resolved. At each reporting period, the Company re-evaluates this associated revenue recognition constraint. Any resulting adjustments are recorded to revenue on a cumulative catch-up basis, and reflected in the financial statements in the period of adjustment.
Regulatory Milestones: The Company utilizes the most likely amount method to estimate the consideration to which it will be entitled and recognizes revenue in the period regulatory approval occurs (the performance obligation is satisfied) as these represent variable consideration. Amounts constrained as variable consideration are included in the transaction price to the extent that it is probable that a significant reversal in the amount of cumulative revenue recognized will not occur when the uncertainty associated with the variable consideration is subsequently resolved. The Company evaluates whether the milestones are considered probable of being reached and not otherwise constrained. Accordingly, due to the inherent uncertainty of achieving regulatory approval, associated milestones are deemed constrained for revenue recognition until achievement.
Royalties: Under the sales-or-usage-based royalty exception the Company recognizes revenue based on the contractual percentage of the licensee’s sale of products to its customers at the later of (i) the occurrence of the related product sales or (ii) the date upon which the performance obligation to which some or all of the royalty has been allocated has been satisfied or partially satisfied. To date, the Company has not recognized any royalty revenue from the China Out-License.
Sales Threshold Milestones: Similar to royalties, applying the sales-or-usage-based royalty exception, the Company recognizes revenue from sales threshold milestones at the later of (i) the period the licensee achieves the one-time annual product sales levels in their territories for which the Company is contractually entitled to a specified lump-sum receipt, or (ii) the date upon which the performance obligation to which some or all of the milestone has been allocated has been satisfied or partially satisfied. To date, the Company has not recognized any sales threshold milestone revenue from the China Out-License.
The Company re-evaluates the measure of progress to each performance obligation in each reporting period as uncertain events are resolved and other changes in circumstances occur.
Other License Fees and Collaboration Revenue
License fees and collaboration revenue also includes revenue recognized from satisfaction of performance obligations under an existing clinical supply agreement. The Company recognizes revenue when a customer obtains control of the promised good or service. Revenue recognized under this arrangement for the year ended December 31, 2023 was $0.2 million. No revenue was recognized under this arrangement for the year ended December 31, 2022.
Cost of Sales
Cost of sales consists of direct and indirect costs related to the manufacturing and distribution of XDEMVY, including raw materials, third-party manufacturing costs, packaging services, freight, third-party royalties payable on the Company’s product sales, net and amortization of capitalized intangible assets associated with XDEMVY. Cost of sales may also include period costs related to certain inventory warehouse and distribution operations and inventory adjustment charges. The Company began capitalizing inventory costs upon FDA approval of XDEMVY in July 2023. Prior to FDA approval of XDEMVY, manufacturing and other inventory costs were recorded to research and development expenses in the Statements of Operations and Comprehensive Loss. Therefore, cost of sales of XDEMVY will reflect a lower average per unit cost until the related inventory is sold.
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
Selling, General and Administrative
Selling, general and administrative costs consist of salaries, benefits, stock-based compensation and other personnel-related costs for our executive, finance, sales and marketing, and other administrative functions. Other selling, general and administrative expenses include sales and marketing costs to support our commercial launch, consulting fees, legal services, rent and other facilities costs, patient assistance donations, and other general operating expenses not otherwise classified as research and development expenses. Advertising costs are expensed as incurred and were $9.4 million for the year ended December 31, 2023.
Research and Development Costs
Research and development costs are expensed as incurred or as certain upfront or milestone payments become contractually due to licensors upon the achievement of clinical or regulatory events. Research and development expenses include internal costs directly attributable to in-development programs, including the costs of salaries, payroll taxes, employee benefit and other employee-related costs (including stock-based compensation expense), license fees, materials, supplies, and the cost of services provided by outside contractors to conduct nonclinical studies, clinical trials and contract manufacturing activities. All costs associated with research and development are expensed as incurred. The Company accrues for these costs based on factors such as estimates of the work completed and in accordance with agreements established with third-party service providers under the service agreements. As it relates to clinical trials, the financial terms of these contracts are subject to negotiations which vary from contract to contract and may result in payment flows that do not match the periods over which materials or services are provided under such contracts. Payments made prior to the receipt of goods or services to be used in research and development are capitalized until the goods or services are received. Such payments are evaluated for current or long-term classification based on when they will be realized. The Company's objective is to reflect the appropriate expense in its financial statements by matching those expenses with the period in which the services and efforts are expended. The Company accounts for these expenses according to the progress of the trial as measured by patient progression and the timing of various aspects of the trial taking into consideration discussions with applicable personnel and outside service providers. The clinical trial accrual is dependent in part upon the timely and accurate reporting of progress and efforts incurred from CROs, contract manufacturers and other third-party vendors. Although estimates are expected to be materially consistent with actual amounts incurred, the Company's understanding of the status and timing of services performed relative to the actual status and timing of services performed can vary and may result in changes in estimates in any particular period. The Company makes significant judgments and estimates in determining the accrued liabilities balance at each reporting period. As actual costs become known, the Company adjusts its accrued liabilities. To date, there have been no material differences between estimates of such expenses and the amounts actually incurred.
The Company has entered into, and may continue to enter into, license agreements to access and utilize certain technology. In each case, the Company evaluates if the license agreement results in the acquisition of an asset or a business. To date, none of the Company's license agreements have been considered an acquisition of a business. For asset acquisitions, the upfront payments to acquire such licenses, as well as any future milestone payments made before product approval that do not meet the definition of a derivative, are immediately recognized as research and development expense in the Statements of Operations and Comprehensive Loss when paid or become payable, provided there is no alternative future use of rights in other research and development projects.
Stock-Based Compensation
The Company recognizes stock-based compensation expense for equity awards granted to employees, consultants, and members of its Board of Directors. Stock option awards are at an exercise price of not less than 100% of the fair market value of common stock on the respective date of grant. The grant date is the date the terms of the award are formally approved by the Company’s Board of Directors or its designee. The Company uses the Black-Scholes option pricing model to estimate the fair value of stock option awards as of the date of grant. The fair value of restricted stock units is representative of the closing market price of the Company's common stock on the date preceding the award grant-date.
Stock awards granted typically have one to four-year service conditions and a contractual term of 10 years. Any performance conditions for vesting are explicitly stated in each award agreement and are associated with clinical, business development, or operational milestones. For stock-based awards that vest subject to the satisfaction of a service requirement, the related expense is recognized on a straight-line basis over each award’s actual or implied vesting period. For stock-based awards that vest subject to a performance condition, the Company recognizes related expense on an accelerated attribution
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
method, if and when it concludes that it is highly probable that the performance condition will be achieved. At each reporting period, the Company reassesses the probability of the achievement of the performance vesting conditions. As applicable, the Company reverses previously recognized expense for unvested awards in the same period of forfeiture.
The measurement of the fair value of stock option awards and recognition of stock-based compensation expense requires assumptions to be estimated by management that involve inherent uncertainties and the application of management’s judgment, including (a) the fair value of the Company’s common stock on the date of the option grant for all awards granted prior to the IPO, (b) the expected term of the stock option until its exercise by the recipient, (c) stock price volatility over the expected term, (d) the prevailing risk-free interest rate over the expected term, and (e) expected dividend payments over the expected term.
All stock-based compensation expense is reported in the accompanying Statements of Operations and Comprehensive Loss within cost of sales, research and development expense or selling, general and administrative expense, based upon the assigned department of the award recipient. The measurement of the fair value of stock option awards and recognition of stock-based compensation expense requires assumptions to be estimated by management that involve inherent uncertainties and the application of management’s judgment, including:
Fair Value of Common Stock — The fair value of the Company’s common stock is based on the closing quoted market price of its common stock as reported by the Nasdaq Global Select Market on the date of the option grant.
Expected Term — The Company’s expected term represents the period that the Company’s stock option awards are expected to be outstanding. Management estimates the expected term of awarded stock options utilizing the simplified method (based on the mid-point between the vesting date and the end of the contractual term) to determine the expected term since the Company does not yet have sufficient exercise history.
Expected Volatility — Prior to 2023, the Company did not have sufficient trading history for its common stock to use its own historical volatility. Management estimated the expected volatility based on a designated peer-group of publicly-traded companies for a look-back period (from the date of grant) that corresponded with the expected term of the awarded stock option. Beginning in January 2023, the Company began using its own historical stock price for expected volatility.
Risk-Free Interest Rate — The Company estimates the risk-free interest rate based upon the U.S. Department of Treasury yield curve in effect at award grant date for the time period that corresponds with the expected term of the awarded stock option.
Dividend Yield — The Company’s expected dividend yield is zero because it has never paid cash dividends and does not expect to for the foreseeable future.
Income Taxes
Income taxes are accounted for using the asset and liability method. Deferred tax assets and liabilities are recorded based on the estimated future tax effects of temporary differences between the tax basis of assets and liabilities and amounts reported in the financial statements, as well as operating losses and tax credit carry forwards using enacted tax rates and laws that are expected to be in effect when the differences are expected to reverse. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period of enactment. Realization of deferred tax assets is dependent upon future earnings, the timing and amount of which are uncertain due to the Company’s historical operating performance and recorded cumulative net losses in prior fiscal periods. A valuation allowance is recorded to reduce deferred tax assets, because based upon a weighting of positive and negative factors, it is more likely than not that these deferred tax assets will not be realized. If/when the Company were to determine that deferred tax assets are realizable, an adjustment to the corresponding valuation allowance would increase the net income in the period that such determination was made.
The Company’s income tax returns are based on calculations and assumptions that are subject to examination by the Internal Revenue Service and other tax authorities. In addition, the calculation of the Company’s tax liabilities involves dealing with uncertainties in the application of complex tax regulations. The Company recognizes liabilities for uncertain tax
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
positions based on a two-step process. The first step is to evaluate the tax position for recognition by determining if the weight of available evidence indicates that it is more likely than not that the position will be sustained on audit, including resolution of related appeals or litigation processes, if any. The second step is to measure the tax benefit as the largest amount that is more than 50% likely of being realized upon settlement. While the Company believes it has appropriate support for the positions taken on its tax returns, the Company regularly assesses the potential outcomes of examinations by tax authorities in determining the adequacy of its provision for income taxes. The Company continually assesses the likelihood and amount of potential revisions and adjusts the income tax provision, income taxes payable and deferred taxes in the period in which the facts that give rise to a revision become known.
Interest and penalties related to unrecognized tax benefits, if any, are recorded as a component of income tax expense.
Net Loss per Share
Basic net loss per share is calculated by dividing the net loss by the weighted-average number of shares of common stock outstanding for the period, without consideration for potential dilutive shares of common stock. Diluted net loss per share is computed by dividing the net loss by the weighted-average number of common stock equivalents outstanding for the period determined using the treasury-stock method and if-converted method as applicable.
Due to net losses in all periods presented, all otherwise potentially dilutive securities are antidilutive, and accordingly, the reported basic net loss per share equals diluted net loss per share.
Comprehensive Loss
Comprehensive loss represents all changes in stockholders’ equity (deficit), except those resulting from distributions to stockholders. For all(i) net loss for the periods presented, comprehensive loss wasand (ii) unrealized gains or losses on the same asCompany's reported net loss.available-for-sale debt securities.
(xv) Recently Issued or Effective Accounting Standards
Recently issued or effective accounting pronouncements that impact, or may have an impact, on the Company’s financial statements have been discussed within the footnote to which each relates. Other recent accounting pronouncements not disclosed in these financial statementsFinancial Statements have been determined by the Company’s management to have no impact, or an immaterial impact, on its current and expected future financial position, results of operations, or cash flows.
3. BALANCE SHEET ACCOUNT DETAILFAIR VALUE MEASUREMENTS
The compositionFinancial assets and liabilities subject to fair value measurements on a recurring basis and the level of select financial statement captions that compriseinputs used in such measurements by major security type are presented in the accompanyingfollowing table:
| | | | | | | | | | | | | | | | | | | | | | | |
| | Year Ended December 31, 2023 |
| Fair Value Measurements |
| | Level 1 | | Level 2 | | Level 3 | | Total |
| Assets: | | | | | | | |
Money market funds(1) | $ | 224,947 | | | $ | — | | | $ | — | | | $ | 224,947 | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| Government-related debt securities | — | | | 2,495 | | | — | | | 2,495 | |
| Common stock in LianBio | 631 | | | — | | | — | | | 631 | |
| Equity warrants (for LianBio shares) | — | | | — | | | 225 | | | 225 | |
| Total assets measured at fair value | $ | 225,578 | | | $ | 2,495 | | | $ | 225 | | | $ | 228,298 | |
(1) This balance sheets are summarized below:
(a) Property and Equipment, net of Accumulated Depreciationincludes cash requirements settled on a nightly basis.
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
“Property | | | | | | | | | | | | | | | | | | | | | | | |
| | Year Ended December 31, 2022 |
| Fair Value Measurements |
| | Level 1 | | Level 2 | | Level 3 | | Total |
| Assets: | | | | | | | |
Money market funds(1) | $ | 64,685 | | | $ | — | | | $ | — | | | $ | 64,685 | |
| U.S. Treasury securities | 69,644 | | | — | | | — | | | 69,644 | |
| Commercial paper | — | | | 60,355 | | | — | | | 60,355 | |
| Corporate debt securities | — | | | 11,521 | | | — | | | 11,521 | |
| Government-related debt securities | — | | | 10,821 | | | — | | | 10,821 | |
| Common stock in LianBio | 371 | | | — | | | — | | | 371 | |
| Equity warrants (for LianBio shares) | — | | | — | | | 108 | | | 108 | |
| Total assets measured at fair value | $ | 134,700 | | | $ | 82,697 | | | $ | 108 | | | $ | 217,505 | |
(1) This balance includes cash requirements settled on a nightly basis.
Money Market Funds and equipment, net” consistsU.S. Treasury Securities
Money market funds and U.S. Treasury securities are highly liquid investments and are actively traded with readily-available market prices that are publicly observable and independently validated as of the following:measurement date. This approach results in the classification of these securities as Level 1 of the fair value hierarchy.
| | | | | | | | | | | |
| | December 31, |
| | 2020 | | 2019 |
| Furniture and fixtures | $ | 294 | | | $ | 5 | |
| Office equipment | 74 | | | 26 | |
| Lab equipment | 173 | | | 92 | |
| Leasehold improvements | 141 | | | 69 | |
| Property and equipment, at cost | 682 | | | 192 | |
| (Less): Accumulated depreciation and amortization | 134 | | | 38 | |
| Property and equipment, net of accumulated depreciation | $ | 548 | | | $ | 154 | |
Commercial paper, corporate debt securities and government-related debt securities were valued using Level 2 inputs that utilized industry standard valuation models, including both income and market-based approaches, for which all significant inputs are observable, either directly or indirectly, to estimate fair value. The Company reviews trading activity and pricing for these investments as of each measurement date.
LianBio Common Stock and Equity Warrants
In March 2021, contemporaneous with the China Out-License transaction, the Company and LianBio executed a warrant agreement for the Company to purchase, in three tranches, common shares in LianBio at an exercise price equal to common stock par value, which converted into warrants of the parent company of LianBio (a pharmaceutical company focused on the Greater China and other Asian markets; Nasdaq: LIAN; any references to common stock or warrants of LianBio shall refer to common stock or warrants of the publicly-traded parent of LianBio) in connection with LianBio's previous Initial Public Offering. The first two tranches were vested, exercised, and converted into 156,746 shares of LianBio common stock as of December 31, 2022 and are recognized at fair value within long-term investments on the Balance Sheets as of December 31, 2023 and 2022. The Company does not believe a reserve for credit losses is required for the LianBio common stock as of December 31, 2023 and 2022. LianBio common stock is classified within Level 1 of the fair value hierarchy, given its publicly reported price on the Nasdaq Global market.
The third warrant tranche remains classified as Level 3 in the fair value hierarchy and is presented within other assets on the accompanying Balance Sheets as of December 31, 2023 and 2022. This warrant tranche remains classified as Level 3 in the fair value hierarchy. The most significant assumptions used in the option pricing valuation model as of each balance sheet date to determine its fair value include observable and unobservable inputs: LianBio common stock volatility (based on the historical volatility of similar companies); the probability of regulatory milestone achievement for vesting; and the application of an assumed discount rate.
Depreciation expense (includedThe estimated fair value of the equity warrants are remeasured each reporting period with adjustments reported within “total operating expenses” inother income (expense) on the accompanying Statements of Operations and Comprehensive Loss) forLoss, until exercised or expired. These equity warrants are valued in the years ended December 31, 2020 and 2019 was $0.1 million anaccompanying Financial Statements as follows:d $37 thousand, respectively.
(b) Accounts Payable and Other Accrued Liabilities
“Accounts payable and other accrued liabilities” consists of the following:
| | | | | | | | | | | |
| | December 31, |
| | 2020 | | 2019 |
| Trade accounts payable and other | $ | 2,237 | | | $ | 456 | |
| Operating lease liability, current portion | 282 | | | 64 | |
| Accrued clinical studies | 1,524 | | | 0 | |
| Employee stock option early exercise liability, current portion | 304 | | | 0 | |
| Accounts payable and other accrued liabilities | $ | 4,347 | | | $ | 520 | |
(c) Other Long-Term Liabilities
“Other long-term liabilities” consists of the following:
| | | | | | | | | | | |
| | December 31, |
| | 2020 | | 2019 |
| Operating lease liability, non-current portion | $ | 549 | | | $ | 100 | |
| Employee stock option early exercise liability, non-current portion | 56 | | | 0 | |
| Other long-term liabilities | $ | 605 | | | $ | 100 | |
4. STOCKHOLDERS’ EQUITY
Authorized Stock
Under the October 2020 Amended and Restated Certificate of Incorporation, the Company is authorized to issue two classes of stock: common and preferred. The total number of shares authorized for issuance is 200.0 million of common shares and 10.0 million of preferred shares.
Preferred Stock Overview
Series A Preferred Stock Issuance
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
In March and May 2018, the Company executed a private placement Series A Stock Purchase Agreement and issued 1.6 million shares of Series A Preferred Stock at $2.3174(all tabular amounts presented in thousands, except share, per share, for net proceedsper unit, and number of $3.6 million, after issuance costs of $0.1 million.years)
Series B Preferred Stock Issuance | | | | | | | | |
| | Value of equity warrants |
| Fair value as of December 31, 2022 | | $ | 108 | |
| Remeasurement of equity warrants | | 117 | |
| | |
| Fair value as of December 31, 2023 | | $ | 225 | |
In December 2019, the Company executed a private placement Series B Stock Purchase Agreement of 6.7 million shares of Series B Preferred Stock at $8.9904 per share for net proceeds of $57.4 million, after issuance costs of $0.2 million. Concurrently, convertible notes issued in May, August, and October 2019 for aggregate proceeds of $2.0 million were converted based on principal and accrued interest, and the Company issued 0.3 million shares of Series B Preferred Stock at its contractual conversion price (see Note 8).
Series C Preferred Stock Issuance
In September 2020, the Company executed a private placement Series C Stock Purchase agreement of 2.9 million shares of Series C Preferred Stock at a purchase price of $14.0003 per share for net proceeds of $39.8 million, after issuance costs of $0.2 million.
On October 20, 2020, upon the closing of the IPO, all outstanding shares of preferred stock were automatically converted into an aggregate 11,107,018 shares of the Company's common stock and $103.2 million of mezzanine equity was reclassified to common stock and additional paid-in capital. As of December 31, 2020, there were 0 shares of preferred stock issued and outstanding. | | | | | | | | |
| | Value of equity warrants |
| Fair value as of December 31, 2021 | | $ | 663 | |
| Remeasurement of equity warrants | | (501) | |
| Recognition of equity warrants | | 103 | |
| Exercise of the second tranche of equity warrants into LianBio common stock | | (157) | |
| Fair value as of December 31, 2022 | | $ | 108 | |
The table belowfair value and amortized cost of cash equivalents and available-for-sale investments by major security type are presented in the following table:
| | | | | | | | | | | | | | | | | | | | | | | |
| December 31, 2023 |
| Amortized cost | | Unrealized gains | | Unrealized losses | | Estimated fair value |
| Cash equivalents: | | | | | | | |
Money market funds(1) | $ | 224,947 | | | $ | — | | | $ | — | | | $ | 224,947 | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| Total cash equivalents | $ | 224,947 | | | $ | — | | | $ | — | | | $ | 224,947 | |
| | | | | | | |
| Marketable securities: | | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| Government-related securities | $ | 2,496 | | | $ | — | | | $ | (1) | | | $ | 2,495 | |
| Total marketable securities | $ | 2,496 | | | $ | — | | | $ | (1) | | | $ | 2,495 | |
| | | | | | | |
| Long-term investments: | | | | | | | |
| Common stock in LianBio | $ | 1,108 | | | $ | — | | | $ | (477) | | | $ | 631 | |
| Total long-term investments | $ | 1,108 | | | $ | — | | | $ | (477) | | | $ | 631 | |
(1) This balance includes preferred stock details as of December 31, 2019.cash requirements settled on a nightly basis.
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| As of December 31, 2019 | | Authorized | | Outstanding | | Net
Carrying
Value | | Liquidation
Preference | | Original
Issue
Price |
| Series A Preferred Stock | | 1,575,030 | | | 1,575,030 | | | $ | 3,564 | | | $ | 3,650 | | | $ | 2.3174 | |
| Series B Preferred Stock | | 6,731,649 | | | 6,674,909 | | | $ | 59,838 | | | $ | 60,010 | | | $ | 8.9904 | |
| Total | | 8,306,679 | | | 8,249,939 | | | $ | 63,402 | | | $ | 63,660 | | | |
Common Stock Overview and Reserve for Future Issuance
Common stockholders have 1 vote for each share of common stock held and are entitled to receive any dividends declared by the Company’s Board of Directors when legally available for distribution, subject to the dividend rights of the holders of Series A, Series B, and Series C preferred stock discussed above. For the years ended December 31, 2020 and 2019, 0 dividends were declared.
As of December 31, 2020 and 2019, the Company had 20.5 million and 2.7 million common shares issued, respectively. At December 31, 2020 and 2019, the Company had 20.3 million and 2.6 million, common shares outstanding, respectively. The following shares of common stock were reserved for issuance:
| | | | | | | | | | | |
| | December 31, |
| | 2020 | | 2019 |
| Preferred Stock outstanding | 0 | | | 8,249,939 | |
| Stock options issued and outstanding | 1,836,739 | | | 297,142 | |
| Stock options reserved for future grant | 9,414,091 | | | 2,150,867 | |
| Total shares of common stock reserved | 11,250,830 | | | 10,697,948 | |
5. STOCK-BASED COMPENSATION
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
| | | | | | | | | | | | | | | | | | | | | | | |
| December 31, 2022 |
| Amortized cost | | Unrealized gains | | Unrealized losses | | Estimated fair value |
| Cash equivalents: | | | | | | | |
Money market funds(1) | $ | 64,685 | | | $ | — | | | $ | — | | | $ | 64,685 | |
| Government-related securities | 4,978 | | | — | | | — | | | 4,978 | |
| Commercial paper | 1,997 | | | — | | | — | | | 1,997 | |
| Total cash equivalents | $ | 71,660 | | | $ | — | | | $ | — | | | $ | 71,660 | |
| | | | | | | |
| Marketable securities: | | | | | | | |
| U.S. Treasury securities | $ | 69,720 | | | $ | 5 | | | $ | (81) | | | $ | 69,644 | |
| Commercial paper | 58,358 | | | — | | | — | | | 58,358 | |
| Corporate debt securities | 11,524 | | | 8 | | | (11) | | | 11,521 | |
| Government-related securities | 5,838 | | | 5 | | | — | | | 5,843 | |
| Total marketable securities | $ | 145,440 | | | $ | 18 | | | $ | (92) | | | $ | 145,366 | |
| | | | | | | |
| Long-term investments: | | | | | | | |
| Common stock in LianBio | $ | 1,231 | | | $ | — | | | $ | (860) | | | $ | 371 | |
| Total long-term investments | $ | 1,231 | | | $ | — | | | $ | (860) | | | $ | 371 | |
(1) This balance includes cash requirements settled on a nightly basis.
As of December 31, 2023, all available-for-sale debt securities had a maturity of 12 months or less. As of December 31, 2022, substantially all available-for-sale debt securities had a maturity of 12 months or less. Three securities had a contractual maturity between one and five years, with an estimated fair market value of $4.6 million and amortized cost of $4.6 million. As of December 31, 2023 and 2022, the Company had one available-for-sale debt security and twenty-four available-for-sale debt securities, respectively, in a continuous gross unrealized loss position for less than one year. As of December 31, 2023 and 2022, unrealized credit losses on these securities were not material. Further, the Company does not intend to sell these investments prior to maturity and it is not more likely than not that the Company will be required to sell these investments before recovery of their amortized cost basis. Accordingly, the Company did not recognize any other-than-temporary impairment losses.
4. BALANCE SHEET ACCOUNT DETAIL
The composition of selected captions within the accompanying Balance Sheets are summarized below:
Inventory
Inventory consists of the following:
| | | | | | | | |
| | December 31, 2023 |
| Raw materials | | $ | 2,533 | |
| Work in progress | | 392 | |
| Finished goods | | 182 | |
| Inventory | | $ | 3,107 | |
Property and Equipment, Net
Property and equipment, net consists of the following:
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
| | | | | | | | | | | |
| | December 31, |
| | 2023 | | 2022 |
| Furniture and fixtures | $ | 1,251 | | | $ | 714 | |
| Office equipment | 660 | | | 197 | |
| Laboratory equipment | 167 | | | 167 | |
| Leasehold improvements | 680 | | | 425 | |
| Property and equipment, at cost | 2,758 | | | 1,503 | |
| (Less): Accumulated depreciation and amortization | (1,290) | | | (546) | |
| Property and equipment, net | $ | 1,468 | | | $ | 957 | |
Depreciation expense for the years ended December 31, 2023 and 2022 was $0.7 million and $0.3 million, respectively.
Accounts Payable and Other Accrued Liabilities
Accounts payable and other accrued liabilities consists of the following:
| | | | | | | | | | | |
| | December 31, |
| | 2023 | | 2022 |
| Trade accounts payable and other | $ | 18,149 | | | $ | 5,498 | |
| Accrued product sales deductions | 4,867 | | | — | |
| Accrued clinical studies | 277 | | | 3,691 | |
| Operating lease liability, current | 398 | | | 721 | |
| Accounts payable and other accrued liabilities | $ | 23,691 | | | $ | 9,910 | |
5. STOCKHOLDERS’ EQUITY
2020 and 2016 Equity Incentive PlanPlans
The Company's Board of Directors and stockholders adopted and approved the Company's 2020 Equity Incentive Plan (the “2020 Plan”) onin October 8, 2020. The 2020 Plan replacesreplaced the Company's 2016 Equity Incentive Plan however,that was earlier adopted in December 2016 (the "2016 Plan"). However, awards outstanding under the 2016 Plan will continue to be governed by their existingits original terms. The number of shares of the Company's common stock that were initially available for issuance under the 2020 Plan equalequaled the initial sum of 9,000,000 shares plus 2,432,980 shares remainingthat were then available for issuance under the 2016 Plan, or issued pursuant to or subject to awards granted under the 2016 Plan. The 2020 Plan provides for the following types of awards: incentive and non-statutory stock options, stock appreciation rights, restricted shares, and restricted stock units.
The number of shares of common sharesstock reserved for issuance under the 2020 Plan are increased automatically on the first business day of each fiscal year, commencing in 2021 and ending in 2030, by a number equal to the lesser of: (i) 4% of the shares of common stock outstanding on the last business day of the prior fiscal year; or (ii) the number of shares determined by the Company's Board of Directors. In general, to the extent that any awards under the 2020 Plan are forfeited, terminate, expire or lapse without the issuance of shares, or if the Company reacquires the shares subject to awards granted under the 2020 Plan, those shares will again become available for issuance under the 2020 Plan, as will shares applied to pay the exercise or purchase price of an award or to satisfy tax withholding obligations related to any award.
PriorEmployee Stock Purchase Plan
Under the terms of the Company's 2020 Employee Stock Purchase Plan ("ESPP"), eligible employees can purchase common stock through scheduled payroll deductions. The purchase price is equal to adopting the 2020 Plan, the Company had 1 active stockholder-approved stock-based compensation plan (the “2016 Plan”), adopted in December 2016, which permitted the grant of incentive stock options, nonqualified stock options, stock awards and certain other awards to its employees, members of its Board of Directors, and consultants.
Stock-based awards are governed by agreements between the Company and the recipients. Incentive stock options and nonqualified stock options may be granted under the 2016 Plan and 2020 Plan at an exerciseclosing price of not less than 100% of the fair market value ofCompany's common stock on the respective date of grant. The grant date is the date the termsfirst or last day of the award are formally approved byoffering period (whichever is less), minus a 15% discount. To determine the Company’s Boardvalue of Directors or its designee.
Through December 31, 2020, all awards issued underESPP expense to be recognized during each offering period, the 2016 Plan wereBlack-Scholes option-pricing model is used, in the form of stock options. These option agreements have service and/or performance conditions for vesting, unless immediately vested on the date of grant. Stock options granted typically have one to four-year service conditions for full vesting. The performance conditions for vesting are explicitly stated in each option agreement and are associated with clinical, business development, or operational milestones.
Stock options must generally be exercised, if at all, no later than 10 years from the date of grant. Upon termination of employment, vested stock options may be exercised within 12 months after the date of termination upon death; six months after the date of termination upon disability; and three months after the date of termination for all other separations.
Stock-Based Compensation Summary
Stock-based compensation expense for the years ended December 31, 2020 and 2019 was as follows:
| | | | | | | | | | | |
| | Year Ended
December 31, |
| | 2020 | | 2019 |
| Research and development | $ | 260 | | | $ | 6 | |
| General and administrative | 579 | | | 12 | |
| Total stock-based compensation | $ | 839 | | | $ | 18 | |
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
Valuation Assumptionswith the discounted employee price. A participant may purchase a maximum of 3,000 shares of common stock during a six-month offering period, not to exceed $25,000 at full market value on the offering date during each ESPP year.
Beginning on January 1, 2021, and each January 1st thereafter, pursuant to the terms of the ESPP, the number of common stock available for issuance under the ESPP is automatically increased by an amount equal to the lesser of (i) one percent of the total number of shares of common stock outstanding on the last day of the year, (ii) 2.5 million shares, or (iii) a number determined by the board of directors.
Common Stock OptionsOutstanding and Reserves for Future Issuance
As of December 31, 2023 and 2022, the Company had 34.2 million and 26.7 million, respectively, of common stock issued and outstanding. Common stockholders have one vote for each share of common stock held and are entitled to receive dividends declared by the Company’s Board of Directors when legally available for distribution, then-subject to the dividend rights of the holders of preferred stock. For the years ended December 31, 2023 and 2022, no dividends were declared.
The Company's outstanding equity awards and shares reserved for future issuance under its 2020 and 2016 Equity Incentive Plans and 2020 Employee Stock Purchase Plan are summarized below:
| | | | | | | | | | | |
| | December 31, |
| | 2023 | | 2022 |
| Common stock awards reserved for future issuance under 2020 and 2016 Equity Incentive Plans | 7,054,222 | | | 8,346,738 | |
| Common stock awards reserved for future issuance under the 2020 Employee Stock Purchase Plan | 2,859,434 | | | 2,663,319 | |
| Stock options issued and outstanding (unvested and vested) under 2020 and 2016 Equity Incentive Plans | 4,760,366 | | | 3,899,342 | |
| Restricted stock units issued and outstanding (unvested) under 2020 Equity Incentive Plan | 1,708,725 | | | 551,258 | |
| Total shares of common stock reserved | 16,382,747 | | | 15,460,657 | |
6. STOCK-BASED COMPENSATION
Stock-Based Compensation Expense
Stock-based compensation expense was recognized in the accompanying Statements of Operations and Comprehensive Loss as follows:
| | | | | | | | | | | |
| | Year Ended
December 31, |
| | 2023 | | 2022 |
| Cost of sales | $ | 190 | | | $ | — | |
| Research and development | 5,833 | | | 3,736 | |
| Selling, general and administrative | 13,807 | | | 9,724 | |
| Total stock-based compensation | $ | 19,830 | | | $ | 13,460 | |
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
The fair value of granted stock options was estimated as of the date of grant using the Black-Scholes option-pricing model, based on the following inputs:
| | | | | | | | | | | |
| | Year Ended
December 31, |
| 2020 | | 2019 |
| Exercise price (estimated fair value per common share on grant date) | $2.01 to $10.99 | | $ | 0.45 | |
| Expected term (in years) (a) | 6.25 | | 5.94 to 6.25 |
| Risk-free interest rate (b) | 0.38% to 0.47% | | 2.4% to 2.5% |
| Expected volatility (c) | 70.1% to 71.2% | | 82.5% to 89.4% |
| Expected dividend yield (d) | 0 | | | 0 | |
| Weighted-average grant-date fair value per stock option | $ | 4.16 | | | $ | 0.34 | |
(a)Determined using the “simplified method” under applicable GAAP (SAB 107); the maximum contractual term of issued stock options is 10 years.
(b)Based upon the U.S. Treasury yields in effect during the period which the options were granted (for a period equaling the stock options’ expected term).
(c)Measured using the volatility of the stock price for the Company’s designated peer group of publicly-traded companies for a period equal to the expected option term.
(d)Tarsus does not expect to declare any cash dividends in the foreseeable future. | | | | | | | | | | | |
| | Year Ended
December 31, |
| 2023 | | 2022 |
| Exercise price | $12.48 to $18.78 | | $12.89 to $20.64 |
| Expected term (in years) | 6.25 | | 6.25 |
| Risk-free interest rate | 3.38% to 4.83% | | 1.63% to 4.21% |
| Weighted average volatility | 69.7% to 73.3% | | 77.3% to 83.0% |
| Dividend yield rate | — | | | — | |
| Weighted-average grant-date fair value per stock option | $ | 15.49 | | | $ | 18.37 | |
Stock Option Activity
Stock option activity during the years ended December 31, 20202023 and 20192022 was as follows:
| | Number of
Shares | | Weighted-
Average
Exercise
Price/Share | | Weighted-
Average
Remaining
Contractual
Term (Years) | | Aggregate Intrinsic Value (1) |
| Outstanding - December 31, 2018 | 267,041 | | | 0.31 | | | 9.51 | | $ | 37 | |
| Number of
Shares | | | Number of
Shares | | Weighted-
Average
Exercise
Price/Share | | Weighted-
Average
Remaining
Contractual
Term (Years) | | Aggregate Intrinsic Value (1) |
| Outstanding - December 31, 2021 | |
| Granted | Granted | 32,793 | | | 0.45 | | |
| Exercised | Exercised | 0 | | |
| Exercised | |
| Exercised | |
| Forfeited | |
| Forfeited | |
| Forfeited | Forfeited | (1,907) | | | 0.45 | | |
| Expired | Expired | (785) | | | 0.45 | | |
| Outstanding - December 31, 2019 | 297,142 | | | 0.32 | | | 8.60 | | $ | 37 | |
| Expired | |
| Expired | |
| Outstanding - December 31, 2022 | |
| Outstanding - December 31, 2022 | |
| Outstanding - December 31, 2022 | |
| Granted | Granted | 1,741,384 | | | 4.16 | | |
| Exercised | Exercised | (197,179) | | | 1.88 | | |
| Exercised | |
| Exercised | |
| Forfeited | |
| Forfeited | |
| Forfeited | Forfeited | (4,608) | | | 0.90 | | |
| Expired | Expired | 0 | | | 0 | |
| Outstanding - December 31, 2020 | 1,836,739 | | | $ | 3.77 | | | 9.15 | | $ | 68,981 | |
| Vested - December 31, 2020 | 428,491 | | | $ | 1.68 | | | 8.47 | | $ | 16,988 | |
| Unvested - December 31, 2020 | 1,408,248 | | | $ | 4.41 | | | 9.36 | | $ | 51,993 | |
| Expired | |
| Expired | |
| Outstanding - December 31, 2023 | |
| Outstanding - December 31, 2023 | |
| Outstanding - December 31, 2023 | |
| Vested - December 31, 2023 | |
| Unvested - December 31, 2023 | |
____________
(1)RepresentsThe aggregate intrinsic value is calculated as the total difference between the estimated stockexercise price of the options and the fair value of the Company's common stock as of December 31, 2020 and the stock option exercise price, multiplied by the number of in-the-money options as of December 31, 2020. The amount of any intrinsic value will change in relation to any increases or decreases in the then-determined fair value of the Company’s common stock.2023.
The total grant-date fair value of options that vested during the years ended December 31, 20202023 and 20192022 was $0.7$26.4 million and $33 thousand,$13.7 million, respectively.
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
The following table summarizes information with respect to stock option grants as ofFor the year ended December 31, 2020:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | As of December 31, 2020 |
| | Outstanding | | Exercisable |
| Exercise Price | | Granted
Stock
Options
Outstanding | | Aggregate
Intrinsic
Value | | Weighted-
Average
Remaining
Contractual
Life (Years) | | Granted
Stock
Options
Exercisable | | Aggregate
Intrinsic
Value | | Weighted-
Average
Remaining
Contractual
Life (Years) |
| $0.0007 | | 83,809 | | | $ | 3,464 | | | 6.91 | | 83,809 | | | $ | 3,464 | | | 6.91 |
| $0.45 | | 195,088 | | | 7,976 | | | 7.86 | | 158,647 | | | 6,547 | | | 7.82 |
| $2.01 | | 1,143,868 | | | 44,982�� | | | 9.33 | | 1,085,376 | | | 44,566 | | | 9.34 |
| $10.99 | | 413,974 | | | 12,559 | | | 9.73 | | 22,158 | | | 883 | | | 9.73 |
| | 1,836,739 | | | $ | 68,981 | | | 9.15 | | 1,349,990 | | | $ | 55,460 | | | 9.01 |
2023, the Company recorded stock-based compensation expense for stock options of $13.0 million. As of December 31, 2020,2023, there was approximately $22.9 million of unrecognized compensation expense of $6.5 million related to unvested stock options, which the Company expects to recognize over a weighted average period of 2.82.1 years.
Early Exercise Feature of CertainRestricted Stock OptionsUnit Activity
The 2016 Plan permits certain option holders to exercise awarded options prior to vesting. Upon early exercise,Restricted stock unit activity during the options become subject to ayear ended December 31, 2023 was as follows:
| | | | | | | | | | | |
| Number of
Shares | | Weighted Average Value per Share |
| Outstanding - December 31, 2021 | 17,251 | | | $ | 27.53 | |
| Granted | 573,009 | | | 17.87 | |
| Vested | (32,914) | | | 24.17 | |
| Forfeited | (6,088) | | | 19.40 | |
| Outstanding - December 31, 2022 | 551,258 | | | $ | 17.78 | |
| Granted | 1,442,677 | | | 15.95 | |
| Vested | (206,813) | | | 17.87 | |
| Forfeited | (78,397) | | | 16.02 | |
| Outstanding - December 31, 2023 | 1,708,725 | | | $ | 16.31 | |
For the year ended December 31, 2023, the Company recorded stock-based compensation expense for restricted stock agreement and remain subjectunits of $6.5 million. As of December 31, 2023, there was approximately $23.3 million of unrecognized compensation expense related to unvested restricted stock units, which the Company expects to recognize over a weighted average period of 3.2 years.
Employee Stock Purchase Plan
Stock-based compensation expense related to the same vesting provisions inESPP was $0.3 million and $0.3 million, respectively, for the corresponding stock option awardyears ended December 31, 2023 and unvested options are subject to repurchase by2022.
7. NET LOSS PER SHARE
The following table sets forth the Company upon termination at the same price exercised. These unvested shares are reported as issued, but not outstanding while subject to repurchase by the Company and are also excluded from thecomputation of basic and diluted net loss per share calculation until the repurchase right lapses upon vesting.share:
The Company initially records a liability for these early exercises that is subsequently reclassified into stockholders’ equity on a pro rata basis as vesting occurs. As of December 31, 2020 the Company has recorded the unvested portion of the exercise proceeds of $0.4 million as a liability from the early exercise in the accompanying Balance Sheets. During the years ended December 31, 2019, the Company repurchased 26,927 of unvested common stock options from a former consultant in connection with termination of his consulting agreement. As of December 31, 2019, all stock options with early exercise features were either vested or immaterial if early exercised and the Company reported 0 corresponding liability in the accompanying Balance Sheets.
6. NET LOSS PER SHARE
Net loss per share attributable to common stockholders was computed by dividing net loss by the weighted-average number of common shares outstanding for the years ended December 31, 2020 and 2019:
| | | | Year Ended December 31, | | Year Ended December 31, |
| | | 2020 | | 2019 | | 2023 | | 2022 |
| Net loss | Net loss | $ | (26,811) | | | $ | (4,670) | |
| Weighted-average shares—basic and diluted | Weighted-average shares—basic and diluted | 6,207,367 | | | 2,362,768 | |
Net loss per share attributable to common
stockholders—basic and diluted | $ | (4.32) | | | $ | (1.98) | |
| Net loss per share—basic and diluted | |
The following outstanding and potentially dilutive securities were excluded from the calculation of diluted net loss per share attributable to common stockholders because their impact under the “treasurytreasury stock method”method and “if-converted method”if-converted method would have been anti-dilutive for the periodseach period presented:
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
| | | | | | | | | | | |
| | Year Ended December 31, |
| | 2020 | | 2019 |
| Stock options, unexercised—vested and unvested | 1,836,739 | | | 297,142 | |
| Series A and Series B Preferred Stock, outstanding | 0 | | | 1,922,491 | |
| Stock options early-exercised and unvested | 179,375 | | | 4,300 | |
| Convertible promissory notes | 0 | | | 93,421 | |
| Total | 2,016,114 | | | 2,317,354 | |
| | | | | | | | | | | |
| | Year Ended December 31, |
| | 2023 | | 2022 |
| Stock options, unexercised—vested and unvested | 4,760,366 | | | 3,899,342 | |
| Restricted stock units—unvested | 1,708,725 | | | 551,258 | |
| Total | 6,469,091 | | | 4,450,600 | |
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
7. FAIR VALUE MEASUREMENTS
The table below summarizes certain financial instruments measured at fair value that are included within the accompanying balance sheets, and their designation among the three fair value measurement categories (see Note 2(xiii)):
| | | | | | | | | | | | | | | | | | | | | | | |
| | Year Ended December 31, 2020 |
| Fair Value Measurements |
| | Level 1 | | Level 2 | | Level 3 | | Total |
| Assets: | | | | | | | |
| Money market funds | $ | 168,129 | | | $ | 0 | | | $ | 0 | | | $ | 168,129 | |
| Total assets measured at fair value | $ | 168,129 | | | $ | 0 | | | $ | 0 | | | $ | 168,129 | |
| | | | | | | | | | | | | | | | | | | | | | | |
| | Year Ended December 31, 2019 |
| Fair Value Measurements |
| | Level 1 | | Level 2 | | Level 3 | | Total |
| Assets: | | | | | | | |
| Money market funds | $ | 57,952 | | | $ | 0 | | | $ | 0 | | | $ | 57,952 | |
| Total assets measured at fair value | $ | 57,952 | | | $ | 0 | | | $ | 0 | | | $ | 57,952 | |
Money Market Funds
Money market fund holdings are included(all tabular amounts presented in cashthousands, except share, per share, per unit, and cash equivalents on the accompanying balance sheets and are classified within Level 1 of the fair value hierarchy because they have readily-available market prices in active markets that are publicly accessible at the measurement date. These money market funds are invested in U.S. Treasury bills and notes, and other obligations issued or guaranteed as to principal and interest by the U.S. Government or its agencies.
Convertible Promissory Notes – Derivative Liabilities
The following table sets forth a summary of the changes in fair value of the bifurcated derivative liability associated with the convertible promissory notes issued and settled during 2019 to certain related parties (see Note 8). The measurement of the derivative liabilities represents a Level 3 financial instrument:
| | | | | |
| Derivative
Liabilities |
Fair value as of December 31, 2018 | $ | 0 | |
Initial fair value of derivative liability upon issuance of May 2019 Notes | 28 | |
Initial fair value of derivative liability upon issuance of August 2019 Notes | 50 | |
Initial fair value of derivative liability upon issuance of October 2019 Notes | 209 | |
Revaluation of derivative liabilities included in other income (expense), net within the Statement of Operations for the year ended December 31, 2019 | 76 | |
Settlement of derivative liabilities through conversion of all Notes | (363) | |
Fair value as of December 31, 2019 | $ | 0 | |
The fair values of the derivative liabilities presented above were estimated at the date of issuance and at subsequent balance sheet dates using a two-step approach to valuation. Management utilized a probability-weighted valuation method and then compared the instrument’s value with-and-without the derivative features in order to estimate their combined fair value, using unobservable inputs, which are classified as Level 3 within the fair value hierarchy. The significant inputs not included in the market and thus represents a Level 3 measurement in the valuation approach included the probability of achieving a settlement that provides the note holders the rights or the obligations to receive cash or a variable number of shares upon the completion of a then-future capital transaction. The convertible notes were issued and settled in full during the year ended December 31, 2019 (see Note 8).
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTSyears)
8. CONVERTIBLE PROMISSORY NOTES PAYABLE
Overview of Notes and Conversion in December 2019
In May 2019, the Company entered into a Note Purchase Agreement (the “May 2019 Purchase Agreement”) with its co-founders and certain other related parties (the “Note Holders”). Under the terms of the May 2019 Purchase Agreement, the Company received cash proceeds of $0.5 million and issued $0.5 million of convertible promissory notes (the “May 2019 Notes”) with a stated maturity of December 2020. These notes bore interest at a rate of 8.0% per annum, compounded annually, and payable at maturity. In the event of a qualified equity financing, the outstanding principal of the May 2019 Notes plus all accrued and previously unpaid interest would, at the option of the holder, either (i) automatically convert into shares of stock issued in the qualified equity financing based on a conversion price equal to 90% of the issuance price paid by these new investors, or (ii) be repaid in full.
In August 2019, the Company amended and restated the May 2019 Purchase Agreement with the Note Holders and received an additional $0.5 million of proceeds and issued new $0.5 million convertible promissory notes to the same parties (the “August 2019 Notes”) with identical terms.
In October 2019, the Company entered into a new Note Purchase Agreement (the “October 2019 Purchase Agreement”) with the Note Holders. Under the terms of the October 2019 Purchase Agreement, the Company received proceeds of $1.0 million and issued $1.0 million of convertible promissory notes (the “October 2019 Notes,” collectively with the May 2019 Notes and the August 2019 Notes, the “Notes”) with a conversion price equal to 80% of the issuance price in a qualified equity financing.
In December 2019, the Company completed an issuance of Series B Preferred Stock (see Note 5). Upon this issuance, the $2.0 million of Note principal value, along with accrued interest, were converted into 0.3 million shares of Series B Preferred Stock under its contractual terms. The Company recorded “loss on extinguishment of convertible notes” (non-cash) of $0.3 million within “other income (expense)” in the accompanying Statements of Operations and Comprehensive Loss for the year ended December 31, 2019.
Embedded Derivative and its Accounting
The Notes allowed for redemption upon a qualified equity financing that was determined to be a contingent redemption feature that was not closely related to the Notes and was thus was required to be bifurcated as a derivative liability on the balance sheet. Based on the features of this derivative, the Company accounted for it as an implied discount in presenting the carrying value of these Notes. This discount was accreted over the term to maturity of the Notes using the effective interest method, resulting in aggregate interest expense recognition (non-cash) of $0.1 million for the year ended December 31, 2019. As the Notes were converted in December 2019, 0 interest expense was recorded for the year ended December 31, 2020.
Changes in the embedded derivatives’ fair value at each reporting period were recognized in the accompanying statements of operations and comprehensive loss within “changes in fair value of derivative liabilities,” resulting in incremental “other expense” recognition (non-cash) of $0.1 million for the year ended December 31, 2019. As the Notes were converted in December 2019, 0 other expense related to changes in fair value of derivative liabilities was recorded for the year ended December 31, 2020.
9. COMMITMENTS & CONTINGENCIES
(a) Facility Leases
OverviewLease Agreements
In the ordinary course of business, the Company enters into lease agreements with unaffiliated third parties for the use of office and laboratoryits facilities and office equipment. As of December 31, 2019,2023, the Company had 1five active facility lease in Irvine, California, that commenced on March 1, 2019 and expires April 30, 2022. This lease has a renewal option at the end of term, for which the Company was not reasonably certain to exercise at the lease commencement. As such, the renewal option
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
was not included in the lease term used to calculate the right-of-use lease asset and lease liability. Prior to March 1, 2019, the Company did not have any material lease arrangements. As of December 31, 2020, the Company recorded an impairment of the operating lease right-of-use asset for this research and development facility of $15 thousand.
The Company entered into 2 additional facility leases that commenced on June 1, 2020 for adjacent administrativeoffice and laboratory suites in Irvine, California. These leases expire onOn May 1, 2023 the Company amended the existing facilities lease, extending the term for three years through January 31, 2024. Both2027.
The below table summarizes the components of these leases included a renewal option attotal lease expense:
| | | | | | | | | | | |
| Year Ended December 31, |
| 2023 | | 2022 |
| Operating lease expense | $ | 705 | | | $ | 569 | |
| Variable lease expense | 391 | | | 244 | |
| Total lease expense | $ | 1,096 | | | $ | 813 | |
As of December 31, 2023, the end of its term, for which the Company was not reasonably certain to exercise at the lease commencement. As such, the renewal option was not included in the lease term used to calculate the right-of-use lease asset and lease liability. In connection with these 2 leases the Company capitalized right-of-use assets along with an accompanying lease liability of $0.7 million.
All of the Company’sCompany's facility leases have minimum annual rent, payable monthly, and carry fixed annual rent increases. Under the arrangements, real estate taxes, certain operating expenses, and common area maintenance are reimbursable to the lessor. These amounts are expensed as incurred, as they are variable in nature and therefore excluded from the measurement of the reported right-of-use asset and liability discussed below. During the years ended December 31, 2020 and 2019, the Company had no sublease arrangements with it as lessor.
Adoption of New Lease Standard, Topic 842
Beginning January 1, 2019, the Company adopted ASU 2016-02, Leases (“Topic 842”). Under this new lease accounting standard, the Company recognized a right-of-use asset and lease liability on the accompanying Balance Sheets for all material leases. Management elected the “modified retrospective approach” with an effective transition date of January 1, 2018 upon adoption and the available practical expedients. The Company also elected to (1) not separate “lease components” from “non-lease components” in the measurement of minimum payments for its leases and (2) not recognize a lease asset and liability for a term of 12 months or less. Lease expense is recognized on a straight-line basis over the expected term of the lease. In March 2019, the Company capitalized a right-of-use asset of $0.2 million along with an accompanying $0.2 million lease liability.
As part of applying Topic 842, the Company determines if an arrangement is or contains a lease and the classification of that lease at contract execution. The classification of leases as “operating” or “finance” leases along with the initial measurement and recognition of the associated right-of-use assets and lease liabilities is performed at the lease commencement date. The measurement of lease liabilities is based on the present value of future lease payments over the lease term. Since an implicit rate for the operating lease for the Company’s Irvine facility was not determinable and was not implicit in the lease, the Company calculated an estimated incremental borrowing rate based upon what it would have to pay to borrow on a collateralized bases over a similar term and amount equal to the lease payments in a similar economic environment. The right-of-use lease asset is based on the corresponding lease liability and adjusted for (i) applicable payments made at or before the commencement date, (ii) initial direct costs incurred, and (iii) tenant incentives provided by the lessor.
Components of Lease Expense
The liability associated with each lease is amortized over the respective lease term using the effective interest rate method. The Company’s right-of-use assets are amortized over the lease term on a straight-line basis to lease expense, as reported on an allocated basis within “research and development” and “general and administrative” expenses in the accompanying Statements of Operations and Comprehensive Loss. For the years ended December 31, 2020 and 2019 the Company recognized $0.2 million and $0.1 million for lease expense, respectively. The Company had $25 thousand of variable lease payments, including non-lease components such as common area maintenance fees recognized as lease expense for the year ended December 31, 2020. There were 0 variable lease payments for the year ended December 31, 2019.
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
Weighted-Average Remaining Lease Term and Applied Discount Rate
The Company had 3 active leases for its Irvine office and laboratory facility, with a remaining lease term of two3.1 years and four months as of December 31, 2019 and a remaining lease term of 1 year and 4 months as of December 31, 2020. The Company had 2 additional facility leases commence on June 1, 2020, with remaining lease terms of 3 years, 1 month as of December 31, 2020. The weighted-average estimated incremental borrowing rate of 10% was utilized to present value future minimum lease payments since an implicit interest rate was not readily determinable for each lease. The weighted average remaining lease term for the Company’s leases as of December 31, 2020 is 2 years, 10 months.
Future Contractual Lease Payments as of December 31, 2020
The below table summarizes the (i) minimum lease payments over the next five years and thereafter, (ii) lease arrangement imputed interest, and (iii) present value of future lease payments:
| | Operating Leases - future payments | December 31, 2020 |
| 2021 | $ | 349 | |
| 2022 | 298 | |
| 2023 | 281 | |
| | December 31, 2023 | | | | December 31, 2023 |
| 2024 | 2024 | 25 | |
| 2025 | 2025 | 0 | |
| 2026 | |
| 2027 | |
| 2028 | |
| Total future lease payments, undiscounted | Total future lease payments, undiscounted | $ | 953 | |
| (Less): Imputed interest | (Less): Imputed interest | (122) | |
| (Less): Tenant improvement allowance | |
| Present value of operating lease payments | Present value of operating lease payments | $ | 831 | |
| Operating lease liability, current | |
| Operating lease liability, noncurrent | |
| Total operating lease liability | |
(b) In-License Agreements for Lotilaner
Elanco In-License Agreement for Skin and Eye Disease or Conditions in Humans
In January 2019, the Company entered intoexecuted a license agreement with Elanco Tiergesundheit AG (“Elanco”), granting it a for exclusive worldwide licenserights to certain intellectual property for the development and commercialization of lotilaner forin the treatment or cure of any eye or skin disease or condition in humans, as amended in June 2022 (the "January 2019"Eye and Derm Elanco Agreement"). The Company has sole financial responsibility for related development, regulatory, and commercialization activities.
In March 2023, a clinical milestone was triggered to Elanco under the Eye and Derm Agreement upon enrollment of the first patient in the Phase 2a Galatea trial, evaluating the potential treatment of rosacea. The Company paid arelated milestone payment of $1.0 million upfront payment at the execution of the January 2019 Agreement, which is reported within “researchwas included in research and development”development expense withinin the accompanying Statements of Operations and Comprehensive Loss for the year ended December 31, 2019. 2023.
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
The Company also paid a requiredhas made cash payments to Elanco under the Eye and Derm Agreement comprised of $1.0 million clinicalupfront upon contract execution in January 2019 and a total of $4.0 million for three specified clinical milestone paymentachievements in the September 2020, as part of an achieved Phase 2b/3 clinical trial milestone forApril 2021, and March 2023, which were all recorded in research and development expense in the treatment of Demodex blepharitis; this amount is reported within “research and development” expense within the accompanying Statements of Operations and Comprehensive Loss for year endedLoss. During 2023, a milestone of $4.0 million was achieved and paid to Elanco upon the first commercial sale of XDEMVY in the U.S., which was recorded as an intangible asset in the accompanying Balance Sheet as of December 31, 2020.
2023. The Company willis amortizing the intangible asset to cost of sales over its useful life of 10 years from the date of the first commercial sale.
As of December 31, 2023, the Company is obligated to make further cash payments to Elanco pursuant toof $2.0 million under the January 2019Eye and Derm Elanco Agreement upon achievement of the last clinical milestone in the treatment of human skin diseases using lotilaner and a maximum of $75.0 million for various clinical milestones for an aggregate maximum of $5.0 million and various commercialcommercial and sales threshold milestones for an aggregate maximumthe treatment of $79.0 million. human skin diseases and the treatment of blepharitis in humans using lotilaner.
In addition, the Company will beis obligated to pay tiered contractual royalties to Elanco in the mid to high single digits of its net sales. If the Company receives certain types of payments from anyits sublicensees, it will be obligated to pay Elanco a variable percentage in the low to mid double-digits of such proceeds, except for territories in whichuntil achievement of the Company achievedfirst applicable regulatory approval priorof a product covered under the license. As a result of the commercialization of XDEMVY, the Company began accruing royalties payable to sublicense execution.Elanco during 2023, which are recorded to cost of sales in the accompanying Statement of Operations and Comprehensive Loss for the year ended December 31, 2023 and accounts payable and other accrued liabilities in the accompanying Balance Sheet as of December 31, 2023. Royalty expense during the year ended December 31, 2023 was $0.7 million.
Elanco In-License Agreement for All Other DiseaseDiseases or Conditions in Humans
In September 2020, the Company executed an expandeda license agreement with Elanco granting it a worldwide license to certain intellectual property for the development and commercialization of lotilaner for the treatment, palliation, prevention, or cure of all other diseases and conditions in humans –(i.e., beyond that of the eye or skinskin), as amended in June 2022 (the “September"All Human Uses Elanco Agreement"). In September 2020, Agreement”).
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
Thethe Company issued ElaElanconco 222,460 shares of its common stock as consideration for the license at the execution of the September 2020 Agreement. Thewith an estimated fair value of these shares was $3.1 million ($14.0003 per share, approximating the issuance price of the Company's Series C preferred stock issuance price – see Note 4) and is reported within “research and development” expense within the accompanying Statements of Operations and Comprehensive Loss for thein September 2020) year ended December 31, 2020. In addition, in March 2021, the Company agreed to issue 187,500 shares of its common stock to Elanco during the second quarter of 2021 to maintain the September 2020 Agreement.
The Company recordedmade cash payments under the transaction as an asset acquisition as substantially allAll Human Uses Elanco Agreement of $0.5 million related to a clinical milestone that was triggered in December 2022 upon enrollment of the fair value of the gross assets acquired were concentrated in a group of similar identifiable assets thus satisfying the requirements of the screen test in ASU 2017-01. The assets acquiredfirst patient in the transaction were measured based onPhase 2a Carpo trial, for the upfront payment to Elanco and the fair valuepotential treatment of the common stock shares issued to Elanco, as the fair value of the consideration given was more readily determinable than the fair value of the assets received. Because the assets have not yet received regulatory approval and have no alternative future use, the fair value attributable to these assets were recorded as research and development expenses.
Lyme disease. The Company willis required to make further cash payments to Elanco under this agreement upon the September 2020 Agreement upon achievement of various clinical milestones forup to an aggregate maximum of $4.5$4.0 million and various commercial and sales threshold milestones for an aggregate maximum of $77.0 million. In addition, the Company will be obligated to pay contractual royalties to Elanco in the single digits of its net sales.product sales, net. If the Company receives certain types of payments from anyits sublicensees, it will also be obligated to pay Elanco a variable percentage in the low to mid double-digits of such proceeds, except for territories in whichuntil achievement of the Company achievedfirst applicable regulatory approval prior to sublicense execution.of a product covered under the license.
(c) Clinical Research Organization and Contract Manufacturer Agreements
We enter into contracts in the normal course of business with clinical research organizations and clinical sites and with contract manufacturers for pre-clinical and clinical drug supply, as well as with various other vendors in operating our business. These contracts generally provide for termination provisions with requisite notice.
(d) Employment Agreements
The Company has entered into employment agreements with 4seven of its named executive officers. These agreements provide for the payment of certain benefits upon separation of employment under specified circumstances, such as termination without cause, or termination in connection with a change in control event or other significant transaction.event.
(e) OtherLitigation Contingencies
From time to time, the Company may be subject to various litigation and related matters arising in the ordinary course of business. The Company is currently not aware of any such matters where there is at least a reasonable possibilityprobability that a material loss, if any, has been or will be incurred for financial statement recognition.
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
Indemnities and Guarantees
The Company has certain indemnity commitments, under which it may be required to make payments to its officers and directors in relation to certain transactions to the maximum extent permitted under applicable laws. The duration of these indemnities varies, and in certain cases, isare indefinite and doesdo not provide for any limitation of maximum payments. The Company has not been obligated to make any such payments to date and no liabilities have been recorded for this contingency in the accompanying balance sheets.Balance Sheets.
9. OUT-LICENSE AGREEMENT
Out-License of TP-03 Commercial Rights in the China Territory in March 2021
In March 2021, the Company entered into the China Out-License agreement with LianBio for its exclusive development and commercialization rights of TP-03 (lotilaner ophthalmic solution, 0.25%) in the China Territory, as defined in the agreement, for the treatment of Demodex blepharitis and Meibomian Gland Disease. LianBio is contractually responsible for all clinical development and commercialization activities and costs within the China Territory.
The Company assessed this arrangement and identified the following material promises under the arrangement: (i) the exclusive license to research, develop, manufacture, commercialize, make, offer for sale, sell, and import TP-03 in the China Territory; and (ii) the research and development services in the form of clinical study materials for the respective Phase 2b/3 trial (Saturn-1) and Phase 3 (Saturn-2) TP-03 trials. The promises to provide research and development services for Saturn-1 and Saturn-2 clinical trials were evaluated and determined to be distinct promises in the contract and each of the two clinical trials are separate performance obligations apart from the promise to provide the license.
The assessment of the initial transaction price for the China Out-License agreement included an analysis of amounts the Company expected to receive, which at contract inception consisted of: (i) the upfront cash payment of $15.0 million; (ii) a second cash payment of $10.0 million; (iii) a $10.0 million milestone that was determined to be within the control of the Company; and (iv) $1.2 million representing the initial fair value of the equity warrant.
The Company accounted for each performance obligation as follows:
Out-License
The Company determined that this license was distinct based on an evaluation of the delivery of the functional license that was in the later stages of development, and it met the criteria for being distinct from the research and development services required under the China Out-License agreement. The Company determined the standalone selling price of this license using a discounted projected sales model and recognized as license fees and collaboration revenue the total allocated transaction price at contract inception, upon delivery of the license.
Research and Development Services
The standalone selling price of these performance obligations was determined using the adjusted market assessment approach. The Company analyzed costs expected to be incurred for each of the clinical trials through completion to estimate the price that a customer would be willing to pay for these services in order to benefit from the clinical trials. The Company determined that LianBio simultaneously benefited from the research and development services that are satisfied over time, as they were able to request and access the clinical trial data at any point through the trial completion. Therefore, the Company recognized the amounts allocated to the respective research and development performance obligations for Saturn-1 and Saturn-2 within license fees and collaboration revenue as the research and development services were provided using an input method, based on the costs incurred for each clinical trial and the total costs expected to be incurred to satisfy each performance obligation. The Company believes this method most faithfully depicted its performance in transferring the promised services during the expected period of time that each clinical trial was ongoing. The Company monitored the expected completion dates for each clinical trial and updated its estimated time to completion at each reporting period, as necessary.
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
In February 2023, a specified milestone event was triggered based upon the signing of an agreement for which the Company has no ongoing obligations, resulting in $2.5 million recognized as license fees and collaboration revenue in the accompanying Statements of Operations for the year ended December 31, 2023. Through December 31, 2023, the Company received aggregate payments from LianBio totaling $82.5 million, comprised of initial consideration of $15.0 million and $67.5 million for the achievement of specified milestones.
As of December 31, 2023 the Company is eligible to receive further consideration from LianBio upon the achievement of additional TP-03 events, including: (i) additional regulatory milestones and one-time payments of up to an aggregate of $22.5 million; (ii) China-Based TP-03 sales threshold milestone payments of up to an aggregate of $100.0 million; (iii) tiered low-to-high-teen royalties for China Territory TP-03 product sales; and (iv) vesting of a LianBio equity warrant upon certain regulatory milestones. On February 13, 2024, LianBio announced its plan to wind down its operations. As of the date of this filing, it is uncertain if and when the Company will receive any royalties or future milestone consideration under the China Out-License, including but not limited to the milestone achievement of an additional drug supply agreement execution.
As part of the China Out-License with LianBio the Company granted Elanco an additional 187,500 shares of the Company's common stock that otherwise would have been issuable no later than the 18-month anniversary of the All Human Uses Elanco Agreement for its continued license exclusivity. These issued shares were valued at $5.5 million, based on the Company's closing stock price of $29.30 per share on the date this issuance became contractually required.
The Company made a contractual payment in the amount of $2.5 million to Elanco following the receipt of $25 million of proceeds from LianBio during the second quarter of 2021. During the fourth quarter of 2022, the Company recognized $0.4 million of cost of license fees and collaboration revenue upon receipt of $10 million of cash proceeds from LianBio for the achievement of a clinical development milestone.
There were no expenses recognized under the China-Out License for the year ended December 31, 2023. During the year ended December 31, 2022, the Company recognized $1.0 million of expense under the China-Out License within cost of license fees and collaboration revenue in the accompanying Statement of Operations and Comprehensive Loss.
10. CREDIT FACILITY AGREEMENT
On February 2, 2022, the Company executed the Credit Facility with Hercules Capital, Inc. ("Hercules") and SVB that expires on February 2, 2027. As security for the obligations under the agreement, the Company granted a continuing security interest in substantially all of the Company's assets, excluding intellectual property on which there is a negative pledge. Concurrent with the execution of the Credit Facility, the Company made a $20.0 million draw.
On January 5, 2023, the Company entered into an amendment to the loan and security agreement (the "First Amendment"). The First Amendment set a maximum interest rate and updated the terms of prepayment under the Credit Facility and other certain specific conditions including an extended period for the Company to drawdown the $25.0 million tranche associated with the New Drug Application ("NDA") submission from March 15, 2023 to March 15, 2024, provided at least $5.0 million was drawn on or before March 15, 2023 and at least an additional $5.0 million was drawn on or before September 15, 2023. The Company did not incur any lender fees as part of this First Amendment.
On August 23, 2023, the Company entered into a second amendment to the loan and security agreement (the "Second Amendment"). The Second Amendment updated the terms of the amount and proportion of the Company’s cash required to be maintained at SVB or affiliates of SVB. The Company did not incur any lender fees as part of this Second Amendment.
On March 15, 2023 and September 15, 2023, respectively, the Company made separate draws of $5.0 million (including SVB's commitment of $1.25 million) from the $25.0 million tranche that became available upon submission of the NDA. As of December 31, 2023, the Credit Facility provides for a remaining aggregate principal amount of up to $125.0 million with tranched availability as follows: $15.0 million currently available related to the Company's NDA submission with the FDA for TP-03 in September 2022; $35.0 million currently available due to the FDA approval of XDEMVY in July 2023; $50.0 million available upon achievement of product sales, net thresholds; and $25.0 million available upon lender approval.
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
Each of these tranches may be drawn down in $5.0 million increments at the Company's election. The Credit Facility requires interest-only payments through February 1, 2026, followed by 12 months of principal amortization, unless extended for one year to its maturity, upon meeting certain contractual conditions. All unpaid amounts under the Credit Facility become due on its February 2, 2027 expiry.
Under the First Amendment, the outstanding principal draws accrue interest at a floating interest rate per annum equal to the greater of either (i) The Wall Street Journal ("WSJ") prime rate plus 4.45% with an aggregate cap of 11.45%, or (ii) 8.45%. At the execution date of the Credit Facility, the WSJ prime rate was 3.25% and increased to 8.50% as of December 31, 2023.
The Company is required to pay a specified fee upon the earlier of (i) February 2, 2027, or (ii) the date the Company prepays, in full or in part, the outstanding principal balance of the Credit Facility ("End of Term Charge"). The current End of Term Charge of $1.4 million was derived by multiplying 4.75% by the $30.0 million outstanding principal balance as of December 31, 2023 and is accreted to interest expense through maturity.
As of December 31, 2023 and 2022, the effective interest rate for the full term of the Credit Facility was 11.96% and 13.61%, respectively. During the year ended December 31, 2023 and 2022, the Company recognized interest expense on the accompanying Statements of Operations and Comprehensive Loss in connection with the Credit Facility as follows:
| | | | | | | | | | | |
| Year Ended December 31, |
| 2023 | | 2022 |
| Interest expense for term loan | $ | 2,961 | | | $ | 1,890 | |
| Accretion of end of term charge | 264 | | | 174 | |
| Amortization of debt issuance costs | 121 | | | 135 | |
| Total interest expense related to term loan | $ | 3,346 | | | $ | 2,199 | |
The carrying value of the Credit Facility consists of principal outstanding less legal and administrative issuance costs that were recorded as a debt discount to the term loan, net and will continue to be accreted to interest expense using the effective interest method during its term. The principal balance of this Credit Facility and related accretion and amortization are reported on a combined basis as term loan, net on the accompanying Balance Sheets as follows:
| | | | | | | | | | | |
| December 31, |
| 2023 | | 2022 |
| Term loan, gross | $ | 30,000 | | | $ | 20,000 | |
| Debt issuance costs | (875) | | | (875) | |
| Accretion of end of term charge | 438 | | | 174 | |
| Accumulated amortization of debt issuance costs | 256 | | | 135 | |
| Term loan, net | $ | 29,819 | | | $ | 19,434 | |
11. RELATED PARTY TRANSACTIONS
The Company has a preexisting consulting agreement with a board member who was appointed in December 2021. This consulting agreement provides for annual cash compensation of approximately $0.2 million and option grants to purchase 45,134 shares of the Company’s common stock, with exercise prices ranging from $2.01 to $34.72 per share. This Consulting Agreement may be terminated by either party with ten days' notice and contains standard confidentiality, indemnification, and intellectual property assignment provisions in favor of the Company.
During the year ended December 31, 2023 and 2022, the Company recorded $0.3 million and $0.3 million, respectively, of selling, general and administrative expenses in the accompanying Statements of Operations and Comprehensive Loss related to this consulting agreement.
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
Sponsorship Activities
In May 2023, a board member of the Company was appointed president of the American Society of Cataract and Refractive Surgery ("ASCRS"), a society dedicated to meeting the needs of anterior segment ophthalmic surgeons.
During the year ended December 31, 2023, the Company recorded $0.4 million of selling, general and administrative expenses in the accompanying Statement of Operations and Comprehensive Loss for sponsorship and event-related activities associated with ASCRS.
12. INCOME TAXES
The components of loss from operations before benefit from income taxes were as follows:
| | | | | | | | | | | |
| Year Ended
December 31, |
| 2023 | | 2022 |
| United States | $ | (135,893) | | | $ | (62,095) | |
| Total | $ | (135,893) | | | $ | (62,095) | |
The benefit from income taxes from operations were as follows:
| | | | | | | | | | | |
| Year Ended
December 31, |
| 2023 | | 2022 |
| Current: | | | |
| Federal | $ | — | | | $ | (4) | |
| State | — | | | — | |
| $ | — | | | $ | (4) | |
| | | |
| Deferred: | | | |
| Federal | — | | | — | |
| State | — | | | — | |
| — | | | — | |
| Total income tax benefit | $ | — | | | $ | (4) | |
A reconciliation of income taxes was computed by applying the federal statutory income tax rate in each period to the pretax loss for the years ended December 31, 20202023 and 2019,2022, and adjusted for certain classes of transactions, as summarized below:
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
| | | | Year Ended
December 31, | | Year Ended
December 31, |
| | | 2020 | | 2019 | | 2023 | | 2022 |
| Expected tax benefit at statutory rate | Expected tax benefit at statutory rate | $ | (5,630) | | | $ | (981) | |
| State income tax, net of federal benefit | State income tax, net of federal benefit | 1 | | | 1 | |
| Permanent items | Permanent items | 5 | | | 4 | |
| Stock-based compensation | |
| Executive compensation | |
| Research and development credits | Research and development credits | (632) | | | (31) | |
| Loss on extinguishment of convertible notes | 0 | | | 54 | |
| Non-deductible interest | 0 | | | 35 | |
| State rate adjustment | |
| Change in fair value of equity warrants and equity securities | |
| Other | Other | 119 | | | 4 | |
| Change in valuation allowance | Change in valuation allowance | 6,138 | | | 915 | |
| Income tax provision | $ | 1 | | | $ | 1 | |
| Income tax benefit | |
Significant components of the deferred tax assets and liabilities at December 31, 2020 and 2019, are presented below:were as follows:
| | | | Year Ended
December 31, | | Year Ended
December 31, |
| | | 2020 | | 2019 | | 2023 | | 2022 |
| Deferred tax assets: | Deferred tax assets: | | | |
| Net operating loss carry forwards | Net operating loss carry forwards | $ | 5,391 | | | $ | 961 | |
| Net operating loss carry forwards | |
| Net operating loss carry forwards | |
| Research and development credit carryforwards | Research and development credit carryforwards | 710 | | | 88 | |
| Capitalized research and development | |
| Intangible assets | Intangible assets | 1,046 | | | 211 | |
| Stock-based compensation | |
| Accruals | |
| Other, net | Other, net | 476 | | | 104 | |
| Total deferred tax assets before valuation allowance | Total deferred tax assets before valuation allowance | 7,623 | | | 1,364 | |
| Less: valuation allowance | (7,475) | | | (1,338) | |
| (Less): Valuation allowance | |
| Total deferred tax assets | Total deferred tax assets | $ | 148 | | | $ | 26 | |
| Deferred tax liabilities, net: | Deferred tax liabilities, net: | | | |
| Operating lease right-of-use assets | Operating lease right-of-use assets | (148) | | | (26) | |
| Operating lease right-of-use assets | |
| Operating lease right-of-use assets | |
| Fixed assets | |
| | Net deferred tax asset | Net deferred tax asset | $ | 0 | | | $ | 0 | |
| Net deferred tax asset | |
| Net deferred tax asset | |
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amount of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. The Company maintains a valuation allowance against its net deferred tax assets due to the uncertainty that such assets will be realized and evaluates the recoverability of its deferred tax assets on at least an annual basis. The Company has determined that its deferred tax assets, with the exception of amounts supported by the reversal of taxable temporary differences, are not realizable. Consequently, the Company has recorded a valuation allowance on deferred tax assets of $7.5$60.3 million and $1.3$25.2 million at December 31, 20202023 and 2019,2022, respectively.
At December 31, 2020,2023, the Company has federal and Californiastate net operating loss carryforwards of approximately $25.3$107.8 million and $4.0$84.0 million, respectively. As a result of the Tax Cuts and Jobs Act of 2017 (the Tax Act)"Tax Act"), for U.S. income tax purposes, net operating losses generated prior to December 31, 2017 can be carried forward for up to 20 years, while net operating losses generated after December 31, 2017 can be carried forward indefinitely, but are limited to 80% utilization against taxable income. The Company’s total federal net operating loss of $25.3$107.8 million includes $45 thousand that will begin to expire in 2037 and $25.2 million that will not expire but will only be able to be offset 80% of future taxable income within each year. The Californiaother state net operating losses will begin to expire in 2037. At December 31, 2020,2023, the Company had federal and Californiaother state research and development tax credits of $0.6$7.4 million and $0.4 million, respectively. The federal research and development tax credits begin to expire in 2037 unless previously utilized, and the California credit carryforwards are available indefinitely.
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
$2.4 million, respectively. The federal research and development tax credits begin to expire in 2040 unless previously utilized, and the other state credit carryforwards begin to expire in 2037.
The Internal Revenue Code (IRC)("IRC") Sections 382 and 383 limit annual use of NOL and research and development credit carryforwards in the event a cumulative change in ownership of more than 50% occurs within a three-year period. The Company has not yet completed an ownership change analysis. If a requisite ownership change occurs, the amount of remaining tax attribute carryforwards available to offset taxable income and income tax expense in future years may be restricted or eliminated. If eliminated, the related asset would be removed from deferred tax assets with a corresponding reduction in the valuation allowance. Due to the existence of the valuation allowance, limitations created by future ownership changes, if any, will not impact the Company’s effective tax rate.
Uncertain tax positions are evaluated based upon the facts and circumstances that exist at each reporting period. Subsequent changes in judgement based upon new information may lead to changes in recognition, derecognition, and measurement. Adjustment may result, for example, upon resolution of an issue with the taxing authorities or expiration of a statute of limitations barring an assessment for an issue. The Company recognizes a tax benefit from an uncertain tax position when it is more-likely-than-not that it will be sustained upon examination by tax authorities. As of December 31, 2020,2023, the Company had gross unrecognized tax benefits of $0.5$3.9 million, NaNnone of which would affect the effective tax rate if recognized. The Company does not anticipate any significant changes in its unrecognized tax benefits over the next 12 months. The Company's policy is to recognize the interest expense and/or penalties related to income tax matters as a component of income tax expense. The Company had no accrual for interest or penalties on its balance sheetthe accompanying Balance Sheets at December 31, 20202023 and has not recognized interest and/or penalties in its statementon the accompanying Statements of operationsOperations for the yearyears ended December 31, 20202023 or 2019.2022.
The following table summarizes the changes to the gross unrecognized tax benefits for the years ended December 31, 2020 and 2019:benefits:
| | | | Year Ended
December 31, | | Year Ended
December 31, |
| | | 2020 | | 2019 | | 2023 | | 2022 |
| Balance at beginning of year | Balance at beginning of year | 33 | | | 20 | |
| Additions related to current year positions | Additions related to current year positions | 217 | | | 13 | |
| Additions related to prior year positions | Additions related to prior year positions | 242 | | | 0 | |
| Decreases related to prior year positions | Decreases related to prior year positions | (4) | | | 0 | |
| Balance at end of year | Balance at end of year | 488 | | | 33 | |
The Company is subject to taxation in the United StatesU.S. Federal jurisdiction and California.various states. All tax years from inception are subject to examination by federal and state tax authorities. The Company’s practice is to recognize interest and penalties related to income tax matters in income tax expense. NaNNo interest or penalties related to income tax matters have been incurred at December 31, 20202023 and 20192022 and the years then ended. Further, the Company is not currently under examination by any federal, state, or local tax authority.
TARSUS PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
11. SUBSEQUENT EVENTS
Out-license of TP-03 Commercial Rights in Greater China in March 2021
On March 26, 2021, the Company executed an out-license agreement with LianBio Ophthalmology Limited ("LianBio"), granting exclusive commercial rights of TP-03 for the treatment of Demodex blepharitis and Meibomian Gland Disease (MGD) within The People’s Republic of China, Macau, Hong Kong, and Taiwan (the "Territory"). The Company is contractually entitled to receive (i) an aggregate $25 million by June 30, 2021, (ii) regulatory and sales milestone receipts totaling $75 million and $100 million, respectively, (iii) tiered royalties in the low double-digits on net sales of TP-03 within the Territory, and (iv) a minority interest in LianBio that vests upon the achievement of certain clinical and regulatory events.
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
Conclusions Regarding the Effectiveness of Disclosure Controls and Procedures
The Company maintainsWe maintain a system of disclosure controls and procedures that are designed to provide reasonable assurance that information required to be disclosed in the reports that the Company fileswe file or submitssubmit under the Securities Exchange Act of 1934, as amended (the "Exchange Act"), is processed, recorded, summarized and reported within the time periods specified in the Securities and Exchange Commission’s rules and forms. These disclosure controls and procedures include, among other processes, controls and procedures designed to ensure that information required to be disclosed in the reports that the Company fileswe file or submitssubmit under the Exchange Act is accumulated and communicated to management, including our Chief Executive Officer and Chief Financial Officer (our principal executive officer and principal financial officer, respectively), as appropriate, to allow for timely decisions regarding required disclosure.
The CompanyOur management carried out an evaluation, under the supervision and with the participation of management, including our Chief Executive Officer and Chief Financial Officer, of the effectiveness of the design and operation of our disclosure controls and procedures (as defined in Rules 13a-15(e) under the Exchange Act) as of December 31, 2020.2023. Based upon that evaluation, the Company’sour Chief Executive Officer and Chief Financial Officer have concluded that as of December 31, 2023, the Company’s disclosure controls and procedures were effective to provide reasonable assurance that information we are required to disclose in reports that we file or submit under the Exchange Act is recorded, processed, summarized, and reported within the time periods specified in SEC rules and forms, and that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as of the end of the period covered by this Annual Report on Form 10-K.appropriate, to allow timely decisions regarding required disclosure.
Management’s Assessment RegardingReport on Internal Control Over Financial Reporting
This Annual Report on Form 10-K does not include a report of management's assessment regardingOur management is responsible for establishing and maintaining adequate internal control over financial reporting, oras defined in Exchange Act Rules 13a-15(f) and 15d-15(f). We maintain internal control over financial reporting designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles.
Under the supervision and with the participation of our Chief Executive Officer and our Chief Financial Officer, our management conducted an attestation reportevaluation of the Company's independent registered public accounting firm due to a transition period establishedeffectiveness of our internal control over financial reporting based on the criteria set forth in “Internal Control—Integrated Framework” issued by rulesthe Committee of Sponsoring Organizations of the Securities and ExchangeTreadway Commission for newly public companies.(2013 framework). Based on this assessment, our management concluded that our internal control over financial reporting was effective at the reasonable assurance level as of December 31, 2023.
Changes in Internal Control over Financial Reporting
During the year ended December 31, 2023, we implemented certain internal controls in connection with our commercial launch. There have beenwere no other changes in our internal control over financial reporting (as defined by Exchange Act Rule 13a-15(f) and 15d-15(f)) that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting during the quarteryear ended December 31, 2020.2023.
Inherent Limitations on Effectiveness of Controls
Our management, including our Chief Executive Officer and Chief Financial Officer, do not expect that our disclosure controls or our internal control over financial reporting will prevent all errors and all fraud. A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, have been detected. These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur because of a simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people or by management override of the controls. The design of any system of controls is also based in part on certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions, over time, controls may become inadequate because of changes in conditions,
or the degree of compliance with policies or procedures may deteriorate. Because of the inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and not be detected.
Item 9B. Other Information.
None.Securities Trading Plans of Directors and Executive Officers
During quarter ended December 31, 2023, none of our officers or directors, as defined in Rule 16a-1(f), informed us of the adoption or termination of a Rule 10b5-1 trading arrangement or a non-Rule 10b5-1 trading arrangement, each as defined in Regulation S-K Item 408.
Item 9C. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections.
Not applicable.
Part III
Item 10. Directors, Executive Officers and Corporate Governance
The information required by this item will be contained in our definitive proxy statement to be filed with the SEC in connection with the Annual Meeting of Stockholders within 120 days after December 31, 20202023 (the Proxy Statement)"Proxy Statement"), and is incorporated in this Annual Report on Form 10-K by reference.
Item 11. Executive Compensation
The information required by this item will be contained in theour Proxy Statement and is incorporated in this Annual Report on Form 10-K by reference.
Item 12. Security Ownership of Certain Beneficial OwnerOwners and Management and Related Stockholder Matters
The information required by this item will be contained in theour Proxy Statement and is incorporated in this Annual Report on Form 10-K by reference.
Item 13. Certain Relationships and Related Transactions, and Director Independence
The information required by this item will be contained in theour Proxy Statement and is incorporated in this Annual Report on Form 10-K by reference.
Item 14. Principal AccountingAccountant Fees and Services
The information required by this item will be contained in theour Proxy Statement and is incorporated in this Annual Report on Form 10-K by reference.
Part IV
Item 15. Exhibits, Financial Statement Schedules
(a) The following documents are filed as part of this report:
(1) Financial Statements (included in Part II of this Annual Report on Form 10-K):
•Report of Independent Registered Public Accounting Firm
•Balance Sheets
•Statements of Operations and Comprehensive Loss
•Statements of Preferred and Common Stock and Stockholder's Equity (Deficit)
•Statements of Cash Flows
•Notes to Financial Statements
(2) Financial Statement Schedules:
All financial statement schedules are omitted because the information is inapplicable or presented in the notes to the financial statements
(b) The following exhibits are included herein or incorporated herein by reference:
INDEX TO EXHIBITS
| | | | | | | | | | | | | | | | | | | | | | | |
| | | Incorporated by Reference |
| Exhibit Number | | Description | Form | File No. | Exhibit | Filing Date | Filed Herewith |
| 3.1 | | | 8-K | 001-39614 | 3.1 | October 20, 2020 | |
| 3.2 | | | 8-K | 001-39614 | 3.2 | October 20, 2020 | |
| 4.1 | | | S-1/A | 333-249076 | 4.1 | October 9, 2020 | |
| 4.2 | | | | | | | X |
| 4.3 | | | S-1/A | 333-249076 | 4.2 | October 9, 2020 | |
| 10.1 | | | S-1/A | 333-249076 | 10.1 | October 9, 2020 | |
| 10.2# | | | S-1 | 333-249076 | 10.2 | September 25, 2020 | |
| 10.3# | | | S-8 | 333-249571 | 99.2 | October 20, 2020 | |
| 10.4# | | | S-8 | 333-249571 | 99.3 | October 20, 2020 | |
| 10.5# | | | S-1/A | 333-249076 | 10.5 | October 9, 2020 | |
| 10.6# | | | S-1 | 333-249076 | 10.6 | September 25, 2020 | |
| 10.7# | | | S-1 | 333-249076 | 10.7 | September 25, 2020 | |
| 10.8# | | | S-1 | 333-249076 | 10.8 | September 25, 2020 | |
| 10.9# | | | S-1 | 333-249076 | 10.9 | September 25, 2020 | |
| 10.10† | | | S-1/A | 333-249076 | 10.10 | October 9, 2020 | |
| 10.11† | | | S-1/A | 333-249076 | 10.11 | October 9, 2020 | |
| 10.12†^ | | | S-1/A | 333-249076 | 10.12 | October 9, 2020 | |
| 10.13 | | | S-1 | 333-249076 | 10.13 | 44099 | |
| 10.14^* | | | S-1 | 333-249076 | 10.14 | September 25, 2020 | |
| 10.15# | | | S-1/A | 333-249076 | 10.15 | October 9, 2020 | |
| 23.1 | | | | | | | X |
| 24.1 | | | | | | | X |
| 31.1* | | | | | | | X |
| 31.2* | | | | | | | X |
| 32.1* | | | | | | | X |
| | | | | | | | | | | | | | | | | | | | | | | |
| | | Incorporated by Reference |
| Exhibit Number | | Description | Form | File No. | Exhibit | Filing Date | Filed Herewith |
| 3.1 | | | 8-K | 001-39614 | 3.1 | October 20, 2020 | |
| 3.2 | | | 8-K | 001-39614 | 3.2 | October 20, 2020 | |
| 4.1 | | | S-1/A | 333-249076 | 4.1 | October 9, 2020 | |
| 4.2 | | | 10-K | 001-39614 | 4.2 | March 14, 2022 | |
| 4.3 | | | S-1/A | 333-249076 | 4.2 | October 9, 2020 | |
| 10.1# | | | S-1/A | 333-249076 | 10.1 | October 9, 2020 | |
| 10.2# | | | S-1 | 333-249076 | 10.2 | September 25, 2020 | |
| 10.3# | | | S-8 | 333-249571 | 99.2 | October 20, 2020 | |
| 10.4# | | | S-8 | 333-249571 | 99.3 | October 20, 2020 | |
| 10.5# | | | S-1/A | 333-249076 | 10.5 | October 9, 2020 | |
| 10.6# | | | S-1 | 333-249076 | 10.6 | September 25, 2020 | |
| 10.7# | | | S-1 | 333-249076 | 10.7 | September 25, 2020 | |
| 10.8# | | | S-1 | 333-249076 | 10.9 | September 25, 2020 | |
| 10.9# | | | S-1 | 333-249076 | 10.13 | September 25, 2020 | |
| 10.10^* | | | S-1 | 333-249076 | 10.14 | September 25, 2020 | |
| 10.11# | | | S-1/A | 333-249076 | 10.15 | October 9, 2020 | |
| 10.12† | | | 10-Q | 001-39614 | 10.1 | May 11, 2021 | |
| 10.13 | | | 10-Q | 001-39614 | 10.1 | May 11, 2022 | |
| 10.14# | | | 10-Q | 001-39614 | 10.2 | May 11, 2022 | |
| 10.15† | | | 10-Q | 001-39614 | 10.1 | August 11, 2022 | |
| 10.16^ | | | 10-Q | 001-39614 | 10.2 | August 11, 2022 | |
| 10.17 | | | 10-K | 001-39614 | 10.18 | March 17, 2023 | |
| 10.18 | | | 10-Q | 001-39614 | 10.18 | November 9, 2023 | |
| 10.19# | | | | | | | X |
| | | | | | | | | | | | | | | | | | | | | | | |
| 10.20# | | | 10-Q | 001-39614 | 10.1 | August 10, 2023 | |
| 10.21# | | | 10-Q | 001-39614 | 10.2 | August 10, 2023 | |
| 23.1 | | | | | | | X |
| 24.1 | | | | | | | X |
| 31.1 | | | | | | | X |
| 31.2 | | | | | | | X |
| 32.1* | | | | | | | X |
| 32.2* | | | | | | | X |
| 97.1 | | | | | | | X |
| 101.INS | | XBRL Instance Document - The instance document does not appear in the interactive data file because its XBRL tags are embedded within the inline XBRL document. | | | | | X |
| 101.SCH | | XBRL Taxonomy Extension Schema Document. | | | | | X |
| 101.CAL | | XBRL Taxonomy Extension Calculation Linkbase Document. | | | | | X |
| 101.DEF | | XBRL Taxonomy Extension Definition Linkbase Document. | | | | | X |
| 101.LAB | | XBRL Taxonomy Extension Label Linkbase Document. | | | | | X |
| 101.PRE | | XBRL Taxonomy Extension Presentation Linkbase Document. | | | | | X |
| 104 | | Cover Page Interactive Data File (formatted as Inline XBRL and contained in Exhibit 101). | | | | | X |
| | | | | |
| ^ | Pursuant to Item 601(a)(5) of Regulation S-K, certain exhibits and schedules have been omitted. The CompanyRegistrant hereby undertakes to furnish supplementally a copy of any omitted exhibit or schedule upon request by the SEC. |
| † | Pursuant to Item 601(b)(10) of Regulation S-K, certain confidential portions of this exhibit have been omitted by means of marking such portions with asterisks as the identified confidential portions (i) are not material and (ii) would likely cause competitive harm to the registrant if publicly disclosed. |
| # | Indicates a management contract or compensatory plan. |
| * | The certifications attached as Exhibits 32.1 and 32.2 that accompany this Annual Report on Form 10-K are not deemed
filed with the SEC and are not to be incorporated by reference into any filing of the Registrant under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended, whether made before or after the date of this Annual Report on Form 10-K, irrespective of any general incorporation language contained in such filing.
|
(c) Financial Statement Schedules. All schedules have been omitted because the information required to be presented in them is not applicable or is shown in the financial statements or related notes.
Item 16. Form 10-K Summary
None.
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, as amended, the registrant has duly caused this Annual Report on Form 10-K to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of Irvine, State of California, on this 31st day of March, 2021.February 27, 2024.
| | |
| Tarsus Pharmaceuticals, Inc. |
|
| /s/ Bobak Azamian, M.D., Ph.D. |
| Bobak Azamian, M.D., Ph.D. |
President, and Chief Executive Officer and Chairman |
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Bobak Azamian, M.D., Ph.D., Leo Greenstein,Jeffrey Farrow, and Bryan Wahl, M.D., and each of them, as his or her true and lawful attorney-in-fact and agent, with full power of substitution and resubstitution, for him or her and in his or her name, place and stead, in any and all capacities, to sign any and all amendments to this Annual Report on Form 10-K, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he or she might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents, or any of them, or their or his substitutes, may lawfully do or cause to be done by virtue thereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, as amended, this Annual Report on Form 10-K has been signed by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| | | | | | | | |
| Signature | Title | Date |
| | |
| /s/ Bobak Azamian, M.D., Ph.D. | President, Chief Executive Officer President and DirectorChairman | March 31, 2021February 27, 2024 |
| Bobak Azamian, M.D., Ph.D. | (Principal Executive Officer) | |
| | |
/s/ Leo M. GreensteinJeffrey Farrow | Chief Financial Officer and Chief Strategy Officer | March 31, 2021February 27, 2024 |
Leo M. GreensteinJeffrey Farrow | (Principal Financial Officer and Principal Accounting Officer) | |
| | |
/s/ Michael Ackermann, Ph.D.Scott Morrison | ChairmanDirector | March 31, 2021February 27, 2024 |
Michael Ackermann, Ph.D.Scott Morrison | | |
| | |
| /s/ Bhaskar Chaudhuri, Ph.D. | Director | March 31, 2021February 27, 2024 |
| Bhaskar Chaudhuri, Ph.D. | | |
| | |
| /s/ Rosemary Crane | Director | February 27, 2024 |
| Rosemary Crane | | |
| | |
| /s/ Andrew Goldberg, M.D. | Director | March 31, 2021February 27, 2024 |
| Andrew Goldberg, M.D. | | |
| | |
| /s/ William J. Link, Ph.D. | Director | March 31, 2021February 27, 2024 |
| William J. Link, Ph.D. | | |
| | |
/s/ Jason Tester | Director | March 31, 2021 |
Jason Tester | | |
| | |
| /s/ Wendy Yarno | Director | March 31, 2021February 27, 2024 |
| Wendy Yarno | | |
| | |
| /s/ Elizabeth Yeu-Lin, M.D. | Director | February 27, 2024 |
| Elizabeth Yeu-Lin, M.D. | | |