UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
______________
FORM 10-K/A
(Amendment No. 1)10-K
(Mark One)
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
| For the fiscal year ended December 31, 2012 |
| Or |
For the fiscal year ended December 31, 2013
Or
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
| For the transition period from ___________ to ___________ |
For the transition period from ___________ to ___________
Commission File No. 000-23143
______________
PROGENICS PHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
| Delaware | 13-3379479 |
(State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification Number) |
______________
777 Old Saw Mill River Road
Tarrytown, NY 10591
(Address of principal executive offices, including zip code)
Registrant's telephone number, including area code: (914) 789-2800
Securities registered pursuant to Section 12(b) of the Act:
Title of each className of each exchange on which registered
Common Stock, par value $0.0013 per share The NASDAQ Stock Market LLC
| Securities registered pursuant to Section 12(b) of the Act: |
| Title of each class | Name of each exchange on which registered |
| Common Stock, par value $0.0013 per share | The NASDAQ Stock Market LLC |
| Securities registered pursuant to Section 12(g) of the Act: | None |
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports) and (2) has been subject to such filing requirements for the past 90 days.
Yes x No ¨o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes.Yes x No ¨o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K.x¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See definition of "large accelerated filer," "accelerated filer" and "smaller reporting company" in Rule 12b-2 of the Act:
Large Accelerated Fileraccelerated filer ¨ | Accelerated Filerfiler x |
Non-accelerated Filerfiler ¨ (Do not check if a smaller reporting company) | Smaller Reporting Companyreporting company ¨ |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes o No x
The aggregate market value of the voting and non-voting stock held by non-affiliates of the Registrantregistrant on June 30, 2012,2013, based upon the closing price of the Common Stock on The NASDAQ Stock Market LLC on that date of $9.78$4.46 per share, was $184,846,567$171,517,805 (1).
(1) | (1) | Calculated by excluding all shares that may be deemed to be beneficially owned by executive officers, directors and five percent stockholders of the Registrant, without conceding that any such person is an "affiliate" of the Registrant for purposes of the Federal securities laws. |
As of March 8, 2013, 51,131,3497, 2014, a total of 69,575,404 shares of Common Stock, par value $.0013 per share, were outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Specified portions of the Registrant's definitive proxy statement to be filed in connection with solicitation of proxies for its 2014 Annual Meeting of Shareholders are hereby incorporated by reference into Part III of this Form 10-K where such portions are referenced.
| | |
| Explanatory Note | 1 |
| PART I | | |
| Item 1. | | 2 |
| Item 1A. | | 12 |
| Item 1B. | | 25 |
| Item 2. | | 25 |
| Item 3. | | 25 |
| Item 4. | | 25 |
| PART II | | |
| Item 5. | | 25 |
| Item 6. | | 27 |
| Item 7. | | 28 |
| Item 7A. | | 39 |
| Item 8. | | 39 |
| Item 9. | | 39 |
| Item 9A. | | 39 |
| Item 9B. | | 41 |
| PART III | | |
| Item 10. | | 242 |
| Item 11. | | 642 |
| Item 12. | | 1842 |
| Item 13. | | 2142 |
| Item 14. | | 2242 |
| PART IV | | |
| Item 15. | | 43 |
| F-1 |
| S-1 |
| E-1 |
PART I
This document and other public statements we make may contain statements that do not relate strictly to historical fact, any of which may be forward-looking statements within the meaning of the U.S. Private Securities Litigation Reform Act of 1995. When we use the words "anticipates," "plans," "expects" and similar expressions, we are identifying forward-looking statements. Forward-looking statements involve known and unknown risks and uncertainties which may cause our actual results, performance or achievements to be materially different from those expressed or implied by forward-looking statements. While it is impossible to identify or predict all such matters, these differences may result from, among other things, the inherent uncertainty of the timing and success of, and expense associated with, research, development, regulatory approval and commercialization of our products and product candidates, including the risks that clinical trials will not commence or proceed as planned; products appearing promising in early trials will not demonstrate efficacy or safety in larger-scale trials; clinical trial data on our products and product candidates will be unfavorable; our products will not receive marketing approval from regulators or, if approved, do not gain sufficient market acceptance to justify development and commercialization costs; competing products currently on the market or in development might reduce the commercial potential of our products; we, our collaborators or others might identify side effects after the product is on the market; or efficacy or safety concerns regarding marketed products, whether or not originating from subsequent testing or other activities by us, governmental regulators, other entities or organizations or otherwise, and whether or not scientifically justified, may lead to product recalls, withdrawals of marketing approval, reformulation of the product, additional pre-clinical testing or clinical trials, changes in labeling of the product, the need for additional marketing applications, declining sales or other adverse events.
We are also subject to risks and uncertainties associated with the actions of our corporate, academic and other collaborators and government regulatory agencies, including risks from market forces and trends; potential product liability; intellectual property, litigation and other dispute resolution, environmental and other risks; the risk that we may not be able to enter into favorable collaboration or other relationships or that existing or future relationships may not proceed as planned; the risk that current and pending patent protection for our products may be invalid, unenforceable or challenged, or fail to provide adequate market exclusivity, or that our rights to in-licensed intellectual property may be terminated for our failure to satisfy performance milestones; the risk of difficulties in, and regulatory compliance relating to, manufacturing products; and the uncertainty of our future profitability.
Risks and uncertainties also include general economic conditions, including interest and currency exchange-rate fluctuations and the availability of capital; changes in generally accepted accounting principles; the impact of legislation and regulatory compliance; the highly regulated nature of our business, including government cost-containment initiatives and restrictions on third-party payments for our products; trade buying patterns; the competitive climate of our industry; and other factors set forth in this document and other reports filed with the U.S. Securities and Exchange Commission (SEC). In particular, we cannot assure you that Relistor® will be commercially successful or be approved in the future in other formulations, indications or jurisdictions, or that any of our other programs will result in a commercial product.
We do not have a policy of updating or revising forward-looking statements and we assume no obligation to update any statements as a result of new information or future events or developments. It should not be assumed that our silence over time means that actual events are bearing out as expressed or implied in forward-looking statements.
Available Information
We file annual, quarterly and current reports, proxy statements and other documents with the SEC under the Securities Exchange Act of 1934. The SEC maintains an Internet website that contains reports, proxy and information statements and other information regarding issuers, including Progenics, that file electronically with the SEC. You may obtain documents that we file with the SEC at http://www.sec.gov, and read and copy them at the SEC's Public Reference Room at 100 F Street NE, Washington, DC 20549. You may obtain information on operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. We also make available our annual, quarterly and current reports and proxy materials on http://www.progenics.com.
Additional information concerning Progenics and its business may be available in press releases or other public announcements and quarterly and current reports and documents filed with the SEC. Information on or accessed through our website is not included in the Company's SEC filings.
In this document, Relistor, a registered trademark, refers to methylnaltrexone – the active ingredient of Relistor -- as it has been and is being developed and commercialized by or in collaboration with Progenics. Subcutaneous Relistor has received regulatory marketing approval for specific indications, and references to Relistor do not imply that any other form or possible use of the drug has received approval. Relistor's approved U.S. label and full U.S. prescribing information is available at www.Relistor.com. Other approved labels for Relistor apply in ex-U.S. markets. Azedra and Onalta are trademarks of our Molecular Insight Pharmaceuticals, Inc. subsidiary.
Progenics Pharmaceuticals develops innovative medicines for oncology. Our clinical development efforts center on late-stage oncology assets. An important focus for Progenics is targeted therapeutics and imaging agents. A number of our candidates target prostate specific membrane antigen (PSMA).
| | | | |
| OUR PRODUCT CANDIDATES | | | | |
| | |
PART IV | | |
Item 15. | Exhibits, Financial Statement Schedules | 23 |
| |
SIGNATURES | S-1 |
EXHIBIT INDEX | E-1 |
EXPLANATORY NOTE
Progenics Pharmaceuticals, Inc. is filing this Amendment No. 1 on Form 10-K/A to its Annual Report on Form 10-K for the year ended December 31, 2012, as originally filed with the U.S. Securities and Exchange Commission March 15, 2013, to amend Part III -- Items 10-14 to include therein the information required by such Items that, in the March 15 filing, was to be incorporated by reference to the Company's definitive proxy statement for its 2013 Annual Meeting of Stockholders to be held June 12, 2013. The Company now expects that the definitive proxy statement will be filed shortly after April 30, 2013, the end of the period referenced in Form 10-K General Instruction G(3) during which such incorporation by reference may occur.
Information contained in this Amendment is identical to information covering the same subject matter included in the Company's preliminary proxy statement for the 2013 Annual Meeting filed with the SEC on April 22, 2013; its presentation here does not change the information contained in that filing.
Amendment No.1 also amends (i) the cover page of the Form 10-K to delete the information under the heading "Documents Incorporated by Reference" and check the box confirming the absence of Regulation S-K Item 405 disclosure, and (ii) Part IV Item 15, which includes the Exhibit Index, to add new officer certifications in accordance with Rule 12b-15 and Rule 13a-14(a) under the U.S. Securities Exchange Act of 1934, as amended.
Progenics is not modifying, and this Amendment does not change, other disclosure presented in its 2012 Form 10-K, including the financial statements contained therein. This Amendment should therefore be read in conjunction with the March 15 filing and our other SEC reports and filings since that date.
This Amendment, together with the Registrant's 2012 Annual Report on Form 10-K filed March 15, 2013, constitutes its Annual Report on Form 10-K for the year ended December 31, 2012.
PART III
Item 10. Directors, Executive Officers and Corporate Governance.
Directors of the Company
Our directors are listed below together with information concerning (i) their principal occupations or employment, including other public-company directorships, during the past five years, and (ii) the particular experience, qualifications, attributes and/or skills of each nominee that led the Board and its Nominating and Corporate Governance Committee to determine that he or she should serve as a director.
| Director Since | | Position with the Company |
Peter J. Crowley (3)(4)(5)
| 2009 | | Chairman |
Paul J. Maddon | 1986 | | Vice Chairman |
Charles A. Baker(1)(2)(3)*
| 1994 | | Director |
Mark R. Baker (4)(5)
| 2009 | | Chief Executive Officer and Director |
Kurt W. Briner*
| 1998 | | Director |
Stephen P. Goff (2)(4)
| 1993In development for | | DirectorStatus |
David A. Scheinberg (1)(4)(5)
| 1996 | | Director |
Nicole S. Williams (1)
PSMA ADC | 2007 | Treatment of prostate cancer | Director | Phase 2 testing in chemotherapy-experienced patients completed; a second cohort in chemotherapy-naïve patients ongoing |
____________
Member of:
(1)Audit Committee.
(2)Nominating and Corporate Governance Committee.
(3)Compensation Committee.
(4)Science and Technology Committee.
(5)Strategy and External Technology Subcommittee.
*As noted in the Company's preliminary proxy statement for its 2013 Annual Meeting of Stockholders, Messrs. Charles Baker and Briner are not standing for re-election at the upcoming 2013 Annual Meeting of Stockholders.
Mr. Crowley, 54, is Operating Partner of MTS Health Partners, L.P., a New York based healthcare merchant bank, and an Operating Partner at JH Partners, a private equity firm. Mr. Crowley retired in 2008 as Managing Director of the Healthcare Investment Banking group at Oppenheimer & Company (formerly CIBC World Markets), which he headed since 1995, with responsibility for public and private financing and advisory services for biotechnology, pharmaceutical, medical device and healthcare services companies. Mr. Crowley serves on the board of directors of the New York Eye and Ear Infirmary, Continuum Hospitals, the Foundation Fighting Blindness and Rye Country Day School. He is also a board member at La Perla, an Italian lingerie company, Ouidad, a hair salon company, and Napastyle, a specialty food and furnishings company. Mr. Crowley holds an M.B.A. in finance from Columbia University Graduate School of Business and a B.A. in economics from Harvard University.
Mr. Crowley brings to the Board deep perspective into U.S. and international capital markets and strategic business trends. As a senior investment banker specializing in the healthcare industry, Mr. Crowley developed financial and analytic capabilities which are key inputs in the development of the Company's strategic direction, the setting of goals for its financial and operational plans, and the oversight of its financial reporting and audit functions. He has extensive knowledge of, and contacts with major participants in, the global biotechnology and pharmaceutical industries, as well as a wealth of experience evaluating the performance of businesses and products in the Company's industry and designing appropriate strategic and financial alternatives for them.
Dr. Maddon, 53, Progenics' Founder and Vice Chairman, has served in various capacities since the Company's inception, including Chairman of the Board, Chief Executive Officer, President and Chief Science Officer. Now a private investor and entrepreneur, he serves as a trustee of Columbia University and as a member of the Board of Advisors of its Medical Center. He also serves as a trustee of the Society for Science & the Public and NYSCI (New York Hall of Science), as well as a director of the Children's Health Fund and the Fund for Public Health in New York. Dr. Maddon serves as a director of Azevan Pharmaceuticals, Inc., a privately-held biotechnology company developing therapeutics to treat disorders of stress, mood, and behavior, as well as an advisor to Nutrition 21, LLC, a privately-held developer and marketer of nutritional products.
Prior to founding Progenics, from 1981 to 1988 Dr. Maddon performed research at the Howard Hughes Medical Institute at Columbia in the laboratory of Dr. Richard Axel. Dr. Maddon holds a B.A. in biochemistry, an M.D., and a Ph.D. in biochemistry and molecular biophysics, all from Columbia.
As a result of Dr. Maddon's long tenure as CEO and CSO of Progenics, he has extensive knowledge of the Company and the biopharmaceutical industry and provides valuable business, leadership and management insights. Dr. Maddon's background reflects significant achievements in and knowledge of business, science and medicine, with a deep understanding of product development and the role of science in business. Through his service as a board member of other organizations, he also brings important insight on emerging technologies, products and markets, and how various areas of research and development, and the Company as a whole, compare against competing alternatives.
Mr. Charles Baker, 80, is a business advisor to biotechnology companies. He is the former Chairman, President and Chief Executive Officer of The Liposome Company, Inc., a position he held from 1989 until the sale of the company in 2000. Mr. Baker has over 45 years of pharmaceutical industry experience and has held senior management positions at Pfizer, Abbott Laboratories and Squibb Corporation. He is a director of Regeneron Pharmaceuticals, Inc., a biotechnology company. Mr. Baker received a B.A. from Swarthmore College and a J.D. from the Columbia University School of Law.
Mr. Baker's decade of service at Liposome has given him special insight into the needs and value drivers of enterprises similar to Progenics in scale and scope. He has extensive experience in product commercialization both in the biotechnology setting and, earlier in his career, at Pfizer, Abbott Laboratories and Squibb Corporation, as well as interacting with fellow board members and understanding the expectations of shareholders. His service as a board member of Regeneron, a company with a business model similar to ours, also contributes to his extensive knowledge and understanding of the biotechnology industry.
Messrs. Charles Baker and Mark Baker are not related.
Mr. Mark Baker, 58, Chief Executive Officer, joined the Company in 2005 as Senior Vice President & General Counsel and Secretary. In 2008, he was appointed Executive Vice President – Corporate, in 2009 became President, and has been CEO since March 2011. From 2003 to 2005, Mr. Baker was Chief Business Officer, Secretary and a director of New York Trans Harbor LLC, a privately-held ferry operation in New York City. From 1997 to 2001, he was Executive Vice President, Chief Legal Officer and Secretary of Continental Grain Company, a privately-held international agri-business and financial concern. Prior thereto, he was a partner and Co-Chairman of the Capital Markets Group of the New York law firm, Dewey Ballantine. Mr. Baker has an A.B. degree from Columbia College and a J.D. from the Columbia University School of Law.
Mr. Baker's qualifications for serving as a director of the Company include his 30 years of business and legal experience, a significant portion of which has been in the life sciences industry. Mr. Baker has been involved in the senior management of the organizations with which he worked before joining the Company, and has extensive experience managing public and private companies, including specific experience with respect to the financial, accounting, audit, human resources, intellectual property, legal, environmental, insurance, scientific and operational aspects of businesses in diverse industries. He has also served as a legal and business advisor to numerous boards of directors of public and private entities.
Mr. Briner, 68, is the former President and Chief Executive Officer of Sanofi Pharma S.A, a position he held from 1988 until his retirement in 2000. He has over 35 years of experience in the pharmaceutical industry, and is a director of Galenica S.A., a European-based pharmaceutical company. He was a director of Novo Nordisk Danmark prior to 2010. Mr. Briner attended Humanistisches Gymnasium in Basel and École de Commerce in Basel and Lausanne.
Mr. Briner's extensive experience in the pharmaceutical industry is a principal qualification for his nomination as a director. Through his service in senior management of large pharmaceutical enterprises and oversight of commercialization programs for pharmaceutical products, Mr. Briner has acquired broad perspective on historical and current trends and developments in the pharmaceutical industry and an appreciation of business organizations and practices in diverse international cultures. From his service as a board member of Novo Nordisk Danmark and Galenica, Mr. Briner also maintains personal and business relationships with key individuals throughout the global pharmaceutical industry.
Dr. Goff, 61, the Higgins Professor in the Departments of Biochemistry and Molecular Biophysics, and Microbiology and Immunology at Columbia University, became a scientific advisor to us in 1988. He received an A.B. in biophysics from Amherst College and a Ph.D. in biochemistry from Stanford University. Dr. Goff performed post-doctoral research at the Massachusetts Institute of Technology in the laboratory of Dr. David Baltimore.
Dr. Goff's reputation and extensive relationships in the medical research community have resulted in his being involved in evaluating cutting-edge research proposals in diverse areas of inquiry, and he brings that experience and knowledge to advising the Company on its research and development initiatives. His long academic career concentrating in infectious disease research has been especially valuable to the Company's efforts in virology and related fields.
Dr. Scheinberg, 57, became a scientific advisor to Progenics in 1994. He has been associated with the Sloan-Kettering Institute for Cancer Research since 1986, where he is the Vincent Astor Chair and Member, Leukemia Service; Chairman, Molecular Pharmacology and Chemistry Program; Chairman, Experimental Therapeutics Center; and Head, Laboratory of Hematopoietic Cancer Immunochemistry. He also holds the positions of Professor of Medicine and of Pharmacology at the Medical College of Cornell University. He received a B.A. from Cornell and an M.D. and a Ph.D. in pharmacology and experimental therapeutics from The Johns Hopkins University School of Medicine. Dr. Scheinberg is a director of ContraFect Corporation, a privately-held biotechnology company.
Dr. Scheinberg's expertise as a leading academic oncologist at Sloan-Kettering and Cornell is exceptionally valuable to the Board and its scientific committees, which he chairs. He evaluates potential research directions and the design and monitoring of resulting programs. His broad knowledge of and contacts in the highest levels of medical research are important to the Company's efforts to advance its research and development initiatives.
Ms. Williams, 68, was elected to our Board in 2007 after retiring in 2006 as Chief Financial Officer of Abraxis Bioscience Inc., a biopharmaceutical company, and President of its Abraxis Pharmaceutical Products division, positions she assumed upon the 2006 merger of American Pharmaceutical Partners, Inc. and American Bioscience Inc. From 2002 to 2006, Ms. Williams was the Executive Vice President and Chief Financial Officer of American Pharmaceutical Partners, as well as President from 2005. Previously, she was Executive Vice President and Chief Financial Officer of R.P. Scherer, Inc., a global drug delivery company. Ms. Williams is President of the Nicklin Capital Group, Inc., a firm she founded in 1999 to invest in and provide consulting to early-stage technology companies in the Midwest. She is a director, Audit Committee chair and Compensation Committee member at Intercept Pharmaceuticals, Inc. (ICPT), a biotechnology company, and previously held the same positions at Orchid Cellmark, Inc., a leading DNA identity testing service company, until its 2011 purchase by LabCorp. Ms. Williams received her Demi-License en Science Politique from the University of Geneva, Switzerland, her License en Science Politique from the Graduate Institute of International Affairs, University of Geneva, Switzerland and her M.B.A. from the Graduate School of Business, University of Chicago. She is a National Association of Corporate Directors (NACD) Board Leadership Fellow.
Ms. Williams' experience gives her special insight into the financial and operational issues that a company in the pharmaceutical industry faces. She brings expertise to the Company in the areas of financial analysis and reporting, internal auditing and controls, and risk management oversight. Her board and audit committee roles at other public companies give her a broad perspective in the areas of financial reporting, and audit and Enterprise Risk Management. Her international training and experience with global corporations helps to guide the Company as its operations and activities have become more global.
Other Information. In 2005, the SEC issued an order against Mr. Crowley arising out of allegations that his former employer violated Section 15B(c)(1) of the U.S. Securities Exchange Act of 1934 and Rule G-37(b) of the Municipal Securities Rulemaking Board, which prohibit a broker, dealer or municipal securities dealer from engaging in municipal securities business with an issuer within two years after any contribution to an official of such issuer. The alleged violations occurred as a result of a 2002 political donation made by Mr. Crowley to the re-election campaign of an official of an issuer with which the employer subsequently engaged in municipal securities business. Mr. Crowley was ordered to cease and desist from causing any violations and any future violations of the above provisions and pay a $25,000 civil money penalty. Mr. Crowley consented to the entry of the order without admitting or denying its findings, except as to the SEC's jurisdiction over him and the subject matter of the proceedings.
Executive and Other Officers
Information concerning our executive and other officers is set forth below. Our executive officers are the CEO and our four Senior Vice Presidents. There are no family relationships between any of our directors and executive officers. None of the organizations identified below with which an officer has previously been employed or associated is a parent, subsidiary or affiliate of the Company. Background information for Mr. Baker is provided in Proposal 1.
| | | Position with the Company | |
John W. Babich1404 | | Imaging agent for prostate cancer | | Phase 2 testing completed |
| | | Senior Vice President | |
Robert J. IsraelAzedra™ | | Treatment of pheochromocytoma and paraganglioma | | Phase 2b registrational trial under Special Protocol Assessment (SPA) |
| | | Senior Vice President, Medical Affairs & Clinical Research | |
William C. OlsonRELISTOR® | | | | |
| | | Senior Vice President, Research & Development | |
Nitya G. RayRelistor-Subcutaneous injection | | Treatment of opioid induced constipation (OIC) in advanced-illness patients receiving palliative care when laxative therapy has not been sufficient | | Marketed in the U.S., E.U., Canada, Australia and elsewhere; licensed to Salix Pharmaceuticals |
| | | Senior Vice President, Manufacturing | |
Ann Marie AssummaRelistor-Subcutaneous injection | | Treatment of OIC in patients with non-cancer pain | | Complete Response Letter received in 2012 on pending sNDA; awaiting meeting date of Advisory Committee convened by FDA |
| | | Vice President, Regulatory Affairs | |
Angelo W. Lovallo, Jr.Relistor-Oral | | Treatment of OIC | | Phase 3 testing completed |
| | | Vice President, Finance and Treasurer | |
Vivien Wong | | | Vice President, Product Development |
David E. Martin | | |
Oncology
Our principal clinical-stage product candidates in oncology are
General Counsel· | PSMA ADC, a fully human monoclonal antibody-drug conjugate designed to deliver a chemotherapeutic agent to cancer cells by targeting the three-dimensional structure of the PSMA protein. In a phase 2 open-label, multicenter clinical trial to assess anti-tumor activity, tolerability and safety, we have completed enrollment in an original cohort of chemotherapy refractory patients with metastatic castration-resistant prostate cancer (mCRPC) and are conducting a second cohort of chemotherapy-naïve patients. |
| · | 1404 (trofolastat), a radio-labeled small molecule which binds PSMA and acts as an imaging agent to diagnose and detect prostate cancer, including metastases in other soft tissue and bone. We have recently completed a global multicentered phase 2 study assessing the diagnostic accuracy of 1404 imaging in men with high-risk prostate cancer scheduled for radical prostectomy. |
| · | Azedra, a phase 2 radiotherapeutic product candidate in development as a treatment for pheochromocytoma -- rare tumors found primarily in the adrenal glands – and related paraganglioma tumors occurring in other tissues. Azedra has Orphan Drug designation and Fast Track status in the U.S. for pheochromocytoma and paraganglioma, with clinical study conducted under a 2009 Special Protocol Assessment (SPA) with the FDA. There is currently no FDA-approved therapeutic for the treatment of pheochromocytoma. |
See Clinical Trial Activities, below, for additional information concerning these studies and those for Relistor.
We have decided to move forward MIP-1095, a PSMA-targeted small molecule radiopharmaceutical originally developed by our Molecular Insight subsidiary, into clinical development, and expects to file an Investigational New Drug (IND) application in the U.S. later this year. Memorial Sloan-Kettering Cancer Center has agreed to serve as a study site for a phase 1 trial of this compound.
While MIP-1095 was being developed by Molecular Insight, it provided the compound to an academic institution in Germany for compassionate use in men with late stage metastatic prostate cancer who had exhausted other available therapies. This use was not in conjunction with any clinical trial. Information from this compassionate use was recently published in the European Journal of Nuclear Medicine and Molecular Imaging. A link to this article is found on Progenics' website.
We continue to consider opportunities for strategic collaborations, out-licenses and other arrangements with biopharmaceutical companies involving proprietary research, development and clinical programs, and may in the future also in-license or acquire additional oncology compounds and/or programs.
Relistor
Relistor, the first approved treatment for opioid-induced constipation (OIC) that addresses its underlying mechanism, decreases the constipating side effects induced by opioid pain medications such as morphine and codeine without diminishing their ability to relieve pain. Relistor subcutaneous injection is approved for sale in the U.S., E.U., Canada, Australia and elsewhere in pre-filled syringes, which are designed to ease preparation and administration for patients and caregivers. Under our License Agreement, Salix is responsible for developing and commercializing Relistor, including completing clinical development necessary to support regulatory marketing approvals for potential new indications (such as OIC in patients with chronic, non-cancer pain) and formulations of the drug (such as an oral formulation of methylnaltrexone, the active ingredient in Relistor). The U.S. Food and Drug Administration in July 2012 issued a Complete Response Letter for the supplemental New Drug Application for Relistor injection for subcutaneous use for the treatment of OIC in adult patients with chronic, non-cancer pain, in which the FDA requested additional clinical data. After an End-of-Review meeting in October of that year, the FDA's Division of Gastroenterology and Inborn Errors Products expressed a concern that there may be a risk associated with the chronic use of mu-opioid receptor antagonists in patients who are taking opioids for chronic pain, and, in order to understand this potential risk, the Division communicated that a very large, well-controlled, chronic administration trial will have to be conducted to assess the safety of any mu-opioid receptor antagonist prior to market approval for the treatment of patients with OIC who are taking opioids for chronic, non-cancer pain. Salix subsequently held discussions with the Division and expressed the view that the post-marketing, clinical and preclinical data currently available for Relistor adequately demonstrate an appropriate and expected safety profile sufficient to permit the approval of the current Relistor sNDA. In response to Salix's formal appeal of the FDA's complete response letter, the FDA informed Salix and Progenics in June 2013 that it will seek input from an Advisory Committee, which it originally scheduled for March 2014. The FDA has recently postponed this meeting and said it will convene in the near future. The FDA has also stated that it will take action under the appeal within 30 days after receiving input from the Committee.
We believe that some of the areas being considered for discussion during the meeting would include the potential for drugs in Relistor's class to cause withdrawal symptoms; the strength of a potential cardiovascular signal seen with another drug in this class of drugs and the available safety data with Relistor in regards to a potential cardiovascular (CV) signal; and the need and timing (pre-approval vs. post-approval) of major adverse cardiac events (MACE) studies with drugs of this class. The FDA has communicated that it is convening the Advisory Committee for the following reasons: (i) the potential cardiovascular safety signal observed in the 12-month safety trial of another peripheral mu-opioid antagonist for the treatment of opioid-induced constipation in patients with chronic non-cancer pain raises concern for this class of drug products; (ii) the RELISTOR supplemental application contains only uncontrolled long-term safety data, and while there is no cardiovascular signal apparent in this data, the lack of a control population does not allow a definitive evaluation to be made to rule out a potential cardiovascular safety signal; and (iii) the FDA needs to provide consistent advice regarding the need for MACE studies to applicants developing drug products in this class for this indication, and for this reason, a broader discussion of the potential for cardiovascular events across the drug class is necessary.
See Risk Factors.
Relistor net sales and related royalties earned during the years 2011-2013 are set forth below. Our recognition of royalty revenue for financial reporting purposes is explained in Management's Discussion and Analysis of Financial Condition and Results of Operations (MD&A) and our financial statements included elsewhere in this document. Royalties are based on net sales reported by commercialization collaborators.
| | First Quarter | | | Second Quarter | | | Third Quarter | | | Fourth Quarter | | | Full Year | |
| | | | | | | | (in thousands) | | | | | | | |
| 2013: | | | |
| Net Sales | | $ | 7,700 | | | $ | 7,900 | | | $ | 4,800 | | | $ | 19,000 | | | $ | 39,400 | |
| Royalties Earned | | | 1,155 | | | | 1,174 | | | | 719 | | | | 2,847 | | | | 5,895 | |
| 2012: | | | |
| Net Sales | | $ | 12,300 | | | $ | 10,800 | | | $ | 4,900 | | | $ | 5,200 | | | $ | 33,200 | |
| Royalties Earned | | | 1,834 | | | | 1,619 | | | | 728 | | | | 782 | | | | 4,963 | |
| 2011: | | | |
| Net Sales | | $ | 3,300 | | | $ | 5,200 | | | $ | 9,700 | | | $ | 8,800 | | | $ | 27,000 | |
| Royalties Earned | | | - | | | | 527 | | | | 1,240 | | | | 1,279 | | | | 3,046 | |
Clinical Trial Activities
Progenics' practice has been to announce commencement and results of all of its significant clinical trials in press releases, medical and scientific meetings and other venues. Following is a summary of current clinical trial activities involving our principal product candidates and Relistor.
PSMA ADC. In our open-label, multicenter U.S. phase 2 trial of PSMA ADC to assess anti-tumor activity and tolerability an initial cohort of chemotherapy experienced participants with metastatic castration-resistant prostate cancer (mCRPC) have received initial doses of drug starting at either 2.5 mg/kg or 2.3 mg/kg; in some cases subsequent doses were adjusted based on tolerability. The study endpoints evaluate the anti-tumor effect of the compound as measured by changes in prostate specific antigen (PSA) levels, number of circulating tumor cells (CTC), pain scores, and tumor size as measured under RECIST criteria. Safety is also being assessed. Progenics has completed enrollment of 83 patients in this cohort and is treating an additional trial cohort of up to 35 chemotherapy naive patients who have progressed on hormonal therapies.
Adverse events (AEs) associated with PSMA ADC observed in this phase 2 study through mid-2013 have been consistent with those observed in the phase 1 study noted below; the most common grade 3 or higher AEs in this study have been neutropenia (grade 4: 6.7% at 2.3 mg/kg and 11.4% at 2.5 mg/kg) and peripheral neuropathy (grade 3 or higher: 6.7% at 2.3 mg/kg and 5.7% at 2.5 mg/kg). Eight patients experienced serious adverse events (SAEs) which included febrile neutropenia, neutropenia, sinus tachycardia, gastrointestinal disorders, fatigue, infections and dehydration. Two deaths occurred after treatment with PSMA ADC. The first, a patient hospitalized ten days following his first dose of study drug (2.5 mg/kg) with febrile neutropenia and E. coli positive blood cultures, progressed despite treatment to septic shock and died; both the investigator and Progenics considered this patient's septic shock as probably related to PSMA ADC. The second patient developed a rash and fever and was hospitalized for neutropenic fever, where further assessment revealed Strep Viridans bacteria (suspected to be from a tunnel catheter) in blood. Approximately two weeks after receiving study drug (also 2.5 mg/kg), this patient was intubated due to hypoxia and respiratory failure, and died three days after being removed from a ventilator. The investigator considered sepsis as unlikely, bacteremia as probably, febrile neutropenia as definitely related to PSMA ADC; Progenics assessed sepsis as probably, bacteremia as unlikely, and febrile neutropenia as definitely related to PSMA ADC. A third patient died before receiving study drug.
The phase 2 trial was undertaken after review and analysis of results from a phase 1 dose-ranging study of the compound in 52 mCRPC patients whose cancer had progressed despite prior treatment with taxane-based chemotherapy regimens.
The initial 12-week clinical trial period of the phase 1 study evaluated up to four intravenous doses of PSMA ADC administered at three-week intervals. Following completion of the four doses, patients were offered, at their physicians' discretion, the option to continue treatment with PSMA ADC for up to an additional 39 weeks. The anti-tumor effect of the compound as measured by changes in PSA levels and number of CTCs was observed in the study across doses ranging from 1.8 mg/kg to 2.8 mg/kg, and durable responses were seen in some of these heavily pre-treated patients. PSMA ADC was generally well tolerated in patients at doses up to and including 2.5 mg/kg. Dose limiting toxicities, primarily neutropenia, were seen at 2.8 mg/kg. The most commonly reported AEs were anorexia and fatigue. Five patients experienced SAEs, two of which resulted in death. One patient dosed at 1.8 mg/kg died from multi-organ failure due to acute pancreatitis: while no data suggested this event was drug-related, a possible relationship could not be definitively ruled out. A second patient died 11 days after receiving study drug at 2.8 mg/kg: septic shock was cited as the cause of death. The investigator considered septic shock as probably related and febrile neutropenia as definitely related to PSMA ADC.
Through mid-2013, 14% of the 95 patients treated with PSMA ADC in these trials have reported SAEs, including acute pancreatitis, sepsis/septic shock (each as noted above), tachycardia, abnormal ECG, atrial fibrillation, chest pain, confusion, deep vein thrombosis, pulmonary embolism, hematuria, increased International Normalized Ratio (INR), leukopenia, metabolic acidosis, myocardial infarction, pneumonia and renal failure. In conducting these studies, Progenics has taken into account SAEs reported to date as treatment-related or possibly treatment-related; based on data currently available to it, the Company is continuing development of PSMA ADC and has not determined what effects, if any, treatment-related SAEs reported to date or that may be reported in the future may have on the development of PSMA ADC going forward. See Risk Factors.
1404. Molecular Insight conducted four phase 1 studies with 1404 to establish proof-of-concept and dosimetry, and to assess a simplified kit preparation as compared to multi-step preparation. It commenced a phase 2 safety and efficacy study in 2012 to assess the diagnostic accuracy of the compound imaging in men with high-risk prostate cancer scheduled for radical prostatectomy (RP) and extended pelvic lymph node dissection (EPLND) compared to histopathology. The primary objective of this phase 2 international multi-center, multi-reader, open-label trial is to assess clinical safety as well as the compound's ability to detect prostate cancer within the prostate gland. Patient enrollment has been completed with 105 participants dosed at 20 centers in the U.S. and Europe. An additional phase 1 study in 14 patients to assess safety and diagnostic accuracy of 1404 whole body and pelvic SPECT/CT imaging and pelvic MRI in normal healthy men without current evidence of prostate cancer has completed dosing. There have to date been no treatment related AEs reported in these studies, other than one mild injection site reaction.
Azedra. In 2006, Molecular Insight commenced a phase 1 study with Azedra in 11 patients with pheochromocytoma / paraganglioma and metastatic carcinoid tumors to assess the safety, radiation dosimetry, and distribution metabolism of a single imaging dose of this compound. Following completion of this study, two dose-finding studies were conducted to determine a maximum tolerated therapeutic dose, and to assess safety, dosimetry and preliminary efficacy of Azedra in 21 patients with pheochromocytoma / paraganglioma and 15 with high-risk neuroblastoma, respectively.
Molecular Insight subsequently commenced a phase 2 study of Azedra under the 2009 SPA regarding the design of this intended registrational phase 2 trial to evaluate the efficacy and safety of the administration of two therapeutic doses of the compound in patients with pheochromocytoma / paraganglioma. The primary objective of this U.S. study is to determine the clinical benefit of Azedra based on the proportion of study participants with a reduction of antihypertensive medication by at least 50% for at least six months. This phase 2 trial has enrolled 41 of the 58 patients required to be treated under the SPA, and the long-term follow up phase of the study is ongoing. To date 49% of patients receiving study drug reported SAEs, the most common of which were hematologic (12%); other SAEs included constipation, dyspnea and myelodysplastic syndrome (each at 4.5%).
This trial was suspended in late 2010 due to lack of funding. Progenics is now resuming this trial, and plans for patient recruitment to continue after making drug supply manufacturing arrangements for the trial.
Relistor. Salix and Progenics completed in 2011 a phase 3 U.S. trial to evaluate the efficacy and safety of oral methylnaltrexone for the treatment of opioid-induced constipation in patients with chronic, non-cancer pain. This 804-patient trial assessed a once-daily oral methylnaltrexone dose of 150, 300 or 450 mg compared to placebo for 12 weeks: patients received one daily dose during the first four weeks and dosing on an as needed basis for the remaining eight weeks. Both the 300 and 450 mg treatment arms demonstrated statistically significant results for the primary endpoint of average proportion of rescue-free bowel movements (RFBMs) per patient within four hours of administration over four weeks of dosing, when compared to the placebo treatment arm. Efficacy was also seen in both the 300 and 450 mg treatment groups for other endpoints, including change in weekly RFBMs from baseline over the first four weeks and one assessing response (responder/non-responder) to study drug during weeks one to four, where "responder" was defined as having three or more RFBMs per week, with an increase of at least one RFBM per week over baseline, for at least three out of the first four weeks. Overall, efficacy of oral methylnaltrexone in this study was comparable to that reported in clinical studies of subcutaneous methylnaltrexone in patients with chronic, non-cancer pain, and the overall observed safety profile seen in patients treated with oral methylnaltrexone was comparable to placebo.
The phase 3 trial was undertaken after review and analysis of results from clinical trials initiated by Progenics' former collaboration partner, Wyeth Pharmaceuticals, and Progenics utilizing formulations of oral methylnaltrexone in patients with chronic, non-cancer pain receiving opioid treatment.
In one of these trials, of the oral formulation utilized in the phase 3 trial described above, 48% of the 25 patients receiving methylnaltrexone tablets laxated within four hours of treatment. In this study, there were no drug related SAEs reported, and the most frequent AEs were nausea and headache (10.8% each); others were vomiting (4.6%), abdominal pain and muscle spasms (3.1% each). Based on the data from this study and other information regarding oral methylnaltrexone, Progenics concluded that the methylnaltrexone tablet was active and generally well tolerated and that its safety, pharmacokinetics and activity profiles were comparable to that of subcutaneous methylnaltrexone, and decided to commence the phase 3 trial described above.
For purposes of the foregoing summary, in general phase 1 trials are initial evaluations of safety in humans which study methods of action and metabolization; phase 2 trials evaluate safety, dosing and activity or efficacy, and continue safety evaluation; and phase 3 trials involve larger scale evaluations of safety, efficacy and dosage.
License and Other Agreements
Following is a summary of significant agreements relating to our pipeline.
Oncology
Our PSMA Development Company LLC subsidiary has a collaboration agreement with Seattle Genetics, Inc. under which SGI has granted us an exclusive worldwide license to its proprietary ADC technology. We have the right to use this technology, which is based in part on technology licensed by SGI from third parties, to link chemotherapeutic agents to our monoclonal antibodies that target prostate specific membrane antigen utilizing technology licensed to us, through Cytogen Corporation, from Sloan-Kettering Institute for Cancer Research. We are responsible for research, product development, manufacturing and commercialization of all products, and are obligated to make maintenance and milestone payments and to pay royalties to SGI and its licensors, as applicable, on a percentage of net sales. The SGI agreement terminates at the latest of (i) the tenth anniversary of the first commercial sale of each licensed product in each country or (ii) the latest date of expiration of patents underlying the licensed products. We may terminate the agreement upon advance written notice; SGI may terminate if we fail to cure a breach of an SGI in-license after written notice; and either party may terminate after written notice upon an uncured breach or in the event of the other party's bankruptcy.
PSMA LLC also has a worldwide exclusive licensing agreement with Abgenix (now Amgen Fremont, Inc.) to use its XenoMouse® technology for generating fully human antibodies to PSMA. We are obligated to make development and commercialization milestone payments with respect to products incorporating an antibody generated utilizing that technology, along with royalties based upon net sales of such products. Abgenix may terminate this agreement for cause, after an opportunity to cure, upon 30 days prior written notice; we have the right to terminate upon 30 days prior written notice. The agreement continues until the later of the expiration of specified patents or seven years from the first commercial sale of eligible products.
We acquired Molecular Insight in January 2013 by purchasing all of its outstanding capital stock for 4,566,210 shares of Progenics common stock (with 500,000 held in an escrow expiring in April 2014) in a private transaction. Under the agreement, we also agreed to pay to the former stockholders potential milestones, in cash or Progenics stock at our option, of up to $23 million contingent upon achieving specified commercialization events and up to $70 million contingent upon achieving specified sales targets relating to the acquired company's products. The agreement contains customary representations and warranties, covenants, and indemnification and other provisions.
In addition to utilizing its own proprietary technology, Molecular Insight has a number of agreements with owners of intellectual property which we use or believe may be useful in research and development of product candidates, including:
| · | A 2012 co-exclusive license agreement with the University of Zurich and the Paul Scherrer Institute for worldwide sublicensable rights to certain intellectual property related to production methodologies relevant to 1404. Under this agreement, we maintain related patent rights and are obligated to pay low single-digit royalties on products using the licensed technology, license maintenance fees creditable against royalties, an annual fee for an option to expand the license's field of use, and clinical and regulatory milestone payments aggregating approximately $1.2 million. The agreement may be terminated by the licensors upon certain material defaults by, and automatically terminates upon certain bankruptcy events relating to Molecular Insight, and may be terminated by us on prior written notice. |
| · | A 2000 exclusive license agreement with The University of Western Ontario for worldwide sublicensable rights to certain intellectual property related to production methodologies relevant to Azedra. Under this agreement, we maintain related patent rights and are obligated to pay low single-digit royalties on products using the licensed technology, minimum annual royalties creditable against royalties and clinical and regulatory milestone payments aggregating approximately $0.2 million. The agreement, which either party may terminate upon certain bankruptcy events or material defaults, continues through the last to expire of the related patent rights. |
| · | A 2006 agreement with Novartis Pharma AG for exclusive rights to certain technology relating to Onalta™, a compound which has been outlicensed to third parties which are obligated to make milestone payments and royalties consistent with Molecular's obligations under this agreement, including royalties and milestone payments, partially creditable against future royalties, totaling $4.6 million upon the attainment of certain regulatory approvals. Either party may terminate this agreement immediately upon the occurrence of bankruptcy proceedings relating to or in the event of an uncured material breach by the other party, and Novartis may terminate in the event Molecular Insight is acquired by a direct competitor of Novartis. |
Relistor
Under our License Agreement, Salix Pharmaceuticals is responsible for further developing and commercializing subcutaneous Relistor worldwide other than Japan, including completing clinical development necessary to support regulatory marketing approvals for potential new indications and formulations, and marketing and selling the product. Salix is marketing Relistor directly through its specialty sales force in the U.S., and outside the U.S., directly through distribution and marketing partners and sublicensing regional companies. Salix is paying us royalties ranging from 15 to 19 percent on its net sales of Relistor as well as 60% of any upfront, milestone, reimbursement or other revenue (net of costs of goods sold and territory-specific research and development expense reimbursement) it receives from sublicensees in respect of any country outside the U.S. We are also eligible to receive (i) a development milestone of up to $40 million upon U.S. marketing approval for subcutaneous Relistor in chronic, non-cancer pain patients (the proposed indication addressed in the Complete Response Letter referred to above), (ii) a development milestone of up to $50 million upon U.S. marketing approval of an oral formulation of Relistor, and (iii) up to $200 million of commercialization milestone payments upon achievement of specified U.S. sales targets. In the event that either marketing approval is subject to a Black Box Warning or Risk Evaluation and Mitigation Strategy (REMS), payment of a substantial portion of specified milestone amounts would be deferred, and subject, to achievement of the first commercialization milestone payable upon annual U.S. sales first exceeding $100 million. See Risk Factors.
We have licensed to Ono Pharmaceutical the rights to subcutaneous Relistor in Japan. We are entitled to receive up to $20 million upon achievement of development milestones as well as royalties and commercialization milestones. Ono has the option to acquire the rights to develop and commercialize other formulations of Relistor in Japan, on terms to be negotiated separately. The license permits termination by either party upon the occurrence of certain events. Progenics in October 2013 commenced an arbitration with Ono, following a communication from Ono that it has determined to discontinue development because of "commercial concerns" that Ono contends would permit it to cease development and terminate the License. Ono's discontinuation of development and/or protracted dispute resolution proceedings could result in reduced or delayed, or in the elimination of, milestone and/or royalty revenue from subcutaneous Relistor development in Japan. Under our License Agreement with Salix, in the event the Ono license terminates or rights thereunder otherwise revert to Progenics, the Salix Agreement automatically without payment by Salix extends to the license grants, territory and other rights provided in the Ono license, as a result of which Salix and not Progenics would receive such rights.
Among the rights we have licensed to Salix are our exclusive rights to develop and commercialize methylnaltrexone, the active ingredient of Relistor, which we in-licensed from the University of Chicago. Our agreement with Chicago provides for an exclusive license to intellectual property in exchange for development and potential commercialization obligations, low single-digit royalties on commercial sales of resulting products and single-digit percentages of milestone and sublicensing revenues, and shared patent policing responsibilities. The Chicago agreement, as amended in connection with our Relistor collaborations, including substantially all of Progenics' payment obligations thereunder, expires by its terms upon the expiration of the last to expire of the patents licensed thereunder, the last-to-expire of which expires in 2017.
Patents and Proprietary Technology
Protection of our intellectual property rights is important to our business. We seek U.S. patent protection for many of our inventions, and generally file patent applications in Canada, Japan, European countries that are party to the European Patent Convention and other countries on a selective basis in order to protect inventions we consider to be important to the development of business in those areas. Generally, patents issued in the U.S. are effective for either (i) 20 years from the earliest asserted filing date, if the application was filed on or after June 8, 1995, or (ii) the longer of 17 years from the date of issue or 20 years from the earliest asserted filing date, if the application was filed prior to that date.
In certain instances, the U.S. patent term can be extended up to a maximum of five years to recapture a portion of the term during which FDA regulatory review was being conducted. The duration of foreign patents varies in accordance with the provisions of applicable local law, although most countries provide for patent terms of 20 years from the earliest asserted filing date and allow patent extensions similar to those permitted in the U.S.
Patents may not enable us to preclude competitors from commercializing drugs in direct competition with our products, and consequently may not provide us with any meaningful competitive advantage. See Risk Factors. We also rely on trade secrets, proprietary know-how and continuing technological innovation to develop and maintain a competitive position in our product areas. We require our employees, consultants and corporate partners who have access to our proprietary information to sign confidentiality agreements.
Information with respect to our current patent portfolio is set forth below.
| Clinical and Research | | Number of Patents | | Expiration | | Number of Patent Applications |
| Candidates; Relistor | | U.S. | | International | | Dates (1) | | U.S. | | International |
| | | | | | | | | | |
| Development programs | | 21 | | 76 | | 2015-2031 | | 15 | | 49 |
| | | | | | | | | | |
| Relistor | | 19 | | 112 | | 2015-2031 | | 23 | | 218 |
| | | | | | | | | | |
| Other | | 1 | | 1 | | 2029 | | 4 | | 19 |
| | | | | | | | | | |
| (1) | Patent term extensions and pending patent applications may extend periods of patent protection. |
With respect to PSMA ADC, currently issued composition-of-matter patents comprising co-owned and in-licensed properties have expiration ranges of 2022 to 2023 in the U.S. and 2022 to 2026 ex-U.S. Corresponding patent applications as well as patent applications directed to methods of use (except for the U.S. patent expiring in 2023) are pending worldwide, which if issued would have expiration ranges from 2022 to 2029. We view all of these patents as significant.
Owned and in-licensed properties relating to 1404 have expiration ranges of 2020 to 2029; we view as most significant the composition-of-matter patent on the compound, as well as technetium-99 labeled forms, which expires in 2029. Owned properties relating to MIP-1095 have expiration ranges of 2027 to 2031; we view as most significant the composition-of-matter patent on this compound, as well as radiolabeled forms, which expires in 2027. Additional U.S. patents are directed to various inventions relating to these product candidates, and corresponding patent applications are pending worldwide.
With regard to our Relistor-related intellectual property, the composition-of-matter patent for the active ingredient of Relistor, methylnaltrexone, was invented in the 1970's and has expired. The University of Chicago, as well as Progenics and its collaborators, have extended the methylnaltrexone patent estate with additional patents and pending patent applications covering various inventions relating to the product. Salix has listed in the FDA Orange Book four U.S. patents relating to subcutaneous Relistor, which have expiration dates ranging from 2017 to 2030, and one patent (expiring in 2024) with Health Canada. A patent issued in September 2013 provides protection for the oral methylnaltrexone product until 2031.
We are aware of intellectual property rights held by third parties that relate to products or technologies we are developing. For example, we are aware of others investigating and developing technologies and drug candidates directed toward PSMA or related compounds as well as methylnaltrexone and other peripheral opioid antagonists, and of patents and applications held or filed by others in those areas. The validity of issued patents, patentability of claimed inventions in pending applications and applicability of any of them to our programs are uncertain and subject to change, and patent rights asserted against us could adversely affect our ability to commercialize or collaborate with others on specific products.
Research, development and commercialization of a biopharmaceutical product often require choosing between alternative development and optimization routes at various stages in the development process. Preferred routes depend upon current -- and may be affected by subsequent -- discoveries and test results and cannot be identified with certainty at the outset. There are numerous third-party patents in fields in which we work, and we may need to obtain license under patents of others in order to pursue a preferred development route of one or more of our product candidates. The need to obtain a license would decrease the ultimate value and profitability of an affected product. If we cannot negotiate such a license, we might have to pursue a less desirable development route or terminate the entire program altogether.
Government Regulation
Progenics and its product candidates are subject to comprehensive regulation by the U.S. FDA and comparable authorities in other countries. Pharmaceutical regulation currently is a topic of substantial interest in lawmaking and regulatory bodies in the U.S. and internationally, and numerous proposals exist for changes in FDA and non-U.S. regulation of pre-clinical and clinical testing, approval, safety, effectiveness, manufacturing, storage, recordkeeping, labeling, marketing, export, advertising, promotion and other aspects of biologics, small molecule drugs and medical devices, many of which, if adopted, could significantly alter our business and the current regulatory structure described below. See Risk Factors.
FDA Regulation. FDA approval, which involves review of scientific, clinical and commercial data, manufacturing processes and facilities, is required before a product candidate may be marketed in the U.S. This process is costly, time consuming and subject to unanticipated delays, and a drug candidate may fail to progress at any point.
None of our product candidates other than Relistor has received marketing approval from the FDA or any other regulatory authority. The process required by the FDA before product candidates may be approved for marketing in the U.S. generally involves:
| · | pre-clinical laboratory and animal tests; |
| · | submission to and favorable review by the FDA of an investigational new drug application before clinical trials may begin; |
| · | adequate and well-controlled human clinical trials to establish the safety and efficacy of the product for its intended indication (animal and other nonclinical studies also are typically conducted during each phase of human clinical trials); |
| · | submission to the FDA of a marketing application; and |
| · | FDA review of the marketing application in order to determine, among other things, whether the product is safe and effective for its intended uses. |
Dr. BabichPre-clinical tests include laboratory evaluation of product chemistry and animal studies to gain preliminary information about a compound's pharmacology and toxicology and to identify safety problems that would preclude testing in humans. Since product candidates must generally be manufactured according to current Good Manufacturing Practices (cGMP), pre-clinical safety tests must be conducted by laboratories that comply with FDA good laboratory practices regulations. Pre-clinical testing is preceded by initial research related to specific molecular targets, synthesis of new chemical entities, assay development and screening for identification and optimization of lead compound(s).
Results of pre-clinical tests are submitted to the FDA as part of an Investigational New Drug application (IND) which must become effective before clinical trials may commence. The IND submission must include, among other things, a description of the sponsor's investigational plan; protocols for each planned study; chemistry, manufacturing and control information; pharmacology and toxicology information and a summary of previous human experience with the investigational drug. Unless the FDA objects to, makes comments or raises questions concerning an IND, it becomes effective 30 days following submission, and initial clinical studies may begin. Companies often obtain affirmative FDA approval, however, before beginning such studies.
Clinical trials involve the administration of the investigational new drug to healthy volunteers or to individuals under the supervision of a qualified principal investigator. Clinical trials must be conducted in accordance with the FDA's Good Clinical Practice requirements under protocols submitted to the FDA that detail, among other things, the objectives of the study, parameters used to monitor safety and effectiveness criteria to be evaluated. Each clinical study must be conducted under the auspices of an Institutional Review Board, which considers, among other things, ethical factors, safety of human subjects, possible liability of the institution and informed consent disclosure which must be made to participants in the trial.
Clinical trials are typically conducted in three sequential phases, which may overlap. During phase 1, when the drug is initially administered to human subjects, the product is tested for safety, dosage tolerance, absorption, metabolism, distribution and excretion. Phase 2 involves studies in a limited population to evaluate preliminarily the efficacy of the product for specific, targeted indications, determine dosage tolerance and optimal dosage and identify possible adverse effects and safety risks.
When a product candidate is found in phase 2 evaluation to have an effect and an acceptable safety profile, phase 3 trials are undertaken in order to further evaluate clinical efficacy and test for safety within an expanded population. Safety studies are conducted in accordance with the FDA's International Conference on Harmonization (ICH) Guidelines. Phase 2 results do not guarantee a similar outcome in phase 3 trials. The FDA may suspend clinical trials at any point in this process if it concludes that clinical subjects are being exposed to an unacceptable health risk.
A New Drug Application, 56,or NDA, is an application to the FDA to market a 1997 foundernew drug. A Biologic License Application, or BLA, is an application to market a biological product. The new drug or biological product may not be marketed in the U.S. until the FDA has approved the NDA or issued a biologic license. The NDA must contain, among other things, information on chemistry, manufacturing and controls; non-clinical pharmacology and toxicology; human pharmacokinetics and bioavailability; and clinical data. The BLA must contain, among other things, data derived from nonclinical laboratory and clinical studies which demonstrate that the product meets prescribed standards of safety, purity and potency, and a full description of manufacturing methods. Supplemental NDAs (sNDAs) are submitted to obtain regulatory approval for additional indications for a previously approved drug, and are reviewed by the FDA in a similar manner.
The results of the pre-clinical studies and clinical studies, the chemistry and manufacturing data, and the proposed labeling, among other things, are submitted to the FDA in the form of an NDA or BLA. The FDA may refuse to accept the application for filing if certain administrative and content criteria are not satisfied, and even after accepting the application for review, the FDA may require additional testing or information before approval of the application, in either case based upon changes in applicable law or FDA policy during the period of product development and FDA regulatory review. The applicant's analysis of the results of clinical studies is subject to review and interpretation by the FDA, which may differ from the applicant's analysis, and in any event, the FDA must deny an NDA or BLA if applicable regulatory requirements are not ultimately satisfied. If regulatory approval of a product is granted, such approval may be made subject to various conditions, including post-marketing testing and surveillance to monitor the safety of the product, or may entail limitations on the indicated uses for which it may be marketed. Product approvals may also be withdrawn if compliance with regulatory standards is not maintained or if problems occur following initial marketing.
Underlying this process are FDA regulations relating to drug approval which are complex and multi-faceted. Two such policies which may apply to some of our current or future product candidates are "Fast Track" approval for new subsidiarydrugs or biologics intended for the treatment of serious or life-threatening conditions which demonstrate the potential to address unmet medical needs, and Orphan Drug designation, available under U.S., E.U. and other laws, for drug candidates intended to treat rare diseases or conditions, and which if approved are granted a period of market exclusivity, subject to various conditions. Orphan Drug designation does not shorten or otherwise convey any advantage in the regulatory approval process.
Both before and after approval is obtained, a product, its manufacturer and the sponsor of the marketing application for the product are subject to comprehensive regulatory oversight. Violations of existing or newly-adopted regulatory requirements at any stage, including the pre-clinical and clinical testing process, the approval process, or thereafter, may result in various adverse consequences, including FDA delay in approving or refusal to approve a product, withdrawal of an approved product from the market or the imposition of criminal penalties against the manufacturer or sponsor. Later discovery of previously unknown problems may result in restrictions on the product, manufacturer or sponsor, including withdrawal of the product from the market.
Regulation Outside the U.S. Whether or not FDA approval has been obtained, approval of a pharmaceutical product by comparable government regulatory authorities abroad must be obtained prior to marketing the product there. The approval procedure varies from country to country, and the time required may be longer or shorter than that required for FDA approval. The requirements for regulatory approval by governmental agencies in other countries prior to commercialization of products there can be rigorous, costly and uncertain, and approvals may not be granted on a timely basis or at all.
In E.U. countries, Canada, Australia and Japan, regulatory requirements and approval processes are similar in principle to those in the U.S. Regulatory approval in Japan requires that clinical trials of new drugs be conducted in Japanese patients. Depending on the type of drug for which approval is sought, there are currently two potential tracks for marketing approval in the E.U. countries: mutual recognition and the centralized procedure. These review mechanisms may ultimately lead to approval in all E.U. countries, but each method grants all participating countries some decision-making authority in product approval. The centralized procedure, which is mandatory for biotechnology derived products, results in a recommendation in all member states, while the E.U. mutual recognition process involves country-by-country approval.
In other countries, regulatory requirements may require additional pre-clinical or clinical testing regardless of whether FDA or European approval has been obtained. This is the case in Japan, where trials are required to involve patient populations which we and our other collaborators have not examined in detail. If a product is manufactured in the U.S., it is also subject to FDA and other U.S. export provisions. In most countries outside the U.S., coverage, pricing and reimbursement approvals are also required, which may affect the profitability of the affected product.
Other Regulation. In addition to regulations enforced by the FDA, we are also subject to regulation under the U.S. Occupational Safety and Health Act, Environmental Protection Act, Toxic Substances Control Act, Resource Conservation and Recovery Act and various other current and potential future U.S. federal, state or local regulations. In addition, Molecular InsightInsight's research is dependent on maintenance of licenses from various authorities permitting the acquisition, use and storage of quantities of radioactive isotopes that are critical for its manufacture and testing of research products. Biopharmaceutical research and development generally involves the controlled use of hazardous materials, chemicals, viruses and various radioactive compounds. Even strict compliance with safety procedures for storing, handling, using and disposing of such materials prescribed by applicable regulations cannot completely eliminate the risk of accidental contaminations or injury from these materials, which may result in liability for resulting legal and regulatory violations as well as damages.
Manufacturing
Under our License Agreement, Salix is responsible for the manufacture and supply, at its expense, of all active pharmaceutical ingredient (API) and finished and packaged products for its Relistor commercialization efforts, including contracting with contract manufacturing organizations (CMOs) for supply of Relistor API and subcutaneous and oral finished drug product. We expect to engage third-party CMOs to manufacture additional clinical trial supplies of our PSMA monoclonal antibody and to continue using CMOs for other portions of the PSMA ADC manufacturing process. We have engaged and are finalizing definitive manufacturing arrangements with third-party CMOs for clinical trial supplies of Azedra and 1404, and may in the future undertake such efforts with respect to other assets and programs. If we are unable to arrange for satisfactory CMO services, we would need to undertake such responsibilities on our own. See Risk Factors.
Sales and Marketing
We from time to time seek strategic collaborations and other funding support for product candidates in our pipeline. We expect that we would market other products for which we obtain regulatory approval through co-marketing, co-promotion, licensing and distribution arrangements with third-party collaborators, and might also consider contracting with professional detailing and sales organizations to perform promotional and/or medical-scientific support functions for them. See Risk Factors.
Competition
Competition in the biopharmaceutical industry is intense and characterized by ongoing research and development and technological change. We face competition from many for-profit companies and major universities and research institutions in the U.S. and abroad. We face competition from companies marketing existing products or developing new products for diseases targeted by our technologies. Many of our competitors have substantially greater resources, experience in conducting pre-clinical studies and clinical trials and obtaining regulatory approvals for their products, operating experience, research and development and marketing capabilities and production capabilities than we do. Our products and product candidates under development may not compete successfully with existing products or products under development by other companies, universities and other institutions. Drug manufacturers that are first in the market with a therapeutic for a specific indication generally obtain and maintain a significant competitive advantage over later entrants and therefore, the speed with which industry participants move to develop products, complete clinical trials, approve processes and commercialize products is an important competitive factor.
Relistor is the first FDA-approved product for any indication involving OIC. We are, however, aware of approved and marketed products, as well as candidates in pre-clinical or clinical development, that target the side effects of opioid pain therapy. Cubist Pharmaceuticals, which acquired Adolor Corporation in 2011, markets ENTEREG® (alvimopan) for the treatment of postoperative ileus, and is in phase 3 testing of a compound for OIC in chronic-pain patients. Sucampo Pharmaceuticals, Inc., in collaboration with Takeda Pharmaceutical Company Limited, markets AMITIZA® (lubiprostone), a selective chloride channel activator, for chronic idiopathic (non-opioid related) constipation, and in April 2013 received FDA approval of this drug for opioid-induced constipation. The FDA has also recently accepted for review, with a PDUFA date of September 14, 2014, an NDA for Naloxegol, an oral peripheral mu-opioid receptor antagonist for patients with OIC being developed by a Nektar Therapeutics-AstraZeneca PLC collaboration, which Progenics acquiredalso has a related combination product in January 2013. He served as Executive Vice President, Chief Scientific Officer, Presidentearly stage development. Shionogi & Co. is developing Naldemedine, another mu-opioid receptor antagonist, for patients with OIC. Theravance, Inc. has completed phase 2 clinical testing of Research and Developmentan oral peripheral mu-opioid antagonist. In Europe, Mundipharma International Limited markets TARGIN® (oxycodone/naloxone), a combination of an opioid and a directorsystemic opioid antagonist. Other prescription as well as over-the-counter (OTC) constipation products are also prescribed first line for OIC.
As to our oncology pipeline, radiation and surgery are two traditional forms of treatment for prostate cancer. If the disease spreads, hormone (androgen) suppression therapy is often used to slow the cancer's progression, but this form of treatment can eventually become ineffective. We are aware of several competitors who are developing or have received approval for alternative treatments for castration-resistant prostate cancer, some of which are directed against PSMA, including Johnson & Johnson subsidiary Janssen Biotech, Inc.'s Zytiga® (abiraterone acetate), approved in 2011 for use in combination with prednisone as a second-line (after treatment with docetaxel) advanced prostate cancer treatment, and later for use with prednisone for metastatic castration-resistant disease before the use of chemotherapy; Medivation, Inc.'s Xtandi® (enzalutamide), approved in August 2012 for patients with metastatic castration-resistant prostate cancer previously treated with docetaxel; and Algeta ASA's Alpharadin® (radium-223 dichloride) (marketed as Xofigo®), submitted in late 2012 for marketing authorization for therapy of bone metastases in prostate cancer patients. A competitive product to 1404 is Jazz Pharmaceuticals' ProstaScint®, which is approved for detection of metastatic prostate cancer or relapsed or high-risk prostate cancer patients.
A significant amount of research in the biopharmaceutical field is carried out at academic and government institutions. An element of our research and development strategy has been to in-license technology and product candidates from academic and government institutions. These institutions are sensitive to the commercial value of their findings and pursue patent protection and negotiate licensing arrangements to collect royalties for use of technology they develop. They may also market competitive commercial products on their own or in collaboration with competitors and compete with us in recruiting highly qualified scientific personnel, which may result in increased costs or decreased availability of technology or product candidates from these institutions to other industry participants.
Competition with respect to our technologies and products is based on, among other things, product efficacy, safety, reliability, method of administration, availability, price and clinical benefit relative to cost; timing and scope of regulatory approval; sales, marketing and manufacturing capabilities; collaborator capabilities; insurance and other reimbursement coverage; and patent protection. Competitive position in our industry also depends on a participant's ability to attract and retain qualified personnel, obtain patent protection or otherwise develop proprietary products or processes, and secure sufficient capital resources for the typically substantial period between technological conception and commercial sales.
Product Liability
The testing, manufacturing and marketing of our product candidates and products involves an inherent risk of product liability attributable to unwanted and potentially serious health effects. To the extent we elect to test, manufacture or market product candidates and products independently, we bear the risk of product liability directly. We maintain product liability insurance coverage in the amount of $10.0 million per occurrence, subject to a deductible and a $10.0 million aggregate limitation. Where local statutory requirements exceed the limits of our existing insurance or local policies of insurance are required, we maintain additional clinical trial liability insurance to meet these requirements. This insurance is subject to deductibles and coverage limitations. The availability and cost of maintaining insurance may change over time.
Human Resources
At December 31, 2013, we had 69 full-time employees, 12 of whom hold Ph.D. degrees and two of whom hold M.D. degrees. At that date, 49 employees were engaged in research and development, medical, regulatory affairs and manufacturing related activities and 20 were engaged in finance, legal, administration and business development. We consider our relations with our employees to be good. None of our employees is covered by a collective bargaining agreement.
The future of our business and operations depends on the success of our Relistor collaborations and our oncology research and development programs.
Our business and operations entail a variety of serious risks and uncertainties and are inherently risky. The research and development programs on which we focus involve novel approaches to human therapeutics. Our principal product candidates are in clinical development, and in some respects involve technologies with which we have limited prior experience. We are subject to the risks of failure inherent in the development of product candidates based on new technologies. There is little precedent for the successful commercialization of products based on our technologies, and there are a number of technological challenges that we must overcome to complete most of our development efforts. We may not be able successfully to develop further any of our product candidates. We and our Relistor and other collaborators must successfully complete clinical trials and obtain regulatory approvals for potential commercial products. Once approved, if at all, commercial product sales are subject to general and industry-specific local and international economic, regulatory, technological and policy developments and trends. The oncology space in which we operate presents numerous significant risks and uncertainties that may be expected to increase to the extent it becomes more competitive or less favored in the commercial healthcare marketplace.
The long-term success of our acquisition of Molecular Insight from 1998will be subject to 2010, and from January to May 2009, as Chairmanall of the Boardrisks and Chief Executive Officer. Prior to foundinguncertainties described in these risk factors. In addition, the estimated fair values of Molecular Insight Dr. Babich was Assistant Professorassets and liabilities reflected in our financial statements do not, given their uniqueness and attendant uncertainties, reflect actual transactions or quoted prices and may not correlate to any future values or results. Such information should not be interpreted or relied upon as indicative of Radiology at Harvard Medical Schoolany future value or results. Our failure to manage successfully any of our product candidates, technologies or programs could have an adverse impact on our business, and Staff Radiopharmaceutical Chemist at Massachusetts General Hospital, Boston, Massachusetts. Prioron the price of our stock.
We are dependent on Salix, Ono and other business partners to joining MGH, he was Principal Scientistdevelop and Headcommercialize Relistor, exposing us to significant risks.
We rely on Salix to complete development and obtain regulatory approvals for additional formulations of and indications for Relistor and, in the Japanese market, we rely on Ono to conduct clinical trials and obtain regulatory approvals. We are and will be dependent upon Salix, Ono and any other business partners with which we may collaborate in the future to perform and fund development, including clinical testing of Relistor, make related regulatory filings and manufacture and market products, including for new indications and in new formulations, in their respective territories. Revenue from the sale of Relistor depends entirely upon the efforts of Salix and its sublicensees, which have significant discretion in determining the efforts and resources they apply to sales of Relistor. Ono will have similar discretion with respect to sales in Japan. Neither may be effective in obtaining approvals for new indications or formulations, marketing existing or future products or arranging for necessary sublicense or distribution relationships. Our business relationships with Salix, Ono and other partners may not be scientifically, clinically or commercially successful. For example, Salix has a variety of marketed products. Salix is not, however, a large diversified pharmaceutical company and does not have resources commensurate with such companies. Salix has its own corporate objectives, which may not be consistent with our best interests, and may change its strategic focus or pursue alternative technologies in a manner that results in reduced or delayed revenue to us. Changes of this nature might also occur if Salix were acquired or if its management changed. We may have future disagreements with Salix or Ono, both of which have significantly greater financial and managerial resources which either could draw upon in the event of a dispute. Such disagreements could lead to lengthy and expensive litigation or other dispute-resolution proceedings as well as extensive financial and operational consequences to us and have a material adverse effect on our business, results of operations and financial condition. In addition, independent actions may be taken by Salix and/or Ono concerning product development, marketing strategies, manufacturing and supply issues, and rights relating to intellectual property, including, as discussed below, Relistor's path forward in light of the Radiopharmaceutical SectionJuly 2012 Complete Response Letter from the FDA.
As noted elsewhere in this document, Progenics in October 2013 commenced an arbitration with Ono under the provisions of the parties' License Agreement for development and commercialization of subcutaneous Relistor in Japan, following a communication from Ono that it has determined to discontinue development because of "commercial concerns" that Ono contends would permit it to cease development and terminate the Agreement. Ono's discontinuation of development and/or protracted dispute resolution proceedings could result in reduced or delayed, or in the elimination of, milestone and/or royalty revenue from subcutaneous Relistor development in Japan. Under our License Agreement with Salix, in the event our Agreement with Ono terminates or rights thereunder otherwise revert to Progenics, the Salix Agreement automatically without payment by Salix extends to the license grants, territory and other rights provided in the Ono Agreement, as a result of which Salix and not Progenics would receive such rights.
As a result of the FDA's Complete Response Letter on Relistor for chronic pain, the Relistor program may be discontinued or otherwise at risk.
As previously announced, and noted elsewhere in this document, the FDA in July 2012 issued a Complete Response Letter in response to Salix's supplemental New Drug Application for Relistor for the treatment of OIC in adult patients with chronic, non-cancer pain. This development may result in Salix and/or Ono taking independent actions concerning product development, marketing strategies or other matters for Relistor, including termination of their efforts to develop and commercialize the drug. At an End-of-Review meeting in October 2012, the FDA's Division of Gastroenterology and Inborn Errors Products has expressed a concern that there may be a risk associated with the chronic use of mu-opioid receptor antagonists in patients who are taking opioids for chronic pain, and, in order to understand this potential risk, the Division communicated that a very large, well-controlled, chronic administration trial will have to be conducted to assess the safety of any mu-opioid receptor antagonist prior to market approval for the treatment of patients with OIC who are taking opioids for chronic, non-cancer pain. In addition, the FDA informed Salix and Progenics in June 2013 that it will seek input from an Advisory Committee, which is now expected to convene in the near future, and at that time expressed our belief that some of the areas being considered for discussion during the meeting would include the potential for drugs in Relistor's class to cause withdrawal symptoms; the strength of a potential cardiovascular signal seen with another drug in this class of drugs and the available safety data with Relistor in regards to a potential cardiovascular signal; and the need and timing (pre-approval vs. post-approval) of major adverse cardiac events (MACE) studies with drugs of this class.
Salix has disclosed in regulatory filings that it might terminate its development program for Relistor subcutaneous injection for treatment of OIC in chronic non-cancer pain patients, and that additional information and additional guidance from the FDA could result in the termination of its oral OIC Relistor development program. As noted in our risk factor on regulatory approvals below, if clinical trials indicate, or regulatory bodies are concerned about, actual or possible serious problems with the safety or efficacy of a product candidate, such as the concerns expressed in the FDA's CRL and as described above, we or our collaborators may stop or significantly slow development or commercialization of affected products. As a result of such concerns, the development programs for subcutaneous and/or oral Relistor for chronic, non-cancer pain patients may be significantly delayed or terminated altogether. In such an event, we could be faced with either further developing and commercializing the drug on our own or with one or more substitute collaborators, either of which paths would subject us to the development, commercialization, collaboration and/or financing risks discussed in these risk factors. Any such significant action adverse to development and commercialization of Relistor could have a material adverse impact on our business, and on the price of our stock.
We are subject to extensive regulation, which can be costly and time consuming, may not lead to marketing approval for our product candidates, and can subject us to unanticipated limitations, restrictions, delays and fines.
Our business, products and product candidates are subject to comprehensive regulation by the FDA and comparable authorities in other countries. These agencies and other entities regulate the pre-clinical and clinical testing, safety, effectiveness, approval, manufacture, labeling, marketing, export, storage, recordkeeping, advertising, promotion and other aspects of our products and product candidates. We cannot guarantee that approvals of product candidates, processes or facilities will be granted on a timely basis, or at all. If we experience delays or failures in obtaining approvals, commercialization of our product candidates will be slowed or stopped.
For example, as described in Business – Clinical Trial Activities, in clinical studies of one of our principal product candidates, PSMA ADC, investigators have reported serious adverse events (SAEs), including three deaths, in a small proportion of patients treated with the drug. Based on data currently available to us, the Company is continuing development of PSMA ADC and has not determined what effects, if any, treatment-related SAEs reported to date or that may be reported in the future may have on the development of PSMA ADC going forward. If, however, we, together with or independently of investigators participating in our clinical trials, or regulators evaluating PSMA ADC were to determine that this candidate cannot safely be administered to patients with sufficient therapeutic effect, we may determine to attempt to reformulate or otherwise change the candidate and/or its administration to alleviate such concerns, which could result in costs and delays that could impair the value of the candidate. If such costs and delays were sufficiently large, we could determine to abandon the PSMA ADC program. Concerns about the safety and/or efficacy of PSMA ADC could also make it more difficult or impossible for us to enter into licensing, collaboration or other arrangements with third parties for further development and commercialization of PSMA ADC. Any of these possibilities could have material adverse effects on Progenics' business, its financial condition, and/or the price of our stock.
Even if we obtain regulatory approval for a product candidate, the approval may include significant limitations on indicated uses for which the product could be marketed or other significant marketing restrictions, such as a Risk Evaluation and Mitigation Strategy (REMS). For example, Relistor is only approved for OIC in patients with advanced illness and not for chronic, non-cancer pain, and our product candidates, if approved at all, may be subject to those or other such limitations and restrictions.
If we or our collaborators violate regulatory requirements at any stage, whether before or after marketing approval is obtained, we or they may be subject to forced removal of a product from the market, product seizure, civil and criminal penalties and other adverse consequences. Under our license agreement with Salix, we are dependent on Salix for compliance with these regulatory requirements as they apply to Relistor. Salix has disclosed that in February 2013 it received a subpoena from the U.S. Attorney's Office for the Southern District of New York requesting documents regarding its sales and promotional practices for Relistor and certain of its other products, that it is in the process of responding to the subpoena and intends to cooperate fully with the subpoena and related government investigation, which has and will continue to increase its legal expenses, and might require management time and attention, and that at the time of its disclosure it cannot predict or determine the timing or outcome of the inquiry or its impact on Salix's financial condition or results of operations.
Our products may face regulatory, legal or commercial challenges even after approval.
Even if a product receives regulatory approval:
| · | It might not obtain labeling claims necessary to make the product commercially viable (in general, labeling claims define the medical conditions for which a drug product may be marketed, and are therefore very important to the commercial success of a product), or may be required to carry Boxed or other warnings that adversely affect its commercial success. |
| · | Approval may be limited to uses of the product for treatment or prevention of diseases or conditions that are relatively less financially advantageous to us than approval of greater or different scope or subject to an FDA-imposed REMS that imposes limits on the distribution or use of the product. |
| · | Side effects (including different or aggravated effects such as SAEs encountered in our PSMA ADC program) identified after the product is on the market might hurt sales or result in mandatory safety labeling changes, additional pre-clinical testing or clinical trials, imposition of a REMS, product recalls or withdrawals from the market. |
| · | Efficacy or safety concerns (including those arising from SAEs heretofore or hereafter encountered in our PSMA ADC program) regarding a marketed product, or manufacturing or other problems, may lead to a recall, withdrawal of marketing approval, reformulation of the product, additional pre-clinical testing or clinical trials, changes in labeling, imposition of a REMS, the need for additional marketing applications, declining sales or other adverse events. These potential consequences may occur whether or not the concerns originate from subsequent testing or other activities by us, governmental regulators, other entities or organizations or otherwise, and whether or not they are scientifically justified. If products lose previously received marketing and other approvals, our business, results of operations and financial condition would be materially adversely affected. |
We or our collaborators will be subject to ongoing FDA obligations and continuous regulatory review, and might be required to undertake post-marketing trials to verify the product's efficacy or safety or other regulatory obligations.
Competing products in development may adversely affect acceptance of our products.
We are aware of a number of products and product candidates described in this Annual Report under Business – Competition which compete or may potentially compete with Relistor. Any of these approved products or product candidates, or others which may be developed in the future may achieve a significant competitive advantage relative to Relistor, and, in any event, the existing or future marketing and sales capabilities of these competitors may impair Salix's and/or Ono's ability to compete effectively in the market.
We are also aware of competitors, including those described under Business – Competition, which are developing alternative treatments for disease targets to which our research and development programs are directed, any of which – or others which may be developed in the future – may achieve a significant competitive advantage relative to any product we may develop.
Developing product candidates will require us to obtain additional financing. Our access to capital funding is uncertain.
We expect to continue to incur significant development expenditures for our product candidates. We do not have committed external sources of funding for most of these projects. Our expenditures will be funded from cash on hand, or we may seek additional external funding for them, most likely through collaborative, license or royalty financing agreements with one or more pharmaceutical or other companies and equity securities issuances in public offerings, private placements or through our January 2014 Sales Agreement with Cantor Fitzgerald & Co., pursuant to which we may sell from time to time up to $50 million of our stock. We may also seek capital through debt financings or government grants or contracts. To the extent we raise additional capital by issuing equity securities in the future, existing stockholders could experience substantial dilution in addition to the dilution experienced as a result of our recent equity offerings and the Molecular Insight acquisition, and new investors could have rights superior to existing stockholders, if securities other than common stock were to be issued. Any debt financing that we are able to obtain may involve operating covenants that restrict our business and significant repayment obligations. To the extent that we raise additional funds through any new collaboration and licensing arrangements, we may be required to relinquish some rights to our technologies or product candidates, or grant licenses on terms that are not favorable to us.
We cannot predict when we will need additional funds, how much we will need, the form any financing may take or whether additional funds will be available at all, especially in light of current conditions in global credit and financial markets. Our need for future funding will depend on numerous factors, such as the availability of new product development projects; the achievement of events identified in our collaboration agreements that trigger payments to us from our collaboration partners, most of which are out of our control and rely entirely on the efforts of our partners; the progress and success of clinical trials and pre-clinical activities (including studies and manufacture of materials) of our product candidates conducted by our collaborators or us; the progress of research programs carried out by us; any changes in the breadth of our research and development programs; the progress of the research and development efforts of our collaborators; our ability to acquire or license other technologies or compounds that we seek to pursue; competing technological and market developments; the costs and timing of obtaining, enforcing and defending our patent and intellectual property rights; the costs and timing of regulatory approvals and filings by us and our collaborators; our ability to manage our growth; and any unforeseen litigation. These factors may be more important with respect to product candidates and programs that involve technologies with which we have limited prior experience, such as those originally developed by Molecular Insight. Insufficient funds may require us to delay, scale back or eliminate some or all of our research and development programs, to lose rights under existing licenses or to relinquish greater or all rights to product candidates at an earlier stage of development or on less favorable terms than we would otherwise choose or may adversely affect our ability to operate as a going concern. We may not be able at the necessary time to obtain additional funding on acceptable terms, or at all. Our inability to raise additional capital on terms reasonably acceptable to us would seriously jeopardize our business.
If we are unable to negotiate collaboration agreements, our cash burn rate could increase and our rate of product development could decrease.
Our ability to generate revenue in the near term depends on the timing of achievement, if any, of certain payment triggering events under our existing collaboration agreements and our ability to enter into additional collaboration agreements with third parties. We may not be successful in negotiating additional collaboration arrangements with pharmaceutical and biotechnology companies to develop and commercialize product candidates and technologies. If we do not enter into new collaboration arrangements, we would have to devote more of our resources to clinical product development and product launch activities and to seeking additional sources of capital to fund those activities. If we were not successful in seeking such capital, our cash burn rate would increase or we would need to take steps to reduce our rate of product development. Our ability to enter into new collaborations may be dependent on many factors, such as the results of clinical trials, competitive factors and the fit of our programs with the risk tolerance of a potential collaborator, including in relation to regulatory issues, the patent portfolio, the clinical pipeline, the stage of the available data, overall corporate goals and financial position. If we are not able to generate revenue under our collaborations when and in accordance with our expectations or the expectations of industry analysts, this failure could harm our business and have an immediate adverse effect on the trading price of our common stock.
Drug development is a long and inherently uncertain process with a high risk of failure at every stage of development.
Drug development is a highly uncertain scientific and medical endeavor, and failure can unexpectedly occur at any stage of clinical development. Typically, there is a high rate of attrition for product candidates in preclinical and clinical trials due to scientific feasibility, safety, efficacy, changing standards of medical care and other variables. The risk of failure increases for our product candidates that are based on new technologies, as well as technologies with which we have limited prior experience, such as those originally developed by Molecular Insight. Pre-clinical studies and clinical trials are long, expensive and highly uncertain processes that can take many years. It will take us, or our collaborators, several years to complete clinical trials and the time required for completing testing and obtaining approvals is uncertain. The start or end of a clinical trial is often delayed or halted due to changing regulatory requirements, manufacturing challenges, required clinical trial administrative actions, slower than anticipated patient enrollment, changing standards of care, availability or prevalence of use of a comparator drug or required prior therapy, clinical outcomes, or our and our partners' financial constraints. The FDA and other U.S. and foreign regulatory agencies have substantial discretion, at any phase of development, to terminate clinical trials, require additional clinical development or other testing, delay or withhold registration and marketing approval and mandate product withdrawals, including recalls. Results attained in early human clinical trials may not be indicative of results in later clinical trials. In addition, many of our investigational or experimental drugs are at an early stage of development, and successful commercialization of early stage product candidates requires significant research, development, testing and approvals by regulators, and additional investment. Our products in the research or pre-clinical development stage may not yield results that would permit or justify clinical testing. Our failure to demonstrate adequately the safety and efficacy of a product under development would delay or prevent marketing approval, which could adversely affect our operating results and credibility. The failure of one or more of our product candidates could have a material adverse effect on our business, financial condition and results of operations.
If we or our collaborators do not obtain regulatory approval for our product candidates on a timely basis, or at all, or if the terms of any approval impose significant restrictions or limitations on use, our business, results of operations and financial condition will be adversely affected. Setbacks in clinical development programs could have a material adverse effect on our business.
Regulatory approvals are necessary to market product candidates and require demonstration of a product's safety and efficacy through extensive pre-clinical and clinical trials. We or our collaborators may not obtain regulatory approval for product candidates on a timely basis, or at all, and the terms of any approval (which in some countries includes pricing approval) may impose significant restrictions, limitations on use or other commercially unattractive conditions. We, our collaborators or regulators may also amend, suspend or terminate clinical trials if we or they believe that the participating subjects are being exposed to unacceptable health risks, and after reviewing trial results, we or our collaborators may abandon projects which we previously believed to be promising for commercial or other reasons unrelated to patient risks. During this process, we may find, for example, that results of pre-clinical studies are inconclusive or not indicative of results in human clinical trials, clinical investigators or contract research organizations do not comply with protocols or applicable regulatory requirements, or that product candidates do not have the desired efficacy or have undesirable side effects or other characteristics that preclude marketing approval or limit their potential commercial use if approved. In such circumstances, the entire development program for that product candidate could be adversely affected, resulting in delays in trials or regulatory filings for further marketing approval and a possible need to reconfigure our clinical trial programs to conduct additional trials or abandon the program involved. Conducting additional clinical trials or making significant revisions to a clinical development plan would lead to delays in regulatory filings. If clinical trials indicate, or regulatory bodies are concerned about, actual or possible serious problems with the safety or efficacy of a product candidate, such as the concerns expressed in the FDA's July 2012 Complete Response Letter or during consideration of the oral Relistor development program, we or our collaborators may stop or significantly slow development or commercialization of affected products. As a result of such concerns, the development programs for subcutaneous and/or oral Relistor for chronic, non-cancer pain patients may be significantly delayed or terminated altogether.
Even if we agree to a path forward with Salix and the FDA, if the results of any future Relistor trials are not satisfactory or we or our collaborators encounter problems enrolling subjects, clinical trial supply issues, setbacks in developing drug formulations, including raw material-supply, manufacturing, stability or other difficulties, or issues complying with protocols or applicable regulatory requirements, the entire development program for Relistor could be adversely affected in a material manner. Such scenarios could also befall our other clinical-stage product candidates. If any of our collaborators breach or terminate its agreement with us or otherwise fail to conduct successfully and in a timely manner the collaborative activities for which they are responsible, the preclinical or clinical development or commercialization of the affected product candidate or research program could be delayed or terminated. We generally do not control the amount and timing of resources that our collaborators devote to our programs or product candidates. We also do not know whether current or future collaboration partners, if any, might pursue alternative technologies or develop alternative products either on their own or in collaboration with others, including our competitors, as a means for developing treatments for the diseases or conditions targeted by our collaborative arrangements. Setbacks of these types could have a material adverse effect on our business, results of operations and financial condition.
We or our collaborators must design and conduct successful clinical trials for our product candidates to obtain regulatory approval. We rely on third parties for conduct of clinical trials, which reduces our control over them and may expose us to conflicts of interest. Clinical trial results may be unfavorable or inconclusive, and often take longer than expected.
We have limited experience in conducting clinical trials, and we rely on or obtain the assistance of others to design, conduct, supervise or monitor some or all aspects of some of our clinical trials, including our ongoing phase 2 trials of PSMA ADC and 1404. We have less control over the timing and other aspects of clinical trials for which we rely on third parties, such as CROs, clinical data management organizations, medical institutions or clinical investigators, than if we conducted them entirely on our own. These third parties may also have relationships with other entities, some of which may be our competitors. In all events, we are responsible for ensuring that each of our clinical trials is conducted in accordance with the general investigational plan and protocols for the trial. The FDA requires us to comply with good clinical practices for conducting and recording and reporting the results of clinical trials to assure that data and reported results are credible and accurate and that the rights, integrity and confidentiality of trial participants are protected. Our reliance on third parties that we do not control does not relieve us of these responsibilities and requirements.
To obtain regulatory approval of drug candidates, we must demonstrate through preclinical studies and clinical trials that they are safe and effective. Adverse or inconclusive clinical trial results concerning any of our drug candidates, or trials which regulators find deficient in scope, design or one or more other material respects, could require additional trials, resulting in increased costs, significant delays in submissions of approval applications, approvals in narrower indications than originally sought, or denials of approval, none of which we can predict. As a result, any projections that we publicly announce of commencement and duration of clinical trials are not certain. We have experienced clinical trial delays in the past as a result of slower than anticipated enrollment and such delays may recur. Delays can be caused by, among other things, deaths or other adverse medical events; regulatory or patent issues; interim or final results of ongoing clinical trials; failure to enroll clinical sites as expected; competition for enrollment from other clinical trials; scheduling conflicts with participating clinicians and institutions; disagreements, disputes or other matters arising from collaborations; our inability to obtain necessary funding; or manufacturing problems.
Under our license agreement, Salix generally has responsibility for conducting Relistor clinical trials, including all trials outside of the U.S. other than Japan, where Ono has that responsibility. In addition, certain clinical trials for our product candidates may be conducted by government-sponsored agencies, and consequently will be dependent on governmental participation and funding. These arrangements expose us to the same considerations we face when contracting with third parties for our own trials.
Our product candidates may not obtain regulatory approvals needed for marketing.
None of our product candidates, other than Relistor for the treatment of OIC in patients with advanced illnesses, has been approved by applicable regulatory authorities for marketing. The process of obtaining FDA and foreign regulatory approvals often takes many years and can vary substantially based upon the type, complexity and novelty of the products involved. We have had only limited experience in filing and pursuing applications and other submissions necessary to gain marketing approvals. Products under development may never obtain marketing approval from the FDA or other regulatory authorities necessary for commercialization.
Even if our product candidates obtain marketing approval, our ability to generate revenue will be diminished if our products are not accepted in the marketplace or our collaboration partners fail to obtain acceptable prices or an adequate level of reimbursement for products from third-party payors or government agencies.
The commercial success of our products will depend upon their acceptance by the medical community and third-party payors as clinically useful, cost effective and safe. Market acceptance of approved products, such as Relistor for patients with advanced illnesses, is affected by the timing of regulatory approvals, product launches and reimbursement programs for existing and expanded uses or generic, over-the-counter or other competitors; price increases for the product and relative prices of competing products; product development efforts for new indications; availability of sufficient commercial quantities of the product; success in arranging for necessary sublicense or distribution relationships; and general and industry-specific local and international economic pressures such as those experienced worldwide over the last five years. If health care providers believe that patients can be managed adequately with alternative, currently available therapies, they may not prescribe our products, especially if the alternative therapies are viewed as more effective, as having a better safety or tolerability profile, as being more convenient to the patient or health care providers or as being less expensive. Third-party insurance coverage may not be available to patients for any products we develop, alone or with collaborators. For pharmaceuticals administered in an institutional setting, the ability of the institution to be adequately reimbursed from government and health administration authorities, private health insurers and other third-party payors could also play a significant role in demand for our products. Significant uncertainty exists as to the reimbursement status of newly-approved pharmaceuticals. Government and other third-party payors increasingly are attempting to contain healthcare costs by limiting both coverage and the level of reimbursement for new drugs and by refusing, in some cases, to provide coverage for uses of approved products for indications for which the FDA has not granted labeling approval. In some foreign markets, pricing and profitability of prescription pharmaceuticals are subject to government control. In the U.S., we expect that there will continue to be a number of federal and state proposals to implement similar government control and that the emphasis on managed care in the U.S. will continue to put pressure on the pricing of pharmaceutical products. Cost control initiatives could decrease the price that our collaborators receive for any products in the future and adversely affect the ability of our collaborators to commercialize our products and our realization of royalties from commercialization. If any of our products do not achieve market acceptance, we will likely lose our entire investment in that product.
Marketplace acceptance depends in part on competition in our industry, which is intense, and competing products in development may adversely affect acceptance of our products.
The extent to which any of our products achieves market acceptance will depend on competitive factors. Competition in the biopharmaceutical industry is intense and characterized by ongoing research and development and technological change. We face competition from many for-profit companies and major universities and research institutions in the U.S. and abroad. We face competition from companies marketing existing products or developing new products for diseases and conditions targeted by our technologies. We are aware of a number of products and product candidates, including those described in this Annual Report under Business – Competition, which compete or may potentially compete with Relistor, PSMA ADC or our other product candidates. For instance, there are product candidates in pre-clinical or clinical development that target the side effects of opioid pain therapy, and a marketed product for the treatment of post-operative ileus could compete with Relistor. We are aware of several competitors, including those described under Business – Competition, which have received approval for or are developing alternative treatments or diagnostics for castration-resistant prostate cancer, some of which are directed against PSMA. Any of these competing approved products or product candidates, or others which may be developed in the future, may achieve a significant competitive advantage relative to Relistor, PSMA ADC, 1404, Azedra, MIP-1095 or other product candidates.
Competition with respect to our technologies and products is based on, among other things, product efficacy, safety, reliability, method of administration, availability, price and clinical benefit relative to cost; timing and scope of regulatory approval; sales, marketing and manufacturing capabilities; collaborator capabilities; insurance and other reimbursement coverage; and patent protection. Competitive disadvantages in any of these factors could materially harm our business and financial condition. Many of our competitors have substantially greater research and development capabilities and experience and greater manufacturing, marketing, financial and managerial resources than we do. These competitors may develop products that are superior to those we are developing and render our products or technologies non-competitive or obsolete. Our products and product candidates under development may not compete successfully with existing products or product candidates under development by other companies, universities and other institutions. Drug manufacturers that are first in the market with a therapeutic for a specific indication generally obtain and maintain a significant competitive advantage over later entrants and therefore, the speed with which industry participants move to develop products, complete clinical trials, approve processes and commercialize products is an important competitive factor. If our product candidates receive marketing approval but cannot compete effectively in the marketplace, our operating results and financial position would suffer.
If we or our collaborators are unable to obtain sufficient quantities of the raw and bulk materials needed to make our product candidates or Relistor, development of our product candidates or commercialization of our approved product could be slowed or stopped.
Salix or Ono may not be able to fulfill manufacturing obligations for Relistor, a key raw material for which grows in Tasmania, either on their own or through third-party suppliers. A delay or disruption of supplies of Relistor would have a material adverse effect on the Relistor franchise, and therefore on our business as a whole. Our existing arrangements with suppliers for our other product candidates may not result in the supply of sufficient quantities of our product candidates needed to accomplish our clinical development programs, and we may not have the right and in any event do not currently have the capability to manufacture these products if our suppliers are unable or unwilling to do so. We currently arrange for supplies of critical raw materials used in production of our product candidates from single sources. We do not have long-term contracts with any of these suppliers. Any delay or disruption in the availability of raw materials would slow or stop product development and commercialization of the relevant product.
Manufacturing resources could limit or adversely affect our ability to commercialize products.
We or our collaborators engage third parties to manufacture our approved product and product candidates. We or our collaborators may not be able to obtain adequate supplies from third-party manufacturers in a timely fashion for development or commercialization purposes, and commercial quantities of products may not be available from contract manufacturers at acceptable costs. Under our license agreement with Salix, Salix is responsible for obtaining supplies of Relistor, including contracting with contract manufacturing organizations for supply of Relistor active pharmaceutical ingredient and subcutaneous and oral finished drug product. These arrangements may not be on terms that are advantageous and, as a result of our royalty and other interests in Relistor's commercial success, will subject us to risks that the counterparties may not perform optimally in terms of quality or reliability. In engaging third parties for these activities, we do not control many aspects of the manufacturing process, including compliance with current Good Manufacturing Practices (cGMP) and other regulatory requirements. In order to commercialize our product candidates successfully, we or our collaborators need to be able to manufacture or arrange for the manufacture of products in commercial quantities, in compliance with regulatory requirements, at acceptable costs and in a timely manner. Manufacture of our product candidates can be complex, difficult to accomplish even in small quantities, difficult to scale-up for large-scale production and subject to delays, inefficiencies and low yields of quality products. The cost of manufacturing some of our product candidates may make them prohibitively expensive. If adequate supplies of any of our product candidates or related materials are not available on a timely basis or at all, our clinical trials could be seriously delayed, since these materials are time consuming to manufacture and cannot be readily obtained from third-party sources. If we were to decide to establish a commercial-scale manufacturing facility in the future, we would require substantial additional funds and be required to hire and train significant numbers of employees and comply with applicable regulations.
Failure of any manufacturer of Relistor or our product candidates to comply with applicable regulatory requirements could subject us to penalties and have a material adverse effect on supplies of our product or products candidates.
Third-party manufacturers are required to comply with cGMP or similar regulatory requirements outside of the U.S. If manufacturers of our product or product candidates cannot successfully manufacture material that conforms to the strict regulatory requirements of the FDA and any applicable foreign regulatory authority, they may not be able to obtain any required approval for their manufacturing facilities. If these facilities are not approved for commercial manufacture, we may need to find alternative manufacturing facilities, which could result in delays of several years in obtaining approval for a product candidate. We do not control the manufacturing process and are completely dependent on our third-party manufacturing partners or contractors for compliance with the applicable regulatory requirements for the manufacture of Relistor and our product candidates. Manufacturers are subject to ongoing periodic unannounced inspections by the FDA and corresponding state and foreign agencies for compliance with cGMP and similar regulatory requirements. Failure of any manufacturer of Relistor or any of our product candidates to comply with applicable cGMP or other regulatory requirements could result in sanctions being imposed on our collaborators or us, including fines, injunctions, civil penalties, delays, suspensions or withdrawals of approvals, operating restrictions, interruptions in supply and criminal prosecutions, any of which could significantly and adversely affect supplies of Relistor or such product candidate and have a material adverse impact on our business, financial condition and results of operations.
We are dependent on patents and other intellectual property rights.
The validity, enforceability and commercial value of our patents and other intellectual property rights are highly uncertain.
We own or have direct or sub-licenses to a number of issued patents. We must obtain, maintain and enforce patent and other rights to protect our intellectual property. The patent position of biotechnology and pharmaceutical firms is highly uncertain and involves many complex legal and technical issues. There are many laws, regulations and judicial decisions that dictate and otherwise influence the manner in which patent applications are filed and prosecuted and in which patents are granted and enforced, all of which are subject to change from time to time. There is no clear policy involving the breadth of claims allowed, or the degree of protection afforded, under patents in this area. In addition, we are aware of others who have patent applications or patents containing claims similar to or overlapping those in our patents and patent applications. Accordingly, patent applications owned by or licensed to us may not result in patents being issued. Even if we own or license a relevant issued patent, we may not be able to preclude competitors from commercializing drugs that may compete directly with one or more of our products or product candidates, in which event such rights may not provide us with any meaningful competitive advantage. In the absence or upon successful challenge of patent protection, drugs may be subject to generic competition, which could adversely affect pricing and sales volumes of the affected products.
It is generally difficult to determine the relative strength or scope of a biotechnology or pharmaceutical patent position in absolute terms at any given time. The issuance of a patent is not conclusive as to its validity or enforceability, which can be challenged in litigation or via administrative proceedings. The license agreements from which we derive or out-license intellectual property provide for various royalty, milestone and other payment, commercialization, sublicensing, patent prosecution and enforcement, insurance, indemnification and other obligations and rights, and are subject to certain reservations of rights. While we generally have the right to defend and enforce patents licensed to or by us, either in the first instance or if the licensor or licensee chooses not to do so, we must usually bear the cost of doing so. Under our license agreement with Salix, Salix generally has the first right to control the defense and enforcement of our Relistor patents. With respect to Japan, Ono has certain limited rights to prosecute, maintain and enforce relevant intellectual property. We may incur substantial costs in seeking to uphold the validity of patents or to prevent infringement. If the outcome of a dispute or contest is adverse to us, third parties may be able to use our patented invention without payment to us. Third parties may also avoid our patents through design innovation.
Patents have a limited life and expire by law.
In addition to uncertainties as to scope, validity, enforceability and changes in law, patents by law have limited lives. Upon expiration of patent protection, our drug candidates and/or products may be subject to generic competition, which could adversely affect pricing and sales volumes of the affected products.
With respect to PSMA ADC, currently issued composition-of-matter patents comprising co-owned and in-licensed properties have expiration ranges of 2022 to 2023 in the U.S. and 2022 to 2026 ex-U.S. Corresponding patent applications as well as patent applications directed to methods of use (except for the U.S. patent expiring in 2023) are pending worldwide, which if issued would have expiration ranges from 2022 to 2029. We view all of these patents as significant.
Owned and in-licensed properties relating to the 1404 product candidate have expiration ranges of 2020 to 2029; we view as most significant the composition-of-matter patent on the compound, as well as technetium-99 labeled forms, which expires in 2029. Additional U.S. patents are directed to various inventions relating to the product candidate, and corresponding patent applications are pending worldwide.
With regard to our Relistor-related intellectual property, the composition-of-matter patent for the active ingredient of Relistor, methylnaltrexone, was invented in the 1970's and has expired. The University, as well as Progenics and its collaborators, have extended the methylnaltrexone patent estate with additional patents and pending patent applications covering various inventions relating to the product. Salix has listed in the FDA Orange Book four U.S. patents relating to subcutaneous Relistor, which have expiration dates ranging from 2017 to 2030, and one patent (expiring in 2024) with Health Canada. A patent issued in September 2013 provides protection for the oral methylnaltrexone product until 2031.
We depend on intellectual property licensed from third parties and unpatented technology, trade secrets and confidential information. If we lose any of these rights, including by failing to achieve milestone requirements or to satisfy other conditions, or if they or data embodying or relevant to them are compromised by disruptions or breaches of information or data security, our business, results of operations and financial condition could be harmed.
Most of our product candidates, including Relistor, incorporate intellectual property licensed from third parties. For example, PSMA ADC utilizes technology licensed to us from Sloan-Kettering Institute offor Cancer Research, through Cytogen Corporation, and Seattle Genetics, Inc. We can lose the right to patents and other intellectual property licensed to us if the related license agreement is terminated due to a breach by us or otherwise. Our ability, and that of our collaboration partners, to commercialize products incorporating licensed intellectual property would be impaired if the related license agreements were terminated. In addition, we are required to make substantial cash payments, achieve milestones and satisfy other conditions, including filing for and obtaining marketing approvals and introducing products, to maintain rights under our intellectual property licenses. Due to the nature of these agreements and the uncertainties of research and development, we may not be able to achieve milestones or satisfy conditions to which we have contractually committed, and as a result may be unable to maintain our rights under these licenses. If we do not comply with our license agreements, the licensors may terminate them, which could result in England. Dr. Babich receivedour losing our rights to, and therefore being unable to commercialize, related products.
We also rely on unpatented technology, trade secrets and confidential information. Third parties may independently develop substantially equivalent information and techniques or otherwise gain access to our technology or disclose our technology, and we may be unable to effectively protect our rights in unpatented technology, trade secrets and confidential information. We require each of our employees, consultants and advisors to execute a B.S. degreeconfidentiality agreement at the commencement of an employment or consulting relationship with us. These agreements may, however, not provide effective protection in the event of unauthorized use or disclosure of confidential information. Any loss of trade secret protection or other unpatented technology rights could harm our business, results of operations and financial condition.
Progenics and other businesses and organizations worldwide, and in particular technology-intensive activities such as biotechnology research and development, are increasingly dependent on critical, complex and interdependent information technology systems, including Internet-based systems, to facilitate or perform basic research and development functions, business processes, internal and external communications, and other critical functions. Progenics relies on such systems for most aspects of its business. The size and complexity of computer, communications and other electronic networked data generation, storage and transfer systems make them potentially vulnerable to breakdown, malicious intrusion, computer viruses and data security breaches by unauthorized third parties, employees or others. Such events may permit unauthorized persons to access, misappropriate and/or destroy sensitive data and result in the impairment or disruption of important business processes, loss of trade secrets or other proprietary intellectual property or public exposure of personal information (including sensitive personal information) of employees, business partners, clinical trial patients, customers and others. Any of the foregoing could have a material adverse effect on our business, prospects, operating results, and financial condition.
If we do not achieve milestones or satisfy conditions regarding some of our product candidates, we may not maintain our rights under related licenses.
We are required to make substantial cash payments, achieve milestones and satisfy other conditions, including filing for and obtaining marketing approvals and introducing products, to maintain rights under our intellectual property licenses. Due to the nature of these agreements and the uncertainties of research and development, we may not be able to achieve milestones or satisfy conditions to which we have contractually committed, and as a result may be unable to maintain our rights under these licenses. If we do not comply with our license agreements, the licensors may terminate them, which could result in our losing our rights to, and therefore being unable to commercialize, related products.
If we infringe third-party patent or other intellectual property rights, we may need to alter or terminate a product development program.
There may be patent or other intellectual property rights belonging to others that require us to alter our products, pay licensing fees or cease certain activities. If our products infringe patent or other intellectual property rights of others, the owners of those rights could bring legal actions against us claiming damages and seeking to enjoin manufacturing and marketing of the affected products. If these legal actions are successful, in addition to any potential liability for damages, we could be required to obtain a license in order to continue to manufacture or market the affected products. We may not prevail in any action brought against us, and any license required under any rights that we infringe may not be available on acceptable terms or at all. We are aware of intellectual property rights held by third parties that relate to products or technologies we are developing. For example, we are aware of other groups investigating PSMA or related compounds, monoclonal antibodies directed at PSMA and targets relevant to PSMA ADC, and methylnaltrexone and other peripheral opioid antagonists, and of patents held, and patent applications filed, by these groups in those areas. While the validity of these issued patents, patentability of these pending patent applications and applicability of any of them to our programs are uncertain, if asserted against us, any related patent or other intellectual property rights could adversely affect our ability to commercialize our products.
Research, development and commercialization of a biopharmaceutical often requires choosing between alternative development and optimization routes at various stages in the development process. Preferred routes depend on subsequent discoveries and test results and cannot be predicted with certainty at the outset. There are numerous third-party patents in our field, and we may need to obtain a license under a patent in order to pursue the preferred development route of one or more of our products or product candidates. The need to obtain a license would decrease the ultimate profitability of the applicable product. If we cannot negotiate a license, we might have to pursue a less desirable development route or terminate the program altogether.
We are dependent upon third parties for a variety of functions. These arrangements may not provide us with the benefits we expect.
We rely on third parties to perform a variety of functions. We are party to numerous agreements which place substantial responsibility on clinical research organizations, consultants and other service providers for the development of our approved product and our product candidates. We also rely on medical and academic institutions to perform aspects of our clinical trials of product candidates. In addition, an element of our research and development strategy has been to in-license technology and product candidates from academic and government institutions in order to minimize investments in early research. We have entered into agreements under which we are now dependent on Ono and Salix for the commercialization and development of Relistor. We may not be able to maintain our relationships with them, or establish new ones for Relistor or other product candidates on beneficial terms. We may not be able to enter new arrangements without undue delays or expenditures, and these arrangements may not allow us to compete successfully. Moreover, if third parties do not successfully carry out their contractual duties, meet expected deadlines or conduct clinical trials in accordance with regulatory requirements or applicable protocols, our product candidates may not be approved for marketing and commercialization or such approval may be delayed. If that occurs, we or our collaborators will not be able, or may be delayed in our efforts, to commercialize our product candidates.
We lack sales and marketing infrastructure and related staff, which will require significant investment to establish and in the meantime may make us dependent on third parties for their expertise in this area.
We have no established sales, marketing or distribution infrastructure. If we receive marketing approval for a pharmaceutical sciencesproduct, significant investment, time and managerial resources will be required to build the commercial infrastructure required to market, sell and support it. Should we choose to commercialize a product directly, we may not be successful in developing an effective commercial infrastructure or in achieving sufficient market acceptance. Alternatively, we may choose to market and sell products through distribution, co-marketing, co-promotion or licensing arrangements with third parties. We may also consider contracting with a third party professional pharmaceutical detailing and sales organization to perform the marketing function for one or more products. To the extent that we enter into distribution, co-marketing, co-promotion, detailing or licensing arrangements for the marketing and sale of product candidates, any revenues we receive will depend primarily on the efforts of third parties. We will not control the amount and timing of marketing resources these third parties devote to our products.
We are exposed to product liability claims, and in the future may not be able to obtain insurance against claims at a reasonable cost or at all.
Our business exposes us to product liability risks, which are inherent in the testing, manufacturing, marketing and sale of pharmaceutical products. We may not be able to avoid product liability exposure. If a product liability claim is successfully brought against us, our financial position may be adversely affected. Under our license agreement with Salix, we are responsible for product liability claims arising out of clinical trials that were conducted under our supervision. We are indemnified by Salix under our license agreement with Salix for product liability exposure arising from St. John's University, an M.S.its marketing and sales of Relistor, and maintain our own product liability insurance coverage in the amount of $10.0 million per occurrence, subject to a deductible and a $10.0 million annual aggregate limitation and other clinical trial or other insurance as required by contract and local laws. Pursuant to our transition agreement with Wyeth Pharmaceuticals, we released Wyeth from its indemnification responsibility for product liability exposure arising from its marketing and sales of Relistor. Product liability insurance for the biopharmaceutical industry is generally expensive, when available at all, and may not be available to us at a reasonable cost in the future. Our current insurance coverage and indemnification arrangements may not be adequate to cover claims brought against us, and are in any event subject to the insuring or indemnifying entity discharging its obligations to us.
We handle hazardous materials and must comply with environmental laws and regulations, which can be expensive and restrict how we do business. If we are involved in a hazardous waste spill or other accident, we could be liable for damages, penalties or other forms of censure.
Our research and development work and manufacturing processes involve the use of hazardous, controlled and radioactive materials. We are subject to federal, state and local laws and regulations governing the use, manufacture, storage, handling and disposal of these materials. Despite procedures that we implement for handling and disposing of these materials, we cannot eliminate the risk of accidental contamination or injury. In the event of a hazardous waste spill or other accident, we could be liable for damages, penalties or other forms of censure. We may be required to incur significant costs to comply with environmental laws and regulations in the future.
If we lose key management and scientific personnel on whom we depend, our business could suffer.
We are dependent upon our key management and scientific personnel, the loss of whom could require us to identify and engage qualified replacements, and could cause our management and operations to suffer in the interim. Competition for qualified employees among companies in the biopharmaceutical industry is intense. Future success in our industry depends in significant part on the ability to attract, retain and motivate highly skilled employees, which we may not be successful in doing.
Heath care reform measures could adversely affect our operating results and our ability to obtain marketing approval of and to commercialize our product candidates.
In the U.S. and some foreign jurisdictions, there have been a number of legislative and regulatory changes and proposed changes regarding the health care system that could prevent or delay marketing approval of our product candidates, restrict or regulate post-approval activities and affect our ability to profitably sell any product candidates for which we obtain marketing approval. Legislative and regulatory proposals have been made to expand post-approval requirements and restrict sales and promotional activities for pharmaceutical products. In addition, increased scrutiny by the U.S. Congress of the FDA's approval process may significantly delay or prevent marketing approval, as well as subject us to more stringent product labeling and post-marketing testing and other requirements. In the U.S., federal legislation has changed the way Medicare covers and pays for pharmaceutical products. Cost reduction initiatives and other provisions of legislation have decreased coverage and reimbursement. Though such legislation applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own reimbursement rates. More recent legislation is intended to broaden access to health insurance, further reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for health care and health insurance industries, and impose new taxes and fees on the health industry and additional health policy reforms. New laws impose significant annual fees on companies that manufacture or import branded prescription drug products, and contain substantial new compliance provisions, which in each case may affect our business practices with health care practitioners. Subject to federal and state agencies issuing regulations or guidance, it appears likely that new laws will continue to pressure pharmaceutical pricing, especially under the Medicare program, and may also increase regulatory burdens and operating costs. We cannot be sure whether additional legislative changes will be enacted, whether the FDA regulations, guidance or interpretations will be changed or what the impact of such changes on the marketing approvals of our product candidates, if any, may be.
Our and/or our collaborators' relationships with customers and third-party payors will be subject to applicable anti-kickback, fraud and abuse and other healthcare laws and regulations, which could expose us or them to criminal sanctions, civil penalties, program exclusion, contractual damages, reputational harm and diminished profits and future earnings.
Health care providers, physicians and third-party payors play a primary role in the recommendation and prescription of any product candidates for which we obtain marketing approval. Our or our collaborators' future arrangements with third-party payors and customers may expose us or them to broadly applicable fraud and abuse and other health care laws and regulations that may constrain the business or financial arrangements and relationships through which we or our collaborators market, sell and distribute our products that obtain marketing approval. Efforts to ensure that business arrangements comply with applicable health care laws and regulations involve substantial costs. It is possible that governmental authorities will conclude that our or our collaborators' business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If such operations are found to be in violation of any of these laws or other applicable governmental regulations, we or the collaborator may be subject to significant civil, criminal and administrative penalties, damages, fines, exclusion from government funded healthcare programs, such as Medicare and Medicaid, and the curtailment or restructuring of related operations. If physicians or other providers or entities involved with our products are found to be not in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs, which may adversely affect us.
We no longer derive significant funds from federal government grants and research contracts.
While we have in the past been awarded federal government grants and research contracts, in particular from the UniversityNational Institutes of Southern California,Health, we do not currently have significant funding from such sources, and a Ph.D. fromdo not expect to apply for such funding in the University of London.foreseeable future.
Our future depends on the proper management of our current and future business operations, including those of Molecular Insight, and their associated expenses.
Our business strategy requires us to manage our business to provide for the continued development and potential commercialization of our proprietary and partnered product candidates. Our strategy also calls for us to undertake increased research and development activities and to manage an increasing number of relationships with partners and other third parties, while simultaneously managing the capital necessary to support this strategy. These tasks are significantly increased as a result of our acquisition of Molecular Insight. If we are unable to manage effectively our current operations and any growth we may experience, our business, financial condition and results of operations may be adversely affected. If we are unable to effectively manage our expenses, we may find it necessary to reduce our personnel-related costs through reductions in our workforce, which could harm our operations, employee morale and impair our ability to retain and recruit talent. Furthermore, if adequate funds are not available, we may be required to obtain funds through arrangements with partners or other sources that may require us to relinquish rights to certain of our technologies, products or future economic rights that we would not otherwise relinquish or require us to enter into other financing arrangements on unfavorable terms.
Progenics has a history of operating losses, as does Molecular Insight, which has also been reorganized under the U.S. Bankruptcy Code.
Progenics has incurred substantial losses throughout its history. A large portion of our revenue has historically consisted of upfront and milestone from licensing transactions. We have reported operating losses for 2013 and 2012 and while we reported operating income for 2011, as a result of a one-time upfront payment from Salix, the timing and amount of any similar transactions in the future is highly unpredictable and uncertain. Without upfront or other such payments, we operate at a loss, due in large part to the significant research and development expenditures required to identify and validate new product candidates and pursue our development efforts. Moreover, we have derived no significant revenue from product sales and have only in the last several years derived revenue from royalties. We may not achieve significant product sales or royalty revenue for a number of years, if ever. We expect to incur net operating losses and negative cash flow from operations in the future, which could increase significantly if we expand our clinical trial programs and other product development efforts, including those attendant to the product candidates and programs originally developed by Molecular Insight. Our ability to achieve and sustain profitability is dependent in part on obtaining regulatory approval for and then commercializing our product candidates, either alone or with others. We may not be able to develop and commercialize products beyond subcutaneous Relistor for OIC in patients with advanced illness. Our operations may not be profitable even if any of our other product candidates under development are commercialized.
Molecular Insight incurred net losses every year from its inception in 1997 and generated no significant revenue from product sales and only limited revenue from licenses. In December 2010, Molecular Insight filed a voluntary petition in the United States Bankruptcy Court for the District of Massachusetts seeking relief under the provisions of Chapter 11 of the U.S. Bankruptcy Code (Case No. 10-23355). It operated its business and managed its properties as a debtor in possession under bankruptcy protection until emerging from bankruptcy in May 2011.
Dr. Israel, 56, Senior Vice President, Medical Affairs & Clinical Research, has been with Progenics since 1994. He previously held oncology positions at Sandoz Pharmaceuticals Corporation and Schering-Plough Corporation. Dr. Israel is a licensed physician and is board certified in internal medicine and medical oncology. He received a B.A. in physics from Rutgers University and an M.D. from the University of Pennsylvania, and completed an oncology fellowship at the Memorial Sloan-Kettering Cancer Center. Dr. Israel has been a consultant to the Solid Tumor Service at Sloan-Kettering.
Our ability to use net operating losses to offset future taxable income is subject to certain limitations.
Dr. Olson, 50,We currently serveshave significant net operating losses (NOLs) that may be used to offset future taxable income. The U.S. Internal Revenue Code limits the amount of taxable income that may be offset annually by NOL carryforwards after a change in control (generally greater than 50% change in ownership) of a loss corporation, and our use of NOL carryforwards may be further limited as Senior Vice President, Research & Development. He alsoa result of any future equity transactions that result in an additional change of control.
Progenics' stock price has been witha history of volatility and may be affected by selling pressure, including in the Company since 1994,event of substantial sales of Progenics stock by former Molecular Insight stockholders. You should consider an investment in Progenics stock as risky and wasinvest only if you can withstand a scientist at Johnson & Johnson and MicroGeneSys, Inc.,significant loss.
Our stock price has a biotechnology company, for five years before joining Progenics. Dr. Olsonhistory of significant volatility. It has served on scientific review committeesvaried between a high of the National Institutes of Health and has co-authored more than 40 articles for peer-reviewed scientific journals. He received a Ph.D. from the Massachusetts Institute of Technology$6.47 and a B.S. from the Universitylow of North Dakota, both$2.53 in chemical engineering.
Dr. Ray, 60, Senior Vice President, Manufacturing, joined Progenics in 2001. Prior to joining Progenics, Dr. Ray served as Director2013 and between a high of Bioprocess Development at Ortec International, and before that held positions of increasing responsibility at Hoffmann-La Roche in the areas of GMP Manufacturing and Process Development, ultimately as Research Leader, Biopharmaceuticals. Early in his career he developed process technology for biopharmaceutical manufacturing at Verax Corporation. Dr. Ray received an M.S. and Ph.D. in chemical and biochemical engineering from Rutgers University$11.34 and a B.S.low of $1.41 in chemical engineering from Jadavpur University, India.
Ms. Assumma, 59, joined Progenics2012. Factors that may have a significant impact on the market price of our common stock include the results of clinical trials and pre-clinical studies undertaken by us or others; delays, terminations or other changes in 2004 and currently servesdevelopment programs; developments in marketing approval efforts, such as Vice President, Regulatory Affairs. She has over 30 years' experience in the pharmaceutical industry and regulatory affairs at companies including Dov Pharmaceuticals, Emisphere Technologies, Bayer and American Cyanamid. Ms. Assumma earned her M.S. in pharmacology from New York Medical College and a B.S. in biology from Fordham University.
Mr. Lovallo, 48, was appointed Treasurer in 2011 and Vice President, Finance this year. Before joining Progenics in 2008 as Senior Director, Financial Reporting, he held Vice President, Financial Reporting positions at MBIA Inc. from 2004 to 2008 and AllianceBernstein from 1998 to 2004. Earlier in his career, he was in accounting policy at Salomon Smith Barney and a senior manager at KPMG LLP. He received a B.B.A. in public accounting from Hofstra University and is a Certified Public Accountant.
Dr. Wong, 56, currently serves as Vice President, Product Development. For three years prior to joining Progenics in 2007, Dr. Wong was Principal at Theritas Pharmaceutical Consultants. From 1989 to 2004, she held positions of increasing responsibility in preclinical development and pharmacology at Emisphere Technologies, Vivoquest, and Regeneron Pharmaceuticals. Dr. Wong has been a co-author on over 30 scientific articles for peer-reviewed journals. She received a B.Sc. in biology from the Mississippi University for Women, a Ph.D. in anatomy and neurobiology from the University of Maryland School of Medicine, and completed a postdoctoral fellowship in neurology at the Albert Einstein College of Medicine.
Mr. Martin, 58, General Counsel, joined Progenics in 2008 as Associate General Counsel after practicing corporate and finance law in New York City for over 25 years including with Loeb & Loeb, AXA Equitable, Dewey Ballantine and in his own special clients practice. He holds a B.A. in mathematics and history from the University of Pennsylvania and a J.D. from the Columbia University School of Law.
Section 16(a) Beneficial Ownership Reporting Compliance
Based solely on a review of the reports under Section 16(a) of the Exchange Act and representations furnished to usFDA's July 2012 Complete Response Letter with respect to the last fiscal year, we believesNDA for Relistor subcutaneous injection for the treatment of OIC in adult patients with chronic, non-cancer pain; developments in collaborator or other business relationships, particularly regarding Relistor, PSMA ADC or other significant products or programs; technological innovation or product announcements by us, our collaborators or our competitors; patent or other proprietary rights developments; governmental regulation; changes in reimbursement policies or health care legislation; safety and efficacy concerns about products developed by us, our collaborators or our competitors; our ability to fund ongoing operations; fluctuations in our operating results; general market conditions; and the reporting of or commentary on such matters by the press and others. At times, our stock price has been volatile even in the absence of significant news or developments. The stock prices of biotechnology companies and securities markets generally have been subject to dramatic price swings in recent years, and financial and market conditions during that eachperiod have resulted in widespread pressures on securities of issuers throughout the persons required to file such reports is in compliance with all applicable filing requirements. We continue to monitorworld economy.
Our stockholders may be diluted, and the effectivenessprice of our policies and procedures designed to ensure compliance with Section 16 reporting requirements.
Codecommon stock may decrease, as a result of Business Ethics and Conduct
We have a Codefuture issuances of Business Ethics and Conduct which is applicable to allsecurities, exercises of our directors, employees and consultants. The Code meets the criteria for a "codeoutstanding stock options, or sales of ethics" under the SEC rules and "code of conduct" under the NASDAQ Marketplace rules. The Code is described in more detail under Item 13 - Certain Relationships and Related Transactions, and Director Independance, and is available on our website at http://www.progenics.com/documents.cfm.
outstanding securities.
Certain Audit Committee Matters
The Company's Audit Committee of the Board of Directors, consisting of Ms. Williams, Chair, Mr. Charles Baker and Dr. Scheinberg, reviewsWe expect to issue additional common stock in public offerings, private placements and/or through our annual financial statements priorJanuary 2014 Sales Agreement with Cantor Fitzgerald & Co., pursuant to their submissionwhich we may sell from time to the SEC, consults with our independent auditors and examines and considers such other matters in relationtime up to the audit$50 million of our financial statementsstock, and in relation to issue options to purchase common stock for compensation purposes. We may issue preferred stock, restricted stock units or securities convertible into or exercisable or exchangeable for our financial affairs, includingcommon stock. All such issuances would dilute existing investors and could lower the selection and retentionprice of our independent auditors. It is responsible for oversightcommon stock. Sales of the Company's internal auditor, who reports directly to it, oversees the worksubstantial numbers of management to identify, assess and monitor risk, and liaises with management and the Board in risk mitigation efforts. The Board has also determined that each memberoutstanding shares of the Audit Committee meets the independence requirements prescribedcommon stock, such as sales by the NASDAQ Marketplace rules, the SEC and the Internal Revenue Service. The Board has determined that Ms. Williams is an "audit committee financial expert" as such term is defined in Item 404(d)(5)former Molecular Insight stockholders of Regulation S-K.
Item 11. Executive Compensation.
Compensation Discussion and Analysis
Progenics is a biotechnology company engaged in drug development and commercialization, focusing on oncology. We compete with biopharmaceutical companies of all sizes to attract employees with the skills and expertise necessary to develop and commercialize drugs and achieve our objectives. Since the funds we can use for compensation are limited, we have worked to develop a compensation program that allows us to attract and retain talented individuals with the essential experience and skills we need at the executive level while being mindful of our limited resources. We have employed a philosophy which combines base salary with bonus compensation and long-term equity incentives in the form primarily of stock options and in the past also in restricted stock. We strive to conserve cash resources by setting base salaries and total cash compensation at what we deem an appropriate level in view of market compensation data in our industry while providing meaningful long-term equity opportunity.
This Compensation Discussion and Analysis (CD&A) outlines, among other things, our compensation philosophy, objectives and processes as they relate to our Named Executive Officers (NEOs) in 2012: Mr. Baker (CEO); Vice President and Treasurer Mr. Lovallo (PFO); Mr. McKinney (CFO until September 30, 2012); Senior Vice Presidents Drs. Israel and Olson; Vice President Ms. Assumma; and Dr. Maddon (Vice Chairman and CSO until March 14, 2012; now a non-employee Vice Chairman).
The Company's Compensation Committee of the Board of Directors makes recommendations concerning salaries and incentive compensation for our employees and consultants, establishes and approves salaries and incentive compensation for our executive officers and other senior employees, administers our stock option plans and otherwise seeks to ensure that our compensation philosophy is consistent with our best interests and is properly implemented. Mr. Crowley is Chair of this Committee.
Overview. Progenics made significant progress in achieving strategic objectives and program development in 2012, but as discussed below two important goals related to our first commercial drug, Relistor®, which we repartnered with Salix Pharmaceuticals in early 2011, were not achieved.
Beginning in late 2011, after a strategic review of our programs and operations and continuing to the present, we have reoriented our strategic focus on the oncology space and devoted significant efforts to enhancing and expanding this area of our existing and prospective business. Our principal efforts in 2012 were to (a) move forward our oncology drug candidate programs, particularly our PSMA ADC product candidate for prostate cancer, and early research in proprietary phosphoinositide 3-kinase (PI3K) inhibitors (which we are seeking to partner); (b) identify and pursue a number of opportunities to expand our oncology pipeline through in-licensing and acquisitions, culminating early this yearunregistered shares received in the acquisition of Molecular Insight; and (c) evaluate internally developed programs and technologies outside our oncology focus for possibleadvancement with other parties,(including shares held in a 15-month escrow which resultedexpires in our partnering our proprietary C.difficile research program and our PRO 140 HIV viral-entry inhibitor.
At the same time, we continued to focus on controlling costs and achievingApril 2014), could also cause a sustainable cash burn rate, and redoubled those efforts after, as previously announced, the U.S. Food and Drug Administration issued a Complete Response Letter for a supplemental New Drug Application for an expanded Relistor indication. We also completed a follow-on public offering of common stockdecline in the fourth quarter of 2012 to provide funding for our R&D and strategic efforts.
The compensationmarket price of our executives reflected the performance of the Company during 2012.stock. We believe overall Company compensation has historically been highly correlatedrequire substantial external funding to Company performance. We have adopted a formal annual incentive plan based on established metrics and administered by the Compensation Committee to provide a framework to determine annual bonus payments to NEOs and other executives. This plan is used to establish an even stronger linkage between compensation and individual and Company performance. As part of that process, we established specific annual goals for the Company and for each NEO, and, as discussed below, determined bonuses against specific target bonuses established for each NEO taking into account how well the Company and the executive performed against the established goals as determined by the Compensation Committee.
The Committee believes that the total compensation earned by the NEOs and approved by the Committee appropriately reflect the performance of the executives and the Company and is consistent with our compensation philosophy.
Executive Compensation Objectives. We seek to achieve the following broad goals in our executive compensation programs and decisions regarding individual compensation:
· | Attract and retain executives critical to our overall success. |
· | Reward executives for contributions to achieving strategic goals that enhance stockholder value. |
· | Foster and maintain a company culture of ownership, creativity and innovation. |
· | Motivate our NEOs to achieve critical long- and short-term development, product and financial milestones set by the Board and management. |
General Compensation Process. The Compensation Committee is responsible for determining the elements and levels of compensation for our NEOs. In doing so, it reviews our corporate performance against financial and corporate achievement measures, assesses individual performance and evaluates recommendations of the CEO regarding compensation for other NEOs.
In assessing Company and individual NEO performance, the Committee has considered the progress of the Company in achieving its strategic goal of being a leading oncology biotechnology company; research, development and commercialization of its product candidates; and the contributions made by each NEO to those efforts. Recent examples of this progress include advancing our PSMA ADC clinical development program; partnering selected proprietary programs and technologies; identifying and executing on opportunities complementary to our focus on oncology; and managing our relationships with collaborators and others.
The process the Committee followed in assessing the NEOs and the Company's performance for 2012 began, as has been the Committee's practice in the past, with meetings in February 2012. These meetings were focused on compensation for the NEOs and were principally focused on approving bonuses relating to 2011 performance and establishing 2012 salary levels. Bonus targets for 2012 performance were established in mid-year. In those meetings, Mr. Baker was invited to make an oral presentation and submit to the Committee written materials regarding the performance of the NEOs and other executive officers, his views regarding the performance of the Company and his assessment of his own performance. These presentations and the Committee's assessment considered activities for 2011 and 2012 as described above. In February and March 2012 meetings, the Committee also considered the possible retirement of Dr. Maddon as CSO, which was implemented when the Company and Dr. Maddon entered into a retirement agreement.
In 2012 the Committee conferred with its then-compensation consultant, Pearl Meyer & Partners, to establish target compensation for the Company's NEOs. Pearl Meyer provided to the Committee a report outlining competitive market compensation data for consideration when determining different levels and mix of compensation. The primary data sources utilized for this purpose were publicly available compensation information of NEOs of a Peer Group of companies within the biotechnology industry, selected by the Committee with the consultant's assistance, that were similar to Progenics in size, stature and state of development. The Peer Group companies, reviewed by the Committee with the consultant's assistance to ensure that they remained relevant and meaningful comparators, consisted of:
Acorda Therapeutics, Inc. | Idenix Pharmaceuticals, Inc. |
Adolor Corp. | Immunomedics Inc. |
Allos Therapeutics Inc. | Infinity Pharmaceuticals Inc. |
Alnylam Pharmaceuticals Inc. | Nektar Therapeutics |
Ariad Pharmaceuticals Inc. | Neurocrine Biosciences, Inc. |
Array Biopharma Inc. | Sangamo Biosciences Inc. |
Dyax Corp. | Sucampo Pharmaceuticals, Inc. |
GTx Inc. | Synta Pharmaceuticals Corp. |
Changes in this year's Peer Group reflect Progenics' decreased market capitalization, as well as the acquisition of several peer group members by other companies. Peer Group data were supplemented with published executive compensation surveys providing position-based compensation data from biotechnology companies similar in size and scope to the Company.
In addition to Mr. Baker's recommendations regarding the other NEOs and its review of market data provided by its consultant, in setting 2012 target compensation opportunities the Committee considered other factors such as the individual's corporate roles and responsibilities, particular experience and expertise, performance and specific duties, the scope of his or her position and the department(s) or group(s) for which he or she had responsibility, the Company's overall corporate financial performance and the progress offinance our research and development programs and strategic initiatives duringmay seek such funding through the year.
Having reviewedissuance and sale of our common stock, which we have recently done in follow-on primary offerings in late 2012, mid-2013 and February 2014. We have recently established a new shelf registration statement, which we used for our most recent offering and may be used to issue up to approximately an additional $110 million of common stock and other securities before any underwriter discounts, commissions and offering expenses. We also have in place registration statements covering shares issuable pursuant to our equity compensation plans, and sales of our securities under them could cause the market data and taken into consideration all other pertinent information, the Committee made decisions regarding 2012 base salaries and target bonuses by majority vote. Mr. Baker neither had a vote nor was present for decisions regarding his own compensation.
The Committee used a similar process at mid-year to determine the long-term incentive elementsprice of our NEO compensation program paid in stock to decline. Sales by existing stockholders or holders of options and restricted stock. The Committee considered Mr. Baker's recommendation for long-term incentive awards to theor other NEOs, and made its own determination of the appropriate long-term incentive awards for Mr. Baker. The Committee determined to make all such awards in respect of 2012 performance in the form of stock options, and issued these grants in the second quarter of 2013.
We believe that each NEO's 2012 compensation package is generally within the competitive range of practices when compared to objective comparative data even where qualitative factorsrights may have influenced our compensation decision.
In addition to providing market compensation information, Pearl Meyer advised the Committee on various aspects of the foregoing process, including Peer Group composition, comparisons of compensation components and levels, and compensation program structure. The Committee retained Pearl Meyer for its services in 2012 directly, although in carrying out assignments, the consultant interacted directly with our management when necessary and appropriate. In addition, in its discretion, Pearl Meyer sought input and feedback from our executives, primarily Mr. McKinney, regarding its work product prior to presentation to the Committee, in order to confirm alignment with the Company's business strategy and to obtain data or information necessary for its work. Pearl Meyer provided the Committee with an annual Compensation Report.
Elements of Compensation. We utilize a compensation strategy in line with that of other companies within the biotechnology industry which includes base salary, annual bonus, long-term incentives and retirement and severance benefits. These elements are designed to reward (i) the level of effort and competence demonstrated in light of the executive's duties and responsibilities (base salary), (ii) decision-making that supports our annual product, development and financial goals (annual bonus), and (iii) a focus on building shareholder value over the long term by making decisions that will not sacrifice long-term prospects for a particular short-term achievement or goal (long-term incentives).
Base Salary. Levels of base salary for our executives in general take into account an individual's role and responsibilities, experience, expertise, individual performance and tenure. The amount is typically at or slightly below the median industry compensation level for the position as shown by appropriate market data provided by the Committee's consultant. Relative to market data for other CEOs, Mr. Baker's salary of $517,500 was below the median of market data. The Committee determined this competitive positioning to be appropriate and reflective of Mr. Baker's tenure as CEO.
Annual Bonus. Beginning in the fourth quarter of each year, the Compensation Committee works collaboratively with senior management to develop corporate goals and objectives tied to strategic plans for the coming year. In 2012, this process was completed in mid-year, and established five corporate strategic and research and development goals and one operational and financial goal, with the weightings noted below, for use in determining cash bonuses:
· | FDA approval of RELISTOR for opioid induced constipation in patients with chronic, non-cancer pain (25%). |
· | Submission and acceptance of a New Drug Application filing with the FDA for approval of an oral formulation of methylnaltrexone (the active ingredient of Relistor) (25%). |
· | Significant enhancement of the Company's pipeline through in-license or acquisition (10%). |
· | Initiation of a phase 2 trial of the Company's PSMA ADC product candidate and advancement of the program to the satisfaction of the Committee (20%).
|
· | Advancement of the Company's research program to the satisfaction of the Committee (10%). |
· | Achievement of a cash burn amount not in excess of the amount provided in the Company's 2012 budget (10%). |
These corporate goals, along with individual goals for each NEO, were used in a program measuring performance over the course of the year. Target bonus amounts were based on a percentage of base salaries and are generally set near the target bonus amounts for comparable positions in our Peer Group. In 2012, a bonus target of 50% of base salary, based entirely on achievement of corporate goals, was established for Mr. Baker. For the other NEOs, each of whom had a target maximum bonus set by Mr. Baker, 75% of the bonus amount was attributed to achievement of corporate goals and 25% to individual goals, in the case of Senior Vice Presidents; for Vice Presidents, the percentages were 67% and 33%, respectively.
Based on the overall percentage of corporate goal achievement determined by the Committee during the first quarter of the following year and the percentage of individual goal achievement of each NEO determined by Mr. Baker and reviewed by the Committee, actual bonus payments may be above or below bonus target amounts in the Committee's discretion based on actual performance. Bonus amounts for NEOs other than Mr. Baker are recommended by Mr. Baker and reviewed by the Committee, and the Committee determines the bonus amount for Mr. Baker. Bonuses are generally paid in the first quarter of the following year.
For 2012, the Committee approved a corporate achievement percentage of 60% of target based on the Company:
· | Not meeting the subcutaneous RELISTOR chronic, non-cancer pain sNDA approval goal. |
· | Not meeting the oral methylnaltrexone NDA submission goal. |
· | Not meeting the significant pipeline enhancement goal in 2012, but meeting the goal in January 2013. |
· | Meeting the goal of initiating a phase 2 trial of PSMA ADC and the advancement criterion. |
· | Meeting the research program advancement goal. |
· | Substantially meeting the 2012 cash burn goal by advancing our strategic focus on oncology and partnering selected proprietary research and development programs while controlling research expense, headcount and other costs. |
For the individual NEOs, the Company's analyses of performance were as follows:
Mr. Baker. Compensation decisions regarding Mr. Baker in 2012 reflected his capacity as CEO and the breadth of his leadership responsibilities. Based entirely on the percentage achievement of the corporate goals and his target bonus of 50% of base salary, Mr. Baker was awarded a bonus of $155,250, equal to 30% of his 2012 annual salary.
Mr. Lovallo. Mr. Lovallo's contributions during 2012 included assuming additional responsibilities as the Company's principal financial officer. His individual goals included facilitating the transition to a new outside audit and tax firm, managing financial components of corporate transactions, and supervising the Company's accounting, treasury and financial reporting functions as PFO. For Mr. Lovallo and the other NEOs other than Mr. Baker, the Compensation Committee accepted Mr. Baker's determination of the NEO's individual contribution component. Based on the percentage achievement of his individual and the corporate goals, and his target bonus of 30% of base salary, Mr. Lovallo was awarded a bonus of $56,120, equal to 22% of his 2012 annual salary.
Mr. McKinney. Mr. McKinney stepped down from his positions with the Company in September and did not receive an annual bonus for 2012.
Dr. Israel. Dr. Israel's contributions during 2012 included his leadership of the effort to achieve the Company's R&D goals during the year. His individual goals included efforts to identify in-license and acquisition candidates, managing medical affairs aspects of our relationships with Relistor collaboration partners, and supporting the change in the Company's strategic focus. Based on the percentage achievement of his individual and the corporate goals, and his target bonus of 30% of base salary, Dr. Israel was awarded a bonus of $83,455, equal to 21% of his 2012 annual salary.
Dr. Olson. Dr. Olson's contributions during 2012 included leadership of the Company's R&D efforts in oncology. His individual goals included investigational goals with respect to specific research projects and contributions to strategic and business development initiatives. Based on the percentage achievement of his individual and the corporate goals, and his target bonus of 30% of base salary, Dr. Olson was awarded a bonus of $65,941, equal to 21% of his 2012 annual salary.
Ms. Assumma. Ms. Assumma's contributions during 2012 included supervision of regulatory compliance. Her individual goals included work on Relistor and PSMA ADC regulatory submissions and strategic and business development initiatives. Based on the percentage achievement of her individual and the corporate goals, and her target bonus of 30% of base salary, Ms. Assumma was awarded a bonus of $61,592, equal to 22% of her 2012 annual salary.
Dr. Maddon. During the first quarter of 2012, Dr. Maddon and the Company set the terms for his retirement from the Company which became effective in June.
In addition to the bonus plan described above, the Committee has the authority to approve discretionary bonuses to any NEO. This discretion was not used in 2012, as the Committee felt that the new bonus plan met its objectives and no further bonuses were required.
Long-term incentives. Long-term incentives may include both non-qualified stock options and restricted stock awards. During 2012, we made option awards to NEOs under our 2005 Stock Incentive Plan, the terms of which are described in this Proxy Statement and our 2012 Annual Report on Form 10-K. Stock options vesting in equal annual installments over three years from the date of grant subject to continued employment were granted at market exercise prices. Mr. Baker was also awarded 33,334 performance-vesting stock options. Long-term incentive awards granted to our NEOs during 2012 and awards outstanding from prior grants are presented in the Grants of Plan-Based Awards in 2012 and Outstanding Equity Awards at Fiscal Year-End tables, below.
Although our long-term incentive grants have typically been time-vested instruments, we have in some years issued performance based stock options to our CEO. The Company's increased reliance on goals-based incentive compensation, however, may be expected to result in the use of fewer performance based awards. Dr. Maddon received several performance grants during his tenure, and Mr. Baker has received performance-based stock option grants which the Committee has used to align more closely his compensation with the business goals established by the Committee and the Board. Vesting of a percentage of Mr. Baker's awards occurs upon the Committee's certification of achievement of defined pre-established performance conditions reflecting significant regulatory, commercial and business development Company goals. The percentages of the award that vest, if every performance condition is achieved, total more than 100% of the award, recognizing that the Company is pursuing multiple goals and that not every performance condition must be achieved in order for his performance to be considered sufficient to justify full vesting of the award. Even if the percentage of the award that vests for the performance conditions actually achieved exceeds 100%, however, he is entitled to only 100% of the award. As of 2012 year-end, 196,875 of Mr. Baker's 2010 and 2011 performance-based options had vested.
Other Considerations. When determining amounts of long-term incentive grants to our NEOs, the Committee compares (i) the value of the grant with the value of comparable grants made to NEOs in our Peer Group; (ii) the number of incentive units granted by position as a percentage of total common shares outstanding, compared with the applicable percentages of comparable grants made to NEOs in our Peer Group; and (iii) the executive's overall equity incentive opportunity. We believe these comparisons provide a meaningful context for comparing the competitive level of our equity based compensation practices to those of companies in our Peer Group and ensure that we are not at a competitive disadvantage in terms of hiring or retaining key executive talent.
We make annual equity awards near the beginning of the year (typically February) to coincide with other compensation decisions. Each NEO received a 2013 grant of stock options in April 2013; we do not expect they will receive another annual grant before 2014. We also award options to newly-hired employees, and may from time to time issue equity-based compensation at other times during the year.
Retirement, Welfare Benefits. We make available to our NEOs retirement and welfare benefits, consisting of partial matching contributions to 401(k) retirement plans and access to medical, dental and other welfare plans, all of which are available to all full-time employees. NEOs also receive reimbursement of premiums for enhanced life and disability insurance, totaling less than $10,000 per NEO, which are the only other perquisites given to our NEOs. This philosophy is consistent with our view that company resources are generally best utilized in research, development and commercialization efforts. The total of retirement and welfare benefits for each NEO is presented in the Summary Compensation Table, below.
Equity Ownership by Executives. We do not currently have a formal stock ownership requirement for executives, but we encourage stock ownership by executives on a voluntary basis and through participation in the Stock Incentive Plan. Our NEOs have vested and unvested stock options and unvested restricted stock as shown in the Outstanding Equity Awards at Fiscal Year-End table.
Employment Agreements. We have no employment agreements with any of our NEOs except Dr. Israel, whose agreement, entered into in 1994, provides for severance of nine months' salary and benefits upon termination without cause, ceasing upon his securing new employment. The agreement contains restrictive covenants against disclosure of confidential business information, solicitation of employees and customers and competition with our business, and establishes our right to inventions and intellectual property. The March, 2011 amendment and March 2012 termination of Dr. Maddon's former employment agreement modified his relationship with the Company consistent with his continuing roles and retirement during that period; provisions of the agreement relating to confidentiality, indemnification and insurance, and certain technical and other matters, continue in effect notwithstanding the termination.
Compensation and Risk. The Company does not believe, given the nature of our activities and the manner in which our employees are compensated, that risks arising from our compensation policies and practices relating to all of our employees are reasonably likely to have a material adverse effect on the Company.
As noted above, Mr. Baker has received performance based options, which vest uponour ability to raise capital and subject to achievement of Company milestones. He could have a financial incentive to accelerate achievement of milestones relevant to these options, even if that was not in the best interests of the Company. To the extent that milestones established in earlier grants cease to reflect the Company's current goals, he may also have a financial incentive to achieve milestones no longer in the best interests of the Company.
The Committee has dealt with these potential conflicts by establishing multiple milestones in the annual incentive plan and for performance-based option grants, as a result of which annual incentive awards and options may be partially earned or become fully vested even if not every milestone is achieved. Further mitigating risk, annual incentives are capped at fixed percentages of target and our CEO will only vest into a fixed amount of options per award regardless of how many goals are met or achieved. The Board of Directors carefully monitors the Company's activities, including those relevant to achievement of respective milestones specified in the annual incentive plan and performance-based option grants, and believes that these awards have not adversely affected the Company or resulted in its taking actions not in the best interests of stockholders. As noted above, the Company's increased reliance on goals-based incentive compensation, however, may be expected to result in the use of fewer performance based awards.
Tax and Accounting Considerations. The compensation paid to our NEOs is generally subject to taxation at ordinary rates. Although we endeavor to structure our compensation packages so that they are not subject to tax penalties (such as additional taxes arising under Section 409A of the Internal Revenue Code), these efforts do not materially affect the terms of compensation arrangements.
Section 162(m) of the IRC limits deduction of compensation paid to eachmarket price of our NEOs to $1,000,000 unless the compensation is "performance-based," as defined in the Code. The Stock Incentive Plan allows the Committee to grant awards that will be exempt from the deduction limits of section 162(m) if certain criteria are satisfied. While the Committee considers the tax and accounting effect of the compensation programs, there may be times when the Committee accepts a less advantageous tax and accounting outcome in order to achieve other goals, such as motivating and retaining executives.
We have designed our stock incentive plans from which long-term incentive awards have been granted to be in compliance with generally-accepted accounting principles in order to avoid additional non-cash compensation charges.
2011 Shareholder Advisory Vote on Executive Compensationcommon stock.
Our principal stockholders votedare able to approve Progenics' 2011exert significant influence over matters submitted to stockholders for approval.
At 2013 year end, our directors and executive compensation by the advisory vote which was partofficers together beneficially owned or controlled approximately six percent of our 2012 Annual Meeting. We believe thatoutstanding common shares, including shares currently issuable upon option exercises, and our executive compensation practices are aligned with Company performancefive largest other stockholders approximately 52 percent. Should these parties choose to act alone or together, they could exert significant influence in determining the outcome of corporate actions requiring stockholder approval and market practice. Stockholders approvedotherwise control our business. This control could, among other things, have the compensationeffect of our 2011 NEOs by 98.6% of the votes cast.
We do not believe any changes in our executive compensation practices were made necessary as a result of the 2012 vote. We use the formal annual incentive plan described above to further align our executive compensation practices with the Company's performance and the interests of our stockholders.
Compensation Committee Report
The Compensation Committee has submitted the following report for inclusion in the Company's Proxy Statement:
Our Committee has reviewed and discussed the Compensation Discussion and Analysis contained in the Company's Proxy Statement with management. Based on our Committee's review of and the discussions with management with respect to the Compensation Discussion and Analysis, our Committee recommended to the Board that the Compensation Discussion and Analysis be included in the Company's Proxy Statement and includeddelaying or incorporated by reference in the Company's 2013 Annual Report on Form 10-K.
The foregoing report is provided by the following directors, who constitute the Committee:
By the Compensation Committee of the Board of Directors,
Peter J. Crowley, Chair
Charles A. Baker
Summary Compensation Table
The table and footnotes below describe the total compensation paid to our CEO, CFO, and three other most-highly compensated NEOs. As reflected in the table and discussed above in the CD&A, we compensate these executive officers with a combination of cash and equity compensation, the latter of which is presented in this table in dollar values (see note 2 and the tables following).
| | Year | | Salary | | | Bonus (1) | | | Stock Awards (2) | | | Option Awards(2) | | | Non-Equity Incentive Plan Compen- sation | | | All Other Compen-sation(3)(4) | | | Total |
| Mr. Baker | | 2012 | | $ | 517,500 | | | $ | 155,250 | | | | - | | | $ | 689,244 | | | | - | | | $ | 15,612 | | | $ | 1,377,606 |
| (CEO; President | | 2011 | | | 487,500 | | | | 312,500 | | | | - | | | | 1,685,158 | | | | - | | | | 37,906 | | | | 2,523,064 |
| before March 3, 2011) | | 2010 | | | 425,000 | | | | 350,000 | | | | - | | | | 883,275 | | | | - | | | | 46,360 | | | | 1,704,635 |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Mr. Lovallo (VP; PFO) | | 2012 | | | 236,025 | | | | 56,120 | | | | - | | | | 63,584 | | | | - | | | | 8,261 | | | | 363,990 |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Mr. McKinney | | 2012 | | | 245,979 | | | | - | | | | - | | | | 331,020 | | | | - | | | | 258,072 | | | | 835,071 |
| (CFO before | | 2011 | | | 313,664 | | | | 137,228 | | | | - | | | | 349,125 | | | | - | | | | 25,493 | | | | 825,510 |
| September 30, 2012 | | 2010 | | | 301,600 | | | | 105,560 | | | | 53,500 | | | | 106,386 | | | | - | | | | 32,370 | | | | 599,416 |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Dr. Israel | | 2012 | | | 397,407 | | | | 83,455 | | | | - | | | | 331,020 | | | | - | | | | 15,448 | | | | 827,330 |
| (SVP) | | 2011 | | | 383,968 | | | | 143,988 | | | | - | | | | 349,125 | | | | - | | | | 27,028 | | | | 904,109 |
| | | 2010 | | | 369,200 | | | | 110,760 | | | | 53,500 | | | | 106,386 | | | | - | | | | 35,150 | | | | 674,996 |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Dr. Olson | | 2012 | | | 314,007 | | | | 65,941 | | | | - | | | | 331,020 | | | | - | | | | 14,140 | | | | 725,108 |
| (SVP) | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Ms. Assumma | | 2012 | | | 279,864 | | | | 61,592 | | | | - | | | | 264,816 | | | | - | | | | 15,607 | | | | 621,879 |
| (VP) | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Dr. Maddon (Vice | | 2012 | | | 257,237 | (5) | | | - | | | | - | | | | 53,451 | | | | - | | | | 1,801,940 | | | | 2,112,628 |
| Chairman; CSO | | 2011 | | | 465,208 | | | | 150,000 | | | | - | | | | 428,077 | | | | - | | | | 13,964 | | | | 1,057,250 |
| before March 14, 2012; | | 2010 | | | 618,000 | | | | 250,000 | | | | - | | | | 405,260 | | | | - | | | | 13,893 | | | | 1,287,153 |
| CEO before March 3, 2011) | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
____________
(1) | Discretionary cash bonus approved by the Compensation Committee. |
(2) | Amount of compensation for each NEO reflects the aggregate grant date fair value for the year presented, in accordance with FASB ASC Topic 718, in respect of awards made for the year presented. The fair values were determined based on the assumptions for calculating the expense amounts as set forth in the notes to our consolidated financial statements included in our Annual Report on Form 10-K for the relevant years (Note 10 – Share-Based Payment Arrangements, in our 2012 Annual Report). The aggregate grant date fair value of the performance-based awards, assuming achievement of the highest level of performance conditions, are (i) $247,888, $345,719 and $762,000 for Mr. Baker's 2012, 2011 and 2010 awards, respectively, and (ii) $440,000 for Dr. Maddon's 2010 award. Additional information on the 2012 awards is included in the Grants of Plan-Based Awards for 2012 and Outstanding Equity Awards at Fiscal Year-End tables, below. |
(3) | Includes the Company's matching 2012 contribution under our 401(k) Plan in the amount of $11,250 for each named executive officer ($11,000 in earlier periods, and except in the case of Mr. Lovallo, for whom such contribution was the only other compensation for 2012). Also includes payments of premiums for enhanced life and disability insurance for Mr. Baker ($4,362), Mr. McKinney ($3,341), Dr. Israel ($4,198), Dr. Olson ($2,890), Ms. Assumma ($4,357), and Dr. Maddon ($1,356). Includes the gross-up for payment of taxes and compensation cost associated with payments pursuant to now-terminated Employee Stock Purchase Plans, computed in accordance with FASB ASC Topic 718, in 2011 and 2010, respectively, to Mr. Baker ($22,331 and $30,710), Mr. McKinney ($10,109 and $18,478), Dr. Israel ($11,687 and $21,257), Dr. Olson ($9,235 and $17,031), and Ms. Assumma ($8,230 in 2011). |
(4) | Includes $243,482 of severance payments to Mr. McKinney, and a $1,789,333 payment to Dr. Maddon pursuant to his Retirement Agreement. |
(5) | Includes $32,750 in non-employee director fees as set forth in the Director Compensation table, below. |
Grants of Plan-Based Awards in 2012
The following table sets forth information regarding grants of compensatory awards we made to our NEOs during 2012.
| Grant Date | | Estimated Future Payouts Under Equity Incentive Plan Awards (#)(1) | | All Other Stock Awards: Number of Shares of Stock or Units (#) | | All Other Option Awards: Number of Securities Underlying Options (#)(2) | | | Exercise or Base Price of Option Awards ($/Share) | | | Grant Date Fair Value of Stock and Option Awards ($)(3)(4) |
| Mr. Baker | 3/1/2012 | | | | | | | 66,666 | | | $ | 9.81 | | | $ | 441,356 |
| 3/1/2012 | | | 33,334 | | | | | | | | $ | 9.81 | | | $ | 247,888 |
| | | | | | | | | | | | | | | | | |
| Mr. Lovallo | 3/1/2012 | | | | | | | | 10,000 | | | $ | 9.81 | | | $ | 63,584 |
| | | | | | | | | | | | | | | | | |
| Mr. McKinney | 3/1/2012 | | | | | | | | 50,000 | | | $ | 9.81 | | | $ | 331,020 |
| | | | | | | | | | | | | | | | | |
| Dr. Israel | 3/1/2012 | | | | | | | | 50,000 | | | $ | 9.81 | | | $ | 331,020 |
| | | | | | | | | | | | | | | | | |
| Dr. Olson | 3/1/2012 | | | | | | | | 50,000 | | | $ | 9.81 | | | $ | 331,020 |
| | | | | | | | | | | | | | | | | |
| Ms. Assumma | 3/1/2012 | | | | | | | | 40,000 | | | $ | 9.81 | | | $ | 264,816 |
____________
(1) | Performance-based equity awards only. |
(2) | Options which vest one-third in each of the three years beginning March 1, 2013. |
(3) | Reported in Summary Compensation Table – Option Awards, above; reflects the aggregate grant date fair value determined, in accordance with FASB ASC Topic 718, using the assumptions for calculating the expense amounts as set forth in Note 10 – Share-Based Payment Arrangements to our consolidated financial statements included in our 2012 Annual Report on Form 10-K. The grant date fair value of Mr. Baker's 2012 performance-based award assumes achievement of the highest level of performance conditions. |
(4) | Dr. Maddon retired in 2012 and did not receive any compensatory awards as an employee in 2012; he received options for service as a non-employee director as set forth in the Director Compensation table, below. |
Outstanding Equity Awards at Fiscal Year-End
The table below (including the notes on the following page) sets forth information regarding unexercised stock option and unvested stock awards held by our NEOs as of December 31, 2012:
| | Option Awards | | Stock Awards |
| | No. of Securities Underlying Unexercised Options (#) Exercisable | | | No. of Securities Underlying Unexercised Options (#) Unexercisable | | | Equity Incentive Plan Awards: No. of Securities Underlying Unexercised Unearned Options (#) | | | Option Exercise Price ($) | | Option Expiration Date | | No. of Shares or Units of Stock That Have Not Vested (#)(1) | | | Market Value of Shares or Units of Stock That Have Not Vested ($) | | Equity Incentive Plan Awards: No. of Unearned Shares, Units or Other Rights That Have Not Vested (#) | Equity Incentive Plan Payout Value of Unearned Shares, Units or Other Rights That Have Not Vested ($) |
| Mr. Baker | | | - | | | | | | | 33,334 | (2) | | $ | 9.81 | | 3/1/2022 | | | | | | | | |
| | | - | | | | 66,666 | (3) | | | | | | $ | 9.81 | | 3/1/2022 | | | | | | | | |
| | | 20,833 | | | | 41,667 | (4) | | | | | | $ | 7.40 | | 7/1/2021 | | | | | | | | |
| | | 46,875 | | | | | | | | 15,625 | (2) | | $ | 7.40 | | 7/1/2021 | | | | | | | | |
| | | 50,000 | | | | 150,000 | (5) | | | | | | $ | 7.66 | | 6/8/2021 | | | | | | | | |
| | | 150,000 | | | | | | | | 50,000 | (2) | | $ | 5.35 | | 7/1/2020 | | | | | | | | |
| | | 83,333 | | | | 41,667 | (6) | | | | | | $ | 5.35 | | 7/1/2020 | | | | | | | | |
| | | 60,000 | | | | | | | | | | | $ | 16.05 | | 7/1/2018 | | | | | | | | |
| | | 10,000 | | | | | | | | | | | $ | 22.01 | | 7/2/2017 | | | | | | | | |
| | | 25,000 | | | | | | | | | | | $ | 24.26 | | 7/3/2016 | | | | | | | | |
| | | 60,000 | | | | | | | | | | | $ | 27.71 | | 2/21/2016 | | | | | | | | |
| | | 50,000 | | | | | | | | | | | $ | 20.02 | | 6/20/2015 | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | |
| Mr. Lovallo | | | - | | | | 10,000 | (3) | | | | | | $ | 9.81 | | 3/1/2022 | | | | | | | | |
| | | 3,000 | | | | 6,000 | (4) | | | | | | $ | 7.40 | | 7/1/2021 | | | | | | | | |
| | | 2,666 | | | | 1,334 | (6) | | | | | | $ | 5.35 | | 7/1/2020 | | | | | | | | |
| | | 3,375 | | | | | | | | | | | $ | 5.33 | | 7/1/2019 | | | | | | | | |
| | | 4,800 | | | | 1,200 | (7) | | | | | | $ | 14.27 | | 9/2/2018 | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | 444 | | | $ | 1,323 | | | |
Mr. McKinney(8) | | | - | | | | 50,000 | | | | | | | $ | 9.81 | | 3/1/2022 | | | | | | | | | | |
| | | 23,333 | | | | 46,667 | | | | | | | $ | 7.40 | | 7/1/2021 | | | | | | | | | | |
| | | 20,000 | | | | 10,000 | | | | | | | $ | 5.35 | | 7/1/2020 | | | | | | | | | | |
| | | 50,000 | | | | | | | | | | | $ | 5.33 | | 7/1/2019 | | | | | | | | | | |
| | | 25,000 | | | | | | | | | | | $ | 16.05 | | 7/1/2018 | | | | | | | | | | |
| | | 10,000 | | | | | | | | | | | $ | 22.01 | | 7/2/2017 | | | | | | | | | | |
| | | 25,000 | | | | | | | | | | | $ | 24.26 | | 7/3/2016 | | | | | | | | | | |
| | | 12,500 | | | | | | | | | | | $ | 21.39 | | 7/1/2015 | | | | | | | | | | |
| | | 25,000 | | | | | | | | | | | $ | 22.68 | | 3/1/2015 | | | | | | | | | | |
| | | 25,000 | | | | | | | | | | | $ | 15.06 | | 7/1/2013 | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | 3,333 | | | $ | 9,932 | | | |
| Dr. Israel | | | | | | | 50,000 | (3) | | | | | | $ | 9.81 | | 3/1/2022 | | | | | | | | | | |
| | | 23,333 | | | | 46,667 | (4) | | | | | | $ | 7.40 | | 7/1/2021 | | | | | | | | | | |
| | | 20,000 | | | | 10,000 | (6) | | | | | | $ | 5.35 | | 7/1/2020 | | | | | | | | | | |
| | | 25,000 | | | | | | | | | | | $ | 5.33 | | 7/1/2019 | | | | | | | | | | |
| | | 20,000 | | | | | | | | | | | $ | 16.05 | | 7/1/2018 | | | | | | | | | | |
| | | 7,000 | | | | | | | | | | | $ | 22.01 | | 7/2/2017 | | | | | | | | | | |
| | | 17,500 | | | | | | | | | | | $ | 24.26 | | 7/3/2016 | | | | | | | | | | |
| | | 10,000 | | | | | | | | | | | $ | 21.39 | | 7/1/2015 | | | | | | | | | | |
| | | 35,000 | | | | | | | | | | | $ | 15.06 | | 7/1/2013 | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | 3,333 | | | $ | 9,932 | | | |
| Dr. Olson | | | - | | | | 50,000 | (3) | | | | | | $ | 9.81 | | 3/1/2022 | | | | | | | | | | |
| | | 23,333 | | | | 46,667 | (4) | | | | | | $ | 7.40 | | 7/1/2021 | | | | | | | | | | |
| | | 20,000 | | | | 10,000 | (6) | | | | | | $ | 5.35 | | 7/1/2020 | | | | | | | | | | |
| | | 50,000 | | | | | | | | | | | $ | 5.33 | | 7/1/2019 | | | | | | | | | | |
| | | 20,000 | | | | | | | | | | | $ | 16.05 | | 7/1/2018 | | | | | | | | | | |
| | | 7,000 | | | | | | | | | | | $ | 22.01 | | 7/2/2017 | | | | | | | | | | |
| | | 17,500 | | | | | | | | | | | $ | 24.26 | | 7/3/2016 | | | | | | | | | | |
| | | 12,500 | | | | | | | | | | | $ | 21.39 | | 7/1/2015 | | | | | | | | | | |
| | | 25,000 | | | | | | | | | | | $ | 15.06 | | 7/1/2013 | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | 3,333 | | | $ | 9,932 | | | |
| Ms. Assumma | | | - | | | | 40,000 | (3) | | | | | | $ | 9.81 | | 3/1/2022 | | | | | | | | | | |
| | | 20,000 | | | | 40,000 | (4) | | | | | | $ | 7.40 | | 7/1/2021 | | | | | | | | | | |
| | | 16,666 | | | | 8,334 | (6) | | | | | | $ | 5.35 | | 7/1/2020 | | | | | | | | | | |
| | | 30,000 | | | | | | | | | | | $ | 5.33 | | 7/1/2019 | | | | | | | | | | |
| | | 10,000 | | | | | | | | | | | $ | 16.05 | | 7/1/2018 | | | | | | | | | | |
| | | 2,400 | | | | | | | | | | | $ | 22.01 | | 7/2/2017 | | | | | | | | | | |
| | | 4,500 | | | | | | | | | | | $ | 24.26 | | 7/3/2016 | | | | | | | | | | |
| | | 4,000 | | | | | | | | | | | $ | 21.39 | | 7/1/2015 | | | | | | | | | | |
| | | 10,000 | | | | | | | | | | | $ | 11.50 | | 8/9/2014 | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | 2,777 | | | $ | 8,275 | | | |
| Dr. Maddon | | | 10,000 | (9) | | | | | | | | | | $ | 7.93 | | 6/13/22 | | | | | | | | | | |
| | | 28,333 | | | | 56,667 | (4) | | | | | | $ | 7.40 | | 7/1/2021 | | | | | | | | | | |
| | | | 100,000 | | | | | | | | | | | $ | 5.35 | | 7/1/2020 | | | | | | | | | | |
| | | | 50,000 | | | | | | | | | | | $ | 5.35 | | 7/1/2020 | | | | | | | | | | |
| | | | 175,000 | | | | | | | | | | | $ | 5.33 | | 7/1/2019 | | | | | | | | | | |
| | | | 75,000 | | | | | | | | | | | $ | 16.05 | | 7/1/2018 | | | | | | | | | | |
| | | | 112,500 | | | | | | | | | | | $ | 22.01 | | 7/2/2017 | | | | | | | | | | |
| | | | 145,000 | | | | | | | | | | | $ | 24.26 | | 7/3/2016 | | | | | | | | | | |
| | | | 75,000 | | | | | | | | | | | $ | 21.39 | | 7/1/2015 | | | | | | | | | | |
| | | | 37,500 | | | | | | | | | | | $ | 16.85 | | 7/1/2014 | | | | | | | | | | |
| | | | 37,500 | | | | | | | | | | | $ | 16.85 | | 7/1/2014 | | | | | | | | | | |
| | | | 112,500 | | | | | | | | | | | $ | 15.06 | | 7/1/2013 | | | | | | | | | | |
| | | | 112,500 | | | | | | | | | | | $ | 15.06 | | 7/1/2013 | | | | | | | | | | |
____________
(1) | Shares vest in equal annual installments commencing one year from date of grant with final vesting no later than June 20, 2013. |
(2) | Performance stock options vest in percentages upon achievement of milestones or share price performance (see CD&A – Elements of Compensation – Long-term incentives, above, for a discussion of the performance criteria). |
(3) | Options vesting over three years beginning March 1, 2013. |
(4) | Options vesting over three years beginning July 1, 2012. |
(5) | Options vesting over four years beginning July 1, 2012. |
(6) | Options vesting over three years beginning July 1, 2011. |
(7) | Options vesting over five years beginning September 2, 2009. |
(8) | Pursuant to Mr. McKinney's agreement with the Company, vested and unexercised options (other than options expiring in 2013) continue to be exercisable, and unvested options continue to be outstanding, through September 30, 2013. |
(9) | Options granted in 2012 for service as a non-employee director. |
Option Exercises and Stock Vested in 2012
The following table sets forth information regarding the exercise and vesting of stock and stock option awards held by our NEOs during 2012:
| | | Option Awards | | Stock Awards |
| | No. of Shares Acquired on Exercise (#) | | Value Realized on Exercise ($) | | No. of Shares Acquired on Vesting (#) | | Value Realized on Vesting ($) |
| Mr. Baker | | - | | - | | - | | - |
| Mr. Lovallo | | - | | - | | 819 | | 7,101 |
| Mr. McKinney | | - | | - | | 3,333 | | 28,897 |
| Dr. Israel | | 7,000 | | 29,842 | | 3,333 | | 28,897 |
| Dr. Olson | | - | | - | | 3,333 | | 28,897 |
| Ms. Assumma | | - | | - | | 2,778 | | 24,085 |
| Dr. Maddon | | - | | - | | 18,750 | | 176,438 |
Potential Payments upon Termination or Change in Control
Progenics has no employment agreements with any of its NEOs other than Dr. Israel, whose agreement dates from 1994. As a result, the only amounts to which other NEOs would be legally entitled upon termination orpreventing a change in control of the Company, wouldadversely affecting our stock price.
Anti-takeover provisions may make removal of our Board and/or management more difficult, discouraging hostile bids for control that may be their vested equity compensation (whichbeneficial to our stockholders.
Our Board is authorized, without further stockholder action, to issue from time to time shares of preferred stock in general ceases vestingone or more designated series or classes. The issuance of preferred stock, as well as provisions in some outstanding stock options that provide for acceleration of exercisability upon termination outside a change of control, and Section 203 and other provisions of the Delaware General Corporation Law could make a takeover or the removal of our Board or management more difficult; discourage hostile bids for control in control context, but vests in full ifwhich stockholders may receive a premium for their shares; and otherwise dilute the employee is terminated without cause duringrights of common stockholders and depress the one-year period following a change in control) and amounts payable from their qualified retirement 401(k) plans.market price of our stock.
Under his employment agreement, Dr. Israel will be entitledItem 1B. Unresolved Staff Comments
We responded to nine months' salary continuation at his current salary and benefits if he is terminated without cause, with the salary continuation subject to termination if he secures new employment (which he is required to seek). He will not be entitled to severance, and all vested and unvested equity compensation will be immediately forfeited, if the Company terminates him for cause (defined as continual failure to perform substantially one's duties, conviction of a felony, habitual drunkenness, excessive absenteeism, dishonesty, unauthorized disclosure of confidential information, continuous conflict of interest or any other reason constituting cause under New York or federal law). His outstanding equity awards vest in full as provided aboveSEC staff comments issued in the event he is terminated without cause following a change in control. The agreement includes one-year non-competition, nondisclosure and two-year non-solicitation covenants, nonethird quarter of 2013 regarding recent Exchange Act filings, substantially all of which affects payments or benefits due. The following table describes potential payments we may be required to provide Dr. Israelwere addressed in the event we terminate his employment, assuming salaryour third quarter Form 10-Q and benefitsresolved as of December 31, 2012:
| Circumstances of Termination by the Company | Cash Severance | | Equity | Benefits Continuation ($) | Gross up of I.R.C. Golden Parachute Excise Tax Resulting from Change in Control ($) | Total ($) |
| Base Salary | | Bonus | | Value of Vested Equity ($) | Value of Accelerated Unvested Equity ($) |
| Multiple | ($) | | Multiple | ($) | |
| For cause | N/A | N/A | | N/A | N/A | | N/A | N/A | N/A | N/A | - |
| Without cause | N/A | 298,055 | | N/A | N/A | | N/A | N/A | 21,712 | N/A | 319,767 |
15
2013. We amended our 2012 Form 10-K to address remaining comments on January 17, 2014 and the Staff informed us on January 22 that it has completed its review.
At December 31, 2013, we occupied approximately 72,900 square feet of laboratory and office space in Tarrytown, New York, pursuant to lease agreements expiring in December 2020 under which we pay rent and facilities charges including utilities, taxes and operating expenses.
Item 3. Legal Proceedings
Progenics is a party to a proceeding brought by a former employee complaining that the termsCompany violated the anti-retaliation provisions of Dr. Maddon's now-terminated employment agreement,the federal Sarbanes-Oxley law by terminating the former employee. The Company believes the former employee's claims are without merit and is contesting the matter vigorously. The federal District Court hearing the case issued last July an order denying our motion for summary judgment dismissing the former employee's complaint, making it likely that the proceeding will continue to trial. Given the inherent uncertainty attendant to the proceeding, it is not possible at this time to estimate the likelihood or potential magnitude of any outcome, and we were required prior to March 14, 2012 to provide compensation to him under certain circumstanceshave accordingly not recorded any associated liability in the event of termination of his employment or a change in controlConsolidated Financial Statements.
Progenics last October commenced an arbitration with Ono under the provisions of the Company. We believedparties' License Agreement for development and commercialization of subcutaneous Relistor in Japan, following a communication from Ono that those arrangements with Dr. Maddon were competitive withit has determined to discontinue development because of "commercial concerns" that Ono contends would permit it to cease development and terminate the arrangements involving executives at similar levels of comparable companiesAgreement. Progenics is not in default under the Agreement, and were fair toOno has neither asserted that Progenics is, nor terminated the Company. The agreement obligated both parties to perform certain responsibilities upon termination of employment, which were described in detail in the Company's Proxy Statement for its 2011 Annual Meeting of Stockholders, when the agreement as then amended was still in effect. The following table describes those potential payments as they might have been payable prior to the termination based on the terms of the now-terminated agreement and the Company's stock price at the agreement's termination date.
Circumstances of Termination(1) | Cash Severance | | Equity | Benefits Continuation ($)(5) | Gross up Of I.R.C. Golden Parachute Excise Tax Resulting From Change in Control ($) | Total ($) |
| Base Salary | | Bonus(2) | Value of Vested Equity ($)(3) | Value of Accelerated Unvested Equity ($)(4) |
| Multiple | ($) | | Multiple | ($) |
| For cause or voluntary termination | N/A | N/A | | N/A | N/A | | 535,764 | N/A | N/A | N/A | 535,764 |
| | | | | | | | | | | |
Death or disability(6) | 1 | 425,000 | | N/A | 366,667 | | 535,764 | N/A | 35,292 | N/A | 1,362,722 |
Without cause or by the executive with good reason(7) | 2 | 850,000 | | 2 | 583,333 | | 535,764 | 1,070,211 | 35,292 | N/A | 3,074,600 |
Without cause or by the executive with good reason following a change in control(8) | 3 | 1,275,000 | | 3 | 800,000 | | 535,764 | 1,070,211 | 52,937 | N/A | 3,733,912 |
____________
| (1) | Calculations in this table utilize the closing price per share of our common stock on the agreement termination date, of $9.26. |
| (2) | Includes, where any amount is shown, bonus multiple calculated using the multiple shown plus a pro-rated bonus of $150,000, representing an estimated pro-rata bonus for the year of termination. |
| (3) | 680,875 of Dr. Maddon's 817,541 outstanding vested stock options were underwater as of the agreement termination date. |
| (4) | Assumed (i) acceleration of vesting at the agreement termination date of all 407,959 unexercisable and unearned stock options, as set forth in the Outstanding Equity Awards at Fiscal Year-End table above, 134,625 of which were underwater at such date, and (ii) acceleration of vesting and sale of all 18,750 outstanding shares of restricted stock at such date, yielding $1,070,211 before taxes, using the closing price of Progenics common stock on that date. |
| (5) | Health and welfare benefits were to continue for 24 or 36 months, depending on the circumstances of termination, and included the employer cost of health, dental, disability and group life insurance. |
| (6) | In this circumstance, Dr. Maddon or his estate would have received one times his base salary for the year of termination and average bonus (calculated using the average of the annual bonuses paid to him in the three years preceding the year of termination), and a pro-rated amount of bonus from the beginning of the year of termination to the date of termination based on an assumed bonus of $150,000. For purposes of this calculation, base salary at the agreement termination date was $425,000, the fair market values of his bonuses for the three years preceding such date were $150,000, $250,000 and $250,000. |
| (7) | In this circumstance, Dr. Maddon would have received cash severance equal to twice the sum of his base salary for the year of termination and the average of the annual bonuses paid to him in the three years preceding the year of termination, together with a pro-rated bonus from the beginning of the year of termination to the date of termination based on an assumed bonus of $150,000, calculated as specified in note (6)Agreement. See Item 1A, Risk Factors. |
| (8) | In this circumstance, Dr. Maddon would have received cash severance equal to three times the sum of base salary and average bonus, together with the pro-rated bonus, as described in note (7). |
Director CompensationItem 4. Not Applicable
PART II
Item 5. Market for Registrant's Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities Price Range of Common Stock
Our common stock is quoted on The NASDAQ Stock Market LLC under the symbol PGNX. The following table sets forth, for the periods indicated, the high and low sales price per share of the common stock, as reported on NASDAQ.
| | | High | | | Low | |
| 2013: | Fourth quarter | | $ | 5.90 | | | $ | 3.45 | |
| Third quarter | | | 6.47 | | | | 4.36 | |
| Second quarter | | | 5.57 | | | | 3.54 | |
| First quarter | | | 5.96 | | | | 2.53 | |
| | | | | | | | | |
| 2012: | Fourth quarter | | $ | 3.30 | | | $ | 1.41 | |
| Third quarter | | | 11.00 | | | | 2.81 | |
| Second quarter | | | 11.34 | | | | 7.44 | |
| First quarter | | | 10.50 | | | | 8.32 | |
On March 7, 2014, the last sale price for our common stock, as reported by The NASDAQ Stock Market LLC, was $4.70. There were approximately 85 holders of record of our common stock as of that date.
Comparative Stock Performance Graph
The graph below compares, for the past five years, the cumulative stockholder return on our common stock with the cumulative stockholder return of (i) the NASDAQ Stock Market (U.S.) Index and (ii) the NASDAQ Pharmaceutical Index, assuming an investment in each of $100 on December 31, 2008.
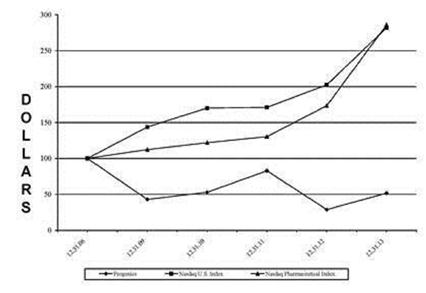
Effective January 2014, NASDAQ OMX, which provides us with total return values for the Comparative Stock Performance Graph, is replacing values prepared by the Center for Research in Security Prices (CRSP) at the University of Chicago "in an effort to provide a greater level of transparency and leverage [its] existing universe of indexes." As a result, our performance graphs going forward will be using the replacement indices provided by NASDAQ OMX Global Indexes: the NASDAQ U.S. Benchmark TR Index and the ICB: 4577 Pharmaceuticals (Subsector) Index. Please note that information regardingfor the aggregate compensationNASDAQ Stock Market (U.S.) and the NASDAQ Pharmaceutical indices is provided only from December 31, 2008 through December 31, 2013, the last day this data was made available to us by NASDAQ OMX. The following graph compares, for the past five years, the cumulative stockholder return on our common stock with the cumulative stockholder return of the replacement indices, assuming an investment in each of $100 on December 31, 2008.
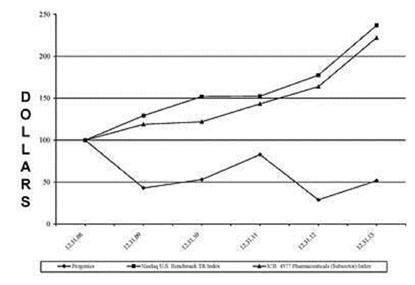
Dividends
Progenics has never paid any dividends, and we paidcurrently anticipate that all earnings, if any, will be retained for development of our business and no dividends will be declared in the foreseeable future.
Item 6. Selected Financial Data
The selected financial data presented below as of December 31, 2013 and 2012 and for each of the three years in the period ended December 31, 2013 are derived from our audited financial statements, included elsewhere herein. The selected financial data presented below with respect to the non-employee membersbalance sheet data as of December 31, 2011, 2010 and 2009 and for each of the two years in the period ended December 31, 2010 are derived from our audited financial statements not included herein. The data set forth below should be read in conjunction with Management's Discussion and Analysis of Financial Condition and Results of Operations and the Financial Statements and related Notes included elsewhere herein.
| | Years Ended December 31, | |
| | 2013 | | | 2012 | | | 2011 | | | 2010 | | | 2009 | |
| | (in thousands, except per share data) | |
| Statement of Operations Data: | | | | | | | | | | | | | | | |
| Revenues: | | | | | | | | | | | | | | | |
| Royalty income | | $ | 5,923 | | | $ | 4,963 | | | $ | 3,046 | | | $ | 1,826 | | | $ | 2,372 | |
| Collaboration revenue | | | 1,595 | | | | 8,525 | | | | 76,764 | | | | 1,413 | | | | 44,351 | |
| Research grants | | | 275 | | | | 488 | | | | 4,810 | | | | 4,573 | | | | 1,968 | |
| Other revenues | | | 69 | | | | 72 | | | | 176 | | | | 140 | | | | 256 | |
| Total revenues | | | 7,862 | | | | 14,048 | | | | 84,796 | | | | 7,952 | | | | 48,947 | |
| Expenses: | | | | | | | | | | | | | | | | | | | | |
| Research and development | | | 33,903 | | | | 31,840 | | | | 53,183 | | | | 50,640 | | | | 49,798 | |
| License fees - research and development | | | 567 | | | | 1,170 | | | | 578 | | | | 1,270 | | | | 1,058 | |
| Royalty expense | | | 624 | | | | 499 | | | | 405 | | | | 241 | | | | 237 | |
| General and administrative | | | 14,809 | | | | 14,706 | | | | 18,248 | | | | 22,832 | | | | 25,106 | |
| Depreciation and amortization | | | 939 | | | | 1,324 | | | | 2,066 | | | | 2,853 | | | | 5,078 | |
| Total expenses | | | 50,842 | | | | 49,539 | | | | 74,480 | | | | 77,836 | | | | 81,277 | |
| Operating (loss) income | | | (42,980 | ) | | | (35,491 | ) | | | 10,316 | | | | (69,884 | ) | | | (32,330 | ) |
| Other income: | | | | | | | | | | �� | | | | | | | | | | |
| Interest income | | | 46 | | | | 60 | | | | 65 | | | | 64 | | | | 1,481 | |
| Gain on sale of marketable securities | | | - | | | | - | | | | - | | | | - | | | | 237 | |
| Total other income | | | 46 | | | | 60 | | | | 65 | | | | 64 | | | | 1,718 | |
| Net (loss) income before provision for income taxes | | | (42,934 | ) | | | (35,431 | ) | | | 10,381 | | | | (69,820 | ) | | | (30,612 | ) |
| Income tax benefit | | | 362 | | | | - | | | | - | | | | 95 | | | | - | |
| Net (loss) income | | $ | (42,572 | ) | | $ | (35,431 | ) | | $ | 10,381 | | | $ | (69,725 | ) | | $ | (30,612 | ) |
| Per share amounts on net (loss) income: | | | | | | | | | | | | | | | | | | | | |
| Basic | | $ | (0.76 | ) | | $ | (1.02 | ) | | $ | 0.31 | | | $ | (2.14 | ) | | $ | (0.98 | ) |
| Diluted | | $ | (0.76 | ) | | $ | (1.02 | ) | | $ | 0.31 | | | $ | (2.14 | ) | | $ | (0.98 | ) |
| | December 31, | |
| | 2013 | | | 2012 | | | 2011 | | | 2010 | | | 2009 | |
| | (in thousands) | |
| Balance Sheet Data: | | | | | | | | | | | | | | | |
| Cash and cash equivalents | | $ | 65,860 | | | $ | 58,838 | | | $ | 70,105 | | | $ | 47,918 | | | $ | 90,903 | |
| Auction Rate and marketable securities | | | 2,208 | | | | 3,240 | | | | 3,332 | | | | 3,608 | | | | 5,293 | |
| Working capital | | | 64,055 | | | | 58,805 | | | | 65,890 | | | | 42,207 | | | | 95,388 | |
| Total assets | | | 114,541 | | | | 76,308 | | | | 80,110 | | | | 62,738 | | | | 113,613 | |
| Deferred revenue - current | | | - | | | | 838 | | | | 204 | | | | - | | | | - | |
| Deferred revenue - long term | | | - | | | | - | | | | 162 | | | | - | | | | - | |
| Other liabilities - long term | | | 28,935 | | | | 1,078 | | | | 1,497 | | | | 1,635 | | | | - | |
| Total stockholders' equity | | | 78,979 | | | | 66,568 | | | | 71,801 | | | | 51,308 | | | | 107,607 | |
Item 7. Management's Discussion and Analysis of Financial Condition and Results of Operations (MD&A)
Overview
General. As discussed in Business, above, we are conducting phase 2 clinical trials of two product candidates for prostate cancer, and resuming a pivotal phase 2 trial of an ultra-orphan radiotherapy candidate for pheochromocytoma. We have also decided to move forward MIP-1095, a compound originally developed by Molecular Insight, into clinical development, and expect to file an IND application in the U.S. later this year.
Our acquisition last year of the privately-held Molecular Insight included the issuance of Progenics common stock in a private transaction not taxable to Progenics, and its agreement to pay potential milestones, in cash or Progenics stock at its option, of up to $23 million, contingent upon achieving specified commercialization events and up to $70 million contingent upon achieving specified sales targets relating to the acquired company's products. As described in Note 2 to the Consolidated Financial Statements, the acquisition was accounted for using the acquisition method of accounting, under which the acquired company's assets and liabilities were recorded at their estimated respective fair values as of the acquisition date in our consolidated financial statements. The difference between the estimated fair value of the acquisition consideration and fair value of the identifiable net assets represents potential future economic benefits arising from combining the companies, and has been recorded as goodwill. The results of operations of the acquired company's business from January 18, 2013, the closing date of the acquisition, the estimated fair market values of the assets acquired and liabilities assumed, and goodwill are included in our consolidated financial statements since the date of the acquisition and are included in the discussion and analysis below.
We have licensed Relistor to Salix Pharmaceuticals, and have partnered other internally-developed or acquired compounds and technologies with third parties. We continue to consider opportunities for strategic collaborations, out-licenses and other arrangements with biopharmaceutical companies involving proprietary research, development and clinical programs, and may in the future also in-license or acquire additional oncology compounds and/or programs.
Our current principal sources of revenue from operations are royalty, commercialization milestone and revenue-sharing payments from Salix's Relistor operations. Royalty and milestone payments from Relistor depend on success in development and commercialization, which is dependent on many factors, such as Salix's efforts, decisions by the FDA and other regulatory bodies such as the Complete Response Letter mentioned in Business and Risk Factors, competition from drugs for the same or similar indications, and the outcome of clinical and other testing of Relistor. As previously reported, Progenics in October 2013 commenced an arbitration with Ono under the provisions of the parties' License Agreement, following a communication from Ono that it has determined to discontinue development of subcutaneous Relistor in Japan because of "commercial concerns" that Ono contends would permit it to cease development and terminate the Agreement. Progenics is not in default under the Agreement, and Ono has neither asserted that Progenics is, nor terminated the Agreement. See Risk Factors.
We fund our operations to a significant extent from capital-raising. During 2013, we completed an underwritten public offering of 9.8 million shares of common stock at a public offering price of $4.40 per share, resulting in net proceeds of approximately $40.1 million, and in early 2014 sold an additional 8.75 million shares at $4.60 per share, for net proceeds of approximately $37.5 million.
Most of our Boardexpenditures are for research and development activities. During 2013, expenses for Oncology, primarily related to PSMA ADC, 1404, Azedra™ and 1095, were $33.5 million compared to $29.1 million in 2012 and $22.2 million in 2011. Expenses for Relistor and Other programs in 2013 were $0.9 million and $0.7 million, respectively, compared to $1.7 million and $2.7 million in 2012 and $23.2 million and $8.8 million in 2011. We expect to incur significant development expenses for our PSMA ADC, 1404, Azedra™ and 1095 product candidates as clinical trials progress, while expenses, and the resulting reimbursement revenue, related to Relistor depend on the amount of research and development work we perform upon request by Salix or Ono.
At December 31, 2013, we held $65,860 in cash and cash equivalents, an increase of $7,022 from $58,838 at 2012 year-end. We expect that this amount, together with the additional 2014 public offering proceeds, will be sufficient to fund operations as currently anticipated beyond one year. We expect to incur operating losses during the near term. At December 31, 2013, cash, cash equivalents and auction rate securities increased $5,990 to $68,068 from $62,078 at December 31, 2012. Mr. Mark Baker did
If we do not realize sufficient royalty or other revenue from Relistor, or are unable to enter into favorable collaboration, license, asset sale, capital raising or other financing transactions, we will have to reduce, delay or eliminate spending on certain programs, and/or take other economic measures.
Relistor has been approved by regulatory authorities in the U.S., countries in the E.U., Canada and Australia since 2008 for treatment of OIC in advanced-illness patients receiving palliative care when laxative therapy has not been sufficient. Salix is responsible for further developing and commercializing Relistor, including completing clinical development necessary to support regulatory marketing approvals for potential new indications (such as chronic pain) and formulations of the drug, such as oral methylnaltrexone. Under our Agreement with Salix, we are eligible to receive (i) a development milestone of up to $40 million upon U.S. marketing approval for subcutaneous Relistor in non-cancer pain patients (the proposed indication addressed in the Complete Response Letter mentioned above), (ii) a development milestone of up to $50 million upon U.S. marketing approval of an oral formulation of Relistor, (iii) up to $200 million of commercialization milestone payments upon achievement of specified U.S. sales targets, (iv) royalties ranging from 15 to 19 percent of net sales by Salix and its affiliates, and (v) 60% of any upfront, milestone, reimbursement or other revenue (net of costs of goods sold, as defined, and territory-specific research and development expense reimbursement) Salix receives from sublicensees outside the U.S. In the event either marketing approval is subject to a Black Box Warning or Risk Evaluation and Mitigation Strategy (REMS), payment of a substantial portion of the milestone amount would be deferred, and subject, to achievement of the first commercialization milestone (payable on annual U.S. sales first exceeding $100 million).
Salix has secured distribution for Relistor in the European territory and has licensed Link Medical Products Pty Limited for distribution in Australia, New Zealand, South Africa and certain other markets in Asia. Salix is continuing efforts to secure additional distribution partners and/or sublicensees in Europe and elsewhere.
Results of Operations (amounts in thousands unless otherwise noted)
| | 2013 | | | 2012 | | | 2011 | | | 2013 vs. 2012 | | | 2012 vs. 2011 | |
| | | | | | | | | | | Percent Change | |
| | | | | | | | | | | | | | | |
| Revenues | | $ | 7,862 | | | $ | 14,048 | | | $ | 84,796 | | | | (44 | %) | | | (83 | %) |
| Expenses | | | (50,842 | ) | | | (49,539 | ) | | | (74,480 | ) | | | 3 | % | | | (33 | %) |
| Operating (loss) income | | | (42,980 | ) | | | (35,491 | ) | | | 10,316 | | | | 21 | % | | | (444 | %) |
| Other income | | | 46 | | | | 60 | | | | 65 | | | | (23 | %) | | | (8 | %) |
| Income tax benefit | | | 362 | | | | - | | | | - | | | | 100 | % | | | N/ | A |
| Net (loss) income | | $ | (42,572 | ) | | $ | (35,431 | ) | | $ | 10,381 | | | | 20 | % | | | (441 | %) |
Revenues:
Sources of revenue during the years indicated below included license and other agreements with Salix and other collaborators, research grants from the National Institutes of Health (NIH) and, to a small extent, sales of research reagents.
| Sources of Revenue | | 2013 | | | 2012 | | | 2011 | | | 2013 vs. 2012 | | | 2012 vs. 2011 | |
| | | | | | | | | | | Percent Change | |
| | | | | | | | | | | | | | | |
| Royalty income | | $ | 5,923 | | | $ | 4,963 | | | $ | 3,046 | | | | 19 | % | | | 63 | % |
| Collaboration revenue | | | 1,595 | | | | 8,525 | | | | 76,764 | | | | (81 | %) | | | (89 | %) |
| Research grants | | | 275 | | | | 488 | | | | 4,810 | | | | (44 | %) | | | (90 | %) |
| Other revenues | | | 69 | | | | 72 | | | | 176 | | | | (4 | %) | | | (59 | %) |
| Total | | $ | 7,862 | | | $ | 14,048 | | | $ | 84,796 | | | | (44 | %) | | | (83 | %) |
Royalty income. During the last three years, we recognized royalty income primarily based on the below net sales of Relistor reported by Salix or a former collaborator, and net sales reported by other licensees.
| | Relistor Net Sales Reported by Collaborators | |
| | Years Ended December 31, | |
| | 2013 | | | 2012 | | | 2011 | |
| U.S. | | $ | 35,000 | | | $ | 29,200 | | | $ | 21,500 | |
| Ex-U.S. | | | 4,400 | | | | 4,000 | | | | 5,500 | |
| Global | | $ | 39,400 | | | $ | 33,200 | | | $ | 27,000 | |
Collaboration revenue:
During 2013, we recognized revenue from upfront and reimbursement payments from partnering arrangements consisting of (i) $676 from amortization of upfront payments for partnering the Company's PRO 140 and C. difficile programs, (ii) $420 from amortization of upfront payment and expense reimbursement for licensing 1404 in Japan, (iii) $295 from amortization of upfront payment and expense reimbursement for licensing Relistor, and (iv) a $189 upfront payment from another licensee.
During 2012, we recognized $7,949 of revenue from upfront and reimbursement payments from PRO 140 and C. difficile partnering. As of December 31, 2012, $676 is recorded in deferred revenue – current.
During 2012 and 2011, we recognized $558 and $75,091, respectively, of revenue from Salix, which includes $204 and $59,634, respectively, from the upfront cash payment under our License Agreement, $225 in 2011 in respect of Salix ex-U.S. sublicensee revenue and $354 and $15,232, respectively, as reimbursement of expenses. As of December 31, 2012, $162 is recorded in deferred revenue – current. During 2011 we recognized $1,630 of revenue from a former collaborator as reimbursement of expenses under a transition agreement; we received no such reimbursement in 2013 or 2012.
During 2013, 2012 and 2011, we recognized $13, $18 and $43, respectively, of reimbursement revenue for activities requested by a collaborator.
Research grants. During the last three years, we recognized the amounts in the above table as revenue from federal government grants from the NIH to support research and development programs. Decreases in grant revenue resulted from lower reimbursable expenses year-to-year. We do not expect to recognize revenue from the NIH in the near future.
Other revenues, primarily from orders for research reagents, decreased as shown in the above table.
Expenses:
Research and Development Expenses include scientific labor, clinical trial costs, supplies, product manufacturing costs, consulting, license fees, royalty payments and other operating expenses. Research and development expenses increased to $35,094 for 2013 from $33,509 for 2012 and decreased from $54,166 for 2011. During 2013, the increase in research and development expenses compared to those in 2012 was primarily due to higher clinical trial costs related to PSMA ADC and 1404, and rent expense, partially offset by lower compensation, contract manufacturing and subcontracting and license fee expenses. See Liquidity and Capital Resources – Uses of Cash, for details of the changes in these expenses by project. Salix reimbursed us for development expenses we incurred related to Relistor; most of these expenses were incurred and reimbursed in 2011. Portions of our expenses during the periods presented were funded through grants from the NIH (see Revenues- Research Grants). The changes in research and development expense, by category of expense, are as follows:
| | 2013 | | | 2012 | | | 2011 | | | 2013 vs. 2012 | | | 2012 vs. 2011 | |
| | | | | | | | | | | Percent change | |
| Salaries and benefits | | $ | 12,481 | | | $ | 15,372 | | | $ | 18,658 | | | | (19 | %) | | | (18 | %) |
2013 vs. 2012 Salaries and benefits decreased due to a decline in average headcount, and reflecting a non-recurring retirement expense of $1,804 incurred in the first quarter of 2012.
2012 vs. 2011 Salaries and benefits decreased due to a decline in average headcount and lower accrued bonus expense, partially offset by the above non-recurring retirement expense and accrued severance expense related to additional headcount reductions in the third quarter.
| | 2013 | | | 2012 | | | 2011 | | | 2013 vs. 2012 | | | 2012 vs. 2011 | |
| | | | | | | | | | | Percent change | |
| Share-based compensation | | $ | 2,524 | | | $ | 4,568 | | | $ | 4,499 | | | | (45 | %) | | | 2 | % |
2013 vs. 2012 Share-based compensation decreased primarily due to lower equity incentives expenses, and reflecting non-recurring 2012 retirement-related option and restricted stock expense noted below.
2012 vs. 2011 Share-based compensation increased primarily due to the acceleration of options and restricted stock expenses of $1,638 resulting from a former senior executive retirement, partially offset by lower restricted stock expenses and elimination of employee stock purchase plan expenses resulting from the 2011 termination of those plans.
For 2011, share-based compensation included restricted stock and option plan expenses from (i) accelerated vesting of outstanding awards to non-management employees in connection with a change in program eligibility and termination of the Company's employee stock purchase plans (the latter of which resulted in a decline in share-based compensation), and (ii) a shift in headcount from general and administrative departments to research and development. See Critical Accounting Policies − Share-Based Payment Arrangements.
| | 2013 | | | 2012 | | | 2011 | | | 2013 vs. 2012 | | | 2012 vs. 2011 | |
| | | | | | | | | | | Percent change | |
| Clinical trial costs | | $ | 8,862 | | | $ | 2,692 | | | $ | 10,099 | | | | 229 | % | | | (73 | %) |
2013 vs. 2012 Clinical trial costs increased primarily due to higher expenses for Oncology ($6,214), partially offset by decreased expenses in Other programs ($27) and Relistor ($17).
2012 vs. 2011 Clinical trial costs decreased primarily due to lower clinical trial activities related to Relistor ($9,132), partially offset by increased expenses in Oncology ($1,700), primarily related to PSMA ADC and Other ($25).
| | 2013 | | | 2012 | | | 2011 | | | 2013 vs. 2012 | | | 2012 vs. 2011 | |
| Laboratory and manufacturing | | | | | | | | | | | Percent change | |
| supplies and equipment | | $ | 632 | | | $ | 592 | | | $ | 4,203 | | | | 7 | % | | | (86 | %) |
2013 vs. 2012 Laboratory and manufacturing supplies and equipment increased by $511 for Other programs, including second quarter impairment losses from the write-off of laboratory equipment in connection with an amendment to the Company's Tarrytown lease, partially offset by lower Oncology expenses ($471), primarily from a decline in lab supplies for PSMA ADC.
2012 vs. 2011 Laboratory and manufacturing supplies and equipment decreased due to lower expenses in (i) Relistor ($2,197), (ii) Oncology ($388), reflecting a decline in manufacturing supplies for PSMA ADC, and (iii) Other ($1,026).
| | 2013 | | | 2012 | | | 2011 | | | 2013 vs. 2012 | | | 2012 vs. 2011 | |
| Contract manufacturing and | | | | | | | | | | | Percent change | |
| subcontractors | | $ | 2,042 | | | $ | 3,111 | | | $ | 6,713 | | | | (34 | %) | | | (54 | %) |
2013 vs. 2012 Contract manufacturing and subcontractors decreased due to lower expenses for Oncology ($571), Other ($353), and Relistor ($145).
2012 vs. 2011 Contract manufacturing and subcontractors decreased due to lower expenses for Relistor ($2,399), reflecting a decrease in purchases of subcutaneous Relistor related products, and Other ($1,596), partially offset by an increase in Oncology ($393).
Expenses in this category relate to the conduct of clinical trials, including manufacture by third parties of drug materials, testing, analysis, formulation and toxicology services, providedand vary as the timing and level of such services are required.
| | 2013 | | | 2012 | | | 2011 | | | 2013 vs. 2012 | | | 2012 vs. 2011 | |
| | | | | | | | | | | Percent change | |
| Consultants | | $ | 905 | | | $ | 330 | | | $ | 1,270 | | | | 174 | % | | | (74 | %) |
2013 vs. 2012 Consultants expense increased primarily due to higher expenses for Oncology ($613), partially offset by lower expenses for Other programs ($27) and Relistor ($11).
2012 vs. 2011 Consultants expense decreased due to lower expenses in 2012 for Relistor regulatory and other activities ($816), Oncology ($18) and Other programs ($106).
Expenses in this category relate to monitoring ongoing clinical trials and reviewing data from completed trials including the preparation of filings and vary as the timing and level of such services are required.
| | 2013 | | | 2012 | | | 2011 | | | 2013 vs. 2012 | | | 2012 vs. 2011 | |
| | | | | | | | | | | Percent change | |
| License fees | | $ | 567 | | | $ | 1,170 | | | $ | 578 | | | | (52 | %) | | | 102 | % |
2013 vs. 2012 License fees decreased due to lower expenses for Oncology ($573) and Other programs ($30).
2012 vs. 2011 License fees increased due to higher expenses for clinical trial initiation in Oncology ($902), partially offset by lower expenses for Relistor ($156) and Other programs ($154).
| | 2013 | | | 2012 | | | 2011 | | | 2013 vs. 2012 | | | 2012 vs. 2011 | |
| | | | | | | | | | | Percent change | |
| Royalty expense | | $ | 624 | | | $ | 499 | | | $ | 405 | | | | 25 | % | | | 23 | % |
2013 vs. 2012 The increase in royalty expense was due to higher net sales of Relistor in 2013.
2012 vs. 2011 The increase in royalty expense was due to higher net sales of Relistor in 2012.
| | 2013 | | | 2012 | | | 2011 | | | 2013 vs. 2012 | | | 2012 vs. 2011 | |
| | | | | | | | | | | Percent change | |
| Other operating expenses | | $ | 6,457 | | | $ | 5,175 | | | $ | 7,741 | | | | 25 | % | | | (33 | %) |
2013 vs. 2012 Other operating expenses increased from 2012 primarily due to increases in rent ($1,131), as a Board member duringresult of lease amendment and termination expenses, travel ($106), other operating expenses ($140) and insurance ($57), partially offset by a decrease in facilities ($152).
2012 vs. 2011 Other operating expenses decreased primarily due to decreases in rent ($1,943), travel ($111), insurance ($55) and other operating expenses ($489), partially offset by increases in expenses for facilities ($32).
General and Administrative Expenses increased to $14,809 for 2013 from $14,706 for 2012 and continues to servedecreased from $18,248 for 2011, as a director without compensation.follows:
| | Fees Earned or Paid in Cash | | | Option Awards(1) | | | All Other Compensation | | | Total | |
| Mr. Briner | | $ | 44,000 | | | $ | 53,451 | | | | - | | | $ | 97,451 | |
| Mr. Charles Baker | | | 68,000 | | | | 53,451 | | | | - | | | | 121,451 | |
| Mr. Crowley | | | 105,500 | | | | 213,804 | | | | - | | | | 319,304 | |
Mr. Dalton(2) | | | 15,250 | | | | - | | | | - | | | | 15,250 | |
| Dr. Goff | | | 62,000 | | | | 53,451 | | | | - | | | | 115,451 | |
Dr. Maddon(2) | | | 32,750 | | | | 53,451 | | | | - | | | | 86,201 | |
| Dr. Scheinberg | | | 74,000 | | | | 53,451 | | | | - | | | | 127,451 | |
| Ms. Williams | | | 71,000 | | | | 53,451 | | | | - | | | | 124,451 | |
| | 2013 | | | 2012 | | | 2011 | | | 2013 vs. 2012 | | | 2012 vs. 2011 | |
| | | | | | | | | | | Percent change | |
| Salaries and benefits | | $ | 4,821 | | | $ | 6,493 | | | $ | 7,228 | | | | (26 | %) | | | (10 | %) |
____________
2013 vs. 2012 Salaries and benefits decreased primarily due to 2012 accrued severance expense resulting from headcount reductions, while the average headcount remained unchanged.
2012 vs. 2011 Salaries and benefits decreased due to a decline in average headcount in the general and administrative departments and lower accrued bonus expense in 2012, partially offset by an increase in accrued severance expense related to additional headcount reductions in the third quarter.
| | 2013 | | | 2012 | | | 2011 | | | 2013 vs. 2012 | | | 2012 vs. 2011 | |
| | | | | | | | | | | Percent change | |
| Share-based compensation | | $ | 1,022 | | | $ | 1,968 | | | $ | 1,863 | | | | (48 | %) | | | 6 | % |
2013 vs. 2012 Share-based compensation decreased due to lower equity incentives expenses, which included restructuring expenses in 2012.
2012 vs. 2011 Share-based compensation increased due to higher stock option expenses in connection with the third quarter 2012 restructuring, partially offset by lower restricted stock and elimination of employee stock purchase plans expenses resulting from the 2011 termination of those plans.
For 2011, share-based compensation reflected accelerated vesting in connection with termination of employee stock purchase plans.
| | 2013 | | | 2012 | | | 2011 | | | 2013 vs. 2012 | | | 2012 vs. 2011 | |
| | | | | | | | | | | Percent change | |
| Consulting and professional fees | | $ | 3,922 | | | $ | 2,362 | | | $ | 4,389 | | | | 66 | % | | | (46 | %) |
2013 vs. 2012 Consulting and professional fees increased due to higher consulting ($697), patent ($457), legal ($256), audit ($101) and other fees ($49).
2012 vs. 2011 Consulting and professional fees decreased due to lower consulting ($1,246), audit ($297), patent ($294), accounting ($91), legal ($63) and other fees ($36).
| | 2013 | | | 2012 | | | 2011 | | | 2013 vs. 2012 | | | 2012 vs. 2011 | |
| | | | | | | | | | | Percent change | |
| Other operating expenses | | $ | 4,325 | | | $ | 3,883 | | | $ | 4,768 | | | | 11 | % | | | (19 | %) |
2013 vs. 2012 Other operating expenses increased due to higher expenses for market research ($158), recruiting ($112), investor relations ($109) and taxes ($108) and other operating expenses ($127), partially offset by a decreases in rent ($172).
2012 vs. 2011 Other operating expenses decreased due to lower expenses for rent ($646), investor relations ($63), taxes ($30) and other operating expenses ($359), partially offset by an increases in recruiting ($137), computer software ($66) and travel ($10).
| | 2013 | | | 2012 | | | 2011 | | | 2013 vs. 2012 | | | 2012 vs. 2011 | |
| | | | | | | | | | | Percent change | |
| Depreciation and amortization | | $ | 939 | | | $ | 1,324 | | | $ | 2,066 | | | | (29 | %) | | | (36 | %) |
2013 vs. 2012 Depreciation and amortization expense decreased primarily due to lower leasehold improvements and machinery and equipment fixed asset balances.
2012 vs. 2011 Depreciation and amortization expense decreased primarily due to lower machinery and equipment fixed asset balances.
Intangible impairment charges and change in contingent consideration liability
2013 As of December 31, 2013 indefinite-lived intangible assets decreased by $919, from $32.3 million to $31.4 million, resulting from our annual impairment testing, with the corresponding expense recorded in the general and administrative expenses in the Consolidated Statements of Operations. This impairment was the result of change in the estimated timing of beginning cash inflows from 2014 to 2018 and an increase in discount rate from 15% to 18% for the Onalta intangible asset. In addition, the fourth quarter review of the contingent consideration liability fair value resulted in a $200 decrease, from $15.9 million to $15.7 million, which has also been recorded in the general and administrative expenses in the Consolidated Statements of Operations. The decrease in contingent consideration liability was due to an increase in the discount period.
Other income:
| | 2013 | | | 2012 | | | 2011 | | | 2013 vs. 2012 | | | 2012 vs. 2011 | |
| | | | | | | | | | | Percent change | |
| Interest income | | $ | 46 | | | $ | 60 | | | $ | 65 | | | | (23 | %) | | | (8 | %) |
2013 vs. 2012 Interest income decreased due to lower average interest rates in 2013 than in 2012, partially offset by increases resulting from higher average balances of cash equivalents.
2012 vs. 2011 Interest income decreased to $60 from $65 over the period.
Interest income, as reported, is primarily the result of investment income from auction rate securities held by us.
Income Taxes:
For 2013, income tax benefit of $362 resulted from the change in the difference between carrying amounts of in-process research and development assets for financial reporting purposes and the amounts used for income tax purposes. For 2012 and 2011, there was no provision for income taxes due to a pre-tax loss for 2012 and the 2011 pre-tax income amount was completely offset by our available net operating loss carry-forwards.
Net (Loss) Income:
2013 net loss was $42,572, compared to net loss of $35,431 for 2012, and net income of $10,381 for 2011.
Liquidity and Capital Resources
We have to date funded operations principally through payments received from private placements of equity securities, public offerings of common stock, collaborations, grants and contracts, royalties, interest on investments and proceeds from the exercise of outstanding options and warrants.
We received in 2013 a $5,000 upfront payment from partnering of the C. difficile program and are eligible to receive future milestone and royalty payments. This receipt resulted in the reversal in 2013 of deferred tax assets and liabilities established in 2012 to reflect the net tax effects of temporary differences between the carrying amounts of certain assets and liabilities for financial reporting purposes and the amounts used for income tax purposes.
We received in 2012 a $3,500 payment upon sale of our PRO 140 program and are eligible to receive future milestone and royalty payments in respect of this asset.
Under the Salix License Agreement, we received in 2011 a $60,000 upfront cash payment and $225 in respect of Salix ex-U.S. sublicensee revenue and are eligible to receive development and commercialization milestone payments plus royalties on net sales and 60% of any upfront, milestone, reimbursement or other revenue (net of costs of goods sold, as defined, and territory-specific research and development expense reimbursement) Salix receives from ex-U.S. sublicensees.
Our expenses and reimbursement revenue related to Relistor have declined substantially since Salix assumed direct responsibility for expenses under third-party contracts we have assigned to it. Under the Salix License Agreement, we are reimbursed for Salix approved full-time equivalents (FTE) and third-party development expenses incurred and paid by us after February 3, 2011.
At December 31, 2013, we held $65,860 in cash and cash equivalents, an increase of $7,022 from $58,838 at December 31, 2012. We expect that this amount will be sufficient to fund operations as currently anticipated beyond one year. In addition, at December 31, 2013 and 2012, our investment in auction rate securities classified as long-term assets on the Consolidated Balance Sheets amounted to $2,208 and $3,240, respectively.
If we do not realize sufficient royalty or other revenue from Relistor or other collaboration, license, asset sale, capital raising or other financing transactions, we will have to reduce, delay or eliminate spending on certain programs, and/or take other economic measures.
Cash used in operating activities for 2013 and 2012 was $36,107 and $34,644, respectively, due to excess of expenditures on our research and development programs and general and administrative costs over cash received from collaborators and government grants. Our cash flow from operating activities was positive for 2011, due to the receipt of a $60,000 Salix upfront payment, $225 in respect of Salix ex-U.S. sublicensee revenue and $16,289 in reimbursement payments from Salix and Wyeth, partially offset by expenditures on our research and development programs and general and administrative costs. See Risk Factors.
In 2013, we completed an underwritten public offering under our 2011 shelf registration statement of 9.8 million shares of common stock at a public offering price of $4.40 per share (including the underwriters' overallotment option), resulting in net proceeds of approximately $40.1 million. In December 2012, we completed a public offering of 12.7 million shares of common stock for net proceeds of approximately $23.3 million. During the first quarter of 2014, we established a $150 million replacement shelf registration statement which we used for our recent underwritten public offering of 8.75 million shares of common stock at a public offering price of $4.60 per share, resulting in net proceeds of approximately $37.5 million. We may utilize this shelf registration for the issuance of up to approximately $110 million of additional common stock and other securities.
Sources of Cash
Operating Activities. During 2013 we received $9,686 under our collaborations, primarily consisting of (i) $5,125 in upfront and reimbursement payments from partnering of the C. difficile program, (ii) $3,952 in royalties and reimbursements from Salix, (iii) payments totaling $224 from out-licenses of other assets, and (iv) $385 in reimbursement payments relating to 1404. During 2012 we received $9,393 under collaborations, out-licenses and sale of assets, consisting of (i) $404 in reimbursement payments under the Salix License Agreement, (ii) $5,461 in royalties from Salix, (iii) $3,500 from the sale of our PRO 140 program and (iv) $28 under another out-license. During 2011 we received $79,998 under our collaborations, consisting of (i) $60,000 Salix upfront cash payment and $225 in respect of Salix ex-U.S. sublicensee revenue, (ii) $14,659 in reimbursement payments under the Salix License Agreement, (iii) $1,767 in royalties from Salix, (iv) $3,317 pursuant to our former collaboration with Wyeth Pharmaceuticals and (v) $30 under another out-license.
We have in the past partially funded research programs through awards from the NIH, which we do not expect to receive in the foreseeable future. For 2013, 2012 and 2011, we received $287, $576 and $5,178, respectively, of revenue from all of our NIH awards.
Changes in Accounts receivable and Accounts payable for 2013, 2012 and 2011 resulted from the timing of receipts from Salix, Wyeth, Ono, Fuji, other partnering transactions, and NIH, and payments made to trade vendors in the normal course of business.
We have no committed external sources of funding or capital other than agreements under which collaborators and licensees have contractual obligations to make payments to us. Other than revenues from Relistor, we expect no significant product revenues in the immediate or near-term future, as it will take significant time to bring any of our current product candidates to the commercial marketing stage.
Investing Activities. Approximately 92% of our $65,860 in cash and cash equivalents at December 31, 2013 was invested in money market funds. Auction rate securities of $2,208 consist of securities collateralized by student loan obligations subsidized by the U.S. government, $1,100 of which was redeemed at par during the first and second quarters of 2013. These auction rate securities are rated investment grade by the Standard & Poor's and Moody's rating agencies and have scheduled maturities greater than ten years. During 2013, we realized $174 of proceeds from sales of fixed assets.
Financing Activities. During 2013, net cash provided by financing activities included $40,078 in net proceeds from the issuance of 9.775 million shares of common stock. In addition, during 2013, 2012 and 2011, we received cash of $71, $306 and $3,726, respectively, from exercise of stock options, sales of common stock in satisfaction of severance obligations (in 2012) and under the now-discontinued employee stock purchase plans (in 2011). The amount of cash we receive from these sources fluctuates commensurate with headcount levels and changes in the common stock price on the grant date for options exercised, and on the sale date for shares sold under the employee stock purchase plans.
Unless we obtain regulatory approval for additional product candidates and/or enter into agreements with corporate collaborators with respect to other proprietary assets, we will be required to fund our operations through sales of common stock or other securities or royalty or other financing agreements. Adequate additional funding may not be available to us on acceptable terms or at all. Our inability to raise additional capital on terms reasonably acceptable to us may seriously jeopardize the future success of our business.
Uses of Cash
Operating Activities. The majority of our cash has been used to advance our research and development programs, including conducting pre-clinical studies and clinical trials, pursuing regulatory approvals for product candidates, filing and prosecuting patent applications and defending patent claims. Included in the 2012 period presented below is $2,073 of cash disbursements incurred in connection with a former senior executive first quarter retirement. For various reasons, including the early stage of certain of our programs, the timing and results of our clinical trials, our dependence in certain instances on third parties, many of which are outside of our control, we cannot estimate the total remaining costs to be incurred and timing to complete all our research and development programs.
Research and development costs incurred by project over the past three years were as follows:
| | 2013 | | | 2012 | | | 2011 | |
| | | |
| Oncology | | $ | 33.4 | | | $ | 29.1 | | | $ | 22.2 | |
| Relistor | | | 0.9 | | | | 1.7 | | | | 23.2 | |
| Other programs | | | 0.8 | | | | 2.7 | | | | 8.8 | |
| Total | | $ | 35.1 | | | $ | 33.5 | | | $ | 54.2 | |
We will require additional funding to continue our research and product development programs, conduct pre-clinical studies and clinical trials, pursue regulatory approvals for our product candidates, file and prosecute patent applications and enforce or defend patent claims, if any, fund other operating expenses, and fund product in-licensing and any possible acquisitions.
Investing Activities. During the past three years, we have spent $137, $767 and $226, respectively, on capital expenditures.
Contractual Obligations
Our funding requirements, both for the next 12 months and beyond, will include required payments under operating leases and fixed and contingent payments under licensing, collaboration and other agreements, including those to which our Molecular Insight subsidiary is a party. The following table summarizes our contractual obligations as of December 31, 2013 for future payments under these agreements, including Molecular obligations:
| | | | | Payments due by Year-end | |
| | Total | | | 2014 | | | | 2015-2016 | | | | 2017-2018 | | | Thereafter | |
| | (in millions) | |
| Operating leases | | $ | 13.9 | | | $ | 1.9 | | | $ | 3.8 | | | $ | 4.0 | | | $ | 4.2 | |
| License, collaboration and other agreements: | | | | | | | | | | | | | | | | | | | | |
| Fixed payments | | | 1.3 | | | | 0.3 | | | | 0.4 | | | | 0.6 | | | | - | |
| Contingent payments (1) | | | 105.7 | | | | - | | | | 2.3 | | | | 7.7 | | | | 95.7 | |
| Total | | $ | 120.9 | | | $ | 2.2 | | | $ | 6.5 | | | $ | 12.3 | | | $ | 99.9 | |
_______________
| (1) | At December 31, 2012,Based on assumed achievement of milestones covered under each agreement, the aggregate numbertiming and payment of stock options outstanding for each of our non-employee directors was as follows: Mr. Charles Baker, 95,000; Mr. Briner, 182,500; Mr. Crowley, 155,000; Mr. Dalton, 80,000; Dr. Goff, 95,000; Dr. Maddon, 10,000; Dr. Scheinberg, 115,357; Ms. Williams, 85,000. In general, options granted to non-employee directors vested immediately on the date of grant and the amounts shown represent the grant date fair value of stock awards made during the fiscal year under FASB ASC Topic 718. The grant date fair value of the 2012 option awards was determined using the assumptions disclosed in Note 10 – Share-Based Payment Arrangements to our consolidated financial statements included in our 2012 Annual Report on Form 10-K.which is highly uncertain. |
(2) | Mr. Dalton did not stand for re-election as director at the 2012 Annual Meeting. Dr. Maddon did not receive any additional compensation for services provided as a Board member prior to his retirement on June 13, 2012, and has been compensated as a non-employee director since that date.
|
We periodically assess the scientific progress and merits of each of our programs to determine if continued research and development is commercially and economically viable. Certain of our programs have been terminated due to the lack of scientific progress and prospects for ultimate commercialization. Because of the uncertainties associated with research and development in these programs, the duration and completion costs of our research and development projects are difficult to estimate and are subject to considerable variation. Our inability to complete research and development projects in a timely manner or failure to enter into collaborative agreements could significantly increase capital requirements and adversely affect our liquidity.
Our cash requirements may vary materially from those now planned because of results of research and development and product testing, changes in existing relationships or new relationships with licensees, licensors or other collaborators, changes in the focus and direction of our research and development programs, competitive and technological advances, the cost of filing, prosecuting, defending and enforcing patent claims, the regulatory approval process, manufacturing and marketing and other costs associated with the commercialization of products following receipt of regulatory approvals and other factors.
The above discussion contains forward-looking statements based on our current operating plan and the assumptions on which it relies. There could be deviations from that plan that would consume our assets earlier than planned.
Off-Balance Sheet Arrangements and Guarantees
We have no obligations under off-balance sheet arrangements and do not guarantee the obligations of any other unconsolidated entity.
Critical Accounting Policies
We prepare our financial statements in conformity with accounting principles generally accepted in the U.S. Our significant accounting policies are disclosed in Note 3 to our financial statements included in this Report. The selection and application of these accounting principles and methods requires us to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenues and expenses, as well as certain financial statement disclosures. We evaluate these estimates on an ongoing basis. We base these estimates on historical experience and on various other assumptions that we believe reasonable under the circumstances. The results of these evaluations form the basis for making judgments about the carrying values of assets and liabilities that are not otherwise readily apparent. While we believe that the estimates and assumptions we use in preparing the financial statements are appropriate, they are subject to a number of factors and uncertainties regarding their ultimate outcome and, therefore, actual results could differ from these estimates.
The critical accounting policies we use and the estimates we make are described below. These are policies and estimates that we believe are the most important in portraying our financial condition and results of operations, and that require our most difficult, subjective or complex judgments, often as a result of the need to make estimates about the effect of matters that are inherently uncertain. We have discussed the development, selection and disclosure of these critical accounting policies and estimates with the Audit Committee of our Board of Directors.
Revenue Recognition. We recognize revenue from all sources based on the provisions of the SEC's Staff Accounting Bulletin (SAB) No. 104 (SAB 104) and ASC 605 Revenue Recognition.
The FASB's ASC 605 Revenue Recognition specifies how to separate deliverables in multiple-deliverable arrangements, and how to measure and allocate arrangement consideration to one or more units of accounting, and provides that the delivered item(s) are separate units of accounting, if (i) the delivered item(s) have value to a collaborator on a stand-alone basis, and (ii), if the arrangement includes a general right of return relative to the delivered item, delivery or performance of the undelivered item(s) is considered probable and substantially in our control. We adopted this update on January 1, 2011.
Royalty revenue is recognized based upon net sales of related licensed products, and is recognized in the period the sales occur, provided that the royalty amounts are fixed or determinable, collection of the related receivable is reasonably assured and we have no remaining performance obligations under the arrangement providing for the royalty.
Amounts not expected to be recognized within one year of the balance sheet date are classified as long-term deferred revenue. The classification of deferred revenue as short-term or long-term is based upon the periods in which we expect to perform our collaboration arrangement obligations including non-reimbursable technical assistance.
Share-Based Payment Arrangements. Our share-based compensation of employees includes non-qualified stock options and restricted stock, which are compensatory under ASC 718 Compensation – Stock Compensation. We account for share-based compensation to non-employees, including non-qualified stock options and restricted stock, in accordance with ASC 505 Equity.
The fair value of each non-qualified stock option award is estimated on the date of grant using the Black-Scholes option pricing model. The model requires input assumptions with respect to (i) expected volatility of our common stock, which is based upon the daily quoted market prices on The NASDAQ Stock Market LLC over a period equal to the expected term, (ii) the period of time over which employees, officers, directors and non-employee consultants are expected to hold their options prior to exercise, (iii) expected dividend yield (zero in our case due to never having paid dividends and not expecting to pay dividends in the future), and (iv) risk-free interest rates for periods within the expected term of the options, which are based on the U.S. Treasury yield curve in effect at the time of grant.
Historical volatilities are based upon daily quoted market prices of our common stock on The NASDAQ Stock Market LLC over a period equal to the expected term of the related equity instruments. We rely only on historical volatility since we believe it is generally viewed as providing the most reliable indication of future volatility. In estimating expected future volatility, we assume it will be consistent with historical; we calculate historical volatility using a simple average calculation; we use available historical data for the length of the option's expected term, and we consistently use a sufficient number of price observations. Since our stock options are not traded on a public market, we do not use implied volatility.
The expected term of options granted represents the period of time that options granted are expected to be outstanding based upon historical data related to exercise and post-termination cancellation activity. The expected term of stock options granted to our Chief Executive Officer (CEO) and non-employee directors, consultants and officers are calculated separately from stock options granted to other employees.
We apply a forfeiture rate to the number of unvested awards in each reporting period in order to estimate the number of awards that are expected to vest. Estimated forfeiture rates are based upon historical data on vesting behavior of employees. We adjust the total amount of compensation cost recognized for each award, in the period in which each award vests, to reflect the actual forfeitures related to that award. Changes in our estimated forfeiture rate will result in changes in the rate at which compensation cost for an award is recognized over its vesting period.
Changes in the assumptions used to compute the fair value of the option awards are likely to affect their fair value and the amount of compensation expense recognized in future periods. A higher volatility, longer expected term and higher risk-free rate increases the resulting compensation expense recognized in future periods as compared to prior periods. Conversely, a lower volatility, shorter expected term and lower risk-free rate decreases such expense recognized in future periods as compared to prior periods.
For Boardperformance stock option awards to our CEO (awarded in 2012 and committee service, our directors are entitled to receive:
a $25,000 annual retainer for Board service ($50,000 for service as Chair)2011 only), an option for 10,000 common shares (40,000 in the casevesting occurs upon achievement of the Chair), and $2,000 per meeting attendance fees;
an annual retainer fee for committee service as (i) Audit Committee Chair ($12,000; currently Ms. Williams); (ii) Compensation Committee Chair ($8,000; currently Mr. Crowley); (iii) Nominating and Corporate Governance Committee Chair ($5,000; currently Mr. Charles Baker); and (iv) Science and Technology Committee and Strategy and External Technology Subcommittee Chair ($2,500 each; currently Dr. Scheinberg); and,
reflecting differing Committee meeting formats and frequencies, (i) per-meeting attendance fees of $1,000 for the Audit, Nominating and Corporate Governance, Compensation, Strategic Review and ad hoc Committees, and $7,500 for the Science & Technology Committee; and (ii) a $10,000 annual retainer for Strategy and External Technology Subcommittee service.
Optionspecified performance-based milestones. The awards, for Board service during 2012 were made under the 2005 Stock Incentive Plan, as described in the CD&A. These non-qualified options generally vest on and expire ten years after the grant date andwhich have an exercise price equal to the closing price of Progenicsour common stock on the date of grant, date (for the 2012 grant, $7.93 per share). Fair values have been determinedare valued using the assumptions for calculatingBlack-Scholes option pricing model: the expense amounts as set forthrelated to these grants is recognized during the period, if any, in Note 10 – Share-Based Payment Arrangements to the consolidated financial statements included in our 2012 Annual Report on Form 10-K, and for the 2012 grant was $5.35 per share.which each performance milestone is achieved.
Compensation Committee InterlocksClinical Trial and Insider Participation.Other Research and Development Expenses. The Compensation Committee's membersClinical trial expenses, which are independent directors. As reportedincluded in Item 13 -- Certain Relationshipsresearch and Related Transactions,development expenses, represent obligations resulting from contracts with various clinical investigators and Director Independence, Progenics in 2012 entered into a financial advisory agreement with MTS Health Partners, L.P., of which Mr. Crowley is a Senior Managing Director and partner, on customary terms and conditions, whereby MTS will receive a monthly retainer of $10,000 during the term of the agreement (which may be terminated by either party on 30 days notice) and $300,000 for MTS' servicesclinical research organizations in connection with conducting clinical trials for our product candidates. Such costs are expensed as incurred, and are generally based on the Company'stotal number of patients in the trial, the rate at which the patients enter the trial and the period over which the clinical investigators and clinical research organizations to provide services. We believe that this method best aligns the efforts expended on a clinical trial with the expenses we record. We adjust our rate of clinical expense recognition if actual results differ from our estimates. In addition to clinical trial expenses, we estimate the amounts of other research and development expenses, for which invoices have not been received at the end of a period, based upon communication with third parties that have provided services or goods during the period. Such estimates are subject to change as additional information becomes available.
Fair Value Measurements. Our available-for-sale investment portfolio consists of money market funds and $2.4 million face amount of auction rate securities (ARS), and is recorded at fair value in the accompanying Consolidated Balance Sheets in accordance with ASC 320 Investments – Debt and Equity Securities. The change in the fair value of these investments is recorded as a component of other comprehensive income (loss). We expect to recover the amortized cost of all of our investments at maturity. We currently do not anticipate having to sell these securities in order to operate our business and we believe that it is not more likely than not that we will be required to sell these securities before recovery of principal. We do not believe the carrying values of our ARS investments are other than temporarily impaired and therefore expect the positions will eventually be liquidated without significant loss. We monitor markets for our investments and consider the impact, if any, of market conditions on the fair market value of our investments.
Valuation of securities is subject to uncertainties that are difficult to predict, such as changes to credit ratings of the securities and/or underlying assets supporting them, default rates applicable to the underlying assets, underlying collateral value, discount rates, counterparty risk, ongoing strength and quality of market credit and liquidity and general economic and market conditions. The valuation of the ARS in our portfolio is based on an internal analysis of timing of expected future successful auctions, collateralization of underlying assets of the security and credit quality of the security.
In –Process Research and Development and Goodwill. In connection with the acquisition of Molecular Insight. Neither Mr. Charles Baker nor Mr. Mark Dalton, who wasInsight, we have established a memberpolicy for accounting for intangible assets, under which in process research and development (IPR&D) and goodwill are initially measured at fair value and capitalized as an intangible asset and an impairment test for these intangibles is performed annually in the fourth quarter, unless impairment indicators require an earlier evaluation. Upon and subject to commercialization of these candidates, the IPR&D will be amortized over the relevant estimated useful life.
Contingent Consideration Liability. The estimated fair value of the Compensation Committee during 2012 before his retirement fromcontingent consideration liability, initially measured and recorded on the Board, had any affiliationacquisition date, is considered to be a Level 3 instrument and is reviewed quarterly, or relationship withwhenever events or circumstances occur that indicate a change in fair value. The contingent consideration liability is recorded at fair value at the Company which requires disclosure under this heading.end of each period.
Item
12. Security Ownership of Certain Beneficial Owners7A. Quantitative and
Management and Related Stockholder Matters.
Security Ownership of Certain Beneficial Owners and Management
The following table sets forth certain information as of March 31, 2013, except as noted, regarding the beneficial ownership of the common stock by (i) each person or group known to us to be the beneficial owner of more than five percent of our common stock outstanding, (ii) each of our directors and executive officers and (iii) all of our directors and executive officers as a group.
| | Shares Beneficially Owned(2) |
Name and Address of Beneficial Owner(1) | | Number | | Percent |
Federated Investors, Inc.(3) Federated Investors Tower Pittsburgh, PA 15222-3779 | | 12,538,268 | | 24.52 % |
FMR LLC(4) 82 Devonshire Street Boston, MA 02109 | | 5,146,395 | | 10.06 % |
Broadfin Capital, LLC(5) 237 Park Avenue, Suite 900 New York, NY 10017 | | 3,651,666 | | 7.14% |
Tudor Investment Corporation and affiliates(6) 1275 King Street Greenwich, CT 06831 | | 2,888,513 | | 5.65% |
Charles A. Baker(7) | | 116,481 | | * |
Mark R. Baker(8) | | 739,403 | | 1.43% |
Kurt W. Briner(9) | | 213,000 | | * |
Peter J. Crowley(10) | | 150,000 | | * |
Stephen P. Goff(11) | | 158,500 | | * |
Paul J. Maddon(12) | | 1,924,890 | | 3.69% |
David A. Scheinberg(13) | | 160,289 | | * |
Nicole S. Williams(14) | | 90,000 | | * |
Angelo W. Lovallo, Jr. (15) | | 20,563 | | * |
Robert A. McKinney(16) | | 276,913 | | * |
Robert J. Israel(17) | | 209,549 | | * |
William C. Olson(18) | | 237,543 | | * |
Ann Marie Assumma (19) | | 128,869 | | * |
All directors and executive officers as a group(20) | | 4,426,000 | | 8.16% |
____________
* Less than one percent.
| (1) | The address of each beneficial owner who is a director or officer of the Company is c/o the Company. |
| (2) | With respect to our directors and executive officers, and except as indicated and/or pursuant to applicable community property laws, each stockholder possesses sole voting and investment power with respect to the shares of common stock listed. The number of shares of common stock beneficially owned includes the shares issuable pursuant to stock options held by the stockholder that are currently exercisable, i.e., within 60 days of March 31, 2013. Shares issuable upon exercise of these options are deemed outstanding for computing the percentage of beneficial ownership of the person holding the options but not of any other person. None of the shares held by our directors and executive officers are pledged as collateral. |
With respect to other stockholders identified above, the percent reported is calculated by dividing (i) the number of shares reported by the stockholder in its Schedule 13G filing as described in the related note by (ii) the aggregate number of our common shares outstanding on March 31, 2013, and differs from the Percent of Class reported in the stockholder's Schedule 13G; it assumes that the stockholder continued to own the number shares reported in its Schedule 13G on March 31, 2013.
| (3) | Based on a Schedule 13G (Amendment No. 8) filed February 12, 2013, which reports that Federated Investors, Inc. is the parent holding company of Federated Equity Management Company of Pennsylvania and Federated Global Investment Management Corp., which act as investment advisers to registered investment companies and separate accounts that own the shares reported. Such document also reports that the investment advisers are wholly owned subsidiaries of FII Holdings, Inc., which is wholly owned subsidiary of Federated Investors, Inc., all of whose outstanding voting stock is held in the Voting Shares Irrevocable Trust for which John F. Donahue, Rhodora J. Donahue and J. Christopher Donahue act as trustees. |
| (4) | Based on a Schedule 13G (Amendment No. 2) filed on February 14, 2013, which reports that Fidelity Management & Research Company (Fidelity), a wholly-owned subsidiary of FMR LLC (FMR) and an investment adviser registered under Section 203 of the Investment Advisers Act of 1940, is the beneficial owner of the shares reported as a result of acting as investment adviser to various investment companies registered under Section 8 of the Investment Company Act of 1940. The ownership of one investment company, Fidelity Select Biotechnology Portfolio, amounted to 4,451,226 shares or 9.919% of Progenics common stock outstanding according to such document. Such document also reports that (i) Edward C. Johnson 3d and FMR, through its control of Fidelity, and the funds each has sole power to dispose of the shares reported owned by the Funds; (ii) members of the family of Edward C. Johnson 3d, Chairman of FMR, are the predominant owners, directly or through trusts, of Series B voting common shares of FMR, representing 49% of the voting power of FMR; (iii) the Johnson family group and all other Series B shareholders have entered into a shareholders' voting agreement under which all Series B voting common shares will be voted in accordance with the majority vote of Series B voting common shares, and accordingly, through their ownership of voting common shares and the execution of the shareholders' voting agreement, members of the Johnson family may be deemed, under the Investment Company Act of 1940, to form a controlling group with respect to FMR; (iv) neither FMR nor Edward C. Johnson 3d, Chairman of FMR, has the sole power to vote or direct the voting of the shares owned directly by the Fidelity Funds, which power resides with the Funds' Boards of Trustees; and (v) Fidelity carries out the voting of the shares under written guidelines established by the Funds' Boards of Trustees. |
| (5) | Based on a Schedule 13G filed on December 10, 2012, which reports that Broadfin Capital, LLC, Broadfin Healthcare Master Fund, Ltd., and Kevin Kotler have shared power to vote or to direct the vote and to dispose or to direct the disposition of the securities reported. |
| (6) | Based on a Schedule 13G (Amendment No. 14) filed February 14, 2013, which reports that the number of reported shares owned by Tudor Investment Corporation (TIC), Tudor Group Holdings LLC (TGH), Paul Tudor Jones, II and/or Mark F. Dalton include shares owned directly by The Tudor BVI Global Portfolio L.P., a limited partnership organized under the laws of the Cayman Islands (Tudor BVI) (1,308,892 shares), Tudor Global Fund L.P. (TGF) (511,176 shares), Tudor Proprietary Trading, L.L.C. (219,107 shares), and/or Tudor Global Trading LLC (TGT) (15,400 shares). Such document also reports that (i) because TIC provides investment advisory services to each of Tudor BVI and TGF and acts as general partner of TGF, TIC may be deemed to beneficially own the shares owned by such entities, and disclaims beneficial ownership of such shares; (ii) TGH holds a majority of the equity interests of TGT and indirectly holds a majority of the membership interests of TPT, and disclaims beneficial ownership of the Shares beneficially owned by each of such entities; (iii) Mr. Dalton, as the sole general partner of D.F. Partners, a New York limited partnership, may be deemed to beneficially own the shares owned by such entity (16,500 shares), and disclaims beneficial ownership of such shares; (iv) Mr. Jones is the Co-Chairman and principal equity owner of TIC and TGH, and disclaims beneficial ownership of the shares beneficially owned, or deemed beneficially owned, by such entities; (v) Mr. Dalton is (1) the Co-Chairman, Chief Executive Officer, President, and an equity owner of TIC and (2) the Co-Chairman, Chief Executive Officer, and an equity owner of TGH, and disclaims beneficial ownership of the shares beneficially owned, or deemed beneficially owned, by such entities; and (vi) the shares reported in such document under Items 5 and 7 of Mr. Jones' cover page include 4,600 shares held directly by Mr. Jones' individual retirement account, and the shares reported in such document under Items 5 and 7 of Mr. Dalton's cover page include 87,500 immediately exercisable options and 78,500 of the shares. |
| (7) | Includes 21,481 shares owned by the Baker Family Limited Partnership and 95,000 issuable upon exercise of currently exercisable options. |
| (8) | Includes 161,140 shares outstanding and 578,263 issuable upon exercise of currently exercisable options. |
| (9) | Includes 33,000 shares outstanding and 180,000 issuable upon exercise of currently exercisable options. |
| (10) | Consists of 150,000 shares issuable upon exercise of currently exercisable options. |
| (11) | Includes 66,000 shares outstanding and 92,500 issuable upon exercise of currently exercisable options. |
| (12) | Includes 854,057 shares outstanding and 1,070,833 issuable upon exercise of currently exercisable options. |
| (13) | Includes 46,182 shares outstanding and 114,107 issuable upon exercise of currently exercisable options. |
| (14) | Includes 5,000 shares outstanding and 85,000 issuable upon exercise of currently exercisable options. |
| (15) | Includes 2,945 shares outstanding, of which 444 are restricted shares subject to forfeiture, and 17,174 shares issuable upon exercise of currently exercisable options. |
| (16) | Includes 41,081 shares outstanding, of which 3,333 are restricted, and 232,499 issuable upon exercise of currently exercisable options. |
| (17) | Includes 31,717 shares outstanding, of which 3,333 are restricted, and 174,499 issuable upon exercise of currently exercisable options. |
| (18) | Includes 42,211 shares outstanding, of which 3,333 are restricted, and 191,999 issuable upon exercise of currently exercisable options. |
| (19) | Includes 15,193 shares outstanding, of which 2,777 are restricted, and 110,899 issuable upon exercise of currently exercisable options. |
| (20) | Includes 1,320,007 shares outstanding, of which 13,220 are restricted, and 3,092,773 issuable upon exercise of currently exercisable options held by directors, NEOs and other executive officers of the Company. |
Equity Compensation Plan Information
The following table sets forth certain information related to our equity compensation plans as of April 17, 2013.
| | Number of shares to be issued upon exercise of outstanding options, warrants and rights(1) | | | Weighted average exercise price of outstanding options, warrants and rights | | | Number of shares remaining available for future issuance (excluding securities reflected in first column)(2) | |
| Equity compensation plans approved by stockholders | | | 5,999,976 | | | $ | 12.26 | | | | 1,979,090 | |
| Equity compensation plans not approved by stockholders | | | - | | | | - | | | | - | |
Total(3) | | | 5,999,976 | | | $ | 12.26 | | | | 1,979,090 | |
____________
1. | The weighted average remaining life of outstanding options is 5.35 years. |
2. | Does not reflect the "fungible share" counting method we use to distinguish between options and stock appreciation rights, on the one hand, and awards other than options and SARs, on the other. |
3. | Does not include 27,460 unvested restricted stock awards outstanding under equity compensation plans. |
Item 13. Certain Relationships and Related Transactions, and Director Independence.
Certain Relationships and Related TransactionsQualitative Disclosures about Market Risk
Our Codeprimary investment objective is to preserve principal. Our money market funds and ARS have interest rates that were variable and totaled $62,572 at December 31, 2013. As a result, we do not believe that these investment balances have a material exposure to interest-rate risk.
At that date, we held approximately $2,208 (3.53% of Business Ethicsassets measured at fair value) carrying amount of ARS, in respect of which we have received all scheduled interest payments. The principal amount of these remaining ARS will not be accessible until the issuer calls or restructures the underlying security, the underlying security matures and Conduct, which our Corporate Governance Guidelines makeis paid or a buyer outside the auction process emerges.
We continue to monitor the market for ARS and consider the impact, if any, of market conditions on the fair market value of these investments. We believe that the failed auctions experienced to date are not a result of the deterioration of the underlying credit quality of these securities, although valuation of them is subject to uncertainties that are difficult to predict, such as changes to credit ratings of the securities and/or the underlying assets supporting them, default rates applicable to all directorsthe underlying assets, underlying collateral value, discount rates, counterparty risk, ongoing strength and employees, includingquality of market credit and liquidity, and general economic and market conditions. We do not believe the carrying values of the ARS that we hold are other than temporarily impaired and therefore expect the positions will eventually be liquidated without significant loss.
The valuation of the ARS we hold is based on an internal analysis of timing of expected future successful auctions, collateralization of underlying assets of the security and credit quality of the security. As a result of re-evaluating the valuation of these securities as of December 31, 2013, we reduced the temporary impairment amount to $192 from $260 at December 31, 2012. A 100 basis point increase to our CEO and PFO, requires all Progenics personnel to actinternal analysis would result in a $24 increase in the best intereststemporary impairment of the Company consistent with their dutythese securities as of loyaltyDecember 31, 2013.
Item 8. Financial Statements and Supplementary Data
See page F-1, Index to it, including by avoiding situationsConsolidated Financial Statements.
Item 9. Changes in and
relationshipsDisagreements With Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
We maintain disclosure controls and procedures that involve actual or potential conflicts of interest. Situationsare designed to ensure that could be perceived as conflicts of interest include related party transactions. The Code requires Progenics personnel who believe they are involved in or become aware of a potential conflict of interest to discuss the matter with the individual's manager and our General Counsel. Our Audit Committee is required and empowered to meet with our management and independent auditors to review all related party transactions that would beinformation required to be disclosed in our annual proxy statement for potential conflicts of interest situationsExchange Act reports is recorded, processed, summarized and on an ongoing basis, approve such transactions. The Audit Committee's policy is to approve only those transactions that arereported within the timelines specified in the best interestsSEC's rules and forms, and that such information is accumulated and communicated to our management, including our Chief Executive Officer (CEO) and Principal Financial Officer (PFO), as appropriate, to allow timely decisions regarding required disclosure. In designing and evaluating the disclosure controls and procedures, management recognized that any controls and procedures, no matter how well designed and operated, can only provide reasonable assurance of achieving the desired control objectives, and in reaching a reasonable level of assurance, management necessarily was required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures. We have a Disclosure Committee consisting of members of our stockholders. In addition,senior management which monitors and implements our Nominating and Corporate Governance Committee is required and empowered to conduct any and all investigations into alleged violationspolicy of our Corporate Governance Guidelines or Code of Business Ethics and Conduct, and presentdisclosing material information concerning the results of such investigations to our Board.Company in accordance with applicable law.
As required by SEC rules,Rule 13a-15(e), we disclosecarried out an evaluation, under the supervision and with the participation of our management, including our CEO and PFO, of the effectiveness of the design and operation of our disclosure controls and procedures as of the end of the period covered by this report. Based on the foregoing, our CEO and PFO concluded that our current disclosure controls and procedures, as designed and implemented, were effective at the reasonable assurance level.
Changes in Internal Control over Financial Reporting
There have been no changes in our Proxy Statement, and are disclosing in this Form 10-K/A, under this heading any relevant transactions. We disclosed in Note 9(e) to our consolidatedinternal control over financial statements included in our 2012 Annual Report on Form 10-K that in December 2012, Progenics entered into a financial advisory agreement with MTS Health Partners, L.P., of which Board Chair Mr. Crowley is a Senior Managing Director and partner, on customary terms and conditions, whereby MTS will receive a monthly retainer of $10,000 during the term of the agreement (which may be terminated by either party on 30 days notice), $300,000 for MTS' services in connection with the Company's acquisition of Molecular Insight, and in connection with other transactions, if any, as to which MTS provides services to the Company, such other amounts as the parties may mutually agree.
Director Independence and Other Qualifications
The Board has determined each of Messrs. Charles Baker, Briner and Crowley, Drs. Goff and Scheinberg and Ms. Williams to be an "independent director"reporting, as such term is defined by NASDAQ Marketplace Rule 5605(a)(2).
The Board has also determinedin the Exchange Act Rules 13a-15(f) and 15d-15(f) during our fiscal quarter ended December 31, 2013 that each member of the Audit Committee, the Compensation Committee and the Nominating and Corporate Governance Committee meets the independence requirements applicablehave materially affected, or are reasonably likely to those committees prescribed by the NASDAQ Marketplace rules, the SEC and the Internal Revenue Service.
With the assistance ofmaterially affect, our legal counsel, the Nominating and Corporate Governance Committee reviewed the applicable legal standards for Board member and Board committee independence and the criteria applied to determine "audit committeeinternal control over financial expert" status, as well as the answers to annual questionnaires completed by each of our directors. On the basis of this review, the Committee delivered a report to the full Board and the Board made its independence and "audit committee financial expert" determinations based upon the Committee's report and each member's review of the information made available to the Committee.reporting.
2139
Management's Report on Internal Control Over Financial Reporting
Item 14. Principal Accounting FeesInternal control over financial reporting is a process designed by, or under the supervision of, our CEO and ServicesPFO and effected by our Board, management and other personnel to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Our management is responsible for establishing and maintaining adequate internal control over financial reporting which includes policies and procedures that:
| · | Pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of assets; |
| |
| · | Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures are being made only in accordance with authorization of management and directors; and |
| |
| · | Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of assets that could have a material effect on the financial statements. |
Fees Billed for Services RenderedBecause of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Management has used the framework set forth in the report entitled Internal Control – Integrated Framework issued by Current and Former Accounting Firms
the Committee of Sponsoring Organizations of the Treadway Commission (1992 framework), known as COSO, to evaluate the effectiveness of our internal control over financial reporting. Management has concluded that our internal control over financial reporting was effective as of December 31, 2013. The following table discloses the fees thateffectiveness of our internal control over financial reporting has been audited by Ernst & Young LLP, (E&Y), ouran independent registered public accounting firm, foras of December 31, 2013 billed oras stated in their report which is expected to bill for professional services rendered to us for 2012, and fees that our 2011 accountant, PricewaterhouseCoopers LLP (PwC), billed for professional services rendered to us for 2011 and 2012:provided below.
| | 2012 | | | 2011 | |
Audit Fees(1) | | $ | 464,000 | | | $ | 587,204 | |
| Audit Related Fees | | | - | | | | 40,650 | (2) |
Tax Fees(3) | | | 15,000 | | | | 108,000 | |
All Other Fees(4) | | | 4,075 | | | | 1,933 | |
____________
| (1) | In connection with (i) the audit of our annual financial statements, including attestation services required under section 404 of the Sarbanes-Oxley Act of 2002 and reviews of quarterly interim financial statements ($319,000 by E&Y and $10,000 by PwC in 2012; $561,204 by PwC in 2011), (ii) filing of registration statements with the SEC ($10,000 by E&Y and $5,000 by PwC in 2012; $20,000 by PwC in 2011), (iii) statutory non-U.S. audit fees ($6,000 by PwC in 2011), and (iv) the Company's 2012 follow-on public offering ($60,000 by E&Y and $60,000 by PwC). |
| (2) | In connection with accounting consultations concerning financial accounting and reporting matters by PwC. |
| (3) | In connection with tax return preparation ($15,000 by E&Y in 2012 and $38,000 by PwC in 2011), and tax advice and consultations regarding IRC Section 382 analysis ($70,000 by PwC in 2011). |
| (4) | For proprietary internet-based services ($2,142 by E&Y and $1,933 by PwC in 2012; $1,933 by PwC in 2011) . |
Pre-Approval of Audit and Non-Audit Services by the Audit Committee
Audit and non-audit services performed for the Company by its independent registered public accounting firm must be pre-approved by the Audit Committee in order to assure that the provision of such services does not impair the accounting firm's independence. In April of each year, the Committee reviews a schedule prepared by the accounting firm of certain types of services to be provided for that year along with projected fees. The Committee reviews the schedule and provides general pre-approval of those types of services. The fee amounts are updated to the extent necessary at regularly scheduled meetings of the Committee. Any additional service proposed to be provided after the annual pre-approval process requires specific pre-approval by the Audit Committee. The Committee may delegate either general or specific pre-approval authority to its chair or any other member(s). The member(s) to whom such authority is delegated must report, for informational purposes only, any pre-approval decisions to the Committee at its next meeting. The Committee approved all services described above during 2012 and 2011.
2240
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders of Progenics Pharmaceuticals, Inc.
We have audited Progenics Pharmaceuticals, Inc.'s internal control over financial reporting as of December 31, 2013, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (1992 framework) (the COSO criteria). Progenics Pharmaceuticals, Inc.'s management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management's Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company's internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company's internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company's internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company's assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Progenics Pharmaceuticals, Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2013, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of Progenics Pharmaceuticals, Inc. as of December 31, 2013 and 2012, and the related consolidated statements of operations, comprehensive (loss) income, stockholders' equity, and cash flows for the years then ended of Progenics Pharmaceuticals, Inc. and our report dated March 13, 2014 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Hartford, Connecticut
March 13, 2014
Item 9B. Other Information
None.
PART III
The information required by the Form 10-K Items listed in the following table will be included under the respective headings specified for such Items in our definitive proxy statement for our 2014 Annual Meeting of Stockholders to be filed with the SEC:
| Item of Form 10-K | Location in 2014 Proxy Statement |
| | |
| Directors, Executive Officers and Corporate Governance | Election of Directors. Executive and Other Officers. Corporate Governance. Code of Business Ethics and Conduct.* Section 16(a) Beneficial Ownership Reporting and Compliance. *The full text of our Code of Business Ethics and Conduct is available on our website (www.progenics.com). |
| | |
| Item 11. | Executive Compensation | Executive Compensation. Compensation Committee Report. Compensation Committee Interlocks and Insider Participation. |
| | |
| Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters | Equity Compensation Plan Information. Security Ownership of Certain Beneficial Owners and Management. |
| | |
| Item 13. | Certain Relationships and Related Transactions, and Director Independence | Certain Relationships and Related Transactions. Affirmative Determinations Regarding Director Independence and Other Matters. |
| | |
| Principal Accounting Fees and Services | Fees Billed for Services Rendered by our Independent Registered Public Accounting Firm. Pre-approval of Audit and Non-Audit Services by the Audit Committee. |
PART IV
Item 15. Exhibits, Financial Statement Schedules
The following documents or the portions thereof indicated are filed as a part of this Annual Report.
| (a) | Documents filed as part of this Annual Report: |
Consolidated Financial Statements of Progenics Pharmaceuticals, Inc.:
Reports of Independent Registered Public Accounting Firms
Consolidated Balance Sheets at December 31, 2013 and 2012
Consolidated Statements of Operations for the years ended December 31, 2013, 2012 and 2011
Consolidated Statements of Comprehensive (Loss) Income for the years ended December 31, 2013, 2012 and 2011
Consolidated Statements of Stockholders' Equity for the years ended December 31, 2013, 2012 and 2011
Consolidated Statements of Cash Flows for the years ended December 31, 2013, 2012 and 2011
Notes to Consolidated Financial Statements
| (b) | Financial Statement Schedules |
Schedule II – Valuation and Qualifying Accounts
Financial statement schedules referred to in Item 12-01 of Regulation S-X and not listed above are inapplicable and therefore have been omitted.
| (c) | Item 601 Exhibits:Exhibits |
Those exhibitsExhibits required to be filed by Item 601 of Regulation S-K are listed in the Exhibit Index immediately following the signature page hereofof this Report and preceding the exhibits filed herewith, which is incorporated herein by this reference.
PROGENICS PHARMACEUTICALS, INC.
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
| Page |
| F-2 |
| Financial Statements: | |
| F-3 |
| F-4 |
| F-5 |
| F-6 |
| F-7 |
| F-8 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders of Progenics Pharmaceuticals, Inc.
We have audited the accompanying consolidated balance sheets of Progenics Pharmaceuticals, Inc. as of December 31, 2013 and 2012, and the related consolidated statements of operations, comprehensive (loss) income, stockholders' equity, and cash flows for the years then ended. Our audits also included the financial statement schedule listed in the Index at Item 15(b). These financial statements and schedule are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements and schedule based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of Progenics Pharmaceuticals, Inc. at December 31, 2013 and 2012, and the consolidated results of its operations and its cash flows for the years then ended, in conformity with U.S. generally accepted accounting principles. Also, in our opinion, the related financial statement schedule, when considered in relation to the basic financial statements taken as a whole, presents fairly in all material respects information set forth therein.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Progenics Pharmaceuticals, Inc.'s internal control over financial reporting as of December 31, 2013, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (1992 framework) and our report dated March 13, 2014 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Hartford, Connecticut
March 13, 2014
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of Progenics Pharmaceuticals, Inc.
In our opinion, the consolidated statements of operations, comprehensive income (loss), of stockholders' equity and of cash flows for the year ended December 31, 2011 present fairly, in all material respects, the results of operations and cash flows of Progenics Pharmaceuticals, Inc. and its subsidiaries for the year ended December 31, 2011, in conformity with accounting principles generally accepted in the United States of America. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements based on our audit. We conducted our audit of these statements in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. We believe that our audit provides a reasonable basis for our opinion.
/s/ PricewaterhouseCoopers LLP
New York, New York
March 15, 2012
PROGENICS PHARMACEUTICALS, INC.
CONSOLIDATED BALANCE SHEETS (amounts in thousands, except for par value and share amounts)
| | December 31, | |
| | 2012 | | | 2012 | |
| Assets | | | | | | |
| Current assets: | | | | | | |
| Cash and cash equivalents | | $ | 65,860 | | | $ | 58,838 | |
| Accounts receivable, net | | | 2,879 | | | | 6,937 | |
| Other current assets | | | 1,943 | | | | 1,692 | |
| Total current assets | | | 70,682 | | | | 67,467 | |
| Auction rate securities | | | 2,208 | | | | 3,240 | |
| Fixed assets, at cost, net of accumulated depreciation and amortization | | | 2,413 | | | | 3,399 | |
| Intangible assets, net (Note 3) | | | 31,379 | | | | - | |
| Goodwill | | | 7,702 | | | | - | |
| Deferred tax assets – long term | | | - | | | | 2,052 | |
| Other assets | | | 157 | | | | 150 | |
| Total assets | | $ | 114,541 | | | $ | 76,308 | |
| | | | | | | | |
| Liabilities and Stockholders' Equity | | | | | | | | |
| Current liabilities: | | | | | | | | |
| Accounts payable and accrued expenses | | $ | 6,512 | | | $ | 5,640 | |
| Deferred tax liability - current | | | - | | | | 2,069 | |
| Deferred revenue - current | | | - | | | | 838 | |
| Other current liabilities | | | 115 | | | | 115 | |
| Total current liabilities | | | 6,627 | | | | 8,662 | |
| Contingent consideration liability | | | 15,700 | | | | - | |
| Deferred tax liability - long term | | | 12,321 | | | | - | |
| Other liabilities | | | 914 | | | | 1,078 | |
| Total liabilities | | | 35,562 | | | | 9,740 | |
| Commitments and contingencies (Note 10) | | | | | | | | |
| Stockholders' equity: | | | | | | | | |
| Preferred stock, $.001 par value; 20,000,000 shares authorized; issued and outstanding – none | | | - | | | | - | |
| Common stock, $.0013 par value; shares authorized - 160,000,000 in 2013 and 80,000,000 in 2012; issued – 61,025,404 in 2013 and 46,765,472 in 2012 | | | 79 | | | | 61 | |
| Additional paid-in capital | | | 548,510 | | | | 493,613 | |
| Accumulated deficit | | | (466,677 | ) | | | (424,105 | ) |
| Accumulated other comprehensive loss | | | (192 | ) | | | (260 | ) |
| Treasury stock, at cost (200,000 shares in 2013 and 2012) | | | (2,741 | ) | | | (2,741 | ) |
| Total stockholders' equity | | | 78,979 | | | | 66,568 | |
| Total liabilities and stockholders' equity | | $ | 114,541 | | | $ | 76,308 | |
The accompanying notes are an integral part of the financial statements.
PROGENICS PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS (amounts in thousands, except net (loss) income per share)
| | Years Ended December 31, | |
| | 2013 | | | 2012 | | | 2011 | |
| Revenues: | | | | | | | | | |
| Royalty income | | $ | 5,923 | | | $ | 4,963 | | | $ | 3,046 | |
| Collaboration revenue | | | 1,595 | | | | 8,525 | | | | 76,764 | |
| Research grants | | | 275 | | | | 488 | | | | 4,810 | |
| Other revenues | | | 69 | | | | 72 | | | | 176 | |
| Total revenues | | | 7,862 | | | | 14,048 | | | | 84,796 | |
| | | | | | | | | | | | |
| Expenses: | | | | | | | | | | | | |
| Research and development | | | 33,903 | | | | 31,840 | | | | 53,183 | |
| License fees – research and development | | | 567 | | | | 1,170 | | | | 578 | |
| Royalty expense | | | 624 | | | | 499 | | | | 405 | |
| General and administrative | | | 14,809 | | | | 14,706 | | | | 18,248 | |
| Depreciation and amortization | | | 939 | | | | 1,324 | | | | 2,066 | |
| Total expenses | | | 50,842 | | | | 49,539 | | | | 74,480 | |
| | | | | | | | | | | | |
| Operating (loss) income | | | (42,980 | ) | | | (35,491 | ) | | | 10,316 | |
| | | | | | | | | | | | |
| Other income: | | | | | | | | | | | | |
| Interest income | | | 46 | | | | 60 | | | | 65 | |
| Total other income | | | 46 | | | | 60 | | | | 65 | |
| | | | | | | | | | | | |
| Net (loss) income before provision for income taxes | | | (42,934 | ) | | | (35,431 | ) | | | 10,381 | |
| | | | | | | | | | | | |
| Income tax benefit | | | 362 | | | | - | | | | - | |
| | | | | | | | | | | | |
| Net (loss) income | | $ | (42,572 | ) | | $ | (35,431 | ) | | $ | 10,381 | |
| | | | | | | | | | | | |
| Net (loss) income per share - basic | | $ | (0.76 | ) | | $ | (1.02 | ) | | $ | 0.31 | |
| Weighted-average shares - basic | | | 55,798 | | | | 34,754 | | | | 33,375 | |
| | | | | | | | | | | | |
| Net (loss) income per share - diluted | | $ | (0.76 | ) | | $ | (1.02 | ) | | $ | 0.31 | |
| Weighted-average shares - diluted | | | 55,798 | | | | 34,754 | | | | 33,494 | |
The accompanying notes are an integral part of the financial statements.
PROGENICS PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF COMPREHENSIVE (LOSS) INCOME (amounts in thousands)
| | Years Ended December 31, | |
| | 2013 | | | 2012 | | | 2011 | |
| Net (loss) income | | $ | (42,572 | ) | | $ | (35,431 | ) | | $ | 10,381 | |
| Other comprehensive income: | | | | | | | | | | | | |
| Net change in unrealized loss on auction rate securities | | | 68 | | | | 8 | | | | 24 | |
| Total other comprehensive income | | | 68 | | | | 8 | | | | 24 | |
| Comprehensive (loss) income | | $ | (42,504 | ) | | $ | (35,423 | ) | | $ | 10,405 | |
The accompanying notes are an integral part of the financial statements.
PROGENICS PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS' EQUITY For the Years Ended December 31, 2013, 2012 and 2011
(amounts in thousands)
| | Common Stock | | | Additional | | | | | | Accumulated Other | | | Treasury Stock | | | | |
| | Shares | | | Amount | | | Paid-In Capital | | | Accumulated Deficit | | | Comprehensive Income (Loss) | | | Shares | | | Amount | | | Total | |
| Balance at December 31, 2010 | | | 33,326 | | | $ | 43 | | | $ | 453,353 | | | $ | (399,055 | ) | | $ | (292 | ) | | | (200 | ) | | $ | (2,741 | ) | | $ | 51,308 | |
| Net loss | | | - | | | | - | | | | - | | | | 10,381 | | | | - | | | | - | | | | - | | | | 10,381 | |
| Other comprehensive income | | | - | | | | - | | | | - | | | | - | | | | 24 | | | | - | | | | - | | | | 24 | |
| Compensation expenses for share-based payment arrangements | | | - | | | | - | | | | 6,362 | | | | - | | | | - | | | | - | | | | - | | | | 6,362 | |
| Forfeitures of restricted stock | | | (38 | ) | | | - | | | | - | | | | - | | | | - | | | | - | | | | - | | | | - | |
| Sale of common stock under employee stock purchase plans and exercise of stock options | | | 758 | | | | 1 | | | | 3,725 | | | | - | | | | - | | | | - | | | | - | | | | 3,726 | |
| Balance at December 31, 2011 | | | 34,046 | | | | 44 | | | | 463,440 | | | | (388,674 | ) | | | (268 | ) | | | (200 | ) | | | (2,741 | ) | | | 71,801 | |
| Net income | | | - | | | | - | | | | - | | | | (35,431 | ) | | | - | | | | - | | | | - | | | | (35,431 | ) |
| Other comprehensive income | | | - | | | | - | | | | - | | | | - | | | | 8 | | | | - | | | | - | | | | 8 | |
| Compensation expenses for share-based payment arrangements | | | - | | | | - | | | | 6,536 | | | | - | | | | - | | | | - | | | | - | | | | 6,536 | |
| Sale of common stock in public offering, net of underwriting discounts and commissions ($1,518) and offering expenses ($434) | | | 12,650 | | | | 17 | | | | 23,331 | | | | - | | | | - | | | | - | | | | - | | | | 23,348 | |
| Forfeitures of restricted stock | | | (6 | ) | | | - | | | | - | | | | - | | | | - | | | | - | | | | - | | | | - | |
| Sale of common stock under employee stock purchase plans and exercise of stock options | | | 75 | | | | - | | | | 306 | | | | - | | | | - | | | | - | | | | - | | | | 306 | |
| Balance at December 31, 2012 | | | 46,765 | | | | 61 | | | | 493,613 | | | | (424,105 | ) | | | (260 | ) | | | (200 | ) | | | (2,741 | ) | | | 66,568 | |
| Net loss | | | - | | | | - | | | | - | | | | (42,572 | ) | | | - | | | | - | | | | - | | | | (42,572 | ) |
| Other comprehensive income | | | - | | | | - | | | | - | | | | - | | | | 68 | | | | - | | | | - | | | | 68 | |
| Compensation expenses for share-based payment arrangements | | | - | | | | - | | | | 3,546 | | | | - | | | | - | | | | - | | | | - | | | | 3,546 | |
| Acquisition of subsidiary, net of issuance costs | | | 4,472 | | | | 6 | | | | 11,214 | | | | - | | | | - | | | | - | | | | - | | | | 11,220 | |
| Sale of common stock in public offering, net of underwriting discounts and commissions ($2,581) and offering expenses ($351) | | | 9,775 | | | | 12 | | | | 40,066 | | | | - | | | | - | | | | - | | | | - | | | | 40,078 | |
| Forfeitures of restricted stock | | | (1 | ) | | | - | | | | - | | | | - | | | | - | | | | - | | | | - | | | | - | |
| Exercise of stock options | | | 14 | | | | - | | | | 71 | | | | - | | | | - | | | | - | | | | - | | | | 71 | |
| Balance at December 31, 2013 | | | 61,025 | | | $ | 79 | | | $ | 548,510 | | | $ | (466,677 | ) | | $ | (192 | ) | | | (200 | ) | | $ | (2,741 | ) | | $ | 78,979 | |
The accompanying notes are an integral part of the financial statements.
PROGENICS PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS (amounts in thousands)
| | Years Ended December 31, | |
| | 2013 | | | 2012 | | | 2011 | |
| Cash flows from operating activities: | | | | | | | | | |
| Net (loss) income | | $ | (42,572 | ) | | $ | (35,431 | ) | | $ | 10,381 | |
| Adjustments to reconcile net (loss) income to net cash (used in) provided by operating activities: | | | | | | | | | | | | |
| Depreciation and amortization | | | 939 | | | | 1,324 | | | | 2,066 | |
| Losses (gains) on sales of fixed assets | | | 204 | | | | (327 | ) | | | - | |
| Intangible impairment charge | | | 919 | | | | - | | | | - | |
| Deferred income tax | | | (362 | ) | | | - | | | | - | |
| Change in contingent consideration liability | | | (200 | ) | | | - | | | | - | |
| Expenses for share-based compensation awards | | | 3,546 | | | | 6,536 | | | | 6,362 | |
| Changes in assets and liabilities: | | | | | | | | | | | | |
| Decrease (increase) in accounts receivable | | | 4,114 | | | | (5,421 | ) | | | 767 | |
| Decrease (increase) in other current assets | | | 336 | | | | (754 | ) | | | 882 | |
| Decrease (increase) in deferred tax and other assets | | | 2,044 | | | | (2,002 | ) | | | 1,050 | |
| (Decrease) in accounts payable and accrued expenses | | | (1,956 | ) | | | (691 | ) | | | (3,352 | ) |
| (Decrease) increase in deferred revenue – current | | | (886 | ) | | | 634 | | | | 204 | |
| (Decrease) increase in deferred tax and other current liabilities | | | (2,069 | ) | | | 2,069 | | | | 3 | |
| (Decrease) increase in deferred revenue - long term | | | - | | | | (162 | ) | | | 162 | |
| (Decrease) in other liabilities | | | (164 | ) | | | (419 | ) | | | (138 | ) |
| Net cash (used in) provided by operating activities | | | (36,107 | ) | | | (34,644 | ) | | | 18,387 | |
| Cash flows from investing activities: | | | | | | | | | | | | |
| Cash acquired in acquisition of subsidiary | | | 1,888 | | | | - | | | | - | |
| Capital expenditures | | | (137 | ) | | | (767 | ) | | | (226 | ) |
| Proceeds from sales of fixed assets | | | 174 | | | | 390 | | | | - | |
| Proceeds from redemption of auction rate securities | | | 1,100 | | | | 100 | | | | 300 | |
| Net cash provided by (used in) investing activities | | | 3,025 | | | | (277 | ) | | | 74 | |
| Cash flows from financing activities: | | | | | | | | | | | | |
| Equity issuance costs in connection with acquisition of subsidiary | | | (45 | ) | | | - | | | | - | |
| Proceeds from public offering of common stock, net of underwriting discounts and commissions and offering expenses | | | 40,078 | | | | 23,348 | | | | - | |
| Proceeds from the exercise of stock options and sale of common stock under the employee stock purchase plans | | | 71 | | | | 306 | | | | 3,726 | |
| Net cash provided by financing activities | | | 40,104 | | | | 23,654 | | | | 3,726 | |
| Net increase (decrease) in cash and cash equivalents | | | 7,022 | | | | (11,267 | ) | | | 22,187 | |
| Cash and cash equivalents at beginning of period | | | 58,838 | | | | 70,105 | | | | 47,918 | |
| Cash and cash equivalents at end of period | | $ | 65,860 | | | $ | 58,838 | | | $ | 70,105 | |
| | | | | | | | | | | | |
| Supplemental disclosure of cash flow information: | | | | | | | | | | | | |
| Contingent consideration liability | | $ | 15,700 | | | $ | - | | | $ | - | |
| Stock acquisition consideration | | $ | 11,265 | | | $ | - | | | $ | - | |
The accompanying notes are an integral part of the financial statements.
1. Organization and Business
Progenics Pharmaceuticals, Inc. ("Progenics," "we" or "us") develops innovative medicines for oncology. Our clinical development efforts center on late-stage oncology assets. We are conducting phase 2 clinical trials of two product candidates for prostate cancer: our therapeutic candidate, PSMA ADC, a fully human monoclonal antibody-drug conjugate (ADC), and 1404 (trofolastat), an imaging agent candidate, and resuming a pivotal phase 2 clinical trial of Azedra™, our ultra-orphan radiotherapy candidate for pheochromocytoma. We have also decided to move forward MIP-1095, a compound originally developed by Molecular Insight, into clinical development, and expect to file an IND application in the U.S. later this year.
We have licensed our first commercial drug, Relistor® (methylnaltrexone bromide) subcutaneous injection for the treatment of opioid induced constipation (OIC), to Salix Pharmaceuticals, Inc., and have partnered other internally-developed or acquired compounds and technologies with third parties. We continue to consider opportunities for strategic collaborations, out-licenses and other arrangements with biopharmaceutical companies involving proprietary research, development and clinical programs, and may in the future also in-license or acquire additional oncology compounds and/or programs.
Our current principal sources of revenue from operations are royalty, commercialization milestone and revenue-sharing payments from Salix's Relistor operations. Royalty and milestone payments from Relistor depend on success in development and commercialization, which is dependent on many factors, such as Salix's efforts, decisions by the FDA and other regulatory bodies such as the July 2012 Complete Response Letter in respect of the Relistor chronic pain sNDA, competition from drugs for the same or similar indications, and the outcome of clinical and other testing of Relistor.
We fund our operations to a significant extent from capital-raising. During 2013, we completed an underwritten public offering of 9.8 million shares of common stock at a public offering price of $4.40 per share, resulting in net proceeds of approximately $40.1 million, and in early 2014 sold an additional 8.75 million shares at $4.60 per share for net proceeds of approximately $37.5 million.
Progenics commenced principal operations in 1988, became publicly traded in 1997 and throughout has been engaged primarily in research and development efforts, establishing corporate collaborations and related activities. Certain of our intellectual property rights are held by wholly owned subsidiaries. All of our operations are conducted at our facilities in Tarrytown, New York. We operate under a single research and development segment.
Funding and Financial Matters. At December 31, 2013, we held $65.9 million in cash and cash equivalents, an increase of $7.1 million from $58.8 million at December 31, 2012. We expect that this amount, together with the additional 2014 public offering proceeds, will be sufficient to fund operations as currently anticipated beyond one year. We may require additional funding in the future, and if we are unable to conclude favorable collaboration, license, asset sale, capital raising or other financing transactions, we will have to reduce, delay or eliminate spending on some current operations, and/or reduce salary and other overhead expenses, to extend our remaining operations. We expect to incur operating losses during the near term. At December 31, 2013, cash, cash equivalents and auction rate securities increased $6.0 million to $68.1 million from $62.1 million at December 31, 2012.
2. Acquisition of Molecular Insight Pharmaceuticals, Inc.
Molecular Insight's operations from January 18, 2013, the date we acquired this subsidiary which significantly expanded the Company's focus on PSMA as an oncology target while broadening the oncology pipeline, are included in the Consolidated Financial Statements. The acquisition consideration included 4,566,210 shares (500,000 of which were placed in an escrow expiring in April 2014) of Progenics common stock in a private transaction not taxable to Progenics. (The closing NASDAQ market price of Progenics' freely transferable common shares on January 18, 2013 was $2.83 per share.) Under the acquisition agreement, Progenics also agreed to pay to the stockholders potential milestones, in cash or Progenics stock at Progenics' option, of up to $23 million contingent upon achieving specified commercialization events and up to $70 million contingent upon achieving specified sales targets relating to all MIP products. Of the 500,000 placed in escrow, shares have been returned to Progenics to date pursuant to financial adjustment provisions of the agreement.
The acquisition was accounted for using the acquisition method of accounting, under which assets and liabilities of the acquired entity are recorded at their respective fair values as of the acquisition date (estimated as described below) and added to those of the acquiring entity. The difference between the estimated fair value of the acquisition consideration paid and fair value of the identifiable net assets represents potential future economic benefits arising from combining Progenics and MIP, taking into account a deferred tax liability related to in process research and development (IPR&D) intangible assets, and has been recorded as goodwill. The results of operations of MIP's business, the estimated fair market values of the assets acquired and liabilities assumed, and goodwill are included in our consolidated financial statements since the date of the acquisition.
During the year ended December 31, 2013, the Company incurred $790 in transaction costs related to the acquisition, which primarily consisted of legal, accounting and valuation-related expenses and reduced additional paid-in capital in the first quarter of 2013 by $45 for acquisition-related equity issuance costs. The transaction costs were recorded in general and administrative expenses in the accompanying consolidated statements of operations. During the year ended December 31, 2013, MIP's business contributed $884 of revenues and $11,379 of net loss.
Purchase Price Allocation: We have accounted for the Molecular Insight acquisition by allocating our estimate of the fair market value of the consideration we paid to the fair values of the assets acquired and liabilities assumed at the effective date of the acquisition, estimated using the valuation models summarized below. Given the uniqueness of and uncertainties attendant to the assets and liabilities, the derived values do not reflect actual transactions or quoted prices. Acquired intangible assets, including goodwill, are not deductible for tax purposes.
| | Amount | |
| Consideration: | | | |
| Progenics common stock consideration paid | | $ | 11,265 | |
| Contingent consideration (pursuant to future milestone obligations) | | | 15,900 | |
| Total consideration | | | 27,165 | |
| | | | |
| Tangible assets acquired and liabilities assumed: | | | | |
| Cash and cash equivalents | | | 1,888 | |
| Accounts receivable | | | 56 | |
| Other current assets | | | 529 | |
| Fixed assets | | | 249 | |
| Accounts payable, accrued expenses and deferred revenue - current | | | (2,876 | ) |
| Deferred tax liability – long term | | | (12,683 | ) |
| Total tangible assets acquired and liabilities assumed | | | (12,837 | ) |
| | | | |
| Intangible assets – in process research and development | | | 32,300 | |
| Total tangible and intangible assets acquired and liabilities assumed | | | 19,463 | |
| | | | |
| Goodwill | | $ | 7,702 | |
Intangible assets and goodwill: In connection with the acquisition of Molecular Insight, in process research and development and goodwill are initially measured at estimated fair value and capitalized as an intangible asset. We perform an impairment test for these intangibles annually in the fourth quarter, unless impairment indicators require an earlier evaluation. Upon and subject to commercialization of the Company's product candidates, the IPR&D will be amortized over its estimated useful life.
We valued as intangible assets the in process research and development projects acquired as follows:
(i) 1404, an imaging agent in phase 2 development, at an estimated fair value of $23.2 million resulting from a probability adjusted discounted cash flow model which includes estimates of significant cash inflows beginning in 2017 and a 18% discount rate;
(ii) Azedra, a small molecule candidate for the treatment of pheochromocytoma and paraganglioma in phase 2b development, and for neuroblastoma in phase 2a development, at an estimated fair value of $4.9 million resulting from a probability adjusted discounted cash flow model which includes estimates of significant cash inflows beginning in 2017 and a 15% discount rate;
(iii) small molecule candidate MIP-1095, in preclinical development for the treatment of prostate cancer, at an estimated fair value of $2.7 million resulting from a probability adjusted discounted cash flow model which includes estimates of significant cash inflows beginning in 2021 and a 20% discount rate; and
(iv) Onalta, a drug candidate in phase 2 development for the treatment of metastatic carcinoid and pancreatic neuroendocrine tumors, at an estimated fair value of $1.5 million resulting from a probability adjusted discounted cash flow model which includes estimates of significant cash inflows beginning in 2014 and a 15% discount rate.
On the acquisition date, we recorded a contingent consideration liability at an estimated fair value of $15.9 million resulting from probability adjusted discounted cash flow and Monte Carlo simulation models which include estimates of significant milestone payments to former MIP stockholders under the acquisition agreement ranging from 2016 to 2022 and risk adjusted discount rates ranging from 10% to 12.5%.
Pro forma financial information (unaudited): The following unaudited pro forma information presents the results of operations of the combined companies for the periods indicated as if the acquisition had been consummated on January 1, 2012, combining the respective historical results of Progenics and MIP for each period. Non-recurring transaction expenses of $790, incurred in the year ended December 31, 2013, are reflected in the pro forma information as if these were incurred in the corresponding 2012 period, due to the pro forma assumption of January 1, 2012 as the date of the acquisition consummation.
| | Three Months Ended December 31, | | | Year Ended December 31, | |
| | 2013 | | | 2012 | | | 2013 | | | 2012 | |
| Revenues | | $ | 2,968 | | | $ | 12,801 | | | $ | 7,867 | | | $ | 18,235 | |
| Net loss | | | (8,551 | ) | | | (3,458 | ) | | | (43,736 | ) | | | (57,877 | ) |
| Basic and diluted loss per share | | | (0.14 | ) | | | (0.08 | ) | | | (0.78 | ) | | | (1.48 | ) |
3. Summary of Significant Accounting Policies
Basis of Presentation
The consolidated financial statements have been prepared on the basis of accounting principles generally accepted in the U.S. (GAAP). The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the amounts reported in the financial statements and the accompanying notes. On an ongoing basis, the Company evaluates its estimates, including but not limited to those related to collectability of receivables, intangible assets and contingencies. As additional information becomes available or actual amounts become determinable, the recorded estimates are revised and reflected in the operating results. Actual results could differ from those estimates.
Consolidation
The consolidated financial statements include the accounts of Progenics and PSMA LLC, as of and for the years ended December 31, 2013, 2012 and 2011 and Molecular from January 18, 2013, the date we acquired this subsidiary. Inter-company transactions have been eliminated in consolidation.
Revenue Recognition
We recognize revenue from all sources based on the provisions of the SEC's Staff Accounting Bulletin (SAB) No. 104 (SAB 104) and ASC 605 Revenue Recognition. Under ASC 605, delivered items are separate units of accounting, provided (i) the delivered items have value to a collaborator on a stand-alone basis, and (ii) if the arrangement includes a general right of return relative to the delivered item, delivery or performance of the undelivered items is considered probable and substantially in our control. A separate update to ASC 605 provides guidance on the criteria that should be met when determining whether the milestone method of revenue recognition is appropriate.
If we are involved in a steering or other committee as part of a multiple-deliverable arrangement, we assess whether our involvement constitutes a performance obligation or a right to participate. For those committees that are deemed obligations, we will evaluate our participation along with other obligations in the arrangement and will attribute revenue to our participation through the period of our committee responsibilities. We recognize revenue for payments that are contingent upon performance solely by our collaborator immediately upon the achievement of the defined event if we have no related performance obligations. Reimbursement of costs is recognized as revenue provided the provisions of ASC 605 are met, the amounts are determinable and collection of the related receivable is reasonably assured.
Amounts received prior to satisfying the above revenue recognition criteria are recorded as deferred revenue. Amounts not expected to be recognized within one year of the balance sheet date are classified as long-term. The estimate of the classification of deferred revenue as short- or long-term is based upon the period in which we expect to perform joint committee services.
Royalty revenue is recognized in the period the sales occur, provided the royalty amounts are fixed or determinable, collection of the related receivable is reasonably assured and we have no remaining performance obligations under the arrangement providing for the royalty.
During the past three years, we also recognized revenue from sales of research reagents and from government research grants, awarded to us by the National Institutes of Health (NIH), which we used in proprietary research programs. NIH grant revenue is recognized as efforts are expended and as related program costs are incurred. We performed work under the NIH grants on a best-effort basis.
Under Molecular's 2013 license of certain research, development and commercialization rights to Onalta™, we received a $0.2 million in upfront payment and are eligible for future milestone and royalty payments. In consideration for the upfront payment, we have delivered relevant know-how (including patent rights), inventory and non-reimbursable services.
In the fourth quarter of 2012, we out-licensed our C. difficile program to MedImmune, LLC for a $5.0 million upfront payment, and the right to receive potential future milestone and royalty payments. In consideration for the upfront payment, we have delivered relevant know-how (including patent rights) and non-reimbursable services.
Under our 2012 agreement with CytoDyn Inc. for our PRO 140 program, we received $3.5 million payment and are eligible for future milestone and royalty payments. In consideration for the upfront payment, we have delivered relevant know-how (including patent rights), inventory and non-reimbursable services.
Under our license agreement, Salix is responsible for further developing and commercializing Relistor worldwide other than Japan. In consideration of the $60.0 million upfront payment from Salix, we have granted Salix an exclusive license of relevant know-how, patent rights and technology, assigned relevant third-party contracts, and served on joint committees provided for in the License Agreement through end of 2013.
These deliverables, which have stand-alone value and represent separate units of accounting, include (i) the exclusive license which was delivered for revenue recognition purposes during the 2011 second quarter, (ii) performing reimbursable development services at Salix's direction during the 2011 second quarter, the period in which we and Salix finalized the development plan, and (iii) joint committee services, which have been performed through 2013. We determined that the license has stand-alone value as the license was delivered to Salix for revenue recognition purposes in the second quarter of 2011 and Salix is responsible for continuing research and development.
We developed a best estimate of selling price for each deliverable as vendor-specific objective evidence and third-party evidence was not available. We allocated the best estimate of selling price, on a relative basis, to each of the three units of accounting as the $60.0 million upfront payment was the only payment from Salix which was fixed and determinable at the inception of the arrangement. As a result, $58.4 million, $1.1 million and $0.5 million was allocated to the license, reimbursable development services and our participation in the joint committees as provided in the License Agreement, respectively. We recognized $58.4 million for the license and relevant know-how, patent rights and technology and $1.1 million for the reimbursable development services, respectively, during the second quarter of 2011, the period in which we delivered these items and performed the development services. We recognized $0.2 million, $0.2 million and $59.6 million during 2013, 2012 and 2011, respectively. At December 31, 2012, the remaining deferred revenue of $0.2 million, pertaining to joint committee services, was recognized as collaboration revenue in 2013, as such activities were performed.
Ono is responsible for developing and commercializing subcutaneous Relistor in Japan, including conducting the clinical development necessary to support regulatory marketing approval. Ono will own the filings and approvals related to subcutaneous Relistor in Japan. In addition to the $15.0 million upfront payment from Ono, we are entitled to receive up to an additional $20.0 million, payable upon achievement by Ono of its development milestones. Ono is also obligated to pay to us royalties and commercialization milestones on sales by Ono of subcutaneous Relistor in Japan. Ono has the option to acquire from us the rights to develop and commercialize in Japan other formulations of Relistor, including intravenous and oral forms, on terms to be negotiated separately. Ono may request us to perform activities related to its development and commercialization responsibilities, beyond our participation in joint committees and specified technology transfer-related tasks, at its expense payable at the time we perform such services. Revenue earned from activities we perform for Ono is recorded in collaboration revenue. See Note 10.
Research and Development Expenses
Research and development expenses include costs directly attributable to the conduct of research and development programs, including the cost of salaries, payroll taxes, employee benefits, materials, supplies, maintenance of research equipment, costs related to research collaboration and licensing agreements, the purchase of in-process research and development, the cost of services provided by outside contractors, including services related to our clinical trials, the full cost of manufacturing drug for use in research, pre-clinical development and clinical trials. All costs associated with research and development are expensed as incurred.
At each period end, we evaluate the accrued expense balance related to these activities based upon information received from the suppliers and estimated progress towards completion of the research or development objectives to ensure that the balance is reasonably stated. Such estimates are subject to change as additional information becomes available.
Use of Estimates
Significant estimates include useful lives of fixed assets, the periods over which certain revenues and expenses will be recognized, including collaboration revenue recognized from non-refundable up-front licensing payments and expense recognition of certain clinical trial costs which are included in research and development expenses, the amount of non-cash compensation costs related to share-based payments to employees and non-employees and the periods over which those costs are expensed, the likelihood of realization of deferred tax assets and the assumptions used in the valuations of in-process research and development and contingent consideration liability.
Patents
As a result of research and development efforts conducted by us, we have applied, or are applying, for a number of patents to protect proprietary inventions. All costs associated with patents are expensed as incurred.
Net (Loss) Income Per Share
We prepare earnings per share (EPS) data in accordance with ASC 260 Earnings Per Share. Basic net (loss) income per share amounts have been computed by dividing net (loss) income by the weighted-average number of common shares outstanding during the period. For 2013 and 2012, we reported net losses and, therefore, potential common shares, amounts of unrecognized compensation expense and windfall tax benefits have been excluded from diluted net loss per share since they would be anti-dilutive. For 2011, we reported net income, and the computation of diluted earnings per share is based upon the weighted-average number of our common shares and dilutive effect, determined using the treasury stock method, of potential common shares outstanding including amounts of unrecognized compensation expense. As of December 31, 2012 and 2011, 28 and 98, respectively, shares of unvested restricted stock outstanding have non-forfeitable rights to dividends; all such shares were vested at the end of December 31, 2013. The allocation of 2013 and 2012 net losses and the 2011 net income to these participating securities pursuant to the two-class method is not material to both basic and diluted earnings per share.
Concentrations of Credit Risk
Financial instruments which potentially subject Progenics to concentrations of risk consist principally of cash, cash equivalents, auction rate securities and receivables. We invest our excess cash in money market funds. We have established guidelines that relate to credit quality, diversification and maturity and that limit exposure to any one issue of securities. We hold no collateral for these financial instruments.
Cash and Cash Equivalents
We consider all highly liquid investments which have maturities of three months or less, when acquired, to be cash equivalents. The carrying amount reported in the balance sheet for cash and cash equivalents approximates its fair value. Cash and cash equivalents subject us to concentrations of credit risk. At December 31, 2013 and 2012, we have invested approximately $60,364 and $56,224, respectively, in cash equivalents in the form of money market funds with one major investment company and held approximately $4,898 and $2,614, respectively, in a single commercial bank.
Accounts Receivable
We estimate the level of accounts receivable which ultimately will be uncollectable based on a review of specific receivable balances, industry experience and the current economic environment. We reserve for affected accounts receivable an allowance for doubtful accounts, which at December 31, 2013 was $7.
Auction Rate Securities
In accordance with ASC 320 Investments – Debt and Equity Securities, investments are classified as available-for-sale. Available-for-sale securities are carried at fair value, with the unrealized gains and losses reported in comprehensive (loss) income. The amortized cost of debt securities is adjusted for amortization of premiums and accretion of discounts to maturity. Such amortization is included in interest income or expense. Realized gains and losses and declines in value judged to be other-than-temporary, if any, on available-for-sale securities are included in other income or expense. In computing realized gains and losses, we compute the cost of its investments on a specific identification basis. Such cost includes the direct costs to acquire the securities, adjusted for the amortization of any discount or premium. The fair value of auction rate securities has been estimated based on a three-level hierarchy for fair value measurements. Interest and dividends on securities classified as available-for-sale are included in interest income (see Note 4).
At December 31, 2013 and 2012, our investment in auction rate securities (recorded as long-term assets in the Consolidated Balance Sheets) amounted to $2,208 and $3,240, respectively. Valuation of securities is subject to uncertainties that are difficult to predict, such as changes to credit ratings of the securities and/or the underlying assets supporting them, default rates applicable to the underlying assets, underlying collateral value, discount rates, counterparty risk, ongoing strength and quality of market credit and liquidity and general economic and market conditions. The valuation of the auction rate securities we hold is based on an internal analysis of timing of expected future successful auctions, collateralization of underlying assets of the security and credit quality of the security. We re-evaluated the valuation of these securities as of December 31, 2013 and the temporary impairment amount decreased $68 from $260 at December 31, 2012 to $192. All income generated from these investments was recorded as interest income (see Note 4).
In-Process Research and Development and Goodwill
The fair values of in-process research and development (IPR&D) acquired in business combinations are capitalized. The Company utilizes the "income method," which applies a probability weighting that considers the risk of development and commercialization to the estimated future net cash flows that are derived from projected sales revenues and estimated costs. These projections are based on factors such as relevant market size, patent protection, historical pricing of similar products and expected industry trends. The estimated future net cash flows are then discounted to the present value using an appropriate discount rate. This analysis is performed for each project independently. These assets are treated as indefinite-lived intangible assets until completion or abandonment of the projects, at which time the assets are amortized over the remaining useful life or written off, as appropriate. IPR&D intangible assets which are determined to have a decline in their fair value are adjusted downward and an expense is recognized as part of the general and administrative expenses in the Consolidated Statements of Operations. These are tested at least annually or when a triggering event occurs that could indicate a potential impairment.
Goodwill represents excess consideration in a business combination over the fair value of identifiable net assets acquired. Goodwill is not amortized, but is subject to impairment testing at least annually or when a triggering event occurs that could indicate a potential impairment. The Company determines whether goodwill may be impaired by comparing the fair value of the reporting unit, calculated as the product of shares outstanding and the share price as of the end of a period, to its carrying value. No goodwill impairment has been recognized as of December 31, 2013. The Company has determined that it has only one reporting unit, which includes the acquired Molecular Insight.
The following table reflects the components of the finite lived intangible assets as of December 31, 2013:
| | Gross Amount | | | Accumulated Amortization | | | Net Carrying Value | |
| Finite lived intangible assets | | $ | 21 | | | $ | 2 | | | $ | 19 | |
| Total | | $ | 21 | | | $ | 2 | | | $ | 19 | |
The weighted-average remaining life of the finite lived intangible assets is five years at December 31, 2013.
Amortization expense is calculated on a straight-line basis over the estimated useful life of the asset. Amortization expense for the period from January 18, 2013 to December 31, 2013 was $2. Estimated amortization expense related to intangible assets existing as of December 31, 2013 is approximately $4 annually for each of the succeeding five years.
The following table summarizes the activity related to the Company's goodwill and indefinite lived IPR&D:
| | Goodwill | | | IPR&D | |
| Balance at January 1, 2013 | | $ | - | | | $ | - | |
| Increase related to MIP acquisition | | | 7,702 | | | | 32,300 | |
| Reclassification to finite lived IPR&D | | | - | | | | (21 | ) |
| Impairment | | | - | | | | (919 | ) |
| Balance at December 31, 2013 | | $ | 7,702 | | | $ | 31,360 | |
Fair Value Measurements
In accordance with ASC 820 Fair Value Measurements and Disclosures, we use a three-level hierarchy for fair value measurements of certain assets and liabilities for financial reporting purposes that distinguishes between market participant assumptions developed from market data obtained from outside sources (observable inputs) and our own assumptions about market participant assumptions developed from the best information available to us in the circumstances (unobservable inputs). We assign hierarchy levels to assets constituting our available-for-sale portfolio and to our contingent consideration liability arising from the MIP acquisition based on our assessment of the transparency and reliability of the inputs used in the valuation. ASC 820 defines the three hierarchy levels as:
| · | Level 1 - Valuations based on unadjusted quoted market prices in active markets for identical securities. |
| |
| · | Level 2 - Valuations based on observable inputs other than Level 1 prices, such as quoted prices for similar assets at the measurement date, quoted prices in markets that are not active or other inputs that are observable, either directly or indirectly. |
| |
| · | Level 3 - Valuations based on unobservable inputs that are significant to the overall fair value measurement, which as noted above involve management judgment. |
Recurring Fair Value Measurements
We believe the carrying amounts of the Company's cash equivalents, accounts receivable, other current assets, other assets (restricted cash providing collateral for a letter of credit securing lease obligations) and accounts payable and accrued expenses approximated their fair values as of December 31, 2013 and 2012, due to their short-term nature; we consider them Level 1 instruments.
The fair value of the contingent consideration liability, consisting of future potential milestone payments related to the MIP acquisition was $15.7 million as of December 31, 2013 and $15.9 million as January 18, 2013, the acquisition date (see Note 2). The fair value of the contingent consideration liability is categorized as a Level 3 instrument, as displayed in Note 4. The Company records the contingent consideration liability at fair value with changes in estimated fair values recorded in general and administrative expenses in the Consolidated Statements of Operations. As of December 31, 2013, we reassessed the fair value of the contingent consideration and recorded a $0.2 million decrease, due to an increase in the discount period, and a corresponding credit in the general and administrative expenses in the fourth quarter of 2013. The December 31, 2013 contingent consideration of $15.7 million results from probability adjusted discounted cash flow and Monte Carlo simulation models which include estimates of significant milestone payments to former MIP stockholders under the acquisition agreement ranging from 2017 to 2022 and risk adjusted discount rates ranging from 10% to 12.5%.
Nonrecurring Fair Value Measurements
The Company's non-financial assets, such as intangible assets and property and equipment, are measured and recorded at fair value on the acquisition date, and if indicators of impairment exist, we assess recoverability by measuring the amount of any impairment by comparing the carrying value of the asset to its then-current estimated fair value (for intangible assets) or to market prices for similar assets (for property and equipment). If the carrying value is not recoverable we record an impairment charge as a general and administrative expense in the Consolidated Statements of Operations. The company reassessed the value of the indefinite lived intangible assets and recorded a non-cash charge to earnings of $919 in the fourth quarter of 2013. This impairment was the result of change in the Level 3 assumptions: (i) the timing of the estimated beginning of cash inflows from 2014 to 2018 and (ii) an increase in discount rate from 15% to 18% for the Onalta intangible asset. In connection with the second quarter amendment of the Company's Tarrytown lease, we recognized impairment losses of $347 on leasehold improvements and machinery and equipment removed from service which are included in Research and development expenses in our accompanying Consolidated Statements of Operations for the year ended December 31, 2013. As a result of closing our biologics pilot facilities in 2011, an impairment loss of $22 was included in Research and development expenses in our accompanying Consolidated Statement of Operations during 2011. No impairments occurred for the year ended December 31, 2012.
Other current assets are comprised of prepaid expenses, interest, deferred tax asset and other receivables of $1,943 and $1,692 at December 31, 2013 and 2012, respectively, which are expected to be settled within one year. Restricted cash of $157 and $150 at December 31, 2013 and 2012, respectively, consists of collateral for a letter of credit securing lease obligations. We believe the carrying value of these assets approximates fair value.
Fixed Assets
Leasehold improvements, furniture and fixtures, and equipment are stated at cost. Furniture, fixtures and equipment are depreciated on a straight-line basis over their estimated useful lives. Leasehold improvements are amortized on a straight-line basis over the life of the lease or of the improvement, whichever is shorter. Costs of construction of long-lived assets are capitalized but are not depreciated until the assets are placed in service.
Expenditures for maintenance and repairs which do not materially extend the useful lives of the assets are charged to expense as incurred. The cost and accumulated depreciation of assets retired or sold are removed from the respective accounts and any gain or loss is recognized in operations. The estimated useful lives of fixed assets are as follows:
| Computer equipment | 3 years |
| Machinery and equipment | 5-7 years |
| Furniture and fixtures | 5 years |
| Leasehold improvements | Earlier of life of improvement or lease |
Deferred Lease Liability and Incentive
Our lease agreements include fixed escalations of minimum annual lease payments and we recognize rental expense on a straight-line basis over the lease terms and record the difference between rent expense and current rental payments as deferred rent. Deferred lease incentive includes a construction allowance from our landlord which is amortized as a reduction to rental expense on a straight-line basis over the lease term. As of December 31, 2013 and 2012, the Consolidated Balance Sheets include the following:
| | 2013 | | | 2012 | |
| Other current liabilities: | | | | | | |
| Deferred lease incentive | | $ | 115 | | | $ | 115 | |
| Total other current liabilities | | $ | 115 | | | $ | 115 | |
| Other liabilities: | | | | | | | | |
| Deferred lease liability | | $ | 224 | | | $ | 273 | |
| Deferred lease incentive | | | 690 | | | | 805 | |
| Total other liabilities | | $ | 914 | | | $ | 1,078 | |
Income Taxes
We account for income taxes in accordance with the provisions of ASC 740 Income Taxes, which requires that we recognize deferred tax liabilities and assets for the expected future tax consequences of events that have been included in the financial statements or tax returns. Under this method, deferred tax assets and liabilities are determined on the basis of the difference between the tax basis of assets and liabilities and their respective financial reporting amounts (temporary differences) at enacted tax rates in effect for the years in which the temporary differences are expected to reverse. A valuation allowance is established for deferred tax assets for which realization is uncertain.
In accordance with ASC 718 Compensation – Stock Compensation and ASC 505 Equity, we have made a policy decision related to intra-period tax allocation, to account for utilization of windfall tax benefits based on provisions in the tax law that identify the sequence in which amounts of tax benefits are used for tax purposes (i.e., tax law ordering).
Uncertain tax positions are accounted for in accordance with ASC 740 Income Taxes, which prescribes a comprehensive model for the manner in which a company should recognize, measure, present and disclose in its financial statements all material uncertain tax positions that we have taken or expect to take on a tax return. ASC 740 applies to income taxes and is not intended to be applied by analogy to other taxes, such as sales taxes, value-add taxes, or property taxes. We review our nexus in various tax jurisdictions and our tax positions related to all open tax years for events that could change the status of our ASC 740 liability, if any, or require an additional liability to be recorded. Such events may be the resolution of issues raised by a taxing authority, expiration of the statute of limitations for a prior open tax year or new transactions for which a tax position may be deemed to be uncertain. Those positions, for which management's assessment is that there is more than a 50 percent probability of sustaining the position upon challenge by a taxing authority based upon its technical merits, are subjected to the measurement criteria of ASC 740. We record the largest amount of tax benefit that is greater than 50 percent likely of being realized upon ultimate settlement with a taxing authority having full knowledge of all relevant information. Any ASC 740 liabilities for which we expect to make cash payments within the next twelve months are classified as "short term." In the event that we conclude that we are subject to interest and/or penalties arising from uncertain tax positions, we will record interest and penalties as a component of income taxes (see Note 13).
Risks and Uncertainties
We have to date relied principally on external funding, collaborations with Salix, Fuji and others, out-licensing and asset sale arrangements, royalty and product revenue to finance our operations. There can be no assurance that our research and development will be successfully completed, that any products developed will obtain necessary marketing approval by regulatory authorities or that any approved products will be commercially viable. In addition, we operate in an environment of rapid change in technology, and we are dependent upon satisfactory relationships with our partners and the continued services of our current employees, consultants and subcontractors. We are also dependent upon Salix, Fuji and Ono fulfilling their manufacturing obligations, either on their own or through third-party suppliers. For 2013, 2012 and 2011, the primary sources of our revenues were Salix, Ono, Fuji, asset out-licensing and disposition, and research grant revenues from the NIH. There can be no assurance that revenues from asset out-licensing and disposition, Salix, Ono and Fuji or from research awards will continue. Substantially all of our accounts receivable at December 31, 2013 and 2012 were from the above-named sources.
Comprehensive (Loss) Income
Comprehensive (loss) income represents the change in net assets of a business enterprise during a period from transactions and other events and circumstances from non-owner sources. Our comprehensive (loss) income includes net (loss) income adjusted for the change in net unrealized gain or loss on auction rate securities. The disclosures required by ASC 220 Comprehensive Income for 2013, 2012 and 2011 have been included in the Consolidated Statements of Comprehensive (Loss) Income. There was no income tax expense/benefit allocated to any component of Other Comprehensive (Loss) Income (see Note 13).
Impact of Recently Adopted Accounting Standards
In February 2013, the FASB issued ASU No. 2013-02, which requires presentation of amounts reclassified out of accumulated other comprehensive income by component. The ASU is effective for reporting periods beginning after December 15, 2012. We adopted this new standard on January 1, 2013 and it had no material impact on our consolidated financial statements.
4. Fair Value Measurements
We record auction rate securities at fair value in the accompanying Consolidated Balance Sheets in accordance with ASC 320 Investments – Debt and Equity Securities. The change in the fair value of these securities is recorded as a component of other comprehensive (loss) income. We also record the contingent consideration liability resulting from the MIP acquisition at fair value in accordance with ASC 820-10-50.
The following tables present our money market funds, included in cash and cash equivalents, and auction rate securities assets and contingent consideration liability measured at fair value on a recurring basis as of the dates indicated, classified by valuation hierarchy:
| | | | | Fair Value Measurements at December 31, 2013 | |
| | Balance at December 31, 2013 | | | Quoted Prices in Active Markets for Identical Assets (Level 1) | | | Significant Other Observable Inputs (Level 2) | | | Significant Unobservable Inputs (Level 3) | |
| Assets: | | | | | | | | | | | | |
| Money market funds | | $ | 60,364 | | | $ | 60,364 | | | $ | - | | | $ | - | |
| Auction rate securities | | | 2,208 | | | | - | | | | - | | | | 2,208 | |
| Total Assets | | $ | 62,572 | | | $ | 60,364 | | | $ | - | | | $ | 2,208 | |
| | | | | | | | | | | | | | | | |
| Liability: | | | | | | | | | | | | | | | | |
| Contingent consideration | | $ | 15,700 | | | $ | - | | | $ | - | | | $ | 15,700 | |
| Total Liability | | $ | 15,700 | | | $ | - | | | $ | - | | | $ | 15,700 | |
| | | | | Fair Value Measurements at December 31, 2012 | |
| | Balance at December 31, 2012 | | | Quoted Prices in Active Markets for Identical Assets (Level 1) | | | Significant Other Observable Inputs (Level 2) | | | Significant Unobservable Inputs (Level 3) | |
| | | | | | | | | | | | |
| Money market funds | | $ | 56,224 | | | $ | 56,224 | | | $ | - | | | $ | - | |
| Auction rate securities | | | 3,240 | | | | - | | | | - | | | | 3,240 | |
| Total | | $ | 59,464 | | | $ | 56,224 | | | $ | - | | | $ | 3,240 | |
At December 31, 2013, we hold $2,208 in auction rate securities which are classified as Level 3. The fair value of these securities includes $2,208 of U.S. government subsidized securities collateralized by student loan obligations, with maturities greater than 10 years. We will not realize cash in respect of the principal amount of these securities until the issuer calls or restructures the security, the security reaches any scheduled maturity and is paid, or a buyer outside the auction process emerges. As of December 31, 2013, we have received all scheduled interest payments on these securities, which, in the event of auction failure, are reset according to the contractual terms in the governing instruments.
The valuation of auction rate securities we hold is based on Level 3 unobservable inputs which consist of our internal analysis of (i) timing of expected future successful auctions or issuer calls of the securities, (ii) collateralization of underlying assets of the security and (iii) credit quality of the security. Significant increases (decreases) in the redemption period or discount rates would result in a significantly lower (higher) fair value measurement. In re-evaluating the valuation of these securities as of December 31, 2013, the temporary impairment amount, the duration of which is greater than 12 months, decreased from $260 at December 31, 2012, to $192, which is reflected as a part of accumulated other comprehensive loss on our accompanying Consolidated Balance Sheets and based on such re-evaluation, we believe that we have the ability to hold these securities until recovery of fair value. Due to the uncertainty related to the liquidity in the auction rate security market and therefore when individual positions may be liquidated, we have classified these auction rate securities as long-term assets on our accompanying Consolidated Balance Sheets. We continue to monitor markets for our investments and consider the impact, if any, of market conditions on the fair market value of our investments. We do not believe the carrying values of our investments are other than temporarily impaired and therefore expect the positions will eventually be liquidated without significant loss.
The estimated fair value of the contingent consideration liability of $15.7 million represents future potential milestone payments to former MIP stockholders. The Company considers this liability a Level 3 instrument (one with significant unobservable inputs) in the fair value hierarchy. The estimated fair value was determined based on probability adjusted discounted cash flow and Monte Carlo simulation models that included significant estimates and assumptions pertaining to commercialization events and sales targets. The most significant unobservable inputs were the probabilities of achieving regulatory approval of the development projects and subsequent commercial success, and discount rates. Significant changes in any of the probabilities of success would result in a significantly higher or lower fair value measurement, respectively. Significant changes in the probabilities as to the periods in which milestones will be achieved would result in a significantly lower or higher fair value measurement, respectively. The Company records the contingent consideration liability at fair value with changes in estimated fair values recorded in general and administrative expenses in the Consolidated Statements of Operations.
The following table presents quantitative information pertaining to the fair value measurement of the Level 3 inputs:
| | Fair Value as of December 31, 2013 | | Valuation Technique | | Unobservable Input | | Range (Weighted Average) | |
| | | | | | | | | |
| Asset: | | | | | | | | | |
| Auction Rate Securities | | $ | 2,208 | | Discounted cash flow model | | Redemption period | | 5 to 15 years (6 years) | |
| | | | | | | Discount rate | | | 0.25% - 3.00% (1.55%) | |
Contingent consideration liability: | | | | | | | | | | | |
| | | | | | | | | | | |
| Azedra commercialization | | $ | 2,300 | | Probability adjusted discounted cash flow model | | Probability of success | | | 40% | |
| | | | | | | Period of milestone expected achievement | | | 2017 | |
| | | | | | | Discount rate | | | 10% | |
| | | | | | | | | | | |
| 1404 commercialization | | $ | 2,000 | | Probability adjusted discounted cash flow model | | Probability of success | | | 31% | |
| | | | | | | Period of milestone expected achievement | | | 2018 | |
| | | | | | | Discount rate | | | 10% | |
| | | | | | | | | | | |
| MIP-1095 commercialization | | $ | 500 | | Probability adjusted discounted cash flow model | | Probability of success | | | 19% | |
| | | | | | | Period of milestone expected achievement | | | 2021 | |
| | | | | | | Discount rate | | | 10% | |
| | | | | | | | | | | |
| Net sales targets | | $ | 10,900 | | Monte-Carlo simulation | | Probability of success | | | 19% - 40% (32.8%) | |
| | | | | | | Period of milestone expected achievement | | | 2018 - 2022 | |
| | | | | | | Discount rate | | | 12.5% | |
| | Fair Value as of December 31, 2012 | | Valuation Technique | | Unobservable Input | | Range (Weighted Average) | |
| | | | | | | | | |
| Asset: | | | | | | | | | |
| Auction Rate Securities | | $ | 3,240 | | Discounted cash flow model | | Redemption period | | 4 to 15 years (5.9 years) | |
| | | | | | | Discount rate | | | 0.125% - 2.102% (0.71%) | |
For those financial instruments with significant Level 3 inputs, the following table summarizes the activities for the periods indicated:
| | Asset – Auction Rate Securities Fair Value Measurements Using Significant Unobservable Inputs (Level 3) | |
| Description | | 2013 | | | 2012 | |
| Balance at beginning of period | | $ | 3,240 | | | $ | 3,332 | |
| Transfers into Level 3 | | | - | | | | - | |
| Total realized/unrealized gains (losses) | | | | | | | | |
| Included in net income (loss) | | | - | | | | - | |
| Included in comprehensive income (loss) | | | 68 | | | | 8 | |
| Settlements | | | (1,100 | ) | | | (100 | ) |
| Balance at end of period | | $ | 2,208 | | | $ | 3,240 | |
| Total amount of unrealized gains (losses) for the period included in other comprehensive loss attributable to the change in fair market value of related assets still held at the reporting date | | $ | - | | | $ | - | |
| | Liability – Contingent Consideration Fair Value Measurements Using Significant Unobservable Inputs (Level 3) | |
| Description | | 2013 | | | 2012 | |
| Balance at beginning of period | | $ | - | | | $ | - | |
| Fair value of contingent consideration – acquisition of Molecular Insight | | | 15,900 | | | | - | |
| Fair value adjustment to contingent consideration included in net loss | | | (200 | ) | | | - | |
| Balance at end of period | | $ | 15,700 | | | $ | - | |
| Changes in unrealized gains or losses for the period included in earnings (or changes in net assets) for liabilities held at the end of the reporting period | | $ | (200 | ) | | $ | - | |
The following tables summarize the amortized cost basis, the aggregate fair value and gross unrealized holding gains and losses at December 31, 2013 and 2012:
| | Amortized | | | Fair | | | Unrealized Holding | |
| 2013: | | Cost Basis | | | Value | | | Gains | | | (Losses) | | | Net | |
| Maturities greater than ten years: | | | | | | | | | | | | | | | |
| Auction rate securities | | $ | 2,400 | | | $ | 2,208 | | | $ | - | | | $ | (192 | ) | | $ | (192 | ) |
| | $ | 2,400 | | | $ | 2,208 | | | $ | - | | | $ | (192 | ) | | $ | (192 | ) |
| | Amortized | | | Fair | | | Unrealized Holding | |
| 2012: | | Cost Basis | | | Value | | | Gains | | | (Losses) | | | Net | |
| Maturities greater than ten years: | | | | | | | | | | | | | | | |
| Auction rate securities | | $ | 2,500 | | | $ | 2,300 | | | $ | - | | | $ | (200 | ) | | $ | (200 | ) |
| Investments without stated maturity dates: | | | | | | | | | | | | | | | | | | | | |
| Auction rate securities | | | 1,000 | | | | 940 | | | | - | | | | (60 | ) | | | (60 | ) |
| | $ | 3,500 | | | $ | 3,240 | | | $ | - | | | $ | (260 | ) | | $ | (260 | ) |
We compute the cost of its investments on a specific identification basis. Such cost includes the direct costs to acquire the securities, adjusted for the amortization of any discount or premium.
The following table shows the gross unrealized losses and fair value of our auction rate securities with unrealized losses that are not deemed to be other-than-temporarily impaired, aggregated by investment category and length of time that individual securities have been in a continuous unrealized loss position, at December 31, 2013 and 2012.
| 2013: | | Less than 12 Months | | | 12 Months or Greater | | | Total | |
| Description of Securities | | Fair Value | | | Unrealized Losses | | | Fair Value | | | Unrealized Losses | | | Fair Value | | | Unrealized Losses | |
| | | | | | | | | | | | | | | | | | |
| Auction rate securities | | $ | - | | | $ | - | | | $ | 2,208 | | | $ | (192 | ) | | $ | 2,208 | | | $ | (192 | ) |
| Total | | $ | - | | | $ | - | | | $ | 2,208 | | | $ | (192 | ) | | $ | 2,208 | | | $ | (192 | ) |
| 2012: | | Less than 12 Months | | | 12 Months or Greater | | | Total | |
| Description of Securities | | Fair Value | | | Unrealized Losses | | | Fair Value | | | Unrealized Losses | | | Fair Value | | | Unrealized Losses | |
| | | | | | | | | | | | | | | | | | |
| Auction rate securities | | $ | - | | | $ | - | | | $ | 3,240 | | | $ | (260 | ) | | $ | 3,240 | | | $ | (260 | ) |
| Total | | $ | - | | | $ | - | | | $ | 3,240 | | | $ | (260 | ) | | $ | 3,240 | | | $ | (260 | ) |
Other-than-temporary impairment analysis on auction rate securities. The unrealized losses on our auction rate securities resulted from an internal analysis of timing of expected future successful auctions, collateralization of underlying assets of the security and credit quality of the security. At December 31, 2013 there was one and at December 31, 2012, there were two securities with a gross unrealized loss position of $192 and $260 ($2,208 and $3,240 of the total fair value), respectively.
The severity of the unrealized losses for auction rate securities at December 31, 2013 and 2012 was 8 percent below amortized cost, and the weighted average duration of the unrealized losses for these securities was 70 and 58 months, respectively.
We have evaluated our individual auction rate securities holdings for other-than-temporary impairment and determined that the unrealized losses as of December 31, 2013 and 2012 are attributable to uncertainty in the liquidity of the auction rate security market. Because we do not intend to sell these securities, and believe it is not more likely than not that we would be required to sell these securities before recovery of principal, we do not consider these securities to be other-than-temporarily impaired at December 31, 2013 and 2012.
5. Accounts Receivable
Our accounts receivable represent amounts due to Progenics from collaborators, royalties, research grants and the sales of research reagents and as of December 31, 2013 and 2012, consisted of the following:
| | 2013 | | | 2012 | |
| Collaborators | | $ | 12 | | | $ | 6,125 | |
| Royalties | | | 2,862 | | | | 781 | |
| Research grants | | | - | | | | 12 | |
| Other | | | 12 | | | | 19 | |
| | | 2,886 | | | | 6,937 | |
| Less, allowance for doubtful accounts | | | (7 | ) | | | - | |
| Total | | $ | 2,879 | | | $ | 6,937 | |
6. Fixed Assets
Fixed assets as of December 31, 2013 and 2012 consisted of the following:
| | 2013 | | | 2012 | |
| Computer equipment | | $ | 2,234 | | | $ | 2,166 | |
| Machinery and equipment | | | 7,091 | | | | 8,031 | |
| Furniture and fixtures | | | 170 | | | | 133 | |
| Leasehold improvements | | | 5,020 | | | | 5,327 | |
| Other | | | 16 | | | | 12 | |
| | | 14,531 | | | | 15,669 | |
| Less, accumulated depreciation and amortization | | | (12,118 | ) | | | (12,270 | ) |
| Total | | $ | 2,413 | | | $ | 3,399 | |
At December 31, 2013 and 2012, $2.0 million and $2.6 million, respectively, of leasehold improvements, net were being amortized over periods of 8.5-10.8 years, under leases with terms through December 31, 2020.
7. Accounts Payable and Accrued Expenses
The carrying value of our accounts payable and accrued expenses approximates fair value, as it represents amounts due to vendors and employees, which will be satisfied within one year. Accounts payable and accrued expenses as of December 31, 2013 and 2012, consisted of the following:
| | 2013 | | | 2012 | |
| Accrued consulting and clinical trial costs | | $ | 2,672 | | | $ | 2,193 | |
| Accrued payroll and related costs | | | 2,123 | | | | 1,552 | |
| Restructuring accrual | | | - | | | | 813 | |
| Legal and professional fees | | | 608 | | | | 774 | |
| Accounts payable | | | 793 | | | | 229 | |
| Other | | | 316 | | | | 79 | |
| Total | | $ | 6,512 | | | $ | 5,640 | |
8. Restructuring
We reduced headcount in the third and fourth quarters of 2011, resulting in a restructuring accrual of $1.3 million for severance and related benefits which were paid through August 2012. We incurred other exit and contract termination costs, including expenses related to a lease amendment and consolidation of employees within reduced facility space. We also reduced headcount in the third quarter of 2012, resulting in a restructuring accrual of $1.9 million which was paid through August 2013, and the first quarter of 2013, resulting in an approximately $1.5 million restructuring accrual which was paid through the end of 2013. During the second quarter of 2013, we incurred other exit and contract termination costs, including lease termination and amendment and consolidation expenses ($1,359).
Activity in the restructuring accrual, which is included in accounts payable and accrued expenses in our Consolidated Balance Sheets, and in research and development and general and administrative expenses in the Consolidated Statements of Operations, is specified below.
| | Severance and Related Benefits | | | Other Exit Costs | | | Contract Termination Costs | | | Total Restructuring Accrual | |
| Balance at December 31, 2010 | | $ | - | | | $ | - | | | $ | - | | | $ | - | |
| Additions, net | | | 1,341 | | | | 8 | | | | 292 | | | | 1,641 | |
| Payments | | | (770 | ) | | | (2 | ) | | | (138 | ) | | | (910 | ) |
| Balance at December 31, 2011 | | | 571 | | | | 6 | | | | 154 | | | | 731 | |
| Additions, net | | | 1,905 | | | | 184 | | | | 3 | | | | 2,092 | |
| Payments | | | (1,663 | ) | | | (190 | ) | | | (157 | ) | | | (2,010 | ) |
| Balance at December 31, 2012 | | | 813 | | | | - | | | | - | | | | 813 | |
| Additions, net | | | 1,492 | | | | 15 | | | | 1,359 | | | | 2,866 | |
| Payments | | | (2,305 | ) | | | (15 | ) | | | (1,359 | ) | | | (3,679 | ) |
| Balance at December 31, 2013 | | $ | - | | | $ | - | | | $ | - | | | $ | - | |
9. Stockholders' Equity
We are authorized to issue 160.0 million shares of Common Stock, par value $.0013, and 20.0 million shares of preferred stock, par value $.001. The Board of Directors has the authority to issue common and preferred shares, in series, with rights and privileges as determined by the Board of Directors. In July 2013, we completed a public offering of 9,775 shares of common stock, with net proceeds of approximately $40.1 million.
10. Commitments and Contingencies
a. Operating Leases
As of December 31, 2013, we leased office, manufacturing and laboratory space, under lease agreements expiring in December 2020.
Rental payments are recognized as rent expense on a straight-line basis over the term of the lease. In addition to rents due under these agreements, we are obligated to pay additional facilities charges, including utilities, taxes and operating expenses.
As of December 31, 2013, future minimum annual payments under all operating lease agreements are as follows:
| Years ending December 31, | | Minimum Annual Payments | |
| 2014 | | $ | 1,841 | |
| 2015 | | | 1,887 | |
| 2016 | | | 1,934 | |
| 2017 | | | 1,983 | |
| 2018 | | | 2,032 | |
| Thereafter | | | 4,218 | |
| Total | | $ | 13,895 | |
Rental expense totaled approximately $3,548, $2,074 and $3,475 for 2013, 2012 and 2011, respectively. For 2013 and 2012, amounts paid exceeded rent expense by $164 and $419, respectively, due to the recognition of lease incentives. For 2011, we recognized rent expense in excess of amounts paid of $63, due to the recognition of escalation clauses and lease incentives. Additional facility charges, including utilities, taxes and operating expenses, for 2013, 2012 and 2011 were approximately $2,330, $2,845 and $4,033, respectively.
b. Licensing, Service and Supply Agreements
Progenics and its subsidiaries have entered into intellectual property-based license and service agreements in connection with product development programs, and have recognized milestone, license and sublicense fees and supply costs, included in research and development expenses, totaling approximately $567, $1,170 and $578 during the last three years, respectively.
| | Paid from inception to December 31, 2013 | | | Future (1) Commitments | | Terms |
| Progenics agreements with: |
| Lonza Sales AG | | $ | 909 | | | $ | 824 | | Annual license fee payments, milestones and royalties, as applicable, in respect of oncology and other products. |
| | | | | | | | | |
| PSMA LLC agreements with: |
| Seattle Genetics, Inc. | | | 4,400 | | | | 13,900 | | Milestone and periodic maintenance payments to use ADC technology to link chemotherapeutic agents to monoclonal antibodies that target prostate specific membrane antigen. ADC technology is based in part on technology licensed by SGI from third parties. |
| Amgen Fremont, Inc. (formerly Abgenix) | | | 1,350 | | | | 5,750 | | Milestones and royalties to use XenoMouse® technology for generating fully human antibodies to PSMA LLC's PSMA antigen. |
| Former member of PSMA LLC | | | 278 | | | | 52,188 | | Annual minimum royalty payments and milestones to use technology related to PSMA. |
| | | | | | | | | |
| | Paid from acquisition date to December 31, 2013 | | | Future (1) Commitments | | Terms |
MIP agreements with: | | | | | | | | | |
| | | | | | | | | |
| University of Zurich and the Paul Scherrer Institute | | | 65 | | | | 1,225 | | Annual maintenance and license fee payments, milestones and royalties in respect of licensed technology related to 1404. |
| University of Western Ontario | | | 4 | | | | 374 | | Annual minimum royalty, administration and milestone payments in respect of licensed technology related to Azedra. |
| Novartis Pharma AG and other interests | | | - | | | | 4,600 | | Milestone and royalty payments in respect of licensed technology related to Onalta. |
| Bayer Schering Pharma AG | | | - | | | | 9,000 | | Milestone and royalty payments in respect of licensed technology related to a MIP asset. |
| | | | | | | | | |
(1) Amounts based on known contractual obligations as specified in the respective license agreements, which are dependent on the achievement or occurrence of future milestones or events and exclude amounts for royalties which are dependent on future sales and are unknown.
We are seeking to out-license or terminate non-germane Molecular Insight licenses and service agreements, as to which we have paid $216 through December 31, 2013, and have future commitments of $3,453, subject to occurrence of future milestones or events.
c. Consulting Agreements
As part of our research and development efforts, we have from time to time entered into consulting agreements with external scientific specialists. These agreements contain various terms and provisions, including fees to be paid by us and royalties, in the event of future sales, and milestone payments, upon achievement of defined events, payable by us. Certain of these scientists are advisors to Progenics, and some have purchased our Common Stock or received stock options which are subject to vesting provisions. We have recognized expenses with regard to the consulting agreements of $39, $8 and $27 for 2013, 2012 and 2011, respectively. Those expenses include the fair value of stock options granted during 2013 and 2011, which were fully vested at grant date, of approximately $7 and $11, respectively. Such amounts of fair value are included in research and development expense for each year presented (see Note 11).
d. Retirement Agreement
On March 14, 2012, Progenics and company founder Paul J. Maddon entered into an agreement providing for his retirement as Chief Science Officer. In connection with Dr. Maddon's retirement and termination of his employment agreement, Progenics agreed to pay him an amount equal to $1,789 and provide other benefits under the agreement.
e. Related Party Agreement
In December 2012, Progenics entered into a financial advisory agreement with MTS Health Partners, L.P., of which the Company's Board Chair is a Senior Managing Director and partner, on customary terms and conditions, whereby, in 2013, MTS has received monthly retainers totaling $55 during the term of the agreement and $300 for MTS' services in connection with the Molecular Insight acquisition described in Note 2. This agreement was terminated in June 2013.
f. Legal Proceedings
Progenics is a party to a proceeding brought by a former employee complaining that the Company violated the anti-retaliation provisions of the federal Sarbanes-Oxley law by terminating the former employee. The Company believes the former employee's claims are without merit and is contesting the matter vigorously. The federal District Court hearing the case issued in July 2013 an order denying our motion for summary judgment dismissing the former employee's complaint, making it likely that the proceeding will continue to trial. Given the inherent uncertainty attendant to the proceeding, it is not possible at this time to estimate the likelihood or potential magnitude of any outcome, and we have accordingly not recorded any associated liability in these Consolidated Financial Statements.
Progenics in October 2013 commenced an arbitration with Ono under the provisions of the parties' License Agreement, following a communication from Ono that it has determined to discontinue development of subcutaneous Relistor in Japan because of "commercial concerns" that Ono contends would permit it to cease development and terminate the Agreement. Under our Agreement with Ono, Ono may cease development of subcutaneous Relistor only if it terminates the License Agreement, which it may do unilaterally only if Progenics is in material default. Progenics is not in default under the Agreement, and Ono has neither asserted that Progenics is, nor terminated the Agreement.
11. Share-Based Payment Arrangements
Our share-based compensation to employees includes non-qualified stock options, restricted stock and shares issued under our Purchase Plans, which are compensatory under ASC 718 Compensation – Stock Compensation. During the second quarter of 2011, we accelerated the vesting of outstanding awards to non-management employees in connection with a change in program eligibility and termination of the Company's employee stock purchase plans. We account for share-based compensation to non-employees, including non-qualified stock options and restricted stock, in accordance with ASC 505 Equity.
Compensation cost for share-based awards will be recognized in our financial statements over the related requisite service periods; usually the vesting periods for awards with a service condition. We have made an accounting policy decision to use the straight-line method of attribution of compensation expense, under which the grant date fair value of share-based awards will be recognized on a straight-line basis over the total requisite service period for the total award.
We have adopted two stock incentive plans, the 1996 Amended Stock Incentive Plan (terminated in 2006) and the 2005 Stock Incentive Plan. Under these Plans as amended, up to 5,000 and 8,450 shares of common stock, respectively, have been reserved for the issuance of awards to employees, consultants, directors and other individuals who render services to Progenics (collectively, Awardees). The Plans contain anti-dilution provisions in the event of a stock split, stock dividend or other capital adjustment as defined. Each Plan provides for the Board or Committee to grant to Awardees stock options, stock appreciation rights, restricted stock, performance awards or phantom stock, as defined (collectively, Awards). The Committee is also authorized to determine the term and vesting of each Award and the Committee may in its discretion accelerate the vesting of an Award at any time. Stock options granted under the Plans generally vest pro rata over three to five years and have terms of ten years. Restricted stock issued under either Plan generally vested annually over three to five years, unless specified otherwise by the Committee. The exercise price of outstanding non-qualified stock options is usually equal to the fair value of our common stock on the date of grant. The exercise price of non-qualified stock options granted from the 2005 Plan and incentive stock options (ISO) granted from the Plans may not be lower than the fair value of our common stock on the dates of grant. At December 31, 2013, 2012 and 2011, all outstanding stock options were non-qualified options. The 2005 Plan will terminate in April 2015; options granted before termination of the Plans will continue under the respective Plans until exercised, cancelled or expired.
We apply a forfeiture rate to the number of unvested awards in each reporting period in order to estimate the number of awards that are expected to vest. Estimated forfeiture rates are based upon historical data on vesting behavior of employees. We adjust the total amount of compensation cost recognized for each award, in the period in which each award vests, to reflect the actual forfeitures related to that award. Changes in our estimated forfeiture rate will result in changes in the rate at which compensation cost for an award is recognized over its vesting period.
Under ASC 718 Compensation – Stock Compensation, the fair value of each non-qualified stock option award is estimated on the date of grant using the Black-Scholes option pricing model, which requires input assumptions noted in the following table. Ranges of assumptions for inputs are disclosed where the value of such assumptions varied during the related period. Historical volatilities are based upon daily quoted market prices of our common stock on The NASDAQ Stock Market LLC over a period equal to the expected term of the related equity instruments. We rely only on historical volatility since it provides the most reliable indication of future volatility. Future volatility is expected to be consistent with historical; historical volatility is calculated using a simple average calculation; historical data is available for the length of the option's expected term and a sufficient number of price observations are used consistently. Since our stock options are not traded on a public market, we do not use implied volatility. For 2013, 2012 and 2011 our expected term was calculated based upon historical data related to exercise and post-termination cancellation activity; accordingly, for grants issued to employees and directors and officers (excluding our former CEO in 2011), we are using expected terms of 5.3 and 7.4 years, 5.4 and 7.4 years and 5.3 and 7.4 years, respectively. The expected term of stock options granted to our former CEO in 2011 was calculated separately from stock options granted to employees and directors and officers, and was 8 years for 2011. The expected term for options granted to non-employees was also calculated separately from stock options granted to employees and directors and officers and was ten years, which is the contractual term of those options. We have never paid dividends and do not expect to pay dividends in the future. Therefore, our dividend rate is zero. The risk-free rate for periods within the expected term of the options is based on the U.S. Treasury yield curve in effect at the time of grant. The following table presents assumptions used in computing the fair value of option grants during 2013, 2012 and 2011:
| | 2013 | | | 2012 | | | 2011 | |
| | | | | | | | | |
| Expected volatility | | | 73% – 90 | % | | | 70% – 85 | % | | | 68% – 78 | % |
| Expected dividends | | Zero | | | Zero | | | Zero | |
| Expected term (years) | | | 5.3 – 10 | | | | 5.3 – 10 | | | | 5.3 – 10 | |
| Weighted average expected term (years) | | | 5.96 | | | | 6.11 | | | | 6.17 | |
| Risk-free rate | | | 0.76% – 2.83 | % | | | 0.57% – 1.71 | % | | | 0.77% – 2.97 | % |
A summary of option activity under the Plans as of December 31, 2013 and changes during the year then ended is presented below:
| Options | | Shares | | | Weighted Average Exercise Price | | | Weighted Average Remaining Contractual Term (Yr.) | | | Aggregate Intrinsic Value | |
| | | | | | | | | | | | |
| Outstanding at January 1, 2013 | | | 5,366 | | | $ | 12.27 | | | | | | | |
| Granted | | | 1,018 | | | | 5.13 | | | | | | | |
| Exercised | | | (14 | ) | | | 5.08 | | | | | | | |
| Forfeited | | | (608 | ) | | | 10.63 | | | | | | | |
| Expired | | | (463 | ) | | | 14.70 | | | | | | | |
| Outstanding at December 31, 2013 | | | 5,299 | | | | 10.89 | | | | 5.82 | | | $ | 304 | |
| Exercisable at December 31, 2013 | | | 3,875 | | | $ | 12.45 | | | | 4.86 | | | $ | 118 | |
The weighted average grant-date fair value of options granted under the Plans during 2013, 2012 and 2011 was $3.74, $6.38 and $5.51, respectively. The total intrinsic value of options exercised during 2013, 2012 and 2011 was $11, $174 and $345, respectively.
The options granted under the Plans, described above, include non-qualified stock options granted to our former CEO on July 3, 2006. For the 2006 award, the requisite service period is the shortest of the explicit or implied service periods and the explicit service period for this award is nine years and 11 months from the grant date. On July 1, 2011 and March 1, 2012, we granted option awards to our CEO which vests on the basis of the achievement of specified performance-based milestones. The options have exercise prices equal to the closing price of our common stock on the dates of grant. The awards are valued using the Black-Scholes option pricing model. The expense related to the grants with performance and market-based milestones will be recognized over the shortest estimated time for the achievement of the performance or market conditions. The awards will not vest unless one of the milestones is achieved or the market condition is met. Changes in the estimate of probability of achievement of any performance or market condition will be reflected in compensation expense of the period of change and future periods affected by the change.
At December 31, 2013, the estimated requisite service periods for the 2006, 2011 and 2012 awards, described above, were 2.5, 1.0 and 1.0 years, respectively. For 2013, 2012 and 2011, the total compensation expense recognized for the performance-based options was $0.1 million, $2.0 million and $0.4 million, respectively.
A summary of the status of our outstanding restricted stock awarded under the Plans as of December 31, 2013 and changes during the year then ended is presented below:
| Restricted Stock Awards | | Shares | | | Weighted Average Grant-Date Fair Value | |
| | | | | | |
| Nonvested at January 1, 2013 | | | 28 | | | $ | 5.35 | |
| Granted | | | - | | | | - | |
| Vested | | | (27 | ) | | | 5.35 | |
| Forfeited | | | (1 | ) | | | 5.35 | |
| Nonvested at December 31, 2013 | | | - | | | $ | - | |
Two employee stock purchase plans (the Purchase Plans), the 1998 Employee Stock Purchase Plan (the Qualified Plan) and the 1998 Non-Qualified Employee Purchase Plan (the Non-Qualified Plan), as amended, provided for the issuance of up to 4,400 and 1,100 shares of common stock, respectively. Issuances of common stock under the Purchase Plans, terminated by the Company during the second quarter of 2011, provided for the grant to all employees of options to use an amount equal to 25% of their quarterly compensation, as such percentage was determined by the Board of Directors prior to the date of grant, to purchase shares of our common stock at a price per share equal to the lesser of the fair market value of the common stock on the date of grant or 85% of the fair market value on the date of exercise. Options were granted automatically on the first day of each fiscal quarter and expired six months after the date of grant. The Qualified Plan was not available to employees owning more than five percent of the common stock and imposed certain other quarterly limitations on option grants. Options under the Non-Qualified Plan were granted to the extent that option grants were restricted under the Qualified Plan.
The fair value of shares purchased under the Purchase Plans was estimated on the date of grant in accordance with ASC 718 Compensation – Stock Compensation, via the same option valuation model used for options granted under the Plans, but with the following assumptions during 2011:
| | 2011 | |
| | | |
| Expected volatility | | | 43% – 51 | % |
| Expected dividends | | zero | |
| Expected term | | 6 months | |
| Risk-free rate | | | 0.06% – 0.22 | % |
Purchases of common stock under the Purchase Plans during 2011 are summarized as follows:
| | Qualified Plan | | | Non-Qualified Plan | |
| | Shares Purchased | | | Price Range | | | Weighted Average Grant-Date Fair Value | | | Shares Purchased | | | Price Range | | | Weighted Average Grant-Date Fair Value | |
| | | | | | | | | | | | | | | | | | |
| 2011 | | | 428 | | | $ | 4.62 – $5.65 | | | $ | 0.88 | | | | 162 | | | $ | 4.62 – $5.65 | | | $ | 0.84 | |
The total compensation expense of shares, granted to both employees and non-employees, under all of our share-based payment arrangements that was recognized in operations during 2013, 2012 and 2011 was:
| | 2013 | | | 2012 | | | 2011 | |
| Recognized as: | | | | | | | | | |
| Research and Development | | $ | 2,524 | | | $ | 4,568 | | | $ | 4,499 | |
| General and Administrative | | | 1,022 | | | | 1,968 | | | | 1,863 | |
| Total | | $ | 3,546 | | | $ | 6,536 | | | $ | 6,362 | |
No tax benefit was recognized related to such compensation cost because of the Company's net operating losses and the related deferred tax assets were fully offset by valuation allowance. Accordingly, no amounts related to windfall tax benefits have been reported in cash flows from operations or cash flows from financing activities for the periods presented.
As of December 31, 2013, there was $4.4 million of total unrecognized compensation cost related to non-vested stock options under the 1996 and 2005 Plans. Those costs are expected to be recognized over a weighted average period of 2.1 years. Cash received from exercises under all share-based payment arrangements for 2013 was $0.1 million. We issue new shares of our common stock upon share option exercises.
In applying the treasury stock method for the calculation of diluted EPS, amounts of unrecognized compensation expense and windfall tax benefits are required to be included in the assumed proceeds in the denominator of the diluted EPS calculation unless they are anti-dilutive. We incurred net losses for 2013 and 2012 and, therefore, such amounts have not been included in the calculations for those periods since they would be anti-dilutive. As a result, basic and diluted EPS are the same for the 2013 and 2012 periods. We reported net income for 2011 and included the dilutive effect of unrecognized compensation expense in the assumed proceeds in the denominator of the diluted EPS calculation. We have made an accounting policy decision to calculate windfall tax benefits/shortfalls, for purposes of diluted EPS calculation, excluding the impact of deferred tax assets. This policy decision will apply when we have net income and windfall tax benefits/shortfalls are realizable.
12. Employee Savings Plan
The terms of the amended and restated Progenics Pharmaceuticals 401(k) Plan (the Amended Plan), among other things, allow eligible employees to participate in the Amended Plan by electing to contribute to the Amended Plan a percentage of their compensation to be set aside to pay their future retirement benefits. During the three years ended December 31, 2013, we matched 50% of those employee contributions that are equal to 5%-8% of compensation and are made by eligible employees to the Amended Plan (the Matching Contribution). In addition, we may also make a discretionary contribution each year on behalf of all participants who are non-highly compensated employees. We made Matching Contributions of approximately $330, $535 and $597 to the Amended Plan for 2013, 2012 and 2011, respectively. No discretionary contributions were made during those years.
13. Income Taxes
We account for income taxes using the liability method in accordance with ASC 740 Income Taxes. Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes.
There is no provision or benefit for federal or state income taxes for 2013, 2012 and 2011, other than $0.4 million income tax benefit in 2013, resulting from the change in the temporary difference between carrying amounts of in-process research and development assets for financial reporting purposes and the amounts used for income tax purposes. We have completed a calculation through March 31, 2011, under Internal Revenue Code Section 382, the results of which indicate that past ownership changes will limit utilization of NOLs in the future. Ownership changes subsequent to March 31, 2011, may further limit the future utilization of net operating loss and tax credit carry-forwards as defined by the federal and state tax codes.
Deferred tax assets and liabilities as of December 31, 2013 and 2012, consisted of the following:
| | 2013 | | | 2012 | |
| Deferred tax assets: | | | | | | |
| Depreciation and amortization | | $ | 6,165 | | | $ | 6,497 | |
| R&E tax credit carry-forwards | | | 5,129 | | | | 11,843 | |
| NYS investment tax credit carry-forwards | | | 1,095 | | | | 1,084 | |
| AMT credit carry-forwards | | | 211 | | | | 211 | |
| Net operating loss carry-forwards | | | 190,263 | | | | 112,966 | |
| Capitalized research and development expenditures | | | 25,231 | | | | 30,884 | |
| Stock compensation | | | 13,826 | | | | 14,436 | |
| Other items | | | 1,097 | | | | 2,193 | |
| Total gross deferred tax assets | | | 243,017 | | | | 180,114 | |
| Less: Valuation allowance | | | (243,017 | ) | | | (178,045 | ) |
| Deferred tax assets | | | - | | | | 2,069 | |
| Deferred tax liability - current | | | - | | | | (2,069 | ) |
| Deferred tax liability – long term | | | (12,321 | ) | | | - | |
| Net deferred tax liability | | $ | (12,321 | ) | | $ | - | |
We do not recognize deferred tax assets considering our history of taxable losses and the uncertainty regarding our ability to generate sufficient taxable income in the future to utilize these deferred tax assets. For 2013 and 2012, we incurred net losses for tax purposes. For 2011, we had income for tax purposes and such amount was offset completely by our available net operating loss carry-forwards. We recognized a full tax valuation against deferred taxes at December 31, 2013. In 2012, we recognized deferred income tax assets, net of a valuation allowance, of $2,069 ($17 in current assets and $2,052 in non-current assets) and in 2013 and 2012 we recognized deferred income tax liabilities of $12,321 and $2,069, respectively to reflect the net tax effects of temporary differences between the carrying amounts of certain assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. The recognition of these deferred income tax assets and liabilities had no effect on our net loss for 2012, however, it resulted in $362 income tax benefit for 2013.
The following is a reconciliation of income taxes computed at the Federal statutory income tax rate to the actual effective income tax provision during 2013, 2012 and 2011:
| | 2013 | | | 2012 | | | 2011 | |
| | | | | | | | | |
| U.S. Federal statutory rate | | | (34.0 | )% | | | (35.0 | )% | | | 35.0 | % |
| State income taxes, net of Federal benefit | | | (4.9 | ) | | | (5.4 | ) | | | 8.0 | |
| Research and experimental tax credit | | | (3.6 | ) | | | - | | | | (4.1 | ) |
| Change in valuation allowance | | | 11.4 | | | | 34.7 | | | | (22.6 | ) |
| Effect of federal tax rate bracket change on valuation allowance | | | 8.7 | | | | - | | | | (34.8 | ) |
| Equity compensation | | | 3.1 | | | | 4.2 | | | | 17.0 | |
| Investment tax credit | | | (0.1 | ) | | | - | | | | (0.1 | ) |
| NOL expiration – Section 382 | | | 18.6 | | | | - | | | | - | |
| Other | | | - | | | | 1.5 | | | | 1.6 | |
| Income tax provision (benefit) | | | (0.8 | )% | | | 0.0 | % | | | 0.0 | % |
As of December 31, 2013, we had available, for tax return purposes, unused federal NOLs of approximately $513.8 million, which will expire in various years from 2018 to 2033, $18.2 million of which were generated from deductions post January 1, 2006 that, when realized, will reduce taxes payable and will increase paid-in-capital and are not reflected in our deferred tax assets above. Additionally, $11.2 million of the valuation allowance relates to NOLs attributable to excess tax deductions for equity compensation pre January 1, 2006. When realized this will also be reflected as an increase to paid-in-capital. Also, we had available, for tax return purposes, unused state NOLs of approximately $453.1 million, which will expire in various years from 2019 to 2033.
We have reviewed our nexus in various tax jurisdictions and our tax positions related to all open tax years for events that could change the status of our ASC 740 Income Taxes liability, if any, or require an additional liability to be recorded. During 2013, 2012 and 2011, we had no unrecognized tax benefits resulting from tax positions during a prior or current period, settlements with taxing authorities or the expiration of the applicable statute of limitations, except for the $0.4 million income tax benefit recognized in 2013, resulting from the change in the difference between carrying amounts of in-process research and development assets for financial reporting purposes and the amounts used for income tax purposes. We have not, as of yet, conducted a study of our research and development credit carry-forwards. Such a study might result in an adjustment to our research and development credit carry-forwards, but until a study is completed and any adjustment is known, no amounts are being presented as an uncertain tax position under ASC 740-10. A full valuation allowance has been provided against the Company's research and development credits and, if an adjustment is required, this adjustment would be offset by an adjustment to the valuation allowance. Thus, there would be no impact to the statements of operations and comprehensive loss if an adjustment was required.
As of December 31, 2013, we are subject to federal and state income tax in the U.S. Open tax years relate to years in which unused net operating losses were generated or, if used, for which the statute of limitation for examination by taxing authorities has not expired. Our open tax years extend back to 1996. No amounts of interest or penalties were recognized in our Consolidated Statements of Operations or Consolidated Balance Sheets as of and for 2013, 2012 and 2011.
Our research and experimental (R&E) tax credit carry-forwards of approximately $5.1 million at December 31, 2013 expire in various years from 2018 to 2033. During 2013, research and experimental tax credit carry-forwards of approximately $20 expired. The American Taxpayer Relief Act of 2012, enacted on January 2, 2013, retroactively reinstated the federal research and development credit for 2012 and extended these credits through 2013.
As of December 31, 2013, we have not recognized any liability for uncertain tax positions, because of our full valuation allowance. We will recognize interest and penalties related to these positions, should such costs be assessed. As of December 31, 2013, we have not recognized interest and penalties. The recognition of unrecognized tax benefits would not affect our effective tax rate because the tax benefit would be offset by an increase in our valuation allowance.
The following is a tabular reconciliation of the total amounts of unrecognized tax benefits for the year ended December 31, 2013.
| | 2013 |
| | |
| Beginning uncertain tax benefits | | $ | 2,661 |
| Current year - increases | | | - |
| Current year - decreases | | | - |
| Settlements | | | - |
| Expired statuses | | | - |
| Ending uncertain tax benefits | | $ | 2,661 |
14. Net Income (Loss) Per Share
Our basic net (loss) income per share amounts have been computed by dividing net (loss) income by the weighted-average number of common shares outstanding during the period. For 2013 and 2012, we reported net losses and, therefore, potential common shares were not included since such inclusion would have been anti-dilutive. For 2011, we reported net income, and the computation of diluted earnings per share is based upon the weighted-average number of our common shares and dilutive effect, determined using the treasury stock method, of potential common shares outstanding. As of December 31, 2012 and 2011, our 28 and 98, respectively, shares of unvested restricted stock with non-forfeitable rights to dividends were outstanding; all such shares were vested at the end of December 31, 2013. The allocation of 2012 net losses and the 2011 net income to these participating securities pursuant to the two-class method is not material to both basic and diluted earnings per share. The calculations of net loss per share, basic and diluted, are as follows:
| | Net (Loss) Income
(Numerator) | | | Weighted Average
Common Shares
(Denominator) | | | Per Share
Amount | |
| 2013: | | | | | | | | | |
| Basic and diluted | | $ | (42,572 | ) | | | 55,798 | | | $ | (0.76 | ) |
| 2012: | | | | | | | | | | | | |
| Basic and diluted | | $ | (35,431 | ) | | | 34,754 | | | $ | (1.02 | ) |
| 2011: | | | | | | | | | | | | |
| Basic | | $ | 10,381 | | | | 33,375 | | | $ | 0.31 | |
| Dilutive effect of stock options | | | - | | | | 66 | | | | | |
| Dilutive effect of restricted stock | | | - | | | | 53 | | | | | |
| Diluted | | $ | 10,381 | | | | 33,494 | | | $ | 0.31 | |
During 2013, 2012 and 2011, anti-dilutive common shares excluded from diluted per share amounts consist of the following:
| | 2013 | | | 2012 | | | 2011 |
| | Weighted
Average
Number | | | Weighted
Average
Exercise Price | | | Weighted
Average
Number | | | Weighted
Average
Exercise Price | | | Weighted
Average
Number | | | Weighted
Average
Exercise Price |
| Options | | | 5,969 | | | $ | 11.54 | | | | 5,947 | | | $ | 12.32 | | | | 4,543 | | | $ | 14.92 |
| Restricted stock | | | - | | | | | | | | 60 | | | | | | | | 45 | | | | |
| Total | | | 5,969 | | | | | | | | 6,007 | | | | | | | | 4,588 | | | | |
15. Unaudited Quarterly Results (unaudited)
Summarized quarterly financial data during 2013 and 2012 are as follows:
| | 2013 Quarter Ended | |
| | March 31 | | | June 30 | | | September 30 | | | December 31 | |
| Revenues | | $ | 2,226 | | | $ | 1,801 | | | $ | 867 | | | $ | 2,968 | |
| Net loss | | | (11,258 | ) | | | (12,263 | ) | | | (10,500 | ) | | | (8,551 | ) |
| Net loss per share - basic and diluted | | | (0.22 | ) | | | (0.24 | ) | | | (0.17 | ) | | | (0.14 | ) |
| | 2012 Quarter Ended | |
| | March 31 | | | June 30 | | | September 30 | | | December 31 | |
Revenues (1) | | $ | 2,226 | | | $ | 1,820 | | | $ | 1,117 | | | $ | 8,885 | |
| Net loss | | | (13,086 | ) | | | (10,720 | ) | | | (11,301 | ) | | | (324 | ) |
| Net loss per share - basic and diluted | | | (0.39 | ) | | | (0.32 | ) | | | (0.33 | ) | | | (0.01 | ) |
_______________
| (1) | Revenues in the fourth quarter of 2012 include $5.0 million and $2.8 million from the MedImmune and CytoDyn Agreements, respectively. |
16. Subsequent Event
During the first quarter of 2014, we established a $150 million shelf registration statement (inclusive of approximately $31.7 million of availability under a superseded registration) which we used for our recent underwritten public offering of 8.75 million shares of common stock at a public offering price of $4.60 per share, resulting in net proceeds of approximately $37.5 million.
Allowance for Doubtful Accounts
| Year ended December 31, | | Beginning Balance | | | Additions Charged to General and administrative expenses | | | Deductions Accounts Written Off During Period | | | Ending Balance | |
| (in thousands) | | | | | | | | | | | | |
| 2013 | | $ | - | | | $ | 7 | | | $ | - | | | $ | 7 | |
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrantregistrant has duly caused this report to be signed on its behalf by the undersigned, hereuntothereunto duly authorized.
| PROGENICS PHARMACEUTICALS, INC. |
| By: | /s/ MARK R. BAKER |
| | Mark R. Baker
Chief Executive Officer and Director (Principal Executive Officer) |
Date: March 13, 2014
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant in the capacities and on the dates indicated.
Signature | | Capacity | Date |
/s/ PETER J. CROWLEY | | Chairman | March 13, 2014 |
| Peter J. Crowley | | | |
| | | |
| /s/ PAUL J. MADDON | | Vice Chairman | March 13, 2014 |
| Paul J. Maddon, M.D., Ph.D. | | | |
| | | |
| /s/ MARK R. BAKER | | Chief Executive Officer and Director | March 13, 2014 |
| Mark R. Baker | | (Principal Executive Officer) | |
| | | |
| /s/ KAREN J. FERRANTE | | Director | March 13, 2014 |
| Karen J. Ferrante, M.D. | | | |
| | | |
| /s/ MICHAEL D. KISHBAUCH | | Director | March 13, 2014 |
| Michael D. Kishbauch | | | |
| | | |
| /s/ DAVID A. SCHEINBERG | | Director | March 13, 2014 |
| David A. Scheinberg, M.D., Ph.D. | | | |
| | | |
| /s/ NICOLE S. WILLIAMS | | Director | March 13, 2014 |
| Nicole S. Williams | | | |
| | | |
| /s/ ANGELO W. LOVALLO, JR. |
| | Angelo W. Lovallo, Jr.
(Duly authorized officer of the Registrant and Vice President, Finance and Treasurer (Principal
| March 13, 2014 |
| Angelo W. Lovallo, Jr. | | (Principal Financial and Accounting Officer)) | |
| | | |
Date: April 30, 2013
S-1
| | |
| Exhibit | | |
| Number * | | Description |
| 3.1(1) | | Amended and Restated Certificate of Incorporation of the Registrant. |
| 3.2(2) | | Amended and Restated By-laws of the Registrant. |
| 4.1(3) | | Specimen Certificate for Common Stock, $0.0013 par value per share, of the Registrant. |
10.1(3) | | Form of Registration Rights Agreement. |
10.2(3) | | 1989 Non-Qualified Stock Option Plan‡ |
10.3(3) | | 1993 Stock Option Plan, as amended‡ |
10.4(3) | | 1993 Executive Stock Option Plan‡ |
| 10.5(4) | | Amended and Restated 1996 Stock Incentive Plan‡ |
| 10.6.3(5) | | Amended 2005 Stock Incentive Plan ‡ |
| 10.6.4(6) | | Form of Non-Qualified Stock Option Award Agreement ‡ |
| 10.6.5(6) | | Form of Restricted Stock Award Agreement ‡ |
| 10.7(7) | | Form of Indemnification Agreement‡ |
10.8(8)10.8.2(8) | | Employment Agreement, dated December 31, 2007, between the Registrant and Dr. Paul J. Maddon‡ |
10.8.1(9) | | First Amendment to Employment Agreement, dated March 31, 2011, between the Registrant and Dr. Paul J. Maddon‡ |
10.8.2(10) | | Retirement Agreement, dated as of March 14, 2012, between the Registrant and Dr. Paul J. Maddon‡ |
| 10.9(3) | | Letter dated August 25, 1994 between the Registrant and Dr. Robert J. Israel‡ |
10.16(11)10.16(9)† | | Development and License Agreement, dated April 30, 1999, between Protein Design Labs, Inc. and the Registrant. |
10.16.1(12)10.16.1(10) | | Letter Agreement, dated November 24, 2003, relating to the Development and License Agreement between Protein Design Labs, Inc. and the Registrant. |
10.18(13) | | Director Stock Option Plan‡ |
10.19(14)10.19(11)† | | Exclusive Sublicense Agreement, dated September 21, 2001, between the Registrant and UR Labs, Inc. |
10.19.1(15)10.19.1(12) | | Amendment to Exclusive Sublicense Agreement, dated September 21, 2001, between the Registrant and UR Labs, Inc. |
10.21.1(16)10.21.1(13) | | Amended and Restated Agreement of Lease, dated October 28, 2009, between BMR-Landmark at Eastview LLC and the Registrant. |
| 10.23 | | Information concerning compensation of the Registrant's non-employee directors is included in the Registrant's proxy material for its 20122013 Annual Meeting of Stockholders and its Current Report on Form 8-K filed on September 12, 2013 and is incorporated herein by reference.‡ |
10.25(17)10.25(14) † | | Option and License Agreement, dated May 8, 1985, by and between the University of Chicago and UR Labs, Inc., as amended by (i) Amendment to Option and License Agreement, dated September 17, 1987, by and between the University of Chicago and UR Labs, Inc., (ii) Second Amendment to Option and License Agreement, dated March 3, 1989, by and among the University of Chicago, ARCH Development Corporation and UR Labs, Inc., and (iii) Letter Agreement Related to Progenics' Relistor In-License dated, December 22, 2005, by and among the University of Chicago, acting on behalf of itself and ARCH Development Corporation, the Registrant, Progenics Pharmaceuticals Nevada, Inc. and Wyeth, acting through its Wyeth Pharmaceuticals Division. |
10.26(18)10.26(15) | | Membership Interest Purchase Agreement, dated April 20, 2006, between the Registrant Inc. and Cytogen Corporation. |
10.27(18)10.27(15) † | | Amended and Restated PSMA/PSMP License Agreement, dated April 20, 2006, by and among the Registrant, Cytogen Corporation and PSMA Development Company LLC. |
10.28(19) | | Consulting Agreement, dated May 1, 1995, between Active Biotherapies, Inc. and Dr. David A. Scheinberg, M.D., Ph.D., as amended on June 13, 1995, as assigned to the Registrant, and as amended on January 1, 2001‡ |
10.29(20)10.29(16) † | | License Agreement, dated as of October 16, 2008, by and among Ono Pharmaceutical Co., Ltd. and the Registrant. |
10.30(20)10.30(16) † | | Partial Termination and License Agreement, dated October 16, 2008, by and among Wyeth, acting through Wyeth Pharmaceuticals Division, Wyeth-Whitehall Pharmaceuticals, Inc. and Wyeth-Ayerst Lederle, Inc. and the Registrant and Progenics Pharmaceuticals Nevada, Inc. |
10.31(20)10.31(16) † | | Consent, Acknowledgment and Agreement, dated as of October 16, 2008, by and among Wyeth, acting through Wyeth Pharmaceuticals Division, Wyeth-Whitehall Pharmaceuticals, Inc. and Wyeth-Ayerst Lederle, Inc., the Registrant and Ono Pharmaceutical Co., Ltd. |
10.32(20)10.32(16) † | | 2008 Agreement Related to Progenics' MNTX In-License, dated October 16, 2008, by and among the University of Chicago, acting on behalf of itself and ARCH Development Corporation, the Registrant, Progenics Pharmaceuticals Nevada, Inc. and Ono Pharmaceutical Co., Ltd. |
10.33(21) | | |
| | |
| 10.34(17) † | | Termination and Transition Agreement, effective as of October 1, 2009, by and among Wyeth, acting through Wyeth Pharmaceuticals Division, Wyeth-Whitehall Pharmaceuticals, Inc., Wyeth-Ayerst Lederle, Inc., and AHP Manufacturing B.V., and the Registrant, Progenics Pharmaceuticals Nevada, Inc. and Excelsior Life Sciences Ireland Limited. |
10.33.1(22) † | | First Amendment to Termination and Transition Agreement, effective as of October 1, 2010, by and among Wyeth LLC, acting through Wyeth Pharmaceuticals Division, Wyeth-Whitehall Pharmaceuticals LLC, Wyeth-Ayerst Lederle LLC, and AHP Manufacturing B.V., and the Registrant, Progenics Pharmaceuticals Nevada, Inc. and Excelsior Life Sciences Ireland Limited. |
10.34(23) † | | Collaboration Agreement, effective June 14, 2005, by and between Seattle Genetics, Inc. and PSMA Development Company, LLC. |
| 10.37(9) † | | License Agreement dated as of February 3, 2011, by and between Salix Pharmaceuticals, Inc., the Registrant, Progenics Pharmaceuticals Nevada, Inc. and Excelsior Life Sciences Ireland Limited. |
| 10.37.1(9) † | | 2010 Agreement Related to Progenics' MNTX In-License, dated February 3, 2011, by and among the University of Chicago, acting on behalf of itself and ARCH Development Corporation, the Registrant, Progenics Pharmaceuticals Nevada, Inc. and Salix Pharmaceuticals, Inc. |
| 10.38(18) † | | Stock Purchase and Sale Agreement, dated January 16, 2013, by and between Molecular Insight Pharmaceuticals, Inc., its Stockholders, the Registrant, and Highland Capital Management, L.P., as Stockholders Representative. |
| 10.39(18) † | | License Agreement, dated September 1, 2012, by and between FUJIFILM RI Pharma Co., Ltd. and Molecular Insight Pharmaceuticals, Inc. |
| 10.40 †† | | License Agreement, dated May 2, 2012, between Molecular Insight Pharmaceuticals, Inc., the University of Zurich and the Paul Scherrer Institute. |
| 10.41(19) | | License Agreement, dated as of December 15, 2000, between Molecular Insight Pharmaceuticals, Inc. and The Board of Governors of the University of Western Ontario. |
| 10.42(19) | | License Agreement, dated as of November 3, 2006, between Molecular Insight Pharmaceuticals, Inc. and Novartis Pharma AG, together with First Amendment, dated January 4, 2007, thereto. |
| 10.43(20) | | Controlled Equity OfferingSM Sales Agreement dated as of January 23, 2014, by and between the Registrant and Cantor Fitzgerald & Co. (filed as Exhibit 1.1 to Registration Statement on Form S-3, Commission File No. 333-193521. |
| 12.1 | | Statement re computation of ratio of earnings (loss) to combined fixed charges and preferred stock dividends. |
| 21.1 | | Subsidiaries of the Registrant. |
| 23.1 | | Consent of PricewaterhouseCoopers LLP. |
| 23.2 | | Consent of Ernst & Young LLP. |
| 31.1 | | Certification of Mark R. Baker, Chief Executive Officer of the Registrant pursuant to 13a-14(a) and Rule 15d-14(a) under the Securities Exchange Act of 1934, as amended. |
31.2 | | Certification of Angelo W. Lovallo, Jr., Senior Executive Director, Financial Reporting & Treasurer of the Registrant pursuant to 13a-14(a) and Rule 15d-14(a) under the Securities Exchange Act of 1934, as amended. |
31.3** | | Certification of Mark R. Baker, Chief Executive Officer of the Registrant pursuant to 13a-14(a) and Rule 15d-14(a) under the Securities Exchange Act of 1934, as amended. |
31.4**31.2 | | Certification of Angelo W. Lovallo, Jr., Vice President, Finance & Treasurer of the Registrant pursuant to 13a-14(a) and Rule 15d-14(a) under the Securities Exchange Act of 1934, as amended. |
| 32.1 | | Certification of Mark R. Baker, Chief Executive Officer of the Registrant pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
| 32.2 | | Certification of Angelo W. Lovallo, Jr., Senior Executive Director, Financial Reporting & Treasurer of the Registrant pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
32.3** | | Certification of Mark R. Baker, Chief Executive Officer of the Registrant pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
32.4** | | Certification of Angelo W. Lovallo, Jr., Senior Executive Director, Financial ReportingVice President, Finance & Treasurer of the Registrant pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
| 101 | | Interactive Data File |
| 101.INS | | XBRL Instance Document |
| 101.SCH | | XBRL Taxonomy Extension Schema |
| 101.CAL | | XBRL Taxonomy Extension Calculation Linkbase |
| 101.LAB | | XBRL Taxonomy Extension Label Linkbase |
| 101.PRE | | XBRL Taxonomy Extension Presentation Linkbase |
| 101.DEF | | XBRL Taxonomy Extension Definition Document |
| | |
| * | | Exhibits footnoted as previously filed have been filed as an exhibit to the document of the Registrant or other registrant referenced in the footnote below, and are incorporated by reference herein. Exhibits not so footnoted and not filed herewith were filed with the Registrant's Annual Report on Form 10-K for the year ended December 31, 2012 filed with the SEC March 15, 2012. |
| | |
**(1) | | Filed herewith. |
| | |
(1) | | Previously filed in QuarterlyCurrent Report on Form 10-Q for the quarter ended8-K filed on June 30, 2011.13, 2013. |
| (2) | | Previously filed in Current Report on Form 8-K filed on March 16, 2012. |
| (3) | | Previously filed in Registration Statement on Form S-1, Commission File No. 333-13627. |
| (4) | | Previously filed in Registration Statement on Form S-8, Commission File No. 333-120508. |
| (5) | | Previously filed in Current Report on Form 8-K filed on November 28, 2012.June 17, 2013. |
| (6) | | Previously filed in Current Report on Form 8-K filed on July 8, 2008. |
| (7) | | Previously filed in Quarterly Report on Form 10-Q for the quarterly period ending March 31, 2007 |
(8) | | Previously filed in Annual Report on Form 10-K for the year ended December 31, 2007. |
(9) | | Previously filed in Quarterly Report on Form 10-Q for the quarter ended March 31, 2011.2007. |
(10)(8) | | Previously filed in Quarterly Report on Form 10-Q for the quarter ended March 31, 2012. |
(11)(9) | | Previously filed in Quarterly Report on Form 10-Q for the quarterly periodquarter ended June 30, 1999. |
(12)(10) | | Previously filed in Annual Report on Form 10-K for the year ended December 31, 2004. |
(13) | | Previously filed in Annual Report on Form 10-K for the year ended December 31, 1999. |
(14)(11) | | Previously filed in Annual Report on Form 10-K for the year ended December 31, 2002. |
(15)(12) | | Previously filed in Current Report on Form 8-K filed on September 20, 2004. |
(16)(13) | | Previously filed in Current Report on Form 8-K filed on November 28, 2012. |
(17)(14) | | Previously filed in Annual Report on Form 10-K for the year ended December 31, 2005. |
(18)(15) | | Previously filed in Quarterly Report on Form 10-Q for the quarterly period endingquarter ended June 30, 2006.2006 |
(19)(16) | | Previously filed in Annual Report on Form 10-K/A for the year ended December 31, 2006. |
(20) | | Previously filed in Annual Report on Form 10-K for the year ended December 31, 2008. |
(21)(17) | | Previously filed in Annual Report on Form 10-K for the year ended December 31, 2009. |
(22) | | Previously filed in Annual Report on Form 10-K for the year ended December 31, 2010. |
(23) | | Previously filed in Amendment No. 2 to Annual Report on Form 10-K/A for the year ended December 31, 2009. |
| (18) | | Previously filed in Quarterly Report on Form 10-Q for the quarter ended March 31, 2013. |
| (19) | | Previously filed in Registration Statement on Form S-1, Commission File No. 333-129570 filed by Molecular Insight Pharmaceuticals, Inc. |
| (20) | | Previously incorporated in Current Report on Form 8-K filed on January 24, 2014 by reference to Exhibit 1.1 to Registration Statement on Form S-3, Commission File No. 333-193521. |
| | |
| | |
| † | | Confidential treatment granted as to certain portions omitted and filed separately with the Commission. |
| †† | | Confidential treatment requested as to certain portions omitted and filed separately with the Commission. |
| ‡ | | Management contract or compensatory plan or arrangement. |
| | |
E-2