Executive Officers
The following table identifies certain information about our executive officers as of April 29, 2022. Our executive officers are appointed by, and serve at the discretion of, our Board.
Name | | Age | | Principal Position | |
Eric I. Richman | | 61 | | Chief Executive Officer and Director | |
Matthias Alder | | 57 | | Chief Operating Officer | |
Salvatore Calabrese | | 52 | | Chief Financial Officer | |
Eric I. Richman Biographical information for Mr. Richman is included above with the director biographies under the caption “Board of Directors.”
Matthias Alder has served as our Chief Operating Officer since October 2021. Prior to joining us, he served as Chief Business Officer at Autolus Therapeutics plc from 2017 to 2021. Prior to then, from 2014 to 2017, Mr. Alder held various executive management positions at Sucampo Pharmaceuticals, Inc., a biopharmaceutical company which was subsequently acquired by Mallinckrodt Pharmaceuticals, including executive vice president of Business DevelopmentStrategic Transactions; Collaboration and Licensing and general counsel and corporate secretary. Prior to 2014, Mr. Alder served as executive vice president of corporate development and legal affairs and corporate secretary at Cytos Biotechnology AG, a biopharmaceutical company focused on the development of targeted immunotherapies, from 2013 to October 2014. From 2006 to 2012, Mr. Alder held various executive management roles at Micromet, Inc., serving as senior vice president for administration, general counsel and secretary at the time of the acquisition of Micromet by Amgen Inc. in 2012. He was also a partner in the Life Sciences Transactions Practice at Cooley LLP from 2000 to 2006, where he represented biotech companies in strategic transactions with pharmaceutical companies. Earlier in his career, Mr. Alder was in-house counsel at Ciba-Geigy and Novartis. Mr. Alder holds law degrees from the University of Basel and the University of Miami and is qualified to practice law in Switzerland and the United States.
Salvatore CalabreseArrangements has served as our Chief Financial Officer since November 2020. Prior to joining us, Mr. Calabrese served as the Chief Financial Officer of Molmed S.p.A., a cell & gene company, from September 2018 to September 2020. Prior to joining Molmed S.p.A, Mr. Calabrese served as General Manager and Site Leader for the Italian Operation of Jazz Pharmaceuticals from 2014 to 2018. From February 2005 to August 2014, he held various positions of increasing responsibility at Gentium S.p.A., most recently holding the position of Chief Operating Officer. From December 2003 until February 2005, he served as Accounting and Finance Manager for Novuspharma, S.p.A., a development stage biopharmaceutical company focused on the discovery and development of cancer drugs and a subsidiary of Cell Therapeutics, Inc. From 1996 until 2003, Mr. Calabrese was employed by PricewaterhouseCoopers as an accountant and was a Manager in Assurance Business Advisory Services at the time of his departure. He earned a B.A. in Economics at the University of Messina and a M.S. in Accounting, Audit and Financial Control at the University of Pavia. He is also listed in the Italian National Audit Register held by the National Justice Department in the Republic of Italy.
There are no family relationships among any of our directors or executive officers.
Corporate Governance
General
Our Board has adopted Corporate Governance Guidelines, a Code of Business Conduct and Ethics and charters for the Audit Committee, Compensation Committee and Nominating and Corporate Governance Committee of our Board to assist the Board in the exercise of its responsibilities and to serve as a framework for the effective governance of the Company. You can access our current committee charters, our Corporate Governance Guidelines and our Code of Business Conduct and Ethics in the “Corporate Governance” section of the “Investors & Media” page of our website located at https://gaintherapeutics.com/investors-media/corporate-governance.html or by writing to our Secretary at our offices at 4800 Montgomery Lane, Suite 220 Bethesda, Maryland 20814.
If we make any substantive amendments to the Code of Business Conduct or we grant any waiver from a provision of the Code of Business Conduct to any executive officer or director, we will promptly disclose the nature of the amendment or waiver on our website.
Independence of the Board of Directors
As required under the Nasdaq Stock Market, or Nasdaq, listing standards, a majority of the members of a listed company’s Board of Directors must qualify as “independent,” as affirmatively determined by the Board of Directors. The Board consults with its counsel to ensure that the Board’s determinations are consistent with relevant securities and other laws and regulations regarding the definition of “independent,” including those set forth in pertinent listing standards of Nasdaq, as in effect from time to time.
Consistent with these considerations, after review of all relevant identified transactions or relationships between each director, or any of his or her family members, and us, our senior management and our independent auditors, the Board has affirmatively determined that the following five directors are independent directors within the meaning of the applicable Nasdaq listing standards: Dr. Goldstein, Mr. Hasler, Ms. Melincoff, Dr. Nicaise and Mr. Riley. In making this determination, the Board found that none of these directors or nominees for director had a material or other disqualifying relationship with us.
Accordingly, a majority of our directors are independent, as required under applicable Nasdaq rules. In making this determination, our Board considered the applicable Nasdaq rules and the current and prior relationships that each non-employee director has with our company and all other facts and circumstances our Board deemed relevant in determining their independence, including their beneficial ownership of our capital stock.
Board Leadership Structure
The Chairman of our Board is Dr. Islam. The primary responsibilities of the Chairman of our Board of Directors are to: work with our Chief Executive Officer, Mr. Richman, to develop board meeting schedules and agendas; provide our Chief Executive Officer feedback on the quality, quantity and timeliness of the information provided to the Board of Directors; develop the agenda for and moderate executive sessions of the independent members of the Board of Directors; preside over board meetings; act as principal liaison between the independent members of the Board of Directors and the Chief Executive Officer; convene meetings of the independent directors as appropriate; and perform other duties as the Board of Directors may determine from time to time. Accordingly, Dr. Islam has substantial ability to shape the work of our Board of Directors.
We believe that separation of the positions of board chair and Chief Executive Officer reinforces the independence of the Board in its oversight of our business and affairs.
Board Diversity
Our Board believes that a diverse board is better able to effectively oversee our management and strategy, and position us to deliver long-term value for our stockholders. Our nominating and corporate governance committee considers diversity, including gender, sexual preference and ethnicity, as adding to the overall mix of perspectives of our Board as a whole. With the assistance of the nominating and corporate governance committee, our Board regularly reviews trends in board composition, including on director diversity.
The table below provides additional diversity information regarding our Board as of April 29, 2022. Each of the categories listed in the below table has the meaning as it is used in Nasdaq Listing Rule 5605(f). While the Board satisfies the minimum objectives of Nasdaq Rule 5605(f)(2) by having at least one director who identifies as female and at least one director who identifies as a member of an Underrepresented Minority (as defined by Nasdaq Rules), the Nominating and Corporate Governance Committee will continue to consider the diversity of the Board in its selection of director nominees.
Board Diversity Matrix (As of April 29, 2022) |
| Total Number of Directors | 7 |
| | Female | Male | Non- Binary | Did Not Disclose Gender |
| Part I: Gender Identity | | | | |
| Directors | 1 | 6 | | |
| Part II: Demographic Background | | | | |
| African American or Black | | | | |
| Alaskan Native or Native American | | | | |
| Asian | | 1 | | |
| Hispanic or Latinx | | | | |
| Native Hawaiian or Pacific Islander | | | | |
| White | 1 | 4 | | |
| Two or More Races or Ethnicities | | 1 | | |
| LGBTQ+ | |
| Did Not Disclose Demographic Background | |
Audit Committee and Audit Committee Financial Expert
We have a separately designated standing Audit Committee of the Board established in accordance with Section 3(a)(58)(A) of the Securities Exchange Act of 1934, as amended, or the Exchange Act, to oversee our corporate accounting and financial reporting processes and audits of our financial statements.
Dr. Goldstein, Mr. Hasler and Mr. Riley serve as the current members of the Audit Committee, with Dr. Goldstein serving as Chair of the Audit Committee. The Board also determined that Dr. Goldstein is an “audit committee financial expert” within the meaning of the SEC regulations and applicable listing standards of Nasdaq. Our Board has determined that each of the members of our Audit Committee satisfies the independence requirements under the listing standards of Nasdaq and Rule 10A-3(b)(1) of the Exchange Act.
Code of Business Conduct and Ethics
We have adopted a Code of Business Conduct and Ethics that applies to all of our officers, directors and employees. The Code of Business Conduct and Ethics is available on our website at https://gaintherapeutics.com/investors-media/corporate-governance.html. If we make any substantive amendments to the Code of Business Conduct or we grant any waiver from a provision of the Code of Business Conduct to any executive officer or director, we will promptly disclose the nature of the amendment or waiver on our website.
Stockholder Communications with the Board
Stockholder communications will be reviewed by one or more employees of the Company designated by the Board, who will determine whether the communication should be presented to the Board. The purpose of this screening is to allow the Board to avoid having to consider irrelevant or inappropriate communications (such as advertisements, solicitations and hostile communications). All communications directed to the Audit Committee in accordance with our Whistleblower Policy for Accounting and Auditing Matters that relate to questionable accounting or auditing matters involving us will be promptly and directly forwarded to the Audit Committee. We also have a corporate ethics hotline to allow complaints related to questionable accounting or auditing matters. All inquiries made through this hotline are immediately directed to the chairman of the Audit Committee.
Stockholder Nomination Process
Our Nominating and Corporate Governance Committee will consider director candidates recommended by stockholders. The Nominating and Corporate Governance Committee does not intend to alter the manner in which it evaluates candidates, based on whether or not the candidate was recommended by a stockholder. Stockholders who wish to recommend individuals for consideration by the Nominating and Corporate Governance Committee to become nominees for election to the Board may do so by delivering a written recommendation to the Nominating and Corporate Governance Committee at the following address: 4800 Montgomery Lane, Suite 220 Bethesda, Maryland 20814 at least 120 days prior to the anniversary date of the mailing of our proxy statement for the last Annual Meeting of Stockholders. Submissions must include the full name of the proposed nominee, a description of the proposed nominee’s business experience for at least the previous five years, complete biographical information, a description of the proposed nominee’s qualifications as a director and a representation that the nominating stockholder is a beneficial or record holder of the Company’s stock and has been a holder for at least one year. Any such submission must be accompanied by the written consent of the proposed nominee to be named as a nominee and to serve as a director if elected.
Item 11. Executive Compensation.
Compensation Overview
This section provides a discussion of the total compensation awarded to, earned by, or paid to, during the years ended December 31, 2021 and 2020: (1) the individual who served as our principal executive officer during the fiscal year ended December 31, 2021 and (2) our two next most highly compensated executive officers who earned more than $100,000 during the fiscal year ended December 31, 2021 and were serving as executive officers as of such date. We refer to these individuals in this prospectus as our named executive officers.
Our named executive officers for 2021 who appear in the Summary Compensation Table are:
| • | Eric I. Richman, our President and Chief Executive Officer; |
| • | Matthias Alder, our Chief Operating Officer; and |
| • | Salvatore Calabrese, our Chief Financial Officer. |
Summary Compensation Table
The following table shows, for the fiscal years ended 2021 and 2020, compensation awarded to or paid to, or earned by, our named executive officers.
Summary Compensation Table
Name and Principal
Position | | Year | | Salary
($)(1) | | | Bonus
($) | | | Stock Awards | | Option Awards
($)(2) | | | Non-Equity Incentive Plan
Compensation
($)(3) | | | All Other
Compensation
($) | | | Total ($) | |
| Eric I. Richman (4) | | 2021 | | | 373,458 | | | | 80,000 | (5) | | — | | | — | | | | 120,000 | | | | 26,707 | (6) | | | 600,165 | |
| Chief Executive Officer | | 2020 | | | 131,250 | | | | 30,000 | (7) | | — | | | 63,843 | | | | 37,500 | (8) | | | 14,540 | | | | 277,133 | |
| Matthias Alder (9) | | | | | | | | | | | | 0 | (10) | | | | | | | | | | | | | | | |
Chief Operating Officer | | 2021 | | | 83,333 | | | | — | | | — | | | 848,826 | | | | 21,875 | (11) | | | — | | | | 954,034 | |
| Salvatore Calabrese | | 2021 | | | 268,000 | (12) | | | 40,000 | (13) | | — | | | — | | | | 37,389 | | | | — | | | | 345,389 | |
| Chief Financial Officer | | 2020 | | | 41,810 | | | | 21,095 | (14) | | — | | | 202,253 | | | | 25,086 | (15) | | | 29,116 | (16) | | | 319,360 | |
(1) | Salary amounts represent actual amounts earned during the periods presented. See “ —Narrative to the Summary Compensation Table—Annual Base Salary” below. |
| |
(2) | In accordance with SEC rules, this column reflects the aggregate grant date fair value of the option awards granted during the periods presented computed in accordance with ASC 718 for stock-based compensation transactions. Assumptions used in the calculation of these amounts are included in Note 15 to our audited consolidated financial statements included in our Annual Report on Form 10-K. These amounts do not reflect the actual economic value that will be realized by the named executive officer upon the vesting of the options, the exercise of the options or the sale of shares of common stock underlying such options. |
| |
(3) | Reflects performance-based cash bonuses awarded to our named executive officers during the periods presented. See “—Non-Equity Incentive Plan Compensation” below for a description of the material terms of the program pursuant to which this compensation was awarded. |
(4) | Mr. Richman was appointed our Chief Executive Officer in July 2020. He also serves as a member of our board of directors but does not receive any additional compensation for his service as a director. |
| |
(5) | Represents a one-time bonus payment in the amount of $80,000 awarded in April 2021 in recognition of the successful completion of our initial public offering. |
| |
(6) | Represents reimbursement of medical, dental and vision benefits. |
| |
(7) | Represents a one-time signing bonus payment in the amount of $30,000. |
| |
(8) | Mr. Richman’s bonus payment was pro-rated for the portion of fiscal year 2020 which he was employed by us. |
| |
(9) | Mr. Alder commenced employment with us on October 15, 2021. |
| |
(10) | The performance-based restricted stock unit award granted to Mr. Alder in December 2021 will vest upon the achievement of the following milestones: (i) the first tranche of 100,000 restricted stock units will vest upon (a) the closing of an exclusive license deal for one or more Company pipeline programs and/or research, development and commercialization of a collaboration deal with a “Validating Industry Partner” (as such term is defined in his employment agreement), and (b) a resulting extension of our cash runway by one year or more with one or more transactions; and (ii) the second tranche of 100,000 restricted stock units will vest upon the initiation (which we define as the dosing of a first patient) of a Phase 1b clinical trial conducted in the United States. The grant date fair value for this award was determined to be $0 under ASC 718 based upon a determination that, as of the grant date, it was not probable that the performance conditions will be achieved. |
| |
(11) | Mr. Alder’s bonus payment was pro-rated for the portion of fiscal year 2021 which he was employed by us. |
| |
(12) | Mr. Calabrese’s 2021 annual compensation was €226,600 which for reporting purposes has been converted into U.S. dollars at the 2021 average exchange rate of $1.18. |
| |
(13) | Represents a one-time bonus payment in the amount of $40,000 awarded in April 2021 in recognition of the successful completion of our initial public offering. |
| |
(14) | Represents a one-time signing bonus of €18,500 ($21,095), which for reporting purposes was converted into U.S. dollars at the exchange rate of $1.14. |
| |
(15) | Mr. Calabrese’s bonus payment of €22,000 ($25,086) was pro-rated for the portion of fiscal year 2020 which he was employed by us, and for reporting purposes was converted into U.S. dollars at the exchange rate of $1.14. |
| |
(16) | Amount reflects consulting fees paid to Mr. Calabrese prior to his appointment as our Chief Financial Officer in November 2020. |
Narrative to Summary Compensation Table
Our Board of Directors reviews compensation annually for all employees, including our named executive officers. In setting executive base salaries and bonuses and granting equity incentive awards, we consider compensation for comparable positions in the market, the historical compensation levels of our executives, individual performance as compared to our expectations and objectives, our desire to motivate our employees to achieve short- and long-term results that are in the best interests of our stockholders and a long-term commitment to our company.
Our Board of Directors has historically determined the compensation of our executives, upon recommendation of the compensation committee. The compensation committee has reviewed and recommended to the board for approval the compensation and other terms of employment of our Chief Executive Officer, and evaluates the Chief Executive Officer’s performance in light of relevant corporate goals and objectives. Our Chief Executive Officer has typically discussed his recommendations for all other executives (other than himself) with the compensation committee and the board. Based on those discussions and its discretion, the Compensation Committee has recommended the compensation of each executive officer to the board, and the Board has then approved.
Annual Base Salary
The annual base salaries of our named executive officers are generally reviewed, determined and approved by the Board periodically upon the recommendation of the compensation committee in order to compensate our named executive officers for the satisfactory performance of duties to our company. Annual base salaries are intended to provide a fixed component of compensation to our named executive officers, reflecting their skill sets, experience, roles and responsibilities. Base salaries for our named executive officers have generally been set at levels deemed necessary to attract and retain individuals with superior talent.
The following table sets forth the annual base salaries for each of our named executive officers for 2021 and 2022, as determined by the Board upon the recommendation of the compensation committee:
| Name | | 2021 Base Salary ($) | | | 2022 Base Salary ($) | |
| Eric I. Richman | | | 400,000 | (1) | | | 420,000 | (2) |
| Matthias Alder | | | 400,000 | | | | 420,000 | |
| Salvatore Calabrese | | | 268,000 | (3) | | | 281,400 | (4) |
(1) | Mr. Richman’s base salary is pro-rated from an annual base salary of $500,000 for his 80% work schedule. |
| |
(2) | Mr. Richman’s base salary is pro-rated from an annual base salary of $525,000 for his 80% work schedule. |
| |
(3) | Mr. Calabrese’s 2021 base salary was set at €226,600, which for reporting purposes has been converted into U.S. dollars at the 2021 average exchange rate of $1.1827. |
| |
(4) | Mr. Calabrese’s 2022 base salary was increased by 5% to €237,930. Assuming the 2021 average exchange rate of $1.1827, Mr. Calabrese’s 2022 base salary would be equivalent to $281,400. |
Bonus
In April 2021, Mr. Richman and Mr. Calabrese were awarded one-time, discretionary bonus payments of $80,000 and $40,000, respectively, in recognition of the successful completion of our initial public offering.
Non-Equity Incentive Plan Compensation
In accordance with the terms of their respective employment agreements, our named executive officers are eligible to receive discretionary annual bonuses of up to a percentage of each executive’s gross base salary based on individual performance, company performance or as otherwise determined appropriate, as determined by the Compensation Committee.
| Name | | 2021 Bonus
Target (%) | | | 2022 Bonus
Target (%) | |
| Eric I. Richman | | | 40 | | | | 50 | |
| Matthias Alder | | | 35 | | | | 40 | |
| Salvatore Calabrese | | | 30 | | | | 35 | |
In April 2022, the Compensation Committee completed an evaluation of our overall performance for 2021 and the named executive officers’ respective contributions in achieving this performance. The Compensation Committee’s review was based on company performance against corporate objectives previously approved by the Board. Following such review, the Compensation Committee determined that the corporate performance objectives were achieved at approximately the 50% achievement level. The Compensation Committee also determined that Mr. Richman had earned 75% of his target bonus for 2021 based on additional achievements and his overall performance, equal to a bonus of $120,000; Mr. Alder had earned 75% of his target bonus for 2021 based on additional achievements and his overall performance, equal to a bonus of $21,875 which was pro-rated for the portion of 2021 which he was employed by us; and Mr. Calabrese had earned 50% of his target bonus for 2021, equal to a bonus of $37,389.
Equity-Based Incentive Awards
Our equity-based incentive awards granted to our named executive officers are designed to align our interests and those of our stockholders with those of our employees and consultants, including our executive officers.
Historically, we have used stock options as an incentive for long-term compensation to our executive officers because the options allow our executive officers to profit from this form of equity compensation only if our stock price increases relative to the option’s exercise price, which exercise price is set at the fair market value of our shares of common stock on the date of grant. However, in December 2021, our Board granted a performance-based restricted stock unit award to Mr. Alder, the vesting of which is based on the achievement of the following milestones: (i) the first tranche of 100,000 restricted stock units will vest upon (a) the closing of an exclusive license deal for one or more Company pipeline programs and/or research, development and commercialization of a collaboration deal with a “Validating Industry Partner” (as such term is defined in his employment agreement), and (b) a resulting extension of our cash runway by one year or more with one or more transactions; and (ii) the second tranche of 100,000 restricted stock units will vest upon the initiation (which we define as the dosing of a first patient) of a Phase 1b clinical trial conducted in the United States. We may consider using restricted stock unit awards again in the future as long-term incentives for our executive officers, in combination with or without stock option awards.
Vesting of equity awards is generally tied to each officer’s continuous service with us and serves as an additional retention measure. We may grant equity awards at such times as our Board or Compensation Committee determines appropriate. Our executives generally are awarded an initial grant in the form of an option in connection with their commencement of employment with us. Additional grants may occur periodically in order to specifically incentivize executives with respect to achieving certain corporate goals or to reward executives for exceptional performance.
All options are granted with an exercise price per share that is no less than the fair market value of our common stock on the date of grant of such award. Our option awards generally vest over a four-year period and may be subject to acceleration of vesting and exercisability under certain termination and change in control events. See “— Outstanding Equity Awards at Fiscal Year-End for further information regarding option and stock awards granted during fiscal 2021.”
Outstanding Equity Awards at Fiscal Year End
The following table sets forth certain information regarding outstanding equity awards granted to our named executive officers that remain outstanding as of December 31, 2021.
| | | | | Option Awards | | | Stock Awards | |
| Name | | Grant Date | | Number of
Securities
Underlying
Unexercised
Options (#)
Exercisable | | | Number of
Securities
Underlying
Unexercised
Options (#)
Unexercisable | | | Equity
Incentive Plan
Awards:
Number of
Securities
Underlying
Unexercised
Unearned
Options (#) | | | Option
Exercise
Price
($) | | | Option
Expiration
Date | | | Equity Incentive
Plan Awards;
Number of
Unearned Shares,
Units or Other
Rights That
Have Not
Vested
(#) | | | Equity Incentive
Plan Awards;
Market or
Payout Value
of Unearned
Shares, Units
or Other
Rights that
Have Not
Vested
($) | |
Eric I. Richman
Chief Executive Officer | | 12/15/20 | | | 10,007 | | | | 16,417 | (3) | | | - | | | | 3.38 | | | | 12/15/2030 | | | | - | | | | - | |
Matthias Alder
Chief Operating Officer | | 12/23/21 | | | 0 | | | | 200,000 | (4) | | | - | | | | 5.86 | | | | 12/23/2031 | | | | - | | | | - | |
| | | 12/23/21 | | | - | | | | - | | | | - | | | | - | | | | - | | | | 200,000 | (5) | | | 1,086,000 | |
Salvatore Calabrese
Chief Financial Officer | | 9/25/20 | | | 34,870 | | | | 48,804 | (6) | | | - | | | | 3.38 | | | | 9/25/2030 | | | | - | | | | - | |
(1) | All of the awards in this table granted to Mr. Richman and Mr. Calabrese were granted under the Gain Therapeutics Inc. 2020 Omnibus Incentive Plan, or the 2020 Plan. In December 2021, the Board amended the 2020 Plan to reserve an additional 1,000,000 shares of our common stock to be used for grants to individuals who were not previously employees of the Company, which we refer to as the “2021 Inducement Award Subplan.” The awards granted to Mr. Alder were granted under the 2021 Inducement Award Subplan to the 2020 Plan. |
| |
(2) | All of the option awards listed in the table with a grant date up to March 17, 2021 were granted with an exercise price per share that is no less than the fair market value of our common stock on the date of grant of such award, as determined in good faith by our Board. All of the option awards granted as of March 17, 2021 onwards are granted with an exercise price per share that is the closing price of our common stock on the date of grant. |
(3) | One-third of the shares underlying this award vested on December 15, 2021, and the remaining shares vested in 24 equal monthly installments thereafter, subject to officer’s continued service through each vesting date. |
| |
(4) | Twenty-five percent of the common stock subject to this award shall vest on October 15, 2022, and the remaining shares vested in 36 equal monthly installments thereafter, subject to the officer’s continued service through each vesting date. |
| |
(5) | The performance-based restricted stock unit award granted to Mr. Alder in December 2021 will vest upon the achievement of the following milestones: (i) the first tranche of 100,000 restricted stock units will vest upon (a) the closing of an exclusive license deal for one or more Company pipeline programs and/or research, development and commercialization of a collaboration deal with a “Validating Industry Partner” (as such term is defined in his employment agreement), and (b) a resulting extension of our cash runway by one year or more with one or more transactions; and (ii) the second tranche of 100,000 restricted stock units will vest upon the initiation (which we define as the dosing of a first patient) of a Phase 1b clinical trial conducted in the United States. The grant date fair value for this award was determined to be $0 under ASC 718 based upon a determination that as of the grant date, it was not probable that the performance conditions will be achieved. The maximum potential fair value as of December 31, 2021 for the performance-based restricted stock unit award, based on achieving the maximum level of performance under the award as of the grant date, was calculated to be $1,086,000, using the closing price of our common stock on December 31, 2021 ($5.43). |
| |
(6) | One-third of the shares underlying this award vested on September 25, 2021, and the remaining shares vested in 24 equal monthly installments thereafter, subject to officer’s continued service through each vesting date.
|
Health and Welfare and Retirement Benefits
Our named executive officers are eligible to participate in benefits available generally to salaried employees, such as participation in our 401(k) plan in the United States, paid time off, and holiday, in each case on the same basis as our other employees.
The Compensation Committee periodically reviews the levels of benefits provided to executive officers to ensure that they remain reasonable and consistent with its compensation philosophy. Our Board may elect to adopt qualified or nonqualified benefit plans in the future, if it determines that doing so is in our best interests.
Perquisites
We generally do not provide significant perquisites or personal benefits to our named executive officers.
Employment Agreements and Severance Benefits
We provide certain of our named executive officers with certain severance protections in their employment agreements in order to attract and retain an appropriate caliber of talent for such positions. Our employment agreements with the named executive officers and the severance provisions set forth therein are summarized below under “—Employment Arrangements with our Named Executive Officers” and “—Potential Payments and Benefits upon Termination or Change in Control.” Our compensation committee intends to periodically review the level of the benefits in these agreements
Employment Arrangements with our Named Executive Officers
We have entered into employment agreements with each of our named executive officers. The agreements set forth the terms and conditions of each executive’s employment with us, including base salary, bonus opportunity, eligibility for employee benefits and certain non-solicitation and non-competition provisions. Our agreements with Mr. Alder and Mr. Calabrese set forth certain severance benefits upon a qualifying termination of employment. Any potential payments and benefits due upon a qualifying termination of employment or a change in control are further described below under “— Potential Payments and Benefits upon Termination or Change in Control.”
The employment of each of our named executive officers may be terminated at any time in accordance with the terms of the respective agreements. In addition, each of our named executive officers has executed a form of our standard proprietary information and inventions agreement. The material terms of each agreement are described below.
Eric I. Richman
We entered into an employment agreement with Mr. Richman in July 2020 in connection with his appointment as our President and Chief Executive Officer for a two-year term. He will devote approximately 80% of his time to the affairs of the Company. Pursuant to his employment agreement, Mr. Richman was initially entitled to an annual base salary of $300,000 and a signing bonus of $30,000, which was paid in July 2020. He is also eligible to receive an annual performance incentive bonus subject to review and adjustment each year by the Board, and for additional equity awards under our equity compensation plans, as may be granted from time to time. In April 2021, his annual base salary was increased to $400,000. As of January 1, 2022, Mr. Richman’s base salary was increased to $420,000 (pro-rated from an annual base salary of $525,000 for his 80% work schedule), with an annual target bonus set at 50% of his annual base salary, subject to review and adjustment each year by the Board.
Matthias Alder
We entered into an employment agreement with Mr. Alder in October 2021 in connection with his employment as our Chief Operating Officer. Under his employment agreement, Mr. Alder was entitled to an initial annual base salary of $400,000 as may be adjusted from time to time by the Compensation Committee of the Board. As of January 1, 2022, Mr. Alder’s base salary was increased to $420,000. He is also eligible to receive an annual performance incentive bonus with a target amount in 2022 set at 40% of his annual base salary and certain severance benefits, as described below under “—Potential Payments and Benefits upon Termination or Change of Control.”
Under the employment agreement, in December 2021, we granted Mr. Alder an option to acquire 200,000 shares of common stock pursuant to the 2020 Plan, with an exercise price of $5.86, as described in “—Outstanding Equity Awards as of December 31, 2021” above. We also granted Mr. Alder a performance-based restricted stock unit award to acquire up to 200,000 shares of common stock which will vest in two tranches upon the achievement of the following milestones: (i) the first tranche of 100,000 restricted stock units will vest upon (a) the closing of an exclusive license deal for one or more Company pipeline programs and/or research, development and commercialization of a collaboration deal with a “Validating Industry Partner” (as such term is defined in his employment agreement), and (b) a resulting extension of our cash runway by one year or more with one or more transactions; and (ii) the second tranche of 100,000 restricted stock units will vest upon the initiation (which we define as the dosing of a first patient) of a Phase 1b clinical trial conducted in the United States. Mr. Alder is also eligible for additional equity awards under our equity compensation plans, as may be granted from time to time.
Salvatore Calabrese
We entered into an employment agreement with Mr. Calabrese in November 2020 in connection with his employment as our Chief Financial Officer. Under his employment agreement, which is with our subsidiary GT Gain Therapeutics SA, Mr. Calabrese was entitled to an annual base salary of €226,600 ($268,000, converted into U.S. dollars using the 2021 average foreign exchange rate of 1.1827) and a signing bonus of €18,500. Mr. Calabrese is also eligible to receive an annual bonus with a target amount in 2022 equal to 35% of his annual base salary and certain severance benefits, as described below under “—Potential Payments and Benefits upon Termination or Change of Control.” As of January 1, 2022, Mr. Calabrese’s base salary was increased by 5% to €237,930. Assuming the 2021 average exchange rate of $1.1827, Mr. Calabrese’s 2022 base salary would be equivalent to $281,400.
Mr. Calabrese is also eligible for additional equity awards under our equity compensation plans, as may be granted from time to time. Under his employment agreement, we also agreed to grant Mr. Calabrese a certain amount of equity representing 1% of our share capital, with anti-dilution protection in the event our share capital increases such Mr. Calabrese shall retain at least 1% of our fully-diluted ownership. On September 25, 2020, we granted Mr. Calabrese an initial option to acquire 83,674 shares of common stock pursuant to the 2020 Plan, with an exercise price of $3.37, as described in “—Outstanding Equity Awards as of December 31, 2021” above.
Potential Payments Upon Termination or Change in Control
Our named executive officers, other than Mr. Richman, are entitled to any severance benefits upon a termination of employment.
Matthias Alder
Mr. Alder’s employment is at-will. In the event of termination of Mr. Alder’s employment by us without “Cause”, by Mr. Alder with “Good Reason” or Mr. Alder’s employment terminates upon death or his “Disability” (as such terms are defined in Mr. Alder’s employment agreement), he would be entitled to receive cash severance equal to twelve (12) months of base salary, or if within three (3) months prior to twenty-four (24) months following a “Change of Control” of the company (as such term is defined in Mr. Alder’s employment agreement), Mr. Alder’s severance period would be increased to twenty four (24) months from the date of termination. We also agreed to pay Mr. Alder an annual cash bonus equal to his pro-rated annual target bonus opportunity for the year in which the termination of employment occurs, or upon a “Change of Control,” Mr. Alder would be eligible to receive (i) an annual cash bonus equal to his pro-rated annual target bonus opportunity for the year in which the termination of employment occurs, and (ii) an annual cash bonus equal to his annual target bonus opportunity that would accrue during the severance (i.e. the year following in which the termination of employment occurs). In addition, if Mr. Alder timely elects to receive continued coverage under our group health care plan pursuant to COBRA, then he will be entitled to receive payment of the employee portion of his COBRA premiums until the earlier of (a) twelve (12) or twenty-four (24) months from his termination date or (b) the date he obtains or becomes eligible for health care coverage from a new employer or otherwise.
Salvatore Calabrese
Under the terms of our employment agreement with Mr. Calabrese, Mr. Calabrese’s employment is terminable by us or Mr. Calabrese upon six months’ notice. If Mr. Calabrese’s employment is terminated within 24 months following a “change in control” either by us without “cause” or by Mr. Calabrese for resigns within 24 months following a change in control” for “good reason” (each term as defined in his employment agreement), Mr. Calabrese is entitled to severance pay equal to the sum of (i) two times his base salary, plus (ii) two times the greater of (x) the average of the bonuses received in the three years prior to his separation date or (y) the target bonus for the year of Mr. Calabrese’s termination, plus (iii) a pro-rated target bonus for the year of Mr. Calabrese’s termination. In addition, all unvested stock options will fully vest and be exercisable for 90 days following Mr. Calabrese’s termination. Mr. Calabrese would also be entitled to reimbursement of any legal expenses incurred in the course of enforcing such payments. Severance is in lieu of any notice pay, and is contingent on Mr. Calabrese’s execution of a waiver and release. Mr. Calabrese’s employment agreement is governed by Swiss law.
Director Compensation
The following table sets forth information regarding compensation earned by or paid to our non-employee directors for the fiscal year ended December 31, 2021. Mr. Richman, our President and Chief Executive Officer, is also a member of our board of directors but does not receive any additional compensation for his service as a director.
| Name | | Fees
Earned or Paid in Cash ($) | | | Option Awards ($)(1) | | | Total ($) | |
| Khalid Islam, Ph.D. | | | 60,000 | | | | 93,500 | | | | 153,500 | |
| Dov Goldstein, M.D. | | | 47,000 | | | | 70,647 | | | | 117,647 | |
| Hans Peter Hasler | | | 41,000 | | | | 70,647 | | | | 111,647 | |
| Gwen Melincoff | | | 35,000 | | | | 70,647 | | | | 105,647 | |
| Claude Nicaise, M.D. | | | 40,000 | | | | 70,647 | | | | 110,647 | |
| Jeffrey Riley | | | 43,000 | | | | 70,647 | | | | 113,647 | |
| (1) | In accordance with SEC rules, this column reflects the aggregate grant date fair value of the option awards granted during the periods presented computed in accordance with ASC 718 for stock-based compensation transactions. Assumptions used in the calculation of these amounts are included in Note 15 to our audited consolidated financial statements included in our Annual Report on Form 10-K. These amounts do not reflect the actual economic value that will be realized by the director upon the vesting of the options, the exercise of the options or the sale of the shares of common stock underlying such options. |
| (2) | The following table provides information regarding the number of shares of common stock underlying options granted to our non-employee directors that were outstanding as of December 31, 2021: |
Name | | Option Awards
Outstanding at
Year-End (#)
| |
Khalid Islam, Ph.D. | | | 32,770 | |
Dov Goldstein, M.D. | | | 19,558 | |
Hans Peter Hasler | | | 19,558 | |
Gwen Melincoff | | | 19,558 | |
Claude Nicaise, M.D. | | | 19,558 | |
Jeffrey Riley | | | 45,982 | |
| | | | |
Non-Employee Director Compensation Policy
We do not have a formal non-employee director compensation policy. Our Board intends to adopt a formal policy in the future, based upon market research and advice from our compensation consultant. We have reimbursed and will continue to reimburse all of our non-employee directors for their travel, lodging and other reasonable expenses incurred in connection with their attendance at Board and committee meetings.
Equity Compensation
On January 22, 2021, we granted an option award to purchase 8,808 shares of common stock with an exercise price of $3.37 to each of our non-executive directors, other than Dr. Islam. On the same date, we granted Dr. Islam an option award to purchase 22,020 shares of common stock with an exercise price of $3.37 in recognition of his service as our chairman. On May 10, 2021, we granted an option award to purchase 10,750 shares of common stock with an exercise price of $10.03 to each of our non-executive directors in recognition of service as a director.
These awards have one year cliff vesting, subject to the director's continued service with us through the applicable vesting date. Notwithstanding any vesting schedule, for each non-employee director who remains in continuous service with us until immediately prior to the closing of a change in control, the shares subject to his or her then-outstanding equity awards that were granted pursuant to directors as director compensation will become fully vested immediatelyprior to the closing of such change in control.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
Security Ownership of Certain Beneficial Owners and Management
The following table sets forth certain information regarding the ownership of our common stock as of March 31, 2022 by:
| ● | each of our named executive officers; |
| ● | all of our executive officers and directors as a group; and |
| ● | each person or entity known by us to be beneficial owners of more than five percent of our common stock. |
We have determined beneficial ownership in accordance with the rules and regulations of the SEC, and the information is not necessarily indicative of beneficial ownership for any other purpose. Except as indicated by the footnotes below, we believe, based on information furnished to us, that the persons and entities named in the table below have sole voting and sole investment power with respect to all shares that they beneficially own, subject to applicable community property laws.
Applicable percentage ownership is based on 11,883,368 shares of common stock outstanding as of March 31, 2022. In computing the number of shares beneficially owned by a person and the percentage ownership of such person, we deemed to be outstanding all shares subject to options held by the person that are currently exercisable, or exercisable or would vest based on service-based vesting conditions within 60 days of March 31, 2022. However, except as described above, we did not deem such shares outstanding for the purpose of computing the percentage ownership of any other person.
Unless otherwise indicated, the address for each beneficial owner listed in the table below is c/o Gain Therapeutics, Inc., 4800 Montgomery Lane, Suite 220, Bethesda, Maryland 20814.
| | | Beneficial Ownership (1) | |
| Name of Beneficial Owner | | Number of Shares | | | Percent of Total | |
| 5% of Greater Stockholders: | | | | | | | | |
| Shawn Milmore Titcomb(2) | | | 729,737 | | | | 6.1 | % |
| TiVenture S.A.(3) | | | 854,572 | | | | 7.2 | |
| 1MM & 1PP AG(4) | | | 880,784 | | | | 7.4 | |
| Named Executive Officers: | | | | | | | | |
| Eric I. Richman(5) | | | 230,247 | | | | 1.9 | |
| Matthias Alder | | | - | | | | * | |
| Sal Calabrese | | | 46,490 | | | | * | |
| Directors: | | | | | | | | |
| Khalid Islam(6) | | | 913,554 | | | | 7.7 | |
| Dov Goldstein | | | 19,558 | | | | * | |
| Hans Peter Hasler | | | 19,558 | | | | * | |
| Gwen Melincoff | | | 19,558 | | | | * | |
| Claude Nicaise | | | 19,558 | | | | * | |
| Jeffrey Riley | | | 34,238 | | | | * | |
| All executive officers and directors as a group (9 persons)(7) | | | 1,302,761 | | | | 11 | % |
*Less than one percent.
| (1) | This table is based upon information supplied by officers, directors and principal shareholders and Schedules 13D and 13G filed with the SEC. Unless otherwise indicated in the footnotes to this table and subject to community property laws where applicable, we believe that each of the stockholders named in this table has sole voting and investment power with respect to the shares indicated as beneficially owned. Applicable percentages are based on 11,883,368 shares outstanding on March 31, 2022, adjusted as required by rules promulgated by the SEC. |
| (2) | Consists of 729,737 shares of common stock held by Shawn Milemore Titcomb, of which 14,829 shares are wholly owned by the Shawn Milemore Titcomb Revocable Trust, over which Mr. Titcomb is the trustee. The address of Shawn Milemore Titcomb is c/o Allele Capital Ptners LLC, 900 N Federal Highway, Suite 400, Boca Raton, FL 33432. |
| (3) | The directors of TiVenture S.A. are Renato Boldini, Sergio Magistri, Fabio Selmoni. Each of these individuals may be deemed to share voting and dispositive power with regard to the common stock directly held by TiVenture S.A. The address for each of these individuals and TiVenture S.A. is c/o Via Pietro Peri 9D, 69100 Lugano, Switzerland. |
| (4) | Consists of shares of our common stock held by 1 MM & 1 PP AG. Dr. Islam, a member of our Board of Directors, is the ultimate shareholder of 1 MM & 1 PP AG. Dr. Islam has the power to vote, acquire, hold and dispose of the shares held by 1 MM & 1 PP AG. The address for 1 MM & 1 PP AG and Khalid Islam is c/o Financial Consulting & Accounting Group GmbH, Flurstrasse 1. Hagendorn, Switzerland 6332. |
| (5) | Consists of (a) 201,991 shares of common stock held by Mr. Richman, (b) 13,427 shares of common stock issuable upon the exercise of options granted to Mr. Richman that are exercisable within 60 days of March 31, 2022, and (c) 14,829 shares of common stock held by the Eric I Richman Living Trust. Mr. Richman is the trustee for the Eric I Richman Living Trust and has sole voting and investment power over the shares held by the Eric I Richman Living Trust. |
| (6) | Consists of (a) 880,784 shares of common stock held by 1 MM & 1 PP AG, of with Dr. Islam is the ultimate shareholder, and (b) 32,770 shares of common stock held by Dr. Islam in his individual name. |
| (7) | Consists of (a) 1,097,604 shares of common stock held by our directors and executive officers, and (b) 205,157 shares of common stock issuable upon the exercise of options granted to our directors and executive officers that are exercisable within 60 days of March 31, 2022. |
Delinquent Section 16(a) Reports
Section 16(a) of the Exchange Act requires our directors and executive officers, and persons who own more than ten percent of a registered class of our equity securities, to file with the SEC initial reports of ownership and reports of changes in ownership of common stock and other equity securities of the Company. Officers, directors and greater than ten percent stockholders are required by SEC regulation to furnish us with copies of all Section 16(a) forms they file.
To our knowledge, based solely on a review of the copies of such reports filed on the SEC’s EDGAR system and written representations that no other reports were required, during the fiscal year ended December 31, 2021, all Section 16(a) filing requirements applicable to our officers, directors, and greater than ten percent beneficial owners were complied with, except that: during fiscal 2021, Mr. Richman timely reported all transactions but reported his ownership incorrectly on his initial report of ownership and a transaction by Mr. Richman was incorrectly reported on a Form 4. These errors were corrected by amending a previously filed Form 3 report and a previously filed Form 4 report.
Securities Authorized for Issuance Under Equity Compensation Plans
The following table provides certain information with respect to our equity compensation plans in effect as of December 31, 2021.
| Plan Category | | Number of securities
to be issued upon
exercise of outstanding options, warrants and rights
(a) | | | Weighted-average
exercise price of outstanding
options, warrants
and rights
(b) | | | Number of securities remaining available for issuance under equity compensation plans (excluding securities reflected in column (a))
(c) | |
| Equity compensation plans approved by security holders | | | | | | | | | | | | |
| 2020 Omnibus Incentive Plan(1) | | | 760,216 | | | | 3.25 | | | | 547,920 | |
| Equity compensation plans not approved by security holders | | | | | | | | | | | | |
| 2021 Inducement Plan(2) | | | 400,000 | | | | 4.24 | | | | 600,000 | |
(1) On September 24, 2020, the Board of Directors adopted the 2020 Omnibus Incentive Plan, or the 2020 Plan. The maximum number of shares to be issued under the 2020 Plan is 1,310,000. The exercise price for a stock option awarded under the 2020 Plan shall not be less than the fair market value of our common stock on the date of the grant.
(2) On December 23, 2021, to facilitate inducement awards to new hires under Nasdaq Listing Rule 5635(c)(4), the Board approved 1,000,000 shares of the Company’s common stock to be used for grants to individuals who were not previously employees of the Company, which we refer to as the “2021 Inducement Plan.” The 2021 Inducement Plan was adopted without stockholder approval in reliance on the exception for “inducement awards” provided by Nasdaq Rule 5635(c)(4).
Item 13. Certain Relationships and Related Transactions, and Director Independence.
Policies and Procedures for Related Party Transactions
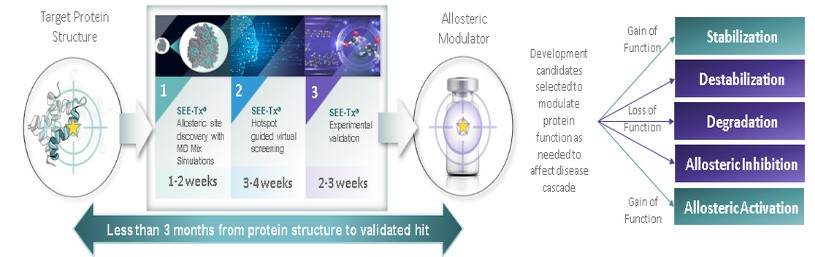
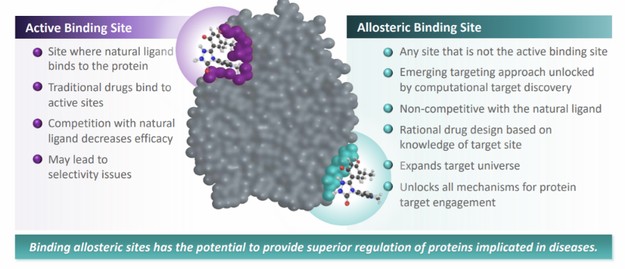
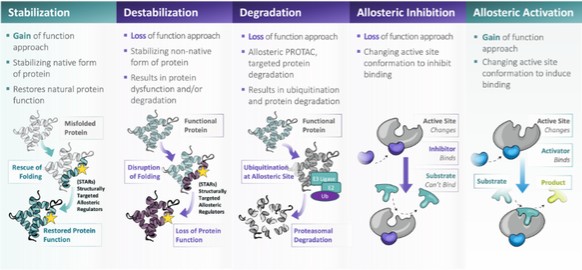
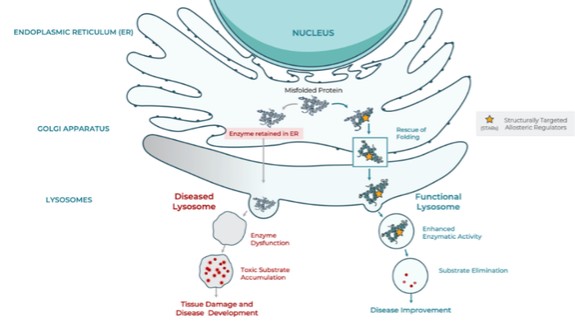
In connection with our initial public offering,business development activities, we enter into collaborative and licensing arrangement with third parties, to use our Board adoptedlicensed SEE-Tx® computational platform technology to discover novel allosteric sites on misfolded proteins and identify proprietary small molecules that bind these sites, potentially restoring protein folding and treating disease. We expect to continue to identify and evaluate collaboration, co-development and licensing opportunities that may be similar to or different from the collaborations and licenses that we have entered into.
Zentalis Pharmaceuticals, Inc.
On April 20, 2021, we entered into a written related-party transaction policy, setting forthmulti-target collaboration agreement, or the policies and proceduresZentalis Agreement, with Zentalis Pharmaceuticals, Inc., or Zentalis, to discover new product candidates for the reviewtreatment of cancer. Under the terms of the agreement, we used our licensed SEE-Tx computational platform technology to identify binding site on target proteins and approval or ratificationdetermine the potential suitability of transactions involving us and “related persons.” Forthese sites as drug targets, as well as the purposes of this policy, “related persons” will include our executive officers, directors, director nominees, and their immediate family members, and stockholders owning five percent or moreprospective therapeutic use of our outstanding common stock and their immediate family members. The policy covers with certain exceptions set forth in Item 404technology for treatment of Regulation S-K underoncology.
During the Securities Act, any transaction, arrangement, or relationship, or any seriescourse of similar transactions, arrangements, or relationships in which we were or are to be a participant, where the amount involved exceeds $120,000 and a related person has or will have a direct or indirect material interest, including, without limitation, purchases of goods or services by or from the related person or entities in which the related person has a material interest, indebtedness, guarantees of indebtedness, and employment by2022, Zentalis informed us of a related person. In reviewing and approving any such transactions, our audit committee is taskedits desire to consider all relevant facts and circumstances, including, but not limitedwind down the collaboration. Based on the results generated during the collaboration, we determined to whethercontinue the transaction is on terms comparable to those that could be obtained in an arm’s length transaction with an unrelated party and the extentresearch activities independently (without support of the related person’s interest in the transaction. All related-party transactions may only be consummated if our audit committee has approved or ratified such transaction in accordance with the guidelines set forth in the policy. Any member of the audit committee who is a related personZentalis) with respect to a transaction under review will not be permitted to participate in the deliberations or vote respecting approval or ratification of the transaction. However, such director may be counted in determining the presence of a quorum at a meeting of the audit committee that considers the transaction.
All of the transactions described in this section were entered into prior to the adoption of this policy. Although we did not have a written policy for the review and approval of transactions with related persons prior to the adoptionapplicable target as one of our related party transaction policy, our board of directors has historically reviewed and approved any transaction where a director or officer had a financial interest, including the transactions described above. Prior to approving such a transaction, the material facts as to a director’s or officer’s relationship or interest in the agreement or transaction were disclosed to our board of directors. Our board of directors took this information into account when evaluating the transaction and in determining whether such transaction was fair to us and in the best interest of all our stockholders.
Certain Related Person Transactions
All share and per share information presented in this section do not reflect our 0.88-for-1 reverse stock split effective March 17, 2021, and the automatic conversion of all of our outstanding shares of convertible preferred stock into shares of our common stock upon the closing of our initial public offering.
The following is a summary of transactions since January 1, 2020, to which we have been a participant in which:
| ● | the amount involved exceeded or will exceed the lesser of (i) $120,000 or (ii) 1% of the average of our total assets as of December 31, 2020 and 2021, and |
| ● | any of our directors, executive officers or holders of more than 5% of any class of our capital stock at the time of such transaction, or any members of their immediate family, had or will have a direct or indirect material interest. |
Corporate Reorganization with GT Gain Therapeutics SA
On July 20, 2020, we engaged in a series of transactions, which we refer to collectively as the “Corporate Reorganization”. Following the Corporate Reorganization, GT Gain Therapeutics SA became a wholly-owned subsidiary of the company. We, GT Gain Therapeutics SA, and the holders of all of the issued and outstanding equity interests of GT Gain Therapeutics SA entered into exchange agreements, dated as of July 20, 2020, pursuant to which the Corporate Reorganization was effected. Following the incorporation of the Company and prior to the Corporate Reorganization, we issued an aggregate of 1,772,736 shares of common stock at par value $0.0001.
In connection with the Corporate Reorganization, each holder of GT Gain Therapeutics SA’s equity interests contributed, transferred, granted, assigned and delivered to us all of its right, title and interest in and to all GT Gain Therapeutics SA equity interests owned by such holder which resulted in us issuing an aggregate of 2,250,000 shares of our common stock and 1,346,390 shares of our Series A preferred stock. The common stock and Series A Preferred Stock issued in connection with the Corporate Reorganization reflects a 10:1 forward-stock split.
The Corporate Reorganization was treated as a recapitalization by us for financial reporting purposes and the historical financial statements of GT Gain Therapeutics SA, adjusted to give effect to the share exchange transaction for all periods presented, are our historical financial statements. The Corporate Reorganization was treated as a “plan of reorganization” under Section 368(a) of the Code. The exchange agreements contained customary representations and warranties and pre- and post-closing covenants of each party and customary closing conditions. A condition to the Corporate Reorganization was the simultaneous closing of the Series B Private Placement, described below.
Private Placements of Securities
Series B Private Placement
own internal programs.
In July 2020, we sold an aggregate of 3,366,999 shares of our Series B convertible preferred stock at a purchase price of $2.97 per share for an aggregate amount of $10 million. In connection with the Series B private placement, we also entered into a preferred stock purchase agreement and warrant agreement with certain investors, including beneficial owners of greater than 5% of our capital stock, members of our Board of Directors and affiliates of members of our Board, pursuant to which we issued and sold to such investors an aggregate of 33,672 shares of our Series B convertible preferred stock at a purchase price of $2.97 per share for aggregate gross proceeds of $100,005.84. Such purchases were made on the same terms as the shares that were sold to other investors in the Series B private placement and not pursuant to any pre-existing contractual rights or obligations. The following table summarizes purchases of our Series B convertible preferred stock and placement agent warrants by related persons:
| Name | | Series B
Convertible Preferred Stock
(#) | | | Aggregate Purchase Price
($) | | | Placement
Agent Warrants
(#) | | | Strike Price
($) | |
| Eric I Richman Living Trust(1) | | | 16,836 | | | | 50,002.92 | | | | - | | | | - | |
| Shawn Milemore Titcomb(2) | | | 16,836 | | | | 50,002.92 | | | | 101,599 | | | | 4.46 | |
(1) Shawn Milemore Titcomb was a holder of more than 5% of our capital stock prior to this offering.
(2) Mr. Richman is our Chief Executive Officer and a member of our board of directors. Mr. Richman is the trustee for the Eric I Richman Living Trust and has sole voting and investment power over the shares held by the Eric I Richman Living Trust. As such, Mr. Richman may be deemed to be the beneficial owner of such shares.
February 2020 Capital Raise
In February 2020, prior to the Corporate Reorganization, GT GainMinoryx Therapeutics, SA sold 35,295 shares of Series A preferred stock, par value CHF 1.0 per share, to certain investors, including beneficial owners of greater than 5% of our capital stock, members of our Board of Directors and affiliates of members of our Board of Directors, pursuant to which we issued and sold to such investors an aggregate of 35,295 shares of our Series A Preferred Stock for aggregate gross proceeds of CHF 1,082,500 or $1,101,898. The following table summarizes purchases of our Series A convertible preferred stock by related persons:
| Name | | Series A Convertible Preferred Stock (#) | | | Aggregate
Purchase
Price (CHF) | | | Aggregate
Purchase
Price ($) | |
| 3B Future Health Fund S.A.(1). | | | 18,748 | | | | 575,000 | | | | 585,304 | |
| VitaTech SA(2) | | | 9,374 | | | | 287,500 | | | | 292,652 | |
| TiVenture SA(3) | | | 7,173 | | | | 220,000 | | | | 223,942 | |
(1) 3B Future Health Fund S.A. was a holder of more than 5% of our capital stock prior to this offering.
(2) VitaTech SA was a holder of more than 5% of our capital stock prior to this offering.
(3) TiVenture S.A. was a holder of more than 5% of our capital stock prior to this offering.
Investor Rights Agreement
In connection with the Corporate Reorganization and our convertible preferred stock financings, we entered into investor rights and stockholder agreements containing registration rights, information rights and rights of first refusal, among other things, with certain holders of our convertible preferred stock and certain holders of our common stock. these stockholder agreements terminated upon the closing of our initial public offering in March 2021, except for the registration rights granted under the investor rights agreement, dated as of July 20, 2020, which provides, among other things, that certain holders of our capital stock, including TiVenture SA, VitaTech SA, 3B Future Health Fund S.A. (f/k/a Helsinn Investment Fund) and Allele Capital Partners, LLC, have the right to demand that we file a registration statement or request that their shares of our capital stock be covered by a registration statement that we are otherwise filing. Lorenzo Leoni and Marianne Bjørdal, former members of our Board of Directors, are affiliated with TiVenture SA and VitaTech SA, and 3B Future Health Fund S.A. (f/k/a Helsinn Investment Fund), respectively. Eric Richman, our Chief Executive Officer and member of our Board of Directors, also has registration rights and is affiliated with Allele Capital Partners, LLC. Khalid Islam, a member of our Board of Directors, and Lorenzo Leoni, a former member of our Board of Directors, also have certain registration rights.
Agreements with Our Directors and Officers
Relationship with MinoryxS.L.
We have entered into a license agreement, dated December 20, 2017 or the Minoryx(the “Minoryx License Agreement,Agreement”), with Minoryx Therapeutics, S.L., a company organized under the laws of Spain or Minoryx,(“Minoryx”), pursuant to which we obtained exclusive worldwide license rights from Minoryx to use and exploit its intellectual property (“IP”), including its SEE-Tx® discovery platform for the identification of non-competitive pharmacological chaperones and exclusive worldwide sublicense rights to certain IP licensed by Minoryx from the University of Barcelona and the Institució Catalana de Recerca i Estudis Avançats. Mr.Under the terms of the Minoryx License Agreement, we have an exclusive, worldwide, royalty-bearing, assignable, transferable license, including the right to license through multiple tiers of sublicense, to Minoryx’s IP to make, have made, use, import, export, offer to sell, have sold, copy, modify, perform, display, create derivative versions of products in the licensed field or otherwise to exploit Minoryx’s IP in the field. Minoryx’s IP includes the SEE-Tx® discovery platform, certain proprietary Minoryx compounds acting as pharmacological chaperones, all patents and pending applications related thereto and Minoryx’s Know-How and Trademark related to the SEE-Tx® platform. We also have an exclusive, worldwide, royalty-bearing, assignable, transferable sublicense, including the right to sublicense through multiple tiers of sublicense, to the IP of Universitat de Barcelona (UB) and Institucio Catalana de Recerca i Estudis Avancats (ICREA) in EP11380102 and know-how and software related thereto, for the purpose of making, having made, using, importing, offering to sell, selling and having sold, copying, modifying, performing, displaying, and creating derivative versions of products in the field. Under the Minoryx License Agreement, products include any product in the field that would infringe the UB/ICREA IP or the Minoryx IP in the absence of the license provided therein. Also, the field encompasses any field of use and commercialization of the UB/ICREA IP or the Minoryx IP. Unless earlier terminated, the Minoryx License Agreement expires upon expiration of the royalty term, which occurs ten years after the first product covered by the licensed IP is commercialized. Khalid Islam, the Chairman of our board of directors and one of our founders, andis currently the Chairman of our Board of Directors, is the chairman of the board of directors of Minoryx, and as such, there may be times when there is a conflict of interest between us and Minoryx.
As consideration for the license grant under thefrom Minoryx, License Agreement, we have agreed to pay Minoryx royalties on a product-by-product basis based on the licensed intellectual propertyIP used by us, ranging from a high single digit to low single digit percentage of net revenues of products during the royalty term commencing on the effective date of the Minoryx License Agreement and continuing until the 10th10th anniversary of the first product commercialization. Upon the expiration of the royalty term for a product or service in a country, the license with respect to the product or service, as the case may be, shall become royalty-free, fully-paid, irrevocable and perpetual.
The Minoryx License Agreement will terminate upon expiration of the royalty term (which is the 10th anniversary of the commercialization of the first product covered by the licensed IP) or by mutual agreement. In addition, each party has the right to terminate the Minoryx License Agreement upon a material breach by the other party that remains uncured. Minoryx has the right to terminate the Minoryx License Agreement on a country-by-country basis if we abandon the technology or use the technology for purposes in violation of law and we fail to cure