(Mark One) (Mark One) | | | ☒ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | For the fiscal year ended December 31, 20172018 | or | | ☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | | For the transition period from to |
Commission File Number 001-33299001-36621
Foamix Pharmaceuticals Ltd. (Exact name of registrant as specified in its charter)
Israel (State or other jurisdiction of incorporation or organization) | | Not Applicable (I.R.S. Employer Identification Number) |
2 Holzman Street, Weizmann Science Park Rehovot 7670402, Israel (Address of principal executive offices, including zip code)
+972-8-9316233 (Registrant’sRegistrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of Each Class: | | Name of Each Exchange on Which Registered: | | Ordinary shares, par value NIS 0.16 per share | | NASDAQNasdaq Global Market |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Yes Yes☐☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act.
Yes ☐No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files).
Yes ☒ No ☐
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of Registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ☐ Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company,, or emerging growth company. See the definitions of “large“large accelerated filer,” “accelerated“accelerated filer,,” “ “smallersmaller reporting company,,” and “emerging“emerging growth company”” in Rule 12b-2 of the Exchange Act.
Large accelerated filer ☐ | Accelerated filer ☒ | Non-accelerated filer ☐ (Do not check if a smaller reporting company) | Smaller reporting company ☐☒ | | | Emerging growth company ☒ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐☒
Indicate by check mark whether the registrant is a shell company (as defined in Exchange Act Rule 12b-2).
Yes ☐No ☒
The aggregate market value of the registrant’sregistrant’s ordinary shares, par value NIS 0.16 per share, held by non-affiliates of the registrant on June 30, 2017,29, 2018, the last business day of the registrant’sregistrant’s most recently completed second fiscal quarter, was approximately $121$179.4 million (based on the closing sales price of the registrant’sregistrant’s ordinary shares on that date). Ordinary shares held by each director and executive officer of the registrant, as well as shares held by each holder of more than 10% of the ordinary shares known to the registrant, have been excluded in that such persons may be deemed to be affiliates. This determination of affiliate status is not necessarily a conclusive determination for other purposes.
The total number of shares outstanding of the registrant’sregistrant’s ordinary shares, par value NIS 0.16 per share, as of February 26, 2018,22, 2019, was 37,551,199.54,365,955.
DOCUMENTS INCORPORATED BY REFERENCE
None ii
EXPLANATORY NOTE
This Amendment No. 1 (this “Amendment”) on Form 10-K/A amends the Annual Report on Form 10-K of Foamix Pharmaceuticals Ltd. for the year ended December 31, 2017 as originally filed with the Securities and Exchange Commission on February 27, 2018 (the “Original Filing”), to correct (i) an inadvertent reversal between the numerical data in the two columns labeled “Bonus” and “Non-Equity Incentive Plan Compensation” in the table titled “Summary Compensation Table” in “Item 11—Executive Compensation”, (ii) a clerical error in the amount of “All Other Compensation” of Mr. Yohan Hazot in the same table, and (iii) an inadvertent omission of the reference to “emerging growth company” in the paragraph in the cover page preceding the list of types of filers.
Except as described above, this Amendment does not amend or otherwise update any other information in the Original Filing. For the convenience of the reader, this Amendment restates in its entirety our Original Filing. This Amendment is presented as of the filing date of the Original Filing and does not reflect events occurring subsequent to the date of the Original Filing.
We have also updated the signature page, the consent of our independent registered public accounting firm in Exhibit 23.1 and the certifications of our Principal Executive Officer and Principal Financial Officer in Exhibits 31 and 32.
FOAMIX PHARMACEUTICALS LTD. FORM 10-K TABLE OF CONTENTS
| | | Page No. | | | | 2 | | | 2 | | | 2023 | | | 4754 | | | 4754 | | | 4754 | | | 4754 | | | | 4755 | | | 4755 | | | 4955 | | | 5056 | | | 5966 | | | F-167 | | | 6168 | | | 6168 | | | 6169 | | | | 6270 | | | 6270 | | | 6876 | | | 7685 | | | 7988 | | | 7989 | | | | 8090 | | | 8090 | | | 8090 | | | 8291 |
iv
DEFINITIONS
Unless otherwise indicated, all references to the “company,“company,” “we,“we,” “us,“us,” “our”“our” and “Foamix”“Foamix” refer to Foamix Pharmaceuticals Ltd. and its subsidiary, Foamix Pharmaceuticals Inc., a Delaware corporation.
References to the “Companies Law”“Companies Law” are to Israel’sIsrael’s Companies Law, 5759-1999, as currently amended;amended;
References to the “Exchange Act”“Exchange Act” are to the Securities Exchange Act of 1934, as amended;amended;
References to the “FDA”“FDA” are to the United States Food and Drug Administration;
References to “NASDAQ”“Nasdaq” are to the NASDAQNasdaq Global Stock Market;Market;
References to “ordinary shares”“ordinary shares” are to our ordinary shares, par value of NIS 0.16 per share;
References to the “SEC”“SEC” are to the United States Securities and Exchange Commission;
References to the “Securities Act”“Securities Act” are to the Securities Act of 1933, as amended; and
References to “U.S. dollars”“U.S. dollars” and “$“$” are to currency of the United States of America, and references to “NIS”“NIS” are to New Israeli Shekels.
USE OF TRADEMARKS
“Foamix”Foamix”, the Foamix logo and other trademarks or service marks of Foamix appearing in this Annual Report on Form 10-K are the property of Foamix. This Form 10-KAnnual Report also contains trade names, trademarks and service marks of others, which are the property of their respective owners. We do not intend our use or display of other companies’companies’ trade names, trademarks or service marks to imply a relationship with, or endorsement or sponsorship of us by, these other companies.
FORWARD-LOOKING STATEMENTS
This annual reportAnnual Report contains express or implied “forward-looking statements”“forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995 and other U.S. Federal securities laws.
These forward-looking statements include, but are not limited to, statements regarding the following matters:
| -· | U.S. Food and Drug Administration, or FDA, approval of, or other regulatory action in the U.S.United States and elsewhere with respect to, our product candidates; |
| -· | the commercial launch of current or future product candidates; |
| -· | our ability to achieve favorable pricing for our product candidates; |
| -· | our expectations regarding the commercial supply of our product candidates; |
| -· | third-party payor reimbursement for our product candidates; |
| -· | our estimates regarding anticipated expenses, capital requirements and needs for additional financing; |
| -· | the patientpotential market size of treatments for any diseases and market adoption of our products by physicians and patients; |
| -· | the timing, cost or other aspects of the commercialization of our product candidates; |
| -· | the completion of, and receiving favorable results of, clinical trials for our product candidates; |
| -· | application for and issuance of patents to us by the United States Patent and Trademark Office, or U.S. PTO,USPTO, and other governmental patent agencies; |
| -· | the timing, costs or results of litigation to protect our intellectual property portfolio; |
| · | development and approval of the use of our product candidates for additional indications; and |
| -· | our expectations regarding licensing, business transactions and strategic operations. |
In some cases, forward-looking statements are identified by terminology such as “may,” “will,” “could,” “should,” “expects,” “plans,” “anticipates,” “believes,” “intends,” “estimates,” “predicts,” “potential,” or “continue” or the negative of these terms or other comparable terminology. Such forward-looking statements involve known and unknown risks, uncertainties and other factors that may cause actual results or performance to differ materially from those projected. These statements are only current predictions and are subject to known and unknown risks, uncertainties, and other factors that may cause our or our industry’s actual results, levels of activity, performance or achievements to be materially different from those anticipated by the forward-looking statements. In addition, historic results of scientific research and clinical and preclinical trials do not guarantee that the conclusions of future research or trials would not be different, and historic results referred to in this Annual Report on Form 10-K may be interpreted differently in light of additional research and clinical and preclinical trials results. The forward-looking statements contained in this annual report are subject to risks and uncertainties, including those discussed under “Item 1A—Risk“Risk Factors” and in our other filings with the Securities and Exchange Commission, or the SEC. Readers are cautioned not to place undue reliance on these forward-looking statements, which speak only as of the date hereof. Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee future results, levels of activity, performance, or achievements. Except as required by law, we are under no duty to (and expressly disclaim any such obligation to) update or revise any of the forward-looking statements, whether as a result of new information, future events or otherwise, after the date of this annual report.
STATEMENTS BY RESEARCH OR FORECAST FIRMS
We do not endorse or adopt any third-partythird party research or forecast firms’firms’ statements or reports referred to in this annual report and assume no responsibility for the contents or opinions represented in such statements or reports, nor for the updating of any information contained therein.
Overview
We are a late clinical-stage specialty pharmaceutical company focused on developing and commercializing our proprietary, innovative and differentiated topical drug candidates for dermatological therapy. Our lead product candidate, FMX101 (4% minocycline foamfoam), is being developed for the treatment of acne, rosacea and other skin conditions. Our lead product candidates, FMX101 for moderate-to-severe acne and our second product candidate, FMX103 (1.5% minocycline foam), is being developed for the treatment of moderate-to-severe papulopustular rosacea,rosacea. Both product candidates are novel topical foam formulations of the antibiotic minocycline. Based onminocycline and were developed using our Molecule Stabilizing Technology, a proprietary foam platform designed to optimize the topical delivery of minocycline, an active pharmaceutical ingredient, or API, that is currently available only in oral form despite its prevalent use in dermatology.
We announced positive top-line results demonstrated infrom both of our Phase II and Phase III clinical trials for each of FMX101 and FMX103 in the second half of 2018. We submitted our Phase II clinical trialnew drug application, or NDA, to the U.S. Food and Drug Administration, or FDA, for FMX101 in December 2018, and expect to submit an NDA for FMX103 in mid-2019. Despite the considerable U.S. market opportunities for acne and rosacea, we believe these product candidates have the potential to provide a fast, effective and well-tolerated treatment for their respective indications, whichmarkets are currently underserved and commonly treated by oral prescription products such as oral minocycline oraland doxycycline and various other non-foamnon-minocycline topical therapies. If approved, we believe FMX101 and FMX103 have the potential to provide a first-in-class, effective and well-tolerated topical treatment for the tens of millions of people who suffer from their respective indications.
Our corporate strategy is to develop and solidify a commercial presence in acne and rosacea by obtaining FDA approval for, and launching our lead product candidates, FMX101 and FMX103, in the United States. We may also enter into partnerships with third parties to reach other geographic territories or therapeutic fields through their respective sales forces and infrastructure. Following these near-term goals, we intend to grow beyond these indications into other dermatological indications, and to diversify our product and commercial development beyond minocycline and the tetracycline class of antibiotics. We are currently investingdeveloping additional foam and other topical products for acne, rosacea and other dermatology indications in vehicle platforms designed to enhance delivery of their respective APIs. We are also evaluating diversifying into synergistic technologies and specialties either on our own or through partnerships.
FMX101 is a product candidate containing micronized minocycline hydrochloride, an antibiotic in the majoritytetracycline class, in a 4% concentration for the treatment of moderate-to-severe acne vulgaris. The API is suspended in our Molecule Stabilizing Technology foam vehicle, an elegant, light-feeling topical foam that is easily spread across wide areas of the skin. In September 2018, we announced our third Phase III clinical trial of FMX101 (Study FX2017-22) met both of its co-primary endpoints, demonstrating a statistically significant reduction in the number of inflammatory lesions and a statistically significant improvement in patients’ Investigator’s Global Assessment, or IGA, scores, a metric commonly used to measure efficacy in acne trials. These positive results followed the results from our initial two Phase III clinical trials of FMX101 that we announced in 2017 wherein both co-primary endpoints were met in one trial (Study FX2014-05) but only one of the two co-primary endpoints showed statistical significance in the other trial (Study FX2014-04). We embarked on our third Phase III trial (Study FX2017-22) following a Type B meeting with the FDA in which the FDA confirmed that replicating the results of our effortsStudy FX2014-05 trial would likely support an efficacy claim for FMX101. In addition to the positive Study FX2017-22 efficacy results, very few safety adverse events (and no treatment-related serious adverse events) were observed both in Study FX2017-22 and resourcesin the 40-week open label safety portion of Studies FX2014-04 and FX20410-05 that we concluded in January 2018.
FMX103 is a product candidate also containing micronized minocycline hydrochloride suspended in our Molecule Stabilizing Technology vehicle, at a lower 1.5% concentration, for the treatment of moderate-to-severe papulopustular rosacea. In November 2018, we announced that both of our Phase III clinical trials for FMX103 (Studies FX2016-11 and FX2016-12) met each of their co-primary endpoints, demonstrating a statistically significant reduction in inflammatory lesion counts and IGA treatment success of approximately 50% from baseline. There were no treatment-related serious adverse events, very few reported adverse events and positive user-experience reports overall in these Phase III clinical trials as well as in the 40-week open label safety extension (Study FX2016-13) that was recently completed in February 2019.
We developed FMX101 and FMX103 using our proprietary Molecule Stabilizing Technology foam-based technology platform that was optimized for delivery of minocycline hydrochloride, a characteristically unstable small molecule, through the skin. We are currently developing in-house a pipeline of other innovative products to advanceenhance our minocycline platform, including FCD105, a product candidate for the treatment of acne vulgaris that combines minocycline with a retinoid and which we anticipate evaluating in a Phase II clinical trial (Study FX2016-40) beginning in mid-2019. We are also currently reviewing potential acquisitions of pipeline products at various stages of development that could be incorporated into our vehicle for optimized delivery.
In addition, we have other proprietary delivery technologies in development that enable topical delivery of other APIs, each having unique pharmacological features and characteristics designed to keep the API stable when delivered and directed to the target site. We are conducting research and are in the early stages of in-house development of FMX110, a topical gel formulation of doxycycline hyclate for the treatment of papulopustular rosacea, and FMX109, a non-tetracycline acne product candidate that contains a combination of nicotinamide and a retinoid for the treatment of moderate-to-severe acne vulgaris. We believe our foam and other topical delivery platforms may offer significant advantages over alternative delivery options and are suitable for multiple application sites across a wide range of conditions.
In addition to our in-house development projects, we have entered into development and license agreements relating to our technology with various pharmaceutical companies, most notably with LEO Pharma A/S, or LEO, who assumed a license agreement we initially entered into with Bayer HealthCare AG, or Bayer. In 2015, Bayer received FDA approval for Finacea® Foam (15% azelaic acid), or Finacea, a prescription foam product for the treatment of rosacea, which utilizes an emulsion-based proprietary foam platform that we licensed to them that is different from our surfactant-free foam platform that supports our lead product candidates. Bayer began selling Finacea in the United States in the third quarter of 2015 and in September 2018, LEO acquired Finacea from Bayer and assumed all rights and responsibilities under our initial license agreement with Bayer. Together with LEO, we are litigating against several generic pharmaceutical companies for alleged infringement of certain of our patents following the generic companies’ submission of abbreviated new drug applications, or ANDAs, to the FDA seeking approval to manufacture and sell generic versions of Finacea. We are committed to defending our intellectual property rights globally, including patents we have licensed to other pharmaceutical companies as part of our collaboration efforts.
We have also out-licensed other foam technology platforms to other third parties to develop branded pharmaceutical products containing different APIs for potential commercialization that are in the early stages of development.
Product Candidates and Pipeline
The following chart provides a summary of the developmental pipeline for our product candidates: Product Candidate | Preclinical | Phase I | Phase III | Milestones Achieved | Anticipated Milestones | | FMX101 (4%) for Moderate to Severe Acne Vulgaris |  | ·Positive top-line results (Study FX2017-22) ·Long-term safety study completed ·NDA submitted | ·PDUFA (Q4 2019) | FMX103 (1.5%) for Moderate to Severe Rosacea | | ·Positive top-line results (Studies FX2016-11 and FX2016-12) | ·Long term safety study completion ·NDA filing (mid 2019) | FCD105 for Moderate-to-Severe Acne Vulgaris | | ·IND meeting request submitted (Q4 2018) | ·Initiation of Phase II clinical trial (mid 2019) | FMX109 for Moderate to Severe Acne Vulgaris | | ·IND for clinical proof of principle submitted (1H 2019) | ·Initiation of Phase II clinical trial (1H 2020) | | FMX110 for Rosacea | | | ·Development candidate selection (mid 2019) |
FMX101 for moderate-to-severe acne
Our lead product candidate, FMX101 (4% minocycline foam), is a novel topical foam formulation of minocycline for the treatment of moderate-to-severe acne. In 2018, we completed our third pivotal Phase III clinical trial (Study 22)FX2017-22) for FMX101 in the U.S. We and announced the first patient enrolled in this trial on August 3, 2017. We expect to havethat we received positive top-line results from this trial in the third quarter of 2018. In March of 2017,January 2018, we announced the resultscompletion of the double-blind stagea long-term safety study that was an extension of our two initial Phase III clinical trials. Statisticaltrials for FMX101. The results from the long-term safety study showed FMX101 to be well-tolerated and to have an acceptable safety profile. On December 21, 2018, we announced the submission of an NDA for FMX101.
We initiated a third Phase III trial for FMX101 (Study FX2017-22) following our announcement in March 2017 of results from our two initial Phase III clinical trials for FMX101. In the previous Phase III clinical trials, statistical significance was demonstratedobserved in both co-primary efficacy endpoints in one study (Study 05)FX2014-05); however, in the other study (Study FX2014-04), however, statistical significance was demonstrated in only one of the co-primary efficacy endpoints in the second study (Study 04).endpoints. Statistical significance was also demonstrated for FMX101 compared to vehicle in the pooled analysis of the co-primary endpoints as well as key secondary endpoints. The third trialendpoints for Studies FX2014-04 and FX2014-05. Study FX2017-22 was initiated following a Type B meeting conducted with the FDA in June of 2017. During this meeting, we confirmed that achieving statistically significant results for FMX101 versus vehicle in both co-primary efficacy endpoints in a third independent clinical trial wouldcould be sufficient for establishing an efficacy claim. A previous Phase II clinical trial of FMX101 also demonstrated clinically and statistically significant resultsclaim in all primary and secondary endpoints. In January 2018, we announced the completion of a long-term safety study that was an extension of our two initial Phase III clinical trials for FMX101. The results from the study showed FMX101 to be well-tolerated and to have an acceptable safety profile.NDA submission. We are also investing significant efforts and resources to advance our two pivotal Phase III clinical trials in the U.S. for FMX103, minocycline foam for moderate-to-severe papulopustular rosacea, after our Phase II clinical trial for FMX103 demonstrated clinically and statistically significant results in all primary and secondary endpoints. We announced the enrollment of the first patient in our Phase III trials on June 12, 2017. We expect to have top-line results from the blinded stage of both trials by the end of the third quarter or in the beginning of the fourth quarter of 2018, and to complete the trials, including a long-term safety extension study, in 2019.
In addition, we successfully completed a Phase II clinical trial with FDX104, our proprietary doxycycline foam for the management of moderate-to-severe rash associated with epidermal growth factor receptor inhibitor (EGFRI) anticancer treatments, and we are currently assessing our various options with regard to this product candidate, including seeking out licensing opportunities for it. We have also successfully completed a Phase II clinical trial of FMX102, our minocycline foam for the treatment of impetigo, including impetigo caused by methicillin-resistant staphylococcus aureus, or MRSA. However, as described in previous reports, we have been contemplating the commercial viability of this product candidate for some time, given its limited market dominated by generic products, and following additional analysis of its potential we have recently decided to discontinue its further development in light of our current priorities and our other ongoing research and development efforts.
We developed FMX101, FMX102, FMX103 and FDX104 using our proprietary technology, which includes our foam-based platforms. This technology enables us to formulate and stabilize a wide variety of drugs and deliver them directly to their target site. We have independently developed a series of proprietary foam platforms, each having unique pharmacological features and characteristics. Our foam platforms may offer significant advantages over alternative delivery options and are suitable for multiple application sites. We believe our proprietary foam-based platform may serve as a foundation in developing a potential pipeline of products across a range of conditions.
Beside our in-house development projects, we have entered into development and license agreements relating to our technology with various pharmaceutical companies such as Bayer HealthCare AG, Mylan N.V. and Actavis Laboratories. Our total revenues from such agreements from our inception through December 31, 2017 were approximately $28.1 million.
In the third quarter of 2015, Bayer began selling Finacea® Foam (azelaic acid) 15%, or Finacea, for the treatment of rosacea in the U.S. Finacea is a prescription foam product which was developed as part of a research and development collaboration between Foamix and Bayer, utilizing one of Foamix’s proprietary foam technology platforms. According to our license agreement with Bayer, we are entitled to royalties and certain contingent payments upon the commercialization of Finacea based on Bayer’s net sales of Finacea. In 2017 we were entitled to receive royalty payments from Bayer in a total amount of $3.5 million for sales of Finacea. In January 2018, we filed a Complaint along with Bayer AG and Bayer HealthCare Pharmaceuticals Inc., alleging patent infringement under the patent laws of the United States arising out of the submission by defendant Teva Pharmaceuticals USA, Inc. of an Abbreviated New Drug Application, or ANDA, to the FDA, seeking approval to manufacture and sell a generic version of Bayer’s Finacea. In February 2018, Bayer and Foamix filed a Complaint alleging patent infringement under the patent laws of the United States arising out of the submission by defendant Perrigo UK FINCO Limited Partnership, or Perrigo, of an ANDA to the FDA, seeking approval to manufacture and sell a generic version of Bayer’s Finacea. See also “Item 1A—Risk Factors—Risks Related to Our Intellectual Property—We have received notice letters of ANDAs submitted for drug products that are generic versions of Finacea and we are involved in lawsuits to protect or enforce our patents, which could be expensive, time consuming and unsuccessful.”
Product Candidates and Pipeline
The following chart provides a summary of the developmental pipeline for our lead product candidates:
Product Candidate
| Preclinical
| Phase II
| Phase III
| Planned Milestones
| FMX101 (4%) for Moderate-Severe Acne | | Study 04 / 05 top-line results announced
Long-term safety study completed
3rd Phase III initiated August 2017
Top-line results – Q3 / Q4 2018
NDA filing – H2 2018
| FMX103 (1.5%) for Moderate-Severe Rosacea | | Phase II completed
Phase III initiated June 2017
Top-line results – end of Q3 or beginning of Q4 2018
NDA filing – 2019
|
FMX101 for moderate-to-severe acne
Our lead product candidate, FMX101, minocycline foam 4%, is a novel topical foam formulation of minocycline for the treatment of moderate-to-severe acne.
Market opportunity
Acne is characterized by areas of scaly red skin, non-inflammatory blackheads and whiteheads, inflammatory lesions, papules and pustules and occasionally boils and scarring. It affects approximately 40 to 50 million people in the U.S.United States alone, of whom approximately 10 million suffer from moderate-to-severe acne. For most people, acne diminishes over time and tends to disappear or decrease by age 25. However, some individuals continue to suffer from acne well into their 30s, 40s and later.
The current U.S. market size for treatment of acne is considerable and estimated at approximately $3 billion, presenting significant unmet needs of patients and healthcare providers to be addressed. We believe that our FMX101 product candidate for this indication, if approved, may provide a new treatment alternative for patients and healthcare providers who are unsatisfied with their current therapies.
Limitations of oral minocycline for acne
Oral minocycline, such as Solodyn, has been widely prescribed for the treatment of moderate-to-severe acne. According to the product label of Solodyn, inflammatory lesions were reduced by 44% at week 12, and a positive effect on the reduction of non-inflammatory acne lesions versus vehicle was not demonstrated. According to its product label, the most common adverse systemic side effects of Solodyn include diarrhea, dizziness, drowsiness, indigestion, lightheadedness, loss of appetite, nausea, sore mouth, throat or tongue and vomiting.
In 2009, the FDA added oral minocycline to its Adverse Event Reporting System, a list of medications under investigation by the FDA, due to its severe side effects. In 2011, we conducted a blind survey of 40 U.S. dermatologists. The results of the survey revealed that 90% of the dermatologists surveyed who prescribed oral minocycline were concerned about its side effects, and 76% of these dermatologists stated they would prefer prescribing a topical minocycline product over an existing oral medication, assuming the topical treatment was safe, effective and approved by the FDA.
FMX101 clinical trials
FMX101 third Phase IIIII clinical trial
We conductedIn June 2017, following the top-line data from Studies FX2014-04 and FX2014-05 as summarized below, we held a randomized, double-blind, dose-ranging, controlled Phase II clinical trial in Israel over 12 weeks with 150 patients between 12 and 25 years old with a mean age of approximately 16.5 years, each presenting with a minimum of 20 inflammatory and 25 non-inflammatory facial acne lesions. The patients were randomly divided into three groups of 50 patients each, with one group receiving a 1% concentration of our minocycline foam, a second group receiving a 4% concentration and a third control group receiving our foam vehicle without minocycline, which we refer to as the vehicle. Each patient received one application daily before bedtime.
The primary efficacy endpoints of the trial were:
| · | the reduction in inflammatory and non-inflammatory lesions (as well as the total counts of facial lesions) over the course of the 12-week treatment period; |
| · | improvement in the investigator’s global assessment, or IGA, based on the uniform graded scale adopted for the trial, ranging from “clear” skin with no inflammatory or non-inflammatory lesions to “severe acne”, as well as safety and tolerability. |
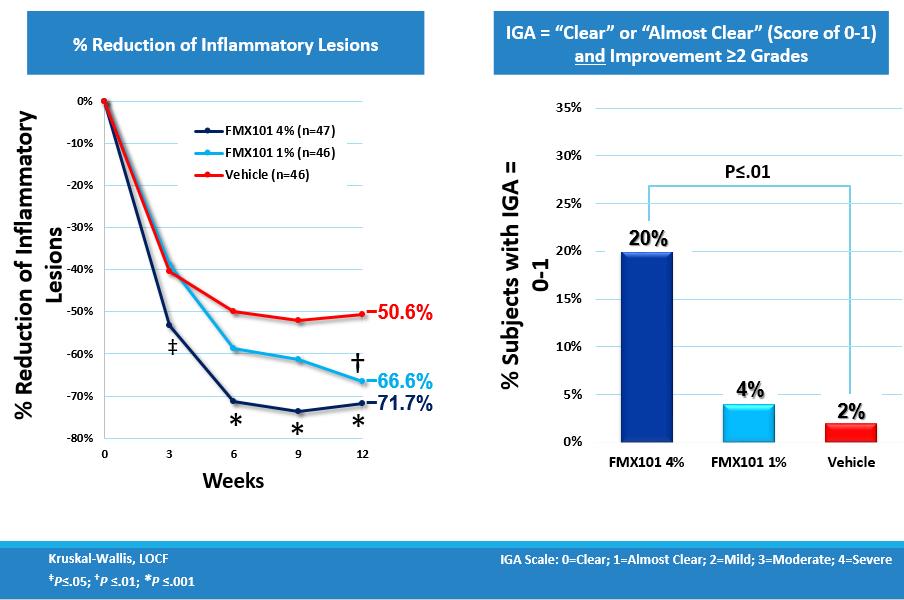
The trial was completed in 2013 and showed a dose-dependent effect that was statistically significant for both primary endpoints of the trial. Notably, the effect on inflammatory lesions became statistically significant in the 4% dosage group versus the vehicle-only treatment group after just three weeks of therapy, and there was an approximate 72% reduction in inflammatory lesions reached in the 4% dosage group after 12 weeks of treatment. Additionally, for the patients in the 4% dosage group, the effect on non-inflammatory lesions also became statistically significant versus the vehicle-only group after twelve weeks of therapy, with approximately a 73% reduction in non-inflammatory lesions.
The percent of patients with IGA improvement of at least two grades and a grading of clear or almost clear (score of 0 or1) at the completion of the trial was 20% in the 4% dosage group, compared with 4% in the 1% dosage group and 2% in the vehicle-only treatment.
The safety and tolerability profile of the drug was also favorable, with no serious adverse events and no reported drug-related systemic side effects. The cases of skin reaction in the trial were few, mild and transient, with all reactions subsiding by week 12 of treatment, and there was similar incidence of skin reaction in all three groups.
The following charts show the reduction of inflammatory lesion count from baseline and over the trial period for the 4% dosage, 1% dosage and vehicle treatment groups and the percentage of patients who met the IGA success criterion:
While we did not file a formal application for an INDType B Meeting with the FDA, during which we confirmed that statistically significant findings from a third study would constitute replication of the Study FX2014-05 results and would be sufficient for establishing an efficacy claim. This affirmed our plans for conducting a third Phase III trial for FMX101.
In August 2017, we announced the dosing of the first patient in connection with the FMX101our third Phase IIIII acne clinical trial, known as Study FX2017-22, a double-blind, vehicle-controlled, multi-center trial conducted at approximately 80 sites throughout the trial was conducted in compliance with the International Conference of Harmonization, or ICH, good clinical practice, or GCP, guidelines and applicable Israel Ministry of Health regulations. The trial protocol complied with the procedures, criteria and endpoints specified by the FDA’s 2005 draft industry guidance for acne trials. Because minocycline, the active ingredient in FMX101, is a well-known drug with an established safety profile, the ethical committee for our Phase II clinical trial and the Israeli Ministry of Health allowed us to conduct the Phase II clinical trial of FMX101 without having first conducted a Phase I clinical trial.United States. We completed a Phase I maximum use pharmacokinetics study of FMX101, intended to characterize the systemic absorption of minocycline after repeated maximum-dose applications in patients with moderate-to-severe acne, and to assess the relative bioavailability of topically-applied FMX101 compared to orally-administered Solodyn (minocycline HCl). The study enrolled 30 patients with moderate-to-severe acne on their face and on two or more other regions (neck, upper chest, upper back or arms) in a single-center, nonrandomized, open-label, active-controlled, two-period and two-treatment evaluation. Each of the patients received a single dose of Solodyn, 1 mg/kg in accordance with its approved instructions for use, and one week later received 4 grams of FMX101 applied topically once daily for 21 days. The study showed that the bioavailability, or systemic exposure, of minocycline following topical FMX101 administration was approximately 700 times lower than that of Solodyn, and that FMX101 was well-tolerated with no serious adverse events being reported.
In addition,February 2018, we completedheld a single-center, non-randomized, open-label study to evaluate multiple dose topical administration of FMX101 4% minocycline foam in 20 subjects with ages ranging from 9 years to 16 years 11 months years of age with moderate to severe acne vulgaris. The objectives were to characterize minocycline pharmacokinetics after administration of FMX101 4% once daily for 7 days under maximal use conditions and to evaluate the safety and tolerability. The levels of minocycline were relatively constant over the entire sampling interval on day 7. The overall average plasma concentration of minocycline, across all age ranges, was comparable with the equivalent adult study. Concentrations tended to be slightly higher in the subjects aged 9 to 11 years (approximately 3.5 ng/mL) and subjects aged 12 to 14 years (approximately 2.5 ng/mL) than the subjects aged 15 years to 16 years 11 months (approximately 1.7 ng/mL). Similar to the study in adult patients, FMX101 was well-tolerated with no serious adverse events being reported.
A series of Phase I human dermal safety studies, were completed in 2016. Results of these trials were as expected and no safety signals were detected.
As part of our overall development plan for FMX101, we have conducted a series of animal safety studies, which revealed no signs of toxicity. We completed a long-term, 39-week dermal toxicity study in mini pigs, which included concentrations of our minocycline foam up to 16%. Results from this study also reflect no toxicity associated with our drug product.
We also held an End of Phase IIType B pre-NDA meeting with the FDA to review the clinical development plan for FMX101, and implemented the comments we received from the FDA regarding overall study design, primary efficacy endpoints, and safety assessments in the FMX101 Phase III program.
FMX101 initial two Phase III clinical trials
Based on the resultsFDA. The purpose of the Phase II clinical trial, as described above, and guidance frommeeting was to discuss the FDA insubmission of a pre-IND505(b)(2) application for FMX101. During the meeting, we conducted two multi-center pivotal Phase III clinical trials indiscussed various chemistry, manufacturing and controls, or CMC, aspects of FMX101, sufficiency of nonclinical toxicology studies, format and other information required for the U.S. for FMX101 (minocycline foam 4%) in moderate-to severe acne, known as Studies 04 and 05.NDA submission.
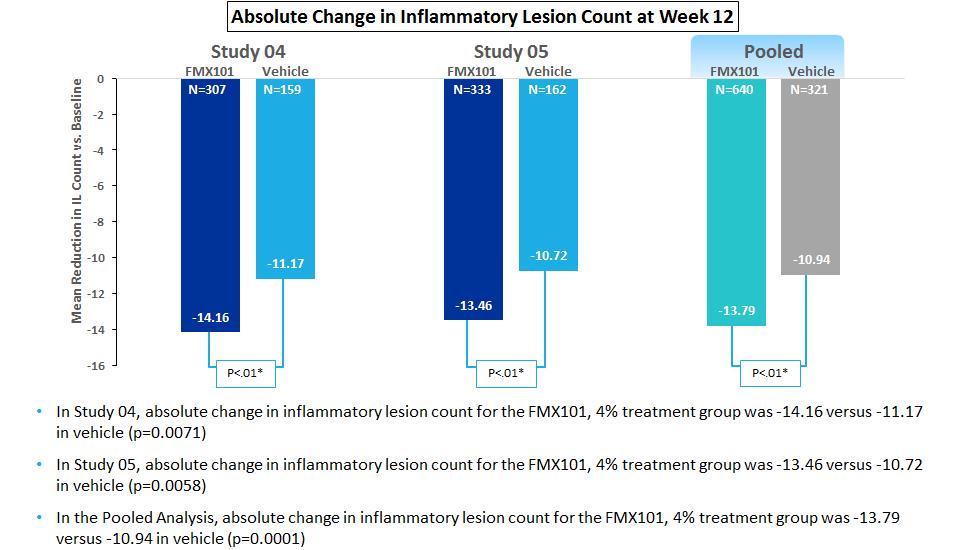
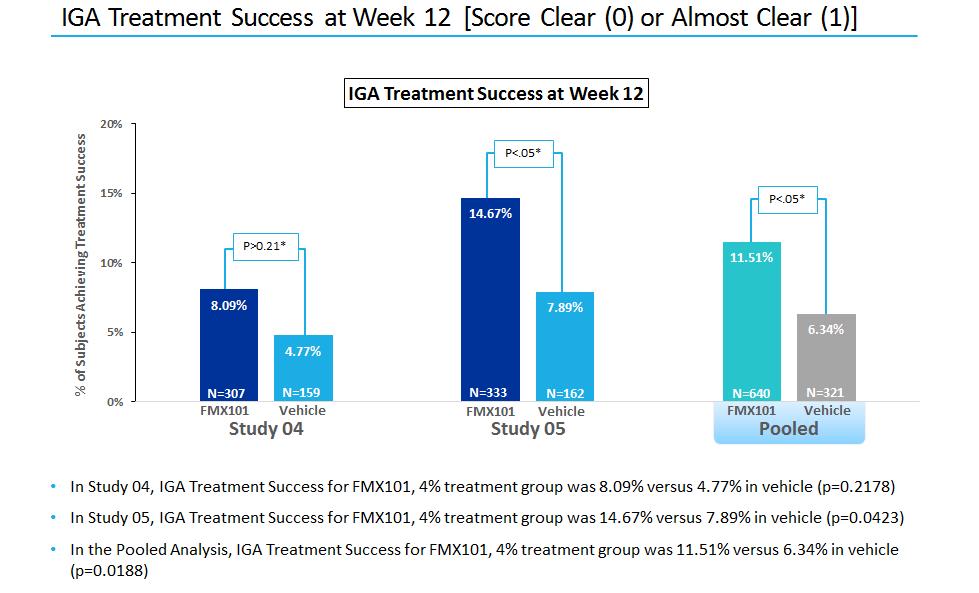
In November 2016May 2018, we completed patient enrollment resulting in an intent-to-treat population of 961a total of 1,507 patientswith moderate-to-severe acne, enrolled betweenwith 1,488 patients in the two trials. Patientsintent-to-treat population in Study FX2017-22. In January 2018, we prospectively excluded 19 patients from the intent-to-treat population due to data integrity issues identified at one clinical trial site following an audit, investigation and termination of the site. We notified the FDA of our audit findings and of the termination of the clinical trial site in February 2018, in advance of our Type B meeting with the FDA. We do not believe that the exclusion of the 19 patients from the intent-to-treat population for Study FX2017-22 is material to our clinical trial results, our clinical trials generally or to our company as a whole. In the Phase III clinical trial, patients were randomized on a 2:1:1 basis, (active compound versus vehicle-only), initially into a 12-week double-blind, multi-center phase where they were treated topically once daily with either FMX101 (minocycline foam 4%) or the respective foam vehicle. vehicle foam. The two co-primaryprimary efficacy endpoints of both trials were: (1) the absolute change from baseline in inflammatory lesion counts in each treatment group at week 12; and (2) the proportion of patients achieving success at week 12 based as defined by an IGA score of 0 “clear” or 1 “almost clear” and at least a two-grade improvement from baseline at week 12; and (2) the mean change from baseline in inflammatory lesion counts in each treatment group at week 12. Safety evaluation includesincluded reported adverse events, assessments of tolerability, clinical laboratory tests and vital signs. Patients who completed the 12-week double-blind portion of the trials had the option to continue in a long-term open-label safety extension, aimed at evaluating the safety of intermittent use of FMX101 for up to nine additional months. On March 27, 2017 we announced top-line data from our two Phase III clinical trials for FMX101. In the intent-to-treat analysis, FMX101 demonstrated statistical significance compared to vehicle on both co-primary endpoints in Study 05 (specifically the absolute reduction in inflammatory lesions at week 12, and investigator global assessment treatment success (IGA0/1) at week 12 compared to baseline). In Study 04, statistical significance was demonstrated for FMX101 compared to vehicle in the co-primary endpoint of absolute reduction in inflammatory lesions. However, statistical significance was not achieved in the co-primary endpoint of IGA0/1.
On May 3, 2017 we provided new data from our two Phase III clinical trials for FMX101, including a pooled analysis of our co-primary endpoints and certain secondary clinical endpoints (absolute reduction of non-inflammatory lesions at week 12; and percent change in inflammatory lesions at weeks 3, 6, 9 and 12). Statistical significance was demonstrated for FMX101 compared to vehicle in the pooled analysis of the co-primary endpoints as well as the secondary endpoints presented.
In August 2018, we received a letter from the FDA’s Division of Dermatology requesting information on the canister, foaming pump and other device constituent parts of our FMX101 product candidate. The letter referred to FMX101 as a combination product and requested information relating to the quality and design control of the device, including (1) the device description documentation, (2) the design control documentation, (3) the traceability documentation and (4) additional considerations related to the biocompatibility and sterility of the product candidate. We have provided the requested information to the FDA as part of our NDA submission using readily available data derived and generated from our Study FX2017-22.
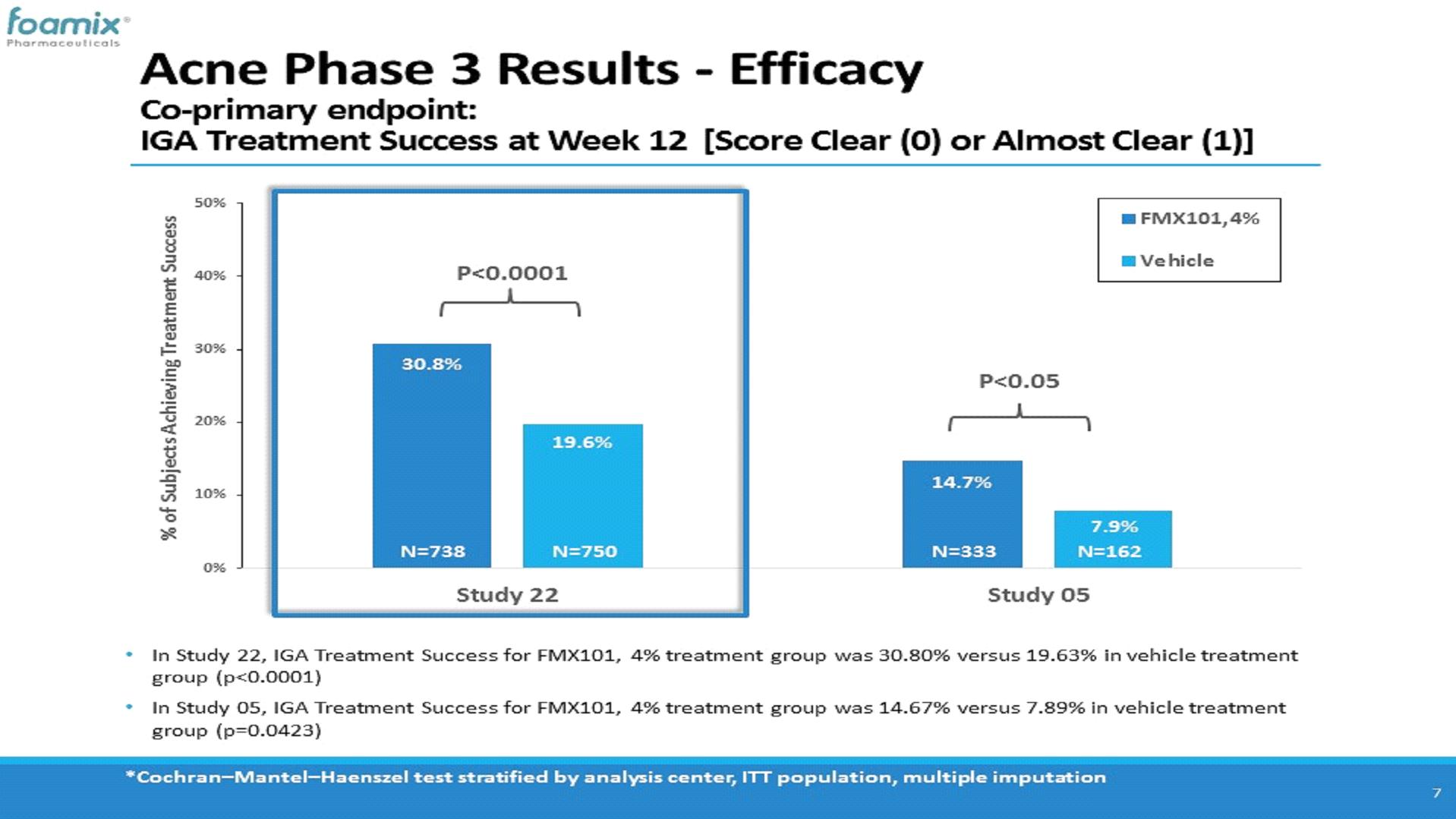
In September 2018, we announced positive top-line results of Study FX2017-22. The study met both co-primary endpoints of (a) absolute change from baseline in inflammatory lesion count at week 12, and (b) investigator global assessment treatment success (IGA 0/1) at week 12, and at least a 2-grade improvement (decrease) from baseline. The mean inflammatory lesion count at baseline was 30.7 and 30.8 for the FMX101 and vehicle treatment groups, respectively. The proportion of subjects with an IGA score of 3 (“moderate”) or 4 (“severe”) at baseline was 84.0% and 16.0%, respectively, in the FMX101 treatment group and 83.5% and 16.5%, respectively, in the vehicle treatment group. The co-primary efficacy assessment showed a statistically significant mean reduction in inflammatory lesion count at week 12 relative to baseline of -16.93 lesions for the FMX101 treatment group and -13.40 lesions for the vehicle treatment group. In addition, in respect of the second co-primary endpoint, the proportion of subjects that achieved IGA treatment success at week 12 was 30.80% for the FMX101 treatment group and 19.63% for the vehicle treatment group.
The following charts show the reduction of inflammatory lesion count from baseline for FMX101 4% and vehicle treatment groups in each of Studies 04 and 05 and on a pooled basisStudy FX2017-22 at week 12:12, and compared to Study FX2014-05, which is the comparison discussed in our Type B meeting with the FDA, in which the FDA confirmed that replicating the results of Study FX2014-05 on such endpoint would likely support an efficacy claim for FMX101:
The following charts show the percentage of patients who met the IGA treatment success criterion at week 12 (defined as at least a 2 grade point reduction from baseline IGA score and a final score of “clear”“clear” (0) or “almost clear”“almost clear” (1)) for FMX101 4% and vehicle treatment groups in each of Studies 04 and 05 and on a pooled basisStudy FX2017-22 at week 12:12, compared to Study FX2014-05, as explained above:
The safety and tolerability of FMX101 thirdwere also evaluated and the safety profile of FMX101 in Study FX2017-22 was found to be consistent with that observed in the two prior Phase III trials. The most commonly reported adverse events in Study FX2017-22 related to upper respiratory tract infections. There were no treatment-related serious adverse events. In these trials, FMX101 was observed to have a generally favorable safety profile and to be generally well tolerated. Based on the efficacy and safety profile observed in clinical studies to date, we believe FMX101, if approved, may present an attractive option for the treatment of moderate-to-severe acne.
In December 2018, we submitted an NDA to the FDA seeking approval for FMX101 for the treatment of inflammatory lesions of non-nodular moderate-to-severe acne vulgaris in patients 9 years of age and older. The submission followed the successful outcomes in our Phase III clinical trial On June 21, 2017, following and incorporated guidance received from the top-lineFDA in our Type B pre-NDA meeting held in February 2018. The NDA submission also incorporated information on chemistry manufacturing and controls, and data from Studies 04 and 05, we held a Type B Meeting withnon-clinical toxicology studies on FMX101. Our application was submitted under the FDA during’s 505(b)(2) regulatory pathway, which we confirmed that statistically significant findings from a third study would constitute replicationpermits the filing of an NDA where at least some of the Study 05 resultsinformation required for approval comes from studies that were not conducted by or for the applicant, and wouldfor which the applicant has not received a right of reference. For additional information see “Risk Factors-Risks Related to Our Business and Industry-If the FDA does not conclude that FMX101 or FMX103 satisfy the requirements under Section 505(b)(2) of the Federal Food Drug and Cosmetic Act, or Section 505(b)(2), or if the requirements for these product candidates under Section 505(b)(2) are not as we expect, the approval pathway for these product candidates will likely take significantly longer, cost significantly more and entail significantly greater complications and risks than anticipated, and in either case may not be sufficient for establishing an efficacy claim. This confirmation reaffirmed our plans for conducting a thirdsuccessful.”
FMX101 initial two Phase III trial for FMX101.clinical trials
On August 3, 2017 we announced the dosingWe initiated Study FX2017-22, as described above, after conducting Studies FX2014-04 and FX2014-05. These trials comprised a total of the first patient in our third Phase III acne clinical trial, known as Study 22, which is designed as a double-blind, vehicle-controlled, multi-center trial conducted at approximately 80 sites throughout the U.S. that will enroll a target number of 1,500961 patients with moderate-to-severe acne. Patients areacne, who were randomized 1:on a 2:1 tobasis (active compound versus vehicle-only), initially into a 12-week double-blind phase, in which they were treated topically once daily with either FMX101 4% dosage or vehicle, with once daily treatment for 12 weeks.the respective foam vehicle. The primarytwo co-primary efficacy endpoints areof both trials were identical to the primary endpoints in Studies 04Study FX2017-22, and 05.evaluated safety (including reported adverse events, assessments of tolerability, clinical laboratory tests and vital signs) and allowed patients who completed the 12-week double-blind portion of the trials to continue in a long-term open-label safety extension, aimed at evaluating the safety of intermittent use of FMX101 for up to nine additional months.
We selected Premier Research International LLC, or Premier Research,The results of these trials demonstrated statistical significance for FMX101 compared to vehicle on both co-primary endpoints in Study FX2014-05 and in the pooled analysis for both trials combined, and in Study FX2014-04 statistical significance was further demonstrated for FMX101 compared to vehicle in the co-primary endpoint of absolute reduction in inflammatory lesions. However, Study FX2014-04 did not demonstrate statistical significance on the co-primary endpoint of improvement in IGA score. Consequently, and as our designated clinical research organization (CRO) for the execution ofexplained above, we embarked on Study FX2017-22, our third multi-center pivotal Phase III trial in acne. Premier Research has significant experience in the execution of global clinical trial programs and is a recognized leader in clinical trial management within the field of dermatology. We expect to have top-line resultsfor FMX101, following confirmation from the trialFDA that replicating the results of Study FX2014-05, in the third quarter of 2018 and to complete the trial by the end of 2018.which both co-primary endpoints were successfully achieved, would likely support an efficacy claim for FMX101.
FMX101 open-label safety extension study
OnIn January 4, 2018, we announced positive safety data for our Phase III open-label safety extension study, evaluating FMX101 in moderate-to-severe acne for a treatment period of up to one year. The open-label safety extension study enrolled a total of 657 patients, all of whom had completed 12 weeks of FMX101 or vehicle treatment in the preceding double-blind phase of Studies 04 and 05.FX2014-04 or FX2014-05. Patients continued open-label treatment with FMX101 for up to an additional 40 weeks.weeks of open-label treatment with FMX101. 291 patients completed a total of 52 weeks on FMX101, therapy, which exceedsis in excess of the subject sample size requirements specified in the regulatory guidance for this type of safety evaluation (ICH E1A, 1995). No serious drug-related adverse events were reported in this comprehensive safety evaluation, which validated earlier data demonstrating that FMX101 appears to be well-tolerated and has an acceptable safety profile. More specifically,The key findings from the study found that:are as follows:
| · | non-dermalNon-dermal adverse events were comparable in type and frequency with those reported during the double-blinded portion of Studies 04FX2014-04 and 05, with theFX2014-05. The most frequently reported treatment-emergent adverse event beingwas nasopharyngitis or common cold. Three(common cold). In the open-label extension part of the study, three patients discontinued the study for non-dermal adverse events, two of them forwhom discontinued due to abdominal pain and one fordue to back pain;pain. No serious drug-related adverse events were reported; |
| · | application-siteApplication site adverse events occurred in less than 2% of patients during the additional 40 weeks of open-label treatment.treatment with FMX101. Four patients discontinued the study for an application-siteapplication site adverse event, –of which two patients fordiscontinued due to worsening of acne, one forpatient discontinued due to contact dermatitis and one patient discontinued for localized facial edema. In the assessment of facial dermal tolerability at week 52, more than 95% of patients had “none”“none” or “mild”“mild” signs and symptoms such as erythema, dryness, hyperpigmentation, peeling and itching, and no severe local tolerability scores were recorded; and |
| · | subjectPatient satisfaction with FMX101 treatment remained high when re-assessed at week 52, which was consistent with scores obtained at the end of the double-blind phase of Studies 04 and 05 at week 12. |
Efficacy was also measured as a secondary endpoint in the open-label study for FMX101, and was based on summary statistics from observed cases. During the study, patients were allowed to discontinue therapy with FMX101 if they believed their acne was under control. Patients could re-start therapy as needed and were also allowed to use other acne medications concomitantly. As a result, no claim of statistical difference was made between any of the treatment arms,arms; however, notable findings were observed:
| · | At week 52, 37.7% of patients from Study 04FX2014-04 had an Investigator’s Global Assessment (IGA)IGA score of 0 (clear) or 1 (almost clear) and 50.3% of subjects from Study 05FX2014-05 had an IGA score of 0 or 1. |
| · | At week 52, patients from Study 04FX2014-04 had a 64.3% reduction in inflammatory lesions and patients from Study 05FX2014-05 had a 78% reduction in inflammatory lesions. |
| · | At week 52, patients from Study 04FX2014-04 had a 52.5% reduction in non-inflammatory lesions and patients from Study 05FX2014-05 had a 59.6% reduction in non-inflammatory lesions. |
Next steps
We expect to develop FMX101 through the FDA’s 505(b)(2) regulatory pathway, which permits the filing of a new drug application where at least some of the information required for approval comes from studies that were not conducted by or for the applicant, and for which the applicant has not received a right of reference. This approach could expedite the development program for FMX101 by potentially decreasing the amount of clinical data that we would need to generate in order to obtain FDA approval. For additional information see “Item 1A—Risk Factors—Risks Related to Our Business and Industry—If the FDA does not conclude that FMX101 or FMX103 satisfy the requirements under Section 505(b)(2) of the Federal Food Drug and Cosmetics Act, or Section 505(b)(2), or if the requirements for this product candidate under Section 505(b)(2) are not as we expect, the approval pathway for this product candidate will likely take significantly longer, cost significantly more and entail significantly greater complications and risks than anticipated, and in either case may not be successful.”
FMX103 for moderate-to-severe papulopustular rosacea
Our product candidate FMX103 minocycline(minocycline foam 1.5%,) is a novel topical foam formulation of minocycline for the treatment of moderate-to-severe papulopustular rosacea.
Market opportunity
Papulopustular rosacea is a chronic skin disease causing inflammatory lesions (papules and pustules) on the face. It can create psychosocial burdens, such as embarrassment, anxiety and low self-esteem that adversely affect quality of life. Rosacea is most frequently seen in adults between 30 and 50 years of age and affects more than 16 million people in the U.S.United States alone. There is no known cure for rosacea and the exact root cause of the disease remains unknown as well, though both genetic and environmental factors are thought to have an impact on its outbreak. Mild papulopustular rosacea is currently treated by topical antimicrobials (such as metronidazole, clindamycin and ivermectin) or azelaic acid, while the mainstaymainstays for the treatment of moderate-to-severe rosacea are systemic antibiotics such as minocycline and doxycycline.
The current U.S. market size for treatment of rosacea is estimated to be approximately $1.0$1 billion, and we believe that our FMX103 product candidate for this indication, if approved, can offer advantages over other currently available products.may provide a new treatment alternative for patients and healthcare providers who are unsatisfied with their current therapies.
FMX103 clinical trials FMX103 Phase II clinical trials
In the third quarter of 2016 we announced positive top-line results from our Phase II trial evaluating FMX103. The double-blind, randomized, vehicle-controlled Phase II trial was conducted in 18 sites in Germany and included 233 patients with moderate-to-severe rosacea. Patients were randomized to receive either one of two doses of FMX103 minocycline foam (3% or 1.5%) or vehicle foam once daily over 12 weeks, followed up by a four-week post-treatment evaluation. The efficacy endpoints were (a) the absolute change in the number of inflammatory lesions – papules and pustules (primary endpoint), and (b) improvement of the IGA (first secondary endpoint). Safety and tolerability were also evaluated. The mean baseline lesion count for all groups ranged from 30.6 to 34.5 and the IGA scores were all moderate (score 3) or severe (score 4) with approximately 50-60% of the subjects having a severe rating.
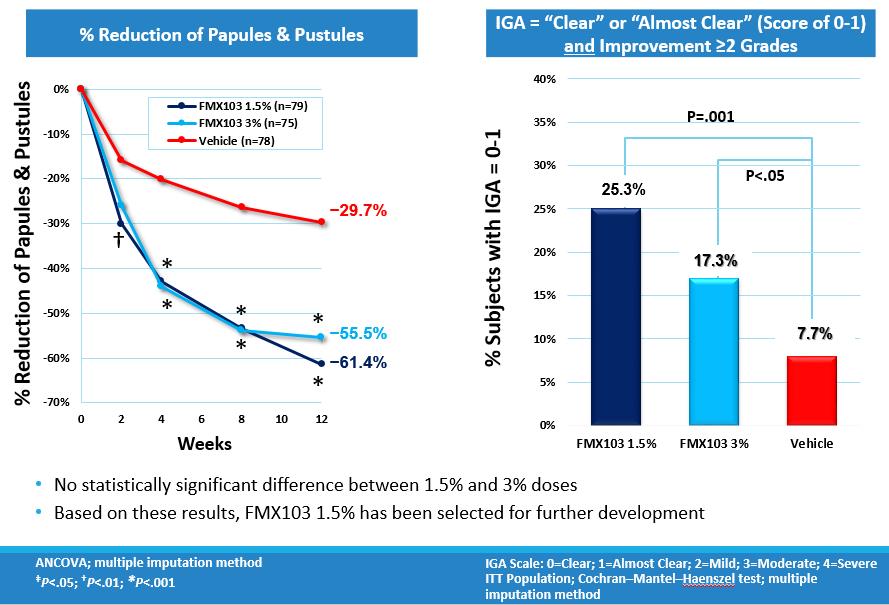
At week 12, statistically significant results were demonstrated in the reduction of inflammatory lesions (papules and pustules) versus vehicle in both the 1.5% and 3% doses of FMX103. The mean reduction in lesion count of each treatment group versus its baseline was 21.1 for the 1.5% dose, 19.9 for the 3% dose and 7.8 for vehicle. The corresponding percent reductions were 61.4% and 55.5% for the FMX103 1.5% and 3% groups, respectively, and 29.7% for the vehicle. The trial further showed a significant improvement in IGA scores. Both the 1.5% and 3% doses of FMX103 were significantly better compared to vehicle alone in reducing the IGA score by two grades and in reaching a “clear” (score=0) or “almost clear” (score=1) rating at week 12 (p<0.01 and p<0.05, respectively). The percentage of subjects achieving IGA success, defined as at least a two-grade point reduction and a rating of clear or almost clear (score of 0 or 1), was 25.3 for the 1.5% dose, 17.3 for the 3% dose, and 7.7 for vehicle. Both the 1.5% and 3% doses were efficacious and there was no statistically significant difference between these two doses.
The following charts show the percent reduction of inflammatory lesions (papules & pustules) and the percentage of patients who met the IGA success criterion:
FMX103 also appeared to be generally safe and well-tolerated. During the Phase II trial, there were no serious adverse events and no drug related systemic adverse events were reported. A few patients overall exhibited treatment-related dermal adverse events (four in the 3% group, five in the vehicle group and none in the 1.5% group). Four patients discontinued the trial due to an adverse event – three in the 3% group and one in the vehicle group.
FMX103 Phase III clinical trials
In December 2016, we conducted a pre-IND meeting with the FDA to confirm that our clinical and non-clinical programs outlined were sufficient to submit an IND and to begin our Phase III clinical trials, utilizing the results of toxicology, pharmacology and human safety studies that were completed for FMX101.
OnIn June 12, 2017, we announced that the first patient had been dosed in our Phase III program to evaluate the efficacy and safety of our topical minocycline foam 1.5% FMX103 for the treatment of moderate-to-severe rosacea. The Phase III program consists of two multi-center trials (referred to as Studies 11FX2016-11 and 12) conducted at approximately 80 sites throughout the U.S.FX2016-12), implementing protocols and endpoints in accordance with the FDA’sFDA’s guidance as provided in the pre-IND meeting. Each trial is expected to enroll approximately 750
Studies FX2016-11 and FX2016-12 were identical, double-blind, randomized, vehicle-controlled studies that enrolled a total of 1,522 patients (Study FX2016-11: 751 patients, Study FX2016-12: 771 patients) with moderate-to-severe papulopustular rosacea across 100 sites in the United States. Patients were randomized 2:1 (1.5% minocycline foam versus vehicle)into a 12-week double-blind vehicle-controlled phase. Patients are randomized on a 2:1 basis (1.5% minocycline foam versus vehicle) andphase where they were treated once daily for 12 weeks inwith either FMX103 minocycline foam (1.5%) or the initial double-blind portions of the trials.respective vehicle foam. The primaryco-primary efficacy endpoints are were: (a) the dichotomized IGA score where treatment success is defined as at least a 2-step improvement resulting in a 0 (clear) or 1 (almost clear) score at week 12 compared to day 0 / baseline, and (b) the absolute change in the inflammatory lesion count at week 12 compared to day 0 / baseline. Safety evaluation will include
In November 2018, we announced positive topline results from these two Phase III clinical trials for FMX103.
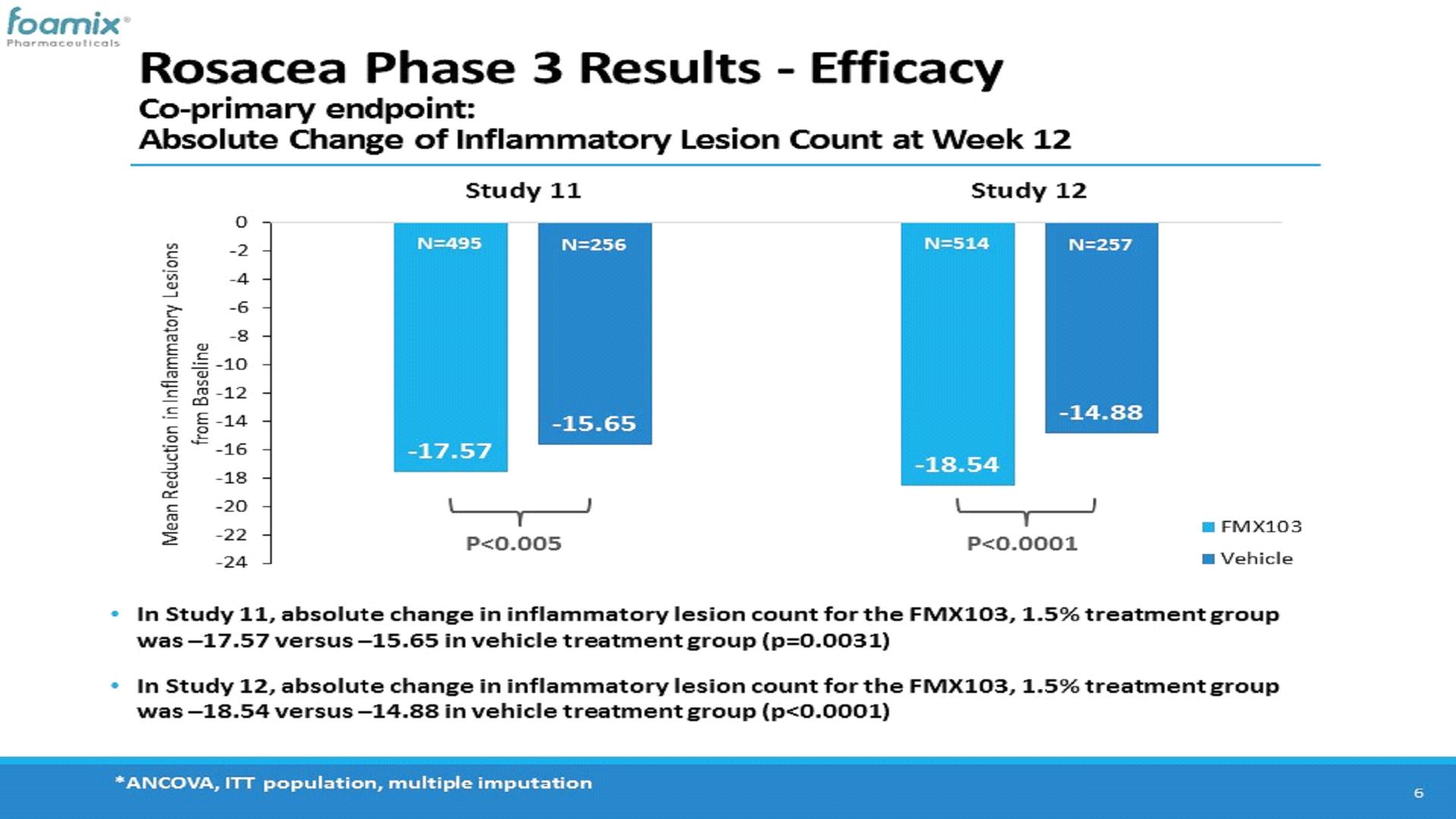
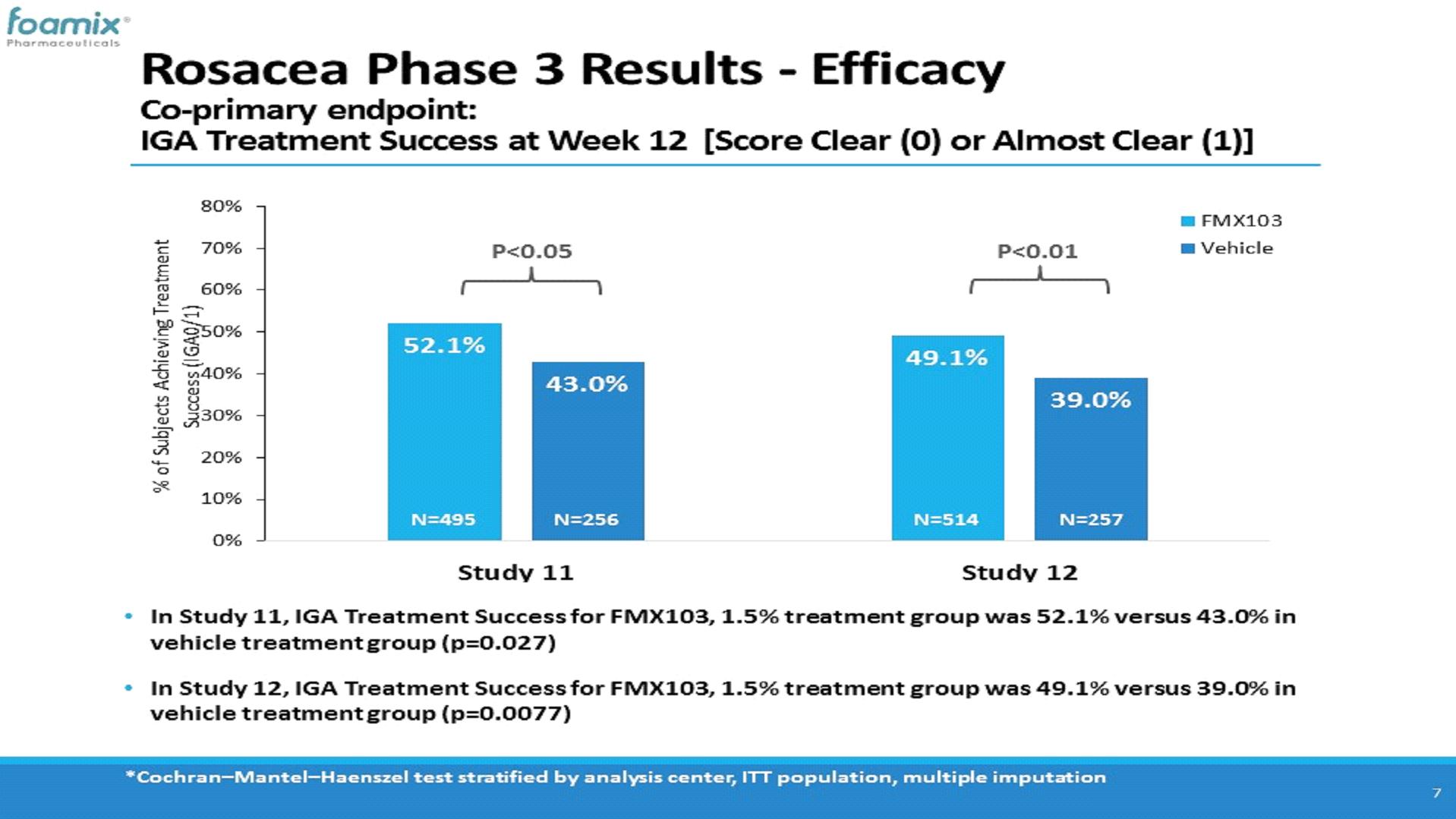
In Study FX2016-11, the mean inflammatory lesion count at baseline was 28.5 and 29.0 for the FMX103 and vehicle treatment groups, respectively, and the proportion of patients with an IGA score at baseline of 3 (“moderate”) or 4 (“severe”) was 89.7% and 10.3%, respectively, in the FMX103 treatment group, and 86.7% and 13.3%, respectively, in the vehicle treatment group. The co-primary efficacy assessment showed a statistically significant mean reduction in inflammatory lesion count at week 12 relative to baseline of -17.57 lesions for the FMX103 treatment group and -15.65 lesions for the vehicle treatment group. In addition, in respect of the second co-primary endpoint, the proportion of subjects that achieved IGA treatment success at week 12 was 52.1% for the FMX103 treatment group and 43.0% for the vehicle treatment group.
In Study FX2016-12, the mean inflammatory lesion count at baseline was 30.0 and 30.2 for the FMX103 and vehicle treatment groups, respectively, and the proportion of patients with an IGA score at baseline of 3 (“moderate”) or 4 (“severe”) was 86.2% and 13.8%, respectively, in the FMX103 treatment group, and 82.9% and 17.1%, respectively, in the vehicle treatment group. The co-primary efficacy assessment showed a statistically significant mean reduction in inflammatory lesion count at week 12 relative to baseline of -18.54 lesions for the FMX103 treatment group and -14.88 lesions for the vehicle treatment group. In addition, in respect of the second co-primary endpoint, the proportion of subjects that achieved IGA treatment success at week 12 was 49.1% for the FMX103 treatment group and 39.0% for the vehicle treatment group.
The following charts show the reduction of inflammatory lesion count from baseline for FMX103 and vehicle treatment groups in each of Studies FX2016-11 and FX2016-12 at week 12:
The following charts show the percentage of patients who met the IGA treatment success criterion at week 12 (defined as at least a 2 grade point reduction from baseline IGA score and a final score of “clear” (0) or “almost clear” (1)) for FMX103 and vehicle treatment groups in each of Studies FX2016-11 and FX2016-12 at week 12:
The safety and tolerability of FMX103 were also evaluated. The most commonly reported adverse events assessmentsin the clinical trials related to upper respiratory tract infections. There were no treatment-related serious adverse events. FMX103 was observed in the clinical trials to have a generally favorable safety profile and appeared to be generally well tolerated. Based on the efficacy and safety profile observed in clinical studies to date, we believe FMX103, if approved, may present an attractive option for the treatment of tolerability, clinical laboratory testsmoderate-to-severe papulopustular rosacea. Studies FX2016-11 and vital signs. The two double-blinded efficacy trials will beFX2016-12 were followed by anstudy FX2016-13 (Study FX2016-13), a 40-week, open-label safety extension study (Study 13) to evaluate the long-term safety of FMX103.The open-label safety study enrolled a total of 505 patients, all of whom had completed 12 weeks of FMX103 or vehicle treatment in the preceding double-blind studies (FX2016-11 or FX2016-12). Patients continued for up to an additional 40 weeks.weeks of open-label treatment with FMX103. As465 patients received FMX103 therapy for at least 26 weeks and 272 patients received FMX103 therapy for a total of 52 weeks. A total of 410 patients completed participation in our third Phase III trial in acne, we selected Premier Research as our designated CRO for the execution of our two Phase III trials in rosacea as well as the open-levelstudy. The key safety extension study. We expect to have top-line resultsfindings from the blinded stage of the trials by the end of the third quarter or in the beginning of the fourth quarter of 2018 and to complete these trials in 2019.study were as follows:
FDX104 for chemotherapy-induced rash
FDX104 is a topical foam formulation of the antibiotic doxycycline for the treatment of severe acne-like rashes induced by chemotherapy.
EGFRI induced-rash and lack of designated treatment
Between 45% and 95% of cancer patients taking epidermal growth-factor receptor inhibitors (EGRFI), such as cetuximab (Erbitux®, Eli Lilly), panitumumab (Vectibix®, Amgen) and erlotinib (Tarceva®, Genentech) are affected by these severe acne-like rashes, which typically occur in cosmetically sensitive areas such as the face and upper trunk. These symptoms can lead to patients modifying their dosage of EGFRI drugs and potentially stop treatment altogether. While there are no approved drugs for the treatment of these rashes, oral doxycycline and minocycline are often used to treat these conditions, but they have significant shortcomings, including systemic side effects such as diarrhea, nausea and skin redness.
FDX104 Phase II clinical trial
In the fourth quarter of 2015, we completed a Phase II clinical trial for FDX104 on patients with metastatic colon cancer who were treated with cetuximab or panitumumab, to prevent the serious rash-like dermal side effects that can be induced by these chemotherapeutic agents. The results showed a statistically significant effect of FDX104 in reducing the severity of these dermal effects, although our assessment of the limited commercial potential of the product in combination with our current priorities has caused us to re-assess our near term efforts with regard to the product.
Twenty-four patients were enrolled and received trial drug in a multicenter, randomized, double-blind, placebo-controlled trial, to evaluate the safety and efficacy of FDX104. Each patient acted as his or her own control by treating one side of the face with FDX104 and the other side with the matching foam vehicle in a blinded and randomized manner. Photographs of the face were taken at each trial visit and were used for the grading of rash severity in a blinded manner by an independent dermatologist at the end of the trial (general rash severity score, or GSS). Rash severity was also performed at each visit by the investigator (modified MASCC EGFR inhibitor papulopustular eruption grading scale, or MESTT). The GSS ratings of rash severity were: none = 0, mild = 1, moderate = 2 and severe = 3. The key findings were:
| · | Non-cutaneous adverse events were comparable in type and frequency with those reported during the double-blinded portion of FX2016-11 and FX2016-12. The severity of rash onmost frequently reported treatment-emergent adverse event was upper respiratory tract infection, or the FDX104 treatment side ofcommon cold (3.8%). 4 patients discontinued the face was overall better than in the vehicle-treated side when analyzed in the ITT population (N=24 patients);study due to a non-application site adverse event – mydriasis, anaemia/leukocytosis, appendicitis and enchondromatosis. No serious drug-related adverse events were reported. |
| · | Cutaneous adverse events occurred in 1% or less of patients during the mean maximal rash severityadditional 40 weeks of open-label treatment with FMX103 with the most frequently reported treatment emergent adverse event being contact dermatitis (1.0%). 2 patients discontinued in the ITT population was 1.33study for an application site adverse event – worsening of rosacea and 1.71 incontact dermatitis. In the FDX104-treatedassessment of facial dermal tolerability at Week 52, more than 95% of patients had “none” or “mild” signs and placebo-treated sides respectively;symptoms (burning/stinging, flushing/blushing, dryness, itching, peeling and hyperpigmentation). The severity of key clinical manifestations of rosacea - erythema and telangiectasia - had both significantly improved when compared to baseline of the preceding double-blind studies. |
| · | 9Subject satisfaction with FMX103 treatment remained high when re-assessed at Week 52 which was consistent with scores obtained at Week 12 (end of double-blind studies). |
Open label efficacy was also assessed throughout the 40-week FMX103 treatment course. The key efficacy findings from the study were:
| · | Mean absolute reduction of inflammatory lesion count when compared to baseline of the 24 patients in the trial (37.5%) developed severe (grade 3) rash during thepreceding double-blinded study on the vehicle-treated side, while only 4(FX2016-11 or FX2016-12) was -23.0 for subjects treated with FMX103 for 52 weeks and –22.5 for subjects treated for 40 weeks. Corresponding mean inflammatory lesion counts at baseline of the 24 patients in the study (16.7%) developed severe rash on the FDX104-treated side.preceding double-blind studies for these groups were 28.8 and 28.7 respectively (all observed cases). |
Comparison of the two treatments (FDX104 | · | The proportion of subjects achieving IGA treatment success at Week 52 defined as at least a 2-step improvement resulting in a 0 (clear) or 1 (almost clear) score compared to baseline of the preceding double-blinded study (FX2016-11 or FX2016-12) was 81.6% for subjects treated with FMX103 for 52 weeks and 76.0% for subjects treated for 40 weeks (all observed cases). |
We intend to submit an NDA for FMX103 in mid-2019.
FMX103 Phase II clinical trials
We initiated Studies FX2016-11 and vehicle) on the prevention of rash based on clinical importance reached statistical significance (p<0.05, Wilcoxon Signed-Rank test). MESTT-based analyses had similar but non-statistically significant results. FDX104 was well-tolerated during the study. No drug-related systemic adverse events were recorded. Local reactions were noted in 5 patients, all were mild and 4 were resolved before the end of the study. While theFX2016-12, as described above, following positive top-line results of the FDX104from our Phase II clinical trial were generally positive, we are assessing our various options with regard to FDX104 in light of our current focus and priorities. This includes seeking out licensing opportunities for the product and discontinuing its further in-house clinical developmentFMX103, announced in the near term so as to better focus on additional research and development efforts.
FMX102 for impetigo
FMX102 is a formulationthird quarter of our minocycline foam currently being developed for the treatment of impetigo.
Impetigo and limitations of current standard of care
Impetigo is a highly contagious bacterial skin infection that primarily afflicts preschool-aged children, and is typically caused by staphylococcus aureus, including methicillin-resistant staphylococcus aureus, or MRSA. It usually results in red sores and lesions on the face, neck, arms and legs.2016. The topical antibiotic Bactroban and other mupirocin-based topical products are the current standard of care for the treatment of impetigo. According to its product label, Bactroban achieves a clinical efficacy rate of between 71% and 96% for impetigo after eight to 12 days of three-times daily treatment. According to the product label for Altabax, the most recently approved topical treatment for impetigo, it achieves a success rate of 89% for impetigo after five days of twice-daily treatment.
FMX102double-blind, randomized, vehicle-controlled Phase II clinical trial
We was conducted in 18 sites in Germany and included 233 patients with moderate-to-severe rosacea who were randomized to receive either one of two doses of FMX103 minocycline foam (3% or 1.5%) or vehicle foam once daily over 12 weeks, followed up by a randomized, double-blindfour-week post-treatment evaluation. The efficacy endpoints were identical to those of Studies FX2016-11 and FX2016-12. Safety and tolerability were also evaluated in the Phase II clinical trial in Israel over seven days with 32 pediatric patients ages two to 15 with at least two impetigo lesions. Of these patients, 32%trial. At week 12, statistically significant results were diagnosed with MRSA infection. The patients were randomly divided into two groups of 16 patients each, with one group receiving a 1% concentration of our minocycline foam and the other group receiving a 4% concentration of our minocycline foam, applied to each patient twice a day. No vehicle-only control was used, as ethical guidelines for pediatric trials in Israel do not permit the use of control groups.
The primary efficacy endpoints of the trial were (a) clinical success, defined as a total absence of treated lesions or certain specific improvementsobserved in the reduction of inflammatory lesions during(papules and pustules) versus vehicle in both the trial1.5% and the continuous absence3% doses of the treated lesions or certain specific improvements in the lesions at follow-up; (b) bacteriological success, measured by elimination of the bacteria in the lesion as shown by a bacterial culture at end of treatment or follow-up or by the lack of any material to culture as a result of the lesion healing; and (c) safety and tolerability.
FMX103. The trial was completedfurther showed a statistically significant improvement in 2012IGA scores. FMX103 also appeared to be generally well-tolerated, with no report of serious adverse events or drug related systemic adverse events. A few patients overall exhibited treatment-related dermal adverse events and showed that approximately 80%four of the patients in both groups met the clinical success criteria after three days of treatment. Clinical response at the end of the treatment (on day 7) was 92% for the 1% dosage and 100% for the 4% dosage, and all patients (100%) showed success by the fourteenth day of the trial. In a post-hoc analysis of the clinical trial results, eleven of the patients in this trial were also diagnosed with methicillin-resistant staphylococcus aureus (MRSA). Bacteriological success was reached in all 11 patients by the end of the trial and no presence of MRSA was detected. The safety and tolerability profile of the drug was favorable, and no drug-related side effects were reported duringthem consequently discontinued the trial.
In October 2015 we held a Pre-IND meeting with the FDA to seek guidance with regards to the preclinical and clinical activities that are required to advance the development program of FMX102. The FDA requested that we conduct a photo-safety study prior to further evaluation of our clinical development plan. We began this photo-safety study in 2016. In 2017, further work on FMX102 was de-prioritized as we focused on our clinical development work for FMX101 and FMX103. Following our further evaluation of the clinical development plan this year and the potential market for FMX102 in the U.S., and considering the treatment currently available for impetigo, we concluded that the business opportunity presented by this product is insufficient to justify further investment of our resources in its advancement at this time, and have discontinued its development.
Development and License Agreements
Parallel to the development of our product candidates, we have entered into development and license agreements with various pharmaceutical companies, including Bayer HealthCare AG,LEO, Mylan N.V. and Actavis Laboratories,plc, combining our emulsion-based foam technology with drugs selected by the licensee to create new product offerings for patients. Each license agreement entitles us to service payments, contingent payments and royalties from sales of any new products that are commercialized. Each agreement is exclusive only to the specific drug that is licensed, leaving us the rights to commercialize and develop products with other drugs for the same indications using our proprietary foam technology while also allowing the licensee to apply the new products to any indication with its specific drug.
In September 2015, Bayer began selling Finacea® Foam (azelaic acid) 15%, ora product based on our foam technology called Finacea in the U.S.United States. Finacea is a prescription topical drug which was developed through a collaboration between Bayer and Foamix. It is the first prescription product developed using our proprietary technology that has been approved by the FDA for sale in the U.S.United States. Bayer listed in the Orange Book several patents that were licensed from Foamixus in connection with the development of Finacea Foam.Finacea. According to our initial license agreement with Bayer, we are entitled to receive royalties and certain contingent payments upon the commercialization of Finacea. On September 4, 2018, LEO acquired Finacea from Bayer. As part of the acquisition, our license agreement with Bayer with respect to Finacea was assigned to LEO. LEO has assumed all of the rights and responsibilities of Bayer under the license agreement as it relates to Finacea, including the payment of royalties to us and rights and obligations related to patent litigation matters. In 20172018, we werereceived (or became entitled to receive) a total of $3.5 million from Bayer in royalties from sales of Finacea. In January 2018Finacea from both Bayer and Foamix filed a complaint alleging patentLEO. Together with LEO, we are litigating against affiliates of Teva, Perrigo and Taro Pharmaceuticals Industries Ltd., or Taro, respectively, for their alleged infringement under the patent laws of the United States that arises outcertain of the submission by defendant Teva of an ANDA to the FDA seeking approval to manufacture and sell a generic version of Bayer’s Finacea Foam. In February 2018 Bayer and Foamix filed a complaint alleging patent infringement under U.S. patent law arising from Perrigo’s our patents following their submission of an ANDA to the FDA seeking approval to manufacture and sell a generic version of Bayer’s Finacea. See also “Item1A—“Risk Factors—RisksFactors-Risks Related to Our Intellectual Property—WeProperty-We have received notice letters of ANDAs submitted for drug products that are generic versions of Finacea Foam and we are involved in lawsuits to protect or enforce our patents, which could be expensive, time consuming and unsuccessful”unsuccessful.”
Our total revenues from all development, collaboration and license agreements from our inception to December 31, 20172018 were approximately $28.1$31.7 million.
Additional Research and Development In addition to FMX101 for the treatment of moderate-to-severe acne, FMX103 for the treatment of moderate-to-severe rosacea and licensed products resulting from our development and license agreements with various pharmaceutical companies, we are developing a series of product candidates for various indications to which we own worldwide rights, and which are all based on formulations and adaptations of our patented, versatile foam platforms or other dosage forms. See “Business–Product Candidates and Pipeline.”
We intend to selectively proceed into clinical trials with these formulations under the FDA’sFDA’s 505(b)(2) regulatory pathway wherever possible,necessary to expedite FDA approval, and according to our identification of unmet needs and potential market opportunities.
Our research and development expenses totaled $64.5 million, $57.8 million and $25.9 million in 2018, 2017 and $10.7 million in 2017, 2016, and 2015, respectively. In the ordinary course of business, we enter into agreements with third parties such as contract research organizations, or CROs, medical institutions, clinical investigators and contract laboratories, to conduct our clinical trials and aspects of our research and preclinical testing. These CROs and other third parties provide us with project management, monitoring, regulatory consulting and investigative services, and their fees are part of our research and development expenses.
Intellectual Property
Our intellectual property and proprietary technology are essential to the development, manufacture, and sale of FMX101 and FMX103 and our future pipeline products.product candidates. We seekare committed to protectprotecting our intellectual property rights, core technologies and other know-how, through a combination of patents, trademarks, trade secrets, non-disclosure and confidentiality agreements, licenses, assignments of invention and other contractual arrangements with our employees, consultants, partners, suppliers, customers and others. Additionally, we rely on our research and development program, clinical trials, know-how and marketing and distribution programs to advance our products.
We submit applications directly or under the Patent Cooperation Treaty, or PCT, which is an international patent law treaty that provides a unified procedure for filing a single initial patent application to seek patent protection for an invention simultaneously in any one of the designated member states. Although a PCT application does not issue as a patent, it allows the applicant to seek protection in any of the member states through national-phase applications.
We also submit applications for a single European patent application covering the EU member states. If granted, the European patent must be validated in each national member state in which the patent is to continue and becomes a bundle of individual national patents.
Our most important patents are several U.S. patents relating to our lead product candidates, FMX101 and FMX103 which are expected to remain in effect until 2030. These patents relate to a composition of matter comprising a claim to a formulation of a tetracycline antibiotic, which can include minocycline or doxycycline, and therefore, may be less protective than patents that claim a new drug. We also have patent applicationspatents claiming compositions of matter, which relate to FMX101 and FMX103, pending in each of the following international markets:Australia, Canada, Great Britain, Israel and Mexico, and patent applications in Canada, the European Union, India and Mexico.
Our other patents granted in the U.S.United States have claims relating to certain formulations of our foam platforms and other technology, including emulsion foams, hydrophobic foams, hydroalcoholic and aqueous foams.
Our FMX101 and FMX103 product candidates are based on a different foam technology platform and different patents than those listed in the Orange Book for the foam technology used in Finacea.
As of December 31, 20172018, we had a patent portfolio of 165175 granted patents in certain countries worldwide, including 6268 granted patents in the U.S.United States. Additionally, as of December 31, 20172018, we had over 40 pending patent applications worldwide, of which over 3025 are pending in the U.S.,United States, describing and claiming our various foam basedfoam-based platforms and other technology. Our other pending applications relate to various foam platforms such as emulsion foam, hydrophobic foam, hydro-alcoholic foam and water-free foams, as well as specialty foams (such as potent solvent foams).foams.
Competition
The medical and pharmaceutical industries in which we operate are intensely competitive and subject to significant technological change and changes in practice. While we believe that our innovative technology, knowledge, experience and resources provide us with competitive advantages, we may face competition from many different sources with respect to FMX101, FMX103 and our other pipeline products or any product candidates that we may seek to develop or commercialize in the future. Possible competitors may include pharmaceutical companies, academic and medical institutions, governmental agencies and public and private research institutions. These prospective competitors have the ability to effectively discover, develop, test and obtain regulatory approvals for products that compete with ours, as well as the ability to effectively commercialize, market and promote approved products, including communicating the effectiveness, safety and value of products to actual and prospective customers and medical staff.
AtIf approved and launched, on-marketed products that could compete with our FMX101 product candidate, if approved, include: (A) oral products such as Solodyn (minocycline, Bausch Health), Doryx (doxycycline, Mayne Pharmaceuticals), Targadox (doxycycline, Journey), Acticlate (docycycline, Almirall), Seysara (sarecycline, Almirall) and (B) topical products such as Epiduo (adapalene + BPO, Galderma), Aczone (dapsone, Almirall), Retin-A (tretinoin, Bausch Health), Onexton-Acanya (clindamycin + BPO, Bausch Health) and Tazorac (tazarotene, Almirall).
On-marketed products that could compete with our FMX103 product candidate include: (A) branded and generic oral products containing minocycline, Oracea (doxycycline, Galderma) and (B) topical products such as all forms of metrogel/metronidazole available as a branded or generic product, Soolantra (ivermectin, Galderma), Finacea (azaleic acid, LEO), Rhofade (oxymetazoline, Allergan), Mirvaso (brimonidine, Galderma).
In addition, new products are currently being developed that may compete with our FMX101 and FMX103 product candidates, if they are approved, including: generic versions of any of the endabove on-marketed products and, specifically for acne: Altreno (tretinoin, Ortho Derm), Seysara (sarecycline, Almirall). In December 2017, Hovione Farmaciencia SA, a private company, announced the commencement of 2014 we became aware that a privately-held active pharmaceutical ingredient and drug product intermediate manufacturer, Hovione, had submitted an IND for Phase I and II clinical trials of a newtrial for its topical gel suspension containing minocycline non-hydrochloride for the treatment of inflammatory skin disease, including acne and rosacea. On December 4 2017, Hovione announced the commencement of a Phase II clinical trial for the treatment of moderate to severe papulopustular rosacea. In 2015 we became aware that another company,October 2018, BioPharmX Corporation (NYSE MKT: BPMX), wasinitiated its Phase IIb clinical trial of BPX-04, a 1% topical minocycline gel formulation for the treatment of moderate-to-severe papulopustular rosacea. BioPharmX is also developing a topical hydrophilicminocycline gel containing minocycline hydrochlorideformulation for the treatment of acne known as BPX-01. BioPharmX announced the results of its Phase IIa andvulgaris, which is currently in Phase IIb clinical trials for BPX-01 in April 2016 and May 2017, respectively, and in November 2017 it announced that it had gained concurrence with FDA on the design of a planned Phase III clinical trial for BPX-01 for treatment of acne. Earlier that year, in September 2017, BioPharmX announced interim results of a feasibility study with BPX-01 for treatment of rosacea, later renamed BPX-04.trials. If ultimately approved and launched in the U.S.,United States, these products would becomecould be direct competitors to FMX101 and FMX103.
Further, we are developing certain topical products with various licensees combining our proprietary technology with a drug selected by the licensee. While the licenses we grant are exclusive with respect to the specific drug which is licensed, our agreements with these licensees allow them to commercialize the licensed developed products for any topical dermatological application, not just for the specific indication for which each product was originally intended. If any such licensed product proves to be effective for moderate-to severe acne, impetigo, rosacea chemotherapy-induced rash, or any other indication that we are pursuing with FMX101, FMX103 or our other product candidates, we may face competition from these licensees, as they are not bound by non-compete restrictions.licensees. Although we believe that FMX101, FMX103 and our other product candidates can outperform the licensed products in the specific indications our product candidates are targeting, such licensed products may nevertheless pose a competitive challenge, as they will have the benefit of our foam technology coupled with the licensees’licensees’ potentially greater resources, experience and brand recognition, extensive marketing channels and other capabilities, and possibly the advantage of entering the market before us.
In addition to products that are currently available, other products may be introduced to treat acne, rosacea impetigo and other skin disorders during the time that we engage in necessary development. Accordingly, if one of our pipeline products is approved, our main challenge in the market would be to convince dermatologists, pediatricians or other physicians seeking alternatives to oral or other existing treatments to use our product instead. While we are still in the preliminary stages, based on our studies, we believe that our pipeline products could be more effective than the current non-topical alternatives and exhibit significantly less adverse side effects.effects based on our studies to date.
Commercialization
We are currently building a commercial infrastructure to support the potential sales of our product candidates in the United States, and may partner with third parties outside the United States to launch our products in other geographic territories or therapeutic classes. Our intent is to build a commercial sales force specifically targeting dermatologists and other healthcare practitioners who diagnose and manage acne and rosacea patients. We also plan to deploy marketing and targeting efforts using data analytics and consumer outreach vehicles that we believe are underutilized. We expect our sales force will be supported by a centralized, internal team who will direct and manage our marketing and sales efforts, and coordinate market access and payor relationships as well as manage the supply chain. In order to be ready to launch our product candidates once approved, we expect to invest significant financial and management resources to build our commercial operations even before our product candidates are approved.
As part of our commercialization planning, we are currently building our internal sales, marketing, market access and distribution infrastructure. Market research, data analysis and strategic launch planning have been conducted on our near-to-market product portfolio. We have established the market entry strategy for FMX101, preliminary physician targeting models, and portfolio strategies to capitalize on infrastructure synergies. FMX103 market entry planning and comprehensive portfolio commercial plans will begin development in the second half of 2019.
We are conducting landscape assessment research on the market access environment for FMX101 to complete a market access strategy. We are also evaluating the optimal price range for FMX101 and FMX103, that will reflect their benefits relative to alternative treatments while remaining affordable to potential customers and reimbursable by governments and third-party payors.
Additionally, continuous efforts are deployed to identify unmet needs in the dermatology market, assess their commercial potential and advise on the prioritization of the development of our future product candidates accordingly.
We have entered into a packaging and supply distribution contract with Sharp Corporation, with whom we have contracted to package and distribute our FMX101 product candidate for commercial distribution, if we receive regulatory approval.
Government Regulation
Our business is subject to extensive government regulation. Regulation by governmental authorities in the U.S.United States and other jurisdictions is a significant factor in the development, manufacture and marketing of our foam delivered treatments and in our ongoing research and development activities.
Product approval process in the U.S.United States
Review and approval of drugs
In the U.S.,United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, or FDCA and implementing regulations and other laws, including the Public Health Service Act.regulations. Drugs require the submission of an NDA, and approval by the FDA prior to being marketed in the U.S.United States. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations requires the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval may subject an applicant to a variety of administrative or judicial sanctions and enforcement actions brought by the FDA, the Department of Justice or other governmental entities. Possible sanctions may include the FDA’sFDA’s refusal to approve pending NDAs, withdrawal of an approval, imposition of a clinical hold, issuance of warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement and civil or criminal penalties.
The process required by the FDA prior to marketing and distributing a drug in the U.S.United States generally involves the following:
| · | completion of laboratory tests, animal studies and formulation studies in compliance with the FDA’sFDA’s good laboratory practice, or GLP, or other applicable regulations; |
| · | submission to the FDA of an application for an investigational new drug application, or IND, designation, which must become effective before human clinical trials may begin; |
| · | approval by an independent institutional review board, or IRB, at each clinical site before each trial may be initiated; |
| · | performance of adequate and well-controlled human clinical trials in accordance with good clinical practice, or GCP, to establish the safety and efficacy of the proposed drug for its intended use; |
| · | preparation and submission to the FDA of an NDA or supplemental NDA; |
| · | satisfactory completion of an FDA advisory committee review, if applicable; |
| · | satisfactory completion of one or more FDA inspections of the manufacturing facility or facilities at which the product or components thereof are produced, to assess compliance with current good manufacturing processes, or cGMP, and to assure that the facilities, methods and controls are adequate to preserve the drug’sdrug’s identity, strength, quality and purity; and |
| · | payment of user fees and FDA review and approval of the NDA. |
Preclinical studies
Preclinical studies include laboratory evaluation, as well as animal studies to assess the potential safety and efficacy of the product candidate. Preclinical safety tests must be conducted in compliance with the FDA’sFDA’s GLP regulations. The results of the preclinical tests, together with manufacturing information and analytical data, are submitted to the FDA as part of an IND which must become effective before clinical trials may be commenced.
Clinical trials in support of an NDA
Clinical trials involve the administration of an investigational product to human subjects under the supervision of qualified investigators in accordance with GCP requirements, which include, among other things, the requirement that all research subjects provide their informed consent in writing before their participation in any clinical trial. Clinical trials are conducted under written trial protocols detailing, among other things, the objectives of the trial, the parameters to be used in monitoring safety, and the effectiveness criteria to be evaluated. A protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA as part of the IND. An IND becomes effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions related to a proposed clinical trial and places the trial on clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin.
In addition, an IRB representing each institution participating in the clinical trial must review and approve the plan for any clinical trial before it commences at that institution, and the IRB must conduct continuing review and reapprove the trial at least annually. The IRB must review and approve, among other things, the trial protocol and informed consent information to be provided to trial subjects. An IRB must operate in compliance with FDA regulations. Information about certain clinical trials must be submitted within specific timeframes to the National Institutes of Health for public dissemination on their ClinicalTrials.gov website.
Clinical trials are typically conducted in three sequential phases, which may overlap or be combined:
| · | Phase I: | The drug is initially introduced into healthy human subjects or patients with the target disease or condition and tested for safety, dosage tolerance, absorption, metabolism, distribution, excretion and, if possible, to gain an early indication of its effectiveness and to determine optimal dosage. |
| · | Phase II: | The drug is administered to a limited patient population to identify possible short-term adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage. |
| · | Phase III: | The drug is administered to an expanded patient population, generally at geographically dispersed clinical trial sites, in well-controlled clinical trials to generate enough data to statistically evaluate the efficacy and safety of the product for approval, to establish the overall risk-benefit profile of the product, and to provide adequate information for the labeling of the product. |
Submission of an NDA to the FDA
The results of the preclinical studies and clinical trials, together with other detailed information, including information on the manufacture, control and composition of the product, are submitted to the FDA as part of an NDA requesting approval to market the product candidate for a proposed indication. Under the Prescription Drug User Fee Act, as amended, applicants are required to pay fees to the FDA for reviewing an NDA. These user fees, as well as the annual fees required for commercial manufacturing establishments and for approved products, can be substantial. The NDA review fee alone exceeded $2,038,000 for fiscal year 2017, subject to certain limited deferrals, waivers and reductions that may be available. The manufacturer or sponsor under an approved NDA are also subject to annual product and establishment user fees, currently exceeding $97,000 per product and $512,000 per establishment for fiscal year 2017. Although these fees were reduced from fiscal year 2016, they are typically increased annually. Each NDA submitted to the FDA for approval is typically reviewed for administrative completeness and reviewability within 45 to 60 days following submission of the application. If found complete, the FDA will “file”“file” the NDA, thus triggering a full review of the application. The FDA may refuse to file any NDA that it deems incomplete or not properly reviewable at the time of submission.
Before approving an NDA, the FDA may inspect the facilities at which the product is manufactured or facilities that are significantly involved in the product development and distribution process, and will not approve the product unless cGMP compliance is satisfactory. The FDA may deny approval of an NDA if applicable statutory or regulatory criteria are not satisfied, or may require additional testing or information, which can delay the approval process. FDA approval of any application may include many delays or may never be granted. If a product is approved, the approval will impose limitations on the indicated uses for which the product may be marketed, may require that warning statements be included in the product labeling, may require that additional studies or trials be conducted following approval as a condition of the approval, may impose restrictions and conditions on product distribution, prescribing or dispensing in the form of a risk management plan,evaluation and mitigation strategies (REMS) , or impose other limitations.
Once a product is approved, marketing the product for other indicated uses or making certain manufacturing or other changes requires FDA review and approval of a supplementsupplemental NDA or a new NDA, which may require additional clinical data and review fees. In addition, further post-marketing testing and surveillance to monitor the safety or efficacy of a product may be required. Also, product approvals may be withdrawn if compliance with regulatory standards is not maintained or if safety or manufacturing problems occur following initial marketing. In addition, new government requirements may be established that could delay or prevent regulatory approval of our product candidatecandidates under development.
505(b)(2) NDAs
As an alternative path to FDA approval for modifications to formulations or uses of productsactive ingredients previously approved by the FDA, an applicant may submit an NDA under Section 505(b)(2) of the Federal Food, Drug and Cosmetic Act, or FDCA. Section 505(b)(2) was enacted as part of the Hatch-Waxman Amendments and permits the filing of an NDA where at least some of the information required for approval comes from FDA’s conclusions of safety or efficacy from studies or trials not conducted by or for the applicant.applicant, and for which the applicant has not received a right of reference If the 505(b)(2) applicant can establish that reliance on FDA’sthe FDA’s previous findings of safety and effectiveness is scientifically appropriate, it may eliminate the need to conduct certain preclinical studies or clinical trials of the new product. The FDA may also require companies to perform additional studies or measurements, including clinical trials, to support the change from the approved reference, or “listed”“listed” drug. The FDA may then approve the new product candidate for all, or some, of the label indications for which the reference drug has been approved, as well as for any new indication sought by the 505(b)(2) applicant.
Notwithstanding the approval of many products by the FDA pursuant to Section 505(b)(2), over the last few years, some pharmaceutical companies and others have objected to the FDA’s interpretation of Section 505(b)(2). If the FDA changes its interpretation of Section 505(b)(2), or if the FDA’s interpretation is successfully challenged in court, this could delay or even prevent the FDA from approving any Section 505(b)(2) NDA that we submit. The Orange Book
In seeking approval for a drug through an NDA, applicants are required to list with the FDA each patent whose claims cover the applicant’s product. Upon approval of a drug, each of the patents listed in the application for the drug is then published in the FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the Orange Book. Drugs listed in the Orange Book can, in turn, be cited by potential generic competitors in support of approval of an ANDA. An ANDA provides for marketing of a drug product that has the same active ingredients in the same strengths and dosage form as the listed drug and has been shown through bioequivalence testing to be bioequivalent to the listed drug. Other than the requirement for bioequivalence testing, ANDA applicants are not required to conduct, or submit results of, pre-clinical or clinical tests to prove the safety or effectiveness of their drug product. Drugs approved in this way are considered to be therapeutically equivalent to the listed drug, are commonly referred to as “generic equivalents” to the listed drug, and can often be substituted by pharmacists under prescriptions written for the original listed drug in accordance with state law.
To the extent that a Section 505(b)(2) NDA relies on clinical trials conducted for a previously approved drug product or the FDA’sFDA’s prior findings of safety and effectiveness for a previously approved drug product, the Section 505(b)(2) applicant must submit patent certifications in its Section 505(b)(2) application with respect to any patents for the previously approved product on which the applicant’sapplicant’s application relies that are listed in the FDA’s Publication of Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the ‘Orange Book’.Orange Book. Specifically, the applicant must certify for each listed patent that, in relevant part, (i) the required patent information has not been filed; (ii) the listed patent has expired; (iii) the listed patent has not expired, but will expire on a particular date and approval is not sought until after patent expiration; or (iv) the listed patent is invalid, unenforceable or will not be infringed by the proposed new product.
A certification that the new product will not infringe the previously approved product’sproduct’s listed patent or that such patent is invalid or unenforceable is known as a Paragraph IV certification. If the applicant does not challenge one or more listed patents through a Paragraph IV certification, the FDA will not approve the Section 505(b)(2) NDA application until all the listed patents claiming the referenced product have expired. Further, the FDA will also not approve, as applicable, a Section 505(b)(2) NDA application until any non-patent exclusivity, such as, for example, five-year exclusivity for obtaining approval of a new chemical entity, three-year exclusivity for an approval based on new clinical trials, or pediatric exclusivity, listed in the Orange Book for the referenced product, has expired.
If the Section 505(b)(2) NDA applicant has provided a Paragraph IV certification to the FDA, the applicant must also send notice of the Paragraph IV certification to the owner of the referenced NDA for the previously approved product and relevant patent holders within 20 days after the Section 505(b)(2) NDA has been accepted for filing by the FDA. The NDA and patent holders may then initiate a patent infringement suit against the Section 505(b)(2) applicant. Under the FDCA, the filing of a patent infringement lawsuit within 45 days of receipt of the notification regarding a Paragraph IV certification automatically prevents the FDA from approving the Section 505(b)(2) NDA until the earliest to occur of 30 months beginning on the date the patent holder receives notice, expiration of the patent, settlement of the lawsuit, or until a court deems the patent unenforceable, invalid or not infringed. Even if a patent infringement claim is not brought within the 45-day period, a patent infringement claim may be brought under traditional patent law, but it does not invoke the 30-month stay.
Moreover, in cases where a Section 505(b)(2) application containing a Paragraph IV certification is submitted after the fourth year of a previously approved drug’s five-year exclusivity period and the patent holder brings suit within 45 days of notice of certification, the 30-month period is automatically extended to prevent approval of the Section 505(b)(2) application until the date that is seven and one-half years after approval of the previously approved reference product. The court also has the ability to shorten or lengthen either the 30 month or the seven and one-half year period if either party is found not to be reasonably cooperating in expediting the litigation.
Exclusivity
In addition to patent protections applicable to a listed drug, a Section 505(b)(2) application may be subject to periods of statutory market exclusivity afforded to an approved new drug. Statutory market exclusivity provides the holder of an approved NDA limited protection from new competition in the marketplace for the innovation represented by its approved drug product, and precludes approval of certain 505(b)(2) applications for prescribed periods of time. Exclusivity is available for new chemical entities, as well as for significant changes in already approved drug products, such as a new use. FDA may refuse to approve a Section 505(b)(2) application to the extent it is subject to the market exclusivity. Notwithstanding the Upon NDA approval of many productsa new chemical entity or NCE, which is a drug that contains no active moiety that has been approved by the FDA pursuant to Section 505(b)(2), over the last fewin any other NDA, that drug receives five years some pharmaceutical companies and others have objected to the FDA’s interpretation of Section 505(b)(2). Ifmarketing exclusivity during which time the FDA changes its interpretation of Sectioncannot receive any ANDA or 505(b)(2), or if the FDA’s interpretation is successfully challenged in court, this could delay or even prevent the FDA from approving any Section 505(b)(2) NDA that we submit.
The FDA regulations and other applicable regulations and policies provide incentives to manufacturers to create modified, non-infringing versions application seeking approval of a drug that references a version of the NCE drug. Certain approvals granted for change(s) to facilitatea drug resulting from new clinical studies that were “essential to approval,” such a new dosage form, strength, route of administration, dosing regimen or indication, are associated with a three-year period of exclusivity. During this three-year exclusivity, the approval ofFDA cannot approve an ANDA or other505(b)(2) application that includes the change(s). Drugs based on an “old antibiotic,” such as our FMX101 and FMX103 product candidates which contain minocycline, must also demonstrate “a significant new use” such as a new indication for generic substitutes.a previously approved antibiotic, and not just refinements in labeling related to previously approved uses, in order to qualify for the three-year exclusivity.
Post-approval requirements
Any drug products for which we receive FDA approval will be subject to continuing regulation by the FDA. Certain requirements include, among other things,inter alia, record-keeping requirements, reporting of adverse experiences with the product, providing the FDA with updated safety and efficacy information on an annual basis or more frequently for specific events, product sampling and distribution requirements, complying with certain electronic records and signature requirements and complying with FDA promotion and advertising requirements. These promotion and advertising requirements include, among others, standards for direct-to-consumer advertising, prohibitions against promoting drugs for uses or patient populations that are not described in the drug’sdrug’s approved labeling, known as “off-label“off-label use,” and other promotional activities, such as those considered to be false or misleading. Failure to comply with FDA requirements can have negative consequences, including the immediate discontinuation of noncomplying materials, adverse publicity, enforcement letters from the FDA, mandated corrective advertising or communications with doctors, and civil or criminal penalties. Such enforcement may also lead to scrutiny and enforcement by other government and regulatory bodies.
Although physicians may prescribe legally available drugs for off-label uses, manufacturers may not encourage, market or promote such off-label uses. As a result, “off-label promotion”“off-label promotion” has formed the basis for litigation under the Federal False Claims Act, or FCA, violations of which are subject to significant civil fines and penalties. In addition, under the federal Physician Payments Sunshine Act, manufacturers of certain prescription products are required to disclose annually to the CenterCenters for Medicaid and Medicare anyServices, or CMS payments or transfers of value made to physicians“covered recipients” and teaching hospitals, in the U.S. under the federal Physician Payment Sunshine Act.and ownership or investment interests held by covered recipients and their immediate family members. Reportable payments and transfers of value may be direct or indirect, in cash or kind, for any reason, and are required to be disclosed even if the paymentstransfers are not related to thean approved product. Failure to fully disclose or notcomply with the Physician Payments Sunshine Act could result in time reporting could lead to penalties up to $1.15 million per year.
The manufacturing of any of our products will be required to comply with applicable FDA manufacturing requirements contained in the FDA’sFDA’s cGMP regulations. The FDA’sFDA’s cGMP regulations require, among other things, quality control and quality assurance, as well as the corresponding maintenance of comprehensive records and documentation. Drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are also required to register their establishments and list any products they make with the FDA and to comply with related requirements in certain states. These entities are further subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP and other laws. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain cGMP compliance.
Discovery of problems with a product after approval may result in serious and extensive restrictions on a product, manufacturer or holder of an approved NDA, as well as lead to potential market disruptions. These restrictions may include recalls, suspension of a product until the FDA is assured that quality standards can be met, and continuing oversight of manufacturing by the FDA under a “consent“consent decree,” which frequently includes the imposition of costs and continuing inspections over a period of many years, as well as possible withdrawal of the product from the market. In addition, changes to the manufacturing process generally require prior FDA approval before being implemented. Other types of changes to the approved product, such as adding new indications and additional labeling claims, are also subject to further FDA review and approval.
The FDA also may require post-marketing testing, or Phase IV testing, as well as risk minimization action plans and surveillanceREMS to monitor the effects of an approved product or place conditions on an approval that could otherwise restrict the distribution or use of our products.
Pediatric trials and exclusivity
Even when not pursuing a pediatric indication, under the Pediatric Research Equity Act of 2003, an NDA or supplement thereto must contain data that are adequate to assess the safety and effectiveness of the drug product for the claimed indications in all relevant pediatric subpopulations, and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. With enactment of the Food and Drug Administration Safety and Innovation Act, or the FDASIA, in 2012, sponsors must also submit pediatric trial plans prior to the assessment data. Those plans must contain an outline of the proposed pediatric trials the applicant plans to conduct, including trial objectives and design, any deferral or waiver requests, and other information required by regulation. The applicant, the FDA, and the FDA’sFDA’s internal review committee must then review the information submitted, consult with each other, and agree upon a final plan. The FDA or the applicant may request an amendment to the plan at any time.
The FDA may, on its own initiative or at the request of the applicant, grant deferrals for submission of some or all pediatric data until after approval of the product for use in adults, or full or partial waivers from the pediatric data requirements. Additional requirements and procedures relating to deferral requests and requests for extension of deferrals are contained in the FDASIA.
Separately, in the event the FDA makes a written request for pediatric data relating to a drug product, an NDA sponsor who submits such data may be entitled to pediatric exclusivity. Pediatric exclusivity is another type of non-patent marketing exclusivity in the U.S.United States and, if granted, provides for the attachment of an additional 6 months of marketing protection to the term of any existing regulatory exclusivity, including the non-patent and orphan exclusivity.
Patent term restoration and extension
A patent claiming a new drug product may be eligible for a limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, commonly referred to as the “Hatch-Waxman“Hatch-Waxman Act,” which permits a patent restoration of up to five years for patent term lost during product development and the FDA regulatory review. The restoration period granted is typically one-half the time between the effective date of an IND and the submission date of an NDA, plus the time between the submission date of aan NDA and the ultimate approval date. Patent term restoration cannot be used to extend the remaining term of a patent past a total of 14 years from the product’sproduct’s approval date. Only one patent applicable to an approved drug product is eligible for the extension, and the application for the extension must be submitted prior to the expiration of the patent in question. A patent that covers multiple drugs for which approval is sought can only be extended in connection with one of the approvals. The U.S. Patent and Trademark OfficeUSPTO reviews and approves the application for any patent term extension or restoration in consultation with the FDA. Since the active pharmaceutical ingredients of our FMX101 FMX102,and FMX103 and FDX104product candidates are already known and marketed in tablet form, the patents supporting these products maywere not be eligible for the said patent term extension, if applicable.extension.
Orphan drug designation18
Orphan drug designation in the U.S. is designed to encourage sponsors to develop drugs intended for rare diseases or conditions. In the U.S., a rare disease or condition is statutorily defined as a condition that affects fewer than 200,000 individuals in the U.S. or that affects more than 200,000 individuals in the U.S. and for which there is no reasonable expectation that the cost of developing and making available the drug for the disease or condition will be recovered from sales of the drug in the U.S.
Orphan drug designation qualifies a company for tax credits and market exclusivity for seven years following the date of the drug’s marketing approval, if granted by the FDA. An application for designation as an orphan product can be made any time prior to the filing of an application for approval to market the product. A drug becomes an orphan when it receives orphan drug designation from the Office of Orphan Products Development, or OOPD, at the FDA based on acceptable confidential requests made under the regulatory provisions. The drug must then go through the new drug approval process like any other drug. Orphan drug designations are decided solely by the OOPD staff, but the OOPD occasionally will request opinions from the Center for Drug Evaluation and Research, especially when dealing with issues such as the appropriateness of the requested indication or the scientific rationale described by the sponsor.
A sponsor may request orphan drug designation of a previously unapproved drug or new orphan indication for an already marketed drug. In addition, a sponsor of a drug that is otherwise the same drug as an already approved orphan drug may seek and obtain orphan drug designation for the subsequent drug for the same rare disease or condition if it can present a plausible hypothesis that its drug may be clinically superior to the first drug. More than one sponsor may receive orphan drug designation for the same drug for the same rare disease or condition, but each sponsor seeking orphan drug designation must file a complete request for designation.
The period of exclusivity begins on the date that the marketing application is approved by the FDA and applies only to the indication for which the drug has been designated. The FDA could approve a second application for the same drug for a different use or a second application for a clinically superior version of the drug for the same use. The FDA cannot, however, approve the same drug made by another manufacturer for the same indication during the market exclusivity period unless it has the consent of the sponsor or the sponsor is unable to provide sufficient quantities.
Review and approval of drug products outside the U.S.United States
In addition to regulations in the U.S.,United States, we will be subject to a variety of foreign regulations governing manufacturing, clinical trials, commercial sales and distribution of our future products. Whether or not we obtain FDA approval for a product candidate, we must obtain approval of the product by the comparable regulatory authorities of foreign countries before commencing clinical trials or marketing in those countries. The approval process varies from country to country, and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country. Under European Union regulatory systems, marketing authorizations may be submitted either under a centralized, decentralized or mutual recognition procedure. The centralized procedure provides for the grant of a single marketing authorization that is valid for all European Union member states. The decentralized procedure includes selecting one “reference member state,” or RMS, and submitting to more than one member state at the same time. The RMS National Competent Authority conducts a detailed review and prepares an assessment report, to which concerned member states provide comment. The mutual recognition procedure provides for mutual recognition of national approval decisions. Under this procedure, the holder of a national marketing authorization may submit an application to the remaining member states post-initial approval. Within 90 days of receiving the applications and assessment report, each member state must decide whether to recognize the approval.
Pharmaceutical coverage, pricing and reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of any product candidates for which we obtain regulatory approval. In the U.S.United States and other markets, sales of any products for which we receive regulatory approval for commercial sale will depend in part on the availability of coverage and adequate reimbursement from third-party payers. Third-party payersthird party payors. Third party payors include government health administrative authorities, managed care providers, private health insurers and other organizations. The process for determining whether a payerpayor will provide coverage for a drug product may be separate from the process for setting the price or reimbursement rate that the payerpayor will pay for the drug product. Third-party payersThird party payors may limit coverage to specific drug products on an approved list, or formulary, which might not include all of the FDA-approved drug products for a particular indication.
Third-party payersThird party payors are increasingly challenging the price and examining the medical necessity and cost-effectiveness of medical products and services, in addition to their safety and efficacy. We may need to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost-effectiveness of FMX101 and FMX103, in addition to the costs required to obtain the FDA approvals. Additionally, FMX101 and FMX103 may not be considered medically necessary or cost-effective. A payer’spayor’s decision to provide coverage for a drug product does not imply that an adequate reimbursement rate will be approved. Adequate third-partythird party reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development.
In March 2010, the President of the U.S.United States signed one of the most significant healthcare reform measures in decades. The healthcare reform law, also known as the Affordable Care Act, or ACA, substantially changes the way healthcare will be financed by both governmental and private insurers, and significantly impacts the pharmaceutical industry. This comprehensive legislative overhaul was expected to extend coverage to approximately 36 million previously uninsured Americans. However, the individual mandate was recently repealed by Congress in The Tax Cuts and Jobs Act of 2017, or the Tax Act, tax reform bill that was signed into law in December 2017.2017 and became effective January 1, 2019. The Joint Committee on Taxation estimates that the repeal will result in over 13 million Americans losing their health insurance coverage over the next ten years and is likely to lead to increases in insurance premiums. On December 14, 2018, a U.S. District Court Judge in the Northern District of Texas ruled that the individual mandate is a critical and inseverable feature of the ACA, and because it was repealed as part of the Tax Act, the remaining provisions of the ACA are invalid as well. While neither the Texas District Court Judge, Trump administration nor CMS have stated that the ruling will have an immediate effect, it is unclear how this decision, subsequent appeals, if any, and other efforts to repeal and replace the ACA will impact the ACA.
The ACA requires the pharmaceutical industry to share in the costs of reform by increasing Medicaid rebates and expanding Medicaid rebates to cover Medicaid managed care programs, among other things. The ACA also includes funding of pharmaceutical costs for Medicare patients in excess of the prescription drug coverage limit and below the catastrophic coverage threshold. UnderThe Bipartisan Budget Act of 2018, or the BBA, among other things, amends the ACA, pharmaceutical companies are obligatedeffective January 1, 2019, to fund 50% of the patient obligation for branded prescription pharmaceuticals inclose this gap, oralso known as the “donut hole.”hole”. Additionally, an excise tax was levied against certain branded pharmaceutical products. The tax is specified by statute to be approximately $3.5 billion in 2017, $4.2 billion in 2018 and $2.8 billion each year thereafter. The tax is to be apportioned to qualifying pharmaceutical companies based on an allocation of their governmental programs as a portion of total pharmaceutical government programs. The Administration is currently looking atexpected to evaluate drug pricing and the Medicare parts B and D programs in terms of policy changes in the next session of Congress.
The Centers for Medicare & Medicaid Services (“CMS”) administerCMS administers the Medicaid drug rebate program, in which pharmaceutical manufacturers pay quarterly rebates to each state Medicaid agency. Generally, for branded prescription drugs marketed under NDAs, as our product candidates are expected to be, manufacturers are required to rebate the greater of 23.1% of the average manufacturer price or the difference between such price and the best price during a specified period. An additional rebate for products marketed under NDAs is payable if the average manufacturer price increases at a rate higher than inflation, and other methodologies apply to new formulations of existing drugs. In addition, the ACA revised certain definitions used for purposes of calculating the rebates, including the definition of “average“average manufacturer price.” The Comprehensive Addiction and Recovery Act of 2016 contains language intended to exempt certain abuse-deterrent formulations of a drug from the definition of line extension for purposes of the program. Various state Medicaid programs have implemented voluntary supplemental drug rebate programs that may provide states with additional manufacturer rebates in exchange for preferred status on a state’sstate’s formulary or for patient populations that are not included in the traditional Medicaid drug benefit coverage.
In the European Union, pricing and reimbursement schemes vary widely from country to country. Some countries provide that drug products may be marketed only after a reimbursement price has been agreed. Some countries may require the completion of additional studies or trials that compare the cost-effectiveness of a particular drug candidate to currently available therapies. For example, the European Union provides options for its member states to restrict the range of drug products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. European Union member states may approve a specific price for a drug product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the drug product on the market. Other member states allow companies to fix their own prices for drug products, but monitor and control company profits. The downward pressure on health care costs in general, and particularly on prescription drugs, has become intense. As a result, increasingly high barriers are being erected to the entry of new products. In addition, in some countries, cross-border imports from low-priced markets exert competitive pressure that may reduce pricing within a country. Any country that has price controls or reimbursement limitations for drug products may not allow favorable reimbursement and pricing arrangements.
Healthcare laws and regulations
Healthcare providers, physicians and third-party payersthird party payors play a primary role in the recommendation and prescription of drug products that are granted marketing approval. Arrangements with healthcare providers, third-party payersthird party payors and other customers are subject to broadly applicable fraud and abuse and other healthcare laws and regulations. Such restrictions under applicable federal and state healthcare laws and regulations include the following:
| · | the federal healthcare Anti-Kickback Statute prohibits, among other things, persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, order or recommendation of, any good or service for which payment may be made, in whole or in part, under a federal healthcare program such as Medicare;Medicare or Medicaid, the term “remuneration” has been broadly interpreted to include anything of value. The intent standard under the federal Anti-Kickback Statute was amended by ACA to a stricter standard such that a person or entity no longer needs to have actual knowledge of the statute or specific intent to violate it, in order to have committed a violation. Moreover, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil FCA; |
| · | the federal False Claims Act imposes civil and criminal false claims laws and civil monetary penalties and provides for civil whistleblower or qui tam actions, againstlaws, including the federal FCA, which prohibit, among other things, individuals or entities forfrom knowingly presenting, or causing to be presented, to the federal government,false, fictitious or fraudulent claims for payment that arefrom Medicare, Medicaid or other federal healthcare programs, knowingly making, using or causing to be made or used a false record or statement material to a false or fraudulent or making a false statementclaim to avoid, decrease or conceal an obligation to pay money to the federal government;government, Private individuals or whistleblowers can bring FCA “qui tam” actions on behalf of the government and may share in amounts recovered; |
| · | the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, imposes criminal and civil liability for, among other things, executing or attempting to execute a scheme to defraud any healthcare benefit program, including any third party payors, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense, and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statements or representations, or making false statements relating to healthcare matters;benefits, items, or services. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it to have committed a violation; |
| · | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH, and itstheir implementing regulations, alsowhich imposes privacy, security transmission and breach reporting obligations including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information;information including PHI, upon “covered entities” subject to the law, such as health plans, healthcare clearinghouses and certain healthcare providers, and their respective business associates that perform services on their behalf that involve individually identifiable health information, including PHI. HITECH also created new tiers of civil monetary penalties, amended HIPAA to make civil and criminal penalties directly applicable to business associates, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce HIPAA laws and seek attorneys’ fees and costs associated with pursuing federal civil actions; and |
| · | the federal false statements statute prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statement in connection with the deliveryPhysician Payments Sunshine Act requires certain manufacturers of or payment for healthcare benefits, items or services; |
| · | the federal transparency requirements under the Health Care Reform Law require manufacturers ofprescription drugs, devices and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program to annually report to the Department of Health and Human ServicesCMS information related to payments and other transfers of value to physicians, dentists, optometrists, podiatrists, chiropractors and teaching hospitals, and physician ownership and investment interests;interests held by physicians and their immediate family members; and |
| · | analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payers,third party payors, including private insurers; state laws that require drug companies to comply with the industry’s voluntary compliance guidelines and the applicable compliance guidance promulgated by the federal government or otherwise restrict payments that may be made to healthcare providers and other potential referral sources; state and local laws that require the licensure of sales representatives; state laws that require drug manufacturers to report information related to drug pricing or payments and other transfers of value to healthcare providers or marketing expenditures and pricing information; data privacy and security laws and regulations in foreign jurisdictions that may be more stringent than those in the United States (such as the European Union, which adopted the General Data Protection Regulation, which became effective in May 2018); state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts; and state laws related to insurance fraud in the case of claims involving private insurers. |
Some stateIf our operations are found to be in violation of any of the healthcare laws require pharmaceutical companiesor regulations described above or any other healthcare regulations that apply to comply with the pharmaceutical industry’s voluntary compliance guidelinesus, we may be subject to penalties, including administrative, civil and criminal penalties, damages, fines, disgorgement, exclusion from participation in government healthcare programs, such as Medicare and Medicaid, imprisonment, additional reporting obligations and oversight if we becomes subject to a corporate integrity agreement or consent decree, reputational harm, diminished profits and future earnings, and the relevant compliance guidance promulgated by the federal government in addition to requiring drug manufacturers to report information related to payments to physicians and other health care providerscurtailment or marketing expenditures. State and foreign laws also govern the privacy and securityrestructuring of health information in some circumstances, manyour operations, any of which differ from each other in significant wayscould adversely affect our ability to operate our business and often are not preempted by HIPAA, thus complicating compliance efforts.pursue our strategy.
Manufacturing, Supply and Production
We do not own or operate manufacturing facilities for the production of our product candidates, nor docandidates; however, we have plans tomay develop our own manufacturing operations in the foreseeable future. We currently rely on third-partythird party contract manufacturers for all of our required raw materials, active ingredients and finished products for our preclinical research and clinical trials, including the Phase III clinical trials for FMX101, FMX103 and our additional product candidates, as applicable. We have contractual relationships for the manufacture of clinical supplies of FMX101 and FMX103, and for commercial supplies if these products are approved. If FMX101, FMX103 or any of our other product candidates are approved by any regulatory agency, we intend to enter into additional agreements with one or more third-partythird party contract manufacturers as secondary manufacturers for the commercial production of these products. We, and our contract manufacturers, are developing the validation processes, methods, tests and or controls suitable for commercial scale manufacturing of our various product candidates and for defining their properties. Changes in manufacturing scale or the manufacturer may require changes to processes, methods, tests and or controls, which may take time to develop, validate and implement.
Development stage and commercial quantities of any products that we develop will need to be manufactured in facilities, and by processes, that comply with the requirements of the FDA and the regulatory agencies of other jurisdictions in which we are seekingseek approval. We currently employ internal and external resources to manage our manufacturing contractors. The relevant manufacturers of our drug products for our current preclinical and clinical trials have advised us that they are compliant with both Good Laboratory Practices, or GLP, and current Good Manufacturing Practices, or cGMP.
Our product candidates, if approved, may not be producible in sufficient commercial quantities, in compliance with regulatory requirements or at an acceptable cost. We and our contract manufacturers are, and will be, subject to extensive governmental regulation in connection with the manufacture of any pharmaceutical products or medical devices.device constituent components. We and our contract manufacturers must ensure that all of the processes, methods and equipment are compliant with cGMP and cGLP for drugs on an ongoing basis, as mandated by the FDA and foreign regulatory authorities, and conduct extensive audits of vendors, contract laboratories and suppliers.
We use, and we intend to continue to use, leading providers of manufacturing services to the global pharmaceutical industry, to scale-up and validate a robust manufacturing process to support commercialization and distribution of our products if approved by the FDA.
Marketing, Sales and Distribution21
Given our stage of development, we do not have any internal sales, marketing or distribution infrastructure. Extensive planning and market research has been conducted on our current product portfolio. Additionally, continuous efforts are deployed to identify unmet needs in the dermatology market, asses their commercial potential and advise on the prioritization of the development of our future product candidates accordingly. We have formed a U.S. subsidiary, Foamix Pharmaceuticals Inc., with key executives to support our clinical development, regulatory affairs, CMC / drug development, quality and commercialization efforts in the U.S.
We are also evaluating the optimal price range for FMX101 and FMX103, that will reflect their benefits relative to alternative treatments while remaining affordable to potential customers and reimbursable by governments and third-party payers.
In the event that we receive regulatory approvals for FMX101 and FMX103 in markets outside of the U.S., we intend, where appropriate, to pursue commercialization relationships, including strategic alliances and licensing, with pharmaceutical companies and other strategic partners, which are equipped to market or sell our products through their well-developed sales, marketing and distribution organizations in such countries.
In addition, we may out-license some or all of our worldwide patent rights to more than one party to achieve the fullest development, marketing and distribution of any products we develop.
Environmental, Health and Safety Matters
We are subject to extensive environmental, health and safety laws and regulations in a number of jurisdictions, primarily Israel, governing, among other things, (i) the use, storage, registration, handling, emission and disposal of chemicals, waste materials and sewage; and (ii) chemical, air, water and ground contamination, air emissions and the cleanup of contaminated sites, including any contamination that results from spills due to our failure to properly dispose of chemicals, waste materials and sewage. Our operations at our Rehovot research and development facility use chemicals and produce waste materials and sewage. Our activities require permits from various governmental authorities including, local municipal authorities, the Ministry of Environmental Protection and the Ministry of Health. The Ministry of Environmental Protection and the Ministry of Health, local authorities and the municipal water and sewage company conduct periodic inspections in order to review and ensure our compliance with the various regulations.
These laws, regulations and permits could potentially require the expenditure by us of significant amounts for compliance or remediation. If we fail to comply with such laws, regulations or permits, we may be subject to fines and other civil, administrative or criminal sanctions, including the revocation of permits and licenses necessary to continue our business activities. In addition, we may be required to pay damages or civil judgments in respect of third-partythird party claims, including those relating to personal injury (including exposure to hazardous substances we use, store, handle, transport, manufacture or dispose of), property damage or contribution claims. Some environmental, health and safety laws allow for strict, joint and several liability for remediation costs, regardless of comparative fault. We may be identified as a responsible party under such laws. Such developments could have a material adverse effect on our business, financial condition and results of operations.
In addition, laws and regulations relating to environmental, health and safety matters are often subject to change. In the event of any changes or new laws or regulations, we could be subject to new compliance measures or to penalties for activities which were previously permitted. For instance, Israeli regulations were promulgated in 2011 relating to the discharge of industrial sewage into the sewer system. These regulations establish new and potentially significant finesfees for discharging forbidden or irregular sewage into the sewage system.
The operations of our subcontractors and suppliers are also subject to various Israeli and foreign laws and regulations relating to environmental, health and safety matters, and their failure to comply with such laws and regulations could have a material adverse effect on our business and reputation, result in an interruption or delay in the development or manufacture of our product candidates, or increase the costs for the development or manufacture of our product candidates.
Employees
As of February 5, 2018,1, 2019, we had a total of 7581 employees, 7180 of whom are full-time employees, 49 of whom were primarily engaged in research and development activities. A total of 118 employees have an M.D. or Ph.D. degree. None of our employees are represented by a labor union, and we consider our employee relations to be good.
Financial and Segment Information
We operate our business as a single segment, as defined by generally accepted accounting principles. Our financial information is included in the consolidated financial statements and the related notes (see “Item 8—notes. See “Financial Statements and Supplementary Data”).Data” for further information.
Legal Proceedings
Currently,From time to time, we are notmay become involved in anylitigation or other legal proceedings other than the complaints filed by Bayer and Foamix in the U.S. in January 2018 against Teva and in February 2018 against Perrigo, alleging patent infringement arising out of ANDA submissions seeking approvalrelating to manufacture and sell a generic version of Bayer’s Finacea Foam prior to the expiry of patents licensed by Foamix to Bayer (see “Item 1A—Risk Factor—Risks Related to Our Intellectual Property—We have received notice letters of ANDAs submitted for drug productsclaims that are generic versions of Finacea Foam and we are involved in lawsuits to protect or enforce our patents, which could be expensive, time consuming and unsuccessful”). We consider such actions to be a part of arising from the ordinary course of our business. We may become parties to additional litigationThere are currently no claims or other legal proceedingsactions pending against us that, we consider to be a part ofin the ordinary courseopinion of our business, and may also become involved inmanagement, are likely to have a material legal proceedings.adverse effect on our business.
Organizational Structure and StatusCorporate Information
Our legal and commercial name is Foamix Pharmaceuticals Ltd. (formerly Foamix Ltd.). We were originally incorporated as a limited liability company under the laws of the State of Israel on January 19, 2003.2003 as Foamix Ltd. We are registered with the Israeli Registrar of Companies. Our registration number is 51-336881-1. Article 3 of our articlesArticles of associationAssociation provides that our objectives are to conduct all types of business as are permitted by law.
Our corporate structure consists of Foamix Pharmaceuticals Ltd. and Foamix Pharmaceuticals Inc., our wholly-owned U.S. subsidiary, which was incorporated on May 6, 2014, under the laws of the State of Delaware, and which is intended to serve as our marketing and sales arm in the U.S.United States.
Effective January 1, 2018, we ceased22
We continue to be a “foreign private issuer” as defined in Rule 3b-4 of the Exchange Act, and became subject to the rules and regulations under the Exchange Act applicable to U.S. domestic issuers. As a result, we are filing an Annual Report on Form 10-K beginning with the fiscal year ended December 31, 2017. Our annual reports for prior years were filed on Form 20-F. We are an “emerging“emerging growth company,” as defined in Section 2(a) of the Securities Act of 1933 and as modified by the JOBS Act. As such, we are eligible to, and intend to, take advantage of certain exemptions from various reporting requirements applicable to other public companies that are not “emerging“emerging growth companies”companies,” such as not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002. We will remain an emerging growth company until the earliest of: (i) the last day of our fiscal year during which we have total annual gross revenues of at least $1.0$1.07 billion; (ii) the last day of our fiscal year following the fifth anniversary of the closing of our initial public offering;offering, specifically, December 31, 2019; (iii) the date on which we have, during the previous three-year period, issued more than $1.0 billion in non-convertible debt; or (iv) the date on which we are deemed to be a “large“large accelerated filer”filer” under the Exchange Act.Act with at least $700 million of equity securities held by non-affiliates. However, in the event we are still a "smaller reporting company," as defined in Rule 12b-2 of the Exchange Act, after our emerging growth company status has terminated, we will be able to continue to take advantage of certain reduced or scaled disclosure requirements for as long as we continue to have smaller reporting company status.
Our principal executive offices are located at 2 Holzman St., Weizmann Science Park, Rehovot 7670402, Israel, and our telephone number is +972-8-9316233. Our website is www.foamix.com. The information contained on, or that can be accessed through, our website does not constitute a part of this formAnnual Report and is not incorporated by reference herein. Foamix Pharmaceuticals Inc., our wholly-owned subsidiary, was incorporated on May 6, 2014 under the laws of the State of Delaware, with the intent to serve as our marketing and sales arm in the U.S.United States. Foamix Pharmaceuticals Inc. has been appointed as our agent for service of process in the United States and is located at 520 U.S. Highway 22, Suite 305, Bridgewater, New Jersey 08807.
ITEM 1A—RISK1A-RISK FACTORS
In conducting our business, we face many risks that may interfere with our business objectives. Some of these risks could materially and adversely affect our business, financial condition and results of operations. In particular, we are subject to various risks resulting from changing economic, political, industry, business and financial conditions. The risks and uncertainties described below are not the only ones we face.
You should carefully consider the following factors and other information in this annual report. If any of the negative events referred to below occur, our business, financial condition and results of operations could suffer. In any such case, the trading price of our ordinary shares could decline.
Risks Related to Our Business and Industry
We are largely dependent on the success of our lead product candidates, FMX101 and FMX103 for the treatment of acne and rosacea.rosacea, respectively.
We have invested a majority of our efforts and financial resources in the research and development of FMX101 for the treatment of moderate-to-severe acne and FMX103 for the treatment of moderate-to-severe papulopustular rosacea, which have both completed Phase IIIII clinical trials. We continue to dedicate our resources toward (i) announcing top-line results from our third pivotal Phase III clinical trialobtaining approval of an NDA by the FDA for FMX101 in the third quarter of 2018 and filing a new drug application, or NDA, with the FDA by the end of the first half of 2019; (ii) announcing top-line results from our two pivotal Phase III clinical trialssubmitting an NDA to the FDA for FMX103 by the end of the third quarter or in the beginning of the fourth quarter of 2018 and completing such trials in 2019;mid-2019; and (iii) advancing our other pipeline candidates. The success of our business depends largely on our ability to fund, execute and complete the development of, obtain regulatory approval for and successfully commercialize FMX101 and FMX103 in a timely manner. If we fail to do so, we may not be able to obtain adequate funding to continue to operate our business.
We have not finalized our Phase III clinical programs for any of our product candidates, nor have we applied forobtained regulatory approvals to market any of our product candidates, and we may be delayed in obtaining or fail to obtain such regulatory approvals and to commercialize our product candidates.
The process of developing, obtaining regulatory approval for and commercializing our product candidates is long, complex, costly and uncertain, and delays or failure can occur at any stage, as evident from our recent experience. In March 2017 we suffered a setback in our Phase III clinical program for FMX101 for treatment of moderate-to-severe acne, following the announcement of mixed top-line results from our two pivotal Phase III clinical trials in this product candidate, known as Studies 04 and 05. Although FMX101 demonstrated statistical significance compared to vehicle on the co-primary endpoint of absolute reduction in inflammatory lesions in both Studies 04 and 05, and further demonstrated statistical significance in the co-primary endpoint of investigator global assessment (IGA) treatment success compared to baseline in Study 04 and on a pooled analysis basis, FMX101 failed to demonstrate statistical significance in the co-primary endpoint of IGA treatment success on a standalone basis in Study 05. Consequently, we were compelled to launch a third pivotal Phase III clinical trial for FMX101, in which we are seeking to replicate the results of Study 04 in a larger patient population and confirm the efficacy of FMX101, in order to proceed with the filing of an NDA for this lead product candidate. stage. Furthermore, the research, testing, manufacturing, labeling, marketing, sale and distribution of drugs are subject to extensive and rigorous regulation by the FDA, and foreign regulatory agencies. These regulations are agency-specific and differ by jurisdiction. We are not permitted to market any of our product candidates in the U.S.United States until we receive approval of aan NDA from the FDA, or in any foreign countries until we receive the requisite approval from the respective regulatory agencies in such countries. To gain approval of an NDA or other equivalent regulatory approval, we must provide the FDA or relevant foreign regulatory authority with clinical data that demonstrates the continued safety purity and potencyefficacy of the product for the intended indication.
Before we can submit an NDA to the FDA or similar applications to foreign regulatory authorities, for FMX101 or FMX103, our leading product candidates, we must complete Phase III clinical trials for them. These clinical trials are substantially broader than our Phase II clinical trials and have required (or will require) us to enlist a considerably larger number of patients in multiple clinics and medical centers. 23
We have not received formal regulatory clearance to file an NDA withmarket FMX101 or FMX103 from the FDA, or from comparable applications to foreign regulatory authorities. Our other product candidates are at earlier stages of development and therefore subject to similar or even greater uncertainty and risk than FMX101 and FMX103.
Although we received positive Phase III clinical trials often produce unsatisfactorytrial results even though prior clinical trials were successful, as we have witnessed in the case of Study 04 for FMX101 as described above. Moreover,and FMX103, the results of those clinical trials may be unsatisfactory to the FDA or foreign regulatory authorities even if we believe those clinical trials to bewere successful. The FDA or applicable foreign regulatory agencies may suspend one or all of our clinical trials or require that we conduct additional clinical, nonclinical, manufacturing, validation or drug product quality studies and submit that data before considering or reconsidering any NDA or similar foreign regulatory application we may submit. Depending on the extent of these additional studies, approval of any applications that we submit may be significantly delayed, or may require us to expend more resources than we have available. It is also possible that additional studies we conduct may not be considered sufficient by the FDA or applicable foreign regulatory agencies to provide regulatory approval.
If any of these outcomes occur, we would not receive approval for FMX101, FMX103 or our other product candidates and may be forced to cease operations.
21Changes in funding for the FDA, the SEC and other government agencies could hinder their ability to hire and retain key leadership and other personnel, prevent new products and services from being developed or commercialized in a timely manner or otherwise prevent those agencies from performing normal functions on which the operation of our business may rely, which could negatively impact our business.
The ability of the FDA to review and approve new products can be affected by a variety of factors, including government budget and funding levels, ability to hire and retain key personnel and accept payment of user fees, and statutory, regulatory, and policy changes. Average review times at the agency have fluctuated in recent years as a result. In addition, government funding of the SEC and other government agencies on which our operations may rely, including those that fund research and development activities is subject to the political process, which is inherently fluid and unpredictable. We have conducted only one Phase II clinical trial relating
Disruptions at the FDA and other agencies may also slow the time necessary for new drugs to each of FMX102, FMX103 and FDX104, in each case outsidebe reviewed and/or approved by necessary government agencies, which would adversely affect our business. For example, over the last several years, including beginning on December 22, 2018, the U.S., government has shut down several times and certain regulatory agencies, such as the results of which may not be predictive of future trial results. Positive results in preclinical testingFDA and early clinical trials do not ensure that later clinical trials will be successful. We sufferedthe SEC, have had to furlough critical FDA, SEC and other government employees and stop critical activities. If a significant setback in our Phase III clinical trials for FMX101 with regard to oneprolonged government shutdown occurs, it could significantly impact the ability of the two co-primary endpointsFDA to timely review and process our regulatory submissions including our NDA which was filed in one of these two trials, and other pharmaceutical companies have had similar experiences in their Phase III clinical trials after demonstrating promising results in preclinical testing and early clinical trials. These setbacks have included negative safety and efficacy observations in later clinical trials, including previously unreported adverse events, though we have not encountered such issues to date.
To date, we have conducted only one Phase II clinical trial for each of FMX102, FMX103 and FDX104 which met their respective primary efficacy and secondary endpoints. Our Phase III clinical trialsDecember 2018 seeking approval for our lead product candidates may not be successful,candidate FMX101 for the treatment of inflammatory lesions of non-nodular moderate-to-severe acne vulgaris in patients 9 years of age and even if they are,older, and such delays could have a material adverse effect on our business. Further, future government shutdowns could impact our ability to access the FDA may not approvepublic markets and obtain necessary capital in order to properly capitalize and continue our NDA for such product candidates, should we be in position to file one, and may not agree that the benefits of such product candidates outweigh its risks, or may raise new concerns regarding our clinical trial designs.operations.
If the FDA does not conclude that FMX101 or FMX103 satisfy the requirements under Section 505(b)(2) of the Federal Food Drug and Cosmetics Act, or Section 505(b)(2), or if the requirements for thisthese product candidatecandidates under Section 505(b)(2) are not as we expect, the approval pathway for this product candidate will likely take significantly longer, cost significantly more and entail significantly greater complications and risks than anticipated, and in either case may not be successful.
We expect to completehave completed our pivotal Phase III trials for FMX101 and we have commenced pivotal Phase III trials for FMX103 under the FDA’sFDA’s 505(b)(2) regulatory pathway. The Drug Price Competition and Patent Term Restoration Act of 1984, also known as the Hatch-Waxman Act, added Section 505(b)(2) to the Federal Food, Drug and Cosmetic Act, or FDCA. Section 505(b)(2) permits the filing of an NDA where at least some of the information required for approval comes from studies that were not conducted by or for the applicant, and for which the applicant has not received a right of reference, which could expedite the development program for FMX101 and FMX103 by potentially decreasing the amount of clinical data that we would need to generate in order to obtain FDA approval. If the FDA does not allow us to pursue the Section 505(b)(2) regulatory pathway, we may need to conduct additional clinical trials, provide additional data and information, and meet additional standards for regulatory approval. If this were to occur, the time and financial resources required to obtain FDA approval for these product candidates, and complications and risks associated with these product candidates, would likely increase significantly. Moreover, inability to pursue the Section 505(b)(2) regulatory pathway could result in new competitive products reaching the market more quickly than our product candidates, which would likely harm our competitive position and prospects. Even if we are allowed to pursue the Section 505(b)(2) regulatory pathway, our product candidates may not receive the requisite approvals for commercialization.
In addition, notwithstanding the approval of certain products by the FDA under Section 505(b)(2) over the last few years, certain competitors and others have objected to the FDA’sFDA’s interpretation of Section 505(b)(2). If the FDA’sFDA’s interpretation of Section 505(b)(2) is successfully challenged, the FDA may be required to change its 505(b)(2) policies and practices, which could delay or even prevent the FDA from approving any NDA that we submit under Section 505(b)(2).In addition, the pharmaceutical industry is highly competitive, and Section 505(b)(2) NDAs are subject to special requirements designed to protect the patent rights of sponsors of previously approved drugs that are referenced in a Section 505(b)(2) NDA. These requirements may give rise to patent litigation and mandatory delays in approval of our NDAs for up to 30 months depending on the outcome of any litigation. It is not uncommon for a manufacturer of an approved product to file a citizen petition with the FDA seeking to delay approval of, or impose additional approval requirements for, pending competing products. If successful, such petitions can significantly delay, or even prevent, the approval of the new product. However, even if the FDA ultimately denies such a petition, the FDA may substantially delay approval while it considers and responds to the petition. In addition, even if we are able to utilize the Section 505(b)(2) regulatory pathway for FMX101 and FMX103, there is no guarantee this would ultimately lead to faster product development or earlier approval.
Moreover, even if theseour product candidates are approved under Section 505(b)(2), the approval may be subject to limitations on the indicated uses for which the product may be marketed or to other conditions of approval, or may contain requirements for costly post-marketing testing and surveillance to monitor the safety or efficacy of the products.
We may not receive regulatory exclusivity for our product candidates under the Hatch-Waxman Act since our lead product candidates are based on an “old antibiotic” and therefore potential competitors may develop generic versions of our product(s) after launch that, if approved, could compete directly with our product(s) sooner that we expect.
Statutory exclusivity provides the holder of an approved NDA limited protection from new competition in the marketplace for the innovation represented by its approved drug product, and precludes approval of certain 505(b)(2) applications and ANDAs, including for a generic version of the drug product, for prescribed periods of time. During the exclusivity period, the FDA may not approve a Section 505(b)(2) application or ANDA to the extent it is subject to exclusivity, or in the case of exclusivity for a new chemical entity may not receive a Section 505(b)(2) application or ANDA. Changes to a drug resulting from new clinical studies (other than bioavailability studies) that were “essential to approval,” and conducted or sponsored by the applicant, such as a new dosage form, strength, route of administration, dosing regimen or indication, are associated with a three-year period of exclusivity during which the FDA cannot approve an ANDA or 505(b)(2) application for the change.
Drugs based on an “old antibiotic,” such as minocycline, are subject to an additional limitation and will not receive three-year exclusivity for a “condition of use” that was approved before October 8, 2008. Prior to 2008, drugs based on an “old antibiotic” were not eligible for Hatch-Waxman Act exclusivity. In 2008, the Q1 Program Supplemental Funding Act of 2008 made drugs containing old antibiotics eligible for three-year exclusivity under certain conditions, but excluded from eligibility for that exclusivity any “condition of use” approved for such drugs before October 8, 2008. The statute does not define “condition of use” but the U.S. District Court has provided recent guidance in Viropharma, Inc. v. Hamburg, 898 F. Supp.2d (District of Columbia, 2012). In Viropharma, the court held that a supplemental new drug approval for the drug Vancocin was not eligible for three-year exclusivity because the supplemental new drug application at issue did not constitute a “significant new use” for the drug. The court held that the “inclusion of more specific dosing information that was within the range specified in the prior label,” “new instructions on monitoring patients’ renal function,” and “new instructions for the continuation of treatment in older patients” did not effect a “significant new use” but rather served only to “refine labeling regarding already approved conditions of use.” The FDA also emphasized that had the company sought approval for a new indication or a new dosing regimen, it would have had to comply with other statutory requirements (including by providing new pediatric data), and that since the company did not have to provide the clinical data, it did not merit the three-year exclusivity for old antibiotics.
We believe that the clinical data submitted for our product candidates will satisfy the exclusivity requirements for old antibiotics. Our Phase III clinical trials for FMX101 provide new clinical (including new pediatric) data that supports a topical route of administration, a new dosing regimen and a significantly lower concentration of minocycline than the prior oral form. FMX103 is indicated for the treatment of moderate-to-severe papulopustular rosacea, which is a new indication for minocycline. While we believe that any clinical data submitted to support FMX101, FMX103 and each of our other pipeline products containing an old antibiotic will provide the required new significant benefits and uses to qualify for the three-year non-patent exclusivity, if the FDA and the U.S. courts do not agree with us, the product candidate would not be protected by three-year exclusivity under the Hatch Waxman Act. While we would continue to be able to enforce our patents against infringement by third-parties, including a 30-month or any further stay or injunction from a court during the pendency of litigation, the FDA could approve an ANDA for a generic version of our product and a company could launch the product at risk, which would allow a generic into the market sooner than we expect. In addition, even if the FDA awards three-year exclusivity to each of these products, its scope will depend on how the FDA defines the exclusivity-protected change. If the FDA defines this change more narrowly than we anticipate, the three-year exclusivity could provide less protection against generic competition than expected.
Because our Phase IIIII clinical trials for the other product candidatesFMX101 and FMX103 were not conducted head-to-head with the current standard of care drugs, the comparison of our results to those of existing drugs, and the conclusions we have drawn from such comparisons, may be inaccurate, and the FDA may require our Phase III trials to be controlled against such drugs.
None of our initial two Phase III clinical trials for FMX101 or Phase II clinical trial forand FMX103 nor our third Phase III trial for FMX101 or our two Phase III trials for FMX103 which are currently under way, were or are being conducted in head-to-head comparison with the drugs considered the current standard of care for the relevant indications, namely Solodyn for moderate-to-severe acne, Bactroban for impetigo,and topical antimicrobials (such as such as Metrogel, generic metronidazole and Finacea) for rosacea and oral doxycycline for chemotherapy induced rash.rosacea. This means that none of the patient groups participating in these trials were or are being treated with the standard of care drugs alongside the groups treated with our product candidates. Instead, we have compared the results of our clinical trials with historical data from prior clinical trials conducted for the standard of care drugs, as presented in their respective product labels.
Direct comparison generally provides more reliable information about how two or more drugs compare, and reliance on indirect comparison for evaluating their relative efficacy or other qualities is problematic due to a lack of objective or validated methods to assess trial similarity. For example, the various trials were likely conducted in different countries with different demographic features and in patients with different baseline conditions and different hygiene standards, among other relevant asymmetries. Therefore, the conclusions we have drawn from comparing the results of our trials with those published in the product labels for these current standardsstandard of care drugs, including conclusions regarding the relative efficacy and expediency of FMX101 FMX102,and FMX103, and FDX104, may be distorted by the inaccurate methodology of the comparison.
The FDA may require the Phase III clinical trials of our product candidates to be controlled against the drugs that are currently considered the standards of care for the treatment of the relevant indications, instead of being controlled against a placebovehicle that does not contain minocycline or against a different dosage of our minocycline foam, as was the case in our Phase II clinical trials. Furthermore, even if the FDA does not impose such a requirement in connection with our Phase III clinical trials, the FDA generally requires adequate, well-controlled head-to-head clinical trials to support comparative claims regarding marketed products. As a result, we may decide to conduct comparative studies of FMX101, FMX103 or any of our other product candidates that are commercialized in the future to support comparative claims used in the marketing of those product candidates. Significant additional time and expense will be required to design and conduct any head-to-head trials.trials, and any such trials may face difficulty in patient enrollment due to their complexity. For example, in the case of FMX101 for moderate-to-severe acne, the standard of care is an oral drug, Solodyn, whereas FMX101 is a topical drug. To conduct a double blinddouble-blind study comparing the two treatments, all patients would need to receive both modalities, with either the oral or topical treatment consisting of a placebo, increasing the complexity and cost. If we are unable to conduct head-to-head trials for one or more of our product candidates, even if such product candidates are approved for marketing in the U.S.,United States, we will not be able to make claims comparing such product candidates to the current standards of care or other competitor products which may negatively impact sales of these products.
Our ability to finance our operations and generate revenues depends on the clinical and commercial success of FMX101, FMX103 and our other product candidates and failure to achieve such success will negatively impact our business.
Our near-term prospects, including our ability to finance our operations and generate revenues, depend on the successful development, regulatory approval and commercialization of FMX101 and FMX103, as well as our other product candidates. The clinical and commercial success of FMX101, FMX103 and our other product candidates depends on a number of factors, many of which are beyond our control, including:
| · | the FDA’sFDA’s and foreign regulatory agencies’agencies’ acceptance of our parameters for regulatory approval relating to FMX101, FMX103 and our other product candidates, including our proposed indications, primary endpoint assessments, primary endpoint measurements and regulatory pathways; |
| · | the FDA’sFDA’s and foreign regulatory agencies’agencies’ acceptance of the number, design, size, conduct and implementation of our clinical trials, our trial protocols and the interpretation of data from preclinical studies or clinical trials; |
| · | the FDA’sFDA’s and foreign regulatory agencies’agencies’ acceptance of the sufficiency of the data we collected from our preclinical studies and early clinical trials of FMX101 and FMX103 to support the submission of an NDA or similar foreign regulatory application without requiring additional preclinical or clinical trials; |
| · | the FDA’sFDA’s and foreign regulatory agencies’agencies’ willingness to schedule an advisory committee meeting in a timely manner to evaluate and decide on the approval of our NDA or similar foreign regulatory application; |
| · | the recommendation of the FDA and foreign regulatory agencies’agencies’ advisory committee to approve our application without limiting the approved labeling, specifications, distribution or use of the products, or imposing other restrictions; |
| · | the FDA’sFDA’s and foreign regulatory agencies’agencies’ willingness to grant separate approvals for adults and children, where we may have successful clinical trial results for childrenadults but not for adults,children, or vice versa; |
| · | the FDA’sFDA’s and foreign regulatory agencies’agencies’ satisfaction with the safety and efficacy of FMX101 and FMX103 or our other product candidates; |
| · | the prevalence and severity of adverse events associated with FMX101, FMX103 and our other product candidates; |
| · | the timely and satisfactory performance by third party contractors of their obligations in relation to our clinical trials, including any future Phase III clinical trials for FMX101 and FMX103;trials; |
| · | our success in educating dermatologists, pediatricians and patients about the benefits, administration and use of FMX101, FMX103 and our other product candidates, if approved; |
| · | our ability to raise additional capital on acceptable terms in order to achieve our goals; |
| · | the availability, perceived advantages, relative cost, safety and efficacy of alternative and competing treatments; |
| · | the effectiveness of our marketing, sales and distribution strategy and operations, as well as that of our current and future licensees; |
| · | our ability to develop, validate and maintain a commercially viable manufacturing process that is compliant with current good manufacturing practices, or cGMP; |
| · | our ability to take advantage of the 505(b)(2) regulatory pathway and obtain regulatory marketing exclusivity for our products, under the Hatch-Waxman Act; |
| · | our ability to obtain protect and enforce our intellectual property rights with respect to FMX101, FMX103 or our other product candidates; |
| · | our ability to bring an action timely for patent infringement arising out of the filing of ANDAs by generic companies seeking approval to market generic versions of our products before the expiry of our patents; and |
| · | our ability to avoid third party claims of patent infringement or intellectual property violations. |
If we fail to achieve these objectives or to overcome the challenges presented above, many of which are beyond our control, in a timely manner, we could experience significant delays or an inability to successfully commercialize our product candidates. Accordingly, we may not be able to generate sufficient revenues through the sale of FMX101, FMX103 or our other product candidates to enable us to continue our business.
We may encounter delays in completing clinical trials for FMX101, FMX103 and our other product candidates and may even be prevented from commencing such trials due to factors that are largely beyond our control.
We have in the past and may in the future experience delays in completing our ongoing clinical trials and in commencing future clinical trials. We have already experienced significant delays in our Phase III clinical program for FMX101 for acne, first due to quality control issues with certain active ingredients supplied to us by a third party and more recently due to insufficient results in one of the co-primary endpoints, namely IGA treatment success, in one of the two initial Phase III trials. We have also experienced a delay in ourtrials, after which we conducted an additional Phase IIIII clinical trial with FMX102 for impetigo that took place in Israel, due to our difficulty in enrolling a sufficient number of pediatric patients with the necessary severity of the disease to participate in the trial. Such difficulties may arise again in future trials for other indications and for our other product candidates.
We rely on contract research organizations, or CROs and clinical trial sites to ensure the proper and timely conduct of our clinical trials. While we have agreements governing the committed activities of our CROs, we have limited influence over their actual performance. A failure of one or more of our clinical trials can occur at any time during the clinical trial process. Clinical trials can be delayed or aborted for a variety of other reasons, including delay or failure to:
| · | obtain regulatory approval to commence a trial; |
| · | reach agreement on acceptable terms with prospective CROs and clinical trial sites, the terms of which may be subject to extensive negotiation and vary significantly among different CROs and trial sites; |
| · | obtain institutional review board, or IRB, approval at each site; |
| · | enlist suitable patients to participate in a trial; |
| · | have patients complete a trial or return for post-treatment follow-up; |
| · | ensure clinical sites observe trial protocol or continue to participate in a trial; |
| · | address any patient safety concerns that arise during the course of a trial; |
| · | address any conflicts with new or existing laws or regulations; |
| · | add a sufficient number of clinical trial sites; or |
| · | manufacture sufficient quantities of the product candidate for use in clinical trials. |
Patient enrollment is also a significant factor in the timing of clinical trials and is affected by many factors, including the size and nature of the patient population, the proximity of patients to clinical sites, the eligibility criteria for the trial, the design of the clinical trial, competing clinical trials and clinicians’clinicians’ and patients’patients’ perceptions as to the potential advantages of the drug being studied in relation to available alternatives, including any new drugs or treatments that may be approved for the indications we are investigating.
We may also encounter delays if a clinical trial is suspended or terminated by us, by the IRBs of the institutions in which such trials are being conducted, by the trial’strial’s data safety monitoring board, by the FDA or by the applicable foreign regulatory authorities. Such authorities may suspend or terminate one or more of our clinical trials due to a number of factors, including our failure to conduct the clinical trial in accordance with relevant regulatory requirements or clinical protocols, inspection of the clinical trial operations or trial site by the FDA or foreign regulatory authorities resulting in the imposition of a clinical hold, unforeseen safety issues or adverse side effects, failure to demonstrate a benefit from using a drug, changes in governmental regulations or administrative actions or lack of adequate funding to continue the clinical trial.
If we experience delays in carrying out or completing any clinical trial of our product candidates, the commercial prospects of our product candidates may be harmed, and our ability to generate product revenues from any of these product candidates will be delayed. In addition, any delays in completing our clinical trials will increase our costs, slow down our product candidate development and approval process and jeopardize our ability to commence product sales and generate revenues. Any of these occurrences may significantly harm our business and financial condition. In addition, many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of our product candidates.
FMX101, FMX103 and other product candidates may produce undesirable side effects that we may not have detected in our Phase II clinical trials or initial Phase III trials. This could prevent us from gaining marketing approval or market acceptance for this product candidate, or from maintaining such approval and acceptance, and could substantially increase commercialization costs and even force us to cease operations.
To date, FMX101 and FMX103 have so farnot been shown in Phase I and Phase II to have noassociated with drug-related systemic side effects and only a few cases of mild and temporary skin reactions have been reported, most of which disappeared on their own within 12 weeks from the beginning of the treatment. FMX101 has further demonstrated thisand FMX103 have both been observed to have a generally favorable safety profile, in its initial Phase III trialswith the most commonly reported adverse events related to upper respiratory tract infections, and extended open-label safety study.there were no treatment-related serious adverse events reported. Nonetheless, in further Phase III clinical trials, which involve large patient populations, and upon commercialization of FMX101, FMX103 or other product candidates, if approved, the clinical exposure of the drugs will be significantly expanded to a wider and more diverse group of patients than those participating in the clinical trials, which may reveal undesirable side effects caused by these products that were not previously observed or reported in the current clinical trials. The FDA and foreign regulatory agency regulations require that we report certain information about adverse medical events if our products may have caused or contributed to those adverse events. The timing of our obligation to report would be triggered by the date on which we become aware of the adverse event as well as the nature of the event. We may fail to report adverse events we become aware of within the prescribed timeframe. We may also fail to appreciate that we have become aware of a reportable adverse event, especially if it is not reported to us as an adverse event or if it is an adverse event that is unexpected or removed in time from the use of our products. If we fail to comply with our reporting obligations, the FDA or a foreign regulatory agency could take action including criminal prosecution, the imposition of civil monetary penalties or seizure of our products.
Additionally, in the event we discover the existence of adverse medical events or side effects caused by one of our product candidates, a number of other potentially significant negative consequences could result, including:
| · | the FDA or foreign regulatory authorities may suspend or withdraw their approval of the product; |
| · | the FDA or foreign regulatory authorities may require the addition of labeling statements, such as warnings or contraindications or distribution and use restrictions; |
| · | the FDA or foreign regulatory authorities may require us to issue specific communications to healthcare professionals, such as letters alerting them to new safety information about our product, changes in dosage or other important information; |
| · | the FDA or foreign regulatory authorities may issue negative publicity regarding the affected product, including safety communications; |
| · | we may be limited with respect to the safety-related claims that we can make in our marketing or promotional materials; |
| · | we may be required to change the way the product is administered, conduct additional preclinical studies or clinical trials or restrict or cease the distribution or use of the product; |
| · | perception of our products by physicians and patients may be adversely affected; and |
| · | we could be sued and held liable for harm caused to patients. |
Any of these events could prevent us from achieving or maintaining market acceptance of the affected product candidate and could substantially increase commercialization costs or even force us to cease operations.
Even if FMX101, FMX103 or our other product candidates receive marketing approval, we may continue to face future developmental and regulatory difficulties. In addition, we are subject to government regulations and we may experience delays in obtaining required regulatory approvals to market our proposed product candidates.
Even if we complete clinical testing and receive approval of any regulatory filing for FMX101, FMX103 or any of our other product candidates, the FDA or applicable foreign regulatory agency may grant approval contingent on the performance of additional costly post-approval clinical trials risk mitigation requirements and surveillance requirementsor REMS to monitor the safety or efficacy of the product, which could negatively impact us by reducing revenues or increasing expenses, and cause the approved product candidate not to be commercially viable. Absence of long-term safety data may further limit the approved uses of our products, if any.
The FDA or applicable foreign regulatory agency may also approve FMX101, FMX103 or any of our other product candidates for a more limited indication or a narrower patient population than we originally requested, or may not approve the labeling that we believe is necessary or desirable for the successful commercialization of our product candidates. Furthermore, any such approved product will remain subject to extensive regulatory requirements, including requirements relating to manufacturing, labeling, packaging, adverse event reporting, storage, advertising, promotion, distribution and recordkeeping.
If we fail to comply with the regulatory requirements of the FDA or other applicable foreign regulatory authorities, or if we discover previously unknown problems with any approved commercial products, manufacturers or manufacturing processes, are discovered, we could be subject to administrative or judicially imposed sanctions or other setbacks, which could require us to take corrective actions, including the following:to:
| · | suspend or impose restrictions on operations, including costly new manufacturing requirements; |
| · | refuse to approve pending applications or supplements to applications; |
| · | suspend any ongoing clinical trials; |
| · | suspend or withdraw marketing approval; |
| · | seek an injunction or impose civil or criminal penalties or monetary fines; |
| · | seize or detain products; |
| · | ban or restrict imports and exports; |
| · | issue warning letters or untitled letters; |
| · | suspend or impose restrictions on operations, including costly new manufacturing requirements; or |
| · | refuse to approve pending applications or supplements to applications. |
In addition, various aspects of our operations are subject to federal, state or local laws, rules and regulations, any of which may change from time to time. Costs arising out of any regulatory developments could be time-consuming and expensive and could divert management resources and attention and, consequently, could adversely affect our business operations and financial performance.
Even if FMX101, FMX103 or our other product candidates receive regulatory approval, they may fail to achieve the broad degree of physician adoption and use and market acceptance necessary for commercial success.
Even if we obtain FDA or foreign regulatory approvals for FMX101, FMX103 or any of our other product candidates, the commercial success of such products will depend significantly on their broad adoption and use by dermatologists, pediatricians and other physicians for approved indications, including, in the case of FMX101 and FMX103, for the treatment of moderate-to-severe acne moderate-to-severand moderate-to-severe rosacea, andrespectively, as well as any other therapeutic indications that we may seek to pursue.
Moreover, if the treatment of acne with FMX101 or rosacea with FMX103 is deemed to be an elective procedure, the cost of which is borne by the patient, it will not be reimbursable through government or private health insurance.
The degree and rate of physician and patient adoption of FMX101, FMX103 and any of our other product candidates, if approved, will depend on a number of factors, including:
| · | the clinical indications for which the product is approved; |
| · | the safety and efficacy of our product as compared to existing therapies for those indications; |
| · | the prevalence and severity of adverse side effects; |
| · | patient satisfaction with the results and administration of our product and overall treatment experience, including relative convenience, ease of use and avoidance of, or reduction in, adverse side effects; |
| · | patient demand for the treatment of moderate-to-severe acne and rosacea or other indications; |
| · | overcoming biases of physicians and patients towards [existing??] topical treatments for moderate-to-severe acne, rosacea or other indications and their willingness to adopt new therapies for these indications; |
| · | the cost of treatment in relation to alternative treatments, the extent to which these costs are covered and adequately reimbursed by third party payors, and patients’patients’ willingness to pay for our products; |
| · | proper training and administration of our products by dermatologists, pediatricians and medical staff; |
| · | the revenues and profitability that our products will offer physicians as compared to alternative therapies; and |
| · | the effectiveness of our sales and marketing efforts, especially the success of any targeted marketing efforts directed toward dermatologists, pediatricians, other physicians, clinics and any direct-to-consumer marketing efforts we may initiate. |
We may decide not to continue developing or commercializing any of our product candidates at any time during development or after approval, which would reduce or eliminate our potential return on investment for those product candidates.
We have in the past and may again in the future decide to discontinue the development of any of our product candidates in our pipeline or not to continue to commercialize any of our product candidates. In December 2017, we discontinued further in-house development of FMX102 for the treatment of impetigo and FMX104 for the treatment of chemotherapy-induced rash due to our reassessment of the limited market opportunities for these products in light of our then-existing limited cash and resources. In the future, we may discontinue development of other product candidates for a variety of reasons, such as the appearance of new technologies that make our product less commercially viable, an increase in competition from generic or other competing products, changes in or failure to comply with applicable regulatory requirements, the discovery of unforeseen side effects during clinical development or after the approved product has been marketed or the occurrence of adverse events at a rate or severity level that is greater than experienced in prior clinical trials. If we discontinue a program in which we have invested significant resources, such as our decision to discontinue further in-house development of FMX102 for the treatment of impetigo and FDX104 for the treatment of chemotherapy induced rash, we will not receive any return on our investment and we will have missed the opportunity to have allocated those resources to other product candidates in our pipeline that may have had potentially more productive uses.
If FMX101, FMX103 or any of our other product candidates are approved for use but fail to achieve the broad degree of physician adoption and market acceptance necessary for commercial success, our operating results and financial condition will be adversely affected.
Our ability to market FMX101 and FMX103, if approved, may be limited to use for the treatment of moderate-to-severe acne or moderate-to-severe rosacea, respectively, in the U.S., and if we want to expand the indications for which we may market FMX101 or the territories in which we may market these products, we will need to obtain additional regulatory approvals, which may not be granted.
We plan to seek regulatory approval in the U.S.United States for FMX101 and FMX103 for the treatment of moderate-to-severe acne and moderate-to-severe rosacea, respectively, in the U.S.respectively. If FMX101 is approved, the FDA will likely restrict our ability to market or advertise FMX101 and FMX103 for other indications, which could limit physician and patient adoption. We may seek to promote and commercialize our products, FMX101 and FMX103 in Europe as well,or in other jurisdictions through third party partnerships, by applying for marketing approval from the European Medicines Agency, or EMA, or in other respective jurisdictions, or we may develop new or additional uses or protocols for FMX101 or FMX103 in the future, but we may not receive the clearances required to do so. If we proceeded to submit for marketing approval in Europeother territories, we or our partners would likely be required to conduct additional clinical trials or studies to support approvals for such additional jurisdictions or indications, which would be time consuming and expensive, and may produce results that do not support regulatory approvals.
Approval procedures vary among countries and can involve additional product testing and additional administrative review periods. The time required to obtain approval in other countries might differ from that required to obtain FDA approval. The marketing approval process in other countries may include all of the risks detailed above regarding FDA approval in the U.S.United States as well as other risks. In particular, in many countries outside the U.S.,United States, it is required that a product receivesreceive pricing and reimbursement approval before the product can be commercialized. This can result in substantial delays in such countries. In other countries, product approval depends on showing superiority to an approved alternative therapy. This may lead to conducting complex clinical trials that require additional significant resources.
None of our products are currently approved for sale in any jurisdiction, including the U.S.United States or any international markets. If we fail to comply with regulatory requirements in the U.S.United States or any international market we decide to enter, or to obtain and maintain required approvals, or if regulatory approvals in the U.S.United States or the relevant international markets are delayed, our target market will be reduced and our ability to realize the full market potential of our products will be harmed.
Marketing approval in one jurisdiction does not ensure marketing approval in another, but a failure or delay in obtaining marketing approval in one jurisdiction may have a negative effect on the regulatory process in others. Failure to obtain a marketing approval in other countries or any delay or setback in obtaining such approval would impair our ability to obtain approval for our product candidates in the United States or to develop foreign markets for FMX101 and FMX103.our products. This would reduce our target market andcould limit the full commercial potential of FMX101, and FMX103.FMX103 or our other products.
If we are not successful in developing, acquiring regulatory approval for and commercializing additional product candidates beyond FMX101 or FMX103, our ability to expand our business and achieve our strategic objectives will be impaired.
Although we will devote a substantial portion of our resources on the continued clinical testing and potential approval of FMX101 for the treatment of moderate-to-severe acne and FMX103 for the treatment of moderate-to-severe rosacea, another key element of our strategy is to discover, develop and commercialize a portfolio of products based on our proprietary foam platforms to serve additional dermatology indications and therapeutic markets. We are seeking to do so through our internal research programs, but our resources are limited, and those that we haveour current programs are primarily geared towards clinical testing and seeking regulatory approval for FMX101 and FMX103. We may also explore strategic collaborations for the development or acquisition of new products and product candidates, but we may not be successful in entering into such relationships. While we have completed two initialthree Phase III clinical trials and commenced a third Phase III clinical trial for our lead product candidate, FMX101, for the treatment of moderate-to-severe acne, and have commenced two Phase III clinical trials for FMX103 for the treatment of rosacea, all of our other potential product candidates remain in earlier stages of development. Research programs to identify product candidates require substantial technical, financial and human resources, regardless of whether any product candidates are ultimately identified. Our research programs may initially show promise in identifying potential product candidates, yet fail to yield product candidates for clinical development for many reasons, including:
| · | the research methodology used may not be successful in identifying potential product candidates; |
| · | competitors may develop alternatives that render our product candidates obsolete or less attractive; |
| · | product candidates we develop may nevertheless be covered by third parties’parties’ patents or other proprietary rights; |
| · | a product candidate may in a subsequent trial be shown to have harmful side effects or other characteristics that indicate it is unlikely to be effective or otherwise does not meet applicable regulatory criteria; |
| · | a product candidate may not be capable of being produced in commercial quantities at an acceptable cost, or at all; |
| · | a product candidate may not be accepted as safe and effective by patients, the medical community or third party payors, if applicable; |
| · | intellectual property rights, such as patents, which are necessary to protect our interests in a product candidate, may be difficult to obtain or unobtainable; |
| · | intellectual property rights, such as patents, may fail to provide adequate protection, may be challenged and one or more claims may be revoked or the patent may be held to be invalid; and |
| · | intellectual property rights of third parties may potentially block our entry into certain markets, or make such entry economically impracticable. |
If we fail to develop and successfully commercialize other product candidates, our business and future prospects may be harmed and our business will be more vulnerable to any problems that we encounter in developing and commercializing FMX101 and FMX103.
Our product candidates, if approved, will face significant competition and our failure to compete effectively may prevent us from achieving significant market penetration and expansion.
If we receive marketing approval, the expected indication of FMX101 will be moderate-to-severe acne and the expected indication of FMX103 will be moderate-to-severe rosacea. The facial aesthetic market in general, and the market for acne treatments in particular, is highly competitive and dynamic, and is characterized by rapid and substantial technological development and product innovations. FMX101, if approved, may face significant competition from other acne products, including oral drugs such as Solodyn, Doryx, Dynacin, Acticlate and Minocin, and topical anti-acne drugs such as Acanya, Ziana, Epiduo, Benzaclin, Aczone and Differin. FMX103, if approved, may face significant competition from other rosacea products, including oral drugs such as Oracea®Oracea®, and topical anti-rosacea drugs such as Metrogel, Soolantra and Finacea, all of which have been approved for marketing and are currently available to consumers. If approved, FMX101 and FMX103 may also compete with non-prescription anti-acne and rosacea products and unapproved and off-label treatments.
There are also several potential competing products currently under development. One of such potential competing products is a new topical gel suspension containing minocycline non-hydrochloride for the treatment of inflammatory skin disease, including acne and rosacea, developed by Hovione, a manufacturer of active pharmaceutical ingredients and drug product intermediates, which product candidate is currently undergoing a Phase II clinical trial for the treatment of moderate-to-severe papulopustular rosacea. Another such potential competing product is a topical hydrophilic gel containing minocycline hydrochloride for the treatment of acne, known as BPX-01, developed by BioPharmX Corporation, for which BioPharmX has completed Phase IIa and Phase IIb clinical trials and has obtained FDA approvalinput for the design of a planned Phase III clinical trial. BioPharmX has also announced interim results ofinitiated a feasibilityPhase IIb study with BPX-01 for a topical minocycline gel named BPX-04 for the treatment of rosacea, later renamed BPX-04.rosacea. If ultimately approved and launched in the U.S., these products would become direct competitors to FMX101 and FMX103.
To compete successfully in the acne and rosacea treatment markets, we will have to demonstrate that FMX101 is safe and effective for the treatment of moderate-to-severe acne and FMX103 is safe and effective for the treatment of moderate-to-severe rosacea, and that they do not infringe the intellectual property rights of any third parties. Competing in the acne and rosacea markets could result in price-cutting, reduced profit margins and loss of market share, any of which would harm our business, financial condition and results of operations.
Due to less stringent regulatory requirements, there are many more acne products and procedures available for use in international markets than are approved for use in the U.S.United States. There are also fewer limitations on the claims that our competitors in international markets can make about the effectiveness of their products and the manner in which they can market them. As a result, if we partner with other companies in these markets and launch our products, we may face more competition in these markets than in the U.S.United States.
In addition, even if we are able to commercialize our product candidates, we may not be able to price them competitively with current standard of care products or their price may drop considerably due to factors outside our control. If this happens or the price of materials and manufacture increases dramatically, our ability to continue to operate our business would be materially harmed and we may be unable to commercialize FMX101 or FMX103 successfully. Other pharmaceutical companies may develop competing products for acne, rosacea and other indications we are pursuing and enter the market ahead of us.
Other pharmaceutical companies are engaged in developing, patenting, manufacturing and marketing healthcare products that compete with those that we are developing. These potential competitors include large and experienced companies that enjoy significant competitive advantages over us, such as greater financial, research and development, manufacturing, personnel and marketing resources, greater brand recognition and more experience and expertise in obtaining marketing approvals from the FDA and foreign regulatory authorities.
Several of these potential competitors are privately-owned companies that are not bound by public disclosure requirements and closely guard their development plans, marketing strategies and other trade secrets. Publicly-traded pharmaceutical companies are also able to maintain a certain degree of confidentiality over their pipeline developments and other sensitive information. As a result, we do not know whether these potential competitors are already developing, or plan to develop, foam-based or other topical treatments for acne, rosacea, impetigo or other indications we are pursuing, and we will likely be unable to ascertain whether such activities are underway in the future. These potential competitors may therefore introduce competing products without our prior knowledge and without our ability to take preemptive measures in anticipation of their commercial launch.
In this regard we became aware at the end of 2014 that an active pharmaceutical ingredient and drug product intermediate manufacturer, Hovione, has submitted an IND for Phase I and II clinical trials of a new topical product containing minocycline for the treatment of inflammatory skin disease including acne and rosacea. Hovione is a privately-held company and we do not know if they have commenced a clinical trial for such new topical minocycline product. Hovione also currently manufactures and supplies Foamix with pharmaceutical-grade minocycline for use in FMX101, FMX103 and other products. Although we have not experienced unique difficulties in procuring minocycline from Hovione, we could experience such difficulties in the future. During 2015 we became aware that another company, BioPharmX Corporation (NYSE MKT: BPMX), is developing a topical hydrophilic gel containing minocycline for the treatment of acne, known as BPX-01, for which BioPharmX has announced Phase IIa clinical trial results. If ultimately approved and commercialized in the U.S., such products would become direct competitors of FMX101, FMX103 and other potential pipeline products.
Furthermore, such potential competitors may enter the market before us, and their products may be designed to circumvent our granted patents and pending patent applications. Potential competitors may also challenge, narrow, invalidate or seek to design around our granted patents or our patent applications, and such patents and patent applications may fail to provide adequate protection for our product candidates. Furthermore, such potential competitors may enter the market before us, and their products may be designed to circumvent our granted patents and pending patent applications.
We have agreements with third party licensees to develop new product candidates for them utilizing our foam technology, and our ability to benefit from such product candidates, or from product candidates that we are developing independently of such licensees, could be impaired or delayed if our licensees’ efforts to develop and commercialize these product candidates are unsuccessful.
In parallel to our core business focused on the development of FMX101, FMX103 and other product candidates, we are pursuing development and license agreements with various pharmaceutical companies for the development and commercialization of product candidates that combine our proprietary technology with the licensees’licensees’ drugs for the treatment of various indications. These license agreements generally provide rights to the licensees for a single active pharmaceutical ingredient, and grant the licensee exclusivity in the development and commercialization of the specific licensed product candidates incorporating such active pharmaceutical ingredient. Our entitlement to contingent payments and royalties from such potential product candidates is therefore dependent upon the licensees’licensees’ performance of their responsibilities and their continued cooperation in developing and commercializing the potential product candidates.
Our licensees may not cooperate with us or perform their obligations under our agreements with them. Furthermore, the obligations of the licensees under such agreements are, for the most part, limited to ‘commercially‘commercially reasonable efforts,’ and they do not face penalties or other repercussions for failing to develop or commercialize the relevant product candidates within the designated timetable other than potentially forfeiting their rights to the relevant product candidate and assigning such rights to us. However, there is no guarantee that we will be able to develop, manufacture or commercialize successfully any such product candidate assigned to us. We cannot control the scope or timing of the resources that will be devoted by our licensees to performing their responsibilities under our agreements with them. Our licensees may choose to pursue alternative technologies in preference to those being developed with us. Several of these agreements may also be terminated for convenience by the licensee. The development and commercialization of these licensed product candidates, as well as the anticipated contingent payments and royalties we hope to generate from them, will be delayed or never obtained if the licensees fail to conduct their responsibilities in a timely manner or in accordance with applicable regulatory requirements, or if they breach their agreements with us. Disputes with our licensees could also impair our reputation or result in development delays, decreased revenues and litigation expenses.
Furthermore, our licensees may encounter adverse outcomes in the process of developing or commercializing their product candidates, and any such outcomes or delays may adversely affect our ability to seek approval for our product candidates or to commercialize our other products. For example, adverse events reported in connection with the use of our licensee’s products that incorporate our technology could negatively affect the perception by regulators, physicians or patients of our platform, which may impair our ability to advance the development of our product candidates and the commercialization of other products.
We have granted several of our licensees the right to commercialize the licensed products for any indication, including acne and rosacea, which may allow them to compete against us using our own technology.
The licenses we granted to several of our licensees, with whom we are developing certain topical products based on our technology and the licensees’licensees’ proprietary drugs, allow them to commercialize the developed products for any topical application, and not only for the specific indication for which each product was originally intended. If any such licensed products prove to be effective for moderate-to-severe acne, rosacea or any other indication that we are pursuing with FMX101, FMX103 or other product candidates, we may face competition from these licensed products, as the licensees are not bound by any non-compete restrictions.products. Healthcare reforms by governmental authorities and related reductions in pharmaceutical pricing, reimbursement and coverage by third-partythird party payors may adversely affect our business. We expect the healthcare industry to face increased limitations on reimbursement, rebates and other payments as a result of healthcare reform, which could adversely affect third-partythird party coverage of our products and how much or under what circumstances healthcare providers will prescribe or administer our products, if approved.
In both the U.S.United States and other countries, sales of our products, if approved for marketing, will depend in part upon the availability ofcoverage and adequate reimbursement from third-partythird party payors, which include governmental authorities, managed care organizations and other private health insurers. Third-partyThird party payors are increasingly challenging the price and examining the cost effectiveness of medical products and services.
Increasing expenditures for healthcare have been the subject of considerable public attention in the U.S.United States Both private and government entities are seeking ways to reduce or contain healthcare costs. Numerous proposals that would effect changes in the U.S. healthcare system have been introduced or proposed in Congress and in some state legislatures, including reducing reimbursement for prescription products and reducing the levels at which consumers and healthcare providers are reimbursed for purchases of pharmaceutical products.
Cost reduction initiatives and changes in coverage implemented through legislation or regulation could decrease utilization of and reimbursement for any approved products, which in turn would affect the price we can receive for those products. Any reduction in reimbursement that results from federal legislation or regulation may also result in a similar reduction in payments from private payors, as private payors often follow Medicare coverage policy and payment limitations in setting their own reimbursement rates. Significant developments that may adversely affect pricing in the U.S.United States include the enactment of federal healthcare reform laws and regulations, including the ACA and the Medicare Prescription Drug Improvement and Modernization Act of 2003. Changes toin the healthcare system enacted as part of healthcare reform in the U.S.,United States, as well as the increased purchasing power of entities that negotiate on behalf of Medicare, Medicaid, and private sector beneficiaries, may result in increased pricing pressure by influencing, for instance, the reimbursement policies of third-partythird party payors. While healthcare reform legislation may have increased the number of patients who are expected to have insurance coverage for our product candidates, provisions such as the assessment of a branded pharmaceutical manufacturer fee and an increase in the amount of rebates that manufacturers pay for coverage of their drugs by Medicaid programs may have an adverse effect on us. It is uncertain how current and future reforms in these areas will influence the future of our business operations and financial condition.
In 2017, a new administration, which had promised to repeal and replace the ACA, took office in the United States. For example, President Trump has signed two Executive Orders and other directives designed to delay the implementation of certain provisions of the ACA or otherwise circumvent some of the requirements for health insurance mandated by the ACA. Concurrently, Congress has considered legislation that would repeal or repeal and replace all or part of the ACA. While Congress has not passed comprehensive repeal legislation, two bills affecting the implementation of certain taxes under the ACA have been signed into law. More recently, in July 2018, CMS published a final rule permitting further collections and payments to and from certain ACA qualified health plans and health insurance issuers under the ACA risk adjustment program in response to the outcome of federal district court litigation regarding the method CMS uses to determine this risk adjustment. On December 14, 2018, a U.S. District Court Judge in the Northern District of Texas ruled that the individual mandate is a critical and inseverable feature of the ACA, and because it was repealed as part of the Tax Act, the remaining provisions of the ACA are invalid as well. While neither the Texas District Court Judge, Trump administration nor CMS have stated that the ruling will have an immediate effect, it is unclear how this decision, subsequent appeals, if any, and other efforts to repeal and replace the ACA will impact the ACA. Although we cannot predict the form of any such replacement of the ACA may take, or the full effect on our business of the enactment of additional legislation pursuant to healthcare and other legislative reform, we believe that legislation or regulations that would reduce reimbursement for, or restrict coverage of, our products could adversely affect how much or under what circumstances healthcare providers will prescribe or administer our products. This could materially and adversely affect our business by reducing our ability to generate revenues, raise capital, obtain additional licensees and market our products. In addition, we believe the increasing emphasis on managed care in the U.S.United States has and will continue to put pressure on the price and usage of pharmaceutical products, which may adversely impact product sales.
Recently there has been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products, which has resulted in several U.S. Congressional inquiries and proposed federal legislation designed to, among other things, bring more transparency to product pricing, reduce the cost of certain products under Medicare, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies. At the state level, individual states in the United States are also increasingly passing legislation and implementing regulations designed to control product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures.
It is likely that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for a pharmaceutical manufacturer’s products or additional pricing pressure.
It will be difficult for us to profitably sell FMX101, FMX103 or our other product candidates if reimbursement for these products is limited by government authorities and third-partythird party payor policies.
In addition to any healthcare reform measures which may affect reimbursement, market acceptance and sales of FMX101, FMX103 and our other product candidates, if approved, will depend on the reimbursement policies of government authorities and third-partythird party payors. Government authorities and third-partythird party payors, such as private health insurers and health maintenance organizations, decide which medications they will pay for, and establish reimbursement levels.
A primary trend in the U.S. healthcare industry and elsewhere is cost containment. Government authorities and these third-partythird party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medications. We cannot be sure that reimbursement will be available for FMX101 or FMX103, or, if reimbursement is available, the level of reimbursement.
Reimbursement may impact the demand for, or the price of, any product for which we obtain marketing approval. In addition, third-partythird party payors are likely to impose strict requirements for reimbursement in order to limit off-label use of a higher priced drug. Reimbursement by a third-partythird party payor may depend upon a number of factors including the third-party payor’sthird party payor’s determination that use of a product is:
| · | a covered benefit under its health plan; |
| · | safe, effective and medically necessary; |
| · | appropriate for the specific patient; |
| · | neither experimental nor investigational. |
Obtaining coverage and reimbursement approval for a product from a government or other third-partythird party payor is a time-consuming and costly process that could require us to provide supporting scientific, clinical and cost effectiveness data for the use of our products to the payor. We may not be able to provide data sufficient to gain acceptance with respect to coverage and reimbursement. We cannot be sure that coverage or adequate reimbursement will be available for our product candidates, if approved. Also, we cannot be sure that reimbursement amounts will not reduce the demand for, or the price of, our future products. If reimbursement is not available, or is available only to limited levels, we may not be able to commercialize our product candidates, profitably or at all, even if approved.
Legislative or regulatory healthcare reforms in the U.S.United States may make it more difficult and costly for us to obtain regulatory clearance or approval of our product candidates and to produce, market, and distribute our products after clearance or approval is obtained.
From time to time, legislation is drafted and introduced in Congress that could significantly change the statutory provisions governing the regulatory clearance or approval, manufacture, and marketing of regulated products or the reimbursement thereof. In addition, FDA regulations and guidance are often revised or reinterpreted by the FDA in ways that may significantly affect our business and our products. Any new regulations or revisions or reinterpretations of existing regulations may impose additional costs or lengthen review times of FMX101, FMX103 or any of our other product candidates. We cannot determine what effect changes in regulations, statutes, legal interpretation or policies, when and if promulgated, enacted or adopted may have on our business in the future. Such changes could, among other things,inter alia, require:
| · | changes to manufacturing methods; |
| · | recall, replacement, or discontinuance of one or more of our products; and |
| · | additional recordkeeping. |
Each of these would likely entail substantial time and cost and could materially harm/adversely affect our business and our financial results.
We will require substantial additional financing to achieve our goals, and a failure to obtain this necessary capital when needed on acceptable terms, or at all, could force us to delay, limit, reduce or terminate our product development, other operations or commercialization efforts.
We anticipate that we will continue to expend substantial resources for the foreseeable future for the clinical development and regulatory approval forand commercialization of FMX101 and FMX103. We also wish to continue the development of other indications and product candidates. However, we may not have sufficient funds to carry out and complete all of these plans, and may need to raise additional funds for such purposes.
These expenditures will include costs associated with research and development, conducting preclinical studies and clinical trials, and manufacturing and supply, as well as marketing and selling any products approved for sale. In addition, other unanticipated costs may arise. Because the outcome of any clinical trial is highly uncertain, we cannot reasonably estimate the actual amounts necessary to successfully complete the development and commercialization of any of our product candidates.
We believe that the net proceeds from our initial and follow-on public offerings will allow us to fund our operating expenses and capital expenditure requirements throughout the Phase III clinical trialsour NDA application and approval process for our lead product candidate, FMX101 forand FMX103, which we expect to announce top-line results in the third quarter of 2018 and to complete by the end of 2018. Such proceeds should also fund our Phase III clinical program for FMX103, for which we expect to announce top-line results by the end of the third quarter or in the beginning of the fourth quarter of 2018 and to complete in 2019. However, our operating plan may change as a result of many factors currently unknown to us, as recently exhibited by our need to commence a third pivotal Phase III clinical trial for FMX101 due to inconclusive results in the co-primary endpoint of IGA treatment success in one of the two initial trials.us. We may therefore need to seek additional capital sooner than planned, through public or private equity or debt financings or other sources, such as strategic collaborations or additional license arrangements. Such financings may result in dilution to shareholders, imposition of debt covenants and repayment obligations or other restrictions that may affect our business. In addition, we may seek additional capital due to favorable market conditions or strategic considerations even if we believe we have sufficient funds for our current or future operating plans.
Our future capital requirements depend on many factors, including:
| · | the results of the clinical trials of our productsproduct candidates; |
| · | the timing of, and the costs involved in, obtaining regulatory approvals for our product candidates; |
| · | the number and characteristics of any additional product candidates we develop or acquire; |
| · | the scope, progress, results and costs of researching and developing our product candidates, and conducting preclinical and clinical trials; |
| · | the cost of commercialization activities if any of our product candidates are approved for sale, including marketing, sales and distribution costs; |
| · | the cost of manufacturing our product candidates and any products we successfully commercialize, and maintaining our related facilities; |
| · | our ability to establish and maintain strategic collaborations, licensing or other arrangements and the terms of and timing of such arrangements; |
| · | the degree and rate of market acceptance of any future approved products; |
| · | the emergence, approval, availability, perceived advantages, relative cost, relative safety and relative efficacy of alternative and competing products or treatments; |
| · | any product liability or other lawsuits related to our products; |
| · | the expenses needed to attract and retain skilled personnel; |
| · | the costs associated with being a public company; |
| · | the costs associated with evaluation of our product candidates; |
| · | the costs associated with evaluation of third party intellectual property; |
| · | the costs associated with obtaining and maintaining licenses; |
| · | the costs associated with obtaining, protecting and enforcing intellectual property, such as costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing patent claims, litigation costs, including the costs of litigation for patent infringement arising out of ANDA submissions by generic companies to manufacture and sell generic products, and the outcome of such litigation; and |
| · | the timing, receipt and amount of sales of, or royalties on, future approved products, if any. |
Additional capital may not be available when we need it, on terms that are acceptable to us or at all. If adequate funds are not available to us on a timely basis, we may be required to:
| · | delay, limit, reduce or terminate preclinical studies, clinical trials or other development activities for FMX101, FMX103 or any of our other product candidates; |
| · | delay, limit, reduce or terminate our research and development activities; or |
| · | delay, limit, reduce or terminate our establishment of manufacturing, sales and marketing or distribution capabilities or other activities that may be necessary to commercialize FMX101, FMX103 or any of our other product candidates. |
If we raise additional capital through marketing and distribution arrangements or other collaborations, strategic alliances or licensing arrangements with third parties, we may have to relinquish certain valuable rights to our product candidates, technologies, future revenue streams or research programs or grant licenses on terms that may not be favorable to us. If we raise additional capital through public or private equity offerings, the ownership interest of our existing shareholders will be diluted and the terms of any new equity securities may have a preference over our ordinary shares. If we raise additional capital through debt financing, we may be subject to covenants limiting or restricting our ability to take specific actions, such as incurring additional debt or making capital expenditures or specified financial ratios, any of which could restrict our ability to commercialize our product candidates or operate as a business.
We have a limited operating history and have incurred significant losses since our inception and we anticipate that we will continue to incur losses for the foreseeable future. We have only fourtwo product candidates that have completed anyPhase III clinical trials and have no sales, which, together with our limited operating history, make it difficult to assess our future commercial viability.
We are a small clinical-stage specialty pharmaceutical company with a limited operating history. Pharmaceutical product development is a highly speculative undertaking and involves a substantial degree of risk. We are not profitable and have incurred losses in each year since we commenced operations in 2003. We have only a limited operating history upon which you can evaluate our business and prospects. In addition, we have limited experience and have not yet demonstrated an ability to successfully overcome many of the risks and uncertainties frequently encountered by companies in new and rapidly evolving fields, particularly in the pharmaceutical industry.
To date, we have not obtained any regulatory approvals for any of our product candidates or generated any revenues from product sales relating to FMX101, FMX103 or any of our other product candidates. We have generated revenues only from service payments, and contingent payments paid towards or in the course of projects carried out under several of our development and license agreements with various pharmaceutical companies. We have also received (and continue to receive) royalty payments with respect to Finacea,®, a prescription foam product that we developed in collaboration with Bayer.Bayer under a license agreement which was assigned to LEO in September 2018.
We continue to incur significant research and development and other expenses related to our ongoing clinical trials and operations. We have recorded a net loss of $74.2 million, $65.7 million $29.3 million and $16.5$29.3 million for the twelve monthsyears ended December 31, 2018, 2017 2016 and 2015,2016, respectively. As of December 31, 20172018, we had an accumulated deficit of $141.3$215,409 million and had a working capital surplus of $59.3$90.7 million. We expect to continue to incur losses for the foreseeable future, and we anticipate these losses will increase substantially as we continue our development of, and seek regulatory approvals for, FMX101, FMX103 and our other product candidates, and begin to commercialize such product candidates.
Our ability to achieve revenues and profitability is dependent on our ability to complete the development of our product candidates, obtain necessary regulatory approvals and successfully manufacture, market and commercialize our products. Even if we achieve profitability in the future, we may not be able to sustain profitability in subsequent periods. Our prior losses, combined with expected future losses, may adversely affect the market price of our ordinary shares and our ability to raise capital and continue operations.
We currently contract with third party subcontractors and suppliers for certain compounds and components necessary to produce FMX101 and FMX103 for clinical trials and expect to continue to do so to support commercial scale production if one or more of such product candidates is approved. This increases the risk that we will not have sufficient quantities of FMX101 and FMX103 or such quantities at an acceptable cost, which could delay, prevent or impair our development or commercialization efforts.
We currently rely on third party subcontractors and suppliers for certain compounds and components necessary to produce FMX101and FMX103 for our clinical trials, including minocycline and other active ingredients, excipients used in the formulation of the foam, delivery apparatus comprising canisters, valves and propellants. We expect to continue to rely on these or other subcontractors and suppliers to support our commercial requirements if FMX101 and FMX103 or any of our other product candidates is approved for marketing by the FDA or foreign regulatory authorities.
Reliance on third party subcontractors and suppliers entails a number of risks, including reliance on the third party for regulatory compliance and quality assurance, the possible breach of the manufacturing or supply agreement by the third party, the possibility that the supply is inadequate or delayed, , the risk that the third party may enter the field and seek to compete and may no longer be willing to continue supplying, and the possible termination or nonrenewal of the agreement by the third party at a time that is costly or inconvenient for us. In addition, third party subcontractors and suppliers may not be able to comply with cGMP or quality system regulation, or QSR, or similar regulatory requirements outside the U.S. If any of these risks transpire, we may be unable to timely retain alternate subcontractors or suppliers on acceptable terms and with sufficient quality standards and production capacity, which may disrupt and delay our clinical trials or the manufacture and commercial sale of our product candidates, if approved.
Our failure or the failure of our third party subcontractors and suppliers to comply with applicable regulations could result in sanctions being imposed on us, including fines, injunctions, civil penalties, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of products, operating restrictions and criminal prosecutions, any of which could significantly and adversely affect supplies of our product candidates that we may develop.
Although we have not experienced unique difficulties in procuring compounds and components for FMX101, FMX103 or any other product candidates, and while we are acting to secure additional suppliers for such compounds and components, we could experience such difficulties in the future.
We will rely on third parties and consultants to assist us in conducting our trials and studies. If these third parties or consultants will not successfully carry out their contractual duties or meet expected deadlines, we may be unable to obtain regulatory approval for or commercialize our product candidates.
We do not have the ability to independently perform all aspects of our preclinical studies and clinical trials. We will relyhave relied on medical institutions, clinical investigators, contract laboratories, collaborative partners and other third parties, such as CROs, to assist us in conducting our Phase III clinical trials for FMX101 and FMX103, and expect to continue doing so with regard to studies and clinical trials for our other product candidates. The third parties with whom we contract for execution of our clinical trials play a significant role in the conduct of these trials and the subsequent collection and analysis of data. However, these third parties are not our employees, and except for contractual duties and obligations, we will have limited ability to control the amount or timing of resources that they devote to our programs.
Although we rely on these third parties to conduct certain aspects of our Phase III clinical trials and other studies and clinical trials, we remain responsible for ensuring that each of our clinical trials and preclinical studies is conducted in accordance with its investigational plan and protocol. Moreover, the FDA and foreign regulatory authorities require us to comply with regulations and standards, commonly referred to as current good clinical practices, or GCPs, for conducting, monitoring, recording and reporting the results of clinical trials to ensure that the data and results are scientifically credible and accurate, and that the trial subjectspatients are adequately informed of the potential risks of participating in clinical trials. We also rely on our consultants to assist us in the execution, including data collection and analysis of our clinical trials.
In addition, the execution of clinical trials and preclinical studies, and the subsequent compilation and analysis of the data produced, will require coordination among these various third parties. In order for these functions to be carried out effectively and efficiently, it will be imperative that these parties communicate and coordinate with one another, which may prove difficult to achieve. Moreover, these third parties may also have relationships with other commercial entities, some of which may compete with us. Our agreement with these third parties may inevitably enable them to terminate such agreements upon reasonable prior written notice under certain circumstances.
If the third parties or consultants that assist us in conducting our clinical trials do not perform their contractual duties or obligations, experience work stoppages, do not meet expected deadlines, terminate their agreements with us or need to be replaced, or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical trial protocols or GCPs, or for any other reason, we may need to conduct additional clinical trials or enter into new arrangements with alternative third parties, which could be difficult, costly or impossible, and our clinical trials may be extended, delayed or terminated or may need to be repeated. If any of the foregoing were to occur, we may not be able to obtain, or may be delayed in obtaining, regulatory approval for the product candidates being tested in such trials, and will not be able to, or may be delayed in our efforts to, successfully commercialize these product candidates.
We have no experience manufacturing our product candidates at full commercial scale. If our product candidates are approved, we intend to outsource our manufacturing to third parties, and will face certain risks associated with such outsourcing.
We have a small-scale integrated research, development and testing facility located at our corporate headquarters in Rehovot, Israel. However, we have not equipped our facility with manufacturing capabilities other than small scale manufacture, and do not currently plan to do so. We do not have experience in manufacturing our product candidates at commercial scale, and ifIf our product candidates are approved, we will outsource all or a significant portion of the manufacturing of our products to third parties, including our drug substances and finished dose forms. Reliance on third parties to manufacture our products entails various risks, including the possibility of increased costs associated with the large- scale production of our products. These risks are similar to those involved in our current use of subcontractors and suppliers for certain compounds and components necessary to produce FMX101, FMX103 and any other of our other product candidates, as explained above.
We have research and development supply arrangements in place with two suppliers for the supply of our API, and with ASM Aerosol-Service AG, for the supply of our finished product. We are currently negotiating commercial supply agreements with our API suppliers and with ASM Aerosol-Service AG. If we are unsuccessful in outsourcingfinalizing our manufacturing tonegotiations with these third parties, who areor if our suppliers breach any of our supply agreements, or do not remain compliant with regulatory requirements, we may encounter delays or additional costs in achieving our commercialization objectives, which could materially damage our business and financial position.
We currently have limitedare building our marketing capabilities and no sales organization.organization to commercialize our products. If our product candidates are approved, and we are unable to establish sales and marketing capabilitiesnot successful in commercializing FMX101, FMX103 or any of our other product candidates, either on our own or through collaborations with one or more third parties, our revenues will suffer and we will be unable to successfully commercialize FMX101, FMX103 or any other of our other product candidates, if approved, or generate product revenues.incur significant additional losses.
We currently have limited marketing capabilities and no sales organization. To commercialize FMX101, FMX103 or any other of our other product candidates, if approved, in the U.S.United States and other jurisdictions we may seek to enter, we must build our marketing, sales, distribution, managerial and other non-technical capabilities or make arrangements with third parties to perform these services, and we may not be successful in doing so. If FMX101 or FMX103 receive regulatory approval, we expect to market them in the U.S.United States through a specialized internal sales force or a combination of our internal sales force and distributors, which will be expensive and time-consuming.
There are significant risks involved in building and managing a sales organization, including our ability to hire, retain and incentivize qualified individuals, generate sufficient sales leads, provide adequate training to sales and marketing personnel and effectively manage a geographically dispersed sales and marketing team. Any failure or delay in the development of our internal sales, marketing and distribution capabilities would adversely impact the commercialization of our product candidates.
We may choose to collaborate with third parties that have direct sales forces and established distribution systems, either to augment our own sales force and distribution systems in the United States or in lieu of our own sales force and distribution systems.systems in other countries. If we are unable to enter into such arrangements on acceptable terms or at all, we may not be able to successfully commercialize FMX101, FMX103 or any of our other product candidates. If we are not successful in commercializing FMX101, FMX103 or any of our other product candidates, either on our own or through collaborations with one or more third parties, our revenues will suffer and we will incur significant additional losses.
To establish our sales and marketing infrastructure and manufacturing capabilities, we will also need to increase the size of our organization, and we may experience difficulties in managing this expansion. As of February 1, 20182019, we had 75two sales and marketing employees. We have hired a Chief Commercial Officer but we will need to continue to expand our managerial, operational, financesales and othermarketing resources in order to manage our operations and clinical trials, continue our development activities and commercialize FMX101, FMX103 and any other product candidates, if approved. Our management, personnel, systems and facilities currently in place may not be adequate to support this future growth. Our need to effectively execute our expansion strategy requires that we: | · | manage our clinical trials effectively; |
| · | identify, recruit, retain, incentivize and integrate additional employees; |
| · | manage our internal development efforts effectively while carrying out our contractual obligations to third parties; and |
| · | continue to improve our operational, financial and management controls, reporting systems and procedures. |
Due to our limited financial resources and our limited experience in managing a company with such anticipated expansion, we may not be able to effectively manage the expansion of our sales and marketing operations or recruit and train additional qualified personnel. The physical expansion of our sales and marketing operations may lead to significant costs and may divert our management and business development resources. Any inability to manage expansion could delay the execution of our development and strategic objectives, or disrupt our operations.
We currently develop our clinical drug products exclusively in one research and development facility and may utilize this facilitylocated in the future to support commercial production if our product candidates are approved.Rehovot, Israel. If this or any future facility or our equipment were to be damaged or destroyed, or if we experience a significant disruption in our operations for any other reason, our ability to continue to operate our business wouldcould be materially harmed.
We currently research and develop our product candidates primarily in our laboratory located in Rehovot, Israel. If this or any future facility were to be damaged, destroyed or otherwise unable to operate, whether due to war, acts of hostility, earthquakes, fire, floods, hurricanes, storms, tornadoes, other natural disasters, employee malfeasance, terrorist acts, power outages or otherwise, or if performance of our research and development facility is disrupted for any other reason, such an event could delay our clinical trials or, if our product candidates are approved and we choose to manufacture all or any part of them internally, jeopardize our ability to manufacture our products as promptly as our prospective customers will likely expect, or possibly at all.trials. If we experience delays in achieving our development objectives, or if we are unable to manufacture an approved product within a timeframe that meets our prospective customers’customers’ expectations, our business, prospects, financial results and reputation could be materially harmed.
Currently, we maintain insurance coverage totaling $1.33$2.4 million against damage to our property and equipment and $5.8$5.4 million in workers compensation coverage, subject to deductibles and other limitations. If we have underestimated our insurance needs with respect to an interruption, or if an interruption is not subject to coverage under our insurance policies, we may not be able to cover our losses.
If product liability lawsuits are brought against us, we may incur substantial liabilities that may not be fully covered by our insurance policies and we may be required to limit commercialization of any of our other products we develop.
We face an inherent risk of product liability as a result of the clinical testing of our product candidates and will face an even greater risk if we commercialize any products. For example, we may be sued if any product we develop allegedly causes injury or is found to be otherwise unsuitable during product testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability and a breach of warranties. Claims could also be asserted under state consumer protection acts. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our products. Even a successful defense would require significant financial and management resources. Regardless of the merits or eventual outcome, liability claims may result in:
| · | decreased demand for FMX101, FMX103 or any of our other product candidates or products we develop; |
| · | injury to our reputation and significant negative media attention; |
| · | withdrawal of clinical trial participants or cancellation of clinical trials; |
| · | costs to defend the related litigation, which may be only partially recoverable even in the event of successful defense; |
| · | a diversion of management’smanagement’s time and our resources; |
| · | substantial monetary awards to trial participants or patients; |
| · | regulatory investigations, product recalls, withdrawals or labeling, marketing or promotional restrictions; |
| · | the inability to commercialize any products we develop. |
Our inability to obtain and maintain sufficient product liability insurance at an acceptable cost and scope of coverage to protect against potential product liability claims could prevent or inhibit the commercialization of FMX101, FMX103 or any other product we may develop. We currently carry generalclinical trials insurance of $10 million and third party liability insurance up to an amount of $10$1.35 million per annum. Although we maintain such insurance, any claim that may be brought against us could result in a court judgment or settlement in an amount that is not covered, in whole or in part, by our insurance or that is in excess of the limits of our insurance coverage. Our insurance policies also have various exclusions and deductibles, and we may be subject to a product liability claim for which we have no coverage. We will have to pay any amounts awarded by a court or negotiated in a settlement that exceed our coverage limitations or that are not covered by our insurance, and we may not have, or be able to obtain, sufficient capital to pay such amounts. Moreover, in the future, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses. If and when we obtain approval for marketing of one or more of FMX101, FMX103 or any other product we may develop, we intend to expand our insurance coverage to include their sale; however, we may be unable to obtain this liability insurance on commercially reasonable terms. If we fail to attract and keep senior management and key scientific personnel, we may be unable to successfully develop FMX101, FMX103 or any of our other product candidates, conduct our clinical trials and commercialize FMX101, FMX103 or any of our other products we develop.
Our success depends in part on our continued ability to attract, retain and motivate highly qualified management, clinical and scientific personnel. We believe that our future success is highly dependent upon the contributions of our senior management, particularly our Chief Executive Officer, as well as our senior technologists and scientists. The loss of services of any of these individuals could delay or prevent the successful development of our product pipeline or completion of our planned clinical trials or the commercialization of FMX101, FMX103 or any of the clinical development of our other product candidates.
Although we have not historically experienced unique difficulties in attracting and retaining qualified employees, we could experience such problems in the future. For example, competition for qualified personnel in the pharmaceutical field is intense due to the limited number of individuals who possess the skills and experience required by our industry. We will need to hire additional personnel as we expand our clinical development and commercial activities. We may not be able to attract and retain quality personnel on acceptable terms, or at all. In addition, to the extent we hire personnel from competitors, we may be subject to allegations that they have been improperly solicited or that they have divulged proprietary or other confidential information, or that their former employers own their research output.
We have incurred, and will continue to incur significant increased costs as a result of operating as a public company in the U.S.,United States, and our management will be required to devote substantial time to new compliance initiatives.
As a public company in the U.S.,United States, we are subject to an extensive regulatory regime, requiring us to maintain various internal controls and to prepare and file periodic and current reports and statements, including reports on the effectiveness of our internal control over financial reporting pursuant to Section 404 of the Sarbanes-Oxley Act of 2002, or Section 404. Complying with these requirements is costly and time consuming. In the event that we are unable to demonstrate compliance with our obligations as a public company in the U.S.United States in a timely manner, or are unable to produce timely or accurate financial statements, we may be subject to sanctions or investigations by regulatory authorities, such as the U.S. Securities and Exchange Commission,SEC, or the NASDAQNasdaq Global Market, and investors may lose confidence in our operating results and the price of our ordinary shares could decline.
Our independent registered public accounting firm was not engaged to perform an audit of our internal control over financial reporting, and as long as we remain an “emerging“emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act, we are exempt from the requirement to have an independent registered public accounting firm perform such audit. Accordingly, no such opinion was expressed. Once we cease to qualify as an “emerging“emerging growth company,” our independent registered public accounting firm will need to attest to our management’s annual assessment of the effectiveness of our internal controls over financial reporting, which will entail additional costs and expenses.
Our business involves the use of hazardous materials and we and our third party manufacturers and suppliers must comply with environmental laws and regulations, which can be expensive and restrict how we do business.
Our research and development activities and our third party subcontractors’subcontractors’ and suppliers’suppliers’ activities involve the controlled storage, use and disposal of hazardous materials owned by us, including minocycline and doxycycline, key components of our product candidates, and other hazardous compounds. We and our manufacturers and suppliers are subject to laws and regulations governing the use, manufacture, storage, handling and disposal of these hazardous materials. We are licensed by the Israeli Ministry of Health to manufacture small batches of product in topical dose form for our Phase I, II and III clinical trials. In some cases, these hazardous materials are stored at our and our subcontractors’subcontractors’ facilities pending their use and disposal.
Despite our efforts, we cannot eliminate the risk of contamination. This could cause an interruption of our commercialization efforts and business operations, environmental damage resulting in costly clean-up and liabilities under applicable laws and regulations governing the use, storage, handling and disposal of these materials and specified waste products. Although we believe that the safety procedures utilized by us and our subcontractors and suppliers for handling and disposing of these materials generally comply with the standards prescribed by these laws and regulations, we cannot guarantee that this is the case or eliminate the risk of accidental contamination or injury from these materials. In such an event, we may be held liable for any resulting damages and such liability could exceed our resources and state or federal or other applicable authorities may curtail our use of certain materials and interrupt our business operations.
Furthermore, environmental laws and regulations are complex, change frequently and have tended to become more stringent. We cannot predict the impact of such changes and cannot be certain of our future compliance.
We may in the future beare subject to various U.S. federal, state and state laws pertaining toforeign health care fraud and abuse laws, including anti-kickback, self-referral, false claims and fraud laws, health information privacy and security, and transparency laws and any violations by us of such laws could result in finessubstantial penalties. Additionally, any challenge to or other penalties.investigation into our practices under these laws could cause adverse publicity and be costly to respond to, and thus could harm our business.
While we do not expect that FMX101, FMX103 or any other of our other product candidates, if approved, will subject us to the variousThere are numerous U.S. federal, state and state laws intended to preventforeign health care fraud and abuse we may in the future becomelaws pertaining to our business, including anti-kickback, false claims and physician transparency laws. Our business practices and relationships with providers, patients and third party payors are subject to suchscrutiny under these laws. The federal anti-kickback statute prohibits the offer, receipt, or payment of remuneration in exchange for orWe may also be subject to induce the referral of patients or the use of products or services that would be paid for in whole or partpatient information privacy and security regulation by Medicare or other federal health care programs. Remuneration has been broadly defined to include anything of value, including cash, improper discounts, and free or reduced price items and services. Many states have similar laws that apply to their state health care programs as well as private payors. Violations of the anti-kickback laws can result in exclusion from federal and state health care programs and substantial civil and criminal penalties.
The federal False Claims Act, or FCA, imposes liability on persons who, among other things, present or cause to be presented false or fraudulent claims for payment by a federal health care program. The FCA has been used to prosecute persons submitting claims for payment that are inaccurate or fraudulent, that are for services not provided as claimed, or for services that are not medically necessary. The FCA includes a whistleblower provision that allows individuals to bring actions on behalf ofboth the federal government, states and share a portion of the recovery of successful claims. Ifforeign jurisdictions in which we conduct our marketing or other arrangements were determinedbusiness. The healthcare laws and regulations that may affect our ability to violate anti-kickback or related laws, including the FCA, then our revenues could be adversely affected, which would likely harm our business, financial condition, and results of operations.operate include:
| · | the U.S. federal Anti-Kickback Statute, which prohibits knowingly and willfully offering, soliciting, receiving, or paying remuneration directly or indirectly, in cash or in kind to induce or reward either the referral of an individual for, or the purchase, order or recommendation of goods or services for which payment may be made in whole or part by Medicare, Medicaid or other federal health care programs. Remuneration has been broadly defined to include anything of value, including cash, improper discounts, and free or reduced price items and services. The intent standard under the federal Anti-Kickback Statute was amended by the ACA to a stricter standard such that a person or entity no longer needs to have actual knowledge of the statute or specific intent to violate it, in order to have committed a violation. Moreover, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil FCA. Additionally, many states have similar laws that apply to their state health care programs as well as private payors. Violations of the federal and state anti-kickback laws can result in exclusion from federal and state health care programs and substantial civil and criminal penalties. |
| · | the federal civil and criminal false claims laws and civil monetary penalties laws, including the False Claims Act, or FCA, prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, false, fictitious or fraudulent claims for payment from Medicare, Medicaid or other federal healthcare programs, and knowingly making, using or causing to be made or used a false record or statement material to a false or fraudulent claim to avoid, decrease or conceal an obligation to pay money to the federal government. Private individuals or “whistleblowers” can bring FCA “qui tam” actions on behalf of the government and may share in recovered amounts. The FCA has been used to prosecute persons submitting claims for payment that are inaccurate or fraudulent, that are for services not provided as claimed, or for services that are not medically necessary. |
| · | the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, imposes criminal and civil liability for, among other things, executing or attempting to execute a scheme to defraud any healthcare benefit program, including any third party payors, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense, and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statements or representations, or making false statements relating to healthcare benefits, items or services. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it to have committed a violation; |
| · | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH, and their implementing regulations, which imposes privacy, security transmission and breach reporting obligations with respect to individually identifiable health information, including PHI, upon “covered entities” subject to the law, such as health plans, healthcare clearinghouses and certain healthcare providers, and their respective business associates that perform services on their behalf that involve individually identifiable health information, including PHI. HITECH also created new tiers of civil monetary penalties, amended HIPAA to make civil and criminal penalties directly applicable to business associates, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce HIPAA laws and seek attorneys’ fees and costs associated with pursuing federal civil actions; and |
| · | the federal Physician Payments Sunshine Act, which requires certain manufacturers of prescription drugs, devices and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program to annually report to CMS information related to payments and other transfers of value to physicians, dentists, optometrists, podiatrists, chiropractors, and teaching hospitals, and ownership and investment interests held by physicians and their immediate family members; and |
| · | analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third party payors, including private insurers; state laws that require drug companies to comply with the industry’s voluntary compliance guidelines and the applicable compliance guidance promulgated by the federal government or otherwise restrict payments that may be made to healthcare providers and other potential referral sources; state and local laws that require the licensure of sales representatives; state laws that require drug manufacturers to report information related to drug pricing or payments and other transfers of value to healthcare providers or marketing expenditures and pricing information; data privacy and security laws and regulations in foreign jurisdictions that may be more stringent than those in the United States (such as the European Union, which adopted the General Data Protection Regulation, which became effective in May 2018); state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts; and state laws related to insurance fraud in the case of claims involving private insurers. |
State and federal authorities have aggressively targeted medical technologypharmaceutical companies for alleged violations of these anti-fraud statutes,fraud and abuse laws based on improper research or consulting contracts with doctors, certain marketing arrangements that rely on volume-based pricing, off-label marketing schemes, and other improper promotional practices. Companies targeted in such prosecutions have paid substantial fines in the hundreds of millions of dollars or more, have been forced to implement extensive corrective action plans, and have often become subject to consent decrees or integrity agreements that severely restrictingrestrict the manner in which they conduct their business. If we becomeDue to the targetbreadth of these laws, the narrowness of statutory exceptions and regulatory safe harbors available, and the range of interpretations to which they are subject, it is possible that some of our current or future practices might be challenged under one or more of these laws. Responding to investigations can be time-and resource-consuming and can divert management’s attention from the business. Any such an investigation or prosecution basedsettlement could increase our costs or otherwise have an adverse effect on our contractual relationships with providersbusiness. Even an unsuccessful challenge or institutions, orinvestigation into our marketingpractices could cause adverse publicity, and promotional practices, we could face similar sanctions, which would materially harm our business.be costly to respond to.
Also, the U.S. Foreign Corrupt Practices Act and similar worldwide anti-bribery laws generally prohibit companies and their intermediaries from making improper payments to government officials for the purpose of obtaining or retaining business. Our internal control policies and procedures may not protect us from reckless or negligent acts committed by our employees, future distributors, licensees or agents. Violations of these laws, or allegations of such violations, could result in fines, penalties or prosecution and have a negative impact on our business, results of operations and reputation.
If our operations are found to be in violation of any of the healthcare laws or regulations described above or any other healthcare regulations that apply to us, we may be subject to penalties, including administrative, civil and criminal penalties, damages, fines, disgorgement, exclusion from participation in government healthcare programs, such as Medicare and Medicaid, imprisonment, additional reporting obligations and oversight if we becomes subject to a corporate integrity agreement or consent decree, reputational harm, diminished profits and future earnings, and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and pursue our strategy.
Unfavorable global economic conditions could adversely affect our business, financial condition or results of operations.
Although we believe the market for acne and rosacea therapies is less vulnerable to unfavorable economic conditions due to the significant discomfort and distress that these conditions inflict, our results of operations could be adversely affected by general conditions in the global economy and in the global financial markets. We currently have very limited visibility regarding the prospects of FMX101, FMX103 or our other product candidates becoming eligible for reimbursement by any government or third party payor and the possible scope of such reimbursement, and we must assume that demand for these product candidates may be tied to discretionary spending levels of our targeted patient population.
A severe or prolonged economic downturn could result in a variety of risks to our business, including weakened demand for FMX101, FMX103 or any of our other product candidates, if approved, and our ability to raise additional capital when needed on acceptable terms, if at all. A weak or declining economy could also strain our suppliers, possibly resulting in supply disruption, or cause our customers to delay making payments for our services.
Any of the foregoing could harm our business and we cannot anticipate all of the ways in which the current economic climate and financial market conditions could adversely impact our business.
Exchange rate fluctuations between the U.S. dollar and the New Israeli shekelShekel may negatively affect our earnings.
The dollar is our functional and reporting currency. However, a significant portion of our operating expenses are incurred in Israeli shekels.NIS. As a result, we are exposed to the risks that the shekelNIS may appreciate relative to the dollar, or, if the shekelNIS instead devalues relative to the dollar, that the inflation rate in Israel may exceed such rate of devaluation of the shekel,NIS, or that the timing of such devaluation may lag behind inflation in Israel. In any such event, the dollar cost of our operations in Israel would increase and our dollar-denominated results of operations would be adversely affected. We cannot predict any future trends in the rate of inflation or deflation in Israel or the rate of devaluation or appreciation of the shekelNIS against the dollar. For example, the rate of appreciation of the shekelNIS depreciated against the dollar by 8.1% in 2018, which depreciation was 11.6% and 1.5%partly offset by inflation at the rate of 0.8% that year, while in 2017 and 2016, respectively,the NIS appreciated 11.6% against the dollar, which appreciation was offsetfurther amplified by inflation in Israel at a rate of 0.4% in 2017 and compounded by deflation at a rate of -0.2% in 2016.that year. If the dollar cost of our operations in Israel increases, our dollar-measured results of operations will be adversely affected. Our operations also could be adversely affected if we are unable to effectively hedge against currency fluctuations in the future.
Risks Related to Our Intellectual Property
If our efforts to obtain, protect or enforce our patents and other intellectual property rights related to FMX101, FMX103 or any of our other product candidates are not adequate, we may not be able to compete effectively and we otherwise may be harmed.
Our commercial success depends in part on our ability to obtain and maintain patent protection and other intellectual property rights and to utilize trade secret protection for our intellectual property and proprietary technologies, our products and their uses, as well as our ability to operate without infringing upon the proprietary rights of others. We rely upon a combination of patents, trade secret protection and confidentiality agreements, assignment of invention agreements and other contractual arrangements to protect the intellectual property related to FMX101, FMX103 and our other development programs. Limitations on the scope of our intellectual property rights may limit our ability to prevent third parties from designing around such rights and competing against us. For example, our patents do not claim a new compound. Rather, the active pharmaceutical ingredients of our products are existing compounds and our granted patents and pending patent applications are directed to, among other things, novel formulations of these existing compounds that are dispensed as a foam. Accordingly, other parties may compete with us, for example, by independently developing or obtaining competing topical formulations that design around our patent claims, or by using formulations from expired patents but which may contain the same active ingredients, or by seeking to invalidate our patents. Moreover, any disclosure to or misappropriation by third parties of our confidential proprietary information, unless we have sufficient patent and/or trade secret protection and we are able to enforce such rights successfully, could enable competitors to quickly duplicate or surpass our technological achievements, eroding our competitive position in our market.
We currently have several granted patents related to FMX101 and FMX103 in the U.S.,United States, which are expected to remain in effect until 2030. These patents relate to a composition of matter comprising a claim to a formulation of a tetracycline antibiotic which can include minocycline or doxycycline, and therefore may be less protective than patents that claim a new drug. We also have patents granted claiming compositions of matter relating to FMX101 and FMX103 in each of the following international markets Australia, Canada, Great Britain, Israel, and Israel,Mexico and we have patent applications claiming compositions of matter relating to FMX101 and FMX103 pending in Canada, the European Union, India and Mexico.
As of December 31, 20172018, we had 165175 granted patents and over 40 patent applications pending worldwide covering our various foam-based platforms and other technology. However, the patent applications that we own or license may fail to result in granted patents in the U.S. or foreign jurisdictions, or if granted the patent claims may fail to prevent a potential infringer from marketing its product or be deemed invalid andor held unenforceable by a court. Competitors and others in the field of topically-administered therapies comprising an active ingredient in foam presentation have created a substantial amount of scientific publications, patents and patent applications and other materials relating to their technologies. Our ability to obtain and maintain valid and enforceable patents depends on various factors, including interpretation of our technology and the prior art and whether the differences between them allow our technology to be patentable. Patent applications and patents granted from them are complex, lengthy and highly technical documents that are often prepared under very limited time constraints and may not be free from errors or words that make their interpretation uncertain. The existence of errors in a patent may have a materially adverse effect on the patent, its scope and its enforceability. Our pending patent applications may not issue, andor the scope of the claims of patent applications that do issue may be too narrow or inadequate to protect our competitive advantage. Also, our granted patents may be subject to challenges or construed in a way that may not provide adequate protection.
Even if these patents do successfully issue, third parties may challenge the validity, enforceability or scope of such granted patents or any other granted patents we own or license, which may result in such patents being narrowed, invalidated or held unenforceable. For example, patents granted by the European Patent Office may be opposed by any person within 9 months from the publication of their grant. Also, patents granted by the U.S. Patent and Trademark Office, or USPTO, may be subject to review, reexamination and other challenges. Changes to the patent laws of the U.S. in 2012 provide additional procedures for third parties to challenge the validity of patents issuing from patent applications including post-grant review, which generally applies to patents first filed after March 15, 2013. A post-grant review petition must be filed on or prior to the date which is 9 months after the patent is granted. The procedures also expand and reform the proceedings for challenging issued patents on grounds of prior art and publications, also known as inter partes review, or IPR. For patents filed after March 15, 2013, a petition for IPR may be filed the later of 9 months after grant of the patent or after a post-grant review proceeding on the patent has terminated. For patents filed prior to March 15, 2013, the rules regarding IPR filing remain unchanged and an IPR petition may be filed any time following issuance of the patent.
Furthermore, efforts to enforce our patents could give rise to challenges to their validity or unenforceability in court proceedings. If the patents and patent applications we hold or pursue with respect to FMX101, FMX103 or any of our other product candidates are challenged, it could put one or more patents at risk of being invalidated, or interpreted narrowly and threaten our competitive advantage for FMX101, FMX103 or any of our other product candidates. Furthermore, even if they are not challenged, our patents and patent applications may not adequately protect our intellectual property or prevent others from designing around our claims. To meet such challenges, which are part of the risks and uncertainties of developing and marketing product candidates, we may need to evaluate third party intellectual property rights and, if appropriate, to seek licenses for such third party intellectual property or to challenge such third party intellectual property, which may be costly and may or may not be successful, which could also have a material adverse effect on the commercial potential for FMX101, FMX103 and any of our other product candidates.
Further, if we encounter delays in our clinical trials or in seeking marketing approval of our products, the period of time during which we could market FMX101, FMX103 or any of our other product candidates under patent protection could be reduced.
Since patent applications in the U.S.United States and most other countries are confidential for a period of time after filing, we cannot be certain that we were the first to (i) file any patent application related to FMX101, FMX103 or any of our other product candidates or (ii) conceive and invent any of the inventions claimed in our patents or patent applications.
Furthermore, for applications filed before March 16, 2013, or patents issuing from such applications, an interference proceeding can be invoked by a third party, or instituted by the USPTO, to determine who was the first to invent any of the subject matter covered by the patent claims of our applications and patents. As of March 16, 2013, the U.S.United States transitioned to a “first-to-file”“first-to-file” system for deciding which party should be granted a patent when two or more patent applications are filed by different parties claiming the same invention. A third party that files a patent application in the USPTO under the new first-to-file system before we did, could therefore be awarded a patent covering an invention of ours even if we had made the invention before it was made by the third party.
The change to “first-to-file”“first-to-file” from “first-to-invent”“first-to-invent” is one of the changes to the patent laws of the U.S.United States. resulting from the Leahy-Smith America Invents Act, or AIA, signed into law on September 16, 2011. Among some of the other changes to the patent laws are changes that limit where a patentee may file a patent infringement suit and providing opportunities for third parties to challenge any issued patent in the USPTO. Because ofUntil recently, a lower evidentiary standard was applied in certain USPTO proceedings compared to the evidentiary standard in U.S. federal court necessary to invalidate a patent claim, a third party could potentially provide evidence in a USPTO proceeding sufficientclaim. Under the new final rule, effective for petitions filed on or after November 13, 2018, the USPTO PTAB is to hold a claim invalid even thoughapply the same evidenceclaim construction standard applied by civil courts under 35 USC §282(b) in IPR, post-grant review, and the transitional program for covered business method patents proceedings. The impact this may be insufficient to invalidatehave in practice on the claim if first presented in a district court action. Accordingly, a third party may attempt to use and outcome of USPTO proceedings is uncertain. Because of lower costs and the USPTO procedures to invalidate our patent claimsfact that would not have been invalidated if first challenged by the third party as a defendant in a district court action. USPTO statistics indicate that a high rate of challenged claims are being invalidated in these USPTO procedures.procedures, they may continue to be a popular and effective means of challenging patents.
Even where patent, trade secret and other intellectual property laws provide protection, costly and time-consuming litigation could be necessary to enforce and determine the scope of our proprietary rights, and the outcome of such litigation would be uncertain. Moreover, any actions we may bring to enforce our intellectual property against our competitors could provoke them to bring counterclaims against us, and our competitors have intellectual property portfolios of their own, some of which are substantial.include substantial patent portfolios. An unfavorable outcome could have a material adverse effect on our business and could result in the challenged patent or one or more of its claims being interpreted narrowly or invalidated, or held not to be infringed, or one or more of our patent applications may be not be granted.
We also rely on trade secret protection and confidentiality agreements to protect our know-how, data and information prior to filing patent applications and during the period before they are published. We additionally rely on trade secret protection and confidentiality agreements to protect proprietary know-how that we consider may be preferably maintained as a trade secret rather than the subject of a patent application. We further rely on trade secret protection and confidentiality agreements to protect proprietary know-how that may not be patentable, processes for which patents may be difficult to obtain or enforce and other elements of our product development processes that involve proprietary know-how, information or technology that is not covered by patents.
In an effort to protect our trade secrets and other confidential information, we incorporate confidentiality provisions in all our employees’employees’ agreements and require our consultants, contractors and licensees to which we disclose such information to execute confidentiality agreements upon the commencement of their relationships with us. These agreements require that confidential information, as defined in the agreement and disclosed to the individual by us during the course of the individual’sindividual’s relationship with us, be kept confidential and not disclosed to third parties for an agreed term. These agreements, however, may not provide us with adequate protection against accidental or improper use or disclosure of confidential information, and these agreements may be breached. Adequate remedies may not exist in the event of unauthorized use or disclosure of our confidential information. A breach of confidentiality could significantly affect our competitive position and we could lose our trade secrets or they could become otherwise known or be independently discovered by our competitors. Also, to the extent that our employees, consultants or contractors use any intellectual property owned by others in their work for us, disputes may arise as to the rights in any related or resulting know-how and inventions. Additionally, others may independently develop the same or substantially equivalent proprietary information and techniques or otherwise gain access to our trade secrets and other confidential information. Any of the foregoing could deteriorate our competitive advantages, undermine the trade secret and contractual protections afforded to our confidential information and have material adverse effects on our business.
Cybersecurity disruptions may impact our business operations if it becomes a target for such activities.
We may be subject to attempted cybersecurity disruptions from a variety of threat actors. If our systems for protecting against cybersecurity disruptions prove to be insufficient, our employees or third parties could be adversely affected. Such cybersecurity disruptions could cause damage or destroy assets, compromise business systems, result in proprietary information and trade secrets being altered, lost or stolen; result in employee or third-party information being compromised, or otherwise disrupt business operations. Significant costs to remedy the effects of such a cybersecurity disruption may be incurred by us, as well as in connection with resulting regulatory actions and litigation, and such disruption may harm relationships with third parties and impact our business reputation.
Changes in U.S. or foreign patent law could diminish the value of patents in general, thereby impairing our ability to protect our products.
As is the case with other companies in the markets in which we participate, our success is heavily dependent on intellectual property, particularly patents. The strength of patents in the pharmaceutical field involves complex legal and scientific questions and in the U.S.United States and many foreign jurisdictions patent policy and case law also continues to evolve and change and the issuance, scope, validity, enforceability and commercial value of our patent rights are highly uncertain. This uncertainty includes changes to the patent laws through eitherone or more of legislative action to change statutory patent law, rule changes and practice directions issued by National Patent Offices, or court action that may reinterpret or expand on existing law in ways affecting the scope or validity of granted patents, or both.patents. Particularly in recent years in the U.S.,United States, there have been several major legislative developments and court decisions that have affected patent laws in significant ways and there may be more developments in the future that may weaken or undermine our ability to obtain patents or to enforce our existing and future patents.
We have agreed to share ownership in certain patents that may result from our development and license agreements with certain major pharmaceutical companies, which may detract from our rights to such patents.
We have agreed with several of the pharmaceutical companies with whom we are developing certain topical products, based on our foam technology and the licensees’licensees’ active ingredients, to jointly own and have an undivided interest in patents that arise from the relevant projects, where the licensee made its own material contributions to the invention. In certain agreements, we have further agreed that inventions achieved exclusively or primarily by the licensees in the course of the development without significant contribution by us will be owned solely by them, and they will be allowed to file patent applications covering such inventions without our participation.
We have granted certain licensees the right to provide input during the prosecution of licensed patent applications. We have further granted certain licensees the primary right to enforce several of our existing patents, which we have licensed to these licensees to allow them to commercialize or continue to commercialize our jointly-developed product, in the event that any infringement of the licensed patents adversely affects the licensees’licensees’ ability to utilize the licenses for the purpose they were granted. Such rights may detract from our rights and title to such patents.patents and we may as a result have little or no control or say in any proceedings concerning them. In addition, any negative proceedings against our technology could impact any or all of our licensees, and we may be considered or found to be contractually responsible for the payment of certain claims and losses as a result of such impact.
If we infringe or are alleged to infringe or otherwise violate intellectual property rights of third parties, our business could be harmed.
Our research, development and commercialization activities may infringe or otherwise violate or be claimed to infringe or otherwise violate patents owned or controlled by other parties. Competitors in the field of topical and oral drugs for the treatment of acne impetigo,or rosacea chemotherapy-induced rash and other indications have developed large portfolios of patents and patent applications relating to our business. In particular, there are patents held by third parties that relate to theformulations with minocycline-based and doxycycline-based products and to methods of treatment with minocycline-based and doxycycline-based products for indications we are pursuing with our product candidates, namely FMX101, FMX103 and any other of our other product candidates. There may be granted patents that could be asserted against us in relation to such product candidates. There may also be granted patents held by third parties that may be infringed or otherwise violated by our other product candidates and activities, and we do not know whether or to what extent we aremay be infringing or otherwise violating third party patents. There may also be third party patent applications that if approved and granted as patents may be asserted against us in relation to FMX101, FMX103 or any of our other product candidates or activities. These third parties could bring claims against us that would cause us to incur substantial expenses and, if successful against us, could cause us to pay substantial damages and legal fees. Further, if a patent infringement suit were brought against us, we could be temporarily or permanently enjoined or otherwise forced to stop or delay research, development, manufacturing or sales of the product candidate that is the subject of the suit.
As a result of patent infringement claims, or to avoid potential claims, we may choose or be required to seek licenses from third parties. These licenses may not be available on acceptable terms, or at all. Even if we are able to obtain a license, the license would likely obligate us to pay license fees or royalties or both, and the rights granted to us might be nonexclusive, which could result in our competitors gaining access to the same intellectual property, or such rights might be restrictive and limit our present and future activities. Ultimately, we or a licensee could be prevented from commercializing a product, or be forced to cease some aspect of our business operations, if, as a result of actual or threatened patent infringement claims, we are unable to enter into licenses on acceptable terms.
There has been and there currently is substantial litigation and other proceedings regarding patent and other intellectual property rights in the pharmaceutical industry. In addition to possible infringement claims against us, we may become a party to other patent litigation and other proceedings, including interference, derivation, review, re-examination or other post-grant proceedings declared or granted by the USPTO and similar proceedings in foreign countries, regarding intellectual property rights with respect to our current or any future products. The cost and burden to us of any patent litigation or other proceeding, even if resolved in our favor, could be substantial. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their substantially greater financial resources. Patent litigation and other proceedings may also absorb significant management time. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings and their outcome could impair our ability to compete in the marketplace and impose a substantial financial burden on us. The occurrence of any of the foregoing could have a material adverse effect on our business, financial condition or results of operations.
Furthermore, several of our employees were previously employed at universities or other pharmaceutical companies, including potential competitors. While we take steps to prevent our employees from using the proprietary information or know-how of others that is not in the public domain or that has not already been independently developed by us earlier, we may be subject to claims that we or these employees have inadvertently or otherwise used or disclosed intellectual property, trade secrets or other proprietary information of any such employee’semployee’s former employer. Litigation may be necessary to defend against these claims and, even if we are successful in defending ourselves, could result in substantial costs to us or be distracting to our management. If we do not succeed with respect to any such claims, in addition to paying monetary damages and possible ongoing royalties, we may lose valuable intellectual property rights or personnel.
If we are unable to protect our trademarks from infringement, our business prospects may be harmed.
We own trademarks that identify “Foamix”,“Foamix” and have registered these trademarks in the U.S.United States and Israel. We also own and have filed applications for trademarks in the United States that represent the proposed commercial name(s) of our drug product candidates. Although we take steps to monitor the possible infringement or misuse of our trademarks, it is possible that third parties may infringe, dilute or otherwise violate our trademark rights. Any unauthorized use of our trademarks could harm our reputation or commercial interests. In addition, our enforcement against third-partythird party infringers or violators may be unduly expensive and time-consuming, and the outcome may be an inadequate remedy.
We may become involved in lawsuits to protect or enforce our patents or other intellectual property or the patents of our licensors, which could be expensive and time-consuming.
Competitors may infringe our intellectual property, including our patents or the patents of our licensors. As a result, we may be required to file infringement claims to stop third party infringement or unauthorized use. This can be expensive and burdensome, particularly for a company of our size, andas well as time-consuming. In addition, in an infringement proceeding, a court may decide that a patent of ours is not valid or is unenforceable, or may refuse to stop the other party from using the technology or method at issue on the grounds that our patent claims do not cover its technology or method or that the factors necessary to grant an injunction against an infringer are not satisfied.
We have received notice letters of ANDAs submitted for drug products that are generic versions of Finacea Foamfoam and we are involved in lawsuits to protect orand enforce our patents, which could beare expensive, time consuming and may be unsuccessful.
Competitors may infringe our patents. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time consuming. The infringing party may deny any infringement or challenge the patents as invalid or unenforceable. Litigation proceedings may also fail, and even if successful, they may result in substantial costs and distraction of our management and other employees.
For example, we and Bayer have received Paragraph IV Certification Notice Letters from each of Teva and Perrigo, were received dated November 20, 2017 and January 4, 2018, respectively in connection with Bayer’s Finacea Foam.Finacea. A Paragraph IV Certification Notice Letter notifies the FDA that one or more patents listed in the FDA’sFDA’s Orange Book is allegedly invalid, unenforceable or will not be infringed by an ANDA product. In the case at hand, boththe letters were directed against several of our U.S. patents and stated that each of Teva and Perrigo had submitted to the FDA an ANDA for an azelaic acid foam composition which is a generic version of Finacea Foam. We and Bayer responded byFinacea.
Complaints were jointly filing complaintsfiled against each of Teva and Perrigo with the U.S. District Court for the District of Delaware, filed January 4, 2018 and February 15, 2018, respectively, asserting, among other things, that each of Teva and Perrigo had infringed our patents, as listed in their Paragraph IV Notice Letters, by seeking FDA approval to manufacture and sell a generic version of Bayer’s Finacea Foam prior to expiration of these patents. Since the complaints were filed within the 45-day period required under the Hatch WaxmanHatch-Waxman Act, we were granted a 30-month stay which precludes a genericTeva from receiving final FDA approval of a generic version of Finacea Foam prior to May 2020.2020 and Perrigo from receiving final FDA approval of a generic version of Finacea prior to July 2020, unless a court enters a judgment that the patents are invalid or not infringed.
We and LEO have also recently received Paragraph IV Certification Notice Letters from Taro, dated December 20, 2018 and January 18, 2019, in connection with Finacea, stating Taro had submitted to the FDA an ANDA for an azelaic acid foam composition which is a generic version of Finacea. A complaint has been filed within the 45- day period, and a 30-month stay will preclude Taro from receiving final FDA approval of a generic version of Finacea prior to June 2021, unless a court enters judgment that the patents are invalid or not infringed. These proceedings should proceed separately from those with Teva and Perrigo.
Pursuant to our licensing agreement with Bayer,LEO, this litigation is in the sole control of Bayer,LEO and we are currently unable to predict its outcome. Potentially,The substitution of Finacea Foam in favor of generic versions is likelyhas the potential to have a negative impact on future commercialization of Finacea Foam and to result in a loss of license revenue. Furthermore, in any infringement proceeding, including the foregoing, a court may decide that a patent of ours, or one or more claims of such patent, is not valid or is unenforceable, or may refuse to stop the other party from using the supposedly infringing technology on the grounds that our patents, or one or more claims of such patents, do not cover such technology. An adverse result in any litigation or defense proceedings could put one or more of our patents at risk of being invalidated or interpreted narrowly and could also put one or more of our pending patent applications at risk of not issuing. There can be no assurance that our product candidates will not be subject to the same risks.
41We have also entered into license agreements with other commercial partners for the development and commercialization of products with active ingredients other than azelaic acid that license one or more of the patents listed in the FDA’s Orange Book for Finacea. Whilst these license agreements are not considered material to our main business, an adverse result may put at risk the development and commercialization of any of these licensed products.
An adverse determination of any litigation or other proceedings could put one or more of our patents at risk of being invalidated or interpreted narrowly and could put our patent applications at risk of not issuing.
Interference, derivation review, or other proceedings brought at the USPTO may be necessary to determine the priority or patentability of inventions with respect to our patent applications or those of our licensors or licensees. Litigation or USPTO proceedings brought by us may fail or may be invoked against us by third parties. Even if we are successful, domestic or foreign, litigation or USPTO or foreign patent office proceedings, may result in substantial costs and distraction to our management. Moreover, proceedings may be appealed and obtaining a final resolution can take a long time and substantial resources. We may not be able, alone or with our licensors or licensees, to prevent misappropriation of our proprietary rights, particularly in countries where the laws may not protect such rights as fully as in the U.S.
Furthermore, because of the substantial amount and extent of discovery required in connection with intellectual property litigation or other proceedings, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation or proceedings. In addition, during the course of this kind of litigation or proceedings, there could be public announcements of the results of hearings, motions or other interim proceedings or developments or public access to related documents. If investors perceive these results to be negative, the market price for our ordinary shares could be significantly harmed.
We may not obtain intellectual property rights or otherwise be able to protect our intellectual property rights throughout the world.
Filing, prosecuting and defending patents on product candidates in all or most countries throughout the world would be prohibitively expensive. We primarily file patent applications in the U.S.,United States, and may file in some other selected jurisdictions on a case-by-case basis. As a result, our intellectual property rights in countries outside the U.S.United States. are generally significantly less extensive than those in the U.S.United States. In addition, the laws of some foreign countries and jurisdictions, particularly of certain developing countries and jurisdictions, do not protect intellectual property rights to the same extent as federal and state laws in the U.S.,United States, and these countries and jurisdictions may limit the scope of what can be claimed, and in some cases may even force us to grant a compulsory license to competitors or other third parties. Consequently, we may not be able to prevent third parties from practicing our inventions outside the U.S.,United States, or from selling or importing products made using our inventions in and into the U.S.United States or other jurisdictions. Competitors may use our technologies in jurisdictions where we have not sought or obtained patent protection to develop their own products and further, may export otherwise infringing products to territories where we have patent protection, but protection and enforcement is not as strong as that in the U.S.United States. These products may compete with our products and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing. Moreover, competitors or others may raise legal challenges to our intellectual property rights or may infringe upon our intellectual property rights, including through means that may be difficult to prevent or detect.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, especially those relating to pharmaceuticals, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly and our patent applications at risk of not issuing and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded, if any, may not be commercially meaningful. Further, third parties may prevail in their claims against us, which could potentially result in the award of injunctions or substantial damages against us. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
In addition, our ability to protect and enforce our intellectual property rights may be adversely affected by unforeseen changes in domestic and foreign intellectual property laws and practice. We have received notices of opposition to our European patent no. 1556009
Two oppositions were filed against the grant of our European patent EP1556009 entitled “Cosmetic and Pharmaceutical Foam”, which relates to an alcohol-free foamable pharmaceutical or cosmetic carrier and its use. The oppositions were filed by Guderma GmbH and Henkel AG & Co. KGaA. We defended the patent at oral proceedings on January 23, 2017 before the Opposition Division of the European Patent Office, or EPO, and the outcome was a favorable interlocutory decision to maintain the patent in amended form. Both of the unsuccessful opponents filed an appeal against this decision, and in response we have restated our position in support of the patent. We await the next communication from the EPO, which is likely to be a summons to oral proceedings at which the outcome of the appeal will be decided. The opposition and appeal documents are available at the online EPO file for the patent. Although the patent involved is believed by Foamix to be of no material interest to our lead product candidates, it may be of interest in relation to one or more of our licensed products. We are unable to predict the outcome of the appeal, which might result in the patent being revoked, but note that it may potentially have a negative impact on the future commercialization of one or more licensed products.
Under applicable employment laws, we may not be able to enforce covenants not to compete.
We generally enter into non-competition agreements as part of our employment agreements with our employees. These agreements generally prohibit our employees, if they cease working for us, from competing directly with us or working for our competitors or clients for a limited period. We may be unable to enforce these agreements under the laws of the jurisdictions in which our employees work and it may be difficult for us to restrict our competitors from benefitting from the expertise our former employees or consultants developed while working for us.
For example, Israeli labor courts place emphasis on freedom of employment and have required employers seeking to enforce non-compete undertakings of a former employee to demonstrate that the competitive activities of the former employee will harm one of a limited number of material interests of the employer which have been recognized by the courts, such as the protection of a company’scompany’s trade secrets or other intellectual property.
Risks Related to Our Ordinary Shares
We do not know whether a market for our ordinary shares will be sustained and as a result it may be difficult for holders of our ordinary shares to sell their shares.
Although our ordinary shares are quoted on the NASDAQNasdaq Global Market, an active trading market for our shares may not be sustained. The lack of an active market may impair the ability of holders of our ordinary shares ability to sell their shares at the time they wish to sell them or at a price that they consider reasonable. The lack of an active market may also reduce the fair market value of our shares, and may cause the trading price of our ordinary shares to be more volatile. An inactive market may also impair our ability to raise capital by selling shares and may impair our ability to acquire other companies by using our shares as consideration.
The market price of our ordinary shares may be subject to fluctuation and holders of our ordinary shares could lose all or part of their investment.
The stock market in general and the market price of our ordinary shares in particular has been, and will likely continue to be, subject to fluctuation, whether due to, or irrespective of, our operating results and financial condition. For example, the price of our shares plummetedfell by more than 40% in a single trading day on March 27, 2017, following the announcement of somewhat mixed top-line results from our first two pivotal Phase III clinical trials for our lead product candidate FMX101 and then increased by more than 40% on September 11, 2018, following the announcement of successful results from our third Phase III clinical for treatment of moderate-to-severe acne.FMX101. The market price of our ordinary shares on the NASDAQNasdaq Global Market may fluctuate as a result of a number of factors, some of which are beyond our control, including, but not limited to:
| · | actual or anticipated variations in our and our competitors’competitors’ results of operations and financial condition; |
| · | market acceptance of our products; |
| · | the mix of products that we sell and related services that we provide; |
| · | the success or failure of our licensees to develop, obtain approval for and commercialize our licensed products, for which we are entitled to contingent payments and royalties; |
| · | changes in earnings estimates or recommendations by securities analysts, if our ordinary shares are covered by analysts; |
| · | development of technological innovations or new competitive products by others; |
| · | announcements of technological innovations or new products by us; |
| · | publication of the results of preclinical or clinical trials for FMX101, FMX103 or our other product candidates; |
| · | failure by us to achieve a publicly announced milestone; |
| · | delays between our expenditures to develop and market new or enhanced products and the generation of sales from those products; |
| · | Thethe filing of ANDAs by generic companies seeking approval to market generic versions of our products and of our licensee’slicensee’s products; |
| · | developments concerning intellectual property rights, including our involvement in litigation brought by or against us, including patent infringement proceedings before national and state courts, and patent opposition and review proceedings before national patent offices; |
| · | regulatory developments and the decisions of regulatory authorities as to the approval or rejection of new or modified products; |
| · | changes in the amounts that we raise in financings or collaboration transactions and spend to develop, acquire or license new products, technologies or businesses; |
| · | changes in our expenditures to promote our products; |
| · | our sale or proposed sale, or the sale by our significant shareholders, of our ordinary shares or other securities in the future; |
| · | changes in key personnel; |
| · | success or failure of our research and development projects or those of our competitors; |
| · | the trading volume of our ordinary shares; and |
| · | general economic and market conditions and other factors, including factors unrelated to our operating performance. |
These factors and any corresponding price fluctuations may materially and adversely affect the market price of our ordinary shares and result in substantial losses being incurred by our investors. In the past, following periods of market volatility, public company shareholders have often instituted securities class action litigation. If we were involved in securities litigation, it could impose a substantial cost upon us and divert the resources and attention of our management from our business.
If equity research analysts do not publish research or reports about our business or if they issue unfavorable commentary or downgrade our ordinary shares, the price of our ordinary shares could decline.
The trading market for our ordinary shares relies in part on the research and reports that equity research analysts publish about us and our business, if at all. We do not have control over these analysts and we do not have commitments from them to write research reports about us. The price of our ordinary shares could decline if no research reports are published about us or our business, or if one or more equity research analysts downgrades our ordinary shares or if those analysts issue other unfavorable commentary or cease publishing reports about us or our business.
Future sales of our ordinary shares could reduce the market price of our ordinary shares.
If our shareholders, particularly our directors and their affiliates or our executive officers, sell a substantial number of our ordinary shares in the public market, the market price of our ordinary shares could decrease significantly. The perception in the public market that our shareholders might sell our ordinary shares could also depress the market price of our ordinary shares and could impair our future ability to obtain capital, especially through an offering of equity securities. In addition, the perception in the public market that we will need to issue new shares to raise capital and our sale of additional ordinary shares or similar securities in order to raise capital might have a similar negative impact on the share price of our ordinary shares. A decline in the price of our ordinary shares might impede our ability to raise capital through the issuance of additional ordinary shares or other equity securities, and may cause holders of our ordinary shares to lose part or all of their investment.
The significant share ownership position of affiliates of our co-founders, Dr. Dov Tamarkin and Meir Eini, may limit your ability to influence corporate matters.50
Tamarkin Medical Innovations Ltd., a company beneficially owned by Dr. Dov Tamarkin, or Tamarkin, our co-founder, beneficially owns or controls, directly or indirectly, 7.0% of our outstanding ordinary shares, and Meir Eini Holdings Ltd., a company beneficially owned by Meir Eini, or Eini, our other co-founder, beneficially owns or controls, directly or indirectly, 7.7% of our outstanding ordinary shares, as of December 31, 2017. Accordingly, Tamarkin and Eini are able to significantly influence, though not independently determine, the outcome of matters required to be submitted to our shareholders for approval, including decisions relating to the election of our board of directors and the outcome of any proposed merger or consolidation of our company. Tamarkin’s and Eini’s interests may not be consistent with those of our other shareholders. In addition, Tamarkin’s and Eini’s significant interest in us may discourage third parties from seeking to acquire control of us, which may adversely affect the market price of our ordinary shares.
We have never paid cash dividends on our share capital, and we do not anticipate paying any cash dividends in the foreseeable future.
We have never declared or paid cash dividends on our share capital, nor do we anticipate paying any cash dividends on our share capital in the foreseeable future. We currently intend to retain all available funds and any future earnings to fund the development and growth of our business. As a result, capital appreciation, if any, of our ordinary shares will be investors’investors’ sole source of gain for the foreseeable future. In addition, Israeli law limits our ability to declare and pay dividends, and may subject our dividends to Israeli withholding taxes. As of January 1, 2018 we are required to report as a U.S. domestic issuer and the benefits of a “foreign private issuer” are no longer available to us, which will likely result in additional costs and expenses for us.
As of January 1, 2018 we have lost our status as a “foreign private issuer” and are required to adjust our disclosure and reporting to comply with the requirements for domestic U.S. companies, given that more than 50% of our outstanding voting securities were owned by residents of the U.S. and more than 50% of our executive officers were U.S. citizens or residents as of June 30, 2017. As a result:
| · | we are required to report on forms that are applicable to U.S. companies, such as Forms 10-K, 10-Q and 8-K, rather than the forms formerly used us, such as Forms 20-F and 6-K; |
| · | we are required to include substantially more information in proxy statements than previously provided; |
| · | we can no longer make use of the shelf registration statement on Form F-3 that was declared effective on March 14, 2017, and will need to file a new registration statement on the relevant form applicable to domestic issuers should we wish to engage in capital raising activities; |
| · | if we engage in capital raising activities, there is a higher likelihood that investors may require us to file resale registration statements with the SEC as a condition to any such financing; and |
| · | we may be required to modify certain of our policies to comply with accepted governance practices associated with U.S. domestic issuers. |
We expect that complying with these additional requirements would increase our legal and audit fees which in turn, could have a material adverse effect on our business, financial condition and results of operations. In addition, as a result of being considered a “domestic issuer” for reporting and disclosure requirements:
| · | we are no longer exempt from certain of the provisions of U.S. securities laws such as (i) Regulation FD, which restricts the selective disclosure of material information, (ii) exemptions for filing beneficial ownership reports under Section 16(a) of the Exchange Act for executive officers, directors and 10% shareholders (Forms 3, 4, and 5), and (iii) the Section 16(b) short swing profit rules; |
| · | we are no longer permitted to disclose compensation information for our executive officers on an aggregate rather than an individual basis, although such exemption may still be available to us as long as we remain an “emerging growth company”; and |
| · | we have lost the ability to rely upon exemptions from NASDAQ corporate governance requirements that are available to foreign private issuers. |
As a foreign private issuer, we are permitted to follow, and had followed through December 31, 2017, certain home country corporate governance practices instead of those otherwise required under the NASDAQ Stock Market for domestic U.S. issuers. For instance, we followed home country practice in Israel with regard to (a) the quorum requirement for shareholder meetings, (b) the lack of need for independent director oversight of director nominations and for a nominating and governance committee; (c) the lack of need for separate executive sessions of independent directors and non-management directors; and (d) the lack of need to obtain shareholder approval for certain dilutive events such as (i) the establishment or amendment of certain equity-based compensation plans and (ii) certain transactions other than a public offering involving issuances of a 20% or more interest in the company.
We are an “emerging growth company” and a “smaller reporting company” and the reduced disclosure requirements applicable to emerging growth companies and smaller reporting companies may make our ordinary shares less attractive to investors. We are an “emerging“emerging growth company,” as defined in the JOBS Act, and we may take advantage of certain exemptions from various requirements that are applicable to other public companies that are not “emerging“emerging growth companies.” Most of such requirements relate to disclosures that we would otherwise be required to make, having ceased to be a foreign private issuer.make. For example, as an emerging growth company, and despite no longer being a foreign private issuer, we will not be required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act for up to five fiscal years after the date of our initial public offering. We will remain an emerging growth company until the earliest of: (i) the last day of our fiscal year during which we have total annual gross revenues of at least $1.0$1.07 billion; (ii) the last day of our fiscal year following the fifth anniversary of the closing of our initial public offering;offering, specifically, December 31, 2019; (iii) the date on which we have, during the previous three-year period, issued more than $1.0 billion in non-convertible debt; or (iv) the date on which we are deemed to be a “large“large accelerated filer”filer” under the Exchange Act.
We are also currently a “smaller reporting company,” as defined in Rule 12b-2 of the Exchange Act. In the event that we are still considered a smaller reporting company when we cease being an emerging growth company, the disclosure we will be required to provide in our SEC filings will still be less than it would be if we were not considered either an emerging growth company or a smaller reporting company. For example, “smaller reporting companies” are able to provide simplified executive compensation disclosures in their filings and have certain other decreased disclosure obligations in their SEC filings, including, among other things, being required to provide only two years of audited financial statements in annual reports.
When we are no longer deemed to be an emerging growth company or a smaller reporting company, we will not be entitled to the exemptions provideddiscussed above. Decreased disclosures in the JOBS Act discussed above.our SEC filings due to our status as an emerging growth company or a smaller reporting company may make it harder for investors to analyze our results of operations and financial prospects. We cannot predict if investors will find our ordinary shares less attractive as a result of our reliance on exemptions under the JOBS Act.these exemptions. If some investors find our ordinary shares less attractive as a result, there may be a less active trading market for our ordinary shares and our share price may be more volatile.
Our U.S. shareholders may suffer adverse tax consequences if we are characterized as a passive foreign investment company.
Generally, if, for any taxable year, 75% or more of our gross income is passive income, or at least 50% of the average quarterly value of our assets (which may be determined in part by the market value of our ordinary shares, which is subject to change) are held for the production of, or produce, passive income, we would be characterized as a passive foreign investment company, or PFIC, for U.S. federal income tax purposes.
Based on certain estimates of our gross income and gross assets, our use of proceeds of our initial public offering, and the nature of our business, we believe that we were not classified as a PFIC for the taxable year ended December 31, 2017,2018, and do not anticipate being classified as a PFIC for the taxable year ending December 31, 2018.2019. Because we currently hold, and expect to continue to hold, a substantial amount of cash and cash equivalents and other passive assets used in our business, and because the value of our gross assets is likely to be determined in large part by reference to our market capitalization, a decline in the value of our ordinary shares may result in our becoming a PFIC. Accordingly, we may be considered a PFIC for any taxable year.
If we are characterized as a PFIC, our U.S. shareholders may suffer adverse tax consequences, including having gains realized on the sale of our ordinary shares treated as ordinary income, rather than as capital gain, the loss of the preferential rate applicable to dividends received on our ordinary shares by individuals who are U.S. Holders (as defined in “Item 10.E. Taxation—U.S.the section titled “U.S. Federal Income Tax Consequences”Considerations” in our annual reportmost recent prospectus supplement filed pursuant to Rule 424(b)(2) on September 14, 2018 under our shelf registration statement on Form 20-F for the year 2016, filed with the SEC on February 21, 2017)S-3 (registration statement no. 333-224084, declared effective April 12, 2018), and having interest charges apply to distributions by us and the proceeds of share sales. Certain elections exist that may alleviate some of the adverse consequences of PFIC status and would result in an alternative treatment (such as mark-to-market treatment) of our ordinary shares; however, we do not intend to provide the information necessary for U.S. holders to make qualified electing fund elections if we are classified as a PFIC. See also “Item 10.E. Taxation—
If a United States person is treated as owning at least 10% of our common shares, such holder may be subject to adverse U.S. federal income tax consequences.
If a U.S. Holder (as defined in the section titled “U.S. Federal Income Tax Consequences—Passive Foreign Investment Company Considerations” in our annual reportmost recent prospectus supplement filed pursuant to Rule 424(b)(2) on September 14, 2018 under our shelf registration statement on Form 20-FS-3 (registration statement no. 333-224084, declared effective April 12, 2018) is treated as owning (directly, indirectly or constructively) at least 10% of the value or voting power of our common shares, such U.S. Holder may be treated as a “United States shareholder” with respect to each “controlled foreign corporation” in our group. Because our group includes at least one U.S. subsidiary, if we were to form or acquire any non-U.S. subsidiaries in the future, they may be treated as controlled foreign corporations. A United States shareholder of a controlled foreign corporation may be required to annually report and include in its U.S. taxable income its pro rata share of “Subpart F income,” “global intangible low-taxed income” and investments in U.S. property by that controlled foreign corporation, regardless of whether that controlled foreign corporation, or we, make any distributions. An individual that is a United States shareholder with respect to a controlled foreign corporation generally would not be allowed certain tax deductions or foreign tax credits that would be allowed to a United States shareholder that is a U.S. corporation. We cannot provide any assurances that we will assist investors in determining whether any non-U.S. subsidiaries that we may form or acquire in the future will be treated as controlled foreign corporations or whether any such investor would be treated as a United States shareholder with respect to any of such controlled foreign corporations. Further, we cannot provide any assurances that we will furnish to any investor information that may be necessary to comply with the reporting and tax paying obligations discussed above. Failure to comply with these reporting obligations may subject a U.S. Holder to significant monetary penalties and may extend the statute of limitations with respect to its U.S. federal income tax return for the year 2016, filedfor which reporting was due. U.S. Holders should consult their tax advisors regarding the potential application of these rules to their investment in our common shares.
Future changes to tax laws could materially adversely affect our company and reduce net returns to our shareholders.
Our tax treatment is subject to the enactment of, or changes in, tax laws, regulations and treaties, or the interpretation thereof, tax policy initiatives and reforms under consideration and the practices of tax authorities in jurisdictions in which we operate. Such changes may include (but are not limited to) the taxation of operating income, investment income, dividends received or (in the specific context of withholding tax) dividends paid. We are unable to predict what tax reform may be proposed or enacted in the future or what effect such changes would have on our business, but such changes, to the extent they are brought into tax legislation, regulations, policies or practices, could affect our financial position and overall or effective tax rates in the future in countries where we have operations, reduce post-tax returns to our shareholders, and increase the complexity, burden and cost of tax compliance.
In addition, on December 22, 2017, U.S. federal tax legislation popularly known as the Tax Cuts and Jobs Act was signed into law, which significantly revised the Internal Revenue Code of 1986, as amended. The overall impact of the Tax Cuts and Jobs Act is uncertain and our business and financial condition could be adversely affected by it or as a result of interpretations thereof. It is uncertain if and to what extent various states will conform to the Tax Cuts and Jobs Act. The impact of this tax reform on holders of our common shares is also uncertain and could be adverse.
Tax authorities may disagree with our positions and conclusions regarding certain tax positions, resulting in unanticipated costs, taxes or non-realization of expected benefits.
A tax authority may disagree with tax positions that we have taken, which could result in increased tax liabilities. For example, the SEC on February 21, 2017.U.S. Internal Revenue Service or another tax authority could challenge our allocation of income by tax jurisdiction and the amounts paid between our affiliated companies pursuant to our intercompany arrangements and transfer pricing policies. Similarly, a tax authority could assert that we are subject to tax in a jurisdiction where we believe we have not established a taxable connection, often referred to as a ‘‘permanent establishment’’ under international tax treaties, and such an assertion, if successful, could increase our expected tax liability in one or more jurisdictions. A tax authority may take the position that material income tax liabilities, interest and penalties are payable by us, in which case we expect that we might contest such assessment. Contesting such an assessment may be lengthy and costly and, if we were unsuccessful in disputing the assessment, the result could increase our anticipated effective tax rate.
Risks Related to Our Operations in Israel
Our headquarters, research and development and other significant operations are located in Israel and, therefore, our results may be adversely affected by political, economic and military instability in Israel.
Our headquarters and research and development facilities are located in Rehovot, Israel. In addition, a significant numbersome of our key employees, officers and directors are residents of Israel. Accordingly, political, economic and military conditions in Israel may directly affect our business. Since the establishment of the State of Israel in 1948, a number of armed conflicts have taken place between Israel, its neighboring countries and other organizations. Any hostilities involving Israel or the interruption or curtailment of trade or transport between Israel and its trading partners could adversely affect our operations and results of operations. Further, our operations could be disrupted by the obligations of personnel to perform military reserve service.
Provisions of Israeli law and our amended and restated articles of association may delay, prevent or otherwise impede a merger with, or an acquisition of, us, even when the terms of such a transaction are favorable to us and our shareholders.
Israeli corporate law regulates mergers, requires tender offers for acquisitions of shares above specified thresholds, requires special approvals for transactions involving directors, officers or significant shareholders and regulates other matters that may be relevant to such types of transactions. For example, a tender offer for all of a company’scompany’s issued and outstanding shares can only be completed if the acquirer receives positive responses from the holders of at least 95% of the issued share capital. Completion of the tender offer also requires approval of a majority of the offerees that do not have a personal interest in the tender offer unless, following consummation of the tender offer, the acquirer would hold at least 98% of the company’scompany’s outstanding shares. Furthermore, the shareholders, including those who indicated their acceptance of the tender offer, may, at any time within six months following the completion of the tender offer, petition an Israeli court to alter the consideration for the acquisition, unless the acquirer stipulated in its tender offer that a shareholder that accepts the offer may not seek such appraisal rights.
Furthermore, Israeli tax considerations may make potential transactions unappealing to us or to our shareholders whose country of residence does not have a tax treaty with Israel exempting such shareholders from Israeli tax. For example, Israeli tax law does not recognize tax-free share exchanges to the same extent as U.S. tax law. With respect to mergers, Israeli tax law allows for tax deferral in certain circumstances but makes the deferral contingent on the fulfillment of a number of conditions, including, in some cases, a holding period of two years from the date of the transaction during which sales and dispositions of shares of the participating companies are subject to certain restrictions. Moreover, with respect to certain share swap transactions, the tax deferral is limited in time, and when such time expires, the tax becomes payable even if no disposition of the shares has occurred.
It may be difficult to enforce a judgment of a U.S. court against us, our officers and directors in Israel or the U.S., to assert U.S. securities laws claims in Israel or to serve process on our officers and directors.
We are incorporated in Israel. A significant number of our executive officers and directors listed in this annual report reside outside of the U.S., and most of our assets and most of the assets of these persons are located outside of the U.S. Therefore, a judgment obtained against us, or any of these persons, including a judgment based on the civil liability provisions of the U.S. federal securities laws, may not be collectible in the U.S. and may not be enforced by an Israeli court. It also may be difficult for you to effect service of process on these persons in the U.S. or to assert U.S. securities law claims in original actions instituted in Israel. Israeli courts may refuse to hear a claim based on an alleged violation of U.S. securities laws reasoning that Israel is not the most appropriate forum in which to bring such a claim. In addition, even if an Israeli court agrees to hear a claim, it may determine that Israeli law and not U.S. law is applicable to the claim. If U.S. law is found to be applicable, the content of applicable U.S. law must be proven as a fact by expert witnesses, which can be a time consuming and costly process. Certain matters of procedure will also be governed by Israeli law.
There is little binding case law in Israel that addresses the matters described above. As a result of the difficulty associated with enforcing a judgment against us in Israel, holders of our ordinary shares may not be able to collect any damages awarded by either a U.S. or foreign court.
The rights and responsibilities of our shareholders are governed by Israeli law, which differs in some material respects from the rights and responsibilities of shareholders of U.S. companies.
The rights and responsibilities of the holders of our ordinary shares are governed by our amended articles of association and by Israeli law. These rights and responsibilities differ in some material respects from the rights and responsibilities of shareholders in U.S.-based companies. In particular, a shareholder of an Israeli company has a duty to act in good faith and in a customary manner in exercising its rights and performing its obligations towards the company and other shareholders, and to refrain from abusing its power in the company, including, among other things, in voting at a general meeting of shareholders on matters such as amendments to a company’scompany’s articles of association, increases in a company’scompany’s authorized share capital, mergers and acquisitions and related party transactions requiring shareholder approval. In addition, a shareholder who is aware that it possesses the power to determine the outcome of a vote at a meeting of the shareholders or to appoint or prevent the appointment of a director or executive officer in the company has a duty of fairness toward the company with regard to such vote or appointment. There is limited case law available to assist us in understanding the nature of this duty or the implications of these provisions. These provisions may be interpreted to impose additional obligations and liabilities on holders of our ordinary shares that are not typically imposed on shareholders of U.S. companies.
ITEM 1B —- UNRESOLVED STAFF COMMENTS
None.
Our facilities in Israel, which house our corporate headquarters and our research and developments laboratories, are located at two sites in the Weizmann Science Park in Rehovot, Israel. Under a Lease Agreement with Gav Yam Lands Ltd., we are leasingWe currently lease approximately 2,199 square meters untilunder a lease that expires in December 31, 2020.
Our executive offices in the U.S.United States are located in Bridgewater, New Jersey. TheWe currently lease agreement was signed in October 2017,approximately 10,000 square feet of office space under a lease that expires in March 2019 and consists31, 2019. We have an option to extend our existing lease for an additional three years on similar conditions. We are currently re-negotiating the lease with our landlord for an additional 5,000 square feet of approximately 929 square metersoffice space in the same location. If we are not able to reach agreement, we will exercise our option to extend our existing lease prior to the expiration of space.our option on March 8, 2019.
We believe that our current office space and facilities in Israel and the U.S. is sufficientUnited States are adequate to meet our anticipatedcurrent needs, and that suitable additional alternative spaces will be available in the future on commercially reasonable terms for the foreseeableour future and is suitable for the conduct of our business.growth.
ITEM 3 —- LEGAL PROCEEDINGS
From time to time, we may become party toinvolved in litigation or other legal proceedings relating to claimsthat we consider to be a part of arising from the ordinary course of our business. Other than the complaints filedThere are currently no claims or actions pending against Teva and against Perrigous that, in the United States (see above “Item 1A—Risk Factor—Risks Relatedopinion of our management, are likely to Our Intellectual Property—We have received notice letters of ANDAs submitted for drug products that are generic versions of Finacea Foam and we are involved in lawsuits to protect or enforcea material adverse effect on our patents, which could be expensive, time consuming and unsuccessful”) we are not currently involved in any legal proceedings. We may become involved in material legal proceedings in the future.business.
ITEM 4 —- MINE SAFETY DISCLOSURES
Not applicable.
ITEM 5 —- MARKET FOR REGISTRANT’S ORDINARY SHARES, RELATED SHAREHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information
Our ordinary shares have been listed on the NASDAQNasdaq Global Market under the symbol “FOMX”“FOMX” since September 17, 2014. Prior to that date, there was no public trading market for our ordinary shares. Our initial public offering was priced at $6.00 per share. The following table sets forth for the periods indicated the high and low sales prices per ordinary share as reported on the NASDAQ Global Market:
| 2017 | | High | | | Low | | | First quarter | | | 11.27 | | | | 4.40 | | | Second quarter | | | 5.11 | | | | 4.03 | | | Third quarter | | | 5.99 | | | | 4.34 | | | Fourth quarter | | | 7.00 | | | | 5.17 | | | | | | | | | | | | | 2016 | | High | | | Low | | | First quarter | | | 8.45 | | | | 5.48 | | | Second quarter | | | 7.67 | | | | 5.70 | | | Third quarter | | | 10.40 | | | | 6.16 | | | Fourth quarter | | | 11.26 | | | | 7.12 | |
Holders
As of February 26, 2018, the closing price per share of our ordinary shares on the NASDAQ Global Market was $6.24. Holders
As of February 26, 2018,22, 2019, we had approximately 615 holders of record of our ordinary shares. ThisThe actual number of holders of our ordinary shares is greater than this number of record holders, as this number does not include the number of persons whose shares are in nominee or in “street name”“street name” accounts through brokers.
Dividends
We have never declared or paid cash dividends to our shareholders and we do not intend to pay cash dividends in the foreseeable future. We intend to reinvest any earnings in developing and expanding our business. Any future determination relating to our dividend policy will be at the discretion of our board of directors and will depend on a number of factors, including future earnings, our financial condition, operating results, contractual restrictions, capital requirements, business prospects, our strategic goals and plans to expand our business, applicable law and other factors that our board of directors may deem relevant.
Pursuant to the Israeli Companies Law, if we do distribute dividends, the distribution amount will be limited to the greater of retained earnings or earnings generated over the previous two years, according to our then last reviewed or audited financial statements, provided that the end of the period to which the financial statements relate is not more than six months prior to the date of the distribution. If we do not meet such criteria, then we may distribute dividends only with court approval. In each case, we are only permitted to distribute a dividend if our board of directors and the court, if applicable, determines that there is no reasonable concern that payment of the dividend will prevent us from satisfying our existing and foreseeable obligations as they become due.
In the event of our liquidation, after satisfaction of liabilities to creditors, our assets will be distributed to the holders of our ordinary shares in proportion to their shareholdings. This right, as well as the right to receive dividends, may be affected by the grant of preferential dividend or distribution rights to the holders of a class of shares with preferential rights that may be authorized in the future.
Payment of dividends may be subject to Israeli withholding taxes. For additional information see “Item 10.E.—Taxation—Taxation of our Shareholders—Taxation of non-Israeli shareholders on receipt of dividends” in our annual report on Form 20-F for the year 2016, filed with the SEC on February 21, 2017.
Securities Authorized for Issuance under Equity Compensation Plans
Set forth below is a table that summarizes compensation plans (including individual compensation arrangements) under which the company’s equity securities are authorized for issuance as of February 27, 2018.
| Plan category | | Number of securities to be issued upon exercise of warrants and rights | | | Weighted-average exercise price of outstanding warrants and rights | | | Number of securities remaining available for future issuance under equity compensation plans(1) | | | Equity compensation plans approved by security holders | | | - | | | | - | | | | - | | | Equity compensation plans not approved by security holders | | | 4,230,101 | | | $ | 5.65 | (2) | | | 2,076,088 | | | Total | | | 4,230,101 | | | $ | 5.65 | | | | 2,076,088 | |
| (1) | Excluding securities reflected in column titled “Number of securities to be issued upon exercise of warrants and rights”. |
| (2) | including restricted share units with an exercise price of $0. |
Our equity compensation plan information required by this item is incorporated by reference to the information in Part III, Item 12 of this report.
Share Performance
The table below compares the three-year cumulative total shareholder return on our ordinary shares with the cumulative total return on the NASDAQ Composite Index and the NASDAQ Biotechnology Index. The period shown commences on December 31, 2014 (the end of the first fiscal year following our initial public offering) and ends on December 31, 2017, the end date of our last fiscal year. The table assumes an investment of $100 on December 31, 2014, and the reinvestment of any dividends. No cash dividends have been declared or paid on our ordinary shares during such period. Shareholder returns over the indicated periods should not be considered indicative of future share prices or shareholder returns.
| | | December 31, 2014* | | | December 31, 2015 | | | December 31, 2016 | | | December 31, 2017 | | | Foamix Pharmaceuticals Ltd. | | | 100.00 | | | | 115.69 | | | | 158.35 | | | | 85.73 | | | NASDAQ Composite Index | | | 100.00 | | | | 105.73 | | | | 113.66 | | | | 145.76 | | | NASDAQ Biotechnology Index | | | 100.00 | | | | 111.42 | | | | 87.26 | | | | 105.64 | |
* $100 invested on December 31, 2014 in shares or index-including reinvestment of dividends.
Below is a graphical depiction of the comparison of the cumulative total return over the years 2015-2017 among Foamix Pharmaceuticals Ltd., NASDAQ Composite Index and NASDAQ Biotechnology Index:
This performance graph shall not be deemed “soliciting material” or to be “filed” with the SEC for purposes of Section 18 of the Exchange Act or otherwise subject to the liabilities under that Section, and shall not be deemed incorporated by reference into any filing of Foamix Pharmaceuticals Ltd. under the Securities Act of 1933.
Recent Sales of Unregistered Securities
None.
Purchases of Equity Securities by the Issuer and Affiliated Purchasers
None.
ITEM 6 —- SELECTED FINANCIAL DATA
Our historical consolidated financial statements are prepared in accordance with generally accepted accounting principles in the United States and are presented in U.S. dollars. The selected historical consolidated financial information as of December 31, 20172018 and 20162017 and for the years ended December 31, 2018, 2017 2016 and 20152016 have been derived from, and should be read in conjunction with, theour consolidated financial statements of Foamix Pharmaceuticals Ltd. and notes thereto appearing elsewhere in this annual report.Annual Report. The selected financial data as of December 31, 2016, 2015 2014 and 20132014 and for the years ended December 31, 20142015 and 20132014 have been derived from our audited consolidated financial statements of the company not included in this annual report.Annual Report.
The information presented below is qualified by the more detailed historical consolidated financial statements set forth in this annual report, and should be read in conjunction with those consolidated financial statements, the notes thereto and the discussion under “Item“Item 7 — Management’s- Management’s Discussion and Analysis of Financial Condition and Results of Operations”Operations” included elsewhere in this annual report.Annual Report.
Consolidated Statement of Operations Data
| | | Year ended December 31, | | | | | 2017 | | | 2016 | | | 2015 | | | 2014 | | | 2013 | | | | | (in thousands of U.S. dollars, except loss per share) | | Statements of operations data: | | | | | | | | | | | | | | | | | Revenues | | $ | 3,669 | | | $ | 5,527 | | | $ | 849 | | | $ | 5,414 | | | $ | 1,404 | | Cost of revenues(1) | | | 13 | | | | 59 | | | | 70 | | | | 527 | | | | 453 | | | Gross profit | | | 3,656 | | | | 5,468 | | | | 779 | | | | 4,887 | | | | 951 | | | Operating expenses: | | | | | | | | | | | | | | | | | | | | | Research and development(1) | | | 57,779 | | | | 25,897 | | | | 10,680 | | | | 3,557 | | | | 1,086 | | Selling, general and administrative(1) | | | 11,491 | | | | 9,221 | | | | 7,029 | | | | 2,964 | | | | 1,221 | | | Total operating expenses | | | 69,270 | | | | 35,118 | | | | 17,709 | | | | 6,521 | | | | 2,307 | | | Operating loss | | | 65,614 | | | | 29,650 | | | | 16,930 | | | | 1,634 | | | | 1,356 | | | Net Loss | | $ | 65,715 | | | $ | 29,336 | | | $ | 16,517 | | | $ | 11,484 | | | $ | 2,431 | | | Loss per share basic and diluted | | | 1.76 | | | | 0.91 | | | | 0.58 | | | | 0.79 | | | | 0.22 | |
_________________________________________________| | | Year ended December 31, | | | | | 2018 | | | 2017 | | | 2016 | | | 2015 | | | 2014 | | | | | (in thousands of U.S. dollars, except loss per share) | | | Statements of operations data: | | | | | | | | | | | | | | | | | Revenues | | $ | 3,595 | | | $ | 3,669 | | | $ | 5,527 | | | $ | 849 | | | $ | 5,414 | | Cost of revenues(1) | | | - | | | | 13 | | | | 59 | | | | 70 | | | | 527 | | | Gross profit | | | 3,595 | | | | 3,656 | | | | 5,468 | | | | 779 | | | | 4,887 | | | Operating expenses: | | | | | | | | | | | | | | | | | | | | | Research and development(1) | | | 64,474 | | | | 57,779 | | | | 25,897 | | | | 10,680 | | | | 3,557 | | Selling, general and administrative(1) | | | 14,013 | | | | 11,491 | | | | 9,221 | | | | 7,029 | | | | 2,964 | | | Total operating expenses | | | 78,487 | | | | 69,270 | | | | 35,118 | | | | 17,709 | | | | 6,521 | | | Operating loss | | | 74,892 | | | | 65,614 | | | | 29,650 | | | | 16,930 | | | | 1,634 | | | Net loss | | $ | 74,163 | | | $ | 65,715 | | | $ | 29,336 | | | $ | 16,517 | | | $ | 11,484 | | | Loss per share basic and diluted | | | 1.70 | | | | 1.76 | | | | 0.91 | | | | 0.58 | | | | 0.79 | |
(1) Includes share-based compensation expenses as follows: | | | Year ended December 31, | | | | | 2017 | | | 2016 | | | 2015 | | | 2014 | | | 2013 | | | | | (in thousands of U.S. dollars) | | | Cost of revenues | | $ | 2 | | | $ | 3 | | | $ | 2 | | | $ | 15 | | | $ | 16 | | | Research and development | | | 1,711 | | | | 1,135 | | | | 588 | | | | 80 | | | | 59 | | | Selling, general and administrative | | | 2,453 | | | | 1,774 | | | | 1,187 | | | | 102 | | | | 430 | | | Total share-based compensation | | $ | 4,166 | | | $ | 2,912 | | | $ | 1,777 | | | $ | 197 | | | $ | 505 | |
| | | Year ended December 31, | | | | | 2018 | | | 2017 | | | 2016 | | | 2015 | | | 2014 | | | | | (in thousands of U.S. dollars) | | | Cost of revenues | | $ | - | | | $ | 2 | | | $ | 3 | | | $ | 2 | | | $ | 15 | | | Research and development | | | 2,054 | | | | 1,711 | | | | 1,135 | | | | 588 | | | | 80 | | | Selling, general and administrative | | | 3,266 | | | | 2,453 | | | | 1,774 | | | | 1,187 | | | | 102 | | | Total share-based compensation | | $ | 5,320 | | | $ | 4,166 | | | $ | 2,912 | | | $ | 1,777 | | | $ | 197 | |
Consolidated Balance Sheet Data
| | | As of December 31, | | | As of December 31, | | | | | 2017 | | | 2016 | | | 2015 | | | 2014 | | | 2013 | | | 2018 | | | 2017 | | | 2016 | | | 2015 | | | 2014 | | | | | (in thousands of U.S. dollars, other than number of shares) | | | (in thousands of U.S. dollars, other than number of shares) | | Balance sheet data: | | | | | | | | | | | | | | | | | | Balance sheet data: | | | | | Cash and investments(1) | | $ | 76,412 | | | $ | 130,988 | | | $ | 103,779 | | | $ | 49,966 | | | $ | 2,308 | | | $ | 99,385 | | | $ | 76,412 | | | $ | 130,988 | | | $ | 103,779 | | | $ | 49,966 | | Working capital(2) | | | 59,276 | | | | 111,730 | | | | 53,091 | | | | 48,757 | | | | 1,144 | | | | 90,699 | | | | 59,276 | | | | 111,730 | | | | 53,091 | | | | 48,757 | | | Total assets | | | 80,254 | | | | 135,635 | | | | 105,245 | | | | 51,277 | | | | 3,086 | | | | 103,731 | | | | 80,254 | | | | 135,635 | | | | 105,245 | | | | 51,277 | | | Total long-term liabilities | | | 1,425 | | | | 379 | | | | 385 | | | | 381 | | | | 4,917 | | | | 1,081 | | | | 1,425 | | | | 379 | | | | 385 | | | | 381 | | | Total shareholders’ equity (capital deficiency) | | | 68,601 | | | | 129,985 | | | | 100,802 | | | | 48,762 | | | | (3,582 | ) | | | Total shareholders’ equity | | | | 92,182 | | | | 68,601 | | | | 129,985 | | | | 100,802 | | | | 48,762 | | | Capital shares | | $ | 1,576 | | | $ | 1,561 | | | $ | 1,284 | | | $ | 954 | | | $ | 471 | | | $ | 2,331 | | | $ | 1,576 | | | $ | 1,561 | | | $ | 1,284 | | | $ | 954 | | | Number of ordinary shares | | | 37,498,128 | | | | 37,167,791 | | | | 30,639,134 | | | | 22,443,934 | | | | 11,408,490 | | | | 54,351,140 | | | | 37,498,128 | | | | 37,167,791 | | | | 30,639,134 | | | | 22,443,934 | |
(1) Cash and investments includes cash and cash-equivalents, restricted cash, bank deposits, marketable securities and restricted marketable securities. | (1) | Cash and investments includes cash and cash-equivalents, restricted cash, bank deposits, marketable securities and restricted marketable securities. |
(2) Working capital is defined as total current assets minus total current liabilities.
ITEM 7 —- MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
You should read the following discussion and analysis of our financial condition and results of operations in conjunction with the financial statements and the notes thereto included elsewhere in this report. The following discussion contains forward-looking statements that reflect our plans, estimates and beliefs. Our actual results could differ materially from those discussed in the forward-looking statements. Factors that could cause or contribute to these differences include those discussed below and elsewhere in this report, particularly in the section entitled “Item 1A—Risk Factors”“Item 1A-Risk Factors”.
Company Overview
We are a late clinical-stage specialty pharmaceutical company focused on developing and commercializing our proprietary, innovative and differentiated topical drug candidates for dermatological therapy. Our lead product candidate, FMX101 (4% minocycline foamfoam), is being developed for the treatment of acne, rosacea and other skin conditions. Our lead product candidates, FMX101 for moderate-to-severe acne and our second product candidate, FMX103 (1.5% minocycline foam), is being developed for the treatment of moderate-to-severe papulopustular rosacea,rosacea. Both product candidates are novel topical foam formulations of the antibiotic minocycline. Based onminocycline and were developed using our Molecule Stabilizing Technology, a proprietary foam platform designed to optimize the topical delivery of minocycline, an active pharmaceutical ingredient, or API, that is currently available only in oral form despite its prevalent use in dermatology.
We announced positive top-line results demonstrated infrom both of our Phase II and Phase III clinical trials for each of FMX101 and FMX103 in the second half of 2018. We submitted our Phase II clinical trialnew drug application, or NDA, to the U.S. Food and Drug Administration, or FDA, for FMX101 in December 2018, and expect to submit an NDA for FMX103 in mid-2019. Despite the considerable U.S. market opportunities for acne and rosacea, we believe these product candidates have the potential of providing a fast, effective and well-tolerated treatment for their respective indications, whichmarkets are currently underserved and commonly treated by oral prescription products such as oral minocycline oraland doxycycline and various non-minocycline topical therapies. If approved, we believe FMX101 and FMX103 have the potential to provide a first-in-class, effective and well-tolerated topical treatment for the tens of millions of people who suffer from their respective indications.
Our corporate strategy is to develop and solidify a commercial presence in acne and rosacea by obtaining FDA approval for, and launching our lead product candidates, FMX101 and FMX103, in the United States. We may also enter into partnerships with third parties to reach other geographic territories or therapeutic fields through their respective sales forces and infrastructure. Following these near-term goals, we intend to grow beyond these indications into other dermatological indications, and to diversify our product and commercial development beyond minocycline and the tetracycline class of antibiotics. We are currently developing additional foam and other topical products for acne, rosacea and other dermatology indications in vehicle platforms designed to enhance delivery of their respective APIs. We are also evaluating diversifying into synergistic technologies and specialties either on our own or through partnerships.
We are currently investingFMX101 is a product candidate containing micronized minocycline hydrochloride, an antibiotic in the majoritytetracycline class, in a 4% concentration for the treatment of moderate-to-severe acne vulgaris. The API is suspended in our efforts and resources to advanceMolecule Stabilizing Technology foam vehicle, an elegant, light-feeling topical foam that is easily spread across wide areas of the skin. In September 2018, we announced our third pivotal Phase III clinical trial of FMX101 (Study 22) for FMX101FX2017-22) met both of its co-primary endpoints, demonstrating a statistically significant reduction in the U.S. We announcednumber of inflammatory lesions and a statistically significant improvement in patients’ Investigator’s Global Assessment, or IGA, scores, a metric commonly used to measure efficacy in acne trials. These positive results followed the first patient enrolled in this trial on August 3, 2017. We expect to have top-line results from this trial in the third quarter of 2018. In March of 2017, we announced the results of the double-blind stage of our initial two initial Phase III clinical trials. Statistical significance was demonstratedtrials of FMX101 that we announced in 2017 wherein both co-primary efficacy endpoints were met in one studytrial (Study 05), however, statistical significance was demonstrated inFX2014-05) but only one of the two co-primary efficacy endpoints showed statistical significance in the second studyother trial (Study 04)FX2014-04). Statistical significance was also demonstrated for FMX101 compared to vehicle in the pooled analysis of the co-primary endpoints as well as key secondary endpoints. TheWe embarked on our third Phase III trial was initiated(Study FX2017-22) following a Type B meeting conducted with the FDA in June of 2017. During this meeting,which the FDA confirmed that achieving statistically significantreplicating the results for FMX101 versus vehicle in both co-primary efficacy endpoints in a third independent clinicalof our Study FX2014-05 trial would be sufficient for establishinglikely support an efficacy claim.claim for FMX101. In addition to the positive Study FX2017-22 efficacy results, very few safety adverse events (and no treatment-related serious adverse events) were observed both in Study FX2017-22 and in the 40-week open label safety portion of Studies FX2014-04 and FX20410-05 that we concluded in January 2018.
FMX103 is a product candidate also containing micronized minocycline hydrochloride suspended in our Molecule Stabilizing Technology A previous Phase II clinical trialvehicle, at a lower 1.5% concentration, for the treatment of FMX101 also demonstrated clinically and statistically significant results in all primary and secondary endpoints. moderate-to-severe papulopustular rosacea. In JanuaryNovember 2018, we announced the completionthat both of a long-term safety study that was an extension of our two initial Phase III clinical trials for FMX101. The resultsFMX103 (Studies FX2016-11 and FX2016-12) met each of their co-primary endpoints, demonstrating a statistically significant reduction in inflammatory lesion counts and IGA treatment success of approximately 50% from the study showed FMX101 to be well-toleratedbaseline. There were no treatment-related serious adverse events, very few reported adverse events and to have an acceptable safety profile. We are also investing significant efforts and resources to advance our two pivotalpositive user-experience reports overall in these Phase III clinical trials as well as in the U.S. for FMX103, minocycline foam for moderate-to-severe papulopustular rosacea, after our Phase II clinical trial for FMX103 demonstrated clinically and statistically significant results in all primary and secondary endpoints. We announced the enrollment of the first patient in our Phase III trials on June 12, 2017. We expect to have top-line results from the blinded stage of both trials by the end of the third quarter or in the beginning of the fourth quarter of 2018 and to complete the trials, including a long-term40-week open label safety extension study,(Study FX2016-13) that was recently completed in February 2019.
In addition,We developed FMX101 and FMX103 using our proprietary Molecule Stabilizing Technology foam-based technology platform that was optimized for delivery of minocycline hydrochloride, a characteristically unstable small molecule, through the skin. We are currently developing in-house a pipeline of other innovative products to enhance our minocycline platform, including FCD105, a product candidate for the treatment of acne vulgaris that combines minocycline with a retinoid and which we successfully completedanticipate evaluating in a Phase II clinical trial with FDX104,(Study FX2016-40) beginning in mid-2019. We are also currently reviewing potential acquisitions of pipeline products at various stages of development that could be incorporated into our proprietary doxycycline foamvehicle for the management of moderate-to-severe rash associated with epidermal growth factor receptor inhibitor (EGFRI) anticancer treatments, and we are currently assessing our various options with regard to this product candidate, including seeking out licensing opportunities for it. We have also successfully completed a Phase II clinical trial of FMX102, our minocycline foam for the treatment of impetigo, including impetigo caused by methicillin-resistant staphylococcus aureus, or MRSA. However, as described in previous reports,optimized delivery.
In addition, we have been contemplating the commercial viabilityother proprietary delivery technologies in development that enable topical delivery of this product candidate for some time, given its limited market dominated by generic products, and following additional analysis of its potential we have recently decided to discontinue its further development in light of our current priorities and our other ongoing research and development efforts. We developed FMX101, FMX102, FMX103 and FDX104 using our proprietary technology, which includes our foam-based platforms. This technology enables us to formulate and stabilize a wide variety of drugs and deliver them directly to their target site. We have independently developed a series of proprietary foam platforms,APIs, each having unique pharmacological features and characteristics. Ourcharacteristics designed to keep the API stable when delivered and directed to the target site. We are conducting research and are in the early stages of in-house development of FMX110, a topical gel formulation of doxycycline hyclate for the treatment of papulopustular rosacea, and FMX109, a non-tetracycline acne product candidate that contains a combination of nicotinamide and a retinoid for the treatment of moderate-to-severe acne vulgaris. We believe our foam and other topical delivery platforms may offer significant advantages over alternative delivery options and are suitable for multiple application sites. We believe our proprietary foam-based platform may serve assites across a foundation in developing a potential pipeline of products across awide range of conditions.
BesideIn addition to our in-house developments,development projects, we have also entered into development and license agreements relating to our technology with various pharmaceutical companies, most notably with LEO Pharma A/S, or LEO, who assumed a license agreement we initially entered into with Bayer HealthCare AG, (formerly, Intendis)or Bayer. In 2015, Bayer received FDA approval for Finacea® Foam (15% azelaic acid), or Bayer, which in the third quarter of 2015 began selling in the U.S.Finacea, a prescription foam product for the treatment of rosacea, known as Finacea® Foam (azelaic acid) 15%, or Finacea, after developing such product in collaboration with Foamix and utilizing Foamix’swhich utilizes an emulsion-based proprietary foam technology platform. Accordingplatform that we licensed to them that is different from our surfactant-free foam platform that supports our lead product candidates. Bayer began selling Finacea in the United States in the third quarter of 2015 and in September 2018, LEO acquired Finacea from Bayer and assumed all rights and responsibilities under our initial license agreement with Bayer,Bayer. Together with LEO, we are entitledlitigating against several generic pharmaceutical companies for alleged infringement of certain of our patents following the generic companies’ submission of abbreviated new drug applications, or ANDAs, to royaltiesthe FDA seeking approval to manufacture and certain contingent payments upon commercialization of Finacea, based on Bayer’s net salessell generic versions of Finacea. In 2017We are committed to defending our intellectual property rights globally, including patents we were entitledhave licensed to receive royalty payments from Bayerother pharmaceutical companies as part of our collaboration efforts.
We have also out-licensed other foam technology platforms to other third parties to develop branded pharmaceutical products containing different APIs for potential commercialization that are in a total amountthe early stages of $3.5 million on account of its sales of Finacea. Our total revenues from such agreements from our inception through December 31, 2017 were approximately $28.1 million.development.
To date, we have not yetonly submitted anyone product candidatescandidate for approval by regulatory authorities and we do not currently have rights, other than those under our license agreement with LEO Pharma A/S, or LEO, to any products that have been approved for marketing in any territory. We have financed our operations primarily through private and public placements of our ordinary shares, convertible loans and through fees, cost reimbursements and royalties received from development and licensing collaborations.our licensees. We have incurred significant losses since our inception in 2003. Our accumulated deficit at December 31, 20172018 was $141.3$215.4 million and our net loss for the year ended December 31, 20172018 was $65.7$74.2 million. A substantial amount of our net losses resulted from costs incurred in connection with our research and development programs and clinical trials and from general and administrative costs associated with our operations. The net losses and negative operating cash flows incurred to date, together with expected future losses, have had, and likely will continue to have, an adverse effect on our shareholders’shareholders’ equity and working capital. The amount of future net losses will depend, in part, on the rate of future growth of our expenses and our ability to generate offsetting revenue, if any. We expect to continue to incur significant expenses and operating losses for the foreseeable future.
We do not expect to generate revenue from product sales unless and until we successfully complete clinical trials and obtain marketing approval from the FDA for one or more of our lead product candidates, FMX101 or FMX103. Accordingly, we anticipate that we will need to raise additional capital in order to complete the development and commercialization of FMX101 and FMX103 and to advance the development of our other product candidates. Until we can generate a sufficient amount of product revenue to finance our cash requirements, we expect to finance our future cash needs primarily through a combination of public and private equity offerings, debt or other structured financings, and entry into strategic collaborations. We may be unable to raise capital when needed or on attractive terms, which would force us to delay, limit, reduce or terminate our development programs or commercialization efforts. We will need to generate significant revenue to achieve and sustain profitability, and we may never be able to do so.
Revenues
To date, we have not generated any revenues from sales of FMX101, FMX103 or any of our other product candidates. We do not expect to commercially launch FMX101 or other product candidates or generate any revenues from sales of any of our product candidates before the end of 2019, until after completing their development and clinical testing and obtaining approvals for theirthe marketing of FMX101 in the U.S.United States. Our ability to generate revenues from sales will depend on the successful commercialization of FMX101, FMX103 and our other product candidates.
As of December 31, 2017,2018, we had generated cumulative revenues of approximately $28.1$31.7 million under development and license agreements, of which approximately $18.3$18.4 million were development service payments, approximately $3.1 million were contingent payments and $6.7$10.2 million were royalty payments. The royalties were paid in relation to Finacea. Our total revenues for the years ended December 31, 2018, 2017 and 2016 and 2015 were $3.6 million, $3.7 million and $5.5 million, and $849,000, respectively. We may become entitled to additional contingent payments, subject to achievement of the applicable clinical results by our other licensees. In light of the current phase of development under these agreements, we do not expect to receive significant payments in the near term, if at all. We are also entitled to additional royalties from net sales or net profits generated by other products to be developed under these agreements, if they are successfully commercialized. In those development and license agreements in which royalties are based on net sales, their rate ranges from 3% to 8.5%, and in the agreement in which royalties are based on net profits, their rate is 6%.
Pursuant to a collaboration agreement with Bayer HealthCare AG, we are entitled to receive royalty payments with respect to Finacea®, a prescription foam product that we developed in collaboration with Bayer. In the year ended December 31, 2017,2018, we were entitled to receive royalty payments in an aggregate amount of $3.5 million.
Cost of Revenues Cost of revenues includes costs and expenses we incur in supplying services to our licensees under our development and license agreements with them. These services include design and development of product prototypes, performance of in-vitro studies and other lab tests, compiling project reports and recommendations and carrying out other tasks related to such efforts. Our services to licensees do not include development work beyond the prototype stage, clinical trials or pursuit of regulatory approval, which are the responsibility and at the expense of each licensee.
Accordingly, our cost of revenues includes payroll and other payments on behalf of the employees and consultants assigned to these projects; laboratory services related to the studies we perform on behalf of the licensees; rent and office maintenance costs related to the use of our facilities and infrastructure, utilities and other overhead services in connection with the projects performed for the licensees.
Our total cost of revenues for the twelve monthsyears ended December 31, 2017 and 2016 were $13 thousand and 2015 were $13,000, $59,000 and $70,000,$59 thousand, respectively. There was no similar cost of revenues for the year ended December 31, 2018, as revenues in 2018 consisted almost entirely of royalties, which do not bear related cost of revenue.
We do not expect substantial changes in cost of revenue unless and until we obtain regulatory approval for our lead product candidates and begin serial production of such products, whether internally or through third party manufacturers, at which point we expect our cost of revenues to grow along with the growth of our sales and inventory needs. Cost of revenues as a percentage of revenues for the twelve months ended December 31, 2017, 2016 and 2015 were 0.4%, 1.1%, and 8.2%, respectively. The decrease in cost of revenues is primarily due to the decrease in new development projects and the increase in royalty payments which do not bear related cost of revenue.
Operating Expenses
Research and development expenses
Research and development activities are, and will continue to be, central to our business. Product candidates in later stages of clinical development generally have higher development costs than those in earlier stages of clinical development, primarily due to the increased size and duration of later-stage clinical trials. We expect to incur significant research and development costs to increase significantly forin the foreseeable future, asassuming our pipeline products progress into clinical trials. However, we do not believe that it is possible at this time to accurately project total program-specific expenses to reach commercialization. There are numerous factors associated with the successful commercialization of any of our product candidates, including future trial design and various regulatory requirements, many of which cannot be determined with accuracy at this time based on our stage of development. Additionally, future commercial and regulatory factors beyond our control will affect our clinical development programs and plans.
Our research and development expenses relate primarily to the development of FMX101.FMX101 and FMX103. From January 1, 2007 until December 31, 2017,2018, we cumulatively spent approximately $104.9$169.4 million on research and development of FMX101, FMX103 and our other product candidates. Our total research and development expenses for the years ended December 31, 2018, 2017 2016 and 20152016 were approximately $64.5 million, $57.8 $25.9million and $10.7$25.9 million, respectively. We charge all research and development expenses to operations as they are incurred. We expect research and development expenses to increasedecrease in the near term due to the ongoingcompletion of the Phase III clinical trials for FMX101 and FMX103.
The successful development of FMX101, FMX103 and additionalour product candidates, excluding FMX101 and FMX103, is highly uncertain. As such, at this time, we cannot reasonably estimate or know the nature, timing and costs of the efforts that will be necessary to complete the remainder of the development of our technology for additional indications. This uncertainty is due to numerous risks and variables associated with developing products, including the uncertainty of:
| · | the scope, rate of progress and expense of our research and development activities; |
| · | the terms and timing of regulatory approvals; and |
| · | our ability to file, prosecute, obtain, maintain, defend and enforce patents and other intellectual property rights and the expense of filing, prosecuting, obtaining, maintaining, defending and enforcing patents and other intellectual property rights; |
| · | the ability to market, commercialize and achieve market acceptance for FMX101 or any other product candidate that we may develop in the future; and |
| · | our ability to identify, evaluate, acquire or in-license intellectual property, if needed, to facilitate the commercialization of our products and technologies. |
A change in the outcome of any of these variables with respect to the development of FMX101, FMX103 or our other product candidates could result in a significant change in the costs and timing associated with their development. For example, if the FDA or foreign regulatory authority were to require us to conduct preclinical studies and clinical trials beyond those which we currently anticipate for the completion of clinical development of our product candidates, or if we experience significant delays in enrollment in any clinical trials, we could be required to expend significant additional time and financial resources and time on the completion of the clinical development.
Research and development expenses consist primarily of:
| · | employee-related expenses, including salaries, benefits and related expenses, including share based compensation expenses; |
| · | expenses incurred under agreements with third parties, including subcontractors, suppliers and consultants that conduct regulatory activities, clinical trials and preclinical studies; |
| · | expenses incurred to acquire, develop and manufacture clinical trial materials; |
| · | facilities, depreciation and other expenses, which include direct and allocated expenses for rent and maintenance of facilities, insurance, and other operating costs; and |
| · | other costs associated with preclinical and clinical activities and regulatory operations. |
We have managed to finance our research and development operations and expenses without the aid of government grants, other than a loan in the amount of approximately $450,000 received from the Israel-U.S. Bi-national Industrial Research and Development Foundation, or BIRD, in 2008, which was fully repaid in 2016. Accordingly, we are not subject to the provisions of the Law for Encouragement of Research and Development in the Industry, 5744-1984, nor to any directives issued by the Israel Innovation Authority, previously known as the Office of the Chief Scientist.Scientist of the Ministry of Economy.
Selling, general and administrative expenses
Our selling, general and administrative expenses consist principally of:
| · | employee-related expenses, including salaries, benefits and related expenses, including share based compensation expenses; |
| · | costs associated with market research and business development activities in preparation for future marketing and sales, including activities intended to select the most promising product candidates for further development and commercialization; |
| · | legal and professional fees for auditors and other consulting expenses not related to research and development activities or to market research or business development activities; |
| · | cost of offices,office spaces, communication and office expenses; |
| · | information technology expenses; |
| · | depreciation of tangible fixed assets related to our general and administrative activities or to our market research and business development activities; and |
| · | costs associated with filing, prosecuting, obtaining and maintaining patents and other intellectual property. |
As part of our growth strategy, we have begun building up our dedicated U.S. marketing and business development team and infrastructure, and we intend to further increase such U.S. infrastructure, as well as expand our marketing effort to new markets. We therefore expect selling and marketing expenses to increase in absolute terms as a percentage of our revenues. Our total selling, general and administrative expenses for the years ended December 31, 2018, 2017 2016 and 20152016 were approximately $14.0 million, $11.5 $9.2million and $7.0$9.2 million, respectively.
Our ability to commercialize FMX101 and FMX103 successfully, if approved, is highly uncertain and depends on a number of factors, including market adoption of our product candidates by physicians and patients, market access uncertainty, our ability to scale to the opportunity and the existence of existing and future products that may compete with ours. As such, at this time, we cannot reasonably estimate or know the nature, timing and costs of the efforts that will be necessary for successful commercialization of our product candidates, if approved.
Financial Income
Financial income consists primarily of gains from interest earned from our bank deposits and financial income on our marketable securities.
Taxes on Income
During 2016 theThe standard corporate tax rate in Israel was 25%during the year 2018 is 23%, which represents a 1% decrease compared to the tax rate during 2017. Effective January 1, 2018 the U.S. Tax cuts and duringJobs Act (Tax Act) reduced the U.S Federal tax by 14% from 35% in 2017 it was 24%.to 21% in 2018.
We have yet to generate taxable income in Israel, as we have historically incurred operating losses resulting in carry forward tax losses totaling approximately $88.6$146.7 million as of December 31, 2017.2018. During 2018 we incurred a carry forward tax loss in our U.S. subsidiary, Foamix Pharmaceuticals Inc. of $0.4 million. We anticipate that we will be able to carry forward these tax losses to future tax years. Accordingly, we do not expect to pay taxes in Israel until we have taxable income after the full utilization of our carry forward tax losses. We provided a full valuation allowance with respect to the deferred tax assets related to these carry forward losses.
During 2018, 2017 2016 and 20152016, we incurred tax expenses of $0.2 million. $1.2 million $387,000 and $39,000,$0.4 million, respectively, related to our U.S. subsidiary, Foamix Pharmaceuticals Inc.
Comparison of the Year Ended December 31, 2018 to the Year Ended December 31, 2017
Revenues
Our total revenues decreased by $74 thousand, or 2%, from $3.7 million in the year ended December 31, 2017 to $3.6 million in the year ended December 31, 2018. The revenues for the year ended December 31, 2018 consist of royalty payments in the amount of $3.5 million and $62 thousand from contingent payments.
Cost of revenues
Our total cost of revenues for the year ended December 31, 2017 was $13 thousand and there was no cost of revenues for the year ended December 31, 2018, as revenues in 2018 consisted almost entirely of royalties, which bear no related cost of revenue.
Research and development expenses
Our research and development expenses for the year ended December 31, 2018 were $64.5 million, representing an increase of $6.7 million, or 11.6%, compared to $57.8 million for the year ended December 31, 2017. The increase in research and development expenses resulted primarily from an increase of $5 million in costs relating predominantly to FMX101 and FMX103 clinical trials; an increase of $0.6 million in consultant expenses and an increase of $0.4 million in payroll and payroll related expenses mostly due to increase in headcount, bonuses and share based compensation expenses.
Selling, general and administrative expenses
Our general and administrative expenses for the year ended December 31, 2018 were $14.0 million, representing an increase of $2.5 million, or 21.7%, compared to $11.5 million for the year ended December 31, 2017. The increase in selling, general and administrative expenses resulted primarily from an increase of $1.1 million in consultant expenses mostly relating to pre-commercialization activities and an increase of $0.7 million in payroll and other payroll-related expenses mostly due to increase in headcount, bonuses and share based compensation expenses.
Operating loss
As a result of the foregoing, our operating loss for the year ended December 31, 2018, was $74.9 million, compared to an operating loss of $65.6 million for the year ended December 31, 2017, an increase of $9.3 million, or 14.2%.
Finance income
In the years ended December 31, 2018 and 2017, our financial income included mostly gains from marketable securities and interest earned on our bank deposits.
The finance expenses (income) by cash and non-cash components are as follows:
| | | Year ended December 31, | | | | | 2018 | | | 2017 | | | | | (in thousands of U.S. dollars) | | | Interest on bank deposits | | $ | (297 | ) | | $ | (532 | ) | | Gain from marketable securities, net | | | (688 | ) | | | (602 | ) | | Total income | | | (985 | ) | | | (1,134 | ) | | Less: | | | | | | | | | | Other expenses | | | 17 | | | | 14 | | | Foreign exchange loss, net | | | 27 | | | | 57 | | | Total expenses | | | 44 | | | | 71 | | | Finance income, net | | $ | (941 | ) | | $ | (1,063 | ) |
Taxes on income
During 2018 and 2017, we did not generate taxable income in Israel and during 2018 we did not generate taxable income in our U.S. subsidiary, Foamix Pharmaceuticals Inc. However, we incurred tax expenses relating to our U.S. subsidiary, in the amount of $0.2 million and $1.2 million for the years ended December 31, 2018 and 2017, respectively. The decrease in tax expenses resulted primarily from a provision for uncertain tax positions recorded in 2017.
Net Loss
Our net loss for the year ended December 31, 2018 was $74.2 million, compared to $65.7 million for the year ended December 31, 2017, an increase of $8.5 million, or 12.9%.
Comparison of the Year endedEnded December 31, 2017 to the Year Ended December 31, 2016
Revenues
Our total revenues decreased by $1.8 million, or 32.7%, from $5.5 million in the year ended December 31, 2016 to $3.7 million in the year ended December 31, 2017. The decrease is mainly due to a decrease of $2.5 million in contingent payments from Buyer,Bayer, that were payable for 2016 due to Bayer’s achievement of certain sale targets during that year, offset by an increase in royalty payments in the amount of $565,000$0.6 million from Bayer for the sales of Finacea® Foam.Finacea.
Cost of revenues
Our total cost of revenues for the years ended December 31, 2017 and 2016 were $13,000was $13 thousand and $59,000,$59 thousand, respectively. The $46,000$46 thousand decrease in cost of revenues resulted primarily from a decrease in the number of our development projects.projects. Cost of revenues as a percentage of revenues for the years ended December 31, 2017 and 2016 was 0.4% and 1.1%, respectively. The decrease in the cost of revenues as a percentage of revenues was primarily due to thea decrease in the number of new development projects and thean increase in royalty payments, which do not bear relatedno cost of revenue.
Research and development expenses
Our research and development expenses for the year ended December 31, 2017 were $57.8 million, representing an increase of $31.9 million, or 123%, compared to $25.9 million for the year ended December 31, 2016. The increase in research and development expenses resulted primarily from an increase of $28.0 million in costs relating predominantly to the FMX101 and FMX103 clinical trials and an increase of $3.0 million in payroll and payroll related expenses primarily due to an increase in headcount.
Selling, general and administrative expenses
Our general and administrative expenses for the year ended December 31, 2017 were $11.5 million, representing an increase of $2.3 million, or 25%, compared to $9.2 million for the year ended December 31, 2016. The increase in selling, general and administrative expenses resulted primarily from an increase of $1.9 million in payroll and other payroll-related expenses mostlymainly due to an increase in headcount and salary raises.
Operating loss
As a result of the foregoing, our operating loss for the year ended December 31, 2017, was $65.6 million, compared to an operating loss of $29.6 million for the year ended December 31, 2016, an increase of $36.0 million, or 122%.
Finance income
In the years ended December 31, 2017 and 2016, our financial income included mostly gains from marketable securities and interest earned on our bank deposits. In the year ended December 31, 2016, those gains were partially offset by expenses on the loan from the BIRD foundation fully repaid during the second quarter of 2016.
The finance expenses (income) by cash and non-cash components are as follows: | | | Year ended December 31, | | | | | 2017 | | | 2016 | | | | | (in thousands of U.S. dollars) | | | Interest on bank deposits | | $ | (532 | ) | | $ | (536 | ) | | Gain from marketable securities, net | | | (602 | ) | | | (401 | ) | | Non-cash foreign exchange profit, net | | | - | | | | (24 | ) | | Total income | | | (1,134 | ) | | | (961 | ) | | Less: | | | | | | | | | | Other expenses | | | 14 | | | | 17 | | | Finance expenses on BIRD loan | | | - | | | | 243 | | | Non-cash foreign exchange loss, net | | | 57 | | | | - | | | Total expenses | | | 71 | | | | 260 | | | Finance income, net | | $ | (1,063 | ) | | $ | (701 | ) |
54| | | Year ended December 31, | | | | | 2017 | | | 2016 | | | | | (in thousands of U.S. dollars) | | | Interest on bank deposits | | $ | (532 | ) | | $ | (536 | ) | | Gain from marketable securities, net | | | (602 | ) | | | (401 | ) | | Non-cash foreign exchange profit, net | | | - | | | | (24 | ) | | Total income | | | (1,134 | ) | | | (961 | ) | | Less: | | | | | | | | | | Other expenses | | | 14 | | | | 17 | | | Finance expenses on BIRD loan | | | - | | | | 243 | | | Non-cash foreign exchange loss, net | | | 57 | | | | - | | | Total expenses | | | 71 | | | | 260 | | | Finance income, net | | $ | (1,063 | ) | | $ | (701 | ) |
Taxes on income
During 2016 and 2017, and 2016, we havedid not generatedgenerate taxable income in Israel. However, we had incurred tax expenses in our U.S. subsidiary, Foamix Pharmaceuticals, Inc., in the amount of $1.2 million and $387,000$0.4 million for the years ended December 31, 2017 and 2016, respectively. The increase in tax expenses resulted from an increase in our provision for uncertain tax positions.
Net Loss
As a result of the foregoing, our net loss for the year ended December 31, 2017 was $65.7 million, compared to $29.3 million for the year ended December 31, 2016, an increase of $36.4 million, or 124%. Comparison of the Year ended December 31, 2016 to the Year Ended December 31, 2015
Revenues
Our total revenues increased by $4.7 million, or 553%, from $849,000 in the year ended December 31, 2015 to $5.5 million in the year ended December 31, 2016. The increase is mainly due to the increase of $2.7 million in royalty payments from Bayer HealthCare AG for the sales of Finacea® Foam, and additional contingent payments totaling $2.5 million, due to Bayer’s achievement of certain sale targets during 2016.
Cost of revenues
Our cost of revenues for the years ended December 31, 2016 and 2015 were $59,000 and $70,000, respectively. The $11,000 decrease in cost of revenues resulted primarily from a decrease in the development projects. Cost of revenues as a percentage of revenues for the years ended December 31, 2016 and 2015 was 1.1% and 8.2%, respectively. The decrease in the cost of revenues as a percentage of revenues was primarily due to the increase in royalty and contingent payments, to which no cost of revenue is related.
Research and development expenses
Our research and development expenses for the year ended December 31, 2016 were $25.9 million, representing an increase of $15.2 million, or 142%, compared to $10.7 million for the year ended December 31, 2015. The increase in research and development expenses resulted primarily from an increase of $12.8 million in costs relating to the FMX101 and FMX103 clinical trials and an increase of $2.2 million in payroll and payroll related expenses due to an increase in the number of R&D employees.
Selling, general and administrative expenses
Our general and administrative expenses for the year ended December 31, 2016 were $9.2 million, representing an increase of $2.2 million, or 31%, compared to $7.0 million for the year ended December 31, 2015. The increase in selling, general and administrative expenses resulted primarily from an increase of $1.3 million in payroll and other payroll-related expenses mainly due to an increase in headcount; an increase of $417,000 in market research expenses; an increase of $225,000 in travel expenses; and an increase of $201,000 in expenses related to the company’s board of directors.
Operating loss
As a result of the foregoing, our operating loss for the year ended December 31, 2016, was $29.6 million, compared to an operating loss of $16.9 million for the year ended December 31, 2015, an increase of $12.7 million, or 75%.
Finance income
In the year ended December 31, 2016, our financial income included mostly gains from marketable securities and interest earned on our bank deposits, partially offset by expenses on the loan from the BIRD foundation fully repaid during the second quarter of 2016. In the year ended December 31, 2015 our financial income included mostly gains from marketable securities and interest earned on our bank deposits.
The finance expenses (income) by cash and non-cash components are as follows:
| | | Year ended December 31, | | | | | 2016 | | | 2015 | | | | | (in thousands of U.S. dollars) | | | Other expenses | | $ | 17 | | | $ | 23 | | | Finance expenses on BIRD loan | | | 243 | | | | * | | | Total expenses | | | 260 | | | | 23 | | | Less: | | | | | | | | | | Interest on bank deposits | | | (536 | ) | | | (289 | ) | | Gain from marketable securities, net | | | (401 | ) | | | (180 | ) | | Non-cash foreign exchange profit, net | | | (24 | ) | | | (6 | ) | | Total income | | | (961 | ) | | | (475 | ) | | Finance income, net | | $ | (701 | ) | | $ | (452 | ) |
________________________
* Less than $1,000
Taxes on income
During 2015 and 2016, we have not generated taxable income in Israel. However, we had incurred tax expenses in our U.S. subsidiary, Foamix Pharmaceuticals, Inc., in the amount of $39,000 and $387,000 for the years 2015 and 2016 respectively.
Net Loss
As a result of the foregoing our loss for the year ended December 31, 2016 was $29.3 million, compared to $16.5 million for the year ended December 31, 2015, an increase of $12.8 million, or 78%.
Liquidity
Since our inception, we have incurred losses from operations and negative cash flows from our operations. For the twelve monthsyear ended December 31, 2018, we incurred a net loss of $74.2 million, which included $68.7 million used for operating activities. For the year ended December 31, 2017, we incurred a net loss of $65.7 million, which included $53.2 million used for operating activity. For the twelve months ended December 31, 2016, we incurred a net loss of $29.3 million, which included $27.4 million used for operating activity.activities.
As of December 31, 20172018 and December 31, 2016,2017, we had a working capital surplus of $59.3$90.7 million and $111.7$59.3 million, respectively, and an accumulated deficit of $215.4 million and $141.3 million, and $75.6 million, respectively.
Our principal source of liquidity as of December 31, 20172018 consisted of cash cash equivalents, restricted cash, bank deposits and marketable securitiesinvestments of $76.4$99.4 million.
In the second quarter of 2014,On April 13, 2018, we completedentered into a private placement of preferred A shares and warrantsSecurities Purchase Agreement with a group of new investors and several of our existing shareholders in two phases, on May 13, 2014 and June 3, 2014, raising a total of $8.2 million, net of issuance costs, in consideration of 1,036,431 preferred shares and 1,061,469 warrantsOrbiMed Partners Master Fund Limited, or OrbiMed, pursuant to purchase preferred shares.
In September 2014, we completed our initial public offering in which we sold 6,700,000 ordinary shares for $6.00 per share raising total net proceeds, after expenses,agreed to issue and sell, in a registered offering, an aggregate of approximately $35.7 million. In October 2014 the underwriters exercised their option to purchase an additional 968,2002,940,000 ordinary shares at a purchase price equivalent to $5.50 per share, for aggregate net proceeds of $6.00 per share.approximately $16.1 million, after deducting offering expenses. The proceeds from the exerciseclosing of the option, netissuance and sale of underwriters’ commission, were approximately $5.4 million, bringing the total net proceeds from the initial public offering, after expenses,these securities took place on April 16, 2018, pursuant to approximately $41.1 million.
In April 2015, we completed a follow-on offering in which we sold 7,419,353 ordinary shares, including the exercise of underwrites option, for $9.30 per share, raising total net proceeds, after expenses, of approximately $64.2 million.
On October 21, 2015, we filed with the Securities and Exchange Commission (the SEC) a “shelf” registration statement on a Form F-3 for the registration of our ordinary shares that we may, from time to time, offer and sell in one or more offerings with an aggregate offering price of up to $150 million. On September 12, 2016 we filed with the SEC an amendment to theeffective shelf registration statement on a Form F-3/A, which became effective on September 23, 2016.S-3.
On September 30, 2016,18, 2018, we completed anotheran additional follow-on offering under our amendedeffective shelf registration statement in which we sold 5,700,00011,670,000 ordinary shares for $9.50$6.00 per share, raising net proceeds, after expenses and underwriter commissions, of approximately $50.4$65.6 million. An additional 300,000 ordinary shares were sold by certain selling shareholders. In October 2016Upon closing of the offering, the underwriters partially exercised the option granted to them in the underwriting agreement and purchased an1,750,500 additional 411,959 ordinary shares at athe per share price of $9.50 per share.the offering. The proceeds from the exercise of the option, net of expenses and underwriter commissions, were approximately $3.7$9.8 million, bringing the total net proceeds from the offering to approximately $54.1$75.4 million. On February 24, 2017, we filed with the SEC a “shelf” registration statement on a Form F-3 for the registration of our ordinary shares that we may, from time to time, offer and sell in one or more offerings with an aggregate offering price of up to $291,936,389, which became effective on March 14, 2017. However, following our transition from foreign private issuer to domestic issuer status beginning January 1, 2018 and the filing of this Annual Report on Form 10-K, we will no longer be able to make use of such shelf registration statement on Form F-3 and will need to file a new registration statement on a form applicable to domestic issuers, such as Form S-3, should we wish to engage in further public offerings of our shares or other instruments.
We anticipate that with our existing cash and investments we will be able to fund our planned operating expenses and capital expenditure requirements for the third Phase III clinical trialthrough mid-2020. These planned expenses include: (a) any pre-commercialization and launch preparations for FMX101, whichassuming we expect to complete by the endreceive regulatory approval, (b) full development and filing of 2018, and for the two Phase III clinical trialsan NDA for FMX103, which we expect to complete bysubmit in mid-2019 and (c) certain pipeline development activities. We expect we will need additional funding to support our operating expenses and capital requirements for 2020 and beyond, including with regard to the endcommercialization of the third quarterany of 2019,our product candidates if regulatory approval is granted, and throughout the NDA filing for FMX101, whichto fund our internal and external research and development efforts. We have based this estimate on assumptions that may prove to be wrong, and we expect to complete by the end of 2019.could use our capital resources sooner than we currently expect.
Foamix Pharmaceuticals Inc., our wholly-owned subsidiary, was incorporated on May 6, 2014 under the laws of the State of Delaware, with the intent to serve as our marketing and sales arm in the U.S. As a result, we do not expect our subsidiary to distribute any dividends, or extend any loans or advances to us in the foreseeable future.
Capital Resources
Overview
To date, we have financed our operations primarily through private and public placements of our ordinary shares, convertible loans and through fees, cost reimbursements and royalties received from our licensees.
From inception through December 31, 2017,2018, we have received net cash proceeds of approximately $187.7$280.1 million from the issuance of ordinary shares, preferred shares, exercise of options and warrants and from convertible loans.
Cash flows
The following table summarizes our statement of cash flows for the years ended December 31, 2018, 2017 December 31, 2016 and December 31, 2015:2016:
| | | Year ended December 31, | | | | | | 2017 | | | 2016 | | | 2015 | | | Year Ended December 31, | | | | | (in thousands of U.S. dollars) | | | 2018 | | | 2017 | | | 2016 | | | Net cash (used in) / provided by: | | | | | | | | | | | (in thousands) | | | Operating activities | | $ | (53,177 | ) | | $ | (27,370 | ) | | $ | (12,498 | ) | | $ | (68,664 | ) | | $ | (53,177 | ) | | $ | (27,370 | ) | | Investing activities | | | 37,755 | | | | (15,018 | ) | | | (78,516 | ) | | | (11,755 | ) | | | 37,755 | | | | (15,018 | ) | | Financing activities | | $ | 140 | | | $ | 55,031 | | | $ | 66,801 | | | $ | 92,374 | | | $ | 140 | | | $ | 55,031 | |
Net cash used in operating activities
The use of cash in all periods resulted primarily from our net losses adjusted for non-cash charges and measurements and changes in components of working capital. Adjustments to net income for non-cash items consists mainly include depreciation and amortization andof share-based compensation.
Net cash used in operating activities was $68.7 million in the year ended December 31, 2018, compared to $53.2 million in the twelve monthsyear ended December 31, 2017 and compared to $27.4 million in the twelve months ended December 31, 2016 and compared to $12.5 of net cash used in operating activities in the twelve monthsyear ended December 31, 2015.2016. The increase was attributable primarily to the increase in company activity mostly related to clinical trials and payroll expenses.
Net cash used in investing activities
The use of cash in investing activities has been primarily related to purchase of marketable securities and investment in bank deposits. Net cash provided by investing activities was $37.8 million in the twelve months ended December 31, 2017, compared to net cash used in investing activities of $15.0 million in the twelve months ended December 31, 2016 and comparedwas primarily related to $78.5 million in the twelve months ended December 31, 2015. The change in investing activities between 2017, 2016 and 2015 was attributable primarily to increase in proceeds from the sale and maturity of marketable securities, andoffset by investment in bank deposits and marketable securities. Net cash used in investing activities was $11.8 million in the decreaseyear ended December 31, 2018, compared to net cash provided by investing activities of $37.8 million in the year ended December 31, 2017 and compared to $15.0 million cash invested.used in the year ended December 31, 2016.
Net cash provided by financing activities
Net cash provided by financing activities was $140,000$92.4 million in the twelve monthsyear ended December 31, 2018, an increase of $92.3 million from $0.1 million in the year ended December 31, 2017. The increase was attributable primarily to (i) the proceeds from the issuance of shares to OrbiMed based on the aforementioned securities purchase agreement, in an aggregate net amount of approximately $16.1 million after deducting offering expenses, (ii) an $0.8 million increase in proceeds from the exercise of warrants, and (iii) the proceeds from the issuance of shares in the follow-on offering which closed on September 18, 2018, of approximately $75.4 million after deducting offering expenses and underwriters’ commissions.
Net cash provided by financing activities was $0.1 million in the year ended December 31, 2017, a decrease of $54.9 million from $55.0 million in the twelve monthsyear ended December 31, 2016. The decrease was attributable primarily to the capital raised in our 2016 follow-on public offering. Net cash provided by financing activities was $55.0 million in the twelve months ended December 31, 2016, a decrease of $11.8 million from $66.8 million in the twelve months ended December 31, 2015. The decrease was attributable primarily to a larger amount of capital raised in the 2015 financing round compared to the 2016 financing round.
Cash and funding sources
The table below summarizes our main sources of financing for the years ended December 31, 2018, 2017 2016 and 2015:2016:
| | | Proceeds from our public offerings(1) | | | Proceeds from issuance of ordinary shares | | | Payments from licensees | | | Total | | | | | (in thousands of U.S. dollars) | | | Year ended December 31, 2017 | | $ | - | | | $ | 161 | | | $ | 5,978 | | | $ | 6,139 | | | Year ended December 31, 2016 | | $ | 54,132 | | | $ | 1,407 | | | $ | 2,575 | | | $ | 58,114 | | | Year ended December 31, 2015 | | $ | 64,202 | | | $ | 2,629 | | | $ | 1,063 | | | $ | 67,894 | |
___________________________| | | Proceeds from our underwritten public offerings(1) | | | Proceeds from our direct public offerings | | | Proceeds from issuance of ordinary shares | | | Payments from licensees | | | Total | | | | | (in thousands of U.S. dollars) | | | 2018 | | $ | 75,356 | | | $ | 16,131 | | | $ | 887 | | | $ | 3,457 | | | $ | 95,831 | | | 2017 | | $ | - | | | $ | - | | | $ | 161 | | | $ | 5,978 | | | $ | 6,139 | | | 2016 | | $ | 54,132 | | | $ | - | | | $ | 1,407 | | | $ | 2,575 | | | $ | 58,114 | |
__________________________ (1) Net of issuance costs.
Our sources of financing in the year ended December 31, 2018 totaled $95.8 million and consisted primarily of $75.4 million of net proceeds from our underwritten follow-on offering which closed in September 2018 and $16.1 million of net proceeds from our registered direct offering to OrbiMed.
Our sources of financing in the year ended December 31, 2017 totaled $6.1 million and consisted primarily of payments from licensees. Our sources of financing in the year ended December 31, 2016 totaled $58.1 million and consisted primarily of $54.1 million of net proceeds from our 2016 follow-on public offering.
We currently have no ongoing material financial commitments, (suchsuch as lines of credit)credit, that may affect our liquidity over the next five years.
Funding requirements
We believe, based on our current business plan,anticipate that with our existing cash and investments we will enable usbe able to fund our planned operating expenses and capital expenditure requirements throughout the completionthrough mid-2020. These planned expenses and expenditures include: (a) full development and filing of our third pivotal Phase III clinical trialan NDA for our lead product candidate FMX101 and our two pivotal Phase III clinical trials for FMX103, the first of which we expect to complete bysubmit in mid-2019, (b) certain pipeline development activities, and (c) any pre-commercialization and launch preparations for FMX101 in anticipation of regulatory approval. We expect we will need additional funding to support our operating expenses and capital requirements for 2020 and beyond, including with regard to the endcommercialization of 2018our product candidates and the other two by the end of the third quarter of 2019,to fund our internal and the fullexternal research and development and NDA submission for FMX101. The full development and NDA submission for FMX103, as well as any future pipeline products, will require us to raise additional funds.efforts. We have based this estimate on assumptions that may prove to be wrong, and we could use our capital resources sooner than we currently expect. Our present and future funding requirements will depend on many factors, including, among other things:inter alia:
| · | the progress, timing and completion of preclinical testing and clinical trials for FMX101, FMX103 or any future pipeline product;product candidates; |
| · | selling, marketing and patent-related activities undertaken in connection with the anticipated commercialization of FMX101, FMX103 and any other product candidates and costs involved in the development of an effective sales and marketing organization; |
| · | the time and costs involved in obtaining regulatory approval for FMX101, FMX103 and our other pipeline products and any delays we may encounter as a result of evolving regulatory requirements or adverse results with respect to any of these products; |
| · | the number of potential new products we identify and decide to develop; |
| · | the costs involved in filing and prosecuting patent applications and obtaining, maintaining and enforcing patents or defending against claims or infringements raised by third parties, and license royalties or other amounts we may be required to pay to obtain rights to third party intellectual property rights; and |
| · | the amount of revenues, if any, we may derive either directly or in the form of royalty payments from future sales of FMX101, FMX103 and any other pipeline product that is commercialized. |
For more informationFurthermore, our operating plan may change as a result of many factors currently unknown to us. We have never before launched a product commercially, and the risks associated withcosts involved in such commercial launch may exceed our future funding needs, see “Item 1A—Risk Factors—Risks Relatedexpectations. We may therefore need to Our Businessseek additional capital sooner than planned, through public or private equity or debt financings or other sources, such as strategic collaborations or additional license arrangements. Such financings may result in dilution to shareholders, imposition of debt covenants and Industry—We will require substantial additional financing to achieverepayment obligations or other restrictions that may affect our goals, and a failure to obtain this necessary capital when needed on acceptable terms, or at all, could force us to delay, limit, reduce or terminate our product development, other operations or commercialization efforts.”business.
Our capital expenditures for 2018, 2017 2016 and 20152016 amounted to $0.6 million, $1.5 million $424,000 and $500,000,$0.4 million, respectively. During 2017,2018, these expenditures were primarily related to laboratory equipment, computers and leasehold improvements and purchase of laboratory equipment.improvements.
Off-Balance Sheet Arrangements
As of December 31, 2017,2018, we did not have any off-balance sheet arrangements. Contractual Obligations
Our significant non-cancelable contractual obligations as of December 31, 2017 are summarized in the following table:
| | | Payments due by period | | | | | Total | | | Less than 1 year | | | 1-3 years | | | 3-5 years | | | More than 5 years | | | Other | | | | | (in thousands of U.S. dollars) | | | | | Operating lease obligations(1) | | $ | 2,322 | | | $ | 865 | | | $ | 1,457 | | | | - | | | | - | | | | | Liability for employee severance benefits(2) | | $ | 437 | | | | - | | | | - | | | | - | | | | - | | | $ | 437 | | | Total | | $ | 2,759 | | | $ | 865 | | | $ | 1,457 | | | | - | | | | - | | | $ | 437 | |
_________________________________________
(1) | Operating lease obligations consist of lease of our facilities and lease of vehicles. |
(2) | The liability is considered long term, however we cannot estimate the exact period in which they will be paid. |
Critical Accounting Policies and Significant Judgments and Estimates
We prepare our consolidated financial statements in accordance with generally accepted accounting principles in the U.S.United States. The preparation of consolidated financial statements also requires us to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenue, expenses and related disclosures. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances. Actual results could differ significantly from the estimates made by our management. To the extent that there are differences between our estimates and actual results, our future financial statement presentation, financial condition, results of operations and cash flows will be affected.
While our significant accounting policies are more fully described in Note 2, Summary“Summary of Significant Accounting Policies,” to the consolidated financial statements included in “Item 8—“Financial Statements and Supplementary Data”Data” of this Annual Report, on Form 10-K, we believe that the following accounting policies arepolicy is the most critical to assist shareholders and investors reading the consolidated financial statements in fully understanding and evaluating our financial condition and results of operations. These policies relateThe policy relates to the more significant areas involving management’smanagement’s judgments and estimates and they requirerequires our most difficult, subjective or complex judgments, often as a result of the need to make estimates about the effect of the matters that are inherently uncertain.
Clinical trial accruals
Clinical trial costs are charged to research and development expense as incurred. We accrue for expenses resulting from obligations under contracts with clinical research organizations, or CROs. The financial terms of these contracts are subject to negotiations, which vary from contract to contract and may result in payment flows that do not match the periods over which materials or services are provided. Our objective is to reflect the appropriate trial expense in the consolidated financial statements by matching the appropriate expenses with the period in which services and efforts are expended. In the event advance payments are made to a CRO, the payments will be recorded as other assets, which will be recognized as expenses as services are rendered. The CRO contracts generally include pass-through fees including, but not limited to, regulatory expenses, investigator fees, travel costs and other miscellaneous costs. We estimate our clinical accruals based on reports from and discussion with clinical personnel and the CRO as to the progress or state of completion of the trials. We estimate accrued expenses as of each balance sheet date in the consolidated financial statements based on the facts and circumstances known at that time. Our clinical trial accrual is dependent, in part, upon the receipt of timely and accurate reporting from the CROs. Equity-based compensation
The fair value of equity-based payment transactions is recognized as an expense over the requisite service period and computed using the Black-Scholes model. We recognize compensation costs for awards that are conditioned only on continued service and which have a graded vesting schedule using the straight-line method based on the multiple-option award approach. When options and restricted share units, or RSUs, are granted as consideration for services provided by consultants and other non-employees, the grant is accounted for based on the fair value of the consideration received or the fair value of the awards issued, whichever is more reliably measurable. The fair value of the awards granted is measured on a final basis at the end of the related service period and is recognized over the related service period using the straight-line method.
Recently Issued Accounting Pronouncements
Certain recently issued accounting pronouncements are discussed in Note 2, Summary“Summary of Significant Accounting Policies,” to the consolidated financial statements included in “Item 8—“Financial Statements and Supplementary Data”Data” of this Annual Report on Form 10-K.Report. ITEM 7A —- QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Market risk is the risk of loss related to changes in market prices, including interest rates, foreign exchange rates and prices of financial instruments, that may adversely impact our financial position, results of operations or cash flows. As of December 31, 20172018 we did not have any financial instruments sensitive to market risk. We, therefore, have little exposure to market risks in the ordinary course of our operations, and such risks are primarily related to changes in foreign currency exchange rates and in interest rates.
Foreign Currency Exchange Risk
The U.S. dollar is our functional and reporting currency. Although a substantial portion of our expenses (mainly salaries and related costs) are denominated in Israeli shekels,NIS, accounting for 11%13%, 21%,11% and 32%21% of our expenses in the years ended December 31, 2018, 2017 2016 and 2015,2016, respectively, almost all our revenues were generated under agreements denominated in U.S. dollars and our proceeds from our public offerings, share issuance and convertible loan agreements, which are the main source of our financing, are denominated in U.S. dollars. Furthermore, while we anticipate that a portion of our expenses, principally salaries and related personnel expenses in Israel, will continue to be denominated in shekels,NIS, we expect to incur an increasing amount of expenses in U.S. dollars as we expand our operations in the U.S. We also have expenses, although to a much lesser extent, in other non-dollar currencies, in particular the Euro.Swiss Franc. Moreover, for the next few years we expect that the substantial majority of our revenues, if any, will be denominated in U.S. dollars from the sale of FMX101, if approved, and potentially other product candidates in the U.S. Having the substantial majority of our revenues denominated in U.S. dollars while having a substantial portion of our expenses denominated in Israeli shekelsNIS and other non-U.S. currencies exposes us to risk associated with exchange rate fluctuations vis-à-vis the U.S. dollar. See “Item 1A—“Risk Factors—RisksFactors-Risks Related to Our Business and Industry—Industry-Exchange rate fluctuations between the U.S. dollar and the New Israeli shekelShekel may negatively affect our earnings.earnings.”
A devaluation of the shekelNIS in relation to the U.S. dollar has the effect of reducing the U.S. dollar amount of our expenses or payables that are payable in shekels,NIS, unless those expenses or payables are linked to the U.S. dollar. Conversely, any appreciation of the shekelNIS in relation to the U.S. dollar has the effect of increasing the U.S. dollar value of our unlinked shekelNIS expenses, which would have a negative impact on our profit margins. In 2017,2018, the value of the shekel appreciatedNIS depreciated in relation to the U.S. dollar by 11.6%8.1%, the effect of which was partially offset by inflation in Israel at athe rate of approximately 0.4%0.8%. In 2016,2017, the value of the shekelNIS appreciated in relation to the U.S. dollar by approximately 1.5%11.6%, the effect of which was compoundedamplified by deflationinflation in Israel at the rate of approximately -0.2%0.4%.
Because exchange rates between the U.S. dollar and the shekelNIS (as well as between the U.S. dollar and other currencies) fluctuate continuously, such fluctuations have an impact on our results and period-to-period comparisons of our results. The effects of foreign currency re-measurements are reported in our statements of operations.
The following table presents information about the changes in the exchange rates of the shekelNIS against the U.S. dollar:
| | Shekel against the U.S. dollar | | NIS against the U.S. dollar | | | 2016 | 1.5% | | | 2017 | 11.6% | | | 11.6 | % | | 2018 | | | | (8.1 | )% |
We will continue to monitor our exposure to currency fluctuations. Since February 2015, we engagehave engaged in currency hedging activities in order to reduce our exposure to currency fluctuations. Instruments that are used to hedge future risks may include foreign currency forward contract, swap contracts and options. These instruments may be used to selectively manage risks, but we may not be fully protected against material foreign currency fluctuations.
Inflation-Related Risks
We do not believe that the rate of inflation in Israel has had a material impact on our business to date, however,financial condition or results of operations during the years ended December 31, 2018 and 2017. However, our costs in Israel will increase if inflation in Israel exceeds the devaluation of the shekelNIS against the U.S. dollar or if the timing of such devaluation lags behind inflation in Israel.
66 60
ITEM 8 —- FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
Financial Statements INDEX OFFOAMIX PHARMACEUTICALS LTD. CONSOLIDATED FINANCIAL STATEMENTS
AS OF DECEMBER 31, 2018
67 FOAMIX PHARMACEUTICALS LTD. CONSOLIDATED FINANCIAL STATEMENTS AS OF DECEMBER 31, 2018 INDEX | | Page | | | | | F-2 | | | | | F-3 | | | | | F-5 | | | | | F-6 | | | | | F-7 | | | | | F-8 | | | | | F-10 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM To the shareholders and the board of directors of FOAMIX PHARMACEUTICALS LTD.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Foamix Pharmaceuticals Ltd. and its subsidiarySubsidiary (the "Company") as of December 31, 20172018 and 2016,2017, and the related consolidated statements of operations, comprehensive loss, changes in shareholders' equity and cash flows for each of the three years in the period ended December 31, 2017,2018, including the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company and its subsidiary as of December 31, 20172018 and 2016,2017, and the results of theirits operations and theirits cash flows for each of the three years in the period ended December 31, 20172018 in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management and Board of Directors.management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”)(PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits of these consolidated financial statements in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion. | Tel-Aviv, Israel | /s/ Kesselman & Kesselman | February 27, 201828, 2019 | Certified Public Accountants (Isr.) | | | A member firm of PricewaterhouseCoopers International Limited |
We have served as the Company's auditor since 2006.
Kesselman & Kesselman, Trade Tower, 25 Hamered Street, Tel-Aviv 6812508, Israel,P.O Box 50005 Tel-Aviv 6150001 Telephone: +972 -3- 7954555, Fax:+972 -3- 7954556, www.pwc.com/il
| | | December 31 | | | December 31 | | | | | 2017 | | | 2016 | | | 2018 | | | 2017 | | | A s s e t s | | | | | | | | | | | | | | CURRENT ASSETS: | | | | | | | | | | | | | | Cash and cash equivalents | | $ | 15,956 | | | $ | 31,190 | | | $ | 27,868 | | | $ | 15,956 | | | Restricted cash | | | 250 | | | | 250 | | | | 250 | | | | 250 | | | Short term bank deposits | | | 19,443 | | | | 38,351 | | | | 24,047 | | | | 19,443 | | | Investment in marketable securities (Note 4) | | | 31,797 | | | | 43,275 | | | | 46,669 | | | | 31,797 | | | Restricted investment in marketable securities (Note 4) | | | 290 | | | | 261 | | | | 268 | | | | 290 | | | Accounts receivable: | | | | | | | | | | | | | | | | | | Trade | | | 996 | | | | 3,236 | | | | 1,066 | | | | 996 | | | Other (Note 11a) | | | 772 | | | | 438 | | | | 999 | | | | 772 | | | TOTAL CURRENT ASSETS | | | 69,504 | | | | 117,001 | | | | 101,167 | | | | 69,504 | | | | | | | | | | | | | | | | | | | | | NON-CURRENT ASSETS: | | | | | | | | | | | | | | | | | | Investment in marketable securities (Note 4) | | | 8,533 | | | | 17,532 | | | | 150 | | | | 8,533 | | | Restricted investment in marketable securities (Note 4) | | | 143 | | | | 129 | | | | 133 | | | | 143 | | | Property and equipment, net (Note 5) | | | 2,042 | | | | 938 | | | | 2,235 | | | | 2,042 | | | Other | | | 32 | | | | 35 | | | | 46 | | | | 32 | | | TOTAL NON-CURRENT ASSETS | | | 10,750 | | | | 18,634 | | | | 2,564 | | | | 10,750 | | | | | | | | | | | | | | | | | | | | | TOTAL ASSETS | | $ | 80,254 | | | $ | 135,635 | | | $ | 103,731 | | | $ | 80,254 | |
The accompanying notes are an integral part of these consolidated financial statements.
FOAMIX PHARMACEUTICALS LTD. CONSOLIDATED BALANCE SHEETS (U.S. dollars in thousands) | | | December 31 | | | | | 2018 | | | 2017 | | | Liabilities and shareholders’ equity | | | | | | | | | | | | | | | | CURRENT LIABILITIES: | | | | | | | | Accounts payable and accruals: | | | | | | | | Trade | | $ | 6,327 | | | $ | 6,436 | | | Deferred revenues | | | - | | | | 62 | | | Other (Note 11b) | | | 4,141 | | | | 3,730 | | | TOTAL CURRENT LIABILITIES | | | 10,468 | | | | | | | | | | | | | | | | LONG-TERM LIABILITIES: | | | | | | | | | | Liability for employee severance benefits (Note 6) | | | 367 | | | | 437 | | Other liabilities | | | | | | | | | | TOTAL LONG-TERM LIABILITIES | | | | | | | | | | TOTAL LIABILITIES | | | | | | | | | COMMITMENTS (Note 7) | | | | | | | | | | SHAREHOLDERS' EQUITY: | | | | | | | | | Ordinary shares, NIS 0.16 par value - authorized: 90,000,000 ordinary shares as of December 31, 2018 and December 31, 2017; issued and outstanding: 54,351,140 and 37,498,128 ordinary shares as of December 31, 2018 and December 31, 2017, respectively | | | 2,331 | | | | 1,576 | | | Additional paid-in capital | | | 305,303 | | | | 208,364 | | | Accumulated deficit | | | (215,409 | ) | | | (141,281 | ) | | Accumulated other comprehensive loss | | | (43 | ) | | | (58 | ) | | TOTAL SHAREHOLDERS' EQUITY | | | | | | | | | | TOTAL LIABILITIES AND SHAREHOLDERS’ EQUITY | | $ | 103,731 | | | $ | 80,254 | |
| | | December 31 | | | | | 2017 | | | 2016 | | | Liabilities and shareholders’ equity | | | | | | | | | | | | | | | | CURRENT LIABILITIES: | | | | | | | | Current maturities of bank borrowing (Note 8b) | | $ | - | | | $ | 20 | | | Accounts payable and accruals: | | | | | | | | | | Trade | | | 6,436 | | | | 2,267 | | | Deferred revenues | | | 62 | | | | - | | | Other (Note 11b) | | | 3,730 | | | | 2,984 | | | TOTAL CURRENT LIABILITIES | | | 10,228 | | | | | | | | | | | | | | | | LONG-TERM LIABILITIES: | | | | | | | | | | Liability for employee severance benefits (Note 6) | | | 437 | | | | 379 | | Other liabilities | | | | | | | | | | TOTAL LONG-TERM LIABILITIES | | | | | | | | | | TOTAL LIABILITIES | | | | | | | | | COMMITMENTS (Note 7) | | | | | | | | | | SHAREHOLDERS' EQUITY: | | | | | | | | | Ordinary Shares, NIS 0.16 par value - authorized: 90,000,000 and 50,000,000 Ordinary Shares as of December 31, 2017 and December 31, 2016, respectively; issued and outstanding: 37,498,128 and 37,167,791 Ordinary Shares as of December 31, 2017 and December 31, 2016, respectively | | | 1,576 | | | | 1,561 | | | Additional paid-in capital | | | 208,364 | | | | 204,052 | | | Accumulated deficit | | | (141,281 | ) | | | (75,566 | ) | | Accumulated other comprehensive loss | | | (58 | ) | | | (62 | ) | | TOTAL SHAREHOLDERS' EQUITY | | | | | | | | | | TOTAL LIABILITIES AND SHAREHOLDERS’ EQUITY | | $ | 80,254 | | | $ | 135,635 | |
The accompanying notes are an integral part of these consolidated financial statements.
| | | Year ended December 31 | | | | | 2018 | | | 2017 | | | 2016 | | REVENUES (Note 11c) | | $ | 3,595 | | | $ | 3,669 | | | $ | 5,527 | | | COST OF REVENUES | | | - | | | | 13 | | | | 59 | | | GROSS PROFIT | | | 3,595 | | | | 3,656 | | | | 5,468 | | | OPERATING EXPENSES: | | | | | | | | | | | | | | Research and development | | | 64,474 | | | | 57,779 | | | | 25,897 | | | Selling, general and administrative | | | 14,013 | | | | 11,491 | | | | 9,221 | | | TOTAL OPERATING EXPENSES | | | | | | | | | | | | | | OPERATING LOSS | | | 74,892 | | | | 65,614 | | | | 29,650 | | FINANCE INCOME, net (Note 11d) | | | (941 | ) | | | (1,063 | ) | | | (701 | ) | | LOSS BEFORE INCOME TAX | | | 73,951 | | | | 64,551 | | | | 28,949 | | INCOME TAX (Note 10) | | | | | | | | | | | | | | NET LOSS FOR THE YEAR | | $ | 74,163 | | | $ | 65,715 | | | $ | 29,336 | | | | | | | | | | | | | | | | | LOSS PER SHARE BASIC AND DILUTED | | $ | 1.70 | | | $ | 1.76 | | | $ | 0.91 | | | | | | | | | | | | | | | | | WEIGHTED AVERAGE NUMBER OF SHARES OUTSTANDING USED IN COMPUTATION OF BASIC AND DILUTED LOSS PER SHARE IN THOUSANDS | | | 43,660 | | | | 37,376 | | | | 32,263 | |
The accompanying notes are an integral part of these consolidated financial statements. FOAMIX PHARMACEUTICALS LTD.
CONSOLIDATED STATEMENTS OF OPERATIONS (U.S. dollars in thousands, except per share data)
| | | Year ended December 31 | | | | | 2017 | | | 2016 | | | 2015 | | REVENUES (Note 11c) | | $ | 3,669 | | | $ | 5,527 | | | $ | 849 | | | COST OF REVENUES | | | 13 | | | | 59 | | | | 70 | | | GROSS PROFIT | | | 3,656 | | | | 5,468 | | | | 779 | | | OPERATING EXPENSES: | | | | | | | | | | | | | | Research and development | | | 57,779 | | | | 25,897 | | | | 10,680 | | | Selling, general and administrative | | | 11,491 | | | | 9,221 | | | | 7,029 | | | TOTAL OPERATING EXPENSES | | | | | | | | | | | | | | OPERATING LOSS | | | 65,614 | | | | 29,650 | | | | 16,930 | | FINANCE INCOME, net (Note 11d) | | | (1,063 | ) | | | (701 | ) | | | (452 | ) | | LOSS BEFORE INCOME TAX | | | 64,551 | | | | 28,949 | | | | 16,478 | | INCOME TAX (Note 10) | | | | | | | | | | | | | | NET LOSS FOR THE YEAR | | $ | 65,715 | | | $ | 29,336 | | | $ | 16,517 | | | | | | | | | | | | | | | | | LOSS PER SHARE BASIC AND DILUTED | | $ | 1.76 | | | $ | 0.91 | | | $ | 0.58 | | | | | | | | | | | | | | | | | WEIGHTED AVERAGE NUMBER OF SHARES OUTSTANDING USED IN COMPUTATION OF BASIC AND DILUTED LOSS PER SHARE IN THOUSANDS | | | 37,376 | | | | 32,263 | | | | 28,229 | |
The accompanying notes are an integral part of these consolidated financial statements.
| | | Year ended December 31 | | | Year ended December 31 | | | | | 2017 | | | 2016 | | | 2015 | | | 2018 | | | 2017 | | | 2016 | | | NET LOSS | | | | | | | | | | | $ | 74,163 | | | $ | 65,715 | | | $ | 29,336 | | | OTHER COMPREHENSIVE LOSS (INCOME): | | $ | 65,715 | | | $ | 29,336 | | | $ | 16,517 | | | | OTHER COMPREHENSIVE INCOME: | | | | | | | | | | | | | | | Net unrealized losses (gains) from marketable securities | | | 5 | | | | (65 | ) | | | 103 | | | | (59 | ) | | | 5 | | | | (65 | ) | | Gains (losses) on marketable securities reclassified into net loss | | | - | | | | 4 | | | | (57 | ) | | | (5 | ) | | | - | | | | 4 | | | Net unrealized losses (gains) on derivative financial instruments | | | (146 | ) | | | (20 | ) | | | 5 | | | | 74 | | | | (146 | ) | | | (20 | ) | | Gains on derivative financial instruments reclassified into net loss | | | 137 | | | | 13 | | | | - | | | | TOTAL OTHER COMPREHENSIVE LOSS (INCOME) | | | (4 | ) | | | (68 | ) | | | 51 | | | | Gains (losses) on derivative financial instruments reclassified into net loss | | | | (60 | ) | | | 137 | | | | 13 | | | TOTAL OTHER COMPREHENSIVE INCOME | | | | (50 | ) | | | (4 | ) | | | (68 | ) | | TOTAL COMPREHENSIVE LOSS | | $ | 65,711 | | | $ | 29,268 | | | $ | 16,568 | | | $ | 74,113 | | | $ | 65,711 | | | $ | 29,268 | |
The accompanying notes are an integral part of these consolidated financial statements
| | | Ordinary shares | | | Additional paid-in capital | | | Accumulated deficit | | | Accumulated other comprehensive loss | | | Total | | | | | Number of shares | | | Amounts | | | Amounts | | | BALANCE AT JANUARY 1, 2015 | | | 22,443,934 | | | | 954 | | | | 77,600 | | | | (29,713 | ) | | | (79 | ) | | | 48,762 | | | CHANGES DURING 2015: | | | | | | | | | | | | | | | | | | | | | | | | | | Comprehensive loss | | | - | | | | - | | | | - | | | | (16,517 | ) | | | (51 | ) | | | (16,568 | ) | | Issuance of Ordinary Shares through a public offering, net of $4.8 million issuance costs (Note 9b) | | | 7,419,353 | | | | 298 | | | | 63,904 | | | | - | | | | - | | | | 64,202 | | | Exercise of warrants (Note 9c) | | | 546,322 | | | | 23 | | | | 2,262 | | | | - | | | | - | | | | 2,285 | | | Exercise of options and restricted share units (Note 9d) | | | 229,525 | | | | 9 | | | | 335 | | | | - | | | | - | | | | 344 | | | Share-based compensation (Note 9d) | | | - | | | | - | | | | 1,777 | | | | - | | | | - | | | | 1,777 | | | BALANCE AT DECEMBER 31, 2015 | | | 30,639,134 | | | | 1,284 | | | | 145,878 | | | | (46,230 | ) | | | (130 | ) | | | 100,802 | | | | | | | | | | | | | | | | | | | | | | | | | | | | | CHANGES DURING 2016: | | | | | | | | | | | | | | | | | | | | | | | | | | Comprehensive income (loss) | | | - | | | | - | | | | - | | | | (29,336 | ) | | | 68 | | | | (29,268 | ) | Issuance of Ordinary Shares through a public offering, net of $3.9 million issuance costs (Note 9b) | | | 6,111,959 | | | | 260 | | | | 53,872 | | | | - | | | | - | | | | 54,132 | | | Exercise of warrants (Note 9c) | | | 257,137 | | | | 10 | | | | 1,285 | | | | - | | | | - | | | | 1,295 | | | Exercise of options and restricted share units (Note 9d) | | | 159,561 | | | | 7 | | | | 105 | | | | - | | | | - | | | | 112 | | | Share-based compensation (Note 9d) | | | - | | | | - | | | | 2,912 | | | | | | | | | | | | 2,912 | | | BALANCE AT DECEMBER 31, 2016 | | | 37,167,791 | | | | 1,561 | | | | 204,052 | | | | (75,566 | ) | | | (62 | ) | | | 129,985 | | | | | | | | | | | | | | | | | | | | | | | | | | | | | CHANGES DURING 2017: | | | | | | | | | | | | | | | | | | | | | | | | | | Comprehensive income (loss) | | | - | | | | - | | | | - | | | | (65,715 | ) | | | 4 | | | | (65,711 | ) | | Exercise of warrants (Note 9c) | | | 191,793 | | | | 8 | | | | (8 | ) | | | - | | | | - | | | | - | | | Exercise of options and restricted share units (Note 9d) | | | 138,544 | | | | 7 | | | | 154 | | | | - | | | | - | | | | 161 | | | Share-based compensation (Note 9d) | | | - | | | | - | | | | 4,166 | | | | - | | | | - | | | | 4,166 | | | BALANCE AT DECEMBER 31, 2017 | | | 37,498,128 | | | $ | 1,576 | | | $ | 208,364 | | | $ | (141,281 | ) | | $ | (58 | ) | | $ | 68,601 | |
| | | Ordinary shares | | | Additional paid-in capital | | | Accumulated deficit | | | Accumulated other comprehensive loss | | | Total | | | | Number of shares | | | Amounts | | | Amounts | | | BALANCE AT JANUARY 1, 2016 | | | 30,639,134 | | | $ | 1,284 | | | $ | 145,878 | | | $ | (46,230 | ) | | $ | (130 | ) | | $ | 100,802 | | | CHANGES DURING 2016: | | | | | | | | | | | | | | | | | | | | | | | | | | Comprehensive income (loss) | | | - | | | | - | | | | - | | | | (29,336 | ) | | | 68 | | | | (29,268 | ) | Issuance of ordinary shares through a public offering, net of $3.9 million issuance costs (Note 9b) | | | 6,111,959 | | | | 260 | | | | 53,872 | | | | - | | | | - | | | | 54,132 | | | Exercise of warrants (Note 9d) | | | 257,137 | | | | 10 | | | | 1,285 | | | | - | | | | - | | | | 1,295 | | | Exercise of options and restricted share units (Note 9e) | | | 159,561 | | | | 7 | | | | 105 | | | | - | | | | - | | | | 112 | | | Share-based compensation (Note 9e) | | | - | | | | - | | | | 2,912 | | | | - | | | | - | | | | 2,912 | | | BALANCE AT DECEMBER 31, 2016 | | | 37,167,791 | | | | 1,561 | | | | 204,052 | | | | (75,566 | ) | | | (62 | ) | | | 129,985 | | | CHANGES DURING 2017: | | | | | | | | | | | | | | | | | | | | | | | | | | Comprehensive income (loss) | | | - | | | | - | | | | - | | | | (65,715 | ) | | | 4 | | | | (65,711 | ) | | Exercise of warrants (Note 9d) | | | 191,793 | | | | 8 | | | | (8 | ) | | | - | | | | - | | | | - | | | Exercise of options and restricted share units (Note 9e) | | | 138,544 | | | | 7 | | | | 154 | | | | - | | | | - | | | | 161 | | | Share-based compensation (Note 9e) | | | - | | | | - | | | | 4,166 | | | | - | | | | - | | | | 4,166 | | BALANCE AT DECEMBER 31, 2017, as previously reported | | | 37,498,128 | | | $ | 1,576 | | | $ | 208,364 | | | $ | (141,281 | ) | | $ | (58 | ) | | $ | 68,601 | | Impact of initial adoption of new accounting standards (Note 4) | | | - | | | | - | | | | - | | | | 35 | | | | (35 | ) | | | - | | | CHANGES DURING 2018: | | | | | | | | | | | | | | | | | | | | | | | | | | Comprehensive income (loss) | | | - | | | | - | | | | - | | | | (74,163 | ) | | | 50 | | | | (74,113 | ) | | Issuance of ordinary shares through a public offering, net of $5.2 million issuance costs (note 9c) | | | 13,420,500 | | | | 599 | | | | 74,757 | | | | - | | | | - | | | | 75,356 | | | Issuance of ordinary shares through a securities purchase agreement, net of $39 issuance costs (note 9b) | | | 2,940,000 | | | | 134 | | | | 15,997 | | | | - | | | | - | | | | 16,131 | | | Exercise of warrants (Note 9d) | | | 178,468 | | | | 8 | | | | 832 | | | | - | | | | - | | | | 840 | | | Exercise of options and restricted share units (Note 9e) | | | 314,044 | | | | 14 | | | | 33 | | | | - | | | | - | | | | 47 | | | Share-based compensation (Note 9e) | | | - | | | | - | | | | 5,320 | | | | - | | | | - | | | | 5,320 | | | BALANCE AT DECEMBER 31, 2018 | | | 54,351,140 | | | $ | 2,331 | | | $ | 305,303 | | | $ | (215,409 | ) | | $ | (43 | ) | | $ | 92,182 | |
The accompanying notes are an integral part of these consolidated financial statements | | | Year ended December 31 | | | Year ended December 31 | | | | | 2017 | | | 2016 | | | 2015 | | | 2018 | | | 2017 | | | 2016 | | | CASH FLOWS FROM OPERATING ACTIVITIES: | | | | | | | | | | | | | | | | | | | | Net Loss | | $ | (65,715 | ) | | $ | (29,336 | ) | | $ | (16,517 | ) | | $ | (74,163 | ) | | $ | (65,715 | ) | | $ | (29,336 | ) | Adjustments required to reconcile net loss to net cash used in operating activities: | | | | | | | | | | | | | | | | | | | | | | | | | | Depreciation and amortization | | | 221 | | | | 143 | | | | 87 | | | | 319 | | | | 221 | | | | 143 | | | Loss from sale and disposal of fixed assets | | | 134 | | | | 16 | | | | 15 | | | | 44 | | | | 134 | | | | 16 | | | Changes in marketable securities and bank deposits, net | | | 97 | | | | 91 | | | | 57 | | | | 201 | | | | 97 | | | | 91 | | Changes in accrued liability for employee severance benefits, net of retirement fund profit | | | 57 | | | | 14 | | | | 35 | | | | (70 | ) | | | 57 | | | | 14 | | | Share-based compensation | | | 4,166 | | | | 2,912 | | | | 1,777 | | | | 5,320 | | | | 4,166 | | | | 2,912 | | | Non-cash finance income, net | | | (47 | ) | | | (1 | ) | | | (8 | ) | | | Non-cash finance expenses (income), net | | | | 43 | | | | (47 | ) | | | (1 | ) | | Changes in operating asset and liabilities: | | | | | | | | | | | | | | | | | | | | | | | | | | Decrease (increase) in trade and other receivable | | | 1,915 | | | | (2,889 | ) | | | 140 | | | | (308 | ) | | | 1,915 | | | | (2,889 | ) | | Decrease (increase) in other non-current assets | | | 4 | | | | - | | | | (1 | ) | | | (14 | ) | | | 4 | | | | - | | | Increase in accounts payable and accruals | | | 5,003 | | | | 1,680 | | | | 1,917 | | | | 238 | | | | 5,003 | | | | 1,680 | | | Increase in other liabilities | | | 988 | | | | | | | | | | | | Increase (decrease) in other liabilities | | | | (274 | ) | | | 988 | | | | - | | | Net cash used in operating activities | | | (53,177 | ) | | | (27,370 | ) | | | (12,498 | ) | | | (68,664 | ) | | | (53,177 | ) | | | (27,370 | ) | | | | | | | | | | | | | | | | | | | | | | | | | | | | CASH FLOWS FROM INVESTING ACTIVITIES: | | | | | | | | | | | | | | | | | | | | | | | | | | Purchase of fixed assets | | | (1,518 | ) | | | (424 | ) | | | (500 | ) | | | (567 | ) | | | (1,518 | ) | | | (424 | ) | | Proceeds from sale of fixed assets | | | 33 | | | | - | | | | - | | | | 10 | | | | 33 | | | | - | | | Investment in bank deposits | | | (17,000 | ) | | | (23,000 | ) | | | (28,000 | ) | | | (39,000 | ) | | | (17,000 | ) | | | (23,000 | ) | | Investment in marketable securities | | | (22,839 | ) | | | (31,700 | ) | | | (72,518 | ) | | | (38,652 | ) | | | (22,839 | ) | | | (31,700 | ) | Proceeds from sale and maturity of marketable securities and
bank deposits | | | 79,079 | | | | 40,106 | | | | 22,502 | | | | 66,454 | | | | 79,079 | | | | 40,106 | | | Net cash provided by (used in) investing activities | | | 37,755 | | | | (15,018 | ) | | | (78,516 | ) | | | (11,755 | ) | | | 37,755 | | | | (15,018 | ) | | | | | | | | | | | | | | | | | | | | | | | | | | | | CASH FLOWS FROM FINANCING ACTIVITIES: | | | | | | | | | | | | | | | | | | | | | | | | | | Proceeds from issuance of ordinary shares through public offerings, net of issuance costs | | | - | | | | 54,132 | | | | 64,202 | | | | Proceeds from issuance of ordinary shares through a public offering, net of $5.2 million issuance costs | | | | 75,356 | | | | - | | | | 54,132 | | | Proceeds from issuance of ordinary shares through a securities purchase agreement, net of $39 issuance costs | | | | 16,131 | | | | - | | | | - | | | Proceeds from exercise of warrants | | | - | | | | 1,295 | | | | 2,285 | | | | 840 | | | | - | | | | 1,295 | | | Proceeds from exercise of options | | | 161 | | | | 112 | | | | 344 | | | | 47 | | | | 161 | | | | 112 | | Payments in respect of BIRD loan | | | - | | | | (476 | ) | | | - | | | | - | | | | - | | | | (476 | ) | | Payments in respect of bank borrowings | | | (21 | ) | | | (32 | ) | | | (30 | ) | | | - | | | | (21 | ) | | | (32 | ) | | Net cash provided by financing activities | | | 140 | | | | 55,031 | | | | 66,801 | | | | 92,374 | | | | 140 | | | | 55,031 | | | | | | | | | | | | | | | | | | | | | | | | | | | | | INCREASE (DECREASE) IN CASH, CASH EQUIVALENTS AND RESTRICTED CASH | | | (15,282 | ) | | | 12,643 | | | | (24,213 | ) | | | 11,955 | | | | (15,282 | ) | | | 12,643 | | | EFFECT OF EXCHANGE RATE ON CASH, CASH EQUIVALENTS AND RESTRICTED CASH | | | 48 | | | | 2 | | | | *- | | | | (43 | ) | | | 48 | | | | 2 | | | | | | | | | | | | | | | | | | | | | | | | | | | | | CASH, CASH EQUIVALENTS AND RESTRICTED CASH AT BEGINNING OF THE YEAR | | | | | | | | | | | | | | | 16,206 | | | | 31,440 | | | | 18,795 | | | | | | | | | | | | | | | | | | | | | | | | | | | | | CASH, CASH EQUIVALENTS AND RESTRICTED CASH AT END OF THE YEAR | | $ | 16,206 | | | $ | 31,440 | | | $ | 18,795 | | | $ | 28,118 | | | $ | 16,206 | | | $ | 31,440 | | | | | | | | | | | | | | | | | | | | | | | | | | | | | Cash and cash equivalents | | $ | 15,956 | | | $ | 31,190 | | | $ | 18,795 | | | $ | 27,868 | | | $ | 15,956 | | | $ | 31,190 | | | Restricted cash | | | 250 | | | | 250 | | | | - | | | | 250 | | | | 250 | | | | 250 | | TOTAL CASH, CASH EQUIVALENTS AND RESTRICTED CASH SHOWN IN STATEMENT OF CASH FLOWS | | $ | 16,206 | | | $ | 31,440 | | | $ | 18,795 | | | $ | 28,118 | | | $ | 16,206 | | | $ | 31,440 | |
FOAMIX PHARMACEUTICALS LTD. CONSOLIDATED STATEMENTS OF CASH FLOWS
(U.S. dollars in thousands) | | | Year ended December 31 | | | | | 2017 | | | 2016 | | | 2015 | | SUPPLEMENTARY INFORMATION ON INVESTING AND FINANCING ACTIVITIES NOT INVOLVING CASH FLOWS: | | | | | | | | | | | Cashless exercise of warrants | | | 8 | | | | - | | | | 4 | | | Exercise of restricted share units | | | 3 | | | | 4 | | | | - | | | Property and equipment purchases included in accounts payable and accruals | | | 1 | | | | 27 | | | | - | | | | | | | | | | | | | | | | | SUPPLEMENTAL DISCLOSURE OF CASH FLOW INFORMATION: | | | | | | | | | | | | | | Cash paid for taxes | | | 478 | | | | 163 | | | | 16 | | | Interest received | | | 1,209 | | | | 1,015 | | | | 921 | | | Interest paid | | | | | | | 239 | | | | *- | |
| | | Year ended December 31 | | | | | 2018 | | | 2017 | | | 2016 | | | SUPPLEMENTARY INFORMATION ON INVESTING AND FINANCING ACTIVITIES NOT INVOLVING CASH FLOWS: | | | | | | | | | | | Cashless exercise of warrants | | | 1 | | | | 8 | | | | - | | | Exercise of restricted share units | | | 10 | | | | 3 | | | | 4 | | | Property and equipment purchases included in accounts payable and accruals | | | - | | | | 1 | | | | 27 | | | | | | | | | | | | | | | | SUPPLEMENTAL DISCLOSURE OF CASH FLOW INFORMATION: | | | | | | | | | | | | | | Cash paid for taxes | | | 587 | | | | 478 | | | | 163 | | | Interest received | | | 1,173 | | | | 1,209 | | | | 1,015 | | | Interest paid | | | | | | | *- | | | | 239 | |
* Represents an amount less than $1. The accompanying notes are an integral part of these consolidated financial statements. | | | December 31, 2016 | | | | | Level 1 | | | Level 2 | | | Total | | | Marketable securities | | $ | 957 | | | $ | 60,240 | | | $ | 61,197 | | | Currency options designated as hedging instruments (current asset) | | | - | | | $ | 2 | | | $ | 2 | |
The Company’s corporate debt securities are traded in markets that are not considered to be active, but are valued based on quoted market prices, broker or dealer quotations, or alternative pricing sources with reasonable levels of price transparency. Accordingly, these assets are categorized as Level 2.
Foreign exchange risk management
The Company purchases and writes non-functional currency options in order to hedge the currency exposure on the Company’s cash flow. The currency hedged items are denominated in New Israeli Shekel (NIS). The purchasing and writing of options is part of a comprehensive currency hedging strategy with respect to salary and rent expenses denominated in NIS. These transactions are at zero cost for periods of up to one year. The counterparties to the derivatives are major banks in Israel. As of December 31, 2017,2018, the total hedged amount was NIS 5.312.8 million.
The derivative asset,liability, in the amount of $11$3 as of December 31, 2017,2018, qualifies as hedge accounting.
As of December 31, 2017,2018, the Company has a lien in the amount of $290$268 on the Company’s marketable securities and a lien in the amount $250 on the Company’s checking account, in respect of bank guarantees granted in order to secure the hedging transactions.
NOTE 4 - MARKETABLE SECURITIES
Marketable securities as of December 31, 2017,2018, and December 31, 2016,2017 consist mainly of debt and mutual funds securities. TheseThe debt securities are classified as available-for-sale and are recorded at fair value. Changes in fair value, net of taxes (if applicable), are reflected in other comprehensive loss. Realized gains and losses on sales of the securities, as well as premium or discount amortization, are included in the consolidated statement of operations as finance income or expenses.
As of January 1, 2018, following the adoption of ASU No. 2016-01, Financial Instruments—Overall (Subtopic 825-10), equity securities with readily determinable fair value are measured at fair value. The changes in the fair value of equity investments are recognized through net income. Adoption of the standard was applied through a cumulative one-time adjustment of $35 to the accumulated deficit.
FOAMIX PHARMACEUTICALS LTD. NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (continued) (U.S. dollars in thousands, except share and per share amounts) NOTE 4 - MARKETABLE SECURITIES (continued):
The following table sets forth the Company’s marketable securities:
| | | December 31 | | | December 31 | | | | | 2017 | | | 2016 | | | 2018 | | | 2017 | | | Israeli mutual funds | | $ | 987 | | | $ | 957 | | | $ | 991 | | | $ | 987 | | | Certificates of deposit | | | 17,206 | | | | 33,350 | | | | 2,773 | | | | 17,206 | | | Government and agency bonds | | | 22,570 | | | | 26,890 | | | U.S Government and agency bonds | | | | 25,215 | | | | 22,570 | | U.S Treasury bills | | | | 18,241 | | | | - | | | Total | | $ | 40,763 | | | $ | 61,197 | | | $ | 47,220 | | | $ | 40,763 | |
FOAMIX PHARMACEUTICALS LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands, except share and per share amounts)
NOTE 4 - MARKETABLE SECURITIES (continued):
At December 31, 20172018 and 2016,2017, the fair value, cost and gross unrealized holding gains and losses of the marketable securities owned by the Company were as follows: | | | December 31, 2017 | | | | | Fair
value | | | Cost or Amortized cost | | | Gross unrealized
holding loss | | | Gross unrealized
holding gains | | | Israeli mutual funds | | $ | 987 | | | $ | 952 | | | $ | - | | | $ | 35 | | | Certificates of deposit | | | 17,206 | | | | 17,243 | | | | 38 | | | | 1 | | | Government and agency bonds | | | 22,570 | | | | 22,638 | | | | 68 | | | | - | | | Total | | $ | 40,763 | | | $ | 40,833 | | | $ | 106 | | | $ | 36 | |
| | | December 31, 2018 | | | | | Fair
value | | | Cost or Amortized cost | | | Gross unrealized
holding loss | | | Gross unrealized
holding gains | | Certificates of deposit | | $ | 2,773 | | | $ | 2,790 | | | $ | 17 | | | $ | - | | U.S Government and agency bonds | | | 25,215 | | | | 25,236 | | | | 22 | | | | 1 | | U.S Treasury bills | | | 18,241 | | | | 18,243 | | | | 3 | | | | 1 | | Total | | $ | 46,229 | | | $ | 46,269 | | | $ | 42 | | | $ | 2 | |
| | | | | | | | | December 31, 2016 | | | December 31, 2017 | | | | | Fair
value | | | Cost or Amortized cost | | | Gross unrealized
holding loss | | | Gross unrealized
holding gains | | | Fair
value | | | Cost or Amortized cost | | | Gross unrealized
holding loss | | | Gross unrealized
holding gains | | | Israeli mutual funds | | $ | 957 | | | $ | 952 | | | $ | - | | | $ | 5 | | | $ | 987 | | | $ | 952 | | | $ | - | | | $ | 35 | | | Certificates of deposit | | | 33,350 | | | | 33,408 | | | | 68 | | | | 10 | | | | 17,206 | | | | 17,243 | | | | 38 | | | | 1 | | | Government and agency bonds | | | 26,890 | | | | 26,901 | | | | 13 | | | | 2 | | | U.S. Government and agency bonds | | | | 22,570 | | | | 22,638 | | | | 68 | | | | - | | | Total | | $ | 61,197 | | | $ | 61,261 | | | $ | 81 | | | $ | 17 | | | $ | 40,763 | | | $ | 40,833 | | | $ | 106 | | | $ | 36 | |
As of December 31, 2017,2018, the unrealized losses attributed to the Company’s marketable securities were primarily due to credit spreads and interest rate movements. The Company has considered factors regarding other than temporary impaired securities and determined that there are no securities with impairment that is other than temporary as of December 31, 2017,2018, and December 31, 2016. During the year ended December 31, 2017 and December 31, 2016 the Company received proceeds of $43,079 and $27,106 upon sale and maturity of marketable securities.2017.
As of December 31, 2017,2018, and December 31, 2016,2017, the Company’s debt securities had the following maturity dates: | | | Market value | | | | | December 31 | | | | | 2018 | | | 2017 | | Due within one year | | $ | 46,079 | | | $ | 31,244 | | 1 to 2 years | | | 150 | | | | 8,380 | | 2 to 3 years | | | - | | | | 152 | | Total | | $ | 46,229 | | | $ | 39,776 | |
| | | Market value | | | | | December 31 | | | | | 2017 | | | 2016 | | | Due within one year | | $ | 31,244 | | | $ | 42,708 | | | 1 to 2 years | | | 8,380 | | | | 14,513 | | | 2 to 3 years | | | 152 | | | | 3,019 | | | Total | | $ | 39,776 | | | $ | 60,240 | |
$433401 and $390$433 of the Company’s marketable securities were restricted as of December 31, 2017,2018, and December 31, 2016,2017, respectively, due to a lien in respect of bank guarantees granted to secure hedging transaction and the Company’s rent agreement. Refer to note 7 and note 3.
NOTE 5 - PROPERTY AND EQUIPMENT
| | | December 31 | | | | | 2017 | | | 2016 | | | Cost: | | | | | | | | Leasehold improvements | | $ | 902 | | | $ | 229 | | | Computers and software | | | 299 | | | | 187 | | | Laboratory equipment | | | 1,257 | | | | 896 | | | Furniture | | | 288 | | | | 96 | | | Vehicles | | | 106 | | | | 198 | | | | | | 2,852 | | | | 1,606 | | | Less: | | | | | | | | | | Accumulated depreciation and amortization | | | 810 | | | | 668 | | | Property and Equipment, net | | $ | 2,042 | | | $ | 938 | |
FOAMIX PHARMACEUTICALS LTD. NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (continued) (U.S. dollars in thousands, except share and per share amounts) NOTE 5 - PROPERTY AND EQUIPMENT(continued):
| | | December 31 | | | | | 2018 | | | 2017 | | | Cost: | | | | | | | | Leasehold improvements | | $ | 978 | | | $ | 902 | | | Computers and software | | | 515 | | | | 299 | | | Laboratory equipment | | | 1,399 | | | | 1,257 | | | Furniture | | | 245 | | | | 288 | | | Vehicles | | | 82 | | | | 106 | | | | | | 3,219 | | | | 2,852 | | | Less: | | | | | | | | | | Accumulated depreciation and amortization | | | 984 | | | | 810 | | | Property and Equipment, net | | $ | 2,235 | | | $ | 2,042 | |
Depreciation and amortization expense totaled $319, $221 $143 and $87$143 for the years ended December 31, 2017,2018, December 31, 2016,2017 and December 31, 2015,2016, respectively. During the years ended December 31, 2018, December 31, 2017 and December 31, 2016, the Company disposed of fixed assets in the net amount of $42, $104 and $16, respectively. Loss from Salessales of fixed assets for the yearyears ended December 31, 2018 and December 31, 2017 was $30.$2 and $30, respectively.
NOTE 6 - EMPLOYEE SEVERANCE BENEFITS
The Company's liability for severance pay for its Israeli employees is calculated pursuant to Israeli severance pay law based on the most recent salary of the employee multiplied by the number of years of employment, as of the balance sheet date, less amounts funded in each employee’s severance fund. Such liability is recorded on the Company’s balance sheet under “Liability for employee severance benefits” as if it were payable at each balance sheet date on an undiscounted basis. The Company partially secures this liability by purchasing insurance policies or establishing dedicated severance accounts within the relevant employees’ pension funds, and making monthly deposits under such policies or into such accounts. The value of these policies is recorded as an asset in the Company's balance sheet.
During 2014, all of the Israeli employees agreed to the terms of Section 14 of the Israeli Severance Pay Law, 1963, according to which all deposits in the pension fund and/or with the insurance company, thereafter, exempt the Company from any additional obligation. These deposits are accounted as defined contribution payments and therefore not recorded on the Company’s balance sheet. Once the employees agreed to the terms of Section 14, all amounts funded on behalf of the employees were released to their full ownership. The liability for employee severance benefits as of December 31, 2017,2018, represents the Company's obligation that has not been secured by deposits to employee severance funds.
The amount of severance payment expenses to Israel employees were $374, $244$592, $569 and $193$407 for the years ended December 31, 2018, December 31, 2017 and December 31, 2016, and December 31, 2015, respectively.
During 2018, the Company expects to deposit approximately $739 with respect to employee’s severance benefits.
Beginning September 2017, the Company has retirement savings plans available to all employees of the Subsidiary, which are intended to qualify as deferred compensation plans under Section 401(k) of the Internal Revenue Code (the “401k“401(k) Plans”). The Company made contributions to these plans401(k) Plans during the yearyears ended December 31, 2018 and December 31, 2017 of approximately $35.$84 and $35, respectively. During 2019, the Company expects to deposit approximately $930 with respect to employee’s severance benefits
FOAMIX PHARMACEUTICALS LTD. NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (continued) (U.S. dollars in thousands, except share and per share amounts)
NOTE 7 - COMMITMENTS
Lease agreement
The Company leases office space for its headquarters and research and development facilities in Israel and the United States of America under several lease agreements. The lease agreements for the facilities in Israel are linked to the Israeli CPI and expire in December 2020. The lease agreement in the United States is due to expire during March 2019.2019; however, the company has the option to renew the lease for an additional three years.
In July 2017, the Company has entered into operating lease agreement in connection with a number of vehicles. The lease periods are generally for three years and the payments are linked to the Israeli CPI. To secure the terms of the lease agreements, the Company has made certain prepayments to the leasing company, representing approximately three months of lease payments. These amounts have been recorded as other non-current assets.
Operating lease expenses for the years ended December 31, 2018, December 31, 2017 and December 31, 2016, and December 31, 2015, are as follows:
| | Year Ended December 31 | | | | 2017 | | | 2016 | | | 2015 | | | Rental expenses | | $ | 645 | | | $ | 358 | | | $ | 352 | | | Vehicles lease expenses | | $ | 22 | | | $ | - | | | $ | - | |
| | | Year Ended December 31 | | | | | 2018 | | | 2017 | | | 2016 | | Office lease expenses | | $ | 772 | | | $ | 645 | | | $ | 358 | | Vehicles lease expenses | | $ | 100 | | | $ | 22 | | | $ | - | |
Future minimum lease commitments under non-cancelable operating lease agreements are as follows:
| 2018 | | $ | 865 | | | | 2019 | | | 756 | | | $ | 746 | | | 2020 and thereafter | | | 701 | | | 2020 | | | | 682 | | 2021 and thereafter | | | | 21 | | | Total | | $ | 2,322 | | | $ | 1,449 | |
The Company has a lien in the amount of $143$133 on the Company’s marketable securities in respect of bank guarantees granted in order to secure the lease agreements.
NOTE 8 - LOANS:
| a. | Loan from the BIRD foundation |
During the second quarter of 2016, the Company repaid the loan received from the Israel United States Binational Industrial Research and Development Foundation (the "BIRD foundation") upon the completion of a certain clinical development. The loan, received in instalments between 2008 and 2011, was denominated in USU.S. dollars and linked to the USU.S. Consumer Price Index.
| b. | Bank Borrowingsborrowings |
During 2014, the Company entered into several finance agreements with a bank in order to finance the purchase of vehicles (hereinafter “the loans”Loans”). The loans areLoans were denominated in NIS and bear interest at a rate per annum equal to Prime minus 0.5%. The loansLoans were fully repaid during 2017.
FOAMIX PHARMACEUTICALS LTD. NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (continued) (U.S. dollars in thousands, except share and per share amounts) NOTE 9 - SHARE CAPITAL:
| a. | Rights of the Company’s ordinary shares |
Each ordinary share is entitled to one vote. The holders of ordinary shares are also entitled to receive dividends whenever funds are legally available, when and if declared by the Board of Directors. Since its inception, the Company has not declared any dividends. FOAMIX PHARMACEUTICALS LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands, except share and per share amounts)
NOTE 9 - SHARE CAPITAL (continued):
In September 2014, the Company completed an IPO, pursuant to which the Company issued 7,668,200 ordinary shares (including underwriters ‘green shoe’), NIS 0.16 par value (“Ordinary Shares”) at $6.00 per share raising a total of approximately $41,100, net of underwriting discounts, commissions and other offering expenses.
On April 20, 2015, the Company completed a follow-on public offering. A total of 7,419,353 ordinary shares were sold at a price of $9.30 per share. Prior to closing, the underwriters fully exercised their option to purchase 967,741 additional ordinary shares. The net proceeds from the sale of shares, after deducting underwriting discounts, commissions and other offering expenses, were approximately $64,202.
On September 30, 2016, the Company completed an additionala public offering in which 5,700,000 ordinary shares were sold at a price of $9.50 per share. On October 28, 2016, the underwriters partially exercised their ‘green shoe’ option and purchased 411,959 additional ordinary shares. The net proceeds, including the underwriters' option, were approximately $54,132, after deducting underwriter’s discounts, commissions and other offering expenses.
On September 18, 2018, the Company completed a public offering in which 11,670,000 ordinary shares were sold at a price of $6.00 per share. Upon closing of the offering, the underwriters exercised their ‘green shoe’ option at full and purchased 1,750,500 additional ordinary shares. The net proceeds, including the underwriters' option, were approximately $75,356, after deducting underwriter’s discounts, commissions and other offering expenses.
| c. | Securities Purchase Agreement |
On April 13, 2018, the Company entered into a Securities Purchase Agreement with an existing investor pursuant to which the Company agreed to issue and sell, in a registered offering, an aggregate of 2,940,000 ordinary shares at a purchase price of $5.50 per share. The net proceeds from the offering were $16,131 after deducting transaction expenses. The closing of the issuance and sale of these shares took place on April 16, 2018.
As of December 31, 2017, and December 31, 2016, the Company’s outstanding warrants were 1,394,558 and 1,807,800, respectively. Each warrant can becould have been exercised for one ordinary share at an exercise price of $5.04 per share or through a cashless exercise. TheDuring the years ended December 31, 2018, December 31, 2017 and December 31, 2016, 1,394,558, 413,242 and 257,137 warrants arewere exercised into 178,468, 191,793 and 257,137 ordinary shares, respectively.
As defined in the warrant agreement dated May and June 2014, the warrants were exercisable until May 13, 2018 (“Expiration date”).2018. On the occasion where thethis date all outstanding warrants are not exercised by the Expiration date, and the fair value of share price on such day is higher than the warrant exercise price, the warrants shall be deemedwere automatically exercised by the warrant holder on a net issuance basis. During the years ended December 31, 2017 and December 31, 2016, 413,242 and 257,137 warrants were exercised into 191,793 and 257,137 ordinary shares, respectively.
| d.e. | Share-based compensation |
In June 2009, the Company’s Board of Directors approved a share option plan and reserved a pool of 1,635,694 ordinary shares for grant to Company employees, consultants, directors and other service providers.
In May 2015, the Company's board of directors approved a new option plan (the "Plan") replacing the previous plan approved in 2009. The Plan includesinitially included a pool of 2,690,694 ordinary shares for grant to Company employees, consultants, directors and other service providers. During the years ended December 31, 2016 and December 31, 2017, a totalthe Board of Directors approved an accumulated increase of 2,900,000 ordinary shares was approved byto the board of directors and registered with the Securities and Exchange Commission. Plan. As of December 31, 2017, 2,076,0882018, 1,179,346 shares remain available for grant under the Plan.
The Plan is designed to enable the Company to grant options to purchase Ordinary Sharesordinary shares and RSUs under various and different tax regimes including, without limitation: (i) pursuant and subject to Section 102 of the Israeli Tax Ordinance or any provision which may amend or replace it and any regulations, rules, orders or procedures promulgated thereunder and to designate them as either grants made through a trustee or not through a trustee; and (ii) pursuant and subject to Section 3(i) of the Israeli Tax Ordinance.
FOAMIX PHARMACEUTICALS LTD. NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (continued) (U.S. dollars in thousands, except share and per share amounts)
NOTE 9 - SHARE CAPITAL (continued):
The fair value of each option granted is estimated using the Black-Scholes option pricing method. The volatility is based on a combination of the Company’s historical volatility, historical volatilities of companies in comparable stages as well as companies in the industry, by statistical analysis of daily share pricing model. The risk-free interest rate assumption is based on observed interest rates appropriate for the expected term of the options granted in dollar terms. The Company’s management uses the contractual term or its expectations, as applicable, of each option as its expected life. The expected term of the options granted is derived from the output of the option pricing model and represents the period of time that granted options are expected to remain outstanding.
Options and RSUs granted to employees and directors: In the three years ended December 31, 2018, December 31, 2017 and December 31, 2016, the Company granted options to employees and non-employees as follows:
| | | Year ended December 31, 20172018 | | | | | Award amount | | | Exercise price range | | | Vesting period | | | | | | Employees: | | | | | | | | | | | | | | Options | | | 1,162,558721,530 | | | | $5.22- $10.314.06- $6.4 | | | 4 years | | | 10 years | | | RSU | | | 350,694201,844 | | | | - | | | 4 years | | | | - | | | | | | | | | | | | | | | | | | | Directors: | | | | | | | | | | | | | | | | | | Options | | | 189,709174,373 | | | | $4.69- $4.765.02- $5.06 | | 4 | 1 years | | | 10 years | | | RSU | | | 19,39714,829 | | | | - | | 4 | 3 years | | | | - | |
| | | Year ended December 31, 2016 | | | | | Award amount | | | Exercise price range | | Vesting period | | | | | Employees: | | | | | | | | | | | | Options | | | 715,310 | | | | $6.04- $8.54 | | 4 years | | 10 years | | | RSU | | | 25,000 | | | | - | | 4 years | | | - | | | | | | | | | | | | | | | | | | Directors: | | | | | | | | | | | | | | | Options | | | 24,000 | | | | $7.09 | | 3 years | | 10 years | | | | | | | | | | | | | | | | | | Consultants: | | | | | | | | | | | | | | | Options | | | 4,800 | | | | $6.34 | | 4 years | | 10 years | |
| | Year ended December 31, 2015 | | | Year ended December 31, 2017 | | | | Award amount | | | Exercise price range | | Vesting period | | | | | Award amount | | | Exercise price range | | | Vesting period | | | | | | Employees: | | | | | | | | | | | | | | | | | | | | | | | | Options | | | 852,501 | | | | $6.77- $10.88 | | 4 years | | 10 years | | | | 1,162,558 | | | | $5.22- $10.31 | | | 4 years | | | 10 years | | | RSU | | | 216,050 | | | | - | | 4 years | | | - | | | | 350,694 | | | | - | | | 4 years | | | | - | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | Directors: | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | Options | | | 27,000 | | | | $10.80-$11.87 | | 3 years | | 10 years | | | | 189,709 | | | | $4.69- $4.76 | | | 4 years | | | 10 years | | | RSU | | | | 19,397 | | | | - | | | 4 years | | | | - | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | Consultants: | | | | | | | | | | | | | | | | | | | Year ended December 31, 2016 | | | | | | Award amount | | | Exercise price range | | | Vesting period | | | | | | Employees: | | | | | | | | | | | | | | | | | | | Options | | | 4,000 | | | | $10.88 | | 4 years | | 10 years | | | | 715,310 | | | | $6.04- $8.54 | | | 4 years | | | 10 years | | | RSU | | | 83,800 | | | | - | | 4 years | | | - | | | | 25,000 | | | | - | | | 4 years | | | | - | | | | | | | | | | | | | | | | | | | | | | Directors: | | | | | | | | | | | | | | | | | | | Options | | | | 24,000 | | | | $7.09 | | | 3 years | | | 10 years | |
FOAMIX PHARMACEUTICALS LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands, except share and per share amounts)
NOTE 9 - SHARE CAPITAL (continued):
The fair value of options and RSUs granted to employeesduring 2018, 2017 and directors during 2017, 2016 was $3,953, $8,510 and 2015 was $8,510, $2,816, and $6,592 respectively. The fair value of options and RSUs granted to consultants during the years ended 2016 and 2015 was $42 and $686, respectively. The fair value of RSUs granted to employees and directors is based on the share price on grant date.date and was computed using the Black-Scholes model. The underlying data used for computing the fair value of the options are as follows: FOAMIX PHARMACEUTICALS LTD. NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (continued) (U.S. dollars in thousands, except share and per share amounts) NOTE 9 - SHARE CAPITAL (continued): | | | Year ended December 31 | | | | | 2018 | | | 2017 | | | 2016 | | Value of ordinary share | | | $4.09-$5.99 | | | | $4.44-$10.12 | | | | $5.9-$8.35 | | Dividend yield | | | 0 | % | | | 0 | % | | | 0 | % | Expected volatility | | | 61%-62.6 | % | | | 58.41%-61.7 | % | | | 60.3%-63.2 | % | Risk-free interest rate | | | 2.75%-2.87 | % | | | 1.97%-2.16 | % | | | 1.25%-1.86 | % | Expected term | | 6 years | | | 6 years | | | 6 years | |
The total unrecognized share-based compensation cost at December 31, 2018 is $8,242, which is expected to be recognized over a weighted average period of two years. Options and RSUs granted to consultants and service providers: During the years ended December 31, 2018 and 2017 there were no grants of options and RSUs to consultants and service providers. In the year ended December 31, 2016, the Company granted options as follows: | | | Year ended December 31, 2016 | | | | | Award amount | | | Exercise price range | | | Vesting period | | | Expiration | | Options | | | 4,800 | | | $ | 6.34 | | | 4 years | | | 10 years | | | | | | | | | | | | | | | | | | | |
The fair value of options and RSUs granted to consultants during the year ended December 31, 2016 was $42. The fair value of options granted to employees and directors on the date of grantduring 2016, was computed using the Black-Scholes model. The underlying data used for computing the fair value of the options are as follows: | | | Year ended December 31 | | | | | 2017 | | | 2016 | | | 2015 | | | Value of ordinary share | | | $4.44-$10.12 | | | | $5.9-$8.35 | | | | $6.96-$11.3 | | | Dividend yield | | | 0 | % | | | 0 | % | | | 0 | % | | Expected volatility | | | 58.41%-61.7 | % | | | 60.3%-63.2 | % | | | 60.1%-64.9 | % | | Risk-free interest rate | | | 1.97%-2.16 | % | | | 1.25%-1.86 | % | | | 1.38%-1.98 | % | | Expected term | | 6 years | | | 6 years | | | 6 years | |
The total unrecognized compensation cost of employee and directors options and RSUs at December 31, 2017 is $9,340 which is expected to be recognized over a weighted average period of 2 years.
The fair value of options granted during 2016 and 2015 to consultants, was computed using the Black-Scholes model. The underlying data used for computing the fair value of the options are as follows:
| | | December 31 | | | | | 2016 | | | 2015 | | | Value of ordinary share | | | $11.1 | | | | $8.11 | | | Dividend yield | | | 0 | % | | | 0 | % | | Expected volatility | | | 64.8 | % | | | 69.4 | % | | Risk-free interest rate | | | 2.38 | % | | | 2.27 | % | | Expected term | | 9 years | | | 10 years | |
| | December 31 | | | | 2016 | | Value of ordinary share | | | $11.1 | | Dividend yield | | | 0 | % | Expected volatility | | | 64.8 | % | Risk-free interest rate | | | 2.38 | % | Expected term | | 9 years | |
F - 25Modification of share-based compensation:
FOAMIX PHARMACEUTICALS LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands, except share and per share amounts)
NOTE 9 - SHARE CAPITAL (continued):
In June, 2017, the Company entered into new agreements with Dr. Dov Tamarkin and Mr. Meir Eini to serve as consultants, pursuant tofollowing their resignation from their roles as Chief Executive Officer and Chief Innovation Officer of the Company (see also Note 13), pursuant to which all options and RSUs previously awarded to Dr. Dov Tamarkin and Mr. Meir Eini willwere to remain outstanding and continue to vest as though they remained employed by the Company through each applicable vesting date. In addition to the new agreements with Dr. Dov Tamarkin and Mr. Meir Eini,above, during 2017, the Company entered into a similar agreement with another employee who has become a consultant to the Company. Pursuant to the agreement all options and RSUs previously awarded to the employee willwere to remain outstanding and continue to vest as though he remained employed by the Company through each applicable vesting date, as long as he remainsremained a consultant to the Company. On May 31, 2018 the consultant’s agreement was terminated and all unvested awards were forfeited. FOAMIX PHARMACEUTICALS LTD. NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (continued) (U.S. dollars in thousands, except share and per share amounts) NOTE 9 - SHARE CAPITAL (continued):
The retention of the options and RSUs was considered a Type III modification for share-based compensation, and, as a result, during the year ended December 31, 2017, the Company reversed all expense previously recorded for these retained awards in the amount of $2,037 and recorded the newadditional compensation expense in the amount of $1,058 over the new requisite service period. On January 1, 2018, the Company and Dr. Tamarkin agreed to terminate the consulting agreement signed in June 2017. Pursuant to the termination, the Board of Directors resolved that all options and RSUs previously granted to Dr. Tamarkin shall continue to vest and may be exercised until their expiration date. The retention of the options and RSUs was considered an additional Type III modification. As a result, on January 1, 2018, the Company remeasured the fair value of all outstanding options and RSUs granted to Dr. Tamarkin and recognized the fair value of the unvested options and RSUs as an immediate expense. The compensation expenses recorded on January 1, 2018, were $239. Following changes in circumstances, including Mr. Eini’s resignation from his position as an observer to the Board of Directors, the Company reassessed the services provided by Mr. Eini and concluded they are not substantive in comparison to the value of the equity awards he received. Therefore, in January 2018, all expenses related to the awards previously granted to Mr. Eini were measured and the unrecognized amount of the fair value was fully recognized. The compensation expenses recorded in January 2018, were $494. On November 19, 2018, the Company entered into a retirement agreement with one of its Directors pursuant to which all options previously awarded will remain outstanding and continue to vest throughout the vesting period subject to shareholders approval. The retention of the options was considered a Type III modification. As a result, on November 19, 2018 the Company reversed all expense previously recorded for the unvested retained awards in the amount of $90 and recorded the new compensation expense in the amount of $47 as an immediate expense. Summary of outstanding and exercisable options and RSUs: The following table summarizes the number of options outstanding for the years ended December 31, 2017,2018, December 31, 20162017 and December 31, 2015,2016, and related information: | | | Employees and directors | | | Consultants and service providers | | | Employees and directors | | | Consultants and service providers | | | | | Number of options | | | USD(1) | | | Number of options | | | USD(1) | | | Number of options | | | USD(1) | | | Number of options | | | USD(1) | | | Outstanding at January 1, 2015 | | | 904,250 | | | $ | 3.28 | | | | 266,875 | | | $ | 2.10 | | | | Granted | | | 879,501 | | | | 7.72 | | | | 4,000 | | | | 10.88 | | | | Exercised | | | (29,525 | ) | | | 1.92 | | | | (150,000 | ) | | | 1.92 | | | Re-designated(2) | | | (27,000 | ) | | | 5.88 | | | | 27,000 | | | | 5.88 | | | | Outstanding at December 31, 2015 | | | 1,727,226 | | | $ | 5.41 | | | | 147,875 | | | $ | 3.24 | | | | Outstanding at January 1, 2016 | | | | 1,727,226 | | | $ | 5.41 | | | | 147,875 | | | $ | 3.24 | | | Granted | | | 739,310 | | | | 6.55 | | | | 4,800 | | | | 6.34 | | | | 739,310 | | | | 6.55 | | | | 4,800 | | | | 6.34 | | | Forfeited | | | (20,000 | ) | | | 6.66 | | | | (15,625 | ) | | | 7.98 | | | | (20,000 | ) | | | 6.66 | | | | (15,625 | ) | | | 7.98 | | | Exercised | | | (69,444 | ) | | | 1.64 | | | | - | | | | - | | | | (69,444 | ) | | | 1.64 | | | | - | | | | - | | | Outstanding at December 31, 2016 | | | 2,377,092 | | | $ | 5.87 | | | | 137,050 | | | $ | 2.81 | | | | 2,377,092 | | | $ | 5.87 | | | | 137,050 | | | $ | 2.81 | | | Granted | | | 1,352,267 | | | | 7.47 | | | | - | | | | - | | | | 1,352,267 | | | | 7.47 | | | | - | | | | - | | | Forfeited | | | (39,213 | ) | | | 7.93 | | | | (8,800 | ) | | | 8.40 | | | | (39,213 | ) | | | 7.93 | | | | (8,800 | ) | | | 8.40 | | | Exercised | | | (61,881 | ) | | | 2.63 | | | | - | | | | - | | | | (61,881 | ) | | | 2.63 | | | | - | | | | - | | Re-designated(3) | | | (252,210 | ) | | | 7.71 | | | | 252,210 | | | | 7.71 | | | Re-designated(2) | | | | (252,210 | ) | | | 7.71 | | | | 252,210 | | | | 7.71 | | | Outstanding at December 31, 2017 | | | 3,376,055 | | | $ | 6.41 | | | | 380,460 | | | $ | 5.93 | | | | 3,376,055 | | | $ | 6.41 | | | | 380,460 | | | $ | 5.93 | | | Granted | | | | 895,903 | | | | 5.61 | | | | - | | | | - | | | Forfeited | | | | (150,240 | ) | | | 7.98 | | | | (41,697 | ) | | | 8.12 | | | Exercised | | | | (24,625 | ) | | | 1.92 | | | | (67,500 | ) | | | 0.05 | | | Outstanding at December 31, 2018 | | | | 4,097,093 | | | $ | 6.20 | | | | 271,263 | | | $ | 7.05 | |
| (1) | Weighted average price per share |
| (2) | Pursuant to change in status of granteegrantees from ‘employee’ and ‘director’ to ‘consultant’ during the reporting period. |
| (3) | Pursuant to change in status of grantees from ‘employee’ and ‘Director’ to ‘consultant’ during the reporting period. |
FOAMIX PHARMACEUTICALS LTD. NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (continued) (U.S. dollars in thousands, except share and per share amounts)
NOTE 9 - SHARE CAPITAL (continued): The following table summarizes the number of RSUs outstanding for the years ended December 31, 2018, December 31, 2017 and December 31, 2016:2016: | | Employees and directors | | | Consultants and service providers | | | | Number of RSUs | | | Outstanding at January 1, 2016 | | | 186,800 | | | | 63,050 | | | Awarded | | | 25,000 | | | | - | | | Vested | | | (69,117 | ) | | | (21,000 | ) | | Outstanding at December 31, 2016 | | | 142,683 | | | | 42,050 | | | Awarded | | | 370,091 | | | | - | | | Forfeited | | | (4,025 | ) | | | (550 | ) | | Vested | | | (43,038 | ) | | | (33,625 | ) | Re-designated(1) | | | (78,120 | ) | | | 78,120 | | | Outstanding at December 31, 2017 | | | 387,591 | | | | 85,995 | |
| | Employees and directors | | | Consultants and service providers | | | | Number of RSUs | | | Outstanding at January 1, 2016 | | | 186,800 | | | | 63,050 | | | Awarded | | | 25,000 | | | | - | | | Vested | | | (69,117 | ) | | | (21,000 | ) | Outstanding at December 31, 2016 | | | 142,683 | | | | 42,050 | | | Awarded | | | 370,091 | | | | - | | | Forfeited | | | (4,025 | ) | | | (550 | ) | | Vested | | | (43,038 | ) | | | (33,625 | ) | Re-designated(1) | | | (78,120 | ) | | | 78,120 | | | Outstanding at December 31, 2017 | | | 387,591 | | | | 85,995 | | | Awarded | | | 216,673 | | | | - | | | Forfeited | | | (11,746 | ) | | | (12,150 | ) | | Vested | | | (161,648 | ) | | | (60,271 | ) | | Outstanding at December 31, 2018 | | | 430,870 | | | | 13,574 | |
| (1) | Pursuant to change in status of grantees from ‘employee’ and ‘Director’‘director’ to ‘consultant’ during the reporting period. |
The following tables summarizes information concerning outstanding and exercisable options as of December 31, 2017:2018:
December 31, 2017 | | | | December 31, 2018 | | December 31, 2018 | | | Options outstanding | Options outstanding | | | Options exercisable | | Options outstanding | | | Options exercisable | | | | | Number of | | | Weighted | | | Number of | | | Weighted | | | | Number of | | | Weighted | | | Number of | | | Weighted | | | | | options | | | average | | | options | | | Average | | | | options | | | average | | | options | | | Average | | | Exercise | | outstanding | | | remaining | | | exercisable | | | Remaining | | | | outstanding | | | remaining | | | exercisable | | | Remaining | | | prices per | | at end of | | | contractual | | | at end of | | | contractual | | | | at end of | | | contractual | | | at end of | | | contractual | | | share (USD) | | year | | | life | | | year | | | Life | | | | year | | | Life | | | year | | | Life | | | 0.048-1.312 | | | 268,125 | | | | 1.90 | | | | 268,125 | | | | 1.90 | | | | | | | | | | | | | | | | | | | | 0.062-1.312 | | | | | 200,625 | | | | 0.90 | | | | 200,625 | | | | 0.90 | | | 1.92 | | | 258,587 | | | | 4.08 | | | | 252,650 | | | | 4.03 | | | | | 227,400 | | | | 2.97 | | | | 227,400 | | | | 2.97 | | | 4.687-4.761 | | | 189,709 | | | | 9.53 | | | | - | | | | - | | | | 5.216-5.879 | | | 810,175 | | | | 8.75 | | | | 253,600 | | | | 6.98 | | | | 4.056-4.760 | | | | | 339,709 | | | | 9.15 | | | | 59,284 | | | | 8.53 | | | 5.022-5.879 | | | | | 1,067,235 | | | | 8.13 | | | | 521,344 | | | | 7.41 | | | 6.04-6.77 | | | 766,010 | | | | 8.02 | | | | 364,154 | | | | 7.83 | | | | | 1,183,803 | | | | 7.86 | | | | 522,254 | | | | 6.88 | | | 7.093-8.545 | | | 757,126 | | | | 7.82 | | | | 365,032 | | | | 7.66 | | | | 7.09-8.544 | | | | | 719,514 | | | | 6.82 | | | | 529,042 | | | | 6.72 | | | 10.217-11.868 | | | 706,783 | | | | 8.85 | | | | 80,719 | | | | 7.59 | | | | | 630,070 | | | | 7.86 | | | | 325,770 | | | | 7.68 | | | | | | 3,756,515 | | | | | | | | 1,584,280 | | | | | | | | | 4,368,356 | | | | | | | | | | | | | |
The aggregate intrinsic value of the total of both the outstanding and exercisable options as of December 31, 2017,2018, is $3,083 and $2,555 respectively.$964.
Share-based compensation expenses:
The following table illustrates the effect of share-based compensation on the statements of operations: | | | Year ended December 31 | | | Year ended December 31 | | | | | 2017 | | | 2016 | | | 2015 | | | 2018 | | | 2017 | | | 2016 | | | Cost of revenues | | $ | 2 | | | $ | 3 | | | $ | 2 | | | $ | - | | | $ | 2 | | | $ | 3 | | | Research and development expenses | | | 1,711 | | | | 1,135 | | | | 588 | | | | 2,054 | | | | 1,711 | | | | 1,135 | | | Selling, general and administrative | | | 2,453 | | | | 1,774 | | | | 1,187 | | | | 3,266 | | | | 2,453 | | | | 1,774 | | | | | $ | 4,166 | | | $ | 2,912 | | | $ | 1,777 | | | $ | 5,320 | | | $ | 4,166 | | | $ | 2,912 | |
FOAMIX PHARMACEUTICALS LTD. NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (continued) (U.S. dollars in thousands, except share and per share amounts) NOTE 10 - INCOME TAX:
The Company is taxed under Israel and the United States of America tax laws:
| 1) | Income from Israel was taxed at the corporate tax rate of 26.5% in 2015, 25% in 2016, and 24% in 2017. Capital gains are subject to capital gain tax, which equals to 25%.2017, and 23% in 2018 and thereafter. |
In December 2016, the Economic Efficiency Law (Legislative Amendments for Implementing the Economic Policy for the 2017 and 2018 Budget Year), 2016 was published, introducing a gradual reduction in corporate tax rate from 25% to 23%. However, the law also included a temporary provision setting the corporate tax rate in 2017 at 24%. As a result, the corporate tax rate in 2017 was 24% and in 2018 and thereafter reduced to 23%.
| 2) | Income ofEffective January 1, 2018, the subsidiary is taxed according toU.S. Tax Cuts and Jobs Act, reduced the federal tax laws in the US and the relevant state laws. The relevant U.S. statutory tax rates forrate from 35% in 2017 and 2016 and 2015 were 35%, 35% and 30%, respectively.to 21%. The relevant state tax rate for 2018, 2017 2016 and 20152016 was 9%. |
The U.S. Tax Cuts and Jobs Act (Tax Act) was enacted on December 22, 2017 and introduces significant changes to U.S. income tax law. Effective in 2018, the Tax Act reduces the U.S. statutory tax rate from 35% to 21% and creates new taxes on certain foreign-sourced earnings and certain related-party payments, which are referred to as the global intangible low-taxed income tax and the base erosion tax, respectively.
Foamix has tax assessments that are considered to be final through tax year 2012.2013.
| c. | Tax benefits under the Law for Encouragement of Industry (Taxation), 1969 |
Foamix believes that it currently qualifies as an "Industrial Company" under the above law. As such it is entitled to certain tax benefits, mainly the right to deduct share issuance costs over three years for tax purposes in the event of a public offering.
Foamix utilizes this tax benefit.
| d. | Losses for tax purposes carried forward to future years |
As of December 31, 2017,2018, Foamix had approximately $88.6$146.7 million of net carry forward tax losses in Israel, which are available to reduce future taxable income with no limited period of use. During 2018 the subsidiary incurred $0.4 million of net carry forward tax losses in the United States to reduce future taxable income with no limited period of use. FOAMIX PHARMACEUTICALS LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands, except share and per share amounts)
NOTE 10 - INCOME TAX (continued):
| e. | Subsidiary tax liability |
During 2018, 2017 2016 and 2015,2016, the US subsidiarySubsidiary incurred a tax expense in the amount of $212, $1,164 $387 and $39,$387, respectively.
| | | December 31, | | | December 31, | | | | | 2017 | | | 2016 | | | 2018 | | | 2017 | | | In respect of: | | | | | | | | | | | | | | Net operating loss carry forward | | $ | 20,385 | | | $ | 11,512 | | | $ | 33,859 | | | $ | 20,385 | | | Research and development | | | 9,856 | | | | 4,147 | | | | 12,932 | | | | 9,856 | | Share based compensation | | | | 957 | | | | 433 | | | Other | | | 608 | | | | 332 | | | | 164 | | | | 175 | | | Less - valuation allowance | | | (30,849 | ) | | | (15,991 | ) | | | (47,912 | ) | | | (30,849 | ) | | Net deferred tax assets | | $ | - | | | $ | - | | | $ | - | | | $ | - | |
FOAMIX PHARMACEUTICALS LTD. NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (continued) (U.S. dollars in thousands, except share and per share amounts) NOTE 10 - INCOME TAX (continued): Realization of deferred tax assets is contingent upon sufficient future taxable income during the period that deductible temporary differences and carry forward losses are expected to be available to reduce taxable income. As the achievement of required future taxable income is not likely, the Company recorded a full valuation allowance. Deferred tax has not been provided on taxes that would apply in the event of disposal of the investments in the subsidiary,Subsidiary, as it is the Company’s intention to hold this investment and not to realize it. Foamix may incur an additional tax liability in the event of an inter-company dividend distribution from its subsidiary;Subsidiary; no additional deferred taxes have been provided, since it is the Company’s policy not to distribute in the foreseeable future, dividends which would result in additional tax liability. Following is a reconciliation of the theoretical tax benefit, assuming all income is taxed at the statutory corporate tax rate applicable to Israeli corporations, and the actual tax expense: | | | Year ended December 31 | | | Year ended December 31 | | | | | 2017 | | | 2016 | | | 2015 | | | 2018 | | | 2017 | | | 2016 | | | Loss before income taxes | | $ | 64,551 | | | $ | 28,949 | | | $ | 16,478 | | | $ | 73,951 | | | $ | 64,551 | | | $ | 28,949 | | | | | | | | | | | | | | | | | | Theoretical tax benefit on the above amount | | | (15,492 | ) | | | (7,237 | ) | | | (4,367 | ) | | | (17,009 | ) | | | (15,492 | ) | | | (7,237 | ) | | Decrease (increase) in tax refund resulting from: | | | | | | | | | | | | | | | | | | | | | | | | | | Reduction and different corporate tax rates | | | 711 | | | | 1,965 | | | | - | | | | (101 | ) | | | 711 | | | | 1,965 | | | Non-deductible expenses and other permanent differences, mainly share based compensation expenses and issuance costs | | | 80 | | | | (491 | ) | | | (585 | ) | | | (84 | ) | | | 80 | | | | (491 | ) | | Uncertain tax position | | | 988 | | | | - | | | | - | | | | (98 | ) | | | 988 | | | | - | | | Net change in valuation allowance | | | 14,858 | | | | 5,777 | | | | 3,973 | | | | 17,063 | | | | 14,858 | | | | 5,777 | | | Other | | | 19 | | | | 373 | | | | 1,018 | | | | 441 | | | | 19 | | | | 373 | | | Actual tax expense | | $ | 1,164 | | | $ | 387 | | | $ | 39 | | | $ | 212 | | | $ | 1,164 | | | $ | 387 | |
| g. | ASC No. 740, Income Taxes, requires significant judgment in determining what constitutes an individualUncertain tax position as well as assessing the outcome of each tax position. Changes in judgment as to recognition or measurement of tax positions can materially affect the estimate of the effective tax rate and consequently, affect the operating results of the Company. positions: |
ASC No. 740, Income Taxes, requires significant judgment in determining what constitutes an individual tax position as well as assessing the outcome of each tax position. Changes in judgment as to recognition or measurement of tax positions can materially affect the estimate of the effective tax rate and consequently, affect the operating results of the Company. The following table summarizes the activity of the Company unrecognized tax benefits:
Balance at January 1, 2017 | | $ | - | | Increase in uncertain tax positions for the current year | | | 988 | | Balance at December 31, 2017 | | $ | 988 | | Decrease in uncertain tax positions for the current year | | | (98 | ) | Balance at December 31, 2018 | | $ | 890 | |
FOAMIX PHARMACEUTICALS LTD. NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (continued) (U.S. dollars in thousands, except share and per share amounts) NOTE 10 - INCOME TAX (continued):
The following table summarizes the activity of the Company unrecognized tax benefits:
| | | Year ended December 31, | | | | | 2017 | | | Balance at January 1, 2017 | | $ | - | | | Increase in uncertain tax positions for the current year | | | 988 | | | Balance at December 31, 2017 | | $ | 988 | |
The Company does not expect unrecognized tax expenses to change significantly over the next 12 months.
| h. | Roll forward of valuation allowance: |
| Balance at January 1, 2015 | | $ | 6,241 | | | | Additions | | | 3,973 | | | | Balance at December 31, 2015 | | $ | 10,214 | | | Balance at January 1, 2016 | | | $ | 10,214 | | | Additions | | | 5,777 | | | | 5,777 | | | Balance at December 31, 2016 | | $ | 15,991 | | | $ | 15,991 | | | Additions | | | 14,858 | | | | 14,858 | | | Balance at December 31, 2017 | | $ | 30,849 | | | $ | 30,849 | | Additions | | | | 17,063 | | Balance at December 31, 2018 | | | $ | 47,912 | |
NOTE 11 - SUPPLEMENTARY FINANCIAL STATEMENT INFORMATION:
Balance sheets: | | | December 31 | | | | | 2018 | | | 2017 | | | a. Account receivable - other: | | | | | | | | Institutions | | $ | 446 | | | $ | 91 | | | Prepaid expenses | | | 450 | | | | 588 | | | Other | | | 103 | | | | 93 | | | | | $ | 999 | | | $ | 772 | |
| a. | Account receivable - other:
|
| Institutions | | $ | 91 | | | $ | 174 | | | Prepaid expenses | | | 588 | | | | 246 | | | Other | | | 93 | | | | 18 | | | | | $ | 772 | | | $ | 438 | |
| b. Accounts payable and accruals - other: | | | | | | | | Accrued expenses | | $ | 351 | | | $ | 1,622 | | | Payroll and related institutions | | | 1,166 | | | | 872 | | | Bonus accrual | | | 2,332 | | | | 1,166 | | | Other | | | 292 | | | | 70 | | | | | $ | 4,141 | | | $ | 3,730 | |
| b. | Accounts payable and accruals - other:
|
| Accrued expenses | | $ | 1,622 | | | $ | 709 | | | Payroll and related institutions | | | 872 | | | | 581 | | | Bonus accrual | | | 1,166 | | | | 1,661 | | | Other | | | 70 | | | | 33 | | | | | $ | 3,730 | | | $ | 2,984 | |
Statements of operations:
In the yearyears ended December 31, 2017 and 2016, the Company’s revenues were driven virtually all from one main customer. Based on the agreement with this customer, the Company is entitled to royalty payments with respect to sales of a product developed by the customer in collaboration with the Company.Company (“the Product”). During 2018, a new customer acquired the Product and assumed all rights and responsibilities under the initial agreement. In the year ended December 31, 2018, virtually all revenues were driven from these two customers. The following table provides a breakdown of the Company’s net revenues:
| | | Year ended December 31 | | | | | 2018 | | | 2017 | | | 2016 | | Development Service Payments | | $ | 62 | | | $ | 140 | | | $ | 63 | | Contingent Payments | | | - | | | | - | | | | 2,500 | | Royalties | | | 3,533 | | | | 3,529 | | | | 2,964 | | Total revenues | | $ | 3,595 | | | $ | 3,669 | | | $ | 5,527 | |
FOAMIX PHARMACEUTICALS LTD. NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (continued) (U.S. dollars in thousands, except share and per share amounts) NOTE 11 - SUPPLEMENTARY FINANCIAL STATEMENT INFORMATION (continued): The following table provides a breakdown of the Company’s net revenues:
| | | Year ended December 31 | | | | | 2017 | | | 2016 | | | 2015 | | | Development Service Payments | | $ | 140 | | | $ | 63 | | | $ | 596 | | | Contingent Payments | | | - | | | | 2,500 | | | | - | | | Royalties | | | 3,529 | | | | 2,964 | | | | 253 | | | Total revenues | | $ | 3,669 | | | $ | 5,527 | | | $ | 849 | |
| d. | Finance income, net:net: |
| | | Year ended December 31 | | | | | 2017 | | | 2016 | | | 2015 | | | Finance expenses: | | | | | | | | | | | Finance expenses on BIRD loan | | | - | | | | 243 | | | | * | | | Foreign exchange losses, net | | | 57 | | | | - | | | | - | | | Other expenses | | | 14 | | | | 17 | | | | 23 | | | Total finance expenses | | | 71 | | | | 260 | | | | 23 | | | | | | | | | | | | | | | | | Finance income: | | | | | | | | | | | | | | Gains from securities, net | | | (602 | ) | | | (401 | ) | | | (180 | ) | | Interest on bank deposits | | | (532 | ) | | | (536 | ) | | | (289 | ) | | Foreign exchange gains, net | | | - | | | | (24 | ) | | | (6 | ) | | Total finance income | | | (1,134 | ) | | | (961 | ) | | | (475 | ) | | | | $ | (1,063 | ) | | $ | (701 | ) | | $ | (452 | ) |
* Represents an amount less than $1.| | | Year ended December 31 | | | | | 2018 | | | 2017 | | | 2016 | | Finance expenses: | | | | | | | | | | | Finance expenses on BIRD loan | | $ | - | | | $ | - | | | $ | 243 | | | Foreign exchange losses, net | | | 27 | | | | 57 | | | | - | | | Other expenses | | | 17 | | | | 14 | | | | 17 | | Total finance expenses | | | 44 | | | | 71 | | | | 260 | | | | | | | | | | | | | | | | Finance income: | | | | | | | | | | | | | | Gains from securities, net | | | (688 | ) | | | (602 | ) | | | (401 | ) | | Interest on bank deposits | | | (297 | ) | | | (532 | ) | | | (536 | ) | | Foreign exchange gains, net | | | - | | | | - | | | | (24 | ) | Total finance income | | | (985 | ) | | | (1,134 | ) | | | (961 | ) | | | | $ | (941 | ) | | $ | (1,063 | ) | | $ | (701 | ) |
NOTE 12 - ENTITY-WIDE DISCLOSURE:
| a. | Net revenues by geographic area were as follows: |
| | | Year ended December 31 | | Year ended December 31 | | | | 2017 | | | 2016 | | | 2015 | | | 2018 | | | 2017 | | | 2016 | | | Germany | | $ | 3,529 | | | $ | 5,464 | | | $ | 253 | | | $ | 2,467 | | | $ | 3,529 | | | $ | 5,464 | | | United States | | | 140 | | | | 14 | | | | 587 | | | | 62 | | | | 140 | | | | 14 | | | France | | | - | | | | 49 | | | | 9 | | | | - | | | | - | | | | 49 | | Denmark | | | | 1,066 | | | | - | | | | - | | | Total revenues | | $ | 3,669 | | | $ | 5,527 | | | $ | 849 | | | $ | 3,595 | | | $ | 3,669 | | | $ | 5,527 | |
| b. | Revenues from principal customers - revenues from single customers that exceed 10% of total revenues in the relevant year: |
| | | Year ended December 31 | | | Year ended December 31 | | | | | 2017 | | | 2016 | | | 2015 | | | 2018 | | | 2017 | | | 2016 | | | Customer A | | $ | - | | | $ | - | | | $ | 86 | | | $ | 2,467 | | | $ | 3,529 | | | $ | 5,464 | | | Customer B | | $ | - | | | $ | 14 | | | $ | 366 | | | $ | 1,066 | | | $ | - | | | $ | - | | | Customer C | | $ | 3,529 | | | $ | 5,464 | | | $ | 253 | | | | Customer D | | $ | - | | | $ | - | | | $ | 135 | | |
FOAMIX PHARMACEUTICALS LTD. NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (continued) (U.S. dollars in thousands, except share and per share amounts)
NOTE 13 - RELATED PARTY TRANSACTIONS
In June 2017, the Company entered into new agreements with Dr. Dov Tamarkin and Mr. Meir Eini pursuant to their resignation from their roles as Chief Executive Officer and Chief Innovation Officer of the Company, respectively, effective as of June 29, 2017. As part of the agreements, as of July 1, 2017, Dr. Tamarkin and Mr. Eini began to serve as consultants to the Company. In addition, Dr. Tamarkin and Mr. Eini will retainretained all outstanding stock options and RSUs as long as they serve as consultants of the Company and arewere entitled to cash severance payments in the total amount of $1,800, out of which approximately $600 was paid as of December 31, 2017.
$1,800. In January 2018, subsequent to balance sheet date, Dr. Tamarkin has reached an agreement with the Company pursuant to which he will discontinuediscontinued his services as Chief Scientific Advisor to the Company in addition toand resigned from his resignation fromposition as a Director on the board of directors.directors of the Company. However, the board agreed that all of options and RSUs previously granted to Dr. Tamarkin will continue to vest and may be exercised until their expiration date (see note 9e). In addition, in May 2018, the shareholders approved an addition, special, one-time cash payment of $193. This cash payment was a conversion of 137,428 options and 45,750 restricted share units included, but not granted, in Dr. Tamarkin’s initial retirement agreement.
In November 2018, the Company entered into a retirement agreement with one of its directors. Pursuant to the agreement and subject to shareholders approval, the director shall receive one-time compensation of $50 and will retain all of his equity awards (see note 9e). Supplemental Financial Information
Unaudited selected quarterly financial results for the years ended December 31, 20172018 and 20162017 were as follows: | | | 2017 | | | 2016 | | | | | First Quarter | | | Second Quarter | | | Third Quarter | | | Fourth Quarter | | | First Quarter | | | Second Quarter | | | Third Quarter | | | Fourth Quarter | | | | | (in thousands of U.S. dollars, other than loss per share) | | | Revenues | | $ | 927 | | | $ | 798 | | | $ | 901 | | | $ | 1,043 | | | $ | 745 | | | $ | 752 | | | $ | 2,544 | | | $ | 1,486 | | | Cost of revenues | | | - | | | | - | | | | 11 | | | | 2 | | | | 31 | | | | 12 | | | | 8 | | | | 8 | | | Gross profit | | | 927 | | | | 798 | | | | 890 | | | | 1,041 | | | | 714 | | | | 740 | | | | 2,536 | | | | 1,478 | | | Operating loss | | | 14,570 | | | | 16,593 | | | | 17,828 | | | | 16,623 | | | | 4,562 | | | | 8,122 | | | | 5,946 | | | | 11,020 | | | Loss per share basic and diluted | | $ | 0.39 | | | $ | 0.44 | | | $ | 0.47 | | | $ | 0.46 | | | $ | 0.15 | | | $ | 0.27 | | | $ | 0.19 | | | $ | 0.29 | |
F- 33| | | 2018 | | | 2017 | | | | | First Quarter | | | Second Quarter | | | Third Quarter | | | Fourth Quarter | | | First Quarter | | | Second Quarter | | | Third Quarter | | | Fourth Quarter | | | Revenues | | $ | 906 | | | $ | 964 | | | $ | 865 | | | $ | 860 | | | $ | 927 | | | $ | 798 | | | $ | 901 | | | $ | 1,043 | | | Cost of revenues | | | - | | | | - | | | | - | | | | - | | | | - | | | | - | | | | 11 | | | | 2 | | | Gross profit | | | 906 | | | | 964 | | | | 865 | | | | 860 | | | | 927 | | | | 798 | | | | 890 | | | | 1,041 | | | Operating loss | | | 25,720 | | | | 18,787 | | | | 15,586 | | | | 14,799 | | | | 14,570 | | | | 16,593 | | | | 17,828 | | | | 16,623 | | | Loss per share basic and diluted | | $ | 0.69 | | | $ | 0.46 | | | $ | 0.38 | | | $ | 0.26 | | | $ | 0.39 | | | $ | 0.44 | | | $ | 0.47 | | | $ | 0.46 | |
ITEM 9 —- CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A —- CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
AsWe maintain “disclosure controls and procedures,” as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act, that are designed to ensure that information required to be disclosed by SEC Rule 13a-15(b), oura company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms, and that such information is accumulated and communicated to the company’s management, including its chief executive officer and chief financial officer, as appropriate to allow timely decisions regarding required disclosure. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures.
Our management, with the participation of our Chief Executive Officer and our Chief Financial Officer, has evaluated the effectiveness of our disclosure controls and procedures (as such term is defined in Rules 13a-15(b)13a-15(e) and 15d-15(e) under the Exchange Act and regulations promulgated thereunder) as of December 31, 2017, or the Evaluation Date.2018. Based on such evaluation, those officers have concluded that, as of the Evaluation Date,December 31, 2018, our disclosure controls and procedures arewere effective in recording, processing, summarizing and reporting, on a timely basis, information required to be included in periodic filings underat the Exchange Act and that such information is accumulated and communicated to management, including our principal executive and financial officers, as appropriate to allow timely decisions regarding required disclosure.reasonable assurance level.
Changes in Internal Control Overover Financial Reporting
There were no changes in our internal control over financial reporting that occurred during the yearfourth quarter ended December 31, 20172018 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Management’s Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over our financial reporting. Internal control over financial reporting is defined in Rule 13a-15(f) or 15d-15(f) under the Exchange Act as a process designed by, or under the supervision of, the company’scompany’s executive and financial officers and effected by the company’scompany’s board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes and includes those policies and procedures that (a) pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of the assets of the company; (b) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (c) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the company’scompany’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Our management assessed the effectiveness of our internal control over financial reporting, as of December 31, 2017.2018. In making this assessment, our management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control-Integrated Framework (2013). Based on our assessment, management believesconcluded that, as of December 31, 2017,2018, our internal control over financial reporting iswas effective based on these criteria.
As an emerging growth company, our auditors were not required to attest to, or report on, our management’s assessment of the effectiveness of our internal control over financial reporting, and therefore such attestation is not included in this an annual report on Form 10-K, in accordance with section 103 of the JOBS Act which amended section 404(b) of the Sarbanes-Oxley Act with regard to emerging growth companies.
ITEM 9B —- OTHER INFORMATION
None.On February 28, 2019, Dr. Dalia Meggido notified the board of directors of her intention to resign from the board of directors and from all committees thereof, effective as of immediately after the Company’s 2019 Annual General Meeting of Shareholders, which is currently expected to take place in April 2019. Dr. Meggido’s decision to resign was not due to any disagreement with the Company on any matter relating to the operations, policies (including accounting or financial policies) or practices of the Company. The Company expresses its appreciation for Dr. Meggido’s service as a member of the Board of Directors.
ITEM 10 —- DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The following table sets forth information relating to our executive officers and directors as of December 31, 2017.2018. Unless otherwise stated, the address for our directors and executive officers is c/o Foamix Pharmaceuticals Ltd., 2 Holzman St., Weizmann Science Park, Rehovot 7670402, Israel.
| Name | | Age | | Position | | Executive Officers | | | | | | David Domzalski | | 5152 | | Chief Executive Officer and Director | | Ilan Hadar, M.B.A. | | 4849 | | Chief Financial Officer, Country Manager (Israel) | | Mutya Harsch | | 44 | | General Counsel and Chief Legal Officer | | Iain A. Stuart, Ph.D. | | 4445 | | Senior Vice President,Chief Research and Development Officer | Yohan HazotMatthew Wiley | | 3447 | | Vice President, Pharmaceutical Development | David Schuz | | 63 | | Vice President, Intellectual Property | Mitchell Shirvan, Ph.D., M.B.A. | | 64 | | Vice President, Innovation & Discovery | Alvin Howard | | 63 | | Vice President, Regulatory Affairs | Russell Elliott, D.Phil. | | 51 | | Vice President Drug Development | Bonnie Pappacena | | 57 | | Vice President, QualityChief Commercial Officer | | Non-Executive Directors | | | | | | Stanley Hirsch, D.Phil. | | 6061 | | Director, Chairman of the Board of Directors, Chairman of Nominating Committee | | Sharon Barbari | | 64 | | Director | | Rex Bright | | 7778 | | Director, Chairman of the Compensation Committee | Darrell Rigel, M.D.Anthony Bruno | | 6762 | | Director | Stanley SternAnna Kazanchyan, M.D. | | 6050 | | Director Chairman of the Audit Committee | Anna Kazanchyan, Ph.D.Dalia Megiddo, M.D., M.B.A. | | 4967 | | Director | | Aharon Schwartz, Ph.D. | | 7576 | | Director | Dalia Megiddo, Ph.D., M.B.A.Stanley Stern | | 6661 | | Director, | Dov Tamarkin, Ph.D | | 62 | | Director Chairman of the Audit Committee |
_____________________________
Our Executive Officers
David Domzalski has served as our Chief Executive Officer since July 2017. Mr. Domzalski has been with Foamixus since April 2014, and previously served as the Presidentpresident of our U.S. subsidiary, Foamix Pharmaceuticals Inc. (U.S. subsidiary).Previously, Mr. Domzalski was the Vice President of Sales and Marketing at LEO Pharm Inc. from 2009 to 2013. On January 1, 2018, Mr. Domzalski was also appointed as a director tomember of our board replacing Dr. Dov Tamarkin. He has 25 years of industry experience, previously holding positions as Vice President Sales and Marketing at LEO Pharma Inc. from 2009 to 2013, Senior Vice President and General Manager at Azur Pharma from 2008 to 2009, and Vice President Sales and Marketing at Warner Chilcott from 2003 to 2008.directors. Mr. Domzalski holds a B.A. in economics and political science from Muhlenberg College, Allentown, Pennsylvania.
Ilan Hadar has served as our Chief Financial Officer since February 2014 and as thewas also appointed our Israel Country Manager, apposition he has held since July 2017. Mr. Hadar has over 20 years of experience in executive financial positions, previously holding positions as Finance Director of the Israeli subsidiary of Pfizer from 2011 to 2013, as Finance Manager, Accounting and Reporting at the Israeli subsidiary of HP from 2007 to 2011, and prior to that as Finance Director of the Israeli subsidiary of BAE Systems. Mr. Hadar holds a B.A. in business administration and economics and an MBAM.B.A. from The Hebrew University of Jerusalem.
Mutya Harsch has served as our General Counsel and Chief Legal Officer since January 2019. She initially joined us in January 2018, and previously served as our General Counsel and Senior Vice President of Legal Affairs from January 2018 to January 2019. Ms. Harsch has over 19 years of legal experience, previously holding positions as Special Counsel, Mergers & Acquisitions at Cooley LLP from 2015 to 2017 and as a corporate lawyer at Davis Polk & Wardwell from 2005 to 2015. Ms. Harsch received her J.D. and B.A. from the University of California at Berkeley.
Iain Stuart, Ph.D. has served as our Chief Research & Development Officer since January 2019, and previously served as our Senior Vice President of Research & Development sincefrom August 2017 to January 2019 and previously served as our Vice President of Clinical Development sincefrom October 2016. Dr. Stuart has over 18 years of clinical development scientific affairs experience in multiple therapeutic classes with the last 8 years being focused exclusively in dermatology.2016 to 2017. Prior to joining Foamix,us, Dr. Stuart served asheld several positions including Vice President of Medical Strategy and Scientific Affairs at LEO Pharma. PriorPharma, Inc. from 2008 to this, Dr. Stuart served as Director, Clinical Operations – The Americas, also at LEO Pharma.2016. Dr. Stuart holds a Ph.D. from Glasgow Caledonian University in Scotland.
Yohan HazotMatthew Wiley has served as our Vice President of Pharmaceutical DevelopmentChief Commercial Officer since August 2017.November 2018. Mr. HazotWiley has been with Foamix since April 2007, and previously served as the Chief Technology Officer from 2014. Prior to this role, he has held various positions of increasing responsibility in product development and intellectual property, including Director of Pharmaceutical Development. Mr. Hazot holds an MSc in Biochemistry & Biotechnology from the National Institute of Applied Sciences, Lyon, France and is the inventor of several patents in the field of pharmaceutical chemistry.
David Schuz David Schuz has served as our Vice President of Intellectual Property since August 2017. Mr. Schuz has led the Intellectual Property function at Foamix since July 2006. Mr. Schuz has overmore than 20 years of industrycommercial experience in the fieldacross a broad range of pharmaceuticals and biotechnology intellectual property, previously holding positions at Biotechnology General Israel Ltd. and Savient Pharmaceuticals, Inc. between 1996 and 2006, including Vice President from 2003.specialty pharmaceutical categories. Prior to joining us, Mr. Schuz holds an LL.M. from the London School Economics; a Certificate in Patent Law, Queen Mary London University; an M. Phil. (Biochemistry), and a Diploma of Pharmacology, both from Cambridge University; and a B.Sc. Hons. (Chemistry) from Manchester University.
Mitchell Shirvan, Ph.D. has served as our Vice President of Innovation and Discovery since August 2017. Dr. Shirvan has been with Foamix since March 2014, where he hasWiley held various leadership positions in innovation and research & development. He has over 20 years of industry experience, previously holding positions as Chief Executive Officer at Macrocure Ltd. from 2008 to 2012. From 1992 until 2008, Dr. Shirvan held variousseveral positions of increasing responsibility at Teva Pharmaceutical Industries, including Senior Director, Strategic Business Planning and Senior Manager, Research & Development. Dr. Shirvan holds a Ph.D. in microbiologyJazz Pharmaceuticals from The Hebrew University of Jerusalem, and an MBA from the University of Bradford.
Alvin Howard has served2012 to 2018, most recently as our Vice President of Regulatory Affairs since April 2014. Mr. Howard has over 30 years of industry experience, previously holding positions as Senior Vice President of Regulatory Affairs in Warner Chilcott from 2005 to 2013, Vice President of Regulatory Affairs at Roberts Pharmaceuticals from 1998 to 2000Marketing and various positions at Solvay Pharmaceuticals from 1990 to 1998. Mr. HowardBusiness Unit Lead for the company’s sleep disorder portfolio. He holds a B.Sc.B.A. in chemistryEnglish from Stillman College, Tuscaloosa, Alabama.
Russell Elliott, D.Phil. has served as our Vice President of Drug Development since May 2016. Dr. Elliott has over 25 years of experience in the pharmaceutical industry, previously holding the position of Vice President Product Development at Stiefel. Prior to this, Dr. Elliott served as Vice President and Head of Center of Excellence for topical formulations at GSK. Dr. Elliott holds a D. Phil from the Oxford University in the U.K.
Bonnie Pappacena has served as our Vice President of Quality since September 2017. Ms. Pappacena has over 30 years of quality and compliance expertise spanning across good clinical practices (GCP), good laboratory practices (GLP) and good manufacturing practices (GMP) for clinical supplies and commercial product and good pharmacovigilance practices (GVP) regulations. Ms. Pappacena has worked primarily in the pharmaceutical industry and her experience includes drug product as well as combination product development. Prior to this, Ms. Pappacena served as Vice President, Quality at G&W Laboratories, Vice President, Quality at Turing Pharmaceuticals and at Acorda Therapeutics. Mrs. Pappacena also held positions of increasing quality responsibility at Schering Plough Research Institute and Lederle (Wyeth). Ms. Pappacena has a B.S./B.A in Psychology/Philosophy from the University of Scranton and an M.S. in Experimental Psychology from VillanovaSyracuse University.
Our Directors Stanley Hirsch, D.Phil. has served as our director since February 2005 and as chairman of the board since May 2016. Dr. Hirsch has over 30 years of experience in executive positions, including director of business development for a privately held group of healthcare companies. He has also served as general manager of two diagnostics development companies. Dr. Hirsch has been CEO atserved as Chief Executive Officer of FuturaGene Ltd., and its predecessor company CBD Technologies Ltd., since 1989,1995, and has also held the position of General Manager of Portman Pharmaceutical Industries. Since the acquisition of FuturaGene Plc by Suzano Pulp and Paper, a major Brazilian industrial public corporation in July 2010, he also holds thehas held a position equivalent position ofto a vice president at Suzano, reporting to the CEO at Suzano. Dr. Hirsch currently serves as a directorchairman of the board of directors of OWC Pharmaceutical Research Crop (OTC: OWCP)Corp, a position he has held since July 2017. Dr. Hirsch holds a D. PhilPhil. in Cell Biology and Immunology from Oxford University, England. Rex Bright has served as our director since our initial public offering that was completed in September 2014. Mr. Bright hasis currently retired, but previously held CEOchief executive officer positions in the health care industry for the past 21over 20 years. Mr. Bright was the co-founder and CEOChief Executive Officer of SkinMedica, a specialty pharmaceutical business that was later acquired by Allergan.Allergan in 2012. Mr. Bright has worked inalso held executive positions for Johnson & Johnson and GlaxoSmithKline. Mr. Bright haspreviously served as a director of RestorGenex Corporation until 2016 when the company was acquired. Mr. Bright holds a B.A. in Business Administration and Marketing from Drury University, Springfield, Missouri.University.
Aharon Schwartz, Ph.D.Anthony Bruno has served as our director since November 2014. Dr. Schwartz retired from Teva Pharmaceutical Industries Ltd. in 2011, where he served in a number of positions from 1975, the most recent being Vice President, Head of Teva Innovative Ventures from 2008. Dr. Schwartz is currently chairman of the board of directors of BiolineRx Ltd. (Nasdaq: BLRX) 2018 and a director of Barcode Ltd., a private company. From January 2013 through November 2017 he formerly served as a director of Alcobra Ltd. (Nasdaq: ADHD)strategic consultant to us from 2014 to 2018. Dr. Schwartz received his Ph.D. in organic chemistry Mr. Bruno is currently retired. He previously in 1978 from the Weizmann Institute of Science, his M.Sc. in organic chemistry from the Technion and his B.Sc. in chemistry and physics from the Hebrew University of Jerusalem. Dr. Schwartz received a second Ph.D. in 2014 from the Hebrew University of Jerusalem in history and philosophy of science.
Anna Kazanchyan, M.D. has served as our director since December 2014. Dr. Kazanchyan founded Saghmos Therapeutics in September 2016a strategic consultant to various healthcare-focused investment funds from 2011 to 2018, and serveswas employed at Warner Chilcott from 2000 to 2011, most recently as its CEOExecutive Vice President, with responsibility for all business development activities including product acquisitions and Chairwoman. She isdivestitures as well as licensing agreements. Mr. Bruno also the founder and Managing Partner since 2004worked at Warner Lambert for 16 years, holding several positions of Primary i-Research, LLC. In addition, Dr. Kazanchyan has beenincreasing strategic responsibility. Mr. Bruno was also an advisor to CEOs of biopharmaceutical companies (start-ups to global companies). Previously, Dr. Kazanchyan was Senior Biotechnology Analyst at Wachovia Securities, and was a member of the biotechnology equity research teams at Goldman Sachs and Citigroup. She received an M.D. from Harvard Medical School andassociate with Shearman & Sterling. Mr. Bruno holds a B.A. in biology, summa cum laude, from Clark University.
Darrell Rigel, M.D. has served as our director since our initial public offering that was completed in September 2014. Dr. Rigel is a Clinical Professor of Dermatology at the New York University Medical School, an Adjunct Clinical Professor of Dermatology at the Mount Sinai School of Medicine, as well as being an Attending at the Tisch and Bellevue Hospitals in New York. Dr. Rigel also served as a director of Sensus Healthcare, Inc. (Nasdaq: SRTS) until April 2017. Dr. Rigel holds an SB in Management Information Sciences and an SM in Management InformationPolitical Science from the Massachusetts Institute of TechnologySyracuse University, and an M.D.a J.D. from theThe George Washington University School of Medicine.Law School.
Stanley Stern has served as our director since our initial public offering that was completed in September 2014. Mr. Stern has 32over 30 years of experience as an investment banker, working primarily for Oppenheimer & Co, in a number of positions including head of investment banking. He also worked for STI Ventures, Salomon Brothers and C.E. Unterberg. In 2013, Mr. Stern founded Alnitak Capital Partners,. LLC. He currently serves as a director of Audiocodes (Nasdaq: AUDC) since December 2012, Sodastream (Nasdaq: SODA) since November 2015, Ormat Technologies, Inc. (NYSE: ORA) since February 2016 and Ekso Bionics Holdings, Inc. (OTCQB: Ekso).since March 2015. He was previously a member of the board of directors of Sodastream International Ltd. from November 2015 to December 2018. Mr. Stern holds a B.A. in Economics and Accounting from City University of New York, Queens College, and an M.B.A. from Harvard University. Anna Kazanchyan, M.D. has served as our director since December 2014. Dr. Kazanchyan founded Saghmos Therapeutics in September 2016 and serves as its Chief Executive Officer and Chairwoman. She is also the founder and Managing Partner since 2004 of Primary i-Research, LLC. Previously, she was SVP, Business Development and Product Development at Ovid Therapeutics, a company focused on rare neurological disorders. Dr. Kazanchyan was previously Senior Biotechnology Analyst at Wachovia Securities, and was a member of the Biotechnology Equity Research teams at Goldman Sachs and Citigroup. She has served as a director of Innovate Biopharmaceuticals from February to June 2018. She received an M.D. from Harvard Medical School and a B.A. in biology, summa cum laude, from Clark University.
Aharon Schwartz, Ph.D. has served as our director since November 2014. Dr. Schwartz retired from Teva Pharmaceutical Industries Ltd. in 2011, where he served in a number of positions of increasing responsibility from 1975 until his retirement, most recently serving as its Vice President, Head of Teva Innovative Ventures from 2008 to 2011. Dr. Schwartz has been the chairman of the board of directors of BiolineRx Ltd. since 2003 and a director of Protalix BioTherapeutics, Inc. since 2015. From January 2013 through November 2017, he served as a director of Alcobra Ltd. Dr. Schwartz received his Ph.D. in organic chemistry in 1978 from the Weizmann Institute of Science, his M.Sc. in organic chemistry from the Technion and his B.Sc. in chemistry and physics from the Hebrew University of Jerusalem. Dr. Schwartz received a second Ph.D. in 2014 from the Hebrew University of Jerusalem in history and philosophy of science.
Dalia Megiddo, M.D. has served as our director since May 2016. On February 28, 2019, Dr. Megiddo submitted her resignation from the board, effective on the date of the Company’s 2019 annual general meeting of shareholders. Dr. Megiddo co-founded a number of pharmapharmaceutical companies, including Alcobra Ltd. (Nasdaq: ADHD), Bioblast-Pharma Ltd. (Nasdaq: ORPN) and Chiasma Ltd. (Nasdaq: CHMA). She currently serves as Managing Partner of Expedio Ventures, and previously ranled other life science investment funds, including Jerusalem Global Ventures and 7-Health. Dr. Megiddo serveshas been serving as a director of Bioblast-Pharma Ltd.Ltd. since 2012 and formerly served as a director of Alcobra Ltd. from 2013 to 2014. Dr. Megiddo holds an M.D. in Medicine from The Hebrew University of Jerusalem, and is a licensed specialist in family medicine. She also holds an M.B.A. degree from the Kellogg Recanati International School of Business (Tel Aviv University and North Western University). Dov Tamarkin, Ph.D.Sharon Barbari has served as our director fromsince January 2003 until his resignation on January 1, 2018. Dr Tamarkin2019. She is one of the founders of the Company and hadcurrently retired. She previously served as our Chiefthe Executive Officer until June 2017. Prior to that, Dr. Tamarkin served as Vice President of R&D at Portman Pharmaceuticals,Finance and Chief Financial Officer of Cytokinetics Inc. from July 2009 to July 2017, and prior to then, she served as Senior Vice President of Finance and Chief Financial Officer from September 2004 through June 2009. From September 2002 to August 2004, Ms. Barbari served as Chief Financial Officer and Senior Vice President of Finance and Administration of InterMune, Inc., a biotech research and developmentbiopharmaceutical company. From January 1998 to June 2002, she served at Gilead Sciences, Inc., a biopharmaceutical company, and held several positions of increasing responsibility including most recently as Senior R&D Manager at Teva Pharmaceutical Industries Ltd., a public Israeli pharmaceutical company. Dr. Tamarkin holds a Ph.D. in chemistry from The Hebrew University of Jerusalem.
Observers toits Vice President and Chief Financial Officer. Ms. Barbari has served on the Board of Directors
Chaim Chizic, who served as our director until May 7, 2015, served as a non-voting observer from that time until his resignation from such position on January 24, 2018. Meir Eini, who served as a director of the company until May 16, 2016, served as a non-voting observer to our board of directordirectors of Sonoma Pharmaceuticals, Inc. since March 2014. Ms. Barbari received a B.S. in Accounting from that time until his resignation on January 28, 2018. In such capacity, Messrs. Chizic and Eini were entitled to attend meetings of the board and to receive all notices and other correspondence and communications sent by us to members of our board of directors. In addition, and until their resignation, they each received compensation equal to the compensation we paid our directors.San Jose State University.
Identification of Certain Significant Employees71
None.
Family Relationships
There are no family relationships among our executive officers and directors.
Section 16(a) Beneficial Ownership Reporting Compliance
In 2018, we were no longer considered a “foreign private issuer” and began filing as a U.S. domestic filer under SEC regulations. As of January 1, 2018, our officers, directors and greater than 10% shareholders began to file forms pursuant to Section 16(a). In connection with this transition, we filed initial beneficial ownership forms on behalf of our executive officers and directors, and changes to beneficial ownership forms on Form 4 for our Section 16 officers and directors. All of our executive officers’ and directors’ Form 3s were filed 9 days late, on January 11, 2018. Form 4s were filed late on January 24, 2018, for the following executive officers: Mr. David Domzalski, Mr. Mitchell Shirvan, Mr. Russell Elliott, Mr. Alvin Howard, Mr. Yohan Hazot, and Mr. David Schuz. In addition, Mr. Meir Eini, who was an observer to our board until he resigned on January 24, 2018, filed a late Form 3 and late Form 4, each dated January 18, 2018; Mr. Chaim Chizic, who was the other observer to our board until he resigned on January 28, 2018, did not file a Form 3 or Form 4. In addition, we filed a Form 4 that was 12 days late on behalf of our director, Dr. Hirsch, in connection with warrants that automatically exercised in accordance with their terms. Also, on December 6, 2018, we filed a Form 4 that was 5 days late on behalf of our director, Dr. Schwartz, who purchased 112,200 ordinary shares over three days. Code of Business Conduct and Ethics
We have adopted a Code of Business Conduct and Ethics applicable to all of our directors and employees, including our Chief Executive Officer, Chief Financial Officer, controller or principal accounting officer,officers, or other persons performing similar functions, which is a “code“code of ethics”ethics” as defined in Item 406 of Regulation S-K. The full text of the Code of Business Conduct and Ethics will beis available on our website at www.foamix.com. Information contained on our website or that can be accessed through our website does not constitute a part of this report and is not incorporated by reference herein. If we make any amendment to the Code of Business Conduct and Ethics or grant any waivers, including any implicit waiver, from a provision of the code of ethics, we will disclose the nature of such amendment or waiver on our website to the extent required by the rules and regulations of the SEC. Under Item 10 of Form 10-K, if a waiver or amendment of the Code of Business Conduct and Ethics applies to our principal executive officer, principal financial officer, principal accounting officer, controller, or persons performing similar functions and that relates to any element of the code of ethics definition enumerated in Item 406 of Regulation S-K, we are required to disclose such waiver or amendment on our website.
Corporate Governance
Nominating directorsand corporate governance committee
Since we ceased to be a foreign private issuer asOur board of January 1, 2018 and in accordance with NASDAQ Stock Market rules, we were required to either appointdirectors has adopted a nominating and corporate governance committee forcharter that sets forth the nominationresponsibilities of our directors or have director nominees recommended for appointment by a majority of the board’s independent directors in a vote in which only independent directors participate. Our board has opted for the first alternative and has established a nominating and governance committee consistent with the rules and regulations of the board.SEC and Nasdaq, including: (a) assisting in identifying, recruiting and, if appropriate, interviewing candidates to fill positions on the board of directors, including persons suggested by shareholders or others, (b) establishing procedures to be followed by shareholders in submitting recommendations for board candidates, if appropriate, (c) reviewing the background and qualifications of individuals being considered as director candidates, while considering the candidate’s experience, skills, expertise, diversity, personal and professional integrity, character, business judgment, time availability in light of other commitments, dedication, conflicts of interest and such other relevant factors that the committee considers appropriate in the context of the needs of the board, (d) recommending the board nominees for election by shareholders or appointment by the board, as the case may be, in a manner consistent with the criteria for selecting directors, as established by the board from time to time, (e) reviewing the suitability for continued service as a director of each board member, when the term of service of the director expires, and when the director has a change in status (including, but not limited to, an employment change) and recommending whether or not the director should be re-nominated, (f) making recommendations to the board regarding the size and composition of each committee; and (g) overseeing the performance of the board as a whole. A copy of the Nominating and Corporate Governance Committee Charter is available on our website at www.foamix.com.
Our nominating and corporate governance committee consists of Dr. Stanley Hirsch, who also serves as chairman of the committee, along with Mr. Stanley Stern and Mr. Rex Bright. Each of the members of our nominating and corporate governance committee is independent under the NASDAQ Stock MarketNasdaq rules.
The nominating and corporate governance committee is responsible for identifying and making recommendations to the board of directors regardingbelieves that candidates for directorships. In addition,director should have certain minimum qualifications, including the committee is responsible for developing our corporate governance policies, as appropriate, overseeing our corporate governance guidelinesability to read and reportingunderstand basic financial statements, being over 21 years of age and making recommendations tohaving the board concerning governance matters. The committee shall exercise such other powershighest personal integrity and authority as are set forth in its charter, which is available at www.foamix.com website, as well as such other powers and authority as shall from time to time be assigned thereto by resolution of the board, to the extent permitted by law. ethics. When considering director candidates, the nominating and corporate governance committee will also generally consider all of theother relevant qualifications of the candidates, including such factors as the candidate’scandidate’s relevant expertise upon which to be able to offer advice and guidance to management, having sufficient time to devote to the affairs of the company, demonstrated excellence in his or her field, having relevant financial or accounting expertise, having the ability to exercise sound business judgment, having the commitment to rigorously represent the long-term interests of our shareholders and whether the board candidates will be independent for purposes of the NASDAQ listing standards,Nasdaq rules, as well as the current needs of the board and the company. At this time, the Nominating and Corporate Governance Committee does not have a policy with regard to the consideration of director candidates recommended by shareholders. The Nominating and Corporate Governance Committee believes that it is in the best position to identify, review, evaluate and select qualified candidates for Board membership, based on the comprehensive criteria for Board membership approved by the Board.
In addition, while the nominating and corporate governance committee does not have a formal policy on director diversity, our independent directors will take into account a broad range of diversity considerations when assessing director candidates, including individual backgrounds and skill sets, professional experiences and other factors that contribute to the board having an appropriate range of expertise, talents, experiences and viewpoints.
Our nominating and corporate governance committee will consider diversity criteria in view of the needs of the board as a whole when making decisions on director nominations. In the case of incumbent directors who have stepped down or whose terms of office are set to expire, the committee will also review, prior to nominating such directors for another term, such directors’directors’ overall service to the company during their term. The committee will conduct any appropriate and necessary inquiries into the backgrounds and qualifications of possible candidates after considering the function and needs of the board of directors. We may, from time to time, engage an executive search firm to assist our nominatingNominating and corporate governance committeeCorporate Governance Committee in identifying and recruiting potential candidates for membership on the board of directors.
External directors Under the Israeli Companies Law, Israeli public companies are generally required to appoint at least two external directors, who need to meet certain criteria and be appointed according to a specific procedure. However, under the Israeli Companies Regulations (Concessions for Companies Whose Shares are Listed for Trading on a Stock Exchange Abroad), 5760-2000, or the Concession Regulations, as amended on April 17, 2016, Israeli public companies whose shares are traded exclusively on a non-Israeli stock exchange, such as ourselves,us, may opt out from such requirement. Accordingly, in August 2016, our board of directors resolved to opt out from the requirement to appoint external directors. In the same resolution, our board also opted out from certain restrictions on the engagement of former external directors and their relatives and affiliates following the end of their tenure. Consequently, and in accordance with the Concession Regulations, our directors who were previously classified as external directors prior to the date on which our board decided to opt out of the requirement to maintain such function may continue to hold their office until the earlier of the expiry of their original three-year term or the second annual shareholder meeting held after such decision was taken, and following such expiry will not be subject to any of the reappointment limitations imposed on external directors by the Israeli Companies Law. Under the Concession Regulations, these concessions will continue to be available to the company so long as (i) its shares are traded on a U.S. stock exchange, including the NASDAQ Global Market;Nasdaq; (ii) it does not have a “controlling shareholder”“controlling shareholder” (as such term is defined under the Israeli Companies Law), and (iii) it complies with the majority board independence requirements and audit committee and compensation committee requirements under U.S. laws applicable to U.S. domestic issuers.
Audit committee
Roles, responsibilities and procedures
Our audit committee provides assistance to our board of directors in fulfilling its legal and fiduciary obligations in matters involving our accounting, auditing, financial reporting, internal control and legal compliance functions by pre-approving the services performed by our independent accountants and reviewing their reports regarding our accounting practices. Our audit committee also oversees the audit efforts of our independent accountants and takes those actions that it deems necessary to satisfy itself that the accountants are independent of management.
Our board of directors has adopted an audit committee charter that sets forth the responsibilities of the audit committee consistent with the rules and regulations of the SEC and the NASDAQ Stock Market rules,Nasdaq, as well as the requirements for such committee under the Israeli Companies Law, including (a) oversight of our independent registered public accounting firm and recommending the engagement, compensation or termination of engagement of our independent registered public accounting firm to the board of directors in accordance with Israeli law; (b) recommending the engagement or termination of our internal auditor; and (c) recommending the terms of audit and non-audit services provided by the independent registered public accounting firm for pre-approval by our board of directors. Under the Israeli Companies Law, our audit committee is responsible for (a) determining whether there aredirectors(d) identifying deficiencies in the business management practices of our company, including, inter alia, in consultation with our internal auditor or the independent auditor, and making recommendations to the board of directors as to improvehow to correct such practices; (b)(e) reviewing and considering the approval of related party transactions; (f) determining whether to approve certain related party transactions are extraordinary or material under the Israeli Companies Law, including transactions in which an office holder has a personal interest and whether such transaction is extraordinary or material“personal interest” under the Israeli Companies Law, – see our Registration Statement on Form F-1 as filed under the Securities Act with the SEC on September 3, 2014, under “Management—Approval of Related Party Transactions under Israeli Law—Fiduciary Duties of Directors and Executive Officers”); (c)whether to approve such transactions; (g) establishing the approval process for certain transactions with a controlling shareholder or in which the controlling shareholder has a personal interest; (d) where the board of directors approves(h) examining and approving the working plan of the internal auditor, examining such working plan beforesubject to any modifications in its submission to the board of directors and proposing amendments thereto; (e)discretion; (i) examining our internal audit controls and internal auditor’sauditor’s performance, including whether the internal auditor has sufficient resources and tools to fulfill his or her responsibilities; (f)(j) examining the scope of our auditor’sauditor’s work and compensation and submitting aits recommendation with respect thereto to our board of directors or shareholders, depending on which of them is considering the appointment of our auditor; and (g)(k) establishing procedures for the handling of employees’employees’ complaints as to the management of our business and the protection to be provided to such employees.
Compositionemployees; and quorum
Following(l) reviewing the decisionour annual audited financial statements and quarterly financial statements with management and the independent auditor, including a review of our boarddisclosures under “Management’s Discussion and Analysis of directors to opt outFinancial Condition and Results of Operations” . A copy of the various restrictions concerning the composition and quorum of the audit committeeAudit Committee Charter is available on our website at www.foamix.com.
A “personal interest” under the Israeli Companies Law includes an interest of any person in an action or transaction of a company, excluding any interest arising solely from holding the composition and quorumCompany’s shares, but including the personal interest of our audit committeesuch person’s spouse, siblings, parents, grandparents, descendants, spouse’s descendants, siblings or parents or the spouse of any of such persons, and the nominationpersonal interest of members toany entity in which such committee are now governed by the regulationsperson or one of the U.S. Securities Exchange Commission,aforementioned relatives of such person serves as a director or SEC, andchief executive officer, owns 5% or more of such entity’s outstanding shares or voting rights or has the NASDAQ Stock Market rules, as set out below.right to appoint one or more directors or the chief executive officer. Further, in the case of a person voting by proxy at a shareholder meeting, “personal interest” includes the personal interest of either the proxy holder or the shareholder granting the proxy, whether or not the proxy holder has discretion how to vote.
Under the NASDAQ Stock MarketIsraeli Companies Law, an extraordinary transaction is defined as any of the following:
| · | a transaction other than in the ordinary course of business; |
| · | a transaction that is not on market terms; or |
| · | a transaction that may have a material impact on a company’s profitability, assets or liabilities. |
Our audit committee may not approve any actions requiring its approval, unless, at the time of the approval, a majority of the committee’s members are present, which majority consists of independent directors.
Composition and quorum
Under Nasdaq rules and SEC regulations, we are required to maintain an audit committee consisting of at least three independent directors, each of whom is financially literate and one of whom has accounting or related financial management expertise. In order for a director to be designated as “independent” under the NASDAQ Stock Market rulesexpertise and SEC regulations, he or she must not have a material relationship with the company that would impair his or her independence, such as a commercial, consulting, legal, accounting or familial relationships, among others. However, ownership of a significant amount of shares or affiliation with a major shareholder should not, in and of itself, preclude the board from determining that a director is independent, nor is the board precluded from appointing its chairman as a member of the audit committee or as chairman of the committee.
In order for a director to be designated as “financially literate” under the NASDAQ Stock Market rules and SEC regulations, he or she are required to have sufficient understanding of the language of accounting and corporate finance to act as effective overseers of the integrity of a company’s financial reporting process and its financial statements, including the selection and oversight of the performance of the external and internal auditors.
In order for a director to qualify as an “audit committee financial expert” under SEC regulations he or she must have educationas such term is defined in Item 407(d)(5) of Regulation S-K of the Securities and experience as chief financial officer, chief accounting officer, controller, public accountant or auditor, or experience in one or more positions that involve the performanceExchange Act of similar functions or in actively supervising such positions. If no audit committee member qualifies, the company must state why its audit committee lacks a financial expert.1934.
Our audit committee consists of Dr. Stanley Stern, (chairman),who also serves as chairman of the committee, Mr. Rex Bright, Dr. Dalia Megiddo (who will continue to serve as a member of our board of directors until our 2019 annual general meeting of shareholders) and Darrell Rigel. EachMs. Sharon Barbari. The board has determined that each of the members of our audit committee is eligible to be classed as an independent director in accordance with SEC regulations and satisfies the independent director requirements under the NASDAQ Stock MarketNasdaq rules. All designated members of our audit committee meet the requirements for financial literacy under the applicable Nasdaq rules of the NASDAQ Stock Market and SEC regulations. However, we do not currently haveOur board has determined that Ms. Barbari is an audit“audit committee financial expert,” as such term is defined by the SEC. Nevertheless, our board of directors has determined in its business judgment that our existing committee members have the ability to oversee our financial statements based on their extensive business backgrounds and that Stanley Stern has “financial sophistication” under the NASDAQ Stock Marketapplicable SEC rules.
Compensation committee
Roles, responsibilities and procedures
Our board of directors has established a compensation committee and adopted a charter setting forth its roles and responsibilities, which include (a) recommending a compensation policy regarding the terms of engagement of office holders, to which we refer as a compensation policy,is recommended to the board of directors for its approval and the subsequentsubsequently to shareholders for their approval, by the shareholders, in accordance with the Israeli Companies Law, and reviewing such policies from time to time, (b) recommending to the board of directors periodic updates to the compensation policy and whether the compensation policy should continue in effect every three years; (c) assessing the implementation of the compensation policy; (d) reviewing and approving the granting of options, restricted share units and other incentive awards to the extent such authority is delegated by the board of directors; (e) reviewing, evaluating and making recommendations regarding the compensation and benefits for non-executive directors, and (f) determining whether to approve and recommend to the board of directors and shareholders to approve transactions with office holders relating to their terms of compensation, as required under the Israeli Companies Law, (g) determining whether changes to the compensation terms of the chief executive officerCEO of the company need notCompany are material and if the changes are required to be brought to the shareholders for approval, (h) overseeing compliance reporting requirements of the shareholders.SEC, (i) determining whether to recommend to the board to adopt a share ownership policy for directors and executive officers, and (j) performing such other activities as may be required. A copy of the Compensation Committee Charter is available on our website at www.foamix.com.
Under the Israeli Companies Law, the compensation policy must be adopted by the board of directors after considering the recommendations of the compensation committee and needs to be further brought before the company’scompany’s shareholders for approval, referred to herein as the Special Majority Approval for Compensation. A Special Majority Approval for Compensation requires shareholder approval by a majority vote of the shares present and voting at a meeting of shareholders called for such purpose, provided that either: (a) such majority includes at least a majority of the shares held by all shareholders who are not controlling shareholders and do not have a personal interest in such compensation arrangement; or (b) the total number of shares of non-controlling shareholders and shareholders who do not have a personal interest in the compensation arrangement and who vote against the arrangement does not exceed 2% of the company’scompany’s aggregate voting rights.
The compensation policy must serve as the basis for decisions concerning the financial terms of employment or engagement of office holders, including exculpation, insurance, indemnification and any monetary payment and obligation of payment in respect of employment or engagement. The compensation policy must relate to certain factors, including advancement of the company’scompany’s objectives, the company’scompany’s business plan and its long-term strategy, and creation of appropriate incentives for office holders. It must also consider, among other things,inter alia, the company’scompany’s risk management, size and the nature of its operations.
The compensation policy must furthermore consider additional factors, as follows: (a) the knowledge, skills, expertise and accomplishments of the relevant office holder; (b) the office holder’sholder’s roles and responsibilities and prior compensation agreements with him or her; (c) the relationshipratio between the terms offered and the average compensation of the other employees of the company, including those employed through manpower companies; (d) the impact of disparities in salary upon work relationships in the company; (e) the possibility of reducing variable compensation at the discretion of the board of directors; (f) as to variable compensation, the possibility of setting a limit on the exercise value of non-cash variable equity-based compensation; and (g) as to severance compensation, the period of service of the office holder, the terms of his or her compensation during such service period, the company’sCompany’s performance during that period of service, the person’sperson’s contribution towards the company’sCompany’s achievement of its goals and the maximization of its profits, and the circumstances under which the person is leaving the company.of termination of service.
The compensation policy must also include the following principles: (a) the link between variable compensation and long-term performance and measurable criteria; (b) the relationshipratio between variable and fixed compensation, and the ceiling for the value of variable compensation; (c) the conditions under which an office holder would be required to repay compensation paid to him or her if it was later shown that the data upon which such compensation was based was inaccurate and was required to be restated in the company’sCompany’s financial statements; (d) the minimum holding or vesting period for variable, equity-based compensation; and (e) maximum limits for severance compensation.severance.
Under the Israeli Companies Law, every three years we are required to re-obtain the approval of our compensation committee, board of directors and shareholders for either the continuation of our existing compensation policy or adoption of a new compensation policy. Our compensation policy was last approved by our shareholders on June 22, 2015,May 8, 2018, after having been recommended by our compensation committee and approved by our board of directors, and will therefore need to be either re-approved, amended, or replaced by a new policy this year.in 2021.
Our compensation committee may conduct or authorize investigations into, or studies of, matters within its scope of responsibilities, and may retain or obtain the advice of a compensation consultant, legal counsel or other advisor in its sole discretion. The compensation committee is directly responsible for the appointment, compensation and oversight of the work of any compensation consultant, legal counsel or other advisor that it retains, at the expense of the Company. The compensation committee may select, or receive advice from, a compensation consultant, legal counsel or other advisor to the compensation committee, other than in-house legal counsel, only after conducting an assessment of, and determining, the advisor’sadvisor’s independence, including whether the advisor’sadvisor’s work has raised any questions of independence or conflicts of interest, taking into consideration the Exchange Act, the factors set forth in theNasdaq rules of the NASDAQ and any other factors that the committee deems relevant.
In determining the compensation of the company’sour chief executive officer and other executive officers, as well as itsour directors and chairman of the board for 2017,2018, including bonus amounts and performance criteria, the compensation committee retained the services of a U.S.-based, compensation consultant, Frederic W. Cook & Co., Inc. (“FW Cook”), to conduct a comparative survey of the compensation of such office holders. The survey examined the publicly-reported cash and equity compensation of chief executive officers and other executive officers, board members and chairmenchairs of the board of 21 comparable U.S. and Israeli pharmaceutical and biotechnology companies.
Based on this survey, the compensation committee set the cash compensation and cash bonuses of our chief executive officer and each of our other executive officers, and the cash compensation of our directors and chairmanChairman of the board, within the range of compensation of similarly-situated officer holders,, with the specific compensation for each office holder varying across such range in accordance with the committee’scommittee’s evaluation of his or her individual performance. Frederic W.
FW Cook & Co., Inc. did not perform any services for the Companyus other than services for the compensation committee. After review and consultation with Frederic W.FW Cook, & Co., Inc., the compensation committee determined that there was no conflict of interest resulting from retaining the consultant in fiscal year 2017.2018.
Composition and quorum Following the Concession Regulations and the decision of our board of directors to avail itself of the right to opt out of the requirements concerning the appointment of a compensation committee under the Israeli Companies Law, the company is only required to comply with the requirements regarding such appointment under the NASDAQ Stock Market rules. Accordingly, the composition and quorum
The members of our compensation committee and the nomination of members to such committee are now governed by the regulationsMr. Rex Bright, who also serves as chairman of the SEC and the NASDAQ Stock Market rules. Under NASDAQ Stock Market rules, we have established and continue to maintain a compensation committee, the members of which are currently Rex Bright (Chairman), Darrel Rigel,Mr. Anthony Bruno, Mr. Stanley Stern and Ms. Anna Kazanchyan. Each member of our compensation committee is independent under the NASDAQ Stock MarketNasdaq rules.
ITEM 11 —- EXECUTIVE COMPENSATION
As a smaller reporting company, and as an emerging growth company, or EGC, and as permitted by Title I of the Jumpstart Our Business Startups Act of 2012, or JOBS Act, we have scaled down our disclosure on executive compensation to omit compensation discussion and analysis and certain tables and items that would otherwise be required in an annual report on Form 10-K of a non-EGC issuer.10-K. Such omitted tables and items include, but are not limited to, the table displaying grants of plan-based awards, the table displaying option exercises and vesting, disclosure on CEO pay ratio and disclosure of compensation policies as related to risk management. Furthermore, our disclosure on compensation focuses only on an individual serving as our principal executive officer or acting in a similar capacity during the last completed fiscal year and the next two most-highly compensated executive officers (and up to two additional individuals no longer serving as executive officers at year-end) (our “named“named executive officers”officers”). In addition, for as long as we remain an EGC, we are not subject to certain governance requirements relating to executive compensation such as holding a “say-on-pay”“say-on-pay” and “say-on-golden-parachute”“say-on-golden-parachute” advisory votes.
Our named executive officers for 2017 consisted of Messrs. David Domzalski, CEO, Ilan Hadar, CFO, Yohan Hazot, Vice President, Pharmaceutical Development, Dov Tamarkin,2018, which include our former CEO,principal executive officer and Meir Eini, former Chief Information Officer.the next two most highly compensated executive officers, are:
| · | David Domzalski, our Chief Executive Officer; |
| · | Ilan Hadar, our Chief Financial Officer and Israel Country Manager; and |
| · | Mutya Harsch, General Counsel and Chief Legal Officer. |
Summary Compensation Table
The following table sets forth all of the compensation awarded to, earned by or paid to our named executive officers during 20162017 and 2017:2018: | Name and Principal Position | | Year | | Salary ($) | | | Bonus ($) | | | Option Awards ($)(3) | | | Non-Equity Incentive Plan Compensation ($) | | | All Other Compen-sation ($) | | | Total ($)(4) | | | David Domzalski, CEO | | 2017 | | | 416,980 | | | | 165,000 | | | | 2,204,155 | | | | - | | | | 10,800 | (5) | | | 2,796,935 | | | 2016 | | | 370,125 | | | | 166,888 | | | | 211,579 | | | | 66,420 | | | | - | | | | 815,012 | | | Ilan Hadar, CFO | | 2017 | | | 342,253 | | | | 119,978 | | | | 1,212,809 | | | | - | | | | 121,815 | (6) | | | 1,796,854 | | | 2016 | | | 239,546 | | | | 103,365 | | | | 207,709 | | | | 71,064 | | | | 81,458 | (6) | | | 703,143 | | | Yohan Hazot, Vice President, Pharmaceutical Development | | 2017 | | | 217,797 | | | | 57,687 | | | | 496,304 | | | | - | | | | 72,453 | (7) | | | 844,241 | | | 2016 | | | 164,558 | | | | 82,185 | | | | 192,959 | | | | - | | | | 61,854 | (7) | | | 501,556 | | Dov Tamarkin, former CEO(1) | | 2017 | | | 210,600 | | | | 86,625 | | | | - | | | | - | | | | 1,092,084 | (8) | | | 1,389,309 | | | 2016 | | | 385,000 | | | | 209,597 | | | | 321,599 | | | | 184,515 | | | | 206,873 | (8) | | | 1,307,584 | | Meir Eini, former Chief Innovation Officer(2) | | 2017 | | | 181,500 | | | | 61,875 | | | | 335,874 | | | | - | | | | 918,008 | (9) | | | 1,497,257 | | | 2016 | | | 300,000 | | | | 142,801 | | | | 339,918 | | | | 51,823 | | | | 158,133 | (9) | | | 992,676 | |
| Name and Principal Position | | Year | | Salary ($) | | | Bonus ($) | | | Share Awards ($)(1) | | | Option Awards ($)(1) | | | Non-Equity Incentive Plan Compensa-tion ($) | | | All Other Compensa-tion ($)(2) | | | Total ($)(3) | | | David Domzalski, CEO | | 2018 | | | 440,000 | | | | 440,000 | (4) | | | 119,788 | | | | 214,424 | | | | - | | | | 11,000 | (7) | | | 1,225,212 | | | 2017 | | | 416,980 | | | | 165,000 | | | | 711,033 | | | | 1,493,122 | | | | - | | | | 10,800 | | | | 2,796,935 | | | Ilan Hadar, CFO and Israel Country Manager | | 2018 | | | 367,026 | | | | 281,750 | (4) | | | 70,071 | | | | 120,848 | | | | - | | | | 145,962 | (5) | | | 985,657 | | | 2017 | | | 342,253 | | | | 119,978 | | | | 421,502 | | | | 791,307 | | | | - | | | | 121,815 | | | | 1,796,855 | | | Mutya Harsch, General Counsel and Chief Legal Officer | | 2018(6) | | | 325,000 | | | | 220,000 | (8) | | | 149,750 | (9) | | | 172,709 | (9) | | | - | | | | 11,000 | (10) | | | 878,459 | |
(1) | Dr. Dov Tamarkin ceased being our CEO on July 1, 2017 and ceased being our employee, at which point he became our chief scientific advisor and continued to serve as our director, until his resignation on January 1, 2018.
|
(2) | Mr. Meir Eini ceased being our chief innovation officer on June 29, 2017, following which date he continued to serve as an advisor to the company and an observer to the board until January 28, 2018.
|
(2) The contributions and benefits referred to in some of the footnotes to this column reflect compensation to our executives residing in Israel that is customarily provided to Israeli executives in the high-tech and bio-pharmaceutical markets. For example, deposits to “severance funds,” are contributions made by us in lieu of its statutory severance pay obligation under Israeli law. Generally, an Israeli company is required to pay, upon termination of an employee, an amount equal to such employee’s last monthly salary for each full year of his or her employment with the company (or a pro-rata portion of such last salary for any part of a year), and the obligation for such severance pay is recorded as a long-term liability on the company's balance sheet. Alternatively, a company may make monthly contributions to a severance fund in the employee's name, in an amount equal to one twelfth (8⅓%) of the employee’s salary for such month. Making such contributions in a consistent manner releases the company from further liability for severance pay to the employee upon his or her actual termination, even if the amount accumulated in the severance fund falls short of the amount of mandatory severance pay to which the employee would have otherwise been entitled under law (which is usually the case, as the average salary during the course of employment is typically lower than the last salary, which reflects the employee’s highest-achieved seniority and rank). At the same time, the employees benefit from the certainty and security of having their severance pay deposited in a fund in their name, and from the right to have the fund released to them both in the event of termination or resignation. An “education fund” is a medium-term savings scheme that takes advantage of a unique tax break granted under Israeli law, whereby a company's contributions to such fund (which, despite its misleading name, may be used by the employee for any purpose), as well as all capital gains accrued on such contributions, are free of tax if (a) the company contributes an amount equal to 7.5% of the employee's salary to such fund, up to a certain limit, and the employee further contributes 2.5% of his salary at his expense, and (b) the fund remains undrawn for a period of at least 6 years from the time of the first contribution. (3) | The value in this column represents the aggregate grant date fair value of our stock option awards computed in accordance with FASB ASC Topic 718. For a discussion of valuation assumptions used in the calculation of these amounts, see Note 9 to our audited financial statements, included in Item 8—Financial Statements and Supplemental Financial Information.”
|
(3) Salary and other compensation of Israeli executive officers for the years ended December 31, 2018 and 2017 are based on an average US$/NIS representative exchange rate of NIS 3.60 per dollar for both years. Bonuses for 2018 and 2017 are based on US$/NIS representative exchange rates of NIS 3.748 and NIS 3.47 per dollar as of December 31, 2018 and 2017, respectively.
(4) Consists of discretionary bonuses as described below under “—General Policies Regarding Cash Bonuses, Equity Awards and Other Benefits to Named Executive Officers.”
(5) Mr. Hadar’s other compensation in 2018 consisted of (i) $30,257 of automobile expenses, (ii) $55,127 of deposits to severance funds, (iii) $27,527 of deposits to an education fund, (iv) $22,269 of gross-up for related taxes, (iv) $10,050 of social security payments and (v) $732 of gross up of other benefit.
(6) Ms. Harsch was not one of our named executive officers for the year ended December 31, 2017.
(7) Consists of employer contributions to Mr. Domzalski’s 401(k) plan.
(4) | Salary and other compensation of Israeli executive officers for the years 2017 and 2016 are based on average US$/NIS representative exchange rates of NIS 3.60 and NIS 3.84 per dollar for 2017 and 2016, respectively. Bonuses for the years 2017 and 2016 are based on US$/NIS representative exchange rates of NIS 3.47 and NIS 3.85 per dollar as of December 31, 2017 and 2016, respectively. |
(8) Consists of (i) discretionary bonuses as described below under “—General Policies Regarding Cash Bonuses, Equity Awards and Other Benefits to Named Executive Officers” and (ii) a signing cash bonus granted to Ms. Harsch upon commencement of her employment with us.
(5) | Solely includes employer contribution to Mr. Domzalski’s 401K plan. |
(9) Consists of a one-off grant of options and restricted share units upon commencement of Ms. Harsch’s employment with us.
(10) Consists of employer contributions to Ms. Harsch’s 401(k) plan.
77 (6) | In 2017, the bulk of Mr. Hadar’s other compensation consisted of $23,979 of automobile expenses, $50,976 deposits to severance funds, $10,153 of gross-up of related tax, $10,027 of social security payments and deposits of $25,669 to an education fund. In 2016, the bulk of such compensation consisted of $16,476 of automobile expenses, $35,342 of deposits to severance funds, $9,414 of social security payments, and deposits of $17,698 to an education fund.
|
(7) | In 2017, the bulk of Mr. Hazot’s other compensation consisted of $12,668 of automobile expenses, $32,299 of deposits to severance funds, $10,027 of social security payments and deposits of $16,335 to an education fund. In 2016, the bulk of such compensation consisted of $11,873 of automobile expenses, $24,181 of deposits to severance funds, $9,463 of social security payments, and deposits of $12,238 to an education fund.
|
(8) | In 2017, the bulk of Dr. Tamarkin’s other compensation consisted of $973,177 of retirement compensation, $17,835 of automobile expenses, $33,862 of deposits to severance funds, $17,835 of gross-up of related tax and deposits of $16,043 to an education fund. In 2016, the bulk of such compensation consisted of $33,432 of automobile expenses, $63,475 of deposits to severance funds, $9,398 of social security payments, $33,432 of gross-up of related tax, and deposits of $30,073 to an education fund.
|
(9) | In 2017, the bulk of Mr. Eini’s other compensation consisted of $820,315 of retirement compensation, $12,668 of automobile expenses, $29,024 of deposits to severance funds, $12,668 of gross-up of related tax and deposits of $13,751 to an education fund. In 2016, the bulk of such compensation consisted of $23,746 of automobile expenses, $54,407 of deposits to severance funds, $9,398 of social security payments, $23,746 of gross-up of related tax, and deposits of $25,777 to an education fund.
|
Outstanding Equity Awards at Fiscal Year-End Table
The table below sets forth information regarding outstanding equity awards held by each of our named executive officers as of December 31, 2017:2018:
| | | | | Option Awards | | | Stock Awards | | | Name | | Grant Date | | Number of Shares Under-lying Unexer-cised Op-tions (#) Exercisa-ble | | | Number of Shares Underlying Unexer-cised Options (#) Un-exercisable | | | Option Exercise Price ($) | | | Option Expiration Date | | | Number of Shares or Units of Shares That Have Not Vested (#) | | | Market Value of Shares or Units of Shares That Have Not Vested ($) | | | David Domzalski | | 06/09/14(1) | | | 14,063 | | | | 4,688 | | | | 7.98 | | | 06/09/24 | | | | - | | | | - | | 05/26/15(2) | | | - | | | | - | | | | - | | | | - | | | | 12,500 | | | | 75,125 | | 11/10/15(3) | | | 118,405 | | | | 118,405 | | | | 7.14 | | | 11/10/25 | | | | - | | | | - | | 03/01/16(4) | | | 26,250 | | | | 33,750 | | | | 6.04 | | | 03/01/26 | | | | 1,406 | | | | 8,452 | | 01/01/17(5) | | | - | | | | 71,369 | | | | 10.22 | | | 01/01/27 | | | | 23,790 | | | | 142,978 | | 08/08/17(6) | | | - | | | | 327,720 | | | | 5.76 | | | 08/08/27 | | | | 81,930 | | | | 492,399 | | | Ilan Hadar | | 03/31/14(7) | | | 4,375 | | | | 1,094 | | | | 1.92 | | | 03/31/24 | | | | - | | | | - | | 11/19/14(8) | | | 19,300 | | | | 4,825 | | | | 5.46 | | | 11/19/24 | | | | - | | | | - | | 01/15/15(9) | | | 12,375 | | | | 5,625 | | | | 6.77 | | | 01/12/25 | | | | - | | | | - | | 06/28/15(10) | | | - | | | | - | | | | - | | | | - | | | | 2,813 | | | | 16,903 | | 11/10/15(11) | | | 101,002 | | | | 101,002 | | | | 7.13 | | | 11/10/25 | | | | - | | | | - | | 03/01/16(12) | | | 26,250 | | | | 33,750 | | | | 6.34 | | | 03/01/26 | | | | 1,406 | | | | 8,452 | | 01/01/17(13) | | | - | | | | 60,389 | | | | 10.31 | | | 01/01/27 | | | | 20,130 | | | | 120,981 | | 08/08/17(14) | | | - | | | | 196,205 | | | | 5.22 | | | 08/08/27 | | | | 49,051 | | | | 294,797 | | | Yohan Hazot | | 02/02/10(15) | | | 6,125 | | | | - | | | | 1.92 | | | 02/01/20 | | | | - | | | | - | | 03/31/14(16) | | | 5,938 | | | | 313 | | | | 1.92 | | | 03/31/24 | | | | | | | | - | | 01/15/15(17) | | | 12,375 | | | | 5,625 | | | | 6.77 | | | 01/12/25 | | | | - | | | | - | | 06/28/15(18) | | | - | | | | - | | | | - | | | | - | | | | 2,813 | | | | 16,903 | | 11/10/15(19) | | | 21,397 | | | | 21,397 | | | | 7.13 | | | 11/10/25 | | | | - | | | | - | | 03/01/16(20) | | | 26,250 | | | | 33,750 | | | | 6.34 | | | 03/01/26 | | | | - | | | | - | | 01/01/17(21) | | | - | | | | 54,899 | | | | 10.31 | | | 01/01/27 | | | | 18,300 | | | | 109,983 | | | Dov Tamarkin | | 12/29/14(22) | | | 24,000 | | | | - | | | | 5.88 | | | 12/29/24 | | | | - | | | | - | | 01/15/15(23) | | | 33,750 | | | | 11,250 | | | | 6.77 | | | 12/28/24 | | | | - | | | | - | | 06/28/15(24) | | | - | | | | - | | | | - | | | | - | | | | 5,625 | | | | 33,806 | | 03/01/16(25) | | | 43,750 | | | | 56,250 | | | | 6.34 | | | 03/01/26 | | | | - | | | | - | | | Meir Eini | | 12/29/14(26) | | | 48,000 | | | | - | | | | 5.88 | | | 12/29/24 | | | | - | | | | - | | 01/15/15(27) | | | 27,000 | | | | 9,000 | | | | 6.77 | | | 12/28/24 | | | | - | | | | - | | 06/28/15(28) | | | - | | | | - | | | | - | | | | - | | | | 5,625 | | | | 33,806 | | 11/15/16(29) | | | - | | | | - | | | | - | | | | - | | | | 20,000 | | | | 120,200 | | 08/09/16(30) | | | 10,938 | | | | 24,063 | | | | 7.09 | | | 08/09/26 | | | | - | | | | - | | 01/01/17(31) | | | - | | | | 60,389 | | | | 10.31 | | | 01/01/27 | | | | 20,130 | | | | 120,981 | |
| Name | | Grant Date | | Number of Shares Underlying Unexercised Options Exercisable | | | Number of Shares Underlying Unexercised Options Un-exercisable | | | Option Exercise Price ($) | | | Option Expiration Date | | | Number of Shares or Units of Shares That Have Not Vested | | | Market Value of Shares or Units of Shares That Have Not Vested ($) | | | David Domzalski | | 06/09/14(1) | | | 18,750 | | | | - | | | | 7.98 | | | 06/09/24 | | | | - | | | | - | | 11/10/15(2) | | | 177,610 | | | | 59,200 | | | | 7.14 | | | 11/10/25 | | | | - | | | | - | | 03/01/16(3) | | | 41,250 | | | | 18,750 | | | | 6.04 | | | 03/01/26 | | | | 783 | | | | 2,811 | | 01/01/17(4) | | | 31,225 | | | | 40,144 | | | | 10.22 | | | 01/01/27 | | | | 13,381 | | | | 48,038 | | 08/08/17(5) | | | 102,412 | | | | 225,308 | | | | 5.76 | | | 08/08/27 | | | | 56,327 | | | | 202,214 | | 08/05/18(6) | | | - | | | | 70,187 | | | | 5.06 | | | 02/27/28 | | | | 23,396 | | | | 83,992 | | | Ilan Hadar | | 03/31/14(7) | | | 5,469 | | | | - | | | | 1.92 | | | 03/31/24 | | | | - | | | | - | | 11/19/14(8) | | | 24,125 | | | | - | | | | 5.46 | | | 11/19/24 | | | | - | | | | - | | 01/15/15(9) | | | 16,875 | | | | 1,125 | | | | 6.77 | | | 01/15/25 | | | | - | | | | - | | 06/28/15(10) | | | - | | | | - | | | | - | | | | - | | | | 568 | | | | 2,039 | | 11/10/15(11) | | | 151,501 | | | | 50,503 | | | | 7.13 | | | 11/10/25 | | | | - | | | | - | | 03/01/16(12) | | | 41,250 | | | | 18,750 | | | | 6.34 | | | 03/01/26 | | | | 783 | | | | 2,811 | | 01/01/17(13) | | | 26,419 | | | | 33,970 | | | | 10.31 | | | 01/01/27 | | | | 11,324 | | | | 40,653 | | 08/08/17(14) | | | 61,314 | | | | 134,891 | | | | 5.22 | | | 08/08/27 | | | | 33,722 | | | | 121,062 | | 02/27/18(15) | | | - | | | | 35,093 | | | | 6.40 | | | 02/27/28 | | | | 11,698 | | | | 41,996 | | | Mutya Harsch | | 02/27/18(16) | | | - | | | | 50,000 | | | | 6.35 | | | 02/27/28 | | | | 25,000 | | | | 89,750 | |
________________________ | (1) | The options vest over a period of four years from June 9, 2014, 25% on each anniversary of such date, ending June 9, 2018; |
(1) The options vested over a period of four years from June 9, 2014, 25% on each anniversary of such date, ending June 9, 2018.
(2) The options vest over a period of four years from November 10, 2015, 25% on the first anniversary of such date and 6.25% every three months thereafter, ending November 10, 2019.
(3) The options vest over a period of four years from March 1, 2016, 25% on the first anniversary of such date and 6.25% every three months thereafter, ending March 1, 2020. The restricted share units vest in equal installments every three months over the vesting period beginning December 1, 2018 and ending March 1, 2020.
(4) The options vest over a period of four years from January 1, 2017, 25% on the first anniversary of such date and 6.25% every three months thereafter, ending January 1, 2021. The restricted share units vest in equal installments every three months over the vesting period beginning October 1, 2018 and ending January 1, 2021.
(5) The options vest over a period of four years from August 8, 2017, 25% on the first anniversary of such date and 6.25% every three months thereafter, ending August 8, 2021. The restricted share units vest in equal installments every three months over the vesting period beginning November 8, 2018 and ending August 8, 2021.
(6) The options and restricted share units vest over a period of four years from February 27, 2018, 25% on the first anniversary of such date and 6.25% every three months thereafter, ending February 27, 2022.
(7) The options vested over a period of four years from March 31, 2014, 20% on such date and 5% every three months thereafter, ending March 31, 2018.
(8) The options vested over a period of four years from November 19, 2014, 20% on such date and 5% every three months thereafter, ending November 19, 2018.
(9) The options vest over a period of four years from January 15, 2015, 25% on the first anniversary of such date and 6.25% every three months thereafter, ending January 15, 2019.
(10) The restricted share units vest on January 15, 2019.
(11) The options vest over a period of four years from November 10, 2015, 25% on the first anniversary of such date and 6.25% every three months thereafter, ending November 10, 2019.
(12) The options vest over a period of four years from March 1, 2016, 25% on the first anniversary of such date and 6.25% every three months thereafter, ending March 1, 2020. The restricted share units vest in equal installments every three months over the vesting period beginning December 1, 2018 and ending March 1, 2020.
(13) The options vest over a period of four years from January 1, 2017, 25% on the first anniversary of such date and 6.25% every three months thereafter, ending January 1, 2021. The restricted share units vest in equal installments every three months over the vesting period beginning October 1, 2018 and ending January 1, 2021. | (2) | The RSUs vest on November 19, 2018; |
(14) The options vest over a period of four years from August 8, 2017, 25% on the first anniversary of such date and 6.25% every three months thereafter, ending August 8, 2021. The restricted share units vest in equal installments every three months over the vesting period beginning November 8, 2018 and ending August 8, 2021. | (3) | The options vest over a period of four years from November 10, 2015, 25% on the first anniversary of such date and 6.25% every three months thereafter, ending November 10, 2019; |
(15) The options and restricted share units vest over a period of four years from February 27, 2018, 25% on the first anniversary of such date and 6.25% every three months thereafter, ending February 27, 2022. | (4) | The options vest over a period of four years from March 1, 2016, 25% on the first anniversary of such date and 6.25% every three months thereafter, ending March 1, 2020. The RSUs vest in equal installments every three months over the vesting period beginning December 1, 2017 and ending March 1, 2020; |
(16) The options and restricted share units vest over a period of four years from February 27, 2018, 25% on the first anniversary of such date and 6.25% every three months thereafter, ending February 27, 2022. | (5) | The options vest over a period of four years from January 1, 2017, 25% on the first anniversary of such date and 6.25% every three months thereafter, ending January 1, 2021; |
| (6) | The options and RSUs vest over a period of four years from August 8, 2017, 25% on the first anniversary of such date and 6.25% every three months thereafter, ending August 8, 2021; |
| (7) | The options vest over a period of four years from March 31, 2014, 20% on such date and 5% every three months thereafter, ending March 31, 2018; |
| (8) | The options vest over a period of four years from November 19, 2014, 20% on such date and 5% every three months thereafter, ending November 19, 2018; |
| (9) | The options vest over a period of four years from January 15, 2015, 25% on the first anniversary of such date and 6.25% every three months thereafter, ending January 15, 2019; |
| (10) | The RSUs vest in equal installments every three months over the vesting period beginning October 15, 2017 and ending January 15, 2019; |
| (11) | The options vest over a period of four years from November 10, 2015, 25% on the first anniversary of such date and 6.25% every three months thereafter, ending November 10, 2019; |
| (12) | The options vest over a period of four years from March 1, 2016, 25% on the first anniversary of such date and 6.25% every three months thereafter, ending March 1, 2020. The RSUs vest in equal installments every three months over the vesting period beginning December 1, 2017 and ending March 1, 2020; |
| (13) | The options and RSUs vest over a period of four years from January 1, 2017, 25% on the first anniversary of such date and 6.25% every three months thereafter, ending January 1, 2021; |
| (14) | The options and RSUs vest over a period of four years from August 8, 2017, 25% on the first anniversary of such date and 6.25% every three months thereafter, ending August 8, 2021; |
| (15) | The options vested over a period of four years from February 2, 2010, 20% on such date and 5% every three months thereafter, ending February 2, 2014; |
| (16) | The options vest over a period of four years from March 31, 2014, 20% on such date and 5% every three months thereafter, and ending March 31, 2018; |
| (17) | The options vest over a period of four years from January 15, 2015, 25% on the first anniversary of such date and 6.25% every three months thereafter, ending January 15, 2019; |
| (18) | The RSUs vest in equal installments every three months over the vesting period beginning October 15, 2017 ending January 15, 2019; |
| (19) | The options vest over a period of four years from November 10, 2015, 25% on the first anniversary of such date and 6.25% every three months thereafter, ending November 10, 2019; |
| (20) | The options vest over a period of four years from March 1, 2016, 25% on the first anniversary of such date and 6.25% every three months thereafter, ending March 1, 2020; |
| (21) | The options and RSUs vest over a period of four years from January 1, 2017, 25% on the first anniversary of such date and 6.25% every three months thereafter, ending January 1, 2021; |
| (22) | The options vested over a period of three years from December 29, 2014, 33.3% on each anniversary of such date, ending December 29, 2017; |
| (23) | The options vest over a period of four years from December 29, 2014, 25% on the first anniversary of such date and 6.25% every three months thereafter, and ending December 29, 2018; |
| (24) | the RSUs vest in equal installments every three months over the vesting period beginning October 15, 2017 and ending January 15, 2019; |
| (25) | The options vest over a period of four years from March 1, 2016, 25% on the first anniversary of such date and 6.25% every three months thereafter, ending March 1, 2020; |
| (26) | The options vested over a period of three years from December 29, 2014, 33.3% on each anniversary of such date, ending December 29, 2017; |
| (27) | The options vest over a period of four years from December 29, 2014, 25% on the first anniversary of such date and 6.25% every three months thereafter, ending December 29, 2018; |
| (28) | The RSUs vest in equal installments every three months over the vesting period beginning January 15, 2018 and ending January 15, 2019; |
| (29) | The RSUs vest on November 15, 2018; |
| (30) | The options vest over a period of four years from August 9, 2016, 25% on the first anniversary of such date and 6.25% every three months thereafter, and ending August 9, 2020; |
| (31) | The options and RSUs vest over a period of four years from January 1, 2017, 25% on the first anniversary of such date and 6.25% every three months thereafter, ending January 1, 2021; |
Employment Arrangements with Our Named Executive Officers
David Domzalski. The terms of Mr. Domzalski’s employment are governed by his employment agreement, our compensation policy, and the Companies Law. Under his employment agreement, effective as of July 1, 2017, Mr. Domzalski’sDomzalski’s annual base salary is currently $440,000 and hewas increased to $560,000 in January 2019 by the board, subject to shareholder approval at our 2019 annual shareholders meeting. Pursuant to our compensation policy, Mr. Domzalski is also eligible to receive an annual target bonus of up to 60% of his annual base salary. His eligibility for such annual target bonus and the amount of such bonus will be subject to the achievement of personal and company performance criteria, as determined by the company,compensation committee and board of directors, and further subject to the terms of the company’sour compensation policy then in effect, as approved by the company’sour shareholders. For the year 2017,2018, Mr. Domzalski’sDomzalski’s eligibility to thereceive his target bonus iswas subject to the following criteria:terms and key performance criteria, which were approved by the shareholders in the 2018 annual general meeting: (a) 30%40% of the bonus is based on the company’s achievement of its annual goals, operating plan, long-range planclinical trial results for FMX101 and financialFMX103 and investor relation measures,regulatory filing for FMX101, (b) 50%10% of the bonus is based on performance measures directly related to Mr. Domzalski’s individual responsibilities, according topipeline objectives, (c) 10% of the following breakdown: (i) clinical trial results – 15%, (ii) meeting clinical trials timelines – 15%, (iii)bonus is based on organizational objectives for commercial development, – 10%; (iv)(d) 20% of the bonus is based on financial key performance indicators, – 10%, and (c)(e) 20% of the bonus is based on an evaluation of Mr. Domzalski’sDomzalski’s overall performance by the compensation committee and the board of directors, based on quantitative and qualitative criteria such as establishing and implementing the company’scompany’s strategy, leadership, entrepreneurship and team collaboration. Furthermore, Mr. Domzalski iswas also eligible to receive an additional special annual bonus of up to 60% of his annual base salary in the event of exceptional performance, as determined by our compensation committee and board of directors. The amount and payment of both the regular bonusMr. Domzalski’s target and the special bonus shall bewas set by the board of directors in its sole and absolute discretion. Following his appointment as CEO,our Chief Executive Officer, Mr. Domzalski was further granted 327,720 options and 81,930 RSUsrestricted share unit awards under the terms of the 2015 Israeli Share Incentive Plan, or the 2015 Plan, and its 2015 US Addendum, and following our 2018 annual general meeting, Mr. Domzalski was granted an additional amount of 70,187 options and 23,396 restricted share units under the terms of the 2015 Plan and its 2015 US Addendum. UponIn the event of termination of his employment without cause, following a 60 days’days’ advance notice (subject to the company’sour right, at itsour election, to reduce such notice period and pay for the remainder of the period in lieu of notice), Mr. Domzalski will receive any earned but unpaid base salary, any incurred but unreimbursed business expenses and any accrued but unused vacation and sick days as of the date of termination of his employment. Additionally, the company shallwe have agreed to (1) continue to pay him his base salary for 12 months following termination (the “Mr. Domzalski’s(“Mr. Domzalski’s severance period”period”); (2) continue to make employer contributions towards our healthcare plan on his behalf for the duration of the Mr. Domzalski’sDomzalski’s severance period, and (3) cause unvested options and restricted share units (RSUs)unit awards held by Mr. Domzalski to become fully vested and exercisable, with the options remaining exercisable for 90 days following the date of termination. Upon termination of his employment without cause in connection with certain change of control events (i.e., merger, acquisition, reorganization, or sale of substantially all assets of the company), Mr. Domzalski’sDomzalski’s severance period shall be extended to 18 months and, in lieu of his entitlement to the regular annual bonus, he shall receive a lump-sum amount equal to 60% of his continued base salary, multiplied by 1.5,, to be paid within 60 days following termination. Ilan Hadar. The terms of Mr. Hadar’s employment are governed by his employment agreement and our shareholder-approved compensation policy. Under his employment agreement, effective as of July 1, 2017, Mr. Hadar’sHadar’s annual base salary is currently $380,733,was $352,188, based on the US$/NIS representative exchange rate of the Bank of Israel as of December 31, 2017,2018, and he is eligible to receive an annual target bonus of up to 50% of his annual base salary. On January 1, 2019, Mr. Hadar’s annual base salary was increased to $385,000. His eligibility for suchhis annual target bonus and the amount of such annual target bonus will be subject to his personal achievements and performance criteria, as determined by the companyboard and subject to the terms of the company’sour compensation policy, as approved by the company’sour shareholders. Furthermore, pursuant to our compensation policy, Mr. Hadar is eligible to receive an additional special annual bonus of up to 50%100% of his annual base salary in the event of exceptional performance, subject to the sole discretion of our compensation committee and board of directors. The amount and payment of both the regular and specialMr. Hadar’s cash bonuses shall be determined by our board of directors at its sole and absolute discretion. Upon termination of his employment without cause and subject to the approval of the board of directors, Mr. Hadar will continue to receive his base salary and welfare benefits for a period of 6 months following the termination date (the “Mr. Hadar’s(“Mr. Hadar’s severance period”period”). Furthermore, the board of directors may (but is not obliged to) cause unvested options held by Mr. Hadar to become fully vested and exercisable, with such options remaining exercisable for 90 days following the date of termination. Upon termination of his employment without cause in connection with certain change of control events (i.e., merger, acquisition, reorganization, or sale of substantially all assets of the parent company), Mr. Hadar’sHadar’s severance period will be extended to 1218 months and during such period he shall remain entitled to his regular annual bonus.
Yohan HazotMutya Harsch. Under his The terms of Ms. Harsch’s employment agreement, effectiveare governed by her employment offer letter and our compensation policy. Effective as of JanuaryNovember 1, 2017, Mr. Hazot’sMs. Harsch’s employment offer letter offered an annual base salary is currently $226,131, based on the US$/NIS representative exchange rate of the Bank$325,000, with an annual target bonus of Israel as of December 31, 2017, and he was eligible by the board in February 22, 2017 to a one-time annual bonus equalup to 40% of hisher annual base salary. UponHer eligibility for such annual target bonus, and the amount of such annual target bonus, will be subject to her achievement of performance targets and milestone criteria, as determined by the CEO, in accordance with our current general bonus plan and in accordance with the terms of our shareholder-approved compensation policy. On January 1, 2019, in connection with her promotion to Chief Legal Officer, Ms. Harsch’s annual base salary was increased to $370,000. Furthermore, under our compensation policy, chief officers are eligible to receive up to a 45% target bonus. In addition, under the compensation policy, the compensation committee and the board may elect to pay Ms. Harsch up to an additional annual cash bonus in the event of exceptional performance, provided that her total annual bonus shall not exceed 67.5% of her annual base salary. Pursuant to her offer letter, Ms. Harsch received 50,000 employee options and 25,000 restricted share units under our 2015 Israeli Share Incentive Plan. Pursuant to her offer letter, upon termination any vestedof her employment without cause and subject to the approval of the board of directors, Ms. Harsch will continue to receive her base salary and benefits for a period of three months plus one month for every year she has served as an employee of the company following the termination date (“Ms. Harsch’s severance period”). Furthermore, pursuant to Ms. Harsch’s option award agreements and the compensation policy,the board of directors may (but is not obliged to) cause unvested options held by Mr. Hazot shall be treated in accordanceMs. Harsch to become fully vested and exercisable, with the company’s 2015 Plan, depending on the circumstances of the termination.
Dov Tamarkin. Under his termination agreement, effective as of July 1, 2017, Dr. Tamarkin received the salary and benefit continuation (as he was entitled under his employment agreement dated August 22, 2014)such options remaining exercisable for a period of 6 months following his termination, which coincided with his contractual notice period. Following his termination, the company also paid Dr. Tamarkin an amount equal to 12 months’ salary and permitted him to retain his company cellphone, laptop and similar equipment for a period of 18 months90 days following the date of his termination. TheUpon termination agreement further provided thatof her employment without cause in connection with certain change of control events (i.e., merger, acquisition, reorganization, or sale of substantially all equity-based compensation previously granted to Dr. Tamarkin underassets of the 2015 Plan and the company’s 2009 Plan will continue to vest, without being conditioned on continued employment or service. Additionally, and subject to shareholder approval, Dr. Tamarkinparent company), Ms. Harsch’s severance period will be granted 137,428 options and 45,750 RSUs under the 2015 Plan, which shall vest over four years, 25% on the first anniversary of the grant date and the remainder in equal installments on a quarterly basis over the following three years, in consideration for his performance during the year 2016 and without being further conditioned on continued employment or service. Following his termination, Dr. Tamarkin became the company’s chief scientific advisor, which further entitled him, until his resignation from such role on January 1, 2018, to certain additional benefits such as secretarial services and reimbursement of his travel, insurance, registration and participation fees and other reasonable expenses with regard to his participation in up to six conferences per year, within a certain budget approved by the chairman of the board. Except for such benefits and expense reimbursements, Dr. Tamarkin was not entitled to any additional compensation for his role as chief scientific advisor.
Meir Eini. Under his termination agreement, effective as of July 1, 2017, Mr. Eini continued to receive his salary and benefit (as he was entitled under his employment agreement dated August 22, 2014) for a period of 6 months following his termination, which coincided with his contractual notice period. Following his termination, Mr. Eini continued to serve as an observer on our board of directors until his resignation on January 28, 2018. During such period, he was entitled to board participation and standard board-member compensation (please see below for the description and quantification of such compensation). Following the termination of his employment, the company paid Mr. Eini a lump-sum cash payment equalextended to 12 months’ of his base salary and allowed Mr. Eini to retain his company cellphone, laptop and similar equipment for a period of 18 months following the termination date. The termination agreement further provided that for 18 months following the date of termination of his employment, Mr. Eini shall provide innovation advisory services to the company, and that during the period in which he provides such services, the equity-based compensation previously granted to him shall continue to vest, but he shall not be entitled to receive any additional compensation.months.
General Policies regardingRegarding Cash Bonuses, Equity Awards and Other Benefits to Named Executive Officers As approved at our 2015 annual general meeting of shareholders, as amended at our 2018 annual general meeting of shareholders, and as required by the Israeli Companies Law, we have adopted a compensation policy regarding the terms of office and employment of our “office holders”“office holders” (as defined under the Israeli Companies Law, which includes directors, the CEO,our Chief Executive Officer, our other executive officers and any other managers directly subordinate to the CEO)Chief Executive officer), including cash compensation, equity-based awards, releases from liability, indemnification and insurance, severance and other benefits. Each of our named executive officers is (or was, while employed by us) an “office holder”“office holder” within the meaning of the Israeli Companies Law. The compensation policy is reviewed from time to time by our compensation committee and board of directors to ensure its appropriateness, and is required to be brought at least once every three years to our shareholders for reassessment and approval. According to the compensation policy, as amended at our 2018 annual general meeting of shareholders, or the Amended Compensation Policy, our short-to-medium term incentive scheme is based on a monetary bonus paid annually or at the end of such longer periods for which targets may be set as part of a multi-year plan, and is designed to reward officers based on theour performance of the company and on their individually-defined results. During the last calendar quarter of each calendar year, the compensation committee and the board of directors determine for each officer the maximum bonus amount and the objectives for receiving such bonus, as well as the formula for calculating the bonus payment upon achievement of such objectives (including minimum thresholds below which no part of the bonus will be payable), which are examined at the end offor the following calendar year or the relevant target period.
The maximumtarget bonus payable upon achievement or overachievement of theperformance objectives is 65%60% of the base salary for the CEO,Chief Executive Officer, 50% of the base salary for the chief innovation officer,Country Manager and Chief Financial Officer, 45% of the base salary for senior vice presidents, executive vice president chief financial officer, chief technology officerand other chief officers, and 40% for our other vice presidents. The objectives for receiving the bonus are intended to be measurable and quantified and may include (but are not limited to) innovation objectives such as introducing new products, entering clinical trials and developing future pipeline products; operating plan targets such as manage corporate operations to the approved annual budget and meet human resources objective; financials objectives such as revenue, EBITDA, cash balance, net profit, market cap and share price; business development objectives such as engaging with new partners or licensees, receiving product marketing approvals or approval of reimbursement schemes, and intellectual property objectives such as submission or grant of new patents.
The compensation policy Amended Compensation Policy allows for (a) a discretionary bonus of up to 20% of the officer’s regular performancesuch officer’s annual cash bonus, based on the evaluation of such officer’sofficer’s performance by the CEO, while the CEO’s performance shall be evaluated by the compensation committee and the board of directors;directors, and (b) aan additional cash bonus which may be granted to an officer in extraordinary circumstances based on special bonus in recognition of an officer’s (including the CEO’s) exceptional contribution to key transactions or events such as mergers, acquisitions, public offerings, major research and development breakthroughs or regulatory approvals, as determined by the compensation committee andCompany, or the boardachievement of directors.major corporate operational goals. The maximum specialadditional cash bonus is 60% of the base salary for the CEO,Chief Executive Officer, 50% of the base salary for the chief innovation officer, 40%Country Manager and Chief Financial Officer, 22.5% of the base salary for senior vice presidents, executive vice president, chief financial officer, chief technology officerand other chief officers, and 30%10% for other vice presidents. Our compensation policyAmended Compensation Policy also includes an equity incentive component designed to retain officers, align officers and shareholders’shareholders’ interests and incentivize achievement of medium-to-long term goals, under which the company may grant officers share options, restricted sharesshare units or any other equity-based compensation (collectively referred to as “equity awards”“equity-based awards”). The equityequity-based awards are determined individually and awarded from time to time according to each officer’sofficer’s performance, skills, qualifications, experience, roles and personal responsibilities. However, the policy caps the annual value of the equityequity-based awards to be granted to each officer at 0.5% of the company’scompany’s issued and outstanding share capital on a fully-diluted basis, as at the grant date, per each year of the vesting period, on a linear basis.period. The equityequity-based awards usually vest over a period of 3 to 4 years, in equal installments,, beginning from the first year anniversary of the grant, and expire after 10 years from the grant date. The exercise price of equityequity-based awards shall be determined in accordance with local tax laws in the territory in which the employee is employed. In Israel, for example, it is the average closing price of our shares during the 30-day30-calendar days period preceding the grant date, while in the U.S.United States, it is the last knownaverage of the closing share price atprices on the thirty (30) business days before the grant date. In special cases, to be setdetermined by the compensation committee and board of directors (and, where required by Israeli law, – by our shareholders), such as major transactions or events, or the Companyachievement of major corporate operational goals, we may grant to itsour officers special equity bonuses.equity-based awards.
Under the compensation policy,Amended Compensation Policy, our executive officers are further entitled to certain fringe benefits that we believe are commonly provided to similarly situated executives in the market in which we compete for talent and therefore are important to our ability to attract and retain top-level executive management. For Israeli officers these benefits include disbursements to a pension fund, provident fund or managers’ insurance policy in accordance with applicable law; maintenance of disability insurance on behalf of the officer, disbursement to an education fund of up to 7.5% of the officer’s monthly salary; convalescence pay according to applicable law;This includes up to 30 days of annual vacation per annum;annum, paid sick leave, in accordance with law, as well as additional benefits such as, but not limited to, a company car and cell phone (including gross-up of related tax), complementarycompany-provided health insurance and meals. Non-Israeli For officers residing in Israel, these benefits may also include contributions to a pension fund, provident fund or managersofficers’ may receive similar, comparableinsurance policy in accordance with Israeli law, maintenance of disability insurance on behalf of the officer, contributions to an education fund of up to 7.5% of the officer’s monthly salary and convalescence pay as required under applicable law. While some of these contributions and benefits are not mandatory under Israeli law, or are provided by us above and beyond the minimum statutory requirement, the nature and amount of the benefits provided to our Israeli officers are customary benefits as applicableand prevalent in the jurisdiction in which they are employed. The amounts paid to our namedIsraeli high-tech and bio-pharmaceutical market, especially among executive officers in 2016 and 2017 in respect of these benefits are reflected above in the “Summary Compensation Table” section under the “All Other Compensation” heading.officers.
Pursuant to the Israeli Companies Law, our arrangements with our officers must generally be consistent with the compensation policy,Amended Compensation Policy, as described above. However, under certain circumstances, we may approve an arrangement that is not consistent with the compensation policy,Amended Compensation Policy, if the arrangement is approved by a majority of our shareholders, provided that (a) the majority includes a majority of the votes cast by shareholders who are present and voting (disregarding (disregarding abstentions) who (i) are not controlling shareholders and (ii) do not have a personal interest in the matter, or (b) the votes cast against the arrangement by shareholders who are not controlling shareholders and who do not have a personal interest in the matter who were present and voted constitute 2% or less of the voting power of the Company (a “special majority”“special majority”). In addition, pursuant to the Israeli Companies Law, the terms of employment of directors further require the approval of the shareholders by a simple majority, and the terms of employment with respect to our CEOChief Executive Officer require the approval of the shareholders by the special majority referenced above. Pursuant to regulations promulgated under the Israeli Companies Law, shareholder approval is not required with respect to terms of employment granted to a director or the CEOChief Executive Officer for the period following his or her appointment until the next annual general meeting of shareholders, provided these terms are (a) approved by the compensation committee and the board of directors, (b) consistent with the compensation policyAmended Compensation Policy and (c) on similar or less favorable terms than those of the person’sperson’s predecessor. In addition, under certain circumstances, shareholder approval is not required with respect to the terms of employment of a candidate for CEOChief Executive Officer if the compensation committee determines that the engagement will be frustrated if the approval is pursued, provided that the terms are consistent with the compensation policy.Amended Compensation Policy.
Under certain circumstances, if the terms of employment of the CEOChief Executive Officer are not approved by the shareholders, where such approval is required, the compensation committee and the board of directors may nonetheless approve such terms. In addition, non-material amendments of the terms of employment of officers who are not directors may be approved by the compensation committee only, and non-material amendments of the terms of employment of officers who are not directors may be approved by the CEO only,alone, provided such amendments are consistent with the compensation policy.Amended Compensation Policy.
Director Compensation
Our board has adopted a compensation policy for non-employee directors, which was most recently amended by the Board, and approved by shareholders as part of our overall compensation policy in May 2018, upon recommendation by the compensation committee. Non-employee directors receive a combination of cash and equity compensation. Cash Compensation
Under the current compensation policy, each non-executive directors, comprising all our directorsdirector, other than Messrs. Stanley Hirsch and Dov Tamarkin,our chairman who receives separate compensation arrangements as described further below, received the following cash compensation for the year ended December 31, 2017:2018:
| · | a fixed annual paymentretainer of $33,000; and |
| · | a per-meeting payment of $1,000 for each board or committee meeting attended in person by the director, a $600 per-meeting payment for meetings held by teleconference or other means of communication, and $500 per each written resolution;resolution. |
| · | reimbursement of business expenses and travel and accommodation expenses incurred in the performance of duties as a member of the board in accordance with the company’s travel and expense policy; and |
Directors also receive reimbursement of business expenses and travel and accommodation expenses incurred in the performance of duties as a member of the board in accordance with our travel and expense policy.
Equity Compensation
In addition to cash compensation, each non-employee director, other than our chairman who receives separate compensation arrangements as described further below, is eligible to receive options and/or restricted share unit awards under our 2015 Plan. In 2018, each of our non-executive directors other than Dr. Hirsch received an equity grant with a fair market value on the date of grant equal to $75,000 in either options or restricted share units, at the director’s election.
As required by the 2015 Plan, the options awarded to our non-executive directors, including Dr. Hirsch, were granted at an exercise price equal to the average market price of our ordinary shares during the 30 days preceding the grant date. Options granted were subject to a 12-month vesting period, with the options vesting ratably in equal quarterly installments. Restricted share units granted were subject to a three-year vesting period vesting ratably in equal yearly installments. The vesting of each tranche of restricted share units or options was conditioned upon the respective director still serving as a director or an executive officer at that time. | · | a grant of a combination of options and RSUs having an aggregate grant date value of $75,000, granted under the 2015 Plan, as defined below. As required by the 2015 Plan, the options were granted at an exercise price equal to the average market price of our ordinary shares during the 30 days preceding the grant date, and both options and RSUs were subjected to a 4-year vesting period with 25% of the options and RSUs vesting upon the first anniversary of the grant date and 6.25% vesting every 3 months thereafter, in each case provided that the respective director is still serving as a director of the company at such time. |
Chairman Compensation Arrangements
Mr.In 2018, our chairman, Dr. Stanley Hirsch, was paid an annual compensation of $150,000 for his services as the chairman of the board, with retroactive effect from May 1, 2016, and was further awarded (i) 50,000 options underwith a fair market value on the 2015 Plan, vesting in the same manner as regular option grantsdate of grant equal to directors described above, and (ii) $37,500 worth of RSUs$150,000 (equal to 6,466 RSUs) and $112,500 worth of additional options (equal to 32,15349,766 options), in each case subject to allthe terms of the 2015 Plan.
Dr. Dov Tamarkin received compensation in his capacity as CEO until June 30, 2017, and thereafter as a board member and a consultant to the company in accordance with the terms of his termination agreement, as described above under “Item 11—Executive Compensation—Potential Payments to Named Executive Officers upon Termination or Change of Control”, and otherwise did not receive additional remuneration for his service as a director.
Mr. David Domzalski, who was appointed as a director effective January 1, 2018, instead of Mr. Tamarkin, does not receive remuneration for his service in that capacity beyond his regular compensation as the company’s CEO.
Messrs. Chaim Chizik and Meir Eini, who served as observers to the board of director throughout the year 2017 and until their resignation on January 24, 2018 and January 28, 2018, respectively, were entitled to the same compensation as our non-executive directors. The compensation paid to Mr. Eini for his service as an observer was in addition to the payments to which he was entitled under his termination agreement, as described above.
Director Compensation Table
The following table showssets forth information regarding the compensation paid toearned for service on our board by our non-executive directors and observes for their board of directors service during the fiscal year ended December 31, 2017:2018. Mr. Domzalski serves as our Chief Executive Officer in addition to being a director but does not receive any additional compensation for his service as a director and accordingly, he is not included in the table.
| Name | | Fees Earned or Paid in Cash ($) | | | Share Awards ($)(4) | | | Option Awards ($)(4) | | | Total ($) | | | Fees Earned or Paid in Cash ($) | | | Share Awards ($)(4)(5) | | | Option Awards ($)(4)(5) | | | Total ($) | | | Stanley Hirsch | | | 150,000 | | | | 37,503 | | | | 287,448 | | | | 474,951 | | | | 150,000 | | | | - | | | | 150,000 | | | | 300,000 | | | Rex Bright | | | 44,700 | | | | - | | | | 75,002 | | | | 119,702 | | | | 46,400 | | | | - | | | | 75,000 | | | | 121,400 | | | Darrell Rigel | | | 44,700 | | | | - | | | | 75,002 | | | | 119,702 | | | Anthony Bruno(1) | | | | 5,725 | | | | - | | | | - | | | | 5,725 | | | Anna Kazanchyan | | | | 42,600 | | | | 75,000 | | | | - | | | | 117,600 | | Dalia Megiddo(2) | | | | 40,600 | | | | - | | | | 75,000 | | | | 115,600 | | Darrell Rigel(3) | | | | 40,200 | | | | - | | | | 75,000 | | | | 115,200 | | | Aharon Schwartz | | | | 40,000 | | | | - | | | | 75,000 | | | | 115,000 | | | Stanley Stern | | | 44,700 | | | | - | | | | 75,002 | | | | 119,702 | | | | 45,800 | | | | - | | | | 75,000 | | | | 120,800 | | | Anna Kazanchyan | | | 41,500 | | | | 75,000 | | | | - | | | | 116,500 | | | | Aharon Schwartz | | | 39,100 | | | | - | | | | 75,000 | | | | 114,100 | | | | Dalia Megiddo | | | 39,700 | | | | - | | | | 75,000 | | | | 114,700 | | | Dov Tamarkin(1) | | | 18,100 | | | | - | | | | - | | | | 18,100 | | | Meir Eini(2) | | | 16,500 | | | | - | | | | - | | | | 16,500 | | | Chaim Chizic(3) | | | 34,000 | | | | - | | | | - | | | | 34,000 | | |
_______________________
(1) Anthony Bruno was appointed as a member of our board of directors effective November 14, 2018.
(2) On February 28, 2019, Dr. Megiddo submitted her resignation from the Board, effective on the date of our 2019 annual general meeting of shareholders.
82 | (1) | Dov Tamarkin served as a director throughout the year 2017 and resigned from the board on January 1, 2018, at which time he was replaced by Dov Domzalski, the incumbent CEO. The amounts listed in this table refer only to Dr. Tamarkin’s compensation as a director. Compensation paid to Dr. Tamarkin as an executive officer is included in the executive compensation table above. |
| (2) | Meir Eini served as an observer throughout the year 2017 and until his resignation on January 28,(3) Darrell Rigel served as a director until his resignation on November 14, 2018. The amounts listed in this table refer only to Mr. Eini’s compensation as an observer. Compensation paid to Mr. Eini as an executive officer or advisor is included in the executive compensation table above. |
| (3) | Chaim Chizic served as an observer throughout the year 2017 and until his resignation on January 24, 2018. |
(4) The value in this column represents the aggregate grant date fair value of our option awards computed in accordance with FASB ASC Topic 718. For a discussion of valuation assumptions used in the calculation of these amounts, see Note 9 to our audited financial statements.(5) The following table below shows the aggregate number of restricted share unit awards and option awards outstanding for each of our non-executive directors as of December 31, 2018: | (4) | The value in this column represents the aggregate grant date fair value of our stock option awards computed in accordance with FASB ASC Topic 718. For a discussion of valuation assumptions used in the calculation of these amounts, see Note 9 to our audited financial statements, included in Item 8—Financial Statements and Supplemental Financial Information.” |
Name | | Share Awards (#) | | | Option Awards (#) | | Stanley Hirsch | | | 4,042 | | | | 158,919 | | Rex Bright | | | - | | | | 73,509 | | Anthony Bruno | | | - | | | | - | | Anna Kazanchyan | | | 22,911 | | | | 11,000 | | Dalia Megiddo | | | - | | | | 70,318 | | Darrell Rigel | | | - | | | | 73,509 | | Aharon Schwartz | | | - | | | | 70,318 | | Stanley Stern | | | - | | | | 73,509 | |
2009 Israeli Share Option Plan
In July 2009, we adopted our 2009 Israeli Share Option Plan, or the 2009 Plan. The 2009 Plan provides for the grant of options to our and our subsidiaries’subsidiaries’ directors, employees, officers, consultants and service providers.
The 2009 Plan is administered by our board of directors or a committee designated by our board of directors, which determines, subject to Israeli law, the grantees of options, the terms of the options, including exercise or purchase prices, vesting schedules, acceleration of vesting, the type of option and the other matters necessary or desirable for, or incidental to the administration of the 2009 Plan. The 2009 Plan provides for the issuance of options under various tax regimes including, without limitation, pursuant to Sections 102 and 3(i) of the Israeli Income Tax Ordinance (New Version) 1961, or the Ordinance. Any options that expire or are canceled for any reason prior to their exercise or relinquishment in full, may once again be granted. If our outstanding shares are changed or exchanged at any time by declaration of a share dividend, share split, combination or exchange of shares, recapitalization or any similar event, then the number, class and kind of shares subject to the 2009 Plan, the options granted under the 2009 Plan and the applicable exercise prices will be proportionally adjusted.
Section 102 of the Ordinance allows employees, directors and officers, who are not controlling shareholders and who are Israeli residents, to receive favorable tax treatment for compensation in the form of shares or options. Section 102 of the Ordinance includes two alternatives for tax treatment involving the issuance of options or shares to a trustee for the benefit of the grantees and also includes an additional alternative for the issuance of options or shares directly to the grantee.
Section 102(b)(2) of the Ordinance, which provides the most favorable tax treatment for grantees, permits the issuance to a trustee under the “capital“capital gains track.” In order to comply with the terms of the capital gains track, all options granted under a specific plan and subject to the provisions of Section 102 of the Ordinance, as well as the shares issued upon exercise of such options and other shares received following any realization of rights with respect to such options, such as share dividends and share splits, must be registered in the name of a trustee selected by the board of directors and held in trust for the benefit of the relevant employee, director or officer. Such tax benefits are granted subject to the trustee not releasing these options or shares to the relevant grantee before the second anniversary of the issuance and deposit of the options with the trustee. However, under this track, we are not allowed to deduct an expense with respect to the issuance of the options or shares.
The 2009 Plan provides that options granted to our employees, directors and officers who are not controlling shareholders and who are considered Israeli residents may qualify for special tax treatment under the “capital“capital gains track”track” provisions of Section 102(b)(2) of the Ordinance. Our Israeli non-employee service providers and controlling shareholders may only be granted options under Section 3(i) of the Ordinance, which does not provide for similar tax benefits.
Options granted under the 2009 Plan are subject to vesting and vest over a four-year period commencing on the date of grant, such that 20% of the granted options are fully vested as of the date of the grant and thereafter 5% of the granted options vest every three months. Options generally expire 10 years from their date of grant. Under the 2009 Plan, in the event of termination of employment or services for reasons of disability or death, the grantee, or in the case of death, his or her legal successor, may exercise options that have vested prior to termination within a period of 12 months after the date of termination. If a grantee’sgrantee’s employment or service is terminated for cause, as defined in the 2009 Plan, all of the grantee’sgrantee’s vested and unvested options expire or forfeited on the date of termination. If a grantee’sgrantee’s employment or service is terminated without cause, the grantee may exercise his or her vested options within 90 days after the date of termination. Any expired or forfeited options prior to their exercise are returned to the option share pool and may be reissued.re-granted.
The 2009 Plan provides that in the event of a merger or consolidation of our company, or a sale of all, or substantially all, of our assets, the unexercised options outstanding may be assumed, or substituted for an appropriate number of shares of each class of shares or other securities as were distributed to our shareholders in connection with such transaction and the exercise price will be appropriately adjusted. If not so assumed or substituted, all unvested options and all vested but unexercised options will expire upon the closing of the transaction. Our board of directors or its designated committee, as applicable, may provide in the option agreement that if the acquirer does not agree to assume or substitute the options, vesting of the options shall be accelerated so that any unvested option or any portion thereof will vest 10 days prior to the closing of the transaction. In the event that the consideration received in such transaction is not solely in the form of ordinary shares of another company, the board of directors or the designated committee, as applicable, may, with the approval of the acquirer, provide that in lieu of the assumption or substitution of the options, the options will be substituted by another type of asset or property, including cash. If the board of directors does not authorize the acceleration of any unvested options, such options shall expire upon closing of thesuch a transaction. If we are voluntarily liquidated or dissolved, option holders will have 10 days to exercise any then-vested options upon receiving notification from us of the liquidation or dissolution.
The board may amend, alter, suspend or terminate the 2009 Plan at any time. However no amendment, alteration, suspension or termination may impair the rights of any option holder under the 2009 Plan unless agreed upon in writing by us and the affected option holder.
Since the adoption of the 2015 Plan, (see below), the company stopped grantingwe no longer grant options under the 2009 Plan.
2015 Israeli Share Incentive Plan
In May 2015, we adopted our 2015 Israeli Share Incentive Plan, or the 2015 Plan. The 2015 Plan provides for the grant of options and RSUsrestricted share units to our and our subsidiaries’subsidiaries’ directors, employees, officers, consultants and service providers, among others. As of the adoption of the 2015 Plan, all new grants of options and RSUsrestricted share units are made pursuant to the 2015 Plan.
The 2015 Plan is administered by our board of directors or a committee designated by our board of directors, which determines, subject to Israeli or U.S. law (as applicable), the grantees of options or RSUsrestricted share units (collectively –“Awards”-“Awards”), the terms of the Awards including exercise prices (with regard to options), vesting schedules, acceleration of vesting, the type of Award and the other matters necessary or desirable for, or incidental to the administration of the 2015 Plan. The 2015 Plan also authorizes our board of directors or a committee designated by our board of directors to allow net‘net’ or ‘cashless’ exercise of options. The 2015 Plan provides for the issuance of Awards under various tax regimes including, without limitation, pursuant to Sections 102 and 3(i) of the Ordinance. Shares underlying any Awards that expire or are canceled for any reason may once again be granted under the 2015 Plan in a form of a new Award. If our outstanding shares are changed or exchanged at any time by declaration of a share dividend, share split, combination or exchange of shares, recapitalization or any similar event, then the number, class and kind of shares subject to the 2015 Plan, the Awards granted under the 2015 Plan and the applicable exercise prices will be proportionally adjusted.
The 2015 Plan provides that Awards granted to our employees, directors and officers who are not controlling shareholders and who are considered Israeli residents may qualify for special tax treatment under the “capital“capital gains track”track” provisions of Section 102(b)(2) of the Ordinance. Our Israeli non-employee service providers and controlling shareholders (if any) may only be granted options under Section 3(i) of the Ordinance, which does not provide for similar tax benefits.
Awards granted under the 2015 Plan are subject to vesting schedules and option Awards generally expire 10 years from their date of grant. Under the 2015 Plan, in the event of termination of employment or services for reasons of disability or death, the grantee, or in the case of death, his or her legal successor, may exercise options that have vested prior to termination within a period of 12 months after the date of termination. If a grantee’sgrantee’s employment or service is terminated for cause, as defined in the 2015 Plan, all of the grantee’sgrantee’s vested and unvested Awards expire on the date of termination. If a grantee’sgrantee’s employment or service is terminated without cause, the grantee may exercise his or her vested Awards within 90 days after the date of termination.
The 2015 Plan provides that in the event of a merger or consolidation of our company, or a sale of all, or substantially all, of our assets, the outstanding Awards may be assumed, or substituted for an appropriate number of Awards denominated in shares of each class of shares or other securities as were distributed to our shareholders in connection with such transaction and the exercise price, if any, will be appropriately adjusted. If not so assumed or substituted, all Awards, including RSUs,restricted share units, will expire upon the closing of the transaction. Our board of directors or its designated committee, as applicable, may provide in the Award agreement that if the acquirer does not agree to assume or substitute the options or RSUs,restricted share units, vesting of any or all of such options and RSUsrestricted share units shall be accelerated so that any unvested option or RSU,restricted share units, or any portion thereof, will vest 10 days prior to the closing of the transaction. In the event that such consideration received in the transaction is not solely in the form of ordinary shares of another company, the board of directors or the designated committee, as applicable, may, with the approval of the acquirer, provide that in lieu of the assumption or substitution of the Awards, including RSUs,restricted share units, the Awards will be substituted by another type of asset or property, including cash. If we are voluntarily liquidated or dissolved, Award holders will have 10 days to exercise any then-vested Awards, including vested RSUs,restricted share units, upon receiving notification from us of the liquidation or dissolution.
The board may amend, alter, suspend or terminate the 2015 Plan at any time. However, no amendment, alteration, suspension or termination may impair the rights with respect to any outstanding Awards unless agreed upon in writing by us and the affected holder of such Awards.
In November 2016 and December 2017, the board of directors approved an increase of 900,000 and 2,000,000 ordinary shares, respectively, in the share reversereserve under the 2015 Plan. As of December 31, 2017, 2,076,0882018, 1,179,346 shares remain available for grant under the 2015 Plan.
As of December 31, 2017,2018, there were a total of 473,586 RSUs444,444 restricted share unit awards and 3,756,5154,368,356 options outstanding under the 2009 and 2015 Plans, collectively. The weighted-average exercise price of outstanding options was $6.36.$6.25. Out of Awards granted, 216,780 RSUs221,919 restricted share unit awards have vested and 310,85092,125 options have been exercised.
ITEM 12 —- SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED SHAREHOLDER MATTERS
The following table summarizes, as of December 31, 2017,2018, (i) the number of shares of our ordinary shares that are issuable under our equity compensation plans upon the exercise of outstanding options, warrants and other rights, (ii) the weighted-average exercise price of such options, warrants and rights, and (iii) the number of securities remaining available for future issuance under our equity compensation plans:
| Plan Category | | Number of Shares to be Issued Upon Exercise of Outstanding Options, Warrants and Rights | | | Weighted-Average Exercise Price of Outstanding Options, Warrants and Rights | | | Number of Shares Remaining Available For Future Issuance Under Equity Compensation Plans(1) | | | Number of Shares to be Issued Upon Exercise of Outstanding Options, Warrants and Rights | | | Weighted-Average Exercise Price of Outstanding Options, Warrants and Rights | | | Number of Shares Remaining Available For Future Issuance Under Equity Compensation Plans(1) | | | Equity compensation plans approved by shareholders | | | - | | | | - | | | | - | | | | - | | | | - | | | | - | | Equity compensation plans not approved by shareholders | | | 4,230,101 | | | $ | 5.65 | (2) | | | 2,076,088 | | | | 4,812,800 | | | | 5.68 | (2) | | | 1,179,346 | | | Total | | | 4,230,101 | | | $ | 5.65 | | | | 2,076,088 | | | | 4,812,800 | | | | 5.68 | | | | 1,179,346 | |
___________________________ | (1) | excluding the shares reflected in the column titled “Number of Shares to be Issued Upon Exercise of Outstanding Options, Warrants and Rights”. |
(1) Excluding the shares reflected in the column titled “Number of Shares to be Issued Upon Exercise of Outstanding Options, Warrants and Rights”. | (2) | including restricted share units with an exercise price of $0. |
(2) Including restricted share unit awards with an exercise price of $0.
(3) For a description of the material features of our equity compensation plans not approved by shareholders, see “Executive Compensation—2009 Israeli Share Option Plan” and “—2015 Israeli Share Incentive Plan.”
Security Ownership of Certain Beneficial Owners and Management
The following table sets forth certain information with respect to the beneficial ownership of our ordinary shares as of February 1, 2018,2019, for (i) each of our named executive officers, (ii) each of our directors, (iii) all of our directors and executive officers as a group, and (iv) each person, or group of affiliated persons, known by us to beneficially own more than 5% of our ordinary shares.
According to our transfer agent, asBeneficial ownership is determined in accordance with the rules of the SEC and generally includes any shares over which a person exercises sole or shared voting or dispositive power. Ordinary shares issuable under options or warrants that are exercisable within 60 days after February 1, 2018, there were 6 record holders2019 are deemed beneficially owned and such shares are used in computing the percentage ownership of our ordinary shares, among whom only Cede & Co. is U.S. holders who beneficially ownthe person holding the options or warrants, but are not deemed outstanding for the purpose of computing the percentage ownership of any other person. The information contained in the aggregate 34,180,791following table is not necessarily indicative of our ordinary shares. None of our shareholders has different voting rights frombeneficial ownership for any other shareholders.
We are not owned or controlled, directly or indirectly, by another corporation or by any foreign government. We are not awarepurpose, and the inclusion of any arrangement that may, at a subsequent date, resultshares in a changethe table does not constitute an admission of controlbeneficial ownership of our company.those shares.
Except as indicated in footnotes to this table, we believe that the shareholders named in this table have sole voting and investment power with respect to all shares shown to be beneficially owned by them, based on information provided to us by such shareholders. Unless otherwise noted below, each beneficial owner’sowner’s address is: c/o 2 Holzman St., Weizmann Science Park, Rehovot 7670402, Israel.
76| Name of Beneficial Owner | | Number of Shares Beneficially Owned | | | Percentage of Shares Beneficially Owned | | | 5% Shareholders: | | | | | | | OrbiMed Capital, LLC(1) | | | 5,195,330 | | | | 9.6 | % | Perceptive Advisors LLC(2) | | | 4,661,824 | | | | 8.6 | % | Great Point Partners, LLC(3) | | | 4,047,561 | | | | 7.4 | % | | Directors and Executive Officers: | | | | | | | | | Stanley Hirsch(4) | | | 286,504 | | | | * | | Rex Bright(5) | | | 53,796 | | | | * | | Stanley Stern(6) | | | 53,796 | | | | * | | Anna Kazanchyan(7) | | | 15,849 | | | | * | | Aharon Schwartz(8) | | | 162,900 | | | | * | | Dalia Megiddo(9) | | | 42,700 | | | | * | | Anthony Bruno(10) | | | 57,125 | | | | * | | | Sharon Barbari | | | - | | | | - | | David Domzalski(11) | | | 497,420 | | | | * | | Ilan Hadar(12) | | | 411,517 | | | | * | | Mutya Harsch(13) | | | 18,750 | | | | * | | All Directors and Executive Officers as a Group (13 Persons)(14): | | | 1,644,459 | | | | 2.95 | % |
| Name of Beneficial Owner | | Number of Shares Beneficially Owned | | | Percentage of Shares Beneficially Owned | | 5% Shareholders (Other than Directors and Executive Officers): | | | | | | | Great Point Partners, LLC(1) | | | 4,781,708 | | | | 12.8 | % | Baker Bros. Advisors (GP) LLC(2) | | | 2,466,702 | | | | 6.6 | % | Directors and Executive Officers: | | | | | | | | | Dov Tamarkin(3) | | | 2,647,941 | | | | 7.0 | % | Meir Eini(4) | | | 2,861,510 | | | | 7.7 | % | Stanley Hirsch(5) | | | 222,172 | | | | * | | Rex Bright(6) | | | 27,000 | | | | * | | Darrell Rigel(7) | | | 27,000 | | | | * | | Stanley Stern(8) | | | 27,000 | | | | * | | Anna Kazanchyan(9) | | | 10,000 | | | | * | | Aharon Schwartz(10) | | | 16,000 | | | | * | | Dalia Megiddo(11) | | | 8,000 | | | | * | | Chaim Chizic(12) | | | 1,215,309 | | | | 3.2 | % | David Domzalski(13) | | | 222,010 | | | | * | | Ilan Hadar(14) | | | 211,232 | | | | * | | Yohan Hazot(15) | | | 104,996 | | | | * | | David Schuz(16) | | | 118,278 | | | | * | | Mitchell Shirvan(17) | | | 84,893 | | | | * | | Alvin Howard(18) | | | 76,679 | | | | * | | Russell Elliott(19) | | | 18,060 | | | | * | | Iain A. Stuart(20) | | | 10,000 | | | | * | | | Bonnie Pappacena | | | - | | | | - | | All Directors and Executive Officers as a Group (19 Persons): | | | 7,908,080 | | | | 20.2 | % |
* Less than 1%
(1) | Based on information contained in Schedule 13G filed with the SEC on February 14, 2018, jointly by Great Point Partners LLC, a limited liability company organized under the laws of the State of Delaware (“Great Point”), Dr. Jeffery R. Jay, M.D., a U.S. citizen (“Dr. Jay”), and Mr. David Kroin, a U.S. citizen (“Mr. Kroin”, and collectively with Dr. Jay and Great Point, in this footnote, the “Reporting Persons”). Pursuant to that Schedule 13G (i) Biomedical Value Fund, L.P. (“BVF”) is the record owner of 1,317,181 ordinary shares (the “BVF Shares”). Great Point is the investment manager of BVF. Each of Dr. Jay, as senior managing member of Great Point, and Mr. Kroin, as special managing member of Great Point, has voting and investment power with respect to the BVF Shares, (ii) Biomedical Offshore Value Fund, Ltd. (“BOVF”) is the record owner of 1,883,662 ordinary shares (the “BOVF Shares”). Great Point is the investment manager of BOVF, and each of Dr. Jay, as senior managing member of Great Point, and Mr. Kroin, as special managing member of Great Point, has voting and investment power with respect to the BOVF Shares, (iii) GEF-SMA, LP (“GEF-SMA”) is the record owner of 1,404,687 ordinary shares (the “GEF-SMA Shares”). Great Point is the investment manager of GEF-SMA and each of Dr. Jay, as senior managing member of Great Point, and Mr. Kroin, as special managing member of Great Point, has voting and investment power with respect to the GEF-SMA Shares, and (iv) Class D Series of GEF-PS, L.P. (“GEF-PS”) is the record owner of 176,178 ordinary shares (the “GEF-PS Shares”). Great Point is the investment manager of GEF-PS and each of Dr. Jay, as senior managing member of Great Point, and Mr. Kroin, as special managing member of Great Point, has voting and investment power with respect to the GEF-PS Shares. Notwithstanding the above, Great Point, Dr. Jay and Mr. Kroin disclaim beneficial ownership of the BVF Shares, the BOVF Shares, the GEF-SMA Shares and the GEF-PS Shares, except to the extent of their respective pecuniary interests. The business address of each of the Reporting Persons is 165 Mason Street, 3rd Floor, Greenwich, CT 06830. |
(1) Based on information contained in Schedule 13G filed with the SEC on February 13, 2019, OrbiMed Capital LLC holds the shares on behalf of other persons who have the right to receive or the power to direct the receipt of dividends from, or proceeds from the sale of, such securities. No one such other person’s interest in the securities whose ownership is reported here relates to more than five percent of the class. Orbimed Capital LLC exercises investment and voting power over the shares through a management committee comprised of Carl L. Gordon, Sven H. Borho and Jonathan T. Silverstein. The business address of each of OrbiMed Capital LLC, Sven H. Borho and Jonathan T. Silverstein is 601 Lexington Avenue, 54th Floor, New York, NY 10022.(2) Based on information contained in Schedule 13G/A filed with the SEC on February 14, 2019, Perceptive Life Sciences Master Fund, Ltd. (the “Master Fund”) directly holds 4,661,824 ordinary shares. Perceptive Advisors LLC (“Perceptive Advisors”) serves as the investment manager to the Master Fund and may be deemed to beneficially own the shares directly held by the Master Fund. Joseph Edelman is the managing member of Perceptive Advisors and may be deemed to beneficially own the securities directly held by the Master Fund. The business address of each of Master Fund, Perceptive Advisors and Mr. Edelman is 51 Astor Place, 10th Floor, New York, NY 10003. (2) | Based on information contained in Schedule 13G filed with the SEC on February 14, 2017 jointly by Baker Bros. Advisors LP, a limited partnership organized under the laws of the State of Delaware (the “Adviser”), Baker Bros Advisors (GP) LLC, a limited liability company organized under the laws of the State of Delaware (the “Adviser GP”), Felix J. Baker, a U.S. citizen and Julian C. Baker, a U.S. citizen (in this footnote, collectively with the Adviser and the Adviser GP, the “Reporting Persons”). Pursuant to that Schedule 13G, 206,624 ordinary shares are held directly by 667 L.P. and 2,260,078 ordinary shares are held by Baker Brothers Life Sciences, L.P. (collectively, the “Funds”). Pursuant to an amended and restated management agreements among the Adviser, the Funds and their respective general partners, the Funds’ respective general partners relinquished to the Adviser all discretion and authority with respect to the investment and voting power of the securities held by the Funds, and the Adviser has complete and unlimited discretion and authority with respect to the Funds' investments and voting power over investments. The business address of each of the Reporting Persons is c/o Baker Bros. Advisors LP, 667 Madison Avenue, 21st Floor, New York, NY 10065. |
(3) Based on information contained in Schedule 13G/A filed with the SEC on February 14, 2019, jointly by Great Point Partners LLC, a limited liability company organized under the laws of the State of Delaware (“Great Point”), Dr. Jeffery R. Jay, M.D. (“Dr. Jay”), and Mr. David Kroin (“Mr. Kroin”, and collectively with Dr. Jay and Great Point, in this footnote, the “Reporting Persons”). Pursuant to that Schedule 13G/A (i) Biomedical Value Fund, L.P. (“BVF”) is the record owner of 1,185,317 ordinary shares (the “BVF Shares”). Great Point is the investment manager of BVF. Each of Dr. Jay, as senior managing member of Great Point, and Mr. Kroin, as special managing member of Great Point, has voting and investment power with respect to the BVF Shares, (ii) Biomedical Offshore Value Fund, Ltd. (“BOVF”) is the record owner of 1,532,332 ordinary shares (the “BOVF Shares”). Great Point is the investment manager of BOVF, and each of Dr. Jay, as senior managing member of Great Point, and Mr. Kroin, as special managing member of Great Point, has voting and investment power with respect to the BOVF Shares, (iii) GEF-SMA, LP (“GEF-SMA”) is the record owner of 1,181,420 ordinary shares (the “GEF-SMA Shares”). Great Point is the investment manager of GEF-SMA and each of Dr. Jay, as senior managing member of Great Point, and Mr. Kroin, as special managing member of Great Point, has voting and investment power with respect to the GEF-SMA Shares, and (iv) Class D Series of GEF-PS, L.P. (“GEF-PS”) is the record owner of 148,492 ordinary shares (the “GEF-PS Shares”). Great Point is the investment manager of GEF-PS and each of Dr. Jay, as senior managing member of Great Point, and Mr. Kroin, as special managing member of Great Point, has voting and investment power with respect to the GEF-PS Shares. The business address of each of the Reporting Persons is 165 Mason Street, 3rd Floor, Greenwich, CT 06830. (3) | Tamarkin Medical Innovation Ltd., an Israeli company controlled by Dr. Dov Tamarkin, our co-founder, former Chief Executive Officer and former director. Consists of (i) 2,523,489 ordinary shares; (ii) 11,795 ordinary shares, held directly by Dr. Tamarkin (iii) 2,094 ordinary shares issuable upon exercise of outstanding warrants at a price of $5.04 per share (iv) 24,000 ordinary shares, held directly by Dr. Tamarkin, issuable upon exercise of outstanding options at a price of $5.88 per share; (v) 36,563 ordinary shares, held directly by Dr. Tamarkin, issuable upon exercise of outstanding options at a price of $6.77 per share and (vi) 50,000 ordinary shares, held directly by Dr. Tamarkin, issuable upon exercise of outstanding options at a price of $6.34 per share. The address of Tamarkin Medical Innovation Ltd. is 537 Har Hila St., Modiin-Maccabim-Reut 7179901, Israel. |
(4) Consists of (i) 8,872 ordinary shares; (ii) 30,807 ordinary shares issuable upon exercise of outstanding options at a price of $4.69 per share; (iii) 182,500 ordinary shares issuable to ZEAS Technology and Science Management Ltd., a company beneficially owned by Stanley Hirsch, upon exercise of outstanding options at a price of $0.62 per share; (iv) 37,325 ordinary shares issuable upon vesting of outstanding options at a price of $5.02 per share, and (v) 27,000 ordinary shares issuable upon vesting of outstanding options at a price of $5.88 per share. (4) | Meir Eini Holdings Ltd., an Israeli company controlled by Meir Eini, our co-founder, former Chief Innovation Officer and former observer to the board. Consists of (i) 2,717,781 ordinary shares; (ii) 17,396 ordinary shares, held directly by Mr. Eini (iii) 20,860 ordinary shares issuable upon exercise of outstanding warrants at a price of $5.04 per share (iv) 48,000 ordinary shares, held directly by Mr. Eini, issuable upon exercise of outstanding options at a price of $5.88 per share; (v) 29,250 ordinary shares, held directly by Mr. Eini, issuable upon exercise of outstanding options at a price of $6.77 per share; (vi) 13,125 ordinary shares, held directly by Mr. Eini, issuable upon exercise of outstanding options at a price of $7.09 per share, and (vii) 15,098 ordinary shares, held directly by Mr. Eini, issuable upon exercise of outstanding options at a price of $10.31 per share. The address of Meir Eini Holdings Ltd. is 2 Hashaked St., Ness-Ziona 7408711, Israel. |
(5) Consists of (i) 8,086 ordinary shares issuable upon exercise of outstanding options at a price of $4.76 per share; (ii) 18,710 ordinary shares issuable upon exercise of outstanding options at a price of $5.06 per share, and (iii) 27,000 ordinary shares issuable upon exercise of outstanding options at a price of $5.88 per share. (5) | Consists of (i) 6,448 ordinary shares; (ii) 6,224 ordinary shares issuable upon exercise of outstanding warrants at a price of $5.04 per share; (iii) 182,500 ordinary shares issuable to ZEAS Technology and Science Management Ltd., a company beneficially owned by Stanley Hirsch, upon exercise of outstanding options at a price of $0.62 per share, and (iv) 27,000 ordinary shares issuable upon vesting of outstanding options at a price of $5.88 per share. |
(6) Consists of (i) 8,086 ordinary shares issuable upon exercise of outstanding options at a price of $4.76 per share; (ii) 18,710 ordinary shares issuable upon exercise of outstanding options at a price of $5.06 per share, and (iii) 27,000 ordinary shares issuable upon exercise of outstanding options at a price of $5.88 per share.
(7) Consists of (i) 4,849 ordinary shares; (ii) 8,000 ordinary shares issuable upon exercise of outstanding options at a price of $5.88 per share, and (iii) 3,000 ordinary shares issuable upon exercise of outstanding options at a price of $10.80 per share.
(8) Consists of (i) 112,200 ordinary shares; (ii) 8,038 ordinary shares issuable upon exercise of outstanding options at a price of $4.69 per share; (iii) 18,662 ordinary shares issuable upon exercise of outstanding options at a price of $5.02 per share, and (vi) 24,000 ordinary shares issuable upon exercise of outstanding options at a price of $11.87 per share.
(9) Consists of (i) 8,038 ordinary shares issuable upon exercise of outstanding options at a price of $4.69 per share; (ii) 18,662 ordinary shares issuable upon exercise of outstanding options at a price of $5.02 per share, and (iii) 16,000 ordinary shares issuable upon exercise of outstanding options at a price of $7.09 per share.
(10) Consists of (i) 41,500 ordinary shares, and (ii) 15,625 ordinary shares issuable upon exercise of outstanding options at a price of $7.98 per share.
(11) Consists of (i) 52,522 ordinary shares; (ii) 18,750 ordinary shares issuable upon exercise of outstanding options at a price of $7.98 per share; (iii) 35,685 ordinary shares issuable upon exercise of outstanding options at a price of $10.22 per share; (iv) 45,000 ordinary shares issuable upon exercise of outstanding options at a price of $6.04 per share; (v) 192,408 ordinary shares issuable upon exercise of outstanding options at a price of $7.14 per share; (vi) 122,894 ordinary shares issuable upon exercise of outstanding options at a price of $5.76 per share; (vii) 17,547 ordinary shares issuable upon exercise of outstanding options at a price of $5.06 per share, and (viii) 12,614 ordinary shares issuable upon vesting of outstanding restricted share units.
(12) Consists of (i) 34,284 ordinary shares; (ii) 5,469 ordinary shares issuable upon exercise of outstanding options at a price of $1.92 per share; (iii) 24,121 ordinary shares issuable upon exercise of outstanding options at a price of $5.46 per share; (iv) 18,000 ordinary shares issuable upon exercise of outstanding options at a price of $6.77 per share; (v) 30,195 ordinary shares issuable upon exercise of outstanding options at a price of $10.31 per share; (vi) 45,000 ordinary shares issuable upon exercise of outstanding options at a price of $6.34 per share; (vii) 164,128 ordinary shares issuable upon exercise of outstanding options at a price of $7.13 per share; (viii) 73,577 ordinary shares issuable upon exercise of outstanding options at a price of $5.22 per share; (ix) 8,773 ordinary shares issuable upon exercise of outstanding options at a price of $6.40 per share, and (x) 7,970 ordinary shares issuable upon vesting of outstanding restricted share units.
(13) Consists of (i) 12,500 ordinary shares issuable upon exercise of outstanding options at a price of $6.35 per share, and (ii) 6,250 ordinary shares issuable upon vesting of outstanding restricted share units.
(6) | Consists of 27,000 ordinary shares issuable upon exercise of outstanding options at a price of $5.88 per share. |
(7) | Consists of 27,000 ordinary shares issuable upon exercise of outstanding options at a price of $5.88 per share. |
(8) | Consists of 27,000 ordinary shares issuable upon exercise of outstanding options at a price of $5.88 per share. |
(9) | Consists of (i) 8,000 ordinary shares issuable upon exercise of outstanding options at a price of $5.88 per share, and (ii) 2,000 ordinary shares issuable upon exercise of outstanding options at a price of $10.80 per share. |
(10) | Consists of 16,000 ordinary shares issuable upon exercise of outstanding options at a price of $11.87 per share. |
(11) | Consists of 8,000 ordinary shares issuable upon exercise of outstanding options at a price of $7.09 per share. |
(12) | Consists of (i) 558,459 ordinary shares; (ii) 266,412 ordinary shares issuable upon exercise of outstanding warrants at a price of $5.04 per share; (iii) 300,938 ordinary shares held by Rosa Alba Commerce & Investments Ltd., a company beneficially owned by Mr. Chizic; (iv) 62,500(14) Includes an aggregate of (i) 259,267 ordinary shares (ii) 1,354,172 ordinary shares issuable upon exercise of outstanding options, and (iii) 31,020 ordinary shares issuable upon vesting of outstanding options at a price of $1.92 per share, and (v) 27,000 ordinary shares issuable upon vesting of outstanding options at a price of $5.88 per share. The company’s disclosure is based on the latest information available to it, as Mr. Chizic did not respond to the company’s recent queries in the matter. To the company’s knowledge, the shares and options listed in sub-clauses (i) and (ii) above are held by Mr. Chizic and his wife in equal parts. |
(13) | Consists of (i) 26,743 ordinary shares; (ii) 14,062 ordinary shares issuable upon exercise of outstanding options at a price of $7.98 per share; (iii) 17,842 ordinary shares issuable upon exercise of outstanding options at a price of $10.22 per share; (iv) 30,000 ordinary shares issuable upon exercise of outstanding options at a price of $6.04 per share; (v) 133,206 ordinary shares issuable upon exercise of outstanding options at a price of $7.14 per share, and (vi) 157 ordinary shares issuable upon vesting of outstanding RSUs. |
(14) | Consists of (i) 12,871 ordinary shares; (ii) 5,469 ordinary shares issuable upon exercise of outstanding options at a price of $1.92 per share; (iii) 20,506 ordinary shares issuable upon exercise of outstanding options at a price of $5.46 per share; (iv) 13,500 ordinary shares issuable upon exercise of outstanding options at a price of $6.77 per share; (v) 15,097 ordinary shares issuable upon exercise of outstanding options at a price of $10.31 per share; (vi) 30,000 ordinary shares issuable upon exercise of outstanding options at a price of $6.34 per share; (vii) 113,627 ordinary shares issuable upon exercise of outstanding options at a price of $7.13 per share, and (viii) 162 ordinary shares issuable upon vesting of outstanding RSUs. |
(15) | Consists of (i) 11,321 ordinary shares (ii) 12,375 ordinary shares issuable upon exercise of outstanding options at a price of $1.92 per share; (iii) 30,000 ordinary shares issuable upon exercise of outstanding options at a price of $6.34 per share; (iv) 13,500 ordinary shares issuable upon exercise of outstanding options at a price of $6.77 per share; (v) 24,071 ordinary shares issuable upon exercise of outstanding options at a price of $7.13 per share, and (vi) 13,725 ordinary shares issuable upon exercise of outstanding options at a price of $10.31 per share, and (vi) 4 ordinary shares issuable upon vesting of outstanding RSUs. |
(16) | Consists of (i) 9,491 ordinary shares; (ii) 65,750 ordinary shares issuable upon exercise of outstanding options at a price of $1.92 per share; (iii) 15,000 ordinary shares issuable upon exercise of outstanding options at a price of $6.34 per share; (iv) 13,500 ordinary shares issuable upon exercise of outstanding options at a price of $6.77 per share; (v) 6,298 ordinary shares issuable upon exercise of outstanding options at a price of $7.13 per share, and (vi) 8,235 ordinary shares issuable upon exercise of outstanding options at a price of $10.31 per share, and (vi) 4 ordinary shares issuable upon vesting of outstanding RSUs. |
(17) | Consists of (i) 9,495 ordinary shares; (ii) 7,656 ordinary shares issuable upon exercise of outstanding options at a price of $1.92 per share; (iii) 20,506 ordinary shares issuable upon exercise of outstanding options at a price of $5.46 per share; (iv) 15,000 ordinary shares issuable upon exercise of outstanding options at a price of $6.34 per share; (v) 13,500 ordinary shares issuable upon exercise of outstanding options at a price of $6.77 per share; (vi) 10,500 ordinary shares issuable upon exercise of outstanding options at a price of $7.13 per share, and (vii) 8,235 ordinary shares issuable upon exercise of outstanding options at a price of $10.31 per share. |
(18) | Consists of (i) 19,754 ordinary shares; (ii) 15,000 ordinary shares issuable upon exercise of outstanding options at a price of $6.04 per share; (iii) 25,688 ordinary shares issuable upon exercise of outstanding options at a price of $7.14 per share; (iv) 9,375 ordinary shares issuable upon exercise of outstanding options at a price of $7.98 per share, and (v) 6,862 ordinary shares issuable upon exercise of outstanding options at a price of $10.22 per share. |
(19) | Consists of (i) 817 ordinary shares; (ii) 13,125 ordinary shares issuable upon exercise of outstanding options at a price of $6.46 per share, and (iii) 4,118 ordinary shares issuable upon exercise of outstanding options at a price of $10.22 per share. |
(20) | Consists of 10,000 ordinary shares issuable upon exercise of outstanding options at a price of $8.54 per share. |
Section 16(a) Beneficial Ownership Reporting Compliance
We were a “foreign private issuer” until January 1, 2018. Therefore, our officers and directors, and persons who owned more than 10% of our shares, were exempt from filing reports of ownership and changes in ownership with the SEC under Section 16(a) of the Exchange Act during the year ended December 31, 2017, in accordance with Rule 3a12-3 under the Exchange Act. As of January 1, 2018, our officers, directors and greater than 10% shareholders are required by SEC regulations to file forms pursuant to Section 16(a). In connection with this transition, we filed initial beneficial ownership forms on behalf of our executive officers and directors, and changes to beneficial ownership forms on Form 4 for certain executive officers whose restricted share units vested shortly after January 1, 2018. All of our executive officers’ and directors’ Form 3s were filed 9 days late, on January 11, 2018. Form 4s were filed late on January 24, 2018, for the following executive officers: Mr. David Domzalski, Mr. Mitchell Shirvan, Mr. Russell Elliott, Mr. Alvin Howard, Mr. Yohan Hazot, and Mr. David Schuz. In addition, Mr. Meir Eini, who was an observer to our board of directors, filed a late Form 3 and late Form 4, each dated January 18, 2018; Mr. Chaim Chizic, another observer to our board of directors, did not file a Form 3 or Form 4. Such non-compliance may result in the imposition of monetary penalties by the SEC on such delinquent insiders, depending on the degree of untimeliness and severity and recurrence of their infractions.units.
ITEM 13 —- CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
Transactions with Related Persons
OnExcept as described below, there have been no transactions since January 1, 2017 in which we have been a participant in which the amount involved exceeded or will exceed the lesser of $120,000 and one percent of the average of our total assets at year end for the last two completed fiscal years, and in which any of our directors, executive officers or holders of more than 5% of our common stock, or any members of their immediate family, had or will have a direct or indirect material interest, other than compensation arrangements which are described under “Executive Compensation” and “Director Compensation.”
Separation Agreement with Dr. Dov Tamarkin
In June 2017, we entered into a separation agreement with Dr. Dov Tamarkin following his termination of service as our Chief Executive Officer. As part of the separation agreement, as of July 1, 2017, Dr. Tamarkin agreed to serve as a consultant to us. In January 2018, we reached an agreement with Dr. Tamarkin pursuant to which he discontinued his services as a consultant to us. In May 2018, following the termination of services of Dr. Tamarkin as a consultant, the separation agreement was amended. Pursuant to the amendment, the 137,428 options and 45,750 restricted share units to which Dr. Tamarkin was entitled to for his services as CEO under the separation agreement were converted into a special, one-time payment of $192,500 in cash. As required under the Israeli Companies Law, this equity conversion was approved by our shareholders at our annual general meeting in 2018.
Consulting Agreement with Anthony Bruno
In August 2018, we terminated our consulting agreement dated as of April 19, 2014, as amended in May 26, 2015, with Mr. Anthony Bruno, who joined our board appointedin November 2018. Pursuant to the consulting agreement, Mr. Bruno provided strategic consulting services to the Company. Upon termination of the consulting agreement, Mr. Bruno’s outstanding, unvested options became immediately exercisable pursuant to the terms of the consulting agreement, which provided for acceleration of the options upon termination. On February 26, 2019, Mr. Bruno’s options expired in accordance with their terms.
Arrangements with Departing Directors
In November 2018, in connection with the retirement of Dr. Darrell Rigel from our CEO, David Domzalski,board of directors and in consideration for his service as a director, we approved: (1) a retirement payment of $50,000 (or approximately $1,000 for every month of service as a director), (2) a waiver of the service requirements for Dr. Rigel’s outstanding but unvested option awards, and (3) the payment of director compensation to fillDr. Rigel for the vacancy created byfull year 2018, even though he retired one month short of the resignationcompletion of Dov Tamarkin and2018, in each case, subject to furtherthe receipt of shareholder approval as required under the Israeli Companies Law.
In February 2019, in connection with the retirement of Dr. Dalia Megiddo from our board of directors, which will become effective upon our 2019 annual shareholders’ meeting, and in consideration for her service as a director, we approved: (1) a retirement payment of $36,000 (or approximately $1,000 for every month of service as a director), and (2) a waiver of the service requirements for Dr. Megiddo’s outstanding but unvested option awards, in each case, subject to shareholder approval as required under the Israeli Companies Law.
Indemnification Agreements with Directors
Our articles of association provide that we may indemnify each of our directors and officers to the fullest extent permitted by Israeli law. Accordingly, we have entered into standard indemnification agreements with each of our shareholdersdirectors, whereby the we have undertaken to indemnify each such director, in advance, for losses, damages, costs or expenses that such director may suffer or incur as a result of his or her actions or omissions in such capacity on behalf of the upcoming annual general meeting. The mainCompany in certain circumstances and events, subject to the terms, of employment of Mr. Domzalski, in his capacity as our CEO, areconditions and limitations set out in “Item 11—Executive Compensation—Potential Payments to Named Executive Officers upon Termination or Change of Control”.the indemnification agreement.
Review, Approval or Ratification of Transactions with Related Persons
The related party transactions described above were reviewed and approved in accordance with the provisions of the Companies Law and the Articles.
Pursuant to the Companies Law, an “office holder” of a company (as defined under the Companies Law, which includes directors, the Chief Executive Officer, other executive officers and any other managers directly subordinate to the Chief Executive Officer), is required to promptly disclose any personal interest that he or she may have, and all related material information known to such person in connection with any existing or proposed transaction of the company.
Under the Israeli Companies Law, our audit committee is responsible for, inter alia, determining whether to approve certain related party transactions, including transactions in which an office holder has a personal interest and whether such transaction is extraordinary or material under the Israeli Companies Law. See our Registration Statement on Form F-1 as filedthe description above in the “Audit Committee-Roles, responsibilities and procedure” section under the Securities Act with“Structure and Practices of the SEC on September 3, 2014, under “Management—Approval of Related Party Transactions under Israeli Law—Fiduciary DutiesBoard of Directors” heading. Notwithstanding, under the Companies Law, a related party transaction with respect to office holders’ terms of office and Executive Officers”;employment, including indemnification, exemption and insurance, does not require the approval of our audit committee, but requires the approval of our compensation committee, as further described below. We do not have a separate policy with respect to related party transactions.
Under the Israeli Companies Law, shareholder approval is required for, among other things:inter alia: (a) transactions with directors concerning the terms of their service or indemnification, exemption and insurance for their service (or for any other position that they may hold at a company), for which approvals of the compensation committee, board of directors and shareholders are all required, (b) extraordinary transactions with controlling shareholders, of publicly held companies, which require the special approval described in the Registration Statement on Form F-1 as filed under the Securities Act with the SEC on September 3, 2014, under “Management—Approval of Related Party Transactions Under Israeli Law—Disclosure of Personal Interests of a Controlling Shareholder and Approval of Certain Transactions,”by Special Majority and (c) terms of employment or other engagement of the controlling shareholder of the company or such controlling shareholder’s relative, by Special Majority. Further, so long as an office holder has disclosed his or her personal interest in a transaction, the board may approve an action by the office holder that would otherwise be deemed a breach of his or her fiduciary duty. However, a company may not approve a transaction or action that is not in the company’s interest or that is not performed by the office holder in good faith. An extraordinary transaction in which an office holder has a personal interest requires approval first by a company
The compensation of, or an undertaking to indemnify or insure, an office holder who is not a director, requires approval first by a company’s compensation committee, then by a company’s board of directors. If such compensation arrangement or an undertaking to indemnify or insure is inconsistent with a company’s compensation policy then in effect, or if the office holder is the chief executive officer (apart from a number of specific exceptions), then such arrangement is further subject to approval by a Special Majority. Arrangements regarding the compensation, indemnification or insurance of a director require the special approval described Registration Statementof the compensation committee, board and shareholders by ordinary majority, in that order, and under certain circumstances, a Special Majority as aforesaid.
Generally, a person who has a personal interest in a matter which is considered at a meeting of the board or the audit committee may not be present at such a meeting or vote on Form F-1 as filed underthat matter unless the Securities Act withchairman of the SEC on September 3, 2014, under “Management—Approvalrelevant committee or board (as applicable) determines that he or she should be present in order to present the transaction that is subject to approval. If a majority of Related Party Transactions Under Israeli Law— Disclosurethe members of the audit committee or the board (as applicable) has a personal interestsinterest in the approval of a controllingtransaction, then all directors may participate in discussions of the audit committee or the board (as applicable) on such transaction and the voting on approval thereof, but shareholder and approval of transactions.” is also required for such transaction.
In addition, under the Israeli Companies Law, a merger requires approval of the shareholders of each of the merging companies.
Director Independence
Our board of directors has determined that following the resignation of Dr. Dov Tamarkin from the board of directors effective January 1, 2018 and his replacement by Dov Domzalski, the incumbent CEO, all of our directors, except for Mr. Domzalski, our Chief Executive Officer, are independent under the NASDAQ Stock MarketNasdaq rules.
ITEM 14 —- PRINCIPAL ACCOUNTANT FEES AND SERVICES
Kesselman & Kesselman (a member firm of Pricewaterhouse Coopers International Limited, or PwC), has served as our principal independent registered public accounting firm for each of the two years ended December 31, 20162017 and 2017.2018.
The following table provides information regarding fees paid by us to PwC for all services, for the years ended December 31, 20162017 and 2017:2018:
| | | Fiscal year ended | | | | | (in thousands of U.S. dollars) | | | | | 2016 | | | 2017 | | Audit Fees(1) | | | 182 | | | | 136 | | | Audit-related Fees | | | - | | | | - | | | Tax Fees | | | - | | | | - | | | All Other Fees | | | - | | | | - | | | | | | | | | | | | | Total Fees | | | 182 | | | | 136 | |
| | | Fiscal year ended December 31, | | | | | 2018 | | | 2017 | | | | | (in thousands of U.S. dollars) | | Audit fees(1) | | $ | 193 | | | $ | 136 | | | Audit-related fees | | | - | | | | - | | | Tax Fees | | | - | | | | - | | | All other fees | | | - | | | | - | | | | | | | | | | | | | Total Fees | | $ | 193 | | | $ | 136 | |
| (1) | Includes professional services rendered in connection with the audit of our annual financial statements, the review of our interim financial statements, fees for the 2016 follow-on offerings and shelf registration statements. |
(1) Includes professional services rendered in connection with the audit of our annual financial statements, the review of our interim financial statements, and fees for the 2018 follow-on offering and shelf registration statement.
Our audit committee is responsible for pre-approvingmust pre-approve audit and non-audit services provided to us by our independent registered public accounting firm. All of the non-audit services provided to us by the independent auditors in following the formation of our audit committee waswere pre-approved by the audit committee. PART IV
ITEM 15 —- EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
(a) Documents Filed as Part of This Report
1. Financial statements.
See Index to Financial Statements under Item 8 of Part II of this Annual Report, on Form 10-K, which listing is incorporated herein by reference.
2. Financial statement schedules.
No schedules are applicable or required, or the information is included in the Consolidated Financial Statementsconsolidated financial statements or Notesnotes thereto.
3. Exhibits. See Item 15(b) below. Each
(b) Exhibits
| | | Incorporation by Reference | | | Exhibit Number | Description Of Document | Form | SEC File No. | Exhibit | Filing Date | Filed Herewith | | | | | | | X | | | F-1/A | 333-198123 | 4.1 | September 3, 2014 | | | | F-1/A | 333-198123 | 10.1 | September 3, 2014 | | | | F-3 | 333-207546 | 10.2 | October 21, 2015 | | | | | | | | X | | | F-1/A | 333-198123 | 10.3 | September 3, 2014 | | | | | | | | X | | | | | | | X | | | | | | | X | | | | | | | X | | | | | | | X | | | | | | | X | | | | | | | X | | | | | | | X | | | | | | | X | | | | | | | X | | | | | | | X |
# Indicates management contract or compensatory plan or arrangement required to be filed has been identified.plan. (b) Exhibits
See the Exhibit Index immediately preceding the signature page of this Annual Report on Form 10-K.
ITEM 16 —- FORM 10-K SUMMARY
Not applicable.
INDEX TO EXHIBITS
| | | | | Incorporation by Reference | | | | Exhibit Number | | Description Of Document | | Form | | SEC File No. | | Exhibit | | Filing Date | | Filed Herewith | | | | | S-8 | | 333-222155 | | 4.1 | | December 19, 2017 | | | | | | | F-1/A | | 333-198123 | | 10.1 | | September 3, 2014 | | | | | | | F-3 | | 333-207546 | | 10.2 | | October 21, 2015 | | | | | | | F-3/A | | 333-207546 | | 10.3 | | September 12, 2016 | | | | | | | F-1/A | | 333-198123 | | 10.3 | | September 3, 2014 | | | | | | | 6-K/A | | 001-36621 | | 99.1 | | May 20, 2015 | | | | | | | | | | | | | | | X | | | | | | | | | | | | | X | | | | | | | | | | | | | X | | | | | | | | | | | | | X | | | | | | | | | | | | | X | | | | | | | | | | | | | X | | | | | | | | | | | | | X | | | | | | | | | | | | | X |
* This exhibit is a management contract or a compensatory plan or arrangement.
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, Foamix Pharmaceuticals Ltd. has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized on March 1, 2018. February 28, 2019.
| | FOAMIX PHARMACEUTICALS LTD. | | | By: | /s//s/ David Domzalski
| | | | David Domzalski Chief Executive Officer |
KNOW ALL MEN AND WOMEN BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints David Domzalski and Stanley Hirsch, and each of them, his or her attorney-in-fact and agent, each with the power of substitution, for him or her in any and all capacities, to sign any and all amendments to this Annual Report on Form 10-K, and to file the same, with exhibits thereto and other documents in connection therewith, with the U.S. Securities and Exchange Commission, hereby ratifying and confirming all that said attorneys-in-fact, or his or her or their substitute or substitutes, may do or cause to be done by virtue thereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, the Registrantregistrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
Signature and Name | Title | Date | | | | | /s/ David Domzalski | Chief Executive Officer (principal executive officer)(Principal Executive Officer) | February 28, 2019 | | David Domzalski | | | | | | | /s/ Ilan Hadar | Chief Financial Officer (principal financial accounting and officer)(Principal Financial Officer | February 28, 2019 | | Ilan Hadar | and Principal Accounting Officer) | | | | | | /s/ Stanley Hirsch | Chairman of the Board of Directors | February 28, 2019 | | Stanley Hirsch | | | | | | | /s/ Sharon Barbari | Director | February 28, 2019 | | Sharon Barbari | | | | | | /s/ Rex Bright | Director | February 28, 2019 | | Rex Bright | | | | | | /s/ Anthony Bruno | Director | February 28, 2019 | | Anthony Bruno | | | | | | /s/ Anna Kazanchyan | Director | February 28, 2019 | | Anna Kazanchyan | | | | | | /s/ Dalia Megiddo | Director | February 28, 2019 | | Dalia Megiddo | | | | | | | /s/ Rex BrightAharon Schwartz | Director | February 28, 2019 | Rex BrightAharon Schwartz | | | | | | | /s/ Darrell Rigel
| Director | | Darrell Rigel | | | | | | /s/ Stanley Stern | Director | February 28, 2019 | | Stanley Stern | | | | | | /s/ Anna Kazanchyan
| Director | | Anna Kazanchyan | | | | | | /s/ Aharon Schwartz
| Director | | Aharon Schwartz | | |
|