
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K/A
Amendment No. 1
| ANNUAL REPORT UNDER SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For fiscal year ended: December 31 2017, 2023
OR
| TRANSITION REPORT UNDER SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from _______________ to _______________
Commission file number: _______________________001-12555
| Protagenic Therapeutics, Inc. |
| (Exact name of registrant as specified in its charter) |
| Delaware | 06-1390025 | |
| (State or other jurisdiction of | (I.R.S. Employer | |
| incorporation or organization) | Identification No.) |
| 149 Fifth Avenue | ||
| New York, New York | 10010 | |
| (Address of principal executive offices) | (Zip Code) |
Registrant’s telephone number, including area code:(212)994-8200
Securities registered under Section 12(b) of the Exchange Act:
| Title of each class | Name of exchange on which registered | |
| Common Stock Purchase Warrant, PTIXW | Nasdaq Capital Market |
Securities registered under Section 12(g) of the Exchange Act:
Common Stock, $0.0001 par value
(Title of class)
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
Yes [ ] No [X]
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Exchange Act. Yes [ ] ☐ No [X] ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Exchange Act during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes [X] ☒ No [ ]☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files).Yes ☒ No ☐
Yes [X] No [ ]
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. [ ]
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated file, smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | Accelerated filer | |
| Non-accelerated filer | Smaller reporting company | |
| Emerging growth company |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. [ ]☐
Indicate by check mark whether the registrant has filed a report and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. Yes ☐ No ☒
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. Yes [ ]☐ No [X]☒
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). Yes ☐ No ☒
The aggregate market value of the voting and non-voting common equity held by non-affiliates of the registrant on June 30, 2017,2023, based on a closing price as reported on the OTCQBNasdaq Capital Market of $2.00$2.03 was approximately $15,230,814.$8,791,847.
As of March 30, 2018,29, 2024, there were 10,261,419 shares of the registrant'sregistrant’s common stock, par value $0.0001, issued and outstanding, and 872,766 shares of the registrant’s Series B Preferred Stock, issued and outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
None.None.
EXPLANATORY NOTE
This Amendment No. 1 on Form 10-K/A (Amendment No. 1) is being filed to amend our Annual Report on Form 10-K for the fiscal year ended December 31, 2017 (Original Filing), filed with the U.S. Securities and Exchange Commission on April 2, 2018 (Original Filing Date). The sole purpose of this Amendment No. 1 is to insert the dates and electronic signatures on the Signature page (page 72) and in the Certifications (Exhibit 31.2 and 32.1).
Except as described above, no material changes have been made to the Original Filing and this Amendment No. 1 does not modify, amend or update in any way any of the financial or other information contained in the Original Filing.
PROTAGENIC THERAPEUTICS, INC.
ANNUAL REPORT ON FORM 10-K
FOR THE YEAR ENDED DECEMBER 31, 20172023
TABLE OF CONTENTS
| 2 |
EXPLANATORY NOTE
Protagenic Therapeutics, Inc. (the “Company”) is filing this Amendment No. 1 (this “Amendment”) to its Annual Report on Form 10-K for the fiscal year ended December 31, 2023, as filed with the Securities and Exchange Commission (the “SEC”) on April 1, 2023 (the “Original Filing”) to correct an inadvertent error in the Original Filing. Exhibit 32.1 (Certification Pursuant To 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes Oxley Act of 2002) was inadvertently omitted from the filed version of the Original Filing. The Company is filing this Amendment solely for the purpose of including Exhibit 32.1.
No attempt has been made in this Amendment to otherwise modify or update the other disclosures presented in the Original Filing. This Amendment does not reflect events occurring after the Original Filing (i.e., those events occurring after April 1, 2024) or modify or update those disclosures that may be affected by subsequent events. Such subsequent matters are addressed in subsequent reports filed with the SEC. Accordingly, this Amendment should be read in conjunction with the Original Filing and the Company’s other filings with the SEC.
| 3 |
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This report on Form 10-K contains forward-looking statements made pursuant to the safe harbor provisions of the Private Securities Litigation Reform Act of 1995 under Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. Forward-looking statements include statements with respect to our beliefs, plans, objectives, goals, expectations, anticipations, assumptions, estimates, intentions and future performance, and involve known and unknown risks, uncertainties and other factors, which may be beyond our control, and which may cause our actual results, performance or achievements to be materially different from future results, performance or achievements expressed or implied by such forward-looking statements. All statements other than statements of historical fact are statements that could be forward-looking statements. You can identify these forward-looking statements through our use of words such as “may,” “can,” “anticipate,” “assume,” “should,” “indicate,” “would,” “believe,” “contemplate,” “expect,” “seek,” “estimate,” “continue,” “plan,” “point to,” “project,” “predict,” “could,” “intend,” “target,” “potential” and other similar words and expressions of the future. The matters discussed in these forward-looking statements are subject to risks, uncertainties and other factors that could cause our actual results to differ materially from those projected, anticipated or implied in the forward-looking statements. As a result, you should not place undue reliance on any forward-looking statements. The most significant of these risks, uncertainties and other factors are described in “Item 1A — Risk Factors” of this Annual Report on Form 10-K. Except to the limited extent required by applicable law, we undertake no obligation to update or revise any forward-looking statements, whether as a result of new information, future events or otherwise.
Risk Factors Summary
Below is a summary of material factors that make an investment in our securities speculative or risky. Importantly, this summary does not address all of the risks and uncertainties that we face. Additional discussion of the risks and uncertainties summarized in this risk factor summary, as well as other risks and uncertainties that we face, can be found under “Risk Factors” in Part I, Item 1A of this Annual Report on Form 10-K. The below summary is qualified in its entirety by that more complete discussion of such risks and uncertainties. You should consider carefully the risks and uncertainties described under “Risk Factors” in Part I, Item 1A of this Annual Report on Form 10-K as part of your evaluation of the risks associated with an investment in our securities.
Risks Related to Our Financial Condition and Capital Requirements
| ● | The Company’s financial statements have been prepared on a going concern basis, and do not include adjustments that might be necessary if the Company is unable to continue as a going concern. | |
| ● | If we continue to incur operating losses and fail to obtain the capital necessary to fund our operations, we will be unable to advance our development programs, complete our clinical trials, or bring products to market, or may be forced to reduce or cease operations entirely. In addition, any capital obtained by us may be obtained on terms that are unfavorable to us, our investors, or both. | |
| ● | Unstable market and economic conditions may have serious adverse consequences on our ability to raise funds, which may cause us to cease or delay our operations. |
Risks Related to Clinical Development and Regulatory Approval
| ● | Our results to date provide no basis for predicting whether any of our product candidates will be safe or effective, or receive regulatory approval. | |
| ● | We may not be able to initiate and complete preclinical studies and clinical trials for our product candidates which could adversely affect our business. | |
| ● | If we experience delays or difficulties in the enrollment of subjects to our clinical trials, our receipt of necessary regulatory approvals could be delayed or prevented, which could materially affect our financial condition. | |
| ● | If the market opportunities for our current and potential future drug candidates are smaller than we believe they are, our ability to generate product revenues will be adversely affected and our business may suffer. |
| 4 |
Risks Related to Our Reliance on Third Parties
| ● | We have no experience in sales, marketing and distribution and may have to enter into agreements with third parties to perform these functions, which could prevent us from successfully commercializing our product candidates. | |
| ● | Data provided by collaborators and other parties upon which we rely have not been independently verified and could turn out to be inaccurate, misleading, or incomplete. | |
| ● | We rely on third parties to conduct our non-clinical studies and our clinical trials. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may be unable to obtain regulatory approval for or commercialize our current product candidates or any future products, on a timely basis or at all, and our financial condition will be adversely affected. |
Risks Related to Commercialization of Our Product Candidates
| ● | We have no experience as a company in commercializing any product. If we fail to obtain commercial expertise, upon product approval by regulatory agencies, our product launch and revenues could be delayed. | |
| ● | We may not be able to gain market acceptance of our product candidates, which would prevent us from becoming profitable. | |
| ● | We may not be able to manufacture our product candidates in clinical or commercial quantities, which would prevent us from commercializing our product candidates. | |
| ● | Disputes under key agreements or conflicts of interest with our scientific advisors or clinical investigators could delay or prevent development or commercialization of our product candidates. |
Risks Related to Our Intellectual Property
| ● | We may not be able to maintain our exclusive worldwide license to use and develop PT00114 which could materially affect our business plan. |
Risks Related to Our Business Operations and Industry
| ● | If we are not able to retain our current senior management team and our scientific advisors or continue to attract and retain qualified scientific, technical and business personnel, our business will suffer. | |
| ● | We may encounter difficulties in managing our growth, which could adversely affect our operations. | |
| ● | Healthcare reform measures could adversely affect our business. | |
| ● | Our business and operations are vulnerable to computer system failures, cyber-attacks or deficiencies in our cyber-security, which could increase our expenses, divert the attention of our management and key personnel away from our business operations and adversely affect our results of operations. | |
| ● | Failure to comply with health and data protection laws and regulations could lead to government enforcement actions (which could include civil or criminal penalties), private litigation or adverse publicity and could negatively affect our operating results and business. | |
| ● | If we, our CROs or our IT vendors experience security or data privacy breaches or other unauthorized or improper access to, use of, or destruction of personal data, we may face costs, significant liabilities, harm to our brand and business disruption. | |
| ● | If we do not comply with laws regulating the protection of the environment and health and human safety, our business could be adversely affected. |
| 5 |
Risks Associated to our Common Stock
| ● | If we fail to comply with the continued minimum closing bid requirements of Nasdaq or other requirements for continued listing, including stockholder equity requirements, our common stock may be delisted and the price of our common stock and our ability to access the capital markets could be negatively impacted. | |
| ● | Our common stock is a “Penny Stock” subject to specific rules governing its sale to investors that could impact its liquidity. | |
| ● | The market price of our common stock may be volatile, which could lead to losses by investors and costly securities litigation. | |
| ● | If we fail to maintain an effective system of internal controls, we may not be able to accurately report our financial results or detect fraud. Consequently, investors could lose confidence in our financial reporting and this may decrease the trading price of our stock. | |
| ● | Investors may experience dilution of their ownership interests because of the future issuance of additional shares of our common stock. | |
| ● | Our common stock is controlled by insiders. | |
| ● | We do not intend to pay dividends for the foreseeable future and may never pay dividends. | |
| ● | Our certificate of incorporation allows for our board to create new series of preferred stock without further approval by our stockholders, which could adversely affect the rights of the holders of our common stock. |
PART I
OverviewItem 1. Business.
Overview
Protagenic Therapeutic, Inc. (together with its subsidiary, “Protagenic,” the “Company,” “we,” “our” or “us”) isare a Delaware corporationbiopharmaceutical company specializing in the discovery and development of therapeutics to treat central nervous system (CNS)stress-related neuropsychiatric and mood disorders. Our missionproprietary, patent-protected, first-in-class lead compound, PT00114, is a synthetic form of Teneurin Carboxy-terminal Associated Peptide (“TCAP”), an endogenous brain signaling peptide that can dampen overactive stress responses. Our preclinical models have demonstrated efficacy of PT00114 in animal models of depression, anxiety, substance abuse & addiction, and PTSD.
PT00114 leverages a completely novel mechanism of action. Protagenic owns exclusive, worldwide rights to provide safePT00114 through its license agreement with the University of Toronto and effective treatments for mood,has an exclusive right to license additional intellectual property generated by Dr. David Lovejoy’s lab at University of Toronto. Additionally, the company is engaged in the research & development of follow-on compounds in the TCAP family. Extensive publications in peer-reviewed scientific journals underline the central role stress plays in the onset and proliferation of neuropsychiatric disorders like depression, anxiety, depressionsubstance abuse & addiction, and neurodegenerative disorders by using novel peptide-base, brain active therapeutics. Our strategy isPTSD. The mechanism of action of TCAP suggests that it counterbalances stress overdrive at the cellular level within the brain’s stress response cascade. TCAP works to develop, testalleviate the harmful behavioral, biochemical, and obtain regulatory approval for various applicationsphysiological effects of these disorders, while simultaneously restoring brain active therapeutics.
Our current business model is designed aroundhealth. This mechanism has been corroborated in preclinical animal models of the further development of these applications, and to obtain thepsychiatric disorders listed above. We completed our preclinical experiments required regulatory approvals to allow for the commercialization of our neuropeptide-based applications and products (see “Governmental Regulation” below). If approval is obtained, we expect to begin a clinical trial in the first half of 2023. We commenced our sales effortsfirst human trial, seeking to prove the safety and anticipate generating revenue through both licensing and direct salesefficacy of our products.PT00114, on September 26, 2023. We believeannounced that the trial has passed its safety milestone, relating to the complete enrollment of the first cohort of patients in the single-dose portion of the Phase I trial, on February 13, 2024. We currently anticipate that we can establish and subsequently strengthen our market positionwill be able to announce the complete results of the single-dose portion of the Phase I trial in the following ways: (i) working to obtain FDA approvalsecond quarter of current and future neuropeptide applications; (ii) investigating foreign markets for the use of our current and future products; (iii) securing relationships with strong partners in our field; (iv) entering into license agreements, strategic partnerships and joint ventures for our various applications; and, (v) continuing our current research into improving our processes, reducing costs and developing new and innovative applications.2024.
We intend to advance our lead drug candidate, PT00114 through Investigational New Drug (IND)-enabling studies, and enter PT00114 into clinical proof-of-concept studies in Treatment-Resistant Depression (TRD) and/or Post-Traumatic Stress Disorder (PTSD) (anticipated clinical start: 2017-2018).
Corporate History
We are currently a Delaware corporation with one subsidiary named Protagenic Therapeutics Canada (2006) Inc., a corporation formed in 2006 under the laws of the Province of Ontario, Canada.
We were previously known as Atrinsic, Inc., a company that was once a reporting company under the Securities Act, but that, in 2012 and 2013, reorganized under Chapter 11 of the United States Bankruptcy Code and emerged from bankruptcy. On February 12, 2016, we acquired Protagenic Therapeutics, Inc. through a reverse merger (see “Corporate History – The Reverse Business Combination (Merger) Transaction”). On June 17, 2016, Protagenic Therapeutics, Inc. (the then wholly-owned subsidiary of Atrinsic, Inc.) was merged with and into Atrinsic, Inc. Atrinsic, Inc. was the surviving corporation in this merger and changed its name from Atrinsic, Inc. to Protagenic Therapeutics, Inc. (see “Corporate History – The Subsidiary Merger”).
The Reverse Business Combination (Merger) Transaction
On February 12, 2016, which we refer to as the Merger Closing Date, we (as Atrinsic, Inc.), Protagenic Therapeutics, Inc. and Protagenic Acquisition Corp., Atrinsic, Inc.’s wholly-owned subsidiary, entered into a merger agreement and completed the merger contemplated by the merger agreement. Pursuant to the merger agreement, on the Merger Closing Date, Protagenic Acquisition Corp. merged with and into Protagenic Therapeutics, Inc., with Protagenic Therapeutics, Inc. remaining as the surviving entity and wholly-owned subsidiary of Atrinsic, Inc. (the “Merger”)
Simultaneously with the Merger, on the Merger Closing Date all of the issued and outstanding shares of Protagenic common stock converted, on a 1-for-1 basis into shares of the Company’s Series B Preferred Stock, par value $0.000001 per share (“Series B Preferred Stock”) (assuming no exercise of dissenters’ rights by any Protagenic stockholder). Also on the Merger Closing Date, all of the issued and outstanding options to purchase shares of Protagenic common stock, and all of the issued and outstanding warrants to purchase shares of Protagenic common stock, converted, on a 1-for-1 basis, into options (the “New Options”) and new warrants (the “New Warrants”) respectively, to purchase shares of our Series B Preferred Stock. The New Options will be administered under Protagenic’s 2006 Employee, Director and Consultant Stock Plan (the “2006 Plan”), which the Company assumed and adopted on the Merger Closing Date in connection with the Merger.
On the Closing Date, (i) the former Protagenic common stock was exchanged for the right to receive 6,612,838 shares of Series B Preferred Stock; (ii) New Options to purchase 1,807,744 shares of Series B Preferred Stock granted under the 2006 Plan, having an average exercise price of approximately $0.87 per share, were issued to optionees pursuant to the assumption of the 2006 Plan; (iii) the holders of options to purchase the common stock of Atrinsic before the Merger (“Predecessor”) were issued options (“Predecessor Options”) to purchase 17,784 shares of Series B Preferred Stock at $1.25 per share; (iv) New Warrants to purchase 3,403,367 shares of Series B Preferred Stock at an average exercise price of approximately $1.05 per share were issued to holders of Protagenic warrants; and (v) 2,775,000 shares of Series B Preferred Stock were issued to investors at a purchase price of $1.25 per share in the Private Offering, as defined below. In addition, warrants (“Predecessor Warrants”) to purchase 295,945 shares of Series B Preferred Stock at $1.25 per share were issued to Strategic Bio Partners, LLC, the designee (the “Designee”) of the holders of Predecessor’s debt, in consideration of the cancellation of debt of $665,000 in principal and $35,000 in interest, and Placement Agent Warrants, as such term is defined below, to purchase 127,346 shares of Series B Preferred Stock were issued to the Placement Agent of the Private Offering. The common stockholders of Predecessor before the Merger retained 25,867 shares of our common stock, par value $0.000001 per share. In addition, upon the effectiveness of the Merger, the holders of the Predecessor’s Series A Preferred Stock exchanged all of the issued and outstanding Series A Preferred Stock for an aggregate of 297,468 shares of Series B Preferred Stock. These shares were issued to the Designee.
The Merger was treated as a recapitalization of Protagenic for financial accounting purposes and the historical financial statements of Protagenic Therapeutics, Inc. are our financial statements as a result of the Merger. The parties to the merger agreement have agreed to take all actions necessary to ensure the Merger is treated as a “plan of reorganization” under Section 368(a) of the Internal Revenue Code of 1986, as amended (the “Code”).
2016 Private Placement
Concurrently with the closing of the Merger, we conducted the first closing of an offering (the “Private Offering”) of our Series B Preferred Stock. At the first closing, we sold 2,775,000 shares of Series B Preferred Stock at a purchase price of $1.25 per share, for which we received total gross consideration of $3,468,750. Of this amount, $350,000 consisted of conversion of outstanding stockholder debt held by Garo H. Armen, our chairman and a member of our board of directors (“Board”), and $150,000 of legal expenses incurred by Strategic Bio Partners LLC, stockholders of the Predecessor, in conjunction with and as allowed by the merger agreement. On March 2, 2016, we completed the second closing of the Private Offering, at which we issued an additional 913,200 shares of Series B Preferred Stock to accredited investors, for total gross proceeds of $1,141,500. On April 15, 2016, we completed the final closing of the Private Offering, at which we issued an additional 420,260 shares of Series B Preferred Stock to accredited investors, for total gross proceeds of $525,325.
| 6 |
As Protagenic transitions into a clinical-stage company, we aim to complete certain key strategic and tactical milestones over the coming two years;
| ● | Rapidly advance our lead product candidate, PT00114, through clinical trials in treatment resistant depression, substance use disorder, generalized anxiety disorder, and/or post-traumatic stress disorder | |
| ● | Develop additional product candidates from the TCAP family to build out a broad pipeline of assets with differentiated features using our unique expertise with this mechanism | |
| ● | Explore efficacy in additional stress-related neuropsychiatric and mood disorders beyond initially targeted indications | |
| ● | Facilitate long-term growth by building a nimble R&D, operational, clinical and commercial team | |
| ● | Proactively assess strategic partnership opportunities including in important international markets |
Continue with our strategy of strengthening our IP position in this important novel field of neuropsychiatry
IND Submission
We paid Katalyst Securities LLC,currently anticipate re-submitting an investigational new drug (IND) application in advance of initiating the Phase IIa portion of our placement agent (the “Placement Agent”) and its selected dealers for the Private Offering a commission of 10%present clinical study, to ascertain whether this portion of the funds raisedstudy may be conducted in the Private Offering from investors introducedUnited States. The Phase I/IIa study, to evaluate the safety, tolerability, and early activity of PT100114 (TCAP) in healthy volunteers and patients with psychiatric illnesses, commenced in the third quarter of 2023. The IND enabling studies, including the preclinical efficacy data generated, as well as the GLP toxicology study, and a summary of the Phase I clinical trial plan, were among the components of this regulatory submission.
Clinical Development
The clinical development program will be led by Dr. Maurizio Fava, MD, PhD, a world-leader in psychiatric disorders, the Psychiatrist-in-Chief of the Massachusetts General Hospital and Slater Family Professor of Psychiatry at Harvard Medical School. Dr. Fava was co-principal investigator of STAR*D, the largest research study ever conducted in depression, has coauthored more than 800 medical journal publications, and is one of the top enrolling psychiatry clinicians in the US. Protagenic’s Phase I/II clinical study was designed by Dr. Fava, who will be the trial’s principal investigator.
We will launch our clinical program with a basket trial designed first to evaluate the safety of TCAP in a small cohort of healthy volunteers, immediately followed by the Placement Agentevaluation of safety, pharmacological and its selected dealers. In addition,clinical activity in cohorts of patients with stress-related neuropsychiatric disorders including, but not limited to depression, addiction, anxiety, and Post-Traumatic Stress Disorder (PTSD). We will be using this study for both safety and preliminary efficacy to prioritize indications for later phase development that would ultimately support a New Drug Application (NDA) and registration. The four indications were chosen for multiple reasons, including the Placement Agent received $15,000 to reimburse it for its expensesmechanism of TCAP in the private Offering,reducing biological stress signals, preclinical evidence of efficacy in animal models of these disorders and the placement Agent and its selected dealers were issued warrants (the “Placement Agent Warrants”) to purchase a number of shares of Series B Preferred Stock equal to 10% of the shares of Series B Preferred Stock sold to investors in the Private Offering who were introduced by the Placement Agent and its selected dealers. The Placement Agent Warrants, which contain a “cashless exercise” provision, are exercisable for a period of five years from the initial closing of the Private Offering at a price of $1.25 per share.
Pursuant to a registration statement declared effective by the SEC on February 8, 2017, we registered the shares of common stock underlying the Series B Preferred Stock and the Placement Agent Warrants issued in the 2016 Private Placement for public resale by the selling stockholders named therein and their assigns. The Company was not required to update and maintain the effectiveness of this registration statement after February 8, 2018.
Debt Exchange
Simultaneous with the Merger and the Private Offering, holders of $665,000 of Atrinsic’s debt accompanied with $35,000 in accrued interest exchanged such debt for five-year warrants of Predecessor (the “Predecessor Warrants”) to purchase 295,945 shares of Series B Preferred Stock at $1.25 per share.
Split-Off Agreements
At the closing of the Merger we had a 51% interest in MomSpot LLC, and the remaining 49% was held by B.E. Global LLC. Barry Eisenberg is the sole owner of B.E. Global LLC and is the Chief Executive Officer of MomSpot LLC. Immediately after the closing of the Merger, we split off our 51% membership interests in MomSpot LLC. The split-off was accomplished through the transfer of all of our membership interests of MomSpot LLC to B.E. Global LLC.
Immediately after the closing of the Merger, we split off all of our equity interest in 29 wholly-owned subsidiaries. The split-off was accomplished through the sale of all equity interestshigh unmet need in these wholly-owned subsidiariespatient populations, which creates significant market opportunity. We believe the basket trial structure offers the most efficient use of capital in early-stage development and will give us insights into which indication we should focus on in advanced clinical trials. Healthy volunteers will be the first cohort and subsequent parallel cohorts will include patients with:
| ● | Major Depressive Disorder (MDD) who have suboptimal response to or poorly tolerated two prior SSRIs / SNRIs | |
| ● | Generalized Anxiety Disorder (GAD) who have suboptimal response to or poorly tolerated two prior SSRIs /SNRIs | |
| ● | Opioid Use Disorder (OUD) who are on treatment with Suboxone and have suboptimal response | |
| ● | Post-Traumatic Stress Disorder (PTSD) who have suboptimal response to or intolerance of sertraline and paroxetine |
The trial will use a classic sequential dose escalation design using cohort replication with initial doses estimated from non-clinical data. The study will assess dose ranging through standard and small cohorts with a rules-based approach for dose, safety, efficacy, and biomarkers. Trial participants will have a maximal 28-day exposure. As this will be the first in human study of TCAP, safety and adverse events will be the primary endpoint. Key secondary endpoints were chosen to Quintel Holdings, Inc.ascertain efficacy in individual conditions and compare drug impact across disparate diseases. All disease cohorts will be measured for Strengths and Difficulties Questionnaire (SDQ), which is a validated broad self-rated outcome measure that has outperformed the clinician-rated Montgomery–Åsberg Depression Rating Scale (MADRS) scale in previous trials. Patients will also be assessed for stress biomarkers via pre- and post-treatment systemic cortisol levels and skin conductance. Each disease cohort (anxiety, depression, PTSD and addiction) will also have disease specific assessments.
Reverse Stock Split
Our stockholders voted at a special meeting held on June 17, 2016 in favor of, and we effectuated, a 1-for-15,463.7183 reverse stock split of our common stock, or the Reverse Split. As a result of the Reverse Split, 400,000,000 shares of common stock were split into 25,867 shares of common stock. Additionally, as a result of the Reverse Split and in accordance with our certificate of designations for our Series B Preferred Stock, our Series B Preferred Stock immediately and automatically converted into our common stock on a 1-for-1 basis other than any Series B Preferred Stock (i) to the extent (but only to the extent) a Series B Preferred Stock holder would beneficially own greater than 9.99% of our common stock (the “Springing Blocker”) and (ii) such holder has notified the Company in writing that it wants the Springing Blocker to apply to such holder. On July 27, 2016, 10,146,000 of the Company’s 11,018,766 outstanding shares of Series B Preferred Stock were eligible to immediately convert into 10,146,000 shares of the Company’s common stock with 872,766 shares of Series B Preferred Stock remaining as a result of one holder exercising the Springing Blocker. As of December 31, 2017, 10,146,000 shares of the Series B Preferred Stock were converted into 10,146,000 shares of common stock on the records of the Company. As of December 31, 2017 872,766 shares of Series B Preferred Stock remained outstanding.
Any Series B Preferred Stock not converted as a result of this provision would automatically convert into common stock as soon as such conversion would not violate the Springing Blocker. Our Series B Preferred Stock will cease to be designated as a separate series of our preferred stock when all of such shares have converted into shares of our common stock.
| 7 |
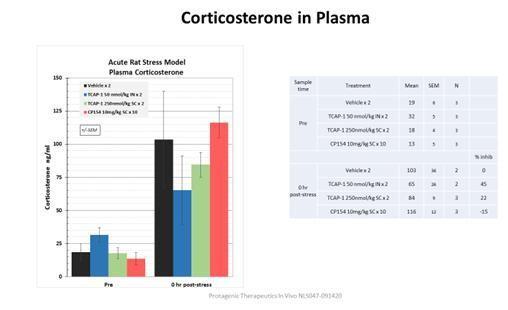
Furthermore, although patient populations and their responses to CNS agents can be highly variable in clinical studies, we attempt to mitigate this by stratifying the initial series of cohorts to select for and control for corticosterone levels to enable the broadest window of effect detection. Preclinical studies of TCAP demonstrate that its beneficial actions are most easily observed in stressed animals, which show elevations of plasma corticosterone levels at baseline before TCAP treatment. Anxious or depressed patients have elevated corticosterone levels, providing an opportunity to identify patients more likely to benefit pharmacologically and potentially clinically. This also provides a useful translational bridge between preclinical behavioral models and human clinical studies and enables flexibility in evaluating routes of administration.
Market for Stress-Related Neuropsychiatric Disorders: Depression, Addiction, Anxiety, and PTSD
Humans living in our modern world, in both developed and developing nations, are being exposed to a multitude of life stressors that are progressively taking a toll on our mental health. The Subsidiary Merger
On June 17, 2016, we merged our wholly-owned subsidiary, Protagenic Therapeutics, Inc.recent COVID-19 has exacerbated both near-term and long-term global impacts of stress-induced disorders on modern society. Stress-related mental, mood and behavioral disorders include, but are not limited to: treatment resistant depression (TRD), withwhich is a subgroup of major depressive disorder (MDD); addiction or substance use disorder (SUD); and intoanxiety, including generalized anxiety disorder (GAD) and post-traumatic stress disorder (PTSD). These disorders are a leading cause of disability worldwide and also a major contributor to suicide. Yet, a majority of these patients are inadequately served by current therapeutic options, which can have limited efficacy, significant side effects and high treatment burden. We believe these stress-related disorders are suitable indications for the Company and we changed our name from Atrinsic, Inc. to Protagenic Therapeutics, Inc. We are the parent companyuse of Protagenic Therapeutics Canada (2006), Inc.,neuropeptide-based drug candidates.
Major depressive disorder (MDD) is highly prevalent and disabling. The lifetime prevalence is approximately 12% with a corporation incorporated in the Provincepast year prevalence of Ontario.
Mood and Anxiety Disorders
An estimated 340 million people worldwide and 40-60 million people7.8% of adults in the United States alonein 2019, translating to over 19 million adults each year. The World Health Organization estimates 264 million people globally suffer from mental disorders including Major Depressive Disorder, or MDD, including TRD, PTSD, Bipolar Disorderdepression, which ranks depression as one of the highest causes of disability and various Anxiety Disorders. The global sales of anxiolytic and antidepressant drugsmortality in the U.S. were estimated to be $69 billion in 2013 and are projected to grow to nearly $77.1 billion by 2018. Yet, up to one-half of mood disorder patients are unresponsive to current treatments. Efficacy of therapy is challenged by non-compliance during the weeks to months required to achieve therapeutic benefit in combination with daily dosing requirements. Major targets in this space include TRD and PTSD, both indications which are highly resistant to available therapies.
Approximately 37% of those suffering from a MDD that do not respond to the current antidepressant medications constitute a separate group of people suffering from TRD. Despite a large patient population and current treatments that leave much room for improvement, the developmental pipelines are sparse and few novel candidates are in development. The serendipitous discoveries of current drug classes, and lack of efficacy have led to shrinkage or extinction of many pharma or small biotech neuroscience research programs. It is in this TRD market that we intend to focus our PT00114 development efforts.
TRD is the type of MDD that does not respond to standard courses of antidepressant medication.world. Stress plays a significant role in this illness thatand affects as many as half of people diagnosed with depression. MDD is characterized by multiple symptoms, potentially including depressed mood, loss of interest or pleasure, change in appetite or weight, sleep disturbance, fatigue or loss of energy, neurocognitive dysfunction, psychomotor agitation or retardation, feelings of worthlessness or excessive guilt, and suicidal ideation and behavior. MDD is highly treatment resistant, with 45-50% of patients who receive initial treatment for MDD not achieving long term remission, generally referred to as Treatment Resistant Depression (TRD). Patients suffering with TRD are at greater risk of hospitalization for their psychiatric illness and are more likely to abuse drugs and alcohol. These patients have a lower long-term quality of life and are at increased risk of attempting suicide. MDD is also highly recurrent and the estimated rate of recurrence over two years is over 40%, which rises to 75% after two episodes within five years.
Treatment guidelines recommend the combination of pharmacotherapy plus psychotherapy, but pharmacotherapy alone and psychotherapy alone are frequently used. For initial pharmacotherapy with antidepressants, selective serotonin reuptake inhibitors (SSRIs) are recommended. However, several classes of antidepressants are available, including serotonin-norepinephrine reuptake inhibitors (SNRIs), atypical antidepressants, and serotonin modulators, with efficacy generally comparable across and within classes. Drug choice is based on multiple factors, including side effect profile, comorbid illnesses, concurrent medications, patient preference, and cost. Physicians typically cycle through multiple generics if the initial response is suboptimal or patients experience AEs. Efficacy of therapy is challenged by non-compliance during the weeks to months required to achieve therapeutic benefit in combination with daily dosing requirements. However, SSRIs can produce significant quality of life side effects that interfere with medication adherence, including sexual dysfunction, gastrointestinal nausea and diarrhea, insomnia and weight gain. As a last resort, this disease is currently managed by invasive treatment, primarily electroconvulsive therapy (ECT). However, the ECT treatment’s side effects and high cost prevent millionswidespread adoption. Several drugs that have launched in recent years validate the market for branded agents in this field, in spite of people from taking advantage of it.their marginal improvements in safety or efficacy.
According to an article titled “Global prevalence of anxiety disorders: a systemic review and meta-regression,” written by AJ Baxter et al., (published inPsychological Medicine in 2013), PTSD affects an estimated 7.7 million adults (3.5%) in the US, with a disproportionately high prevalence in war veterans. Therapeutic approaches include cognitive therapy in combination with antidepressants, such as selective serotonin reuptake inhibitors (SSRIs). In addition to the vulnerabilities noted above for antidepressant-related treatments, PTSD patients often present with co-morbidities such as addictions or dependencies, which make therapeutic case management difficult.
Protagenic Research
PT00114 is the first known example of a new class of brain-targeted therapies based on a newly-described and highly conserved family of neuropeptides that regulate stress-induced mood and addictive behaviors. PT00114 is believed to act via a novel mechanism of action and is therefore expected to provide an extremely attractive therapeutic and commercial profile, especially for those patients who are not fully responsive to or compliant with current interventions. Based on preclinical data, we believe that PT00114 is well differentiated from other drug candidates on the basis of having: Dual activity on stress- and addiction-related pathways (as present in TRD and PTSD); Blood-brain barrier permeability; Rapid onset of action and long duration of therapeutic effects; Restoration of normalcy in stress, anxiety and addiction disorders; No adverse effects with little to no accumulation; Good safety and tolerability profiles; Convenient dosing route and schedule; High potency/low dose; and, Ease of chemical synthesis.
We believe that optimal cellular energy metabolism is fundamental to the biology of the brain, and clinical manifestation of aberrant energy metabolism often manifests in debilitating neurological disorders. PT00114’s ability in preclinical models to enhance glucose mobilization and utilization in the brain, maintain energy homeostasis, inhibit stress-related pathways and protect cells from oxidative damage suggests potential therapeutic benefits in a range of indications involving both acute and chronic neurological injury. Potential applications include traumatic brain injury, stroke recovery, and neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease and ALS, among others.
| 8 |
Technology
PT00114Generalized anxiety disorder (GAD) is one of the most common mental disorders in both community and clinical settings. In the United States, the estimated lifetime prevalence of GAD is 5.7%, corresponding to 18 million and 9 million individuals, respectively. GAD is characterized by excessive and persistent worrying that causes significant distress or impairment on most days and is hard to control. Other symptoms can include apprehensiveness, irritability, increased fatigue and muscular tension. GAD is also associated with increased rates of substance abuse, posttraumatic stress disorder, and obsessive-compulsive disorder. GAD is a syntheticpotentially chronic illness, with symptom severity fluctuating over time. A 12-year study of treated patients showed approximately 60% of patients had symptoms resolve, but around one-half of those subsequently relapsed.
Pharmacotherapy for GAD is primarily selective-serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs), which are mildly efficacious. Clinical trials for different SSRIs and SNRIs have shown approximately the same effectiveness, with response rates of approximately 60- 70% for the drug and 40% for placebo. However, SSRIs can produce significant quality of life side effects that interfere with medication adherence, including sexual dysfunction, gastrointestinal nausea and diarrhea, insomnia and weight gain. Thus, choice of agent is often dependent on the patient’s side effect profile for individual drugs. Benzodiazepines are efficacious and can reduce emotional and somatic symptoms within hours. However, concerns about dependence risk has contributed to a decline in their use. Buspirone has similar efficacy to benzodiazepines without the risk of dependence but has a time to onset of approximately four weeks. As the majority of these agents are now available as generics, the worldwide market for GAD therapies was expected to reach $1.8 billion in 2023 and with an anticipated forecasted value of $4.3 billion by 2033 (https://www.futuremarketinsights.com/reports/generalized-anxiety-disorder-treatment-market).
Post-traumatic stress disorder (PTSD) is one of the most common psychiatric disorders, with an estimated past-year and lifetime prevalence of 4.7% and 6.1%, translating to 11.5M adults in the US each year. PTSD develops in some patients following exposure to a traumatic event involving actual or threatened injury to themselves or others, such as war, natural disasters, rape or assault. Symptoms can be severe, chronic and disabling, which can include intrusive thoughts, nightmares and flashbacks of past traumatic events, avoidance of reminders of trauma, hypervigilance, and sleep disturbance, all of which lead to significant occupational and social impairment. Currently, PTSD is treated with psychotherapy and/or pharmacotherapy, with psychotherapy as the recommended primary intervention. Logistics and cost often limit access to psychotherapy, which results in many patients needing to rely on pharmacotherapy. Guidelines for pharmacotherapy recommend first-line treatment with sertraline and paroxetine, selective serotonin reuptake inhibitors (SSRI) antidepressants, as these are the only approved medications for PTSD. However, these only treat one aspect of symptomology and efficacy is limited, with fewer than 30% of patients experiencing remission. The side effect profile of these agents results in significant rates of discontinuation, particularly the severe effects such as suicidality and sexual dysfunction. Serotonin-norepinephrine reuptake inhibitors (SNRI) and second-generation antipsychotics are used off-label in some patients, but efficacy is sporadic, and side-effects can make these undesirable therapeutic options. As all of these options are currently generic, branded commercial sales for PTSD is almost non-existent. Given the size of the potential addressable population and limited therapeutic options available, a therapy with a superior therapeutic index could achieve significant market penetration and sales.
Substance use disorders (SUDs) are highly prevalent. According to the 2020 National Survey on Drug Use and Health (NSDUH), 40.3 million Americans, aged 12 or older, had a substance use disorder (SUD) in the past year. The majority of SUDs involve alcohol use disorder (14 million), followed by illicit drug use disorder (8 million). Illicit drug use and nonmedical use of medications alone or in combination with alcohol are associated with a substantial proportion of emergency department visits in the United States. Pharmacologic options to treat SUDs typically have limited efficacy, high treatment burden, with suboptimal side-effect profiles, ultimately leading to limited uptake and high remaining unmet medical need. 40- 60% of patients who receive SUD care experience chronic or relapsing disease course.
The incidence of opioid use disorder (OUD) and overdose deaths have reached epidemic proportions. Opioid use disorder is typically a chronic, relapsing illness, associated with significantly increased rates of morbidity and mortality. Opioid use disorder can be related to misuse of pharmaceutical opioids, heroin, or other opioids such as fentanyl and its analogues. In 2021, 3.3% of those 12 or older in the US were estimated to have used heroin at some point in their lives, translating 9.2 million people (https://www.samhsa.gov/data/sites/default/files/reports/rpt39443/2021NSDUHFFRRev010323.pdf)., Worldwide, an estimated 60 million people engaged in non-medical opioid use in 2021 (https://www.unodc.org/res/WDR-2023/WDR23_Exsum_fin_DP.pdf, https://www.unodc.org/res/wdr2022/MS/WDR22_Booklet_3.pdf ). Correspondingly, overdose deaths involving opioids in the US increased from an estimated 70,029 in 2020 to 80,816 in 2021, representing a 15% increase.
| 9 |
Unmet needs are particularly high in OUD. First-line treatment for most patients is medication-assisted treatment, consisting of pharmacotherapy with an opioid agonist or antagonist in combination with psychotherapy. Pharmacotherapy can include an opioid agonist (methadone or buprenorphine) and/or an opioid antagonist (e.g. naltrexone). Guidelines for mild opioid use disorder suggest first-line treatment with long-acting injectable naltrexone (e.g. Vivitrol) administered monthly. Guidelines for moderate to severe opioid use disorder suggest initial use of buprenorphine (e.g. Suboxone) due to the higher risk of lethal overdose with methadone. Treatment can allow patients to return to a productive lifestyle but has low success rates and can be extremely burdensome. These therapies require patients remain on maintenance treatment with an opioid agonist for many years as they are physically dependent upon the medications. A minority may be tapered off after a few years, with the taper itself taking several months to years.
The worldwide market for OUD therapies was valued at $3.5 billion in 2023 (https://www.thebusinessresearchcompany.com/report/opioid-use-disorder-oud-global-market-report#:~:text=It%20is%20distributed%20by%20hospital,and%20stores%2C%20and%20online%20pharmacies.&text=The%20opioid%20use%20disorder%20(OUD)%20market%20size%20has%20grown%20rapidly,(CAGR)%20of%2011.3%25. ) and projected to reach $8.4 billion by 2033 (https://www.futuremarketinsights.com/reports/opioid-use-disorder-treatment-market ).
The treatment burden and side effect profile of these therapies is substantial. Buprenorphine is classified as a schedule III controlled substance in the United States, with use limited to certified and specially trained physicians. Side effects include sedation, headache, nausea, constipation, insomnia, and sweating. Death is possible if buprenorphine is taken in combination with other substances, especially benzodiazepines and alcohol. Methadone is highly regulated in the United States, where it is classified as a schedule II drug. Only licensed opioid treatment programs or inpatient hospital units are permitted to dispense. Typical side effects of methadone include constipation, drowsiness, sweating, peripheral edema, reduced libido, and erectile dysfunction, with some patients experiencing severe adverse effects including cardiac arrhythmias, hyperalgesia, and overdose.
Alcohol use disorder (AUD) is extraordinarily prevalent. Approximately 30% of adults in the United States use alcohol in an unhealthy manner and may need some form of intervention. The 2019 United States National Survey on Drug Use and Health estimated that of Americans over the natural peptide sequence TCAP-1.age of 12 in the past 30 days, 24% reported binge drinking (five or more drinks on one occasion) and 6% reported heavy drinking (five or more drinks on each of five or more days). The National Institute on Alcohol Abuse and Alcoholism (NIAAA) reports 28% of US adults exceed thresholds for risky use alcohol consumption, with 19% exceeding the daily limit and 9% exceeding both the daily and weekly limits. Rates of diagnosable AUD by DSM-5 criteria from the third National Epidemiologic Survey on Alcohol and Related Conditions showed that 29% had met criteria for an alcohol use disorder in their lifetime and 14% met criteria for a current alcohol use disorder. Worldwide, the World Health Organization estimates that 5% of adults (>283 million people) had alcohol use disorder within the prior 12 months.
AUD is responsible for significant mortality and morbidity. Excessive alcohol consumption is the third leading preventable cause of death in the United States directly causing approximately 85,000 deaths per year, roughly 10% of deaths among working age adults. Nearly 5% of all deaths worldwide (approximately three million each year) have been attributed to alcohol use with 5% of those specifically due to AUD. The economic cost of excessive alcohol use in the United States is estimated to be $249 billion in 20101 by the CDC. Therapeutic unmet needs are significant for AUD and the condition is frequently untreated. Psychosocial interventions can be effective for treatment but up to 70% of individuals return to heavy drinking. For patients who met DSM-IV criteria for alcohol abuse, 46% were in remission, 24% continued to meet abuse criteria, and 30% met criteria for alcohol dependence in the future. For patients who met DSM-IV criteria for alcohol dependence, 39% were in remission, 15% met criteria for abuse only, and 46% continued to meet dependence criteria.
1As of March 2023, these are the most recent data released by the CDC.
| 10 |
TCAP-1
Several medications can be used to treat AUD, which can lead to reduced heavy drinking and increased days of abstinence. For most patients treated with moderate to severe alcohol use disorder, guidelines recommend first-line treatment with naltrexone (e.g. Vivitrol), an opioid antagonist. Vivitrol is an extended-release injectable naltrexone that allows for once monthly dosing that was approved in 2006. Vivitrol is priced at $~1,738/month (https://www.drugs.com/price-guide/vivitrol ) and 2023 worldwide sales have grown to $400.4 million (https://investor.alkermes.com/news-releases/news-release-details/alkermes-plc-reports-financial-results-fourth-quarter-and-year-3). The manufacturer projects Vivitrol sales will increase to $410 – 430 million in 2024 (https://investor.alkermes.com/news-releases/news-release-details/alkermes-plc-reports-financial-results-fourth-quarter-and-year-3), with patent expiry in 2029 (https://www.fdanews.com/articles/192221-alkermes-grants-amneal-generic-rights-for-vivitrol). Acamprosate (e.g. Campral) is recommended for those in whom naltrexone is contraindicated, such as those taking opioids or with acute hepatitis. Campral (Acamprosate) was approved by the FDA in 2004 and reached peak worldwide sales of $87M in 2008. Acamprosate is currently only available as generic in the US, but is still sold as branded Campral ex-US. Given the overall prevalence of AUD, these relatively low sales numbers indicate the vast majority of patients with AUD are not treated with pharmacotherapy.
Teneurin Carboxy-terminal Associated Peptide (TCAP) as a Therapy
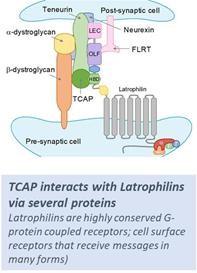
Our approach to treating stress-related neuropsychiatric and mood disorders is based on research into brain mechanisms conducted over the last 15 years in the laboratory of the company’s scientific founder, Dr. David Lovejoy, from the University of Toronto. TCAP was discovered in a genome-wide search for proteins related to corticotropin releasing factor (CRF), a keyan endogenous brain peptide known to be the central mechanism coupling external stress to psychological, behavioral, and endocrine responses. Dr. Lovejoy and his colleagues discovered and characterized Teneurin Carboxy-terminal Associated Peptide (TCAP); their further work revealed that TCAP is of ancient evolutionary origin and plays a central role in maintaining healthy brain structure and function in the face of the negative effects of stress. Although four TCAP peptides were discovered, only TCAP-1 is expressed independent of a larger Teneurin protein and is the primary focus of our development (PT00114).
TCAP reverses the impact of stress on the Hypothalamic-Pituitary-Adrenal (HPA) axis, the endocrine and behavioral control system which connects environmental stress to behavioral responses via brain levels of Corticotropin Releasing Factor (CRF) and blood levels of the stress hormone cortisol. Stress elevates CRF, which in stress response. While TCAP-1turn elevates cortisol levels. Studies have demonstrated that TCAP counteracts stress, it does so bythe effects of either endogenous or pharmacologically-administered CRF via a non-CRF receptor pathway and unlike direct CRF antagonists it does not exhibitin the brain, that is believed to be evolved over millions of years as a homeostasis-related pathway. There has been strong interest in the pharmaceutical industry for decades to develop drug candidates that block the negative effects of CRF by attempting to directly antagonize the CRF receptor, however clinical results to date with prior CRF receptor antagonists have been disappointing. Because TCAP counteracts the action of CRF by activating separate receptors instead of directly blocking CRF receptors, we believe it is a superior approach to alleviating stress-related neuropsychiatric disorders; TCAP-1 acts by binding to Latrophilin-1 and Latrophilin-3, G-protein-coupled receptors (GPCRs) expressed on nerve cells in animal models studiedthe extended amygdala, the region of the brain involved in memory, emotion, and fear. TCAP acts through these receptors to date.block the effects of CRF and potentially other stress mediators such as Arginine-Vasopressin (AVP). Due to differences in the mechanism of action, TCAP is expected to be efficacious in clinical settings in which earlier studies with CRF receptor antagonists were not. We believe this novel mechanism of action can provide an attractive therapeutic profile for patients who are not fully responsive to currently available therapies.
| 11 |
PT00114 inhibits
| |
Two key effects of TCAP may contribute to its pharmacological activity in reversing or preventing stress-induced behavioral distortions. In settings of stress and stress (CRF)-induced actionsdepression, the activity of specific neural circuits can be diminished compared to the levels of activity observed in clinically-relevant gold-standard animal models of anxiety, depression and addiction at concentrations several magnitudes below current front-line therapeutics. These beneficial effects are maintained for as long as three weeks after treatment. PT00114 promotes neuronal process development, spine density, axon fasciculation and branching in neurons.
PT00114healthy brain tissue. After administration, TCAP crosses the blood brain barrier and concentrates in regions of the brain associated with the regulation of mood disorders. Preliminary toxicity assessment (non-GLP) indicates no clear or significant adverseAdministered TCAP can lead to increases in activity in some of the neuronal circuitry implicated in depression, demonstrated by increases in the utilization of glucose, a surrogate for cell activity. The fact that the pharmacological effects although further toxicity testingof TCAP persist after the drug has been cleared aligns with findings that TCAP applied to neurons in culture stabilizes dendritic spines, structures that sprout from the surface of neurons and can form synapses with other neurons to create functional circuitry. Stress and the associated rise in CRF have been reported to cause loss of synapses in animal models. The fact that the pharmacological actions of TCAP persist for weeks are consistent with its producing lasting changes in neuronal function by changing patterns of gene expression and thus creating relatively stable changes in neuronal function. In a number of these models, a single subcutaneous dose of TCAP will prevent the behavioral consequences of stress encountered three weeks later. This is required.especially notable since the administered dose of TCAP is eliminated from the plasma within hours of administration.
PT00114Our lead compound is highly soluble and shows excellent stabilitya 41-residue peptide synthetic TCAP-1, which we have designated PT00114. In addition, we have a portfolio of earlier stage neuropeptides targeting the TCAP pathway that are in several storage conditions.preclinical evaluation. The initial dosage form is intended as a subcutaneous injection but is also amenable to other routes of administration.administration including sublingually or intra-nasally. This affords a range of target product profiles and opportunities for lifecycle management.
Business plan / Proposed next steps
The Company’s business plan calls for the following processes during 2018 and 2019:
Preclinical Efficacy Data
Historically, muchWhile many of the preclinical efficacy data regarding specific therapeutic benefitsinitial studies of PT00114TCAP had been generated in the lab of our Chief Technology Officer, Dr. David Lovejoy, atwe have designed several preclinical studies over the Universitylast four years to validate the safety and efficacy of Toronto. The Company recognizes that to fully validate its business proposal, and persuade potential corporate partners of target-disease efficacy, additional preclinical efficacy data from unaffiliated research organizations would be valuable. Hence, the Company has engaged twoPT00114, for which we hired multiple independent contract research organizations (CROs) to conduct these studies. In preclinical testsrodent models, administration of PT00114 results in reproducible, dose-dependent reversal of a range of stress-induced behavioral distortions, including depression, stress-exacerbated anxiety, excessive startle, drug seeking, and opioid withdrawal. Stress-induced anxiety was measured by an elevated plus maze, an open field with stressed animals, and acoustic startle in CRF-treated animals. Depression was measured by tail suspension and forced swim. Stress-induced changes in tube-restrained rodents were used as a well-validated model for anxiety and depression, as well as alleviationsub-acute stress. Notably, PT00114 was found to be pharmacologically active in stressed rodents but relatively inactive in non-stressed rodents.
In studies conducted with Charles River Laboratories in Kuopio, Finland, PT00114 showed beneficial effects in Chronic Social Defeat, a murine model of drug addictive behavior.
Process Development and Manufacturing
stress-induced behavioral dysfunction that has features of depression. In parallelthis model, male mice are placed in cages along with older, dominant male mice. This results in progressively more “resigned” behaviors in the Company’s external CRO research studies,mice experiencing this domineering exposure. This results in a series of behaviors in the Company is pursuing good manufacturing practices (cGMP) synthesis of PT00114. The Company obtained enough TCAP in July 2017 to supply its Phase I human clinical trials anticipated to begin in 2019. The Company intends to secure at least two supplier relationships for sourcing synthesized human PT00114.
Preclinical Safety & Toxicology
A key partcowed mice, termed Chronic Social Defeat. PT00114 reverses many of the Company’s preclinical studies for IND readiness iscomponent behaviors typically measured in this model, suggesting that it reverses the toxicology testingnegative effects of PT00114 in two animal species. Because these toxicology tests will be carried out with a drug concentration that is a multiple of the intended concentrationstress in the eventual marketable drug, the Company plans to commence its safety and toxicology testing only after receiving a confirmatory positive result from the latest external CRO efficacy tests. This means toxicology testing could begin as soon as the third quarter of 2018.“defeated” animals.
Pursue Strategic Partnership
The Company believes it would be to its advantage to secure a collaboration with a pharma/biopharma company with a presence in neurological and psychiatric diseases and/or addiction. Therefore, it plans to use the preclinical efficacy data generated during 2017 and the first quarter 2018 as a point of instigation with potential pharma/biopharma corporate partners.
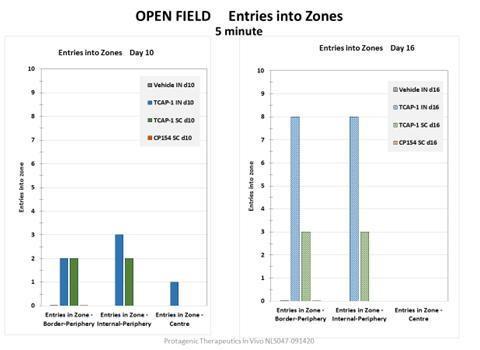
CompilePT00114 demonstrated efficacy in a variable chronic stress model that has features of anxiety and File INDPTSD. In an open field assessment, mice or rats are stressed by being placed in a tube for several hours, then placed in an open box where their movement is observed for 20 minutes. Control animals exhibit stress response behavior by not moving around much and staying near the edges of the box. Animal receiving PT00114 at the end of the stress condition moved around the open field. Animals receiving multiple administrations of a control small molecule CRH antagonist did not venture into the open field, indicating they were stressed. These results are also reflected in blood cortisol levels, where control mice had increased cortisol levels, which were reduced by treatment with PT00114, but not by the small molecule CRF antagonist.
| 13 |
Stress plays a central role in a broad range of addictions, including alcohol and opioids. The most important corporate goal for whichability of PT00114 to blunt excessive stress may be able to provide non-dependence forming treatment of addictions. A series of studies conducted at Porsolt Laboratories in Lavel France support the Company is deployingpotential utility of PT00114 as a treatment to help people defeat opioid addiction. In rats addicted to opioids, administering CRF models environmental stress, causing them to frantically seek opioids. PT00114 reduces the working capital it raisedopioid seeking behavior in 2016 is the compilationresponse to CRF administration. Further studies conducted by Porsolt following EMEA guidelines demonstrated that on its own, PT00114 was not addictive and submission of an investigational new drug (IND) applicationrats did not develop dependence to the FDA.peptide after chronic administration.
PT00114 has also demonstrated pre-clinical efficacy in a murine model of opioid withdrawal called the Saleens test. In this test, mice are addicted to opioids and the animals are then administered the opioid antagonist naloxone, which immediately blocks opioid action and triggers profound stress and opioid withdrawal. This manifests as a behavioral stress response with the mice jumping up to six inches into the air over 70 times in a 20-minute observation period. Administering PT00114 at three different time points within the experiment – before the naloxone-driven withdrawal, before the period of opioid addiction, or up to three weeks before the induced withdrawal – results in a reproducible, dose-dependent restoration to non-stressed behavior and reduced jumping. Significantly, this is not accompanied by any evidence of sedation or reduced activity. This effect appears independent of the opioid used as PT00114 ameliorates this withdrawal-triggered jumping stress behavior in mice experiencing withdrawal from both fentanyl and morphine.
Preclinical Safety and Toxicology
Preclinical safety data for PT00114 demonstrates a robust profile in both rats and non-human primates. As the mechanism is unique and TCAP is a prerequisitepart of healthy brain signaling, we believe PT00114 will have a differentiated side effect profile relative to begin Phase I human testingexisting antidepressant and antipsychotic agents. A key aspect of the TCAP mechanism is that it does not completely block the perception of and responses to stress; it rather protects against stress overload. Some perception of environmental stress and a proportionate response to that stress is adaptive behavior and it is not desirable to completely block stress responses. Unlike benzodiazepines that can cause sedation and are prone to dependence, TCAP prevents the maladaptive response to environmental stress without sedation and without developing dependence.
We have completed non-GLP Dose-Range-Finding (DRF) toxicology studies of PT00114 administered subcutaneously daily for any indication.five days in rats and non-human primates. The preclinical efficacy datadoses tested were substantially above the anticipated clinical doses and were well tolerated and safe, with no dose-limiting toxicities observed at doses at least 50-fold higher than anticipated clinical exposures. No major changes in hematology or clinical chemistries were seen, including prolactin levels or testosterone levels, changes in which may impact libido. Distinct from SSRI’s, there was no impact on ambulation, sedation or weight gain. Importantly, further studies conducted following EMEA guidelines, demonstrated that on its own PT00114 was not addictive and rats did not develop dependence to the peptide after chronic administration. The in life 28-day GLP toxicology testing in both the rats and non-human primate have been completed. There have been no changes in clinical chemistries or pathology that would prompt a stop in the program and the therapeutic margin if large. The final audited reports are currently being generatedcompiled.
Process Development and Manufacturing
We currently do not own any manufacturing facilities and rely on 3rd party contract manufacturers for synthesis of PT00114. We have sufficient PT00114 synthesized under current Good Manufacturing Practices (“cGMP”) conditions to complete GLP toxicology studies and Phase 1 human clinical trials. This material is currently undergoing requisite stability and accelerated stability testing. PT00114 is highly soluble and has shown excellent preliminary stability in several storage conditions, with the material being stable for at two external CROs, as well as the toxicology test results that the Company plans to obtain, and a specific plan and protocol for a Phase I trial,least 12 months.
The initial dosage form developed will be among the componentsa subcutaneous injection. Because PT00114 is also amenable to other routes of this key regulatory submission anticipatedadministration including sublingually or intra-nasally, we will be doing preliminary process work to develop these formulations, and anticipate using one of these dosage forms in early 2019.later stage clinical studies.
| 14 |
Initiate Phase I Clinical Studies
One the Company’s IND application has been filed, the next major milestone is anticipated to be an approval by the Company’s FDA review team that the Phase I trial protocol proposed in the IND application is acceptable to begin. The Company believes that this may be achieved in the second half of 2019.
Technology License Agreement
On July 31, 2005, the Company had entered into a Technology License Agreement (“License Agreement”) with the University of Toronto (the “University” or “UT”) pursuant to which the University agreed to license to the Company patent rights and other intellectual property, among other things (the “Technologies”). The Technology License Agreement was amended on February 18, 2015 and currently does not provide for an2015. Unless earlier terminated, the term of this License Agreement shall terminate on the expiration date.or invalidity of the last issued Patent in the License Agreement
Pursuant to the License Agreement and its amendment, the Company obtained an exclusive worldwide license to make, have made, use, sell and import products based upon the Technologies, or to sublicense the Technologies in accordance with the terms of the License Agreement and amendment. In consideration, the Company agreed to pay to the University a royalty payment of 2.5% of net sales of any product based on the Technologies. If the Company elects to sublicense any rights under the License Agreement and amendment, the Company agrees to pay to the University 10% of any up-front sub-license fees for any sub-licenses that occurred on or after September 9, 2006, and, on behalf of the sub-licensee, 2.5% of net sales by the sub-licensee of all products based on the Technologies. The Company had no sales revenue for the year ended December 31, 20172023 and therefore was not subject to paying any royalties.
In the event the Company fails to provide the University with semi-annual reports on the progress or fails to continue to make reasonable commercial efforts towards obtaining regulatory approval for products based on the Technologies, the University may convert our exclusive license into a non-exclusive arrangement. Interest on any amounts owed under the License Agreement and amendment will be at 3% per annum. All intellectual property rights resulting from the Technologies or improvements thereon will remain the property of the other inventors and/or Dr. David Lovejoy (“The Professor”) at the University, and/or the University, as the case may be. The Company has agreed to pay all out-of- pocketout-of-pocket filing, prosecution and maintenance expenses in connection with any patents relating to the Technologies. In the case of infringement upon any patents relating to the Technologies, the Company may elect, at its own expense, to bring a cause of action asserting such infringement. In such a case, after deducting any legal expenses the Company may incur, any settlement proceeds will be subject to the 2.5% royalty payment owed to the University under the License Agreement and amendment.
The patent applications were made in the name of the ProfessorDr. Lovejoy and other inventors, but the Company’s exclusive, worldwide rights to such patent applications are included in the License Agreement and its amendment with the University. The Company maintains exclusive licensing agreements and it currently controls the six intellectual patent properties.
Sales and Marketing
We currently have no sales, marketing or distribution capabilities. In order to commercially market PT00114 and any product candidates we develop in the future, we would either need to develop an internal sales team and marketing department or collaborate with third parties who have sales and marketing capabilities. As we commenced clinical trials in the third quarter of 2023, we expect to seek a Market Access expert or consultancy to better understand clinician and payor dynamics in the therapeutic areas we are focused on, so that, as we begin later stage studies, we are working on a deeper commercial assessment in parallel. We have done some high-level benchmarking of pricing based on the current landscape of approved and available therapies for psychiatric disorders we are targeting, both in the generics and on-patent realms.
ManufacturingCompetition
We currently do not own any manufacturing facilities, nor have we entered into any agreements with contract manufacturer for the production of PT00114. Currently we synthesize all the PT00114 we use in our development activities.
Competition
The pharmaceutical and biotechnology industries are highly competitive and characterized by rapidly evolving technology and intense research and development efforts. We expect to compete with companies, including major international pharmaceutical companies and other institutions that have substantially greater financial, research and development, marketing and sales capabilities and have substantially greater experience in undertaking preclinical and clinical testing of products, obtaining regulatory approvals and marketing and selling biopharmaceutical products. We will face competition based on, among other things, product efficacy and safety, the timing and scope of regulatory approvals, product ease of use and price.
Major depressive disorder patients that do not respond to the current antidepressant medications constitute a separate group of TRD. Despite a large patient population and current treatments that leave much room for improvement, the developmental pipelines are sparse and few novel candidates are in development. The serendipitous discoveries of current drug classes, side effects and lack of efficacy have led to shrinkage or extinction of many pharma or small biotech neuroscience research programs. According to a May 10, 2016 Zion Research report, the current global depression drug market was valued at approximately $14.5 billion in 2014, and is expected to generate $16.8 billion by the end of 2020. The pharmaceutical addiction market is very large but has not yet been quantified because no successful drug has been launched to treat victims. We intend to launch PT00114 into either the TRD, anti-anxiety, or pharmaceutical addiction markets.
| 15 |
Set forth below is a discussion of competitive factors for each of the current drug classes commercially available for TRD, and the competitive advantages that we believe PT00114 may offer. The basis for our beliefs regarding the competitive advantages that PT00114 may offer over its competitors is our own pre-clinical animal studies. We acknowledge that these beliefs and conclusions about competitive advantages must be regarded as theoretical until such time as we have human clinical data that supports and re-affirms the results seen in the pre-clinical animal studies.
Opioid receptor modulators
Opioid receptor modulators have the potential to be non-addictive therapeutic drugs for TRD. Competitors include ALKS 5461 (from Alkermes) isTRD but have a fixed combinationhigh likelihood of buprenorphineabuse and samidorphan being developed as a therapy for TRD. Buprenorphine is a mu opioid receptor partial agonist as well as an antagonist of the kappa-opioid receptor (KOR), while samidorphan is an antagonist of mu opioid receptors that essentially works to block the buprenorphine from binding to the mu-receptor. The combination of these mechanisms may result in attenuation of the mu agonist effects of buprenorphine, potentially making this a non-addictive therapy. ALKS 5461 is in phase III as a once-daily therapy administered as a sublingual tablet. It is well tolerated and treatment effects were evident after one week of dosing.thus regulatory restrictions. We believe that our competitive advantage is that PT00114 targets a different receptor system therefore it is not likely to have a clinical overlap with opioid receptor modulators.
Atypical Antipsychotics with antidepressant effects (dopamine receptor modulators)
Brexpiprazole (from(Rexulti from Otsuka) is a dopamine (D2 receptor) partial stimulator (agonist) approved as an oral adjunctive TRD therapy. Its side effects include suicidal risk, weight gain and restlessness. Cariprazine (from Gedeon Richter)(Vraylar from AbbVie) is an oral dopamine D2 and D3 receptor antagonist approved for schizophrenia and bipolar disorder in development for TRD. The most common side effects reported were extrapyramidal symptoms, the urge to move (akathisia), indigestion (dyspepsia), vomiting, drowsiness (somnolence) and restlessness. We believe that our competitive advantage is that PT00114, due to its low toxicity profile, will be clinically preferable to these antipsychotic drugs.
Ketamine-like TRD drugsKetamine and Esketamine
Drugs that act in a mechanism similar to Ketamine such asand Esketamine (Spratavo nasal spray (fromfrom Johnson and& Johnson) is the S(+) enantiomer of the drug ketamine actsact primarily as a non-competitive NMDA receptor antagonist, but is also a dopamine reuptake inhibitor. As of December 2017, the CompanyAlthough ketamine is used off-label and Esketamine was awaiting phase III clinical trial resultsrecently approved for treatment-resistant depression (TRD). This class of candidates is generatingTRD, limitations and concerns around use limit uptake in a lot of excitement but uncertainty due to their use history will be a compounding factor.broader population. We believe that our competitive advantage is that the toxicity profile is likely to be less favorable when compared with PT00114.
GABA receptor modulators
GABA receptors, when bound by inhibitory neurotransmitters found throughout the brain, act as a brake on nerve activity. Sage Therapeutics is developing multiple compounds that target this mechanism and more candidates are expected to come from this therapeutic class that may present a competitive challenge for PT00114.
NMDA receptor modulators
The N-methyl-D-aspartate (or “NMDA”) receptor is a molecule that appears on the surface of neurons. When “activated” by a drug that binds with it, the NMDA receptor is a potential natural way to counteract TRD. A drug called GLYX 13, an amidated tetrapeptide (with the amino acid sequence Thr-Pro-Pro-Thr-NH2) is a glycine-site functional partial agonist of the NMDA receptor discovered at Northwestern University, now being developed by Naurex/Allergan, in Phase III U.S. clinical trials. It will be administered by intravenous injection and has a rapid onset. Phase II results have shown that GLYX 13 treatment reduces depression scores in patients with TRD, with no psychotomimetic side effects common to other NMDA receptor modulators. The major peptide candidate in this group GLYX13 shows a better tolerance profile and even IV dosing once weekly is not a deterrent enough in the clinic so PT00114 peptide with possible subcutaneous delivery would be a much more preferable clinical option. The development of the tetrapeptide and entry into the trials demonstrated room and willingness to accept peptide based therapies in TRD. More candidates are expected to come from this therapeutic class that may present a competitive challenge for PT00114.
Another of Naurex’s small molecule candidates, NRX-1074, is an orally active therapy based on GLYX13, in preclinical stages. L-4-Chlorokynurenine, AV-101 (from VistaGen Therapeutics) is a fast acting, orally active small molecule glycine binding site NMDA receptor antagonist. A NIH-funded phase II trial in major depressive disorder has been initiated in the US. CERC-301 (Cerecor) is an orally-active, selective NMDA receptor subunit 2B (NR2B) antagonist which is in phase II an adjunctive therapy for TRD.
PT00114’s Competitive Advantages/DisadvantagesAdvantages
We believeOur preclinical data and the corroborated mechanism of action of PT00114 will be ableindicates its advantages as compared to compete against each of these drugs based on its core advantages:current approved therapies:
| ● | PT00114 | |
| ● | PT00114’s effects are |
| 16 |
| ● | ||
| PT00114 is rapidly cleared | ||
| ● | PT00114 naturally crosses the blood brain barrier | |
| ● | PT00114 is an L-isomer, a naturally modified peptide, | |
| ● | PT00114 | |
| ● | PT00114 will be manufactured by standard solid phase chemistry, which is less expensive than manufacturing processes required by other TRD | |
The main competitive disadvantage that PT00114 will have relative to other antidepressant drugs is that it will have fewer marketing resources behind it, assuming that the Company consummates a partnership with a large pharmaceutical company during its commercial marketing phase. Beyond this marketing resources disadvantage, the Company acknowledges that PT00114 may have efficacy disadvantages that we are not yet aware of since the drug has not yet been tested in humans. Extrapolating the early results obtained in rodent studies, PT00114 appears to be more effective and with few or no side effects, but this must be treated as an unknown since no human studies have yet been performed, and a new competitive disadvantage could be discovered during the clinical trial phase.
Although we believe PT00114’s advantages will allow it to compete effectively against other antidepressant drugs in the TRD market, many of our competitors and potential competitors have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, clinical trials, obtaining regulatory approvals and marketing approved products than we do. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller or early stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These third parties compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to our programs or advantageous to our business.
Intellectual Property
We believe that patents, trademarks, copyrights and other proprietary rights are important to our business. We also rely on trade secrets, know-how, continuing technological innovations and licensing opportunities to develop and maintain our competitive position. We seek to protect our intellectual property rights by a variety of means, including obtaining patents, maintaining trade secrets and proprietary know-how, and technological innovation to operate without infringing on the proprietary rights of others and to prevent others from infringing on our proprietary rights. Our policy is to seek to protect our proprietary position by, among other methods, actively seeking patent protection in the United States and foreign countries.
As of December 31, 2017,2023, we have four patents issued by the Governments of the United States, Canada, European Union (validated in Germany, France and Great Britain) and Australia and three patent applications pending worldwide includingon our original platform technology, all of which have expired aside from the one granted in the U.S. SomeUnited States. The patent applications were made in the name of Dr. David A. Lovejoy and inventors, but the Company’s exclusive, worldwide rights to such patent applications are included in the License Agreement with UT. OtherWe have eight further issued patents and five pending patent applications were made with various Company personnel as inventors and allin related technology that the company has rights have been assigned to the Company.in or own.
Our success will depend in part on our ability to maintain our proprietary position through effective patent claims and their enforcement against our competitors. Although we believe our patent applications provide a competitive advantage, the patent positions of companies like ours are generally uncertain and involve complex legal and factual questions. We do not know whether any of our patent applications will result in the issuance of any patents. Those patents that may be issued in the future or those acquired by us may be challenged, invalidated or circumvented, and the rights granted under any issued patent may not provide us with proprietary protection or competitive advantages against competitors with similar technology. In particular, we do not know if competitors will be able to design variations on our treatment methods to circumvent our current and anticipated patent claims. Furthermore, competitors may independently develop similar technologies or duplicate any technology developed by us. Because of the extensive time required for the development, testing and regulatory review of a potential product, it is possible that, before any of our products can be commercialized or marketed, any related patent claim may expire or remain in force for only a short period following commercialization, thereby reducing the advantage of the patent.
We also rely upon trade secrets, confidentiality agreements, proprietary know-how and continuing technological innovation to remain competitive, especially where we do not believe patent protection is appropriate or obtainable. We continue to seek ways to protect our proprietary technology and trade secrets, including entering into confidentiality or license agreements with our employees and consultants, and controlling access to and distribution of our technologies and other proprietary information. While we use these and other reasonable security measures to protect our trade secrets, our employees or consultants may unintentionally or willfully disclose our proprietary information to competitors.
| 17 |
Our commercial success will depend in part on our ability to operate without infringing upon the patents and proprietary rights of third parties. It is uncertain whether the issuance of any third partythird-party patents would require us to alter our products or technology, obtain licenses or cease certain activities. Our failure to obtain a license to technology that we may require to discover, develop or commercialize our future products may have a material adverse impact on us. One or more third-party patents or patent applications may conflict with patent applications to which we have rights. Any such conflict may substantially reduce the coverage of any rights that may issue from the patent applications to which we have rights. If third parties prepare and file patent applications in the United States that also claim technology to which we have rights, we may have to participate in interference proceedings in the USPTO to determine priority of invention.
We may collaborate in the future with other entities on research, development and commercialization activities. Disputes may arise about inventorship and corresponding rights in know-how and inventions resulting from the joint creation or use of intellectual property by us and our subsidiaries, collaborators, partners, licensors and consultants. As a result, we may not be able to maintain our proprietary position.
As of December 31, 2017,2023, we controlledown or have rights in the following intellectual property:
| COUNTRY | SERIAL# | |||||||||||||||||||
| PATENT# |
| STATUS | |||||||||||||||||
| UNITED STATES | 11/01/2004 | 10/510,959 | 01/03/2012 | 8,088,889 | ISSUED | |||||||||||||||
| A METHOD FOR REGULATING NEURITE GROWTH* | ||||||||||||||||||||
| COUNTRY | FILED | SERIAL# | ISSUED | PATENT# | STATUS | |||||||||||||||
| UNITED STATES | 06/19/2012 (Continuation) | 13/527,414 | 08/01/2017 | 9,718,857 | ISSUED | |||||||||||||||
| A METHOD FOR MODULATING INSULIN-INDEPENDENT GLUCOSE TRANSPORT USING TENEURIN C-TERMINAL ASSOCIATED PEPTIDE (TCAP)* | ||||||||||||||||||||
| COUNTRY | FILED | SERIAL# | ISSUED | PATENT# | STATUS | |||||||||||||||
| CANADA | 07/21/2015 | 2,955,410 | 02/20/2024 | 2,955,410 | ISSUED | |||||||||||||||
| GREAT BRITAIN | 07/21/2015 | 1702638.6 | 07/21/2020 | 2543996 | ISSUED | |||||||||||||||
| UNITED STATES | 07/21/2015 (PCT)/ 01/17/2017(371c) | 15/326,735 | 04/14/2020 | 10,617,736 | ISSUED | |||||||||||||||
| COMPOSITIONS, METHODS AND USES FOR ENHANCING MUSCLE FUNCTION* | ||||||||||||||||||||
| COUNTRY | FILED | SERIAL# | ISSUED | PATENT# | STATUS | |||||||||||||||
| US | 09/26/2017(PCT) / 03/25/2019(371c) | 16/336,334 | 09/20/2022 | 11,446,355 | ISSUED | |||||||||||||||
| CA | 09/26/2017 | 3,038,169 | ||||||||||||||||||
|
| |||||||||||||||||||
| PENDING | ||||||||||||||||||||
| COMPOSITIONS, METHODS AND USES FOR TREATING POST-TRAUMATIC STRESS DISORDER * | ||||||||||||||||||||
| COUNTRY | FILED | SERIAL# | ISSUED | PATENT# | STATUS | |||||||||||||||
| UNITED STATES | 10/12/2018(PCT) /04/10/2020(371c) | 11,426,444 | 08/30/2022 | 11,426,444 | ISSUED | |||||||||||||||
| CANADA | 04/14/2020 | 3,079,724 | PENDING | |||||||||||||||||
|
| |||||
|
|
| ||||
| ||||||
|
| COMPOSITIONS, METHODS AND USES OF A TENEURIN C-TERMINAL ASSOCIATED PEPTIDE-1 (TCAP-1) FOR TREATING OPIOID ADDICTION | ||||||||||||||||||||
| COUNTRY | FILED | SERIAL# | ISSUED | PATENT# | STATUS | |||||||||||||||
| CANADA | 3/13/2019 | 3,093,841 | PENDING | |||||||||||||||||
| UNITED STATES | 3/13/2019(PCT) / 9/11/2010 (371c) | 16/980,176 | PENDING | |||||||||||||||||
| EUROPE(UPC + Switzerland, Spain, Great Britain and Ireland) | 3/13/2019 | 19712494.4 | 02-08-2023 | 3765056 | ISSUED | |||||||||||||||
| HONG KONG (Extended EP Application. Registration for Grant filed Feb. 1, 2024)) | 3/13/2019 | 62021035260.0 | PENDING | |||||||||||||||||
In the future we may file additional patent applications based on proprietary formulations and novel compounds.
Governmental Regulation
Our technologies are subject to extensive government regulation, principally by FDA and state and local authoritiescompounds in the United States and by comparable agencies in foreign countries. Governmental authorities in the United States extensively regulate the preclinical and clinical testing, safety, efficacy, research, development, manufacturing, labeling, storage, record-keeping, advertising, promotion, export, marketing and distribution, among other things, of pharmaceutical products under various federal laws including the Federal Food, Drug and Cosmetic Act, or FFDCA, and under comparable laws by the states and in most foreign countries.TCAP family.
Properties
The Company has not commenced its FDA approval application process, and does not plancurrently own any real property. The Company leases office space for its principal executive office located at 149 Fifth Avenue, Suite 500, New York, New York 10010.
Legal Proceedings
From time to launch the FDA application process until 2022 or 2023. We cannot commence the FDA application process until we have obtained clinical human data on PT00114 in three phases of trials, none of which have been initiated. Similarly, the Company will be required to obtain regulatory approval in every country or region outside the United States into which it plans to sell its drug products. We may seek approval from authorities outside the United States such as the European Union CE Mark and Japanese Ministry of Health. As of December 31, 2017, the Company has not launched the approval application process for any region in the world because of its lack of clinical human data on PT00114.
Domestic Regulation
In the United States, the FDA, under the FFDCA, the Public Health Service Act and other federal statutes and regulations, subject pharmaceutical and biologic products to rigorous review. If we do not comply with applicable requirements,time we may be fined, the government may refuse to approve our marketing applications or allow us to manufacture or market our products or product candidates, and we may be criminally prosecuted. The FDA also has the authority to discontinue or suspend manufacture or distribution, require a product withdrawal or recall or revoke previously granted marketing authorizations, if we fail to comply with regulatory standards or if we encounter problems following initial marketing.
FDA Approval Process
To obtain approval of a new product from the FDA, we must, among other requirements, submit data demonstrating the product’s safety and efficacy as well as detailed information on the manufacture and composition of the product candidate. In most cases, this entails extensive laboratory tests and preclinical and clinical trials. This testing and the preparation of necessary applications and processing of those applications by the FDA are expensive and typically take many years to complete. The FDA may deny our applications or may not act quickly or favorablynamed in reviewing these applications, and we may encounter significant difficulties or costs in our efforts to obtain FDA approvals that could delay or preclude us from marketing any products we may develop. The FDA also may require post-marketing testing and surveillance to monitor the effects of approved products or place conditions on any approvals that could restrict the commercial applications of these products. Regulatory authorities may withdraw product approvals if we fail to comply with regulatory standards or if we encounter problems following initial marketing. With respect to patented products or technologies, delays imposed by the governmental approval process may materially reduce the period during which we will have the exclusive right to exploit the products or technologies.
The process required by the FDA before a new drug or biologic may be marketedclaims arising in the United States generally involvesordinary course of business. Currently, no legal proceedings, government actions, administrative actions, investigations or claims are pending against us or involve us that, in the following:
Preclinical tests include laboratory evaluation of product chemistry formulation and stability, as well as studies to evaluate toxicity. The results of preclinical testing, together with manufacturing information and analytical data, are submitted to the FDA as part of an IND application. The FDA requires a 30-day waiting period after the filing of each IND application before clinical trials may begin, in order to ensure that human research subjects will not be exposed to unreasonable health risks. At any time during this 30-day period or at any time thereafter, the FDA may halt proposed or ongoing clinical trials, or may authorize trials only on specified terms. The IND application process may become extremely costly and substantially delay developmentopinion of our products. Moreover, positive results of preclinical tests will not necessarily indicate positive results in clinical trials.
The sponsor typically conducts human clinical trials in three sequential phases, which may overlap. These phases generally include the following:
Phase I: The product is usually first introduced into healthy humans or, on occasion, into patients, and is tested for safety, dosage tolerance, absorption, distribution, excretion and metabolism.
Phase II: The product is introduced into a limited patient population to:
Phase III: These are commonly referred to as pivotal studies. If a product is foundmanagement, could reasonably be expected to have an acceptable safety profilea material adverse effect on our business and to be potentially effective in Phase II clinical trials, new clinical trials will be initiated to further demonstrate clinical efficacy, optimal dosage and safety within an expanded and diverse patient population at geographically-dispersed clinical study sites.financial condition.
If the FDA does ultimately approve the product, it may require post-marketing testing, including potentially expensive Phase IV studies, to monitor its safety and effectiveness.Subsidiary
Clinical trials must meet requirements for Institutional Review Board, or IRB, oversight, informed consent and the FDA’s Good Clinical Practices. Prior to commencement of each clinical trial, the sponsor must submit to the FDA a clinical plan, or protocol, accompanied by the approval of the committee responsible for overseeing clinical trials at one of the clinical trial sites. The FDA and the IRB at each institution at which a clinical trial is being performed may order the temporary or permanent discontinuation of a clinical trial at any time if it believes that the clinical trial is not being conducted in accordance with FDA requirements or presents an unacceptable risk to the clinical trial patients.
The sponsor must submit to the FDA the results of the preclinical and clinical trials, together with, among other things, detailed information on the manufacturing and composition of the product, in the form of an NDA, or, in the case of a biologic, a BLA. Once the submission has been accepted for filing, the FDA has 180 days to review the application and respond to the applicant. The review process is often significantly extended by FDA requests for additional information or clarification. The FDA may refer the BLA to an advisory committee for review, evaluation and recommendation as to whether the application should be approved, but the FDA is not bound by the recommendation of an advisory committee.
It is possible that our product candidates will not successfully proceed through this approval process or that the FDA will not approve them in any specific period of time, or at all. The FDA may deny or delay approval of applications that do not meet applicable regulatory criteria, or if the FDA determines that the clinical data do not adequately establish the safety and efficacy of the product. Satisfaction of FDA pre-market approval requirements for a new biologic is a process that may take several years and the actual time required may vary substantially based upon the type, complexity and novelty of the product or disease. The FDA reviews these applications and, when and if it decides that adequate data are available to show that the product is both safe and effective and that other applicable requirements have been met, approves the drug or biologic for marketing. Government regulation may delay or prevent marketing of potential products for a considerable period of time and impose costly procedures upon our activities. Success in early stage clinical trials does not assure success in later stage clinical trials. Data obtained from clinical activities is not always conclusive and may be susceptible to varying interpretations that could delay, limit or prevent regulatory approval. Upon approval, a product candidate may be marketed only for those indications approved in the BLA or NDA and may be subject to labeling and promotional requirements or limitations, including warnings, precautions, contraindications and use limitations, which could materially impact profitability. Once approved, the FDA may withdraw the product approval if compliance with pre- and post-market regulatory standards is not maintained or if safety, efficacy or other problems occur after the product reaches the marketplace.
The FDA may, during its review of an NDA or BLA, ask for additional test data. If the FDA does ultimately approve the product, it may require post-marketing testing, including potentially expensive Phase IV studies, to monitor the safety and effectiveness of the product. In addition, the FDA may, in some circumstances, impose restrictions on the use of the product, which may be difficult and expensive to administer and may require prior approval of promotional materials.
We have not yet begun the preparation of our IND application to begin Phase I clinical trials. We anticipate doing so in late 2018. We also have not begun to prepare our application for FDA approval which we anticipate will be in 2022 or 2023. The process of collecting the clinical data needed to complete our IND application is the focus of all of our working capital, and is expected to consume all of our available capital resources over the next eighteen months. The expenditures necessary to make progress along our IND program are expected to keep our operations in a cash flow negative state for the entire period from now until and after our IND application in 2019. To maintain our liquidity, we should endeavor to obtain an influx of cash from a non-revenue source in mid-2018, from either an up-front payment from a large pharmaceutical partner or an equity financing.
Ongoing FDA Requirements
Before approving an NDA or BLA, the FDA will inspect the facilities at which the product is manufactured and will not approve the product unless the manufacturing facilities are in compliance with the FDA’s current Good Manufacturing Practices, or cGMP, requirements which govern the manufacture, holding and distribution of a product. Manufacturers of biologics also must comply with the FDA’s general biological product standards. Following approval, the FDA periodically inspects drug and biologic manufacturing facilities to ensure continued compliance with the cGMP requirements. Manufacturers must continue to expend time, money and effort in the areas of production, quality control, record keeping and reporting to ensure full compliance with those requirements. Failure to comply with these requirements subjects the manufacturer to possible legal or regulatory action, such as suspension of manufacturing, seizure of product, voluntary recall of product, withdrawal of marketing approval or civil or criminal penalties. Adverse experiences with the product must be reported to the FDA and could result in the imposition of marketing restrictions through labeling changes or market removal. Product approvals may be withdrawn if compliance with regulatory requirements is not maintained or if problems concerning safety or efficacy of the product occur following approval.
The labeling, advertising, promotion, marketing and distribution of a drug or biologic product also must be in compliance with FDA and FTC requirements which include, among others, standards and regulations for direct-to-consumer advertising, industry-sponsored scientific and educational activities, and promotional activities involving the internet. The FDA and FTC have very broad enforcement authority, and failure to abide by these regulations can result in penalties, including the issuance of a Warning Letter directing the company to correct deviations from regulatory standards, a requirement that future advertising and promotional materials be pre-cleared by the FDA and enforcement actions that can include seizures, injunctions and criminal prosecution.
Manufacturers are also subject to various laws and regulations governing laboratory practices, the experimental use of animals and the use and disposal of hazardous or potentially hazardous substances in connection with their research. In each of the above areas, the FDA has broad regulatory and enforcement powers, including the ability to levy fines and civil penalties, suspend or delay issuance of approvals, seize or recall products and deny or withdraw approvals.
HIPAA Requirements
Other federal legislation may affect our ability to obtain certain health information in conjunction with our research activities. The Health Insurance Portability and Accountability Act of 1996, or HIPAA, mandates, among other things, the adoption of standards designed to safeguard the privacy and security of individually identifiable health information. In relevant part, the U.S. Department of Health and Human Services, or HHS, has released two rules mandating the use of new standards with respect to such health information. The first rule imposes new standards relating to the privacy of individually identifiable health information. These standards restrict the manner and circumstances under which covered entities may use and disclose protected health information so as to protect the privacy of that information. The second rule released by HHS establishes minimum standards for the security of electronic health information. While we do not believe we are directly regulated as a covered entity under HIPAA, the HIPAA standards impose requirements on covered entities conducting research activities regarding the use and disclosure of individually identifiable health information collected in the course of conducting the research. As a result, unless they meet these HIPAA requirements, covered entities conducting clinical trials for us may not be able to share with us any results from clinical trials that include such health information.
In addition to the statutes and regulations described above, we are also subject to regulation under the Occupational Safety and Health Act, the Environmental Protection Act, the Toxic Substances Control Act, the Resource Conservation and Recovery Act and other present and potential future federal, state and local regulations.
Research and Development
Our research and development efforts with respect to the formulations of PT00114 as our first potential product are exclusively conducted under premises of UT, Ontario, Canada. Much of our scientific research and discovery work is performed by Dr. David A. Lovejoy, our Chief Science Advisor and Dr. Dalia Barsyte, our Chief Technology Officer. These activities are funded by us under our Sponsored Research agreements with UT. We intend in the future to raise capital in distinct phases, matched to relevant scientific developments. The Company has financed completion of its preclinical proof of principle studies and the solidification of its intellectual property position through private offerings of its securities. In addition, the proceeds of bridge loans from the Company’s Chairman were used to fund research, development and the general operating activities of the Company. We anticipate that we will require additional financing through IND-enabling studies, and to support entry into clinical proof-of-concept studies in Treatment-Resistant Depression (TRD) and/or Post-Traumatic Stress Disorder (PTSD). As we develop new product candidates, we may be required to conduct additional scientific, preclinical and as well as clinical studies. We currently have no commitments to provide us with any such additional funding.
We incurred approximately $717,452 and $417,866 for research and development activities for the years ended December 31, 2017 and 2016, respectively.
The Company derives income from scientific research and experimental development tax credits/and or refunds issued by the Canada Revenue Agency for qualified expenditures. The credits are recognized when the refund is issued. The amounts received are reinvested into the Company’s scientific research, experimental development and operational works conducted in Canada.
Subsidiary
Protagenic Therapeutics Canada (2006) Inc. (“PTI Canada”) was incorporated in 2006 in the Province on Ontario, Canada. PTI Canada is a wholly-owned subsidiary of Protagenic. It provides operational support and assistance for the implementation of corporate and operational activities conducted in Canada. It also oversees and supports research and development activities conducted under auspices of UT. PTI Canada has three directors: Garo H. Armen (Chairman), Alexander K. Arrow and Vigen Nazarian. PTI Canada also has one part-time consultant, Robert Ziroyan. PTI Canada also benefits through tax incentive programs provided by the governments of Canada and the Province of Ontario. We derived incomecredits from Canadian research and development tax credits for the years ended December 31, 20172023 and 20162022 of $0 and $56,085,$0, respectively.
Employees
We currently have threeone full time and two part-time employees. We also engage consultants and temporary employees from time to time to provide services that relate to our research and development activities as well as for general administrative and accounting services. We believe that our current personnel are capable of meeting our operating requirements in the near term. We expect that as our business grows we may hire additional personnel to handle the increased demands on our operations, preclinical and clinical activities.
FacilitiesCorporate and Available Information
Our principal office is located at 149 Fifth Avenue, Suite 500, New York, New York 10010, in a conference room of Agenus, Inc. We utilize our principal office for quarterly board meetings and our annual shareholder meeting. Our personnel and consultants all work remotely, the Company’s basic science laboratory work is conducted in the Lovejoy Lab at the University of Toronto, and its preclinical efficacy work is conducted at CROs. Hence the Company does not have the need for a day-to-day physical office location other than a mailing address and conference room facility for meetings. For that reason, the Agenus conference room suits its purposes without imposing any inconveniences upon Agenus. Dr. Armen, our Executive Chairman, is also the Chairman and Chief Executive Officer of Agenus Inc.
Legal Matters
From time to time we may be named in claims arising in the ordinary course of business. Currently, no legal proceedings, government actions, administrative actions, investigations or claims are pending against us or involve us that, in the opinion of our management, could reasonably be expected to have a material adverse effect on our business and financial condition.
We anticipate that we will expend significant financial and managerial resources in the defense of our intellectual property rights in the future if we believe that our rights have been violated. We also anticipate that we will expend significant financial and managerial resources to defend against claims that our products and services infringe upon the intellectual property rights of third parties.
Reports to Security Holders
Our principal offices are located at 149 Fifth Avenue, New York, New York 10010. Our web address iswww.protagenic.com.
We make available, free of charge through our website, our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to those reports filed or furnished pursuant to Section 13(a) of the Securities Exchange Act of 1934, as amended, as soon as reasonably practicable after we electronically file or furnish such materials to the Securities and Exchange Commission, or SEC. In addition, you may read and copy any materials we file with the SEC at its Public Reference Room at 100 F Street, NE, Washington, DC 20549, on official business days during the hours of 10:00 am to 3:00 pm. You may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. The SEC maintains an Internet site,www.sec.govthat contains reports, proxy and information statements, and other information that we file electronically with the SEC. All statements made in any of our securities filings, including all forward-looking statements or information, are made as of the date of the document in which the statement is included, and we do not assume or undertake any obligation to update any of those statements or documents unless we are required to do so by law.
| 19 |
Item 1A. Risk Factors.
An investment in our common stock is speculative and illiquid and involves a high degree of risk including the risk of a loss of your entire investment. You should carefully consider the risks and uncertainties described below and the other information contained in this report before purchasing shares of our common stock. The risks set forth below are not the only ones facing us. Additional risks and uncertainties may exist that could also adversely affect our business, operations and prospects. If any of the following risks actually materialize, our business, financial condition, prospects and/or operations could suffer. In such event, the value of our common stock could decline, and you could lose all or a substantial portion of the money that you pay for our common stock.
Risks Related to Our Financial Condition and Capital Requirements
The Company’s financial statements have been prepared on a going concern basis, and do not include adjustments that might be necessary if the Company is unable to continue as a going concern.
The Company’s consolidated financial statements have been prepared on a going concern basis, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. As of December 31, 2023, the Company had incurred significant operating losses since inception, and continues to generate losses from operations, and has an accumulated deficit of $30,777,872. Based on its cash resources as of December 31, 2023, the Company has sufficient resources to fund its operations until the end of the third quarter of 2024. The consolidated financial statements included in this report do not include any adjustments relating to the recoverability and classification of asset amounts or the classification of liabilities that might be necessary should the Company be unable to continue as a going concern.
We have a history of losses and expect that losses may continue in the future.
We have generated net losses since we began operations, including $5,000,497 and $3,555,505 for the years ended December 31, 2023 and December 31, 2022, respectively. As of December 31, 2023, we had an accumulated deficit of $30,777,872. We have no approved products and have generated no product revenue. We expect that product development, preclinical and clinical programs will increase losses significantly over the next five years. In order to achieve profitability, we will need to generate significant revenue. We cannot be certain that we will generate sufficient revenue to achieve profitability. We anticipate that we will continue to generate operating losses and negative cash flow from operations and our current cash position is sufficient to fund our current business plan until the third quarter of 2024. We cannot be certain that we will ever achieve, or if achieved, maintain profitability. If our revenue grows at a slower rate than we anticipate or if our product development, marketing and operating expenses exceed our expectations or cannot be adjusted accordingly, our business, results of operation and financial condition will be materially adversely affected, and we may be unable to continue operations.
We will not be able to generate product revenue unless and until one of our product candidates successfully completes clinical trials and receives regulatory approval. As our most advanced product candidates are at an early proof-of-concept stage, we do not expect to receive revenue from any product candidate for the foreseeable future. We may seek to obtain revenue from collaboration or licensing agreements with third parties. We currently have no such agreements which will provide us with material, ongoing future revenue and we may never enter into any such agreements. Even if we eventually generate revenues, we may never be profitable, and if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis.
Risk Related
We need to obtain financing in order to continue our operations.
On a prospective basis, we will require both short-term financing for operations and long-term capital to fund our expected growth. We have no existing bank lines of credit and have not established any definitive sources for additional financing. Additional financing may not be available to us, or if available, then it may not be available upon terms and conditions acceptable to us. If adequate funds are not available, then we may be required to delay, reduce or eliminate product development or clinical programs. Our inability to take advantage of opportunities in the industry because of capital constraints may have a material adverse effect on our business and our prospects. If we fail to obtain the capital necessary to fund our operations, we will be unable to advance our development programs and complete our clinical trials.
In addition, our research and development expenses could exceed our current expectations. This could occur for many reasons, including:
| ● | some or all of our product candidates fail in clinical or preclinical studies and we are forced to seek additional product candidates; | |
| ● | our product candidates require more extensive clinical or preclinical testing than we currently expect; | |
| ● | we advance more of our product candidates than expected into costly later stage clinical trials; | |
| ● | we advance more preclinical product candidates than expected into early stage clinical trials; | |
| ● | we are required, or consider it advisable, to acquire or license rights from one or more third parties; or | |
| ● | we determine to acquire or license rights to additional product candidates or new technologies. |
While we expect to seek additional funding through public or private financings, we may not be able to obtain financing on acceptable terms, or at all. In addition, the terms of our financings may be dilutive to, or otherwise adversely affect, holders of our common stock. We may also seek additional funds through arrangements with collaborators or other third parties. These arrangements would generally require us to relinquish rights to some of our technologies, product candidates or products, and we may not be able to enter into such agreements, on acceptable terms, if at all. If we are unable to obtain additional funding on a timely basis, we may be required to curtail or terminate some or all of our development programs, including some or all of our product candidates.
If we continue to incur operating losses and fail to obtain the capital necessary to fund our operations, we will be unable to advance our development programs, complete our clinical trials, or bring products to market, or may be forced to reduce or cease operations entirely. In addition, any capital obtained by us may be obtained on terms that are unfavorable to us, our investors, or both.
Developing a new drug and conducting clinical trials and the regulatory review processes involves substantial costs. We have projected cash requirements for the near term based on a variety of assumptions, but some or all of such assumptions are likely to be incorrect and/or incomplete, possibly materially in an adverse direction. Our actual cash needs may deviate materially from those projections, changes in market conditions or other factors may increase our cash requirements, or we may not be successful even in raising the amount of cash we currently project will be required for the near term. We will need to raise additional capital in the future; the amount of additional capital needed will vary as a result of a number of factors, including without limitation the following:
| ● | receiving less funding than we require; | |
| ● | higher than expected costs to manufacture our product candidates; | |
| ● | higher than expected costs for preclinical testing; | |
| ● | an increase in the number, size, duration, and/or complexity of our clinical trials; | |
| ● | slower than expected progress in developing PT00114, or other product candidates, including without limitation, additional costs caused by program delays; | |
| ● | higher than expected costs associated with attempting to obtain regulatory approvals, including without limitation additional costs caused by additional regulatory requirements or larger clinical trial requirements; | |
| ● | higher than expected personnel, consulting or other costs, such as adding personnel or industry expert consultants or pursuing the licensing/acquisition of additional assets; and | |
| ● | higher than expected costs to protect our intellectual property portfolio or otherwise pursue our intellectual property strategy. |
| 21 |
When we attempt to raise additional financing, there can be no assurance that we will be able to secure such additional financing in sufficient quantities or at all. We may be unable to raise additional capital for reasons including, without limitation, our operational and/or financial performance, investor confidence in us and the biopharmaceutical industry, credit availability from banks and other financial institutions, the status of current projects, and our prospects for obtaining any necessary regulatory approvals. Potential investors’ capital investments may have shifted to other opportunities with perceived greater returns and/or lower risk thereby reducing capital available to us, if available at all.
In addition, any additional financing might not be available, and even if available, may not be available on terms favorable to us or our then-existing investors. We will seek to raise funds through public or private equity offerings, debt financings, corporate collaboration or licensing arrangements, mergers, acquisitions, sales of intellectual property, or other financing vehicles or arrangements. To the extent that we raise additional capital by issuing equity securities or other securities, our then-existing investors will experience dilution. If we raise funds through debt financings or bank loans, we may become subject to restrictive covenants, our assets may be pledged as collateral for the debt, and the interests of our then-existing investors would be subordinated to the debt holders or banks. In addition, our use of and ability to exploit assets pledged as collateral for debt or loans may be restricted or forfeited. To the extent that we raise additional funds through collaboration or licensing arrangements, we may be required to relinquish significant rights (including without limitation intellectual property rights) to our Companytechnologies or product candidates, or grant licenses on terms that are not favorable to us. If we are not able to raise needed funding under acceptable terms or at all, then we will have to reduce expenses, including the possible options of curtailing operations, abandoning opportunities, licensing or selling off assets, reducing costs to a point where clinical development or other progress is impaired, or ceasing operations entirely.
We have a limited operating history, expect to incur significant operating losses, and have a high risk of never being profitable.
We commenced operations in February 2016 through a reverse merger and have a limited operating history of less than five years. Therefore, there is limited historical financial or operational information upon which to evaluate our Businessperformance. Our prospects must be considered in light of the uncertainties, risks, expenses, and difficulties frequently encountered by companies in their early stages of operations. Many if not most companies in our industry at our stage of development never become profitable and are acquired or go out of business before successfully developing any product that generates revenue from commercial sales or enables profitability.
As of December 31, 2023, we have incurred an accumulated deficit of $30,777,872. We expect to continue to incur substantial operating losses over the next several years for the clinical development of our current and future licensed or purchased product candidates.
The amount of future losses and when, if ever, we will become profitable are uncertain. We do not have any products that have generated any revenues from commercial sales, and do not expect to generate revenues from the commercial sale of products in the near future, if ever. Our ability to generate revenue and achieve profitability will depend on, among other things, successful completion of the development of our product candidates; obtaining necessary regulatory approvals from the FDA and international regulatory agencies; establishing manufacturing, sales, and marketing arrangements with third parties; obtaining adequate reimbursement by third-party payers; and raising sufficient funds to finance our activities. If we are unsuccessful at some or all of these undertakings, our business, financial condition, and results of operations are expected to be materially and adversely affected.
As a recently established public reporting company, we are subject to SEC reporting and other requirements, which will lead to increased operating costs in order to meet these requirements.
| 22 |
Unstable market and economic conditions may have serious adverse consequences on our ability to raise funds, which may cause us to cease or delay our operations.
From time to time, global and domestic credit and financial markets have experienced extreme disruptions, including severely diminished liquidity and credit availability, declines in consumer confidence, declines in economic growth, increases in unemployment rates, and uncertainty about economic stability. Our financing strategy will be adversely affected by any such economic downturn, volatile business environment and continued unpredictable and unstable market conditions. If the equity and credit markets deteriorate, it may make a debt or equity financing more difficult to complete, costlier, and more dilutive. Failure to secure any necessary financing in a timely manner and on favorable terms will have a material adverse effect on our business strategy and financial performance, and could require us to cease or delay our operations.
Risks Related to our Discovery,Clinical Development and Commercialization of New MedicinesRegulatory Approval
Our results to date provide no basis for predicting whether any of our product candidates will be safe or effective, or receive regulatory approval.
The Company’s proprietary portfolio of five new neuropeptide hormones are in various stages of research and preclinical evaluation and their risk of failure is high. It is impossible to predict when or if any of our neuropeptide hormones will prove effective or safe in humans or will receive regulatory approval. These compounds may not demonstrate in patients the chemical and pharmacological properties ascribed to them in laboratory studies, and they may interact with human biological systems or other drugs in unforeseen, ineffective or harmful ways. If we are unable to discover or successfully develop drugs that are effective and safe in humans, we will not have a viable business.
We may not be able to initiate and complete preclinical studies and clinical trials for our product candidates which could adversely affect our business.
We must successfully initiate and complete extensive preclinical studies and clinical trials for our product candidates before we can receive regulatory approval. Preclinical studies and clinical trials are expensive and will take several years to complete and may not yield results that support further clinical development or product approvals. Conducting clinical studies for any of our drug candidates for approval in the United States requires filing an IND and reaching agreement with the FDA on clinical protocols, finding appropriate clinical sites and clinical investigators, securing approvals for such studies from the independent review board at each such site, manufacturing clinical quantities of drug candidates, supplying drug product to clinical sites and enrolling sufficient numbers of participants. We cannot guarantee that we will be able to successfully accomplish all of the activities necessary to initiate and complete clinical trials.
As a result, our preclinical studies and clinical trials may be extended, delayed or terminated, and we may be unable to obtain regulatory approvals or successfully commercialize our products.
NoneThe drug development and approval process is uncertain, time-consuming and expensive.
The process of our product candidates has received regulatory approvals. If we are unable to obtainobtaining and maintaining regulatory approvals for new therapeutic products is lengthy, expensive and uncertain. It also can vary substantially based on the type, complexity, and novelty of the product. We must provide the FDA and foreign regulatory authorities with preclinical and clinical data demonstrating that our products are safe and effective before they can be approved for commercial sale. Clinical development, including preclinical testing, is a long, expensive and uncertain process. It may take us several years to market onecomplete our testing, and failure can occur at any stage of testing. Any preclinical or moreclinical test may fail to produce results satisfactory to the FDA. Preclinical and clinical data can be interpreted in different ways, which could delay, limit or prevent regulatory approval. Negative or inconclusive results from a preclinical study or clinical trial, adverse medical events during a clinical trial or safety issues resulting from products of our product candidates, our business may be adversely affected.
Allthe same class of our product candidates are in early stages of development, and we do not expect our product candidatesdrug could cause a preclinical study or clinical trial to be commercially available for several years,repeated or a program to be terminated, even if at all. Our product candidatesother studies or trials relating to the program are subject to strict regulation bysuccessful.
| 23 |
The regulatory authorities in the United Statesapproval process is costly and in other countries. We cannot market any product candidate until we have completed all necessary preclinical studieslengthy and clinical trials and have obtained the necessary regulatory approvals. We do not know whether regulatory agencies will grant approval for any of our product candidates. Even if we complete preclinical studies and clinical trials successfully, we may not be able to successfully obtain all required regulatory approvals.
The preclinical development, clinical trials, manufacturing, marketing and labeling of pharmaceuticals are all subject to extensive regulation by numerous governmental authorities and agencies in the United States and other countries. We must obtain regulatory approvalsapproval for each of our product candidates before marketing or we mayselling any of them. It is not receive approvalspossible to make claims aboutpredict how long the approval processes of the FDA or any other applicable federal or foreign regulatory authority or agency for any of our products that we believe towill take or whether any such approvals ultimately will be necessary to effectively market our products. Datagranted. The FDA and foreign regulatory agencies have substantial discretion in the drug approval process, and positive results in preclinical testing or early phases of clinical studies offer no assurance of success in later phases of the approval process. Generally, preclinical and clinical testing of products can take many years and require the expenditure of substantial resources, and the data obtained from preclinical studiesthese tests and clinical trials are subjectcan be susceptible to varying interpretations that could delay, limit or prevent regulatory approval. If we encounter significant delays in the regulatory process that result in excessive costs, this may prevent us from continuing to develop our product candidates. Any delay in obtaining, or failure to obtain, approvals could adversely affect the marketing of our products and our ability to generate product revenue. The risks associated with the approval process include:
| ● | failure of our product candidates to meet a regulatory agency’s requirements for safety, efficacy and quality; | |
| ● | limitation on the indicated uses for which a product may be marketed; | |
| ● | unforeseen safety issues or side effects; and | |
| ● | governmental or regulatory delays and changes in regulatory requirements and guidelines. |
Even if we receive regulatory approvals for marketing our product candidates, if we fail to comply with continuing regulatory requirements, we could lose our regulatory approvals, and our business would be adversely affected.
The FDA continues to review products even after they receive initial approval. If we receive approval to commercialize any product candidates, the manufacturing, marketing and sale of these drugs will be subject to continuing regulation, including compliance with quality systems regulations, good manufacturing practices, adverse event requirements, and prohibitions on promoting a product for unapproved uses. Enforcement actions resulting from our failure to comply with government and regulatory requirements or inadequate manufacturing processes are examplescould result in fines, suspension of approvals, withdrawal of approvals, product recalls, product seizures, mandatory operating restrictions, criminal prosecution, civil penalties and other problemsactions that could impair the manufacturing, marketing and sale of our potential products and our ability to conduct our business.
Even if we are able to obtain regulatory approvals for any of our product candidates, if they exhibit harmful side effects after approval, our regulatory approvals could be revoked or otherwise negatively impacted, and we could be subject to costly and damaging product liability claims.
Even if we receive regulatory approval for our product candidates, we will have tested them in only a small number of patients during our clinical trials. If our applications for marketing are approved and more patients begin to use our product, new risks and side effects associated with our products may be discovered. As a result, regulatory authorities may revoke their approvals; we may be required to conduct additional clinical trials, make changes in labeling of our product, reformulate our product or make changes and obtain new approvals for our and our suppliers’ manufacturing facilities. We might have to withdraw or recall our products from the marketplace. We may also experience a significant drop in the potential sales of our product if and when regulatory approvals for such product are obtained, experience harm to our reputation in the marketplace or become subject to lawsuits, including class actions. Any of these results could decrease or prevent approval. any sales of our approved product or substantially increase the costs and expenses of commercializing and marketing our product.
If we experience delays or difficulties in the enrollment of subjects to our clinical trials, our receipt of necessary regulatory approvals could be delayed or prevented, which could materially affect our financial condition.
Identifying, screening and enrolling patients to participate in clinical trials of our product candidates is critical to our success, and we may not be able to identify, recruit, enroll and dose a sufficient number of patients with the required or desired characteristics to complete our clinical trials in a timely manner. The timing of our clinical trials depends on our ability to recruit patients to participate as well as to subsequently dose these patients and complete required follow-up periods.
In addition, we may encounterexperience enrollment delays related to increased or rejections due to additional government regulation from future legislation, administrative action unforeseen regulatory, legal and logistical requirements at certain clinical trial sites. These delays could be caused by reviews by regulatory authorities and contractual discussions with individual clinical trial sites. Any delays in enrolling and/or changesdosing patients in our planned clinical trials could result in increased costs, delays in advancing our product candidates, delays in testing the FDA policy. Even ifeffectiveness of our product candidates or in termination of the FDA approves a product, the approval will be limited to those indications covered in the approval.clinical trials altogether.
| 24 |
Outside
Patient enrollment may be affected if our competitors have ongoing clinical trials with products for the same indications as our product candidates, and patients who would otherwise be eligible for our clinical trials instead enroll in our competitors’ clinical trials. Patient enrollment may also be affected by other factors, including:
| ● | coordination with clinical research organizations to enroll and administer the clinical trials; | |
| ● | coordination and recruitment of collaborators and investigators at individual sites; | |
| ● | size of the patient population and process for identifying patients; | |
| ● | design of the clinical trial protocol; | |
| ● | eligibility and exclusion criteria; | |
| ● | perceived risks and benefits of the product candidates under study; | |
| ● | availability of competing commercially available therapies and other competing products’ clinical trials; | |
| ● | time of year in which the trials are initiated or conducted; | |
| ● | severity of the diseases under investigation; | |
| ● | ability to obtain and maintain subject consents; | |
| ● | ability to enroll and treat patients in a timely manner; | |
| ● | risk that enrolled subjects will drop out before completion of the trials; | |
| ● | proximity and availability of clinical trial sites for prospective patients; | |
| ● | ability to monitor subjects adequately during and after treatment; and | |
| ● | patient referral practices of physicians. |
Our inability to enroll a sufficient number of patients for clinical trials would result in significant delays and could require us to abandon one or more clinical trials altogether. Enrollment delays in these clinical trials may result in increased development costs for our product candidates, which could materially affect our financial condition.
New federal laws or regulations on drug importation could make lower cost versions of our future products available, which could adversely affect our revenues, if any.
The prices of some drugs are lower in other countries than in the United States because of government regulation and market conditions. Under current law, importation of drugs into the United States is generally not permitted unless the drugs are approved in the United States and the entity that holds that approval consents to the importation. Various proposals have been advanced to permit the importation of drugs from other countries to provide lower cost alternatives to the products available in the United States. In addition, the MMA requires the Secretary of Health and Human Services to promulgate regulations for drug re-importation from Canada into the United States under some circumstances, including when the drugs are sold at a lower price than in the United States.
If the laws or regulations are changed to permit the importation of drugs into the United States in circumstances that are currently not permitted, such a change could have an adverse effect on our abilitybusiness by making available lower priced alternatives to our future products.
Failure to obtain regulatory and pricing approvals in foreign jurisdictions could delay or prevent commercialization of our products abroad.
If we succeed in developing any products, we intend to market any of our potential products is dependent upon receiving marketingthem in the European Union and other foreign jurisdictions. In order to do so, we must obtain separate regulatory approvals and comply with numerous and varying regulatory requirements. The approval procedure varies among countries and can involve additional testing. The time required to obtain approval abroad may differ from the appropriate regulatory authorities. Thesethat required to obtain FDA approval. The foreign regulatory approval processesprocess may include all of the risks associated with obtaining FDA approval and additional risks associated with requirements particular to those foreign jurisdictions where we will seek regulatory approval of our products. We may not obtain foreign regulatory approvals on a timely basis, if at all. Approval by the FDA does not ensure approval process described above.by regulatory authorities in other countries, and approval by one foreign regulatory authority does not ensure approval by regulatory authorities in other foreign countries or by the FDA. We and our collaborators may not be able to file for regulatory approvals and may not receive necessary approvals to commercialize our products in any market outside the United States. The failure to obtain these approvals could materially adversely affect our business, financial condition and results of operations.
| 25 |
It is uncertain whether product liability insurance will be adequate to address product liability claims, or that insurance against such claims will be affordable or available on acceptable terms in the future.
Clinical research involves the testing of new drugs on human volunteers pursuant to a clinical trial protocol. Such testing involves a risk of liability for personal injury to or death of patients due to, among other causes, adverse side effects, improper administration of the new drug, or improper volunteer behavior. Claims may arise from patients, clinical trial volunteers, consumers, physicians, hospitals, companies, institutions, researchers, or others using, selling, or buying our products, as well as from governmental bodies. In addition, product liability and related risks are likely to increase over time, in particular upon the commercialization or marketing of any products by us or parties with which we enter into development, marketing, or distribution collaborations. Although we are contracting for general liability insurance in connection with our ongoing business, there can be no assurance that the amount and scope of such insurance coverage will be appropriate and sufficient in the event any claims arise, that we will be able to secure additional coverage should we attempt to do so, or that our insurers would not contest or refuse any attempt by us to collect on such insurance policies. Furthermore, there can be no assurance that suitable product liability insurance (at the clinical stage and/or commercial stage) will continue to be available on terms acceptable to us or at all, or that, if obtained, the insurance coverage will be appropriate and sufficient to cover any potential claims or liabilities.
If the market opportunities for our current and potential future drug candidates are smaller than we believe they are, our ability to generate product revenues will be adversely affected and our business may suffer.
Our understanding of the number of people who suffer from stress-related indications, including, but not limited to: treatment resistant depression (“TRD”), which is a subgroup of major depressive disorder (“MDD”); addiction, recidivism, or substance use disorder (“SUD”); anxiety, including generalized anxiety disorder (“GAD”), and post-traumatic stress disorder (“PTSD”) is based upon estimates. These estimates may prove to be incorrect, and new studies may demonstrate or suggest a lower estimated incidence or prevalence of this condition. The number of patients in the U.S. or elsewhere may turn out to be lower than expected, may not be otherwise amenable to PT00114 treatment, or treatment-amenable patients may become increasingly difficult to identify and access, all of which would adversely affect our business prospects and financial condition.
Risks Related to Our Reliance on Third Parties
We may not be able to obtain and maintain the third party relationships that are necessary to develop, commercialize and manufacture some or all of our product candidates.
We expect to depend on collaborators, partners, licensees, clinical research organizations, manufacturers and other third parties to support our discovery efforts, to formulate product candidates, to conduct clinical trials for some or all of our product candidates, to manufacture clinical and commercial scale quantities of our product candidates and products and to market, sell, and distribute any products we successfully develop.
We cannot guarantee that we will be able to successfully negotiate agreements for or maintain relationships with collaborators, partners, licensees, clinical investigators, manufacturers and other third parties on favorable terms, if at all. If we are unable to receiveobtain or maintain these agreements, we may not be able to clinically develop, formulate, manufacture, obtain regulatory approvals for or commercialize our product candidates, which will in turn adversely affect our business.
We expect to expend substantial management time and effort to enter into relationships with third parties and, if we successfully enter into such relationships, to manage these relationships. In addition, substantial amounts of our expenditures will be paid to third parties in these relationships. However, we cannot control the amount or timing of resources our contract partners will devote to our research and development programs, product candidates or potential product candidates, and we cannot guarantee that these parties will fulfill their obligations to us under these arrangements in a timely fashion, if at all.
| 26 |
We have no experience in sales, marketing and distribution and may have to enter into agreements with third parties to perform these functions, which could prevent us from successfully commercializing our product candidates.
We currently have no sales, marketing or distribution capabilities. To commercialize our product candidates, we must either develop our own sales, marketing and distribution capabilities, which will be expensive and time consuming, or make arrangements with third parties to perform these services for us. If we decide to market any of our products on our own, we will have to commit significant resources to developing a marketing and sales force and supporting distribution capabilities. If we decide to enter into arrangements with third parties for performance of these services, we may find that they are not available on terms acceptable to us, or at all. If we are not able to establish and maintain successful arrangements with third parties or build our own sales and marketing infrastructure, we may not be unableable to commercialize our product candidates which would adversely affect our business and financial condition.
Data provided by collaborators and other parties upon which we rely have not been independently verified and could turn out to be inaccurate, misleading, or incomplete.
We rely on third-party vendors, scientists, and collaborators to provide us with significant data and other information related to our projects, clinical trials, and business. We do not independently verify or audit all of such data (including possibly material portions thereof). As a result, such data may be inaccurate, misleading, or incomplete.
In certain cases, we may need to rely on a single supplier for a particular manufacturing material or service, and any interruption in or termination of service by such supplier could delay or disrupt the commercialization of our products.
We rely on third-party suppliers for the materials used to manufacture our compounds. Some of these materials may at times only be available from one supplier. Any interruption in or termination of service by such single source suppliers could result in a delay or disruption in manufacturing until we locate an alternative source of supply. There can be no assurance that we would be successful in locating an alternative source of supply or in negotiating acceptable terms with such prospective supplier.
We rely on third parties to conduct our non-clinical studies and our businessclinical trials. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may fail.be unable to obtain regulatory approval for or commercialize our current product candidates or any future products, on a timely basis or at all, and our financial condition will be adversely affected.
We do not have the ability to independently conduct non-clinical studies and clinical trials. We rely on medical institutions, clinical investigators, contract laboratories, collaborative partners and other third parties, such as contract research organizations or clinical research organizations, to conduct non-clinical studies and clinical trials on our product candidates. The third parties with whom we contract for execution of our non-clinical studies and clinical trials play a significant role in the conduct of these studies and trials and the subsequent collection and analysis of data. However, these third parties are not our employees, and except for contractual duties and obligations, we have limited ability to control the amount or timing of resources that they devote to our programs.
Although we rely on third parties to conduct our non-clinical studies and clinical trials, we remain responsible for ensuring that each of our non-clinical studies and clinical trials is conducted in accordance with its investigational plan and protocol. Moreover, the FDA, EMA and other foreign regulatory authorities require us to comply with regulations and standards, including some regulations commonly referred to as good clinical practices (“GCPs”), for conducting, monitoring, recording and reporting the results of clinical trials to ensure that the data and results are scientifically credible and accurate, and that the trial subjects are adequately informed of the potential risks of participating in clinical trials.
In addition, the execution of non-clinical studies and clinical trials, and the subsequent compilation and analyses of the data produced, requires coordination among various parties. In order for these functions to be carried out effectively and efficiently, it is imperative that these parties communicate and coordinate with one another. Moreover, these third parties may also have relationships with other commercial entities, some of which may compete with us. Under certain circumstances, these third parties may be able to terminate their agreements with us upon short notice. If the third parties conducting our clinical trials do not perform their contractual duties or obligations, experience work stoppages, do not meet expected deadlines, terminate their agreements with us or need to be replaced, or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical trial protocols or GCPs, or for any other reason, we may need to enter into new arrangements with alternative third parties, which could be difficult, costly or impossible, and our clinical trials may be extended, delayed or terminated or may need to be repeated. If any of the foregoing were to occur, we may not be able to obtain, on a timely basis or at all, regulatory approval for or to commercialize the product candidate being tested in such trials, and as a result, our financial condition will be adversely affected.
| 27 |
Risks Related to Commercialization of Our Product Candidates
We have no experience as a company in commercializing any product. If we fail to obtain commercial expertise, upon product approval by regulatory agencies, our product launch and revenues could be delayed.
As a company, we have never obtained regulatory approval for, or commercialized, any product. Accordingly, we have not yet begun to build out any sales or marketing capabilities. If we are unable to establish, or contract for, effective sales and marketing capabilities, or if we are unable to enter into agreements with third parties to commercialize our product candidates on favorable terms or on any reasonable terms at all, we may not be able to effectively generate product revenues once our product candidates are approved for marketing. If we fail to obtain commercial expertise or capabilities, upon drug approval, our product launch and subsequent revenues could be delayed and /or fail to reach their commercial potential.
We may not be able to gain market acceptance of our product candidates, which would prevent us from becoming profitable.
We cannot be certain that any of our product candidates will gain market acceptance among physicians, patients, healthcare payers, pharmaceutical companies or others. Demonstrating the safety and efficacy of our product candidates and obtaining regulatory approvals will not guarantee future revenue. Sales of medical products largely depend on the reimbursement of patients’ medical expenses by government healthcare programs and private health insurers. Governments and private insurers closely examine medical products to determine whether they should be covered by reimbursement and if so, the level of reimbursement that will apply. We cannot be certain that third party payers will sufficiently reimburse sales of our products, or enable us to sell our products at profitable prices. Similar concerns could also limit the reimbursement amounts that health insurers or government agencies in other countries are prepared to pay for our products. In many countries where we plan to market our products, including Europe and Canada, the pricing of prescription drugs is controlled by the government or regulatory agencies. Regulatory agencies in these countries could determine that the pricing for our products should be based on prices of other commercially available drugs for the same disease, rather than allowing us to market our products at a premium as new drugs. Sales of medical products also depend on physicians’ willingness to prescribe the treatment, which is likely to be based on a determination by these physicians that the products are safe, therapeutically effective and cost-effective. We cannot predict whether physicians, other healthcare providers, government agencies or private insurers will determine that our products are safe, therapeutically effective and cost effective relative to competing treatments.
We may not be able to manufacture our product candidates in clinical or commercial quantities, which would prevent us from commercializing our product candidates.
To date, our product candidates have been manufactured in small quantities by us and third party manufacturers for preclinical studies. If any of our product candidates is approved by the FDA or other regulatory agencies for commercial sale, we will need to manufacture it in larger quantities and we intend to use third party manufacturers for commercial quantities. Our third party manufacturers may not be able to successfully increase the manufacturing capacity for any of our product candidates in a timely or efficient manner, or at all. If we are unable to successfully increase the manufacturing capacity for a product candidate, the regulatory approval or commercial launch of that product candidate may be delayed or there may be a shortage in the supply of the product candidate. Our failure or the failure of our third party manufacturers to comply with the FDA’s good manufacturing practices and to pass inspections of the manufacturing facilities by the FDA or other regulatory agencies could seriously harm our business.
We may not be able to obtain and maintain the third party relationships that are necessary to develop, commercialize and manufacture some or all of our product candidates.
We expect to depend on collaborators, partners, licensees, clinical research organizations, manufacturers and other third parties to support our discovery efforts, to formulate product candidates, to conduct clinical trials for some or all of our product candidates, to manufacture clinical and commercial scale quantities of our product candidates and products and to market, sell, and distribute any products we successfully develop.
We cannot guarantee that we will be able to successfully negotiate agreements for or maintain relationships with collaborators, partners, licensees, clinical investigators, manufacturers and other third parties on favorable terms, if at all. If we are unable to obtain or maintain these agreements, we may not be able to clinically develop, formulate, manufacture, obtain regulatory approvals for or commercialize our product candidates, which will in turn adversely affect our business.
We expect to expend substantial management time and effort to enter into relationships with third parties and, if we successfully enter into such relationships, to manage these relationships. In addition, substantial amounts of our expenditures will be paid to third parties in these relationships. However, we cannot control the amount or timing of resources our contract partners will devote to our research and development programs, product candidates or potential product candidates, and we cannot guarantee that these parties will fulfill their obligations to us under these arrangements in a timely fashion, if at all.
We have no experience in sales, marketing and distribution and may have to enter into agreements with third parties to perform these functions, which could prevent us from successfully commercializing our product candidates.
We currently have no sales, marketing or distribution capabilities. To commercialize our product candidates, we must either develop our own sales, marketing and distribution capabilities, which will be expensive and time consuming, or make arrangements with third parties to perform these services for us. If we decide to market any of our products on our own, we will have to commit significant resources to developing a marketing and sales force and supporting distribution capabilities. If we decide to enter into arrangements with third parties for performance of these services, we may find that they are not available on terms acceptable to us, or at all. If we are not able to establish and maintain successful arrangements with third parties or build our own sales and marketing infrastructure, we may not be able to commercialize our product candidates which would adversely affect our business and financial condition.
We may fail to select or capitalize on the most scientifically, clinically or commercially promising or profitable product candidates.
We have limited technical, managerial and financial resources to determine which of our product candidates should proceed to initial clinical trials, later stage clinical development and potential commercialization. We may make incorrect determinations. Our decisions to allocate our research and development, management and financial resources toward particular product candidates or therapeutic areas may not lead to the development of viable commercial products and may divert resources from better opportunities. Similarly, our decisions to delay or terminate drug development programs may also be incorrect and could cause us to miss valuable opportunities.
We may not be able to maintainaccepted for reimbursement or properly reimbursed by third-party payers.
The successful commercialization of any products we might develop will depend substantially on whether the costs of our exclusive worldwide license to useproducts and develop PT00114 which could materially affect our business plan.
On July 21, 2005, we entered into the License Agreement with UT pursuant to which UT agreed to license to us patent rightsrelated treatments are reimbursed at acceptable levels by government authorities, private healthcare insurers, and other intellectual property related to PT00114, among other things. The Technology License Agreement was amended on February 18, 2015. There is no expiration date to this agreementthird-party payers, such as long as we continue to provide UT with progress reports every six months and make ongoing progress toward development of the drug.
Pursuant to the License Agreement, we obtained an exclusive worldwide license to make, have made, use, sell and import products basedhealth maintenance organizations. Reimbursement rates may vary, depending upon the Technologies,third-party payer, the type of insurance plan, and other similar or to sublicensedissimilar factors. If our products do not achieve adequate reimbursement, then the Technologies in accordance with the termsnumber of the License Agreement. In the event we fail to provide UT with semi-annual reports on our progress or fail to continue to make reasonable commercial efforts towards obtaining regulatory approval for products based on the Technologies, UT may convert our exclusive license into a non-exclusive one. In such a case, we would lose our competitive advantage in the development of treatments based on PT00114.
If we are not able to retain our current senior management team and our scientific advisors or continue to attract and retain qualified scientific, technical and business personnel, our business will suffer.
We are dependent on the membersphysician prescriptions of our management team and our scientific advisors for our business success. An important element of our strategy is to take advantage of the research and development expertise of our current management and to utilize the unique expertise of our scientific advisors. We do not have any employment agreements with our executive officers. The loss of any one of our executive officers or key scientific consultants, including, in particular, Garo Armen, Ph.D., Chairman of the Board, and Dr. David A. Lovejoy, our Chief Scientific Advisor, could result in a significant loss in the knowledge and experience that we, as an organization, possess and could cause significant delays, or outright failure, in the development and further commercialization of our product candidates.
To grow, we will eventually need to hire a significant number of qualified commercial, scientific and administrative personnel. However, there is intense competition for human resources, including management in the technical fields in which we operate, and weproducts may not be ablesufficient to attract and retain qualified personnel necessary formake our products profitable.
Comparative effectiveness research demonstrating benefits of a competitor’s product could adversely affect the successful development and commercializationsales of our product candidates. Our inabilityIf third-party payers do not consider our products to attractbe cost-effective compared to other available therapies, they may not cover our products as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow us to sell our products on a profitable basis.
Adequate third-party reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in the product development of that product. In addition, in the U.S. there is a growing emphasis on comparative effectiveness research, both by private payers and by government agencies. To the extent other drugs or therapies are found to be more effective than our products, payers may elect to cover such therapies in lieu of our products or reimburse our products at a lower rate.
New federal legislation may increase the pressure to reduce prices of pharmaceutical products paid for by Medicare, which could adversely affect our revenues, if any.
The Medicare Prescription Drug Improvement and Modernization Act of 2003, or MMA, expanded Medicare coverage for drug purchases by the elderly and disabled beginning in 2006. The new employeeslegislation uses formularies, preferred drug lists and similar mechanisms that may limit the number of drugs that will be covered in any therapeutic class or reduce the reimbursement for some of the drugs in a class. More recently, the Patient Protection and Affordable Care Act of 2010 also contained certain provisions with the potential to retain existing employeesaffect pricing of pharmaceutical products.
As a result of the expansion of legislation, including recent healthcare insurance legislation, and the expansion of federal coverage of drug products, we expect that there will be additional pressure to contain and reduce costs. These cost reduction initiatives could limitdecrease the coverage and price that we receive for our growthproducts in the future and could seriously harm our business.
We have not entered into an employment agreement with Dr. David A. Lovejoy, our Chief Scientific Advisor.
Dr. David A. Lovejoy is a key contributor While the MMA applies only to our Company due to his roledrug benefits for Medicare beneficiaries, private payers often follow Medicare coverage policy and payment limitations in setting their own reimbursement systems, and any limits on or reductions in reimbursement that occur in the development of PT00114 and his continued roleMedicare program may result in the development of our products as our Chief Scientific Advisor. We have not entered into an employment agreement with Dr. Lovejoy. If Dr. Lovejoy elects to discontinue his service as our Chief Scientific Advisor, the development of our products and our overall business plan could be materially affected.similar limits on or reductions in payments from private payers.
Disputes under key agreements or conflicts of interest with our scientific advisors or clinical investigators could delay or prevent development or commercialization of our product candidates.
Any agreements we have or may enter into with third parties, such as collaboration, license, formulation supplier, manufacturing, clinical research organization or clinical trial agreements, may give rise to disputes regarding the rights and obligations of the parties. Disagreements could develop over rights to ownership or use of intellectual property, the scope and direction of research and development, the approach for regulatory approvals or commercialization strategy. We intend to conduct research programs in a range of therapeutic areas, but our pursuit of these opportunities could result in conflicts with the other parties to these agreements who may be developing or selling pharmaceuticals or conducting other activities in these same therapeutic areas. Any disputes or commercial conflicts could lead to the termination of our agreements, delay progress of our product development programs, compromise our ability to renew agreements or obtain future agreements, lead to the loss of intellectual property rights or result in costly litigation.
We collaborate with outside scientific advisors and collaborators at academic and other institutions that assist us in our research and development efforts. Our scientific advisors are not our employees and may have other commitments that limit their availability to us. If a conflict of interest between their work for us and their work for another entity arises, we may lose their services.
| 29 |
WeOur competitors and potential competitors may encounter difficultiesdevelop products and technologies that make ours less attractive or obsolete.
Many companies, universities, and research organizations developing competing product candidates have greater resources and significantly greater experience in managingfinancial, research and development, manufacturing, marketing, sales, distribution, and technical regulatory matters than we have. In addition, many competitors have greater name recognition and more extensive collaborative relationships. Our competitors could commence and complete clinical testing of their product candidates, obtain regulatory approvals, and begin commercial-scale manufacturing of their products faster than we are able to for our growth, whichproducts. They could develop products that would render our product candidates, and those of our collaborators, obsolete and noncompetitive. If we are unable to compete effectively against these companies, then we may not be able to commercialize our product candidates or achieve a competitive position in the market. This would adversely affect our operations.
Our ability to manage our operationsgenerate revenues.
Competition in the biotechnology and growth effectively depends uponpharmaceutical industries may result in competing products, superior marketing of other products and lower revenues or profits for us.
There are many companies that are seeking to develop products and therapies for the continual improvementtreatment of mood, anxiety and neurodegenerative disorders. Many of our procedures, reporting systems,competitors have substantially greater financial, technical, human and operational, financial,other resources than we do and management controls.may be better equipped to develop, manufacture and market technologically superior products. In addition, many of these competitors have significantly greater experience than we do in undertaking preclinical testing and human clinical studies of new pharmaceutical products and in obtaining regulatory approvals of human therapeutic products. Accordingly, our competitors may succeed in obtaining FDA approval for superior products.
Other risks and uncertainties include:
| ● | our ability to successfully complete preclinical and clinical development of our products and services | |
| ● | our ability to manufacture sufficient amounts of products for development and commercialization activities | |
| ● | our ability to obtain, maintain and successfully enforce adequate patent and other proprietary rights protection of our products and services | |
| ● | the scope, validity and enforceability of patents and other proprietary rights held by third parties and their impact on our ability to commercialize our products and services | |
| ● | the accuracy of our estimates of the size and characteristics of the markets to be addressed by our products and services, including growth projections | |
| ● | market acceptance of our products and services | |
| ● | our ability to identify new patients for our products and services | |
| ● | the accuracy of our information regarding the products and resources of our competitors and potential competitors | |
| ● | the content and timing of submissions to and decisions made by the US Food and Drug Administration (FDA) and other regulatory agencies | |
| ● | our ability to obtain reimbursement for our products and services from third-party payors, and the extent of such coverage | |
| ● | our ability to establish and maintain strategic license, collaboration and distribution arrangements | |
| ● | the continued funding of our collaborations and joint ventures, if any are ultimately established | |
| ● | the possible disruption of our operations due to terrorist activities and armed conflict, including as a result of the disruption of operation of our subsidiaries and our customers, suppliers, distributors, couriers, collaborative partners, licensees and clinical trial sites. |
Positive or timely results from preclinical studies and early clinical trials do not ensure positive or timely results in late-stage clinical trials or product approval by the FDA or any other regulatory authority. Product candidates that show positive preclinical or early clinical results often fail in later stage clinical trials. Data obtained from preclinical and clinical activities is susceptible to varying interpretations, which could delay, limit, or prevent regulatory approvals.
We have limited experience in conducting the clinical trials required to obtain regulatory approval. We may not be able to implement improvementsconduct clinical trials at preferred sites, enlist clinical investigators, enroll sufficient numbers of participants, or begin or successfully complete clinical trials in an efficienta timely fashion, if at all. Any failure to perform may delay or timely mannerterminate the trials. Our current clinical trials may be insufficient to demonstrate that our potential products will be active, safe, or effective. Additional clinical trials may be required if clinical trial results are negative or inconclusive, which will require us to incur additional costs and may discover deficiencies in existing systems and controls.significant delays. If we do not meet these challenges,receive the necessary regulatory approvals, we may be unable to take advantage of market opportunities, execute our business strategies or respond to competitive pressures which in turn may slow our growth or give rise to inefficiencies that would increase our losses.
We may acquire additional technology and complementary businesses in the future. Acquisitions involve many risks, any one of which could materially harm our business, including the diversion of management’s attention from core business concerns, failure to exploit acquired technologies, or the loss of key employees from either our business or the acquired business.
Company Risks
We have a history of losses and expect that losses may continue in the future.
We have generated net losses since we began operations, including $2,259,636 and $2,275,826 for the years ended December 31, 2017 and December 31, 2016, respectively. As of December 31, 2017, we had an accumulated deficit of $10,841,759. We have no approved products and have generated no product revenue. We expect that product development, preclinical and clinical programs will increase losses significantly over the next five years. In order to achieve profitability, we will need to generate significant revenue. We cannot be certain that we will generate sufficient revenue to achieve profitability. We anticipate that we will continue to generate operating losses and negative cash flow from operations and our current cash position is sufficient to fund our current business plan at least until the third quarter of 2018. We cannot be certain that we will ever achieve, or if achieved, maintain profitability. If our revenue grows at a slower rate than we anticipate or if our product development, marketing and operating expenses exceed our expectations or cannot be adjusted accordingly, our business, results of operation and financial condition will be materially adversely affected and we may be unable to continue operations.
We will not be able to generate product revenue unlessrevenues and until one of our product candidates successfully completes clinical trials and receives regulatory approval. As our most advanced product candidates are at an early proof-of-concept stage, we domay not expectbecome profitable.
| 30 |
Risks Related to receive revenue from any product candidate for the foreseeable future. Our Intellectual Property
We may seek to obtain revenue from collaboration or licensing agreements with third parties. We currently have no such agreements which will provide us with material, ongoing future revenue and we may never enter into any such agreements. Even if we eventually generate revenues, we may never be profitable, and if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis.
We needmaintain our exclusive worldwide license to obtain financing in order to continue our operations.
On a prospective basis, we will require both short-term financing for operationsuse and long-term capital to fund our expected growth. We have no existing bank lines of credit and have not established any definitive sources for additional financing. We believe that cash on hand provided by the investors in the Private Offering will be sufficient to meet our short-term financial requirements for approximately twelve months. However, we will require additional funds if we want to fully implement our business plan and proceed with submission of an IND/CTA application. Additional financing may not be available to us, or if available, then it may not be available upon terms and conditions acceptable to us. If adequate funds are not available, then we may be required to delay, reduce or eliminate product development or clinical programs. Our inability to take advantage of opportunities in the industry because of capital constraints may have a material adverse effect on our business and our prospects. If we fail to obtain the capital necessary to fund our operations, we will be unable to advance our development programs and complete our clinical trials.
In addition, our research and development expensesdevelop PT00114 which could exceed our current expectations. This could occur for many reasons, including:
While we expect to seek additional funding through public or private financings, we may not be able to obtain financing on acceptable terms, or at all. In addition, the terms of our financings may be dilutive to, or otherwise adverselymaterially affect holders of our common stock. We may also seek additional funds through arrangements with collaborators or other third parties. These arrangements would generally require us to relinquish rights to some of our technologies, product candidates or products, and we may not be able to enter into such agreements, on acceptable terms, if at all. If we are unable to obtain additional funding on a timely basis, we may be required to curtail or terminate some or all of our development programs, including some or all of our product candidates.
We currently do not have sufficient cash to fully implement our business plan.
WeOn July 21, 2005, we entered into the License Agreement with University of Toronto, or UT, pursuant to which UT agreed to license to us patent rights and other intellectual property related to PT00114, among other things. The Technology License Agreement was amended on February 18, 2015. Unless earlier terminated, the term of this agreement shall terminate on the expiration or invalidity of the last issued Patent in the Agreement
Pursuant to the License Agreement, we obtained an exclusive worldwide license to make, have experienced a lackmade, use, sell and import products based upon the Technologies, or to sublicense the Technologies in accordance with the terms of adequate capital resources causing usthe License Agreement. In the event we fail to be unable to fully implementprovide UT with semi-annual reports on our business plan. We believe that we need to raiseprogress or otherwise obtain additional financing beyond the Private Offering in order to satisfy our existing obligations and fully implement our business plan. We do not expect to have positive cash flow until the middle of 2018 or longer. If we are not successful in obtaining additional financing, we will not be able to fully implement our business plan and we may not be ablefail to continue our operations.
We have a limited operating history and a history of operating losses, and expect to incur significant additional operating losses.
We began our business in September 2004 and have a limited operating history. Though we have enlisted the assistance of pharmaceutical and academic experts, our lack of experience may cause us to encounter unforeseen problems that could have a material adverse effect on our business and financial condition. As well, there is limited historical financial information upon which to base an evaluation of our performance.
The Company’s financial statements have been prepared on a going concern basis, and do not include adjustments that might be necessary if the Company is unable to continue as a going concern.
The Company’s consolidated financial statements have been prepared on a going concern basis, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. As of December 31, 2017, the Company had incurred significant operating losses since inception, and continues to generate losses from operations, and has an accumulated deficit of $10,841,759. These matters raise substantial doubt about the Company’s ability to continue as a going concern. The consolidated financial statements incorporated in this annual report do not include any adjustments relating to the recoverability and classification of asset amounts or the classification of liabilities that might be necessary should the Company be unable to continue as a going concern.
The drug development andmake reasonable commercial efforts towards obtaining regulatory approval process is uncertain, time-consuming and expensive.
The process of obtaining and maintaining regulatory approvals for new therapeutic products is lengthy, expensive and uncertain. It also can vary substantially based on the type, complexity, and noveltyTechnologies, UT may convert our exclusive license into a non-exclusive one. In such a case, we would lose our competitive advantage in the development of the product. We must provide the FDA and foreign regulatory authorities with preclinical and clinical data demonstrating that our products are safe and effective before they can be approved for commercial sale. Clinical development, including preclinical testing, is a long, expensive and uncertain process. It may take us several years to complete our testing, and failure can occur at any stage of testing. Any preclinical or clinical test may fail to produce results satisfactory to the FDA. Preclinical and clinical data can be interpreted in different ways, which could delay, limit or prevent regulatory approval. Negative or inconclusive results from a preclinical study or clinical trial, adverse medical events during a clinical trial or safety issues resulting from products of the same class of drug could cause a preclinical study or clinical trial to be repeated or a program to be terminated, even if other studies or trials relating to the program are successful.treatments based on PT00114.
We have to sustain and further build our intellectual property rights.
If we fail to sustain and further build our intellectual property rights, competitors will be able to take advantage of our research and development efforts to develop competing products. If we are not able to protect our proprietary technology, trade secrets, and know-how, our competitors may use our inventions to develop competing products. Protagenic has obtained worldwide exclusive rights to PT00114 and related technology that was developed at the UniversityUT. As of Toronto. The Company currently hasDecember 31, 2023, we have four patents issued by the Governments of the United States, Canada, European Union (validated in Germany, France and Australia. AsGreat Britain) and Australia on our original platform technology, all of December 31, 2017, sixwhich have expired aside from the one in the United States. The patent applications were made in the name of Dr. David A. Lovejoy and inventors, but the Company’s exclusive, worldwide rights to such patent applications are pending. included in the License Agreement with UT. We have eight issued patents (Canada, Great Britain, Europe (GPC and additionally validated in Switzerland, Great Britain, Ireland and Spain) and the United States and five pending patent applications in related technology that the company has rights in or own.
However, our patents and patent applications, even if granted, may not protect us against our competitors. Our patent positions, and those of other pharmaceutical and biotechnology companies, are generally uncertain and involve complex legal, scientific and factual questions. The standards which the United States Patent and Trademark Office uses to grant patents, and the standards which courts use to interpret patents, are not always applied predictably or uniformly and can change, particularly as new technologies develop. Consequently, the level of protection, if any, that will be provided by our patents if we attempt to enforce them, and they are challenged, is uncertain. In addition, the type and extent of patent claims that will be issued to us in the future is uncertain. Any patents that are issued may not contain claims that permit us to stop competitors from using similar technology.
In addition to our patentable technology, we also rely on unpatented technology, trade secrets, and confidential information. We may not be able to effectively protect our rights to this technology or information. Other parties may independently develop substantially equivalent information and techniques or otherwise gain access to or disclose our technology. We generally require each of our employees, consultants, collaborators, and certain contractors to execute a confidentiality agreement at the commencement of an employment, consulting, collaborative, or contractual relationship with us. However, these agreements may not provide effective protection of our technology or information or, in the event of unauthorized use or disclosure, they may not provide adequate remedies.
Our patent position is generally uncertain and involves complex legal and factual questions. In addition, the laws of some foreign countries do not protect proprietary rights to the same extent as the laws of the United States, and other biotechnology companies have encountered significant problems in protecting and defending their proprietary rights in foreign jurisdictions. Whether filed in the United States or abroad, our patent applications may be challenged or may fail to result in issued patents. In addition, any future patents we obtain may not be sufficiently broad to prevent others from practicing our technologies or from developing or commercializing competing products. Furthermore, others may independently develop or commercialize similar or alternative technologies or drugs, or design around our patents. Our patents may be challenged, invalidated or fail to provide us with any competitive advantages. We may not have the funds available to protect our patents or other technology; such protection is costly and can result in further litigation expenses.
| 31 |
If we do not obtain or we are unable to maintain adequate patent or trade secret protection for our products in the United States, competitors could duplicate them without repeating the extensive testing that we will be required to undertake to obtain approval of the products by the FDA. Regardless of any patent protection, under the current statutory framework the FDA is prohibited by law from approving any generic version of any of our products for three years after it has approved our product. Upon the expiration of that period, or if that time period is altered, the FDA could approve a generic version of our product unless we have patent protection sufficient for us to block that generic version. Without sufficient patent protection, the applicant for a generic version of our product would be required only to conduct a relatively inexpensive study to show that its product is bioequivalent to our product and may not have to repeat the studies that we will need to conduct to demonstrate that the product is safe and effective. In the absence of adequate patent protection in other countries, competitors may similarly be able to obtain regulatory approval in those countries of products that duplicate our products.
We have to comply with our obligations in our intellectual property licenses with third parties.
If we fail to comply with our obligations in our intellectual property licenses with third parties, we could lose license rights that are important to our business.We are a party to the License Agreement with UT under which we receive the right to practice and use important third party patent rights. We may enter into additional licenses in the future. Our existing licenses impose, and we expect future licenses will impose, various diligences, milestone payment, royalty, insurance and other obligations on us. If we fail to comply with these obligations, the licensor may have the right to terminate the license, in which event we might not be able to market any product that is covered by the licensed patents.
We may need to resort to litigation to enforce or defend our intellectual property rights, including any patents issued to us. If a competitor or collaborator files a patent application claiming technology also invented by us, in order to protect our rights, we may have to participate in an expensive and time consuming interference proceeding before the United States Patent and Trademark Office. We cannot guarantee that our product candidates will be free of claims by third parties alleging that we have infringed their intellectual property rights. Third parties may assert that we are employing their proprietary technologies without authorization and they may resort to litigation to attempt to enforce their rights. Third parties may have or obtain patents in the future and claim that the use of our technology or any of our product candidates infringes their patents. We may not be able to develop or commercialize combination product candidates because of patent protection others have. Our business will be harmed if we cannot obtain a necessary or desirable license, can obtain such a license only on terms we consider to be unattractive or unacceptable, or if we are unable to redesign our product candidates or processes to avoid actual or potential patent or other intellectual property infringement. Obtaining, protecting and defending patent and other intellectual property rights can be expensive and may require us to incur substantial costs, including the diversion of management and technical personnel. An unfavorable ruling in patent or intellectual property litigation could subject us to significant liabilities to third parties, require us to cease developing, manufacturing or selling the affected products or using the affected processes, require us to license the disputed rights from third parties, or result in awards of substantial damages against us.
There can be no assurance that we would prevail in any intellectual property infringement action, will be able to obtain a license to any third party intellectual property on commercially reasonable terms, successfully develop non-infringing alternatives on a timely basis, or license non-infringing alternatives, if any exist, on commercially reasonable terms. Any significant intellectual property impediment to our ability to develop and commercialize our products could seriously harm our business and prospects.
Patent litigation or other litigation in connection with our intellectual property rights may lead to publicity that may harm our reputation and the value of our common stock may decline.
During the course of any patent litigation, there may be public announcements of the results of hearings, motions, and other interim proceedings or developments in the litigation. If securities analysts or investors regard these announcements as negative, the value of our common stock may decline. General proclamations or statements by key public figures may also have a negative impact on the perceived value of our intellectual property.
Protecting and defending against intellectual property claims may have a material adverse effect on our business.
From time to time, we may receive notice that others have infringed on our proprietary rights or that we have infringed on the intellectual property rights of others. There can be no assurance that infringement or invalidity claims will not materially adversely affect our business, financial condition or results of operations. Regardless of the validity or the success of the assertion of claims, we could incur significant costs and diversion of resources in protecting or defending against claims, which could have a material adverse effect on our business, financial condition or results of operations. We may not have the funds or resources available to protect our intellectual property.
Our competitorsIntellectual property disputes could require us to spend time and potential competitorsmoney to address such disputes and could limit our intellectual property rights.
The biopharmaceutical industry has been characterized by extensive litigation regarding patents and other intellectual property rights, and companies have employed intellectual property litigation and USPTO post-grant proceedings to gain a competitive advantage. We may develop productsbecome subject to infringement claims or litigation arising out of patents and technologies that make ours less attractive or obsolete.
Many companies, universities, and research organizations developing competing product candidates have greater resources and significantly greater experience in financial, research and development, manufacturing, marketing, sales, distribution, and technical regulatory matters than we have. In addition, many competitors have greater name recognition and more extensive collaborative relationships. Our competitors could commence and complete clinical testing of their product candidates, obtain regulatory approvals, and begin commercial-scale manufacturing of their products faster than we are able to for our products. They could develop products that would render our product candidates, and thosepending applications of our collaborators, obsoletecompetitors, or additional interference proceedings declared by the USPTO to determine the priority and noncompetitive. Ifpatentability of inventions. The defense and prosecution of intellectual property suits, USPTO proceedings, and related legal and administrative proceedings are costly and time-consuming to pursue, and their outcome is uncertain. Litigation may be necessary to enforce our issued patents, to protect our trade secrets and know-how, or to determine the enforceability, scope, and validity of the proprietary rights of others. An adverse determination in litigation or USPTO post-grant and interference proceedings to which we are unablemay become a party could subject us to compete effectively against these companies, then wesignificant liabilities, require us to obtain licenses from third parties, or restrict or prevent us from selling our products in certain markets. Even if a given patent or intellectual property dispute were settled through licensing or similar arrangements, our costs associated with such arrangements may be substantial and could include the payment by us of large fixed payments and ongoing royalties. Furthermore, the necessary licenses may not be ableavailable on satisfactory terms or at all. Even where we have meritorious claims or defenses, the costs of litigation may prevent us from pursuing these claims or defenses and/or may require extensive financial and personnel resources to commercializepursue these claims or defenses. In addition, it is possible there may be defects of form in our product candidates or achieve a competitive position in the market. This would adversely affect our ability to generate revenues.
Competition in the biotechnologycurrent and pharmaceutical industries mayfuture patents that could result in competing products, superior marketing ofour inability to defend the intended claims. Intellectual property disputes arising from the aforementioned factors, or other products and lower revenues or profits for us.factors, may materially harm our business.
There are many companies that are seeking to develop products and therapies for the treatment of mood, anxiety and neurodegenerative disorders. Many of our competitors have substantially greater financial, technical, human and other resources than we do and may be better equipped to develop, manufacture and market technologically superior products. In addition, many of these competitors have significantly greater experience than we do in undertaking preclinical testing and human clinical studies of new pharmaceutical products and in obtaining regulatory approvals of human therapeutic products. Accordingly, our competitors may succeed in obtaining FDA approval for superior products.
Other risks and uncertainties include:
Positive or timely results from preclinical studies and early clinical trials do not ensure positive or timely results in late stage clinical trials or product approval by the FDA or any other regulatory authority. Product candidates that show positive preclinical or early clinical results often fail in later stage clinical trials. Data obtained from preclinical and clinical activities is susceptible to varying interpretations, which could delay, limit, or prevent regulatory approvals.
We have limited experience in conducting the clinical trials required to obtain regulatory approval. We may not be able to conduct clinical trialsenforce our intellectual property rights throughout the world.
The laws of some foreign countries do not protect intellectual property rights to the same extent as the laws of the U.S. Companies have encountered significant problems in protecting and defending intellectual property rights in certain foreign jurisdictions. The legal systems of some countries, particularly developing countries, do not favor the enforcement of patents and other intellectual property protection, especially those relating to life sciences. This could make it difficult for us to stop the infringement of our patents or the misappropriation of our other intellectual property rights. For example, many foreign countries have compulsory licensing laws under which a patent owner must grant licenses to third parties. In addition, many countries limit the enforceability of patents against third parties, including government agencies or government contractors. In these countries, patents may provide limited or no benefit.
Proceedings to enforce our patent rights in foreign jurisdictions, whether or not successful, could result in substantial costs and divert our efforts and attention from other aspects of our business. Furthermore, while we intend to protect our intellectual property rights in our expected significant markets, we cannot ensure that we will be able to initiate or maintain similar efforts in all jurisdictions in which we may wish to market PT00114 or any future products. Accordingly, our efforts to protect our intellectual property rights in such countries may be inadequate. In addition, changes in the law and legal decisions by courts in the U.S. and foreign countries may affect our ability to obtain and enforce adequate intellectual property protection for our products and technology.
| 33 |
Changes to the patent law in the U.S. and other jurisdictions could diminish the value of patents in general, thereby impairing our ability to protect our product candidates.
As is the case with other biopharmaceutical companies, our success is heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the biopharmaceutical industry involves both technological and legal complexity. Therefore, obtaining and enforcing biopharmaceutical patents is costly, time consuming and inherently uncertain. In addition, the U.S. has recently enacted and is currently implementing wide ranging patent reform legislation. The U.S. Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. In addition to increasing uncertainty with regard to our ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents once obtained. Depending on future actions by the U.S. Congress, the federal courts and the United States Patent and Trademark Office (“USPTO”), as well as other jurisdictions around the world, the laws and regulations governing patents could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce our existing patents and patents that we might obtain in the future.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
The USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other provisions during the patent process. There are situations in which noncompliance can result in abandonment or lapse of a patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, competitors might be able to enter the market earlier than would otherwise have been the case.
If we fail to comply with our obligations under any license, collaboration or other intellectual property-related agreements, we may be required to pay damages and could lose intellectual property rights that may be necessary for developing, commercializing and protecting our current or future technologies or drug candidates or we could lose certain rights to grant sublicenses.
Any license, collaboration or other intellectual property-related agreements impose, and any future license, collaboration or other intellectual property-related agreements we enter into are likely to impose, various development, commercialization, funding, milestone, royalty, diligence, sublicensing, insurance, patent prosecution and enforcement or other obligations on us. If we breach any of these obligations, or use the intellectual property licensed to us in an unauthorized manner, we may be required to pay damages and the licensor may have the right to terminate the license. In spite of our best efforts, any of our future licensors might conclude that we have materially breached our license agreements and might therefore terminate the license agreements, thereby removing our ability to develop and commercialize products and technologies covered by these license agreements. Any license agreements we enter into may be complex, and certain provisions in such agreements may be susceptible to multiple interpretations. The resolution of any contract interpretation disagreement that may arise could narrow what we believe to be the scope our rights to the relevant intellectual property or technology, or increase what we believe to be our financial or other obligations under the relevant agreement, either of which could have a material adverse effect on our business, financial condition, results of operations, and prospects.
We may seek to obtain licenses from licensors in the future, however, we may be unable to obtain any such licenses at preferred sites, enlist clinical investigators, enroll sufficient numbers of participants,a reasonable cost or begin or successfully complete clinical trials in a timely fashion,on reasonable terms, if at all. In addition, if any of our future licensors terminate any such license agreements, such license termination could result in our inability to develop, manufacture and sell products that are covered by the licensed technology or could enable a competitor to gain access to the licensed technology. Any failureof these events could have a material adverse effect on our competitive position, business, financial condition, results of operations, and ability to performachieve profitability.
Furthermore, we may delaynot have the right to control the preparation, filing, prosecution, maintenance, enforcement and defense of patents and patent applications that we license from third parties. Therefore, we cannot be certain that these patents and patent applications will be prepared, filed, prosecuted, maintained, enforced and defended in a manner consistent with the best interests of our business. If our future licensors fail to prosecute, maintain, enforce and defend patents we may in-license, or terminate the trials. Our current clinical trialslose rights to licensed patents or patent applications, our license rights may be insufficientreduced or eliminated. In such circumstances, our right to demonstratedevelop and commercialize any of our products or drug candidates that is the subject of such licensed rights could be materially adversely affected. In certain circumstances, our potential products will be active, safe,licensed patent rights are subject to our reimbursing our licensors for their patent prosecution and maintenance costs.
| 34 |
Moreover, our licensors may own or effective. Additional clinical trialscontrol intellectual property that has not been licensed to us and, as a result, we may be requiredsubject to claims, regardless of their merit, that we are infringing, misappropriating or otherwise violating the licensor’s intellectual property rights and the amount of any damages or future royalty obligations that would result, if clinical trial results are negativeany such claims were successful, would depend on the technology and intellectual property we use in products that we successfully develop and commercialize, if any. Therefore, even if we successfully develop and commercialize products, due to such obligations, we may be unable to achieve or inconclusive, which will require usmaintain profitability.
Risks Related to incur additional costsOur Business Operations and significant delays. Industry
If we are not able to retain our current senior management team and our scientific advisors or continue to attract and retain qualified scientific, technical and business personnel, our business will suffer.
We are dependent on the members of our management team and our scientific advisors for our business success. An important element of our strategy is to take advantage of the research and development expertise of our current management and to utilize the unique expertise of our scientific advisors. We do not receivehave any employment agreements with our executive officers. The loss of any one of our executive officers or key scientific consultants, including, in particular, Garo Armen, Ph.D., Chairman of the necessary regulatory approvals,Board, and Dr. David A. Lovejoy, our Chief Scientific Advisor, could result in a significant loss in the knowledge and experience that we, as an organization, possess and could cause significant delays, or outright failure, in the development and further commercialization of our product candidates.
To grow, we will not be ableeventually need to generate product revenueshire a significant number of qualified commercial, scientific and may not become profitable.
The regulatory approval processadministrative personnel. However, there is costly and lengthyintense competition for human resources, including management in the technical fields in which we operate, and we may not be able to successfully obtain all required regulatory approvals.
The preclinicalattract and retain qualified personnel necessary for the successful development clinical trials, manufacturing, marketing and labeling of pharmaceuticals are all subject to extensive regulation by numerous governmental authorities and agencies in the United States and other countries. We must obtain regulatory approval for eachcommercialization of our product candidates before marketingcandidates. Our inability to attract new employees or selling any of them. It is not possible to predict how long the approval processes of the FDA or any other applicable federal or foreign regulatory authority or agency for any ofretain existing employees could limit our products will take or whether any such approvals ultimately will be granted. The FDAgrowth and foreign regulatory agencies have substantial discretionharm our business.
We may encounter difficulties in the drug approval process, and positive results in preclinical testing or early phases of clinical studies offer no assurance of success in later phases of the approval process. Generally, preclinical and clinical testing of products can take many years and require the expenditure of substantial resources, and the data obtained from these tests and trials can be susceptible to varying interpretations that could delay, limit or prevent regulatory approval. If we encounter significant delays in the regulatory process that result in excessive costs, this may prevent us from continuing to developmanaging our product candidates. Any delay in obtaining, or failure to obtain, approvalsgrowth, which could adversely affect our operations.
Our ability to manage our operations and growth effectively depends upon the marketingcontinual improvement of our productsprocedures, reporting systems, and our abilityoperational, financial, and management controls. We may not be able to generate product revenue. The risks associated with the approval process include:
Even if we receive regulatory approvals for marketing our product candidates, if we fail to comply with continuing regulatory requirements, we could lose our regulatory approvals,implement improvements in an efficient or timely manner and our business would be adversely affected.
The FDA continues to review products even after they receive initial approval.may discover deficiencies in existing systems and controls. If we receive approval to commercialize any product candidates, the manufacturing, marketing and sale ofdo not meet these drugs will be subject to continuing regulation, including compliance with quality systems regulations, good manufacturing practices, adverse event requirements, and prohibitions on promoting a product for unapproved uses. Enforcement actions resulting from our failure to comply with government and regulatory requirements could result in fines, suspension of approvals, withdrawal of approvals, product recalls, product seizures, mandatory operating restrictions, criminal prosecution, civil penalties and other actions that could impair the manufacturing, marketing and sale of our potential products and our ability to conduct our business.
Even if we are able to obtain regulatory approvals for any of our product candidates, if they exhibit harmful side effects after approval, our regulatory approvals could be revoked or otherwise negatively impacted, and we could be subject to costly and damaging product liability claims.
Even if we receive regulatory approval for our product candidates, we will have tested them in only a small number of patients during our clinical trials. If our applications for marketing are approved and more patients begin to use our product, new risks and side effects associated with our products may be discovered. As a result, regulatory authorities may revoke their approvals;challenges, we may be requiredunable to conduct additional clinical trials, make changestake advantage of market opportunities, execute our business strategies or respond to competitive pressures which in labeling ofturn may slow our product, reformulategrowth or give rise to inefficiencies that would increase our product or make changes and obtain new approvals for our and our suppliers’ manufacturing facilities. We might have to withdraw or recall our products from the marketplace. losses.
We may also experience a significant dropacquire additional technology and complementary businesses in the potential salesfuture. Acquisitions involve many risks, any one of which could materially harm our product if and when regulatory approvals for such product are obtained, experience harmbusiness, including the diversion of management’s attention from core business concerns, failure to exploit acquired technologies, or the loss of key employees from either our reputation inbusiness or the marketplace or become subject to lawsuits, including class actions. Any of these results could decrease or prevent any sales of our approved product or substantially increase the costs and expenses of commercializing and marketing our product.acquired business.
Healthcare reform measures could adversely affect our business.
The efforts of governmental and third-party payers to contain or reduce the costs of healthcare may adversely affect the business and financial condition of pharmaceutical companies. In the United States and in foreign jurisdictions there have been, and we expect that there will continue to be, a number of legislative and regulatory proposals aimed at changing the healthcare system. For example, in some countries other than the United States, pricing of prescription drugs is subject to government control, and we expect proposals to implement similar controls in the United States to continue. The pendency or approval of such proposals could result in a decrease in our common stock value or limit our ability to raise capital or to enter into collaborations or license rights to our products.
New federal legislation may increase the pressure to reduce prices of pharmaceutical products paid for by Medicare, which could adversely affect our revenues, if any.
The Medicare Prescription Drug Improvement and Modernization Act of 2003, or MMA, expanded Medicare coverage for drug purchases by the elderly and disabled beginning in 2006. The new legislation uses formularies, preferred drug lists and similar mechanisms that may limit the number of drugs that will be covered in any therapeutic class or reduce the reimbursement for some of the drugs in a class. More recently, the Patient Protection and Affordable Care Act of 2010 also contained certain provisions with the potential to affect pricing of pharmaceutical products.
As a result of the expansion of legislation, including recent healthcare insurance legislation, and the expansion of federal coverage of drug products, we expect that there will be additional pressure to contain and reduce costs. These cost reduction initiatives could decrease the coverage and price that we receive for our products in the future and could seriously harm our business. While the MMA applies only to drug benefits for Medicare beneficiaries, private payers often follow Medicare coverage policy and payment limitations in setting their own reimbursement systems, and any limits on or reductions in reimbursement that occur in the Medicare program may result in similar limits on or reductions in payments from private payers.
New federalOur business and operations are vulnerable to computer system failures, cyber-attacks or deficiencies in our cyber-security, which could increase our expenses, divert the attention of our management and key personnel away from our business operations and adversely affect our results of operations.
Despite the implementation of security measures, our internal computer systems, and those of third parties on which we rely, are to damage from: computer viruses; malware; natural disasters; terrorism; war; telecommunication and electrical failures; cyber-attacks or cyber-intrusions over the Internet; attachments to emails; persons inside our organization; or persons with access to systems inside our organization. The risk of a security breach or disruption, particularly through cyber-attacks or cyber intrusion, including by computer hackers, foreign governments, and cyber terrorists, has generally increased as the number, intensity and sophistication of attempted attacks and intrusions from around the world have increased. If such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our product development programs. For example, the loss of clinical trial data from completed or ongoing or planned clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach was to result in a loss of or damage to our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur material legal claims and liability, and damage to our reputation, and the further development of our product candidates could be delayed. We could be forced to expend significant resources in response to a cyber security breach, including repairing system damage, increasing cyber security protection costs by deploying additional personnel and protection technologies, paying regulatory fines and resolving legal claims and regulatory actions, all of which would increase our expenses, divert the attention of our management and key personnel away from our business operations and adversely affect our results of operations.
Failure to comply with health and data protection laws and regulations could lead to government enforcement actions (which could include civil or regulations on drug importationcriminal penalties), private litigation or adverse publicity and could make lower cost versionsnegatively affect our operating results and business.
We and our current and any of our future products available,collaborators may be subject to federal, state and foreign data protection laws and regulations (i.e., laws and regulations that address privacy and data security). In the U.S., numerous federal and state laws and regulations, including federal health information privacy laws (e.g., the Health Insurance Portability and Accountability Act (“HIPAA”), as amended by the Health Information Technology for Economic and Clinical Health Act (“HITECH”)), state data breach notification laws, state health information privacy laws and federal and state consumer protection laws (e.g., Section 5 of the Federal Trade Commission Act), that govern the collection, use, disclosure and protection of health-related and other personal information could apply to our operations or the operations of our collaborators. In addition, we may obtain health information from third parties (including research institutions from which we obtain clinical trial data) that are subject to privacy and security requirements under HIPAA, as amended by HITECH, or other privacy and data security laws. Depending on the facts and circumstances, we could adversely affectbe subject to criminal penalties if we knowingly obtain, use, or disclose individually identifiable health information maintained by a HIPAA-covered entity in a manner that is not authorized or permitted by HIPAA.
International data protection laws, including Regulation 2016/679, known as the General Data Protection Regulation (“GDPR”) may also apply to health-related and other personal information obtained outside of the U.S. The GDPR went into effect on May 25, 2018. The GDPR introduced new data protection requirements in the EU, as well as potential fines for non-compliant companies of up to the greater of €20 million or 4% of annual global revenue. The regulation imposes numerous new requirements for the collection, use, storage and disclosure of personal information, including more stringent requirements relating to consent and the information that must be shared with data subjects about how their personal information is used, the obligation to notify regulators and affected individuals of personal data breaches, extensive new internal privacy governance obligations and obligations to honor expanded rights of individuals in relation to their personal information (e.g., the right to access, correct and delete their data). In addition, the GDPR includes restrictions on cross-border data transfers. The GDPR increased our revenues, if any.
The pricesresponsibility and liability in relation to personal data that we process where such processing is subject to the GDPR, and we may be required to put in place additional mechanisms to ensure compliance with the GDPR, including as implemented by individual countries. Further, the United Kingdom’s vote in favor of some drugs are lower in other countries thanexiting the EU, often referred to as Brexit, has created uncertainty with regard to data protection regulation in the United States because of government regulationKingdom. In particular, it is unclear how data transfers to and market conditions. Under current law, importation of drugs intofrom the United States is generally not permitted unlessKingdom will be regulated.
| 36 |
In addition, California recently enacted the drugs are approvedCalifornia Consumer Privacy Act (“CCPA”), which creates new individual privacy rights for California consumers (as defined in the United Stateslaw) and places increased privacy and security obligations on entities handling personal data of consumers or households. The CCPA will require covered companies to provide new disclosure to consumers about such companies’ data collection, use and sharing practices, provide such consumers new ways to opt-out of certain sales or transfers of personal information, and provide consumers with additional causes of action. The CCPA goes into effect on January 1, 2020, and the entity that holds that approval consentsCalifornia Attorney General may bring enforcement actions for violations beginning July 1, 2020. The CCPA was amended on September 23, 2018, and it remains unclear what, if any, further modifications will be made to this legislation or how it will be interpreted. As currently written, the CCPA may impact our business activities and exemplifies the vulnerability of our business to the importation. Various proposalsevolving regulatory environment related to personal data and protected health information.
Compliance with U.S. and international data protection laws and regulations could require us to take on more onerous obligations in our contracts, restrict our ability to collect, use and disclose data, or in some cases, impact our ability to operate in certain jurisdictions. Failure to comply with U.S. and international data protection laws and regulations could result in government enforcement actions (which could include civil or criminal penalties), private litigation or adverse publicity and could negatively affect our operating results and business.
If we, our CROs or our IT vendors experience security or data privacy breaches or other unauthorized or improper access to, use of, or destruction of personal data, we may face costs, significant liabilities, harm to our brand and business disruption.
In connection with our drug research and development efforts, we or our CROs may collect and use a variety of personal data, such as names, mailing addresses, email addresses, phone numbers and clinical trial information. Although we have been advancedextensive measures in place to permitprevent the importationsharing and loss of drugspatient data in our clinical trial processes associated with our developed technologies and drug candidates, any failure to prevent or mitigate security breaches or improper access to, use of, or disclosure of our clinical data or patients’ personal data could result in significant liability under state (e.g., state breach notification laws), federal (e.g., HIPAA, as amended by HITECH), and international laws (e.g., the GDPR). Any failure to prevent or mitigate security breaches or improper access to, use of, or disclosure of our clinical data or patients’ personal data may cause a material adverse impact to our reputation, affect our ability to conduct new studies and potentially disrupt our business. We may also rely on third-party IT vendors to host or otherwise process some of our data and that of users, and any failure by such IT vendor to prevent or mitigate security breaches or improper access to or disclosure of such information could have similarly adverse consequences for us. If we are unable to prevent or mitigate the impact of such security or data privacy breaches, we could be exposed to litigation and governmental investigations, which could lead to a potential disruption to our business.
If we do not comply with laws regulating the protection of the environment and health and human safety, our business could be adversely affected.
Our research and development and drug candidates and future commercial manufacturing may involve the use of hazardous materials and various chemicals. We currently do not maintain a research laboratory, but we engage third-party research organizations and manufacturers to conduct our preclinical studies, clinical trials and manufacturing. These third-party laboratories and manufacturers are subject to federal, state and local laws and regulations governing the use, manufacture, storage, handling and disposal of these hazardous materials. We must rely on the third parties’ procedures for storing, handling and disposing of these materials in their facilities to comply with the relevant guidelines of the states in which they operate and the Occupational Safety and Health Administration of the U.S. Department of Labor. Although we believe that their safety procedures for handling and disposing of these materials comply with the standards mandated by applicable regulations, the risk of accidental contamination or injury from other countriesthese materials cannot be eliminated. If an accident occurs, this could result in significant delays in our development. We are also subject to numerous environmental, health and workplace safety laws and regulations. Although we maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees, this insurance may not provide lower cost alternatives to the products availableadequate coverage against potential liabilities. Additional federal, state and local laws and regulations affecting our operations may be adopted in the United States. In addition, the MMA requires the Secretaryfuture. We may incur substantial costs to comply with, and substantial fines or penalties if we violate, any of Health and Human Services to promulgate regulations for drug re-importation from Canada into the United States under some circumstances, including when the drugs are sold at a lower price than in the United States.
If thethese laws or regulations are changed to permit the importation of drugs into the United States in circumstances that are currently not permitted, such a change could have an adverse effect on our business by making available lower priced alternativesregulations.
| 37 |
Risks Associated to our future products.
Failure to obtain regulatory and pricing approvals in foreign jurisdictions could delay or prevent commercialization of our products abroad.
If we succeed in developing any products, we intend to market them in the European Union and other foreign jurisdictions. In order to do so, we must obtain separate regulatory approvals and comply with numerous and varying regulatory requirements. The approval procedure varies among countries and can involve additional testing. The time required to obtain approval abroad may differ from that required to obtain FDA approval. The foreign regulatory approval process may include all of the risks associated with obtaining FDA approval and additional risks associated with requirements particular to those foreign jurisdictions where we will seek regulatory approval of our products. We may not obtain foreign regulatory approvals on a timely basis, if at all. Approval by the FDA does not ensure approval by regulatory authorities in other countries, and approval by one foreign regulatory authority does not ensure approval by regulatory authorities in other foreign countries or by the FDA. We and our collaborators may not be able to file for regulatory approvals and may not receive necessary approvals to commercialize our products in any market outside the United States. The failure to obtain these approvals could materially adversely affect our business, financial condition and results of operations.
Risks Related to Our Common Stock and Liquidity Risks
Our common stock is a “Penny Stock” subject to specific rules governing its sale to investors that could impact its liquidity.
The SEC has adopted Rule 15g-9 which establishes the definition of a “penny stock,” for the purposes relevant to our common stock, as any equity security that has a market price of less than $5.00 per share or with an exercise price of less than $5.00 per share, subject to certain exceptions. For any transaction involving a penny stock, unless exempt, the rules require that a broker or dealer approve a person’s account for transactions in penny stocks; and the broker or dealer receive from the investor a written agreement to the transaction, setting forth the identity and quantity of the penny stock to be purchased.
In order to approve a person’s account for transactions in penny stocks, the broker or dealer must obtain financial information and investment experience objectives of the person; and make a reasonable determination that the transactions in penny stocks are suitable for that person and the person has sufficient knowledge and experience in financial matters to be capable of evaluating the risks of transactions in penny stocks.
The broker or dealer must also deliver, prior to any transaction in a penny stock, a disclosure schedule prescribed by the SEC relating to the penny stock market, which, in highlight form sets forth the basis on which the broker or dealer made the suitability determination, and states that the broker or dealer received a signed, written agreement from the investor prior to the transaction.
Generally, brokers may be less willing to execute transactions in securities subject to the “penny stock” rules. This may make it more difficult for investors sell shares of our common stock.
Disclosure also has to be made about the risks of investing in penny stocks in both public offerings and in secondary trading and about the commissions payable to both the broker-dealer and the registered representative, current quotations for the securities and the rights and remedies available to an investor in cases of fraud in penny stock transactions. Finally, monthly statements have to be sent disclosing recent price information for the penny stock held in the account and information on the limited market in penny stocks.
There is no recent trading activity in our common stock and there is no assurance that an active market will develop in the future.
Although our common stock is currently quoted on the OTCBQ (an interdealer electronic quotation system operated by OTC Markets Group, Inc.) under the symbol “PTIX”, trading of our common stock may be extremely sporadic. For example, several days may pass before any shares may be traded. As a result, an investor may find it difficult to dispose of, or to obtain accurate quotations of the price of our common stock. There can be no assurance that a more active market for our common stock will develop, or if one should develop, there is no assurance that it will be sustained. This severely limits the liquidity of our common stock, and would likely have a material adverse effect on the market price of our common stock and on our ability to raise additional capital.
The market price of our common stock may be volatile, and you could lose all or part of your investment.
The market price of our common stock is likely to be highly volatile and could be subject to wide fluctuations in response to various factors, some of which are beyond our control.
The market price of our common stock may fluctuate substantially and will depend on a number of factors many of which are beyond our control and may not be related to our operating performance. These fluctuations could cause you to lose all or part of your investment in our common stock since you might be unable to sell your shares at or above the price you pay for the shares. Factors that could cause fluctuations in the market price of our common stock include, but are not necessarily limited to, the following:
| ● | price and volume fluctuations in the overall stock market from time to time; | |
| ● | ||
| volatility in the market prices and trading volumes of pharmaceutical and biotechnology stocks; | ||
| ● | ||
| changes in operating performance and stock market valuations of other pharmaceutical and biotechnology companies generally, or those in our industry in particular; | ||
| ● | ||
| sales of shares of our common stock by us or our stockholders; | ||
| ● | ||
| failure of securities analysts to maintain coverage of us, changes in financial estimates by securities analysts who follow our | ||
| ● | ||
| the financial projections we may provide to the public, any changes in those projections or our failure to meet those projections; | ||
| ● | ||
| announcements by us or our competitors of new products or services; | ||
| ● | ||
| the public’s reaction to our press releases, other public announcements and filings with the SEC; | ||
| ● | ||
| rumors and market speculation involving us or other companies in our industry; | ||
| ● | ||
| actual or anticipated changes in our operating results or fluctuations in our operating results; | ||
| 38 |
| ● | actual or anticipated developments in our business, our competitors’ businesses or the competitive landscape generally; |
| ● | litigation involving us, our industry or both, or investigations by regulators into our operations or those of our competitors; | |
| ● | ||
| developments or disputes concerning our intellectual property or other proprietary rights; | ||
| ● | ||
| announced or completed acquisitions of businesses or technologies by us or our competitors; | ||
| ● | ||
| new laws or regulations or new interpretations of existing laws or regulations applicable to our business; | ||
| ● | ||
| changes in accounting standards, policies, guidelines, interpretations or principles; | ||
| ● | ||
| any significant change in our management; and | ||
| ● | ||
| general economic conditions and slow or negative growth of our markets. |
In addition, in the past, following periods of volatility in the overall market and the market price of a particular company’s securities, securities class action litigation has often been instituted against these companies. This litigation, if instituted against us, could result in substantial costs and a diversion of our management’s attention and resources.
Because we became public by means of a reverse business combination (merger) we may not be able to attract the attention of brokerage firms.
Additional risks may exist since we became public through a “reverse business combination (merger).” Securities analysts of brokerage firms may not provide coverage of us since there is little incentive to brokerage firms to recommend the purchase of our common stock. No assurance can be given that brokerage firms will want to conduct any secondary offerings on our behalf in the future.
FINRA sales practice requirements may also limit your ability to buy and sell our common stock, which could depress the price of our shares.
FINRA rules require broker-dealers to have reasonable grounds for believing that an investment is suitable for a customer before recommending that investment to the customer. Prior to recommending speculative low-priced securities to their non-institutional customers, broker-dealers must make reasonable efforts to obtain information about the customer’s financial status, tax status and investment objectives, among other things. Under interpretations of these rules, FINRA believes that there is a high probability such speculative low-priced securities will not be suitable for at least some customers. Thus, FINRA requirements make it more difficult for broker-dealers to recommend that their customers buy our common stock, which may limit your ability to buy and sell our shares once publicly traded, have an adverse effect on the market for our shares, and thereby depress our share price.
Compliance with the reporting requirements of federal securities laws can be expensive.
We are a public reporting company in the United States, and accordingly, subject to the information and reporting requirements of the Exchange Act and other federal securities laws, and certain compliance obligations of the Sarbanes-Oxley Act. The costs of preparing and filing annual and quarterly reports and other information with the SEC and furnishing audited reports to stockholders are substantial.
Applicable regulatory requirements, including those contained in and issued under the Sarbanes-Oxley Act of 2002, may make it difficult for us to retain or attract qualified officers and directors, which could adversely affect the management of its business and its ability to obtain or retain listing of our common stock.
We may be unable to attract and retain those qualified officers, directors and members of board committees required to provide for effective management because of the rules and regulations that govern publicly held companies, including, but not limited to, certifications by principal executive officers. The enactment of the Sarbanes-Oxley Act has resulted in the issuance of a series of related rules and regulations and the strengthening of existing rules and regulations by the SEC, as well as the adoption of new and more stringent rules by the stock exchanges. The perceived increased personal risk associated with these changes may deter qualified individuals from accepting roles as directors and executive officers.
Further, some of these changes heighten the requirements for board or committee membership, particularly with respect to an individual’s independence from the corporation and level of experience in finance and accounting matters. We may have difficulty attracting and retaining directors with the requisite qualifications. If we are unable to attract and retain qualified officers and directors, the management of our business and our ability to obtain or retain listing of our shares of common stock on any stock exchange (assuming we elect to seek and are successful in obtaining such listing) could be adversely affected.
We may have undisclosed liabilities and any such liabilities could harm our business, prospects, financial condition and results of operations.
Even though our pre-merger assets and liabilities were transferred in the split-off of MomSpot LLC (of which we owned a 51% interest) and 29 wholly-owned subsidiaries, we may be liable for any or all of such liabilities although we are unaware of any. Such liabilities that survived the Merger could harm our revenues, business, prospects, financial condition and results of operations upon our acceptance of responsibility for such liabilities.
If we fail to maintain an effective system of internal controls, we may not be able to accurately report our financial results or detect fraud. Consequently, investors could lose confidence in our financial reporting and this may decrease the trading price of our stock.
We must maintain effective internal controls to provide reliable financial reports and detect fraud. We have been assessing our internal controls to identify areas that need improvement and will continue to monitor internal controls to improve them. Failure to implement these changes to our internal controls or any others that it identifies as necessary to maintain an effective system of internal controls could harm our operating results and cause investors to lose confidence in our reported financial information. Any such loss of confidence would have a negative effect on the trading price of our stock.
As of December 31, 2017, management has completed an effective assessment of the Company’s internal controls over financial reporting based on the 2013 Committee of Sponsoring Organizations (“COSO”) framework. Management has concluded that, during the period covered by this report,year-ended December 31, 2023, our internal controls and procedures were not effective to detect the inappropriate application of U.S. GAAP. Management identified the following material weaknesses and concluded that theset forth below in our internal controlscontrol over financial reporting was not effective.reporting.
| 39 |
1. We lack the necessary corporate accounting resources to maintain adequate segregation of duties. We currently rely heavily our Executive Chairman, for most every key financial duty and he has access to materially all of our financial information. Such a lack of segregation of duties is typical in a company with limited resources. Although the Company's Executive Chairman and Board of Directors review the financial statements and would most likely discover any misappropriation of funds, this cannot be assured by the existing system.
| 1. | We lack the necessary corporate accounting resources to maintain adequate segregation of duties; and | |
| 2. | We did not perform an effective risk assessment or monitor internal controls over financial reporting. |
The price of our common stock may become volatile, which could lead to losses by investors and costly securities litigation.
The trading price of our common stock is likely to be highly volatile and could fluctuate in response to factors such as:
| ● | actual or anticipated variations in our operating results; | |
| ● | ||
| announcements of developments by us or our competitors; | ||
| ● | ||
| the timing of IDE and/or NDA approval, the completion and/or results of our clinical | ||
| ● | ||
| regulatory actions regarding our |
| ● | announcements by us or our competitors of significant acquisitions, strategic partnerships, joint ventures or capital commitments; | |
| ● | ||
| adoption of new accounting standards affecting our industry; | ||
| ● | ||
| additions or departures of key personnel; | ||
| ● | ||
| introduction of new products by us or our competitors; | ||
| ● | ||
| sales of our common stock or other securities in the open market; and | ||
| ● | ||
| other events or factors, many of which are beyond our control. |
The stock market is subject to significant price and volume fluctuations. In the past, following periods of volatility in the market price of a company’s securities, securities class action litigation has often been initiated against such a company. Litigation initiated against us, whether or not successful, could result in substantial costs and diversion of our management’s attention and resources, which could harm our business and financial condition.
Investors may experience dilution of their ownership interests because of the future issuance of additional shares of our common stock.
In the future, we may issue additional authorized but previously unissued equity securities, resulting in the dilution of the ownership interests of our present stockholders. We may also issue additional shares of our common stock or other securities that are convertible into or exercisable for our common stock in connection with hiring or retaining employees, future acquisitions, future sales of our securities for capital raising purposes, or for other business purposes and some of these issuances may be at a price (or exercise prices) below the price at which shares of our common stock is currently quoted on the OTCQB.NASDAQ Capital Market. The future issuance of any such additional shares of common stock may create downward pressure on the trading price of our common stock.
Our common stock is controlled by insiders
Our officers and directors beneficially own approximately 30%35% of our outstanding shares of common stock. Such concentrated control of our common stock may adversely affect the price of our common stock. Investors who acquire our common stock may have no effective voice in the management of our operations. Sales by our insiders or affiliates, along with any other market transactions, could affect the market price of our common stock.
We do not intend to pay dividends for the foreseeable future and may never pay dividends.
We have paid no dividends on our common stock to date and it is not anticipated that any dividends will be paid to holders of our common stock in the foreseeable future. While our future dividend policy will be based on the operating results and capital needs of our business, it is currently anticipated that any earnings will be retained to finance our future expansion and for the implementation of our business plan. As an investor, you should take note of the fact that a lack of a dividend can further affect the market value of our stock, and could significantly affect the value of any investment.
| 40 |
There can be no assurance that we will ever provide liquidity to our investors through a sale of our Company.
While acquisitions of pharmaceutical companies like ours are not uncommon, potential investors are cautioned that no assurances can be given that any form of merger, combination, or sale of our Company will take place or that any merger, combination, or sale, even if consummated, would provide liquidity or a profit for our investors. You should not invest in our Company with the expectation that we will be able to sell the business in order to provide liquidity or a profit for our investors.
Our certificate of incorporation allows for our Boardboard to create new series of preferred stock without further approval by our stockholders, which could adversely affect the rights of the holders of our common stock.
Our Boardboard of Directors (“Board”)directors has the authority to issue shares of our preferred stock, with such relative rights and preferences as the Boardboard of directors may determine, without further stockholder approval. As a result, our Boardboard of directors could authorize the issuance of a series of preferred stock that would grant to holders special and unique rights, including without limitation, a preferred right to our assets upon liquidation, a right to receive dividend payments before dividends are distributed to the holders of common stock and the right to convert into our common stock at a price more favorable then the price at which you acquired our common stock. The issuance of any preferred stock could decrease the value of your common stock and relative voting power of our common stock or result in dilution to our existing stockholders.
In addition, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, or DGCL, which may, unless certain criteria are met, prohibit large stockholders, in particular those owning 15% or more of the voting rights on our common stock, from engaging in certain business combinations with us for a prescribed period of time.
Item 1B. Unresolved Staff Comments.
Not applicable.
Item 1C. Cybersecurity.
Not applicable.Risk Management and Strategy
We have developed and implemented a cybersecurity risk management program intended to protect the confidentiality, integrity, and availability of our critical systems and information. Our cybersecurity risk management program includes a cybersecurity incident response plan. We design and assess our program based on ISO 27002 standards. This does not imply that we meet any particular technical standards, specifications, or requirements, only that we use the ISO 27002 as a guide to help us identify, assess, and manage cybersecurity risks relevant to our business. We engage external resources that contribute to, and provide independent evaluation of, our existing cybersecurity practices and organizational risk assessment systems. We use established processes designed to identify, assess, and manage third-party service provider risks when third parties handle, possess, process, and store the Company’s material information. Our cybersecurity risk management program includes (i) a policy designed to help identify material cybersecurity risks to our critical systems, information, products, services, and our broader enterprise information technology environment; (ii) the use of external service providers to manage, assess, test and otherwise assist with aspects of our security controls; and (iii) a cybersecurity incident response plan that includes procedures for responding to cybersecurity incidents. We have not identified risks from known cybersecurity threats, including as a result of any prior cybersecurity incidents, that have materially affected or are reasonably likely to materially affect us, including our operations, business strategy, results of operations, or financial condition. However, there can be no assurance that our cybersecurity prevention and mitigation efforts will always be successful, and it is possible that cybersecurity threats could have a material adverse effect on our business, operations, or financial condition in the future.
Governance
Our board of directors administers its cybersecurity risk oversight function through its audit committee. The audit committee is responsible for overseeing our policies, practices and assessments with respect to cybersecurity, and provides periodic updates to our board of directors. The audit committee receives periodic updates from management and our external third party information technology consultant regarding the effectiveness of the systems and processes we have implemented designed to safeguard our information assets and operational integrity from cyber threats, protect employee information from unauthorized access or attack, as well as secure our networks and systems, and regarding other cybersecurity matters, including the results from cybersecurity systems testing and any recent cybersecurity incidents and related responses. Our audit committee is also notified between such updates as soon as practicable regarding significant new cybersecurity threats or incidents. The audit committee also receives a report on cybersecurity matters and related risk exposures periodically from our Chief Financial Officer.
Item 2. Properties.
OurThe Company does not currently own any real property. The Company leases office space for its principal offices areexecutive office located at 149 Fifth Avenue, Suite 500, New York, New York 10010, in a conference room of Agenus, Inc. We utilize our principal office for quarterly board meetings and our annual shareholder meeting on a month to month basis at a nominal value. Our personnel and consultants all work remotely, the Company’s basic science laboratory work is conducted in the Lovejoy Lab at the University of Toronto, and its preclinical efficacy work is conducted at CROs. Hence the Company does not have the need for a day-to-day physical office location other than a mailing address and conference room facility for meetings. For that reason, the Agenus conference room suits its purposes without imposing any inconveniences upon Agenus. Dr. Armen, our Executive Chairman, is also the Chairman and Chief Executive Officer of Agenus Inc.10010.
From time to time we may be named in claims arising in the ordinary course of business. As of December 31, 2017,2023, no legal proceedings, government actions, administrative actions, investigations or claims are pending against us or involve us that, in the opinion of our management, could reasonably be expected to have a material adverse effect on our business, and financial condition,
We anticipate that we will expend significant financial and managerial resources in the defenseresults of our intellectual property rights in the future if we believe that our rights have been violated. We also anticipate that we will expend significant financial and managerial resources to defend against claims that our products and services infringe upon the intellectual property rights of third parties.operations.
Item 4. Mine Safety Disclosures.
Not applicable.
| 41 |
Not applicable.
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
Market Information
Our common stock is currently available for trading in the over-the-counter market and is quoted on the OTCQBNasdaq Capital Market under the symbol “PTIX.” There has been very limited market for our common stock and trading volume has been negligible. There is no guarantee that an active trading market will develop in our common stock.
Trades in our common stock may be subject to Rule 15g-9 of the Exchange Act, which imposes requirements on broker/dealers who sell securities subject to the rule to persons other than established customers and accredited investors. For transactions covered by the rule, broker/dealers must make a special suitability determination for purchasers of the securities and receive the purchaser’s written agreement to the transaction before the sale.
The SEC also has rules that regulate broker/dealer practices in connection with transactions in “penny stocks.” Penny stocks generally are equity securities with a price of less than $5.00 (other than securities listed on certain national exchanges, provided that the current price and volume information with respect to transactions in that security is provided by the applicable exchange or system). The penny stock rules require a broker/dealer, before effecting a transaction in a penny stock not otherwise exempt from the rules, to deliver a standardized risk disclosure document prepared by the SEC that provides information about penny stocks and the nature and level of risks in the penny stock market. The broker/dealer also must provide the customer with current bid and offer quotations for the penny stock, the compensation of the broker/dealer and its salesperson in the transaction, and monthly account statements showing the market value of each penny stock held in the customer’s account. The bid and offer quotations, and the broker/dealer and salesperson compensation information, must be given to the customer orally or in writing before effecting the transaction, and must be given to the customer in writing before or with the customer’s confirmation. These disclosure requirements may have the effect of reducing the level of trading activity in the secondary market for shares of our common stock. As a result of these rules, investors may find it difficult to sell their shares.
Our common stock was quoted on the OTC Pink under the symbol “ATRN” prior to July 27, 2016 and then under the symbol “PTIX” between July 27, 2016 and October 16, 2016. Commencing on October 17, 2016, our common stock is listed in the OTCQB under the symbol “PTIX”. The following table sets forth, for the periods indicated and as reported on the OTC Markets,Nasdaq Capital Market, the high and low bid prices for our common stock. Such quotations reflect inter-dealer prices, without retail mark-up, markdown or commission and may not necessarily represent actual transactions.
| High | Low | ||||||
| 2016 (1) | |||||||
| First Quarter(1) | $ | * | $ | * | |||
| Second Quarter(1) | $ | * | $ | * | |||
| Third Quarter(2) | $ | 250.00 | $ | 1.67 | |||
| Fourth Quarter(3) | $ | 128.85 | $ | 128.85 | |||
| 2017(3) | |||||||
| First Quarter(3) | $ | 128.85 | $ | 1.06 | |||
| Second Quarter(3) | $ | 2.20 | $ | 2.00 | |||
| Third Quarter(3) | $ | 2.00 | $ | 1.75 | |||
| Fourth Quarter(3) | $ | 2.20 | $ | 1.75 | |||
| High | Low | |||||||
| 2022(1) | ||||||||
| First Quarter (1) | $ | 5.80 | $ | 3.20 | ||||
| Second Quarter (1) | $ | 3.68 | $ | 2.60 | ||||
| Third Quarter (1) | $ | 3.20 | $ | 2.28 | ||||
| Fourth Quarter (1) | $ | 2.60 | $ | 1.40 | ||||
| 2023(1) | ||||||||
| First Quarter (1) | $ | 2.36 | $ | 1.38 | ||||
| Second Quarter (1) | $ | 2.21 | $ | 1.67 | ||||
| Third Quarter (1) | $ | 2.19 | $ | 1.74 | ||||
| Fourth Quarter (1) | $ | 1.79 | $ | 0.74 | ||||
* Less than $0.01 per share
| (1) | |
| The high and low bid prices for this quarter were reported by the | |
Holders
Holders
As of March 30, 2018,April 1, 2024, there are approximately 4313,000 record holders of our common stock and one holderzero holders of our Series B Preferred Stock.
Dividend Policy
We have never paid or declared any cash dividends on our common stock.stock, and we do not anticipate paying any cash dividends on our common stock in the foreseeable future. We anticipate that we willintend to retain all available funds and any future earnings to support operationsfund the development and to finance the growth and developmentexpansion of our business. Therefore, we do not expect to pay cash dividends in the foreseeable future, if at all. Any future determination to pay dividends will be at the discretion of our Boardboard of directors and will depend onupon a number of factors, including our financial condition, results of operations, capital requirementsfinancial condition, future prospects, contractual restrictions, restrictions imposed by applicable law and other factors that our Boardboard of directors deems relevant. In addition,
Reverse Stock Split
On March 22, 2023, the termsCompany effectuated a 1 for 4 reverse stock split (the “Reverse Split”). The Company’s stock began trading on a split-adjusted basis effective on the Nasdaq Stock Market on March 22, 2023. There was no change to the number of any future debt or credit financings may preclude us from paying dividends.authorized shares of the Company’s common stock. All share and per share information in these financial statements are adjusted to reflect the Reverse Split.
Recent Sales of Unregistered Securities
NoneDuring the year ended December 31, 2023, $520,867 in principal and interest were converted to 104,173 shares of the Company’s common stock.
Item 6. Selected Financial Data.[Reserved]
Not applicable.
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
TheYou should read the following discussion and analysis of our financial condition and results of operations should be read together with our consolidated financial statements and the related notes included at the end of this report. This discussion and other financial information included elsewhere in this report. Someparts of the information contained in this discussion and analysis or set forth elsewhere in this report including information with respect to our plans and strategy for our business, includescontain forward-looking statements that involve risks and uncertainties. See “Cautionary Note Regarding Forward-Looking Statements.” You should reviewuncertainties such as statements of our plans, objectives, expectations and intentions. As a result of many factors, including those factors set forth in the “Risk Factors”factors” section of this report, for a discussion of important factors that could causeour actual results tocould differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis.
| 42 |
The discussion and analysis of our financial condition and results of operations are based on Protagenic’s financial statements, which Protagenic has prepared in accordance with U.S. generally accepted accounting principles. The preparation of these financial statements requires Protagenic to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements, as well as the reported revenues and expenses during the reporting periods. On an ongoing basis, Protagenic evaluates such estimates and judgments, including those described in greater detail below. Protagenic bases its estimates on historical experience and on various other factors that Protagenic believes are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
Historical Background
The Company was originally incorporated in Delaware on February 3, 1994 under the name Millbrook Acquisition Corp. The Company was the successorWe expect to The Millbrook Press Inc., which was a wholly-owned subsidiary of Antia Publishing Company, a Delaware corporation, which in turn was a wholly-owned subsidiary of Groupe de la Cite International, a Societe Anonyme organized under French law. In February 1994, the founders of the Company effected a management buyout by forming the Company which purchased substantially all of the assets of The Millbrook Press Inc.
For the next 10 years, the Company was involved in a variety of attempts at creating a profitable, sustainable business model in the publishingcontinue to incur significant expenses and retail sectors, none of which succeeded. On February 6, 2004, the Company filed a voluntary petition for relief under Chapter 11 of the Bankruptcy Code with the United States Bankruptcy Courtminimal positive net cash flows from operations or negative net cash flows from operations for the District of Connecticutforeseeable future, and changed its name to MPLC, Inc.those expenses and losses may fluctuate significantly from quarter-to-quarter and year-to-year. We anticipate that our expenses will fluctuate substantially as we:
Also in 2004, a biotechnology company called Protagenic Therapeutics, Inc. (referred to herein as “Protagenic”) was organized under the name Protagenic Therapeutics, Inc., with the goal of commercializing novel peptide drugs that had been discovered in the laboratory of Dr. David Lovejoy (“The Professor”) at the University of Toronto (UofT). The Company specializes in the discovery and development of therapeutics to treat central nervous system (CNS) disorders. PTI’s mission is to provide safe and effective treatments for mood, anxiety, depression and neurodegenerative disorders by using novel peptide-base, brain active therapeutics. The Company’s strategy is to develop, test and obtain regulatory approval for various applications of these brain active therapeutics.
| ● | continue our ongoing preclinical studies, clinical trials and our product development activities for our pipeline of product candidates; | |
| ● | seek regulatory approvals for any product candidates that successfully complete clinical trials; | |
| ● | continue research and preclinical development and initiate clinical trials of our other product candidates; | |
| ● | seek to discover and develop additional product candidates either internally or in partnership with other pharmaceutical companies; | |
| ● | adapt our regulatory compliance efforts to incorporate requirements applicable to marketed products; | |
| ● | maintain, expand and protect our intellectual property portfolio; and | |
| ● | incur additional legal, accounting and other expenses in operating as a public company. |
On January 31, 2007, MPLC, Inc. entered into an exchange agreement with New Motion, Inc., a Delaware corporation formed in March 2005 (“New Motion”), the stockholders of New Motion (the “Stockholders”), and Trinad Capital Master Fund. On May 2, 2007, the Company changed its corporate name to New Motion, Inc.
As part of a corporate re-branding strategy, on June 25, 2009, New Motion, Inc. changed its name to Atrinsic, Inc. Its new corporate image was as a provider of “digital advertising and marketing services,” primarily digital music, casual games, interactive contests, and communities/lifestyles.
Meanwhile, from 2006 through 2014, Prior Protagenic sponsored fundamental research & development work in the Professor’s lab at the UofT aimed at demonstrating the efficacy of Prior Protagenic’s lead drug candidate, known as Teneurin C-Terminus Associated Peptide, or TCAP-1, and elucidating its mechanism of action. This research resulted in a detailed understanding of the peptide and its actions on neurons. Prior Protagenic’s worldwide exclusive technology license agreement with the UofT gave it sole rights to commercialize and eventually sell drugs related to the TCAP family of proteins.
By early 2015, it became clear that Prior Protagenic needed an influx of working capital in order to meet its goal of completing the process of applying for an Investigational New Drug (IND) application with the U.S. Food and Drug Administration (FDA) to sell a commercial version of TCAP-1. To secure this capital, management of Prior Protagenic chose to pursue a reverse merger and financing strategy, with the help of a placement agent. This resulted in the introduction of Prior Protagenic to the former Atrinsic, Inc., and the consummation of a memorandum of understanding (MOU) in mid-2015 that the two companies would merge and conduct an equity financing, with the former Prior Protagenic shareholders owning approximately 80% of the pre-financing entity, and the former Atrinsic, Inc. shareholders owning the other 20%.
On February 12, 2016, Protagenic Therapeutics, Inc. merged into Protagenic Acquisition Corp., a wholly-owned subsidiary of Atrinsic, Inc., and raised gross proceeds of approximately $4.6 million in the 2016 Private Placement exclusive of $500,000 of debt that was converted as part of the offering. All previous lines of business of Atrinsic, Inc. where theretofore dropped in favor of the field of neurologic drug development. On June 17, 2016, Protagenic Therapeutics, Inc. was merged with and into Atrinsic. Atrinsic was the surviving corporation in this merger and changed its name from Atrinsic to Protagenic Therapeutics, Inc.
Results of Operations
We are a development stage company currently performing clinical trials to obtain FDAFood and Drug Administration (“FDA”) approval and commercialization of our product.
During the year ended December 31, 2017,2023, we incurred a loss from operations of $2,365,324$4,526,974 as compared to $2,233,501$3,557,788 for the year ended December 31, 2016.2022. The increase in the loss is due to an increase in research and development expense of $299,586$1,730,628 from $417,866$1,589,239 for the year ended December 31, 20162022 to $717,452$3,319,867 for the year ended December 31, 2017, and an increase2023, offset by a decrease in general and administrative expenses of $236,406$761,442 from $1,411,466$1,968,549 for the year ended December 31, 20162022 to $1,647,872$1,207,107 for the year ended December 31, 20172023. The increase in research and development expense is due to additional cost related to the Company’s continued research and development efforts. The decrease in general and administrative expenses was due to lower stock compensation expense in the current year.
Liquidity and Capital Resources
Since our inception, we have incurred significant operating losses. We have not yet commercialized any of our product candidates and we do not expect to generate revenue from sales of any product candidates for several years, if at all. To date, we have primarily financed our operations through the public offering of our equity securities and the private placement of our convertible securities.
| 43 |
In June 2021, the SEC declared effective a shelf registration statement filed by us. This shelf registration statement allows us to issue any combination of our common stock, preferred stock, debt securities, warrants, or units from time to time for an increaseaggregate initial offering price of up to $100.0 million. In July 2021, we entered into an At Market Issuance Agreement, or the ATM Agreement, with B. Riley Securities, Inc. and EF Hutton, division of Benchmark Investments, LLC, or the Sales Agents, under which we may issue and sell from time to time up to $10.0 million of our common stock through or to the Sales Agents, as agent or principal. Any sale of shares of our common stock under the Sales Agreement will be made under our shelf registration statement on Form S-3. Sales of our common stock under the Sales Agreement are made at market prices by any method that is deemed to be an “at the market offering” as defined in Rule 415(a)(4) under the Securities Act of 1933, as amended. The Company has not yet sold any shares under the ATM Agreement. Therefore, as of December 31, 2023, $10.0 million of our common stock remained available for sale under the Sales Agreement.
Operating activities used $3,703,776 and $1,993,814 in cash for the years ended December 31, 2023 and 2022, respectively. The use of cash in operating activities during the year ended December 31, 2023, primarily comprised of $5,000,497 net loss, $666,828 in stock compensation expense.expense, a decrease in prepaid expenses and other current assets of $87,086, amortization of debt discount of $85,770, and a $6,326 decrease of accounts payable and accrued expenses, which included payments to legal and accounting professionals, payments to consultants, and other administrative expenses.
LiquidityInvesting activities provided $4,775,482 and used $1,596,974 in cash for the years ended December 31, 2023 and 2022, respectively. The cash provided by investing activities during the year ended December 31, 2023 consisted of $7,689,507 from the sale of marketable securities, $2,764,250 in the purchase of marketable securities, and $149,775 in the purchase in fixed assets.
We continually project anticipated cash requirements, predominantly from the ongoing funding requirements of our neuropeptide drug development program. The majority of these expenses relate to paying external vendors such as Contract Research Organizations (CROs) and peptide synthesizer companies. They could also include business combinations, capital expenditures, and new drug development working capital requirements. As of December 31, 2017,2023, we had cash of $399,687$1,287,893 and working capital of $1,218,290. $3,544,785.
We anticipate furtherthat losses inwill continue for the development of our business.foreseeable future. Based on our current forecast and budget, Management believesoperating plans, we believe that our cash resources will be sufficient to fund ourits operations anticipated capital expenditures and working capital at least until approximately the end of the third quarter of 2018. Absent generation of sufficient revenue from the execution of the Company’s business plan,2024. In order to continue our operations beyond our forecasted runway we will need to obtain debt or equity financing byraise additional capital, and we have no committed sources of additional capital at this time. The forecast of cash resources is forward-looking information that involves risks and uncertainties, and the third quarteractual amount of 2018.
As reflected in the consolidated financial statements, the Company had an accumulated deficit at December 31, 2017, a net lossour expenses could vary materially and net cash used in operating activities for the year ended. These factors raise substantial doubt about the Company’s ability to continueadversely as a going concern.
Operating activities used $1,380,089result of a number of factors. We have based our estimates on assumptions that may prove to be wrong, and $1,140,319 in cash for the years ended December 31, 2017 and 2016, respectively. The use of cash in operating activities during the years ended December 31, 2017 and 2016, was primarily usedour expenses could prove to fund our net loss.
Ourbe significantly higher than we currently anticipate. Management does not know whether additional financing activities provided no cash for the year ended December 31, 2017.
Within the next two years,will be on terms favorable or acceptable to us when needed, if at all. If adequate additional funds are not available when required, or if we are likely to seek additional financingunsuccessful in two ways:
(1) by approaching large pharmaceutical companies who may be interested in licensing the commercial rights to our lead drug candidate, PT00114,entering into partnership agreements for a non-core indication or in a non-core geographic region (such as an indication other than anxiety, depression, or addictive behavior therapy, or a region of the world other than North America or Europe). If we are successful in striking a partnership with a large pharmaceutical company in this way, we may receive an up-front licensing fee that could be significant.
(2) In the absence of a licensing opportunity with a large pharmaceutical partner, we may undertake an equity financing in mid-2018, in order to raise $5-10 million in working capital to fund our first two phases of clinical trials. In this event, Management would aim to disclose additional pre-clinical research results prior to or coincident with this possible equity financing, because this would represent a developmental milestone that could increase the value of the Company’s equity in the view of future investors.
The anticipated impact on our cash position of either of these financing options could be to provide enough working capital to fund Phase I and potentially Phase IIa clinical trials. The anticipated impact on the Company’s liquidity and operations is that the Company would be able to continue operating beyond early 2019 that its current cash position provides for.
Recent Developments
In 2017, we initiated and completed several key pre-clinical research studies. The results of some of these studies comprise the basisfurther development of our outreachproduct candidates, management may need to potential corporate partners.curtail its development efforts and planned operations.
Plan of Operations
Business Overview
The Company is in its developmental stage, with encouraging but not conclusive evidence that its lead drug candidate, PT00014, may be effective as an anti-anxiety and/or anti-depression drug. It is focused on confirming the efficacy of this drug candidate, along with performing the other preclinical steps needed to progress along the pathway to bring this drug candidate into human clinical trials and eventually, to the global market to provide a new pharmaceutical for patients suffering from anxiety or treatment-resistant depression.
Our anticipated timeline for reaching the significant milestones in our plan of operations and the costs associated with our plan are set forth in the table below:
| Estimated Cost | ||||
| 1Q 2018 | ||||
| Completion of ELISA tests | $ | 45,000 | ||
| Complete Custom antibodies as an alternative to ELISA | $ | 21,000 | ||
| 2Q 2018 | ||||
| Complete Stability and Formulation | $ | 85,000 | ||
| Write our first IND application | $ | 80,000 | ||
| 3Q 2018 | ||||
| Possibly Complete toxicology studies in two species | $ | 850,000 | * | |
| 4Q 2018 | ||||
| Submit our first IND application | $ | 60,000 | ||
| 1Q 2019 | ||||
| Begin dosing healthy volunteers in Phase I trial | $ | 175,000 | ||
*This expenditure may depend on a successful capital raising event.
If we are able to successfully develop our drug, PT00114, and obtain FDA approval, we could then begin marketing and selling it in the United States and generate revenue. FDA approval to begin commercial sales is the singular gating item that will allow us to begin generating sales revenue in the U.S., so it will have an enormous impact on our business plan and our financial condition. It is anticipated that the sale of our drug will allow the Company to generate enough sales revenue to support all of our operations and to generate a profit. However, given the stage of development, even if FDA Approval is obtained, it iswe do not anticipatedanticipate generating any revenue from sales prior to 2023.2026.
| 44 |
Development Milestones (upcoming developmental milestones)Currently Anticipated
Upcoming development milestones include confirming efficacy of our lead drug candidateRecent communications with the U.S. FDA has resulted in an animal model in afollowing revised guidance for clinical research organization (CRO), conducting toxicology testing in two animal species, and filing an Investigational New Drug (IND) application to begin human clinical trials.timelines.
| ● | The Company in the process of refiling its IND application for PT00114 addressing the questions raised by regulators. | |
| ● | Anticipate Q2 2024 Public availability of results from single dose portion of Phase I study for PT00114 Anticipate Q2 2024 Commence multiple dose portion of Phase I study for PT00114 | |
| ● | Anticipate Q2 2024 Public availability of results from multiple dose portion of Phase I study for PT00114 | |
| ● | Anticipate Q3 2024 : Initiation of Phase IIa study for PT00114 |
Human Resources (current state of employees and future plans towards employeesemployees)
The Company has threetwo part-time employees: David Hogg, PhD, a Research Technician, Garo H. Armen, PhD, the Executive Chairman, and Alexander K. Arrow, MD, the Chief Financial Officer.Officer, and one full-time employee, Lauren Mueller, PhD, a Senior Research Scientist. The Company also has threesix paid consultants: Andrew Slee, PhD, Chief Operating Officer, Robert S. Stein, MD, PhD, Chief Medical Officer, Dalia Barsyte, PhD, Chief Technology Officer,Scientific Advisor, David Lovejoy, PhD, Chief Scientific Officer,Advisor, and Christina Fam Faragalla, Director of Project Management.Zack Armen, Strategic Advisor.
Financing – Capital Needs
The Company anticipates that it will need to raise additional capital in the next year or so to support its R&D activities as it prepares to commence human clinical trials.
Over the next two years, we anticipate conducting the following research and development activities at the following estimated costs and expense:
| Basic Science of TCAP-1 | $ | 110,000 | ||
| Efficacy Studies | $ | 320,000 | ||
| Toxicology Studies | $ | 200,000 | ||
| Stability and Formulation | $ | 85,000 | ||
| Custom antibodies as an alternative to ELISA | $ | 37,000 | ||
| Tagged antibodies | $ | 104,000 | ||
| Antibody purification | $ | 24,000 | ||
| Clinical consultants | $ | 20,000 | ||
| Medical Writing and IND application compilation | $ | 79,000 | ||
| Technical Infrastructure | $ | 11,000 | ||
| Total R&D not including personnel | $ | 980,000 |
Off Balance Sheet Arrangements
We have no material off-balance sheet arrangements that are likely to have a current or future effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital resources, or capital expenditures.
Critical accounting policies and estimates
Our discussion and analysis of financial condition and results of operations are based upon our consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States of America (“GAAP”). The notes to the consolidated financial statements contained in this Annual Report describe our significant accounting policies used in the preparation of the consolidated financial statements. The preparation of these financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting periods. Actual results could differ from those estimates. We continually evaluate our critical accounting policies and estimates.
We believe the critical accounting policies listed below reflect significant judgments, estimates and assumptions used in the preparation of our consolidated financial statements.
Principles of consolidation
The consolidated financial statements include the accounts of Atrinsic, Inc., and its wholly owned subsidiary, Protagenic Acquisition Corp, and Protagenic Therapeutics, Inc., which merged with and into Protagenic Acquisition Corp, on February 12, 2016, as well as Protagenic Therapeutics’ wholly-owned Canadian subsidiary, PTI Canada. All significant intercompany balances and transactions have been eliminated in consolidation.
Foreign Currency Translation and Transactions.The assets and liabilities of our foreign subsidiary PTI Canada are translated into U.S. dollars from the functional currency using the exchange rate in effect at the balance sheets date. Additionally, the accounts on the statements of operations are translated using exchange rates approximating average rates prevailing during the years. Equity accounts are translated at historical exchange rates. Translation adjustments that arise from translating its financial statements from the local currency to the U.S. dollar are accumulated and reflected as a separate component of stockholders’ equity (deficit). The current year effects of the transaction adjustments are included on the statement of operations as a realized gain (loss) on foreign transaction exchange.
Use of Estimates. The preparation of financial statements in conformity with U.S. Generally Accepted Accounting Principles (“GAAP”) requires management to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenue and expenses as well as the disclosure of contingent assets and liabilities. Management continually evaluates its estimates and judgments including those related to accruals, contingencies, valuation allowance for deferred tax assets, and valuation of stock options and warrants. Management bases its estimates and judgments on historical experience and other factors that are believed to be reasonable in the circumstances. Actual results may differ from those estimates. Macroeconomic conditions may directly, or indirectly through our business partners and vendors, impact our financial performance and available resources. Such conditions may, in turn, impact the aforementioned estimates and assumptions.
Fair Value Measurements.Accounting Standards Codification ASC 820, “Fair Value Measurements and Disclosure,” defines fair value as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date, not adjusted for transaction costs. ASC 820 also establishes a fair value hierarchy that prioritizes the inputs to valuation techniques used to measure fair value into three broad levels giving the highest priority to quoted prices in active markets for identical assets or liabilities (Level 1) and the lowest priority to unobservable inputs (Level 3).
The three levels are described below:
Level 1 Inputs – Unadjusted quoted prices in active markets for identical assets or liabilities that is accessible by the Company;
Level 2 Inputs – Quoted prices in markets that are not active or financial instruments for which all significant inputs are observable, either directly or indirectly;
Level 3 Inputs – Unobservable inputs for the asset or liability including significant assumptions of the Company and other market participants.
Derivative Liability. The Company evaluates its options, warrants or other contracts, if any, to determine if those contracts or embedded components of those contracts qualify as derivatives to be separately accounted for in accordance with ASC 815-10-05-4 and 815-40-25. The result of this accounting treatment is that the fair value of the embedded derivative is marked-to-market each balance sheet date and recorded as either an asset or a liability. In the event that the fair value is recorded as a liability, the change in fair value is recorded in the consolidated statement of operations as other income or expense. Upon conversion, exercise or cancellation of a derivative instrument, the instrument is marked to fair value at the date of conversion, exercise or cancellation and then the related fair value is reclassified to equity.
The classification of derivative instruments, including whether such instruments should be recorded as liabilities or as equity, is re-assessed at the end of each reporting period. Equity instruments that are initially classified as equity that become subject to reclassification are reclassified to liability at the fair value of the instrument on the reclassification date. Derivative instrument liabilities will be classified in the balance sheet as current or non-current based on whether or not net-cash settlement of the derivative instrument is expected within 12 months of the balance sheet date.
Basic and Diluted Net (Loss) per Common Share.Basic (loss) per common share is computed by dividing the net (loss) by the weighted-average number of shares of common stock outstanding for each period. Diluted (loss) per share is computed by dividing the net (loss) by the weighted-average number of shares of common stock outstanding plus the dilutive effect of shares issuable through the common stock equivalents. Potentially dilutive securities consisting of options and warrants aggregating 8,265,723 as of December 31, 2017, including common shares issuable under the conversion feature of the preferred shares, options and warrants issued in the Private Offering closing and merger transactions were not included in the calculation of weighted-average shares of common stock outstanding as they were determined to be anti-dilutive.
Recently Issued Accounting Pronouncements
In January 2016, the FASB issued ASU No. 2016-01, “Financial Instruments - Overall (Subtopic 825-10): Recognition and Measurement of Financial Assets and Financial Liabilities”. The update addresses certain aspects of recognition, measurement, presentation and disclosure of financial instruments. For public business entities, the amendments in this update are effective for fiscal years beginning after December 15, 2017, including interim periods within those fiscal years. Early adoption is permitted only for certain portions of the ASU related to financial liabilities. The Company is currently evaluating the impact of the provisions of this new standard on the consolidated financial statements.None
In February 2016, the FASB issued ASU No. 2016-02, Leases. The main provisions of ASU No. 2016-02 require management to recognize lease assets and lease liabilities for all leases. ASU 2016-02 retains a distinction between finance leases and operating leases. The classification criteria for distinguishing between finance leases and operating leases are substantially similar to the classification criteria for distinguishing between capital leases and operating leases in the previous leases guidance. The result of retaining a distinction between finance leases and operating leases is that under the lessee accounting model, the effect of leases in the statement of comprehensive income and the statement of cash flows is largely unchanged from previous GAAP. The amendments in this ASU are effective for fiscal years beginning after December 15, 2018, including interim periods within those fiscal years. The Company is currently assessing the impact of this ASU on the Company’s consolidated financial statements.
In August 2016, the FASB issued ASU 2016-15,“Statement of Cash Flows (Topic 230): Classification of Certain Cash Receipts and Cash Payments” (“ASU 2016-15”). ASU 2016-15 will make eight targeted changes to how cash receipts and cash payments are presented and classified in the statement of cash flows. ASU 2016-15 is effective for fiscal years beginning after December 15, 2017. The new standard will require adoption on a retrospective basis unless it is impracticable to apply, in which case it would be required to apply the amendments prospectively as of the earliest date practicable.
In October 2016, the FASB issued ASU 2016-16, “Income Taxes (Topic 740): Intra-Entity Transfers of Assets Other than Inventory”, which eliminates the exception that prohibits the recognition of current and deferred income tax effects for intra-entity transfers of assets other than inventory until the asset has been sold to an outside party. The updated guidance is effective for annual periods beginning after December 15, 2019, including interim periods within those fiscal years. Early adoption of the update is permitted. The Company is currently assessing the impact of this ASU on the Company’s consolidated financial statements.
In November 2016, the FASB issued ASU 2016-18, “Statement of Cash Flows (Topic 230)”, requiring that the statement of cash flows explain the change in the total cash, cash equivalents, and amounts generally described as restricted cash or restricted cash equivalents. This guidance is effective for fiscal years, and interim reporting periods therein, beginning after December 15, 2017 with early adoption permitted. The provisions of this guidance are to be applied using a retrospective approach which requires application of the guidance for all periods presented.
In December 2016, the FASB issued ASU 2016-20, “Technical Corrections and Improvements to Topic 606, Revenue from Contracts with Customers”. The amendments in this Update affect the guidance in Update 2014-09, which is not yet effective. The effective date and transition requirements for the amendments are the same as the effective date and transition requirements for Topic 606 (and any other Topic amended by Update 2014-09). Accounting Standards Update No. 2015-14,Revenue from Contracts with Customers (Topic 606): Deferral of the Effective Date, defers the effective date of Update 2014-09 by one year.
In September 2017, the Financial Accounting Standards Board (FASB) issued Accounting Standards Update (ASU) No. 2017-13,Revenue Recognition (Topic 605), Revenue from Contracts with Customers (Topic 606), Leases (Topic 840), and Leases (Topic 842).The new standards, among other things, provide additional implementation guidance with respect to Accounting Standards Codification (ASC) Topic 606 and ASC Topic 842. ASU 2017-13 is effective for annual reporting periods beginning after December 15, 2017, including interim reporting periods within that reporting period. The Company is currently evaluating the impact of the new standard, but does not expect it to have a material impact on its implementation strategies or its consolidated financial statements upon adoption.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk.
Not applicable.
Item 8. Financial Statements and Supplementary Data.
See pages F-1 through F-21F-20 following the Exhibit Index of this Annual Report on Form 10-K.
Item 9. Changes in and Disagreements Withwith Accountants on Accounting and Financial Disclosure.
On May 18, 2017, we engaged MaloneBailey LLP (“Malone”) as our principal independent registered public accounting firm, and effective May 18, 2017, we dismissed Marcum LLP (“Marcum”) as Protagenic Therapeutics, Inc.’s principal independent registered public accounting firm. The decision to dismiss Marcum and to appoint Malone was approved by our board of directors.None.
| 45 |
Marcum’s report on our consolidated financial statements for the fiscal years ended December 31, 2016 and 2015 did not contain an adverse opinion or disclaimer of opinion, or qualification or modification as to uncertainty, audit scope, or accounting principles.
During our two most recent fiscal years ended December 31, 2016 and 2015 and in the subsequent interim period through the date of dismissal, there were no reportable events as described in Item 304(a)(1)(v) of Regulation S-K between the Company and Marcum on any matter of accounting principles or practices, financial statement disclosure, or auditing scope or procedure, which disagreements, if not resolved to the satisfaction of Marcum, would have caused Marcum to make reference to the subject matter of the disagreements in connection with its reports on the consolidated financial statements for such fiscal years, or (2) reportable events, except that Marcum advised the Company of material weaknesses related to difficulty in accounting for complex accounting transactions due to an insufficient number of accounting personnel with experience in that area and limited segregation of duties within the Company’s accounting and financial reporting functions.
During our two most recent fiscal years ended December 31, 2016 and 2015 and in the subsequent interim period through the date of appointment, we have not consulted with Malone regarding either the application of accounting principles to a specified transaction, either completed or proposed, or the type of audit opinion that might be rendered on our financial statements, nor has Malone provided to us a written report or oral advice that Malone concluded was an important factor considered by us in reaching a decision as to the accounting, auditing or financial reporting issue. In addition, during such periods, we have not consulted with Malone regarding any matter that was either the subject of a disagreement (as defined in Item 304(a)(1)(iv) and the related instructions) or a reportable event (as described in Item 304(a)(1)(v) of Regulation S-K).
Item 9A. Controls and Procedures.
Assessment of the Effectiveness of Internal Controls over Financial Reporting
Under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting using the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control-Integrated Framework published in 2013. Based on its evaluation, our management concluded that our internal control over financial reporting was not effective as of the end of the period covered by this Annual Report on Form 10-K.
(a) Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting to provide reasonable assurance regarding the reliability of our financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles in the United States of America. Internal control over financial reporting includes those policies and procedures that:
(i) pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets;
(ii) provide reasonable assurance that the transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with the authorization of management and/or our Board of Directors; and
(iii) provide reasonable assurance regarding the prevention or timely detection of any unauthorized acquisition, use or disposition of our assets that could have a material effect on our financial statements.
Due to its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate due to changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
As of December 31, 2017,2023, management has completed a proper evaluation, risk assessment and monitoring of the Company'sCompany’s internal controls over financial reporting based on the 2013 Committee of Sponsoring Organizations (COSO) framework. Management concluded that, during the period covered by this report, that our internal controls and procedures were not effective to detect the inappropriate application of US GAAP. Management identified the following material weaknesses and concluded that the internal controls over financial reporting waswere not effective.
| 1. | We lack the necessary corporate accounting resources to maintain adequate segregation of duties. We currently rely heavily on our Executive Chairman, for | |
| 2. | Limited level of multiple reviews in connection with the financial reporting process. |
This annual report does not include an attestation report by our independent registered public accounting firm regarding internal control over financial reporting. As we are neither a large accelerated filer nor an accelerated filer, our management’s report was not subject to attestation by our registered public accounting firm pursuant to rules of the Securities and Exchange Commission that permit us to provide only management’s report in this annual report.
| 46 |
(b) Evaluation of Disclosure Controls and Procedures
Pursuant to Rule 13a–15(b) under the Exchange Act, the Company carried out an evaluation, with the participation of the Company'sCompany’s management, including the Company'sCompany’s Board of Directors, the Chief Executive Officer and the Chief ExecutiveFinancial Officer, of the effectiveness of the Company'sCompany’s disclosure controls and procedures (as defined under Rule 13a–15(e) under the Exchange Act) as of the end of the period covered by this Report. Based upon that evaluation, the Company'sCompany’s management concluded that the Company'sCompany’s disclosure controls and procedures were not effective to ensure that information required to be disclosed by the Company in the reports that the Company files or submits under the Exchange Act, is recorded, processed, summarized and reported, within the time periods specified in the SEC'sSEC’s rules and forms, and that such information is accumulated and communicated to the Company'sCompany’s management to allow timely decisions regarding required disclosure due to the following:
| 1. | We do not have sufficient segregation of duties within accounting functions, which is a basic internal control. Due to our size and nature, segregation of all conflicting duties may not always be possible and may not be economically feasible. However, to the extent possible, the initiation of transactions, the custody of assets and the recording of transactions should be performed by separate individuals. Management evaluated the impact of our failure to have segregation of duties on our assessment of our disclosure controls and procedures and has concluded that the control deficiency that resulted represented a material weakness. | |
| 2. | Limited level of multiple reviews among those tasked with preparing the financial statements. |
1. Lack of Segregation of Duties; Management is aware that there is a lack of segregation of accounting duties as a result of limited personnel.
(c) Changes in Internal Control over Financial Reporting
During the quarter ended December 31, 2017,2023, the Company analyzed and documenting accounting policies and procedures. In addition, management implemented certain policies and procedures but concluded that material weaknesses still exist and that such controls are not effective under the COSO framework. These material weaknesses could result in a material misstatement to the annual or interim consolidated financial statements that would not be prevented or detected.
Remediation Plan
To address the material weakness described above, we have engaged an independent third party to enhance our segregation of duties.
Since we remain a small Company, with limited segregation of duties, the third party has identified certain areas where we can layer in added controls and procedures. Management intends to implement such controls and procedures in the future.
Limitations on the Effectiveness of Controls. Our management, including our Chief Executive Officer and Chief Financial Officer, does not expect that our disclosure controls and procedures or our internal control over financial reporting will prevent all errors and all fraud. A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within our Company have been detected.
(c) Changes in Internal Control over Financial Reporting
Other than as discussed above, there were no changes in our internal controls over financial reporting (as defined in the Exchange Act Rules 13a-15(f) and 15d-15(f)) that occurred during the quarter ended December 31, 2023, that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
None.Nasdaq Deficiency Notice
On Tuesday, November 21, 2023, Protagenic Therapeutics, Inc. (“the Company”) received a deficiency letter (the “Notification Letter”) from the Nasdaq Listing Qualifications (“Nasdaq”) stating that it is not in compliance with the minimum bid price requirements set forth in Nasdaq Listing Rule 5550(a)(2) for continued listing on The Nasdaq Capital Market. Nasdaq Listing Rule 5550(a)(2) requires listed securities to maintain a minimum bid price of $1.00 per share, and Nasdaq Listing Rule 5810(c)(3)(A) provides that a failure to meet the minimum bid price requirement exists if the deficiency continues for a period of 30 consecutive business days. Based on the closing bid price of the Company’s common stock for the 30 consecutive business days prior to the date of the Notification Letter, the Company no longer meets the minimum bid price requirement. The Notification Letter had no immediate effect on the listing or trading of the Company’s common stock on the Nasdaq Capital Market. The common stock continued to trade on the Nasdaq Capital Market under the symbol “PTIX”.
On March 19, 2024, the Company received written notice from the Nasdaq Listing Qualifications Department (the “Staff”) that the Company has regained compliance with Nasdaq Listing Rule 5550(a)(2) (the “Minimum Bid Price Requirement”) by maintaining a minimum closing bid price of the Company’s common stock of at least $1.00 per share for the last ten consecutive trading days, from March 5, 2024 to March 18, 2024, and that this matter is now closed. As previously reported, on November 21, 2023, Nasdaq notified the Company that, the closing bid price for the Company’s common stock had been below the minimum $1.00 per share requirement for 30-consecutive business in violation of the Minimum Bid Price Requirement.
Rule 10b5-1 Plans
During the period from October 1, 2023 to December 31, 2023, none of our directors or officers (as defined in Rule 16a-1(f) under the Exchange Act) adopted or terminated any Rule 10b5-1 trading arrangement (as defined in Item 408(a)(1)(i) of Regulation S-K) or any non-Rule 10b5-1 trading arrangement (as defined in Item 408(c) of Regulation S-K).
Item 9C. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections.
[Not applicable].
PART III
Item 10. Directors, Executive Officers and Corporate Governance.
Executive Officers and Directors
The following sets forth certain information with respect to our executive officers and directors.
| Name | Age | Position(s) | |||
| Garo H. Armen | 71 | Executive Chairman of the Board of Directors | |||
| Alexander K. Arrow | 53 | Chief Financial Officer | |||
| Robert B. Stein | 73 | Director, Chief Medical Officer | |||
| 74 | |||||
| 59 | Director | ||||
| 66 | Director | ||||
| Brian Corvese | 66 | Director | |||
| Jennifer Buell | 49 | Director |
Garo H. Armen, PhD, Executive Chairman, is one of our founders and joined us in September 2004. Garo H.Dr. Armen is Chairman and Chief Executive Officer of Agenus Inc., a biotechnology company he co-founded in 1994. From mid-2002 through 2004, he also served as Chairman of the Board of directors of the biopharmaceutical company Elan Corporation, plc, which he successfully restructured. Prior to Agenus Inc., Dr. Armen established Armen Partners, a money management firm specializing in biotechnology and pharmaceutical companies, and was the architect of the widely publicized creation of the Immunex Lederle oncology business in 1993. Earlier, he was a senior vice president of research at Dean Witter Reynolds, having begun his career on Wall Street as an analyst and investment banker at EF Hutton. In 2002, Dr. Armen founded the Children of Armenia Fund, a nonprofit organization dedicated to significantly rebuilding and revitalizing impoverished rural Armenian towns to provide immediate and sustainable benefits to children and youth. He received the Ellis Island Medal of Honor in 2004 for his humanitarian efforts, and received the Sabin Humanitarian Award from the Sabin Vaccine Institute in 2006 for his achievements in biotechnology and progressing medical research. Dr. Armen was also the Ernst & Young 2002 New York City Biotechnology Entrepreneur of the Year, and received a Wings of Hope Award in 2005 from The Melanoma Research Foundation for his ongoing commitment to the melanoma community. Dr. Armen received a PhD in physical chemistry from the Graduate Center, City University of New York, after which he worked as a research fellow at Brookhaven National Laboratories in Long Island, NY.
| 48 |
Alexander K. Arrow, M.D., CFA, –ChiefChief Financial Officer. Dr. Arrow became our Chief Financial Officer in February 2016. Dr. Arrow is also the Chief ExecutiveFinancial Officer of Strateos, Inc., a company that provides drug discovery automation solutions to the pharmaceutical industry, and serves on the Board of Insightful Instruments, Inc., an ophthalmology company developing a novel tool for refractive (vision correction) surgery. From 2019 through 2022 he was the Chief Financial Officer of Carlsmed, Inc, a spinal implant manufacturer that combined personalized medicine and 3D printing with lumbar fusion implants. He previously served on the Boards of Zelegent, Inc., a clinical-stage start-up medical device company preparing to launchselling a minimally invasiveminimally-invasive snoring alleviation tool. From January 2015 through December 2015, Dr. Arrow also served as a directortool, Paragonix Technologies, the supplier of the leading solid organ transportation device and acting Chief Operating Officer of Neumedicines, Inc., a clinical-stage private biotechnology company developing protein therapeutics that address unmet clinical and societal needs in Oncology, Hematology and Immunology. Dr. Arrow servesserved as a director of Gel-e, Inc., a wound-care company with an FDA-cleared hemostatic patch product, BioLx, Inc., a start-up developing an advanced surgical mask, and Rindex Medical, Inc., a developmental-stage company, 30% owned by the Cleveland Clinic, which is developing a diagnostic technology for use in cardiovascular intensive care units. Previously, Dr. Arrow served on the board and was theas Chairman of both the Audit Committee and Compensation CommitteeCommittees of Biolase, Inc. (NASDAQ: BIOL) from July 2010 through February 2014, and served as the President and Chief Operating Officer of Biolase, Inc. from June 2013 through December 2014.Officer. Biolase, Inc. is a medical device manufacturer and the leading providermanufacturer of lasers to the global dentistry industry. From July 2012 to June 2013 Dr. Arrowdental lasers. Before Biolase, he was the Chief Medical and Strategic Officer of Circuit Therapeutics, Inc., a company seeking to realize commercial potential in the field of optogenetics. From December 2007 through June 2012, Dr. Arrow was the Chief Financial Officer of Arstasis, Inc., a cardiology device manufacturer. From 2002 to 2007, Dr. Arrowhe headed medical technology equity research at the global investment bank Lazard Capital markets, LLC. Dr. Arrow spent two years 1999-2001 as Chief Financial Officer of the Patent & License Exchange later renamed PLX Systems, Inc., and three years as the publishing life sciences research analyst at Wedbush Morgan Securities. In 1996, Dr. Arrow was a surgical resident at the UCLA Medical Center. Dr. Arrow received his CFA in 1999. He was awarded an M.D. from Harvard Medical School in 1996 and a B.A. in Biophysics,magna cum laude, from Cornell University in 1992.
Khalil Barrage, Director, joined us in July, 2007. Mr. Khalil Barrage has served as a Managing Director of The Invus Group, LLC since 2003, in charge of the Public Equities Group that he set up in September 2003. Invus manages over $3B of capital, with a primary focus is on private equity investments, biotechnology and health care. In addition, Invus manages a fund-of-funds liquid alternative investment and, most recently, the newly established public equities portfolio activity. Mr. Barrage is a value investor. He started his career in 1988 with The Olayan Group, a multibillion private group. He was in charge of the group’s U.S. public equities portfolio, overseeing more than $2 billion of assets. Mr. Barrage holds a BA from American University of Beirut.
Robert B. Stein, PhD. MD, PhD., Director, Chief Medical Officer, joined us effective the closing of the Merger in February, 2016. Dr. Robert B. Stein is Chief Scientific Officerretired as President of R&D at Agenus Inc. in April 2017. He continues as Senior Advisor, R&D for both Agenus, Inc. and its cell therapy partially-owned subsidiary MiNK Therapeutics (Nasdaq: INKT). Dr. Robert B. Stein leadslead Agenus’ Research, Preclinical Development and Translational Medicine functions. He helps shape clinical development strategy for vaccines and adjuvants. Additionally, he’s leadinghe lead integration of the 4-Antibody, acquisition,PhosImmune, and Xoma Pilot Plant acquisitions, which includes the company’s fully human antibody drug discovery and optimization technology platform, and portfolio of immune checkpoint antibody programs. Over his 3035 years of experience in the biopharmaceutical industry he played a pivotal role in bringing to the market Sustiva®, Fablyn®, Viviant®, PanRetin®, TargRetin®, Promacta® and Eliquis®. Prior to joining Agenus, he held executive management positions at Ligand Pharmaceuticals, DuPont Merck, Incyte Pharmaceuticals, Roche Palo Alto and KineMed. Dr. Stein began his career at Merck, Sharp and Dohme. He holds an MD and a PhD in Physiology & Pharmacology from Duke University. Dr. Stein filed a personal voluntary bankruptcy petition under Chapter 7 in August of 2012 and the bankruptcy was discharged in May 2013.
Andrew Slee, PhD, Chief Operating Officer. Dr. Andy Slee joined us in April 2016. During his 37-year pharmaceutical career, Mr. Slee has taken several drugs from inception through all their pre-clinical and early clinical testing. During the past 37 years, he has worked for Preclinical CROs, immune-oncology companies and natural product companies focusing on anti-infectives, cancer, CNS, diabetes and inflammatory diseases. Spreading his influence beyond a single company, he created and ran his own Contract Research Organization (CRO), VivoSource Laboratories, which for ten years from 2003 to 2013 provided preclinical proof of concept catering to biopharmaceutical companies. For the 18 years before that, Mr. Slee shepherded multiple pharma targets in several therapeutic areas from inception onward at DuPont Pharmaceuticals. He is a graduate of Syracuse University and Leeds University.
| 49 |
Joshua Silverman,Khalil Barrage, Director, joined us effective the closing of the Merger in February 2016.July, 2007. Mr. Silverman is the Co–founder and Managing Member of Parkfield Funding LLC, and is a former Principal and Managing Partner of Iroquois Capital Management, LLC. Mr. Silverman served as Co–Chief Investment Officer of Iroquois from 2003 until July 2016. From 2000 to 2003, Mr. Silverman served as Co–Chief Investment Officer of Vertical Ventures, LLC, a merchant bank. He also serves as the Chairman of the Board of Neurotrope, Inc. (Nasdaq: NTRP). Prior to forming Iroquois, Mr. Silverman was a Director of Joele Frank, a boutique consulting firm specializing in mergers and acquisitions. Previously, Mr. Silverman served as Assistant Press Secretary to The President of The United States. Mr. Silverman received his B.A. from Lehigh University in 1992. In the past five years, Mr. SilvermanKhalil Barrage has served as a directorManaging Director of MGT Capital Investments, Inc.The Invus Group, LLC since 2003, in charge of the Public Equities Group that he set up in September 2003. Invus manages over $3B of capital, with a primary focus is on private equity investments, biotechnology and National Holdings Corporation,health care. In addition, Invus manages a fund-of-funds liquid alternative investment and, most recently, the newly established public equities portfolio activity. Mr. Barrage is a value investor. He started his career in 2016, became1988 with The Olayan Group, a directormultibillion private group. He was in charge of WPCS International, Inc.the group’s US public equities portfolio, overseeing more than $2 billion of assets. Mr. Barrage holds a BA from American University of Beirut.
Brian J. Corvese, Director, joined us on July 28, 2017, filling the open board seat vacated by Gregory H. Ekizian. Since 1999, Mr. Corvese has been the President and Founder of Vencor Capital (“Vencor”), a private equity firm with telecommunications and technology investments in the Middle East and Mediterranean regions. Prior to working at Vencor, Mr. Corvese worked on investments in the U.S. and global equity markets as a Managing Director and partner at Soros Fund Management, the largest hedge fund in the world at the time. From 1988 to 1996, Mr. Corvese was a partner at Chancellor Capital Management (“Chancellor”), a $25 billion money management firm. While at Chancellor, Mr. Corvese was a Portfolio Manager with responsibility for investments made in basic industries, restructurings, and special situations, corporate governance investments, as well as founded and managed his own hedge fund. From 1981 to 1988, Mr. Corvese was with Drexel Burnham Lambert (“Drexel”) as an equity analyst following the chemical and specialty chemical industries and participated in a significant number of merger and acquisition activities. While at Drexel, Mr. Corvese was a member of the top chemical and specialty chemical research team, as ranked by Institutional Investor. Mr. Corvese currently serves on the board of directors of Agenus Inc. and the National Telecommunications Corporation, based in Cairo, Egypt. Mr. Corvese earned degrees in finance and political science from The University of Rhode Island and attended New York University Graduate School. With over 30 years of experience in the financial industry, Mr. Corvese brings substantial financial expertise to our Board.
Timothy Wright, Director, joined us on November 23, 2022, filling the open board seat vacated by Joshua Silverman. Mr. Wright was the Chief Executive Officer of MiMedX Group, Inc., a position he held from May 2019 through September 2022. MiMedX is an advanced wound care and emerging therapeutic biologics company. He is currently the senior advisor to the Wake Forest Institute of Regenerative Medicine and a Director of BIORG , a human organoid development company. Mr. Wright also currently serves on the board of directors of Agenus Inc., which he has served on since 2006. Mr. Wright also serves as a Partner at Signal Hill Advisors, LLC, a position he has held since February 2011. In addition, Mr. Wright serves as Chairman of The Ohio State University Comprehensive Cancer Center Drug Development Institute and Director of the Ohio State University Innovation Foundation. Mr. Wright previously held several executive roles at Covidien (now Medtronic), Teva Pharmaceuticals Industries Ltd., DuPont Merck, Elan Bio-Pharmaceuticals, M2Gen Corp. and Curaxis Pharmaceuticals Corporation. As our Lead Director, Mr. Wright brings 32 years of experience on boards of companies in North America, Europe, Asia and Japan.
Jennifer Buell, PhD, Director, joined our board in July 2020. Dr. Buell is the President and Chief Operating Officer of Agenus, Inc., where she has previously served as served as the Head of Global R&D operations, Head of Research, and Chief Communications and External Affairs Officer. She is also the president of Agenus’ cell therapy partially-owned subsidiary MiNK Therapeutics, Inc, (Nasdaq: INKT). With 20 years of biopharmaceutical R&D experience, Dr. Buell has extensive knowledge in advancing discovery candidates through development and experience communicating with external stakeholders including regulators, investors, and collaborators. She has a proven record of success in R&D leadership, most recently at Agenus, where she led high performing teams in advancing candidates into the clinic and delivered on key alliance collaborations. Prior to joining Agenus, Dr. Buell held leadership positions in R&D operations at Bristol-Myers Squibb and later was responsible for Program and Alliance Management at Harvard Clinical Research Institute (Baim), where she was involved in the development strategy and operations for a portfolio of industry and government sponsored clinical programs. Dr. Buell obtained her PhD in Cellular, Biochemical, and Molecular Biochemistry with an MS in Biostatistics from Tufts University in Boston.
Consultants and Advisors
Dalia Barsyte PhD, Chief Technology Advisor. Dr. Dalia Barsyte received her PhD in molecular and cellular biology from the University of Manchester, UK. She did the postdoctoral training at the University of Manchester and Ontario Cancer Institute, and currently is a scientist at the University of Toronto, Structural Genomics Consortium, where she has been employed since 2009. Dr. Barsyte is an inventor on one of the key Protagenic patents and author of over 50 scientific publications in oncology and neuroscience. Dr. Barsyte’s scientific interests include exploring chemical biology in therapeutic target validation through peptide or small molecule chemical probe compounds as well as novel in vitro models of disease based on patient derived cell culture.
David A. Lovejoy, PhD, Chief Scientific Advisor, is one of our founders and joined us in September 2004. He holds a PhD in Neuroendocrinology from the University of Victoria (Victoria, BC) and spent three years at the Clayton Foundation Laboratories for Peptide Biology at the Salk Institute (San Diego, CA) as a postdoctoral fellow. Dr. Lovejoy took his first academic appointment at the University of Manchester (Manchester, UK), one of the United Kingdom’s top-ranking research universities. He joined the University of Toronto (Toronto, Ontario) in 2000 and is currently Professor of Neuroendocrinology in the Department of Cell and Systems Biology at the University of Toronto. He is the author of more than 210 scientific publications including 3three books in the field and an Associate Editor for a scientific journal and is inventor or co-inventor on all of our intellectual property.
Andrew Slee, DevelopmentDalia Barsyte PhD, Scientific Advisor. Andy Slee joined usDr. Dalia Barsyte received her PhD in April 2016 During his career, he has taken several drugsmolecular and cellular biology from inception through all their pre-clinicalthe University of Manchester, UK. She did the postdoctoral training at the University of Manchester and early clinical testing. During the past five years he has worked for Preclinical CROs, immune-oncology companiesOntario Cancer Institute, and natural product companies focusing on anti-infectives, cancer, CNS, diabetes and inflammatory diseases Spreading his influence beyond a single company, he created and ran his own Contract Research Organization (CRO), VivoSource Laboratories, which for ten years from 2003 to 2013 provided preclinical proof of concept catering to biopharmaceutical companies. For the 18 years before that, Mr. Slee shepherded multiple pharma targets in several therapeutic areas from inception onward at DuPont Pharmaceuticals. Hecurrently is a graduatescientist at the University of Syracuse UniversityToronto, Structural Genomics Consortium, where she has been employed since 2009. Dr. Barsyte is an inventor on one of the key Protagenic patents and Leeds University
Christina Faragalla, Directorauthor of Project Management, joined usover 50 scientific publications in June 2016. Ms. Faragalla is responsible for managing communicationoncology and timelinesneuroscience. Dr. Barsyte’s scientific interests include exploring chemical biology in the Company’s development projects,therapeutic target validation through peptide or small molecule chemical probe compounds as well as beingnovel in vitro models of disease based on patient derived cell culture.
Zack Armen, Strategic Advisor. Mr. Armen became involved with Protagenic in Fall 2018, and brings experience in strategic finance and life sciences venture investing to the Company’s primary interface with its Contract Research Organizations (CROs). Priorcompany through roles at Goldman Sachs, Flagship Pioneering, CiBO Technologies, and his current role as Director of Corporate Development at Valo Health.
Mark Berg, Strategic Consultant. Mr. Berg became a strategic consultant to working with the Company, Ms. Faragalla servedProtagenic in roles both on the sponsorJanuary 2022. He brings several decades of perspective regarding publicly-traded biotechnology companies’ perceptions by investors.
Director Independence
Each of Messrs. Corvese, Wright, and CRO side. From 2010-2014 she worked with large Pharma clients overseeing late phase CNS programs at PAREXEL. From 2014-2016, she transitioned to exclusively serving emerging biotech clientsBarrage are “independent” members of our board of directors as “independence” is defined in early development while at at Novella Clinical, a division of Quintiles CRO, running several first-in-man clinical trials. She is an expert in Global Clinical Operations, SOP Development and Harmonization, Translational medicine, POC to Early and Late phase drug development, IND to NDA to large registries and post marketing trials. She holds a MS Clinical Research Administration from George Washington University, and a BS in Biology from Rutgers College.Nasdaq Marketplace Rule 5605(a)(2).
Family Relationships
There are no family relationships between or among the directors, executive officers or persons nominated or chosen by our stockholders or us to become directors or executive officers. There is one family relationship between Strategic Advisor Zack Armen and our Executive Chairman, Garo Armen (Garo Armen is Zack Armen’s father).
Former Voting Agreement
On February 12, 2016, the Company and certain of its stockholders (currently(then representing approximately 43% of the Company’s issued and outstanding common stock), including Messrs.Drs. Armen, Arrow and former director Mr. Greg Ekizian and former shareholder Strategic Bio Partners, LLC, entered into a voting agreement whereby these stockholders agreed to vote in favor of setting and maintaining the size of the Board at five directors (unless increased by the Board), the election of one director designated by Strategic Bio Partners, LLC (Mr. Silverman) and the election of four directors designated by Mr. GaroDr. Armen (so long as Mr. GaroDr. Armen is an officer or director of the Company). The term of the voting agreement runs untilterminated on February 12, 2019 unless terminated earlier by a vote of at least 90% of the stockholders party to the agreement or the consummation of a firm commitment underwritten public offering of the Company’s common stock resulting in proceeds to the Company of at least $20 million.2019.
Involvement in Certain Legal Proceedings
To our knowledge, during the past ten years, none of our directors, executive officers, promoters, control persons, or nominees has:
been convicted in a criminal proceeding or been subject to a pending criminal proceeding (excluding traffic violations and other minor offenses);
Exceptexcept as set forth above with respect to Dr. Stein, had any bankruptcy petition filed by or against the business or property of the person, or of any partnership, corporation or business association of which he was a general partner or executive officer, either at the time of the bankruptcy filing or within two years prior to that time;
| 51 |
been subject to any order, judgment, or decree, not subsequently reversed, suspended or vacated, of any court of competent jurisdiction or federal or state authority, permanently or temporarily enjoining, barring, suspending or otherwise limiting, his involvement in any type of business, securities, futures, commodities, investment, banking, savings and loan, or insurance activities, or to be associated with persons engaged in any such activity;
been found by a court of competent jurisdiction in a civil action or by the SEC or the Commodity Futures Trading Commission to have violated a federal or state securities or commodities law, and the judgment has not been reversed, suspended, or vacated;
been the subject of, or a party to, any federal or state judicial or administrative order, judgment, decree, or finding, not subsequently reversed, suspended or vacated (not including any settlement of a civil proceeding among private litigants), relating to an alleged violation of any federal or state securities or commodities law or regulation, any law or regulation respecting financial institutions or insurance companies including, but not limited to, a temporary or permanent injunction, order of disgorgement or restitution, civil money penalty or temporary or permanent cease-and-desist order, or removal or prohibition order, or any law or regulation prohibiting mail or wire fraud or fraud in connection with any business entity; or
been the subject of, or a party to, any sanction or order, not subsequently reversed, suspended or vacated, of any self-regulatory organization (as defined in Section 3(a)(26) of the Exchange Act), any registered entity (as defined in Section 1(a)(29) of the Commodity Exchange Act), or any equivalent exchange, association, entity or organization that has disciplinary authority over its members or persons associated with a member.
Code of Business Conduct and Ethics
On February 24, 2017, we adopted a written Code of Business Conduct and Ethics. Guidelines on Significant Governance Issues, and Process for Security Holder Communications with Directors, each of which is attachedfiled as an exhibit hereto.to this annual report.
Board Committees
Our Boardboard of Directorsdirectors has established threefive standing committees: an Audit Committee, a Compensation Committee, and a Nominating and Corporate Governance Committee, a Science Committee and a Clinical and Regulatory Committee. Each of these committees will operate under a charter that has been approved by our Boardboard of Directors.directors.
Audit Committee.The Audit Committee will oversee and monitor our financial reporting process and internal control system, review and evaluate the audit performed by our registered independent public accountants and reports to the Board any substantive issues found during the audit. The Audit Committee will be directly responsible for the appointment, compensation and oversight of the work of our registered independent public accountants. The Audit Committee will review and approve all transactions with affiliated parties. The Audit Committee shall be comprised on two or more independent directors who shall be appointed annually and subject to removal by the Board at any time. Each member of the Audit Committee shall meet the independence requirements of The NASDAQ Stock Market, LLC, and SEC regulations, as well as any other applicable requirements. On June 20, 2017, Gregory K. Ekizian, a director of the CompanyMessrs. Corvese (Committee Chairperson), Wright, and the Chairman ofBarrage comprise the Audit Committee, each of the Company’s Board of Directors, notified the Company that he was resigning from the Board effective immediately. On July 25, 2017, our Board appointed Brian Corvese to the Audit Committee, whom meets the independence requirements. In addition, the Board also designated Brian Corvese as an “audit committee financial expert,” as that term is defined by the NASDAQNSADAQ Listing Rules and SEC regulations.
Compensation Committee.The Compensation Committee will provide advice and make recommendations to the Boardboard in the areas of employee salaries, benefit programs and director compensation. The Compensation Committee will also review the compensation of our President, Chief Executive Officer, and other officers and make recommendations in that regard to the Boardboard as a whole. The Compensation Committee shall be comprised on three or more directors who shall be appointed annually and subject to removal by the Board at any time. The Compensation Committee must have at least two members, and must consist solely of independent directors. Our Board appointed Messrs. Barrage, (Committee Chairperson) and Corvese, and Dr. Stein toWright comprise the Compensation Committee and are all of whom are independentindependent.
| 52 |
Nominating and Corporate Governance Committee.The Nominating and Corporate Governance Committee will nominate individuals to be elected to the full Boardboard by our stockholders. The Nominating and Corporate Governance Committee will determine the slate of director nominees for election to the Board, to identify and recommend candidates to fill vacancies occurring between annual stockholder meetings, to review the Company’s policies and programs that relate to matters of corporate responsibility, including public issues of significance to the Company and its stockholders. The Nominating and Corporate Governance Committee shall be comprised of threeon two or more directors who shall be appointed annually and subject to removal by the Board at any time. Each member of the Nominating and Corporate Governance Committee may or may not meet the independence requirements of The NASDAQ Stock Market, LLC and SEC regulations. Messrs. Wright (Committee Chairperson), and Mr. Corvese comprise the Nominating and Corporate Governance Committee.
Science Committee. The Science Committee will meet regularly to review the strategic direction being taken by Management with respect to developing the Company’s scientific assets. A key function of the Science Committee is to ensure that the Company is targeting disease indications for its drug candidates that take full advantage of the drug candidates’ potential, within the constraints of the working capital available to the Company. This process is expected to continually necessitate difficult choices concerning how many disease targets to pursue. The Science Committee will be directly responsible for the appointment, compensation and oversight of the Company’s top scientific staff. The Science Committee will review and approve all major contractual agreements with contract research organizations. Each member of the Science Committee may or may not meet the independence requirements of The NASDAQ Stock Market, LLC and SEC regulations Drs. Stein (Committee Chairperson), Buell, and Armen comprise the Science Committee.
Clinical and Regulatory Committee: The Clinical and Regulatory committee will meet at least once per year to review progress of the clinical trial programs of the Company. The Clinical and Regulatory committee was created in July 2020 and Dr. Jennifer Buell was appointed as its chair. Each member of the Clinical and Regulatory Committee may or may not meet the independence requirements of The NASDAQ Stock Market, LLC and SEC regulations. Drs. Buell (Chair), Armen, and Stein comprise the Clinical and Regulatory Committee.
Limitation of Directors Liability and Indemnification
The Delaware General Corporation Law authorizes corporations to limit or eliminate, subject to certain conditions, the personal liability of directors to corporations and their stockholders for monetary damages for breach of their fiduciary duties. Our certificate of incorporation limits the liability of our directors to the fullest extent permitted by Delaware law.
We have director and officer liability insurance to cover liabilities our directors and officers may incur in connection with their services to us, including matters arising under the Securities Act. Our certificate of incorporation and bylaws also provide that we will indemnify our directors and officers who, by reason of the fact that he or she is one of our officers or directors of our Company,company, is involved in any action, suit or proceeding, whether civil, criminal, administrative or investigative related to their board role with the Company.company.
We have entered into indemnification agreements with each of our directors and executive officers. It is anticipated that future directors and officers will enter into an Indemnification Agreement with us in substantially similar form. The Indemnification Agreement provides, among other things, that we will indemnify and hold harmless each person subject to an Indemnification Agreement (each, an “Indemnified Party”) to the fullest extent permitted by applicable law from and against all losses, costs, liabilities, judgments, penalties, fines, expenses and other matters that may result or arise in connection with such Indemnified Party serving in his or her capacity as a director of ours or serving at our direction as a director, officer, employee, fiduciary or agent of another entity. The Indemnification Agreement further provides that, upon an Indemnified Party’s request, we will advance expenses to the Indemnified Party to the fullest extent permitted by applicable law. Pursuant to the Indemnification Agreement, an Indemnified Party is presumed to be entitled to indemnification and we have the burden of proving otherwise. The Indemnification Agreement also requires us to maintain in full force and effect directors’ liability insurance on the terms described in the Indemnification Agreement. If indemnification under the Indemnification Agreement is unavailable to an Indemnified Party for any reason, we, in lieu of indemnifying the Indemnified Party, will contribute to any amounts incurred by the Indemnified Party in connection with any claim relating to an indemnifiable event in such proportion as is deemed fair and reasonable in light of all of the circumstances to reflect the relative benefits received or relative fault of the parties in connection with such event.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to our directors, officers, and controlling persons pursuant to the foregoing provisions, or otherwise, we have been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment of expenses incurred or paid by a director, officer or controlling person in a successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, we will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to the court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue.
There is no pending litigation or proceeding involving any of our directors, officers, employees or agents in which indemnification will be required or permitted. We are not aware of any threatened litigation or proceeding that may result in a claim for such indemnification.
Item 11. Executive Compensation.
The following table sets forth information regarding each element of compensation that we paid or awarded to our named executive officers and for fiscal years ended December 31, 20172023 and 2016.2022.
Summary Compensation Table
| Name and Principal Position | Year | Salary | Bonus ($) | Stock Awards ($) | Option Awards ($) | Non-Equity Incentive Plan Compensation ($) | Deferred Compensation ($) | All Other Compensation ($) | Total Compensation ($) | ||||||||||||||||||||
| Garo H. Armen, Chairman | 2017 | N/A | N/A | N/A | $ | 312,500 | (3) | N/A | N/A | N/A | $ | 312,500 | |||||||||||||||||
| 2016 | N/A | N/A | N/A | $ | 580,000 | (1) | N/A | N/A | N/A | $ | 580,000 | ||||||||||||||||||
| Robert Ziroyan, Chief Operating Officer and Interim President (6) | 2017 | $ | 13,168 | $ | 0 | $ | 0 | $ | 0 | $ | 0 | $ | 0 | $ | 0 | $ | 13,168 | ||||||||||||
| 2016 | $ | 16,861 | $ | 0 | $ | 0 | $ | 0 | $ | 0 | $ | 0 | $ | 2,869 | (2) | $ | 19,730 | ||||||||||||
| Alexander K. Arrow, Chief Financial Officer | 2017 | $ | 125,000 | $ | 0 | $ | 0 | $ | 93,750 | (4) | $ | 0 | $ | 0 | N/A | $ | 218,750 | ||||||||||||
| 2016 | $ | 106,552 | $ | 0 | $ | 0 | $ | 277,400 | (5) | $ | 0 | $ | 0 | N/A | $ | 383,952 | |||||||||||||
| Name and Principal Position | Year | Salary | Bonus ($) | Stock Awards ($) | Option Awards ($) | Non-Equity Incentive Plan Compensation ($) | Non- Qualified Deferred Compensation Earnings ($) | All Other Compensation ($) | Total Compensation ($) | |||||||||||||||||||||||||||
| Garo H. Armen, | 2023 | 0 | 0 | 0 | $ | 0 | 0 | 0 | 0 | $ | 0 | |||||||||||||||||||||||||
| Chairman | 2022 | 0 | 0 | 0 | $ | 0 | 0 | 0 | 0 | $ | 0 | |||||||||||||||||||||||||
| Alexander K. Arrow, | 2023 | $ | 150,000 | $ | 0 | $ | 0 | $ | 0 | $ | 0 | $ | 0 | 0 | $ | 150,000 | ||||||||||||||||||||
| Chief Financial Officer | 2022 | $ | 150,000 | $ | 0 | $ | 0 | $ | 0 | $ | 0 | $ | 0 | 0 | $ | 150,000 | ||||||||||||||||||||
Employment Arrangements with Officers and Directors
Dr. Alexander Arrow, our Chief Financial Officer, receives base compensation of $125,000$150,000 per year for his part-time work for us. In addition,us, an increase from the $125,000 he received until July 1, 2021, except for an 18-month period from February 2019 through August 2020 during which he received zero cash salary and three grants totaling 88,541 options in lieu of cash salary. From 2016 through 2020, cumulatively, Dr. Arrow received 100,00025,000 options under the 2006 Plan as a sign-on bonus when he joined us and 140,000three grants totaling 335,000 incentive options in the aggregate under the 2016 plan on April 15, 2016. These options have anwith exercise priceprices of $1.25$5.00 and $7.00 per share, a ten-year term and vest over a three-year period in 35 monthly installments of 2,778 shares and a final installment of 2,770 shares and 3,889 shares and a final installment of 3,885 shares, respectively. On October 16, 2017, we granted Dr. Arrow another ten-year option to purchase 75,000 shares of our common stock at an exercise price of $1.75 per share, which vest in 35 monthly installments of 2,083 shares and a final installment of2,095.share. The terms of Dr. Arrow’s option grants also include full vesting acceleration upon a change of control. Drs. Arrow
Consulting Agreements
Andrew Slee, PhD, Chief Operating Officer. In December 2020, we entered into a consulting agreement with Dr. Slee to act as our Chief Operating Officer. We granted Dr. Slee (i) 25,000 options on April 15, 2016, at an exercise price of $5.00 per option, (ii) 18,750 options on October 16, 2017, at an exercise price of $7.00 per option, (iii) 18,750 options on July 18, 2020, at an exercise price of $7.00 per option, (iv) 37,500 options on February 13, 2020, at an exercise price of $7.00 per option, and Armen are the only currently two executive officers(v) 12,500 options on February 25, 2021, at an exercise price of the Company.$22.40.
Consultancy Agreements
Dalia Barsyte PhD, Chief Technology Officer.Scientific Advisor. Our subsidiary, Protagenic Therapeutics Canada (2006) Inc., entered into a consulting agreement with Dr. Dalia Barsyte. Dr. Barsyte is responsible for overseeing i) design and development of ELISA assays for measuring TCAP, ii) evaluation of TCAP exposure biomarker assay, iii) development of pipeline peptides, iv) development of clinically compatible formulations for TCAP, as well as all of the bench research and development of formulation and extraction methods. Her consulting agreement is effective through December 2017. She is compensated at the rate of up to $3,000 (Canadian) per month, if she works at least 20 hours on behalf of the Company. As well, we have granted Dr. Barsyte 10,0002,500 shares of our common stock and ten-year options to purchase 150,00037,500 shares of our common stock. Options to purchase 100,00025,000 shares of common stock, at an exercise price of $1.00$4.00 per share, have fully vested; the options to purchase the remaining 50,00012,500 shares of common stock, at an exercise price of $1.25$5.00 per share, vested in March 2016. On October 16, 2017, we granted Dr. Barsyte another ten-year option to purchase 20,0005,000 shares of our common stock at an exercise price of $1.75$7.00 per share. On February 13, 2020, we granted Dr. Barsyte ten-year option to purchase 2,500 shares of our common stock at an exercise price of $7.00 per share.
| 54 |
Robert B. Stein, PhD, MD, Director, Chief Medical Officer. We entered into a consulting agreement with Dr. Stein effective January 2015.2015, and amended and restated this consulting agreement in December 2020 to appoint Dr. Stein as our Chief Medical Officer. Dr. Stein is responsible for providing us with technical and advisory services related to our research and development efforts. The consulting agreement is effective through January 2020. On January 23, 2015, we granted Dr. Stein ten-year options to purchase 200,00050,000 shares of our common stock, at an exercise price of $1.25$5.00 per share.share (the “January Options”). The options vest in increments of 1.667% per month on the first day of each calendar month following January 2015, such that the shares shall beOptions are fully vested on January 23, 2020, provided Dr. Stein remains a consultant to us. On October 16, 2017, wevested. We granted Dr. Stein a ten-year grant to purchase another 200,000 shares of our common stock,(i) 10,000 options on April 15, 2016, at an exercise price of $1.75$5.00 per share.option, (ii) 50,000 options on October 16, 2017, at an exercise price of $7.00 per option, (iii) 37,500 options on February 13, 2020, at an exercise price of $5.00 per option, and (iv) 12,500 options on February 25, 2021, at an exercise price of $22.40 per option.
Christina Faragalla, DirectorJennifer Buell, PhD, Clinical and Regulatory Development Advisor. We entered into a consulting agreement with Dr. Buell effective February 2021, providing for her to do three things: (1) advise the Company’s clinical and regulatory development plan to support the Company’s lead product candidate, PT00114, in support of Project Managementan IND application to the U.S. Food and Drug Administration and demonstration of safety and clinical activity in early phase clinical trials, (2) develop a panel of experts to prepare a clinical development plan and operational plan that would enable the evaluation of safety and clinical activity of the companies lead therapeutic, PT00114, and (3) determine the fastest development pathway of PT00114 in four key indications as defined by the Company Management. We granted Dr. Buell 50,000 nonstatutory stock options (“NSOs”) on February 25, 2021 at an exercise price of $22.40 per share, and 36,250 options on July 18, 2020 at an exercise price of $7.00 per share.
Mark Berg, Strategic Advisor. We entered into a consulting agreement with Ms. FaragallaMr. Berg effective June 2016, via her consultancy entitled Lotus Clinical Consulting. She is compensated atJanuary 2022, providing for him to provide strategic consulting services to the rate $100 per hour invoices, subject to a 12-month capcompany, none of $100,000. In addition,which involves direct contact with investors. We granted Mr. Berg 12,500 nonstatutory stock options (“NSOs”) on OctoberJanuary 26, 2016, we granted Ms. Faragalla ten-year options to purchase 25,000 shares of our common stock,2022 at an exercise price of $1.25$4.84 per share. On October 16, 2017, we granted Ms. Faragalla another ten-year option to purchase 30,000 shares of our common stock, at an exercise price of $1.75 per share.
Outstanding Equity Awards at Fiscal Year End
The following table summarizes the equity awards made to our named executive officers that were outstanding at December 31, 2017.
| Name | No. of Securities Underlying Unexercised Options (#) Exercisable | No. of Securities Underlying Unexercised Options (#) Unexercisable | Option Exercise Price | Option Expiration Date | |||||||
| Garo H. Armen (1) | 284,725 | 215,275 | $ | 1.25 | April 15, 2026 | ||||||
| Garo H. Armen (2) | 17,361 | 232,639 | $ | 1.75 | October 16, 2027 | ||||||
| Robert Ziroyan (3) | 100,000 | — | $ | 1.00 | March 30, 2021 | ||||||
| Robert Ziroyan (3) | 50,000 | — | $ | 1.00 | March 1, 2024 | ||||||
| Robert Ziroyan (3) | 75,000 | — | $ | 1.25 | March 9, 2025 | ||||||
| Alexander K. Arrow (4) | 58,333 | 41,666 | $ | 1.25 | February 12, 2026 | ||||||
| Alexander K. Arrow (4) | 79,725 | 60,275 | $ | 1.25 | April 15, 2026 | ||||||
| Alexander K. Arrow (5) | 5,208 | 69,782 | $ | 1.75 | October 16, 2027 | ||||||
For Drs. Armen and Arrow, following a qualified Change of Control, a resignation for Good Reason, or an involuntary termination other than For Cause, 100% of the executives’ then-unvested options shall become immediately vested.
Director Compensation
During fiscal year 2017 we compensated directors who were not employees of the Company.
| Name | Fees earned or paid in cash | Stock awards | Option awards (1) | Non-equity incentive plan compensation | Nonqualified deferred compensation earnings | All other compensation | Total | |||||||||||||||||||||
| Garo H. Armen | $ | 312,500 | $ | 312,500 | ||||||||||||||||||||||||
| Khalil Barrage | $ | 243,750 | $ | 243,750 | ||||||||||||||||||||||||
| Joshua Silverman | $ | 56,250 | $ | 56,250 | ||||||||||||||||||||||||
| Robert Stein | $ | 250,000 | $ | 250,000 | ||||||||||||||||||||||||
| Brian Corvese | $ | 118,750 | $ | 118,750 | ||||||||||||||||||||||||
| Greg Ekizian | $ | 0 | $ | 0 | ||||||||||||||||||||||||
Going forward, on April 15 of each fiscal year, we plan to grant each non-employee directors will receivedirector an option under the 2016 Plan to purchase 40,00010,000 shares of common stock, as well as an option to purchase 5,0001,250 shares for each committee which they chair. No additional options shall be granted for serving on a committee without being its chair. All options will be granted at fair market value, as defined in the 2016 Plan, on the date of grant, and will vest over a three-year period in equal monthly installments. Vesting will accelerate in certain circumstances, such as a change of control of the Company, and unvested options will terminate upon the cessation of an individual’s service to us as a director.
Non-employee directors may be reimbursed for their reasonable expenses in attending Board and committee meetings.
We entered into aan amended and restated consulting agreement during fiscal year 2020 with Robert B. Stein, PhD, MD, under which is described above.we issued 137,500 options on February 13, 2020, at an exercise price of $5.00 per option. During fiscal year 2021 with Robert B. Stein, PhD, MD, under which we issued 12,500 options on February 20, 2021, at an exercise price of $22.40 per option.
| 55 |
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
Equity Compensation Plans
Equity Compensation Plan Information
| Plan category | (a) No. of securities to be issued upon exercise of outstanding options, warrants and rights | (b) Weighted-average exercise price of outstanding options, warrants and rights | (c) No. of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a) | (a) No. of securities to be issued upon exercise of outstanding options, warrants and rights | (b) Weighted-average exercise price of outstanding options, warrants and rights | (c) No. of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a) | ||||||||||||||||
| Equity compensation plans approved by security holders | 6,453,887 | $ | 1.18 | 2,148,300 | 2,300,032 | $ | 12.34 | 1,279,181 | ||||||||||||||
| Equity compensation plans not approved by security holders | 0 | 0 | 0 | 0 | 0 | 0 | ||||||||||||||||
| Total | 6,453,887 | $ | 1.18 | 2,148,300 | 2,300,032 | $ | 12.34 | 1,279,181 | ||||||||||||||
In connection with the Merger, we adopted Protagenic’s 2006 Employee, Director and Consultant Stock Plan (the “2006 Plan”).
On June 17, 2016, our stockholders adopted our 2016 Equity Compensation Plan and, as a result, we terminated the 2006 Plan. We will not grant any further awards under the 2006 Plan. All outstanding grants under the 2006 Plan will continue in effect in accordance with the terms of the particular grant and the 2006 Plan.
2006 Employee, Director and Consultant Stock Plan
The following description of the pertinent terms of the 2006 Plan is a summary and is qualified in its entirety by the full text of the 2006 Plan.
Administration.The administrator (the “Administrator”) of the 2006 Plan is the Board of Directors, except to the extent the Board of Directors delegates its authority to the Compensation committee (the “Committee”) of the Board, in which case the Committee shall be the Administrator. Subject to the provisions of the 2006 Plan, the Administrator is authorized to:
provided, however, that all such interpretations, rules, determinations, terms and conditions shall be made and prescribed in the context of preserving the tax status under Section 422 of the Code of those options which are designated as ISOs. Subject to the foregoing, the interpretation and construction by the Administrator of any provisions of the 2006 Plan or of any award granted under it shall be final.
If permissible under applicable law, the Board of Directors or the Committee may allocate all or any portion of its responsibilities and powers to any one or more of its members and may delegate all or any portion of its responsibilities and powers to any other person selected by it. Any such allocation or delegation may be revoked by the Board of Directors or the Committee at any time.
Terms and Conditions of Options. Options granted under the 2006 Plan may be either “incentive stock options” that are intended to meet the requirements of Section 422 of the Code or “nonqualified stock options” that do not meet the requirements of Section 422 of the Code. The Administrator will determine the exercise price of options granted under the 2006 Plan. The exercise price of stock options may not be less than the fair market value per share of our common stock on the date of grant (or 110% of fair market value in the case of incentive stock options granted to a ten-percent stockholder).
If on the date of grant the common stock is listed on a stock exchange or national market system, the fair market value will generally be the closing sale price on the date of grant. If the common stock is not traded on a stock exchange or national market system on the date of grant, the fair market value will generally be the mean between the bid and the asked price for the common stock at the close of trading in the over-the-counter market for the trading day on which common stock was traded immediately preceding the applicable date. If no such prices are available, the fair market value shall be determined in good faith by the Administrator.
No option intended to qualify as an ISO may be exercisable for more than ten years from the date of grant (five years in the case of an incentive stock option granted to a ten-percent stockholder). Options granted under the 2006 Plan will be exercisable at such time or times as the Administrator prescribes at the time of grant. No employee may receive incentive stock options that first become exercisable in any calendar year in an amount exceeding $100,000.
Generally, the exercise price of an option may be paid (a) in cash or by certified bank check, (b) at the discretion of the Administrator, through delivery of shares of our common stock held for at least six months having a fair market value equal to the purchase price, (c) at the discretion of the Administrator, by delivery of the grantee’s personal note, for full, partial or no recourse, bearing interest payable not less than annually at market rate on the date of exercise and at no less than 100% of the applicable Federal rate, as defined in Section 1274(d) of the Code, with or without the pledge of such shares as collateral, or (d) at the discretion of the Administrator, in accordance with a cashless exercise program established with a securities brokerage firm, and approved by the Administrator, or (e) at the discretion of the Administrator, by any combination of the above methods.
| 56 |
No option may be transferred other than by will or by the laws of descent and distribution, and during a recipient’s lifetime an option may be exercised only by the recipient. The Administrator will determine the extent to which a holder of a stock option may exercise the option following termination of service with us.
The Administrator will determine the extent to which a holder of a stock option may exercise the option following termination of service with us.
Effect of Certain Corporate Transactions. If the Company is to be consolidated with or acquired by another entity in a merger, sale of all or substantially all of the Company’s assets other than a transaction to merely change the state of incorporation (a “Corporate Transaction”), the Administrator or the board of directors of any entity assuming the obligations of the Company hereunder (the “Successor Board”), shall, as to outstanding options, either (i) make appropriate provision for the continuation of such options by substituting on an equitable basis for the Shares then subject to such options either the consideration payable with respect to the outstanding shares of common stock in connection with the Corporate Transaction or securities of any successor or acquiring entity (provided, that, at the discretion of the Administrator, all unvested options shall be made fully or partially exercisable for purposes of this Subparagraph upon the closing of the Corporate Transaction);options; or (ii) upon written notice to the participants, provide that all options must be exercised (either to the extent then exercisable or, at the discretion of the Administrator, all options being made fully or partially exercisable), within a specified number of days of the date of such notice, at the end of which period the options shall terminate; or (iii) terminate all options in exchange for a cash payment equal to the excess of the fair market value of the shares of common stock subject to such options (either to the extent then exercisable or, at the discretion of the Administrator, all options being made fully or partially exercisable) over the exercise price thereof.
Tax Withholding. As and when appropriate, we shall have the right to require each optionee purchasing shares of common stock and each grantee receiving an award of shares of common stock under the 2006 Plan to pay any federal, state or local taxes required by law to be withheld.
2016 Equity Compensation Plan
The following description of the principal terms of the 2016 Plan is a summary and is qualified in its entirety by the full text of the 2016 Plan.
Administration.The 2016 Plan is administered by the Compensation Committee of our Board of Directors, provided that the entire Board of Directors may act in lieu of the Compensation Committee on any matter, subject to certain requirements set forth in the 2016 Plan. The Compensation Committee may grant options to purchase shares of our common stock, stock appreciation rights, stock units, restricted shares of our common stock, performance shares, performance units, incentive bonus awards, other cash-based awards and other stock-based awards. The Compensation Committee also has broad authority to determine the terms and conditions of each option or other kind of award, and adopt, amend and rescind rules and regulations for the administration of the 2016 Plan. Subject to applicable law, the Compensation Committee may authorize one or more reporting persons (as defined in the 2016 Plan) or other officers to make awards (other than awards to reporting persons, or other officers whom the Compensation Committee has specifically authorized to make awards). No awards may be granted under the 2016 Plan on or after the ten-year anniversary of the adoption of the 2016 Plan by our Board of Directors, but awards granted prior to such tenth anniversary may extend beyond that date.
Eligibility.Awards may be granted under the 2016 Plan to any person who is an employee, officer, director, consultant, advisor or other individual service provider of the Company or any subsidiary, or any person who is determined by the Compensation Committee to be a prospective employee, officer, director, consultant, advisor or other individual service provider of the Company or any subsidiary.
Shares Subject to the 2016 Plan. The aggregate number of shares of common stock proposed to be available for issuance in connection with options and awards granted under the 2016 Plan is 3,000,000750,000 shares. Incentive Stock Options may, but need not be, granted with respect to all of the shares available for issuance under the 2016 Plan;provided,,however,, that the maximum aggregate number of shares of common stock which may be issued in respect of Incentive Stock Options (after giving effect to any increases pursuant to the “evergreen” provisions of the 2016 Plan discussed below) shall not exceed 6,000,0001,500,000 shares, subject to adjustment in the event of stock, splits and similar transactions. If any award granted under the 2016 Plan payable in shares of common stock is forfeited, cancelled, or returned for failure to satisfy vesting requirements, otherwise terminates without payment being made, or if shares of common stock are withheld to cover withholding taxes on options or other awards, the number of shares of common stock as to which such option or award was forfeited, or which were withheld, will be available for future grants under the 2016 Plan.
| 57 |
In addition, the 2016 Plan contains an “evergreen” provision allowing for an annual increase in the number of shares of our common stock available for issuance under the 2016 Plan on January 1 of each year during the period beginning January 1, 2017, and ending on (and including) January 1, 2026. The annual increase in the number of shares shall be equal to (i) five point five percent (5.5%) of the total number of shares of common stock outstanding on December 31st of the preceding calendar year, or (ii) with respect to shares of common stock which may be issued under the 2016 Plan other than in respect to Incentive Stock Options, the difference between (x) eighteen percent (18%) of the total number of shares of common stock outstanding on December 31st of the preceding calendar year, and (y) the total number of shares of common stock reserved under the 2016 Plan on December 31st of such preceding calendar year (including shares subject to outstanding awards, issued pursuant to awards or available for future awards) if such amount is greater than the amount determined in (i) immediately above; provided, however, that our Board may act prior to the first day of any calendar year to provide that there shall be no increase such calendar year, or that the increase shall be a lesser number of shares of common stock than would otherwise occur. On January 1, 2017, 564,3782019, and 2020, each year 141,095 shares of common stock were added to the 2016 Plan pursuant to this evergreen provision. On January 1, 2018, another 564,3782021, 142,457 shares of common stock were added to the 2016 Plan pursuant to this evergreen provision. On January 1, 2022, 184,260 additional shares of common stock are available for issuance under the 2016 Plan as a result of operation of the evergreen provision: (a) 141,070 shares resulting from operation of the evergreen provision in 2019, which were never previously registered and (b) 43,191 shares resulting from operation of the evergreen provision in 2022. On January 1, 2023, 184,594 additional shares of common stock are available for issuance under the 2016 Plan as a result of operation of the evergreen provision resulting from operation of the evergreen provision in 2022.
Terms and Conditions of Options. Options granted under the 2016 Plan may be either “incentive stock options” that are intended to meet the requirements of Section 422 of the Code or “nonqualified stock options” that do not meet the requirements of Section 422 of the Code. The Compensation Committee will determine the exercise price of options granted under the 2016 Plan. The exercise price of stock options may not be less than the fair market value, on the date of grant, per share of our common stock issuable upon exercise of the option (or 110% of fair market value in the case of incentive options granted to a ten-percent stockholder).
If on the date of grant the common stock is listed on a stock exchange or national market system, the fair market value shall generally be the closing sale price as of such date, or if there were no trades recorded on such date, then the most recent date preceding such date on which trades were recorded. If on the date of grant the common stock is traded in an over-the-counter market, the fair market will generally be the average of the closing bid and asked prices for the shares of common stock as of such date, or, if there are no closing bid and asked prices for the shares of common stock on such date, then the average of the bid and asked prices for the shares of common stock on the most recent date preceding such date on which such closing bid and asked prices are available. If the common stock is not listed on a national securities exchange or national market system or traded in an over-the-counter market, the fair market value shall be determined by the Compensation Committee in a manner consistent with Section 409A of the Code. Notwithstanding the foregoing, if on the date of grant the common stock is listed on a stock exchange or is quoted on a national market system, or is traded in an over-the-counter market, then solely for purposes of determining the exercise price of any grant of a stock option or the base price of any grant of a stock appreciation right, the Compensation Committee may, in its discretion, base fair market value on the last sale before or the first sale after the grant, the closing price on the trading day before or the trading day of the grant, the arithmetic mean of the high and low prices on the trading day before or the trading day of the grant, or any other reasonable method using actual transactions of the common stock as reported by the exchange or market on which the common stock is traded. In addition, the determination of fair market value also may be made using any other method permitted under Treasury Regulation section 1.409A-1(b)(5)(iv).
No option may be exercisable for more than ten years from the date of grant (five years in the case of an incentive stock option granted to a ten-percent stockholder). Options granted under the 2016 Plan will be exercisable at such time or times as the Compensation Committee prescribes at the time of grant. No employee may receive incentive stock options that first become exercisable in any calendar year in an amount exceeding $100,000. The Compensation Committee may, in its discretion, permit a holder of a nonqualified stock option to exercise the option before it has otherwise become exercisable, in which case the shares of our common stock issued to the recipient will continue to be subject to the vesting requirements that applied to the option before exercise.
Generally, the option price may be paid in cash or by bank check, or such other means as the Compensation Committee may accept. As set forth in an award agreement or otherwise determined by the Compensation Committee, in its sole discretion, at or after grant, payment in full or part of the exercise price of an option may be made (a) in the form of shares of common stock that have been held by the participant for such period as the Compensation Committee may deem appropriate for accounting purposes or otherwise, valued at the fair market value of such shares on the date of exercise; (ii) by surrendering to the Company shares of common stock otherwise receivable on exercise of the option; (iii) by a cashless exercise program implemented by the Compensation Committee in connection with the 2016 Plan; and/or (iv) by such other method as may be approved by the Compensation Committee and set forth in an award agreement.
No option may be transferred other than by will or by the laws of descent and distribution, and during a recipient’s lifetime an option may be exercised only by the recipient or the recipient’s guardian or legal representative. However, the Compensation Committee may permit the transfer of a nonqualified stock option, share-settled stock appreciation right, restricted stock award, performance share or share-settled other stock-based award either (a) by instrument to the participant’s immediate family (as defined in the 2016 Plan), (b) by instrument to an inter vivos or testamentary trust (or other entity) in which the award is to be passed to the participant’s designated beneficiaries, or (c) by gift to charitable institutions. The Compensation Committee will determine the extent to which a holder of a stock option may exercise the option following termination of service.
Stock Appreciation Rights. The Compensation Committee may grant stock appreciation rights independent of or in connection with an option. The Compensation Committee will determine the terms applicable to stock appreciation rights. The base price of a stock appreciation right will be determined by the Compensation Committee, but will not be less than 100% of the fair market value of a share of our common stock with respect to the date of grant of such stock appreciation right. The maximum term of any SAR granted under the 2016 Plan is ten years from the date of grant. Generally, each SAR stock appreciation right will entitle a participant upon exercise to an amount equal to:
| ● | the excess of the fair market value of a share of common stock on the date of exercise of the stock appreciation right over the base price of such stock appreciation right, multiplied by | |
| ● | the number of shares as to which such stock appreciation right is exercised. |
Payment may be made in shares of our common stock, in cash, or partly in common stock and partly in cash, all as determined by the Compensation Committee.
Restricted Stock and Stock Units. The Compensation Committee may award restricted common stock and/or stock units under the 2016 Plan. Restricted stock awards consist of shares of stock that are transferred to a participant subject to restrictions that may result in forfeiture if specified conditions are not satisfied. Stock units confer the right to receive shares of our common stock, cash, or a combination of shares and cash, at a future date upon or following the attainment of certain conditions specified by the Compensation Committee. The Compensation Committee will determine the restrictions and conditions applicable to each award of restricted stock or stock units, which may include performance-based conditions. Dividends with respect to restricted stock may be paid to the holder of the shares as and when dividends are paid to stockholders or at the times of vesting or other payment of the restricted stock award. Stock unit awards may be granted with dividend equivalent rights, which may be accumulated and may be deemed reinvested in additional stock units, as determined by the Compensation Committee in its discretion. If any dividend equivalents are paid while a stock unit award is subject to restrictions, the dividend equivalents shall be subject to the same restrictions on transferability as the underlying stock units, unless otherwise set forth in an award agreement. Unless the Compensation Committee determines otherwise, holders of restricted stock will have the right to vote the shares.
Performance Shares and Performance Units. The Compensation Committee may award performance shares and/or performance units under the 2016 Plan. Performance shares and performance units are awards which are earned during a specified performance period subject to the attainment of performance criteria, as established by the Compensation Committee. The Compensation Committee will determine the restrictions and conditions applicable to each award of performance shares and performance units.
Incentive Bonus Awards. The Compensation Committee may award Incentive Bonus Awards under the 2016 Plan. Incentive Bonus Awards may be based upon the attainment of specified levels of Company or subsidiary performance as measured by pre-established, objective performance criteria determined at the discretion of the Compensation Committee. Incentive Bonus Awards will be paid in cash or common stock, as set forth in an award agreementagreement.
Other Stock-Based and Cash-Based Awards. The Compensation Committee may award other types of equity-based or cash-based awards under the 2016 Plan, including the grant or offer for sale of unrestricted shares of our common stock and payment in cash or otherwise of amounts based on the value of shares of common stock.
Section 162(m) Compliance. If stock or cash-based awards are intended to satisfy the conditions for deductibility under Section 162(m) of the Code as “performance-based compensation,” the performance criteria will be selected from among the following, which may be applied to our Company as a whole, any subsidiary or any division or operating unit thereof: (a) pre-tax income; (b) after-tax income; (c) net income; (d) operating income or profit; (e) cash flow, free cash flow, cash flow return on investment, net cash provided by operations, or cash flow in excess of cost of capital; (f) earnings per share; (g) return on equity; (h) return on sales or revenues; (i) return on invested capital or assets; (j) cash, funds or earnings available for distribution; (k) appreciation in the fair market value of the common stock; (l) operating expenses; (m) implementation or completion of critical projects or processes; (n) return on investment; (o) total return to stockholders; (p) dividends paid; (q) net earnings growth; (r) related return ratios; (s) increase in revenues; (t) the Company’s published ranking against its peer group of pharmaceutical companies based on total stockholder return; (u) net earnings; (v) changes (or the absence of changes) in the per share or aggregate market price of the common stock; (w) number of securities sold; (x) earnings before or after any one or more of the following items: interest, taxes, depreciation or amortization, as reflected in the Company’s financial reports for the applicable period; (y) total revenue growth; (z) economic value created; (aa) operating margin or profit margin; (bb) share price or total stockholder return; (cc) cost targets, reductions and savings, productivity and efficiencies; (dd) strategic business criteria, consisting of one or more objectives based on meeting objectively determinable criteria: specified market penetration, geographic business expansion, progress with research and development activities, investor satisfaction, employee satisfaction, human resources management, supervision of litigation, information technology, and goals relating to acquisitions, divestitures, joint ventures and similar transactions, and budget comparisons; (ee) objectively determinable personal or professional objectives, including any of the following performance goals: the implementation of policies and plans, the negotiation of transactions, the development of long term business goals, formation of joint ventures, research or development collaborations, and the completion of other corporate transactions, and (ff) any combination of, or a specified increase or improvement in, any of the foregoing.
At the end of the performance period established in connection with any award, the Compensation Committee will determine the extent to which the performance goal or goals established for such award have been attained, and shall determine, on that basis, the number of performance shares or performance units included in such award that have been earned and as to which payment will be made. The Compensation Committee will certify in writing the extent to which it has determined that the performance goal or goals established by it for such award have been attained.
With respect to awards intended to be performance-based compensation under Section 162(m) of the Code, no participant of the 2016 Plan may receive in any one fiscal year (a) options or stock appreciation rights relating to more than 1,000,000250,000 shares of our common stock, and (b) stock units, restricted shares, performance shares, performance units or other stock-based awards that are denominated in shares of common stock relating to more than 1,000,000250,000 shares of our common stock in the aggregate. The maximum dollar value payable to any participant for a fiscal year of the Company with respect to stock units, performance units or incentive bonus awards or other stock-based awards that may be settled in cash or other property (other than common stock) is $1,500,000.
| 60 |
Effect of Certain Corporate Transactions. The Compensation Committee may, at the time of the grant of an award, provide for the effect of a change in control (as defined in the 2016 Plan) on any award, including (i) accelerating or extending the time periods for exercising, vesting in, or realizing gain from any award, (ii) eliminating or modifying the performance or other conditions of an award, (iii) providing for the cash settlement of an award for an equivalent cash value, as determined by the Compensation Committee, or (iv) such other modification or adjustment to an award as the Compensation Committee deems appropriate to maintain and protect the rights and interests of participants upon or following a change in control. The Compensation Committee may, in its discretion and without the need for the consent of any recipient of an award, also take one or more of the following actions contingent upon the occurrence of a change in control: (a) cause any or all outstanding options and stock appreciation rights to become immediately exercisable, in whole or in part; (b) cause any other awards to become non-forfeitable, in whole or in part; (c) cancel any option or stock appreciation right in exchange for a substitute option; (d) cancel any award of restricted stock, stock units, performance shares or performance units in exchange for a similar award of the capital stock of any successor corporation; (e) redeem any restricted stock, stock unit, performance share or performance unit for cash and/or other substitute consideration with a value equal to the fair market value of an unrestricted share of our common stock on the date of the change in control; (f) cancel any option or stock appreciation right in exchange for cash and/or other substitute consideration based on the value of our common stock on the date of the change in control, and cancel any option or stock appreciation right without any payment if its exercise price exceeds the value of our common stock on the date of the change in control; (g) cancel any stock unit or performance unit held by a participant affected by the change in control in exchange for cash and/or other substitute consideration with a value equal to the fair market value per share of common stock on the date of the change in control, or (h) make such other modifications, adjustments or amendments to outstanding awards as the Compensation Committee deems necessary or appropriate.
Amendment, Termination. The 2016 Equity Compensation Plan will remain in effect until March 2026, or, if earlier, when awards have been granted covering all available shares under the 2016 Plan or the 2016 Plan is otherwise terminated by the Board. The Board may amend the terms of awards in any manner not inconsistent with the 2016 Plan, provided that no amendment shall adversely affect the rights of a participant with respect to an outstanding award without the participant’s consent. In addition, our Board of Directors may at any time amend, suspend, or terminate the 2016 Plan, provided that (i) no such amendment, suspension or termination shall materially and adversely affect the rights of any participant under any outstanding award without the consent of such participant and (ii) to the extent necessary and desirable to comply with any applicable law, regulation, or stock exchange rule, the 2016 Plan requires us to obtain stockholder consent. Stockholder approval is required for any plan amendment that increases the number of shares of common stock available for issuance under the 2016 Plan or changes the persons or classes of persons eligible to receive awards.
Tax Withholding.The Company has the power and right to deduct or withhold, or require a participant to remit to the Company, the minimum statutory amount to satisfy federal, state, and local taxes, domestic or foreign, required by law or regulations to be withheld.
Recoupment Policy.Awards granted under the 2016 Plan will be subject to any provisions of applicable law providing for the recoupment or clawback of incentive compensation, such as provisions imposed pursuant to the Dodd-Frank Wall Street Reform and Consumer Protection Act; the terms of any Company recoupment, clawback or similar policy in effect at the time of grant of the award; and any recoupment, clawback or similar provisions that may be included in the applicable award agreement.
Federal Income Tax Consequences.The following is a brief summary of the U.S. federal income tax consequences applicable to awards granted under the 2016 Plan based on the federal income tax laws in effect on the date of this report. This summary is not intended to be exhaustive and does not address all matters relevant to a particular participant based on his or her specific circumstances. The summary expressly does not discuss the income tax laws of any state, municipality, or non-U.S. taxing jurisdiction, or the gift, estate, excise (including the rules applicable to deferred compensation under Code Section 409A), or other tax laws other than federal income tax law. The following is not intended or written to be used, and cannot be used, for the purposes of avoiding taxpayer penalties. Because individual circumstances may vary, the Company advises all participants to consult their own tax advisor concerning the tax implications of awards granted under the 2016 Plan.
| 61 |
A recipient of a stock option or stock appreciation right will not have taxable income upon the grant of the stock option or stock appreciation right. For non-statutory stock options and stock appreciation rights, the participant will recognize ordinary income upon exercise in an amount equal to the difference between the fair market value of the shares and the exercise price on the date of exercise. Any gain or loss recognized upon any later disposition of the shares generally will be a capital gain or loss.
The acquisition of shares upon exercise of an incentive stock option will not result in any taxable income to the participant, except, possibly, for purposes of the alternative minimum tax. The gain or loss recognized by the participant on a later sale or other disposition of such shares will either be long-term capital gain or loss or ordinary income, depending upon whether the participant holds the shares for the legally-required period (two years from the date of grant and one year from the date of exercise). If the shares are not held for the legally-required period, the participant will recognize ordinary income equal to the lesser of (i) the difference between the fair market value of the shares on the date of exercise and the exercise price, or (ii) the difference between the sales price and the exercise price, and the balance of the gain, if any, will be afforded capital gain treatment.
For awards of stock grants, the participant will not have taxable income upon the receipt of the award (unless the participant elects to be taxed at the time of the stock is granted rather than when it becomes vested). The stock grants will generally be subject to tax upon vesting as ordinary income equal to the fair market value of the shares at the time of vesting less the amount paid for such shares (if any).
A participant is not deemed to receive any taxable income at the time an award of restricted stock units is granted. When vested restricted stock units (and dividend equivalents, if any) are settled and distributed, the participant will recognize ordinary income equal to the amount of cash and/or the fair market value of shares received less the amount paid for such restricted stock units (if any).
If the participant is an employee or former employee, the amount a participant recognizes as ordinary income in connection with any award is subject to withholding taxes (not applicable to incentive stock options) and the Company is allowed a tax deduction equal to the amount of ordinary income recognized by the participant. In addition, Code Section 162(m) contains special rules regarding the federal income tax deductibility of compensation paid to the Company’s chief executive officer and to certain of the Company’s other executive officers. The general rule is that annual compensation paid to any of these specified executives will be deductible only to the extent that it does not exceed $1,000,000. However, the Company can preserve the deductibility of certain compensation in excess of $1,000,000 if such compensation qualifies as “performance-based compensation” by complying with certain conditions imposed by the Code Section 162(m) rules (including the establishment of a maximum number of shares with respect to which awards may be granted to any one employee during one fiscal year).
Option Grants and Stock Awards
As of December 31, 2017,2023, we had outstanding stock options to purchase 3,566,2991,357,466 shares at an average exercise price of approximately $1.20$7.39 per share. Included in the total outstanding stock options were 0 stock options granted under the 2006 Plan in 20172023 and 1,103,0000 nonqualified stock options granted under the 2016 Plan in 20172023 to our executive officers and others at an exercise price of $1.25 per share.officers.
All awards to be made under the 2016 Plan are discretionary, subject to the terms of the 2016 Plan. Therefore, the benefits and amounts that will be received or allocated under the 2016 Plan are generally not determinable at this time. The equity grant program for our non-employee directors is described under the Compensation of Directors section in this proxy statement. The following table summarizes these 2016-20172016 awards to our named executive officers, under the 2016 Plan, all executive officers and the non-executive officer employees and consultants.
New Plan Benefits Table
| Name and Position | Number of Units (options) | |||
| Garo H. Armen, Executive Chairman | 750,000 | (1) | ||
| Alexander K. Arrow, Chief Financial Officer | 215,000 | (2) | ||
| Non-Executive Director Group | 825,000 | (3) | ||
| Non-Executive Officer Employee/Consultant Group | 371,300 | (4) | ||
Outstanding Equity Awards at Fiscal Year End
The following table summarizes the equity awards made to our named executive officers that were outstanding at December 31, 2017.2023.
| Name | No. of Securities Underlying Unexercised Options (#) Exercisable | No. of Securities Underlying Unexercised Options (#) Unexercisable | Option Exercise Price | Option Expiration Date | No. of Securities Underlying Unexercised Options (#) Exercisable | No. of Securities Underlying Unexercised Options (#) Unexercisable | Option Exercise Price | Option Expiration Date | ||||||||||||||||||||
| Garo H. Armen (1) | 284,725 | 215,275 | $ | 1.25 | April 15, 2026 | 125,000 | - | $ | 5.00 | April 15, 2026 | ||||||||||||||||||
| Garo H. Armen (2) | 17,361 | 232,639 | $ | 1.75 | October 16, 2027 | 62,500 | - | $ | 7.00 | October 16, 2027 | ||||||||||||||||||
| Robert Ziroyan (3) | 100,000 | — | $ | 1.00 | March 30, 2021 | |||||||||||||||||||||||
| Robert Ziroyan (3) | 50,000 | — | $ | 1.00 | March 1, 2024 | |||||||||||||||||||||||
| Robert Ziroyan (3) | 75,000 | — | $ | 1.25 | March 9, 2025 | |||||||||||||||||||||||
| Garo H. Armen (3) | 75,000 | - | $ | 7.00 | February 13, 2030 | |||||||||||||||||||||||
| Alexander K. Arrow (4) | 58,333 | 41,667 | $ | 1.25 | February 12, 2026 | 25,000 | - | $ | 5.00 | February 12, 2026 | ||||||||||||||||||
| Alexander K. Arrow (4) | 79,725 | 60,275 | $ | 1.25 | April 15, 2026 | 35,000 | - | $ | 5.00 | April 15, 2026 | ||||||||||||||||||
| Alexander K. Arrow (5) | 5,208 | 69,782 | $ | 1.75 | October 16, 2027 | 18,750 | - | $ | 7.00 | October 16, 2027 | ||||||||||||||||||
| Alexander K. Arrow (6) | 10,417 | - | $ | 4.00 | February 1, 2029 | |||||||||||||||||||||||
| Alexander K. Arrow (7) | 30,000 | - | $ | 7.00 | February 13, 2030 | |||||||||||||||||||||||
| Alexander K. Arrow (8) | 46,874 | - | $ | 7.00 | February 13, 2030 | |||||||||||||||||||||||
| Alexander K. Arrow (9) | 31,250 | - | $ | 7.00 | July 18, 2030 | |||||||||||||||||||||||
| Andrew Slee (10) | 25,000 | - | $ | 5.00 | April 15, 2026 | |||||||||||||||||||||||
| Andrew Slee (11) | 18,750 | - | $ | 7.00 | October 16, 2027 | |||||||||||||||||||||||
| Andrew Slee (12) | 34,375 | 3,125 | $ | 7.00 | February 13, 2030 | |||||||||||||||||||||||
| Andrew Slee (13) | 15,625 | 3,125 | $ | 7.00 | July 18, 2030 | |||||||||||||||||||||||
| Andrew Slee (14) | 8,854 | 3,646 | $ | 22.40 | February 25, 2031 | |||||||||||||||||||||||
| Robert B. Stein (15) | 50,000 | - | $ | 5.00 | January 22, 2030 | |||||||||||||||||||||||
| Robert B. Stein (16) | 10,000 | - | $ | 5.00 | April 15, 2026 | |||||||||||||||||||||||
| Robert B. Stein (17) | 50,000 | - | $ | 7.00 | October 16, 2027 | |||||||||||||||||||||||
| Robert B. Stein (18) | 37,500 | - | $ | 7.00 | February 13, 2030 | |||||||||||||||||||||||
| Robert B. Stein (19) | 8,854 | 3,646 | $ | 22.40 | February 25, 2031 | |||||||||||||||||||||||
| (1) | Dr. Armen was granted a |
| (2) | Dr. Armen was granted a |
| Dr. Armen was granted a 75,000 share option grant on February 13, 2020. | |
| (4) | Dr. Arrow was granted a |
| (5) | Dr. Arrow was granted a |
| (6) | Dr. Arrow was granted a 10,417 share option grant on February 1, 2019. |
| (7) | Dr. Arrow was granted a 30,000 share option grant on February 13, 2020. |
| (8) | Dr. Arrow was granted a 46,874 share option grant on February 13, 2020. |
| (9) | Dr. Arrow was granted a 31,250 share option grant on July 18, 2020. |
| (10) | Dr. Slee was granted a 25,000 shares option grant on April 15, 2016. |
| (11) | Dr. Slee was granted a 18,750 shares option grant on October 16, 2017. |
| (12) | Dr. Slee was granted a 37,500 shares option grant on February 13, 2020. |
| (13) | Dr. Slee was granted a 18,750 shares option grant on July 18, 2020. |
| (14) | Dr. Slee was granted a 12,500 shares option grant on February 25, 2021. |
| (15) | Dr. Stein was granted a 50,000 shares option grant on January 22, 2015. |
| (16) | Dr. Stein was granted a 10,000 shares option grant on April 15, 2016. |
| (17) | Dr. Stein was granted a 50,000 shares option grant on October 16, 2017. |
| (18) | Dr. Stein was granted a 37,500 shares option grant on February 13, 2020. |
| (19) | Dr. Stein was granted a 12,500 shares option grant on February 25, 2021. |
For Drs. Armen and Arrow, following a qualified Change of Control, a resignation for Good Reason, or an involuntary termination other than For Cause, 100% of the executives’ then-unvested options shall become immediately vested.
Security Ownership of Certain Beneficial Owners and Management
The following table summarizessets forth information regarding the beneficial ownersownership of the Common Stock as of April 1, 2024, unless otherwise indicated, by (1) each person known by the Company to be the beneficial owner of more than 5% of the Company’s voting securities and the securitiesoutstanding shares of common stock, (2) each director of the Company, beneficially(3) the Company’s current executive officers, and (4) all current directors and executive officers of the Company as a group. The persons and entities named in the table have sole voting and investment power with respect to all such shares owned by them, unless otherwise indicated.
| Name and address of Beneficial Owner* | Amount and Nature of Beneficial Ownership | Percent of Class | ||||||
| Garo H. Armen(1) | 1,000,989 | (2) | 21 | % | ||||
| Robert B. Stein(1) | 158,906 | (3) | 3 | % | ||||
| Khalil Barrage(1) | 196,255 | (4) | 4 | % | ||||
| Alexander K. Arrow(1) | 244,773 | (5) | 5 | % | ||||
| Brian J. Corvese(1) | 54,479 | (6) | 1 | % | ||||
| David A. Lovejoy | 144,275 | (7) | 3 | % | ||||
| Jennifer Buell(1) | 76,563 | (8) | 2 | % | ||||
| Andrew Slee(1) | 110,859 | (9) | 2 | % | ||||
| All directors and executive officers as a group (7 persons) | 1,846,991 | (10) | 35 | % | ||||
* Address for each party listed in the Company’s directors and officers as of March 27, 2017.above table is c/o Protagenic Therapeutics, Inc., 149 Fifth Avenue, Suite 500, New York, NY 10010.
Name and address of Beneficial Owner | Amount of Beneficial Ownership | Percent of Beneficial Ownership | ||||||
| Garo H. Armen(1) | 4,205,632 | (2) | 33 | |||||
| Robert B. Stein(1) | 256,667 | (3) | 2 | |||||
| Khalil Barrage(1) | 263,750 | (4) | 2 | |||||
| Alexander K. Arrow(1) | 305,277 | (5) | 2 | |||||
| Larry N. Feinberg 808 North St., Greenwich, CT 06831 | 800,000 | (6) | 6 | |||||
Brian J. Corvese(1) | 39,585 | (7) | * | |||||
| David A. Lovejoy | 558,906 | (8) | 4 | |||||
| Josh Silverman(1) | 63,750 | (9) | 1 | |||||
Strategic Bio Partners LLC(10) 777 Third Avenue 30th Floor New York, NY 10017 | 2,193,413 | (11) | 17 | |||||
| All directors and executive officers as a group (6 persons) | 5,134,661 | (12) | ||||||
* Less than 1%
(1) Executive officer and/or director.
(2) Includes warrants738,489 shares of common stock. Also includes options to purchase 1,253,367266,667 shares of common stock at an exercise price of approximately $1.00$1.74, $5.00 or $7.00 per share. Includes 2,296,012 shares held in the name of Dr. Armen and 250,000 shares held in the name of the Garo H. Armen IRA, as to which Dr. Armen has sole voting and dispositive power. Also includes options to purchase 406,253 shares of common stock at an exercise price of $1.25 per share. Does not include options to purchase 343,747245,833 shares that are not exercisable within 60 days of the date of this report.
(3) Represents options to purchase 256,667158,906 shares of common stock at an exercise price of $1.25$1.74, $5.00, $7.00, or $22.40 per share. Does not include options to purchase 183,33376,094 shares in the aggregate that are not exercisable within 60 days of the date of this report.
(4) Includes 50,000150,880 shares of common stock and options to purchase 213,75045,375 shares of common stock at an exercise price of $1.25$1.74, or $14.60 per share. Does not include options to purchase 26,25022,125 shares in the aggregate that are not exercisable within 60 days of the date of this report.
(5) Includes 100,00045,815 shares held in the name of Dr. Arrowcommon stock and 18,260 shares held in the name of the Alexander K. Arrow IRA, as to which Dr. Arrow has sole voting and dispositive power. Also includesIncludes options to purchase 187,017198,958 shares of common stock at an exercise price of $1.25$1.74, $4.00, $5.00 or $7.00 per share. Does not include options to purchase 127,98398,333 shares of common stock in the aggregate that are not exercisable within 60 days of the date of this report.
(6) Includes 200,000 shares of common stock held in the name of Mr. Feinberg and warrantsoptions to purchase 600,00054,479 shares of common stock at an exercise price of $1.00$7.00 per share. Does not include options to purchase 43,021 shares that are not exercisable within 60 days of the date of this report.
(7) Includes 37,200 shares of common stock and options to purchase 107,075 shares of common stock in the aggregate with an exercise price ranging from $4.00, $5.00, or $7.00 per share.
(8) Includes options to purchase 39,58576,568 shares of common stock at an exercise price of $1.75$1.74, $7.00 or $22.40 per share. Does not include options to purchase 55,41784,688 shares of common stock that are not exercisable within 60 days of the date of this report.
(8)(9) Includes 148,800options to purchase 110,859 shares of common stock held in the name of Dr. Lovejoy and options to purchase 410,106 shares of common stock in the aggregate withat an exercise price ranging from $1.00 to $1.25of $1.74, $5.00, $7.00, or $22.40 per share. Does not include options to purchase 123,193106,641 shares of common stock that are not exercisable within 60 days of the date of this report.
(9)(10) Includes options to purchase 63,750911,807 shares of common stock at an exercise price of $1.25 per share. Does not include options to purchase 26,250 shares of common stock that are not exercisable within 60 days of the date of this report.stock.
(10) Hudson Bay Master Fund Ltd. (the “Managing Member”) is the managing member of Strategic Bio Partners, LLC (“SBP”). Pursuant to SBP’s Limited Liability Company Operating Agreement, the Managing Member has delegated to Hudson Bay Capital Management LP (“HBC”) full and sole investment discretion and voting control of SBP’s portfolio securities. Sander Gerber is the managing member of Hudson Bay Capital GP LLC, which is the general partner of HBC. Each of SBP, the Managing Member and Sander Gerber disclaims beneficial ownership over these securities.
(11) SBP also holds shares of Series B Preferred Stock convertible into common stock and Predecessor Warrants to purchase common stock. However, the Series B Preferred and the Predecessor Warrants are subject to a “Beneficial Ownership Cap” limitation pursuant to which the holder thereof does not have the right to convert Series B Preferred Stock or exercise the Predecessor Warrants to the extent that such exercise would result in beneficial ownership by the holder thereof, or any of its affiliates and any other persons or entities whose beneficial ownership of common stock would be aggregated with the holder’s for purposes of Section 13(d) of the Exchange Act, of more than 9.99% of the total number of shares of common stock issued and outstanding immediately after giving effect to such conversion or exercise. Disregarding the Beneficial Ownership Cap, SBP would own 2,193,413 shares of common stock, including the shares underlying Series B Preferred Stock and Predecessor Warrants.
(12) Includes warrants to purchase 1,628,367 shares of common stock and options to purchase 1,167,020 shares of common stock.
Item 13. Certain Relationships and Related Transactions, and Director Independence.
Certain Relationships and Related Party Transactions
Other than compensation arrangements for our named executive officers and directors, we describe below each transaction or series of similar transactions, since January 1, 2016, to which we were a party or will be a party, in which:
| ● | the amounts involved exceeded or will exceed $120,000; and | |
| ● | any of our directors, executive officers or holders of more than 5% of our capital stock, or any member of the immediate family of the foregoing persons, had or will have a direct or indirect material interest. |
Compensation arrangements for our named executive officers and directors are described in Item 11, Executive Compensation.
Our principal offices are located at 149 Fifth Avenue, Suite 500, New York, New York 10010, in a conference room of Agenus, Inc. We utilize our principal office for quarterly board meetings and our annual shareholder meeting at no cost. Our personnel and consultants all work remotely, the Company’s basic science laboratory work is conducted in the Lovejoy Lab at the University of Toronto, and its preclinical efficacy work is conducted at CROs. Hence the Company does not have the need for a day-to-day physical office location other than a mailing address and conference room facility for meetings. For that reason, the Agenus conference room suits its purposes without imposing any inconveniences upon Agenus. Dr. Armen, our Executive Chairman, is also the Chairman and Chief Executive Officer of Agenus Inc.
Transactions with Predecessor Shareholders
Split-Off
At the closing of the Merger we had a 51% interest in MomSpot LLC, and the remaining 49% was held by B.E. Global LLC. Barry Eisenberg is the sole owner of B.E. Global LLC and is the Chief Executive Officer of MomSpot LLC. Immediately after the closing of the Merger, we split off our 51% membership interests in MomSpot LLC. The split-off was accomplished through the transfer of all of our membership interests of MomSpot LLC to B.E. Global LLC having nominal value of nominal considerations via a split off agreement.
Secured Convertible Notes/Predecessor Warrants
Between February 11, 2014 and December 9, 2015, Atrinsic issued secured convertible promissory notes (the “Secured Convertible Notes”) in the aggregate principal amount of $665,000 and $35,000 in interest to two of its stockholders, of which Secured Convertible Notes in the aggregate principal amount of $332,500 were issued to Iroquois Master Fund Ltd. (“IMF”). Josh Silverman, who became one of our directors upon the closing of the Merger, is an affiliate of IMF. The Secured Convertible Notes, as revised and amended, had a maturity date of August 31, 2016 and bore interest at the rate of 5.0% per annum, payable at maturity. The outstanding principal and accrued interest of each Secured2019-2020 Convertible Note was convertible, subject to a 4.99% beneficial ownership cap), into shares of Atrinsic’s common stock at an initial conversion price of $5.00 per share (subject to adjustment), at the option of the respective holders. IMF exchanged the Secured Convertible Notes that it held for 147,972 Predecessor Warrants, which Predecessor Warrants were issued to the Designee at the closing of the Merger, and the instruments by which the Secured Convertible Notes were secured were simultaneously terminated.Offering
Transactions Relating to Protagenic
Garo H. Armen our Chairman and principal stockholder, purchased shares of Series B Preferred StockKhalil Barrage invested $200,000 and $200,000, respectively, in the Private Offering in exchange for the cancellation of $350,000 of loans made by him, plus accrued and unpaid interest on these loans.
During 2013 and 2012, Mr. Armen made loans to us in the amount of $310,000. The proceeds of the loans were used to fund research, development and general operating activity of Protagenic. The loans accrued interest at the rate of 10% per annum. In February 2013, in connection with a capital raise by Protagenic, the loans and accrued interest thereon, totaling $317,789, were converted into Protagenic warrants to purchase 953,367 shares of Protagenic common stock at an exercise price of $1.00 per share. Other than with respect to the payment of the purchase price for the securities by the conversion of debt, Mr. Armen participated in this capital raise on the same terms as all other investors.
From April 15, 2015 through October 29, 2015, Mr. Armen made five loans to Protagenic. The proceeds of the loans were used to fund research, development and general operating activity of Protagenic. The loans accrued interest at the rate of 10% per annum. Principal and accrued interest on these loans, totaling approximately $350,000, were converted into shares of Series B Preferred Stock in the Private Offering at a price of $1.25 per share.
On December 21, 2015, Dr. Alexander K. Arrow purchased 60,000 shares of common stock of Protagenic from Mark Berg at a per share purchase price equal to $0.50 for an aggregate purchase price of $30,000. In addition, Dr. Arrow purchased 58,260 shares of Series B Preferred Stock in the PrivateConvertible Note Offering on the same terms as all other investors.Investors. These options were converted as of December 31, 2023.
Effective December 23, 2015,Zack Armen
During the latter part of 2018 and the first quarter of 2019, Zack Armen, the son of our Executive Chairman, Garo H. Armen, Ph.D., assisted us in the development of slide deck presentations and summaries, video editing, and forecasting and market size projections that were incorporated into presentations to investors and others. We have included these presentations in various Current Reports on Form 8-K which we filed with the Securities and Exchange Commission. On June 17, 2019, the Compensation and Audit Committees of the Board authorized the issuance to Mr. Zack Armen entered into an additional loan agreement with Protagenic pursuant to which he agreed to loan Protagenic up to $150,000. The loansof 6,250 stock options under this Agreement accrued interest at the rate of 10% per year. The principal2016 Plan in consideration for his services. These options vested in their entirety on issuance, have a ten-year term and interest on these loans is convertible into common stockare exercisable at a price of $1.25$7.00 per share. On December 23, 2015, Protagenic borrowed $37,628February 21, 2020 Mr. Zack Armen was also issued an additional 12,500 stock options that vest over 48 months and are exercisable at a price of the $150,000 available Borrowings under the agreement.$7.00 per share. On July 18, 2020 Mr. Zack Armen was also issued an additional 7,500 stock options that vest over 48 months and are exercisable at a price of $7.00 per share.
Effective June 17, 2016, the Board of Directors determined that it was in the best interest of the Company to convert the last remaining portion of debt owed to Mr. Armen into equity, per the terms of the loan agreements. The sum total of remaining debt and accumulated interest as of December 31, 2017 was $0.
Merger Transaction
On February 12, 2016, which we refer to as the Merger Closing Date, Atrinsic, Inc., Protagenic Therapeutics, Inc. and Protagenic Acquisition Corp., Atrinsic, Inc.’s wholly-owned subsidiary, entered into a merger agreement and completed the merger contemplated by the merger agreement. Pursuant to the merger agreement, on the Merger Closing Date, Protagenic Acquisition Corp. merged with and into Protagenic Therapeutics, Inc., with Protagenic Therapeutics, Inc. remaining as the surviving entity and wholly-owned subsidiary of Atrinsic, Inc. On June 17, 2016, we merged our wholly-owned subsidiary Protagenic Therapeutics, Inc. with and into the Company and we changed our name from Atrinsic, Inc. to Protagenic Therapeutics, Inc.
While we believe that all of these agreements and arrangements are in the best interests of our Company, related parties of the Placement Agent may derive material benefits as the result of these transactions. In addition, related parties of the Placement Agent will have a continuing substantial interest in our Company and will derive substantial benefits from any success of our Company.
Policies and Procedures for Related Party Transactions
We have adopted a policy that our executive officers, directors, nominees for election as a director, beneficial owners of more than 5% of any class of our common stock, any members of the immediate family of any of the foregoing persons and any firms, corporations or other entities in which any of the foregoing persons is employed or is a partner or principal or in a similar position or in which such person has a 5% or greater beneficial ownership interest, which we refer to collectively as related parties, are not permitted to enter into a transaction with us without the prior consent of our Board of Directors acting through the audit committee or, in certain circumstances, the chairman of the audit committee. Any request for us to enter into a transaction with a related party, in which the amount involved exceeds $100,000 and such related party would have a direct or indirect interest must first be presented to our audit committee, or in certain circumstances the chairman of our audit committee, for review, consideration and approval. In approving or rejecting any such proposal, our audit committee, or the chairman of our audit committee, is to consider the material facts of the transaction, including, but not limited to, whether the transaction is on terms no less favorable than terms generally available to an unaffiliated third party under the same or similar circumstances, the extent of the benefits to us, the availability of other sources of comparable products or services and the extent of the related party’s interest in the transaction.
| 65 |
Director Independence
We are not currently listed on any national securities exchange or in an inter-dealer quotation system that has a requirement that the Board of Directors be independent. However, in evaluating the independence of our members and the composition of the committees of our Board of Directors, our Board utilizes the definition of “independence” as that term is defined by applicable listing standards of the NASDAQ Stock Market and SEC rules, including the rules relating to the independence standards of an audit committee and the non-employee director definition of Rule 16b-3 promulgated under the Exchange Act.
Our Board of Directors expects to continue to evaluate its independence standards and whether and to what extent the composition of the Board and its committees meets those standards. We ultimately intend to appoint such persons to our Board and committees of our Board as are expected to be required to meet the corporate governance requirements imposed by a national securities exchange. Therefore, we intend that a majority of our directors will be independent directors of which at least one director will qualify as an “audit committee financial expert,” within the meaning of Item 407(d)(5) of Regulation S-K, as promulgated by the SEC.
We believe that Messrs. Barrage, Corvese, Wright, and SilvermanDr. Buell are each an “independent” director as that term is defined by the NASDAQ Stock Market, Inc. Marketplace Rules and SEC Regulations. In addition, the Board also designated Brian Corvese as an “audit committee financial expert,” as that term is defined by the NASDAQ Listing Rules and SEC regulations.
With regard to Mr. Silverman’sWright’s independent status, the Board considered the fact that he is an ex-CEO of one of the institutional funds (Iroquois Asset Management) that is a 50% ownercurrent CEO of a limited liabilitypublicly-traded biopharmaceutical company (MiMedX Group, Inc.) and that he is as a Partner at an investment firm (Signal Hill Advisors, LLC). He also serves on the Board of Directors of Agenus, Inc., a publicly-traded company for which owns just under 10% ofDr. Armen is the Company’s common stock. The Board noted that Mr. Silverman is no longer the CEO of Iroquois Asset Management,Chaiman and as such, he does not represent a major single shareholder.CEO.
With regard to Mr. Corvese’s independent status, the Board considered the fact that he has no business relationship with the Company.
With regard to Mr. Barrage’s independent status, the Board considered the fact that he has no business relationship with the Company.
With regard to Dr. Buell’s independent status, the Board considered the fact that she has no business relationship with the Company except her consulting role assisting with clinical trial design and that she reports to Dr. Armen in the course of her primary roles, as president of Agenus, Inc and MiNK Therapeutics, Inc, and is therefore not considered “independent.”.
Dr. Stein, a member of the Compensation Committee,Science and Clinical & Regulatory Committees, serves as our Chief Medical Officer, and is therefore not considered “independent.”
Dr. Armen, a member of the Science and Clinical & Regulatory Committees, serves as our Executive Chairman, and is therefore not considered “independent.”
Our principal offices are located at 149 Fifth Avenue, Suite 500, New York, New York 10010, in a conference room of Agenus, Inc. We utilize our principal office for quarterly board meetings and our annual shareholder meeting on a month to month basis at a nominal value. Dr. Armen, our Executive Chairman, is also the Chairman and Chief Executive Officer of Agenus Inc.
| 66 |
Item 14. Principal Accounting Fees and Services.
The following table sets forth the fees for services provided and reasonably expected to be billed by Malone Bailey LLP. The following is a summary of the fees billed to the Company for professional services rendered for the fiscal years ended December 31, 20172023 and 2016.2022.
| Fiscal Year 2017 | Fiscal Year 2016 | Fiscal Year 2023 | Fiscal Year 2022 | |||||||||||||
| Audit fees | $ | 30,000 | $ | - | $ | 100,000 | $ | 85,000 | ||||||||
| Audit-related fees | $ | - | $ | - | $ | - | $ | - | ||||||||
| Tax Fees | $ | - | $ | - | $ | - | $ | - | ||||||||
| All other fees | $ | - | $ | - | $ | - | $ | - | ||||||||
| Total | $ | 30,000 | $ | - | $ | 100,000 | $ | 85,000 | ||||||||
The following table sets forth the fees for services provided and reasonably expected to be billed by Marcum LLP. The following is a summary of the fees billed to the Company for professional services rendered for the fiscal years ended December 31, 2017 and 2016.
| Fiscal Year 2017 | Fiscal Year 2016 | |||||||
| Audit fees | $ | 17,500 | $ | 88,500 | ||||
| Audit-related fees | $ | - | $ | 15,458 | ||||
| Tax Fees | $ | - | $ | - | ||||
| All other fees | $ | - | $ | - | ||||
| Total | $ | 17,500 | $ | 103,958 | ||||
Audit Fees:For the fiscal years ended December 31, 20172023 and 2016,2022, the aggregate audit fees billed by our independent auditors were for professional services rendered for audits and quarterly reviews of our consolidated financial statements, and assistance with reviews of registration statements and documents filed with the SEC.
Audit-Related Fees:Audit-related fees are for assurance and other activities not explicitly related to the audit of our financial statements.
Tax Fees:For the fiscal years ended December 31, 20172023 and 2016,2022, there were no tax fees, respectively.
All Other Fees:For the fiscal years ended December 31, 20172023 and 2016,2022, there were $0 and $0,no other fees, respectively
Audit Committee Pre-Approval Policies and Procedures.The Audit Committee oversees and monitors our financial reporting process and internal control system, reviews and evaluates the audit performed by our registered independent public accountants and reports to the Board any substantive issues found during the audit. The Audit Committee is directly responsible for the appointment, compensation and oversight of the work of our registered independent public accountants. The Audit Committee convenes on a quarterly basis to approve each quarterly filing, and an annual basis to review the engagement of the Company’s external auditor.
The Audit Committee has considered whether the provision of Audit-Related Fees, Tax Fees, and all other fees as described above is compatible with maintaining Marcum LLP’s and Malone BaileyMaloneBailey, LLP’s independence and has determined that such services for fiscal years 20162023 and 2017,2022, respectively, were compatible. All such services were approved by the Audit Committee pursuant to Rule 2-01 of Regulation S-X under the Exchange Act to the extent that rule was applicable.
Item 15. Exhibits, Financial Statement Schedules.
(a) List of Documents filed as part of this Report
(1) Consolidated Financial Statements
The financial statements and related notes, together with the reports of Marcum LLP and Malone Bailey LLP appear at pages F-1 through F-21F-20 following the Exhibit List as required by Part II, Item 8 “Financial Statements and Supplementary Data” of this Form 10-K.
(2) Financial Statement Schedules.
Schedules are omitted because they are either not required, not applicable, or the information is otherwise included.
| 67 |
(3) Exhibits
The Company has filed with this report or incorporated by reference herein certain exhibits as specified below pursuant to Rule 12b-32 under the Exchange Act. See Exhibit Index following the signature page to this report for a complete list of documents filed with this report.
| 68 |
| * | Filed herewith |
| ** | Designates management contracts and compensation plans |
| † | Furnished herewith |
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
PROTAGENIC THERAPEUTICS, INC. | ||||
| Date: | April | By: | /s/Garo H. Armen | |
| Garo H. Armen | ||||
Chairman
Duly Authorized Officer) | ||||
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Garo H. Armen as the undersigned’s true and lawful attorney-in-fact and agent, with full power of substitution and resubstitution, for the undersigned and in the undersigned’s name, place, and stead, in any and all capacities, to sign any and all amendments to this report, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorney-in-fact and agent full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as the undersigned might or could do in person, hereby ratifying and confirming that said attorney-in-fact and agent or his substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, as amended, this report has been signed below by the following persons in the capacities and on the dates indicated.
| April 23 ,2024 | |||||
| /s/ Alexander K. Arrow | |||||
| Chief Financial Officer (Principal Financial Officer) |
PROTAGENIC THERAPEUTICS, INC. AND SUBSIDIARY
CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED
DECEMBER 31, 20172024 AND 20162023
| F-1 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the stockholdersStockholders and boardBoard of directorsDirectors of
Protagenic Therapeutics, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheetsheets of Protagenic Therapeutics, Inc. (theand its subsidiary (collectively, the “Company”) as of December 31, 2017,2023 and 2022, and the related consolidated statements of operations and comprehensive loss, changes in stockholders’ equity, (deficit), and cash flows for the yearyears then ended, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2017,2023 and 2022, and the results of itstheir operations and itstheir cash flows for the yearyears then ended, in conformity with accounting principles generally accepted in the United States of America.
Going Concern Matter
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 2 to the financial statements, the Company has suffered recurring losses from operations and has no revenue; these conditions raisenegative cash flows from operations that raises substantial doubt about its ability to continue as a going concern. Management'sManagement’s plans in regard to these matters are also described in Note 2. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audit.audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our auditaudits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our auditaudits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the entity’sCompany’s internal control over financial reporting. Accordingly, we express no such opinion.
Our auditaudits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our auditaudits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audit providesaudits provide a reasonable basis for our opinion.
/s/ MaloneBailey, LLPCritical Audit Matters
www.malonebailey.com
We have served asCritical audit matters are matters arising from the Company’s auditor since 2017.
Houston, Texas
April 2, 2018
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Audit Committee of the
Board of Directors and Shareholders
of Protagenic Therapeutics, Inc.
We have audited the accompanying consolidated balance sheet of Protagenic Therapeutics, Inc. (the “Company”) as of December 31, 2016, and the related consolidated statement of operations and comprehensive loss, stockholders’ equity and cash flow for the year then ended. The consolidated financial statement is the responsibility of the Company’s management. Our responsibility is to express an opinion on the financial statement based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statement are free of material misstatement. The Company is not required to have, nor were we engaged to perform, ancurrent period audit of its internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements assessingthat were communicated or required to be communicated to the accounting principles usedaudit committee and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believethat: (1) relate to accounts or disclosures that our audit provides a reasonable basis for our opinion.
In our opinion,are material to the financial statements referred to above present fairly, in all material respects, the consolidated financial position of Protagenic Therapeutics, Inc., as of December 31, 2016 and the consolidated results of its operations and its cash flow for the year then ended in conformity with accounting principles generally accepted in the United States of America.(2) involved our especially challenging, subjective, or complex judgments. We determined that there are no critical audit matters.
| /s/ MaloneBailey, LLP | |
| www.malonebailey.com | |
| We have served as the Company’s auditor since 2017. | |
| Houston, Texas | |
April 1, 2024 |
| F-2 |
//s// Marcum LLP
Marcum LLP
New York, NY
April 17, 2017
PROTAGENIC THERAPEUTICS, INC., AND SUBSIDIARIES
SUBSIDIARY
CONSOLIDATED BALANCE SHEETS
| December 31, 2023 | December 31, 2022 | |||||||
| ASSETS | ||||||||
| CURRENT ASSETS | ||||||||
| Cash | $ | 1,287,893 | $ | 215,189 | ||||
| Marketable securities | 2,768,119 | 7,763,517 | ||||||
| Prepaid expenses | 144,025 | 56,939 | ||||||
| TOTAL CURRENT ASSETS | 4,200,037 | 8,035,645 | ||||||
| Equipment - net | 123,332 | 1,775 | ||||||
| TOTAL ASSETS | $ | 4,323,369 | $ | 8,037,420 | ||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||
| CURRENT LIABILITIES | ||||||||
| Accounts payable and accrued expenses | $ | 439,757 | $ | 669,704 | ||||
| Accounts payable and accrued expenses - related party | 215,495 | 105,928 | ||||||
| Accounts payable and accrued expenses | 215,495 | 105,928 | ||||||
| PIK convertible notes payable, net of debt discount | - | 150,591 | ||||||
| PIK convertible notes payable, net of debt discount - related parties | - | 193,639 | ||||||
| PIK convertible notes payable, net of debt discount | - | 193,639 | ||||||
| TOTAL CURRENT LIABILITIES | 655,252 | 1,119,862 | ||||||
| TOTAL LIABILITIES | 655,252 | 1,119,862 | ||||||
| STOCKHOLDERS’ EQUITY | ||||||||
| Preferred stock, $ par value; shares authorized; shares issued and outstanding in the following classes: | - | - | ||||||
| Preferred stock; par value $; shares authorized; issued and outstanding | - | - | ||||||
| Series B convertible preferred stock, $ par value; shares authorized; and shares issued and outstanding at December 31, 2023, and December 31, 2022 | - | - | ||||||
| Preferred stock value | - | - | ||||||
| Common stock, $ par value, shares authorized, and shares issued and outstanding at December 31, 2023, and December 31, 2022 | 444 | 434 | ||||||
| Additional paid-in-capital | 34,559,091 | 33,371,406 | ||||||
| Accumulated deficit | (30,777,872 | ) | (25,777,375 | ) | ||||
| Accumulated other comprehensive loss | (113,546 | ) | (676,907 | ) | ||||
| TOTAL STOCKHOLDERS’ EQUITY | 3,668,117 | 6,917,558 | ||||||
| TOTAL LIABILITIES AND STOCKHOLDERS’ EQUITY | $ | 4,323,369 | $ | 8,037,420 | ||||
| December 31, 2017 | December 31, 2016 | |||||||
| ASSETS | ||||||||
| CURRENT ASSETS | ||||||||
| Cash and cash equivalents | $ | 399,687 | $ | 3,100,398 | ||||
| Marketable securities | 1,285,753 | - | ||||||
| Prepaid expenses | 94,542 | 60,417 | ||||||
| TOTAL CURRENT ASSETS | 1,779,982 | 3,160,815 | ||||||
| EQUIPMENT - NET | 1,022 | 1,097 | ||||||
| TOTAL ASSETS | $ | 1,781,004 | $ | 3,161,912 | ||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||
| CURRENT LIABILITIES | ||||||||
| Accounts payable and accrued expenses | 135,854 | 167,987 | ||||||
| Derivative liability | 425,838 | 516,870 | ||||||
| TOTAL CURRENT LIABILITIES | 561,692 | 684,857 | ||||||
| COMMITMENTS AND CONTINGENCIES | ||||||||
| STOCKHOLDERS’ EQUITY | ||||||||
| Preferred stock, $0.000001 par value; 20,000,000 shares authorized; 872,766 shares issued and outstanding in the following classes: | ||||||||
| Preferred stock; par value $0.000001; 2,000,000 shares authorized; none issued and outstanding | - | - | ||||||
| Series B convertible preferred stock, $0.000001 par value; 18,000,000 shares authorized; 872,766 shares issued and outstanding at December 31, 2017, and December 31, 2016, respectively | 1 | 1 | ||||||
| Common stock, $.0001 par value, 100,000,000 shares authorized, 10,261,419 shares issued and outstanding at December 31, 2017, and 10,257,078 shares issued and outstanding at December 31, 2016 | 1,026 | 1,026 | ||||||
| Additional paid-in-capital | 12,227,849 | 11,239,786 | ||||||
| Accumulated deficit | (10,841,759 | ) | (8,582,123 | ) | ||||
| Accumulated other comprehensive loss | (167,805 | ) | (181,635 | ) | ||||
| TOTAL STOCKHOLDERS’ EQUITY | 1,219,312 | 2,477,055 | ||||||
| TOTAL LIABILITIES AND STOCKHOLDERS’ EQUITY | $ | 1,781,004 | $ | 3,161,912 | ||||
See accompanying notes to the consolidated financial statements
| F-3 |
PROTAGENIC THERAPEUTICS, INC., AND SUBSIDIARIES
SUBSIDIARY
CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
| 2023 | 2022 | |||||||
| For the years ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| OPERATING AND ADMINISTRATIVE EXPENSES | ||||||||
| Research and development | $ | 3,319,867 | $ | 1,589,239 | ||||
| General and administrative | 1,207,107 | 1,968,549 | ||||||
| TOTAL OPERATING AND ADMINISTRATIVE EXPENSES | 4,526,974 | 3,557,788 | ||||||
| LOSS FROM OPERATIONS | (4,526,974 | ) | (3,557,788 | ) | ||||
| OTHER INCOME | ||||||||
| Interest income | 264,476 | 185,790 | ||||||
| Interest expense | (107,682 | ) | (137,456 | ) | ||||
| Realized loss on marketable securities | (630,317 | ) | (46,051 | ) | ||||
| TOTAL OTHER INCOME | (473,523 | ) | 2,283 | |||||
| LOSS BEFORE TAX | (5,000,497 | ) | (3,555,505 | ) | ||||
| INCOME TAX EXPENSE | - | - | ||||||
| NET LOSS | $ | (5,000,497 | ) | $ | (3,555,505 | ) | ||
| COMPREHENSIVE LOSS | - | - | ||||||
| Other Comprehensive Loss - net of tax | ||||||||
| Net unrealized gain (loss) on marketable securities | 16,848 | (421,738 | ) | |||||
| Reclassification of realized losses on debt securities | 489,120 | - | ||||||
| Foreign exchange translation income (loss) | 57,393 | (6,820 | ) | |||||
| TOTAL COMPREHENSIVE LOSS | $ | (4,437,136 | ) | $ | (3,984,063 | ) | ||
| Net loss per common share - Basic and Diluted | $ | ) | $ | ) | ||||
| Weighted average common shares - Basic and Diluted | ||||||||
| For the year ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| REVENUE | $ | - | $ | - | ||||
| OPERATING AND ADMINISTRATIVE EXPENSES | ||||||||
| Research and development | 717,452 | 417,866 | ||||||
| General and administrative | 1,647,872 | 1,411,466 | ||||||
| Goodwill impairment | - | 404,169 | ||||||
| TOTAL OPERATING AND ADMINISTRATIVE EXPENSES | 2,365,324 | 2,233,501 | ||||||
| LOSS FROM OPERATIONS | (2,365,324 | ) | (2,233,501 | ) | ||||
| OTHER (EXPENSE) INCOME | ||||||||
| Interest income | 13,890 | 907 | ||||||
| Interest expense - stockholder | - | (7,162 | ) | |||||
| Realized loss on foreign exchange transactions | - | (6,625 | ) | |||||
| Realized gain on marketable securities | 766 | - | ||||||
| Change in fair value of derivative liability | 91,032 | (29,445 | ) | |||||
| TOTAL OTHER (EXPENSE) INCOME | 105,688 | (42,325 | ) | |||||
| NET LOSS | $ | (2,259,636 | ) | $ | (2,275,826 | ) | ||
| COMPREHENSIVE LOSS | ||||||||
| Other Comprehensive Income - net of tax | ||||||||
| Net unrealized gain (loss) on marketable securities | (1,426 | ) | - | |||||
| Foreign exchange translation gain (loss) | 15,256 | (32,984 | ) | |||||
| TOTAL COMPREHENSIVE LOSS | $ | (2,245,806 | ) | $ | (2,308,810 | ) | ||
| Net loss per share - Basic and Diluted | $ | (0.22 | ) | $ | (0.43 | ) | ||
| Weighted average common shares - Basic and Diluted | 10,261,419 | 5,306,035 | ||||||
See accompanying notes to the consolidated financial statements
| F-4 |
PROTAGENIC THERAPEUTICS, INC., AND SUBSIDIARIES
SUBSIDIARY
CONSOLIDATED STATEMENTSTATEMENTS OF CHANGE IN STOCKHOLDERS’ EQUITY (DEFICIT)
For the Fiscal Year Ended December 31, 20162023 and 20172022
| Series B Convertible | Additional | Accumulated Other | Stockholders’ | |||||||||||||||||||||||||||||||||||||
| Preferred Stock | Common Stock | Paid-in- | Accumulated | Treasury Stock | Comprehensive | (Deficit) | ||||||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Capital | (Deficit) | Shares | Amount | Loss | Equity | |||||||||||||||||||||||||||||||
| BALANCE - January 1, 2016 | - | - | 7,612,838 | $ | 761 | $ | 5,886,971 | $ | (6,306,297 | ) | (1,000,000 | ) | $ | (100,000 | ) | $ | (148,651 | ) | $ | (667,216 | ) | |||||||||||||||||||
| Merger: | ||||||||||||||||||||||||||||||||||||||||
| Atrinsic shares converted | 297,468 | 1 | 25,867 | 3 | 63,381 | 63,385 | ||||||||||||||||||||||||||||||||||
| Protagenic shares converted | 6,612,838 | 6 | (7,612,838 | ) | (761 | ) | (99,245 | ) | 1,000,000 | 100,000 | - | |||||||||||||||||||||||||||||
| Private offerings, net of expenses | 4,108,460 | 4 | 4,761,793 | 4,761,797 | ||||||||||||||||||||||||||||||||||||
| Foreign currency translation (loss) | (32,984 | ) | (32,984 | ) | ||||||||||||||||||||||||||||||||||||
| Stock compensation - stock options | 546,134 | 546,134 | ||||||||||||||||||||||||||||||||||||||
| Conversion of series B Preferred Stock | (10,146,000 | ) | (10 | ) | 10,146,000 | 1,015 | (1,005 | ) | - | |||||||||||||||||||||||||||||||
| Conversion of Bridge loan | 60,211 | 6 | 75,259 | 75,265 | ||||||||||||||||||||||||||||||||||||
| Stock options converted to common | 25,000 | 2 | 6,498 | 6,500 | ||||||||||||||||||||||||||||||||||||
| Net loss | (2,275,826 | ) | (2,275,826 | ) | ||||||||||||||||||||||||||||||||||||
| BALANCE - December 31, 2016 | 872,766 | 1 | 10,257,078 | 1,026 | 11,239,786 | (8,582,123 | ) | - | - | (181,635 | ) | 2,477,055 | ||||||||||||||||||||||||||||
| Unrealized gain (loss) on marketable securities | (1,426 | ) | (1,426 | ) | ||||||||||||||||||||||||||||||||||||
| Foreign currency translation gain | 15,256 | 15,256 | ||||||||||||||||||||||||||||||||||||||
| Stock compensation - stock options | 888,281 | 888,281 | ||||||||||||||||||||||||||||||||||||||
| Adjustment to common stock | 4,341 | - | - | - | ||||||||||||||||||||||||||||||||||||
| Modification of warrants | 99,782 | 99,782 | ||||||||||||||||||||||||||||||||||||||
| Net loss | (2,259,636 | ) | (2,259,636 | ) | ||||||||||||||||||||||||||||||||||||
| BALANCE -December 31, 2017 | 872,766 | $ | 1 | 10,261,419 | $ | 1,026 | $ | 12,227,849 | $ | (10,841,759 | ) | $ | - | $ | - | $ | (167,805 | ) | $ | 1,219,312 | ||||||||||||||||||||
| Shares | Amount | Shares | Amount | Capital | (Deficit) | Loss | Equity | |||||||||||||||||||||||||
| Series B Convertible Preferred Stock | Common Stock | Additional Paid-in- | Accumulated | Accumulated Other Comprehensive | Stockholders’ | |||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Capital | (Deficit) | Loss | Equity | |||||||||||||||||||||||||
| BALANCE -December 31, 2021 | - | $ | - | 4,302,403 | $ | 432 | $ | 32,411,742 | $ | (22,221,870 | ) | $ | (248,349 | ) | $ | 9,941,955 | ||||||||||||||||
| Foreign currency translation loss | - | - | - | - | - | - | (6,820 | ) | (6,820 | ) | ||||||||||||||||||||||
| Unrealized loss on marketable securities | - | - | - | - | - | - | (421,738 | ) | (421,738 | ) | ||||||||||||||||||||||
| Stock compensation - stock options | - | - | - | - | 844,248 | - | - | 844,248 | ||||||||||||||||||||||||
| Stock compensation - warrants | - | - | - | - | 20,433 | - | - | 20,433 | ||||||||||||||||||||||||
| Conversion of notes and interest | - | - | 18,912 | 2 | 94,983 | - | - | 94,985 | ||||||||||||||||||||||||
| Net loss | - | - | - | - | - | (3,555,505 | ) | - | (3,555,505 | ) | ||||||||||||||||||||||
| BALANCE -December 31, 2022 | - | $ | - | 4,321,315 | $ | 434 | $ | 33,371,406 | $ | (25,777,375 | ) | $ | (676,907 | ) | $ | 6,917,558 | ||||||||||||||||
| BALANCE | - | $ | - | 4,321,315 | $ | 434 | $ | 33,371,406 | $ | (25,777,375 | ) | $ | (676,907 | ) | $ | 6,917,558 | ||||||||||||||||
| Foreign currency translation gain | - | - | - | - | - | - | 57,393 | 57,393 | ||||||||||||||||||||||||
| Unrealized gain on marketable securities | - | - | - | - | - | - | 16,848 | 16,848 | ||||||||||||||||||||||||
| Reclassification of realized losses on debt securities | - | - | - | - | - | - | 489,120 | 489,120 | ||||||||||||||||||||||||
| Stock compensation - stock options | - | - | - | - | 666,828 | - | - | 666,828 | ||||||||||||||||||||||||
| Conversion of notes and interest | - | - | 104,173 | 10 | 520,857 | - | - | 520,867 | ||||||||||||||||||||||||
| Rounding from reverse split | - | - | 9,644 | - | - | - | - | - | ||||||||||||||||||||||||
| Net loss | - | - | - | - | - | (5,000,497 | ) | - | (5,000,497 | ) | ||||||||||||||||||||||
| BALANCE -December 31, 2023 | - | $ | - | 4,435,132 | $ | 444 | $ | 34,559,091 | $ | (30,777,872 | ) | $ | (113,546 | ) | $ | 3,668,117 | ||||||||||||||||
| BALANCE | - | $ | - | 4,435,132 | $ | 444 | $ | 34,559,091 | $ | (30,777,872 | ) | $ | (113,546 | ) | $ | 3,668,117 | ||||||||||||||||
See accompanying notes to the consolidated financial statements
| F-5 |
PROTAGENIC THERAPEUTICS, INC., AND SUBSIDIARIES
SUBSIDIARY
CONSOLIDATED STATEMENTS OF CASH FLOWS
| For the year ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| CASH FLOWS FROM OPERATING ACTIVITIES | ||||||||
| Net Loss | $ | (2,259,636 | ) | $ | (2,275,826 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities | ||||||||
| Depreciation expense | 148 | 534 | ||||||
| Goodwill impairment | - | 404,169 | ||||||
| Stock based compensation | 888,281 | 546,134 | ||||||
| Accretion to bridge loan | - | 7,162 | ||||||
| Legal fees satisfied through issuance of Series B preferred stock | - | 150,000 | ||||||
| Change in fair value of the derivative liability | (91,032 | ) | 29,445 | |||||
| Gain on sale of marketable securities | (766 | ) | - | |||||
| Modification of warrants | 99,782 | - | ||||||
| Changes in operating assets and liabilities | ||||||||
| Prepaid expenses | (34,125 | ) | (60,417 | ) | ||||
| Other receivables | - | 6,230 | ||||||
| Accounts payable and accrued expenses | 17,259 | 52,250 | ||||||
| NET CASH USED IN OPERATING ACTIVITIES | (1,380,089 | ) | (1,140,319 | ) | ||||
| CASH FLOWS USED IN INVESTING ACTIVITIES | ||||||||
| Sale of marketable securities | 2,145,000 | - | ||||||
| Purchase of marketable securities | (3,431,414 | ) | - | |||||
| NET CASH USED IN INVESTING ACTIVITIES | (1,286,414 | ) | - | |||||
| CASH FLOWS FROM FINANCING ACTIVITIES | ||||||||
| Proceeds from conversion of stock options | - | 6,500 | ||||||
| Proceeds from bridge loan | - | 19,000 | ||||||
| Proceeds from issuance of Series B Preferred Stock | - | 4,258,438 | ||||||
| NET CASH PROVIDED BY FINANCING ACTIVITIES | - | 4,283,938 | ||||||
| Effect of exchange rate on cash and cash equivalents | (34,208 | ) | (46,564 | ) | ||||
| NET INCREASE (DECREASE) IN CASH AND CASH EQUIVALENTS | (2,700,711 | ) | 3,097,055 | |||||
| CASH AND CASH EQUIVALENTS, BEGINNING OF PERIOD | 3,100,398 | 3,343 | ||||||
| CASH AND CASH EQUIVALENTS, END OF PERIOD | $ | 399,687 | $ | 3,100,398 | ||||
| SUPPLEMENTAL DISCLOSURES OF CASH FLOW INFORMATION | ||||||||
| Cash paid for interest expense | $ | - | $ | - | ||||
| Cash paid for income taxes | $ | - | $ | - | ||||
| NONCASH TRANSACTIONS | ||||||||
| Unrealized loss on marketable securities | $ | 1,427 | $ | - | ||||
| Debt settled with issuance of Series B preferred stock | $ | - | $ | 425,265 | ||||
| Reclassification of warrants to derivative liabilities from equity | $ | - | $ | 487,425 | ||||
| Shares issued in connection with reverse business combination | $ | - | $ | 404,169 | ||||
| Accrued liabilities paid through the issuances of Series B preferred stock | $ | - | $ | 150,000 | ||||
| Series B Preferred stock converted to common stock | $ | - | $ | 10 | ||||
| 2023 | 2022 | |||||||
| For the year ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| CASH FLOWS FROM OPERATING ACTIVITIES | ||||||||
| Net Loss | $ | (5,000,497 | ) | $ | (3,555,505 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities | ||||||||
| Depreciation expense | 28,218 | 30 | ||||||
| Stock-based compensation | 666,828 | 864,681 | ||||||
| Realized loss on sale of marketable securities | 630,317 | 46,051 | ||||||
| Amortization of debt discount | 85,770 | 110,797 | ||||||
| Changes in operating assets and liabilities | ||||||||
| Prepaid expenses | (87,086 | ) | 631,728 | |||||
| Accounts payable and accrued expenses | (27,326 | ) | (91,596 | ) | ||||
| NET CASH USED IN OPERATING ACTIVITIES | (3,703,776 | ) | (1,993,814 | ) | ||||
| CASH FLOWS FROM INVESTING ACTIVITIES | ||||||||
| Proceeds from sale of marketable securities | 7,689,507 | 1,632,901 | ||||||
| Purchase of marketable securities | (2,764,250 | ) | (34,122 | ) | ||||
| Purchase of fixed assets | (149,775 | ) | (1,805 | ) | ||||
| NET CASH PROVIDED BY INVESTING ACTIVITIES | 4,775,482 | 1,596,974 | ||||||
| Effect of exchange rate changes on cash | 998 | 70,858 | ||||||
| NET CHANGE IN CASH | 1,072,704 | (325,982 | ) | |||||
| CASH, BEGINNING OF THE PERIOD | 215,189 | 541,171 | ||||||
| CASH, END OF THE PERIOD | $ | 1,287,893 | $ | 215,189 | ||||
| SUPPLEMENTAL DISCLOSURES OF CASH FLOW INFORMATION | ||||||||
| Cash paid for interest expense | $ | - | $ | - | ||||
| Cash paid for income taxes | $ | - | $ | - | ||||
| NONCASH FINANCING AND INVESTING TRANSACTIONS | ||||||||
| Shares issued for conversion of notes and interest | $ | 520,867 | $ | 94,985 | ||||
| Unrealized gain or loss on marketable securities | $ | 16,848 | $ | 421,738 | ||||
See accompanying notes to the consolidated financial statements
| F-6 |
PROTAGENIC THERAPEUTICS, INCINC. & SUBSIDIARIES
SUBSIDIARY
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
December 31, 20172023
NOTE 1 – ORGANIZATION AND NATURE OF BUSINESS
Company Background
Protagenic Therapeutics, Inc. (“we,” “our,” “Protagenic” or “the Company”), formerly known as Atrinsic, Inc., is a Delaware corporation with one subsidiary named Protagenic Therapeutics Canada (2006) Inc. (“PTI Canada”), a corporation formed in 2006 under the laws of the Province of Ontario, Canada.
The Company was previously known as Atrinsic, Inc.,We are a biopharmaceutical company that was once a reporting company underspecializing in the Securities Act, but that, in 2012discovery and 2013, reorganized under Chapter 11development of the United States Bankruptcy Codetherapeutics to treat stress-related neuropsychiatric and emerged from bankruptcy. mood disorders.
Reverse Stock Split
On February 12, 2016,March 22, 2023, the Company acquired Protagenic Therapeutics, Inc. througheffectuated a 1 for 4 reverse merger. On June 17, 2016, Protagenic Therapeutics, Inc.stock split (the then wholly-owned subsidiary of Atrinsic, Inc.“Reverse Split”) was merged with and into Atrinsic, Inc. Atrinsic, Inc. was the surviving corporation in this merger and changed its name from Atrinsic, Inc. to Protagenic Therapeutics, Inc.
Reverse Business Combination (Merger)
On February 12, 2016 (“Closing Date”), Protagenic Acquisition Corp., a wholly-owned subsidiary of the Company (which at the time was named Atrinsic, Inc.), merged (the “Merger”) with and into Prior Protagenic. Prior Protagenic was the surviving corporation of the Merger. As a result of the Merger, the Company acquired the business of prior Protagenic and will continue the existing business operations of Prior Protagenic as a wholly-owned subsidiary. On June 17, 2016, Prior Protagenic merged with and into the Company with the Company as the surviving corporation in the merger. Immediately thereafter, the Company changed its name from Atrinsic, Inc. to Protagenic Therapeutics, Inc.
On the Closing Date, all of the issued and outstanding (6,612,838) shares of Prior Protagenic common. The Company’s stock converted,began trading on a 1 for 1split-adjusted basis into shares of the Company’s Series B Convertible Preferred Stock, par value $0.000001 per share (“Series B Preferred Stock”). Alsoeffective on the Closing Date, all of the issued and outstanding options to purchase shares of Prior Protagenic common stock, and all of the issued and outstanding warrants to purchase shares of Prior Protagenic common stock, converted,Nasdaq Stock Market on a 1 for 1 basis, into options (the “New Options”) and warrants (the “New Warrants”) respectively, to purchase shares of Series B Preferred Stock. New Options to purchase 1,807,744 shares of Series B Preferred Stock, having an average exercise price of approximately $0.87 per share, were issued to Prior Protagenic optionees. New Warrants to purchase 3,403,367 shares of Series B Preferred Stock at an average exercise price of approximately $1.03 per share were issued to holders of Prior Protagenic warrants.
The common stockholders of Atrinsic, Inc. before the Merger (“Predecessor”) retained 25,867 shares of common stock, par value $0.0001 per share (the “Common Stock”). Upon the effectiveness of the Merger, the holders of the Predecessor’s Series A Preferred Stock exchanged all of the issued and outstanding Series A Preferred Stock for an aggregate of 297,468 shares of Series B Preferred Stock. In addition, the holders of options to purchase Predecessor common stock were issued options (“Predecessor Options”) to purchase 17,784 shares of Series B Preferred Stock at $1.25 per share. Warrants (“Predecessor Warrants”) to purchase 295,945 shares of Series B Preferred Stock at $1.25 per share were issued to Strategic Bio Partners, LLC, the designee (the “Designee”) of the holders of the Predecessor’s debt in consideration of the cancellation of such debt amounting to $665,000 in principal and $35,000 in interest.
The Merger is being accounted for as a “Reverse Business Combination,” and Prior Protagenic is deemed to be the accounting acquirer in the merger. Consequently, the assets and liabilities and the historical operations that will be reflected in the financial statements priorMarch 22, 2023. There was no change to the Merger will be thosenumber of Prior Protagenic, and the consolidated financial statements after completion of the Merger will include the assets and liabilities of Prior Protagenic, historical operations of Prior Protagenic and combined operations of Prior Protagenic, Predecessor and the Company from the Closing Date of the Merger. Further, as a result of the issuance of the shares of Series B Preferred Stock pursuant to the Merger, a change in control of the Company occurred as of the date of consummation of the Merger.
The Merger will be treated as a recapitalization of the Company for financial accounting purposes. The historical financial statements of Predecessor before the Merger will be replaced with the historical financial statements of Prior Protagenic before the Merger in all future filings with the Securities and Exchange Commission (the “SEC”).
At the closing of the Merger, Predecessor had a 51% interest in MomSpot, and the remaining 49% was held by B.E. Global LLC. Barry Eisenberg is the sole owner of B.E. Global LLC and is the Chief Executive Officer of MomSpot LLC. Immediately after the closing of the Merger, the Company split off its 51% membership interests in MomSpot. The split-off was accomplished through the transfer of all of its membership interests of MomSpot, having nominal value, to B.E. Global LLC via a split off agreement for nominal consideration.
Immediately after the closing of the Merger, the Company also split off all of its equity interest in 29 wholly-owned subsidiaries of Predecessor. The split-off was accomplished through the sale of all equity interests in these wholly-owned subsidiaries to Quintel Holdings, Inc. for nominal considerations via a split off agreement. These entities had nominal value.
Private Offering
Concurrently and a condition of the closing of the Merger, the Company conducted the first closing of an offering (the “Private Offering”) of our Series B Preferred Stock. At the first closing, we sold 2,775,000 shares of Series B Preferred Stock at a purchase price of $1.25 per share, for which we received total gross consideration of $3,468,750. Of this amount, $350,000 consisted of the conversion of outstanding stockholder debt held by Garo H. Armen, our Chairman and a member of our Board of Directors, and $150,000 of legal expenses incurred by Strategic Bio Partners, LLC, on behalf of the stockholders of Predecessor, in conjunction with and as permitted under the terms of the Merger. On March 2, 2016, we completed the second closing of the Private Offering, at which we sold an additional 913,200 shares of Series B Preferred Stock, for total gross proceeds of $1,141,500. On April 15, 2016 we completed the final closing of the Private Offering, at which we issued an additional 420,260 shares of Series B Preferred Stock to accredited investors, for total gross proceeds of $525,325. The Company paid commissions, legal and miscellaneous fees aggregating $373,778 associated with these closings. We also issued Placement Agent Warrants to purchase 127,346 shares of Series B Preferred Stock valued at $146,641 using a Black-Scholes model at an exercise price of $1.25 per share to the Placement Agent and its selected dealers. The Company determined the fair value of the binomial lattice model and the Black-Scholes valuation model to be materially the same. For all three closings, the Company issued 4,108,460 shares of Series B Preferred Stock and raised total gross proceeds of $4,635,575 and total net proceeds of $4,261,797 (or total gross proceeds of $5,135,575 and total net proceeds of $4,761,797, including the conversion of the $350,000 in principal of stockholder debt, and $150,000 of legal expenses incurred by the Predecessor’s stockholders.
Debt Exchange
Simultaneous with the Merger and the Private Offering, holders of $665,000 of Predecessor debt accompanied with $35,000 in accrued interest exchanged such debt for Predecessor Warrants to purchase 295,945 shares of Series B Preferred Stock at $1.25 per share. The Predecessor Warrants were valued at $340,784 (see Note 6).
Reverse Stock Split
On June 17, 2016, the Company held a Special Meeting of Stockholders (the “Special Meeting”). At the Special Meeting, the Company’s stockholders approved a third amendment and restatement (the “Third Amendment and Restatement”) to the Company’s Amended and Restated Certificate of Incorporation, effective July 27, 2016 (the “Effective Time”), to effect a one-for-15,463.7183 reverse split of the Company’s common stock (the “Reverse Stock Split”). Pursuant to the Reverse Stock Split, at the Effective Time, each 15,463.7183 shares of common stock owned by a stockholder were combined into one new share of common stock, with any fractional shares that would otherwise be issuable as a result of the Reverse Stock Split being rounded up to the nearest whole share. The Third Amendment and Restatement also effected (i) a reduction in the Company’s authorized shares of common stock from 100 billion shares to 100 million shares, (ii) an increase in the par value of the Company’s common stock from $0.000001 per share to $0.0001 per share and (iii) a reduction in the Company’s authorized shares of preferred stock from 5 billion shares to 20 million shares.
As a result of the Reverse Split, 400,000,000 shares of common stock were split into 25,867 shares of common stock. Additionally, as a result of the Reverse Split and in accordance with our certificate of designations for our Series B Preferred Stock, our Series B Preferred Stock immediately and automatically converted into our common stock on a 1-for-1 basis other than any Series B Preferred Stock (i) to the extent (but only to the extent) a Series B Preferred Stock holder would beneficially own greater than 9.99% of our common stock (the “Springing Blocker”) and (ii) such holder has notified the Company in writing that it wants the Springing Blocker to apply to such holder. On July 27, 2016, 10,146,000 of the Company’s 11,018,766 outstanding shares of Series B Preferred Stock were eligible to immediately convert into 10,146,000 shares of the Company’s common stock with 872,766 shares of Series B Preferred Stock remaining as a result of one holder exercising the Springing Blocker. As of December 31, 2016, 10,146,000 shares of the Series B Preferred Stock were converted into 10,146,000 shares of common stock on the records of the Company.
Any Series B Preferred Stock not converted as a result of this provision would automatically convert into common stock as soon as such conversion would not violate the Springing Blocker. Our Series B Preferred Stock will cease to be designated as a separate series of our preferred stock when all of such shares have converted into shares of our common stock.
All share and per share amounts forinformation in these financial statements are adjusted to reflect the common stock have been retroactively restated to give effect to the reverse split.Reverse Split.
NOTE 2 -– LIQUIDITY AND GOING CONCERN
As shown in the accompanying consolidated financial statements, the Company incurred a net loss of $2,259,636 and $2,275,826 for the years ended December 31, 2017 and 2016, respectively. The Company has incurred significant recurring losses since inception resulting in an accumulated deficit of $10,841,759 as of December 31, 2017. The net loss presented for the twelve months is attributed to an increase in research and development expense and an increase in stock compensation expense. The net loss present for the prior period was attributed to goodwill impairment, an increase in professional fees as related to the Merger, and an increase in stock compensation expense.deficit. The Company anticipates further losses in the development of its business. The Company also had a net working capital of $1,218,290 at December 31, 2017 as a result of the Merger and simultaneous financings. Based on its current forecast and budget, Management believes that itsnegative cash resources will be sufficient to fund its operations at least until the third quarter of 2018. Absent generation of sufficient revenue from the execution of the Company’s business plan, it will need to obtain debt or equity financing by the fourth quarter of 2018.
As reflected in the consolidated financial statements, the Company had an accumulated deficit at December 31, 2017, a net loss and net cashflows used in operating activities for the year ended.operations. These factors raise substantial doubt about the Company’s ability to continue as a going concern.
Based on its cash resources and positive working capital as of December 31, 2023, the Company does not have sufficient resources to fund its operations past end of the third quarter of 2024. The positive working capital as of December 31, 2023 was due to funds raised by the Company from its equity offering during the year ended December 31, 2021. Absent generation of sufficient revenue from the execution of the Company’s business plan, the Company will need to obtain debt or equity financing by the third quarter of 2024. Because the Company has insufficient resources on hand to fund operations through the next twelve months from the date these consolidated financial statements are available to be issued, the Company believes that there is substantial doubt in its ability to continue as a going concern. These financial statements do not include any adjustments to the recoverability and classification of recorded asset amounts and classification of liabilities that might be necessary should the Company be unable to continue as a going concern.
NOTE 3 - SUMMARY OF SIGNFICANTSIGNIFICANT ACCOUNTING POLICIES
Basis of presentation
The Company’s consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America (“U.S. GAAP”) and the rules and regulations of the Securities and Exchange Commission (“SEC”).
The consolidated financial statements include the accounts of Protagenic Therapeutics, Inc., and its wholly-owned Canadian subsidiary, PTI Canada. All significant intercompany balances and transactions have been eliminated in consolidation.
Principles of consolidation
The consolidated financial statements include the accounts of Atrinsic, Inc., and its wholly owned subsidiary, Protagenic Acquisition Corp, and Protagenic Therapeutics, Inc., which merged with and into Protagenic Acquisition Corp, on February 12, 2016, as well as Protagenic Therapeutics’ wholly-owned Canadian subsidiary, PTI Canada. All significant intercompany balances and transactions have been eliminated in the consolidated financial statements.
Use of estimates
The preparation of the consolidated financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenue and expense during the reporting period. Actual results could differ from those estimates. Significant estimates underlying the consolidated financial statements include the allocation of the fair value of acquired assets and liabilities associated with the Merger, income tax provisions, impairment of goodwill, valuation of stock options and warrants and assessment of deferred tax asset valuation allowance.
Reclassification
| F-7 |
Certain prior period amounts have been reclassified to conform to current period presentation.
Concentrations of Credit Risk
The Company maintains its cash accounts at financial institutions which are insured by the Federal Deposit Insurance Corporation. At times, the Company may have deposits in excess of federally insured limits. As of December 31, 2023, the Company has bank balances that exceed the federally insured limits. The Company has not experienced losses on these accounts and management believes, based upon the quality of the financial institutions, that the credit risk with regard to these deposits is not significant.
Funds held in the Company’s marketable securities are not insured.
Cash and Cash Equivalents
The Company considers all highly liquid investments with an original maturity of three months or less when purchased to be cash equivalents. As of December 31, 2017,2023 and 2016,December 31, 2022 the Company did not have any cash equivalents.
EquipmentMarketable Securities
The Company accounts for marketable debt securities, the only type of securities it owns, in accordance with the FASB Accounting Standards Codification 320, Investments – Debt and Equity Securities (“ASC 320”).
Pursuant to ASC 320-10-35-1, investments in debt securities that are classified as available for sale shall be measured subsequently at fair value in the consolidated balance sheets at each balance sheet date. Unrealized holding gains and losses for available-for-sale securities (including those classified as current assets) shall be excluded from earnings and reported in other comprehensive income until realized.
During the year ended December 31, 2023 the Company purchased $2,764,250 and sold $7,689,507 in marketable securities with a realized loss of $630,317, which includes a $489,120 reclassification of realized losses from other comprehensive income, and an unrealized gain of $16,848. As of December 31, 2023 and December 31, 2022, the Company owned marketable securities with a total fair value of $2,768,119 and $7,763,517, respectively.
Equipment
Equipment is stated at cost less accumulated depreciation. Cost includes expenditures for computer equipment and lab equipment. Maintenance and repairs are charged to expense as incurred. When assets are sold, retired, or otherwise disposed of, the cost and accumulated depreciation are removed from the accounts and any resulting gain or loss is reflected in operations. The cost of equipment is depreciated using the straight-line method over the estimated useful lives of the related assets which is 3 years.three years. Depreciation expense was not material $28,218 and $30 for the years ended December 31, 20172023 and 2016.2022.
Marketable Securities
The Company accounts for marketable debt and equity securities, available for sale, in accordance with sub-topic 320-10 of the FASB Accounting Standards Codification (“Sub-topic 320-10”).
Pursuant to Paragraph 320-10-35-1, investments in debt securities that are classified as available for sale and equity securities that have readily determinable fair values that are classified as available for sale shall be measured subsequently at fair value in the consolidated balance sheets at each balance sheet date. Unrealized holding gains and losses for available-for-sale securities (including those classified as current assets) shall be excluded from earnings and reported in other comprehensive income until realized except an available-for-sale security that is designated as being hedged in a fair value hedge, from which all or a portion of the unrealized holding gain and loss of shall be recognized in earnings during the period of the hedge, pursuant to paragraphs 815-25-35-1 through 815-25-35-4.
During the year ended December 31, 2017 the Company purchased $3,431,414 and sold $2,145,000 in marketable securities with a realized gain of $766 and an unrealized loss of $1,427. As of December 31, 2017, the Company owns marketable securities with a total value of $1,285,753.
As of December 31, 2017, the marketable securities have maturity dates ranging from January 4, 2018 to February 22, 2018.
Goodwill
Goodwill represents the excess of the purchase price over the fair value of the assets acquired and liabilities assumed. The Company is required to perform impairment reviews annually and more frequently in certain circumstances. The Company performs the annual assessment on December 31.
In accordance with ASC 350–20 “Goodwill”, the Company is able to make a qualitative assessment of whether it is more likely than not that a reporting unit’s fair value is less than its carrying amount before applying the two–step goodwill impairment test. If the Company concludes that it is more likely than not that the fair value of a reporting unit is not less than its carrying amount it is not required to perform the two–step impairment test for that reporting unit.
Atrinsic’s assets and liabilities acquired in the Merger had a minimal value therefore the Company recorded the fair value of shares given to predecessor stockholders as goodwill. Immediately subsequent to the merger the Company fully impaired the goodwill, in as the predecessor business had limited operations.
The allocation of the consideration transferred is as follows:
| Shares issued in connection with Merger: | ||||
| Atrinsic 25,867 shares Common stock | $ | 32,334 | ||
| Atrinsic Series A preferred stock as converted to Series B preferred stock, 297,468 shares | 371,835 | |||
| Total value of shares issued to Atrinsic on Merger | 404,169 | |||
| Fair value of net assets identified | - | |||
| Goodwill | 404,169 | |||
| Net value of consideration | $ | - |
Goodwill impairment for the year ended December 31, 2016 was $404,169. As of December 31, 2017, the goodwill was $0.
Fair Value Measurements
ASC 820, “Fair Value Measurements and Disclosure,” defines fair value as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date, not adjusted for transaction costs. ASC 820 also establishes a fair value hierarchy that prioritizes the inputs to valuation techniques used to measure fair value into three broad levels giving the highest priority to quoted prices in active markets for identical assets or liabilities (Level 1) and the lowest priority to unobservable inputs (Level 3).
The three levels are described below:
Level 1 Inputs – Unadjusted quoted prices in active markets for identical assets or liabilities that is accessible by the Company;
Level 2 Inputs – Quoted prices in markets that are not active or financial instruments for which all significant inputs are observable, either directly or indirectly;
| F-8 |
Level 3 Inputs – Unobservable inputs for the asset or liability including significant assumptions of the Company and other market participants.
The carrying amount of the Company’s financial assets and liabilities, such as cash, accounts payable and accrued expenses approximate their fair value because of the short term maturity of those instruments.
Transactions involving related parties cannot be presumed to be carried out on an arm’s-length basis, as the requisite conditions of competitive, free-market dealings may not exist. Representations about transactions with related parties, if made, shall not imply that the related party transactions were consummated on terms equivalent to those that prevail in arm’s-length transactions unless such representations can be substantiated.
The assets or liability’s fair value measurement within the fair value hierarchy is based upon the lowest level of any input that is significant to the fair value measurement. The following table provides a summary of financial instruments that are measured at fair value on a recurring basis as of December 31, 2017.2023.
SCHEDULE OF FAIR VALUE ASSETS AND LIABILITIES MEASURED ON RECURRING BASIC
| Carrying | Fair Value Measurement Using | |||||||||||||||||||
| Value | Level 1 | Level 2 | Level 3 | Total | ||||||||||||||||
| Marketable securities | 1,285,753 | — | 1,285,753 | — | 1,285,753 | |||||||||||||||
| Derivative warrants liabilities | $ | (425,838 | ) | $ | — | $ | — | $ | (425,838 | ) | $ | (425,838 | ) | |||||||
| Carrying | Fair Value Measurement Using | |||||||||||||||||||
| Value | Level 1 | Level 2 | Level 3 | Total | ||||||||||||||||
| Marketable securities | $ | 2,768,119 | $ | 2,768,119 | $ | — | $ | — | $ | 2,768,119 | ||||||||||
The following table provides a summary of financial instruments that are measured at fair value on a recurring basis as of December 31, 2016.2022.
| Carrying | Fair Value Measurement Using | |||||||||||||||||||
| Value | Level 1 | Level 2 | Level 3 | Total | ||||||||||||||||
| Derivative warrants liabilities | $ | (516,870 | ) | $ | — | $ | — | $ | (516,870 | ) | $ | (516,870 | ) | |||||||
| Carrying | Fair Value Measurement Using | |||||||||||||||||||
| Value | Level 1 | Level 2 | Level 3 | Total | ||||||||||||||||
| Marketable securities | $ | 7,763,517 | $ | 7,763,517 | $ | — | $ | — | $ | 7,763,517 | ||||||||||
The table below provides a summary of the changes in fair value, including net transfers in and/or out, of all financial assets and liabilities measured at fair value on a recurring basis using significant unobservable inputs (Level 3) during the year ended December 31, 2017:
Fair Value Measurement Using Level 3 Inputs | ||||
| Total | ||||
| Balance, December 31, 2016 | $ | 516,870 | ||
| Change in fair value of derivative warrants liabilities | (91,032 | ) | ||
| Balance, December 31, 2017 | $ | 425,838 | ||
The fair value of the derivative feature of the 127,346 and 295,945 warrants to the placement agent of the private offering and to Strategic Bio Partners for debt cancellation, respectively on the issuance dates and at the balance sheet date were calculated using a Black-Scholes option model valued with the following assumptions:
| February 12, 2016 | December 31, 2016 | December 31, 2017 | ||||||||||
| Exercise price | $ | 1.25 | $ | 1.25 | 1.25 | |||||||
| Risk free interest rate | 1.20 | % | 1.93 | % | 1.98 | % | ||||||
| Dividend yield | 0.00 | % | 0.00 | % | 0.00 | % | ||||||
| Expected volatility | 156 | % | 219 | % | 144 | % | ||||||
| Contractual term | 5.0 years | 4.25 years | 3.15 Years | |||||||||
Risk-free interest rate: The Company uses the risk-free interest rate of a U.S. Treasury Note with a similar expected term on the date of the grant.
Dividend yield: The Company uses a 0% expected dividend yield as the Company has not paid dividends to date and does not anticipate declaring dividends in the near future.
Volatility: The Company calculates the expected volatility of the stock price based on the corresponding volatility of the Company’s peer group stock price for a period consistent with the warrants’ expected term.
Expected term: The Company’s expected term is based on the remaining contractual maturity of the warrants.
During the year ended December 31, 2017 and 2016, the Company marked the derivative feature of the warrants to fair value and recorded a loss of $91,032 and a gain of $29,445 relating to the change in fair value, respectively.
Derivative Liability
The Company evaluates its options, warrants or other contracts, if any, to determine if those contracts or embedded components of those contracts qualify as derivatives to be separately accounted for in accordance with ASC 815-10-05-4 and 815-40-25. The result of this accounting treatment is that the fair value of the embedded derivative is marked-to-market each balance sheet date and recorded as either an asset or a liability. In the event that the fair value is recorded as a liability, the change in fair value is recorded in the consolidated statement of operations as other income or expense. Upon conversion, exercise or cancellation of a derivative instrument, the instrument is marked to fair value at the date of conversion, exercise or cancellation and then the related fair value is reclassified to equity.
The classification of derivative instruments, including whether such instruments should be recorded as liabilities or as equity, is re-assessed at the end of each reporting period. Equity instruments that are initially classified as equity that become subject to reclassification are reclassified to liability at the fair value of the instrument on the reclassification date. Derivative instrument liabilities will be classified in the balance sheet as current or non-current based on whether or not net-cash settlement of the derivative instrument is expected within 12 months of the balance sheet date.
Stock-Based Compensation
The Company accounts for stock basedstock-based compensation costs under the provisions of ASC 718, “Compensation—Stock Compensation”, which requires the measurement and recognition of compensation expense related to the fair value of stock basedstock-based compensation awards that are ultimately expected to vest. Stock based compensation expense recognized includes the compensation cost for all stock basedstock-based payments granted to employees, officers, non-employees, and directors based on the grant date fair value estimated in accordance with the provisions of ASC 718. ASC.ASC 718 is also applied to awards modified, repurchased, or canceledcancelled during the periods reported.
If any award granted under the Company’s 2016 Equity Compensation Plan (the “2016 Plan”) payable in shares of common stock is forfeited, cancelled, or returned for failure to satisfy vesting requirements, otherwise terminates without payment being made, or if shares of common stock are withheld to cover withholding taxes on options or other awards, the number of shares of common stock as to which such option or award was forfeited, or which were withheld, will be available for future grants under the 2016 Plan. The companyCompany recognizes the impact of forfeitures when they occur.
The Company accounts for warrants and options issued to non-employees under ASC 505-50,Equity – Equity Based Payments to Non-Employees, using the Black-Scholes option-pricing model. The value of such non-employee awards unvested are re-measured over the vesting terms at each reporting date.
Basic and Diluted Net (Loss) per Common Share
Basic (loss) per common share is computed by dividing the net (loss) by the weighted average number of shares of common stock outstanding for each period. Diluted (loss) per share is computed by dividing the net (loss) by the weighted average number of shares of common stock outstanding plus the dilutive effect of shares issuable through the common stock equivalents. The effect of dilution on net loss becomes anti-dilutive and therefore is not reflected on the consolidated statements of operations and comprehensive loss.
| Potentially Outstanding Dilutive Common Shares | ||||||||
| For the Year Ended December 31, 2017 | For the Year Ended December 31, 2016 | |||||||
| Conversion Feature Shares | ||||||||
| Common shares issuable under the conversion feature of preferred shares | 872,766 | 872,766 | ||||||
| Stock Option | 3,566,299 | 2,484,445 | ||||||
| Warrant | 3,826,658 | 3,826,658 | ||||||
| Total potentially outstanding dilutive common shares | 8,265,723 | 7,183,869 | ||||||
| Potentially Outstanding Dilutive Common Shares | ||||||||
| For the Year Ended December 31, 2023 | For the Year Ended December 31, 2022 | |||||||
| Conversion Feature Shares | ||||||||
| Stock Options | 1,357,466 | 1,357,466 | ||||||
| Warrants | 942,566 | 1,537,158 | ||||||
| Convertible Notes | - | 86,000 | ||||||
| Total potentially outstanding dilutive common shares | 2,300,032 | 2,980,624 | ||||||
| F-9 |
Research and Development
Research and development expenses are charged to operations as incurred.
Foreign Currency Translation
The Company follows Section 830-10-45 of the FASB Accounting Standards CodificationASC 830, Foreign Currency Matters (“Section 830-10-45”ASC 830”) for foreign currency translation to translate the financial statements of the foreign subsidiary from the functional currency, generally the local currency, into U.S. Dollars. SectionASC 830-10-45 sets out the guidance relating to how a reporting entity determines the functional currency of a foreign entity (including of a foreign entity in a highly inflationary economy), re-measures the books of record (if necessary), and characterizes transaction gains and losses. Pursuant to SectionASC 830-10-45, the assets, liabilities, and operations of a foreign entity shall be measured using the functional currency of that entity. An entity’s functional currency is the currency of the primary economic environment in which the entity operates; normally, that is the currency of the environment, or local currency, in which an entity primarily generates and expends cash.
The functional currency of each foreign subsidiary is determined based on management’s judgment and involves consideration of all relevant economic facts and circumstances affecting the subsidiary. Generally, the currency in which the subsidiary transacts a majority of its transactions, including billings, financing, payroll and other expenditures, would be considered the functional currency, but any dependency upon the parent and the nature of the subsidiary’s operations must also be considered. If a subsidiary’s functional currency is deemed to be the local currency, then any gain or loss associated with the translation of that subsidiary’s financial statements is included in accumulated other comprehensive income. However, if the functional currency is deemed to be the U.S. Dollar, then any gain or loss associated with the re-measurement of these financial statements from the local currency to the functional currency would be included in the consolidated statements of incomeoperations and comprehensive income (loss). If the Company disposes of foreign subsidiaries, then any cumulative translation gains or losses would be recorded into the consolidated statements of incomeoperations and comprehensive income (loss). If the Company determines that there has been a change in the functional currency of a subsidiary to the U.S. Dollar, any translation gains or losses arising after the date of change would be included within the statementconsolidated statements of incomeoperations and comprehensive income (loss).loss.
Based on an assessment of the factors discussed above, the management of the Company determined the relevantits subsidiary’s local currency (i.e. the Canadian dollar) to be the functional currency for its foreign subsidiary.
Recent Accounting Pronouncements
In June 2016, the Financial Accounting Standards Board (FASB) issued ASU No. 2016-13, Financial Instruments - Credit Losses (Topic 326) Measurement of Credit Losses on Financial Instruments (ASU 2016-13), which requires an entity to utilize a new impairment model known as the current expected credit loss (CECL) model to estimate its lifetime “expected credit loss” and record an allowance that, when deducted from the amortized cost basis of the financial assets and certain other instruments. ASU 2016-13 requires a cumulative effect adjustment to the balance sheet as of the beginning of the first reporting period in which the guidance is effective. In November 2019, the FASB issued ASU 2019-10, Financial Instruments—Credit Losses (Topic 326), Derivatives and Hedging (Topic 815) and Leases (Topic 842): Effective Dates, which defers the effective date of ASU 2016-13 to fiscal years beginning after December 15, 2022 for all entities except SEC reporting companies that are not smaller reporting companies. ASU 2016-13 became effective for the Company beginning January 1, 2023. The adoption of this ASU did not have a material effect on the Company’s financial statements.
Recent Accounting Pronouncements
In January 2016,August 2020, the FASB issued ASU No. 2016-01, “Financial Instruments - Overall (Subtopic 825-10): Recognition2020-06, which simplifies the guidance on the issuer’s accounting for convertible debt instruments by removing the separation models for convertible debt with a cash conversion feature and Measurementconvertible instruments with a beneficial conversion feature. As a result, entities will not separately present in equity an embedded conversion feature in such debt and will account for a convertible debt instrument wholly as debt, unless certain other conditions are met. The elimination of Financial Assetsthese models will reduce reported interest expense and Financial Liabilities”. The update addresses certain aspectsincrease reported net income for entities that have issued a convertible instrument that is within the scope of recognition, measurement, presentationASU 2020-06. Also, ASU 2020-06 requires the application of the if-converted method for calculating diluted earnings per share and disclosure of financial instruments. For public business entities, the amendments in this update are effectivetreasury stock method will be no longer available. ASU 2020-06 is applicable for fiscal years beginning after December 15, 2017, including interim periods within those fiscal years. Early2023, with early adoption is permitted only for certain portions of the ASU related to financial liabilities. The Company is currently evaluating the impact of the provisions of this new standard on the consolidated financial statements.
In February 2016, the FASB issued ASU No. 2016-02, Leases. The main provisions of ASU No. 2016-02 require management to recognize lease assets and lease liabilities for all leases. ASU 2016-02 retains a distinction between finance leases and operating leases. The classification criteria for distinguishing between finance leases and operating leases are substantially similar to the classification criteria for distinguishing between capital leases and operating leases in the previous leases guidance. The result of retaining a distinction between finance leases and operating leases is that under the lessee accounting model, the effect of leases in the statement of comprehensive income and the statement of cash flows is largely unchanged from previous GAAP. The amendments in this ASU are effective forno earlier than fiscal years beginning after December 15, 2018, including interim periods within those fiscal years.2020. The Company is currently assessingbelieves the impactadoption of this ASU on the Company’s consolidated financial statements.
In August 2016, the FASB issued ASU 2016-15,“Statement of Cash Flows (Topic 230): Classification of Certain Cash Receipts and Cash Payments” (“ASU 2016-15”). ASU 2016-15 will make eight targeted changes to how cash receipts and cash payments are presented and classified in the statement of cash flows. ASU 2016-15 is effective for fiscal years beginning after December 15, 2017. The new standard will require adoption on a retrospective basis unless it is impracticable to apply, in which case it would be required to apply the amendments prospectively as of the earliest date practicable. The Company is currently assessing the impact of this ASU on the Company’s consolidated financial statements.
In October 2016, the FASB issued ASU 2016-16, “Income Taxes (Topic 740): Intra-Entity Transfers of Assets Other than Inventory”, which eliminates the exception that prohibits the recognition of current and deferred income tax effects for intra-entity transfers of assets other than inventory until the asset has been sold to an outside party. The updated guidance is effective for annual periods beginning after December 15, 2019, including interim periods within those fiscal years. Early adoption of the update is permitted. The Company is currently assessing the impact of this ASU on the Company’s consolidated financial statements.
In November 2016, the FASB issued ASU 2016-18, “Statement of Cash Flows (Topic 230)”, requiring that the statement of cash flows explain the change in the total cash, cash equivalents, and amounts generally described as restricted cash or restricted cash equivalents. This guidance is effective for fiscal years, and interim reporting periods therein, beginning after December 15, 2017 with early adoption permitted. The provisions of this guidance are to be applied using a retrospective approach which requires application of the guidance for all periods presented. The Company is currently assessing the impact of this ASU on the Company’s consolidated financial statements.
In December 2016, the FASB issued ASU 2016-20, “Technical Corrections and Improvements to Topic 606, Revenue from Contracts with Customers”. The amendments in this Update affect the guidance in Update 2014-09, which is not yet effective. The effective date and transition requirements for the amendments are the same as the effective date and transition requirements for Topic 606 (and any other Topic amended by Update 2014-09). Accounting Standards Update No. 2015-14,Revenue from Contracts with Customers (Topic 606): Deferral of the Effective Date, defers the effective date of Update 2014-09 by one year.
In September 2017, the Financial Accounting Standards Board (FASB) issued Accounting Standards Update (ASU) No. 2017-13,Revenue Recognition (Topic 605), Revenue from Contracts with Customers (Topic 606), Leases (Topic 840), and Leases (Topic 842).The new standards, among other things, provide additional implementation guidance with respect to Accounting Standards Codification (ASC) Topic 606 and ASC Topic 842. ASU 2017-13 is effective for annual reporting periods beginning after December 15, 2017, including interim reporting periods within that reporting period. The Company is currently evaluating the impact of the new standard, but does not expect it to have a material impact on its implementation strategies or its consolidated financial statements upon adoption.statements.
NOTE 4 - ACCOUNTS PAYABLE AND ACCRUED EXPENSES
Accounts payable and accrued expenses consist of the following at:
SCHEDULE OF ACCOUNTS PAYABLE AND ACCRUED EXPENSES
December 31, 2017 |
December 31, 2016 | December 31, 2023 | December 31, 2022 | |||||||||||||
| Accounting | $ | 36,750 | $ | 36,750 | ||||||||||||
| Research and development | 498,366 | 557,934 | ||||||||||||||
| Legal | $ | - | $ | 1,190 | 6,334 | 25,462 | ||||||||||
| Accounting | 161 | - | ||||||||||||||
| Patent expense | - | 37,142 | ||||||||||||||
| Research and development | 124,728 | 116,255 | ||||||||||||||
| Other | 10,965 | 13,400 | 113,802 | 155,486 | ||||||||||||
| $ | 135,854 | $ | 167,987 | |||||||||||||
| Total | $ | 655,252 | $ | 775,632 | ||||||||||||
NOTE 5 - DERIVATIVE LIABILITIES– NOTE PAYABLE AND CONVERTIBLE NOTE PAYABLE (PIK NOTES)
Upon closingConvertible Notes Payable
During the years ended December 31, 2023 and 2022, the Company amortized $79,409 and $110,797 of the private placement transactionsdebt discount, respectively. At December 31, 2023 and December 31, 2022, the Company had an unamortized debt discount of 0 and $79,409, respectively.
As of December 31, 2023 and December 31, 2022, the Company owes $0 and $230,000 on February 12, 2016,the outstanding Convertible Notes, respectively. These convertible notes that were outstanding during the year had a maturity date of November 6, 2023.
On November 6, 2023, five notes with a total principal of $230,000 and accrued interest of $48,966 was converted into shares of common stock.
Convertible Notes Payable – Related Parties
During the years ended December 1, 2023 and 2022, the Company amortized $6,361 and $7,490 of the debt discount, respectively. At December 31, 2023 and December 31, 2022, the Company had an unamortized debt discount of $0 and $6,361, respectively.
As of December 31, 2023 and December 31, 2022, the Company owes $0 and $200,000 on the outstanding Convertible Notes, respectively. These convertible notes that were outstanding during the year had a maturity date of November 6, 2023.
On November 6, 2023, three notes with a total principal of $200,000 and accrued interest of $41,901 was converted into shares of common stock.
| F-11 |
NOTE 6 - STOCKHOLDERS’ EQUITY
Common Stock
During the year ended December 31, 2023, the Company issued 127,346 and 295,945 warrants, shares of common stock for rounding of shares related to the placement agent ofReverse Split.
During the private offering and to Strategic Bio Partners for debt cancellation, respectively, to purchase the Company’s Series B Preferred Stock with an exercise price of $1.25 and a five-year term. The warrants have a cashless exercise feature that requiresyears ended December 31, 2022, the Company to classifyissued shares of common stock for the warrants as a derivative liability.conversion of notes and interest.
NOTE 6 - STOCKHOLDERS’ EQUITY (DEFICIT)
Stock-Based Compensation
In connection with the consummation of the Merger completed on February 12, 2016, weThe Company adopted the pre-merger Protagenic Therapeutics, Inc.’s 2006an Employee, Director and Consultant Stock Plan (the “2006 Plan”). Onon June 17, 2016 our stockholders adopted our 2016 Equity Compensation Plan (the “2016 Plan”) and, as a result, we terminated the 2006 Plan. We will not grant any further awards under the 2006 Plan. All outstanding grants under the 2006 Plan will continue in effect in accordance with the terms of the particular grant and the 2006 Plan.
. Pursuant to the 2016 Plan, the Company’s Compensation Committee may grant awards to any employee, officer, director, consultant, advisor or other individual service provider of the Company or any subsidiary. On January 1, 2017, pursuantDue to an annual “evergreen” provision contained in the 2016 Plan, the number of shares reserved for future grants was increased by 564,378 shares. and in 2023 and 2022, respectively. As a result of this increase,these increases, as of January 1, 2017,December 31, 2023 and December 31, 2022, the aggregate number of shares of common stock available for awards under the 2016 Plan is 2,712,678.was shares and shares, respectively. Options issued under the 2016 Plan are exercisable for up to 10ten years from the date of issuance.
There were 3,566,299 options outstanding as of December 31, 2017. 2023. During the year ended December 31, 2023, the Company issued options.
There were options outstanding as of December 31, 2022. The fair value of each stock option granted during the year ended December 31, 2022 was estimated using the Black-Scholes assumptions and or factors as follows:
| Exercise price | $ | |||
| Expected dividend yield | 0 | % | ||
| Risk free interest rate | % | |||
| Expected life in years | ||||
| Expected volatility | % |
The following is an analysis of the stock option grant activity under the Plan:
| Weighted Average | Weighted Average | |||||||||||
| Number | Exercise Price | Remaining Life | ||||||||||
| Stock Options | ||||||||||||
| Outstanding January 1, 2016 | 1,707,744 | $ | 0.84 | 6.45 | ||||||||
| Granted | 1,308,300 | $ | 1.25 | 10.35 | ||||||||
| Expired | (506,599 | ) | $ | 0.26 | ||||||||
| Converted | (25,000 | ) | $ | 0.26 | ||||||||
| Outstanding December 31, 2016 | 2,484,445 | $ | 1.18 | 9.82 | ||||||||
| Granted | 1,103,000 | $ | 1.68 | 8.96 | ||||||||
| Expired | (21,146 | ) | $ | 1.00 | ||||||||
| Outstanding December 31, 2017 | 3,566,299 | $ | 1.33 | 8.05 | ||||||||
SCHEDULE OF SHARE-BASED COMPENSATION, STOCK OPTIONS, ACTIVITY
| Number | Weighted Average Exercise Price | Weighted Average Remaining Life | ||||||||||
| Stock Options | ||||||||||||
| Outstanding December 31, 2021 | 1,380,216 | $ | 7.36 | |||||||||
| Granted | 12,500 | 4.84 | ||||||||||
| Expired | (35,250 | ) | 6.09 | - | ||||||||
| Exercised | - | - | - | |||||||||
| Outstanding December 31, 2022 | 1,357,466 | $ | 7.39 | |||||||||
| Granted | - | - | - | |||||||||
| Expired | - | - | - | |||||||||
| Exercised | - | - | - | |||||||||
| Outstanding December 31, 2023 | 1,357,466 | $ | 7.39 | |||||||||
| Nonvested Shares | Shares | Weighted-Average Grant-Date Fair Value | ||||||
| Nonvested at January 1, 2017 | 1,211,463 | $ | 1.25 | |||||
| Granted | 1,103,000 | $ | 1.68 | |||||
| Vested | (800,456 | ) | $ | 1.36 | ||||
| Forfeited | (21,146 | ) | $ | 1.00 | ||||
| Nonvested at December 31, 2017 | 1,492,861 | $ | 1.54 | |||||
SCHEDULE OF SHARE-BASED COMPENSATION NONVESTED SHARES
| Nonvested Options | Options | Weighted-Average Exercise Price | ||||||
| Nonvested at December 31, 2021 | 202,583 | $ | ||||||
| Granted | 12,500 | 4.84 | ||||||
| Vested | (96,896 | ) | 10.44 | |||||
| Forfeited | - | - | ||||||
| Nonvested at December 31, 2022 | 118,187 | $ | 13.07 | |||||
| Granted | - | - | ||||||
| Vested | (68,355 | ) | 11.87 | |||||
| Forfeited | - | - | ||||||
| Nonvested at December 31, 2023 | 49,832 | $ | 14.72 | |||||
As of December 31, 2017,2023, the Company had 3,566,299 shares issuable under options outstanding at a weighted average exercise price of $1.33$ and an intrinsic value of $181,537.$.
| F-12 |
The total number of options granted during the year ended December 31, 20172023 and 20162022 was 1,103,000 and 1,308,300,, respectively. The exercise price for these options was $1.25$ per share or $1.75 per share.
The Company recognized compensation expense related to options issued of $888,281$ and $546,134 during$ for the years ended December 31, 20172023 and 2016,2022, respectively, in which $ and $ is included in general and administrative expenses.expenses and $ and $ in research and development expenses, respectively. For the yearyears ended December 31, 2017, $635,4002023 and 2022, $ and $ of the stock compensation was related to employeeemployees and $281,568$ and $ was related to non-employees.non-employees, respectively.
As of December 31, 2017,2023, the unamortized stock option expense was $1,796,263$ with $1,039,638$ being related to employees and $756,625$ being related to non-employees. As of December 31, 2017,2023, the weighted average period for the unamortized stock compensation to be recognized is 8.19 years.
On October 16, 2017, the Board granted 953,000 options to employees, consultants and Board members. These options shall have 10-year expiration dates, 12 to 48 month vesting cycles, and a strike price of $1.75 per share.
Warrants:
In connection with the Merger, all of the issued and outstanding warrants to purchase shares of Prior Protagenic common stock, converted, on a 1 for 1 basis, into new warrants (the “New Warrants”) to purchase shares of our Series B Preferred Stock.
Simultaneous with the Merger and the Private Offering, New Warrants to purchase 3,403,367 shares of Series B Preferred Stock at an average exercise price of approximately $1.05 per share were issued to holders of Prior Protagenic warrants; additionally, holders of $665,000 of our debt and $35,000 of accrued interest exchanged such debt for five-year warrants to purchase 295,945 shares of Series B Preferred Stock at $1.25 per share. Placement Agent Warrants to purchase 127,346 shares of Series B Preferred Stock at an exercise price of $1.25 per share were issued in connection with the Private offering. These warrants to purchase 423,291 shares of Series B Preferred Stock have been recorded as derivative liabilities. See Note 5.
A summary of warrant issuances are as follows:
| Weighted Average | Weighted Average | |||||||||||
| Number | Exercise Price | Remaining Life | ||||||||||
| Warrants | ||||||||||||
| Outstanding January 1, 2017 | 3,826,658 | $ | 1.05 | 5.61 | ||||||||
| Granted | - | - | - | |||||||||
| Outstanding December 31, 2017 | 3,826,658 | $ | 1.05 | 4.69 | ||||||||
SUMMARY OF WARRANT
| Number | Weighted Average Exercise Price | Weighted Average Remaining Life | ||||||||||
| Warrants | ||||||||||||
| Outstanding December 31, 2021 | 1,533,158 | $ | 13.52 | |||||||||
| Granted | 4,000 | 5.00 | ||||||||||
| Expired | - | - | - | |||||||||
| Exercised | - | - | - | |||||||||
| Outstanding December 31, 2022 | 1,537,158 | $ | 13.49 | |||||||||
| Granted | - | - | - | |||||||||
| Expired | (594,592 | ) | 4.00 | - | ||||||||
| Exercised | - | - | - | |||||||||
| Outstanding December 31, 2023 | 942,566 | $ | 19.47 | |||||||||
As of December 31, 20172023, the Company had 3,826,658 shares issuable under warrants outstanding at a weighted average exercise price of $1.05$ and an intrinsic value of $763,342.$.
During the year ended December 31, 2017, the expiration date for the 100,000 share warrant held by a former consultant was extended by 3 years from January 2, 2017 to January 2, 2020. Related to this warrant modification aThe Company recognized compensation expense related to warrants issued of $99,782 was recorded.
NOTE 7 – INCOME TAXES
The components of loss before income taxes are as follows:
| 2017 | 2016 | |||||||
| Domestic | (2,134,722 | ) | (2,182,114 | ) | ||||
| Foreign | (124,914 | ) | (93,712 | ) | ||||
| Loss before income taxes | (2,259,636 | ) | (2,275,826 | ) | ||||
The Company had no income tax expense due to operating losses incurred for the years ended December 31, 2017$ and 2016.
For the years ended December 31, 2017 and 2016, a reconciliation of the Company’s effective tax rate to the statutory U.S. Federal rate is as follows:
| 2017 | 2016 | |||||||
| Income taxes at Federal statutory rate | (34.0 | )% | (34.0 | )% | ||||
| State income taxes, net of Federal income tax effect | (8.4 | )% | (16.0 | )% | ||||
| Perm difference | 0.0 | % | (7.0 | )% | ||||
| Foreign tax rate differential | (0.2 | )% | (0.2 | )% | ||||
| Change in valuation allowance | 42.6 | )% | 50.4 | % | ||||
| Other | 0.0 | )% | 6.8 | % | ||||
| Income tax provision | 0.0 | )% | 0.0 | % | ||||
In December 2017, the Tax Cuts and Jobs Act (the “Tax Act”) was signed into law. The Tax Act makes changes to U.S. tax law, including a reduction in the corporate tax rate from 35% to 21%. As a result of the enacted law, the Company was required to revalue deferred tax assets and liabilities at the enacted rate. Due to the timing of the enactment and the complexity involved in applying the provisions of the Tax Act, the Company has made reasonable estimates of the effects in its consolidated financial statements as of December 31, 2017. As the Company collects and prepares necessary data and interprets the Tax Act and any additional guidance issued by the U.S. Treasury Department, the IRS, the SEC, and other standard-setting bodies, it may make adjustments to the provisional amounts. The accounting for the tax effects of the Tax Act will be completed in 2018.
The tax effects of temporary differences that give rise to the Company’s deferred tax assets and liabilities are as follows:
| 2017 | 2016 | |||||||
| U.S. net operating loss carryforwards | 2,168,000 | 1,899,000 | ||||||
| Stock compensation | 472,000 | 449,000 | ||||||
| Canadian Provincial income tax losses | 123,000 | 116,000 | ||||||
| Canadian Provincial scientific investment tax credits | - | 56,000 | ||||||
| 2,763,000 | 2,520,000 | |||||||
| Valuation allowance | (2,763,000 | ) | (2,520,000 | ) | ||||
| Net deferred tax assets | - | - | ||||||
As of December 31, 2017 and 2016, the Company had federal net operating loss carryforwards (“NOL”) of approximately $5,287,000 and $4,338,000, respectively. The losses expire beginning in 2024. The Company has not performed a detailed analysis to determine whether an ownership change under IRC Section 382 has occurred. The effect of an ownership change would be the imposition of annual limitation on the use of NOL carryforwards attributable to periods before the change Any limitation may result in expiration of a portion of the NOL before utilization. As of December 31, 2017 and 2016, the Company had state and local net operating loss carryforwards of approximately $5,272,000 and $4,331,000, respectively, to reduce future state tax liabilities also through 2035.
As of December 31, 2017 and 2016, the Company had Canadian NOL of approximately $1,002,000 and $771,000, respectively. The Canadian losses expire in stages beginning in 2026. As of December 31, 2017 and 2016, the Company also has unclaimed Canadian federal scientific research and development investment tax credits, which are available to reduce future federal taxes payable of approximately $0 and $56,000 respectively.
As a result of losses and uncertainty of future profit, the net deferred tax asset has been fully reserved. The net change in the valuation allowance$ during the years ended December 31, 20172023 and 2016 was an increase of $243,000 and $1,148,000,2022, respectively.
Foreign earnings are assumed to be permanently reinvested. U.S. Federal income taxes have not been provided on undistributed earnings of our foreign subsidiary.
The Company recognizes interest and penalties related to uncertain tax positions in selling, general and administrative expenses. The Company has not identified any uncertain tax positions requiring a reserve as of December 31, 2017 and 2016.
The Company is required to file U.S. federal and state income tax returns. These returns are subject to audit by tax authorities beginning with the year ended December 31, 2013.
NOTE 87 - COLLABORATIVE AGREEMENTS
The Company and the University of Toronto a stockholder of the Company (the “University”) entered into an agreement effective December 14, 2004 (the “Research Agreement”) for the performance of a research project titled “Evidence for existence of TCAP receptors in neurons” (the “Project”). The Research Agreement expired on March 31, 2013.
The Company and the University entered into an agreement effective April 1, 2014 (the “New Research Agreement”) for the performance of a research project titled “Teneurin C-terminal Associated Peptide (“TCAP”) mediated stress attenuation in vertebrates: Establishing the role of organismal and intracellular energy and glucose regulation and metabolism” (the “New Project”). The New Project is to perform research related to work done by Dr. David A. Lovejoy, a professor at the University and stockholder of the Company, (the “Professor”) in regard to TCAP mediated stress attenuation in vertebrates: Establishing the role of organismal and intracellular energy and glucose regulation and metabolism. In addition to the New Research Agreement, the ProfessorDr. Lovejoy entered into an agreement with the University in order to commercialize certain technologies. The New Research Agreement expired on March 30, 2016. In February 2017, the New Research Agreement was extended to December 31, 2016 which allows2017. The extension allowed for further development of the technologies and use of their applications. Upon expirationOn April 10, 2018, the agreement was amended and the research agreement has been further extended to December 31, 2023. As of the dated of this filing, this agreement payments to the University and research support from the University will suspend until an agreement can be made.has not been extended.
Prior to January 1, 2016, the University has been granted 25,000 stock options which are fully vested at the exercise price of $1.00$ exercisable over a 10 period which endsended on April 1, 2022. As of December 31, 2016,2023, Dr. David Lovejoy of the ProfessorUniversity has been granted 483,299 stock options, of which 297,190 are fully vested atand have expired. These have an exercise price of $1.00$4.00, $5.00 or $7.00 and are exercisable over a period ranging from 10 orto 13 year periods which end either on March 30, 2021, December 1, 2022, April 15, 2026 or on March 1, 2027. On October 16, 2017, the profession was granted 20,000 stock options which vest monthly for 48 months, has an exercise price of $1.75 and expires on October 16, 2027years.
The sponsorship research and development expenses pertaining to the Research Agreements were $93,919$0 and $65,252$28,645 for the years ended December 31, 20172023 and 2016,2022, respectively.
NOTE 10 - LICENSING AGREEMENTS
On July 31, 2005, the Company had entered into a Technology License Agreement (“License Agreement”) with the University pursuant to which the University agreed to license to the Company patent rights and other intellectual property, among other things (the “Technologies”). The Technology License Agreement was amended on February 18, 2015 and currently does not provide for an expiration date.
Pursuant to the License Agreement and its amendment, the Company obtained an exclusive worldwide license to make, have made, use, sell and import products based upon the Technologies, or to sublicense the Technologies in accordance with the terms of the License Agreement and amendment. In consideration, the Company agreed to pay to the University a royalty payment of 2.5% of net sales of any product based on the Technologies. If the Company elects to sublicense any rights under the License Agreement and amendment, the Company agrees to pay to the University 10% of any up-front sub-license fees for any sub-licenses that occurred on or after September 9, 2006, and, on behalf of the sub-licensee, 2.5% of net sales by the sub-licensee of all products based on the Technologies. The Company had no sales revenue for the years ended December 31, 2017 and 2016 and therefore was not subject to paying any royalties.
In the event the Company fails to provide the University with semi-annual reports on the progress or fails to continue to make reasonable commercial efforts towards obtaining regulatory approval for products based on the Technologies, the University may convert our exclusive license into a non-exclusive arrangement. Interest on any amounts owed under the License Agreement and amendment will be at 3% per annum. All intellectual property rights resulting from the Technologies or improvements thereon will remain the property of the other inventors and/or the Professor, and/or the University, as the case may be. The Company has agreed to pay all out-of- pocket filing, prosecution and maintenance expenses in connection with any patents relating to the Technologies. In the case of infringement upon any patents relating to the Technologies, the Company may elect, at its own expense, to bring a cause of action asserting such infringement. In such a case, after deducting any legal expenses the Company may incur, any settlement proceeds will be subject to the 2.5% royalty payment owed to the University under the License Agreement and amendment.
The patent applications were made in the name of the Professor and other inventors, but the Company’s exclusive, worldwide rights to such patent applications are included in the License Agreement and its amendment with the University. The Company maintains exclusive licensing agreements and it currently controls the six intellectual patent properties.
| F-13 |
NOTE 118 - COMMITMENTS AND CONTINGENCIES
ConsultingLicensing Agreements
On July 31, 2005, the Company had entered into a Technology License Agreement (“License Agreement”) with the University pursuant to which the University agreed to license to the Company patent rights and other intellectual property, among other things (the “Technologies”). The Technology License Agreement was amended on February 18, 2015 and currently does not provide for an expiration date.
Pursuant to the License Agreement and its amendment, the Company obtained an exclusive worldwide license to make, have made, use, sell and import products based upon the Technologies, or to sublicense the Technologies in accordance with the terms of the License Agreement and amendment. In consideration, the Company agreed to pay to the University a royalty payment of 2.5% of net sales of any product based on the Technologies. If the Company elects to sublicense any rights under the License Agreement and amendment, the Company agrees to pay to the University 10% of any up-front sub-license fees for any sub-licenses that occurred on or after September 9, 2006, and, on behalf of the sub-licensee, 2.5% of net sales by the sub-licensee of all products based on the Technologies. The Company had an employment agreement with a former officer (the “Former Officer”) which expired onno sales revenue for the years ended December 31, 2015. The employment agreement indicated2023 and 2022 and therefore was not subject to paying any royalties.
In the event the Company fails to provide the University with semi-annual reports on the progress or fails to continue to make reasonable commercial efforts towards obtaining regulatory approval for products based on the Technologies, the University may convert our exclusive license into a salary of $6,489non-exclusive arrangement. Interest on any amounts owed under the License Agreement and amendment will be at 3% per month plus a bonus, including healthcare benefits. The Former Officer was also granted 75,000 stock options, valued at $64,223 usingannum. All intellectual property rights resulting from the Black-Scholes calculation of which $53,519 was expensed in 2015.
UponTechnologies or improvements thereon will remain the expirationproperty of the employment agreement,other inventors and/or Dr. Lovejoy, and/or the University, as the case may be. The Company has agreed to pay all out-of-pocket filing, prosecution and Former Officer entered into a consulting agreementmaintenance expenses in its place, which provides thatconnection with any patents relating to the Technologies. In the case of infringement upon any patents relating to the Technologies, the Company may retainelect, at its own expense, to bring a cause of action asserting such infringement. In such a case, after deducting any legal expenses the Former Officer as a consultant on an as-needed basis. As a consultant, the Former Officer is responsible for Canadian financial reporting, data compilation, and document retrieval services, reportingCompany may incur, any settlement proceeds will be subject to the Chief Financial Officer, and to endeavor to secure Canadian non-dilutive grant funding for the Company. The Former Officer has been granted 250,000 stock options in total, 25,000 of which expired unexercised. The remaining 225,000 are fully vested, at exercise prices of $1.00 and $1.25, with certain options expiring on March 30, 2021, March 1, 2024 and March 9, 2025. Either party may terminate the agreement either (a) immediately at any time upon written notice2.5% royalty payment owed to the other partyUniversity under the License Agreement and amendment.
The patent applications were made in the eventname of a breach ofDr. Lovejoy and other inventors, but the agreement byCompany’s exclusive, worldwide rights to such patent applications are included in the other party which cannot be cured (i.e. breach ofLicense Agreement and its amendment with the confidentiality obligations) or (b) at any time without cause upon not less than fifteen (15) days’ prior written notice to the other party. Upon expiration or termination, neither the Company nor Former Officer will have any further obligations under the consulting agreement.
University. The Company has accrued $0 tomaintains exclusive licensing agreements and it currently controls the Former Officer for research and development projects and paid the equivalent in U.S. dollars of $13,168 during the year ended December 31, 2017.five intellectual patent properties.
Consulting Agreement
PTI Canada entered into a consulting agreement with a stockholder of the Company (the “Consultant”) which, which as amended, expires on December 31, 2017. Pursuant to the consulting agreement, the Consultant is responsible for overseeing i) design and development of enzyme-linked immunosorbent assay (“ELISA”), assays for measuring TCAP, ii) evaluation of TCAP exposure biomarker assay, iii) development of pipeline peptides, and iv) development of clinically compatible formulations for TCAP, as well as all of the bench research and development of formulation and extraction methods. The Consultant has been granted 150,000 stock options, which are fully vested at exercise prices of $1.00 and $1.25, exercisable over 10 year periods which end either on March 30, 2021 or March 1, 2025. The Consultant is paid the Canadian equivalent of approximately US$2,370 per month. Either party may terminate the consulting agreement either (a) immediately at any time upon written notice to the other party in the event of a breach of the agreement by the other party which cannot be cured (i.e. breach of the confidentiality obligations) or (b) at any time without cause upon not less than fifteen (15) days’ prior written notice to the other party. Upon expiration or termination, neither the Company nor Consultant will have any further obligations under the consulting agreement.
The Company has accrued $0 to pay the Consultant for research and development projects during the year ended December 31, 2017 and paid $25,449 during the year ended December 31, 2017.
Legal Proceedings
From time to time we may be named in claims arising in the ordinary course of business. Currently, no legal proceedings, government actions, administrative actions, investigations or claims are pending against us or involve us that, in the opinion of our management, could reasonably be expected to have a material adverse effect on our business and financial condition.
NOTE 9 – RELATED PARTY TRANSACTIONS
The Company is provided free office space consisting of a conference room by the Company Executive Chairman, Dr. Armen. The Company does not pay any rent for the use of this space. This space is used for quarterly board meetings and our annual shareholder meeting.
During the year ended December 31, 2021, the Company engaged Agenus Inc., a related party, to perform research and development services. Agenus Inc. is a related party due to the Company’s Director and Chairman of the Board being the CEO and Chairman of the Board for Agenus Inc. The Company incurred $149,509 and $105,928 in expenses related to these services during the years ended December 31, 2023 and 2022, respectively. As of December 31, 2023 and December 31, 2022, the outstanding balance owed to Agenus Inc. is $150,296 and $105,928, respectively.
During the year ended December 31, 2022, the Company engaged CTC North, GmbH (“CTC”) to perform research and development services. CTC is a related party due to the Company’s Director and Chairman of the Board being the CEO and Chairman of the Board for Agenus Inc, CTC’s parent company. The total commitment for this agreement is $1.3 million. The Company incurred $106,754 and $105,801 in expenses related to these services during the years ended December 31, 2023 and 2022, respectively. As of December 31, 2023 and December 31, 2022, there is $65,199 and $0 owed to CTC in connection with this agreement, respectively.
| F-14 |
NOTE 10 – INCOME TAXES
The components of loss before income taxes are as follows:
SCHEDULE OF LOSS BEFORE INCOME TAX
| 2023 | 2022 | |||||||
| Domestic | (4,998,066 | ) | (3,522,834 | ) | ||||
| Foreign | (2,431 | ) | (32,671 | ) | ||||
| Loss before income taxes | (5,000,497 | ) | (3,555,505 | ) | ||||
The Company had no income tax expense due to operating losses incurred for the years ended December 31, 2023 and 2022.
For the years ended December 31, 2023 and 2022, a reconciliation of the Company’s effective tax rate to the statutory U.S. Federal rate is as follows:
SCHEDULE OF EFFECTIVE INCOME TAX RATE RECONCILIATION
| 2023 | 2022 | |||||||
| Income taxes at Federal statutory rate | (21.0 | )% | (21.0 | )% | ||||
| State income taxes, net of Federal income tax effect | (8.9 | )% | (8.8 | )% | ||||
| Perm difference | 0.0 | % | 0.0 | % | ||||
| Foreign tax rate differential | (0.1 | )% | (0.2 | )% | ||||
| Change in valuation allowance | 30.0 | % | 30.0 | % | ||||
| Other | 0.0 | % | 0.0 | % | ||||
| Income tax provision | 0.0 | % | 0.0 | % | ||||
The tax effects of temporary differences that give rise to the Company’s deferred tax assets and liabilities are as follows:
SCHEDULE OF DEFERRED TAX ASSETS AND LIABILITIES
| 2023 | 2022 | |||||||
| U.S. net operating loss carryforwards | 4,940,000 | 3,620,000 | ||||||
| Stock compensation | 2,131,000 | 1,931,000 | ||||||
| Canadian Provincial income tax losses | - | 7,000 | ||||||
| Deferred tax assets, gross | 7,071,000 | 5,558,000 | ||||||
| Valuation allowance | (7,071,000 | ) | (5,558,000 | ) | ||||
| Net deferred tax assets | - | - | ||||||
As of December 31, 2023 the Company had federal net operating loss carryforwards (“NOL”) of approximately $15.2 million. The 2017 Tax Cuts and Jobs Act (“TCJA”) will generally allow losses incurred after 2017 to be carried over indefinitely, but will generally limit the net operating loss deduction to the lesser of the net operating loss carryover or 80% of a corporation’s taxable income (subject to Section 382 of the Internal Revenue Code of 1986, as amended). Also, there will be no carryback for losses incurred after 2017. Losses incurred prior to 2018 will generally be deductible to the extent of the lesser of a corporation’s net operating loss carryover or 100% of a corporation’s taxable income and be available for twenty years from the period the loss was generated. The federal net operating losses generated prior to 2018 of $0.1 million will expire at various dates through 2037. As of December 31, 2023 and 2022, the Company had state and local net operating loss carryforwards of approximately $13,975,000 and $9,568,000, respectively, to reduce future state tax liabilities also through 2035.
| F-15 |
As of December 31, 2023 and 2022, the Company had Canadian NOL of approximately $1,415,000 and $1,413,000 respectively. The Canadian losses expire in stages beginning in 2026. As of December 31, 2023 and 2022, the Company also has unclaimed Canadian federal scientific research and development investment tax credits, which are available to reduce future federal taxes payable of approximately $0 and $0 respectively.
As a result of losses and uncertainty of future profit, the net deferred tax asset has been fully reserved. The net change in the valuation allowance during the years ended December 31, 2023 and 2022 was an increase of $1,513,000 and $137,000, respectively.
Foreign earnings are assumed to be permanently reinvested. U.S. Federal income taxes have not been provided on undistributed earnings of our foreign subsidiary.
The Company recognizes interest and penalties related to uncertain tax positions in selling, general and administrative expenses. The Company has not identified any uncertain tax positions requiring a reserve as of December 31, 2023 and 2022.
The Company is required to file U.S. federal and state income tax returns. These returns are subject to audit by tax authorities beginning with the year ended December 31, 2018.
NOTE 12 - 11 – SUBSEQUENT EVENTS
On January 24, 2018,8, 2024, the company entered into a consulting agreement (the “Agreement”) with NeuroAssets Sàrl (“Consultant”), a Swiss company. Under the Agreement, Consultant will provide us with advisory services relating to introductions and presentations to pharmaceutical companies who could potentially become our corporate partners. The Agreement may be terminated by either party at any time upon notice. The Company plans to pay Consultant $5,000 per month until such time as the Agreement is terminated.
The Agreement also provided for the grant ofissued options to Consultant. Accordingly, on February 20, 2018, the Compensation Committee ofpurchase the Company’s Board of Directors approved a grant of 200,000common stock to employees and consultants. These options under our 2016 Equity Compensation Plan. The options vest over 48 months in equal monthly installments with the first monthly vesting event scheduled to occur on March 20, 2018, have a term of 10 years and are exercisable at a price of $1.75 per share. The vesting of the options will accelerate if a corporate partnership results from an introduction made by Consultant.
During the first quarter the company granted 80,000 stock options to four consultants. 50,000 of these options vest immediately and the remaining 30,000 options vest monthly over 48 months, have an exercise price of $1.75,$ and have a term of expire in years from issuance. These options vest over months.
On March 25, 2024, the Company issued options to purchase the Company’s common stock to employees and consultants. These options have an exercise price of $ and expire in years from issuance. These options vest between and months with options to vest upon achievement of certain performance conditions.
On February 12, 2024, the Company entered into a consulting agreement. As part of this agreement the Company agrees to pay $5,000 per month and issue options to purchase the Company’s common stock. These options have an exercise price of $ and expire in years from issuance. These options vest over .
| F-16 |