UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
_____________________
Form 10-Q
_____________________
|
|
|
| (Mark One) |
|
☑
| þQUARTERLY REPORT PURSUANT TO SECTION 13 OR 15(d) |
|
| OF THE SECURITIES EXCHANGE ACT OF 1934 |
|
For the Quarterly Period Ended SeptemberJune 30, 20172021
or
|
|
|
☐
| ¨TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) |
|
| OF THE SECURITIES EXCHANGE ACT OF 1934
For the Transition Period from ___________ to ___________ |
|
Commission File Number: 000-29959
_______________
Pain Therapeutics,Cassava Sciences, Inc.
(Exact name of registrant as specified in its charter)
|
|
|
|
Delaware
| Delaware | 91-1911336 |
|
(State or other jurisdiction of
| (Stateorotherjurisdictionof | (I.R.S.Employer |
|
incorporation or organization)
| incorporationororganization) | IdentificationNumber) |
|
7801 N. Capital of Texas Highway, Suite 260, Austin, TX 78731
(512) 501-2444
(Address, including zip code, of registrant'sregistrant’s principal executive offices and
telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
0 |
|
|
|
|
Title of each class |
| Trading Symbol(s) |
| Name of each exchange on which registered |
Common Stock, $0.001 par value |
| SAVA |
| NASDAQ Capital Market |
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes☑þ No☐¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes☑þ No☐¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, smalla smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act. (Check one):
|
|
| Large accelerated filer ☐Accelerated Filer ¨ | Accelerated filer ☐Filer ¨ |
| Non-accelerated filer ☐Filer þ | Smaller reporting company ☑Reporting Company þ |
|
| Emerging growth company ☐Growth Company ¨ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act .☐Act.¨
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes☐¨ No☑þ
Indicate the number of shares outstanding of each of the issuer's issuer’s classes of common stock, as of the latest practicable date.
|
|
|
|
|
|
|
|
| Common Stock, $0.001 par value | 6,595,50940,014,695
|
|
|
| Shares Outstanding as of October 27, 2017August 2, 2021 |
|
PAIN THERAPEUTICS,CASSAVA SCIENCES, INC.
TABLE OF CONTENTS
PART I. FINANCIAL INFORMATION
Item 1. Financial Statements
| | | | |
|
|
|
|
|
|
| | | | |
|
|
|
|
|
|
PAIN THERAPEUTICS, INC. | |
CASSAVA SCIENCES, INC. | | CASSAVA SCIENCES, INC. |
| | | | |
|
|
|
|
|
|
BALANCE SHEETS | |
(in thousands, except share and per share data) | |
CONDENSED BALANCE SHEETS | | CONDENSED BALANCE SHEETS |
(Unaudited, in thousands, except share and par value data) | | (Unaudited, in thousands, except share and par value data) |
| | | | |
|
| September 30, | December 31, |
|
|
| 2017 | 2016 | June 30, 2021 |
|
| December 31, 2020 |
|
| | | | |
|
|
|
|
|
|
ASSETS | ASSETS | ASSETS |
Current assets: | | | | |
|
|
|
|
|
|
Cash and cash equivalents | $ | 11,916 | $ | 16,615 | $ | 278,254 |
| $ | 93,506 |
|
Marketable securities | | — | | 2,099 | |
Other current assets | | 288 | | 356 | |
Prepaid expenses and other current assets | |
| 1,304 |
|
| 488 |
|
Total current assets | | 12,204 | | 19,070 |
| 279,558 |
|
| 93,994 |
|
Operating lease right-of-use assets | |
| 252 |
|
| 295 |
|
Property and equipment, net | | 173 | | 232 |
| 75 |
|
| 11 |
|
Other assets | |
| 1,420 |
|
| — |
|
Total assets | $ | 12,377 | $ | 19,302 | $ | 281,305 |
| $ | 94,300 |
|
| | | | |
|
|
|
|
|
|
LIABILITIES AND STOCKHOLDERS' EQUITY | LIABILITIES AND STOCKHOLDERS' EQUITY | LIABILITIES AND STOCKHOLDERS' EQUITY |
Current liabilities: | | | | |
|
|
|
|
|
|
Accounts payable | $ | 712 | $ | 303 | $ | 1,912 |
| $ | 911 |
|
Accrued development expense | | — | | 27 |
| 2,462 |
|
| 719 |
|
Accrued compensation and benefits | | 298 | | 335 |
| 120 |
|
| 83 |
|
Operating lease liabilities, current | |
| 93 |
|
| 58 |
|
Other current liabilities | |
| 50 |
|
| 94 |
|
Total current liabilities | | 1,010 | | 665 |
| 4,637 |
|
| 1,865 |
|
Noncurrent liabilities | | — | | — | |
Operating lease liabilities, non-current | |
| 188 |
|
| 235 |
|
Total liabilities | | 1,010 | | 665 |
| 4,825 |
|
| 2,100 |
|
Commitments and contingencies | | | | | |
Commitments and contingencies (Notes 6 and 8) | |
| |
|
| |
|
Stockholders' equity: | | | | |
|
|
|
|
|
|
Preferred stock | | — | | — | |
Common stock | | 7 | | 7 | |
Preferred stock, $0.001 par value; 10,000,000 shares authorized, NaN issued and outstanding | |
| — |
|
| — |
|
Common stock, $0.001 par value; 120,000,000 shares authorized; 40,011,894 and 35,237,987 shares issued and outstanding at June 30, 2021 and December 31, 2020, respectively | |
| 40 |
|
| 35 |
|
Additional paid-in capital | | 166,342 | | 164,118 |
| 460,012 |
|
| 267,086 |
|
Accumulated other comprehensive income | | — | | — | |
Accumulated deficit | | (154,982) | | (145,488) |
| (183,572) |
|
| (174,921) |
|
Total stockholders' equity | | 11,367 | | 18,637 |
| 276,480 |
|
| 92,200 |
|
Total liabilities and stockholders' equity | $ | 12,377 | $ | 19,302 | $ | 281,305 |
| $ | 94,300 |
|
| | | | |
|
|
|
|
|
|
See accompanying notes to condensed financial statements.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CASSAVA SCIENCES, INC. |
|
|
|
|
|
|
|
|
|
|
|
|
|
CONDENSED STATEMENTS OF OPERATIONS |
(Unaudited, in thousands, except per share data) |
|
|
| Three months ended |
| Six months ended |
|
| June 30, |
| June 30, |
|
| 2021 |
| 2020 |
| 2021 |
| 2020 |
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
Research and development, net of grant reimbursement |
| $ | 3,901 |
| $ | 591 |
| $ | 6,430 |
| $ | 1,135 |
General and administrative |
|
| 1,237 |
|
| 818 |
|
| 2,241 |
|
| 1,596 |
Gain on sale of property and equipment |
|
| — |
|
| (246) |
|
| — |
|
| (346) |
Total operating expenses |
|
| 5,138 |
|
| 1,163 |
|
| 8,671 |
|
| 2,385 |
Operating loss |
|
| (5,138) |
|
| (1,163) |
|
| (8,671) |
|
| (2,385) |
Interest income |
|
| 13 |
|
| 27 |
|
| 20 |
|
| 99 |
Net loss |
| $ | (5,125) |
| $ | (1,136) |
| $ | (8,651) |
| $ | (2,286) |
Net loss per share, basic and diluted |
| $ | (0.13) |
| $ | (0.05) |
| $ | (0.22) |
| $ | (0.09) |
Shares used in computing net loss per share, basic and diluted |
|
| 39,953 |
|
| 24,779 |
|
| 38,843 |
|
| 24,630 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| | | | | | | | | | | |
| | | | | | | | | | | |
PAIN THERAPEUTICS, INC. |
| | | | | | | | | | | |
STATEMENTS OF OPERATIONS |
(in thousands, except per share data) |
| | | | | | | | | | | |
| Three months ended | | Nine months ended |
| September 30, | | September 30, |
| 2017 | | 2016 | | 2017 | | 2016 |
Operating expenses: | | | | | | | | | | | |
Research and development | $ | 1,619 | | $ | 2,657 | | $ | 6,071 | | $ | 7,841 |
General and administrative | | 977 | | | 884 | | | 3,455 | | | 4,573 |
Total operating expenses | | 2,596 | | | 3,541 | | | 9,526 | | | 12,414 |
Operating loss | | (2,596) | | | (3,541) | | | (9,526) | | | (12,414) |
Interest income | | 6 | | | 23 | | | 33 | | | 86 |
Net loss | $ | (2,590) | | $ | (3,518) | | $ | (9,493) | | $ | (12,328) |
Net loss per share, basic and diluted | $ | (0.40) | | $ | (0.54) | | $ | (1.45) | | $ | (1.89) |
Shares used in computing net loss per share, basic and diluted | | 6,538 | | | 6,535 | | | 6,537 | | | 6,515 |
| | | | | | | | | | | |
See accompanying notes to condensed financial statements.
|
|
|
|
|
|
|
|
|
|
|
|
CASSAVA SCIENCES, INC. |
|
|
|
|
|
|
CONDENSED STATEMENTS OF CASH FLOWS |
(Unaudited, in thousands) |
|
|
|
|
|
|
|
|
| Six months ended June 30, |
| 2021 |
| 2020 |
Cash flows from operating activities: |
|
|
|
|
|
Net loss | $ | (8,651) |
| $ | (2,286) |
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
|
Stock-based compensation |
| 665 |
|
| 522 |
Depreciation and amortization |
| 9 |
|
| 20 |
Gain on sale of property and equipment |
| — |
|
| (346) |
Changes in operating assets and liabilities: |
|
|
|
|
|
Prepaid and other assets |
| (2,236) |
|
| 82 |
Operating lease right-of-use assets and liabilities |
| 31 |
|
| — |
Accounts payable |
| 1,001 |
|
| (37) |
Accrued development expense |
| 1,743 |
|
| (22) |
Accrued compensation and benefits |
| 37 |
|
| 26 |
Other current liabilities |
| (44) |
|
| 5 |
Net cash used in operating activities |
| (7,445) |
|
| (2,036) |
Cash flows from investing activities: |
|
|
|
|
|
Purchase of property and equipment |
| (73) |
|
| — |
Proceeds from sale of property and equipment |
| — |
|
| 360 |
Net cash (used in) provided by investing activities |
| (73) |
|
| 360 |
Cash flows from financing activities: |
|
|
|
|
|
Proceeds from exercise of stock options |
| 1,749 |
|
| — |
Proceeds from exercise of common stock warrants |
| 692 |
|
| 3,849 |
Proceeds from registered direct offering, net of issuance costs |
| 189,825 |
|
| — |
Net cash provided by financing activities |
| 192,266 |
|
| 3,849 |
Net increase in cash and cash equivalents |
| 184,748 |
|
| 2,173 |
Cash and cash equivalents at beginning of period |
| 93,506 |
|
| 23,081 |
Cash and cash equivalents at end of period | $ | 278,254 |
| $ | 25,254 |
|
|
|
|
|
|
| | | | | | | | | | | | |
| | | | | | | | | | | | |
PAIN THERAPEUTICS, INC. |
| | | | | | | | | | | | |
STATEMENTS OF COMPREHENSIVE INCOME (LOSS) |
(in thousands) |
| | | | | | | | | | | | |
| | Three months ended | | Nine months ended |
| | September 30, | | September 30, |
| | 2017 | | 2016 | | 2017 | | 2016 |
Net loss | | $ | (2,590) | | $ | (3,518) | | $ | (9,493) | | $ | (12,328) |
Other comprehensive income (loss): | | | | | | | | | | | | |
Net unrealized gains (losses) on marketable securities | | | — | | | 1 | | | — | | | 1 |
Comprehensive loss | | $ | (2,590) | | $ | (3,517) | | $ | (9,493) | | $ | (12,327) |
| | | | | | | | | | | | |
See accompanying notes to condensed financial statements.
Cassava Sciences, Inc.
| | | | | |
| | | | | |
PAIN THERAPEUTICS, INC. |
| | | | | |
STATEMENTS OF CASH FLOWS |
(in thousands) |
| | | | | |
| Nine months ended |
| September 30, |
| 2017 | | 2016 |
Cash flows from operating activities: | | | | | |
Net loss | $ | (9,493) | | $ | (12,328) |
Adjustments to reconcile net loss to net cash used in operating activities: | | | | | |
Non-cash stock-based compensation | | 2,223 | | | 3,489 |
Depreciation and amortization | | 51 | | | 41 |
Non-cash net interest income | | (2) | | | (3) |
Changes in operating assets and liabilities: | | | | | |
Other current assets | | 68 | | | (18) |
Accounts payable | | 417 | | | 49 |
Accrued development expense | | (27) | | | (150) |
Accrued compensation and benefits | | (37) | | | (283) |
Net cash used in operating activities | | (6,800) | | | (9,203) |
Cash flows from investing activities: | | | | | |
Purchases of property and equipment | | — | | | (76) |
Purchases of marketable securities | | (399) | | | (2,046) |
Sales of marketable securities | | 400 | | | — |
Maturities of marketable securities | | 2,100 | | | 1,500 |
Net cash provided (used in) investing activities | | 2,101 | | | (622) |
Cash flows from financing activities: | | | | | |
Cash used for statutory taxes for net exercise of Performance Awards | | — | | | (214) |
Deferred financing costs | | — | | | (46) |
Net cash used in financing activities | | — | | | (260) |
Net decrease in cash and cash equivalents | | (4,699) | | | (10,085) |
Cash and cash equivalents at beginning of period | | 16,615 | | | 31,299 |
Cash and cash equivalents at end of period | $ | 11,916 | | $ | 21,214 |
| | | | | |
See accompanying notes to condensed financial statements.
PAIN THERAPEUTICS, INC.
Notes to Condensed Financial Statements
(Unaudited)
Note 1. General and Liquidity
Pain Therapeutics,Cassava Sciences, Inc. (the “Company”) discovers and develops proprietary drugspharmaceutical product candidates that may offer significant improvements to patients and healthcare professionals. WeThe Company generally focus our drugfocuses its discovery and product development efforts on disorders of the nervous system.
In the course of our development activities, we have sustained cumulative operating losses. There are no assurances that additional financing will be available on favorable terms, or at all.
We have prepared theThe accompanying unaudited condensed financial statements of Pain Therapeutics, Inc.the Company have been prepared in accordance with accounting principles generally accepted accounting principlesin the United States (“GAAP”) for interim financial information and pursuant to the instructions to the Quarterly Report on Form 10-Q and Article 10 of Regulation S-X. Accordingly, the condensed financial statements do not include all of the information and footnotes required by generally accepted accounting principlesGAAP for complete financial statements. In ourthe opinion of management of the Company, all adjustments, consisting of normal recurring adjustments, considered necessary for a fair presentation have been included. Operating results for the three and ninesix months ended SeptemberJune 30, 20172021 are not necessarily indicative of the results that may be expected for any other interim period or for the year 20172021. For further information, refer to the consolidated financial statements and footnotes thereto included in our annual reportthe Company’s Annual Report on Form 10-K for the year ended December 31, 2016. 2020.
On November 16, 2016, we receivedCoronavirus Disease 2019 (COVID-19)
The widespread outbreak of a letter fromnovel infectious disease called Coronavirus Disease 2019, or COVID-19, has not significantly impacted the Listing Qualifications staffCompany’s operations or financial condition as of Nasdaq (the “Staff”) notifying us that, forAugust 4, 2021. However, this pandemic has created a dynamic and uncertain situation in the previousnational economy. The Company continues to closely monitor the latest information to make timely, informed business decisions and public disclosures regarding the potential impact of pandemic on its operations and financial condition. The scope of pandemic is unprecedented and its long-term impact on the Company’s operations and financial condition cannot be reasonably estimated at this time.
Liquidity
The Company has incurred significant net losses and negative cash flows since inception, and as a result has an accumulated deficit of $183.6 million at June 30, consecutive business days,2021. The Company expects its cash requirements to be significant in the bid price for our common stock had closed below the minimum $1.00 per share requirement (the “Minimum Price Requirement”) under NASDAQ’s Listing Rule 5450(a)(1) for continued listing onfuture. The Nasdaq Global Market. In accordance with Nasdaq Listing Rule 5810(c)(3)(A), if during the 180 calendar days following the date of the notification, or prior to May 15, 2017, the closing bid price of our common stock is at or above $1.00 for a minimum of 10 consecutive business days, the Staff will provide us with written confirmation of compliance. On May 24, 2017, we received a letter from the Staff indicating that we had regained compliance with the $1.00 minimum closing bid requirement following completion of the reverse stock split described below.
On May 4, 2017, following stockholder approval, our board of directors approved a reverse stock split ratio of 7-for-1. On May 4, 2017, we filed with the Secretary of State of the State of Delaware a Certificate of Amendmentamount and timing of the Company’s Amendedfuture cash requirements will depend on regulatory and Restated Certificatemarket acceptance of Incorporationits product candidates and the resources it devotes to effectresearching and developing, formulating, manufacturing, commercializing and supporting its products. The Company may seek additional funding through public or private financing in the 7-for-1 reverse stock split of our outstanding shares of common stock. The number of outstanding shares of common stockfuture, if such funding is available and on terms acceptable to the date ofCompany. There are no assurances that additional financing will be available on favorable terms, or at all. However, management believes that the reverse split was reduced from 46.1 millioncurrent working capital position will be sufficient to 6.6 million shares. Our common stock began trading onmeet the NASDAQ Global Market on a split-adjusted basis whenCompany’s working capital needs for at least the market opened for trading on May 10, 2017. As a result, all common stock share amounts included in these condensed consolidated financial statements have been retroactively reduced by a factor of seven, and all common stock per share amounts have been increased by a factor of seven, with the exception of our common stock par value.next 12 months.
We have evaluated subsequent events through the date of filing this Form 10-Q.
Note 2. Significant Accounting Policies
Use of Estimates
We makeThe Company makes estimates and assumptions in preparing ourits condensed financial statements in conformity with accounting principles generally accepted in the United States of America.GAAP. These estimates and assumptions affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the condensed financial statements and the reported amount of revenue earned and expenses incurred during the reporting period. The Company evaluates its estimates on an ongoing basis, including those estimates related to manufacturing agreements and research collaborations. Actual results could differ from these estimates and assumptions.
Cash and Cash Equivalents Marketable Securities and Concentration of Credit Risk
We investThe Company invests in cash equivalents and marketable securities. We consider highly-liquidcash equivalents. The Company considers highly liquid financial instruments with original maturities of three months or less to be cash equivalents. Our marketable securitiesHighly liquid investments that are considered
cash equivalents include interest-bearing financial instruments, generally consistingmoney market accounts and funds, certificates of corporate or governmentdeposits, and U.S. Treasury securities.
Our investment policy allows for investments in marketable securities with active secondary or resale markets, establishes diversification The Company maintains its cash and credit quality requirements and limits investments by maturity and issuer. We maintain our investmentscash equivalents at one financial institution.
A changeFair Value Measurements
The Company recognizes financial instruments in prevailing interest rates may causeaccordance with the authoritative guidance on fair value measurements and disclosures for financial assets and liabilities. This guidance defines fair value, establishes a framework for measuring fair value in accordance with GAAP, and expands disclosures about fair value measurements. The guidance also establishes a three-tier fair value hierarchy, which prioritizes the inputs used in measuring fair value. These tiers include:
Level 1 includes quoted prices in active markets.
Level 2 includes significant observable inputs, such as quoted prices for identical or similar securities, or other inputs that are observable and can be corroborated by observable market data for similar securities. The Company uses market pricing and other observable market inputs obtained from third-party providers. It uses the bid price to establish fair value where a bid price is available. The Company does not have any financial instruments where the fair value of the investment to fluctuate. We don’t recognize an impairment charge related to this type of fluctuation because the fluctuation is temporary and eliminatedbased on Level 2 inputs.
Level 3 includes unobservable inputs that are supported by the time an investment matures. We would recognize an impairment charge if and when we determine that a decline inlittle or no market activity. The Company does not have any financial instruments where the fair value belowis based on Level 3 inputs.
If a financial instrument uses inputs that fall in different levels of the amortized costhierarchy, the instrument will be categorized based upon the lowest level of an investmentinput that is other-than-temporary. We consider various factors in determining whethersignificant to recognize an impairment charge, including any adverse changes in the investees’ financial condition, how long the fair value has been below the amortized costcalculation. The fair value of cash and whether it is more likely than not that we would elect to or be required to sell the marketable security before its anticipated recovery.
We may elect to sell marketable securities before they mature. We hold these investments as “available for sale” and include these investments in our Balance Sheets as current assets, even though the contractual maturity of a particular investment may be beyond one year.
Fair Value Measurements
We report our cash equivalents and marketable securities at fair value aswas based on Level 1 Level 2 or Level 3 using the following inputs:inputs at June 30, 2021 and December 31, 2020.
| ·
| | Level 1 includes quoted prices in active markets. We base the fair value of money market funds and U.S. treasury securities on Level 1 inputs.
|
| ·
| | Level 2 includes significant observable inputs, such as quoted prices for identical or similar investments, or other inputs that are observable and can be corroborated by observable market data for similar securities. We use market pricing and other observable market inputs obtained from third-party providers. We use the bid price to establish fair value where a bid price is available. We base the fair value of our marketable securities on Level 2 inputs.
|
| ·
| | Level 3 includes unobservable inputs that are supported by little or no market activity. We do not have any investments where the fair value is based on Level 3 inputs.
|
We include unrealized gains or losses on our investments as Accumulated other comprehensive loss in the Stockholders’ equity section of our Balance Sheets. We include changes in net unrealized gains or losses in our Statements of Comprehensive Income. We would recognize significant realized gains and losses on a specific identification basis as other income in our Statements of Operations.
Proceeds from Grants
During the ninethree months ended SeptemberJune 30, 20172021 and 2016, we2020, the Company received reimbursements totaling $0.9 million and $1.3$1.1 million respectively, pursuant to a grant from the National Institutes of Health or(“NIH”) research grants, respectively. During the six months ended June 30, 2021 and 2020, the Company received reimbursements totaling $1.5 million and $2.4 million pursuant to NIH that we recordedresearch grants, respectively. The Company records the proceeds from these grants as a reductionreductions to ourits research and development expenses.
Non-cash Stock-based Compensation
We recognizeThe Company recognizes non-cash expense for the fair value of all stock options and other share-based awards. We useThe Company uses the Black-Scholes option valuation model (“Black-Scholes”) to calculate the fair value of stock options, using the single-option award approach and straight-line attribution method. For all options granted, to employees and directors, we recognizeit recognizes the resulting fair value as expense on a straight-line basis over the vesting period of each respective stock option, generally four years. For options granted to non-employees, we remeasure the fair value expense using Black-Scholes each reporting period.
We haveThe Company has granted share-based awards that vest upon achievement of certain performance criteria or (“Performance Awards. We multiplyAwards”). The Company multiplies the number of Performance Awards by the fair market value of ourits common stock on the date of grant to calculate the fair value of each award. We estimateIt estimates an implicit service period for achieving performance criteria for each award. We recognizeThe Company recognizes the resulting fair value as expense over the implicit service period when we concludeit concludes that achieving the performance criteria is probable. WeIt periodically reviewreviews and updateupdates as appropriate ourits estimates of implicit service periods and conclusions on achieving the performance criteria. Performance Awards vest and common stock is issued upon achievement of the performance criteria.
Net Loss per Share
We computeThe Company computes basic net loss per share on the basis of the weighted-average number of common shares outstanding for the reporting period. We compute dilutedDiluted net loss per share is computed on the basis of the weighted-average number of common shares outstanding plus potential dilutive common shares outstanding using the treasury-stock method. Potential dilutive common shares consist of outstanding common stock options.options and warrants. There is no difference between the Company’s net loss and comprehensive loss.
We includeThe Company included the following in the calculation of basic and diluted net loss per share (in thousands, except per share data):
| | | | | | | | | | | | | |
|
|
|
|
|
|
|
|
|
|
|
|
| | | | | | | | | | | | | |
|
|
|
|
|
|
|
|
|
|
|
|
| | Three months ended | | Nine months ended | |
| Three months ended |
| Six months ended |
| | September 30, | | September 30, | |
| June 30, |
| June 30, |
| | 2017 | | 2016 | | 2017 | | 2016 | |
| 2021 |
| 2020 |
| 2021 |
| 2020 |
Numerator: | | | | | | | | | | | | | |
|
|
|
|
|
|
|
|
|
|
|
|
Net loss | | $ | (2,590) | | $ | (3,518) | | $ | (9,493) | | $ | (12,328) | |
| $ | (5,125) |
| $ | (1,136) |
| $ | (8,651) |
| $ | (2,286) |
Denominator: | | | | | | | | | | | | | |
|
|
|
|
|
|
|
|
|
|
|
|
Shares used in computing net loss per share, basic and diluted | | | 6,538 | | | 6,535 | | | 6,537 | | | 6,515 | |
|
| 39,953 |
|
| 24,779 |
|
| 38,843 |
|
| 24,630 |
Net loss per share, basic and diluted | | $ | (0.40) | | $ | (0.54) | | $ | (1.45) | | $ | (1.89) | |
| $ | (0.13) |
| $ | (0.05) |
| $ | (0.22) |
| $ | (0.09) |
Dilutive common shares excluded from net loss per share, diluted | | | 2,436 | | | 2,432 | | | 2,414 | | | 2,565 | | |
| | | | | | | | | | | | | |
|
|
|
|
|
|
|
|
|
|
|
|
Dilutive common stock options excluded from net loss per share, diluted | |
|
| 2,129 |
|
| 2,294 |
|
| 2,163 |
|
| 2,177 |
Common stock warrants excluded from net loss per share, diluted | |
|
| — |
|
| 1,427 |
|
| — |
|
| 1,427 |
| |
|
|
|
|
|
|
|
|
|
|
|
|
WeThe Company excluded common stock options and warrants outstanding from the calculation of net loss per share, diluted, because the effect of including options and warrants outstanding would have been anti-dilutive.
Income TaxesFair Value of Financial Instruments
Financial instruments include accounts payable and accrued liabilities. The estimated fair value of certain financial instruments may be determined using available market information or other appropriate valuation methodologies. However, considerable judgment is required in interpreting market data to develop estimates of fair value; therefore, the estimates are not necessarily indicative of the amounts that could be realized or would be paid in a current market exchange. The effect of using different market assumptions and/or estimation methodologies may be material to the estimated fair value amounts. The carrying amounts of accounts payable and accrued liabilities are at cost, which approximates fair value due to the short maturity of those instruments.
We make
Research Contract Costs and Accruals
The Company has entered into various research and development contracts with research institutions and other third-party vendors. These agreements are generally cancelable, and related payments are recorded as research and development expenses as incurred. The Company records accruals for estimated ongoing research costs. When evaluating the adequacy of the accrued liabilities, the Company analyzes progress of the studies including the phase or completion of events, invoices received and contracted costs. Significant judgments and estimates and judgmentsare made in determining the needaccrued balances at the end of any reporting period. Actual results could differ from the Company’s estimates. The Company’s historical accrual estimates have not been materially different from actual costs.
Incentive Bonus Plan
In 2020, the Company established the 2020 Cash Incentive Bonus Plan (the “Plan”) to incentivize Plan participants. Awards under the Plan are accounted for as liability awards under Accounting Standards Codification (ASC) 718 “Stock-based Compensation”. The fair value of each potential Plan award will be determined once a provisiongrant date occurs and will be remeasured each reporting period. Compensation expense associated with the Plan will be recognized over the expected achievement period for each Plan award, when a Performance Condition is considered probable of being met. See Note 8 for further discussion of the Plan.
Leases
The Company recognizes assets and liabilities that arise from leases. For operating leases, the Company is required to recognize a right-of-use asset and a lease liability, initially measured at the present value of the lease payments during the lease term, in the condensed balance sheets. The Company elected the short-term lease recognition exemption for all leases that qualify. This means, for those leases that qualify, the Company does not recognize right-of-use assets or lease liabilities. As the Company`s leases do not provide an implicit rate, it uses its incremental borrowing rate based on the information available at the commencement date in determining the present value of lease payments. Lease expense for lease payments is recognized on a straight-line basis over the lease term.
Income Taxes
The Company accounts for income taxes includingunder the estimationasset and liability method. Deferred tax assets and liabilities are recognized for the estimated future tax consequences attributable to differences between the financial statement carrying amounts of our taxable income or loss for each full fiscal year.
We haveexisting assets and liabilities and their respective tax bases. Deferred tax balances are adjusted to reflect tax rates based on currently enacted tax laws, which will be in effect in the years in which the temporary differences are expected to reverse. The Company has accumulated significant deferred tax assets that reflect the tax effects of net operating loss and tax credit carryovers and temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Realization of certain deferred tax assets is dependent upon future earnings. We areThe Company is uncertain about the timing and amount of any future earnings. Accordingly, we offsetthe Company offsets these deferred tax assets with a valuation allowance.
We mayThe Company accounts for uncertain tax positions in accordance with ASC 740, “Income Taxes”, which clarifies the accounting for uncertainty in tax positions. These provisions require recognition of the impact of a tax position in the future determineCompany’s condensed financial statements only if that certain deferredposition is more likely than not of being sustained upon examination by taxing authorities, based on the technical merits of the position. Any interest and penalties related to uncertain tax assetspositions will likely be realized, in which case we will reduce our valuation allowance in the period in which such determination is made. If the valuation allowance is reduced, we may recognizereflected as a benefit fromcomponent of income taxes in our Statement of Operations in that period.tax expense.
We classify interest recognized pursuant to our deferred tax assets as interest expense, when appropriate.
Note 3. Cash, Cash EquivalentsPrepaid and Marketable SecuritiesOther Assets
Prepaid and Assets Measuredother assets at Fair Value
Our cash, cash equivalents and marketable securities at SeptemberJune 30, 20172021 and December 31, 2016 were as follows2020 consisted of the following (in thousands):
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| Cash, Cash Equivalents and Marketable Securities |
| Amortized Cost | | Unrealized Gains | | Unrealized Losses | | Estimated Fair Value | | Accrued Interest | | Total Value |
September 30, 2017 | | | | | | | | | | | | | | | | | |
Cash | $ | 451 | | $ | — | | $ | — | | $ | 451 | | $ | — | | $ | 451 |
Cash equivalents | | 11,465 | | | — | | | — | | | 11,465 | | | — | | | 11,465 |
Commercial paper | | — | | | — | | | — | | | — | | | — | | | — |
| $ | 11,916 | | $ | — | | $ | — | | $ | 11,916 | | $ | — | | $ | 11,916 |
Reported as: | | | | | | | | | | | | | | | | | |
Cash and cash equivalents | $ | 11,916 | | | — | | | — | | $ | 11,916 | | $ | — | | $ | 11,916 |
Marketable securities | | — | | | — | | | — | | | — | | | — | | | — |
| $ | 11,916 | | $ | — | | $ | — | | $ | 11,916 | | $ | — | | $ | 11,916 |
Maturities: | | | | | | | | | | | | | | | | | |
Matures in one year or less | $ | 11,916 | | $ | — | | $ | — | | $ | 11,916 | | $ | — | | $ | 11,916 |
Matures one to three years | | — | | | — | | | — | | | — | | �� | — | | | — |
| $ | 11,916 | | $ | — | | $ | — | | $ | 11,916 | | $ | — | | $ | 11,916 |
| | | | | | | | | | | | | | | | | |
December 31, 2016 | | | | | | | | | | | | | | | | | |
Cash | $ | 1,434 | | $ | — | | $ | — | | $ | 1,434 | | $ | — | | $ | 1,434 |
Cash equivalents | | 12,783 | | | — | | | — | | | 12,783 | | | — | | | 12,783 |
Commercial paper | | 4,497 | | | — | | | — | | | 4,497 | | | — | | | 4,497 |
| $ | 18,714 | | $ | — | | $ | — | | $ | 18,714 | | $ | — | | $ | 18,714 |
Reported as: | | | | | | | | | | | | | | | | | |
Cash and cash equivalents | $ | 16,615 | | | — | | | — | | $ | 16,615 | | $ | — | | $ | 16,615 |
Marketable securities | | 2,099 | | | — | | | — | | | 2,099 | | | — | | | 2,099 |
| $ | 18,714 | | $ | — | | $ | — | | $ | 18,714 | | $ | — | | $ | 18,714 |
Maturities: | | | | | | | | | | | | | | | | | |
Matures in one year or less | $ | 18,714 | | $ | — | | $ | — | | $ | 18,714 | | $ | — | | $ | 18,714 |
Matures one to three years | | — | | | — | | | — | | | — | | | — | | | — |
| $ | 18,714 | | $ | — | | $ | — | | $ | 18,714 | | $ | — | | $ | 18,714 |
| | | | | | | | | | | | | | | | | |
|
|
|
|
|
|
| June 30, 2021 |
| December 31, 2020 |
Prepaid insurance | $ | — |
| $ | 457 |
Contract research organization deposit |
| 1,166 |
|
| — |
Other |
| 138 |
|
| 31 |
Total prepaid expenses and other current assets | $ | 1,304 |
| $ | 488 |
|
|
|
|
|
|
Contract research organization deposit | $ | 1,420 |
| $ | — |
Total other assets | $ | 1,420 |
| $ | — |
|
|
|
|
|
|
We did not realize any material gains or losses on our investments in marketable securities during the nine months ended September 30, 2017 and 2016. To date we have not recorded any impairment charges on marketable securities related to other-than-temporary declines in market value.
Our assets measured at fair value on a recurring basis are as follows (in thousands):
| | | | | | | | | | | |
| | | | | | | | | | | |
| Level 1 | | Level 2 | | Level 3 | | Total |
September 30, 2017 | | | | | | | | | | | |
Cash and cash equivalents | $ | 11,916 | | $ | — | | $ | — | | $ | 11,916 |
Commercial paper | | — | | | — | | | ��— | | | — |
| $ | 11,916 | | $ | — | | $ | — | | $ | 11,916 |
December 31, 2016 | | | | | | | | | | | |
Cash and cash equivalents | $ | 14,217 | | $ | — | | $ | — | | $ | 14,217 |
Commercial paper | | — | | | 4,497 | | | — | | | 4,497 |
| $ | 14,217 | | $ | 4,497 | | $ | — | | $ | 18,714 |
| | | | | | | | | | | |
The transfers between Level 1 and Level 2 during the nine months ended September 30, 2017 was primarily due to the sale of the marketable securities.
Note 4. Stockholders’ Equity and Stock-Based Compensation Expense
Stockholders’ equity activity in 2017Equity Activity during the Six Months Ended June 30, 2021 and 2020
During the ninesix months ended SeptemberJune 30, 2017, our2021 and 2020, the Company’s common stock outstanding and stockholders’ equity
(in thousands) changed as follows:
| | | | |
| Common Stock | | Stockholders' equity
(in thousands) |
Balance at December 31, 2016 | 6,591,705 | | $ | 18,637 |
Non-cash stock-related compensation for: | | | | |
Stock options for employees | — | | | 2,212 |
Stock options for non-employees | — | | | 11 |
Issuance of common stock pursuant to 7 for 1 Reverse Stock Split | 3,804 | | | — |
Net loss | — | | | (9,493) |
Balance at September 30, 2017 | 6,595,509 | | $ | 11,367 |
| | | | |
|
|
|
|
|
| Common Stock |
| Stockholders' equity
(in thousands) |
Balance at December 31, 2019 | 21,841,810 |
| $ | 22,099 |
Stock-based compensation for: |
|
|
|
|
Stock options for employees | — |
|
| 261 |
Stock options for non-employees | — |
|
| 9 |
Proceeds from exercise of common stock warrants | 2,888,092 |
|
| 3,613 |
Net loss | — |
|
| (1,150) |
Balance at March 31, 2020 | 24,729,902 |
| $ | 24,832 |
|
|
|
|
|
Stock-based compensation for: |
|
|
|
|
Stock options for employees | — |
|
| 249 |
Stock options for non-employees | — |
|
| 3 |
Proceeds from exercise of common stock warrants | 189,431 |
|
| 236 |
Net loss | — |
|
| (1,136) |
Balance at June 30, 2020 | 24,919,333 |
| $ | 24,184 |
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2020 | 35,237,987 |
| $ | 92,200 |
Stock-based compensation for: |
|
|
|
|
Stock options for employees | — |
|
| 249 |
Stock options for non-employees | — |
|
| 1 |
Proceeds from exercise of common stock warrants | 554,019 |
|
| 692 |
Exercise of stock options | 135,015 |
|
| 1,746 |
Proceeds from registered direct offering of common stock | 4,081,633 |
|
| 189,825 |
Net loss | — |
|
| (3,526) |
Balance at March 31, 2021 | 40,008,654 |
| $ | 281,187 |
|
|
|
|
|
Stock-based compensation for: |
|
|
|
|
Stock options for employees | — |
|
| 410 |
Stock options for non-employees | — |
|
| 5 |
Exercise of stock options | 3,240 |
|
| 3 |
Net loss | — |
|
| (5,125) |
Balance at June 30, 2021 | 40,011,894 |
| $ | 276,480 |
|
|
|
|
|
2021 Registered Direct Offering
On February 12, 2021, the Company completed a common stock offering pursuant to which certain investors purchased 4,081,633 shares of common stock at a price of $49.00 per share. Net proceeds of the offering were approximately $189.8 million after deducting offering expenses.
At-the-Market Common Stock optionOffering
In March 2020, the Company established an at-the-market offering program (“ATM”) to sell, from time to time, shares of Company common stock having an aggregate offering price of up to $100 million in transactions pursuant to a shelf registration statement that was declared effective by the U.S. Securities and Exchange Commission (the “SEC”) on May 5, 2020. The Company is obligated to pay a commission of 3.0% of the gross proceeds from the sale of shares of common stock in the offering. The Company is not obligated to sell any shares in the offering.
There were 0 common stock sales under the ATM during the three and six months ended June 30, 2021 and 2020.
Common Stock Warrants
In August 2018, the Company issued warrants to purchase up to an aggregate of 9.1 million shares of its common stock in conjunction with an offering of its common stock.
The Company did 0t receive any proceeds from exercise of common stock warrants during the three months ended June 30, 2021. During the three months ended June 30, 2020, the Company received proceeds of $0.2 million from the exercise of 0.2 million shares pursuant to warrants.
During the six months ended June 30, 2021, the Company received proceeds of $0.7 million from the exercise of 0.6 million shares pursuant to warrants. During the six months ended June 30, 2020, the Company received proceeds of $3.8 million from the exercise of 3.1 million shares pursuant to warrants.
There were 0 remaining common stock warrants outstanding as of June 30, 2021.
Stock Option and Performance Award activityActivity in 20172021
During the ninesix months ended SeptemberJune 30, 2017,2021, stock options and unvested Performance Awards outstanding under our 2008 Equity Incentive Planthe Company’s stock option plans changed as follows:
| | | | | |
| | | | | |
| | Stock Options | | | Performance Awards |
Outstanding as of December 31, 2016 | | 2,435,249 | | | 222,060 |
Granted | | 653,210 | | | — |
Vested Performance Awards | | — | | | — |
Forfeited/expired | | (417,059) | | | (14,723) |
Outstanding as of September 30, 2017 | | 2,671,400 | | | 207,337 |
| | | | | |
|
| Stock Options |
|
| Performance Awards |
Outstanding as of December 31, 2020 |
| 2,817,504 |
|
| 138,055 |
Options granted |
| 85,000 |
|
| — |
Options exercised |
| (234,994) |
|
| — |
Options forfeited/canceled |
| (9,338) |
|
| — |
Outstanding as of June 30, 2021 |
| 2,658,172 |
|
| 138,055 |
|
|
|
|
|
|
The weighted average exercise price of options outstanding at SeptemberJune 30, 20172021 was $18.92.$11.40. As outstanding options vest over the current remaining vesting period of 3.22.2years, we expectthe Company expects to recognize non-cashstock-based compensation expense of $5.2$6.7 million. If and when outstanding Performance Awards vest, we wouldthe Company will recognize non-cashstock-based compensation expense of $4.4$2.3 million over the implicit service period.
During the three months ended June 30, 2021, there were 71,596 stock options exercised. Of the stock options exercised, 68,356 stock options were net settled in satisfaction of the exercise price, with 0 cash proceeds received. Cash proceeds to the Company totaled $3,000 during the three months ended June 30, 2021.
During the six months ended June 30, 2021, there were 234,994 stock options exercised. Of the stock options exercised, 98,739 stock options were net settled in satisfaction of the exercise price, with 0 cash proceeds received. Cash proceeds to the Company totaled $1,749,000 during the six months ended June 30, 2021.
There were 0 stock options exercised during the three and six months ended June 30, 2020.
Subsequent to June 30, 2021, there were 2,801 stock options exercised for cash proceeds of $57,000.
Stock-based Compensation Expense in 20172021
During the three and ninesix months ended SeptemberJune 30, 20172021 and 2016, our non-cash stock-related2020, the Company’s stock-based compensation expenses wereexpense was as follows (in thousands):
| | | | | | | | | | | | |
| | | | | | | | | | | | |
| | Three months ended | | Nine months ended |
| | September 30, | | September 30, |
| | 2017 | | 2016 | | 2017 | | 2016 |
Research and development | | | | | | | | | | | | |
Vesting of stock options | | $ | 286 | | $ | 318 | | $ | 887 | | $ | 997 |
Vesting of Performance Awards | | | — | | | — | | | — | | | 430 |
| | | 286 | | | 318 | | | 887 | | | 1,427 |
General and administrative | | | | | | | | | | | | |
Vesting of stock options | | | 402 | | | 527 | | | 1,336 | | | 1,658�� |
Vesting of Performance Awards | | | — | | | — | | | — | | | 404 |
| | | 402 | | | 527 | | | 1,336 | | | 2,062 |
Total non-cash stock-based compensation expenses | | | | | | | | | | | | |
Vesting of stock options | | | 688 | | | 845 | | | 2,223 | | | 2,655 |
Vesting of Performance Awards | | | — | | | — | | | — | | | 834 |
| | $ | 688 | | $ | 845 | | $ | 2,223 | | $ | 3,489 |
| | | | | | | | | | | | |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Three months ended |
| Six months ended |
|
|
| June 30, |
| June 30, |
|
|
| 2021 |
| 2020 |
| 2021 |
| 2020 |
|
Research and development |
| $ | 295 |
| $ | 111 |
| $ | 415 |
| $ | 226 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
General and administrative |
|
| 120 |
|
| 141 |
|
| 250 |
|
| 296 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total stock-based compensation expense |
| $ | 415 |
| $ | 252 |
| $ | 665 |
| $ | 522 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Capital on Demand Sales Agreement2018 Equity Incentive Plan
In December 2015, we entered intoJanuary 2018, the Company’s Board of Directors (the “Board”) approved the Company’s 2018 Omnibus Incentive Plan (the “2018 Plan”). The Board or a Capital on Demand™ Sales Agreementdesignated committee of the Board is responsible for administration of the 2018 Plan and determines the terms and conditions of each option granted, consistent with JonesTrading Institutional Services, or the ATM Agreement, relatingterms of the 2018 Plan. The Company’s employees, directors, and consultants are eligible to receive awards under the offering2018 Plan, including grants of stock options and Performance Awards. Share-based awards generally expire 10 years from the date of grant. The 2018 Plan provides for issuance of up to 10.0 million1,000,000 shares of our Common Stockcommon stock, par value $0.001 per share, subject to adjustment as provided in “at the market” offerings. We did not issue any2018 Plan.
When stock options or Performance Awards are exercised net of the exercise price and taxes, the number of shares underof stock issued is reduced by the ATM Agreement duringnumber of shares equal to the nine monthsamount of 2017 or 2016. In March 2017,taxes owed by the ATM Agreement expiredaward recipient and was not renewed.that number of shares are cancelled. The Company then uses its cash to pay tax authorities the amount of statutory taxes owed by and on behalf of the award recipient.
Note 5. Income Taxes
WeThe Company did not provide for income taxes during the three and ninesix months ended SeptemberJune 30, 20172021, because we haveit has projected a net loss for the full year 2017.2021 for which any benefit will be offset by an increase in the valuation allowance. There was also 0 provision for income taxes for the three and six months ended June 30, 2020.
Note 6. Commitments
We conduct ourRight-of-use Asset and Liability
The Company has a non-cancelable operating lease for approximately 6,000 square feet of office space in Austin, Texas that expires on April 30, 2024. The Company also has a short-term lease agreement for an additional 3,600 square feet of office space in Austin, Texas that expires on April 30, 2022. Future lease payments as of June 30, 2021 are as follows (in thousands):
|
|
|
|
|
|
|
|
|
|
Future lease payments |
| 2021 | 2022 | 2023 | 2024 | Total future lease payments | Less: imputed interest |
| Total |
Operating leases | $ | 50 | 102 | 107 | 36 | 295 | (14) | $ | 281 |
Short-term operating lease | $ | 32 | 21 | — | — | 53 | — | $ | 53 |
Rent expense for the three months ended June 30, 2021 and 2020 totaled $34,000 and $25,000, respectively.
Rent expense for the six months ended June 30, 2021 and 2020 totaled $57,000 and $50,000, respectively.
Cash paid for operating lease liabilities during the three months ended June 30, 2021 and 2020 totaled $27,000 and $25,000, respectively. Cash paid for operating lease liabilities during the six months ended June 30, 2021 and 2020 totaled $27,000 and $50,000, respectively.
Other Commitments
The Company conducts its product research and development programs through a combination of internal and collaborative programs that include, among others, arrangements with universities, contract research organizations and clinical research sites. We haveThe Company has contractual arrangements with these organizations that are cancelable. OurThe Company’s obligations under these contracts are largely based on services performed. The Company also had non-cancellable commitments for the manufacture of simufilam totaling approximately $1.9 million at June 30, 2021.
We have a non-cancelable operating lease for approximately 6,000 square feet of office space in Austin, Texas that expires in December 2017. Minimum lease payments are as follows (in thousands):
| | | | | |
| | | | | |
| 2017 | | Total |
Minimum lease payments | $ | 148 | | $ | 148 |
| | | | | |
Note 7. Sale of Property and Equipment
There were no sales of property and equipment during the three and six months ended June 30, 2021.
During the three months ended June 30, 2020, the Company sold surplus manufacturing equipment to a third party and received proceeds totaling $260,000. During the six months ended June 30, 2020, the Company sold surplus manufacturing equipment to a third party and received proceeds totaling $360,000. The original cost of the property and equipment was $892,000 and accumulated depreciation was $878,000, resulting a gain on sale of property and equipment of $246,000 and $346,000, respectively, during the three and six months ended June 30, 2020.
Note 8. 2020 Cash Incentive Bonus Plan
On August 26, 2020, the Board approved the Plan. The Plan was established to promote the long-term success of the Company by creating an “at-risk” cash bonus program that rewards Plan participants with additional cash compensation in lockstep with significant increases in the Company’s market capitalization. The Plan is considered “at-risk” because Plan participants will not receive a cash bonus unless the Company’s market capitalization increases significantly and certain other conditions specified in the Plan are met. Specifically, Plan participants will not be paid any cash bonuses unless (1) the Company completes a merger or acquisition transaction that constitutes a sale of ownership of the Company or its assets (a Merger Transaction) or (2) the Compensation Committee of the Board (the Compensation Committee) determines the Company has sufficient cash on hand, as defined in the Plan. Because of the inherent discretion and uncertainty regarding these requirements, the Company has concluded that a Plan grant date has not occurred as of June 30, 2021.
Plan participants will be paid all earned cash bonuses in the event of a Merger Transaction.
The Company’s market capitalization for purposes of the Plan is determined based on either (1) the Company’s closing price of one share on the Nasdaq Capital Market multiplied by the total issued and outstanding shares and options to purchase shares of the Company, or (2) the aggregate consideration payable to security holders of the Company in a Merger Transaction. This constitutes a market condition under applicable accounting guidance.
The Plan triggers a potential cash bonus each time the Company’s market capitalization increases significantly, up to a maximum $5 billion in market capitalization. The Plan specifies 14 incremental amounts between $200 million and $5 billion (each increment, a “Valuation Milestone”). Each Valuation Milestone triggers a potential cash bonus award in a pre-set amount defined in the Plan. Each Valuation Milestone must be achieved and maintained for no less than 20 consecutive trading days for Plan participants to be eligible for a potential cash bonus award. Approximately 58% of each cash bonus award associated with a Valuation Milestone is subject to adjustment and approval by the Compensation Committee. Any amounts not awarded by the Compensation Committee are no longer available for distribution.
If the Company were to exceed a $5 billion market capitalization for no less than 20 consecutive trading days, all Valuation Milestones would be deemed achieved, in which case cash bonus awards would range from a minimum of $139.1 million up to a hypothetical maximum of $322.3 million. Payment of cash bonuses is deferred until such time as (1) the Company completes a Merger Transaction, or (2) the Compensation Committee determines the Company has sufficient cash on hand to render payment (each, a “Performance Condition”), neither of which may ever occur. Accordingly, there can be no assurance that Plan participants will ever be paid a cash bonus that is awarded under the Plan, even if the Company’s market capitalization increases significantly.
The Plan is accounted for as a liability award. The fair value of each Valuation Milestone award will be determined once a grant date occurs and will be remeasured each reporting period. Compensation expense associated with the Plan will be recognized over the expected achievement period for each of the 14 Valuation Milestones, when a Performance Condition is considered probable of being met.
On October 13, 2020, the Company achieved the first Valuation Milestone. Subsequently, the Compensation Committee approved a potential cash bonus award of $7.3 million in total for all Plan participants, subject to future satisfaction of a Performance Condition.
During the six months ended June 30, 2021, the Company achieved 10 Valuation Milestones triggering potential Company obligations to all Plan participants from a minimum of $81.0 million up to a hypothetical maximum of $195.0 million, to be determined by the Compensation Committee. However, 0 compensation expense has been recorded since no grant date has occurred and no Performance Conditions are considered probable of being met. There is no continuing service requirement for Plan participants once the Compensation Committee approves a cash bonus award.
Subsequent to June 30, 2021, the Company achieved one additional Valuation Milestone triggering potential Company obligations to all Plan participants from a minimum of $12.7 million up to a hypothetical maximum of $30.0 million, to be determined by the Compensation Committee and contingent upon future satisfaction of a Performance Condition.
NaN actual cash payments were authorized or made to participants under the Plan through June 30, 2021.
Note 9. Recently Issued Accounting Pronouncements
We reviewed recentlyIn December 2019, the FASB issued Accounting Standards Update (“ASU”) No. 2019-12, Income Taxes (Topic 740) Simplifying Accounting for Income Taxes, as part of its initiative to reduce complexity in the accounting pronouncementsstandards. The guidance amended certain disclosure requirements that had become redundant, outdated or superseded. Additionally, this guidance amends accounting for the interim period effects of changes in tax laws or rates, and have adopted or plan to adopt those that are applicable to us. We do not expectsimplifies aspects of the accounting for franchise taxes. The guidance is effective for annual periods beginning after December 15, 2020, including interim periods therein. The adoption of these pronouncements toASU 2019-12 in the first quarter of 2021 did not have a material impact on ourthe Company’s condensed financial position, resultsstatements.
Note 10. Subsequent Event
On August 4, 2021, the Company completed the purchase of a two-building office complex in Austin, Texas, which will serve as its new corporate headquarters. This property is intended to accommodate the Company’s anticipated significant growth and expansion of its operations orin the coming years. Company management expects to be hands-off with regards to property management. Maintenance, physical facilities, leasing, property management and other key responsibilities around property ownership will all be assumed by professional real-estate managers under long-term contract with the Company. The property purchase price was $21.9 million, exclusive of closing costs, funded with cash flows. on hand. The office complex measures approximately 90,000 rentable square feet. The property is currently 59% leased, before the effect of the Company occupying approximately 25% of the property in the near future. The seller is an independent third party not affiliated with the Company.
Item 2. Management’s Discussion and Analysis of Financial Condition and Results of Operations
This discussion and analysis should be read in conjunction with ourCassava Sciences, Inc.’s (the “Company,”, “we,” “us,” or “our”) condensed financial statements and accompanying notes included elsewhere in this quarterly report.Quarterly Report on Form 10-Q. Operating results are not necessarily indicative of results that may occur in future periods.
This quarterly reportQuarterly Report on Form 10-Q contains certain statements that are considered forward-looking statements within the meaning of the Private Securities Reform Act of 1995. We intend that such statements be protected by the safe harbor created thereby. Forward-looking statements relate to expectations, beliefs, projections, future plans and strategies, anticipated events or trends and similar expressions concerning matters that are not historical facts. In some cases, you can identify forward-looking statements by terms such as “anticipate,” “believe,” “could,” “estimate,” “expect,” “intend,” “may,” “plan,” “potential,” “should,” “will” and “would” or the negatives of these terms or other comparable terminology.
The forward-looking statements are based on our beliefs, assumptions and expectations of our future performance, taking into account all information currently available to us. Forward-looking statements involve risks and uncertainties and our actual results and the timing of events may differ significantly from the results discussed in the forward-looking statements. Examples of such forward-looking statements include, but are not limited to statements about:
our intention to initiate a pivotal Phase 3 clinical program with simufilam in Alzheimer’s disease, the anticipated scope of Phase 3 studies and our estimated timeline for doing so; our reliance on third-party contractors to make drug supply on a large-scale for our Phase 3 clinical program, or their ability to do so on-time or on-budget; limitations around the interpretation of cognitive results from a long-term open-label study design, as compared to efficacy results from a fully completed, randomized controlled study design; the expected rate of cognitive decline over time in untreated Alzheimer’s patients; the ability of the Alzheimer's Disease Assessment Scale-Cognitive Subscale (ADAS-cog), Alzheimer’s Disease Cooperative Study – Activities of Daily Living (ADCS-ADL), Neuropsychiatric Inventory (NPI), CANTAB or other clinical scales to assess cognition or health in our trials of Alzheimer’s disease; announcements or plans regarding any future interim analyses of our open-label study of simufilam and our estimated timeline for doing so; any significant changes we have made, or anticipate making, to the design of an on-going open-label study of simufilam; announcements regarding an End-of-Phase 2 meeting with the U.S. Food and Drug Administration (FDA); our ability to initiate, conduct or analyze additional clinical and non-clinical studies with our product candidates targeted at Alzheimer’s disease and other neurodegenerative diseases; the interpretation of results from our Phase 2 clinical studies; our estimated timeline for publishing in a peer-reviewed technical journal clinical results of our Phase 2b study of simufilam; our plans to further develop SavaDx, our investigational blood-based diagnostic, and our estimated timeline for doing so; the safety, efficacy, or potential therapeutic benefits of our product candidates; | ·
| | actions and studies that are needed to potentially support obtaining approval of the New Drug Application, or NDA for REMOXY (oxycodone capsules CII) with label claims on routes of abuse, by the U.S. Food and Drug Administration, or FDA;
|
| ·
| | our plans to rely on third parties, including Durect Corporation, or Durect, Noramco, Inc., or Noramco, and Mallinckrodt, Inc., or Mallinckrodt, to supply us with excipients and active pharmaceutical ingredients and to manufacture REMOXY;
|
| ·
| | discussions with potential strategic partners for the development and commercialization of REMOXY;
|
| ·
| | expectations with regard to the data, materials and information provided to us by Pfizer Inc., or Pfizer, related to Pfizer’s development activities with respect to REMOXY;
|
| ·
| | the outcome of research and development activities, including, without limitation, development activities for FENROCK™ and potential formulation of additional dosage forms of our drug candidates;
|
| ·
| | the potential benefits of our drug candidates;
|
| ·
| | the utility of protection of our intellectual property;
|
| ·
| | expected future sources of revenue and capital and increasing cash needs;
|
| ·
| | potential competitors or competitive products;
|
| ·
| | market acceptance of our drug candidates and potential drug candidates;
|
| ·
| | expectations regarding trade secrets, technological innovations, licensing agreements and outsourcing of certain business functions;
|
| ·
| | expenses increasing, interest income decreasing or fluctuations in our operating results;
|
| ·
| | operating losses and anticipated operating and capital expenditures;
|
| ·
| | expected uses of capital resources;
|
| ·
| | expectations regarding the issuance of shares of common stock to employees pursuant to equity compensation awards net of employment taxes;
|
| ·
| | anticipated hiring and development of our internal systems and infrastructure;
|
| ·
| | the sufficiency of our current resources to fund our operations over the next twelve months; and
|
| ·
| | assumptions and estimates used for our disclosures regarding stock-based compensation.
|
the utility of protection, or the sufficiency, of our intellectual property;
our potential competitors or competitive products;
expected future sources of revenue and capital and increasing cash needs;
our use of Clinical Research Organizations (CROs) to conduct clinical studies of our product candidates;
expectations regarding trade secrets, technological innovations, licensing agreements and outsourcing of certain business functions;
our expenses increasing or fluctuations in our financial or operating results;
our operating losses and anticipated operating and capital expenditures;
expectations regarding the issuance of shares of common stock to employees pursuant to equity compensation awards, net of employment taxes;
the development and maintenance of our internal information systems and infrastructure;
our need to hire additional personnel and our ability to attract and retain such personnel;
existing regulations and regulatory developments in the United States and other jurisdictions;
our need to expand the size and scope of our physical facilities;
the sufficiency of our current resources to continue to fund our operations;
the accuracy of our estimates regarding expenses, capital requirements, and needs for additional financing;
assumptions and estimates used for our disclosures regarding stock-based compensation; and
the long-term impact of COVID-19, a novel coronavirus first detected in 2019, on our operations and financial condition.
Such forward-looking statements and our business involve risks and uncertainties, including, but not limited to those risks and uncertainties relating to:the following:
| ·
| | difficulties or delays in completing development activities needed to potentially obtain regulatory approval of the NDA for REMOXY, including the potential for requests by the FDA for additional data which may require an extended period of time to obtain and submit;
|
| ·
| | having or obtaining sufficient resources for the successful development and commercialization of REMOXY;
|
| ·
| | the quantity, quality or sufficiency of the data, materials and information transferred to us by Pfizer regarding the REMOXY development program;
|
| ·
| | continuing discussions with potential strategic partners for the development and commercialization of REMOXY;
|
| ·
| | the successful development of other drug candidates, independently as well as pursuant to our other collaboration agreements, and the continuation of such agreements;
|
| ·
| | difficulties or delays in development, testing, clinical trials (including patient enrollment), regulatory authorization or approval, production and commercialization of our drug candidates;
|
| ·
| | unexpected adverse side effects or inadequate therapeutic efficacy of our drug candidates that could slow or prevent product approval (including the risk that current and past results of clinical trials are not indicative of future results of clinical trials) or potential post-approval market acceptance;
|
| ·
| | the uncertainty of protection of our intellectual property rights or trade secrets;
|
| ·
| | potential infringement of the intellectual property rights of third parties;
|
| ·
| | pursuing in-license and acquisition opportunities;
|
| ·
| | maintenance or third party funding of our collaboration and license agreements;
|
| ·
| | legislation or regulatory actions affecting product pricing, reimbursement or access;
|
| ·
| | significant breakdown or interruption of our information technology and infrastructure;
|
| ·
| | significant issues that may arise related to outsourcing certain preclinical studies, clinical trials and formulation and manufacturing activities;
|
| ·
| | hiring and retaining personnel; and
|
| ·
| | our financial position and our ability to obtain additional financing if necessary.
|
In addition, such statementsWe are subject to the risks and uncertainties discussed in the "Risk Factors" sectionearly stages of clinical drug development and elsewherehave a limited operating history in this document.our business targeting Alzheimer’s disease and no products approved for commercial sale.
We have incurred significant net losses in each period since our inception and anticipate that we will continue to incur net losses for the foreseeable future.
Research and development of biopharmaceutical products is a highly uncertain undertaking and involves a substantial degree of risk and our business is heavily dependent on the successful development of our product candidates.
OverviewWe may need to obtain substantial additional financing to complete the development and any commercialization of our product candidates.
We may not be successful in our efforts to continue to develop product candidates or commercially successful products.
Pain Therapeutics, Inc. develops proprietary drugs that offer significant improvementsWe may not be successful in our efforts to patientsexpand indications for product candidates.
We are concentrating a substantial portion of our research and healthcare professionals. We generally focus our drug development efforts on disordersthe diagnosis and treatment of Alzheimer’s disease, an area of research that has recorded many clinical failures.
We may encounter substantial delays in our clinical trials or may not be able to conduct or complete our clinical trials on the timelines we expect, if at all.
Our clinical trials may fail to demonstrate evidence of the nervous system.
Our expertise consists of developing new drug candidatessafety and guiding these through various regulatory and development pathways in preparation for their eventual commercialization. By necessity, the conduct of drug development is complex, lengthy, expensive and risky. The FDA has not yet established the safety or efficacy of our drugproduct candidates, which would prevent, delay, or limit the scope of regulatory approval and the commercialization of our product candidates.
We may be unable to protect our intellectual property rights or trade secrets.
For over a decade, we have pioneered technology, tools and techniques that enableWe may be subject to third-party claims of intellectual property infringement.
We may not succeed in our maintenance or pursuit of licensing rights or third-party intellectual property necessary for the development of Abuse-Deterrent Formulations,our product candidates.
Enacted or ADFs. ADFsfuture legislation or regulatory actions may adversely affect our product pricing, or limit the reimbursement we may receive for our products.
A significant breakdown, security breach or interruption affecting our internal computer systems, or those used by our third-party research collaborators, may compromise the confidentiality of our financial or proprietary information, result in material disruptions of our products and operations and adversely affect our reputation.
We may be unsuccessful at hiring and retaining qualified personnel.
Adverse circumstances caused by disease epidemics or pandemics, such as Coronavirus Disease 2019, or COVID-19, a novel coronavirus first detected in 2019;
Please also refer to the section entitled “Risk Factors” in our Annual Report on Form 10-K for the fiscal year ended December 31, 2020, as such risk factors may be amended, updated or modified periodically in our reports filed with the U.S. Securities and Exchange Commission (the “SEC”) for further information on these and other risks affecting us.
We caution you not to place undue reliance on forward-looking statements because our future results may differ materially from those expressed or implied by them. We do not intend to update any forward-looking statement, whether written or oral, relating to the matters discussed in this Quarterly Report on Form 10-Q, except as required by law.
Our research programs in neurodegeneration benefit from longstanding scientific and financial support from the National Institutes of Health (“NIH”). The contents of this Quarterly Report on Form 10-Q are intendedsolely our responsibility and do not necessarily represent any official views of NIH.
Overview
Cassava Sciences, Inc. is a clinical-stage biotechnology company based in Austin, Tx. Our mission is to make opioid drugs difficult to abuse yet provide steady pain relief when used appropriately by patients. ADFs are intended to helpdetect and treat neurodegenerative diseases, such as Alzheimer’s disease. Our novel science is based on stabilizing – but not removing – a critical protein in the fight against prescription drug abuse.brain.
Opioid drugs,Over the past 10 years, we have combined state-of-the-art technology with new insights in neurobiology to develop novel solutions for Alzheimer’s disease and other neurodegenerative diseases. Our strategy is to leverage our unique scientific/clinical platform to develop a first-in-class program for treating neurodegenerative diseases, such as oxycodone, are an importantAlzheimer’s.
We currently have two clinical-stage biopharmaceutical assets under development:
our lead therapeutic product candidate, called simufilam, is a novel treatment option for patients with severe chronic pain. However, misuse, abuseAlzheimer’s disease; and diversion
our lead investigational diagnostic product candidate, called SavaDx, is a novel way to detect the presence of these prescription drugs remainsAlzheimer’s disease from a serious, persistent problem. Nearly 19,000 people died from opioid overdosesmall sample of blood, possibly years before the overt appearance of clinical symptoms.
Our scientific approach for the treatment of Alzheimer’s disease seeks to simultaneously improve both neurodegeneration and neuroinflammation. We believe our ability to improve multiple vital functions in 2014, accordingthe brain represents a new, different and crucial approach to the NIH’s National Institute on Drug Abuse.address Alzheimer’s disease.
REMOXY
Our lead drugtherapeutic product candidate, is called REMOXY. REMOXYsimufilam, is a proprietary abuse-deterrent, oral formulationsmall molecule (oral) drug. Simufilam targets an altered form of oxycodone (CII). a protein called filamin A (FLNA) in the Alzheimer’s brain. Published studies have demonstrated that the altered form of FLNA causes neuronal dysfunction, neuronal degeneration and neuroinflammation.
We developed REMOXYbelieve simufilam improves brain health by reverting altered FLNA back to make oxycodone difficultits native, healthy conformation, thus countering the downstream toxic effects of altered FLNA. We have generated and published experimental and clinical evidence of improved brain health with simufilam. Importantly, simufilam is not dependent on clearing amyloid from the brain. Since simufilam has a unique mechanism of action, we believe its potential therapeutic effects may be additive or synergistic with that of other therapeutic candidates aiming to abuse yet provide 12 hourstreat neurodegeneration.
Simufilam has demonstrated a multitude of steady pain relief when used appropriatelybeneficial effects in animal models of disease, including normalizing neurotransmission, decreasing neuroinflammation, suppressing neurodegeneration, and restoring memory and cognition.
Simufilam and SavaDx were both discovered and designed in-house and were characterized by patients.
In particular, REMOXY’s thick, sticky, high-viscosity formulation may deter unapproved routes of drug administration, such as injection, snorting or smoking. REMOXY targets the multi-billion-dollar marketplace for long-acting formulations of oxycodone.our academic collaborators during research activities that were conducted from approximately 2008 to date. We own exclusive, worldwide rights to REMOXY.
In March 2016, we resubmittedthese drug assets and related technologies, without royalty obligations to the FDA the NDAany third party. Our patent protection with respect to simufilam and use of simufilam for REMOXY. In April 2016, the FDA determined that the NDA for REMOXY was sufficiently complete to permit a substantive review. On May 19, 2016, we announced that the FDA planned to hold an Advisory Committee meeting to review the NDA for REMOXY. On July 1, 2016, we announced that the FDA had determined that an Advisory Committee meeting for REMOXY was unnecessaryAlzheimer’s disease and would not be held.
In September 2016, we received a Complete Response Letter, or CRL from the FDA on the resubmission of NDA for REMOXY. The CRL informed us that the NDA for REMOXY could not be approved in its present formother neurodegenerative disease currently runs through 2033 and specifies additional actionsincludes six issued patents and data that are needed for drug approval. The CRL focuses on the abuse-deterrent properties of REMOXYrelated patent filings and proposed drug labeling. The CRL makes no mention of clinical safety, drug efficacy, manufacturing, stability, bioequivalence or any other issues from a prior Complete Response Letter.
The CRL focuses on the actions and studies that are needed in order to obtain approval of REMOXY. In conducting the following studies, we expect to generally compare REMOXY with one or more commercially available oxycodone ER drug product:
| ·
| | To support a potential drug label claim against abuse by injection: Repeat an injectability/syringeability study using thin films of drug, smaller volumes of solvents, additional mixed solvents and alternative extraction methods and syringe filter.
|
| ·
| | To support a potential drug label claim against abuse by snorting: Conduct an intranasal abuse potential study in human volunteers.
|
applications. In addition, we had proposedhave patent protection with respect to simufilam for use in treating certain cancers that runs through 2034.
We currently have no patents or patent applications with respect to SavaDx, which is protected in the REMOXY a label claim against abuseUnited States by chewing. Our proposal was based on clinical results of an oral human abuse potential study that met all four co-primary endpoints with statistical significancetrade secrets, know-how and that also met several, but not all, secondary endpoints. The CRL asks us to submit a revised proposed label to indicate results of this study do not support a label claim against abuse by chewing. other proprietary rights technology.
On February 13, 2017, we met with the FDA regarding REMOXY. During this meeting, an agreement was reached on additional studies that are needed for REMOXY’s regulatory approval. Following completion of these studies, we intend to have a pre-NDA meeting with the FDA, followed by resubmission of the REMOXY NDA, with an anticipated priority review, under Section 505(b)(2) of the Federal Food, Drug and Cosmetic Act.
The NDA includes five dosage strengths of REMOXY (5, 10, 20, 30, 40 mg). The proposed indication for REMOXY is “for the management of pain severe enough to require daily, around-the-clock, long-term opioid treatment and for which alternative treatment options are inadequate.”
The safety and clinical efficacy of REMOXY is supported by multiple studies, including a successful Phase III efficacy program conducted under a FDA Special Protocol Assessment, or SPA.
Alzheimer's Disease and PTI-125
PTI-125 is an oral, small molecule, early-stage drug candidate that was designed in-house and characterized by outside collaborators. We plan to develop PTI-125 as a treatment for Alzheimer’s Disease.
Alzheimer’s Disease (AD)disease is a progressive brainneurodegenerative disorder that slowly destroysaffects cognition, function and behavior. There are no disease-modifying drug therapies to treat the disease. As of 2020, there were approximately 50 million people worldwide living with dementia, a figure expected to increase to 150 million by 2050 and the annual global cost of dementia is now above $1 trillion, according to Alzheimer’s Disease International, a charitable organization. According to the non-profit Alzheimer's Association, Alzheimer’s disease is expected to nearly triple in the U.S. between now and 2050. If this occurs, there is potential for Alzheimer's disease to cause a major financial drain on the national economy.
Phase 2a Study
In 2019, we completed a small, first-in-patient, clinical-proof-of-concept, open-label Phase 2a study of simufilam in the U.S., with substantial support from the National Institute on Aging (NIA), a division of the NIH. Treatment with simufilam for 28 days significantly improved key biomarkers of Alzheimer’s pathology, neurodegeneration and neuroinflammation (p<0.001). Biomarkers effects were seen in all patients in both cerebrospinal fluid (CSF) and plasma.
Phase 2b Study
In September 2020, we announced final results of a Phase 2b study with simufilam in Alzheimer’s disease. In this clinical study funded by the NIH, Alzheimer’s patientstreated with 50 mg or 100 mg of simufilam twice-daily for 28 days showed statistically significant (p<0.05) improvements in CSF biomarkers of disease pathology,neurodegeneration and neuroinflammation, versusAlzheimer’s patients who took placebo. In addition, Alzheimer’s patients treated with simufilam showed improvements in validated tests of episodic memory and thinking skills, and eventuallyspatial working memory,versus patients on placebo (Effect Size 17-46%). Cognitive improvements correlated most strongly (R2=0.5) with decreases in levels of P-tau181.
Clinical Strategy Around Open-label Study
Much of the abilityvalue of our open-label study is to carry out the simplest tasks. Theregain data to support simufilam’s long-term safety profile in patients. Interim efficacy data from an open-label study has limitations compared to efficacy data from a fully completed, large, randomized controlled clinical trial, or from a fully enrolled open-label study.
We believe there is logic to conducting an open-label study prior to conducting a large, expensive Phase 3 clinical testing program. First, this is a standard clinical method of demonstrating drug safety. Second, we believe that if an experimental drug for Alzheimer’s shows no treatment benefits in a well-designed open-label study, then there is no approvedchance that drug therapywill succeed in Phase 3 clinical testing. Of course, the opposite is not true: encouraging treatment effects in an open-label study is not proof of drug efficacy, nor can encouraging treatment effects predict clinical success in a Phase 3 program.
In short, we believe a well-designed, open-label study is an exercise in prudent risk-management. Clinical results may serve as a tool to reverse, or even halt,help inform and manage the courseinherent risks and uncertainties of AD. PTI-125 has been showndrug development prior to significantly improve AD neuropathologyundertaking a large, expensive Phase 3 clinical testing program.
Open-label Study Strategy
In March 2020, we initiated a long-term, open-label study to evaluate simufilam in mouse models of the disease and in post-mortem brain tissuepatients with Alzheimer’s disease. This study is funded by a research grant award from AD patients, including receptor dysfunctions, neuroinflammation, tau hyperphosphorylation, insulin resistance and plaques and tangles that are hallmarks of AD. To date, the underlying science for PTI-125 has been published in Journal of Neuroscience, Neurobiology of Aging, Journal of Biological Chemistry, PLOS-One and other peer-reviewed scientific journals.
In June 2017, we announced that the National Institutes of Health (NIH). The study is intended to monitor the long-term safety and tolerability of simufilam 100 mg twice-daily for 12 or more months. Another study objective is to measure changes in cognition using ADAS-Cog, a standard test of cognition in Alzheimer’s disease. The study protocol has pre-specified cognition measurements at 6, 9 and 12 months. This study also uses the Neuropsychiatric Inventory (NPI) to assess the presence and severity of dementia-related behavior. ADAS-Cog and NPI scales are both widely used clinical tools in trials of Alzheimer’s disease.
In June 2021, the open-label study reached its target enrollment of 150 subjects with mild-to-moderate Alzheimer’s disease. By physician and patient request, clinical sites may continue to enroll additional subjects up through the initiation of the Company’s Phase 3 pivotal program of simufilam.
In February 2021, we announced results of a preplanned interim analysis of our open-label study with simufilam. This interim analysis summarized clinical data in the first 50 patients who have completed at least 6 months of drug treatment. Patients’ cognition and behavior scores both improved following six months of simufilam treatment, with no safety issues. Six months of simufilamtreatment improved cognition scores by 1.6 points on ADAS-Cog11, a 10% mean improvement from baseline to month 6. In these same patients, simufilam also improved dementia-related behavior, such as anxiety, delusions and agitation, by 1.3 points on the Neuropsychiatric Inventory, a 29% mean improvement from baseline to month 6.
In July 2021, we announced results of another preplanned interim analysis of our open-label study with simufilam. This interim analysis summarized clinical data on the first 50 patients who have completed at least 9 months of drug treatment.Patients’ cognition and behavior scores both improved following six months of simufilam treatment, with no safety issues. Nine months of simufilam treatment improved cognition scores by 3.0 points on ADAS-Cog11, an 18% mean improvement from baseline to month 9 (p<0.001). Figure 1.
Figure 1. Cognition Results
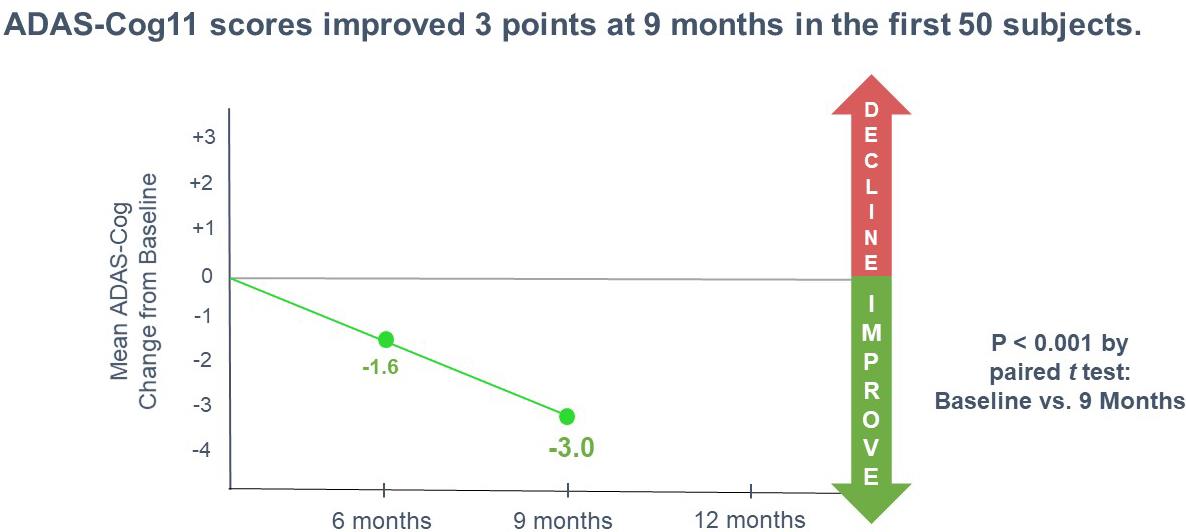
Insert A ADAS-Cog11 scores improved 3 points at 9 months in the first 50 subjects. Mean ADAS-Cog Change from Baseline -4 -3 -2 -1 0 +1 +2 +3 -1.6 -3.0 DECLINE IMPROVE P < 0.001 by paired t test: Baseline vs. 9 Months 6 months 9 months 12 months CASSAVA sciences
Alzheimer’s is a progressive disease. Cognition will always decline over time. Historical controls indicate that in patients with mild-to-moderate Alzheimer’s disease, cognition scores decline 4+ points on ADAS-Cog over 9 months, as reported by the science literature, and, more recently, by Biogen, Inc. In 2020, Biogen reported a 5.2-point decline over 18 months on ADAS-Cog in placebo patients with early Alzheimer’s disease in two Phase 3 studies with their drug, aducanumab.
Despite an expectation of cognitive decline, simufilam improved ADAS-Cog scores in 66% of patients at 9 months. An additional 22% of patients declined less than an expected 4+ point decline at 9 months. Cognition outcomes suggest simufilam’s treatment effects were broad-based. Figure 2.
Figure 2. Individual Patient Changes in ADAS-Cog (N=50)
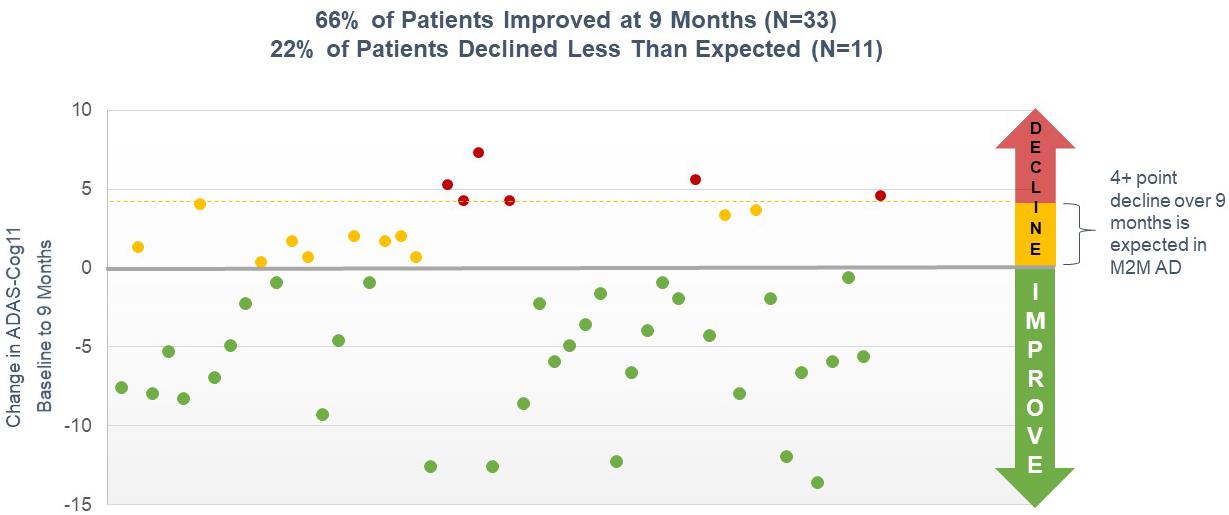
Insert B 66% of Patients Improved at 9 Months (N=33) 22% of Patients Declined Less Than Expected (N=11) Change in ADAS-Cog11 Baseline to 9 Months -15 -10 -5 0 5 10 DECLINE IMPROVE 4+ point decline over 9 months is expected in M2M AD CASSAVA sciences
Alzheimer’s is often accompanied by behaviors disorders, such as anxiety, agitation or delusions. These may become more frequent as disease progresses. Simufilam reduced dementia-related behavior at 9 months on the Neuropsychiatric Inventory (NPI), a clinical tool widely used to measure changes in dementia-related behavior.
At baseline, 34% of study subjects had awardedno neuropsychiatric symptoms.
At month 6, 38% of study subjects had no neuropsychiatric symptoms.
At month 9, over 50% of study subjects had no neuropsychiatric symptoms.
The safety profile of simufilam in the interim analysis is consistent with prior human studies. There were no drug-related serious adverse events. Adverse events were mild and transient.
In July 2021, we also announced positive biomarker data from our open-label study. Biomarkers are objective biological data. There are no placebo effects.
A key objective of this bioanalysis was to measure changes in levels of biomarkers in patients before and after 6 months of treatment with open label simufilam. Biomarker data were analyzed from cerebrospinal fluid (CSF) collected from 25 patients with mild-to-moderate Alzheimer’s disease who are enrolled in the open-label study and who agreed to undergo a lumbar puncture at baseline and again after 6 months of treatment. All bioanalyses were conducted blind by an outside lab.
Simufilam robustly improved all measured CSF biomarkers.
Cerebrospinal fluid (CSF) biomarkers of disease pathology, t-tau and p-tau181, decreased 38% and 18%, respectively (both p<0.00001). CSF biomarkers of neurodegeneration, neurogranin and Nfl, decreased 72% and 55%, respectively (both p<0.00001). CSF biomarkers of neuroinflammation, sTREM2 and YKL-40, decreased 65% and 44% (both p<0.00001). All p-values are baseline vs. 6-month levels by paired t-test. Figure 3.
Core markers of Alzheimer’s pathology are total tau (T-tau), phosphorylated tau (P-tau181), and amyloid beta42 (Aβ42). In Alzheimer’s, tau levels are elevated and Aβ42 is low.
T-tau decreased 38% (p<0.00001)
P-tau181 decreased 18% (p<0.00001)
CSF Aβ42 increased 84% (p<0.00001)
Elevated CSF levels of two proteins, neurogranin (Ng) and neurofilament Light Chain (NfL) indicate neurodegeneration.
Ng decreased 72% (p<0.00001)
NfL decreased 55% (p<0.00001)
Elevated levels of marker YKL-40 indicate neuroinflammation.
YKL-40 decreased 44% (p<0.00001)
sTREM2 is a biomarker of microglia-induced neuroinflammation that has commanded substantial recent attention from researchers for its role in Alzheimer’s and frontotemporal dementia.
sTREM2 decreased 65% (p<0.00001)
HMGB1 protein, is a damage-related protein sometimes called a ‘danger molecule’ because it triggers additional neuroinflammation and loss of neurons.
HMGB1 decreased 53% (p<0.00001)
Figure 3. Significant Decreases in CSF Biomarkers at Month 6
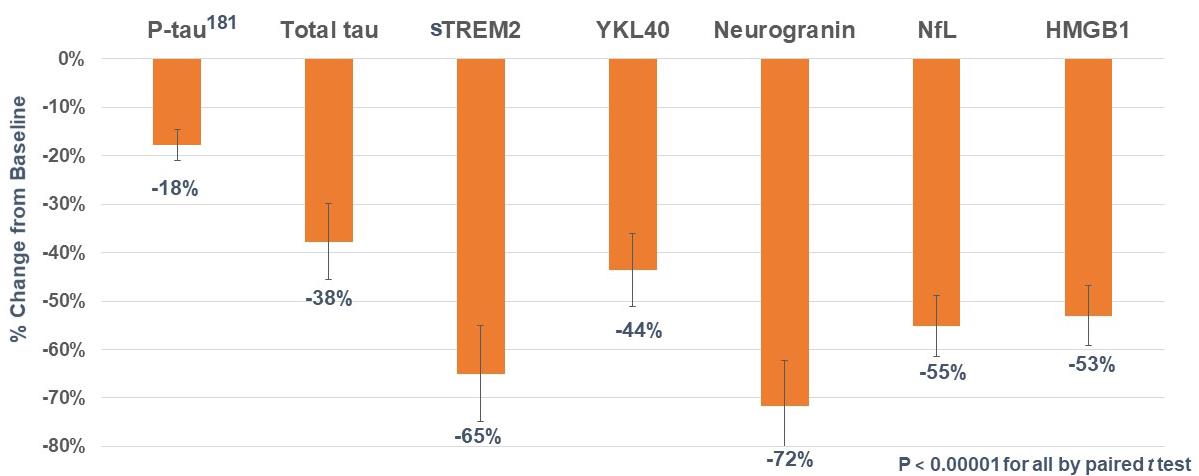
Insert D % Change from Baseline -80% -70% -60% -50% -40% -30% -20% -10% 0% P-tau181 Total tau sTREM2 YKL40 Neurogranin NfL HMGB1 -18% -38% -65% -44% -72% -55% -53% P < 0.0001 for all by paired t test CASSAVA sciences
Cognition Maintenance Study
In May 2021, we initiated a double-blind, randomized, placebo-controlled study in patients with mild-to-moderate Alzheimer’s disease. Patients who have completed at least one year of open-label treatment with simufilam qualify to enroll in the Cognition Maintenance Study (CMS). Study subjects in the CMS are randomized (1:1) to simufilam or placebo for six months. The CMS is designed to compare simufilam’s effects on cognition in Alzheimer’s patients
who continue with drug treatment versus patients who discontinue drug treatment. Figure 4. The target enrollment for the CMS is 100 subjects or more; as of mid-June 2021, approximately 30 subjects were enrolled.
Figure 4. Cognition Maintenance Study Design

End-of-Phase 2 (EOP2) Meeting with FDA
In January 2021, we held an End-of-phase 2 (EOP2) meeting for simufilam with the U.S Food and Drug Administration (FDA). The purpose of this EOP2 was to gain general agreement around key elements of a pivotal Phase 3 program to treat Alzheimer’s disease dementia. FDA attendees included Robert Temple, MD, Deputy Center Director for Clinical Science and Senior Advisor in the Office of New Drugs; Billy Dunn, MD, Director, Office of Neuroscience; Eric Bastings, MD, Director, Division of Neurology, and others.
In February 2021, we announced the successful completion of our EOP2 meeting. Official meeting minutes confirm that we and FDA are aligned on key elements of a Phase 3 clinical program for simufilam. FDA has agreed that the completed Phase 2 program, together with an upcoming and well-defined Phase 3 clinical program, are sufficient to show evidence of clinical efficacy for simufilam in Alzheimer’s disease. There is also agreement that the use of separate clinical scales to assess cognition (ADAS-cog1) and function (ADCS-ADL2) are appropriate co-primary endpoints of efficacy. A clinical scale that combines cognition and function, such as iADRS3, is a secondary efficacy endpoint.
Agreements reached during the EOP2 meeting show a clear path forward for advancing simufilam into Phase 3 studies. As a result, we expect to initiate a pivotal Phase 3 program with simufilam in Alzheimer’s disease in the second half of 2021.
Phase 3 Drug Supply
In March 2021, we announced we had entered into a drug supply agreement with Evonik Industries AG for simufilam. Under the agreement, Evonik will supply us an approximately $1.7 millionwith large-scale, clinical-grade quantities of simufilam. Evonik is one of the world’s largest contract development and manufacturing organizations for pharmaceutical ingredients.
_____________________________
1 ADAS-Cog = The Alzheimer’s Disease Assessment Scale – Cognitive Subscale, a measure of cognition
2 ADCS-ADL = Alzheimer’s Disease Cooperative Study – Activities of Daily Living, a measure of health function
3 iADRS = integrated Alzheimer’s Disease Rating Scale, a composite measure of cognition and health function
Phase 3 Clinical Program
We plan to initiate a Phase 3 program of simufilam in Alzheimer’s disease. The Phase 3 program consists of two large, double-blind, randomized, placebo-controlled studies in patients with mild-to-moderate Alzheimer’s disease dementia. In June 2021, we announced the selection of Premier Research International as our clinical research organization (CRO) to help conduct the Phase 3 clinical program of simufilam for Alzheimer’s disease.
We expect to initiate the Phase 3 program in Q4 2021. Figure 5.
Figure 5. Phase 3 Program Overview
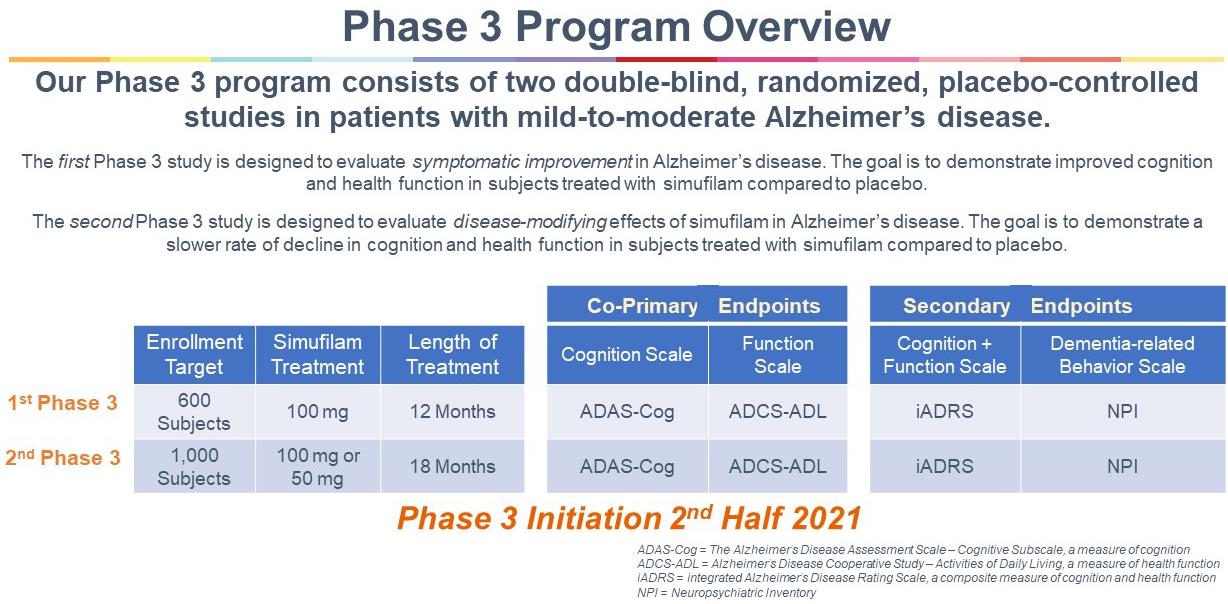
SavaDx
Our diagnostic effort, called SavaDx, is a clinical-stage program focused on detecting the presence of Alzheimer’s disease from a small sample of blood, possibly years before the overt appearance of clinical symptoms. We are developing SavaDx as a fast, accurate and quantitative blood-based investigational biomarker/diagnostic to detect and monitor Alzheimer's disease. The goal is to make the detection of Alzheimer’s disease as simple as getting a blood test. There is no patent protection for SavaDx in the U.S., but we believe this product candidate is protected by trade secrets, know-how and other proprietary rights technology. The SavaDx program is substantially funded by a research grant award from the National Institutes of Health (NIH).
In blinded studies, SavaDx detected >10-fold differences between patients with Alzheimer’s and age-matched normal controls or young cognitively intact subjects (N=232).
In July 2021, we announced positive clinical data with SavaDx when used to study PTI-125measure plasma levels of altered filamin A before and after simufilam treatment in patients with Alzheimer’s disease. In a Phase 2b randomized, controlled trial sponsored by the National Institutes of Health (NIH), simufilam significantly reduced plasma levels of altered filamin A in Alzheimer’s Disease. This NIH research grant has enabled us to begin testing PTI-125patients treated for 28 days. Plasma levels of p-tau181 also dropped significantly in human subjects. these same patients.
In September 2017, we announced that the NIH had awarded an approximately $1.8 million research grant to support innovative technology we have developed to diagnose Alzheimer’s disease with a simple blood test.
Our NIH grants for PTI-125 are multi-year, milestone-based awards. NIH has no obligation to continue funding for PTI-125 beyond the termsSimufilam 100 mg and 50 mg reduced plasma levels of the two grant awards.
Pain Therapeutics owns worldwide commercial rights to PTI-125altered filamin A by 48% (p=0.003) and related technology, without royalty or milestone obligations to any third-parties.
FENROCK™
FENROCK44% (p=0.02) respectively, versus placebo. Additionally, simufilam 100 mg and 50 mg reduced plasma levels of p-tau181 by 17% (p=0.01) and 15% (p=0.02) respectively, versus placebo. Plasma p-tau181 is a transdermal patchbiomarker that containsis known to be elevated in Alzheimer’s disease.
Impact of COVID-19 on our Business
In these times of pandemic, our top priorities are to protect the prescriptionhealth, well-being, and safety of our employees and partners, while still focusing on the key drivers of our business. Despite COVID-19, we believe we remain on-track to achieve our major strategic objectives for 2021 with simufilam. We have not experienced major disruptions across our drug fentanylmanufacturing operations or supply of materials. Our broad spectrum of technical consultants, scientific advisors and service providers continue to manage severe painprovide timely services. We have adapted flexible business practices, such as remote work arrangements and incorporates novel abuse-deterrent technology. FENROCKtemporary travel restrictions, to insure we continue to operate safety and cautiously while also meeting our public health responsibilities. We recognize the pandemic has created a dynamic and uncertain situation in the national economy. We continue to closely monitor the latest information to make timely, informed business decisions and public disclosures regarding the potential impact of pandemic on our operations. However, the scope of pandemic is an early-stage drug candidate that was designed in-houseunprecedented and characterized by outside collaborators.its long-term impact on our operations cannot be reasonably estimated at this time.
In September 2017, we announced that the National Institute on Drug Abuse (NIDA).had awarded us a research and development grant of approximately $2.2 million. This grant award provides us with a path forward to develop FENROCK, a drug candidate for severe pain. Financial Overview
Our NIH grant for FENROCK is a multi-year, milestone-based award. NIH has no obligation to continue funding for FENROCK beyond the term of the grant award.
Pain Therapeutics owns worldwide commercial rights to FENROCK and related technology, without royalty or milestone obligations to any third-parties.
We have yet to generate any revenues from product sales. We have an accumulated deficit of $155$183.6 million at SeptemberJune 30, 2017.2021. These losses have resulted principally from costs incurred in connection with research and development activities, salaries and other personnel-related costs and general corporate expenses. Research and development activities include costs of preclinical and clinical trials as well as clinical supplies associated with our drugproduct candidates. Salaries and other personnel-related costs include non-cash stock-based compensation associated with stock options and other equity awards granted to employees and non-employees. Our operating results may fluctuate substantially from period to period as a result of the timing of reimbursement from NIH grants, preclinical activities, enrollment rates of clinical trials for our drugproduct candidates and our need for clinical supplies.
We expect to continue to use significant cash resources in our operations for the next several years. Our cash requirements for operating activities and capital expenditures may increase substantially in the future as we:
| ·
| | conduct preclinical and clinical trials for our drug candidates;
|
| ·
| | seek regulatory approvals for our drug candidates;
|
| ·
| | develop, formulate, manufacture and commercialize our drug candidates;
|
| ·
| | implement additional internal systems and develop new infrastructure;
|
| ·
| | acquire or in-license additional products or technologies, or expand the use of our technology;
|
| ·
| | maintain, defend and expand the scope of our intellectual property; and
|
| ·
| | hire additional personnel.
|
initiate a large-scale drug manufacturing campaign for simufilam;
plan to initiate a Phase 3 clinical program with simufilam;
conduct other preclinical and clinical studies for our product candidates;
plan to seek regulatory approvals for our product candidates;
develop, formulate, manufacture and commercialize our product candidates;
implement additional internal systems and develop new infrastructure;
acquire or in-license additional products or technologies, or expand the use of our technology;
maintain, defend and expand the scope of our intellectual property
hire additional personnel; and
expand our office facilities to accommodate growth in personnel and R&D activities.
Product revenue will depend on our ability to receive regulatory approvals for, and successfully market, our drugproduct candidates. If our development efforts result in regulatory approval and successful commercialization of our drugproduct candidates, we will generate revenue from direct sales of our drugs and/or, if we license our drugs to future collaborators, from the receipt of license fees and royalties from sales of licensed products. We conduct our research and development programs through a combination of internal and collaborative programs. We rely on arrangements with universities, our collaborators, contract research organizations and clinical research sites for a significant portion of our product development efforts.
We focus substantially all of our research and development efforts in the area of neurology. The following table summarizes expenses by categorywhich have been reduced for researchreimbursements received for NIH grants (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Three months ended |
| Six months ended |
|
|
| June 30, |
| June 30, |
|
|
| 2021 |
| 2020 |
| 2021 |
| 2020 |
|
Research and development expenses - gross |
| $ | 4,787 |
| $ | 1,688 |
| $ | 7,892 |
| $ | 3,574 |
|
Less: Reimbursement from NIH grants |
|
| 886 |
|
| 1,097 |
|
| 1,462 |
|
| 2,439 |
|
Research and development expenses - net |
| $ | 3,901 |
| $ | 591 |
| $ | 6,430 |
| $ | 1,135 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development efforts (in thousands):
| | | | | | | | | | | | |
| | | | | | | | | | | | |
| | Three months ended | | Nine months ended |
| | September 30, | | September 30, |
| | 2017 | | 2016 | | 2017 | | 2016 |
Compensation | | $ | 697 | | $ | 479 | | $ | 2,138 | | $ | 2,838 |
Contractor fees and supplies | | | 789 | | | 1,814 | | | 3,410 | | | 4,346 |
Other common costs | | | 133 | | | 364 | | | 523 | | | 657 |
| | $ | 1,619 | | $ | 2,657 | | $ | 6,071 | | $ | 7,841 |
| | | | | | | | | | | | |
expenses include compensation, contractor fees and supplies as well as allocated common costs. Contractor fees and supplies generally include expenses for preclinical studies and clinical trials and
costs for formulation and manufacturing activities. Other common costs include the allocation of common costs such as facilities. During the three months ended June 30, 2021 and 2020, we received $0.9 million and $1.1 million from NIH research grants, respectively. During the six months ended June 30, 2021 and 2020, we received $1.5 million and $2.4 million from NIH research grants, respectively. These reimbursements were recorded as a reduction to our research and development expenses.
Our technology has been applied across certain of our drugproduct candidates. Data, know-how, personnel, clinical results, research results and other matters related to the research and development of any one of our drugproduct candidates also relate to, and further the development of, our other drugproduct candidates. As a result, costs allocated to a specific drug candidate may not necessarily reflect the actual costs surrounding research and development of that drugproduct candidate due to cross application of the foregoing.
Estimating the dates of completion of clinical development, and the costs to complete development, of our drugproduct candidates would be highly speculative, subjective and potentially misleading. Pharmaceutical productsproduct candidates take a significant amount of time to research, develop and commercialize. The clinical trial portion of the development of a new drug alone usually spans several years. We expect to reassess our future research and development plans based on our review of data we receive from our current research and development activities. The cost and pace of our future research and development activities are linked and subject to change.
Critical Accounting Policies
The preparation of our condensed financial statements in accordance with United Statesaccounting principles generally accepted accounting principlesin the United States requires us to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenues, expenses and interest income in our condensed financial statements and accompanying notes. We evaluate our estimates on an ongoing basis, including those estimates related to agreements and research collaborations and investments.collaborations. We base our estimates on historical experience and various other assumptions that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions. The following items in our condensed financial statements require significant estimates and judgments:
| ·
| | Stock-based compensation. We recognize non-cash expense for the fair value of all stock options and other share-based awards. We use the Black-Scholes option valuation model to calculate the fair value of stock options, using the single-option award approach and straight-line attribution method. For options granted to employees and directors, we recognize the resulting fair value as expense on a straight-line basis over the vesting period of each respective stock option, generally four years. For options granted to non-employees, we remeasure the fair value expense using Black-Scholes each reporting period.
|
Research Contracts and Accruals. We have entered into various research and development contracts with research institutions and other third-party vendors. Related payments are recorded as research and development expenses as incurred. We record accruals for estimated ongoing research costs. When evaluating the adequacy of the accrued liabilities, we analyze progress of the studies including the phase or completion of events, invoices received and contracted costs. Significant judgments and estimates are made in determining the accrued balances at the end of any reporting period. Actual results could differ from our estimates. Our historical accrual estimates have not been materially different from actual costs.
2020 Cash Incentive Bonus Plan. In 2020, we established the 2020 Cash Incentive Bonus Plan (the “Plan”) to incentivize Plan participants. Awards under the Plan are accounted for as liability awards under ASC 718, “Stock-based Compensation”. The fair value of each potential Plan award will be determined once a grant date occurs and will be remeasured each reporting period. Compensation expense associated with the Plan will be recognized over the expected achievement period for each Plan award, when a Performance Condition is considered probable of being met.
The Plan was established to promote the long-term success of the Company by creating an “at-risk” cash bonus program that rewards Plan participants with additional cash compensation in lockstep with significant increases in our market capitalization. The Plan is considered “at-risk” because Plan participants will not receive a cash bonus unless our market capitalization increases significantly and (1) we complete a merger or acquisition transaction that constitutes a sale of ownership of the Company or its assets (a Merger Transaction) or (2) the Compensation Committee of the Board (the Compensation Committee) determines the Company has sufficient cash on hand, as defined in the Plan, to render payment (each, a “Performance Condition”), neither of which may ever occur. Because of the inherent discretion and uncertainty regarding these requirements, we have concluded that a Plan grant date has not occurred as of June 30, 2021. No actual cash payments were authorized or made to participants under the Plan through June 30, 2021.
Stock-based Compensation. We recognize non-cash expense for the fair value of all stock options and other share-based awards. We use the Black-Scholes option valuation model to calculate the fair value of stock options, using the single-option award approach and straight-line attribution method. For all options granted, we recognize the resulting fair value as expense on a straight-line basis over the vesting period of each respective stock option, generally four years.
We have granted share-based awards that vest upon achievement of certain performance criteria, or Performance Awards. We multiply the number of Performance Awards by the fair market value of our common stock on the date of grant to calculate the fair value of each award. We estimate an implicit service period for achieving performance criteria for each award. We recognize the resulting fair value as expense over the implicit service period when we conclude that achieving the performance criteria is probable. We periodically review and update as appropriate our estimates of implicit service periods and conclusions on achieving the performance criteria. Performance Awards vest and common stock is issued upon achievement of the performance criteria.
| ·
| | Income Taxes. We make estimates and judgments in determining the need for a provision for income taxes, including the estimation of our taxable income or loss for each full fiscal year.We have accumulated significant deferred tax assets that reflect the tax effects of net operating loss and tax credit carryovers and temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Realization of deferred tax assets is dependent upon future earnings, if any. We are uncertain as to the timing and amount of any future earnings. Accordingly, we offset these deferred tax assets with a valuation allowance. We may in the future determine that our deferred tax assets will likely be realized, in which case we will reduce our valuation allowance in the quarter in which such determination is made. If the valuation allowance is reduced, we may recognize a benefit from income taxes in our statement of operations in that period. We classify interest recognized in connection with our tax positions as interest expense, when appropriate.
|
Income Taxes.We account for income taxes under the asset and liability method. Deferred tax assets and liabilities are recognized for the estimated future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases. Deferred tax balances are adjusted to reflect tax rates based on currently enacted tax laws, which will be in effect in the years in which the temporary differences are expected to reverse. We have accumulated significant deferred tax assets that reflect the tax effects of net operating loss and tax credit carryovers and temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Realization of certain deferred tax assets is dependent upon future earnings. We are uncertain about the timing and amount of any future earnings. Accordingly, we offset these deferred tax assets with a valuation allowance.
We account for uncertain tax positions in accordance with ASC 740, “Income Taxes”, which clarifies the accounting for uncertainty in tax positions. These provisions require recognition of the impact of a tax position in our condensed financial statements only if that position is more likely than not of being sustained upon examination by taxing authorities, based on the technical merits of the position. Any interest and penalties related to uncertain tax positions will be reflected as a component of income tax expense.
Results of Operations – Three and nine months ended SeptemberSix Months Ended June 30, 20172021 and 2016 2020
Research and Development Expense
Research and development expense consistsexpenses consist primarily of costs of drug development work associated with our drugproduct candidates, including:
| ·
| | clinical supplies and related formulation and design costs, and
|
| ·
| | compensation and other personnel-related expenses.
|
Pre-clinical testing,
clinical trials,
clinical supplies and related formulation and design costs, and
compensation and other personnel-related expenses.
Research and development expenses forwere $3.9 million and $0.6 million during the three months ended SeptemberJune 30, 2017 decreased2021 and 2020, respectively. This 560% increase was due primarily to $1.6 million, respectively, from $2.7 million forcosts related to the same periodmanufacturing of clinical trial supplies in 2016, primarily dueanticipation of launching a Phase 3 clinical program in simufilam, costs of an on-going open-label study in simufilam, as well as increased personnel costs compared to a decrease in REMOXY related expenses and the receipt of grant funding from the National Institutes of Health, recorded as a reduction in research and development expenses.prior year.
Research and development expenses forwere $6.4 million and $1.1 million during the ninesix months ended SeptemberJune 30, 2017 decreased2021 and 2020, respectively. The 467% increase was due primarily to $6.1 million, respectively, from $7.8 million for the same period in 2016, primarily due to decreases in REMOXY related expenses and compensation expensescosts related to Performance Awardsthe manufacturing of clinical trial supplies in the nine months ended September 30, 2017anticipation of launching a Phase 3 clinical program in simufilam, costs of an on-going open-label study in simufilam, increased personnel costs, as well as a decrease in grant funding received from NIH compared to the same period in 2016.
Research and development expenses included non-cash stock-related compensation expenses of $0.3 million in both threeprior year. During the six months ended SeptemberJune 30, 20172021 and 2016. Non-cash stock-related compensation expenses were $0.92020, we received $1.5 million in the nine months ended September 30, 2017 compared to $1.4and $2.4 million for the same period in 2016.from research grants from NIH, respectively.
We expect research and development expensesexpense to fluctuate over the next several yearsincreasesignificantly in future periods as we continue our development efforts. We expectto hire new personnel, manufacture drug supply, continue our development efforts to resultand launch a Phase 3 clinical program in our drug candidates progressing through various stages of clinical trials. Our research and development expenses may fluctuate from period to period due to the timing and scope of our development activities and the results of clinical trials and preclinical studies. We also expect non-cash equity-related expenses to increase in the future.simufilam.
General and Administrative Expense
General and administrative expense consists primarily of compensation and other general corporate expenses. General and administrative expenses slightly increasedconsist of personnel costs, allocated expenses and other expenses for outside professional services, including legal, human resources, audit and accounting services. Personnel costs consist of salaries, bonus, benefits and stock-based compensation. Allocated expenses consist primarily of facility costs. We incur expenses associated with operating as a public company, including expenses related to $1.0compliance with the rules and regulations of the SEC and Nasdaq, additional insurance expenses, additional audit expenses, investor relations activities, Sarbanes-Oxley compliance expenses and other administrative expenses and professional services.
General and administrative expenses were $1.2 million inand $0.8 million during the three months ended SeptemberJune 30, 2017 from $0.9 million for2021 and 2020, respectively. The 51% increase was due primarily to higher annual shareholder meeting and insurance costs in 2021 compared to the same period in 2016. prior year.
General and administrative expenses forwere $2.2 million and $1.6 million during the ninesix months ended SeptemberJune 30, 2017 decreased 2021 and 2020, respectively. The 40% increase was due primarily to $3.5 million respectively, from $4.6 million for the same periodhigher annual shareholder meeting and insurance costs in 2016, primarily due to decreases in compensation expense related to Performance Awards and the departure of our CFO in March 2017 in the nine months ended September 30, 2017 as2021 compared to the same period in 2016. prior year.
General and administrative expenses included non-cash stock-related compensation expenses of $0.4 million in the three months ended September 30, 2017 compared to $0.5 million for the same period in 2016. Non-cash stock-related compensation expenses were $1.3 million in the nine months ended 2017 compared to $2.1 million for the same period in 2016.
We expect our general and administrative expenses to increase over the next several years in connection with support of pre-commercialization and commercialization activities for our drug candidates. The increase may fluctuate from period to periodfuture periods due to higher operating costs such as insurance, office space and information technology related expenses.
Gain on Sale of Property and Equipment
There were no sales of property and equipment during the timingthree and scope of these activities and the results of clinical trials and preclinical studies. We also expect non-cash equity-related expenses to increase in the future.six months ended June 30, 2021.
Interest Income
Interest income forDuring the three months ended SeptemberJune 30, 2017 was $6,000 compared2020, we sold surplus manufacturing equipment to$23,000 for the same period in 2016. an independent third party and received proceeds totaling $260,000. During the ninesix months ended SeptemberJune 30, 2017,2020, we sold surplus manufacturing equipment to an independent third party and received proceeds totaling $360,000.
We do not expect any future gains on sales of property and equipment.
Interest Income
Interest income was $13,000 and $27,000 during the three months ended June 30, 2021 and 2020, respectively. Interest income was $20,000 and $99,000 during the six months ended June 30, 2021 and 2020, respectively. The decrease in interest income was $33,000due to lower interest rates, which more than offset additional interest from increases in our cash balances compared to $86,000 the same periodprior periods.
We expect interest income to decrease in 2016. The2021 compared to 2020 due to decreases in both periods were primarily due to a lower prevailing interest rate on our investments.rates.
Liquidity and Capital Resources
Since inception, we have financed our operations primarily through public and private stock offerings, payments received under collaboration agreements and interest earned on our investments.cash and cash equivalents balances. We intend to continue to use our capital resources to fund research and development activities, capital expenditures, working capital requirements and other general corporate purposes. As of SeptemberJune 30, 2017,2021, cash and cash equivalents were $11.9$278.3 million.
2021 Registered Direct Offering
On February 12, 2021, we completed a common stock offering pursuant to which certain investors purchased 4,081,633 shares of common stock at a price of $49.00 per share. Net proceeds of the offering were approximately $189.8 million after deducting offering expenses.
Common Stock Warrants
In August 2018, we issued warrants to purchase up to an aggregate of 9.1 million shares of common stock in conjunction with an offering of our common stock.
We did not receive any proceeds from exercise of common stock warrants during the three months ended June 30, 2021. During the three months ended June 30, 2020, we received proceeds of $0.2 million from the exercise of 0.2 million shares pursuant to warrants.
During the six months ended June 30, 2021, we received proceeds of $0.7 million from the exercise of 0.6 million shares pursuant to warrants. During the six months ended June 30, 2020, we received proceeds of $3.8 million from the exercise of 3.1 million shares pursuant to warrants.
There were no remaining common stock warrants outstanding as of June 30, 2021.
At-the-Market Common Stock Offering
In March 2020, we established an at-the-market offering program (“ATM”) to sell, from time to time, shares of our common stock having an aggregate offering price of up to $100 million in transactions pursuant to a shelf registration statement that was declared effective by the SEC on May 5, 2020. We are obligated to pay a commission of 3.0% of the gross proceeds from the sale of shares of common stock in the offering. We are not obligated to sell any shares in the offering.
There were no common stock sales under the ATM during the three and six months ended June 30, 2021 and 2020.
NIH Research Grant Awards
Our programs have been supported by NIH under multiple research grant awards. Strong, long-term support from NIH has allowed us to advance our two lead product candidates, simufilam and SavaDx, into clinical development.
In May 2021, we were awarded a new research grant award from NIH of up to $2.7 million to support clinical readiness activities in support of an upcoming Phase 3 program with simufilam. In April 2020, we were awarded a research grant from NIH of up to $2.5 million. In March 2020, we were awarded a supplemental research funding grant from NIH of up to $374,000. These non-dilutive research grants are intended to strengthen our clinical program of simufilam, our investigational drug to treat Alzheimer’s disease. All of our NIH research grant awards are paid out on a reimbursement basis and require milestone-based technical progress.
2020 Cash Incentive Bonus Plan Obligations
On August 26, 2020, the Board approved the 2020 Cash Incentive Bonus Plan (the Plan). The Plan was established to promote the long-term success of the Company by creating an “at-risk” cash bonus program that rewards Plan participants with additional cash compensation in lockstep with significant increases in the Company’s market capitalization. The Plan is considered “at-risk” because Plan participants will not receive a cash bonus unless the Company’s market capitalization increases significantly and certain other conditions specified in the Plan are met. Specifically, Plan participants will not be paid any cash bonuses unless (1) the Company completes a merger or acquisition transaction that constitutes a sale of ownership of the Company or its assets (a Merger Transaction) or (2) the Compensation Committee determines the Company has sufficient cash on hand, as defined in the Plan. Plan participants will be paid all earned cash bonuses in the event of a Merger Transaction.
The Company’s market capitalization, including all outstanding stock options, was $89.4 million at the inception of the Plan on August 26, 2020. If the Company were to exceed a $5 billion market capitalization for no less than 20 consecutive trading days, and conditions noted above for payment are met, all Plan milestones would be deemed achieved, in which case total cash bonus awards would range from a minimum of $139.1 million up to a hypothetical maximum of $322.3 million.
The Company’s potential financial obligation to plan participants at June 30, 2021 totaled $7.3 million, based upon the achievement of one Plan milestone in the Company’s market capitalization in 2020. No actual cash bonus payments have been made to any Plan participant, as the Company has not yet satisfied all the conditions necessary for amounts to be paid under the Plan. During the six months ended June 30, 2021, the Company’s market capitalization increased substantially. These increases triggered the achievement of 10 additional Plan milestones.
Collectively, the achievement of such milestones could trigger potential Company obligations to Plan participants ranging from a minimum of $81.0 million up to a hypothetical maximum of $195.0 million, with exact amounts to be determined by the Compensation Committee and contingent upon future satisfaction of a Performance Condition.
Subsequent to June 30, 2021, the Company achieved one additional Valuation Milestone triggering potential Company obligations to all Plan participants from a minimum of $12.7 million up to a hypothetical maximum of $30.0 million, to be determined by the Compensation Committee and contingent upon future satisfaction of a Performance Condition.
No actual cash payments have been made to participants under the Plan as of June 30, 2021, or through the filing date of this Form 10-Q.
Use of Cash
Net cash used in operating activities during the nine months ended September 30, 2017 was $6.8 million compared to $9.2$7.4 million for the same periodsix months ended June 30, 2021, resulting primarily from the net loss reported of $8.7 million and an increase in 2016. The decreaseprepaid and other assets of $2.2 million, partially offset by an increase in accrued development expense of $1.7 million and accounts payable of $1.0 million, as well as stock-based compensation expense of $0.7 million.
Net cash used in operating activities was $2.0 million for the six months ended June 30, 2020, resulting primarily due to lower researchfrom the net loss reported of $2.3 million and development expensesa gain on sale of property and equipment of $0.3 million, partially offset by stock-based compensation expense of $0.5 million.
Net cash used in investing activities during the ninesix months ended SeptemberJune 30, 2017 compared to the same period in 2016, offset in part by changes in other balance sheet accounts.2021 was $73,000 for purchase of property and equipment.
Net cash provided by investing activities during the ninesix months ended SeptemberJune 30, 20172020 was $2.1million compared to $0.6 million net cash used by investing activities$360,000 for proceeds received from the same period in 2016. Investing activities in both 2017sale of property and 2016 consisted primarily of purchases, sales and maturities of marketable securities.equipment.
Net cash usedprovided by financing activities during the ninesix months ended SeptemberJune 30, 20162021 was $0.3$192.3 million, which resultedconsisting of $189.8 million proceeds from cash used to pay statutory taxes for netour registered direct offering of common stock in February 2021, $1.7 million from exercise of vested Performance Awards.stock options and $0.7 million proceeds from exercise of common stock warrants.
RealizationNet cash provided by financing activities during the six months ended June 30, 2020 was $3.8 million, resulting from proceeds from exercise of our other deferred tax assets is dependent on future earnings, if any. common stock warrants.
Leases
We are uncertain about the timing and amountlease approximately 6,000 square feet of any future earnings. Accordingly, we offset these net deferred tax assets with a valuation allowance.
We haveoffice space pursuant to a non-cancelable operating lease for approximately 6,000in Austin, TX that expires in April 2024. We also lease an additional 3,600 square feet of office space in Austin, Texas that expires on April 30, 2022.
On August 4, 2021, we completed the purchase of a two-building office complex in December 2017. Minimum lease payments areAustin, Texas, which will serve as follows (in thousands):
| | | | | |
| | | | | |
| 2017 | | Total |
Minimum lease payments | $ | 148 | | $ | 148 |
| | | | | |
We have license agreements that require usour new corporate headquarters. This property is intended to make milestone payments upon the successful achievement of milestones, including clinical milestones. Our license agreements also require us to pay certain royalties toaccommodate our licensors if we succeed in fully commercializing products under these license agreements. All of these potential future payments are cancelable as of September 30, 2017. Our formulation agreement with Durect Corporation, or Durect, obligates us to make certain milestone payments upon achieving clinical milestonesanticipated significant growth and regulatory milestones and pay royalties on related drug sales.
Our employees have Performance Awards that vest upon certain conditions. If these Performance Awards vest, we may issue the employees sharesexpansion of our common stock netoperations in the coming years. Company management expects to be hands-off with regards to property management. Maintenance, physical facilities, leasing, property management and other key responsibilities around property ownership will all be assumed by professional real-estate managers under long-term contract with the Company. The property purchase price was $21.9 million, exclusive of statutory employment taxes. This net issuance would resultclosing costs, funded with cash on hand. The office complex measures approximately 90,000 rentable square feet. The property is currently 59% leased, before the effect of the Company occupying approximately 25% of the property in fewer sharesthe near future. The seller is an independent third party not affiliated with the Company.
Other Commitments
We had non-cancellable commitments for the manufacture of common stock issued and uses our cash to pay these taxes on behalf of employees. The usesimufilam totaling approximately $1.9 million at June 30, 2021.
of cash could be higher or lower, depending on the fair value of our common stock on the date the Performance Awards vest.
We have an accumulated deficit of $155$183.6 million at Septemberas of June 30, 2017.2021. We expect our cash requirements to be significant in the future. The amount and timing of our future cash requirements will depend on regulatory and market acceptance of our drug candidates, the resources we devote to researching and developing, formulating, manufacturing,
commercializing and supporting our products and other corporate needs. We believe that our current resources shouldwill be sufficient to fund our operations for at least the next 12 months. We may seek additional future funding through public or private financing within this timeframe,in the future, if such funding is available and on terms acceptable to us. However, there are no assurances that additional financing will be available on favorable terms, or at all.
Off-balance Sheet Arrangements
As of September 30, 2017, we did not have any relationships with unconsolidated entities or financial partnerships, such as entities often referred to as structured finance or special purpose entities, which would have been established for the purpose of facilitating off-balance sheet arrangements or other contractually narrow or limited purposes. In addition, we do not engage in trading activities involving non-exchange traded contracts. Therefore, we are not materially exposed to financing, liquidity, market or credit risk that could arise if we had engaged in these relationships. We do not have relationships or transactions with persons or entities that derive benefits from their non-independent relationship with us or our related parties.
Item 3. Quantitative and Qualitative Disclosures About Market Risk
The primary objectivePer Item 305(e) of our cash investment activitiesRegulation S-K, the information called for by this Item 3 is to preserve principal while at the same time maximizing the income we receive from our investments without significantly increasing risk. Some of the securities that we invest in may be subject to market risk. This means that a change in prevailing interest rates may cause the principal amount of the investment to fluctuate. For example, if we hold a security that was issued with a fixed interest rate at the then-prevailing rate and the interest rate later rises, we expect the fair value of our investment will decline. A hypothetical 50 basis point increase in interest rates would not affect the fair value of securities at September 30, 2017. To minimize risk, we intend to maintain our portfolio of cash equivalents and marketable securities in a variety of securities, including commercial paper, government and non-government debt securities and/or money market funds that invest in such securities. We are not aware of holdings of derivative financial or commodity instruments.required.
As of September 30, 2017, our investments consisted of money market accounts with variable market rates of interest. We believe our credit risk is immaterial. We measure our cash equivalents and marketable securities at fair value on a recurring basis and have significant observable inputs where there are identical or comparable assets in the market to use in establishing our fair value measurements. We use significant observable inputs that include but are not limited to benchmark yields, reported trades, broker/dealer quotes and issuer spreads. We consider these inputs to be Level 2 inputs. Generally, the types of instruments we invest in are not traded on a market such as the NASDAQ Global Market, which we would consider to be Level 1 inputs. We do not have any investments that would require inputs considered to be Level 3. We use the bid price to establish fair value where a bid price is available.
Item 4. Controls and Procedures
Evaluation of disclosure controls and procedures. Our management, evaluated, with the participation of our Chief Executive Officer and our Chief Financial Officer, evaluated the effectiveness of our disclosure controls and procedures as of the end of the period covered by this Quarterly Report on Form 10-Q. Based on this evaluation, our Chief Executive Officer (as PrinciplePrincipal Executive Officer) and our Chief Financial Officer and(as Principal Financial Officer)has have concluded that our disclosure controls and procedures are effective to ensure that information we are required to disclose in reports that we file or submit under the Securities Exchange Act of 1934, as amended (the “Exchange Act”), is recorded, processed, summarized and reported within the time periods specified in the Securities and Exchange Commission, or SEC,SEC’s rules and forms and that such information is accumulated and communicated to management as appropriate to allow timely decisions regarding required disclosures.
Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives, and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on the evaluation of our disclosure controls and procedures as of June 30, 2021, our Chief Executive Officer and Chief Financial Officer concluded that, as of such date, our disclosure controls and procedures were effective at the reasonable assurance level.
Changes in internal control over financial reporting. As previously announced,on February 14, 2017, Peter S. Roddy resigned, effective March 9, 2017, as Vice President, Chief Financial Officer and Secretary. Remi Barbier,There has been no change in our internal control over financial reporting (as defined in Rule 13a-15(f) under the Exchange Act) identified during the three months ended June 30, 2021 that has material affected, or is reasonable likely to materially affect, our internal control over financial reporting.
President and Chief Executive Officer, has assumed the role of Principal Financial Officer until such time as a new Chief Financial Officer is appointed.
PART II – OTHER INFORMATION
Item 1. Legal Proceedings
None.
Item 1A. Risk Factors
Our future operating resultsThere have been no material changes to our risk factors from those disclosed under “Risk Factors” in Part I, Item 1A of our 2020 Annual Report on Form 10-K. The risks and uncertainties described in our 2020 Annual Report on Form 10-K are not the only ones we face. Additional risks and uncertainties not presently known to us or that we currently deem immaterial may vary substantially from anticipated results due to a number of factors, many of which are beyond our control. The following discussion highlights some of these factors and the possible impact of these factors on future results of operations. You should carefully consider these factors before making an investment decision. If any of the following factors actually occur,also materially adversely affect our business, financial condition or results of operations could be harmed. In that case, the price of our common stock could decline, and you could experience losses on your investment in our common stock.operations.
Clinical and Regulatory Risks
If we fail to obtain the necessary regulatory approvals, or if such approvals are limited, we and our collaborators will not be allowed to commercialize our drug candidates, and we will not generate product revenues.
Satisfaction of all regulatory requirements for commercialization of a drug candidate typically takes many years, is dependent upon the type, complexity and novelty of the drug candidate, and requires the expenditure of substantial resources for research and development. In December 2008, we received from the FDA a Complete Response Letter for the NDA for REMOXY. In this Complete Response Letter, the FDA indicated that additional non-clinical data was required to support the approval of REMOXY. However, the FDA did not request or recommend additional clinical efficacy studies prior to approval. In March 2009, King Pharmaceuticals, Inc., or King, assumed sole responsibility for the regulatory approval of REMOXY. In December 2010, King resubmitted the NDA for REMOXY. In June 2011, we and Pfizer announced that King received a Complete Response Letter from the FDA in response to King’s resubmission of the REMOXY NDA. The FDA’s Complete Response Letter raised concerns related to, among other matters, the Chemistry, Manufacturing, and Controls section of the NDA for REMOXY. Certain drug lots showed inconsistent release performance during in vitro testing. Pfizer completed work designed to address the June 2011 Complete Response Letter. On April 21, 2015, we announced that we resumed responsibility for REMOXY under the terms of a letter agreement with Pfizer. The letter agreement was entered into within the scope of the previously disclosed provisions of the Collaboration Agreement between us and Pfizer relating to the return of REMOXY.
We believe Pfizer has now transferred to us its data, materials, capital equipment and other assets related to REMOXY. Pfizer and the FDA had discussed and agreed to a regulatory plan to refile the NDA for REMOXY. The FDA agreed that we may follow this plan for the NDA for REMOXY.
In March 2016, we resubmitted to the FDA the NDA for REMOXY. In April 2016, the FDA determined that the NDA for REMOXY was sufficiently complete to permit a substantive review. On May 19, 2016, we announced that the FDA planned to hold an Advisory Committee meeting to review the NDA for REMOXY. On July 1, 2016, we announced that the FDA had determined that an Advisory Committee meeting for REMOXY was unnecessary and would not be held.
In September 2016, we received a CRL from the FDA on the resubmission of the NDA for REMOXY. The CRL informs that the NDA for REMOXY could not be approved in its present form and specifies additional actions and data that are needed for drug approval. The CRL focuses on the abuse-deterrent properties of REMOXY and proposed drug labeling.
On February 13, 2017, we met with the FDA on the resubmission of NDA for REMOXY. During this meeting, an agreement was reached on additional studies that are needed for REMOXY’s regulatory approval. Following completion of these studies, we intend to have a pre-NDA meeting with the FDA, followed by resubmission of the
REMOXY NDA, with an anticipated priority review, under Section 505(b)(2) of the Federal Food, Drug, and Cosmetic Act.
There can be no assurance that the FDA will approve an NDA for REMOXY or that the FDA will not require submission of additional clinical or non-clinical data. Obtaining data from such studies (even if completed) that is insufficient to support approval of REMOXY, or any adverse decisions by the FDA (including any decision by the FDA to require additional clinical or non-clinical data) may significantly delay or prevent the potential approval of REMOXY.
Our research and clinical approaches may not lead to drugs that the FDA considers safe for humans and effective for indicated uses we are studying. The FDA may require additional studies, in which case we or our collaborators would have to expend additional time and resources and would likely delay the date of potentially receiving regulatory approval. The approval process may also be delayed by changes in government regulation, future legislation or administrative action or changes in FDA policy that occur prior to or during our regulatory review. Delays in obtaining regulatory approvals would:
| ·
| | delay commercialization of, and product revenues from, our drug candidates; and
|
| ·
| | diminish the competitive advantages that we may have otherwise enjoyed, which would have an adverse effect on our operating results and financial condition.
|
Even if we or our collaborators comply with all FDA regulatory requirements, our drug candidates may never obtain regulatory approval. If we or our collaborators fail to obtain regulatory approval for any of our drug candidates we will have fewer commercial products, if any, and corresponding lower product revenues, if any. Even if our drug candidates receive regulatory approval, such approval may involve limitations on the indications and conditions of use or marketing claims for our products. Further, later discovery of previously unknown problems or adverse events could result in additional regulatory restrictions, including withdrawal of products. The FDA may also require us or our collaborators to commit to perform lengthy Phase IV post-approval clinical efficacy or safety studies. Our expending additional resources on such trials would have an adverse effect on our operating results and financial condition.
In jurisdictions outside the United States, we must receive marketing authorizations from the appropriate regulatory authorities before commercializing our drugs. Regulatory approval processes outside the United States generally include all of the aforementioned requirements and risks associated with FDA approval.
If we are unable to design, conduct and complete preclinical and clinical trials successfully, our drug candidates will not be able to receive regulatory approval.
In order to obtain FDA approval for any of our drug candidates, we must submit to the FDA an NDA that demonstrates with substantive evidence that the drug candidate is both safe and effective in humans for its intended use. This demonstration requires significant research and animal tests, which are referred to as preclinical studies, as well as human tests, which are referred to as clinical trials.
Preclinical studies may not provide results we believe are sufficient to support the filing of an IND. Success in early preclinical studies does not ensure success in later preclinical or clinical studies. The FDA may disagree with the design of our preclinical studies or our interpretations of data from preclinical studies. The FDA may not accept an IND for our product candidate and may require additional preclinical studies to support the filing of an IND.
Success in preclinical studies and early clinical trials does not ensure that later clinical trials will be successful. Results from Phase I clinical programs may not support moving a drug candidate to Phase II or Phase III clinical trials. Phase III clinical trials may not demonstrate the safety or efficacy of our drug candidates. Results of later clinical trials may not replicate the results of prior clinical trials and preclinical studies. Even if the results of Phase III clinical trials are positive, we or our collaborators may have to commit substantial time and additional resources to conducting further preclinical studies and clinical trials before obtaining FDA approval for any of our drug candidates.
Clinical trials are very expensive and difficult to design and implement, in part because they are subject to rigorous requirements. The clinical trial process also consumes a significant amount of time. Furthermore, if patients in clinical trials suffer drug-related adverse reactions during the course of such clinical trials, or if we, our collaborators or the FDA believe that participating patients are being exposed to unacceptable health risks, such clinical trials will have to be suspended or terminated. Failure can occur at any stage of the clinical trials, and we or our collaborators could encounter problems that cause abandonment or repetition of clinical trials.
Clinical trials with REMOXY and our potential future clinical trials for other drug candidates for treatment of pain measure clinical symptoms, such as pain and physical dependence, that are not biologically measurable. The success in these clinical trials depends on reaching statistically significant changes in patients’ symptoms based on clinician-rated scales. Due in part to a lack of consensus on standardized processes for assessing clinical outcomes, these scores may or may not be reliable, useful or acceptable to regulatory agencies.
In addition, completion of clinical trials can be delayed by numerous factors, including:
| ·
| | delays in identifying and agreeing on acceptable terms with prospective clinical trial sites;
|
| ·
| | slower than expected rates of patient recruitment and enrollment;
|
| ·
| | unanticipated patient dropout rates;
|
| ·
| | increases in time required to complete monitoring of patients during or after participation in a clinical trial; and
|
| ·
| | unexpected need for additional patient-related data.
|
Any of these delays could significantly impact the timing, approval and commercialization of our drug candidates and could significantly increase our overall costs of drug development.
Even if clinical trials are completed as planned, their results may not support expectations or intended marketing claims. The clinical trials process may fail to demonstrate that our drug candidates are safe and effective for indicated uses. Such failure would cause us to abandon a drug candidate and could delay development of other drug candidates.
Clinical trial designs that were discussed with regulatory authorities prior to their commencement may subsequently be considered insufficient for approval at the time of application for regulatory approval.
We discuss with and obtain guidance from regulatory authorities on certain of our clinical development activities. With the exception of our SPA, these discussions are not binding obligations on the part of regulatory authorities.
Regulatory authorities may revise previous guidance or decide to ignore previous guidance at any time during the course of our clinical activities or after the completion of our clinical trials. Even with successful clinical safety and efficacy data, including such data from a clinical trial conducted pursuant to an SPA, we or our collaborators may be required to conduct additional, expensive clinical trials to obtain regulatory approval.
Developments by competitors may establish standards of care that affect our ability to conduct our clinical trials as planned.
We have conducted clinical trials of our drug candidates comparing our drug candidates to both placebo and other approved drugs. Changes in standards related to clinical trial design could affect our ability to design and conduct clinical trials as planned. For example, regulatory authorities may not allow us to compare our drug candidates to placebo in a particular clinical indication where approved products are available. In that case, both the cost and the amount of time required to conduct a clinical trial could increase.
The U.S. Drug Enforcement Agency, or DEA, limits the availability of the active ingredients in certain of our current drug candidates and, as a result, quotas for these ingredients may not be sufficient to complete clinical trials, or to meet commercial demand, or may result in clinical delays.
The DEA regulates chemical compounds as Schedule I, II, III, IV or V substances, with Schedule I substances considered to present the highest risk of substance abuse and Schedule V substances the lowest risk. Certain active ingredients in our current drug candidates, such as oxycodone, are listed by the DEA as Schedule II under the Controlled Substances Act of 1970. Consequently, their manufacture, research, shipment, storage, sale and use are subject to a high degree of oversight and regulation. For example, all Schedule II drug prescriptions must be signed by a physician, physically presented to a pharmacist and may not be refilled without a new prescription. Furthermore, the amount of Schedule II substances that can be obtained for clinical trials and commercial distribution is limited by the DEA and quotas for these substances may not be sufficient to complete clinical trials or meet commercial demand. There is a risk that DEA regulations may interfere with the supply of the drugs used in clinical trials for our product candidates, and, in the future, the ability to produce and distribute our products in the volume needed to meet commercial demand.
Conducting clinical trials of our drug candidates or potential commercial sales of a drug candidate may expose us to expensive product liability claims, and we may not be able to maintain product liability insurance on reasonable terms or at all.
The risk of product liability is inherent in the testing of pharmaceutical products. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit or terminate testing of one or more of our drug candidates. Our inability to obtain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization of our drug candidates. We currently carry clinical trial insurance but do not carry product liability insurance. If we successfully commercialize one or more of our drug candidates, we may face product liability claims, regardless of FDA approval for commercial manufacturing and sale. We may not be able to obtain such insurance at a reasonable cost, if at all. Even if our agreements with any current or future corporate collaborators entitle us to indemnification against product liability losses, such indemnification may not be available or adequate should any claim arise.
If our drug candidates receive regulatory approval, we and our collaborators will be subject to ongoing FDA obligations and continued regulatory review, such as continued safety reporting requirements, and we and our collaborators may also be subject to additional FDA post-marketing obligations or new regulations, all of which may result in significant expense and limit our and our collaborators’ ability to commercialize our potential drugs.
Any regulatory approvals that our drug candidates receive may also be subject to limitations on the indicated uses for which the drug may be marketed or contain requirements for potentially costly post-marketing follow-up studies. In addition, if the FDA approves any of our drug candidates, the labeling, packaging, adverse event reporting, storage, advertising, promotion and record keeping for the drug will be subject to extensive regulatory requirements. The subsequent discovery of previously unknown problems with the drug, including but not limited to adverse events of unanticipated severity or frequency, or the discovery that adverse events previously observed in preclinical research or clinical trials that were believed to be minor actually constitute much more serious problems, may result in restrictions on the marketing of the drug, and could include withdrawal of the drug from the market.
The FDA’s policies may change and additional government regulations may be enacted that could prevent or delay regulatory approval of our drug candidates. We cannot predict the likelihood, nature or extent of adverse government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are not able to maintain regulatory compliance, we may be subject to fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution. Any of these events could prevent us from marketing our drugs and our business could suffer.
We may not be able to successfully develop or commercialize FENROCK, a proprietary abuse-deterrent transdermal pain patch (fentanyl), designed to prevent common methods of abuse of fentanyl.
We have no history of developing transdermal patches. We do not know whether any of our planned development activities for FENROCK will result in approval of such drug candidate by the FDA, or, if FENROCK is approved, it will be a commercially viable product.
Risks Relating to our Collaboration Agreements
If Pfizer did not transfer to us all data and documentation or the quality of the data and documentation transferred is insufficient, our ability to achieve approval of the NDA for REMOXY will be negatively impacted and our business will suffer.
In April 2015, we announced that we resumed responsibility for REMOXY under the terms of a letter agreement with Pfizer. The letter agreement was entered into within the scope of the previously disclosed provisions of the Collaboration Agreement between us and Pfizer relating to the return of REMOXY.
We believe Pfizer has transferred to us data, materials, capital equipment and other assets related to REMOXY. In preparing to resubmit the NDA for REMOXY, we may find that there are additional data, materials or agreements that Pfizer should have transferred to us. If Pfizer did not meet its obligations to transfer all such materials or if the quality of the data and documentation transferred is insufficient, we would be significantly delayed in our ability to achieve FDA approval of the NDA for REMOXY, and may need to conduct further development activities or clinical
trials to prepare any potential resubmission. As a result, any further development, regulatory approval and product introduction for REMOXY would be delayed or prevented and our business would suffer.
If outside collaborators fail to devote sufficient time and resources to drug development programs related to our product candidates, or if their performance is substandard, regulatory submissions and introductions for our products may be delayed.
We rely on Durect as the sole-source provider of certain components of REMOXY. Durect’s failure for any reason to provide these components could result in delays or failures in product testing or delivery, cost overruns or other problems that could materially harm our business.
We depend on independent investigators and collaborators, such as universities and medical institutions, to conduct our clinical trials under agreements with us. These investigators and collaborators are not our employees, and we cannot control the amount or timing of resources that they devote to our programs. They may not assign as great a priority to our programs or pursue them as diligently as we would if we were undertaking such activities ourselves. If these investigators or collaborators fail to devote sufficient time and resources to our drug development programs, or if their performance is substandard, the approval of our regulatory submissions and our introductions of new drugs will be delayed or prevented.
Our collaborators may also have relationships with other commercial entities, some of which may compete with us. If outside collaborators assist our competitors to our detriment, the approval of our regulatory submissions will be delayed and the sales from our products, if any are commercialized, will be less than expected.
If we fail to enter into or maintain collaboration agreements and licenses for REMOXY and other drugs designed to reduce potential risks of unintended use, we may have to reduce or delay our drug candidate development.
Our plan for developing, manufacturing and commercializing REMOXY currently requires us to successfully maintain our license from Durect. If we are unable to meet the obligations necessary to maintain our license with Durect for one or more potential products we may lose the rights to utilize Durect’s technology for such potential products, our potential future revenues may suffer and we may have to reduce or delay development of our other drug candidates. In addition, we expect to seek a new corporate collaborator with respect to REMOXY. If we do not enter into a new collaboration with respect to the continued development and potential commercialization of REMOXY, we will be required to undertake and fund such activities ourselves and may need to seek additional capital (which may not be available on acceptable terms, if at all), personnel or other resources. If we are not successful in such efforts, development and commercialization of REMOXY and our other drug candidates would be delayed or prevented, and our business would suffer.
We may not succeed at in-licensing drug candidates or technologies to expand our product pipeline.
We may not successfully in-license drug candidates or technologies to expand our product pipeline. The number of such candidates and technologies is limited. Competition among large pharmaceutical companies and biopharmaceutical companies for promising drug candidates and technologies is intense because such companies generally desire to expand their product pipelines through in-licensing. If we fail to carry out such in-licensing and expand our product pipeline, our potential future revenues may suffer.
Our collaborative agreements may not succeed or may give rise to disputes over intellectual property, disputes concerning the scope of collaboration activities or other issues.
Our collaborative agreements with third parties, such as our license agreement with Durect, are generally complex and contain provisions that could give rise to legal disputes, including potential disputes concerning ownership of intellectual property under collaborations or disputes concerning the scope of collaboration activities. Such disputes can delay or prevent the development of potential new drug products, or can lead to lengthy, expensive litigation or arbitration. Other factors relating to collaborative agreements may adversely affect our business, including:
| ·
| | the development of parallel products by our collaborators or by a competitor;
|
| ·
| | arrangements with collaborative partners that limit or preclude us from developing certain products or technologies;
|
| ·
| | premature termination of a collaborative or license agreement; or
|
| ·
| | failure by a collaborative partner to provide required funding, to devote sufficient resources to the development of or legal defense of our potential products or to provide data or other information to us as required by our collaborative agreements.
|
Risks Relating to Commercialization
We currently have no in-house capabilities to commercialize our drug products and if we are unable to develop our own sales, marketing and distribution capabilities, or if we are not successful in contracting with third parties for these services on favorable terms, or at all, our product revenues could be disappointing.
We currently have no sales, marketing or distribution capabilities. We have not established commercial strategies regarding any of our product candidates, including REMOXY. In order to commercialize our products, if any approved by the FDA, we will either have to develop such capabilities internally or collaborate with third parties who can perform these services for us.
If we decide to commercialize any of our drugs ourselves, we may not be able to
| ·
| | hire and retain the necessary experienced personnel;
|
| ·
| | build sales, marketing and distribution operations in a cost effective manner which are capable of successfully launching new drugs;
|
| ·
| | obtain access to adequate numbers of physicians to prescribe our products; or
|
| ·
| | generate sufficient product revenues.
|
In addition, establishing such operations on our own will take time and involve significant expense. If our commercial operations lack complementary products, we may not be able to compete in a cost effective manner with competitors with more products to sell. If we engage third-party collaborators to perform any commercial operations, our future revenues may depend significantly upon the performance of those collaborators.
If we decide to enter into new co-promotion or other licensing arrangements with third parties, we may be unable to locate acceptable collaborators because the number of potential collaborators is limited and because of competition from others for similar alliances with potential collaborators. Even if we are able to identify one or more acceptable new collaborators, we may not be able to enter into any collaborative arrangements on favorable terms, or at all.
In addition, due to the nature of the market for our drug candidates, it may be necessary for us to license all or substantially all of our drug candidates to a single collaborator, thereby eliminating our opportunity to commercialize these other products independently. If we enter into any such new collaborative arrangements, our revenues are likely to be lower than if we marketed and sold our products ourselves.
In addition, any revenues we receive would depend upon our collaborators’ efforts which may not be adequate due to lack of attention or resource commitments, management turnover, change of strategic focus, business combinations or other factors outside of our control. Depending upon the terms of our collaboration, the remedies we have against an under-performing collaborator may be limited. If we were to terminate the relationship, it may be difficult or impossible to find a replacement collaborator on acceptable terms, or at all.
If physicians and patients do not accept and use our drugs, we will not achieve sufficient product revenues and our business will suffer.
Even if the FDA approves our drugs, physicians and patients may not accept and use them. Acceptance and use of our drugs will depend on a number of factors including:
| ·
| | when the drug is launched into the market and related competition;
|
| ·
| | perceptions by members of the healthcare community, including physicians, about the safety and effectiveness of our drugs, and, in particular, the effectiveness of REMOXY in reducing potential risks of unintended use;
|
| ·
| | perceptions by physicians regarding the cost benefit of REMOXY in reducing potential risks of unintended use;
|
| ·
| | published studies demonstrating the cost-effectiveness of our drugs relative to competing products;
|
| ·
| | availability of reimbursement for our products from government or healthcare payers;
|
| ·
| | our or our collaborators’ ability to implement a risk management plan prior to the distribution of any Schedule II drug; and
|
| ·
| | effectiveness of marketing and distribution efforts by us and other licensees and distributors.
|
Because we expect to rely on sales generated by our current lead drug candidates for substantially all of our revenues for the foreseeable future, the failure of any of these drugs to find market acceptance would harm our business and could require us to seek additional financing.
The science of abuse-deterrence is relatively new.
The analytical, clinical, and statistical methods for evaluating abuse-deterrent technologies and study results are new and rapidly evolving. Although we believe the FDA will take a flexible, adaptive approach to the evaluation and labeling of potentially abuse-deterrent products, such as REMOXY, we cannot be certain that our interpretation of abuse-deterrent data for REMOXY is consistent with the views of the FDA. In our opinion, the FDA has limited data correlating the potentially abuse-deterrent properties of certain opioid drug products, such as REMOXY, with actual reduction in abuse or adverse events associated with abuse. In addition, the FDA has stated it is not able to provide specific guidance on the magnitude of effect that would be sufficient to support any particular type of label claim for abuse-deterrence.
In the United States, our ability to market and promote REMOXY and its abuse-deterrent features will be determined by FDA-approved labeling.
The commercial success of REMOXY and certain of our other product candidates will depend upon our ability to obtain FDA-approved labeling describing their abuse-deterrent features. Our failure to achieve FDA approval of product labeling containing such information will prevent us from advertising and promoting the abuse-deterrent features of our product candidates in order to differentiate them from other similar products. This would make our products less competitive in the market.
Abuse-deterrent label claims for our products may not be broad enough to demonstrate a substantial benefit to health care providers and patients.
FDA approval is required in order to make claims that a product has an abuse-deterrent effect. In April 2015, the FDA published final guidance with regard to the evaluation and labeling of abuse-deterrent opioids. The guidance provides direction as to the studies and data required for obtaining abuse-deterrent claims in a product label. FDA guidance describes three categories of pre-market studies that may lead to an abuse-deterrent claim:
Category 1 – laboratory manipulation and extraction studies;
Category 2 – pharmacokinetic studies; and
Category 3 – human abuse potential studies.
According to FDA guidance, label claims for abuse-deterrence should describe the product’s specific abuse-deterrent properties as well as the specific routes of abuse that the product has been developed to deter. When data predict or show that a product’s potentially abuse-deterrent properties can be expected to, or actually do, result in a significant reduction in that product’s abuse potential, these data, together with an accurate characterization of what the data mean, may be included in product labeling.
If a product is approved by the FDA to include such claims in its label, the applicant may use the approved labeling information about the abuse-deterrent features of the product in its marketing efforts to physicians.
Although we intend to provide data to the FDA to support approval of abuse-deterrence label claims for REMOXY, there can be no assurance that REMOXY or any of our other product candidates will receive FDA-approved labeling that describes the abuse-deterrent features of such products. The FDA may find that our studies and data do not support abuse-deterrent labeling or that our product candidates do not provide substantial abuse-deterrence because, for example, their deterrence mechanisms do not address the way they are most likely to be abused. Further, the FDA is not required to follow its guidance and could change this guidance, which could require us to conduct additional studies or generate additional data. If the FDA does not approve abuse-deterrent labeling, we will not be able to promote such products based on their abuse-deterrent features and our business may suffer.
Even if we do receive FDA approval for abuse-deterrent claims, the claims may not be broad enough to demonstrate a substantial benefit to health care providers and patients. For instance, the claims may not encompass the more common forms of abuse for products like our product candidates. Moreover, continued investigation in Phase IV studies following product approval, if required, is expensive and may not support the continued use of abuse-deterrent claims.
If we cannot compete successfully for market share against other drug companies, we may not achieve sufficient product revenues and our business will suffer.
The market for our drug candidates is characterized by intense competition and rapid technological advances. If our drug candidates receive FDA approval, they will compete with a number of existing and future drugs and therapies developed, manufactured and marketed by others. Existing or future competing products may provide greater therapeutic convenience or clinical or other benefits for a specific indication than our products, or may offer comparable performance at a lower cost. If our products are unable to capture and maintain market share, we may not achieve sufficient product revenues and our business will suffer.
We and our collaborators will compete for market share against fully integrated pharmaceutical companies or other companies that are collaborating with larger pharmaceutical companies, academic institutions, government agencies and other public and private research organizations. Many of these competitors have drugs already approved or drug candidates in development that will or may compete against our approved drug candidates. In addition, many of these competitors, either alone or together with their collaborative partners, operate larger research and development programs and have substantially greater financial resources than we do, as well as significantly greater experience in:
| ·
| | conducting preclinical testing and human clinical trials;
|
| ·
| | obtaining FDA and other regulatory approvals of drugs;
|
| ·
| | formulating and manufacturing drugs; and
|
| ·
| | launching, marketing, distributing and selling drugs.
|
If we fail to obtain acceptable prices or an adequate level of reimbursement for our products from healthcare payers, our ability to generate product revenues will be diminished.
Our ability to commercialize drugs we (alone or with other collaborators) may develop will depend in part on the extent to which reimbursement can be obtained for such drugs from:
| ·
| | government and health administration authorities;
|
| ·
| | private health maintenance organizations and health insurers; and
|
| ·
| | other healthcare payers.
|
Significant uncertainty exists as to the reimbursement status of newly approved healthcare products. Healthcare payers, including Medicare, health maintenance organizations and managed care organizations, are challenging the prices charged for medical products and services and/or are seeking pharmacoeconomic data to justify formulary acceptance and reimbursement practices. We currently have not generated pharmacoeconomic data on any of our drug candidates. Government and other healthcare payers increasingly are attempting to contain healthcare costs by limiting both coverage and the level of reimbursement for drugs, and by refusing, in some cases, to provide coverage for uses of approved products for disease indications for which the FDA has or has not granted labeling approval. Adequate third-party insurance coverage may not be available to patients for any products we discover and develop, alone or with collaborators. If government and other healthcare payers do not provide adequate coverage and reimbursement levels for our products, market acceptance of our drug candidates could be limited.
Enacted and future legislation may increase the difficulty and cost for us to commercialize our product candidates and may reduce the prices we are able to obtain for our product candidates.
Legislative and regulatory changes and future changes regarding the healthcare system could prevent or delay marketing approval of our product candidates, restrict or regulate post-approval activities or affect our ability to profitably sell any product candidates for which we obtain marketing approval.
In the United States, the Medicare Prescription Drug, Improvement, and Modernization Act of 2003, or the Medicare Modernization Act, established the Medicare Part D program and provided authority for limiting the number of drugs that will be covered in any therapeutic class thereunder. The Medicare Modernization Act, including its cost reduction initiatives, could limit the coverage and reimbursement rate that we receive for any of our approved products. Private payors may follow Medicare coverage policies and payment limitations in setting their own reimbursement rates resulting in similar limits in payments from private payors.
The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or collectively, the Affordable Care Act, among other things, imposes a significant annual fee on companies that manufacture or import branded prescription drug products. It also contains substantial provisions intended to, among other things, broaden access to health insurance, reduce or constrain the growth of health care spending, enhance remedies against healthcare fraud and abuse, add new transparency requirements for the healthcare and health insurance industries, and impose additional health policy reforms, any of which could have a material adverse effect on our business. A significant number of provisions are not yet, or have only recently become, effective, but the Affordable Care Act may result in downward pressure on pharmaceutical pricing, especially under the Medicare program, and may also increase our regulatory burdens and operating costs.
The Affordable Care Act, as well as other healthcare reform measures that have been and may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we receive for any approved product, and could seriously harm our future revenues. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may compromise our ability to generate revenue, attain profitability or commercialize our products.
The Affordable Care Act is a highly complex piece of legislation that continues to evolve. We do not and cannot understand or anticipate the full impact and potential implications of the Affordable Care Act on our business or on our drugs.
Even if we are able to commercialize any of our product candidates, our products may become subject to unfavorable pricing regulations or third-party coverage and reimbursement policies, which could have a material adverse effect on our business.
Our ability to commercialize any products successfully will depend in part on the extent to which coverage and adequate reimbursement for these products and related treatments will be available from government health administration authorities, private health insurers and other organizations. Government authorities and third-party payors, such as private health insurers and health maintenance organizations, determine which medications they will cover and establish reimbursement levels. A primary trend in the U.S. healthcare industry and elsewhere is cost containment. Government authorities and other third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medications. Increasingly, third-party payors are requiring that drug companies provide them with predetermined discounts from list prices and are challenging the prices charged for medical products. We cannot be sure that coverage and reimbursement will be available for any product that we commercialize and, if reimbursement is available, what the level of reimbursement will be and whether it will be satisfactory. Coverage and reimbursement may impact the demand for, or the price of, any product candidate for which we obtain marketing approval. If coverage and reimbursement are not available or reimbursement is available only to limited levels, we may not be able to successfully commercialize any product candidate for which we obtain marketing approval.
There may be significant delays in obtaining coverage and reimbursement for newly approved drugs, and coverage may be more limited than the purposes for which the drug is approved by the FDA or foreign regulatory authorities. Moreover, eligibility for coverage and reimbursement does not imply that any drug will be paid for in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution. Interim reimbursement levels for new drugs, if applicable, may also be insufficient to cover our costs and may only be temporary. Reimbursement rates may vary according to the use of the drug and the clinical setting in which it is used, may be based on reimbursement levels already set for lower cost drugs and may be incorporated into existing payments for other services. Net prices for drugs may be reduced by mandatory discounts or rebates required by government healthcare programs or private payors and by any future relaxation of laws that presently restrict imports of drugs from countries where they may be sold at lower prices than in the United States. Third-party payors often rely upon Medicare coverage policy and payment limitations in setting their own reimbursement policies. Our inability to promptly obtain coverage and profitable reimbursement rates from both government-funded and private payors for any approved
products that we develop could have a material adverse effect on our operating results, our ability to raise capital needed to commercialize products and our overall financial condition.
Public concern over the abuse of opioids, including law enforcement concerns over diversion of opioid and regulatory efforts to combat abuse, could decrease the potential market for our product candidates.
Media stories regarding prescription drug abuse and the diversion of opioids and other controlled substances are commonplace. Law enforcement and regulatory agencies may apply policies that seek to limit the availability of opioids. Such efforts may inhibit our ability to commercialize our product candidates. Aggressive enforcement and unfavorable publicity regarding, for example, the use or misuse of oxycodone or other opioid drugs; the limitations of ADFs; the ability of drug abusers to discover previously unknown ways to abuse our products; public inquiries and investigations into prescription drug abuse; litigation; or regulatory activity regarding sales, marketing, distribution or storage of opioid drugs could have a material adverse effect on our reputation. Such negative publicity could reduce the potential size of the market for our product candidates and decrease the revenues we are able to generate from their sale. To the extent opioid abuse becomes less prevalent or less urgent of a public health issue, regulators and third party payers may not be willing to pay a premium for ADFs of opioids.
Efforts by the FDA and other regulatory bodies to combat abuse of opioids may negatively impact the market for our product candidates. For example, on September 10, 2013, the FDA announced its intention to effect labeling changes to all approved long-acting opioid formulations. In particular, the FDA announced its intention to update the indication for long-acting opioid formulations so that long-acting opioid formulations will be indicated only for the management of pain severe enough to require daily, around-the-clock, long-term opioid treatment and for which alternative treatment options are inadequate. On April 16, 2014, the FDA updated these indications. It is possible that such changes could reduce the number of prescriptions for opioids written by physicians and negatively impact the potential market for our product candidates.
If the FDA or other applicable regulatory authorities approve generic products with abuse-deterrent claims that compete with any of our product candidates, it could reduce our sales of those product candidates.
Once an NDA, including a Section 505(b)(2) application, is approved, the product covered thereby becomes a "listed drug" which can, in turn, be cited by potential competitors in support of approval of an abbreviated NDA, or ANDA. Potential competitors may create modified, non-infringing versions of a drug to facilitate the approval of an ANDA or other application for generic substitutes. These competitors might only be required to conduct a relatively inexpensive study to show that their product has the same active ingredients, dosage form, strength, route of administration, and conditions of use, or labeling, as our products and that the generic product is absorbed in the body at the same rate and to the same extent as, or is bioequivalent to, our products. These generic equivalents would be significantly less costly than ours to bring to market and companies that produce generic equivalents are generally able to offer their products at lower prices. Thus, after the introduction of a generic competitor, a significant percentage of the sales of any branded product are typically lost to the generic product. Accordingly, competition from generic equivalents to our products would substantially limit our ability to generate revenues and therefore to obtain a return on the investments we have made in our product candidates.
Our relationships with customers and payors will be subject to applicable anti-kickback, fraud and abuse, transparency, and other healthcare laws and regulations, which could expose us to criminal sanctions, civil penalties, exclusion from government healthcare programs, contractual damages, reputational harm, administrative burdens, and diminished profits and future earnings.
Healthcare providers, physicians and payors play a primary role in the recommendation and prescription of any product candidates for which we may obtain marketing approval. Our future arrangements with payors and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we market, sell and distribute any product candidates for which we may obtain marketing approval. Even though we do not and will not control referrals of healthcare services or bill directly to Medicare, Medicaid or other third-party payors, federal and state healthcare laws and regulations pertaining to fraud and abuse and patients' rights are and will be applicable to our business. Restrictions under applicable federal, state and foreign healthcare laws and regulations may affect our ability to operate and expose us to areas of risk, including:
| ·
| | the federal Anti-Kickback Statute, which prohibits, among other things, knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward
|
either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made under federal and state healthcare programs such as Medicare and Medicaid. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation;
|
| ·
| | the federal False Claims Act, which imposes criminal and civil penalties, including through civil whistleblower or qui tam actions, against individuals or entities for knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government. In addition, the government may assert that a claim including items and services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act;
|
| ·
| | the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which imposes criminal and civil liability for executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute to defraud any healthcare benefit program or specific intent to violate it in order to have committed a violation;
|
| ·
| | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, or HITECH, and its implementing regulations, which also imposes obligations on certain covered entity healthcare providers, health plans, and healthcare clearinghouses as well as their business associates that perform certain services involving the use or disclosure of individually identifiable health information, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information;
|
| ·
| | federal laws requiring drug manufacturers to report information related to payments and other transfers of value made to physicians and other healthcare providers, as well as ownership or investment interests held by physicians and their immediate family members, including under the federal Open Payments program, commonly known as the Sunshine Act, as well as other state and foreign laws regulating marketing activities; and
|
| ·
| | state and foreign equivalents of each of the above laws, including state anti-kickback and false claims laws, which may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental payors, including private insurers; state laws which require pharmaceutical companies to comply with the pharmaceutical industry's voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government or otherwise restricting payments that may be made to healthcare providers; and state and foreign laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts.
|
Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations will involve substantial costs. Nonetheless, it is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, imprisonment, exclusion from participation in government funded healthcare programs, such as Medicare and Medicaid, and the curtailment or restructuring of our operations.
Government agencies may establish and promulgate usage guidelines that could limit the use of our drug candidates.
Government agencies, professional and medical societies, and other groups may establish usage guidelines that apply to our drug candidates. These guidelines could address such matters as usage and dose, among other factors. Application of such guidelines could limit the clinical use or commercial appeal of our drug candidates.
Risks Relating to our Intellectual Property
Our ability to commercialize our drug candidates will depend on our ability to sell such products without infringing the patent or proprietary rights of third parties. If we are sued for infringing the intellectual property rights of third parties, such litigation will be costly and time consuming and an unfavorable outcome would have a significant adverse effect on our business.
Our ability to commercialize our drug candidates will depend on our ability to sell such products without infringing the patents or other proprietary rights of third parties. Intellectual property rights in the areas of controlled-release technology and pharmaceutical ingredients are complicated and are continuously evolving. Holders of patent rights in these areas may allege that the commercialization of REMOXY or our other drug candidates infringes such patent rights. While we believe that we would have valid defenses to any claim of infringement, there can be no assurance that these or other third-party patents will not limit our ability to commercialize REMOXY or our other drug candidates.
In addition, because patent applications are published some time after filing, and because applications can take several years to issue, there may be currently pending third-party patent applications that are unknown to us, which may later result in issued patents. If a third-party claims that we infringe on its patents or other proprietary rights, we could face a number of issues that could seriously harm our competitive position, including:
| ·
| | infringement claims that, with or without merit, can be costly and time consuming to litigate, can delay the regulatory approval process and can divert management’s attention from our core business strategy;
|
| ·
| | substantial damages for past infringement which we may have to pay if a court determines that our products or technologies infringe upon a competitor’s patent or other proprietary rights;
|
| ·
| | a court order prohibiting us from commercializing our products or technologies unless the holder licenses the patent or other proprietary rights to us, which such holder is not required to do;
|
| ·
| | if a license is available from a holder, we may have to pay substantial royalties or grant cross licenses to our patents or other proprietary rights; and
|
| ·
| | redesigning our process so that it does not infringe the third-party intellectual property rights, which may not be possible, or which may require substantial time and expense including delays in bringing our own products to market. Such actions could harm our competitive position and our ability to generate revenue and could result in increased costs.
|
If we are unable to protect our intellectual property, our competitors could develop and market products with similar features that may reduce demand for our drug candidates.
Our success, competitive position and potential future revenues will depend in part on our ability to protect our intellectual property. If we or our collaborators fail to file, prosecute, obtain or maintain certain patents, our competitors could market products that contain features and clinical benefits similar to those of our products, and demand for our products could decline as a result.
We and our collaborators have filed patent applications in the United States and select international jurisdictions to protect our intellectual property. The coverage sought in a patent application can be denied or significantly reduced before or after the patent is issued. Consequently, we do not know whether any of our pending applications will result in the issuance of patents, or if any existing or future patents will provide significant protection or commercial advantage or will be circumvented by others. There can be no assurance that patents will issue from our pending or future patent applications or, if issued, that such patents will be of commercial benefit to us, afford us adequate protection from competing products, or not be challenged or declared invalid. Thus, if these patent applications do not result in issued patents or result in a patent that is challenged by others, the duration or scope of our patent rights may be limited and our future revenues could be lower as a result.
We may be involved in challenges to our intellectual property. If our competitors are able to successfully challenge the validity or scope of our patent rights, based on the existence of prior art or otherwise, they might be able to market products that contain features and clinical benefits similar to those of our drug candidates, and demand for our drug candidates could decline as a result. An adverse outcome of a challenge to our intellectual property could result in loss of claims of patents or other intellectual property rights that pertain to certain drugs we currently have under development and could have a material adverse impact on our future revenues.
We intend to file additional patent applications relating to our technology, products and processes. We may direct our collaborators to file additional patent applications relating to the licensed technology or we may do so ourselves. However, our competitors may challenge, invalidate or circumvent any of our current or future patents. These patents may also fail to provide us with meaningful competitive advantages.
We may become involved in expensive litigation or other legal proceedings related to our existing intellectual property rights, including patents.
We expect that we will rely upon patents, trade secrets, know-how, continuing technological innovations and licensing opportunities to develop and maintain our competitive position. While we use confidentiality agreements with our employees, consultants and certain of our contractors, if trade secrets or other confidential information is made public, our business may be harmed and our legal remedies may be limited or insufficient. Others may independently develop substantially equivalent proprietary information or be issued patents that may prevent the sale of our products or know-how or require us to license such information and pay significant fees or royalties in order to produce our products.
Our technology could infringe upon claims of patents owned by others. If we were found to be infringing on a patent held by another, we might have to seek a license to use the patented technology. In that case, we might not be able to obtain such a license on terms acceptable to us, or at all. If a legal action were to be brought against us or our licensors, we could incur substantial defense costs, and any such action might not be resolved in our favor. If such a dispute were to be resolved against us, we could have to pay the other party large sums of money and our use of our technology and the testing, manufacture, marketing or sale of one or more of our proposed products could be restricted or prohibited.
If we are unable to protect the confidentiality of our intellectual property, the value of our intellectual property could be materially adversely affected and our business would be harmed.
We seek to protect our intellectual property, in part, by confidentiality agreements with our employees, consultants, scientific advisors, contractors and collaborators. However, there can be no assurance that our intellectual property will not be disclosed or that competitors will not otherwise gain access to our intellectual property or independently develop substantially equivalent intellectual property. For example, if our confidential information were disclosed in violation of our confidentiality agreements, we may not be able to obtain adequate remedies for such breaches. We also seek to protect our intellectual property by maintaining physical security of our premises and physical and electronic security of our information technology systems, but it is possible that these security measures could be breached. If any of our intellectual property were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent such competitor from using that intellectual property to compete with us, which could harm our business.
Risks Relating to our Business and Strategy
If we are not successful in attracting and retaining qualified personnel, we could experience delays in completing necessary clinical trials, in the regulatory approval process or in formulating, manufacturing, marketing and selling our potential products.
We depend on the services of our key personnel, including Remi Barbier, our Chairman, President and Chief Executive Officer. On February 14, 2017, Peter S. Roddy resigned, effective March 9, 2017, as Vice President, Chief Financial Officer and Secretary. Remi Barbier, President and Chief Executive Officer, has assumed the role of Principal Financial Officer until such time as a new Chief Financial Officer is appointed.
The loss of key personnel, including members of executive management as well as key bioengineering, product development, and technical personnel, could disrupt our operations and have an adverse effect on our business. We will need to hire additional qualified personnel with expertise in clinical research, preclinical testing, government regulation, formulation and manufacturing and sales and marketing. We compete for qualified individuals with numerous biopharmaceutical companies, universities and other research institutions. Competition for such individuals is intense, and our search for such personnel may not be successful. Attracting and retaining qualified personnel is critical to our success.
We have employees whose equity ownership in the Company could result in a substantial increase in personal wealth if the fair value of our common stock increases. Over time, this increase in personal wealth may make it more challenging to retain these employees.
If third-party manufacturers of our drug candidates fail to devote sufficient time and resources to our concerns, or if their performance is substandard, our clinical trials and product introductions may be delayed and our costs may be higher than expected.
We have no manufacturing facilities and have limited experience in drug product development and commercial manufacturing. We lack the resources and expertise to formulate, manufacture or test the technical performance of our drug candidates. We rely on and expect to continue to rely on a limited number of experienced personnel and a small number of contract manufacturers and other vendors to formulate, test, supply, store and distribute drug supplies for our preclinical studies and clinical trials. Our reliance on a limited number of vendors exposes us to the following risks, any of which could delay our clinical trials, and, consequently, FDA approval of our drug candidates and commercialization of our products, result in higher costs, or deprive us of potential product revenues:
| ·
| | Contract commercial manufacturers, their sub-contractors or other third parties we rely on and expect to rely on, may encounter difficulties in achieving the volume of production needed to satisfy preclinical and clinical needs or commercial demand, may experience technical issues that impact quality or compliance with applicable and strictly enforced regulations governing the manufacture of pharmaceutical products, and may experience shortages of qualified personnel to adequately staff production operations.
|
| ·
| | Our contract manufacturers could default on their agreements with us to provide supplies or meet our requirements for commercialization of our products.
|
| ·
| | For certain of our drug candidates, the use of alternate manufacturers may be difficult because the number of potential manufacturers that have the necessary governmental licenses to produce narcotic products is limited. Additionally, the FDA and the DEA must approve any alternative manufacturer of our products before we may use the alternative manufacturer to produce our supplies.
|
| ·
| | It may be difficult or impossible for us to find a replacement manufacturer on acceptable terms quickly, or at all. Our contract manufacturers and vendors may not perform as agreed or may not remain in the contract manufacturing business for the time required to successfully produce, store and distribute our products.
|
| ·
| | If any contract manufacturer makes improvements in the manufacturing process for our products, we may not own, or may have to share, the intellectual property rights to such innovation.
|
We may not be able to successfully develop or commercialize potential drug candidates for indications other than pain.
Our research and development activities include development of potential drug candidates for indications other than pain. We have no history of developing such drug candidates. We do not know whether any of our planned development activities will result in marketable products. We do not anticipate that our drug candidates in these areas will reach the market for at least several years, if at all.
Our employees and consultants are generally subject to confidentiality or other agreements with their former employers and they may inadvertently or otherwise violate those agreements.
Many of our employees and consultants were previously employed at universities or biotechnology or pharmaceutical companies. While we require our employees and consultants to honor any agreements they may have entered into prior to working with us, we may be subject to claims that we inadvertently or otherwise used or disclosed trade secrets or other confidential information belonging to former employers. Failure to defend such claims could result in loss of valuable rights or personnel, which in turn could harm or prevent commercialization of our drug candidates. Successful defense against such claims can be expensive and might distract us from executing our strategies.
Law enforcement concerns over diversion of opioids and social issues around abuse of opioids may make the regulatory approval process and commercialization of our drug candidates very difficult.
Law enforcement agencies or regulatory agencies may apply policies that seek to limit the availability of opioids. Such efforts may adversely affect the regulatory approval and commercialization of our drug candidates.
Developments by competitors may render our products or technologies obsolete or non-competitive.
Alternative technologies and products are being developed to improve or replace the use of opioids for pain management, several of which are in clinical trials or are awaiting approval from the FDA. In addition, the active ingredients in nearly all opioid drugs are available in generic form. Drug companies that sell generic opioid drugs represent substantial competition. Many of these organizations competing with us have substantially greater capital resources, larger research and development staffs and facilities, greater experience in drug development and in obtaining regulatory approvals and greater manufacturing and marketing capabilities than we do. Our competitors may market less expensive or more effective drugs that would compete with our drug candidates or reach market with competing drugs before we are able to reach market with our drug candidates. These organizations also compete with us to attract qualified personnel and partners for acquisitions, joint ventures or other collaborations.
Business interruptions could limit our ability to operate our business.
Our operations, as well as those of our collaborators on which we depend, are vulnerable to damage or interruption from computer viruses, human error, natural disasters, electrical and telecommunication failures, international acts of terror and similar events. We have not established a formal disaster recovery plan and our back-up operations and our business interruption insurance may not be adequate to compensate us for losses we may suffer. A significant business interruption could result in losses or damages incurred by us and require us to cease or curtail our operations.
Cyber-attacks or other failures in telecommunications or information technology systems could result in information theft, data corruption and significant disruption of our business operations.
We use information technology, computer systems and networks to process, transmit and store electronic information in connection with our business activities. Cyber incidents, including deliberate attacks and attempts to gain unauthorized access to computer systems and networks, have increased in frequency, scope and sophistication in every industry. These threats pose a risk to the security of our systems and networks and the confidentiality, availability and integrity of our data, and may cause a disruption in our operations, harm our reputation and increase our stock trading risk. There can be no assurance that we will be successful in preventing cyber-attacks or successfully mitigating their effects. Similarly, there can be no assurance that our third-party collaborators, distributors and other contractors and consultants will be successful in protecting our data that is stored on their systems. Any cyberattack or destruction or loss of data could have a material adverse effect on our business and prospects. In addition, we may suffer reputational harm or face litigation or adverse regulatory action as a result of cyber-attacks or other data security breaches and may incur significant additional expense to implement further data protection measures.
Unfavorable media coverage of opioid pharmaceuticals could negatively affect our business.
Opioid drug abuse receives a high degree of media coverage. Unfavorable publicity regarding, for example, the use or misuse of oxycodone or other opioid drugs, the limitations of ADFs, public inquiries and investigations into prescription drug abuse, litigation or regulatory activity could adversely affect our reputation. Such negative publicity could have an adverse effect on the potential size of the market for our drug candidates and decrease revenues and royalties, which would adversely affect our business and financial results.
Risks Relating to Manufacturing
We do not own any manufacturing facilities and we rely on third-party commercial drug manufacturers for drug supply.
We do not own any manufacturing facilities. We plan to continue to outsource formulation, manufacturing and related activities.
We rely on a limited number of third-party suppliers to formulate, manufacture, fill, label, ship or store all of our drug candidates. These suppliers must comply with current good manufacturing practices, or GMP, regulations enforced by the FDA and other government agencies and DEA regulations, and are subject to ongoing periodic unannounced inspection, including preapproval inspections by the FDA and DEA and corresponding state and foreign
government agencies to ensure strict compliance with GMP and other government regulations and corresponding foreign standards. These manufacturers may subsequently be stopped from producing, storing, shipping or testing our drug products due to their non-compliance with federal, state or local regulations. We do not have control over our suppliers’ compliance with these regulations and standards.
If REMOXY is approved, our commercial suppliers may encounter difficulties in achieving high volumes of production to satisfy commercial demands.
We cannot control decisions by our suppliers that affect their ability or willingness to continue to supply us on acceptable terms, or at all.
We may not be able to replace a commercial supplier on commercially reasonable terms, or at all. Replacing any of our commercial suppliers would be expensive and time consuming.
Failure by any of our suppliers to perform as expected could delay or prevent commercialization of REMOXY or result in shortages, cost overruns, or other problems and would materially harm our business.
We will rely on Durect as the sole source of certain excipients in REMOXY. Durect has limited experience manufacturing pharmaceutical products and maintaining GMP-compliant operations. We currently do not have a long-term commercial supply agreement in place with Durect. We expect that we and Durect will negotiate a supply agreement for these excipients. We may not be able to establish a commercial supply agreement on acceptable terms, or at all.
If we receive marketing approval for and commercially launch REMOXY, Durect may need to materially expand its manufacturing capacity. Durect may not be able to increase its manufacturing capacity for REMOXY in a timely or economic manner, or at all. Moreover, significant scale up of manufacturing will require additional validation studies, which are subject to FDA review and approval. If Durect is unable to successfully increase the manufacturing capacity, at an acceptable cost or otherwise, and we are unable to establish alternative manufacturing capabilities, there may be a shortage in supply, which would harm our future revenues and cause our business to suffer.
If Durect fails to supply excipients to us, they may be in breach of their supply obligations. With or without a commercial supply agreement, Durect’s failure for any reason to supply these excipients, including failure resulting from Durect relying on sole source providers, could delay or prevent commercialization of REMOXY or result in shortages, delays, unexpected costs or other problems and would materially harm our business.
We expect to rely on Noramco as the sole source of the oxycodone in REMOXY. We currently do not have a long-term commercial supply agreement in place with Noramco. Effective July 1, 2016, Noramco is owned by SK Capital Partners, a private investment firm. We expect to negotiate with Noramco a commercial supply agreement to supply us with oxycodone. We may not be able to establish a commercial supply agreement on acceptable terms, or at all. Until we have a commercial supply agreement in place with Noramco, we expect to obtain oxycodone from Noramco via purchase orders. There can be no assurance that Noramco will accept our purchase orders on acceptable terms, or at all. With or without a commercial supply agreement, Noramco’s failure for any reason to supply us with oxycodone could delay or prevent commercialization of REMOXY or result in shortages, cost overruns or other problems that would materially harm our business.
We will need to identify a third-party to manufacture commercial supplies of REMOXY. Without a commercial manufacturer, we may not be able to commercialize REMOXY. To date, we have not identified such third-party manufacturer and we may never find viable source of commercial supplies of REMOXY. We expect to rely on such third-party as a sole-source drug product manufacturer of REMOXY pursuant to a supply agreement. In addition to drug product manufacturing, this third-part manufacturer will need to be responsible for sourcing excipients in REMOXY other than those provided by the Durect Agreement. Failure for any reason to manufacture and supply of REMOXY could delay or prevent commercialization of REMOXY or result in shortages, product recalls or withdrawals, delays or failures in product testing or delivery, cost overruns or other problems that would materially harm our business.
If we cannot formulate and scale-up additional dosage forms of REMOXY, the commercial opportunity for REMOXY might be diminished.
We plan to formulate and scale-up additional dosage forms of REMOXY. We may not be able to successfully complete our formulation or scale-up activities or we may determine that the commercial opportunity for REMOXY in certain dosage forms is too limited to warrant further investment. If we are unsuccessful in our formulation or scale-up activities with REMOXY, our future revenue may be less than expected and our operations may suffer.
We rely solely on Durect to provide us with certain components of drug candidates and will continue to rely on Durect as the sole-source providerof these components.
We rely on Durect as the sole-source provider of certain components of REMOXY and other drug candidates designed to reduce the potential risks of unintended use, and will rely solely on Durect to produce commercial supplies of these components. Durect’s failure for any reason to provide these components or to achieve and maintain satisfactory manufacturing standards could result in product recalls or withdrawals, delays or failures in product testing or delivery, cost overruns or other problems that could materially harm our business.
Durect may encounter manufacturing difficulties involving production yields, quality control and quality assurance. Durect is subject to ongoing periodic unannounced inspection by the FDA and corresponding state and foreign agencies to ensure strict compliance with government regulations and corresponding foreign standards. We cannot control Durect’s compliance with these regulations and standards.
If we receive marketing approval for and commercially launch REMOXY, Durect may need to materially expand its manufacturing capacity. Durect may not be able to increase its manufacturing capacity for REMOXY in a timely or economic manner, or at all. Moreover, significant scale up of manufacturing will require additional validation studies, which are subject to FDA review and approval. If Durect is unable to successfully increase the manufacturing capacity for such components of REMOXY, at an acceptable cost or otherwise, and we are unable to establish alternative manufacturing capabilities, commercialization of REMOXY may be delayed, prevented or impaired or there may be a shortage in supply, which would harm our future revenues and cause our business to suffer.
We expect to rely on Noramco as the sole source of the oxycodone in REMOXY.
We expect to rely on Noramco as the sole source of the oxycodone in REMOXY. We expect we and Noramco will negotiate a supply agreement to supply us with the oxycodone in REMOXY. Noramco’s operation is subject to regulation by the DEA and the Controlled Substances Act. Noramco’s failure for any reason to manufacture and supply us with the oxycodone in REMOXY could result in shortages, cost overruns or other problems that could materially harm our business.
Risks Relating to our Financial Position and Need for Financing
Our operating history may make it difficult for you to evaluate our business to date and to assess its future viability.
Our operations from our inception to date have been limited to organizing and staffing our company, acquiring, developing and securing our technology, undertaking preclinical studies and clinical trials of our drug candidates and forming collaborations. We have not yet demonstrated our ability to obtain regulatory approval, formulate and manufacture our drug candidates on a commercial scale or conduct sales and marketing activities. Consequently, any predictions you make about our future success or viability may not be as accurate as they could be if we had a longer operating history.
We have a history of losses and expect to incur substantial losses and negative operating cash flows for the foreseeable future.
Although we were profitable in some years in the past based on payments received pursuant to collaboration agreements and interest income, we have yet to generate any revenues from product sales. We have an accumulated
deficit of $155 million at September 30, 2017. Even if we succeed in developing and commercializing one or more of our drug candidates, we expect to continue to use significant cash resources in our operations for the foreseeable future.
We anticipate that our expenses will increase substantially in the foreseeable future as we:
| ·
| | continue to conduct preclinical studies and clinical trials for our drug candidates, including drug development activities related to FENROCK;
|
| ·
| | seek regulatory approvals for our drug candidates;
|
| ·
| | develop, formulate, manufacture and commercialize our drug candidates;
|
| ·
| | implement additional internal systems and develop new infrastructure;
|
| ·
| | acquire or in-license additional products or technologies, or expand the use of our technology;
|
| ·
| | maintain, defend and expand the scope of our intellectual property; and
|
| ·
| | hire additional personnel.
|
We will need to generate significant revenues to achieve and maintain profitability. If we or our collaborators cannot successfully develop, obtain regulatory approval for and commercialize our drug candidates, we will not be able to generate such revenues or achieve profitability in the future. Our failure to achieve or maintain profitability would have a material adverse impact on the market price of our common stock.
If we cannot raise additional capital on acceptable terms, we may be unable to complete planned clinical trials of any or some of our drug candidates or to pursue attractive business opportunities.
We have funded all of our operations and capital expenditures with the proceeds from our public and private stock offerings, payments received under collaboration agreements and interest earned on our investments. We expect that our current cash, cash equivalents and marketable securities will be sufficient to meet our working capital and capital expenditure needs for at least the next twelve months. However, we may elect to raise additional funds within such twelve-month period or need to raise additional funds thereafter and additional financing may not be available on favorable terms, if at all. Even if we succeed in selling additional securities to raise funds, our existing stockholders’ ownership percentage would be reduced and new investors may demand rights, preferences or privileges senior to those of existing stockholders. If we raise additional capital through debt financing, if available, such financings may involve covenants that restrict our business activities. If we raise additional capital through strategic alliance and license arrangements, we may have to trade our rights to our technology, intellectual property or drug candidates to others in such arrangements on terms that may not be favorable to us.
If we determine that we need to raise additional funds and we are not successful in doing so, we may be unable to complete the clinical development of some or all of our drug candidates or to seek or obtain FDA approval of our drug candidates. We then could be forced to discontinue product development, enter into a relationship with an additional strategic partner earlier than currently intended, reduce sales and marketing efforts or forego attractive business opportunities.
Risks Relating to an Investment in our Common Stock
Our stock price has been volatile and could experience a sudden decline in value.
Our common stock has experienced significant price and volume fluctuations and may continue to experience volatility in the future. You may not be able to sell your shares quickly or at the latest market price if trading in our stock is not active or the volume is low. The following factors, in addition to other risk factors described in this section, may have a significant impact on the market price of our common stock:
| ·
| | results of or delays in efforts to seek regulatory approval for REMOXY, and in preclinical studies and clinical trials for our other drug candidates;
|
| ·
| | publicity regarding products under development by us or others, including with respect to actual or potential medical results relating to such matters;
|
| ·
| | announcements of technological innovations or new commercial products by us or others;
|
| ·
| | developments in patent or other proprietary rights by us or others;
|
| ·
| | comments or opinions by securities analysts or major stockholders;
|
| ·
| | adverse media coverage related to opioid pharmaceuticals;
|
| ·
| | future sales of our common stock by existing stockholders;
|
| ·
| | developments with respect to potential merger and acquisition activity of companies with whom we have strategic alliances or other agreements;
|
| ·
| | regulatory developments or changes in regulatory guidance enacted by applicable governmental or other authorities;
|
| ·
| | litigation or threats of litigation;
|
| ·
| | economic and other external factors or other disaster or crises;
|
| ·
| | the departure of any of our officers, directors or key employees;
|
| ·
| | period-to-period fluctuations in financial results; and
|
| ·
| | limited daily trading volume.
|
Changing laws, regulations and standards relating to corporate governance and public disclosure, including the Sarbanes-Oxley Act of 2002, the Dodd-Frank Act of 2010, SEC regulations and the rules of The NASDAQ Stock Market LLC, or Nasdaq, create uncertainty for public companies.
If we fail to continue to meet all applicable listing requirements, our common stock may be delisted from The NASDAQ Global Market, which could have an adverse impact on the liquidity and market price of our common stock.
Our common stock is currently listed on The NASDAQ Global Market, which has qualitative and quantitative listing criteria. If we are unable to meet any of the NASDAQ listing requirements in the future, such as the corporate governance requirements, the minimum closing bid price requirement, the minimum market value of listed securities requirement, total assets and total revenues, net income from continuing operations, the number of publicly held shares or any other NASDAQ requirements. NASDAQ could determine to delist our common stock. A delisting of our common stock could adversely affect the market liquidity of our common stock, decrease the market price of our common stock, adversely affect our ability to obtain financing for the continuation of our operations and result in the loss of confidence in our company. On November 16, 2016, we received notice from the Listing Qualifications Department of the NASDAQ Stock Market (the “Staff”) indicating that, for the previous 30 consecutive business days, the bid price for our common stock closed below the minimum $1.00 per share required for continued inclusion on The NASDAQ Global Market. The notification letter stated that we would be afforded 180 calendar days, or until May 15, 2017, to regain compliance with the minimum bid price requirement. In order to regain compliance, on May 4, 2017, our board of directors and stockholders approved the Charter Amendment to effect the Reverse Stock Split. On May 10, 2017, our common stock began trading on The NASDAQ Global Market on a split-adjusted basis at a ratio of 1 share for 7. On May 24, 2017, we received a letter from the Staff indicating that we had regained compliance with the $1.00 minimum closing bid price requirement. Despite effecting the Reverse Stock Split, there can be no assurance that the market price per share of our common stock will remain in excess of the $1.00 minimum closing bid price requirement in the future. The continuing effect of the Reverse Stock Split on the market price of our common stock cannot be predicted with any certainty, and the history of similar stock split combinations for companies in like circumstances is varied.
Anti-takeover provisions in our charter documents and Delaware law may prevent or delay removal of incumbent management or a change of control.
Anti-takeover provisions of our amended and restated certificate of incorporation and amended and restated bylaws and Delaware law may have the effect of deterring or delaying attempts by our stockholders to remove or replace management, engage in proxy contests and effect changes in control. The provisions of our charter documents include:
| ·
| | a classified board so that only one of the three classes of directors on our board of directors is elected each year;
|
| ·
| | elimination of cumulative voting in the election of directors;
|
| ·
| | procedures for advance notification of stockholder nominations and proposals;
|
| ·
| | the ability of our board of directors to amend our bylaws without stockholder approval; and
|
| ·
| | the ability of our board of directors to issue up to 10,000,000 shares of preferred stock without stockholder approval upon the terms and conditions and with the rights, privileges and preferences as our board of directors may determine.
|
In addition, as a Delaware corporation, we are subject to Delaware law, including Section 203 of the Delaware General Corporation Law. In general, Section 203 prohibits a Delaware corporation from engaging in any business combination with any interested stockholder for a period of three years following the date that the stockholder became an interested stockholder unless certain specific requirements are met as set forth in Section 203.
These provisions, alone or together, could have the effect of deterring or delaying changes in incumbent management, proxy contests or changes in control.
Our share ownership is concentrated, and our officers, directors and principal stockholders can exert significant control over matters requiring stockholder approval.
Due to their combined stock holdings, our officers, directors and principal stockholders (stockholders holding greater than 5% of our common stock) acting collectively may have the ability to exercise significant influence over matters requiring stockholder approval including the election of directors and approval of significant corporate transactions. In particular, Remi Barbier, our founder, Chairman of the Board of Directors, President and Chief Executive Officer, owns or controls a significant amount of the voting power of our outstanding capital stock. This concentration of ownership may delay or prevent a change in control of the Company and may make some transactions, including but not limited to any merger, consolidation, or sale of substantially all of our assets, more difficult or impossible to complete without the support of key stockholders.
Publicly available information regarding stockholders’ ownership may not be comprehensive because the SEC does not require certain large stockholders to publicly disclose their stock ownership positions.
If the fair value of our stock increases and outstanding Performance Awards vest, we expect to use substantial amounts of cash to fund employee tax liabilities.
We have Performance Awards outstanding. If these Performance Awards vest, we expect to issue our employees shares of our common stock net of statutory employment taxes. This net issuance results in fewer shares issued and uses our cash to fund these taxes. The use of cash could be substantially higher, depending on the fair value of our common stock on the date the Performance Awards vest. If our use of cash to fund these taxes is substantial, our cash balance could substantially decline and our stock price could also decline.
We may in the future seek to fund the cash used for Performance Awards through the sale of our common stock. However, we may not be successful in selling shares of our common stock to fund the cash used for Performance Awards. If the number of shares we sell to fund the cash used for Performance awards is significant, our stock price could decline.
Volatility in the stock prices of other companies may contribute to volatility in our stock price.
The stock market in general, Nasdaq and the market for technology companies in particular, have experienced significant price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of those companies. Further, there has been particular volatility in the market prices of securities of life sciences companies. These broad market and industry factors may seriously harm the market price of our common stock, regardless of our operating performance. In the past, following periods of volatility in the market price of a company’s securities, securities class action litigation has often been instituted. A securities class action suit against us could result in substantial costs, potential liabilities and the diversion of management’s attention and resources.
Our operating results may fluctuate from quarter to quarter and this fluctuation may cause our stock price to decline.
Our quarterly operating results have fluctuated in the past and are likely to fluctuate in the future. Factors contributing to these fluctuations include, among other items, the timing and enrollment rates of clinical trials for our drug candidates, our need for clinical supplies and the valuation of stock-based compensation. Thus, quarter-to-quarter
comparisons of our operating results may not be not indicative of what we might expect in the future. As a result, in some future quarters our clinical, financial or operating results may not meet the expectations of securities analysts and investors and could result in a decline in the price of our stock.
If securities or industry analysts publish inaccurate or unfavorable research about our business or product candidates, our stock price could decline.
Securities or industry analysts publish research and reports about our business or product candidates. An analyst’s conclusions regarding prospects for product candidates in the biopharmaceutical industry can include judgments based on the limited publicly-available data. If one or more analysts issues unfavorable research about our business or our product candidates, including a downgrade of our common stock, the price of our stock may decline.
There may not be an active, liquid trading market for our common stock.
There is no guarantee that an active trading market for our common stock will be maintained on Nasdaq. Investors may not be able to sell their shares quickly or at the latest market price if trading in our stock is not active.
Item 2. Unregistered Sales of Equity Securities and Use of Proceeds
None.
Item 3. Defaults Upon Senior Securities
None.
Item 4.
Mine Safety Disclosures
Not applicable.
Item 5.
Other Information
None.
None.
Item 6. Exhibits
The following exhibits have been filed with this report:
| | | | | | | | | | | |
|
|
|
|
|
|
|
|
|
|
| | | | | | | | | | | |
|
|
| Incorporated by |
|
|
| | | | | | | | | | | |
|
|
| Reference |
|
|
| | | | | Incorporated by
Reference | | | |
Exhibit
No. | | Description | | Form | | Filing
Date | | Exhibit
No. | | Filed
Herewith | |
Exhibit | |
| Description |
|
|
| Filing |
| Exhibit |
| Filed |
No. | | | | | Form |
| Date |
| No. |
| Herewith |
3.1 | | | Amended and Restated Certificate of Incorporation. | | 10-Q | | 7/29/2005 | | 3.1 | | |
| Amended and Restated Certificate of Incorporation. | | 10-Q |
| 7/29/2005 |
| 3.1 |
|
|
3.2 | | | Amended and Restated Bylaws. | | 10-Q | | 4/24/2013 | | 3.2 | | |
| Certificate of Amendment of Restated Certificate of Incorporation. |
| 8-K |
| 5/8/2017 |
| 3.1 |
|
|
3.3 | |
| Certificate of Amendment of Restated Certificate of Incorporation. |
| 10-K |
| 3/29/2019 |
| 3.3 |
|
|
3.4 | |
| Amended and Restated Bylaws of Cassava Sciences, Inc. |
| 8-K |
| 12/11/2020 |
| 3.1 |
|
|
4.1 | | | Specimen Common Stock Certificate. | | 10-Q | | 7/29/2005 | | 4.1 | | |
| Specimen Common Stock Certificate. |
| 10-Q |
| 8/12/2019 |
| 4.1 |
|
|
10.1 | |
| Form of Securities Purchase Agreement, dated February 10, 2021, by and between Cassava Sciences, Inc. and the purchasers named therein. |
| 8-K |
| 2/12/2021 |
| 10.1 |
|
|
10.2* | |
| Master Services Agreement between Cassava Sciences, Inc. and Evonik Corporation, dated February 22, 2021. |
| 8-K |
| 3/11/2021 |
| 10.1 |
|
|
10.3* | |
| Master Services Agreement between Cassava Sciences, Inc. and Premier Research International LLC, dated June 11, 2021 |
|
|
|
|
|
|
| X |
31.1 | | | Certification of Principal Executive Officer and Principal Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | | | | | | | | X |
| Certification of Principal Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
|
|
|
|
|
|
| X |
31.2 | |
| Certification of Principal Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
|
|
|
|
|
|
| X |
32.1 | | | Certification of the Chief Executive Officer (as Principal Executive Officer and Principal Financial Officer) pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | | | | | | | | X |
| Certification of the Chief Executive Officer and the Chief Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
|
|
|
|
|
|
| X |
101.INS | | | XBRL Instance Document. | | | | | | | | X |
| XBRL Instance Document. |
|
|
|
|
|
|
| X |
101.SCH | | | XBRL Taxonomy Extension Schema Document. | | | | | | | | X |
| XBRL Taxonomy Extension Schema Document. |
|
|
|
|
|
|
| X |
101.CAL | | | XBRL Taxonomy Extension Calculation Linkbase Document. | | | | | | | | X |
| XBRL Taxonomy Extension Calculation Linkbase Document. |
|
|
|
|
|
|
| X |
101.DEF | | | XBRL Taxonomy Extension Definition Linkbase Document. | | | | | | | | X |
| XBRL Taxonomy Extension Definition Linkbase Document. |
|
|
|
|
|
|
| X |
101.LAB | | | XBRL Taxonomy Extension Labels Linkbase Document. | | | | | | | | X |
| XBRL Taxonomy Extension Labels Linkbase Document. |
|
|
|
|
|
|
| X |
101.PRE | | | XBRL Taxonomy Extension Presentation Linkbase Document. | | | | | | | | X |
| XBRL Taxonomy Extension Presentation Linkbase Document. |
|
|
|
|
|
|
| X |
| | | | | | | | | | | | |
104. | |
| Cover Page Interactive Data File – the cover page interactive data file does not appear in the Interactive Data File because its XBRL tags are embedded within the Inline XBRL document. |
|
|
|
|
|
|
| X |
*Confidential portions of this document have been redacted as permitted by applicable regulations.
SIGNATURES
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
|
|
|
| Pain Therapeutics, Inc.
|
|
| (Registrant)Cassava Sciences, Inc.
|
|
| (Registrant) |
|
|
| /s/ REMI BARBIER |
|
| Remi Barbier, |
| Chairman of the Board of Directors, |
| President and Chief Executive Officer |
Date: August 4, 2021
|
|
Date: November 1, 2017
|
|
|
|
|
|
|
| /s/ ERIC J. SCHOEN |
|
| Eric J. Schoen, |
| Chief Financial Officer |
Date: August 4, 2021
|
|