On March 27, 2020, President Trump signed into law the Coronavirus“Coronavirus Aid, Relief, and Economic Security Act (“CARESCARES”) Act”). was signed into law. The CARES Act, among other things, includes provisions relating to refundable payroll tax credits, deferment of employer side social security payments, net operating loss carryback periods, alternative minimum tax credit refunds, modifications to the net interest deduction limitations, increased limitations on qualified charitable contributions, and technical corrections to tax depreciation methods for qualified improvement property. We continueRegulatory guidance has indicated that public companies are ineligible to examineparticipate in certain of the impact thatloan programs provided by the CARES Act may have on our business. Currently, we are unableAct. There is substantial uncertainty with respect to determine the impactscope, content or timing of any further economic stimulus programs to address the economic downturn related to the COVID-19 outbreak. We do not expect that the CARES Act will have a material impact on our financial condition, results of operation,operations, or liquidity.
Clinical Development Programs
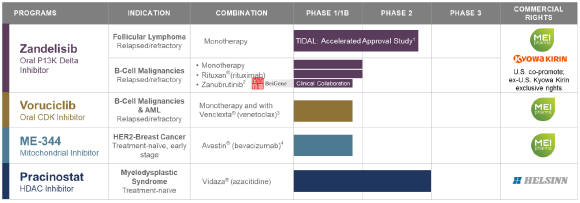
Our approach to building our pipeline is to license promising cancer agents and build value in programs through development, commercialization and strategic partnerships, as appropriate. Our drug candidate pipeline includes:
Zandelisib (f/k/a ME-401,ME-401), an oral PI3Kphosphatidylinositol d3-kinase (“PI3K”) delta inhibitor;
Voruciclib, an oral CDKcyclin-dependent kinase (“CDK”) inhibitor;
ME-344, a mitochondrial inhibitor targeting the OXPHOSoxidative phosphorylation (“OXPHOS”) complex; and
Pracinostat, an oral HDAChistone deacetylase (“HDAC”) inhibitor.
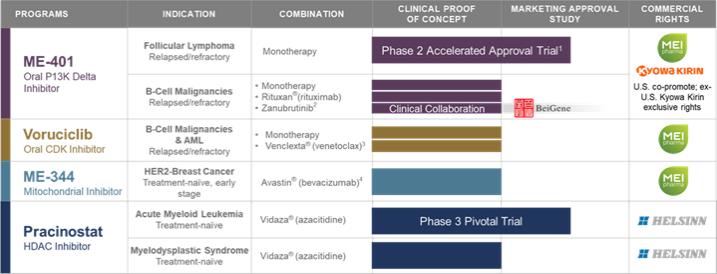
1. Phase 2 study intended to support an accelerated approval marketing application with FDA.
2. Study arm conducted under clinical collaboration with BeiGene, Ltd.
3. Initiation of clinical studies in combination with venetoclax is subject to FDA approval.
4. Investigator-initiated trial; completed.
Zandelisib (f/k/a ME-401): PI3Kd Inhibitor in a Phase 2 |
|
|
|
ME-401: PI3Kd Inhibitor in a Phase 2 Trial Intended to Support an Accelerated Approval in Relapsed or Refractory Follicular Lymphoma
ME-401Zandelisib is an oral, once-daily, selective PI3Kd inhibitor in clinical development for the treatment ofB-cell malignancies. In March 2020, the U.S. FDA granted zandelisib Fast Track designation. In April 2020, we entered into a global license, development and commercialization agreement to further develop and commercializeME-401 zandelisib with Kyowa Kirin Co., Ltd. (KKC)(“KKC”). MEI and KKC willco-develop andco-promoteME-401co-promote zandelisib in the U.S., with MEI recording all revenue from U.S. sales. KKC has exclusive commercialization rights outside of the U.SU.S.
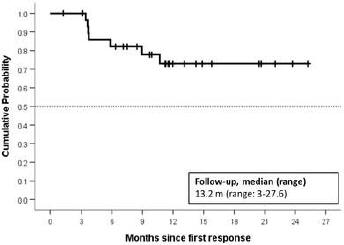
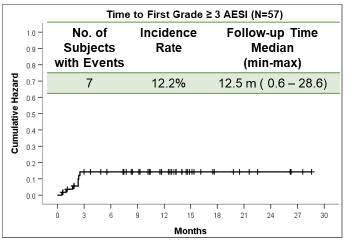
We are conducting twomultiple ongoing studies evaluatingME-401. The first is zandelisib including TIDAL (Trials of PI3K DeltA inNon-Hodgkin’s Lymphoma), a Phase 2 clinical trial evaluatingME-401 zandelisib as a monotherapy for the treatment of adults with relapsed or refractory (“r/r”) follicular lymphoma (“FL”) after failure of at least two prior systemic therapies including chemotherapy and an anti-CD20 antibody. Subject to the results, upon completion of TIDAL, we are planning a submission with the FDA to support an accelerated approval of a marketing application under 21 CFR Part 314.500, Subpart H. The second isWe are also conducting amulti-arm, open-label, Phase 1b dose escalationfinding and expansion trial evaluatingME-401 zandelisib as a monotherapy and in combination with other therapies in patients with relapsed or refractoryB-cell malignancies. Also, in 2019, KKCOther initiated astudies include Phase 1 studyand Phase 2 studies being conducted by KKC evaluatingME-401 zandelisib as a monotherapy in patients in Japan with indolentB-cell malignancy.malignancy pursuant to our agreement with KKC (as described below).