ITEM 2. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS.
The following discussion should be read in conjunction with our unaudited condensed consolidated financial statements and notes thereto included in this Quarterly Report on Form 10-Q, and our audited consolidated financial statements and notes thereto for the year ended December 31, 20212022 included in our 20202022 Form 10-K. This discussion contains forward-looking statements reflecting our current expectations that involve risks and uncertainties. See “Note Regarding Forward-Looking Statements” for a discussion of the uncertainties, risks and assumptions associated with these statements. Our actual results and the timing of events could differ materially from those expressed or implied by the forward-looking statements due to important factors and risks including, but not limited to, those set forth below under “Risk Factors” and elsewhere herein, and those identified under Part I, Item 1A of our 20212022 Form 10-K.
Overview
We are a diversified clinical-stage company developing therapeutics in areas of high unmet need. As a result of the Acquisition of Theriva Biologics, S.L. (“VCN”, formerly named VCN Biosciences, S.L.), described in more detail below, we began transitioning our strategic focus to oncology through the development of VCN’s new oncolytic adenovirus platform designed for intravenous and intravitreal delivery to trigger tumor cell death, improve access of co-administered cancer therapies to the tumor, and promote a robust and sustained anti-tumor response by the patient’s immune system. Prior to the Acquisition, of VCN, our focus was on developing therapeutics designed to treat gastrointestinal (GI) diseases which included. our lead clinical development candidates: (1) SYN-004 (ribaxamase) which is designed to degrade certain commonly used intravenous (IV) beta-lactam antibiotics within the GI tract to prevent microbiome damage, thereby preventing overgrowth and infection by pathogenic organisms such Clostridioides difficile infection (CDI), overgrowth of pathogenic organisms, the emergence of antimicrobial resistance (AMR) and vancomycin resistant Enterococci (VRE), and reducing the incidence and severity of acute graft-versus-host-disease (aGVHD) in allogeneic hematopoietic cell transplant (HCT) recipients, and (2) SYN-020, a recombinant oral formulation of the enzyme intestinal alkaline phosphatase (IAP) produced under cGMP conditions and intended to treat both local GI and systemic diseases. Upon consummation of the Acquisition of VCN, described in more detail below, we are transitioning our strategic focus to oncology, through the development of new oncolytic adenovirus products designed for intravenous and intravitreal delivery to trigger tumor cell death, improve access of co-administered cancer therapies to the tumor, and promote a robust and sustained anti-tumor response by the patient’s immune system.
As part of our strategic transformation into an oncology focused company, we are exploring value creation options around our SYN-020SYN-004 and SYN-004SYN-020 assets. SYN-004 and SYN-020 both have significant potential opportunity in non-oncology related indications. Advancement of these products may be better achieved through out-licensing or partnering and we will explore opportunities for both SYN-004 and SYN-020 moving forward.
Acquisition of VCN Biosciences, S.LOur Current Product Pipeline
On March 10, 2022, pursuant to the terms of the Share Purchase Agreement (“Purchase Agreement”) we entered into with VCN and the shareholders of VCN Biosciences S.L.(the “Sellers”), we completed our acquisition of all the outstanding shares of VCN (the “VCN Shares”) from the shareholders of VCN. Pursuant to the Purchase Agreement, as consideration for the purchase of the VCN Shares of capital stock, we paid $4,700,000 (the “Closing Cash Consideration”) to Grifols Innovation and New Technologies Limited (“Grifols”), the owner of approximately 86% of the equity of VCN, and issued to the remaining Sellers 26,395,303 shares of our common stock, $.001 par value (the “Closing Shares”), representing 19.99% of the outstanding shares of our common stock on December 14, 2021, the date of the Purchase Agreement. As additional consideration for the purchase of the VCN Shares held by Grifols, we also agreed to make certain milestone payments to Grifols. Pursuant to the terms of the Purchase Agreement we loaned VCN $417,000 to help finance the costs of certain of VCN’s research and development activities. In addition, at Closing VCN and Grifols entered into a sublease agreement for the sublease by VCN of the laboratory and office space as well as a transitional services agreement. We agreed as a post- Closing covenant to commit to fund VCN’s research and development programs, including but not limited to VCN-01 PDAC phase 2 trial, VCN-01 RB trial and necessary G&A within a budgetary plan of approximately $27.8 million.
VCN is a private, clinical-stage biopharmaceutical company developing new oncolytic adenoviruses for the treatment of cancer. VCN’s lead product candidate, VCN-01, is being studied in clinical trials for pancreatic cancer and retinoblastoma. VCN-01 is designed to be administered systemically, intratumorally or intravitreally, either as a monotherapy or in combination with standard of care, to treat a wide variety of cancer indications. VCN-01 is designed to replicate selectively and aggressively within tumor cells, and to degrade the tumor stroma barrier that serves as a significant physical and immunosuppressive barrier to cancer treatment, Degrading the tumor stroma has been shown to improve access to the tumor by the virus and additional therapies such as chemo- and immuno-therapies. Importantly, degrading the stroma exposes tumor antigens, turning “cold” tumors “hot” and enabling a sustained anti-tumor immune response. VCN has the rights to four exclusive patents for proprietary technologies, as well as technologies developed in collaboration with the Virotherapy Group of the Catalan Institute of Oncology (ICO-IDIBELL) and with Hospital Sant Joan de Deu (HSJD), with a number of additional patents pending.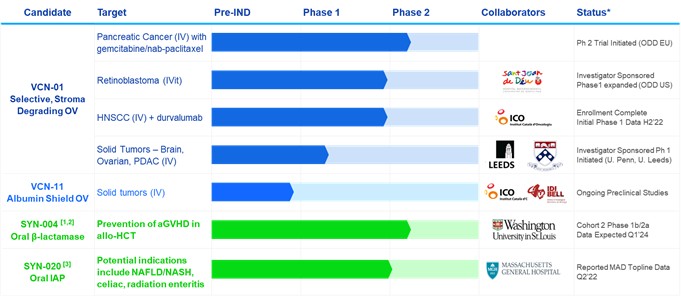
2624
