UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-Q
|
| | | | |
| ☑ | QUARTERLY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the quarterly period ended June 30, 20192020
or
|
| | | | |
| ☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from ___________ to ___________
Commission File Number: 001-36257
RETROPHIN, INC.
(Exact name of registrant as specified in its charter)
|
| | | | | | | | | | | | | |
| Delaware | | 27-4842691 | |
| (State or other jurisdiction of incorporation or organization) | | (I.R.S. Employer Identification No.) | |
3721 Valley Centre Drive,, Suite 200
San Diego,, CA92130
(Address of Principal Executive Offices)
(888) (888) 969-7879
(Registrant's Telephone number including area code)
|
| | | | | | | |
| N/A | |
| Former name, former address and former fiscal year, if changed since last report | |
Securities registered pursuant to Section 12(b) of the Act:
|
| | | | | | | | | | | | | |
| Title of each class | Trading Symbol(s) | Name of each exchange on which registered | |
| Common Stock, par value $0.0001 per share | RTRX | The Nasdaq Global Market | |
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☑ No ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ☑ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and "emerging growth company" in Rule 12b-2 of the Exchange Act.
|
| | | | | | | | | | |
| Large accelerated filer | ☑ | Accelerated filer | ☐ |
| Non-accelerated filer | ☐ | Smaller reporting company | ☐ |
| | Emerging growth company | ☐ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☑
The number of shares of outstanding common stock, par value $0.0001 per share, of the Registrant as of August 5, 2019July 29, 2020 was 42,954,151.
RETROPHIN, INC.
Form 10-Q
For the Fiscal Quarter Ended June 30, 20192020
TABLE OF CONTENTS
FORWARD-LOOKING STATEMENTS
This report contains forward-looking statements regarding our business, financial condition, results of operations and prospects. Words such as “expects,” “anticipates,” “intends,” “plans,” “believes,” “seeks,” “estimates” and similar expressions or variations of such words are intended to identify forward-looking statements, but are not deemed to represent an all-inclusive means of identifying forward-looking statements as denoted in this report. Additionally, statements concerning future matters are forward-looking statements.
Although forward-looking statements in this report reflect the good faith judgment of our management, such statements can only be based on facts and factors currently known by us. Consequently, forward-looking statements are inherently subject to risks and uncertainties and actual results and outcomes may differ materially from the results and outcomes discussed in or anticipated by the forward-looking statements. Factors that could cause or contribute to such differences in results and outcomes include, without limitation, those specifically addressed under the headings “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” in our annual reportAnnual Report on Form 10-K for the fiscal year ended December 31, 20182019 (the "2018"2019 10-K"), and in this quarterly reportQuarterly Report on Form 10-Q and information contained in other reports that we file with the Securities and Exchange Commission (the “SEC”).10-Q. You are urged not to place undue reliance on these forward-looking statements, which speak only as of the date of this report.
In addition, statements that "we believe" and similar statements reflect our beliefs and opinions on the relevant subject. These statements are based upon information available to us as of the date of this quarterly reportQuarterly Report on Form 10-Q, and while we believe such information forms a reasonable basis for such statements, such information may be limited or incomplete, and our statements should not be read to indicate that we have conducted an exhaustive inquiry into, or review of, all potentially available relevant information. These statements are inherently uncertain and you are cautioned to not unduly rely upon these statements.
We file reports with the SEC.Securities and Exchange Commission ("SEC"). The SEC maintains a website (www.sec.gov) that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC, including us.
We undertake no obligation to revise or update any forward-looking statements in order to reflect any event or circumstance that may arise after the date of this report, except as required by law. Readers are urged to carefully review and consider the various disclosures made throughout the entirety of this quarterly report, which are designed to advise interested parties of the risks and factors that may affect our business, financial condition, results of operations and prospects.
PART I - FINANCIAL INFORMATION
Item 1. Financial Statements
RETROPHIN, INC. AND SUBSIDIARIES
CONDENSED CONSOLIDATED BALANCE SHEETS
(in thousands, except par value and share amounts)
| | | | June 30, 2019 | | December 31, 2018 | | June 30, 2020 | | December 31, 2019 |
| Assets | (unaudited) | | |
| Assets | (unaudited) | | |
| Current assets: | |
| | |
| Current assets: | | | |
| Cash and cash equivalents | $ | 75,657 |
| | $ | 102,873 |
| Cash and cash equivalents | $ | 237,170 | | | $ | 62,436 | |
| Marketable securities | 350,243 |
| | 368,668 |
| |
| Available-for-sale debt securities, at fair value (amortized cost $218,596, allowance for credit losses of $0 as of June 30, 2020; amortized cost $335,206, allowance for credit losses of $0 as of December 31, 2019) | | Available-for-sale debt securities, at fair value (amortized cost $218,596, allowance for credit losses of $0 as of June 30, 2020; amortized cost $335,206, allowance for credit losses of $0 as of December 31, 2019) | 220,206 | | | 336,088 | |
| Accounts receivable, net | 15,451 |
| | 12,662 |
| Accounts receivable, net | 14,077 | | | 18,048 | |
| Inventory, net | 5,050 |
| | 5,619 |
| Inventory, net | 6,286 | | | 6,082 | |
| Prepaid expenses and other current assets | 9,164 |
| | 4,140 |
| Prepaid expenses and other current assets | 7,714 | | | 5,015 | |
| Prepaid taxes | 1,450 |
| | 1,716 |
| |
| Tax receivable | | Tax receivable | 20,109 | | | 1,395 | |
| | Total current assets | 457,015 |
| | 495,678 |
| Total current assets | 505,562 | | | 429,064 | |
| Property and equipment, net | 3,015 |
| | 3,146 |
| Property and equipment, net | 2,930 | | | 2,891 | |
| Other non-current assets | 12,702 |
| | 7,709 |
| Other non-current assets | 13,895 | | | 14,709 | |
| Investment-equity | 15,000 |
| | 15,000 |
| |
| | Intangible assets, net | 158,906 |
| | 186,691 |
| Intangible assets, net | 155,371 | | | 157,200 | |
| Goodwill | 936 |
| | 936 |
| Goodwill | 936 | | | 936 | |
| | Total assets | $ | 647,574 |
| | $ | 709,160 |
| Total assets | $ | 678,694 | | | $ | 604,800 | |
| Liabilities and Stockholders' Equity | |
| | |
| Liabilities and Stockholders' Equity | | | |
| Current liabilities: | |
| | |
| Current liabilities: | | | |
| Accounts payable | $ | 12,519 |
| | $ | 6,954 |
| Accounts payable | $ | 10,198 | | | $ | 26,614 | |
| Accrued expenses | 50,557 |
| | 49,695 |
| Accrued expenses | 44,379 | | | 51,745 | |
| | Other current liabilities | 8,417 |
| | 6,165 |
| Other current liabilities | 7,356 | | | 8,590 | |
| Business combination-related contingent consideration | 19,094 |
| | 19,350 |
| Business combination-related contingent consideration | 8,000 | | | 8,500 | |
| 2019 Convertible debt | — |
| | 22,457 |
| |
| | Total current liabilities | 90,587 |
| | 104,621 |
| Total current liabilities | 69,933 | | | 95,449 | |
| 2025 Convertible debt | 199,891 |
| | 195,091 |
| |
| Convertible debt | | Convertible debt | 210,009 | | | 204,861 | |
| Other non-current liabilities | 22,102 |
| | 17,545 |
| Other non-current liabilities | 19,507 | | | 20,894 | |
| | Business combination-related contingent consideration, less current portion | 57,905 |
| | 73,650 |
| Business combination-related contingent consideration, less current portion | 60,600 | | | 62,400 | |
| | Total liabilities | 370,485 |
| | 390,907 |
| Total liabilities | 360,049 | | | 383,604 | |
| Stockholders' Equity: | |
| | |
| Stockholders' Equity: | | | |
| Preferred stock $0.0001 par value; 20,000,000 shares authorized; 0 issued and outstanding as of June 30, 2019 and December 31, 2018 | — |
| | — |
| |
| Common stock $0.0001 par value; 100,000,000 shares authorized; 42,899,318 and 41,389,524 issued and outstanding as of June 30, 2019 and December 31, 2018, respectively | 4 |
| | 4 |
| |
| Preferred stock $0.0001 par value; 20,000,000 shares authorized; 0 issued and outstanding as of June 30, 2020 and December 31, 2019 | | Preferred stock $0.0001 par value; 20,000,000 shares authorized; 0 issued and outstanding as of June 30, 2020 and December 31, 2019 | — | | | — | |
| Common stock $0.0001 par value; 100,000,000 shares authorized; 50,902,874 and 43,088,921 issued and outstanding as of June 30, 2020 and December 31, 2019, respectively | | Common stock $0.0001 par value; 100,000,000 shares authorized; 50,902,874 and 43,088,921 issued and outstanding as of June 30, 2020 and December 31, 2019, respectively | 5 | | | 4 | |
| Additional paid-in capital | 625,999 |
| | 589,795 |
| Additional paid-in capital | 758,945 | | | 636,910 | |
| Accumulated deficit | (349,695 | ) | | (270,017 | ) | Accumulated deficit | (441,704) | | | (416,444) | |
| Accumulated other comprehensive income (loss) | 781 |
| | (1,529 | ) | |
| Accumulated other comprehensive income | | Accumulated other comprehensive income | 1,399 | | | 726 | |
| Total stockholders' equity | 277,089 |
| | 318,253 |
| Total stockholders' equity | 318,645 | | | 221,196 | |
| Total liabilities and stockholders' equity | $ | 647,574 |
| | $ | 709,160 |
| Total liabilities and stockholders' equity | $ | 678,694 | | | $ | 604,800 | |
The accompanying notes are an integral part of these condensed consolidated financial statements.
RETROPHIN, INC. AND SUBSIDIARIES
CONDENSED CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
(in thousands, except share and per share amounts)
(unaudited)
| | | | Three Months Ended June 30, | | Six Months Ended June 30, | | Three Months Ended June 30, | | | Six Months Ended June 30, | |
| | 2019 | | 2018 | | 2019 | | 2018 | | 2020 | | 2019 | | 2020 | | 2019 |
| Net product sales | $ | 44,707 |
| | $ | 41,337 |
| | $ | 84,277 |
| | $ | 79,769 |
| Net product sales | $ | 48,430 | | | $ | 44,707 | | | $ | 96,199 | | | $ | 84,277 | |
| Operating expenses: | |
| | |
| | | | | Operating expenses: | | | | |
| Cost of goods sold | 979 |
| | 1,178 |
| | 1,996 |
| | 2,791 |
| Cost of goods sold | 1,494 | | | 979 | | | 2,864 | | | 1,996 | |
| Research and development | 37,934 |
|
| 34,460 |
|
| 71,377 |
|
| 59,096 |
| Research and development | 30,790 | | | 37,934 | | | 61,038 | | | 71,377 | |
| Selling, general and administrative | 38,970 |
|
| 25,100 |
|
| 71,639 |
|
| 51,568 |
| Selling, general and administrative | 34,971 | | | 38,970 | | | 68,110 | | | 71,639 | |
| Change in fair value of contingent consideration | 3,353 |
| | 2,159 |
| | 6,522 |
| | 5,786 |
| Change in fair value of contingent consideration | 4,286 | | | 3,353 | | | 2,363 | | | 6,522 | |
| Impairment of L-UDCA IPR&D intangible asset | — |
| | — |
| | 25,500 |
| | — |
| |
| Write off of L-UDCA IPR&D intangible asset | | Write off of L-UDCA IPR&D intangible asset | — | | | — | | | — | | | 25,500 | |
| Write off of L-UDCA contingent consideration | — |
| | — |
| | (18,000 | ) | | — |
| Write off of L-UDCA contingent consideration | — | | | — | | | — | | | (18,000) | |
| | Total operating expenses | 81,236 |
| | 62,897 |
| | 159,034 |
| | 119,241 |
| Total operating expenses | 71,541 | | | 81,236 | | | 134,375 | | | 159,034 | |
| Operating loss | (36,529 | ) |
| (21,560 | ) |
| (74,757 | ) |
| (39,472 | ) | Operating loss | (23,111) | | | (36,529) | | | (38,176) | | | (74,757) | |
| Other income (expenses), net: | |
| | |
| | | | | Other income (expenses), net: | | | | | | | |
| Other income (expense), net | 125 |
|
| (403 | ) |
| (177 | ) |
| (282 | ) | Other income (expense), net | 426 | | | 125 | | | 235 | | | (177) | |
| Interest income | 2,589 |
|
| 858 |
|
| 5,408 |
|
| 1,655 |
| Interest income | 1,316 | | | 2,589 | | | 3,291 | | | 5,408 | |
| Interest expense | (4,817 | ) |
| (1,057 | ) |
| (9,682 | ) |
| (2,212 | ) | Interest expense | (4,634) | | | (4,817) | | | (9,521) | | | (9,682) | |
| | Total other expense, net | (2,103 | ) | | (602 | ) | | (4,451 | ) | | (839 | ) | Total other expense, net | (2,892) | | | (2,103) | | | (5,995) | | | (4,451) | |
| Loss before income taxes | (38,632 | ) | | (22,162 | ) | | (79,208 | ) | | (40,311 | ) | Loss before income taxes | (26,003) | | | (38,632) | | | (44,171) | | | (79,208) | |
| Income tax expense | (69 | ) |
| (167 | ) |
| (470 | ) |
| (396 | ) | |
| Income tax (expense) benefit | | Income tax (expense) benefit | (65) | | | (69) | | | 18,911 | | | (470) | |
| Net loss | $ | (38,701 | ) | | $ | (22,329 | ) | | $ | (79,678 | ) | | $ | (40,707 | ) | Net loss | $ | (26,068) | | | $ | (38,701) | | | $ | (25,260) | | | $ | (79,678) | |
| | | |
|
|
|
|
|
| | | | | | | | |
| Basic and diluted net loss per common share: | $ | (0.92 | ) |
| $ | (0.56 | ) |
| $ | (1.91 | ) |
| $ | (1.03 | ) | |
| Basic and diluted weighted average common shares outstanding: | 41,957,860 |
|
| 40,061,045 |
|
| 41,685,599 |
|
| 39,641,334 |
| |
| Basic and diluted net loss per common share | | Basic and diluted net loss per common share | $ | (0.58) | | | $ | (0.92) | | | $ | (0.57) | | | $ | (1.91) | |
| | Basic and diluted weighted average common shares outstanding | | Basic and diluted weighted average common shares outstanding | 44,763,843 | | | 41,957,860 | | | 43,943,370 | | | 41,685,599 | |
| | Comprehensive loss: | |
| | |
| | | | | Comprehensive loss: | | | | |
| Net loss | $ | (38,701 | ) | | $ | (22,329 | ) | | $ | (79,678 | ) | | $ | (40,707 | ) | Net loss | $ | (26,068) | | | $ | (38,701) | | | $ | (25,260) | | | $ | (79,678) | |
| Foreign currency translation | (91 | ) | | 2 |
| | 27 |
| | 24 |
| Foreign currency translation | (247) | | | (91) | | | (56) | | | 27 | |
| Unrealized gain (loss) on marketable securities | 815 |
| | 375 |
| | 2,284 |
| | (161 | ) | |
| Unrealized gain on debt securities | | Unrealized gain on debt securities | 3,146 | | | 815 | | | 729 | | | 2,284 | |
| Comprehensive loss | $ | (37,977 | ) | | $ | (21,952 | ) | | $ | (77,367 | ) | | $ | (40,844 | ) | Comprehensive loss | $ | (23,169) | | | $ | (37,977) | | | $ | (24,587) | | | $ | (77,367) | |
The accompanying notes are an integral part of these condensed consolidated financial statements.
RETROPHIN, INC. AND SUBSIDIARIES
CONDENSED CONSOLIDATED STATEMENTS OF CASH FLOWS
(unaudited, in thousands) | | | | For the Six Months Ended June 30, | | For the Six Months Ended June 30, | |
| | 2019 | | 2018 | | 2020 | | 2019 |
| Cash Flows From Operating Activities: | | | | Cash Flows From Operating Activities: | | | |
| Net loss | $ | (79,678 | ) | | $ | (40,707 | ) | Net loss | $ | (25,260) | | | $ | (79,678) | |
| Adjustments to reconcile net loss to net cash used in operating activities: | |
| | |
| Adjustments to reconcile net loss to net cash used in operating activities: | | | |
| Depreciation and amortization | 9,742 |
| | 8,991 |
| Depreciation and amortization | 11,507 | | | 9,742 | |
| | Non-cash interest expense | 798 |
| | 767 |
| Non-cash interest expense | 893 | | | 798 | |
| (Accretion) amortization of discounts/premiums on investments, net | (580 | ) | | 647 |
| |
| Amortization (accretion) of discounts/premiums on investments, net | | Amortization (accretion) of discounts/premiums on investments, net | 337 | | | (580) | |
| Amortization of debt discount and issuance costs | 4,933 |
| | 325 |
| Amortization of debt discount and issuance costs | 5,147 | | | 4,933 | |
| | Provision for Inventory | (358 | ) | | 378 |
| Provision for Inventory | 662 | | | (358) | |
| Share based compensation | 11,947 |
| | 10,160 |
| |
| ESPP Expense | 345 |
| | — |
| |
| | Share-based compensation | | Share-based compensation | 11,474 | | | 11,947 | |
| ESPP expense | | ESPP expense | 390 | | | 345 | |
| Change in fair value of contingent consideration | (11,478 | ) | | 5,786 |
| Change in fair value of contingent consideration | 2,363 | | | (11,478) | |
| Payments related to change in fair value of contingent consideration | (2,767 | ) | | (5,563 | ) | Payments related to change in fair value of contingent consideration | (8,674) | | | (2,767) | |
| Impairment of IPR&D intangible assets | 25,500 |
| | — |
| |
| | Write off of IPR&D intangible assets | | Write off of IPR&D intangible assets | — | | | 25,500 | |
| | Unrealized foreign currency transaction gain (loss) | 14 |
| | 214 |
| Unrealized foreign currency transaction gain (loss) | (222) | | | 14 | |
| Other | 60 |
| | 199 |
| Other | 496 | | | 60 | |
| Changes in operating assets and liabilities, net of business acquisitions: | |
| | |
| |
| Changes in operating assets and liabilities: | | Changes in operating assets and liabilities: | | | |
| Accounts receivable | (2,396 | ) | | 1,431 |
| Accounts receivable | 3,979 | | | (2,396) | |
| Inventory | 918 |
| | (433 | ) | Inventory | (875) | | | 918 | |
| Tax receivable | | Tax receivable | (18,714) | | | — | |
| Other current and non-current operating assets | (10,101 | ) | | 637 |
| Other current and non-current operating assets | (2,508) | | | (10,101) | |
| Accounts payable and accrued expenses | 6,305 |
| | (2,811 | ) | Accounts payable and accrued expenses | (16,407) | | | 6,305 | |
| Other current and non-current operating liabilities | 7,195 |
| | 2,485 |
| Other current and non-current operating liabilities | (196) | | | 7,195 | |
| Net cash used in operating activities | (39,601 | ) | | (17,494 | ) | Net cash used in operating activities | (35,608) | | | (39,601) | |
| Cash Flows From Investing Activities: | |
| | |
| Cash Flows From Investing Activities: | | | |
| Purchase of fixed assets | (21 | ) | | (601 | ) | Purchase of fixed assets | (518) | | | (21) | |
| Cash paid for intangible assets | (7,347 | ) | | (11,389 | ) | Cash paid for intangible assets | (8,532) | | | (7,347) | |
| Proceeds from the sale/maturity of marketable securities | 115,998 |
| | 63,565 |
| |
| Purchase of marketable securities | (94,812 | ) | | (29,519 | ) | |
| Cash paid for investments - equity | — |
| | (15,000 | ) | |
| Proceeds from the sale/maturity of debt securities | | Proceeds from the sale/maturity of debt securities | 153,146 | | | 115,998 | |
| Purchase of debt securities | | Purchase of debt securities | (36,743) | | | (94,812) | |
| | Net cash provided by investing activities | 13,818 |
| | 7,056 |
| Net cash provided by investing activities | 107,353 | | | 13,818 | |
| Cash Flows From Financing Activities: | |
| | |
| Cash Flows From Financing Activities: | | | |
| Payment of acquisition-related contingent consideration | (1,722 | ) | | (7,947 | ) | Payment of acquisition-related contingent consideration | (6,101) | | | (1,722) | |
| Payment of guaranteed minimum royalty | (1,034 | ) | | (1,000 | ) | Payment of guaranteed minimum royalty | (1,050) | | | (1,034) | |
| Payment of other liability | — |
| | (500 | ) | |
| Proceeds from exercise of warrants | — |
| | 2,140 |
| |
| | Proceeds from exercise of stock options | 317 |
| | 6,890 |
| Proceeds from exercise of stock options | 431 | | | 317 | |
| Other financing activities | 1,005 |
| | 800 |
| |
| Net cash (used in) provided by financing activities | (1,434 | ) | | 383 |
| |
| | Proceeds from issuance of common stock, net of issuance costs | | Proceeds from issuance of common stock, net of issuance costs | 108,644 | | | — | |
| | Proceeds from issuances under employee stock purchase plan | | Proceeds from issuances under employee stock purchase plan | 1,098 | | | 1,005 | |
| Net cash provided by (used in) financing activities | | Net cash provided by (used in) financing activities | 103,022 | | | (1,434) | |
| Effect of exchange rate changes on cash | 1 |
| | (34 | ) | Effect of exchange rate changes on cash | (33) | | | 1 | |
| Net change in cash and cash equivalents | (27,216 | ) | | (10,089 | ) | Net change in cash and cash equivalents | 174,734 | | | (27,216) | |
| Cash and cash equivalents, beginning of year | 102,873 |
| | 99,394 |
| Cash and cash equivalents, beginning of year | 62,436 | | | 102,873 | |
| Cash and cash equivalents, end of period | $ | 75,657 |
| | $ | 89,305 |
| Cash and cash equivalents, end of period | $ | 237,170 | | | $ | 75,657 | |
The accompanying notes are an integral part of these condensed consolidated financial statements.
RETROPHIN, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF STOCKHOLDERS' EQUITY
(unaudited, in thousands, except share amounts)
| | | | Three Months Ended June 30, 2019 | | Three Months Ended June 30, 2018 | | Three Months Ended June 30, 2020 | | | Three Months Ended June 30, 2019 | |
| | Common Stock | | Additional Paid in Capital | | Accumulated Other Comprehensive Income (Loss) | | Accumulated
Deficit | | Total
Stockholders'
Equity | | Common Stock | | Additional Paid in Capital | | Accumulated Other Comprehensive Income (Loss) | | Accumulated
Deficit | | Total
Stockholders'
Equity | | Common Stock | | | Additional Paid in Capital | | Accumulated Other Comprehensive Income (Loss) | | Accumulated
Deficit | | Total
Stockholders'
Equity | | Common Stock | | | Additional Paid in Capital | | Accumulated Other Comprehensive Income (Loss) | | Accumulated
Deficit | | Total
Stockholders'
Equity |
| | Shares | Amount | | Shares | Amount | | | Shares | Amount | | | | | | | | | | Shares | Amount | | | | | | | | |
| Balance - March 31 | 41,438,020 |
| $ | 4 |
| | $ | 596,644 |
| | $ | 57 |
| | $ | (310,994 | ) | | $ | 285,712 |
| | 39,873,285 |
| $ | 4 |
| | $486,717 | | $ | (1,529 | ) | | $ | (185,717 | ) | | $ | 299,475 |
| Balance - March 31 | 43,153,215 | | $ | 4 | | | $ | 642,880 | | | $ | (1,500) | | | $ | (415,636) | | | $ | 225,748 | | | 41,438,020 | | $ | 4 | | | $ | 596,644 | | | $ | 57 | | | $ | (310,994) | | | $ | 285,712 | |
| Share based compensation | | | | 5,577 |
| | | | | | 5,577 |
| | | | | 5,372 |
| | | | | | 5,372 |
| Share based compensation | | | 5,760 | | | | | | | 5,760 | | | | | 5,577 | | | | | | | 5,577 | |
| Issuance of common shares under the equity incentive plan and proceeds from exercise | 100,151 |
| | | 12 |
| | | | | | 12 |
| | 198,630 |
| — |
| | 2,633 |
| | | | | | 2,633 |
| |
| Exercise of warrants | | | | | | | | | | — |
| | 255,445 |
| — |
| | 1,533 |
| | | | | | 1,533 |
| |
| Unrealized gain on marketable securities | | | | | | 815 |
| | | | 815 |
| | | | | | | 375 |
| | | | 375 |
| |
| Issuance of common stock under the equity incentive plan and proceeds from exercise | | Issuance of common stock under the equity incentive plan and proceeds from exercise | 177,115 | | | 371 | | | 371 | | | 100,151 | | | 12 | | | 12 | |
| Equity offering | | Equity offering | 7,475,000 | | 1 | | 108,643 | | | 108,644 | | |
| Unrealized gain on debt securities | | Unrealized gain on debt securities | | 3,146 | | | 3,146 | | | | 815 | | | 815 | |
| Foreign currency translation adjustments | | | | | | (91 | ) | | | | (91 | ) | | | | | | | 2 |
| | | | 2 |
| Foreign currency translation adjustments | | (247) | | | (247) | | | (91) | | | (91) | |
| Issuance of common stock from maturity of the 2019 Convertible debt outstanding | 1,297,343 |
| | | 22,590 |
| | | | | | 22,590 |
| | | | | | | | | | | — |
| Issuance of common stock from maturity of the 2019 Convertible debt outstanding | | 1,297,343 | | | 22,590 | | | 22,590 | |
| ESPP stock purchase and expense | 63,804 |
| — |
| | 1,176 |
| | | | | | 1,176 |
| | 43,161 |
| — |
| | 928 |
| | | | | | 928 |
| ESPP stock purchase and expense | 97,544 | | | 1,291 | | | 1,291 | | | 63,804 | | | 1,176 | | | 1,176 | |
| Net loss | | | | | | | | (38,701 | ) | | (38,701 | ) | | | | | | | | | (22,329 | ) | | (22,329 | ) | Net loss | | (26,068) | | | (26,068) | | | (38,701) | | | (38,701) | |
| Balance - June 30 | 42,899,318 |
| $ | 4 |
| | $ | 625,999 |
| | $ | 781 |
| | $ | (349,695 | ) | | $ | 277,089 |
| | 40,370,521 |
| $ | 4 |
| | $ | 497,183 |
| | $ | (1,152 | ) | | $ | (208,046 | ) | | $ | 287,989 |
| Balance - June 30 | 50,902,874 | | $ | 5 | | | $ | 758,945 | | | $ | 1,399 | | | $ | (441,704) | | | $ | 318,645 | | | 42,899,318 | | $ | 4 | | | $ | 625,999 | | | $ | 781 | | | $ | (349,695) | | | $ | 277,089 | |
| | Six Months Ended June 30, 2019 | | Six Months Ended June 30, 2018 | | | | | | | | | | | | | | | | | | | | |
| | | Six Months Ended June 30, 2020 | | | Six Months Ended June 30, 2019 | |
| | | Common Stock | | | Additional Paid in Capital | | Accumulated Other Comprehensive Income (Loss) | | Accumulated
Deficit | | Total
Stockholders'
Equity | | Common Stock | | | Additional Paid in Capital | | Accumulated Other Comprehensive Income (Loss) | | Accumulated
Deficit | | Total
Stockholders'
Equity |
| | | Shares | Amount | | | | | | | | | | Shares | Amount | | | | | | | | |
| Balance - December 31 | 41,389,524 |
| $ | 4 |
| | $ | 589,795 |
| | $ | (1,529 | ) | | $ | (270,017 | ) | | $ | 318,253 |
| | 39,373,745 |
| $ | 4 |
| | $ | 471,800 |
| | $ | (1,015 | ) | | $ | (177,655 | ) | | $ | 293,134 |
| Balance - December 31 | 43,088,921 | | $ | 4 | | | $ | 636,910 | | | $ | 726 | | | $ | (416,444) | | | $ | 221,196 | | | 41,389,524 | | $ | 4 | | | $ | 589,795 | | | $ | (1,529) | | | $ | (270,017) | | | $ | 318,253 | |
| Adoption of ASU 2017-11 - reclassification of derivative liability of warrants with down round provisions | | | | | | | | | | — |
| | | | | 5,394 |
| | | | 10,316 |
| | 15,710 |
| |
| Share based compensation | | | | 11,947 |
| | | | | | 11,947 |
| | | | | 9,915 |
| | | | | | 9,915 |
| Share based compensation | | | | 11,474 | | | | | | | 11,474 | | | | | 11,947 | | | | | | | 11,947 | |
| Issuance of common shares under the equity incentive plan and proceeds from exercise | 148,647 |
| | | 317 |
| | | | | | 317 |
| | 529,558 |
| — |
| | 6,890 |
| | | | | | 6,890 |
| |
| Exercise of warrants | | | | | | | | | | — |
| | 424,057 |
| — |
| | 2,140 |
| | | | | | 2,140 |
| |
| Unrealized gain (loss) on marketable securities | | | | | | 2,284 |
| | | | 2,284 |
| | | | | | | (161 | ) | | | | (161 | ) | |
| Issuance of common stock under the equity incentive plan and proceeds from exercise | | Issuance of common stock under the equity incentive plan and proceeds from exercise | 241,409 | | | 431 | | | 431 | | | 148,647 | | | 317 | | | 317 | |
| Equity offering | | Equity offering | 7,475,000 | | 1 | | 108,643 | | | 108,644 | | |
| Unrealized gain on debt securities | | Unrealized gain on debt securities | | | 729 | | | 729 | | | 2,284 | | | 2,284 | |
| Foreign currency translation adjustments | | | | | | 26 |
| | | | 26 |
| | | | | | | 24 |
| | | | 24 |
| Foreign currency translation adjustments | | | (56) | | | (56) | | | 26 | | | 26 | |
| Issuance of common stock from maturity of the 2019 Convertible debt outstanding | 1,297,343 |
| — |
| | 22,590 |
| | | | | | 22,590 |
| | | | | | | | | | | — |
| Issuance of common stock from maturity of the 2019 Convertible debt outstanding | | | | 1,297,343 | | | 22,590 | | | | | 22,590 | |
| ESPP stock purchase and expense | 63,804 |
| — |
| | 1,350 |
| | | | | | 1,350 |
| | 43,161 |
| | | 1,044 |
| | | | | | 1,044 |
| ESPP stock purchase and expense | 97,544 | | | 1,487 | | | 1,487 | | | 63,804 | | | 1,350 | | | 1,350 | |
| Net loss | | | | | | | | (79,678 | ) | | (79,678 | ) | | | | | | | | | (40,707 | ) | | (40,707 | ) | Net loss | | | (25,260) | | | (25,260) | | | (79,678) | | | (79,678) | |
| Balance - June 30 | 42,899,318 |
| $ | 4 |
| | $ | 625,999 |
| | $ | 781 |
| | $ | (349,695 | ) | | $ | 277,089 |
| | 40,370,521 |
| $ | 4 |
| | $ | 497,183 |
| | $ | (1,152 | ) | | $ | (208,046 | ) | | $ | 287,989 |
| Balance - June 30 | 50,902,874 | | $ | 5 | | | $ | 758,945 | | | $ | 1,399 | | | $ | (441,704) | | | $ | 318,645 | | | 42,899,318 | | $ | 4 | | | $ | 625,999 | | | $ | 781 | | | $ | (349,695) | | | $ | 277,089 | |
|
The accompanying notes are an integral part of these condensed consolidated financial statements.
RETROPHIN, INC. AND SUBSIDIARIES
NOTES TO THE UNAUDITED CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
NOTE 1. DESCRIPTION OF BUSINESS
Organization and Description of Business
Retrophin, Inc. (“we”, “our”, “us”, “Retrophin” and the “Company”) refers to Retrophin, Inc., a Delaware corporation, as well as our direct and indirect subsidiaries. Retrophin is a fully integrated biopharmaceutical company headquartered in San Diego, California, focused on identifying, developing and delivering life-changing therapies to people living with rare diseases. We regularly evaluate and, where appropriate, act on opportunities to expand our product pipeline through licenses and acquisitions of products in areas that will serve patients with serious or rare diseases and that we believe offer attractive growth characteristics.
On January 30, 2020, the World Health Organization (“WHO”) declared that the recent novel coronavirus (COVID-19) outbreak was a global health emergency, which prompted national governments to begin putting actions in place to slow the spread of COVID-19. In March 2020, the WHO classified the COVID-19 outbreak as a pandemic. The outbreak of COVID-19 has resulted in travel restrictions, quarantines, “stay-at-home” and “shelter-in-place” orders and extended shutdown of certain businesses around the world. While the impact of the COVID-19 pandemic did not have a material adverse effect on our financial position or results of operations for the three and six months ended June 30, 2020, these governmental actions and the widespread economic disruption arising from the pandemic have the potential to materially impact our business and influence our business decisions. The extent and duration of the pandemic is unknown, and the future effects on our business are uncertain and difficult to predict. The Company is developingcontinuing to monitor the following pipeline products:
The Company is developing fosmetpantotenate (RE-024), a novel small molecule, as a potential treatment for pantothenate kinase-associated neurodegeneration (“PKAN”). PKAN is a genetic neurodegenerative disorder that is typically diagnosedevents and circumstances surrounding the COVID-19 pandemic, which may require adjustments to the Company’s estimates and assumptions in the first decade of life.future.
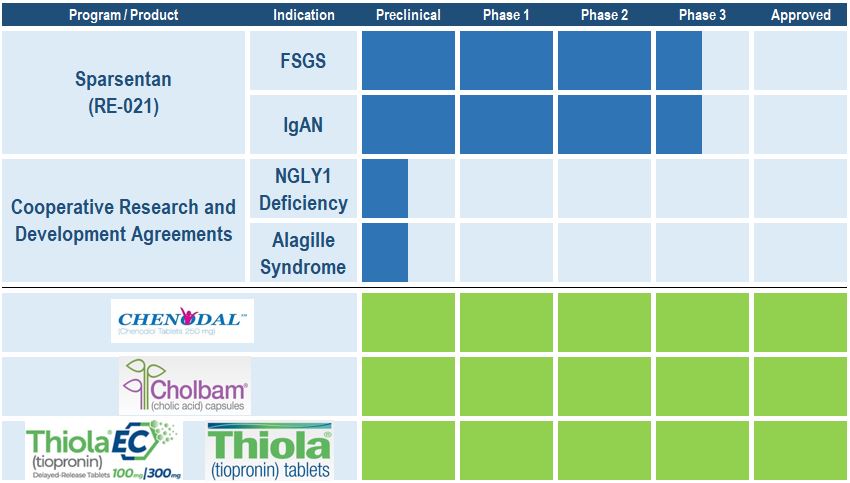
Clinical Programs:
Sparsentan, also known as RE-021, is an investigational product candidate with a dual mechanism of action, a potent angiotensin receptor blocker (“ARB”) and selective endothelin receptor antagonist (“ERA”), with in vitro selectivity toward endothelin receptor type A.A, and a potent angiotensin receptor blocker (“ARB”). Sparsentan is currently being evaluated in two pivotal Phase 3 clinical studies in the following indications:
•Focal segmental glomerulosclerosis ("FSGS") is a rare kidney disease characterized by proteinuria where the glomeruli become progressively scarred. FSGS is a leading cause of end-stage renal disease.
•Immunoglobulin A nephropathy ("IgAN") is an immune-complex-mediated glomerulonephritis characterized by hematuria, proteinuria, and variable rates of progressive renal failure. IgAN is the most common primary glomerular disease.
Chenodal® has been recognized as the standard of care for cerebrotendinous xanthomatosis (“CTX”) patients for more than three decades but is not currently labeled for this indication. In September 2017,January 2020, we randomized the Company entered intofirst patients in a three-way Phase 3 clinical trial to evaluate the effects of Chenodal in adult and pediatric patients with CTX. The pivotal study, known as the RESTORE study, is intended to support a new drug application (“NDA”) submission for marketing authorization of Chenodal for CTX in the United States.
Cooperative Research and Development AgreementAgreements ("CRADA"CRADAs"):
The Company is a participant in two CRADAs, which form a multi-stakeholder approach to pool resources with leading experts, and incorporates the patient perspective early in the identification and development process. Retrophin has partnered with the National Institutes of Health’s National Center for Advancing Translational Sciences ("NCATS") and leading patient advocacy foundationorganizations, NGLY1.org to collaborate on research effortsand Alagille Syndrome Alliance ("ALGSA"), aimed at the identification of potential small molecule therapeutics for NGLY1 deficiency.
In June 2019, the Company announced that it has entered into a three-way CRADA with NCATSdeficiency and patient advocacy foundation Alagille Syndrome Alliance ("ALGSA") to collaborate on research efforts aimed at the identification and development of potentially novel therapeutics for Alagille syndrome, ("ALGS").respectively. There are no treatment options currently approved for these diseases.
The Company sells the following threeApproved products:
•Chenodal (chenodiol tablets) is approved in the United States for the treatment of patients suffering from gallstones in whom surgery poses an unacceptable health risk due to disease or advanced age. Chenodal has been the standard of care for cerebrotendinous xanthomatosis ("CTX")CTX patients for more than three decades and the Company is currently pursuing adding this indication to the label.decades.
Cholbam•Cholbam® (cholic acid capsules) is approved in the United States for the treatment of bile acid synthesis disorders due to single enzyme defects and is further indicated for adjunctive treatment of patients with peroxisomal disorders.
•Thiola® and Thiola EC® (tiopronin tablets) isare approved in the United States for the prevention of cystine (kidney) stone formation in patients with severe homozygous cystinuria. On June 28, 2019, the Company announced that the U.S. Food and Drug Administration ("FDA") approved 100 mg and 300 mg tablets
NOTE 2. BASIS OF PRESENTATION AND SIGNIFICANT ACCOUNTING POLICIES
The accompanying unaudited condensed consolidated financial statements should be read in conjunction with the audited consolidated financial statements and notes thereto included in the 20182019 10-K filed with the SEC on February 26, 2019.24, 2020. The accompanying condensed consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States (“GAAP”) for interim financial information, the instructions for Form 10-Q and the rules and regulations of the SEC. Accordingly, since they are interim statements, the accompanying condensed consolidated financial statements do not include all of the information and notes required by GAAP for annual financial statements, but reflect all adjustments consisting of normal, recurring adjustments, that are necessary for a fair presentation of the financial position, results of operations and cash flows for the interim periods presented. Interim results are not necessarily indicative of the results that may be expected for any future periods. The December 31, 20182019 balance sheet information was derived from the audited financial statements as of that date. Certain reclassifications have been made to the prior period consolidated financial statements to conform to the current period presentation.
A summary of the significant accounting policies applied in the preparation of the accompanying condensed consolidated financial statements follows:
Principles of Consolidation
The unaudited condensed consolidated financial statements represent the consolidation of the accounts of the Company and its subsidiaries in conformity with GAAP. All intercompany accounts and transactions have been eliminated in consolidation.
Revenue Recognition
The Company recognizes revenue when its customer obtains control of promised goods or services, in an amount that reflects the consideration which the entity expects to receive in exchange for those goods or services. To determine revenue recognition for arrangements that an entity determines are within the scope of Topic 606, the entity performs the following five steps: (i) identify the contract(s) with a customer; (ii) identify the performance obligations in the contract; (iii) determine the transaction price; (iv) allocate the transaction price to the performance obligations in the contract; and (v) recognize revenue when (or as) the entity satisfies a performance obligation. The Company only applies the five-step model to contracts when it is probable that the entity will collect substantially all the consideration it is entitled to in exchange for the goods or services it transfers to the customer. See Note 3 for further discussion.
Research and Development Expenses
Research and development expenses are comprised of salaries and bonuses, benefits, non-cash share-based compensation, license fees, costs paid to third-party contractors to perform research, conduct clinical trials and pre/non-clinical trials, develop drug materials, and associated overhead expenses and facilities. We also incur indirect costs that are not allocated to specific programs because such costs benefit multiple development programs and allow us to increase our pharmaceutical development capabilities. These consist of internal shared resources related to the development and maintenance of systems and processes applicable to all of our programs.
Clinical Trial Expenses
Our clinical trials are conducted pursuant to contracts with contract research organizations ("CROs") that support conducting and managing clinical trials. The financial terms and activities of these agreements vary from contract to contract and may result in uneven expense levels. Generally, these agreements set forth activities that drive the recording of expenses such as start-up, initiation activities, enrollment, treatment of patients, or the completion of other clinical trial activities.
Expenses related to clinical trials are accrued based on our estimates and/or representations from service providers regarding work performed, including actual level of patient enrollment, completion of patient studies and progress of the clinical trials, and completion of patient studies.trials. Other incidental costs related to patient enrollment or treatment are accrued when reasonably certain. If the amounts we are obligated to pay under our clinical trial agreements are modified (for instance, as a result of changes in the clinical trial protocol or scope of work to be performed), we adjust our accruals accordingly on a prospective basis. Revisions to our contractual payment obligations are charged to expense in the period in which the facts that give rise to the revision become reasonably certain.
We currently have three Phase 3 clinical trials in process that are in varying stages of activity, with ongoing non-clinical support trials. As such, clinical trial expenses will vary depending on all the factors set forth above and may fluctuate significantly from quarter to quarter.
Adoption of New Accounting Standards
In February 2016, the FASB issued ASU No. 2016-02, Leases. The new standard establishes a right-of-use ("ROU") model that requires a lessee to record a ROU asset and a lease liability on the balance sheet for all leases with terms longer than 12 months. Leases will be classified as either finance or operating, with classification affecting the pattern of expense recognition in the income statement. The new standard is effective for fiscal years beginning after December 15, 2018, including interim periods within those fiscal years. See Note 6 for further discussion.
Recently Issued Accounting Pronouncements
From time to time, new accounting pronouncements are issued by the FASB or other standard setting bodies. Unless otherwise noted, the Company believes that the impact of recently issued standards that are not yet effective will not have a material impact on its consolidated financial position or results of operations upon adoption.
In June 2016, the FASB issued ASU No. 2016-13, Financial Instruments - Credit Losses (Topic 326): Measurement of Credit Losses on Financial Instruments. Topic 326 amends guidance on reporting credit losses for assets held at amortized cost basis and available-for-sale debt securities. For assets held at amortized cost basis, Topic 326 eliminates the probable initial recognition threshold in current GAAP and, instead, requires an entity to reflect its current estimate of all expected credit losses. The allowance for credit losses is a valuation account that is deducted from the amortized cost basis of the financial assets to present the net amount expected to be collected. For available-for-sale debt securities, credit losses should be measured in a manner similar to current GAAP, however Topic 326 will require that credit losses be presented as an allowance rather than as a write-down. This ASU update affects entities holding financial assets and net investment in leases that are not accounted for at fair value through net income. The amendments affect loans, debt securities, trade receivables, net investments in leases, off balance sheet credit exposures, reinsurance receivables, and any other financial assets not excluded from the scope that have the contractual right to receive cash. This update is effective for fiscal years beginning after December 15, 2019, including interim periods within those fiscal years. The Company's adoption of this standard has had an immaterial impact on our condensed consolidated financial statements, and there was no recorded impact to our opening accumulated deficit balance for the cumulative-effect adjustment. As of June 30, 2019,2020, the Company held $350.2$14.1 million in trade receivables and $220.2 million in available-for-sale debt securities. If adoptedExpected credit losses on our trade receivables are estimated to be immaterial and the Company has 0 recorded allowances for expected credit losses on the available-for-sale debt securities held as of June 30, 2019, this ASU update would not have a material impact on the Company's financial statements.2020. See Note 16 and Note 4 for further discussion.
NOTE 3. REVENUE RECOGNITION
Product Revenue, Net
The Company sells ChenodalProduct sales consist of Bile Acid products (Chenodal and Cholbam (Kolbam), which are aggregated as bile acidCholbam) and Tiopronin products (Thiola and Thiola through direct-to-patient distributors.EC). The Company sells its products through direct-to-patient distributors worldwide, with more than 95% of the revenue generated in North America.
Revenues from product sales are recognized when the customer obtains control of the Company’s product, which occurs upon delivery to the customer. The Company receives payments from its product sales based on terms that generally are within 30 days of delivery of product to the patient.
Deductions from Revenue
Revenues from product sales are recorded at the net sales price, which includes provisions resulting from discounts, rebates and co-pay assistance that are offered to customers, health care providers, payors and other indirect customers relating to the Company’s sales of its products. These provisions are based on the amounts earned or to be claimed on the related sales and are classified as a reduction of accounts receivable (if the amount is payable to a customer) or as a current liability (if the amount is payable to a party other than a customer). Where appropriate, these reserves take into consideration the Company’s historical experience, current contractual and statutory requirements and specific known market events and trends, industry data and forecasted customer buying and payment patterns.trends. Overall, these reserves reflect the Company’s best estimates of the amount of consideration to which it is entitled based on the terms of the contract. If actual results in the future vary from the Company’s provisions, the Company will adjust the provision, which would affect net product revenue and earnings in the period such variances become known. Our historical experience is that such adjustments have been immaterial.
Government Rebates: We calculate the rebates that we will be obligated to provide to government programs and deduct these estimated amounts from our gross product sales at the time the revenues are recognized. Allowances for government rebates and discounts are established based on actual payer information, which is reasonably estimated at the time of delivery, and the government-mandated discounts applicable to government-funded programs. Rebate discounts are included in other current liabilities in the accompanying consolidated balance sheets.
Commercial Rebates: We calculate the rebates that we incur due to contracts with certain commercial payors and deduct these amounts from our gross product sales at the time the revenues are recognized. Allowances for commercial rebates are established based on actual payer information, which is reasonably estimated at the time of delivery. Rebate discounts are included in other current liabilities in the accompanying consolidated balance sheets.
Prompt Pay Discounts: We offer discounts to certain customers for prompt payments. We accrue for the calculated prompt pay discount based on the gross amount of each invoice for those customers at the time of sale.
Product Returns: Consistent with industry practice, we offer our customers a limited right to return product purchased directly from the Company, which is principally based upon the product’s expiration date. Generally, shipments are only made upon a patient prescription thus returns are minimal.
Co-pay Assistance: We offer a co-pay assistance program, which is intended to provide financial assistance to qualified commercially insured patients with prescription drug co-payments required by payors. The calculation of the accrual for co-pay assistance is based on an identification of claims and the cost per claim associated with product that has been recognized as revenue.
The following table summarizes net product revenues for the three and six months ended June 30, 20192020 and 20182019 (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | |
| Three Months Ended June 30, | | | | Six Months Ended June 30, | | |
| 2020 | | 2019 | | 2020 | | 2019 |
| Bile acid products | $ | 21,573 | | | $ | 20,929 | | | $ | 43,854 | | | $ | 39,319 | |
| Tiopronin products | 26,857 | | | 23,778 | | | 52,345 | | | 44,958 | |
| Total net product revenue | $ | 48,430 | | | $ | 44,707 | | | $ | 96,199 | | | $ | 84,277 | |
|
| | | | | | | | | | | | | | | |
| | Three Months Ended June 30, | | Six Months Ended June 30, |
| | 2019 | | 2018 | | 2019 | | 2018 |
| Bile acid products | $ | 20,929 |
| | $ | 18,594 |
| | $ | 39,319 |
|
| $ | 37,102 |
|
| Thiola | 23,778 |
| | 22,743 |
| | 44,958 |
|
| 42,667 |
|
| Total net product revenue | $ | 44,707 |
| | $ | 41,337 |
| | $ | 84,277 |
| | $ | 79,769 |
|
NOTE 4. MARKETABLEDEBT SECURITIES
The Company's marketabledebt securities as of June 30, 20192020 and December 31, 20182019 were comprised of available-for-sale marketablecorporate and govenrment debt securities. These securities which are carried at fair value, with the unrealized gains and losses reported in accumulated other comprehensive income (loss). Realized gains and losses and declines in value judged, unless an impairment is determined to be other-than-temporary, if any, on available-for-sale securities are included in other incomethe result of credit-related factors or expense. Interest and dividends on securities classified as available-for-sale are included in interest income.the Company intends to sell the security or it is more likely than not that the Company will be required to sell the security before recovery. The amortized cost of debt securities is adjusted for amortization of premiums and accretion of discounts to maturity. Such amortization and accretion is included in interest income. Realized gains and losses and declines in value that are determined to be the result of credit losses, if any, on available-for-sale securities are included in other income or expense. Unrealized losses that are determined to be credit-related are also recorded as an allowance against the amortized cost basis. The cost of securities sold is based on the specific identification method. Interest and dividends on securities classified as available-for-sale are included in interest income. All available-for-sale securities are classified as current assets, even if the maturity when acquired by the Company is greater than one year due to the ability to liquidate within the next 12 months.
During the six months ended June 30, 2019,2020, investment activity for the Company included $116.0$153.3 million in maturities and $94.8$36.7 million in purchases, all relating to debt based marketable securities.
MarketableDebt securities consisted of the following (in thousands):
| | | | June 30, 2019 | | December 31, 2018 | | June 30, 2020 | | December 31, 2019 |
| Commercial paper | $ | 43,675 |
| | $ | 59,255 |
| Commercial paper | $ | — | | | $ | 17,152 | |
| Corporate debt securities | 306,568 |
| | 299,413 |
| Corporate debt securities | 210,205 | | | 306,436 | |
| Securities of government sponsored entities | — |
| | 10,000 |
| Securities of government sponsored entities | 10,001 | | | 12,500 | |
| Total marketable securities: | $ | 350,243 |
| | $ | 368,668 |
| |
| Total debt securities: | | Total debt securities: | $ | 220,206 | | | $ | 336,088 | |
The following is a summary of short-term debt securities classified as available-for-sale as of June 30, 2020 (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Remaining Contractual Maturity
(in years) | | Amortized Cost | | Unrealized Gains | | Unrealized Losses | | Aggregate Estimated Fair Value |
| Commercial paper | Less than 1 | | $ | — | | | $ | — | | | $ | — | | | $ | — | |
| Corporate debt securities | Less than 1 | | 132,352 | | | 770 | | | — | | | 133,122 | |
| | | | | | | | | |
| Total maturity less than 1 year | | | 132,352 | | | 770 | | | — | | | 133,122 | |
| Corporate debt securities | 1 to 2 | | 76,244 | | | 848 | | | (9) | | | 77,083 | |
| Securities of government-sponsored entities | 1 to 2 | | 10,000 | | | 1 | | | — | | | 10,001 | |
| Total maturity 1 to 2 years | | | 86,244 | | | 849 | | | (9) | | | 87,084 | |
| Total available-for-sale securities | | | $ | 218,596 | | | $ | 1,619 | | | $ | (9) | | | $ | 220,206 | |
The following is a summary of short-term marketable securities classified as available-for-sale as of June 30, 2019 (in thousands): |
| | | | | | | | | | | | | | | | | |
| | Remaining Contractual Maturity
(in years) | | Amortized Cost | | Unrealized Gains | | Unrealized Losses | | Aggregate Estimated Fair Value |
| Commercial paper | Less than 1 | | $ | 43,632 |
| | $ | 43 |
| | $ | — |
| | $ | 43,675 |
|
| Corporate debt securities | Less than 1 | | 178,776 |
| | 315 |
| | (49 | ) | | 179,042 |
|
| Total maturity less than 1 year | | | 222,408 |
| | 358 |
| | (49 | ) | | 222,717 |
|
| Corporate debt securities | 1 to 2 | | 126,833 |
| | 700 |
| | (7 | ) | | 127,526 |
|
| Total maturity 1 to 2 years | | | 126,833 |
| | 700 |
| | (7 | ) | | 127,526 |
|
| Total available-for-sale securities | | | $ | 349,241 |
| | $ | 1,058 |
| | $ | (56 | ) | | $ | 350,243 |
|
The following is a summary of short-term marketabledebt securities classified as available-for-sale as of December 31, 20182019 (in thousands):
|
| | | | | | | | | | | | | | | | | |
| | Remaining Contractual Maturity (in years) | | Amortized Cost | | Unrealized Gains | | Unrealized Losses | | Aggregate Estimated Fair Value |
| Commercial paper | Less than 1 | | $ | 59,313 |
| | $ | — |
| | $ | (58 | ) | | $ | 59,255 |
|
| Corporate debt securities | Less than 1 | | 149,824 |
| | — |
| | (604 | ) | | 149,220 |
|
| Total maturity less than 1 year | | | 209,137 |
| | — |
| | (662 | ) | | 208,475 |
|
| Corporate debt securities | 1 to 2 | | 150,813 |
| | 18 |
| | (638 | ) | | 150,193 |
|
| Securities of government-sponsored entities | 1 to 2 | | 9,997 |
| | 4 |
| | (1 | ) | | 10,000 |
|
| Total maturity 1 to 2 years | | | 160,810 |
| | 22 |
| | (639 | ) | | 160,193 |
|
| Total available-for-sale securities | | | $ | 369,947 |
| | $ | 22 |
| | $ | (1,301 | ) | | $ | 368,668 |
|
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Remaining Contractual Maturity
(in years) | | Amortized Cost | | Unrealized Gains | | Unrealized Losses | | Aggregate Estimated Fair Value |
| Commercial paper | Less than 1 | | $ | 17,136 | | | $ | 16 | | | $ | — | | | $ | 17,152 | |
| Corporate debt securities | Less than 1 | | 191,770 | | | 582 | | | (10) | | | 192,342 | |
| | | | | | | | | |
| Total maturity less than 1 year | | | 208,906 | | | 598 | | | (10) | | | 209,494 | |
| Corporate debt securities | 1 to 2 | | 113,799 | | | 351 | | | (56) | | | 114,094 | |
| Securities of government-sponsored entities | 1 to 2 | | 12,501 | | | — | | | (1) | | | 12,500 | |
| Total maturity 1 to 2 years | | | 126,300 | | | 351 | | | (57) | | | 126,594 | |
| Total available-for-sale securities | | | $ | 335,206 | | | $ | 949 | | | $ | (67) | | | $ | 336,088 | |
The primary objective of the Company’s investment portfolio is to enhance overall returns while preserving capital and liquidity. The Company’s investment policy limits interest-bearing security investments to certain types of instruments issued by institutions with primarily investment grade credit ratings and places restrictions on maturities and concentration by asset class and issuer. All available-for-sale securities are held in current assets regardless of contractual maturities exceeding one year, as the Company has the ability to sell them within the next twelve months.
The Company reviews the available-for-sale investmentsdebt securities for other-than-temporary declines in fair value below the cost basis each quarter and whenever events or changes in circumstances indicate that thequarter. For any security whose fair value is below its amortized cost basis, of an asset may not be recoverable. This evaluation is based on a number of factors, including the length of time and the extent to which the fair value has been below the cost basis and adverse conditions related specifically to the security, including any changes to the credit rating of the security, and the intentCompany first evaluates whether it intends to sell the impaired security, or whether the Company will otherwise be more likely than not be required to sell the security before recovery. If either are true, the amortized cost basis of the security is written down to its fair value at the reporting date. If neither circumstance holds true, the Company assesses whether any portion of the unrealized loss is a result of a credit loss. Any amount deemed to be attributable to credit loss is recognized in the income statement, with the amount of the loss limited to the difference between fair value and amortized cost and recorded as an allowance for credit losses. The portion of the unrealized loss related to factors other than credit losses is recognized in other comprehensive income (loss).
The following is a summary of available-for-sale debt securities in an unrealized loss position with no credit losses reported as of June 30, 2020 (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Less Than 12 Months | | | | 12 Months or Greater | | | | Total | | |
| Description of Securities | | Fair Value | | Unrealized Losses | | Fair Value | | Unrealized Losses | | Fair Value | | Unrealized Losses |
| Commercial paper | | $ | — | | | $ | — | | | $ | — | | | $ | — | | | $ | — | | | $ | — | |
| Corporate debt securities | | 5,015 | | | 9 | | | — | | | — | | | 5,015 | | | 9 | |
| Securities of government-sponsored entities | | — | | | — | | | — | | | — | | | — | | | — | |
| Total | | $ | 5,015 | | | $ | 9 | | | $ | — | | | $ | — | | | $ | 5,015 | | | $ | 9 | |
The following is a summary of available-for-sale debt securities in an unrealized loss position with no credit losses reported as of December 31, 2019 (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Less Than 12 Months | | | | 12 Months or Greater | | | | Total | | |
| Description of Securities | | Fair Value | | Unrealized Losses | | Fair Value | | Unrealized Losses | | Fair Value | | Unrealized Losses |
| Commercial paper | | $ | — | | | $ | — | | | $ | — | | | $ | — | | | $ | — | | | $ | — | |
| Corporate debt securities | | 74,151 | | | 64 | | | 7,509 | | | 2 | | | 81,660 | | | 66 | |
| Securities of government-sponsored entities | | 5,000 | | | 1 | | | — | | | — | | | 5,000 | | | 1 | |
| Total | | $ | 79,151 | | | $ | 65 | | | $ | 7,509 | | | $ | 2 | | | $ | 86,660 | | | $ | 67 | |
As of June 30, 2020 and December 31, 2019, the Company does not intend to sell these investments and it is not more likely than not that the Company will be required to sell the investments before recovery of itstheir amortized cost basis. The assessment of whether a security is other-than-temporarily impaired could changeCompany does not believe the unrealized losses incurred during the period are due to credit-related factors. Liquidity issues that arose from economic circumstances surrounding the COVID-19 pandemic have begun to ease and unrealized losses observed in the future duefirst quarter 2020 have been substantially recovered. The credit ratings of the securities held remain of the highest quality, and while certain securities in the portfolio may be downgraded momentarily, the Federal Reserve has allowed institutions to new developments or changes in assumptions relatedcontinue to any particular security. As of June 30, 2019 and December 31, 2018,issue debt where there is need, with the government itself purchasing such securities. Moreover, the Company believedcontinues to receive payments of interest and principal as they become due, and our expectation is that those payments will continue to be received timely. Uncertainty surrounding the cost basis for available-for-sale investments was recoverableCOVID-19 pandemic, as well as other factors unknown to us at this time, may cause actual results to differ and require adjustments to the Company’s estimates and assumptions in all material respects.the future.
NOTE 5. FUTURE ACQUISITION RIGHT AND JOINT DEVELOPMENT AGREEMENT
Censa Pharmaceuticals Inc.
In December 2017, the Company entered into a Future Acquisition Right and Joint Development Agreement (the “Option Agreement”) with Censa, which became effective in January 2018. The Company made an upfront payment of $10.0 million, agreed to fund certain development activities of Censa’s CNSA-001 program which were expected to be approximately $19.9 million through proof of concept, and paid $5.0 million related to a development milestone for the right, but not the obligation, to acquire Censa (the “Option”) on the terms and subject to the conditions set forth in a separate Agreement and Plan of Merger. The Company capitalized the upfront and milestone payments and expensed the development funding as incurred. The Company treated the upfront payment and milestone payment, both of which were consideration for the Option, as a cost-method investment with a carrying value of $15.0 million as of June 30, 2019.
Ifand expensed the Company had exercised the Option, the Company would have acquired Censa for an additional $65.0 million, which would have been reduced by up to $2.8 million of development funding ("creditable"), paid as a combination of 20% in cash and 80% in shares of the Company’s common stock, valued at a fixed price of $21.40 per share; provided, however, that Censa could have elected on behalf of its equity holders to receive the
upfront consideration in 100% cash if the average price per share of the Company’s common stock for the ten trading days ending on the date the Company provided notice of interest to exercise the Option was less than $19.26. In addition, if the Company had exercised the Option and acquired Censa, the Company would have been required to make further cash payments to Censa’s equity holders of up to an aggregate of $25.0 million if the CNSA-001 program had achieved specified development and commercial milestones.
The Company determined that Censa was a variable interest entity ("VIE"), and concluded that the Company was not the primary beneficiary of the VIE. As such, the Company did not consolidate Censa’s results into its consolidated financial statements.
The following table presents the Company’s development funding roll-forward through June 30, 2019 and its effect on the upfront consideration if the Company exercised the Option (in thousands):
|
| | | |
| | June 30, 2019 |
| Non-creditable development funding commitment | $ | 17,091 |
|
| Development funding creditable against purchase option | 2,831 |
|
| Total development funding | 19,922 |
|
| Development funding paid through June 30, 2019 | 19,308 |
|
| Development funding payable | $ | 614 |
|
In August 2019, following a strategic review of the CNSA-001 program in patients with phenylketonuria (PKU), the Company made the decision to decline to exercise its option to acquire Censa Pharmaceuticals and accordingly discontinue its joint development program for CNSA-001. The Company expects to impairwrote off the $15$15.0 million long term investment on the balance sheet during the third quarter of 2019.investment.
NOTE 6. LEASES
As of January 1, 2019 the Company adopted ASU No. 2016-02, Leases, using a modified retrospective basis method under which prior comparative periods are not restated.
The new standard establishes an ROU model that requires a lessee to record a ROU asset and a lease liability on the balance sheet for all leases with terms longer than 12 months. Leases will be classified as either finance or operating, with classification affecting the pattern of expense recognition in the income statement. In addition, the FASB issued ASU No. 2018-10, Codification Improvements to Topic 842, ASU No. 2018-11, Targeted Improvements, and ASU No. 2018-20, Narrow-Scope Improvements for Lessors, to clarify and amend the guidance in ASU No. 2016-02. The Company has elected the following as practical expedients from within these ASUs: 1) an entity need not reassess whether any expired or existing contractsare or contain leases, 2) an entity need not reassess the lease classification for any expired or existing leases, and 3) an entity need not reassess initial direct costsfor any existing leases.
As of January 1, 2019June 30, 2020, the Company had a singletwo operating leaseleases with Kilroy Realty, L.P. (the "Landlord") for its office space located in San Diego, California. The Company currently occupies the office space subject to the first lease, was originally signed in July 2016 wasand later amended in July 2017 and isMarch 2019. In April 2019, the Company entered into a second office lease with the Landlord, which was subsequently amended in May 2020, for approximately 45,000 square feet of office space in an adjacent buildings.building located in San Diego, California. The termCompany plans to consolidate its corporate headquarters into the new lease space beginning in late 2020 and into early 2021.
As a condition of the originalnew lease, isthe Company has been granted an existing premises continuation option to continue leasing all or a portion of the currently occupied office space, in addition to the new lease space. On February 7, years, 7 months, and is coterminous for all2020, the Company elected to forgo this continuation option by notifying the Landlord of its intention to vacate the premises of its existing lease. The abandonment of this leased office space, occurringwhich was originally scheduled to expire in July 2024. Under2024, coincides with the termsCompany’s expected occupancy of the new office lease space in the adjacent building per the office lease effective April 2019. The Company estimates that it will fully vacate the existing leases in the first quarter of 2021, through which time the Company is obligated to pay all base rent, operating expenses and other obligations due under the existing lease.
Coinciding with the notice delivered to the Landlord, the Company recorded an adjustment to the ROU asset and lease liability in February 2020 to reflect the impending expiration of the existing lease. The remeasurement of the lease liability resulted in a $7.0 million adjustment to the lease liability and ROU asset. A revised timing estimate resulted in the Company will pay base annual rent (subjectrecording an additional adjustment, resulting in a reduction of $0.5 million to an annual fixed percentage increase), plus property taxesthe related lease liability and other normal and necessary expenses, such as utilities, repairs, security and maintenance. Certain incentives were includedROU asset in the current quarter. The remeasured straight-line expense will be amortized over the revised lease including approximately $2.3 million in tenant improvement allowancesterm along with acceleration of related lease incentives.
The ROU asset and seven months of rent abatement. Thelease liability related to the new office lease will be established when the Company is granted access to the premises and has the rightability to extenddirect its use, which is expected to occur in phases over the remainder of 2020 as certain spaces become available, the timing of which may be impacted due to circumstances surrounding the COVID-19 pandemic. As such, the ROU asset for the new lease will be established prior to the extinguishment of the ROU asset and lease liability for five years.the existing lease, resulting in an overlap of ROU assets during the interim period between inception of the new lease and
the Company's remaining minimumexpiration of the old lease. In June 2020, the Landlord delivered possession of a portion of the new lease paymentsspace for the purpose of construction of leasehold improvements. Coinciding with our ability to direct the use of that space, and unamortized lease incentives were approximately $14.0 million and $1.8 million, respectively. Usingutilizing a discount rate equal to our borrowing rate of 7.7%6.7%, the Company has established a ROU asset totaling $8.9 million and a remaining termlease liability totaling $8.8 million. The ROU asset and lease liability are offset by lease incentives associated with tenant improvement allowances totaling $2.0 million. Establishment of 5 years, 7 months, the Company determined the ROU asset and lease liability as of adoption were $7.9 million and $11.3 million, respectively. There was no cumulative adjustment to our beginning accumulated deficit balance.
In March 2019, the company amended the existing office lease to add approximately 16,000 square feet of office space in adjacent buildings; 5,700 square feet has been occupied as of June 30, 2019. The total additional space is expected to be utilized through August 2020 and has future minimum lease payments of approximately $1.0 million. The Company determined the ROU asset and lease liability were each $0.4 million for the remaining spaces under the new lease space that has commenced and is occupied as of June 30, 2019.
On April 23, 2019, the Company entered into an office lease with an effective date of April 12, 2019 with Kilroy Realty, L.P. (the "Landlord") for the lease of 77,242 square feetwill occur upon delivery of the building located at 3611 Valley Centre Drive, San Diego, California. The Company expects to use the premises as its new principal corporate offices and plans to consolidate its corporate headquarters into the premises from the current location of multiple suites in adjacent buildings at 3721 and 3661 Valley Centre Drive, San Diego, California. Under the terms of the lease, the Company will have the one time right of first offer on the suites it currently occupies and a general right of first offer to lease additional space from the Landlord in the development. The commencement date of the lease is expected to be October 1, 2020. respective spaces.
The initial term of the new lease is 7 years, 7 months, and the Landlord has granted the Company an option to extend the term of the lease by a period of 5 years. The measurement of the lease term occurs from the planned occupancy date of the primary spaces estimated to be delivered in the latter part of 2020. The aggregate base rent due over the initial term under the terms of the lease is approximately $49.5 million, which consists of $13.0 million for the space delivered in June 2020, and $36.5 million.million for the primary spaces yet to be delivered.
Following is a schedule of the future minimum rental commitments for our operating lease, excluding commitments related to rental spaces not yet delivered, reconciled to the lease liability and ROU assets as of June 30, 20192020 (in thousands):
| | | | | |
| June 30, 2020 |
| 2020 | $ | 1,207 | |
| 2021 | 960 | |
| 2022 | 1,543 | |
| 2023 | 1,590 | |
| 2024 | 1,637 | |
| Thereafter | 6,435 | |
| Total undiscounted future minimum payments | 13,372 | |
| Lease incentives payable by lessor | (1,983) | |
| Present value discount | (3,121) | |
| Total lease liability | 8,268 | |
| Unamortized lease incentives, less incentives payable by lessor | (404) | |
| Cash payments in excess of straight-line lease expense | (952) | |
| Total ROU asset | $ | 6,912 | |
|
| | | |
| | June 30, 2019 |
|
| 2019 | $ | 1,318 |
|
| 2020 | 2,613 |
|
| 2021 | 2,486 |
|
| 2022 | 2,560 |
|
| 2023 | 2,637 |
|
| Thereafter | 1,585 |
|
| Total undiscounted future minimum payments | 13,199 |
|
| Present value discount | (2,278 | ) |
| Total lease liability | 10,921 |
|
| Lease incentives | (1,703 | ) |
| Straight line lease expense in excess of cash payments | (1,622 | ) |
| Total ROU asset | $ | 7,596 |
|
As of June 30, 2019, the current and non-current portions of2020, the ROU asset wereof $6.9 million was recorded to the Condensed Consolidated Balance Sheets as non-current Other Assets as follows (in thousands):.
|
| | | |
| | June 30, 2019 |
| Prepaid expenses and other current assets | $ | 2,170 |
|
| Other non-current assets | 5,426 |
|
| Total ROU asset | $ | 7,596 |
|
As of June 30, 2019,2020, the current and non-current portions of the lease liability were recorded to the Condensed Consolidated Balance Sheets as follows (in thousands):
| | | | | |
| June 30, 2020 |
| Other current liabilities | $ | 1,379 | |
| Other non-current liabilities | 6,889 | |
| Total lease liabilities | $ | 8,268 | |
|
| | | |
| | June 30, 2019 |
| Other current liabilities | $ | 2,674 |
|
| Other non-current liabilities | 8,247 |
|
| Total lease liabilities | $ | 10,921 |
|
For the three and six months ended June 30, 2020 and 2019, the Company recorded a $0.2 million credit to expense and 0, respectively, and $0.6 million and $1.3 million, respectively, in expense related to operating leases, respectively.leases.
NOTE 7. FAIR VALUE MEASUREMENTS
Financial Instruments and Fair Value
The Company accounts for financial instruments in accordance with ASC 820, Fair Value Measurements and Disclosures (“ASC 820”). ASC 820 establishes a fair value hierarchy that prioritizes the inputs to valuation techniques used to measure fair value. The hierarchy gives the highest priority to unadjusted quoted prices in active markets for identical assets or liabilities (Level 1 measurements) and the lowest priority to unobservable inputs (Level 3 measurements). The three levels of the fair value hierarchy under ASC 820 are described below:
Level 1 – Unadjusted quoted prices in active markets that are accessible at the measurement date for identical, unrestricted assets or liabilities;
Level 2 – Quoted prices in markets that are not active or financial instruments for which all significant inputs are observable, either directly or indirectly; and
Level 3 – Prices or valuations that require inputs that are both significant to the fair value measurement and unobservable.
The valuation techniques used to measure the fair value of the Company’s marketabledebt securities and all other financial instruments, all of which have counter-parties with high credit ratings, were valued based on quoted market prices or model driven valuations using significant inputs derived from or corroborated by observable market data. Based on the fair value hierarchy, the Company classified marketabledebt securities within Level 2.
The Company acquired 2 businesses, related to the Cholbam and Chenodal products, whose purchase price included potential future payments that are contingent on the achievement of certain milestones and percentages of future net sales derived from the products acquired. The Company recorded contingent consideration liabilities at their fair value on the acquisition date and revalues them at the end of each reporting period. In estimating the fair value of the Company’s contingent consideration, the Company used theuses a Monte Carlo SimulationSimulation. The determination of the contingent consideration liabilities requires significant judgements including the appropriateness of the valuation model asand reasonableness of June 30, 2019estimates and December 31, 2018.assumptions included in the forecasts of future net sales and the discount rates applied to such forecasts. Changes in these estimates and assumptions could have a significant impact on the fair value of the contingent consideration liabilities. Based on the fair value hierarchy, the Company classified the fair value measurement of contingent consideration within Level 3.3 because valuation inputs are based on projected revenues discounted to a present value.
Financial instruments with carrying values approximating fair value include cash and cash equivalents, accounts receivable, and accounts payable, due to their short-term nature. As of June 30, 2019,2020, the estimated fair value of the Company's 2.5% Convertible Senior Notes due 2025 was $249.0
$225.9 million, considering factors such aswhich was estimated utilizing market conditions, prepaymentquotations, and make-whole provisions, variability in pricing from multiple lenders and the term of the debt.are considered Level 2.
The following table presents the Company’s assets and liabilities, measured and recognized at fair value on a recurring basis, classified under the appropriate level of the fair value hierarchy as of June 30, 20192020 (in thousands): | |
| As of June 30, 2019 | | As of June 30, 2020 | |
| Total carrying and estimated fair value | | Quoted prices in active markets
(Level 1) | | Significant other observable inputs (Level 2) | | Significant unobservable inputs (Level 3) | | Total carrying and estimated fair value | | Quoted prices in active markets
(Level 1) | | Significant other observable inputs (Level 2) | | Significant unobservable inputs (Level 3) |
| Assets: | | | | | | | | Assets: | | | | | | | |
| Cash and Cash Equivalents | $ | 75,657 |
| | $ | 43,836 |
| | $ | 31,821 |
| | $ | — |
| Cash and Cash Equivalents | $ | 237,170 | | | $ | 237,170 | | | $ | — | | | $ | — | |
| Marketable securities, available-for-sale | 350,243 |
| | — |
| | 350,243 |
| | — |
| |
| Debt securities, available-for-sale | | Debt securities, available-for-sale | 220,206 | | | — | | | 220,206 | | | — | |
| Total | $ | 425,900 |
| | $ | 43,836 |
| | $ | 382,064 |
| | $ | — |
| Total | $ | 457,376 | | | $ | 237,170 | | | $ | 220,206 | | | $ | — | |
| Liabilities: | | | | | | | | Liabilities: | | | | | | | |
| Business combination-related contingent consideration | $ | 76,999 |
| | $ | — |
| | $ | — |
| | $ | 76,999 |
| Business combination-related contingent consideration | $ | 68,600 | | | $ | — | | | $ | — | | | $ | 68,600 | |
| Total | $ | 76,999 |
| | $ | — |
| | $ | — |
| | $ | 76,999 |
| Total | $ | 68,600 | | | $ | — | | | $ | — | | | $ | 68,600 | |
The following table presents the Company’s assets and liabilities, measured and recognized at fair value on a recurring basis, classified under the appropriate level of the fair value hierarchy as of December 31, 20182019 (in thousands):
|
| | | | | | | | | | | | | | | |
| | As of December 31, 2018 |
| | Total carrying and estimated fair value | | Quoted prices in active markets (Level 1) | | Significant other observable inputs (Level 2) | | Significant unobservable inputs (Level 3) |
| Assets: | | | | | | | |
| Cash and Cash Equivalents | $ | 102,873 |
| | $ | 62,978 |
| | $ | 39,895 |
| | $ | — |
|
| Marketable securities, available-for-sale | 368,668 |
| | — |
| | 368,668 |
| | — |
|
| Total | $ | 471,541 |
| | $ | 62,978 |
| | $ | 408,563 |
| | $ | — |
|
| Liabilities: | | | | | | | |
| Business combination-related contingent consideration | 93,000 |
| | — |
| | — |
| | 93,000 |
|
| Total | $ | 93,000 |
| | $ | — |
| | $ | — |
| | $ | 93,000 |
|
| | | | | | | | | | | | | | | | | | | | | | | |
| As of December 31, 2019 | | | | | | |
| Total carrying and estimated fair value | | Quoted prices in active markets
(Level 1) | | Significant other observable inputs (Level 2) | | Significant unobservable inputs (Level 3) |
| Assets: | | | | | | | |
| Cash and Cash Equivalents | $ | 62,436 | | | $ | 62,436 | | | $ | — | | | $ | — | |
| Debt securities, available-for-sale | 336,088 | | | — | | | 336,088 | | | — | |
| Total | $ | 398,524 | | | $ | 62,436 | | | $ | 336,088 | | | $ | — | |
| Liabilities: | | | | | | | |
| Business combination-related contingent consideration | 70,900 | | | — | | | — | | | 70,900 | |
| Total | $ | 70,900 | | | $ | — | | | $ | — | | | $ | 70,900 | |
The following table sets forth a summary of changes in the estimated fair value of the Company's business combination-related contingent consideration for the six months ended June 30, 20192020 (in thousands):
|
| | | |
| | Fair Value Measurements of Acquisition-Related Contingent Consideration
(Level 3) |
| Balance at January 1, 2019 | $ | 93,000 |
|
| L-UDCA write-off | (18,000 | ) |
| Changes in the fair value of contingent consideration | 6,522 |
|
| Contractual payments included in accrued liabilities at June 30, 2019 | (2,269 | ) |
| Contractual payments | (2,253 | ) |
| Foreign currency impact | (1 | ) |
| Balance at June 30, 2019 | $ | 76,999 |
|
| | | | | |
| Fair Value Measurements of Acquisition-Related Contingent Consideration
(Level 3) |
| Balance at January 1, 2020 | $ | 70,900 | |
| Changes in the fair value of contingent consideration | 2,363 | |
| Contractual payments | $ | (2,125) | |
| Contractual payments included in accrued liabilities at June 30, 2020 | (2,424) | |
| Foreign currency impact | (114) | |
| Balance at June 30, 2020 | $ | 68,600 | |
The key assumptions included in the calculations for contingent consideration were the future number of patients in treatment, projected revenues, discount rate, and the timing of payments. The present value of the expected payments considers the time at which the obligations are expected to be settled and a discount rate that reflects the risk associated with the performance payments.
DuringFor the three and six months ended June 30, 2019,2020, the Company recognized $3.4incurred charges of $4.3 million and $6.5$2.4 million, respectively, in operating expenseexpenses on the Condensed Consolidated StatementStatements of Operations and Comprehensive Loss for the change in fair value of the contingent consideration liabilities.
For the three months ended June 30, 2020, the change in fair value of contingent consideration is due to the timing of future payments and changes in market driven discount rates.
For the six months ended June 30, 2020, the change in fair value of contingent consideration is due to the timing of future payments and changes in market driven discount rates, offset by a change in revenue projections.
For the three and six months ended June 30, 2019, the Company incurred charges of $3.4 million and $6.5 million, respectively, in operating expenses on the Condensed Consolidated Statements of Operations and Comprehensive Loss for the change in fair value of the contingent consideration liabilities.
For the six months ended June 30, 2019, $4.3 million and $2.2$6.5 million of the charges were related to the increase in contingent consideration liabilities for the products Chenodal and Cholbam, respectively. In each case, thebile acid products. The value increased due to passage of time. In addition,During the first quarter of 2019, the Company made a portfolio decision not to pursue further development of its product candidate L-UDCA. The related contingent consideration of $18.0 million was accordingly considered fully written off. See Note 17 for further discussion.
During the three and six months ended June 30, 2018, the Company incurred charges of $2.2 million and $5.8 million, respectively, in operating expenses on the Condensed Consolidated Statements of Operations and Comprehensive Loss for the change in fair value of the contingent consideration liabilities. For the six months ended June 30, 2018, $1.9 million, $1.4 million, and $2.5 million of the charges were related to the
increase in contingent consideration liabilities for the products Chenodal and Cholbam and product candidate L-UDCA, respectively. In each case, the value increased due to passage of time.
NOTE 8. INTANGIBLE ASSETS
As of June 30, 2019,2020, the net book value of amortizable intangible assets was approximately $158.9$155.4 million.
The following table sets forth amortizable intangible assets as of June 30, 20192020 and December 31, 20182019 (in thousands):
| | | | | | | | | | | |
| June 30, 2020 | | December 31, 2019 |
| Finite-lived intangible assets | $ | 254,443 | | | $ | 245,802 | |
| Less: accumulated amortization | (99,072) | | | (88,602) | |
| Net carrying value | $ | 155,371 | | | $ | 157,200 | |
|
| | | | | | | |
| June 30, 2019 | | December 31, 2018 |
| Finite-lived intangible assets | $ | 237,427 |
| | $ | 255,643 |
|
| Less: accumulated amortization | (78,521 | ) | | (68,952 | ) |
| Net carrying value | $ | 158,906 |
| | $ | 186,691 |
|
During the first quarter of 2019, the Company made a portfolio decision not to pursue further development of L-UDCA, acquired in 2016. The related in-progress research and development intangible asset ("IPR&D") of $25.5 million was accordingly considered fully impaired and written off. As of December 31, 2018, the value of the IPR&D was $25.5 million. See Note 17 for further discussion.
The following table summarizes amortization expense for the three and six months ended June 30, 20192020 and 20182019 (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | |
| Three Months Ended June 30, | | | | Six Months Ended June 30, | | |
| 2020 | | 2019 | | 2020 | | 2019 |
| Research and development | $ | 289 | | | $ | 289 | | | $ | 578 | | | $ | 574 | |
| Selling, general and administrative | 4,996 | | | 4,567 | | | 9,881 | | | 9,013 | |
| Total amortization expense | $ | 5,285 | | | $ | 4,856 | | | $ | 10,459 | | | $ | 9,587 | |
|
| | | | | | | | | | | | | | | |
| Three Months Ended June 30, | | Six Months Ended June 30, |
| 2019 | | 2018 | | 2019 | | 2018 |
| Research and development | $ | 289 |
| | $ | 289 |
| | $ | 574 |
| | $ | 392 |
|
| Selling, general and administrative | 4,567 |
| | 4,206 |
| | 9,013 |
| | 8,302 |
|
| Total amortization expense | $ | 4,856 |
| | $ | 4,495 |
| | $ | 9,587 |
| | $ | 8,694 |
|
NOTE 9. CONVERTIBLE NOTES PAYABLE
Convertible Senior Notes Due 2025
On September 10, 2018, the Company completed its registered underwritten public offering of $276.0 million aggregate principal amount of 2.50% Convertible Senior Notes due 2025 ("2025 Notes"), and entered into a base indenture and supplemental indenture agreement ("2025 Indenture") with respect to the 2025 Notes. The 2025 Notes will mature on September 15, 2025 ("Maturity Date”), unless earlier repurchased, redeemed, or converted. The 2025 Notes are senior unsecured obligations of the Company and bear interest at an annual rate of 2.50%, payable semi-annually in arrears on March 15 and September 15 of each year. The first payment was made onyear, commencing March 15, 2019.
The composition of the Company’s 2025 Notes are as follows (in thousands):
|
| | | | | | | |
| | June 30, 2019 | | December 31, 2018 |
| 2.50% convertible senior notes due 2025 | $ | 276,000 |
| | $ | 276,000 |
|
| Unamortized debt discount | (70,484 | ) | | (74,836 | ) |
| Unamortized debt issuance costs | (5,625 | ) | | (6,073 | ) |
| Total 2025 Notes, net of unamortized debt discount and debt issuance costs | $ | 199,891 |
| | $ | 195,091 |
|
| | | | | | | | | | | |
| | June 30, 2020 | | December 31, 2019 |
| 2.50% convertible senior notes due 2025 | $ | 276,000 | | | $ | 276,000 | |
| Unamortized debt discount | (61,265) | | | (65,963) | |
| Unamortized debt issuance costs | (4,726) | | | (5,176) | |
| Total 2025 Notes, net of unamortized debt discount and debt issuance costs | $ | 210,009 | | | $ | 204,861 | |
The net proceeds from the issuance of the 2025 Notes were approximately $267.2 million, after deducting commissions and the offering expenses payable by the Company. A portion of the net proceeds from the 2025 Notes was used by the Company to repurchase $23.4 million aggregate principal amount of its then-outstanding 4.5% senior convertible notes due in 2019 in privately-negotiated transactions.
Holders may convert their 2025 Notes at their option only in the following circumstances: (1) during any calendar quarter commencing after the calendar quarter ending on December 31, 2018 (and only during such calendar quarter), if the last reported sale price per share of the Company’s common stock for each of at least 20 trading days, whether or not consecutive, during the period of 30 consecutive trading days ending on, and including, the last trading day of the immediately preceding calendar quarter exceeds 130% of the conversion price on the applicable trading day; (2) during the five consecutive business days immediately after any 10 consecutive trading day period (“measurement period”) if the trading price per $1,000 principal amount of 2025 Notes for each trading day of the measurement period was less than 98% of the product of the last reported sale price per share of the Company’s common stock on such trading day and the conversion rate on such trading day; (3) upon the occurrence of certain corporate events or distributions on the Company’s common
stock; (4) if the Company calls the 2025 Notes for redemption; and (5) at any time from, and including, May 15, 2025 until the close of business on the scheduled trading day immediately before the Maturity Date. The Company will settle conversions by paying or delivering, as applicable, cash, shares of the Company’s common stock, or a combination of cash and shares of the Company’s common stock, at the Company’s election, based on the applicable conversion rate.
The initial conversion rate for the 2025 Notes is 25.7739 shares of the Company’s common stock per $1,000 principal amount of 2025 Notes, which represents an initial conversion price of approximately $38.80 per share. If a “make-whole fundamental change” (as defined in the 2025 Indenture) occurs, then the Company will, in certain circumstances, increase the conversion rate for a specified period of time.
The 2025 Notes will be redeemable, in whole or in part, at the Company’s option at any time, and from time to time, on or after September 15, 2022 and, in the case of any partial redemption, on or before the 40th scheduled trading day before the Maturity Date, at a cash redemption price equal to the principal amount of the 2025 Notes to be redeemed, plus accrued and unpaid interest, if any, to, but excluding, the redemption date, but only if the last reported sale price per share of the Company’s common stock exceeds 130% of the conversion price on each of at least 20 trading days during the 30 consecutive trading days ending on, and including, the trading day immediately before the date the Company sends the related redemption notice. If a fundamental change (as defined in the 2025 Indenture) occurs, then, subject to certain exceptions, holders may require the Company to repurchase their 2025 Notes at a cash repurchase price equal to the principal amount of the 2025 Notes to be repurchased, plus accrued and unpaid interest, if any, to, but excluding, the fundamental change repurchase date.
In the event of conversion, holders would forgo all future interest payments, any unpaid accrued interest and the possibility of further stock price appreciation. Upon the receipt of conversion requests, the settlement of the 2025 Notes will be paid pursuant to the terms of the 2025 Indenture. In the event that all of the 2025 Notes are converted, the Company would be required to repay the $276.0 million principal amount and any conversion premium in any combination of cash and shares of its common stock at the Company’s option. In addition, calling the 2025 Notes for redemption will constitute a “make whole fundamental change."
The 2025 Notes are the Company’s general unsecured obligations that rank senior in right of payment to all of its indebtedness that is expressly subordinated in right of payment to the 2025 Notes, and equal in right of payment to the Company’s unsecured indebtedness.
The 2025 Notes are currently classified on the Company’s Condensed Consolidated Balance Sheets at June 30, 20192020 as long-term debt.
Under ASC 470-20, Debt with Conversion and Other Options, an entity must separately account for the liability and equity components of convertible debt instruments (such as the 2025 Notes) that may be settled entirely or partially in cash upon conversion, in a manner that reflects the issuer’s economic interest cost. The liability component of the instrument is valued in a manner that reflects the market interest rate for a similar nonconvertible instrument at the date of issuance. The initial carrying value of the liability component was $198.6 million. The equity component of $77.4 million, representing the conversion option, was determined by deducting the fair value of the liability component from the par value of the 2025 Notes and is recorded in additional paid-in capital on the consolidated balance sheet at the issuance date. That equity component is treated as a discount on the liability component of the 2025 Notes, which is amortized over the seven yearseven-year term of the 2025 Notes using the effective interest rate method. The equity component is not re-measured as long as it continues to meet the conditions for equity classification. The Company allocated the total transaction costs of approximately $8.8 million related to the issuance of the 2025 Notes to the liability and equity components of the 2025 Notes based on their relative values. Transaction costs attributable to the liability component are amortized to interest expense over the seven-year term of the 2025 Notes, and transaction costs attributable to the equity component are netted with the equity component in stockholders’ equity.
The effective interest rate on the liability components of the 2025 Notes for the period from the date of issuance through June 30, 20192020 was 7.7%. The following table sets forth total interest expense recognized related to the 2025 Notes (in thousands):
|
| | | | | | | | | | | | | | | |
| | Three Months Ended June 30, | | Six Months Ended June 30, |
| | 2019 | | 2018 | | 2019 | | 2018 |
| Contractual interest expense | $ | 1,725 |
| | $ | — |
| | $ | 3,450 |
| | $ | — |
|
| Amortization of debt discount | 2,197 |
| | — |
| | 4,352 |
| | — |
|
| Amortization of debt issuance costs | 224 |
| | — |
| | 448 |
| | — |
|
| Total interest expense for the 2025 Notes | $ | 4,146 |
| | $ | — |
| | $ | 8,250 |
| | $ | — |
|
| | | | | | | | | | | | | | | | | | | | | | | |
| Three Months Ended June 30, | | | | Six Months Ended June 30, | | |
| 2020 | | 2019 | | 2020 | | 2019 |
| Contractual interest expense | $ | 1,725 | | | $ | 1,725 | | | $ | 3,450 | | | $ | 3,450 | |
| Amortization of debt discount | 2,371 | | | 2,197 | | | 4,698 | | | 4,352 | |
| Amortization of debt issuance costs | 225 | | | 224 | | | 449 | | | 448 | |
| Total interest expense for the 2025 Notes | $ | 4,321 | | | $ | 4,146 | | | $ | 8,597 | | | $ | 8,250 | |
The 2025 Notes do not contain any financial or operating covenants or any restrictions on the payment of dividends, the issuance of other indebtedness or the issuance or repurchase of securities by the Company. The 2025 Indenture contains customary events of default with respect to the 2025 Notes, including thatfailure to pay (for more than 30 days) interest when due and certain types of bankruptcy or insolvency involving the Company. Upon an event of default involving certain types of bankruptcy insolvency, 100% of the outstanding principal and accrued and unpaid interest on the 2025 Notes will automatically become due and payable, and upon certainother events of default, the trustee under the 2025 Indenture or the holders of at least 25% of the outstanding principal amount of the 2025 Notes may declare 100% of the principal and accrued and unpaid interest on the 2025 Notes will automatically becometo be immediately due and payable.
Senior Convertible Notes Due 2019
On May 29, 2014, the Company entered into a Note Purchase Agreement relating to a private placement by the Company of $46.0 million aggregate principal amount of 4.50% senior convertible notes due in 2019 (the “2019 Notes”) which were convertible into shares of the Company’s common stock at an initial conversion price of $17.41 per share. The conversion price was subject to customary anti-dilution protection. The 2019 Notes bore interest at a rate of 4.5% per annum, payable semiannually in arrears on May 15 and November 15 of each year. The 2019 Notes had a maturity date of May 30, 2019 and there were no contractual payments due prior to that date.
In September 2018, the Company used part of the net proceeds from the issuance of the 2025 Notes to repurchase $23.4 million aggregate principal amount of the 2019 Notes in privately-negotiated transactions for approximately $40.2 million in cash. The partial repurchase of the 2019 Notes resulted in a $17.0 million loss on early extinguishment of debt in September 2018.
In May 2019, the remaining $22.6 million outstanding principal amount of 2019 Notes was converted by the holders thereof into approximately 1.3 million shares of common stock.
The composition of the Company’s 2019 Notes at December 31, 2018 was as follows (in thousands):
|
| | | | |
| | | December 31, 2018 |
| 4.50% senior convertible notes due 2019 | | $ | 22,590 |
|
| Unamortized debt discount | | (125 | ) |
| Unamortized debt issuance costs | | (8 | ) |
| Total 2019 Notes, net of unamortized debt discount and debt issuance costs | | $ | 22,457 |
|
NOTE 10. ACCRUED EXPENSES
Accrued expenses at June 30, 20192020 and December 31, 20182019 consisted of the following (in thousands):
| | | | | | | | | | | |
| June 30, 2020 | | December 31, 2019 |
| Government rebates payable | $ | 7,817 | | | $ | 6,584 | |
| Compensation related costs | 10,011 | | | 14,045 | |
| Accrued royalties and contingent consideration | 7,432 | | | 7,272 | |
| Research and development | 8,369 | | | 16,067 | |
| Selling, general and administrative | 7,321 | | | 3,552 | |
| | | |
| Miscellaneous accrued | 3,429 | | | 4,225 | |
| Total accrued expenses | $ | 44,379 | | | $ | 51,745 | |
|
| | | | | | | |
| | June 30, 2019 | | December 31, 2018 |
| Government rebates payable | $ | 6,024 |
| | $ | 8,464 |
|
| Compensation related costs | 10,122 |
| | 10,446 |
|
| Accrued royalties and contingent consideration | 6,912 |
| | 6,805 |
|
| Research and development | 21,011 |
| | 16,515 |
|
| Selling, general and administrative | 3,422 |
| | 2,990 |
|
| Miscellaneous accrued | 3,066 |
| | 4,475 |
|
| Total accrued expenses | $ | 50,557 |
| | $ | 49,695 |
|
NOTE 11. NET LOSS PER COMMON SHARE
Basic and diluted net loss per common share is calculated by dividing net loss applicable to common stockholders by the weighted-average number of common shares outstanding during the period, without consideration of common stock equivalents.period. The Company’s potentially dilutive shares, which include outstanding stock options, restricted stock units, warrants, and shares issuable upon conversion of the 2019 Notes and 2025 Notes, are considered to be common stock equivalents and are onlynot included in the calculation of diluted net loss per share whenbecause their effect is dilutive.anti-dilutive.
Basic and diluted net loss per share is calculated as follows (net loss amounts are stated in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Three Months Ended June 30, | | | | | | | | | | |
| 2020 | | | | | | 2019 | | | | |
| Shares | | Net Loss | | Loss per common share | | Shares | | Net Loss | | Loss per common share |
| Basic and diluted loss per share | 44,763,843 | | | $ | (26,068) | | | $ | (0.58) | | | 41,957,860 | | | $ | (38,701) | | | $ | (0.92) | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| Six Months Ended June 30, | | | | | | | | | | |
| 2020 | | | | | | 2019 | | | | |
| Shares | | Net Loss | | Loss per common share | | Shares | | Net Loss | | Loss per common share |
| Basic and diluted loss per share | 43,943,370 | | | $ | (25,260) | | | $ | (0.57) | | | 41,685,599 | | | $ | (79,678) | | | $ | (1.91) | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
|
| | | | | | | | | | | | | | | | | | | | | |
| | Three Months Ended June 30, |
| | 2019 | | 2018 |
| | Shares | | Net Loss | | EPS | | Shares | | Net Loss | | EPS |
| Basic and diluted loss per share | 41,957,860 |
| | $ | (38,701 | ) | | $ | (0.92 | ) | | 40,061,045 |
| | $ | (22,329 | ) | | $ | (0.56 | ) |
|
| | | | | | | | | | | | | | | | | | | | | |
| | Six Months Ended June 30, |
| | 2019 | | 2018 |
| | Shares | | Net Loss | | EPS | | Shares | | Net Loss | | EPS |
| Basic and diluted loss per share | 41,685,599 |
| | $ | (79,678 | ) | | $ | (1.91 | ) | | 39,641,334 |
| | $ | (40,707 | ) | | $ | (1.03 | ) |
The following common stock equivalents have been excluded because they were anti-dilutive (in thousands):anti-dilutive:
| | | | | | | | | | | | | | | | | | | | | | | |
| Three Months Ended June 30, | | | | Six Months Ended June 30, | | |
| 2020 | | 2019 | | 2020 | | 2019 |
| Restricted stock units | 1,517,338 | | | 672,859 | | | 1,437,383 | | | 611,446 | |
| Convertible debt | 7,113,402 | | | 7,891,920 | | | 7,113,402 | | | 8,151,426 | |
| Options | 8,479,710 | | | 7,947,569 | | | 8,327,753 | | | 7,801,606 | |
| | | | | | | |
| Total anti-dilutive shares | 17,110,450 | | | 16,512,348 | | | 16,878,538 | | | 16,564,478 | |
|
| | | | | | | | | | | |
| | Three Months Ended June 30, | | Six Months Ended June 30, |
| | 2019 | | 2018 | | 2019 | | 2018 |
| Restricted stock units | 650 |
| | 350 |
| | 600 |
| | 250 |
|
| Convertible debt | 7,900 |
| | 2,600 |
| | 8,200 |
| | 2,600 |
|
| Options | 7,950 |
| | 7,200 |
| | 7,800 |
| | 7,100 |
|
| Warrants | — |
| | 450 |
| | — |
| | 550 |
|
| Total anti-dilutive shares | 16,500 |
| | 10,600 |
| | 16,600 |
| | 10,500 |
|
NOTE 12. COMMITMENTS AND CONTINGENCIES
Research Collaboration and Licensing Agreements
As part of the Company's research and development efforts, the Company enters into research collaboration and licensing agreements with unrelated companies, scientific collaborators, universities, and consultants. These agreements contain varying terms and provisions which include fees and milestones to be paid by the Company, services to be provided, and ownership rights to certain proprietary technology developed under the agreements. Some of these agreements contain provisions which require the Company to pay royalties, in the event the Company sells or licenses any proprietary products developed under the respective agreements.
Legal Proceedings
In August 2017, Martin Shkreli, the Company’s former Chief Executive Officer, was convicted on securities fraud charges following investigations by the U.S. Attorney for the Eastern District of New York and the U.S. Securities and Exchange Commission.charges. Mr. Shkreli appealed hisShkreli's conviction to the United States Court of Appeals for the Second Circuit, and in July 2019, the Court of Appealswas subsequently affirmed his conviction.upon appeal. In connection with the trial and appeal proceedings, the Company advanced a portion of Mr. Shkreli’s legal fees, of which $3.8 million was reimbursed by its directors’ and officers’ insurance carriers. Pending the outcome of Mr. Shkreli's appeal, the insurance carriers reserved their rights to assert that certain of the advanced funds pertain to claims excluded from coverage under the relevant insurance policy and are therefore recoverable by the carriers, and therefore the final amount of the reimbursement from the insurance carriers is not currently estimable.
As previously disclosed, in August 2015, the Company filed a lawsuit in federal district court for the Southern District of New York against Mr. Shkreli, asserting that he breached his fiduciary duty of loyalty during his previous tenure as the Company’s Chief Executive Officer and a member of its Board of Directors. Mr. Shkreli subsequently initiated a JAMS arbitration against the Company claiming that the Company breached his December 2013 employment agreement. The Company asserted counterclaims in the arbitration that were substantially similar to the claims it previously asserted in the federal lawsuit. In February 2019, Mr. Shkreli amended his arbitration demand to add a claim relating to an October 2014 summary separation proposal. After the arbitration panel determined that Mr. Shkreli could not bring claims against Stephen Aselage, Margaret Valeur-Jensen, and Gary Lyons before JAMS for lack of jurisdiction, Mr. Shkreli brought suit against them in federal court for the Southern District of New York in May 2019 (SDNY Action). The hearing on the merits before the JAMS arbitration panel was scheduled to commence in June 2019.
In advance of the scheduled arbitration hearing on the above-referenced claims and counterclaims, the Company and Mr. Shkreli reached a comprehensive settlement covering all of the disputes between them and Mr. Aselage, Dr. Valeur-Jensen, and Mr. Lyons as of such time and on June 17, 2019, the Company and Mr. Shkreli entered into a Settlement Agreement memorializing such terms. Under the terms of the Settlement Agreement, each of the parties’ pending claims and counterclaims was dismissed with prejudice (and Mr. Shkreli dismissed, with prejudice, the SDNY Action), and the parties, including Retrophin on behalf of Mr. Aselage, Dr. Valeur-Jensen, and Mr. Lyons, granted one another a broad mutual release of claims. Mr. Shkreli also agreed to extinguish his rights to future advancement of legal fees and indemnification by the Company (other than for certain pending litigation), and to abide by certain standstill restrictions and the Company made an undisclosed cash payment to Mr. Shkreli.
In October 2018, Spring Pharmaceuticals, LLC (Spring) filed a lawsuit against the Company, Martin Shkreli, Mission Pharmacal Company and Alamo Pharma Services, Inc. in the United States District Court for the Eastern District of Pennsylvania alleging that the Company violated various federal and state antitrust and unfair competition laws by allegedly refusing to sell samples of the Thiola® brand drug so that Spring can conduct the bioequivalence testing needed to submit an ANDA to the FDA for approval to market a generic version of the product. Spring is seekingThe lawsuit sought injunctive relief and damages. The Company intendsIn December 2019, the Court granted the Company's motion to vigorously defend against Spring’s claims. In January 2019,dismiss the lawsuit, but granted Spring permission to amend its complaint. On February 10, 2020, Spring filed an amended complaint. Spring's amended complaint sought injunctive relief and damages. On April 1, 2020, the Company filed a motion to dismiss the lawsuit. Inamended complaint. On April 2019, the Court stayed the Company's motion to dismiss to allow for discovery limited to the question22, 2020, Spring filed a notice of whether Spring has standing to sue, which has been largely completed.voluntary dismissal, without prejudice. No amounts have been accrued related to this matter and the outcome cannot be determined.
The Company is not aware of any other proceedings or claims that could have, individually or in the aggregate, a material adverse effect on its results of operations or financial condition.
NOTE 13. SHARE BASEDSHARE-BASED COMPENSATION
Restricted Shares
Service Based Restricted Stock Units
The following table summarizes the Company’s service based restricted stock unit activity during the six months ended June 30, 2019:2020:
|
| | | | | | |
| | Number of Restricted Stock Units | | Weighted Average Grant Date Fair Value |
| Unvested December 31, 2018 | 400,426 |
| | $ | 24.95 |
|
| Granted | 395,015 |
| | 18.89 |
|
| Vested | (124,120 | ) | | 24.25 |
|
| Forfeited/canceled | (13,126 | ) | | 23.26 |
|
| Unvested June 30, 2019 | 658,195 |
| | $ | 21.48 |
|
| | | | | | | | | | | |
| | Number of Restricted Stock Units | | Weighted Average Grant Date Fair Value |
| Unvested December 31, 2019 | 763,728 | | | $ | 19.64 | |
| Granted | 637,113 | | | 15.51 | |
| Vested | (173,117) | | | 21.25 | |
| Forfeited/canceled | (68,414) | | | 18.58 | |
| Unvested June 30, 2020 | 1,159,310 | | | $ | 17.19 | |
At June 30, 2019,2020, unamortized stock compensation for service based restricted stock units was $12.9$16.9 million, with a weighted-average recognition period of 3.32.6 years.
Performance Based Restricted Stock Units
The following table summarizes the Company’s performance based restricted stock unit activity during the six months ended June 30, 2019:2020:
|
| | | | | | |
| | Number of Restricted Stock Units | | Weighted Average Grant Date Fair Value |
| Unvested December 31, 2018 | 226,750 |
| | $ | 21.54 |
|
| Granted | 80,000 |
| | 21.32 |
|
| Vested | — |
| | — |
|
| Forfeited/canceled | — |
| | — |
|
| Unvested June 30, 2019 | 306,750 |
| | $ | 21.48 |
|
| | | | | | | | | | | |
| | Number of Restricted Stock Units | | Weighted Average Grant Date Fair Value |
| Unvested December 31, 2019 | 233,500 | | | $ | 20.90 | |
| Granted | 150,000 | | | 15.46 | |
| Vested | (33,250) | | | 25.75 | |
| Forfeited/canceled | (31,125) | | | 17.56 | |
| Unvested June 30, 2020 | 319,125 | | | $ | 18.16 | |
At June 30, 2019,2020, unamortized stock compensation for performance based restricted stock units was $1.9 million, with a weighted-average recognition period of 1.01.4 years.
Stock Options
The following table summarizes stock option activity during the six months ended June 30, 2019:2020:
|
| | | | | | | | | | |
| | Shares Underlying Options | | Weighted Average Exercise Price | | Weighted Average Remaining Contractual Life (years) | | Aggregate Intrinsic Value (in thousands) |
| Outstanding at December 31, 2018 | 7,277,337 |
| | $18.55 | | 6.94 | | $ | 40,650 |
|
| Granted | 1,188,625 |
| | $20.03 | | | | |
| Exercised | (24,527 | ) | | $12.92 | | | | |
| Forfeited/canceled | (852,000 | ) | | $11.06 | | | | |
| Outstanding at June 30, 2019 | 7,589,435 |
| | $19.64 | | 7.13 | | $ | 21,722 |
|
| | | | | | | | | | | | | | | | | | | | | | | |
| | Shares Underlying Options | | Weighted Average Exercise Price | | Weighted Average Remaining Contractual Life (years) | | Aggregate Intrinsic Value (in thousands) |
| Outstanding at December 31, 2019 | 7,371,733 | | | $19.52 | | 6.63 | | $ | 4,906 | |
| Granted | 1,390,125 | | | $15.48 | | | | |
| Exercised | (35,042) | | | $12.31 | | | | |
| Forfeited/canceled | (377,953) | | | $18.96 | | | | |
| Outstanding at June 30, 2020 | 8,348,863 | | | $18.90 | | 6.79 | | $ | 28,920 | |
At June 30, 2019,2020, unamortized stock compensation for stock options was $34.1$31.5 million, with a weighted-average recognition period of 2.92.8 years.
At June 30, 2019,2020, outstanding options to purchase 4.85.3 million shares of common stock were exercisable with a weighted-average exercise price per share of $18.62.$19.20.
Share BasedShare-Based Compensation
The following table sets forth total stock-basedshare-based compensation for the three and six months ended June 30, 20192020 and 20182019 (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | |
| | Three Months Ended June 30, | | | | Six Months Ended June 30, | | |
| | 2020 | | 2019 | | 2020 | | 2019 |
| Research and development | $ | 2,332 | | | $ | 1,896 | | | $ | 4,458 | | | $ | 3,566 | |
| Selling, general & administrative | 3,622 | | | 3,852 | | | 7,406 | | | 8,702 | |
| Total | $ | 5,954 | | | $ | 5,748 | | | $ | 11,864 | | | $ | 12,268 | |
|
| | | | | | | | | | | | | | | |
| | Three Months Ended June 30, | | Six Months Ended June 30, |
| | 2019 | | 2018 | | 2019 | | 2018 |
| Research and development | $ | 1,896 |
| | $ | 1,582 |
| | $ | 3,566 |
| | $ | 2,989 |
|
| Selling, general & administrative | 3,852 |
| | 3,844 |
| | 8,702 |
| | 7,046 |
|
| Total | $ | 5,748 |
| | $ | 5,426 |
| | $ | 12,268 |
| | $ | 10,035 |
|
Exercise
NOTE 14. INCOME TAXES
On March 27, 2020, the Coronavirus Aid, Relief, and Economic Security Act (CARES Act) was enacted in response to the COVID-19 pandemic. The CARES Act, among other things, permits NOL carryforwards generated in taxable years beginning after December 31, 2017, to offset 100% of Warrants
taxable income for taxable years beginning before January 1, 2021, and 80% of taxable income in taxable years beginning after December 31, 2020. In addition, the CARES Act allows net operating losses incurred in taxable years beginning after December 31, 2017, and before January 1, 2021, to be carried back to each of the five preceding taxable years to generate a refund of previously paid income taxes. The Company issued 255,445 and 424,057 shareshas recorded a discrete income tax benefit of common stock upon the exercise of outstanding warrants$18.9 million related to this legislation during the three and six months ended June 30, 2018, respectively, for cash received by the Company in the amount of $1.5 million and $2.1 million, respectively. As of June 30, 2019 and December 31, 2018, there were no warrants outstanding or exercisable.2020.
NOTE 14. INCOME TAXES
The following table summarizes our effective tax rate and income tax expense for the three and six months ended June 30, 2019 and 2018 (dollarsin millions):
|
| | | | | | | | | | | | | | | |
| | Three Months Ended June 30, | | Six Months Ended June 30, |
| | 2019 | | 2018 | | 2019 | | 2018 |
| Effective tax rate | (0.2 | )% | | (0.8 | )% | | (0.6 | )% | | (1.0 | )% |
| Income tax expense | $ | (0.1 | ) | | $ | (0.2 | ) | | $ | (0.5 | ) | | $ | (0.4 | ) |
NOTE 15. INVENTORY
Inventory, net of reserves, consisted of the following at June 30, 20192020 and December 31, 20182019 (in thousands):
|
| | | | | | | |
| | June 30, 2019 | | December 31, 2018 |
| Raw materials | $ | 4,220 |
| | $ | 4,689 |
|
| Finished goods | 830 |
| | 930 |
|
| Total inventory | $ | 5,050 |
| | $ | 5,619 |
|
| | | | | | | | | | | |
| June 30, 2020 | | December 31, 2019 |
| Raw materials | $ | 3,037 | | | $ | 2,713 | |
| Finished goods | 3,249 | | | 3,369 | |
| Total inventory | $ | 6,286 | | | $ | 6,082 | |
The inventory reserve was $2.0$3.6 million and $1.8$3.1 million at June 30, 20192020 and December 31, 2018,2019, respectively.
NOTE 16. ACCOUNTS RECEIVABLE
Accounts receivable, net of reserves for prompt pay discounts and doubtful accounts,expected credit losses, was $15.5$14.1 million and $12.7$18.0 million at June 30, 20192020 and December 31, 2018,2019, respectively. The total reserves for both periods were immaterial.
The Company's adoption of ASU No. 2016-13, Financial Instruments - Credit Losses, included an assessment of our aged trade receivables balances and their underlying credit risk characteristics. Our evaluation of past events, current conditions, and reasonable and supportable forecasts about the future resulted in an expectation of immaterial credit losses.
NOTE 17. DISPOSITIONS
In June 2016, the Company acquired certain rights to its product candidate L-UDCA for $0.5 million cash. At the same time the Company established a related non-cash asset of $25.5 million for IPR&D and a liability of $25.0 million for contingent consideration related net sales royalties and milestones. As a result of our quarterly valuation update process during 2016 and 2017, the contingent liability was decreased by $2.3 million and $5.7 million, respectively, and increased by $1.0 million during 2018. The resulting balance of the L-UDCA contingent liability at December 31, 2018 was $18.0 million.
During the first quarter of 2019, the Company elected to discontinue development of the L-UDCA program, resulting in the write off of the intangible asset of $25.5 million originally recorded in 2016, and the reversal of associated contingent consideration of $18.0 million. This resulted in a net $7.5 million non-cash charge to first quarter operations.
NOTE 18. SUBSEQUENT EVENTEQUITY OFFERINGS
Underwritten Public Offering of Common Stock
In August 2019, following a strategic review of the CNSA-001 program in patients with phenylketonuria (PKU),June 2020, the Company madesold an aggregate of 7.5 million shares of its common stock in an underwritten public offering, at a price to the decisionpublic of $15.50 per share. The net proceeds to declinethe Company from the offering, after deducting the underwriting discounts and offering expenses, was $108.6 million.
At-the-Market Equity Offering Program
In February 2020, the Company entered into an Open Market Sale Agreement ("ATM Agreement") with Jefferies LLC, as agent (“Jefferies”), pursuant to exercisewhich the Company may offer and sell, from time to time through Jefferies, shares of its optioncommon stock having an aggregate offering price of up to acquire Censa Pharmaceuticals$100.0 million. Shares will be sold pursuant to the Company's effective registration statement on Form S-3 (Registration Statement No. 333-227182), as previously filed with the Securities and accordingly discontinue its joint development program for CNSA-001. TheExchange Commission. Through the period ended June 30, 2020, the Company expects to impairhas not sold any shares under the $15 million long term investment on the balance sheet during the third quarter of 2019.ATM Agreement.
Item 2. Management’s Discussion and Analysis of Financial Condition and Results of Operations
The following discussion and analysis of our financial condition and results of operations should be read in conjunction with our unaudited condensed consolidated financial statements and related notes included in this Quarterly Report on Form 10-Q and the audited financial statements and notes thereto as of and for the year ended December 31, 20182019 and the related Management’s Discussion and Analysis of Financial Condition and Results of Operations, both of which are contained in our Annual Report on Form 10-K for the year ended December 31, 2018,2019, filed with the Securities and Exchange Commission (SEC) on February 26, 2019.24, 2020. Past operating results are not necessarily indicative of results that may occur in future periods.
Forward-Looking Statements
The information in this In addition, see the discussion contains forward-lookingunder the heading “Forward-Looking Statements” immediately preceding the consolidated financial statements and information within the meaningincluded under Part I of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended, or the Exchange Act, which are subject to the “safe harbor” created by those sections. These forward-looking statements include, but are not limited to, statements concerning our strategy, future operations, future financial position, future revenues, projected costs, prospects and plans and objectives of management. The words “anticipates,” “believes,” “estimates,” “expects,” “intends,” “may,” “plans,” “projects,” “will,” “would” and similar expressions are intended to identify forward-looking statements, although not all forward-looking statements contain these identifying words. We may not actually achieve the plans, intentions or expectations disclosed in our forward-looking statements and you should not place undue reliance on our forward-looking statements. These statements are inherently uncertain and you are cautioned not to unduly rely upon these statements. Actual results or events could differ materially from the plans, intentions and expectations disclosed in the forward-looking statements that we make. These forward-looking statements involve risks and uncertainties that could cause our actual results to differ materially from those in the forward-looking statements, including, without limitation, the risks set forth in Part II, Item IA, “Risk Factors” in this Quarterly Report on Form 10-Q and in our other filings with the SEC. The forward-looking statements are applicable only as of the date on which they are made, and we do not assume any obligation to update any forward-looking statements.10-Q.
Overview
We are a biopharmaceutical company headquartered in San Diego, California, focused on identifying, developing and delivering life-changing therapies to people living with rare diseases.
Uncertainty Related to the COVID-19 Pandemic
In March 2020, the World Health Organization ("WHO") classified the COVID-19 outbreak as a pandemic. While the impact of the COVID-19 pandemic did not have a material adverse effect on our financial position or results of operations for the three and six months ended June 30, 2020, we have been monitoring the developments and assessing areas where there is potential for our business to be impacted. Our labor force is currently working remotely, which could, among other things, negatively impact our ability to conduct research and development activities, engage in sales-related initiatives, or efficiently conduct day-to-day operations, and we are aware that other businesses with which we engage are likely operating under similar restrictions and experiencing disruptions in their own operations, which may create obstacles in the coordination of business activities. Circumstances arising from the pandemic have slowed and could continue to slow the pace of enrollment in our clinical trials or otherwise hinder patient’s abilities to comply with the clinical trial protocols and could ultimately delay the availability of results and analysis of outcomes. Disruptions in the supply chain could negatively impact our ability to source materials or manufacture and distribute product. While we do not currently anticipate a material reduction in demand for our commercialized products, we could experience a decrease in new patient identification and increased requests for patient assistance due to increased levels of unemployment, either of which would negatively impact our revenues and hinder our cash flows. Similarly, we could face challenges with regard to healthcare programs, including access and changes in coverage. Growth in revenue could also be impeded by these factors. The financial markets have been subject to significant volatility that could impact our ability to enter into, modify, and negotiate favorable terms and conditions relative to equity and debt financing activities. We currently have $237.2 million in cash and cash equivalents and $220.2 million in available-for-sale securities as of June 30, 2020, which we believe provides sufficient capital to fund our operations for at least the next twelve months and withstand the anticipated near-term consequences of the pandemic, although liquidity constraints and access to capital markets could warrant changes to our investment strategy. While we have not yet experienced a material impact to date, the full magnitude of the pandemic cannot be measured at this time, and therefore any of the aforementioned circumstances, as well as other factors, may cause our results of operations to vary substantially from year to year and quarter to quarter.
Our Product Candidates and Approved Products
We are developing*Chenodal is indicated for the following pipeline products:
Fosmetpantotenate
We are developing fosmetpantothenate, a novel small molecule, as a potential treatment for pantothenate kinase-associated neurodegeneration (“PKAN”). PKANof patients suffering from gallstones and is a genetic neurodegenerative disorder that is typically diagnosedalso in the first decade of life. Symptoms of PKAN include dystonia, dysarthria, rigidity, retinal degeneration, and severe digestive problems. PKAN is estimated to affect up to 5,000 patients worldwide. There are currently no viable treatment options for patients with PKAN. Fosmetpantotenate is a phosphopantothenate replacement therapy that aims to restore levels of this key substrate in PKAN patients. Certain international health regulators have approved the initiation of dosing fosmetpantotenate in PKAN patients under physician-initiated studies in accordance with local regulations in their respective countries.
In 2015 and 2016 we filed a U.S. Investigational New Drug application ("IND"), completed the Phase I clinical trials and obtained both orphan drug and fast track designation in the United States. Additionally, we received orphan drug designation in the European Union and reached an agreement with the FDA under the Special Protocol Assessment ("SPA") process for a Phase 3 clinical trialdevelopment for PKAN.treatment of cerebrotendinous xanthomatosis ("CTX").
In the fourth quarter of 2018, the pivotal Phase 3 FORT Study of fosmetpantotenate in PKAN completed patient enrollment. The FORT Study is designed to be registration-enabling in the U.S. and Europe, and we expect top-line data to become available during the third quarter of 2019.
Product Candidate Development Activities:
Sparsentan
Sparsentan, also known as RE-021, is an investigational product candidate with a dual mechanism of action, a potent angiotensin receptor blocker (“ARB”) and selective endothelin receptor antagonist (“ERA”), with in vitro selectivity toward endothelin receptor type A. We have securedA and a license to sparsentan from Ligand Pharmaceuticals, Inc. and Bristol-Myers Squibb (who referred to it as DARA)potent angiotensin receptor blocker (“ARB”). Sparsentan is currently being evaluated in two pivotal Phase 3 clinical studies in the following indications:
•Focal segmental glomerulosclerosis ("FSGS"), a leading cause of end-stage renal disease and nephrotic syndrome (“NS”). There are currently no FDAUnited States Food and Drug Administration ("FDA") approved pharmacologic treatments for FSGS and off-label treatments are limited to ACE/ARBs, steroids, and immunosuppressant agents, which are effective in only a subset of patients. Every year approximately 5,400 patients are diagnosed with FSGS and we estimate that there are up to 40,000 FSGS patients in the United States and a similar number in Europe with approximately half of them being candidates for sparsentan. In 2015 and 2016 we receivedSparsentan has orphan drug designation in the United States and European Union andUnion. In 2016, we generated positive data from our Phase 2 DUET study of sparsentan for the treatment ofin FSGS. In the second quarter of 2018, we announced the initiation of the Phase 3 DUPLEX Study of sparsentan in FSGS. The Company continues to enroll patients with FSGS and enrollment continues. Thisin the pivotal Phase 3 DUPLEX Study, is designed to includea global, randomized, multicenter, double-blind, parallel-arm, active-controlled, clinical trial evaluating the safety and efficacy of sparsentan in approximately 300 patients. The DUPLEX Study protocol provides for an interimunblinded analysis of modifiedat least 190 patients to be performed after 36 weeks of treatment to evaluate the interim efficacy endpoint - the proportion of patients achieving a FSGS partial remission of proteinuria. We expect thatproteinuria endpoint (FPRE), which is defined as urine protein-to-creatinine ratio (Up/C) ≤1.5 g/g and a >40 percent reduction in Up/C from baseline, at week 36. While the confirmatory endpoint of the study is the change in slope of estimated glomerular filtration rate (eGFR) after 108 weeks of treatment, successful achievement of thisthe interim 36-week proteinuria endpoint willis expected to serve as the basis for submission of a New Drug Application ("NDA") for sparsentan for the treatment of FSGS under the Subpart H accelerated approval pathway in the United StatesU.S. and Conditional Marketing Authorization ("CMA") consideration in Europe. The DUPLEX Study protocol also provides for a sample size re-assessment to support the confirmatory endpointportion of the study will compare changes in slope of estimated glomerular filtration rate ("eGFR"). Top-linestudy. At this time, topline data from the interim36-week proteinuria endpoint analysis are expected to become available in the first halfquarter of 2021.2021, and we are monitoring the potential impact the evolving COVID-19 pandemic in conjunction with the outcome of the planned sample size reassessment may have on this timing.
•Immunoglobulin A nephropathy ("IgAN") is characterized by hematuria, proteinuria, and variable rates of progressive renal failure. With an estimated prevalence of more than 100,000 people in the United States and greater numbers in Europe and Asia, IgAN is the most common primary glomerular disease. Most patients are diagnosed between the ages of 16 and 35, with up to 40% progressing to end stage renal disease within 15 years. There are currently no FDA approved treatments for IgAN. The current standard of care is renin-angiotensin-aldosterone system ("RAAS") blockade with immunosuppression also being commonly used for patients with significant proteinuria or rapidly progressive glomerulonephritis. In the fourth quarter of 2018, we announced that the first patient had been dosed in the PROTECT Study, a global, randomized, multicenter, double-blind, parallel-arm, active-controlled pivotal Phase 3 clinical trial evaluating the long-term nephroprotective potentialsafety and efficacy of sparsentan in patients with IgAN, and
the study continues to enroll. The PROTECT Study protocol provides for thean unblinded analysis of at least 280 patients to be performed after 36 weeks of treatment of IgAN. Theto evaluate the primary efficacy endpoint in the PROTECT Study is– the change in proteinuria (urine protein-to-creatinine ratio) at Week 36 from baseline after 36 weeks of treatment. We expect that successfulbaseline. Successful achievement of thisthe proteinuria endpoint will serve as the basis foris expected to support submission of an NDA for sparsentan for the treatment of IgAN under the Subpart H accelerated approval pathway in the U.S. and, as well as an application for CMA consideration in Europe. Secondary efficacy endpoints include the rate of change in eGFR from baseline to four weeks post-cessationfollowing the initiation of randomized treatment over 58-week and 110-week periods, as well as the rate of change in eGFR over 52-week and 104-week periods following the first six weeks of randomized treatment. Top-linetreatment in approximately 380 patients. At this time, topline data from the primary36-week proteinuria endpoint analysis are expected to become available in the firstsecond half of 2022.2021 and we are monitoring the potential impact the evolving COVID-19 pandemic may have on this timing.
NGLY1 Collaboration
NGLY1 deficiency, is an extremely rare genetic disorder believed to be caused by a deficiency in an enzyme called N-glycanase-1, which is encoded by the gene NGLY1. The condition is characterized by symptoms such as developmental delays, seizures, complex hyperkinetic movement disorders, diminished reflexes and an inability to produce tears. There are no approved therapeutic options for NGLY1 deficiency, and current therapeutic strategies are limited to symptom management.Chenodal
In September 2017,January 2020, we entered intorandomized the first patients in a three-way Phase 3 clinical trial to evaluate the effects of Chenodal in adult and pediatric patients with cerebrotendinous xanthomatosis (“CTX”). The pivotal study, known as the RESTORE study, is intended to support a new drug application (“NDA”) submission for marketing authorization of Chenodal for CTX in the United States. Chenodal has also been the standard of care for CTX patients for more than three decades but is not currently labeled for this indication.
Cooperative Research and Development AgreementAgreements ("CRADA"CRADAs"):
We are a participant in two CRADAs, which form a multi-stakeholder approach to pool resources with leading experts, and incorporates the patient perspective early in the identification and development process. We have partnered with the National Institutes of Health’s National Center for Advancing Translational Sciences ("NCATS") and patientleading patients advocacy foundationorganizations, NGLY1.org to collaborate on research effortsand Alagille Syndrome Alliance ("ALGSA"), aimed at the identification of potential small molecule therapeutics for NGLY1 deficiency.
Alagille Collaboration
deficiency and Alagille syndrome, ("ALGS"), is a rare genetic disorder that can affect the liver, heart, skeleton, eyes and kidneys. Symptoms and severity of ALGS can vary greatly from one person to another. Symptoms often develop during the first three months of life and include interrupted bile flow (cholestasis), jaundice, poor weight gain and growth, and severe itching (pruritis). ALGS is an autosomal dominant disorder or can occur spontaneously. According to the National Organization for Rare Disorders ("NORD"), the estimated incidence of ALGS is between 1 in 30,000 and 1 in 45,000 births.respectively. There are no treatment options currently no approved therapies for ALGS.these diseases.
In June 2019, we announced that we entered into a three-way CRADA with NCATS and patient advocacy foundation Alagille Syndrome Alliance ("ALGSA") to collaborate on research efforts aimed at the identification and development of potential therapeutics for ALGS.
We sell the followingThe Company has three approved products:
Chenodal® (chenodiol tablets)
Chenodal is a synthetic oral form of chenodeoxycholic acid, a naturally occurring primary bile acid synthesized from cholesterol in the liver, indicated for the treatment of radiolucent stones in well-opacifying gallbladders in patients in whom selective surgery would be undertaken except for the presence of increased surgical risk due to systemic disease or age.
Chenodal administration is known to reduce biliary cholesterol and the dissolution of radiolucent gallstones through suppression of hepatic synthesis of cholesterol, cholic acid and deoxycholic acid in the bile pool. Chenodal was first approved by the U.S. Food and Drug Administration ("FDA")FDA in 1983 for the management of gallstones but its marketing was later discontinued due to lack of commercial success. In 2009, Nexgen Pharma Inc.'s Abbreviated New Drug Application ("ANDA")ANDA for Chenodal was approved by the FDA for the treatment of gallstones. Chenodal is manufactured under this ANDA. In 2010, Chenodal was granted orphan drug designation for the treatment of cerebrotendinous xanthomatosis (“CTX”),CTX, a rare autosomal recessive lipid storage disease. We acquired Chenodal in March 2014.
While Chenodal is not labeled for CTX, it has been used as the standard of care for over three decades. We are working to obtain FDA approval of Chenodal for the treatment of CTX.CTX and initiated a Phase 3 clinical trial for this indication in January 2020. The prevalence of CTX is estimated in the literature to be as high as 1 in 70,000 in the overall population. Pathogenesis of CTX involves deficiency of the enzyme 27-hydroxylase (encoded by the gene CYP27A1), a rate-limiting enzyme in the synthesis of
primary bile acids, including chenodeoxycholic acid ("CDCA"),CDCA, from cholesterol. The disruption of primary bile acid synthesis in CTX leads to toxic accumulation of cholesterol and cholestanol in most tissues. Most patients present with intractable diarrhea, premature cataracts, tendon xanthomas, atherosclerosis, and cardiovascular disease in childhood and adolescence. Neurological manifestations of the disease, including dementia and cognitive and cerebellar deficiencies, emerge during late adolescence and adulthood. Oral administration of CDCA has been shown to normalize primary bile acid synthesis in patients with CTX.
Cholbam® (cholic acid capsules)
The FDA approved Cholbam (cholic acid capsules) in March 2015, the first FDA approved treatment for pediatric and adult patients with bile acid synthesis disorders due to single enzyme defects, and for adjunctive treatment of patients with peroxisomal disorders (including Zellweger spectrum disorders). The effectiveness of Cholbam has been demonstrated in clinical trials for bile acid synthesis disorders and the adjunctive treatment of peroxisomal disorders. An estimated 200 to 300 patients are current candidates for therapy.
Kolbam,Cholbam was previously marketed in Europe under the branded name Kolbam. Sales of CholbamKolbam in Europe is indicatedwere not a material component of our business, and in EuropeJuly 2020, we withdrew our European marketing approval application for the treatment of certain inborn errors of primary bile acid synthesis, encompassing select single enzyme defects, in infants from one month of age for continuous lifelong treatment through adulthood.Kolbam.
Thiola®and Thiola EC® (tiopronin)
Thiola isand Thiola EC are approved by the FDA for the treatment of cystinuria, a rare genetic cystine transport disorder that causes high cystine levels in the urine and the formation of recurring kidney stones. The resulting long-term damage can cause loss of kidney function in addition to substantial pain and loss of productivity associated with renal colic and stone passage. The prevalence of cystinuria in the United States is estimated to be 10,000 to 12,000, indicating that there may be as many as 4,000 to 5,000 affected individuals with cystinuria in the United States that would be candidates for Thiola.
In June 2019, we announced that the FDA approved 100 mg and 300 mg tablets of THIOLA®Thiola EC, (tiopronin), a new enteric-coated formulation of THIOLA® (tiopronin). THIOLAThiola. Thiola EC became available to patients in July 2019 and the original formulation also remains available to patients.
Pipeline portfolio changes:
During the first quarter of 2019, the Company elected to discontinue development of the L-UDCA program, resulting in removing the intangible asset of $25.5 million, originally recorded in 2016, and the related $18.0 million in contingent liability. This resulted in a net $7.5 million non-cash charge to first quarter operations. In June 2016, the Company acquired certain rights to its product candidate L-UDCA for $0.5 million cash. At the same time the Company established a related non-cash asset of $25.5 million and liability of $25.0 million for IPR&D and contingent consideration (deferred financing) related net sales royalties and milestones. As a result of our quarterly valuation update process during 2016 and 2017, the contingent liability was decreased by $2.3 million and $5.7 million, respectively, and increased by $1.0 million during 2018. The resulting balance of the L-UDCA contingent liability at December 31, 2018 was $18.0 million.
In August 2019, following a strategic review of the CNSA-001 program in patients with phenylketonuria (PKU), the Company made the decision to decline to exercise its option to acquire Censa Pharmaceuticals and accordingly discontinue its joint development program for CNSA-001. The Company expects to impair the $15 million long term investment on the balance sheet during the third quarter of 2019.
Results of Operations
Results of operations for the three and six months ended June 30, 20192020 compared to the three and six months ended June 30, 2018.2019.
Net Product Sales:
The following table provides information regarding net product sales (in thousands):
| | | | Three Months Ended June 30, | | Six Months Ended June 30, | | Three Months Ended June 30, | | | Six Months Ended June 30, | |
| | 2019 | | 2018 | | Change | | 2019 | | 2018 | | Change | | 2020 | | 2019 | | Change | | 2020 | | 2019 | | Change |
| Net product revenues by product: | | | | | | | | | | | | Net product revenues by product: | | | | | | | | | | | |
| Bile acid products | $ | 20,929 |
| | $ | 18,594 |
| | $ | 2,335 |
| | $ | 39,319 |
| | $ | 37,102 |
| | $ | 2,217 |
| Bile acid products | $ | 21,573 | | | $ | 20,929 | | | $ | 644 | | | $ | 43,854 | | | $ | 39,319 | | | $ | 4,535 | |
| Thiola | 23,778 |
| | 22,743 |
| | 1,035 |
| | 44,958 |
| | 42,667 |
| | 2,291 |
| |
| Tiopronin products | | Tiopronin products | 26,857 | | | 23,778 | | | 3,079 | | | 52,345 | | | 44,958 | | | 7,387 | |
| Total net product revenues | $ | 44,707 |
| | $ | 41,337 |
| | $ | 3,370 |
| | $ | 84,277 |
| | $ | 79,769 |
| | $ | 4,508 |
| Total net product revenues | $ | 48,430 | | | $ | 44,707 | | | $ | 3,723 | | | $ | 96,199 | | | $ | 84,277 | | | $ | 11,922 | |
The sales increase for the three and six months ended June 30, 20192020 compared to the three and six months ended June 30, 20182019 was due to increased patient counts, as well as the normal fluctuations in timing of new patient starts and prescription refills.
Operating Expenses:
The following table provides information regarding operating expenses (in thousands):
| | | | Three Months Ended June 30, | | Six Months Ended June 30, | | Three Months Ended June 30, | | | Six Months Ended June 30, | |
| | 2019 | | 2018 | | Change | | 2019 | | 2018 | | Change | | 2020 | | 2019 | | Change | | 2020 | | 2019 | | Change |
| Cost of goods sold | $ | 979 |
| | $ | 1,178 |
| | $ | (199 | ) | | $ | 1,996 |
| | $ | 2,791 |
| | $ | (795 | ) | Cost of goods sold | $ | 1,494 | | | $ | 979 | | | $ | 515 | | | $ | 2,864 | | | $ | 1,996 | | | $ | 868 | |
| Research and development | 37,934 |
| | 34,460 |
| | 3,474 |
| | 71,377 |
| | 59,096 |
| | 12,281 |
| Research and development | 30,790 | | | 37,934 | | | (7,144) | | | 61,038 | | | 71,377 | | | (10,339) | |
| Selling, general and administrative | 38,970 |
| | 25,100 |
| | 13,870 |
| | 71,639 |
| | 51,568 |
| | 20,071 |
| Selling, general and administrative | 34,971 | | | 38,970 | | | (3,999) | | | 68,110 | | | 71,639 | | | (3,529) | |
| Change in fair value of contingent consideration | 3,353 |
| | 2,159 |
| | 1,194 |
| | 6,522 |
| | 5,786 |
| | 736 |
| Change in fair value of contingent consideration | 4,286 | | | 3,353 | | | 933 | | | 2,363 | | | 6,522 | | | (4,159) | |
| Impairment of L-UDCA IPR&D intangible asset | — |
| | — |
| | — |
| | 25,500 |
| | — |
| | 25,500 |
| |
| Write off of L-UDCA IPR&D intangible asset | | Write off of L-UDCA IPR&D intangible asset | — | | | — | | | — | | | — | | | 25,500 | | | (25,500) | |
| Write off of L-UDCA contingent consideration | — |
| | — |
| | — |
| | (18,000 | ) | | — |
| | (18,000 | ) | Write off of L-UDCA contingent consideration | — | | | — | | | — | | | — | | | (18,000) | | | 18,000 | |
| | $ | 81,236 |
| | $ | 62,897 |
| | $ | 18,339 |
| | $ | 159,034 |
| | $ | 119,241 |
| | $ | 39,793 |
| |
| | | | $ | 71,541 | | | $ | 81,236 | | | $ | (9,695) | | | $ | 134,375 | | | $ | 159,034 | | | $ | (24,659) | |
Research and development expenses
We make significant investments in research and development in support of our development programs. Research and development costs are expensed as incurred and include salaries and bonuses, benefits, non-cash share basedshare-based compensation, license fees, costs paid to third-party contractors to perform research, conduct clinical trials, and develop drug materials, and associated overhead expenses and facility costs.
For the three and six months ended June 30, 20192020 as compared to the three and six months ended June 30, 2018, the Company increased its2019, our research and development expenses decreased by $3.5$7.1 million and $12.3$10.3 million, respectively, which is due to increased clinical trial expenses related to our three on-going Phase 3 studies,the discontinuation of the fosmetpantotenate program in PKAN and sparsentan in FSGS and IgAN.PKAN.
Selling, general and administrative expenses
Selling, general and administrative expenses include salaries and bonuses, benefits, non-cash share basedshare-based compensation, professional fees, rent, depreciation and amortization, travel, insurance, business development, sales and marketing programs, and other operating expenses.
For the three and six months ended June 30, 20192020 as compared to the three and six months ended June 30, 2018, the Company increased its2019, our selling, general and administrative expenses decreased by $13.9$4.0 million and $20.1$3.5 million, respectively,respectively. This decrease is primarily due to compensation expense, primarily as a result of a larger employee base, and increasedlower legal expenses.
Change in the valuation of contingent consideration
For the three and sixmonths ended June 30, 2020 as compared to the three months ended June 30, 2019, as compared to the three and six months ended June 30, 2018,the change in fair value of contingent consideration is due to timing of payments for Chenodal and Cholbam.changes in market driven discount rates.
The following table summarizesFor the Company'ssix months ended June 30, 2020 as compared to the six months ended June 30, 2019, the change in valuationfair value of contingent consideration (is due to a change in thousands):
|
| | | | | | | | | | | | | | | | | | | | | | | |
| | Three Months Ended June 30, | | Six Months Ended June 30, |
| | 2019 | | 2018 | | Change | | 2019 | | 2018 | | Change |
| Chenodal | $ | 2,741 |
| | $ | 1,107 |
| | $ | 1,634 |
| | $ | 4,280 |
| | $ | 1,908 |
| | $ | 2,372 |
|
| Cholbam | 612 |
| | (458 | ) | | 1,070 |
| | 2,242 |
| | 1,378 |
| | 864 |
|
| L-UDCA | — |
| | 1,500 |
| | (1,500 | ) | | — |
| | 2,500 |
| | (2,500 | ) |
| Change in fair value of contingent consideration | $ | 3,353 |
| | $ | 2,149 |
| | $ | 1,204 |
| | $ | 6,522 |
| | $ | 5,786 |
| | $ | 736 |
|
Write off of L-UDCA Contingent Consideration and impairment of L-UDCA IPR&D intangible assets
In June 2016, the Company acquired certain rights to its product candidate L-UDCA for $0.5 million cash. At the same time the Company established a related non-cash asset of $25.5 million and liability of $25.0 million for IPR&D and contingent consideration (deferred financing) related net sales royalties and milestones. As a result of our quarterly valuation update process during 2016 and 2017, the contingent liability was decreased by $2.3 million and $5.7 million, respectively, and increased by $1.0 million during 2018. The resulting balance of the L-UDCA contingent liability at December 31, 2018 was $18.0 million.revenue projections.
During the first quarter of 2019, the Companywe elected to discontinue development of the L-UDCA program, resulting in removingthe removal of the intangible asset of $25.5 million, which was originally recorded in 2016, and the reversal of associated contingent consideration of $18.0 million. This resulted in a net $7.5 million non-cash charge to first quarter operations.operations in 2019.
Other Income (Expenses):
The following table provides information regarding other income (expenses), net (in thousands):
| | | | Three Months Ended June 30, | | Six Months Ended June 30, | | Three Months Ended June 30, | | | Six Months Ended June 30, | |
| | 2019 | | 2018 | | Change | | 2019 | | 2018 | | Change | | 2020 | | 2019 | | Change | | 2020 | | 2019 | | Change |
| Other income (expense), net | $ | 125 |
| | $ | (403 | ) | | $ | 528 |
| | $ | (177 | ) | | $ | (282 | ) | | $ | 105 |
| Other income (expense), net | $ | 426 | | | $ | 125 | | | $ | 301 | | | $ | 235 | | | $ | (177) | | | $ | 412 | |
| Interest income | 2,589 |
| | 858 |
| | 1,731 |
| | 5,408 |
| | 1,655 |
| | 3,753 |
| Interest income | 1,316 | | | 2,589 | | | (1,273) | | | 3,291 | | | 5,408 | | | (2,117) | |
| Interest expense | (4,817 | ) | | (1,057 | ) | | (3,760 | ) | | (9,682 | ) | | (2,212 | ) | | (7,470 | ) | Interest expense | (4,634) | | | (4,817) | | | 183 | | | (9,521) | | | (9,682) | | | 161 | |
| | $ | (2,103 | ) | | $ | (602 | ) | | $ | (1,501 | ) | | $ | (4,451 | ) | | $ | (839 | ) | | $ | (3,612 | ) | |
| | | | $ | (2,892) | | | $ | (2,103) | | | $ | (789) | | | $ | (5,995) | | | $ | (4,451) | | | $ | (1,544) | |
The change in the Company'sour other income (expenses) for the three and six months ended June 30, 20192020 as compared to the three and six months ended June 30, 20182019 of $1.5$0.8 million and $3.6$1.5 million, respectively, is primarily due to the additionallower average balances in investments in debt securities and lower interest from the Company's 2.50% Convertible Senior Notes due in 2025 ("2025 Notes") issued in September 2018.rates.
Income Tax Expense:Benefit (Provision):
For the three and six months ended June 30, 2019,2020, we recognized an income tax expensebenefit of $0.1$18.9 million, and $0.5 million, representing an effective tax rate forprimarily related to provisions of the year of 0.2%.CARES Act concerning net operating loss carrybacks. Under GAAP, quarterly effective tax rates may vary significantly depending on the actual operating results in the various tax jurisdictions, and significant transactions, as well as changes in the valuation allowance related to the expected recovery of deferred tax assets.
At June 30, 2019,2020, we had no unrecognized tax benefits. We did not recognize any interest or penalties related to unrecognized tax benefits during the six months ended June 30, 2019.2020.
Liquidity and Capital Resources
We believe that our available cash and short-term investments as of the date of this filing will be sufficient to fund our anticipated level of operations for at leastbeyond the next 12 months. Management believes that our operating results will vary from quarter to quarter and year to year depending upon various factors including revenues, general and administrative expenses, and research and development expenses.
The CompanyWe had the following financial performancebalances at June 30, 20192020 and December 31, 20182019 (in thousands):
| | | | June 30, 2019 | | December 31, 2018 | | June 30, 2020 | | December 31, 2019 |
| Cash & Cash Equivalents | $ | 75,657 |
| | $ | 102,873 |
| Cash & Cash Equivalents | $ | 237,170 | | | $ | 62,436 | |
| Marketable securities | 350,243 |
| | 368,668 |
| |
| Debt securities | | Debt securities | 220,206 | | | 336,088 | |
| Accumulated Deficit | (349,695 | ) | | (270,017 | ) | Accumulated Deficit | (441,704) | | | (416,444) | |
| Stockholders' Equity | 277,089 |
| | 318,253 |
| Stockholders' Equity | 318,645 | | | 221,196 | |
| | | | | |
| Net Working Capital* | $ | 366,428 |
| | $ | 391,057 |
| Net Working Capital* | $ | 435,629 | | | $ | 333,615 | |
| Net Working Capital Ratio** | 5.05 |
| | 4.74 |
| Net Working Capital Ratio** | 7.23 | | | 4.50 | |
* Current assets less current liabilities.
**Current assets divided by current liabilities.
Future Minimum Rental Commitments
We have future minimum rental commitments totaling $13.4 million arising from our operating leases. These commitments consist of $1.4 million in aggregate base rent for our existing office lease estimated to expire in January 2021 and $12.8 million in aggregate base rent through August 2028, less seven months of rent abatement totaling $0.9 million for the new office lease space delivered in June 2020. Excluded from these rental commitments is one month of advance rent totaling $0.1 million, and lease incentives in the form of tenant improvements allowances totaling $2.0 million for the office space delivered in June 2020. Our future minimum rental commitments will increase by $34.0 million upon taking possession of the remainder of the new office lease space, which is expected to occur in the latter part of 2020. These additional commitments consist of $36.5 million in aggregate base rent through August 2028, less seven months of rent abatement totaling $2.5 million for the new office lease space not yet delivered. Excluded from these additional rental commitments is lease incentives in the form of tenant improvements allowances totaling $5.8 million for the office space not yet delivered. See additional discussion of our operating leases within Note 6 of the financial statement disclosures.
Underwritten Public Offering of Common Stock
In June 2020, the Company sold an aggregate of 7.5 million shares of its common stock in an underwritten public offering, at a price to the public of $15.50 per share. The net proceeds to the Company from the offering, after deducting the underwriting discounts and offering expenses, was $108.6 million.
At-the-Market Equity Offering Program
In February 2020, we entered into an Open Market Sale Agreement ("ATM Agreement") with Jefferies LLC, as agent (“Jefferies”), pursuant to which we may offer and sell, from time to time through Jefferies, shares of our common stock having an aggregate offering price of up to $100.0 million. Shares will be sold
pursuant to our effective registration statement on Form S-3 (Registration Statement No. 333-227182), as previously filed with the Securities and Exchange Commission. Through the period ended June 30, 2020, we have not sold any shares under the ATM Agreement.
Convertible Notes Payable
Convertible Senior Notes Due 2025
On September 10, 2018, the Companywe completed itsa registered underwritten public offering of $276.0 million aggregate principal amount of the2.50% Convertible Senior Notes due 2025 Notes,("2025 Notes"), and entered into a base indenture and supplemental indenture agreement ("2025 Indenture") with respect to the 2025 Notes. The 2025 Notes will mature on September 15, 2025 ("Maturity Date”), unless earlier repurchased, redeemed, or converted. The 2025 Notes are senior unsecured obligations of the Companyours and bear interest at an annual rate of 2.50%, payable semi-annually in arrears on March 15 and September 15 of each year. The first payment was madeyear, beginning on March 15, 2019.
The composition of the Company’sour 2025 Notes are as follows (in thousands):
| | | | June 30, 2019 | | December 31, 2018 | | June 30, 2020 | | December 31, 2019 |
| 2.50% convertible senior notes due 2025 | $ | 276,000 |
| | $ | 276,000 |
| 2.50% convertible senior notes due 2025 | $ | 276,000 | | | $ | 276,000 | |
| Unamortized debt discount | (70,484 | ) | | (74,836 | ) | Unamortized debt discount | (61,265) | | | (65,963) | |
| Unamortized debt issuance costs | (5,625 | ) | | (6,073 | ) | Unamortized debt issuance costs | (4,726) | | | (5,176) | |
| Total 2025 Notes, net of unamortized debt discount and debt issuance costs | $ | 199,891 |
| | $ | 195,091 |
| Total 2025 Notes, net of unamortized debt discount and debt issuance costs | $ | 210,009 | | | $ | 204,861 | |
The net proceeds from the issuance of the 2025 Notes were approximately $267.2 million, after deducting commissions and the offering expenses payable by the Company.us. A portion of the net proceeds from the 2025 Notes were used by the Companyus to repurchase $23.4 million aggregate principal amount of itsour then-outstanding 4.50% Senior Convertible Notes due in 2019 ("2019 Notes") in privately-negotiated transactions for approximately $40.2 million in cash.transactions.
Holders may convert their 2025 Notes at their option only in the following circumstances: (1) during any calendar quarter commencing after the calendar quarter ending on December 31, 2018 (and only during such calendar quarter), if the last reported sale price per share of the Company’s common stock for each of at least 20 trading days, whether or not consecutive, during the period of 30 consecutive trading days ending on, and including, the last trading day of the immediately preceding calendar quarter exceeds 130% of the conversion price on the applicable trading day; (2) during the five consecutive business days immediately after any 10 consecutive trading day period (such 10 consecutive trading day period, the “measurement period”) if the trading price per $1,000 principal amount of 2025 Notes for each trading day of the measurement period was less than 98% of the product of the last reported sale price per share of the Company’s common stock on such trading day and the conversion rate on such trading day; (3) upon the occurrence of certain corporate events or distributions on the Company’s common stock; (4) if the Company calls the 2025 Notes for redemption; and (5) at any time from, and including, May 15, 2025 until the close of business on the scheduled trading day immediately before the Maturity Date. The Company will settle conversions by paying or delivering, as applicable, cash, shares of the Company’s common stock, or a combination of cash and shares of the Company’s common stock, at the Company’s election, based on the applicable conversion rate.
The initial conversion rate for the 2025 Notes is 25.7739 shares of the Company’sour common stock per $1,000 principal amount of 2025 Notes, which represents an initial conversion price of approximately $38.80 per share. If a “make-whole fundamental change” (as defined in the 2025 Indenture) occurs, then the Companywe will, in certain circumstances, increase the conversion rate for a specified period of time.
The 2025 Notes will be redeemable, in whole or in part, at the Company’s option at any time, and from time to time, on or after September 15, 2022 and, in the case of any partial redemption, on or before the 40th scheduled trading day before the Maturity Date, at a cash redemption price equal to the principal amount of the 2025 Notes to be redeemed, plus accrued and unpaid interest, if any, to, but excluding, the redemption date but only if the last reported sale price per share of the Company’s common stock exceeds 130% of the conversion price on each of at least 20 trading days during the 30 consecutive trading days ending on, and including, the trading day immediately before the date the Company sends the related redemption notice. If a fundamental change (as defined in the 2025 Indenture) occurs, then, subject to certain exceptions, holders may require the Company to repurchase their 2025 Notes at a cash repurchase price equal to the principal amount of the 2025 Notes to be repurchased, plus accrued and unpaid interest, if any, to, but excluding, the fundamental change repurchase date.
In the event of conversion, holders would forgo all future interest payments, any unpaid accrued interest and the possibility of further stock price appreciation. Upon the receipt of conversion requests, the settlement of the 2025 Notes will be paid pursuant to the terms of the 2025 Indenture. In the event that all of the 2025 Notes are converted, the Company would be required to repay the $276.0 million in principal value and any conversion premium in any combination of cash and shares of its common stock (at the Company’s option).
Convertible Senior Notes Due 2019
On May 29, 2014, the Company entered into a Note Purchase Agreement relating to a private placement by the Company of $46.0 million aggregate principal amount of 2019 Notes which were convertible into shares of the Company’s common stock at an initial conversion price of $17.41 per share. The conversion price was subject to customary anti-dilution protection. The 2019 Notes bore interest at a rate of 4.5% per annum, payable semiannually in arrears on May 15 and November 15 of each year. The maturity date of the 2019 Notes was on May 30, 2019, and there were no contractual payments due prior to that date.
In September 2018, the Company used part of the net proceeds from the issuance of the 2025 Notes to repurchase $23.4 million aggregate principal amount of the 2019 Notes in privately-negotiated transactions for approximately $40.2 million in cash. The partial repurchase of the 2019 Notes resulted in a $17.0 million loss on early extinguishment of debt in September 2018.
In May 2019, the remaining $22.6 million outstanding principal amount of 2019 Notes was converted by the holders thereof into approximately 1.3 million shares of common stock.
The composition of the 2019 Notes at December 31, 2018 was as follows (in thousands):
|
| | | | |
| | | December 31, 2018 |
| 4.50% senior convertible notes due 2019 | | $ | 22,590 |
|
| Unamortized debt discount | | (125 | ) |
| Unamortized debt issuance costs | | (8 | ) |
| Total 2019 Notes, net of debt discount and debt issuance costs | | $ | 22,457 |
|
Cash Flows from Operating Activities
Cash used in operating activities was $35.6 million for the six months ended June 30, 2020 compared to cash used of $39.6 million for the six months ended June 30, 2019 compared to cash used of $17.5 million for the six months ended June 30, 2018.2019. The increasechange in cash used in operations is attributabledue to increasedlower R&D expenses from the discontinuation of the fosmetpantotenate program in PKAN and lower legal expenses.expenses, offset by changes in net working capital.
Cash Flows from Investing Activities
Cash provided by investing activities for the six months ended June 30, 20192020 was $13.8$107.4 million, compared to cash provided of $7.1$13.8 million for the six months ended June 30, 2018.2019. The increase ischange was due to timing differences associated with the purchasessale and sales and maturitiesmaturity of our available-for-sale investments as well as changes in our portfolio-mix between cash equivalents and short-term and long-term investment holdings, offset by our $15.0 million investment in Censa in the first quarterthat were not reinvested.
Cash Flows from Financing Activities
Cash usedprovided by financing activities for the six months ended June 30, 20192020 was $1.4$103.0 million compared to cash providedused of $0.4$1.4 million for the six months ended June 30, 2018.2019. The changeincrease in cash provided was due to lower contingent consideration paymentsthe underwritten public offering of our common stock in 2019, offset by lower proceeds from the exercise of stock options and warrants.June 2020.
Funding Requirements
We believe that our available cash and short-term investments as of the date of this filing will be sufficient to fund our anticipated level of operations for at least the next 12 months. This belief is based on many factors, some of which are beyond our control. Factors that may affect financing requirements include, but are not limited to:
•increases or decreases in revenue growth offrom our marketed products;products, including decreases in revenue resulting from the COVID-19 pandemic, if any;
•the rate of progress and cost of our clinical trials, preclinical studies and other discovery and research and development activities;activities, including any delays resulting from the COVID-19 pandemic;
•the timing of, and costs involved in, seeking and obtaining marketing approvals for our products, and in maintaining quality systems standards for our products;
•debt service obligations on the 2025 Notes;
•our ability to manufacture sufficient quantities of our products to meet expected demand;
•the costs of preparing, filing, prosecuting, maintaining and enforcing any patent claims and other intellectual property rights, litigation costs and the results of litigation;
•our ability to enter into collaboration, licensing or distribution arrangements and the terms and timing of these arrangements;
•the potential need to expand our business, resulting in additional payroll and other overhead expenses;
•the potential in-licensing of other products or technologies; and
•the emergence of competing technologies or other adverse market or technological developments.
Future capital requirements will also depend on the extent to which we acquire or invest in additional complementary businesses, products and technologies.
Other Matters
Adoption of New Accounting Standards
See Note 2 to our unaudited Condensed Consolidated Financial Statements in this report for a discussion of adoption of new accounting standards.
Recently Issued Accounting Pronouncements
See Note 2 to our unaudited Condensed Consolidated Financial Statements in this report for a discussion of recently issued accounting pronouncements.
Off Balance Sheet Arrangements
None.
In December 2017, the Company entered into a Future Acquisition RightTable of Joint Development Agreementwith Censa, which became effective in January 2018. The Company determined that Censa is a variable interest entity ("VIE") and concluded that the Company is not the primary beneficiary of the VIE. As such, the Company did not consolidate Censa’s results into its consolidated financial statements.ContentsIn August 2019, following a strategic review of the CNSA-001 program evaluating CNSA-001 in patients with phenylketonuria (PKU), the Company made the decision to decline to exercise its option to acquire Censa Pharmaceuticals and accordingly discontinue its joint development program for CNSA-001. The Company expects to impair the $15 million long term investment on the balance sheet during the third quarter of 2019. See Note 5 to the Unaudited Condensed Consolidated Financial Statements.
Item 3. Quantitative and Qualitative Disclosures about Market Risk
We invest our excess cash and marketabledebt securities primarily in United States government backed securities, asset-backed securities and debt instruments of financial institutions and corporations with investment-grade credit ratings. These instruments have various short and long-term maturities, not exceeding two years. We do not utilize derivative financial instruments, derivative commodity instruments, or other market risk sensitive instruments, positions or transactions. Accordingly, we believe that, while the instruments held are subject to changes in the financial standing of the issuer of such securities, we are not subject to any material risks arising from changes in interest rates, foreign currency exchange rates, commodity prices, equity prices or other market changes that affect market risk sensitive investments. A hypothetical 1% adverse move in interest rates along the entire interest rate yield curve would decrease our available-for-sale marketabledebt securities as of June 30, 2020 by approximately $2.8$1.1 million if the Company were to sell the securities.
Item 4. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
We maintain disclosure controls and procedures that are designed to ensure that information required to be disclosed in our reports required by the Exchange Act of 1934, as amended, is recorded, processed, summarized and reported within the timelines specified in the SEC’s rules and forms, and that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure. In designing and evaluating the disclosure controls and procedures, management recognized that any controls and procedures, no matter how well designed and operated, can only provide reasonable assurance of achieving the desired control objectives, and in reaching a reasonable level of assurance, management necessarily was required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures.
As required by SEC Rule 13a-15(b), we carried out an evaluation, under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, of the effectiveness of the design and operation of our disclosure controls and procedures as of the end of the quarter covered by this report. Based on the foregoing, our Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures were effective at the reasonable assurance level.
Changes in Internal Control over Financial Reporting
An evaluation was also performed under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, of any change to our internal control over financial reporting that occurred during the quarter covered by this report and that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting. Our evaluation did not identify significant changes in our internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) under the Securities Exchange Act of 1934) that occurred during the quarter ended June 30, 2019,2020, that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
PART II - OTHER INFORMATION
Item 1. Legal Proceedings
The information required by this Item is incorporated herein by reference to the Notes to the Unaudited Condensed Consolidated Financial Statements--Note 12 Commitments and Contingencies: Legal Proceedings in Part I, Item 1, of this Quarterly Report on Form 10-Q.
Item 1A. Risk Factors
The following risk factors do not reflect any material changes to the risk factors set forth in our Annual Report on Form 10-K for the fiscal year ended December 31, 2018,2019, other than the revisions to the risk factors set forth below with an asterisk (*) next to the title. The following information sets forth risk factors that could cause our actual results to differ materially from those contained in forward-looking statements we have made in this Quarterly Report on Form 10-Q and those we may make from time to time. If any of the following risks actually occur, our business, operating results, prospects or financial condition could be harmed. Additional risks not presently known to us, or that we currently deem immaterial, may also affect our business operations.
Risks Related to the Development of our Product Candidates
* Our clinical trials may fail to demonstrate the safety and efficacy of our product candidates, including sparsentan, which could prevent or significantly delay their regulatory approval.
Before obtaining regulatory approval for the sale of any of our current or future product candidates, including sparsentan, we must subject these product candidates to extensive preclinical and clinical testing to demonstrate their safety and efficacy for humans. Clinical trials are expensive, time-consuming and may take years to complete.
We may experience numerous unforeseen events during, or as a result of, preclinical or nonclinical testing and the clinical trial process that could delay or prevent our ability to obtain regulatory approval or commercialize our product candidates, including:
•our preclinical or nonclinical tests or clinical trials may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional preclinical testing or clinical trials or we may abandon projects that we expect to be promising;
•regulators may require us to conduct studies of the long-term effects associated with the use of our product candidates;
•regulators or institutional review boards may not authorize us to commence a clinical trial or conduct a clinical trial at a prospective trial site;
•the FDA or any non-United States regulatory authority may impose conditions on us regarding the scope or design of our clinical trials or may require us to resubmit our clinical trial protocols to institutional review boards for re-inspection due to changes in the regulatory environment;
•the number of patients required for our clinical trials may be larger than we anticipate or participants may drop out of our clinical trials at a higher rate than we anticipate;
•our third-party contractors or clinical investigators may fail to comply with regulatory requirements or fail to meet their contractual obligations to us in a timely manner;
•we might have to suspend or terminate one or more of our clinical trials if we, regulators or institutional review boards determine that the participants are being exposed to unacceptable health risks;
•regulators or institutional review boards may require that we hold, suspend or terminate clinical research for various reasons, including noncompliance with regulatory requirements;
•the cost of our clinical trials may be greater than we anticipate;
•the supply or quality of our product candidates or other materials necessary to conduct our clinical trials may be insufficient or inadequate or we may not be able to reach agreements on acceptable terms with prospective clinical research organizations; and
•the effects of our product candidates may not be the desired effects or may include undesirable side effects or the product candidates may have other unexpected characteristics.
These risks and uncertainties impact all of our clinical programs.programs that we pursue and have been amplified by the recent COVID-19 pandemic, as described below. We will only obtain regulatory approval to commercialize a product candidate if we can demonstrate to the satisfaction of the FDA, and in the case of foreign commercialization, to the applicable foreign regulatory authorities, in well-designed and conducted clinical trials, that our product candidates are safe and effective and otherwise meet the appropriate standards required for approval for a particular indication.
Our product development costs will also increase if we experience delays in testing or approvals. We do not know whether any preclinical tests or clinical trials will be initiated as planned, will need to be restructured or will be completed on schedule, if at all. Significant preclinical or clinical trial delays also could shorten the patent protection period during which we may have the exclusive right to commercialize our product candidates. Such delays could allow our competitors to bring products to market before we do and impair our ability to commercialize our products or product candidates.
If we are required to conduct additional clinical trials or other testing of our product candidates beyond those that we currently contemplate, if we are unable to successfully complete our clinical trials or other testing, if the results of these trials or tests are not positive or are only modestly positive or if there are safety concerns, we may:
•be delayed in obtaining, or may not be able to obtain, marketing approval for one or more of our product candidates;
•obtain approval for indications that are not as broad as intended or entirely different than those indications for which we sought approval; and
•have the product removed from the market after obtaining marketing approval.
Our product candidatesWe are intendeddeveloping in Phase 3 clinical trials sparsentan to treat PKAN, FSGS and IgAN, each of which is a rare disease. Given that these development candidates are still undergoing required testing, we may not be able to initiate or continue clinical trials if we are unable to locate a sufficient number of eligible patients willing and able to participate in the clinical trials required by the FDA or foreign regulatory agencies. In addition, as other companies and researchers may be concurrently developing therapies for the same or similar indications that we are focused on, we could face competition for a limited number of patients, investigators and clinical trial sites willing to participate in clinical trials. Our inability to enroll and maintain a sufficient number of patients for any of our current or future clinical trials would result in significant delays or may require us to abandon one or more clinical trials altogether.
In January 2020, we randomized the first patients in a Phase 3 clinical trial to evaluate the effects of Chenodal in adult and pediatric patients with CTX. The pivotal study, known as the RESTORE study, is intended to support an NDA submission for marketing authorization of Chenodal for CTX in the United States. While Chenodal has been used as the standard of care for CTX for over three decades, it is not labeled for CTX and as such we cannot market this drug candidate for the treatment of CTX unless and until it receives FDA approval for this indication. If we experience delays in obtaining approval or if we fail to obtain approval of Chenodal for the treatment of CTX, our business, financial condition and results of operations could be adversely affected.
Success in preclinical testing and early clinical trials does not ensure that later clinical trials will be successful, and initial results from a clinical trial do not necessarily predict final results.
Success in preclinical testing and early clinical trials does not ensure that later clinical trials will be successful, and initial results from a clinical trial do not necessarily predict final results. For example, there can be no assurance that thealthough we observed favorable responses we have seen with the physician-initiated treatment of fosmetpantotenate in PKAN patients outside the United States, will translate to positive data in the Phase 3 clinical trialFORT Study evaluating the safety and efficacy of fosmetpantotenate orcompared to placebo in patients with PKAN did not meet its primary endpoint, did not demonstrate a difference between treatment groups, and did not meet its secondary endpoint. In addition, there can be no assurance that the positive results from the DUET study of sparsentan in FSGS will be repeated in the Phase 3 clinical trial. Similarly, there can be no assurance that our clinical experience with sparsentan in FSGS will translate to favorable data in IgAN, which patient population has
not previously been treated with sparsentan prior to the Phase 3 trial currently being conducted. We cannot assure that any current or future clinical trials of fosmetpantotenate and/or sparsentan will ultimately be successful.
Before obtaining regulatory approval to conduct clinical trials of our product candidates, we must conduct extensive preclinical tests to demonstrate the safety of our product candidates in animals. Preclinical testing is expensive, difficult to design and implement, and can take many years to complete. In addition,
during the clinical development process, additional nonclinical toxicology studies are routinely conducted concurrently with the clinical development of a product candidate. If any of our product candidates show unexpected findings in concurrent toxicology studies, we could experience potentially significant delays in, or be required to abandon, development of that product candidate. A failure of one or more of our nonclinical studies can occur at any stage of testing.
* Communications and/or feedback from the FDA related to our current or planned future clinical trials does not guarantee any particular outcome from regulatory review for such clinical trials.
Communications and/or feedback from the FDA related to our current or future clinical trials does not guarantee any particular outcome from regulatory review for such clinical trials. For example, although we have obtained a Special Protocol Assessment ("SPA") agreement from the FDA for the Phase 3 clinical trial of fosmetpantotenate for the treatment of PKAN, this agreement does not guarantee any particular outcome from regulatory review. The SPA is intended to provide assurance that if the agreed upon clinical trial protocols are followed and the clinical trial endpoints are achieved, the data may serve as the primary basis for an efficacy claim in support of an NDA. However, a SPA is not a guarantee of an approval of a product candidate or any permissible claims about the product candidate. In particular, a SPA agreement is not binding on the FDA if previously unrecognized public health concerns arise during the performance of the clinical trial, if other new scientific concerns regarding product candidate safety or efficacy arise or if the sponsoring company fails to comply with the agreed upon clinical trial protocols. Moreover, a SPA does not address all of the variables and details that may go into planning for or conducting a clinical trial, and changes in the protocol for a clinical trial can invalidate a SPA or require that the FDA agree in writing to the modified protocol. In addition, while a SPA addresses the requirements for submission of an NDA, the results of the related clinical trial may not support FDA approval. There can be no assurance that the Phase 3 clinical trial for fosmetpantotenate will demonstrate that fosmetpantotenate is safe and effective for treating PKAN or that the data will support an application for approval by the FDA. During the third quarter of 2019, we plan to un-blind the data from our ongoing pivotal Phase 3 trial of fosmetpantontenate. If the results of this trial are not positive, or are not viewed sufficiently favorably, the market price of our common stock could decline significantly. In addition, following the initial 24 week study period, patients in this trial are eligible to participate in an open label extension. Even if the initial un-blinded results from the trial are viewed positively, it is possible that negative consequences of the treatment could be observed in the future, which could adversely impact the regulatory and commercial prospects of fosmetpantontenate and our business may suffer.
In addition, in 2018 we initiated the following Phase 3 clinical trials of sparsentan: 1) a single Phase 3 clinical trial designed to serve as the basis for an NDA and MAA filing for sparsentan for the treatment of FSGS (the “DUPLEX Study”), and 2) a single Phase 3 clinical trial designed to serve as the basis for an NDA and MAA filing for sparsentan for the treatment of IgAN (the “PROTECT Study”). We are conducting the DUPLEX Study and the PROTECT Study under the Subpart H pathway for potential accelerated approval in the US,United States, and in the EUEurope we plan to pursue potential Conditional Marketing Authorization, in both jurisdictions based on change in proteinuria. Recognition of change in proteinuria as a surrogate endpoint in kidney disease is a relatively new regulatory development, and, as the field continues to evolve, new learnings may impact regulatory viewpoints. We expect that the FDA’s and EMA’s determination as to whether the sufficiency of the data supports an accelerated approval in either jurisdiction will be made during the application review process. There can be no assurance that even if we achieve statistical significance on the interim or primary endpoints for the DUPLEX Study and/or the PROTECT Study, as applicable, that the FDA or EMA will deem that sufficient to grant accelerated approval or Conditional Marketing Authorization.
Although we received feedback from the FDA at an End of Phase 2 meeting for the sparsentan FSGS program during which the FDA communicated that it was open to accepting a substantial treatment effect on proteinuria in the DUPLEX Study as a basis for accelerated approval pursuant to Subpart H of the FDA regulations and although we subsequently gained alignment that our statistical modeling supported initiating a Phase 3 trial that proceeds on the Subpart H pathway, there can be no guarantee that the data generated from the study will be sufficient to serve as the basis for an NDA filing, including an NDA under Subpart H for accelerated approval. In addition, our statistical modeling that supports proceeding with the Duplex Study on the Subpart H pathway is based on data from other FSGS studies. To the extent that the model population is not representative of the Duplex Study population, the FDA may not agree that the new results continue to support a Subpart H pathway. Furthermore, even if sparsentan is granted accelerated approval for FSGS, there can be no assurance that the post-marketing confirmatory data will support full approval of sparsentan as a treatment for FSGS.
Also, although we have reached agreement with the FDA regarding the initiation of the PROTECT Study and the trial began in December 2018, we continue to have regulatory interactions regarding certain details of the study. For example, in conjunction with ongoing FDA dialogue, in May 2020 we adopted a measurement of the rate of change in eGFR over the 110-week period following the initiation of randomized treatment as the confirmatory endpoint of the study, and increased the total sample size from 280 patients to 380 while maintaining the sample size for the primary endpoint at 280 patients. There can be no assurance that the study will proceed as planned and there can be no guarantee that the data generated from the study will be sufficient to serve as the basis for an NDA filing, including an NDA under Subpart H for accelerated approval or support Conditional Marketing Authorization in the EU. Furthermore, even if sparsentan is granted accelerated approval for IgAN, there can be no assurance that the post-marketing confirmatory data will support full approval of sparsentan as a treatment for IgAN.
In addition, because both the DUPLEX Study and PROTECT Study are evaluating the same compound for the treatment of chronic kidney diseases and utilizing similar endpoints, the risk of success or failure for the two studies may, depending on the outcomes of the studies, end up being correlated.
* An extended delay in the rate of enrollment in our ongoing DUPLEX or PROTECT Studies, as a result of the COVID-19 pandemic or otherwise, may delay our timelines for un-blinding and announcing top line results from the interim endpoints, and could delay our planned NDA filings under the Subpart H approval pathways.
* In both our DUPLEX and PROTECT studies, we plan to unblind the 36 week proteinuria endpoint data from the initial 190 patients and 280 patients, respectively, and if supported by the totality of the data for each study, submit an NDA under Subpart H for accelerated approval and file for Conditional Marketing Authorization in the EU for sparsentan for the treatment of FSGS and IgAN, respectively. We currently intend to either substantially or fully enroll each of the studies before unblinding and announcing top-line results from the proteinuria analysis from each such study, so as to reduce the risk of bias in the conduct of the ongoing study or jeopardizing completion of enrollment. If the rate of enrollment is slower than we had anticipated, due to the COVID-19 pandemic or otherwise, our current timelines for unblinding and announcing top-line results from the studies could be delayed. In addition, we have recently increased the sample size for the confirmatory portion of the PROTECT Study from 280 patients to 380 patients, which will increase the number of patients to be enrolled in the PROTECT Study in accordance with the above sequencing of events and could further exacerbate any such delays. Separately, the protocol for the DUPLEX Study calls for an unblinded sample size re-assessment for the confirmatory portion of the study to be conducted by an independent data monitoring committee at a pre-specified juncture in the study based on protocol specified criteria. If the sample size for the confirmatory portion of the DUPLEX study were to be increased, this occurrence, in conjunction with timeline impacts an extended COVID-19 pandemic could have on the study, may further exacerbate any delay in the timeline for unblinding the interim data. Any such delays would also push back our planned NDA submission timelines for the particular study.
Interim, topline and preliminary data from our clinical trials that we announce or publish from time to time may change as more patient data become available and are subject to audit and verification procedures that could result in material changes in the final data.
From time to time, we may publicly disclose preliminary or topline or interim data from our clinical studies, which is based on a preliminary analysis of then-available data, and the results and related findings and conclusions are subject to change following a more comprehensive review of the data related to the particular study. We also make assumptions, estimations, calculations and conclusions as part of our analyses of data, and we may not have received or had the opportunity to fully and carefully evaluate all data. As a result, the topline results that we report may differ from future results of the same studies, or
different conclusions or considerations may qualify such results, once additional data have been received and fully evaluated. Topline data also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary data we previously published. As a result, topline data should be viewed with caution. From time to time, we may also disclose interim data from our clinical trials. Interim data from clinical trials that we may complete are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment and dosing continues and more patient data become available. Adverse differences between preliminary or interim data and final or confirmatory data could significantly harm our business prospects.
Further, others, including regulatory agencies, may not accept or agree with our assumptions, estimates, calculations, conclusions or analyses or may interpret or weigh the importance of data differently, which could impact the value of the particular program, the approvability or commercialization of the particular product candidate or product and our company in general. In addition, the information we choose to publicly disclose regarding a particular study or clinical trial is based on what is typically extensive information, and you or others may not agree with what we determine is the material or otherwise appropriate information to include in our disclosure, and any information we determine not to disclose may ultimately be deemed significant with respect to future decisions, conclusions, views, activities or otherwise regarding a particular drug, drug candidate or our business. If the topline data that we report differ from actual results, or if others, including regulatory authorities, disagree with the conclusions reached, our ability to obtain approval for, and commercialize, our product candidates may be harmed, which could harm our business, operating results, prospects or financial condition.
Even if we receive regulatory approval for any product candidates, we will be subject to ongoing regulatory obligations and continued regulatory review, which may result in significant additional expense.
Any regulatory approvals that we receive for any product candidates may be subject to significant limitations on the approved indicated uses for which the product may be marketed or to the conditions of approval, or contain requirements for potentially costly post-marketing testing, including Phase 4 clinical trials, and surveillance to monitor the safety and efficacy of the product candidate.
In addition, if the FDA or a comparable foreign regulatory authority approves any product candidates, those products will be subject to extensive and ongoing regulatory requirements, including for the manufacturing, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion, import, export, recordkeeping, conduct of potential post-marketing studies and post-market submission requirements. These requirements include submissions of safety and other post-marketing information and reports, registration, as well as continued compliance with current good manufacturing practices and good clinical practices, for any clinical trials that we conduct post-approval. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, undesirable side effects caused by the product, problems encountered by our third-party manufacturers or manufacturing processes, or failure to comply with regulatory requirements, either before or after product approval, may result in, among other things:
•restrictions on the marketing, manufacturing, or distribution of the product;
•requirements to include additional warnings on the label;
•requirements to create or enhance a medication guide outlining the risks to patients;
•withdrawal of the product from the market;
•voluntary or mandatory product recalls;
•requirements to change the way the product is administered or for us to conduct additional clinical trials;
•fines, warning or untitled letters or holds on clinical trials;
•refusal by the FDA to approve pending applications or supplements to approved applications filed by us or our strategic partners, or suspension or revocation of product license approvals;
•product seizure or detention, or refusal to permit the import or export of products;
•injunctions or the imposition of civil or criminal penalties; and
•harm to our reputation.
For example, we have certain post-marketing requirements and commitments associated with Cholbam. Further, we face risks relating to the post marketing obligations and commercial acceptance of Cholbam, which was approved by the FDA on March 17, 2015. If the regulatory approval for Chenodal, Cholbam and/or Thiola are withdrawn for any reason, it would have a material adverse impact on our sales and profitability.
* The independent clinical investigators and contract research organizations that we rely upon to conduct our clinical trials may not be diligent, careful or timely, and may make mistakes, in the conduct of our trials.
We depend on independent clinical investigators and contract research organizations (“CROs”) to conduct our clinical trials under agreements with us. The CROs play a significant role in the conduct of our clinical trials. Failure of the CROs to meet their obligations could adversely affect clinical development of our product candidates. The independent clinical investigators are not our employees and we cannot control the timing or amount of resources they devote to our studies. If their performance is substandard, it could delay or prevent approval of our FDA applications. Moreover, these independent investigators and CROs may also have relationships with other commercial entities, some of which may compete with us. If independent investigators and CROs allocate their resources to assist our competitors at our expense, it could harm our competitive position. In response to the COVID-19 pandemic, we have engaged or intend to engage providers of home health and remote monitoring services to assist with the ongoing conduct of our clinical trials in an effort to mitigate disruption caused by COVID-19 related issues. The introduction of new third parties into our ongoing clinical trials increases the risks associated with our dependence on third parties, including the risk that substandard performance by, or competing interests of, such third parties could have a negative impact on our clinical trials. Furthermore, there is no guarantee that the utilization of such home health providers or remote monitoring services will be successful in mitigating disruptions to our clinical trials caused by the COVID-19 pandemic.
Risks Related to the Commercialization of Our Products
The commercial success of Chenodal, Cholbam and Thiola depends on them being considered to be effective drugs with advantages over other therapies.
The commercial success of our products Chenodal, Cholbam and Thiola depends on them being considered to be effective drugs with advantages over other therapies. A number of factors, as discussed in greater detail below, may adversely impact the degree of acceptance of these products, including their efficacy, safety, price and benefits over competing therapies, as well as the coverage and reimbursement policies of third-party payers, such as government and private insurance plans.
If unexpected adverse events are reported in connection with the use of any of these products, physician and patient acceptance of the product could deteriorate and the commercial success of such product could be adversely affected. We are required to report to the FDA events associated with our products relating to death or injury. Adverse events could result in additional regulatory controls, such as a requirement for costly post-approval clinical studies or revisions to our approved labeling which could limit the indications or patient population for a product or could even lead to the withdrawal of a product from the market.
*We are subject to generic competition, and recent developments relating to generic competition for pharmaceutical products could cause our product sales and business to be negatively impacted.
Under the Hatch-Waxman Amendments of the Federal Food, Drug, and Cosmetic Act, a pharmaceutical manufacturer may file an ANDA, seeking approval of a generic copy of an approved innovator product or an NDA under Section 505(b)(2) that relies on the FDA’s prior findings of safety and effectiveness in approving the innovator product. A Section 505(b)(2) NDA may be for a new or improved version of the original innovator product. Certain of our products, including Thiola, are subject to immediate competition from compounded and generic entrants, as the ANDA and NDA for these drug products have no remaining or current patent or nonpatentnon-patent exclusivity.
There have been a number of recent regulatory and legislative initiatives designed to encourage generic competition for pharmaceutical products, including expedited review procedures for generic manufacturers and incentives designed to spur generic competition of branded drugs. In particular, the FDA and the U.S. Federal Trade Commission (“FTC”) have been focused on brand companies’ denial of drug supply to potential generic competitors for testing, andtesting. In December 2019, the U.S. Congress has been consideringCREATES Act was enacted, which provides a legislatively defined private right of action under which generic companies couldcan bring suit against companies who refuse access to product for the bioequivalence testing needed to support approval of a generic product.
In October 2018, we were named as a defendantWe are currently in a lawsuit brought by Spring Pharmaceuticals, LLC (“Spring”), alleging that we refused to sell samplesthe process of Thiola in order for Spring to conduct bioequivalence studies. In addition, we are currently responding to a civil investigative demand received from the FTC in July 2019, requesting information related to the marketing, sale, distribution and pricing of our products, including Thiola. At this time, the FTC has not initiated any claim or proceeding against the Companyus relating to these matters.
We cannot currently predict the specific outcome or impact on our business of such regulatory and legislative initiatives, litigation or investigation. However, it is our policy, which is in compliance with the CREATES Act, to evaluate requests for samples of our branded products, and to provide samples in response to bona fide requests from qualified third parties, including generic manufacturers, subject to specified conditions. We have provided and are in the process of providing samples to Spring and certain generic manufacturers, the provision of which we expect to be complete in the third quarter of 2019.manufacturers.
If a generic version of Thiola, Chenodal or any of our other current or future products is approved, sales of that product likely would be negatively impacted, which could have a material adverse impact on our sales and profitability. Both the original formulation of Thiola and Thiola EC are subject to generic competition, and a generic version of either formulation could have a material adverse impact on sales of Thiola EC. In addition, the defense of litigation and response to investigation requests could result in substantial costs, reputational impact, and the diversion of management attention and resources.
*Changes in reimbursement practices of third-party payers, or patients' access to insurance coverage, could affect the demand for our products andand/or the prices at which they are sold.
The business and financial condition of healthcare-related businesses will continue to be affected by efforts of governments and third-party payers to contain or reduce the cost of healthcare through various means. In the United States and some foreign jurisdictions, there have been a number of legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing approval for fosmetpantotenate and sparsentan, or any other product candidate that we develop, restrict or regulate post-approval activities and affect our ability to profitably sell fosmetpantotenate and sparsentan, or any other product candidate for which we obtain marketing approval.
Our products are sold to patients whose healthcare costs are met by third-party payers, such as government programs, private insurance plans and managed-care programs. These third-party payers are increasingly attempting to contain healthcare costs by limiting both coverage and the level of reimbursement for medical products and services. Levels of reimbursement, if any, may be decreased in the future, and future healthcare reform legislation, regulations or changes to reimbursement policies of third partythird-party payers may otherwise adversely affect the demand for and price levels of our products, which could have a material adverse effect on our sales and profitability.
Economic, social, and congressional pressure may result in individuals and government entities increasingly seeking to achieve cost savings through mechanisms that limit coverage or payment for our products. For example, state Medicaid programs are increasingly requesting manufacturers to pay supplemental rebates and requiring prior authorization for use of drugs. Managed care organizations continue to seek price discounts and, in some cases, to impose restrictions on the coverage of particular drugs. Government efforts to reduce Medicaid expenses may lead to increased use of managed care organizations by Medicaid programs. This may result in managed care organizations influencing prescription decisions for a larger segment of the population and a corresponding constraint on prices and reimbursement for our products.
In addition, patients’ access to employer sponsored insurance coverage may be negatively impacted by the COVID-19 pandemic or other economic factors that result in increased rates of unemployment. To the extent patients taking our approved therapies become unemployed and experience a reduction to, or increased costs associated with, their insurance coverage, demand for our products could decline, which could have a material adverse effect on our sales
and profitability, either as a result of decreased sales of our products and/or increased provision by us of free product to uninsured or commercially insured patients. The extent and duration of this potential impact on our business is currently unknown.
* We are dependent on third parties to manufacture and distribute our pharmaceutical products who may not fulfill their obligations.
We have no manufacturing capabilities and rely on third party manufacturers who are sole source suppliers for manufacturing of Chenodal, Cholbam and Thiola. The facilities used by our third partythird-party manufacturers must be approved by the FDA, or in the case of Kolbam in the European Union, the European Medicines Agency.FDA. Our dependence on third parties for the manufacture of our products may harm our profit margin on the sale of products and our ability to deliver products on a timely and competitive basis. If our third partythird-party manufacturers are unable to manufacture to specifications or in compliance with applicable regulatory requirements, our ability to commercialize our products will be adversely impacted and could affect our ability to gain market acceptance for our products and negatively impact our revenues.
We currently have no in-house distribution channels for Chenodal, Cholbam or Thiola and we are dependent on a third-party distributor, Dohmen Life Sciences Services, an Eversana Company, to distribute such products. We rely on this distributor for all of our proceeds from sales of Chenodal, Cholbam and Thiola in the United States. The outsourcing of our distribution function is complex, and we may experience difficulties that could reduce, delay or stop shipments of such products. If we encounter such distribution problems, and we are unable to quickly enter into a similar agreement with another distributor on substantially similar terms, distribution of Chenodal, Cholbam and/or Thiola could become disrupted, resulting in lost revenues, provider dissatisfaction, and/or patient dissatisfaction.
Governments outside the United States tend to impose strict price controls and reimbursement approval policies, which may adversely affect our prospects for generating revenue.
In some countries, particularly European UnionEU countries and EFTA member states, the pricing of prescription pharmaceuticals is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take considerable time (6 to 12 months or longer) after the receipt of marketing approval for a product. To obtain reimbursement or pricing approval in some countries, we may be required to conduct a clinical trial that compares the cost effectiveness of our product candidate to other available therapies. If reimbursement of our products is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, our prospects for generating revenue outside of the United States, if any, could be adversely affected and our business may suffer.
If we are unable to establish sales and marketing capabilities or enter into agreements with third parties to market and sell our product candidates outside the United States, we may be unable to generate product revenue outside of the United States.
We may not be able to rely on orphan drug exclusivity for Cholbam/KolbamCholbam or any of our products.
Regulatory authorities in some jurisdictions, including the United States and Europe, may designate drugs for relatively small patient populations as orphan drugs. We have obtained orphan designation for Cholbam/KolbamCholbam in the United States, and the European Union.which expires in March 2022. Generally, if a product with an orphan drug designation subsequently receives the first marketing approval for the indication for which it has such designation, that product is entitled to a period of marketing exclusivity, which precludes the applicable regulatory authority from approving another marketing application for the same drug for the same indication for that time period. The applicable period is seven years in the United States and ten years in Europe.Europe or, in the case of orphan drugs for which a pediatric investigation plan has been completed, 12 years. Even though we have been awarded orphan drug exclusivity for Cholbam in the United States, we may not be able to maintain it. For example, if a competitive product that contains the same active moiety and treats the same disease as our product is shown to be clinically superior to our product, any orphan drug exclusivity we have obtained will not block the approval of such competitive product and we may effectively lose orphan drug exclusivity. Similarly, if a competitive product that contains the same active moiety and treats the same disease as our product candidate is approved for orphan drug exclusivity before our product candidate, we may not be able to obtain approval for our product candidate until the expiration of the competitive product’s orphan drug exclusivity unless our product candidate is shown to be clinically superior to the competitive product.
If we are unable to maintain an effective and specialized sales force, we will not be able to commercialize our products successfully.
In order to successfully commercialize our products, we have built a specialized sales force. Factors that may hinder our ability to successfully market and commercially distribute our products include:
•inability of sales personnel to obtain access to or convince adequate numbers of physicians to prescribe our products;
•inability to recruit, retain and effectively manage adequate numbers of effective sales personnel;
•lack of complementary products to be offered by sales personnel, which may put us at a competitive disadvantage relative to companies that have more extensive product lines; and
•unforeseen delays, costs and expenses associated with maintaining our sales organization.
If we are unable to maintain our sales force for our products, we may not be able to generate sufficient product revenue.
We will need to continue to expend significant time and resources to train our sales forces to be credible, persuasive and compliant in discussing our products with the specialists treating the patients indicated under the product’s label. In addition, if we are unable to effectively train our sales force and equip them with effective marketing materials our ability to successfully commercialize our products could be diminished, which would have a material adverse effect on our business, results of operations and financial condition
Risks Related to our Products and Product Candidates
*Our products may not achieve or maintain expected levels of market acceptance or commercial success.
The success of our products is dependent upon achieving and maintaining market acceptance. Commercializing products is time consuming, expensive and unpredictable. There can be no assurance that we will be able to, either by ourselves or in collaboration with our partners or through our licensees, successfully commercialize new products or current products or gain market acceptance for such products. New product candidates that appear promising in development may fail to reach the market or may have only limited or no commercial success.
Further, the discovery of significant problems with a product similar to one of our products that implicate (or are perceived to implicate) an entire class of products could have an adverse effect on sales of the affected products. Accordingly, new data about our products, or products similar to our products, could negatively impact demand for our products due to real or perceived side effects or uncertainty regarding efficacy and, in some cases, could result in product withdrawal.
Our current products and any products that we bring to the market, including fosmetpantotenate and sparsentan, if they receive marketing approval, may not gain market acceptance by physicians, patients, third-party payers, and others in the medical community. If these products do not achieve
an adequate level of acceptance, we may not generate significant product revenue and we may not become profitable. The degree of market acceptance of our current products and product candidates, if approved for commercial sale, will depend on a number of factors, including:
•the prevalence and severity of any side effects, including any limitations or warnings contained in a product’s approved labeling;
•the efficacy and potential advantages over alternative treatments;
•the pricing of our product candidates;
•relative convenience and ease of administration;
•the willingness of the target patient population to try new therapies and of physicians to prescribe these therapies;
•the strength of marketing and distribution support and timing of market introduction of competitive products;
•publicity concerning our products or competing products and treatments; and
•sufficient third-party insurance coverage and reimbursement.
Even if a potential or current product displays a favorable efficacy and safety profile in preclinical and clinical trials, market acceptance of the product will not be known until after it is launched. Our efforts to educate patients, the medical community, and third-party payers on the benefits of our product may require significant resources and may never be successful. Such efforts to educate the marketplace may require more resources than are required by the conventional marketing technologies employed by our competitors.
If the market opportunities for our products and product candidates are smaller than we believe they are, our revenues may be adversely affected and our business may suffer.
Certain of the diseases that our current and future product candidates are being developed to address, such as PKAN, PKU, FSGS and IgAN, are relatively rare. Our projections of both the number of people who have these diseases, as well as the subset of people with these diseases who have the potential to benefit from treatment with our product candidates, may not be accurate.
Currently, most reported estimates of the prevalence of PKAN and FSGS are based on studies of small subsets of the population of specific geographic areas, which are then extrapolated to estimate the prevalence of the diseases in the broader world population. As new studies are performed the estimated prevalence of these diseases may change. There can be no assurance that the prevalence of PKAN and FSGS in the study populations accurately reflect the prevalence of these diseases in the broader world population. If our estimates of the prevalence of PKAN, PKU, FSGS or IgAN or of the number of patients who may benefit from treatment with fosmetpantotenate and sparsentan prove to be incorrect, the market opportunities for our product candidates may be smaller than we believe they are, our prospects for generating revenue may be adversely affected and our business may suffer.
Our product candidates may cause undesirable side effects or have other properties that could delay or prevent their regulatory approval or commercialization.
Undesirable side effects caused by our product candidates could interrupt, delay or halt clinical trials and could result in the denial of regulatory approval by the FDA or other regulatory authorities for any or all targeted indications, and in turn prevent us from commercializing our product candidates and generating revenues from their sale.
In addition, if any of our product candidates receive marketing approval and we or others later identify undesirable side effects caused by the product:
•regulatory authorities may require the addition of restrictive labeling statements;
•regulatory authorities may withdraw their approval of the product; and
•we may be required to change the way the product is administered or conduct additional clinical trials.
Any of these events could prevent us from achieving or maintaining market acceptance of the affected product or could substantially increase the costs and expenses of commercializing the product candidate, which in turn could delay or prevent us from generating significant revenues from its sale or adversely affect our reputation.
We do not currently have patent protection for certain of our products and product candidates.commercial products. If we are unable to obtain and maintain protection for the intellectual property relating to our technology and products, the value of our technology and products will be adversely affected.
Our success will depend in large part on our ability to obtain and maintain protection in the United States and other countries for the intellectual property covering, or incorporated into, our technology and products. The patent situation in the field of biotechnology and pharmaceuticals generally is highly uncertain and involves complex legal, technical, scientific and factual questions. We do not have, and do not expect to obtain, patent protection for Thiola,
Chenodal or Cholbam. Additionally, although we have several pending U.S. patent applications directed to Thiola EC and/or its use for treating cystinuria, we do not know whether any of these patent applications will result in a granted patent covering Thiola EC or its use for treating cystinuria. More generally, we may not be able to obtain additional issued patents relating to our technology or products. Even if issued, patents issued to us or our licensors may be challenged, narrowed, invalidated, held to be unenforceable or circumvented, which could limit our ability to stop competitors from marketing similar products or reduce the term of patent protection we may have for our products. Changes in either patent laws or in interpretations of patent laws in the United States and other countries may diminish the value of our intellectual property or narrow the scope of our patent protection. Fosmetpantotenate is covered by our U.S. Patent No. 8,673,883, which was granted in 2014 and expires in 2033. In addition, our U.S. Patent No. 9,181,286, which was granted on November 10,
2015 and expires in 2033, covers the use of fosmetpantotenate for the treatment of PKAN, and our U.S. Patent No. 9,629,862, which was granted on April 25, 2017 and also expires in 2033, covers pharmaceutical compositions that contain fosmetpantotenate. SparsentanOur product candidate sparsentan is covered by U.S. Patent No. 6,638,937, which expiresexpired in 2019 and to which we have an exclusive license. In addition, U.S. Patent No. 9,662,312, to which we also have an exclusive license and which was granted on May 30, 2017 and expires in 2030, covers the use of sparsentan for treating glomerulosclerosis, including FSGS. And U.S. Patent No.9,993,461,No. 9,993,461, to which we also have an exclusive license and which was granted on June 12, 2018 and expires in 2030, covers the use of sparsentan for treating IgA nephropathy as well as glomerulosclerosis, including FSGS.
For products we develop based on a new chemical entity not previously approved by the FDA, we expect that in addition to the protection afforded by our patent filings that we will be able to obtain either five years regulatory exclusivity via the provisions of the FDC Act and possibly seven years regulatory exclusivity via the orphan drug provisions of the FDC Act. In addition, we may be able to obtain up to five years patent term extension (to compensate for regulatory approval delay) for a patent covering such a product.
The degree of future protection for our proprietary rights is uncertain, and we cannot ensure that:
•we or our licensors were the first to make the inventions covered by each of our pending patent applications;
•we or our licensors were the first to file patent applications for these inventions;
•others will not independently develop similar or alternative technologies or duplicate any of our technologies;
•any patents issued to us or our licensors that provide a basis for commercially viable products will provide us with any competitive advantages or will not be challenged by third parties;
•we will develop additional proprietary technologies that are patentable;
•we will file patent applications for new proprietary technologies promptly or at all;
•the claims we make in our patents will be upheld by patent offices in the United States and elsewhere;
•our patents will not expire prior to or shortly after commencing commercialization of a product; and
•the patents of others will not have a negative effect on our ability to do business.
We have negotiated a license agreement with Ligand Pharmaceuticals for the rights to sparsentan which we are initially developing for the treatment of FSGS and IgAN. This license subjects us to various commercialization, reporting and other obligations. If we were to default on our obligations, we could lose our rights to sparsentan. We have obtained a U.S. and European patent covering the use of sparsentan for treating glomerulosclerosis, including FSGS, and a second U.S. patent covering both the use of sparsentan for treating IgAN and the use of sparsentan for treating glomerulosclerosis, including FSGS. However, we cannot be certain that we will be able to obtain patent protection for various other potential indications for sparsentan, or whether, if granted, we would be able to enforce such patents.
Our patents also may not afford us protection against competitors with similar technology. Because patent applications in the United States and many other jurisdictions are typically not published until 18 months after filing, or in some cases not at all, and because publications of discoveries in the scientific literature often lag behind the actual discoveries, neither we nor our licensors can be certain that we or they were the first to make the inventions claimed in our or their issued patents or pending patent applications, or that we or they were the first to file for protection of the inventions set forth in these patent applications. If a third party has also filed a United States patent application prior to the effective date of the relevant provisions of the America Invents Act (i.e. before March 16, 2013) covering our product candidates or a similar invention, we may have to participate in an adversarial proceeding, known as an interference, declared by the USPTO to determine priority of invention in the United States. The costs of these proceedings could be substantial, and it is possible that our efforts could be unsuccessful, resulting in a loss of our United States patent position.
We cannot assure you that third parties will not assert patent or other intellectual property infringement claims against us with respect to technologies used in our products. If patent infringement suits were brought against us, we may be unable to commercialize some of our products which could severely harm our business. Litigation proceedings, even if not successful, could result in substantial costs and harm our business.
We expect to rely on orphan drug status to develop and commercialize certain of our product candidates, but our orphan drug designations may not confer marketing exclusivity or other expected commercial benefits.
We expect to rely on orphan drug exclusivity for fosmetpantotenate and sparsentan and potential future product candidates that we may develop. Orphan drug status currently confers seven years of marketing exclusivity in the United States under the FDC Act, and up to ten years of marketing exclusivity in Europe for a particular product in a specified indication.indication or, in the case of orphan drugs for which a pediatric investigation plan has been completed, 12 years. The FDA and EMA have granted orphan designation for Chenodal fosmetpantotenate and sparsentan for the treatment of CTX PKAN and FSGS, respectively. While we have been granted these orphan designations, we will not be able to rely on these designations to exclude other companies from manufacturing or selling these molecules for the same indication beyond these time frames. Furthermore, any marketing exclusivity in Europe can be reduced from ten years to six years if the initial designation criteria have significantly changed since the market authorization of the orphan product.
For any product candidate for which we have been granted orphan drug designation in a particular indication, it is possible that another company also holding orphan drug designation for the same product candidate will receive marketing approval for the same indication before we do. If that were to happen, our applications for that indication may not be approved until the competing company's period of exclusivity expires. Even if we are the first to obtain marketing authorization for an orphan drug indication in the United States, there are circumstances under which a competing product may be approved for the same
indication during the seven-year period of marketing exclusivity, such as if the later product is shown to be
clinically superior to our orphan product, or if the later product is deemed a different product than ours. Further, the seven-year marketing exclusivity would not prevent competitors from obtaining approval of the same product candidate as ours for indications other than those in which we have been granted orphan drug designation, or for the use of other types of products in the same indications as our orphan product.
*Any drugs we develop may become subject to unfavorable pricing regulations, third-party reimbursement practices or healthcare reform initiatives, thereby harming our business.
In March 2010, President Obama signed the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010 (collectively, the "PPACA"), a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for healthcare and health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms. The PPACA revised the definition of “average manufacturer price” for reporting purposes, which could increase the amount of Medicaid drug rebates to states. The PPACA also increased the mandated Medicaid rebate from 15.1% to 23.1% of the average manufacturer price, expanded the rebate to Medicaid managed care utilization and increased the types of entities eligible for the federal 340B drug discount program. Further, the law imposes a significant annual fee on companies that manufacture or import certain branded prescription drug products. There have beenremain judicial, Congressional, and political challenges to certain aspects of the PPACA, as well as recent efforts by the Trump administration to repeal or replace certain aspects of the PPACA. Since January 2017, President Trump has signed two Executive Orders and other directives designed to delay the implementation of certain provisions of the PPACA or otherwise circumvent some of the requirements for health insurance mandated by the PPACA. Concurrently, Congress has considered legislation that would repeal or repeal and replace all or part of the PPACA. While Congress has not passed comprehensive repeal legislation, bills affecting the implementation of certain taxes under the PPACA have been signed into law. For example, legislation enacted in 2017, informally titled the Tax Cuts and Jobs Act (the "Tax Act"), includes a provision which repealed, effective January 1, 2019, the tax-based shared responsibility payment imposed by the PPACA on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate”. In addition, onthe 2020 federal spending package eliminated, effective January 22, 2018, President Trump signed a continuing resolution on appropriations for fiscal year 2018 that delayed1, 2020, the implementation of certain PPACA-mandated fees, including the so-called “Cadillac” tax on certain high costhigh-cost employer-sponsored insurance plans, the annual fee imposed on certain health insurance providers based on market share,coverage and the medical device excise tax on non-exempt medical devices.and, effective January 1, 2021, also eliminates the health insurer tax. The Bipartisan Budget Act of 2018 ("BBA"), among other things, amendsamended the PPACA, effective January 1, 2019, to increase the discount that is owed by pharmaceutical manufacturers who participate in Medicare Part D from 50 percent to 70 percent, and closesclosed the coverage gap in most Medicare drug plans, commonly referred to as the “donut hole”. In December 2018, the Centers for Medicare & Medicaid Services ("CMS") published a new final rule permitting further collections and payments to and from certain PPACA qualified health plans and health insurance issuers under the PPACA risk adjustment program in response to the outcome of federal district court litigation regarding the method CMS uses to determine the risk adjustment. On April 27, 2020, the United States Supreme Court reversed a federal circuit court decision that previously upheld Congress' denial of $12 billion in "risk corridor" funding. On December 14, 2018, a Texas U.S. District Court Judge ruled that the PPACA is unconstitutional in its entirety because the “individual mandate” was repealed by Congress as part of the Tax Cuts and Jobs Act. WhileAdditionally, on December 18, 2019, the Texas U.S. Court of Appeals for the 5th Circuit upheld the District Court Judge, as well as the Trump administration and CMS, have statedruling that the ruling will have no immediate effect pending appealindividual mandate was unconstitutional and remanded the case back to the District Court to determine whether remaining provisions of the decision, itPPACA are invalid as well. On March 2, 2020, the United States Supreme Court granted the petitions for writs of certiorari to review this case, and has allotted one hour for oral arguments, which are expected to occur in the fall. It is unclear how this decision, subsequent appeals,such litigation and other efforts to repeal and replace the PPACA will impact the PPACA and our business.
In addition, other legislative changes have been proposed and adopted since the PPACA was enacted. For example, in August 2011, the President signed into law the Budget Control Act of 2011, which, among other things, created the Joint Select Committee on Deficit Reduction to recommend to Congress proposals in spending reductions. The Joint Select Committee on Deficit Reduction did not achieve a targeted deficit reduction of at least $1.2 trillion for fiscal years 2012 through 2021, triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions to Medicare payments to providers of up to 2% per fiscal year, which went into effect beginning on April 1, 2013 and, due to subsequent legislative amendments, including the BBA, will stay in effect through 20272030 unless additional Congressional action is taken. The CARES Act suspended the 2% Medicare sequester from May 1, 2020 through December 31, 2020, while also extending it by one year, through 2030. Additionally, in January 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, reduced Medicare payments to several providers, including hospitals and imaging centers.
If we are unable to obtain coverage and adequate reimbursement from governments or third-party payers for any products that we may develop or if we are unable to obtain acceptable prices for those products, our prospects for generating revenue and achieving profitability will suffer.
Our prospects for generating revenue and achieving profitability will depend heavily upon the availability of coverage and adequate reimbursement for the use of our approved product candidates from governmental and other third-party payers, both in the United States and in other markets. Reimbursement by a third-party payer may depend upon a number of factors, including the third-party payer’s determination that use of a product is:
•a covered benefit under its health plan;
•safe, effective and medically necessary;
•appropriate for the specific patient;
•cost-effective; and
•neither experimental nor investigational.
Obtaining reimbursement approval for a product from each government or other third-party payer is a time consuming and costly process that could require us to provide supporting scientific, clinical and cost effectiveness data for the use of our products to each payer. We may not be able to provide data sufficient to gain acceptance with respect to reimbursement or we might need to conduct post-marketing studies in order to demonstrate the cost-effectiveness of any future products to such payers’ satisfaction. Such studies might require us to commit a significant amount of management time and financial and other resources. Even when a payer determines that a product is eligible for reimbursement, the payer may impose coverage limitations that preclude payment for some uses that are approved by the FDA or non-United States regulatory authorities. Also prior authorization for a product may be required. In addition, there is a risk that full reimbursement may not be available for high-priced products.
Moreover, eligibility for coverage does not imply
that any product will be reimbursed in all cases or at a rate that allows us to make a profit or even cover our costs. Interim payments for new products, if applicable, may also not be sufficient to cover our costs and may not be made permanent.
A primary trend in the United States healthcare industry and elsewhere is toward cost containment. We expect the changes made by PPACA, other legislation impacting the Medicare program and the 340B program, and the increasing emphasis on managed care to continue to put pressure on pharmaceutical product pricing. As these concerns continue to grow over the need for tighter oversight, there remains the possibility that the Heath Resources and Services Administration or another agency under the HHS will propose regulations or that Congress will explore changes to the 340B program through legislation. For example, on November 30, 2018, HRSA published its final rule regarding the calculation of 340B ceiling price and imposition of civil monetary penalties on manufacturers for knowingly and intentionally overcharging covered entities, which became effective on January 1, 2019. Pursuant to the final rule, after January 1, 2019, manufacturers must calculate 340B program ceiling prices on a quarterly basis. Moreover, manufacturers could be subject to a $5,000 penalty for each instance where they knowingly and intentionally overcharge a covered entity under the 340B program.
Further, there has been increasing legislative and enforcement interest in the United States with respect to drug pricing practices, including several recent U.S. congressional inquiries and federal and state legislation designed to, among other things, increase drug pricing transparency, expedite generic competition, review relationships between pricing and manufacturer patient assistance programs, and reform government program drug reimbursement methodologies. At the federal level, the Trump administration’s budget proposalproposals for fiscal year 2019 contains further2021 includes a $135 billion allowance to support legislative proposals seeking to reduce drug price control measuresprices, increase competition, lower out-of-pocket drug costs for patients, and increase patient access to lower-cost generic and biosimilar drugs. On March 10, 2020, the Trump administration sent “principles” for drug pricing to Congress, calling for legislation that could be enacted during the 2019 budget process or inwould, among other future legislation, including, for example, measures to permitthings, cap Medicare Part D plansbeneficiary out-of-pocket pharmacy expenses, provide an option to negotiate the price of certain drugs undercap Medicare Part B, to allow some states to negotiate drug prices under Medicaid,D beneficiary monthly out-of-pocket expenses, and to eliminate cost sharing for generic drugs for low-income patients.place limits on pharmaceutical price increases. Further, the Trump administration previously released a "Blueprint" to lower drug prices and reduce out of pocket costs of drugs that contains additionalcontained proposals to increase manufacturer competition, increase the negotiating power of certain federal healthcare programs, incentivize manufacturers to lower the list price of their products and reduce the out of pocket costs of drug products paid by consumers. The HHS has started solicitingsolicited feedback on some of these measures and, at the same time, is immediately implementinghas implemented others under its existing authority. For example, in May 2019, CMS issued a final rule to allow Medicare Advantage Plans the option to use step therapy for Part B drugs beginning January 1, 2020. The final rule codified CMS’s policy change that was effective January 1, 2019. Although a number of these existing measures, and other potential proposals, may require additional authorization to become effective, Congress and the Trump administration have each indicated that it will continue to seek new legislative and/or administrative measures to control drug costs. At the state level, legislatures are increasingly passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing.
Any reduction in reimbursement from Medicare, Medicaid or other government-funded programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability or commercialize our drugs. Additionally, we are currently unable to predict what additional legislation or regulation, if any, relating to the healthcare industry may be enacted in the future or what effect recently enacted federal legislation or any such additional legislation or regulation would have on our business.
It is also possible that additional governmental action is taken to address the COVID-19 pandemic. For example, on April 18, 2020, CMS announced that Qualified Health Plan issuers under the PPACA may suspend activities related to the collection and reporting of quality data that would have otherwise been reported between May and June 2020 given the challenges healthcare providers are facing responding to the COVID-19 virus.
* We face potential product liability exposure far in excess of our limited insurance coverage.
The use of any of our potential products in clinical trials, and the sale of any approved products, may expose us to liability claims. These claims might be made directly by consumers, health care providers, pharmaceutical companies or others selling our products. We have obtained limited product liability insurance coverage for our clinical trials in the amount of $10 million per occurrence and $10$25 million in the aggregate. However, our insurance may not reimburse us or may not be sufficient to reimburse us for any expenses or losses we may suffer. Moreover, insurance coverage is becoming increasingly expensive, and we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses due to liability. We intend to expand our insurance coverage to include the sale of commercial products if we obtain marketing approval for product candidates in development, but we may be unable to obtain commercially reasonable product liability insurance for any products approved for marketing. On occasion, juries have awarded large judgments in class action lawsuits based on drugs that had unanticipated side effects. A successful product liability claim or series of claims brought against us would decrease our cash reserves and could cause our stock price to fall.
We face substantial competition, which may result in others discovering, developing or commercializing products before or more successfully than we do. Our operating results will suffer if we fail to compete effectively.
Several of our competitors have substantially greater financial, research and development, distribution, manufacturing and marketing experience and resources than we do and represent substantial long-term competition for us. Other companies may succeed in developing and marketing products that are more effective and/or less costly than any products that may be developed and marketed by us, or that are commercially accepted before any of our products. Factors affecting competition in the pharmaceutical and drug industries vary, depending on the extent to which a competitor is able to achieve a competitive advantage based on its proprietary technology and ability to market and sell drugs. The industry in which we compete is characterized by extensive research and development efforts and rapid technological progress. Although we believe that our orphan drug status for Cholbam and proprietary position with respect to fosmetpantotenate and sparsentan may give us a competitive advantage, new developments are expected to continue and there can be no assurance that discoveries by others will not render such potential products noncompetitive. Furthermore, competitors could enter the market with generic versions of our products.
Our competitive position also depends on our ability to enter into strategic alliances with one or more large pharmaceutical and contract manufacturing companies, attract and retain qualified personnel, develop effective proprietary products, implement development and marketing
plans, obtain patent
protection, secure adequate capital resources and successfully sell and market our approved products. There can be no assurance that we will be able to successfully achieve all of the foregoing objectives.
Use of third parties to manufacture and distribute our products and product candidates may increase the risk that we will not have sufficient quantities of our product and product candidates or such quantities at an acceptable cost, and clinical development and commercialization of our product and product candidates could be delayed, prevented or impaired.
We do not own or operate manufacturing facilities for clinical or commercial production of our products.products or product candidates. We have limited personnel with experience in drug manufacturing and we lack the resources and the capabilities to manufacture any of our product candidates on a clinical or commercial scale. We outsource all manufacturing and packaging of our preclinical, clinical, and commercial products to third parties. The manufacture of pharmaceutical products requires significant expertise and capital investment, including the development of advanced manufacturing techniques and process controls. Manufacturers of pharmaceutical products often encounter difficulties in production, particularly in scaling up initial production and in maintaining required quality control. These problems include difficulties with production costs and yields and quality control, including stability of the product candidate.
We do not currently have any agreements with third-party manufacturers for the long-term commercial supply of any of our development stage product candidates.candidates, including sparsentan. We may be unable to enter into agreements for commercial supply with third-party manufacturers, or may be unable to do so on acceptable terms. Even if we enter into these agreements, the manufacturers of each product candidate will be single source suppliers to us for a significant period of time. Reliance on third-party manufacturers entails risks to which we may not be subject if we manufactured our product candidates or products ourselves, including:
•reliance on the third party for regulatory compliance and quality assurance;
•limitations on supply availability resulting from capacity and scheduling constraints of the third parties;
•impact on our reputation in the marketplace if manufacturers of our products fail to meet the demands of our customers;
•the possible breach of the manufacturing agreement by the third party because of factors beyond our control; and
•the possible termination or nonrenewal of the agreement by the third party, based on its own business priorities, at a time that is costly or inconvenient for us.
The failure of any of our contract manufacturers to maintain high manufacturing standards could result in injury or death of clinical trial participants or patients using our products. Such failure could also result in product liability claims, product recalls, product seizures or withdrawals, delays or failures in testing or delivery, cost overruns or other problems that could seriously harm our business or profitability.
Our contract manufacturers will be required to adhere to FDA regulations setting forth cGMP. These regulations cover all aspects of the manufacturing, testing, quality control and recordkeeping relating to our product candidates and any products that we may commercialize. Our manufacturers may not be able to comply with cGMP regulations or similar regulatory requirements outside the United States. Our manufacturers are subject to unannounced inspections by the FDA, state regulators and similar regulators outside the United States. Our failure, or the failure of our third-party manufacturers, to comply with applicable regulations could result in sanctions being imposed on us, including fines, injunctions, civil penalties, failure of regulatory authorities to grant marketing approval of our product candidates, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of product candidates or products, operating restrictions and criminal prosecutions, any of which could significantly and adversely affect regulatory approval and supplies of our product candidates.
Our product and any products that we may develop may compete with other product candidates and products for access to manufacturing facilities. There are a limited number of manufacturers that operate under cGMP regulations and that are both capable of manufacturing for us and willing to do so. If the third parties that we engage to manufacture products for our developmental or commercial products should cease to continue to do so for any reason, we likely would experience interruptions in cash flows and/or delays in advancing our clinical trials while we identify and qualify replacement suppliers, and we may be unable to obtain replacement supplies on terms that are favorable to us. Later relocation to another manufacturer will also require notification, review and other regulatory approvals from the FDA and other regulators and will subject our production to further cost and instability in the availability of our product candidates. In addition, if we are not able to obtain adequate supplies of our product candidates, or the drug substances used to manufacture them, it will be more difficult for us to sell our products and to develop our product candidates. This could greatly reduce our competitiveness.
Our current and anticipated future dependence upon others for the manufacture of our product candidates may adversely affect our future profit margins and our ability to develop product candidates and commercialize any products that obtain regulatory approval on a timely and competitive basis.
Materials necessary to manufacture our products and product candidates may not be available on commercially reasonable terms, or at all, which may delay the development and commercialization of our products and product candidates.
We rely on the manufacturers of our products and product candidates to purchase from third-party suppliers the materials necessary to produce the compounds for our preclinical and clinical studies and rely on these other manufacturers for commercial distribution if we obtain marketing approval for any of our product candidates. Suppliers may not sell these materials to our manufacturers at the time we need them or on commercially reasonable terms and all such prices are susceptible to fluctuations in price and availability due to transportation costs, government regulations, price controls, and changes in economic climate or other foreseen circumstances. We do not have any control over the process or timing of the acquisition of these materials by our manufacturers. Moreover, we currently do not have any agreements for the commercial production of these
materials. If our manufacturers are unable to obtain these materials for our preclinical and clinical studies, product testing and potential regulatory approval of our product candidates would be delayed, significantly impacting our ability to develop our product candidates. If our manufacturers or we are unable to purchase these materials after regulatory approval has been obtained for our product candidates, the commercial launch of our product candidates would be delayed or there would be a shortage in supply, which would materially affect our ability to generate revenues from the sale of our product candidates.
Risks Related to Our Business
* The COVID-19 pandemic could materially adversely affect our business, results of operations and financial condition.
In March 2020, the WHO declared the COVID-19 outbreak a pandemic, which continues to spread throughout the world. The COVID-19 pandemic is impacting domestic and worldwide economic activity, including global financial markets. The COVID-19 pandemic also poses the risk that we or our clinical trial subjects, employees, contractors, collaborators and vendors may be prevented from conducting certain clinical trials or other business activities for an indefinite period of time, including due to travel restrictions, quarantines, “stay-at-home” and "shelter-in-place" orders or shutdowns that have been or may be requested or mandated by governmental authorities. In addition, the COVID-19 pandemic could impact personnel at third-party manufacturing facilities in the United States and other countries, including China, or the availability or cost of materials, which could potentially disrupt the supply chain for our commercial products, our product candidates or the comparator products in our ongoing clinical trials.
The timelines and conduct of our ongoing clinical trials may be affected by the COVID-19 pandemic. For example, we have experienced a reduction in the rates of patient enrollment in our ongoing clinical trials as a result of the pandemic. Clinical site initiation and patient enrollment may be further delayed due to prioritization of hospital resources toward the COVID-19 pandemic and patients’ ability or willingness to participate in clinical trials. For those patients who are enrolled and desire to continue in the clinical trials, some patients may not be able or willing to comply with clinical trial protocols if quarantines or governmental orders impede patient movement or interrupt healthcare services. Similarly, we may face increased challenges with the ability to recruit and retain patients and principal investigators and site staff who, as healthcare providers, may have heightened exposure to COVID-19, which could adversely impact our clinical trial operations, timelines and outcomes. In addition, we rely on independent clinical investigators, contract research organizations (CROs) and other third-party service providers to assist us in managing, monitoring and otherwise carrying out our clinical trials, and the pandemic may affect their ability to devote sufficient time and resources to our programs or to travel to sites to perform work for us. While we remain in close contact with our CROs, clinical sites and suppliers to attempt to assess the impacts that COVID-19 may have on our clinical trials and projected timelines and we are in the process of implementing appropriate mitigating measures in accordance with recent FDA guidance in an effort to ensure the ongoing safety of the patients in our clinical trials and the continued collection of high quality data, there is no guarantee that such efforts will be successful. As challenging as conducting clinical trials is during normal times, the risks, operational challenges and costs of conducting clinical trials has increased substantially during the pandemic.
Beginning the week of March 16, 2020, substantially all of our workforce began working from home either all or substantially all of the time as a result of applicable stay-at-home and shelter-in-place orders. The effects of these orders and our related work-from-home policies may negatively impact productivity, disrupt our business and delay our development programs, regulatory and commercialization timelines, the magnitude of which will depend, in part, on the length and severity of the restrictions and other limitations on our ability to conduct our business in the ordinary course. In addition, if and when the applicable orders are lifted and our employees return to the office, although we plan to take precautions to avoid the spread of COVID-19 among our employees, we cannot guarantee our workforce will not face an outbreak that could adversely impact our operations
While the long-term economic impact and the duration of the COVID-19 pandemic may be difficult to predict, the widespread pandemic has resulted in, and may continue to result in, significant disruption of global financial markets, which could reduce our ability to access capital and could negatively affect our liquidity and the liquidity and stability of markets for our common stock and convertible notes. In addition, a further market correction, recession or depression resulting from the spread of COVID-19 could materially adversely affect our business and the value of our common stock and convertible notes.
Moreover, the COVID-19 pandemic continues to rapidly evolve, and the extent to which the COVID-19 pandemic may impact our business, results of operations and financial position will depend on future developments, which are highly uncertain and cannot be predicted with confidence, such as the ultimate geographic spread of the disease, the duration of the pandemic, travel restrictions and social distancing in the United States and other countries, business closures or business disruptions, and the effectiveness of actions taken in the United States and other countries to contain and treat the disease.
Our limited operating history makes it difficult to evaluate our future prospects, and our profitability in the future is uncertain.
We face the problems, expenses, difficulties, complications and delays, many of which are beyond our control, associated with any business in its early stages and have a limited operating history on which an evaluation of our prospects can be made. Such prospects should be considered in light of the risks, expenses and difficulties frequently encountered in the establishment of a business in a new industry, characterized by a number of market entrants and intense competition, and in the shift from development to commercialization of new products based on innovative technologies.
We have experienced significant growth over the past threefive years in the number of our employees and the scope of our operations. We have added sales and marketing, compliance and legal functions in addition to expansion of all functions to support a commercial organization. To manage our anticipated future growth, we must continue to implement and improve our managerial, operational and financial systems, expand our facilities and continue to recruit and train additional qualified personnel. Due to our limited resources, we may not be able to effectively manage the expansion of our operations or recruit and train additional qualified personnel. The physical expansion of our operations may lead to significant costs and may divert our management and business development resources. Any inability on the part of our management to manage growth could delay the execution of our business plans or disrupt our operations.
Factors that may inhibit our efforts to commercialize our products without strategic partners or licensees include:
•our inability to recruit and retain adequate numbers of effective sales and marketing personnel;
•the inability of sales personnel to obtain access to or educate adequate numbers of physicians to prescribe our products;
•the lack of complementary products to be offered by our sales personnel, which may put us at a competitive disadvantage against companies with broader product lines;
•unforeseen costs associated with expanding our own sales and marketing team for new products or with entering into a partnering agreement with an independent sales and marketing organization; and
•efforts by our competitors to commercialize competitive products.
Moreover, though we generate revenues from product sales arrangements, we may incur significant operating losses over the next several years. Our ability to achieve profitable operations in the future will depend in large part upon successful in-licensing of products approved by the FDA, selling and manufacturing these products, completing development of our products, obtaining regulatory approvals for these products, and bringing these products to market. The likelihood of the long-term success of our company must be considered in light of the expenses, difficulties and delays frequently encountered in the development and commercialization of new drug products, competitive factors in the marketplace, as well as the regulatory environment in which we operate.
In addition, we may encounter unforeseen expenses, difficulties, complications, delays and other known and unknown factors.
* We will likely experience fluctuations in operating results and could incur substantial losses.
We expect that our operating results will vary significantly from quarter-to-quarter and year-to-year as a result of investments in research and development, specifically our clinical and preclinical development activities. We have not completed development of any drugs and we anticipate that our expenses will increase substantially as we:
continue our ongoing clinical development of fosmetpantotenate for the treatment of PKAN;
•continue the open label portion of DUET and conduct the planned Phase 3 trials of sparsentan indications;sparsentan;
launch Thiola EC;
•continue the research and development of additional product candidates;
•expand our sales and marketing infrastructure to commercialize our current products and any new products for which we may obtain regulatory approval; and
•expand operational, financial, and management information systems and personnel, including personnel to support product development efforts and our obligations as a public company.
Furthermore, the extent of the ultimate impact of the COVID- 19 pandemic on our operational and financial performance will depend on various developments, including the duration and spread of the pandemic, and its impact on potential customers, employees, and vendors, all of which cannot be reasonably predicted at this time.
To attain and sustain profitability, we must succeed in developing and commercializing drugs with significant market potential. This will require us to be successful in a range of challenging activities, including the discovery of product candidates, successful completion of preclinical testing and clinical trials of our product candidates, obtaining regulatory approval for these product candidates and manufacturing, marketing and selling those products for which we may obtain regulatory approval. We are only in the preliminary stages of these activities. We may not be successful enough in these activities to generate revenues that are substantial enough to achieve profitability. Even if we do achieve profitability, we may not be able to
sustain or increase profitability on a quarterly or annual basis. Our failure to become or remain profitable could depress the market price of our common stock and could impair our ability to raise capital, expand our business, diversify our product offerings or continue our operations. A decline in the market price of our common stock may also cause a loss of a part or all of your investment.
Negative publicity regarding any of our products could impair our ability to market any such product and may require us to spend time and money to address these issues.
If any of our products or any similar products distributed by other companies prove to be, or are asserted to be, harmful to consumers and/or subject to FDA enforcement action, our ability to successfully market and sell our products could be impaired. Because of our dependence on patient and physician perceptions, any adverse publicity associated with illness or other adverse effects resulting from the use or misuse of our products or any similar products distributed by other companies could limit the commercial potential of our products and expose us to potential liabilities.
We may not have sufficient insurance to cover our liability in any current or future litigation claims either due to coverage limits or as a result of insurance carriers seeking to deny coverage of such claims.
We face a variety of litigation-related liability risks. Our certificate of incorporation, bylaws, other applicable agreements, and/or Delaware law require us to indemnify (and advance expenses to) our current and past directors and officers and employees from reasonable expenses related to the defense of any action arising from their service to us, including circumstances under which indemnification is otherwise discretionary. While our directors and officers are included in a director and officer liability insurance policy, which covers all our directors and officers in some circumstances, our insurance coverage does not cover all of our indemnification obligations and may not be adequate to cover any indemnification or other claims against us. In addition, the underwriters of our present coverage may seek to avoid coverage in certain circumstances based upon the terms of the respective policies. If we incur liabilities that exceed our coverage under our directors and officers insurance policy or incur liabilities not covered by our insurance, we would have to self-fund any indemnification amounts owed to our directors and officers and employees in which case our results of operations and financial condition could be materially adversely affected. Further, if D&O insurance becomes prohibitively expensive to maintain in the future, we may be unable to renew such insurance on economic terms or unable renew such insurance at all. The lack of D&O insurance may make it difficult for us to retain and attract talented and skilled directors and officers to serve our company, which could adversely affect our business
* We may need substantial funding and may be unable to raise capital when needed, which would force us to delay, reduce or eliminate our product development programs or commercialization efforts.
We expect our general and research and development expenses to increase in connection with our ongoing and planned activities, particularly as we conduct Phase 3 clinical trials of fosmetpantotenate and sparsentan, and conduct any other later-stage clinical trials of our product candidates. In addition, subject to obtaining regulatory approval of any of our product candidates, we expect to incur significant commercialization expenses for product sales and marketing, securing commercial quantities of product from our manufacturers, and product distribution. We currently have no additional commitments or arrangements for any additional financing to fund the research and development and commercial launch of our product candidates. General market conditions resulting from the
ongoing issues arising from the COVID-19 pandemic, as well as market conditions affecting companies in the life sciences industry in general, may make it difficult for us to seek financing from the capital markets on attractive terms, or at all.
Management believes our ability to continue our operations depends on our ability to sustain and grow revenue, results of operations and our ability to access capital markets when necessary to accomplish our strategic objectives. Management believes that we may incur losses in the immediate future. We expect that our operating results will vary significantly from quarter-to-quarter and year-to-year as a result of investments in research and development, specifically our clinical and preclinical development activities. We expect to finance our cash needs from cash on hand and results of operations, and depending on results of operations we may either need additional equity or debt financing, or need to enter into strategic alliances on products in development to continue our operations until we can achieve sustained profitability and positive cash flows from operating activities. Additional funds may not be available to us when we need them on terms that are acceptable to us, or at all. If adequate funds are not available to us on a timely basis, we may be required to reduce or eliminate research development programs or commercial efforts.
Our future capital requirements will depend on many factors, including:
•the progress and results of our pre-clinical and clinical studies of fosmetpantotenate, sparsentan for FSGS and Igan, Chenodal for CTX, and any other drug candidates;
•the costs, timing and outcome of regulatory review of our product candidates;
•debt service obligations on the 2025 Notes;
•the number and development requirements of other product candidates that we pursue;
•the costs of commercialization activities, including product marketing, sales and distribution;
•the emergence of competing technologies and other adverse market developments;
•the costs of preparing, filing and prosecuting patent applications and maintaining, enforcing and defending intellectual property related claims;
•the extent to which we acquire or invest in businesses, products and technologies; and
•our ability to establish collaborations and obtain milestone, royalty or other payments from any such collaborators.
The market price for shares of our common stock may be volatile and purchasers of our common stock could incur substantial losses.
The price of our stock is likely to be volatile. The stock market in general, and the market for biotechnology companies in particular, have experienced extreme volatility that has often been unrelated to the operating performance of particular companies. The market price for our common stock may be influenced by many factors, including:
•results of clinical trials of our product candidates or those of our competitors;
•our entry into or the loss of a significant collaboration;
•regulatory or legal developments in the United States and other countries, including changes in the health care payment systems;
•our ability to obtain and maintain marketing approvals from the FDA or similar regulatory authorities outside the United States;
•variations in our financial results or those of companies that are perceived to be similar to us;
•changes in the structure of healthcare payment systems;
•market conditions in the pharmaceutical and biotechnology sectors and issuance of new or changed securities analysts’ reports or recommendations;
•general economic, industry and market conditions;
•results of clinical trials conducted by others on drugs that would compete with our product candidates;
•developments or disputes concerning patents or other proprietary rights;
•public concern over our product candidates or any products approved in the future;
•litigation;
•communications from government officials regarding health care costs or pharmaceutical pricing;
•future sales or anticipated sales of our common stock by us or our stockholders; and
•the other factors described in this “Risk Factors” section.
In addition, the stock markets, and in particular, the Nasdaq Global Market, have experienced extreme price and volume fluctuations that have affected and continue to affect the market prices of equity securities of many pharmaceutical companies. The realization of any of the above risks or any of a broad range of other risks, including those described in these “Risk Factors” could have a dramatic and material adverse impact on the market price of our common stock.
We may be unable to successfully integrate new products or businesses we may acquire.
We intend to expand our product pipeline by pursuing acquisition of pharmaceutical products. If an acquisition is consummated, the integration of the acquired business, product or other assets into our company may also be complex and time- consuming and, if such businesses, products and assets are
not successfully integrated, we may not achieve the anticipated benefits, cost-savings or growth opportunities. Potential difficulties that may be encountered in the integration process include the following:
•integrating personnel, operations and systems, while maintaining focus on producing and delivering consistent, high quality products;
•coordinating geographically dispersed organizations;
•distracting employees from operations;
•retaining existing customers and attracting new customers; and
•managing inefficiencies associated with integrating the operations of the Company.
Furthermore, these acquisitions and other arrangements, even if successfully integrated, may fail to further our business strategy as anticipated, expose us to increased competition or challenges with respect to our products or geographic markets, and expose us to additional liabilities associated with an acquired business, product, technology or other asset or arrangement. Any one of these challenges or risks could impair our ability to realize any benefit from our acquisitions or arrangements after we have expended resources on them.
If we are unable to maintain an effective and specialized sales force, we will not be able to commercialize our products successfully.
In order to successfully commercialize our products, we have built a specialized sales force. Factors that may hinder our ability to successfully market and commercially distribute our products include:
inability of sales personnel to obtain access to or convince adequate numbers of physicians to prescribe our products;
inability to recruit, retain and effectively manage adequate numbers of effective sales personnel;
lack of complementary products to be offered by sales personnel, which may put us at a competitive disadvantage relative to companies that have more extensive product lines; and
unforeseen delays, costs and expenses associated with maintaining our sales organization.
If we are unable to maintain our sales force for our products, we may not be able to generate sufficient product revenue.
We will need to continue to expend significant time and resources to train our sales forces to be credible, persuasive and compliant in discussing our products with the specialists treating the patients indicated under the product’s label. In addition, if we are unable to effectively train our sales force and equip them with effective marketing materials our ability to successfully commercialize our products could be diminished, which would have a material adverse effect on our business, results of operations and financial condition.
Product liability lawsuits against us could cause us to incur substantial liabilities and to limit commercialization of any products that we may develop.
Our business exposes us to potential liability risks inherent in the research, development, manufacturing and marketing of pharmaceutical products. If any of our product candidates in clinical trials or commercialized products harm people we may be subject to costly and damaging product liability claims. We have clinical trial insurance and commercial product liability coverage. However, this insurance may not be adequate to cover all claims. We may be exposed to product liability claims and product recalls, including those which may arise from misuse or malfunction of, or design flaws in, such products, whether or not such problems directly relate to the products and services we have provided. If we cannot successfully defend ourselves against claims that our product candidates or products caused injuries, we will incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
•decreased demand for any product candidates or products that we may develop;
•damage to our reputation;
•regulatory investigations that could require costly recalls or product modifications;
•withdrawal of clinical trial participants;
•costs to defend the related litigation;
•substantial monetary awards to trial participants or patients, including awards that substantially exceed our product liability insurance, which we would then be required to pay from other sources, if available, and would damage our ability to obtain liability insurance at reasonable costs, or at all, in the future;
•loss of revenue;
•the diversion of management’s attention from managing our business; and
•the inability to commercialize any products that we may develop.
A successful product liability claim or a series of claims brought against us could cause our stock price to fall and, if judgments exceed our insurance coverage, could decrease our available cash and adversely affect our business.
* We aremay become involved in certain litigation matters, any of which could result in substantial costs, divert management's attention and otherwise have a material adverse effect on our business, operating results or financial condition.
We areFrom time to time we may become involved in certain litigation matters, including those described in Note 12 of the Consolidated Financial Statements included in this report. Although we intend to vigorously defend our interests in each matter, there is no guarantee that we will be successful and we may have to pay damages awards or otherwise may enter into settlement arrangements in connection with such matters. Any such payments or settlement arrangements could have material adverse effects on our business, operating results or financial condition. Even if we are successful in defending our interests in each matter, litigation with respect to such matters could result in substantial costs and significant adverse impact on our reputation and divert management's attention and resources, which could have a material adverse effect on our business, operating results or financial condition.
We are subject to significant ongoing regulatory obligations and oversight, which may result in significant additional expense and may limit our commercial success.
We are subject to significant ongoing regulatory obligations, such as safety reporting requirements and additional post-marketing obligations, including regulatory oversight of the promotion and marketing of our products. In addition, the manufacture, quality control, labeling, packaging, safety surveillance, adverse event reporting, storage and recordkeeping for our products are subject to extensive and ongoing regulatory requirements. If we become aware of previously unknown problems with any of our products, a regulatory agency may impose restrictions on our products, our contract manufacturers or us. If we, our products and product candidates, or the manufacturing facilities for our products and product candidates fail to comply with applicable regulatory requirements, a regulatory agency, including the FDA, may send enforcement letters, mandate labeling changes, suspend or withdraw regulatory approval, suspend any ongoing clinical trials, refuse to approve pending applications or supplements filed by us, suspend or impose restrictions on manufacturing operations, request a recall of, seize or detain a product, seek criminal prosecution or an injunction, or impose civil or criminal penalties or monetary fines. In such instances, we could experience a significant drop in the sales of the affected products, our product revenues and reputation in the marketplace may suffer, and we could become the target of lawsuits.
We are also subject to regulation by national, regional, state and local agencies, including but not limited to the FDA, CMS, Department of Justice, the Federal Trade Commission, the HHS Office of Inspector General and other regulatory bodies. The FDC Act, Social Security Act, Public Health Service Act and other federal and state statutes and regulations govern to varying degrees the research, development, manufacturing and commercial activities relating to prescription pharmaceutical products, including preclinical testing, clinical research, approval, production, labeling,
sale, distribution, post-market surveillance, advertising, dissemination of information, promotion, marketing, and pricing to government purchasers and government health care programs. Our manufacturing partners are subject to many of the same requirements.
Companies may not promote drugs for “off-label” uses-that is, uses that are not described in the product’s labeling and that differ from those approved by the FDA or other applicable regulatory agencies. However, a company may share truthful and not misleading information that is otherwise consistent with the product’s labeling. A company that is found to have improperly promoted off-label uses may be subject to significant liability, including civil and administrative remedies as well as criminal sanctions. In addition, management’s attention could be diverted from our business operations and our reputation could be damaged.
The federal health care program Anti-Kickback statute prohibits, among other things, knowingly and willfully offering, paying, soliciting, or receiving remuneration to induce or in return for purchasing, leasing, ordering or arranging for the purchase, lease or order of any health care item or service reimbursable under Medicare, Medicaid or other federally financed healthcare programs. This statute has been interpreted broadly to apply to arrangements that pharmaceutical companies have with prescribers, purchasers and formulary managers, among others. Further, the PPACA, among other things, amends the intent requirement of the federal anti-kickback statute so that a person or entity no longer needs to have actual knowledge of this statute or specific intent to violate it. In addition, the PPACA provides that the government may assert that a claim including items or services resulting from a violation of the federal anti-kickback statute constitutes a false or fraudulent claim for purposes of the civil False Claims Act. Although there are a number of statutory exceptions and regulatory safe harbors under the federal anti-kickback statute protecting certain common manufacturer business arrangements and activities from prosecution, the exceptions and safe harbors are drawn narrowly and an arrangement must meet all of the conditions specified in order to be fully protected from scrutiny under the federal anti-kickback statute. We seek to comply with the exceptions and safe harbors whenever possible, but our practices, such as our patient assistance programs and prompt pay discounts with certain customers, may not in all cases meet all of the criteria for protection from anti-kickback liability and may be subject to scrutiny.
The federal false claims laws, including the federal False Claims Act, prohibit any person or entity from knowingly presenting, or causing to be presented, a false claim for payment to the federal government, or knowingly making, or causing to be made, a false statement to get a false claim paid. Additionally, the civil monetary penalties statute imposes penalties against any person or entity that, among other things, is determined to have presented or caused to be presented a claim to a federal health program that the person knows or should know is for an item or service that was not provided as claimed or is false or fraudulent. Many pharmaceutical and other health care companies have been investigated and have reached substantial financial settlements with the federal government under the federal False Claims Act for a variety of alleged marketing activities, including providing free product to customers with the expectation that the customers would bill federal programs for the product; providing consulting fees, grants, free travel, and other benefits to physicians to induce them to prescribe the company’s products; and inflating prices reported to private price publication services, which may be used by states to set drug payment rates under government health care programs. Companies have been prosecuted for causing false claims to be submitted because of the marketing of their products for unapproved uses. Pharmaceutical and other health care companies have also been prosecuted on other legal theories of Medicare and Medicaid fraud.
Legislative and regulatory proposals have been made to expand post-approval requirements and restrict sales and promotional activities for pharmaceutical products. It is not clear whether additional legislative changes will be enacted, or whether the FDA regulations, guidance or interpretations will be changed, or what the impact of such changes on the marketing approvals of any Retrophin products, if any, may be. In addition, increased scrutiny by the U.S. Congress of the FDA’s approval process may significantly delay or prevent marketing approval, as well as subject Retrophin to more stringent product labeling and post-marketing testing and other requirements.
We also could become subject to government investigations and related subpoenas. Such subpoenas are often associated with previously filed qui tam actions, or lawsuits filed under seal under the federal False Claims Act. Qui tam actions are brought by private plaintiffs suing on behalf of the federal government for alleged violations of the federal False Claims Act. The time and expense associated with responding to such subpoenas, and any related qui tam or other actions, may be extensive, and we cannot predict the results of our review of the responsive documents and underlying facts or the results of such actions. Responding to government investigations, defending any claims raised, and any resulting fines, restitution, damages and penalties, settlement payments or administrative actions, as well as any related actions brought by stockholders or other third parties, could have a material impact on our reputation, business and financial condition and divert the attention of our management from operating our business.
The number and complexity of both federal and state laws continues to increase, and additional governmental resources are being added to enforce these laws and to prosecute companies and individuals who are believed to be violating them. In particular, the PPACA includes a number of provisions aimed at strengthening the government’s ability to pursue anti-kickback and false claims cases against pharmaceutical manufacturers and other healthcare entities, including substantially increased funding for healthcare fraud enforcement activities, enhanced investigative powers, amendments to the federal False Claims Act that make it easier for the government and whistleblowers to pursue cases for alleged kickback and false claim violations and for payments made on or after August 1, 2013, public reporting of certain payments and transfers of value by certain pharmaceutical manufacturers to physicians and teaching hospitals nationwide. We anticipate that government scrutiny of pharmaceutical sales and marketing practices will continue for the foreseeable future and subject us to the risk of further government investigations and enforcement actions. Responding to a government investigation or enforcement action would be expensive and time-consuming, and could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
The U.S. Foreign Corrupt Practices Act, and similar worldwide anti-bribery laws generally prohibit companies and their intermediaries from making improper payments to government officials for the purpose of obtaining or retaining business. Our policies mandate compliance with these anti-bribery laws. We operate in parts of the world that have experienced governmental corruption to some degree and in certain circumstances, strict compliance with antibribery laws may conflict with local customs and practices or may require us to interact with doctors and hospitals, some of which may be state controlled, in a manner that is different than in the United States. We cannot assure that our internal control policies and
procedures will protect us from reckless or criminal acts committed by our employees or agents. Violations of these laws, or allegations of such violations, could disrupt our business and result in criminal or civil
penalties or remedial measures, any of which could have a material adverse effect on our business, financial condition and results of operations and could cause the market value of our common stock to decline.
The federal Health Insurance Portability and Accountability Act of 1996 ("HIPAA"), created new federal criminal statutes that prohibit, among other actions, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payers, and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Like the federal anti-kickback statute, the PPACA amended the intent standard for certain healthcare fraud provisions under HIPAA such that a person or entity no longer needs to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
Additionally, the federal Physician Payments Sunshine Act within the PPACA, and its implementing regulations, require that certain manufacturers of drugs, devices, biologicals and medical supplies to report annually information related to certain payments or other transfers of value made or distributed to physicians and teaching hospitals, or to entities or individuals at the request of, or designated on behalf of, physicians, as defined by such law and teaching hospitals and to report annually certain ownership and investment interests held by physicians and their immediate family members. Moreover, the Drug Supply Chain Security Act imposes new obligations on manufacturers of pharmaceutical products related to product tracking and tracing. We are not sure whether additional legislative changes will be enacted, or whether the current regulations, guidance or interpretations will be changed, or what the impact of such changes on our business, if any, may be.
Also, many states have similar fraud and abuse statutes or regulations, including state anti-kickback and false claims laws, that apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payer. Further, certain states require implementation of commercial compliance programs and marketing codes, compliance with the pharmaceutical industry’s voluntary compliance guidelines, and compliance with the applicable compliance guidance promulgated by the federal government. Other various state level requirements include restricting payments or the provision of other items of value that may be made to healthcare providers and other potential referral sources; restricting various marketing practices; requiring prescription drug companies to report expenses relating to the marketing and promotion of drug products; requiring the posting of information relating to clinical studies and their outcomes; requiring the registration of sales representatives; requiring the reporting of certain information related to drug pricing; and requiring drug manufacturers to track and report information related to payments, gifts, compensation, and other items of value to physicians and other healthcare providers.
In addition, we may be subject to data privacy and security regulation by both the federal government and the states in which we conduct our business. HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act ("HITECH"), and their respective implementing regulations, imposes specified requirements relating to the privacy, security and transmission of individually identifiable health information. Among other things, HITECH, through its implementing regulations, makes certain of HIPAA’s privacy and security standards directly applicable to business associates, defined as a person or organization, other than a member of a covered entity’s workforce, that creates, receives, maintains or transmits protected health information for or on behalf of a covered entity for a function or activity regulated by HIPAA. International data protection laws also impose strict obligations on the ability to process health related and other personal information of citizens of member states, including in relation to collection, analysis and transfer. The EU General Data Protection Regulation was officially adopted in April 2016 and has been in effect since May 2018. The EU General Data Protection Regulation introduced new data protection requirements in the European Union, as well as substantial fines for breaches of the data protection rules. The EU General Data Protection Regulation will increase our responsibility and liability in relation to personal data that we control and/or process, and we may be required to put in place additional mechanisms to ensure compliance with the new EU data protection rules. For example, on July 16, 2020, the Court of Justice of the European Union issued a decision that declared the "Privacy Shield" framework invalid, and will also result in additional compliance obligations for companies that implement standard contractual clauses to ensure a valid basis for the transfer of personal data outside of the European Economic Area ("EEA") to the United States and certain other countries. We are evaluating the implications of the decision for transfers of personal data from the EEA and will assess alternate transfer options and limitations as additional regulatory guidance becomes available. Additionally, California recently enacted legislation known as the California Consumer Privacy Act (the “CCPA”), which creates new individual privacy rights for consumers (as that word is broadly defined in the law) and places increased privacy and security obligations on entities handling personal data of consumers or households. When it goes into effectThe CCPA, which became effective on January 1, 2020, the CCPA will requirerequires covered companies to provide new disclosures to California consumers, provideprovides such consumers new ways to opt-out of certain sales of personal information, and allowallows for a new cause of action for data breaches. Legislators have stated that amendments will be proposed to the CCPA before it goes into effect, but it remains unclear what, if any, modifications will be made to this legislation or how it will be interpreted. As currently written, theThe CCPA will likely impact (possibly significantly) our business activities and exemplifies the vulnerability of our business to not only cyber threats but also the evolving regulatory environment related to personal data and protected health information.
If we or any of our partners fail to comply with applicable regulatory requirements, we or they could be subject to a range of regulatory actions that could affect our or our partners' ability to commercialize our products and could harm or prevent sales of the affected products, or could substantially increase the costs and expenses of commercializing and marketing our products. Any threatened or actual government enforcement action could also generate adverse publicity and require that we devote substantial resources that could otherwise be used in other aspects of our business. Compliance with applicable federal and state laws is difficult and time consuming, and companies that violate them may face substantial penalties. The potential sanctions include significant criminal fines, civil monetary penalties, administrative penalties, disgorgement, exclusion from participation in federal health care programs, individual imprisonment, injunctions, recall or seizure of products, total or partial suspension of production, reputational harm, administrative burdens, additional oversight and reporting obligations if we become subject to a corporate integrity agreement or similar agreement to resolve allegation of non-compliance with these laws, diminished profits and future earnings, and the curtailment or restructuring of our operations, and other sanctions. Because of the breadth of these laws, it is possible that some of our business activities could be subject to challenge under one or more of these laws. Such a challenge, irrespective of the underlying merits of the challenge or the ultimate outcome of the matter, could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
If we are not able to obtain and maintain required regulatory approvals, we will not be able to commercialize our products, and our ability to generate revenue will be materially impaired.
Our product candidates, once approved, and the activities associated with their manufacture, marketing, distribution, and sales are subject to extensive regulation by the FDA and other regulatory agencies in the United States and by comparable authorities in other countries. Failure to adhere to regulations set out by these bodies for one or more of our commercial products could prevent us from commercializing the product candidate in the jurisdiction of the regulatory authority. We have only limited experience in meeting the regulatory requirements incumbent on the sale of drugs in the United States and elsewhere, and expect to rely on third-parties to assist us in these processes. If these third parties fail to adequately adhere to the regulations governing drug distribution and promotion we may be unable to sell our products, which could have a material effect on our ability to generate revenue.
Our product candidates and the activities associated with their development and commercialization, including testing, manufacture, safety, efficacy, recordkeeping, labeling, storage, approval, advertising, promotion, sale and distribution, are subject to comprehensive regulation by the FDA and other regulatory agencies in the United States and by comparable authorities in other countries. Failure to obtain regulatory approval for a product candidate will prevent us from commercializing the product candidate in the jurisdiction of the regulatory authority. We have only limited experience in filing and prosecuting the applications necessary to obtain regulatory approvals and expect to rely on third-party contract research organizations to assist us in this process.
Securing FDA approval requires the submission of extensive preclinical and clinical data and supporting information to the FDA for each therapeutic indication to establish the product candidate’s safety and efficacy. Securing FDA approval also requires the submission of information about the product manufacturing process to, and successful inspection of manufacturing facilities by, the FDA. Our future products may not be effective, may be only moderately effective or may prove to have undesirable or unintended side effects, toxicities or other characteristics that may preclude our obtaining regulatory approval or prevent or limit commercial use.
Our product candidates may fail to obtain regulatory approval for many reasons, including:
•our failure to demonstrate to the satisfaction of the FDA or comparable regulatory authorities that a product candidate is safe and effective for a particular indication;
•the results of clinical trials may not meet the level of statistical significance required by the FDA or comparable regulatory authorities for approval;
•our inability to demonstrate that a product candidate’s benefits outweigh its risks;
•our inability to demonstrate that the product candidate presents an advantage over existing therapies;
•the FDA’s or comparable regulatory authorities’ disagreement with the manner in which we interpret the data from preclinical studies or clinical trials;
•failure of the third-party manufacturers with which we contract for clinical or commercial supplies to satisfactorily complete an FDA pre-approval inspection of the facility or facilities at which the product is manufactured to assess compliance with the FDA’s cGMP regulations to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity; and
•a change in the approval policies or regulations of the FDA or comparable regulatory authorities or a change in the laws governing the approval process.
The process of obtaining regulatory approvals is expensive, often takes many years, if approval is obtained at all, and can vary substantially based upon a variety of factors, including the type, complexity and novelty of the product candidates involved. Changes in regulatory approval policies during the development period, changes in or the enactment of additional statutes or regulations, or changes in regulatory review for each submitted product application may cause delays in the approval or rejection of an application. The FDA and non-United States regulatory authorities have substantial discretion in the approval process and may refuse to accept any application or may decide that our data is insufficient for approval and require additional preclinical, clinical or other studies. In addition, varying interpretations of the data obtained from preclinical and clinical testing could delay, limit or prevent regulatory approval of a product candidate. Any regulatory approval we ultimately obtain may be limited or subject to restrictions or post approval commitments that render the approved product not commercially viable. Any FDA or other regulatory approval of our product candidates, once obtained, may be withdrawn, including for failure to comply with regulatory requirements or if clinical or manufacturing problems follow initial marketing.
* Changes in tax laws or regulations that are applied adversely to us or our customers may have a material adverse effect on our business, cash flow, financial condition or results of operations.
New income, sales, use or other tax laws, statutes, rules, regulations or ordinances could be enacted at any time, which could affect the tax treatment of our domestic and foreign earnings. Any new taxes could adversely affect our domestic and international business operations, and our business and financial performance. Further, existing tax laws, statutes, rules, regulations or ordinances could be interpreted, changed, modified or applied adversely to us. For example, the Tax Act enacted many significant changes to the U.S. tax laws and significantly revised the Internal Revenue Code of 1986, as amended. Future guidance from the Internal Revenue Service and other tax authorities with respect to the Tax Act may affect us, and certain aspects of the Tax Act could be repealed or modified in future legislation. For example, the CARES Act modified certain tax provisions of the Tax Act. In addition, it is uncertain if and to what extent various states will conform to the Tax Act, the CARES Act, or any newly enacted federal tax legislation. Changes in corporate tax rates, the realization of net deferred tax assets relating to our operations, the taxation of foreign earnings, and the deductibility of expenses under the Tax Act or future reform legislation could have a material impact on the value of our deferred tax assets, could result in significant one-time charges, and could increase our future U.S. tax expense.
Our effective tax rate may fluctuate, and we may incur obligations in tax jurisdictions in excess of accrued amounts.
Our effective tax rate is derived from a combination of applicable tax rates in the various places that we operate. In preparing our financial statements, we estimate the amount of tax that will become payable in each of such places. Nevertheless, our effective tax rate may be different than experienced in the past due to numerous factors, including passage of the Tax Act, changes in the mix of our profitability from state to state, the results of examinations and audits of
our tax filings, our inability to secure or sustain acceptable agreements with tax authorities, changes in accounting for income taxes and changes in tax laws. Any of these factors could cause us to experience an effective tax rate significantly different from previous periods or our current expectations and may result in tax obligations in excess of amounts accrued in our financial statements.
* Our ability to use net operating loss carryforwards and certain other tax attributes to offset future taxable income and taxes may be subject to limitations.
As of December 31, 2019, we had federal net operating loss, or NOL, carryforwards of $113.6 million. On March 27, 2020, the Coronavirus Aid, Relief, and Economic Security Act (CARES Act) was enacted in response to the COVID-19 pandemic. The CARES Act, among other things, permits NOL carryovers and carrybacks to offset 100% of taxable income for taxable years beginning before 2021. In addition, the CARES Act allows net operating losses incurred in 2018, 2019, and 2020 to be carried back to each of the five preceding taxable years to generate a refund of previously paid income taxes. The Company has recorded an income tax benefit of $18.9 million related to this legislation.
Under the Tax Act, as modified by the CARES Act, our federal NOLs generated in tax years beginning after December 31, 2017, may be carried forward indefinitely, but the deductibility of such federal NOLs in tax years beginning after December 31, 2020, is limited to 80% of taxable income. In addition, under the CARES Act, NOLs generated in tax years beginning after December 31, 2017, and before January 1, 2021, may be carried back to each of the five tax years preceding the tax years of such loss. It is uncertain if and to what extent various states will conform to the Tax Act or the CARES Act. In addition, under Sections 382 and 383 of the Internal Revenue Code of 1986, as amended, and corresponding provisions of state law, if a corporation undergoes an “ownership change,” which is generally defined as a greater than 50% change, by value, in its equity ownership over a three-year period, the corporation’s ability to use its pre-change NOL carryforwards and other pre-change U.S. tax attributes (such as research tax credits) to offset its post-change income or taxes may be limited. It is possible that we have experienced an ownership change in the past. In addition, we may experience ownership changes in the future as a result of subsequent shifts in our stock ownership, some of which may be outside of our control.
As a result, our federal NOL carryforwards may be subject to a percentage limitation if used to offset income in tax years following an ownership change. Furthermore, while we believe based on input from our accountants that we are entitled to a refund from the carryback of post-2017 federal NOLs, there is no guarantee that the IRS will agree or that the refund will be received rapidly. In addition, it is possible that we have in the past undergone, and in the future may undergo, additional ownership changes that could limit our ability to use all of our pre-change NOL carryforwards and other pre-change tax attributes (such as research tax credits) to offset our post-change income or taxes. Similar provisions of state tax law may also apply to limit our use of accumulated state tax attributes. In addition, at the state level, there may be periods during which the use of NOL carryforwards is suspended or otherwise limited, which could accelerate or permanently increase state taxes owed. As a result, we may be unable to use all or a material portion of our NOL carryforwards and other tax attributes, which would harm our future operating results by effectively increasing our future tax obligations.
Our internal computer systems, or those of our CROs or other contractors and vendors who host our applications or consultants, may fail or suffer security breaches, which could result in a material disruption of our product development programs.
Despite the implementation of security measures, our internal computer systems and those of our CROs and other contractors or vendors who host our applications and those of our consultants are vulnerable to damage or disruption from computer viruses, software bugs, unauthorized access including cyber-attack, natural disasters, terrorism, war, and telecommunication, equipment and electrical failures. While we have not, to our knowledge, experienced any significant system failure, accident or security breach to date, if such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our programs. For example, the loss of clinical trial data from completed or ongoing clinical trials for any of our product candidates could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach results in a loss of or damage to our data or applications, or inappropriate
disclosure or theft of confidential or proprietary information, we could incur liability, the further development of our product candidates could be delayed, our competitive position could be compromised, or our business reputation could be harmed.
* Changes in tax laws or regulations that are applied adversely to us or our customers may have a material adverse effect on our business, cash flow, financial condition or results of operations.
New income, sales, use or other tax laws, statutes, rules, regulations or ordinances could be enacted at any time, which could affect the tax treatment of our domestic and foreign earnings. Any new taxes could adversely affect our domestic and international business operations, and our business and financial performance. Further, existing tax laws, statutes, rules, regulations or ordinances could be interpreted, changed, modified or applied adversely to us. For example, on December 22, 2017, U.S. federal income tax legislation was signed into law (H.R. 1, “An Act to provide for reconciliation pursuant to titles II and V of the concurrent resolution on the budget for fiscal year 2018”), informally titled the Tax Cuts and Jobs Act, that significantly revised the Internal Revenue Code of 1986, as amended. The Tax Cuts and Jobs Act, among other things, contains significant changes to corporate taxation, including reduction of the corporate tax rate from a top marginal rate of 35% to a flat rate of 21%, limitation of the tax deduction for interest expense to 30% of adjusted taxable income (except for certain small businesses), limitation of the deduction for net operating losses generated in taxable years beginning after December 31, 2017 to 80% of current year taxable income, and elimination of most carrybacks of net operating losses arising in taxable years ending after December 31, 2017, one-time taxation of offshore earnings at reduced rates regardless of whether they are repatriated, elimination of U.S. tax on foreign earnings (subject to certain important exceptions), immediate deductions for certain new investments instead of deductions for depreciation expense over time, and modifying or repealing many business deductions and credits including reducing the business tax credit for certain clinical testing expenses incurred in the testing of certain drugs for rare diseases or conditions.
Notwithstanding the reduction in the corporate income tax rate, the overall impact of the Tax Cuts and Jobs Act is uncertain and our business and financial condition could be adversely affected. In addition, it is unknown if and to what extent various states will conform to the Tax Cuts and Jobs Act. The impact of the Tax Cuts and Jobs Act on holders of our common stock is likewise uncertain and could be adverse. We urge our stockholders to consult with their legal and tax advisors with respect to the Tax Cuts and Jobs Act and the potential tax consequences of investing in or holding our common stock.
Our effective tax rate may fluctuate, and we may incur obligations in tax jurisdictions in excess of accrued amounts.
Our effective tax rate is derived from a combination of applicable tax rates in the various places that we operate. In preparing our financial statements, we estimate the amount of tax that will become payable in each of such places. Nevertheless, our effective tax rate may be different than experienced in the past due to numerous factors, including passage of the Tax Cuts and Jobs Act, changes in the mix of our profitability from state to state, the results of examinations and audits of our tax filings, our inability to secure or sustain acceptable agreements with tax authorities, changes in accounting for income taxes and changes in tax laws. Any of these factors could cause us to experience an effective tax rate significantly different from previous periods or our current expectations and may result in tax obligations in excess of amounts accrued in our financial statements.
Our ability to use net operating losses to offset future taxable income may be subject to limitations.
As of December 31, 2018, we had federal net operating loss, or NOL, carryforwards of $36.5 million. Our federal NOL carryforwards generated in tax years ending on or prior to December 31, 2017, are only permitted to be carried forward for 20 years under applicable U.S. tax law. Under the Tax Cuts and Jobs Act, our federal NOLs generated in tax years ending after December 31, 2017, may be carried forward indefinitely, but the deductibility of such federal NOLs generated in tax years beginning after December 31, 2017, is limited. It is uncertain if and to what extent various states will conform to the Tax Cuts and Jobs Act. In addition, under Sections 382 and 383 of the Internal Revenue Code of 1986, as amended, and corresponding provisions of state law, if a corporation undergoes an “ownership change,” which is generally defined as a greater than 50% change, by value, in its equity ownership over a three-year period, the corporation’s ability to use its pre-change NOL carryforwards and other pre-change U.S. tax attributes (such as research tax credits) to offset its post-change income or taxes may be limited. It is possible that we have experienced an ownership change limitation in the past. In addition, we may experience ownership changes in the future as a result of subsequent shifts in our stock ownership, some of which may be outside of our control.
As a result, our pre-2018 NOL carryforwards may expire prior to being used, and our NOL carryforwards generated in 2018 and thereafter will be subject to a percentage limitation. In addition, it is possible that we have in the past undergone, and in the future may undergo, additional ownership changes that could limit our ability to use all of our pre-change NOLs and other pre-change tax attributes (such as research tax credits) to offset our post-change income or taxes. Similar provisions of state tax law may also apply to limit our use of accumulated state tax attributes. In addition, at the state level, there may be periods during which the use of NOLs is suspended or otherwise limited, which could accelerate or permanently increase state taxes owed. As a result, we may be unable to use all or a material portion of our NOLs and other tax attributes, which would harm our future operating results by effectively increasing our future tax obligations.
We are dependent on information technology systems, infrastructure and data, which exposes us to data security risks.
We are dependent upon our own or third-party information technology systems, infrastructure and data, including mobile technologies, to operate our business. The multitude and complexity of our computer systems may make them vulnerable to service interruption or destruction, disruption of data integrity, malicious intrusion, or random attacks. Likewise, data privacy or security incidents or breaches by employees or others may pose a risk that sensitive data, including our intellectual property, trade secrets or personal information of our employees, patients, customers or other business partners may be exposed to unauthorized persons or to the public. Cyber-attacks are increasing in their frequency, sophistication and intensity. Cyber-attacks could include the deployment of harmful malware, denial-of-service, social engineering and other means to affect service reliability and threaten data confidentiality, integrity and availability. Our business partners face similar risks and any security breach of their systems
could adversely affect our security posture. A security breach or privacy violation that leads to disclosure or modification of or prevents access to patient information, including personally identifiable information or protected health information, could harm our reputation, compel us to comply with federal and/or state breach notification laws and foreign law equivalents, subject us to mandatory corrective action, require us to verify the correctness of database contents and otherwise subject us to litigation or other liability under laws and regulations that protect personal data, any of which could disrupt our business and/or result in increased costs or loss of revenue. Moreover, the prevalent use of mobile devices that access confidential information increases the risk of data security breaches, which could lead to the loss of confidential information, trade secrets or other intellectual property. While we have invested, and continue to invest, in the protection of our data and information technology infrastructure, there can be no assurance that our efforts will prevent service interruptions, or identify breaches in our systems, that could adversely affect our business and operations and/or result in the loss of critical or sensitive information, which could result in financial, legal, business or reputational harm to us. In addition, our liability insurance may not be sufficient in type or amount to cover us against claims related to security breaches, cyber-attacks and other related breaches.
Changes in funding for the FDA, the SEC and other government agencies could hinder their ability to hire and retain key leadership and other personnel, prevent new products and services from being developed or commercialized in a timely manner or otherwise
prevent those agencies from performing normal functions on which the operation of our business may rely, which could negatively impact our business.
The ability of the FDA to review and approve new products can be affected by a variety of factors, including government budget and funding levels, ability to hire and retain key personnel and accept payment of user fees, and statutory, regulatory, and policy changes. Average review times at the agency have fluctuated in recent years as a result. In addition, government funding of the SEC and other government agencies on which our operations may rely, including those that fund research and development activities is subject to the political process, which is inherently fluid and unpredictable.
Disruptions at the FDA and other agencies may also slow the time necessary for new drugs to be reviewed and/or approved by necessary government agencies, which would adversely affect our business. For example, over the last several years, the U.S. government has shut down several times and certain regulatory agencies, such as the FDA and the SEC, have had to furlough critical FDA, SEC and other government employees and stop critical activities. If a prolonged government shutdown occurs, or if resource constraints continue to arise from the COVID-19 pandemic, it could significantly impact the ability of the FDA to timely review and process our regulatory submissions, which could have a material adverse effect on our business. Further, future government shutdowns could impact our ability to access the public markets and obtain necessary capital in order to properly capitalize and continue our operations.
The withdrawal of the United Kingdom, from the European Union, commonly referred to as “Brexit,” may adversely impact our ability to obtain regulatory approvals of our product candidates in the European Union, result in restrictions or imposition of taxes and duties for importing our product candidates into the European Union, and may require us to incur additional expenses in order to develop, manufacture and commercialize our product candidates in the European Union.
Following the result of a referendum in 2016, the United Kingdom left the European Union on January 31, 2020, commonly referred to as Brexit. Pursuant to the formal withdrawal arrangements agreed between the United Kingdom and the European Union, the United Kingdom will be subject to a transition period until December 31, 2020, or the Transition Period, during which EU rules will continue to apply. Negotiations between the United Kingdom and the European Union are expected to continue in relation to the customs and trading relationship between the United Kingdom and the European Union following the expiry of the Transition Period.
Since a significant proportion of the regulatory framework in the United Kingdom applicable to our business and our product candidates is derived from EU directives and regulations, Brexit, following the Transition Period, could materially impact the regulatory regime with respect to the development, manufacture, importation, approval and commercialization of our product candidates in the United Kingdom or the European Union. For example, as a result of the uncertainty surrounding Brexit, the EMA relocated to Amsterdam from London. Following the Transition Period, the United Kingdom will no longer be covered by the centralized procedures for obtaining EU-wide marketing authorization from the EMA and, unless a specific agreement is entered into, a separate process for authorization of drug products, including our product candidates, will be required in the United Kingdom, the potential process for which is currently unclear. Any delay in obtaining, or an inability to obtain, any marketing approvals, as a result of Brexit or otherwise, would prevent us from commercializing our product candidates in the United Kingdom or the European Union and restrict our ability to generate revenue and achieve and sustain profitability. In addition, we may be required to pay taxes or duties or be subjected to other hurdles in connection with the importation of our product candidates into the European Union, or we may incur expenses in establishing a manufacturing facility in the European Union in order to circumvent such hurdles. If any of these outcomes occur, we may be forced to restrict or delay efforts to seek regulatory approval in the United Kingdom or the European Union for our product candidates, or incur significant additional expenses to operate our business, which could significantly and materially harm or delay our ability to generate revenues or achieve profitability of our business. Any further changes in international trade, tariff and import/export regulations as a result of Brexit or otherwise may impose unexpected duty costs or other non-tariff barriers on us. These developments, or the perception that any of them could occur, may significantly reduce global trade and, in particular, trade between the impacted nations and the United Kingdom.
* Business disruptions could seriously harm future revenue and financial condition and increase our costs and expenses.
Our operations, and those of our third-party manufacturers, CROs and other contractors and consultants, could be subject to earthquakes, power shortages, telecommunications failures, water shortages, floods, hurricanes, typhoons, fires, extreme weather conditions, medical epidemics and other natural or man-made disasters or business interruptions, for which we are predominantly self-insured. The occurrence of any of these business disruptions could seriously harm our operations and financial condition and increase our costs and expenses.
Our corporate headquarters are located in San Diego, California, an area prone to wildfires and earthquakes. These and other natural disasters could severely disrupt our operations, and have a material adverse effect on our business, results of operations, financial condition and prospects. If a natural disaster, power outage or other event occurred that prevented us from using all or a significant portion of our headquarters, that damaged critical infrastructure, such as the manufacturing facilities of our third-party contract manufacturers, or that otherwise disrupted operations, it may be difficult or, in certain cases, impossible for us to continue our business for a substantial period of time. Any disaster recovery and business continuity plans we have in place may prove inadequate in the event of a serious disaster or similar event. We may incur substantial expenses as a result of the limited nature of our disaster recovery and business continuity plans, which, could have a material adverse effect on our business.
In addition, we rely on third-party manufacturers, some of whom are located in China, to manufacture API for certain of our product candidates, including sparsentan. Any disruption in production or inability of our manufacturers in China to produce or ship adequate quantities to meet our needs, whether as a result of a natural disaster or other causes (such as the COVID-19 pandemic), could impair our ability to operate our business on a day-to-day basis and to continue our research and development of our product candidates. In addition, we are exposed to the possibility of product supply disruption and increased costs in the event of changes in the policies of the United States or Chinese governments (such as tariffs on chemical intermediates we use that are manufactured in China), political unrest or unstable economic conditions in China. Any recall of the manufacturing lots or similar action regarding our API used in clinical trials could delay the trials or detract from the integrity of the trial data and its potential use in future regulatory filings. In addition, manufacturing interruptions or failure to comply with regulatory requirements by any of these manufacturers could significantly delay clinical development of potential products and reduce third-party or clinical researcher interest and support of proposed trials. These interruptions or failures could also impede commercialization of our product candidates and impair our competitive position.
Risks Related to our Indebtedness and Investments
* Our indebtedness could adversely affect our financial condition.
As of June 30, 2019,2020, we had approximately $276 million of total debt outstanding, classified as long term. As a result of our indebtedness, a portion of our cash flow will be required to pay interest and principal on the 2025 Notes if the notes are not converted to shares of common stock prior to maturity. We may not generate sufficient cash flow from operations or have future borrowings available to enable us to repay our indebtedness or to fund other liquidity needs.
Our indebtedness pursuant to the 2025 Notes could have important consequences. For example, it could:
•make it more difficult for us to satisfy our obligations with respect to any other debt we may incur in the future;
•increase our vulnerability to general adverse economic and industry conditions;
•require us to dedicate a substantial portion of our cash flow from operations to payments on our indebtedness and related interest, thereby reducing the availability of our cash flow to fund working capital, capital expenditures and other general corporate purposes;
•limit our flexibility in planning for, or reacting to, changes in our business and the industry in which we operate;
•increase our cost of borrowing;
•place us at a competitive disadvantage compared to our competitors that may have less debt; and
•limit our ability to obtain additional financing for working capital, capital expenditures, acquisitions, debt service requirements or general corporate purposes.
We expect to use cash flow from operations and outside financings to meet our current and future financial obligations, including funding our operations, debt service and capital expenditures. Our ability to make these payments depends on our future performance, which will be affected by financial, business, economic and other factors, many of which we cannot control. Our business may not generate sufficient cash flow from operations in the future, which could result in our being unable to repay indebtedness, or to fund other liquidity needs. If we do not generate sufficient cash from operations, we may be forced to reduce or delay our business activities and capital expenditures, sell assets, obtain additional debt or equity capital or restructure or refinance all or a portion of our debt, including the 2025 Notes, on or before maturity. We cannot make any assurances that we will be able to accomplish any of these alternatives on terms acceptable to us, or at all. In addition, the terms of existing or future indebtedness may limit our ability to pursue any of these alternatives.
We may be unable to raise the funds necessary to repurchase the 2025 Notes for cash following a fundamental change, or to pay any cash amounts due upon conversion, and our future indebtedness may limit our ability to repurchase the 2025 Notes or pay cash upon their conversion.
Noteholders may require us to repurchase their 2025 Notes following a fundamental change at a cash repurchase price generally equal to the principal amount of the 2025 Notes to be repurchased, plus accrued and unpaid interest to, but excluding, the fundamental change repurchase date. In addition, upon conversion, we will satisfy part or all of our conversion obligation in cash unless we elect to settle conversions solely in shares of our common stock.
We may not have enough available cash or be able to obtain financing at the time we are required to repurchase the 2025 Notes or pay the cash amounts due upon conversion of the 2025 Notes. In addition, applicable law, regulatory authorities and the agreements governing our future indebtedness may restrict our ability to repurchase the 2025 Notes or pay the cash amounts due upon conversion of the 2025 Notes. Our failure to repurchase the 2025 Notes or to pay the cash amounts due upon conversion of the 2025 Notes when required will constitute a default under the base and supplemental indentures that will govern the 2025 Notes, which we refer to collectively as the “indenture.” We may not have sufficient funds to satisfy all amounts due under the other indebtedness and the 2025 Notes.
A default under the 2025 Notes may have a material adverse effect on our financial condition.
If an event of default under the 2025 Notes occurs, the principal amount of the 2025 Notes, plus accrued and unpaid interest (including additional interest, if any) may be declared immediately due and payable, subject to certain conditions set forth in the indenture governing such notes. Events of default include, but are not limited to:
•failure to pay (for more than 30 days) interest when due;
•failure to pay principal when due;
•failure to deliver shares of common stock upon conversion of a 2025 Notes;
•failure to provide notice of a fundamental change;
•acceleration on our other indebtedness in excess of $10 million (other than indebtedness that is non-recourse to us); or
•certain types of bankruptcy or insolvency involving us.
Accordingly, the occurrence of a default under the 2025 Notes, unless cured or waived, may have a material adverse effect on our results of operations.
Provisions of the 2025 Notes could discourage an acquisition of us by a third party.
Certain provisions of the 2025 Notes could make it more difficult or more expensive for or prevent a third party to acquire us. Upon the occurrence of certain transactions constituting a fundamental change, holders of the 2025 Notes will have the right, at their option, to require us to repurchase all of their 2025 Notes or any portion of the principal amount of such Notes in integral multiples of $1,000. We may also be required to increase the conversion rate for conversions in connection with certain fundamental changes.
Conversion of the Notes may dilute the ownership interest of existing stockholders, including holders who had previously converted their 2025 Notes.
To the extent we issue shares of common stock upon conversion of the 2025 Notes, the conversion of some or all of the 2025 Notes will dilute the ownership interests of existing stockholders. Any sales in the public market of shares of the common stock issuable upon such conversion could adversely affect prevailing market prices of shares of our common stock. In addition, the existence of the 2025 Notes may encourage short selling by market participants because the conversion of the 2025 Notes could depress the price of shares of our common stock.
Item 2. Unregistered Sales of Equity Securities and Use of Proceeds
None.
Item 3. Defaults Upon Senior Securities
None.
Item 4. Mine Safety Disclosures
Not applicable.
Item 5. Other Information
None.
Item 6. Exhibits
(a) Exhibits
|
| | | | |
| 3.1 | |
| 3.2 | |
| 3.3 | |
| 4.1 | |
| 4.2 | |
| 4.3 | |
| 4.4 | |
| 4.5 | |
| 10.1 | |
| 10.2 | |
10.210.3 | |
10.3 | |
10.4 | |
10.531.1 | |
31.1 | |
| 31.2 | |
| 32.1 | |
| 32.2 | |
| 101.INS | Inline XBRL Instance Document |
| 101.SCH | Inline XBRL Taxonomy Extension Schema Document |
| 101.CAL | Inline XBRL Taxonomy Extension Calculation Linkbase Document |
| 101.DEF | Inline XBRL Taxonomy Extension Definition Linkbase Document |
| 101.LAB | Inline XBRL Taxonomy Extension Label Linkbase Document |
| 101.PRE | Taxonomy Extension Presentation Linkbase Document |
| 104 | The cover page to this Quarterly Report on Form 10-Q has been formatted in Inline XBRL |
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, as amended, the registrant caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| | | | | | | | | | | |
| Date: July 30, 2020 | RETROPHIN, INC. | | |
| | | |
Date: August 6, 2019 | RETROPHIN, INC. |
| By: | | | |
| By: | /s/ Eric M. Dube | |
| | Name: | Eric M. Dube |
| | Title: | Chief Executive Officer |
| | | |
| By: | /s/ Laura Clague | |
| | Name: | Laura Clague |
| | Title: | Chief Financial Officer |