Overview
We are a biopharmaceutical company with peptide-based new chemical entities rusfertide and JNJ-2113 (formerly PN-235) in differentadvanced Phase 3 stages of development, both derived from our proprietary discovery technology platform. Our clinical programs fall into two broad categories of diseases;diseases: (i) hematology and blood disorders, and (ii) inflammatory and immunomodulatory (“I&I”) diseases.
Our Product Pipeline
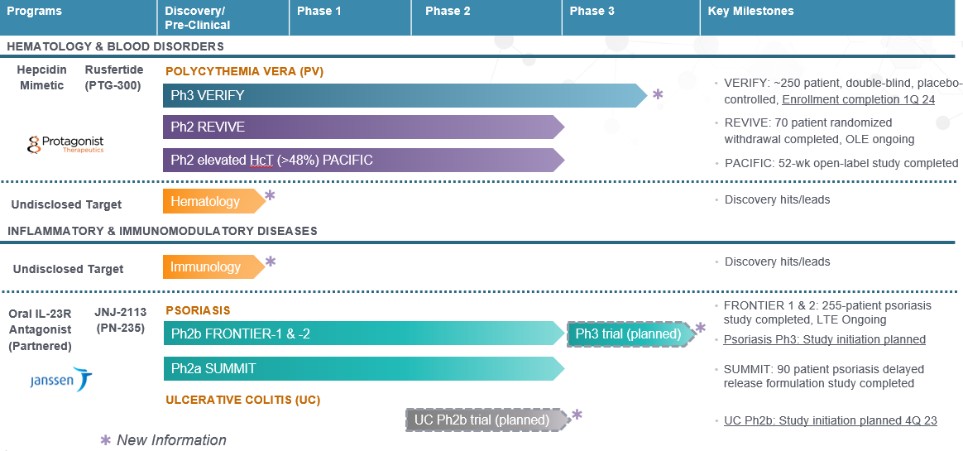
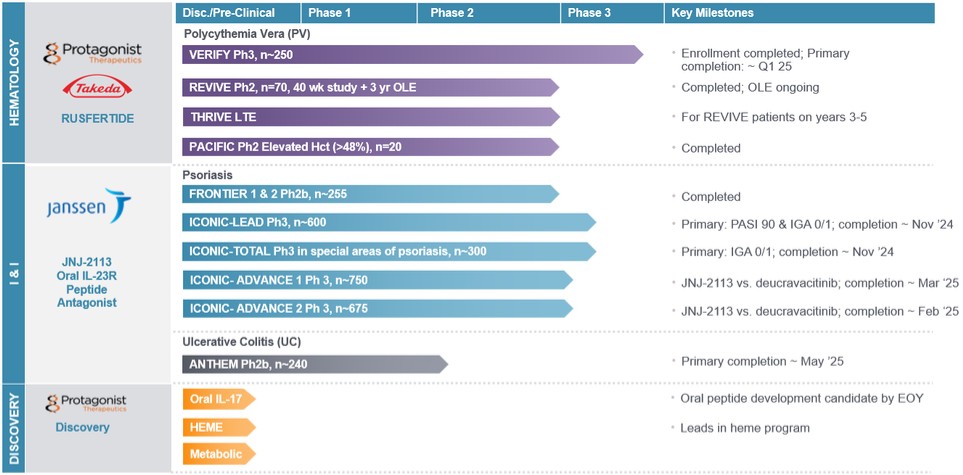
Rusfertide
Our most advanced clinical asset, rusfertide (generic name for PTG-300), is anRusfertide, our injectable hepcidin mimetic partnered with Takeda Pharmaceuticals USA, Inc. (“Takeda”), is in development for the potential treatment of erythrocytosis, iron overloadpolycythemia vera (“PV”). We have initiated VERIFY (ClinicalTrials.gov identifier NCT05210790), a global double-blind, placebo-controlled Phase 3 clinical trial of rusfertide in PV for approximately 250 patients. The trial evaluates the efficacy, symptom burden and other blood disorderssafety of once-weekly, subcutaneously self-administered rusfertide in patients with uncontrolled hematocrit who are phlebotomy dependent despite standard of care treatment. The trial enrolled patients across North and is wholly owned. Hepcidin is a key hormone in regulating iron equilibriumSouth America, Europe, Asia and is criticalAustralia. Enrollment for the VERIFY trial has been completed and we expect to announce top-line data for the proper development of red blood cells. Rusfertide mimicstrial’s 32-week primary efficacy endpoint by the effectend of the natural hormone hepcidin, but with greater potency, solubility and stability. Data fromfirst quarter of 2025, potentially leading to a New Drug Application (“NDA”) filing in the fourth quarter of 2025. By the end of 2024, we expect to receive the results of our rusfertide Phase 2 clinical trials presented at medical conferences in 2021 and 2022 provided evidence regardingongoing two-year study evaluating the carcinogenicity potential of rusfertide for managing hematocrit, reducing thrombotic risk and improving iron deficiency symptoms. Rusfertide has a unique mechanism of action in the potential treatment of the blood disorder polycythemia vera (“PV”), which may enable itwhen administered once weekly to specifically decrease and maintain hematocrit levels within the range of recommended clinical guidelines without causing the iron deficiency that can occur with frequent phlebotomy. rats.
Our rusfertide Phase 2 clinical trials include the following:
| ● | REVIVE, a Phase 2 proof of concept (“POC”) trial, was initiated in the fourth quarter of 2019. We completed enrollment of patients in the first quarter of 2022 and 70 patients were enrolled through the end of the randomized withdrawal portion of the trial, which was completed during the first quarter of 2023 and |
| ● | THRIVE, a Phase 2 long-term extension trial for REVIVE patients on years three through five of treatment; and |
19