UNITED STATES SECURITIES AND EXCHANGE COMMISSION Washington, D.C. 20549
FORM 10-Q
(Mark One) ☒ QUARTERLY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 For the quarterly period ended March 31, 2021
or
☐ TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the transition period from to
Commission File Number: 001-40323
Recursion Pharmaceuticals, Inc.
For the quarterly period ended March 31, 2024 or ☐TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 For the transition period from to Commission File Number: 001-40323
RECURSION PHARMACEUTICALS, INC. (Exact name of registrant as specified in its charter)
| | | | | | | | | | | | | | | | | | | Delaware | | | 46-4099738 | | (State or other jurisdiction of incorporation or organization) | | (I.R.S. (I.R.S. Employer Identification No.)
| 41 S Rio Grande Street Salt Lake City, UT 84101 (Address of principal executive offices) (Zip code) (385) 269 - 0203 (Registrant’s telephone number, including area code) |
41 S Rio Grande Street
Salt Lake City, UT 84101
(Address of principal executive offices) (Zip code)
(385) 269 - 0203
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| | | | | | | | | | Securities registered pursuant to Section 12(b) of the Act: | | Title of each class | Trading symbol(s) | Name of each exchange on which registered | | Class A Common Stock, par value $0.00001 | RXRX | Nasdaq Global Select Market |
| | | Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☐ Nox
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes x No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and "emerging growth company" in Rule 12b-2 of the Exchange Act.
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes x No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and "emerging growth company" in Rule 12b-2 of the Exchange Act. |
| | | | | | | | | | | | | | | | Large accelerated filer | ☐x | | Non-accelerated filer | x☐ | | Accelerated filer | ☐ | | Smaller reporting company | ☐ | | | | Emerging growth company | x☐ |
| | | If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
As of April 30, 2024, there were 230,273,797 and 7,389,871 of the registrant’s Class A and B common stock outstanding, respectively. |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No x
As of April 30, 2021, there were 168,320,745 of the registrant’s Class A and B common stock, par value $0.00001 per share, outstanding.
TABLE OF CONTENTS | | | | | | | | | | | Page | | | | | Item 1. | | | | Item 2. | | | | Item 3. | | | | Item 4. | | | | | | | | | | Item 1. | | | | Item 1A. | | | | Item 2. | | | | Item 5. | | | | Item 6. | | | | | |
SUMMARY RISK FACTORS
Our business is subject to numerous risks and uncertainties that you should consider before investing in our company. These risks are described more fully in the section titled “Risk Factors” in this
Cautionary Note Regarding Forward-Looking Statements This Quarterly Report on Form 10-Q. These risks include, but are not limited to, the following:
•We are a clinical-stage biotechnology company with a limited operating history.
•We have incurred significant operating losses since our inception and anticipate that we will incur continued losses for the foreseeable future.
•Our mission is broad and expensive to achieve and we will need to raise substantial additional funding.
•We have no products approved for commercial sale and have not generated any revenue from product sales.
•We or our current and future collaborators may never successfully develop and commercialize drug products, which would negatively affect our results of operation10-Q contains “forward-looking statements” about us and our ability to continue our business operations.
•Our quarterly and annual operating results may fluctuate significantly in the future due to a variety of factors, many of which are outside of our control and may be difficult to predict.
•Our approach to drug discovery is unique and may not lead to successful drug products, for reasons including but not limited to potential challenges identifying mechanisms of action for our candidates.
•Our drug candidates are in preclinical or clinical development, which are lengthy and expensive processes with uncertain outcomes and the potential for substantial delays.
•Although we intend to explore other therapeutic opportunities, in addition to the drug candidates that we are currently developing, we may fail to identify viable new drug candidates for clinical development for a number of reasons. If we fail to identify additional viable potential drug candidates, our business could be materially harmed.
•Our business and operations would suffer in the event of computer system failures, cyber-attacks, or deficiencies in our cybersecurity or the cybersecurity of third parties, suppliers, or service providers.
•If we are not able to develop new solutions and enhancements to our platform that keep pace with technological developments, our business and results of operations would be harmed.
•Defects or disruptions in our platform could result in diminishing our value and prospects.
•A pandemic, epidemic, or outbreak of an infectious disease, such as COVID-19, or other force majeure events, may materially and adversely affect our business and our financial results and could cause a disruption to the development of our drug candidates.
•If we fail to sufficiently manage and improve our technical hardware infrastructure we may experience errors, delays and other performance problems.
•We are subject to regulatory and operational risks associated with the physical and digital infrastructure at both our internal facilities and those of our external service providers and suppliers.
•We may seek to establish additional collaborations for clinical development or commercialization of our drug candidates, and, if we are not able to establish them on commercially reasonable terms, or at all, we may have to alter our development and commercialization plans.
•If we are unable to adequately protect and enforce our intellectual property and proprietary technology or obtain and maintain patent protection for our technology and products, or if the scope of the patent protection obtained is not sufficiently broad, our competitors could develop and commercialize technology and products similar or identical to ours, and our ability to successfully commercialize our technology and products may be impaired.
•If we are unable to protect the confidentiality of our trade secrets and know-how, our business and competitive position may be harmed.
•If we fail to comply with our obligations in the agreements under which we collaborate with and/or license intellectual property rights from third parties, or otherwise experience disruptions to our business relationships with our collaborators or licensors, we could lose rights that are important to our business.
Special Note Regarding Forward-Looking Information
This report contains “forward-looking statements”industry within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. All statements other than statements of historical facts contained in this Quarterly Report, including statements regarding our future results of operations and financial position, business strategy, development plans, planned preclinical studies, and clinical trials, future results of clinical trials, expected research and development costs, regulatory strategy, timing, and likelihood of success, as well as plans and objectives of management for future operations, are forward-looking statements. In some cases, you can identify forward-looking statements by terms such as “may,” “will,” “should,” “would,” “expect,” “plan,” “anticipate,” “could,” “intend,” “target,” “project,” “contemplate,” “believe,” “estimate,”
“predict, “predict,” “potential,” or “continue” or the negative of these terms or other similar expressions. Forward-looking statements contained in this report may include but are not limited to, statements about:without limitation those regarding:
•our research and development programs and programs; •the initiation, timing, progress, results, and cost of our current and future preclinical and clinical studies, including statements regarding the design of, and the timing of initiation and completion of, studies or trials and related preparatory work, as well as the period during which the results of the trialsstudies will become available; •the ability of our clinical trials to demonstrate the safety and efficacy of our drug candidates, and other positive results; •the ability and willingness of our third-party strategic collaborators to continue research and development activities relating to our development candidates and investigational medicines; •future agreements with third parties in connection with the commercialization of our investigational medicines and any other approved product; •the timing, scope, andor likelihood of regulatory filings and approvals, including the timing of Investigational New Drug applications and final approval by the U.S. Food and Drug Administration, or FDA, of our current drug candidates and any other future drug candidates, includingas well as our ability to maintain any such approvals; •the timing, scope, or likelihood of foreign regulatory filings and approvals, including our ability to maintain any such approvals; •the size of the potential market opportunity for our drug candidates, including our estimates of the number of patients who suffer from the diseases we are targeting;targeting and potential annual sales; •our ability to identify viable new drug candidates for clinical development and the accelerating rate at which we expect to identify such candidates, whether through an inferential approach or otherwise; •our expectation that the assets that will drive the most value for us are those that we will identify in the future using our datasets and tools; •our ability to develop and advance our current drug candidates and programs into, and successfully complete, clinical studies; •our ability to reduce the time or cost or increase the likelihood of success of our research and development relative to the traditional drug discovery paradigm; •our ability to improve, and the rate of improvement in, our infrastructure, datasets, biology, and technology tools and drug discovery platform, orand our ability to realize benefits from such improvements; •our expectations related to the performance and benefits of our BioHive-1 supercomputer;BioHive supercomputer, including our planned expansion of the BioHive supercomputer capabilities; •our ability to realize a return on our investment of resources and cash in our drug discovery collaborations; •our ability to integrate acquired businesses with our existing programs and platform and realize a return on acquired assets; •our ability to leverage datasets acquired through licenses with third parties, including with Tempus, into increased machine learning capabilities, novel genetic associations and mechanisms, innovative therapeutics, or other beneficial outcomes; •our ability to derive value from our Recursion OS by licensing subsets of data and key tools; •the ability to construct and apply more and increasingly sophisticated foundation models and large language models across biology, chemistry and translation and to use these models to drive new, better programs into clinical development both in our own pipeline and with our current and future partners at scale; •our ability to scale like a technology company, including scaling our Recursion OS, and to add more programs to our pipeline each year than in the prior;year; •our ability to successfully compete in a highly competitive market; •our manufacturing, commercialization and marketing capabilities and strategy;strategies; •our plans relating to commercializing our drug candidates, if approved, including the geographic areas of focus and sales strategy; •our expectations regarding the approval and use of our drug candidates in combination with other drugs; •the rate and degree of market acceptance and clinical utility of our current drug candidates, if approved, and other drug candidates we may develop; •our competitive position and the success of competing therapiesapproaches that are or may become available;
•our estimates of the number of patients that we will enroll in our clinical trials and the timing of their enrollment; •the beneficial characteristics, safety, efficacy and therapeutic effects of our drug candidates; •our plans relating to thefor further development of our drug candidates, including additional indications we may pursue; •our ability to adequately protect and enforce our intellectual property and proprietary technology, including the scope of protection we are able to establish and maintain for intellectual property rights covering our current drug candidates and other drug candidates we may develop, obtainingreceipt of patent protection, the extensions of existing patent terms where available, the validity of intellectual property rights held by third parties, the protection of our trade secrets, and our ability not to infringe, misappropriate or otherwise violate any third-party intellectual property rights; •the impact of any current or future intellectual property litigationdisputes and our ability to defend against claims of infringement, misappropriation, or other violations of any third-party intellectual property rights; •our ability to keep pace with new technological developments; •our ability to utilize third-party open source software and cloud-based infrastructure, on which we are dependent; •the adequacy of our insurance policies and the scope of their coverage;
•the potential impact of a pandemic, epidemic, or outbreak of an infectious disease, such as COVID-19, or natural disaster, global political instability or warfare, and the effect of such outbreak or natural disaster, global political instability or warfare on our business and financial results; •our ability to maintain our technical operations infrastructure to avoid errors, delays, or cybersecurity breaches; •our continued reliance on third parties to conduct additional clinical trials of our drug candidates, and for the manufacture of our drug candidates for preclinical studies and clinical trials; •our ability to obtain and negotiate favorable terms of, any collaboration, licensing, or other arrangements that may be necessary or desirable to research, develop, manufacture, or commercialize our platform and drug candidates; •the pricing and reimbursement of our current drug candidates and other drug candidates we may develop, if approved; •our estimates regarding expenses, future revenue, capital requirements and needsneed for additional financingfinancing; •our financial performance; •the period over which we estimate our existing cash and cash equivalents will be sufficient to fund our future operating expenses and capital expenditure requirements; •our ability to raise substantial additional funding; •the impact of current and future laws and regulations, and our ability to comply with all regulations that we are, or may become, subject to; •the need to hire additional personnel and our ability to attract and retain such personnel; •the impact of any current or future litigation, which may arise during the ordinary course of business and be costly to defend; •the need to raise additional capital may cause dilution to our expectations regarding the period during which we will qualify as an emerging growth company under the JOBS Act;stockholders, restrict our operations, require us to relinquish rights to our technologies or drug candidates, and divert management’s attention from our core business; •our anticipated use of our existing resources and the net proceeds from thisour initial public offering; and •other risks and uncertainties, including those listed underin the captionsection titled “Risk Factors.”
We have based these forward-looking statements largely on our current expectations and projections about our business, the industry in which we operate, and financial trends that we believe may affect our business, financial condition, results of operations and prospects, and theseprospects. These forward-looking statements are not guarantees of future performance or development. These forward-looking statements speak only as of the date of this report and are subject to a number of risks, uncertainties and assumptions described in the section titled “Risk Factors” and elsewhere in this report. Because forward-looking statements are inherently subject to risks and uncertainties, some of which cannot be predicted or quantified, you should not rely on these forward-looking statements as predictions of future events. The events and circumstances reflected in our forward-looking statements may not be achieved or occur and actual results could differ materially from those projected in the forward-looking statements. Except as required by applicable law, we do not planundertake no obligation to publicly update or revise any forward-looking statements contained herein, until after we distribute this report, whether as a result of any new information, future events, or otherwise.
In addition, statements that “we believe” and similar statements reflect our beliefs and opinions on the relevant subject. These statements are based upon information available to us as of the date of this report, and whilereport. While we believe such information forms a reasonable basis for such statements, suchthe information may be limited or incomplete, and our statements should not be read to indicate that we have conducted an exhaustive inquiry into, or review of, all
potentially available relevant information. These statements are inherently uncertain and you are cautioned not to unduly rely upon them.
PART I - FINANCIAL INFORMATION
Item 1. Financial Statements.
Recursion Pharmaceuticals, Inc. Condensed Consolidated Balance Sheets (unaudited) (in thousands, except share and per share amounts) | | | | March 31, | December 31, | | March 31, | December 31, | | | | 2021 | 2020 | | 2024 | 2023 | | Assets | Assets | | Assets | | | Current assets | Current assets | | Current assets | | | Cash and cash equivalents | Cash and cash equivalents | $ | 214,088 | | $ | 262,126 | | | Restricted cash | Restricted cash | 5,042 | | 5,041 | | | Accounts receivable | 71 | | 156 | | | Other receivables | | | Other current assets | Other current assets | 2,621 | | 2,155 | | | Total current assets | Total current assets | 221,822 | | 269,478 | | | | Restricted cash, non-current | | | Restricted cash, non-current | | | Restricted cash, non-current | | | Property and equipment, net | Property and equipment, net | 44,642 | | 25,967 | | | Operating lease right-of-use assets | | | Intangible assets, net | Intangible assets, net | 2,414 | | 2,490 | | | Other non-current assets | 3,065 | | 650 | | | Goodwill | | | Other assets, non-current | | | Total assets | Total assets | $ | 271,943 | | $ | 298,585 | | | | Liabilities, convertible preferred stock and stockholders’ deficit | | | Liabilities and stockholders’ equity | | | Liabilities and stockholders’ equity | | | Liabilities and stockholders’ equity | | | Current liabilities | | | Current liabilities | | | Current liabilities | Current liabilities | | | Accounts payable | Accounts payable | $ | 3,125 | | $ | 1,074 | | | Accounts payable | | | Accounts payable | | | Accrued expenses and other liabilities | Accrued expenses and other liabilities | 11,085 | | 10,485 | | | Current portion of unearned revenue | 10,000 | | 10,000 | | | Current portion of notes payable | 2,145 | | 1,073 | | | Current portion of lease incentive obligation | 499 | | 467 | | | Unearned revenue | | | Notes payable | | | Operating lease liabilities | | | Total current liabilities | Total current liabilities | 26,854 | | 23,099 | | | | Deferred rent | 2,750 | | 2,674 | | | Unearned revenue, net of current portion | 14,167 | | 16,667 | | | Notes payable, net of current portion | 10,339 | | 11,414 | | | Lease incentive obligation, net of current portion | 2,552 | | 2,708 | | | Unearned revenue, non-current | | | Unearned revenue, non-current | | | Unearned revenue, non-current | | | Notes payable, non-current | | | Operating lease liabilities, non-current | | | Deferred tax liabilities | | | Total liabilities | Total liabilities | 56,662 | | 56,562 | | | Commitments and contingencies (Note 6) | 0 | | Convertible preferred stock (series A, A-1, B, C, and D), $0.00001 par value; 121,434,713 shares authorized as of March 31, 2021 and December 31, 2020; 112,088,065 shares issued and outstanding as of March 31, 2021 and December 31, 2020; Liquidation preference of $450,850 as of March 31, 2021 and December 31, 2020 | 448,312 | | 448,312 | | | Stockholders’ deficit | | Common stock, $0.00001 par value; 188,400,000 shares authorized as of March 31, 2021 and December 31, 2020; 24,036,725 and 22,314,685 shares issued and outstanding as of March 31, 2021 and December 31, 2020, respectively | 0 | | 0 | | | Commitments and contingencies (Note 7) | | | Commitments and contingencies (Note 7) | | | Commitments and contingencies (Note 7) | | | | | Stockholders’ equity | | | Stockholders’ equity | | | Stockholders’ equity | | | Common stock, $0.00001 par value; 2,000,000,000 shares (Class A 1,989,032,117 and Class B 10,967,883) authorized as of March 31, 2024 and December 31, 2023; 237,508,682 shares (Class A 230,043,061, Class B 7,464,871 and Exchangeable 750) and 234,270,384 shares (Class A 226,264,764, Class B 7,544,871 and Exchangeable 460,749) issued and outstanding as of March 31, 2024 and December 31, 2023, respectively | | | Common stock, $0.00001 par value; 2,000,000,000 shares (Class A 1,989,032,117 and Class B 10,967,883) authorized as of March 31, 2024 and December 31, 2023; 237,508,682 shares (Class A 230,043,061, Class B 7,464,871 and Exchangeable 750) and 234,270,384 shares (Class A 226,264,764, Class B 7,544,871 and Exchangeable 460,749) issued and outstanding as of March 31, 2024 and December 31, 2023, respectively | | | Common stock, $0.00001 par value; 2,000,000,000 shares (Class A 1,989,032,117 and Class B 10,967,883) authorized as of March 31, 2024 and December 31, 2023; 237,508,682 shares (Class A 230,043,061, Class B 7,464,871 and Exchangeable 750) and 234,270,384 shares (Class A 226,264,764, Class B 7,544,871 and Exchangeable 460,749) issued and outstanding as of March 31, 2024 and December 31, 2023, respectively | | | Additional paid-in capital | Additional paid-in capital | 11,287 | | 7,312 | | | Accumulated deficit | Accumulated deficit | (244,318) | | (213,601) | | | Total stockholders’ deficit | (233,031) | | (206,289) | | | Total stockholders’ equity | | | | Total liabilities, convertible preferred stock and stockholders’ deficit | $ | 271,943 | | $ | 298,585 | | | Total liabilities and stockholders’ equity | | | Total liabilities and stockholders’ equity | | | Total liabilities and stockholders’ equity | |
See the accompanying notes to these condensed condensed consolidated financial statements.
Recursion Pharmaceuticals, Inc. Condensed Consolidated Statements of Operations and Comprehensive Loss (unaudited) (in thousands, except share and per share amounts)
| | Three months ended | | March 31, | | 2021 | 2020 | | Three months ended
March 31, | | | Three months ended
March 31, | | 2024 | | | 2024 | 2023 | | Revenue | Revenue | | | Operating revenue | | | Operating revenue | | | Operating revenue | | | Grant revenue | Grant revenue | $ | 62 | | $ | 60 | | | Operating revenue | 2,500 | | 0 | | | Total revenue | Total revenue | 2,562 | | 60 | | | | Operating expenses | | | Operating costs and expenses | | | Operating costs and expenses | | | Operating costs and expenses | | | Cost of revenue | | | Cost of revenue | | | Cost of revenue | | | Research and development | Research and development | 24,109 | | 12,842 | | | General and administrative | General and administrative | 8,937 | | 5,561 | | | Total operating expenses | 33,046 | | 18,403 | | | Total operating costs and expenses | | | | Loss from operations | Loss from operations | (30,484) | | (18,343) | | | Other loss, net | (233) | | (81) | | | Loss from operations | | | Loss from operations | | | Other income, net | | | Loss before income tax benefit | | | Income tax benefit | | | Net loss and comprehensive loss | Net loss and comprehensive loss | $ | (30,717) | | $ | (18,424) | | | | Per share data | Per share data | | | Net loss per share, basic and diluted | $ | (1.33) | | $ | (0.85) | | | Weighted average shares of common stock, basic and diluted | 23,035,623 | | 21,639,891 | | | Per share data | | | Per share data | | | Net loss per share of Class A, B and Exchangeable common stock, basic and diluted | | | Net loss per share of Class A, B and Exchangeable common stock, basic and diluted | | | Net loss per share of Class A, B and Exchangeable common stock, basic and diluted | | | Weighted-average shares (Class A, B and Exchangeable) outstanding, basic and diluted | |
See the accompanying notes to these condensed consolidated financial statements.
Recursion Pharmaceuticals, Inc. Condensed Consolidated Statements of Stockholders’ Equity (unaudited) (in thousands, except share amounts)
| | | | | | | | | | | | | | | | | | | Common Stock | Additional Paid-in-Capital | Accumulated Deficit | Stockholders’ Equity | | (Class A, B and Exchangeable) | | Shares | Amount | | Balance as of December 31, 2023 | 234,270,384 | | $ | 2 | | $ | 1,431,056 | | $ | (967,622) | | $ | 463,436 | | | Comprehensive loss | — | | — | | — | | (91,373) | | (91,373) | | | Stock option exercises and other | 2,317,083 | | — | | 2,088 | | — | | 2,088 | | | Stock-based compensation | — | | — | | 16,127 | | — | | 16,127 | | | Common stock sales issuances, net of issuance costs | 921,215 | | — | | 10,873 | | — | | 10,873 | | | Balance as of March 31, 2024 | 237,508,682 | | $ | 2 | | $ | 1,460,144 | | $ | (1,058,995) | | $ | 401,151 | |
| | | | | | | | | | | | | | | | | | | Common Stock | Additional Paid-in-Capital | Accumulated Deficit | Stockholders’ Equity | | (Class A, B and Exchangeable) | | Shares | Amount | | Balance as of December 31, 2022 | 191,022,864 | | $ | 2 | | $ | 1,125,360 | | $ | (639,556) | | $ | 485,806 | | | Comprehensive loss | — | | — | | — | | (65,327) | | (65,327) | | | Stock option exercises and other | 1,207,990 | | — | | 882 | | — | | 882 | | | Stock-based compensation | — | | — | | 8,814 | | — | | 8,814 | | | Balance as of March 31, 2023 | 192,230,854 | | $ | 2 | | $ | 1,135,056 | | $ | (704,883) | | $ | 430,175 | |
See the accompanying notes to these condensed consolidated financial statements.
Recursion Pharmaceuticals, Inc. Condensed Consolidated Statements of Convertible Preferred Stock and Stockholders’ DeficitCash Flows (unaudited) (in thousands, except share amounts) | | | | | | | | | | | | | | | | | | | | | | | | | Convertible Preferred Stock | Common Stock | Additional Paid-in-Capital | Accumulated Deficit | Stockholders’ Deficit | | Shares | Amount | Shares | Amount | | Balance as of December 31, 2020 | 112,088,065 | | $ | 448,312 | | 22,314,685 | | $ | 0 | | $ | 7,312 | | $ | (213,601) | | $ | (206,289) | | | Net loss | — | | — | | — | | — | | — | | (30,717) | | (30,717) | | | Stock option exercises and other | — | | — | | 1,722,040 | | — | | 2,154 | | — | | 2,154 | | | Stock-based compensation | — | | — | | — | | — | | 1,821 | | — | | 1,821 | | | Balance as of March 31, 2021 | 112,088,065 | | $ | 448,312 | | 24,036,725 | | $ | 0 | | $ | 11,287 | | $ | (244,318) | | $ | (233,031) | |
| | | | | | | | | | | | | | | | | | | | | | | | | Convertible Preferred Stock | Common Stock | Additional Paid-in-Capital | Accumulated Deficit | Stockholders’ Deficit | | Shares | Amount | Shares | Amount | | Balance as of December 31, 2019 | 75,189,517 | | $ | 201,109 | | 21,637,609 | | $ | 0 | | $ | 2,330 | | $ | (126,595) | | $ | (124,265) | | | Net loss | — | | — | | — | | — | | — | | (18,424) | | (18,424) | | | Stock option exercises | — | | — | | 14,668 | | — | | 16 | | — | | 16 | | | Stock-based compensation | — | | — | | — | | — | | 1,286 | | — | | 1,286 | | | Balance as of March 31, 2020 | 75,189,517 | | $ | 201,109 | | 21,652,277 | | $ | 0 | | $ | 3,632 | | $ | (145,019) | | $ | (141,387) | |
| | | | | | | | | | Three months ended
March 31, | | | 2024 | 2023 | | Cash flows from operating activities | | | | Net loss | $ | (91,373) | | $ | (65,327) | | | Adjustments to reconcile net loss to net cash used in operating activities: | | | | Depreciation and amortization | 7,377 | | 3,728 | | | Stock-based compensation | 16,127 | | 8,814 | | | Asset impairment | 108 | | 1,169 | | | Lease expense | 2,472 | | 1,988 | | | Other, net | (560) | | 716 | | | Changes in operating assets and liabilities: | | | | Other receivables and assets | (1,040) | | (317) | | | Unearned revenue | (13,655) | | (12,134) | | | Accounts payable | 1,168 | | (339) | | | Accrued development expense | (273) | | 676 | | | Accrued expenses and other current liabilities | (18,857) | | (9,846) | | | Operating lease liabilities | (3,794) | | (2,444) | | | Net cash used in operating activities | (102,300) | | (73,316) | | | | | | Cash flows from investing activities | | | | Purchases of property and equipment | (6,653) | | (5,175) | | | Purchase of an intangible asset | — | | (165) | | | Net cash used in investing activities | (6,653) | | (5,340) | | | | | | Cash flows from financing activities | | | | Proceeds from issuance of common shares, net of issuance costs | 10,873 | | — | | | Proceeds from equity incentive plans | 3,050 | | 1,946 | | | Repayment of long-term debt | (26) | | (24) | | | Net cash provided by financing activities | 13,897 | | 1,922 | | | | | | Effect of exchange rate changes on cash, cash equivalents and restricted cash | (219) | | (2) | | | | | | Net change in cash, cash equivalents and restricted cash | (95,275) | | (76,734) | | | Cash, cash equivalents and restricted cash, beginning of period | 401,425 | | 559,112 | | | Cash, cash equivalents and restricted cash, end of period | $ | 306,150 | | $ | 482,378 | | | | | | Supplemental schedule of non-cash investing and financing activities | | | | Accrued property and equipment | $ | 80 | | $ | 244 | | | Right-of-use asset additions and modifications | 3,266 | | 3,520 | | | Financed equipment purchase | — | | 1,214 | | | | | | Supplemental schedule of cash flow information | | | | Cash paid for operating leases | $ | 3,955 | | $ | 2,444 | |
See the accompanying notes to these condensed consolidated financial statements.
Recursion Pharmaceuticals, Inc.
Condensed Consolidated Statements of Cash Flows (unaudited)
(in thousands)
| | | | | | | | | | Three months ended | | March 31, | | | 2021 | 2020 | | Cash flows from operating activities | | | | Net loss | $ | (30,717) | | $ | (18,424) | | | Adjustments to reconcile net loss to net cash used in operating activities: | | | | Depreciation and amortization | 1,402 | | 937 | | | Stock-based compensation | 1,821 | | 1,265 | | | Other, net | 101 | | 22 | | | Changes in operating assets and liabilities: | | | | Accounts receivable | 85 | | 82 | | | Other assets | (2,915) | | (132) | | | Unearned revenue | (2,500) | | 0 | | | Accounts payable | 2,051 | | 625 | | | Accrued development expense | (1,216) | | (400) | | | Accrued expenses, deferred rent and other current liabilities | 1,133 | | (1,792) | | | Net cash used in operating activities | (30,755) | | (17,817) | | | | | | Cash flows from investing activities | | | | Purchases of property and equipment | (19,416) | | (684) | | | Net cash used in investing activities | (19,416) | | (684) | | | | | | Cash flows from financing activities | | | | Proceeds from exercise of stock options | 2,154 | | 16 | | | Repayment of long-term debt | (20) | | (19) | | | Proceeds from convertible notes | 0 | | 6,000 | | | Net cash provided by financing activities | 2,134 | | 5,997 | | | | | | Net change in cash, cash equivalents and restricted cash | (48,037) | | (12,504) | | | Cash, cash equivalents and restricted cash, beginning of period | 267,167 | | 75,171 | | | Cash, cash equivalents and restricted cash, end of period | $ | 219,130 | | $ | 62,667 | | | | | | Supplemental disclosure of non—cash investing and financing information | | | | Accrued property and equipment | $ | 705 | | 0 | | | Deferred issuance costs | 2,997 | | 0 | | | | | | Supplemental disclosure of cash flow information | | | | Cash paid for interest | $ | 254 | | $ | 306 | |
See the accompanying notes to these condensed consolidated financial statements.
Recursion Pharmaceuticals, Inc. Notes to Condensed Consolidated Financial Statements (unaudited) Note 1. Description of the Business
Recursion Pharmaceuticals, Inc. (Recursion, the Company, we or we)our) was originally formed as a limited liability company on November 4, 2013 under the name Recursion Pharmaceutical,Pharmaceuticals, LLC. In September 2016, wethe Company converted to a Delaware corporation and subsequently changed ourits name to Recursion Pharmaceuticals, Inc.
Recursion is a biotechnologyclinical stage TechBio company that combines automation, artificial intelligence, machine learning, in vivo validation capabilitiesdecoding biology to industrialize drug discovery. The Recursion Operating System (OS), a platform built across diverse technologies, enables the Company to map and a highly cross-functional team to discover novel medicines that expand our collective understanding of biology. Recursion’s rich, relatable databasenavigate trillions of biological images generated in-house onand chemical relationships within the Company’s robotics platform enables advanced machine learning approaches to reveal drug candidates, mechanismsRecursion Data Universe, one of action, novel chemistry,the world’s largest proprietary biological and potential toxicity, with the eventual goalchemical datasets. The Company integrates physical and digital components as iterative loops of decodingatoms and bits scaling wet lab biology and advancing new therapeutics that radically improve people’s lives.chemistry data organized into virtuous cycles with computational tools to rapidly translate in silico hypotheses into validated insights and novel chemistry.
As of March 31, 2021,2024, the Company had an accumulated deficit of $244.3 million.$1.1 billion. The Company expects to incur substantial operating losses in future periods and will require additional capital to advance its drug candidates. The Company does not expect to generate significant revenue until the Company successfully completes significant drug development milestones with its subsidiaries or in collaboration with third parties, which the Company expects will take a number of years. In order to commercialize its drug candidates, the Company or its partners need to complete clinical development and comply with comprehensive regulatory requirements. The Company is subject to a number of risks and uncertainties similar to those of other companies of the same size within the biotechnology industry, such as the uncertainty of clinical trial outcomes, uncertainty of additional funding and a history of operating losses.
The Company has funded its operations to date primarily through the issuance of convertible preferred stock and the issuance of Class A common stock (see Note 7, “Convertible Preferred8, “Common Stock” for additional information) anddetails). Additionally, the Company has received payments from its strategic partnerships (see Note 9, “Collaborative Development Contracts” for additional details). Recursion will likely be required to raise additional capital. As of March 31, 2021,2024, the Company did not have any unconditional outstanding commitments for additional funding. If the Company is unable to access additional funds when needed, it may not be able to continue the development of its products or the Company could be required to delay, scale back or abandon some or all of its development programs and other operations. The Company’s ability to access capital when needed is not assured and, if not achieved on a timely basis, could materially harm its business, financial condition and results of operations.
In April 2021, the Company completed an Initial Public Offering (IPO). See Note 16, “Subsequent Events” for additional details.
The CompanyRecursion believes that the net proceeds from the IPO, together with the Company’s existing cash and cash equivalents and borrowings available to us will be sufficient to fund the Company’s operating expenses and capital expenditures for at least the next 12 months.
Note 2. Basis of Presentation
Basis of Presentation
The unaudited interim condensed consolidated financial statements of Recursion have been prepared pursuant to the rules and regulations of the U.S. Securities and Exchange Commission (SEC). Accordingly, certain information and footnote disclosures normally included in annual financial statements prepared in accordance with generally accepted accounting principles in the United States (U.S. GAAP) have been condensed or omitted. These unaudited interim condensed consolidated financial statements should be read in conjunction with the Company’s audited consolidated financial statements and notes for the year ended December 31, 2020 included in the Company’s final prospectus dated as of April 15, 2021 and filed with the Securities and Exchange Commission (SEC) pursuant to Rule 424(b)(4) on April 16, 2020.2023.
In April 2021, the Company completed a 1.5-for-1 forward stock split of common and convertible preferred stock. All shares presented within these condensed consolidated financial statements were adjusted to reflect the forward stock split for all periods presented. See Note 16, “Subsequent Events” for additional details.
It is management’s opinion that these condensed consolidated financial statements include all normal and recurring adjustments necessary for a fair presentation of the Company’s financial position, operating results and cash flows. Revenuesstatements. Revenue and net loss for any interim period are not necessarily indicative of future or annual results. Certain reclassifications were made to conform the prior period interim condensed consolidated financial statements to the current period presentation.
Emerging Growth Company
The Company is an emerging growth company (EGC), as defined by the Jumpstart Our Business Startups Act of 2012 (the JOBS act). The JOBS Act, exempts EGCs from being required to comply with new or revised financial accounting standards until private companies are required to comply. Recursion has elected to use the extended transition period for new or revised financial accounting standards. However, the Company may adopt certain new or revised accounting standards early. This may make comparisons of the Company’s financial statements with other public companies difficult because of the potential differences in accounting standards used.
Recursion may remain an EGC until December 31, 2026 although if we: (1) become a “large accelerated filer;” (2) have annual gross revenues of $1.07 billion or more in any fiscal year; or (3) issue more than $1.0 billion of non-convertible debt over a three-year period, the Company would cease to be an EGC as of December 31 of the applicable year.
Recent Accounting Pronouncements
In February 2016,March 2024, the Financial Accounting Standards Board (FASB)SEC issued ASU 2016-02, Leases -Topic 842 (ASU 2016-02)rule 33-11275, The Enhancement and Standardization of Climate-Related Disclosures for Investors. Under Topic 842,The new rule requires Recursion to provide certain disclosures in the footnotes to the
financial statements of climate-related information. These disclosures include the impact of severe weather and other natural conditions on the Company’s consolidated balance sheet and statement of operations, to the extent they are material. Recursion will also need to disclose a rollforward of the beginning and ending balances of its carbon offsets and renewable energy credits or certificates (RECs), if they are a material component of meeting the Company’s climate-related targets and goals. Additionally, the Company will need to disclose whether and, if so, how severe weather events and other natural conditions and disclosed climate-related targets or transition plans materially affected estimates and assumptions in the financial statements.
The standard’s effective dates, if adopted, will be required to recognize a lease liability and a right-of-use asset for all leases (withphased in depending on the exception of short-term leases) atdisclosure requirement starting the commencement date of each lease. ASU 2016-02 is effective for annual and interim periods beginning on or afterperiod ending December 15, 2021 and early adoption is permitted. The Company must adopt31, 2025. In April 2024, the standard using the modified retrospective approach either: (1) asSEC voluntarily stayed implementation of the earliest period presented and through the comparative periods in the entity’s financial statements or (2) as of the effective date of ASC 842, with a cumulative-effect adjustment to equity. The Company expects the adoption to materially increase assets and liabilities on the Condensed Consolidated Balance Sheets related to those leases classified as operating and not recognized on the Balance Sheets under current GAAP.new climate-related disclosure requirements pending judicial review. The Company is continuing to evaluatecurrently evaluating the effect that ASU 2016-02impact this rule will have on its consolidated financial statements and related disclosures.
In December 2023, the Financial Accounting Standards Board (FASB) issued Accounting Standards Update (ASU) No. 2023-9, Income Taxes (Topic 740). The Company will adopt the new standard updates disclosure requirements for Accounting Standards Codification (ASC) 740 primarily by requiring additional information in the income tax rate reconciliation and additional disclosures about income taxes paid. This standard will be effective for Recursion starting the annual period ending December 31, 2025. Early adoption is permitted for annual financial statements that have not yet been issued. The amendments can be applied on January 1, 2022.a prospective or retrospective basis. The adoption of this standard will not impact Recursion’s consolidated balance sheet and statement of operations.
In November 2023, the FASB issued ASU No. 2023-7, Segment Reporting (Topic 280). The standard requires new disclosures related to ASC 280 including: disclosing significant segment expenses by category; requiring all the ASC 280 disclosures for Companies with a single reportable segment and; requiring an increased frequency of the ASC 280 disclosures. Recursion must apply the amendments retrospectively to each prior reporting period presented. This standard will be effective for Recursion starting the annual period ending December 31, 2024. Early adoption is permitted. The adoption of this standard will not impact Recursion’s consolidated balance sheet and statement of operations.
Note 3. Supplemental Financial Information
Tempus agreement
In November 2023, Recursion entered into a five-year agreement with Tempus Labs, Inc. (Tempus) to purchase access to their records of patient-centric multimodal oncology data and use rights for therapeutic development purposes. This data will be used to improve the training of Recursion’s artificial intelligence and machine learning models and is expected to accelerate Recursion’s drug discovery process. Recursion is making annual payments, ranging between $22.0 million and $42.0 million, up to $160.0 million in aggregate, to Tempus in cash or equity at the Company’s option. The equity value is determined by using the seven-trading day period dollar volume-weighted average price (VWAP) for Recursion Class A common stock ending on the day immediately preceding the date that is five business days prior to the payment date.
Recursion is expensing the record purchases as “Research and Development” expenses in the Condensed Consolidated Statements of Operations as the records are purchased. To the extent that the Recursion payments to Tempus are greater than or less than the records purchased amount, Recursion records the applicable amount to “Other Current Assets” or “Accrued Expenses and Other Liabilities” on the Condensed Consolidated Balance Sheet, respectively. As of March 31, 2024, Recursion had recorded $16.0 million within “Other Current Assets” on the Consolidated Balance Sheet related to the Tempus agreement.
Property and Equipment
| | March 31, | December 31, | | March 31, | | | March 31, | December 31, | | (in thousands) | (in thousands) | 2021 | 2020 | (in thousands) | 2024 | 2023 | | Lab equipment | Lab equipment | $ | 21,234 | | $ | 19,701 | | | Leasehold improvements | Leasehold improvements | 13,792 | | 13,792 | | | Office equipment | Office equipment | 18,994 | | 1,075 | | | Construction in progress | Construction in progress | 2,016 | | 1,361 | | | Property and equipment, gross | Property and equipment, gross | 56,036 | | 35,929 | | | Less: Accumulated depreciation | Less: Accumulated depreciation | (11,394) | | (9,962) | | | Property and equipment, net | Property and equipment, net | $ | 44,642 | | $ | 25,967 | |
Depreciation expense on property and equipment was $1.4$4.0 million and $1.0$3.6 million during the three months ended March 31, 20212024 and 2020, respectively. 2023, respectively
For$1.2 million during the three months ended March 31, 20212024 and 2023, respectively, related to construction projects for leasehold improvements as the Company purchasedno longer intended to use them. The impairment was recorded in “General and Administrative” in the Condensed Consolidated Statements of Operations.
For the three months ended March 31, 2023, the Company initiated and completed a Dell EMCproject to upgrade the BioHive supercomputer accessories and parts for $17.9 million.$1.7 million. The purchasesupercomputer was classified as office equipment in the above table.
Accrued Expenses and Other Liabilities
| | March 31, | December 31, | | March 31, | | | March 31, | December 31, | | (in thousands) | (in thousands) | 2021 | 2020 | (in thousands) | 2024 | 2023 | | Accrued compensation | Accrued compensation | $ | 1,800 | | $ | 3,085 | | | Accrued development expenses | Accrued development expenses | 1,073 | | 2,289 | | | Accrued administrative expenses | 2,487 | | 10 | | | Accrued early discovery expenses | | | Accrued construction | | | Materials received not invoiced | | | Accrued other expenses | Accrued other expenses | 5,725 | | 5,101 | | | Accrued expense and other liabilities | Accrued expense and other liabilities | $ | 11,085 | | $ | 10,485 | |
Interest Expense, net
| | | | | | | | | | Three months ended March 31, | | (in thousands) | 2021 | 2020 | | Interest expense | $ | 249 | | $ | 301 | | | Interest income | (16) | | (220) | | | Interest expense, net | $ | 233 | | $ | 81 | |
Interest expense primarily relates to the Midcap and tenant improvement allowance notes (see Note 5, “Notes Payable” for additional details on the notes.) Interest expense is included in Other loss, net on the Condensed Consolidated Statements of Operations and Comprehensive Loss.
The Company capitalizes certain legal, professional, accounting and other third-party fees that are directly associated with debt and equity financings. There was $3.0 million of capitalized issuance costs on the Condensed Consolidated Balance Sheet on March 31, 2021 related to the planned IPO. See Note 16, “Subsequent Events” for additional information on the IPO.
Note 4. Goodwill and Intangible Assets
Goodwill
The carrying amount of Goodwill was $801 thousand as of March 31, 2021. There were no changes to the carrying amount of goodwill during the three months ended March 31, 2021. There was no Goodwill balance outstanding during the three months ended March 31, 2020. As of March 31, 2021, there were no reductions in Goodwill relating to impairment losses.
Intangible Assets, Net
The following table summarizes intangible assets:
| | | | | | | | | | | | | | | | | | | | | | March 31, 2021 | December 31, 2020 | | (in thousands) | Gross carrying amount | Accumulated Amortization | Net carrying amount | Gross carrying amount | Accumulated Amortization | Net carrying amount | | Definite-lived intangible asset | $ | 911 | | $ | (202) | | $ | 709 | | $ | 911 | | $ | (127) | | $ | 784 | | | Indefinite-lived intangible asset | 904 | | — | | 904 | | 904 | | — | | 904 | |
Amortization expense was $76 thousand during the three months ended March 31, 2021. There was 0 amortization expense during the three months ended March 31, 2020. Amortization expense was included in research and development in the Condensed Consolidated Statements of Operations and Comprehensive Loss. No definite-lived intangible asset impairment charges were recorded during the three months ended March 31, 2021. There were no intangible asset balances outstanding during the three months ended March 31, 2020.
The indefinite-lived intangible asset represents the Recursion domain name that the Company purchased. No indefinite-lived intangible asset impairment charges were recorded during the three months ended March 31, 2021.
Note 5. Notes Payable
Midcap Financial
In September 2019,January 2023, the Company entered into a new Credit and Security Agreement with Midcap Financial Trust (Midcap), and the other lenders party thereto (the Midcap loan agreement). The Midcap loanfinancing agreement provides for a term loan facility that includes: i) an initial tranche of $11.9 million; and ii) a second tranche of up to $15.0borrowing $1.9 million which if drawn would result in a maximum outstanding amount of $26.9 million. The Company used a portionas part of the proceeds from the initial tranche to fully repaysupercomputer upgrade project. The debt will be repaid over a previously outstanding term loan, the Pacific Western Bank (Pacific) loan, for $11.2 million. Proceeds from the term loans may be used for general corporate purposes.three-year period at a 7% interest rate. As of March 31, 2021 and December 31, 2020,2024, the outstanding principal balance under the Midcap loan agreement was $11.9 million.
Interest on the Midcap loan accrues on the principal amount outstanding at a floating per annum rate equal to the LIBOR rate (floor of 2.00%) plus 5.75% and is payable monthly in arrears. The Company is required to make interest-only payments from September 2019 to September 2021, and thereafter, 36 monthly principal payments of $330 thousand plus interest. The interest only period will be extended an additional 12 months under certain conditions.
The Company may voluntarily prepay the Midcap term loan, subject to certain minimum repayment requirements and prepayment fees. The Midcap term loan is subject to a mandatory prepayment under certain conditions.
The debt is secured against substantially all of the Company assets. The Midcap loan agreement includes standard affirmative and restrictive covenants, including covenants limiting the ability of the Company and its subsidiaries, among other things, to dispose of assets, grant certain licenses, make investments, merger or consummate acquisitions, incur debt, grant liens and make dividends or distributions, in each case subject to certain exceptions. The loan agreement also includes standard events of default, including, subject to grace periods in certain instances, payment defaults, breaches of covenants, breaches of representations and warranties, cross-defaults with certain other indebtedness, insolvency and bankruptcy defaults, change of control of the Company or any subsidiary, and a material adverse change in the business, operations or conditions of the Company. Upon the occurrence of an event of default, Midcap may declare all outstanding obligations immediately due and payable, increase the applicable interest rate by 2% and take such other actions as set forth in the Credit and Security Agreement. As of March 31, 2021 and 2020, the Company was in compliance with all debt covenants.
In 2019, the Company paid fees of approximately $298 thousand in connection with the origination of the Midcap loan agreement. These fees were deferred and recorded as a direct deduction from the carrying value of the loan payable and are being amortized to interest expense over the remaining term of the agreement.
Pacific Western
In May 2018, Pacific issued a standby letter of credit of $3.8 million for the benefit of the Company’s landlord, securing certain Company obligations relating to tenant improvements. As of March 31, 2021 and December 31, 2020, the outstanding letter of credit was $3.8 million, for which the Company held $4.0 million as of March 31, 2021 and December 31, 2020, of restricted cash as collateral.
Convertible Notes
In March 2020, the Company issued convertible promissory notes for an aggregate principal amount of $6.0 million. Under certain conditions, the principal would convert to an amount of equity with a fair value that exceeded the amount of the notes’ principal on the conversion date. This feature of the notes was accounted for separately at fair value as a derivative liability. Changes in the fair value of the derivative were recorded in other loss, net in the Condensed Consolidated Statements of Operations and Comprehensive Loss and were immaterial during the three months ended March 31, 2020.
In September 2020, these notes and additional notes that were issued in April 2020 converted to 1,203,231 shares of Series D Preferred Stock. Upon conversion of the notes, the Company recorded the $1.6 million fair value of the derivative liability as equity on the Condensed Consolidated Balance Sheet.
Notes Payable for Tenant Improvement Allowance$616 thousand.
In 2018, the Company borrowed $992 thousand, which was available as part of the station 41a lease from our landlord to be usedagreement for use on tenant improvements (see Note 6, “Commitments and Contingencies” for additional details.) Under the terms of the lease, theimprovements. The note will be repaid over a 10 year10-year period at an 8% interest rate. Notes payable for As of March 31, 2024, the Midcap loan agreement and tenant improvement allowance consisted of the following:outstanding balance was $510 thousand.
| | | | | | | | | | March 31, | December 31, | | (in thousands) | 2021 | 2020 | | Current portion of notes payable | $ | 2,145 | | $ | 1,073 | | | Long-term portion of notes payable | 10,522 | | 11,615 | | | Less: unamortized issuance costs | (183) | | (201) | | | Notes payable, net | $ | 12,484 | | $ | 12,487 | |
Interest Income, net
| | | | | | | | | | Three months ended
March 31, | | (in thousands) | 2024 | 2023 | | Interest income | $ | 4,048 | | $ | 4,660 | | | Interest expense | (20) | | (19) | | | Interest income, net | $ | 4,028 | | $ | 4,641 | |
For the three months ended March 31, 2024 and 2023, interest income primarily related to earnings on cash and cash equivalents in money market funds. Interest income was included in “Other income, net” on the Condensed Consolidated Statements of Operations.
Note 4. Acquisitions
Valence Discovery Inc.
On May 16, 2023, Recursion acquired all of the outstanding equity interests in Valence Discovery Inc. (Valence), a privately-held machine learning (ML) / artificial intelligence (AI) digital chemistry company. The integration of Valence’s AI-based chemistry engine into Recursion’s operating system will allow Recursion to expand its technology-enabled drug discovery process. This will accelerate Recursion’s digital chemistry capabilities and its drug discovery process.
The acquisition of Valence was accounted for as a business combination using the acquisition method of accounting. The aggregate upfront consideration for the acquisition of Valence consisted of 2.2 million shares of Recursion Class A common stock, 4.4 million shares of a subsidiary of Recursion, exchangeable for shares of Recursion’s Class A common stock, 792 thousand shares issuable upon exercise of stock options held by Valence equity award holders and deferred liabilities for additional consideration. An insignificant number of the aforementioned shares of consideration had not yet been issued as of March 31, 2024.The final number of shares to be issued has not yet been finalized and so are subject to change.
The following table summarizes total consideration:
| | | | | | | (in thousands) | | | Fair value of Recursion Class A common stock | $ | 11,096 | | | Fair value of Exchangeable stock | 22,473 | | | Fair value of equity awards issued to Valance equity award holders | 1,933 | | | Deferred liabilities for additional consideration | 396 | | | Total consideration | $ | 35,898 | |
The following table summarizes the fair value of assets acquired and liabilities assumed as of the acquisition date:
| | | | | | | (in thousands) | | | Cash | $ | 4,235 | | | Other receivables | 536 | | | Intangible asset - technology | 15,000 | | | Accounts payable and accrued liabilities | (872) | | | Deferred income taxes | (3,265) | | | Total identifiable net assets | 15,634 | | | Goodwill | 20,264 | | | Total assets acquired and liabilities assumed | $ | 35,898 | |
The intangible asset related to Valence’s ML and AI digital chemistry platform. The estimated fair value of the intangible asset was determined using a cost approach. This valuation technique provides the fair value of an asset based on estimates of the total costs to develop the technology. Significant inputs used to determine the total cost includes the length of time required and service hours performed by Company employees. The technology intangible asset is being amortized on a straight-line basis over its four-year useful life.
Goodwill was calculated as the excess of the consideration transferred over the net assets recognized. The goodwill recognized represents the assembled workforce and expected synergies, including the ability to: (i) leverage Valence’s digital chemistry platform across Recursion’s business; (ii) leverage Valence’s ML and AI capabilities; (iii) integrate Recursion’s data and operating system into Valence’s platform; and (iv) accelerate Recursion’s pipeline. Goodwill was also impacted by the establishment of a deferred tax liability for the acquired identifiable intangible assets which have no tax basis. The goodwill is not deductible for tax purposes.
Recursion’s condensed consolidated statement of operations during the three months ended March 31, 2024 included no net revenue and a $2.9 million operating loss associated with Valence’s operations. As the acquisition occurred in May 2023, the Company is still finalizing the allocation of the purchase price to the individual assets acquired and liabilities assumed. The allocation of the purchase price included in the current period balance sheet is based on the best estimate of management and is preliminary and subject to change. The primary area subject to change relates to the valuation of other receivables. To assist management in the allocation, the Company engaged external specialists. The Company will finalize the amounts recognized as the information necessary to complete the analysis is obtained. The Company expects to finalize these amounts as soon as possible but no later than one year from the acquisition date.
Cyclica Inc.
On May 25, 2023, Recursion acquired all of the outstanding equity interests in Cyclica Inc. (Cyclica), a privately-held Company that has built a digital chemistry software suite which enables mechanism of action deconvolution and generative chemistry suggestions based on desired targets. Cyclica’s platform is expected to enhance the optimization of Recursion’s compounds for efficacy while minimizing liabilities through generative machine learning approaches.
The acquisition of Cyclica was accounted for as a business combination using the acquisition method of accounting. The aggregate upfront consideration for the acquisition of Cyclica consisted of 5.8 million shares of Recursion Class A common stock, cash payments, 1.0 million shares issuable upon exercise of stock options held by Cyclica equity award holders and deferred liabilities for additional consideration. Approximately 182 thousand of the aforementioned shares of Class A common stock consideration had not yet been issued as of March 31, 2024.
The following table summarizes total consideration:
| | | | | | | (in thousands) | | | Fair value of Recursion Class A common stock | $ | 49,915 | | | Cash | 6,505 | | | Fair value of equity awards issued to Cyclica equity award holders | 3,852 | | | Deferred liabilities for additional consideration | 344 | | | Total consideration | $ | 60,617 | |
The following table summarizes the fair value of assets acquired and liabilities assumed as of the acquisition date:
| | | | | | | (in thousands) | | | Cash | $ | 2,429 | | | Restricted cash | 1,685 | | | Other receivables | 741 | | | Investments | 1,000 | | | Other current assets | 385 | | | Intangible assets - technology | 28,000 | | | Accounts payable and accrued liabilities | (579) | | | Unearned revenue | (1,754) | | | Deferred income taxes | (2,075) | | | Other liabilities, current | (66) | | | Other liabilities, non-current | (139) | | | Total identifiable net assets | 29,627 | | | Goodwill | 30,990 | | | Total assets acquired and liabilities assumed | $ | 60,617 | |
The intangible assets are related to Cyclica’s digital chemistry platforms. The estimated fair value of the intangible assets were determined using a cost approach. This valuation technique provides the fair value of an asset based on estimates of the total costs to develop the technology. Significant inputs used to determine the total cost includes the length of time required and service hours performed by Company employees. The technology intangible assets are being amortized on a straight-line basis over their three-year useful lives.
Goodwill was calculated as the excess of the consideration transferred over the net assets recognized. The goodwill recognized represents the assembled workforce and expected synergies, including the ability to: (i) leverage Cyclica’s digital chemistry platform across Recursion’s business; (ii) leverage Cyclica’s ML and AI capabilities; (iii) integrate Recursion’s data and operating system into Cyclica’s platform; and (iv) accelerate Recursion’s pipeline. Goodwill was also impacted by the establishment of a deferred tax liability for the acquired identifiable intangible assets. The goodwill is not deductible for tax purposes.
Recursion’s condensed consolidated statement of operations during the three months ended March 31, 2024 included immaterial net revenue and a $3.8 million operating loss associated with Cyclica’s operations. As the acquisition occurred in May 2023, the Company is still finalizing the allocation of the purchase price to the individual assets acquired and liabilities assumed. The allocation of the purchase price included in the current period balance sheet is based on the best estimate of management and is preliminary and subject to change. The primary area subject to change relates to the valuation of the other receivables. To assist management in the allocation, the Company engaged external specialists. The Company will finalize the amounts recognized as the information necessary to complete the analysis is obtained. The Company expects to finalize these amounts as soon as possible but no later than one year from the acquisition date.
Pro forma financial information
The following table presents information regarding the Company’s debt principal repayment obligationsunaudited pro forma combined results of operations of Recursion, Valence and Cyclica as of March 31, 2021:if the acquisitions had occurred on January 1, 2022:
| | | | | | | (in thousands) | Amount | | 2021 | $ | 1,053 | | | 2022 | 4,052 | | | 2023 | 4,059 | | | 2024 | 3,059 | | | 2025 | 112 | | | Thereafter | 346 | | | Total debt principal payments | $ | 12,681 | |
| | | | | | | (in thousands) | Three months ended March 31, 2023 | | Net revenue | $ | 12,155 | | | Net loss | (73,514) | |
The unaudited pro forma financial information was prepared using the acquisition method of accounting and was based on the historical financial information of Recursion, Valence and Cyclica. In order to reflect the occurrence of the acquisitions on January 1, 2022 as required, the unaudited pro forma financial information includes adjustments to reflect the incremental amortization expense to be incurred based on the fair values of the identifiable intangible assets acquired and the additional stock compensation expense associated with the issuance of equity compensation related to the acquisitions. The unaudited pro forma financial information is not necessarily indicative of what the consolidated results of operations would have been had the acquisitions been completed on January 1, 2022. In addition, the unaudited pro forma financial information is not a projection of the future results of operations of the combined company nor does it reflect the expected realization of any cost savings or synergies associated with the acquisitions.
Note 6. Commitments and Contingencies
Lease Obligations5. Leases
The Company has entered into various long-term real estate leases primarily related to office, research and development (R&D) and operating activities. The Company’s leases have remaining terms from under 1 to 9 years and some of those leases include options that provide Recursion with the ability to extend the lease term for five years. The options are included in the lease term when it is reasonably certain that the option will be exercised.
For the three months ended March 31, 20212024 and 2020, total rent expense was $1.32023, Recursion entered into lease modifications resulting in a decrease to the right-of-use asset and lease liability of $3.1 million and $988 thousand, respectively.an increase to the right-of-use asset and lease liability $3.5 million, respectively. The following Komas, Station 41 and Milpitas leases are classified as operating leases.modifications had no impact to the Condensed Consolidated Statements of Operations.
Komas LeaseIn January 2024
In August 2016,, the Company entered into a new facilities lease agreement for office space in London, England with theapproximately 6,792 square feet (the “London Lease”). The right of use and payments beginning in began January 2017. The term2024 when the control of the asset was obtained. The London Lease term is 5 years with a five-year renewal option. The London Lease includes provisions for escalating rent payments. Total fixed payments are expected to be approximately $7.9 million, additionally there will be variable expenses including building service charges related to the lease.
In February 2024, the Company entered into a lease agreement for the supercomputer for the exclusive use of physical space in a data center of approximately 1,851 square feet (the “Data Center Lease”). The right of use is 7 years. Thisexpected to begin in the second quarter of 2024 and the Data Center Lease term is 5 years with a five-year renewal option. The lease includes provisions for escalating rent payments. Rent expense is recognized on a straight-line basis overTotal fixed lease payments are expected to be approximately $13.0 million with additional variable expenses, including utilities and tax expenses. The Company did not control the termspace or any of the lease. This lease included an allowance for tenant improvements. Tenant improvements were recordedassets being constructed as property and equipment and are being depreciated over the term of the lease. In conjunction with the allowance for tenant improvements, the Company recorded a lease incentive obligation of $847 thousand which is being amortized over the term of the lease as a reduction to rent expense. As of March 31, 2021, the related unamortized lease incentive obligation was $343 thousand.
Station 41 Lease2024
In August 2017, the Company entered a new facilities lease, with the and therefore no right of use beginning in December 2017 and payments beginning in June 2018. The term of theasset or lease is 10 years, with 1 five-year renewal option exercisable by the Company. This lease includes provisions for escalating rent payments. Rent expense is recognized straight-
line over the term of the lease. This lease included an allowance for tenant improvements of $4.0 million, the full balance of whichliability was drawn in 2017. Tenant improvements were recorded as property and equipment and are being depreciated over the remaining term of the lease. In conjunction with the allowance for tenant improvements, the Company recorded a leasehold obligation, which is being amortized over the term of the lease as a reduction to rent expense. As of March 31, 2021, the related unamortized lease incentive obligation was $2.7 million.
In 2018, the Company elected to draw an additional tenant improvement loan of $992 thousand offered in the Station 41 lease. This loan is incorporated into, and acts to increase the base rent over the remaining life of the lease. The increase in rent includes a charge for interest, which accrues on the principal amount outstanding at a rate equal to 8%. The Company accounts for this additional tenant improvement loan as a oote payable on the Condensed Consolidated Balance Sheets with the current portion included in the Current PortionSheet as of Notes Payable.March 31, 2024.
In 2019, the Company amended the Station 41 Lease to include additional space in the conjoining unit with the right to use the new space beginning in June 2020 for an additional 7 years. This amendment for the extra space includes provisions for escalating rent payments. Rent expense is recognized straight-line over the termThe components of the lease.lease cost were:
In January 2021, the company amended the Station 41 Lease, increasing the leased square footage by an additional 91,478 square feet. This amendment includes provisions for escalating rent, has a 10 year term and additional total minimum payments of $32.4 million. This lease included a tenant improvement allowance of up to approximately $10.1 million. | | | | | | | | | | Three months ended
March 31, | | (in thousands) | 2024 | 2023 | | Operating lease cost | $ | 2,472 | | $ | 1,998 | | | Variable lease cost | 538 | | 657 | | | Short-term lease cost | 40 | | — | | | Lease cost | $ | 3,050 | | $ | 2,655 | |
Milpitas Lease
In August 2019, the Company entered a new facilitiesThe remaining lease with the right of useterm and payments beginning in August 2019. The term of the lease is 9 years. This lease includes provisions for escalating rent payments. Rent expense is recognized on a straight-line basis over the term of the lease.discount rate were:
Future Minimum Lease Payments | | | | | | | (in thousands) | March 31, 2024 | | Operating leases | | | Weighted-average remaining lease term (years) | 6.4 | | Weighted-average discount rate | 7.7 | % |
Future minimum commitments
Maturities of operating lease liabilities as of March 31, 2021 under the Company’s lease agreements are as follows:2024 were:
| | | | | | | (in thousands) | Amount | | 2021 | $ | 2,911 | | | 2022 | 4,963 | | | 2023 | 7,344 | | | 2024 | 7,371 | | | 2025 | 7,560 | | | Thereafter | 33,214 | | | Total Minimum Payments | $ | 63,363 | |
| | | | | | | (in thousands) | Operating leases | | Remainder of 2024 | $ | 7,300 | | | 2025 | 10,748 | | | 2026 | 10,882 | | | 2027 | 11,281 | | | 2028 | 8,850 | | | Thereafter | 16,948 | | | Total lease payments | 66,009 | | | Less: imputed interest | (16,161) | | | Present value of lease liabilities | $ | 49,848 | |
Note 6. Goodwill and Intangible Assets
Goodwill
There were no changes to the carrying amount of goodwill during the three months ended March 31, 2024 and 2023. No goodwill impairment was recorded during the three months ended March 31, 2024 and 2023.
Intangible Assets, Net
The following table summarizes intangible assets:
| | | | | | | | | | | | | | | | | | | | | | | | | March 31, 2024 | | December 31, 2023 | | (in thousands) | Gross carrying amount | Accumulated Amortization | Net carrying amount | | Gross carrying amount | Accumulated Amortization | Net carrying amount | | Definite-lived intangible assets | $ | 44,426 | | $ | (12,336) | | $ | 32,090 | | | $ | 44,426 | | $ | (8,969) | | $ | 35,457 | | | Indefinite-lived intangible assets | 986 | | — | | 986 | | | 986 | | — | | 986 | | | Intangible assets, net | $ | 45,412 | | $ | (12,336) | | $ | 33,076 | | | $ | 45,412 | | $ | (8,969) | | $ | 36,443 | |
Amortization expense was $3.4 million and $152 thousand during the three months ended March 31, 2024 and 2023, respectively. The increase in amortization expense was due to the intangible assets purchased in the acquisitions, (see Note 4, “Acquisitions” for additional details). Amortization expense was included in Research and Development in the Condensed Consolidated Statements of Operations. No indefinite-lived intangible asset impairment charges were recorded during the three months ended March 31, 2024 and 2023.
Note 7. Commitments and Contingencies
Contract Obligations
In the normal course of business, the Company enters into contracts with clinical research organizations, drug manufacturers and other vendors for preclinical and clinical research studies, research and development supplies and other services and products for operating purposes. These contracts generally provide for termination on notice and therefore are cancellable contracts.
Indemnification
The Company has agreed to indemnify its officers and directors for certain events or occurrences, while the officer or director is or was serving at the Company’s request in such capacity. The Company purchases directors and officers liability insurance coverage that provides for corporate reimbursements ofreimbursement to the Company for covered obligations that limitsand this is intended to limit the Company’s exposure and enablesenable it to recover a portion of potential futureany amounts paid.it pays under its
indemnification obligations. The Company had no liabilities recorded for these agreements as of March 31, 20212024 and December 31, 20202023, as no amounts in excess of insurance coverage are probable or estimable.
Employee Agreements
The Company has signed employment agreements with certain key employees pursuant to which, if their employment is terminated by the Company following a change of control of the Company, the employees are entitled to receive certain benefits, including accelerated vesting of equity incentives.
Legal Matters
The Company is not currently a party to any material litigation or other material legal proceedings. The Company may, from time to time, be involved in various legal proceedings arising in the normal course of business, and anbusiness. An unfavorable resolution of any of these matterssuch matter could materially affect the Company’s future financial position, results of operations or cash flows.
Note 7. Convertible Preferred StockIn February 2021, the Company entered into a lease agreement for laboratory and office space (the Industry Lease) with Industry Office SLC, LLC (the landlord). In March 2023, the Company sent a letter to the landlord detailing numerous construction delays and irregularities, deficiencies and deviations from applicable structural drawings and/or non-conforming conditions with applicable building codes. On June 23, 2023, the landlord filed a lawsuit against the Company (Industry Office SLC, LLC v. Recursion Pharmaceuticals, Inc., Case No. 230904627) in the Third District Court for Salt Lake County, State of Utah (the Court), alleging anticipatory repudiation and breach of contract. The Plaintiff seeks monetary damages and attorney’s fees. In July 2023, the Company filed a motion to dismiss. In September 2023, Recursion was granted its motion to dismiss, and the Court provided the landlord until October 23, 2023, to amend and re-file the dismissed complaint. On October 23, 2023, the landlord filed an amended complaint again alleging anticipatory repudiation, breach of contract and breach of the implied covenant of good faith and fair dealing (the Amended Complaint) and seeks monetary damages and attorney’s fees. In November 2023, the Company filed a motion to dismiss the Amended Complaint. The Court set a hearing on the Company’s motion to dismiss in May 2024. As ofMarch 31, 2024, the Company had no liability recorded for these events as an unfavorable outcome was not probable.
In connection with the Industry Lease, in September 2023, the Company filed claims in the Court against the landlord alleging, among other things, breach of contract and fraudulent misrepresentation (the Counterclaims). In October 2023, the landlord filed an answer and denied the Company’s allegations asserted in the Counterclaims. The Company has issued preferred stock as part of various financing events. No new convertible preferred stock was issued during three months ended March 31, 2021 and 2020. As of March 31, 2021 and 2020, there were no cumulative dividends owed orthe landlord are currently engaged in arrearsdiscovery. On October 27, 2023, the Company filed a motion for partial judgment on the preferred stock.
Convertible Preferred Stock consistedpleadings, seeking judgment on one of the following as of March 31, 2021 and December 31, 2020:
| | | | | | | | | | | | | | | | | | | | | | | | | (in thousands except share data) | Preferred Shares Authorized | Preferred Shares Issued and Outstanding | Carrying Value | Liquidation Preferences | Shares of Common Stock Issuable Upon Conversion | | Series A | 30,078,402 | | 29,965,754 | | $ | 21,281 | | $ | 21,281 | | 29,965,754 | | | Series A-1 | 4,975,521 | | 4,975,520 | | 0 | | 0 | | 4,975,520 | | | Series B | 21,497,667 | | 21,471,898 | | 59,913 | | 60,000 | | 21,471,898 | | | Series C | 18,956,354 | | 18,776,345 | | 119,915 | | 122,058 | | 22,286,298 | | | Series D | 45,926,769 | | 36,898,548 | | 247,203 | | 247,511 | | 36,898,548 | | | Total convertible preferred stock | 121,434,713 | | 112,088,065 | | $ | 448,312 | | $ | 450,850 | | 115,598,018 | |
In April 2021, all outstanding shares of convertible preferred stock converted into common stock as part of the IPO. See Note 16, “Subsequent Events” for additional details.
Balance Sheet Classification
its four counterclaims. The Company’s convertible preferred stock is classified outside of stockholders’ deficitCourt set a hearing on the Condensed Consolidated Balance Sheets becauseCompany’s motion for partial judgment on the holderspleadings in May 2024. The Company is unable to estimate the possible amount or range of such shares have liquidation rights inamounts associated with the event of a deemed liquidation that, in certain situations, are not solely within the control of the Company and would require the redemption of the then-outstanding convertible preferred stock. The convertible preferred stock is not redeemable, except in the event of a deemed liquidation event.Counterclaims.
Note 8. Common stockStock
Each share of Class A common stock entitles the holder to one vote per share and each share of Class B common stock entitles the holder to 10 votes per share on all matters submitted to a vote of the Company’s stockholders. Common stockholders are entitled to receive dividends, as may be declared by the Company’s boardBoard of directors.Directors. As of March 31, 20212024 and December 31, 2020,2023, no dividends had been declared.
At-The-Market Offering
In August 2023, the Company entered into an Open Market Sales Agreement (the “Sales Agreement”) with Jefferies LLC (the “Sales Agent”), to provide for the offering, issuance and sale of up to an aggregate amount of $300.0 million of its Class A common stock from time to time in “at-the-market” (ATM) offerings. As of March 31, 20212024, an amount of $208.2 million remained available for future sales under the Sales Agreement. For the three months ended March 31, 2024, the Company has sold 921 thousand shares and received net proceeds of $10.9 million under the agreement. Recursion is not required to sell additional shares under the Sales Agreement. The Company will pay the Sales Agent a commission of up to 3% of the aggregate gross proceeds received from all sales of Class A common stock. The Sales Agreement continues until the earlier of selling all shares available under the Sales Agreement or terminated by written notice from either of the parties. The ATM Offering is being made under a prospectus supplement dated August 8, 2023, and related prospectus filed with the Securities and Exchange
Commission pursuant to our automatically effective shelf registration statement on Form S-3ASR (Registration No. 333-264845).
NVIDIA Private Placement
In July 2023, Recursion entered into a Stock Purchase Agreement for a private placement with NVIDIA Corporation (2023 Private Placement), pursuant to which the Company sold an aggregate of 7.7 million shares of the Company’s Class A common stock at a price of $6.49 per share for net proceeds of approximately $49.9 million.
Valence Acquisition Exchangeable Shares
In May 2023, in connection with the acquisition of Valence, the Company entered into an agreement to issue up to 5.9 million shares of Class A common stock (the “Exchange Shares”), that may be issued upon exchange, retraction or redemption of exchangeable shares of a subsidiary of Recursion. Each exchangeable share of the subsidiary of Recursion entitles the holder to exchange those shares on a one-for-one basis for Recursion’s Class A common stock. The shares are entitled to receive dividends economically equivalent to dividends declared by Recursion, are non-voting and are subject to customary adjustments for stock splits or other reorganizations. In addition, the Company may require all outstanding exchangeable shares to be exchanged into an equal number of Class A common stock upon the occurrence of certain events and at any time following the seventh anniversary of the closing of the Valence acquisition. The exchangeable shares are substantially the economic equivalent of the Class A shares and classified as common stock within the Company’s stockholders’ equity. The Company’s calculation of weighted-average shares outstanding includes the exchangeable shares. As of March 31, 2024, 4.2 million Exchangeable shares have been redeemed for Class A shares.
Registration Rights Agreements
Tempus agreement In November 2023, in connection with the Tempus Agreement, the Company agreed to prepare and file a registration statement (or a prospectus supplement to an effective registration statement on Form S-3ASR that will become automatically effective upon filing with the SEC pursuant to Rule 462(e)) with the SEC, for resale of the shares of Class A common stock issued or issuable under the Tempus Agreement. A prospectus supplement to a registration statement (File No. 333-264845) was subsequently filed in December 2023 to register shares issued to Tempus for the initial license fee under the Tempus Agreement for resale.
After registration of any shares issued to Tempus under the Tempus Agreement, the Company has agreed to use commercially reasonable efforts to keep such registration statement effective until such date that all shares issued to Tempus covered by such registration statement have been sold or are able to be publicly sold by relying on Rule 144 of the Securities Act without registration.
NVIDIA Private Placement In July 2023, in connection with the 2023 Private Placement with NVIDIA, the Company entered into a Registration Rights Agreement providing for the registration for resale of the shares of Class A common stock issued in such transaction. A prospectus supplement to a registration statement (File No. 333-264845) was subsequently filed in August 2023 to register the resale of the shares of Class A common stock issued to NVIDIA. The Company has agreed to use commercially reasonable efforts to keep the registration statement continuously effective until such date that all registrable securities under the agreement have been sold. In the event the holders cannot sell their shares due to certain circumstances causing the registration statement to be ineffective, the Company must pay each holder of shares outstanding on the date and each month thereafter 1% of the aggregate purchase price with the maximum payable amount of 5% of the aggregate purchase price. As of March 31, 2020,2024, there were 188,400,000was no accrued liability related to this agreement, as it was not probable that a payment would be required.
Acquisitions In May 2023, in connection with the acquisition of Valence, the Company entered into a Registration Agreement providing for the registration for resale of the shares of Class A common stock and Exchange Shares issued or issuable in such transaction. A registration statement on Form S-3ASR (File No. 333-272281) was filed to register
the shares for resale by the holders. The registration statement must remain effective for a period of not less than three years.
In May 2023, in connection with the acquisition of Cyclica, the Company entered into a Registration Agreement providing for the registration for resale of the shares of Class A common stock issued in such transaction. A prospectus supplement to a registration statement (File No. 333-264845) was subsequently filed in June 2023 to register the shares for resale by the holders. The registration statement must be continuously effective until the earlier of the date that all shares have been sold thereunder or are able to be publicly sold by relying on Rule 144 of the Securities Act without registration.
2022 Private Placement In October 2022, in connection with the 2022 Private Placement, the Company entered into a Registration Rights Agreement providing for the registration for resale of the shares of Class A common stock issued in such transaction. A prospectus supplement to a registration statement (File No. 333-264845) was subsequently filed in October 2022 to register the resale of the shares of Class A common stock by the Purchasers. The agreement must remain effective until registrable securities covered by the agreement have been publicly sold by the holders or all shares cease to be registrable securities. In the event the holders cannot sell their shares due to certain circumstances causing the agreement to be ineffective, the Company must pay each holder of shares outstanding on the date and each month thereafter 1% of the aggregate purchase price paid by the holder without limit until the agreement is cured. As of March 31, 2024, there was no accrued liability related to this agreement, as it was not probable that a payment would be required.
Class A and B Common Shares Authorization
In April 2021, the Company’s Board of Directors authorized two classes of common stock, authorized,Class A and Class B. The rights of which 24,036,725the holders of Class A and 22,314,685 shares were outstanding, respectively.B common stock are identical, except with respect to voting and conversion. Each share of Class A common stock is entitled to one vote per share. Each share of Class B common stock is entitled to 10 votes per share and is convertible at any time into one share of Class A common stock.
Additionally,All Class B common stock is held by Christopher Gibson, Ph.D., the Company has reserved the followingCompany’s Chief Executive Officer (CEO), or his affiliates. As of March 31, 2024, Dr. Gibson and his affiliates held outstanding shares of Class B common stock representing approximately 25% of the voting power of the Company’s outstanding shares. This voting power may increase over time as Dr. Gibson vests in and exercises equity awards outstanding. If all the exchangeable equity awards held by Dr. Gibson had been fully vested, exercised and exchanged for issuanceshares of Class B common stock as of March 31, 2021:
| | | | | | | Shares | Conversion of series A preferred stock | 29,965,754 | | Conversion of series A-1 preferred stock | 4,975,520 | | Conversion of series B preferred stock | 21,471,898 | | Conversion of series C preferred stock | 22,286,298 | | Conversion of series D preferred stock | 36,898,548 | | Conversion of series A warrants | 112,647 | | Conversion of series B warrants | 25,762 | | Conversion of series C warrants | 213,646 | | 2016 equity incentive plan | 25,686,958 | | Key personnel incentive plan | 1,601,566 | | Total shares of common stock reserved for issuance | 143,238,597 | |
2024, Dr. Gibson and his affiliates would hold approximately 26% of the voting power of the Company’s outstanding shares. As a result, Dr. Gibson will be able to significantly influence any action requiring the approval of Recursion stockholders, including the election of the Board of Directors; the adoption of amendments to the Company’s certificate of incorporation and bylaws; and the approval of any merger, consolidation, sale of all or substantially all of the Company’s assets, or other major corporate transaction.
Note 9. Collaborative and Other Research and Development Contracts
Bayer AGRoche and Genentech
Description In August 2020, the CompanyDecember 2021, Recursion entered into a Research Collaborationcollaboration and Option Agreement (the Bayer Agreement)license agreement with Bayer AG (Bayer)Roche and Genentech (collectively referred to as Roche). Recursion is constructing, using the Company’s imaging technology and proprietary machine-learning algorithms, unique maps of the inferred relationships amongst perturbation phenotypes in a given cellular context with the goal to discover and develop therapeutic small molecule programs in a gastrointestinal cancer indication and in key areas of neuroscience. Roche and Recursion will collaborate to select certain novel inferences with respect to small molecules or targets generated from the Phenomaps for further validation and optimization as collaboration programs. Roche and Recursion may also combine sequencing datasets from Roche with Recursion’s Phenomaps and collaborate to generate new algorithms to produce multi-modal maps from which additional collaboration programs may be initiated. For every collaboration program that successfully identifies potential therapeutic small molecules or validates a five-year term pursuanttarget, Roche will have an option to whichobtain an exclusive license to develop and commercialize such potential therapeutic small molecules or to exploit such target in the Company and Bayer may initiate approximately 10 research projects related to fibrosis across multiple organ systems, including lung, liver, and heart. Under the agreement, the Company contributed compounds from our proprietary library and Bayer contributed compounds from its proprietary library and will contribute scientific expertise throughout the collaboration.applicable exclusive field.
Pricing In January 2022, Recursion received a $150.0 million non-refundable upfront payment from the Company’s collaboration with Roche. Recursion is eligible for additional milestone payments based on performance progress of $30.0the collaboration. Each of the Phenomaps requested by Roche and created by Recursion may be subject to either an initiation fee, acceptance fee or both. Such fees could exceed $250.0 million for 16 accepted Phenomaps. In addition, for a period of time after Roche’s acceptance of certain Phenomaps, Roche will have the option to obtain, subject to payment of an exercise fee, rights to use outside the collaboration the raw images generated in the course of creating those Phenomaps. If Roche exercises its external use option for all 12 eligible Phenomaps, Roche’s associated exercise fee payments to Recursion could exceed $250.0 million. Under the collaboration, Roche may initiate up to 40 programs, each of which, was recordedif successfully developed and commercialized, could yield more than $300.0 million in development, commercialization and net revenue milestones for Recursion, as unearned revenuewell as tiered royalties on the Condensed Consolidated Balance Sheet. The Companynet revenue.
Accounting This agreement represents a transaction with a customer and therefore is accounted for in accordance with ASC 606. Recursion has determined that it has three performance obligations, one related to gastrointestinal cancer and two in neuroscience. These performance obligation under the agreement, which is to performobligations are for performing research and development services for Bayer. RecursionRoche to identify targets and medicines. The performance obligations also include potential licenses related to the intellectual property. The Company concluded that licenses within the contract are not distinct from the research and development services as they are interrelated due to the fact that the research and development services significantly impact the potential licenses. Any additional services are considered customer options and will be considered as separate contracts for accounting purposes.
The Company has determined the transaction price to be $150.0 million, comprised of the $30.0 million upfront paymentpayment. Recursion will fully constrain the amounts of variable consideration to be received from potential milestones considering the stage of development and allocated the amountrisks associated with the remaining development required to achieve each milestone. Recursion will re-evaluate the transaction price each reporting period.
The transaction price was allocated to the single performance obligation.obligations based on the estimated relative stand-alone selling price of each performance obligation as determined using an expected cost plus margin approach. The Company is recognizing therecognizes revenue over time using a cost-based input method, based on labor costs incurred relative to total expected costs to perform the research and development services. Recursion determined that this method provides a faithful depiction of the transfer of control to the customer. This method of recognizing revenue requires the Company to make estimates of the total costs to provide the services required under the performance obligation.obligations. Significant inputs used to determine the total costs included the length of time required, service hours performed by Company employees and materials costs. A significant change in these estimates could have a material effect on the timing and amount of revenue recognized in future periods. Recursion has estimated the completion of the performance obligations by 2026.
For the three months ended March 31, 2021, the Company recognized $2.5 million of revenue resulting from the collaboration. There is $10.0 million and $14.2 million of current and non-current unearned revenue, respectively, remaining as of March 31, 2021. The allocation of unearned revenue between current and non-current is based on Recursion’s estimates of when the Company expects to incur the related costs.Additional Revenue Disclosures
Under each research project,Of the Company will work with Bayer to identify potential candidates for development. Under the agreement, Bayer has the first option for licenses to potential candidates. Each such license could potentially result in option exercise fees and development and commercial milestones paid to the Company with an aggregate value of up to approximately $100.0 million (for an option on a lead series) or up to approximately $120.0 million (for an option on a development candidate), as well as tiered royalties for each such license, ranging from low- to mid-single digit percentages of sales, depending on commercial success.
The National Institute of Health
During the year ended December 31, 2018, the Company was awarded a grant by the National Institute of Health, which included potential funding of $1.4 million. Revenuerevenue recognized related to this grant during the three months ended March 31, 20212024 and 20202023, primarily all of it was $62 thousandincluded in the unearned revenue balance as of December 31, 2023 and $60 thousand,2022, respectively. Revenue recognized was from upfront payments received at the inception of the related contracts, which decreased the initial unearned revenue recognized. As of March 31, 2021, $395 thousand2024, the Company had $6.5 million of the potential funding remained.costs incurred to fulfill a contract on its Condensed Consolidated Balance Sheet within “Other Current Assets.”
AsUnearned revenue was classified as short-term and long-term on the Condensed Consolidated Balance Sheets based on the Company’s estimate of March 31, 2021 and December 31, 2020,revenue that will be recognized during the Company had $62 thousand and $140 thousand of outstanding receivables, respectively with the National Institute of Health, which was deemed to be collectible.
Note 10. Stock-Based Compensation
Stock Options
Key Personnel Incentive Plan
In November 2013, the Company adopted the Key Personnel Incentive Plan ( the KPI Plan). The KPI Plan provides for the grant of restricted units and non-statutory option awards to employees, non-employee directors and consultants of the Company. As of March 31,April 2021, and December 31, 2020, there were 0 shares of common stock available for grant under the KPI Plan.
The KPI Plan provides for the early exercise of options. Upon exercise, such option holder receives common stock of the Company, subject to a lapsing right of repurchase. Upon termination of such individual, the Company may exercise its right to repurchase any unvested shares for the exercise price paid by the option holder.
2016 Equity Incentive Plan
In August 2016, the Board of Directors and the stockholders of the Company adopted the 20162021 Equity Incentive Plan. Under the 2016 Plan 25,686,958 shares of common stock were reserved.(the 2021 Plan). The Company may grant stock options, to purchase commonrestricted stock units (RSUs), stock appreciation rights, restricted stock awards and other forms of stock-based compensation. As of March 31, 2024, 18.9 million shares of Class A common stock were available for grant.
The following table presents the classification of stock-based compensation expense for employees and non-employees within the Condensed Consolidated Statements of Operations:
| | | | | | | | | | Three months ended
March 31, | | (in thousands) | 2024 | 2023 | | Cost of revenue | $ | 766 | | $ | 1,011 | | | Research and development | 7,666 | | 2,683 | | | General and administrative | 7,077 | | 4,578 | | | Total | $ | 15,509 | | $ | 8,272 | |
Stock Options
Stock options are primarily granted to executive leaders at the Company, generally vest over four years and expire no later than 10 years from the date of grant.
As of March 31, 2021, 3,684,798 shares of common stock are available for grant. Stock option activity during the three months ended March 31, 20212024 was as follows:
| | | | | | | | | | | | | | | | (in thousands except share data) | Shares | Weighted-Average Exercise Price | Weighted-Average Remaining Contractual Life (In Years) | Aggregate Intrinsic Value | | Outstanding as of 12/31/2020 | 20,937,443 | | $ | 1.85 | | 8.5 | $ | 12,956 | | | Granted | 1,849,311 | | 4.44 | | | | | Cancelled | 384,540 | | 2.33 | | | | | Exercised | 1,722,027 | | 1.25 | | | 4,455 | | | Outstanding as of March 31, 2021 | 20,680,187 | | $ | 2.14 | | 8.5 | $ | 50,346 | | | Exercisable as of March 31, 2021 | 6,365,727 | | $ | 1.20 | | 6.9 | $ | 21,450 | | | Non-vested options as of March 31, 2021 | 14,314,460 | | $ | 2.55 | | 9.2 | $ | 28,897 | |
| | | | | | | | | | | | | | | | (in thousands except share and per share amounts) | Shares | Weighted-Average Exercise Price | Weighted-Average Remaining Contractual Life (in years) | Aggregate Intrinsic Value | | Outstanding as of December 31, 2023 | 14,957,617 | | $ | 6.13 | | 7.0 | $ | 72,416 | | | Granted | 2,558,102 | | 10.09 | | | | | Cancelled | (178,609) | | 12.70 | | | | | Exercised | (1,303,878) | | 2.80 | | | 11,575 | | | Outstanding as of March 31, 2024 | 16,033,232 | | $ | 6.99 | | 7.3 | $ | 64,131 | | | Exercisable as of March 31, 2024 | 8,909,647 | | $ | 5.49 | | 6.2 | $ | 51,959 | |
The fair value of options granted to employees is calculated on the grant date using the Black-Scholes option valuation model. The weighted-average grant-date fair values of stock options granted during the three months ended March 31, 20212024 and 20202023 were $2.63$6.41 and $1.33,$5.32, respectively.
The following weighted-average assumptions were used to calculate the grant-date fair value of employee stock options:
| | Three months ended March 31, | | Three months ended March 31, | | | Three months ended March 31, | | | | 2021 | 2020 | | 2024 | 2023 | | Expected term (in years) | Expected term (in years) | 6.1 | 6.2 | Expected term (in years) | 6.3 | | Expected volatility | Expected volatility | 66% | 65% | Expected volatility | 65 | % | 64 | % | | Expected dividend yield | Expected dividend yield | 0 | | Risk-free interest rate | Risk-free interest rate | 0.78% | 1.33% | Risk-free interest rate | 4.2 | % | 3.5 | % |
For the three months endedAs of March 31, 2021, the Company granted 150,000 shares2024, $43.9 million of unrecognized compensation cost related to stock options with a performanceis expected to be recognized as expense over approximately the next three years.
RSUs
Equity awards granted to employees primarily consist of RSUs and service condition that had agenerally vest over four years. The weighted-average grant-date fair value of $358 thousand. NaN expense related to these options was recordedRSUs generally is determined based on the number of units granted and the quoted price of Recursion’s common stock on the date of grant.
The following table summarizes Recursion’s RSU activity during the three months ended March 31, 2021 as the performance condition was not considered probable.2024:
13 | | | | | | | | | | Stock units | Weighted-average grant date fair value | | Outstanding as of December 31, 2023 | 15,223,764 | | $ | 8.39 | | | Granted | 1,840,877 | | 10.46 | | | Vested | (1,023,464) | | 8.60 | | | Forfeited | (409,634) | | 7.99 | | | Outstanding as of March 31, 2024 | 15,631,543 | | $ | 8.63 | |
For the three months ended March 31, 2020, the Company granted 1,500,000 shares of stock options with performance,The fair market conditions and service conditions. At grant date, the Company estimated that the fair value of the optionsRSUs vested was approximately $2.0 million. NaN expense related to these options was recorded$13.0 million during the three months ended March 31, 2021 and 2020 as the performance conditions were not considered probable.
During the years ended December 31, 2020 and 2017, the Company granted options to purchase 120,000 and 330,000 shares, respectively, of common stock to non-employee consultants. These options were granted in exchange for consulting services and vest over a period that approximates the term of the services to be provided by the Company. The fair value of the options granted prior to 2020 were remeasured in each period until they were fully vested. Following the adoption of ASU 2018-07 on January 1, 2020, the fair value of options granted to non-employees were no longer remeasured subsequent to the grant date. The fair value of each option on the date of grant was calculated using the Black-Scholes option model. There were 0 grants to non-employee consultants during the three months ended March 31, 2021 or 2020.
The following table presents the classification of stock-based compensation expense for employees and non-employees within the Condensed Consolidated Statements of Operations and Comprehensive Loss:
| | | | | | | | | | Three months ended March 31, | | (in thousands) | 2021 | 2020 | | Research and development | $ | 628 | | $ | 688 | | | General and administrative | $ | 1,070 | | $ | 577 | | | Total | $ | 1,698 | | $ | 1,265 | |
2024. As of March 31, 2021, there was $22.02024, $124.8 million of unamortized stock-basedunrecognized compensation cost related to unvested stock options whichRSUs is expected to be recognized as expense over a weighted average period of 2.98approximately the next three years.
Warrants
In connection with the execution of the December 2016 Pacific loan agreement (see Note 5, “Notes Payable” for additional details), the Company issued Pacific fully vested warrants to purchase Series A Preferred Stock. In May 2017, the Company drew on additional borrowing capacity under the Pacific loan agreement, this required the Company to issue additional fully vested warrants. These Series A warrants remained outstanding as of March 31, 2021.
In July 2018, the Company drew on additional borrowing capacity under an amended agreement. This required the Company to issue fully vested warrants to purchase Series B Preferred Stock. These warrants remained outstanding as of March 31, 2021.
In January 2020, the Company issued warrants to purchase 180,000 shares of Series C Preferred Stock at a purchase price of $6.50 per share as part of a services agreement. The warrants vest ratably over 18 months. These warrants remained outstanding and 139,999 were vested and exercisable as of March 31, 2021. The grant date fair value was $4.10 per share. As of March 31, 2021, there was $158 thousand of unamortized cost related to the unvested warrants which is expected to be recognized over four months.
The following tables summarize the Series A and B warrants outstanding as of March 31, 2021:
| | | | | | | | | | | | | | | | | | | (in thousands except share data) | | | | | | | Series A | Grant Date | Number of Warrants | Exercise Price | Fair Value as of March 31, 2021 | Fair Value as of December 31, 2020 | | 2017 Warrants | 12/19/2016 | 84,486 | $ | 0.71 | | $ | 56,232 | | $ | 58,000 | | | 2018 Warrants | 5/27/2017 | 28,161 | $ | 0.71 | | $ | 18,743 | | $ | 19,000 | |
| | | | | | | | | | | | | | | | | | | (in thousands except share data) | | | | | | | Series B | Grant Date | Number of Warrants | Exercise Price | Fair Value as of March 31, 2021 | Fair Value as of December 31, 2020 | | 2019 Warrants | 7/9/2018 | 25,762 | $ | 2.79 | | $ | 45,499 | | $ | 48,000 | |
The fair value of the warrants was calculated using the Black-Scholes option-pricing model with the following weighted average assumptions:
| | | | | | | | | | Three months ended March 31, | | | 2021 | 2020 | | Expected term (in years) | 6.16 | 7.12 | | Expected volatility | 66% | 66% | | Expected dividend yield | 0 | 0 | | Risk-free interest rate | 1.20% | 0.70% |
The FASB has issued accounting guidance on the classification of freestanding warrants and other similar instruments on shares that are redeemable (either puttable or mandatorily redeemable). The guidance requires liability classification for certain warrants issued that are exercisable into convertible preferred stock. The initial fair values of Series A and B warrants were recorded as debt issuance costs, which resulted in a reduction in the carrying value of the debt and subsequent accretion. The Company remeasures the Series A and B warrants on each Condensed Condensed Consolidated Balance Sheet date. The change in the valuation is recorded in the Condensed Consolidated Statements of Operations and Comprehensive Loss.
The Series C warrants compensation expense is being recorded ratably over the requisite service period based on the award’s fair value at the date of grant in general and administrative expense. These warrants were classified as equity as they were issued to non-employees for services and the convertible preferred stock is not redeemable, except in the event of a deemed liquidation event, which is not considered probable.
The following is a summary of the changes in the Company’s Series A and B warrant liability balance during the three months ended March 31, 2021 and 2020:
| | | | | | (in thousands) | | Balance as of December 31, 2019 | $ | 128 | | Net decrease in fair value of warrants | (10) | | Balance as of March 31, 2020 | $ | 118 | | | | Balance as of December 31, 2020 | $ | 125 | | Net decrease in fair value of warrants | (5) | | Balance as of March 31, 2021 | $ | 120 | |
Note 11. Employee Benefit Plans
The Company has an employee benefit plan under Section 401(k) of the Internal Revenue Code. The plan allows employees to make contributions up to a specified percentage of their compensation. The Company is currently contributing up to 4% of employee base salary, by matching 100% of the first 4% of annual base salary contributed
by each employee. Employer expenses were approximately $281 thousand and $199 thousand during the three months ended March 31, 2021 and 2020, respectively.
Note 12. Income Taxes
The Company did 0tnot record any U.S. income tax expense during the three months ended March 31, 20212024 and 2020.2023. The Company has historically incurred operating losses and maintains a full valuation allowance against its U.S. net deferred tax assets. Valuation allowances are recorded whenForeign taxes were insignificant during the expected realization of the deferred tax assets does not meet a “more likely than not” criterion. Realization of the Company’s deferred tax assets are dependent upon the generation of future taxable income, the amountthree months ended March 31, 2024 and timing of which are uncertain.2023.
Net operating loss carryforwardslosses (NOLs) and tax credit carry-forwards are subject to review and possible adjustment by the Internal Revenue Service (IRS)(“IRS”) and may become subject to annual limitationslimitation due to ownership changes that have occurred previously or that could occur in the future under Section 382 of the Internal Revenue Code.Code, as amended and similar state provisions. These ownership changes may limit the amount of carryforwards that can be utilized annually to offset future taxable income. In general, an ownership change, as defined by Section 382, results from transactions increasing the ownership of certain shareholders or public groups in the stock of a corporation by more than 50% over a three-year period. As of March 31, 2024, the Company was not limited on its NOLs and tax credit carry-forwards. The Company has not conducted a studywill continue to assess whether a change of control has occurred or whether there have been multiplemonitor future ownership changes of control since inception due to the significant complexity and cost associated with such a study. Any limitation may result in expiration of a portion of the NOLs or research and development tax credit carryforwards before utilization. Further, until a study is completed and any limitation is known, no amounts are being presented as an uncertain tax position.for potential Section 382 limitations.
The Company files income tax returns in the United States, Canada, United Kingdom, Utah, California and California.Massachusetts. The Company is not currently under examination in any of these jurisdictions. The Company is subject to income tax examinations on all federal returns since the 20182017 tax return.
Note 13.12. Net Loss Per Share
For the three months ended March 31, 2024 and 2023, Recursion issued certain convertible preferred stock that was concluded to be participating securities. Due to the presence of participating securities, Recursion calculatescalculated net loss per share using the more dilutive of the treasuryClass A, Class B and Exchangeable common stock orusing the two-class method. Basic net loss per share is computed using the weighted-average number of shares outstanding during the period. Diluted net loss per share is computed using the weighted-average number of shares and the effect of potentially dilutive securities outstanding during the period. Potentially dilutive securities consist of stock options and other contingently issuable shares. For periods presented in which the Company reports a net loss, all potentially dilutive shares are anti-dilutive and as such are excluded from the losses are not allocated tocalculation. For the participating securities. Asthree months ended March 31, 2024 and 2023, the Company reported a net loss and therefore basic and diluted loss per share were the same.
The rights, including the liquidation and dividend rights, of the holders of the Company’s Class A, Class B and the Exchangeable common stock are identical, except with respect to voting. As a result, the undistributed earnings for each period are allocated based on the contractual participation rights of the Class A, Class B and the Exchangeable common stock as if the earnings for the period had been distributed. As the liquidation and dividend rights are identical, the undistributed earnings are allocated on a proportionate basis and the resulting amount per share for Class A, Class B and the Exchangeable common stock was the same during the three months ended March 31, 20212024 and 2020, diluted net loss per share is the same as basic net loss per share, since dilutive common shares are not assumed to have been issued if their effect is anti-dilutive.2023.
The following table setstables set forth the computation of basic and diluted net loss per share:share of Class A, Class B and Exchangeable common stock:
| | Three months ended March 31, | | Three months ended March 31, | | | Three months ended March 31, | | (in thousands, except share amounts) | (in thousands, except share amounts) | 2021 | 2020 | (in thousands, except share amounts) | 2024 | 2023 | | Numerator: | Numerator: | | | Net loss | Net loss | $ | (30,717) | | $ | (18,424) | | | Net loss | | | Net loss | | | Denominator: | Denominator: | | | Weighted average common shares outstanding | Weighted average common shares outstanding | 23,035,623 | | 21,639,891 | | | Weighted average common shares outstanding | | | Weighted average common shares outstanding | | | Net loss per share, basic and diluted | Net loss per share, basic and diluted | $ | (1.33) | | $ | (0.85) | |
The Company excluded the following potential common shares from the computation of diluted net loss per share for the periods indicated because including them would have had an anti-dilutive effect:
| | | | | | | | | | Three months ended March 31, | | | 2021 | 2020 | | Convertible preferred stock | 115,598,018 | | 78,699,470 | | | Options to purchase common stock | 4,706,313 | | 3,373,642 | | | Warrants | 122,358 | | 115,029 | | | Total | 120,426,689 | | 82,188,141 | |
| | | | | | | | | | Three months ended March 31, | | | 2024 | 2023 | | Stock based compensation | 11,159,250 | | 8,534,876 | | | Tempus agreement | 5,143,690 | | — | | | Total | 16,302,940 | | 8,534,876 | |
Note 14.13. Fair Value Measurements
The fair value hierarchy consists of the following three levels:
•Level 1 — Valuations based on unadjusted quoted prices in active markets for identical assets that the company has the ability to access; •Level 2 — Valuations based on quoted prices for similar instruments in active markets, quoted prices for identical or similar instruments in markets that are not active and model-based valuations in which all significant inputs are observable in the market; and •Level 3 — Valuations using significant inputs that are unobservable in the market and include the use of judgment by the company's management about the assumptions market participants would use in pricing the asset or liability.
As of March 31, 2021 and December 31, 2020, cash and cash equivalents (including restricted cash) included bank deposits held in checking and savings accounts. The Company is required to maintain a cash balance in a collateralized account to secure the Company’s credit cards. Additionally, the Company holds restricted cash related to an outstanding letter of credit issued by Pacific Western Bank,J.P. Morgan, which was securingobtained to secure certain Company obligations relating to tenant improvements. As of March 31, 2021 and December 31, 2020, cash restricted for the letters of credit was $4.0 million. The remainingRecursion also holds restricted cash related to the Company’s credit cards.
The Company measures the Series Aa Bill and B Preferred Stock warrant liabilities at fair value using the Black-Scholes option-pricing model. See Note 10, “Stock-based Compensation” for details on the valuation of the warrant liabilities and a reconciliation of the balance.Melinda Gates Foundation grant.
The following tables summarize the Company’s assets and liabilities that are measured at fair value on a recurring basis:
| | Basis of fair value measurement | | | Basis of fair value measurement | | | | Basis of fair value measurement | | (in thousands) | (in thousands) | March 31, 2021 | Level 1 | Level 2 | Level 3 | (in thousands) | March 31, 2024 | Level 1 | Level 2 | Level 3 | | Assets | Assets | | | Cash and equivalents | $ | 214,088 | | $ | 214,088 | | $ | 0 | | $ | 0 | | | Cash equivalents: | | | Cash equivalents: | | | Cash equivalents: | | | Money market funds | | | Money market funds | | | Money market funds | | | Restricted cash | Restricted cash | 5,042 | | 5,042 | | 0 | | 0 | | | Total assets | Total assets | $ | 219,130 | | $ | 219,130 | | $ | 0 | | $ | 0 | | | Liabilities | | | Warrant liability | $ | 120 | | $ | 0 | | $ | 0 | | $ | 120 | | | Total liabilities | $ | 120 | | $ | 0 | | $ | 0 | | $ | 120 | |
| | | | | | | | | | | | | | | | | Basis of fair value measurement | | (in thousands) | December 31, 2020 | Level 1 | Level 2 | Level 3 | | Assets | | | | | | Cash and equivalents | $ | 262,126 | | $ | 262,126 | | $ | 0 | | $ | 0 | | | Restricted cash | 5,041 | | 5,041 | | 0 | | 0 | | | Total assets | $ | 267,167 | | $ | 267,167 | | $ | 0 | | $ | 0 | | | Liabilities | | | | | | Warrant liability | $ | 125 | | $ | 0 | | $ | 0 | | $ | 125 | | | Total liabilities | $ | 125 | | $ | 0 | | $ | 0 | | $ | 125 | |
| | | | | | | | | | | | | | | | | Basis of fair value measurement | | (in thousands) | December 31, 2023 | Level 1 | Level 2 | Level 3 | | Assets | | | | | | Cash equivalents: | | | | | | Money market funds | $ | 322,653 | | $ | 322,653 | | $ | — | | $ | — | | | Restricted cash | 9,860 | | 9,860 | | — | | — | | | Total assets | $ | 332,513 | | $ | 332,513 | | $ | — | | $ | — | |
In addition to the financial instruments that are recognized at fair value on the Condensed Consolidated Balance Sheets,Sheet, the Company has certain financial instruments that are recognized at amortized cost or some basis other than fair value. The carrying amount of these amountsinstruments are considered to be representative of their approximate fair values.
The following tables summarize the Company’s assets and liabilitiesfinancial instruments that are not measured at fair value:
| | Book values | Fair values | | Book values | | | Book values | | Fair values | | (in thousands) | (in thousands) | March 31, 2021 | December 31, 2020 | March 31, 2021 | December 31, 2020 | (in thousands) | March 31, 2024 | December 31, 2023 | | March 31, 2024 | December 31, 2023 | | Liabilities | Liabilities | | | Current portion of notes payable | Current portion of notes payable | $ | 2,145 | | $ | 1,073 | | $ | 2,145 | | $ | 1,073 | | | Current portion of notes payable | | | Current portion of notes payable | | | Notes payable, net of current portion | Notes payable, net of current portion | 10,339 | | 11,414 | | 10,339 | | 11,414 | | | Total liabilities | Total liabilities | $ | 12,484 | | $ | 12,487 | | $ | 12,484 | | $ | 12,487 | |
Note 15. Related Party Transactions
On December 5, 2017, the Company entered into a loan agreement with its Chief Executive Officer (CEO) to provide a loan of $595 thousand. The loan had a seven-year term. As of March 31, 2021, 0 amount remained outstanding on the loan. As of March 31, 2020, the outstanding balance of $595 thousand was recorded on the Condensed Consolidated Balance Sheets within other non-current assets.
Note 16. Subsequent Events
Initial Public Offering
On April 20, 2021, the Company closed its IPO and issued 27,878,787 shares of its common stock at a price of $18.00 per share for approximate net proceeds of $462.6 million, after deducting underwriting discounts and commissions of $35.1 million and expenses of $4.1 million. In connection with the IPO, all shares of Series A, B, C and D convertible preferred stock converted into 115,598,018 shares of Class A common stock.
Stock Split
In April 2021, the Board of Directors approved a 1.5-for-1 forward stock split of the Company’s common and convertible preferred stock. Each shareholder of record on April 9, 2021 received 1.5 shares for each then-held share. The split proportionally increased the authorized shares and did not change the par values of the Company’s stock. The split affected all stockholders uniformly and did not affect any stockholder's ownership percentage of the Company's shares of Common Stock. All shares and per share amounts presented within these condensed consolidated financial statements were adjusted to reflect the forward stock split for all periods presented.
Equity Plans
In April 2021, the Company’s Board of Directors adopted, and its stockholders approved, the 2021 Equity Incentive Plan (the “2021 Plan”), which became effective in connection with the closing of the Company’s IPO. A total of 16,186,000 shares of the Company’s Class A common stock have been reserved for issuance under the 2021 Plan in addition to any shares of Class A common stock outstanding under the 2016 Plan that expire are withheld by the Company for payment of an exercise price or for satisfying tax withholding obligations, or are forfeited to the Company due to failure to vest, subject to an addition of a maximum of 19,479,146 shares. The number of shares of Class A common stock reserved for future issuance under the 2021 Plan will also be increased pursuant to provisions for annual automatic evergreen increases.
In April 2021, the Company’s Board of Directors adopted, and its stockholders approved, the 2021 Employee Stock Purchase Plan (the “2021 ESPP”), which became effective in connection with the closing of the Company’s IPO. A total of 3,238,000 shares of the Company’s Class A common stock have been reserved for issuance under the 2021 ESPP. In addition, the number of shares reserved for future issuance under the 2021 ESPP will be increased for annual automatic evergreen increases.
Class A and B Common Shares Authorization
In April 2021, the Company’s Board of Directors authorized two classes of common stock, Class A and Class B. The rights of the holders of Class A and B common stock are identical, except with respect to voting and conversion. Each share of Class A common stock is entitled to one vote per share. Each share of Class B common stock is entitled to ten votes per share and is convertible at any time into one share of Class A common stock.
All Class B common stock is held by Christopher Gibson, Ph.D., our Chief Executive Officer, or his affiliate. Dr. Gibson and his affiliate hold outstanding shares of Class B common stock representing approximately 38% of the voting power of the Company’s outstanding shares. This voting power may increase over time as Dr. Gibson vests in and exercises equity awards outstanding. If all the equity awards held by Dr. Gibson had been fully vested and exercised and exchanged for shares of Class B common stock as of the date of the IPO, Dr. Gibson and his affiliate would hold approximately 42% of the voting power of the Company’s outstanding shares. As a result, Dr. Gibson will be able to significantly influence any action requiring the approval of Recursion stockholders, including the election of the board of directors, the adoption of amendments to the Company’s certificate of incorporation and bylaws, and the approval of any merger, consolidation, sale of all or substantially all of the Company’s assets, or other major corporate transaction.
Item 2. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
The following is a discussion and analysis of the financial condition of Recursion Pharmaceuticals, Inc. (Recursion, the Company, we, us or we) as of March 31, 2021 and December 31, 2020our) and the results of operations during the three months ended March 31, 2021 and 2020.operations. This commentary should be read in conjunction with the unaudited Condensed Consolidated Financial Statements and accompanying notes appearing in Item 1, “Financial Statements” and the Company’s audited consolidated financial statements and accompanying notes and "Management's Discussion and Analysis of Financial Condition and Results of Operations" included in the final prospectusAnnual Report on Form 10-K for our initial public offering (IPO), which was filed with the Securities and Exchange Commission (SEC), pursuant to Rule 424(b)(4) on April 16, 2020 (the Final Prospectus).year ended December 31, 2023. This discussion, particularly information with respect to our future results of operations or financial condition, business strategy and plans and objectives of management for future operations, includes forward-looking statements that involve risks and uncertainties as described under the heading "Special Note"Note About Forward-Looking Statements" in this Quarterly Report on Form 10-Q. You should review the disclosure under the heading "Risk Factors" in this Quarterlythe Annual Report on Form 10-Q10-K for a discussion of important factors that could cause our actual results to differ materially from those anticipated in these forward-looking statements. We assume no obligation to revise or publicly release any revision to any forward-looking statements contained in this Quarterly Report on Form 10-Q, unless required by law.
Investors and others should note that we announce material financial and other information to our investors using our investor relations website (https://ir.recursion.com/), SEC filings, press releases, public conference calls and webcasts. We use these channels as well as social media and blogs to communicate with our stakeholders and the public about our company, our services and other issues. It is possible that the information we post on social media and blogs could be deemed to be material information. Therefore, we encourage investors, the media and others interested in our company to review the information we post on the social media channels and blogs listed on our investor relations website. Information contained in, or that can be accessed through, our website is not a part of, and is not incorporated into, this report.
Overview
We areRecursion is a clinical-stage biotechnologyleading clinical stage TechBio company decoding biology by integrating technological innovations across biology, chemistry, automation, data science and engineering to radically improve the lives of patients and industrialize drug discovery. Central to our mission is the Recursion Operating System (Recursion OS)(OS), a platform built across diverse technologies that combines anenables us to map and navigate trillions of biological, chemical and patient-centric relationships across over 50 petabytes of proprietary data. We frame this integration of the physical and digital components as iterative loops, where scaled ‘wet-lab’ biology, chemistry and patient-centric experimental data are organized by ‘dry-lab’ computational tools in order to identify, validate and translate therapeutic insights. We believe Recursion’s unbiased, data-driven approach to understanding biology will bring more, new and better medicines at higher scale and lower cost to patients.
There are three key value-drivers at Recursion: •An expansive pipeline of internally developed clinical and preclinical programs focused on precision oncology and genetically driven rare diseases with significant unmet need and market opportunities that could potentially exceed $1 billion in annual sales in some cases •Transformational partnerships with leading biopharma and technology companies to map and navigate intractable areas of biology, identify novel targets and develop potential new medicines by using advanced infrastructure layercomputational and data resources •An industry-leading dataset intentionally designed to capitalize on computational tools and accelerate value created through our pipeline, partnerships and technology products
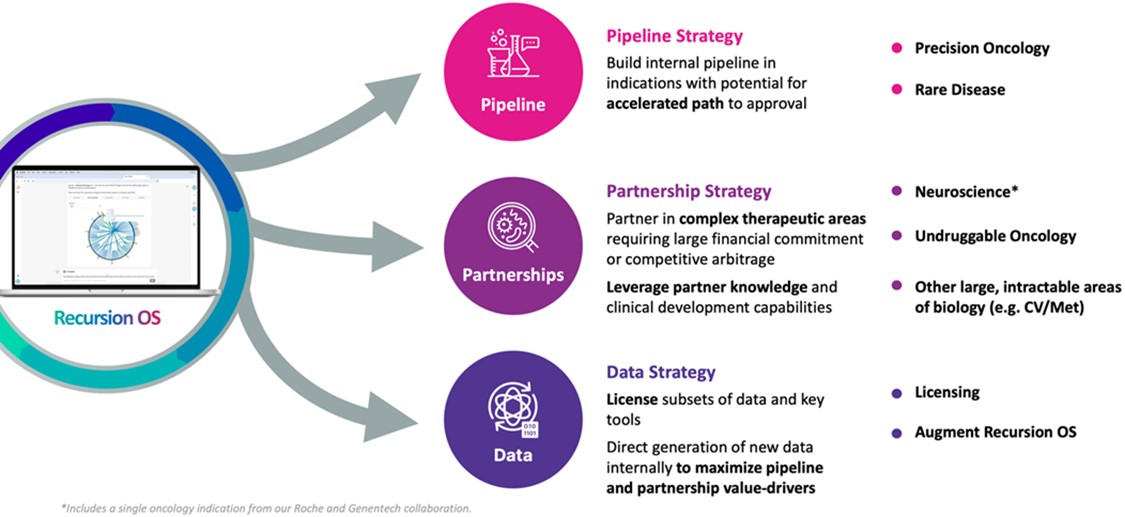
We drive value by scaling and leveraging the Recursion OS to generate, what we believe is oneaggregate and integrate over 50 petabytes of data spanning large language model derived disease relevance and target-compound relationships, predicted protein-ligand binding interactions for ~36 billion compounds, over 250 million total staining and multi-timepoint live-cell (brightfield) phenomics experiments, over 1 million whole transcriptomics experiments, tens of thousands of ADME experiments using our automated DMPK module, InVivomics and multimodal precision oncology patient data. This dataset has been curated using over 50 human cell types, our cell manufacturing facility which has produced over 1 trillion hiPSC-derived neuronal cells since 2022, our in-house chemical library of over 1.7 million compounds, an in silico library of over 1 trillion small molecules and other capabilities. We have built proprietary software applications and AI/ML models within the world’s largestRecursion OS which predict and fastest-growing proprietarynavigate over 6 trillion biological and chemical datasets,relationships. With our approach and the Recursion Map, a suiteour team of custom software, algorithmic and machine learning toolsover 500 Recursionauts that we use to explore foundational biology unconstrained by human bias, navigate to new biological insights, and accelerate programs. The combination of wet-lab biology and in silico tools in our closed-loop system accelerates our drug discovery process and differentiates us from others within the industry. Similarly, ouris balanced team ofbetween life scientists and computational and technical experts, createswe endeavor to turn drug discovery into a search problem, where we map and navigate biology in an environment where empirical data, statistical rigor,unbiased manner in order to translate insights into more, new and creative thinking are broughtbetter medicines at higher scale and lower cost to bear on every decision. Thus far, we have leveraged our Recursion OS to create three value drivers: i) advancement of 37 internally-developed programs focused on areas of significant unmet need, several of which have market opportunities in excess of $1.0 billion in annual sales, ii) strategic partnerships with leading biopharmaceutical companies, and iii) Induction Labs, a growth engine created to explore new extensions of the Recursion OS both within and beyond therapeutics. The number of programs we are advancing has more than doubled in size since 2019. Although we cannot provide any guarantee that we will achieve similar development timelines with future product candidates, we believe we will be able to continue accelerating the pace of program additions in the future. As such, we are a biotechnology company scaling more like a technology company.patients.
Integrating technological innovations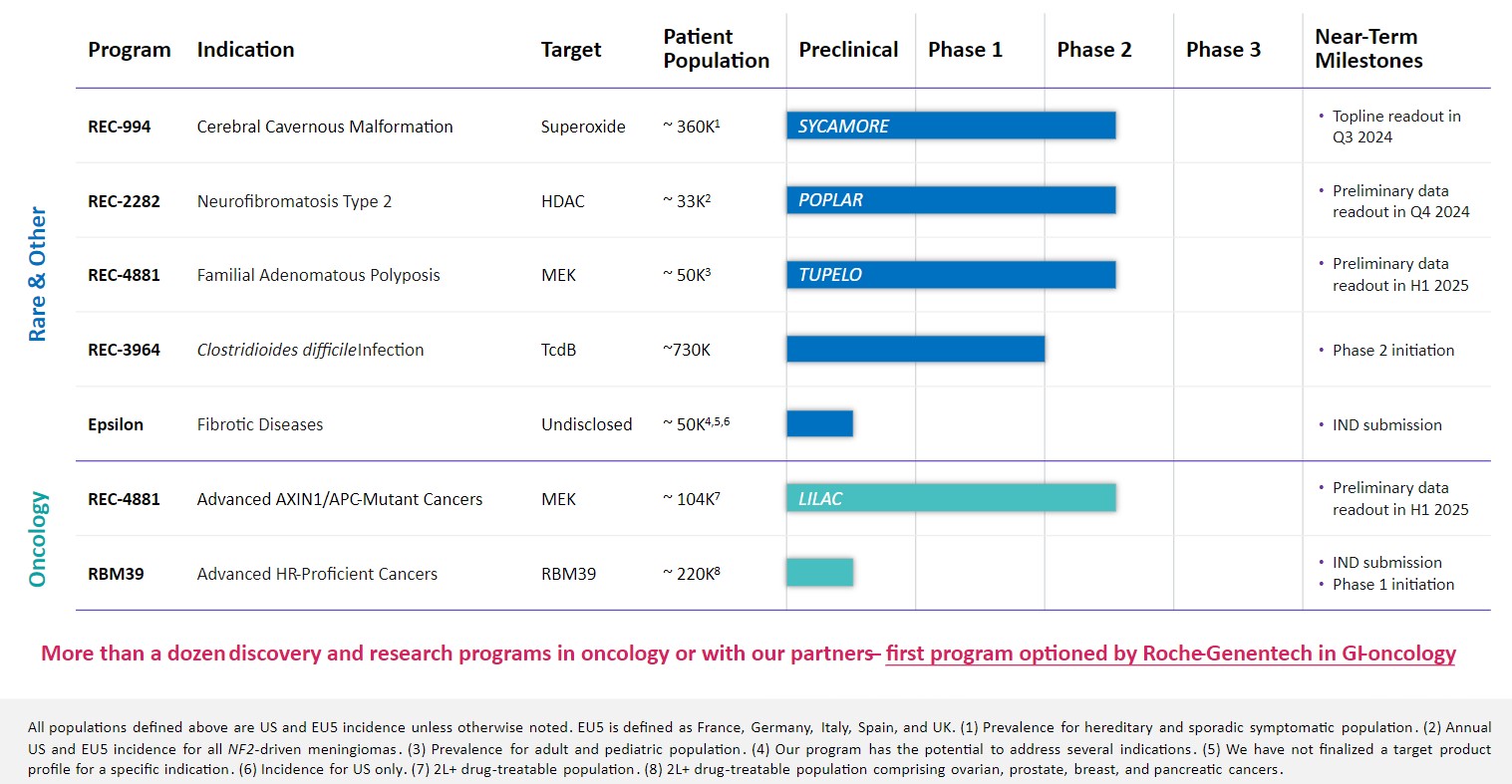
Summary of Business Highlights
Pipeline •Cerebral Cavernous Malformation (CCM) (REC-994): Our Phase 2 SYCAMORE clinical trial is a randomized, double-blind, placebo-controlled study of two doses of REC-994 in participants with CCM. The primary endpoint of the study is safety and tolerability. Secondary and exploratory endpoints, including clinician measured outcomes, imaging of CCM lesions, patient reported outcomes and selected biomarkers, will be evaluated. This trial was fully enrolled in June 2023 with 62 participants, where the vast majority of participants who completed 12 months of treatment have entered the long-term extension study. We expect to share Phase 2 data in Q3 2024. •Neurofibromatosis Type 2 (NF2) (REC-2282): Our adaptive Phase 2/3 POPLAR clinical trial is a randomized, two part study of REC-2282 in participants with progressive NF2-mutated meningiomas. Part 1 of the study is ongoing and is exploring two doses of REC-2282 in approximately 23 adults and 9 adolescents, with enrollment in adults expected to complete in Q2 2024. We expect to share Phase 2 safety and preliminary efficacy data in Q4 2024. •Familial Adenomatous Polyposis (FAP) (REC-4881):Our Phase 1b/2 TUPELO clinical trial is an open label, multicenter, two part study of REC-4881 in participants with FAP. Part 1 is complete and enrollment in Part 2 has commenced. We expect to share Phase 2 safety and preliminary efficacy data in H1 2025. •AXIN1or APCMutant Cancers (REC-4881): Our Phase 2 LILAC clinical trial is an open label, multicenter study of REC-4881 in participants with unresectable, locally advanced or metastatic cancer with AXIN1 or APC mutations. This study was initiated at the end of 2023 with the first participant dosed in Q1 2024. Since that time, multiple participants are now enrolled. We expect to share Phase 2 safety and preliminary efficacy data in H1 2025. •Clostridioides difficile Infection (REC-3964): REC-3964 is a first-in-class C. difficile toxin inhibitor and the first new chemical entity developed by Recursion, with promising preclinical efficacy data seen in relevant models (superiority versus bezlotoxumab). Full Phase 1 data from our healthy volunteers study will be presented at the World Congress on Infectious Diseases in Paris in June 2024. We expect to initiate a randomized Phase 2 study in patients at high risk for C. difficile infection recurrence in 2024. •Advanced HR-Proficient Cancers (RBM39): RBM39 is a novel CDK12-adjacent target identified by the Recursion OS. We intend to position our lead candidate as a single agent for the potential treatment of advanced HR-proficient cancers including ovarian and other solid tumors.We expect to submit an IND in H2 2024 and anticipate initiating a Phase 1 open label study of our lead candidate in participants with relapsed/refractory cancer. The primary endpoint of the study will be safety and tolerability. Secondary endpoints will explore pharmacokinetics and preliminary signs of anti-tumor activity. •Undisclosed Indication in Fibrosis (Target Epsilon): This program originated under our initial fibrosis collaboration with Bayer and we have since in-licensed from Bayer all rights to this program. We are advancing our lead candidate through IND-enabling studies with IND submission expected in the near-term.
Platform •Supercomputer Expansion: We worked with our partner NVIDIA to design and build BioHive-2, our next generation supercomputer with over 500 H100 GPUs. We have nearly completed the build out of BioHive-2 and began performance benchmarking tests. We believe that the performance of our supercomputer may place BioHive-2 in the top 50 of the next TOP500 list, making it one of the most powerful supercomputers in the world across any industry and the most powerful supercomputer owned and operated by any biopharma company. These computational resources, paired with Recursion’s vast datasets and data generation capabilities, enable the construction of Recursion’s large foundation models for biology, chemistry automation, data science and engineeringcausal patient outcomes. •Whole-Genome Transcriptomics Map: We continue to focus on key technologies that enhance our ability to generate, extract and validate novel insights for therapeutic advancements. Over the past year, we have scaled our transcriptomics technology in order to industrialize the discovery of therapeutics has required usvalidate phenotypic-insights and relate to raise significant capital and adopt a long-term approach to capital allocation that balances near-term risks and long-term value creation. Ofpatient-derived RNA sequencing data. In April, we announced sequencing our 37 internally developed programs, we have four drug candidates1 millionth transcriptome. We believe that we expect will be entering clinical trialsare one of the largest transcriptomics sequencers in the next fourworld and are advancing the development of a whole-genome knockout transcriptomics map, which we expect to fivecomplete in the coming quarters. OurSuch platform capabilities are important for curating scaled datasets that are relatable and provide a more complete understanding of biology, chemistry and patient outcomes.
•Active Learning: We have been applying active learning approaches to predict where our OS should generate and enrich biological and chemical datasets via phenotypic and ADME compound profiling across existing and new cellular contexts. These capabilities enable Recursion to rapidly growing teamconstruct multiomics maps that are enriched for areas of biology and chemistry that may be of high value for translating insights into therapeutic programs. We believe that such approaches enable Recursion to more than 200 employees is balanced between life scientists (approximately 40% of employees)rapidly expand its data moat and computational and technical expects (approximately 35% of employees).see active learning capabilities as an important step towards autonomous drug discovery.
From inception through March 31,Partnerships
•Helix Collaboration: Recursion entered into a multi-year agreement with Helix to access hundreds of thousands of de-identified records including Helix’s Exome+(R) genomic data and data from longitudinal health records. Recursion plans to use this data to train causal AI models and design biomarker and patient stratification strategies across broad disease areas. The Helix dataset expands Recursion’s integration of real-world patient data and complements Recursion’s access to Tempus’ oncology data. •Transformational Collaborations: We continue to advance efforts to discover potential new therapeutics with our strategic partners in the areas of undruggable oncology (Bayer) as well as neuroscience and a single indication in gastrointestinal oncology (Roche-Genentech). In the near-term, there is the potential for option exercises associated with partnership programs, option exercises associated with map building initiatives or data sharing and additional partnerships in large, intractable areas of biology or technological innovation.
Financing and Operations
We were incorporated in November 2013. In April 2021, we haveclosed our Initial Public Offering (IPO) and issued 27.9 million shares of Class A common stock at a price of $18.00 per share, raising net proceeds of $462.4 million. Prior to our IPO, we had raised approximately $448.9$448.9 million in equity financing from investors in addition to $30.0$30.0 million in an upfront payment from our strategic partnershipcollaboration with Bayer. Bayer AG (Bayer). In January 2022, we received an upfront payment of $150.0 million from our collaboration with Roche. See Note 9, “Collaborative Development Contracts” to the Condensed Consolidated Financial Statements for additional information on the collaboration with Roche. In October 2022, we issued 15.3 million shares of our Class A common stock at a purchase price of $9.80 per share in the 2022 private placement to qualified institutional buyers and institutional accredited investors for net proceeds of $143.7 million, after deducting fees and offering costs of $6.6 million. In July 2023, we issued an aggregate of 7.7 million shares of our Class A common stock at a purchase price of $6.49 per share in the 2023 Private Placement with NVIDIA Corporation for net proceeds of approximately $49.9 million. In August 2023, we entered into an Open Market Sales Agreement with Jefferies LLC to provide for the offering, issuance and sale of up to an aggregate amount of $300.0 million of its Class A common stock of which $208.2 million remain available for future sales. The Company has sold 12.9 million shares and received net proceeds of $89.1 million under the agreement. See Note 8, “Common Stock” to the Condensed Consolidated Financial Statements for additional information on the Sales Agreement.
We use the capital we have raised to fund operationsoperating and investing activities across platform research operations, drug discovery, clinical development, digital and other infrastructure, creation of our portfolio of intellectual property and administrative support. We do not have any products approved for commercial sale and have not generated any revenues from product sales. We had cash and cash equivalents of $214.1$296.3 million as of March 31, 2021.2024. Based on our current operating plan, we believe that our cash and cash equivalents will be sufficient to fund our operations for at least the next twelve months.
Since inception, we have incurred significant operating losses. Our net losses were $30.7$91.4 million and $18.4$65.3 million during the three months ended March 31, 2024 and 2023, respectively. As of March 31, 2024, our accumulated deficit was $1.1 billion.
We anticipate that we will need to raise additional financing in the future to fund our operations, including the potential commercialization of any approved product candidates. Until such time, if ever, as we can generate significant product revenue, we expect to finance our operations with our existing cash and cash equivalents, any future equity or debt financings and upfront, milestone and royalty payments, if any, received under current or future license or collaboration agreements. We may not be able to raise additional capital on terms acceptable to us or at all. If we are unable to raise additional capital when desired, our business, results of operations and financial condition may be adversely affected.
Results of Operations
The following table summarizes our results of operations:
| | | | | | | | | | | | | | | | (in thousands, except percentages) | Three months ended March 31, | Change | | 2024 | 2023 | $ | % | | Revenue | | | | | | Operating revenue | $ | 13,491 | | $ | 12,134 | | $ | 1,356 | | 11 | % | | Grant revenue | 303 | | — | | 303 | | n/m | | Total revenue | 13,794 | | 12,134 | | 1,659 | | 14 | % | | | | | | | Operating costs and expenses | | | | | | Cost of revenue | 11,166 | | 12,448 | | (1,282) | | (10) | % | | Research and development | 67,560 | | 46,677 | | 20,883 | | 45 | % | | General and administrative | 31,408 | | 22,874 | | 8,534 | | 37 | % | | Total operating costs and expenses | 110,134 | | 81,999 | | 28,135 | | 34 | % | | | | | | | Loss from operations | (96,340) | | (69,865) | | (26,476) | | 38 | % | | Other income, net | 4,188 | | 4,538 | | (351) | | (8) | % | | Loss before income tax benefit | (92,152) | | (65,327) | | (26,827) | | 41 | % | | Income tax benefit | 779 | | — | | 779 | | n/m | | Net loss | $ | (91,373) | | $ | (65,327) | | $ | (26,048) | | 40 | % |
Summary
Our financial performance during the three months ended March 31, 20212024 compared to the prior period included an increase in research and 2020, respectively. As of March 31, 2021, our accumulated deficit was $244.3 million. We expect todevelopment costs across all development stages as we continue to incur significant expensesexpand and operating losses for the
foreseeable future. In addition, we anticipate that our expenses will increase significantly in connection with our ongoing activities, as we:
•continueupgrade our platform, research and drug discovery and clinical development efforts;
•continue to invest in the scale and scope of our platform research capabilities in order to identify novel biology and therapeutics;
•continue to invest in expansions of the modality capabilities across our platform including large molecules and RNA therapeutics;
•invest in or acquire companies or intellectual property that achieves our platform objectives;
•accelerate investments in mechanisms to significantly expand our total addressable markets through Induction Labs;
•utilize our platform to identify and validate additional therapeutic candidates, technologies, and business opportunities;
•initiate additional preclinical studies or clinical or other trials for our product candidates, including under our collaboration agreements;
•continue or expand the scope of our clinical trials for our product candidates;
•conduct the above and below development activities on an extensive pipeline of therapeutic candidates across diverse areas of biology;
•establish agreements with contract research organizations (CROs) and contract manufacturing organizations (CMOs) in connection withadvance our preclinical studiespipeline and progress through our various clinical trials;
•change or add to internal manufacturing capacity or capability;
•change or add additional suppliers;
•seek regulatory approval for our therapeutic candidates;
•seek marketing approvals and reimbursement for our therapeutic candidates;
•establish a sales, marketing, and distribution infrastructure to commercialize any products for which we may obtain marketing approval;
•acquire or in-license other therapeutic candidates and technologies;
•make milestone or other payments under any in-license agreements;
•maintain, protect, defend, enforce, and expand our intellectual property portfolio;
•add additional infrastructure to our quality control, quality assurance, legal, compliance, and other groups to support our operations as we progress our therapeutics candidates toward commercialization;
•add additional infrastructure to support our operations as a public company and our product development and future commercialization efforts, including expansion of company sites;
•attract and retain world-class talent, including in competitive areas; and
•experience any delays or encounter issues with any of the above.
Components of Operating Results
Revenues
To date, our business generates revenue from two sources: i) grant revenue and ii) operating revenue.
Grant Revenue—We recognize grant revenue in the period in which the revenue is earned in accordance with the associated grant agreement, which is the period in which corresponding reimbursable expenses under the grant agreement are incurred. Grant revenue was generated from grants awarded by the National Institute of Health.trials.
Revenue Operating Revenue
The following table summarizes our components of revenue: | | | | | | | | | | | | | | | | Three months ended March 31, | Change | | (in thousands, except percentages) | 2024 | 2023 | $ | % | | Revenue | | | | | | Operating revenue | $ | 13,491 | | $ | 12,134 | | $ | 1,356 | | 11 | % | | Grant revenue | 303 | | — | | 303 | | n/m | | Total revenue | $ | 13,794 | | $ | 12,134 | | $ | 1,659 | | 14 | % |
—
Operating revenue is primarily generated through funded research and development agreements derived from strategic alliances such as our strategic partnership with Bayer.alliances. We are entitled to receive variable consideration as certain milestones are achieved. The timing of revenue recognition is not directly correlated to the timing of cash receipts.
For the three months ended March 31, 2024, the increase in revenue compared to prior period was due to revenue recognized from our partnership with Roche, as our mix of work on the three performance obligations shifted towards higher cost processes including the progression of work related to one of our neuroscience performance obligations.
Cost of Revenue
The following table summarizes our cost of revenue:
| | | | | | | | | | | | | | | | (in thousands, except percentages) | Three months ended March 31, | Change | | 2024 | 2023 | $ | % | | Total cost of revenue | $ | 11,166 | | $ | 12,448 | | $ | (1,282) | | (10) | % |
Cost of revenue consists of the Company’s costs to provide services for drug discovery required under performance obligations with partnership customers. These primarily include materials costs, service hours performed by our employees and depreciation of property and equipment.
For the three months ended March 31, 2024, the decrease in cost of revenue compared to prior period was due to our strategic partnership with Bayer which was completed in 2023.
Research and Development
The following table summarizes our components of research and development expense:
| | | | | | | | | | | | | | | | (in thousands, except percentages) | Three months ended March 31, | Change | | 2024 | 2023 | $ | % | | Research and development expense | | | | | | Platform | $ | 25,914 | | $ | 18,492 | | $ | 7,422 | | 40 | % | | Discovery | 16,642 | | 13,505 | | 3,137 | | 23 | % | | Clinical | 16,597 | | 11,522 | | 5,075 | | 44 | % | | Stock based compensation | 7,995 | | 2,833 | | 5,162 | | >100% | | Other | 412 | | 325 | | 87 | | 27 | % | | Total research and development expense | $ | 67,560 | | $ | 46,677 | | $ | 20,883 | | 45 | % |
Research and development expenses account for a significant portion of our operating expenses. We recognize research and development expenses as they are incurred. Research and development expenses compriseconsist of costs incurred in performing research and development activities including:
•costcosts to develop and operate our platform; •costs of discovery efforts leadingwhich may lead to development candidates; •clinical development costs for our programs;
•costs associated with discovery as well as clinical development efforts,candidates, including research materials and external research;
•costs for clinical development of our investigational products; •costs for materials and supply costssupplies associated with the manufacture of drug substance and drug productactive pharmaceutical ingredients, investigational products for preclinical testing and clinical trials; •personnel-related expenses, including salaries, benefits, bonuses and stock-based compensation for employees engaged in research and development functions; •costs associated with operating our digital infrastructure; and •facilities, depreciation and amortization, insurance and other direct and allocated expenses incurred as a result of research and development activities.
We monitor researchactivities, including those for facilities, depreciation, amortization and development expenses directly associated with our clinical assets to some degree at the program level, however, indirect costs associated with clinical development and the balance of our research and development expenses are not tracked at the program or candidate level.insurance.
We recognize expenses associated with third-party contracted services based on the completion of activities as specified in the applicable contracts.they are incurred. Upon termination of contracts with third parties, our financial obligations are generally limited to costs incurred or committed to date. Any advance payments for goods or services to be used or rendered in future research and product development activities pursuant to a contractual arrangement are classified as prepaid expenses until such goods or services are rendered.
Significant components of research and development expense include the following allocated by development phase: Platform, which refers primarily to expenses related to screening of product candidates through hit
identification; Discovery, which refers primarily to expenses related to hit identification through development of candidates; and Clinical, which refers primarily to expenses related to development of candidates and beyond.
For the three months ended March 31, 2024, the increase in research and development expenses compared to the prior period was across all development phases as we continue to expand and upgrade our platform, including our chemical technology, machine learning and transcriptomics platform. Our discovery costs increased as we advanced our preclinical pipeline including our work on Target Epsilon. Our clinical costs grew as we continued to progress through our various clinical trials.
General and Administrative Expense
The Company expensesfollowing table summarizes our general and administrative expense:
| | | | | | | | | | | | | | | | (in thousands, except percentages) | Three months ended March 31, | Change | | 2024 | 2023 | $ | % | | Total general and administrative expense | $ | 31,408 | | $ | 22,874 | | $ | 8,534 | | 37 | % |
We expense general and administrative costs as incurred. General and administrative expenses consist primarily of salaries,salaries; including employee benefits and stock-based compensation, and outsourced labor for personnel in executive, finance, human resources, legal and other corporate administrative functions.compensation. General and administrative expenses also include facilities, depreciation, information technology, professional fees for auditing and tax, legal fees incurred relating tofor corporate and patent matters professional fees incurred for accounting, auditing, tax and administrative consulting services, insurance costs, facilities and depreciation expenses.
Recursion expects that ourFor the three months ended March 31, 2024, the increase in general and administrative expenses willexpense compared to prior period was primarily driven by an increase in the future to support personnelsalaries and wages of $3.9 million and increases in researchsoftware and development and to support our operations as we increase our research and development activities and activities related to the potential commercialization of our initial drug candidates REC-4881, REC-3599, REC-2282, and REC-994. The Company also expects to incur increased expenses associated with operating as a public company, including costs of accounting, audit, legal, regulatory, and tax-related services associated with maintaining compliance with exchange listing and Securities and Exchange Commission, or SEC, requirements, director and officer insurance costs, and investor and public relations costs.depreciation expense.
Other Income, Net
OtherThe following table summarizes our components of other income, net primarily consists of interest earned on our cash and cash equivalents and interest expense incurred under our loan agreements.
Results of Operationsnet:
Comparison of | | | | | | | | | | | | | | | | (in thousands, except percentages) | Three months ended March 31, | Change | | 2024 | 2023 | $ | % | | Interest income | $ | 4,048 | | $ | 4,660 | | $ | (612) | | (13.1) | % | | Interest expense | (20) | | (19) | | (1) | | 7.6 | % | | Other | 160 | | (103) | | 263 | | 256.1 | % | | Other income, net | $ | 4,188 | | $ | 4,538 | | $ | (350) | | (7.7) | % |
n/m = Not meaningful
For the three months ended March 31, 2021 and 2020
The following table summarizes2024, the Company’s results of operations:
| | | | | | | | | | | | | | | | | Three months ended March 31, | Change | | (in thousands, except percentages) | 2021 | 2020 | $ | % | | Revenue | | | | | | Grant revenue | $ | 62 | | $ | 60 | | $ | 2 | | 3.5 | % | | Operating revenue | 2,500 | | — | | 2,500 | | n/m | | Total revenue | 2,562 | | 60 | | 2,502 | | >100% | | | | | | | Operating expenses | | | | | | Research and development | 24,109 | | 12,842 | | 11,267 | | 87.7 | % | | General and administrative | 8,937 | | 5,561 | | 3,376 | | 60.7 | % | | Total operating expenses | 33,046 | | 18,403 | | 14,643 | | 79.6 | % | | | | | | | Loss from operations | (30,484) | | (18,343) | | (12,141) | | 66.2 | % | | Other loss, net | (233) | | (81) | | (152) | | >100% | | Net loss and comprehensive loss | $ | (30,717) | | $ | (18,424) | | $ | (12,293) | | 66.7 | % |
n / m = Not meaningful
Revenue
The following table summarizes the components of revenue recognized during the three months ended March 31, 2021 and 2020:
| | | | | | | | | | | | | | | | Three months ended March 31, | Change | | (in thousands, except percentages) | 2020 | 2019 | $ | % | | Revenue | | | | | | Grant revenue | $ | 62 | | $ | 60 | | $ | 2 | | 3.5 | % | | Operating revenue | 2,500 | | — | | 2,500 | | n/m | | Total revenue | $ | 2,562 | | $ | 60 | | $ | 2,502 | | >100% |
Revenue increased by $2.5 million, or >100%, to $2.6 million during the three months ended March 31, 2021 in interest income compared to $60 thousand during the three months ended March 31, 2020. The increase in revenue was due to revenue recognized from our strategic partnership with Bayer entered into in August 2020.
Research and Development
The following table summarizes the components of research and development expense during the three months ended March 31, 2021 and 2020:
| | | | | | | | | | | | | | | | Three months ended March 31, | Change | | (in thousands, except percentages) | 2021 | 2020 | $ | % | | Research and development expenses | | | | | | Platform | $ | 10,532 | | $ | 6,319 | | $ | 4,212 | | 66.7 | % | | Discovery | 7,739 | | 4,047 | | 3,692 | | 91.2 | % | | Clinical | 2,955 | | 1,323 | | 1,632 | | >100% | | Stock based compensation | 628 | | 688 | | (60) | | (8.7) | % | | Other | 2,255 | | 465 | | 1,789 | | >100% | | Total research and development expenses | $ | 24,109 | | $ | 12,842 | | $ | 11,265 | | 87.7 | % |
Significant components of research and development expense include the following: Platform, which refers primarily to expensesprior period related to screening through hit identification; Discovery, which refers primarily to expenses related to hit identification through development candidate; and Clinical, which refers primarily to expenses related to development candidate and beyond.
Research and development expenses increased by $11.3 million, or 87.7%, to $24.1 million during the three months ended March 31, 2021 compared to $12.8 million during the three months ended March 31, 2020. The increase in research and development expenses was due to an increased number of experiments screened on the platform, an increased number of pre-clinical assets being validated and increased clinical costs as studies progress.
General and Administrative Expenses
The following table summarizes the components of general and administrative expense during the three months ended March 31, 2021 and 2020:
| | | | | | | | | | | | | | | | Three months ended March 31, | Change | | (in thousands, except percentages) | 2021 | 2020 | $ | % | | Total general and administrative expenses | $ | 8,937 | | $ | 5,561 | | $ | 3,376 | | 60.7 | % |
General and administrative expenses increased by $3.4 million, or 60.7%, to $8.9 million during the three months ended March 31, 2021 compared to $5.6 million during the three months ended March 31, 2020. The increase in general and administrative expenses was due to growth in size of the Company’s operations including an increase in salaries and wages of $1.2 million, human resources costs, facilities costs, finance costs and other administrative costs associated with operating a growth-stage Company.
Other loss, net
The following table summarizes the components of Other loss, net during the three months ended March 31, 2021 and 2020:
| | | | | | | | | | | | | | | | | Three months ended March 31, | Change | | (in thousands, except percentages) | 2021 | 2020 | $ | % | | Interest expense | $ | 249 | | $ | 301 | | $ | (52) | | (17.2) | % | | Interest income | (16) | | (220) | | 204 | | (92.8) | % | | Other loss, net | $ | 233 | | $ | 81 | | $ | 152 | | >100% |
Other loss, net increased by $152 thousand to $233 thousand during the three months ended March 31, 2021 compared to $81 thousand during the three months ended March 31, 2020. The increase in Other loss, net was primarily due to a decrease in interest earned from the Company’s checking accounts.earnings on cash and cash equivalents in money market funds.
Liquidity and Capital Resources
Sources of Liquidity
The Company hasWe have not yet commercialized any products and doesdo not expect to generate revenue from the sales of any product candidates for at least several years. Cash and cash equivalents totaled $214.1$296.3 million and $391.6 million as of March 31, 20212024 and $262.1 million as of December 31, 2020.2023, respectively.
The Company hasWe have incurred operating losses and experienced negative operating cash flows and Recursion anticipateswe anticipate that the Company will continue to incur losses for at least the foreseeable future. Our net loss totaled $30.7was $91.4 million and $65.3 million during the three months ended March 31, 20212024 and $18.42023, respectively. As of March 31, 2024 and December 31, 2023, we had an accumulated deficit of $1.1 billion and $967.6 million, respectively.
We have financed our operations through the private placements of preferred stock and Class A common stock issuances. As of March 31, 2024, we have received net proceeds of $448.9 million from the sale of preferred stock and $745.0 million from Class A common stock issuances. See Note 8, “Common Stock” to the Condensed Consolidated Financial Statements for additional details on Class A common stock issuances. Additionally, as of March 31, 2024, we have received proceeds of $183.0 million from our strategic partnerships. See Note 9, “Collaborative Development Contracts” to the Condensed Consolidated Financial Statements for additional details on the Roche partnership.
Cash Flows
The following table is a summary of the Condensed Consolidated Statements of Cash Flows for each of the periods presented below:
| | | | | | | | | | Three months ended
March 31, | | (in thousands) | 2024 | 2023 | | Cash used in operating activities | $ | (102,300) | | $ | (73,316) | | | Cash used in investing activities | (6,653) | | (5,340) | | | Cash provided by financing activities | 13,897 | | 1,922 | |
Operating Activities Cash used by operating activities increased during the three months ended March 31, 2020. As of March 31, 2021 and December 31, 2020, Recursion had an accumulated deficit of $244.3 million and $213.6 million, respectively.
To date, Recursion has financed the Company’s operations primarily through private placements of preferred stock. Through March 31, 2021, the Company has received gross proceeds of $448.9 million from sales of our preferred stock. In April 2021, the Company completed an IPO receiving an approximate net proceeds of $462.6 million. See Note 16, “Subsequent Events” to the Condensed Consolidated Financial Statements for additional detail.
Over September and October 2020, the Company received a $30.0 million upfront payment from the Company’s strategic partnership with Bayer.
Midcap Credit and Security Agreement
In September 2019, we entered into a Credit and Security Agreement with Midcap Financial Trust (Midcap), which we refer to as our Credit Agreement. The Credit Agreement includes: i) an initial term loan in an aggregate principal amount of $11.9 million; and ii) a second tranche term loan, which if drawn would result in an aggregate outstanding maximum principal amount of $26.9 million. The second tranche will become available to be drawn upon the achievement of certain drug development milestones. We are required to make interest-only payments from September 2019 to September 2021, and thereafter, 36 monthly principal payments of $330 thousand plus interest commencing in October 2021 and continuing until the maturity date in September 2024. The interest-only period will be extended an additional 12 months upon achievement of certain fundraising related milestones. Interest accrues on the principal amount outstanding at a floating per annum rate equal to the LIBOR (floor of 2.00%) rate plus 5.75%.
The debt is secured against all of our assets. The Credit Agreement includes standard affirmative and restrictive covenants and standard events of default, including payment defaults, breaches of covenants following any applicable cure period, a material impairment in the perfection or priority of Midcap’s security interest or in the value of the collateral and a material adverse change in our business, operations, or conditions. Upon the occurrence of an event of default and following any applicable cure periods, Midcap may declare all outstanding obligations immediately due and payable and take such other actions as set forth in the Credit Agreement. As of March 31, 2021, the Company was in compliance with all debt covenants under the Credit Agreement. In 2019, we paid fees of approximately $298 thousand in connection with the origination of the Credit Agreement. These fees were deferred and recorded2024 as a direct deduction from the carrying valueresult of the loan payable and are amortized to interest expense over the remaining term of the Credit Agreement.
Cash Flows
The following table sets forth the primary sources and uses of cash and cash equivalents for each of the periods presented below:
| | | | | | | | | | Three months ended March 31, | | (in thousands) | 2021 | 2020 | | Cash used in operating activities | $ | (30,755) | | $ | (17,817) | | | Cash used in investing activities | (19,416) | | (684) | | | Cash provided by financing activities | 2,134 | | 5,997 | | | Net decrease in cash and cash equivalents | $ | (48,037) | | $ | (12,504) | |
Operating Activities
Net cash used in operating activities was $30.8 million during the three months ended March 31, 2021. Net cash used in operating activities increased from the three months ended March 31, 2020 due to higher costs incurred for research and development and general and administrative due to the Company’s growth as well asexpansion and upgraded capabilities.
Cash used by operating activities increased during the timing of working capital cash flows for the three months ended March 31, 2021. Net cash used in operating activities was $17.82023 as a result of an upfront payment of $150.0 million from our strategic partnership with Roche received during the three months ended March 31, 2020. Cash used in operating activities increased from the three months ended March 31, 2019 due to higher costs incurred for research and development and general and administrative due to the Company’s growth which was partially offset by the timing of working capital cash flows for the three months ended March 31, 2020.2022.
Investing Activities Net cashCash used inby investing activities was $19.4 million during the three months ended March 31, 2021. Cash used in investing activities was2024 consisted primarily for the purchase of a Dell EMC supercomputer as well as accessoriesproperty and partsequipment purchases of $6.7 million, which included $2.9 million for a total of $17.9 million.project to upgrade the BioHive supercomputer and $2.7 million for lab equipment purchases.
Net cashCash used inby investing activities was $684 thousandduring the three months ended March 31, 2023 consisted primarily of property and equipment purchases of $5.2 million, which included $1.7 million for a project to upgrade the BioHive supercomputer and $2.3 million for lab equipment purchases.
Financing Activities Cash provided by financing activities during the three months ended March 31, 2020. Cash used in investing activities was 2024 primarily for the purchaseincluded proceeds of lab equipment and leasehold improvements.$10.9 million from common stock issuances. Financing inflows also included proceeds from equity incentive plans of $3.0 million.
Financing Activities
Net cash Cash provided by financing activities was $2.1 million during the three months ended March 31, 2021. Cash provided by financing activities2023 primarily consisted of $2.2 million ofincluded proceeds from the exerciseequity incentive plans of stock options.
Net cash provided by financing activities was $6.0 million during the three months ended March 31, 2020, which consisted primarily of $6.0 million of proceeds from the issuance of convertible notes. See Note 5, “Notes Payable” to the Condensed Consolidated Financial Statements for additional detail on the convertible notes.
Future Funding Requirements
Since inception, the Company has incurred significant operating losses. Given our broad and ambitious mission, we expect to continue to incur significant expenses and operating losses for the foreseeable future.
The Company believes that the net proceeds from the IPO, together with the Company’s existing cash, and cash equivalents and borrowings available to us will be sufficient to fund the Company’s operating expenses and capital expenditures for at least the next 12 months. The Company’s assumptions that may be incorrect and we could exhaust our available capital resources sooner than we expect.
Recursion does not expect to generate significant revenue from out-licensing transactions, development milestones, or royalties until successfully completing significant drug development milestones, whether on our own or in collaboration with third parties, which Recursion expects will take a number of years. In order to commercialize the Company’s drug candidates, we or our partners need to complete clinical development and comply with comprehensive regulatory requirements. Recursion is subject to a number of risks and uncertainties similar to those of other companies of the same size within the biotechnology industry, such as uncertainty of clinical trial outcomes, uncertainty of additional funding, and history of operating losses.
Developing pharmaceutical products, including conducting preclinical studies and clinical trials, is a time-consuming, expensive, and uncertain process that takes years to complete, and we may never generate the necessary data or results required to obtain marketing approval for any product candidates or generate revenue from the sale of any product candidate for which we may obtain marketing approval. In addition, our drug candidates, if approved, may not achieve commercial success. Our commercial revenues, if any, will be derived from sales of drugs that we do not expect to be commercially available for many years, if ever. Accordingly, we will need to obtain substantial additional funds to achieve our business objectives.
Adequate additional funds may not be available to us on acceptable terms, or at all. We do not currently have any committed external source of funds. To the extent that we raise additional capital through the sale of equity or convertible debt securities, your ownership interest may be diluted, and the terms of these securities may include liquidation or other preferences and anti-dilution protections that could adversely affect your rights as a common stockholder. Additional debt or preferred equity financing, if available, may involve agreements that include restrictive covenants that may limit our ability to take specific actions, such as incurring debt, making capital expenditures, or declaring dividends, which could adversely impact our ability to conduct our business, and may require the issuance of warrants, which could potentially dilute your ownership interest further.
If we raise additional funds through collaborations, strategic alliances, or licensing arrangements with third parties, we may have to relinquish valuable rights to our technology, future revenue streams, research programs, or product candidates or grant licenses on terms that may not be favorable to us. If we are unable to raise additional funds through equity or debt financings or collaborations, strategic alliances, or licensing arrangements with third parties when needed, we may be required to delay, limit, reduce, and/or terminate our product development programs or any future commercialization efforts or grant rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves.
Critical Accounting Estimates and Policies
A summary of the Company’s significant accounting estimates and policies is included in Note 2, “Summary of Significant Accounting Policies” in our Final Prospectus.2023 Annual Report. There have beenwere no significant changes in the company’sCompany’s application of its critical accounting policies during the three months ended March 31, 2021. 2024
Impact of the COVID-19 Pandemic
In March 2020, the World Health Organization declared the outbreak of novel coronavirus disease, or COVID-19, as a pandemic. The COVID-19 pandemic is evolving, and to date has led to the implementation of various responses, including government-imposed quarantines, travel restrictions and other public health safety measures. COVID-19 has caused market volatility and uncertainty around the world in various industries and, as a result, we expect our operations may also be affected. The Company is closely monitoring the impact of the pandemic of COVID-19 on all aspects of Recursion’s business. The extent to which COVID-19 ultimately impacts our operations and financial position will depend on future developments, which are highly uncertain and cannot be predicted with confidence, such as the duration of the outbreak, new information that may emerge concerning the severity of COVID-19 or the effectiveness of actions to contain COVID-19 or treat its impact, among others. In addition, recurrences or additional waves of COVID-19 cases could cause other widespread or more severe impacts depending on where infection rates are highest.
The Company has not incurred any significant impairment losses in the carrying values of our assets as a result of the pandemic and we are not aware of any specific related event or circumstance that would require us to revise our estimates reflected in our audited consolidated financial statements.
Emerging Growth Company
The Company is an emerging growth company (EGC), as defined by the Jumpstart Our Business Startups Act of 2012 (the JOBS act). The JOBS Act, exempts EGCs from being required to comply with new or revised financial accounting standards until private companies are required to comply. Recursion as elected to use the extended transition period for new or revised financial accounting standards during the period in which we remain an EGC. However, the Company may adopt certain new or revised accounting standards early. This may make comparisons
of the Company’s financial statements with other public companies difficult because of the potential differences in accounting standards used.
Recursion may remain an EGC until December 31, 2026 although if we: (1) become a “large accelerated filer;” (2) have annual gross revenues of $1.07 billion or more in any fiscal year; or (3) issue more than $1.0 billion of non-convertible debt over a three-year period, the Company would cease to be an EGC as of December 31 of the applicable year.
Recently Issued and Adopted Accounting Pronouncements
Refer toSee Note 2, “Basis of Presentation” in Item 1 of this Quarterly Report on Form 10-Q for information regarding recently issued and adopted accounting pronouncements.
Item 3. Quantitative and Qualitative Disclosures About Market Risk.
The Company is subjectInterest Rate Risk
We are exposed to market risk associated with changing interest rates on our variable rate note issued under our Credit Agreement with Midcap; the interest accrues on the principal amount outstanding at a floating per annum rate equalrelated to the LIBOR rate plus 5.75% with a LIBOR floor of 2.00%. The interest rates applicable to our variable rate note may rise and increase the amount of interest expense. We do not purchase or hold any derivative instruments to protect against the effects of changes in interest rates. As of March 31, 2021 and December 31, 2020 the outstanding balance on the debt issued under our Credit Agreement with Midcap was $11.9 million.
The Company’s cash and cash equivalents consist primarily of highly liquid investments in money market funds and cash on hand and have an original maturity date of 90 days or less. The fair valuerates of our cash and cash equivalents. As of March 31, 2024, our cash and cash equivalents would not be significantlyprimarily consisted of money market funds. Our primary exposure to market risk is interest income sensitivity, which is affected by eitherchanges in U.S. interest rates. A hypothetical 100 basis point decrease in interest rates as of as of March 31, 2024, would have an insignificant effect on net loss in the ensuring year.
Foreign Currency Exchange Risk
Our employees and our operations are primarily located in the United States and Canada and our expenses are generally denominated in U.S. and Canadian dollars. We also have entered into a limited number of contracts with vendors for research and development services that have underlying payment obligations denominated in foreign currencies. We are subject to foreign currency transaction gains or losses on our contracts denominated in foreign currencies. To date, foreign currency transaction gains and losses have not been material to our financial statements, and we do not have a formal hedging program with respect to foreign currency. A 10% increase or decrease in interestcurrent exchange rates due mainly towould have an insignificant effect on our financial results during the short-term nature of these instruments.three months ended March 31, 2024 and 2023.
Item 4. Controls and Procedures.
The Company has established disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended or the(the Exchange Act) designed to ensure that information required to be disclosed in the reports that the Company files or submits under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms and is accumulated and communicated to management, including the principal executive officer (our Chief Executive Officer) and principal financial officer (our Chief Financial Officer), to allow timely decisions regarding required disclosure. In addition, the design of disclosure controls and procedures must reflect the fact that there are resource constraints and that management is required to apply judgment in evaluating the benefits of possible controls and procedures relative to their costs.
Evaluation of Disclosure Controls and Procedures
Our management has evaluated, with the participation of our Chief Executive Officer and Chief Financial Officer, the effectiveness of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act) as of the end of the period covered by this Quarterly Report on Form 10-Q.report. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives andas management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Our disclosure controls and procedures have been designed to provide reasonable assurance of achieving their objectives. Based on that evaluation, our Chief Executive Officer and Chief Financial Officer have concluded that, as of March 31, 2021,2024, our disclosure controls and procedures were effective.ineffective due to the material weakness in internal controls related to our revenue and unearned revenue process including the operating effectiveness of management review controls over the estimated costs and time to completion and controls to validate the completeness and accuracy of information used to calculate revenue and unearned revenue related to its license agreement disclosed in Part II, Item 9A of our Annual Report on Form 10-K for the year ended December 31, 2023.
Remediation of Material Weakness
The following remediation actions have been taken as of March 31, 2024:
•Improvement of documentation procedures regarding specific inquiries related to the cost model used for revenue recognition and the resulting responses •Improvement of documentation for the review of changes in cost model due to responses from inquiries •Provided additional documentation for internal reports to validate and support completeness and accuracy of reports
•Improvement of documentation of these processes was done with the input of our third-party consultants who continue to be involved in the design and enhancement of the revenue recognition policies and procedures
While significant progress has been made to enhance our internal controls over financial reporting, we are still in the process of implementing and testing these remediated processes, procedures and controls. We believe the above actions will be effective in remediating the material weakness described above. However, the material weakness cannot be considered remediated until controls operate for a sufficient period of time and management has concluded, through testing, that these controls are operating effectively. As such, we were unable to conclude that the material weakness has been remediated as of March 31, 2024.
Changes in Internal Control Over Financial Reporting
There has beenAs of March 31, 2024, management is in the process of integrating the internal controls of the acquired businesses into Recursion's existing operations as part of planned integration activities. With the exception of the steps taken to remediate the material weakness as described above, there were no changeother changes in our internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act) that occurred during the quarter ended March 31, 20212024 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
As a result of the COVID-19 pandemic, many of the Company’s employees are working remotely. Recursion has not identified any material changes in our internal control over financial reporting as a result of these changes to the working environment, in part because our internal control over financial reporting was designed to operate in a remote working environment. The Company is continually monitoring and assessing the COVID-19 situation to determine any potential impacts on the design and operating effectiveness of our internal controls over financial reporting.
PART II - OTHER INFORMATION
Item 1. Legal Proceedings.
The Company may, from time to time, be involved in various legal proceedings arising in the normal course of business. An unfavorable resolution of any such matter could materially affect the Company’s future financial position, results of operations or cash flows. For more information inpertaining to legal proceedings, see Part I, Item 1, Note 67, “Commitments and Contingencies,” which is incorporated herein by reference.
Item 1A. Risk Factors.
RISK FACTORS
You should carefully consider the risks and uncertainties described below together with all of the other information containedInvesting in this Quarterly Report on Form 10-Q and our other public filings with the SEC. The risks described below are not the only risks facing our company. The occurrence of any of the following risks, or of additional risks and uncertainties not presently known to us or that we currently believe to be immaterial, could cause our business, prospects, operating results, and financial condition to suffer materially.
Risks Related to Our Financial Position and Need for Additional Capital
We are a clinical-stage biotechnology company with a limited operating history.
We are a clinical-stage biotechnology company with a limited operating history. Biopharmaceutical product development is a highly speculative undertaking andcommon stock involves a substantialhigh degree of risk. SinceFor a detailed discussion of the risks that affect our inceptionbusiness. Please refer to the sections titled “Risk Factors” in November 2013, we have focused substantially allPart I, Item 1A. of our efforts and financial resources on building our drug discovery platform and developing our initial drug candidates. We have no products approved for commercial sale and therefore have never generated any revenue from drug product sales, and we do not expect to generate any revenue from drug product sales in the foreseeable future. We have not obtained regulatory approvals to market any of our drug candidates and there is no assurance that we will obtain regulatory approvals to market and sell drug products in the future.
We have incurred significant operating losses since our inception and anticipate that we will incur continued losses for the foreseeable future.
We have incurred net losses in each year since our inception. Our net losses were $30.7 million and $18.4 million for the three months ended March 31, 2021 and 2020, respectively. We had an accumulated deficit of $244.3 million as of March 31, 2021. Substantially all of our operating losses have resulted from costs incurred in connection with research and development efforts, including clinical studies, and from general and administrative costs associated with our operations. We expect our operating expenses to significantly increase as we continue to invest in research and development efforts and the commencement and continuation of clinical trials of our existing and future drug candidates. In addition, if we obtain marketing approval for any drug candidates, we will incur significant sales, marketing, and outsourced-manufacturing expenses. We continue to incur additional costs associated with operating as a public company. As a result, we expect to continue to incur significant and increasing operating losses for the foreseeable future. Our prior losses, combined with expected future losses, have had and will continue to have an adverse effect on our stockholders’ deficit and working capital. Because of the numerous risks and uncertainties associated with developing pharmaceutical products and new technologies, we are unable to predict the extent of any future losses or when we will become profitable, if at all. Even if we do become profitable, we may not be able to sustain or increase our profitability on a quarterly or annual basis.
Our mission is broad and expensive to achieve and we will need to raise substantial additional funding. If we are unable to raise capital when needed, we would be forced to delay, reduce, or eliminate at least some of our product development programs, business development plans, strategic investments, or potential commercialization efforts.
We have ambitious plans to decode biology and deliver new drugs to the patients that need them. Our mission is broad, expensive to achieve and will require additional capital in the future. In addition, the development of pharmaceutical products is capital-intensive. We have four clinical stage programs and 33 additional programs in various stages of preclinical development. We expect our expenses to increase in connection with our ongoing
activities, particularly as we continue the research and development of, initiate clinical trials of, and potentially seek marketing approval for, our drug candidates, and add to our pipeline what we believe will be an accelerating number of additional programs. In addition, depending on the status of potential regulatory approval, or if we obtain marketing approval for any current or future drug candidates, we could expect to incur significant expenses related to product sales, marketing, manufacturing and distribution. We may also need to raise additional funds sooner if we choose to pursue additional indications and/or geographies for our drug candidates or otherwise expand more rapidly than we presently anticipate. Furthermore, , we have incurred, and expect to continue to incur, additional costs associated with operating as a public company. Accordingly, we will need to obtain substantial additional funding in connection with our continuing operations. If we are unable to raise capital when needed or on attractive terms, we would be forced to delay, reduce, or eliminate certain of our research and development programs or potential future commercialization efforts.
We expect that the net proceeds from our initial public offering completed on April 20, 2021, together with our existing cash and cash equivalents, borrowings available to us and short-term investments as of the date of this Quarterly Report on Form 10-Q, will be sufficient to fund our operating expenses and capital expenditures for at least the next 12 months. Our future capital requirements will depend on and could increase significantly as a result of many factors, including:
• the impact of any business interruptions to our operations, including the timing and enrollment of participants in our planned clinical trials, or to operations of our manufacturers, suppliers, or other vendors resulting from the COVID-19 pandemic or a similar public health crisis or other force majeure event;
• the scope, progress, results and costs of our current and future clinical trials and additional preclinical research for our programs;
• the number of future drug candidates that we pursue and their development requirements;
• the costs, timing, and outcome of regulatory review of our drug candidates;
• our ability to establish and maintain collaborations on favorable terms, if at all;
• the success of any collaborations that we may enter into with third parties;
• the extent to which we acquire or invest in businesses, products, and technologies, including entering into licensing or collaboration arrangements for drug candidates;
• the costs of preparing, filing, and prosecuting patent applications, maintaining, protecting, and enforcing our intellectual property rights and defending intellectual property-related claims;
• our headcount growth and associated costs as we expand our business operations and our research and development activities; and
• the costs of operating as a public company.
Identifying potential drug candidates and conducting preclinical development testing and clinical trials is a time-consuming, expensive, and uncertain process that takes years to complete, and we may never generate the necessary data or results required to obtain marketing approval and achieve product sales. In addition, our drug candidates, if approved, may not achieve commercial success. We anticipate that our commercial revenues, if any, will be derived from sales of products that we do not expect to be commercially available for many years, if at all. Accordingly, we will need to continue to rely on additional financing to achieve our business objectives.
Any additional fundraising efforts may divert our management from day-to-day activities, which may adversely affect our ability to develop and commercialize our drug candidates and technologies, and we can provide no assurance that such funding will be available on terms that are acceptable to us, or at all.
Until such time, if ever, as we can generate substantial revenues, we expect to finance our cash needs through a combination of private and public equity offerings, debt financings, strategic collaborations, strategic alliances, and licensing arrangements. We do not have any committed external source of funds. Disruptions in the financial markets in general and more recently due to the COVID-19 pandemic may make equity and debt financing more difficult to obtain and may have a material adverse effect on our ability to meet our fundraising needs. We cannot guarantee that future financing will be available in sufficient amounts or on terms acceptable to us, if at all. Moreover, the terms of any financing may adversely affect the holdings or the rights of our stockholders and the issuance of additional securities, whether equity or debt, by us, or the possibility of such issuance, may cause the market price of our shares to decline. To the extent that we raise additional capital through the sale of Class A common stock or securities convertible or exchangeable into Class A common stock, our stockholders’ ownership interest will be diluted, and the terms of those securities may include liquidation or other preferences that materially adversely affect our stockholders’ rights as a common stockholder. The incurrence of indebtedness would result in
increased fixed payment obligations and we may be required to agree to certain restrictive covenants, such as limitations on our ability to incur additional debt, limitations on our ability to acquire, sell or license intellectual property rights and other operating restrictions that could adversely impact our ability to make capital expenditures, declare dividends, or otherwise conduct our business. We could also be required to seek funds through arrangements with collaborators or otherwise at an earlier stage than otherwise would be desirable and we may be required to relinquish rights to some of our technologies or drug candidates, future revenue streams, or research programs or otherwise agree to terms unfavorable to us. If we are unable to obtain funding on a timely basis, we may be required to significantly curtail, delay or discontinue one or more of our research or development programs or the commercialization of any drug candidate or be unable to expand our operations or otherwise capitalize on our business opportunities, as desired, which could materially affect our business, financial condition, and results of operations.
We have no products approved for commercial sale and have not generated any revenue from product sales.
Our ability to become profitable depends upon our ability to generate substantial revenue in an amount necessary to offset our expenses. To date, we have not generated any revenue from our drug candidates or technologies, other than limited grant revenues, milestone payments from Takeda Pharmaceutical Company Limited and a technology access fee from Bayer, and we do not expect to generate any revenue from the sale of products in the near future. We do not expect to generate significant revenue unless and until we progress our drug candidates through clinical trials and obtain marketing approval of, and begin to sell one or more of our drug candidates, or otherwise receive substantial licensing or other payments. Our ability to generate revenue depends on a number of factors, including, but not limited to, our ability to:
• successfully complete preclinical studies;
• have Investigational New Drug, or IND, applications approved by the U.S. Food Drug Administration, or FDA, allowing us to commence clinical trials;
• successfully enroll subjects in, and complete, clinical trials;
• receive regulatory approvals from applicable regulatory authorities;
• initiate and successfully complete all safety and other studies required to obtain U.S. and foreign marketing approval for our drug candidates;
• establish commercial manufacturing capabilities or make arrangements with third-party manufacturers for clinical supply and commercial manufacturing;
• obtain and maintain patent and trade secret protection or regulatory exclusivity for our drug candidates;
• launch commercial sales of our drug candidates, if and when approved, whether alone or in collaboration with others;
• obtain and maintain acceptance of the drug candidates, if and when approved, by patients, the medical community, and third-party payors;
• effectively compete with other therapies;
• obtain and maintain healthcare coverage and adequate reimbursement;
• protect and enforce our intellectual property rights and defend against intellectual property claims;
• take temporary precautionary measures to help minimize the impact of the COVID-19 pandemic or other force majeure event on our business; and
• maintain a continued acceptable safety profile of the drug candidates following approval.
If we do not achieve one or more of these factors in a timely manner or at all, we could experience significant delays or an inability to successfully commercialize our drug candidates, which would materially harm our business. If we do not receive regulatory approvals for our drug candidates, we may not be able to continue our operations.
We or our current and future collaborators may never successfully develop and commercialize drug products, which would negatively affect our results of operation and our ability to continue our business operations.
We may not succeed in producing drug candidates that can be commercialized. To achieve success with our drug candidates, we or our current or future collaborators must develop, and eventually commercialize, a drug product or drug products that generate significant revenue. We currently generate revenues primarily from our collaboration relationships and expect to continue to derive most of our revenue from these relationships until such time as our or our collaborators’ drug development and commercialization efforts are successful, if ever.
Achieving success in drug development will require us or our current or future collaborators to be effective in a range of challenging activities, including completing preclinical testing and clinical trials of drug candidates, obtaining regulatory approval for these drug candidates, and manufacturing, marketing, and selling any products for which we or they may obtain regulatory approval. We and our current drug discovery collaborators are only in the preliminary stages of most of these activities. We and they may never succeed in these activities and, even if we do, we may never generate revenues that are significant enough to achieve profitability, or even if our collaborators do, we may not receive option fees, milestone payments, or royalties from them that are significant enough for us to achieve profitability. Because of the intense competition in the market for our data solutions and the numerous risks and uncertainties associated with biopharmaceutical product development, we are unable to accurately predict when, or if, we will be able to achieve or sustain profitability.
Even if we achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our failure to become and remain profitable would eventually depress the value of our company and could impair our ability to raise capital, expand our business, maintain our research and development efforts, develop a pipeline of drug candidates, enter into collaborations, or even continue our operations. A decline in the value of our company could also cause our Class A common stock to decline substantially and our stockholders to lose all or part of their value in our Class A common stock.
Our quarterly and annual operating results may fluctuate significantly in the future due to a variety of factors, many of which are outside of our control and may be difficult to predict.2023 Annual Report.
The amountrisk factors set forth below represent new risk factors or those containing changes to the similarly titled risk factor included in the sections titled “Risk Factors” in Part I, Item 1A. of our future losses is uncertain and our quarterly and annual operating results may fluctuate significantly or may fall below the expectations of investors or securities analysts, each of which may cause our stock price to fluctuate or decline. The reasons our quarterly and annual operating results may fluctuate include the following: • the cost to continue to maintain, develop, and integrate technological advancements;
• the timing, quality, regulatory compliance, and success or failure of clinical trials for our drug candidates or competing drug candidates, or any other change in the competitive landscape of our industry, including consolidation among our competitors or partners;
• our ability to successfully recruit and retain subjects, sites, and staff for clinical trials, and any delays caused by difficulties in such efforts;
• our ability to obtain marketing approval for our drug candidates, and the timing and scope of any such approvals we may receive;
• the timing and cost of, and level of investment in, research and development activities relating to our drug candidates, which may change from time to time;
• the timing, complexity, and cost of manufacturing our drug candidates, which may vary depending on the quantity of production and the terms of our agreements with manufacturers;
• our ability to attract, hire, and retain qualified personnel, including highly specialized scientists, clinicians, and engineers;
• expenditures that we will or may incur to develop additional drug candidates;
• the level of demand for our drug candidates should they receive approval, which may vary significantly;
• the risk/benefit profile, cost, and reimbursement policies with respect to our drug candidates, if approved, and existing and potential future therapeutics that compete with our drug candidates;
• the changing and volatile U.S. and global economic environments, including as a result of the COVID-19 pandemic and terrorism; and
• future accounting pronouncements or changes in our accounting policies.2023 Annual Report.
The cumulative effects of these and other factors could result in large fluctuations and unpredictability in our quarterly and annual operating results. As a result, comparing our operating results on a period-to-period basis may not be meaningful. This variability and unpredictability could also result in our failing to meet the expectations of industry or financial analysts or investors for any period. If our revenue or operating results fall below the expectations of analysts or investors or below any forecasts we may provide to the market, or if the forecasts we provide to the market are below the expectations of analysts or investors, the price of our Class A common stock could decline substantially. Such a stock price decline could occur even when we have met any previously publicly stated guidance we may have provided or provide in the future.RISKS RELATED TO OUR RELIANCE ON THIRD PARTIES
If we engage in future acquisitions or strategic partnerships, this may increase our capital requirements, dilute our stockholders’ equity, cause us to incur debt or assume contingent liabilities, and subject us to other risks.
We may engage in various acquisitions and strategic partnerships in the future, including by licensing or acquiring complementary products, intellectual property rights, technologies, or businesses. Any acquisition or strategic partnership may entail numerous risks, including:
• increased operating expenses and cash requirements;
• the assumption of indebtedness or contingent liabilities;
• the issuance of our equity securities which would result in dilution to our stockholders’ equity;
• assimilation of operations, intellectual property, products, and drug candidates of an acquired company, including difficulties associated with integrating new personnel;
• the diversion of our management’s attention from our existing product programs and initiatives in pursuing such an acquisition or strategic partnership;
• retention of key employees, the loss of key personnel, and uncertainties in our ability to maintain key business relationships;
• risks and uncertainties associated with the other party to such a transaction, including the prospects ofThird parties that party and their existing products or drug candidates and regulatory approvals; and
• our inability to generate revenue from acquired intellectual property, technology, and/or products sufficient to meet our objectives or even to offset the associated transaction and maintenance costs.
In addition, if we undertake such a transaction, we may issue dilutive securities, assume, or incur debt obligations, incur large one-time expenses, and acquire intangible assets that could result in significant future amortization expense.
Risks Related to the Discovery and Development of Drug Candidates
Our approach to drug discovery is unique and may not lead to successful drug products, for reasons including but not limited to challenges identifying mechanisms of action for our candidates.
We image cells and use cell morphology to understand how a diseased cell responds to drugs and when it appears normal. Biology is complex. If studying the shape, structure, form, and size of cells does not prove to be an accurate way to better understand diseases or does not lead to the biological insights, viable drug candidates, or products we anticipate, our drug discovery platform may not be useful or may not lead to successful drug products or we may have to pivot to a new business model, any of which could have an adverse effect on our reputation and results of operations. If the mechanism of action of a drug candidate is unknown, it may be more difficult to: i) choose the best lead to optimize from an efficacy standpoint and ii) avoid potential off-target side effects of the candidate that could affect safety. Such uncertainty could make it more difficult to form partnerships with larger pharmaceutical companies, as the expenses involved in late-phase clinical trials increase the level of risk related to potential efficacy and/or safety concerns, and may pose challenges to IND and/or NDA approval by the FDA or other regulatory agencies.
Our drug candidates are in preclinical or clinical development, which are lengthy and expensive processes with uncertain outcomes and the potential for substantial delays. We cannot give any assurance that any of our drug candidates will be successful in clinical trials or receive regulatory approval, which approval is necessary before they can be commercialized.
Our drug candidates are in preclinical or clinical development, which are lengthy and expensive processes with uncertain outcomes and the potential for substantial delays. We have not yet demonstrated our ability to complete clinical development, obtain regulatory approvals, manufacture a commercial scale product, or arrange for a third party to do so on our behalf, or conduct sales and marketing activities necessary for successful commercialization. We currently have four clinical-stage drug candidates focused on rare, monogenic diseases with no known established regulatory precedent. We anticipate filing IND applications with the FDA for Phase 2 studies and beginning such studies for all four drug candidates within the next four to five quarters. We may not be able to file such INDs or INDs for any other drug candidates on the timelines we expect, if at all, and any such delays could impact any additional product development timelines. For example, we may experience manufacturing delays with preclinical and clinical studies. Moreover, we cannot be sure that submission of an IND will result in the FDA allowing further clinical trials to begin, or that, once begun, issues will not arise that require us to suspend or terminate clinical trials. Commencing each of these clinical trials is subject to finalizing the trial design based on discussions with the FDA and other regulatory authorities. Any guidance we receive from the FDA or other regulatory authorities is subject to change. These regulatory authorities could change their positions at any time,
including their positions on the acceptability of our trial designs or the clinical endpoints or populations selected, which may require us to complete additional clinical trials or impose stricter approval conditions than we currently expect. Successful completion of our clinical trials is a prerequisite to submitting a New Drug Application, or NDA, to the FDA and a Marketing Authorization Application, or MAA, to the European Medicines Agency, or EMA, or Medicines and Healthcare Products Regulatory Agency, or MHRA, for each drug candidate and, consequently, the ultimate approval and commercial marketing of each drug candidate. We do not know whether any of our future clinical trials will begin on time or ever be completed on schedule, if at all.
If we are required to conduct additional clinical trials or other testing of our drug candidates beyond those that we currently contemplate, if we are unable to successfully complete clinical trials of our drug candidates or other testing, if the results of these trials or tests are not positive or are only modestly positive or if there are safety concerns, we may:
• be delayed in obtaining marketing approvals for our drug candidates;
• not obtain marketing approvals at all;
• obtain approvals for indications or patient populations that are not as broad as intended or desired or that impose label restrictions or warnings or risk mitigation requirements;
• be subject to post-marketing testing requirements; or
• have products removed from the market after obtaining marketing approval.
Clinical development is a lengthy and expensive process, with an uncertain outcome.
It is impossible to predict when or if any of our drug candidates will prove effective and safe in humans or will receive regulatory approval. To obtain marketing approval from regulatory authorities for the sale of any drug candidate, we must complete preclinical studies and then conduct extensive clinical trials to demonstrate the safety and efficacy of our drug candidates in humans. We may accelerate from cell models in our drug discovery platform directly to patients without validating results through animal studies, or validate them in animal studies at the same time as we conduct Phase 1 clinical trials. This approach could pose additional risks to our success because the effect of certain of our drug candidates on diseases has not been tested in animals prior to testing in humans. Clinical testing is expensive, difficult to design and implement, can take many years to complete, and is uncertain as to outcome. A failure of one or more clinical trials can occur at any stage of testing. The outcome of preclinical development testing and early clinical trials may not be predictive of the success of later clinical trials, and interim results of a clinical trial do not necessarily predict final results. Moreover, preclinical and clinical data are often susceptible to varying interpretations and analyses, and many companies that have believed their drug candidates performed satisfactorily in preclinical studies and clinical trials have nonetheless failed to obtain marketing approval of their drug candidates or adequate payor reimbursement for approved products. Our preclinical studies and future clinical trials may not be successful.
From time to time, we may publish interim top-line or preliminary data from our clinical trials. Interim data from clinical trials are subject to the risk that one or more of the clinical outcomes may materially change as enrollment of participants continues and more data become available. Preliminary or top-line data also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary data we previously published. As a result, interim and preliminary data should be viewed with caution until the final data are available. Adverse differences between preliminary or interim data and final data could significantly harm our business prospects.
We may incur additional costs or experience delays in completing, or ultimately be unable to complete, the development and commercialization of our drug candidates.
We may experience delays in completing our preclinical studies and initiating or completing clinical trials, and we may experience numerous unforeseen events during, or as a result of, any future clinical trials that we could conduct that could delay or prevent our ability to receive marketing approval or commercialize our drug candidates, including:
• regulators or Institutional Review Boards, or IRBs, or ethics committees may not authorize us or our investigators to commence a clinical trial or conduct a clinical trial at a prospective trial site;
• we may experience delays in reaching, or fail to reach, agreement on acceptable terms with prospective trial sites and prospective Contract Research Organizations, or CROs, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites;
• clinical trials of our drug candidates may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional preclinical studies or clinical trials or we may decide to abandon product development programs;
• the number of participants required for clinical trials of our drug candidates may be larger than we anticipate, enrollment in these clinical trials may be slower than we anticipate or participants may drop out of these clinical trials or fail to return for post-treatment follow-up at a higher rate than we anticipate;
• our third-party contractors may fail to comply with regulatory requirements or to meet their contractual obligations to us in a timely manner, or at all, or may deviate from the clinical trial protocol or drop out of the trial, which may require that we add new clinical trial sites or investigators;
• we may elect to, or regulators or IRBs or ethics committees may require us or our investigators to, suspend or terminate clinical research for various reasons, including noncompliance with regulatory requirements or a finding that the participants are being exposed to unacceptable health risks;
• the cost of clinical trials of our drug candidates may be greater than we anticipate;
• the supply or quality of our drug candidates or other materials necessary to conduct clinical trials of our drug candidates may be insufficient or inadequate;
• delays in the manufacturing of our drug candidates; and
• our drug candidates may have undesirable side effects or other unexpected characteristics, causing us or our investigators, regulators, or IRBs or ethics committees to suspend or terminate the trials, or reports may arise from preclinical or clinical testing of other therapies that raise safety, efficacy, or other concerns about our drug candidates.
We could encounter delays if a clinical trial is suspended or terminated by us, by the IRBs or ethics committees of the institutions at which such trials are being conducted, by the Data Safety Monitoring Board for such a trial, or by the FDA or other regulatory authorities. Regulatory authorities may impose a suspension or termination or clinical hold due to a number of factors, such as failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols, inspection of the clinical trial operations or trial site by the FDA or other regulatory authorities, unforeseen safety issues or adverse side effects, failure to demonstrate a benefit from using a product, changes in governmental regulations or administrative actions, or lack of adequate funding to continue the clinical trial. Many of the factors that could cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of our drug candidates. Further, the FDA may disagree with our clinical trial design and our interpretation of data from clinical trials or may change the requirements for approval even after it has reviewed and commented on the design for our clinical trials.
Our product development costs will also increase if we experience delays in testing or regulatory approvals. We do not know whether any of our future clinical trials will begin as planned, or whether any of our current or future clinical trials will need to be restructured or will be completed on schedule, if at all. Significant preclinical study or clinical trial delays, including those caused by the COVID-19 pandemic, also could shorten any periods during which we may have the exclusive right to commercialize our drug candidates or allow our competitors to bring products to market before we do and impair our ability to successfully commercialize our drug candidates and may harm our business and results of operations. Any delays in our preclinical or future clinical development programs may harm our business, financial condition, and prospects significantly.
If we experience delays or difficulties in the enrollment of patients in clinical trials, our receipt of necessary regulatory approvals could be delayed or prevented.
We may not be able to initiate or continue clinical trials for current or future drug candidates if we are unable to locate and enroll a sufficient number of eligible participants in these trials as required by the FDA or similar regulatory authorities outside the United States. Our ability to enroll eligible participants may be limited or may result in slower enrollment than we anticipate. In addition, competitors may initiate or have ongoing clinical trials for drug candidates that treat the same indications as our current or future drug candidates, and participants who would otherwise be eligible for our clinical trials may instead enroll in our competitors’ clinical trials. Furthermore, our ability to enroll participants may be delayed by the evolving COVID-19 pandemic and we do not know the extent and scope of such delays at this point.
In addition to the competitive trial environment, the eligibility criteria of our planned clinical trials will further limit the pool of available study participants as we will require that participants have specific characteristics, such as rare diseases connected to our drug candidates, which also may make enrollment challenging. Additionally, the process of finding potential participants may prove costly. We also may not be able to identify, recruit, and enroll a sufficient
number of participants to complete our clinical studies because of the perceived risks and benefits of the drug candidates under study, the availability and efficacy of competing therapies and clinical trials, the proximity and availability of clinical trial sites for prospective participants, and the referral practices of physicians. If people are unwilling to participate in our studies for any reason, the timeline for recruiting participants, conducting studies, and obtaining regulatory approval of potential products may be delayed.
Clinical trial enrollment may be affected by other factors including:
• the severity of the disease under investigation;
• the eligibility criteria for the clinical trial in question;
• the availability of an appropriate genomic screening test;
• the perceived risks and benefits of the drug candidate under study;
• the efforts to facilitate timely enrollment in clinical trials;
• the referral practices of physicians;
• the ability to monitor participants adequately during and after the trial;
• the proximity and availability of clinical trial sites for prospective participants;
• factors we may not be able to control, such as current or potential pandemics that may limit the availability of participants, principal investigators, study staff, or clinical sites, such as the outbreak of COVID-19;
• referral practices of physicians;
• ability to monitor participants adequately during and after the trial;
• ability to recruit clinical trial investigators with the appropriate competencies and experience;
• our ability to maintain participant informed consent and privacy; and
• the risk that enrolled participants will not complete a clinical trial.
Our planned clinical trials or those of our potential future collaborators may reveal significant adverse events not seen in our preclinical or nonclinical studies and may result in a safety profile that could inhibit regulatory approval or market acceptance of any of our drug candidates.
Before obtaining regulatory approvals for the commercial sale of any products, we must demonstrate through lengthy, complex, and expensive preclinical studies and clinical trials that our drug candidates are both safe and effective for use in each target indication. Clinical testing is expensive and can take many years to complete, and its outcome is inherently uncertain. Failure can occur at any time during the clinical trial process. The results of preclinical studies and early clinical trials of our drug candidates may not be predictive of the results of later-stage clinical trials. In addition, initial success in clinical trials may not be indicative of results obtained when such trials are completed. There is typically an extremely high rate of attrition from the failure of drug candidates proceeding through clinical trials. Drug candidates in later stages of clinical trials also may fail to show the desired safety and efficacy profile despite having progressed through nonclinical studies and initial clinical trials. A number of companies in the biopharmaceutical industry have suffered significant setbacks in advanced clinical trials due to lack of efficacy or unacceptable safety issues, notwithstanding promising results in earlier trials. Most drug candidates that commence clinical trials are never approved as products and there can be no assurance that any of our current or future clinical trials will ultimately be successful or support further clinical development of any of our drug candidates.
We may develop future drug candidates, in combination with one or more disease therapies. The uncertainty resulting from the use of our drug candidates in combination with other disease therapies may make it difficult to accurately predict side effects in future clinical trials.
As is the case with many treatments for rare diseases and other conditions, there have been, and it is likely that there may be, side effects associated with the use of our drug candidates. If significant adverse events or other side effects are observed in any of our current or future drug candidates, we may have difficulty recruiting participants in our clinical trials, they may drop out of our trials, or we may be required to abandon the trials or our development efforts of one or more drug candidates altogether. We, the FDA or other applicable regulatory authorities, or an IRB may suspend or terminate clinical trials of a drug candidate at any time for various reasons, including a belief that subjects in such trials are being exposed to unacceptable health risks or adverse side effects. Some potential therapeutics developed in the biotechnology industry that initially showed therapeutic promise in early-stage trials have later been found to cause side effects that prevented their further development. Even if the side effects do not preclude the product from obtaining or maintaining marketing approval, undesirable side effects may inhibit market
acceptance of the approved product due to its tolerability versus other therapies. Any of these developments could materially harm our business, financial condition, and prospects.
We may in the future conduct clinical trials for our drug candidates outside the United States, and the FDA and similar foreign regulatory authorities may not accept data from such trials.
We may in the future choose to conduct additional clinical trials outside the United States, including in Australia, Europe, Asia, or other foreign jurisdictions. FDA acceptance of trial data from clinical trials conducted outside the United States may be subject to certain conditions. In cases where data from clinical trials conducted outside the United States are intended to serve as the sole basis for marketing approval in the United States, the FDA will generally not approve the application on the basis of foreign data alone unless i) the data are applicable to the United States population and United States medical practice; ii) the trials were performed by clinical investigators of recognized competence and iii) the data may be considered valid without the need for an on-site inspection by the FDA or, if the FDA considers such an inspection to be necessary, the FDA is able to validate the data through an on-site inspection or other appropriate means. Additionally, the FDA’s clinical trial requirements, including large enough size of trial populations and statistical powering, must be met. Many foreign regulatory bodies have similar approval requirements. In addition, such foreign trials would be subject to the applicable local laws of the foreign jurisdictions where the trials are conducted. There can be no assurance that the FDA or any similar foreign regulatory authority will accept data from trials conducted outside of the United States or the applicable jurisdiction. If the FDA or any similar foreign regulatory authority does not accept such data, it would result in the need for additional trials, which would be costly and time-consuming and delay aspects of our business plan, and which may result in our drug candidates not receiving approval or clearance for commercialization in the applicable jurisdiction.
Following the United Kingdom’s departure from the EU on January 31, 2020, and the end of the a “transition period” on December 31, 2020, the EU and the United Kingdom have entered into a trade and cooperation agreement which governs certain aspects of their future relationship, including by ensuring tariff-free trade for certain goods and services. Since the regulatory framework for pharmaceutical products in the United Kingdom relating to quality, safety and efficacy of pharmaceutical products, clinical trials, marketing authorization, commercial sales and distribution of pharmaceutical products is derived from EU directives and regulations, Brexit will materially impact the future regulatory regime which applies to products and the approval of drug candidates in the United Kingdom. Longer term, the United Kingdom is likely to develop its own legislation that diverges from that in the EU.
The incidence and prevalence for target patient populations of our drug candidates have not been established with precision. If the market opportunities for our drug candidates are smaller than we estimate or if any approval that we obtain is based on a narrower definition of the patient population, our revenue and ability to achieve profitability will be adversely affected, possibly materially.
Even if approved for commercial sale, the total addressable market for our drug candidates will ultimately depend upon, among other things, the diagnosis criteria included in the final label, if our drug candidates are approved for these indications, acceptance by the medical community and patient access, product pricing and reimbursement. The number of patients targeted by our drug candidates may turn out to be lower than expected, patients may not be otherwise amenable to treatment with our products, or new patients may become increasingly difficult to identify or gain access to, all of which would adversely affect our results of operations and our business. We may not be successful in our efforts to identify additional drug candidates. Due to our limited resources and access to capital, we must prioritize development of certain drug candidates, which may prove to be the wrong choice and may adversely affect our business.
Although we intend to explore other therapeutic opportunities, in addition to the drug candidates that we are currently developing, we may fail to identify viable new drug candidates for clinical development for a number of reasons. If we fail to identify additional potential drug candidates, our business could be materially harmed.
Research programs to pursue the development of our existing and planned drug candidates for additional indications and to identify new drug candidates and disease targets require substantial technical, financial, and human resources whether or not they are ultimately successful. For example, pursuant to our Research Collaboration and Option Agreement with Bayer AG, or the Bayer Agreement, we collaborate with Bayer AG, or Bayer, to develop various projects related to fibrosis. There can be no assurance that we will find potential targets using this approach, that any such targets will be tractable, or that such clinical validations will be successful. Our
research programs may initially show promise in identifying potential indications and/or drug candidates, yet fail to yield results for clinical development for a number of reasons, including:
• the research methodology used may not be successful in identifying potential indications and/or drug candidates, including as a result of the limited patient sample represented in our databases and the validity of extrapolating based on insights from a particular cellular context that may not apply to other more relevant cellular contexts;
• potential drug candidates may, after further study, be shown to have harmful adverse effects or other characteristics that indicate they are unlikely to be effective products; or
• it may take greater human and financial resources than we will possess to identify additional therapeutic opportunities for our drug candidates or to develop suitable potential drug candidates through internal research programs, thereby limiting our ability to develop, diversify and expand our product portfolio.
Because we have limited financial and human resources, we will have to prioritize and focus on certain research programs, drug candidates and target indications while forgoing others. As a result, we may forgo or delay pursuit of opportunities with other drug candidates or for other indications that later prove to have greater commercial potential or a greater likelihood of success. Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities.
Accordingly, there can be no assurance that we will ever be able to identify additional therapeutic opportunities for our drug candidates or to develop suitable potential drug candidates through internal research programs, which could materially adversely affect our future growth and prospects. We may focus our efforts and resources on potential drug candidates or other potential programs that ultimately prove to be unsuccessful.
If we are not able to obtain, or if there are delays in obtaining, required regulatory approvals for our drug candidates, we will not be able to commercialize, or will be delayed in commercializing, our drug candidates, and our ability to generate revenue will be materially impaired.
Our drug candidates and the activities associated with their development and commercialization, including their design, testing, manufacture, safety, efficacy, recordkeeping, labeling, storage, approval, advertising, promotion, sale, distribution, import and export are subject to comprehensive regulation by the FDA and other regulatory agencies in the United States and by comparable authorities in other countries. Before we can commercialize any of our drug candidates, we must obtain marketing approval. Currently, all of our drug candidates are in development, and we have not received approval to market any of our drug candidates from regulatory authorities in any jurisdiction. It is possible that our drug candidates, including any drug candidates we may seek to develop in the future, will never obtain regulatory approval. We have only limited experience in filing and supporting applications to regulatory authorities and expect to rely on CROs and/or regulatory consultants to assist us in this process. Securing regulatory approval requires the submission of extensive preclinical and clinical data and supporting information to the various regulatory authorities for each therapeutic indication to establish the drug candidate’s safety and efficacy. Securing regulatory approval also requires the submission of information about the product manufacturing process to, and inspection of manufacturing facilities by, the relevant regulatory authority. Our drug candidates may not be effective, may be only moderately effective or may prove to have undesirable or unintended side effects, toxicities or other characteristics that may preclude our obtaining marketing approval or prevent or limit commercial use. In addition, regulatory authorities may find fault with our manufacturing process or facilities or that of third-party contract manufacturers. We may also face greater than expected difficulty in manufacturing our drug candidates.
The process of obtaining regulatory approvals, both in the United States and abroad, is expensive and often takes many years. If the FDA or a comparable foreign regulatory authority requires that we perform additional preclinical or clinical trials, approval, if obtained at all, may be delayed. The length of such a delay varies substantially based upon a variety of factors, including the type, complexity and novelty of the drug candidates involved. Changes in marketing approval policies during the development period, changes in or the enactment of additional statutes or regulations, or changes in regulatory review for each submitted NDA, 510(k), Premarket Approval Application, or PMA, or equivalent application types, may cause delays in the approval or rejection of an application. The FDA and comparable authorities in other countries have substantial discretion in the approval process and may refuse to accept any application or may decide that our data are insufficient for approval and require additional preclinical, clinical, or other studies. Our drug candidates could be delayed in receiving, or fail to receive, regulatory approval for many reasons, including the following:
• the FDA or comparable foreign regulatory authorities may disagree with the design or implementation of our clinical trials;
• we may not be able to enroll a sufficient number of patients in our clinical studies;
• we may be unable to demonstrate to the satisfaction of the FDA or comparable foreign regulatory authorities that a drug candidate is safe and effective for its proposed indication or a related companion diagnostic is suitable to identify appropriate patient populations;
• the results of clinical trials may not meet the level of statistical significance required by the FDA or comparable foreign regulatory authorities for approval;
• we may be unable to demonstrate that a drug candidate’s clinical and other benefits outweigh its safety risks;
• the FDA or comparable foreign regulatory authorities may disagree with our interpretation of data from preclinical studies or clinical trials;
• the data collected from clinical trials of our drug candidates may not be sufficient or of sufficient quality to support the submission of an NDA or other submission or to obtain regulatory approval in the United States or elsewhere;
• the FDA or comparable foreign regulatory authorities may find deficiencies with or fail to approve the manufacturing processes or facilities of third-party manufacturers with which we contract for clinical and commercial supplies; and
• the approval policies or regulations of the FDA or comparable foreign regulatory authorities may significantly change such that our clinical or manufacturing data are insufficient for approval.
Even if we were to obtain approval, regulatory authorities may approve any of our drug candidates for fewer or more limited indications than we request, thereby narrowing the commercial potential of the drug candidate. In addition, regulatory authorities may grant approval contingent on the performance of costly post-marketing clinical trials or may approve a drug candidate with a label that does not include the labeling claims necessary or desirable for the successful commercialization of that drug candidate. Any of the foregoing scenarios could materially harm the commercial prospects for our drug candidates.
If we experience delays in obtaining approval or if we fail to obtain approval of our drug candidates, the commercial prospects for our drug candidates may be harmed and our ability to generate revenues will be materially impaired.
We may never realize return on our investment of resources and cash in our drug discovery collaborations.
We conduct drug discovery activities for or with collaborators who are engaged in drug discovery and development. These collaborators include pre-commercial biotechnology companies and large pharmaceutical companies. When we engage in drug discovery with these collaborators, we typically provide the benefit of our platform and platform experts who identify molecules that have activity against one or more specified targets. In consideration, we have received equity investments, upfront fees, and/or the right to receive option fees, cash milestone payments upon the achievement of specified development, regulatory, commercial sales milestones for the drug discovery targets, and potential royalties.
We may never realize a longer-term return on our investment of resources and cash in our drug discovery collaborations. Clinical drug development involves a lengthy and expensive process, with an uncertain outcome. Our drug discovery collaborators may incur additional costs or experience delays in completing, or ultimately be unable to complete, the development and commercialization of any drug candidates. In addition, our ability to realize returns from our drug discovery collaborations is subject to the following risks:
• drug discovery collaborators have significant discretion in determining the amount and timing of efforts and resources that they will apply to our collaborations and may not perform their obligations as expected;
• drug discovery collaborators may not pursue development or commercialization of any drug candidates for which we are entitled to option fees, milestone payments, or royalties or may elect not to continue or renew development or commercialization programs based on results of clinical trials or other studies, changes in the collaborator’s strategic focus or available funding, or external factors, such as an acquisition, that divert resources or create competing priorities;
• drug discovery collaborators may delay clinical trials for which we are entitled to milestone payments;
• we may not have access to, or may be restricted from disclosing, certain information regarding our collaborators’ drug candidates being developed or commercialized and, consequently, may have limited
ability to inform our stockholders about the status of, and likelihood of achieving, milestone payments or royalties under such collaborations;
• drug discovery collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with any drug candidates and products for which we are entitled to milestone payments or royalties and the collaborator may believe that the competitive products are more likely to be successfully developed or can be commercialized under terms that are more economically attractive;
• drug candidates discovered in drug discovery collaborations with us may be viewed by our collaborators as competitive with their own drug candidates or products, which may cause our collaborators to cease to devote resources to the commercialization of any such drug candidates;
• existing drug discovery collaborators and potential future drug discovery collaborators may begin to perceive us to be a competitor more generally, particularly as we advance our internal drug discovery programs, and therefore may be unwilling to continue existing collaborations with us or to enter into new collaborations with us;
• a drug discovery collaborator may fail to comply with applicable regulatory requirements regarding the development, manufacture, distribution, or marketing of a drug candidate or product, which may impact our ability to receive milestone payments;
• disagreements with drug discovery collaborators, including disagreements over intellectual property or proprietary rights, contract interpretation, or the preferred course of development, might cause delays or terminations of the research, development, or commercialization of drug candidates for which we are eligible to receive milestone payments, or might result in litigation or arbitration;
• drug discovery collaborators may not properly obtain, maintain, enforce, defend or protect our intellectual property or proprietary rights or may use our proprietary information in such a way as to potentially lead to disputes or legal proceedings that could jeopardize or invalidate our or their intellectual property or proprietary rights or expose us and them to potential litigation;
• drug discovery collaborators may infringe, misappropriate, or otherwise violate the intellectual property or proprietary rights of third parties, which may expose us to litigation and potential liability; and
• drug discovery collaborations may be terminated prior to our receipt of any significant value from the collaboration.
Our drug discovery collaborations may not lead to development or commercialization of drug candidates that results in our receipt of option fees, milestone payments, or royalties or other payments in a timely manner, or at all. For example, we may be over-reliant on our partners to provide information for molecules that we in-license. The molecules that we in-license may not be well protected because the composition of matter patents that once protected them have expired. Moreover, we may have difficulty obtaining the quality and quantity of active pharmaceutical ingredient, or API, or be able to ensure the stability of the molecule, all of which is needed to conduct clinical trials or bring a drug candidate to market. For those molecules that we are attempting to repurpose for other indications, our partners may have not have sufficient data, poor quality data or be able to help us interpret data, any of which could cause our collaboration to fail.
If any drug discovery collaborations that we enter into do not result in the successful development and commercialization of drug products that result in option fees, milestone payments, or royalties or other payments to us, we may not receive return on the resources we have invested in such drug discovery collaborations. Moreover, even if a drug discovery collaboration initially leads to the achievement of milestones that result in payments to us, it may not continue to do so.
We face substantial competition, which may result in others discovering, developing, or commercializing products before or more successfully than we do.
The development and commercialization of new products in the biopharmaceutical and related industries is highly competitive. There are other companies focusing on technology-enabled drug discovery to identify and develop NCEs and KCEs. Some of these competitive companies are based on scientific approaches that are the same as or similar to our approach, and others are based on entirely different approaches. These companies include divisions of large pharmaceutical companies and biotechnology companies of various sizes. We face competition with respect to our current drug candidates and will face competition with respect to any drug candidates that we may seek to develop or commercialize in the future, from major pharmaceutical companies, specialty pharmaceutical companies and biotechnology companies worldwide. Potential competitors also include academic institutions, government agencies, and other public and private research organizations that conduct research, seek patent
protection, and establish collaborative arrangements for research, development, manufacturing, and commercialization.
Any drug candidates that we successfully develop and commercialize will compete with currently approved therapies and new therapies that may become available in the future from segments of the pharmaceutical, biotechnology and other related industries that pursue new therapeutics. Key product features that would affect our ability to effectively compete with other therapeutics include the efficacy, safety, and convenience of our products. We believe principal competitive factors to our business include, among other things, the accuracy of our computations and predictions, ability to integrate experimental and computational capabilities, ability to successfully transition research programs into clinical development, ability to raise capital, and the scalability of the platform, pipeline, and business.
Many of the companies that we compete against or against which we may compete in the future have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. Mergers and acquisitions in the pharmaceutical, biotechnology and diagnostic industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient recruitment for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs. In addition, we cannot predict whether our current competitive advantages and our software tools, will remain in place and evolve appropriately as barriers to entry in the future. If not, other companies may be able to more directly or effectively compete with us.
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient or are less expensive than any products that we or our collaborators may develop. Our competitors also may obtain FDA or other regulatory approval for their products more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we or our collaborators are able to enter the market. The key competitive factors affecting the success of all of our drug candidates, if approved, are likely to be their efficacy, safety, convenience, price, the level of generic competition and the availability of reimbursement from government and other third-party payors.
Because we have multiple programs and drug candidates in our development pipeline and are pursuing a variety of target indications and treatment modalities, we may expend our limited resources to pursue a particular drug candidate and fail to capitalize on development opportunities or drug candidates that may be more profitable or for which there is a greater likelihood of success.
We currently focus on the development of drug candidates regardless of the treatment modality or the particular target indication. Because we have limited financial and personnel resources, we may forgo or delay pursuit of opportunities with potential target indications or drug candidates that later prove to have greater commercial potential than our current and planned development programs and drug candidates. Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities. Our spending on current and future research and development programs and other future drug candidates for specific indications may not yield any commercially viable future drug candidates.
We and our collaborators may not achieve projected discovery and development milestones and other anticipated key events in the time frames that we or they announce, which could have an adverse impact on our business and could cause our stock price to decline.
From time to time, we expect that we will make public statements regarding the expected timing of certain milestones and key events, such as the commencement and completion of preclinical and clinical studies in our internal drug discovery programs as well developments and milestones under our collaborations. Our collaborators, such as Bayer, have also made public statements regarding expectations for the development of programs under collaboration with us and may in the future make additional statements about their goals and expectations for collaborations with us. The actual timing of these events can vary dramatically due to a number of factors such as delays or failures in our or our current and future collaborators’ drug discovery and development programs, the amount of time, effort, and resources committed by us and our current and future collaborators, and the numerous
uncertainties inherent in the development of drugs. As a result, there can be no assurance that our or our current and future collaborators’ programs will advance or be completed in the time frames we or they announce or expect. If we or any collaborators fail to achieve one or more of these milestones or other key events as planned, our business could be materially adversely affected, and the price of our Class A common stock could decline.
Risks Related to our Platform and Data
Our business and operations would suffer in the event of computer system failures, cyber-attacks, or deficiencies in our or third parties’ cybersecurity.
We are increasingly dependent upon information technology systems, infrastructure, and data to operate our business. In the ordinary course of business, we collect, store, and transmit confidential information (including but not limited to intellectual property, proprietary business information, personal information, and other confidential information). It is critical that we do so in a secure manner to maintain the confidentiality and integrity of this information. We also have outsourced elements of our operations to third parties, and as a result we manage a number of third-party vendors and other contractors and consultants who have access to our confidential information.
Given our limited operating history, we are still in the process of implementing our internal security and business continuity measures and developing our information technology infrastructure. Our internal computer systems and those of current and future third parties on which we rely may fail and are vulnerable to damage from computer viruses and unauthorized access. Our information technology and other internal infrastructure systems, including corporate firewalls, servers, data center facilities that we colocate in, lab equipment, leased lines, and connection to the Internet, face the risk of breakdown or other damage or interruption from service interruptions, system malfunction, natural disasters, terrorism, war and telecommunication and electrical failures, as well as security breaches from inadvertent or intentional actions by our employees, contractors, consultants, business partners, and/or other third parties, or from cyber-attacks by malicious third parties (including the deployment of harmful malware, ransomware, denial-of-service attacks, social engineering and other means to affect service reliability and threaten the confidentiality, integrity and availability of information), each of which could compromise our system infrastructure or lead to the loss, destruction, alteration, disclosure, or dissemination of, or damage or unauthorized access to, our data or data that is processed or maintained on our behalf, or other assets.
If such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our development programs and our business operations, and could result in financial, legal, business, and reputational harm to us. For example, one of our primary differentiators is our proprietary technical information and biological and chemical data. The loss, corruption, unavailability of, or damage to our data would interfere with and undermine the insights we draw from our platform, which could result in the waste of resources on insights based on flawed premises. In addition, the loss or corruption of, or other damage to, clinical trial data from completed or future clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. Likewise, we rely on third parties for the manufacture of our drug candidates or any future drug candidates and to conduct clinical trials, and similar events relating to their systems and operations could also have a material adverse effect on our business and lead to regulatory agency actions. The risk of a security breach or disruption, particularly through cyber-attacks or cyber intrusion, including by computer hackers, foreign governments, and cyber terrorists, has generally increased as the number, intensity, and sophistication of attempted attacks and intrusions from around the world have increased. Sophisticated cyber attackers (including foreign adversaries engaged in industrial espionage) are skilled at adapting to existing security technology and developing new methods of gaining access to organizations’ sensitive business data, which could result in the loss of proprietary information, including trade secrets. We may not be able to anticipate all types of security threats, and we may not be able to implement preventive measures effective against all such security threats. For example, third parties have in the past and may in the future illegally pirate our software and make that software publicly available on peer-to-peer file sharing networks or otherwise. The techniques used by cyber criminals change frequently, may not be recognized until launched, and can originate from a wide variety of sources, including outside groups such as external service providers, organized crime affiliates, terrorist organizations, or hostile foreign governments or agencies. In addition, in response to the ongoing COVID-19 pandemic, the majority of our workforce is currently working remotely. This could increase our cybersecurity risk, create data accessibility concerns, and make us more susceptible to communication disruptions.
Any security breach or other event that leads to loss, damage, or unauthorized access to, or use, alteration, or disclosure or dissemination of, personal information, including personal information regarding clinical trial subjects, contractors, directors, or employees, our intellectual property, proprietary business information, or other confidential or proprietary information, could harm our reputation directly, enable competitors to compete with us more effectively, compel us to comply with federal and/or state breach notification laws and foreign law equivalents, subject us to mandatory corrective action and otherwise subject us to liability under laws and regulations that protect the privacy and security of personal information, which could result in significant legal and financial exposure and reputational damage that could potentially have an adverse effect on our business. Notifications and follow-up actions related to a security incident could impact our reputation, and we could incur substantial costs, including legal and remediation costs, in connection with these measures and otherwise in connection with any actual or suspected security breach. We expect to incur significant costs in an effort to detect and prevent security incidents and otherwise implement our internal security and business continuity measures, and actual, potential, or anticipated attacks may cause us to incur increasing costs, including costs to deploy additional personnel and protection technologies, train employees, and engage third-party experts and consultants. We may face increased costs and find it necessary or appropriate to expend substantial resources in the event of an actual or perceived security breach.
The costs related to significant security breaches or disruptions could be material and our insurance policies may not be adequate to compensate us for the potential losses arising from any such disruption in, or failure or security breach of, our systems or third-party systems where information important to our business operations or commercial development is stored or processed. In addition, such insurance may not be available to us in the future on economically reasonable terms, or at all. Further, our insurance may not cover all claims made against us and could have high deductibles in any event, and defending a suit, regardless of its merit, could be costly and divert management attention. Furthermore, if the information technology systems of our third-party vendors and other contractors and consultants become subject to disruptions or security breaches, we may have insufficient recourse against such third parties and we may have to expend significant resources to mitigate the impact of such an event, and to develop and implement protections to prevent future events of this nature from occurring.
If we are not able to develop new solutions and enhancements to our platform that keep pace with technological developments, our business and results of operations would be harmed.
Our ability to increase revenue depends in large part on our ability to enhance and improve our platform. The success of any enhancement to our platform depends on several factors, including the generation of additional biological and chemical data, innovation in hardware solutions, increased computational storage and processing capacity and development of more advanced algorithms. Any new enhancement that we develop may not be introduced in a timely or cost-effective manner, may contain errors, vulnerabilities or bugs, or may not achieve the functionality necessary to generate significant revenue. If we are unable to successfully develop new innovations, enhance our existing platform, or otherwise gain market acceptance, our reputation, business, results of operations, and financial condition would be harmed. Our success also depends on our ability to identify important and emerging use cases and quickly develop new and effective innovations to address those use cases.
We have invested and expect to continue to invest in research and development efforts that further enhance our platform. Such investments may affect our operating results, and, if the return on these investments is lower or develops more slowly than we expect, our revenue and operating results may suffer.
We have invested and expect to continue to invest in research and development efforts that further enhance our platform. These investments may involve significant time, risks, and uncertainties, including the risk that the expenses associated with these investments may affect our margins and operating results and that such investments may not generate sufficient revenues to offset liabilities assumed and expenses associated with these new investments. The software industry changes rapidly as a result of technological and product developments, which may render our solutions less effective. We believe that we must continue to invest a significant amount of time and resources in our platform to maintain and improve our competitive position. If we do not achieve the benefits anticipated from these investments, if the achievement of these benefits is delayed, our business, operating results and prospects may be materially adversely affected.
Defects or disruptions in our platform could result in diminishing our value and prospects.
Our platform depends upon the continuous, effective, and reliable operation of our software, hardware, databases, and related tools and functions and the integrity of our data. Our proprietary software tools, hardware, and data sets are inherently complex and may contain defects or errors. Errors may result from the interface of our proprietary software and hardware tools with our data or third-party systems and data, which we did not develop. The risk of errors is particularly significant when new software or hardware is first introduced or when new versions or enhancements of existing software or hardware are implemented. We have from time to time found defects in our software and hardware, and new errors in our existing software and hardware may be detected in the future. Any errors, defects, disruptions, or other performance problems with our software, hardware, or data sets could hurt our ability to gather valuable insights that drive our drug discoveries. Furthermore, our platform may produce an incomplete data set lacking in coverage which could result in a material adverse effect on our ability to discover new drug candidates. Such discovery is dependent on the integrity and completeness of our data. The occurrence of any of these events could result in diminishing value of our platform and data and have a material adverse effect on our business, operating results and prospects.
We rely upon third-party providers of cloud-based infrastructure to host our platforms. Any disruption in the operations of these third-party providers, limitations on capacity, or interference with our use could adversely affect our business, financial condition, and results of operations.
We outsource substantially all of the technological infrastructure relating to our hosted platform to third-party hosting services, such as Google Cloud and Amazon Web Services, or AWS. We have no control over any of these third parties, and while we attempt to reduce risk by minimizing reliance on any single third party or its operations, we cannot guarantee that such third-party providers will not experience system interruptions, outages or delays, or deterioration in their performance. We need to be able to access our computational platform at any time, without interruption or degradation of performance. Our hosted platform depends on protecting the virtual cloud infrastructure hosted by third-party hosting services by maintaining its configuration, architecture, features, and interconnection specifications, as well as protecting the information stored in these virtual data centers, which is transmitted by third-party Internet service providers. We have experienced, and expect that in the future we may again experience interruptions, delays and outages in service and availability from time to time due to a variety of factors, including infrastructure changes, human or software errors, website hosting disruptions and capacity constraints. Any limitation on the capacity of our third-party hosting services could adversely affect our business, financial condition, and results of operations. In addition, any incident affecting our third-party hosting services’ infrastructure that may be caused by cyber-attacks, natural disasters, fire, flood, severe storm, earthquake, power loss, telecommunications failures, terrorist or other attacks, and other disruptive events beyond our control could negatively affect our cloud-based solutions. A prolonged service disruption affecting our cloud-based solutions could damage our reputation or otherwise harm our business. We may also incur significant costs for using alternative equipment or taking other actions in preparation for, or in reaction to, events that damage the third-party hosting services we use.
In the event that our service agreements with our third-party hosting services are terminated, or there is a lapse of service, elimination of services or features that we utilize, interruption of Internet service provider connectivity, or damage to such facilities, we could experience interruptions in access to the our platform as well as significant delays and additional expense in arranging or creating new facilities and services and/or re-architecting our hosted software solutions for deployment on a different cloud infrastructure service provider, which could adversely affect our business, financial condition, and results of operations.
If our security measures are breached or unauthorized access to our other data is otherwise obtained, our data may be perceived as not being secure and we may incur significant liabilities.
We use a set of proprietary tools to generate, analyze, and derive novel insights from our data. As a result, unauthorized access to or security breaches of our data, as a result of third-party action, employee or contractor error, malfeasance, or otherwise could result in the loss or corruption of, or other damage to information, claims and litigation, indemnity obligations, damage to our reputation, and other liability. Our collaborators and other third parties we work with may also suffer similar security breaches of data that we rely on. Because the techniques used to obtain unauthorized access or sabotage systems change frequently and generally are not identified until they are launched against a target, we and those we collaborate with may be unable to anticipate these techniques or implement adequate preventative measures. In addition, if our employees or contractors fail to adhere to practices we have established to maintain a firewall between our internal drug discovery team and our teams that work with external individuals, including our collaborators, or if the technical solutions we have adopted to maintain the firewall
malfunction, our collaborators may lose confidence in our ability to maintain the confidentiality of their intellectual property, we may have trouble attracting new collaborators, we may be subject to breach of contract claims by our collaborators, and we may suffer reputational and other harm as a result. Any or all of these issues could result in reputational damage or subject us to third-party lawsuits or other action or liability, which could adversely affect our operating results. Our insurance may not be adequate to cover losses associated with such events, and in any case, such insurance may not cover all of the types of costs, expenses, and losses we could incur to respond to and remediate a security breach. For more information see “Risk Factors—Our business and operations would suffer in the event of computer system failures, cyber-attacks or deficiencies in our or third parties’ cyber security.”
Our solutions utilize third-party open source software, and any failure to comply with the terms of one or more of these open source software licenses could adversely affect our business, subject us to litigation, or create potential liability.
Our solutions include software licensed by third parties under any one or more open source licenses, including the Apache 2.0 License, MIT, BSD variants, and others, and we expect to continue to incorporate open source software in our solutions in the future. Moreover, we cannot ensure that we have effectively monitored our use of open source software, or validated the quality or source of such software, or that we are in compliance with the terms of the applicable open source licenses or our current policies and procedures. There have been claims against companies that use open source software in their products and services asserting that the use of such open source software infringes the claimants’ intellectual property rights. As a result, we could be subject to suits by third parties claiming that what we believe to be licensed open source software infringes such third parties’ intellectual property rights. Additionally, if an author or other third party that distributes such open source software were to allege that we had not complied with the conditions of one or more of these licenses, we could be required to incur significant legal expenses defending against such allegations and could be subject to significant damages and required to comply with onerous conditions or restrictions on these solutions, which could disrupt the distribution and sale of these solutions. Litigation could be costly for us to defend, have a negative effect on our business, financial condition, and results of operations, or require us to devote additional research and development resources to change our solutions. Furthermore, these third-party open source providers could experience service outages, data loss, privacy breaches, cyber-attacks, and other events relating to the applications and services they provide that could diminish the utility of these services and which could harm our business as a result.
Use of open source software may entail greater risks than use of third-party commercial software, as open source licensors generally do not provide warranties or other contractual protections regarding infringement claims or the quality of the code, including with respect to security vulnerabilities where open source software may be more susceptible. In addition, certain open source licenses require that source code for software programs that interact with such open source software be made available to the public at no cost and that any modifications or derivative works to such open source software continue to be licensed under the same terms as the open source software license. The terms of various open source licenses to which we are subject have not been interpreted by courts in the relevant jurisdictions, and there is a risk that such licenses could be construed in a manner that imposes unanticipated conditions or restrictions on our ability to market or provide our software and data. By the terms of certain open source licenses, we could be required to release the source code of our proprietary software, and to make our proprietary software available under open source licenses, if we combine our proprietary software with open source software in a certain manner. In the event that portions of our proprietary software are determined to be subject to an open source license, we could be required to publicly release the affected portions of our source code, re-engineer all or a portion of our solutions, or otherwise be limited in the licensing of our solutions, each of which could reduce or eliminate the value of our solutions. Disclosing our proprietary source code could allow our competitors to create similar products with lower development effort and time and ultimately could result in a loss of sales. Furthermore, any such re-engineering or other remedial efforts could require significant additional research and development resources, and we may not be able to successfully complete any such re-engineering or other remedial efforts. Any of these events could create liability for us and damage our reputation, which could have a material adverse effect on our revenue, business, results of operations, and financial condition and the market price of our shares.
Risks Related to Our Operations/Commercialization
Our insurance policies are expensive and protect us only from some business risks, which leaves us exposed to significant uninsured liabilities.
We do not carry insurance for all categories of risk that our business may encounter and insurance coverage is becoming increasingly expensive. For example, we can only obtain insurance for the loss of our data that would partially compensate us for its loss. We do not know if we will be able to maintain existing insurance with adequate levels of coverage in the future, and any liability insurance coverage we acquire in the future may not be sufficient to reimburse us for any expenses or losses we may suffer. If we obtain marketing approval for any drug candidates that we or our collaborators may develop, we intend to acquire insurance coverage to include the sale of commercial products, but we may be unable to obtain such insurance on commercially reasonable terms or in adequate amounts. The coverage or coverage limits currently maintained under our insurance policies may not be adequate. If our losses exceed our insurance coverage, our financial condition would be adversely affected. Clinical trials or regulatory approvals for any of our drug candidates could be suspended, which could adversely affect our results of operations and business, including by preventing or limiting the development and commercialization of any drug candidates that we or our collaborators may identify. Additionally, operating as a public company will make it more expensive for us to obtain directors and officers liability insurance. If we do not have adequate levels of directors and officers liability insurance, it may be more difficult for us to attract and retain qualified individuals to serve on our board of directors.
A pandemic, epidemic, or outbreak of an infectious disease, such as COVID-19, may materially and adversely affect our business and our financial results and could cause a disruption to the development of our drug candidates.
Public health crises such as pandemics or similar outbreaks could adversely impact our business. In early 2020, a novel strain of a virus named SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2), or coronavirus, which causes COVID-19 has spread to most countries across the world and all 50 states within the U.S. including Utah and specifically Salt Lake City, where our primary office and laboratory space is located. The coronavirus pandemic is evolving, and to date has led to the implementation of various responses, including government-imposed quarantines, travel restrictions and other public health safety measures. The extent to which the coronavirus impacts our operations or those of our third-party partners, including our preclinical studies or clinical trial operations, will depend on future developments, which are highly uncertain and cannot be predicted with confidence, including the duration of the outbreak, new information that will emerge concerning the severity of coronavirus infection and the actions to contain the coronavirus or treat its impact, among others. The continued spread of COVID-19 globally, or the evolution of a new variant of COVID-19 that is more contagious, has more severe effects or is resistant to treatments or vaccinations, could adversely impact our preclinical or clinical trial operations in the U.S., including our ability to recruit and retain trial participants as well as principal investigators and site staff who, as healthcare providers, may have heightened exposure to COVID-19 if an outbreak occurs in their geography. For example, similar to other biopharmaceutical companies, we may experience delays in initiating preclinical and clinical studies, protocol deviations, enrolling our clinical trials, or dosing of patients in our clinical trials as well as in activating new trial sites. COVID-19 or any variants may also affect employees of third-party CROs located in affected geographies that we rely upon to carry out our clinical trials. In addition, as a result of medical complications associated with the diseases of the patients we seek to enroll and treat in our trials, the patient populations that our lead and other drug candidates target may be particularly susceptible to COVID-19 or any variants, which may make it more difficult for us to identify individuals able to enroll in our current and future clinical trials and may impact the ability of those enrolled to complete any such trials. Any negative impact COVID-19 or any variants has on enrollment or the execution of our drug trials could cause costly delays, which could adversely affect our ability to obtain regulatory approval for and to commercialize our drug candidates, increase our operating expenses, and have a material adverse effect on our financial results.
Additionally, timely enrollment in planned clinical trials is dependent upon clinical trial sites which could be adversely affected by global health issues, such as pandemics. We plan to conduct clinical trials for our drug candidates in geographies which are currently being affected by the coronavirus. Some factors from the coronavirus outbreak that will delay or otherwise adversely affect enrollment in the clinical trials of our drug candidates, as well as our business generally, include:
• the potential diversion of healthcare resources away from the conduct of clinical trials to focus on pandemic concerns, including the attention of physicians serving as our clinical trial investigators, hospitals serving as our clinical trial sites and hospital staff supporting the conduct of our prospective clinical trials;
• limitations on travel that could interrupt key trial and business activities, such as clinical trial site initiations and monitoring, domestic and international travel by employees, contractors or patients to clinical trial sites, including any government-imposed travel restrictions or quarantines that will impact
the ability or willingness of patients, employees or contractors to travel to our clinical trial sites or secure visas or entry permissions, a loss of face-to-face meetings and other interactions with potential partners, any of which could delay or adversely impact the conduct or progress of our prospective clinical trials;
• the potential negative effect on the operations of our third-party manufacturers;
• interruptions in global shipping affecting the transport of clinical trial materials, such as tissue samples, investigational drug product and comparator drugs and other supplies used in our studies; and
• business disruptions caused by potential workplace, laboratory and office closures and an increased reliance on employees working from home, disruptions to or delays in ongoing laboratory experiments and operations, staffing shortages, travel limitations or mass transit disruptions, any of which could adversely impact our business operations or delay necessary interactions with local regulators, ethics committees and other important agencies and contractors.
We have taken temporary precautionary measures intended to help minimize the risk of the coronavirus to our employees, including temporarily permitting certain employees to work remotely, suspending all non-essential travel worldwide for our employees and discouraging employee attendance at industry events and in-person work-related meetings, which could negatively affect our business. We cannot presently predict the scope and severity of the planned and potential shutdowns or disruptions of businesses and government agencies, such as the Securities and Exchange Commission, or the SEC, or FDA.
These and other factors arising from the coronavirus could worsen in countries that are already afflicted with the coronavirus or could continue to spread to additional countries. Any of these factors, and other factors related to any such disruptions that are unforeseen, could have a material adverse effect on our business and our results of operation and financial condition. Further, uncertainty around these and related issues could lead to adverse effects on the economy of the United States and other economies, which could impact our ability to raise the necessary capital needed to develop and commercialize our drug candidates.
If we fail to sufficiently manage and improve our technical hardware infrastructure we may experience errors, delays and other performance problems.
We have experienced significant growth in the complexity of our data and the software tools that our hardware infrastructure supports. In addition, we need to properly manage and improve our technological hardware infrastructure in order to support changes in hardware and software parameters and the evolution of our tools. We have experienced, and may in the future experience, disruptions, outages, failures and other performance problems with our software tools or hardware infrastructure. These types of problems may be caused by a variety of factors, including infrastructure changes, human, mechanical, or software errors, viruses, security attacks, and fraud. In some instances, we may not be able to identify the cause or causes of these problems within an acceptable period of time or at all. If we do not accurately predict and identify our infrastructure requirements and failures, including acquisition of newer infrastructure, our team may experience performance problems that may cause delays in our research and development programs, which could adversely affect our business, financial condition, results of operations, and prospects.
Even if any drug candidates we develop receive marketing approval, they may fail to achieve the degree of market acceptance by physicians, patients, healthcare payors, and others in the medical community necessary for commercial success.
The commercial success of our drug candidates will depend upon their degree of market acceptance by physicians, patients, third-party payors, and others in the medical community. Even if any drug candidates we may develop receive marketing approval, they may nonetheless fail to gain sufficient market acceptance by physicians, patients, healthcare payors, and others in the medical community. The degree of market acceptance of any drug candidates we may develop, if approved for commercial sale, will depend on a number of factors, including:
• the efficacy and safety of such drug candidates as demonstrated in pivotal clinical trials and published in peer-reviewed journals;
• the potential and perceived advantages compared to alternative treatments, including any similar generic treatments;
• the ability to offer these products for sale at competitive prices;
• the ability to offer appropriate patient access programs, such as co-pay assistance;
• convenience and ease of dosing and administration compared to alternative treatments;
• the clinical indications for which the drug candidate is approved by FDA or comparable regulatory agencies;
• product labeling or product insert requirements of the FDA or other comparable foreign regulatory authorities, including any limitations, contraindications or warnings contained in a product’s approved labeling;
• restrictions on how the product is distributed;
• the timing of market introduction of competitive products;
• publicity concerning these products or competing products and treatments;
• the strength of marketing and distribution support;
• favorable third-party coverage and sufficient reimbursement; and
• the prevalence and severity of any side effects or adverse events.
Sales of medical products also depend on the willingness of physicians to prescribe the treatment, which is likely to be based on a determination by these physicians that the products are safe, therapeutically effective and cost effective. In addition, the inclusion or exclusion of products from treatment guidelines established by various physician groups and the viewpoints of influential physicians can affect the willingness of other physicians to prescribe the treatment. We cannot predict whether physicians, physicians’ organizations, hospitals, other healthcare providers, government agencies or private insurers will determine that any product we may develop is safe, therapeutically effective and cost effective as compared with competing treatments. If any drug candidates we develop do not achieve an adequate level of acceptance, we may not generate significant product revenue, and we may not become profitable.
If, in the future, we are unable to establish sales and marketing capabilities or enter into agreements with third parties to sell and market any drug candidates we may develop, we may not be successful in commercializing those drug candidates if and when they are approved.
We do not have a sales or marketing infrastructure and have little experience in the sale, marketing, or distribution of pharmaceutical products. To achieve commercial success for any approved product for which we retain sales and marketing responsibilities, we must either develop a sales and marketing organization, sales and marketing software solutions, or outsource these functions to third parties. In the future, we may choose to build a focused sales, marketing, and commercial support infrastructure to market and sell our drug candidates, if and when they are approved. We may also elect to enter into collaborations or strategic partnerships with third parties to engage in commercialization activities with respect to selected drug candidates, indications or geographic territories, including territories outside the United States, although there is no guarantee we will be able to enter into these arrangements even if the intent is to do so.
There are risks involved with both establishing our own commercial capabilities and entering into arrangements with third parties to perform these services. For example, recruiting and training a sales force or reimbursement specialists is expensive and time consuming and could delay any product launch. If the commercial launch of a drug candidate for which we recruit a sales force and establish marketing and other commercialization capabilities is delayed or does not occur for any reason, we would have prematurely or unnecessarily incurred these commercialization expenses. This may be costly, and our investment would be lost if we cannot retain or reposition commercialization personnel.
Factors that may inhibit our efforts to commercialize any approved product on our own include:
• the inability to recruit and retain adequate numbers of effective sales, marketing, reimbursement, customer service, medical affairs, and other support personnel;
• the inability of sales personnel or software tools to obtain access to physicians or persuade adequate numbers of physicians to prescribe any future approved products;
• the inability of reimbursement professionals to negotiate arrangements for formulary access, reimbursement, and other acceptance by payors;
• the inability to price products at a sufficient price point to ensure an adequate and attractive level of profitability;
• restricted or closed distribution channels that make it difficult to distribute our products to segments of the patient population;
• the lack of complementary products to be offered by sales personnel, which may put us at a competitive disadvantage relative to companies with more extensive product lines; and
• unforeseen costs and expenses associated with creating an independent commercialization organization.
If we enter into arrangements with third parties to perform sales, marketing, commercial support, and distribution services, our product revenue or the profitability of product revenue may be lower than if we were to market and sell any products we may develop internally. In addition, we may not be successful in entering into arrangements with third parties to commercialize our drug candidates or may be unable to do so on terms that are favorable to us or them. We may have little control over such third parties, and any of them may fail to devote the necessary resources and attention to sell and market our products effectively or may expose us to legal and regulatory risk by not adhering to regulatory requirements and restrictions governing the sale and promotion of prescription drug products, including those restricting off-label promotion. If we do not establish commercialization capabilities successfully, either on our own or in collaboration with third parties, we will not be successful in commercializing our drug candidates, if approved.
As our drug development pipeline increases and matures, the increased demand for clinical and commercial supplies from our facilities and third parties may impact our ability to operate. We will require increased capacity across our entire supply chain. Furthermore, we rely on many service providers, including those that provide manufacturing or testing services, all of whom have inherent risks in their operations that may adversely impact our operations.
We currently utilize, and expect to continue to utilize, third parties to, among other things, manufacture raw materials, components, parts, and consumables, and to perform quality testing, including such materials for our automated robotics platform. If the field of technology-enabled drug discovery continues to expand, we may encounter increasing competition for these materials and services. Demand for third-party manufacturing or testing facilities may grow at a faster rate than their existing capacity, which could disrupt our ability to find and retain third-party suppliers and manufacturers capable of producing sufficient quantities of such raw materials, components, parts, and consumables required for our drug candidates and to maintain our automated robotics platform. The use of service providers and suppliers could expose us to risks, including, but not limited to:
• termination or non-renewal of supply and service agreements with third parties in a manner or at a time that is costly or damaging to us;
• disruptions to the operations of these suppliers and service providers caused by conditions unrelated to our business or operations, including the bankruptcy of the supplier or service provider or force majeure events, such as the COVID-19 pandemic; and
• inspections of third-party facilities by regulatory authorities that could have a negative outcome and result in delays to or termination of their ability to supply our requirements.
Additional risks to our automated robotics platform include reliance on third-party equipment and instrument suppliers and consumable and reagent suppliers. The failure of third-party suppliers to fulfill our needs could adversely affect our ability to continue to operate our drug discovery platform and generate new insights that lead to successful drug candidates.
We are subject to regulatory and operational risks associated with the physical and digital infrastructure at both our internal facilities and those of our external service providers.
Our facilities in Salt Lake City, Utah have not been reviewed or pre-approved by any regulatory agency, nor has the facility been inspected by any Federal regulatory agency such as the FDA. An inspection by the FDA could disrupt our ability to generate data and develop drug candidates. Our laboratory facilities are designed to incorporate a significant level of automation of equipment with integration of several digital systems to improve efficiency of research operations. We have attempted to achieve a high level of digitization for a research operation relative to industry standards. While this is meant to improve operational efficiency, this may pose additional risk of equipment malfunction and even overall system failure or shutdown due to internal or external factors including, but not limited to, design issues, system compatibility, or potential cybersecurity breaches. This may lead to delay in potential drug candidate identification or shutdown of our facility. Any disruption in our data generation capabilities could cause delays in advancing new drug candidates into our pipeline, advancing existing programs, or enhancing the capabilities of our platform, including expanding our data, the occurrence of which could have a material adverse effect on our business, financial condition, results of operations, and prospects.
In the future, we may manufacture drug substances or products for preclinical and clinical use at our facilities and we have limited prior manufacturing experience.
If, in the future, we decide to produce drug substances or products, we will have no prior experience producing it at our facilities for preclinical and clinical use. We could incur delays in implementing the full operational state of the facility, causing delays to preclinical or clinical supply or need to rely on third-party service providers, resulting in unplanned expenses.
As we expand our development and commercial capacity, we may establish manufacturing capabilities inside the Salt Lake City footprint or expand to other locations or geographies, which may lead to regulatory delays or prove costly. If we fail to select the correct location, complete the construction in an efficient manner, recruit the appropriate personnel, and generally manage our growth effectively, the development and production of our investigational medicines could be delayed or curtailed. Additional investments may be needed if changes in our manufacturing process lead to required changes in the facility’s infrastructure.
Our current operations are located in Utah and California; and we or the third parties upon whom we depend may be adversely affected by natural disasters and our business continuity and disaster recovery plans may not adequately protect us from a serious disaster.
Our current operations are located in Salt Lake City, Utah and Milpitas, California. Any unplanned event, such as flood, fire, explosion, earthquake, extreme weather condition, medical epidemics, including any potential effects from the current global spread of COVID-19, power shortage, telecommunication failure or other natural or man-made accidents or incidents that result in us being unable to fully utilize our facilities, or the manufacturing facilities of our third-party contract manufacturers, may have a material and adverse effect on our ability to operate our business and have significant negative consequences on our financial and operating conditions. Loss of access to these facilities may result in increased costs, delays in the development of our drug candidates or interruption of our business operations. Natural disasters or pandemics such as the COVID-19 outbreak could further disrupt our operations, and have a material and adverse effect on our business, financial condition, results of operations and prospects. For example, we have instituted a temporary work from home policy for non-essential office personnel and it is possible that this could have a negative impact on the execution of our business plans and operations, especially because we rely on validating some of the drug discovery biology in our wet lab. Furthermore, our wet lab houses the robots used to produce our dataset that builds the Recursion Data Universe which is a key means by which we conduct drug candidate discovery. If a natural disaster, power outage or other event occurred that prevented us from using all or a significant portion of our headquarters or the datacenter where we collocate our GPU cluster, or damaged critical infrastructure or our robots, such as our research facilities or the manufacturing facilities of our third-party contract manufacturers, or that otherwise disrupted operations, it may be difficult or, in certain cases, impossible, for us to continue our business for a substantial period of time. The disaster recovery and business continuity plans we have in place may prove inadequate in the event of a serious disaster or similar event.
Furthermore, we do not have a disaster recovery and business continuity plan for systems related to chemistry. We may incur substantial expenses as a result of the limited nature of our disaster recovery and business continuity plans, which could have a material adverse effect on our business. As part of our risk management policy, we maintain insurance coverage at levels that we believe are appropriate for our business. However, in the event of an accident or incident at these facilities, we cannot assure our investors that the amounts of insurance will be sufficient to satisfy any damages and losses. If our facilities or the manufacturing facilities of our third-party contract manufacturers are unable to operate because of an accident or incident or for any other reason, even for a short period of time, any or all of our research and development programs may be harmed. Any business interruption may have a material and adverse effect on our business, financial condition, results of operations and prospects.
Furthermore, our facilities in Salt Lake City, Utah are located in a busy downtown area. Although we believe we have taken the necessary steps to ensure our operations are safe to the surrounding area, there could be a risk to the public if we were to conduct hazardous material research, including use of flammable chemicals and materials, at our facilities. To date, we have not received any complaints from the public associated with our operations. From time to time, we also hold public events in our Salt Lake City facilities. We have protocols in place to protect our facilities and the confidential information and assets inside; however, it is difficult to secure certain portions of our facilities and security of our confidential and proprietary information could be compromised. Despite the steps we have taken, the surrounding community may still perceive our facility as unsafe, which could have a material and adverse effect on our reputation and operations.
If we fail to comply with environmental, health and safety, or other laws and regulations, we could become subject to fines or penalties or incur costs that could have a material adverse effect on the success of our business.
We are subject to numerous environmental, health and safety, and other laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment and disposal of hazardous materials and wastes. Our operations involve the use of hazardous and flammable materials, including chemicals and biological and radioactive materials. Our operations also produce hazardous waste products. We generally contract with third parties for the disposal of these materials and wastes. We cannot eliminate the risk of contamination or injury from these materials. In the event of contamination or injury resulting from our use of hazardous materials, we could be held liable for any resulting damages, and any liability could exceed our resources. We also could incur significant costs associated with civil or criminal fines and penalties.
Although we maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees resulting from the use of hazardous materials, this insurance may not provide adequate coverage against potential liabilities. We do not maintain insurance for environmental liability or toxic tort claims that may be asserted against us in connection with our storage or disposal of biological, hazardous or radioactive materials.
Our ability to utilize our net operating loss carryforwards and certain other tax attributes may be limited.
Under Section 382 of the Internal Revenue Code of 1986, as amended, or the Code, if a corporation undergoes an “ownership change,” which is generally defined as a greater than 50% change by value in the ownership of its equity over a three-year period, our ability to use our pre-change net operating loss carryforwards and certain other pre-change tax attributes to offset its post-change income could be subject to an annual limitation. Such annual limitation could result in the expiration of a portion of the net operating loss carryforward before utilization. If not utilized the carryforwards will begin to expire in 2036. We may have experienced such ownership changes in the past, and we may experience ownership changes in the future as a result of our initial public offering or subsequent shifts in our stock ownership, some of which are outside of our control; however, we have not determined whether an ownership change has occurred. As of December 31, 2020, we had federal net operating loss carryforwards of approximately $193.8million, and our ability to utilize those net operating loss carryforwards could be limited by enacted legislation or an “ownership change” as described above, which could result in increased tax liability to us.
If our estimates or judgments relating to our critical accounting policies prove to be incorrect or financial reporting standards or interpretations change, our results of operations could be adversely affected.
The preparation of financial statements in conformity with generally accepted accounting principles in the United States, or U.S. GAAP, requires management to make estimates and assumptions that affect the amounts reported in the consolidated financial statements and accompanying notes. We base our estimates on historical experience, known trends and events, and various other factors that we believe to be reasonable under the circumstances, as provided in “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Critical Accounting Policies and Use of Estimates.” The results of these estimates form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Significant assumptions and estimates used in preparing our consolidated financial statements include stock-based compensation and valuation of our equity investments in early-stage biotechnology companies. Our results of operations may be adversely affected if our assumptions change or if actual circumstances differ from those in our assumptions, which could cause our results of operations to fall below the expectations of securities analysts and investors, resulting in a decline in the trading price of our Class A common stock.
Additionally, we regularly monitor our compliance with applicable financial reporting standards and review new pronouncements and drafts thereof that are relevant to us. As a result of new standards, changes to existing standards and changes in their interpretation, we might be required to change our accounting policies, alter our operational policies, and implement new or enhance existing systems so that they reflect new or amended financial reporting standards, or we may be required to restate our published financial statements. Such changes to existing standards or changes in their interpretation may have an adverse effect on our reputation, business, financial position, and profit.
Risks Related to Our Reliance on Third Parties
We expect to rely on third parties to conduct our clinical trials and some aspects of our research and preclinical testing and those third partiesor conduct our clinical trials may not perform satisfactorily including failing to meet deadlines for the completion of such trials, research, or testing.their agreements may be terminated.
We currently rely, and expect to continue to rely, on third parties such as clinical research organizations, clinical data management organizations, medical institutions, and clinical investigators, to conduct some aspects of research and preclinical testing and clinical trials. The third parties include CROs, clinical data management organizations, medical institutions, and principal investigators. Any of these third parties may fail to fulfill their contractual obligations, including by not meeting deadlines for the completion of research, testing, or trials, or we or they may terminate their engagements with us or be unable to fulfill their contractual obligations.us. If any of our relationships with these third parties terminate, we may not be able to enter into arrangements with alternative third parties on commercially reasonable terms, or at all. If we need to enter into alternative arrangements, it wouldsuch negotiations could delay product development activities. Termination of our relationships with foreign third parties can also occur if U.S. legislation, sanctions, trade restrictions, or other U.S. and foreign regulatory requirements, prohibitions or restrictions, limit or prevent our ability to enter into arrangements with such foreign third parties. For example, we currently rely on foreign CROs and CDMOs, including an affiliate/subsidiary of WuXi AppTec Co., Ltd. (WuXi AppTec) in China that has been listed as a biotechnology company “of concern” in proposed U.S. legislation known as the BIOSECURE Act (recently introduced in the U.S. House of Representatives as H.R. 7085 and in the U.S. Senate as S.3558). While the BIOSECURE Act as currently proposed would restrict purchasing of services or products from WuXI AppTec and other companies of concern in China, it would only impact U.S. companies that contract with or receive funding from the U.S. government, which means that our company would not be directly impacted by the BIOSECURE Act. However, passage of the BIOSECURE Act could potentially lead the way for further legislation, sanctions, or restrictions that could potentially impact our third-party arrangements with companies such as WuXi AppTec which could delay or impact clinical trials and consequently delay or obstruct regulatory approval of our drug candidates.
Our reliance on these third parties for research and development activities reduces our control over these activities, but does not relieve us of our responsibilities. For example, we remain responsible for ensuring that each of our respective clinical trials is conducted in accordance with the general investigational plan and protocols for the trial, andas well as applicable legal, regulatory, and scientific standards,standards. We also are required to register ongoing clinical trials and our reliancepost the results of completed clinical trials on third parties does not relieve us of our regulatory responsibilities.a government-sponsored database within certain timeframes. In addition, the FDA and comparable foreign regulatory authorities require compliance with good clinical practices or GCP(GCP) guidelines for conducting, recording, and reporting the results of clinical trials to assure that data and reported results are credible, reproducible, and accurate, and that the rights, integrity, and confidentiality of trial participants are protected. Regulatory authorities enforce GCP compliance through periodic inspections of trial sponsors, principal investigators, and trial sites.
If we or any of thesethe third parties fail to comply with applicable GCP regulations, some or all of the clinical data generated in our clinical trials may be deemed unreliable, and the FDA or comparable foreign regulatory authorities may require us to perform additional nonclinical or clinical trials or to enroll additional patients before approving our marketing applications. We cannot be certain that, upon inspection, such regulatory authorities will determine that any ofIn addition, if we or the third parties fail to comply with our clinical trials complies with the GCP regulations. For any violations ofstated protocols or applicable laws and regulations during the conduct of clinical trials, we or the third parties could be subject to untitled and warning letters or
enforcement action that may includeactions by the FDA and comparable foreign regulatory authorities, which could result in civil penalties up to and includingor criminal prosecution. We also are required to register ongoing clinical trials and post the results of completed clinical trials on a government-sponsored database within certain timeframes. Failure to do so can result in fines,prosecution, as well as adverse publicity and civil and criminal sanctions.that harms our business.
IfWe also will not be able to obtain, or may be delayed in obtaining, marketing approvals for any drug candidates we may develop if these third parties do not successfully carry out their contractual duties, meet expected deadlines, or conduct clinical trials in accordance with regulatory requirements or our stated protocols or regulatory requirements. As a result, we will not be able to obtain, or may be delayed in obtaining, marketing approvals for any drug candidates we may develop and will not be able to, or may be delayed in our efforts to, successfully commercialize our medicines. Our failure or the failure of these third parties to comply with applicable regulatory requirements or our stated protocols could also subject us to enforcement action.
We contract with third parties for the manufacture of our drug candidates for preclinical development, clinical testing, and expect to continue to do so for commercialization. This reliance on third parties increases the risk that we will not have sufficient quantities of our drug candidates or products or such quantities at an acceptable cost, which could delay, prevent or impair our development or commercialization efforts.
We do not currently own or operate any manufacturing facilities or personnel, although we are in the process of securing a facility to establish production capabilities for preclinical animal studies and early human clinical trials. We rely, and could expect to continue to rely, on third parties for the manufacture of many of our drug candidates for preclinical development and clinical testing, as well as for the commercial manufacture of our products if any of our drug candidates receive marketing approval. This reliance on third parties increases the risk that we will not have sufficient quantities of our drug candidates or products or such quantities at an acceptable cost or quality, which could delay, prevent or impair our development or commercialization efforts.
The facilities used by our contract manufacturers to manufacture our drug candidates must be inspected by the FDA pursuant to pre-approval inspections that will be conducted after we submit our marketing applications to the FDA. We do not control the manufacturing process of, and will be completely dependent on, our contract manufacturers for compliance with current good manufacturing practice guidelines, or cGMP, in connection with the manufacture of our drug candidates in the near to intermediate term or possibly long term. If our contract manufacturers cannot successfully manufacture material that conforms to our specifications and the strict regulatory requirements of the
FDA or others, they will not be able to pass regulatory inspections and/or maintain regulatory compliance for their manufacturing facilities. In addition, we have no control over the ability of our contract manufacturers to maintain adequate quality control, quality assurance and qualified personnel. If the FDA or a comparable foreign regulatory authority finds deficiencies with or does not approve these facilities for the manufacture of our drug candidates or if it finds deficiencies or withdraws any such approval in the future, we may need to find alternative manufacturing facilities, which would significantly impact our ability to develop, obtain regulatory approval for or market our drug candidates, if approved. Further, our failure, or the failure of our third- party manufacturers, to comply with applicable regulations could result in sanctions being imposed on us, including clinical holds, fines, injunctions, civil penalties, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of drug candidates or products, if approved, operating restrictions and criminal prosecutions, any of which could significantly and adversely affect our business and supplies of our drug candidates. Our third-party manufacturers may be subject to third-party litigation which could disrupt our supply chain, result in liability and harm our business, including the need to increase prices in connection with the commercialization of future drug candidates.
We may be unable to establish any agreements with third-party manufacturers or to do so on acceptable terms. Even if we are able to establish agreements with third-party manufacturers, reliance on third-party manufacturers entails additional risks, including:
• reliance on the third party for regulatory compliance and quality assurance;
• the possible breach of the manufacturing agreement by the third party;
• the possible misappropriation of our proprietary information, including our trade secrets and know-how; and
• the possible termination or nonrenewal of the agreement by the third party at a time that is costly or inconvenient for us.
Our drug candidates and any products that we may develop may compete with other drug candidates and approved products for access to manufacturing facilities or capacity. There are a limited number of manufacturers that operate under cGMP regulations and that might be capable of manufacturing for us.
Any performance failure on the part of our existing or future manufacturers could delay clinical development or marketing approval. If our current contract manufacturers cannot perform as agreed, we may be required to replace such manufacturers. We may incur added costs and delays in identifying and qualifying any such replacement.
Our current and anticipated future dependence upon others for the manufacture of our drug candidates or products may adversely affect our future profit margins and our ability to commercialize any products that receive marketing approval on a timely and competitive basis.
We also expect to rely on other third parties to store and distribute drug supplies for our clinical trials. Any performance failure on the part of our distributors could delay clinical development or marketing approval of any drug candidates we may develop or commercialization of our medicines, producing additional losses and depriving us of potential product revenue.
The third parties upon whom we rely for certain equipment and the supply of the active pharmaceutical ingredients used in our drug candidates are our only source of supply, and the loss of any of these suppliers could significantly harm our business.
Certain of our specialized equipment and the active pharmaceutical ingredients, or API, used in our drug candidates are supplied to us from single-source suppliers. Our ability to successfully develop our drug candidates, and to ultimately supply our commercial products in quantities sufficient to meet the market demand, depends in part on our ability to obtain equipment and the API for these products in accordance with regulatory requirements and in sufficient quantities for clinical testing and commercialization. We do not currently have arrangements in place for a redundant or second-source supply of any such equipment or API in the event any of our current suppliers of such equipment or API ceases their operations for any reason. We are also unable to predict how changing global economic conditions or potential global health concerns such as the COVID-19 pandemic will affect our third-party suppliers and manufacturers. Any negative impact of such matters on our third-party suppliers and manufacturers may also have an adverse impact on our results of operations or financial condition.
For all of our drug candidates, we intend to identify and qualify additional vendors and manufacturers to provide such equipment or API prior to submission of an NDA to the FDA and/or an MAA to the EMA. We are not certain,
however, that our single-source suppliers will be able to meet our demand for their products, either because of the nature of our agreements with those suppliers, our limited experience with those suppliers or our relative importance as a customer to those suppliers. It may be difficult for us to assess their ability to timely meet our demand in the future based on past performance. While our suppliers have generally met our demand for their products on a timely basis in the past, they may subordinate our needs in the future to their other customers.
Establishing additional or replacement suppliers for certain equipment and the API used in our drug candidates, if required, may not be accomplished quickly. If we are able to find a replacement supplier, such replacement supplier would need to be qualified and may require additional regulatory inspection or approval, which could result in further delay. While we seek to maintain adequate inventory of the API used in our drug candidates, any interruption or delay in the supply of components or materials, or our inability to obtain such API from alternate sources at acceptable prices in a timely manner could impede, delay, limit or prevent our development efforts, which could harm our business, results of operations, financial condition and prospects.
We may seek to establish additional collaborations for clinical development or commercialization of our drug candidates, and, if we are not able to establish them on commercially reasonable terms, or at all, we may have to alter our development and commercialization plans.
Our product development programs and the potential commercialization of our drug candidates will require substantial additional cash to fund expenses. For some of our drug candidates, we may decide to collaborate with additional pharmaceutical and biotechnology companies for the development and potential commercialization of those drug candidates. In the near term, the value of our company will depend in part, on the number of and the quality of the collaborations that we create.
Potential collaborators may reject collaborations based upon their assessment of our financial, regulatory or intellectual property position. Whether we reach a definitive agreement for a collaboration will depend, among other things, upon our assessment of the collaborator’s resources and expertise, the terms and conditions of the proposed collaboration and the proposed collaborator’s evaluation of a number of factors. Those factors may include the design or results of clinical trials, the likelihood of approval by the FDA or similar regulatory authorities outside the United States, the potential market for the subject drug candidate, the costs and complexities of manufacturing and delivering such drug candidate to patients, the potential of competing products, the existence of uncertainty with respect to our ownership of technology, which can exist if there is a challenge to such ownership without regard to the merits of the challenge and industry and market conditions generally. The collaborator may also consider alternative drug candidates or technologies for similar indications that may be available to collaborate on and whether such a collaboration could be more attractive than the one with us for our drug candidate. The terms of any additional collaborations or other arrangements that we may establish may not be favorable to us.
Collaborative relationships with third parties could cause us to expend significant resources and incur substantial business risk with no assurance of financial return. Management of our relationships with collaborators will require:
• significant time and effort from our management team;
• coordination of our marketing and research and development programs with the marketing and research and development priorities of our collaborators; and
• effective allocation of our resources to multiple projects.
If we are unable to establish or maintain such strategic collaborations on terms favorable to us in the future, our research and development efforts and potential to generate revenue may be limited.
We may also be restricted under collaboration agreements from entering into future agreements on certain terms with potential collaborators. Collaborations are complex and time-consuming to negotiate and document. In addition, the significant number of recent business combinations among large pharmaceutical companies has resulted in a reduced number of potential future collaborators.
We may not be able to negotiate additional collaborations on a timely basis, on acceptable terms, or at all. If we are unable to do so, we may have to curtail the development of the drug candidate for which we are seeking to collaborate, reduce or delay its development program or one or more of our other development programs, delay its potential commercialization or reduce the scope of any sales or marketing activities, or increase our expenditures and undertake development or commercialization activities at our own expense. If we elect to increase our expenditures to fund development or commercialization activities on our own, we may need to obtain additional
capital, which may not be available to us on acceptable terms or at all. If we do not have sufficient funds, we may not be able to further develop our drug candidates or bring them to market and generate product revenue. Even if we successfully establish new collaborations, these relationships may never result in the successful development or commercialization of drug candidates or the generation of sales revenue. The success of our collaboration arrangements will depend heavily on the efforts and activities of our collaborators. Collaborators generally have significant discretion in determining the efforts and resources that they will apply to these collaborations. To the extent that we enter into collaborative arrangements, the related product revenues are likely to be lower than if we directly marketed and sold products. Such collaborators may also consider alternative drug candidates or technologies for similar indications that may be available to collaborate on and whether such a collaboration could be more attractive than the one with us for any future drug candidate. Disagreements between parties to a collaboration arrangement regarding clinical development or commercialization matters can lead to delays in the development process or commercialization of the applicable drug candidate and, in some cases, the termination of the collaboration arrangement. These disagreements can be difficult to resolve if neither of the parties has final decision-making authority. Collaborations with pharmaceutical or biotechnology companies or other third parties often are terminated or allowed to expire by the other party. Any such termination or expiration would adversely affect us financially and could harm our business reputation. If we were to become involved in arbitration or litigation with any of our collaborators it would consume time and divert management resources away from operations, damage our reputation and impact our ability to enter into future collaboration agreements and may result in substantial payments from us to our collaborators to settle any disputes.
Risks Related to Our Intellectual Property
If we are unable to adequately protect and enforce our intellectual property and proprietary technology or obtain and maintain patent protection for our technology and products or if the scope of the patent protection obtained is not sufficiently broad, our competitors could develop and commercialize technology and products similar or identical to ours, and our ability to successfully commercialize our technology and products may be impaired.
Our commercial success will depend in part on our ability to obtain, maintain, protect and enforce our proprietary and intellectual property rights in the United States and other countries for our drug candidates, and our core technologies, including our phenomic platform, preclinical and clinical assets, composition of matter, methods of use and formulation patents and related know-how. We seek to protect our proprietary and intellectual property position by, among other methods, filing patent applications in the United States and abroad related to our proprietary technology, inventions and improvements that are important to the development and implementation of our business. However, the patent process is expensive, time consuming and complex, and we may not be able to apply for patents on certain aspects of our technology and products in a timely fashion, at a reasonable cost, in all jurisdictions or at all, and any potential patent coverage we obtain may not be sufficient to prevent substantial competition. In addition, we also rely on trade secrets, know-how and continuing technological innovation to develop and maintain our proprietary and intellectual property position. We do not own or in-license any issued patents with respect to certain of our programs, including our REC-3599 product candidate, our lead molecules for the treatment of C. difficile colitis (REC-163964, REC-164014, and REC-164067), our lead molecules for the treatment of neuroinflammation (REC-648455, REC-648597, and REC-648677), our lead molecules for the treatment of Batten disease (REC-648190, REC-259618, and REC-648647), or the lead molecules for the treatment of CMT2A (REC-64810, REC-648458, REC-1262, and REC-150357), REC-64151 for the treatment of STK11-mutant immune checkpoint resistance in non-small cell lung cancer and MYC inhibitory molecules for the treatment of solid and hematological malignancies we can provide no assurance that any of our current or future patent applications will result in issued patents or that any issued patents will provide us with any competitive advantage.
The patent position of biotechnology and pharmaceutical companies generally is highly uncertain, involves complex legal and factual questions and has in recent years been the subject of much litigation.
The degree of patent protection we require to successfully commercialize our drug candidates may be unavailable or severely limited in some cases and may not adequately protect our rights or permit us to gain or keep any competitive advantage. We cannot provide any assurances that any of our pending patent applications will issue, or that any of our pending patent applications that mature into issued patents will include claims with a scope sufficient to protect our drug candidates from competition. In addition, the laws of foreign countries may not protect our rights to the same extent as the laws of the United States, and many companies have encountered significant challenges in establishing and enforcing their proprietary rights outside of the United States. Furthermore, patents have a
limited lifespan. In the United States, the natural expiration of a patent is generally twenty years after its first effective non-provisional filing date. Various extensions may be available; however, the life of a patent, and the protection it affords, is limited. Given the amount of time required for the development, testing and regulatory review of new drug candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. As a result, our owned patent portfolio and any patent portfolio we may license in the future may not provide us with adequate and continuing patent protection sufficient to exclude others from commercializing products similar or identical to our drug candidates, including generic versions of such products.
Other parties have developed technologies that may be related or competitive to our own, and such parties may have filed or may file patent applications, or may have received or may receive patents, claiming inventions that may overlap or conflict with those claimed in our own patent. Publications of discoveries in scientific literature often lag behind the actual discoveries, and patent applications in the United States and other jurisdictions are typically not published until 18 months after filing, or in some cases not at all. Therefore, we cannot know with certainty whether we or our licensors were the first to make the inventions claimed in our owned or licensed patents or pending patent applications, or that we or our licensors were the first to file for patent protection of such inventions. As a result, the issuance, scope, validity, enforceability and commercial value of our patent rights cannot be predicted with any certainty.
In addition, the patent prosecution process is expensive, time-consuming and complex, and we may not be able to file, prosecute, maintain, enforce or license all necessary or desirable patent applications at a reasonable cost or in a timely manner. Further, with respect to most of the pending patent applications covering our drug candidates, prosecution has yet to commence. Patent prosecution is a lengthy process, during which the scope of the claims initially submitted for examination by the U.S. Patent and Trademark Office, or USPTO, may be significantly narrowed by the time they issue, if at all. It is also possible that we will fail to identify patentable aspects of our research and development output in time to obtain patent protection. Moreover, in some circumstances, we do not have the right to control the preparation, filing and prosecution of patent applications, or to maintain the patents, covering technology that we license from third parties. We may also require the cooperation of our licensors and collaborators to enforce any licensed patent rights, and such cooperation may not be provided. Therefore, these patents and applications may not be prosecuted and enforced in a manner consistent with the best interests of our business.
We currently own a number of U.S. provisional patent applications. U.S. provisional patent applications are not eligible to become issued patents until, among other things, we file a non-provisional patent application within 12 months of filing one or more of our related provisional patent applications. With regard to such U.S. provisional patent applications, if we do not timely file any non-provisional patent applications, we may lose our priority dates with respect to our provisional patent applications and any patent protection on the inventions disclosed in our provisional patent applications. Further, in the event that we do timely file non-provisional patent applications relating to our provisional patent applications, we cannot predict whether any such patent applications will result in the issuance of patents or if such issued patents will provide us with any competitive advantage.
Even if we acquire patent protection that we expect should enable us to maintain such competitive advantage, third parties may challenge the validity, enforceability or scope thereof, which may result in such patents being narrowed, invalidated or held unenforceable. The issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability, and our owned and licensed patents may be challenged in the courts or patent offices in the United States and abroad. For example, we may be subject to a third-party submission of prior art to the USPTO challenging the priority of an invention claimed within one of our patents, which submissions may also be made prior to a patent’s issuance, precluding the granting of any of our pending patent applications. Further, inadvertent or intentional public disclosures of our inventions prior to the filing of a patent application have precluded, and in the future may preclude us from obtaining patent protection in certain jurisdictions. We may become involved in opposition, derivation, reexamination, inter parties review, post-grant review or interference proceedings challenging our patent rights or the patent rights of others from whom we have obtained licenses to such rights. Such proceedings could result in revocation of or amendment to our patents in such a way that they no longer cover our technology or drug candidates.
Competitors may claim that they invented the inventions claimed in our issued patents or patent applications prior to us, or may file patent applications before we do. Competitors may also claim that we are infringing on their patents and that we therefore cannot practice our technology as claimed under our patents, if issued. Competitors may also contest our patents, if issued, by showing the patent examiner that the invention was not original, was not novel or
was obvious. In litigation, a competitor could claim that our patents, if issued, are not valid for a number of reasons. If a court agrees, we would lose our rights to those challenged patents.
In addition, we may in the future be subject to claims by our former employees or consultants asserting an ownership right in our patents or patent applications, as a result of the work they performed on our behalf. Although we generally require all of our employees, consultants and advisors and any other third parties who have access to our proprietary know-how, information or technology to assign or grant similar rights to their inventions to us, we cannot be certain that we have executed such agreements with all parties who may have contributed to our intellectual property, nor can we be certain that our agreements with such parties will be upheld in the face of a potential challenge, or that they will not be breached, for which we may not have an adequate remedy. A loss of exclusivity, in whole or in part, could allow others to compete with us and harm our business.
An adverse determination in any such submission or proceeding may result in loss of exclusivity or freedom to operate or in patent claims being narrowed, invalidated or held unenforceable, in whole or in part, which could limit our ability to stop others from using or commercializing similar or identical technology and products, without payment to us, or could limit the duration of the patent protection covering our technology and drug candidates. Such challenges may also result in our inability to manufacture or commercialize our drug candidates without infringing third-party patent rights. In addition, if the breadth or strength of protection provided by our patents and patent applications is threatened, it could dissuade companies from collaborating with us to license, develop or commercialize current or future drug candidates.
Even if they are unchallenged, our owned patent portfolio and any patent portfolio we may license in the future may not provide us with any meaningful protection or prevent competitors from designing around our patent claims to circumvent our owned or licensed patents by developing similar or alternative technologies or products in a non-infringing manner. For example, a third party may develop a competitive product that provides benefits similar to one or more of our drug candidates but that has a different composition that falls outside the scope of our patent protection. If the patent protection provided by the patents and patent applications we hold or pursue with respect to our drug candidates is not sufficiently broad to impede such competition, our ability to successfully commercialize our drug candidates could be negatively affected, which would harm our business.
Obtaining and maintaining patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
The USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application and prosecution process. In addition, periodic maintenance fees, renewal fees, annuity fees and various other governmental fees on patents and/or patent applications often must be paid to the USPTO and foreign patent agencies over the lifetime of the patent and/or patent application. While an unintentional lapse can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which non-compliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Non-compliance events that could result in abandonment or lapse of a patent or patent application include, but are not limited to, failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. If we fail to maintain the patents and patent applications covering our products or procedures, we may not be able to stop a competitor from marketing products that are the same as or similar to our drug candidates, which would have a material adverse effect on our business.
If we are unable to protect the confidentiality of our trade secrets and know-how, our business and competitive position may be harmed.
In addition to the protection afforded by patents, we rely upon unpatented trade secret protection, unpatented know-how and continuing technological innovation to develop and maintain our competitive position. With respect to curating our data and our library of small molecules generally, we consider trade secrets and know-how to be our primary intellectual property. We seek to protect our proprietary technology and processes, in part, by entering into confidentiality agreements with our collaborators, scientific advisors, employees and consultants, and invention assignment agreements with our consultants and employees. We may not be able to prevent the unauthorized disclosure or use of information which we consider to be confidential, our technical know-how or other trade secrets
by the parties to these agreements, however, despite the existence generally of confidentiality agreements and other contractual restrictions. Monitoring unauthorized uses and disclosures is difficult, and we do not know whether the steps we have taken to protect our proprietary technologies will be effective. If any of the collaborators, scientific advisors, employees and consultants who are parties to these agreements breaches or violates the terms of any of these agreements, we may not have adequate remedies for any such breach or violation, and we could lose our trade secrets as a result. For example, if one of our employees publicly discloses information that we believe to be confidential or a trade secret we may be unable to protect it in the future. Even where remedies are available, enforcing a claim that a party illegally disclosed or misappropriated our trade secrets, like patent litigation, is expensive and time consuming, and the outcome is unpredictable. In addition, courts outside the United States are sometimes less willing to protect trade secrets.
Our trade secrets could otherwise become known or be independently discovered by our competitors. Competitors could purchase our drug candidates and attempt to replicate some or all of the competitive advantages we derive from our development efforts, willfully infringe our intellectual property rights, design around our protected technology or develop their own competitive technologies that fall outside of our intellectual property rights. If any of our trade secrets were to be lawfully obtained or independently developed by a competitor or other third party, we would have no right to prevent them, or those to whom they communicate it, from using that technology or information to compete with us.
Third parties may initiate legal proceedings alleging that we are infringing, misappropriating or otherwise violating their intellectual property or proprietary rights, the outcome of which would be uncertain and could have a material adverse effect on the success of our business.
Our commercial success depends upon our ability and the ability of our collaborators to develop, manufacture, market and sell our drug candidates and use our proprietary technologies without infringing, misappropriating or otherwise violating the intellectual property or proprietary rights of third parties. The biotechnology and pharmaceutical industries are characterized by extensive and frequent litigation regarding patents and other intellectual property rights. For example, we sublicense CRISPR-Cas9 gene editing technology from a licensed vendor, which provides critical tools upon which portions of our drug discovery process relies, but there are ongoing disputes between third parties, which we are not party to, regarding the ownership of and licensing rights related to such technology. CRISPR-Cas9 gene editing is a field that is highly active for patent filings. In November 2018, it was reported that 211 patent families and 1835 patent family members worldwide referenced CRISPR or Cas in the title, abstracts or claims. The extensive patent filings related to CRISPR and Cas make it difficult for us to assess the full extent of relevant patents and pending applications that may cover CRISPR-Cas9. There may be third-party patents or pending patent applications with claims that may issue in the future, covering our use of CRISPR-Cas9. We may need access to such patents in order to continue using CRISPR-Cas9, however we cannot be certain that such patents will be available for license on commercially reasonable terms. We may in the future become party to, or threatened with, adversarial proceedings or litigation regarding intellectual property rights with respect to our drug candidates and technology, including interference proceedings before the USPTO. Our competitors or other third parties may assert infringement claims against us, alleging that our products or technologies are covered by their patents. Given the vast and continually-increasing number of patents in our field of technology, we cannot be certain that we do not infringe existing patents or that we will not infringe patents that may be granted in the future. Many companies have filed, and continue to file, patent applications related to artificial intelligence and deep learning, technology-aided drug discovery, CRISPR, high-throughput screening, and combinations of any or all of these fields. Some of these patent applications have already been allowed or issued, and others may issue in the future. Since these areas are competitive and of strong interest to pharmaceutical and biotechnology companies, there will likely be additional patent applications filed and additional patents granted in the future, as well as additional research and development programs expected in the future. If a patent holder believes our product or drug candidate infringes on its patent, the patent holder may sue us even if we have received patent protection for our technology. Moreover, we may face patent infringement claims from non-practicing entities that have no relevant product revenue and against whom our owned patent portfolio and any patent portfolio we may license in the future may thus have no deterrent effect. If any such claim or proceeding is brought against us, our collaborators or our third-party service providers, our development, manufacturing, marketing, sales and other commercialization activities could be similarly adversely affected. Even if we believe third-party intellectual property claims are without merit, there is no assurance that a court would find in our favor on questions of infringement, validity, enforceability, or priority. A court of competent jurisdiction could hold that third-party patents asserted against us are valid, enforceable, and infringed, which could materially and adversely affect our ability to develop, manufacture, market, sell and commercialize any of our drug candidates or technology. In order to successfully challenge the validity of
any such U.S. patent in federal court, we would need to overcome a presumption of validity. As this burden is a high one requiring us to present clear and convincing evidence as to the invalidity of any such U.S. patent claim, there is no assurance that a court of competent jurisdiction would invalidate the claims of any such U.S. patent.
If we are found to infringe a third party’s patent or other intellectual property rights, we could be required to obtain a license from such third party to continue developing and marketing our drug candidates and technology. We may choose to obtain a license, even in the absence of an action or finding of infringement. In either case, we may not be able to obtain any required license on commercially reasonable terms or at all. Even if we were able to obtain such a license, it could be granted on non-exclusive terms, thereby providing our competitors and other third parties access to the same technologies licensed to us, and it could require us to make substantial licensing, royalty and other payments. Without such a license, we could be forced, including by court order, to cease developing and commercializing the infringing technology or drug candidates. In addition, we could be found liable for monetary damages, including treble damages and attorneys’ fees if we are found to have willfully infringed such third-party patent or other intellectual property rights. A finding of infringement could prevent us from commercializing our drug candidates or force us to cease some of our business operations, which could materially harm our business. If we lose a foreign patent lawsuit, alleging our infringement of a competitor’s patents, we could be prevented from marketing our products in one or more foreign countries, which would have a materially adverse effect on our business.
We may be subject to damages resulting from claims that we or our employees have wrongfully used or disclosed alleged trade secrets of our competitors or are in breach of non-competition or non-solicitation agreements with our competitors or claims asserting ownership of what we regard as our own intellectual property.
We could in the future be subject to claims that we or our employees have inadvertently or otherwise used or disclosed alleged trade secrets or other proprietary information of former employers or competitors. Although we try to ensure that our employees and consultants do not use the intellectual property, proprietary information, know-how or trade secrets of others in their work for us, we may in the future be subject to claims that we caused an employee to breach the terms of his or her non-competition or non-solicitation agreement, or that we or these individuals have, inadvertently or otherwise, used or disclosed the alleged trade secrets or other proprietary information of a former employer or competitor. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and could be a distraction to management. If our defenses to these claims fail, in addition to requiring us to pay monetary damages, a court could prohibit us from using technologies or features that are essential to our drug candidates, if such technologies or features are found to incorporate or be derived from the trade secrets or other proprietary information of the former employers. An inability to incorporate such technologies or features would have a material adverse effect on our business, and may prevent us from successfully commercializing our drug candidates. In addition, we may lose valuable intellectual property rights or personnel as a result of such claims. Moreover, any such litigation or the threat thereof may adversely affect our ability to hire employees or contract with independent sales representatives. A loss of key personnel or their work product could hamper or prevent our ability to commercialize our drug candidates, which would have an adverse effect on our business, results of operations and financial condition.
In addition, while it is our policy to require our employees and contractors who may be involved in the conception or development of intellectual property to execute agreements assigning such intellectual property to us, we may be unsuccessful in executing such an agreement with each party who, in fact, conceives or develops intellectual property that we regard as our own. The assignment of intellectual property rights may not be self-executing, or the assignment agreements may be breached, and we may be forced to bring claims against third parties, or defend claims that they may bring against us, to determine the ownership of what we regard as our intellectual property. Such claims could have an adverse effect on our business, results of operations and financial condition.
We may become involved in lawsuits to protect or enforce our patents and other intellectual property rights, which could be expensive, time consuming and unsuccessful.
Competitors and other third parties may infringe, misappropriate or otherwise violate our patents and other intellectual property rights. To counter infringement or unauthorized use, we may be required to file infringement claims. A court may disagree with our allegations, however, and may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover it. Further, such third parties could counterclaim
that we infringe their intellectual property or that a patent we have asserted against them is invalid or unenforceable. In patent litigation in the United States, defendant counterclaims and inter partes reviews challenging the validity, enforceability or scope of asserted patents are commonplace. In addition, third parties may initiate legal proceedings against us to assert such challenges to our intellectual property rights. The outcome of any such proceeding is generally unpredictable. Grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements, including lack of novelty, obviousness or non-enablement. Patents may be unenforceable if someone connected with prosecution of the patent withheld relevant information from the USPTO or made a misleading statement during prosecution. It is possible that prior art of which we and the patent examiner were unaware during prosecution exists, which could render any patents that may issue invalid. Moreover, it is also possible that prior art may exist that we are aware of but do not believe is relevant to our future patents, should they issue, but that could nevertheless be determined to render our patents invalid.
An adverse result in any litigation proceeding could put one or more of our patents at risk of being invalidated or interpreted narrowly. If a defendant were to prevail on a legal assertion of invalidity or unenforceability of our patents covering one of our drug candidates, we would lose at least part, and perhaps all, of the patent protection covering such drug candidate or technology. Competing products may also be sold in other countries in which our patent coverage might not exist or be as strong.
Intellectual property litigation could cause us to spend substantial resources and distract our personnel from their normal responsibilities.
Litigation or other legal proceedings relating to intellectual property claims, with or without merit, is unpredictable and generally expensive and time consuming and is likely to divert significant resources from our core business, including distracting our technical and management personnel from their normal responsibilities. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments and if securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our Class A common stock. Such litigation or proceedings could substantially increase our operating losses and reduce the resources available for development activities or any future sales, marketing or distribution activities.
We may not have sufficient financial or other resources to adequately conduct such litigation or proceedings. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their greater financial resources and more mature and developed intellectual property portfolios. Accordingly, despite our efforts, we may not be able to prevent third parties from infringing upon or misappropriating or from successfully challenging our intellectual property rights. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could have a material adverse effect on our ability to compete in the marketplace.
We may not be able to effectively prosecute and enforce our intellectual property rights throughout the world.
Filing, prosecuting and defending patents on our drug candidates in all countries throughout the world would be prohibitively expensive. The requirements for patentability may differ in certain countries, particularly in developing countries. Moreover, our ability to protect and enforce our intellectual property rights may be adversely affected by unforeseen changes in foreign intellectual property laws. Patent protection must ultimately be sought on a country-by-country basis, which is an expensive and time-consuming process with uncertain outcomes. Accordingly, we may choose not to seek patent protection in certain countries, and we will not have the benefit of patent protection in such countries. Additionally, the patent laws of some foreign countries, including some jurisdictions of significant commercial interest, do not afford intellectual property protection to the same extent as the laws of the United States, particularly with regard to software technologies and methods of treatment involving existing drugs. Many companies have encountered significant problems in protecting and defending intellectual property rights in certain foreign jurisdictions. The legal systems of some countries, particularly developing countries, do not favor the enforcement of patents and other intellectual property rights. This could make it difficult for us to stop the infringement of our patents, if obtained, or the misappropriation of our other intellectual property rights. For example, many foreign countries have compulsory licensing laws under which a patent owner must grant licenses to third parties and/or which limit the enforceability of patents against third parties, including government agencies or
government contractors. Consequently, we may not be able to prevent third parties from practicing our inventions in all countries outside the United States and, in those foreign countries, patents may provide limited or no benefit. In addition, we and our licensors may have limited remedies in those foreign countries if patents are infringed or if we or our licensors are compelled to grant a license to a third party, which could materially diminish the value of those patents and could limit our potential revenue opportunities.
Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and, further, may export otherwise infringing products to territories where we have patent protection, if our ability to enforce our patents to stop infringing activities is inadequate. These products may compete with our drug candidates, and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing. Proceedings to enforce our patent rights in foreign jurisdictions, whether or not successful, could result in substantial costs and divert our efforts and resources from other aspects of our business. Furthermore, while we intend to protect our intellectual property rights in the major markets for our drug candidates, we cannot ensure that we will be able to initiate or maintain similar efforts, or obtain similar patent scope, in all jurisdictions in which we may wish to market our drug candidates. Accordingly, our efforts to protect our intellectual property rights in such countries may be inadequate to obtain a significant commercial advantage from the intellectual property that we own or license.
If we do not obtain patent term extension and data exclusivity for any drug candidates we may develop, our business may be materially harmed.
Depending upon the timing, duration and specifics of any FDA marketing approval of any drug candidates we may develop, one or more of our U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Action of 1984, or Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a patent extension term of up to five years as compensation for patent term lost during the FDA regulatory review process. A patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval; only one patent may be extended and only those claims covering the approved drug, a method for using it or a method for manufacturing it may be extended. However, we may not be granted an extension because of, for example, failing to exercise due diligence during the testing phase or regulatory review process, failing to apply within applicable deadlines, failing to apply prior to expiration of relevant patents or otherwise failing to satisfy applicable requirements. Moreover, the applicable time period or the scope of patent protection afforded could be less than we request. If we are unable to obtain patent term extension or term of any such extension is less than we request, our competitors may obtain approval of competing products following our patent expiration, and our business, financial condition, results of operations and prospects could be materially harmed.
We may need to license certain intellectual property from third parties, and such licenses may not be available or may not be available on commercially reasonable terms.
A third party may hold intellectual property, including patent rights, that are important or necessary to the development of our products. It may be necessary for us to use the patented or proprietary technology of third parties to commercialize our products, in which case we would be required to obtain a license from these third parties on commercially reasonable terms, or our business could be harmed, possibly materially. For example, when we explore repurposing molecules owned by our collaboration partners or other third parties, we in-license the rights to use those molecules for our use. If we were not able to obtain a license, or were not able to obtain a license on commercially reasonable terms or with sufficient breadth to cover the intended use of third-party intellectual property, our business could be materially harmed or we may become involved in disputes.
If we fail to comply with our obligations in the agreements under which we collaborate with or license intellectual property rights from third parties, or otherwise experience disruptions to our business relationships with our collaborators or licensors, we could lose rights that are important to our business.
We license certain intellectual property that is important to our business, and in the future we may enter into additional agreements that provide us with licenses to valuable intellectual property or technology. We expect our future license agreements will impose various development, diligence, commercialization, and other obligations on us in order to maintain the licenses. In spite of our efforts, a future licensor might conclude that we have materially breached our obligations under such license agreements and seek to terminate the license agreements, thereby removing or limiting our ability to develop and commercialize products and technology covered by these license
agreements. If these in-licenses are terminated, or if the underlying patent rights licensed thereunder fail to provide the intended exclusivity, competitors or other third parties would have the freedom to seek regulatory approval of, and to market, products identical to ours and we may be required to cease our development and commercialization of certain of our drug candidates. Any of the foregoing could have a material adverse effect on our competitive position, business, financial conditions, results of operations, and prospects.
Moreover, disputes may arise regarding intellectual property subject to a licensing agreement, including:
• the scope of rights granted under the license agreement and other interpretation-related issues;
• the extent to which our technology and processes infringe on intellectual property of the licensor that is not subject to the licensing agreement;
• the sublicensing of patent and other rights under our collaborative development relationships;
• our diligence obligations under the license agreement and what activities satisfy those diligence obligations;
• the inventorship and ownership of inventions and know-how resulting from the joint creation or use of intellectual property by our licensors and us and our partners; and
• the priority of invention of patented technology.
The agreements under which we may license intellectual property or technology from third parties may be complex, and certain provisions in such agreements may be susceptible to multiple interpretations. The resolution of any contract interpretation disagreement that may arise could narrow what we believe to be the scope of our rights to the relevant intellectual property or technology, or increase what we believe to be our financial or other obligations under the relevant agreement, either of which could have a material adverse effect on our business, financial condition, results of operations, and prospects. Moreover, if disputes over intellectual property that we have licensed prevent or impair our ability to maintain our licensing arrangements on commercially acceptable terms, we may be unable to successfully develop and commercialize the affected drug candidates, which could have a material adverse effect on our business, financial conditions, results of operations, and prospects.
These and similar issues may arise with respect to our collaboration agreements, such as the Bayer Agreement. Our collaboration with Bayer is one of our key collaborations, and there can be no assurance that this collaboration will continue past the current term, on favorable terms or at all, or that at any time while the collaboration is in effect the parties will operate under the agreement without disputes. Possible disputes may involve ownership or control of intellectual property rights, negotiations of licensing agreements resulting from the collaboration, exclusivity obligations, diligence and payment obligations, for example.
Some of our intellectual property has been discovered through government funded programs and thus may be subject to federal regulations such as “march-in” rights, certain reporting requirements and a preference for U.S.-based companies, and compliance with such regulations may limit our exclusive rights and our ability to contract with non-U.S. manufacturers.
Our intellectual property rights may be subject to a reservation of rights by one or more third parties. For example, certain intellectual property rights that we have licensed have been generated through the use of U.S. government funding and are therefore subject to certain federal regulations. As a result, the U.S. government may have certain rights to intellectual property embodied in our current or future processes and related products and services pursuant to the Bayh-Dole Act of 1980, or the Bayh-Dole Act. These U.S. government rights include a non-exclusive, non-transferable, irrevocable worldwide license to use inventions for any governmental purpose. In addition, the U.S. government has the right, under certain limited circumstances, to require the licensor to grant exclusive, partially exclusive or non-exclusive licenses to any of these inventions to a third party if it determines that (1) adequate steps have not been taken to commercialize the invention and achieve practical application of the government-funded technology, (2) government action is necessary to meet public health or safety needs, (3) government action is necessary to meet requirements for public use under federal regulations or (4) we fail to meet requirements of federal regulations (also referred to as “march-in rights”). The U.S. government also has the right to take title to these inventions if we or our licensors fail to disclose the invention to the government or fail to file an application to register the intellectual property within specified time limits. These rights may permit the government to disclose our confidential information to third parties. In addition, our rights in such inventions may be subject to certain requirements to manufacture products embodying such inventions in the United States. Intellectual property generated under a government funded program is also subject to certain reporting requirements, compliance with which may require us to expend substantial resources. To the extent any of our future owned or licensed intellectual property is also generated through the use of U.S. government funding, the provisions of the Bayh-Dole Act may
similarly apply. Any exercise by the government of such rights could have a material adverse effect on our competitive position, business, results of operations and financial condition.
Changes to the patent law in the United States and other jurisdictions could diminish the value of patents in general and may impact the validity, scope or enforceability of our patent rights, thereby impairing our ability to protect our drug candidates.
As is the case with other biopharmaceutical companies, our success is heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the biopharmaceutical industry involve both technological and legal complexity and is therefore costly, time consuming, and inherently uncertain. Our patent rights, their associated costs, and the enforcement or defense of such patent rights may be affected by developments or uncertainty in the patent statute, patent case law or USPTO rules and regulations. Changes in either the patent laws or interpretation of the patent laws could increase the uncertainties and costs surrounding the prosecution of patent applications and the enforcement or defense of our issued patents. For example, in March 2013, under the Leahy-Smith America Invents Act, or the America Invents Act, the United States transitioned from a “first to invent” to a “first-to-file” patent system. Under a “first-to-file” system, assuming that other requirements for patentability are met, the first inventor to file a patent application generally will be entitled to a patent on an invention regardless of whether another inventor had made the invention earlier. A third party that files a patent application in the USPTO after March 2013, but before us could therefore be awarded a patent covering an invention of ours even if we had made the invention before it was made by such third party. This will require us to be cognizant going forward of the time from invention to filing of a patent application. Since patent applications in the United States and most other countries are confidential for a period of time after filing or until issuance, we cannot be certain that we or our licensors were the first to either file any patent application related to our technology or drug candidates or invent any of the inventions claimed in our or our licensor’s patents or patent applications. The America Invents Act also includes a number of other significant changes to U.S. patent law, including provisions that affect the way patent applications will be prosecuted, allowing third party submission of prior art and establishing a new post-grant review system including post-grant review, inter partes review, and derivation proceedings. Because of a lower evidentiary standard in USPTO proceedings compared to the evidentiary standard in United States federal courts necessary to invalidate a patent claim, a third party could potentially provide evidence in a USPTO proceeding sufficient for the USPTO to hold a claim invalid even though the same evidence would be insufficient to invalidate the claim if first presented in a district court action. Accordingly, a third party may attempt to use the USPTO procedures to invalidate our patent claims that would not have been invalidated if first challenged by the third party as a defendant in a district court action. The effects of these changes are currently unclear as the USPTO continues to promulgate new regulations and procedures in connection with the America Invents Act and many of the substantive changes to patent law, including the “first-to-file” provisions, only became effective in March 2013. In addition, the courts have yet to address many of these provisions and the applicability of the act and new regulations on the specific patents discussed in this filing have not been determined and would need to be reviewed. However, the America Invents Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents.
The U.S. Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. Additionally, there have been recent proposals for additional changes to the patent laws of the United States and other countries that, if adopted, could impact our ability to obtain patent protection for our proprietary technology or our ability to enforce rights in our proprietary technology. Depending on future actions by the U.S. Congress, the U.S. courts, the USPTO and the relevant law-making bodies in other countries, the laws and regulations governing patents could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce any patents that we may obtain in the future.
Intellectual property rights do not necessarily address all potential threats.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations and may not adequately protect our business or permit us to maintain our competitive advantage. For example:
• others may be able to make products that are similar to our drug candidates or utilize similar technology but that are not covered by the claims of the patents that we license or may own;
• others may be able to duplicate or utilize similar technology in a manner that infringes our patents but is undetectable, or done in a jurisdiction where we cannot secure or enforce patent rights;
• we or our licensors or collaborators, might not have been the first to make the inventions covered by the issued patent or pending patent application that we license or own now or in the future;
• we or our licensors or collaborators, might not have been the first to file patent applications covering certain of our or their inventions;
• others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our owned or licensed intellectual property rights;
• it is possible that our present or future pending patent applications (whether owned or licensed) will not lead to issued patents;
• issued patents that we hold rights to may be held invalid or unenforceable, including as a result of legal challenges by our competitors or other third parties;
• our competitors or other third parties might conduct research and development activities in countries where we do not have patent rights and then use the information learned from such activities to develop competitive products for sale in our major commercial markets;
• we may not develop additional proprietary technologies that are patentable;
• the patents of others may harm our business; and
• we may choose not to file a patent in order to maintain certain trade secrets or know-how, and a third party may subsequently file a patent covering such intellectual property.
Should any of these events occur, they could have a material adverse effect on our business, financial condition, results of operations and prospects.
If our trademarks and trade names are not adequately protected, then we may not be able to build name recognition in our markets of interest and our business may be adversely affected.
Our registered or unregistered trademarks or trade names may be challenged, infringed, circumvented or declared generic or determined to be infringing on other marks. We may not be able to protect our rights to these trademarks and trade names, which we need to build name recognition among potential collaborators or customers in our markets of interest. At times, competitors may adopt trade names or trademarks similar to ours, thereby impeding our ability to build brand identity and possibly leading to market confusion. In addition, there could be potential trade name or trademark infringement claims brought by owners of other trademarks or trademarks that incorporate variations of our registered or unregistered trademarks or trade names. Over the long term, if we are unable to establish name recognition based on our trademarks and trade names, then we may not be able to compete effectively and our business may be adversely affected. We may license our trademarks and trade names to third parties, such as distributors. Though these license agreements may provide guidelines for how our trademarks and trade names may be used, a breach of these agreements or misuse of our trademarks and trade names by our licensees may jeopardize our rights in or diminish the goodwill associated with our trademarks and trade names. Our efforts to enforce or protect our proprietary rights related to trademarks, trade names, trade secrets, know-how, domain names, copyrights or other intellectual property may be ineffective and could result in substantial costs and diversion of resources and could adversely affect our business, financial condition, results of operations and prospects.
We license patent rights from third-party owners. If such owners do not properly or successfully obtain, maintain, or enforce the patents underlying such licenses, our competitive position and business prospects may be harmed.
We are a party to license agreements that give us rights to third-party intellectual property that is necessary or useful for our business. For example, we have obtained licenses from third parties to patent rights covering a number of our clinical drug candidates and licenses (implied or explicit) from certain other parties for technology used in our drug discovery efforts. We may enter into additional license agreements to third-party intellectual property in the future.
Our success will depend in part on the ability of our licensors to obtain, maintain, and enforce patent protection for our licensed products. Our licensors may not successfully prosecute the patent applications we license. Even if patents issue in respect of these patent applications, our licensors may fail to maintain these patents, may determine not to pursue litigation against other companies that are infringing these patents, or may pursue such litigation less aggressively than we would. Without protection for the intellectual property we license, other
companies might be able to offer substantially identical products for sale, which could adversely affect our competitive business position and harm our business prospects.
Our current proprietary position for certain drug candidates depends upon our owned or in-licensed patent filings covering components of such drug candidates, manufacturing-related methods, formulations and/or methods of use, which may not adequately prevent a competitor or other third party from using the same drug candidate for the same or a different use.
Composition of matter patent protection is generally considered to be desirable for drug products because it provides protection without regard to any particular method of use or manufacture or formulation. For some of the molecules that we in-license from our collaboration partners, we cannot rely on composition of matter patent protection as the term on those patents has expired or is approximately expired.
Method of use patents protect the use of a product for the specified method and formulation patents cover formulations to deliver therapeutics. While we file applications covering method of use for our programs at appropriate times in the development process, we cannot be certain that claims in any future patents issuing from these applications will cover all commercially-relevant applications of molecules in competing uses. These types of patents do not prevent a competitor or other third party from developing, marketing or commercializing a similar or identical product for an indication that is outside the scope of the patented method or from developing a different formulation that is outside the scope of the patented formulation. Moreover, with respect to method of use patents, even if competitors or other third parties do not actively promote their product for our targeted indications or uses for which we may obtain patents, physicians may recommend that patients use these products off-label, or patients may do so themselves. Although off-label use may infringe or contribute to the infringement of method of use patents, the practice is common and this type of infringement is difficult to prevent or enforce. Consequently, we may not be able to prevent third parties from practicing our inventions in the United States or abroad. Additionally, some commercially-relevant jurisdictions do not allow for patents covering a new method of use of an otherwise-known molecule.
Risks Related to Government Regulation
Even if we receive regulatory approval for any of our drug candidates, we will be subject to ongoing regulatory obligations and continued regulatory review, which may result in significant additional expense. Additionally, our drug candidates, if approved, could be subject to post-market study requirements, marketing and labeling restrictions, and even recall or market withdrawal if unanticipated safety issues are discovered following approval. In addition, we may be subject to penalties or other enforcement action if we fail to comply with regulatory requirements.
The FDA may not approve any of our drug candidates derived from our platform given our novel approach to drug discovery and may elect to inspect our automated robotics platform used to generate our data. However, if the FDA or a comparable foreign regulatory authority approves any of our drug candidates, the manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion and recordkeeping for the product will be subject to extensive and ongoing regulatory requirements. These requirements include submissions of safety and other post-marketing information and reports, establishment registration and listing, as well as continued compliance with cGMPs and GCPs for any clinical trials that we conduct post-approval. Any regulatory approvals that we receive for our drug candidates may also be subject to limitations on the approved indicated uses for which the product may be marketed or to the conditions of approval, or contain requirements for potentially costly post-marketing studies, and surveillance to monitor the safety and efficacy of the product. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with our third-party manufacturers or manufacturing processes, or failure to comply with regulatory requirements, may result in, among other things:
• restrictions on the marketing or manufacturing of the product, withdrawal of the product from the market, or voluntary or mandatory product recalls;
• clinical trial holds
• fines, warning letters or other regulatory enforcement action;
• refusal by the FDA to approve pending applications or supplements to approved applications filed by us;
• product seizure or detention, or refusal to permit the import or export of products; and
• injunctions or the imposition of civil or criminal penalties.
The FDA’s policies may change, and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our drug candidates. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained, which would adversely affect our business, prospects and ability to achieve or sustain profitability.
We may seek orphan drug designation for certain of our drug candidates, and we may be unsuccessful or may be unable to maintain the benefits associated with orphan drug designation, including the potential for market exclusivity.
As part of our business strategy, we may seek orphan drug designation for certain of our drug candidates, and we may be unsuccessful. Regulatory authorities in some jurisdictions, including the United States and Europe, may designate drugs for relatively small patient populations as orphan drugs. Under the Orphan Drug Act, the FDA may designate a drug as an orphan drug if it is a drug intended to treat a rare disease or condition, which is generally defined as a patient population of fewer than 200,000 individuals annually in the United States, or a patient population of 200,000 or more in the United States where there is no reasonable expectation that the cost of developing the drug will be recovered from sales in the United States. In the United States, orphan drug designation entitles a party to financial incentives such as opportunities for grant funding towards clinical trial costs, tax advantages and user-fee waivers.
Similarly, in Europe, the European Commission, upon the recommendation of the EMA’s Committee for Orphan Medicinal Products, grants orphan drug designation to promote the development of drugs that are intended for the diagnosis, prevention or treatment of life-threatening or chronically debilitating conditions affecting not more than 5 in 10,000 persons in Europe and for which no satisfactory method of diagnosis, prevention, or treatment has been authorized (or the product would be a significant benefit to those affected). Additionally, designation is granted for drugs intended for the diagnosis, prevention, or treatment of a life-threatening, seriously debilitating or serious and chronic condition and when, without incentives, it is unlikely that sales of the drug in Europe would be sufficient to justify the necessary investment in developing the drug. In Europe, orphan drug designation entitles a party to financial incentives such as reduction of fees or fee waivers.
Generally, if a drug with an orphan drug designation subsequently receives the first marketing approval for the indication for which it has such designation, the drug is entitled to a period of marketing exclusivity, which precludes the FDA or the EMA from approving another marketing application for the same drug and indication for that time period, except in limited circumstances. The applicable period is seven years in the United States and ten years in Europe. The European exclusivity period can be reduced to six years if a drug no longer meets the criteria for orphan drug designation or if the drug is sufficiently profitable so that market exclusivity is no longer justified.
Even if we obtain orphan drug exclusivity for a drug, that exclusivity may not effectively protect the drug from competition because different drugs can be approved for the same condition. Even after an orphan drug is approved, the FDA can subsequently approve a different drug for the same condition if the FDA concludes that the later drug is clinically superior in that it is shown to be safer, more effective or makes a major contribution to patient care. In addition, a designated orphan drug may not receive orphan drug exclusivity if it is approved for a use that is broader than the indication for which it received orphan designation. Moreover, orphan drug exclusive marketing rights in the United States may be lost if the FDA later determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantity of the drug to meet the needs of patients with the rare disease or condition or if another drug with the same active part of the molecule is determined to be safer, more effective, or represents a major contribution to patient care. Orphan drug designation neither shortens the development time or regulatory review time of a drug nor gives the drug any advantage in the regulatory review or approval process. While we may seek orphan drug designation for our drug candidates, we may never receive such designations. Even if we do receive such designations, there is no guarantee that we will enjoy the benefits of those designations.
Obtaining and maintaining regulatory approval of our drug candidates in one jurisdiction does not mean that we will be successful in obtaining regulatory approval of our drug candidates in other jurisdictions.
We may also submit marketing applications in other countries. Regulatory authorities in jurisdictions outside of the United States have requirements for approval of drug candidates with which we must comply prior to marketing in those jurisdictions. For example, our trials consist of small patient populations to date and some international
regulatory filings may require larger patient populations. Obtaining foreign regulatory approvals and compliance with foreign regulatory requirements could result in significant delays, difficulties and costs for us and could delay or prevent the introduction of our products in certain countries. If we fail to comply with the regulatory requirements in international markets and/or receive applicable marketing approvals, our target market will be reduced and our ability to realize the full market potential of our drug candidates will be harmed.
Obtaining and maintaining regulatory approval of our drug candidates in one jurisdiction does not guarantee that we will be able to obtain or maintain regulatory approval in any other jurisdiction, while a failure or delay in obtaining regulatory approval in one jurisdiction may have a negative effect on the regulatory approval process in others. For example, even if the FDA grants marketing approval of a drug candidate, comparable regulatory authorities in foreign jurisdictions must also approve the manufacturing, marketing and promotion of the drug candidate in those countries. Approval procedures vary among jurisdictions and can involve requirements and administrative review periods different from, and greater than, those in the United States, including additional nonclinical studies or clinical trials as clinical trials conducted in one jurisdiction may not be accepted by regulatory authorities in other jurisdictions. In short, the foreign regulatory approval process involves all of the risks associated with FDA approval. In many jurisdictions outside the United States, a drug candidate must be approved for reimbursement before it can be approved for sale in that jurisdiction. In some cases, the price that we may intend to charge for our products will also be subject to approval.
Laws and regulations governing any international operations we may have in the future may preclude us from developing, manufacturing and selling certain products outside of the United States and require us to develop and implement costly compliance programs.
If we expand our operations outside of the United States, we must dedicate additional resources to comply with numerous laws and regulations in each jurisdiction in which we plan to operate. The Foreign Corrupt Practices Act, or FCPA, prohibits any U.S. individual or business from paying, offering, authorizing payment or offering of anything of value, directly or indirectly, to any foreign official, political party or candidate for the purpose of influencing any act or decision of the foreign entity in order to assist the individual or business in obtaining or retaining business. The FCPA also obligates companies whose securities are listed in the United States to comply with certain accounting provisions requiring the company to maintain books and records that accurately and fairly reflect all transactions of the corporation, including international subsidiaries, and to devise and maintain an adequate system of internal accounting controls for international operations.
Compliance with the FCPA is expensive and difficult, particularly in countries in which corruption is a recognized problem. In addition, the FCPA presents particular challenges in the pharmaceutical industry, because, in many countries, hospitals are operated by the government, and doctors and other hospital employees are considered foreign officials. Certain payments to hospitals in connection with clinical trials and other work have been deemed to be improper payments to government officials and have led to FCPA enforcement actions.
Various laws, regulations and executive orders also restrict the use and dissemination outside of the United States, or the sharing with certain non-U.S. nationals, of information classified for national security purposes, as well as certain products and technical data relating to those products. If we expand our presence outside of the United States, it will require us to dedicate additional resources to comply with these laws, and these laws may preclude us from developing, manufacturing, or selling certain products and drug candidates outside of the United States, which could limit our growth potential and increase our development costs.
The failure to comply with laws governing international business practices may result in substantial civil and criminal penalties and suspension or debarment from government contracting. The Securities and Exchange Commission, or SEC, also may suspend or bar issuers from trading securities on U.S. exchanges for violations of the FCPA’s accounting provisions.
We are subject to certain U.S. and foreign anti-corruption, anti-money laundering, export control, sanctions, and other trade laws and regulations. We can face serious consequences for violations.
Among other matters, U.S. and foreign anti-corruption, anti-money laundering, export control, sanctions, and other trade laws and regulations, which are collectively referred to as Trade Laws, prohibit companies and their employees, agents, clinical research organizations, legal counsel, accountants, consultants, contractors, and other partners from authorizing, promising, offering, providing, soliciting, or receiving directly or indirectly, corrupt or
improper payments or anything else of value to or from recipients in the public or private sector. Violations of Trade Laws can result in substantial criminal fines and civil penalties, imprisonment, the loss of trade privileges, debarment, tax reassessments, breach of contract and fraud litigation, reputational harm, and other consequences. We have direct or indirect interactions with officials and employees of government agencies or government-affiliated hospitals, universities, and other organizations. We also expect our non-U.S. activities to increase in time. We plan to engage third parties for clinical trials and/or to obtain necessary permits, licenses, patent registrations, and other regulatory approvals and we can be held liable for the corrupt or other illegal activities of our personnel, agents, or partners, even if we do not explicitly authorize or have prior knowledge of such activities.
We may seek priority review designation for one or more of our other drug candidates, but we might not receive such designation, and even if we do, such designation may not lead to a faster regulatory review or approval process.
If the FDA determines that a drug candidate offers a treatment for a serious condition and, if approved, the product would provide a significant improvement in safety or effectiveness, the FDA may designate the drug candidate for priority review. A priority review designation means that the goal for the FDA to review an application is six months, rather than the standard review period of ten months. We may request priority review for our drug candidates. The FDA has broad discretion with respect to whether or not to grant priority review status to a drug candidate, so even if we believe a particular drug candidate is eligible for such designation or status, the FDA may decide not to grant it. Moreover, a priority review designation does not necessarily result in an expedited regulatory review or approval process or necessarily confer any advantage with respect to approval compared to conventional FDA procedures. Receiving priority review from the FDA does not guarantee approval within the six-month review cycle or at all.
Breakthrough therapy designation and fast track designation by the FDA, even if granted for any of our drug candidates, may not lead to a faster development, regulatory review or approval process, and each designation does not increase the likelihood that any of our drug candidates will receive marketing approval in the United States.
We may seek a breakthrough therapy designation for some of our drug candidates. A breakthrough therapy is defined as a drug that is intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition, and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. For drugs that have been designated as breakthrough therapies, interaction and communication between the FDA and the sponsor of the trial can help to identify the most efficient path for clinical development while minimizing the number of patients placed in ineffective control regimens. Drugs designated as breakthrough therapies by the FDA may also be eligible for priority review and accelerated approval. Designation as a breakthrough therapy is within the discretion of the FDA. Accordingly, even if we believe one of our drug candidates meets the criteria for designation as a breakthrough therapy, the FDA may disagree and instead determine not to make such designation. In any event, the receipt of a breakthrough therapy designation for a drug candidate may not result in a faster development process, review or approval compared to therapies considered for approval under conventional FDA procedures and does not assure ultimate approval by the FDA. In addition, even if one or more of our drug candidates qualify as breakthrough therapies, the FDA may later decide that such drug candidates no longer meet the conditions for qualification or decide that the time period for FDA review or approval will not be shortened.
We may seek fast track designation for some of our drug candidates. If a drug is intended for the treatment of a serious or life-threatening condition and the drug demonstrates the potential to address unmet medical needs for this condition, the drug sponsor may apply for fast track designation. The FDA has broad discretion whether or not to grant this designation, so even if we believe a particular drug candidate is eligible for this designation, we cannot assure you that the FDA would decide to grant it. Even if we do receive fast track designation, we may not experience a faster development process, review or approval compared to conventional FDA procedures. The FDA may withdraw fast track designation if it believes that the designation is no longer supported by data from our clinical development program. Fast track designation alone does not guarantee qualification for the FDA’s priority review procedures.
The FDA, the EMA and other regulatory authorities may implement additional regulations or restrictions on the development and commercialization of our drug candidates, and such changes can be difficult to predict.
The FDA, the EMA and regulatory authorities in other countries have each expressed interest in further regulating small molecule pharmaceuticals. Agencies at both the federal and state level in the United States, as well as the U.S. Congressional committees and other governments or governing agencies, have also expressed interest in further regulating the small molecule pharmaceutical industry. Such action may delay or prevent commercialization of some or all of our drug candidates. Adverse developments in clinical trials of products conducted by others may cause the FDA or other oversight bodies to change the requirements for approval of any of our drug candidates. These regulatory review agencies and committees and the new requirements or guidelines they promulgate may lengthen the regulatory review process, require us to perform additional studies or trials, increase our development costs, lead to changes in regulatory positions and interpretations, delay or prevent approval and commercialization of our drug candidates or lead to significant post-approval limitations or restrictions. As we advance our drug candidates, we will be required to consult with these regulatory agencies and comply with applicable requirements and guidelines. If we fail to do so, we may be required to delay or discontinue development of such drug candidates. These additional processes may result in a review and approval process that is longer than we otherwise would have expected. Delays as a result of an increased or lengthier regulatory approval process or further restrictions on the development of our drug candidates can be costly and could negatively impact our ability to complete clinical trials and commercialize our current and future drug candidates in a timely manner, if at all.
Healthcare legislative reform measures may have a material adverse effect on our business and results of operations.
The U.S. and many foreign jurisdictions have enacted or proposed legislative and regulatory changes affecting the healthcare system that could prevent or delay marketing approval of our current or future drug candidates or any future drug candidates, restrict or regulate post-approval activities and affect our ability to profitably sell a product for which we obtain marketing approval. Changes in regulations, statutes or the interpretation of existing regulations could impact our business in the future by requiring, for example: i) changes to our manufacturing arrangements, ii) additions or modifications to product labeling, iii) the recall or discontinuation of our products or iv) additional record-keeping requirements. If any such changes were to be imposed, they could adversely affect the operation of our business. In the U.S., there have been and continue to be a number of legislative initiatives to contain healthcare costs. For example, in March 2010, the Affordable Care Act, or the ACA, was passed, which substantially changed the way healthcare is financed by both governmental and private insurers, and significantly impacted the U.S. pharmaceutical industry. The ACA, among other things, subjects biological products to potential competition by lower-cost biosimilars, addresses a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected, increases the minimum Medicaid rebates owed by manufacturers under the Medicaid Drug Rebate Program and extends the rebate program to individuals enrolled in Medicaid managed care organizations, establishes annual fees and taxes on manufacturers of certain branded prescription drugs, and creates a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 70% (increased pursuant to the Bipartisan Budget Act of 2018, effective as of 2019) point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D. Since then, the ACA risk adjustment program payment parameters have been updated annually.
Members of the U.S. Congress have expressed intent to pass legislation or adopt executive orders to fundamentally change or repeal parts of the ACA. While Congress has not passed repeal legislation to date, the TCJA, repealed, effective January 1, 2019, the tax-based shared responsibility payment imposed by the ACA on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate.” On December 14, 2018, a federal district court in Texas ruled the individual mandate is a critical and inseverable feature of the ACA, and therefore, because it was repealed as part of the TCJA, the remaining provisions of the ACA are invalid as well, and on December 18, 2019, the Fifth Circuit U.S. Court of Appeals held that the individual mandate is unconstitutional, and remanded the case to the lower court to reconsider its earlier invalidation of the full ACA. The Supreme Court of the United States granted certiorari on March 2, 2020, and heard oral arguments on the case on November 10, 2020, and the case is expected to be decided sometime in 2021. Pending review, the ACA remains in effect, but it is unclear at this time what effect the latest ruling will have on the status of the ACA. Litigation and legislation over the ACA are likely to continue, with unpredictable and uncertain results. We will continue to evaluate the effect that the ACA and its possible repeal and replacement has on our business.
Further, on January 20, 2017, an Executive Order was signed directing federal agencies with authorities and responsibilities under the ACA to waive, defer, grant exemptions from, or delay the implementation of any provision of the ACA that would impose a fiscal burden on states or a cost, fee, tax, penalty or regulatory burden on individuals, healthcare providers, health insurers, or manufacturers of pharmaceuticals or medical devices. On October 13, 2017, an Executive Order was signed terminating the cost-sharing subsidies that reimburse insurers under the ACA. The Trump administration has concluded that cost-sharing reduction, or CSR, payments to insurance companies required under the ACA have not received necessary appropriations from Congress and announced that it will discontinue these payments immediately until those appropriations are made. The loss of the CSR payments is expected to increase premiums on certain policies issued by qualified health plans under the ACA. Several state Attorneys General filed suit to stop the administration from terminating the subsidies, but their request for a restraining order was denied by a federal judge in California on October 25, 2017. The loss of the cost share reduction payments is expected to increase premiums on certain policies issued by qualified health plans under the ACA. Further, on June 14, 2018, the U.S. Court of Appeals for the Federal Circuit ruled that the federal government was not required to pay to third-party payors more than $12 billion in ACA risk corridor payments that they argued were owed to them. The effects of this gap in reimbursement on third-party payors, the viability of the ACA marketplace, providers, and potentially our business, are not yet known.
Moreover, on January 22, 2018, a continuing resolution on appropriations for fiscal year 2018 was approved that delayed the implementation of certain ACA-mandated fees, including the so called “Cadillac” tax on certain high cost employer-sponsored insurance plans, the annual fee imposed on certain health insurance providers based on market share, and the medical device excise tax on non-exempt medical devices; however on December 20, 2019, the Further Consolidated Appropriations Act (H.R. 1865) was signed into law, which repeals the Cadillac tax, the health insurance provider tax, and the medical device excise tax. It is impossible to determine whether similar taxes could be instituted in the future. The Bipartisan Budget Act of 2018, also amended the ACA, effective January 1, 2019, by increasing the point-of-sale discount that is owed by pharmaceutical manufacturers who participate in Medicare Part D and closing the coverage gap in most Medicare drug plans, commonly referred to as the “donut hole.” CMS published a final rule permitting further collections and payments to and from certain ACA qualified health plans and health insurance issuers under the ACA risk adjustment program in response to the outcome of federal district court litigation regarding the method CMS uses to determine this risk adjustment. In addition, CMS has recently published a final rule that would give states greater flexibility, starting in 2020, in setting benchmarks for insurers in the individual and small group marketplaces, which may have the effect of relaxing the essential health benefits required under the ACA for plans sold through such marketplaces. Other legislative changes have been proposed and adopted in the U.S. since the ACA was enacted. In August 2011, the Budget Control Act of 2011, among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions of Medicare payments to providers up to 2% per fiscal year, and, due to subsequent legislative amendments, will remain in effect through 2029 unless additional Congressional action is taken. The American Taxpayer Relief Act of 2012 among other things, reduced Medicare payments to several providers, including hospitals, imaging centers and treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
There has been increasing legislative and enforcement interest in the U.S. with respect to specialty drug pricing practices. Specifically, there have been several recent U.S. Congressional inquiries and proposed federal and state legislation designed to, among other things, bring more transparency to drug pricing, reduce the cost of prescription drugs under Medicare, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drugs. At the federal level, the Trump administration’s budget for fiscal years 2019 and 2020 contain further drug price control measures that could be enacted during the budget process or in other future legislation, including, for example, measures to permit Medicare Part D plans to negotiate the price of certain drugs under Medicare Part B, to allow some states to negotiate drug prices under Medicaid, and to eliminate cost sharing for generic drugs for low income patients. Additionally, the Trump administration released a “Blueprint” to lower drug prices and reduce out of pocket costs of drugs that contains additional proposals to increase manufacturer competition, increase the negotiating power of certain federal healthcare programs, incentivize manufacturers to lower the list price of their products and reduce the out of pocket costs of drug candidates paid by consumers. The U.S. Department of Health and Human Services, or HHS, has already started the process of soliciting feedback on some of these measures and, at the same time, is immediately implementing others under its existing authority. For example, in May 2019, CMS issued a final rule to allow Medicare Advantage Plans the option of using step therapy, a type of prior authorization, for Part B drugs beginning
January 1, 2020. This final rule codified CMS’s policy change that was effective January 1, 2019. In addition, in September 2020, an Executive Order was issued directing the Secretary of Health and Human Services to pursue implementation of two new payment models under which Medicare would test whether paying no more than the “most-favored-nation” price for certain included drugs and biological products covered under Part B and Part D, respectively, would mitigate poor clinical outcomes and increased Medicare expenditures associated with high drug costs. If implemented, the “most-favored-nation” price would generally reflect the lowest price, after certain adjustments, for a pharmaceutical product sold in an economically comparable member country of the Organization for Economic Co-operation and Development. Congress has also continued to conduct inquiries into the prescription drug industry’s pricing practices. While several proposed reform measures will require Congress to pass legislation to become effective, Congress has indicated that it will continue to seek new legislative and/or regulatory measures to address prescription drug costs. At the state level, legislatures are increasingly passing legislation and states are implementing regulations designed to control spending on, and patient out-of-pocket costs for, drug products. Implementation of cost containment measures or other healthcare reforms that affect the pricing and/or availability of drug products may impact our ability to generate revenue, attain or maintain profitability, or commercialize products for which we may receive regulatory approval in the future.
Further, on May 30, 2018, the Right to Try Act, was signed into law. The law, among other things, provides a federal framework for certain patients to access certain investigational new drug candidates that have completed a Phase 1 clinical trial and that are undergoing investigation for FDA approval. Under certain circumstances, eligible patients can seek treatment without enrolling in clinical trials and without obtaining FDA permission under the FDA expanded access program. There is no obligation for a pharmaceutical manufacturer to make its drug candidates available to eligible patients as a result of the Right to Try Act.
At the state level, individual states are increasingly aggressive in passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. In addition, regional health care authorities and individual hospitals are increasingly using bidding procedures to determine what pharmaceutical products and which suppliers will be included in their prescription drug and other health care programs. These measures could reduce the ultimate demand for our products, once approved, or put pressure on our product pricing.
We expect that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for our current or future drug candidates or additional pricing pressures.
Our revenue prospects could be affected by changes in healthcare spending and policy in the U.S. and abroad.
We operate in a highly regulated industry and new laws, regulations or judicial decisions, or new interpretations of existing laws, regulations or decisions, related to healthcare availability, the method of delivery or payment for healthcare products and services could negatively impact our business, operations and financial condition.
There have been, and likely will continue to be, legislative and regulatory proposals at the foreign, federal, and state levels directed at broadening the availability of healthcare and containing or lowering the cost of healthcare. We cannot predict the initiatives that may be adopted in the future, including repeal, replacement, or significant revisions to the ACA. The continuing efforts of the government, insurance companies, managed care organizations and other payors of healthcare services to contain or reduce costs of healthcare and/or impose price controls may adversely affect:
• the demand for our current or future drug candidates, if we obtain regulatory approval;
• our ability to set a price that we believe is fair for our products;
• our ability to obtain coverage and reimbursement approval for a product;
• our ability to generate revenue and achieve or maintain profitability;
• the level of taxes that we are required to pay; and
• the availability of capital.
Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors, which may adversely affect our results of operations and future profitability.
Our relationships with healthcare providers, other customers, and third-party payors will be subject to applicable anti-kickback, fraud and abuse and other healthcare laws and regulations, which could expose us to criminal sanctions, civil penalties, exclusion from government healthcare programs, contractual damages, reputational harm and diminished profits and future earnings.
Although we do not currently have any products on the market, once we begin commercializing our drug candidates, we will be subject to additional healthcare statutory and regulatory requirements and enforcement by the federal government and the states and foreign governments in which we conduct our business. Healthcare providers, physicians and third-party payors play a primary role in the recommendation and prescription of any drug candidates for which we obtain marketing approval. Our future arrangements with third-party payors and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we market, sell and distribute our drug candidates for which we obtain marketing approval. Restrictions under applicable federal and state healthcare laws and regulations include the following:
• the federal Anti-Kickback Statute prohibits, among other things, persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made under federal and state healthcare programs such as Medicare and Medicaid. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation;
• the federal civil and criminal false claims and civil monetary penalties laws, including the federal False Claims Act, or FCA, imposes criminal and civil penalties, including through civil whistleblower or qui tam actions, against individuals or entities for knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government. In addition, the government may assert that a claim including items and services resulting from a violation of the federal Anti-Kickback Statute constitutes a false of fraudulent claim for purposes of the False Claims Act;
• the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, imposes criminal and civil liability for executing a scheme to defraud any healthcare benefit program, or knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statement in connection with the delivery of or payment for healthcare benefits, items or services; similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation;
• the federal physician payment transparency provisions, sometimes referred to as the “Sunshine Act” under the Affordable Care Act, require manufacturers of drugs, devices, biologics and medical supplies that are reimbursable under Medicare, Medicaid, or the Children’s Health Insurance Program to report to the Department of Health and Human Services information related to transfers of value made to licensed physicians (currently defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals, as well as ownership and investment interests of such physicians and their immediate family members. Effective January 1, 2022, these reporting obligations will extend to include transfers of value made to certain non-physician providers such as physician assistants and nurse practitioners;
• HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009 and its implementing regulations, imposes obligations on certain covered entity healthcare providers, health plans, and healthcare clearinghouses as well as their business associates that perform certain services involving the use or disclosure of individually identifiable health information, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information; and
• analogous state laws and regulations, such as state anti-kickback and false claims laws may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers. Some state laws require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government in addition to requiring drug manufacturers to report information related to payments to physicians and other health care providers or marketing expenditures. Further, many state laws governing the privacy and security of health information in certain circumstances, differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts.
Efforts to ensure that our future business arrangements with third parties comply with applicable healthcare laws and regulations could involve substantial costs and may require us to undertake or implement additional policies or measures. We may face claims and proceedings by private parties, and claims, investigations and other proceedings by governmental authorities, relating to allegations that our business practices do not comply with current or future statutes, regulations or case law involving applicable fraud and abuse, privacy or data protection, or other healthcare laws and regulations, and it is possible that courts or governmental authorities may conclude that we have not complied with them, or that we may find it necessary or appropriate to settle any such claims or other proceedings. In connection with any such claims, proceedings, or settlements, we may be subject to significant civil, criminal and administrative penalties, damages, fines, other damages, imprisonment, exclusion from government funded healthcare programs, such as Medicare and Medicaid, and the curtailment or restructuring of our operations. If any of the physicians or other providers or entities with whom we expect to do business is found not to be in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs.
Compliance with global privacy and data security requirements could result in additional costs and liabilities to us or inhibit our ability to collect and process data globally, and the failure to comply with such requirements could subject us to significant fines and penalties, which may have a material adverse effect on our business, financial condition, or results of operations.
The regulatory framework for the collection, use, safeguarding, sharing, transfer, and other processing of information worldwide is rapidly evolving and is likely to remain uncertain for the foreseeable future. Globally, virtually every jurisdiction in which we operate has established its own data security and privacy frameworks with which we must comply. For example, the collection, use, disclosure, transfer, or other processing of personal data regarding individuals in the European Union, including personal health data and employee data, is subject to the European Union General Data Protection Regulation, or the GDPR, which took effect across all member states of the European Economic Area, or EEA, in May 2018. The GDPR is wide-ranging in scope and imposes numerous requirements on companies that process personal data, including requirements relating to processing health and other sensitive data, obtaining consent of the individuals to whom the personal data relates, providing information to individuals regarding data processing activities, implementing safeguards to protect the security and confidentiality of personal data, providing notification of data breaches, and taking certain measures when engaging third-party processors. The GDPR informs our obligations with respect to any clinical trials conducted in the EEA by expanding the definition of personal data to include coded data and requiring changes to informed consent practices and more detailed notices for clinical trial subjects and investigators. In addition, the GDPR imposes strict rules on the transfer of personal data to countries outside the European Union, including the United States and, as a result, increases the scrutiny that such rules should apply to transfers of personal data from any clinical trial sites located in the EEA to the United States. The GDPR also permits data protection authorities to require destruction of improperly gathered or used personal information and/or impose substantial fines for violations of the GDPR, which can be up to four percent of global revenues or 20 million Euros, whichever is greater, and confers a private right of action on data subjects and consumer associations to lodge complaints with supervisory authorities, seek judicial remedies, and obtain compensation for damages resulting from violations of the GDPR. In addition, the GDPR provides that European Union member states may make their own further laws and regulations limiting the processing of personal data, including genetic, biometric, or health data.
Given the breadth and depth of its obligations, complying with the GDPR’s requirements is rigorous and time intensive and requires significant resources and assessment of our technologies, systems and practices, as well as those of any third-party collaborators, service providers, contractors, or consultants that process or transfer personal data collected in the European Union.
Further, the United Kingdom exited the EU effective January 31, 2020, subject to a transition period that ended December 31, 2020. Brexit and ongoing developments in the United Kingdom have created uncertainty with regard to the regulation of data protection in the United Kingdom and could result in the application of new data privacy and protection laws and standards to our operations in the United Kingdom and our handling of personal data of individuals located in the United Kingdom. The United Kingdom has implemented legislation that substantially implements the GDPR, and the European Commission and the United Kingdom government announced a EU-UK Trade and Cooperation Agreement on December 24, 2020, providing for a temporary free flow of personal data between the EU and the United Kingdom, but it remains to be seen how the United Kingdom’s withdrawal from the EU will impact the manner in which United Kingdom data protection laws or regulations will develop and how data
transfers to and from the United Kingdom will be regulated and enforced by the UK Information Commissioner’s Office, EU data protection authorities, or other regulatory bodies in the longer term.
In the United States, a broad variety of laws and regulations relating to privacy and data security may be applicable to our activities. New laws also are being considered at both the state and federal levels, and state legislatures such as California have already passed and enacted privacy legislation. For example, the California Consumer Privacy Act, or CCPA, which became effective on January 1, 2020, creates individual privacy rights for California consumers and increases the privacy and security obligations of entities handling certain personal data. The CCPA, among other things, requires covered companies to provide new disclosures to California consumers, and afford such consumers new abilities to opt out of certain sales of personal information, access and require deletion of their personal information, and receive detailed information about how their personal information is used. The CCPA has been amended on multiple occasions and additional regulations of the California Attorney General came into effect on August 14, 2020. However, aspects of the CCPA and its interpretation remain unclear. The effects of the CCPA are significant and may require us to modify our data processing practices and policies and to incur substantial costs and expenses in an effort to comply. Failure to comply with the CCPA may result in attorney general enforcement action and damage to our reputation. The CCPA provides for civil penalties for violations, as well as a private right of action for data breaches that is expected to increase data breach litigation. Moreover, a ballot initiative from privacy rights advocates intended to augment and expand the CCPA called the California Privacy Rights Act, or CPRA, was approved by California voters in the November 2020 election. The CPRA imposes additional obligations relating to consumer data on companies doing business in California beginning January 1, 2022, with implementing regulations expected on or before July 1, 2022, and enforcement beginning July 1, 2023. The CPRA significantly modifies the CCPA, including by expanding consumers’ rights with respect to certain sensitive personal information, potentially resulting in further uncertainty and requiring us to incur additional costs and expenses in an effort to comply, as we may need to modify or augment our existing practices. The CPRA also creates a new state agency that will be vested with authority to implement and enforce the CCPA and the CPRA. New legislation proposed or enacted in a number of states impose, or have the potential to impose additional obligations on companies that collect, store, use, retain, disclose, transfer and otherwise process confidential, sensitive and personal information, and will continue to shape the data privacy environment nationally. State laws are changing rapidly and there is discussion in Congress of a new federal data protection and privacy law to which we would become subject if it is enacted. In addition, all 50 states have laws including obligations to provide notification of security breaches of computer databases that contain personal information to affected individuals, state officers and others.
The myriad international and U.S. privacy and data breach laws are not consistent, and compliance in the event of a widespread data breach is difficult and may be costly. Moreover, states have been frequently amending existing laws, requiring attention to changing regulatory requirements. In addition to government regulation, privacy advocates and industry groups have and may in the future propose self-regulatory standards from time to time. These and other industry standards may legally or contractually apply to us, or we may elect to comply with such standards. We expect that there will continue to be new proposed laws and regulations concerning data privacy and security, and we cannot yet determine the impact such future laws, regulations and standards may have on our business. With the GDPR, CCPA, CPRA, and other laws, regulations and other obligations relating to privacy and data protection imposing new and relatively burdensome obligations, and with substantial uncertainty over the interpretation and application of these and other obligations, we may face challenges in addressing their requirements, putting in place additional compliance mechanisms and making necessary changes to our policies and practices, and may incur significant costs and expenses in an effort to do so.
We make public statements about our use and disclosure of personal information through our privacy policy, information provided on our website and press statements. Although we endeavor to comply with our public statements and documentation, we may at times fail to do so or be alleged to have failed to do so. We may be subject to potential government or legal action if such policies or statements are found to be deceptive, unfair or misrepresentative of our actual practices. In addition, from time to time, concerns may be expressed about whether our technology compromises the privacy of our customers and others. While we believe that we comply with industry standards and applicable laws and industry codes of conduct relating to privacy and data protection in all material respects, there is no assurance that we will not be subject to claims that we have violated applicable laws or codes of conduct, that we will be able to successfully defend against such claims or that we will not be subject to significant fines and penalties in the event of non-compliance. Additionally, to the extent multiple state-level laws are introduced with inconsistent or conflicting standards and there is no federal law to preempt such laws, compliance with such laws could be difficult to achieve and we could be subject to fines and penalties in the event of non-
compliance. Furthermore, enforcement actions and investigations by regulatory authorities related to data security incidents and privacy violations continue to increase.
In addition, if third parties we work with, such as vendors or service providers, violate applicable laws or regulations or our policies, such violations may also put our data at risk and could in turn have an adverse effect on our business. Any failure or perceived failure by us or our service providers to comply with our applicable policies or notices relating to privacy or data protection, our contractual or other obligations to third parties, or any of our other legal obligations relating to privacy or data protection, may result in governmental investigations or enforcement actions, litigation, claims and other proceedings, and could result in significant fines, penalties, and other liability. Additionally, defending against any claims, litigation, regulatory proceedings, or other proceedings can be costly, time-consuming and may require significant financial and personnel resources. Therefore, even if we are successful in defending against any such actions or proceedings that may be brought against us, our business may be impaired, and we may suffer reputational and other harm.
Our employees, independent contractors, consultants, and vendors may engage in misconduct or other improper activities, including non-compliance with regulatory standards and requirements and insider trading laws, which could cause significant liability for us and harm our reputation.
We are exposed to the risk of fraud or other misconduct by our employees, independent contractors, KOLs, CROs, consultants, and vendors. Misconduct by these parties could include intentional, reckless and/or negligent conduct that causes us to fail to comply with FDA regulations or similar regulations of comparable foreign regulatory authorities, provide accurate information to the FDA or comparable foreign regulatory authorities, comply with manufacturing standards, comply with federal and state healthcare fraud and abuse laws and regulations and similar laws and regulations established and enforced by comparable foreign regulatory authorities, report financial information or data accurately, or disclose unauthorized activities to us. Employee misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. This could include violations of HIPAA, other U.S. federal and state law, and requirements of non-U.S. jurisdictions, including the European Union Data Protection Directive. We are also exposed to risks in connection with any insider trading violations by employees or others affiliated with us, including inadvertent violations such as a sale of pledged shares by a lender when the pledgor is in possession of material nonpublic information. It is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws, standards, regulations, guidance, or codes of conduct. Additionally, we are subject to the risk that a person could allege such fraud or other misconduct, even if none occurred and our employees may, from time to time, bring lawsuits against us for employment issues, including injury, discrimination, wage and hour disputes, sexual harassment, hostile work environment, or other employment issues. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of civil, criminal and administrative penalties, damages, monetary fines, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, contractual damages, reputational harm, diminished profits and future earnings, and curtailment of our operations, any of which could adversely affect our ability to operate our business and our results of operations.
Risks Relating to Employee Matters and Managing Growth
Our future success depends on our ability to retain key executives and experienced scientists and to attract, retain and motivate qualified personnel.
We are highly dependent on the research and development, clinical and business development expertise of Christopher Gibson, our Chief Executive Officer, Tina Marriott Larson, our Chief Operating Officer and President, Michael Secora, our Chief Financial Officer, Shafique Virani, our Chief Corporate Development Officer, and Ramona Doyle, our Chief Medical Officer, as well as the other principal members of our management, scientific, technological and clinical team. Although we have entered into employment letter agreements with our executive officers, each of them may terminate their employment with us at any time or not be able to perform the services we need in the future. We do not maintain “key person” insurance for any of our executives or other employees. In addition, we rely on our employees to help operate and repair our robots and consultants and advisors, including scientific and clinical advisors, to assist us in formulating our research and development and commercialization strategy. Our consultants and advisors may be employed by employers other than us and may have commitments
under consulting or advisory contracts with other entities that may limit their availability to us. If we are unable to continue to attract and retain high quality personnel, our ability to pursue our growth strategy will be limited.
Recruiting and retaining qualified scientific, clinical, manufacturing and sales and marketing personnel will also be critical to our success. The loss of the services of our executive officers or other key employees, including temporary loss due to illness, could impede the achievement of our research, development and commercialization objectives and seriously harm our ability to successfully implement our business strategy. Furthermore, replacing executive officers and key employees may be difficult and may take an extended period of time because of the limited number of individuals in our industry with the breadth of skills and experience required to successfully develop, gain regulatory approval of, and commercialize products. Competition to hire from this limited pool is intense, and we may be unable to hire, train, retain or motivate these key personnel on acceptable terms given the competition among numerous pharmaceutical and biotechnology companies for similar personnel. We also experience competition for the hiring of scientific and clinical personnel from universities and research institutions. Failure to succeed in clinical trials may make it more challenging to recruit and retain qualified scientific personnel.
Our presence in Salt Lake City, where we are headquartered, may limit our ability to hire talent. Some of the employees we may want to hire in the future will reside in the greater San Francisco, New York, San Diego or Boston metro areas and may not want to relocate to Salt Lake City. Many of the other pharmaceutical companies that we compete against for qualified personnel have greater financial and other resources, different risk profiles and a longer history in the industry than we do. They also may provide more diverse opportunities and better chances for career advancement. Some of these characteristics may be more appealing to high-quality candidates than what we have to offer. If we are unable to continue to attract and retain high-quality personnel, the rate and success with which we can discover and develop drug candidates and our business will be limited.
We expect to expand our development and regulatory capabilities and potentially implement sales, marketing, and distribution capabilities, and as a result, we may encounter difficulties in managing our growth, which could disrupt our operations.
As of March 31, 2021, we had 218 full-time employees. We expect to experience significant growth in the number of our employees and the scope of our operations, particularly as we function as a public company and in the areas of product development, regulatory affairs and, if any of our drug candidates receives marketing approval, sales, marketing and distribution. To manage our anticipated future growth, we must continue to implement and improve our managerial, operational, and financial systems, expand our facilities and continue to recruit and train additional qualified personnel. Due to our limited financial resources and the limited experience of our management team in managing a company with such anticipated growth, we may not be able to effectively manage the expansion of our operations or recruit and train additional qualified personnel. The expansion of our operations may lead to significant costs and may divert our management and business development resources. Any inability to manage growth could delay the execution of our business plans or disrupt our operations.
We may acquire additional businesses or products, form strategic alliances, or create joint ventures with third parties that we believe will complement or augment our existing business. If we acquire businesses with promising markets or technologies, we may not be able to realize the benefit of acquiring such businesses if we are unable to successfully integrate them with our existing operations and company culture. We may encounter numerous difficulties in developing, manufacturing, and marketing any new products resulting from a strategic alliance or acquisition that delay or prevent us from realizing their expected benefits or enhancing our business. We cannot assure you that, following any such acquisition, we will achieve the expected synergies to justify the transaction.
Product liability lawsuits against us could cause us to incur substantial liabilities and could limit commercialization of any drug candidates that we may develop.
We face an inherent risk of product liability exposure related to the testing of drug candidates in human clinical trials and will face an even greater risk if we commercially sell any medicines that we may develop. If we cannot successfully defend ourselves against claims that our drug candidates or medicines caused injuries, we could incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
• decreased demand for any drug candidates or therapeutics that we may develop;
• injury to our reputation and significant negative media attention;
• withdrawal of clinical trial participants;
• significant costs to defend the related litigation;
• substantial monetary awards to trial participants or patients;
• loss of revenue; and
• the inability to commercialize our drug candidates.
Although we maintain product liability insurance, including coverage for clinical trials that we sponsor, it may not be adequate to cover all liabilities that we may incur. We anticipate that we will need to increase our insurance coverage as we commence additional clinical trials and if we successfully commercialize any drug candidates. The market for insurance coverage is increasingly expensive, and the costs of insurance coverage will increase as our clinical programs increase in size. We may not be able to maintain insurance coverage at a reasonable cost or in an amount adequate to satisfy any liability that may arise.
Risks Related to the Securities Markets and Ownership of Our Class A Common Stock
The dual-class structure of our common stock affects the concentration of voting power, which limits our Class A common stockholders’ ability to influence the outcome of matters submitted to our stockholders for approval, including the election of our board of directors, the adoption of amendments to our certificate of incorporation and bylaws, and the approval of any merger, consolidation, sale of all or substantially all of our assets, or other major corporate transaction.
Our Class A common stock offered in our initial public offering has one vote per share, and our Class B common stock has 10 votes per share. Based on 168,320,745 shares of our Class A common stock and 9,467,883 shares of our Class B common stock outstanding as of April 30, 2021. Dr. Gibson and his affiliate hold all of the issued and outstanding shares of our Class B common stock and approximately 37.3% of the voting power of our outstanding capital stock, which voting power may increase over time as Dr. Gibson exercises or vests in equity awards outstanding as of April 30, 2021. If all such equity awards held by Dr. Gibson had been exercised or vested and exchanged for shares of Class B common stock as of April 30, 2021, Dr. Gibson and his affiliate would hold 40.8% of the voting power of our outstanding capital stock. As a result, Dr. Gibson will be able to significantly influence any action requiring the approval of our stockholders, including the election of our board of directors, the adoption of amendments to our certificate of incorporation and bylaws, and the approval of any merger, consolidation, sale of all or substantially all of our assets, or other major corporate transaction. Dr. Gibson may have interests that differ from our Class A common stockholders and may vote in a way with which our Class A stockholders disagree with and which may be adverse to our Class A stockholders’ interests. The concentrated control may have the effect of delaying, preventing, or deterring a change in control of our company, could deprive our stockholders of an opportunity to receive a premium for their capital stock as part of a sale in our company, and, thus, may affect the market price of our Class A common stock.
Future transfers by the holders of Class B common stock will generally result in those shares automatically converting into shares of Class A common stock, subject to limited exceptions, such as certain transfers for estate planning. Transfers or exchanges of shares of Class B common stock may result in the issuance of additional shares of Class A common stock and such issuances will be dilutive to holders of our Class A common stock. In addition, each share of Class B common stock will convert automatically into one share of Class A common stock upon the earliest of (i) April 16, 2028, (ii) the date specified by written consent or agreement of the holders of 662⁄3% of our then outstanding shares of Class B common Stock, (iii) nine months after Dr. Gibson ceases to hold any positions as an officer or director of the Company, or (iv) nine months after the death or disability of Dr. Gibson. We refer to the date on which such final conversion of all outstanding shares of Class B common stock pursuant to the terms of amended and restated certificate of incorporation occurs as the Final Conversion Date.
Our principal stockholders and management own a significant percentage of our stock and will be able to exert significant control over matters subject to stockholder approval.
As of April 30, 2021, our executive officers, directors, holders of 5% or more of our capital stock and their respective affiliates beneficially owned approximately 61.6% of our voting power. These stockholders, acting together, may be able to impact matters requiring stockholder approval. For example, they may be able to impact elections of directors, amendments of our organizational documents or approval of any merger, sale of assets or other major corporate transaction. This may prevent or discourage unsolicited acquisition proposals or offers for our common stock that investors may feel are in their best interest as one of our stockholders. The interests of this group of stockholders may not always coincide with each investor’s interests or the interests of other stockholders and they may act in a manner that advances their best interests and not necessarily those of other stockholders, including
seeking a premium value for their common stock, and might affect the prevailing market price for our common stock.
We are an “emerging growth company” as defined in the JOBS Act and will be able to avail ourselves of reduced disclosure requirements applicable to emerging growth companies, which could make our Class A common stock less attractive to investors and adversely affect the market price of our Class A common stock.
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act. We will remain an emerging growth company until the earlier of i) the last day of the fiscal year in which we have total annual gross revenues of $1.07 billion or more; ii) the last day of the fiscal year following the fifth anniversary of the date of the completion of our initial public offering; iii) the date on which we have issued more than $1 billion in nonconvertible debt during the previous three years; or iv) the date on which we are deemed to be a large accelerated filer under the rules of the Securities and Exchange Commission, which means the market value of our Class A common stock that is held by non-affiliates exceeds $700 million as of the prior June 30th. For so long as we remain an emerging growth company, we are permitted and intend to rely on exemptions from certain disclosure requirements that are applicable to other public companies that are not emerging growth companies. These exemptions include:
• not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002, or Section 404;
• not being required to comply with any requirement that may be adopted by the Public Company Accounting Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements;
• providing only two years of audited financial statements in addition to any required unaudited interim financial statements and a correspondingly reduced “Management’s Discussion and Analysis of Financial Condition and Results of Operations” disclosure;
• reduced disclosure obligations regarding executive compensation; and
• exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. In this Quarterly Report, we have not included all of the executive compensation-related information that would be required if we were not an emerging growth company.
We may choose to take advantage of some, but not all, of the available exemptions. We have taken advantage of reduced reporting burdens in this Quarterly Report. In particular, we have provided only two years of audited financial statements and have not included all of the executive compensation information that would be required if we were not an emerging growth company. We cannot predict whether our Class A common stockholders will find it less attractive if we rely on these exemptions. If some of our Class A common stockholders find it less attractive, as a result, there may be a less active trading market for our Class A common stock and our stock price may be more volatile.
In addition, the JOBS Act provides that an emerging growth company can take advantage of an extended transition period for complying with new or revised accounting standards. This allows an emerging growth company to delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have elected to use the extended transition period for new or revised accounting standards during the period in which we remain an emerging growth company; however, we may adopt certain new or revised accounting standards early.
Although we are still evaluating the JOBS Act, we currently intend to take advantage of some, but not all, of the reduced regulatory and reporting requirements that will be available to us so long as we qualify as an “emerging growth company.” We have elected to avail ourselves of this exemption and, therefore, we are not subject to the same new or revised accounting standards as other public companies that are not emerging growth companies. As a result, changes in rules of U.S. generally accepted accounting principles or their interpretation, the adoption of new guidance or the application of existing guidance to changes in our business could significantly affect our financial position and results of operations. In addition, our independent registered public accounting firm will not be required to provide an attestation report on the effectiveness of our internal control over financial reporting so long as we qualify as an “emerging growth company,” which may increase the risk that material weaknesses or significant deficiencies in our internal control over financial reporting go undetected. Likewise, so long as we qualify as an “emerging growth company,” we may elect not to provide you with certain information, including certain
financial information and certain information regarding compensation of our executive officers, that we would otherwise have been required to provide in filings we make with the SEC, which may make it more difficult for investors and securities analysts to evaluate our company. We cannot predict if our Class A common stockholders will find it less attractive because we may rely on these exemptions. If our Class A common stockholders find it less attractive, as a result, there may be a less active trading market for our Class A common stock, and our stock price may be more volatile and may decline.
The price of our Class A common stock may be volatile and fluctuate substantially, which could result in substantial losses for holders of our Class A common stock.
Our stock price is likely to be volatile. The stock market in general and the market for biotechnology companies in particular have experienced extreme volatility that has often been unrelated to the operating performance of particular companies. As a result of this volatility, holders of our Class A common stock may not be able to sell their Class A common stock at or above the price they originally paid for it. The market price for our Class A common stock may be influenced by many factors, including:
• the success of competitive products or technologies;
• results of clinical trials of our drug candidates or those of our competitors;
• regulatory or legal developments in the United States and other countries;
• developments or disputes concerning patent applications, issued patents or other proprietary rights;
• the recruitment or departure of key personnel;
• the level of expenses related to any of our drug candidates or clinical development programs;
• the results of our efforts to discover, develop, acquire or in-license additional drug candidates or products;
• actual or anticipated changes in estimates as to financial results, development timelines or recommendations by securities analysts;
• variations in our financial results or those of companies that are perceived to be similar to us;
• changes in the structure of healthcare payment systems;
• market conditions in the pharmaceutical and biotechnology sectors;
• general economic, industry and market conditions; and
• the other factors described in this “Risk Factors” section.
Sales of a substantial number of shares of our Class A common stock in the public market could cause our stock price to fall.
The market price of our Class A common stock could decline at any time as a result of sales of a large number of shares of our Class A common stock in the market. These sales, or the perception that these sales could occur, may also depress the market price of our Class A common stock.
As of April 30, 2021, we had 158,852,862 shares of our Class A common stock and 9,467,883 shares of our Class B common stock outstanding. Of these shares, the 27,878,787 shares of Class A common stock were sold in our initial public offering and may be resold in the public market immediately, unless such shares are purchased by our affiliates. The remaining 130,974,075 shares of Class A common stock, or 82.5% of our outstanding shares of Class A common stock, and all shares of our Class B common stock (and any shares of Class A common stock into which they are converted) are currently prohibited or otherwise restricted from being sold in the public market under securities laws, market standoff agreements entered into by our stockholders with us, or lock-up agreements entered into by our stockholders with the underwriters.
The lock-up agreements pertaining to our initial public offering are set to expire on October 13, 2021. The representatives of the underwriters in our initial public offering, however, may, in their sole discretion, permit our officers, directors and other stockholders who are subject to these lock-up agreements to sell shares prior to the expiration of the lock-up agreements. After the lock-up agreements expire, based upon the number of shares of Class A common stock issued and issuable upon exchange of shares of Class B common stock, on an as-converted basis, outstanding as of April 30, 2021, up to an additional 140,441,958 shares of Class A common stock will be eligible for sale in the public market, 44.0% of which shares are held by directors, executive officers and other affiliates and will be subject to certain limitations of Rule 144 under the Securities Act of 1933, as amended, or the Securities Act. Approximately 926,250 shares of our Class B common stock that are beneficially owned by Christopher Gibson, Ph.D., our Chief Executive Officer and a member of our board of directors, are not subject to a lock-up agreement and have been pledged to secure his obligations under a line of credit with UBS Credit Corp., or
UBS. If he defaults on his repayment obligations under the line of credit, UBS or any designee of UBS may exercise its rights to sell shares pledged to cover the amount due thereunder. Any transfers or sales of such pledged shares may cause the price of our Class A common stock to decline.
As of April 30, 2021 42,531,759 shares of Class A common stock that are either subject to outstanding options and warrants or reserved for future issuance under our equity compensation plans will become eligible for sale in the public market to the extent permitted by the provisions of various vesting schedules, the lock-up agreements, and Rule 144 and Rule 701 under the Securities Act. If these additional shares of Class A common stock are sold, or if it is perceived that they will be sold, in the public market, the market price of our Class A common stock could decline.
As of March 31, 2021, the holders of approximately 135,870,793 shares of our Class A common stock issued and issuable upon conversion of Class B common stock will be entitled to rights with respect to the registration of their shares under the Securities Act, subject to the lock-up agreements described above. Registration of these shares under the Securities Act would result in the shares becoming freely tradable without restriction under the Securities Act, except for shares purchased by affiliates. Any sales of securities by these stockholders could have a material adverse effect on the market for our Class A common stock.
We have broad discretion in how we use the proceeds from our initial public offering and may not use these proceeds effectively, which could affect our results of operations and cause our stock price to decline.
We will have considerable discretion in the application of the net proceeds from our initial public offering. We intend to use the net proceeds from our initial public offering to fund our drug discovery platform, pursue strategic collaborations and advance our drug candidates through clinical development efforts. We also intend to use the proceeds from our initial public offering to expand our infrastructure and facilities to support our development efforts, to fund new and ongoing research activities and new drug candidates and for working capital and other general corporate purposes, which may include funding for the hiring of additional personnel, capital expenditures and the costs of operating as a public company. Investors will be relying upon management’s judgment with only limited information about our specific intentions for the use of the balance of the net proceeds from our initial public offering. We may use the net proceeds from our initial public offering for purposes that do not yield a significant return or any return at all for our stockholders. In addition, pending their use, we may invest the net proceeds from our initial public offering in a manner that does not produce income or that loses value.
Because we do not anticipate paying any cash dividends on our capital stock in the foreseeable future, capital appreciation, if any, will be your sole source of gain.
We have never declared or paid cash dividends on our capital stock. We currently intend to retain all of our future earnings, if any, to finance the growth and development of our business. In addition, the terms of any existing or future debt agreements may preclude us from paying dividends. As a result, capital appreciation, if any, of our Class A common stock will be the Class A common stockholders’ sole source of gain for the foreseeable future.
We have increased costs as a result of operating as a public company, and our management will be required to devote substantial time to new compliance initiatives.
As a public company, and particularly after we are no longer an “emerging growth company,” we will incur significant legal, accounting, and other expenses that we did not incur as a private company. In addition, the Sarbanes-Oxley Act of 2002 and rules subsequently implemented by the Securities and Exchange Commission and the Nasdaq Stock Market have imposed various requirements on public companies, including establishment and maintenance of effective disclosure and financial controls and corporate governance practices. Our management and other personnel will need to devote a substantial amount of time to these compliance initiatives. Moreover, these rules and regulations will increase our legal and financial compliance costs and will make some activities more time-consuming and costly. For example, we expect that these rules and regulations may make it more difficult and more expensive for us to obtain director and officer liability insurance.
Pursuant to Section 404, we will be required to furnish a report by our management on our internal control over financial reporting, including an attestation report on internal control over financial reporting issued by our independent registered public accounting firm. However, while we remain an emerging growth company, we will not be required to include an attestation report on internal control over financial reporting issued by our independent registered public accounting firm. To achieve compliance with Section 404 within the prescribed period, we will be
engaged in a process to document and evaluate our internal control over financial reporting, which is both costly and challenging. In this regard, we will need to continue to dedicate internal resources, potentially engage outside consultants and adopt a detailed work plan to assess and document the adequacy of internal control over financial reporting, continue steps to improve control processes as appropriate, validate through testing that controls are functioning as documented and implement a continuous reporting and improvement process for internal control over financial reporting. Despite our efforts, there is a risk that neither we nor our independent registered public accounting firm will be able to conclude within the prescribed timeframe that our internal control over financial reporting is effective as required by Section 404. This could result in an adverse reaction in the financial markets due to a loss of confidence in the reliability of our financial statements. In addition, if we are not able to continue to meet these requirements, we may not be able to remain listed on the Nasdaq Stock Market.
Provisions in our amended and restated certificate of incorporation and amended and restated bylaws and Delaware law might discourage, delay, or prevent a change in control of our company or changes in our management and, therefore, depress the market prices of our Class A common stock.
Our amended and restated certificate of incorporation and amended and restated bylaws contain provisions that could depress the market prices of our Class A common stock by acting to discourage, delay or prevent a change in control of our company or changes in our management that the stockholders of our company may deem advantageous. These provisions, among other things:
• establish a classified board of directors so that not all members of our board are elected at one time;
• permit only the board of directors to establish the number of directors and fill vacancies on the board;
• authorize the issuance of “blank check” preferred stock that our board could use to implement a stockholder rights plan (also known as a “poison pill”);
• eliminate the ability of our stockholders to call special meetings of stockholders;
• prohibit stockholder action by written consent, which requires all stockholder actions to be taken at a meeting of our stockholders;
• prohibit cumulative voting;
• authorize our board of directors to amend the bylaws;
• establish advance notice requirements for nominations for election to our board or for proposing matters that can be acted upon by stockholders at annual stockholder meetings; and
• require a super-majority vote of stockholders to amend some provisions described above.
In addition, Section 203 of the General Corporation Law of the State of Delaware, or DGCL, prohibits a publicly-held Delaware corporation from engaging in a business combination with an interested stockholder, generally a person which together with its affiliates owns, or within the last three years has owned, 15% of our voting stock, for a period of three years after the date of the transaction in which the person became an interested stockholder, unless the business combination is approved in a prescribed manner.
Any provision of our amended and restated certificate of incorporation, amended and restated bylaws or Delaware law that has the effect of delaying or preventing a change in control could limit the opportunity for our stockholders to receive a premium for their shares of our capital stock and could also affect the price that some investors are willing to pay for our Class A common stock.
Our amended and restated bylaws in effect provide that the Court of Chancery of the State of Delaware and the federal district courts of the United States of America will be the exclusive forums for substantially all disputes between us and our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers or employees.
Our amended and restated bylaws filed in connection with our initial public offering provide that the Court of Chancery of the State of Delaware or, if the Court of Chancery does not have jurisdiction, another State court in Delaware or the federal district court for the District of Delaware, is the exclusive forum for the following, except for any claim as to which such court determines that there is an indispensable party not subject to the jurisdiction of such court, and the indispensable party does not consent to the personal jurisdiction of such court within 10 days following such determination, which is vested in the exclusive jurisdiction of a court or forum other than such court or for which such court does not have subject matter jurisdiction:
• any derivative action or proceeding brought on our behalf;
• any action asserting a claim of breach of fiduciary duty;
• any action asserting a claim against us arising under the DGCL, our amended- and restated certificate of incorporation or our amended and restated bylaws; and
• any action asserting a claim against us that is governed by the internal-affairs doctrine.
This provision would not apply to suits brought to enforce a duty or liability created by the Exchange Act or any other claim for which the U.S. federal courts have exclusive jurisdiction. Our amended and restated bylaws further provide that the federal district courts of the United States of America will be the exclusive forum for resolving any complaint asserting a cause of action arising under the Securities Act.
These exclusive-forum provisions may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, officers or other employees, and may result in increased costs to stockholders of bringing a claim, each of which may discourage lawsuits against us and our directors, officers and other employees. Any person or entity purchasing or otherwise acquiring any interest in any of our securities shall be deemed to have notice of and consented to these provisions. There is uncertainty as to whether a court would enforce such provisions, and the enforceability of similar choice of forum provisions in other companies’ charter documents has been challenged in legal proceedings. It is possible that a court could find these types of provisions to be inapplicable or unenforceable, and if a court were to find either exclusive-forum provision in our amended and restated bylaws to be inapplicable or unenforceable in an action, we may incur additional costs associated with resolving the dispute in other jurisdictions, which could seriously harm our business.
We may be subject to securities litigation, which is expensive and could divert management attention.
The market price of our Class A common stock may be volatile. The stock market in general, and the Nasdaq Stock Market and biotechnology companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies. In the past, companies that have experienced volatility in the market price of their stock have been subject to securities class action litigation. We may be the target of this type of litigation in the future. Securities litigation against us could result in substantial costs and divert our management’s attention from other business concerns, which could seriously harm our business.
Our actual operating results may differ significantly from any guidance that we provide.
From time to time, we may provide guidance in our quarterly earnings conference calls, quarterly earnings releases, or otherwise, regarding our future performance that represents our management’s estimates as of the date of release. This guidance, which would include forward-looking statements, would be based on projections prepared by our management. Neither our registered public accountants nor any other independent expert or outside party would compile or examine the projections. Accordingly, no such person would express any opinion or any other form of assurance with respect to the projections.
Projections are based upon a number of assumptions and estimates that, while presented with numerical specificity, are inherently subject to significant business, economic, and competitive uncertainties and contingencies, many of which are beyond our control and are based upon specific assumptions with respect to future business decisions, some of which will change. The principal reason that we would release guidance is to provide a basis for our management to discuss our business outlook with analysts and investors. We do not accept any responsibility for any projections or reports published by any such third parties.
Guidance is necessarily speculative in nature, and it can be expected that some or all of the assumptions underlying any guidance furnished by us will not materialize or will vary significantly from actual results. Accordingly, our guidance would be only an estimate of what management believes is realizable as of the date of release. Actual results may vary from our guidance and the variations may be material.
As a a public company, we are obligated to develop and maintain proper and effective internal controls over financial reporting. Any failure to maintain the adequacy of these internal controls may adversely affect investor confidence in our company and, as a result, the value of our Class A common stock.
Our chief financial officer has only been the chief financial officer of a publicly traded company since our initial public offering and our chief executive officer has only been the chief executive officer of a publicly traded company since our initial public offering. Neither has been involved in the long term operations of a public company. Pursuant to
Section 404 of the Sarbanes-Oxley Act, we will be required to furnish a report by our management on our internal control over financial reporting beginning with our second filing of an Annual Report on Form 10-K with the SEC. This assessment will need to include disclosure of any material weaknesses identified by our management in our internal control over financial reporting. However, our independent registered public accounting firm will not be required to attest to the effectiveness of our internal control over financial reporting until our first annual report required to be filed with the SEC following the date we are no longer an emerging growth company. At such time as we are required to obtain auditor attestation, if we then have a material weakness, we would receive an adverse opinion regarding our internal control over financial reporting from our independent registered accounting firm.
To achieve compliance with Section 404 within the prescribed period, we will be engaged in a process to document and evaluate our internal control over financial reporting, which is both costly and challenging. In this regard, we will need to continue to dedicate internal resources, including through hiring additional financial and accounting personnel, potentially engage outside consultants and adopt a detailed work plan to assess and document the adequacy of internal control over financial reporting, continue steps to improve control processes as appropriate, validate through testing that controls are functioning as documented, and implement a continuous reporting and improvement process for internal control over financial reporting. During our evaluation of our internal control, if we identify one or more material weaknesses in our internal control over financial reporting, we will be unable to assert that our internal control over financial reporting is effective. We cannot assure you that there will not be material weaknesses or significant deficiencies in our internal control over financial reporting in the future. Any failure to maintain internal control over financial reporting could severely inhibit our ability to accurately report our financial condition, or results of operations. If we are unable to conclude that our internal control over financial reporting is effective, or if our independent registered public accounting firm determines we have a material weakness or significant deficiency in our internal control over financial reporting, we could lose investor confidence in the accuracy and completeness of our financial reports, the market price of shares of our Class A common stock could decline, and we could be subject to sanctions or investigations by the Nasdaq Stock Market, the SEC, or other regulatory authorities. Failure to remedy any material weakness in our internal control over financial reporting, or to implement or maintain other effective control systems required of public companies, could also restrict our future access to the capital markets.
General Risks
Unfavorable global economic conditions could adversely affect our business, financial condition or results of operations.
Our results of operations could be adversely affected by general conditions in the global economy and in the global financial markets. For example, in 2008, the global financial crisis caused extreme volatility and disruptions in the capital and credit markets and the current COVID-19 pandemic has caused significant volatility and uncertainty in U.S. and international markets. A severe or prolonged economic downturn could result in a variety of risks to our business, including weakened demand for our drug candidates and impaired ability to raise additional capital when needed on acceptable terms, if at all. A weak or declining economy could also strain our suppliers or possibly result in supply disruption. Any of the foregoing could harm our business and we cannot anticipate all of the ways in which the current economic climate and financial market conditions could adversely impact our business.
Current and future litigation against us, which may arise in the ordinary course of our business, could be costly and time consuming to defend.
We are periodically subject to claims that arise in the ordinary course of business, such as claims brought by our collaborators or suppliers in connection with commercial disputes, employment claims made by our current or former employees, or claims brought by third parties for failure to adequately protect their personal data. Third parties may in the future assert intellectual property rights to technologies that are important to our business and demand back royalties or demand that we license their technology. Litigation may result in substantial costs and may divert management’s attention and resources, which may seriously harm our business, overall financial condition and operating results. Insurance may not cover such claims, may not be sufficient for one or more of such claims and may not continue to be available on terms acceptable to us. A claim brought against us that is uninsured or underinsured could result in unanticipated costs and management distraction, negatively affecting our business, financial condition and results of operations.
If securities analysts do not publish research or reports about our business or if they publish negative evaluations of our stock, the price of our stock could decline.
The trading market for our Class A common stock relies, in part, on the research and reports that industry or financial analysts publish about us or our business. If only a small number of analysts maintain coverage of us, the trading price of our stock would likely decrease. If an analyst covering our stock downgrade their evaluations of our stock, the price of our stock could decline. If one or more of these analysts cease to cover our stock, we could lose visibility in the market for our stock, which in turn could cause our stock price to decline.
Item 2. Unregistered Sales of Equity Securities and Use of Proceeds.
(a) Sales of Unregistered Securities
Stock Option Exercises For the three months ended March 31, 2021,2024, we issued 1,722,02738 thousand shares of our Class A common stock to our employees, directors, advisors and consultants upon the exercise of stock options outstanding under our 2016 Equity Incentive and Key Personnel Incentive Stock PlansPlan for aggregate consideration of $2.2 million.approximately $10 thousand. The shares of Class A common stock issued upon the exercise of stock options were issued pursuant to written compensatory plans or arrangements with our employees, directors, advisors and consultants, in reliance on the exemption provided by Rule 701 promulgated under the Securities Act of 1933, as amended, or pursuant to Section 4(a)(2) underof the Securities Act of 1933, as amended, relative to transactions by an issuer not involving any public offering, to the extent an exemption from such registration was required. All recipients either received adequate information about our company or had access, through employment or other relationships, to such information.
Stock Option Grants
ForItem 5. Other Information.
On March 1, 2024, Terry-Ann Burrell, a member of our Board of Directors, adopted a Rule 10b5-1 trading arrangement that is intended to satisfy the three months ended March 31, 2021, we issuedaffirmative defense of Rule 10b5-1(c) for the sale of up to employees, directors, advisors and consultants, options to purchase an aggregate of 1,849,311211,290 shares of ourthe Company’s Class A common stock at a weighted-average exercise price of $4.44 per share in reliance on the exemption provided by Rule 701 promulgated under the Securities Act, or pursuant to Section 4(a)(2) under the Securities Act, relative to transactions by an issuer not involving any public offering, to the extent an exemption from such registration was required.
(b) Use of Proceeds from Public Offering of Class A Common Stockuntil May 30, 2025.
On April 15, 2021,March 1, 2024, Tina Marriott, President and Chief Operating Officer, adopted a Rule 10b5-1 trading arrangement that is intended to satisfy the Registration Statements on Form S-1 (File No. 333-254576)affirmative defense of Rule 10b5-1(c) for our initial public offeringthe sale of ourup to 672,000 shares of the Company’s Class A common stock was declared effective byuntil June 27, 2025.
On March 1, 2024, Michael Secora, Chief Financial Officer, adopted a Rule 10b5-1 trading arrangement that is intended to satisfy the SEC. Sharesaffirmative defense of ourRule 10b5-1(c) for the sale of up to 1,259,955 shares of the Company’s Class A common stock began trading on the Nasdaq Global Market on April 16, 2021. The offering closed on April 20, 2021.
The underwriters of our initial public offering were Goldman Sachs & Co. LLC, J.P. Morgan, BofA Securities, SVB Leerink, Allen & Company LLC and KeyBanc Capital Markets.
We paid to the underwriters of our initial public offering an underwriting discount totaling approximately $35.1 million. In addition, we incurred expenses of approximately $4.1 million which, when added to the underwriting discount, amount to total expenses of approximately $39.2 million. Thus, the net offering proceeds, after deducting underwriting discounts and offering expenses, were approximately $462.6 million. No offering expenses were paid directly or indirectly to any of our directors or officers (or their associates) or persons owning 10.0% or more of any class of our equity securities or to any other affiliates.
We are holding a significant portion of the balance of the net proceeds in bank deposits held in checking accounts. There has been no material change in the planned use of proceeds from our IPO from those that were described in the final prospectus filed pursuant to Rule 424(b) under the Securities Act and other periodic reports previously filed with the SEC.
(c) Issuer Purchases of Equity Securities
None.
Item 6. Exhibits.
Exhibit Index: | | | Incorporated by Reference | | | | Incorporated by Reference | | Exhibit number | Exhibit number | Description | Form | File No. | Exhibit No. | Filing Date | Filed / Furnished Herewith | Exhibit number | Description | Form | File No. | Exhibit No. | Filing Date | Filed / Furnished Herewith | | 3.1 | 3.1 | | 8-K | 001-40323 | 3.1 | April 21, 2021 | | | 3.2 | 3.2 | | 8-K | 001-40323 | 3.2 | April 21, 2021 | | | 3.2 | | | 3.2 | | | 4.1 | | | 4.1 | | | 4.1 | 4.1 | | S-1/A | 333-254576 | 4.1 | April 15, 2021 | | | 4.2 | 4.2 | | S-1/A | 333-254576 | 4.2 | April 15, 2021 | | | 10.1+ | | | X | | 10.2+ | | | X | | 4.2 | | | 4.2 | | 10.1+ | | 10.1+ | | 10.1+ | | | | | | | X | 10.2+ | | 10.2+ | | | | | | X | | 31.1 | 31.1 | | | X | 31.1 | | | | | | X | | 31.2 | 31.2 | | | X | 31.2 | | | | | | X | | 32.1* | 32.1* | | | X | 32.1* | | | | | | X | | 101.INS | 101.INS | XBRL Instance Document | | X | 101.INS | XBRL Instance Document | | | | | X | | 101.SCH | 101.SCH | XBRL Taxonomy Extension Schema Document | | X | 101.SCH | XBRL Taxonomy Extension Schema Document | | | | | X | | 101.CAL | 101.CAL | XBRL Taxonomy Extension Calculation Linkbase Document | | X | 101.CAL | XBRL Taxonomy Extension Calculation Linkbase Document | | | | | X | | 101.DEF | 101.DEF | XBRL Taxonomy Extension Definition Linkbase Document | | X | 101.DEF | XBRL Taxonomy Extension Definition Linkbase Document | | | | | X | | 101.LAB | 101.LAB | XBRL Taxonomy Extension Label Linkbase Document | | X | 101.LAB | XBRL Taxonomy Extension Label Linkbase Document | | | | | X | | 101.PRE | 101.PRE | XBRL Taxonomy Extension Presentation Linkbase Document | | X | 101.PRE | XBRL Taxonomy Extension Presentation Linkbase Document | | | | | X | | 104 | 104 | Cover Page Interactive Data File (formatted as Inline XBRL and contained in Exhibit 101) | | X | 104 | Cover Page Interactive Data File (formatted as Inline XBRL and contained in Exhibit 101) | | | | | X | | | + | | | + | | | + | | Indicates a management contract or compensatory plan. | | * | * | The certifications furnished in Exhibit 32.1 hereto are deemed to accompany this Quarterly Report on Form 10-Q and will not be deemed “filed” for purposes of Section 18 of the Securities Exchange Act of 1934, as amended. Such certifications will not be deemed to be incorporated by reference into any filings under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended, except to the extent that the Registrant specifically incorporates it by reference. | * | The certifications furnished in Exhibit 32.1 hereto are deemed to accompany this Quarterly Report on Form 10-Q and will not be deemed “filed” for purposes of Section 18 of the Securities Exchange Act of 1934, as amended. Such certifications will not be deemed to be incorporated by reference into any filings under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended, except to the extent that the Registrant specifically incorporates it by reference. | | + | Indicated management contract or compensatory plan. | |
SIGNATURES
Pursuant to the requirements of the Securities Act of 1934, the registrant has duly caused this registration statementreport to be signed on its behalf by the undersigned thereunto duly authorized on May 12, 2021.9, 2024.
| | | | | | | | | | RECURSION PHARMACEUTICALS, INC. | | | | By: | | | By: | /s/ Christopher Gibson | | | Christopher Gibson | | | Chief Executive Officer | | | (Principal Executive Officer) | | | | | By: | | /s/ Michael Secora | | | Michael Secora | | | Chief Financial Officer | | | (Principal Financial and Accounting Officer) | | | |
|