QuickLinksUNITED STATES
-- Click here to rapidly navigate through this documentSECURITIES AND EXCHANGE COMMISSION
FORM 20-F
Registration Statement Pursuant to Section 12(b) or 12(g) of The Securities Exchange Act of 1934 |
OR
Annual Report Pursuant to Section 13 or 15(d) of The Securities Exchange Act of 1934 for the fiscal year ended December 31, |
OR
Transition Report Pursuant to Section 13 or 15(d) of The Securities Exchange Act of 1934 |
OR
Shell Company Report Pursuant to Section 13 or 15(d) of The Securities Exchange Act of 1934 |
Commission file number 0-30752
AETERNA ZENTARIS INC.
(Exact Name of Registrant as Specified in its Charter)
Not Applicable
(Translation of Registrant’s Name into English)
Canada
(Jurisdiction of Incorporation)
1405 du Parc-Technologique Blvd.
Quebec City, Quebec
Canada, G1P 4P5
(Address of Principal Executive Offices)
Dennis Turpin
Telephone: 418-652-8525
E-mail: dturpin@aezsinc.com
1405 du Parc-Technologique Blvd.
Quebec City, Quebec
Canada, G1P 4P5
(Name, Telephone, E-mail and Address of Company Contact Person)
Securities registered or to be registered pursuant to Section 12(b) of the Act:
Title of Each Class | Name of Each Exchange on Which Registered | |||
Common Shares | Nasdaq Global Market | |||
| Toronto Stock Exchange |
Securities registered or to be registered pursuant to Section 12(g) of the Act:NONE
Securities for which there is a reporting obligation pursuant to Section 15(d) of the ACT:NONE
Indicate the number of outstanding shares of each of the issuer'sissuer’s classes of capital or common stock as at the close of the period covered by the annual report: 83,429,914104,762,096 common shares as at December 31, 2010.2011.
Indicate by check mark whether the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Yes o¨ No ýx
If this report is an annual or transition report, indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934.
Yes o¨ No ýx
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Yes ýx No o¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files).
Yes o¨ No ýx
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer or, or a non-accelerated filer. See definitions of "accelerated filer"“accelerated filer” and "large“large accelerated filer"filer” in Rule 12b-2 of the Exchange Act. (Check one):
Large accelerated filer | Accelerated filer | Non-accelerated filer |
Indicate by check mark which basis of accounting the registrant has used to prepare the financial statements included in this filing:
US GAAP | International Financial Reporting Standards as issued by the International Accounting Standards Board | Other |
If "other"“other” has been checked in response to the previous question, indicate by check mark which financial statement item the registrant has elected to follow.
Item 17 o¨ Item 18 ý¨
If this is an annual report, indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act).
Yes o¨ No ýx
Basis of Presentation
General
Except where the context otherwise requires, all references in this annual report on Form 20-F ("(“Form 20-F"20-F”) to the "Company"“Company”, "Aeterna“Aeterna Zentaris Inc."”, "we"“we”, "us"“us”, "our"“our” or similar words or phrases are to Aeterna Zentaris Inc. and its subsidiaries, taken together. In this annual report, references to "$"“$” and "US$"“US$” are to United States dollars, references to “CAN$” are to Canadian dollars and references to "CAN$"“EUR” are to Canadian dollars.euros. Unless otherwise indicated, the statistical and financial data contained in this annual report are presented as at December 31, 2010.2011.
This annual report on Form 20-F also contains certain information regarding products or product candidates that may potentially compete with our products and product candidates, and such information has been primarily derived from information made publicly available by the companies developing such potentially competing products and product candidates and has not been independently verified by Aeterna Zentaris Inc.
Forward-Looking Statements
This annual report contains forward-looking statements made pursuant to the safe harbor provisions of the U.S. Securities Litigation Reform Act of 1995. Forward-looking statements involve known and unknown risks and uncertainties, which could cause the Company'sCompany’s actual results to differ materially from those in the forward-looking statements. Such risks and uncertainties include, among others, the availability of funds and resources to pursue our research and development (“R&D&D”) projects, the successful and timely completion of clinical studies, the ability of the Company to take advantage of business opportunities in the pharmaceutical industry, uncertainties related to the regulatory process and general changes in economic conditions. Investors should consult the Company'sCompany’s quarterly and annual filings with the Canadian and U.S. securities commissions for additional information on risks and uncertainties relating to the forward-looking statements. Investors are cautioned not to rely on these forward-looking statements. The Company does not undertake to update these forward-looking statements and we disclaim any obligation to update any such factors or to publicly announce the result of any revisions to any of the forward-lookingforwards-looking statements contained herein to reflect future results, events or developments except if we are required to do so by a governmental authority or applicable law.
Page
| 1 | ||||||||||
Item 1. | 1 | |||||||||
1 | ||||||||||
B. Advisors | 1 | |||||||||
C. Auditors | 1 | |||||||||
Item 2. | 1 | |||||||||
| 1 | ||||||||||
| 1 | ||||||||||
Item 3. | 1 | |||||||||
| 1 | |||||||||
C. Reasons for the offer and | ||||||||||
| ||||||||||
| 5 | ||||||||||
Item 4. | ||||||||||
| Information on the Company | 21 | ||||||||
A. | History and development of the Company | 21 | ||||||||
B. | Business overview | |||||||||
| 61 | ||||||||||
| 61 | ||||||||||
Item 4A. | ||||||||||
Item 5. | ||||||||||
| ||||||||||
| Operating and Financial Review and Prospects | |||||||||
Item 6. | Directors, Senior Management and Employees | |||||||||
| 93 | ||||||||||
B. Compensation | 96 | |||||||||
| 113 | ||||||||||
D. Employees | 114 | |||||||||
| 114 | ||||||||||
Item 7. | ||||||||||
| Major Shareholders and Related Party Transactions | |||||||||
A. | Major shareholders | |||||||||
B. | Related party transactions | |||||||||
C. | Interests of experts and counsel | |||||||||
Item 8. | Financial Information | |||||||||
A. | Consolidated statements and other financial information | |||||||||
| 115 | ||||||||||
Item 9. | ||||||||||
| ||||||||||
| 116 | ||||||||||
C. Markets | 116 | |||||||||
| 116 | ||||||||||
E. Dilution | 117 | |||||||||
| 117 | ||||||||||
Item 10. | ||||||||||
| 117 | ||||||||||
| 117 | ||||||||||
| 127 | |||||||||
E. Taxation | 130 | ||||||||
| 136 | |||||||||
| 136 | |||||||||
| 136 | |||||||||
| 137 | |||||||||
Item 11. | |||||||||
Item 12. | |||||||||
| |||||||||
| 139 | |||||||||
| |||||||||
| |||||||||
| A. | Debt securities | ||||||||
| |||||||||
|
| ||||||||
| 139 | ||||||||
| 139 | |||||||||
| 140 | |||||||||
Item 13. | Defaults, Dividend Arrearages and Delinquencies | 140 | |||||||
Item 14. | Material Modification to the Rights of Security Holders and Use of Proceeds | ||||||||
Item 15. | Controls and Procedures | ||||||||
Item 16A. | Audit Committee Financial Expert | ||||||||
Item 16B. | Code of Ethics | ||||||||
Item 16C. | Principal Accountant Fees and Services | ||||||||
A. | Audit Fees | ||||||||
B. | Audit-related Fees | ||||||||
C. | Tax Fees | ||||||||
D. | All Other Fees | ||||||||
E. | Audit Committee Pre-Approval Policies and Procedures | ||||||||
Item 16D. | Exemptions from the Listing Standards for Audit Committees | ||||||||
Item 16E. | Purchases of Equity Securities by the Issuer and Affiliated Purchasers | ||||||||
Item 16F. | Changes in | ||||||||
Item 16G. | Corporate Governance | ||||||||
Item 16H. | Mine Safety Disclosure | 143 | |||||||
Item 17. | |||||||||
Item 18. | Financial Statements | ||||||||
Item 19. | Exhibits | ||||||||
Item 1. Identity of Directors, Senior Management and Advisers
A. Directors and senior management
Not applicable.
Not applicable.
Not applicable.
Item 2. Offer Statistics and Expected Timetable
Not applicable.
B. Method and expected timetable
Not applicable.
The consolidated statement of comprehensive loss data set forth in this Item 3.A with respect to the years ended December 31, 2011 and 2010, and the consolidated statement of financial position data as at December 31, 2011 and 2010, have been derived from the audited consolidated financial statements listed in Item 18, which have been prepared in accordance with International Financial Reporting Standards (“IFRS”), as issued by the International Accounting Standards Board (“IASB”). The consolidated statement of operations data set forth in this Item 3.A with respect to the years ended December 31, 2010, 2009, 2008 and 2008,2007, and the consolidated balance sheet data as at December 31, 20102009, 2008 and 2009,2007, have been derived from the audited consolidated financial statements listed in Item 18, which have been prepared in accordance with Canadian generally accepted accounting principles ("Canadian GAAP"), except as otherwise described therein. The consolidated statement of operations data set forth in this Item 3.A with respect to the years ended December 31, 2007 and 2006, and the consolidated balance sheet data as at December 31, 2008, 2007 and 2006, have been derived from otherour previous consolidated financial statements not included herein, and have beenwhich were prepared in accordance with Canadian GAAP, except as otherwise described therein. The selected financial data should be read in conjunction with our audited consolidated financial statements and the related notes included elsewhere in this annual report, and "Item“Item 5. — Operating and Financial Review and Prospects"Prospects” of this annual report.
Consolidated Statements of Operations Data
Comprehensive Loss
(in thousands of US dollars, except share and per share data)
Derived from financial statements prepared in accordance with IFRS
| Years ended December 31, | ||||||||
| 2011 | 2010 | |||||||
| $ | $ | |||||||
Revenues | ||||||||
Sales and royalties | 31,306 | 24,857 | ||||||
License fees and other | 4,747 | 2,846 | ||||||
| 36,053 | 27,703 | |||||||
Operating expenses | ||||||||
Cost of sales | 27,560 | 18,700 | ||||||
Research and development costs, net of refundable tax credits and grants | 24,517 | 21,257 | ||||||
Selling, general and administrative expenses | 16,170 | 12,552 | ||||||
| 68,247 | 52,509 | |||||||
Loss from operations | (32,194) | (24,806) | ||||||
Finance income | 6,239 | 1,800 | ||||||
Finance costs | (8) | (5,445) | ||||||
Net finance income (costs) | 6,231 | (3,645) | ||||||
Loss before income taxes | (25,963) | (28,451) | ||||||
Income tax expense | (1,104) | - | ||||||
Net loss | (27,067) | (28,451) | ||||||
Other comprehensive (loss) income: | ||||||||
Foreign currency translation adjustments | (789) | 1,001 | ||||||
Actuarial gain (loss) on defined benefit plans | (1,335) | 191 | ||||||
Comprehensive loss | (29,191) | (27,259) | ||||||
Net loss per share | ||||||||
Basic and diluted | (0.29) | (0.38) | ||||||
Weighted average number of shares outstanding | ||||||||
Basic and diluted | 94,507,988 | 75,659,410 | ||||||
Consolidated Statements of Operations Data
(in thousands of US dollars, except share and per share data)
Derived from financial statements prepared in accordance with Canadian GAAP
| | Years Ended December 31, | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2010 | 2009 | 2008 | 2007 | 2006 | ||||||||||||
| | $ | $ | $ | $ | $ | ||||||||||||
Revenues | 27,703 | 63,237 | 38,478 | 42,068 | 38,799 | ||||||||||||
Operating expenses | |||||||||||||||||
Cost of sales, excluding depreciation and amortization | 18,700 | 16,501 | 19,278 | 12,930 | 11,270 | ||||||||||||
Research and development costs | 20,546 | 44,217 | 57,448 | 39,248 | 27,422 | ||||||||||||
Research and development tax credits and grants | (687) | (403) | (343) | (2,060) | (1,564) | ||||||||||||
Selling, general and administrative expenses | 11,875 | 16,040 | 17,325 | 20,403 | 16,478 | ||||||||||||
Depreciation and amortization | |||||||||||||||||
Property, plant and equipment | 1,005 | 3,285 | 1,515 | 1,562 | 2,816 | ||||||||||||
Intangible assets | 1,492 | 7,555 | 5,639 | 4,004 | 6,148 | ||||||||||||
Impairment of long-lived assets held for sale | — | — | — | 735 | — | ||||||||||||
| 52,931 | 87,195 | 100,862 | 76,822 | 62,570 | |||||||||||||
Loss from operations | (25,228) | (23,958) | (62,384) | (34,754) | (23,771) | ||||||||||||
Other income (expenses) | |||||||||||||||||
Unrealized gain on held-for-trading financial instrument | 687 | — | — | — | — | ||||||||||||
Interest income | 207 | 349 | 868 | 1,904 | 1,441 | ||||||||||||
Interest expense | |||||||||||||||||
Long-term debt and convertible term loans | — | — | — | (85) | (1,270) | ||||||||||||
Other | (26) | (5) | (118) | — | (163) | ||||||||||||
Foreign exchange gain (loss) | 1,170 | (1,110) | 3,071 | (1,035) | 319 | ||||||||||||
Loss on disposal of long-lived assets held for sale | — | — | (35) | — | — | ||||||||||||
Loss on disposal of equipment | (28) | — | (44) | (28) | — | ||||||||||||
Gain on disposal of long-term investment | — | — | — | — | 409 | ||||||||||||
| 2,010 | (766) | 3,742 | 756 | 736 | |||||||||||||
Share in the results of an affiliated company | — | — | — | — | 1,575 | ||||||||||||
Loss before income taxes from continuing operations | (23,218) | (24,724) | (58,642) | (33,998) | (21,460) | ||||||||||||
Income tax (expense) recovery | — | — | (1,175) | 1,961 | 29,037 | ||||||||||||
Net (loss) earnings from continuing operations | (23,218) | (24,724) | (59,817) | (32,037) | 7,577 | ||||||||||||
Net (loss) earnings from discontinued operations | — | — | — | (259) | 25,813 | ||||||||||||
Net (loss) earnings for the year | (23,218) | (24,724) | (59,817) | (32,296) | 33,390 | ||||||||||||
Net (loss) earnings per share from continuing operations | |||||||||||||||||
Basic | (0.31) | (0.43) | (1.12) | (0.61) | 0.14 | ||||||||||||
Diluted | (0.31) | (0.43) | (1.12) | (0.61) | 0.14 | ||||||||||||
Net earnings per share from discontinued operations | |||||||||||||||||
Basic | — | — | — | — | 0.50 | ||||||||||||
Diluted | — | — | — | — | 0.48 | ||||||||||||
Net (loss) earnings per share | |||||||||||||||||
Basic | (0.31) | (0.43) | (1.12) | (0.61) | 0.64 | ||||||||||||
Diluted | (0.31) | (0.43) | (1.12) | (0.61) | 0.62 | ||||||||||||
Weighted average number of shares | |||||||||||||||||
Basic | 75,659,410 | 56,864,484 | 53,187,470 | 53,182,803 | 52,099,290 | ||||||||||||
Diluted | 75,659,410 | 56,864,484 | 53,187,470 | 53,182,803 | 52,549,260 | ||||||||||||
| Years Ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| $ | $ | $ | ||||||||||
Revenues | 63,237 | 38,478 | 42,068 | |||||||||
|
| |||||||||||
Operating expenses | ||||||||||||
Cost of sales, excluding depreciation and amortization | 16,501 | 19,278 | 12,930 | |||||||||
Research and development costs | 44,217 | 57,448 | 39,248 | |||||||||
Research and development tax credits and grants | (403 | ) | (343 | ) | (2,060) | |||||||
Selling, general and administrative expenses | 16,040 | 17,325 | 20,403 | |||||||||
Depreciation and amortization | ||||||||||||
Property, plant and equipment | 3,285 | 1,515 | 1,562 | |||||||||
Intangible assets | 7,555 | 5,639 | 4,004 | |||||||||
Impairment of long-lived assets held for sale | — | — | 735 | |||||||||
|
| |||||||||||
| 87,195 | 100,862 | 76,822 | ||||||||||
|
| |||||||||||
Loss from operations | (23,958 | ) | (62,384 | ) | (34,754) | |||||||
|
| |||||||||||
Other income (expenses) | ||||||||||||
Interest income | 349 | 868 | 1,904 | |||||||||
Interest expense | (5 | ) | (118 | ) | (85) | |||||||
Foreign exchange gain (loss) | (1,110 | ) | 3,071 | (1,035) | ||||||||
Loss on disposal of long-lived assets held for sale | — | (35 | ) | — | ||||||||
Loss on disposal of equipment | — | (44 | ) | (28) | ||||||||
|
| |||||||||||
| (766 | ) | 3,742 | 756 | |||||||||
|
| |||||||||||
Loss before income taxes from continuing operations | (24,724 | ) | (58,642 | ) | (33,998) | |||||||
Income tax (expense) recovery | — | (1,175 | ) | 1,961 | ||||||||
|
| |||||||||||
Net loss from continuing operations | (24,724 | ) | (59,817 | ) | (32,037) | |||||||
Net loss from discontinued operations | — | — | (259) | |||||||||
|
| |||||||||||
Net loss for the year | (24,724 | ) | (59,817 | ) | (32,296) | |||||||
|
| |||||||||||
Net loss per share from continuing operations | ||||||||||||
Basic and diluted | (0.43 | ) | (1.12 | ) | (0.61) | |||||||
Net loss per share | ||||||||||||
Basic and diluted | (0.43 | ) | (1.12 | ) | (0.61) | |||||||
Weighted average number of shares | ||||||||||||
Basic and diluted | 56,864,484 | 53,187,470 | 53,182,803 | |||||||||
TableConsolidated Statements of ContentsOperations Data
(in thousands of US dollars, except share and per share data)
Derived from reconciliation to US GAAP
| | Years Ended December 31, | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2010 | 2009 | 2008 | 2007 | 2006 | |||||||||||
| | $ | $ | $ | $ | $ | |||||||||||
Net (loss) earnings for the year | (29,165) | (16,794) | (56,070) | (37,428) | 34,262 | |||||||||||
Of which: | ||||||||||||||||
Net (loss) earnings from continuing operations | (29,165) | (16,794) | (56,070) | (36,415) | 8,449 | |||||||||||
Net (loss) earnings from discontinued operations | — | — | — | (1,013) | 25,813 | |||||||||||
Net (loss) earnings per share from continuing operations | ||||||||||||||||
Basic | (0.39) | (0.30) | (1.05) | (0.68) | 0.16 | |||||||||||
Diluted | (0.39) | (0.30) | (1.05) | (0.68) | 0.16 | |||||||||||
Net (loss) earnings per share from discontinued operations | ||||||||||||||||
Basic | — | — | — | (0.02) | 0.50 | |||||||||||
Diluted | — | — | — | (0.02) | 0.49 | |||||||||||
Net (loss) earnings per share | ||||||||||||||||
Basic | (0.39) | (0.30) | (1.05) | (0.70) | 0.66 | |||||||||||
Diluted | (0.39) | (0.30) | (1.05) | (0.70) | 0.65 | |||||||||||
Weighted average number of shares | ||||||||||||||||
Basic | 75,659,410 | 56,864,484 | 53,187,470 | 53,182,803 | 52,099,290 | |||||||||||
Diluted | 75,659,410 | 56,864,484 | 53,187,470 | 53,182,803 | 52,549,260 | |||||||||||
| Years ended December 31, | ||||||||||||
| 2009* | 2008* | 2007* | ||||||||||
| $ | $ | $ | ||||||||||
Net loss for the year | (16,794 | ) | (56,070 | ) | (37,428 | ) | ||||||
Of which: | ||||||||||||
Net loss from continuing operations | (16,794 | ) | (56,070 | ) | (36,415 | ) | ||||||
Net loss from discontinued operations | — | — | (1,013 | ) | ||||||||
Net loss per share from continuing operations | ||||||||||||
Basic and diluted | (0.30 | ) | (1.05 | ) | (0.68 | ) | ||||||
Net loss per share from discontinued operations | ||||||||||||
Basic and diluted | — | — | (0.02 | ) | ||||||||
Net loss per share | ||||||||||||
Basic and diluted | (0.30 | ) | (1.05 | ) | (0.70 | ) | ||||||
Weighted average number of shares | ||||||||||||
Basic and diluted | 56,864,484 | 53,187,470 | 53,182,803 | |||||||||
TableConsolidated Statement of ContentsFinancial Position Data
Consolidated Balance Sheet Data
(in thousands of US dollars)
Derived from financial statements prepared in accordance with IFRS for 2011 and 2010, and Canadian GAAP for 2009, 2008 and 2007
| As at December 31, | ||||||||||||||||||||||||||||||||||||
| | As at December 31, | 2011 | 2010 | 2009* | 2008* | 2007* | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2010 | 2009 | 2008 | 2007 | 2006 | $ | $ | $ | $ | $ | ||||||||||||||||||||||||||
| | $ | $ | $ | $ | $ | |||||||||||||||||||||||||||||||
Cash and cash equivalents | 31,998 | 38,100 | 49,226 | 10,272 | 8,939 | 46,881 | 31,998 | 38,100 | 49,226 | 10,272 | ||||||||||||||||||||||||||
Short-term investments | 1,934 | — | 493 | 31,115 | 51,550 | — | 1,934 | — | 493 | 31,115 | ||||||||||||||||||||||||||
Working capital | 30,688 | 29,745 | 39,554 | 37,325 | 85,413 | 42,254 | 29,444 | 29,745 | 39,554 | 37,325 | ||||||||||||||||||||||||||
Restricted cash | 827 | 878 | — | — | — | 806 | 827 | 878 | — | — | ||||||||||||||||||||||||||
Total assets | 76,574 | 86,262 | 108,342 | 123,363 | 223,491 | 75,369 | 61,448 | 86,262 | 108,342 | 123,363 | ||||||||||||||||||||||||||
Long-term debt and payable | 90 | 143 | 172 | — | 687 | |||||||||||||||||||||||||||||||
Warrant liability short-term | 42 | 955 | * | * | * | |||||||||||||||||||||||||||||||
Warrant liability long-term | 9,162 | 13,412 | * | * | * | |||||||||||||||||||||||||||||||
Long-term payable | 29 | 90 | 143 | 172 | — | |||||||||||||||||||||||||||||||
Share capital | 60,149 | 41,203 | 30,566 | 30,566 | 168,466 | 101,884 | 60,900 | 41,203 | 30,566 | 30,566 | ||||||||||||||||||||||||||
Shareholders' equity | 12,439 | 9,226 | 21,475 | 88,591 | 178,879 | |||||||||||||||||||||||||||||||
Shareholders’ (deficiency) equity | (4,546 | ) | (17,575 | ) | 9,226 | 21,475 | 88,591 | |||||||||||||||||||||||||||||
| * | We adopted IFRS in 2011 with a transition date of January 1, 2010. The selected financial information for the years ended December 31, 2009, 2008 and 2007 is derived from financial statements that were presented in accordance with Canadian GAAP and has not been restated in accordance with IFRS. Consequently, the selected financial information for the years ended December 31, 2009, 2008 and 2007 may not be comparable with the corresponding selected financial information for the years ended December 31, 2011 and 2010. Please refer to “Critical Accounting Policies, Estimates and Judgments” for the policy differences between Canadian GAAP and IFRS. |
US GAAP
| | As at December 31, | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2010 | 2009 | 2008 | 2007 | 2006 | |||||||||||
| | $ | $ | $ | $ | $ | |||||||||||
Cash and cash equivalents | 31,998 | 38,100 | 49,226 | 10,272 | 8,939 | |||||||||||
Short-term investments | 1,934 | — | 493 | 31,115 | 51,550 | |||||||||||
Working capital | 29,733 | 29,745 | 39,554 | 37,325 | 85,413 | |||||||||||
Restricted cash | 827 | 878 | — | — | — | |||||||||||
Total assets | 74,853 | 84,116 | 100,001 | 109,182 | 209,143 | |||||||||||
Warrant liability, short-term | 955 | — | — | — | — | |||||||||||
Warrant liability, long-term | 13,412 | 1,351 | — | — | — | |||||||||||
Long-term debt and payable | 90 | 143 | 172 | — | 687 | |||||||||||
Share capital | 52,318 | 33,226 | 22,589 | 22,589 | 160,489 | |||||||||||
Shareholders' (deficiency) equity | (3,649) | 5,729 | 13,134 | 74,410 | 169,704 | |||||||||||
TableConsolidated Statement of ContentsFinancial Position Data
B. Capitalization(in thousands of US dollars)
Derived from financial statements prepared in accordance with IFRS for 2011 and indebtedness2010, and US GAAP for 2009, 2008 and 2007
| As at December 31, | ||||||||||||||||||||
| 2011 | 2010 | 2009* | 2008* | 2007* | ||||||||||||||||
| $ | $ | $ | $ | $ | ||||||||||||||||
Cash and cash equivalents | 46,881 | 31,998 | 38,100 | 49,226 | 10,272 | |||||||||||||||
Short-term investments | — | 1,934 | — | 493 | 31,115 | |||||||||||||||
Working capital | 42,254 | 29,444 | 29,745 | 39,554 | 37,325 | |||||||||||||||
Restricted cash | 806 | 827 | 878 | — | — | |||||||||||||||
Total assets | 75,369 | 61,448 | 84,116 | 100,001 | 109,182 | |||||||||||||||
Warrant liability, short-term | 42 | 955 | * | * | * | |||||||||||||||
Warrant liability, long-term | 9,162 | 13,412 | 1,351 | * | * | |||||||||||||||
Long-term payable | 29 | 90 | 143 | 172 | — | |||||||||||||||
Share capital | 101,884 | 60,900 | 33,226 | 22,589 | 22,589 | |||||||||||||||
Shareholders’ (deficiency) equity | (4,546 | ) | (17,575 | ) | 5,729 | 13,134 | 74,410 | |||||||||||||
| * | We adopted IFRS in 2011 with a transition date of January 1, 2010. The selected financial information for the years ended December 31, 2009, 2008 and 2007 is derived from financial statements that were presented in accordance with Canadian GAAP and has not been restated in accordance with IFRS. Consequently, the selected financial information for the years ended December 31, 2009, 2008 and 2007 may not be comparable with the corresponding selected financial information for the years ended December 31, 2011 and 2010. Please refer to “Critical Accounting Policies, Estimates and Judgments” for the policy differences between Canadian GAAP and IFRS. |
| B. | Capitalization and indebtedness |
Not applicable.
C. Reasons for the offer and use of proceeds
| C. | Reasons for the offer and use of proceeds |
Not applicable.
D. Risk factors
| D. | Risk factors |
Risks Relating to Us and Our Business
Investments in biopharmaceutical companies are generally considered to be speculative.
The prospects for companies operating in the biopharmaceutical industry may generally be considered to be uncertain, given the very nature of the industry and, accordingly, investments in biopharmaceutical companies should be considered to be speculative.
We have a history of operating losses and we may never achieve or maintain operating profitability.
Our product candidates remain at the development stage, and we have incurred substantial expenses in our efforts to develop products. Consequently, we have incurred recurrent operating losses and, as disclosed in our audited consolidated financial statements as at December 31, 20102011 and December 31, 20092010 and for the years ended December 31, 2010, 20092011 and 2008,2010, we had an accumulated deficit of $150.8$189.0 million as at December 31, 2010.2011. Our operating losses have adversely impacted, and will continue to adversely impact, our working capital, total assets and shareholders' equity.shareholders’ deficiency. We do not expect to reach operating profitability in the immediate future, and our expenses are likely to increase as we continue to expand our research and development ("R&D")&D and clinical study programs and our sales and marketing activities and seek regulatory approval for our product candidates. Even if we succeed in developing new commercial products, we expect to incur additional operating losses for at least the next several years. If we ultimately do not ultimately generate sufficient revenue from commercialized products and achieve or maintain operating profitability, an investment in our securities could result in a significant or total loss.
Our clinical trials may not yield results which will enable us to obtain regulatory approval for our products, and a setback in any of our clinical trials would likely cause a drop in the price of our securities.
We will only receive regulatory approval for a product candidate if we can demonstrate in carefully designed and conducted clinical trials that the product candidate is both safe and effective. We do not know whether our pending or any future clinical trials will demonstrate sufficient safety and efficacy to obtain the requisite regulatory approvals or will result in marketable products. Unfavorable data from those studies could result in the withdrawal of marketing approval for approved products or an extension of the review period for developmental products. For example, our partner Keryx is conducting phase 3 trials of perifosine for the treatment of colorectal cancer and multiple myeloma. The enrollment of the trial in colorectal cancer has been completed and results of this trial are expected in the near future. If the results of this clinical trial are unfavorable, it would likely adversely affect our and our partners’ perifosine development programs. In addition, unfavorable results in this trial could adversely affect our stock price. Clinical trials are inherently lengthy, complex, expensive and uncertain processes and have a high risk of failure. It typically takes many years to complete testing, and failure can occur at any stage of testing. Results attained in pre-clinicalpreclinical testing and early clinical studies, or trials, may not be indicative of results that are obtained in later studies.
None of our product candidates has to date received regulatory approval for its intended commercial sale. We cannot market a pharmaceutical product in any jurisdiction until it has completed rigorous pre-clinicalpreclinical testing and clinical trials and passed such jurisdiction'sjurisdiction’s extensive regulatory approval process. In general, significant research and development and clinical studies are required to demonstrate the safety and efficacy of our product candidates before we can submit regulatory applications. Pre-clinicalPreclinical testing and clinical development are long, expensive and uncertain processes. Preparing, submitting and advancing applications for regulatory approval is complex, expensive and time-consuming and entails significant uncertainty. Data obtained from pre-clinicalpreclinical and clinical tests can be interpreted in different ways, which could delay, limit or prevent regulatory approval. It may take us
many years to complete the testing of our product candidates and failure can occur at any stage of this process. In addition, we have limited experience in conducting and managing the clinical trials necessary to obtain regulatory approval in the United States, in Canada and abroad and, accordingly, we may encounter unforeseen problems and delays in the approval process. Though we may engage a clinicalcontract research organization (a “CRO”) with experience in conducting regulatory trials, errors in the conduct, monitoring and/or auditing could invalidate the results from a regulatory perspective. Even if a product candidate is approved by the United States Food and Drug Administration ("FDA"(the “FDA”), the Canadian Therapeutic Products Directorate or any other regulatory authority, we may not obtain approval for an indication whose market is large enough to recoup our investment in that product candidate. In addition, there can be no assurance that we will ever obtain all or any required regulatory approvals for any of our product candidates.
We are currently developing our product candidates based on R&D activities, pre-clinicalpreclinical testing and clinical trials conducted to date, and we may not be successful in developing or introducing to the market these or any other new products or technology. If we fail to develop and deploy new products successfully and on a timely basis, we may become non-competitive and unable to recoup the R&D and other expenses we incur to develop and test new products.
Interim results of pre-clinicalpreclinical or clinical studies do not necessarily predict their final results, and acceptable results in early studies might not be obtained in later studies. Safety signals detected during clinical studies and pre-clinicalpreclinical animal studies may require us to do additional studies, which could delay the development of the drug or lead to a decision to discontinue development of the drug. Product candidates in the later stages of clinical development may fail to show the desired safety and efficacy traits despite positive results in initial clinical testing. Results from earlier studies may not be indicative of results from future clinical trials and the risk remains that a pivotal program may generate efficacy data that will be insufficient for the approval of the drug, or may raise safety concerns that may prevent approval of the drug. Interpretation of the prior pre-clinicalpreclinical and clinical safety and efficacy data of our product candidates may be flawed and there can be no assurance that safety and/or efficacy concerns from the prior data were overlooked or misinterpreted, which in subsequent, larger studies appear and prevent approval of such product candidates.
Furthermore, we may suffer significant setbacks in advanced clinical trials, even after promising results in earlier studies. Based on results at any stage of clinical trials, we may decide to repeat or redesign a trial or discontinue development of one or more of our product candidates. Further, actual results may vary once the final and quality-controlled verification of data and analyses has been completed. If we fail to adequately demonstrate the safety and efficacy of our products under development, we will not be able to obtain the required regulatory approvals to commercialize our product candidates.
Clinical trials are subject to continuing oversight by governmental regulatory authorities and institutional review boards and:
| — | must meet the requirements of these authorities; |
| — | must meet requirements for informed consent; and |
| — | must meet requirements for good clinical practices. |
We may not be able to comply with these requirements in respect of one or more of our product candidates.
In addition, we rely on third parties, including Contract Research Organizations ("CROs")CROs and outside consultants, to assist us in managing and monitoring clinical trials. Our reliance on these third parties may result in delays in completing, or in failing to complete, these trials if one or more third parties fails to perform with the speed and level of competence we expect.
A failure in the development of any one of our programs or product candidates could have a negative impact on the development of the others. Setbacks in any phase of the clinical development of our product candidates would have an adverse financial impact (including with respect to any agreements and partnerships that may exist between us and other entities), could jeopardize regulatory approval and would likely cause a drop in the price of our securities.
If we are unable to successfully complete our clinical trial programs, or if such clinical trials take longer to complete than we project, our ability to execute our current business strategy will be adversely affected.
Whether or not and how quickly we complete clinical trials is dependent in part upon the rate at which we are able to engage clinical trial sites and, thereafter, the rate of enrollment of patients, and the rate at which we collect, clean, lock and analyze the clinical trial database. Patient enrollment is a function of many factors, including the design of the protocol, the size of the patient population, the proximity of patients to and availability of clinical sites, the eligibility criteria for the study, the perceived risks and benefits of the drug under study and of the control drug, if any, the efforts to facilitate timely enrollment in clinical trials, the patient referral practices of physicians, the existence of competitive clinical trials, and whether existing or new drugs are approved for the indication we are studying. Certain clinical trials are designed to continue until a pre-determined number of events have occurred to the patients enrolled. Trials such as thisSuch trials are subject to delays stemming from patient withdrawal and from lower than expected event rates and may also incur increased costs if enrollment is increased in order to achieve the desired number of events. If we experience delays in identifying and contracting with sites and/or in patient enrollment in our clinical trial programs, we may incur additional costs and delays in our development programs, and may not be able to complete our clinical trials on a cost-effective or timely basis. In addition, conducting multi-national studies adds another level of complexity and risk as we are subject to events affecting countries outside Canada.North America. Moreover, negative or inconclusive results from the clinical trials we conduct or adverse medical events could cause us to have to repeat or terminate the clinical trials. Accordingly, we may not be able to complete the clinical trials within an acceptable time frame, if at all. If we or any third party have difficulty enrolling a sufficient number of patients to conduct our clinical trials as planned, we may need to delay or terminate ongoing clinical trials.
Additionally, we have never filed a new drug application ("NDA"New Drug Application (“NDA”), or similar application for approval in the United States or in any country for our current product candidates, which may result in a delay in, or the rejection of, our filing of an NDA or similar application. During the drug development process, regulatory agencies will typically ask questions of drug sponsors. While we endeavor to answer all such questions in a timely fashion, or in the NDA filing, some questions may not be answered by the time we file our NDA. Unless the FDA waives the requirement to answer any such unanswered questions, submission of an NDA may be delayed or rejected.
We are and will continue to be subject to stringent ongoing government regulation for our products and, our product candidates, even if we obtain regulatory approvals, for the latter.our product candidates.
The manufacture, marketing and sale of our products and product candidates are and will continue to be subject to strict and ongoing regulation, even if regulatory authorities approve any of the latter.our product candidates. Compliance with such regulation will be expensive and consume substantial financial and management resources. For example, an approval for a product may be conditioned on our agreement to conduct costly post-marketing follow-up studies to monitor the safety or efficacy of the products. In addition, as a clinical experience with a drug expands after approval because the drug is used by a greater number and more diverse group of patients than during clinical trials, side effects or other problems may be observed after approval that were not observed or anticipated during pre-approval clinical trials. In such a case, a regulatory authority could restrict the indications for which the product may be sold or revoke the product'sproduct’s regulatory approval.
We and our contract manufacturers are and will continue to be required to comply with applicable current Good Manufacturing Practice ("cGMP"(“cGMP”) regulations for the manufacture of our products. These regulations include requirements relating to quality assurance, as well as the corresponding maintenance of rigorous records and documentation. Manufacturing facilities must be approved before we can use them in the commercial manufacturing of our products and are subject to subsequent periodic inspection by regulatory authorities. In addition, material changes in the methods of manufacturing or changes in the suppliers of raw materials are subject to further regulatory review and approval.
If we, or any future marketing collaborators or contract manufacturers, fail to comply with applicable regulatory requirements, we may be subject to sanctions including fines, product recalls or seizures and related publicity requirements, injunctions, total or partial suspension of production, civil penalties, suspension or withdrawals of previously granted regulatory approvals, warning or untitled letters, refusal to approve pending applications for marketing approval of new products or of supplements to approved applications, import or export bans or restrictions, and criminal prosecution and penalties. Any of these penalties could delay or prevent the promotion, marketing or sale of our products and product candidates.
If our products do not gain market acceptance, we may be unable to generate significant revenues.
Even if our products are approved for commercialization, they may not be successful in the marketplace. Market acceptance of any of our products will depend on a number of factors including, but not limited to:
| — | demonstration of clinical efficacy and safety; |
| — | the prevalence and severity of any adverse side effects; |
| — | limitations or warnings contained in the product’s approved labeling; |
| — | availability of alternative treatments for the indications we target; |
| — | the advantages and disadvantages of our products relative to current or alternative treatments; |
| — | the availability of acceptable pricing and adequate third-party reimbursement; and |
| — | the effectiveness of marketing and distribution methods for the products. |
If our products do not gain market acceptance among physicians, patients, healthcare payers and others in the medical community, which may not accept or utilize our products, our ability to generate significant revenues from our products would be limited and our financial conditions will be materially adversely affected. In addition, if we fail to further penetrate our core markets and existing geographic markets or successfully expand our business into new markets, the growth in sales of our products, along with our operating results, could be negatively impacted.
Our ability to further penetrate our core markets and existing geographic markets in which we compete or to successfully expand our business into additional countries in Europe, Asia or elsewhere is subject to numerous factors, many of which are beyond our control. Our products, if successfully developed, may compete with a number of drugs and therapies currently manufactured and marketed by major pharmaceutical and other biotechnology companies. Our products may also compete with new products currently under development by others or with products which may be less expensive than our products. We cannot assure you that our efforts to increase market penetration in our core markets and existing geographic markets will be successful. Our failure to do so could have an adverse effect on our operating results and would likely cause a drop in the price of our securities.
We maywill likely require significant additional financing, and we may not have access to sufficient capital.
We maywill likely require additional capital to pursue planned clinical trials, regulatory approvals, as well as further R&D and marketing efforts for our product candidates and potential products. Except as otherwise described in this annual report, we do not anticipate generating significant revenues from operations in the near future and we currently have no committed sources of capital.
We may attempt to raise additional funds through public or private financings, collaborations with other pharmaceutical companies or financing from other sources. Additional funding may not be available on terms which are acceptable to us. If adequate funding is not available to us on reasonable terms, we may need to delay, reduce or eliminate one or more of our product development programs or obtain funds on terms less favorable than we would otherwise accept. To the extent that additional capital is raised through the sale of equity securities or securities convertible into or exchangeable for equity securities, the issuance of those securities couldwould result in dilution to our shareholders. Moreover, the incurrence of debt financingindebtedness could result in a substantial portion of our future operating cash flow, if any, being dedicated to the payment of principal and interest on such indebtedness and could impose restrictions on our operations. This could render us more vulnerable to competitive pressures and economic downturns.
We anticipate that our existing working capital, including the proceeds from any sale and anticipated revenues, will be sufficient to fund our development programs, clinical trials and other operating expenses for the near future.more than 12 months following year-end. However, our future capital requirements are substantial and may increase beyond our current expectations depending on many factors including:
| — | the duration and results of our clinical trials for our various product candidates going forward; |
| — | unexpected delays or developments in seeking regulatory approvals; |
| — | the time and cost involved in preparing, filing, prosecuting, maintaining and enforcing patent claims; |
| — | other unexpected developments encountered in implementing our business development and commercialization strategies; |
| — | the outcome of litigation, if any; and |
| — | further arrangements, if any, with collaborators. |
In addition, global economic and market conditions as well as future developments in the credit and capital markets may make it even more difficult for us to raise additional financing in the future.
A substantial portion of our future revenues may be dependent upon our agreements with Keryx Biopharmaceuticals, Inc. and Yakult Honsha Co. Ltd
We currently expect that a substantial portion of our future revenues may be dependent upon our strategic partnerships with Keryx Biopharmaceuticals, Inc. ("Keryx"(“Keryx”) for North America and Yakult Honsha Co. Ltd ("Yakult"Ltd. (“Yakult”) for Japan. Under these strategic partnerships, Keryx and Yakult have significant development and commercialization responsibilities with respect to the development and sale of perifosine.perifosine in their respective territories. If Keryx or Yakult were to terminate their agreements with us, fail to meet their obligations or otherwise decrease their level of efforts, allocation of resources or other commitments under their respective agreements, our future revenues and/or prospects could be negatively impacted and the development and commercialization of perifosine would be interrupted. In addition, if either Keryx or Yakult dodoes not achieve some or any of their respective development, regulatory and commercial milestones or if they do not achieve certain net sales thresholds as set forth in the agreements, we will not fully realize the expected economic benefits of thesesuch agreements. Further, the achievement of certain of the milestones under these strategic partnership agreements will depend on factors that are outside of our control and most are not expected to be achieved for several years, if at all. Any failure
to successfully maintain our strategic partnership agreements could materially and adversely affect our ability to generate revenues.
If we are unsuccessful in increasing our revenues and/or raising additional funding, we may possibly cease to continue operating as we currently do.
Although our audited consolidated financial statements as at December 31, 20102011 and December 31, 20092010 and for the years ended December 31, 2010, 20092011 and 20082010 have been prepared on a going concern basis, which contemplates the realization of assets and liquidation of liabilities during the normal course of operations, our ability to continue as a going concern is dependent on the successful execution of our business plan, which will require an increase in revenue and/or additional funding to be provided by potential investors as well as non-traditional sources of financing. Although we stated in our audited consolidated financial statements as at December 31, 20102011 and December 31, 20092010 and for the years ended December 31, 2010, 20092011 and 20082010 that management believed that the Company had, as at December 31, 2010,2011, sufficient financial resources to fund planned expenditures and other working capital needs for at least, but not limited to, the 12-month period following such date, there can be no assurance that managementmanagement’s assumptions will be able to reiterate such beliefnot change in our future financial statements.
WeSince our inception, we have had sustainedincurred losses, accumulated deficits and negative cash flows from operations since our inception.operations. We expect that this will continue throughout 2011.2012.
Additional funding may be in the form of debt or equity or a hybrid instrument depending on the needs of the investor. In light of present and future global economic and credit market conditions, we may not be able to raise additional cash resources through these traditional sources of financing. Although we are also pursuing non-traditional sources of financing with third parties, the global credit market crisis has alsomarkets may adversely affectedaffect the ability of potential third parties to pursue such transactions. We do not believe that the ability to access capital markets or these adverse conditions are likely to improve significantly in the near future.transactions with us. Accordingly, as a result of the foregoing, we continue to review traditional sources of financing, such as private and public debt or various equity financing alternatives, as well as other alternatives to enhance shareholder value including, but not limited to, non-traditional sources of financing, such as alliances with strategic partners, the sale of assets or licensing of our technology or intellectual property, a combination of operating and related initiatives or a substantial reorganization of our business. If we do not raise additional capital, we do not expect our operations to generate sufficient cash flow to fund our obligations as they come due.
There can be no assurance that we will achieve profitability or positive cash flows or be able to obtain additional funding or that, if obtained, they will be sufficient, or whether any other initiatives will be successful, such that we may continue as a going concern. There arecould be material uncertainties related to certain adverse conditions and events that could cast significant doubt on our ability to remain a going concern.
We may not achieve our projected development goals in the time-frames we announce and expect.
We set goals and make public statements regarding the timing of the accomplishment of objectives material to our success, such as the commencement, enrollment and completion of clinical trials, anticipated regulatory submission and approval dates and time of product launch. The actual timing of these events can vary dramatically due to factors such as delays or failures in our clinical trials, the uncertainties inherent in the regulatory approval process and delays in achieving manufacturing or marketing arrangements sufficient to commercialize our products. There can be no assurance that our clinical trials will be completed, that we will make regulatory submissions or receive regulatory approvals as planned or that we will be able to adhere to our current schedule for the launch of any of our products. If we fail to achieve one or more of these milestones as planned, the price of our securities would likely decline.
If we fail to obtain acceptable prices or adequate reimbursement for our products, our ability to generate revenues will be diminished.
The ability for us and/or our partners to successfully commercialize our products will depend significantly on our ability to obtain acceptable prices and the availability of reimbursement to the patient from third-party payers, such as governmental and private insurance plans. These third-party payers frequently require companies to provide predetermined discounts from list prices, and they are increasingly challenging the prices charged for pharmaceuticals and other medical products. Our products may not be considered cost-effective, and reimbursement to the patient may not be available or sufficient to allow us or our partners to sell our products on a competitive basis. It may not be possible to negotiate favorable reimbursement rates for our products.
In addition, the continuing efforts of third-party payers to contain or reduce the costs of healthcare through various means may limit our commercial opportunity and reduce any associated revenue and profits. We expect proposals to implement similar government control to continue. In addition, increasing emphasis on managed care will continue to put pressure on the pricing of pharmaceutical and biopharmaceutical products. Cost control initiatives could decrease the price that we or any current or potential collaborators could receive for any of our products and could adversely affect our profitability. In addition, in the United States, in Canada and in many other countries, pricing and/or profitability of some or all prescription pharmaceuticals and biopharmaceuticals are subject to government control.
If we fail to obtain acceptable prices or an adequate level of reimbursement for our products, the sales of our products would be adversely affected or there may be no commercially viable market for our products.
Competition in our targeted markets is intense, and development by other companies could render our products or technologies non-competitive.
The biomedical field is highly competitive. New products developed by other companies in the industry could render our products or technologies non-competitive. Competitors are developing and testing products and technologies that would compete with the products that we are developing. Some of these products may be more effective or have an entirely different approach or means of accomplishing the desired effect than our products. We expect competition from biopharmaceutical and pharmaceutical companies and academic research institutions to increase over time. Many of our competitors and potential competitors have substantially greater product development capabilities and financial, scientific, marketing and human resources than we do. Our competitors may succeed in developing products earlier and in obtaining regulatory approvals and patent protection for such products more rapidly than we can or at a lower price.
We may not obtain adequate protection for our products through our intellectual property.
We rely heavily on our proprietary information in developing and manufacturing our product candidates. Our success depends, in large part, on our ability to protect our competitive position through patents, trade secrets, trademarks and other intellectual property rights. The patent positions of pharmaceutical and biopharmaceutical firms, including Aeterna Zentaris, are uncertain and involve complex questions of law and fact for which
important legal issues remain unresolved. Applications for patents and trademarks in Canada, the United States and in other foreign territories have been filed and are being actively pursued by us. Pending patent applications may not result in the issuance of patents and we may not be able to obtain additional issued patents relating to our technology or products. Even if issued, patents to us or our licensors may be challenged, narrowed, invalidated, held to be unenforceable or circumvented, which could limit our ability to stop competitors from marketing similar products or limit the length of term of patent protection we may have for our products. Changes in either patent laws or in interpretations of patent laws in the United States and other
countries may diminish the value of our intellectual property or narrow the scope of our patent protection. The patents issued or to be issued to us may not provide us with any competitive advantage or protect us against competitors with similar technology. In addition, it is possible that third parties with products that are very similar to ours will circumvent our patents by means of alternate designs or processes. We may have to rely on method of use and new formulation protection for our compounds in development, and any resulting products, which may not confer the same protection as claims to compounds per se.
In addition, our patents may be challenged by third parties in patent litigation, which is becoming widespread in the biopharmaceutical industry. There may be prior art of which we are not aware that may affect the validity or enforceability of a patent claim. There may also may be prior art of which we are aware, but which we do not believe affects the validity or enforceability of a claim, which may, nonetheless, ultimately be found to affect the validity or enforceability of a claim. No assurance can be given that our patents would, if challenged, be held by a court to be valid or enforceable or that a competitor'scompetitor’s technology or product would be found by a court to infringe our patents. Our granted patents could also be challenged and revoked in opposition or nullity proceedings in certain countries outside the United States. In addition, we may be required to disclaim part of the term of certain patents.
Patent applications relating to or affecting our business have been filed by a number of pharmaceutical and biopharmaceutical companies and academic institutions. A number of the technologies in these applications or patents may conflict with our technologies, patents or patent applications, and any such conflict could reduce the scope of patent protection which we could otherwise obtain. Because patent applications in the United States and many other jurisdictions are typically not published until eighteen months after their first effective filing date, or in some cases not at all, and because publications of discoveries in the scientific literature often lag behind actual discoveries, neither we nor our licensors can be certain that we or they were the first to make the inventions claimed in our or their issued patents or pending patent applications, or that we or they were the first to file for protection of the inventions set forth in these patent applications. If a third party has also filed a patent application in the United States covering our product candidates or a similar invention, we may have to participate in an adversarial proceeding, known as an interference, declared by the United States Patent and Trademark Office to determine priority of invention in the United States. The costs of these proceedings could be substantial and it is possible that our efforts could be unsuccessful, resulting in a loss of our U.S. patent position.
In addition to patents, we rely on trade secrets and proprietary know-how to protect our intellectual property. If we are unable to protect the confidentiality of our proprietary information and know-how, the value of our technology and products could be adversely affected. We seek to protect our unpatented proprietary information in part by requiring our employees, consultants, outside scientific collaborators and sponsored researchers and other advisors to enter into confidentiality agreements. These agreements provide that all confidential information developed or made known to the individual during the course of the individual'sindividual’s relationship with us is to be kept confidential and not disclosed to third parties except in specific circumstances. In the case of our employees, the agreements provide that all of the technology which is conceived by the individual during the course of employment is our exclusive property. These agreements may not provide meaningful protection or adequate remedies in the event of unauthorized use or disclosure of our proprietary information. In addition, it is possible that third parties could independently develop proprietary information and techniques substantially similar to ours or otherwise gain access to our trade secrets. If we are unable to protect the confidentiality of our proprietary information and know-how, competitors may be able to use this information to develop products that compete with our products and technologies, which could adversely impact our business.
We currently have the right to use certain technology under license agreements with third parties. Our failure to comply with the requirements of material license agreements could result in the termination of such agreements, which could cause us to terminate the related development program and cause a complete loss of our investment in that program.
As a result of the foregoing factors, we may not be able to rely on our intellectual property to protect our products in the marketplace.
We may infringe the intellectual property rights of others.
Our commercial success depends significantly on our ability to operate without infringing the patents and other intellectual property rights of third parties. There could be issued patents of which we are not aware that our products or methods may be found to infringe, or patents of which we are aware and believe we do not infringe but which we may ultimately be found to infringe. Moreover, patent applications and their underlying discoveries are in some cases maintained in secrecy until patents are issued. Because patents can take many years to issue, there may be currently pending applications of which we are unaware that may later result in issued patents that our products or methods are found to infringe. Moreover, there may be published pending applications that do not currently include a claim covering our products or methods but which nonetheless provide support for a later drafted claim that, if issued, our products or methods could be found to infringe.
If we infringe or are alleged to infringe intellectual property rights of third parties, it will adversely affect our business. Our research, development and commercialization activities, as well as any product candidates or products resulting from these activities, may infringe or be accused of infringing one or more claims of an issued patent or may fall within the scope of one or more claims in a published patent application that may subsequently issue and to which we do not hold a license or other rights. Third parties may own or control these patents or patent applications in the United States and abroad. These third parties could bring claims against us or our collaborators that would cause us to incur substantial expenses and, if successful against us, could cause us to pay substantial damages. Further, if a patent infringement suit were brought against us or our collaborators, we or they could be forced to stop or delay research, development, manufacturing or sales of the product or product candidate that is the subject of the suit.
The biopharmaceutical industry has produced a proliferation of patents, and it is not always clear to industry participants, including us, which patents cover various types of products. The coverage of patents is subject to interpretation by the courts, and the interpretation is not always uniform. In the event of infringement or violation of another party'sparty’s patent or other intellectual property rights, we may not be able to enter into licensing arrangements or make other arrangements at a reasonable cost. Any inability to secure licenses or alternative technology could result in delays in the introduction of our products or lead to prohibition of the manufacture or sale of products by us or our partners and collaborators.
Patent litigation is costly and time consuming and may subject us to liabilities.
Our involvement in any patent litigation, interference, opposition or other administrative proceedings will likely cause us to incur substantial expenses, and the efforts of our technical and management personnel will be significantly diverted. In addition, an adverse determination in litigation could subject us to significant liabilities.
We may not obtain trademark registrations.
We have filed applications for trademark registrations in connection with our product candidates in various jurisdictions, including the United States. We intend to file further applications for other possible trademarks for our product candidates. No assurance can be given that any of our trademark applications will be registered in the United States or elsewhere, or that the use of any registered or
unregistered trademarks will confer a competitive advantage in the marketplace. Furthermore, even if we are successful in our trademark registrations, the FDA and regulatory authorities in other countries have their own process for drug nomenclature and their own views concerning appropriate proprietary names. The FDA and other regulatory authorities also have the power, even after granting market approval, to request a company to reconsider the name for a product because of evidence of confusion in the marketplace. No assurance can be given that the FDA or any other regulatory authority will approve of any of our trademarks or will not request reconsideration of one of our trademarks at some time in the future. The loss, abandonment, or cancellation of any of our trademarks or trademark applications could negatively affect the success of the product candidates to which they relate.
Our revenues and expenses may fluctuate significantly, and any failure to meet financial expectations may disappoint securities analysts or investors and result in a decline in the price of our securities.
We have a history of operating losses. Our revenues and expenses have fluctuated in the past and are likely to do so in the future. These fluctuations could cause our share price to decline. Some of the factors that could cause our revenues and expenses to fluctuate include but are not limited to:
| — | the inability to complete product development in a timely manner that results in a failure or delay in receiving the required regulatory approvals to commercialize our product candidates; |
| — | the timing of regulatory submissions and approvals; |
| — | the timing and willingness of any current or future collaborators to invest the resources necessary to commercialize our product candidates; |
| — | the revenue available from royalties derived from our strategic partners; |
| — | licensing fees revenues; |
| — | tax credits and grants (R&D); |
| — | the outcome of litigation, if any; |
| — | changes in foreign currency fluctuations; |
| — | the timing of achievement and the receipt of milestone payments from current or future collaborators; and |
| — | failure to enter into new or the expiration or termination of current agreements with collaborators. |
Due to fluctuations in our revenues and expenses, we believe that period-to-period comparisons of our results of operations are not necessarily indicative of our future performance. It is possible that in some future quarter or quarters, our revenues and expenses will be above or below the expectations of securities analysts or investors. In this case, the price of our securities could fluctuate significantly or decline.
We will not be able to successfully commercialize our product candidates if we are unable to make adequate arrangements with third parties for such purposes.
We currently have a lean sales and marketing staff. In order to commercialize our product candidates successfully, we need to make arrangements with third parties to perform some or all of these services in certain territories.
We contract with third parties for the sales and marketing of our products. Our revenues will depend upon the efforts of these third parties, whose efforts may not be successful. If we fail to establish successful marketing and sales capabilities or to make arrangements with third parties for such purposes, our business, financial condition and results of operations will be materially adversely affected.
If we had to resort to developing a sales force internally, the cost of establishing and maintaining a sales force would be substantial and may exceed its cost effectiveness. In addition, in marketing our products, we would likely compete with many companies that currently have extensive and well-funded marketing and sales operations. Despite our marketing and sales efforts, we may be unable to compete successfully against these companies.
We are currently dependent on strategic partners and may enter into future collaborations for the research, development and commercialization of our product candidates. Our arrangements with these strategic partners may not provide us with the benefits we expect and may expose us to a number of risks.
We are dependent on, and rely upon, strategic partners to perform various functions related to our business, including, but not limited to, the research, development and commercialization of some of our product candidates. Our reliance on these relationships poses a number of risks.
We may not realize the contemplated benefits of such agreements nor can we be certain that any of these parties will fulfill their obligations in a manner which maximizes our revenue. These arrangements may also require us to transfer certain material rights or issue our equity, voting or other securities to corporate partners, licensees and others. Any license or sublicense of our commercial rights may reduce our product revenue.
These agreements also create certain risks. The occurrence of any of the following or other events may delay product development or impair commercialization of our products:
| — | not all of our strategic partners are contractually prohibited from developing or commercializing, either alone or with others, products and services that are similar to or competitive with our product candidates and, with respect to our strategic partnership agreements that do contain such contractual prohibitions or restrictions, prohibitions or restrictions do not always apply to our partners’ affiliates and they may elect to pursue the development of any additional product candidates and pursue technologies or products either on their own or in collaboration with other parties, including our competitors, whose technologies or products may be competitive with ours; |
| — | our strategic partners may under-fund or fail to commit sufficient resources to marketing, distribution or other development of our products; |
| — | we may not be able to renew such agreements; |
| — | our strategic partners may not properly maintain or defend certain intellectual property rights that may be important to the commercialization of our products; |
| — | our strategic partners may encounter conflicts of interest, changes in business strategy or other issues which could adversely affect their willingness or ability to fulfill their obligations to us (for example, pharmaceutical companies historically have re-evaluated their priorities following mergers and consolidations, which have been common in recent years in this industry); |
| — | delays in, or failures to achieve, scale-up to commercial quantities, or changes to current raw material suppliers or product manufacturers (whether the change is attributable to us or the supplier or manufacturer) could delay clinical studies, regulatory submissions and commercialization of our product candidates; and |
| — | disputes may arise between us and our strategic partners that could result in the delay or termination of the development or commercialization of our product candidates, resulting in litigation or arbitration that could be time-consuming and expensive, or causing our strategic partners to act in their own self-interest and not in our interest or those of our shareholders or other stakeholders. |
In addition, our strategic partners can terminate our agreements with them for a number of reasons based on the terms of the individual agreements that we have entered into with them. If one or more of these agreements were to be terminated, we would be required to devote additional resources to developing and commercializing our product candidates, seek a new partner or abandon this product candidate which would likely cause a drop in the price of our securities.
We have entered into important strategic partnership agreements relating to certain of our product candidates for various indications. Detailed information on our research and collaboration agreements is available in our various reports and disclosure documents filed with the Canadian securities regulatory authorities and filed with or furnished to the United States Securities and Exchange Commission ("SEC"(“SEC”), including the documents incorporated by reference ininto this Annual Report on Form 20-F. See, for example, Note 255 to our audited consolidated balance sheetsfinancial statements as at December 31, 2011 and December 31, 2010 and 2009 and our audited consolidated statements of operations, changes in shareholders' equity, accumulated other comprehensive income and deficit, comprehensive loss and cash flows for each of the years in the three-year period ended December 31, 2011 and 2010 included in this Annual Report on Form 20-F.
We have also entered into a variety of collaborative licensing agreements with various universities and institutes under which we are obligated to support some of the research expenses incurred by the university laboratories and pay royalties on future sales of the products. In turn, we have retained exclusive rights for the worldwide exploitation of results generated during the collaborations.
In particular, we have entered into an agreement with the Tulane Educational Fund ("Tulane"(“Tulane”), which provides for the payment by us of single-digit royalties on future worldwide net sales of cetrorelix, and including Cetrotide®Cetrotide®. Tulane is also entitled to receive a low double-digit participation payment on any lump-sum, periodic or other cash payments received by us from sub-licensees (see Note 25 to our audited consolidated balance sheets as at December 31, 2010 and 2009 and our audited consolidated statements of operations, changes in shareholders' equity, accumulated other comprehensive income and deficit, comprehensive loss and cash flows for each of the years in the three-year period ended December 31, 2010 included in this Annual Report on Form 20-F).sub-licensees.
We rely on third parties to conduct, supervise and monitor our clinical trials, and those third parties may not perform satisfactorily.
We rely on third parties such as CROs, medical institutions and clinical investigators to enroll qualified patients and conduct, supervise and monitor our clinical trials. Our reliance on these third parties for clinical development activities reduces our control over these activities. Our reliance on these third parties, however, does not relieve us of our regulatory responsibilities, including ensuring that our clinical trials are conducted in accordance with Good Clinical Practice guidelines and the investigational plan and protocols contained in an Investigational New Drug (“IND”) application, or comparable foreign regulatory submission. Furthermore, these third parties may also have relationships with other entities, some of which may be our competitors. In addition, they may not complete activities on schedule, or may not conduct our pre-clinicalpreclinical studies or clinical trials in accordance with regulatory requirements or our trial design. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, our efforts to obtain regulatory approvals for, and commercialize, our product candidates may be delayed or prevented.
In carrying out our operations, we are dependent on a stable and consistent supply of ingredients and raw materials.
There can be no assurance that we, our contract manufacturers or our partners, will be able, in the future, to continue to purchase products from our current suppliers or any other supplier on terms similar to current terms or at all. An interruption in the availability of certain raw materials or
ingredients, or significant increases in the prices paid by us for them, could have a material adverse effect on our business, financial condition, liquidity and operating results.
The failure to perform satisfactorily by third parties upon which we rely to manufacture and supply products may lead to supply shortfalls.
We rely on third parties to manufacture and supply marketed products. We also have certain supply obligationsvis-à-vis to our licensing partners who are responsible for the marketing of thesuch products. To be successful, our products have to be manufactured in commercial quantities in compliance with quality controls and regulatory requirements. Even though it is our objective to minimize such risk by introducing alternative suppliers to ensure a constant supply at all times, we cannot guarantee that we will not experience supply shortfalls and, in such event, we may not be able to perform our obligations under contracts with our partners.
We are subject to intense competition for our skilled personnel, and the loss of key personnel or the inability to attract additional personnel could impair our ability to conduct our operations.
We are highly dependent on our management and our clinical, regulatory and scientific staff, the loss of whose services might adversely impact our ability to achieve our objectives. Recruiting and retaining qualified management and clinical, scientific and regulatory personnel is critical to our success. Competition for skilled personnel is intense, and our ability to attract and retain qualified personnel may be affected by such competition.
Our strategic partners'partners’ manufacturing capabilities may not be adequate to effectively commercialize our product candidates.
Our manufacturing experience to date with respect to our product candidates consists of producing drug substance for clinical studies. To be successful, these product candidates have to be manufactured in commercial quantities in compliance with regulatory requirements and at acceptable costs. Our strategic partners'partners’ current manufacturing facilities have the capacity to produce projected product requirements for the foreseeable future, but we will need to increase capacity if sales continue to grow. Our strategic partners may not be able to expand capacity or to produce additional product requirements on favorable terms. Moreover, delays associated with securing additional manufacturing capacity may reduce our revenues and adversely affect our business and financial position. There can be no assurance that we will be able to meet increased demand over time.
We are subject to the risk of product liability claims, for which we may not have or be able to obtain adequate insurance coverage.
The sale and use of our products, in particular our biopharmaceutical products, involve the risk of product liability claims and associated adverse publicity. Our risks relate to human participants in our clinical trials, who may suffer unintended consequences, as well as products on the market whereby claims might be made directly by patients, healthcare providers or pharmaceutical companies or others selling, buying or using our products. We manage our liability risks by means of insurance. We maintain liability insurance covering our liability for our pre-clinicalpreclinical and clinical studies and for our pharmaceutical products already marketed. However, we may not have or be able to obtain or maintain sufficient and affordable insurance coverage, including coverage for potentially very significant legal expenses, and without sufficient coverage any claim brought against us could have a materially adverse effect on our business, financial condition or results of operations.
Our business involves the use of hazardous materials which requires usand, as such, we are subject to comply with environmental and occupational safety laws regulating the use of such materials. If we violate these laws, we could be subject toincur significant fines or liabilities or suffer other adverse consequences.
Our discovery and development processes involve the controlled use of hazardous and radioactive materials. We are subject to federal, provincial and local laws and regulations governing the use,
manufacture, storage, handling and disposal of such materials and certain waste products. The risk of accidental contamination or injury from these materials cannot be completely eliminated. In the event of an accident or a failure to comply with environmental or occupational safety laws, we could be held liable for any damages that result, and any such liability could exceed our resources. We may not be adequately insured against this type of liability. We may be required to incur significant costs to comply with environmental laws and regulations in the future, and our operations, business or assets may be materially adversely affected by current or future environmental laws or regulations.
Legislative actions, new accounting pronouncements and higher insurance costs are likely to impact our future financial position or results of operations.
Changes in financial accounting standards or implementation of accounting standards may cause adverse, unexpected revenue or expense fluctuations and affect our financial position or results of operations. New pronouncements and varying interpretations of pronouncements have occurred with greater frequency and are expected to occur in the future, and we may make or be required to make changes in our accounting policies in the
future. Compliance with changing regulations of corporate governance and public disclosure, notably with respect to internal controls over financial reporting, may result in additional expenses. Changing laws, regulations and standards relating to corporate governance and public disclosure are creating uncertainty for companies such as ours, and insurance costs are increasing as a result of this uncertainty.
We are subject to additional reporting requirements under applicable Canadian securities laws and the Sarbanes-Oxley Act in the United States. We can provide no assurance that we will at all times in the future be able to report that our internal controls over financial reporting are effective.
As a public company, we are required to comply with Section 404 of the Sarbanes-Oxley Act ("(“Section 404"404”) and National Instrument 52-109 — Certification of Disclosure in Issuers'Issuers’ Annual and Interim Filings, and we are required to obtain an annual attestation from our independent auditors regarding our internal control over financial reporting. In any given year, we cannot be certain as to the time of completion of our internal control evaluation, testing and remediation actions or of their impact on our operations. Upon completion of this process, we may identify control deficiencies of varying degrees of severity under applicable SEC and Public Company Accounting Oversight Board rules and regulations. As a public company, we are required to report, among other things, control deficiencies that constitute material weaknesses or changes in internal controls that, or that are reasonably likely to, materially affect internal controls over financial reporting. A "material weakness"“material weakness” is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of the company'sCompany’s annual consolidated financial statements will not be prevented or detected on a timely basis. If we fail to comply with the requirements of Section 404, Canadian requirements or report a material weakness, we might be subject to regulatory sanction and investors may lose confidence in our consolidated financial statements, which may be inaccurate if we fail to remedy such material weakness.
It is possible that we may be a passive foreign investment company, which could result in adverse tax consequences to U.S. investors.
Adverse U.S. federal income tax rules apply to "U.S. Holders"“U.S. Holders” (as defined in "Item“Item 10.E — Taxation — CertainMaterial U.S. Federal Income Tax Consideration"Considerations” in this Annual Report on Form 20-F) that directly or indirectly hold common shares or warrants of a passive foreign investment company ("PFIC"(“PFIC”). We will be classified as a PFIC for U.S. federal income tax purposes for a taxable year if (i) at least 75 percent of our gross income is "passive income"“passive income” or (ii) at least 50 percent of the average value of our assets, including goodwill (based on annual quarterly average), is attributable to assets which produce passive income or are held for the production of passive income.
We believe that we were not a PFIC for the 20102011 taxable year. However, since the fair market value of our assets may be determined in large part by the market price of our common shares, which is likely to fluctuate, and the composition of our income and assets will be affected by how, and how quickly, we spend any cash that is raised in any financing transaction,transaction. Thus no assurance can be provided that we will not be classified as a PFIC for the 20112012 taxable year and for any future taxable year.
PFIC characterization could result in adverse U.S. federal income tax consequences to U.S. Holders. In particular, absent certain elections, a U.S. Holder would be subject to U.S. federal income tax at ordinary income tax rates, plus a possible interest charge, in respect of a gain derived from a disposition of our common shares, as well as certain distributions by us. If we are treated as a PFIC for any taxable year, a U.S. Holder may be able to make an election to "mark“mark to market"market” common shares each taxable year and recognize ordinary income pursuant to such election based upon increases in the value of the common shares. However, a mark-to-market election is not available to be made in respect of a warrant.
Under recently enacted U.S. tax legislation and subject to future guidance, if we arethe Company is a PFIC, U.S. Holders will be required to file for returns due after March 18, 2010, an annual information return with the Internal Revenue Service (“IRS”) (on IRS Form 8621, which PFIC shareholders will be required to file with their income tax or information returns) relating to their ownership of our common shares. Although expected, no guidancePursuant to Notice 2011-55, the IRS has suspended this new
filing requirement for U.S. Holders that are not otherwise required to file the current version of the IRS Form 8621 until the IRS releases a subsequent revision of IRS Form 8621, modified to reflect the recently enacted U.S. tax legislation. Guidance has not yet been issued about such return, including onregarding the information required to be reportedincluded on such return,form. This new filing requirement is in addition to any pre-existing reporting requirements that apply to a U.S. Holder’s interest in a PFIC (which the form of the return, or the due date of the return.recently enacted tax legislation and IRS Notice 2011-55 do not affect).
For a more detailed discussion of the potential tax impact of us being a PFIC, see "Item“Item 10.E — Taxation — CertainMaterial U.S. Federal Income Tax Considerations"Considerations” in this Annual Report on Form 20-F.
We will report under International Financial Reporting Standards for our interim and annual consolidated financial statements for the financial year ending December 31, 2011.
Effective January 1, 2011, the Accounting Standards Board of the Canadian Institute of Chartered Accountants require that Canadian publicly accountable enterprises adopt International Financial Reporting Standards ("IFRS"), as issued by the International Accounting Standards Board. We are thus required to report under IFRS for our interim and annual consolidated financial statements for the financial year ending December 31, 2011.
IFRS uses a conceptual framework that is similar to Canadian generally accepted accounting principles; however, we have identified certain differences that will result in changes to some of our accounting policies. We are currently in the process of preparing our first interim unaudited financial statements in accordance with IFRS, and the notes to such financial statements will explain in detail the specific impact of IFRS on our financial statements. Additional information on our conversion to IFRS is provided under "Item 5. — Operating and Financial Review and Prospects" included in this Annual Report on Form 20-F.
We may incur losses associated with foreign currency fluctuations.
Our operations are in many instances conducted in currencies other than the euro, our functional currency. Fluctuations in the value of currencies could cause us to incur currency exchange losses. We do not currently employ a hedging strategy against exchange rate risk. We cannot assert with any assurance that we will not suffer losses as a result of unfavorable fluctuations in the exchange rates between the United States dollar, the euro, the Canadian dollar and other currencies. For more information, see "Item“Item 11. — Quantitative and Qualitative Disclosures About Market Risk"Risk” in this Annual Report on Form 20-F.
We may not be able to successfully integrate acquired businesses.
Future acquisitions may not be successfully integrated. The failure to successfully integrate the personnel and operations of businesses which we may acquire in the future with ours could have a material adverse effect on our operations and results.
Risks Related to our Securities
Our share price is volatile, which may result from factors outside of our control. If our common shares were to be delisted from the NASDAQ Global Market (“NASDAQ”) or TSX,the Toronto Stock Exchange (the “TSX”), investors may have difficulty in disposing of our common shares held by them.
Our common shares are currently listed and traded only on NASDAQ and TSX. Our valuation and share price since the beginning of trading after our initial listings, first in Canada and then in the United States, have had no meaningful relationship to current or historical financial results, asset values, book value or many other criteria based on conventional measures of the value of shares.
During the year ended December 31, 2010,2011, the closing price of our common shares ranged from $0.79$1.43 to $2.09$2.58 on NASDAQ and from C$0.801.41 to C$2.142.51 per share on TSX. Our share price may be affected by developments directly affecting our business and by developments out of our control or unrelated to us. The stock market generally, and the biopharmaceutical sector in particular, are vulnerable to abrupt changes in investor sentiment. Prices of shares and trading volumevolumes of companies in the biopharmaceutical industry can swing dramatically in ways unrelated to, or that bear a disproportionate relationship to, operating performance. Our share price and trading volume may fluctuate based on a number of factors including, but not limited to:
| — | clinical and regulatory developments regarding our product candidates; |
| — | delays in our anticipated development or commercialization timelines; |
| — | developments regarding current or future third-party collaborators; |
| — | other announcements by us regarding technological, product development or other matters; |
| — | arrivals or departures of key personnel; |
| — | governmental or regulatory action affecting our product candidates and our competitors’ products in the United States, Canada and other countries; |
| — | developments or disputes concerning patent or proprietary rights; |
| — | actual or anticipated fluctuations in our revenues or expenses; |
| — | general market conditions and fluctuations for the emerging growth and biopharmaceutical market sectors; and |
| — | economic conditions in the United States, Canada or abroad. |
Our listing on both NASDAQ and TSX may increase price volatility due to various factors, including different ability to buy or sell our common shares, different market conditions in different capital markets and different trading volumes. In addition, low trading volume may increase the price volatility of our common shares. A thin trading market could cause the price of our common shares to fluctuate significantly more than the stock market as a whole.
In the past, following periodsA period of large price declinesdecline in the public market price of a company's securities,our common shares could increase the risk that securities class action litigation has often beencould be initiated against that company.us. Litigation of this type could result in substantial costs and diversion of management'smanagement’s attention and resources, which would adversely affect our business. Any adverse determination in litigation could also subject us to significant liabilities.
We must meet continuing listing requirements to maintain the listing of our common shares on NASDAQ and TSX. For continued listing, NASDAQ requires, among other things, that listed securities maintain a minimum closing bid price of not less than $1.00 per share.
If we are unsuccessful in maintaining the NASDAQ'sNASDAQ’s minimum bid price or other requirements in the future and are unable to subsequently regain compliance within the applicable grace period, our common shares will be subject to delisting from the NASDAQ Global Market.delisting. Should we receive a delisting notification, we may appeal to the Listing Qualifications Panel or apply to transfer the listing of our common shares to the NASDAQ Capital Market if we satisfy at such time all of the initial listing standards onof the NASDAQ Capital Market, other than compliance with the minimum closing bid price requirement. If the application to the NASDAQ Capital Market iswere approved, then we willwould have an additional 180-day grace period in order to regain compliance with the minimum bid price requirement while listed on the NASDAQ Capital Market. There can be no assurance that we will meet the requirements for continued listing on the NASDAQ Global Market or whether ouran application to the NASDAQ Capital Market willwould be approved or that any appeal would be granted by the Listing Qualifications Panel.
We do not intend to pay dividends in the near future.
To date, we have not declared or paid any dividends on our common shares. We currently intend to retain our future earnings, if any, to finance further research and the expansion of our business. As a result, the return on an investment in our securities will, for the foreseeable future, depend upon any future appreciation in value. There is no guarantee that our securities will appreciate in value or even maintain the price at which shareholders have purchased their securities.
Item 4. Information on the Company
| A. | History and development of the Company |
Aeterna Zentaris Inc. is a late-stage drug development company specialized in oncology and endocrine therapy.
We were incorporated on September 12, 1990 under theCanada Business Corporations Act (the "CBCA"“CBCA”) and continue to be governed by the CBCA. Our registered office is located at 1405 du Parc-Technologique Blvd., Quebec City, Quebec, Canada G1P 4P5, our telephone number is (418) 652-8525 and our website is www.aezsinc.com. None of the documents or information found on our website shall be deemed to be included in or incorporated by reference into this annual report.
On December 30, 2002, we acquired Zentaris AG, a biopharmaceutical company based in Frankfurt, Germany. Zentaris was a spin-off of Degussa AG and Asta Medica GmbH, a former pharmaceutical company. With this acquisition, the Company changed its risk profile and inherited an extensive and robust product pipeline with capabilities from drug discovery to commercialization with a particular focus on endocrine therapy and oncology. As part of the acquisition, we also inheritedacquired a very experienced pharmaceutical team along with a network of strategic pharmaceutical partners. The total consideration paid for the acquisition of Zentaris was $51.9 million, net of cash and cash equivalents acquired of $2.3 million, of which an amount of $26.7 million was paid in cash and the remaining amount of $25.2 million as a balance of purchase price.
In May 2004, we changed our name to Aeterna Zentaris Inc. and on May 11, 2007, Zentaris GmbH was renamed Aeterna Zentaris GmbH ("(“AEZS GmbH"GmbH”). AEZS GmbH is our principal operating subsidiary.
On April 6, 2005, our former subsidiary Atrium Biotechnologies Inc. (now Atrium Innovations Inc.) ("Atrium"(“Atrium”), completed its initial public offering in Canada and began trading on the TSX under the ticker symbol "ATB."“ATB”.
Throughout 2006, as part of a thorough, strategic planning process, our management and Board of Directors (the "Board"“Board”) made the decision to spin off Atrium in two phases. On September 19, 2006,
we initiated the first phase, a secondary offering in which we sold 3,485,000 Subordinate Voting Shares of Atrium at a price of CAN$15.80 per share. This secondary offering closed on October 18, 2006, generating net proceeds of nearly $45 million to Aeterna Zentaris. With this transaction closed, our remaining interest in Atrium was 11,052,996 Subordinate Voting Shares representing 36.1% of its issued and outstanding shares. Therefore, we no longer had a controlling interest in Atrium as at October 18, 2006.
The second phase was to distribute our remaining interest in Atrium to our shareholders concurrently with a reduction of the stated capital of our common shares.
On December 15, 2006, our shareholders approved a reduction of the stated capital of our common shares in an amount equal to the fair market value of our remaining interest in Atrium by way of a special distribution in kind to all our shareholders. This special distribution was completed on January 2, 2007. For each common share held as at the record date of December 29, 2006, our shareholders received 0.2078824 Subordinate Voting Shares of Atrium.
In May 2007, we opened an office in the United States, located at 20 Independence Boulevard, Warren, New Jersey 07059-2731. The Company moved this office to a new location in December 2011 at 25 Mountainview Blvd., Suite 203, Basking Ridge, NJ 07920.
We currently have three wholly-owned direct and indirect subsidiaries, Aeterna Zentaris GmbH ("(“AEZS Germany"Germany”), based in Frankfurt, Germany, Zentaris IVF GmbH, a direct wholly-owned subsidiary of AEZS Germany based in Frankfurt, Germany, and Aeterna Zentaris, Inc., based in Warren,Basking Ridge, New Jersey in the United States.
From the formation of Atrium as our subsidiary in 1999 until the distribution of our remaining interest in Atrium on January 2, 2007, Atrium did not declare or pay any dividends to its shareholders. Since the disposition of our entire interest in Atrium, we have not had access to the liquidity or cash flows generated by Atrium.
Our current drug development strategy focuses mainly on our late-stage compounds perifosine (Phase 3and AEZS-108 (zoptarelin doxorubicin) in multiple myeloma and colorectal cancer) and our Phase 2 programoncology, AEZS-130 (macimorelin) in multiple cancers, AEZS-108 (we recently completed with success a Phase 2 trial in endometrial and ovarian cancer and in clinical development in bladder and prostate cancer) and AEZS-130 (Solorel®) (Phase 3 as diagnostic test for adult growth hormone deficiency),endocrinology, as well as on strategic and targeted earlier-stage compounds, as depicted in the chart reproduced under the heading, "Our“Our Product Pipeline"Pipeline”.
Our common shares are listed for trading on the TSX under the trading symbol "AEZ"“AEZ” and on the NASDAQ under the trading symbol "AEZS."“AEZS”.
The Company'sCompany’s agent for service of process and SEC matters in the United States is its wholly-owned subsidiary, Aeterna Zentaris, Inc., located at 20 Independence Boulevard, Warren, New Jersey 07059-2731.
Table of Contents25 Mountainview Blvd., Suite 203, Basking Ridge, NJ 07920.
There have been no public takeover offers by third parties with respect to the Company or by the Company in respect of other companies'companies’ shares during the last or current fiscal year.
| B. | Business overview |
We are a late-stage drug development company specialized in oncology and endocrine therapy.
Our pipeline encompasses compounds at all stages of development, from drug discovery through to marketed products. The highest development priorities in oncology are ourthe completion of Phase 3 programtrials with perifosine in colorectal cancer (“CRC”) and in multiple myeloma and colorectal cancer, combined with our Phase 2 program in multiple cancers,(“MM”), as well as the further advancement of AEZS-108, for which we recentlyhave successfully completed with success a Phase 2 trial in advanced endometrial and advanced ovarian cancer. We are planning for the initiation of a pivotal program with AEZS-108 in endometrial cancer and also a Phase 2 trial in triple-negative breast cancer. AEZS-108 is also in development in other cancer indications, including castration- and taxane-resistant prostate cancer, as well as refractory bladder and castration refractory prostate cancer. In endocrinology, our lead program is our Phase 3 trial with AEZS-130 (Solorel®) as a GH stimulation test for the diagnosis of GH deficiency
Our pipeline also encompasses other earlier-stage programs in adults. We are advancing this Phase 3 trial with a Special Protocol Assessment ("SPA") obtained from the FDA.
Additionally, we are advancingoncology. AEZS-112, an oral anticancer agent which involves three mechanisms of action (tubulin, topoisomerase II and angiogenesis inhibition) inhas completed a Phase 1 trial in advanced solid tumors and lymphoma. Additionally, several novel targeted potential anti-cancer candidates such as AEZS-120, a live recombinant oral tumor vaccine candidate, as well as severalour phosphoinositide 3-kinase (“PI3K”)/Erk inhibitors AEZS-129, AEZS-131, AEZS-132 and their respective follow-up compounds are currently in preclinical programsdevelopment.
Our lead program in endocrinology, a Phase 3 trial under a Special Protocol Assessment (“SPA”) obtained from the FDA with novel targeted potential development candidates.AEZS-130 as an oral diagnostic test for adult growth hormone deficiency (“AGHD”), has been completed with positive results. We are planning to file an NDA for the registration of AEZS-130 in the United States, subject to a successful pre-NDA meeting with the FDA. Furthermore, AEZS-130 is in a Phase 2A trial for the treatment of cancer cachexia.
Recent Developments
For a complete description of our recent corporate and pipeline developments, refer to "Item“Item 5. — Operating and Financial Review and Prospects — Highlights"Highlights”.
Our Business Strategy
Our primary business strategy is to advance, with the collaboration of our strategic partners, our product development pipeline with a focus on our flagship product candidates in oncology and endocrinology. In addition, we also continue to advance certain other clinical and pre-clinicalpreclinical programs as described below.below, some of which are conducted through grants from various governmental agencies. Our vision is to become a fully-integrated specialty biopharmaceutical company.
Our product pipeline
Pipeline table
Status of our drug pipeline as at March 27, 2012 | ||||||||||
| Discovery | Preclinical | Phase 1 | Phase 2 | Phase 3 | Commercial | |||||
~120,000 compound library | AEZS-120 Prostate cancer vaccine (oncology) AEZS-129, 131, 132 and 136; Erk & PI3K inhibitors (oncology) AEZS-137 (Disorazol Z) (oncology) AEZS-125 (LHRH- Disorazol Z) (oncology) | AEZS-112 (oncology) | Perifosine • Multiple cancers AEZS-108 • Endometrial cancer • Triple-negative breast cancer • Ovarian cancer • Castration-and taxane-resistant - prostate cancer • Refractory bladder cancer Ozarelix • Prostate cancer AEZS-130 • Therapeutic in cancer cachexia | Perifosine • Refractory advanced colorectal cancer • Multiple myeloma AEZS-130 • Diagnostic in adult growth hormone deficiency (endocrinology) | Cetrotide® (in vitro fertilization) | |||||
Partners | ||||||||||
Perifosine: Keryx North America Handok Korea Yakult Japan Hikma Middle East/ North Africa Ozarelix: Spectrum World (ex-Japan for oncology indications, ex-Korea and ex-other Asian countries for BPH indication) Handok Korea and other Asian countries for BPH indication Nippon Kayaku Japan for oncology indications | Perifosine: Keryx North America Handok Korea Yakult Japan Hikma Middle East/ North Africa | Cetrotide®: Merck Serono World except Japan Nippon Kayaku / Shionogi Japan | ||||||||
Oncology
Our highest priorities in oncology are ourwith perifosine, to complete the two Phase 3 program with perifosinetrials in multiple myelomaCRC and colorectal cancer, combined within MM, and to continue our Phase 2 program in multiple cancers, as well as the further advancement of AEZS-108, for which recentlywe successfully completed with success a Phase 2 trialtrials in advanced endometrial and advanced ovarian cancer. AEZS-108 is also in development in other cancer indications, including refractory bladder and castration refractory prostate cancer.
Perifosine
Perifosine is a novel, oral anticancer treatment that inhibits Akt activation in the PI3K pathway. Perifosine, in combination with chemotherapeutic agents, is currently in Phase 3 studies for the treatment of multiple myeloma,metastatic colorectal cancer (“mCRC”), MM and in Phase 2 studies for the treatment of other cancers, andcancers. We believe perifosine is the most advanced anti-canceranticancer compound of its class in late-stage development. Perifosine as monotherapy is also being explored in other indications.chronic lymphocytic leukemia (“CLL”). The FDA has granted perifosine orphan-drug designation in multiple myelomaMM and in neuroblastoma and Fast Track designations in both multiple myelomaMM and refractory advanced colorectal cancer.CRC. Additionally, an agreement was reached with the FDA to conduct the Phase 3 trials in both of these indications under ana SPA. Perifosine has also been granted Orphan Medicinal Product designation from the European Medicine Agency ("EMA"(“EMA”) in multiple myeloma,MM, and has received positive Scientific Advice from the EMA for both the multiple myelomaMM and advanced colorectal cancerCRC programs, with ongoing Phase 3 trials for these indications expected to be sufficient for registration in Europe.
Table of Contents Perifosine rights have been licensed to Keryx for North America, to Yakult for Japan, to Handok Pharmaceuticals Co. Ltd. (“Handok”) for Korea and to Hikma Pharmaceuticals PLC (“Hikma”) for the Middle East and North Africa (“MENA”) region.
AEZS-108
AEZS-108 represents a new targeting concept in oncology leading to personalized medicine using a cytotoxic peptide conjugate which is a hybrid molecule composed of a synthetic peptide carrier and doxorubicin. The design of AEZS-108 allows for the specific binding and selective uptake of the cytotoxic conjugate by luteinizing hormone-releasing hormone releasing hormone ("LHRH"(“LHRH”)-receptor-positive receptor-positive tumors. Phase 2 trials in advanced endometrial cancer and advanced ovarian cancer have been completed with success.successfully completed. AEZS-108 is also in development in other cancer indications, including refractory bladder and castration refractorycastration-and taxane-resistant prostate cancer. A pivotal trial in endometrial cancer and also a Phase 2 trial in triple-negative breast cancer are expected to be initiated in 2012. We have obtained orphan-drug status for AEZS-108 in advanced ovarian cancer from the FDA and from the Committee for Orphan Medicinal Products (“COMP”) of the EMA. An IND in the United States is in place for the treatment of prostate, bladder and triple-negative breast cancer. We own the worldwide rights to AEZS-108 and also have a collaboration agreement with Ventana Medical Systems, Inc. (“Ventana”), a member of the Roche Group, to develop a companion diagnostic for the immunohistochemical determination of LHRH-receptor expression, for AEZS-108.
Endocrinology
In endocrinology, aside from Cetrotide®Cetrotide®, we reactivated thecompleted a Phase 3 trial with AEZS-130, (Solorel®) as anwhich would be the first oral growth hormone ("GH") stimulationdiagnostic test for the diagnosis of adult growth hormone deficiency ("AGHD").AGHD.
AEZS-130/Solorel®AEZS-130
AEZS-130,/Solorel® (macimorelin), a ghrelin agonist, is aan orally available novel synthetic small molecule that stimulates the secretion of growth hormone. The product is currently inWe completed a Phase 3 for use astrial under a simple oral diagnostic test for AGHD. Solorel®SPA obtained from the FDA and after conclusion of a successful pre-NDA meeting with the FDA, we plan to file a NDA in the United States. AEZS-130 has been granted orphan-drug designation by the FDA. In addition to the diagnostic indication, we believe that AEZS-130 based on the results of Phase 1 studies, has potential applicationsapplication for the treatment of cachexia, a condition frequently associated with severe chronic diseases such as cancer, chronic obstructive pulmonary disease and Acquired Immune Deficiency Syndrome or AIDS.(“AIDS”). Furthermore, the FDA agreed to allow for the initiation of a physician sponsored IND Phase 2A trial in cancer induced cachexia. The study is currently conducted under a cooperative research and development agreement (“CRADA”) with the Michael E. DeBakey Veterans Affairs Medical Center which will be funding the study.
Clinical and Preclinical Programs
Additionally, we are advancing AEZS-112 in Phase 1, AEZS-112, an oral anticancer agent which involves three mechanisms of action, (tubulin, topoisomerastopoisomerase II and angiogenesicangiogenesis inhibition), as well as several preclinical programs with targeted potential development candidates. Among the targets for whichagents that we expect to proposereach the clinical development candidatesstage in the coming years are: AEZS-120 (prostate(oral prostate cancer vaccine), AEZS-127 (erucylphosphocholine derivatives), AEZS-129, AEZS-131 and AEZS-132 or their respective follow-up compounds (Erk and PI3K inhibitors), AEZS-115 (non-peptide LHRH antagonists) and AEZS-123 (ghrelin receptor antagonist).
We also continue to perform targeted drug discovery activities from which we are able to derive pre-clinicalpreclinical candidates. This drug discovery includes high throughput screening systems and a library of more than 120,000 compounds.
We are currently in a stage in which some of our products and product candidates are being further developed or marketed jointly with strategic partners.
Table of Contentspartners or with fundings from governmental organizations.
Our product pipeline
Pipeline table
| 1.1 | SIGNAL TRANSDUCTION INHIBITORS |
ONCOLOGY
SIGNAL TRANSDUCTION INHIBITORS
Perifosine
Perifosine is a novel, oral anticancer treatment that inhibits Akt activation in the PI3K pathway.
Perifosine is an alkylphosphocholine compound with structural similarity to phospholipids, which are the main constituents of cellular membranes, and it is an active ingredient with anti-tumorantitumor capacities. In tumor cells, perifosine has demonstrated interactions with vital signal transduction mechanisms and induction of programmed cell death (apoptosis).
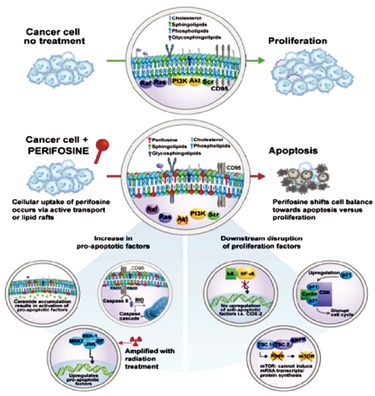
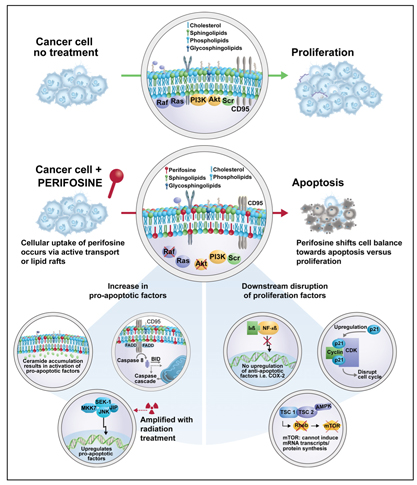
Perifosine exerts a marked cytotoxic effect in animal and human tumor cell lines. The most sensitive cancer cell lines were larynx carcinoma, breast, small cell lung, prostate and colon. Based on thein vitro trials, the mode of action of perifosine appears to be fundamentally different from that of currently available cytotoxics.
Pharmacodynamic data have demonstrated that perifosine possesses anti-tumorantitumor activity, including tumor models that are resistant to currently available agents for cancer therapy. This activity is based on a direct and relatively specific action on tumors.
In preclinical and clinical Phase 1 trials (solid tumors), this orally administered agent has been found to have good tolerability. Five Phase 1 trials have been conducted on perifosine, including one trial of perifosine in combination with radiotherapy.
Based on findings in various tumor models, the U.S. National Cancer Institute (“NCI”), along with our North American partner, Keryx, investigated additional dosage regimens of perifosine in oncology patients. A number of screening Phase 2 studies examined perifosine as a single agent or in combination in several tumor types. Encouraging results lead to further development in specific indications.
Perifosine, in combination with chemotherapeutic agents, is currently in Phase 3 studies for the treatment of multiple myeloma, colorectal cancerCRC and MM, and in Phase 2 studies for the treatment of other cancers, and we believe is the most advanced anti-canceranticancer compound of its class in late-stage development.
Perifosine as monotherapy is also being explored in other indications.CLL. The FDA has granted perifosine orphan-drug designation in multiple myelomaMM and in neuroblastoma and Fast Track designations in both multiple myeloma and refractory advanced colorectal cancer.CRC and MM. Additionally, an agreement was reached with the FDA to conduct the Phase 3 trials in both of these indications under ana SPA. Perifosine has also been granted Orphan Medicinal Product designation from the EMA in multiple myeloma,MM, and has received positive Scientific Advice from the EMA for both the multiple myelomaadvanced CRC and advanced colorectal cancerMM programs, with ongoing Phase 3 trials for these indications expected to be sufficient for registration in Europe. Perifosine rights have been licensed to Keryx for North America, to Handok for Korea, and recently to Yakult for Japan.Japan and Hikma for the MENA region.
On July 12, 2011, the European Patent Office (“EPO”) granted a patent for the use of alkylphosphocholines, more specifically perifosine, in the preparation of a medicament for the treatment of benign and malignant tumours, prior to and/or during the treatment with approved antitumor antimetabolites such as 5-fluorouracil (“5-FU”) and capecitabine. Filed on July 29, 2003, the patent (EP #1 545 553) entitled,Use of Alkyl Phosphocholines in Combination with Anti-Tumours Medicaments, became effective as of July 13, 2011, following its announcement in the European Patent Bulletin, and will expire on July 28, 2023.
1.1.1.1 Perifosine — Anti-cancerCRC
Phase 2 trial – perifosine + capecitabine
On October 5, 2011, we announced that a manuscript, entitledRandomized Placebo-Controlled Phase 2 Trials of Perifosine Plus Capecitabine as Second- or Third-Line Therapy in Patients with Metastatic Colorectal Cancer, had been published in the October 3, 2011 online edition of theJournal of Clinical Oncology (“JCO”).
This randomized, double-blind, placebo-controlled study was conducted at 11 centers across the United States. Patients with 2nd or 3rd line mCRC were randomized to receive capecitabine (Xeloda®), an approved drug for mCRC, at a dose of 825 mg/m2 BID (total daily dose of 1,650 mg/m2) on days 1 - 14 every 21 days, plus either perifosine or placebo at 50 daily. Treatment was continued until progression. The study enrolled a total of 38 patients, 34 of which were third-line or greater. Of the 38 patients enrolled, 35 were evaluable for response (20 patients on the perifosine + capecitabine arm and 15 patients on the placebo + capecitabine arm). Of the three patients on the placebo + capecitabine arm not evaluable for response, 2 patients were not evaluable due to toxicity (days 14, 46) and 1 patient was not evaluable due to a new malignancy on day 6. All patients in the perifosine + capecitabine arm were evaluable for response.
The patients in the study were heavily pretreated, with the arms well-balanced in terms of prior treatment regimens. The median number of prior treatment regimens for all 38 patients was two (range 1-5), with prior treatment regimens as follows: 89% of the patients received FOLFIRI (irinotecan + 5-FU + leucovorin); 74% FOLFOX (oxaliplatin + 5-FU + leucovorin); 66% were previously treated with both FOLFIRI and FOLFOX; 79% received Avastin®; and 50% Erbitux®. Prior treatment with single agent capecitabine was excluded.
The primary endpoints of this study were to measure 1) time to progression (“TTP”), 2) overall response rate (“ORR”), defined as the percentage of patients achieving a complete response (“CR”) or partial response (“PR”) by Response Evaluation Criteria in Solid Tumors (“RECIST”), and 3) clinical benefit rate (“CBR”) defined as the percentage of patients on treatment for greater than three months with at least stable disease (“SD”). Safety of perifosine + capecitabine vs. placebo + capecitabine in this patient population was evaluated as a secondary endpoint.
Best response and median TTP of perifosine + capecitabine vs. placebo + capecitabine were as follows:
Group |
N* |
CR |
PR
|
ORR |
SD > 12 |
CBR |
Median TTP | |||||||||||||||||||
| Perifosine + capecitabine | 20 | 1(5%) | 3(15%) | 4(20%) | 11 weeks (55%) | 15(75%) | 27.5 weeks (95% CI**, 12.1 to 48.1) | |||||||||||||||||||
| Placebo + capecitabine | 15 | 0 | 1 (7%) | 1(7%) | 5 weeks (33%) | 6(40%) | 10.1 weeks (95% CI, 6.6 to 13.0) | |||||||||||||||||||
* N = Number of patients
** CI = Confidence interval
Perifosine — Multiple myeloma ("MM"+ capecitabine more than doubled TTP vs. placebo + capecitabine with a statistically significant p-value <.001. In addition, perifosine + capecitabine more than doubled the ORR and almost doubled the CBR vs. placebo + capecitabine.
Although not a primary endpoint in the study, overall survival (“OS”) was analyzed with results as follows:
In June
Group | Median OS | |
Perifosine + capecitabine | 17.7 months (95% CI, 8.5 to 24.6) | |
Placebo + capecitabine | 7.6 months (95% CI, 5.0 to 16.3) |
Of notable interest were data showing a highly statistically significant benefit in median OS (more than doubling) and December 2007, preliminary positive PhaseTTP for the subset of patients who were refractory to a 5-FU chemotherapy-based treatment regimen. 5-FU is a core component of the standard of care FOLFIRI and FOLFOX regimens, and capecitabine is a 5-FU pro-drug. These results are shown below:
Group | 5-FU Ref* | > SD (min 12 weeks) | Median TTP | Median OS | ||||||
| Perifosine + capecitabine | 14(70%) | 1 PR /8 SD (64%) | 17.6 weeks (95% CI, 12 to 36) | 15.1 months (95% CI, 7.2 to 22.3) | ||||||
| Capecitabine | 11(73%) | 0 PR /3 SD (27%) | 9.0 weeks (95% CI, 6.6 to 11.0) | 6.5 months (95% CI, 4.8 to 10.9) | ||||||
| * | Ref= refractory |
All 38 patients were evaluable for safety. The perifosine + capecitabine combination was well-tolerated with Grade 3 and Grade 4 adverse events of > 10% incidence for perifosine + capecitabine arm versus capecitabine arm as follows: anemia (15% vs. 0%), fatigue (0% vs. 11%), abdominal pain (5% vs. 11%) and hand-foot syndrome (30% vs. 0%). Of note, incidence of Grade 1 and Grade 2 hand-foot syndrome was similar in both the perifosine + capecitabine and capecitabine arms (25% vs. 22%, respectively). Hand-foot syndrome is a reported
adverse event with capecitabine monotherapy. Patients who remained on treatment longer in the Phase 2 study had a greater chance to develop hand-foot syndrome as illustrated by a median time to onset of Grade 3 and Grade 4 hand-foot syndrome in the perifosine + capecitabine arm of 19 weeks.
Based on the data, in which the combination of perifosine + capecitabine demonstrated statistical significance with respect to median OS and median time to tumor progression, the investigators concluded that the perifosine + capecitabine combination showed promising clinical activity compared to single-agent capecitabine, and that the difference in clinical outcome seen with the addition of perifosine was impressive.
Phase 3 trial and regulatory milestones
On February 3, 2010, we announced that our partner Keryx had reached an agreement with the FDA on a SPA for the Phase 3 Xeloda® + Perifosine Evaluation in Colorectal Cancer Treatment (“X-PECT”) trial for perifosine were presented in patients with relapsed/refractory MM.mCRC.
On April 5, 2010, our partner Keryx was granted Fast Track designation by the FDA for the Phase 3 X-PECT registration trial.
On April 8, 2010, we announced that our partner Keryx initiated a randomized (1:1), double-blind Phase 3 X-PECT trial comparing the efficacy and safety of perifosine + capecitabine (Xeloda®) vs. placebo + capecitabine in approximately 430 patients with refractory mCRC. Patients must have failed available therapy including 5-FU, oxaliplatin (Eloxatin®), irinotecan, bevacizumab (Avastin®) and, if K-Ras wild-type, failed therapy with prior cetuximab (Erbitux®) or panitumumab (Vectibix®). For oxaliplatin-based therapy, failure of therapy also includes patients who discontinued due to toxicity. The primary endpoint is OS, with secondary endpoints including ORR (CR + PR), progression-free survival (“PFS”) and safety. Approximately 70 U.S. sites are participating in the study and un-blinding of the study will be triggered by 360 events of death. Dr. Johanna Bendell, Director of GI Oncology Research for the Sarah Cannon Research Institute, Nashville, Tennessee, leads the Phase 3 investigational team. Patient recruitment for this study was completed in July 2011.
On June 29, 2010, we announced that we had received positive Scientific Advice from the EMA regarding the Phase 3 X-PECT trial for the development of perifosine in refractory advanced CRC. The Scientific Advice from the EMA indicates that the ongoing study, in conjunction with safety data generated from other clinical studies with perifosine, is considered sufficient to provide all data necessary to support a marketing authorization of perifosine in advanced CRC. We do not intend to initiate any additional studies with perifosine for this indication. Therefore, for the development of perifosine in both MM and CRC, we believe that the planned North American clinical program, sponsored by our partner Keryx, is now sufficient for approval in Europe and in many countries in the rest of the world, where we hold rights for our compound.
On July 27, 2011, we announced completion of patient recruitment for the ongoing Phase 3 trial with perifosine in refractory advanced CRC, involving over 465 patients from 65 sites in the United States. This Phase 3 X-PECT trial is a randomized (1:1), double-blind trial comparing the efficacy and safety of perifosine + capecitabine vs. placebo + capecitabine. The primary endpoint is OS, with secondary endpoints including ORR (CR + PR), PFS and safety. Approximately 360 events of death will trigger the unblinding of the study.
On August 31, 2011, an independent Data demonstrated clinical activitySafety Monitoring Board (“DSMB”) completed an interim safety and futility analysis of the Phase 3 X-PECT study of perifosine in patients with refractory advanced CRC and recommended that the study continue to completion, as planned.
On January 3, 2012, we announced that our Japanese partner, Yakult, had initiated a Phase 1/2 trial in Japan to assess the safety and efficacy of perifosine in combination with bortezomiba chemotherapeutic agent, capecitabine, in patients with refractory advanced CRC. The primary endpoint of the Phase 1 portion of the trial is the safety profile of perifosine in combination with capecitabine. The primary endpoint of the Phase 2 portion is efficacy (Disease Control Rate). The initiation of this trial on December 27, 2011 triggered a milestone receivable of $2.6 million (according to the agreement signed in March 2011 for perifosine in Japan).
Competitors for Perifosine in CRC
Products on the market:
Surgery is often the main treatment for early stage CRC. When CRC metastasizes (spreads to other parts of the body such as the liver), chemotherapy is commonly used. Treatment of patients with recurrent or advanced CRC depends on the location of the disease. Currently, there are seven approved drugs for patients with mCRC: 5-FU, capecitabine (Xeloda®), irinotecan (Camptosar®), oxaliplatin (Eloxatin®), bevacizumab (Avastin®), cetuximab (Erbitux®), and dexamethasone,panitumumab (Vectibix®). Depending on the stage of the cancer, two or more of these types of treatment may be combined at the same time, such as FOLFOX (5-FU; leucovorin; oxaliplatin) and FOLFIRI (5-FU; leucovorin; irinotecan), or used after one another. Bevacizumab, a vascular endothelial growth factor (“VEGF”) monoclonal antibody, is commonly administered with lenalidomide (Revlimid®chemotherapy. Typically, patients who fail 5-FU, oxaliplatin, irinotecan, and bevacizumab-containing therapies, and who have wild-type KRAS status receive epidermal growth factor receptor (“EGFR”) + dexamethasone.monoclonal antibody therapy with either cetuximab or panitumumab. Once patients progress on these agents, there are no further standard treatment options.
Product / mode of action | Company | Sales | ||
| Avastin® (bevacizumab) / a humanized monoclonal antibody targeting vascular endothelial growth factor | Manufactured by Genentech/Roche | According to Decision Resources (Jan. 2012), a research and advisory firm focusing on pharmaceutical and healthcare issues (“Decision Resources (January 2012)”) the 2010 G7* sales were estimated to be $2.3 billion | ||
| Oxaliplatin / platinum agent | Sanofi’s Eloxatin/Eloxatine, Yakult Honsha’s Elplat, generics | According to Decision Resources (January 2012), the 2010 G7* sales were estimated to be $2.0 billion | ||
| Erbitux® (cetuximab) / a chimeric monoclonal antibody that specifically blocks EGFR | Manufactured and distributed in North America: Bristol-Myers Squibb Co. and Eli Lilly and Co. Distributed in the rest of the world by Merck KGaA. | According to Decision Resources (January 2012), the 2010 G7* sales were estimated to be $1.1 billion | ||
| Xeloda® (capecitabine) / oral fluoropyrimidine which generates fluorouracil preferentially in tumor tissues by enzymatic cascade | Manufactured by Roche | According to Decision Resources (January 2012), the 2010 G7* sales were estimated to be $636 million | ||
| Vectibix® (panitumumab) / a recombinant, human IgG2 kappa monoclonal antibody that binds specifically to the human EGFR | Manufactured by Amgen | According to Decision Resources (January 2012), the 2010 G7* sales were estimated to be $266.1 million |
| * | G7 = United States, France, Germany, Italy, Spain, UK and Japan. |
Products in Phase 3 development:
According to Decision Resources (January 2012), the most promising emerging therapies for CRC are the following: perifosine, regorafenib, aflibercept and ramucirumab.
Product / mode of action* | Company* | Development Status* | ||
| Regorafenib (BAY 73-4506) / oral multi-kinase inhibitor targeting angiogenic, stromal and oncogenic kinases | Bayer HealthCare Pharmaceuticals / Onyx Pharmaceuticals, Inc. | Phase 3 completed | ||
| Aflibercept (ZaltrapTM) / intravenous angiogenesis inhibitor | Sanofi / Regeneron Pharmaceuticals | Regulatory applications for marketing approval submitted (FDA and EMA) | ||
| Ramucirumab (IMC-1121B) / intravenous fully human immunoglobulin G1 Mab targeted to VEGF-2 | ImClone Systems (a subsidiary of Eli Lilly) | Phase 3 |
* Source: Company’s Website
Market Data — CRC
According to the American Cancer Society, CRC is the third most common cancer in both men and women in the United States. An estimated 103,170 cases of colon and 40,290 cases of rectal cancer are expected to occur in 2012 and nearly 51,690 people will die from the disease.
According to Decision Resources (January 2012), 368,000 patients will be diagnosed in Stage II-IV in the G7 market in 2014. In 2010, the total market for stage IV-second-line CRC treatments accounted for 25% of major-market sales ($1.8 billion) and 5% ($360 million) for the total market for the third-line CRC treatment.
Treatable pool stage IV patient population*
| ||||
| 3rd line in 2014 | 2nd line in 2014 | |||
USA | 28,320 | 41,650 | ||
EU-5 | 48,070 | 74,480 | ||
Japan | 19,750 | 35,270 | ||
* According to Decision Resources (January 2012).
1.1.1.2 Perifosine — MM
Phase 1/2 trial – Perifosine in combination with Revlimid® (lenalidomide) + dexamethasone
In December 2008, our partner Keryx presented final results of the Phase 1 clinical trial in which patients with relapsed or refractory MM were administered a combination of perifosine + lenalidomide and+ dexamethasone. Four cohorts of³6 patients each were enrolled and perifosine dose was 50 or 100 mg (daily), lenalidomide dose was 15 or 25 mg for days 1 to 21 and dexamethasone dose was 20 mg (for days 1-4; 9-12; and 17-20 for 4 cycles, followed bythen 20 mg for days 1-4) in 28-day cycles. To limit dexamethasone-related toxicities, the protocol was amended to use weekly dexamethasone (40 mg), applying to cohorts 3, 4, and the Maximal Tolerated Dose ("MTD"maximal tolerated dose (“MTD”) cohort. Dose Limiting Toxicity ("DLT"limiting toxicity (“DLT”) was defined as grade (G)Grade 3 non-hematologic toxicity, G4Grade 4 neutropenia for 5 days and/or neutropenic fever, or platelets <25,000/mm3 on >1 occasion despite transfusion. Response was assessed by modified EBMT criteria. To be enrolled, patients had to have received at least one but no more than four prior therapies. Patients refractory to lenalidomide/dexamethasone were excluded. 32 patients (17 men and 15 women,
median age 61 years old, range 37-80) were enrolled; 6 patients in cohort 1 (perifosine 50 mg, lenalidomide 15 mg, dexamethasone 20 mg); 6 patients in cohort 2 (perifosine 50 mg, lenalidomide 25 mg, dexamethasone 20 mg); 8 patients in cohort 3 (perifosine 100 mg, lenalidomide 15 mg, dexamethasone 40mg/40 mg/week); 6 patients in cohort 4 (perifosine 100 mg, lenalidomide 25 mg, dexamethasone 40 mg/week) and 6 patients at MTD (Cohort 4). Median prior lines of treatment was 2 (range 1-4). Prior therapy included dexamethasone (94%), thalidomide (83%), bortezomib (47%), and stem cell transplant (47%). 37% of patients had progressed on prior thalidomide/dexamethasone. Two patients did not complete one full cycle (non-compliance and adverse event not related to study drugs — both in cohort 3) and were not included in the safety and efficacy analysis. Of the 30 patients evaluable for safety, the most common (³10%) gradeGrade 1 / 2 events included nausea (13%); diarrhea (17%); weight loss (17%); upper respiratory infection (23%); fatigue (30%); thrombocytopenia (20%); neutropenia (20%); hypophosphatemia (23%); increased creatinine (23%); anemia (36%); hypercalcemia (47%). Grade 3 / 4 adverse events³5% included neutropenia (20%); hypophosphatemia (17%); thrombocytopenia (13%); anemia (10%), fatigue (7%). There was one reported DLT in cohort 3 (nausea). Lenalidomide was reduced in 8 patients, perifosine reduced in
8 patients and dexamethasone reduced in 6 patients. All 30 patients in the analysis were evaluable for response, with best response as follows:
| ||||||||||
| ||||||||||
| ||||||||||
| ||||||||||
| ||||||||||
| ||||||||||
| ||||||||||
Patients have tolerated the treatment regimen of perifosine + lenalidomide + dexamethasone well with manageable toxicity, and with encouraging clinical activity demonstrated by an overall response rate ("ORR")ORR (> PR) of 50%.
Updated results of this Phase 1 study were presented in February 2009 at the 12th International Multiple Myeloma Meeting by our partner Keryx. Results indicated that Perifosineperifosine in combination with lenalidomide (Revlimid®(Revlimid®) + dexamethasone continues to be well tolerated, with a median progression-free survivalPFS in responding patients of 10.9 months. Median overall survivalOS still was not reached and was at 17 months at time of analysis.
Also inOn December 2008 during the meeting of6, 2010 at the American Society of Hematology, Hematology’s (“ASH”) 52nd annual meeting in Orlando, Florida, we announced final positive results for this Phase 1 trial. The final data showed a 73% objective response rate (minimal response or better) with a 50% PR or better, a median PFS of 10.8 months, and a median duration for OS of 30.6 months. The myeloma investigators concluded that perifosine in combination with lenalidomide (Revlimid®) + dexamethasone was well tolerated even at the highest doses used, and demonstrated encouraging clinical activity and survival.
Best Response (N = 30 pts) |
N (%) |
Duration on Tx (months) Median (range)
|
³ PR |
³ MR | ||||||||
Near complete response (nCR)
| 4(13%) | 32+, 32+, 28, 6 |
|
50% |
|
|
73% |
| ||||
Very good partial response (VGPR)
| 3(10%) | 35, 7, 4 | ||||||||||
Partial response (PR)
| 8(27%) | Median 5.5 (4 – 29) | ||||||||||
Minimal reponse (MR)
| 7(23%) | Median 12 (2 – 34) | ||||||||||
Stable disease (SD)
| 6(20%) | Median 3 (2 – 30) | ||||||||||
Progressive disease (PD)
| 2(7%) | 9, 4 weeks | ||||||||||
Phase 1/2 trial – Perifosine in combination with Velcade® (bortezomib) + dexamethasone
Keryx presented preliminary results of a Phase 1/2 multicenter trial of perifosine + bortezomib (Velcade®(Velcade®) in patients with relapsed or relapsed/refractory MM who were previously relapsed from or refractory to bortezomib ± dexamethasone.dexamethasone in December 2008 during the meeting of the ASH and in February 2009 at the 12th International Multiple Myeloma Meeting. Final results were presented during the ASH meeting in December 2009. The Phase 1 stage of the study enrolled a total of 18 patients in 4 cohorts of 3 patients each with dosing of perifosine
50 mg or 100 mg (daily) and bortezomib 1.0 or 1.3 mg/m2 (on day 1, 4, 8, 11) in 21-day cycles. The selected dose for Phase 2 was perifosine 50 mg once daily + bortezomib 1.3 mg/m2 (on day 1, 4, 8, 11) in 21-day cycles, with a planned enrollment of 64 patients. Dexamethasone 20 mg (on day of and after each bortezomib dose) could be added in patients with progressive disease ("PD").PD. For the Phase 1 portion, DLT was defined as any grade (G)Grade 3 non-hematologic toxicity, G4Grade 4 neutropenia for 5 day and/or neutropenic fever, or platelets <10,000/mm3 on more than one occasion despite transfusion. Response was assessed by modified EBMT and Uniform criteria. A total
On October 13, 2011, we announced that the manuscript, entitledPerifosine Plus Bortezomib and Dexamethasone in patients with Relapsed/Refractory Multiple Myeloma Previously Treated with Bortezomib: Results of 76 patients havea Multicenter Phase 1/2 Trial, had been enrolled (18 patients in Phase 1 and 58 patients in Phase 2) comprised of 45 men and 31 women, median age 63 years old, (range 41-89). 84% of patients had relapsed/refractory MM, with a median of 6 lines of prior treatment (range 2-13). Prior therapy included bortezomib (100%), dexamethasone (95%), thalidomide (79%), lenalidomide (71%) and stem cell transplant (57%). 63 patients have completed at least one cycle and were evaluable for safety (13 patients are currently not evaluable; 3 were removed in cycle 1 and 10 are too early in their treatment). Most common (>10%) grade 1 / 2 events were nausea, diarrhea, fatigue and myelosuppression, which were manageable with supportive care and growth factors. Grade 3 / 4 adverse events >5% included thrombocytopenia (40%); lymphopenia (36%); neutropenia (21%); anemia (14%); hyponatremia (13%); leukopenia (11%); proteinuria (8%), and upper respiratory infection (6%). No deep vein thrombosis has been seen, and only one worsening peripheral neuropathy from grade 1 to 3 has been reported to date. Two patients had perifosine reduced to 50 mg (nausea, fatigue)published in the Phase 1 cohort, and 7 patients had bortezomib dose reductions primarily due to hematologic
toxicity. 57 patients had completed at least 2 cycles and were evaluable for response, with best response to perifosine + bortezomib (+/ - dexamethasone) as follows:
| | | CR | PR | MR | ORR | SD | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| All Patients: Best Response | N=57 | 2 | 4% | 7 | 12% | 14 | 25% | 23 | 40% | 23 | 40% | |||||||||||||||||||||||
Perifosine + bortezomib | 57 | 1 | 2% | 5 | 9% | 8 | 14% | 14 | 24% | 17 | 30% | |||||||||||||||||||||||
With dexamethasone added* | 31 | 1 | 2% | 2 | 3% | 6 | 11% | 9 | 16% | 6 | 11% | |||||||||||||||||||||||
(* as a subsetOctober 10, 2011 online edition of the evaluable population)
9Journal of 76 patients (12%) rapidly progressed without response or stable disease ("SD")Clinical Onclogy (JCO), including 6 patients in whom dexamethasone was also added. As at August 2008, the median time to progression ("TTP") for patients achieving³PR is 34 weeks, and for all patients achieving³MR is 33 weeks. Perifosine inwhich Phase 1/2 combination with bortezomib (+/- dexamethasone) was generally well tolerated and is active in a heavily pre-treated bortezomib-exposed patient population, with an ORR of 40%, including an ORR of 37% and a median TTP of 9.25 months in responding but previously bortezomib-refractory patients.
Updated data for the effectactivity of perifosine in combination with bortezomib (Velcade®) +/- dexamethosone were reported at the 12th International Multiple Myeloma Meeting in February 2009 by our partner Keryx. treatment of advanced MM patients was reported.
Eighty-four patients were enrolled in a combined Phase I/II1/2 study (18 patients in the Phase I1 component and 66 patients in the Phase II2 component). At the time of this analysis, 73 patients were evaluable for response. Median prior lines of therapy was 5 (range 1 - 13), including 74 (88%) with relapsed/refractory MM. Sixty-one patients (73%) were bortezomib refractory and 43 (51%) were refractory to bortezomib with dexamethasone. The median number of prior treatments was five (range, one to 13 treatments). Prior therapies received at any time point included bortezomib (100%; 50%), with patients receiving a median of the patients were previously treated with at least 2two (range, one to four) prior bortezomib-based therapies and 81% were previously treated with bortezomib + dexamethasone);regimens; dexamethasone (98%); lenalidomide (Revlimid®(Revlimid®) and/or(76%); thalidomide (Thalomid) (99%(Thalomid®) (75%); and prior stem cell transplant (57%(58%). No unexpected adverse events have been seen. Toxicities were manageable with supportive care and/or dose reductions as required.
Best response (MR or better) and stable disease (no progression for 3 months) to either perifosine + bortezomib (+/- dexamethasone) for patients previously relapsed from or refractory to prior bortezomib (Velcade®) treatment was as follows:
| Evaluable Patients | CR | PR | MR | ORR | SD> 3 mos | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Bortezomib relapsed (n=20) | 2 | 10% | 6 | 30% | 3 | 15% | 11 | 55% | 9 | 45% | |||||||||||||||||||||
Bortezomib refractory (n=53) | 1 | 2% | 6 | 11% | 10 | 19% | 17 | 32% | 24 | 45% | |||||||||||||||||||||
All evaluable patients (n=73) | 3 | 4% | 12 | 16% | 13 | 18% | 28 | 38% | 33 | 45% | |||||||||||||||||||||
Patients who had previously relapsed on a bortezomib-based treatment had a median TTP of 8.5 months. The median TTP for all 73 evaluable study patients (both bortezomib relapsed and refractory) was 6.4 months. As stated in Keryx's February 26, 2009 press release, there were 16 patients who remained at the time on active treatment.
Updated efficacy and safety data as well as new survival data on the clinical activity of perifosine in combination with bortezomib (Velcade®) +/- dexamethasone in patients with relapsed/refractory multiple myeloma were presented by our partner Keryx during the American Society of Hematology ("ASH") meeting in December 2009. Of the 73 evaluable patients, 53 patients (73%) were previously refractory to bortezomib (defined as progression on or within 60 days of treatment to a bortezomib-based regimen), including 44 patients who were refractory to the combination of
bortezomib + dexamethasone.. Twenty evaluable patients (27%) were relapsed to a prior bortezomib-based regimen. Best response for all 73 evaluable patients was as follows:
| Evaluable Patients | CR/nCR* | PR | MR | ORR | SD** | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
All Evaluable Patients (n=73) | 3 | 4% | 13 | 18% | 14 | 19% | 30 | 41% | 30 | 41% | |||||||||||||||||||||
Bortezomib relapsed (n=20) | 2 | 10% | 7 | 35% | 4 | 20% | 13 | 65% | 7 | 35% | |||||||||||||||||||||
Bortezomib refractory (n=53) | 1 | 2% | 6 | 11% | 10 | 19% | 17 | 32% | 23 | 43% | |||||||||||||||||||||
Evaluable Patients
|
CR /nCR*
|
PR
|
MR
|
ORR
|
SD**
| |||||||||||||||||||||||||||||||||||
All Evaluable Patients (N = 73) | 3 | 4% | 13 | 18% | 14 | 19% | 30 | 41% | 30 | 41% | ||||||||||||||||||||||||||||||
Bortezomib relapsed (N = 20) | 2 | 10% | 7 | 35% | 4 | 20% | 13 | 65% | 7 | 35% | ||||||||||||||||||||||||||||||
Bortezomib refractory (N = 53) | 1 | 2% | 6 | 11% | 10 | 19% | 17 | 32% | 23 | 43% | ||||||||||||||||||||||||||||||
| * | nCR = Near Complete Response is defined as meeting the criteria for CR (non-detectable monoclonal protein by serum and urine), except with detectable monoclonal protein by immunofixation. |
| ** | SD = Stable Disease for a minimum of 3 months. |
Approximately 60% (45 / 73) of patients demonstrated progression (or SD for 4 cycles) at some point in their treatment and received 20 mg dexamethasone, four times per week, in addition to perifosine + bortezomib. Responses occurred both with patients taking perifosine in combination with bortezomib and with patients receiving the combination + dexamethasone.
Best response for each group was as follows:
| Best Response | CR/nCR | PR | MR | ORR | SD | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Perifosine + bortezomib (n=73) | 2 | 3% | 10 | 14% | 6 | 8% | 18 | 25% | 19 | 26% | |||||||||||||||||||||
Dexamethasone added (n=45) | 1 | 2% | 6 | 13% | 10 | 23% | 17 | 38% | 14 | 31% | |||||||||||||||||||||
Five patients achieved an initial response on perifosine + bortezomib alone, and subsequently responded again with the addition of dexamethasone. Three additional patients achieved stable disease on perifosine + bortezomib alone, and subsequently achieved stable disease again with the addition of dexamethasone.
Best Response
|
CR /nCR
|
PR
|
MR
|
ORR
|
SD
| |||||||||||||||||||||||||||||||||||
Perifosine + bortezomib (N = 73) | 2 | 3% | 10 | 14% | 6 | 8% | 18 | 25% | 19 | 26% | ||||||||||||||||||||||||||||||
Dexamethasone added (N = 45) | 1 | 2% | 6 | 13% | 10 | 22% | 17 | 38% | 14 | 31% | ||||||||||||||||||||||||||||||
Reported for the first time was median Progression-Free Survival ("PFS")PFS and Overall Survival ("OS")OS data for all evaluable patients, as follows:
Evaluable Patients | Median PFS* | Median OS** | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
All Evaluable Patients | 6.4 months | 25 months | |||||||||
| 16.3, 31.1)*** | |||||||||||
| * | Median PFS and median TTP were identical, as no patient deaths occurred prior to progression. |
| ** | Kaplan Meier methodology was used to determine OS figures. |
| *** | Results from the abstractPerifosine Plus Bortezomid and Dexamethasone in Relapsed/Refractory Multiple Myeloma Patients Previously Treated with Bortezomib: Final Results of a Phase 1/2 Trial (abstract # 815) were presented as an oral presentation at the ASH Annual Meeting and Exposition (Dec. 2011) in San Diego, California. |
NR = Not Reached
Of particular interest was the comparison of evaluable patients who were previously refractory and the patients who were relapsed to a bortezomib-based regimen. Median PFS and OS for bortezomib relapsed vs. refractory was as follows:
Bortezomib Relapsed vs. Refractory | Median PFS* | Median OS** | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
Bortezomib relapsed | 8.8 months | ||||||||||
| *** | |||||||||||
Bortezomib refractory | 5.7 months | 22.5 months | |||||||||
| 14.2, 31.1)*** | |||||||||||
Table of ContentsNR = Not Reached
| * | Median PFS and median TTP were identical, as no patient deaths occurred prior to progression. |
| ** | Kaplan Meier methodology was used to determine OS figures. |
| *** | Results from the abstractPerifosine Plus Bortezomid and Dexamethasone in Relapsed/Refractory Multiple Myeloma Patients Previously Treated with Bortezomib: Final Results of a Phase 1/2 Trial (abstract # 815) were presented as an oral presentation at the ASH Annual Meeting and Exposition (Dec. 2011) in San Diego, California. |
No unexpected adverse events have been observed. ToxicitiesTherapy was generally well-tolerated, and toxicities, including gastrointestinal side-effects and fatigue, proved manageable. No treatment-related mortality was seen. The investigators concluded the data reported for both safety and efficacy in this patient population were manageable with supportive care.encouraging for the continued study of perifosine.
Phase 3 trial and regulatory milestones
In August 2009, we announced that our partner Keryx reported that it had reached an agreement with the FDA regarding an SPA on the design of a Phase 3 trial for perifosine, in relapsed or relapsed/refractory multiple myelomaMM patients previously treated with bortezomib (Velcade®(Velcade®). The SPA provided agreement that the Phase 3 study design adequately addresses objectives in support of a regulatory submission.
In September 2009, we announced that our partner Keryx reported that it had received orphan-drug designation for perifosine from the FDA for the treatment of multiple myeloma.MM. Orphan-drug designation is granted by the FDA Office of Orphan Drug Products to novel drugs or biologics that treat a rare disease or condition affecting fewer than 200,000 patients in the U.S.United States. The designation provides the drug developer with a seven-year period of U.S. marketing exclusivity if the drug is the first of its type approved for the specified indication or if it demonstrates superior safety, efficacy or a major contribution to patient care versus another drug of its type previously granted the designation for the same indication.
On December 2, 2009, we announced that the FDA had granted Fast Track designation for perifosine for the treatment of relapsed/refractory multiple myeloma.MM. The Fast Track program of the FDA is designed to facilitate the development and expedite the review of new drugs that are intended to treat serious or life-threatening conditions and that demonstrate the potential to address unmet medical needs. Fast Track designated drugs ordinarily qualify for priority review, thereby expediting the FDA review process.
On December 16, 2009, we announced that our partner Keryx initiated a Phase 3 trial for perifosine entitled, "AA Phase 3 Randomized Study to Assess the Efficacy and Safety of Perifosine Added to the Combination of Bortezomib (Velcade®(Velcade®) and Dexamethasone in Multiple Myeloma Patients Previously Treated with Bortezomib"Bortezomib. The randomized (1:1), double-blind trial powered at 90%, will enroll approximately 400 patients with relapsed or
relapsed/refractory multiple myelomaMM (patients can be relapsed from and refractory to all non-bortezomib based therapies, however, patients can only be relapsed (progressed > 60 days after discontinuing therapy) from prior bortezomib-based therapies. Patients must have been previously treated with both bortezomib (Velcade®(Velcade®) and an immunomodulatory agent (Revlimid®(Revlimid® or Thalidomid®Thalidomid®) and previously treated with one to four prior lines of therapy. Enrolled patients are randomized to bortezomib (Velcade®(Velcade®) at 1.3 mg/m2 days 1, 4, 8 and 11 every 21 days in combination with dexamethasone 20 mg on the day of and day after bortezomib (Velcade®(Velcade®) treatment, and either perifosine 50 mg daily or placebo. The primary endpoint is progression-free survivalPFS and secondary endpoints include overall response rate, overall survivalORR, OS and safety.
As stated by our partner Keryx, it is expected that the study will be completed during the second half of 2012. Approximately 265 events (defined as disease progression or death) will trigger the un-blinding of the data.
In March 2010, we announced that we had received a positive opinion for orphan medicinal product designation for perifosine from the Committee for Orphan Medicinal Products ("COMP")COMP of the European Medicines Agency,EMA, for the treatment of multiple myeloma.MM. Orphan medicinal product designation is granted by the European Commission, following a positive opinion from the COMP, to a medicinal product that is intended for the diagnosis, prevention or treatment of a life-threatening or a chronically debilitating condition affecting not more than five in 10,000 persons in the European Community when the application for designation is submitted.
Orphan medicinal product designation provides the sponsor with access to the Centralized Procedure for the application for marketing authorization, protocol assistance, up to a 100% reduction in fees related to a marketing authorization application, pre-authorizationpreauthorization inspection and post-authorization activities, and could provide ten years of market exclusivity in the EU, once approved for the treatment of multiple myeloma.
On April 15, 2010, we received Positivepositive Scientific Advice (“PSA”) from the EMA for the Phase 3 registration trial with perifosine in multiple myeloma,MM, therefore indicating that the data from the ongoing trial are expected to be sufficient for product registration in Europe.
On December 6, 2010 at the ASH's 52nd annual meeting in Orlando, Florida, we announced updated positive Phase 1 results of perifosine in combination with lenalidomide (Revlimid®) + dexamethasone in patients with relapsed or refractory multiple myeloma. The final data showed a 73% objective response rate (minimal response or better) with a 50% PR or better, a median Progression-Free Survival of 10.8 months, and a median duration for Overall Survival of 30.6 months. The myeloma investigators concluded that perifosine in combination with lenalidomide (Revlimid®) + dexamethasone was well tolerated even at the highest doses used, and demonstrated encouraging clinical activity and survival.
Competitors for Perifosine in Multiple Myeloma IndicationMM
Products on the market
Major products available on the market for the treatment of multiple myelomaMM are the following:
Velcade® (bortezomib — manufactured by Millenium: The Takeda Oncology Company), a proteasome inhibitor approved in combination with melphalan (Alkeran® — Manufactured by Celgene) and prednisone as a 1st-line treatment and as a monotherapy for 2nd-line treatment in both the U.S. and the EU Millennium reported, according to Takeda's 2010 Annual Report, approximately $0.5 billion in global Velcade® estimated sales in 2009 (Velcade® is co-developed by Millennium Pharmaceuticals, Inc. and Johnson & Johnson Pharmaceutical Research & Development, L.L.C. Millennium is responsible for commercialization of Velcade® in the U.S., Janssen-Cilag is responsible for commercialization in Europe and the rest of the world. Janssen Pharmaceutical K.K. is responsible for commercialization in Japan).
Caelyx®/Doxil® (pegylated liposomal doxorubicin — Manufactured by Schering Plough), a topoisomerase II inhibitor and DNA intercalating agent, is approved as a 2nd-line treatment in combination with Velcade® in patients with advanced multiple myeloma.
Thalomid® (thalidomide — Manufactured by Celgene), an antiangiogenic compound has been approved by the FDA for use in combination with dexamethasone for the treatment of patients with newly diagnosed multiple myeloma. The Australian Therapeutic Goods Administration (TGA) approved a supplemental filing granting Thalomid® marketing approval for use in combination with melphalan and prednisone for patients with untreated multiple myeloma or ineligible for high-dose chemotherapy, and also granted Thalomid® marketing approval in combination with dexamethasone for induction therapy prior to high-dose chemotherapy with autologous stem cell rescue, for the treatment of patients with untreated multiple myeloma. In addition, Thalomid® was granted full marketing authorization by the European Commission ("EC") for use in combination with melphalan and prednisone as a treatment for patients with newly diagnosed multiple myeloma. Internationally, Thalomid® is also distributed under mandatory risk-management distribution programs tailored to meet local competent authorities' specifications to help ensure the safe and appropriate distribution and use of Thalomid®. According to Celgene's 2009 Annual Report, Thalomid® sales were down 13.4% to approximately $436.9 million in 2009.
Revlimid® (lenalidomide — Manufactured by Celgene): Revlimid® is an oral immunomodulatory drug approved by the FDA and a number of other regulatory agencies in Europe, Latin America, Middle East and Asia/Pacific for treatment in combination with dexamethasone for multiple myeloma patients who have received at least one prior therapy and in Australia and New Zealand in combination with dexamethasone for the treatment of patients whose disease has progressed after one therapy. Revlimid® is distributed internationally under mandatory risk-management distribution programs tailored to meet local competent authorities' specifications to help ensure the safe and appropriate distribution and use of Revlimid®.
Revlimid® continues to be evaluated in numerous clinical trials worldwide either alone or in combination with one or more other therapies in the treatment of a broad range of hematological malignancies, including multiple myeloma, myelodysplastic syndromes ("MDS"), non-Hodgkin's lymphoma ("non-HL"), chronic lymphocytic leukemia ("CLL"), other cancers and other diseases. According to Celgene's 2009 Annual Report, Revlimid® sales were up 28.8% to approximately $1.7 billion in 2009.
Products in Phase 3 development:
Panobinostat (LBH5893) — Novartis: Panobinostat is a highly potent pan-deacetlyase inhibitor (pan-DACi) developed by Novartis. Panobistat's mechanism of action involves disrupting aggresome function, promoting accumulation of cytotoxic misfolded protein aggregates and triggering of myeloma cell death. Combination of pan-DAC and protease inhibition by co-treatment with panobinostat and bortezomib as demonstrated synergistic cytotoxicityin vitro andin vivo in preclinical experiments. Clinical experience in advanced multiple myeloma patients treated by oral panobinostat and i.v. bortezomib +/- dexamethasone showed efficacy and manageable toxicity profile. Panobinostat is currently in Phase 3 trial in patients with relapsed multiple myeloma in combination with bortezomib.
Idamycin (Idarubicin) — Pfizer: Idarubicin is an oral anthracyclines and an analogue of daunorubicin (but 5 to 6 times more potent than daunorubicin) developed by Pfizer. The mechanism of action of anthracyclines is poorly understood and cytotoxicity is generally attributed to intercalation of the drug into DNA and inhibition of DNA topoisomerase II activity resulting in double and single strand DNA breaks. Idarubicin is already approved in Canada for Acute lymphocytic leukemia in adults and children as a second-line treatment and in Acute non-lyphocytic leukemia in adults as a front-line treatment or for refractory/relapsed disease. Idarubicin is currently in Phase 3 clinical trial for patients with Stage I or Stage II multiple myeloma in combination with dexamethasone.
Zolinza (vorinostat — MK0683) — Merck: Zolanza is an oral histone deacetylase (HDAC) inhibitor developed by Merck. Zolinza works by inhibiting the enzymatic activity of HDAC1, HDAC2, HDAC3 (Class 1) and HDAC6 (Class II). Inhibition of HDAC may result in anti-cancer effects since HDAC inhibitors, like zolinza, have the ability to induce antiproliferative effects including cyto-differentiation, cell cycle growth arrest or apoptosis in various cancer cell lines. The exact mechanism of the anticancer effect of Zolinza has not been fully characterized. Phase 1 results showed early anti-tumor activity in patients with releaspsed and/or refractory multiple myeloma when zolenza was administered in combination with bortezomib, including in patients previously treated with and no longer responding to bortezomib. A Phase 3 randomized, double-blind, placebo-controlled trial of zolinza in combination with bortezomib in patients with relapsed and/or refractory multiple myeloma is currently enrolling patients. Pulmonary embolism and deep vein thrombosis have been reported as adverse reactions following treatment with zolinza.
Carfilzomib — Onyx Pharmaceuticals: Carfilzomib is the first in a new class of selective, irreversible proteasome inhibitors being developed by Proteolix (now part of Onyx Pharmaceuticals) for the treatment of hematologic malignancies and solid tumors. Carfilzomib produces specific and sustained inhibition of the proteasome, leading to apoptosis in cancer cells with minimal off-target effects. In Phase 1 and Phase 2 clinical trials, carfilzomib has demonstrated single-agent activity in hematologic malignancies and solid tumors, including multiple myeloma, Waldenstrom's macroglobulinemia, mantle cell lymphoma and renal cell carcinoma. Carfilzomib was generally well tolerated and toxicities were manageable. A Phase 3 international randomized trial evaluating the efficacy of carfilzomib in combination with lenalidomide and dexamethasone versus lenalidomide and dexamethasone as a potential treatment option for patients with relapsed multiple myeloma was started in March 2010. Orphan Drug designation was granted by EMA in June 2008 for the treatment of multiple myeloma.
Market Data — Multiple Myeloma
Multiple myeloma is the second most common blood cancer in United States and constitutes approximately 1% of all cancers.
According to Decision Resources January 2011, about 131,190 diagnosed prevalent cases (men and women) occurred in multiple myeloma in 2010 in the major markets comprising the U.S., Europe (G5) and Japan. The number of diagnosed incident cases was estimated at 44,780 in 2010 for the total major markets.
Perifosine — Colon Cancer
In June 2009, results of a randomized Phase 2 study of perifosine in combination with capecitabine versus capecitabine alone in patients with second- or third-line metastatic colon cancer were presented during the American Society of Clinical Oncology ("ASCO") meeting.
This randomized, double-blind, placebo-controlled study was conducted at 11 centers across the United States. Patients with 2nd or 3rd line metastatic colon cancer were randomized to receive capecitabine (Xeloda®), an approved drug for metastatic colon cancer, at a dose of 825 mg/m2 BID (total daily dose of 1650 mg/m2) on days 1 - 14 every 21 days, plus either perifosine or placebo at 50 mg daily. Treatment was continued until progression. The study enrolled a total of 38 patients, 34 of which were third-line or greater. Of the 38 patients enrolled, 35 were evaluable for response (20 patients on the perifosine + capecitabine arm and 15 patients on the placebo + capecitabine arm). Of the three patients on the placebo + capecitabine arm not evaluable for response, 2 patients were not evaluable due to toxicity (days 14, 46) and 1 patient was not evaluable due to a new malignancy on day 6. All patients in the perifosine + capecitabine arm were evaluable for response.
The patients in the study were heavily pre-treated, with the arms well-balanced in terms of prior treatment regimens. The median number of prior treatment regimens for all 38 patients was two (range 1-5), with prior treatment regimens as follows: 91% of the patients received FOLFIRI (Irinotecan + 5FU + Leucovorin); 74% FOLFOX (Oxaliplatin + 5FU + Leucovorin); 63% were previously treated with both FOLFIRI and FOLFOX; 77% received Avastin; and 43% Erbitux®. Prior treatment with single agent capecitabine was excluded.
The primary endpoints of this study were to measure 1) TTP, 2) ORR, defined as the percentage of patients achieving a Complete Response ("CR") or Partial Response ("PR") by Response Evaluation Criteria in Solid Tumors ("RECIST"), and 3) Clinical Benefit Rate ("CBR") defined as the percentage of patients on treatment for greater than three months with at least SD. Safety of perifosine + capecitabine vs. placebo + capecitabine in this patient population was evaluated as a secondary endpoint. Perifosine in combination with capecitabine was well tolerated with hand/foot syndrome (14%) and anemia (11%) as the highest reported grade3/4 adverse events.
Best response and median time to progression of perifosine + capecitabine vs. placebo + capecitabine were as follows:
| Group | N | CR N(%) | PR N(%) | ORR N(%) | SD > 12 wks N(%) | CBR N(%) | Median TTP (wks) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Perifosine + capecitabine | 20 | 1 (5% | ) | 3 (15% | ) | 4 (20% | ) | 11 (55% | ) | 15 (75% | ) | 28.9 weeks {95% CI (13, 48.1)} | ||||||||||
Placebo + capecitabine | 15 | 0 | 1 (7% | ) | 1 (7% | ) | 5 (33% | ) | 6 (40% | ) | 11 weeks {95% CI (9, 15.9)} | |||||||||||
Perifosine + capecitabine more than doubled time to progression vs. placebo + capecitabine with a statistically significant p-value = 0.0006. In addition, perifosine + capecitabine more than doubled the ORR and almost doubled the CBR vs. placebo + capecitabine.
Although not a primary endpoint in the study, overall survival was analyzed with results as follows:
| ||||||||
| Development Status* | |||||||
| Takeda/Janssen-Cilag/Janssen | According to Decision Resources (January 2012), the 2010 G7** sales were estimated to be $1.14 billion | |||||||
| Caelyx®/Doxil® (pegylated liposomal doxorubicin) / topoisomerase II inhibitor and DNA intercalating agent | Janssen (J&J): Doxil® / Merck & Co.: Caelyx | According to Decision Resources (January 2012), the 2010 G7** sales were estimated to be $36.2 million | ||||||
| Thalomid® (thalidomide) / antiangiogenic compound | Celgene Corporation | According to Decision Resources (January 2012), the 2010 G7** sales were estimated to be $383.9 million | ||||||
| Revlimid® (lenalidomide) / oral immunomodulatory drug | Celgene Corporation | According to Decision Resources (January 2012), the 2010 G7** sales were estimated to be $1.94 billion | ||||||
*
**
Products in the perifosine + capecitabine patient group is ongoing with 10 of the 20 patients in this arm still alive.
Updated results of this Phase 2 study were presented in January 2010 during the ASCO Gastrointestinal Cancers symposium. The primary endpoint of this study was to measure TTP. ORR, defined as CR+PR by RECIST, and OS were measured as a secondary endpoint. Updated results demonstrated a statistically significant advantage in the combination arm of perifosine + capecitabine for TTP and OS, as well as for the percentage of patients achieving SD lasting 12 or more weeks or better, as compared to the capecitabine arm. The perifosine + capecitabine arm demonstrated a greater than 60% improvement in OS, a more than doubling of median TTP, and almost a doubling of the percentage of patients achieving SD or better. In addition, the ORR was 20% (including one CR, and durable responses) in the perifosine + capecitabine arm vs 7% in the capecitabine arm. The updated efficacy results for all evaluable patients are as follows:
| ||||||||||||||||
| Development Status* | |||||||||||||||
- NDA submitted to the FDA - Phase 2 and Phase 3 | ||||||||||||||||
- Phase 3 - Phase 2 completed | ||||||||||||||||
Of notable interest, and for the first time presented, were data showing a highly statistically significant benefit in median OS (more than doubling) and TTP for the subset of patients who were refractory to a 5-FU (Fluorouracil) chemotherapy-based treatment regimen. 5-FU is a core component of the standard of care FOLFIRI and FOLFOX regimens, and capecitabine is a 5-FU pro-drug. These results are shown below:
| |||||||||||||
| Zolinza (vorinostat — MK0683) / | Phase 3 completed | ||||||||||||
| |||||||||||||
| * | Source: Company’s Website |
All 38 patients were evaluable for safety. The perifosine + capecitabine combination was well-tolerated with Grade 3 and Grade 4 adverse events of > 10% incidence for perifosine + capecitabine arm versus capecitabine arm as follows: anemia (15% vs. 0%), fatigue (0% vs. 11%), abdominal pain (5% vs. 11%) and hand-foot syndrome (30% vs. 0%). Of note, incidence of Grade 1 and Grade 2 hand-foot syndrome was similar in bothMarket Data — MM
MM is the perifosine + capecitabine and capecitabine arms (25% vs. 22%, respectively). Hand-foot syndrome is a reported adverse event with capecitabine monotherapy. Patients who remained on treatment longer in the Phase 2 study had a greater chance to develop hand-foot syndrome as illustrated by a median time to onset of Grade 3 and Grade 4 hand-foot syndrome in the perifosine + capecitabine arm of 19 weeks.
On February 3, 2010, we announced that our partner Keryx had reached an agreement with the FDA on an SPA for the Phase 3 X-PECT trial for perifosine in patients with refractory metastatic colorectal cancer.
On April 5, 2010, our partner Keryx was granted Fast Track designation by the FDA for the Phase 3 X-PECT registration trial.
On April 8, 2010, we announced that our partner Keryx initiated a randomized (1:1), double-blind Phase 3 X-PECT trial comparing the efficacy and safety of perifosine + capecitabine (Xeloda®) vs. placebo + capecitabine in approximately 430 patients with refractory metastatic colorectal cancer. Patients must have failed available therapy including 5-fluorouracil, oxaliplatin (Eloxatin®), irinotecan, bevacizumab (Avastin®) and, if K-Ras wild-type, failed therapy with prior cetuximab (Erbitux®) or panitumumab (Vectibix®). For oxaliplatin-based therapy, failure of therapy also includes patients who discontinued due to toxicity. The primary endpoint is overall survival, with secondary endpoints including overall response rate (complete responses + partial responses), progression-free survival and safety. Approximately 70 U.S. sites are participating in the study. Enrollment is expected to take approximately 12 months, with study completion expected by the end of 2011. Dr. Johanna Bendell, Director of GI Oncology Research for the Sarah Cannon Research Institute, Nashville, Tennessee, leads the Phase 3 investigational team.
On June 8, 2010, Phase 2 results were reported at the ASCO annual meeting, confirming a statistically significant improvement in both time to tumor progression and overall survival with perifosine, in combination with capecitabine in the treatment of advanced metastatic colorectal cancer. The perifosine + capecitabine arm demonstrated a greater than 60% improvement in overall survival, a more than doubling of median time to progression, and almost a doubling of the percentage of patients achieving stable disease or better. In addition, the overall response rate was 20% (including one complete response, and durable responses) in the perifosine + capecitabine arm versus 7% in the capecitabine arm. Of notable interest were the patients who were previously refractory to a 5-FU based regimen. The perifosine + capecitabine arm again demonstrated a statistically significant increase in both time to progression and overall survival, as compared to the capecitabine arm. As for safety, the perifosine + capecitabine arm was well tolerated.
On June 29, 2010, we announced that we had received positive Scientific Advice from the EMA regarding the Phase 3 X-PECT trial for the development of perifosine in refractory advanced colorectal cancer. The Scientific Advice from the EMA indicates that the ongoing study, in conjunction with safety data generated from other clinical studies with perifosine, is considered sufficient to provide all data necessary to support a marketing authorization of perifosine in advanced colorectal cancer. We do not intend to initiate any additional studies with perifosine for this indication. Therefore, for the development of perifosine in both multiple myeloma and colorectalsecond most common blood cancer we believe that the planned North American clinical program, sponsored by our partner Keryx, is now sufficient for approval in Europe and in many countries in the rest of the world, where we hold rights for our compound.
Competitors for Perifosine in colon cancer indication
Products on the market:
Standard 1st-line therapies for treatment of colon cancer are usually the FOLFOX (5-fluorouracil; leucovorin; oxaliplatin) or the FOLFIRI (5-fluorouracil; leucovorin; irinotecan) combination.
Current therapies also include:
Xeloda® (Capecitabine — Manufactured by Roche) is an oral fluoropyrimidine which generates fluorouracil preferentially in tumor tissues by enzymatic cascade and is used in 1st or 2nd-line setting for treatment of metastatic colorectal or colon cancer in monotherapy and also in combination with any chemotherapy in all lines with or without Avastin. According to Roche's 2010 Annual Report, sales of
Xeloda for colorectal, stomach and breast cancer increased 17% to approximately 1.4 billion Swiss francs in 2010.
Avastin® (Bevacizumab, a humanized monoclonal antibody targeting vascular endothelial growth factor — manufactured by Genentech/Roche) is also used in 1st or 2nd-line treatment of metastatic colorectal cancer combined with available Standard therapy FOLFOX. According to Roche's 2010 Annual Report, sales of Avastin® for advanced colorectal, breast, lung and kidney cancer, and for relapsed glioblastoma (a type of brain tumour), rose 9% to approximately 6.5 billion Swiss francs in 2010.
Erbitux® (Cetuximab) is a chimeric monoclonal antibody that specifically blocks the epidermal growth factor receptor (EGFR). Cetuximab is indicated for the treatment of patients with EGFR-expressing, KRAS wild-type metastatic colorectal cancer in combination with Standard chemotherapy FOLFIRI, and in patients who have failed oxaliplatin- and irinotecan-based therapy. Erbitux® is manufactured and distributed in North America by ImClone and Bristol-Myers Squibb, while in the rest of the world distribution is by Merck KGaA. According to Merck's 2009 Annual Report, sales of Erbitux® increased by 23% to €697 million, or approximately $968 million, compared to 2008. On March 29, 2010, Merck Serono, a division of Merck KGaA announced that Erbitux® granted extended use in Japan for first-line-treatment for mCRC patients with KRAS wild-type tumors. In the United Kingdom, the National Institute for Health and Clinical Excellence (NICE) recommended in June the use of Erbitux® in combination with chemotherapy as a first-line treatment for patients with metastatic colorectal cancer who have met specific additional criteria — improving the possibility of potentially curative surgery.
Vectibix® (Panitumumab) is a recombinant, human IgG2 kappa monoclonal antibody manufactured by Amgen that binds specifically to the human epidermal growth factor receptor (EGFR). Vectibix® is indicated as a single agent for the treatment of EGFR-expressing, metastatic colorectal carcinoma with disease progression on or following fluoropyrimidine-, oxaliplatin-, and irinotecan-containing chemotherapy regimens. FDA approval was achieved in September 2006. There are 2 boxed warnings for Vectibix®: dermatologic toxicity and infusion reactions. According to Amgen's 2009 Annual Report Vectibix® worldwide sales for the year 2009 were $233 million. In 2009, Amgen announced that primary endpoint of extending progression-free survival was met in Phase 3 studies evaluating Vectibix® in combination with FOLFOX or FOLFIRI. Based on these study results, Amgen is planning to file for regulatory approval in the United States and Europe for first- and second-line treatment in patients with KRAS wild-type metastatic colorectal cancer.
Product in Phase 3 development:
Aflibercept — Sanofi + Regeneron: Aflibercept is an anti-angiogenesis inhibitor with a unique mechanismconstitutes approximately 1% of action being developed by Sanofi and Regeneron. This fusion protein binds all forms of Vascular Endothelial Growth Factor-A ("VEGF-A"cancers (source: NCI), as well as VEGF-B and placental growth factor ("PIGF"), additional angiogenic growth factors that appear to play a role in tumor angiogenesis and inflammation. Aflibercept has been shown to bind VEGF-A, VEGF-B, and PlGF with higher affinity than their natural receptors. The following clinical studies are currently ongoing and are fully enrolled:
Aptocine — Light Sciences Oncology: Aptocine is a water-soluble drug targeted by a single-use, disposable drug activator included with the drug. Aptocine has three mechanisms of action: direct tumor cytotoxicity, apoptosis caused by vascular shutdown and potential anti-tumor immune stimulation. Enrollment of a Phase 3 trial for aptocine in metastatic colorectal cancer is nearly completed. This Phase 3 trial is a 450-patient trial, conducted primarily at sites in Europe and India, to assess the progression-free survival and overall survival of patients treated with Aptocine + chemotherapy versus chemotherapy alone.
Brivanib — Bristol-Myers Squibb: Brivanib, developed by Bristol-Myers Squibb, is an oral prodrug of BMS-540215, a dual tyrosine kinase inhibitor of VEGFR and FGFR signalling. Brivanib strongly binds to and inhibits VEGFR2, a tyrosine kinase receptor expressed almost exclusively on vascular endothelial cells. The inhibition of VEGFR2 may result in inhibition of tumor angiogenesis, inhibition of tumor cell growth, and tumor regression. Brivanib is currently in Phase 3 randomized trial investigating Brivanib Alaninate in combination with cetuximab (Erbitux®) vs. placebo in combination with cetuximab (Erbitux®) in patients with K-RAS tumors previously treated with combination chemotherapy for metastatic colorectal carcinoma. It is not yet known whether giving brivanib together with cetuximab is more effective than cetuximab alone in treating patients with metastatic colorectal cancer.
OncoVax® — Vaccinogen: OncoVax® is an autologous tumour cell vaccine and prepared for each patient using the patient's own surgically removed tumor. The active specific immunotherapy falls within the classification of Advanced Therapeutic Medicinal Product (ATMP). The patient received the first of four vaccinations several weeks after surgery. The vaccine consists of a portion of the tumor cells that has been thawed and combined with a proprietary formulation of BCG that serves as an immunogenic enhancer. This formulation is also used for the 2nd inoculation. The 3rd and the final booster inoculations are prepared the same way but without the addition of BCG. Phase 3a results demonstrated efficacy of Oncovax® in Stage II colon cancer patients with a statistically significant increased 5-year overall survival rate and increased recurrence-free survival by log-rank analysis. OncoVax® currently has a marketing authorization from Swissmedic, Switzerland's medical authority, in the category of "procedes therapeutiques". A pre-submission meeting to request Scientific Advice from the EMA for submission of a Conditional Marketing Autorization was done in December 2009.
Ramucirumab — Eli Lilly + ImClone: Ramucirumab is an anti-VEGFR2 antibody blocking the binding of VEGF to its receptor. Ramucirumab is currently being tested for 2nd-line treatment in metastatic colorectal cancer in combination with FOLFIRI.
Market Data — Colon Cancer
According to the American Cancer Society, colorectal cancer is the third most common form of cancerDecision Resources – Patient Data Base (March 2012), 49,840 patients will be diagnosed in the United States, excluding skin cancers. ItG7 market in 2014.
| Treatable pool relapsed/refractory patient population* | ||||
| 3rd line in 2014 | 2nd line in 2014 | |||
USA | 10,080 | 14,830 | ||
EU-5 | 11,840 | 17,410 | ||
Japan | 2,860 | 4,200 | ||
| * | According to Decision Resources – Patient Data Base (March 2012). |
1.1.1.3 Perifosine – Other indications
Perifosine is estimatedalso studied in different indications including gliomas, RCC, sarcoma, Hodgkin’s lymphoma (“HL”), neuroblastoma and other indications. As a monotherapy, perifosine is being explored in CLL.
On April 4, 2011, we announced that over 142,570 peopletwo posters on perifosine were diagnosed with some formpresented at the 102nd annual meeting of colorectalthe AACR at the Orange County Convention Center in Orlando, Florida. Perifosine demonstrated antitumor activity in several gastric cancer with over 51,370 patients dying from colorectal cancercell lines. Furthermore, perifosine enhanced the antitumor activity of 5-FU in 2010. Surgery is often the main treatment for early stage colorectal cancer. When colorectal cancer metastasizes (spreads to other parts of the body such ascell lines, including 5-FU resistant cell lines. 5-FU is the liver), chemotherapy is commonly used. Treatment of patients with recurrent or advanced colorectal cancer depends on the locationactive metabolite of the disease. Chemotherapy regimens (i.e. FOLFOX or FOLFIRI either with or without bevacizumab) have been shown to increase survival rates in patients with metastatic/advanced colorectal cancer. Currently, there are sevenprodrug Xeloda, which is approved drugs for patients with metastatic colorectal cancer: 5-fluorouracil (5-FU), capecitabine (Xeloda®), irinotecan (Camptosar®), oxaliplatin (Eloxatin®), bevacizumab (Avastin®), cetuximab (Erbitux®), and panitumumab (Vectibix®). Depending on the stage of the cancer, two or more of these types of treatment may be combined at the same time, such as FOLFOX and FOLFIRI, or used after one another. Bevacizumab, a VEGF monoclonal antibody, is commonly
administered with chemotherapy. Typically, patients who fail 5-FU, oxaliplatin, irinotecan, and bevacizumab-containing therapies, and who have wild-type KRAS status receive EGFR monoclonal antibody therapy with either cetuximab or panitumumab. Once patients progress on these agents, there are no further standard treatment options.
Perifosine — Waldenstrom's Macroglobulinemia ("WM")
Results of a Phase 2 study on perifosine in patients with WM were presented in June 2008 at ASCO and in December 2008 during the ASH meeting. Thirty-six patients were evaluable for response. Perifosine showed clinical activity as a single agent in patients with relapsed/refractory WM, with an ORR (partial response ["PR"] + minimal response ["MR"]) of thirteen patients (36%). PR occurred in 2 patients (6%), with a median duration of response of 9+ and 18+ months, MR occurred in 11 patients (30%), with a median duration of response of 7 months (2-21+ months). SD occurred in 21 patients (58%) and progressive disease ["PD"] in 2 patients (6%) at 2 and 4 months. The most common adverse events were GI toxicities (nausea, vomiting and diarrhea) with grade 1 and 2 in 36% of the patients. Grade 3 and 4 events included anemia (9%) and leucopenia (9%). Grade 3 arthritis occurred in 9% of the patients; was considered likely related to therapy, (especially in rapidly responding patients), and reversed with symptomatic treatment as well as dose reduction. Dose reductions to 100 mg occurred in a total of 36% of the patients and were otherwise due to GI toxicity or cytopenias. Perifosine monotherapy induces a prolonged time to progression in relapsed or refractory WM, with a promising response rate of 36%, stabilization of disease in 58% of patients, and manageable toxicity, as well as the convenience of oral administration. Future clinical trials in combination with rituximab are planned.
In January 2010, we announced that an article entitled "Clinical and Translational Studies of a Phase II Trial of the Novel Oral Akt Inhibitor Perifosine in Relapsed or Relapsed/Refractory Waldenstrom's Macroglobulinemia," reporting Phase 2 data demonstrating the single agent activity of perifosine for the treatment of advanced Waldenstrom's Macroglobulinemia, appearedgastric cancer in many countries including Japan.
Perifosine also markedly enhanced the February 1, 2010 issueantitumor activity of the Journalcellular TRAIL based treatment and was able to overcome TRAIL resistance bothin vitro andin vivo. The results are in line with other studies demonstrating the synergistic effects of Clinical Cancer Research. Dr. Irene Ghobrial, Assistant Professor of Medicine, Bing Centerperifosine with cytotoxic drugs, including bortezomib and 5-FU.
On December 12, 2011, we reported encouraging preclinical data for Waldenstrom's Macroglobulinemia at Dana-Farber Cancer Institute, ledperifosine in HL. In vitrodata from HL cell lines showed that perifosine combined with sorafenib induced increased apoptosis, while in vivo data for the Phase 2 study, in which 37 patients were treated with perifosine 150 mg dailysame combination treatment for 6 cycles. In this study, 41% of the patients had 3 or more lines of prior therapy and 78% had 2 or more prior lines of therapy. Such prior therapies include nucleoside analogues, bortezomib, alkylating agents and rituximab, which are not approved for, but are often used in the treatment of Waldenstrom's. Stable or responding patients were allowed to continue therapy until progression. Of the 37 patients, 4 achieved a partial response (11%), 9 achieved a minimal response (24%), and 20 showed stable disease (54%). Overall, 89% (33/37) of patients treated with single agent perifosine were reported to have stable disease or better, while 11% (4 patients) demonstrated progression. The median progression-freeHL significantly increased survival in mice. Data were presented during the ASH Annual Meeting and Exposition, in San Diego, California.
The study was 12.6 months (90% C.I. (10.2, 22.7))conducted to investigate, in vitro andin vivo, with a median overall survivalthe activity and mechanism(s) of 26 months (90% C.I. (26 — upper limit not reached)). Perifosine was generally well tolerated with gastrointestinal symptoms and fatigue reported as the most common adverse events related to therapy.
Perifosine — Renal Cell Carcinoma ("RCC")
In June 2006, we announced positive data of perifosine in patients with advanced RCC. Keryx disclosed results from an interim analysis performed at the end of the first year of accrual, from a Phase 2, multi-center trial of perifosine that included multiple types of tumor and the results of the RCC group met protocol requirements for expansion of this cohort. Of the 13 patients with RCC, seven were evaluable for response. Three of them (43%) had a partial response and an additional two patients (29%) achieved long-term stable disease. Two patients (29%) had progressive disease. Results of a Phase 1 multicenter trialaction of perifosine in combination with sorafenib for patientsby using three HL cell lines (HD-MyZ, L-540, HDLM-2). In thein vitro experiments perifosine/sorafenib treatment resulted in synergistic cell growth inhibition and cell death induction in HD-MyZ and L-540 cell lines, but not in the HDLM-2 cell line. Cell cycle arrest, down-modulating of the MAPK and PI3K/Akt pathways as well as caspase-independent cell death was observed, which was associated with advanced cancers including RCC were disclosed by Keryxsevere mitochondrial dysfunction. Further, expression of genes involved in June 2007 duringamino acid metabolism, cell cycle, DNA replication and cell death was shown to be modulated. In addition, overexpression of the ASCO meetingtribbles homologue 3 [TRIB3] was observed.
In vivo, perifosine/sorafenib treatment significantly increased survival in the HD-MyZ model (45 vs. 81 days, as compared to controls), with 25% tumor-free mice at the end of the 200-day observation period. In the L-540 model, subcutaneous tumor volume was also reduced as compared to controls (by 42%), perifosine (by 35%) or sorafenib (by 46%) alone. In HD-MyZ and L-540 but not HDLM-2, the combined treatment induced an increase in tumor necrosis (2- to 8-fold, P£.0001) and in November 2007. The trial was designedtumor apoptosis (2- to accrue 3-6 patients in each2.5-fold, P£.0001). In 2 of four cohorts. Response by
RECIST criteria was a secondary endpoint. Perifosine was escalated from 50 mg once per day to 50 mg three times per day; 3 HL cell lines, perifosine/sorafenib dose was escalated from 400 mg once per day to 400 mg twice per day; and sunitinib dose was escalated from 25 mg to 50 mg once per day for 4 weeks ofcombination treatment out of 6. DLT was defined as grade (G) 3 non-hematologic or G4 hematologic toxicity. MTD was the dose below that at which 2 out of 6 patients experienced a DLT.
For the combination perifosine + sorafenib, 20 patients were enrolled (12 males / 8 females, median age 64 (range 44-87)) with a median number of 2 prior therapies (range 1-4). Three patients were not evaluable due to rapid disease progression. Diagnosis was as follows; RCC (11 pts), sarcoma (5), colorectal (2), hepatocellular (1) and neuroendocrine (1). 17 patients were evaluable for toxicity: no drug-related Grade 4 adverse events (AE) were seen. Suspected DLT of hand-foot syndrome was seen in cohort 4 and additional patients were enrolled. There was noinduced potent antitumor effects. A significant increase in hand-foot syndrome compared to sorafenib alone. Of interest, 6/9 evaluable RCC patients (67%) had SD >12 weeks (median 26 weeks, range 12-62+). One hepatocellular patient had SDmitochondrial injury and apoptosis and a marked reduction of cell viability were observed in thein vitroexperiments.In vivocombination treatment increased survival and inhibited tumor growth.
On December 13, 2011, we reported encouraging clinical data for 36 weeks. The combination of perifosine + sorafenib was well tolerated with no increased hand-foot syndrome compared to sorafenib alone. Six out of 9 RCC patients (67%) achieved SD up to 62+ weeks.
For the combination perifosine + sunitinib, 14 patients (8 males / 6 females; media range 62 years old, range 28-81) were enrolled. Disease type was as follows: RCC (3), Sarcoma (3), Other (8). Six patients were evaluable for response. After 2 treatment cycles, one patient had a PR, 3 patients showed a SD and 2 patients had disease progression (PD). In the sub-group RCC, three out of three patients were evaluable for response: one patient had a PR, 1 patient showed a SD and 1 patient had a PD. Results indicated that patients to date have tolerated well the treatment combination of perifosine + sunitinib with no unexpected toxicities and clinical activity has been noted within the first 3 cohorts with 4 of 6 (67%) evaluable patients with advanced cancer achieving at least SD for more than 6 months.
Results from aan ongoing Phase 2 trial of perifosineclinical study in patients with advanced RCC who have failed tyrosine kinase inhibitors (TKI) were also presented atrefractory/relapsed HL during the ASCO meetingASH Annual Meeting and Exposition.
Preliminary response data showed that perifosine combined with sorafenib significantly increased median PFS in June 2009 by our partner Keryx. refractory/relapsed HL patients with high phosphorylation levels of Erk and Akt as compared to patients with low baseline phosphorylation levels of Erk and Akt.
The goalstudy evaluates phosphorylation levels of this multi-centerErk (pErk) and Akt (pAkt) in circulating lymphocytes from patients enrolled in two consecutive Phase 2 trial was to determine thetrials evaluating activity and safety and efficacy of perifosine in patients with advanced RCC refractory to VEGFR TKI.
The study enrolled a total of 50 patients, of which 46 patients were evaluable for response. Evaluable patients were defined as those who had greater than 7 days of treatment. The primary endpoint of this study was clinical benefit, defined as response rate (RECIST), and PFS in RCC patients who failed a prior VEGF receptor inhibitor (sunitinib or sorafenib). Safety of perifosine in this patient population was evaluated as a secondary endpoint. The best response to single-agent perifosine was as follows:
| ||||||||||
The median PFS for all 46 patients was 12.5 weeks [95% CI (11.9, 19)]. The median overall survival has not been reached with 33 of 46 patients (72%) still alive.
Also of interest was the patient subgroup who had failed both a VEGF receptor inhibitor (sunitinib or sorafenib) and an mTOR inhibitor (either everolimus or temsirolimus). For this group, the best response and median PFS to single agent perifosine was as follows:
| ||||||||||||||
Three patients out of the group of patients previously treated with and failed both a VEGF and an mTOR inhibitor remain on active treatment, now out 5, 9 and 17 months.
Updated clinical results of this Phase II study of perifosine as a single-agent treatment for advanced metastatic RCC were presented in September 2009 at the 8th International Kidney Cancer Symposium. Those updated data included results from a subgroup of patients who failed both a VEGF receptor inhibitor (sunitinib or sorafenib) and an mTOR inhibitor (temsirolimus or everolimus). Evaluable patients (n=16) were defined as those who had greater than 7 days of treatment (2 additional patients withdrew consent within 7 days). Patients received 100 mg of perifosine daily until progression or unacceptable toxicity. The primary endpoint of this study was clinical benefit, defined as response rate (CR / PR by RECIST) or percent of patients progression-free for at least 3 months. Median PFS and overall survival were also analyzed for efficacy. Safety was a secondary endpoint. Perifosine was well-tolerated with the most common adverse events being gastrointestinal discomfort and fatigue. Best response to single agent perifosine was as follows:
| ||||||||||
Perifosine — Sarcoma
In June 2007, our partner Keryx presented results of Phase 1 and 2 studies for the treatment of patients with advanced sarcoma at the ASCO meeting. The dose schedules in the Phase 1 trials were weekly 100-800 mg or loading dose 300-1,800 mg on Day 1 followed by 50-150 mg daily for Days 2-21 every 28 days or loading dose 400-900 mg and daily 50-100 mg continuously. In the Phase 2 trial, doses were loading dose 900 mg on Day 1 and 150 mg daily for days 2-21 every 28 days; loading dose 900 mg and 100 mg daily continuously; 50 mg daily continuously without a loading dose; and 900-1,500 mg weekly. 145 patients with sarcoma were entered into studies and were assessed for CBR. Partial responses were seen, in one patient each, with chondrosarcoma, extra-skeletal myxoid chondrosarcoma, leiomyosarcoma and a desmoid tumor. At lower doses with 52 patients fully evaluable for CBR, the CBR was 52% with four partial responses and 23 stable diseases at³ 4 months. At higher doses with 30 patients fully evaluable for CBR, CBR was 53% with 16 stable diseases at³ 4 months. Toxicities were mainly gastrointestinal and/or fatigue. The percentage of patients with grade 0 nausea, vomiting, diarrhea and fatigue for lower dose perifosine (76 patients) was 46%, 49%, 38% and 55%, respectively, compared to 26%, 32%, 20%, and 58% for higher dose perifosine (69 patients). The proportion of patients with grade 2+ nausea, vomiting, diarrhea and fatigue was 20%, 13%, 15%, and 21% for lower dose perifosine and 49%, 35%, 42%, and 25% for higher dose perifosine.
In November 2007, our partner Keryx announced positive preliminary Phase 2 data of perifosine in patients with chemo-insensitive sarcoma. Data demonstrated the tolerability and clinical activity of perifosinesorafenib as a single agent or in combination with an overall clinical benefit of 40% (stable disease > 3 months)perifosine in
patients with relapsed/refractory rare sarcomas. Perifosine was well tolerated with the most common grade 1 & 2 adverse events reported as nausea, vomiting, diarrhea and fatigue.
Perifosine — Gliomas
In November 2007, our partner Keryx announced early results of a Phase 2 trial of perifosine as a single agent for the treatment of recurrent malignant gliomas (malignant glioblastoma and malignant anaplastic gliomas). Twenty-fiveHL patients. Four patients with advanced malignant gliomas were treated with sorafenib alone at a loading dose level of 600400 mg (150BID and twenty-one patients received a 4-week treatment with perifosine alone at a dose level of 50 mg x4)BID, followed by a 100perifosine/sorafenib combination therapy with 50 mg daily doseBID and 400 mg BID, respectively. Circulating lymphocytes were evaluated for their phosphorylation levels of perifosine. The median progression free survivalErk and overall survival in the anaplastic glioma group was nine weeks (range 2-50 weeks) and 49 weeks, respectively. Toxicity was minimal with the following reported events: one grade 1 nausea, one grade 1 diarrhea, one grade 2 pain, and one grade 4 gout exacerbation. The study was designed to enroll at least 12 evaluable malignant glioblastoma patients and at least 10 evaluable malignant anaplastic gliomas patients. If at least one patient achieves six month progression free survival, the study would continue to enroll an additional subset of patients. Therefore, the malignant glioblastoma arm has been halted and the malignant anaplastic gliomas arm will continue to enroll.
Perifosine — Neuroblastoma
On April 20, 2010 at the American Association for Cancer Research's ("AACR") annual meeting, we presented preclinical data that demonstrated that single agent perifosine targets activation of Akt, in neuroblastoma cells and xenografts, significantly inhibited tumor growthin vivo and improved the survival of mice bearing neuroblastoma tumors.
On May 17, 2010, we announced the publication of an article in the May 12, 2010 issue of theJournalorder to assess predictive value of the National Cancer Institutephosphokinase levels for therapy responses.
entitled "In VitroClinical response data showed that baseline pErk andIn Vivo Inhibition pAkt levels were significantly higher in responsive patients, as compared to unresponsive patients. The pErk and pAkt levels measured after 60 days of Neuroblastoma Tumor Cell Growth by AKT Inhibitor Perifosine", demonstrating the single agent activity of perifosine in neuroblastoma tumor preclinical models.
On June 7, 2010, we announced that Phase 1 data for perifosine in recurrent pediatric solid tumors had been presented in the pediatric solid tumor poster discussion session held at the 46th annual ASCO meeting in Chicago. This study, conducted by the Memorial Sloan-Kettering Cancer Center pediatric group, marks the first time that perifosine has been administered in a pediatric patient setting.
This Phase 1 study of perifosine for recurrent pediatric solid tumors is a single center, open-label, dose-escalating study to assess safety, tolerability, pharmacokinetics ("PK"), and to identify any DLT of single agent perifosine in pediatric patientstherapy with any solid tumor that has failed standard therapy. Eleven patients (4 males, 7 females), at a median age of 13 years (5-18) were treated in this study to date. The following tumor types have been treated thus far: high-grade glioma (5), medulloblastoma (2), neuroblastoma (3), and ependymoma (1). Most patients were heavily pretreated with a median of three prior lines of therapy. Cohorts of three patients were treated at three dose levels of perifosine after a loading dose on day 1, and taking into account the drug's long half-life (t1/2 100 hours). No DLTs were observed at any of the three dose levels; dose level 4 is currently open for accrual. PK data thus far suggest similar drug absorption by pediatric patients relative to adult patients treated with single agent perifosine.
Of particular interest are the early signs of clinical activity observed in two of the three patients with Stage IV refractory neuroblastoma. Both patients were refractory to prior treatments upon entering the study and achieved stable disease for 48 weeks and 55+ weeks (ongoing). The investigators concluded that perifosine is well-tolerated in children with recurrent solid tumors and that these early signals of activity warrant further investigation in patients with advanced neuroblastoma and select brain tumors. Previously, perifosine has been shown to target activation of Akt in neuroblastoma cells and xenografts and to significantly inhibit tumor growthin vivo and improve the survival of mice bearing neuroblastoma tumors.
On July 14, 2010, our partner Keryx was granted orphan-drug designation by the FDA for perifosine for the treatment of neuroblastoma, a cancer of the nervous system affecting mostly children and infants for which there are no FDA-approved therapies.
Perifosine — Other indications
On March 2, 2006, our partner Keryx announced the initiation of a corporate-sponsored Phase 2 trial, multi-cancer, clinical program to evaluate perifosine as a treatment for leukemia. Dr. Frank Giles, Professor, Department of Leukemia, at the MD Anderson Cancer Center in Houston, TX, is the principal investigator. This Phase 2 trial will assess the objective response rate and evaluate the pharmacokinetics and safety and tolerability of perifosine as a single agent in relapsed or refractory acute myeloid leukemia, acute lymphocytic leukemia, CLL, high-risk myelodysplastic syndrome and chronic myeloid leukemia in the blastic phase.
In November 2006, our partner Keryx presented intermediary results of the Phase 2 study of imatinib + perifosine in patients with imatinib-resistant gastrointestinal stromal tumor ("GIST"). The primary endpoint of this study is to evaluate the efficacy and toxicity of the combination imatinib and perifosine in patients with imatinib-resistant GIST. To date, 16 patients have been enrolled in the current study. Of the 12 patients with evaluable disease, there were two partial responses by Choi criteria (17% objective response rate ("ORR")) and one partial response by RECIST criteria (8% objective response rate). Grade 3 and 4 adverse events were rare and included fatigue, myalgias, ocular toxicity and nausea/emesis. The early data from the current study suggest that the addition of perifosine to imatinib is well-tolerated and may have efficacy in the treatment of patients with imatinib-resistant GIST.
Updated results of this trial were presented in June 2009 by our partner Keryx during the ASCO meeting. Patients with Kit (+) advanced GIST who have progressed on imatinib were eligible. Patients continued their current dose of imatinib and were randomized to one of two dosing schedules of perifosine (Arm A: 100 mg p.o. qd × 28 + imatinib or Arm B: 900 mg [300 mg p.o. tid] qweekly + qd imatinib). A Bayesian approach was utilized to assess a target response rate of 20% with an unacceptable toxicity rate of 15% or less. Response was measured every 8 weeks by RECIST and Choi criteria. The primary endpoint was to determine the efficacy of perifosine with imatinib in patients with advanced GIST who progressed while receiving imatinib. 41 patients were enrolled from August 2005 to July 2008. After 1 patient exclusion and 2 cross-overs, 22 patients were in Arm A and 18 patients in Arm B. Median age was 58 (range, 32-82), 51% were male, and median ECOG performance status was 1. The most common primary site of disease and metastasis was the stomach (29%) and liver (66%), respectively. KIT genotype was available for 22 patients (54%); 5(12%) WT, 13(32%) exon 11 mutations, and 4(10%) exon 9 mutations. The median number of cycles was 2 (range, 1-24). By Choi and RECIST, 30 patients (73%) and 36 patients (87%) were available for response, respectively. No CR was identified but the PR rate was 4/36 (11%) by Choi (4 PR, 9 SD) and 0/36 (0%) by RECIST (16 SD). 4/5 (80%) of patients with WT KIT appeared to benefit (Choi: 1 PR, 3 SD; RECIST: 4 SD). Median PFS and OS for 40 patients were 2.2 months and 18.3 months. No difference in PFS was noted for the 2 schedules. Toxicity was assessed in 39 patients; 46 grade 3 events and 4 grade 4 events (ALT elevation, blurred vision, fatigue, and mood alteration) were noted. The most common grade 3 event was fatigue (20%). Three patients (7%) were removed from the study for toxicity (Arm A:1 patient, Arm B:2 patients).
On July 14, 2009, our partner Keryx announced the initiation of a Phase 1 clinical study to evaluate perifosine as a single agent treatment for recurrent solid tumors in pediatric patients. This single-center open-label study, fully funded by an external grant provided by a private organization, will be conducted at Memorial Sloan-Kettering Cancer Center in New York City. Oren Becher, MD, Instructor, Department of Pediatrics, in coordination with Eric Holland, MD, PhD, Director of the Brain Tumor group at Memorial Sloan-Kettering Cancer Center, will act as the study's Principal
Investigator. Perifosine is being evaluated as a single-agent in pediatric patients with any solid tumor that has failed standard therapy. Patients up to 18 years of age with a performance status of greater than 40% are eligible for this study. The study was designed as a dose escalation study to determine the MTD of perifosine alone in recurrent/progressive pediatric tumors. A standard 3+3 dose escalation design will be employed with 3 to 6 patients at each dose level. All patients will receive perifosine at a loading dose on the first day, followed by a maintenance dose to start on day two until progression of disease. A minimum of 4 and a maximum of 24 patients will be required to complete the study.
On October 8, 2009, our partner Keryx announced the initiation of a Phase 2 clinical study to evaluate perifosine as a single agent treatment for relapsed or refractory CLL and Small Lymphocytic Lymphoma (SLL). This externally funded Phase 2 study was designed by Daphne Friedman, M.D., Instructor and Principal Investigator, in coordination with J. Brice Weinberg, Professor, and Mark Lanasa, Assistant Professor, Divisions of Medical Oncology and Hematology, Duke University Medical Center, and is open for enrollment at Duke University. The single-center, open-label, study entitled, "Phase 2 Trial of Perifosine in Patients with Relapsed or Refractory Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma", will enroll approximately 30 patients. Perifosine will be given orally at a dose of 50 mg twice daily, for a total of six 28-day cycles. The patients will be formally restaged upon completion of the trial. Overall Response Rate is the primary endpoint with overall survival, progression-free survival and safety as secondary endpoints. Correlative studies will also be conducted and evaluated as a secondary endpoint.
On November 17, 2010, at the EORTC-NCI-AACR Symposium on Molecular Targets and Cancer Therapeutics in Berlin, Germany, we presented an abstract on perifosine combined with antimetabolitessorafenib were significantly reduced in responsive patients. The median baseline value of pErk and pAkt efficiently discriminated responsive and unresponsive patients which induces synergistic effects on cytotoxicity and apoptosis in human colon, multiple myeloma, breast, renal, and liver tumor cell lines.
On December 6, 2010 at the ASH's 52nd annual meeting in Orlando, Florida, we announced positive safety and tolerability Phase 2 datawas associated with a significantly improved median PFS for perifosine in patients with advanced lymphoma.baseline pErk³
43% and/or pAkt >23%. Based on these data, the correlation of baseline pErk and pAkt levels with objective responses and time to tumor progression will need to be validated in prospective studies. In the first Phase 2 study related to CLL, 12conclusion, refractory/relapsed HL patients with advanced CLL began treatment with single agent perifosine at 50 mg BID. The patients on the study were heavily pre-treated having had a medianincreased baseline levels of four prior lines of therapy with 75% of patients classified as Rai Stage IV. One patient achieved a partial response (5 months on treatment)pErk and 5 additional patients achieved stable disease (median duration of 4.25 months), for an overall 50% clinical benefit rate (PR + SD). Perifosine was well tolerated with minimal dose modifications.
In the second study presented, 26 patients were enrolled in a Phase 2 study with advanced lymphoma (6 non-Hodgkin's lymphoma, 4 CLL, 1 Waldenstrom's Macroglobulinemia and 15 HL). 73% of patients were previously refractory to their prior therapy, with 85% of patients having had 4 or more prior therapies. Perifosine (50 mg BID) was started as a single agent for 28 days; after 28 days, patients achieving PR or better were continued on single agent perifosine. Patients achieving less than a PR were given the combination of perifosine (50 mg BID) + sorafenib (Nexavar®) at 400 mg BID. All of the 4 CLL patients in this study achieved a partial response on single-agent perifosine within one month of treatment and remained on perifosine single agent. Response durations for each of the 4 patients were 4, 8, 9+ and 12 months. The remaining 22 patients were administered the combination with sorafenib, where 5 of the 15 (33%) HL patients achieved a partial response with a median response duration of 9 months. An additional 6 patients receiving the combination (40%) achieved stable disease. The combination was well tolerated with no unexpected safety events. The investigators concluded that perifosine in combination with sorafenib has significant anti-lymphoma activity in relapsed/refractory HL, and that perifosine as a single agent induced prolonged responses in high-risk, heavily pretreated CLL patients.
Perifosine — Radio-enhancer
A proof-of-concept Phase 1 study of perifosine in combination with radiotherapy conducted by the National Cancer Institute of the Netherlands was completed in 2004. Results from this trial were presented at ASCO 2004. A total of 21 radiotherapy-naïve patients, of whom 17 had advanced non-small cell lung cancer ("NSCLC") and 14 had become refractory to prior chemotherapy, received oral perifosine doses ranging from 50 mg to 200 mg/day concurrently with standard doses of radiotherapy. The trial datapAkt demonstrated an acceptable safety and tolerability profile, with 150 mg/day established as the dose recommended for use in subsequent clinical trials. Also demonstrated was preliminary evidence of anti-tumor activity at all dosage levels, including complete or partial responses (complete disappearance and decreased tumor size, respectively), or stable disease, with a median follow-up for responders of eight months. Importantly, in the cohort of 10 patients who wereincreased PFS when treated with 150 mg/day, the established dose recommended for use in subsequent clinical trials, there were three complete responses, three partial responses and four patients with stable disease.
On September 22, 2005, we announced the initiation of a multi-center Phase 2 randomized, double-blind, placebo-controlled trial with perifosine in combination with radiotherapy for NSCLC. Patients received perifosine 150 mg daily for five to six weeks and were followed for at least 12 months. The primary endpoint of this trial was the extent and duration of local control, i.e., the absence of tumor recurrence or progression in the area that has been irradiated. The trial was conducted in collaboration with the Netherlands Cancer Institute. The lead investigator is Marcel Verheij, M.D., Ph.D., of the Department of Radiation Oncology / Division of Cellular Biochemistry, at the Netherlands Cancer Institute in Amsterdam. We announced completion of recruitment of 160 patients with inoperable Stage III NSCLC on November 14, 2007.
We disclosed preliminary results for this European multi-center Phase 2 trial in NSCLC in June 2009. Starting one week before the onset of a 4-week course of radiotherapy (51 Gy in 17 fractions), 177 patients with non-metastatic but inoperable NSCLC, mainly Stage III, received a 5-week course of 150 mg perifosine daily or placebo. After end of radiotherapy, patients were followed up to determine the time to tumor recurrence or progression in the area that had been irradiated, the so-called "local control". The primary endpoint of this trial was the extent and duration of local control, specifically the proportion of patients with absence of recurrence or progression 12 months after the end of treatment. The study was planned under the basic assumption that radiotherapy alone would result in a 35% local control rate, one year after end of therapy in the placebo group. It was hypothesized that the addition of perifosine would sensitize tumor cells to the tumor-killing effect of the radiotherapy, leading to a 15% higher rate of local control. Secondary efficacy parameters included the times to loco-regional or distant/systemic failure, the tumor response rate, and overall survival. Safety investigations included the monitoring of clinical laboratory, electrocardiograms, lung function, and adverse events.
In all, 22 study sites in The Netherlands, Bulgaria, Romania, Macedonia, and Belarus participated in this trial. A total of 177 patients were randomized and treated, of whom only 26 reached the milestone of one year post-treatment follow-up without disease relapse or progression, 14 of 95 patients (14.7%) in the perifosine and 12 of 82 patients (14.6%) in the placebo control group. No difference between treatment groups could be shown for local, loco-regional and overall disease control. Also, the tumor response rate, as assessed after the end of the radiotherapy, was not different between the groups.
In contrast to the lack of an observed local effect, patients in the perifosine group, particularly the subgroup of patients who entered the study without prior chemotherapy, showed a trend towards longer survival than patients of the placebo control group despite the short duration of treatment (5-week course of 150 mg perifosine daily).
Table of Contentssorafenib.
There were no safety signals that would lead to an amendment of the current safety data or risk benefit assessments of perifosine. The type and severity of side effects were in the expected range. Following these neutral results and an unchanged safety profile, we announced that we will concentrate our efforts for perifosine on the disease targets of both multiple myeloma and metastatic colon cancer.
1.1.1.4 Partners for perifosine
A Cooperative Research and Development Agreement ("CRADA")CRADA was put in place with the National InstituteInstitutes of Health/the National Cancer InstituteNCI in May 2000.
A cooperation and license agreement was signed in September 2002 with Access Oncology, Inc. ("AOI"(“AOI”), for the use of perifosine as an anti-canceranticancer agent covering the United States, Canada and Mexico. In January 2004, AOI was acquired by Keryx, which is pursuing the clinical development of perifosine under the same conditions as AOI. The agreement, in particular, provides us free access to all data from Keryx and its partner'spartners’ studies, as well as milestone payments and scale-up royalties (from high single to low double digit) to be paid to us on future net sales of perifosine in the United States, Canada and Mexico.
In April 2009 we entered into an agreement to out-license the rights of perifosine to Handok in South Korea.
On March 9, 2011, we announced that we had entered into an agreement with Yakult for the development, manufacture and commercialization of perifosine in all human uses, excluding leishmaniasis, in Japan. Under the terms of this agreement, Yakult made an initial up-front payment to us of €6 million ($8.3(approximately $8.4 million). Also per the agreement, we will beare entitled to receive in addition, up to a total of €44 million ($60.9(approximately $57.1 million) upon achieving certain pre-established milestones, including clinical and regulatory events in Japan. Furthermore, we will be supplying perifosine to Yakult on a cost-plus basis and we will be entitled to receive double-digit royalties on future net sales of perifosine in the Japanese market. Yakult will be responsible for the development, registration and commercialization of perifosine in Japan.
On November 23, 2011, we announced the signing of an exclusive commercialization and licensing agreement with Hikma for the registration and marketing of perifosine for the MENA region. Under the terms of the agreement, we received an upfront payment of $0.2 million and are entitled to receive up to a total of $1.8 million upon achieving certain pre-established milestones. Furthermore, we will be supplying perifosine to Hikma on a cost-plus basis and are entitled to receive double-digit royalties on future net sales of perifosine in the MENA region. Hikma will be responsible for the registration and commercialization of perifosine in the MENA region. We own rest of the world rights to perifosine.
AEZS-127 erucylphosphocholine
On January 6, 2005, we announced the initiation of preclinical development of erucylphosphocholine (AEZS-127), an analog of perifosine which is suitable for i.v. administration. Like perifosine, AEZS-127 belongs to a new class of compounds based on alkylphosphocholines. AEZS-127 possesses distinctive reduced haemolytic activity thus allowing for i.v. injection.
On January 6, 2005, we also licensed to Keryx certain rights to develop and market AEZS-127 in North America, South Africa, Israel, Australia and New Zealand while keeping rights for the rest of the world. According to the agreement with Keryx, the preclinical development costs of AEZS-127 are shared between Keryx (50%) and us (50%). In the fourth quarter of 2008, we repatriated all rights for AEZS-127 from Keryx.
In 2006, studies for acute toxicity and dose range finding of erucylphosphocholine were actively pursued. The 4-week toxicity studies in rats and dogs as well as the safety pharmacology package was completed in 2007. These preclinical data are a prerequisite for the performance of a Phase 1 clinical study.
1.1.2 Erk/PI3K inhibitors and dual kinase inhibitors
In addition to our activities with alkylphosphocholines, we are screening small molecules for activity as agonists and antagonists to lipid-protein signaling interactions, which are seen as new and potentially important therapeutic targets.
We are focusing our efforts on single and dual inhibitors of Ras-Raf-Mek-Erk and PI3K-Akt pathways. The Ras-Raf-Mek-ErkRas/Raf/Mek/Erk and the PI3K-AktPI3K/Akt signaling pathways are constitutively activatedprime targets for drug discovery in many cancer types,proliferative diseases such as cancer. The results of research to date indicate that both the MAPK and influence both tumor development and progression.
Boththe PI3K signaling pathways represent promising therapeutic targetsintervention points for the clinical treatment of malignant tumors. We have now identified a new compound class with inhibitory activity against both the Erk and PI3K
kinases. These small molecules inhibit the kinases at nanomolar concentrations in a dose-dependent manner by competing directly at the ATP binding site. In a broad kinase panel, the molecules are very selective against other kinases. In cellular experiments the compounds inhibit the activation of downstream targets Akt and Rsk1, and can stop the proliferation of various human cancer cell lines. Moreover, a new generation of aniline-substituted pyridopyrazine-urea derivative shows highly selective PI3K inhibition. We are currently performingin vivo studies with front-runner compounds in four mouse xenograft models (HCT116, U87, A549 and PC3) as well as pharmacokinetic studies in rodents using an oral pre-formulation. On the basis of these studies, AEZS-126 was selected as a preclinical development candidate forin vivo pharmacology and pharmacokinetic studies.
AEZS-126
The firstOur multi-parameter optimization program for kinase inhibitor selectivity, cellular efficacy, physicochemical andin vitro andin vivo data for AEZS-126 were presented in April 2009 at the AACR meeting. The first poster, entitled, "AEZS-126, a new orally bioavailable PI3K inhibitor with antitumor effects", focuses on ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) and safety profiling of the compound, as well asin vivo pharmacokinetic experiments and mouse xenograft antitumor studies. Results indicated that AEZS-126 was identified as a potent inhibitor of class I PI3Ks in biochemical and cellular assays and demonstrated favorable properties in earlyin vitro ADMET screening including microsomal stability, plasma stability and screening against a large safety profile composed of receptors, enzymes and cardiac ion-channels. During the course ofin vivo pharmacokinetic experiments and mouse xenograft antitumor studies, the oral bioavailability in mice was determined to be about 60%, leading to micromolar plasma levels which are well above the nanomolar IC50 valuesin vitro studies. Significant antitumor activity was observed at 30 mg/kg daily oral administration in Hct116 and A549 models. These data suggest that AEZS-126 is a promising compound for clinical intervention of the PI3K/Akt pathway in human tumors.
The 2nd poster, entitled "In vitro profiling of the potent and selective PI3K inhibitor, AEZS-126", outlines the keyin vitro characteristics of this compound thathas led to its selectionthe identification of small molecular compounds with a unique kinase selectivity profile. Our kinase research program comprises the investigation of different compounds forin vivo development. AEZS 126 inhibits PI3Ka with an IC50 value of 10nM single Erk inhibition, single PI3K inhibition and proved to be a potent inhibitor of Akt phosphorylation in cellular assays. Mode-of-action studies showed that AEZS-126 acts as an ATP competitive compound. Thein vitro antiproliferative activity against different human tumor cell lines (MDA-MB 468, U87, Hct116, PC-3, A549 and others) was determined, with EC50 values in the nanomolar range. Based on those results presenting a favorablein vitro pharmacologic profile for AEZS-126, furtherin vivo profiling experiment will be performed.dual Erk/PI3K kinase inhibition.
1.1.2.1 AEZS-129
On April 21, 2009, we presented two posters on AEZS-129, a promising compound for clinical intervention of the PI3K/Akt pathway in human tumors, at the AACR Annual Meeting.In vivo andin vitro data showed significant antitumor activity and a favorablein vitro pharmacologic profile which could lead to furtherin vivo profiling.
On November 17, 2010, we presented a poster on encouraging preclinical results for AEZS-129, a novel orally active compound with anti-tumorantitumor effects, at the 22nd EORTC-NCI-AACR Symposium on Molecular Targets and Cancer Therapeutics in Berlin, Germany. AEZS-129 has been identified as a highly potent and selective inhibitor of PI3K. The compound inhibits the PI3K/Akt signaling pathway bothin vitro andin vivo and leads to growth inhibition of tumor cells. The compound was well tolerated during the 4 week treatment period and showed substantial tumor growth inhibition in different mouse xenograft tumor models.
TableOn March 22, 2011, we presented preclinical results for AEZS-129 at the Informa Life Sciences Protein Kinases Congress in Berlin, Germany. AEZS-129 was identified as a potent inhibitor of Contentsclass I PI3Ks lacking activity against mTOR. Lack of mTOR activity is considered to potentially lead to a better safety profile. In biochemical and cellular assays, AEZS-129 demonstrated favorable properties in earlyin vitro ADMET screening, including
microsomal stability, plasma stability and screening against a safety profile composed of receptors, enzymes and cardiac ion-channels.In vitro, the compound was shown to be a selective ATP-competitive inhibitor of PI3K with a broad antiproliferative activity against a broad panel of tumor cell lines.In vivo, AEZS-129 showed excellent plasma exposure and significant tumor growth inhibition in several tumor xenografts models, including A-549 (lung), HCT-116 (colon) and Hec1B (endometrium). These data suggest that AEZS-129 is a promising compound for clinical intervention of the PI3K/Akt pathway in human tumors.
1.1.2.2 AEZS-131
On March 22, 2011, we presented preclinical results for AEZS-131 at the Informa Life Sciences Protein Kinases Congress in Berlin, Germany. AEZS-131 was established as a small molecular compound that inhibits Erk in the low nanomolar (“nM”) range and shows an excellent selectivity profile. Further characterization experiments revealed an ATP-competitive mode of action and the potent inhibition of the cellular downstream target Rsk1 in tumor cells. The frontrunner, AEZS-131, produces cell cycle arrest in G1 and results in growth inhibition of cancer cells. Furthermore, the potential of combination therapy of AEZS-131 with inhibitors of the PI3K pathway was addressed and the analysis of combination effects on tumor cell proliferation has been presented. These results support the evaluation of selective Erk inhibitors as antiproliferative agents either as monotherapy or in combination with inhibitors of the PI3K/Akt pathway.
On April 5, 2011, we announced results for AEZS-131 at the AACR’s annual meeting. Results showed that AEZS-131 is an orally active small molecular compound that selectively inhibits Erk 1/2 with an IC50 of 4nM, blocks cellular Rsk-1 phosphorylation, modulates downstream cellular substrate activation, arrests tumor cells in G1 and inhibits the growth of multiple human tumor cell lines in the nM range. In in vivo pharmacokinetic studies, AEZS-131 showed a favorable PK profile. Antitumor activity was studied in in vivo mouse xenograft experiments utilizing the HCT 116 CRC model. AEZS-131 significantly inhibited tumor growth and was well tolerated at daily doses up to 120 mg/kg. Focus on inhibition of downstream kinase Erk 1/2 activity as a therapeutic target may be attractive because the pharmacologic inhibition of Erk 1/2 reverses Ras and Raf activation in cells which also demonstrate resistance to common Raf inhibitors, such as PLX-4720/4032.
AEZS-132On August 30, 2011, we presented preclinical data on AEZS-131 at the American Chemical Society National Meeting in Denver, Colorado. Data showed that thein vitro antiproliferative efficacy proved to be excellent in diverse human tumor cell lines and that GI50 values in the low nM range were obtained.In vivo antitumor activity was studied in a mouse xenograft experiment utilizing the human HCT-116 colon cancer model. Up to 74% inhibition of tumor growth was achived with daily oral doses of 30 – 120 mg/kg. Our medicinal chemistry programs are supported by X-ray crystallography and modeling towards the optimization of pyrido[2,3-b]pyrazines as novel series of kinase inhibitors.
On December 9, 2011, we announced positive preclinical data in triple-negative breast cancer (“TNBC”) for AEZS-131. Data showed that AEZS-131 selectively inhibits Erk at low nM concentrations and induces G1 arrest. Accordingly, the cytotoxic effect of AEZS-131 was most pronounced in TNBC cell lines with mutations in the MAPK pathway. Data were presented at the CTRC - AACR San Antonio Breast Cancer Symposium, in San Antonio, Texas. AEZS-131 was tested to check for selectivity, inhibition of Rsk-phosphorylation (cellular substrate of Erk), mode of action and cleavage of PARP. Cytotoxic efficacy was evaluated in a selection of TNBC cell lines, with or without mutations in the MAPK signal transduction pathway, by MTT assay. The study showed that AEZS-131 selectively inhibited Erk with an IC50<4nM. Phosphorylation of Rsk-1, the cellular substrate of Erk, was inhibited with an IC50 of 158 nM. AEZS-131 induced cell cycle arrest in G1 dose-dependently and cleavage of PARP. EC50 values were below 1µM for cell lines with mutations in the MAPK pathway. TNBC cell lines without mutations in the MAPK pathway were less responsive.
1.1.2.3 AEZS-132
On April 20, 2010, at the AACR'sAACR’s annual meeting we presented data on our dual Erk/PI3K inhibitors and on our selective Erk inhibitors. Data supported further evaluation of selective Erk inhibitors as antiproliferative agents, either as monotherapy or in combination with inhibitors of the PI3K/Akt pathway. Other data resulted in the identification of AEZS-132, a unique dual inhibitor of PI3K and Erk with a favourable pharmacology and ADMET profile, for further evaluation as an antitumor agent.
On November 17, 2010, at the EORTC-NCI-AACR meeting, we presented a poster on AEZS-132, the first-in-class dual PI3K/Erk inhibitor being selected as the optimized lead compound for further development. The compound is a unique orally active low molecular weight dual PI3K/Erk inhibitor derived from Aeterna Zentaris'Zentaris’ medicinal chemistry program. Due to its dual PI3K and Erk inhibition, a broad anti-tumorantitumor activity is expected in tumors with over-activation of both pathways. AEZS-132 demonstrated prolonged plasma exposure when given orally in mice. Significant tumor inhibition resulted from mouse xenograft models with human colon, endometrium and lung tumors.
TUMOR TARGETING CYTOTOXIC CONJUGATES AND CYTOTOXICSOn March 22, 2011, we presented preclinical results for AEZS-132 at the Informa Life Sciences Protein Kinases Congress in Berlin, Germany. AEZS-132 is a unique dual inhibitor of PI3K and Erk in the nM range and exerts high selectivity against other serine threonine and tyrosine kinases. AEZS-132 is also an ATP-competitive inhibitor, with a broad antiproliferative profilein vitro, a favorable safety profile and beneficial ADME properties.In vivo pharmacokinetic experiments showed plasma profiles expected to result in positive antitumor efficacy, and led to significant antitumor activity in mouse xenograft models, including HCT-116 (colon), A-549 (lung), and Hec1B (endometrium). Cellular inhibition of the downstream targets p-Akt and p-Rsk was confirmed within thein vivo tumor studies. In summary, we hope that AEZS-132, by targeting the PI3K and MAPK pathways, will be especially suited to treat tumors with over-activation of both pathways.
| 1.2 | TUMOR TARGETING CYTOTOXIC CONJUGATES AND CYTOTOXICS |
Cytotoxic conjugates
In view of the non-specific toxicity of most chemotherapeutic agents against normal cells, targeting such drugs to cancerous tissue offers a potential benefit for patients with advanced or metastatic tumors. Targeted cytotoxic peptide conjugates are hybrid molecules composed of a cytotoxic moiety linked to a peptide carrier which binds to receptors on tumors. Cytotoxic conjugates are designed to achieve differential delivery, or targeting, of the cytotoxic agent to cancer vs. normal cells.
Our cytotoxic conjugates represent a novel oncological strategy to control and reduce toxicity and improve the effectiveness of cytotoxic drugs. The development strategy was to create targeted conjugates with high cytotoxic activity based on doxorubicin, an approved and commercialized product or 2-pyrrolino-doxorubicin which is 500 to 1,000 times more active than the parent compound. We are exploring several candidates in which doxorubicin or 2-pyrrolino-doxorubicin are coupled to the peptide carriers targeting LHRH (AEZS-108 & AN-207), somatostatin (AN-238 & AN-162) or bombesin (AN-215) receptors. These conjugates are less toxic and more effectivein vivo than the respective radicals in inhibiting tumor growth in LHRH receptor positive models of human ovarian, mammary or prostatic cancer.
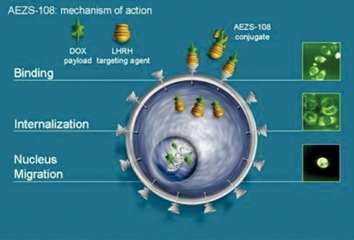
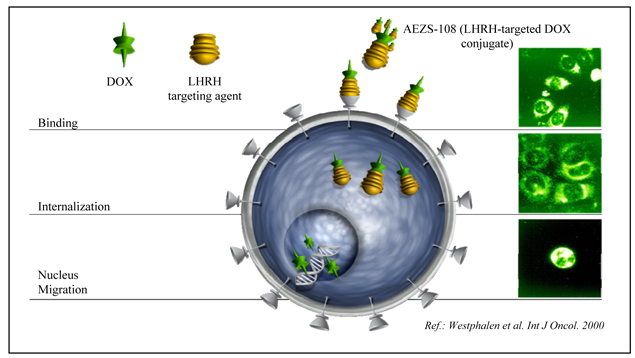
In AEZS-108, the most advanced of theour cytotoxic conjugates, doxorubicin is chemically linked to an LHRH agonist, a modified natural hormone with affinity for the LHRH receptor. This design allows
for the specific binding and selective uptake of the cytotoxic conjugate by LHRH receptor positivereceptor-positive tumors. Potential benefits of this targeted approach include a more favorable safety profile with lower incidence and severity of side effects, as normal tissues are spared from toxic effects of doxorubicin. In addition, the targeted approach may enable treatment of LHRH receptor positivereceptor-positive cancers that have become refractory to doxorubicin which has been administered in its non-targeted form.
In preclinical studies conducted to date in several animal models of LHRH receptor positivereceptor-positive human cancer cell lines, AEZS-108 anti-tumorantitumor activity and tolerability were shown to be superior to that of doxorubicin. As would be expected, AEZS-108 was not active or was significantly less active than doxorubicin in LHRH receptor negativereceptor-negative cancer cell lines. On January 18, 2005, we announced the initiation of a company-sponsored Phase 1 dose-ranging study with the targeted anti-canceranticancer agent AEZS-108.
In June 2006 and on November 27, 2006, we announceddisclosed positive Phase 1 results forregarding AEZS-108 in patients with gynaecological and breast cancers whichcancers. Data showed that the compound has acompound’s good safety profile and no dose-limiting toxicities. Eight patients received AEZS-108 by i.v. infusion.established the maximum tolerated dose at 267 mg/m2, which is equimolar to a doxorubicin dose of 77 mg/m2. Infusion was well tolerated at all dosages without supportive treatment. Pharmacokinetic analyses showed dose-dependent plasma levels of AEZS-108 and only minor (10-20%) release of doxorubicin. Stabilization of disease was observed in one out of eight patients in the ongoing Phase 1 study.
On November 27, 2006, we disclosed additional positive Phase 1 results regarding AEZS-108 in patients with gynaecological and breast cancers. Further data showed the compound's good safety profile and established the maximum tolerated dose at 267 mg/m2, which is equimolar to a doxorubicin dose of 77 mg/m2. This dose will be the recommended dose for a Phase 2 trial. The Phase 1 open-label, multi-center,multicenter, dose-escalation, safety and pharmacokinetic study conducted in Europe included 17 patients suffering from breast, endometrial and ovarian cancers with proven LHRH receptor status. Evidence of anti-tumorantitumor activity was found at 160 mg/m2 and 267 mg/m2 doses of AEZS-108, where 7 out of 13 patients showed signs of tumor response, including 3 patients with complete or partial responses. TheRecommended dose for Phase 2 trials will focus on advanced or recurrent ovarian and endometrial cancers, two forms of cancer where LHRH receptors are highly expressed. Recommended dose will betrial was 267 mg/m2 given once every three weeks.weeks (source: Emons et al. Dose escalation and pharmacokinetic study of AEZS-108 (AN-152), an LHRH agonist linked to doxorubicin, in women with LHRH receptor-positive tumors. Gynecol Oncol. 2010 Dec;119(3):457-61).
AEZS-108 — Ovarian and Endometrial Cancer
| 1.2.1 | AEZS-108 — Ovarian and Endometrial Cancer |
In 2007, a Phase 2 open-label, non-comparative, multicenter two indication trial stratified with two stages Simon Design was prepared. The study involved 82 patients with up to 41 patients with either a diagnosis of platinum-resistant ovarian cancer (stratum A) or disseminated endometrial cancer (stratum B). Under coordination by Prof. Günter Emons, MD,M.D., Chairman of the Department of Obstetrics & Gynaecology at the University of Göttingen, Germany, this open-label, multi-centermulticenter and multi-nationalmultinational Phase 2 study "AGO-GYN 5"“AGO-GYN 5” was being conducted by the German AGO Study Group (Arbeitsgemeinschaft Gynäkologische Onkologie / Gynaecological Oncology Working Group), in cooperation with clinical sites in Europe. InAs part of a personalized healthcare approach, the study selected patients with tumors expressing LHRH receptors, anthe key element in the targeting mechanism of AEZS-108. An i.v. infusion of AEZS-108 (267 mg/m2) was administered over a period of 2 hours, every Day 1 of a 21-day (3-week) cycle. The proposed duration of the study treatment was 6 courses of a 3-week cycles. The study was performed with 14 centers of the German Gynaecological Oncology Working Group, in cooperation with 3 clinical sites in Europe. The primary efficacy endpoint at the end of Stage II was defined as 5 or more patients with partial or complete tumor responses according to RECIST and/or Gynaecologic Cancer Intergroup (GCIG)(“GCIG”) guidelines. Secondary endpoints included time to progression,TTP, survival, toxicity, as well as adverse effects. On February 12, 2008, we reported that the treatment of first patients had commenced in this Phase 2 trial. In October 2008, we announced that we had entered the second stage of patient recruitment for the Phase 2 trial in platinum-resistant ovarian cancer indication. This decision was taken following the report of two partial responsesPR among patients with a diagnosis of platinum-resistant ovarian cancer. The second stage of patient recruitment for the
endometrial cancer indication was reached in November 2008 and was based on the report of one complete responseCR and two partial responsesPR among 14 patients with a diagnosis of disseminated endometrial cancer.
In November 2009, we announced preliminary positive efficacy data from this Phase 2 study in patients with platinum-resistant and taxane-pretreated ovarian cancer. In a personalized healthcare approach, the study selectedcancer and in patients with tumors expressing LHRH receptors,advanced or recurrent endometrial cancer. Of the key element in the targeting mechanism of AEZS-108. All 42 patients with LHRH-receptor positiveLHRH receptor-positive ovarian cancer and the 43 patients with LHRH receptor-positive endometrial cancer who entered study AGO-GYN, 5 had completed their study treatment. A preliminary evaluation showed that the studyboth indications met itsthe primary efficacy endpoint of 5 or more responders in 41 evaluable patients. Responders, as well as patients with stable disease after completion of treatment with AEZS-108, were to be followed to assess the duration of progression-free survivalPFS and, ultimately, overall survival.OS.
In November 24, 2009, we announced positive efficacy data from the Phase 2 study with the targeted cytotoxic peptide conjugate, AEZS-108, in patients with advanced or recurrent endometrial cancer. A preliminary evaluation showed that the study AGO-GYN 5 had met its predefined primary efficacy endpoint of 5 or more responder patients with endometrial cancer. Responders, as well as patients with stable disease after completion of treatment with AEZS-108, were to be followed to assess the duration of progression-free survival and, ultimately, overall survival.
On May 6, 2010, we announced that we had received orphan drug designation from the FDA for AEZS-108 for the treatment of ovarian cancer. Orphan drug designation is granted by the FDA's Office of Orphan Products Development to novel drugs or biologics that treat a rare disease or condition affecting fewer than 200,000 patients in the U.S. The designation provides a drug developer with a seven-year period of U.S. marketing exclusivity if the drug is the first of its type approved for the specified indication or if it demonstrates superior safety or efficacy versus another drug of its type previously granted the designation for the same indication.
On May 17, 2010, we announced that we had received a positive opinion for orphan medicinal product designation from the COMP of the EMA for AEZS-108 for the treatment of ovarian cancer. Orphan medicinal product designation is granted by the European Commission, following a positive opinion from the COMP, to a medicinal product that is intended for the diagnosis, prevention or treatment of a life-threatening or a chronically debilitating condition affecting not more than five in 10,000 persons in the Community when the application for designation is submitted. Orphan medicinal product designation provides the sponsor with access to the Centralized Procedure for the application for marketing authorization, protocol assistance, up to a 100% reduction in fees related to a marketing authorization application, pre-authorization inspection and post-authorization activities, and could provide ten years of market exclusivity in the European Union for AEZS-108, once approved for the treatment of ovarian cancer.
On June 7, 2010, Prof. Günter Emons, Chairman, Department of Obstetrics & Gynaecology Georg-August University Göttingen, Germany, presented positive efficacy and safety data for AEZS-108 in ovarian cancer at the ASCO Annual Meeting. The poster (abstract #5035) entitled "PhasePhase 2 study of AEZS-108, a targeted cytotoxic LHRH analog, in patients with LHRH receptor-positive platinum resistant ovarian cancer" (G. Emons, S. Tomov, P. Harter, J. Sehouli, P. Wimberger, A. Staehle, L. C. Hanker, F. Hilpert, P. Dall and C. Gruendker, for the AGO Study Group)cancer, details the use of AEZS-108 in women with histologically confirmed taxane-pretreated platinum-resistant/refractory LHRH receptor-positive advanced (FIGO III or IV) or recurrent ovarian cancer. Patients received a recommended dose of 267 mg/m2 by intravenous infusion over 2 hours, with retreatment every 3 weeks, for up to 6 courses. Response rate (RECIST and/or GCIG criteria) was defined as the primary endpoint. Secondary endpoints were safety, time-to-progression and overall survival.
Forty-two patients with platinum-resistant ovarian cancer entered the study. Efficacy included partial responsePR in 5 patients (11.9%) and stable disease for more than 12 weeks in 11 patients
(26.2%). Based on those data, a CBR of 38% can be estimated. Median time to progressionTTP and overall survivalOS were 3.5 months (104 days) and 15.6 months (475 days), respectively. Overall survivalOS compares favourably with data from Doxil® and Topotecantopotecan (8-9 months). In all, tolerability of AEZS-108 was good and commonly allowed retreatment as scheduled. Only one patient (2.4%) had a dose reduction, and overall, 25 of 170 (14.7%) courses were given with a delay, including cases in which delay was not related to toxicity. Severe (Grade 3 or 4) toxicity was mainly restricted to rapidly reversible hematologic toxicity (leukopenia / neutropenia) associated with fever in 3 cases. Good tolerability of AEZS-108 was also reflected with only a few patients with non-hematological toxicities of gradeGrade 3 (none with Grade 4), including single cases (2.4%) each of nausea, constipation, poor general condition, and an enzyme elevation. No cardiac toxicity was reported.
On November 18, 2010, Prof. Günter Emons of the Department of Obstetrics & Gynaecology Georg-August at the University of Göttingen (Germany)September 14, 2011, we presented positive data for thefinal Phase 2 ofefficacy and safety data for AEZS-108 in advanced endometrial cancer at the EORTC-NCI-AACR symposiumEuropean Society of Gynecological Oncology in Berlin, Germany. The studyMilan, Italy. Data showed encouraging results as AEZ-108 was usedthat AEZS-108, administered as a single agent.
Of 43 patients treated with AEZS-108 in this study, 39 were evaluable for efficacy. Responses confirmed by independent review included 2 patients with complete response (CR; 5.1%), 10 patients with partial response (PR; 25.6%), and 17 patients with stable disease (SD; 43.6%). Based on those data, an overall response rate (ORR = CR+PR) of 30.8% and a clinical benefit rate (CBR = CR+PR+SD) of 74.4% can be estimated. Responses were also achieved in patients with prior chemotherapy, 1 CR, 1 PR and 2 SDs in 8 of the patients pre-treated with platinum/taxane regimens. Median time to progression and overall survival were 7 months (30 weeks) and 14.3 months (62 weeks), respectively. Conclusions from this trial were as follows:
The overall survival after single agent AEZS-108that OS is similar to that reported for modern triple combination chemotherapy, but was achieved with lower toxicity.
Competitors for AEZS-108 in Ovarian Cancer Indication The primary endpoint was the response rate as defined by the RECIST. Secondary endpoints included safety, TTP and OS.
Products on the market:
Major products available on the marketIn all, of 43 patients treated with AEZS-108, 39 were evaluable for the treatment of resistant or refractory ovarian cancers:
Doxil® (pegylated liposomal doxorubicin — manufacturedefficacy. Efficacy confirmed by Schering Plough) a topoisomerase II inhibitorindependent response review included 2 CR, 10 PR, and DNA intercalating, is approved for 2nd-line treatment in women with advanced ovarian cancer who have failed a 1st-line platinum based chemotherapy regimen.
Gemzar® (Gemcitabine — manufactured by Eli Lilly) is a deoxycytidine analogue, a pyrimidine antimetabolite related to cytarabine. The drug exhibits cell phase specificity, killing cells undergoing DNA synthesis. Gemcitabine is a prodrug and is metabolized intra cellularly to the active di-phosphate and tri-phosphate nucleosides. A small increase in the share of gemcitabine and carboplatin combination second line treatment in platinum sensitive population who has suffered paclitaxel-associated neurological toxicity from 1st-line treatment is expected. A decrease in sales of gemcitabine of under $17 million in 2009 to under $9 million in 2019 is expected.
Yondelis® (Trabectedin — manufactured by PharmaMar/Centocor Ortho Biotech's Yondelis) is a tris, tetrahydroisoquinoline alkaloid which binds to the major groove of DNA, generating the formation of lethal DNA strands which causes cell cycle arrest and apoptosis. Trabectedin is used in 2nd-line therapy in combination with Doxil® in relapsed platinum sensitive ovarian cancer. The drug was approved in Europe in September 2009. European sales of Trabectedin are estimated to be under $22 million in 2019 as the drug has a poor toxicity profile.
Hycamtin® (Topotecan — manufactured by GlaxoSmithKline) is a topoisomerase I inhibitor used for the treatment of17 patients with metastatice carcinomaSD. Based on those data, the estimated ORR (ORR = CR+PR) was 30.8% and the CBR (CBR = CR+PR+SD) was 74.4%. Responses in patients previously treated with chemotherapy included 1 CR, 1 PR and 2 SDs in 8 of the ovarypatients with prior use of platinum/taxane regimens. Median TTP and OS were 7 months and 13.7 months, respectively.
Overall, tolerability of AEZS-108 was good and commonly allowed retreatment as scheduled. Severe (Grade 3 or 4) toxicity was mainly restricted to rapidly reversible leukopenia and neutropenia, associated with fever in only 1 patient who had been treated only 3 weeks after failurea surgery. Good tolerability of 1st-line or subsequent therapy. The drugAEZS-108 was approvedalso reflected by the FDA in 2007.
Products in Phase 3 development:a low rate of severe non hematological possibly drug-related adverse events which included single cases each of nausea, diarrhea, fatigue, general health deterioration, creatinine elevation, and blood potassium decrease. No cardiac toxicity was reported.
Farletuzumab (MORAb-003 — manufactured by Esai/Morphotek) is a folate receptor inhibitor targeting folate receptor alpha which is over-expressed on a number of epithelial-derived cancers such as ovarian cancer. A Phase 3 study of MORAb-003 in subjects with platinum sensitive ovarian cancer in first relapse started in 2009 and is currently recruiting patients. MORAb-003 received orphan drug designation by the FDA. Farletuzumab is expected to be approved for 2nd-line treatment of platinum sensitive ovarian cancer in the U.S. and Europe in 2013 and Japan in 2015. Approval for platinum resistant and refractory ovarian cancer is estimated for 2016 in U.S. and Europe.
Avastin® (Bevacizumab — manufactured by Roche/Genentech) is a humanized monoclonal antibody targeting vascular endothelial growth factor. Off label sales of bevacizumab in the 3rd-line treatment will remain small but almost stable over the period 2009 to 2019 increasing from $1.4 million to $1.7 million.
Market Data — Ovarian Cancer
According to Decision Resources February 2011, the number of ovarian cancer drug treatable populations in the major markets (U.S., Europe — G5 and Japan) was mentioned as being 117,466 for the year 2010 and the number of total incident cases was mentioned as being 57,340 for 2010.
Competitors for AEZS-108 in Endometrial Cancer Indication
At present, there is no approved drug product for the treatment of advanced and recurrent metastatic endometrial cancer in the U.S.United States and Europe. There is also no systemic therapy approved in the U.S.United States and Europe for treating advanced or recurrent endometrial cancer.
Letrozol — Novartis: Letrozol is a non-steroidal aromatase inhibitor which completed Phase 2 clinical development in the treatment of advanced or recurrent hormone receptor positive endometrial cancer.
A Phase 2 study was performed in collaboration with Sanofi Aventis investigating how well carboplatin and docetaxel followed by radiation therapy works in treating patients with Stage III, Stage IV or recurrent endometrial cancer.
XL-147 — Exelixis: XL-147 is a potent and highly selective inhibitor of the class I PI3K family of lipid kinases which targets the PI3K/PTEN pathway. The drug is currently in phase 2 development for treatment of advanced or recurrent endometrial cancer.
Product / mode of action* | Company* | Development Status* | ||
| Ixabepilone / microtubule inhibitor | Bristol-Myers Squibb | Phase 3 | ||
| Letrozole / non-steroidal aromatase inhibitor | Novartis | - Phase 2 completed - Phase 3 (University of California, Davis) | ||
| XL-147 (Exelixis) / potent and highly selective inhibitor of the class I PI3K family of lipid kinases which targets the PI3K/PTEN pathway | Sanofi | Phase 2 | ||
| * | Source: Company’s Website and www.clinicaltrials.gov |
Market Data — Endometrial Cancer
According to the American Cancer Society, endometrial cancer is the most common invasive gynecologic cancer in women in the United States, with an estimated 43,47047,130 new cases expected to occur in 2012. This disease primarily affects postmenopausal women at an average age of 60 years at diagnosis. In the United States, it is estimated that approximately 8,010 women will die of endometrial cancer were expectedin 2012.
According to be diagnosedDatamonitor Healthcare (March 2010), a research and advisory firm focusing on therapeutic, strategic and ehealth market analysis and competitive intelligence, the incidence of endometrial cancer in 2010the seven major pharmaceutical markets was 92,180 patients and 7,950 deaths were expected during the same year.
AEZS-108 — Prostate and Bladder Canceris forecasted to reach about 98,500 cases by 2019.
| 1.2.2 | AEZS-108 — Prostate and Bladder Cancer |
In May 2009, we announced at the ASCO meeting the results supporting the evaluation of AEZS-108 in prostate cancer. Expression of LHRH receptors was determined using immunohistochemistry and the intensity was graded on a scale from zero to 3. The expression was
analyzed in three cohorts of patients: (1) 47 men with localized prostate cancer treated with radical prostatectomy with no hormone therapy, (2) 61 men with localized prostate cancer treated with neoadjuvant LHRH agonists for varying duration prior to prostatectomy, and (3) 22 men with metastatic prostate cancer who received a palliative transurethral resection of the prostate after clinical progression. In the final cohort, 15 men were treated with castration and 7 were treated with LHRH agonists. 45 of 47 hormone naïve samples (95.7%) demonstrated LHRH receptor expression. Statistical analysis revealed a correlation between strong receptor expression and higher pathologic tumor stage as well as shorter overall survival.OS. 60 of 61 samples treated with neoadjuvant LHRH agonist therapy (98.4%) demonstrated LHRH receptor expression. All 22 samples from patients with metastatic disease demonstrated LHRH receptor expression. The majority of these samples demonstrated moderate to strong intensity. LHRH receptors are expressed on prostate cancers cells of hormone naïve and castrated patients. The expression of these receptors appears to persist despite prolonged treatment with LHRH agonists. The new results show continued expression of LHRH receptors in prostate cancer specimens after prolonged use of LHRH agonists. These data provide further support to the investigation of the drug in hormone-refractory prostate cancer, a major genitourinary cancer indication in male patients.
On May 12, 2010, we announced that the FDA had approved our Investigational New Drug application ("IND")IND application for AEZS-108 in LHRH receptor-positive urothelial (bladder) cancer. Following this approval from the FDA, this trial will be conducted by Dr. Gustavo Fernandez at the Sylvester Comprehensive Cancer Center at the University of Miami'sMiami’s Miller School of Medicine, and will include up to 64 patients, male and female, with advanced LHRH receptor-positive urothelial (bladder) cancer. The study will be conducted in two parts: first, a dose-finding part in up to 12 patients; subsequently, a selected dose will be studied for its effect on progression-free survival.PFS.
On August 5, 2010, we announced that the NIH had awarded Dr. Jacek Pinski, Associate Professor of Medicine at the Norris Comprehensive Cancer Center of the University of Southern California, a grant of approximately $1.5 million over three years to conduct a Phase 1/2 study in refractory prostate cancer with AEZS-108. The study, entitled "AA Phase I/II Trial of AN-152 [AEZS-108] in Castration- and Taxane-Resistant Prostate Cancer"Cancer, will enroll up to 55 patients and will be conducted in two portions: an abbreviated dose-escalation followed by a single arm, Simon Optimum two-stage design Phase 2 study using the dose selected in the Phase 1 portion. The primary objective of the Phase 2 portion is to evaluate the clinical benefit of AEZS-108 in men with castration- and taxane-resistant metastatic prostate cancer, for which the presence of LHRH receptors has been confirmed.
On December 14, 2010, we announced the initiation of aboth Phase 1/2 trials.
On September 26, 2011, we announced positive interim data for the Phase 1 portion of our Phase 1/2 trial with AEZS-108 in castration refractorycastration-and taxane-resistant prostate cancer conductedat the ESMO meeting, Stockholm, Sweden. This is a single arm study with a Phase 1 lead-in to a Phase 2 clinical trial. The primary endpoint of the Phase 1 portion is safety. The primary objective of the Phase 2 portion is to evaluate the clinical benefit of AEZS-108 for these patients. Twelve patients entered the study: 3 patients each received AEZS-108 at the lower dose levels of 160 and and 210 mg/m2, and 6 patients at 267 mg/m2. Data on 10 patients were presented as 2 patients were too early for evaluation. AEZS-108 was generally well tolerated and there were no dose limiting toxicities so far. The only Grade 3 and 4 toxicities were hematologic in nature. At the time, there were three Grade 4 toxicities and six Grade 3 toxicities. Signs of therapeutic activity included 5 patients with PSA regression. Three out of 4 evaluable patients with radiologic evaluable disease achieved stable disease per RECIST. The Phase 2 extension is planned after completion of the toxicity assessment in the final dose level of the Phase 1 portion of the study. In correlative studies, drug internalization was demonstrated for the first time in captured circulating tumor cells of patients, thus validating the principle of targeted tumor therapy with AEZS-108 in a clinical setting.
On February 3, 2012, we reported positive updated results for the Phase 1 portion of our ongoing Phase 1/2 study in castration- and taxane-resistant prostate cancer (“CRPC”) with AEZS-108. This is a single-arm study with a Phase 1 lead-in portion (testing 3 dose levels) to a Phase 2 clinical trial. The primary endpoint of the Phase 1 portion is safety. The primary objective of the Phase 2 portion is to evaluate the clinical benefit of AEZS-108 for these patients. Data were presented by Dr. Jacek Pinski, M.D., Ph.D., Associate Professor of Medicine at the Norris Comprehensive Cancer Center asof the University of Southern California, during a poster session at the American Society of Clinical Oncology Genitourinary Cancers Symposium which is being held in San Francisco. Prior interim data on this study were presented at the European Society of Medical Oncology Congress in September 2011. The trial is being supported by a three-year US$1.6 million grant from the National Institutes of Health to Dr. Pinski.
The results were based on 13 patients that were treated on 3 dose levels: 3 at 160 mg/m2, 3 at 210 mg/m2, and 7 at 267 mg/m2. Overall, AEZS-108 was well astolerated among this group of heavily pretreated older patients. To date, there have been 2 dose limiting toxicities; both were cases of asymptomatic Grade 4 neutropenia at the 267 mg/m2 dose level and both patients fully recovered. The Grade 3 and 4 toxicities were primarily hematologic. There has been minimal non-hematologic toxicity, most frequently fatigue and alopecia. Despite the low doses of AEZS-108 in the first cohorts, there was some evidence of antitumor activity. One patient received 8 cycles (at 210 mg/m2) due to continued benefit. Among the 5 evaluable patients with measurable disease, 4 achieved stable disease. At the time of submission of the abstract, a decrease in PSA was noted in 6 patients. Six of 13 (46%) treated patients received at least 5 cycles of therapy with no evidence of disease progression at 12 weeks. Correlation studies on circulating tumor cells (“CTCs”) demonstrated the uptake of AEZS-108 into the targeted tumor. After completion of 3 additional patients at the 210 mg/m2 dose level, the study is expected to be extended into the Phase 1/2 trial in refractory bladder cancer conductedportion.
1.2.3 AEZS-108 — Triple-Negative Breast Cancer
On October 25, 2011, we announced that the FDA had completed the review of the IND application filed by Dr. Gustavo Fernandez atAlberto J. Montero M.D. of the Sylvester Comprehensive Cancer Center.Center and concluded that Dr. Montero may proceed with the initiation of a randomized Phase 2 trial in chemotherapy refractory triple-negative (ER/PR/HER2-negative) LHRH receptor-positive metastatic breast cancer with AEZS-108. This open-label, randomized, two-arm, multicenter Phase 2 study will involve up to 74 patients who will be randomized in a 1:1 ratio into one of the two treatment arms: AEZS-108 (267 mg/m2 every 21 days) [Arm A] or standard single agent cytotoxic chemotherapy [Arm B].
The primary study endpoint will be PFS. Secondary endpoints will include ORR and OS. The study will also evaluate AEZS-108’s toxicity profile and the patients’ quality of life relative to conventional cytotoxic chemotherapy used in the control arm of this study.
1.2.4 AEZS-108 - Companion diagnosticDiagnostic Tool
On June 28, 2010,January 5, 2012, we announced that we had concluded anentered into a collaboration agreement, dated December 19, 2011, with Almac's Diagnostics division for AEZS-108, aimed at determining LHRH receptor expression through the development ofVentana, to develop a companion diagnostic tool. Selection for treatment with AEZS-108 is determined on the basisimmunohistochemical determination of LHRH receptor expression, currently measured immunohistochemically.for AEZS-108. In humans, LHRH receptors are expressed in a significant proportion of endometrial, ovarian, endometrial, breast, bladder, prostate and pancreatic tumors. This stateAEZS-108 specifically targets LHRH receptors and could therefore prove to be more efficient in treating patients with these types of LHRH receptor-positive cancers.
1.2.5 AEZS-137 (Disorazol Z) / AEZS-125 (LHRH-Disorazol Z)
In search of new antitumor agents, we found that disorazol Z (AEZS-137), isolated from the myxobacteriumSorangium cellulosum, possesses remarkable, non-specific cytotoxicity in a panel of different tumor cell lines. Inhibition of tubulin polymerization, cell cycle arrest and efficient induction of apoptosis, have been identified as modes of action. AEZS-137 is thus a bacterially produced compound with cytotoxic activity in the sub-nanomal range.
In order to obtain a specifically acting antitumor agent, we have linked disorazol Z to [D-Lys6] LHRH. The resulting conjugate, AEZS-125, has been characterized with respect toin vitro andin vivo antitumor activity. In CD 1 nu/nu mice xenografted with the LHRH receptor-positive, human ovarian carcinoma cell line OVCAR-3, we have shown tumor suppression by single administration of AEZS-125 in doses as low as 45 nmol/kg (0.1 mg/kg). Proof of concept for this approach is the far more efficient tumor suppression obtained with AEZS-125 in comparison to equimolar doses of disorazol Z itself. The results were published during the 99th AACR Annual Meeting in April 2008 in San Diego, California.
On March 24, 2011, we were awarded a $1.5 million grant from the German Ministry of Education and Research to develop, up to the clinical stage, cytotoxic conjugates of the art companion diagnostic toolproprietary cytotoxic compound AEZS-137 and peptides targeting G-protein coupled receptors, including the LHRH receptors. The compounds being developed will allow us to develop improved methods of selectingcombine the most appropriate patients to be treatedtargeting principle successfully employed in Phase 2 with AEZS-108 in order to enhance the efficiency of our clinical trials(doxorubicin and help usLHRH receptor targeting agent) with the novel cytotoxic disorazol Z. Furthermore, diagnostic tools systematically assessing the receptor expression in tumor specimens will be developed to allow the future developmentselection of AEZS-108patients and tumor types with the highest chance of benefiting from this personalized medicine approach. The grant will be payable as a partial reimbursement of qualifying expenditures over a three-year period. The qualified project will be performed with Morphisto GmbH and the Helmholts Institute in Saarbrücken, Germany, which will receive additional funding of approximately US$0.7 million. Researchers from the departments of Gynecology and Obstetrics at both the University of Göttigen and the University of Würzburg, Germany, will also be part of the collaboration.
On November 16, 2011, we announced the presentation of a poster at the AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics on encouraging preclinical data for AEZS-137. Data showed that AEZS-137 possesses outstanding cytotoxicity in a numberhighly diverse panel of 60 different LHRH expressingtumor cell lines, and also underlined the identification of important aspects of this novel natural compound’s mechanism of action. AEZS-137 has been identified as a tubulin binding agent with highly potent antitumor properties. Cell cycle analysis revealed that AEZS-137 arrested cells in the G2/M phase and subsequently induced apoptosis with remarkable potency, as shown by subnanomolar EC50 values. Currently, experiments are under way to determine the tubulin binding site for disorazol Z and to identify further mechanisms of action of this novel highly potent agent. To expand our AEZS-108 technology platform, we aim to evaluate the utility of disorazol Z as a cytotoxic component in a drug-targeting approach utilizing GPCR ligands as the targeting moieties for the treatment of GPCR over-expressing cancers.
TUBULIN INHIBITORS / VASCULAR TARGETING AGENTS
AEZS-112 — Development of a Low Molecular Weight Tubulin Inhibitor with Anti-Angiogenic Properties
| 1.3 | TUBULIN INHIBITORS / VASCULAR TARGETING AGENTS |
| 1.3.1 | AEZS-112 — Development of a Low Molecular Weight Tubulin Inhibitor with Antiangiogenic Properties |
Tubulin is a protein found in all cells that plays an important role during cell division, in that it helps to transmit genetic information to the daughter cells. Inhibition of this process leads to the death of the affected cell. The anti-tumor agentantitumor agents taxol and vincristine, which are widely used in cancer therapy, are based on this principle. Both compounds are expensive natural substances and cause severe side effects when used in humans.
We are currently identifying and developing novel tubulin inhibitors which, compared with currently used products, exhibit improved efficacy in animal models, improved efficacy, have a more acceptable side effect profile, an incomplete or no cross-resistance and are administered orally.
AEZS-112 is a drug development candidate with an excellenta favorable safety and tolerability profile showing excellentin vivo activity in various tumor models including mammary, colon, melanoma and leukemia cancers after oral administration.at acceptable and very well tolerated doses administered orally once weekly. This compound acts through three mechanisms of action. Strong anti-canceranticancer activity is combined with pro-apoptoticproapoptotic and anti-angiogenicantiangiogenic properties. AEZS-112 inhibits the polymerization of cancer tubulin rather than bovine brain tubulin, it destroys the mitotic spindle of the cancer cells and it inhibits topoisomerase II activity. AEZS-112 arrests the cancer cells in the G2M phase at a nanomolarnM concentration and induced apoptosis. AEZS-112 is not cross-resistant to cisplatin, vincristine and doxorubicine in cell lines resistant to these drugs. Given orally once weekly, AEZS-112 proved to be a potent inhibitor ofin vivo tumor growth in melanoma, mammary, colon, lung, renal as well as in leukemia cancers at acceptable and very well tolerated doses. Furthermore AEZS-112 showed favorable safety and toxicity profiles. No findings with respect to cardiotoxicity and neurotoxicology parameters could be observed during the toxicological evaluation in mice, rats and dogs. With this profile of activity, AEZS-112 is a promising candidate for further clinical development.
On January 8, 2007, we announced the initiation of a Phase 1 trial for AEZS-112 in patients with solid tumors and lymphoma. This open-label, dose-escalation, multi-center,multicenter, intermittent treatment Phase 1 trial is being conducted in the United States with Daniel D. Von Hoff, MD,M.D., Senior Investigator at the Translational Genomics Research Institute in Phoenix, AZ, as the lead investigator. The trial includes up to 50 patients with advanced solid tumors and lymphoma who have either failed standard therapy or for whom no standard therapy exists. Patients will receive a once-a-week oral administration of AEZS-112 for three consecutive weeks, followed by a one-week period without treatment. The cycles will be repeated every four weeks based on tolerability and response, basically planned for up to four cycles, but allowing for continuation in case of potential benefit for the patient. The starting dose of AEZS-112 in this study is 13 mg/week, with doubling of doses in subsequent cohorts in the absence of significant toxicity. Primary endpoint of the Phase 1 trial focuses on determining the safety and tolerability of AEZS-112 as well as establishing the recommended Phase 2 dose and regimen. Secondary endpoints are aimed at establishing the pharmacokinetics and determining the efficacy based on standard response criteria.
Results of this Phase 1 study were presented in April 2009 at the AACR meeting. In part I, 22 patients (12 men / 10 women) were studied on 7 dose levels ranging from 13 to 800 mg/week. In all, 62 treatment cycles were administered. In part II, the weekly dose was split into 3 doses taken 8 hours apart. Ultimately, 22 patients (12 men / 10 women) were studied on 5 dose levels ranging from 120 to 600 (= 200 x 3) mg/week. As at April 1, 2009, 62 treatment cycles were administered (mean 3.2/patient) and treatment werewas ongoing in 8 patients. SD for more than 12 weeks was observed in 16 patients; 4 more patients were ongoing at less than 12 weeks. Prolonged courses of SD ranging from 20 to 39+ weeks were observed in 9 patients with the following primary cancer types: trachea (39+), tongue (30+), thyroid (29+), prostate and melanoma (28), non-small cell lung cancer (26+), pancreas and 2x colorectal (20). Except for one patient with a background of gastrointestinal problems (GI)(“GI”) who had dose-limiting GI reactions and electrolyte loss at a dose of 200x3mg/200 x 3 mg/week, no clinically relevant
drug-related adverse events or changes in laboratory parameters were observed. AEZS-112 was shown to be metabolically stable in human plasma. As predicted by pharmacokinetic modelling based on data from part I of
the study, the split-dose scheme leads to a higher Cmax and trough values after administration of comparable doses. Those preliminary results showed that a maximum tolerated dose for weekly dosing has not been defined so far. However, prolonged courses of stable disease in both parts of the study are an encouraging observation.
Completion of this Phase 1 trial was announced on September 21, 2009. Stable disease with time to failure ranging from 20 to 60+ weeks was achieved in 12 patients with various cancer types, including melanoma and cancers of the colon/rectum, lung, pancreas, prostate, tongue, trachea and thyroid. In several of these patients, the duration of stabilization exceeded the duration of disease control on previous treatment regimens. Except for a dose-limiting gastrointestinal reaction in a patient with pre-existing GI problems, no clinically relevant drug-related adverse events or changes in laboratory safety parameters were observed.
IMMUNOTHERAPY / VACCINESDuring the year 2011, we have developed a higher concentrated oral formulation of AEZS-112 in order to improve patient compliance. Clinical protocol is in preparation to continue to advance AEZS-112 with this new formulation.
| 1.4 | IMMUNOTHERAPY / VACCINES |
| 1.4.1 | AEZS-120 |
Cellular proteins expressed by oncogenes have been recognized as a major cause of tumor development. One of the central oncoproteins involved in cancer formation are the Raf proteins. Based on these proteins, new unique therapeutic strategies, new predictive animal models and new development products have been generated to efficiently combat cancer. These consist of virulence attenuated, genetically modified bacteria expressing tumor antigens, including oncoproteins or enzymes. Such bacteria are used for vaccination as well as tumor targeting and delivery of antitumoral compounds towards the tumor tissues. Therefore, this new vaccine approach exploits the ability of bacteria to induce potent immune responses as well as direct these responses against malignancies. The immunogenicity of the vaccine will be further enhanced by the capacity of bacteria to colonize tumor tissues. This property will be used to transport substances, e.g. proteins, into the tumor tissue, which are capable of converting non-toxic pro-drugs into active drugs. The use of bacterial carriers for therapeutic vaccination against tumors and the concept of bacterial tumor targeting will be further developed with the Julius-Maximilians-University of Würzburg, including the highly recognized researchers Prof. Dr. Ulf R. Rapp, who is a member of our Scientific Advisory Board, and Prof. Dr. Werner Goebel. Prof. Rapp is a known expert in the field of cell and tumor biology and Prof. Goebel is a pioneer in the field of vaccines based on recombinant bacteria.
The preclinical proof of principle has already been shown in a transgenic animal model and is supported by several patent applications that we have filed. The most advanced products are bacterial tumor vaccines which are based on the approved human vaccine strainSalmonella typhi Ty21a. The principle of these recombinant vaccine strains is the secretion of the tumor antigen using a so-called Type I secretion machinery derived fromEscherichia coli. To date, two different vaccine strains have been generated up to GMP scale production — a melanoma vaccine encompassing a mutated form of the oncogene B-Raf, which is present in more than 65% of melanomas, and a prostate cancer vaccine strain expressing and secreting PSA. For both vaccines, the preclinical proof of principle has been demonstrated in distinct animal models and the immunogenicity could be further enhanced compared to our already published strains (patent application filed in November 2006).2006.
In 2007, the PSA vaccine (AEZS-120) was selected as the first preclinical development candidate of an anti-tumorantitumor vaccine. In September 2007, scientific advice fromAEZS-120 is a live recombinant oral tumor vaccine candidate based on Salmonella typhi Ty21a as a carrier strain. Salmonella typhi Ty21a is an approved oral typhoid vaccine which has been safely applied in more than 350 million doses. The principle of AEZS-120 is based on the Paul Ehrlich Institute,recombinant expression of prostate specific antigen fused to the German health authorityB subunit of cholera toxin and a secretion signal in the presence of the Escherichia coli type I hemolysin secretion system. The proprietary system allows the secretion of the antigen together with an immunological adjuvant which has been demonstrated to be required for vaccines,optimal induction of CD8 T-cell responses by recombinant Salmonella based bacterial vaccines. The proof-of-concept was sought andalready demonstrated for the preclinical development program presented by us wasmouse homologue of AEZS-120 in principle accepted.a mouse tumor challenge model.
A grant application was filed in Germany and was approved in 2008.2008 for a preclinical development program for AEZS-120 with our research collaborators, the Department of Medical Radiation Biology and Cell Research, and the Department of Microbiology of the University of Würzburg, Germany. In accordance with this grant, 50% of our preclinical development costs and 100% of those of our university partner will be reimbursed by the German Ministry of Science and Education.
On July 20, 2011, we reached a key milestone in this non-clinical development program of AEZS-120 which encompassed the full development of a GMP process, including GMP production and quality testing of a clinical batch, as well as a non-clinical safety and toxicology package. Once this non-clinical program will have been completed and subject to a positive review by regulatory authorities, we aim to start Phase 1 clinical development in 2012. AEZS-120 has been developed through a research collaboration with the Department of Medical Radiation Biology and Cell Research, and the Department of Microbiology of the University of Würzburg, Germany. The preclinical developmentcollaboration was funded with a total of $890,000 for us and manufacture$870,000 for the university partner by the German Ministry of materialEducation and Research (BMBF) for clinical trial was initiated in 2008 and is still ongoing.
ENDOCRINOLOGY
Growth hormone secretagogue
AEZS-130/Solorel® (macimorelin) ("ghrelin agonist")
Growth hormone secretagogues ("GHS") represent a new class of pharmacological agents that directly stimulate GH secretion from the pituitary gland without the involvement of growth hormone-releasing hormone (GH-RH) or somatostatin. We believe that there is currently no GHS on the pharmaceutical market. Since GH is a potent regulator of lipid, sugar and protein metabolism, the potential clinical uses of GHS are numerous. They include growth retardation in children and treatment of cachexia in AIDS patients, which are currently the only approved uses of therapy of GH. The administration of GH, which has to be injected every day, is cumbersome. Therefore, we believe that there would be a demand for new orally active drugs like GHS.
three years. As part of our universitythe collaboration, we accessed new peptidomimetic compounds with GH secretagogue properties. The lead development candidate, a melanoma vaccine based on the recombinant expression of a modified B-Raf protein has been generated.
| 2.0 | ENDOCRINOLOGY |
| 2.1 | AEZS-130 — ORAL GHRELIN AGONIST |
AEZS-130, a ghrelin agonist, is a novel peptidomimetic GHS with potentorally active small molecule that stimulates the secretion of growth hormone by binding the ghrelin receptor (GHSR-1a). It can be used in both endocrinology and selective GH-releasing activity in humans.oncology indications. In endocrinology, we have completed a Phase 3 trial for its use as a simple oral diagnostic test for AGHD. AEZS-130 underwent limited clinical pharmacology testsworks by stimulating a patient’s growth hormone secretion, which normally only occurs during sleep, after which a healthcare provider will measure how well the body responds to that demonstratedstimulation based on the patient’s growth hormone levels over a potent stimulationperiod of the GH secretion after oral administration in human volunteers. This producttime. Low growth hormone levels, despite giving an effective stimulating agent, confirm a diagnosis of AGHD. AEZS-130 has been licensed to Ardana Bioscience Ltd. ("Ardana") (ARD-07), which initiated an open, randomized, placebo-controlled Phase 1 dose-ranging study in April 2004. Thirty-six healthy subjects were included in this study to receive eithergranted orphan-drug designation by the reference hormone GH-RH by i.v. route or one of the following dose levels of AEZS-130: 0.005, 0.05 or 0.5 mg/kg by oral route. AEZS-130 at the dose of 0.5 mg/kg orally caused an increase inFDA for use as a diagnostic test for growth hormone release equivalentdeficiency. We own the worldwide rights to thatAEZS-130 which, if approved, would become the first orally administered diagnostic test for AGHD. In oncology, the FDA has allowed for the initiation of a physician sponsored IND Phase 2A trial with AEZS-130 in cancer induced by GH-RH i.v. The compound was well toleratedcachexia, a disease which leads to significant weight loss and no other hormones showeddiminished functional performance. Since ghrelin agonists such as AEZS-130 have been shown to stimulate food intake and increase body weight in rats and mice, AEZS-130 could lead to better quality of life for patients with cancer induced cachexia. Ghrelin agonists have been in clinical trials for over a significant modification after any dose of AEZS-130.decade and have demonstrated good safety and efficacy profiles.
In June 2006, Ardana presented results regarding AEZS-130 at the 2006 ENDO Convention. These results referred to the Phase 1 trial regarding the stimulating effects of AEZS-130 on growth hormone following both oral and intra-duodenal administration in healthy males. This study showed that AEZS-130 was well tolerated by the 36 volunteers enrolled and no adverse events were reported. Administration of AEZS-130 either orally or via intra-duodenal infusion results in increased levels of growth hormone in the blood. This stimulation of growth hormone appears to be selective as no other hormones/analytes that were measured (cortisol, ghrelin, prolactin, insulin, glucose and ACTH (adrenocorticotropic hormone)) were affected in a dose-dependent or statistically significant way by administration of AEZS-130 either orally or via intra-duodenal infusion.
In May 2007, Ardana gained orphan drug designation for AEZS-130 (Solorel®) as a diagnostic test for growth hormone deficiency in adults. The clinical development and toxicology programs for this indication were ongoing and Ardana announced the commencement in the United States of the planned pivotal registration study and the enrolment of the first patient in August 2007.
In June 2008, Ardana announced that the company stopped its operations and entered into voluntary administration. Consequently, the clinical study of AEZS-130 (Solorel®) as a diagnostic test for AGHD was suspended.
We announced the recovery of worldwide rights from Ardana for the compound AEZS-130 in the third quarter of 2008. In June 2009, we reported that, after regaininghaving regained from Ardana in the prior year the worldwide rights to the growth hormone secretagogue, AEZS-130, we had entered into an agreement with the administrators of Ardana to acquire all Ardana assets relating to AEZS-130 for $232,000. These assets include development data, inventory of compound, regulatory authorizations, including IND and orphan drug status as a diagnostic test granted in the United States, as well as a patent application protecting the use of AEZS-130 (Solorel®) for the diagnostic of growth hormone secretion deficiency.
During the same month, the first clinical data relating to the use of AEZS-130 (Solorel®) as a simple diagnostic test for AGHD were presented at the ENDO 2009 meeting by the main investigators Dr G. Merriam and Dr B.M.K. Biller. Data showed that in adult growth hormone deficient patients,
the responses to the orally administered AEZS-130 (Solorel®) compound were comparable to currently validated agents and clearly separated patients from normal control subjects.
In October 2009, we announced that we had initiated activities intended to complete the clinical development of AEZS-130 (Solorel®) which could be the first oral diagnostic test approved for growth hormone deficiency ("GHD"(“GHD”). Aeterna ZentarisWe had already assumed the sponsorship of the IND and discussed with the FDA the best way to complete the ongoing Phase 3 clinical trial and subsequently file a New Drug ApplicationNDA for approval of AEZS-130 (Solorel®) as a diagnostic test for AGHD.
The pivotal Phase 3 trial is designed to investigate the safety and efficacy of the oral administration of AEZS-130 (Solorel®) as a growth hormone stimulation diagnostic test. It was accepted by the FDA that for the ongoing part of the study, AEZS-130 is not tested against a comparator drug, as Geref®Geref® has been removed from the market.
Oral administration of AEZS-130 (Solorel®) offers more convenience and simplicity over the current GHD tests used, requiring either i.v. or i.m. administration. Additionally, AEZS-130 (Solorel®) may demonstrate a more favorable safety profile than existing diagnostic tests, some of which may be inappropriate for certain patient populations e.g. diabetes mellitus or renal failure, and have demonstrated a variety of side effects which AEZS-130 (Solorel®) has not thus far. These factors may be limiting the use of GHD testing and may enable AEZS-130 (Solorel®) to become the diagnostic test of choice for GHD. AEZS-130 (Solorel®) has been granted Orphan Drug Designation for the diagnosis of growth hormone deficiency by the FDA, and Aeterna Zentaris is now the sponsor of this orphan designation.
On June 21, 2010, we presented positive data at the 92nd ENDO Meeting on AEZS-130 for diagnostic and therapeutic use. The preclinical data showed that AEZS-130 is a potent and safe oral synthetic GH-releasing compound with potential utility as a diagnostic test for growth hormone deficiencies. In addition to the diagnostic indication, we believe that, based on the results of Phase 1 studies, AEZS-130 (Solorel®) has potential applications for the treatment of cachexia, a condition frequently associated with severe chronic diseases such as cancer, chronic obstructive pulmonary disease and Acquired Immune Deficiency Syndrome, or AIDS.
On July 14, 2010, we announced the presentation of a poster on AEZS-130, (Solorel®), entitled "Use Use of the Orally Active Ghrelin Mimetic AEZS-130 as a Simple Test for the Diagnosis of Growth Hormone (GH) Deficiency (GHD) in adults (AGHD)." Merriam G.R., Yuen K., Bonert V., Dobs A, Garcia J., Kipnes M., Molitch M., Swerdloff R., Wang C., Cook D., Altemose I. and Biller B. This poster was presented at the Seventh International Congress of Neuroendocrinology, in Rouen, France.
On October 5, 2010, we announced at the Fifth International Congress of the Growth Hormone Research Society and the Insulin-like Growth Factors Society, after the interim Phase 3 analysis, that AEZS-130 (Solorel®) demonstrated the potential to provide a simple, well tolerated and safe oral diagnostic test for AGHD.
On December 20, 2010, we announced we had reached agreement with the FDA on an SPA for AEZS-130, (Solorel®), enabling the Company to complete the ongoing registration study required to gain approval as a diagnostic test for AGHD.
Study Design
The SPA agreement has resulted in a modification to the original study, but does not alter the basic study design so that the completed portion of the study will work with the new part of the study to provide one complete Phase 3 study.
Original Study
The completedfirst part of the study conducted by our former partner, Ardana, was a two-way crossovercross-over study and included 42 patients with confirmed AGHD or multiple pituitary hormone deficiencies and a low insulin-like growth factor-I. A
control group of 10 subjects without AGHD were matched to patients for age, gender, body mass index and (for females) estrogen status.
Each patient received two dosing regimens in random order, while fasting, at least 1 week apart. One regimen consisted of a 1 µg/kg (max. 100 µg) dose of GHRH (Geref Diagnostic®Diagnostic®, Serono) with 30 g of ARG (ArGine®(ArGine®, Pfizer) administered intravenously over 30 minutes; the other regimen was a dose of 0.5 mg/kg body weight of Solorel®AEZS-130 given in an oral solution of 0.5 mg/ml.
Completion of the study will be accomplished with the following revisions/additions to the current protocol:
| — | an additional 30 normal controls subjects will be enrolled to match the AGHD patients from the original cohort; |
| — | further, an additional 20 subjects will be enrolled — 10 AGHD patients and 10 matched normal control subjects; |
| — | the above will bring the database to approximately 100 patients; |
| — | all subjects will receive a dose of 0.5 mg/kg body weight of AEZS-130; and |
| — | as a secondary endpoint, the protocol will require that at least 8 of the 10 newly enrolled AGHD patients be correctly classified by a pre-specified peak GH threshold level. |
On July 26, 2011, we announced the completion of the Phase 3 study on AEZS-130 as a first oral diagnostic test for AGHD and the decision to meet with the FDA for the future filing of a NDA for the registration of AEZS-130 in the United States.
On August 30, 2011, we announced favorable top-line results of our completed Phase 3 study with AEZS-130 as a first oral diagnostic test for AGHD. Results showed that AEZS-130 reached its primary endpoint demonstrating >90% area-under-the-curve (“AUC”) of the Receiver Operating Characteristic (“ROC”) curve, which determines the level of specificity and sensitivity of the product. Importantly, the primary efficacy parameters show that the study achieved both specificity and sensitivity at a level of 90% or greater. In addition, 8 of the 10 newly enrolled AGHD patients bewere correctly classified by a pre-specified peak GH threshold level.
On November 28, 2011, we announced that the FDA had completed the review of an IND application filed by Jose M. Garcia, M.D., Ph.D., Assistant Professor, Division of Diabetes Endocrinology and Metabolism, Departments of Medicine and Molecular and Cell Biology, Baylor College of Medicine and Michael E. DeBakey Veterans Affairs Medical Center, in Houston Texas and concluded that Dr. Garcia may proceed with the initiation of a Phase 2A trial to assess the safety and efficacy of repeated doses of AEZS-130, in patients with cancer cachexia. Cachexia, which is characterized by diminished appetite and food intake in cancer patients, is defined as an involuntary weight loss of at least 5% of the pre-illness body weight over the previous 6 months.
On March 8, 2012, we announced that the Michael E. DeBakey Veterans Affairs Medical Center, in Houston, Texas, initiated a Phase 2A trial assessing the safety and efficacy of repeated doses of AEZS-130 in patients with cancer cachexia. The study is conducted under a CRADA between the Company and the Michael E. DeBakey Veterans Affairs Medical Center which is funding the study.
CompetitionThis is a double-blind, randomized, placebo-controlled Phase 2A trial to test the effects of different doses of the ghrelin agonist, AEZS-130, in 18 to 26 patients with cancer-cachexia. AEZS-130 will be provided by Aeterna Zentaris. The study will involve 3 sequential groups receiving differing doses of AEZS-130. Each dose group will have 6 patients who will receive AEZS-130 and 2-4 patients who will receive placebo. After analysis of safety and efficacy at each dose level vs. placebo, a decision will be taken either to decrease or increase the dose. If there are major safety issues, the study will be stopped. For this study, adequate efficacy will be defined as a³0.8 kg of body weight gain or a³50 ng/mL increase in plasma IGF-1 levels. The primary objective of the study is to evaluate the safety and efficacy of repeated oral administration of AEZS-130 at different doses daily for AEZS-130 (Solorel®)1 week in view of developing a treatment for cachexia. The following parameters will be recorded to assess efficacy during the study: change of body weight, change of IGF-1 plasma levels, and change of quality of life score (Anderson Symptom Assessment Scale, FACIT-F). Other secondary objectives will include food intake, and changes in the following: appetite, muscle strength, energy expenditure, reward from food and functional brain connectivity.
Competitors for AEZS-130 (Solorel®)
Competitors for AEZS-130 as a diagnostic test for AGHD are principally the diagnostic tests currently performed by endocrinologists.
Most commonly used diagnostics tests for GHD are:
| — | measurement of blood levels of Insulin Growth Factor (“IGF”)-1, which is often used as the first test when GHD is suspected. However, this test is not used to definitively rule out GHD as many growth hormone deficient patients show normal IGF-1 levels; |
| — | Insulin Tolerance Test (“ITT”), which is considered to be the “gold standard” for GH secretion provocative tests but requires constant monitoring and is contra-indicated in patients with seizure disorders, with cardiovascular disease and in brain injured patients and elderly patients. ITT is administered i.v.; |
| — | GHRH + Arginine test, which is an easier test to perform in an office setting and has a very good safety profile but is considered to be costly to administer compared to ITT and Glucagon. This test is contra-indicated in patients with renal failure. GHRH + Arginine is approved in the EU and has been proposed to be the best alternative to ITT, but it is no longer available in the United States. This test is administered i.v.; and |
| — | glucagon test, which is simple to perform and is considered relatively safe by endocrinologists but is contraindicated in malnourished patients and patients who have not eaten for more than 48 hours. Since there is a suspicion that this test may cause hypoglycemia, it may not be appropriate in diabetic populations. This test is administered i.m. |
Oral administration of AEZS-130 offers more convenience and simplicity over the current GHD tests used, requiring either i.v. or i.m. administration. Additionally, AEZS-130 may demonstrate a more favorable safety profile but is considered tothan existing diagnostic tests, some of which may be costly to administer compared to ITTinappropriate for certain patient populations e.g. diabetes mellitus or renal failure, and Glucagon. This test is contra-indicated in patients with renal failure. GHRH + Arginine is approved inhave demonstrated a variety of side effects which AEZS-130 has not thus far. These factors may be limiting the EU and has been proposed to be the best alternative to ITT, but it is not any longer available in the U.S. This test is administered i.v.; and
Ghrelin receptor ligands
Ghrelin is a peptide predominantly produced by the stomach. Apart from a potent GH-releasing action, ghrelin has other activities including stimulation of lactotroph and corticotroph function, influence on the pituitary gonadal axis, stimulation of appetite, control of energy balance, influence on sleep and behavior, control of gastric motility and acid secretion, and influence on pancreatic exocrine and endocrine function as well as on glucose metabolism. The recent discovery of ghrelin and its receptors opens up new opportunities for the development of drugs that will treat metabolic disorders. There is indeed a possibility that ghrelin analogs, acting as either agonists or antagonists, might have a clinical impact without affecting GH levels. The use of ghrelin antagonists as appetite suppressants or inhibitorsGHD testing and may enable AEZS-130 to become the diagnostic test of lipogenesis could open up new opportunitieschoice for the treatment of obesity and associated
diseases (e.g. diabetes, cardiovascular diseases). The use of ghrelin agonists could have therapeutic benefits which are expected to offer hope for cachexic or anorexic patients.GHD.
In 2004, we signed a research and license collaboration agreement with Le Centre National de la Recherche Scientifique and University Montpellier I and II, France, acting in their own names, as well as in the name and on behalf of the Laboratoire des Aminoacides, Peptides et Protéines (LAPP) (UMR 5810), directed by Dr. Jean Martinez, for the synthesis and characterization of new chemical entities acting as ghrelin receptor ligands. Market Data — AGHD
According to the agreement, we have the worldwide rights to develop and exploit the new compounds for any indication. Compounds with the most potent affinity for the ghrelin receptors will be investigated further through an international network of academic investigators with expertiseHormone Foundation, in the field of endocrinology in order to identify clinical development candidates.United States, about 35,000 adults have GHD, with about 6,000 newly diagnosed each year (source: Hormone Foundation’s Website).
Additionally, we also signed a research contract with the Department of Experimental and Environmental Medicine of the University of Milan, Italy, under the direction of Prof. Vittorio Locatelli, for the pharmacological characterization of potentially ghrelin receptor ligands.
| 2.2 | LHRH ANTAGONISTS |
In August 2005, we filed a first patent application to protect a series of new chemical entities characterized as ghrelin receptor ligands.
| 2.2.1 | Cetrorelix |
In May 2006, we signed a research project agreement with the University of Montreal. This research project will focus on the characterization of ghrelin receptor ligands on fat tissue. This project is led by Huy Ong, Professor at the Faculty of Pharmacy, at the University of Montreal.
In August 2006, we also initiated a research collaboration with the Centre de recherche de l'Hôpital Laval (Québec) under the direction of Dr. Denis Richard. This research collaboration will focus on the pharmacological characterization of ghrelin receptor ligandsin vivo (e.g. the effects in diet-induced obesity models).
In October 2006, we presented for the first time ourin vivo data on the capacity of ghrelin antagonists of selectively inhibiting food intake. This study, using a rat model, outlined the capacity of ghrelin antagonists' ability to inhibit appetite without affecting growth hormone secretion and represents evidence that ghrelin antagonist compounds can selectively inhibit food intake. It further supports the hope that ghrelin antagonist compounds have the potential to be useful for the treatment of obesity.
In 2007 and 2008, we presented at scientific meetings preclinical candidates having the interesting property to decrease body weight gain and fat accumulation in diet induced obesity models. The ongoing work will focus on the improvement of oral bioavailability.
In July 2009, new data supporting the use of AEZS-123 (JMV-2959), a ghrelin receptor antagonist, for the treatment of alcohol dependence that involved ghrelin were published. Data were published in the renowned American scientific journal, Proceedings of the National Academy of Sciences ("PNAS"). Data show that mice treated with ghrelin increase their alcohol consumption. When ghrelin's actions are blocked by administering ghrelin receptor antagonists such as AEZS-123, mice no longer show preference for an alcohol-associated environment — in other words, alcohol is no longer able to produce its addictive effects that include reward searching behaviour (akin to craving in alcoholic patients). The work, coordinated by Aeterna Zentaris, emerged from an international collaboration between the research groups of Prof. Suzanne Dickson and Prof. Jörgen Engel who performed the pharmacology work at the Sahlgrenska Academy, Gothenburg, Sweden, and the research group of Prof. Jean Martinez who synthesized the tested compound AEZS-123 at the Institut des biomolécules Max Mousseron, Montpellier, France.
LHRH ANTAGONISTS
Cetrorelix
Cetrorelix is a peptide-based active substance which was developed in cooperation with Nobel Laureate Professor Andrew Schally presently of the United States Veterans Administration-Miami, University of Miami, and formerly of Tulane University in New Orleans. This compound is a luteinising hormone releasing hormone (LHRH, also known as GnRH) antagonist that blocks the pituitary LHRH receptors resulting in a rapid decrease of sexual hormone levels. Moreover, cetrorelix allows the LHRH receptors on the pituitary gland to be blocked gradually. Conversely, the side effects usually associated with the use of agonists and resulting from total hormone withdrawal can be avoided in conditions that do not require a castrating degree of hormone withdrawal. Therefore, in contrast to treatment with agonists, LHRH antagonists permit dose-dependent hormone suppression which is of critical importance for the tolerability of hormonal therapy.
2.2.1.1 Cetrorelix In Vitro Fertilization (COS/ART) Cetrotide® Cetrorelix is the first LHRH antagonist which was approved for therapeutic use as part of fertilization programs in Europe and was launched on the market under the trade name Cetrotide®Cetrotide®Cetrotide® (cetrorelix acetate) in 1999. In women who undergo controlled ovarian stimulation for recovery of ovocytes for subsequent fertilization, Cetrotide®Cetrotide® helps prevent premature ovulation. LHRH is a naturally occurring hormone produced by the brain to control the secretion of LH and, therefore, final egg maturation and ovulation. Cetrotide®Cetrotide® is designed to prevent LH production by the pituitary gland and to delay the hormonal event, known as the "LH surge"“LH surge” which could cause eggs to be released too early in the cycle, thereby reducing the opportunity to retrieve the eggs for the assisted reproductive techniques procedure.
In comparison with LHRH agonists that require a much longer pre-treatment, the use of our LHRH antagonist, Cetrotide®Cetrotide®, permits the physician to interfere in the hormone regulation of the women undergoing treatment much more selectively and within a shorter time.
The effectiveness of Cetrotide®Cetrotide® has been examined in five clinical trials (two Phase 2 and three Phase 3 trials). Two dose regimens were investigated in these trials: either a single dose per treatment cycle or multiple dosing. In the Phase 2 studies, a single dose of 3 mg was established as the minimal effective dose for the inhibition of premature LH surges with a protection period of at least four days. When Cetrotide®Cetrotide® is administered in a multi-dose regimen, 0.25 mg was established as the minimal effective dose. The extent and duration of LH suppression was found to be dose-dependent. In the Phase 3 program, efficacy of the single 3 mg dose regimen and the multiple 0.25 mg dose regimen was established separately in two controlled studies utilizing active comparators. A third non-comparative study evaluated only the multiple 0.25 mg dose regimen of Cetrotide®Cetrotide®. In the five Phase 2 and Phase 3 trials, 184 pregnancies were reported out of a total of 732 patients (including 21 pregnancies following the replacement of frozen-thawed embryos). In these studies, drug-related side effects were limited to a low incidence of injected site reactions; however, none of them was serious — such as an allergic type of reaction — or required withdrawal from treatment. In addition, no drug-related allergic reactions were reported from these clinical studies.
Cetrotide®Cetrotide® is the only LHRH antagonist that is available in two dosing regimens. With an immediate onset of action, Cetrotide®Cetrotide® permits precise control — a single dose (3 mg), which controls the LH surge for up to four days, or a daily dose (0.25 mg) given over a short period of time (usually five to seven days). The treatment with Cetrotide®Cetrotide® can be accomplished during a one-month cycle with a simplified, more convenient and shorter treatment requiring fewer injections than LHRH agonists.
Cetrotide®Cetrotide® is marketed in a 3 mg and a 0.25 mg subcutaneous injection as cetrorelix acetate by Merck Serono in the United States and Europe. Approval for Cetrotide® in Japan was gained in
April 2006. In September 2006, we announced the launch of Cetrotide®Cetrotide® in Japan forin vitro fertilization. Cetrotide®Cetrotide® is marketed in Japan by our partner Shionogi. We receive revenue from the supply of Cetrotide®Cetrotide® to our Japanese partners. The market competitor is ganirelix (Antagon™/Orgalutran®Orgalutran®) from Schering-Plough (Organon) indicated for the inhibition of premature LH surges in women undergoing controlled ovarian hyperstimulation.
Partners for Cetrotide®Cetrotide®
In August 2000, we entered into a commercialization agreement with Merck Serono for Cetrotide®Cetrotide®. Under the terms of this agreement, we granted an exclusive license to Merck Serono to commercialize Cetrotide®Cetrotide® for IVF/COS/ART worldwide ex-Japan and we are entitled to receive fixed and sales royalties from Merck Serono. The Japanese rights for this indication are held by Shionogi whereby, according to a commercialization agreement, we received transfer pricing from Shionogi.
In DecemberNovember 2008, we sold our rights to royalties on future sales of Cetrotide®Cetrotide® covered by our license agreement with Merck Serono for $52.5 million to Cowen Healthcare Royalty Partners ("CHRP"(“CHRP”) less transaction costs of $1.0 million, resulting in initial net proceeds to us of $51.5 million. In addition, upon net sales of Cetrotide®Cetrotide® having reached a specified level in 2010, we received an additional payment of $2.5 million from CHRP in February 2011. Furthermore, under the terms of the agreement, we agreed to make a one-time cash payment to CHRP in an amount ranging from $5 million up to a maximum of $15 million in the event cetrorelix is approved for sale by the European regulatory authorities in an indication other thanin vitro fertilization. The amount which would be due to CHRP will be higher the earlier the product receives European regulatory approval. Since cetrorelix development has been terminated, we do not expect to make this one-time cash payment to CHRP.
Clinical Development Overview of Cetrorelix in Benign Prostatic Hyperplasia ("BPH"), Endometriosis and Uterine Myoma
Cetrorelix in BPH
BPH is a hormone-driven enlargement of the male prostate gland. The prostate is located directly at the vesicle outlet in the male surrounding the first part of the urethra. The enlargement puts pressure on the urethra, causing difficulty in urinating. BPH is classified into three stages according to symptoms: 1) the irritant phase, where the patient suffers dysuria (pain when urinating) and nocturia (the urge to urinate during the night); 2) residual urine occurring in the bladder thus increasing problems during urinating; and 3) overflow of the bladder. These can result in formation of bladder stones, congestion of urine and engorged kidneys which can in turn lead to life-threatening kidney damage.
BPH clinical trials
On August 17, 2009, we reported Phase 3 results for our North American efficacy trial Z-033 (including certain sites in Europe) and safety trial Z-041 in BPH, with cetrorelix. The study Z-033 failed to achieve its primary endpoint, being an improvement in International Prostate Symptom Score ("IPSS") as compared to placebo, and it demonstrated no clear differences in overall efficacy with all 3 groups showing an improvement in IPSS of approximately 4 points that was maintained throughout the 52 weeks. There was a slight advantage in favor of the main active treatment arm (Arm A) up to Week 46 of the follow-up, which was no longer demonstrated at Week 52. These differences did not achieve statistical significance. Furthermore, a statistically significant effect on the IPSS, as compared to placebo, was seen in a sub-group of patients with large prostate glands (greater than 50 cm3) on entry to the study. Tolerability of cetrorelix in study Z-033 was very good, as evidenced by the absence of major differences to placebo with regard to both clinical adverse events and changes in laboratory parameters.
On December 7, 2009, we reported the Phase 3 results for cetrorelix from the European efficacy trial Z-036, involving 420 patients. Study Z-036 did not reach its primary endpoint. There were no clear differences in overall efficacy, with all 3 groups (including placebo) showing an improvement in IPSS of approximately 6 points that was maintained throughout the 52 weeks. There was observation of an improvement in uroflow, both maximum and mean, and in residual volumedevelopment has been terminated in all treatment groups. These favorable changes are reflected indications other thanin an overall improvement in Qualityvitro fertilization (COS/ART). Furthermore, we focus on our manufacturing on behalf of Life measures. Furthermore, a favorable trend on the IPSS, as compared to placebo, was seen in a sub-group of patients with large prostate glands (greater than 50 cm3) on entry to the study. Cetrorelix was well tolerated, there were no relevant differences to placebo with regard to both clinical adverse events or changes in laboratory parameters with the exception of the anticipated hormonal changes.
On December 18, 2009, following the unsuccessful results of our Phase 3 program in BPH with cetrorelix, we announced the termination of our agreement with sanofi dated March 5, 2009, for the development, commercialization and licensing of cetrorelix in BPH for the U.S. market. Termination of the agreement took effect as at January 9, 2010.
Cetrorelix in endometriosis and uterine myoma
There is no active program ongoing at present.
Partners for Cetrorelix
We previously licensed cetrorelix to Solvay worldwide (ex-Japan) for all indications with the exception of IVF/COS/ART, which rights belong to Merck Serono and Japanese rights are held by Shionogi. In the BPH indication, for which we regained exclusive worldwide (ex-Japan) rights, Japanese rights are held by Shionogi. On May 8, 2007, we and Solvay announced the termination of the license and cooperation agreement for cetrorelix for all remaining indications, including endometriosis, effective on that date, as a result of which we regained exclusive worldwide (ex-Japan) rights for cetrorelix in all indications without any financial compensation payable to Solvay.
On March 22, 2007, we announced that Nippon Kayaku had terminated its development agreement pertaining to cetrorelix pamoate to focus solely in oncology.
We signed a license and cooperation agreement for the commercialization of cetrorelix (BPH indication) with Handok for the Korean market during the third quarter of 2008.
On March 5, 2009, we entered into a development, commercialization and license agreement with sanofi-aventis for the development, registration and marketing of cetrorelix in BPH for the U.S. market. Under the terms of the agreement, sanofi-aventis made an initial upfront payment to us of $30.0 million. Following the announcement of the negative results for the efficacy trial in North America (study Z-033) and in Europe (study Z-036), we announced the termination of our agreement with sanofi-aventis dated March 5, 2009 for the development, commercialization and licensing of cetrorelix in BPH for the U.S. market. Termination of the agreement was effective January 9, 2010.
Following the negative Phase 3 results for cetrorelix in BPH, our Japanese partner, Shionogi, also agreed with the Company to cease the development of cetrorelix in this indication.
Ozarelix
| 2.2.2 | Ozarelix |
Ozarelix is a modified LHRH antagonist which is a linear decapeptide sequence. Ozarelix is a fourth-generation LHRH antagonist aiming at extendeddesigned to extend the suppression of testosterone levels, thatwhich does not require a sophisticated depot formulation for long-lasting activity.
On August 12, 2004, we entered into a licensing and collaboration agreement with Spectrum Pharmaceuticals, Inc. ("Spectrum"(“Spectrum”), for ozarelix and its potential to treat hormone-dependent cancers as well as benign proliferative disorders, such as BPH and endometriosis for all potential indications in North America (including Canada and Mexico) and India while keeping the rights for the rest of the
world. In addition, Spectrum is entitled to receive 50% of upfront and milestone payments and royalties received from our Japanese partner, Nippon Kayaku, that are generated in the Japanese market for oncological indications. In November 2010, this agreement with Spectrum was amended. Under the terms of the amended agreement, Spectrum is entitled to use the Company'sour patent rights and know-how to develop, use, make, have made, sell, offer for sale, have sold, import, export and export, commercialize ozarelix in all worldwide territories except Japan, Korea, Indonesia, Malaysia, the Philippines and Singapore. Under the terms of the amended agreement, Spectrum granted, as further consideration, 326,956 shares of its common stock, with an equivalent fair value of approximately $1,263,000, as an upfront nonrefundable license fee payment to the Company.us. Also per the amended agreement, the Companywe will be entitled to receive a total of approximately $22,765,000 in cash payments, as well as approximately $670,000 of Spectrum'sSpectrum’s common stock, upon achieving certain regulatory milestones in various markets. Furthermore, the Companywe will be entitled to receive royalties (scale-up royalties from high single to low double-digit) on future net sales of ozarelix products in the named territories.
During the third quarter of 2008, we entered into a commercialization agreement with Handok for ozarelix (BPH indication) for the Korean market.
On January 27, 2010, Spectrum announced that it had terminated its development program with ozarelix in BPH. Consequently, an impairment loss of approximately $1,422,000 was recorded as part of amortization expense, and all corresponding unamortized deferred revenues related to the use of ozarelix, totalling approximately $1,606,000, were fully recognized in the 2009our consolidated statement of operations.operations for the year ended December 31, 2009.
BPH Clinical Trials
In October 2006, we announced positive and highly statistically significant Phase 2 results for ozarelix in BPH. The primary efficacy endpoint of improving clinical symptoms of BPH at week 12, as measured by significant changes in IPSS, was achieved at all dosage regimens. Secondary efficacy parameters such as uroflow, residual urinary volume, quality of life and circulating testosterone levels were also measured and showed good results. The outcome of the trial demonstrated an excellent safety profile with ozarelix as patients had no serious side effects. The erectile function was also not affected at any dose regimens.
On May 23, 2007 and September 5, 2007, Spectrum disclosed detailed Phase 2 results for ozarelix in BPH at two medical conferences. Results indicate that ozarelix was well tolerated and demonstrated statistically significant as well as clinically meaningful efficacy in the treatment of LUTS secondary to BPH.
On January 3, 2007, Spectrum announced the FDA's acceptance of an IND for ozarelix in BPH. Spectrum initiated a Phase 2b study in January 2007. On April 22, 2008, our partner Spectrum released the nine-month Phase 2b results for ozarelix. Spectrum indicated that ozarelix demonstrated sufficient clinical activity to justify its continued development in BPH. Based on these results, Spectrum initiated in September 2008 the recruitment of 860 patients for a new BPH study. In January 2010, Spectrum Pharmaceuticals announced the discontinuation of ozarelix development in BPH, stating that the mixed results of their Phase 2b study and the announced negative results of our Phase 3 registrational trial of cetrorelix in BPH does not support continued development of ozarelix in this indication.
2.2.2.1 Prostate Cancer Clinical Trials
In August 2006, we announced positive Phase 2 results for ozarelix in hormone-dependent inoperable prostate cancer. This open-label, randomized-controlled dose-finding trial enrolled 64 patients receiving different IM dosage regimens of ozarelix to assess its safety and efficacy. The study achieved its primary endpoint of defining a tolerable dosage regimen of ozarelix that would ensure continuous suppression of testosterone at castration level for a three-month test period. A
secondary efficacy endpoint aimed at assessing tumor response as determined by a 50% or greater reduction of serum PSA level, compared to baseline, was also achieved. The best results regarding the primary endpoint of continuous suppression were obtained with a dose of 130 mg per cycle where all patients remained suppressed to castration until at least day 85. In patients with continuous testosterone suppression below castration level, tumor response as measured by PSA levels was 97%. Following these results, we, in collaboration with Spectrum, initiated an additional Phase 2 study in European centers to verify and optimize the findings derived from the cohort of patients having received 130 mg of ozarelix per cycle.
On August 3, 2006, we announced a licensing and collaboration agreement with Nippon Kayaku for ozarelix. Under the terms of the agreement, we granted Nippon Kayaku an exclusive license to develop and market ozarelix for all potential oncological indications in Japan. In return, we received an upfront payment upon signature and are eligible to receive payments upon achievement of certain development and regulatory milestones, in addition to low double-digit royalties on potential net sales. Spectrum is entitled to receive 50% of the upfront, milestone payments and royalties received from Nippon Kayaku.
Non-Peptide LHRH AntagonistsOur partner, Spectrum, is currently recruiting patients in Phase 2 trial for the treatment of prostate cancer. This is an international, multicenter, open-label, randomized study assessing the safety and efficacy of a monthly dosing regimen of ozarelix versus goserelin depot in men with prostate cancer (source: www.clinicaltrials.gov).
As outlined above, the LHRH receptor plays an important role in a number of benign and malignant tumors. Our drug discovery unit searches for small, non-peptide molecules which have the same effect on the receptor. Their advantage lies in the potential for oral administration.
AEZS-115 is a new orally bioavailable LHRH antagonist with LHRH-receptor binding affinity in the nanomolar range which is developed for hormone therapy of endocrinological disorder and of benign and malignant tumors. The compound demonstrates excellent selectivity to LHRH-receptor and has advanced to a preclinical stage where thein vivo activity has been confirmed. Major advantages are the dose-dependent reduction of sexual hormones without flare-up effect whereas no decrease down to castration level is necessary and therefore side effects are reduced.
In January 2006, we regained the exclusive worldwide rights to develop and commercialize AEZS-115 from Solvay. Attractivein vivo activity of this orally available peptidomimetic LHRH-antagonist was demonstrated with a single, oral administration (20mg/kg) in rats which led to efficient and revocable suppression of plasma testosterone levels for up to 12 hours. Furthermore, a repeat of the dosing of AEZS-115 increased the suppression time without accumulation in the plasma.
In 2007, an oral formulation was selected and pharmacokinetic data were obtained.
First preclinical results were presented at the 2008 San Antonio Breast Cancer Symposium on December 12, 2008 and showed substantial anti-tumor activity of AEZS-115 in human ovarian and breast cancer cell lines, as evidenced by exposure of human cell lines SKOV3, Ovcar 3 (human ovarian cancer cell lines) and MDA-MB 468 (human breast cancer cell line) to increasing concentrations of AEZS-115, peptidic GnRH-antagonist cetrorelix and GnRH-agonist Triptorelin (1, 10, and 100 µM) for 48 days. The number of viable cells was determined by crystal violet staining as well as by ATP-dependent luminometric assays. Results showed that both GnRH-antagonists dose-dependently inhibited growth of all three cell lines, while GnRH-agonist Triptorelin showed marginal growth inhibition. Cell growth was inhibited by 40-60% following exposure to a concentration of 10 µM of AEZS-115 and by 60-80% when cells were exposed to 100 µM. Inhibition with cetrorelix at 100 µM ranged from 20-40%, while only minor effects on cell growth were seen at 10µM. Optimization is ongoing.
RAW MATERIALS
Raw materials and supplies are generally available in quantities adequate to meet the needs of our business. We are dependent on third-party manufacturers for the pharmaceutical products that we market. An interruption in the availability of certain raw materials or ingredients, or significant
increases in the prices paid by us for them, could have a material adverse effect on our business, financial condition, liquidity and operating results.
DISTRIBUTION
We currently have a lean sales and marketing staff. In order to commercialize our product candidates successfully, we need to make arrangements with third parties to perform some or all of these services in certain territories.
We contract with third parties for the sales and marketing of our products. We are currently dependent on strategic partners and may enter into future collaborations for the research, development and commercialization of our product candidates. Our arrangements with these strategic partners may not provide us with the benefits we expect and may expose us to a number of risks.
REGULATORY COMPLIANCE
Governmental authorities in Canada, the United States, Europe and other countries extensively regulate the preclinical and clinical testing, manufacturing, labeling, storage, record keeping, advertising, promotion, export, marketing and distribution, among other things, of our product candidates. Under the laws of the United States, the countries of the European Union, and other countries, we and the institutions whereat which we sponsor research are subject to obligations to ensure that our clinical trials are conducted in accordance with GCP guidelines and the investigational plan and protocols contained in an Investigational New DrugIND application, or comparable foreign regulatory submission. The Japanese regulatory process for approval of new drugs is similar to the FDA approval process described below except that Japanese regulatory authorities request bridging studies to verify that foreign clinical data are applicable to Japanese patients and also require the tests to determine appropriate dosages for Japanese patients to be conducted on Japanese patient volunteers. Due to these requirements, delays of two to three years in introducing a drug developed outside of Japan to the Japanese market are possible. Set forth below is a brief summary of the material government regulations affecting the Company in the major markets in which we intend to market our products.
Canada
In Canada, the Canadian Therapeutic Products Directorate is the Canadian federal authority that regulates pharmaceutical drugs and medical devices for human use. Prior to being given market authorization, a manufacturer must present substantive scientific evidence of a product'sproduct’s safety, efficacy and quality as required by theFood and Drugs Act and other legislation and regulations. The requirements for the development and sale of pharmaceutical drugs in Canada are substantially similar to those in the United States, which are described below.
United States
In the United States, the FDA under the Federal Food, Drug, and Cosmetic Act, the Public Health Service Act and other federal statutes and regulations, subject pharmaceutical products to rigorous review.
In order to obtain approval of a new product from the FDA, we must, among other requirements, submit proof of safety and efficacy as well as detailed information on the manufacture and composition of the product. In most cases, this proof entails extensive preclinical, clinical, and laboratory tests. Before approving a new drug or marketing application, the FDA also typically conducts pre-approval inspections of the company, its contract research organizations and/or its clinical trial sites to ensure that clinical, safety, quality control, and other regulated activities are compliant with Good Clinical Practices, or GCP, or Good Laboratory Practices, or GLP, for specific non-clinical toxicology studies. Manufacturing facilities used to produce a product are also subject to ongoing inspection by the FDA. The FDA may also require confirmatory trials, post-marketing testing, and extra surveillance to monitor the effects of approved products, or place conditions on any approvals that could restrict the
commercial applications of these products. Once approved, the labeling, advertising, promotion, marketing, and distribution of a drug or biologic product must be in compliance with FDA regulatory requirements.
The first stage required for ultimate FDA approval of a new biologic or drug involves completion of preclinical studies and the submission of the results of these studies to the FDA. This, together with proposed clinical protocols, manufacturing information, analytical data, and other information in an IND, must become effective before human clinical trials may commence. Preclinical studies involve laboratory evaluation of product characteristics and animal studies to assess the efficacy and safety of the product. The FDA regulates preclinical studies under a series of regulations called the current GLP regulations. If the sponsor violates these regulations, the FDA may require that the sponsor replicate those studies.
After the IND becomes effective, a sponsor may commence human clinical trials. The sponsor typically conducts human clinical trials in three sequential phases, but the phases may overlap. In Phase 1 trials, the sponsor tests the product in a small number of patients or healthy volunteers, primarily for safety at one or more doses. Phase 1 trials in cancer are often conducted with patients who have end-stage or metastatic cancer. In Phase 2, in addition to safety, the sponsor evaluates the efficacy of the product in a patient population somewhat larger than Phase 1 trials. Phase 3 trials typically involve additional testing for safety and clinical efficacy in an expanded population at geographically dispersed test sites. The sponsor must submit to the FDA a clinical plan, or "protocol,"“protocol”, accompanied by the approval of the institutions participating in the trials, prior to commencement of each clinical trial. The FDA may order the temporary or permanent discontinuation of a clinical trial at any time. In the case of product candidates for cancer, the initial human testing may be done in patients with the disease rather than in healthy volunteers. Because these patients are already afflicted with the target disease, such studies may provide results traditionally obtained in Phase 2 studies. Accordingly, these studies are often referred to as "Phase“Phase 1/2"2” studies. Even if patients participate in initial human testing and a Phase 1/2 study is carried out, the sponsor is still responsible for obtaining all the data usually obtained in both Phase 1 and Phase 2 studies.
The sponsor must submit to the FDA the results of the preclinical and clinical testing, together with, among other things, detailed information on the manufacture and composition of the product, in the form of a new drug applicationNDA or, in the case of a biologic, a BLA. In a process that can take a year or more, the FDA reviews this application and, when and if it decides that adequate data are available to show that the new compound is both safe and effective for a particular indication and that other applicable requirements have been met, approves the drug or biologic for marketing. The amount of time taken for this approval process is a function of a number of variables, including the quality of the submission and studies presented and the potential contribution that the compound will make in improving the treatment of the disease in question.
Orphan-drug designation is granted by the FDA Office of Orphan Drug Products to novel drugs or biologics that treat a rare disease or condition affecting fewer than 200,000 patients in the U.S. The designation provides the drug developer with a seven-year period of U.S. marketing exclusivity if the drug is the first of its type approved for the specified indication or if it demonstrates superior safety, efficacy or a major contribution to patient care versus another drug of its type previously granted the designation for the same indication. We holdhave been granted orphan drug designations for perifosine in multiple myelomathe MM and for the treatment of neuroblastoma andindications, for AEZS-108 for the treatment of advanced ovarian cancer as well asand for AEZS-130 (Solorel™) for the diagnosis of growth hormone deficiency.
Under the Hatch-Waxman Act, newly-approved drugs and indications may benefit from a statutory period of non-patent data exclusivity. The Hatch-Waxman Act provides five-year data exclusivity to the first applicant to gain approval of an NDA for a new chemical entity, or NCE, meaning that the FDA has not previously approved any other drug containing the same active pharmaceutical ingredient, or active moiety. Although protection under the Hatch-Waxman Act will not prevent the submission or
approval of another full NDA, such an NDA applicant would be required to conduct its own preclinical and adequate, well-controlledwell controlled clinical trials to demonstrate safety and effectiveness.
The Hatch-Waxman Act also provides three years of data exclusivity for the approval of new and supplemental NDAs, including Section 505(b)(2) applications, for, among other things, new indications, dosage forms, routes of administration, or strengths of an existing drug, or for a new use, if new clinical investigations that were conducted or sponsored by the applicant are determined by the FDA to be essential to the approval of the application. This exclusivity, which is sometimes referred to as clinical investigation exclusivity, would not prevent the approval of another application if the applicant has conducted its own adequate, well-controlled clinical trials demonstrating safety and efficacy, nor would it prevent approval of a generic product that did not incorporate the exclusivity-protected changes of the approved drug product.
The labeling, advertising, promotion, marketing, and distribution of a drug or biologic product must be in compliance with FDA regulatory requirements. Failure to comply with applicable requirements can lead to the FDA demanding that production and shipment cease and, in some cases, that the manufacturer recall products, or to enforcement actions that can include seizures, injunctions, and criminal prosecution. These failures can also lead to FDA withdrawal of approval to market a product.
European Union
Medicines can be authorized in the European Union by using either the centralized authorization procedure or national authorization procedures.
Centralized procedure
The European Union has implemented a centralized procedure coordinated by the EMA for the approval of human medicines, which results in a single marketing authorization issued by the European Commission that is valid across the European Union, as well as Iceland, Liechtenstein and Norway. The centralized procedure is compulsory for human medicines that are derived from biotechnology processes, such as genetic engineering, contain a new active substance indicated for the treatment of certain diseases, such as HIV/AIDS, cancer, diabetes, neurodegenerative disorders or autoimmune diseases and other immune dysfunctions, and designated orphan medicines. For medicines that do not fall within these categories, an applicant has the option of submitting an application for a centralized marketing authorization to the EMA, as long as the medicine concerned is a significant therapeutic, scientific or technical innovation, or if its authorization would be in the interest of public health.
National authorization procedures
There are also two other possible routes to authorize medicinal products in several European Union countries, which are available for investigational drug products that fall outside the scope of the centralized procedure:
Decentralized procedure. Using the decentralized procedure, an applicant may apply for simultaneous authorization in more than one European Union country of medicinal products that have not yet been authorized in any European Union country and that do not fall within the mandatory scope of the centralized procedure.
The application will be reviewed by a selected Reference Member State ("RMS"(“RMS”). The Marketing Authorization granted by the RMS will then be recognized by the other Member States involved in this procedure.
Mutual recognition procedure. In the mutual recognition procedure, a medicine is first authorized in one European Union Member State, in accordance with the national procedures of that country. Following this, further marketing authorizations can be sought from other European Union
countries in a procedure whereby the countries concerned agree to recognize the validity of the original, national marketing authorization.
For more information about the regulatory risks associated with the Company'sCompany’s business operations, see "Item“Item 3. — Key Information — Risk Factors"Factors”.
DRUG DISCOVERY
There is an increasing demand on the world market for active substances. Our internal drug discovery unit provides an important prerequisite for the provision of new patented active substances, which can then be developed further or licensed to third parties.
Our drug discovery unit concentrates on the search for active substances for innovative targets which open the door to the introduction of new therapeutic approaches. Further, this unit searches for new active substances having improved properties for clinically validated targets for which drugs are already being used in humans and which produce inadequate effects, cause severe side effects, are not economical or are not available in a patient-friendly form.
To this end, we possess an original substance library for the discovery of active compounds with a comprehensive range of promising natural substances which can serve as models for the construction of synthetic molecules. The initial tests involve 120,000 samples from our internal substance library in the form of high-throughput screening. The hits, i.e.“hits”, which are the first active compounds found in the library, are tested further and built up specifically into potential lead structures. Based on two to three lead structures, they are then optimized in a further step to potential development candidates.
INTELLECTUAL PROPERTY — PATENTS
We believe that we have a solid intellectual property portfolio that covers compounds, manufacturing processes, compositions and methods of medical use for our lead drugs and drug candidates. Our patent portfolio consists of aboutapproximately 50 owned and in licensedin-licensed patent families (issued, granted or pending in the United States, Europe and other jurisdictions). Independent from the original patent expiry date additional exclusivity is possible in the United States, Europe and several other countries by data protection for new chemical entities, by orphan drug designation, or by patent term extension respective supplementary protection certificate.
In the United States, the patent term of a patent that covers an FDA-approved drug may also be eligible for patent term extension, which permits patent term restoration as compensation for the patent term lost during the FDA regulatory review process. The Drug Price Competition and Patent Term Restoration Act of 1984, or the Hatch-Waxman Act, permits a patent term extension of up to five years beyond the expiration of the patent. The length of the patent term extension is related to the length of time the drug is under regulatory review. Patent extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval and only one patent applicable to an approved drug may be extended. Similar provisions are available in Europe and other foreign jurisdictions to extend the term of a patent that covers an approved drug. In the future, if and when our pharmaceutical products receive FDA approval, we expect to apply for patent term extensions on patents covering those products. While we anticipate that any such applications for patent term extensions will likely be granted, we cannot predict the precise length of the time for which such patent terms would be extended in the United States, Europe or other jurisdictions. If we are not able to secure patent term extensions on patents covering our products for meaningful periods of additional time, we may not achieve or sustain profitability, which would adversely affect our business.
Of the issued or granted patents, the protective rights described below form the core of our patent portfolio with regard to our lead drugs and drug candidates.
Perifosine:
| — | U.S. patent 6,172,050 provides protection in the United States for the compound perifosine and other related alkyl phospholipid derivatives, pharmaceutical compositions comprising the compounds as well as their medical use for the treatment of tumors. This U.S. patent expires in July 2013. A patent term extension of up to five years may be possible. |
| — | European patent 0 579 939 provides protection in European countries for the compound perifosine and other related alkyl phospholipid derivatives, pharmaceutical compositions comprising the compounds as well as their medical use for the treatment of tumors. This European patent expires in June 2013. A patent term extension of up to five years by Supplementary Protection Certificates (“SPC”) may be possible. |
| — | Japanese patent 3 311 431 provides protection in Japan for the compound perifosine and other related alkyl phospholipid derivatives. This Japanese patent expires in July 2013. A patent term extension of up to five years may be possible. |
| — | Perifosine combined with antimetabolites: |
| – | U.S., European and Japanese patent applications have been filed, comprising the combination of perifosine with an antimetabolite for treating tumour diseases; |
| – | U.S. patent application 10/632,187, filed July 2003 and further continuation applications US 12/751,454, US 12/751,608 and US 12/798,267 are still pending. Granted patents will be expire in July 2023; |
| – | European patent 1 545 553 expires in July 2023; |
| – | Japanese patent application 2004-525354, filed July 2003 is still pending. Granted patent will expire in July 2023. |
AEZS-108:
| — | U.S. patent 5,843,903 provides protection in the United States for the compound AEZS-108 and other related targeted cytotoxic anthracycline analogs, pharmaceutical compositions comprising the compounds as well as their medical use for the treatment of cancer. This U.S. patent expires in November 2015. A patent term extension of up to five years may be possible. |
| — | European patent 0 863 917 B1 provides protection in Europe for the compound AEZS-108 and other related targeted cytotoxic anthracycline analogs, pharmaceutical compositions comprising the compounds as well as their medical use for the treatment of tumors. This European patent expires in November 2016. A patent term extension of up to five years may be possible. |
| — | Japanese patent 3 987 575 provides protection in Japan for the compound AEZS-108 and other related targeted cytotoxic anthracycline analogs, pharmaceutical compositions comprising the compounds as well as their medical use for the treatment of tumors. This Japanese patent expires in November 2016. A patent term extension of up to five years may be possible. |
AEZS-130:
| — | U.S. patent 6,861,409 protects the compound AEZS-130 and U.S. patent 7,297,681 protects other related growth hormone secretagogue compounds, each also protecting pharmaceutical compositions comprising the compounds as well as their medical use for elevating the plasma level of growth hormone. This U.S. patent 6,861,409 expires in August 2022. A patent term extension of up to five years may be possible. |
| — | European patent 1 289 951 protects the compound AEZS-130 and European patent 1 344 773 protects other related growth hormone secretagogue compounds, pharmaceutical compositions comprising the compounds as well as their medical use for elevating the plasma level of growth hormone. This European patent 1 289 951 expires in June 2021. A patent term extension of up to five years by SPC may be possible. |
| — | Japanese patent 3 522 265 protects the compound AEZS-130 and pharmaceutical compositions comprising the compounds as well as their medical use for elevating the plasma level of growth hormone. This Japanese patent expires in June 2021. A patent term extension of up to five years may be possible. |
Cetrotide®Cetrotide®:
AEZS-112:
| — | European patent 0 299 402 provides protection in European countries for the compound cetrorelix and other LHRH antagonists. This patent will expire in July 2013 pursuant to granted requests for SPC. |
| — | U.S. patent 5,198,533 provided protection in U.S.A. for the compound cetrorelix per se. This patent expired in extended status in October 2010. |
| — | Japanese patent 2 944 669 provides protection in Japan for the compound cetrorelix and other LHRH antagonists. This patent will expire in July 2013 pursuant to granted requests for patent term extension. |
| — | U.S. patent 6,828,415 protects a method for preparing sterile lyophilizate formulations of cetrorelix. It specifically protects the lyophilization process used to manufacture Cetrotide®. This U.S. patent will expire in December 2021. |
| — | European patent 0 611 572 protects a method for preparing sterile lyophilizate formulations of cetrorelix. It specifically protects the lyophilization process used to manufacture Cetrotide®. This patent will expire in February 2014. |
| — | Japanese patent 4 033 919 protects a method for preparing sterile lyophilizate formulations of cetrorelix. It specifically protects the lyophilization process used to manufacture Cetrotide®. This patent will expire in February 2014. |
| — | U.S. patent 7,790,686 protects an aqueous injectable solution of the compound cetrorelix or other LHRH antagonists in an organic, pharmaceutically acceptable acid. This patent will expire in October 2023. |
| — | European patent 1 448 221 protects an aqueous injectable solution of the compound cetrorelix or other LHRH antagonists in an organic, pharmaceutically acceptable acid. This patent will expire in November 2022. |
Ozarelix:
| — | U.S. patent 6,627,609 provides protection in the United States for the compound ozarelix and related third-generation LHRH antagonists and pharmaceutical compositions comprising them. This U.S. patent will expire in March 2020. A patent term extension of up to five years may be possible. |
| — | European patent 1 163 264 provides protection in Europe for the compound ozarelix and related third-generation LHRH antagonists and pharmaceutical compositions comprising them. This European patent will expire in March 2020. A SPC of up to five years may be possible. |
| — | Japanese patent 3 801 867 provides protection in Japan for the compound ozarelix and related third-generation LHRH antagonists and pharmaceutical compositions comprising them. This Japanese patent will expire in March 2020. A patent term extension of up to five years may be possible. |
The table below lists some of our issued or granted patents in the United States, Europe and Europe:Japan:
| Patent No. | Title | Country | Expiry Date | |||||||||
Perifosine | ||||||||||||
U.S. 6,172,050 | Phospholipid derivatives | United States | 2013-07-07 | |||||||||
EP 0 579 939 | Phospholipid derivatives | Germany, United Kingdom, France, Switzerland and others | 2013-06-03 | |||||||||
JP 3 311 431 | Phospholipid derivatives | Japan | 2013-07-08 | |||||||||
EP 1 545 553 | Alkylphosphocholines combined with antimetabolites | Europe | 2023-07-28 | |||||||||
AEZS-108 | ||||||||||||
U.S. 5,843,903 | Targeted cytotoxic anthracycline analogs | United States | 2015-11-27 | |||||||||
EP 0 863 917 | Targeted cytotoxic anthracycline analogs | Europe | 2016-11-14 | |||||||||
JP 3 987 575 | Targeted cytotoxic anthracycline analogs | Japan | 2016-11-14 | |||||||||
AEZS-130 | ||||||||||||
U.S. 6,861,409 | Growth hormone secretagogues | United States | 2022-08-01 | |||||||||
EP 1 289 951 | Growth hormone secretagogues | Germany, United Kingdom, France, Switzerland and others | 2021-06-13 | |||||||||
JP 3 522 265 | Growth hormone secretagogues | Japan | 2021-06-13 | |||||||||
Cetrotide® | ||||||||||||
EP 0 299 402 | LHRH antagonists | Germany, United Kingdom, France, Switzerland and others | 2013-07-10* | |||||||||
EP 0 611 572 | Process to prepare a cetrorelix lyophilised composition | Germany, United Kingdom, France, Switzerland and others | 2014-02-04 | |||||||||
U.S. 6,828,415 | Oliogopeptide lyophilisate, their preparation and use | United States | 2021-12-07 | |||||||||
U.S. 6,716,817 | Method of treatment of female infertility | United States | 2014-02-22 | |||||||||
U.S. 6,863,891 | Oligopeptide lyophilisate, their preparation and use | United States | 2014-02-22 | |||||||||
U.S. 6,867,191 | Preparation and use of oligopeptide lyophilisate for gonad protection | United States | 2014-02-22* | |||||||||
U.S. 7,605,121 | Oligopeptide lyophilisate, their preparation and use | United States | 2014-02-22 | |||||||||
U.S. 7,790,686 | Injection solution of an LHRH antagonist | United States | ||||||||||
AEZS-112 | ||||||||||||
U.S. 7,365,081 | Indole derivatives and their use as medicaments | United States | ||||||||||
EP 1 309 585 | Indole derivatives and their use as medicaments | Germany, United Kingdom, France, Switzerland and others | 2021-07-26 | |||||||||
| Title | Country | Expiry Date | |||||||
Ozarelix | |||||||||
U.S. 6,627,609 | LHRH antagonists having improved solubility properties | United States | 2020-03-14 | ||||||
EP 1 163 264 | LHRH antagonists having improved solubility properties | Germany, United Kingdom, France, Switzerland and others | 2020-03-11 | ||||||
JP 3 801 867 | LHRH antagonists having improved solubility properties | Japan | 2020-03-11 |
| * |
| C. |
C. Organizational structure
The following chart presents our corporate structure, the jurisdiction of incorporation of our direct and indirect subsidiaries and the percentage of shares that we held in those subsidiaries as at December 31, 2010.
D. Property, plants and equipment2011.
| D. | Property, plants and equipment |
Our corporate head office and facilities are located in Quebec City, Province of Quebec, Canada. The following table sets forth information with respect to our main facilities as at March 24, 2011.27, 2012.
Location | Use of space | Square Footage | Type of interest | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
1405 du Parc Technologique Blvd., Quebec City (Quebec), Canada | Fully occupied for management, R&D and administration | 4,400 | Leased | |||||||||||||||
203, Basking Ridge, NJ 07920 | Leased | |||||||||||||||||
Weismüllerstr. 50 D-60314 Frankfurt-am-Main, Germany | Fully occupied for management, R&D, business development and administration | 46,465 | Leased | |||||||||||||||
| Item 4A. | Unresolved Staff Comments |
None.
None.
Item 5. Operating and Financial Review and Prospects
| Item 5. | Operating and Financial Review and Prospects |
Highlights
Perifosine
| § | March 9, 2011: We announced that we had entered into an agreement with Yakult Honsha Co. Ltd. (Tokyo: 2267) (“Yakult”) for the development, manufacture and commercialization of perifosine in all human uses, excluding leishmaniasis, in Japan. Under the terms of the agreement, we received an initial $8.4 million upfront payment and are entitled to receive additional payments of up to $57.1 million upon achieving certain pre-established milestones including clinical and regulatory events in Japan. Furthermore, we will be supplying perifosine to Yakult on a cost-plus-basis and be entitled to receive double-digit royalties on future net sales of perifosine in Japan. Yakult will be responsible for the development, registration and commercialization of perifosine in Japan. |
| § | April 4, 2011: We announced that two posters on our lead anticancer agent, perifosine, had been presented at the 102nd annual meeting of the American Association for Cancer Research (“AACR”) held at the Orange County Convention Center in Orlando, Florida. The first poster demonstrated perifosine’s antitumor activity in several gastric cancer cell lines. Furthermore, perifosine enhanced the antitumor activity of 5-fluorouracil (“5-FU”) in parts of the cell lines - including 5-FU resistant cell lines. As for the second poster, perifosine markedly enhanced the antitumor activity of the cellular TRAIL based treatment and was able to overcome TRAIL resistance bothin vitro andin vivo. The results are in line with other studies demonstrating the synergistic effects of perifosine with cytotoxic drugs, including bortezomib and 5-FU. |
| § | July 12, 2011: We announced that the European Patent Office had granted a patent for the use of alkylphosphocholines, more specifically perifosine, in combination with antitumor anti-metabolites, which include among others 5-FU and capecitabine in the preparation of a medicament for the treatment of benign and malignant tumors. This patent will expire on July 28, 2023. |
| § | July 27, 2011: We announced the completion of patient recruitment (over 465 patients) for the ongoing Phase 3 trial with perifosine in refractory advanced colorectal cancer (“CRC”). |
| § | August 31, 2011: We announced the completion of the pre-specified safety and futility interim analysis by the Data Safety Monitoring Board (“DSMB”) for the Phase 3 trial with perifosine in refractory advanced colorectal cancer. The DSMB recommended that the trial proceed as planned. |
| § | October 5, 2011: We announced that an article on perifosine had been published in the October 2011 online issue of theJournal of Clinical Oncology(“JCO”), in which positive Phase 2 results in metastatic colorectal cancer were reported. |
| § | October 13, 2011: We announced that an article on perifosine had been published in the October 2011 online issue of the JCO, in which positive Phase 1/2 results in multiple myeloma were reported. |
| § | November 23, 2011: We announced the signing of an exclusive commercialization and licensing agreement with Hikma Pharmaceuticals PLC (“Hikma”) (LSE: HIK) (NASDAQ Dubai: HIK) for the registration and marketing of perifosine for the Middle East and North Africa (“MENA”) region. Under the terms of the agreement, we received an upfront payment of $0.2 million and are entitled to receive up to a total of $1.8 million upon achieving certain pre-established milestones. Furthermore, we will be supplying perifosine to Hikma on a cost-plus-basis and are entitled to receive double-digit royalties on future net sales of perifosine in the MENA region. Hikma will be responsible for the registration and commercialization of perifosine in the MENA territory. |
| § | December 12, 2011: We announced encouraging preclinical data for perifosine in Hodgkin’s lymphoma (“HL”). In vitrodata from HL cell lines showed that perifosine combined with sorafenib induced increased apoptosis, while in vivo data for the same combination treatment for HL significantly increased survival in mice. Data were presented during the American Society of Hematology (“ASH”) Annual Meeting and Exposition held in San Diego, California. |
| § | December 13, 2011: We announced encouraging clinical data for an ongoing Phase 2 clinical study in patients with refractory/relapsed HL. Preliminary response data showed that perifosine combined with sorafenib significantly increased median progression free survival (“PFS”) in refractory/relapsed HL patients with high phosphorylation levels of Erk and Akt, as compared to patients with low baseline phosphorylation levels of Erk and Akt. Data were presented during the ASH Annual Meeting and Exposition held in San Diego, California. |
| § | On January 3, 2012: We announced that our Japanese partner, Yakult had initiated a Phase 1/2 trial in Japan to assess the safety and efficacy of perifosine, in combination with a chemotherapeutic agent, capecitabine, in patients with refractory advanced CRC. The initiation of this trial on December 27, 2011 triggered a milestone receivable of $2.6 million. |
AEZS-108
| § | September 14, 2011: We presented positive final Phase 2 efficacy and safety data for AEZS-108 (zoptarelin doxorubicin) in advanced endometrial cancer at the 17th International Meeting of the European Society of Gynecological Oncology in Milan, Italy. Furthermore, a Parallel Scientific Advice process was granted by and initiated with the United States Food and Drug Administration (the “FDA”) and the European Medicines Agency (“EMA”), with the aim of establishing a pivotal program in endometrial cancer. |
| § | September 26, 2011: We announced positive interim data for the Phase 1 portion of our Phase 1/2 trial with AEZS-108 in castration- and taxane-resistant prostate cancer (“CRPC”) at the European Society of Medical Oncology (“ESMO”) meeting in Stockholm, Sweden. |
| § | October 25, 2011: We announced that the FDA had agreed to allow for the initiation by Alberto J. Montero, M.D., of the Sylvester Comprehensive Cancer Center of an Investigational New Drug (“IND”) Phase 2 trial in triple-negative breast cancer (“TNBC”) with AEZS-108. |
| § | On January 5, 2012: We announced an agreement, dated December 19, 2011, with Ventana Medical Systems, Inc., a member of the Roche Group, to develop a companion diagnostic for the immunohistochemical determination of LHRH-receptor expression, for AEZS-108. |
AEZS-130/Solorel®AEZS-120
| § | July 20, 2011: We announced that we had reached a key milestone in the non-clinical development of AEZS-120, our oral prostate cancer vaccine candidate. The program encompassed the full development of a Good Manufacturing Practice (“GMP”) process as well as a safety and toxicology package. |
®AEZS-129
| § | On March 22, 2011: We presented preclinical results for AEZS-129 at the Informa Life Sciences Protein Kinases Congress in Berlin, Germany. AEZS-129 was identified as a potent inhibitor of class I phosphoinositide 3-kinase’s (“PI3K”) lacking activity against mTOR. Data suggest that AEZS-129 is a promising compound for clinical intervention of the PI3K/Akt pathway in human tumors. |
, a ghrelin agonist for diagnostic and therapeutic use.AEZS-130
| § | July 26, 2011: We announced the completion of the Phase 3 study for AEZS-130 (macimorelin) as a first oral diagnostic test for adult growth hormone deficiency (“AGHD”). |
| § | August 30, 2011: We reported favorable top-line results for the completed Phase 3 study for AEZS-130 as a first oral diagnostic test for AGHD. We also announced our intention to meet with the FDA for the future filing of a New Drug Application (“NDA”). The results showed that AEZS-130 reached its primary endpoint demonstrating >90% area-under-the-curve (“AUC”) of the Receiver Operating Characteristic (“ROC”) curve, which determines the level of specificity and sensitivity of the product. |
| § | November 28, 2011: We announced that the FDA agreed to allow for the initiation by Jose M. Garcia, M.D., Ph.D., of Baylor College of Medicine and Michael E. DeBakey of Veterans Affairs Medical Center of an IND Phase 2A trial to assess the safety and efficacy of repeated doses of AEZS-130 in cancer-cachexia. |
| § | On March 8, 2012: We announced that the Michael E. DeBakey of Veterans Affairs Medical Center, in Houston, Texas, had initiated a Phase 2A trial with AEZS-130 in patients with cancer-cachexia. |
AEZS-131
| § | March 22, 2011: We presented preclinical results for AEZS-131 at the Informa Life Sciences Protein Kinases Congress in Berlin, Germany. AEZS-131 was established as a small molecular compound that inhibits Erk in the low nanomolar (“nM���) range and shows an excellent selectivity profile. Results support the evaluation of selective Erk inhibitors as antiproliferative agents either as monotherapy or in combination with inhibitors of the PI3K/Akt pathway. |
| § | April 5, 2011: We announced that a poster on our highly selective Erk 1/2 inhibitor anticancer compound, AEZS-131, had been presented at the 102nd annual meeting of the AACR. Results showed that AEZS-131 selectively inhibits Erk 1/2 with an IC50 of 4nM, blocks cellular Rsk-1 phosphorylation, modulates downstream cellular substrate activation, arrests tumor cells in G1 and inhibits the growth of multiple human tumor cell lines in the nanomolar range. In in vivo pharmacokinetic studies, AEZS-131 showed a favorable PK profile. |
| § | August 30, 2011: We announced preclinical results demonstrating antitumor activity in different human tumor cell lines for AEZS-131 at the American Chemical Society National Meeting in Denver, Colorado. |
| § | December 9, 2011: We announced positive preclinical data in TNBC for AEZS-131 at the 34th Annual San Antonio Breast Cancer Symposium. Data showed that AEZS-131 selectively inhibits Erk at low nanomolar concentrations and induces G1 arrest. Accordingly, the cytotoxic effect of AEZS-131 was most pronounced in TNBC cell lines with mutations in the MAPK pathway. |
July 14, 2010: Presentation at the Seventh International Congress of Neuroendocrinology in Rouen, France, of an abstract on Solorel®, an oral synthetic ghrelin receptor agonist, as a diagnostic test for Adult Growth Hormone ("GH") Deficiency ("AGHD").AEZS-132
| § | On March 22, 2011: We presented preclinical results for AEZS-132 at the Informa Life Sciences Protein Kinases Congress in Berlin, Germany. In summary, AEZS-132 is a unique dual kinase inhibitor targeting the PI3K and MAPK pathways, expected to be especially suited to treat tumors with over-activation of both pathways. |
AEZS-137
| § | March 24, 2011: We announced that we had been awarded a grant of $1.5 million from the German Ministry of Education and Research to develop, up to the clinical stage, cytotoxic conjugates of the proprietary cytotoxic compound AEZS-137 (disorazol Z) and peptides targeting G-protein coupled receptors, including luteinizing hormone releasing hormone (“LHRH”) receptors. |
| § | November 16, 2011: We announced the presentation of a poster on encouraging preclinical data for AEZS-137 at the AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics, held at the Moscone Center West, in San Francisco. AEZS-137 has been identified as a tubulin binding agent with highly potent antitumor properties. Cell cycle analysis revealed that AEZS-137 arrested cells in the G2/M phase and subsequently induced apoptosis with remarkable potency, as shown by subnanomolar EC50 values. |
Corporate developmentsDevelopments
| § | On February 22, 2011: We entered into an “At-the-Market” (“ATM”) sales agreement with MLV & Co. LLC (formerly McNicoll, Lewis & Vlak LLC) (“MLV”), under which we were able to sell up to 12.5 million of our common shares through ATM issuances on the NASDAQ Global Market (the “NASDAQ”) for aggregate gross proceeds not to exceed $19.8 million. On April 29, 2011, we completed the drawdowns from this ATM, bringing the total aggregate gross proceeds to $19.8 million. |
| § | On February 28, 2011: We announced that we had received a net sales royalty milestone of $2.5 million from Cowen Healthcare Royalty Partners L.P. (“Cowen”). This milestone was payable pursuant to the sale, in December 2008, to Cowen of our rights to royalties on future net sales of Cetrotide®. |
| § | On June 29, 2011: We entered into an additional ATM Sales Agreement (the “June ATM Sales Agreement”) with MLV, under which we could, at our discretion, from time to time during the 24-month term of the agreement, sell up to a maximum of 9.5 million of our common shares through ATM issuances on NASDAQ for aggregate gross proceeds not to exceed $24.0 million. On December 7, 2011, we completed the drawdowns from this ATM, bringing the total aggregate gross proceeds to $17.7 million. |
Subsequent to time during the 24-month term of the agreement, sell up to a maximum of 12.5 million of our common shares through ATM issuances on the Nasdaq for aggregate gross proceeds not to exceed $19.8 million. On March 10, 2011, we issued
Table of ContentsAt-The-Market issuance program
approximately 1.7 million shares under this agreement for gross proceeds of approximately $3.2 million.
| § | On January 23, 2012, the Company, pursuant to its existing ATM sales agreement dated June 29, 2011, with MLV, was commencing a new ATM issuance program (“January 2012 ATM Sales Agreement”) under which it may, at its discretion, from time to time during the term of the sales agreement, sell up to a maximum of 10.4 million of its common shares through ATM issuances on NASDAQ up to an aggregate amount of $16.0 million. |
| § | From January 23, 2012 through March 15, 2012, the Company issued a total of 3.6 million common shares under the January 2012 ATM Sales Agreement for aggregate gross proceeds of $6.4 million, less cash transaction costs of $0.2 million and previously deferred transaction costs of $56,000. |
On February 28, 2011, we announced that we had received a net sales royalty milestone of $2.5 million from Cowen Healthcare Royalty Partners L.P. ("Cowen"). This milestone was payable pursuant to the sale, in December 2008, to Cowen of our rights to royalties on future net sales of Cetrotide®.AEZS-108
| § | On February 3, 2012: We reported positive updated results for the Phase 1 portion of our ongoing Phase 1/2 study in CRPC with AEZS-108. Data, presented during a poster session at the American Society of Clinical Oncology Genitourinary Cancers Symposium in San Francisco, showed that AEZS-108 was well tolerated and demonstrated early evidence of antitumor activity in men with CRPC. |
Introduction
This Management'sManagement’s Discussion and Analysis ("(“MD&A"&A”) provides a review of the results of operations, financial condition and cash flows of Aeterna Zentaris Inc. for the year ended December 31, 2010.2011. In this MD&A, "Aeterna Zentaris"“Aeterna Zentaris”, the "Company"“Company”, "we"“we”, "us"“us”, "our"“our” and the "Group"“Group” mean Aeterna Zentaris Inc. and its subsidiaries. This discussion should be read in conjunction with the information contained in the Company'sCompany’s consolidated financial statements and related notes as at December 31, 20102011 and December 31, 20092010 and for the years ended December 31, 2010, 20092011 and 2008.2010. Our consolidated financial statements have been prepared in accordance with International Financial Reporting Standards (“IFRS”) as issued by the International Accounting Standards Board (“IASB”).
All amounts in this MD&A are presented in US dollars, except for share, option and warrant data, per share and per warrant data and as otherwise noted.
Adoption of International Financial Reporting Standards (“IFRS”)
In 2008, the Canadian Accounting Standards Board confirmed that all publicly accountable enterprises must adopt IFRS in place of Canadian generally accepted accounting principles ("(“Canadian GAAP"GAAP”) for financial information, which differ in certain respects from United States generally accepted accounting principles ("US GAAP").The recognition, measurements and disclosure differences as they relate to the company are described in note 25 tobeginning on January 1, 2011 (for entities with a calendar year-end). As such, our 2010 consolidated financial statements included elsewhereas at December 31, 2011 and December 31, 2010 and for the years ended December 31, 2011 and 2010 have been prepared in accordance with IFRS, as issued by the IASB, applicable to the preparation of annual financial statements.
Additionally, our consolidated statement of financial position as at January 1, 2010 and our comparative consolidated financial statements for 2010 have been adjusted to reflect our adoption of IFRS on a retrospective basis, effective on January 1, 2010 (the “Transition Date”). Consequently, 2010 comparative financial information presented in this annual report.MD&A reflects the consistent, retrospective application of IFRS.
These updated figures were reflected in our first quarter report to shareholders.
Our transition to IFRS resulted in a decrease of shareholders’ equity (deficiency) on the IFRS consolidated statement of financial position as at December 31, 2010 of approximately $30.0 million and an increase in 2010 comprehensive loss of approximately $4.1 million.
Our transition to IFRS also resulted in a decrease in shareholders’ equity (deficiency) of $20.1 million, a decrease in total assets of $17.7 million and an increase in total liabilities of $2.4 million, as at January 1, 2010. The decrease in shareholders’ equity (deficiency) was primarily a result of the impairment of Cetrotide® asset, the derecognition of deferred transaction costs and the liability accounting for share purchase warrants.
A complete description of our transition to IFRS, including reconciliations of previously reported Canadian GAAP information, is provided in note 29 to our consolidated financial statements as at December 31, 2011 and December 31, 2010 and for the years ended December 31, 2011 and 2010.
In this MD&A, we also include comparative financial information derived from our consolidated financial statements as at December 31, 2010 and 2009 and for each of the years then ended which, prior to January 1, 2011, were prepared in accordance with Canadian GAAP.
About Forward-Looking Statements
This document contains forward-looking statements, which reflect our current expectations regarding future events. Forward-looking statements may include words such as "anticipate"“anticipate”, "believe"“assuming”, "could"“believe”, "expect"“could”, "foresee"“expect”, "goal"“foresee”, "guidance"“goal”, "intend"“guidance”, "may"“intend”, "objective"“may”, "outlook"“objective”, "plan"“outlook”, "seek"“plan”, "should"“seek”, "strive"“should”, "target"“strive”, “target” and "will"“will”.
Forward-looking statements involve risks and uncertainties, many of which are discussed in this MD&A. Results or performance may differ significantly from expectations. For example, the results of current clinical trials cannot be foreseen, nor can changes in policy or actions taken by regulatory authorities such as the FDA, the EMA,European Medicines Agency (“EMA”), the Therapeutic Products Directorate of Health Canada or any other organization responsible for enforcing regulations in the pharmaceutical industry. Additionally, expected adjustments related to our conversion to International Financial Reporting Standards ("IFRS"), discussed below, that likely will impact various components of our future earnings and are referred to in our discussion of future expectations are unaudited, may not be complete and are subject to further review.
Given these uncertainties and risk factors, readers are cautioned not to place undue reliance on any forward-looking statements. We disclaim any obligation to update any such factors or to publicly announce any revisions to any of the forward-looking statements contained herein to reflect future results, events or developments, unless required to do so by a governmental authority or by applicable law.
About Material Information
This MD&A includes information that we believe to be material to investors after considering all circumstances, including potential market sensitivity. We consider information and disclosures to be material if they result in, or reasonably would be expected to result in, a significant change in the market price or value of our securities, or where it is likely that a reasonable investor would consider the information and disclosures to be important in making an investment decision.
The Company is a reporting issuer under the securities legislation of all of the provinces of Canada, and its securities are registered with the United States Securities and Exchange Commission. The Company is therefore required to file or providefurnish continuous disclosure information such as interim and annual financial statements, MD&As, proxy circulars, annual reports on Form 20-F, material change reports and press releases with the appropriate securities regulatory authorities. Copies of these documents may be obtained free of charge upon request from the Company'sCompany’s Investor Relations department or on the Internet at the following addresses:www.aezsinc.com,,www.sedar.com andwww.sec.gov. www.sec.gov.
Company Overview
Aeterna Zentaris Inc. (Nasdaq:(NASDAQ: AEZS and TSX: AEZ) is a late-stage drug development company specializedspecializing in oncology and endocrine therapy. Our pipeline encompasses compounds at all stages of development, from drug discovery through to marketed products. The highest development priorities in oncology are ourthe completion of Phase 3 programtrials with perifosine in multiple myeloma and colorectal cancer combined with our Phase 2 programand in multiple cancers,myeloma, as well as the further advancement of AEZS-108, for which recentlywe have successfully completed with success a Phase 2 trial in advanced endometrial and advanced ovarian cancer. We are planning for the initiation of a pivotal program with AEZS-108 in endometrial cancer and also a Phase 2 trial in triple-negative breast cancer. AEZS-108 is also in development in other cancer indications, including castration- and taxane-resistant prostate cancer, as well as refractory bladder and castration refractory prostate cancer. In endocrinology, our lead program is our Phase 3 trial with AEZS-130 (Solorel®) as a GH stimulation test for the diagnosis of GH deficiency
Our pipeline also encompasses other earlier-stage programs in adults. We are advancing this Phase 3 trial with an SPA obtained from the FDA.
Additionally, we are advancingoncology. AEZS-112, an oral anticancer agent which involves three mechanisms of action (tubulin, topoisomerase II and angiogenesis inhibition) inhas completed a Phase 1 trial in advanced solid tumors and lymphoma. Additionally, several novel targeted potential anti-cancer candidates such as AEZS-120, a live recombinant oral tumor vaccine candidate, as well as severalour PI3K/Erk inhibitors AEZS-129, AEZS-131, AEZS-132 and their respective follow-up compounds are currently in preclinical programsdevelopment.
Our lead program in endocrinology, a Phase 3 trial under a Special Protocol Assessment (“SPA”) obtained from the FDA with novel targeted potential development candidates.
AEZS-130 as an oral diagnostic test for AGHD, has been completed with positive results. We are currently atplanning to file an NDA for the registration of AEZS-130 in the United States, subject to a stagesuccessful pre-NDA meeting with the FDA. Furthermore, AEZS-130 is in which somea Phase 2A trial for the treatment of our products and product candidates are being further developed or marketed jointly with strategic partners.cancer-cachexia.
Key Developments for the year endedYear Ended December 31, 20102011
Drug Development
Status of our drug pipeline as at March | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Discovery | Preclinical | Phase 1 | Phase 2 | Phase 3 | Commercial | |||||||||||||||
~120,000 compound library | AEZS-120 Prostate cancer vaccine (oncology) AEZS-129, 131, 132 and 136; Erk/PI3K inhibitors (oncology) AEZS-137 (Disorazol Z) (oncology) AEZS-125 (LHRH-Disorazol Z) (oncology) | AEZS-112 (oncology) | Perifosine § Multiple cancers § AEZS-108 § Endometrial cancer § Triple-negative breast cancer § Ovarian cancer § Castration- and taxane-resistant prostate cancer § Refractory bladder cancer Ozarelix § Prostate cancer AEZS-130 § Therapeutic in cancer-cachexia | Perifosine § Refractory advanced colorectal cancer § Multiple myeloma AEZS-130 § Diagnostic in adult growth hormone deficiency (endocrinology) | Cetrotide® (in vitro fertilization) | |||||||||||||||
| Partners | ||||||||||||||||||||
Perifosine: Keryx North America Handok Korea Yakult Japan
Middle East/North Africa Ozarelix: Spectrum World (ex-Japan for oncology indications, ex-Korea and ex-other Asian countries for BPH indication) Handok Korea and other Asian countries for BPH indication Nippon Kayaku Japan for oncology indications | Perifosine: Keryx North America Handok Korea Yakult Japan
Middle East/North Africa |
Japan | ||||||||||||||||||
Perifosine
Perifosine is a novel, oral anticancer treatment that inhibits Akt activation in the PI3K pathway. Perifosine, in combination with chemotherapeutic agents, is currently in Phase 3 studies for the treatment of multiple myeloma, colorectal cancer and multiple myeloma, as well as in Phase 2 studies for the treatment of other cancers, and we believe is the most advanced anti-cancer compound of its class in late-stage development. Perifosine as monotherapy also is being explored in other indications. The FDA has granted perifosine orphan-drug designation in multiple myeloma and in neuroblastoma and Fast Track designations in both multiple myeloma and refractory advanced colorectal cancer.cancer and multiple myeloma. Additionally, an agreement was reached with the FDA to conduct the Phase 3 trials in both of these indications under ana SPA. Perifosine has also been granted Orphan Medicinal Product designation from the EMA in multiple myeloma, and has received positive Scientific Advice frombased on discussions with the EMA, for bothwe believe the multiple myeloma and advanced colorectal cancer programs, with ongoing Phase 3 trials for these indications expected toof perifosine in the colorectal cancer and multiple myeloma programs would be sufficient for registration in Europe.Europe for these indications, assuming positive data from the Phase 3 trials. Perifosine rights have been licensed to Keryx Biopharmaceuticals, Inc. (“Keryx”) for North America, to Handok Pharmaceuticals Co. Ltd. for Korea, to Yakult for Japan and to Handok for Korea.
On January 25, 2010, we announced that Keryx, our partner and licensee in North America, had reported a statistically significant benefit in survival from updated results of a Phase 2 study of perifosine in the treatment of advanced metastatic colorectal cancer. Results showed improvement in
both time to tumor progression and overall survival in the perifosine + capecitabine arm, versus the placebo + capecitabine arm. Of notable interest, andHikma for the first time presented, were data showing a statistically significant benefit in median overall survival (15.3 months vs. 6.8 months — p=0.0088) and time to progression (18 weeks vs. 10 weeks — p=0.0004) for the subset of patients who were refractory to a 5-FU (Fluorouracil) chemotherapy-based treatment regimen.MENA region.
On January 29, 2010, we announced the publication in the February 2010 issue of theJournal of Clinical Cancer Research of positive Phase 2 results for perifosine as a single agent for the treatment of advanced Waldenstrom's macroglobulinemia. The data demonstrated a 35% overall response rate with a median progression-free survival of 12.6 months in patients with relapsed or relapsed/refractory Waldenstrom's macroglobulinemia.
On February 3, 2010, we announced that Keryx had reached an agreement with the FDA on an SPA for the Phase 3 X-PECT trial of perifosine in refractory advanced colorectal cancer, in addition to the earlier SPA agreement for the Phase 3 trial in multiple myeloma.
On March 1, 2010, we disclosed that the Committee for Orphan Medicinal Products of the EMA had issued a positive opinion for orphan medicinal product designation for perifosine for the treatment of multiple myeloma.
On April 5, 2010, our partner, Keryx, was granted Fast Track designation by the FDA for the Phase 3 X-PECT registration trial.
On April 8, 2010, our partner, Keryx announced the initiation of a Phase 3 X-PECT registration trial with perifosine in refractory advanced colorectal cancer. The Phase 3 trial is being conducted pursuant to a SPA with the FDA. Approximately 40 to 50 U.S. sites will participate in the study.
On April 15, 2010, we received Positive Scientific Advice from the EMA for the Phase 3 program with perifosine in multiple myeloma, therefore indicating that the data from the ongoing trial are expected to be sufficient for product registration in Europe.
On April 20, 2010, at the AACR's annual meeting we presented data on our dual Erk/PI3K inhibitors and on our selective Erk inhibitors. Data supported further evaluation of selective Erk inhibitors as antiproliferative agents, either as monotherapy or in combination with inhibitors of the PI3K/Akt pathway. Other data resulted in the identification of AEZS-132, a unique dual inhibitor of PI3K and Erk with a favourable pharmacology and ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) profile for further evaluation as an antitumor agent. At that same meeting, preclinical data in neuroblastoma for perifosine were also presented. Data demonstrated that single agent perifosine targets activation of Akt in neuroblastoma cells and xenografts, significantly inhibited tumor growthin vivo and improved the survival of mice bearing neuroblastoma tumors.
On May 17, 2010, we announced the publication of an article in the May 12, 2010 issue of theJournal of the National Cancer Institute entitled "In Vitro andIn Vivo Inhibition of Neuroblastoma Tumor Cell Growth by AKT Inhibitor Perifosine," demonstrating the single agent activity of perifosine in neuroblastoma tumor preclinical models.
On June 7, 2010, we announced that Phase 1 data for perifosine in recurrent pediatric solid tumors had been presented in the pediatric solid tumor poster discussion session held at the 46th annual ASCO meeting in Chicago. This study, conducted by the Memorial Sloan-Kettering Cancer Center pediatric group, marks the first time that perifosine has been administered in a pediatric patient setting.
This Phase 1 study of perifosine for recurrent pediatric solid tumors is a single center, open-label, dose-escalating study to assess safety, tolerability, pharmacokinetics ("PK"), and to identify any dose limiting toxicity ("DLT") of single agent perifosine in pediatric patients with any solid tumor that has failed standard therapy. Eleven patients (4 males, 7 females), at a median age of 13 years (5-18) were treated in this study to date. The following tumor types were treated thus far: high-grade glioma (5),
medulloblastoma (2), neuroblastoma (3), and ependymoma (1). Most patients were heavily pretreated with a median of three prior lines of therapy. Cohorts of three patients were treated at three dose levels of perifosine after a loading dose on day 1, and taking into account the drug's long half-life (t1/2 100 hours). No DLTs were observed at any of the three dose levels; dose level 4 is currently open for accrual. PK data thus far suggest similar drug absorption by pediatric patients relative to adult patients treated with single agent perifosine.
Of particular interest are the early signs of clinical activity observed in two of the three patients with Stage 4 refractory neuroblastoma. Both patients were refractory to prior treatments upon entering the study and achieved stable disease for 48 weeks and 55+ weeks (ongoing). The investigators concluded that perifosine is well tolerated in children with recurrent solid tumors and that these early signals of activity warrant further investigation in patients with advanced neuroblastoma and select brain tumors. Previously, perifosine has been shown to target activation of Akt in neuroblastoma cells and xenografts and to significantly inhibit tumor growthin vivo and improve the survival of mice bearing neuroblastoma tumors.
On June 8, 2010, we reported Phase 2 results at the ASCO annual meeting, confirming a statistically significant improvement in both time to tumor progression and overall survival with perifosine, in combination with capecitabine (Xeloda®), in the treatment of advanced metastatic colorectal cancer.
In this randomized, double-blind, placebo-controlled study, conducted at 11 centers across the United States, heavily pre-treated patients with second- or third-line metastatic colorectal cancer were randomized to receive capecitabine (Xeloda®) at 825 mg/m2 BID (total daily dose of 1650 mg/m2) on days 1 - 14, every 21 days, plus either perifosine or placebo at 50 mg daily. Of the 38 patients enrolled, 35 patients were evaluable for response (20 patients on the perifosine + capecitabine arm and 15 patients on the placebo + capecitabine arm). The perifosine + capecitabine arm demonstrated a greater than 60% improvement in overall survival, a more than doubling of median time to progression, and almost a doubling of the percentage of patients achieving stable disease or better. In addition, the overall response rate was 20% (including one complete response, and durable responses) in the perifosine + capecitabine arm versus 7% in the capecitabine arm. Of notable interest were the patients who were previously refractory to a 5-FU based regimen. The perifosine + capecitabine arm again demonstrated a statistically significant increase in both time to progression and overall survival, as compared to the capecitabine arm. As for safety, the perifosine + capecitabine arm was well tolerated.
On June 29, 2010, we announced that we had received positive Scientific Advice from the EMA regarding the Phase 3 X-PECT trial for the development of perifosine in refractory advanced colorectal cancer. The Scientific Advice from the EMA indicates that the ongoing study, in conjunction with safety data generated from other clinical studies with perifosine, is considered sufficient to provide all data necessary to support a marketing authorization of perifosine in advanced colorectal cancer. We do not intend to initiate any additional studies with perifosine for this indication. Therefore, for the development of perifosine in both multiple myeloma and colorectal cancer, we believe that the planned North American clinical program, sponsored by our partner Keryx, is now sufficient for approval in Europe and in many countries in the rest of the world, where we hold rights for our compound.
On July 14, 2010, our partner, Keryx, was granted orphan-drug designation by the FDA for perifosine for the treatment of neuroblastoma, a cancer of the nervous system affecting mostly children and infants for which there are no FDA-approved therapies.
On November 17, 2010, at the EORTC-NCI-AACR Symposium on Molecular Targets and Cancer Therapeutics in Berlin, Germany, we presented an abstract on perifosine combined with antimetabolites which induces synergistic effects on cytotoxicity and apoptosis in human colon, multiple myeloma, breast, renal, and liver tumor cell lines.
On December 6, 2010 at the ASH's 52nd annual meeting in Orlando, we announced positive safety and tolerability Phase 2 data for perifosine in patients with advanced CLL and HL, as well as positive Phase 1 results of perifosine in combination with lenalidomide (Revlimid®) + dexamethasone in patients with relapsed or refractory multiple myeloma.
In the first Phase 2 study related to CLL, 12 patients with advanced CLL began treatment with single agent perifosine at 50 mg BID. Patients on study were heavily pre-treated having had a median of four prior lines of therapy with 75% of patients classified as Rai stage IV. One patient achieved a partial response (5 months on treatment) and 5 additional patients achieved stable disease (median duration of 4.25 months), for an overall 50% clinical benefit rate (PR + SD). Perifosine was well tolerated with minimal dose modifications.
In the second study presented, 26 patients were enrolled in a Phase 2 study with advanced lymphoma (6 non-HL, 4 CLL, 1 Waldenstrom's Macroglobulinemia and 15 HL). 73% of patients were previously refractory to their prior therapy, with 85% of patients having had 4 or more prior therapies. Perifosine (50 mg BID) was started as a single agent for 28 days; after 28 days, patients achieving partial response (PR) or better were continued on single agent perifosine. Patients achieving less than a PR were given the combination of perifosine (50 mg BID) plus sorafenib (Nexavar®) at 400 mg BID. All of the 4 CLL patients in this study achieved a partial response on single-agent perifosine within one month of treatment and remained on perifosine single agent. Response durations for each of the 4 patients were 4, 8, 9+ and 12 months. The remaining 22 patients were administered the combination with sorafenib, where 5 of the 15 (33%) HL patients achieved a partial response with a median response duration of 9 months. An additional 6 patients receiving the combination (40%) achieved stable disease.
The combination was well tolerated with no unexpected safety events.
The investigators concluded that perifosine in combination with sorafenib has significant anti-lymphoma activity in relapsed/refractory HL, and that perifosine as a single agent induced prolonged responses in high-risk, heavily pretreated CLL patients.
With regard to multiple myeloma, the final data set from the Phase 1 study of perifosine + lenalidomide (Revlimid®) + dexamethasone were also presented during the ASH meeting. The final data showed a 73% objective response rate (minimal response or better) with a 50% PR or better, a median Progression-Free Survival of 10.8 months, and a median duration for Overall Survival of 30.6 months. The myeloma investigators concluded that perifosine in combination with lenalidomide + dexamethasone was well tolerated even at the highest doses used, and demonstrated encouraging clinical activity and survival.
AEZS-108
AEZS-108 has been studied in gynecological cancers and has been shown to be effective and well tolerated in advanced endometrial and ovarian cancers. Positive Phase 2 results for ovarian cancer were disclosed in June 2010 at the annual ASCO meeting, while positive Phase 2 results for endometrial cancer were presented at the EORTC-NCI-AACR International Symposium on Molecular Targets and Cancer Therapeutics in November 2010. In addition to the ongoing Phase 1/2 studies in refractory bladder cancer and castration refractory prostate cancer, we intend to initiate discussions with the FDA and the EMA in an effort to reach an agreement on a protocol for a registration trial in endometrial cancer.
On May 6, 2010, we announced that we had received orphan drug designation from the FDA for AEZS-108 for the treatment of ovarian cancer. Orphan drug designation is granted by the FDA's Office of Orphan Products Development to novel drugs or biologics that treat a rare disease or condition affecting fewer than 200,000 patients in the US. The designation provides a drug developer with a
seven-year period of U.S. marketing exclusivity if the drug is the first of its type approved for the specified indication or if it demonstrates superior safety or efficacy versus another drug of its type previously granted the designation for the same indication.
On May 12, 2010, we announced that the FDA had approved our IND application for AEZS-108 in LHRH receptor-positive urothelial (bladder) cancer. Following this approval from the FDA, this trial will be conducted at the Sylvester Comprehensive Cancer Center at the University of Miami's Miller School of Medicine, and will include up to 64 patients, male and female, with advanced LHRH receptor-positive urothelial (bladder) cancer. The study will be conducted in two parts: first, a dose-finding part in up to 12 patients; subsequently, a selected dose will be studied for its effect on progression-free survival.
On May 17, 2010, we announced that we had received a positive opinion for orphan medicinal product designation from the COMP of the EMA, for AEZS-108 for the treatment of ovarian cancer. Orphan medicinal product designation is granted by the European Commission, following a positive opinion from the COMP, to a medicinal product that is intended for the diagnosis, prevention or treatment of a life-threatening or a chronically debilitating condition affecting not more than five in 10,000 persons in the Community when the application for designation is submitted. Orphan medicinal product designation provides the sponsor with access to the Centralized Procedure for the application for marketing authorization, protocol assistance, up to a 100% reduction in fees related to a marketing authorization application, pre-authorization inspection and post-authorization activities, and could provide ten years of market exclusivity in the European Union for AEZS-108, once approved for the treatment of ovarian cancer.
On June 7, 2010, Prof. Günter Emons, Chairman, Department of Obstetrics & Gynaecology Georg-August University Göttingen, Germany, presented positive efficacy and safety data for AEZS-108 in ovarian cancer at the ASCO Annual Meeting. The poster (abstract #5035), entitled "Phase 2 study of AEZS-108, a targeted cytotoxic LHRH analog, in patients with LHRH receptor-positive platinum resistant ovarian cancer" (G. Emons, S. Tomov, P. Harter, J. Sehouli, P. Wimberger, A. Staehle, L. C. Hanker, F. Hilpert, P. Dall and C. Gruendker, for the AGO Study Group), details the use of AEZS-108 in women with histologically confirmed taxane-pretreated platinum-resistant/refractory LHRH receptor-positive advanced (FIGO III or IV) or recurrent ovarian cancer. Patients received a recommended dose of 267 mg/m2 by intravenous infusion over 2 hours, with retreatment every 3 weeks, for up to 6 courses. Response rate (RECIST and/or GCIG criteria) was defined as the primary endpoint. Secondary endpoints were safety, time-to-progression and overall survival.
42 patients with platinum-resistant ovarian cancer entered the study. Efficacy included partial response in 5 patients (11.9%) and stable disease for more than 12 weeks in 11 patients (26.2%). Based on those data, a Clinical Benefit Rate of 38% can be estimated. Median time to progression and overall survival were 3.5 months (104 days) and 15.6 months (475 days), respectively. Overall survival compares favourably with data from Doxil and Topotecan (8-9 months). In all, tolerability of AEZS-108 was good and commonly allowed retreatment as scheduled. Only one patient (2.4%) had a dose reduction, and overall, 25 of 170 (14.7%) courses were given with a delay, including also cases in which delay was not related to toxicity. Severe (Grade 3 or 4) toxicity was mainly restricted to rapidly reversible hematologic toxicity (leukopenia / neutropenia) associated with fever in 3 cases. Good tolerability of AEZS-108 was also reflected with only a few patients with non-hematological toxicities of grade 3 (none with Grade 4), including single cases (2.4%) each of nausea, constipation, poor general condition, and an enzyme elevation. No cardiac toxicity was reported.
On June 28, 2010, we announced that we had concluded an agreement with Almac's Diagnostics division for AEZS-108, aimed at determining LHRH receptor expression through the development of a companion diagnostic tool. Selection for treatment with AEZS-108 is determined on the basis of LHRH receptor expression, currently measured immunohistochemically. In humans, LHRH receptors
are expressed in ovarian, endometrial, breast, bladder, prostate and pancreatic tumors. This state of the art companion diagnostic tool will allow us to develop improved methods of selecting the most appropriate patients to be treated with AEZS-108 in order to enhance the efficiency of our clinical trials and help us with the future successful development of AEZS-108 in a number of different LHRH expressing cancers.
On August 5, 2010, we announced that the NIH had awarded Dr. Jacek Pinski, Associate Professor of Medicine at the Norris Comprehensive Cancer Center of the University of Southern California, a grant of approximately $1.5 million over three years to conduct a Phase 1/2 study in refractory prostate cancer with AEZS-108. The study, entitledA Phase I/II Trial of AN-152 [AEZS-108] in Castration- and Taxane-Resistant Prostate Cancer, will enrol up to 55 patients and will be conducted in two portions: an abbreviated dose-escalation followed by a single arm, Simon Optimum two-stage design Phase 2 study using the dose selected in the Phase 1 portion. The primary objective of the Phase 2 portion is to evaluate the clinical benefit of AEZS-108 in men with castration- and taxane-resistant metastatic prostate cancer, for which the presence of LHRH receptors has been confirmed.
On November 18, 2010, Prof. Günter Emons of the Department of Obstetrics & Gynaecology Georg-August at the University of Göttingen (Germany) presented positive data for the Phase 2 of AEZS-108 in Advanced Endometrial Cancer at the EORTC-NCI-AACR symposium in Berlin, Germany. The study showed encouraging results as AEZ-108 was used as a single agent.
Of 43 patients treated with AEZS-108 in this study, 39 were evaluable for efficacy. Responses confirmed by independent review included 2 patients with complete response (CR; 5.1%), 10 patients with partial response (PR; 25.6%), and 17 patients with stable disease (SD; 43.6%). Based on those data, an overall response rate (ORR = CR+PR) of 30.8% and a clinical benefit rate (CBR = CR+PR+SD) of 74.4% can be estimated. Responses were also achieved in patients with prior chemotherapy, 1 CR, 1 PR and 2 SDs in 8 of the patients pre-treated with platinum/taxane regimens. Median time to progression and overall survival were 7 months (30 weeks) and 14.3 months (62 weeks), respectively. Conclusions from this trial were as follows:
The overall survival after single agent AEZS-108 is similar to that reported for modern triple combination chemotherapy, but was achieved with lower toxicity.
On December 14, 2010, we announced the initiation of a Phase 1/2 trial in castration refractory prostate cancer conducted by Dr. Jacek Pinski at the Norris Comprehensive Cancer Center, as well as a Phase 1/2 trial in refractory bladder cancer conducted by Dr. Gustavo Fernandez at the Sylvester Comprehensive Cancer Center.
AEZS-130/Solorel®
AEZS-130/Solorel® (macimorelin), a ghrelin agonist, is a novel synthetic small molecule that stimulates the secretion of growth hormone. The product is currently in Phase 3 for use as a simple oral diagnostic test for AGHD. Solorel® has been granted orphan-drug designation by the FDA.
On June 21, 2010, we presented positive data at the 92nd ENDO Meeting and Expo on AEZS-130 for diagnostic and therapeutic use. The preclinical data showed that AEZS-130 is a potent and safe oral synthetic GH-releasing compound with potential utility as a diagnostic test for growth hormone deficiencies. In addition to the diagnostic indication, we believe that, based on the results of Phase 1 studies, AEZS-130 (Solorel®) has potential applications for the treatment of cachexia, a condition frequently associated with severe chronic diseases such as cancer, chronic obstructive pulmonary disease and Acquired Immune Deficiency Syndrome, or AIDS.
On July 14, 2010, we announced the presentation of a poster on Solorel®, entitledUse of the Orally Active Ghrelin Mimetic AEZS-130 as a Simple Test for the Diagnosis of Growth Hormone (GH) Deficiency (GHD) in adults (AGHD), Merriam G.R., Yuen K., Bonert V., Dobs A, Garcia J., Kipnes M., Molitch M., Swerdloff R., Wang C., Cook D., Altemose I. and Biller B. This poster was presented at the Seventh International Congress of Neuroendocrinology, in Rouen, France.
On October 5, 2010, we announced at the Fifth International Congress of the Growth Hormone Research Society and the Insulin-like Growth Factors Society, after the interim Phase 3 analysis of the orphan drug AEZS-130, that it demonstrated the potential to provide a simple, well tolerated and safe oral diagnostic test for AGHD. Solorel® has been granted orphan drug designation by the FDA as a diagnostic test.
Corporate developments
On April 20, 2010, we completed a registered direct offering of 11,111,111 units, with each unit consisting of one common share and a warrant to purchase 0.40 of a common share, at a price of $1.35 per unit (the "April 2010 Offering"). Total proceeds raised upon completion of the April 2010 Offering amounted to $15.0 million less cash transaction costs of approximately $1.3 million. The securities described above were offered by us pursuant to a shelf prospectus dated March 12, 2010 and a prospectus supplement dated April 15, 2010.
We granted warrants (the "April 2010 Investor Warrants") to the investors who participated in the April 2010 Offering. Each April 2010 Investor Warrant entitles the holder to purchase 0.40 of a common share at an exercise price of $1.50 per share. The April 2010 Investor Warrants are exercisable between October 20, 2010 and October 20, 2015, and, upon complete exercise, would result in the issuance of an aggregate of 4,444,444 common shares.
We estimated the fair value attributable to the April 2010 Investor Warrants of $3.6 million as at the date of grant by applying the Black-Scholes pricing model, to which the following additional assumptions were applied: a risk-free annual interest rate of 2.56%, expected volatility of 87.3%, an expected term of 5 years, a dividend yield of 0.0% and an issue-date market share price of $1.24. Transaction costs allocated to the April 2010 Investor Warrants amounted to approximately $0.3 million.
On June 21, 2010, we completed a registered direct offering of 8,805,964 units, with each unit consisting of one common share and a warrant to purchase 0.50 of a common share, at a price of $1.3725 per unit (the "June 2010 Offering"). Total proceeds raised upon completion of the June 2010 Offering amounted to $12.1 million, less cash transaction costs of approximately $0.8 million. The securities described above were offered by us pursuant to a shelf prospectus dated March 12, 2010 and a prospectus supplement dated June 15, 2010.
We granted warrants (the "June 2010 Investor Warrants") to the investors who participated in the June 2010 Offering. Each June 2010 Investor Warrant entitles the holder to purchase a common share at an exercise price of $1.3725 per share. The June 2010 Investor Warrants are exercisable between June 21, 2010 and June 21, 2015, and, upon complete exercise, would result in the issuance of an aggregate of 4,402,982 common shares.
We estimated the fair value attributable to the June 2010 Investor Warrants of $3.5 million as at the date of grant by applying the Black-Scholes pricing model, to which the following additional assumptions were applied: a risk-free annual interest rate of 2.05%, expected volatility of 89.3%, an expected term of 5 years, a dividend yield of 0.0% and an issue-date market share price of $1.18. Transaction costs allocated to the June 2010 Investor Warrants amounted to approximately $0.2 million.
We also granted warrants (the "June 2010 Compensation Warrants") to the sole placement agent (and to certain of its designated representatives) engaged in connection with the June 2010 Offering. Each June 2010 Compensation Warrant entitles the holder to purchase a common share at an exercise price of $1.7156 per share. The June 2010 Compensation Warrants are exercisable between June 15, 2010 and June 15, 2015, and, upon complete exercise, would result in the issuance of 264,178 common shares.
We estimated the fair value attributable to the June 2010 Compensation Warrants of $0.2 million as at the date of grant by applying the Black-Scholes pricing model, to which the following additional assumptions were applied: a risk-free annual interest rate of 2.04%, expected volatility of 89.4%, an expected term of 5 years, a dividend yield of 0.0% and an issue-date market share price of $1.18. The initial fair value of the June 2010 Compensation Warrants has been accounted for as additional transaction costs, since the instruments were granted to the sole placement agent as part of the terms of the underlying engagement and in recognition of the efforts made in connection with the June 2010 Offering.
On June 23, 2009, we completed a registered direct offering of 5,319,149 units, with each unit consisting of one common share and a warrant to purchase 0.35 of a common share at a price of $1.88 per unit (the "June 2009 Offering"). Total proceeds raised through the June 2009 Offering amounted to $10.0 million, less cash and non-cash transaction costs of $1.6 million. The purchasers in this offering were comprised of institutional investors, and the securities described above were offered by us pursuant to a shelf prospectus dated September 27, 2007 and a prospectus supplement dated June 18, 2009.
We granted a total of 5,319,149 warrants (the "June 2009 Investor Warrants") to the institutional investors who participated in the June 2009 Offering. Each June 2009 Investor Warrant entitles the holder to purchase 0.35 of a common share at an exercise price of $2.06 per share. The June 2009 Investor Warrants are exercisable between September 23, 2009 and December 23, 2011, and, upon complete exercise, would result in the issuance of an aggregate of 1,861,702 common shares of the Company.
We estimated the fair value attributable to the June 2009 Investor Warrants of $1.6 million as at the date of grant by applying the Black-Scholes pricing model, to which the following additional assumptions were applied: a risk-free annual interest rate of 1.74%, expected volatility of 90.6%, an expected term of 2.5 years, dividend yield of 0.0% and an issue-date market share price of $1.75. Transaction costs allocated to the June 2009 Investor Warrants amounted to approximately $0.2 million.
We granted a total of 820,668 warrants (the "June 2009 Compensation Warrants") to the sole placement agent and its designated representatives engaged in connection with the June 2009 Offering. Each June 2009 Compensation Warrant entitles the holder to purchase 0.35 of a common share at an exercise price of $2.35 per share. The June 2009 Compensation Warrants are exercisable between December 23, 2009 and December 23, 2011, and, upon complete exercise, would result in the issuance of 287,234 common shares of the Company.
We estimated the fair value attributable to the June 2009 Compensation Warrants of $0.2 million as at the date of grant by applying the Black-Scholes pricing model, to which the following additional assumptions were applied: a risk-free annual interest rate of 1.74%, expected volatility of 90.6%, an expected term of 2.5 years, an expected dividend yield of 0.0% and an issue-date market share price of
$1.75. The initial fair value of the June 2009 Compensation Warrants has been accounted for as additional transaction costs, since the instruments were granted to the sole placement agent as part of the terms of the underlying engagement and in recognition of the efforts made in connection with the June 2009 Offering.
On October 23, 2009, we completed a registered direct offering of 4,583,335 units, with each unit consisting of one common share and a warrant to purchase 0.40 of a common share, at a price of $1.20 per unit (the "October 2009 Offering"). Total proceeds raised through the October 2009 Offering amounted to $5.5 million, less cash transaction costs of approximately $0.4 million. The purchasers in this offering were new and existing institutional investors, and the securities described above were offered by us pursuant to a shelf prospectus dated September 27, 2007 and a prospectus supplement dated October 19, 2009.
We granted a total of 4,583,335 warrants (the "October 2009 Investor Warrants") to the institutional investors who participated in the October 2009 Offering. Each October 2009 Investor Warrant entitles the holder to purchase 0.40 of a common share at an exercise price of $1.25 per share. The October 2009 Investor Warrants are exercisable between October 23, 2009 and October 23, 2014, and, upon complete exercise, would result in the issuance of an aggregate of 1,833,334 common shares.
We estimated the fair value attributable to the October 2009 Investor Warrants of $1.3 million as of the date of grant by applying the Black-Scholes pricing model, to which the following additional assumptions were applied: a risk-free annual interest rate of 2.46%, expected volatility of 84.3%, an expected term of 5 years, dividend yield of 0.0% and an issue-date market share price of $1.09. Transaction costs allocated to the October 2009 Investor Warrants amounted to approximately $0.1 million.
We granted a total of 320,832 warrants (the "October 2009 Compensation Warrants") to the sole placement agent engaged in connection with the October 2009 Offering. Each October 2009 Compensation Warrant entitles the holder to purchase 0.40 of a common share at an exercise price of $1.50 per share. The October 2009 Compensation Warrants are exercisable between April 23, 2010 and October 23, 2012, and, upon complete exercise, would result in the issuance of 128,333 common shares.
We estimated the fair value attributable to the October 2009 Compensation Warrants of $86,653 as at the date of grant by applying the Black-Scholes pricing model, to which the following additional assumptions were applied: a risk-free annual interest rate of 1.57%, expected volatility of 103.4%, an expected term of 3 years, dividend yield of 0.0% and an issue-date market share price of $1.09. The initial fair value of the October 2009 Compensation Warrants has been accounted for as additional transaction costs, since the instruments were granted to the sole placement agent as part of the terms of the underlying engagement and in recognition of the efforts made in connection with the October 2009 Offering.
The terms of all aforementioned warrants are substantially the same, with the exception of the exercise price and contractual period of exercise, as discussed above. In particular, all warrants may be exercised, at the option of the holder, by cash payment of the exercise price or, upon the existence of certain conditions, by "cashless exercise", which means that in lieu of paying the aggregate exercise price for the shares being purchased upon exercise of the warrants in cash, the holder would receive the number of shares underlying the warrants equal to the quotient obtained by applying a formula, as defined by the terms of each warrant. We will not receive additional proceeds to the extent that warrants are exercised by cashless exercise.
The exercise price and number of common shares issuable on exercise of all outstanding warrants may be adjusted in certain circumstances, including stock dividends or splits, subsequent rights offerings, pro-rata distributions and pursuant to transactions involving the merger or consolidation of the Company with another entity or other Fundamental Transaction, as defined in the warrants.
Additionally, and notwithstanding anything to the contrary, in the event of any type of Fundamental Transaction, as defined in the warrants, the Company or any successor entity shall, at our option, have the right to require the holders thereof to exercise the warrants, or, at the holder's option, purchase the warrants from the holders by paying the holders an amount of cash equivalent to the Black-Scholes value, as defined, of the remaining unexercised portion of the warrants on the date of the consummation of an aforementioned Fundamental Transaction.
Subsequent to year-end
On February 22, 2011, we entered into an "At-the-Market" ("ATM") sales agreement, under which we may, at our discretion, from time to time during the 24-month term of the agreement, sell up to a maximum of 12.5 million of our common shares through ATM issuances on the Nasdaq for aggregate gross proceeds not to exceed $19.8 million, being the amount remaining available for distribution, as at February 22, 2011, under our current registration statement on Form F-3. The common shares will be sold at market prices prevailing at the time of a sale of the common shares, and, as a result, prices may vary.
On March 10, 2011, we issued approximately 1.7 million common shares in connection with the aforementioned ATM agreement, for gross proceeds of approximately $3.2 million.
On March 9, 2011, we announced that we had entered into an agreement with Yakult for the development, manufacture and commercialization of perifosine in all human uses, excluding leishmaniasis, in Japan. Under the terms of this agreement, Yakult will makemade an initial, non-refundable gross upfront payment to us of €6.0 million (approximately $8.3$8.4 million). Also per the agreement, we will beare entitled to receive in addition, up to a total of €44.0 million (approximately $60.9$57.1 million) upon achieving certain pre-established milestones, including the occurrence of certain clinical and regulatory events in Japan. As at December 31, 2011, following the achievement of a milestone, we have revenue and trade receivables recorded for an additional amount of €2.0 million (approximately $2.6 million) in connection with the agreement. Furthermore, we will be entitled to receive double-digit royalties on future net sales of perifosine in the Japanese market.
On November 23, 2011, we entered into an agreement with Hikma for the development, and commercialization of perifosine in all oncology uses. Under the terms of this agreement, Hikma made an initial, non-refundable gross upfront payment to us of $0.2 million. Also per the agreement, we are entitled to receive up to a total of $1.8 million upon achieving certain pre-established milestones, including the occurrence of certain regulatory events in certain countries in the MENA region. Furthermore, we will be entitled to receive double-digit royalties on future net sales of perifosine in the MENA market.
We have alsosubstantial continuing involvement in the aforementioned arrangements, including the use of commercially reasonable efforts to develop, apply for and obtain relevant regulatory approval for, manufacture and commercialize perifosine outside Japan and the MENA region, which will facilitate the ultimate commercialization process within Japan and the MENA region. Additionally, we are contractually obligated to ensure a stable supply of perifosine and related trial products to Yakult and Hikma throughout the ongoing development process and will maintain relevant patent rights over the term of the arrangements. Lastly, per the terms of the aforementioned agreements, we have agreed to supply perifosine, on a cost-plus basis, to Yakult and Hikma following regulatory approvals.
We have deferred the non-refundable license fees and we are amortizing the related payments as revenue on a cost-plus-basis.straight-line basis over the duration of our continuing involvement, which approximates the estimated life cycle of the product that is currently under development and the expected period over which Yakult and Hikma will derive value from the use of, and access to, the underlying licenses.
In determining the period over which license revenues are to be recognized, and in addition to due consideration of our continuing involvement, as discussed above, we considered the remaining expected life of applicable patents as the most reasonable basis for estimating the underlying product’s life cycle. However, we may adjust the amortization period based on appropriate facts and circumstances not yet known, including, but not limited to, the extension(s) of patents, the granting of new patents, the economic lives of competing products and other events that would significantly change the duration of our continuing involvement and performance obligations or benefits expected to be derived by Yakult and Hikma.
Future milestones will be recognized as revenue individually and in full upon the actual achievement of the related milestone, given the substantive nature of each milestone. Lastly, upon initial commercialization and sale of the developed product, we will recognize royalty revenues as earned, based on the contractual percentage applied to the actual net sales achieved by Yakult and Hikma, as per the aforementioned agreements.
We were required to remit to the Japanese tax authorities $0.8 million of the gross proceeds received from Yakult and will be required to remit approximately €0.2 million (approximately $0.26 million) when the milestone receivable will be paid by Yakult. These amounts, which were withheld at source, were recognized as income tax expense in our consolidated statement of comprehensive loss.
On April 4, 2011, we announced that two posters on perifosine were presented at the 102nd annual meeting of the AACR at the Orange County Convention Center in Orlando, Florida. Perifosine demonstrated antitumor activity in several gastric cancer cell lines. Furthermore, perifosine enhanced the antitumor activity of 5-FU in parts of the cell lines, including 5-FU resistant cell lines. 5-FU is the active metabolite of the prodrug Xeloda, which is approved for the treatment of advanced gastric cancer in many countries including Japan.
Perifosine also markedly enhanced the antitumor activity of the cellular TRAIL based treatment and was able to overcome TRAIL resistance bothin vitro andin vivo. The results are in line with other studies demonstrating the synergistic effects of perifosine with cytotoxic drugs, including bortezomib and 5-FU.
On July 12, 2011, we announced that the European Patent Office had granted a patent for the use of alkylphosphocholines, more specifically perifosine (octadecyl 1,1-dimethylpiperidino-4-yl phosphate), in the preparation of a medicament for the treatment of benign and malignant tumors, prior to and/or during the treatment with approved antitumor anti-metabolites such as 5-FU and capecitabine. Filed on July 29, 2003, the patent (EP #1 545 553), entitledUse of Alkyl Phosphocholines in Combination with Anti-Tumour Medicaments, became effective as of July 13, 2011 and will expire on July 28, 2023.
On July 27, 2011, we announced completion of patient recruitment for the ongoing Phase 3 trial with perifosine in refractory advanced colorectal cancer, involving over 465 patients from 65 sites in the United States. This Phase 3 X-PECT (Xeloda® + Perifosine Evaluation in Colorectal Cancer Treatment) trial is a randomized (1:1), double-blind trial comparing the efficacy and safety of perifosine + capecitabine vs. placebo + capecitabine. The primary endpoint is overall survival, with secondary endpoints including overall response rate (complete + partial responses), progression-free survival and safety. Approximately 360 events of death will trigger the unblinding of the study.
On August 31, 2011, we announced that the DSMB for the Phase 3 X-PECT study of perifosine in patients with refractory advanced colorectal cancer had completed a pre-specified interim analysis for safety and futility. The DSMB recommended that the Phase 3 study continue to completion, as planned.
On October 5, 2011, we announced that a manuscript, entitledRandomized Placebo-Controlled Phase 2 Trial of Perifosine Plus Capecitabine as Second- or Third-Line Therapy in Patients with Metastatic Colorectal Cancer, had been published in the October 3, 2011 online edition of theJournal of Clinical Onclogy (“JCO”), in which Phase 2 activity of perifosine in the treatment of patients with refractory advanced CRC was reported. The publication highlighted the efficacy and safety data on the 38 CRC patients participating in this Phase 2, randomized, multicenter study, comparing perifosine plus capecitabine (P-CAP) to placebo plus capecitabine. Based on the data, in which the combination of P-CAP demonstrated statistical significance with respect to median overall survival and median time to tumor progression, the investigators concluded that the P-CAP combination showed promising clinical activity compared to single-agent capecitabine, and that the difference in clinical outcome seen with the addition of perifosine was impressive.
Efficacy data from this study had been previously presented in June 2010 at the 46th Annual Meeting of the American Society of Clinical Oncology.
On October 13, 2011, we announced that the manuscript, entitledPerifosine Plus Bortezomib and Dexamethasone in patients with Relapsed/Refractory Multiple Myeloma Previously Treated with Bortezomib: Results of Operationsa Multicenter Phase 1/2 Trial, had been published in the October 10, 2011 online edition of the JCO, in which Phase 1/2 combination activity of perifosine in the treatment of advanced multiple myeloma patients was reported. Results showed that the Overall Response Rate was 65% for bortezomib-relapsed patients and 32% for bortezomib-refractory patients. Median PFS was 6.4 months, with a median PFS of 8.8 months in the bortezomib-relapsed population. Therapy was generally well-tolerated, and toxicities, including gastrointestinal side-effects and fatigue, proved manageable. No treatment-related mortality was seen. The investigators concluded the data reported for both safety and efficacy in this patient population were encouraging for the continued study of perifosine. Data from this study had been previously presented at the 2009 ASH conference.
Quarterly Consolidated Results of Operations Information(On December 12, 2011, we reported encouraging preclinical data for perifosine in thousands, except for per share data)HL. In vitro
| | Quarters ended | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | December 31, 2010 | September 30, 2010 | June 30, 2010 | March 31, 2010 | ||||||||||
| | $ | $ | $ | $ | ||||||||||
Revenues | 9,971 | 5,726 | 5,584 | 6,422 | ||||||||||
Loss from operations | (4,211 | ) | (6,088 | ) | (7,589 | ) | (7,340 | ) | ||||||
Net loss | (2,741 | ) | (10,147 | ) | (4,450 | ) | (5,880 | ) | ||||||
Net loss per share | ||||||||||||||
Basic and diluted | (0.03 | ) | (0.12 | ) | (0.06 | ) | (0.09 | ) | ||||||
| | Quarters ended | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | December 31, 2009 | September 30, 2009 | June 30, 2009 | March 31, 2009 | ||||||||||
| | $ | $ | $ | $ | ||||||||||
Revenues | 40,182 | 8,565 | 8,379 | 6,111 | ||||||||||
Earnings (loss) from operations | 11,511 | (9,789 | ) | (12,238 | ) | (13,442 | ) | |||||||
Net earnings (loss) | 12,032 | (11,288 | ) | (13,080 | ) | (12,388 | ) | |||||||
Net earnings (loss) per share | ||||||||||||||
Basic and diluted | 0.19 | (0.19 | ) | (0.24 | ) | (0.23 | ) | |||||||
Net earnings (loss) per share are (is) based on each reporting period's weighted average number of shares outstanding, which may differ on a quarter-to-quarter basis. As such, the sum of the quarterly net earnings (loss) per share amounts may not equal year-to-date net loss per share.
Fourth Quarter 2010 Results
Revenues were $10.0 million for the quarter ended December 31, 2010, compared to $40.2 milliondata from HL cell lines showed that perifosine combined with sorafenib induced increased apoptosis, while in vivo data for the same quartercombination treatment for HL significantly increased survival in 2009. mice. Data were presented during the ASH Annual Meeting and Exposition, in San Diego, California.
The significant decreasestudy was conducted to investigate, in revenues is due primarily to our having recognized, vitro andin December 2009,vivo, the remaining unamortized portion, or approximately $30.4 million,activity and mechanism(s) of action of perifosine in combination with sorafenib by using three HL cell lines (HD-MyZ, L-540, HDLM-2). In thein vitro experiments perifosine/sorafenib treatment resulted in synergistic cell growth inhibition and cell death induction in HD-MyZ and L-540 cell lines, but not in the HDLM-2 cell line. Cell cycle arrest, down-modulating of the upfront payment received from sanofi-aventis U.S L.L.C. ("sanofi"),MAPK and PI3K/Akt pathways as discussed below, partly offset by the increase in 2010 royalties attributable to the contingent payment of $2.5 million due from Cowen, alsowell as discussed below. Additionally, the decrease is attributable to the recognition of remaining deferred revenues, amounting to approximately $1.8 million,caspase-independent cell death was observed, which was associated with agreements related to the usesevere mitochondrial dysfunction. Further, expression of ozarelix, an intangible asset thatgenes involved in amino acid metabolism, cell cycle, DNA replication and cell death was deemedshown to be fully impairedmodulated. In addition, overexpression of the tribbles homologue 3 [TRIB3] was observed.
In vivo, perifosine/sorafenib treatment significantly increased survival in December 2009.
Net research and development ("R&D") expenses were $5.1 million for the quarter ended December 31, 2010,HD-MyZ model (45 vs. 81 days, as compared to $10.6 million for the same quarter in 2009. The comparative decrease in R&D expenses primarily results from the progressive completion throughcontrols), with 25% tumor-free mice at the end of 2009the 200-day observation period. In the L-540 model, subcutaneous tumor volume was also reduced as compared to controls (by 42%), perifosine (by 35%) or sorafenib (by 46%) alone. In HD-MyZ and L-540 but not HDLM-2, the combined treatment induced an increase in tumor necrosis (2- to 8-fold, P £.0001) and in tumor apoptosis (2- to 2.5-fold, P £.0001). In 2 of 3 HL cell lines, perifosine/sorafenib combination treatment induced potent antitumor effects. Striking increase of mitochondrial injury and apoptosis, and marked reduction of cell viability was observed in thein vitroexperiments.In vivocombination treatment increased survival and inhibited tumor growth.
On December 13, 2011, we reported encouraging clinical data for an ongoing Phase 2 clinical study in patients with refractory/relapsed HL. Preliminary response data showed that perifosine combined with sorafenib significantly increased median PFS in refractory/relapsed HL patients with high phosphorylation levels of Erk and Akt, as compared to patients with low baseline phosphorylation levels of Erk and Akt. Data were presented during the ASH Annual Meeting and Exposition, in San Diego, California.
The study evaluated phosphorylation levels of Erk (pErk) and Akt (pAkt) in circulating lymphocytes from patients enrolled in two consecutive Phase 2 trials evaluating activity and safety of sorafenib as a single agent or in combination with perifosine in relapsed/refractory HL patients. Four patients were treated with sorafenib alone at a dose level of 400mg BID and twenty-one patients received a 4-week treatment with perifosine alone at a dose level of 50mg BID, followed by a perifosine/sorafenib combination therapy with 50mg BID and 400mg BID, respectively. Circulating lymphocytes were evaluated for their phosphorylation levels of Erk and Akt, in order to assess predictive value of the phosphokinase levels for therapy responses.
Clinical response data showed that baseline pErk and pAkt levels were significantly higher in responsive patients, as compared to unresponsive patients. The pErk and pAkt levels measured after 60 days of therapy with perifosine combined with sorafenib were significantly reduced in responsive patients. The median baseline value of pErk and pAkt efficiently discriminated responsive and unresponsive patients which was associated with a significantly improved median PFS for patients with baseline pErk³43% and/or pAkt >23%. Based on these data, the correlation of baseline pErk and pAkt levels with objective responses and time to tumor progression will need to be validated in prospective studies.
On January 3, 2012, we announced that our Japanese partner, Yakult, had initiated a Phase 1/2 trial in Japan to assess the safety and efficacy of perifosine in combination with chemotherapeutic agent, capecitabine, in patients with refractory advanced CRC. The primary endpoint of the Phase 1 portion of the trial is the safety profile of perifosine in combination with capecitabine. The primary endpoint of the Phase 2 portion is efficacy (Disease Control Rate). This trial is sponsored by Yakult and its initiation on December 27, 2011 triggered a milestone payment which is payable in the first quarter of 2012 of approximately €2 million from Yakult to Aeterna Zentaris under the agreement signed in March 2011 for perifosine in Japan described above.
AEZS-108
On September 14, 2011, we presented positive final Phase 2 efficacy and safety studiesdata for AEZS-108 (zoptarelin doxorubicin) in advanced endometrial cancer at the European Society of Gynecological Oncology in Milan, Italy. Data showed that AEZS-108, administered as a single agent at a dosage of 267 mg/m2 every 3 weeks was active, well tolerated and that overall survival is similar to that reported for modern triple combination chemotherapy, but was achieved with lower toxicity. The primary endpoint was the response rate as defined by the Response Evaluation Criteria in Solid Tumors (“RECIST”). Secondary endpoints included safety, time-to-progression (“TTP”) and overall survival (“OS”).
In all, of 43 patients treated with AEZS-108, 39 were evaluable for efficacy. Efficacy confirmed by independent response review included 2 complete responses (“CR”), 10 partial responses (“PR”), and 17 patients with disease stabilization (“SD”). Based on those data, the estimated Overall Response Rate (“ORR” = CR+PR) was 30.8% and the Clinical Benefit Rate (“CBR”) (CBR = CR+PR+SD) was 74.4%. Responses in patients previously treated with chemotherapy included 1 CR, 1 PR and 2 SDs in 8 of the patients with prior use of platinum/taxane regimens. Median TTP and OS were 7 months and 13.7 months, respectively.
Overall, tolerability of AEZS-108 was good and commonly allowed retreatment as scheduled. Severe (Grade 3 or 4) toxicity was mainly restricted to rapidly reversible leukopenia and neutropenia, associated with fever in only 1 patient who had been treated only 3 weeks after a surgery. Good tolerability of AEZS-108 was also reflected by a low rate of severe non hematological possibly drug-related adverse events which included single cases each of nausea, diarrhea, fatigue, general health deterioration, creatinine elevation, and blood potassium decrease. No cardiac toxicity was reported.
On September 26, 2011, we announced positive interim data for the Phase 1 portion of our Phase 31/2 trial with AEZS-108 in castration- and taxane-resistant prostate cancer at the ESMO meeting in Stockholm, Sweden. This is a single arm study with a Phase 1 lead-in to a Phase 2 clinical trial. The primary endpoint of the Phase 1 portion is safety. The primary objective of the Phase 2 portion is to evaluate the clinical benefit of AEZS-108 for these patients. Data showed that AEZS-108 was well tolerated at all dose levels and early evidence of antitumor activity was observed even at low dose level. The Phase 2 extension is planned after completion of the toxicity assessment in the final dose level of the Phase 1 portion of the study. Furthermore, data from correlative studies demonstrated for the first time the internalization of AEZS-108 in circulating tumor cells of patients.
On October 25, 2011, we announced that the FDA had completed the review of the IND application filed by Alberto J. Montero, M.D., Assistant Professor, Department of Medicine, Division of Hematology/Oncology, Sylvester Comprehensive Cancer Center at the University of Miami Miller School of Medicine and concluded that Dr. Montero may proceed with the initiation of a randomized Phase 2 trial in chemotherapy refractory triple-negative (ER/PR/HER2-negative) LHRH receptor-positive metastatic breast cancer with AEZS-108. This will be an open-label, randomized, two-arm, multicenter Phase 2 study involving up to 74 patients. The primary study endpoint will be PFS. Secondary endpoints will also include overall response rate, and overall survival. The study will also evaluate AEZS-108’s toxicity profile and patients’ quality of life relative to conventional cytotoxic chemotherapy used in the control arm of this study.
On January 5, 2012, we announced that we had entered into a collaboration agreement with Ventana Medical Systems, Inc., a member of the Roche Group, to develop a companion diagnostic for the immunohistochemical determination of LHRH-receptor expression, for AEZS-108. In humans, LHRH receptors are expressed in a significant proportion of endometrial, ovarian, breast, bladder, prostate and pancreatic tumors. AEZS-108 specifically targets LHRH receptors and could, therefore, prove to be more efficient in treating patients with these types of LHRH-receptor positive cancers.
AEZS-120
On July 20, 2011, we announced that we had successfully reached a key milestone in the non-clinical development of our live recombinant prostate cancer vaccine candidate, AEZS-120, for oral administration. The program encompassed the full development of a GMP process, including GMP production and quality testing of a clinical batch, as well as a non-clinical safety and toxicology package, as previously agreed with the regulatory authorities. Subject to a positive review by the regulatory authorities, we aim to start Phase 1 clinical development in 2012. AEZS-120 has been developed through a research collaboration with the Department of Medical Radiation Biology and Cell Research, and the Department of Microbiology of the University of Würzburg, Germany. The collaboration was funded with a total of $890,000 for cetrorelixthe Company and $870,000 for the university partner by the German Ministry of Education and Research (BMBF) for a period of three years. As part of the collaboration, a melanoma vaccine based on the recombinant expression of a modified B-Raf protein has been generated.
AEZS-129
On March 22, 2011, we presented preclinical results for AEZS-129 at the Informa Life Sciences Protein Kinases Congress in benign prostatic hyperplasia ("BPH")Berlin, Germany. AEZS-129 was identified as a potent inhibitor of class I PI3Ks lacking activity against mTOR. Lack of mTOR activity is considered to potentially lead to a better safety profile. In biochemical and cellular assays, AEZS-129 demonstrated favorable properties in earlyin vitro ADMET screening, including microsomal stability, plasma stability and screening against a safety profile composed of receptors, enzymes and cardiac ion-channels.In vitro, the compound was shown to be a selective ATP-competitive inhibitor of PI3K with a broad antiproliferative activity against a broad panel of tumor cell lines.In vivo, AEZS-129 showed excellent plasma exposure and significant tumor growth inhibition in several tumor xenografts models, including A-549 (lung), HCT-116 (colon) and Hec1B (endometrium). The decreaseThese data suggest that AEZS-129 is also explained by a comparatively lower overall volumepromising compound for clinical intervention of R&D expenses, most notably given the fact that most costs related to our ongoingPI3K/Akt pathway in human tumors.
AEZS-130
On July 26, 2011, we announced the completion of the Phase 3 programstudy on AEZS-130 as a first oral diagnostic test for AGHD and the decision to meet with perifosine are borne by our North American partner, Keryx.
Selling, general and administrative ("SG&A") expenses were $3.1 millionthe FDA for the quarter ended December 31, 2010, compared to $6.2 millionfuture filing of an NDA for the same quarterregistration of AEZS-130 in 2009. The decrease in SG&A expenses is predominantly related to the expensing, in December 2009,United States.
On August 30, 2011, we announced favorable top-line results of the remaining unamortized portion, or approximately $3.0 million, of the royalty paid to the Tulane Educational Fund ("Tulane") in connection with the agreement entered into with, and subsequently terminated by, sanofi, as discussed below.
Depreciation and amortization expenses for the quarter ended December 31, 2010 amounted to $0.6 million, compared to $8.1 million for the same quarter in 2009. The comparative decrease is attributable to the fact that, in December 2009, and following our announcements that our secondcompleted Phase 3 study with cetrorelix in BPH had notAEZS-130 as the first oral diagnostic test for AGHD. The results showed that AEZS-130 reached its primary endpoint demonstrating >90% AUC of the ROC curve, which determines the level of specificity and that sanofi had decidedsensitivity of the product.
The first part of the study, conducted by our former partner, Ardana, was a two-way crossover study involving 42 patients with confirmed AGHD or multiple pituitary hormone deficiencies and a low insulin-like growth factor-I. A control group of 10 subjects without AGHD were matched to terminatepatients for age, gender, body mass index and (for females) estrogen status. Each patient received two dosing regimens in random order, while fasting, at least 1 week apart. One regimen consisted of a 1 µg/kg (max. 100 µg) dose of GHRH (Geref Diagnostic®, Serono) with 30 g of ARG (Ar-Gine®, Pfizer) administered intravenously over 30 minutes; the related development, commercialization and license agreement (discussed below), we recognizedother regimen was a dose of 0.5 mg/kg body weight of AEZS-130 given in an impairment charge equivalentoral solution of 0.5 mg/ml. As a result of the SPA reached with the FDA in order to complete the trial, the second part of the study contained the following revisions/additions to the remaining carrying valuefirst protocol:
| § | An additional 30 normal control subjects were to be enrolled to match the AGHD patients from the original cohort; |
| § | Further, an additional 20 subjects were to be enrolled: 10 AGHD patients and 10 matched normal control subjects; |
| § | The above brought the database to ~100 subjects; |
| § | All subjects received a dose of 0.5 mg/kg body weight of AEZS-130; |
| § | As a secondary endpoint, the protocol required that at least 8 of the 10 newly enrolled AGHD patients be correctly classified by a pre-specified peak GH threshold level. |
The parameters of cetrorelix,the study were achieved as agreed to with the FDA under our SPA. Importantly, the primary efficacy parameters showed that the study achieved both specificity and sensitivity at a level of 90% or greater. In addition, 8 of the 10 newly enrolled AGHD patients were correctly classified by a pre-specified peak GH threshold level. The use of AEZS-130 was shown to be safe and well tolerated overall throughout the completion of this trial.
The Company is planning for the filing of an NDA for the registration of AEZS-130 in the United States as a diagnostic test for AGHD.
On November 28, 2011, we announced that the FDA had completed the review of an IND application filed by Jose M. Garcia, M.D., Ph.D., Assistant Professor, Division of Diabetes Endocrinology and Metabolism, Departments of Medicine and Molecular and Cell Biology, Baylor College of Medicine and the Michael E. DeBakey Veterans Affairs Medical Center, in Houston Texas and concluded that Dr.Garcia may proceed with the initiation of a Phase 2A trial to assess the safety and efficacy of repeated oral administration of AEZS-130 at different doses daily for 1 week in view of developing a treatment for cancer-cachexia. Cachexia, which is characterized by diminished appetite and food intake in cancer patients, is defined as an involuntary weight loss of at least 5% of the pre-illness body weight over the previous six months.
On March 8, 2012, we announced that the Michael E. DeBakey Veterans Affairs Medical Center, in Houston, Texas, initiated a Phase 2A trial assessing the safety and efficacy of repeated doses of AEZS-130 in patients with cancer-cachexia. The study is conducted under a CRADA between the Michael E. DeBakey Veterans Affairs Medical Center, which is funding the study, and us. This is a double-blind, randomized, placebo-controlled Phase 2A trial to test the effects of different doses of the ghrelin agonist, AEZS-130, in 18 to 26 patients with cancer-cachexia. AEZS-130 will be provided by us. The study will involve 3 sequential groups receiving differing doses of AEZS-130. Each dose group will have 6 patients who will receive AEZS-130 and 2-4 patients who will receive a placebo. After analysis of safety and efficacy at each dose level vs. placebo, a decision will be taken either to decrease or increase the dose. If there are major safety issues, the study will be stopped. For this study, adequate efficacy will be defined as a³0.8 kg of body weight gain or a³50 ng/mL increase in plasma IGF-1 levels. The primary objective of the study is to evaluate the safety and efficacy of repeated oral administration of AEZS-130 at different doses daily for 1 week in view of developing a treatment for cachexia. The following parameters will be recorded to assess efficacy during the study: change of body weight, change of IGF-1 plasma levels, and change of quality of life score (Anderson Symptom Assessment Scale, FACIT-F). Other secondary objectives will include food intake, and changes in the following: appetite, muscle strength, energy expenditure, reward from food and functional brain connectivity.
AEZS-131
On March 22, 2011, we presented preclinical results for AEZS-131 at the Informa Life Sciences Protein Kinases Congress in Berlin, Germany. AEZS-131 was established as a small molecular compound that inhibits Erk in the low nanomolar range and shows an excellent selectivity profile. Further characterization experiments revealed an ATP-competitive mode of action and the potent inhibition of the cellular downstream target Rsk1 in tumor cells. The frontrunner, AEZS-131, produces cell cycle arrest in G1 and results in growth inhibition of cancer cells. Furthermore, the potential of combination therapy of AEZS-131 with inhibitors of the PI3K pathway was
addressed and the analysis of combination effects on tumor cell proliferation has been presented. These results support the evaluation of selective Erk inhibitors as antiproliferative agents either as monotherapy or in combination with inhibitors of the PI3K/Akt pathway.
On April 5, 2011, we announced that a poster on our highly selective Erk 1/2 inhibitor anticancer compound, AEZS-131, had been presented at the 102nd annual meeting of the AACR. Results showed that AEZS-131 selectively inhibits Erk 1/2 with an IC50 of 4nM, blocks cellular Rsk-1 phosphorylation, modulates downstream cellular substrate activation, arrests tumor cells in G1 and inhibits the growth of multiple human tumor cell lines in the nanomolar range. In in vivo pharmacokinetic studies, AEZS-131 showed a favorable PK profile. Antitumor activity was studied in in vivo mouse xenograft experiments utilizing the HCT 116 colon cancer model. AEZS-131 significantly inhibited tumor growth and was well tolerated at daily doses up to 120 mg/kg. Focus on inhibition of downstream kinase Erk 1/2 activity as a therapeutic target may be attractive because the pharmacologic inhibition of Erk 1/2 reverses Ras and Raf activation in cells which also demonstrate resistance to common Raf inhibitors, such as PLX-4720/4032.
On August 30, 2011, we announced that a poster on our novel orally active anticancer Erk inhibitor, which includes AEZS-131, was presented at the American Chemical Society National Meeting in Denver, Colorado. Thein vitroantiproliferative efficacy proved to be excellent in diverse human tumor cell lines. GI50 values in the low nanomolar range were obtained.In vivoantitumor activity was studied in a mouse xenograft experiment utilizing the human HCT-116 colon cancer model. Up to 74% inhibition of tumor growth was achieved with daily oral doses of 30 -120 mg/kg. Our medicinal chemistry programs are supported by X-ray crystallography and modeling towards the optimization of pyrido[2,3-b]pyrazines as novel series of kinase inhibitors.
On December 9, 2011, we announced positive preclinical data in TNBC for AEZS-131 at the 34th Annual San Antonio Breast Cancer Symposium. AEZS-131 was tested to check for selectivity, inhibition of Rsk-phosphorylation (cellular substrate of Erk), mode of action and cleavage of PARP. Cytotoxic efficacy was evaluated in a selection of TNBC cell lines, with or without mutations in the MAPK signal transduction pathway, by MTT assay. The study showed that AEZS-131 selectively inhibited ERK with an IC50<4nM. Phosphorylation of Rsk-1 was inhibited with an IC50 of 158 nM. AEZS-131 induced cell cycle arrest in G1 dose-dependently and cleavage of PARP. EC50 values were below 1µM for cell lines with mutations in the MAPK pathway. TNBC cell lines without mutations in the MAPK pathway were less responsive.
AEZS-132
On March 22, 2011, we presented preclinical results for AEZS-132 at the Informa Life Sciences Protein Kinases Congress in Berlin, Germany. AEZS-132 is a unique dual inhibitor of PI3K and Erk in the nanomolar range and exerts high selectivity against other serine threonine and tyrosine kinases. AEZS-132 is also an ATP-competitive inhibitor, with a broad antiproliferative profilein vitro, a favorable safety profile and beneficial ADME properties.In vivo pharmacokinetic experiments showed plasma profiles expected to result in positive antitumor efficacy, and led to significant antitumor activity in mouse xenograft models, including HCT-116 (colon), A-549 (lung), and Hec 1B (endometrium). Cellular inhibition of the downstream targets p-Akt and p-Rsk was confirmed within thein vivo tumor studies. In summary, AEZS-132 is a unique dual kinase inhibitor targeting the PI3K and MAPK pathways, expected to be especially suited to treat tumors with over-activation of both pathways.
AEZS-137
On March 24, 2011, we announced that we had been awarded a $1.5 million grant from the German Ministry of Education and Research to develop, up to the clinical stage, cytotoxic conjugates of the proprietary cytotoxic compound AEZS-137 (disorazol Z) and peptides targeting G-protein coupled receptors, including the LHRH receptors. The compounds being developed will combine the targeting principle successfully employed in Phase 2 with AEZS-108 (doxorubicin and LHRH receptor targeting agent) with the novel cytotoxic disorazol Z. Furthermore, diagnostic tools systematically assessing the receptor expression in tumor specimens will be developed to allow the future selection of patients and tumor types with the highest chance of benefitting from this personalized medicine approach. The grant will be payable as a partial reimbursement of qualifying
expenditures over a three-year period. The qualified project will be performed with Morphisto GmbH and the Helmholtz Institute in Saarbrücken, Germany, which will receive additional funding of approximately $3.9 million, as$0.7 million. Researchers from the department of Gynecology and Obstetrics at both the University of Göttingen and Würzburg, Germany, will also be part of amortization expense. Further,the collaboration.
On November 16, 2011, we announced encouraging preclinical data for AEZS-137 from a poster presentation at the AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics, in January 2010, Spectrum Pharmaceuticals Inc., to whom we have granted an exclusive license to developSan Francisco. The poster showed that AEZS-137 possesses outstanding cytotoxicity in a highly diverse panel of 60 different tumor cell lines, and commercialize ozarelix for all potential indications in all worldwide territories, excluding certain Asian markets, announcedalso underlined the identification of important aspects of this novel natural compound’s mechanism of action. AEZS-137 has been identified as a tubulin binding agent with highly potent antitumor properties. Cell cycle analysis revealed that it had terminated its development program with ozarelix in BPH. Consequently, we recognized an impairment charge of approximately $1.4 million as part of amortization expenseAEZS-137 arrested cells in the fourth quarterG2/M phase and subsequently induced apoptosis with remarkable potency, as shown by subnanomolar EC50 values.
Corporate Developments
At-the-Market Sales Agreement
On February 22, 2011, we entered into an ATM sales agreement with MLV, under which we were able to sell up to 12.5 million of 2009.our common shares through ATM issuances on NASDAQ for aggregate gross proceeds not to exceed $19.8 million. The ATM sales agreement provided that common shares were to be sold at market prices prevailing at the time of sale, and, as a result, prices may have varied. From February 20, 2011 and up to April 29, 2011, we issued a total of 10.0 million of our common shares for aggregate gross proceeds of $19.8 million, less cash transaction costs of $0.6 million and previously deferred transaction costs of $0.2 million.
Net (loss) earnings amountedOn June 29, 2011, we entered into a second ATM, the June ATM Sales Agreement with MLV, under which we could, at our discretion, from time to ($2.7 million), or ($0.03) per basictime during the 24-month term of the agreement, sell up to 9.5 million of our common shares through ATM issuances on NASDAQ for aggregate gross proceeds not to exceed $24.0 million. The June ATM Sales Agreement, similar to the February ATM Sales Agreement, provided that common shares were to be sold at market prices prevailing at the time of sale, and, diluted share,as a result, prices may have varied. From June 29, 2011 and up to December 7, 2011, under the June ATM agreement, we issued a total of 9.5 million of our common shares for aggregate gross proceeds of $17.7 million, less cash transactions costs of $0.6 million and previously deferred transaction costs of $0.2 million.
Gross proceeds raised under both ATM sales agreements totalled $37.5 million for the quarteryear ended December 31, 2011.
Cowen Royalty Sales Milestone Payment
On February 28, 2011, we announced that we had received a net sales milestone of $2.5 million from Cowen Healthcare Royalty Partners, L.P. (“Cowen”). This milestone was payable pursuant to the previously announced sale to Cowen of Aeterna Zentaris’ rights to royalties on future net sales of Cetrotide®, covered by the Company’s license agreement with Merck Serono, and was contingent on 2010 comparednet sales of Cetrotide® reaching a specified level.
Subsequent to $12.0Year-End
At-The-Market Issuance Program
On January 23, 2012, the Company, pursuant to its existing ATM sales agreement dated June 29, 2011, with MLV, was commencing a new ATM issuance program (“January 2012 ATM Sales Agreement”) under which it may, at its discretion, from time to time during the term of the sales agreement, sell up to a maximum of 10.4 million or $0.19 per basicof its common shares through ATM issuances on NASDAQ up to an aggregate amount of $16.0 million.
From January 23, 2012 through March 15, 2012, the Company issued a total of 3.6 million common shares under the January 2012 ATM Sales Agreement for aggregate gross proceeds of $6.4 million, less cash transaction costs of $0.2 million and diluted share,previously deferred transaction costs of $56,000.
AEZS-108
On February 3, 2012, we reported positive updated results for the same quarterPhase 1 portion of our ongoing Phase 1/2 study in 2009.CRPC with AEZS-108. This is a single-arm study with a Phase 1 lead-in portion (testing 3 dose levels) to a Phase 2 clinical trial. The significant quarter-over-quarterprimary endpoint of the Phase 1 portion is safety. The primary objective of the Phase 2 portion is to evaluate the clinical benefit of AEZS-108 for these patients. Data were presented by Jacek Pinski, M.D., Ph.D., Associate Professor of Medicine at the Norris Comprehensive Cancer Center of the University of Southern California, during a poster session at the American Society of Clinical Oncology Genitourinary Cancers Symposium in San Francisco. The trial is being supported by a three-year $1.6 million grant from the National Institutes of Health to Dr. Pinski.
The results were based on 13 patients who have been treated on 3 dose levels: 3 at 160 mg/m2, 3 at 210 mg/m2, and 7 at 267 mg/m2. Overall, AEZS-108 has been well tolerated among this group of heavily pre-treated older patients. To date, there have been 2 dose limiting toxicities; both were cases of asymptomatic Grade 4 neutropenia at the 267 mg/m2 dose level and both patients fully recovered. The Grade 3 and 4 toxicities were primarily hematologic. There has been minimal non-hematologic toxicity, most frequently fatigue and alopecia.
Despite the low doses of AEZS-108 in the first cohorts, there was some evidence of antitumor activity. One patient received 8 cycles (at 210 mg/m2) due to continued benefit. Among the 5 evaluable patients with measurable disease, 4 achieved stable disease. At the time of submission of the abstract, a decrease in net earningsPSA was noted in 6 patients. Six of 13 (46%) treated patients have received at least 5 cycles of therapy with no evidence of disease progression at 12 weeks. Correlative studies on circulating tumor cells (“CTC”) have demonstrated the uptake of AEZS-108 into the targeted tumor. After completion of 3 additional patients at the 210 mg/m2 dose level, the study is largely attributableexpected to be extended into the significant decrease in license fee revenues, partly offset by lower comparative R&D expenses and by decreased SG&A expenses and depreciation and amortization charges, as discussed above.Phase 2 portion.
Largely given the presence, in our fourth quarter 2010 revenues, of the $2.5 million in additional, non-recurring royalty consideration payable by Cowen (discussed below), and excluding any impact of
foreign exchange gains or losses, we expect that the net loss for the first quarter of 2011 will increase, as compared to the fourth quarter of 2010.
Consolidated Statements of OperationsComprehensive Loss Information(in thousands, except for share and per share data)
| | | Years ended December 31, | Years ended December 31, | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (in thousands, except share and per share data) | 2011 | 2010 | 2009* | ||||||||||||||||||||
| | | 2010 | 2009 | 2008 | $ | $ | $ | ||||||||||||||||
| | | $ | $ | $ | |||||||||||||||||||
Revenues | Revenues | ||||||||||||||||||||||
Sales and royalties | Sales and royalties | 24,857 | 20,957 | 29,462 | 31,306 | 24,857 | 20,957 | ||||||||||||||||
License fees and other | License fees and other | 2,846 | 42,280 | 9,016 | 4,747 | 2,846 | 42,280 | ||||||||||||||||
| 36,053 | 27,703 | 63,237 | |||||||||||||||||||||
| 27,703 | 63,237 | 38,478 | |||||||||||||||||||||
Operating expenses | Operating expenses | ||||||||||||||||||||||
Cost of sales, excluding depreciation and amortization | 18,700 | 16,501 | 19,278 | ||||||||||||||||||||
Research and development costs, net | 19,859 | 43,814 | 57,105 | ||||||||||||||||||||
Cost of sales | 27,560 | 18,700 | 16,501 | ||||||||||||||||||||
Research and development costs, net of refundable tax credits and grants | 24,517 | 21,257 | 43,814 | ||||||||||||||||||||
Selling, general and administrative expenses | Selling, general and administrative expenses | 11,875 | 16,040 | 17,325 | 16,170 | 12,552 | 16,040 | ||||||||||||||||
Depreciation and amortization | |||||||||||||||||||||||
Property, plant and equipment | 1,005 | 3,285 | 1,515 | ||||||||||||||||||||
Intangible assets | 1,492 | 7,555 | 5,639 | ||||||||||||||||||||
Depreciation and amortization* | * | * | 10,840 | ||||||||||||||||||||
| 52,931 | 87,195 | 100,862 | 68,247 | 52,509 | 87,195 | ||||||||||||||||||
Loss from operations | Loss from operations | (25,228 | ) | (23,958 | ) | (62,384 | ) | (32,194) | (24,806) | (23,958) | |||||||||||||
Other income (expenses) | |||||||||||||||||||||||
Unrealized gain on held-for-trading financial instrument | 687 | — | — | ||||||||||||||||||||
Interest income | 207 | 349 | 868 | ||||||||||||||||||||
Interest expense | (26 | ) | (5 | ) | (118 | ) | |||||||||||||||||
Foreign exchange gain (loss) | 1,170 | (1,110 | ) | 3,071 | |||||||||||||||||||
Other | (28 | ) | — | (79 | ) | ||||||||||||||||||
| 2,010 | (766 | ) | 3,742 | ||||||||||||||||||||
Finance income | 6,239 | 1,800 | 349 | ||||||||||||||||||||
Finance costs | (8) | (5,445) | (1,115) | ||||||||||||||||||||
Net finance income (costs) | 6,231 | (3,645) | (766) | ||||||||||||||||||||
Loss before income taxes | Loss before income taxes | (23,218 | ) | (24,724 | ) | (58,642 | ) | (25,963) | (28,451) | (24,724) | |||||||||||||
Income tax expense | Income tax expense | — | — | (1,175 | ) | (1,104) | - | - | |||||||||||||||
Net loss | Net loss | (23,218 | ) | (24,724 | ) | (59,817 | ) | (27,067) | (28,451) | (24,724) | |||||||||||||
Other comprehensive (loss) income: | |||||||||||||||||||||||
Foreign currency translation adjustments | (789) | 1,001 | (1,335) | ||||||||||||||||||||
Actuarial gain (loss) on defined benefit plans | (1,335) | 191 | - | ||||||||||||||||||||
Comprehensive loss | (29,191) | (27,259) | (26,059) | ||||||||||||||||||||
Net loss per share | Net loss per share | ||||||||||||||||||||||
Basic and diluted | (0.31 | ) | (0.43 | ) | (1.12 | ) | |||||||||||||||||
Weighted average number of shares | |||||||||||||||||||||||
Basic and diluted | 75,659,410 | 56,864,484 | 53,187,470 | ||||||||||||||||||||
Basic and diluted | (0.29) | (0.38) | (0.43) | ||||||||||||||||||||
Weighted average number of shares outstanding | |||||||||||||||||||||||
Basic and diluted | 94,507,988 | 75,659,410 | 56,864,484 | ||||||||||||||||||||
| * | We adopted IFRS in 2011 with a transition date of January 1, 2010. The selected financial information for the years ended December 31, 2009, 2008 and 2007 is derived from financial statements that were presented in accordance with Canadian GAAP and has not been restated in accordance with IFRS. Consequently, the selected financial information for the years ended December 31, 2009, 2008 and 2007 may not be comparable with the corresponding selected financial information for the years ended December 31, 2011 and 2010. Please refer to “Critical Accounting Policies, Estimates and Judgments” for the policy differences between Canadian GAAP and IFRS. |
Revenues
Revenues are derived primarily from sales and royalties as well as from license fees. Sales are derived from the manufacturing of Cetrotide®Cetrotide® (cetrorelix acetate solution for injection), marketed globally (ex-Japan) by ARES Trading S.A. (“Merck Serono”) for reproductive health assistance forin vitro fertilization, as well as from active pharmaceutical ingredients.
Royalties are derived from Cetrotide® and, prior to the fourth quarter of 2008, were payable by our partner, ARES Trading S.A. ("Merck Serono"). Beginning on October 1, 2008, royalty revenues derivedindirectly from Merck Serono'sSerono’s net sales of Cetrotide® are recognized viaCetrotide® and represent the periodic amortization, under the units-of-revenue method, of the proceeds received in connection with the 2008 sale in December 2008to Cowen Healthcare Royalty Partners L.P. of the underlying future royalty stream to Cowen. Per the terms of the related purchase and sale agreement, we received net proceeds of $51.5 million from Cowen and were entitled to receive an additional payment of $2.5 million contingent on 2010 net sales of Cetrotide® reaching a specified level. This additional consideration was earned in 2010 and received in February 2011, and the corresponding amount has been recorded as royalty revenues in our consolidated statement of operations.stream.
License fees are derived from non-periodicinclude periodic milestone payments, research and development (“R&D&D”) contract fees and the amortization of upfront payments and amortization thereof, received from our licensing partners. Significant license fee revenues have resulted from our agreement with sanofi, related to the development, commercialization and licensing of cetrorelix in BPH, entered into in March 2009 and terminated in January 2010 following our announcement that our second Phase 3 study with the compound had not reached its primary endpoint.
Sales and royalties increasedwere $31.3 million for the year ended December 31, 2011, compared to $24.9 million for the year ended December 31, 2010, compared2010. This increase is largely related to $21.0 million and $29.5 millioncomparatively higher deliveries of Cetrotide® to Merck Serono.
Despite our original expectations for each ofonly slight sales volume increases for the years ended December 31, 2009 and 2008, respectively. In addition to the recognition, in 2010, of the additional royalty consideration payable by Cowen (discussed above), 2010year 2011, our sales and royalties were positively impacted bysubstantially higher than our original expectations due to an unforeseen increase in Cetrotide® sales to Merck Serono.
Sales and royalties in non-Japanese markets,2012 are expected to stabilize, as compared to 2009.the amounts recorded in 2011, since we expect that the volume of Cetrotide® sales will stabilize in 2012.
The decrease from 2008 to 2009 is mainly related to lower royaltyLicense fees and other revenues having been recognized in 2009 in connection with our agreement with Merck Serono. Amortization of the proceeds received from Cowenwere $4.7 million for the year ended December 31, 2009 was lower than the royalty revenues generated and payable directly by Merck Serono during 2008. Additionally, sales volumes of Cetrotide® were slightly lower during the year ended December 31, 2009, as2011, compared to 2008.
Excluding the impact of foreign exchange rate fluctuations, sales and royalties are expected to decrease in 2011 to between approximately $19.0 million and $21.0 million, largely given the future absence of the non-recurring contingent consideration payable by Cowen, as discussed above.
License fee and other revenues totalled $2.8 million for the year ended December 31, 2010,2010. This increase is mainly due to the recording of a milestone payment from Yakult with respect to the initiation of a Phase 1/2 trial with perifosine in CRC in Japan during the last quarter of the year 2011.
During 2012, we expect to provide more R&D services, compared to $42.3 million and $9.0 million2011, related to perifosine to our license partner for eachNorth America, Keryx. We expect the results of the years ended December 31, 2009 and 2008, respectively. The significant decrease from 2009 to 2010, as well asongoing Phase 3 trial of perifosine in CRC in the significant increase from 2008 to 2009, is almost exclusively attributable tosecond quarter of 2012. If the upfront payment received and recognized in 2009 from sanofi, as well asdata from the full recognition oftrial is positive, we expect to receive development and regulatory milestone payments from our various license partners. With these additional revenues, we expect that our license fees and other previously deferred revenues associated with ozarelix, another BPH-related compound that was deemed impairedwill increase significantly in December 2009.
License fee revenues in 2011 are expected2012, as compared to be largely similar to the amounts recorded during 2010, excluding any revenue associated with the licensing agreement entered into with Yakult, discussed above.2011.
Operating Expenses
Cost of sales increased to $18.7was $27.6 million (75% of sales and royalties) for the year ended December 31, 2010 from $16.52011, compared to $18.7 million (79% of sales and royalties) and $19.3 million (65% of sales and royalties) for each of the yearsyear ended December 31, 2009 and 2008, respectively. Excluding2010. This increase is largely attributable to the impact
Tablecomparative increase in volume of Contents
sales of the additional contingent royalty consideration earned and payable by Cowen (discussed above),Cetrotide® to Merck Serono, as discussed above. Additionally, cost of sales as a percentage of sales and royalties increased to approximately 88.03% for the year ended December 31, 2010 was approximately 84%, representing a consistently comparable increase from 2009. Changes in margin2011, compared to 75.23% for the year ended December 31, 2010. Our lower margins are dependent both upon our sales mix (including dosing level) and upon geographic coverage, both of which tend to vary on a period-to-period basis.
The decrease from 2008 to 2009 is largely attributable to the absencea decrease of Impavido® sales during the first three months$3.4 million of 2009, compared to the same periodroyalties in 2008. We sold our rights related to the manufacture, production, distribution, marketing, sale and use of that compound in March, 2008. The increase in cost of sales as a percentage of sales and royalties from 2008 to 2009 is largely attributable to the comparative decrease in royalty revenues, as discussed above.
We expect cost of sales as a percentage of sales and royalties to remain within the range of 80% to 85% during 2011, as compared to 2010.
R&D costs, net of refundable tax credits and grants include: employee compensation and fringe benefits; third-party costs; building rental, service and maintenance; and other expenses, as shown in the first table below. Third-party R&D costs consist of, among other expenses, external studies and collaborative work performed by contract research organizations, laboratory supplies and services, active pharmaceutical ingredient (or raw material) costs and patent protection expenses. These third-party costs are tracked by product or project, as shown in the second table below. All other R&D costs, including Company employee compensation and benefits, are not charged to specific products or projects, since the number of clinical and preclinical product candidates or development projects tends to vary from period to period and since internal resources are utilized across multiple projects over any given period of time.
Net R&D costs, were $19.9$24.5 million for the year ended December 31, 2010,2011, compared to $43.8 million and $57.1$21.3 million for each of the yearsyear ended December 31, 2009 and 2008, respectively.2010. The decrease from 2009 to 2010comparative increase is primarilymainly attributable to the winding down and termination of development activities related to cetrorelixan increase in BPH subsequent to our announcements that our related Phase 3 studies had not reached their primary endpoints in 2009. The decrease is also explained by a comparatively lower overall volume of R&D expenses, most notably given the fact that mostthird-party costs related to our ongoing Phase 3 program with perifosine are borne by our North American partner, Keryx.
The decrease in R&D costs from 2008 to 2009 is largely attributable to a lower volume of expenses having been incurred in 2009 related toconnection with the continued advancement during the first nine months of 2009, followed by the winding down of our developmentperifosine, AEZS-130 and Erk/PI3K compounds (AEZS-129, AEZS-131, AEZS-132)-related activities linked to cetrorelix in BPH.
The following table summarizes our net R&D costs by nature of expense:
| | Years ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| | 2010 | 2009 | 2008 | |||||||
| | $ | $ | $ | |||||||
| | (in thousands) | |||||||||
Employee compensation and fringe benefits | 9,153 | 10,845 | 14,088 | |||||||
Third-party costs | 8,138 | 28,925 | 39,142 | |||||||
Facilities rent and maintenance | 1,773 | 1,891 | 1,883 | |||||||
Other costs | 1,482 | 2,556 | 2,335 | |||||||
R&D tax credits and grants | (687) | (403) | (343) | |||||||
| 19,859 | 43,814 | 57,105 | ||||||||
| Years ended December 31, | ||||||||
| (in thousands) | 2011 | 2010 | ||||||
| $ | $ | |||||||
Employee compensation and fringe benefits | 10,028 | 9,226 | ||||||
Third-party costs | 10,244 | 8,138 | ||||||
Facilities rent and maintenance | 1,835 | 1,773 | ||||||
Other costs* | 2,793 | 2,807 | ||||||
R&D refundable tax credits and grants | (383) | (687) | ||||||
| 24,517 | 21,257 | |||||||
| * | including depreciation and amortization charges. |
The following table summarizes primary third-party R&D costs, by product candidate, incurred by the Company during the years ended December 31, 2010, 20092011 and 2008.2010.
| | | | Years ended December 31, | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Product | Status | Indication | 2010 | 2009 | 2008 | ||||||||||||||||||
| | | | $ | % | $ | % | $ | % | |||||||||||||||
| | | | (in thousands, except percentages) | ||||||||||||||||||||
Cetrorelix | Phase 3* | BPH* | 2,046 | 25.1 | 23,812 | 82.3 | 27,246 | 69.6 | |||||||||||||||
AEZS-130 (Solorel®) | Phase 3 | Endocrinology (diagnosis of AGHD) | 865 | 10.6 | 592 | 2.0 | — | — | |||||||||||||||
Perifosine | Phases 2 and 3 | Oncology | 968 | 11.9 | 304 | 1.1 | 2,426 | 6.2 | |||||||||||||||
Ozarelix | Phase 2* | BPH* | — | — | 366 | 1.3 | 254 | 0.7 | |||||||||||||||
AEZS-108 | Phase 2 | Oncology | 2,089 | 25.7 | 409 | 1.4 | 1,300 | 3.3 | |||||||||||||||
AEZS-112 | Phase 1 | Oncology | 259 | 3.2 | 430 | 1.5 | 996 | 2.5 | |||||||||||||||
AEZS-129, AEZS-131, AEZS-132, Erk PI3K | Preclinical | Endocrinology and oncology | 923 | 11.4 | 1,151 | 4.0 | 1,991 | 5.1 | |||||||||||||||
AEZS-123 / Ghrelin receptor | Preclinical | Endocrinology and oncology | — | — | 530 | 1.8 | 1,224 | 3.1 | |||||||||||||||
AEZS-115 / LHRH antagonist | Preclinical | Endocrinology and oncology | — | — | 235 | 0.8 | 1,047 | 2.7 | |||||||||||||||
AEZS-120 / Vaccine | Preclinical | Oncology | 149 | 1.8 | 403 | 1.4 | 27 | — | |||||||||||||||
Other | Preclinical | Oncology and endocrinology | 839 | 10.3 | 693 | 2.4 | 2,631 | 6.8 | |||||||||||||||
| 8,138 | 100.0 | 28,925 | 100.0 | 39,142 | 100.0 | ||||||||||||||||||
| (in thousands, except percentages) | Years ended December 31, | |||||||||||||||||||
| Product | Status | Indication | 2011 | 2010 | ||||||||||||||||
| $ | % | $ | % | |||||||||||||||||
Perifosine | Phases 2 and 3 | Oncology | 3,726 | 36.4 | 968 | 11.9 | ||||||||||||||
AEZS-130 | Phase 3 | Endocrinology (diagnosis of AGHD) | 1,156 | 11.3 | 865 | 10.6 | ||||||||||||||
Cetrorelix | Phase 3* | BPH* | - | - | 2,046 | 25.1 | ||||||||||||||
AEZS-108 | Phase 2 | Oncology | 1,652 | 16.1 | 2,089 | 25.7 | ||||||||||||||
AEZS-112 | Phase 1 | Oncology | 538 | 5.3 | 259 | 3.2 | ||||||||||||||
AEZS-129, AEZS-131 and AEZS-132; Erk/PI3K | Preclinical | Oncology | 1,860 | 18.2 | 923 | 11.4 | ||||||||||||||
Other | Oncology and endocrinology | 1,312 | 12.7 | 988 | 12.1 | |||||||||||||||
| 10,244 | 100.0 | 8,138 | 100.0 | |||||||||||||||||
| * | Development activities terminated in the last quarter of 2009 and beginning of 2010. |
We expect that our totalnet R&D expensescosts for 2011 will2012 to increase, as compared to 2010, largely2011, as we continue to focus on the development of perifosine and AEZS-108. In particular, we plan to pursue the ongoing Phase 3 study with perifosine in multiple myeloma, as well as initiate, with AEZS-108, a pivotal trial in endometrial cancer and support the ongoing Phase 1/2 studies in connection with triple-negative breast cancer and other indications.
We expect, excluding the impact of unforeseen foreign exchange rate fluctuations, that net R&D costs will total between $30.0 million and $32.0 million for the twelve-month period ending December 31, 2012, due to the expected incurrence of higher third-party costs associated with the advancement ofadditional expenses discussed above. We note, however, that our Phase 3 trial with perifosine in the multiple myeloma indication,R&D estimates may be revised as we continue to advance our development activities and more specifically in connection with the clinical and validation initiatives related toas new information becomes available. We also expect that study. However, most of these expected third-party costsmany perifosine-related development activities will be reimbursedsponsored by our North American license partner, Keryx, and those reimbursementsJapanese partner, Yakult, and that we will be recorded as license feecontinue to seek government grants for our earlier-stage projects.
Selling, general and other revenues.
administrative (“SG&A&A”) expenses decreased to $11.9 were $16.2 million for the year ended December 31, 2010,2011, compared to $16.0 million and $17.3$12.6 million for each of the yearsyear ended December 31, 2009 and 2008, respectively. The decrease from 2009 to 2010 is related primarily to2010. SG&A expenses were higher during the absence, in 2010, of the royalty paid to Tulane, amounting to approximately $3.0 million, as noted above, to euro-to-US dollar exchange rate fluctuations, largelyyear 2011 mainly due to the comparativerecognition of impairment losses and due to the initiation of pre-launch and marketing efforts related to the potential commercialization of perifosine in Europe.
During the year 2011, we recognized an impairment loss ($1.1 million) following impairment testing that was performed on our Cetrotide® asset. The impairment loss was recognized predominantly to take into account management’s lower trend estimates related to the commercialization of Cetrotide®, due to changes in the competitive environment in the Japanese market.
In addition, during the year 2011, following the relocation of one of the Company’s offices, we recognized an impairment loss on property plant and equipment (leasehold improvements and furniture and fixtures ($0.3 million)) and an additional onerous lease provision ($0.2 million).
Furthermore, we initiated our pre-launch and marketing efforts related to the potential marketing by the Company of perifosine in Europe. During the year 2011, we incurred approximately $0.9 million in connection with the preparation of a launch plan, including market research, pricing expectations and forecast calculations.
We expect that our SG&A expenses will slightly decrease in 2012, as compared to 2011, as costs return to more normalized operating levels, despite our expected inclusion of certain investments in perifosine’s European pre-launch and marketing efforts.
Net finance income (costs), comprised predominantly of net foreign exchange gains (losses), the change in fair value of our warrant liability and the gain on our short-term investment, for the year ended December 31, 2011, totalled $6.2 million, compared to ($3.6 million) for the year ended December 31, 2010, as presented below.
| Years ended December 31, | ||||||||
| (in thousands) | 2011 | 2010 | ||||||
| $ | $ | |||||||
Finance income | ||||||||
Net gains due to changes in foreign currency exchange rates | 2,197 | 932 | ||||||
Change in fair value of warrant liability | 2,533 | - | ||||||
Interest income | 231 | 181 | ||||||
Gain on held-for-trading financial instrument | 1,278 | 687 | ||||||
| 6,239 | 1,800 | |||||||
Finance costs | ||||||||
Change in fair value of warrant liability | - | (5,437) | ||||||
Unwinding of discount | (8) | (8) | ||||||
| (8) | (5,445) | |||||||
| 6,231 | (3,645) | |||||||
The significant increase in net finance income, as compared to the same period in 2010, is mainly due to the change in fair value of our warrant liability. That change results from the periodic “mark-to-market” revaluation, via the application of the Black-Scholes option pricing model, of currently outstanding share purchase warrants. The Black-Scholes “mark-to-market” warrant valuation most notably has been impacted by the market price of our common shares, which, on NASDAQ, has fluctuated from $0.81 as at January 4, 2010 to $1.72 as at December 31, 2010, and $1.54 as at December 31, 2011.
Additionally, our net finance income increased during the year 2011 from the same period in 2010 due to higher foreign exchange gains, which in turn resulted primarily from the overall substantial weakening, in 2010during 2011, of the euro against the US dollar, as compared to an overall lower weakening of the euro against the US dollar within the 2010 period. As a result, during the twelve-month period ended December 31, 2011, we recorded foreign exchange gains on transactions and on cash and cash equivalent balances denominated in US dollars.
The increase in our net finance income during the year ended December 31, 2011, as compared to the continued implementationyear ended December 31, 2010, also included gains on our short-term investment, which was sold during the year 2011.
It can be noted that the gains or losses resulting from the periodic mark-to-market valuation of generalour share purchase warrants did not result in any cash receipt or cash disbursement during the three-month or twelve-month periods ended December 31, 2011 and administrative cost-saving measures.2010.
The decrease from 2008 to 2009 is related to comparative euro-to-US dollar exchange rate fluctuations and to the absence in 2009 of certain non-recurring corporate expenses due to cost-saving measures that were implemented beginning in the second quarter of 2008, despite the additional selling expenses charged during 2009 as pertaining to the royalty paid to Tulane, as discussed above.
Depreciation and amortization expenses decreased to a combined $2.5Income tax expense was $1.1 million for the year ended December 31, 2010,2011, as compared to $10.8 million and $7.2 millionnil for eachthe same period in 2010. The increase consists of the years ended December 31, 2009 and 2008, respectively. The significant decrease from 2009 to 2010 is attributable primarilyforeign withholding taxes related to the absence, in 2010, of impairment charges.Yakult upfront and milestone license fees revenues discussed above.
The increase in depreciation and amortization expenses from 2008 to 2009 is attributable to the impairment charges related to cetrorelix, ozarelix and certain items of property, plant and equipment utilized exclusively in the development activities related to cetrorelix, as discussed above. This year-over-year increase was offset in large proportion by the impairment charge of $2.4 million
recorded in 2008 related to teverelix, an intangible asset that was deemed impaired in the fourth quarter of 2008.
Loss from operations amounted to $25.2 million Net lossfor the year ended December 31, 2010,2011 was $27.1 million, or $0.29 per basic and diluted share, compared to $24.0 million and $62.4 million for each of the years ended December 31, 2009 and 2008, respectively. Our loss from operations from 2009 to 2010 increased mainly due to the absence, in 2010, of licence fee revenues commensurate with the significant amounts recognized in 2009 and despite the comparative reductions in net R&D costs, SG&A expenses and depreciation and amortization charges.
The significant decrease from 2008 to 2009 in loss from operations is due to the significant year-over-year increase in license fee revenues, associated mainly with agreements for cetrorelix and ozarelix, combined with lower comparative R&D and SG&A expenses, partly offset by increased depreciation and amortization expenses and by lower comparative margins on Cetrotide® sales.
Other income (expenses) are comprised primarily of foreign exchange gains and losses, which result from the impact of changes in the value of currencies such as the US dollar and the Canadian dollar ("CAN$") against the Group's functional currency, the euro. Foreign exchange gains and losses are recorded upon settlement or revaluation of non-euro-denominated balances.
Foreign exchange gain (loss) amounted to $1.2 million for the year ended December 31, 2010, compared to ($1.1 million) and $3.1 million for each of the years ended December 31, 2009 and 2008, respectively.
During the first six months of 2010, the euro weakened progressively against the US dollar, losing approximately 14.2% of its value since December 31, 2009. During that time period, we recorded significant foreign exchange gains, mainly as a result of the periodic revaluation of our US dollar-denominated cash and cash equivalents, a significant portion of which resulted from the two registered direct offerings completed in April and June 2010, as noted above. However, the euro recovered partially during the second half of 2010, strengthening approximately 9.4% against the US dollar since June 30, 2010. As a result, we recorded substantial foreign exchange losses on transactions and balances denominated in US dollars during that period. Overall, the net depreciation in the euro against the US dollar during the twelve months ended December 31, 2010 amounted to approximately 6.6%, resulting in a net foreign exchange gain of $1.2 million.
On a comparative basis, the euro strengthened almost consistently against the US dollar on average during the twelve months ended December 31, 2009 by approximately 13.0%. As a result, net foreign exchange losses recorded were significantly higher during the year ended December 31, 2009, as compared to 2010, where we posted a net foreign exchange gain.
The year-end conversion rates from the euro and Canadian dollar to the US dollar can be summarized as follows:
| | As at December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
1 US dollar equivalent to: | 2010 | 2009 | 2008 | |||||||
| | $ | $ | $ | |||||||
Euro | 0.7468 | 0.7007 | 0.7145 | |||||||
Canadian dollar | 0.9946 | 1.0510 | 1.2180 | |||||||
Income tax expense of $1.2 million for the year ended December 31, 2008 is largely attributable to a minimum tax payable in Germany due to the tax accounting ramifications of the sale of future royalties to Cowen, referred to above.
In 2011, we do not expect to record any significant income tax recovery or expense in our foreign or domestic entities.
Net loss was $23.2$28.5 million, or $0.31$0.38 per basic and diluted share for the year ended December 31, 2010,2010.
The decrease in net loss for the year ended December 31, 2011, as compared to $24.7the year ended December 31, 2010, is largely due to the significant increase in license fees revenues, as well as net finance income, partly offset by lower margin contribution from Cetrotide®, higher net R&D costs and SG&A expenses, combined with the recording of income tax expense of $1.1 million in 2011, as discussed above.
Quarterly Consolidated Results of Operations Information
The following is a summary of selected consolidated financial information derived from our unaudited interim period consolidated financial statements for each of the eight most recently completed quarters.
(in thousands, except for per share data)
| Quarters ended | ||||||||||||||||
December 31, 2011 | September 30, 2011 | June 30, 2011 | March 31, 2011 | |||||||||||||
| $ | $ | $ | $ | |||||||||||||
Revenues | 12,627 | 9,514 | 6,523 | 7,389 | ||||||||||||
Loss from operations | (8,688) | (8,244) | (7,971) | (7,291) | ||||||||||||
Net (loss) income | (7,519) | 1,078 | (10,569) | (10,057) | ||||||||||||
Net (loss) income per share | ||||||||||||||||
Basic and diluted | (0.07) | 0.01 | (0.12) | (0.12) | ||||||||||||
| Quarters ended | ||||||||||||||||
December 31, 2010 | September 30, 2010 | June 30, 2010 | March 31, 2010 | |||||||||||||
| $ | $ | $ | $ | |||||||||||||
Revenues | 9,971 | 5,726 | 5,584 | 6,422 | ||||||||||||
Loss from operations | (4,003) | (5,456) | (7,950) | (7,397) | ||||||||||||
Net loss | (6,610) | (9,920) | (6,176) | (5,745) | ||||||||||||
Net loss per share* | ||||||||||||||||
Basic and diluted | (0.08) | (0.12) | (0.08) | (0.09) | ||||||||||||
| * | Net income (loss) per share is based on each reporting period’s weighted average number of shares outstanding, which may differ on a quarter-to-quarter basis. As such, the sum of the quarterly net income (loss) per share amounts may not equal year-to-date net loss per share. |
In the last eight quarters, revenues have increased, when compared quarter over quarter for each of the corresponding periods in 2011 vs. 2010, mainly due to the higher deliveries of Cetrotide® to Merck Serono, which increase have started in the fourth quarter of 2010, the recording of a contingent payment received from Cowen in the fourth quarter of 2010 as well as the recording of a milestone payment from Yakult in the fourth quarter of 2011.
In the last eight quarters, net (loss) income have been impacted by revenues, as mentioned above, by the increased level of net R&D costs in connection with the advancement of perifosine, AEZS-130 and Erk/PI3K compounds, by the recognition of impairment losses in the third and fourth quarters of 2011, by the initiation of pre-launch and marketing efforts related to the potential commercialization of perifosine in Europe in the third and fourth quarters of 2011, as well as by the foreign exchange gain or loss, the change in fair value of our warrant liability and the gain on our short-term investment.
Fourth Quarter 2011 Results
Revenues
Revenueswere $12.6 million for the quarter ended December 31, 2011, compared to $10.0 million for the same quarter in 2010. The increase in revenues is due primarily to comparative higher deliveries of Cetrotide® to Merck Serono ($3.6 million) and to the recording of a milestone payment ($2.6 million) from Yakult with respect to the initiation of a Phase 1/2 trial with perifosine in CRC in Japan, partly offset by the lower royalties in 2011 attributable to the contingent payment of $2.5 million from Cowen recorded in December 2010.
Operating expenses
Cost of saleswas $8.1 million for the quarter ended December 31, 2011, compared to $5.4 million for the same quarter in 2010. This increase is largely attributable to the comparative increase in volume of sales of Cetrotide® to Merck Serono, as discussed above. Additionally, cost of sales as a percentage of sales and royalties increased to approximately 88% for the quarter ended December 31, 2011, compared to 63% for the same period in 2010. Our lower margins are largely attributable to a lower contribution of royalties to total sales and royalties in 2011, as compared to 2010, as a result of a $3.4 million royalty milestone payment recorded in 2010 from Cowen.
R&D costs, net of refundable tax credits and grantswere $7.8 million for the quarter ended December 31, 2011, compared to $5.4 million for the same quarter in 2010. The comparative increase in R&D expenses primarily results from the increase in third-party costs incurred in connection with the advancement of perifosine, AEZS-108 and Erk/PI3K compounds (AEZS-129, AEZS-131, AEZS-132)-related activities.
Selling, general and administrative (“SG&A”) expenseswere $5.4 million for the quarter ended December 31, 2011, compared to $3.2 million for the same quarter in 2010. The increase in SG&A expenses is mainly related to the recognition of an impairment loss on property, plant and equipment (leasehold improvements and furniture and fixtures ($0.3 million)) and an additional onerous lease provision ($0.2 million), combined with the pre-launch and marketing efforts related to the potential commercialization of perifosine in Europe ($0.5 million) and with the increase in foreign exchange loss ($0.6 million).
Net finance income (costs), comprised predominantly of net foreign exchange gains, the change in fair value of our warrant liability and the gain on our short-term investment (2010 only), for the quarter ended December 31, 2011, totalled $1.4 million, compared to ($2.6 million) for the same period in 2010, as presented below.
| (in thousands) | Three months December 31, 2011 | Three months December 31, 2010 | ||||||
| $ | $ | |||||||
Finance income | ||||||||
Net gains due to changes in foreign currency exchange rates | 1,118 | 276 | ||||||
Change in fair value of warrant liability | 221 | - | ||||||
Interest income | 95 | 70 | ||||||
Gain on held-for-trading financial instrument | - | 687 | ||||||
| 1,434 | 1,033 | |||||||
Finance costs | ||||||||
Change in fair value of warrant liability | - | (3,638) | ||||||
Unwinding of discount | (2) | (2) | ||||||
| (2) | (3,640) | |||||||
| 1,432 | (2,607) | |||||||
Net loss amounted to $7.5 million, or $0.43 per basic and diluted share, and $59.8 million, or $1.12$0.07 per basic and diluted share, for each of the yearsquarter ended December 31, 20092011, compared to $6.6 million, or $0.08 per basic and 2008, respectively.diluted share, for the same quarter in 2010. The decreaseincrease in our 2010 net loss as compared to 2009, is mainly attributable to a reduction in netthe higher comparative R&D costs, lowerand SG&A expenses, and lower depreciation and amortization charges, as well as to higher net foreign exchange gains, as discussed above,partly offset by the significant reduction of licence fee revenuesincrease in net finance income and a lowerby higher gross margin onrelating to sales of Cetrotide®Cetrotide®.
The significant decreasePrimarily, given the presence in net lossour 2011 fourth quarter revenues of the $2.6 million milestone receivable from 2008 to 2009 is dueYakult with respect to the significant year-over-year increaseinitiation of a Phase 1/2 trial with perifosine in license fee revenues, associated mainly with agreements for cetrorelixCRC in Japan, and ozarelix, combined with lower comparative R&D, SG&A and income tax expenses, partly offset by lower comparative sales and royalties and increased depreciation and amortization expenses andexcluding any impact of foreign exchange losses.
Wegains or losses, as well as change in fair value of warrant liability, we expect that ourthe net loss for the year 2011first quarter of 2012 will increase, as compared to 2010, primarily due to higher expected comparative R&D costs and excluding the impacts related to foreign exchange gains or losses, our licensing agreement entered into with Yakult, discussed above, and our conversion to IFRS.fourth quarter of 2011.
Consolidated Balance SheetStatement of Financial Position Information
| | As at December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| | 2010 | 2009 | 2008 | |||||||
| | $ | $ | $ | |||||||
| | (in thousands) | |||||||||
Cash and cash equivalents | 31,998 | 38,100 | 49,226 | |||||||
Short-term investments | 1,934 | — | 493 | |||||||
Accounts receivable and other current assets | 10,243 | 10,913 | 12,005 | |||||||
Restricted cash | 827 | 878 | — | |||||||
Property, plant and equipment, net | 3,096 | 4,358 | 6,682 | |||||||
Other long-term assets | 28,476 | 32,013 | 39,936 | |||||||
Total assets | 76,574 | 86,262 | 108,342 | |||||||
Accounts payable and other current liabilities | 13,427 | 19,211 | 22,121 | |||||||
Current portion of long-term payable | 60 | 57 | 49 | |||||||
Long-term payable | 90 | 143 | 172 | |||||||
Non-financial long-term liabilities* | 50,558 | 57,625 | 64,525 | |||||||
Total liabilities | 64,135 | 77,036 | 86,867 | |||||||
Shareholders' equity | 12,439 | 9,226 | 21,475 | |||||||
Total liabilities and shareholders' equity | 76,574 | 86,262 | 108,342 | |||||||
2010 compared to 2009
| As at December 31, | ||||||||||||
| (in thousands) | 2011 | 2010 | 2009* | |||||||||
| $ | $ | $ | ||||||||||
Cash and cash equivalents | 46,881 | 31,998 | 38,100 | |||||||||
Short-term investment | - | 1,934 | - | |||||||||
Trade and other receivables and other current assets | 13,258 | 9,877 | 10,913 | |||||||||
Restricted cash | 806 | 827 | 878 | |||||||||
Property, plant and equipment, net | 2,512 | 3,096 | 4,358 | |||||||||
Other non-current assets | 11,912 | 13,716 | 32,013 | |||||||||
Total assets | 75,369 | 61,448 | 86,262 | |||||||||
Payables and other current liabilities | 17,784 | 13,350 | 19,211 | |||||||||
Long-term payable (current and non-current portions) | 88 | 150 | 200 | |||||||||
Warrant liability (current and non-current portions) | 9,204 | 14,367 | - | |||||||||
Non-financial non-current liabilities** | 52,839 | 51,156 | 57,625 | |||||||||
Total liabilities | 79,915 | 79,023 | 77,036 | |||||||||
Shareholders’ (deficiency) equity | (4,546 | ) | (17,575 | ) | 9,226 | |||||||
Total liabilities and shareholders’ (deficiency) equity | 75,369 | 61,448 | 86,262 | |||||||||
| * | We adopted IFRS in 2011 with a transition date of January 1, 2010. The selected financial information for the years ended December 31, 2009, 2008 and 2007 is derived from financial statements that were presented in accordance with Canadian GAAP and has not been restated in accordance with IFRS. Consequently, the selected financial information for the years ended December 31, 2009, 2008 and 2007 may not be comparable with the corresponding selected financial information for the years ended December 31, 2011 and 2010. Please refer to “Critical Accounting Policies, Estimates and Judgments” for the policy differences between Canadian GAAP and IFRS. |
| ** | Comprised mainly of non-current portion of deferred revenues, employee future benefits and provision. |
The decreaseincrease in cash and cash equivalents as at December 31, 2010,2011, as compared to December 31, 2009,2010, is due to the receipt of net proceeds pursuant to drawdowns made in connection with the February and June ATM Sales Agreements, as discussed above, to the proceeds from the sale of our short-term investment, to the receipt of the upfront license payment in connection with our development, commercialization and licensing agreement entered into with Yakult, as noted above, as well as to the receipt of net proceeds from the exercise of share purchase warrants. These increases were partially offset by recurring disbursements and other variations in components of our working capital, as well ascapital.
Trade and other receivables and other current assets increased from December 31, 2010 to December 31, 2011 mainly due to the comparative weakeningincrease in 2010trade receivable in connection with the higher deliveries of Cetrotide® to Merck Serono for an amount of approximately $2.9 million and due to the euro against the US dollar. However, this decrease is significantlyperifosine inventory to be sold to our partner Keryx for an amount of approximately $0.7 million which in turn were partly offset by the receiptimpact of approximately $25.0 millionforeign exchange rate fluctuations.
Other non-current assets decreased from December 31, 2010 to December 31, 2011 primarily due to the net reduction in the carrying value of net proceeds in connection with two registered direct offerings, completed in April and June 2010.
Other long-term assets, comprised ofour identifiable intangible assets, goodwill and deferred charges, decreased in 2010 due to euro-to-US dollar exchange rate fluctuation impacts and due to recurring amortization chargeswhich was negatively impacted by the impairment loss recognized on those assets (excluding goodwill).Cetrotide®, as discussed above.
The decrease in accounts payablePayables and other current liabilities resultsincreased from December 31, 2010 to a large degree from settlements of trade payables, income tax liability and other accrued expenses, including balances representing certain residual obligations related to former cetrorelix activities.
Non-financial long-term liabilities decreased from 2009 to 2010December 31, 2011 mainly due to an increase of approximately $2.4 million in trade accounts payable and accrued liabilities in connection with the higher deliveries of Cetrotide® and with the higher level of R&D expenses resulting from the advancement of perifosine, AEZS-130 and Erk/PI3K compounds, which includes an increase due to the perifosine R&D inventory received and payable for an amount of approximately $1.3 million, to the increase in current portion of deferred revenues for an amount of $1.3 million, and to the increase in accrued salaries for an amount of approximately $0.5 million which in turn were partly offset by the impact of foreign exchange rate fluctuations.
Our warrant liability was lower as at December 31, 2011, as compared to December 31, 2010, predominantly due to the change in fair value pursuant to the periodic “mark-to-market” revaluation of the underlying outstanding share purchase warrants, as well as following various warrant exercises during the twelve-month period ended December 31, 2011.
Non-financial liabilities were higher as at December 31, 2011, as compared to December 31, 2010, mainly as a result of the deferral of the upfront payment received in connection with our development, commercialization and licensing agreement entered into with Yakult, partially offset by the recurring amortization of deferred revenuesrevenues.
The net decrease in shareholders’ deficiency as well as due to euro-to-US dollar exchange rate fluctuation impacts.
The increase in shareholders' equity fromat December 31, 20092011, as compared to December 31, 2010, is predominantly attributable to an increase in share capital and warrants, following the completionissuance of common shares pursuant to the registered direct offerings discussed above, partlyaforementioned drawdowns made in connection with the February and June ATM Sales Agreements, partially offset by the increase in our deficit due to the net loss for the year ended December 31, 2010.
2009 compared to 2008
The decrease in cash2011 and cash equivalents as at December 31, 2009, compared to December 31, 2008 is due primarily to recurring cash flows used in operating activities and by the reduction of currently available cash due to a transfer of funds to a restricted account, as discussed below, largely offset by the receipt of proceeds from sanofi and to the receipt of net proceeds of approximately $14.3 million in connection with two registered direct offerings.
The decrease in property, plant and equipment as at December 31, 2009, compared to December 31, 2008 is due largely to the impairment charge that was taken against certain items utilized exclusively in the development activities related to cetrorelix, as discussed above.
The decrease in other long-term assets primarily includes the reduction to intangible assets, which in turn was attributable to the impairment charges taken on cetrorelix and ozarelix, as discussed above. Additionally, the reduction is attributable to deferred charges amounting to approximately $0.7 million, which were deferred in 2008, but which were included as a reduction to share capital in connection with the June 2009 Offering, as discussed above.
The reduction in non-financial long-term liabilities is attributable mainly to deferred revenues, which in 2009 were lower following both the ongoing amortization of the proceeds received from Cowen and the full recognition of previously deferred amounts associated with license and development agreements related to the use of ozarelix, as discussed above.
The decrease in shareholders' equity from 2008 to 2009 is attributable to the increase in consolidated deficit due to the 2009 net loss and to the decrease in accumulated other comprehensive income, offsetwhich in large proportion by the increase in share capital and warrants following the two registered direct offerings discussed above.turn is comprised of cumulative translation adjustments.
Financial Liabilities, Obligations and Commitments
We have certain contractual leasing obligations and commercialpurchase obligation commitments. CommercialPurchase obligation commitments mainly include R&D services and manufacturing agreements related to the production of Cetrotide®
CetrotideTable of Contents®
and to other R&D programs. The following table summarizestables summarize future cash requirements with respect to these obligations.
| | | Payments due by period | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Carrying amount | Less than 1 year | 1 to 3 years | 4 to 5 years | After 5 years | |||||||||||
| | $ | $ | $ | $ | $ | |||||||||||
| | (in thousands) | |||||||||||||||
Operating leases | 10,397 | 2,150 | 3,809 | 3,642 | 796 | |||||||||||
Commercial commitments | 17,887 | 9,167 | 8,720 | — | — | |||||||||||
Long-term payable | 150 | 60 | 90 | — | — | |||||||||||
| 28,434 | 11,377 | 12,619 | 3,642 | 796 | ||||||||||||
Per our agreement with Cowen, discussed above, we may be required to remit quarterly make-wholeFuture minimum lease payments related to royalty rate reductions that could materialize between Cowen and Merck Serono. No make-wholefuture minimum sublease payments were paid or became payable during 2010 or 2009, nor do we expectexpected to be required to remit any such make-wholereceived under non-cancellable operating leases (subleases), as well as future payments in the future.connection with utility service agreements, as at December 31, 2011 are as follows:
| (in thousands) | Minimum lease payments | Minimum sub-lease payments | Utilities | |||||||||
| $ | $ | $ | ||||||||||
Less than 1 year | 1,690 | (226 | ) | 609 | ||||||||
1 – 3 years | 3,128 | (451 | ) | 793 | ||||||||
4 – 5 years | 2,172 | (451 | ) | 496 | ||||||||
More than 5 years | 373 | (244 | ) | - | ||||||||
Total | 7,363 | (1,372 | ) | 1,898 | ||||||||
Also per our agreement with Cowen, we have agreed to make a one-time cash payment to Cowen in the event that cetrorelix is approvedService and manufacturing commitments given, which consist of R&D service agreements and manufacturing agreements for sale by European regulatory authorities in an indication other thanCetrotide®in vitro fertilization. Such a payment could range from $5.0 million to a maximum of $15.0 million. No one-time cash amount was paid or became payable during 2010 or 2009, nor do we expect to be required to make such a payment in the future, particularly given the fact that our development activities related to cetrorelix have been terminated., are as follows:
| (in thousands) | As at December 31, 2011 | |||
| $ | ||||
Less than 1 year | 10,760 | |||
1 – 3 years | 3,855 | |||
4 – 5 years | - | |||
More than 5 years | - | |||
Total | 14,615 | |||
Outstanding Share Data
As at March 24, 2011,27, 2012, there were 85,265,033108,787,366 common shares issued and outstanding, and 6,807,463as well as 7,497,634 stock options outstanding. WarrantsShare purchase warrants outstanding as at March 24, 2011 represent27, 2012 represented a total of 12,795,8858,752,868 equivalent common shares.
Capital disclosuresDisclosures
Our objective in managing capital, primarily composed of shareholders’ deficiency and cash and cash equivalents, is to ensure sufficient liquidity to fund our R&D activities, SG&A expenses, working capital and capital expenditures.
We endeavourHistorically, our priority has been to manageoptimize our liquidity to minimize dilution to our shareholders. Non-dilutive activities have includedby non-dilutive sources, including the sale of non-core assets, and rights to future royalties, the collection of investment tax credits and grants, interest income, licensing fees, serviceand related services and royalties. More recently, however, we have raised additional capital throughvia registered direct offerings as discussed above.and drawdowns related to the February and June ATM Sales Agreements, and we expect to continue to raise capital via drawdowns related to the January 2012 ATM Sales Agreement.
At December 31, 2011, cash and cash equivalents amounted to $46.9 million. The Company believes that its cash position will be sufficient to finance its operations and capital needs for at least the next twelve months.
Our capital management objective remains the same as that of previous years.periods. The policy on dividends is to retain cash to keep funds available to finance the activities required to advance our product development pipeline.
We are not subject to any capital requirements imposed by any regulators or any other external source.
It is important to note that historical expenditure patterns of expenditures cannot be taken as an indication of future expenditures. The amount and timing of expenditures and availability of capital resources vary substantially from period to period, depending on the level of research and development activity being undertaken at any onegiven time and on the availability of funding from investors and prospective commercial partners.
Liquidity, Cash Flows and Capital Resources
We endeavour to finance ourOur operations and capital expenditures have been financed mainly through cash flows from product sales, license fee revenuesoperating activities and other non-dilutive activities. However, we have also completedactivities, except for the registered direct offerings completed during the year ended December 31, 20092010 and, more recently, in April and June 2010,the drawdowns related to our various ATM Sales Agreements, as noteddiscussed above.
Our cash and cash equivalents and short-term investment amounted to $33.9$46.9 million as at December 31, 2010,2011, as compared to $38.1$32.0 million as at December 31, 2009. Possible additional operating losses and/or possible investments in complementary businesses or products may require additional financing.2010. As at December 31, 2010,2011, cash and cash equivalents, of the Company included CAN$0.5 million and €1.4denominated in euro, amounted to €10.3 million.
Based on our assessment, which took into account current cash levels, the completion of the registered direct offerings discussed above, as well as our strategic plan and corresponding budgets for 2011 and projections for 2012 and 2013,forecasts, we believe that we have sufficient liquidity and financial resources to fund planned expenditures and other working capital needs for at least, but not limited to, the 12-monthtwelve-month period fromfollowing the balance sheet date of December 31, 2010.2011.
We may endeavour to secure additional financing, as required, through strategic alliance arrangements or through other non-dilutive activities, as well as via the issuance of new share capital pursuant to, for example, the ATM sales agreement referred to above.capital.
The variations in our liquidity by activity are explained below.
Operating Activities
Cash flows used in operating activities amounted to $31.1totalled $26.2 million for the year ended December 31, 2010,2011, as compared to $24.1 million and $1.3$31.7 million for each of the yearsyear ended December 31, 2009 and 2008, respectively.2010. The net increasedecrease in cash used in operating activities from 2009 to 2010 is attributabledue in large part to the fact thatreceipt, during the year 2011, of nearly $8.4 million in connection with our 2009 operating cash flows had been positively impacteddevelopment, commercialization and licensing agreement entered into with Yakult, as discussed above, as well as to lower trade accounts payable settlements, partially offset by the cash proceeds received from sanofi as an upfront payment on our former cetrorelix-related licensing and development agreement. Similar cash payments were not received during 2010. Additionally, settlements of ourlower trade accounts receivable were lower in 2010, as compared to 2009. However, the decrease in cash provided by operating activities in 2010 was partially offset by a significant reduction in cash R&D and other prepaid expenditures.settlements.
The significant increase in cash used in operating activities from 2008 to 2009 is attributable to the receipt of net cash proceeds of $51.5 million in 2008 from Cowen, compared to the lower cash proceeds of $30.0 million from sanofi in 2009, as discussed above. Also, operating cash payments for prepaid expenses and accounts payable were higher during 2009 as compared to 2008.
We expect net cash used in operating activities to decreaseincrease during 2011,2012, as compared to 2010.2011, as we increase our investment in perifosine and in AEZS-108, as discussed above.
Financing Activities
Net cashCash flows provided by (used in) financing activities amountedincreased to $25.4$38.6 million for the year ended December 31, 2010,2011, as compared to $14.2$26.0 million and ($1.2 million) for each of the years ended December 31, 2009 and 2008, respectively. The increases in net cash provided by financing activities from 2008 to 2009 and from 2009 to 2010 are attributable almost entirely to the registered direct offerings discussed above.
Investing Activities
Cash (used in) provided by investing activities amounted to ($0.1 million) for the year ended December 31, 2010,2010. The increase is primarily due to the receipt of higher net proceeds from offerings such as the drawdowns related to the February and June ATM Sales Agreements, discussed above, as well as to the increase in proceeds received following the exercise of share purchase warrants.
Investing Activities
Cash flows provided by investing activities reached $2.5 million for the year ended December 31, 2011, as compared to ($1.1 million) and $42.0cash flows used in investing activities of nearly $0.05 million for eachthe year ended December 31, 2010. The increase in cash provided by investing activities is due to the increase in cash proceeds received on the sale of our short-term investment, partly offset by cash disbursements made in connection with the purchases of laboratory and other equipment used in ongoing R&D activities.
Critical Accounting Policies, Estimates and Judgments
As noted above, our consolidated financial statements as at December 31, 2011 and December 31, 2010 and for the years ended December 31, 20092011 and 2008, respectively.
The significant decrease from 2008 to 2009 relates in large proportion to the sale and maturity of short-term investments as well as to the disposals of the building and land in Quebec City and of Impavido®, in 2008. Also, as discussed above, during 2009, we transferred approximately $0.9 million to a restricted cash account. Changes to restricted cash balances, including any interest earned thereon, are reported in the statement of cash flows as investing activities.
Critical Accounting Policies and Estimates
Our consolidated financial statements are2010 have been prepared in accordance with Canadian GAAP. A summaryIFRS. Note 29 “Transition to IFRS” to our consolidated financial statements as at December 31, 2011 and December 31, 2010 and for the years ended December 31, 2011 and 2010 discusses the impact of the transition to IFRS on our reported financial position, financial performance and cash flows, including the nature and effect of significant and pertinent measurement and disclosure differences betweenchanges in accounting policies from those used in our previous Canadian and US GAAP is provided in note 24 to our 2010 consolidated financial statements. Thestatements as at December 31, 2010 and for the year then ended. Comparative figures for 2010 have been adjusted to give effect to these changes.
These consolidated financial statements were approved by our Board of Directors for issue on March 27, 2012.
Additionally, the preparation of consolidated financial statements in accordance with generally accepted accounting principlesIFRS often requires management to make estimates about and apply assumptions or subjective judgment to future events and other matters that affect the reported amounts of our assets, liabilities, revenues, expenses and liabilitiesrelated disclosures. Assumptions, estimates and the disclosures of contingent assets and liabilities at the date of the financial statements, as well as the reported amounts of revenues and expenses during the reporting years. Significant estimatesjudgments are generally made in connection with the calculation of revenues, inventory and research and development expenses, as well as in determining the allowance for doubtful accounts, valuation allowance for future income tax assets, the useful lives of property, plant and equipment and intangible assets with finite lives, the valuation of intangible assets and goodwill, the fair value of stock options and warrants granted, employee future benefits and certain accrued liabilities. We base our estimatesbased on historical experience, whereexpectations, current trends and other factors that management believes to be relevant at the time at which our consolidated financial statements are prepared. Management reviews, on a regular basis, the Company’s accounting policies, assumptions, estimates and on various other assumptionsjudgments in order to ensure that the consolidated financial statements are presented fairly and in accordance with IFRS.
Critical accounting estimates and judgments are those that have a significant risk of causing material adjustment and are often applied to matters or outcomes that are inherently uncertain and subject to change. As such, management cautions that future events often vary from forecasts and expectations and that estimates routinely require adjustment.
A summary of those areas where we believe to be reasonable under the circumstances. Actual results could differ from those estimates.
The following section summarizes our critical accounting policies and other policies that requireaffect the most significant judgmentjudgments and estimates used in the preparation of our consolidated financial statements.
Revenue Recognition and Deferred Revenues
We are currentlystatements can be found in a phase in which potential products are being further developed or marketed jointly with strategic partners. Existing licensing agreements usually foresee one-time payments (upfront payments), payments for research and development services in the form of cost reimbursements, milestone payments and royalty receipts for licensing and marketing product candidates. Revenues associated with those multiple-element arrangements are allocatednote 3 to the various elements based on their relative fair value.
Agreements containing multiple elements are divided into separate units of accounting if certain criteria are met, including whether the delivered element has stand-alone value to the customer and whether there is objective and reliable evidence of the fair value of the undelivered obligation(s). The consideration received is allocated among the separate units based on each unit's fair value and the applicable revenue recognition criteria are applied to each of the separate units.
License fees representing non-refundable payments received upon the execution of license agreements are recognized as revenue upon execution of the license agreements when we have no significant future performance obligations and when collectibility of the fees is assured. Upfront payments received at the beginning of licensing agreements are not recorded as revenue when received but are amortized based on the progress to the related research and development work. This progress is based on estimates of total expected time or duration to complete the work, which is compared to
the period of time incurred to date in order to arrive at an estimate of the percentage of revenue earned to date.
Milestone payments, which are generally based on developmental or regulatory events, are recognized as revenue when the milestones are achieved, collectibility is assured, and when there are no significant future performance obligations in connection with the milestones.
Royalty revenue, based on a percentage of sales of certain declared products sold by third parties, is recorded when we have fulfilled the terms in accordance with the contractual agreement and have no future obligations, the amount of the royalty fee is determinable and collection is reasonably assured.
Proceeds received in connection with the sale of rights to future royalties are deferred and recognized over the life of the license agreement pursuant to the "units-of-revenue" method, as discussed above.
Revenues from sales of products are recognized when title passes to customers, which is at the time goods are shipped, when there are no future performance obligations, when the purchase price is fixed and determinable, and when collection is reasonably assured.
Impairment of Long-Lived Assets and Goodwill
Property, plant and equipment and intangible assets with finite lives are reviewed for impairment when events or circumstances indicate that carrying values may not be recoverable. Impairment exists when the carrying value of the asset is greater than the undiscounted future cash flows expected to be provided by the asset. The amount of impairment loss, if any, is the excess of its carrying value over its fair value, which in turn is determined based upon discounted cash flows or appraised values, depending of the nature of assets.
Goodwill, which represents the excess of the purchase price over the fair values of the net assets of entities acquired at the respective dates of acquisition, is tested for impairment annually, or more frequently if events or changes in circumstances indicate that the carrying value of the reporting unit to which the goodwill is assigned may exceed the fair value of the reporting unit.
In the event that the carrying amount of a reporting unit, including goodwill, exceeds its fair value, an impairment loss is recognized in an amount equal to the excess. Fair value of goodwill is estimated in the same way as goodwill is determined at the date of the acquisition in a business combination, that is, the excess of the fair value of the reporting unit over the fair value of the identifiable net assets of the reporting unit.
Income Taxes
We operate in multiple jurisdictions, and our earnings are taxed pursuant to the tax laws of these jurisdictions. Our effective tax rate may be affected by changes in, or interpretations of, tax laws in any given jurisdiction, utilization of net operating losses and tax credit carry-forwards, changes in geographical mix of income and expense, and changes in management's assessment of matters, such as the ability to realize future tax assets. As a result of these considerations, we must estimate our income taxes in each of the jurisdictions in which we operate. This process involves estimating our actual current tax exposure, together with assessing temporary differences resulting from differing treatment of items for tax and accounting purposes. These differences result in future tax assets and liabilities, which are included in our consolidated balance sheet. We must then assess the likelihood that our future tax assets will be recovered from future taxable income and establish a valuation allowance if, based on available information, it is more likely than not that some or all of the future income tax assets will not be realized.
Significant management judgment is required in determining our provision for income taxes, our income tax assets and liabilities, and any valuation allowance recorded against our net income tax assets. The valuation allowance is based on our estimates of taxable income by jurisdiction in which we operate and the period over which our income tax assets will be recoverable. In the event that actual results differ from these estimates or we adjust these estimates in future periods, we may need to amend our valuation allowance, which could materially impact our financial position and results of operations.
Stock-Based Compensation Costs
We account for all forms of employee stock-based compensation using the fair value-based method. This method requires that we make estimates about the risk-free interest rate, the expected volatility of our shares and the expected life of the awards.
International Financial Reporting Standards
We are currently finalizing our evaluation of the impacts that likely will result from preparing our consolidated financial statements in accordance with IFRS. The adoption of IFRS will have an impact on our consolidated financial statements as well as on certain operational and performance measures, beginning on January 1,at December 31, 2011 and on a retrospective comparative basis beginning on January 1,December 31, 2010 and for the years ended December 31, 2011 and 2010.
As previously disclosed, we have developed a formal plan for IFRS conversion and the related transition from current standards. To date, we have completed a full diagnostic, in which all existing international standards were examined in comparison with corresponding Canadian guidance, and significant differences between IFRS and Canadian GAAP were documented in order to plan for more detailed analysis, which has been the focus of our conversion project's solutions development phase and which is substantially complete.
Accounting Standards Not Yet Adopted
The key elementsthe first part of our conversion plana three-part project to replace IAS 39,Financial Instruments: Recognition and related status are provided below.
|
| |||
|
| |||
Our solutions development activities completed to date have allowed us to conclude that the adoption of certain international standards will result in a significant change to current accounting policies, reported financial statement amounts or disclosures. With the exception of IFRS 1, selected
areas of international guidance examined to date that are relevant to our business, and the corresponding expected impact that likely will result from the application thereof, are presented below.
| ||
| ||
| ||
| ||
It should be noted that the differences outlined above are not a complete list of topics that are relevant to our business. As such, as we finalize our solutions development activities, we may identify
other areas that could result in significant quantitative or qualitative impacts upon IFRS adoption or thereafter in comparison to currently applied Canadian GAAP. Additionally, the expected adjustments provided above are unaudited.
As we continue to analyze any potential quantitative adjustments and policy decisions that need to be made upon full conversion to IFRS, we have reached some key preliminary conclusions related to the application of IFRS 1.
IFRS 1 provides authoritative guidance for use in the conversion of a setderecognition of financial instruments. IFRS 9 applies to financial statements (and interimfor annual periods beginning on or after January 1, 2015, with early adoption permitted. We currently are evaluating any impact that this new guidance may have on our consolidated financial reports for part of that period) from another basis of accounting to IFRS. The basicstatements.
In May 2011, the IASB issued IFRS 10,Consolidated Financial Statements (“IFRS 10”), which builds on existing principles by identifying the concept of IFRS 1 is thatcontrol as the adoption of IFRS should be applied retrospectively, meaning thatdetermining factor in whether an entity should present its firstbe included within the consolidated financial statements usingof a parent company. IFRS as if10 also provides additional guidance to assist in the determination of control where this is difficult to assess. IFRS had been applied10 applies to financial statements for annual periods beginning on or after January 1, 2013, with early adoption permitted. We currently are evaluating any impact that this new guidance may have on our consolidated financial statements.
In May 2011, the IASB issued IFRS 11,Joint Arrangements(“IFRS 11”), which enhances accounting for joint arrangements, particularly by focusing on the rights and effective from the dateobligations of the entity's inception. However, duearrangement, rather than the arrangement’s legal form. IFRS 11 also addresses inconsistencies in the reporting of joint arrangements by requiring a single method to account for interests in jointly controlled entities and prohibits proportionate consolidation. IFRS 11 applies to financial statements for annual periods beginning on or after January 1, 2013, with early adoption permitted. We currently are evaluating any impact that this new guidance may have on our consolidated financial statements.
In May 2011, the factIASB issued IFRS 12,Disclosure of Interests in Other Entities(“IFRS 12”), which is a comprehensive standard on disclosure requirements for all forms of interests in other entities, including joint arrangements, associates, special purpose vehicles and other off-balance sheet vehicles. IFRS 12 applies to financial statements for annual periods beginning on or after January 1, 2013, with early adoption permitted. We currently are evaluating any impact that full retrospective applicationthis new guidance may have on our consolidated financial statements.
In May 2011, the IASB issued IFRS 13,Fair ValueMeasurement (“IFRS 13”), which defines fair value, sets out in a single IFRS a framework for measuring fair value and requires disclosures about fair value measurements. IFRS 13 does not determine when an asset, a liability or an entity’s own equity instrument is unlikelymeasured at fair value. Rather, the measurement and disclosure requirements of IFRS 13 apply when another IFRS requires or permits the item to be achievablemeasured at fair value (with limited exceptions). IFRS 13 applies to financial statements for annual periods beginning on or after January 1, 2013, with early adoption permitted. We currently are evaluating any impact that this new guidance may have on our consolidated financial statements.
In June 2011, the IASB amended IAS 1,Presentation of Financial Statements (“IAS 1”), to change the disclosure of items presented in a cost-effective manner, IFRS 1 offers certain optional exemptions to first-time preparers of IFRS financial statements. Any, all or none of these exemptionsother comprehensive income into two groups, based on whether those items may be taken.recycled to profit or loss in the future. The amendments to IAS 1 apply to financial statements for annual periods beginning after July 1, 2012, with early adoption permitted. We currently are evaluating any impact that this new guidance may have on our consolidated financial statements.
Presented below
In June 2011, the IASB issued an amended version of IAS 19,Employee Benefits (“IAS 19”), including the elimination of the option to defer the recognition of actuarial gains and losses (known as the “corridor method”), the streamlining of the presentation of changes in assets and liabilities arising from defined benefit plans and the enhancement of the disclosure requirements for defined benefit plans, including additional information about the characteristics of defined benefit plans and the risks to which entities are exposed through participation in those plans. The amendments to IAS 19 apply to financial statements for annual periods beginning on or after January 1, 2013, with early adoption permitted. We currently are evaluating any impact that this new guidance may have on our preliminary conclusions with respect to some key IFRS 1 optional exemptions, as applicable to our business.consolidated financial statements.
Outlook for 20112012
Perifosine
We expect to continue the development of perifosine in collaboration with our partner, Keryx, who is responsible, in accordance with the terms of our license agreement, for the development and registration of perifosine in North America. We have access to all corresponding data at no additional cost; hence, we expect to benefit from current development activities in order to achieve registration in territories excluding North America.
Our primary focus willcontinues to be on the advancement of the ongoing Phase 3 registration studies in both refractory advanced colorectal cancer and multiple myeloma, in conformity withmyeloma. We expect the SPA received by Keryx from the FDA. Furthermore, we have obtained positive scientific advice from the EMA relative to a development and regulatory pathway so as to extend the reachresults of perifosine to European territories for the refractory advanced colorectal cancer and multiple myeloma indications. Consequently, we are not expecting to invest in any additional trials in Europe in refractory advanced
colorectal cancer or multiple myeloma, since the EMA does not require that any studies be performed in addition to the studies currently in progress. For the ongoing Phase 3 studytrial of perifosine in multiple myeloma, we will contributeCRC in the second quarter of 2012.
We expect to the recruitment of patients outside the US andcontinue to other aspects of the ongoing study; however, our partner Keryx will reimburse us for most of the corresponding costs.
Additionally, we will advanceinvest in the preparation of our regulatory filingsmarketing and our commercialization strategy ex-North America. Further, we will continue to accumulate Phase 1 and 2 resultspre-launch activities of perifosine in multiple indications and weEurope.
AEZS-108
We expect to initiate with the collaboration of Keryx, an additional clinical trial in CLL.
AEZS-108
We expect to define our regulatory strategy for endometrial cancer with both the FDA and the EMA, with the goal of initiating a multi-center pivotal study in that indication.
Additional proof-of-concept and investigator-drivenendometrial cancer, as well as a multi-center Phase 2 study in triple-negative breast cancer. We also expect to pursue our Phase 1/2 studies such asin castration- and taxane-resistant prostate cancer with the ongoing studyUniversity of Southern California, and in refractory bladder cancer performed with the University of Miami and in castration refractory prostate cancer with University of Southern California, will continue to progress.
We also expect to initiate further proof-of-concept studies in patients with LHRH receptor-positive cancers such as pancreatic and triple negative breast cancer.Miami.
AEZS-130 (Solorel®)
We expect to completeFollowing the ongoingcompletion and positive Phase 3 study and file an NDAresults for Solorel®AEZS-130 as a diagnostic for AGHD in the United States.States, we expect, after a successful receipt of pre-NDA meeting with the FDA, to file an NDA.
We also expect to startpursue our ongoing Phase 2A study with AEZS-130 as a proof-of-concept study in cancer-induced cachexia.treatment for cancer-cachexia.
Revenue expectationsExpectations
Revenues are expected to decrease slightlyincrease in 2011,2012, as compared to 2010, excluding any revenue associated with2011, given our expectation for potential increased license fees revenues from our partners and despite the licensing agreement entered into with Yakult,expected stabilization of our sales and royalties, as discussed above.
Cost reduction and development focusOperating Expense Expectations
During 2011,2012, we expect to continue to focus our R&D efforts on our later-stage compounds, including perifosine AEZS-108 and Solorel®. Earlier-stage projects will be associated with grants, R&D credits or collaboration agreements.AEZS-108. With our focused strategy, and given our expectations related to R&D investments made on behalf of Keryx, we cannow expect an increase of our R&D expenses to total between $21.0$30 million and $23.0$32 million for the whole of 2011,2012, as compared to $20.5$24.5 million in 2010.2011. However, certain perfosine-related R&D expenses will be reimbursed by our partner, Keryx, as mentioned above.Keryx.
With regard to our SG&A expenses, in light of the continuous cost-saving measures already in place, we now expect to slightly reduce our SG&A costs for the whole of 2011, as compared to 2010.
We expect that our overall operating burn in 20112012 will decrease,increase, as compared to 2010,2011, due most notably to the receipt in 2011 of €6.0$8.4 million (approximately $8.3 million) in connection with our licensing agreement entered into with Yakult.Yakult and our expected increase in R&D investments, as discussed above, partly offset by increased license fees from our partners.
Financial Risk Factors and Other Instruments
Fair Value
The change in fair value of our warrant liability results from the periodic “mark-to-market” revaluation, via the application of the Black-Scholes option pricing model, of currently outstanding share purchase warrants. The Black-Scholes valuation is impacted, among other inputs, by the market price of our common shares. As a result, the change in fair value of the warrant liability, which is reported as finance income (costs) in our consolidated statements of comprehensive loss, has been and may continue in future periods to be materially affected most notably by changes in our common share price, which has ranged from $1.43 to $2.58 on NASDAQ during the year ended December 31, 2011.
Assuming the following variations of the market price of our common shares over a 12-month period:
Market price variations of -10% and +10%
If these variations were to occur, the impact on our net loss for warrant liability held at December 31, 2011 would be as follows:
| (in thousands) | Carrying amount | -10% | +10% | |||||||||
| $ | $ | $ | ||||||||||
Warrant liability* | 9,204 | 1,155 | (1,174) | |||||||||
|
|
|
| |||||||||
Total impact on net loss – decrease/(increase) | 1,155 | (1,174) | ||||||||||
|
|
|
| |||||||||
| * | Includes current and non-current portions. |
Foreign Currency Risk
Since we operate internationally, we are exposed to currency risks as a result of potential exchange rate fluctuations related to non-intragroup transactions. In particular, fluctuations in the US dollar and
Canadian dollar exchange rates against the euro could have a potentially significant impact on our results of operations.
The following variations are reasonably possible over a 12-month period:
Foreign exchange rate variations of -5% (depreciation of the EUR) and +5% (appreciation of the EUR) against the US$, from a year-end rate of EUR1 = US$1.2972.
If these variations were to occur, the impact on the Company’s net loss for each category of financial instruments held at December 31, 2011 would be as follows:
| Balances denominated in US$ | ||||||||||||
| (in thousands) | Carrying amount | -5% | +5% | |||||||||
| $ | $ | $ | ||||||||||
Cash and cash equivalents | 33,669 | 1,683 | (1,683) | |||||||||
Warrant liability* | 9,204 | (460) | 460 | |||||||||
|
|
|
| |||||||||
Total impact on net loss – decrease/(increase) | 1,223 | (1,223) | ||||||||||
|
|
|
| |||||||||
| * | Includes current and non-current portions. |
For the year ended December 31, 2010,2011, we were not a party to any forward-exchange contracts, and no forward-exchange contracts were outstanding as at March 22, 2011.27, 2012.
Liquidity Risk
Liquidity risk is the risk that we will not be able to meet our financial obligations as they become due. As indicated in the Capital Disclosures section above, we manage this risk through the management of our capital structure. We also manage liquidity risk by continuously monitoring actual and projected cash flows. The Board of Directors reviews and approves our operating and capital budgets, as well as any material transactions out of the ordinary course of business. We have adopted an investment policy in respect of the safety and preservation of our capital to ensure our liquidity needs are met. The instruments are selected with regard to the expected timing of expenditures and prevailing interest rates.
Credit Risk
Financial instruments that potentially subject the Company to concentrations of creditCredit risk consist primarily of cash and cash equivalents, restricted cash and accounts receivable. Cash and cash equivalents and restricted cash balances are maintained with high-credit quality financial institutions. Also, no accounts receivable balance due to the Company that is past due as at December 31, 2010 is significant. Consequently, management considers the risk of non-performance relatedan unexpected loss if a customer or counterparty to a financial instrument fails to meet its contractual obligations. We regularly monitor credit risk exposure and take steps to mitigate the likelihood of this exposure resulting in losses. Our exposure to credit risk currently relates to cash and cash equivalents, to trade and other receivables and to restricted cash. We invest our available cash in amounts that are readily convertible to known amounts of cash and deposit our cash balances with financial institutions that have a minimum rating of “A3”.
As at December 31, 2011, trade accounts receivable to be minimal.for an amount of approximately $7,415,000 were with two external customers or partners.
As at December 31, 2011, no trade accounts receivable were past due or impaired.
Generally, we do not require collateral or other security from customers for trade accounts receivable; however, credit is extended following an evaluation of creditworthiness. In addition, we perform ongoing credit reviews of all our customers and establish an allowance for doubtful accounts when accounts are determined to be uncollectible.
Related Party Transactions and Off-Balance Sheet Arrangements
We did not enter into transactions with any related parties during the year ended December 31, 2010.2011.
As at December 31, 2010,2011, we did not have any interests in variable interest entities or any other off-balance sheet arrangements.
Item 6. Directors, Senior Management and Employees
A. Directors and senior management
| Item 6. | Directors, Senior Management and Employees |
| A. | Directors and senior management |
The following table sets forth information about our directors and corporate officers as at March 24, 2011.27, 2012.
Name and Place of Residence | Position with Aeterna Zentaris | ||||
|---|---|---|---|---|---|
Aubut, Marcel | Director | ||||
Quebec, Canada | |||||
Blake, Paul | Senior Vice President and Chief Medical Officer | ||||
Pennsylvania, United States | |||||
Dorais, José P. | Director | ||||
Quebec, Canada | |||||
Engel, Juergen | President and Chief Executive Officer and Director | ||||
Alzenau, Germany | |||||
Ernst, Juergen | Chairman of the Board and Director | ||||
Brussels, Belgium | |||||
Lapalme, Pierre | Director | ||||
Quebec, Canada | |||||
Limoges, Gérard | Director | ||||
Quebec, Canada | |||||
Métivier, Amélie | Assistant Secretary | ||||
Quebec, Canada | |||||
Meyers, Michael | Director | ||||
New York, United States | |||||
Pelliccione, Nicholas | Senior Vice President, Regulatory Affairs and | ||||
New York, United States | Quality Assurance | ||||
Seeber, Matthias | Senior Vice President, Administration and Legal Affairs | ||||
Frankfurt, Germany | |||||
Shapiro, Elliot | Corporate Secretary | ||||
Quebec, Canada | |||||
Turpin, Dennis | Senior Vice President and Chief Financial Officer | ||||
Quebec, Canada | |||||
There are no family relationships among any of the directors or executive officers of the Company and its subsidiaries. The following is a brief biography of each of our directors and senior officers.
Marcel Aubut has served as a director on our Board since 1996. Mr. Aubut is a managing partner of Heenan Blaikie Aubut LLP, a law firm. The countless companies and boards with which Marcel Aubut has been involved with over the years demonstrate his versatility and, above all, his vast experience in the world of business. These include, among others, Atomic Energy of Canada, Olymel L.P. (Olybro), Boralex Power Income Fund, Triton Electronik, Whole Foods Market Canada, Hydro-Québec (Executive Committee), Purolator Courier Ltd., Tremblant Resort, Cinar Inc., La Laurentienne générale, La Laurentienne vie, Investors Group Inc., Transforce Inc., Intra Continental Insurers Ltd., the National Hockey League Pension Society, Boréal Assurances Agricoles Inc.,
Entreprises Premier CDN Ltée, Les Industries Amisco Ltée, Donohue Matane Inc., La Société de développement du Loisir et du Sport du Québec, the Canadian Olympic Committee, the Canadian Olympic Foundation, member of VANOC'sVANOC’s Audit Committee, Governance and Ethics Committee and Observer Team, Sodic Québec Inc., Innovatech Québec, Textile Dionne, Canada'sCanada’s Sports Hall of Fame, the Committee for the 2002 Quebec City Olympic Games Bid, the Committee for the 2015 Toronto Pan American Games Bid, la Fondation Nordiques, etc. He has also presided over the establishment of numerous industrial projects in the greater region of Quebec City.
Paul Blake was appointed our Senior Vice President and Chief Medical Officer in August 2007. Prior to joining us, Dr. Blake was Chief Medical Officer of Avigenics, Inc. since January 2007. In 2005, he was Senior Vice President, Clinical Research and Regulatory Affairs at Cephalon, Inc. before being promoted to Executive Vice President, Worldwide Medical & Regulatory Operations. From 1992 to 1998, he held the position of Senior Vice President and Medical Director, Clinical Research and Development at SmithKline Beecham Pharmaceuticals (now GSK). Dr. Blake earned a medical degree from the London University, Royal Free Hospital. He was elected Fellow of the American College of Clinical Pharmacology, Fellow of the Faculty of Pharmaceutical Medicine, Royal College of Physicians in the UK, and he is a Fellow of the Royal College of Physicians in the UK. Dr. Blake is also a Director of Oxford BioMedica (non-executive) and member of its remuneration committee.
José P. Dorais has served as a director on our Board since 2006. Mr. Dorais is a partner of Miller Thomson Pouliot LLP where he mainly practices administrative, corporate, business and international trade law. Over his 35-year career, he has worked in both the private and public sectors; in the latter he acted as Secretary to the Minister of Justice and as Secretary of the consulting committee on the Free Trade Agreement for the Quebec Provincial Government. Mr. Dorais has been a member of numerous boards of directors, including the Société des Alcools du Québec, Biochem Pharma and St-Luc Hospital in Montreal. He is now a member of the Board of Alliance Films the Société Générale de FinancementInc., Investissement Québec and Chairman of the Board of Recyc-Québec.Foster Wheeler Énergie Inc. He holds a law degree from the University of Ottawa and is a member of the Quebec Bar.Barreau du Québec.
Juergen Engel was appointed President and Chief Executive Officer, effective September 1, 2008, after having up to such time served as our Executive Vice President and Chief Scientific Officer. He became a director on our Board in 2003. Dr. Engel has been Managing Director of AEZS Germany, the Company'sCompany’s principal operating subsidiary, since the beginning of 2001. Before that, he was in charge of all research and development activities of ASTA Medica AG. He is member of the Advisory Board of GIG, Berlin and ElexoPharm, Saarbrücken. He served as a member of the Board of Directors of Isotechnika Pharma Inc until February 2011.
Juergen Ernst was appointed Chairman of the Board, effective August 13, 2007, after having been Interim President and Chief Executive Officer from April 11, 2008 until August 31, 2008. He has served as a director on our Board since 2005. A seasoned executive with more than 20 years of pharmaceutical industry expertise mainly in the field of corporate development and pharmaceutical product marketing, Mr. Ernst was worldwide General Manager, Pharmaceutical Sector of Solvay S.A., before retiring in 2004. He has served as a director of Pharming Group N.V., Leiden, Netherlands since April 15, 2009.
Pierre Lapalme has served as a director on our Board since December 2009. Mr. Lapalme has over the course of his career held numerous senior management positions in various global life sciences companies. He is former Senior Vice-President, Sales and Marketing for Ciba-Geigy (which subsequently became Novartis) and former Chief Executive Officer and Chairman of the Board of Rhone-Poulenc Pharmaceuticals Inc. in Canada and in North America, as well as Executive Vice-President and Chief Executive Officer of Rhone-Poulenc-Rorer Inc. North America (now sanofi-aventis), where he supervised the development, manufacturing and sales of prescription products in North and Central America. Mr. Lapalme served on the Board of the National
Pharmaceutical Council USA and was a Board member of the Pharmaceutical Manufacturers Association of Canada, where he
played a leading role in reinstituting patent protection for pharmaceuticals. Until recently, he was Board member and Chairman of the Board of Sciele Pharma Inc. which was acquired by Shionogi and Co. Ltd. Mr. Lapalme is currently Chairman of the Board of Biomarin Inc., Chairman of the Board of Pediapharm Inc. and, Board member of Algorithme Pharma Inc. and Insy’s Therapeutics Inc., a Phoenix Arizona based specialty pharma Co. He studied at the University of Western Ontario and at INSEAD, France.
Gérard Limoges has served as a director on our Board since 2004. Mr. Limoges served as the Deputy Chairman of Ernst & Young LLP Canada until his retirement in September 1999. After a career of 37 years with Ernst & Young, Mr. Limoges has been devoting his time as a director of a number of companies. Mr. Limoges began his career with Ernst & Young in Montreal in 1962. After graduating from the Management Faculty of Université de Montréal (HEC Montréal) in 1966, he wrote the CICA exams the same year (Honors: Governor General'sGeneral’s Gold Medal for the highest marks in Canada and Gold Medal of the Ordre des Comptables Agréés du Québec). He became a chartered accountant in 1967 and partner of Ernst & Young in 1971. After practicing as auditor since 1962 and partner since 1971, he was appointed Managing Partner of the Montreal Office in 1979 and Chairman for Quebec in 1984 when he also joined the National Executive Committee. In 1992, he was appointed Vice-chairman of Ernst & Young Canada and the following year, Deputy Chairman of the Canadian firm. After retirement from public practice at the end of September 1999, he was appointed Trustee of the School board of Greater Montreal (1999), member of the Quebec Commission on Health Care and Social Services (2000-2001) and special advisor to the Rector of the University de Montreal and affiliate schools (2000-2003). Mr. Limoges, is currently participating, at the request of the Board of the University of Montreal, has participated in the selection of the Dean of the Faculty of Medicine.Medicine in 2011. Mr. Limoges is a board member or trustee and chairman of the audit committees of the following public companies: Aeterna Zentaris Inc. (TSX and NASDAQ), Atrium Innovations Inc. (TSX), Hartco Inc. (TSX), and Hart Stores Inc. (TSX) and Homburg Canada Real Estate Investment Trust.(TSXV). He is also a board member of various private companies and charities. Mr. Limoges became an FCA in 1984 and received the Order of Canada in 2002.
Pierre MacDonald has served as a director on our Board since November 2000. Mr. MacDonald is President and CEO of MacD Consult Inc., a management consulting firm in international finance and marketing, based in Montreal. He served as the Senior Vice President for Eastern Canada for Bank of Montreal, a position which involved the review and evaluation of the financial statements and creditworthiness of borrowers in a wide variety of industries. In December 1995, he was elected to the National Assembly of Quebec and became Minister of International Trade and Technology. He was also named Vice Chairman of the Treasury Board of the Government of Quebec. He also served as the Chairman of the Audit Committee of Teleglobe Inc. for six years. Mr. MacDonald received Bachelor of Arts, Bachelor of Commerce and Master of Commerce degrees from Laval University in Québec.
Amélie Métivier, Assistant Secretary. Ms. Métivier has served as our Assistant Secretary since April 2009. In addition, Ms. Métivier is currently a lawyer at the law firm of Ogilvy RenaultNorton Rose Canada LLP with a business law and transaction-oriented practice, where she has worked since 2003. She is a member of theBarreau du Québec since 2006, and holds an LL.B. (2004) degree from Université de Montréal.
Michael Meyers, M.P.H. is a co-founding member, Chief Executive Officer and Chief Investment Officer of Arcoda Capital Management LP ("Arcoda"(“Arcoda”), a private investment fund manager. Prior to founding Arcoda in 2007, Mr. Meyers was a Partner and Portfolio Manager of two other money management firms located in New York. Between 2000 and 2003 Mr. Meyers was a Managing Director, Partner and Director of a life sciences venture capital firm located in New York and Zurich, Switzerland. Between 1997 and 2000, Mr. Meyers was Director, Biotechnology and Pharmaceutical Investment Banking at Merrill Lynch & Co. Between 1993 and 1997, Mr. Meyers was Vice President, Health Care Investment Banking at Cowen & Company. Prior to Cowen & Company, Mr. Meyers was Special Assistant to the Chief Executive Officer of St. Barnabas Hospital System. Mr. Meyers began his career as a Biotechnology and Medical Device Research Associate at Hambrecht & Quist in New York. Mr. Meyers holds an M.P.H. in Health Policy and Management from Columbia University and an A.B.
in Biology from Brandeis University in Massachusetts. Mr. Meyers has also served on the Board of Directors of six companies at various times.
Nicholas J. Pelliccione was appointed our Senior Vice President, Regulatory Affairs and Quality Assurance in May 2007. In previous roles, Dr. Pelliccione has been responsible for the clinical/preclinical and CMC regulatory aspects of new drugs in the oncology, anti-infectives, cytokines and cardiovascular therapy areas, leading to several approvals. He served as Senior Vice President, Regulatory and Pharmaceutical Sciences at Chugai Pharma USA from May 2005 until March 2007. Prior to his experience at Chugai, Dr. Pelliccione spent more than 15 years at Schering Plough Corporation holding positions with increasing responsibility from Manager of Regulatory Affairs, Oncology to, prior to his departure, Vice President, Global Regulatory Affairs, Chemistry, Manufacturing and Controls. Dr. Pelliccione holds a Ph.D. in Biochemistry from Mount Sinai School of Medicine, New York and a BS in Chemistry from Polytechnic University.
Matthias Seeber was appointed our Senior Vice President, Administration and Legal Affairs in December 2008. Mr. Seeber served as Managing Director of AEZS Germany since July 2003 up to his most recent appointment. Prior to that, he had assumed the position of Investor Relations Manager of Altana AG, following several years in the banking industry with Deka Investment Management and Dresdner Bank AG. Mr. Seeber is a member of the Deutsche Vereinigung für Finanzanalyse und Asset Management (DVFA/CEFA). He obtained his M.B.A. from George Mason University Graduate School of Business Administration in the United States.
Elliot Shapiro was appointed our Corporate Secretary in April 2009. In addition, Mr. Shapiro is currently a partner and a lawyer at the law firm of Ogilvy RenaultNorton Rose Canada LLP with a business law and transaction-oriented practice, where he has worked since 1999. He has been a member of theBarreau du Québec since 2000. Mr. Shapiro holds B.C.L. (1999), LL.B. (1999) and B.A. (1993) degrees from McGill University.
Dennis Turpin was appointed our Senior Vice President and Chief Financial Officer in August 2007. Prior to that, he served as our Vice President and Chief Financial Officer since June 1999. Mr. Turpin joined Aeterna Zentaris in August 1996 as Director of Finance. Prior to that, he was Director in the tax department at Coopers Lybrand, now PricewaterhouseCoopers, from 1988 to 1996 and worked as an auditor from 1985 to 1988. Mr. Turpin earned his Bachelor'sBachelor’s degree in Accounting from Laval University in Québec. He obtained his license in accounting in 1985 and became a chartered accountant in 1987.
B. Compensation
| B. | Compensation |
Our executive officers are generally paid in their home country’s currency. Unless otherwise indicated, all directors’ and executive compensation information included in this document is presented in US dollars and, to the extent a director or officer has been paid in a currency other than US dollars (Canadian dollars or euros), the amounts have been converted from such person’s home country currency to US dollars based on the following average exchange rates: for the financial year ended December 31, 2011: €1.000 = US$1.3919 and CAN$1.000 = US$1.0111; for the financial year ended December 31, 2010: €1.000 = US$1.326 and CAN$1.000 = US$0.970; and for the financial year ended December 31, 2009: €1.000 = US$1.388 and CAN$1.000 = US$0.876.
| 1. | Compensation of Outside Directors |
The compensation paid to the Company'sCompany’s directors is designed to (i) attract and retain the most qualified people to serve on the Board and its committees, (ii) align the interests of the Company'sCompany’s directors with those of its shareholders, and (iii) provide appropriate compensation for the risks and responsibilities related to being an effective director. This compensation is recommended to the Board by the Corporate Governance, Nominating and Human Resources Committee (the "Governance Committee"“Governance Committee”). During the most recently completed financial year, theThe Governance Committee wasis composed of three (3) directors, each of whom is independent, namely Messrs. Pierre MacDonald, José P. Dorais (Chair), Juergen Ernst and Juergen Ernst.Gérard Limoges. One of the members of the Governance Committee, Juergen Ernst, is Chairman of the Board.
The Board has adopted a formal mandate for the Governance Committee, which is available on our website at www.aezsinc.com. The mandate of the Governance Committee provides that it is responsible for (i) assisting the Board in developing our approach to corporate governance issues, (ii) proposing new Board nominees, (iii) assessing the effectiveness of the Board and its committees,
their respective chairs and individual directors and (iv) making recommendations to the Board with respect to directors'directors’ compensation.
In light of prevailing economic and market conditions, as well as the various ongoing cost-saving measures implemented by the Company in the past two years, the Governance Committee recommended and the Board approved reduction to directors' and committee members' retainers and attendance fees, such reductions having taken effect as at January 1, 2010.
We did not retainemploy the services of any external compensation consultant in or with respect to the financial year ended December 31, 2010.2011.
Annual Retainers and Attendance Fees
Annual retainers and attendance fees are paid on a quarterly basis to the members of the Board who are not employees of the Company or its subsidiaries ("(“Outside Directors"Directors”) as described in the table below.
| Type of Compensation | Annual compensation for the year (in units of home country currency) | |||
|---|---|---|---|---|
Chairman’s Retainer | 45,000 | |||
Vice | 15,000 | |||
Board Retainer | 15,000 | |||
Board Meeting Attendance Fees | 1,000 per meeting | |||
Audit Committee Chair Retainer | 15,000 | |||
Audit Committee Member Retainer | 4,000 | |||
Audit Committee Meeting Attendance Fees | 1,000 | |||
Governance Committee Chair Retainer | 12,000 | |||
Governance Committee Member Retainer | 2,000 | |||
Governance Committee Meeting Attendance Fees | 1,000 | |||
| (1) | There is currently no Vice Chairman of the Board. |
All amounts in the above table are paid to Board and committee members in their home country currency.
The President and Chief Executive Officer is the only member of the Board who is not an Outside Director. Therefore, he is not compensated in his capacity as a director. The Chairman is an Outside Director and is compensated as such. Outside Directors are reimbursed for travel and other out-of-pocket expenses incurred in attending Board or committee meetings.
Outstanding Option-Based Awards and Share-Based Awards
The following table shows all awards outstanding to each Outside Director up to the end of the financial year ending and as at December 31, 2010:2011:
| | Option-based Awards | Share-based Awards | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Name | Issuance Date (mm-dd-yyyy) | Number of Securities Underlying Unexercised Options(1) (#) | Option Exercise Price (CAN$) | Option Expiration Date (mm-dd-yyyy) | Value of Unexercised In-the- money Options(2) (CAN$) | Issuance Date (mm-dd-yyyy) | Number of Shares or Units of Shares that have Not Vested (#) | Market or Payout Value of Share-based Awards that have Not Vested ($) | |||||||||||||||||
Aubut, Marcel | 12-04-2001 | 5,000 | 6.18 | 12-31-2011 | — | — | — | — | |||||||||||||||||
| 12-16-2002 | 15,000 | 3.68 | 12-15-2012 | — | — | — | — | ||||||||||||||||||
| 12-11-2003 | 30,000 | 1.74 | 12-10-2013 | — | — | — | — | ||||||||||||||||||
| 12-14-2004 | 15,000 | 5.83 | 12-13-2014 | — | — | — | — | ||||||||||||||||||
| 12-13-2005 | 15,000 | 3.53 | 12-12-2015 | — | — | — | — | ||||||||||||||||||
| 01-04-2007 | 5,000 | 4.65 | 01-03-2017 | — | — | — | — | ||||||||||||||||||
| 12-11-2007 | 25,000 | 1.82 | 12-10-2017 | — | — | — | — | ||||||||||||||||||
| 12-08-2008 | 15,000 | 0.55 | 12-08-2018 | 17,550 | — | — | — | ||||||||||||||||||
| 12-09-2009 | 20,000 | 0.95 | 12-08-2019 | 15,400 | — | — | — | ||||||||||||||||||
| 12-08-2010 | 30,000 | 1.52 | 12-07-2020 | 6,000 | |||||||||||||||||||||
Dorais, José P. | 12-08-2010 | 30,000 | 1.52 | 12-07-2020 | 6,000 | — | — | — | |||||||||||||||||
Ernst, Juergen | 02-25-2005 | 15,000 | 5.09 | 02-24-2015 | — | — | — | — | |||||||||||||||||
| 12-13-2005 | 15,000 | 3.53 | 12-12-2015 | — | — | — | — | ||||||||||||||||||
| 01-04-2007 | 5,000 | 4.65 | 01-03-2017 | — | — | — | — | ||||||||||||||||||
| 12-11-2007 | 25,000 | 1.82 | 12-10-2017 | — | — | — | — | ||||||||||||||||||
| 11-14-2008 | 100,000 | 0.65 | 11-13-2018 | 107,000 | — | — | — | ||||||||||||||||||
| 12-08-2008 | 15,000 | 0.55 | 12-08-2018 | 17,550 | — | — | — | ||||||||||||||||||
| 12-09-2009 | 20,000 | 0.95 | 12-08-2019 | 15,400 | — | — | — | ||||||||||||||||||
| 12-08-2010 | 30,000 | 1.52 | 12-07-2020 | 6,000 | — | — | — | ||||||||||||||||||
Lapalme, Pierre | 12-09-2009 | 20,000 | 0.95 | 12-08-2019 | 15,400 | — | — | — | |||||||||||||||||
| 12-08-2010 | 30,000 | 1.52 | 12-07-2020 | 6,000 | — | — | — | ||||||||||||||||||
Limoges, Gérard | 12-14-2004 | 15,000 | 5.83 | 12-13-2014 | — | — | — | — | |||||||||||||||||
| 12-13-2005 | 15,000 | 3.53 | 12-12-2015 | — | — | — | — | ||||||||||||||||||
| 01-04-2007 | 5,000 | 4.65 | 01-03-2017 | — | — | — | — | ||||||||||||||||||
| 12-11-2007 | 25,000 | 1.82 | 12-10-2017 | — | — | — | — | ||||||||||||||||||
| 12-08-2008 | 15,000 | 0.55 | 12-08-2018 | 17,550 | — | — | — | ||||||||||||||||||
| 12-09-2009 | 20,000 | 0.95 | 12-08-2019 | 15,400 | — | — | — | ||||||||||||||||||
| 12-08-2010 | 30,000 | 1.52 | 12-07-2020 | 6,000 | — | — | — | ||||||||||||||||||
| | Share-based Awards | Share-based Awards | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Name | Issuance Date (mm-dd-yyyy) | Number of Securities Underlying Unexercised Options(1) (#) | Option Exercise Price (CAN$) | Option Expiration Date (mm-dd-yyyy) | Value of Unexercised In-the- money Options(2) (CAN$) | Issuance Date (mm-dd-yyyy) | Number of Shares or Units of Shares that have Not Vested (#) | Market or Payout Value of Share-based Awards that have Not Vested ($) | |||||||||||||||||
MacDonald, Pierre | 12-04-2001 | 5,000 | 6.18 | 12-31-2011 | — | — | — | — | |||||||||||||||||
| 12-16-2002 | 24,000 | 3.68 | 12-15-2012 | — | — | — | — | ||||||||||||||||||
| 12-11-2003 | 30,000 | 1.74 | 12-10-2013 | — | — | — | — | ||||||||||||||||||
| 12-14-2004 | 15,000 | 5.83 | 12-13-2014 | — | — | — | — | ||||||||||||||||||
| 12-13-2005 | 15,000 | 3.53 | 12-12-2015 | — | — | — | — | ||||||||||||||||||
| 01-04-2007 | 5,000 | 4.65 | 01-03-2017 | — | — | — | — | ||||||||||||||||||
| 12-11-2007 | 25,000 | 1.82 | 12-10-2017 | — | — | — | — | ||||||||||||||||||
| 12-08-2008 | 15,000 | 0.55 | 12-08-2018 | 17,550 | — | — | — | ||||||||||||||||||
| 12-09-2009 | 20,000 | 0.95 | 12-08-2019 | 15,400 | — | — | — | ||||||||||||||||||
| 12-08-2010 | 30,000 | 1.52 | 12-07-2020 | 6,000 | — | — | — | ||||||||||||||||||
Name | Option-based Awards | Share-based Awards | ||||||||||||||
Issuance Date (mm-dd- yyyy) | Number of Securities Underlying Unexercised Options(1) (#) | Option Exercise Price (CAN$ or | Option Expiration (mm-dd- yyyy) | Value of Unexercised Options(2) (CAN$ or US$) | Issuance Date (mm-dd- yyyy) | Number of Shares or Units of that have Not Vested (#) | Market or Payout Value of Share- Awards that have ($) | |||||||||
Aubut, Marcel | 12-16-2002 | 15,000 | CAN$3.68 | 12-15-2012 | — | — | — | — | ||||||||
| 12-11-2003 | 30,000 | CAN$1.74 | 12-10-2013 | — | — | — | — | |||||||||
| 12-14-2004 | 15,000 | CAN$5.83 | 12-13-2014 | — | — | — | — | |||||||||
| 12-13-2005 | 15,000 | CAN$3.53 | 12-12-2015 | — | — | — | — | |||||||||
| 01-04-2007 | 5,000 | CAN$4.65 | 01-03-2017 | — | — | — | — | |||||||||
| 12-11-2007 | 25,000 | CAN$1.82 | 12-10-2017 | — | — | — | — | |||||||||
| 12-08-2008 | 15,000 | CAN$0.55 | 12-08-2018 | CAN$15,150 | — | — | — | |||||||||
| 12-09-2009 | 20,000 | CAN$0.95 | 12-08-2019 | CAN$12,200 | — | — | — | |||||||||
| 12-08-2010 | 30,000 | CAN$1.52 | 12-07-2020 | CAN$1,200 | — | — | — | |||||||||
| 12-07-2011 | 50,000 | US$1.74 | 12-06-2021 | — | — | — | — | |||||||||
Dorais, José P. | 12-08-2010 | 30,000 | CAN$1.52 | 12-07-2020 | CAN$1,200 | — | — | — | ||||||||
| 12-07-2011 | 50,000 | US$1.74 | 12-06-2021 | — | — | — | — | |||||||||
Ernst, Juergen | 02-25-2005 | 15,000 | CAN$5.09 | 02-24-2015 | — | — | — | — | ||||||||
| 12-13-2005 | 15,000 | CAN$3.53 | 12-12-2015 | — | — | — | — | |||||||||
| 01-04-2007 | 5,000 | CAN$4.65 | 01-03-2017 | — | — | — | — | |||||||||
| 12-11-2007 | 25,000 | CAN$1.82 | 12-10-2017 | — | — | — | — | |||||||||
| 11-14-2008 | 100,000 | CAN$0.65 | 11-13-2018 | CAN$91,000 | — | — | — | |||||||||
| 12-08-2008 | 15,000 | CAN$0.55 | 12-08-2018 | CAN$15,150 | — | — | — | |||||||||
| 12-09-2009 | 20,000 | CAN$0.95 | 12-08-2019 | CAN$12,200 | — | — | — | |||||||||
| 12-08-2010 | 30,000 | CAN$1.52 | 12-07-2020 | CAN$1,200 | — | — | — | |||||||||
| 12-07-2011 | 50,000 | US$1.74 | 12-06-2021 | — | — | — | — | |||||||||
Lapalme, Pierre | 12-09-2009 | 20,000 | CAN$0.95 | 12-08-2019 | CAN$12,200 | — | — | — | ||||||||
| 12-08-2010 | 30,000 | CAN$1.52 | 12-07-2020 | CAN$1,200 | — | — | — | |||||||||
| 12-07-2011 | 50,000 | US$1.74 | 12-06-2021 | — | — | — | — | |||||||||
Limoges, Gérard | 12-14-2004 | 15,000 | CAN$5.83 | 12-13-2014 | — | — | — | — | ||||||||
| 12-13-2005 | 15,000 | CAN$3.53 | 12-12-2015 | — | — | — | — | |||||||||
| 01-04-2007 | 5,000 | CAN$4.65 | 01-03-2017 | — | — | — | — | |||||||||
| 12-11-2007 | 25,000 | CAN$1.82 | 12-10-2017 | — | — | — | — | |||||||||
| 12-08-2008 | 15,000 | CAN$0.55 | 12-08-2018 | CAN$15,150 | — | — | — | |||||||||
| 12-09-2009 | 20,000 | CAN$0.95 | 12-08-2019 | CAN$12,200 | — | — | — | |||||||||
| 12-08-2010 | 30,000 | CAN$1.52 | 12-07-2020 | CAN$1,200 | — | — | — | |||||||||
| 12-07-2011 | 50,000 | US$1.74 | 12-06-2021 | — | — | — | — | |||||||||
Meyers, Michael | 05-27-2011 | 20,000 | US$2.36 | 05-26-2021 | — | — | — | — | ||||||||
12-07-2011 | 40,000 | US$1.74 | 12-06-2021 | — | — | — | — | |||||||||
| (1) | The number of securities underlying unexercised options represent all awards outstanding as at December 31, 2011. |
| (2) | “Value of unexercised in-the-money options” at financial year-end is calculated based on the difference between the closing prices of the common shares on the TSX or NASDAQ, as applicable, on the last trading day of the fiscal year (December 30, 2011) of CAN$1.56 and US$1.54, respectively, and the exercise price of the options, multiplied by the number of unexercised options. |
See "Summary“Summary of the Stock Option Plan"Plan” below for more details on the Stock Option Plan (as defined below).
Total Compensation of Outside Directors
The table below summarizes the total compensation earned by the Outside Directors during the financial year ended December 31, 20102011 (all amounts are in US dollars):
| | Fees earned ($) | | | | | | | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Share- based Awards ($) | Option- based Awards(2) ($) | Non-Equity Incentive Plan Compensation ($) | Pension Value ($) | All Other Compensation(3) ($) | | |||||||||||||||||||
| | Total ($) | ||||||||||||||||||||||||
| Name | Retainer(1) | Attendance(1) | |||||||||||||||||||||||
Aubut, Marcel | 14,550 | 3,395 | — | 35,793 | — | — | — | 53,738 | |||||||||||||||||
Byorum, Martha(4) | 6,942 | 2,000 | — | — | — | — | — | 8,942 | |||||||||||||||||
Dorais, José P. | 16,490 | 6,305 | — | 35,793 | — | — | — | 58,588 | |||||||||||||||||
Ernst, Juergen | 82,212 | 7,956 | — | 35,793 | — | — | 2,652 | 128,613 | |||||||||||||||||
Lapalme, Pierre(5) | 18,430 | 7,275 | — | 35,793 | — | — | 1,940 | 63,438 | |||||||||||||||||
Laurin, Pierre(6) | 6,025 | 2,910 | — | — | — | — | — | 8,935 | |||||||||||||||||
Limoges, Gérard | 29,100 | 8,245 | — | 35,793 | — | — | 1,940 | 75,078 | |||||||||||||||||
MacDonald, Pierre(7) | 30,070 | 5,820 | — | 35,793 | — | — | — | 71,683 | |||||||||||||||||
Martin, Gerald J.(8) | 5,481 | 2,000 | — | — | — | — | — | 7,481 | |||||||||||||||||
| Name | Fees earned ($) | Share- based | Option- based | Non-Equity Incentive Plan | Pension Value | All Other Compensation(3) | Total | |||||||||
| Awards | Awards(2) | Compensation | ||||||||||||||
| Retainer(1) | Attendance(1) | |||||||||||||||
| ($) | ($) | ($) | ($) | ($) | ($) | |||||||||||
Aubut, Marcel | 15,166 | 4,044 | — | 63,000 | — | — | — | 82,210 | ||||||||
Dorais, José P.(4) | 27,298 | 8,088 | — | 63,000 | — | — | 506 | 98,892 | ||||||||
Ernst, Juergen | 86,299 | 9,743 | — | 63,000 | — | — | 5,568 | 164,610 | ||||||||
Lapalme, Pierre | 19,210 | 9,099 | — | 63,000 | — | — | — | 91,309 | ||||||||
Limoges, Gérard(5) | 32,354 | 11,122 | — | 63,000 | — | — | — | 106,476 | ||||||||
MacDonald, Pierre(6) | 11,883 | 3,033 | — | — | — | — | 506 | 15,422 | ||||||||
Meyers, Michael(7) | 13,250 | 4,500 | — | 85,800 | — | — | 500 | 104,050 | ||||||||
| (1) | These amounts represent the portion paid in cash to the Outside Directors and are paid in each director’s home country currency. |
| (2) | The value of option-based awards represents the closing price of the common shares on NASDAQ at the date of grant (US$1.74 for options granted on December 7, 2011 and US$2.36 for options granted to Michael Meyers on May 27, 2011) multiplied by the Black-Scholes factor as at such date (72.41% for options granted on December 7, 2011 and 75.00% for options granted to Michael Meyers on May 27, 2011) and the number of stock options granted on such date. |
| (3) | These amounts represent fees paid in cash for special tasks or overseas travelling and are also paid in each director’s home country currency. |
| (4) | José P. Dorais was appointed Chairman of the Governance Committee on May 18, 2011. |
| (5) | Gérard Limoges has been a member of the Governance Committee since May 18, 2011. |
| (6) | Pierre MacDonald was a director of the Company from 2001 to May 18, 2011. He did not stand for re-election as director at the annual and special meeting of shareholders held on May 18, 2011 and therefore ceased to be a director as of such date. |
| (7) | Michael Meyers was appointed director on March 22, 2011 and has been a member of the Audit Committee since May 18, 2011. |
During the financial year ended December 31, 2010,2011, the Company paid an aggregate amount of $261,738$262,169 to all of its Outside Directors for services rendered in their capacity as directors, excluding reimbursement of out-of-pocket expenses and the value of option-based awards granted in 2010. Outside Directors are paid in their home country currency and are reimbursed for travel and other out-of-pocket expenses incurred while attending Board or committee meetings.2011.
| 2. | Compensation of Executive Officers |
The mandate of the Governance Committee provides that it is responsible for taking all reasonable measures to ensure that appropriate human resources systems and procedures, such as hiring policies, competency profiles, training policies and compensation structures are in place so that we can attract, motivate and retain the quality of personnelsenior management required to meet our business objectives.
The Governance Committee also assists the Board in discharging its responsibilities relating to executive and other human resources hiring, assessment, compensation and succession planning matters.
Thus, the Governance Committee recommends the appointment of senior officers, including the terms and conditions of their appointment and termination, and reviews the evaluation of the performance of oursuch senior officers, including recommending their compensation.compensation and overseeing risk identification and management in relation to executive compensation policies and practices. The Board, which includes the members of the Governance Committee, reviews the Chief Executive Officer's corporate goals and objectives that are set annually and evaluates histhe Chief Executive Officer’s performance and compensation in light of such goals and objectives.
The Governance Committee recognizes that the industry and competitive environment in which the Company operates requires a balanced level of risk-taking to promote and achieve the performance expectations required to provide its shareholders with a fair return. However, the executive compensation program should not encourage senior executives in taking excessive risk. In this regard, the Governance Committee recommends the implementation of compensation methods that tie a portion of senior executive compensation to each of the short-term and longer-term performance of the Company and each executive officer and that take into account the advantages and risks associated with such compensation methods. The Governance Committee is also responsible for creating compensation policies that are intended to reward the creation of shareholder value while reflecting a balance between the short-term and longer-term performance of the Company and of each executive officer.
Compensation Discussion & Analysis
Compensation Philosophy and Objectives
The Company'sCompany’s executive compensation program is designed to attract, motivate and retain high performing senior executives, encourage and reward superior performance and align the executives'executives’ interests with those of our shareholders by:
| — | providing the opportunity for an executive to earn compensation that is competitive with the compensation received by executives employed by a group of comparable North American companies; |
| — | providing executives with an equity-based incentive plan, namely a stock option plan; |
| — | aligning employee compensation with company corporate objectives; and |
| — | attracting and retaining highly qualified individuals in key positions. |
Risk Assessment of Executive Compensation Program
The Board, through the Governance Committee, oversees the implementation of compensation methods that tie a portion of executive compensation to each of the short-term and longer-term performance of the Company and of each executive officer and that take into account the advantages and risks associated with such compensation methods. In addition, the Board oversees the creation of compensation policies that are intended to reward the creation of shareholder value while reflecting a balance between the short-term and longer-term performance of the Company and of each executive officer.
The Governance Committee has considered in general terms the concept of risk as it relates to the Company’s executive compensation program.
Base salaries are fixed in amount to provide a steady income to the executive officers regardless of share price and thus do not encourage or reward risk-taking to the detriment of other important business metrics. The variable compensation elements (annual bonuses and stock options) are designed to reward both short and long-term performance. For short-term performance, an annual bonus is awarded based on the level of attainment of specific operational and corporate goals that the Governance Committee believes to be challenging, yet does not encourage unnecessary or excessive risk-taking (for 2011, such operational and corporate goals are listed in the table below). While the Company’s bonus payments are generally based on annual results, their value is generally capped and represents only a portion of each individual’s overall total compensation opportunities. Finally, a significant portion of the compensation provided to the Company’s executive officers is in the form of equity awards under the long-term incentive compensation plan that further align executives’ interests with those of shareholders. The Governance Committee believes that these awards do not encourage unnecessary or excessive risk-taking since the ultimate value of the awards is tied to the Company’s share price, and since grants under the long-term incentive compensation plan are subject to long-term vesting schedules to help ensure that executives generally have significant value tied to long-term share price performance.
The Governance Committee believes that the variable compensation elements (annual bonuses and stock options) represent a percentage of overall compensation that is competitivesufficient to motivate the Company’s executive officers to produce superior short-term and long-term corporate results, while the fixed compensation element (base salary) is also sufficient to discourage executive officers from taking unnecessary or excessive risks. The Governance Committee and the Board also generally have the discretion to adjust annual bonuses and stock option grants based on individual performance and any other factors it may determine to be appropriate in the circumstances.
Such factors may include, where necessary or appropriate, the level of risk-taking a particular executive officer may have engaged in during the preceding year.
Based on the foregoing, the Governance Committee has not identified any risks associated with the Company’s executive compensation receivedprogram that are reasonably likely to have a material adverse effect on the Company. The Governance Committee believes that the Company’s executive compensation program does not encourage or reward any unnecessary or excessive risk-taking behaviour.
The Board, based on the Governance Committee’s recommendation, set goals for the Company at the end of 2010, which constitute the performance objectives for the Chief Executive Officer and the Company’s other executive officers. The performance objectives are not established for individual executive officers but rather by executives employedfunction(s) exercised within the Company, for many of which the Named Executive Officers are responsible.
In December 2011, the Governance Committee determined that the Company’s executive officers met or exceeded each of the objectives set forth in the table below as follows:
Objectives for 2011 | Results for 2011 | |
| Clinical | ||
Advancement of product pipeline • Perifosine • AEZS-130 • AEZS-108 | • Completed recruitment of metastatic colorectal Phase 3 study (468 patients) • Supported Phase 3 study in MM by opening several centres outside North America • Reported positive Phase 3 data as diagnostic in AGHD • Reported positive final Phase 2 data in endometrial cancer • Initiated Phase 1/2 study in refractory bladder cancer in the United States | |
| Regulatory | ||
| Optimize regulatory strategy with lead authorities | • Obtained parallel scientific advice for a pivotal study for AEZS-108 in endometrial cancer by both the Food and Drug Administration (FDA) and the EMA | |
| Business Development/Alliance Management: | ||
| Expand partner portfolio for Perifosine in non-core territories | • Signed license agreements with Yakult (for Japan) and Hikma (for MENA region) | |
| Ensure steady supply of Cetrotide® to Merck Serono | • Exceeded pre-defined threshold of Cetrotide® sales triggering further milestone payment by Cowen Healthcare Royalty Partners, L. P. | |
| Manage alliances with existing drug development and commercialization partners actively | • Continued to maintain excellent relations with alliance partners | |
| Financial | ||
| Pursue dilutive and non-dilutive financing alternatives to ensure year-end 2011 cash and cash equivalents in excess of $30 million | • Completed two successful At-the-Market Financings (ATMs) generating proceeds in excess of $36 million • Closed further licensing agreements generating significant upfront payments • Cash and cash equivalents as of December 31, 2011 totalled $46.9 million |
| Investor Relations | ||
| Expand North American research analyst coverage | • Initiation of coverage by several North American institutions including, among others, Oppenheimer & Co Inc., JMP Securities and Needham & Company LLC. | |
| Continue to present at strategic healthcare conferences | • Presented at several strategic healthcare and partnership conferences and continued to build the Company’s investor base | |
| Human Resources | ||
| Maintain high level of motivation at all levels (and sites) of the Company | • Executed effective HR policy comprising both monetary (compensation) and non-monetary (training, etc.) components to ensure both short- and long-term commitment of personnel while generally aligning employee’s and shareholders’ interests • Maintained HR-attrition policy while progressing development pipeline (active headcount virtually unchanged as compared to the prior year) |
The determination of individual performance does not involve quantitative measures using a groupmathematical calculation in which each individual performance objective is given a numerical weight. Instead, the Governance Committee’s determination of comparable North American companies;
While the Company has not formally adopted a stock option plan;
Tablethe Company’s Corporate Secretary each trade relating to the Company’s securities, which would include the entering into of Contentsany such financial instrument or transaction, hedge, swap or forward contract.
Benchmarking
In order to attain our objectives of providing market competitive compensation opportunities, our executive compensation plan, based on a study provided by AON Consulting (and updated annually), is benchmarked against market compensation data gathered from organizations of comparable size andand/or stage of development of other companies with which we competethat the Company competes for executive talent (the "Reference Group"“Reference Group”). We did not, however, pay AON Consulting any fee or other remuneration in 2010.2011. An overview of the characteristics of the Reference Group is provided in the following table:
(In millions of US$)
| (In millions of US$) | Aeterna Zentaris | Survey Reference Group | ||
|---|---|---|---|---|
Location | North America and Europe | North America | ||
Industries | Biopharmaceutical | Biopharmaceutical | ||
Revenues | 63.24(1) | 54.67(2) | ||
Market Capitalization | 104.76 | 309.62 | ||
Net Loss | 24.72(1) | 45.15(2) | ||
| Aeterna Zentaris | Survey Reference Group | |||
Location | North America and Europe | North America | ||
Industries | Biopharmaceutical | Biopharmaceutical | ||
Revenues Last fiscal year | 27.70(1) | 57.02(2) | ||
Market Capitalization As at October 31, 2011 | 175.07 | 290.34 | ||
Net Loss Last fiscal year | 23.22(1) | 19.82(2) |
| (1) | For the year ended December 31, 2010, as presented in our 2010 audited consolidated financial statements, which were presented in conformity with Canadian GAAP. |
| (2) | The Reference Group for the financial year ended December 31, 2011 was selected in October 2011 and these data are based on their most recently completed fiscal year at such time. |
The Reference Group used in respect of the financial year ended December 31, 20102011 was composed of the following companies: Acadia Pharmaceuticals Inc.; Acorda Therapeutics Inc.; Array Biopharma Inc.; Caraco Pharmaceutical Labs; BioSanté Pharmaceuticals, Inc.; Cell Therapeutics Inc.; Enzon Pharmaceuticals Inc.; Genomic Health Inc.; Ista Pharmaceuticals Inc.; Ligand Pharmaceuticals;Pharmaceuticals Inc.; Neurocrine Biosciences Inc.; Nps Pharmaceuticals Inc.; OncoGenex Pharmaceuticals Inc.; Onyx Pharmaceuticals, Inc.; Salix Pharmaceuticals Ltd; Savient Pharmaceuticals Inc.; and Xoma Ltd.
Positioning
The Company'sCompany’s compensation policy is for executive compensation to be generally aligned with the 50th percentile of the Reference Group. The Governance Committee uses discretion and judgment when determining compensation levels as they apply to a specific executive officer. Individual compensation may be positioned above or below median, based on individual experience and performance or other criteria deemed important by the Governance Committee. The total cash target payment for ourthe Company’s executive officers generally falls within the market 50th percentile competitive range.range of the Reference Group.
Compensation Elements
An executive compensation policy has been established to acknowledge and reward the contributions of the executive officers to our success and to ensure competitive compensation, in order that we may benefit from the expertise required to pursue our objectives.
Our executive compensation policy is comprised of both fixed and variable components. The variable components include equity and non-equity incentive plans. Each compensation component is intended to serve a different function, but all elements are intended to work in concert to maximize both company and individual performance by establishing specific, competitive operational and corporate goals and by providing financial incentives to employees based on their level of attainment of these goals.
Our current executive compensation program is comprised of the following four basic components:
| (i) | base salary; |
| (ii) | non-equity incentives – consisting of a cash bonus linked to both individual and corporate performance; |
| (iii) | long-term compensation – consisting of our stock option plan established for the benefit of our directors, executive officers and employees (the “Stock Option Plan”); and |
| (iv) | other elements of compensation – consisting of benefits, perquisites and retirement benefits. |
Base Salary
Salaries of our executive officers are established based on a comparison with competitive benchmark positions. The starting point to determine executive base salaries is the median of executive salaries in the Reference Group.
In determining individual base salaries, the Governance Committee takes into consideration individual circumstances that may include the scope of an executive'sexecutive’s position, the executive'sexecutive’s relevant competencies or experience and retention risk. The Governance Committee also takes into consideration the fulfillment of our corporate objectives as well as the individual performance of the executive.
Short-Term Non-Equity Incentive Compensation
The short-term non-equity incentive compensation plan sets out the allocation of incentive awards based on the financial results and the advancement of ourthe Company’s product developmentpipeline and strategic objectives. These objectives are set at the beginning of each financial year as part of the annual review of corporate strategies.
In the case of executive officers, a program is designed to maximize both corporate and individual performance by establishing specific operational, clinical, regulatory, financial and financialcorporate goals and to provide financial incentives to executive officers based on their level of attainment of these goals. The granting of cash incentives requires the approval of both the Governance Committee and the Board and is based upon an assessment of each individual'sindividual’s performance, as well as the performance of the Company. The underlying objectives are set at the end of each financial year as part of the annual review of corporate strategies.
For the financial year ended December 31, 2010,2011, the Governance Committee recommended, and the Board approved, in the best interests of the Company, given its financial situation, albeit improved as compared to the end of 2009, and also taking into consideration the ongoing cost saving measures implemented by the Company in the past two years, that, to the extent earned, a maximum of 50%70% of an executive officers'officers’ maximum bonus be paid in cash in respect of the year 20102011 (paid in 2011)early 2012), while the remainder of any earned bonus be payable in the form of stock options (as had been done in 2010 as to 50% of any earned bonus and in 2009 as to 100% of any earned bonus) on a dollar-for-dollar basis,, whereby US$1.00 generally equals one stock option, as adjusted based on each senior executive'sexecutive’s individual performance. Theperformance, such that, on a dollar-for-dollar basis, the US$ or C$ amount of the non-cash bonus was determined to be paid in the form of stock options to vest in equal one-third tranches at six-month intervals, with the first one-third to vest on the six-month anniversary of the date of grant, in order to allow these grants to serve their purpose as partial replacement for annual cash bonuses. For the Company'sCompany’s European executives, the number of options was determined to be "grossed-up"“grossed-up” by a multiple of 1.35 to reflect the then prevailing US$ to € exchange rate. See "Summary“Summary of the Stock Option Plan"Plan” below for more details on the Stock Option Plan.
In making decisions related to the short-term non-equity incentive compensation for the Named Executive Officers, other than the President and Chief Executive Officer, the Governance Committee concluded as follows, based on the goals and results for 2011, as described in Section “Risk Assessment of Executive Compensation Program”.
Mr. Turpin’s 2011 goals were aligned with the Company’s overall objectives, with an emphasis on supporting attainment of the financial objectives. Based upon results achieved, the Governance Committee determined that Mr. Turpin’s individual performance exceeded expectations as follows: completed two successful At-the-Market Financings (ATMs) generating proceeds in excess of $36 million as a consequence of which the Company completed 2011 with cash and cash equivalents exceeding significantly the budget amount, and contributed significantly to the achievement of the Company ‘s various goals set in the area of Investor Relations. The Compensation Committee determined that Mr. Turpin’s contributions to the achievement of the Company’s goals merited a cash bonus in an amount of $80,509 and an equity-based bonus in an amount of $34,125 paid through the granting of 34,125 stock options based on 2011 performance.
Mr. Seeber’s 2011 goals were aligned with the Company’s overall objectives, with an emphasis on the achievement of the Company’s strategic milestones. Based upon results achieved, the Governance Committee determined that Mr. Seeber’s individual performance contributed significantly to the achievement of the Company’s various strategic milestones, including in respect of the various goals set in the areas of Business Development/Alliance Management and Human Resources. The Governance Committee determined that Mr. Seeber’s contributions to the achievement of the Company’s goals merited a cash bonus in an amount of $82,818 and an equity-based bonus in an amount of $34,425 paid through the granting of 34,425 stock options based on 2011 performance.
Dr. Blake’s 2011 goals were aligned with the Company’s overall objectives, with an emphasis on overseeing and supporting the attainment of the Company’s clinical and regulatory objectives. Based upon results achieved, the Governance Committee determined that Mr. Blake’s individual performance exceeded expectations, particularly in respect of the advancement of the product pipeline and the optimization of the Company’s regulatory strategy with lead authorities. The Governance Committee determined that Mr. Blake’s contributions to the achievement of the Company’s goals merited a cash bonus in an amount of $89,670 and an equity-based bonus in an amount of $38,430 paid through the granting of 38,430 stock options based on 2011 performance.
Mr. Pellicione’s 2011 goals were aligned with the Company’s overall objectives, with an emphasis on overseeing and supporting the attainment of the Company’s clinical and regulatory objectives. Based upon results achieved, the Governance Committee determined that Mr. Pellicione’s individual performance exceeded expectations, particularly in respect of the advancement of the product pipeline and the optimization of the Company’s regulatory strategy with lead authorities. The Governance Committee determined that Mr. Pellicione’s contributions to the achievement of the Company’s goals merited a cash bonus in an amount of $77,739 and an equity-based bonus in an amount of $33,316 paid through the granting of 33,316 stock options based on 2011 performance.
For the financial year ended December 31, 2010,2011, cash bonuses paid to all of our executive officers under our short-term non-equity incentive compensation plan represented 49%70% of the target payout established by the Governance Committee under such plan. Similarly, stock options granted to our executive officers under the same short-term non-equity incentive compensation plan, in partial
replacement of the remainder of any earned cash bonuses, represented 69.5%30% of the target payout previously established by the Governance Committee.
The total number of stock options granted for 2010 as partial replacement for the remainder of any earned cash bonuses was relatively high since they included an additional bonus award made to the President and Chief Executive Officer and also due to the 1.35 "gross-up" to the base number of options granted to European executives to reflect the US$ to € exchange rate.
Long-termLong-Term Equity Compensation Plan of Executive Officers
The long-term component of the compensation of the Company'sCompany’s executive officers is based exclusively on the Stock Option Plan, which permits the award of a number of options that varies in accordance withbased on the contribution of the officers and their responsibilities. To encourage retention and focus management on developing and successfully implementing the continuing growth strategy of the Company, stock options generally vest over a period of three years, with the first one-third to vest on the one-year anniversary of the date of grant; however, as mentioned in the section above, the vesting schedule for certain of the options granted to senior executives in the financial years ended December 2011, 2010 and 2009 was accelerated from three years to 18 months since the grants were intentedintended to serve as a partial replacement for a certain portion of earned cash bonuses. Stock options are usually granted to executive officers in December of each year.
For the financial year ended December 31, 2011, the Governance Committee recommended, and the Board approved, in the best interests of the Company, that stock options be granted to executive officers under the long-term equity compensation plan, such stock options to vest according to the vesting schedule set forth in the Company’s Stock Option Plan. The Governance Committee considered the performance of the Company and each executive in determining the appropriate level of equity incentive opportunity granted to each executive officer under the long-term equity compensation program in 2011. The Company and the Governance Committee believe that an equitable granting of stock options to senior executives is an important element of the Company’s overall compensation philosophy with a view to ensuring that senior management works to ensure the long-term success and viability of the Company’s business, particularly in light of the length of time required for a biopharmaceutical company such as Aeterna Zentaris to advance its product pipeline from pre-clinical to clinical through the commercialization stages of development.
Based on the foregoing, the Governance Committee approved stock option awards as part of the long-term incentive compensation plan to each NEO on December 7, 2011 as follows:
| Named Executive Officer | Stock Options | |||
| Number granted | Exercise price | |||
Engel, Juergen | 200,000 | US$1.74 | ||
Turpin, Dennis | 70,000 | US$1.74 | ||
Seeber, Matthias | 70,000 | US$1.74 | ||
Blake, Paul | 70,000 | US$1.74 | ||
Pelliccione, Nicholas J. | 70,000 | US$1.74 | ||
Summary of the Stock Option Plan
We established the Stock Option Plan in order to attract and retain directors, executive officers and employees, who will be motivated to work towards ensuring the success of the Company. The Board has full and complete authority to interpret the Stock Option Plan, to establish applicable rules and regulations applying to it and to make all other determinations it deems necessary or useful for the administration of the Stock Option Plan, provided that such interpretations, rules, regulations and determinations are consistent with the rules of all stock exchanges and quotation systems on which our securities are then traded and with all relevant securities legislation.
Individuals eligible to participate under the Stock Option Plan will beare determined from time to time by either the Board or the Governance Committee.
Options granted under the Stock Option Plan may be exercised at any time within a maximum period of ten years following the date of their grant (the "Outside“Outside Expiry Date"Date”). The Board or the Governance Committee, as the case may be, designates, at its discretion, the individuals to whom stock options are granted under the Stock Option Plan and determines the number of common shares covered by each of such optionsoption grants, the grant date, the exercise price of each option, the expiry date, the vesting schedule and any other matter relating thereto, in each case in accordance with the applicable rules and regulations of the regulatory authorities. The price at which the common shares may be purchased may not be lower than the greater of the closing prices of the common shares on the TSX and the Nasdaqor NASDAQ, as applicable, on the last trading day preceding the date of grant of the option. Options granted under the Stock Option Plan generally vest in equal tranches over a three-year period (one-third each year, starting on the first anniversary of the grant date) or as otherwise determined by the Board or the Governance Committee, as the case may be, although, as described above, under section “Short-Term Non-Equity Incentive Compensation”, a certain number of stock options granted to executivesexecutive officers in the past two yearssince 2009 have an accelerated vesting schedule of 18 months.
Unless the Board or the Governance Committee decides otherwise, option holders cease to be entitled to exercise their options under the Stock Option Plan: (i) immediately, in the event an option holder who is an officer or employee resigns or voluntarily leaves his or her employment with the Company or one of its subsidiaries or the employment with the Company or one of its subsidiaries is terminated with cause and, in the case of an optionee who is a non-employee director of the Company or one of its subsidiaries, the date on which such optionee ceases to be a member of the relevant board
of directors; (ii) six months following the date on which employment is terminated as a result of the death of an option holder who is an officer or employee and, in the case of an optionee who is a non-employee director of the Company or one of its subsidiaries, six months following the date on which such optionee ceases to be a member of the relevant board of directors by reason of death; (iii) 30 days following the date on which an option holder'sholder’s employment with the Company or any of its subsidiaries is terminated for a reason other than those mentioned in (i) or (ii) above including, without limitation, upon the disability, long-term illness, retirement or early retirement of the option holder; and (iv) where the option holder is a service supplier, 30 days following the date on which such option holder ceases to act as such, for any cause or reason (each, an "Early“Early Expiry Date"Date”).
The Stock Option Plan also provides that, if the expiry date of an option(s) (whether an Early Expiry Date or an Outside Expiry Date) occurs during a "blackout period"“blackout period” or within the seven business days immediately after a blackout period imposed by the Company, the expiry date will be automatically extended to the date that is seven business days after the last day of the blackout period. For the purposes of the foregoing, "blackout period"“blackout period” means the period during which trading in the Company'sCompany’s securities is restricted in accordance with its corporate policies.
Option holders may not assign their options (nor any interest therein) other than by will or in accordance with the applicable laws of estates and succession.
In the event that, at any time, an offer to purchase is made to holders of all our common shares, notice of such offer shall be given by the Company to each optionee and all unexercised options will become exercisable immediately at their respective exercise prices, but only to the extent necessary to enable optionees to tender their common shares in response to such offer.
The Stock Option Plan currently provides that the following amendments may be made to the Stock Option Planplan upon approval of each of the Board and our shareholders as well as receipt of all required regulatory approvals:
| — | any amendment to Section 3.2 of the Stock Option Plan (which sets forth the limit on the number of options that may be granted to insiders) that would have the effect of permitting, without having to obtain shareholder approval on a “disinterested vote” at a duly convened shareholders’ meeting, the grant of any option(s) under the Stock Option Plan otherwise prohibited by Section 3.2; |
| — | any amendment to the number of securities issuable under the Stock Option Plan (except for certain permitted adjustments, such as in the case of stock splits, consolidations or reclassifications); |
| — | any amendment which would permit any option granted under the Stock Option Plan to be transferable or assignable other than by will or in accordance with the applicable laws of estates and succession; |
| — | the addition of a cashless exercise feature, payable in cash or securities, which does not provide for a full deduction of the number of underlying securities from the Stock Option Plan reserve; |
| — | the addition of a deferred or restricted share unit component or any other provision which results in employees receiving securities while no cash consideration is received by the Company; |
| — | with respect to any option holder whether or not such option holder is an “insider” and except in respect of certain permitted adjustments, such as in the case of stock splits, consolidations or reclassifications: |
| - | any reduction in the exercise price of any option after the option has been granted, or |
| - | any cancellation of an option and the re-grant of that option under different terms; |
| — | any extension to the term of an option beyond its Outside Expiry Date to an option holder who is an “insider” (except for extensions made in the context of a “blackout period”); |
| — | any amendment to the method of determining the exercise price of an option granted pursuant to the Stock Option Plan; |
| — | the addition of any form of financial assistance or any amendment to a financial assistance provision which is more favourable to employees; and |
| — | any amendment to the foregoing amending provisions requiring Board, shareholder and regulatory approvals. |
The Stock Option Plan further currently provides that the following amendments may be made to the Stock Option Plan upon approval of the Board and upon receipt of all required regulatory approvals, but without shareholder approval:
| — | amendments of a “housekeeping” or clerical nature or to clarify the provisions of the Stock Option Plan; |
| — | amendments regarding any vesting period of an option; |
| — | amendments regarding the extension of an option beyond an Early Expiry Date in respect of any option holder, or the extension of an option beyond the Outside Expiry Date in respect of any option holder who is a “non-insider” of the Company; |
| — | adjustments to the number of issuable common shares underlying, or the exercise price of, outstanding options resulting from a split or a consolidation of the common shares, a reclassification, the payment of a stock dividend, the payment of a special cash or non-cash distribution to our shareholders on a pro rata basis provided such distribution is approved by our shareholders in accordance with applicable law, a recapitalization, a reorganization or any other event which necessitates an equitable adjustment to the outstanding options in proportion with corresponding adjustments made to all outstanding common shares; |
| — | discontinuing or terminating the Stock Option Plan; and |
| — | any other amendment which does not require shareholder approval under the terms of the Stock Option Plan. |
The maximum number of common shares issuable under the Stock Option Plan is fixed at 11.4% of the issued and outstanding common shares at any given time, which, as at March 24, 2011,27, 2012, represented 9,720,21312,401,760 common shares. There are currently 6,807,4637,497,634 options outstanding under the Stock Option Plan representing 7.98%approximately 6.9% of all issued and outstanding common shares. Under the Stock Option Plan, (i) the number of securities issued to insiders, at any time, or issuable within any one-year period, under all of the Company'sCompany’s security-based compensation arrangements, cannot exceed 10% of the Company'sCompany’s issued and outstanding securities and (ii) no single option holder may hold options to purchase, from time to time, more than 5% of the Company'sCompany’s issued and outstanding common shares.
Outstanding Option-Based Awards and Share-Based Awards
The following table shows all awards outstanding to each of our Company'sCompany’s President and Chief Executive Officer, the Chief Financial Officer and our three (3) other most highly compensated executive officers of the Company during the most recently completed financial year (collectively, the "Named“Named Executive Officers"Officers”) as at December 31, 2010:2011:
| | Option-based Awards | Share-based Awards | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Name | Issuance Date (mm-dd-yyyy) | Number of Securities Underlying Unexercised Options(1) (#) | Option Exercise Price (CAN$) | Option Expiration Date (mm-dd-yyyy) | Value of Unexercised In-the- money Options(2) (CAN$) | Issuance Date | Number of Shares or Units of shares that have Not Vested (#) | Market or Payout Value of Share-based Awards that have Not Vested ($) | |||||||||||||||||
Engel, Juergen | 02-20-2003 | 60,000 | 2.43 | 12-31-2012 | — | — | — | — | |||||||||||||||||
| 12-11-2003 | 60,000 | 1.74 | 12-10-2013 | — | — | — | — | ||||||||||||||||||
| 12-14-2004 | 100,000 | 5.83 | 12-13-2014 | — | — | — | — | ||||||||||||||||||
| 12-13-2005 | 50,000 | 3.53 | 12-12-2015 | — | — | — | — | ||||||||||||||||||
| 01-04-2007 | 50,000 | 4.65 | 01-03-2017 | — | — | — | — | ||||||||||||||||||
| 12-11-2007 | 50,000 | 1.82 | 12-10-2017 | — | — | — | — | ||||||||||||||||||
| 11-14-2008 | 200,000 | 0.65 | 11-13-2018 | 214,000 | — | — | — | ||||||||||||||||||
| 12-08-2008 | 75,000 | 0.55 | 12-08-2018 | 87,750 | — | — | — | ||||||||||||||||||
| 12-09-2009 | 165,000 | 0.95 | 12-08-2019 | 127,050 | — | — | — | ||||||||||||||||||
| 12-08-2010 | 222,750 | 1.52 | 12-07-2020 | 44,550 | — | — | — | ||||||||||||||||||
Turpin, Dennis | 12-04-2001 | 30,000 | 6.18 | 12-04-2011 | — | — | — | — | |||||||||||||||||
| 11-01-2002 | 90,000 | 3.94 | 10-31-2012 | — | — | — | — | ||||||||||||||||||
| 12-16-2002 | 50,000 | 3.68 | 12-15-2012 | — | — | — | — | ||||||||||||||||||
| 12-11-2003 | 60,000 | 1.74 | 12-10-2013 | — | — | — | — | ||||||||||||||||||
| 12-14-2004 | 90,000 | 5.83 | 12-13-2014 | — | — | — | — | ||||||||||||||||||
| 12-13-2005 | 50,000 | 3.53 | 12-12-2015 | — | — | — | — | ||||||||||||||||||
| 01-04-2007 | 50,000 | 4.65 | 01-03-2017 | — | — | — | — | ||||||||||||||||||
| 12-11-2007 | 50,000 | 1.82 | 12-10-2017 | — | — | — | — | ||||||||||||||||||
| 12-09-2009 | 115,000 | 0.95 | 12-08-2019 | 88,550 | — | — | — | ||||||||||||||||||
| 12-08-2010 | 56,850 | 1.52 | 12-07-2020 | 11,370 | — | — | — | ||||||||||||||||||
Blake, Paul | 07-27-2007 | 45,000 | 3.05 | (3) | 07-26-2017 | — | — | — | — | ||||||||||||||||
| 12-11-2007 | 50,000 | 1.82 | (3) | 12-10-2017 | — | — | — | — | |||||||||||||||||
| 12-08-2008 | 50,000 | 0.55 | 12-08-2018 | 58,500 | — | — | — | ||||||||||||||||||
| 12-09-2009 | 110,000 | 0.95 | 12-08-2019 | 84,700 | — | — | — | ||||||||||||||||||
| 12-08-2010 | 64,050 | 1.52 | 12-07-2020 | 12,810 | — | — | — | ||||||||||||||||||
| Option-based Awards | Share-based Awards | |||||||||||||||
| Name | Issuance Date (mm-dd-yyyy) | Number of Securities Underlying Unexercised Options(1) | Option Exercise | Option Expiration (mm-dd-yyyy) | Value of Unexercised Options(2) | Issuance Date | Number of Shares or Units of Vested | Market or Value of Awards that | ||||||||
| (#) | (CAN$ or US$) | (CAN$ or US$) | (#) | ($) | ||||||||||||
Engel, Juergen | 02-20-2003 | 60,000 | CAN$2.43 | 12-31-2012 | — | — | — | — | ||||||||
12-11-2003 | 60,000 | CAN$1.74 | 12-10-2013 | — | — | — | — | |||||||||
12-14-2004 | 100,000 | CAN$5.83 | 12-13-2014 | — | — | — | — | |||||||||
12-13-2005 | 50,000 | CAN$3.53 | 12-12-2015 | — | — | — | — | |||||||||
01-04-2007 | 50,000 | CAN$4.65 | 01-03-2017 | — | — | — | — | |||||||||
12-11-2007 | 50,000 | CAN$1.82 | 12-10-2017 | — | — | — | — | |||||||||
11-14-2008 | 200,000 | CAN$0.65 | 11-13-2018 | CAN$182,000 | — | — | — | |||||||||
12-08-2008 | 75,000 | CAN$0.55 | 12-08-2018 | CAN$75,750 | — | — | — | |||||||||
Number of Securities Underlying Unexercised Options(1) Option Exercise Option Expiration (mm-dd-yyyy) Value of Unexercised Options(2) Number of Shares or Units of Vested Market or Value of Awards that 12-08-2010 12-07-2011 Turpin, Dennis 11-01-2002 12-16-2002 12-11-2003 12-14-2004 12-13-2005 01-04-2007 12-11-2007 12-09-2009 12-08-2010 12-07-2011 Blake, Paul 12-11-2007 12-08-2008 12-09-2009 12-08-2010 12-07-2011 Seeber, Matthias 12-11-2003 12-14-2004 12-13-2005 01-04-2007 12-11-2007 12-08-2008 12-09-2009 12-08-2010 12-07-2011 Pelliccione, Nicholas J. 12-11-2007 12-08-2008 12-09-2009 12-08-2010 12-07-2011 Seeber, Matthias Pelliccione, Nicholas J. There are no vested share-based awards Table of ContentsOption-based Awards Share-based Awards Name Issuance Date
(mm-dd-yyyy)
Price
Date
In-the-money Issuance
Date
shares that
have Not
Payout
Share-based
have Not
Vested(#) (CAN$ or
US$) (CAN$ or
US$) (#) ($) 12-09-2009 165,000 CAN$0.95 12-08-2019 CAN$100,650 — — — 222,750 CAN$1.52 12-07-2020 CAN$8,910 — — — 267,000 US$1.74 12-06-2021 — — — — 90,000 CAN$0.94 10-31-2012 CAN$55,800 — — — 50,000 CAN$3.68 12-15-2012 — — — — 60,000 CAN$1.74 12-10-2013 — — — — 90,000 CAN$5.83 12-13-2014 — — — — 50,000 CAN$3.53 12-12-2015 — — — — 50,000 CAN$4.65 01-03-2017 — — — — 50,000 CAN$1.82 12-10-2017 — — — — 115,000 CAN$0.95 12-08-2019 CAN$70,150 — — — 56,850 CAN$1.52 12-07-2020 CAN$2,274 — — — 104,125 US$1.74 12-06-2021 — — — — 07-27-2007 45,000 US$3.05 07-26-2017 — — — — 50,000 US$1.82 12-10-2017 — — — — 50,000 CAN$0.55 12-08-2018 CAN$50,500 — — — 110,000 CAN$0.95 12-08-2019 CAN$67,100 — — — 64,050 CAN$1.52 12-07-2020 CAN$2,562 — — — 108,430 US$1.74 12-06-2021 — — — — 02-20-2003 15,000 CAN$2.43 12-31-2012 — — — — 45,000 CAN$1.74 12-10-2013 — — — — 50,000 CAN$5.83 12-13-2014 — — — — 40,000 CAN$3.53 12-12-2015 — — — — 30,000 CAN$4.65 01-03-2017 — — — — 25,000 CAN$1.82 12-10-2017 — — — — 30,000 CAN$0.55 12-08-2018 CAN$30,300 — — — 46,667 CAN$0.95 12-08-2019 CAN$28,467 — — — 51,975 CAN$1.52 12-07-2020 CAN$2,079 — — — 104,425 US$1.74 12-06-2021 — — — — 05-07-2007 25,000 US$3.96 05-06-2017 — — — — 50,000 US$1.82 12-10-2017 — — — — 20,000 CAN$0.55 12-08-2018 CAN$20,200 — — — 60,000 CAN$0.95 12-08-2019 CAN$36,600 — — — 50,000 CAN$1.52 12-07-2020 CAN$2,000 — — — 103,316 US$1.74 12-06-2021 — — — — Option-based Awards Share-based Awards Name Issuance
Date
(mm-dd-yyyy) Number of
Securities
Underlying
Unexercised
Options(1)
(#) Option
Exercise
Price
(CAN$) Option
Expiration
Date
(mm-dd-yyyy) Value of
Unexercised
In-the-
money
Options(2)
(CAN$) Issuance
Date Number of
Shares or
Units of shares
that have Not
Vested
(#) Market or
Payout
Value of
Share-based
Awards that
have Not
Vested
($) 02-20-2003 15,000 2.43 12-31-2012 — — — — 12-11-2003 45,000 1.74 12-10-2013 — — — — 12-14-2004 50,000 5.83 12-13-2014 — — — — 12-13-2005 40,000 3.53 12-12-2015 — — — — 01-04-2007 30,000 4.65 01-03-2017 — — — — 12-11-2007 25,000 1.82 12-10-2017 — — — — 12-08-2008 30,000 0.55 12-08-2018 35,100 — — — 12-09-2009 85,000 0.95 12-08-2019 65,450 — — — 12-08-2010 51,975 1.52 12-07-2020 10,395 — — — 05-07-2007 25,000 3.96 (3) 05-06-2017 — — — — 12-11-2007 50,000 1.82 (3) 12-10-2017 — — — — 12-08-2008 20,000 0.55 12-08-2018 23,400 — — — 12-09-2009 60,000 0.95 12-08-2019 46,200 — — — 12-08-2010 50,000 1.52 12-07-2020 10,000 — — — that have not yet been paid out or distributed.(1)(1) The number of securities underlying unexercised options represents all awards outstanding at December 31, 2011. (2) “Value of unexercised in-the-money options” at financial year-end is calculated based on the difference between the closing prices of the common shares on the TSX or NASDAQ, as applicable, on the last trading day of the year (December 30, 2011) of CAN$1.56 and US$1.54, respectively, and the exercise price of the options, multiplied by the number of unexercised options. outstanding at December 31, 2010.(2)"Value of unexercised in-the-money options" at financial year-end is calculated based on the difference between the closing price of the common shares on the TSX on the last trading day of the year (December 31, 2010) of CAN$1.72 and the exercise price of the options, multiplied by the number of unexercised options.(3)These amounts are expressed in US dollars.
Incentive Plan Awards —– Value Vested or Earned During the Year
The following table shows the incentive plan awards value vested or earned for each Named Executive Officer for the financial year endingended December 31, 2010.2011.
| Name | Option-based awards – Value vested during the year(1) | Share-based awards – Value vested during the year | Non-equity incentive plan compensation - Value earned during the year | |||
| ($) | ($) | ($) | ||||
Engel, Juergen | 251,776 | — | 160,764 | |||
Turpin, Dennis | 73,461 | — | 80,509 | |||
Seeber, Matthias | 83,158 | — | 82,818 | |||
Blake, Paul | 92,499 | — | 89,670 | |||
Pelliccione, Nicholas J. | 52,840 | — | 77,739 |
| (1) | ||||||||||
| ||||||||||
| ||||||||||
| ||||||||||
| ||||||||||
| ||||||||||
| vesting date, based on the difference between the closing price of the common shares on the TSX or NASDAQ, as applicable, and the exercise price on such vesting date. | ||||||||||
Other Forms of Compensation
Benefits and Perquisites
Our executive employee benefits program also includes life, medical, dental and disability insurance. Perquisites consist of a car allowance and human resources counselling. These benefits and perquisites are designed to be competitive overall with equivalent positions in comparable North American organizations in the life sciences industry.
Pension Plan
One of our Named Executive Officers, namely Dr. Juergen Engel, the President and Chief Executive Officer, participates in a non-contributory defined benefit pension plan. Benefits payable under this plan correspond to 40% of the executive officer'sofficer’s average salary of the last twelve months during the first five working years after initial participation in this plan and increase by 0.4% for each additional year of employment.
As the normal retirement age is 65 years, first payments under the pension plan were made to Dr. Engel as atof September 1, 2010. The following table shows total annual pension benefits payable to Dr. Engel pursuant to this plan. Upon the death of a participant, the surviving spouse and/or children of the participant will be entitled to a benefit equal to 60% of the benefits to which such participant was entitled. All benefits payable under this plan are in addition to German governmental social security benefits.
As at December 31, 2010, Dr. Engel had 34 years and 4 months of credited service in the aforementioned non-contributory defined benefit pension plan.
Defined Benefit Plans Table as at December 31, 20102011
| | Number of years of credited service (#) | Annual benefits payable | | | | | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Accrued obligation at start of year(1) ($) | Compensatory change(1) ($) | Non- compensatory change(1) ($) | Accrued obligation at year end(1)(2) ($) | |||||||||||||||||
| Name | At year end(1) ($) | At age 65(1) ($) | |||||||||||||||||||
Juergen Engel | 34 | 201,508 | 201,508 | 3,355,592 | 34,605 | — | 3,323,028 | ||||||||||||||
Number of years of credited service | Annual benefits payable | Accrued obligation at start of year | Compensatory change | Non- compensatory change | Accrued obligation at year end | |||||||||
At year end | At age 65 | |||||||||||||
| Name | ||||||||||||||
| (#) | ($)(1) | ($)(1) | ($) | ($)(1) | ($) | ($)(1)(2) | ||||||||
Engel, Juergen | 34 | 202,755 | 202,755 | 3,323,028 | 549,986 | (103,977) | 3,569,399 | |||||||
| (1) | By way of exception to other currency conversions in this Circular, all amounts in the above table have been converted from euros to US$ based on the exchange rate on December 31, 2011, which was €1.000 = US$1.2972. |
| (2) | The figure in the column “Accrued obligation at year end” was further reduced by an amount of $199,638 representing mandatory pension payments made to Dr. Engel during 2011. |
Employer Contribution to Employees'Employees’ Retirement Plan
In 2008, the Board approved a plan whereby we would contribute to our employees'employees’ retirement plans both in Canada (RRSP) and the United States (401(k)) to the extent of 50% of the employee'semployee’s contribution up to a maximum of $7,750 annually for employees under 50 years old. The plan also includes a contribution for employees over 50 years old up to a maximum of $10,250 for Canadian employees and $11,000 for those in the United States. Employees based in Frankfurt, Germany alreadyalso benefit from certain employer contributions into the employees'employees’ pension funds (DUPK/RUK). Our executive officers, including the Named Executive Officers, are eligible to participate in the aforementioned employer-contribution plans to the same extent and in the same manner as all of our other employees.
Summary Compensation Table
The Summary Compensation Table set forth below shows compensation information for the Named Executive Officers for services rendered in all capacities during the financial years ended December 31, 2011, 2010 2009 and 2008. Our executive officers are generally paid in their home country's currency. All amounts in the Summary Compensation Table below are in US dollars and have been converted from the Named Executive Officers' home country currency to US dollars based on the following average exchange rates for the financial year ended December 31, 2010: €1.00 = US$1.326; and CAN$1.00 = US$0.970; for the financial year ended December 31, 2009: €1.00 = US$1.388; and CAN$1.00 = US$0.876; and for the financial year ended December 31, 2008: €1.00 = US$1.464; and CAN$1.00 = US$0.937.2009.
| | | | | | Non-equity incentive plan compensation | | | | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Name and principal position | Years | Salary ($) | Share based awards ($) | Option based awards(1) ($) | Annual incentive plan ($) | Long-term incentive plans ($) | Pension Value ($) | All other compensation(2) ($) | Total compensation ($) | |||||||||||||||||||
| Engel, Juergen | 2010 | 419,348 | (3) | — | 265,763 | 109,395 | — | 34,605 | 68,593 | (4) | 897,704 | |||||||||||||||||
| President and CEO | 2009 | 458,040 | — | 79,497 | — | — | 431,110 | 3,209 | (5) | 971,856 | ||||||||||||||||||
| 2008 | 405,925 | (6) | — | 67,777 | 248,093 | (7)(8) | — | 473,277 | 3,366 | (5) | 1,198,438 | |||||||||||||||||
| Turpin, Dennis | 2010 | 309,978 | (3) | — | 67,828 | 55,169 | — | — | 7,518 | (9) | 440,493 | |||||||||||||||||
| Senior Vice President | 2009 | 284,700 | — | 55,407 | — | — | — | — | 340,107 | |||||||||||||||||||
| and CFO | 2008 | 317,352 | — | — | 30,000 | (8) | — | — | 95,780 | (10) | 443,132 | |||||||||||||||||
| Blake, Paul | 2010 | 359,876 | (3) | — | 76,418 | 64,050 | — | — | 11,000 | (11) | 511,344 | |||||||||||||||||
| Senior Vice President and | 2009 | 366,000 | — | 52,998 | — | — | — | 10,250 | (11) | 429,248 | ||||||||||||||||||
| Chief Medical Officer | 2008 | 355,250 | — | 10,788 | 135,000 | (8) | — | — | 10,250 | (11) | 511,288 | |||||||||||||||||
| Seeber, Matthias | 2010 | 288,162 | (3) | — | 62,011 | 51,051 | — | — | 38,217 | (5) | 439,441 | |||||||||||||||||
| Senior Vice President, | 2009 | 360,748 | — | 55,407 | — | — | — | 54,300 | (5) | 416,455 | ||||||||||||||||||
| Administration and | 2008 | 307,372 | — | 6,473 | 120,755 | (7) | — | — | 61,881 | (5) | 496,481 | |||||||||||||||||
| Legal Affairs | ||||||||||||||||||||||||||||
| Pelliccione, Nicholas J. | 2010 | 311,992 | (3) | — | 59,655 | 50,001 | — | — | 11,000 | (11) | 432,648 | |||||||||||||||||
| Senior Vice President | 2009 | 317,300 | — | 28,908 | — | — | — | 8,250 | (11) | 354,458 | ||||||||||||||||||
| Regulatory Affairs and | 2008 | 317,300 | — | 4,315 | 70,000 | (8) | — | — | 10,250 | (11) | 401,865 | |||||||||||||||||
| Quality Assurance | ||||||||||||||||||||||||||||
Non-equity incentive plan compensation | ||||||||||||||||||
Name and principal position | Years | Salary | Share based awards | Option based awards(1) | Pension Value | All other compensation(2) | Total compensation | |||||||||||
Annual incentive plan | Long-term incentive plans | |||||||||||||||||
| ($) | ($) | ($) | ($) | �� | ($) | ($) | ($) | ($) | ||||||||||
Engel, Juergen President and CEO | 2011 | 505,260 | — | 336,420 | 160,764 | — | 590,136 | 214,212(3) | 1,806,792 | |||||||||
| 2010 | 419,348(4) | — | 265,763 | 109,395 | — | 34,605 | 68,593(5) | 897,704 | ||||||||||
| 2009 | 458,040 | — | 79,497 | — | — | 431,110 | 3,209(6) | 971,856 | ||||||||||
Turpin, Dennis Senior Vice President and CFO | 2011 | 332,434 | — | 131,198 | 80,509 | — | — | 5,056 | 549,197 | |||||||||
| 2010 | 309,978(4) | — | 67,828 | 55,169 | — | — | 7,518(7) | 440,493 | ||||||||||
| 2009 | 284,700 | — | 55,407 | — | — | — | — | 340,107 | ||||||||||
Seeber, Matthias Senior Vice President, Administration and Legal Affairs | 2011 | 342,512 | — | 131,576 | 82,818 | — | — | 56,966(6) | 613,872 | |||||||||
| 2010 | 288,162(4) | — | 62,011 | 51,051 | — | — | 38,217(6) | 439,441 | ||||||||||
| 2009 | 306,748 | — | 55,407 | — | — | — | 54,300(6) | 416,455 | ||||||||||
Blake, Paul Senior Vice President and Chief Medical Officer | 2011 | 370,223 | — | 136,622 | 89,670 | — | — | 11,000(8) | 607,515 | |||||||||
| 2010 | 359,876(4) | — | 76,418 | 64,050 | — | — | 11,000(8) | 511,344 | ||||||||||
| 2009 | 366,000 | — | 52,998 | — | — | — | 10,250(8) | 429,248 | ||||||||||
Pelliccione, Nicholas J. Senior Vice President Regulatory Affairs and Quality Assurance | 2011 | 321,062 | — | 130,178 | 77,739 | — | — | 11,000(8) | 539,979 | |||||||||
| 2010 | 311,992(4) | — | 59,655 | 50,001 | — | — | 11,000(8) | 432,648 | ||||||||||
| 2009 | 317,300 | — | 28,908 | — | — | — | 8,250(8) | 354,458 | ||||||||||
| (1) | The value of the option-based awards represents the closing price of the common shares on NASDAQ at the date of grant (US$1.74 for options granted on December 7, 2011, CAN$1.52 equivalent to US$1.47 for options granted on December 8, 2010, and CAN$0.95 equivalent to US$0.83 for options granted on December 9, 2009) multiplied by the Black-Scholes factor as at such date (72.414% for options granted on December 7, 2011, 80.921% for options granted on December 8, 2010, and 57.895% for options granted on December 9, 2009) and the number of stock options granted on such dates. |
| (2) | “All Other Compensation” represents perquisites and other personal benefits which, in the aggregate, amount to $50,000 or more, or are equivalent to 10% or more of a Named Executive Officer’s total salary for the financial year ended December 31, 2011. The type and amount of each perquisite, the value of which exceeds 25% of the total value of perquisites, is separately disclosed for each Named Executive Officer, if applicable. In the case of the President and CEO, “All Other Compensation” also includes mandatory pension payments paid to him commencing in 2010. See note (3) and note (5) below. |
| (3) | Represents mandatory pension payments made to Dr. Engel during 2011. |
| (4) | In 2010, the Named Executive Officers agreed to a voluntary reduction in salary between May 1, 2010 and August 31, 2010. |
| (5) | Represents DUPK/RUK (Germany) employer contributions to Dr. Engel’s retirement savings plans from January 1, 2010 to August 31, 2010. The reported amount also includes $67,169 in mandatory pension payments made to Dr. Engel after attaining age 65 commencing on September 1, 2010 for the remainder of 2010. See Section “Pension Plan”, above. |
| (6) | Represents DUPK/RUK (Germany) employer contributions to Dr. Engel’s and Mr. Seeber’s retirement savings plans. |
| (7) | Represents RRSP employer contribution to Mr. Turpin’s retirement savings plan. |
| (8) | Represents 401(k) employer contributions to Messrs. Blake’s and Pelliccione’s retirement savings plans. |
decided to voluntarily disclose the amounts of such employer contributions to the Named Executive Officers in the above table in order to provide fulsome disclosure. The reported amount also includes $67,169 in mandatory pension payments made to Dr. Engel after age 65 after September 1, 2010. See Section 6.5.2, "Pension Plan", above.
Compensation of the Chief Executive Officer
The compensation of the President and Chief Executive Officer is governed by the Company'sCompany’s executive compensation policy described in section entitled, "CompensationSection “Compensation of Executive Officers"Officers”, under Item 6 and the President and Chief Executive Officer participates together with the other Named Executive Officers in all of ourthe Company’s incentive plans.
Dr. Engel'sEngel’s total earned salary for 20102011 was $419,348,$505,260, which places him approximately 12.1% below1.03 times the 50th percentile in relation to the companies in the Reference Group.
The
Based upon the results attained versus established goals for fiscal 2011, as described in Section “Short-Term Non-Equity Incentive Compensation” the Governance Committee determined that the individual performance of Prof. Dr. Engel exceeded expectations in a number of key strategic areas, including in respect of the various goals set in the areas of Business Development and Investor Relations. See Section “Risk Assessment of Executive Compensation Program” above for more details on such goals.
Based on the foregoing, the Governance Committee recommended, and the Board approved, in the best interests of the Company given its financial situation, and in light of the Company’s and the CEO’s performance in 2011 as well as the cost-saving measures implemented byfact that in the Company,previous two years, the payment of cash bonuses was limited, that a maximum of 50%70% of the CEO'sCEO’s bonus under the short-term incentive plan be paid in cash in respect of the year 2010,2011 (paid in early 2012), while the remainder of the bonus be payable in the form of stock options based on his individual performance. Thus, Prof. Dr. Engel received 50%70% of his target cash bonus for 2010,2011, amounting to $109,395,$160,764, for his performance in the context of the Company's objectives.
In addition, the PresidentCompany’s objectives and Chief Executive Officer was awarded a grant of 222,75067,000 stock options on December 8, 20107, 2011 (at an exercise price of CAN$1.52)US$1.74) for his exceptional performance in leading the Company to attain its objectives in 2010.2011. The terms of such stock options grant provide for accelerated vesting conditions, in order to allow these grantsstock options to serve their purpose as a partial replacement for
Table the remaining 30% of Contents
annual cash bonuses.the CEO’s 2011 bonus that would otherwise have been paid in cash. See "SummarySection “Short-Term Non-Equity Incentive Compensation” above for more details on such grants.
For the financial year ended December 31, 2011, the Governance Committee also recommended, and the Board approved, that 200,000 stock options be granted to the President and Chief Executive Officer under the long-term equity compensation plan, such stock options to vest in accordance with the vesting schedule set forth in the Company’s Stock Option Plan. See Section “Long-Term Equity Compensation Plan of Executive Officers—Summary of the Stock Option Plan" on page 115Plan”, for more details on thea complete description of our Stock Option Plan.
C. Board Practices
| C. | Board Practices |
Our Articles provide that our Board shall be composed of a minimum of five and a maximum of fifteen directors. Directors are elected annually by our shareholders, but the directors may from time to time appoint one or more directors, provided that the total number of directors so appointed does not exceed one-third of the number of directors elected at the last annual meeting of shareholders. Each elected director will remain in office until termination of the next annual meeting of the shareholders or until his or her successor is duly elected or appointed, unless his or her post is vacated earlier. For information regarding Dr. Engel'sEngel’s employment agreement with the Company, which provides for benefits on termination of his employment, see "Item“Item 10.C — Material Contracts."Contracts”. None of the other directors are party to any directors'directors’ service contracts with the Company providing for benefits on termination of employment.
Committees of the Board of Directors
Audit Committee
Our Board has established an Audit Committee and a Governance Committee.
The Audit Committee assists the Board in fulfilling its oversight responsibilities. The Audit Committee reviews the financial reporting process, the system of internal control, the audit process, and the Company'sCompany’s process for monitoring compliance with laws and regulations and with our Code of Ethical Conduct. In performing its duties, the Audit Committee will maintain effective working relationships with the Board, management, and the external auditors. To effectively perform his or her role, each committee member will obtain an understanding of the detailed responsibilities of committee membership as well as the Company'sCompany’s business, operations and risks.
The function of the Audit Committee is oversight and while it has the responsibilities and powers set forth in its charter (incorporated by reference to Exhibit 11.2), it is neither the duty of the committee to plan or to conduct audits or to determine that the Company'sCompany’s financial statements are complete, accurate and in accordance with generally accepted accounting principles, nor to maintain internal controls and procedures.
The current members of the Audit Committee are Pierre Lapalme, Gérard Limoges and Pierre MacDonald.Michael Meyers.
Governance Committee
The mandate of the Governance Committee provides that it is responsible for taking all reasonable measures to ensure that appropriate human resources systems and procedures, such as hiring policies, competency profiles, training policies and compensation structures are in place so that the Company can attract, motivate and retain the quality of personnel required to meet its business objectives.
The Governance Committee also assists the Board in discharging its responsibilities relating to executive and other human resources hiring, assessment, compensation and succession planning matters.
Thus, the Governance Committee recommends the appointment of senior officers, including the terms and conditions of their appointment and termination, and reviews the evaluation of the performance of our senior officers, including recommending their compensation.compensation and overseeing risk identification and management in relation to executive compensation policies and practices. The Board, which includes the members of the Governance Committee, reviews the Chief Executive Officer'sOfficer’s corporate
goals and objectives and evaluates his or her performance and compensation in light of such goals and objectives.
The current members of the Governance Committee are Juergen Ernst, José P. Dorais and Pierre MacDonald.
D. EmployeesGérard Limoges.
| D. | Employees |
As at March 1, 2011,2012, we had a total of 87.889 Full Time Equivalents ("FTE"(“FTE”) (as compared to 87.8 at March 1, 2011 and 99 at March 1, 2010 and 109 at March 1, 2009)2010), of which 74.175.3 are based in Frankfurt, Germany, 5 in New Jersey, United States, and 8.7 in Quebec City, Canada. Of these, 5253 are involved in discovery, preclinical, clinical and pharmaceutical development, 9 are involved in regulatory affairs, quality assurance and intellectual property, and 27 are involved in business operations, communications, finance, information technology, human resources, project management and legal affairs. Following the negative results of our Phase 3 efficacy studies for cetrorelix in BPH in 2009, we applied for and were granted approval by the German Ministry of Labor to implement the so-called "Kurzarbeit" ("short hours") regime for a one-year term. "Short hours" is a system offered by the German Ministry of Labor for companies undergoing economic distress as a result of an unexpected event which causes temporary overcapacity in the work force. Under the system, we are allowed to selectively reduce the working hours of our employees thereby reducing the labor costs for the company accordingly. The employees affected by "short hours" are compensated for the salary shortfall by the German Government. The initial approval obtained by the German Ministry of Labor in 2009 was valid and implemented until August 31, 2010. In light of our improved financial condition as well as the workforce reductions seen during such time, we did not apply for a further one-year renewal of "short hours" at the end of 2010.
We have agreements with all of our employees covering confidentiality and loyalty, non-competition, and assignment to the Company of all intellectual property rights developed during the employment period. Some of our employees based in Frankfurt, Germany, were previously represented by the Chemical Union of Germany. As such, their compensation was largely driven by the outcome of the negotiations between the Chemical Union and the Association of Employers for the chemical industry which was then binding for all German companies in the industry. In collaboration with the German works council and upon consent of each such "tariff-employee", we terminated our voluntary commitment to the collective bargaining agreement (Tarifvertrag) effective May 31, 2010. All HR-relevant aspects are since then subject to individual negotiations between employer and employee and free of any binding tariff impositions.
E. Share ownership
| E. | Share ownership |
The information in the table below is provided as at March 24, 2011:
| Name | No. of common shares owned or held | Percent(1) | No. of stock options held(2) | No. of currently exercisable options | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
Marcel Aubut | 112,500 | * | 175,000 | 126,667 | ||||||||
Paul Blake | 65,350 | * | 319,050 | 201,668 | ||||||||
José P. Dorais | 0 | * | 30,000 | 0 | ||||||||
Juergen Engel | 89,779 | * | 1,032,750 | 663,334 | ||||||||
Juergen Ernst | 58,850 | * | 225,000 | 143,334 | ||||||||
Pierre Lapalme | 0 | * | 50,000 | 6,667 | ||||||||
Gérard Limoges | 9,000 | * | 125,000 | 76,667 | ||||||||
Pierre MacDonald | 26,500 | * | 184,000 | 135,667 | ||||||||
Michael Meyers | 408,862 | (3) | * | 0 | 0 | |||||||
Nicholas J. Pelliccione | 25,000 | * | 205,000 | 128,334 | ||||||||
Matthias Seeber | 0 | * | 371,975 | 271,667 | ||||||||
Dennis Turpin | 15,250 | * | 641,850 | 546,667 | ||||||||
Total | 811,091 | 0.95 | 3,359,625 | 2,300,672 | ||||||||
Item 7. Major Shareholders and Related Party Transactions
A. Major shareholders27, 2012:
| Name | No. of common shares owned or held | Percent(1) | No. of stock options held(2) | No. of currently options | ||||
Marcel Aubut | 112,500 | * | 220,000 | 143,334 | ||||
Paul Blake | 70,350 | * | 427,480 | 297,700 | ||||
José P. Dorais | 0 | * | 80,000 | 10,000 | ||||
Juergen Engel | 117,779 | * | 1,299,750 | 958,500 | ||||
Juergen Ernst | 58,850 | * | 275,000 | 198,334 | ||||
Pierre Lapalme | 0 | * | 100,000 | 23,334 | ||||
Gérard Limoges | 9,000 | * | 175,000 | 98,334 | ||||
Michael Meyers | 0 | * | 60,000 | 0 | ||||
Nicholas J. Pelliccione | 27,750 | * | 308,316 | 188,334 | ||||
Matthias Seeber | 0 | * | 438,067 | 316,317 | ||||
Dennis Turpin | 21,250 | * | 715,975 | 592,900 | ||||
Total | 417,479 | 0.38 | 4,099,588 | 2,827,087 |
| * | Less than 1% |
| (1) | Based on 108,787,366 common shares outstanding as at March 27, 2012. |
| (2) | For information regarding option expiration dates and exercise price refer to the tables included under Item 6.B. |
| Item 7. | Major Shareholders and Related Party Transactions |
| A. | Major shareholders |
We are not directly or indirectly owned or controlled by another corporation or by any foreign government.
Based on filings with the Securities and Exchange CommissionSEC and the Canadian securities regulatory authorities, as at March 24, 2011,27, 2012, there are no persons/entities who beneficially owned, directly or indirectly, or exercised control or direction over our common shares carrying more than 5% of the voting rights attached to all our common shares.
United States Shareholders
As at December 31, 2010,2011, there were a total of 234219 holders of record of our common shares, of which 9 were registered with addresses in the United States holding in the aggregate approximately 37.42%50.02% of our outstanding common shares. We believe that the number of beneficial owners of our common shares is substantially greater than the number of record holders, because a large portion of our common shares are held in broker "street names."
B. Related party transactions“street names”.
| B. | Related party transactions |
None
C. Interests of experts and counsel
| C. | Interests of experts and counsel |
Not applicable.
A. Consolidated statements and other financial information
| Item 8. | Financial Information |
| A. | Consolidated statements and other financial information |
The financial statements filed as part of this annual report are presented under "Item“Item 18. — Financial Statements"Statements”.
Valuation and qualifying accounts (in thousands of US dollars)
| B. | Significant changes |
Valuation allowance on future income tax assets
| | Years ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| | 2010 | 2009 | 2008 | |||||||
Balance — Beginning of year | $ | 50,350 | $ | 36,581 | $ | 23,289 | ||||
Change in valuation allowance | 8,426 | 9,959 | 17,554 | |||||||
Impact of foreign exchange rate changes | (651 | ) | 3,810 | (4,262 | ) | |||||
Balance — End of year | $ | 58,125 | $ | 50,350 | $ | 36,581 | ||||
Export Sales
Export and domestic sales in thousands of US dollars and as percentage of total sales as follows:
| | Years ended December 31, | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2010 | 2009 | 2008 | ||||||||||||||||
Export Sales | $ | 27,703 | 100.00% | $ | 63,237 | 100.00% | $ | 38,145 | 99.13% | ||||||||||
Domestic Sales | — | 0.00% | — | 0.00% | 333 | 0.87% | |||||||||||||
| $ | 27,703 | 100.00% | $ | 63,237 | 100.00% | $ | 38,478 | 100.00% | |||||||||||
Dividend Policy
Since our incorporation, we have not paid any dividends, and we do not anticipate paying any dividends in the foreseeable future.
B. Significant changes
No significant changes occurred since the date of our annual consolidated financial statements included elsewhere in this annual report.
Item 9. The Offering and Listing
A. Offer and listing details
| Item 9. | The Offering and Listing |
| A. | Offer and listing details |
Not Applicable, except for Item 9A(4).
Our common shares are listed and posted for trading on NASDAQ under the symbol "AEZS"“AEZS” and on the TSX under the symbol "AEZ"“AEZ”. The following table indicates, for the relevant periods, the high and low closing prices of our common shares on NASDAQ and on the TSX:
| | NASDAQ (US$) | TSX (CAN$) | ||||||
|---|---|---|---|---|---|---|---|---|
| | High | Low | High | Low | ||||
2010 | 2.09 | 0.79 | 2.14 | 0.80 | ||||
2009 | 2.83 | 0.46 | 3.11 | 0.57 | ||||
2008 | 1.80 | 0.40 | 1.85 | 0.44 | ||||
2007 | 4.36 | 1.46 | 5.10 | 1.47 | ||||
2006 | 7.46 | 4.05 | 8.60 | 4.68 | ||||
2008 | ||||||||
Fourth quarter | 0.60 | 0.40 | 0.72 | 0.44 | ||||
Third quarter | 1.36 | 0.59 | 1.42 | 0.61 | ||||
Second quarter | 1.80 | 1.00 | 1.85 | 1.01 | ||||
First quarter | 1.73 | 0.77 | 1.78 | 0.75 | ||||
2009 | ||||||||
Fourth quarter | 1.25 | 0.80 | 1.40 | 0.83 | ||||
Third quarter | 2.83 | 0.89 | 3.11 | 0.97 | ||||
Second quarter | 2.35 | 0.89 | 2.63 | 1.06 | ||||
First quarter | 0.97 | 0.46 | 1.25 | 0.57 | ||||
2010 | ||||||||
Fourth quarter | 1.96 | 1.22 | 1.98 | 1.22 | ||||
Third quarter | 1.40 | 0.95 | 1.44 | 1.00 | ||||
Second quarter | 2.09 | 0.80 | 2.14 | 0.80 | ||||
First quarter | 0.93 | 0.79 | 0.99 | 0.81 | ||||
Most recent 6 months | ||||||||
March 2011(1) | 2.09 | 1.69 | 2.03 | 1.68 | ||||
February 2011 | 1.82 | 1.56 | 1.80 | 1.55 | ||||
January 2011 | 1.77 | 1.55 | 1.77 | 1.54 | ||||
December 2010 | 1.96 | 1.41 | 1.98 | 1.44 | ||||
November 2010 | 1.45 | 1.22 | 1.49 | 1.22 | ||||
October 2010 | 1.32 | 1.22 | 1.33 | 1.24 | ||||
September 2010 | 1.40 | 1.03 | 1.44 | 1.08 | ||||
B. Plan of distribution
| NASDAQ (US$) | TSX (CAN$) | |||||||
| High | Low | High | Low | |||||
2011 | 2.58 | 1.43 | 2.51 | 1.41 | ||||
2010 | 2.09 | 0.79 | 2.14 | 0.80 | ||||
2009 | 2.83 | 0.46 | 3.11 | 0.57 | ||||
2008 | 1.80 | 0.40 | 1.85 | 0.44 | ||||
2007 | 4.36 | 1.46 | 5.10 | 1.47 | ||||
2009 | ||||||||
Fourth quarter | 1.25 | 0.80 | 1.40 | 0.83 | ||||
Third quarter | 2.83 | 0.89 | 3.11 | 0.97 | ||||
Second quarter | 2.35 | 0.89 | 2.63 | 1.06 | ||||
First quarter | 0.97 | 0.46 | 1.25 | 0.57 | ||||
2010 | ||||||||
Fourth quarter | 1.96 | 1.22 | 1.98 | 1.22 | ||||
Third quarter | 1.40 | 0.95 | 1.44 | 1.00 | ||||
Second quarter | 2.09 | 0.80 | 2.14 | 0.80 | ||||
First quarter | 0.93 | 0.79 | 0.99 | 0.81 | ||||
2011 | ||||||||
Fourth quarter | 1.78 | 1.43 | 1.80 | 1.47 | ||||
Third quarter | 2.35 | 1.43 | 2.26 | 1.41 | ||||
Second quarter | 2.58 | 1.82 | 2.51 | 1.75 | ||||
First quarter | 2.00 | 1.55 | 1.93 | 1.54 | ||||
Most recent 6 months | ||||||||
March 2012(1) | 2.15 | 1.69 | 2.13 | 1.67 | ||||
February 2012 | 1.83 | 1.58 | 1.84 | 1.58 | ||||
January 2012 | 1.71 | 1.56 | 1.78 | 1.58 | ||||
December 2011 | 1.78 | 1.51 | 1.80 | 1.54 | ||||
November 2011 | 1.76 | 1.46 | 1.80 | 1.50 | ||||
October 2011 | 1.75 | 1.43 | 1.72 | 1.47 | ||||
September 2011 | 1.99 | 1.48 | 1.98 | 1.54 | ||||
| (1) | Up to and including March 26, 2012 |
| B. | Plan of distribution |
Not applicable.
C. Markets
| C. | Markets |
Our common shares are listed and posted for trading on NasdaqNASDAQ under the symbol "AEZS"“AEZS” and on the TSX under the symbol "AEZ"“AEZ”.
On January 21, 2010, we announced that we had received a notification from NASDAQ regarding the failure by the Company to comply with NASDAQ's minimum bid price requirements. However, we regained compliance with these requirements on April 23, 2010. Nevertheless, if we fail to meet any of NASDAQ's continued listing requirements, our common shares may be delisted. Any delisting of our common shares may adversely affect a shareholder's ability to dispose, or obtain quotations as to the market value, of such shares.
| D. | Selling shareholders |
Not applicable.
D. Selling shareholders
| E. | Dilution |
Not applicable.
E. Dilution
| F. | Expenses of the issuer |
Not applicable.
F. Expenses of the issuer
| Item 10. | Additional Information |
| A. | Share capital |
Not applicable.
Item 10. Additional Information
| B. | Memorandum and articles of association |
A. Share capital
Not applicable.
B. Memorandum and articles of association
The Company is governed by its restated articles of incorporation (the "Restated“Restated Articles of Incorporation"Incorporation”) under theCanada Business Corporations Act (the "CBCA"“CBCA”) and by its bylaws (the "bylaws"“bylaws”). The Company'sCompany’s Restated Articles of Incorporation are on file with the Corporations Directorate of Industry Canada under Corporation Number 264271-9. The Restated Articles of Incorporation do not include a stated purpose and do not place any restrictions on the business that the Company may carry on.
Inspection Rights of Shareholders
Under the CBCA, shareholders are entitled to be provided with a copy of the list of registered shareholders of the Company. In order to obtain the shareholder list, the Company must be provided with an affidavit including, among other things, a statement that the list will only be used for the purposes permitted by the CBCA. These permitted purposes include an effort to influence the voting of shareholders of the Company, an offer to acquire securities of the Company and any other matter relating to the affairs of the Company. The Company is entitled to charge a reasonable fee for the provision of the shareholder list and must deliver that list no more than ten days after receipt of the affidavit described above.
Under the CBCA, shareholders of the Company have the right to inspect certain corporate records, including its Restated Articles of Incorporation and bylaws and minutes of meetings and resolutions of the shareholders. Shareholders have no statutory right to inspect minutes of meetings and resolutions of directors of the Company. Shareholders of the Company have the right to certain financial information respecting the Company. In addition to the annual and quarterly financial statements required to be filed under applicable securities laws, under the CBCA the Company is
required to place before every annual meeting of shareholders its audited comparative annual financial statements. In addition, shareholders have the right to examine the financial statements of each of our subsidiaries and any other corporate entity whose accounts are consolidated in the financial statements of the Company.
Directors
The minimum number of directors of the Company is five and the maximum number is fifteen. In accordance with the Company's bylaws and the CBCA, a majority of its directors must be residents of Canada. In order to serve as a director, a person must be a natural person at least 18 years of age, of sound mind, not bankrupt, and must not be prohibited by any court from holding the office of director. For as long as the Company is a company that publicly distributes its securities, at least two-thirds of its directors must not be officers or employees of the Company or its subsidiaries. None of the Restated Articles of Incorporation, the bylaws and the CBCA imposeimposes any mandatory retirement requirements for directors.
The directors are elected by a majority of the votes cast at the annual meeting at which an election of directors is required, to hold office until the election of their successors except in the case of resignations or if their offices become vacant by death or otherwise. Subject to the provisions of the Company'sCompany’s bylaws, all directors may, if still qualified to serve as directors, stand for re-election. The Board is not replaced at staggered intervals but is elected annually.
UnderThere is no provision in the Company'sCompany’s bylaws and theor its Restated Articles of Incorporation that requires that a director of the Company need notmust be a shareholder.
The directors are entitled to remuneration as shall from time to time be determined by the Board or by a committee to which the Board may delegate the power to do so. Under the mandate of the Company'sCompany’s Governance Committee, such committee, comprised of a majority of independent directors, is tasked with making recommendations to the Board concerning director remuneration.
The Company's bylaws provide that a director shall promptly disclose to the Company any interest he or she has in any undertaking or association that is likely to place him or her in a situation of conflict of interest, as well as the rights he or she may assert against the Company, indicating, should such be the case, the nature and value thereof. Likewise, the CBCA and the Company's bylaws provideprovides that a director who is a party to, or who is a director or officer of, or has a material interest in, any person who is a party to a material contract or transaction or proposed material contract or transaction with the Company must disclose to the Company the nature and extent of his or her interest at the time and in the manner provided by the CBCA, or request that same be entered in the minutes of the meetings of the Board, even if such contract, in connection with the normal business activity of the Company, does not require the approval of either the directors or the shareholders. At the request of the president or any director, the director placed in a situation of conflict of interest must leave the meeting while the Board discusses the matter. The CBCA and the Company's bylaws prohibitprohibits such a director from voting on any resolution to approve the contract or transaction unless the contract or transaction:
| — | relates primarily to his or her remuneration as a director, officer, employee or agent of the Company or an affiliate; |
| — | is for indemnity or insurance for director’s liability as permitted by the CBCA; or |
| — | is with an affiliate of the Company. |
The CBCA provides that the Board may, on behalf of the Company and without authorization of its shareholders:
| — | borrow money upon the credit of the Company; |
| — | issue, reissue, sell or pledge debt obligations of the Company; |
| — | give a guarantee on behalf of the Company to secure performance of an obligation of any person; and |
| — | mortgage, hypothecate, pledge or otherwise create a security interest in all or any property of the Company, owned or subsequently acquired, to secure any obligation of the Company. |
The shareholders have the ability to restrict such powers through the Company'sCompany’s Restated Articles of Incorporation or bylaws (or through a unanimous shareholder agreement), but no such restrictions are in place.
The CBCA prohibits the giving of a guarantee to any shareholder, director, officer or employee of the Company or of an affiliated corporation or to an associate of any such person for any purpose or to any person for the purpose of or in connection with a purchase of a share issued or to be issued by the Company or its affiliates, where there are reasonable grounds for believing that the Company is or, after giving the guarantee, would be unable to pay its liabilities as they become due, or the realizable value of the Company'sCompany’s assets in the form of assets pledged or encumbered to secure a guarantee, after giving the guarantee, would be less than the aggregate of the Company'sCompany’s liabilities and stated capital of all classes. These borrowing powers may be varied by the Company'sCompany’s bylaws or its Restated Articles of Incorporation. However, the Company'sCompany’s bylaws and Restated Articles of Incorporation do not contain any restrictions on or variations of these borrowing powers.
Pursuant to the CBCA, the directors of the Company manage and administer the business and affairs of the Company and exercise all such powers and authority as the Company is authorized to exercise pursuant to the CBCA, the Restated Articles of Incorporation and the bylaws. The general duties of a director or officer of the Company under the CBCA are to act honestly and in good faith with a view to the best interests of the Company and to exercise the care, diligence and skill that a reasonably prudent person would exercise in comparable circumstances. Any breach of these duties may lead to liability to the Company and its shareholders for breach of fiduciary duty. In addition, a breach of certain provisions of the CBCA, including the improper payment of dividends or the improper purchase or redemption of shares, will render the directors who authorized such action liable to account to the Company for any amounts improperly paid or distributed.
The Company'sCompany’s bylaws provide that the Board may, from time to time, appoint from amongst their number committees of the Board, and delegate to any such committee any of the powers of the Board except those which pursuant to the CBCA a committee of the Board has no authority to exercise. As such, the Board has two standing committees: the Audit Committee and the Governance Committee.
Subject to the limitations provided by the CBCA, the Company'sCompany’s bylaws provide that the Company shall, to full extent provided by law, indemnify a director or an officer of the Company, a former director or officer of the Company or a person who acts or acted at the Company'sCompany’s request as a director or officer of a body corporate of which the Company is or was a shareholder or creditor, and his or her heirs and legal representatives, against all costs, losses, charges and expenses, including an amount paid to settle an action or satisfy a judgment, reasonably incurred by him or her in respect of any civil, criminal or administrative action or proceeding to which he or she is made a party by reason of having been a director or officer of the Company or such body corporate, provided:
| (a) | he or she acted in good faith in the best interests of the Company; and |
| (b) | in the case of a criminal or an administrative action or proceeding that is enforced by a monetary penalty, he or she had reasonable grounds to believe that his or her conduct was lawful. |
The directors of the Company are authorized to indemnify from time to time any director or other person who has assumed or is about to assume in the normal course of business any liability for the
Company or for any corporation controlled by the Company, and to secure such director or other person against any loss by the pledge of all or part of the movable or immovable property of the Company through the creation of a hypothec or any other real right in all or part of such property or in any other manner.
Share Capitalization
Our authorized share capital structure consists of an unlimited number of shares of the following classes (all classes are without nominal or par value): common shares; and first preferred shares (the "First“First Preferred Shares"Shares”) and second preferred shares (the "Second“Second Preferred Shares"Shares” and, together with the First Preferred Shares, the "Preferred Shares"“Preferred Shares”), both issuable in series. As at March 24, 2011,27, 2012, there were 85,265,033108,787,366 common shares outstanding. No Preferred Shares of the Company have been issued to date. The Company has also issued warrants to acquire common shareshares in connection with certain equity financings.
Common Shares
The holders of the common shares are entitled to one vote for each common share held by them at all meetings of shareholders, except meetings at which only shareholders of a specified class of shares are entitled to vote. In addition, the holders are entitled to receive dividends if, as and when declared by the Company'sCompany’s Board of Directors on the common shares. Finally, the holders of the common shares are entitled to receive the remaining property of the Company upon any liquidation, dissolution or winding-up of the affairs of the Company, whether voluntary or involuntary. Shareholders have no liability to further capital calls as all shares issued and outstanding are fully paid and non-assessable.
Preferred Shares
The First and Second Preferred Shares are issuable in series with rights and privileges specific to each class. The holders of Preferred Shares are generally not entitled to receive notice of or to attend or vote at meetings of shareholders. The holders of First Preferred Shares are entitled to preference and priority to any participation of holders of Second Preferred Shares, common shares or shares of any other class of shares of the share capital of the Company ranking junior to the First Preferred Shares with respect to dividends and, in the event of the liquidation of the Company, the distribution of its property upon its dissolution or winding-up, or the distribution of all or part of its assets among the shareholders, to an amount equal to the value of the consideration paid in respect of such shares outstanding, as credited to the issued and paid-up share capital of the Company, on an equal basis, in proportion to the amount of their respective claims in regard to such shares held by them. The holders of Second Preferred Shares are entitled to preference and priority to any participation of holders of common shares or shares of any other class of shares of the share capital of the Company ranking junior to the Second Preferred Shares with respect to dividends and, in the event of the liquidation of the Company, the distribution of its property upon its dissolution or winding-up, or the distribution of all or part of its assets among the shareholders, to an amount equal to the value of the consideration paid in respect of such shares outstanding, as credited to the issued and paid-up share capital of the Company, on an equal basis, in proportion to the amount of their respective claims in regard to such shares held by them.
Our Board of Directors may, from time to time, provide for additional series of Preferred Shares to be created and issued, but the issuance of any Preferred Shares is subject to the general duties of the directors under the CBCA to act honestly and in good faith with a view to the best interests of the Company and to exercise the care, diligence and skill that a reasonably prudent person would exercise in comparable circumstances.
Shareholder Actions
The CBCA provides that shareholders of the Company may, with leave of a court, bring an action in the name of and on behalf of the Company for the purpose of prosecuting, defending or discontinuing an action on behalf of the Company. In order to grant leave to permit such an action, the CBCA provides that the court must be satisfied that the directors of the Company were given adequate notice of the application, the shareholder is acting in good faith and that it appears to be in the Company'sCompany’s best interests that the action be brought.
Shareholder Rights Plan
Objectives and Background of the Shareholder Rights Plan
The fundamental objectives of the Company'sCompany’s Shareholder Rights Plan (the "Rights Plan"“Rights Plan”) are to provide adequate time for our Board and shareholders to assess an unsolicited take-over bid for the Company, to provide the Board with sufficient time to explore and develop alternatives for maximizing shareholder value if a take-over bid is made, and to provide shareholders with an equal opportunity to participate in a take-over bid.
The Rights Plan encourages a potential acquiror who makes a take-over bid to proceed either by way of a "Permitted Bid"“Permitted Bid”, as described below, which requires a take-over bid to satisfy certain minimum standards designed to promote fairness, or with the concurrence of our Board. If a take-over bid fails to meet these minimum standards and the Rights Plan is not waived by the Board, the Rights Plan provides that holders of common shares, other than the acquiror, will be able to purchase additional common shares at a significant discount to market, thus exposing the person acquiring common shares to substantial dilution of its holdings.
Summary of the Rights Plan
The following is a summary of the principal terms of the Rights Plan, which summary is qualified in its entirety by reference to the terms thereof. Capitalized terms not otherwise defined in this summary shall have the meaning ascribed to such terms in the Shareholder Rights Plan Agreement which sets forth the Rights Plan. The Rights Plan is filed as an exhibit to this annual report on Form 20-F.
Operation of the Rights Plan
Pursuant to the terms of the Rights Plan, one right was issued in respect of each common share outstanding at 5:01 p.m. on March 29, 2010 (the "Effective Date"“Effective Date”). In addition, one right will be issued for each additional common share issued after the Record Time and prior to the earlier of the Separation Time (as defined below) and the Expiration Time (as defined below). The rights have an initial exercise price equal to the Market Price (as defined below) of the common shares as determined at the Separation Time, multiplied by five, subject to certain anti-dilution adjustments (the "Exercise Price"“Exercise Price”), and they are not exercisable until the Separation Time. Upon the occurrence of a Flip-in Event, each right will entitle the holder thereof, other than an Acquiring Person or any other person whose rights are or become void pursuant to the provisions of the Rights Plan, to purchase from the Company, effective at the close of business on the eighth trading day after the Stock Acquisition Date, upon payment to the Company of the Exercise Price, common shares having an aggregate Market Price equal to twice the Exercise Price on the date of consummation or occurrence of such Flip-in Event, subject to certain anti-dilution adjustments.
Definition of Market Price
Market Price is generally defined in the Rights Plan, on any given day on which a determination must be made, as the volume weighted average trading price of the common shares for the five consecutive trading days (i.e. days on which the TSX is open for the transaction of business, subject to certain exceptions), through and including the trading day immediately preceding such date of determination, subject to certain exceptions.
Trading of Rights
Until the Separation Time (or the earlier termination or expiration of the rights), the rights trade together with the common shares and are represented by the same share certificates as the common shares or an entry in the Company'sCompany’s securities register in respect of any outstanding common shares. From and after the Separation Time and prior to the Expiration Time, the rights are evidenced by rights certificates and trade separately from the common shares. The rights do not carry any of the rights attaching to the common shares such as voting or dividend rights.
Separation Time
The rights will separate from the common shares to which they are attached and become exercisable at the time (the "Separation Time"“Separation Time”) of the close of business on the eighth business day after the earliest to occur of:
| 1. | the first date (the “Stock Acquisition Date”) of a public announcement of facts indicating that a person has become an Acquiring Person; and |
| 2. | the date of the commencement of, or first public announcement of the intention of any person (other than the Company or any of its subsidiaries) to commence a take-over bid or a share exchange bid for more than 20% of the outstanding common shares of the Company other than a Permitted Bid or a Competing Permitted Bid (as defined below), so long as such take-over bid continues to satisfy the requirements of a Permitted Bid or a Competing Permitted Bid), as the case may be. |
The Separation Time can also be such later time as may from time to time be determined by the Board, provided that if any such take-over bid expires, or is cancelled, terminated or otherwise withdrawn prior to the Separation Time, without securities deposited thereunder being taken up and paid for, it shall be deemed never to have been made and if the Board determines to waive the application of the Rights Plan to a Flip-in Event, the Separation Time in respect of such Flip-in Event shall be deemed never to have occurred.
From and after the Separation Time and prior to the Expiration Time, each right entitles the holder thereof to purchase one common share upon payment to the Company of the Exercise Price.
Flip-in Event
The acquisition by a person (an "Acquiring Person"“Acquiring Person”), including others acting jointly or in concert with such person, of more than 20% of the outstanding common shares, other than by way of a Permitted Bid, a Competing Permitted Bid or in certain other limited circumstances described in the Rights Plan, is referred to as a "Flip-in Event"“Flip-in Event”.
In the event that, prior to the Expiration Time, a Flip-in Event which has not been waived occurs (see "Waiver“Waiver and Redemption"Redemption” below), each right (other than those held by or deemed to be held by the Acquiring Person) will thereafter entitle the holder thereof, effective as at the close of business on the eighth trading day after the Stock Acquisition Date, to purchase from the Company, upon payment of the Exercise Price and otherwise exercising such right in accordance with the terms of the Rights
Plan, that number of common shares having an aggregate Market Price on the date of consummation or occurrence of the Flip-in Event equal to twice the Exercise Price, for an amount in cash equal to the Exercise Price (subject to certain anti-dilution adjustments described in the Rights Plan).
A bidder may enter into Lock-up Agreements with the Company'sCompany’s shareholders ("(“Locked-up Persons"Persons”) who are not affiliates or associates of the bidder and who are not, other than by virtue of entering into such agreement, acting jointly or in concert with the bidder, whereby such shareholders agree to tender their common shares to the take-over bid (the "Lock-up Bid"“Lock-up Bid”) without the bidder being deemed to beneficially own the common shares deposited pursuant to the Lock-up Bid. Any such agreement must include a provision that permits the Locked-up Person to withdraw the common shares to tender to another take-over bid or to support another transaction that will either provide greater consideration to the shareholder than the Lock-up Bid or provide for a right to sell a greater number of shares than the Lock-up Bid contemplates (provided that the Lock-up Agreement may require that such greater number exceed the number of shares under the Locked-up Bid by a specified percentage not to exceed 7%).
The Lock-up Agreement may require that the consideration under the other transaction exceed the consideration under the Lock-up Bid by a specified amount. The specified amount may not be greater than 7%. For greater certainty, a Lock-up Agreement may contain a right of first refusal or require a period of delay (or other similar limitation) to give a bidder an opportunity to match a higher price in another transaction as long as the limitation does not preclude the exercise by the Locked-up Person of the right to withdraw the common shares during the period of the other take-over bid or transaction.
The Rights Plan requires that any Lock-up Agreement be made available to the Company and the public. The definition of Lock-up Agreement also provides that under a Lock-up Agreement, no "break up"“break up” fees, "topping"“topping” fees, penalties, expenses or other amounts that exceed in aggregate the greater of (i) 21/2%2% of the price or value of the aggregate consideration payable under the Lock-up Bid, and (ii) 50% of the amount by which the price or value of the consideration received by a Locked-up Person under another take-over bid or transaction exceeds what such Locked-up Person would have received under the Lock-up Bid, can be payable by such Locked-up Person if the Locked-up Person fails to deposit or tender common shares to the Lock-up Bid or withdraws common shares previously tendered thereto in order to deposit such common shares to another take-over bid or support another transaction.
Permitted Bid Requirements
The requirements of a Permitted Bid include the following:
manner in which the common shares are to be voted or direct whether the common shares are to be tendered to a take-over bid (the "Independent Shareholders"), have been deposited or tendered to the take-over bid and not withdrawn;
| 1. | the take-over bid must be made by means of a take-over bid circular; |
| 2. | the take-over bid must be made to all holders of common shares wherever resident, on identical terms and conditions, other than the bidder; |
| 3. | the take-over bid must not permit common shares tendered pursuant to the bid to be taken up or paid for: |
| a) | prior to the close of business on a date which is not less than 60 days following the date of the bid, and |
| b) | then only if at such date more than 50% of the then outstanding common shares held by shareholders other than any other Acquiring Person, the bidder, the bidder’s affiliates or associates, persons acting jointly or in concert with the bidder and any employee benefit plan, deferred profit-sharing plan, stock participation plan or trust for the benefit of employees of the Company or any of its subsidiaries, unless the beneficiaries of such plan or trust direct the manner in which the common shares are to be voted or direct whether the common shares are to be tendered to a take-over bid (the “Independent Shareholders”), have been deposited or tendered to the take-over bid and not withdrawn; |
| 4. | the take-over bid must allow common shares to be deposited, unless the take-over bid is withdrawn, at any time up to the close of business on the date that the common shares are to be first taken up and paid for; |
| 5. | the take-over bid must allow common shares to be withdrawn until taken up and paid for; and |
| 6. | if more than 50% of the then outstanding common shares held by Independent Shareholders are deposited or tendered to the take-over bid within the 60-day period and not withdrawn, the bidder must make a public announcement of that fact and the take-over bid must remain open for deposits and tenders of common shares for not less than ten days from the date of such public announcement. |
A Permitted Bid need not be a bid for all outstanding common shares not held by the bidder, i.e., a Permitted Bid may be a partial bid. The Rights Plan also allows a competing Permitted Bid (a "Competing“Competing Permitted Bid"Bid”) to be made while a Permitted Bid is in existence. A Competing Permitted Bid must satisfy all the requirements of a Permitted Bid other than the requirement set out in clause 3(a) above and must not permit common shares tendered or deposited pursuant to the bid to be taken up or paid for prior to the close of business on a date which is earlier than 35 days (or such longer minimum period of days that the bid must be open for acceptance after the date of the bid under applicable Canadian provincial securities legislation) and the 60th day after the earliest date on which any other Permitted Bid or Competing Permitted Bid that is then in existence was made.
Waiver and Redemption
The Board may, prior to the occurrence of a Flip-in Event, waive the dilutive effects of the Rights Plan in respect of, among other things, a particular Flip-in Event resulting from a take-over bid made by way of a take-over bid circular to all holders of common shares of the Company. In such an event, such waiver shall also be deemed to be a waiver in respect of any other Flip-in Event occurring under a take-over bid made by way of a take-over bid circular to all holders of common shares prior to the expiry of the first mentioned take-over bid.
The Board may, with the approval of a majority of Independent Shareholders (or, after the Separation Time has occurred, holders of rights, other than rights which are void pursuant to the provisions of the Rights Plan or which, prior to the Separation Time, are held otherwise than by Independent Shareholders), at any time prior to the occurrence of a Flip-in Event which has not been waived, elect to redeem all, but not less than all, of the then outstanding rights at a price of $0.00001 each, appropriately adjusted as provided in the Rights Plan (the "Redemption Price"“Redemption Price”).
Where a take-over bid that is not a Permitted Bid or Competing Permitted Bid is withdrawn or otherwise terminated after the Separation Time has occurred and prior to the occurrence of a Flip-in Event, the Board may elect to redeem all the outstanding rights at the Redemption Price without the consent of the holders of the common shares or the rights and reissue rights under the Rights Plan to holders of record of common shares immediately following such redemption. Upon the rights being so redeemed and reissued, all the provisions of the Rights Plan will continue to apply as if the Separation Time had not occurred, and the Separation Time will be deemed not to have occurred and the Company shall be deemed to have issued replacement rights to the holders of its then outstanding common shares.
Amendment to the Rights Plan
The Rights Plan may be amended to correct any clerical or typographical error or to make such changes as are required to maintain the validity of the Rights Plan as a result of any change in any applicable legislation, regulations or rules thereunder, without the approval of the holders of the common shares or rights. Prior to the Separation Time, the Company may, with the prior consent of the holders of common shares, amend, vary or delete any of the provisions of the Rights Plan in order to effect any changes which the Board, acting in good faith, considers necessary or desirable. The Company may, with the prior consent of the holders of rights, at any time after the Separation Time and before the Expiration Time, amend, vary or delete any of the provisions of the Rights Plan.
Protection Against Dilution
The Exercise Price, the number and nature of securities which may be purchased upon the exercise of rights and the number of rights outstanding are subject to adjustment from time to time to prevent dilution in the event of stock dividends, subdivisions, consolidations, reclassifications or other changes in the outstanding common shares,pro rata distributions to holders of common shares and other circumstances where adjustments are required to appropriately protect the interests of the holders of rights.
Fiduciary Duty of Board
The Rights Plan will not detract from or lessen the duty of the Board to act honestly and in good faith with a view to the best interests of the Company and its shareholders. The Board will continue to have the duty and power to take such actions and make such recommendations to the Company'sCompany’s shareholders as are considered appropriate.
Exemptions for Investment Advisors
Fund managers, investment advisors (for fully-managed accounts), trust companies (acting in their capacities as trustees and administrators), statutory bodies whose business includes the management of funds, and administrators of registered pension plans are exempt from triggering a Flip-in Event, provided that they are not making, or are not part of a group making, a take-over bid.
Term
The Rights Plan will expire (the "Expiration Time"“Expiration Time”) on the earlier of the first annual meeting of shareholders of the Company following March 29, 2016, being the sixth anniversary of the Effective Date (subject to the approval of the resolution by the shareholders at the Meeting and reconfirmation at the first annual meeting of shareholders of the Company following March 29, 2013 (being the third anniversary of the Effective Date)) and the time at which the right to exercise rights shall terminate pursuant to the provisions of the Rights Plan pertaining to the redemption of rights and the waiver of the application of the Rights Plan, after which time it will automatically terminate.
Action Necessary to Change Rights of Shareholders
In order to change the rights of its shareholders, the Company would need to amend its Restated Articles of Incorporation to effect the change. Such an amendment would require the approval of holders of two-thirds of the issued and outstanding shares cast at a duly called special meeting. For certain amendments such as those creating a class of Preferred Shares, a shareholder is entitled under the CBCA to dissent in respect of such a resolution amending the Restated Articles of Incorporation and, if the resolution is adopted and the Company implements such changes, demand payment of the fair value of its shares.
Disclosure of Share Ownership
In general, under applicable securities regulation in Canada, a person or company who beneficially owns, or who exercises control or direction over directly or indirectly, voting securities of a reporting issuerissuer; voting securities of an issuer or a combination of both, carrying more than ten percent of the voting rights attached to all the issuer'sissuer’s outstanding voting securities is an insider and must, within ten days of becoming an insider, file a report in the required form effective the date on which the person became an insider, disclosing any direct or indirect beneficial ownership of, or control or direction over, securities of the reporting issuer.
Additionally, securities regulation in Canada provides for the filing of a report by an insider of a reporting issuer whose holdings change, which report must be filed within five days from the day on which the change takes place.
Section 13 of theUnited States Securities Exchange Act of 1934 (the "Exchange Act"“Exchange Act”) imposes reporting requirements on persons who acquire beneficial ownership (as such term is defined in the Rule 13d-3 under the Exchange Act) of more than five percent of a class of an equity security registered under Section 12 of the Exchange Act. The Company'sCompany’s common shares are so registered. In general, such persons must file, within ten days after such acquisition, a report of beneficial ownership with the SEC containing the information prescribed by the regulations under Section 13 of the Exchange Act. This information is also required to be sent to the issuer of the securities and to each exchange where the securities are traded.
Meeting of Shareholders
An annual meeting of shareholders is held each year for the purpose of considering the financial statements and reports, electing directors, appointing auditors and fixing or authorizing the Board to fix their remuneration and for the transaction of other business as may properly come before a meeting of shareholders. Any annual meeting may also constitute a special meeting to take cognizance and dispose of any matter of which a special meeting may take cognizance and dispose. Under the bylaws, the presidentChief Executive Officer or the President of the Company has the power to call a meeting of shareholders.
The CBCA provides that the holders of not less than 5% of the outstanding voting shares of the Company may requisition the directors of the Company to call a meeting of shareholders for the purpose stated in the requisition. Except in limited circumstances, including where a meeting of shareholders has already been called and a notice of meeting already given or where it is clear that the primary purpose of the requisition is to redress a personal grievance against the Company or its directors, officers or shareholders, the directors of the Company, on receipt of such requisition, must call a meeting of shareholders. If the directors fail to call a meeting of shareholders within twenty-one days after receiving the requisition, any shareholder who signed the requisition may call the meeting of shareholders and, unless the shareholders resolve otherwise at the meeting, the Company shall reimburse the shareholders for the expenses reasonably incurred by them in requisitioning, calling and holding the meeting of shareholders.
The CBCA also provides that, except in limited circumstances, a resolution in writing signed by all of the shareholders entitled to vote on that resolution at a meeting of shareholders is as valid as if it had been passed at a meeting of shareholders.
A quorum of shareholders is present at an annual or special meeting of shareholders, regardless of the number of persons present in person at the meeting, if the holder or holders of shares representing at least 10% (subject to ratification at the Company's annual and special meeting of shareholders to be held on May 18, 2011) of the outstanding voting shares at such meeting are present in person or represented in accordance with the Company'sCompany’s bylaws. In the case where the CBCA, the Restated Articles of Incorporation or the bylaws of the Company require or permit the vote by class of holders
of a given class of shares of the share capital of the Company, the quorum at any meeting will be one or more persons representing 10% of the outstanding shares of such class.
Notice of the time and place of each annual or special meeting of shareholders must be given not less than 21 days, nor more than 50 days, before the date of each meeting to each director, to the auditor and to each shareholder entitled to vote thereat. If the address of any shareholder, director or auditor does not appear in the books of the Company, the notice may be sent to such address as the person sending the notice may consider to be most likely to reach such shareholder, director or auditor promptly. Every person who, by operation of the CBCA, transfers or by any other means whatsoever, becomes entitled to any share, shall be bound by every notice given in respect of such share which, prior to the entry of his or her name and address on the register of the Company, is given to the person whose name appears on the register at the time such notice is sent. Notice of meeting of shareholders called for any other purpose other than consideration of the financial statements and auditor'sauditor’s report, election of directors and reappointment of the incumbent auditor, must state the nature of the business in sufficient detail to permit the shareholder to form a reasoned judgment on and must state the text of any special resolution or bylaw to be submitted to the meeting.
Limitations on Right to Own Securities
Neither Canadian law nor the Company'sCompany’s Restated Articles of Incorporation or bylaws limit the right of a non-resident to hold or vote common shares, other than as provided in theInvestment Canada Act (the "Investment Act"“Investment Act”). The Investment Act prohibits implementation of certain direct reviewable investments by an individual, government or agency thereof, corporation, partnership, trust or joint venture that is not a "Canadian,"“Canadian”, as defined in the Investment Act (a "non-Canadian"“non-Canadian”), unless, after review, the minister responsible for the Investment Act is satisfied or is deemed to be satisfied that the investment is likely to be of net benefit to Canada. An investment in the common shares of the Company by a non-Canadian (other than a "WTO Investor,"“WTO Investor”, as defined below) would be reviewable under the Investment Act if it were an investment to acquire direct control of the Company, and the book value of the assets of the Company were CAN$5 million or more (provided that immediately prior to the implementation of the investment the Company was not controlled by WTO Investors). Subject to the Amendments (as defined below), an investment in common shares of the Company by a WTO Investor would be reviewable under the Investment Act if it were an investment to acquire direct control of the Company and the value of the assets of the Company equalled or exceeded CAN$312330 million (for 2011)2012). A non-Canadian, whether a WTO Investor or otherwise, would be deemed to acquire control of the Company for purposes of the Investment Act if he or she acquired a majority of the common shares of the Company. The acquisition of less than a majority, but at least one-third of the shares, would be presumed to be an acquisition of control of the Company, unless it could be established that the Company was not controlled in fact by the acquirer through the ownership of the shares. In general, an individual is a WTO Investor if he or she is a "national"“national” of a country (other than Canada) that is a member of the World Trade Organization ("(“WTO Member"Member”) or has a right of permanent residence in a WTO Member. A corporation or other entity will be a "WTO Investor"“WTO Investor” if it is a "WTO“WTO Investor-controlled entity,"entity”, pursuant to detailed rules set out in the Investment Act. The United States is a WTO Member. Certain transactions involving the common shares would be exempt from the Investment Act, including: (a) an acquisition of the shares if the acquisition were made in the ordinary course of that person'sperson’s business as a trader or dealer in securities; (b) an acquisition of control of the Company in connection with the realization of a security interest granted for a loan or other financial assistance and not for any purpose related to the provisions of the Investment Act; and (c) an acquisition of control of the Company by reason of an amalgamation, merger, consolidation or corporate reorganization, following which the ultimate direct or indirect control in fact of the Company, through the ownership of voting interests, remains unchanged.
The Canadian Federal Government adopted certain amendments (the "Amendments"“Amendments”) to the Investment Act in 2009. Some of the Amendments, which came into force on February 6, 2009, introduce a national security test and review process, authorizing the Canadian Minister of Industry to review investments that "could“could be injurious
to national security,"security”, regardless of the size of the transaction. Some of the other Amendments will come into force on a day to be fixed by order of the Canadian Governor in Council, including the increase to the thresholds that trigger governmental review for WTO Investors. Therefore, the thresholds for the review of direct acquisitions of control by WTO Investors would increase from the current CAN$312330 million (based on book value) to CAN$600 million (to be based on the "enterprise value"“enterprise value” of the Canadian business) for the two years after such Amendments come into force, to CAN$800 million in the following two years and then to CAN$1 billion for the next two years. Thereafter, the thresholds are to be adjusted to account for inflation. A number of the Amendments still require additional definition and details, which will be set forth in regulations promulgated under the Investment Act.
There are no limits on the rights of non-Canadians to exercise voting rights on their common shares of the Company.
C. Material contracts
| C. | Material contracts |
Other than as disclosed herein under "Shareholder“Shareholder Rights Plan"Plan” and below, and except for contracts entered into in the ordinary course of business, there are no material contracts to which the Company or any of its subsidiaries is a party other than the employment agreements and change of control agreements with our executive officers as described below.party.
Employment Agreements
The Company and/or its subsidiaries have entered into employment agreements (the "Employment Agreements"“Employment Agreements”) with each of the Named Executive Officers. The Employment Agreements provide that we will pay the Named Executive Officers a base salary and an annual bonus and that such executives will be eligible to receive grants of stock options which will be reviewed annually in accordance with our policies. The Employment Agreements have an indefinite term. However, in addition to his Employment Agreement, Dr. Engel had previously entered into a service contract in his prior capacity as Managing Director with AEZS GmbH, our Company’s principal operating subsidiary, which service contract has an indefinite term. Each of the Employment Agreements provides that, if we terminate the employment of a Named Executive Officer without cause, then the executive will be entitled to receive, in the case of Dr. Engel, a lumpsumlump-sum payment, less statutory deductions, of the equivalent of 24 months of his then applicable base salary, an amount equivalent to twice the annual bonus received for the most recently completed year and an amount equivalent to twelve months of the cost of the other benefits to which he is entitled. In the case of Mr. Turpin, the lump sumlump-sum payment will be equivalent to 18 months of his then applicable base salary, 1.5 times the annual bonus of the preceding year and 18 months of the value of the other benefits to which he is entitled. In the case of Dr. Blake and Messrs. Pelliccione and Seeber, they are entitled to receive, upon termination of employment without cause, a lump sumlump-sum payment equivalent to twelve months of their then applicable base salaries, an amount equivalent to the annual bonus received for the preceding year and twelve months of the value of the other benefits to which they are entitled.
Furthermore, each Named Executive Officer shall not, directly or indirectly, solicit any of our customers for the purpose or intent of selling them any products which are similar to or otherwise competing with our products; nor shall any Named Executive Officer induce, entice or otherwise attempt to directly or indirectly hire or engage any of our employees, for a period equal to one year following such executive'sexecutive’s termination of employment with the Company.
Pursuant to the Employment Agreements, each of the Named Executive Officers is also entitled to certain payments (the "Change“Change of Control Payments"Payments”) in the event (i) a "Change“Change of Control"Control” occurs and (ii) during the twelve-month period following the Change of Control, either the Company terminates the employment of the executive "without Cause"“without Cause” or if the executive terminates his or her employment "for“for Good Reason"Reason”.
The Change of Control Payments are as follows:
| — | for Dr. Engel and Mr. Seeber, (i) the equivalent of 24 months of their then prevailing annual base salaries, (ii) an amount equivalent to twice the annual bonus, if any, which the executive would have been entitled to receive in the year during which the Change of Control occurred, and (iii) an amount equivalent to 24 months of the value of the benefits which were in force at the time of termination of the executive’s employment, calculated on a yearly basis, including car allowance, but excluding operating costs; |
| — | for Mr. Turpin, the Change of Control Payment would be the same as in the context of a termination of employment described above, except that the 1.5 multiple of his bonus payment would be based on his potential bonus for the year in which the Change of Control occurs as opposed to his actual bonus received for the preceding financial year; and |
| — | for Dr. Blake and Mr. Pelliccione (i) the equivalent of 18 months of their then prevailing annual base salaries, (ii) an amount equivalent to 1.5 times the annual bonus, if any, which the executive would have been entitled to receive in the year during which the Change of Control occurred, and (iii) an amount equivalent to 18 months of the value of the benefits which were in force at the time of termination of the executive’s employment, calculated on a yearly basis, including car allowance, but excluding operating costs. |
All Change of Control Payments described above are subject to applicable statutory withholdings. In addition, any outstanding stock options held by a Named Executive Officer are unaffected by the change of control provisions included in the Employment Agreements and, in the event of a Change of Control followed by termination of employment within twelve months, such stock options will be treated in accordance with the applicable provisions of the Stock Option Plan described elsewhere in this annual report.
For the purposes of the Employment Agreements (including the annexes and schedules thereto):
| — | a “Change of Control” shall be deemed to have occurred in any of the following circumstances: (i) subject to certain exceptions, upon the acquisition by a person (or one or more persons who are affiliates of one another or who are acting jointly or in concert) of a beneficial interest in securities of the Company representing in any circumstance 50% or more of the voting rights attaching to the then outstanding securities of the Company; (ii) upon a sale or other disposition of all or substantially all of the Company’s assets; (iii) upon a plan of liquidation or dissolution of the Company; or (iv) if, for any reason, including an amalgamation, merger or consolidation of the Company with or into another company, the individuals who, as at the date of the relevant Employment Agreement, constituted the Board (and any new directors whose appointment by the Board or whose nomination for election by the Company’s shareholders was approved by a vote of at least two-thirds of the directors then still in office who either were directors as at the date of the relevant Employment Agreement or whose appointment or nomination for election was previously so approved) cease to constitute a majority of the members of the Board; |
| — | termination of employment by the Company “for Cause” includes (but is not limited to) (i) if the executive commits any fraud, theft, embezzlement or other criminal act of a similar nature, and (ii) if the executive is guilty of serious misconduct or willful negligence in the performance of his duties; and |
| — | termination of employment by the executive officer for “Good Reason” means the occurrence, without the executive’s express written consent, of any of the following acts: (i) a material reduction of the executive’s total compensation (including annual base salary plus annual bonus, benefits and number of stock options) as in effect on the date of the relevant Employment Agreement or as same may be increased from time to time; (ii) a material reduction or change in the executive’s duties, authority, responsibilities, accountability or a change in the business or corporate structure of the Company which materially affects his or her authority, compensation or ability to perform duties or responsibilities (such as shifting from a policy-making to a policy-implementation position); (iii) a forced relocation; or (iv) a material change in the terms and conditions of the change of control provisions included in the relevant Employment Agreement. |
Other Material Contracts
Cetrotide®
In DecemberNovember 2008, we signed a definitive agreement to sell to CHRP our rights to royalties on future sales of Cetrotide®Cetrotide® covered by our license agreement with Merck Serono. This license agreement was signed in 2000 and amended in 2003 and granted Merck Serono exclusive rights to market, distribute and sell Cetrotide®Cetrotide® worldwide, with the exception of Japan, in the field ofin vitro fertilization. On closing, we received $52.5 million from CHRP (less transaction costs of $1.0 million) and, upon net sales of Cetrotide®Cetrotide® having reached a specified level in 2010, we received an additional payment of $2.5 million from CHRP in February 2011. Under the terms of the agreement, if cetrorelix is approved for sale by the European regulatory authorities in an indication other thanin vitro fertilization, we have agreed to make a one-time cash payment to CHRP in an amount ranging from $5 million up to $15 million.
Perifosine
We are party to a license and collaboration agreement with Keryx. Under the terms of this agreement, Keryx undertakes, at its own cost, all development activities necessary to obtain regulatory and marketing approvals of perifosine, a signal transduction inhibitor, for all uses in the United States, Canada and Mexico. The agreement provides for, among other things, the availability of data generated by both parties free of charge. In September 2002, we received an upfront payment of approximately $0.5 million and are eligible to receive payments of up to an aggregate of $18.3 million upon Keryx'sKeryx’s successful achievement of clinical development and regulatory milestones, in addition to scale-up royalties (from high single to low double-digit) on future net sales in the United States, Canada and Mexico. The license and collaboration agreement terminates upon the later of the expiration of all underlying patent rights or ten years from the first commercial sale of perifosine in any of the covered territories. In addition, Keryx may, at its option, terminate the agreement with respect to a country in the covered territories or as to the entire agreement at any time by giving written notice to us. Upon such termination, the license shall terminate and Keryx shall pay us any accrued amounts payable under the agreement. The agreement may also be terminated for material breach by the other party and in the certain events of bankruptcy or insolvency of the other party."
On March 8, 2011, we entered into an agreement with Yakult for the development, manufacture and commercialization of perifosine in all human uses, excluding leishmaniasis, in Japan. Under the terms of this agreement, Yakult will makehas made an initial non-refundable upfront payment to us of €6.0 million (approximately $8.3$8.4 million). Also per the agreement, we will be entitled to receive up to a total of €44.0 million (approximately $60.9$57.1 million) upon achieving certain pre-established milestones, including clinical and regulatory events in Japan. Furthermore, we will be entitled to receive double-digit royalties on future net sales of perifosine in the Japanese market. We have also agreed to supply perifosine to Yakult on a cost-plus-basis. In addition, under the agreement, we agreed to use commercially reasonable efforts to develop, manufacture and commercialize perifosine outside of Japan for specific indications, and Yakult is responsible for the development, registration and
commercialization of perifosine in Japan. Either party may terminate the agreement for the material, uncured breach by the other party and in certain events of bankruptcy or insolvency of the other party.
D. Exchange controls
| D. | Exchange controls |
Canada has no system of exchange controls. There are no exchange restrictions on borrowing from foreign countries or on the remittance of dividends, interest, royalties and similar payments, management fees, loan repayments, settlement of trade debts or the repatriation of capital.
E. Taxation
| E. | Taxation |
THE FOLLOWING SUMMARY IS OF A GENERAL NATURE ONLY AND IS NOT INTENDED TO BE, NOR SHOULD IT BE CONSTRUED TO BE, LEGAL OR TAX ADVICE TO ANY PARTICULAR HOLDER. CONSEQUENTLY, HOLDERS ARE URGED TO CONSULT THEIR OWN TAX ADVISORS FOR ADVICE AS TO THE TAX CONSEQUENCES OF AN INVESTMENT IN THE COMMON SHARES HAVING REGARD TO THEIR PARTICULAR CIRCUMSTANCES.
Material Canadian Income Tax Considerations
The following summary describes the principal Canadian federal income tax consequencesconsiderations to a purchaserholder who acquires common shares (a "holder"“holder”) and who, for the purposes of the Canadian federal Income Tax Act, R.S.C. 1985, as amended (the "Tax Act"“Tax Act”), and at all relevant times, deals at arm'sarm’s length with, and is not affiliated with, the CorporationCompany or a subsequent purchaser of such common shares and holds their common shares as capital property. Common shares will generally be considered to be capital property to a holder for purposes of the Tax Act unless either the holder holds such common shares in the course of carrying on a business of trading or dealing in securities, or the holder has held or acquired such common shares in a transaction or transactions considered to be an adventure in the nature of trade.
This summary is not applicable to a holder (i) that is a “financial institution”, as defined in the Tax Act for purposes of the mark-to-market rules, (ii), that is a “specified financial institution”, as defined in the Tax Act, (iii) an interest in which iswould be a "tax“tax shelter investment"investment” as defined in the Tax Act, or to(iv) that has made a holder which is a "financial institution" as defined infunctional currency reporting election for purposes of the Tax Act subject to the "mark-to-market" rules set out therein.Act. Such holders should consult their own tax advisors.
This summary is based upon the current provisions of the Tax Act and the regulations promulgated thereunder (the "Regulations"“Regulations”) and the Company'sCompany’s understanding of the current published administrative practicespolicies and policiesassessing practices of the Canada Revenue Agency ("CRA"(“CRA”). It also takes into account all proposed amendments to the Tax Act and the Regulations publicly released by the Minister of Finance (Canada) ("Tax Proposals") prior to the date hereof (“Tax Proposals”), and assumes that all such Tax Proposals will be enacted as currently proposed. No assurance can be given that the Tax Proposals will be enacted in the form proposed or at all. This summary does not otherwise take into account or anticipate any changes in law or administrative or assessing practice or policy of the CRA, whether by way of legislative, regulatory, judicial or administrative action or interpretation, nor does it address any provincial, local, territorial or foreign tax considerations.
Holders Not Resident in Canada
The following discussion applies to a holder of common shares who, at all relevant times, for purposes of the Tax Act and any applicable income tax treaty or convention,, is neither resident nor deemed to be resident in Canada and does not, and is not deemed to, use or hold common shares in carrying on a business or part of a business in Canada (a "Non-Resident holder"“Non-Resident holder”). In addition, this discussion does not apply to an insurer who carries on an insurance business in Canada and elsewhere or to an authorized foreign bank (as defined in the Tax Act).
Disposition of Common Shares
A Non-Resident holder generally will not be subject to tax under the Tax Act in respect of any capital gain realized by such Non-Resident holder on a disposition or deemed disposition of common shares unless such shares constitute
"taxable “taxable Canadian property"property” (as defined in the Tax Act) of the Non-Resident holder at the time of disposition and the holdergain is not entitledexempt from tax pursuant to relief underthe terms of an applicable income tax treaty or convention.
Common shares will generally not constitute taxable Canadian property to a Non-Resident holder at a particular time provided that (a)As long as the common shares are listed on a designated stock exchange(which (which currently includes the NASDAQ and the TSX) and (b) at the time of their disposition, the common shares generally will not constitute taxable Canadian property of a Non-Resident holder, unless at any time during the 60-month period that endsimmediately preceding the
disposition: (i) the Non-Resident holder, persons with whom the Non-Resident holder did not deal at arm’s length, or the time the common shares are disposed of, both (i)Non-Resident holder together with all such persons, owned 25% or more of the issued shares of any class or series of shares of the Company were not owned bycorporation; and did not belong to one or any combination of the Non-Resident holder and persons with whom the Non-Resident holder did not deal at arm's length, and (ii) not more than 50% of the fair market value of the common shares of the corporation was derived directly or indirectly from one or any combination of real or immovable property situated in Canada, Canadian“Canadian resource properties, timberproperties” ( as defined in the Tax Act), “timber resource properties andproperties” (as defined in the Tax Act) or options in respect of, or interests in, or for civil law rights in, such properties,property whether or not thesuch property exists.
A Non-Resident holder’s capital gain (or capital loss) in respect of common shares that constitute or are deemed to constitute taxable Canadian property ( and are not “treaty-protected property” as defined for purposes of the Tax Act) will generally be computed in the manner described below under the heading“Holders Resident in Canada — Disposition of Common Shares”.
If the common shares were to cease being listed on the NASDAQ, the TSX or another "recognized“recognized stock exchange"exchange”, a Non-Resident holder who disposes of common shares that are taxable Canadian property may be required to fulfill the requirements of section 116 of the Tax Act. An exemption from such requirements is available on the disposition of "treaty-protected property", which is
Non-Resident holders whose common shares are taxable Canadian property any income or gain on the disposition of which is exempt fromshould consult their own tax under Part I of the Tax Act as a result of an applicable income tax treaty or convention.advisors.
Taxation of Dividends on Common Shares
Dividends paid or credited or deemed to be paid or credited on the common shares to a Non-Resident holder will beby the corporation are subject to Canadian withholding tax at athe rate of 25%. Such withholding tax may be unless reduced by virtue of the provisionsterms of an incomeapplicable tax treaty or convention between Canada and the country of which the Non-Resident holder is a resident. convention.
Under the Canada-UnitedCanada — United States Income Tax Convention (1980) (the "Convention"“Convention”), as amended, the rate of withholding tax in respect ofon dividends paid or deemed dividends beneficially owned bycredited to a residentNon-Resident holder who is the beneficial owner of the United Statesdividends, is resident in the U.S. for purposes of the Convention and entitled to the benefits of the Convention (a “U.S. holder”) is generally reducedlimited to 15% of the gross amount of the dividend (or 5% in the case of a U.S. holder that is a company beneficially owning at least 10% of the corporation’s voting shares). Non-Resident holders should consult their own tax advisors.
Holders Resident in Canada
The following discussion applies to a holder of common shares who, at all relevant times, for purposes of the Tax Act, and any applicable income tax treaty or convention, is or is deemed to be resident in Canada (a "Canadian holder"“Canadian holder”). Certain Canadian holders whose common shares might not otherwise qualify as capital property may, in certain circumstances, treat the common shares and every other "Canadian security"“Canadian security” (as defined in the Tax Act) owned by the Canadian holder as capital property by making an irrevocable election provided by subsection 39(4) of the Tax Act.
Taxation of Dividends on Common Shares
Dividends received or deemed to behave been received on the common shares will be included in a Canadian holder'sholder’s income for purposes of the Tax Act. Such dividends received or deemed to behave been received by a Canadian holder that is an individual (other than certain trusts) will be subject to the gross-up and dividend tax credit rules generally applicable under the Tax Act in respect of dividends received on shares of taxable Canadian corporations. Generally, a dividend will be eligible for the enhanced gross-up and dividend tax credit if the recipient receives written notice from the corporation designatingdesignates the dividend as an "eligible dividend"“eligible dividend” (within the meaning of the Tax Act). in accordance with the provisions of the Tax Act. There may be limitations on the ability of the Company to designate dividends as eligible dividends. A Canadian holder that is a corporation will be required to include such dividends in computing its income and will generally be entitled to deduct the amount of such dividends in computing its taxable income. A Canadian holder that is a "private corporation"“private corporation” or a "subject corporation"“subject corporation” (as such terms are defined in the Tax Act), may be liable under Part IV of the Tax Act to pay a refundable tax of 331/3% on
dividends received or deemed to behave been received on the common shares to the extent such dividends are deductible in computing the holder'sholder’s taxable income.
Disposition of Common Shares
A disposition, or a deemed disposition, of a common share by a Canadian holder will generally give rise to a capital gain (or a capital loss) equal to the amount by which the proceeds of disposition of the share, net of any reasonable costs of disposition, exceed (or are less than) the adjusted cost base of the share to the holder. Such capital gain (or capital loss) will be subject to the treatment described below under "Taxation“Taxation of Capital Gains and Capital Losses"Losses”.
Additional Refundable Tax
A Canadian holder that is a "Canadian-controlled“Canadian-controlled private corporation"corporation” (as such term is defined in the Tax Act) may be liable to pay an additional refundable tax of 62/3% 2/3% on certain investment income including amounts in respect of "Taxable“Taxable Capital Gains"Gains”, as defined below.
Taxation of Capital Gains and Capital Losses
In general, one half of any capital gain (a "Taxable“Taxable Capital Gain"Gain”) realized by a Canadian holder in a taxation year will be included in the holder'sholder’s income in the year. Subject to and in accordance with the provisions of the Tax Act, one half of any capital loss (an "Allowable“Allowable Capital Loss"Loss”) realized by a Canadian holder in a taxation year must be deducted from Taxable Capital Gains realized by the holder in the year and Allowable Capital Losses in excess of Taxable Capital Gains may be carried back and deducted in any of the three preceding taxation years or carried forward and deducted in any subsequent taxation year against net Taxable Capital Gains realized in such years. The amount of any capital loss realized by a Canadian holder that is a corporation on the disposition or deemed disposition of a common share may be reduced by the amount of dividends received or deemed to behave been received by it on such common share (or on a share for which the common share has been substituted) to the extent and under the circumstances prescribed by the Tax Act. Similar rules may apply where a corporation is a member of a partnership or a beneficiary of a trust that owns common shares, directly or indirectly, through a partnership or a trust. A Taxable Capital Gain realized and taxable dividends received or deemed to have been received by a Canadian holder who is an individual (including a trust, other than certain specified trusts) may give rise to liability for alternative minimum tax.
CertainMaterial U.S. Federal Income Tax Considerations
The following discussion is a summary of certainmaterial U.S. federal income tax consequences applicable to the ownership and disposition of common shares by a U.S. Holder (as defined below), but does not purport to be a complete analysis of all potential U.S. federal income tax effects. This summary is based on the Internal Revenue Code of 1986, as amended (the "Code"“Code”), U.S. Treasury regulations promulgated thereunder, Internal Revenue Service ("IRS")IRS rulings and judicial decisions in effect on the date hereof. All of these are subject to change, possibly with retroactive effect, or different interpretations.
This summary does not address all aspects of U.S. federal income taxation that may be relevant to particular U.S. Holders in light of their specific circumstances (for example, U.S. Holders subject to the alternative minimum tax provisions of the Code) or to holders that may be subject to special rules under U.S. federal income tax law. This summary also does not address the tax consequences of holding, exercising or disposing of warrants in the Company. If the Company is a "passive foreign investment company" ("PFIC"),PFIC, as defined below, U.S. Holders of its warrants will be subject to adverse tax rules and will not be able to make the mark-to-market or the QEF election described below with respect to such warrants. U.S. Holders of warrants should consult their tax advisors with regard to the U.S. federal income tax consequences of holding, exercising or disposing of warrants in the Company, including in the situation in which the Company is classified as a PFIC.
This summary also does not discuss any aspect of state, local or foreign law, or estate or gift tax law as applicable to U.S. Holders. U.S. Holders should consult their tax advisors about the potential application of such laws and the application of the U.S. federal income tax rules summarized below to their particular situation. In addition, this discussion is limited to U.S. Holders holding common shares as capital assets. For purposes of this summary, "U.S. Holder"“U.S. Holder” means a beneficial holder of common shares who or that for U.S. federal income tax purposes is:
| — | an individual citizen or resident of the United States; |
| — | a corporation or other entity classified as a corporation for U.S. federal income tax purposes created or organized in or under the laws of the United States, any state thereof or the District of Columbia; |
| — | an estate, the income of which is subject to U.S. federal income taxation regardless of its source; or |
| — | a trust, if a court within the United States is able to exercise primary supervision over the administration of such trust and one or more “U.S. persons” (within the meaning of the Code) have the authority to control all substantial decisions of the trust, or if a valid election is in effect for it to be treated as a U.S. person for U.S. federal income tax purposes. |
If a partnership or other entity or arrangement classified as a partnership for U.S. federal income tax purposes holds common shares, the U.S. federal income tax treatment of a partner generally will depend on the status of the partner and the activities of the partnership. This summary does not address the tax consequences to any such partner. Such a partner should consult its own tax advisor as to the tax consequences of the partnership owning and disposing of common shares.
Dividends
Subject to the PFIC rules discussed below, any distributions paid by the Company out of current or accumulated earnings and profits (as determined for U.S. federal income tax purposes), before reduction for any Canadian withholding tax paid with respect thereto, will generally be taxable to a U.S. Holder as foreign source dividend income, and will not be eligible for the dividends received deduction generally allowed to corporations. Distributions in excess of current and accumulated earnings and profits will be treated as a non-taxable return of capital to the extent of the U.S. Holder'sHolder’s adjusted tax basis in the common shares and thereafter as capital gain. U.S. Holders should consult their own tax advisors with respect to the appropriate U.S. federal income tax treatment of any distribution received from the Company.
For taxable years beginning before January 1, 2013, dividends paid by the Company should be taxable to a non-corporate U.S. Holder at the special reduced rate normally applicable to long term capital gains, provided that certain conditions are satisfied. A U.S. Holder will not be able to claim the reduced rate if the Company is treated as a PFIC for the taxable year in which the dividend is paid or the preceding year. See "Passive“Passive Foreign Investment Company Considerations"Considerations” below.
Under current law, payments of dividends by the Company to non-Canadian investors are generally subject to a 25 percent Canadian withholding tax. The rate of withholding tax applicable to U.S. Holders that are eligible for benefits under the Canada-United States Tax Convention (the "Convention"“Convention”) is reduced to a maximum of 15 percent. This reduced rate of withholding will not apply if the dividends received by a U.S. Holder are effectively connected with a permanent establishment of the U.S. Holder in Canada. For U.S. federal income tax purposes, U.S. Holders will be treated as having received the amount of Canadian taxes withheld by the Company, and as then having paid over the withheld taxes to the Canadian taxing authorities. As a result of this rule, the amount of dividend income included in gross income for U.S. federal income tax purposes by a
U.S. Holder with respect to a payment of dividends may be greater than the amount of cash actually received (or receivable) by the U.S. Holder from the Company with respect to the payment.
A U.S. Holder will generally be entitled, subject to certain limitations, to a credit against its U.S. federal income tax liability, or a deduction in computing its U.S. federal taxable income, for Canadian income taxes withheld by the Company.
U.S. Holders For purposes of the foreign tax credit limitation, dividends paid by the Company generally will constitute foreign source income in the “passive category income” basket. The foreign tax credit rules are complex and prospective purchasers should consult their tax advisors concerning the foreign tax credit implications of the payment of Canadian taxes.
Dividends paid in Canadian dollars will be included in the gross income of a U.S. Holder in a U.S. dollar amount calculated by reference to the exchange rate in effect on the date the U.S. Holder receives the dividend, regardless of whether such Canadian dollars are actually converted into U.S. dollars at that time. Gain or loss, if any, realized on a sale or other disposition of the Canadian dollars will generally be U.S. source ordinary income or loss to a U.S. Holder.
The Company generally does not pay any dividends and does not anticipate paying any dividends in the foreseeable future.
Sale or Other Taxable Disposition
Subject to the PFIC rules discussed below, upon a sale or other taxable disposition of common shares, a U.S. Holder generally will recognize capital gain or loss for U.S. federal income tax purposes equal to the difference, if any, between the amount realized on the sale or other taxable disposition and the U.S. Holder'sHolder’s adjusted tax basis in the common shares.
This capital gain or loss will be long-term capital gain or loss if the U.S. Holder'sHolder’s holding period in the common shares exceeds one year. For taxable years beginning before January 1, 2013, the rates of taxation for long-term capital gains of non-corporate U.S. Holders are reduced compared to such rates thereafter provided that certain conditions are satisfied. The deductibility of capital losses is subject to limitations. Any gain or loss will generally be U.S. source for U.S. foreign tax credit purposes.
Passive Foreign Investment Company Considerations
A foreign corporation will be classified as a PFIC for any taxable year in which, after taking into account the income and assets of the corporation and certain subsidiaries pursuant to applicable "look-through rules,"“look-through rules”, either (i) at least 75% of its gross income is "passive income"“passive income” or (ii) at least 50% of the average value of its assets is attributable to assets which produce passive income or are held for the production of passive income.
The Company believes it was not a PFIC for the 20102011 taxable year. However, since the fair market value of the Company'sCompany’s assets may be determined in large part by the market price of the common shares, which is likely to fluctuate, and the composition of the Company'sCompany’s income and assets will be affected by how, and how quickly, the Company spends any cash that is raised in any financing transaction,transaction. Thus, no assurance can be provided that the Company will not be classified as a PFIC for the 20112012 taxable year and for any future taxable year.
If the Company is classified as a PFIC for any taxable year during which a U.S. Holder owns common shares, the U.S. Holder, absent certain elections (including the mark-to-market election described below), will generally be subject to adverse rules (regardless of whether the Company continues to be classified as a PFIC) with respect to (i) any "excess distributions"“excess distributions” (generally, any distributions received by the U.S. Holder on the common shares in a taxable year that are greater than 125% of the average annual distributions received by the U.S. Holder in the three preceding taxable years or, if shorter, the U.S. Holder'sHolder’s holding period for the common shares) and (ii) any gain realized on the sale or other disposition of the common shares.
Under these adverse rules (a) the excess distribution or gain will be allocated ratably over the U.S. Holder'sHolder’s holding period, (b) the amount allocated to the current taxable year and any taxable year prior to the first taxable year in which the Company is classified as a PFIC will be taxed as ordinary income, and (c) the amount allocated to each of the other taxable years during which the Company was classified as a PFIC will be subject to tax at the highest rate of tax in effect for the applicable class of taxpayer for that year and an interest charge will be imposed with respect to the resulting tax attributable to each such other taxable year.
U.S. Holders can avoid the interest charge described above by making a mark-to-market election with respect to the common shares, provided that common shares are "marketable."“marketable”. The common shares will be marketable if they are regularly traded on a qualified exchange or other market. For this purpose, the common shares generally will be considered to be regularly traded during any calendar year during which they are traded, other than inde minimis quantities, on at least 15 days during each calendar quarter. The common shares are currently listed and regularly traded on NASDAQ, which constitutes a qualified exchange. If the common shares were delisted from the NASDAQ and were not traded on another qualified exchange for the requisite time period described above, the mark-to-market election would not be available.
A U.S. Holder that makes a mark-to-market election must include in gross income, as ordinary income, for each taxable year an amount equal to the excess, if any, of the fair market value of the U.S. Holder'sHolder’s common shares at the close of the taxable year over the U.S. Holder'sHolder’s adjusted tax basis in the common shares. An electing U.S. Holder may also claim an ordinary loss deduction for the excess, if any, of the U.S. Holder'sHolder’s adjusted tax basis in the common shares over the fair market value of the common shares at the close of the taxable year, but this deduction is allowable only to the extent of any net mark-to-market gains for prior taxable years. A U.S. Holder that makes a mark-to-market election generally will adjust such U.S. Holder'sHolder’s tax basis in the common shares to reflect the amount included in gross income or allowed as a deduction because of such mark-to-market election. Gains from an actual sale or other disposition of the common shares will be treated as ordinary income, and any losses incurred on a sale or other disposition of the common shares will be treated as ordinary losses to the extent of any net mark-to-market gains for prior taxable years.
A mark-to-market election will be effective for the taxable year for which the election is made and all subsequent taxable years. The election cannot be revoked without the consent of the IRS unless the common shares cease to be marketable, in which case the election is automatically terminated. If the Company is classified as a PFIC for any taxable year in which a U.S. Holder owns common shares but before a mark-to-market election is made, the interest charge rules described above will apply to any mark-to-market gain recognized in the year the election is made.
In some cases, a shareholder of a PFIC can avoid the interest charge and the other adverse PFIC consequences described above by making a qualified electing fund ("QEF"(“QEF”) election to be taxed currently on its share of the PFIC'sPFIC’s undistributed income. If theThe Company is classified as a PFIC, it does not, however, expect to provide the information regarding its income that would be necessary in order for a U.S. Holder to make a QEF election with respect to common shares.shares if the Company is classified as a PFIC.
If the Company is classified as a PFIC, a U.S. Holder of common shares will generally be treated as owning stock owned by the Company in any direct or indirect subsidiaries that are also PFICs and will be subject to similar adverse rules with respect to distributions to the Company by, and dispositions by the Company of the stock of such subsidiaries. A mark-to-market election is not permitted for the shares of any subsidiary of the Company that is also classified as a PFIC.
If the Company is classified as a PFIC and then ceases to be so classified, a U.S. Holder may make an election (a "deemed“deemed sale election"election”) to be treated for U.S. federal income tax purposes as having sold such U.S. Holder'sHolder’s common shares on the last day of the taxable year of the Company
during which it was a PFIC. A U.S. Holder that made a deemed sale election would then cease to be treated as owning a stock in a PFIC by reason of ownership of common shares in the Company. However, gain recognized as a result of making the deemed sale election would be subject to the adverse rules described above.
Under recently enacted U.S. tax legislation and subject to future guidance, if the Company is a PFIC, U.S. Holders will be required to file for returns due after March 18, 2010, an annual information return with the IRS (on IRS Form 8621, which PFIC shareholders will be required to file with their income tax or information returns) relating to their ownership of common shares. Pursuant to Notice 2011-55, the IRS has suspended this new filing
requirement for U.S. Holders that are not otherwise required to file the current version of the IRS Form 8621 until the IRS releases a subsequent revision of IRS Form 8621, modified to reflect the recently enacted U.S. tax legislation. Guidance has not yet been issued regarding the information required to be included on such form.
This new filing requirement is in addition to any pre-existing reporting requirements that appliedapply to a U.S. Holder'sHolder’s interest in a PFIC (which the recently enacted tax legislation doesand IRS Notice 2011-55 do not affect). No additional guidance has yet been issued about the annual information return required under the recently enacted legislation, including on the information required to be reported on such return, the form of the return, or the due date for the return.
U.S. Holders should consult their tax advisors regarding the potential application of the PFIC regime and any reporting obligations to which they may be subject under that regime.
Information Reporting and Backup Withholding
The proceeds of a sale or other disposition of common shares, as well as dividends paid or deemed paid with respect to common shares by a U.S. payor, generally will be reported to the IRS and to the U.S. Holder as required under applicable regulations. Backup withholding tax may apply to these payments if the U.S. Holder fails to timely provide in the appropriate manner an accurate taxpayer identification number or otherwise fails to comply with, or establish an exemption from, such backup withholding tax requirements. Certain U.S. Holders are not subject to the information reporting or backup withholding tax requirements described herein. U.S. Holders should consult their tax advisors as to their qualification for exemption from backup withholding tax and the procedure for obtainingestablishing an exemption.
Backup withholding tax is not an additional tax. U.S. Holders generally will be allowed a refund or credit against their U.S. federal income tax liability for amounts withheld, provided the required information is timely furnished to the IRS.
Subject to specified exceptions and future guidance, recently enacted U.S. tax legislation generally requires a U.S. Holder (that is an individual or, to the extent provided in future guidance, a domestic entity) to report to the IRS on IRS Form 8938 such U.S. Holder'sHolder’s interests in stock or securities issued by a non-U.S. person (such as the Company) for taxable years beginning after March 18, 2010. Although expected, no guidance on this reporting requirement has yet been issued. U.S. Holders should consult their tax advisors regarding the information reporting obligations that may arise from their acquisition, ownership or disposition of common shares.
F. Dividends and paying agents
| F. | Dividends and paying agents |
Not applicable.
G. Statement by experts
| G. | Statement by experts |
Not applicable.
| H. | Documents on display |
H. Documents on display
In addition to placing our audited comparative annual financial statements before every annual meeting of shareholders as described above, we are subject to the information requirements of the Securities Exchange Act of 1934, as amended. In accordance with these requirements, we file and furnish reports and other information with the SEC. These materials, including this annual report on Form 20-F and the exhibits thereto, may be inspected and copied at the SEC'sSEC’s Public Reference Room at 100 F Street, N.E., Room 1580, Washington, D.C. 20549. The public may obtain information on the operation of the SEC'sSEC’s Public Reference Room by calling the SEC in the United States at 1-800-SEC-0330. The SEC also maintains a website at www.sec.gov that contains reports, proxy statements and other information regarding registrants that file electronically with the SEC. The Company'sCompany’s annual reports and some of the other information submitted by the Company to the SEC may be accessed through this website. In addition, material filed by the Company can be inspected on the Canadian Securities Administrators'Administrators’ electronic filing system, SEDAR, accessible at the website www.sedar.com. This material
includes the Company'sCompany’s Management Information Circular for its annual meeting to be held on May 18, 20119, 2012 to be furnished to the SEC on Form 6-K, which provides information including directors'directors’ and officers'officers’ remuneration and indebtedness and principal holders of securities. Additional financial information is provided in our audited annual financial statements for the year ended December 31, 20102011 and our MD&A relating to these statements included elsewhere in this annual report. These documents are also accessible on SEDAR (www.sedar.com) and on EDGAR (www.sec.gov).
I. Subsidiary information
| I. | Subsidiary information |
The subsidiaries of the Company are set forth under "Item“Item 4C. — Organizational Structure"Structure”.
Item 11. Quantitative and Qualitative Disclosures About Market Risk
| Item 11. | Quantitative and Qualitative Disclosures About Market Risk |
We have not entered into any forward currency contracts or other financial derivatives to hedge foreign exchange risk. We are therefore subject to foreign currency transaction and translation gains and losses.
Fair value
The Company has establishedclassifies its financial instruments in the following classifications for itscategories: “Financial assets at fair value through profit or loss (“FVTPL”)”; “Loans and receivables”; “Financial liabilities at FVTPL”; and “Other financial instruments:liabilities”.
| — | Financial assets at FVTPL are comprised exclusively of the Company’s short-term investment. |
| — | The Company’s loans and receivables are comprised of cash and cash equivalents, trade and other receivables and restricted cash. |
| — | Financial liabilities at FVTPL are currently comprised of the Company’s warrant liability. |
| — | Other financial liabilities include trade accounts payable and accrued liabilities, long-term payable and other long-term liabilities. |
The carrying values of all of the aforementioned financial instruments, excluding cash and cash equivalents, short-term investment, and restricted cash and warrant liability which are stated at fair value, approximate their fair values due to their short-term maturity or to the prevailing interest rates of these instruments, which are comparable to those of the market.
Financial risk managementfactors
DisclosuresThe following provides disclosures relating to the nature and extent of the Company'sCompany’s exposure to risks arising from financial instruments, including credit risk, liquidity risk foreign currencyand market risk (share price risk and interest rate risk,currency risk), and how the Company manages those risks, are presented below.
Table of Contentsrisks.
a)(a) Credit risk
Credit risk is the risk of an unexpected loss if a customer or counterparty to a financial instrument fails to meet its contractual obligations. The Company regularly monitors its credit risk exposure and takes steps to mitigate the likelihood of these exposuresthis exposure resulting in actual loss.
Financial instruments that potentially subject the Companylosses. The Company’s exposure to concentrations of credit risk consist primarily of cash and cash equivalents, restricted cash and accounts receivable. Cash and cash equivalents and restricted cash balances are maintained with high-credit quality financial institutions. Also, no accounts receivable balance due to the Company that is past due as at December 31, 2010 is significant. Consequently, management considers the risk of non-performance relatedcurrently relates to cash and cash equivalents, to trade and other receivables and to restricted cash. The Company invests its available cash in amounts that are readily convertible to known amounts of cash and deposits its cash balances with financial institutions that have a minimum rating of “A3”.
As at December 31, 2011, trade accounts receivable for an amount of approximately $7,415,000 were with two external customers or partners.
As at December 31, 2011, no trade accounts receivable were past due or impaired.
Generally, the Company does not require collateral or other security from customers for trade accounts receivable; however, credit is extended following an evaluation of creditworthiness. In addition, the Company performs ongoing credit reviews of all its customers and establishes an allowance for doubtful accounts when accounts are determined to be minimal.uncollectible.
The maximum exposure to credit risk approximates the amount recognized on the statement of financial position.
Liquidity risk
Liquidity risk is the risk that the Company will not be able to meet its financial obligations as they become due. As indicated in the capital disclosures section (see “Item 5 — Operating and Financial Review and Prospects”) the Company manages this risk through the management of its capital structure. It also manages liquidity risk by continuously monitoring actual and projected cash flows. The Board of Directors reviews and approves the Company’s operating and capital budgets, as well as any material transactions out of the ordinary course of business. The Company has adopted an investment policy in respect of the safety and preservation of its capital to ensure the Company’s liquidity needs are met. The instruments are selected with regard to the expected timing of expenditures and prevailing interest rates.
b) Foreign Currency(b) Market risk
Share price risk
The change in fair value of our warrant liability results from the periodic “mark-to-market” revaluation, via the application of the Black-Scholes option pricing model, of currently outstanding share purchase warrants. The Black-Scholes valuation is impacted, among other inputs, by the market price of our common shares. As a result, the change in fair value of the warrant liability, which is reported as finance income (costs) in our consolidated statements of comprehensive loss, has been and may continue in future periods to be materially affected most notably by changes in our common share price, which has ranged from $1.43 to $2.58 on NASDAQ and from C$1.41 to C$2.51 on the TSX during the twelve months ended December 31, 2011. Assuming the following variations of the market price of the Company’s common shares over a 12-month period:
| — | Market price variations of -10% and +10% |
If these variations were to occur, the impact on the Company’s net loss for warrant liability held at December 31, 2011 would be as follows:
Carrying | -10% | +10% | ||||
| $ | $ | $ | ||||
Warrant liability* | 9,204 | 1,155 | (1,174) | |||
|
| |||||
Total impact on net loss – decrease/(increase) | 1,155 | (1,174) | ||||
|
| |||||
| *Includes | current and non-current portions. |
Foreign currency risk
Since the Company operates internationally, it is exposed to currency risks as a result of potential exchange rate fluctuations related to non-intragroup transactions. FluctuationsIn particular, fluctuations in the US dollar ("US$") andexchange rates against the EUR exchange rates could have a potentially significant impact on the Company'sCompany’s results of operations. The following variations are reasonably possible over a 12-month period:
| — | Foreign exchange rate variation of -5% (depreciation of the EUR) and +5% (appreciation of the EUR) against the US$, from a year-end rate of EUR1 = US$1.2972. |
If these variations were to occur, the impact on the Company's consolidatedCompany’s net loss for each category of financial instruments held at December 31, 20102011 would be as follows:
| | | Balances denominated in US$ | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| | Carrying amount | |||||||||
| | -5% | +5% | ||||||||
| | $ | $ | $ | |||||||
Assets | ||||||||||
Cash and cash equivalents | 29,555 | 1,478 | (1,478 | ) | ||||||
Total impact on consolidated net loss — | 1,478 | (1,478 | ) | |||||||
c) Liquidity risk
Liquidity risk is the risk that the Company will not be able to meet its financial obligations as they become due. The Company manages liquidity risk through the management of its capital structure and financial leverage. The Company also manages liquidity risk by continuously monitoring actual and projected cash flows. The Board of Directors reviews and approves the Company's operating and capital budgets and reviews any material transactions outside of the normal course of business.
| Balances denominated in US$ | ||||||||||||
| Carrying amount | -5% | +5% | ||||||||||
| $ | $ | $ | ||||||||||
Cash and cash equivalents | 33,669 | 1,683 | (1,683) | |||||||||
Warrant liability* | 9,204 | (460) | 460 | |||||||||
|
|
|
| |||||||||
Total impact on net loss – decrease/(increase) | 1,223 | (1,223) | ||||||||||
|
|
|
| |||||||||
The Company's investment policy ensures the safety and preservation of its principal, as outlined above, to ensure the Company's liquidity needs are met.
| * | Includes current and non-current portions. |
Item 12. Description of Securities Other than Equity Securities
| Item 12. | Description of Securities Other than Equity Securities |
| A. |
Not applicable.
| B. | Warrants and rights |
Not applicable.
| C. | Other securities |
Not applicable.
| D. | American depositary shares |
Not applicable.
Not applicable.
None. None. Under the supervision of and with the participation of the The The Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate. Management conducted an evaluation of the effectiveness of the There have been no changes in our internal control over financial reporting during the year ended December 31, The Board of the Registrant has determined that the Registrant has at least one audit committee financial expert (as defined in paragraph (b) of Item 16A to Form 20-F). The name of the audit committee financial expert of the Registrant is Mr. Gérard Limoges, FCA, the Audit On March 29, 2004, the Board adopted a (All amounts are in US dollars) During the financial years ended December 31, 2011 and 2010, During the financial years ended December 31, 2011 and 2010, During the financial years ended December 31, 2011 and 2010, During the financial years ended December 31, 2011 and 2010, Under applicable Canadian securities regulations, the Registrant is required to disclose whether its Audit Committee has adopted specific policies and procedures for the engagement of non-audit services and to prepare a summary of these policies and procedures. The Audit Committee Charter (included as Exhibit 11.2 to this annual report on Form 20-F) provides that it is such For each of the years ended December 31, During the financial year ended on December 31, None. Item 16E. None. None.B. Warrants and rightsItem 13. Defaults, Dividend Arrearages and Delinquencies Not applicable.C. Other securitiesItem 14. Material Modification to the Rights of Security Holders and Use of Proceeds Not applicable.Item 15. Controls and Procedures D. American depositary shares Not applicable. Item 13. Defaults, Dividend Arrearages and Delinquencies None. Item 14. Material Modification to the Rights of Security Holders and Use of Proceeds None. Item 15. Controls and ProceduresRegistrant'sRegistrant’s management, including the Chief Executive Officer and Chief Financial Officer, we have conducted an evaluation pursuant to Rule 13a-15, promulgated under the Securities Exchange Act of 1934, as amended, of the effectiveness of our disclosure controls and procedures as at December 31, 2010.2011. Based on that evaluation, the Chief Executive Officer and Chief Financial Officer have concluded that these disclosure controls and procedures were effective as at December 31, 2010.2011.Management'sManagement’s Annual Report on Internal Control over Financial ReportingRegistrant'sRegistrant’s management is responsible for establishing and maintaining adequate internal control over financial reporting. The Registrant'sRegistrant’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with GAAP.IFRS.Registrant'sRegistrant’s internal control over financial reporting includes those policies and procedures that: (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the Registrant'sRegistrant’s assets; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with GAAP,IFRS, and that receipts and expenditures of the Registrant are being made only in accordance with authorizations of the Registrant'sRegistrant’s management; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the Registrant'sRegistrant’s assets that could have a material effect on the financial statements.Registrant'sRegistrant’s internal control over financial reporting based on the criteria established in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on this evaluation, management concluded that the Registrant'sRegistrant’s internal control over financial reporting was effective as at December 31, 2010.Table of Contents2011.
Attestation Report of the Independent Auditors SeeThe effectivemess of the reportRegistrant’s internal control over financial reporting as of December 31, 2011, has been audited by PricewaterhouseCoopers LLP, independent auditors, as stated in their report which is included under "Item“Item 18. — Financial Statements"Statements”.
Changes in Internal Control over Financial Reporting20102011 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.The design of any system of controls and procedures is based in part upon certain assumptions about the likelihood of certain events. There can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions, including conditions that are remote.Item 16A. Audit Committee Financial Expert Item 16A. Audit Committee Financial ExpertCommittee'sCommittee’s Chairman. The Commission has indicated that the designation of Mr. Limoges as the audit committee financial expert of the Registrant does not: (i) make Mr. Limoges an "expert"“expert” for any purpose, including without limitation for purposes of Section 11 of the Securities Act of 1933, as amended, as a result of this designation; (ii) impose any duties, obligations or liability on Mr. Limoges that are greater than those imposed on him as a member of the Audit Committee and the Board in the absence of such designation; or (iii) affect the duties, obligations or liability of any other member of the Audit Committee or the Board. The other members of the Audit committee are Mr. Pierre Lapalme and Mr. Pierre MacDonald,Michael Meyers, each of whom, along with Mr. Limoges, is independent, as that term is defined in the NASDAQ listing standards. For a description of their respective education and experience, please refer to "Item“Item 6. — Directors, Senior Management and Employees"Employees”.Item 16B. Code of Ethics "Code“Code of Ethical Conduct"Conduct”, which has been amended by the Board on November 3, 2004, December 13, 2005, March 2, 2007 and March 10, 2009. The December 13, 2005 amendment incorporates changes to the duty to report violations consistent with applicable laws. The Registrant has selected an independent third party supplier to provide a confidential and anonymous communication channel for reporting concerns about possible violations to the Registrant'sRegistrant’s Code of Ethical Conduct as well as financial and/or accounting irregularities or fraud. A copy of the Code of Ethical Conduct, as amended, is included as Exhibit 11.1 to this annual report and is also available on the Registrant'sRegistrant’s Web site at www.aezsinc.com under the Investors — Governance tab. The Code of Ethical Conduct is a "code“code of ethics"ethics” as defined in paragraph (b) of Item 16B to Form 20-F. The Code of Ethical Conduct applies to all of the Registrant'sRegistrant’s employees, directors and officers, including the Registrant'sRegistrant’s principal executive officer, principal financial officer, and principal accounting officer or controller, or persons performing similar functions, and includes specific provisions dealing with integrity in accounting matters, conflicts of interest and compliance with applicable laws and regulations. The Registrant will provide this document without charge to any person or company upon request to the Chief Financial Officer of the Registrant, at its head office at 1405 du Parc-Technologique Boulevard, Quebec City, Quebec, G1P 4P5, Canada. Item 16C. Principal Accountant Fees and ServicesItem 16C. Principal Accountant Fees and Services A. Audit FeesA. Audit Fees and 2009, the Registrant'sRegistrant’s principal accountant, PricewaterhouseCoopers LLP, billed $605,614$650,460 and $435,710,$605,614, respectively, for the audit of the Registrant'sRegistrant’s annual consolidated financial statements and for services rendered in connection with the Registrant'sRegistrant’s statutory and regulatory filings.B. Audit-related FeesB. Audit-related Fees and 2009, the Registrant'sRegistrant’s principal accountant, PricewaterhouseCoopers LLP, billed $131,833$76,822 and $44,485,$131,833, respectively, for audit or attest services not required by statute or regulation, for accounting consultations on proposed transactions, for the review of prospectuses and prospectus supplements, including the delivery of customary consent and comfort letters in connection therewith, as well as evaluations of accounting policy decisions and expected adjustments related to the Registrant'sRegistrant’s transition to IFRS.C. Tax FeesC. Tax Fees and 2009, the Registrant'sRegistrant’s principal accountant, PricewaterhouseCoopers LLP, billed $58,476$50,282 and $63,819,$58,476, respectively, for services related to tax compliance, tax planning and tax advice.D. All Other FeesD. All Other Fees and 2009, the Registrant'sRegistrant’s principal accountant, Pricewaterhouse CoopersPricewaterhouseCoopers LLP, billed $Nil and $4,164,$Nil, respectively, for services not included in audit fees, audit-related fees and tax fees.E. Audit Committee Pre-Approval Policies and ProceduresE. Audit Committee Pre-Approval Policies and Procedures committee'scommittee’s responsibility to approve all audit engagement fees and terms as well as reviewing policies for the provision of non-audit services by the external auditors and, when required, the framework for pre-approval of such services. The Audit Committee delegates to its Chairman the pre-approval of such non-audit fees. The pre-approval by the Chairman is then presented to the Audit Committee at its first scheduled meeting following such pre-approval.20102011 and 2009,2010, none of the non-audit services provided by the Registrant'sRegistrant’s external auditor were approved by the Audit Committee pursuant to the "de“de minimis exception"exception” to the pre-approval requirement for non-audit services.2010,2011, only full-time, permanent employees of the Registrant'sRegistrant’s principal accountant, PricewaterhouseCoopers LLP, performed audit work on the Registrant'sRegistrant’s financial statements.Item 16D. Exemptions from the Listing Standards for Audit CommitteesExemptions from the Listing Standards for Audit Committees None. Purchases of Equity Securities by the Issuer and Affiliated PurchasersPurchases of Equity Securities by the Issuer and Affiliated Purchasers Item 16F. Changes in Registrant’s Certifying Accountant
| Item 16G. | Corporate Governance |
Item 16F. Changes in Registrant's Certifying Accountant
None.
Item 16G. Corporate Governance
The Registrant is in compliance with the corporate governance requirements of the NASDAQ except as described below. The Registrant is not in compliance with the NASDAQ requirement that a quorum for a meeting of the holders of the common stock of the Registrant be no less than 331/3%3% of such outstanding shares. The by-laws of the Registrant provide that a quorum for purposes of any meeting of shareholders of the Registrant consists of at least 210%10% of the outstanding voting shares(subject to ratification at the Registrant's next annual and special meeting of shareholders to be held on May 18, 2011).shares. The Registrant receivedbenefits from an exemption from NASDAQ from this quorum requirement because the quorum provided for in the by-laws of the Registrant is consistent with generally accepted business practices in Canada, the Registrant'sRegistrant’s country of domicile, and with the TSX, the home country exchange on which the Registrant'sRegistrant’s voting shares are traded.
In addition, the Registrant follows certain of its home country practices in lieu of compliance with the NASDAQ requirements that: (i) independent directors of the Registrant have regularly scheduled meetings at which only independent directors are present ("(“executive sessions"sessions”); (ii) the compensation of the chief executive officer and the other executive officers of the Registrant be determined, or recommended to the Registrant'sRegistrant’s Board for determination, by a compensation committee comprised solely of independent directors; and (iii) the director nominees be selected, or recommended for selection by the Registrant'sRegistrant’s Board, by a nominations committee comprised solely of independent directors. The Chairman of the Board of the Registrant from time to time ensures that directors hold meetings at which senior management is not present, and the Registrant'sRegistrant’s Corporate Governance, Nominating and Human Resources Committee, which serves as the Registrant'sRegistrant’s compensation and nominations committee, is comprised of fourthree members, foureach of whom are independent directors.is independent. In accordance with applicable current NASDAQ requirements, the Registrant has in the past provided to NASDAQ letters from outside counsel certifying that these practices are not prohibited by the Registrant'sRegistrant’s home country law.
| Item 16H. | Mine Safety Disclosure |
None.
| Item 17. | Financial Statements |
We have elected to provide financial statements pursuant to Item 18.
| Item 18. | Financial Statements |
The financial statements appear on pages 155145 through 198.205.
Table of ContentsConsolidated Financial Statements
AETERNA ZENTARIS INC.CONSOLIDATED FINANCIAL STATEMENTS
As at December 31, 20102011 and December 31, 20092010 and for the years ended
December 31, 2010, 20092011 and 2008
2010
(in thousands of US dollars)
March 27, 2012
To the Shareholders of
Aeterna Zentaris Inc.
We have completed integrated audits of Aeterna Zentaris Inc. and its subsidiaries'subsidiaries’ 2011 and 2010 2009 and 2008 consolidated financial statements and their internal control over financial reporting as at December 31, 2010.2011. Our opinions, based on our audits, are presented below.
Report on the consolidated financial statements
We have audited the accompanying consolidated financial statements of Aeterna Zentaris Inc. and its subsidiaries, which comprise the consolidated balance sheetsstatements of financial position as at December 31, 2011, December 31, 2010 and December 31, 2009January 1, 2010 and the consolidated statementsstatement of operations and comprehensive loss, accumulated other comprehensive income and deficit, changes in shareholders'shareholders’ equity and cash flows for each of the years in the three-year period ended December 31, 2011 and December 31, 2010, and the related notes, includingwhich comprise a summary of significant accounting policies. We have also audited the financial statement schedules, Valuationpolicies and Qualifying Accounts and Export Sales, in Item 8.A. of this Annual Report on Form 20-F.other explanatory information.
Management'sManagement’s responsibility for the consolidated financial statements
Management is responsible for the preparation and fair presentation of these consolidated financial statements and financial statement schedules in accordance with Canadian generally accepted accounting principlesInternational Financial Reporting Standards as issued by the International Accounting Standards Board and for such internal control as management determines is necessary to enable the preparation of consolidated financial statements and financial statement schedules that are free from material misstatement, whether due to fraud or error.
Auditor'sAuditor’s responsibility
Our responsibility is to express an opinion on these consolidated financial statements and financial statement schedules based on our audits. We conducted our audits in accordance with Canadian generally accepted auditing standards and the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform an audit to obtain reasonable assurance about whether the consolidated financial statements and financial statement schedules are free from material misstatement. Canadian generally accepted auditing standards also require that we comply with ethical requirements.
An audit involves performing procedures to obtain audit evidence, on a test basis, about the amounts and disclosures in the consolidated financial statements. The procedures selected depend on the auditor'sauditor’s judgment, including the assessment of the risks of material misstatement of the consolidated financial statements, whether due to fraud or error. In making those risk assessments, the auditor considers internal control relevant to the company'scompany’s preparation and fair presentation of the consolidated financial statements in order to design audit procedures that are appropriate in the circumstances. An audit also includes evaluating the appropriateness of accounting principles and policies used and the reasonableness of accounting estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements.
We believe that the audit evidence we have obtained in our audits is sufficient and appropriate to provide a basis for our audit opinion on the consolidated financial statements.
Opinion
In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of Aeterna Zentaris Inc. and its subsidiaries as at December 31, 2010 and
2011, December 31, 20092010 and the results of their operationsJanuary 1, 2010 and its financial performance and its cash flows for each of the years in the three-year period ended December 31, 2011 and December 31, 2010 in accordance with Canadian generally accepted accounting principles. Furthermore, in our opinion,International Financial Reporting Standards as issued by the financial statement schedules, Valuation and Qualifying Accounts and Export Sales, in Item 8.A. of this Annual Report on Form 20-F present fairly, in all material respects, the information set forth therein when read in conjunction with the related consolidated financial statements.International Accounting Standards Board.
Report on internal control over financial reporting
We have also audited Aeterna Zentaris Inc. and its subsidiaries'subsidiaries’ internal control over financial reporting as at December 31, 2010,2011, based on criteria established inInternal Control —– Integrated Framework, issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO).
Management'sPricewaterhouseCoopers LLP/s.r.l./s.e.n.c.r.l., Chartered Accountants
1250 René-Lévesque Boulevard West, Suite 2800, Montréal, Quebec, Canada H3B 2G4
T: +1 514 205 5000, F: +1 514 876 1502, www.pwc.com/ca
“PwC” refers to PricewaterhouseCoopers LLP, an Ontario limited liability partnership, which is a member firm of PricewaterhouseCoopers International Limited, each member firm of which is a separate legal entity.
Management’s responsibility for internal control over financial reporting
Management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the section entitled, "Management's“Management’s Annual Report on Internal Control over Financial Reporting"Reporting” appearing on Page 151140 of this Annual Report on Form 20-F.
Auditor'sAuditor’s responsibility
Our responsibility is to express an opinion on the company'scompany’s internal control over financial reporting based on our audit. We conducted our audit of internal control over financial reporting in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects.
An audit of internal control over financial reporting includes obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we consider necessary in the circumstances.
We believe that our audit provides a reasonable basis for our audit opinion on the company'scompany’s internal control over financial reporting.
Definition of internal control over financial reporting
A company'scompany’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company'scompany’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company'scompany’s assets that could have a material effect on the financial statements.
Inherent limitations
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions or that the degree of compliance with the policies or procedures may deteriorate.
Opinion
In our opinion, Aeterna Zentaris Inc. and its subsidiaries maintained, in all material respects, effective internal control over financial reporting as at December 31, 20102011, based on criteria established inInternal Control —– Integrated Framework, issued by COSO.
Montréal, Québec, Canada
Chartered accountant auditor permit No. 21323 |
March 22, 2011
Place de la Cité, Tour Cominar2640 Laurier Boulevard, Suite 1700Québec, QuebecCanada G1V 5C2
TableConsolidated Statements of ContentsFinancial Position
AETERNA ZENTARIS INC.CONSOLIDATED BALANCE SHEETS
(in thousands of US dollars)
| | As at December 31, | |||||||
|---|---|---|---|---|---|---|---|---|
| | 2010 | 2009 | ||||||
| | $ | $ | ||||||
ASSETS | ||||||||
Current assets | ||||||||
Cash and cash equivalents | 31,998 | 38,100 | ||||||
Short-term investment (note 21) | 1,934 | — | ||||||
Accounts receivable | ||||||||
Trade | 4,555 | 2,444 | ||||||
Other | 748 | 992 | ||||||
Income taxes | 118 | 113 | ||||||
Inventory (note 7) | 3,311 | 4,415 | ||||||
Prepaid expenses and other current assets | 1,511 | 2,949 | ||||||
| 44,175 | 49,013 | |||||||
Restricted cash (note 8) | 827 | 878 | ||||||
Property, plant and equipment (note 9) | 3,096 | 4,358 | ||||||
Deferred charges and other long-term assets (note 10) | 4,384 | 4,733 | ||||||
Intangible assets (note 11) | 14,478 | 17,034 | ||||||
Goodwill (note 12) | 9,614 | 10,246 | ||||||
| 76,574 | 86,262 | |||||||
LIABILITIES | ||||||||
Current liabilities | ||||||||
Accounts payable and accrued liabilities (note 13) | 9,382 | 11,919 | ||||||
Income taxes | — | 965 | ||||||
Deferred revenues (note 5) | 4,045 | 6,327 | ||||||
Current portion of long-term payable (note 6) | 60 | 57 | ||||||
| 13,487 | 19,268 | |||||||
Deferred revenues (note 5) | 39,052 | 45,919 | ||||||
Long-term payable (note 6) | 90 | 143 | ||||||
Employee future benefits (note 14) | 11,324 | 11,640 | ||||||
Other long-term liabilities | 182 | 66 | ||||||
| 64,135 | 77,036 | |||||||
Commitments and contingencies (note 22) | ||||||||
SHAREHOLDERS' EQUITY | ||||||||
Share capital (note 15) | 60,149 | 41,203 | ||||||
Warrants (note 15) | 9,493 | 2,899 | ||||||
Other capital | 80,785 | 79,943 | ||||||
Deficit | (150,756 | ) | (127,538 | ) | ||||
Accumulated other comprehensive income | 12,768 | 12,719 | ||||||
| 12,439 | 9,226 | |||||||
| 76,574 | 86,262 | |||||||
Basis of presentation (note 2) | ||||||||
Subsequent events (note 24) | ||||||||
| December 31, | December 31, | January 1, | ||||||||||
| 2011 | 2010 | 2010 | ||||||||||
| $ | $ | $ | ||||||||||
ASSETS | ||||||||||||
Current assets | ||||||||||||
Cash and cash equivalents (note 6) | 46,881 | 31,998 | 38,100 | |||||||||
Short-term investment (note 7) | - | 1,934 | - | |||||||||
Trade and other receivables (note 8) | 8,325 | 5,421 | 3,549 | |||||||||
Inventory (note 9) | 3,456 | 3,311 | 4,415 | |||||||||
Prepaid expenses and other current assets | 1,477 | 1,145 | 2,407 | |||||||||
| 60,139 | 43,809 | 48,471 | ||||||||||
Restricted cash (note 10) | 806 | 827 | 878 | |||||||||
Property, plant and equipment (note 11) | 2,512 | 3,096 | 4,358 | |||||||||
Other non-current assets | 830 | 803 | 528 | |||||||||
Identifiable intangible assets (note 12) | 1,769 | 3,299 | 4,127 | |||||||||
Goodwill (note 13) | 9,313 | 9,614 | 10,246 | |||||||||
| 75,369 | 61,448 | 68,608 | ||||||||||
LIABILITIES | ||||||||||||
Current liabilities | ||||||||||||
Payables and accrued liabilities (note 14) | 12,257 | 10,260 | 11,845 | |||||||||
Current portion of deferred revenues (note 5) | 5,310 | 4,045 | 6,327 | |||||||||
Income taxes (notes 5 and 22) | 259 | - | 965 | |||||||||
Current portion of long-term payable | 59 | 60 | 57 | |||||||||
| 17,885 | 14,365 | 19,194 | ||||||||||
Deferred revenues (note 5) | 39,242 | 39,052 | 45,919 | |||||||||
Warrant liability (note 15) | 9,162 | 13,412 | 1,664 | |||||||||
Long-term payable | 29 | 90 | 143 | |||||||||
Employee future benefits (note 19) | 12,880 | 11,533 | 12,021 | |||||||||
Provision and other non-current liabilities (note 16) | 717 | 571 | 507 | |||||||||
| 79,915 | 79,023 | 79,448 | ||||||||||
SHAREHOLDERS’ DEFICIENCY | ||||||||||||
Share capital (note 17) | 101,884 | 60,900 | 41,524 | |||||||||
Other capital | 82,327 | 81,091 | 79,943 | |||||||||
Deficit | (188,969) | (160,567) | (132,307) | |||||||||
Accumulated other comprehensive (loss) income | 212 | 1,001 | - | |||||||||
| (4,546) | (17,575) | (10,840) | ||||||||||
| 75,369 | 61,448 | 68,608 | ||||||||||
Approved by the Board of DirectorsSubsequent event (note 30)
The accompanying notes are an integral part of these consolidated financial statements.
Approved by the Board of Directors
 |  | |||
Juergen Ernst Director | Gérard Limoges Director |
Consolidated Statements of ContentsChanges in Shareholders’ Deficiency
AETERNA ZENTARIS INC.CONSOLIDATED STATEMENTS OF CHANGES IN SHAREHOLDERS' EQUITY
For the years ended December 31, 2010, 20092011 and 2008
2010
(in thousands of US dollars, except share data)
| | Common shares | Share capital | Warrants | Other capital | Deficit | Accumulated other comprehensive income | Total | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | (number of) | $ | $ | $ | $ | $ | $ | |||||||||||||||
Balance — December 31, 2007 | 53,187,470 | 30,566 | — | 79,306 | (42,997 | ) | 21,716 | 88,591 | ||||||||||||||
Net loss | — | — | — | — | (59,817 | ) | — | (59,817 | ) | |||||||||||||
Foreign currency translation adjustments | — | — | — | — | — | (7,655 | ) | (7,655 | ) | |||||||||||||
Variation in the fair value of short-term investments, net of income taxes | — | — | — | — | — | (7 | ) | (7 | ) | |||||||||||||
Stock-based compensation costs | — | — | — | 363 | — | — | 363 | |||||||||||||||
Balance — December 31, 2008 | 53,187,470 | 30,566 | — | 79,669 | (102,814 | ) | 14,054 | 21,475 | ||||||||||||||
Net loss | — | — | — | — | (24,724 | ) | — | (24,724 | ) | |||||||||||||
Issuances pursuant to registered direct offerings, net of transaction costs (note 15) | 9,902,484 | 10,637 | 2,899 | — | — | — | 13,536 | |||||||||||||||
Foreign currency translation adjustments | — | — | — | — | — | (1,332 | ) | (1,332 | ) | |||||||||||||
Variation in the fair value of short-term investments, net of income taxes | — | — | — | — | — | (3 | ) | (3 | ) | |||||||||||||
Stock-based compensation costs | — | — | — | 274 | — | — | 274 | |||||||||||||||
Balance — December 31, 2009 | 63,089,954 | 41,203 | 2,899 | 79,943 | (127,538 | ) | 12,719 | 9,226 | ||||||||||||||
Net loss | — | — | — | — | (23,218 | ) | — | (23,218 | ) | |||||||||||||
Issuances pursuant to registered direct offerings, net of transaction costs (note 15) | 19,917,075 | 18,181 | 6,807 | — | — | — | 24,988 | |||||||||||||||
Issuance pursuant to the exercise of warrants (note 15) | 298,817 | 609 | (213 | ) | — | — | — | 396 | ||||||||||||||
Issuance pursuant to the exercise of stock options (note 15) | 124,068 | 156 | — | (44 | ) | — | — | 112 | ||||||||||||||
Foreign currency translation adjustments | — | — | — | — | — | 49 | 49 | |||||||||||||||
Stock-based compensation costs | — | — | — | 886 | — | — | 886 | |||||||||||||||
Balance — December 31, 2010 | 83,429,914 | 60,149 | 9,493 | 80,785 | (150,756 | ) | 12,768 | 12,439 | ||||||||||||||
Common shares (number of)* | Share capital | Other capital | Deficit | Accumulated other comprehensive income (loss) | Total | |||||||||||||||||||
| $ | $ | $ | $ | $ | ||||||||||||||||||||
Balance – January 1, 2010 | 63,089,954 | 41,524 | 79,943 | (132,307) | - | (10,840) | ||||||||||||||||||
Net loss | - | - | - | (28,451) | - | (28,451) | ||||||||||||||||||
Other comprehensive income: | ||||||||||||||||||||||||
Foreign currency translation adjustments | - | - | - | - | 1,001 | 1,001 | ||||||||||||||||||
Actuarial gain on defined benefit plans | - | - | - | 191 | - | 191 | ||||||||||||||||||
Comprehensive loss | - | - | - | (28,260) | 1,001 | (27,259) | ||||||||||||||||||
Issuances pursuant to registered direct offerings, net of transaction costs (note 17) | 19,917,075 | 18,391 | - | - | - | 18,391 | ||||||||||||||||||
Issuance pursuant to the exercise of warrants (note 15) | 298,817 | 829 | - | - | - | 829 | ||||||||||||||||||
Issuance pursuant to the exercise of stock options (note 17) | 124,068 | 156 | (44) | - | - | 112 | ||||||||||||||||||
Share-based compensation costs | - | - | 1,192 | - | - | 1,192 | ||||||||||||||||||
Balance – December 31, 2010 | 83,429,914 | 60,900 | 81,091 | (160,567) | 1,001 | (17,575) | ||||||||||||||||||
Net loss | - | - | - | (27,067) | - | (27,067) | ||||||||||||||||||
Other comprehensive loss: | ||||||||||||||||||||||||
Foreign currency translation adjustments | - | - | - | - | (789) | (789) | ||||||||||||||||||
Actuarial loss on defined benefit plans | - | - | - | (1,335) | - | (1,335) | ||||||||||||||||||
Comprehensive loss | - | - | - | (28,402) | (789) | (29,191) | ||||||||||||||||||
Share issuances in connection with “At-the-Market” drawdowns (note 17) | 19,464,548 | 35,881 | - | - | - | 35,881 | ||||||||||||||||||
Share issuances pursuant to the exercise of warrants (note 15) | 1,707,286 | 4,861 | - | - | - | 4,861 | ||||||||||||||||||
Share issuances pursuant to the exercise of stock options (note 17) | 160,348 | 242 | (97) | - | - | 145 | ||||||||||||||||||
Share-based compensation costs | - | - | 1,333 | - | - | 1,333 | ||||||||||||||||||
Balance – December 31, 2011 | 104,762,096 | 101,884 | 82,327 | (188,969) | 212 | (4,546) | ||||||||||||||||||
*Issued and paid in full
The accompanying notes are an integral part of these consolidated financial statements.
TableConsolidated Statements of ContentsComprehensive Loss
AETERNA ZENTARIS INC.CONSOLIDATED STATEMENTS OF ACCUMULATED OTHER COMPREHENSIVEINCOME AND DEFICIT(in thousands of US dollars)
| | As at December 31, | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2010 | 2009 | 2008 | ||||||||
| | $ | $ | $ | ||||||||
Accumulated other comprehensive income | |||||||||||
Foreign currency translation adjustments | 12,768 | 12,719 | 14,051 | ||||||||
Change in fair market value of short-term investments, net of income taxes | — | — | 3 | ||||||||
Accumulated other comprehensive income | 12,768 | 12,719 | 14,054 | ||||||||
Deficit | (150,756 | ) | (127,538 | ) | (102,814 | ) | |||||
Total accumulated other comprehensive income and deficit | (137,988 | ) | (114,819 | ) | (88,760 | ) | |||||
AETERNA ZENTARIS INC.CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
For the years ended December 31,
2011 and 2010
(in thousands of US dollars, except share and per share data)
| | 2010 | 2009 | 2008 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | $ | $ | $ | ||||||||||
Revenues | |||||||||||||
Sales and royalties | 24,857 | 20,957 | 29,462 | ||||||||||
License fees and other | 2,846 | 42,280 | 9,016 | ||||||||||
| 27,703 | 63,237 | 38,478 | |||||||||||
Operating expenses | |||||||||||||
Cost of sales, excluding depreciation and amortization (note 7) | 18,700 | 16,501 | 19,278 | ||||||||||
Research and development costs | 20,546 | 44,217 | 57,448 | ||||||||||
Research and development tax credits and grants | (687 | ) | (403 | ) | (343 | ) | |||||||
Selling, general and administrative expenses | 11,875 | 16,040 | 17,325 | ||||||||||
Depreciation and amortization | |||||||||||||
Property, plant and equipment (note 9) | 1,005 | 3,285 | 1,515 | ||||||||||
Intangible assets (note 11) | 1,492 | 7,555 | 5,639 | ||||||||||
| 52,931 | 87,195 | 100,862 | |||||||||||
Loss from operations | (25,228 | ) | (23,958 | ) | (62,384 | ) | |||||||
Other income (expenses) | |||||||||||||
Unrealized gain on held-for-trading financial instrument (note 21) | 687 | — | — | ||||||||||
Interest income | 207 | 349 | 868 | ||||||||||
Interest expense | (26 | ) | (5 | ) | (118 | ) | |||||||
Foreign exchange gain (loss) | 1,170 | (1,110 | ) | 3,071 | |||||||||
Loss on disposal of long-lived assets (note 6) | — | — | (35 | ) | |||||||||
Loss on disposal of equipment | (28 | ) | — | (44 | ) | ||||||||
| 2,010 | (766 | ) | 3,742 | ||||||||||
Loss before income taxes | (23,218 | ) | (24,724 | ) | (58,642 | ) | |||||||
Income tax expense (note 17) | — | — | (1,175 | ) | |||||||||
Net loss | (23,218 | ) | (24,724 | ) | (59,817 | ) | |||||||
Other comprehensive income (loss): | |||||||||||||
Foreign currency translation adjustments | 49 | (1,332 | ) | (7,655 | ) | ||||||||
Variation in fair market value of short-term investments classified as available-for-sale, net of income taxes | — | (3 | ) | (7 | ) | ||||||||
Comprehensive loss | (23,169 | ) | (26,059 | ) | (67,479 | ) | |||||||
Net loss per share | |||||||||||||
Basic and diluted | (0.31 | ) | (0.43 | ) | (1.12 | ) | |||||||
Weighted average number of shares (note 19) | |||||||||||||
Basic and diluted | 75,659,410 | 56,864,484 | 53,187,470 | ||||||||||
| Years ended December 31, | ||||||||
| 2011 | 2010 | |||||||
| $ | $ | |||||||
Revenues | ||||||||
Sales and royalties | 31,306 | 24,857 | ||||||
License fees and other | 4,747 | 2,846 | ||||||
|
| |||||||
| 36,053 | 27,703 | |||||||
|
| |||||||
Operating expenses (note 18) | ||||||||
Cost of sales | 27,560 | 18,700 | ||||||
Research and development costs, net of refundable tax credits and grants | 24,517 | 21,257 | ||||||
Selling, general and administrative expenses (notes 11 and 12) | 16,170 | 12,552 | ||||||
|
| |||||||
| 68,247 | 52,509 | |||||||
|
| |||||||
Loss from operations | (32,194 | ) | (24,806) | |||||
|
| |||||||
Finance income (note 20) | 6,239 | 1,800 | ||||||
Finance costs (note 20) | (8 | ) | (5,445) | |||||
|
| |||||||
Net finance income (costs) | 6,231 | (3,645) | ||||||
| �� |
|
| ||||||
Loss before income taxes | (25,963 | ) | (28,451) | |||||
Income tax expense (notes 5 and 22) | (1,104 | ) | - | |||||
|
| |||||||
Net loss | (27,067 | ) | (28,451) | |||||
Other comprehensive (loss) income: | ||||||||
Foreign currency translation adjustments | (789 | ) | 1,001 | |||||
Actuarial gain (loss) on defined benefit plans | (1,335 | ) | 191 | |||||
|
| |||||||
Comprehensive loss | (29,191 | ) | (27,259) | |||||
|
| |||||||
Net loss per share (note 26) | ||||||||
Basic and diluted | (0.29 | ) | (0.38) | |||||
|
| |||||||
Weighted average number of shares outstanding (note 26) | ||||||||
Basic and diluted | 94,507,988 | 75,659,410 | ||||||
|
| |||||||
The accompanying notes are an integral part of these consolidated financial statements.
TableConsolidated Statements of ContentsCash Flows
AETERNA ZENTARIS INC.CONSOLIDATED STATEMENTS OF CASH FLOWS
For the years ended December 31,
2011 and 2010
(in thousands of US dollars)
| | 2010 | 2009 | 2008 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| | $ | $ | $ | ||||||||
Cash flows from operating activities | |||||||||||
Net loss | (23,218 | ) | (24,724 | ) | (59,817 | ) | |||||
Items not affecting cash and cash equivalents | |||||||||||
Depreciation and amortization | 2,497 | 10,840 | 7,154 | ||||||||
Stock-based compensation costs | 886 | 274 | 363 | ||||||||
Non-cash consideration received in connection with an amended licensing agreement (note 21) | (1,263 | ) | — | — | |||||||
Unrealized gain on held-for-trading financial instrument (note 21) | (687 | ) | — | — | |||||||
Inventory write-down (note 7) | — | — | 726 | ||||||||
Employee future benefits | 400 | 1,365 | 984 | ||||||||
Amortization of deferred charges and other long-term assets | 380 | 1,819 | 729 | ||||||||
Amortization of deferred revenues | (5,873 | ) | (13,506 | ) | (6,213 | ) | |||||
Loss on disposal of long-lived assets held for sale | — | — | 35 | ||||||||
Loss on disposal of property, plant and equipment | 28 | — | 44 | ||||||||
Foreign exchange loss (gain) on items denominated in foreign currencies | (1,170 | ) | 1,098 | (3,801 | ) | ||||||
Amortization of prepaid expenses and other non-cash items | 4,587 | 15,192 | 10,179 | ||||||||
Changes in operating assets and liabilities (note 16) | (7,687 | ) | (16,496 | ) | 48,345 | ||||||
Net cash used in operating activities | (31,120 | ) | (24,138 | ) | (1,272 | ) | |||||
Cash flows from financing activities | |||||||||||
Proceeds from issuance of common shares and warrants, net of cash transaction costs of $2,098 in 2010 and $1,244 in 2009 (note 15) | 24,988 | 14,256 | — | ||||||||
Repayment of long-term debt and long-term payable | (59 | ) | (51 | ) | (784 | ) | |||||
Proceeds from the exercise of warrants (note 15) | 396 | — | — | ||||||||
Proceeds from the exercise of stock options (note 15) | 112 | — | — | ||||||||
Deferred share issue expenses | — | — | (408 | ) | |||||||
Net cash provided by (used in) financing activities | �� | 25,437 | 14,205 | (1,192 | ) | ||||||
Cash flows from investing activities | |||||||||||
Purchase of short-term investments | — | — | (1,664 | ) | |||||||
Proceeds from sale and maturity of short-term investments | — | 553 | 30,027 | ||||||||
Increase in restricted cash (note 8) | — | (866 | ) | — | |||||||
Purchases of property, plant and equipment | (82 | ) | (510 | ) | (1,147 | ) | |||||
Net proceeds from sale of long-lived assets held for sale | — | — | 14,854 | ||||||||
Proceeds from sale of property, plant and equipment | 32 | 6 | — | ||||||||
Purchases of amortizable intangible assets | — | (280 | ) | (67 | ) | ||||||
Net cash (used in) provided by investing activities | (50 | ) | (1,097 | ) | 42,003 | ||||||
Effect of exchange rate changes on cash and cash equivalents | (369 | ) | (96 | ) | (585 | ) | |||||
Net change in cash and cash equivalents | (6,102 | ) | (11,126 | ) | 38,954 | ||||||
Cash and cash equivalents — Beginning of year | 38,100 | 49,226 | 10,272 | ||||||||
Cash and cash equivalents — End of year | 31,998 | 38,100 | 49,226 | ||||||||
Cash and cash equivalents components: | |||||||||||
Cash | 12,922 | 33,100 | 13,256 | ||||||||
Cash equivalents | 19,076 | 5,000 | 35,970 | ||||||||
| 31,998 | 38,100 | 49,226 | |||||||||
| Years ended December 31, | ||||||||
| 2011 | 2010 | |||||||
| $ | $ | |||||||
Cash flows from operating activities | ||||||||
Net loss | (27,067 | ) | (28,451) | |||||
Items not affecting cash and cash equivalents | ||||||||
Change in fair value of warrant liability (note 15) | (2,533 | ) | 5,437 | |||||
Depreciation, amortization and impairment (notes 11 and 12) | 2,876 | 1,573 | ||||||
Share-based compensation costs (note 17) | 1,333 | 1,192 | ||||||
Non-cash consideration received in connection with an amended licensing agreement | - | (1,263) | ||||||
Gain on held-for-trading financial instrument (note 7) | (1,278 | ) | (687) | |||||
Employee future benefits (note 19) | 492 | 249 | ||||||
Amortization of deferred revenues | (5,840 | ) | (5,873) | |||||
Foreign exchange gain on items denominated in foreign currencies | (1,955 | ) | (965) | |||||
(Gain) loss on disposal of property, plant and equipment | (26 | ) | 28 | |||||
Amortization of prepaid and other non-cash items | 4,207 | 4,587 | ||||||
Changes in operating assets and liabilities (note 21) | 3,548 | (7,539) | ||||||
|
| |||||||
Net cash used in operating activities | (26,243 | ) | (31,712) | |||||
|
| |||||||
Cash flows from financing activities | ||||||||
Proceeds from issuances of common shares, net of cash transaction costs of $1,204 in 2011 and $1,506 in 2010 (note 17) | 36,250 | 25,580 | ||||||
Proceeds from the exercise of share purchase warrants (note 15) | 2,222 | 396 | ||||||
Proceeds from the exercise of stock options (note 17) | 145 | 112 | ||||||
Repayment of long-term payable | (61 | ) | (59) | |||||
|
| |||||||
Net cash provided by financing activities | 38,556 | 26,029 | ||||||
|
| |||||||
Cash flows from investing activities | �� | |||||||
Proceeds from the sale of short-term investment (note 7) | 3,242 | - | ||||||
Purchase of identifiable intangible assets (note 12) | (69 | ) | - | |||||
Purchases of property, plant and equipment (note 11) | (736 | ) | (82) | |||||
Disposals of property, plant and equipment (note 11) | 26 | 32 | ||||||
|
| |||||||
Net cash provided by (used in) investing activities | 2,463 | (50) | ||||||
Effect of exchange rate changes on cash and cash equivalents | 107 | (369) | ||||||
|
| |||||||
Net change in cash and cash equivalents | 14,883 | (6,102) | ||||||
Cash and cash equivalents – Beginning of the year | 31,998 | 38,100 | ||||||
|
| |||||||
Cash and cash equivalents – End of the year | 46,881 | 31,998 | ||||||
|
| |||||||
Cash and cash equivalents components (note 6): | ||||||||
Cash | 15,112 | 12,922 | ||||||
Cash equivalents | 31,769 | 19,076 | ||||||
|
| |||||||
| 46,881 | 31,998 | |||||||
|
| |||||||
The accompanying notes are an integral part of these consolidated financial statements.
Table of ContentsNotes to Consolidated Financial Statements
AETERNA ZENTARIS INC.NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
As at December 31, 20102011 and December 31, 2009
2010 and for the years ended December 31, 2010, 20092011 and 2008
2010
(tabular amounts in thousands of US dollars, except share/option/warrant and
per share/option/warrant data and as otherwise noted)
| 1 | Summary of business, reporting entity and basis of preparation |
Summary of business
1. INCORPORATION AND NATURE OF ACTIVITIES
Aeterna Zentaris Inc. ("(“Aeterna Zentaris"Zentaris” or the "Company"“Company”), incorporated under theCanada Business Corporations Act, is a late-stage global biopharmaceutical company specializedspecializing in oncology and endocrine therapy with expertise in drug discovery, development and commercialization. The Company'sCompany’s pipeline encompasses compounds at all stages of development, from drug discovery through marketed products.
The Company benefits from strategic collaborators and licensee partners to contribute to the development of its pipeline of product candidates and to establish commercial activities in specific territories.
The highest development priorities in oncology are the completion of Phase 3 trials with perifosine in colorectal cancer and in multiple myeloma as well as the further advancement of AEZS-108, for which the Company has successfully completed a Phase 2 trial in advanced endometrial and advanced ovarian cancer. The Company is currently planning for the initiation of a pivotal program with AEZS-108 in endometrial cancer. AEZS-108 is also in development in other cancer indications, including castration and taxane-resistant prostate cancer, refractory bladder cancer, as well as triple-negative breast cancer.
The Company’s pipeline also encompasses other earlier-stage programs in oncology. AEZS-112, an oral anticancer agent which involves three mechanisms of action (tubulin, topoisomerase II and angiogenesis inhibition) has completed a Phase 1 trial in advanced solid tumors and lymphoma. Additionally, several targeted potential anti-cancer candidates such as AEZS-120, a live recombinant oral tumor vaccine candidate, as well as our PI3K/Erk inhibitors AEZS-129, AEZS-131, AEZS-132 and their respective follow-up compounds are currently in preclinical development.
The Company’s lead program in endocrinology, a Phase 3 trial under a Special Protocol Assessment obtained from the FDA with AEZS-130 as an oral diagnostic test for Growth Hormone Deficiency in adults, has been completed with positive results. The Company expects to file a New Drug Application for the registration of AEZS-130 in the United States. Furthermore, AEZS-130 is in development for the treatment of cancer cachexia.
2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Basis of presentationReporting entity
The accompanying consolidated financial statements have beeninclude the accounts of Aeterna Zentaris Inc., an entity incorporated under theCanada Business Corporations Act, and its wholly-owned subsidiaries (collectively referred to as the “Group”). Aeterna Zentaris Inc. is the parent company of the Group and represents the ultimate parent of the Group.
The Company currently has three wholly-owned direct and indirect subsidiaries, Aeterna Zentaris GmbH (“AEZS Germany”), based in Frankfurt, Germany, Zentaris IVF GmbH, a direct wholly-owned subsidiary of AEZS Germany based in Frankfurt, Germany, and Aeterna Zentaris, Inc., based in Basking Ridge, New Jersey in the United States.
The address of the Company is 1405, du parc-Technologique Blvd, Québec, Canada, G1P 4P5.
The Company’s common shares are listed on both the Toronto Stock Exchange and the NASDAQ.
Aeterna Zentaris Inc.
Notes to Consolidated Financial Statements
As at December 31, 2011 and December 31, 2010 and for the years ended December 31, 2011 and 2010
(tabular amounts in thousands of US dollars, except share/option/warrant and per share/option/warrant data and as otherwise noted)
Basis of preparation
(a) Statement of compliance
The accompanying consolidated financial statements represent the first annual financial statements of the Company and its subsidiaries prepared in accordance with International Financial Reporting Standards (“IFRS”), as issued by the International Accounting Standards Board (“IASB”). The accompanying consolidated financial statements were also prepared in accordance with IFRS 1,First-time Adoption of International Financial Accounting Standards (“IFRS 1”). The first date at which IFRS was applied was January 1, 2010 (the “Transition Date”).
Prior to January 1, 2011, the Company’s consolidated financial statements were prepared in accordance with Canadian generally accepted accounting principles ("Canadian GAAP"(“GAAP”). In these consolidated financial statements, the term “Canadian GAAP” refers to Canadian GAAP as applicable prior to the adoption of IFRS.
Subject to certain transition exceptions and exemptions, discussed in note 29, the Company has consistently applied the same accounting policies in the preparation of its opening IFRS statement of financial position at January 1, 2010 throughout all periods presented, as if these policies had always been in effect. Note 29 discusses the impact of the transition to IFRS on the Company’s reported financial position, financial performance and cash flows, including the nature and effect of significant changes in accounting policies from those used in the Company’s consolidated financial statements for the year ended December 31, 2010 prepared in accordance with Canadian GAAP.
These consolidated financial statements differ in certain respects from thosewere approved by the Company’s Board of Directors for issue on March 27, 2012.
The accompanying consolidated financial statements were prepared on a going concern basis, under the historical cost convention, except for held-for-trading financial assets and of the warrant liability derivatives, which are measured at fair value through profit or loss (“FVTPL”).
The preparation of financial statements in accordance with United States generally accepted principles ("US GAAP"). The recognition, measurementIFRS requires the use of certain critical accounting estimates and disclosure differences as they relatethe exercise of management’s judgment in applying the Company’s accounting policies. Areas involving a high degree of judgment or complexity and areas where assumptions and estimates are significant to the CompanyCompany’s consolidated financial statements are described in note 25, "Differences between Canadian and US GAAP".
Evaluation of going concern, results of operations and management's plans
The Canadian Institute of Chartered Accountants' ("CICA") Handbook Section 1400,General Standards of Financial Statement Presentation, requires management to make an assessment of an entity's ability to continue as a going concern, taking into account all available information about the future, which is at least, but is not limited to, 12 months from the balance sheet dates. Disclosure is required of material uncertainties related to events or conditions that may cast significant doubt upon the entity's ability to continue as a going concern. Management's assessment took into account current cash levels, the completion of the registered direct offerings discussed in note 15 and the "At-the-Market" ("ATM") sales agreement entered into on February 22, 2011 discussed in note 24, as well as the Company's strategic plan and corresponding budgets for 2011 and projections for 2012 and 2013. As a result3.
(b) Principles of this assessment, management believes that the Company has sufficient financial resources to fund planned expenditures and other working capital needs for at least, but not limited to, the 12-month period following the balance sheet date of December 31, 2010.
Basis of consolidation
These consolidated financial statements include all companiesentities in which the Company, directly or indirectly, holds more than 50% of the voting rights or over which it exercises control. CompaniesEntities are included in the consolidation from the date that control is transferred toobtained by the Company, while companies soldentities are excludeddeconsolidated from the consolidation from the date that control ceases. The purchase method of accounting is used to account for acquisitions. All intercompany balances and transactions are eliminated on consolidation.
Accounting estimatesAeterna Zentaris Inc.
The preparation of financial statements in conformity with Canadian generally accepted accounting principles requires managementNotes to make estimates and assumptions that affect the amounts reported in the financial statements. Those estimates and assumptions also affect the disclosure of contingencies at the date of the financial statements as well as the reported amounts of revenues and expenses during the reported years. Significant estimates are generally made in connection with the calculation of revenues, inventory and research and development expenses, as well as in determining the allowance for doubtful accounts, valuation allowance for future income tax assets, the useful lives of property, plant and equipment and intangible assets with finite lives, the valuation of intangible assets and goodwill, the fair value of stock options and warrants granted, employee future benefits and certain accrued liabilities. The Company bases its estimates on historical experience, where relevant, and on various other assumptions that management believes to be reasonable under the circumstances. Actual results could differ from those estimates.
Table of ContentsConsolidated Financial Statements
AETERNA ZENTARIS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
As at December 31, 20102011 and December 31, 2009
2010 and for the years ended December 31, 2010, 20092011 and 2008
2010
(tabular amounts in thousands of US dollars, except share/option/warrant and
per share/option/warrant data and as otherwise noted)
2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
(c) Foreign currency translation
Reporting currency
The Company usesaccompanying consolidated financial statements are presented in thousands of US dollars, which is the US dollar as its reportingCompany’s presentation currency.
Assets and liabilities of Group entities are translated from euro at the Companyperiod end rates of exchange, and the results of its self-sustaining subsidiaries whosetheir operations are translated from euro at average rates of exchange for the period. The resulting translation adjustments are included in accumulated other comprehensive income within shareholders’ deficiency.
Items included in the financial statements of the Group’s entities are measured using the currency of the primary economic environment in which the entities operate (the “functional currency”), which is the euro (“EUR”). Foreign currency transactions are translated into the functional currency is other than the US dollar are translated using the exchange rate in effectrates prevailing at the balance sheet date. Revenues and expenses are translated atdates of the average rate in effect during the year. Translationtransaction. Foreign exchange gains and losses resulting from the settlement of such transactions and from the translation of monetary assets and liabilities not denominated in euro are included in other comprehensive income (loss)recognized in the consolidated statement of comprehensive loss.
Foreign currency transactions and remeasurement
The financial statements of integrated foreign operations and transactions denominated in currencies other than the functional currency are remeasured into the functional currency using the temporal method. Under this method, monetary assets and liabilities are remeasured to their functional currency at the exchange rate in effect on the date of the balance sheet. Non-monetary assets and liabilities are remeasured at historical rates, unless such assets and liabilities are carried at market, in which case, they are remeasured using the exchange rate in effect on the date of the balance sheet. Revenues and expenses are remeasured at the monthly average exchange rate. Transaction gains and losses resulting from such remeasurementthat relate to cash and cash equivalents, the short-term investment and the warrant liability are reflectedpresented within finance income or finance costs in the consolidated statement of operations.comprehensive loss. All other foreign exchange gains and losses are presented in the consolidated statement of comprehensive loss within operating expenses.
Effective January 1, 2009, due to a change in economic facts and circumstances, the Company and its US subsidiary adopted the euro ("EUR") as their functional currency. This change did not result in any significant impact on the Company's consolidated financial statements.
| 2 | Summary of significant accounting policies |
Cash and cash equivalents
Cash and cash equivalents consist of unrestricted cash on hand and balances with banks, as well as short-term interest-bearing deposits (including a money market account) that are readily convertible to known amounts of cash and which are subject to an insignificant risk of changes in value.
Short-term investment
In accordance with the requirements of CICA Handbook Section 3855,Financial Instruments, the Company'sThe Company’s short-term investment representingrepresents shares of a publicly-heldpublicly held company and is classified as "held-for-trading"held-for-trading and carriedstated at fair value. Any changes in fair value are recognized in the consolidated statement of operations.
Inventory
Inventory is valued at the lower of cost and net realizable value, which is defined as the estimated selling price in the ordinary course of business less the estimated costs of completion and the estimated costs necessary to make the sale.selling costs. Cost is determined on a first-in, first-out basis. The cost of finished goods and work in progress includes raw materials, labour and manufacturing overhead under the absorption costing method.
Restricted cash
Restricted cash includesis comprised of a bank deposit, related to the Company'sa long-term operating lease obligation in Germany, as discussed in note 8.that cannot be used for current purposes.
Table of ContentsAeterna Zentaris Inc.
AETERNA ZENTARIS INC.Notes to Consolidated Financial Statements
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
As at December 31, 20102011 and December 31, 2009
2010 and for the years ended December 31, 2010, 20092011 and 2008
2010
(tabular amounts in thousands of US dollars, except share/option/warrant and
per share/option/warrant data and as otherwise noted)
2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
Property, plant and equipment and depreciation
Property,Items of property, plant and equipment are recorded at cost, net of related government grants and accumulated depreciation.depreciation and impairment charges. Depreciation is calculated using the following methods, annual rates and periods:period:
| Methods | Annual rates and period | ||||
Equipment | Declining balance and straight-line | 20% | |||
Furniture and fixtures | Declining balance and straight-line | 10% and 20% | |||
Computer equipment | Straight-line | 25% and 331/3% | |||
Leasehold improvements | Straight-line | Remaining lease term | |||
Deferred charges
The Company has deferred directDepreciation expense is allocated to the appropriate expense categories to which the underlying items of property, plant and incremental costs associated with its transaction to sell its future rights to a royalty streamequipment relate, and are accounted for as discussed in note 5.is recorded under the captions selling, general and administrative expenses, and research and development (“R&D”) on the statement of comprehensive income.
IntangibleIdentifiable intangible assets
IntangibleIdentifiable intangible assets with finite useful lives consist of in-process research and development,R&D, acquired in business combinations, patents and trademarks, technology and other. In-process R&D acquired in business combinations are recognised at fair value at the acquisition date. Patents and trademarks are comprised of costs, including professional fees incurred in connection with the filing of patents and the registration of trademarks for product marketing and manufacturing purposes, net of related government grants, impairment losses, where applicable, and accumulated amortization. IntangibleIdentifiable intangible assets with finite useful lives are amortized on a straight-line basis over their estimated useful lives of eight to fifteen years for in-process research and developmentR&D and patents and ten years for trademarks. Amortization expense is allocated to the appropriate expense categories to which the underlying identifiable intangible assets relate, and recorded under the captions selling, general and administrative expenses, and R&D on the statement of comprehensive income.
Goodwill
Goodwill represents the excess of the purchase price over the fair values of the net assets of entities acquired at their respective dates of acquisition. Goodwill is not amortized butcarried at cost less accumulated impairment losses. Goodwill is testedallocated to each cash-generating unit (“CGU”) or group of CGUs that are expected to benefit from the related business combination.
Impairment of assets
Items of property, plant and equipment and identifiable intangible assets with finite lives subject to depreciation or amortization, respectively, are reviewed for impairment annually, or more frequently ifwhenever events or changes in circumstances indicate that the carrying amounts of the assets may not be recoverable. Management is required to assess at each reporting date whether there is any indication that an asset may be impaired. Where such an indication exists, the asset’s recoverable amount is compared to its carrying value, and an impairment loss is recognized for the amount by which the asset’s carrying amount exceeds its recoverable amount. The recoverable amount is the higher of an asset’s fair value less costs to sell and value in use. For the purpose of
Aeterna Zentaris Inc.
Notes to Consolidated Financial Statements
As at December 31, 2011 and December 31, 2010 and for the years ended December 31, 2011 and 2010
(tabular amounts in thousands of US dollars, except share/option/warrant and per share/option/warrant data and as otherwise noted)
assessing impairment, assets are grouped at the lowest levels for which there are separately identifiable cash flows, or CGU. In determining value in use of a given asset or CGU, estimated future cash flows are discounted to their present value using a pre-tax discount rate that reflects current market assessments of the time value of money and the reporting unitrisks specific to the asset. Impairment losses are allocated to the appropriate expense categories to which the underlying identifiable intangible assets relate, and recorded under the captions selling, general and administrative expenses, and R&D on the statement of comprehensive income.
Items of property, plant and equipment and amortizable identifiable intangible assets with finite lives that suffered impairment are reviewed for possible reversal of the impairment if there has been a change, since the date of the most recent impairment test, in the estimates used to determine the impaired asset’s recoverable amount. However, an asset’s carrying amount, increased due to the reversal of a prior impairment loss, must not exceed the carrying amount that would have been determined, net of depreciation or amortization, had the original impairment not occurred.
Goodwill is not subject to amortization and instead is tested for impairment annually or more often if there is an indication that the CGU to which the goodwill has been allocated may be impaired. Impairment is assigned may exceeddetermined for goodwill by assessing whether the carrying value of a CGU, including the allocated goodwill, exceeds its recoverable amount, which is the higher of fair value of the reporting unit.
less costs to sell and value in use. In the event that the carrying amount of a reporting unit, including goodwill exceeds its fair value,recoverable amount, an impairment loss is recognized in an amount equal to the excess. Fair value ofImpairment losses related to goodwill is estimatedare not subsequently reversed.
Share purchase warrants
Share purchase warrants are classified as liabilities, since the Company does not have the unconditional right to avoid delivering cash to the holders in the same way as goodwillfuture. The warrant liability is determinedinitially measured at the date of the acquisition in a business combination, that is, the excess of the fair value, and any subsequent changes in fair value are recognized as gains or losses through profit or loss. Any transaction costs related to the share purchase warrants are expensed as incurred.
The warrant liability is classified as non-current, unless the underlying share purchase warrants are expected to expire or be settled within 12 months from the end of a given reporting period.
Employee benefits
Salaries and other short-term benefits
Salaries and other short-term benefit obligations are measured on an undiscounted basis and are recognized in the reporting unitconsolidated statement of comprehensive loss over the fair value of the identifiable net assets of the reporting unit.
Impairment of long-lived assets
Property, plant and equipment and intangible assets with finite lives are reviewed for impairment when eventsrelated service period or circumstances indicate that carrying values may not be recoverable. Impairment exists when the carrying valueCompany has a present legal or constructive obligation to make payments as a result of past events and when the asset or asset group is greater than the undiscounted future cash flows expected toamount payable can be provided by the asset or asset group. The amount of impairment loss, if any, is the excess of its carrying value over its fair value, which in turn is determined based upon discounted cash flows or appraised values, depending on the nature of assets.
Table of Contentsestimated reliably.
AETERNA ZENTARIS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
As at December 31, 2010 and December 31, 2009and for the years ended December 31, 2010, 2009 and 2008(tabular amounts in thousands of US dollars, except share/option/warrant andper share/option/warrant data and as otherwise noted)
2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
Employee futurePost-employment benefits
The Company'sCompany’s subsidiary in Germany maintains defined contribution and unfunded defined benefit plans, as well as other benefit plans for its employees. Its obligations are accrued under employeeFor defined benefit pension plans and other post-employment benefits, net periodic pension expense is actuarially determined on an annual basis using the related costs. In this regard, the following policies have been adopted:
Aeterna Zentaris Inc.
Notes to Consolidated Financial Statements
As at December 31, 2011 and December 31, 2010 and for the years ended December 31, 2011 and 2010
(tabular amounts in thousands of US dollars, except share/option/warrant and per share/option/warrant data and as otherwise noted)
The employee future benefits liability is recognized at its present value, which is determined by discounting the estimated future cash outflows using interest rates of high-quality corporate bonds that are denominated in the currency in which the benefits will be paid and that have terms to maturity approximating the terms of the related future benefit liability. Actuarial gains and losses that arise in calculating the present value of the defined benefit obligation are recognized in other comprehensive loss, net of tax, in the deficit in the consolidated statement of financial position in the year in which the actuarial gains and losses arise and without recycling to the consolidated statement of comprehensive loss in subsequent periods.
For defined contribution plans, expenses are recorded in the consolidated statement of comprehensive loss as incurred—namely, over the period that the related employee service is rendered.
Termination benefits
Termination benefits are recognized in the consolidated statement of comprehensive loss when the Company is demonstrably committed, without the realistic possibility of withdrawal, to a formal detailed plan either to terminate employment before the normal retirement date or to provide termination benefits as a result of an offer made to encourage voluntary redundancy. Termination benefit liabilities expected to be settled after 12 months from the end of a given reporting period are discounted to their present value, where material.
Financial instruments
The Company classifies its financial instruments in the following categories: “Financial assets at FVTPL”; “Loans and receivables”; “Financial liabilities at FVTPL”; and “Other financial liabilities”.
Financial assets and liabilities are offset, and the net amount is reported in the consolidated statement of financial position, when there is a legally enforceable right to offset the recognized amounts and there is an intention to settle on a net basis or realize the asset and settle the liability simultaneously.
(a) Classification
Financial assets at fair value through profit or loss
Financial assets at FVTPL are financial assets held for trading. Fair value is defined as the amount at which the financial assets could be exchanged in a current transaction between willing parties, other than in a forced or liquidation sale. A financial asset is classified as at FVTPL if the instrument is acquired or received as consideration principally for the purpose of selling in the short-term. Financial assets at FVTPL are classified as current assets if expected to be settled within 12 months from the end of a given reporting period; otherwise, the assets are classified as non-current.
Financial assets at FVTPL are comprised exclusively of the Company’s short-term investment.
Loans and receivables
Loans and receivables are non-derivative financial assets with fixed or determinable payments that are not quoted in an active market. Loans and receivables are included in current assets, except for instruments with maturities greater than 12 months after the end of a given reporting period or where restrictions apply that limit the Company from using the projected unit credit methodinstrument for current purposes, which are classified as non-current assets.
Aeterna Zentaris Inc.
Notes to Consolidated Financial Statements
As at December 31, 2011 and benefit method proratedDecember 31, 2010 and for the years ended December 31, 2011 and 2010
(tabular amounts in thousands of US dollars, except share/option/warrant and per share/option/warrant data and as otherwise noted)
The Company’s loans and receivables are comprised of cash and cash equivalents, trade and other receivables and restricted cash.
Financial liabilities at fair value through profit or loss
Financial liabilities at FVTPL are financial liabilities held for trading. A financial liability is classified as at FVTPL if the instrument is acquired or incurred principally for the purpose of selling or repurchasing in the short-term or where the Company does not have the unconditional right to avoid delivering cash or another financial asset to the holders in certain circumstances. Financial liabilities at FVTPL are classified as current liabilities if expected or potentially required to be settled within 12 months from the end of a given reporting period; otherwise, the liabilities are classified as non-current.
Financial liabilities at FVTPL are currently comprised of the Company’s warrant liability.
Other financial liabilities
Other financial liabilities include trade accounts payable and accrued liabilities, long-term payable and other long-term liabilities.
(b) Recognition and measurement
Financial assets at fair value through profit or loss
Financial assets at FVTPL are recognized on lengththe settlement date, which is the date on which the asset is delivered to the Company. Financial assets at FVTPL are initially recognized at fair value, and transaction costs are expensed immediately in the consolidated statement of servicecomprehensive loss. Financial assets at FVTPL are derecognized when the rights to receive cash flows from the underlying investment have expired or have been transferred and management's best estimatewhen the Group has transferred substantially all risks and rewards of salary escalation, retirement agesownership. Gains and losses arising from changes in the fair value of employeesfinancial assets at FVTPL are presented in the consolidated statement of comprehensive loss within finance income or finance costs in the period in which they arise.
Loans and employee turnover;receivables
Loans and receivables are recognized on the net actuarial gainsettlement date and are measured initially at fair value and subsequently at amortized cost using the effective interest rate method.
Financial liabilities at fair value through profit or loss associated with
Financial liabilities at FVTPL are recognized on the benefit obligationsettlement date. Financial liabilities at FVTPL are initially recognized at fair value, and transaction costs are expensed immediately in the consolidated statement of comprehensive loss. Gains and losses arising from changes in the fair value of financial liabilities at FVTPL are presented in the consolidated statement of comprehensive loss within finance income or finance costs in the period in which they arise.
Other financial liabilities
Financial instruments classified as “Other financial liabilities” are measured initially at fair value and subsequently at amortized cost using the effective interest rate method.
Aeterna Zentaris Inc.
Notes to Consolidated Financial Statements
As at December 31, 2011 and December 31, 2010 and for the years ended December 31, 2011 and 2010
(tabular amounts in thousands of US dollars, except share/option/warrant and per share/option/warrant data and as otherwise noted)
| (c) | Impairment |
Financial assets measured at amortized cost are reviewed for impairment at each reporting date. Where there is objective evidence that impairment exists for a financial asset measured at amortized cost, an impairment charge equivalent to the difference between the asset’s carrying amount and the present value of estimated future cash flows is recorded in the consolidated statement of operations as it arises.
For defined contribution plans,comprehensive loss. The expected cash flows exclude future credit losses that have not been incurred and are discounted at the pension expenses recordedfinancial asset’s original effective interest rate.
Impairment charges of financial assets carried at amortized cost are reversed if, in the consolidated statement of operations isa subsequent period, the amount of contributionthe impairment loss decreases and the decrease can be related objectively to an event occurring after the impairment was recognized. However, the reversal cannot result in a carrying amount of the financial asset that exceeds what the amortized cost would have been had the impairment not been recognized at the date the impairment is reversed.
Share capital
The Company has authorized an unlimited number of common shares (being voting and participating shares) with no par value, as well as an unlimited number of preferred, first and second ranking shares, issuable in series, with rights and privileges specific to each class, with no par value.
Common shares are classified as equity. Incremental costs that are directly attributable to the issue of common shares and stock options are recognized as a deduction from equity, net of any tax effects.
Provisions
Provisions represent liabilities to the Company for which the amount or timing is uncertain. Provisions are recognized when the Company has a present legal or constructive obligation as a result of past events, when it is probable that an outflow of resources will be required to paysettle the obligation and where the amount can be reliably estimated. Provisions are not recognized for services rendered by employees.future operating losses.
Deferred revenues
Deferred revenues relateProvisions are made for any contracts, including lease arrangements, which are deemed onerous. A contract is onerous if the unavoidable costs of meeting the obligations under the contract exceed the economic benefits expected to be received under it. Provisions for onerous contracts are measured at the present value of the lower of the expected cost of terminating the contract and the expected net cost of continuing with the contract. Present value is determined based on expected future cash flows that are discounted at a pre-tax rate that reflects current market assessments of the time value of money and the risks specific to the unamortized portionliability. The unwinding of the cashdiscount is recognized in finance costs.
Revenue recognition
Sales of products
Revenues from the sale of goods are recognized when the Company has transferred to the buyer the significant risks and rewards of ownership of the goods (which is at the time the goods are shipped), when the Company retains neither continuing managerial involvement to the degree usually associated with ownership nor effective control over the goods sold, when the amount of revenues can be measured reliably, when it is probable that the economic benefits associated with the transaction will flow to the Company and when the costs incurred or to be incurred in respect of the transaction can be measured reliably.
Aeterna Zentaris Inc.
Notes to Consolidated Financial Statements
As at December 31, 2011 and December 31, 2010 and for the years ended December 31, 2011 and 2010
(tabular amounts in thousands of US dollars, except share/option/warrant and per share/option/warrant data and as otherwise noted)
Royalty revenues
The Company has deferred recognition of proceeds received in connection withDecember 2008 from Cowen Healthcare Royalty Partners L.P. (“Cowen”) relating to the Company's sale of futureCompany’s rights to royalties on future sales of Cetrotide® covered by a royalty stream. Thoselicense agreement with ARES Trading S.A. (“Merck Serono”) in which the latter had been granted worldwide marketing, distribution and selling rights, except in Japan, for Cetrotide®, a compound used forin vitro fertilization. Royalties on Merck Serono’s net sales of Cetrotide®, formerly payable to the Company, are now payable directly to Cowen.
The Company recognized the proceeds are recognizedreceived from Cowen as royalty revenue based onrevenues over the "units-of-revenue"life of the underlying royalty sale arrangement, pursuant to the “units-of-revenue” method. Under that method, as discussed in note 5. Also included in deferredperiodic royalty revenues are upfrontcalculated as the ratio of the remaining deferred revenue amount to the total estimated remaining royalties that Merck Serono is expected to pay to Cowen over the term of the underlying arrangement multiplied by the royalty payments received primarily in connection with license cooperation agreements. Those payments are recognized asdue to Cowen for the period.
Licensing revenues as discussed below.
Revenue recognitionand multiple element arrangements
The Company is currently in a phase in which certain potential products are being further developed or marketed jointly with strategic partners. Existing licensing agreements usually foresee one-time payments (upfront payments), payments for research and developmentR&D services in the form of cost reimbursements, milestone payments and royalty receipts for licensing and marketing product candidates. Revenues associated with those multiple-element arrangements are allocated to the various elements based on their relative fair value.
Agreements containing multiple elements are divided into separate units of accounting if certain criteria are met, including whether the delivered element has stand-alone value to the customer and whether there is objective and reliable evidence of the fair value of the undelivered obligation(s). The consideration received is allocated among the separate units based on each unit'sunit’s fair value, and the applicable revenue recognition criteria are applied to each of the separate units.
License fees representing non-refundable payments received at the time of signature of license agreements are recognized as revenue upon signature of the license agreements when the Company has no significant future performance obligations and collectibility of the fees is assured.probable. Upfront payments received at the beginning of licensing agreements are not recordeddeferred and recognized as revenue when received but are amortized based on a systematic basis over the progress toperiod during which the related researchservices are rendered and development work. This progress is based on estimates of total expected time or duration to complete the work, which is compared to the period of time incurred to date in order to arrive at an estimate of the percentage of revenue earned to date.all obligations are performed.
Milestone payments
Milestone payments, which are generally based on developmental or regulatory events, are recognized as revenue when the milestones are achieved, collectibility is assured, and when there arethe Company has no significant future performance obligations in connection with the milestones.
Royalty revenue, based on a percentageCost of sales
Cost of certain declared productssales represents the cost of goods sold by third parties, is recorded when the Company has fulfilled the terms in accordance with the contractual agreement, has no future obligations,and represents almost exclusively the amount of inventory recognized as an expense during the royalty fee is determinable and collection is reasonably assured.year. This amount includes the cost of raw materials, supplies, manufacturing fees as well as write-down of inventories.
The Company defers recognition of proceeds received in connection with the sale of rights to future royalties (see note 5) and recognizes these deferred revenues over the life of the license agreement, pursuant to the "units-of-revenue" method.
Table of ContentsAeterna Zentaris Inc.
AETERNA ZENTARIS INC.Notes to Consolidated Financial Statements
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
As at December 31, 20102011 and December 31, 2009
2010 and for the years ended December 31, 2010, 20092011 and 2008
2010
(tabular amounts in thousands of US dollars, except share/option/warrant and
per share/option/warrant data and as otherwise noted)
2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
Revenues from sales of products are recognized when title passes to customers, which is at the time goods are shipped, when there are no future performance obligations, when the purchase price is fixed and determinable and when collection is reasonably assured.
Stock-basedShare-based compensation costs
The Company operates an equity-settled share-based compensation plan under which the Company receives services from employees as consideration for equity instruments of the Company.
The Company accounts for all forms of employee stock-based compensation using the fair value-based method.
The fair Fair value of stock options is determined onat the date of grant using the Black-Scholes option pricing model, and stock-based compensation costswhich includes estimates of the number of awards that are recognized ratably, for all tranches,expected to vest over the vesting period. Where granted share options vest in installments over the vesting period of(defined as graded vesting), the optionsCompany treats each installment as a separate share option grant. Share-based compensation expense is recognized over the vesting period, or as specified vesting conditions are satisfied, and credited to Other Capital.
Any consideration received by the Company in connection with the exercise of stock options is credited to Share Capital. Any Other Capital component of the stock-based compensation is transferred to Share Capital upon the issuance of shares.
Income taxesCurrent and deferred income tax
The Company followstax expense for the liability methodperiod comprises current and deferred tax. Tax is recognized in profit or loss, except that a change attributable to an item of accounting for income taxes. Under this method,or expense recognized as other comprehensive income (loss) or directly in equity (deficit) is also recognized directly in other comprehensive income (loss) or directly in equity (deficit). Management periodically evaluates positions taken in tax returns with respect to situations in which applicable tax regulation is subject to interpretation. It establishes provisions where appropriate on the basis of amounts expected to be paid to the tax authorities.
The current income tax charge is calculated on the basis of tax rates and laws that have been enacted or substantively enacted by the reporting date in the countries where the company’s subsidiaries operate and generate taxable income.
Deferred income tax is recognized on temporary differences (other than temporary differences associated with unremitted earnings from foreign subsidiaries and associates to the extent that the investment is essentially permanent in duration, or temporary differences associated with the initial recognition of goodwill) arising between the tax bases of assets and liabilities and their carrying amounts in the consolidated financial statements and on unused tax losses or R&D non-refundable tax credits in the group. Deferred income tax is determined using tax rates and laws that have been enacted or substantively enacted by the reporting date.
Deferred income tax assets are recognized only to the extent that it is probable that future taxable profit will be available against which the temporary differences can be utilised.
Deferred income tax assets and liabilities are determined based onoffset when there is a legally enforceable right to offset current tax assets against current tax liabilities and when the temporary differences between the carrying amounts and tax bases of the assets and liabilities. Futuredeferred income taxtaxes assets and liabilities are measured using substantively enactedrelate to income taxes levied by the same taxation authority on either the same taxable entity or different taxable entities where there is an intention to settle the balances on a net basis.
Aeterna Zentaris Inc.
Notes to Consolidated Financial Statements
As at December 31, 2011 and enacted tax rates expected to apply inDecember 31, 2010 and for the years ended December 31, 2011 and 2010
(tabular amounts in which the differences are expected to reverse.thousands of US dollars, except share/option/warrant and per share/option/warrant data and as otherwise noted)
The Company establishes a valuation allowance against future income tax assets if, based on available information, it is more likely than not that some or all of the future income tax assets will not be realized.
Research and development costs
Research costs are expensed as incurred. Development costs are expensed as incurred except for those which meet generally accepted criteria for deferral, in which case, the costs are capitalized and amortized to operations over the estimated period of benefit. No costs have been deferred during any of the periods presented.
Research and development refundable tax credits and grants
The Company'sCompany’s German subsidiary is entitled to receive research grants from the German Federal Ministry of Education and Research. Funding is earned on qualified projects, and corresponding expenses are reimbursed at a rate of 50% of eligible base amounts.
TaxRefundable R&D tax credits and grants are accounted for using the cost reduction method. Accordingly, refundable R&D tax credits and grants are recorded as a reduction of the related expenses or capital expenditures in the period the expenses are incurred, provided that the Company has reasonable assurance the refundable R&D tax credits or grants will be realized.
LossNet loss per share
Basic net loss per share is calculated using the weighted average number of common shares outstanding during the year.
Diluted net loss per share is calculated based on the weighted average number of common shares outstanding during the year, plus the effects of dilutive common share equivalents, such as stock options and share purchase warrants. This method requires that diluted net loss per share be calculated using the treasury stock method, as if all common share equivalents had been exercised at the beginning of the reporting period, or period of issuance, as the case may be, and that the funds obtained thereby were used to purchase common shares of the Company at the average trading price of the common shares during the period.
| 3 | Critical accounting estimates and judgments |
The preparation of consolidated financial statements in accordance with IFRS often requires management to make estimates about and apply assumptions or subjective judgment to future events and other matters that affect the reported amounts of the Company’s assets, liabilities, revenues, expenses and related disclosures. Assumptions, estimates and judgments are based on historical experience, expectations, current trends and other factors that management believes to be relevant at the time at which the Company’s consolidated financial statements are prepared. Management reviews, on a regular basis, the Company’s accounting policies, assumptions, estimates and judgments in order to ensure that the consolidated financial statements are presented fairly and in accordance with IFRS.
Critical accounting estimates and judgments are those that have a significant risk of causing material adjustment and are often applied to matters or outcomes that are inherently uncertain and subject to change. As such, management cautions that future events often vary from forecasts and expectations and that estimates routinely require adjustment.
The following discusses the most significant accounting estimates and judgments that the Company has made in the preparation of the consolidated financial statements.
Table of ContentsAeterna Zentaris Inc.
AETERNA ZENTARIS INC.Notes to Consolidated Financial Statements
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
As at December 31, 20102011 and December 31, 2009
2010 and for the years ended December 31, 2010, 20092011 and 2008
2010
(tabular amounts in thousands of US dollars, except share/option/warrant and
per share/option/warrant data and as otherwise noted)
Revenue recognition
3. NEW ACCOUNTING STANDARDS AND PRONOUNCEMENTSRoyalty revenues calculated under the “units-of-revenue” method are dependent upon certain assumptions, including expected future third-party net sales of Cetrotide®. Any future changes in the assumptions utilized to determine the amortization of deferred revenues could result in a change in the timing of the Company’s royalty revenue recognition.
BeginningManagement’s assessments related to the recognition of revenues related to arrangements containing multiple elements are based on January 1, 2011,estimates and assumptions. Judgment is necessary to identify separate units of accounting and to allocate related consideration to each separate unit of accounting. Where deferral of upfront payments or license fees is deemed appropriate, subsequent revenue recognition is often determined based upon certain assumptions and estimates, including arrangement-specific cash flow projections (discounted using appropriate interest rates), the Company will ceaseCompany’s continuing involvement in the arrangement, the benefits expected to prepare its consolidated financial statements in accordance with Canadian GAAP. For periods beginning on January 1, 2011, the Company will apply International Financial Reporting Standards ("IFRS") as issuedbe derived by the International Accounting Standards Board ("IASB") ascustomer and expected patent lives. To the Company's primary basis of accounting. Consequently, the following guidance will never be applied by the Company.
Not yet adopted
In December 2009, the Emerging Issues Committee ("EIC")extent that any of the Canadian institute of Chartered Accountants ("CICA") issued abstract EIC-175, "Multiple Deliverable Revenue Arrangements" ("EIC-175"), which requires a vendor to allocate arrangement consideration at the inception of an arrangement to all deliverables using the relative selling price method. EIC-175 also changes the level of evidenceassumptions or estimates change, future operating results could be affected.
Fair value of the standalone selling price required to separate deliverables when more objective evidenceshort-term investment, warrant liability and stock options
Determining the fair value of the sellingshort-term investment, warrant liability and stock options requires judgment related to the choice of a pricing model, the estimation of stock price volatility and the expected term of the underlying instruments. Any changes in the estimates or inputs utilized to determine fair value could result in a significant impact on the Company’s future operating results, liabilities or other components of shareholders’ deficiency. Fair value assumptions used are described in notes 7, 15 and 17.
Identifiable intangible assets and goodwill impairment
The values associated with identifiable intangible assets with finite lives and goodwill are determined by applying significant estimates and assumptions, including those related to cash flow projections, economic risk, discount rates and asset useful lives.
Valuations performed in connection with post-acquisition assessments of impairment of identifiable intangible assets are based on estimates that include risk-adjusted future cash flows, which are discounted using appropriate interest rates. Projected cash flows are based on business forecasts, trends and expectations and are therefore inherently judgmental. Future events could cause the assumptions utilized in impairment assessments to change, resulting in a potentially significant effect on the Company’s future operating results due to increased impairment charges, or reversals thereof, or adjustments to amortization charges.
The annual impairment assessment related to goodwill is not available. Givenbased on estimates that are derived from current market capitalization and on other factors, including assumptions related to recent industry-specific market analyses. Future events, including a significant reduction in the requirementCompany’s share price, could cause the assumptions utilized in the impairment tests to usechange, resulting in a potentially adverse effect on the relative selling price methodCompany’s future results due to increased impairment charges.
Aeterna Zentaris Inc.
Notes to Consolidated Financial Statements
As at December 31, 2011 and December 31, 2010 and for the years ended December 31, 2011 and 2010
(tabular amounts in thousands of allocating arrangement consideration, EIC-175 prohibitsUS dollars, except share/option/warrant and per share/option/warrant data and as otherwise noted)
Employee future benefits
The determination of expense and obligations associated with employee future benefits requires the use of assumptions, such as the residual method. EIC-175 may be applied prospectivelydiscount rate to measure obligations, the projected age of employees upon retirement, the expected rate of future compensation and estimated employee turnover. Because the determination of the cost and obligations associated with employee future benefits requires the use of various assumptions, there is applicable to revenue arrangements with multiple deliverables entered into or materially modifiedmeasurement uncertainty inherent in the actuarial valuation process. Actual results will differ from results that are estimated based on the aforementioned assumptions.
Income taxes
The estimation of income taxes includes evaluating the recoverability of deferred tax assets based on an assessment of Group entities’ ability to utilize the underlying future tax deductions against future taxable income prior to expiry of those deductions. Management assesses whether it is probable that some or all of the deferred income tax assets will not be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income, which in turn is dependent upon the successful commercialization of the Company’s products. To the extent that management’s assessment of any Group entity’s ability to utilize future tax deductions changes, the Company would be required to recognize more or fewer deferred tax assets, and future income tax provisions or recoveries could be affected.
| 4 | Recent accounting pronouncements |
In November 2009 and October 2010, the IASB issued IFRS 9,Financial Instruments (“IFRS 9”), which represents the completion of the first part of a three-part project to replace IAS 39,Financial Instruments: Recognition and Measurement, with a new standard. Per the new standard, an entity choosing to measure a liability at fair value will present the portion of the change in its fair value due to changes in the entity’s own credit risk in the other comprehensive income or loss section of the entity’s statement of comprehensive loss, rather than within profit or loss in case where the fair value option is taken for financial liabilities. Additionally, IFRS 7 Amendment includes revised guidance related to the derecognition of financial instruments. IFRS 9 applies to financial statements for annual fiscal periodperiods beginning on or after January 1, 2011,2015, with early adoption permitted. The Company currently is evaluating any impact that this new standard may have on the Company’s consolidated financial statements.
4. DEVELOPMENT, COMMERCIALIZATION AND LICENSE AGREEMENTIn May 2011, the IASB issued IFRS 10,Consolidated Financial Statements (“IFRS 10”), which builds on existing principles by identifying the concept of control as the determining factor in whether an entity should be included within the consolidated financial statements of a parent company. IFRS 10 also provides additional guidance to assist in the determination of control where this is difficult to assess. IFRS 10 applies to financial statements for annual periods beginning on or after January 1, 2013, with early adoption permitted. The Company currently is evaluating any impact that this new guidance may have on the Company’s consolidated financial statements.
In May 2011, the IASB issued IFRS 11,Joint Arrangements(“IFRS 11”), which enhances accounting for joint arrangements, particularly by focusing on the rights and obligations of the arrangement, rather than the arrangement’s legal form. IFRS 11 also addresses inconsistencies in the reporting of joint arrangements by requiring a single method to account for interests in jointly controlled entities and prohibits proportionate consolidation. IFRS 11 applies to financial statements for annual periods beginning on or after January 1, 2013, with early adoption permitted. The Company currently is evaluating any impact that this new guidance may have on the Company’s consolidated financial statements.
Aeterna Zentaris Inc.
Notes to Consolidated Financial Statements
As at December 31, 2011 and December 31, 2010 and for the years ended December 31, 2011 and 2010
(tabular amounts in thousands of US dollars, except share/option/warrant and per share/option/warrant data and as otherwise noted)
In May 2011, the IASB issued IFRS 12,Disclosure of Interests in Other Entities(“IFRS 12”), which is a comprehensive standard on disclosure requirements for all forms of interests in other entities, including joint arrangements, associates, special purpose vehicles and other off-balance sheet vehicles. IFRS 12 applies to financial statements for annual periods beginning on or after January 1, 2013, with early adoption permitted. The Company currently is evaluating any impact that this new guidance may have on the Company’s consolidated financial statements.
In May 2011, the IASB issued IFRS 13,Fair ValueMeasurement (“IFRS 13”), which defines fair value, sets out in a single IFRS a framework for measuring fair value and requires disclosures about fair value measurements. IFRS 13 does not determine when an asset, a liability or an entity’s own equity instrument is measured at fair value. Rather, the measurement and disclosure requirements of IFRS 13 apply when another IFRS requires or permits the item to be measured at fair value (with limited exceptions). IFRS 13 applies to financial statements for annual periods beginning on or after January 1, 2013, with early adoption permitted. The Company currently is evaluating any impact that this new guidance may have on the Company’s consolidated financial statements.
In June 2011, the IASB amended IAS 1,Presentation of Financial Statements (“IAS 1”), to change the disclosure of items presented in other comprehensive income into two groups, based on whether those items may be recycled to profit or loss in the future. The amendments to IAS 1 apply to financial statements for annual periods beginning after July 1, 2012, with early adoption permitted. The Company currently is evaluating any impact that this new guidance may have on the Company’s consolidated financial statements.
In June 2011, the IASB issued an amended version of IAS 19,Employee Benefits (“IAS 19”), including the elimination of the option to defer the recognition of actuarial gains and losses (known as the “corridor method”), the streamlining of the presentation of changes in assets and liabilities arising from defined benefit plans and the enhancement of the disclosure requirements for defined benefit plans, including additional information about the characteristics of defined benefit plans and the risks to which entities are exposed through participation in those plans. The amendments to IAS 19 apply to financial statements for annual periods beginning on or after January 1, 2013, with early adoption permitted. The Company currently is evaluating any impact that this new standard may have on the Company’s consolidated financial statements.
| 5 | Development, commercialization and license agreement |
On March 4, 2009,8, 2011, the Company and sanofi-aventis U.S L.L.C. ("sanofi") entered into a development, commercialization and licensean agreement (the "sanofi Agreement"with Yakult Honsha Co. Ltd. (“Yakult”) for the development, registrationmanufacture and marketingcommercialization of cetrorelixperifosine in benign prostatic hyperplasia ("BPH") for the United States market.all human uses, excluding leishmaniasis, in Japan. Under the terms of the sanofi Agreement, sanofithis agreement, Yakult made an upfrontinitial, non-refundable license feegross upfront payment to the Company of $30,000,000.€6,000,000 (approximately $8,412,000). Also per the sanofi Agreement,agreement, the Company would have beenis entitled to receive certain paymentsup to a total of €44,000,000 (approximately $57,075,000) upon achieving certain pre-established milestones, including the occurrence of certain clinical and regulatory events in Japan. As at December 31, 2011, following the achievement of a milestone, the Company has revenue and commercial milestones as well as escalatingtrade receivables recorded for an additional amount of €2,000,000 (approximately $2,594,000) in connection with the agreement. Furthermore, the Company will be entitled to receive double-digit royalties on future net sales of cetrorelix for BPHperifosine in the United States.Japanese market.
On November 23, 2011, the Company entered into an agreement with Hikma Pharmaceuticals LLC. (“Hikma”) for the development and commercialization of perifosine in all oncology uses in certain countries in the Middle East and North Africa (“MENA”). Under the terms of this agreement, Hikma made an initial, non-refundable gross upfront payment to the Company of $200,000. Also per the agreement,
Aeterna Zentaris Inc.
Notes to Consolidated Financial Statements
As with similar priorat December 31, 2011 and December 31, 2010 and for the years ended December 31, 2011 and 2010
(tabular amounts in thousands of US dollars, except share/option/warrant and per share/option/warrant data and as otherwise noted)
the Company will be entitled to receive up to a total of $1,800,000 upon achieving certain pre-established milestones, including the occurrence of certain regulatory events in certain countries in the MENA region. Furthermore, the Company will be entitled to receive double-digit royalties on future net sales of perifosine in the MENA market.
The Company has substantial continuing involvement in the aforementioned arrangements, including the use of commercially reasonable efforts to develop, apply for and obtain relevant regulatory approval for, manufacture and commercialize perifosine outside Japan and MENA region, which will facilitate the ultimate commercialization process within Japan and MENA region. Additionally, the Company will ensure a stable supply of perifosine and related trial products to Yakult and Hikma throughout the ongoing development process and will maintain relevant patent rights over the term of the arrangements. Lastly, per the terms of the aforementioned agreements, the Company has agreed to supply perifosine, on a cost-plus basis, to Yakult and Hikma following regulatory approvals.
The Company has applied the provisions of the EIC's Abstract No. 142, "Revenue Arrangements with Multiple Deliverables",IAS 18, Revenue, and hadhas determined that all deliverables and performance obligations contemplated by the sanofi Agreementagreements with Yakult and Hikma should be accounted for as a single unit of accounting, limited to amounts that wereare not contingent upon the delivery of additional items or the meeting of other specified performance conditions which wereare not known, probable or estimable at the time at which the sanofi Agreement wasagreements with Yakult and Hikma were entered into.
On December 18, 2009,The Company has deferred the non-refundable license fees and followingis amortizing the Company's announcementrelated payments as revenue on a straight-line basis over the duration of the Company’s continuing involvement in the arrangements, which approximate the estimated life cycle of the product that its second Phase 3 study with cetrorelixis currently under development and the expected period over which Yakult and Hikma will derive value from the use of, and access to, the underlying licenses.
In determining the period over which license revenues are to be recognized, and in BPH had not reached its primary endpoint,addition to due consideration of the Company’s continuing involvement, as discussed above, the Company disclosed that it had received notice from sanofi to terminateconsidered the sanofi Agreement. As a result,remaining expected life of applicable patents as the most reasonable basis for estimating the underlying product’s life cycle. However, the Company fullymay adjust the amortization period based on appropriate facts and circumstances not yet known, including, but not limited to, the extension(s) of patents, the granting of new patents, the economic lives of competing products and other events that would significantly change the duration of the Company’s continuing involvement and performance obligations or benefits expected to be derived by Yakult and Hikma.
Future milestones will be recognized as revenue individually and in full upon the actual achievement of the related milestone, given the substantive nature of each milestone. Lastly, upon initial commercialization and sale of the developed product, the Company will recognize royalty revenues as earned, based on the contractual percentage applied to the actual net sales achieved by Yakult and Hikma, as per the aforementioned upfront payment, asagreement.
The Company was required to remit to the culminationJapanese tax authorities $841,000 of the earnings process was deemedgross proceeds received from Yakult and will be required to remit €200,000 (approximately $259,000) when the milestone receivable will be complete.
The sanofi Agreement also stipulated that certain development expenses incurredpaid by Yakult. These amounts, which were (will be) withheld at the Company, including those costs associated with an open label extension study andsource, were recognized as income tax expense in the consolidated statement of comprehensive loss in accordance with the establishmentprovision of a supply arrangement, were reimbursable, upIAS 12, Income Taxes.
Aeterna Zentaris Inc.
Notes to predetermined maximum amounts or limits, by sanofi. Total revenues were recognized on a gross basis and as corresponding reimbursable costs were incurred. During the year ended December 31, 2009, revenues recognized in connection with the reimbursable development activities amounted to approximately $2,135,000, and corresponding expenses totalled approximately $2,814,000 for the same period.Consolidated Financial Statements
As a result of entering into the sanofi Agreement, the Company paid a royalty to the Tulane Educational Fund ("Tulane") pursuant to a license agreement whereby the Company obtained licenses to use Tulane's patents to develop, manufacture, market and distribute various compounds, including cetrorelix. This royalty, amounting to $3,000,000, was charged in full to selling expenses during the year ended December 31, 2009 as a result of sanofi's decision to terminate the sanofi Agreement.
AETERNA ZENTARIS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
As at December 31, 20102011 and December 31, 2009
2010 and for the years ended December 31, 2010, 20092011 and 2008
2010
(tabular amounts in thousands of US dollars, except share/option/warrant and
per share/option/warrant data and as otherwise noted)
5. SALE OF CETROTIDE® ROYALTY STREAM
| 6 | Cash and cash equivalents |
In June 2003, the Company amended certain provisions
| As at December 31, 2011 | As at December 31, 2010 | As at January 1, 2010 | ||||
| $ | $ | $ | ||||
Cash on hand and balances with banks | 15,112 | 12,922 | 33,100 | |||
Short-term interest-bearing deposits | 31,769 | 19,076 | 5,000 | |||
| 46,881 | 31,998 | 38,100 | ||||
| 7 | Short-term investment |
The short-term investment consisted of shares of a license and supply agreement with ARES Trading S.A. ("Merck Serono")publicly held company. The underlying shares were sold in whichJuly 2011.
The change in the latter was granted worldwide marketing, distribution and selling rights, except in Japan, for Cetrotide®, a compound used forin vitro fertilization (referred toCompany’s short-term investment can be summarized as follows:
| Years ended December 31, | ||||||||
| 2011 | 2010 | |||||||
| $ | $ | |||||||
Balance at January 1 | 1,934 | - | ||||||
Addition of short-term investment | - | 1,263 | ||||||
Change in fair value of shares | 1,278 | 687 | ||||||
Change in value attributable to foreign exchange rate changes | 30 | (16 | ) | |||||
Sale of short term investment | (3,242 | ) | - | |||||
Balance at December 31 | - | 1,934 | ||||||
The fair value of the License Agreement). Under the License Agreement, Merck Serono agreed to pay certain lump-sum payments to the Company each calendar year up to and includingshort-term investment as at December 31, 2010 as well as certain variable royalties throughwas discounted via the expiry dateapplication of the Company's underlying patent rights.
In November 2008, the Company entered into a purchase and sale agreement ("PSA") with Cowen Healthcare Royalty Partners L.P. ("Cowen") relating to the Company's rights to royalties on future sales of Cetrotide® covered by the License Agreement.
In connectionBlack-Scholes pricing model with the PSA, which was effective for royalty determination purposes on October 1, 2008following inputs and finalized in December 2008, the Company received $52,500,000 from Cowen, less certain transaction costs of $1,000,000 that had been advanced by Cowen to certain third-party firms and institutions on the Company's behalf, resulting in net proceeds of $51,500,000. Under the terms of the PSA, the Company is entitled to an additional payment of $2,500,000 contingent on 2010 net sales of Cetrotide® reaching a specified level. This additional consideration was earned in 2010, and the corresponding amount has been recorded as royalty revenues in the accompanying consolidated statement of operations.assumptions:
Number of shares | 326,956 | |||||
Market-value price per share | $6.87 | |||||
Expected dividend yield | 0% | |||||
Expected volatility | 36.2% | |||||
Risk-free annual interest rate | 0.3% | |||||
Expected life (years) | 0.34 | |||||
Discount per share | $0.98 |
Per the PSA, if cetrorelix, the active compound in Cetrotide®, is approved for sale by European regulatory authorities in an indication other thanin vitro fertilization, the Company has agreed to make a one-time cash payment to Cowen in an amount ranging from $5,000,000 up to a maximum of $15,000,000. The amount which may be due to Cowen will be higher in proportion to the timing of the product's receiving European regulatory approval; that is, the earlier the product receives regulatory approval, the higher the amount payable to Cowen will be. No payment was made or became payable during 2010 or 2009.
Also per the PSA, for each calendar quarter in which a royalty rate reduction — defined as the actual reduction by Merck Serono, for any calendar quarter(s), of the rate appliedBlack-Scholes valuation methodology uses “Level 2” inputs in calculating variable royalties under the License Agreement,fair value, as defined in IFRS 7,Financial Instruments: Disclosures (“IFRS 7”), and as discussed in note 24.
Aeterna Zentaris Inc.
Notes to amounts less than pre-established percentages — has occurred or is continuing, the Company will pay Cowen a quarterly make-whole payment in an amount equal to the lesser of (i) the variable royalties in respect of such quarter that would have been received by Cowen if the aforementioned royalty rate reduction had not occurred or been continuing, and (ii) the difference of $15,000,000 less Cowen's net reduction payments, as defined. No make-whole payments were paid or became payable during 2010 or 2009.Consolidated Financial Statements
Pursuant to the aforementioned transactions, the Company has certain obligations in the royalty arrangement, including the supply of Cetrotide® to Merck Serono, the payment of royalties to a third party under the License Agreement, overseeing Merck-Serono's compliance with the License Agreement, cooperation in handling any adverse claims or litigation involving the License Agreement and monitoring and defending any patent or trademark infringement.
The Company has recorded the proceeds, as per the provisions of guidance now codified as the United States Financial Accounting Standards Board's ("FASB")Accounting Standards Codification ("ASC") Topic 470, Debt, as deferred revenues, which are recognizable as royalty revenues over the life of the License Agreement under the "units-of-revenue" method. Under that method, periodic royalty revenues are calculated by multiplying the ratio of the remaining deferred revenue amount to the total estimated remaining royalties that Merck Serono is expected to pay to Cowen over the term of the underlying arrangement by the royalty payments due to Cowen for the period.
Transaction costs incurred in connection with the aforementioned PSA were comprised of fees charged by the investment banking institution that was engaged to monetize the royalty stream, transaction costs charged by Cowen (discussed above), legal expenses and other professional fees directly related to the consummation of the PSA. The Company has recognized and will continue to recognize royalty expenses in each period based on the transaction costs, which have been capitalized as deferred charges in the accompanying balance sheets (see note 10), in the same manner and over the same period in which the related deferred revenues are recognized as royalty revenues.
AETERNA ZENTARIS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
As at December 31, 20102011 and December 31, 2009
2010 and for the years ended December 31, 2010, 20092011 and 2008
2010
(tabular amounts in thousands of US dollars, except share/option/warrant and
per share/option/warrant data and as otherwise noted)
5. SALE OF CETROTIDE® ROYALTY STREAM (Continued)
| 8 | Trade and other receivables |
During the years ended December 31, 2010, 2009 and 2008, the Company recorded approximately $7,981,000, $5,686,000 and $1,355,000, respectively, as royalty revenues. During the years ended December 31, 2010, 2009 and 2008, the Company recorded $504,000, $522,000 and $124,000, respectively, as royalty expense, which is included in selling, general and administrative expenses in the accompanying consolidated statements of operations.
| As at December 31, 2011 | As at December 31, 2010 | As at January 1, 2010 | |||||||||||||
| $ | $ | $ | |||||||||||||
Trade accounts receivable | 7,716 | 4,555 | 2,444 | ||||||||||||
Value added tax | 439 | 595 | 729 | ||||||||||||
Other | 170 | 271 | 376 | ||||||||||||
| 8,325 | 5,421 | 3,549 | |||||||||||||
6. LOSS ON DISPOSAL OF LONG-LIVED ASSETS
| 9 | Inventory |
On June 26, 2008, the Company sold its Quebec City building and land for a gross amount of $7,061,000. The net proceeds received amounted to $6,545,000, resulting in a loss on sale of $810,000.
| As at December 31, 2011 | As at December 31, 2010 | As at January 1, 2010 | |||||||||||||
| $ | $ | $ | |||||||||||||
Raw materials | 1,608 | 1,899 | 2,998 | ||||||||||||
Work in progress | 1,848 | 1,412 | 1,417 | ||||||||||||
| 3,456 | 3,311 | 4,415 | |||||||||||||
| 10 | Restricted cash |
In connection with the salesupport of the Quebec City buildingCompany’s long-term operating lease obligation in Germany and land, the Company entered into a long-term lease agreement with the principal tenant of the building. As part of the agreement, the Company agreed to pay the principal tenant CAN$300,000 as an incentive and service fee. This fee is included in the additional loss on sale, and the resulting payable is non interest-bearing and is due in bi-annual instalments of CAN$30,000 through January 2013.
On March 1, 2008, the Company entered into a definitive purchase and sale agreement with respect to all rights related to the manufacture, production, distribution, marketing, sale and/or use of Impavido® (miltefosine) with Paladin Labs Inc., for an aggregate purchase price of approximately $9,200,000, payable in cash, subject to certain post-closing purchase price adjustments. The transaction, which closed on March 31, 2008, generated net cash proceeds of $8,309,000, resulting in a gain of $775,000.
7. INVENTORY
| | As at December 31, | |||||||
|---|---|---|---|---|---|---|---|---|
| | 2010 | 2009 | ||||||
| | $ | $ | ||||||
Raw materials | 1,899 | 2,998 | ||||||
Work in progress | 1,412 | 1,417 | ||||||
| 3,311 | 4,415 | |||||||
For the years ended December 31, 2010, 2009 and 2008, cost of sales, as presented in the accompanying consolidated statements of operations, represents almost exclusively the amount of inventory recognized as an expense during the year.
In December 2008, the Company wrote down certain inventory items, consisting predominantly of raw materials, to their estimated net realizable values. The adjustment, which amounted to approximately $726,000, was recorded as an additional cost of sales in the accompanying consolidated statement of operations.
8. RESTRICTED CASH
In July 2009, following a mutual agreement between landlord and tenant, in replacement of a related bank guarantee, the Company transferred approximately $866,000$806,000 to a restricted cash account in support of its long-term lease obligation in Germany (see also note 22).account. The fixed amount, including any interest earned thereon, is restricted for as long as the underlying lease arrangement has not expired and therefore cannot be utilized for current purposes as at December 31, 2010.2011.
| 11 | Property, plant and equipment |
Components of the Company’s property, plant and equipment are summarized below.
| Cost | |||||||||||||||||||||||||
| Equipment | Furniture and fixtures | Computer equipment | Leasehold improvements | Total | |||||||||||||||||||||
| $ | $ | $ | $ | $ | |||||||||||||||||||||
At January 1, 2010 | 9,941 | 1,653 | 1,851 | 1,232 | 14,677 | ||||||||||||||||||||
Additions | 76 | - | 6 | - | 82 | ||||||||||||||||||||
Disposals | (455 | ) | - | (5 | ) | - | (460 | ) | |||||||||||||||||
Impact of foreign exchange rate changes | (623 | ) | (102 | ) | (114 | ) | (76 | ) | (915 | ) | |||||||||||||||
At December 31, 2010 | 8,939 | 1,551 | 1,738 | 1,156 | 13,384 | ||||||||||||||||||||
Additions | 684 | - | 46 | 6 | 736 | ||||||||||||||||||||
Disposals | (96 | ) | - | (2 | ) | - | (98 | ) | |||||||||||||||||
Impact of foreign exchange rate changes | (330 | ) | (49 | ) | (58 | ) | (37 | ) | (474 | ) | |||||||||||||||
At December 31, 2011 | 9,197 | 1,502 | 1,724 | 1,125 | 13,548 | ||||||||||||||||||||
Table of ContentsAeterna Zentaris Inc.
AETERNA ZENTARIS INC.Notes to Consolidated Financial Statements
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
As at December 31, 20102011 and December 31, 2009
2010 and for the years ended December 31, 2010, 20092011 and 2008
2010
(tabular amounts in thousands of US dollars, except share/option/warrant and
per share/option/warrant data and as otherwise noted)
9. PROPERTY, PLANT AND EQUIPMENT
| Accumulated depreciation | |||||||||||||||||||||||||
| Equipment | Furniture and fixtures | Computer equipment | Leasehold improvements | Total | |||||||||||||||||||||
| $ | $ | $ | $ | $ | |||||||||||||||||||||
At January 1, 2010 | 6,831 | 1,382 | 1,678 | 428 | 10,319 | ||||||||||||||||||||
Disposals | (395 | ) | - | (5 | ) | - | (400 | ) | |||||||||||||||||
Impairment loss | - | - | - | - | - | ||||||||||||||||||||
Recurring depreciation expense | 736 | 54 | 102 | 113 | 1,005 | ||||||||||||||||||||
Impact of foreign exchange rate changes | (423 | ) | (84 | ) | (103 | ) | (26 | ) | (636 | ) | |||||||||||||||
At December 31, 2010 | 6,749 | 1,352 | 1,672 | 515 | 10,288 | ||||||||||||||||||||
Disposals | (96 | ) | - | (2 | ) | - | (98 | ) | |||||||||||||||||
Impairment loss | - | 134 | - | 178 | 312 | ||||||||||||||||||||
Recurring depreciation expense | 728 | 39 | 50 | 101 | 918 | ||||||||||||||||||||
Impact of foreign exchange rate changes | (255 | ) | (47 | ) | (56 | ) | (26 | ) | (384 | ) | |||||||||||||||
At December 31, 2011 | 7,126 | 1,478 | 1,664 | 768 | 11,036 | ||||||||||||||||||||
| | As at December 31, | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2010 | 2009 | ||||||||||||
| | Cost | Accumulated depreciation | Cost | Accumulated depreciation | ||||||||||
| | $ | $ | $ | $ | ||||||||||
Equipment | 8,939 | 6,749 | 9,941 | 6,831 | ||||||||||
Furniture and fixtures | 1,551 | 1,352 | 1,653 | 1,382 | ||||||||||
Computer equipment | 1,738 | 1,672 | 1,851 | 1,678 | ||||||||||
Leasehold improvements | 1,156 | 515 | 1,232 | 428 | ||||||||||
| 13,384 | 10,288 | 14,677 | 10,319 | |||||||||||
Less: | ||||||||||||||
Accumulated depreciation | 10,288 | 10,319 | ||||||||||||
Net amount | 3,096 | 4,358 | ||||||||||||
| Carrying amount | |||||||||||||||||||||||||
| Equipment | Furniture and fixtures | Computer equipment | Leasehold improvements | Total | |||||||||||||||||||||
| $ | $ | $ | $ | $ | |||||||||||||||||||||
At January 1, 2010 | 3,110 | 271 | 173 | 804 | 4,358 | ||||||||||||||||||||
At December 31, 2010 | 2,190 | 199 | 66 | 641 | 3,096 | ||||||||||||||||||||
At December 31, 2011 | 2,071 | 24 | 60 | 357 | 2,512 | ||||||||||||||||||||
FollowingDepreciation of $918,000 ($1,005,000 in 2010) is included in the Company's announcement that its second Phase 3 study with cetrorelixstatement of comprehensive loss: $824,000 ($902,000 in BPH did not reach its primary endpoint2010) in R&D and $94,000 ($103,000 in 2010) in selling, general and administrative expenses.
During the subsequent terminationyear ended December 31, 2011, an impairment loss was recognized based on the results of impairment testing of the sanofi Agreement, as discussed in note 4,Company’s Leasehold improvements and Furniture and fixtures assets. The impairment loss was recognized predominantly to take into account the Company determinedrelocation of one of the Company’s offices.
Following management’s analyses, which demonstrated that certain items of property, plant and equipment, utilized exclusively in the development activities related to cetrorelix, were no longer recoverable. As a result, an impairment charge, which was determined by applying a present value model, representing the full remaining carrying value of these assets as summarized below,was no longer recoverable, an impairment charge of approximately $312,000, was recorded as additional depreciation expense, which in December 2009turn is included within general and administrative expenses in the accompanying consolidated statement of operations.comprehensive loss.
| |||||
| |||||
| |||||
10. DEFERRED CHARGES AND OTHER LONG-TERM ASSETS
| | As at December 31, | |||||||
|---|---|---|---|---|---|---|---|---|
| | 2010 | 2009 | ||||||
| | $ | $ | ||||||
Royalty sale transaction expenses (note 5) | 3,580 | 4,205 | ||||||
Other | 804 | 528 | ||||||
| 4,384 | 4,733 | |||||||
Table of ContentsAeterna Zentaris Inc.
AETERNA ZENTARIS INC.Notes to Consolidated Financial Statements
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
As at December 31, 20102011 and December 31, 2009
2010 and for the years ended December 31, 2010, 20092011 and 2008
2010
(tabular amounts in thousands of US dollars, except share/option/warrant and
per share/option/warrant data and as otherwise noted)
11. INTANGIBLE ASSETS
| 12 | Identifiable intangible assets |
Identifiable intangible assets with finite useful lives consist entirely of in-process R&D costs, patents and trademarks. The changes in the carrying value of the Company'sCompany’s identifiable intangible assets consisting entirely of in-process research and development costs, patents and trademarks, iswith finite useful lives are summarized below.
| Year ended December 31, 2011 | Year ended December 31, 2010 | |||||||||||||||||||||||
| Cost $ | Accumulated amortization $ | Carrying value $ | Cost $ | Accumulated amortization $ | Carrying value $ | |||||||||||||||||||
Balances – At January 1 | 39,141 | (35,842) | 3,299 | 41,715 | (37,588) | 4,127 | ||||||||||||||||||
Additions | 69 | - | 69 | - | - | - | ||||||||||||||||||
Impairment loss | - | (1,093) | (1,093) | - | - | - | ||||||||||||||||||
Recurring amortization expense | - | (553) | (553) | - | (568) | (568) | ||||||||||||||||||
Impact of foreign exchange rate changes | (1,228) | 1,275 | 47 | (2,574) | 2,314 | (260) | ||||||||||||||||||
|
|
|
| |||||||||||||||||||||
Balances – At December 31 | 37,982 | (36,213) | 1,769 | 39,141 | (35,842) | 3,299 | ||||||||||||||||||
|
|
|
| |||||||||||||||||||||
| | As at December 31, | |||||||
|---|---|---|---|---|---|---|---|---|
| | 2010 | 2009 | ||||||
| | $ | $ | ||||||
Historical cost | 39,141 | 41,715 | ||||||
Less: accumulated amortization | 24,663 | 24,681 | ||||||
| 14,478 | 17,034 | |||||||
FollowingAmortization of $553,000 ($568,000 in 2010) is included in the Company's announcement that its second Phase 3 study with cetrorelixstatement of comprehensive loss: $507,000 ($481,000 in BPH did not reach its primary endpoint2010) in R&D and $46,000 ($87,000 in 2010) in selling expenses.
During the subsequent terminationyear ended December 31, 2011, an impairment loss was recognized based on the results of impairment testing of the sanofi Agreement, as discussedCompany’s Cetrotide® asset. The impairment loss was recognized predominantly to take into account management’s lower trend estimates related to the commercialization of Cetrotide®, due to changes in note 4, the Companycompetitive environment in the Japanese market.
Management determined that the carryingrecoverable amount of Cetrotide®, as at September 30, 2011, was equivalent to the asset’s value in use, as defined by IAS 36,Impairment of Assets. Value in use was determined by applying a discount rate of 20%—a pre-tax rate that reflects both current market assessments of the time value of cetrorelix was no longer recoverable. Asmoney and the risks specific to the asset for which the future cash flow estimates have not been adjusted—to the estimated future cash flows deemed to be attributable to the use of Cetrotide®. Management has projected cash flows over a result,period of 11 years, which is until the end of the expected life of Cetrotide® and has used an impairment charge, representingaverage growth rate of 1.7 % for the fullyears 1 to 6 and an average growth rate of -9.7% for the years 7 to 11 to extrapolate cash flow projections. More specifically, the discount rate represents an estimate of a reasonable return that would be expected by an investor in an arm’s length monetization transaction in respect of future Cetrotide®-derived cash flows.
Following management’s analyses, which demonstrated that the remaining carrying value of the intangible asset, orCetrotide® was no longer recoverable, an impairment charge of approximately $3,854,000,$1,093,000, was recorded during the year ended December 31, 2011 as additional amortization expense, which in December 2009turn is included within selling expenses in the accompanying consolidated statement of operations.
Management also determined that ozarelix — another luteinizing hormone-releasing ("LHRH") antagonist that, despite its different formulation, works on the same mechanism of action as cetrorelix — was impaired. Additionally, on January 27, 2010, Spectrum Pharmaceuticals, Inc. ("Spectrum"), to whom the Company has granted an exclusive license to develop and commercialize ozarelix for all potential indications in all worldwide territories, excluding certain Asian markets, announced that it had terminated its development program with ozarelix in BPH. Consequently, an impairment loss of approximately $1,422,000 was recorded as part of amortization expense, and all corresponding unamortized deferred revenues related to the use of ozarelix, totalling approximately $1,606,000, were fully recognized in the 2009 consolidated statement of operations.
In June 2008, Ardana Bioscience Ltd. ("Ardana"), to whom the Company had granted an exclusive license for the development and commercialization of teverelix, an LHRH antagonist, communicated that it was entering into voluntary administration, and, consequently, clinical studies and future development efforts were suspended. The Company subsequently terminated the aforementioned agreement, upon which the cash recoverability of teverelix exclusively had depended. Given these facts, the Company determined that teverelix was impaired, and consequently, an impairment charge to amortize the full remaining carrying value of the intangible asset, or approximately $2,362,000, was recorded as additional amortization expense in the 2008 consolidated statement of operations, and the asset was written off. Additionally, the remaining balance of deferred revenues related to the use of teverelix, amounting to approximately $1,047,000, was fully recognized in the 2008 consolidated statement of operations.
Expected amortization expense for intangible assets, excluding any potential impairment charges, for each of the next five fiscal years is shown below.
| | $ | ||||
|---|---|---|---|---|---|
2011 | 1,505 | ||||
2012 | 1,505 | ||||
2013 | 1,505 | ||||
2014 | 1,189 | ||||
2015 | 1,178 | ||||
Table of Contentscomprehensive loss.
AETERNA ZENTARIS INC.Aeterna Zentaris Inc.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)Notes to Consolidated Financial Statements
As at December 31, 20102011 and December 31, 2009
2010 and for the years ended December 31, 2010, 20092011 and 2008
2010
(tabular amounts in thousands of US dollars, except share/option/warrant and
per share/option/warrant data and as otherwise noted)
12. GOODWILL
| 13 | Goodwill |
Goodwill has been allocated to the only CGU of the Group.
The change in the carrying value is as follows:
| Cost | Accumulated impairment loss | Carrying amount | ||||||||
|
| |||||||||
| $ | $ | $ | ||||||||
Balance as at January 1, 2010 | 10,246 | - | 10,246 | |||||||
Impact of foreign exchange rate changes | (632) | - | (632) | |||||||
|
| |||||||||
Balance as at December 31, 2010 | 9,614 | - | 9,614 | |||||||
Impact of foreign exchange rate changes | (301) | - | (301) | |||||||
|
| |||||||||
Balance as at December 31, 2011 | 9,313 | - | 9,313 | |||||||
|
| |||||||||
| 14 | |||||
| |||||
| |||||
| |||||
| |||||
| |||||
As at December 31, | As at December 31, | As at January 1, 2010 | ||||||||||
|
| |||||||||||
| $ | $ | $ | ||||||||||
Trade accounts payable | 8,062 | 6,388 | 8,152 | |||||||||
Salaries, employment taxes and benefits | 1,958 | 1,427 | 587 | |||||||||
Current portion of warrant liability | 42 | 955 | - | |||||||||
Accrued R&D costs | 1,056 | 615 | 1,883 | |||||||||
Accrued Cetrotide® services and deliveries | 160 | 221 | 65 | |||||||||
Other | 979 | 654 | 1,158 | |||||||||
|
| |||||||||||
| 12,257 | 10,260 | 11,845 | ||||||||||
|
| |||||||||||
13. ACCOUNTS PAYABLE AND ACCRUED LIABILITIES
| | As at December 31, | |||||||
|---|---|---|---|---|---|---|---|---|
| | 2010 | 2009 | ||||||
| | $ | $ | ||||||
Trade payables | 6,388 | 8,152 | ||||||
Salaries and employee benefits | 1,427 | 587 | ||||||
Other accrued liabilities | 1,567 | 3,180 | ||||||
| 9,382 | 11,919 | |||||||
14. EMPLOYEE FUTURE BENEFITS
The Company's subsidiary in Germany provides unfunded defined benefit pension plans and unfunded postemployment benefit plans for some groups of employees. Provisions for pension obligations are established for benefits payable in the form of retirement, disability and surviving dependent pensions.
The following table provides a reconciliation of the changes in the aforementioned plans' accrued benefit obligations:
| | Pension benefit plans | Other benefit plans | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2010 | 2009 | 2008 | 2010 | 2009 | 2008 | ||||||||||||||
| | $ | $ | $ | $ | $ | $ | ||||||||||||||
Obligation — Beginning of year | 10,768 | 9,177 | 8,390 | 872 | 915 | 794 | ||||||||||||||
Current service cost | 217 | 205 | 216 | 51 | 51 | 47 | ||||||||||||||
Interest cost | 546 | 507 | 473 | 44 | 50 | 44 | ||||||||||||||
Actuarial (gain) loss | (191 | ) | 773 | 544 | (4 | ) | (19 | ) | 230 | |||||||||||
Benefits paid | (173 | ) | (102 | ) | (89 | ) | (84 | ) | (140 | ) | (163 | ) | ||||||||
Effect of foreign currency exchange rate changes | (668 | ) | 208 | (357 | ) | (54 | ) | 15 | (37 | ) | ||||||||||
Obligation — End of year | 10,499 | 10,768 | 9,177 | 825 | 872 | 915 | ||||||||||||||
Expenses recognized | 572 | 1,485 | 1,233 | 91 | 82 | 321 | ||||||||||||||
Table of ContentsAeterna Zentaris Inc.
AETERNA ZENTARIS INC.Notes to Consolidated Financial Statements
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
As at December 31, 20102011 and December 31, 2009
2010 and for the years ended December 31, 2010, 20092011 and 2008
2010
(tabular amounts in thousands of US dollars, except share/option/warrant and
per share/option/warrant data and as otherwise noted)
14. EMPLOYEE FUTURE BENEFITS (Continued)
| 15 | Warrant liability |
The significant actuarial assumptions adopted to determinechange in the Company's accrued benefit obligations areCompany’s warrant liability can be summarized as follows:
| Years ended December 31, | ||||||||
| 2011 | 2010 | |||||||
|
| |||||||
| $ | $ | |||||||
Balance – Beginning of the year | 14,367 | 1,664 | * | |||||
Share purchase warrants granted during the year (note 17) | - | 7,341 | ||||||
Share purchase warrants exercised during the year | (2,638) | (429) | ||||||
Change in fair value of share purchase warrants | (2,533) | 5,437 | ||||||
Change in value attributable to foreign exchange rate changes | 8 | 354 | ||||||
|
| |||||||
| 9,204 | 14,367 | |||||||
Less: current portion | (42) | (955) | ||||||
|
| |||||||
Balance – End of the year | 9,162 | 13,412 | ||||||
|
| |||||||
| ||||||||
| | Pension benefit plans | Other benefit plans | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Actuarial assumptions | 2010 | 2009 | 2008 | 2010 | 2009 | 2008 | ||||||||||||||
| | % | % | % | % | % | % | ||||||||||||||
Discount rate for expenses | 5.10 | 5.30 | 5.60 | 5.10 | 5.30 | 5.60 | ||||||||||||||
Discount rate for liabilities | 5.10 | 5.30 | 5.60 | 5.10 | 5.30 | 5.60 | ||||||||||||||
Pension benefits increase | 2.00 | 2.00 | 2.00 | 2.00 | 2.00 | 2.00 | ||||||||||||||
Rate of compensation increase | 2.75 to 3.75 | 2.75 | 2.75 to 3.75 | 2.75 | 2.75 | 2.75 | ||||||||||||||
The last actuarial reports give effect * Includes current portion of $0 and non-current portion of $1,664.
A summary of the activity related to the pensionCompany’s share purchase warrants is provided below.
| Years ended December 31, | ||||||||||||||||
| 2011 | 2010 | |||||||||||||||
|
| |||||||||||||||
| Number | Weighted average exercise price (US$) | Number | Weighted average exercise price (US$) | |||||||||||||
Balance - Beginning of the year | 12,923,390 | 1.53 | 4,110,603 | 1.70 | ||||||||||||
Granted | - | - | 9,111,604 | 1.44 | ||||||||||||
Exercised | (1,707,286) | 1.30 | (298,817) | 1.32 | ||||||||||||
Expired | (2,148,936) | 2.10 | - | - | ||||||||||||
|
| |||||||||||||||
Balance - End of the year | 9,067,168 | 1.44 | 12,923,390 | 1.53 | ||||||||||||
|
| |||||||||||||||
Aeterna Zentaris Inc.
Notes to Consolidated Financial Statements
As at December 31, 2011 and postemployment benefit obligationsDecember 31, 2010 and for the years ended December 31, 2011 and 2010
(tabular amounts in thousands of US dollars, except share/option/warrant and per share/option/warrant data and as otherwise noted)
The following table summarizes the share purchase warrants outstanding and exercisable as at December 31, 2010. 2011:
| Warrants outstanding and currently exercisable | ||||||||
Exercise price (US$) | Number | Weighted average remaining contractual life (years) | Total intrinsic value | |||||
1.25 | 733,334 | 2.81 | 213 | |||||
1.37 | 3,496,879 | 3.47 | 586 | |||||
1.50 | 4,572,777 | 3.72 | 183 | |||||
1.72 | 264,178 | 3.46 | - | |||||
|
| |||||||
| 9,067,168 | 3.54 | 982 | ||||||
|
| |||||||
The next actuarial reports are planned for December, 2011.
In accordance withtable presented below shows the inputs and assumptions usedapplied to the Black-Scholes option pricing model in order to determine the fair value of warrants outstanding as at December 31, 2011.
October 2009 Investor | October 2009 Compensation Warrants | April 2010 Investor | June 2010 Investor | June 2010 Compensation Warrants | ||||||||||
Number of equivalent shares | 733,334 | 128,333 | 4,444,444 | 3,496,879 | 264,178 | |||||||||
Market-value per share price | 1.54 | 1.54 | 1.54 | 1.54 | 1.54 | |||||||||
Exercise price | 1.25 | 1.50 | 1.50 | 1.37 | 1.72 | |||||||||
Risk-free annual interest rate | (a | ) | 0.34% | 0.12% | 0.55% | 0.47% | 0.47% | |||||||
Expected volatility | (b | ) | 95.18% | 56.85% | 99.25% | 98.95% | 99.13% | |||||||
Expected life (years) | (c | ) | 2.81 | 0.81 | 3.80 | 3.47 | 3.46 | |||||||
Expected dividend yield | (d | ) | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | |||||||
| ||||||||||||||
| (a) | Based on United States Treasury Government Bond interest rates with a term that is consistent with the expected life of the warrants. |
| (b) | Based on the historical volatility of the Company’s stock price over the most recent period consistent with the expected life of the warrants, as well as on future expectations. |
| (c) | Based upon time to expiry from the reporting period date. |
| (d) | The Company has not paid dividends nor intends to pay dividends in the foreseeable future. |
The Black-Scholes valuation methodology uses “Level 2” inputs in calculating fair value, as defined in IFRS 7 and as discussed in note 24.
Aeterna Zentaris Inc.
Notes to Consolidated Financial Statements
As at December 31, 2011 and December 31, 2010 the future benefits expected to be paid can be presented as follows:
| | $ | ||||
|---|---|---|---|---|---|
2011 | 341 | ||||
2012 | 471 | ||||
2013 | 493 | ||||
2014 | 505 | ||||
2015 | 558 | ||||
2016 through 2020 | 3,063 | ||||
| 5,431 | |||||
Cash required in the next year to fund the plans will approximate the amount of expected benefits.
Total expensesand for the Company's defined contribution plan in its German subsidiary amounted to approximately $257,260 for the yearyears ended December 31, 2010 ($334,400 for 20092011 and $344,2372010
(tabular amounts in 2008).thousands of US dollars, except share/option/warrant and per share/option/warrant data and as otherwise noted)
| 16 | Provision and other non-current liabilities |
As at December 31, 2011 | As at December 31, 2010 | As at January 1, 2010 | ||||
| ||||||
| $ | $ | $ | ||||
Onerous lease provision (see below) | 619 | 441 | 491 | |||
Other | 187 | 183 | 66 | |||
| ||||||
| 806 | 624 | 557 | ||||
| ||||||
Current | 89 | 53 | 50 | |||
Non-current | 717 | 571 | 507 | |||
| ||||||
| 806 | 624 | 557 | ||||
| ||||||
15. SHARE CAPITALOnerous lease provision*
Year ended | ||
| $ | ||
Balance at January 1, 2011 | 441 | |
Additional provision recognised | 189 | |
Utilization of provision | (62) | |
Effect of change in the discount rate | 43 | |
Unwinding of discount | 8 | |
Balance at December 31, 2011 | 619 | |
Current portion | (89) | |
Non-current portion at December 31, 2011 | 530 | |
| * | The provision for onerous lease contract represents the present value of the future lease payments that the Company is presently obligated to make under non-cancellable onerous operating lease contract, less revenue expected to be earned on the lease, including estimated future sub-lease revenue. The estimate may vary as a result of changes in the utilisation of the leased premises and sub-lease arrangement. The unexpired term of the lease is six years. On December 1st 2011, following the relocation of one of the Company’s offices, the lease arrangement became fully onerous. As such, an additional provision was recognised. |
| 17 | Share capital |
The Company has authorized an unlimited number of common shares (being voting and participating shares) with no par value, as well as an unlimited number of preferred, first and second ranking shares, issuable in series, with rights and privileges specific to each class.class, with no par value.
Common shares issued in connection with “At-the-Market” (“ATM”) drawdowns
February ATM Sales Agreement
On February 22, 2011, the Company entered into an ATM sales agreement (the “February ATM Sales Agreement”), under which the Company was able, at its discretion and from time to time during the 24-month term of the agreement, to sell up to 12,500,000 of its common shares through ATM issuances on the Nasdaq Stock Market for aggregate gross proceeds not to exceed $19,750,831. The February ATM Sales Agreement provided that common shares were to be sold at market prices prevailing at the time of sale, and, as a result, prices may have varied.
Aeterna Zentaris Inc.
Notes to Consolidated Financial Statements
As at December 31, 2011 and December 31, 2010 and for the years ended December 31, 2011 and 2010
(tabular amounts in thousands of US dollars, except share/option/warrant and per share/option/warrant data and as otherwise noted)
The Company issued a total of 9,964,548 common shares under the February ATM Sales Agreement for aggregate gross proceeds of $19,749,945, less cash transaction costs of $592,498 and previously deferred transaction costs of $207,000.
June ATM Sales Agreement
On June 29, 2011, the Company entered into an additional ATM sales agreement (the “June ATM Sales Agreement”), under which the Company was able, at its discretion, from time to time during the 24-month term of the agreement, sell up to 9,500,000 of its common shares through ATM issuances on the Nasdaq Stock Market for aggregate gross proceeds not to exceed $24,000,000. The June ATM Sales Agreement provided that common shares were to be sold at market prices prevailing at the time of sale, and, as a result, prices may have varied.
The Company issued a total of 9,500,000 common shares under the June ATM Sales Agreement for aggregate gross proceeds of $17,705,416, less cash transaction costs of $612,357 and previously deferred transaction costs of $162,407.
Common shares issued in connection with registered direct offerings
a)
On April 20, 2010, the Company completed a registered direct offering of 11,111,111 units, with each unit consisting of one common share and a warrant to purchase 0.40 of a common share, at a price of $1.35 per unit (the "April“April 2010 Offering"Offering”). Total proceeds raised upon completion of the April 2010 Offering amounted to $15,000,000, less cash transaction costs of approximately $1,315,000. The securities described above were offered by the Company pursuant to a shelf prospectus dated March 12, 2010 and a prospectus supplement dated April 15, 2010.
AETERNA ZENTARIS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
As at December 31, 2010 and December 31, 2009and for the years ended December 31, 2010, 2009 and 2008(tabular amounts in thousands of US dollars, except share/option/warrant andper share/option/warrant data and as otherwise noted)
15. SHARE CAPITAL (Continued)
The Company granted warrants (the "April“April 2010 Investor Warrants"Warrants”) to the investors who participated in the April 2010 Offering. Each April 2010 Investor Warrant entitles the holder to purchase 0.40 of a common share at an exercise price of $1.50 per share. The April 2010 Investor Warrants are exercisable between October 20, 2010 and October 20, 2015, and, upon complete exercise, would result in the issuance of an aggregate of 4,444,444 common shares.
The Company estimated the fair value attributable to the April 2010 Investor Warrants of $3,639,815 as of the date of grant by applying the Black-Scholes pricing model, to which the following additional assumptions were applied: a risk-free annual interest rate of 2.56%, expected volatility of 87.3%, an expected term of 5 years, a dividend yield of 0.0% and an issue-date market share price of $1.24. Transaction costs allocated to the April 2010 Investor Warrants amounted to approximately $309,000.
b)
On June 21, 2010, the Company completed a registered direct offering of 8,805,964 units, with each unit consisting of one common share and a warrant to purchase 0.50 of a common share, at a price of $1.3725 per unit (the "June“June 2010 Offering"Offering”). Total proceeds raised upon completion of the June 2010 Offering amounted to $12,086,186, less cash transaction costs of approximately $783,000. The securities described above were offered by the Company pursuant to a shelf prospectus dated March 12, 2010 and a prospectus supplement dated June 15, 2010.
Aeterna Zentaris Inc.
Notes to Consolidated Financial Statements
As at December 31, 2011 and December 31, 2010 and for the years ended December 31, 2011 and 2010
(tabular amounts in thousands of US dollars, except share/option/warrant and per share/option/warrant data and as otherwise noted)
The Company granted warrants (the "June“June 2010 Investor Warrants"Warrants”) to the investors who participated in the June 2010 Offering. Each June 2010 Investor Warrant entitles the holder to purchase a common share at an exercise price of $1.3725 per share. The June 2010 Investor Warrants are exercisable between June 21, 2010 and June 21, 2015, and, upon complete exercise, would result in the issuance of an aggregate of 4,402,982 common shares.
The Company estimated the fair value attributable to the June 2010 Investor Warrants of $3,502,572 as of the date of grant by applying the Black-Scholes pricing model, to which the following additional assumptions were applied: a risk-free annual interest rate of 2.05%, expected volatility of 89.3%, an expected term of 5 years, a dividend yield of 0.0% and an issue-date market share price of $1.18. Transaction costs allocated to the June 2010 Investor Warrants amounted to approximately $223,000.
The Company also granted warrants (the "June“June 2010 Compensation Warrants"Warrants”) to the sole placement agent (and to certain of its designated representatives) engaged in connection with the June 2010 Offering. Each June 2010 Compensation Warrant entitles the holder to purchase a common share at an exercise price of $1.7156 per share. The June 2010 Compensation Warrants are exercisable between June 15, 2010 and June 15, 2015, and, upon complete exercise, would result in the issuance of 264,178 common shares.
The Company estimated the fair value attributable to the June 2010 Compensation Warrants of $198,609 as of the date of grant by applying the Black-Scholes pricing model, to which the following additional assumptions were applied: a risk-free annual interest rate of 2.04%, expected volatility of 89.4%, an expected term of 5 years, a dividend yield of 0.0% and an issue-date market share price of $1.18. The initial fair value of the June 2010 Compensation Warrants has been accounted for as additional transaction costs, since the instruments were granted to the sole placement agent as part of the terms of the underlying engagement and in recognition of the efforts made in connection with the June 2010 Offering.
On June 23, 2009, the Company completed a registered direct offering of 5,319,149 units, with each unit consisting of one common share and a warrant to purchase 0.35 of a common share at a price of $1.88 per unit (the "June 2009 Offering"). Total proceeds raised through the June 2009 Offering amounted to $10,000,000, less cash and non-cash transaction costs of $1,554,000. The purchasers in this offering were comprised of institutional investors, and the securities described above were offered by the Company pursuant to a shelf prospectus dated September 27, 2007 and a prospectus supplement dated June 18, 2009.
AETERNA ZENTARIS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
As at December 31, 2010 and December 31, 2009and for the years ended December 31, 2010, 2009 and 2008(tabular amounts in thousands of US dollars, except share/option/warrant andper share/option/warrant data and as otherwise noted)
15. SHARE CAPITAL (Continued)
The Company granted a total of 5,319,149 warrants (the "June 2009 Investor Warrants") to the institutional investors who participated in the June 2009 Offering. Each June 2009 Investor Warrant entitles the holder to purchase 0.35 of a common share at an exercise price of $2.06 per share. The June 2009 Investor Warrants are exercisable between September 23, 2009 and December 23, 2011, and, upon complete exercise, would result in the issuance of an aggregate of 1,861,702 common shares of the Company.
The Company estimated the fair value attributable to the June 2009 Investor Warrants of $1,620,998 as of the date of grant by applying the Black-Scholes pricing model, to which the following additional assumptions were applied: a risk-free annual interest rate of 1.74%, expected volatility of 90.6%, an expected term of 2.5 years, dividend yield of 0.0% and an issue-date market share price of $1.75. Transaction costs allocated to the June 2009 Investor Warrants amounted to approximately $247,000.
The Company granted a total of 820,668 warrants (the "June 2009 Compensation Warrants") to the sole placement agent and its designated representatives engaged in connection with the June 2009 Offering. Each June 2009 Compensation Warrant entitles the holder to purchase 0.35 of a common share at an exercise price of $2.35 per share. The June 2009 Compensation Warrants are exercisable between December 23, 2009 and December 23, 2011, and, upon complete exercise, would result in the issuance of 287,234 common shares of the Company.
The Company estimated the fair value attributable to the June 2009 Compensation Warrants of $234,251 as of the date of grant by applying the Black-Scholes pricing model, to which the following additional assumptions were applied: a risk-free annual interest rate of 1.74%, expected volatility of 90.6%, an expected term of 2.5 years, an expected dividend yield of 0.0% and an issue-date market share price of $1.75. The initial fair value of the June 2009 Compensation Warrants has been accounted for as additional transaction costs, since the instruments were granted to the sole placement agent as part of the terms of the underlying engagement and in recognition of the efforts made in connection with the June 2009 Offering.
On October 23, 2009, the Company completed a registered direct offering of 4,583,335 units, with each unit consisting of one common share and a warrant to purchase 0.40 of a common share, at a price of $1.20 per unit (the "October 2009 Offering"). Total proceeds raised through the October 2009 Offering amounted to $5,500,002, less cash transaction costs of approximately $410,000. The purchasers in this offering were new and existing institutional investors, and the securities described above were offered by the Company pursuant to a shelf prospectus dated September 27, 2007 and a prospectus supplement dated October 19, 2009.
The Company granted a total of 4,583,335 warrants (the "October 2009 Investor Warrants") to the institutional investors who participated in the October 2009 Offering. Each October 2009 Investor Warrant entitles the holder to purchase 0.40 of a common share at an exercise price of $1.25 per share. The October 2009 Investor Warrants are exercisable between October 23, 2009 and October 23, 2014, and, upon complete exercise, would result in the issuance of an aggregate of 1,833,334 common shares.
The Company estimated the fair value attributable to the October 2009 Investor Warrants of $1,302,259 as of the date of grant by applying the Black-Scholes pricing model, to which the following additional assumptions were applied: a risk-free annual interest rate of 2.46%, expected volatility of 84.3%, an expected term of 5 years, dividend yield of 0.0% and an issue-date market share price of $1.09. Transaction costs allocated to the October 2009 Investor Warrants amounted to approximately $97,000.
The Company granted a total of 320,832 warrants (the "October 2009 Compensation Warrants") to the sole placement agent engaged in connection with the October 2009 Offering. Each October 2009 Compensation Warrant entitles the holder to purchase 0.40 of a common share at an exercise price of $1.50 per share. The October 2009 Compensation Warrants are exercisable between April 23, 2010 and October 23, 2012, and, upon complete exercise, would result in the issuance of 128,333 common shares.
AETERNA ZENTARIS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
As at December 31, 2010 and December 31, 2009and for the years ended December 31, 2010, 2009 and 2008(tabular amounts in thousands of US dollars, except share/option/warrant andper share/option/warrant data and as otherwise noted)
15. SHARE CAPITAL (Continued)
The Company estimated the fair value attributable to the October 2009 Compensation Warrants of $86,653 as of the date of grant by applying the Black-Scholes pricing model, to which the following additional assumptions were applied: a risk-free annual interest rate of 1.57%, expected volatility of 103.4%, an expected term of 3 years, dividend yield of 0.0% and an issue-date market share price of $1.09. The initial fair value of the October 2009 Compensation Warrants has been accounted for as additional transaction costs, since the instruments were granted to the sole placement agent as part of the terms of the underlying engagement and in recognition of the efforts made in connection with the October 2009 Offering.
The terms of all aforementioned warrants are substantially the same, with the exception of the exercise price and contractual period of exercise, as discussed above. In particular, all warrants may be exercised, at the option of the holder, by cash payment of the exercise price or, upon the existence of certain conditions, by "cashless exercise", which means that in lieu of paying the aggregate exercise price for the shares being purchased upon exercise of the warrants in cash, the holder would receive the number of shares underlying the warrants equal to the quotient obtained by applying a formula, as defined by the terms of each warrant. The Company will not receive additional proceeds to the extent that warrants are exercised by cashless exercise.
The exercise price and number of common shares issuable on exercise of all outstanding warrants may be adjusted in certain circumstances, including stock dividends or splits, subsequent rights offerings, pro-rata distributions and pursuant to transactions involving the merger or consolidation of the Company with another entity or other Fundamental Transaction, as defined in the warrants.
Additionally, and notwithstanding anything to the contrary, in the event of any type of Fundamental Transaction, as defined in the warrants, the Company or any successor entity shall, at the Company's option, have the right to require the holders thereof to exercise the warrants, or, at the holder's option, purchase the warrants from the holders by paying the holders an amount of cash equivalent to the Black-Scholes value, as defined, of the remaining unexercised portion of the warrants on the date of the consummation of an aforementioned Fundamental Transaction.
The Black-Scholes pricing model uses "Level 2" inputs in calculating fair value, as defined by CICA Handbook Section 3862, which establishes a fair value hierarchy that prioritizes the inputs used to measure fair value. The hierarchy gives the highest priority to unadjusted quoted prices in active markets for identical assets or liabilities (Level 1 measurement) and the lowest priority to unobservable inputs (Level 3 measurement). Level 2 inputs are those which are either directly or indirectly observable as of the reporting date and include financial instruments that are valued using models or other valuation methodologies, such as the Black-Scholes pricing model.
Share purchase warrants
A summary of the activity related to the Company's share purchase warrants for the years ended December 31, 2010 and 2009 is provided below.
| | Years ended December 31, | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2010 | 2009 | ||||||||||||
| | Number | Weighted average exercise price (US$) | Number | Weighted average exercise price (US$) | ||||||||||
Balance — Beginning of year | 4,110,603 | 1.70 | — | — | ||||||||||
Granted | 9,111,604 | 1.44 | 4,110,603 | 1.70 | ||||||||||
Exercised | (298,817 | ) | 1.32 | — | — | |||||||||
Balance — End of year | 12,923,390 | 1.53 | 4,110,603 | 1.70 | ||||||||||
Share purchase warrants exercisable — End of year | 12,923,390 | 1.53 | 3,982,270 | 1.71 | ||||||||||
AETERNA ZENTARIS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
As at December 31, 2010 and December 31, 2009and for the years ended December 31, 2010, 2009 and 2008(tabular amounts in thousands of US dollars, except share/option/warrant andper share/option/warrant data and as otherwise noted)
15. SHARE CAPITAL (Continued)
The following table summarizes the share purchase warrants outstanding and exercisable as at December 31, 2010:
| | Warrants outstanding and currently exercisable | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
Exercise price (US$) | Number | Weighted average remaining contractual life (years) | Global intrinsic value | ||||||||
1.25 | 1,716,667 | 3.81 | 807 | ||||||||
1.37 | 4,220,832 | 4.47 | 1,477 | ||||||||
1.50 | 4,572,777 | 4.72 | 1,006 | ||||||||
1.72 | 264,178 | 4.45 | — | ||||||||
2.06 | 1,861,702 | 0.98 | — | ||||||||
2.35 | 287,234 | 0.98 | — | ||||||||
| 12,923,390 | 3.89 | 3,290 | |||||||||
Shareholder rights plan
Effective March 29, 2010, the Company adopted a shareholder rights plan (the "Rights Plan"“Rights Plan”). The Rights Plan was approved by the Board of Directors on March 22, 2010 and ratified on May 13, 2010 by the Company'sCompany’s shareholders. The rights issued to the shareholders under the Rights Plan will be exercisable, under certain conditions, only when a person or entity, including related parties, acquires or announces his/her or its intention to acquire more than twenty (20) percent of the outstanding common shares of the Company (as such shares may be redesignated or reclassified) without complying with the "permitted bid"“permitted bid” provisions of the Rights Plan or without approval of the Company'sCompany’s Board of Directors. Should such an acquisition occur, each right would, upon exercise, entitle a holder, other than the person pursuing the acquisition together with any related parties, to purchase common shares of the Company at a fifty (50) percent discount to the market price of the Company'sCompany’s shares at that time.
Aeterna Zentaris Inc.
Notes to Consolidated Financial Statements
As at December 31, 2011 and December 31, 2010 and for the years ended December 31, 2011 and 2010
(tabular amounts in thousands of US dollars, except share/option/warrant and per share/option/warrant data and as otherwise noted)
Stock option planoptions
In December 1995, the Company'sCompany’s Board of Directors adopted a stock option plan (the "Stock“Stock Option Plan"Plan”) for its directors, senior executives, employees and other collaborators who provide services to the Company. The total number of common shares that may be issued under the Stock Option Plan cannot exceed 11.4% of the total number of issued and outstanding common shares at any given time.
Options granted under the Stock Option Plan expire after a maximum period of ten years following the date of grant. Options granted under the Stock Option Plan generally vest over a three-year period. However, 524,468 of the options granted in 2011 and 883,525 of the options granted in 2010 and 1,288,422 of the options granted in 2009 vest over a period of 18 months.
The following tables summarizetable summarizes the activity under the Stock Option Plan.Company’s stock option plan.
| Years ended December 31, | ||||||||||||||||||||||||
| 2011 | 2010 | |||||||||||||||||||||||
| Canadian Dollar Options | US Dollar Options | Canadian Dollar Options | US Dollar Options | |||||||||||||||||||||
| Number | Weighted average exercise price (CAN$) | Number | Weighted average exercise price (US$) | Number | Weighted average exercise price (CAN$) | Number | Weighted average exercise price (US$) | |||||||||||||||||
Balance - Beginning of the year | 6,558,679 | 2.55 | 293,334 | 2.83 | 5,920,588 | 2.72 | 293,334 | 2.83 | ||||||||||||||||
Granted | 15,000 | 1.93 | 1,434,468 | 1.75 | 1,088,525 | 1.51 | - | - | ||||||||||||||||
Exercised | (160,348 | ) | 0.89 | - | - | (124,068 | ) | 0.90 | - | - | ||||||||||||||
Forfeited | (46,666 | ) | 1.54 | - | - | (74,699 | ) | 0.95 | - | - | ||||||||||||||
Expired | (178,333 | ) | 6.18 | - | - | (251,667 | ) | 3.22 | - | - | ||||||||||||||
|
|
|
|
|
|
|
| |||||||||||||||||
Balance - End of the year | 6,188,332 | 2.50 | 1,727,802 | 1.93 | 6,558,679 | 2.55 | 293,334 | 2.83 | ||||||||||||||||
|
|
|
|
|
|
|
| |||||||||||||||||
| CAN$ options outstanding as at December 31, 2011 | ||||||||||||
Exercise price (CAN$) | Number | Weighted average contractual life (years) | Weighted average (CAN$) | Total intrinsic value (CAN$) | ||||||||
0.55 to 0.95 | 1,776,974 | 7.55 | 0.82 | 1,319 | ||||||||
0.96 to 1.56 | 1,048,525 | 8.14 | 1.50 | 63 | ||||||||
1.57 to 1.82 | 944,500 | 3.05 | 1.77 | - | ||||||||
1.83 to 3.54 | 797,500 | 2.34 | 3.21 | - | ||||||||
3.55 to 4.92 | 775,333 | 1.98 | 4.08 | - | ||||||||
4.93 to 8.88 | 845,500 | 2.07 | 5.96 | - | ||||||||
|
| |||||||||||
| 6,188,332 | 4.85 | 2.50 | 1,382 | |||||||||
|
| |||||||||||
Table of ContentsAeterna Zentaris Inc.
AETERNA ZENTARIS INC.Notes to Consolidated Financial Statements
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
As at December 31, 20102011 and December 31, 2009
2010 and for the years ended December 31, 2010, 20092011 and 2008
2010
(tabular amounts in thousands of US dollars, except share/option/warrant and
per share/option/warrant data and as otherwise noted)
15. SHARE CAPITAL (Continued)
Canadian dollar denominated awards
| | Years ended December 31, | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | | | | | 2008 | |||||||||||||||
| | 2010 | 2009 | ||||||||||||||||||
| | | Weighted average exercise price (CAN$) | ||||||||||||||||||
| | Number | Weighted average exercise price (CAN$) | Number | Weighted average exercise price (CAN$) | Number | |||||||||||||||
Balance — Beginning of year | 5,920,588 | 2.72 | 4,490,759 | 3.28 | 4,136,092 | 3.83 | ||||||||||||||
Granted | 1,088,525 | 1.51 | 1,448,422 | 0.95 | 735,000 | 0.59 | ||||||||||||||
Exercised | (124,068 | ) | 0.90 | — | — | — | — | |||||||||||||
Forfeited | (74,699 | ) | 0.95 | (15,000 | ) | 0.55 | (165,000 | ) | 3.41 | |||||||||||
Expired | (251,667 | ) | 3.22 | (3,593 | ) | 1.73 | (215,333 | ) | 4.51 | |||||||||||
Balance — End of year | 6,558,679 | 2.55 | 5,920,588 | 2.72 | 4,490,759 | 3.28 | ||||||||||||||
Options exercisable — End of year | 4,802,382 | 3.04 | 3,898,844 | 3.66 | 3,462,441 | 3.91 | ||||||||||||||
| | Options outstanding as at December 31, 2010 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Exercise price (CAN$) | Number | Weighted average remaining contractual life (years) | Weighted average exercise price (CAN$) | Global intrinsic value (CAN$) | ||||||||||
0.55 to 0.95 | 1,943,988 | 8.56 | 0.82 | 1,744 | ||||||||||
0.96 to 1.72 | 1,063,525 | 9.15 | 1.50 | 234 | ||||||||||
1.73 to 1.82 | 969,500 | 4.19 | 1.77 | — | ||||||||||
1.83 to 3.54 | 782,500 | 3.20 | 3.23 | — | ||||||||||
3.55 to 4.92 | 775,333 | 2.98 | 4.08 | — | ||||||||||
4.93 to 8.88 | 1,023,833 | 2.70 | 6.00 | — | ||||||||||
| 6,558,679 | 5.80 | 2.55 | 1,978 | |||||||||||
| | Options exercisable as at December 31, 2010 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Exercise price (CAN$) | Number | Weighted average remaining contractual life (years) | Weighted average exercise price (CAN$) | Global intrinsic value (CAN$) | ||||||||||
0.55 to 0.95 | 1,222,882 | 8.54 | 0.82 | 1,106 | ||||||||||
0.96 to 1.72 | 53,334 | 4.72 | 1.39 | 18 | ||||||||||
1.73 to 1.82 | 944,500 | 4.05 | 1.77 | — | ||||||||||
1.83 to 3.54 | 782,500 | 3.20 | 3.23 | — | ||||||||||
3.55 to 4.92 | 775,333 | 2.98 | 4.08 | — | ||||||||||
4.93 to 8.88 | 1,023,833 | 2.70 | 6.00 | — | ||||||||||
| 4,802,382 | 4.60 | 3.04 | 1,124 | |||||||||||
AETERNA ZENTARIS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
As at December 31, 2010 and December 31, 2009and for the years ended December 31, 2010, 2009 and 2008(tabular amounts in thousands of US dollars, except share/option/warrant andper share/option/warrant data and as otherwise noted)
15. SHARE CAPITAL (Continued)
US dollar denominated awards
| | Years ended December 31, | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2010 | 2009 | 2008 | |||||||||||||||||
| | Number | Weighted average exercise price (US$) | Number | Weighted average exercise price (US$) | Number | Weighted average exercise price (US$) | ||||||||||||||
Balance — Beginning of year | 293,334 | 2.83 | 313,334 | 2.76 | 870,000 | 2.79 | ||||||||||||||
Forfeited | — | — | (20,000 | ) | 1.78 | (556,666 | ) | 2.80 | ||||||||||||
Balance — End of year | 293,334 | 2.83 | 293,334 | 2.83 | 313,334 | 2.76 | ||||||||||||||
Options exercisable — End of year | 293,334 | 2.83 | 233,336 | 2.93 | 176,669 | 3.08 | ||||||||||||||
| | Options outstanding and currently exercisable | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Exercise price (US$) | Number | Weighted average remaining contractual life (years) | Weighted average exercise price (US$) | Global intrinsic value (US$) | ||||||||||
1.82 to 1.87 | 115,000 | 6.94 | 1.82 | — | ||||||||||
1.88 to 3.96 | 178,334 | 6.33 | 3.48 | — | ||||||||||
| 293,334 | 6.57 | 2.83 | — | |||||||||||
| CAN$ options exercisable as at December 31, 2011 | ||||||||||||
Exercise price (CAN$) | Number | Weighted average contractual life (years) | Weighted average (CAN$) | Total intrinsic value (CAN$) | ||||||||
0.55 to 0.95 | 1,723,646 | 7.54 | 0.81 | 1,287 | ||||||||
0.96 to 1.56 | 692,353 | 7.73 | 1.49 | 48 | ||||||||
1.57 to 1.82 | 944,500 | 3.05 | 1.77 | - | ||||||||
1.83 to 3.54 | 782,500 | 2.20 | 3.23 | - | ||||||||
3.55 to 4.92 | 775,333 | 1.98 | 4.08 | - | ||||||||
4.93 to 8.88 | 845,500 | 2.07 | 5.96 | - | ||||||||
|
| |||||||||||
| 5,763,832 | 4.55 | 2.57 | 1,335 | |||||||||
|
| |||||||||||
| US$ options outstanding as at December 31, 2011 | ||||||||||||
Exercise price (US$) | Number | Weighted average contractual life (years) | Weighted average (US$) | Total intrinsic value (US$) | ||||||||
1.74 to 1.78 | 1,414,468 | 9.93 | 1.74 | - | ||||||||
1.79 to 1.87 | 115,000 | 5.94 | 1.82 | - | ||||||||
1.88 to 3.96 | 198,334 | 5.74 | 3.37 | - | ||||||||
|
| |||||||||||
| 1,727,802 | 9.19 | 1.93 | - | |||||||||
|
| |||||||||||
| US$ options exercisable as at December 31, 2011 | ||||||||||||
Exercise price (US$) | Number | Weighted average contractual life (years) | Weighted average (US$) | Total intrinsic value (US$) | ||||||||
1.79 to 1.87 | 115,000 | 5.94 | 1.82 | - | ||||||||
1.88 to 3.96 | 178,334 | 5.33 | 3.48 | - | ||||||||
|
| |||||||||||
| 293,334 | 5.57 | 2.83 | - | |||||||||
|
| |||||||||||
As at December 31, 2010,2011, the total compensation cost related to unvested Canadian Dollar stock options not yet recognized amounted to $1,320,958$146,641 ($853,9241,014,958 in 2009)2010). This amount is expected to be recognized over a weighted average period of 0.98 year (1.02 years in 2010).
As at December 31, 2011, the total compensation cost related to unvested US Dollar stock options not yet recognized amounted to $1,322,752 ($0 in 2010). This amount is expected to be recognized over a weighted average period of 1.48 years.
Aeterna Zentaris Inc.
Notes to Consolidated Financial Statements
As at December 31, 2011 and December 31, 2010 and for the years (1.44 yearsended December 31, 2011 and 2010
(tabular amounts in 2009).thousands of US dollars, except share/option/warrant and per share/option/warrant data and as otherwise noted)
The Company settles stock options exercised through the issuance of common shares from treasury.
Fair value input assumptions for Canadian Dollar Options granted
The table below shows the assumptions, or weighted average parameters, applied to the Black-Scholes option pricing model in order to determine stock-based compensation costs over the life of the awards.
| | Years ended December 31, | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2010 | 2009 | 2008 | ||||||||
Expected dividend yield(a) | 0.0% | 0.0% | 0.0% | ||||||||
Expected volatility(b) | 78.7% | 130.9% | 60.0% | ||||||||
Risk-free annual interest rate(c) | 3.1% | 1.2% | 1.98% | ||||||||
Expected life (years)(d) | 9.26 | 1.50 | 3.04 | ||||||||
Weighted average grant date fair value | CAN$ | 1.20 | CAN$ | 0.55 | CAN$ | 0.25 | |||||
| Years ended December 31, | ||||||||||
|
| |||||||||
| 2011 | 2010 | |||||||||
Expected dividend yield | (a) | 0.0% | 0.0% | |||||||
Expected volatility | (b) | 81.0% | 84.5% | |||||||
Risk-free annual interest rate | (c) | 1.8% | 2.6% | |||||||
Expected life (years) | (d) | 6.82 | 6.07 | |||||||
Weighted average grant date fair value | CAN$1.40 | CAN$1.09 | ||||||||
| (a) | The Company has not paid dividends nor intends to pay dividends in the foreseeable future. |
| (b) | Based on the historical volatility of the Company’s stock price over the most recent period consistent with the expected life of the stock options, as well as on future expectations. |
| (c) | Based on Canadian Government Bond interest rates with a term that is consistent with the expected life of the stock options. |
| (d) | Based upon historical data related to the exercise of stock options, on post-vesting employment terminations and on future expectations related to exercise behaviour. |
Table of ContentsFair value input assumptions for US Dollar Options granted
The table below shows the assumptions, or weighted average parameters, applied to the Black-Scholes option pricing model in order to determine stock-based compensation costs over the life of the awards.
Year ended December 31, 2011 | ||||||
Expected dividend yield | (a) | 0.0% | ||||
Expected volatility | (b) | 81.6% | ||||
Risk-free annual interest rate | (c) | 1.4% | ||||
Expected life (years) | (d) | 6.82 | ||||
Weighted average grant date fair value | US$1.27 | |||||
| (a) | The Company has not paid dividends nor intends to pay dividends in the foreseeable future. |
| (b) | Based on the historical volatility of the Company’s stock price over the most recent period consistent with the expected life of the stock options, as well as on future expectations. |
| (c) | Based on United States Treasury Government Bond interest rates with a term that is consistent with the expected life of the stock options. |
| (d) | Based upon historical data related to the exercise of stock options, on post-vesting employment terminations and on future expectations related to exercise behaviour. |
The Black-Scholes pricing models referred above use “Level 2” inputs in calculating fair value, as defined by IFRS 7, and as discussed in note 24.
AETERNA ZENTARIS INC.Aeterna Zentaris Inc.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)Notes to Consolidated Financial Statements
As at December 31, 20102011 and December 31, 2009
2010 and for the years ended December 31, 2010, 20092011 and 2008
2010
(tabular amounts in thousands of US dollars, except share/option/warrant and
per share/option/warrant data and as otherwise noted)
15. SHARE CAPITAL (Continued)
| 18 | Operating expenses |
| Years ended December 31, | ||||||||
| 2011 | 2010 | |||||||
|
| |||||||
| $ | $ | |||||||
Subcontractor fees | 25,667 | 15,907 | ||||||
Raw material purchases | 2,154 | 1,897 | ||||||
Change in inventory | (261) | 896 | ||||||
|
| |||||||
Cost of sales | 27,560 | 18,700 | ||||||
|
| |||||||
Salaries, employment taxes and short-term benefits | 13,029 | 11,885 | ||||||
Post-employment benefits | 864 | 665 | ||||||
Termination benefits | 182 | 45 | ||||||
Share-based compensation costs | 1,333 | 1,192 | ||||||
|
| |||||||
Total employee benefits expenses | 15,408 | 13,787 | ||||||
|
| |||||||
Goods and services(1) | 20,334 | 17,316 | ||||||
Lease payments(2), net of sublease payments of $179,000 | 2,153 | 1,825 | ||||||
Refundable tax credits and grants | (383) | (687) | ||||||
Depreciation and amortization | 1,471 | 1,573 | ||||||
Impairment losses | 1,405 | - | ||||||
Operating foreign exchange (gains) loss | 299 | (5) | ||||||
|
| |||||||
Total operating expenses | 68,247 | 52,509 | ||||||
|
| |||||||
| (1) | Goods and services include third-party R&D costs, laboratory supplies, royalty expenses, professional fees, marketing services, insurance as well as travel expenses. |
| (2) | Lease expense also includes changes in the onerous lease provision (note 16), except for the unwinding of discount. |
Aeterna Zentaris Inc.
Notes to Consolidated Financial Statements
As at December 31, 2011 and December 31, 2010 and for the years ended December 31, 2011 and 2010
(tabular amounts in thousands of US dollars, except share/option/warrant and per share/option/warrant data and as otherwise noted)
| 19 | Employee future benefits |
The Company’s subsidiary in Germany provides unfunded defined benefit pension plans and unfunded post-employment benefit plans for some groups of employees. Provisions for pension obligations are established for benefits payable in the form of retirement, disability and surviving dependent pensions.
The following table provides a reconciliation of the changes in the aforementioned plans’ accrued benefit obligations:
| Pension benefit plans | Other benefit plans | |||||||||||||||
|
| |||||||||||||||
2011 $ | 2010 $ | 2011 $ | 2010 $ | |||||||||||||
Obligation – Beginning of year | 10,492 | 10,767 | 1,041 | 1,254 | ||||||||||||
Current service cost | 185 | 217 | 206 | 254 | ||||||||||||
Interest cost | 555 | 546 | 54 | 63 | ||||||||||||
Actuarial (gain) loss | 1,335 | (191) | 46 | (370) | ||||||||||||
Benefits paid | (354) | (173) | (196) | (86) | ||||||||||||
Effect of foreign currency exchange rate changes | (444) | (674) | (40) | (74) | ||||||||||||
|
| |||||||||||||||
Obligation – End of year | 11,769 | 10,492 | 1,111 | 1,041 | ||||||||||||
|
| |||||||||||||||
Amount recognized | ||||||||||||||||
In comprehensive loss | (740) | (763) | (306 | ) | 53 | |||||||||||
In other comprehensive (loss) income | (1,335) | 191 | - | - | ||||||||||||
The cumulative amount of actuarial losses recognized in other comprehensive (loss) income as at December 31, 2011 is approximately $1,144,000 (cumulative actuarial gain of approximately $191,000 as at December 31, 2010).
The significant actuarial assumptions adopted to determine the Company’s accrued benefit obligations are as follows:
| Pension benefit plans | Other benefit plans | |||||||
| ||||||||
Actuarial assumptions | 2011 % | 2010 % | 2011 % | 2010 % | ||||
Discount rate | 4.20 | 5.10 | 4.20 | 5.10 | ||||
Pension benefits increase | 2.00 | 2.00 | 2.00 | 2.00 | ||||
Rate of compensation increase | 2.75 to 3.75 | 2.75 to 3.75 | 2.75 | 2.75 | ||||
The last actuarial reports give effect to the pension and post-employment benefit obligations as at December 31, 2011. The next actuarial reports are planned for December 31, 2012.
Aeterna Zentaris Inc.
Notes to Consolidated Financial Statements
As at December 31, 2011 and December 31, 2010 and for the years ended December 31, 2011 and 2010
(tabular amounts in thousands of US dollars, except share/option/warrant and per share/option/warrant data and as otherwise noted)
In accordance with the assumptions used as at December 31, 2011, the future benefits expected lifeto be paid can be presented as follows:
| $ | ||||
2012 | 680 | |||
2013 | 745 | |||
2014 | 702 | |||
2015 | 692 | |||
2016 | 644 | |||
2017 through 2020 | 3,148 | |||
| 6,611 | ||||
Cash required in the next year to fund the plans will approximate the amount of expected benefits.
Total expenses for the Company’s defined contribution plan in its German subsidiary amounted to approximately $312,984 for the year ended December 31, 2011 ($257,260 for 2010).
| 20 | Finance income and finance costs |
Components of the stock options,Company’s finance income and finance costs can be summarized as wellfollows:
| Years ended December 31, | ||||||||
| 2011 | 2010 | |||||||
| $ | $ | |||||||
Finance income | ||||||||
Net gains due to changes in foreign currency exchange rates | 2,197 | 932 | ||||||
Net change in fair value of warrant liability | 2,533 | - | ||||||
Interest income | 231 | 181 | ||||||
Gain on held-for-trading financial instrument | 1,278 | 687 | ||||||
| 6,239 | 1,800 | |||||||
Finance costs | ||||||||
Net change in fair value of warrant liability | - | (5,437) | ||||||
Unwinding of discount | (8) | (8) | ||||||
| (8) | (5,445) | |||||||
| 6,231 | (3,645) | |||||||
Aeterna Zentaris Inc.
Notes to Consolidated Financial Statements
As at December 31, 2011 and December 31, 2010 and for the years ended December 31, 2011 and 2010
(tabular amounts in thousands of US dollars, except share/option/warrant and per share/option/warrant data and as on future expectations.
| 21 | Supplemental disclosure of cash flow information |
| Years ended December 31, | ||||||||
| 2011 | 2010 | |||||||
| $ | $ | |||||||
Changes in operating assets and liabilities | ||||||||
Trade and other receivables | (3,332) | (2,029) | ||||||
Inventory | (261) | 896 | ||||||
Prepaid expenses and other current assets | (4,068) | (3,344) | ||||||
Other non-current assets | (456) | (315) | ||||||
Payables and accrued liabilities | 2,970 | (1,956) | ||||||
Provision and other non-current liabilities | (24) | 109 | ||||||
Deferred revenues | 8,614 | - | ||||||
Income taxes | 105 | (900) | ||||||
| 3,548 | (7,539) | |||||||
During the expected life of the stock options.
The Black-Scholes pricing model referred above uses "Level 2" inputs in calculating fair value, as defined by CICA Handbook Section 1862, except for certain options granted in 2009 and 2010, to whichyear ended December 31, 2011, the Company estimatedpaid approximately $841,000 in income taxes, as discussed in note 5.
| 22 | Income taxes |
| Years ended December 31, | ||||||||
2011 $ | 2010 $ | |||||||
Current: | (1,104) | - | ||||||
Deferred: | ||||||||
Origination and reversal of temporary differences | 9,017 | 7,632 | ||||||
Change in enacted tax rates | (104) | (272) | ||||||
Adjustments in respect of prior years | 3,428 | (176) | ||||||
Change in unrecognized tax assets | (12,341) | (7,184) | ||||||
| - | - | |||||||
Income tax expense | (1,104) | - | ||||||
Aeterna Zentaris Inc.
Notes to Consolidated Financial Statements
As at December 31, 2011 and December 31, 2010 and for the expected life using a "Level 3" input.years ended December 31, 2011 and 2010
16. SUPPLEMENTAL DISCLOSURE OF CASH FLOW INFORMATION
(tabular amounts in thousands of US dollars, except share/option/warrant and per share/option/warrant data and as otherwise noted)
| | Years ended December 31, | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2010 | 2009 | 2008 | |||||||||
| | $ | $ | $ | |||||||||
Changes in operating assets and liabilities | ||||||||||||
Accounts receivable | (2,029 | ) | 1,098 | 4,353 | ||||||||
Inventory | 896 | (864 | ) | 1,171 | ||||||||
Prepaid expenses and other current assets | (3,344 | ) | (14,478 | ) | (10,234 | ) | ||||||
Deferred charges and other long-term assets | (315 | ) | (472 | ) | (4,689 | ) | ||||||
Accounts payable and accrued liabilities | (2,104 | ) | (1,969 | ) | (1,089 | ) | ||||||
Other long-term liabilities | 109 | 66 | — | |||||||||
Income taxes | (900 | ) | 123 | 775 | ||||||||
Deferred revenues | — | — | 58,058 | |||||||||
| (7,687 | ) | (16,496 | ) | 48,345 | ||||||||
17. INCOME TAXES
The reconciliation of the combined Canadian federal and Quebec provincial income tax rate to the income tax expense is provided below:
| | Years ended December 31, | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2010 | 2009 | 2008 | ||||||||
Combined federal and provincial statutory income tax rate | 29.9% | 30.9% | 30.9% | ||||||||
| Years ended December 31, | ||||||||
| 2011 | 2010 | |||||||
Combined Canadian federal and provincial statutory income tax rate | 28.4% | 29.9% | ||||||
| Years ended December 31, | ||||||||
2011 $ | 2010 $ | |||||||
Income tax recovery based on statutory income tax rate | 7,290 | 8,500 | ||||||
Change in unrecognized tax assets | (12,341) | (7,184) | ||||||
Permanent difference attributable to the use of local currency for tax reporting | 378 | 1,232 | ||||||
Permanent difference attributable to net change in fair value of warrant liability | 661 | (1,761) | ||||||
Stock-based compensation costs | (441) | (357) | ||||||
Difference in statutory income tax rate of foreign subsidiaries | 893 | 391 | ||||||
Permanent difference attributable to unrealized foreign exchange gain/loss | (32) | (159) | ||||||
Change in enacted rates used | (104) | (272) | ||||||
Expiry of loss carryforwards | - | (164) | ||||||
Foreign witholding tax | (1,104) | - | ||||||
Adjustments in respect of prior years | 3,428 | (176) | ||||||
Other | 268 | (50) | ||||||
| (1,104) | - | |||||||
The decrease in the applicable tax rates is mainly due to the reduction of the federal income tax rate in 2011 from 18% to 16.5%.
Income tax expense of $1,104,000 for the year ended December 31, 2011 represents current taxation in the form of foreign jurisdiction tax withholdings on payments pursuant to a licensing agreement (note 5).
Deferred income tax assets are recognized to the extent that the realization of the related tax benefit through reversal of temporary differences and future taxable profits is probable. If income tax assets had been booked in 2011, an amount of $12,341,000 would have been credited to the income statement, $425,000 to other comprehensive income and $334,000 to equity. The remaining variation in unrecognized tax assets of $2,610,000 is due to exchange rate differences.
Table of ContentsAeterna Zentaris Inc.
AETERNA ZENTARIS INC.Notes to Consolidated Financial Statements
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
As at December 31, 20102011 and December 31, 2009
2010 and for the years ended December 31, 2010, 20092011 and 2008
2010
(tabular amounts in thousands of US dollars, except share/option/warrant and
per share/option/warrant data and as otherwise noted)
17. INCOME TAXES (Continued)
| | Years ended December 31, | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2010 | 2009 | 2008 | ||||||||
| | $ | $ | $ | ||||||||
Income tax recovery based on statutory income tax rate | 6,942 | 7,640 | 18,120 | ||||||||
Change in valuation allowance | (8,426 | ) | (9,959 | ) | (17,554 | ) | |||||
Permanent difference attributable to the use of local currency for tax reporting | 1,341 | 1,727 | — | ||||||||
Minimum tax attributable to German subsidiary | — | — | (1,175 | ) | |||||||
Stock-based compensation costs | (265 | ) | (85 | ) | (112 | ) | |||||
Share issue expenses not affecting earnings | 606 | 354 | — | ||||||||
Difference in statutory income tax rate of foreign subsidiaries | 425 | 222 | 576 | ||||||||
Permanent difference attributable to unrealized foreign exchange gain/loss | (159 | ) | (291 | ) | 494 | ||||||
Change in enacted rates used | (280 | ) | (89 | ) | (985 | ) | |||||
Expiry of loss carryforwards | (164 | ) | — | — | |||||||
Other | (20 | ) | 481 | (539 | ) | ||||||
| — | — | (1,175 | ) | ||||||||
Income tax expense for the year ended December 31, 2008 is entirely foreign in nature and represents current taxation.
Loss before income taxes
Loss before income taxes is attributable to the Company'sCompany’s tax jurisdictions as follows:
| | Years ended December 31, | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2010 | 2009 | 2008 | ||||||||
| | $ | $ | $ | ||||||||
Germany | (20,950 | ) | (20,335 | ) | (52,730 | ) | |||||
Canada | (2,191 | ) | (4,200 | ) | (5,103 | ) | |||||
United States | (77 | ) | (189 | ) | (809 | ) | |||||
| (23,218 | ) | (24,724 | ) | (58,642 | ) | ||||||
| Years ended December 31, | ||||||||
2011 $ | 2010 $ | |||||||
Germany | (25,246) | (19,763) | ||||||
Canada | (290) | (8,664) | ||||||
United States | (427) | (24) | ||||||
| (25,963) | (28,451) | |||||||
Significant components of deferred tax assets and liabilities:
As at December 31, | As at December 31, | As at January 1, | ||||||||||
2011 $ | 2010 $ | 2010 $ | ||||||||||
Deferred tax assets | ||||||||||||
Long-term: | ||||||||||||
Operating losses carried forward | 224 | 1,531 | 1,383 | |||||||||
Research and development costs | - | 169 | 226 | |||||||||
| 224 | 1,700 | 1,609 | ||||||||||
Deferred tax liabilities | ||||||||||||
Deferred revenues | - | 1,083 | 1,089 | |||||||||
Property, plant and equipment | 197 | 243 | 262 | |||||||||
Warrant liability | - | 169 | 188 | |||||||||
Other | 27 | 205 | 70 | |||||||||
| 224 | 1,700 | 1,609 | ||||||||||
Deferred tax assets (liabilities), net | - | - | - | |||||||||
Table of ContentsAeterna Zentaris Inc.
AETERNA ZENTARIS INC.Notes to Consolidated Financial Statements
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
As at December 31, 20102011 and December 31, 2009
2010 and for the years ended December 31, 2010, 20092011 and 2008
2010
(tabular amounts in thousands of US dollars, except share/option/warrant and
per share/option/warrant data and as otherwise noted)
17. INCOME TAXES (Continued)
Significant components of future incomeunrecognized deferred tax assets and liabilities are as follows:
| | As at December 31, | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| | 2010 | 2009 | |||||||
| | $ | $ | |||||||
Future income tax assets | |||||||||
Current | |||||||||
Deferred revenues | 65 | 713 | |||||||
Inventory | 352 | 73 | |||||||
Other | 9 | 108 | |||||||
| 426 | 894 | ||||||||
Long-term | |||||||||
Operating losses carried forward | 35,275 | 27,461 | |||||||
Intangible assets | 10,825 | 12,328 | |||||||
Research and development costs | 11,103 | 10,484 | |||||||
Employee future benefits | 1,167 | 1,014 | |||||||
Property, plant and equipment | 1,237 | 702 | |||||||
Share issue expenses | 715 | 374 | |||||||
Other | 138 | — | |||||||
| 60,460 | 52,363 | ||||||||
Valuation allowance | (58,125 | ) | (50,350 | ) | |||||
| 2,335 | 2,013 | ||||||||
| 2,761 | 2,907 | ||||||||
Future income tax liabilities | |||||||||
Current | |||||||||
Prepaid expenses and other current assets | — | 175 | |||||||
Other | 292 | — | |||||||
| 292 | 175 | ||||||||
Long-term | |||||||||
Deferred charges and other long-term assets | 1,143 | 1,343 | |||||||
Deferred revenues | 1,083 | 1,089 | |||||||
Property, plant and equipment | 243 | 262 | |||||||
Other | — | 38 | |||||||
| 2,469 | 2,732 | ||||||||
| 2,761 | 2,907 | ||||||||
Future income tax assets (liabilities), net | — | — | |||||||
As at December 31, | As at December 31, | As at January 1, | ||||||||||
2011 $ | 2010 $ | 2010 $ | ||||||||||
Deferred tax assets | ||||||||||||
Inventory | 740 | 352 | 262 | |||||||||
Deferred revenues | 507 | 65 | 713 | |||||||||
Other | - | 9 | 108 | |||||||||
| 1,247 | 426 | 1,083 | ||||||||||
Long term: | ||||||||||||
Operating losses carried forward | 38,665 | 33,745 | 26,078 | |||||||||
Intangible assets | 13,170 | 14,394 | 16,448 | |||||||||
Research and development costs | 12,310 | 10,934 | 10,257 | |||||||||
Unused tax credits | 10,667 | 7,158 | 6,738 | |||||||||
Employee future benefits | 1,534 | 1,233 | 1,136 | |||||||||
Property, plant and equipment | 1,301 | 1,237 | 702 | |||||||||
Share issue expenses | 813 | 715 | 374 | |||||||||
Deferred revenues | 589 | - | - | |||||||||
Onerous lease provision | 186 | 151 | 174 | |||||||||
Other | 139 | 138 | 1 | |||||||||
| 79,374 | 69,705 | 61,908 | ||||||||||
Unrecognized deferred tax assets | 80,621 | 70,131 | 62,991 | |||||||||
As at December 31, 2011, amounts and expiry dates of tax attributes to be deferred for which no deferred tax asset was recognized were as follow:
| Canada | ||||||||
Federal $ | Provincial $ | |||||||
2015 | 4,719 | - | ||||||
2028 | 10,962 | 10,079 | ||||||
2029 | 6,520 | 6,496 | ||||||
2030 | 5,586 | 5,666 | ||||||
2031 | 2,606 | 2,606 | ||||||
| 30,393 | 24,747 | |||||||
Aeterna Zentaris Inc.
Notes to Consolidated Financial Statements
As at December 31, 2011 and December 31, 2010 and for the years ended December 31, 2011 and 2010
(tabular amounts in thousands of US dollars, except share/option/warrant and per share/option/warrant data and as otherwise noted)
The Company has estimated non-refundable research and development tax credits of approximately $7,158,000$10,667,000 which can be carried forward to reduce Canadian federal income taxes payable and which expire at dates ranging from 20212018 to 2030. No tax benefit has been accounted for in connection with those credits.
AETERNA ZENTARIS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
As at December 31, 2010 and December 31, 2009and for the years ended December 31, 2010, 2009 and 2008(tabular amounts in thousands of US dollars, except share/option/warrant andper share/option/warrant data and as otherwise noted)
17. INCOME TAXES (Continued)
As at December 31, 2010, the Company had available operating losses in Canada, expiring as follows:
| | Canada | |||||||
|---|---|---|---|---|---|---|---|---|
| | Federal | Provincial | ||||||
| | $ | $ | ||||||
2014 | 9,768 | — | ||||||
2015 | 6,913 | 32 | ||||||
2028 | 12,339 | 11,437 | ||||||
2029 | 6,667 | 6,642 | ||||||
2030 | 5,712 | 5,691 | ||||||
| 41,399 | 23,802 | |||||||
Furthermore, the Company has availableunrecognized tax assets in respect of operating losses in Germany amountingand in the United States. The losses amount to approximately $80,924,000,$106,521,789 in Germany, for which there is no expiry date as well asand to $791,333 in the United States, totalling $896,772 and expiringwhich expire as follows:
| | United States | ||||
|---|---|---|---|---|---|
| | $ | ||||
2027 | 123 | ||||
2028 | 596 | ||||
2029 | 178 | ||||
| 897 | |||||
| United States | ||||
| $ | ||||
2027 | 17 | |||
2028 | 596 | |||
2029 | 178 | |||
|
| |||
| 791 | ||||
|
| |||
The operating loss carryforwards and the tax credits claimed are subject to review, and potential adjustment, by tax authorities.
18. SEGMENT INFORMATIONOther deductible temporary differences for which tax assets have not been booked are not subject to a time limit, except for share issue expenses which are amortizable over 5 years.
| 23 | Capital disclosures |
The Company operates in one single operating segment, being the biopharmaceutical segment.
Information by geographic region
Revenues by geographic region are detailed as follows:
| | Years ended December 31, | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2010 | 2009 | 2008 | |||||||||
| | $ | $ | $ | |||||||||
United States | 9,902 | 41,434 | 2,987 | |||||||||
Europe | ||||||||||||
Switzerland | 15,907 | 12,728 | 24,928 | |||||||||
United Kingdom | 13 | 35 | 3,823 | |||||||||
Other | 126 | 743 | 874 | |||||||||
Japan | 1,684 | 4,717 | 4,029 | |||||||||
Other | 71 | 3,580 | 1,837 | |||||||||
| 27,703 | 63,237 | 38,478 | ||||||||||
AETERNA ZENTARIS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
As at December 31, 2010 and December 31, 2009and for the years ended December 31, 2010, 2009 and 2008(tabular amounts in thousands of US dollars, except share/option/warrant andper share/option/warrant data and as otherwise noted)
18. SEGMENT INFORMATION (Continued)
Revenues have been allocated to geographic regions based on the country of residence of the Company's customers or partners.
Companies representing 10% or more of the Company's revenues in any of the last three fiscal years are as follows:
| | Years ended December 31, | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2010 | 2009 | 2008 | ||||||||
| | % | % | % | ||||||||
Company 1 | 58 | 21 | 66 | ||||||||
Company 2 | 29 | 9 | — | ||||||||
Company 3 | 1 | 54 | — | ||||||||
Company 4 | — | — | 10 | ||||||||
Net long-lived assets by geographic region are detailed as follows:
| | As at December 31, | |||||||
|---|---|---|---|---|---|---|---|---|
| | 2010 | 2009 | ||||||
| | $ | $ | ||||||
Germany | 26,758 | 31,016 | ||||||
United States | 381 | 551 | ||||||
Canada | 49 | 71 | ||||||
| 27,188 | 31,638 | |||||||
Long-lived assets consist of property, plant and equipment, intangible assets and goodwill.
19. LOSS PER SHARE
The following table sets forth data relating to the computation of basic and diluted net loss per share.
| | Years ended December 31, | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2010 | 2009 | 2008 | ||||||||
| | $ | $ | $ | ||||||||
Net loss | (23,218 | ) | (24,724 | ) | (59,817 | ) | |||||
Basic weighted average number of shares outstanding | 75,659,410 | 56,864,484 | 53,187,470 | ||||||||
Dilutive effect of stock options | 326,478 | 310,556 | 18,315 | ||||||||
Diluted weighted average number of shares outstanding | 75,985,888 | 57,175,040 | 53,205,785 | ||||||||
Items excluded from the calculation of diluted net loss per share because the exercise price was greater than the average market price of the common shares or due to their anti-dilutive effect | |||||||||||
Warrants (number of equivalent shares) | 12,923,390 | 4,110,603 | — | ||||||||
Stock options | 4,999,656 | 5,493,922 | 4,069,093 | ||||||||
| 17,923,046 | 9,604,525 | 4,069,093 | |||||||||
AETERNA ZENTARIS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
As at December 31, 2010 and December 31, 2009and for the years ended December 31, 2010, 2009 and 2008(tabular amounts in thousands of US dollars, except share/option/warrant andper share/option/warrant data and as otherwise noted)
19. LOSS PER SHARE (Continued)
For the years ended December 31, 2010, 2009 and 2008, diluted net loss per share was the same as basic net loss per share, since the effect of the assumed exercise of stock options and warrants (2010 and 2009 only) to purchase common shares is anti-dilutive. Accordingly, diluted net loss per share for these years was calculated using the basic weighted average number of shares outstanding.
20. CAPITAL DISCLOSURES
The Company'sCompany’s objective in managing capital, primarily composed of shareholders' equityshareholders’ deficiency and cash and cash equivalents, is to ensure sufficient liquidity to fund research and developmentR&D activities, general and administrative expenses, working capital and capital expenditures.
The Company has endeavouredCompany’s priority is to optimize its liquidity needs by non-dilutive sources, including the sale of non-core assets, and rights to future royalties, investment tax credits and grants, interest income, licensing serviceand related services and royalties. More recently, however, the Company has raised additional capital via the registered direct offerings and drawdowns related to the Company’s ATM sales agreement, as discussed in note 15.notes 17 and 30.
At December 31, 2011, cash and cash equivalents amounted to US$46,881,000. The Company believes that its cash position will be sufficient to finance its operations and capital needs for at least, but not limited to the next twelve months.
The capital management objective of the Company remains the same as that of previous years.periods. The policy on dividends is to retain cash to keep funds available to finance the activities required to advance the Company'sCompany’s product development pipeline.
The Company is not subject to any capital requirements imposed by any regulators or by any other external source.
21. FINANCIAL INSTRUMENTS AND FINANCIAL RISK MANAGEMENTAeterna Zentaris Inc.
Notes to Consolidated Financial Statements
As at December 31, 2011 and December 31, 2010 and for the years ended December 31, 2011 and 2010
(tabular amounts in thousands of US dollars, except share/option/warrant and per share/option/warrant data and as otherwise noted)
| 24 | Financial instruments and financial risk management |
Financial assets (liabilities) as at December 31, 2011, December 31, 2010 and January 1, 2010 are presented below.
| December 31, 2011 | Loans and receivables | Financial liabilities at FVTPL | Other financial liabilities | Total | ||||||||||||
| $ | $ | $ | $ | |||||||||||||
Cash and cash equivalents | 46,881 | - | - | 46,881 | ||||||||||||
Trade and other receivables (note 8) | 8,325 | - | - | 8,325 | ||||||||||||
Restricted cash (note 10) | 806 | - | - | 806 | ||||||||||||
Payables and accrued liabilities (note 14) | - | - | (12,126) | (12,126) | ||||||||||||
Long-term payable* | - | - | (88) | (88) | ||||||||||||
Warrant liability (note 15)* | - | (9,204) | - | (9,204) | ||||||||||||
Other non-current liabilities (note 16) | - | - | (187) | (187) | ||||||||||||
| 56,012 | (9,204) | (12,401) | 34,407 | |||||||||||||
* Includes current and non-current portions.
| December 31, 2010 | Financial asset at FVTPL | Loans and receivables | Financial liabilities at FVTPL | Other financial liabilities | Total | |||||||||||||||
| $ | $ | $ | $ | $ | ||||||||||||||||
Cash and cash equivalents | - | 31,998 | - | - | 31,998 | |||||||||||||||
Short-term investment (note 7) | 1,934 | - | - | - | 1,934 | |||||||||||||||
Trade and other receivables (note 8) | - | 5,421 | - | - | 5,421 | |||||||||||||||
Restricted cash (note 10) | - | 827 | - | - | 827 | |||||||||||||||
Payables and accrued liabilities (note 14) | - | - | - | (9,305) | (9,305) | |||||||||||||||
Long-term payable* | - | - | - | (150) | (150) | |||||||||||||||
Warrant liability (note 15)* | - | - | (14,367) | - | (14,367) | |||||||||||||||
Other non-current liabilities (note 16) | - | - | - | (183) | (183) | |||||||||||||||
| 1,934 | 38,246 | (14,367) | (9,638) | 16,175 | ||||||||||||||||
*Includes current and non-current portions.
Aeterna Zentaris Inc.
Notes to Consolidated Financial Statements
As at December 31, 2011 and December 31, 2010 and for the years ended December 31, 2011 and 2010
(tabular amounts in thousands of US dollars, except share/option/warrant and per share/option/warrant data and as otherwise noted)
| January 1, 2010 | Loans and receivables | Financial liabilities at FVTPL | Other financial liabilities | Total | ||||||||||||
| $ | $ | $ | $ | |||||||||||||
Cash and cash equivalents | 38,100 | - | - | 38,100 | ||||||||||||
Trade and other receivables (note 8) | 3,549 | - | - | 3,549 | ||||||||||||
Restricted cash (note 10) | 878 | - | - | 878 | ||||||||||||
Payables and accrued liabilities (note 14) | - | - | (11,845) | (11,845) | ||||||||||||
Long-term payable* | - | - | (200) | (200) | ||||||||||||
Warrant liability (note 15) | - | (1,664) | - | (1,664) | ||||||||||||
Other non-current liabilities (note 16) | - | - | (66) | (66) | ||||||||||||
| 42,527 | (1,664) | (12,111) | 28,752 | |||||||||||||
*Includes current and non-current portions.
Fair value
The Company has established the following classifications for its financial instrumentsBlack-Scholes valuation methodology uses “Level 2” inputs in accordance with CICA Handbook Section 3862:
The carrying values of all of the aforementioned financial instruments, excluding cash and cash equivalents, short-term investment and restricted cash, which are stated atcalculating fair value, approximate their fair values due to their short-term maturity or to the prevailing interest rates of these instruments, which are comparable to those of the market.
Fair value for the Company's financial instruments was established pursuant to the provisions of Section 3862,as defined in IFRS 7, which establishes a hierarchy that prioritizes the inputs used to measure fair value. The hierarchy gives the highest priority to unadjusted quoted prices in active markets for identical assets or liabilities (Level 1 measurement) and the lowest priority to unobservable inputs (Level 3 measurement). The input levels discussed in Section 3862IFRS 7 are:
| Level 1 | Unadjusted quoted prices in active markets for identical assets or liabilities. | ||||
| Level 2 – | Inputs other than quoted prices included within Level 1 that are observable for | ||||
| Level 3 – | Inputs for | ||||
The carrying values of Contents
AETERNA ZENTARIS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
As at December 31, 2010the Company’s cash and December 31, 2009
cash equivalents, trade and forother receivables, restricted cash, payables, provisions, accrued liabilities, long-term payable and other long-term liabilities approximate their fair values due to their short-term maturities or to the years ended December 31, 2010, 2009 and 2008(tabular amounts in thousandsprevailing interest rates of US dollars, except share/option/warrant andper share/option/warrant data and as otherwise noted)
21. FINANCIAL INSTRUMENTS AND FINANCIAL RISK MANAGEMENT (Continued)
The following table sets forth the Company's financial assets subjectrelated instruments, which are comparable to fair value measurements as at December 31, 2010:
| | Level 1 | Level 2 | Level 3 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| | $ | $ | $ | ||||||||
Cash | 12,922 | — | — | ||||||||
Cash equivalents | — | 19,076 | — | ||||||||
Short-term investment | — | 1,934 | — | ||||||||
Restricted cash | — | 827 | — | ||||||||
| 12,922 | 21,837 | — | |||||||||
The short-term investment presented above consiststhose of shares of a publicly held company. The underlying shares are restricted for resale until May 5, 2011.the market.
Financial risk managementfactors
DisclosuresThe following provides disclosures relating to the nature and extent of the Company'sCompany’s exposure to risks arising from financial instruments, including credit risk, liquidity risk foreign currencyand market risk (share price risk and interest rate risk,currency risk), and how the Company manages those risks, are presented below.risks.
a)Aeterna Zentaris Inc.
Notes to Consolidated Financial Statements
As at December 31, 2011 and December 31, 2010 and for the years ended December 31, 2011 and 2010
(tabular amounts in thousands of US dollars, except share/option/warrant and per share/option/warrant data and as otherwise noted)
| (a) | Credit risk |
Credit risk is the risk of an unexpected loss if a customer or counterparty to a financial instrument fails to meet its contractual obligations. The Company regularly monitors its credit risk exposure and takes steps to mitigate the likelihood of these exposuresthis exposure resulting in actual loss.
Financial instruments that potentially subject the Companylosses. The Company’s exposure to concentrations of credit risk consist primarily of cash and cash equivalents, restricted cash and accounts receivable. Cash and cash equivalents and restricted cash balances are maintained with high-credit quality financial institutions. Also, no accounts receivable balance due to the Company that is past due as at December 31, 2010 is significant. Consequently, management considers the risk of non-performance relatedcurrently relates to cash and cash equivalents (note 6), to trade and other receivables (note 8) and to restricted cash (note 10). The Company invests its available cash in amounts that are readily convertible to known amounts of cash and deposits its cash balances with financial institutions that have a minimum rating of “A3”.
As at December 31, 2011, trade accounts receivable for an amount of approximately $7,415,000 were with Company 1 and Company 3 (note 28).
As at December 31, 2011, no trade accounts receivable were past due or impaired.
Generally, the Company does not require collateral or other security from customers for trade accounts receivable; however, credit is extended following an evaluation of creditworthiness. In addition, the Company perform ongoing credit reviews of all its customers and establish an allowance for doubtful accounts when accounts are determined to be minimal.uncollectible.
The maximum exposure to credit risk approximates the amount recognized on the statement of financial position.
| (b) | Liquidity risk: |
Liquidity risk is the risk that the Company will not be able to meet its financial obligations as they become due. As indicated in the capital disclosures section (note 23), the Company manages this risk through the management of its capital structure. It also manages liquidity risk by continuously monitoring actual and projected cash flows. The Board of Directors reviews and approves the Company’s operating and capital budgets, as well as any material transactions out of the ordinary course of business. The Company has adopted an investment policy in respect of the safety and preservation of its capital to ensure the Company’s liquidity needs are met. The instruments are selected with regard to the expected timing of expenditures and prevailing interest rates.
| (c) | Market risk |
| Share | price risk |
The change in fair value of the Company’s warrant liability, which is measured at FVTPL, results from the periodic “mark-to-market” revaluation, via the application of the Black-Scholes option pricing model, of currently outstanding share purchase warrants. The Black-Scholes valuation is impacted, among other inputs, by the market price of the Company’s common shares. As a result, the change in fair value of the warrant liability, which is reported as finance income (costs) in the accompanying consolidated statements of comprehensive loss, has been and may continue in future periods to be materially affected most notably by changes in the Company’s common share price, which on the Nasdaq market, has ranged from $1.43 to $2.58 during the year ended December 31, 2011. Assuming the following variations of the market price of the Company’s common shares over a 12-month period:
| — | Market price variations of -10% and +10% |
b)Aeterna Zentaris Inc.
Notes to Consolidated Financial Statements
As at December 31, 2011 and December 31, 2010 and for the years ended December 31, 2011 and 2010
(tabular amounts in thousands of US dollars, except share/option/warrant and per share/option/warrant data and as otherwise noted)
If these variations were to occur, the impact on the Company’s net loss for warrant liability held at December 31, 2011 would be as follows:
| Carrying amount | -10% | +10% | ||||||||||
|
|
|
|
|
| |||||||
| $ | $ | $ | ||||||||||
Warrant liability* | 9,204 | 1,155 | (1,174) | |||||||||
|
|
|
| |||||||||
Total impact on net loss – decrease/(increase) | 1,155 | (1,174) | ||||||||||
|
|
|
| |||||||||
*Includes current and non-current portions.
Foreign Currencycurrency risk
Since the Company operates internationally, it is exposed to currency risks as a result of potential exchange rate fluctuations related to non-intragroup transactions. FluctuationsIn particular, fluctuations in the US dollar ("US$") andexchange rates against the EUR exchange rates could have a potentially significant impact on the Company'sCompany’s results of operations. The following variations are reasonably possible over a 12-month period:
AETERNA ZENTARIS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
As at December 31, 2010 and December 31, 2009and for the years ended December 31, 2010, 2009 and 2008(tabular amounts in thousands of US dollars, except share/option/warrant andper share/option/warrant data and as otherwise noted)
21. FINANCIAL INSTRUMENTS AND FINANCIAL RISK MANAGEMENT (Continued)
| — | Foreign exchange rate variations of -5% (depreciation of the EUR) and +5% (appreciation of the EUR) against the US$, from a year-end rate of EUR1 = US$1.2972. |
If these variations were to occur, the impact on the Company's consolidatedCompany’s net loss for each category of financial instruments held at December 31, 20102011 would be as follows:
| | | Balances denominated in US$ | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| | Carrying amount | ||||||||||
| | -5% | +5% | |||||||||
| | $ | $ | $ | ||||||||
Assets | |||||||||||
Cash and cash equivalents | 29,555 | 1,478 | (1,478 | ) | |||||||
Total impact on consolidated net loss — (increase)/decrease | 1,478 | (1,478 | ) | ||||||||
| Balances denominated in US$ | ||||||||||||
|
| |||||||||||
| Carrying amount | -5% | +5% | ||||||||||
| $ | $ | $ | ||||||||||
Cash and cash equivalents | 33,669 | 1,683 | (1,683) | |||||||||
Warrant liability* | 9,204 | (460) | 460 | |||||||||
|
|
|
| |||||||||
Total impact on net loss – decrease/(increase) | 1,223 | (1,223) | ||||||||||
|
|
|
| |||||||||
Liquidity risk is the risk that the Company will not be able to meet its financial obligations as they become due. The Company manages liquidity risk through the management of its capital structure
*Includes current and financial leverage, as outlined in note 20. The Company also manages liquidity risk by continuously monitoring actual and projected cash flows, as discussed in note 2. The Board of Directors reviews and approves the Company's operating and capital budgets and reviews any material transactions outside of the normal course of business.non-current portions.
| 25 | Commitments, contingencies and guarantee |
The Company's investment policy ensures the safety and preservation of its principal, as outlined above, to ensure the Company's liquidity needs are met.
| | Carrying Amount | 2011 | 2012-2013 | After 2013 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | $ | $ | $ | $ | ||||||||||
Accounts payable and accrued liabilities | 9,382 | 9,382 | — | — | ||||||||||
Long-term payable | 150 | 60 | 90 | — | ||||||||||
Other long-term liabilities | 182 | — | — | 182 | ||||||||||
| 9,714 | 9,442 | 90 | 182 | |||||||||||
22. COMMITMENTS, CONTINGENCIES AND GUARANTEE
In addition to the long-term payable discussed in note 6, the Company is committed to various operating leases for its premises plus service and manufacturing contracts, with payments expected as follows:contracts.
| | Minimum Lease Commitments | Service & Manufacturing Commitments | Total Commitments | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| | $ | $ | $ | ||||||||
2011 | 2,150 | 9,167 | 11,317 | ||||||||
2012 | 1,926 | 5,163 | 7,089 | ||||||||
2013 | 1,883 | 3,557 | 5,440 | ||||||||
2014 | 1,818 | — | 1,818 | ||||||||
2015 | 1,824 | — | 1,824 | ||||||||
Thereafter | 796 | — | 796 | ||||||||
| 10,397 | 17,887 | 28,284 | |||||||||
Table of ContentsAeterna Zentaris Inc.
AETERNA ZENTARIS INC.Notes to Consolidated Financial Statements
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
As at December 31, 20102011 and December 31, 2009
2010 and for the years ended December 31, 2010, 20092011 and 2008
2010
(tabular amounts in thousands of US dollars, except share/option/warrant and
per share/option/warrant data and as otherwise noted)
22. COMMITMENTS, CONTINGENCIES AND GUARANTEE (Continued)
As discussed in note 5,Future minimum lease payments and future minimum sublease payments expected to be received under non-cancellable operating leases (subleases), as well as future payments in connection with the PSA entered into with Cowen, the Company has agreed to make a one-time cash payment to Cowen in the event that cetrorelix is approved for sale by European regulatory authorities in an indication other thanin vitro fertilization. Such a payment, which is not probable or reasonably estimableutility service agreements, as at December 31, 2010, could range from $5,000,000 to2011 are as follows:
| Minimum lease payments | Minimum sub-lease payments | Utilities | ||||||||
| $ | $ | $ | ||||||||
Less than 1 year | 1,690 | (226) | 609 | |||||||
1 – 3 years | 3,128 | (451) | 793 | |||||||
4 – 5 years | 2,172 | (451) | 496 | |||||||
More than 5 years | 373 | (244) | - | |||||||
Total | 7,363 | (1,372) | 1,898 | |||||||
The Company has a maximum of $15,000,000. Also as discussedleasing arrangement in note 5, the Company could also be required to pay Cowen a quarterly make-whole payment.
Rent expense for operating leases,Germany under which may have escalating rentals over theit rents laboratory, storage and office space. The original term of the lease, are recorded on a straight-line basis overten years (or March 2016), is automatically renewable for two further five-year periods if not terminated otherwise by the termCompany within 12 months prior to original expiry. Under the terms of the lease.arrangement, the minimum lease payment may be increased or lowered proportionally to the fluctuation in consumer price index for Germany if that change is more than 5% on an accumulated basis. The rent expense underterms of the operating leaseslease arrangement for the periods ended December 31, 2010, 2009 and 2008 was approximately $1,899,000, $2,133,000 and $1,700,647, respectively.ten years following automatic renewal are the same as per the original agreement.
In October 2007, the Company entered into a $100,000 letter of credit agreement in favour of its landlord in the United States with respect to the Company'sCompany’s long-term lease obligation. In August 2009 and November 2011, the amount of the letter of credit was reduced to $75,000 and $50,000, respectively, as per the original landlord-tenant agreement, and is payable to the landlord in the event that the Company fails to perform any of its obligations under the related lease agreement.
Service and manufacturing commitments given, which consist of R&D service agreements and manufacturing agreements for Cetrotide®, are as follows:
| December 31, 2011 | |||||||
| $ | |||||||
Less than 1 year | 10,760 | ||||||
1 – 3 years | 3,855 | ||||||
4 – 5 years | - | ||||||
More than 5 years | - | ||||||
Total | 14,615 | ||||||
Contingencies
In the normal course of operations, the Company may become involved in various claims and legal proceedings related to, for example, contract terminations, employee-related and other matters. No contingent liabilities have been accrued as at December 31, 20102011 or 2009,2010, nor are there any known disputes pending against the Company that could significantly impact the Company'sCompany’s consolidated financial statements.
23. COMPARATIVE FIGURES
To conform to the presentation adopted in the current year, certain amounts from the prior year have been reclassified.
24. SUBSEQUENT EVENTS
On February 22, 2011, the Company entered into an ATM sales agreement, under which the Company may, at its discretion, from time to time during the 24-month term of the agreement, sell up to a maximum of 12,500,000 of its common shares through ATM issuances on the NASDAQ Stock Market for aggregate gross proceeds not to exceed $19,750,831, being the amount remaining available for distribution, as at February 22, 2011, under the Company's current registration statement on Form F-3. The common shares will be sold at market prices prevailing at the time of a sale of the common shares, and, as a result, prices may vary.
On March 10, 2011, the Company issued 1,663,064 common shares in connection with the aforementioned ATM agreement, for gross proceeds of approximately $3,240,000.
On March 8, 2011, the Company entered into an agreement with Yakult Honsha Co. Ltd. ("Yakult") for the development, manufacture and commercialization of perifosine in all human uses, excluding leishmaniasis, in Japan. Under the terms of this agreement, Yakult will make an initial non-refundable upfront payment to the Company of €6,000,000 (approximately $8,300,000). Also per the agreement, the Company will be entitled to receive up to a total of €44,000,000 (approximately $60,900,000) upon achieving certain pre-established milestones, including clinical and regulatory events in Japan. Furthermore, the Company will be entitled to receive double-digit royalties on future net sales of perifosine in the Japanese market.
25. DIFFERENCES BETWEEN CANADIAN AND US GAAP
The Company is required to reconcile its financial statements for significant measurement differences between Canadian GAAP and US GAAP, as well as provide additional significant disclosures required under US GAAP and Regulation S-X of the Securities and Exchange Commission in the United States ("SEC"). As such, presented below are the significant quantitative differences between Canadian and US GAAP, as well as other significant disclosures required under US GAAP
Table of ContentsAeterna Zentaris Inc.
AETERNA ZENTARIS INC.Notes to Consolidated Financial Statements
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
As at December 31, 20102011 and December 31, 2009
2010 and for the years ended December 31, 2010, 20092011 and 2008
2010
(tabular amounts in thousands of US dollars, except share/option/warrant and
per share/option/warrant data and as otherwise noted)
25. DIFFERENCES BETWEEN CANADIAN AND US GAAP (Continued)
and Regulation S-X of the SEC, not already provided in the accompanying consolidated financial statements and notes thereto.
| 26 | Net loss per share |
The following summarytable sets out the material adjustmentsforth pertinent data relating to the Company's reported net loss,computation of basic and diluted net loss per share attributable to common shareholders.
| Years ended December 31, | ||||||||
| 2011 | 2010 | |||||||
| $ | $ | |||||||
Net loss | (27,067) | (28,451) | ||||||
Basic weighted average number of shares outstanding | 94,507,988 | 75,659,410 | ||||||
Dilutive effect of stock options | 1,143,749 | 326,478 | ||||||
Dilutive effect of share purchase warrants | 1,697,418 | - | ||||||
Diluted weighted average number of shares outstanding | 97,349,155 | 75,985,888 | ||||||
Items excluded from the calculation of diluted net loss per share because the exercise price was greater than the average market price of the common shares or due to their anti-dilutive effect | ||||||||
Stock options | 3,681,864 | 4,999,656 | ||||||
Warrants (number of equivalent shares) | - | 12,923,390 | ||||||
For the years ended December 31, 2011 and shareholders' equity that would be made to conform with US GAAP:
Reconciliation of2010, the diluted net loss per share was the same as the basic net loss per share, since the effect of the assumed exercise of stock options and warrants to US GAAP
| | | Years ended December 31, | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | | 2010 | 2009 | 2008 | |||||||||||
| | | $ | $ | $ | |||||||||||
Net loss under Canadian GAAP | (23,218 | ) | (24,724 | ) | (59,817 | ) | |||||||||
Variation in warrant liability, including amortization of transaction costs | (a | ) | (6,370 | ) | 1,557 | — | |||||||||
Amortization of in-process research and development costs | (b | ) | 423 | 6,373 | 3,747 | ||||||||||
Net loss under US GAAP | (29,165 | ) | (16,794 | ) | (56,070 | ) | |||||||||
Net loss per share | |||||||||||||||
Basic and diluted | (0.39 | ) | (0.30 | ) | (1.05 | ) | |||||||||
Weighted average number of shares (note 19) under US GAAP | |||||||||||||||
Basic and diluted | 75,659,410 | 56,864,484 | 53,187,470 | ||||||||||||
Reconciliationpurchase common shares is anti-dilutive. Accordingly, the diluted net loss per share for these periods was calculated using the basic weighted average number of shareholders' equity to conform to US GAAPshares outstanding.
The following summary sets outweighted average number of shares is influenced most notably by share issuances made in connection with financing activities, such as ATM drawdowns and registered direct offerings, which resulted in the significant differences betweenissuance of a total of 19,464,548 and 19,917,075 common shares (see note 17) during the Company's reported shareholders' equity under Canadian GAAP as comparedyears ended December 31, 2011 and 2010, respectively. See also note 30.
Aeterna Zentaris Inc.
Notes to US GAAP.
| | | As at December 31, 2010 | As at December 31, 2009 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| | | $ | $ | ||||||||
Shareholders' equity in accordance with Canadian GAAP | 12,439 | 9,226 | |||||||||
Net impact of liability accounting for warrants | (a | ) | (14,367 | ) | (1,351 | ) | |||||
In-process research and development costs | (b | ) | (1,721 | ) | (2,146 | ) | |||||
Shareholders' (deficiency) equity in accordance with US GAAP | (3,649 | ) | 5,729 | ||||||||
Table of ContentsConsolidated Financial Statements
AETERNA ZENTARIS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
As at December 31, 20102011 and December 31, 2009
2010 and for the years ended December 31, 2010, 20092011 and 2008
2010
(tabular amounts in thousands of US dollars, except share/option/warrant and
per share/option/warrant data and as otherwise noted)
| 27 | Compensation of key management |
Compensation awarded to key management* included:
| 2011 | 2010 | |||||||||
| $ | $ | |||||||||
Salaries and short-term employee benefits | 2,886 | 2,524 | ||||||||
Post-employment benefits | 684 | 114 | ||||||||
Termination benefits | - | - | ||||||||
Share-based compensation cost | 936 | 554 | ||||||||
| 4,506 | 3,192 | |||||||||
* Key management includes the Company’s directors and members of the Executive Committee.
| 28 | Segment information |
The Company operates in a single operating segment, being the biopharmaceutical segment.
25. DIFFERENCES BETWEEN CANADIAN AND US GAAP (Continued)
Balance sheetsGeographical information
The following table summarizesCompany is domiciled in Canada and derives all its revenues from its operating subsidiaries domiciled in Germany.
Revenues by geographical area are detailed as follows:
| Years ended December 31, | ||||||||||
| 2011 | 2010 | |||||||||
| $ | $ | |||||||||
United States | 5,492 | 9,902 | ||||||||
Switzerland | 24,977 | 15,907 | ||||||||
Japan | 5,472 | 1,684 | ||||||||
Other | 112 | 210 | ||||||||
| 36,053 | 27,703 | |||||||||
Revenues have been allocated to geographic regions based on the significantcountry of residence of the Company’s external customers or partners.
Aeterna Zentaris Inc.
Notes to Consolidated Financial Statements
As at December 31, 2011 and December 31, 2010 and for the years ended December 31, 2011 and 2010
(tabular amounts in thousands of US dollars, except share/option/warrant and per share/option/warrant data and as otherwise noted)
Non-current assets* by geographical area are detailed as follows:
As at December 31, 2011 | As at December 31, 2010 | As at January 1, 2010 | ||||||||||
| $ | $ | $ | ||||||||||
Germany | 13,557 | 15,579 | 18,109 | |||||||||
United States | - | 381 | 551 | |||||||||
Canada | 36 | 49 | 71 | |||||||||
| 13,593 | 16,009 | 18,731 | ||||||||||
* Non-current assets exclude financial instruments and other non-current assets.
Companies information about major customers representing 10% or more of the Company’s revenues in any of the last two years are as follows:
| Years ended December 31, | ||||||||
| 2011 | 2010 | |||||||
| $ | $ | |||||||
Company 1 | 24,977 | 15,907 | ||||||
Company 2 | 4,556 | 7,990 | ||||||
Company 3 | 3,657 | - | ||||||
| 29 | Transition to IFRS |
The accompanying consolidated financial statements were prepared as described in note 1 and reflect the relevant provisions of IFRS 1. IFRS 1 is based on the principle that the adoption of IFRS should be applied retrospectively. Retrospective application necessitates that comparative financial information be provided, and, as a result, the first date at which the Company has applied IFRS was January 1, 2010. However, IFRS 1 offers certain optional exemptions and mandatory exceptions to the retrospective application of IFRS to first-time preparers of IFRS financial statements. Those exemptions and exceptions, which are relevant to the Company, are discussed in turn below.
Optional IFRS exemptions
Business combinations
IFRS 1 allows first-time preparers to elect not to restate any business combinations that have occurred prior to the Transition Date in accordance with IFRS 3,Business Combinations (as revised in 2008) (“IFRS 3”). Retrospective application would require that all business combinations that occurred prior to an entity’s date of transition to IFRS be restated, and any goodwill arising on such business combinations would be adjusted from its carrying value as determined under Canadian GAAP.
Aeterna Zentaris Inc.
Notes to Consolidated Financial Statements
As at December 31, 2011 and December 31, 2010 and for the years ended December 31, 2011 and 2010
(tabular amounts in thousands of US dollars, except share/option/warrant and per share/option/warrant data and as otherwise noted)
The Company has elected to apply this exemption and has not restated any prior business combinations. Consequently, IFRS 3 is applicable only to business combinations occurring after the Transition Date. There have been no business combinations since the Transition Date, and, as a result, the Company will apply the provisions of IFRS 3 to future transactions, if any.
Currency translation differences between relevant
Full retrospective application of IFRS would require an entity to determine the cumulative foreign currency translation differences, as per the provisions of IAS 21,The Effects of Changes in Foreign Exchange Rates, from the date that a subsidiary or an equity-method investee was acquired. IFRS 1 permits a first-time adopter to reset any cumulative translation differences that existed at the date of transition to IFRS to zero. The Company has elected to reset its cumulative translation adjustment balance sheet itemsto zero on January 1, 2010, with a corresponding adjustment to the Company’s Transition Date deficit.
Mandatory IFRS exception
Accounting estimates
IFRS 1 requires that estimates under IFRS at the date of transition should be consistent with estimates made for the same date under previous GAAP, after applying any adjustments to reflect differences in accounting policies, unless there is objective evidence that those estimates were made in error. As such, a first-time adopter cannot use hindsight in order to create or revise any accounting estimates. Estimates previously made by the Company under Canadian GAAP have not been revised, except where necessary to reflect any differences in accounting policies.
Reconciliation of Canadian GAAP to IFRS
IFRS 1 requires a first-time adopter of IFRS to reconcile shareholders’ equity (deficiency), comprehensive income (loss) and cash flows for prior periods beginning on the date of transition to IFRS. The Company’s first-time adoption of IFRS did not have any impact on previously reported cash flows from operating activities, financing activities or investing activities. Reconciliations of shareholders’ equity (deficiency) as at January 1, 2010 and December 31, 2010 and comprehensive loss for the year December 31, 2010 are provided below.
Reconciliation of shareholders’ equity (deficiency) | As at 2010 | As at 2010 | ||||||||||
| $ | $ | |||||||||||
Shareholders’ equity under Canadian GAAP | 12,439 | 9,226 | ||||||||||
IFRS adjustments attributable to: | ||||||||||||
Impairment of Cetrotide® asset | (a | ) | (11,179) | (12,907) | ||||||||
Derecognition of deferred transaction costs | (b | ) | (3,947) | (4,747) | ||||||||
Liability accounting for share purchase warrants | (c | ) | (14,367) | (1,664) | ||||||||
Onerous lease provision | (d | ) | (312) | (367) | ||||||||
Termination benefit adjustment | (e | ) | (209) | (381) | ||||||||
Shareholders’ deficiency under IFRS | (17,575) | (10,840) | ||||||||||
Aeterna Zentaris Inc.
Notes to Consolidated Financial Statements
As at December 31, 2011 and December 31, 2010 and for the years ended December 31, 2011 and 2010
(tabular amounts in thousands of US dollars, except share/option/warrant and per share/option/warrant data and as otherwise noted)
| Reconciliation of comprehensive loss | Year ended December 31, 2010 | |||||||
| $ | ||||||||
Comprehensive loss under Canadian GAAP | (23,169) | |||||||
IFRS adjustments attributable to: | ||||||||
Impairment of Cetrotide® asset | (a | ) | 924 | |||||
Derecognition of deferred transaction costs | (b | ) | 504 | |||||
Liability accounting for share purchase warrants | (c | ) | (6,368) | |||||
Onerous lease provision | (d | ) | 52 | |||||
Termination benefit adjustment | (e | ) | 152 | |||||
Share-based compensation | (f | ) | (306) | |||||
Foreign currency translation adjustments to shareholders’ deficiency | 952 | |||||||
Comprehensive loss under IFRS | (27,259) | |||||||
Explanatory notes
In addition to the IFRS 1 exemptions discussed above, the following section discusses the changes in accounting policies that resulted in the adjustments shown in the preceding reconciliations.
| (a) | Impairment of assets |
Under Canadian GAAP, property, plant and equipment and intangible assets with finite lives were reviewed for impairment whenever events or circumstances indicated that the carrying values of those assets may not be recoverable. Impairments were deemed to exist when the carrying value of the asset or asset group was greater than the undiscounted future cash flows expected to be provided by the asset or asset group. The amount of impairment loss, if any, was equivalent to the excess of the asset’s or asset group’s carrying value over fair value, which in turn was determined based upon discounted cash flows or appraised values, depending on the nature of assets.
Under IFRS, once an indication of impairment is identified, similar to Canadian GAAP, an entity is required to make a formal estimate of recoverable amount. However, unlike Canadian GAAP, the carrying amount of an asset that is subject to impairment testing under IFRS is compared to the higher of fair value less costs to sell or value in use. Where the recoverable amount of an asset subject to impairment testing is compared to the asset’s value in use, any future cash flows expected to be provided by the asset are discounted, unlike Canadian GAAP.
As a result of the change in measurement methodology, the Company recognized an additional impairment charge, amounting to $12,907,000 as of the Transition Date related to the intangible asset, Cetrotide®, since the carrying amount of that asset exceeded its recoverable amount. The Company has adjusted related amortization charges in the Company’s comparative consolidated statement of comprehensive loss for the year ended December 31, 2010.
Aeterna Zentaris Inc.
Notes to Consolidated Financial Statements
As at December 31, 2011 and December 31, 2010 and for the years ended December 31, 2011 and 2010
(tabular amounts in thousands of US GAAP.
| | | As at December 31, 2010 | As at December 31, 2009 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | | As Reported | US GAAP | As Reported | US GAAP | ||||||||||||
| | | $ | $ | $ | $ | ||||||||||||
Warrant liability, long-term and short-term portions | (a | ) | — | 14,367 | * | — | 1,351 | ||||||||||
Intangible assets | (b | ) | 14,478 | 12,757 | 17,034 | 14,888 | |||||||||||
| (b) | Deferred transaction costs |
(a)
InUnder Canadian GAAP, transaction costs incurred in connection with the registered direct offerings thatCompany’s December 2008 sale to Cowen of the Company’s rights to royalties on future sales of Cetrotide® were completedcapitalized as deferred charges and were being amortized based on the “units-of-revenue” method and over the same period in April and June 2010 and in June and October 2009,which the related deferred revenues were recognized as discussed in note 15,royalty revenues.
Under IFRS, the related transaction costs have been charged to expense as incurred.
As a result, the Company issued warrantshas derecognized the remaining unamortized portion of the deferred transaction costs, amounting to purchase common shares to$4,747,000, as of the institutional investors who participatedTransition Date and has reversed any related amortization expense in the offerings and, as partCompany’s comparative consolidated statements of most, but not all, of these offerings, tocomprehensive loss for the sole placement agent and its designated representatives.year ended December 31, 2010.
| (c) | Accounting for share purchase warrants |
Under Canadian GAAP, the Company hashad classified and iswas accounting for all of its outstanding common share purchase warrants as equity, on the basis that thesethe warrants dodid not embody a contractual obligation on the Company to deliver cash or another financial asset to the holder of thesethose warrants. The conditionalAll share purchase warrants issued in connection with the Company’s registered direct offerings contain a written put option, that arisesarising upon the occurrence of a Fundamental Transaction, as defined in all outstanding warrants and including a change in control,control. The exercise of the put options was not considered to be probable at any reporting date under Canadian GAAP.
Under IFRS, financial instruments that have terms whereby the CICA's Emerging Issues Committee Abstract No. 70,Presentationissuer of a Financial Instrument Labelledthe instrument does or could in the future not have the unconditional right to avoid delivering cash must be classified as a Share When a Future Event or Circumstance May Affect the Issuer's Obligations. Under US GAAP, the Company has determinedliability and measured at FVTPL. Additionally, IFRS requires that the common share purchase warrants are within the scope the FASB's ASC Topic 480,Distinguishing Liabilities from Equity ("Topic 480"), and as such has classified and is accounting for these instruments as a liability. Topic 480 states thatany transaction costs on financial instruments which containcarried at FVTPL be expensed immediately, and not included in the initial measurement of the instrument.
As a writtenresult of the presence of the aforementioned put option, even ifoptions, and despite the fact that the repurchase feature is conditional on a defined contingency, shouldthe share purchase warrants are required to be classified as a liability ifunder IFRS, since such a contingency ultimately could result in the transfer of assets by the issuer.
The total warrantCompany. As a result, all share purchase warrants are classified as a liability would, for US GAAP purposes, be carriedand are measured at fair value, or $1,664,000 as of the Transition Date, and any periodic changes in fair value wouldare recognized as gains or losses through profit or loss. Transaction costs related to the FVTPL instruments were expensed under IFRS.
| (d) | Onerous lease provision |
Under Canadian GAAP, there is no single standard that provides guidance related to the identification or accounting for provisions, nor are provisions specifically defined. Additionally, onerous contracts are not specifically referred to under Canadian GAAP. Implicit references to onerous arrangements are derived from broader principles underlying the definition of a contingency or liability and are oftentimes limited in practice to an entity’s exit or restructuring activities.
Under IFRS, accounting for onerous contracts, defined as contracts in which the unavoidable costs of meeting the obligations under the contract exceed the economic benefits expected to be reflected within other income (expenses) inreceived under it, is explicitly prescribed. Furthermore, the consolidated statement of operations. There were no common share purchase warrants issued or outstanding during 2008.Company is required to determine, at each reporting date, whether any onerous contracts exist, and, if the Company has a contract that is deemed to be onerous, the present obligation under that contract must be recognized and measured as a provision.
Table of ContentsAeterna Zentaris Inc.
AETERNA ZENTARIS INC.Notes to Consolidated Financial Statements
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
As at December 31, 20102011 and December 31, 2009
2010 and for the years ended December 31, 2010, 20092011 and 2008
2010
(tabular amounts in thousands of US dollars, except share/option/warrant and
per share/option/warrant data and as otherwise noted)
The Company determined that it had a lease arrangement that was partially onerous as of the Transition Date, and as a result, a provision of $367,000 was recognized at that date.
| (e) | Employee benefits |
Actuarial gains and losses
25. DIFFERENCES BETWEEN CANADIAN AND USUnder Canadian GAAP, (Continued)
The table presented below showsall actuarial gains and losses arising in the assumptions appliedcalculation of the Company’s defined benefit obligation have been recorded in profit or loss as incurred.
Under IFRS, actuarial gains and losses may be recognized, as incurred, in other comprehensive income or loss, provided that the Company does so for all of its defined benefit plans and all of its actuarial gains and losses.
Management has chosen to recognize actuarial gains and losses as incurred directly in other comprehensive loss beginning on the Transition Date. As a result, the Company adjusted its employee benefit expenses to remove the net actuarial gains, amounting to $191,000 for the year ended December 31, 2010. Instead, net actuarial gains has been recorded directly in other comprehensive loss, net of tax, in the deficit in the consolidated statement of financial position as at December 31, 2010, without recycling to the Black-Scholes pricing modelconsolidated statement of comprehensive loss in ordersubsequent periods.
Termination benefits
Certain Group employees in Germany are eligible to determineparticipate in a partial retirement program, which is composed of a full-time service period and an inactive period, where the employee receives 50% of his or her salary for each year as well as an annual bonus during the entire partial retirement period.
Under Canadian GAAP, the annual bonus to be paid in the inactive period is recognized as expense on a pro rata basis over the full-time service period.
Under IFRS, the entire bonus element of the partial retirement arrangement is recognized as an expense immediately when the related partial retirement agreement is established.
As a result, the Company has recognized an additional liability, amounting to $381,000, to reflect the aggregate bonus element of the partial retirement arrangement as of the Transition Date.
| (f) | Share-based payments |
Under Canadian GAAP, for share-based awards with graded vesting, the Company recognizes the fair value of warrants outstanding asthe award (all tranches) on a straight-line basis over the underlying vesting period. Additionally, forfeitures of awards are not estimated at December 31, 2010.
| | June 2009 Investor Warrants | June 2009 Compensation Warrants | October 2009 Investor Warrants | October 2009 Compensation Warrants | April 2010 Investor Warrants | June 2010 Investor Warrants | June 2010 Compensation Warrants | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Number of equivalent shares | 1,861,702 | 287,234 | 1,716,667 | 128,333 | 4,444,444 | 4,220,832 | 264,178 | ||||||||||||||||
Market-value per share price | $ | 1.72 | $ | 1.72 | $ | 1.72 | $ | 1.72 | $ | 1.72 | $ | 1.72 | $ | 1.72 | |||||||||
Exercise price | $ | 2.06 | $ | 2.35 | $ | 1.25 | $ | 1.50 | $ | 1.50 | $ | 1.37 | $ | 1.72 | |||||||||
Risk-free annual interest rate | 0.29% | 0.29% | 1.43% | 0.56% | 1.91% | 1.75% | 1.74% | ||||||||||||||||
Expected volatility | 83.0% | 83.0% | 99.3% | 113.0% | 92.2% | 94.6% | 94.7% | ||||||||||||||||
Expected life (years) | 1.0 | 1.0 | 3.8 | 1.8 | 4.8 | 4.5 | 4.5 | ||||||||||||||||
Expected dividend yield | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | ||||||||||||||||
The Black-Scholes valuation methodology uses "Level 2" inputsthe date of grant and therefore are not included in calculating fair value, as defined in ASC Topic 820,Fair Value Measurements and Disclosures ("Topic 820"), which establishes a fair value hierarchy that prioritizes the inputs used to measurecalculation of the grant-date fair value. The hierarchy gives the highest priorityInstead, all forfeitures of awards are recognized as they occur.
Aeterna Zentaris Inc.
Notes to unadjusted quoted prices in active markets for identical assets or liabilities (Level 1 measurement) and the lowest priority to unobservable inputs (Level 3 measurement). Level 2 inputs are those which are either directly or indirectly observable as of the reporting date and include financial instruments that are valued using models or other valuation methodologies, such as the Black-Scholes pricing model.
Under US GAAP, prior to the issuance of ASC Topic 805,Business Combinations, in-process research and development acquired in a business combination is written off at the time of acquisition.Consolidated Financial Statements
Under Canadian GAAP, in-process research and development acquired in a business combination is capitalized and amortized over its estimated useful life.
Statements of cash flows
For each of the years ended December 31, 2010, 2009 and 2008, there are no significant differences between the statements of cash flows under Canadian GAAP as compared to US GAAP.
New accounting standards and pronouncements
ASC Topic 810,Consolidation ("Topic 810")
In June 2009, the FASB amended the consolidation guidance in Topic 810 for variable-interest entities. Amendments include the elimination of the exemption for qualifying special purpose entities, a new approach for determining who should consolidate a variable-interest entity and changes to when it is necessary to reassess who should consolidate a variable-interest entity. This guidance is effective for years beginning after November 15, 2009, for interim periods within those years, and for interim and annual reporting periods thereafter. The Company adopted this guidance on January 1, 2010, and there has been no impact on the Company's consolidated financial statements.
ASC Topic 860,Transfers and Servicing ("Topic 860")
AETERNA ZENTARIS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
As at December 31, 20102011 and December 31, 2009
2010 and for the years ended December 31, 2010, 20092011 and 2008
2010
(tabular amounts in thousands of US dollars, except share/option/warrant and
per share/option/warrant data and as otherwise noted)
25. DIFFERENCES BETWEEN CANADIAN AND US GAAP (Continued)
In June 2009, the FASB amended Topic 860 to remove the conceptUnder IFRS, each tranche of a qualifying special-purpose entityshare-based award with graded vesting is treated as a separate award, and the exception from applying Topic 810resulting compensation expense is recognized for each tranche over its distinct vesting period. Furthermore, expected forfeitures of awards are required to qualifying special-purpose entities. This guidance containedbe estimated at the date of grant and therefore are factored into the determination of fair value.
The Company has adjusted its periodic share-based compensation expense for underlying awards in Topic 860 must be applied asorder to reflect the change in policy related to graded vesting and estimated expected forfeitures. These adjustments have resulted in an increase in share-based compensation costs of $306,000 for the year ended December 31, 2010.
Reconciliations of consolidated financial statements
Presented below are reconciliations of the beginning of each reporting entity's first annual reporting period that begins after November 15, 2009, for interim periods within that first annual reporting period and for interim and annual reporting periods thereafter. Topic 860 must be applied to transfers occurring on or after the effective date. The Company adopted this guidance on January 1, 2010, and there has been no impact on the Company's consolidated financial statements.
Accounting Standards Update ("ASU") 2010-06,Fair Value Measurements and Disclosures (Topic 820): Improving Disclosures about Fair Value Measurements ("ASU 2010-06")
The FASB recently issued ASU 2010-06 to enhance the usefulness of fair value measurements. The amended guidance requires both the disaggregation of information in certain existing disclosures, as well as the inclusion of more robust disclosures about valuation techniques and inputs to recurring and nonrecurring fair value measurements. ASU 2010-06 amends the disclosures about fair value measurements in Topic 820 and is effective for interim and annual reporting periods beginning after December 15, 2009, except for disaggregation requirements for the reconciliation disclosure of Level 3 measurements, which is effective for fiscal years beginning after December 15, 2010 and for interim periods within those years. The Company adopted this guidance on January 1, 2010, and there has been no impact on the Company's consolidated financial statements.
As discussed in note 3, beginning on January 1, 2011, the Company will cease to prepare itsCompany’s consolidated financial statements previously prepared under Canadian GAAP to the consolidated financial statements prepared in accordance with Canadian GAAP and instead will apply IFRS as issued by the IASB as the Company's primary basis of accounting. Additionally, the Company will ceaseIFRS.
Aeterna Zentaris Inc.
Notes to reconcile its financial statements to US GAAP for periods beginning on January 1, 2011. As a result, the following guidance will never be applied by the Company.Consolidated Financial Statements
ASC Topic 605,Revenue Recognition ("Topic 605")
In October 2009, the FASB amended Topic 605 to include a consensus ratified by the FASB's Emerging Issues Task Force relating to multiple-deliverable revenue arrangements. These amendments significantly change certain guidance pertaining to revenue arrangements with multiple deliverables and modify the separation criteria of Topic 605 by eliminating the criterion for objective and reliable evidence of fair value for the undelivered products or services. The amendments also eliminate the use of the residual method of allocation and require instead that arrangement consideration be allocated at the inception of the arrangement to all deliverables based on their relative selling price. This guidance is effective for revenue arrangements entered into or materially modified in fiscal years beginning on or after June 15, 2010. Early adoption is permitted.
ASU 2010-13,Compensation — Stock Compensation (Topic 718): Effect of Denominating the Exercise Price of a Share-Based Payment Award in the Currency of the Market in Which the Underlying Equity Security Trades ("ASU 2010-13")
ASU 2010-13, issued in April 2010, addresses the classification of a share-based payment award with an exercise price denominated in the currency of a market in which the underlying equity security trades. Topic 718,Compensation — Stock Compensation, is amended to clarify that a share-based payment award with an exercise price denominated in the currency of a market in which a substantial portion of the entity's equity securities trades shall not be considered to contain a condition that is not a market, performance, or service condition. Therefore, such an award is not to be classified as a liability if it otherwise qualifies as equity classification. The amendments in ASU 2010-13 are effective for fiscal years, and interim periods within those fiscal years, beginning on or after December 15, 2010.
ASU 2010-17,Revenue Recognition — Milestone Method (Topic 605): Milestone Method of Revenue Recognition ("ASU 2010-17")
ASU 2010-17, issued in April 2010, provides guidance on defining a milestone under Topic 605 and determining when it may be appropriate to apply the milestone method of revenue recognition for research or development transactions.
AETERNA ZENTARIS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
As at December 31, 20102011 and December 31, 2009
2010 and for the years ended December 31, 2010, 20092011 and 2008
2010
(tabular amounts in thousands of US dollars, except share/option/warrant and
per share/option/warrant data and as otherwise noted)
25. DIFFERENCES BETWEEN CANADIAN AND US GAAP (Continued)
Consideration that is contingent on achievementReconciliation of a milestone in its entirety may be recognizedConsolidated Statement of Financial Position as revenue in the period in which the milestone is achieved only if the milestone is judged to meet certain criteria to be considered substantive. The amendments in ASU 2010-17 are effective on a prospective basis for milestones achieved in fiscal years, and interim periods within those years, beginning on or after June 15, 2010. Early adoption is permitted.
ASU 2010-28,Intangibles — Goodwill and Other (Topic 350): When to Perform Step 2 of the Goodwill Impairment Test for Reporting Units with Zero or Negative Carrying Amounts ("ASU 2010-28")
In December 2010, the FASB issued ASU 2010-28, which modifies goodwill impairment testing for reporting units with a zero or negative carrying amount. Under the new guidance, an entity must consider whether it is more likely than not that goodwill impairment exists for each reporting unit with a zero or negative carrying amount, and, if it is more likely than not that goodwill impairment exists, the second step of the goodwill impairment test in ASC Topic 350,Intangibles — Goodwill and Other: Goodwill, must be performed to measure the amount of goodwill impairment loss, if any. The amended guidance in ASU 2010-28 is effective for public entities for fiscal years, and for interim periods within those years, beginning after December 15, 2010, with early adoption prohibited.
ASU 2010-29,Business Combinations (Topic 805): Disclosure of Supplementary Pro Forma Information for Business Combinations ("ASU 2010-29")
In December 2010, the FASB issued ASU 2010-29, which clarifies how public entities disclose supplemental pro forma information for business combinations that occur during the current year. Under the amended guidance, a public entity that presents comparative financial statements must disclose the revenue and earnings of the combined entity as though the business combination(s) that occurred during the current year had occurred as of the beginning of the prior annual reporting period. ASU 2010-29 also amends Topic 805 to require public entities to provide a description of the nature and amount of any material, nonrecurring pro forma adjustments directly attributable to business combinations that are included in reported pro forma revenue and earnings. The amended guidance in ASU 2010-29 is effective prospectively for business combinations for which the acquisition date is on or after the beginning of the first annual reporting period beginning on or after December 15, 2010. Early adoption is permitted.
Other disclosures
Research and development tax credits
Under Canadian GAAP, all research and development tax credits are recorded as a reduction of costs in the consolidated statements of operations, while under US GAAP, non-refundable tax credits that reduce current income taxes payable are recorded in income taxes. Non-refundable tax credits of $nil were recognized in each of the three years endedat December 31, 2010 2009 and 2008.
However, aggregate future income tax assets related to the unrecognized tax credits for prior years totalled approximately $7,158,000 in 2010, $6,738,000 in 2009 and $5,742,000 in 2008 for US GAAP purposes, though a valuation allowance corresponding to the same amounts would have been established in each of these years.
Canadian GAAP accounts | Canadian GAAP balances | IFRS adjustments | IFRS reclassifications | IFRS balances | IFRS accounts | |||||||||||||
| $ | $ | $ | $ | |||||||||||||||
ASSETS | ASSETS | |||||||||||||||||
Current assets | Current assets | |||||||||||||||||
Cash and cash equivalents | 31,998 | - | - | 31,998 | Cash and cash equivalents | |||||||||||||
Short-term investment | 1,934 | - | - | 1,934 | Short-term investment | |||||||||||||
Accounts receivable | ||||||||||||||||||
Trade | 4,555 | - | 866 | 5,421 | Trade and other receivables | |||||||||||||
Other | 748 | - | (748) | - | ||||||||||||||
Income taxes | 118 | - | (118) | - | ||||||||||||||
Inventory | 3,311 | - | - | 3,311 | Inventory | |||||||||||||
Prepaid expenses and other current assets | 1,511 | (366) | - | 1,145 | Prepaid expenses and other current assets | |||||||||||||
|
|
|
|
|
|
|
| |||||||||||
| 44,175 | (366) | - | 43,809 | |||||||||||||||
Restricted cash | 827 | - | - | 827 | Restricted cash | |||||||||||||
Property, plant and equipment | 3,096 | - | - | 3,096 | Property, plant and equipment | |||||||||||||
Deferred charges and other long-term assets | 4,384 | (3,581) | - | 803 | Deferred charges and other non-current assets | |||||||||||||
Intangible assets | 14,478 | (11,179) | - | 3,299 | Identifiable intangible assets | |||||||||||||
Goodwill | 9,614 | - | - | 9,614 | Goodwill | |||||||||||||
|
|
|
|
|
|
|
| |||||||||||
| 76,574 | (15,126) | - | 61,448 | |||||||||||||||
|
|
|
|
|
|
|
| |||||||||||
LIABILITIES | LIABILITIES | |||||||||||||||||
Current liabilities | Current liabilities | |||||||||||||||||
Accounts payable and accrued liabilities | 9,382 | 878 | - | 10,260 | Payables and accrued liabilities | |||||||||||||
Deferred revenues | 4,045 | - | - | 4,045 | Current portion of deferred revenues | |||||||||||||
Current portion of long-term payable | 60 | - | - | 60 | Current portion of long-term payable | |||||||||||||
|
|
|
|
|
|
|
| |||||||||||
| 13,487 | 878 | - | 14,365 | |||||||||||||||
Deferred revenues | 39,052 | - | - | 39,052 | Deferred revenues | |||||||||||||
| - | 13,412 | - | 13,412 | Warrant liability | ||||||||||||||
Long-term payable | 90 | - | - | 90 | Long-term payable | |||||||||||||
Employee future benefits | 11,324 | 209 | - | 11,533 | Employee future benefits | |||||||||||||
Other long-term liability | 182 | 389 | - | 571 | Provision and other non-current liabilities | |||||||||||||
|
|
|
|
|
|
|
| |||||||||||
| 64,135 | 14,888 | - | 79,023 | |||||||||||||||
|
|
|
|
|
|
|
| |||||||||||
SHAREHOLDERS’ EQUITY | SHAREHOLDERS’ DEFICIENCY | |||||||||||||||||
Share capital | 60,149 | 751 | - | 60,900 | Share capital | |||||||||||||
Warrants | 9,493 | (9,493) | - | - | ||||||||||||||
Other capital | 80,785 | 306 | - | 81,091 | Other capital | |||||||||||||
Deficit | (150,756) | (9,811) | - | (160,567) | Deficit | |||||||||||||
Accumulated other comprehensive income | 12,768 | (11,767) | - | 1,001 | Accumulated other comprehensive income | |||||||||||||
|
|
|
|
|
|
|
| |||||||||||
| 12,439 | (30,014) | - | (17,575) | |||||||||||||||
|
|
|
|
|
|
|
| |||||||||||
| 76,574 | (15,126) | - | 61,448 | |||||||||||||||
|
|
|
|
|
|
|
| |||||||||||
AETERNA ZENTARIS INC.Aeterna Zentaris Inc.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)Notes to Consolidated Financial Statements
As at December 31, 20102011 and December 31, 2009
2010 and for the years ended December 31, 2010, 20092011 and 2008
2010
(tabular amounts in thousands of US dollars, except share/option/warrant and
per share/option/warrant data and as otherwise noted)
25. DIFFERENCES BETWEEN CANADIAN AND US GAAP (Continued)
Long-lived assets
Under US GAAP, long-lived assets by geographic region only consistReconciliation of property, plant and equipment which are detailed as follows:
| | As at December 31, | |||||||
|---|---|---|---|---|---|---|---|---|
| | 2010 | 2009 | ||||||
| | $ | $ | ||||||
Germany | 2,666 | 3,736 | ||||||
United States | 381 | 551 | ||||||
Canada | 49 | 71 | ||||||
| 3,096 | 4,358 | |||||||
Research and collaboration agreements
As partConsolidated Statement of its strategy to enhance development capabilities and to partially fund capital requirements, the Company has entered into research and development collaboration agreements with several pharmaceutical companies. Pursuant to these collaboration arrangements, the Company often receives upfront payments, license fees and milestone payments and has the potential to receive royalty payments in the future. Upfront payments are typically non-refundable, received upon the signature of an agreement, or shortly thereafter, and are amortized over the estimated corresponding research and development period. License fees typically are contractually obligated payments that the Company receives and uses to fund research and development activities over the term of the collaboration and include milestone payments, as well as contract services. Milestone payments are contingent payments that are made upon the achievement of specified milestones, such as at the time of selection of candidates for drug development, the commencement or termination of clinical trials or the receipt of regulatory approvals and achievement of a certain level of sales. If drugs are successfully developed and commercialized as a result of collaboration agreements, the Company will receive royalty payments based upon a percentage of net sales of those drugs developed under the collaboration. Finally, contract service fees relate to research and development activities performed by the Company on behalf of the counterparty to the related arrangement and for which the Company has the right to receive compensation.
Ardana
In 2002, the Company granted an exclusive license to Ardana for the development and commercialization of teverelix, an LHRH antagonist, for all therapeutic uses worldwide with the exception of Japan, Korea and Taiwan. On April 2, 2004, Ardana acquired full worldwide rights and was assigned the intellectual property rights relating to teverelix and the underlying microcrystalline suspension technology for the use thereof. As discussed in note 11, this agreement was terminated by the Company in light of Ardana's having entered into voluntary administration.
Revenues recognized under this agreementComprehensive Loss for the year ended December 31, 2008 were approximately $3,621,000, and corresponding research and development costs incurred under the agreement were approximately $61,000.
Keryx Biopharmaceuticals, Inc.2010
The Company is party
Canadian GAAP accounts | Canadian GAAP balances | IFRS adjustments | IFRS reclassifications | IFRS balances | IFRS accounts | |||||||||||||
| $ | $ | $ | $ | |||||||||||||||
Revenues | Revenues | |||||||||||||||||
Sales and royalties | 24,857 | - | - | 24,857 | Sales and royalties | |||||||||||||
License fees and other | 2,846 | - | - | 2,846 | License fees and other | |||||||||||||
|
|
|
|
|
|
|
| |||||||||||
| 27,703 | - | - | 27,703 | |||||||||||||||
|
|
|
|
|
|
|
| |||||||||||
Operating expenses | Operating expenses | |||||||||||||||||
Cost of sales, excluding depreciation and amortization | 18,700 | - | - | 18,700 | Cost of sales | |||||||||||||
Research and development costs, net of tax credits and grants | 19,859 | 1,398 | - | 21,257 | Research and development costs, net of refundable tax credits and grants | |||||||||||||
Selling, general and administrative expenses | 11,875 | 682 | (5) | 12,552 | Selling, general and administrative expenses | |||||||||||||
Depreciation and amortization | ||||||||||||||||||
Property, plant and equipment | 1,005 | (1,005) | - | - | ||||||||||||||
Intangible assets | 1,492 | (1,492) | - | - | ||||||||||||||
|
|
|
|
|
|
|
| |||||||||||
| 52,931 | (417) | (5) | 52,509 | |||||||||||||||
|
|
|
|
|
|
|
| |||||||||||
Loss from operations | (25,228) | 417 | 5 | (24,806) | Loss from operations | |||||||||||||
|
|
|
|
|
|
|
| |||||||||||
Other income | ||||||||||||||||||
Unrealized gain on held-for-trading financial instrument | 687 | - | (687) | - | ||||||||||||||
Interest income | 181 | - | 4,860 | 5,041 | Finance income | |||||||||||||
Foreign exchange gain | 1,170 | 1,091 | (2,261) | - | ||||||||||||||
Loss on disposal of equipment | (28) | - | 28 | - | Finance costs | |||||||||||||
| - | (6,741) | (1,945) | (8,686) | Net finance income (costs) | ||||||||||||||
|
|
|
|
|
|
|
| |||||||||||
| 2,010 | (5,650) | (5) | (3,645) | |||||||||||||||
|
|
|
|
|
|
|
| |||||||||||
Net loss | (23,218) | (5,233) | (28,451) | Net loss | ||||||||||||||
Other comprehensive income: | Other comprehensive income: | |||||||||||||||||
Foreign currency translation adjustments | 49 | 952 | - | 1,001 | Foreign currency translation adjustments | |||||||||||||
| - | 191 | - | 191 | Actuarial gain on defined benefit plans, net of tax | ||||||||||||||
|
|
|
|
|
|
|
| |||||||||||
Comprehensive loss | (23,169) | (4,090) | - | (27,259) | Comprehensive loss | |||||||||||||
|
|
|
|
|
|
|
| |||||||||||
Net loss per share | Net loss per share | |||||||||||||||||
Basic and diluted | (0.31) | (0.07) | - | (0.38) | Basic and diluted | |||||||||||||
|
|
|
|
|
|
|
| |||||||||||
Weighted average number of shares | Weighted average number of shares | |||||||||||||||||
Basic and diluted | 75,659,410 | - | - | 75,659,410 | Basic and diluted | |||||||||||||
|
|
|
|
|
|
|
| |||||||||||
Aeterna Zentaris Inc.
Notes to a license and collaboration agreement with Keryx Biopharmaceuticals, Inc. ("Keryx"). Under the terms of this agreement, Keryx undertakes, at its own cost, all development activities necessary to obtain regulatory and marketing approvals of perifosine, a signal transduction inhibitor, for all uses in the United States, Canada and Mexico. The agreement provides for, among other things, the availability of data generated by both parties free of charge. In September 2002, the Company received an upfront payment of approximately $500,000 and is eligible to receive payments of
Table of ContentsConsolidated Financial Statements
AETERNA ZENTARIS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
As at December 31, 20102011 and December 31, 2009
2010 and for the years ended December 31, 2010, 20092011 and 2008
2010
(tabular amounts in thousands of US dollars, except share/option/warrant and
per share/option/warrant data and as otherwise noted)
Reconciliation of Consolidated Statement of Financial Position as at January 1, 2010
25. DIFFERENCES BETWEEN CANADIAN AND US GAAP (Continued)
| Canadian GAAP accounts | Canadian GAAP balances | IFRS adjustments | IFRS reclassifications | IFRS balances | IFRS accounts | |||||||||||||
| $ | $ | $ | $ | |||||||||||||||
ASSETS | ASSETS | |||||||||||||||||
Current assets | Current assets | |||||||||||||||||
Cash and cash equivalents | 38,100 | - | - | 38,100 | Cash and cash equivalents | |||||||||||||
Accounts receivable | ||||||||||||||||||
Trade | 2,444 | - | 1,105 | 3,549 | Trade and other receivables | |||||||||||||
Other | 992 | - | (992) | - | ||||||||||||||
Income taxes | 113�� | - | (113) | - | ||||||||||||||
Inventory | 4,415 | - | - | 4,415 | Inventory | |||||||||||||
Prepaid expenses and other current assets | 2,949 | (542) | - | 2,407 | Prepaid expenses and other current assets | |||||||||||||
|
|
|
|
|
|
|
| |||||||||||
| 49,013 | (542) | - | 48,471 | |||||||||||||||
Restricted cash | 878 | - | - | 878 | Restricted cash | |||||||||||||
Property, plant and equipment | 4,358 | - | - | 4,358 | Property, plant and equipment | |||||||||||||
Deferred charges and other long-term assets | 4,733 | (4,205) | - | 528 | Deferred charges and other non-current assets | |||||||||||||
Intangible assets | 17,034 | (12,907) | - | 4,127 | Identifiable intangible assets | |||||||||||||
Goodwill | 10,246 | - | - | 10,246 | Goodwill | |||||||||||||
|
|
|
|
|
|
|
| |||||||||||
| 86,262 | (17,654) | - | 68,608 | |||||||||||||||
|
|
|
|
|
|
|
| |||||||||||
LIABILITIES | LIABILITIES | |||||||||||||||||
Current liabilities | Current liabilities | |||||||||||||||||
Accounts payable and accrued liabilities | 11,919 | (74) | - | 11,845 | Payables and accrued liabilities | |||||||||||||
Deferred revenues | 6,327 | - | - | 6,327 | Current portion of deferred revenues | |||||||||||||
Income taxes | 965 | - | - | 965 | Income taxes | |||||||||||||
Current portion of long-term payable | 57 | - | - | 57 | Current portion of long-term payable | |||||||||||||
|
|
|
|
|
|
|
| |||||||||||
| 19,268 | (74) | - | 19,194 | |||||||||||||||
Deferred revenues | 45,919 | - | - | 45,919 | Deferred revenues | |||||||||||||
| - | 1,664 | - | 1,664 | Warrant liability | ||||||||||||||
Long-term payable | 143 | - | - | 143 | Long-term payable | |||||||||||||
Employee future benefits | 11,640 | 381 | - | 12,021 | Employee future benefits | |||||||||||||
Other long-term liability | 66 | 441 | - | 507 | Provision and other non-current liability | |||||||||||||
|
|
|
|
|
|
|
| |||||||||||
| 77,036 | 2,412 | - | 79,448 | |||||||||||||||
|
|
|
|
|
|
|
| |||||||||||
SHAREHOLDERS’ EQUITY | SHAREHOLDERS’ DEFICIENCY | |||||||||||||||||
Share capital | 41,203 | 321 | - | 41,524 | Share capital | |||||||||||||
Warrants | 2,899 | (2,899) | - | - | ||||||||||||||
Other capital | 79,943 | - | - | 79,943 | Other capital | |||||||||||||
Deficit | (127,538) | (4,769) | - | (132,307) | Deficit | |||||||||||||
Accumulated other comprehensive income | 12,719 | (12,719) | - | - | ||||||||||||||
|
|
|
|
|
|
|
| |||||||||||
| 9,226 | (20,066) | - | (10,840) | |||||||||||||||
|
|
|
|
|
|
|
| |||||||||||
| 86,262 | (17,654) | - | 68,608 | |||||||||||||||
|
|
|
|
|
|
|
| |||||||||||
up to an aggregate of $18,300,000 upon Keryx's successful achievement of clinical development and regulatory milestones, in addition to scale-up royalties (from high single to low double-digit) on future net sales in the United States, Canada and Mexico.
Revenues recognized under the agreement with Keryx for the years ended December 31, 2010, 2009 and 2008 were approximately $521,000, $128,000 and $410,000, respectively.
Corresponding research and development costs incurred under the agreement for the years ended December 31, 2010, 2009 and 2008 were approximately $1,334,000, $154,000, and $448,000, respectively.
Nippon Kayaku Co. Ltd.Aeterna Zentaris Inc.
In 2006, the Company entered into a licensing and collaboration agreement with Nippon Kayaku Co. Ltd. ("Nippon Kayaku"). Under the terms of the agreement, Nippon Kayaku was granted an exclusive licenseNotes to develop and market ozarelix, an LHRH antagonist, for all potential oncological indications in Japan. In return, the Company received approximately $1,900,000 as an upfront payment upon signature. The agreement provides for, among other things, the availability of data generated by both parties free of charge. The Company is entitled to receive payments of up to an aggregate of approximately $23,800,000 upon Nippon Kayaku's successful achievement of clinical development, regulatory milestones and a certain level of sales, in addition to low double-digit royalties on potential net sales. In turn, as indicated below regarding the related agreement, Spectrum is entitled to receive fifty percent of any upfront, milestone payments and royalties received from any research and collaboration agreement signed by the Company for the development and commercialization of ozarelix in Japan.Consolidated Financial Statements
Revenues recognized under the agreement for the years ended December 31, 2010, 2009 and 2008 were $nil, approximately $882,000 and $445,000 respectively. Corresponding research and development costs incurred under the agreement for the years ended December 31, 2010, 2009 and 2008 were $nil, approximately $397,000 and $nil, respectively.
Shionogi and Co.
In 1995, the Company entered into a research and collaboration agreement with Shionogi and Co. ("Shionogi"). The Company granted Shionogi a license to develop, use, commercialize and manufacture cetrorelix in Japan and for all human indications. Under the agreement, Shionogi is responsible, at its own cost, for all activities necessary to obtain regulatory and marketing approvals for cetrorelix. The agreement provides, among other things, availability of data generated by both parties free of charge. Upon signature of this agreement, the Company received approximately $1,400,000 as an upfront payment and was eligible to receive milestone payments of up to an aggregate of approximately $7,100,000 upon Shionogi's successful achievement of clinical development and regulatory milestones. To date, the Company has received approximately $5,800,000 of these milestone payments. Since the development of cetrorelix is completed inin vitro fertilization ("IVF"), Control Ovarian Stimulation ("COS") and Assisted Reproductive Technology ("ART") in Japan, and given the other events related to cetrorelix in BPH, discussed in notes 4 and 11, the Company does not expect to receive any additional milestone payments or any other development revenues under this agreement.
Revenues recognized under the agreement with Shionogi for the years ended December 31, 2010, 2009 and 2008 were approximately $86,000, $2,262,000, and $1,000, respectively.
Corresponding research and development costs incurred under the agreement for the years ended December 31, 2010, 2009 and 2008 were approximately $11,000, $838,000, and $13,000, respectively.
Spectrum
In 2004, the Company entered into a licensing and collaboration agreement with Spectrum for ozarelix, an LHRH antagonist. Under the terms of the agreement, the Company granted Spectrum an exclusive license to develop and commercialize ozarelix for all potential indications in North America and India. The agreement provided, among other
AETERNA ZENTARIS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
As at December 31, 20102011 and December 31, 2009
2010 and for the years ended December 31, 2010, 20092011 and 2008
2010
(tabular amounts in thousands of US dollars, except share/option/warrant and
per share/option/warrant data and as otherwise noted)
25. DIFFERENCES BETWEEN CANADIAN AND US GAAP (Continued)
| 30 | Subsequent events |
things, availability of data generated by both parties free of charge. Upon signature of this agreement,On January 23, 2012, the Company, received approximately $2,400,000 as an upfront payment, ofpursuant to its existing ATM sales agreement dated June 29, 2011, with MLV, was commencing a new ATM issuance program (“January 2012 ATM Sales Agreement”) under which approximately $1,200,000 was paid in cash andit may, at its discretion, from time to time during the balance paid through the issuance of sharesterm of the capitalsales agreement, sell up to a maximum of Spectrum.
As discussed in note 11, on January 27, 2010, Spectrum announced that it had terminated its development program with ozarelix in BPH. Also as discussed in note 11, management determined that ozarelix was fully impaired, and, consequently, all remaining deferred revenues related to the use of ozarelix, including those deferred revenues related to the agreement with Spectrum, were fully recognized in the Company's 2009 consolidated statement of operations.
In November 2010, the aforementioned agreement with Spectrum was amended. Under the terms of the amended agreement, Spectrum is entitled to use the Company's patent rights and know-how to develop, use, make, have made, sell, offer for sale, have sold, import and export, commercialize ozarelix in all worldwide territories except Japan, Korea, Indonesia, Malaysia, the Philippines and Singapore. Under the terms of the amended agreement, Spectrum granted, as further consideration, 326,956 shares10,400,000 of its common stock, withshares through ATM issuances on the NASDAQ up to an equivalent fair valueaggregate amount of approximately $1,263,000, as an upfront nonrefundable license fee payment to the Company.$16,000,000.
Also per the amended agreement,From January 23, 2012 through March 15, 2012, the Company will be entitled to receiveissued a total of approximately $22,765,000 in cash payments, as well as approximately $670,000 of Spectrum's3,565,470 common stock, upon achieving certain regulatory milestones in various markets. Furthermore, the Company will be entitled to receive royalties (scale-up royalties from high single to low double-digit) on future net sales of ozarelix products in the named territories.
Revenues recognizedshares under the agreements with SpectrumJanuary 2012 ATM Sales Agreement for the years ended December 31, 2010, 2009aggregate gross proceeds of $6,397,045, less cash transaction costs of $191,911 and 2008 were approximately $1,219,000, $860,000 and $678,000, respectively. Corresponding research and developmentpreviously deferred transaction costs incurred under the agreements for the years ended December 31, 2010, 2009 and 2008 were approximately $74,000, $109,000 and $255,000, respectively.of $56,000.
| Item 19. | Exhibits |
TulaneExhibit Index
In 2002, the Company signed license agreements with Tulane with regard to various compounds, including cetrorelix. Under the agreements, the Company obtained exclusive worldwide licenses to use Tulane's patents to develop, manufacture, market and distribute these compounds.
The agreements provide for the payment by the Company of single-digit royalties on future worldwide net sales for all indications except BPH, for which the payment of low single-digit royalties would be required. Tulane is entitled to receive a low double-digit royalty on any lump sum, periodic or other cash payments received by the Company from sub-licensees.
Costs incurred under the agreements with Tulane for the years ended December 31, 2010, 2009 and 2008 were approximately $2,249,000, $4,703,000 and $2,030,000, respectively. The expense recognized in 2009 predominantly includes the royalty paid by the Company in connection with the sanofi Agreement, which was subsequently terminated, as discussed in note 4.
| 1.1 | Restated Certificate of Incorporation and Restated Articles of Incorporation of the Registrant (incorporated by reference to Exhibit | |
1.2 | By-Law One adopted by the | |
2 | Amended and Restated Shareholder Rights Plan Agreement between the Registrant and Computershare Trust Company of Canada dated as at March 29, 2010 (incorporated by reference to Exhibit 99.1 to the | |
4.1 | Stock Option Plan of the Registrant (incorporated by reference to Exhibit 4.1 of the | |
4.2 | Employment Agreement dated July 18, 2007 between Paul Blake, M.D. and the Registrant (incorporated by reference to Exhibit 4.2 of the | |
4.3 | Service Contract dated December 5, 2007 between Aeterna Zentaris GmbH and Prof. Juergen Engel, Ph.D. Consent of the | |
4.4 | Amendment #1 to Service Contract dated September 1, 2008 between Aeterna Zentaris GmbH and Prof. Juergen Engel, Ph.D. (incorporated by reference to Exhibit 4.4 of the | |
4.5 | Amendment #2 to Service Contract dated August 30, 2010 between Aeterna Zentaris GmbH and Prof. Juergen Engel, Ph.D. (incorporated by reference to Exhibit 4.5 of the Registrant’s annual report on Form 20-F for the financial year ended December 31, 2010 filed with the Commission on March 31, 2011) | |
4.6 | Employment Agreement dated September 1, 2008 between the Registrant and Prof. Juergen Engel, Ph.D. (incorporated by reference to Exhibit 4.5 of the | |
4.7 | Employment Agreement dated May 7, 2007 between the Registrant and Nicholas J. Pelliccione (incorporated by reference to Exhibit 4.7 of the | |
4.8 | Service Contract dated May 18, 2006 among Aeterna Zentaris GmbH, the Registrant and Matthias Seeber (incorporated by reference to Exhibit 4.7 of the | |
4.9 | Amendment #1 to Service Contract dated December 9, 2008 among Aeterna Zentaris GmbH, the Registrant and Matthias Seeber (incorporated by reference to Exhibit 4.8 of the |
| 4.10 | Amendment to Amended Employment Agreement dated as at June 20, 2007 among the Registrant, Aeterna Zentaris, Inc. and Dennis Turpin (incorporated by reference to Exhibit 4.8 of the | |
Purchase Agreement by and among Aeterna Zentaris IVF GmbH, Aeterna Zentaris GmbH, the Registrant and Cowen Healthcare Royalty Partners L.P. dated November 11, 2008 (incorporated by reference to the | ||
License and Cooperation Agreement for Perifosine by and between Zentaris AG and AOI Pharma, Inc. dated September 18, 2002 (incorporated by reference to Exhibit 99.1 to the |
Addendum agreement to License and Cooperation Agreement for Perifosine by and between Zentaris AG and AOI Pharma, Inc. dated December 3, 2003 (incorporated by reference to Exhibit 99.2 to the | |||
First Amendment to License and Cooperation Agreement for perifosine by and between Aeterna Zentaris GmbH and AOI Pharma Inc., dated November 29, 2007 (incorporated by reference to Exhibit 99.3 to the | |||
| Development, Commercialization and Licensing Agreement for perifosine by and between Aeterna Zentaris GmbH and Yakult Honsha Co., Ltd dated March 8, 2011 (incorporated by reference to Exhibit 99.1 to the | |||
8.1 | Subsidiaries of the Registrant | ||
11.1 | Code of Ethical Conduct of the Registrant (incorporated by reference to Exhibit 11.1 of the | ||
11.2 | Audit Committee Charter of the Registrant (incorporated by reference to Exhibit 11.2 of the Registrant’s annual report on Form 20-F for the financial year ended December 31, 2010 filed with the Commission on March 31, 2011) | ||
12.1 | Certification of the Principal Executive Officer pursuant to §302 of the Sarbanes-Oxley Act of 2002 | ||
12.2 | Certification of the Principal Financial Officer pursuant to §302 of the Sarbanes-Oxley Act of 2002 | ||
13.1 | Certification of the Principal Executive Officer pursuant to 18 U.S.C. Section 1350 as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | ||
13.2 | Certification of the Principal Financial Officer pursuant to 18 U.S.C. Section 1350 as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | ||
15.1 | Consent of the Independent Auditors |
| † | Confidential treatment has been granted for certain portions of this exhibit, which portions have been omitted and filed separately with the U.S. Securities and Exchange Commission. |
| †† | Confidential treatment has been requested for certain portions of this exhibit, which portions have been omitted and filed separately with the U.S. Securities and Exchange Commission. |
Table of ContentsSIGNATURES
The registrant hereby certifies that it meets all of the requirements for filing on Form 20-F and that it has duly caused and authorized the undersigned to sign this annual report on its behalf.
| AETERNA ZENTARIS INC. | ||
/s/ Dennis Turpin | ||
Dennis Turpin, CA | ||
Senior Vice President and Chief Financial Officer | ||
|
Date: March 27, 2012