| ○ | Surgical debridement predominantly includes tangential excision, a procedure in which a surgeon amputates the entire dead tissue mass, layer after layer, down to healthy, viable tissue. The excision is extended into healthy intact tissue to make sure that no trace of the eschar remains, resulting in up to an estimated 30-50% of healthy tissue being excised during this procedure. Other methods include dermabrasion, in which a mechanically powered, hand-held rotating abrading cylinder is used to slowly scrape off tissue, and hydro surgery, in which a high-pressure flow of water abrades the tissue. These alternative methods have attempted to limit the trauma associated with tangential excision, but entail spray of contaminated eschar or take a significantly longer time to complete than tangential excision. |
| ○ | The benefits of surgical eschar removal are that it is usually fast and effective. Disadvantages include the significant trauma of the procedure, associated blood loss, risk of surgery in delicate areas of the body such as hands, added costs, and, most importantly, the loss of viable tissue that necessitates additional surgical procedures for harvesting skin from healthy donor sites and autografting. |
| ○ | Due to the disadvantages of surgery in extensive burns some surgeons limit their debriding surgery to only a part of the affected area in a single session (15-30% TBSA in most centers), thus delaying full debridement by days. After several days, complications related to eschar contamination may begin and some of the benefits of the earlier debridement may not be realized. On the other hand, when excising burns immediately, all suspected necrotic tissue will be excised, inevitably resulting in over-excision, especially in “indeterminate” burns, as after surgical excision, the remaining skin often no longer has any spontaneous healing potential and will heal only by autografting. |
| • | Non-surgical debridement |
| ○ | Non-surgical debridement includes many different treatment options that do not require direct surgical removal of the skin to remove eschar. With non-surgical debridement, the eschar is naturally, but slowly, removed by contaminant microorganisms, tissue autolysis, or self-decomposition, and the inflammatory process that may lead to serious local and systemic complications. In seeking to facilitate such natural processes, topical medication, anti-microbial agents, enzymes and biological/chemical applications are often applied onto the eschar. |
| ○ | The benefits of this approach are that it is non-surgical, reduces trauma to the patient and is easier to apply. Disadvantages include numerous dressing changes and mechanical scraping with limited debridement efficacy. This prolongs the eschar removal process, which may lead to death of the tissue surrounding the initial burn wound, causing partial-thickness wounds to transform into full-thickness wounds and forming granulation tissue that may develop into heavy scars. |
As demonstrated in our clinical trials, NexoBrid combines the advantages of surgical and non-surgical debridement modalities by providing rapid and effective eschar removal while not harming viable tissues.tissue. This allows for earlier direct visual assessment of the burn wound in order to formulate proper treatment.
Market Opportunity Severe burns require specialized care in hospitals or burn centers. Approximately 100,000160,000 patients with severe burns are hospitalized every year in the United States and Europe. The prevalence of patients with severe burns is even higher in emerging economies. For example, approximately 400,000 patients are hospitalized every year with burns in India according to a study conducted by IMS Health. The severe burn patients are predominantly treated by specialists in approximately 250 burn centers in Europe and the United States, as well as at burn units of large hospitals in Europe. We believe these patients can benefit from NexoBrid’s effective and selective, non-surgical eschar removal. In addition to our current marketing of NexoBrid in Europe and the United States, we have signed local distribution agreements for distribution of NexoBrid in Europe, Latin America, certain Asia-Pacific countries, members of the Commonwealth of Independent States (“CIS”), and the Middle East and we plan to target additional markets in these territories by leveraging our approved registration file for additional regional marketing authorizations. In addition to the market opportunities for NexoBrid discussed above, we believe that NexoBrid has the potential to play a critical role in the event of a burn mass casualty incident (“MCI”BMCI”), which is generally defined as any incident in which emergency medical services resources, such as personnel and equipment, are overwhelmed by the number and severity of casualties. A variety of public emergencies may give rise to an MCI,a BMCI, such as terrorist attacks, natural disasters, fires and explosions. One example of an MCIa BMCI is a mass burn casualty disaster, which is defined by the American Burn Association as a catastrophic event in which the number of burn victims exceeds the capacity of the local burn center to provide optimal care. If a significant number of burn victims arrive at a burn center following an event, some victims may go untreated until the bottleneck is resolved. The use of non-surgical means that are capable of providing rapid eschar removal without harming healthy tissues,tissue, particularly during public health emergencies, could potentially reduce the time, labor and resource burdens associated with the current standard-of-care, thereby enabling the treatment of more patients. In the event of a mass burn casualty disaster, healthcare professionals can use NexoBrid to begin treatment at the patient’s bedside without the need for a surgical team and facilities. NexoBrid has demonstrated in clinical studies, with statistical significance, its ability to non-surgically and rapidly remove eschar in a single four-hour application. Once the acute treatment has been completed, the wound can be covered with available means and further managed once the MCIBMCI is under control and the bottlenecks resolved. NexoBrid has been recognized by BARDA as a medical countermeasure for treatment of burns in the event of a MCI.BMCI. Additionally, during the war in Israel, the entire non-U.S. NexoBrid inventory has been deployed to hospitals in Israel and the military, to effectively treat those affected by the war. BARDA Contracts In In September 2015, we were awarded the First BARDA Contract for treatment of thermal burn injuries, whichinjuries. This contract was valued atamended several times over the years to extend its term until September 2024 (the Company is pursuing an extension of the contract) and its total value, up to $112 million. In July 2017 and in May 2019, BARDA expanded its commitment by an aggregate supplementala total amount of $41 million.$165 million as of the end of 2022. In March 2020, BARDA further expanded its commitment by additional $5.5 million to support emergency readiness for NexoBrid deployment upon request of use of NexoBrid in mass casualty situations and in February 2022May 2023 BARDA expanded its awarded contract by providing supplemental funding of $9$10 million to support a $3 million replenishment of expired product previously procured for emergency preparedness, the NexoBrid BLA resubmissionpediatric indication sBLA submission to the FDA, and enrollment of an additional 50 patients in the continuous expanded access program (collectively the "First BARDA Contract")treatment protocol (NEXT). The First BARDA Contract is our primary contract with BARDAprovided funding and relates totechnical support for the advancement ofpivotal U.S. Phase 3 clinical study (DETECT), the development and manufacturing,randomized, controlled pivotal clinical trial for use in the pediatric population (CIDS), the marketing approval registration process for NexoBrid as well as its procurement and availability under the procurement of NexoBrid as a medical countermeasure as part ofexpanded access treatment protocol (NEXT) in the U.S. preparedness for mass casualty events. UnderThe total amount of the First BARDA Contract BARDA provided technical assistance and a totalis comprised of up to $91$110 million to support research and development activities and up to $65 million to procure NexoBrid for U.S. emergency preparedness. As of December 31, 2023, the Company has received approximately $88 million in funding for NexoBrid development activities required to achieve U.S. marketing approval fromin the FDA. These activities includeaggregate, under the NexoBrid Phase 3 (DETECT) studyFirst BARDA Contract, and subsequent requirements for BLA submission, the ongoing Phase 3 pediatric (CIDS) study and the NexoBrid expanded access treatment protocol (NEXT). In January 2020, BARDA committed an additional $16.5 million to procurefor procurement of NexoBrid as part of the HHS mission to build national preparedness for public health medical emergencies. The contract further includes a $10 million option to fund development of other potential NexoBrid indications and an option to procure additional NexoBrid valued at up to $50 million.U.S. emergency preparedness. The first BARDA Contract may be terminated by BARDA at any time at BARDA’s discretion. In September 2018, we were awarded the secondan additional, separate BARDA contract (the "Second“Second BARDA Contract"Contract”), which is an additional, separate contract to develop NexoBrid for the treatment of Sulfur Mustard injuries as part of BARDA’s preparedness for mass casualty events. The Second BARDA Contract provides approximately $12 million of funding to support research and development activities up to pivotal studies in animals under the U.S. FDA Animal Rule, and contains options for BARDA to provide additional funding of up to $31$29 million for additional development activities, animal pivotal studies, and the BLA submission for licensure of NexoBrid for the treatment of Sulfur Mustardsulfur mustard injuries. The Second BARDA Contract was expired in 2023. As of December 31, 2021,2023, the Company has received approximately $70$4.4 million of funding from the Second BARDA contract. DOD and MTEC contracts In February, 2023, the Company was entered in funding into a contract with the aggregate, from BARDA underU.S. Department of Defense (DoD), through the two contracts,Medical Technology Enterprise Consortium (MTEC), for development and an additional $14.6 million for procurementproduction of a new, temperature-stable formulation of NexoBrid, positioning it as the first-line non-surgical solution for U.S. emergency preparedness.treating severe burn injuries in pre-hospital settings. The contract provides funding up to $2.7 million. Each BARDADuring 2023, the DOD through MTEC and directly through MTEC awarded the Company additional funding of $10.3 million to advance this development of a new temperature stable formulation of NexoBrid.
The MTEC contract may be terminated by BARDAMTEC at any time at BARDA’sMTEC’s discretion. NexoBrid Clinical History NexoBrid, our innovative biopharmaceutical product, has received marketing authorizationauthorizations from the EMAU.S., European Commission and the Israeli, Argentinean, South Korean, Russian, Peruvian, Chilean, Taiwanese, Ukrainian, other Eurasian states, and United Arab Emirates, Japanese and Indian Ministries of Health for the removal of eschar in adults with deep partial- and full-thickness thermal burns. The active ingredient of NexoBrid is a concentrate of proteolytic enzymes enriched in bromelain extracted from the pineapple stems. Proteolysis is a breakdown of proteins into smaller building blocks, polypeptides or amino acids. Our research and development strategy is centered around our validated proteolytic enzyme platform technology, focused on next-generation bio-active therapies for burn and wound care and biological medicinal products for tissue repair. OurFor each indication, our research and development team further developeddevelops and optimizedoptimizes our enzymatic platform technology, creating unique and differentiated products meeting separate needs based on the specific indication, which is the basis for NexoBrid, EscharEx and all other pipeline product candidates. One vial of NexoBrid containing 2 grams of concentrate of proteolytic enzymes enriched in bromelain is sufficient for treating a burn wound area of 1% total body surface area (TBSA)(“TBSA”). We developed NexoBrid to fulfill the previously unmet need for a non-surgical effective and selective debriding agent that combines the efficacy and speed of surgery with the non-invasiveness of non-surgical methods. NexoBrid enhances the ability of physicians to conduct an earlier direct visual assessment of the burn depth to reach an informed decision on further treatment as well as to reduce the surgical burden and achieve a favorable long-term patient outcome. NexoBrid has been investigated in hundreds of patients across 22 countries and four continents in nineten completed Phase 2II and Phase 3III and post-marketing clinical studies. While we are marketing our product for the removal of eschar in burn wounds under the name “NexoBrid,” in clinical trials the product has been referred to as “Debridase” and “Debrase.”
The following table sets forth information regarding the completed clinical trials of NexoBrid:
| | Trial 1 | Trial 2 | Trial 3 | Trial 4 | Trial 5 | Trial 6 | Trial 7 | Trial 8 | Trial 9 | Trial 1 | Trial 2 | Trial 3 | Trial 4 | Trial 5 | Trial 6 | Trial 7 | Trial 8 | Trial 9 | Trial 10 | | Study Type | Retrospective Phase 2 Investigator initiated | Dose range Phase 2 | Prospective Phase 2 IND/FDA | Phase 2 IND/FDA | Phase 3 EMA | Phase 3b EMA | Phase 2 EMA | Post approval safety study EMA | Phase 3 IND/FDA | Retrospective Phase II Investigator initiated | Dose range Phase II | Prospective Phase II IND/FDA | Phase II IND/FDA | Phase III EMA | Phase IIIb EMA | Phase II EMA | Post approval safety study EMA | Phase III IND/FDA | Phase III IND/FDA | | Design | Data collected from files of patients treated with NexoBrid | Parallel, controlled, observer-
blind, randomized, single-center | Parallel, controlled, observer-
blind, three-arm, randomized, multi-center | Parallel, controlled, open label, three-arm, randomized, single-center | Parallel, controlled, open label, two-arm, randomized, multi-center | Parallel, controlled, blinded, two-arm, multi-center | Open label,
single-arm,
multi-center | Observational retrospective data collection | Parallel, controlled, open label, three-arm, randomized, multi-center | Data collected from files of patients treated with NexoBrid | Parallel, controlled, observer-
blind, randomized, single-center | Parallel, controlled, observer-
blind, three-arm, randomized, multi-center | Parallel, controlled, open label, three-arm, randomized, single-center | Parallel, controlled, open label, two-arm, randomized, multi-center | Parallel, controlled, blinded, two-arm, multi-center | Open label,
single-arm,
multi-center | Observational retrospective data collection | Parallel, controlled, open label, three-arm, randomized, multi-center | multicenter, multinational, randomized, controlled, open-label study in children | | Main Objectives | Safety and efficacy | Comparison of efficacy and safety | Safety and efficacy | Safety | Safety Efficacy | Long-term scar assessment Quality of life | Safety and pharmacokinetics Efficacy | Effectiveness of the risk minimization activities | Safety Efficacy | Safety and efficacy | Comparison of efficacy and safety | Safety and efficacy | Safety | Safety Efficacy | Long-term scar assessment Quality of life | Safety and pharmacokinetics Efficacy | Effectiveness of the risk minimization activities | Safety Efficacy | Safety Efficacy | | Wound Types | Deep partial/full thickness thermal burns | Deep partial /full thickness thermal burns | Deep partial /full thickness thermal burns | Deep partial /full thickness thermal burns | Deep partial/ full thickness thermal burns | Scar formation | Deep partial/full thickness thermal burns | Burns which were treated with NexoBrid in the market | Deep partial/ full thickness thermal burns | Deep partial/full thickness thermal burns | Deep partial /full thickness thermal burns | Deep partial /full thickness thermal burns | Deep partial /full thickness thermal burns | Deep partial/ full thickness thermal burns | Scar formation | Deep partial/full thickness thermal burns | Burns which were treated with NexoBrid in the market | Deep partial/ full thickness thermal burns | Deep partial/ full thickness thermal burns | | Number of Patients | 154 | 20 | 140 | 30 | 182 | 89 | 36 | 160 | 175 | 154 | 20 | 140 | 30 | 182 | 89 | 36 | 160 | 175 | 145 | | Study Length | 1985-2000 | 2002-2005 | 2003-2004 | 2006-2007 | 2006-2009 | 2011 | 2009-2015 | 2017-2019 | 2015-2020 | 1985-2000 | 2002-2005 | 2003-2004 | 2006-2007 | 2006-2009 | 2011 | 2009-2015 | 2017-2019 | 2015-2020 | 2015-2023 | | Location | Israel | Israel | International | United States | International | International | Europe | International | Israel | Israel | International | United States | International | International | International | Europe | International | International |
Recent completedCompleted clinical trials
U.S. Phase 3 Study – DETECT study (Trial 9)
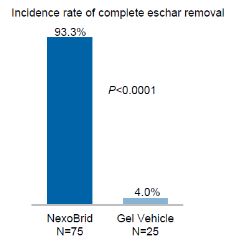
The DETECT study is a prospective, multicenter, multinational, randomized, controlled, assessor blinded Phase 3 study, performed in subjects with thermal burns, to evaluate the efficacy and safety of NexoBrid compared to Gel Vehicle and compared to SOC in 175 hospitalized patients with severe burns of up to 30% TBSA randomized in a 3:1:3 ratio, with 12-month and 24-month follow-ups. The study involved 44 burn centers. The study objectives were to evaluate the efficacy and safety of NexoBrid by removing burn eschar earlier and reducing surgical burden and related blood loss in hospitalized patients with severe burns. Complete eschar removal was the primary endpoint of the study and was tested against the Gel Vehicle control arm. The primary analysis was based on whether complete eschar removal was achieved in all target wounds of a patient. The analysis compared all randomized patients to the NexoBrid arm to all randomized patients to the Gel Vehicle control arm. Secondary endpoints included reduction in the need for surgical eschar removal (surgical burden), earlier eschar removal, and blood loss, which were tested against the SOC control arm. All secondary endpoints were analyzed and compared all patients randomized to the NexoBrid arm to all patients randomized to the SOC control arm. The study met its primary endpoint with statistical significance. Patients treated with NexoBrid demonstrated a significantly higher incidence of complete eschar removal compared with patients treated with the Gel Vehicle (NexoBrid: 93.3% (70/75) vs. Gel Vehicle: 4.0% (1/25), p<0.00011).
 The study included secondary endpoints that were all met with statistical significance and provided further insight on several efficacy parameters: (i) Patients treated with NexoBrid demonstrated shorter time to achieve complete eschar removal compared with patients treated with SOC (median time - NexoBrid: 1 day vs. SOC: 3.8 days, p<0.00012); (ii) Patients treated with NexoBrid demonstrated a significantly lower incidence of surgical eschar removal compared with patients treated with SOC (NexoBrid: 4.0% (3/75) vs. SOC: 72.0% (54/75), p<0.00013); (iii) and Patients treated with NexoBrid incurred significantly lower blood loss during the eschar removal procedure compared with patients treated with SOC (mean volume – NexoBrid: 14.2 ml vs. SOC: 814.5 ml, p<0.00014). In addition, Patients treated with NexoBrid had a non-inferior time to complete wound closure compared with patients treated with SOC (p=0.00035). The study Data Safety Monitoring Board ("DSMB") concluded after all patients had been treated that the overall safety profile of NexoBrid in the study is consistent with the safety data known from previous studies.
1 Fisher's exact test
2 Generalized Wilcoxon-Gehan test
3 Logistic regression model - Wald test
4 Wilcoxon test pooled using Rubin's rules
5 Accelerated failure time model
* Kaplan-Meier analysis
The twelve- and twenty four-month patients’ follow-up safety data of cosmesis, function and quality of life were found to be comparable across all study arms, and no new safety signals were observed.
The study also serves to address our post approval commitment to EMA. This study is funded by BARDA. See “—BARDA Contracts” above.
Ongoing clinical trialstrails
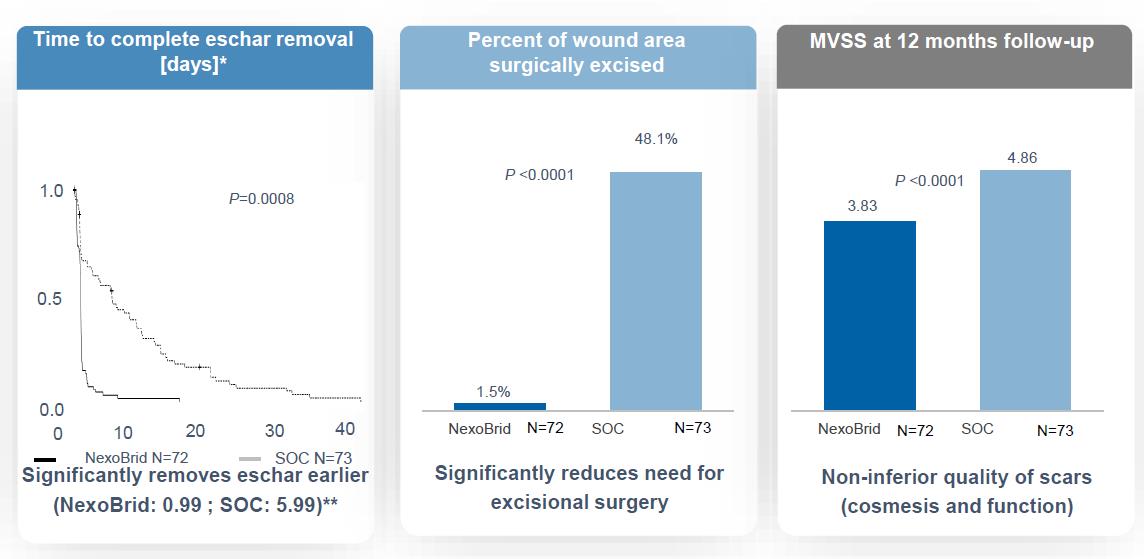
Pediatric investigational plan –- CIDS study The CIDS study iswas a Phase 3,III, multicenter, multinational, randomized, controlled, open-label study in children with thermal burns. The study objectives arewere to evaluate the efficacy and safety of treatment with NexoBrid compared with SOC in hospitalized children with severe thermal burns of 1% to 30% total body surface area (TBSA).TBSA. We expanded this study also to United States burn centers, following approval of the study protocol by the FDA. The study is underwaywas conducted in accordance with a study design endorsed by the FDA and the EMA as part of the agreed Pediatric Investigational Plan (“PIP”) to support extension of the indication to pediatric patients. The CIDS study includesincluded pediatric patients of all ages, from newborn to eighteen years of age, offering NexoBrid to this important and sensitive group of patients. The primary endpoints evaluate early eschar removal, surgical burden, and cosmesis and function with a 12-month follow-up. The European Medicines Agency (“EMA”) endorsed the study design as part of the agreed-upon Pediatric Investigational Plan (“PIP”) to support the indication label expansion to include pediatric patients. The primary endpoints included early eschar removal, reduction of wound area surgically excised (surgical need) and non-inferiority cosmesis and function at twelve months follow-up from wound closure. Secondary endpoints included reduction in the need for surgical excision for eschar removal (surgical need), blood loss, reduction of the need for autograft in DPT wounds and non-inferiority in cosmesis and function at twenty-four months follow-up from wound closure. Additional extended long term cosmesis and function assessment at more than 30 months from wound closure was added to the protocol. Non-inferiority of the time to complete wound closure and other standard safety measurements were also compared with the SOC control arm.
The study was expanded to include burn centers in the United States following agreement with the FDA, under the same protocol with alignment to the U.S. phase 3Phase III study (DETECT) protocol for the adult population. The non-inferiority of cosmesis and function at twelve months and twenty-four months from wound closure were defined as safety measurements. In addition, reduction in surgical need was measured only by reduction in incidence of surgical excision for eschar removal. In July 2021, we announced positive top-line results, which include acute phase and 12-month follow-up data analysis. The study enrolled 145 pediatric patients, from newborn to eighteen years of age, randomized to either NexoBrid or SOC at a ratio of 1:1, across 36 burn centers worldwide. ThePhase III study met allits three primary endpoints with a high degree of statistical significance, as well as certain secondary endpoints.significance. NexoBrid demonstrated a significant reduction in the time to achieve complete eschar removal and a significant reduction in the wound area requiring surgical excision (surgical need) while demonstrating non-inferiority to standard-of-careSOC in quality of scars.scars as measured by MVSS. The study also met certain secondary endpoints showing statistically significant reduction in the incidence of surgical excision and a reduction in the need for autograft in deep partial thickness burns, as well as a favorable trend in the reduction of blood loss during the eschar removal process. In addition, the study showed thatconfirmed NexoBrid wasto be safe and well-tolerated. The long-term follow-upwell-tolerated for cosmesisall ages.
Based on these results, we received in December 2023 the European Commission approval for the removal of eschar in deep partial- and function, quality of lifefull-thickness thermal burns for all ages. Additionally, In September 2023, the Supplemental BLA for pediatric indication was accepted for review by the U.S. Food and safety measurements is ongoing, and data isDrug Administration (FDA), with a decision expected in the firstsecond half of 2023. This study is funded by BARDA. See “—BARDA Contracts” above.2024. 42Ongoing clinical trials
Expanded access treatment protocol (NEXT)(“NEXT”) The NEXT protocol, which we initiated in October 2019, is an open-label, single-arm treatment protocol which allows for the treatment of up to 200250 burn patients with deep partial- and full-thickness thermal burns up to 30 percent of total body surface area.30% TBSA. In September 2020, the FDA agreed to allow the NEXT protocol to be expanded to include pediatric as well as adult burn patients. The NEXT protocol is being funded by BARDA. See “—BARDA“BARDA Contracts” above. NEXT has been designed to be consistent with current real-life burn treatment practices in the U.S. and up to 30 U.S. burn centers are anticipated to participate. We received FDA concurrence that patients can be treated under the NEXT protocol in a burn MCI that is not a declared national emergency. We have provided documents for consideration byFollowing the FDA supporting the usecommercial availability of NexoBrid in a declared national medical emergency contingent upon the FDA issuance of an Emergency Use Authorization (EUA). The EUAU.S., NEXT is a mechanism by which the FDA can allow an unapproved medical product that qualifies as a mass casualty medical countermeasurescheduled to be used in a public health emergency.closed during the first half of 2024.
Wound Care Our second innovative product candidate, EscharEx, a phase III-ready biologic, is a bio-active therapeutic product under developmentmultimodal debridement therapy for debridementthe treatment of chronic wounds, with significant potential advantages over the $360 million dominant product and other hard-to-heal wounds. EscharEx is complementaryan opportunity to expand the large number of existing advanced wound healing therapies, which require a clean wound bed in order to heal the wound. EscharEx active substance (API)market. It is a concentrate of proteolytic enzymes enriched in bromelain and as such, benefits from the wealth of existing development data on NexoBrid. The mechanism of action offor topical, easy to use daily applications. In several Phase II trials, EscharEx is mediated by the proteolytic enzymes that cleaves and removes the necrotic tissue and prepare the wound bed for healing. In two Phase 2 studies that we conducted, EscharExwas shown to be well-tolerated, and demonstrated safetyits positive efficacy results in debridement and efficacypromotion of granulation tissue in the debridement of chronic and othervarious hard-to-heal wounds, inwith only a few daily applications. InEscharEx’s mechanism of action is mediated by proteolytic enzymes that may cleave to and remove the U.S, we are conducting a Phase 2 clinical study with the second generation EscharEx,necrotic tissue, preparing wound bed for the treatment of venous leg ulcers (VLUs). The study is built on the positive data from the completed Phase 2 study of the first-generation EscharEx. The study is designed to assess the safety and efficacy of EscharEx compared to gel vehicle (placebo control) and non-surgical standard-of-care (either enzymatic or autolytic debridement). Topline results announced in January 2022 demonstrated that the study met its primary endpoint, demonstrating that patients treated with EscharEx had a statistically significant higher incidence of complete debridement compared to the gel vehicle, with a p-value of 0.004.healing. Chronic and Other Hard-to-Heal Wounds The chronic and other hard-to-heal wound market consists of a broader addressable population of more than 14 million patients in Europe and the United States alone suffering from chronic wounds such as VLUs, Diabetic Foot Ulcers (DFUs),DFUs, pressure ulcers and additional patients suffering from surgical/traumatic hard-to-heal wounds. Chronic and other hard-to-heal wounds represent a $25 billion burden to the U.S. healthcare system. Chronic and hard-to-heal wounds are caused by impairment in the biochemical and cellular healing processes due to local or systemic conditions and generally can take several weeks to heal, if not longer. Such wounds can lead to significant morbidity, including pain, infection, impaired mobility, hospitalization, reduced productivity, amputation and mortality. In each of the various wound types, the presence of the eschar and/or other devitalized tissue is a frequent cause for “chronification” of wounds and the removal of escharthis non-viable material is the key step to commence healing. EscharThe non-viable material needs to be removed to prevent further deterioration of the wound that may result in additional adverse patient outcomes. If not effectively treated, these wounds can lead to potentially severe complications including further infection, osteomyelitis, fasciitis, amputation and mortality. Most advanced wound care therapies, including negative pressure wound therapy, such as V.A.C. Therapy, and skin substitutes such as Apligraf and Dermagraft and human amniotic tissue products, are complementary to our lead product candidate, EscharEx, as these products require a clean wound bed to effectively heal a wound. Four common chronic and other hard-to-heal wounds are:
| • | Venous leg ulcers. VLUs develop as a result of vascular insufficiency, or the inability for the vasculature of the leg to return blood back toward the heart properly. Based on our comprehensive market research study on EscharEx that involved more than 200 healthcare professionals in the U.S. and Europe, which was last updated in 2019,2022, the VLU overall prevalence is approximately 3.3 million (1% of total U.S. population). Furthermore, the annual incidence of VLUs in the U.S. alone, is approximately 960,000 (accounting for 45% recurrence), of which approximately 690,000 undergo debridement in a given year. These ulcers usually form on the sides of the lower leg, above the ankle and below the calf, and are slow to heal and often recur if preventative steps are not taken. The risk of VLUs can increase as a result of a blood clot forming in the deep veins of the legs, obesity, smoking, lack of physical activity or work that requires many hours of standing. |
| • | Diabetic foot ulcers. Diabetes can lead to a reduction in blood flow, which can cause patients to lose sensation in their feet and may prevent them from noticing injuries, sometimes leading to the development of DFUs, which are open sores or ulcers on the feet that may take several weeks to heal, if ever. Based on our comprehensive market research study conducted in 2015 on EscharEx that involved more than 200 healthcare professionals in the U.S. and Europe and, which was updated in 2019,2022, there arewere estimated 31 million diabetics in the United States in 2019 (9.4% of the U.S. population). The annual incidence of DFUs in the United States alone is approximately 990,000 (accounting for 45% recurrence), of which approximately 820,000 undergo debridement in a given year. |
| • | Pressure ulcers. Pressure ulcers form, also known as a result of pressure sores, or bed sores, which are injuries to the skin or the tissue beneath the skin. Constantskin caused by pressure on an area of skin reduces blood supplyapplied to the areaskin and over time can causesubsequent death of the skin to break down and form an open ulcer.tissue as a result of the reduced blood supply. These often occur in patients who are hospitalized or confined to a chair or bed, and usually form over bony areas, where there is little cushion between the bone and the skin, such as lower partsheels, elbows, the sacral area, and back of the body.head. Annually, 2.5 million pressure ulcers are treated in the United States in acute care facilities alone. |
| • | Surgical/traumatic wounds. Surgical wounds form as a result of various types of surgical procedures such as investigative or corrective, minor or major, open (traditional) or minimal access surgery, elective or emergency, and incisions (simple cuts) or excision (removal of tissue), among others. Traumatic wounds form as a result of cuts, lacerations or puncture wounds, which have caused damage to the skin and underlying tissue. Severe traumatic wounds may require surgical intervention to close the wound and stabilize the patient. Surgical/traumatic hard-to-heal wounds develop for various reasons, such as local surgical complications, suboptimal closure techniques, presence of foreign materials, exposed bones or tendons and infection. In the United States, millions receive post-surgical wound care annually. |
Market Opportunity Currently, surgery (sharp debridement) is generally considered a first-line option. Sharp debridement is an effective method to debride a wound, however,wound. However, this method requires surgically skilled physicians performing surgery with patients under, anesthesia, which in elderly patients with various co-morbidities is accompanied with a higher risk of local and systemic complications. Surgery may also involve hemorrhage which could be more difficult to control due to a high incidence of use of anticoagulants in this population. Surgery on wounds may very easily become infected, with the infection propagating to surrounding soft and boney tissues ending in life threatening major complication or amputation. Very often even minor, limited sharp debridement exposes other sensitive tissue, such as tendons, deep vessels/nerves and bones that may become infected or may be severely damaged, necessitating additional, more extensive debridement or even amputation. Due to these limitations, chronic wounds are treated by conservative methods, whilewith autolytic and enzymatic debridement arebeing the most commonly-used non-sharp methods. This includes a collagenase-based enzymatic debriding ointment, hydrogels and other topical dressings, which require numerous application sessions and a long time (6-8 weeks) to achieve a clean wound bed, if they achieve this at all. Thus, there is an unmet medical need for a non-surgical rapid and effective debridement agent for the outpatient setting, nurseryall care facilities and patients home.settings. Given the high demand for an effective non-surgical debridement technique outside of wound care clinic settings and the clinical data generated to date, EscharEx has the potential to expand the current use of enzymatic debridement across all sites of care and achieve substantial market share. As documented in the Phase 2II study described below, EscharEx significantly improved the rate of complete debridement after few once-daily applications, thus potentially facilitating wound debridement without the need for surgery. Based on our comprehensive market research study on EscharEx that involved more than 200 healthcare professionals in the U.S. and Europe, which was last updated in 2022, the EscharEx TAM for VLUs and DFUs is estimated at approximately $2 billion in the U.S. This market research and physician feedback suggests potential market share for EscharEx at approximately 30%. Due to traditional drug pricing disparities between the U.S. and countries within the EU, we expect the TAM for the market in the EU to be less than that of the U.S., but to be roughly similar on a unit volume basis.
EscharEx Clinical History EscharEx is a topical agent being developed for debridement of chronic and other hard-to-heal wounds, in order to fulfill an unmet need for a non-surgical rapid and effective debridement mean.option. EscharEx is based on the same active substance as NexoBrid but differs in other aspects, such as in formulation and presentation. Based on our current pre-clinical studies, the second generation EscharEx demonstrated even higher potency in lower doses, which could further contribute to EscharEx’s potential efficacy and tolerability. This advanced generation of EscharEx has been designed in accordance with the current treatment workflow and reimbursement programs, providing a non-surgical easy-to-use, potent product for daily application, which we believe will enhance patient compliance and improve quality of care. Based on the feedback received from different stakeholders, we believe that our second generation EscharEx can better address the unmet medical need for a non-surgical rapid and effective product, particularly in the outpatient setting, where the majority of patients are treated, and has a greater potential to achieve substantial market share. Second generation EscharEx is more differentiated from NexoBrid, which further limits the chances for competition between the two products.
Non-clinical safety studies performed with NexoBrid support EscharEx development, and we have already completed successfullysuccessful bridging toxicology studies. In a pre-IND meeting the FDA stated that existing toxicology data for EscharEx, including cross-referenced NexoBrid data, could be sufficient to support initiation of clinical studies in the product. The Following discussions with the regulatory authorities, we aligned the Phase III study protocol with EMA and the FDA, also stated thatand are on track to submit a final protocol in the first half of 2024. 216 patients will be treated globally across 40 sites with either EscharEx or a gel vehicle placebo, with an interim assessment to be performed once 67% of participants complete the study. Study initiation is expected in the second generationhalf of 2024. Additionally, we established research collaborations with 3M, Mölnlycke and MIMEDX to support the EscharEx formulation, manufacturing process and controls were sufficient to initiate dosing in Humans.Phase III clinical study. Completed clinical trials We completed a first Phase 2II feasibility study in Israel for chronic and other hard-to-heal wound technology.wounds. In January 2017 we completed and announced the final results of a second Phase 2II prospective study in Israel and Europe. In November 2017, we announced the final results of a second cohort of the second Phase 2II study. Based on the completed studies, we believe that our product candidate may be effective for debridement of chronic and other hard-to-heal wounds. First Phase 2II feasibility study—Israelstudy-Israel This first Phase 2II feasibility study was conducted in Israel to study the efficacy of our technology on chronic and other hard-to-heal wounds. The study assessed 24 patients at two sites. The results showed that our technology was effectivedemonstrated positive efficacy results in debriding various chronic and other hard-to-heal wound etiologies, such as VLUs, DFUs, VLUs, pressure sores and trauma on diseased skin. Second Phase 2 study—Israel/II study-Israel/E.U. –- First Cohort This second Phase 2II study was a prospective, controlled, assessor-blinded, randomized, multi-center Phase 2II study in Israel and Europe. The study objectives were to evaluate the efficacy and safety of EscharEx in comparison to the Gel Vehicle1 at a ratio of 2:1 for the treatment of a variety of chronic and other hard-to-heal wounds in three etiologies,etiologies: DFUs, VLUs and post-surgical or traumatic hard-to-heal wounds.
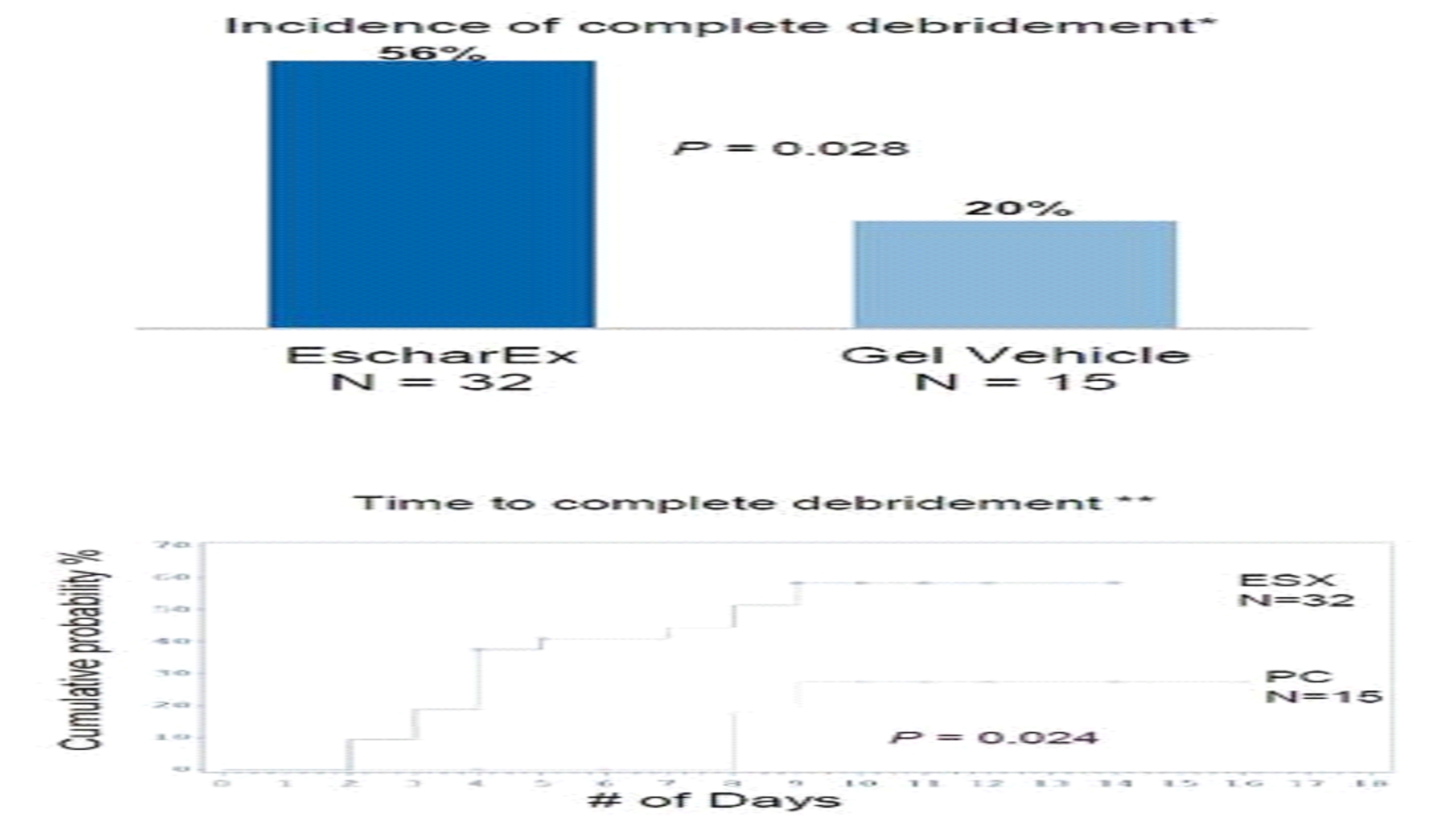
The primary endpoint assessed incidence of complete non-viable tissue removal (debridement) at the end of the debridement period (within up to 10 daily applications) and the secondary endpoints assessed various efficacy and safety endpoints, including wound bed preparation and wound healing. In January 2017 we reported final results of the first cohort of 73 patients. The average wound age in the EscharEx arm was more than double (72.8 weeks) that of the gel vehicle group (30.8 weeks). The average wound size was 33.6 cm2cm2 in the EscharEx arm vs. 25.8 cm2cm2 in the gel vehicle group. Despite the larger wounds and that wounds treated with EscharEx were older than wounds treated with gel vehicle (72.8 vs. 30.8 weeks), the study met its primary endpoint. EscharEx demonstrated a statistically significantsignificantly higher incidence of complete debridement at the end of the debridement period. Patients treated with EscharEx demonstrated a higher incidence of complete debridement (55% or 27/49) compared with patients treated with the hydrogel6hydrogel6 vehicle (29% or 7/24) with p=0.047. 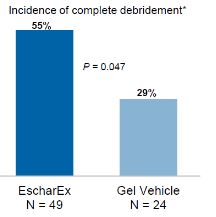 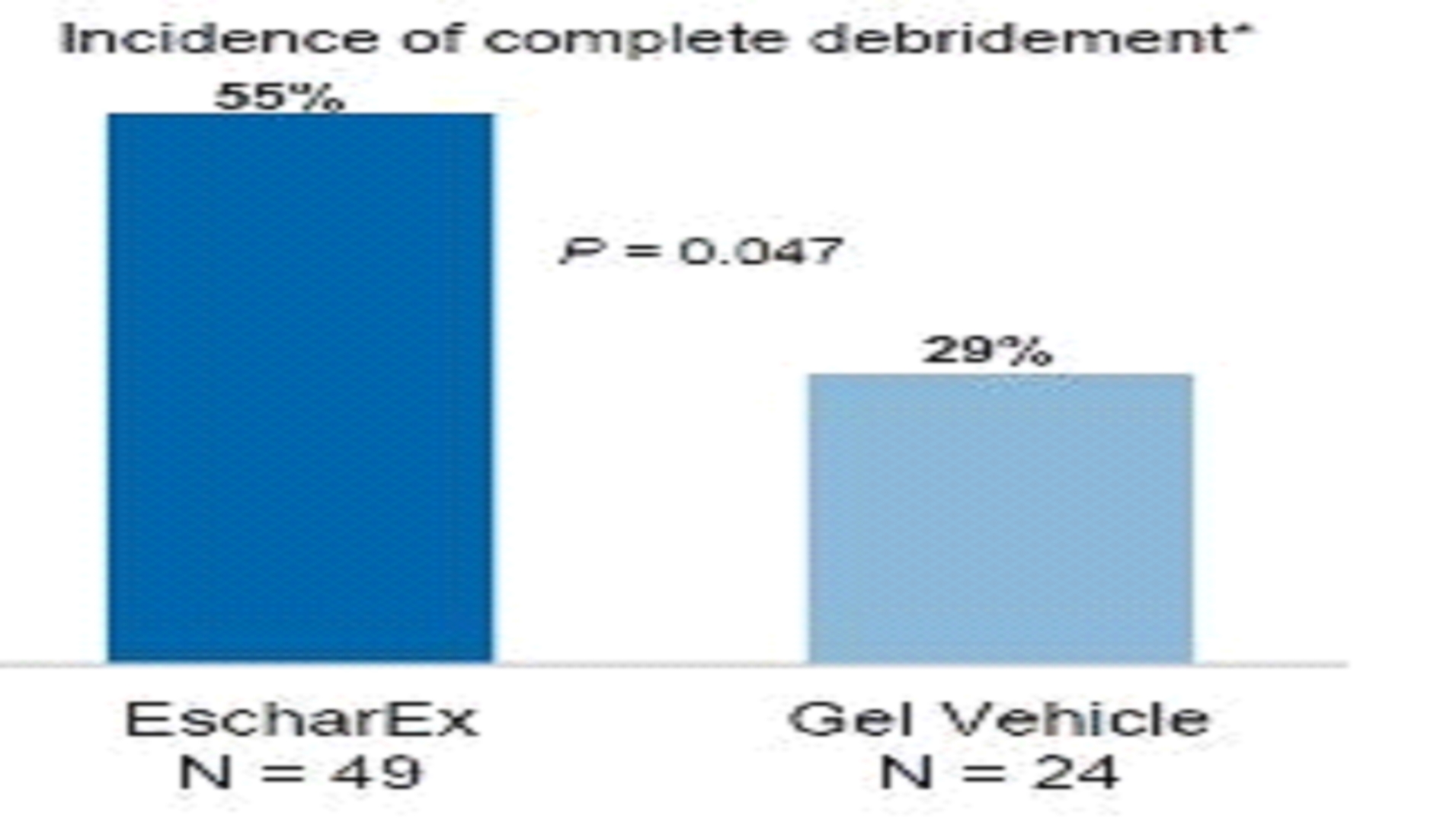 *w/i 10 daily applications Predefined sub-group analyses showed that 50% of patients with DFUs treated with EscharEx (8/16) achieved complete debridement at the end of the debridement period compared with 14.3% of patients with DFUs treated with hydrogel vehicle (1/7). In addition, 62.5% of patients with VLUs treated with EscharEx (10/16) achieved complete debridement at the end of the debridement period compared with 25% of patients with VLUs treated with hydrogel vehicle (2/8). Post hoc analysis showed that 56.3% of patients with DFUVLU or VLUDFU in the EscharEx group had complete debridement at the end of the debridement period compared with 20.0% in hydrogel vehicle group (p=0.028).
The study included secondary endpoints that provide further insight into number of efficacy and safety parameters. The secondary endpoint of time to complete debridement demonstrated a clear trend (p=0.075) that strongly suggests that not only iswas there a difference in the incidence of debridement, as confirmeddemonstrated by the primary endpoint, but that debridement occurred earlier in the group treated by EscharEx. The advantage in time to complete debridement was corroborated by the statistically significant post hoc result in the subgroup of patients with DFUsVLUs or VLUsDFUs that were treated with EscharEx (p=0.024).
Post hoc analysis showed that of patients who achieved complete debridement in the EscharEx group, 93% (25/27) completed the debridement within 7 days (4-5 applications on average).
6 Hydrogel is not a true sham placebo as it is a common and widely used treatment for the debridement of chronic wounds.
The overall patient demographics were comparable across both arms. No deleterious effect on wound healing was observed and no material differences were found in reported adverse events. The overall safety wasdata were comparable between the arms.
Second Phase 2 study—Israel/II study-Israel/E.U. –- Second Cohort After successfully completing the first cohort of the study which included 73 patients recruited in 15 clinical sites, we initiated a second cohort of patients to demonstrateevaluate safety and tolerability over extended periods of application to further support the product’s convenient application. In this second cohort, we recruited 38 patients from two etiologies, either DFUsVLUs or VLUs,DFUs, over extended periods of application (24-72 hours) with up to eight applications, randomizing the patients to two study arms EscharEx or gel vehicle at a ratio of 2:1. The second cohort of the study included 38 patients. The primary objective was to assess safety. EscharEx met its primary safety endpoint in this cohort, and the overall patient demographics and wound baseline characteristics were comparable across the arms in the second cohort. No related systemic adverse events were reported and adverse events related to local application were mild to moderate, reversible and resolved during the trial. Vital signs, pain scores, infection rates, laboratory parameters and blood loss were comparable between the two arms of the trial. Overall, no material safety concerns were identified. Ongoing clinical trials
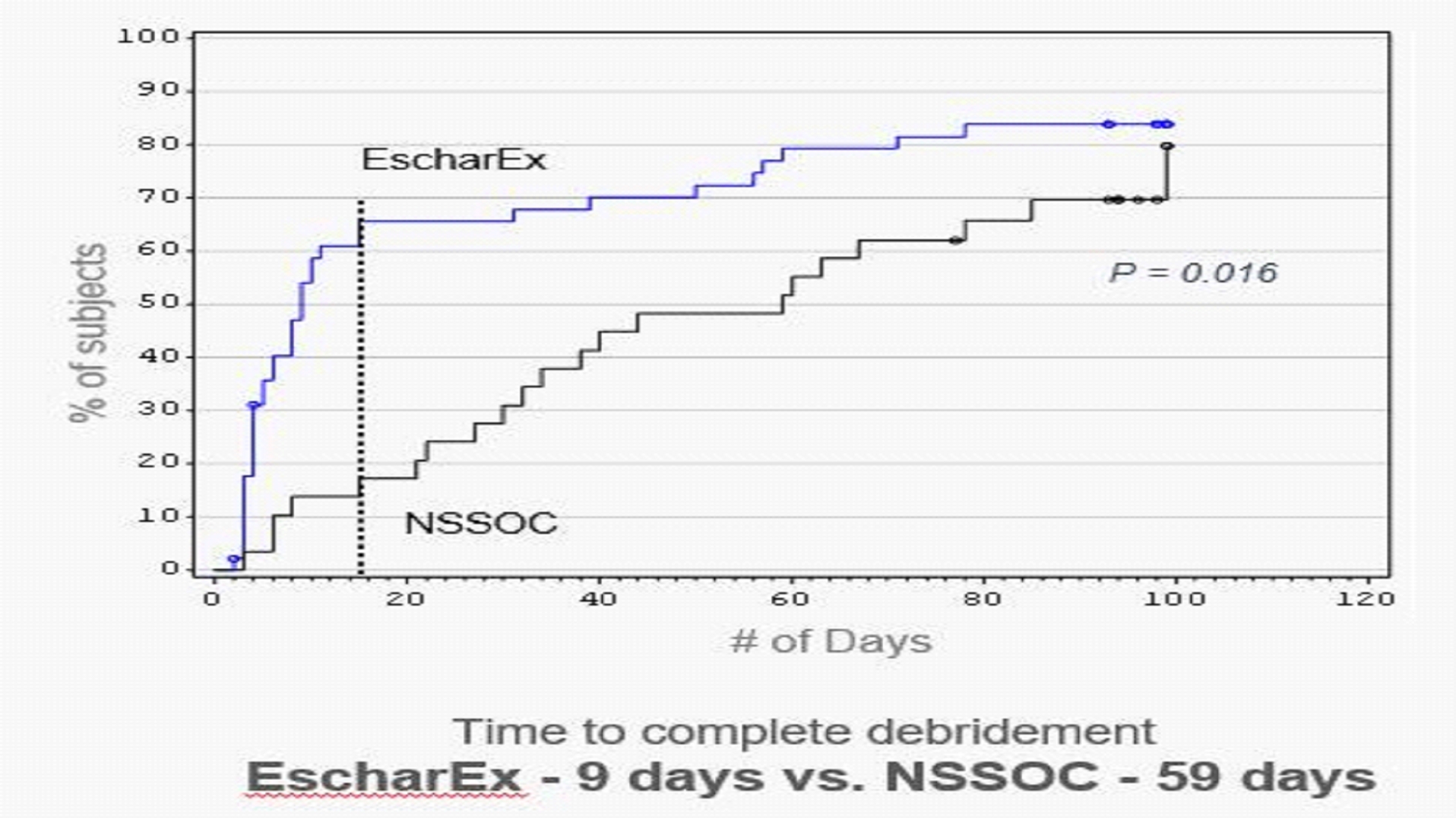
EscharEx U.S. Phase 2II Study in Venous Leg Ulcer (VLU) Patients In December 2019, we initiated a U.S. Phase 2II adaptive design clinical study of EscharEx for the treatment of venous leg ulcers (VLUs).VLUs. The study iswas a multicenter, prospective, randomized, placebo-controlled, adaptive design study, evaluating the safety and efficacy of EscharEx in debridement of VLUs compared to gel vehicle (placebo control) and non-surgical standard-of-care of either enzymatic or autolytic debridement.debridement (NSSOC). The study enrolled 120 patients, with 119 treated at approximately 20 clinical sites, primarily in the United States.U.S. Study participants were randomized totreated with either EscharEx (n=46), gel vehicle placebo control (n=43), or non-surgical standard-of-care at a ratio of 3:3:2,(n=30), with a three-month follow-up. The single primary endpoint was incidence of complete debridement (non-viable tissue removal), clinically assessed, within up to 8 treatment applications during the assessment period (up to 8 treatment applications within(within 14 days), compared to gel vehicle placebo control. Secondary and exploratory endpoints assessassessed time to achieve complete debridement, reduction of pain, reduction of wound area, granulation tissue and wound quality of life, enabling evaluation of clinical benefits compared to both gel vehicle and non-surgical standard-of-care.NSSOC. Incidence and time to achieve wound closure will bewere assessed as safety measurements. In JanuaryMay 2022 we announced positive toplineour results from this study. These topline results showed that theThe study met its primary endpoint with a high degree of statistical significance, demonstrating that patients treated with EscharEx had a statistically significant higher incidence of complete debridement compared to the gel vehicle. The study randomized 120 patients, of which 119 patients were treated by either EscharEx (n=46), a gel vehicle (n=43), or a non-surgical standard-of-care consisting of either enzymatic or autolytic debridement (n=30). Patients treated with EscharEx demonstrated a statistically significant higher incidence of complete debridement during the 14-day measurement period within up to 8 applications compared to patients treated with gel vehicle (EscharEx: 63% (29/46) vs. gel vehicle: 30% (13/43), p-value=0.004). EscharEx efficacy superiorityresults remained statistically significant compared to gel vehicle also after adjusting for pre-specified covariates ascribed to patient baseline characteristics, wound size, wound age and age, regions,region. 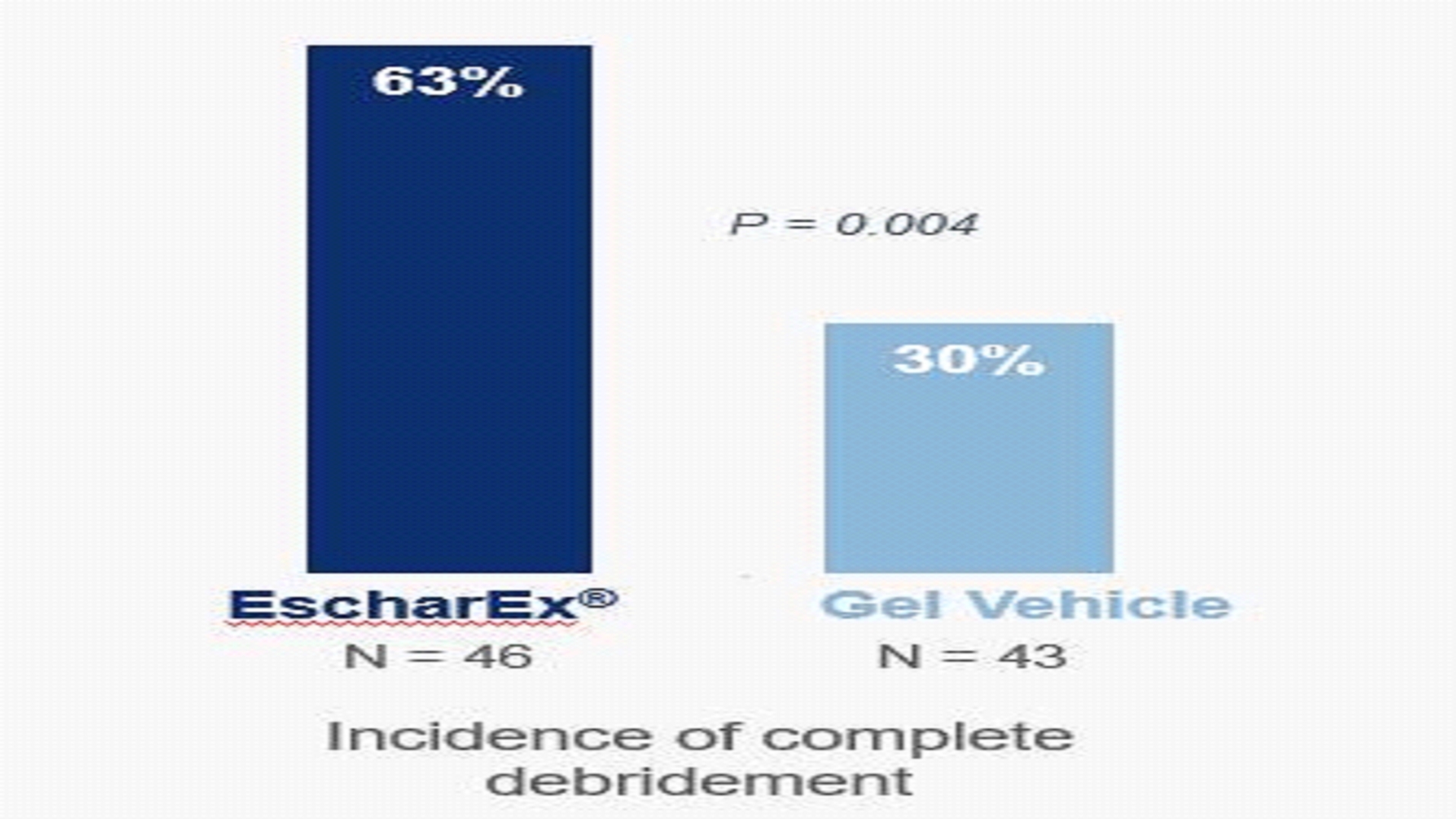
The study met key secondary and sites. Incidenceexploratory endpoints. Patients treated with EscharEx had a statistically significant higher incidence of complete debridement, of the non-surgical standard-of-care arm, during the same 14-day measurement period, wascompared to patients treated by non-surgical standard-of-care ("NSSOC") (EscharEx: 63% (29/46) vs. NSSOC: 13% (4/30)) and the time to achieve complete debridement was significantly shorter. Estimated median time to complete debridement was 9 days for patients treated with EscharEx and 59 days for patients treated with NSSOC (p-value=0.016). On average, complete debridement was achieved after 3.6 applications of EscharEx compared to 12.8 applications with NSSOC. Patients treated with EscharEx demonstrated significantly higher incidence of greater than 75% granulation tissue at the end of the treatment period compared to gel vehicle (p-value <0.0001). Favorable trends were observed in wound area reduction and reduction of pain compared to gel vehicle. 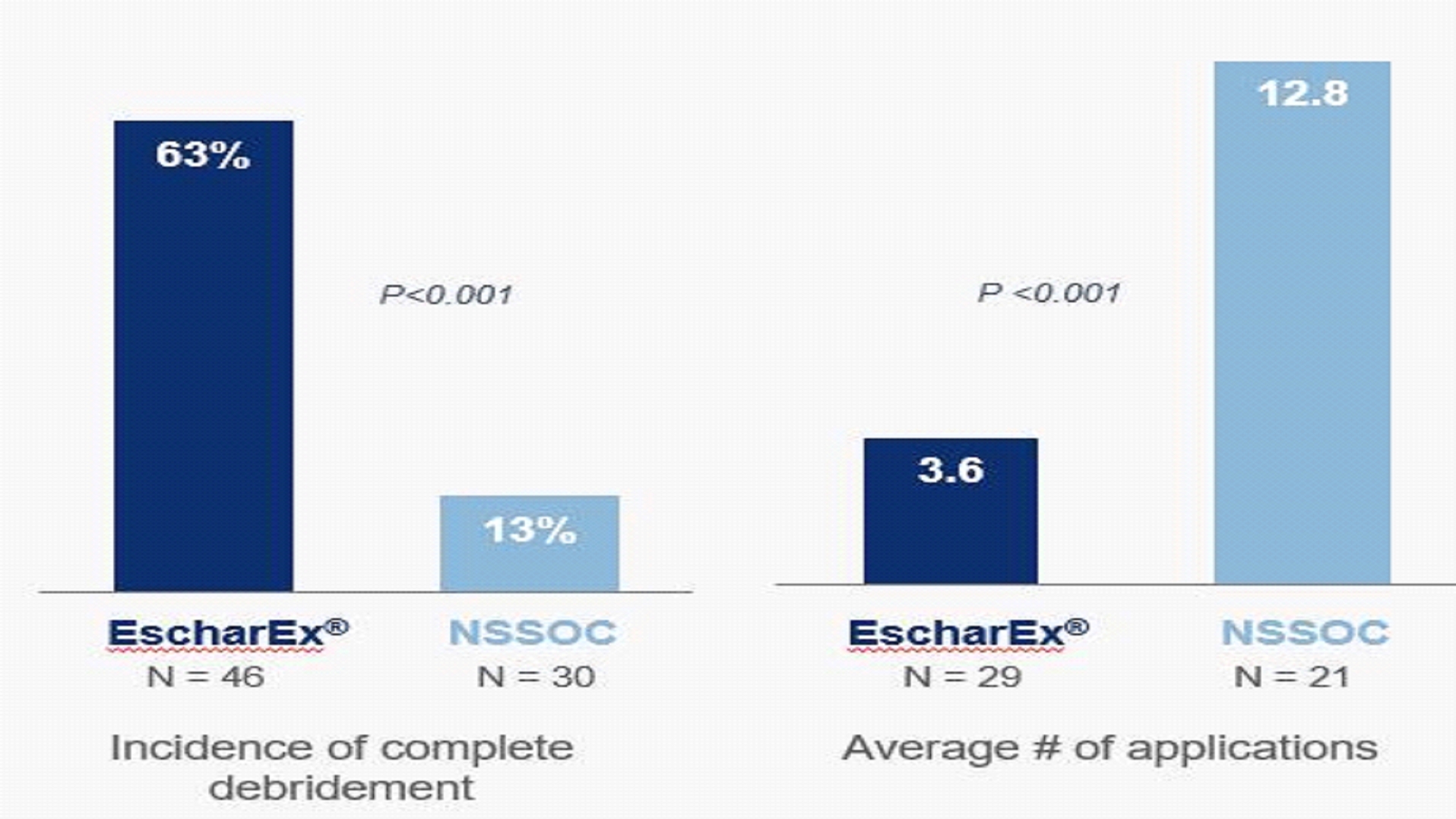
In addition, the Independent Data Monitoring Committee reviewedstudy showed that EscharEx was well tolerated, and the overall safety results were comparable between the arms as assessed by the data of allsafety monitoring board. Importantly, there were no observed deleterious effects on wound closure and no material differences in reported adverse events. Estimated time to complete wound closure was 64 days for patients treated with EscharEx compared to 78 days for patients treated with NSSOC. Post-hoc analyses from this study assessed the incidence and no safety concerns were identifiedtime to wound bed completely covered with granulation tissue. The incidence of achieving complete debridement and complete cover of the wound bed with granulation tissue (i.e., wound bed preparation, WBP) during the daily treatment period was 50.0% for EscharEx vs. 25% for the Gel Vehicle (p-value= 0.01) and 10% for NSSOC (p-value< 0.0001). The estimated median time to achieve WBP was 11 days for EscharEx vs. 85 days for the Gel Vehicle (p-value= 0.002) and 63 days for the NSSOC (p-value= 0.0106). Furthermore, it was shown that patients reaching WBP in the study population.are 4 times more likely to achieve wound closure (p=0004). Post-hoc analyses from this study assessed the incidence and time to complete debridement, complete granulation, and wound closure in patients treated with EscharEx (n=46) compared to a sub-group of patients who were treated with SANTYL (n=8). Baseline characteristics (age, gender, wound age, wound size) were comparable in both groups. The incidence of complete debridement during the daily treatment period (the first two weeks of the study) was well-tolerated63.0% (95% CI=47.5-76.8) for EscharEx vs. 0% for SANTYL; p=0.001. The estimated median time to achieve complete debridement during the study was 9 days (95% CI=5-15 days) for EscharEx vs. not achieved for SANTYL (95% CI=22-Not Applicable); p=0.023. The incidence of achieving complete debridement and complete cover of the wound bed with granulation tissue (i.e., wound bed preparation, WBP) during the daily treatment period was 50.0% (95% CI = 34.9%-65.1%) for EscharEx vs. 0% for SANTYL; p=0.015. The incidence of achieving WBP throughout the study was 78.3% (95% CI = 63.6-89.1) for EscharEx vs. 37.5% for SANTYL (95% CI=8.5-75.5); p=0.03. The estimated median time to achieve WBP was 11 days (95% CI =7-50 days) for EscharEx vs. not achieved for SANTYL (95% CI=22-Not Applicable); p=0.014. 15 of the 46 patients (32.6%) treated with EscharEx completely closed their wounds during the study, compared to 2 out of 8 patients (25%) treated with SANTYL (NSS). In those patients who achieved complete wound closure, the average time to wound closure was 48.4 days (SD=23.5) for EscharEx vs. 76.0 days (SD=2.8) for SANTYL; p=0.05. Patient reported applicational pain was comparable in both groups. The safety results and overall safety wasincidence of adverse wound reactions were comparable between the arms. No differences were found in reported adverse events and no serious adverse event was related to study treatment. Patient baseline characteristics were comparable across all study arms. Patient follow-up is ongoing and additional data, including secondary and exploratory endpoints as well as additional safety measurements, which will allow further evaluation of clinical benefits, is expected in the second quarter of 2022. EscharEx Pharmacology Study In December 2021,May 2022, we announced positive initial dataresults from sevenour U.S. Phase II pharmacology study of EscharEx for debridement of lower leg ulcers. The study was a prospective, open label, single-arm study, conducted at three U.S. clinical sites. The study evaluated the maximum fifteen patients in our ongoing open-label, phase 2clinical performance, safety, and pharmacology studyeffect of EscharEx in the debridement of lower leg ulcers (VLUs and DFUs). Based on this initial data,The study evaluated the safety and efficacy of debridement as measured by incidence of, and time to complete debridement. In addition, the study evaluated the pharmacological effects of EscharEx as measured by the changes from baseline to end of treatment period in (1) wound biofilm presence in wound biopsies, (2) bacterial burden measured by MolecuLight® fluorescence images, and (3) biomarkers of wound healing and inflammation in wound fluid. Twelve patients with either VLUs or DFUs were enrolled in the study. Patients were treated with up to eight daily applications of EscharEx and then continued follow-up for 2 weeks. Punch biopsies and wound fluids were collected prior to the first, and after the last treatment. Biofilm presence was analyzed from wound biopsies. Wound fluids were analyzed to evaluate biomarkers of wound healing and inflammation, i.e., MMPs, cytokines, chemokines, growth factors and HNE. Fluorescent imaging was used during treatment to measure wound size and bacterial load. Fluorescent imaging was also utilized to identify the highest fluorescence area to obtain the biopsy. EscharEx demonstrated safe and effective debridement of lower leg ulcers withinwith a few daily applications. In addition, evaluation of wounds’ tissue samples (biopsies) and fluorescence images, indicated reduction of wound area, biofilm and bacterial loadbioburden following the treatment with EscharEx. We expect to share the full data set from this study in the first half of 2022. 47Seventy percent of patients achieved complete debridement during the course of treatment within up to 8 applications. On average, complete debridement was achieved after 3.9 applications of EscharEx. Additionally, an average reduction of 35% in wound size was achieved by the end of the 2-week follow-up period. In all patients that were positive for biofilm at baseline, the biofilm was reduced substantially to single individual microorganisms or completely removed by the end of treatment. Seven patients had positive red fluorescence (indicative of bacteria) at baseline and average red fluorescence was reduced from 1.69 cm2 pre-treatment to 0.60 cm2 post treatment. Biomarker analysis from wound fluid safety data showed that EscharEx was well-tolerated.
The development of EscharEx for the debridement of chronic and other hard-to-heal wound indications is in Phase 2 studies, and there is no certainty that EscharEx will achieve all of the objectives of the trials as required or that the FDA will allow at this stage to initiate further studies or that we will successfully complete the development to obtain a marketing authorization for EscharEx. MediWound currently expects to request an end-of-Phase 2 meeting with the FDA in the second half of 2022, to discuss program results and the potential Phase 3 pivotal plan for EscharEx. See “ITEM 3.D. Risk Factors—DevelopmentFactors-Development and commercialization of NexoBrid and EscharEx in the United States and our pipeline product candidates worldwide requires successful completion of the regulatory approval process, and may suffer delays or fail.”
Non-Melanoma Skin Cancer MW005 is a topically applied biological product candidate for the treatment of non-melanoma skin cancers, based on the same active substance of NexoBrid and EscharEx, a concentrate of proteolytic enzymes enriched in bromelain. The clinical development plan of MW005 is supported by the results from several toxicological and other preclinical studies, as well as vast clinical experience from NexoBrid and EscharEx, which share the same active substance.API. We launched a new clinical program to evaluate our drug product candidate MW005 in patients with non-melanoma skin cancer.
Non-melanoma Skin Cancers
CancersIn July 2023, we announced the final results of this study. Fifteen patients were treated with MW005 and completed the skin are by farstudy. Results showed MW005 was well-tolerated, with a high level of patient compliance. Based on clinical assessments, eleven out of fifteen patients achieved complete clearance of their BCCs; the most common of all types of cancer with about approximately 5.4 million basal and squamous cell skin cancers are diagnosed each year in the US. The numbermajority of these cancers has been increasing for many years due to combination of better skin cancer detection, people getting more sun exposure, and people living longer.
Basal cell carcinomas - basal cell carcinoma (BCC) starts in the basal cell layer, which is the lower part of the epidermis. If not removed completely, basal cell carcinoma can come back (recur) in the same place on the skin. People who havepatients also had basal cell skin cancers are also more likely to get new ones in other places. BCCs are uncontrolled and abnormal growths that arise in the basal cells of the skin and the tumors primarily affect photoexposed areas, most commonly in the head, and infrequently appear on per genital and genitalia regions. The main cause of BCC is chronic ultraviolet (UV) exposure. BCC is the most common form of skin cancer, accounting for 75-80% of all skin cancers
Squamous cell carcinomas - Squamous cell carcinomas (SCC) start in the flat cells in the upper (outer) part of the epidermis
Actinic keratosis - Actinic keratosis (AK), also known as solar keratosis, is a pre-cancerous skin condition caused by too much exposure to the sun. People who have them usually develop more than one. A small percentage of AKs may turn into squamous cell skin cancer.
Bowen disease - Bowen disease (squamous cell carcinoma in situ), is the earliest form of squamous cell skin cancer
Market opportunity
Basal cell carcinoma is a non-melanoma skin cancer that arises from the basal layer of epidermis and its appendages and is the most diagnosed skin cancer in the US (~4.3 million cases annually).
Under existing standard of care, low-risk patients are treated with tumor resection via either standard surgical excision or Mohs micrographic surgery. Recurrence rates for these sharp methods of tumor removal are low (~5% at 5 years), and procedure is considered straightforward with limited patient downtime or side effects. Topical products (5-FU and Imiquimod) are used primarily in superficial lesions, but have limited use and are reserved for surgery ineligible patients. Drawbacks include longer treatment duration (>6 weeks), low efficacy (~14% at 5 years), and side effects such as scarring, skin-site reactions, and fatigue/flu-like illness. High-risk patients are also primarily treated with surgery; surgery-ineligible patients are treated with oral hedgehog pathway inhibitors, which are effective in the short-term, but have high recurrence rates / safety concerns. There is a need for more effective, safer topical products in low-risk superficial basal cell carcinoma for surgery-ineligible patients (e.g., site of tumor is challenging for excision or may result in cosmetic issues) or for patients for whom surgery is not appropriate (e.g., older / frail patients, or those with challenges in seeking pre and post-surgical appointments) and current topical agents may be avoided due to long treatment durations and because they result in an unpleasant treatment process for patients.
MW005 Clinical History
Ongoing clinical trials
U.S. Phase I/II Study in basal cell carcinoma Patients
In July 2021, we initiated a U.S. phase I/II study of MW005 for the treatment of low-risk basal cell carcinoma (BCC). The phase I/II open-label, randomized clinical study in BCC is designed to evaluate safety and tolerability of MW005 using different schedules of administration, as well as provide a preliminary evaluation of efficacy as measured by the percentage of target lesion with complete histological clearance. The trial will enroll up to 32 patients, comprised of 2 cohorts of 16 patients each, with histologically confirmed superficial or nodular BCC and will be conducted at three leading clinical centers in the U.S. We expect data to be available in the first half of 2022.complete clearance.
Although we have conducted preclinical trials, the development of MW005 for non-melanoma skin cancer indications is still in its preliminary phase and there is no certainty that it will achieve all the aims of the trials as required and/or successfully complete the approval process for such indication. See “ITEM 3.D. Risk Factors—DevelopmentFactors-Development and commercialization of NexoBrid and EscharEx in the United States and our pipeline product candidates worldwide requires successful completion of the regulatory approval process, and may suffer delays or fail.”
Research and Development Our research and development strategy is centered around our validated proteolytic enzyme platform technology, focused on next-generation protein-based therapies for burn and wound care, and for tissue repair, which underlies NexoBrid and EscharEx, into additional product candidates for high-value indications. For more information regarding our research and development expenses, see “ITEM 5.C. Research and Development, Patents and Licenses, etc.”
Pre-Clinical Clinical Studies We conduct clinical studies and preclinical studies to support the efficacy and safety of our products and their ingredients and to extend and validate their benefits for human health. Preclinical studies allow us to substantiate the safety of our products and obtain preliminarily indications of their pharmacological and safety profile. As of the date hereof, we hadhave conducted more than 50 non-GLP8non-GLP and GLP preclinical studies. All pre-clinical safety and toxicology studies were conducted according to the principles of Good Laboratory Practices (“GLP”), and twelvethirteen clinical studies, according to the principles of Good Clinical Practices (“GCP”), for NexoBrid, EscharEx and our pipeline product candidates. As a result, we have developed significant experience in planning, designing, executing, analyzing and publishing clinical studies. Our research and development team manages our clinical studies and coordinates the project planning, trial design, execution, outcome analyses and clinical study report submission. During the design, execution and analyses of our studies, our research and development team consults with key opinion leaders and top-tier consultants in the relevant field of research to optimize both design and execution, as well as to strengthen the scientific, medical and regulatory compliance level of the investigational plan. Our clinical studies have been conducted in collaboration with leading medical and research centers throughout the world.
Manufacturing, Supply and Production We operate a manufacturing facility in Yavne, Israel, in a building that we sub-lease from Clal Life Sciences L.P., with 31 employees as of December 31, 2021.Israel. This facility allows us to manufacture sterile biopharmaceutical products, such as NexoBrid. The facility meetsis designed to meet current cGMP requirements and similar foreign requirements, as certified by each of the EMA,U.S., EU member states competent authorities, the Israeli Ministry of Health, and South Korean ministry of health and Japanese ministry of health. Our facility is subject to audits for reassessment of cGMP compliance and similar foreign requirements, which are preformedperformed periodically by regulatory authorities and was re-approved as cGMP-compliant for an additional three yearsyear term as of the audit date, until 2023.2025. Additionally, as we seekpart of the regulatory approval process for NexoBrid our plant was inspected in the United States2022 by the FDA will need to inspect our plantand the Pharmaceuticals and Medical Devices Agency (“PMDA”) of Japan to confirm it meets all regulatory requirements. In addition, other regional applicable authorities may also need to inspect our plant to confirm it meets all regulatory requirements in order to obtain marketing authorization in these jurisdictions. Applicable changes in our production processes for NexoBrid must be approved by the EMA and similar authorities in other jurisdictions. While we believe that our current manufacturing capacity at the facility is sufficient to meet the expected near-term commercial Our global demand for NexoBrid we are planning to scale-upsurpasses the current manufacturing capabilities. We are currently seeking to expand our manufacturing capabilities in order to increase our capacity subject to BLA approval,manufacture NexoBrid and future product candidates and satisfy near term demand. The new GMP-compliant state-of-the-art manufacturing facility is projected to be completed by mid-2024, with full-scale manufacturing currently expected to commence in 2023.2025. We expect the cost will be approximately $8-10$12.7 million.
The starting material used by us in the manufacturing of NexoBrid and our other product candidates is bromelain SP, which is derived from pineapple plant stems. We have entered into an agreement with CBC, dated January 11, 2001, as amended on February 28, 2010, pursuant to which CBC uses proprietary methods to manufacture bromelain SP and supplies us with this intermediate drug substance in bulk quantities. According to the terms of the agreement, CBC shall not, and shall not permit related companies or a third party to, manufacture, use, supply or sell the raw materials for the use or production of a product directly or indirectly competing with any of our products. Our supply agreement with CBC has no fixed expiration date and can be voluntarily terminated by us, with at least six months’ advance written notice, or by CBC, with at least 24 months’ advance written notice. Upon obtaining bromelain SP from CBC, we further process it into the drug substance and then into the drug product to finally create the powder form of NexoBrid. The necessary inactive ingredients contained in NexoBrid, or the excipients, are readily available and generally sold to us by multiple suppliers. In addition to this powder, we manufacture a sterile gel substance by combining water for injections produced by us at our facility and additional excipients.
Marketing, Sales and Distribution We commercialize globally NexoBrid via multiple sales channels: Europe In Europe and Israel, we sell NexoBrid, primarily through our own sales force consisting of a marketing team of specialized and knowledgeable sales representatives in Europe, focusing on leading burn centers and Key Opinion Leaders (KOL) management. We have obtained national reimbursement for NexoBrid in Belgium, Italy, and ItalyGreece and we continue to locally execute our market access strategy for most of Europe to obtain procurement by burn centers and hospitals as part of their budget, or under local, regional or national reimbursement, depending on the specific process required in each country. We believe that additional burn units in large hospitals as well as smaller hospitals will follow the treatment trends once established by the burn centers. See “—Government“-Government Legislation and Regulation—PharmaceuticalRegulation-Pharmaceutical Coverage, Pricing and Reimbursement.” Furthermore, we are establishing additional distribution channels through local partners to extend outreach in EU (Sweden, Finland, the Baltic states, France, Switzerland, (Romandie region), Greece, Malta, Bulgaria Cyprus, Portugal, the Netherlands and Luxemburg)Cyprus,), where NexoBrid is already approved for marketing as part of the European marketing authorization. On November 2023, we expanded NexoBrid’s European market presence by establishing a collaboration with PolyMedics Innovations (PMI) for the promotion of NexoBrid in Germany, Austria, Belgium, the Netherlands and Luxembourg. In addition to receiving marketing authorization for NexoBrid in the European Union, key opinion leaders in the burn care field worldwide are already aware of NexoBrid’s efficiency in removing eschar due to hundreds of scientific presentations and several award winningaward-winning abstracts at international and national conferences and about 100120 peer-reviewed papers. North America Vericel License and Supply Agreements
On May 6, 2019, we entered into exclusive license and supply agreements with Vericel to commercialize NexoBrid in all countries of North America (which we refer to as the “Territory”).
NexoBrid is currently under registration stage in the U.S., and pursuant to the terms of the License Agreement described below, we will continue to conduct all clinical and other activities described in the development plan to support the BLA resubmission with the FDA under the supervision of a Central Steering Committee comprised of members of each of our Company and Vericel.
License Agreement. We entered into a license agreement (the “License Agreement”) withthe Vericel License Agreement pursuant to which we granted Vericel an exclusive license, with the right to grant sublicenses, to develop and commercialize NexoBrid and any improvements of NexoBrid (the “Licensed Product”) in the Territory. Pursuant to the terms of the Vericel License Agreement, Vericel will have exclusive control regarding the commercialization of Licensed Products in the Territory and must use commercially reasonable efforts to commercialize Licensed Products within the Territory. We and Vericel have made customary representations and warranties and have agreed to certain customary covenants, including confidentiality and indemnification. Within 10 days of signing the Vericel License Agreement, Vericel paid us an upfront fee of $17.5 million (the “Upfront Payment”). Vericel is obligated to pay us $7.5 million and upon the U.S. regulatory approval of the BLA for NexoBrid andVericel paid us $7.5 million. Vericel is obligated to pay us up to $125 million, in the aggregate, upon attainment of certain sales milestones. The first sales milestone of $7.5 million is triggered when annual net sales of the Licensed Products in the Territory exceed $75 million. Vericel is also obligated to pay us tiered royalties on net sales of Licensed Products at rates ranging from mid-high single-digit to mid-teen percentages, subject to certain customary reductions, as well as a percentage of gross profits on committed purchases by BARDA and a royalty on additional purchases bysales to BARDA. The royalties will expire on a product-by-product and country-by-country basis upon the latest to occur of (i) twelve years following the first commercial sale of such Licensed Product in such country, (ii) the earliest date on which there are no valid claims of MediWound patent rights covering such Licensed Product in such country, and (iii) the expiration of the regulatory exclusivity period for such Licensed Product in such country (the “Royalty Term”). Such royalties are subject to reduction in the event that (a) Vericel must license additional third-party intellectual property in order to develop, manufacture or commercialize a Licensed Product, or (b) biosimilar competition occurs with respect to the Licensed Product in any country within the Territory. After the expiration of the applicable royalties for the Licensed Product in any country within the Territory, the license for such Licensed Product in such country would become a fully paid-up, royalty-free, perpetual and irrevocable license. The Vericel License Agreement expires on the date of expiration of all royalty obligations due under the agreement unless earlier terminated in accordance with its terms. Either party may terminate the agreement upon the failure of the other party to comply with its material obligations under the agreement if that failure is not remedied within certain specified cure periods or in the event of a party’s insolvency. In addition, Vericel may terminate the agreement upon 150 daysa 150-day written notice to us.
Supply Agreement. On May 6, 2019, concurrently with our entry into the License Agreement, we entered into a supply agreement with Vericel (the “Supply Agreement”) with Vericel pursuant to which we are obligated to supply Vericel with NexoBrid for sale in the Territory on an exclusive basis for the first five years of the term of the Supply Agreement. The Supply Agreement requires us to take steps to ensure that our manufacturing capacity meets Vericel’s demand for NexoBrid. In addition, after the exclusivity period or upon supply failure, Vericel will be permitted to establish an additional or alternate source of supply. Pursuant to the Supply Agreement, we will supply NexoBrid to Vericel based on Vericel’s fixed orders on a unit price basis. After a specified period, the unit price, on an annual basis, may be increased based on the United States Producer Price Index for Chemical Manufacturing published by the Bureau of Labor Statistics. The Supply Agreement’s initial term is five years (the “Initial Term”), with Vericel required to provide us with notice regarding whether it plans to extend the Initial Term for an additional two years by the third anniversary of the Supply Agreement. In May 2022, Vericel notified us on its election to extend the Initial Term for an additional 2 years until 2026. After the Initial Term and optional two-year extension, Vericel, at its sole discretion, may choose to extend the Supply Agreement’s term for additional one-year periods for a potential total term of fifteen years. In September 2023, NexoBrid was launched in the U.S. by Vericel. The Supply Agreement will automatically terminate upon the expiration or termination of the License Agreement. Either party may terminate the Supply Agreement upon the failure of the other party to comply with its material obligations under the Supply Agreement if such failure is not remedied within certain specified cure periods. After the Initial Term, Vericel may terminate the Supply Agreement upon 12 months’ prior written notice to us, and we may terminate the Supply Agreement upon 36 months prior written notice to Vericel. BARDA Pursuant to the First BARDA Contract, BARDA has initiatedcompleted the procurement of NexoBrid valued at $16.5 million, for emergency stockpile as part of the HHS mission to build national preparedness for public health medical emergencies. BARDA purchased inventory is being managed by MediWound under vendor managed inventory. As of December 31, 2021, the Company has received $14.6 million for procurement of NexoBrid for U.S. emergency preparedness. Under our exclusive license and supply agreements with Vericel, we will equally split the gross profits on the initial procurement and receive a double-digit royalty on any additional future BARDA purchases of NexoBrid. Please see “Vericel License and Supply Agreements” above. Other International Markets In other international markets, we sell NexoBrid through local distributors with which we have distribution agreements, focusing on Asia Pacific, EMEA CEE and LATAM.CEE. We have signed local distribution agreements for distribution in Argentina, Russia, South Korea, Colombia, Mexico, Peru, Chile, Ecuador, Panama, India, Bangladesh, Sri Lanka, Turkey, Japan, Australia, New-Zealand, Singapore, Ukraine, Taiwan and United Arab Emirates. Our distributors in Argentina,Russia, South Korea, Russia, Peru, Chile, Taiwan, Japan, India, United Arab Emirates and Eurasian countries have obtained marketing authorization. Our additional distributors have filed or are in the process of filing for market authorization in their respective territories and are expected to launch NexoBrid after receipt of local regulatory approval, which may take a year or more to be granted, and, consequently, may occur in certain markets during 2022.2023. We have launched NexoBrid in Argentina, South Korea, Russia, Taiwan ChileJapan and United Arab EmiratesIndia and expect additional launches following receipt of local marketing authorizations. We plan to enter other international markets through collaboration with local distributors and leverage our approved registration file in Europe to obtain regional marketing authorizations. For a breakdown of our consolidated revenues by geographic markets and by categories of operations for the years ended December 31, 20202022 and 2021,2023, please see “Item 5.A Operating and Financial Review and Prospects—OperatingProspects-Operating Results.”
Intellectual Property Our intellectual property and proprietary technology are important to the development, manufacture and sale of NexoBrid, EscharEx and our future pipeline product candidates. We seek to protect our intellectual property, core technologies and other know-how through a combination of patents, trademarks, trade secrets, non-disclosure and confidentiality agreements, licenses, assignments of invention and other contractual arrangements with our employees, consultants, partners, suppliers, customers and others. Additionally, we rely on our research and development program, clinical trials, know-how and marketing and distribution programs to advance our products and product candidates. As of December 31, 2021,2023, we had been granted a total of 6488 patents and have 2719 pending patent applications. The family of patents that covers NexoBrid specifically includes 35 granted patents worldwide. EscharEx is covered by 715 patents and 244 national phase applications. The main patents for our proteolytic enzyme technology which underlies NexoBrid, EscharEx and our current pipeline product candidates have been issued in Europe, the United States and other international markets. Our patents which cover NexoBrid claim specific mixtures of proteolytic enzymes, methods of producing such mixtures and methods of treatment using such mixtures. Although the protection achieved is significant for NexoBrid, EscharEx and our pipeline product candidates, when looking at our patents’ ability to block competition, the protection offered by our patents may be, to some extent, more limited than the protection provided by patents which claim chemical structures which were previously unknown. Absent patent-term extensions, the NexoBrid patents are nominally set to expire in 2025 and in 2029 in the United States. The NexoBrid patents issued in Europe and in other foreign jurisdictions are nominally set to expire in 2025. The patents and the national phase applications relating to EscharEx, if the national phase applications are granted, will expire on January 30, 2037, absent any patent-term adjustment and/or extensions.
While our policy is to obtain patents by application, license or otherwise, to maintain trade secrets and to seek to operate without infringing on the intellectual property rights of third parties, technologies related to our business have been rapidly developing in recent years. Additionally, patent applications that we may file or license from third parties may not result in the issuance of patents, and our issued patents and any issued patents that we may receive in the future may be challenged, invalidated or circumvented. For example, we cannot predict the extent of claims that may be granted or enforceable in our patents nor can we be certain of the priority of inventions covered by pending third‑party patent applications filed in the U.S. If third parties prepare and file patent applications that also claim technology or therapeutics to which we have rights, we may have to participate in proceedings to determine priority of invention, which could result in substantial costs to us, even if the eventual outcome is favorable to us. Moreover, because of the extensive time required for clinical development and regulatory review of a product we may develop, it is possible that, before NexoBrid can be commercialized in additional jurisdictions and/or before any of our future products can be commercialized, related patents will expire a short period following commercialization, thereby reducing the advantage of such patent. Loss or invalidation of certain of our patents, or a finding of unenforceability or limited scope of certain of our intellectual property rights, could have a material adverse effect on us. See “ITEM 3.D. Risk Factors —- Our success depends in part on our ability to obtain and maintain protection for the intellectual property relating to, or incorporated into, our technology and products.” In addition to patent protection, we also rely on trade secrets, including unpatented know‑how, technology innovation, drawings, technical specifications and other proprietary information in attempting to develop and maintain our competitive position. We also rely on protection available under trademark laws, and we currently hold various registered trademarks, including “MediWound,” “NexoBrid” and “EscharEx” in various jurisdictions, including the United States, the European Union and Israel. Klein License Agreement In September 2000, we signed an exclusive license agreement, as amended in June 2007, with Mark Klein, a third party, for use of certain patents and intellectual property (the “Klein License Agreement”). Under the Klein License Agreement, we received an exclusive license to use the third party’s patents and intellectual property to develop, manufacture, market and commercialize NexoBrid and its pipeline product candidates for the treatment of burns and other wounds. The claims of such patents are directed to a process of preparing a mixture of escharase and proteolytic enzymes and cover the underlying proteolytic mixture of escharase and proteolytic enzymes prepared by that specific process. Pursuant to the Klein License Agreement, we are obligated to keep accounting records related to the sales of NexoBrid and its pipeline product candidates and pay royalties as discussed below. The Klein License Agreement may be terminated by Mark Klein, subject to notice and dispute resolution provisions of the Klein License Agreement, in the event of our breach, bankruptcy petition, insolvency or failure to achieve a development milestone within six months of a target date. We have already achieved all development milestones under the Klein License Agreement.
As consideration forunder the Klein License Agreement, we paid an aggregate amount of $1.0 million following the achievement of certain development milestones. In addition, we undertook to pay royalties of 1.5‑2.5% from revenues, 10% of royalties received from sublicensing and 2% of lump‑sum payments received from sublicensing up to $1 million and 4% above $1 million, in each case relating to products based on the licensed patents and intellectual property, for a term of 10‑15 years, as applicable, from the date of the first commercial delivery in a major country. In addition, under the Klein License Agreement, we agreed to pay a one‑time lump‑sum amount of $1.5 million upon reaching aggregate revenues of $100 million from the sale of such products.
Competition NexoBrid received orphan drug status in the European Union on July 31, 2002 and in the United States on August 20, 2003 for debridement of deep partial‑ and full‑thickness burns in hospitalized patients. In the United States and the European Union, a sponsor that develops an orphan drug has marketing exclusivity for seven years post‑approval by the FDA and for ten years post‑approval by the EMA, respectively. The exclusive marketing rights in both regions are subject to certain exceptions, including the development of a clinically significant benefit over the prevalent SOC. Once the market exclusivity for our orphan indication expires in a given jurisdiction, subject to other protections such as patents, we could face competition from other companies that may attempt to develop other products for the same indication.
The medical, biotechnology and pharmaceutical industries are intensely competitive and subject to significant technological change and changes in practice. While we believe that our innovative technology, knowledge, experience and scientific resources provide us with competitive advantages, we may face competition from many different sources with respect to NexoBrid, EscharEx, MW005 and our existing pipeline product candidates or any product candidates that we may seek to develop or commercialize in the future. Possible competitors may include medical practitioners, pharmaceutical and wound care companies, academic and medical institutions, governmental agencies and public and private research institutions, among others. Any product that we successfully develop and commercialize will compete with existing therapies and new therapies that may become available in the future. In addition, we face competition from the current SOC. The current SOC for eschar removal in severe burns is surgery, where eschar removal can be performed by tangential excision, dermabrasion or hydro jet, or non‑surgical alternatives, such as applying topical medications to the eschar to facilitate the natural healing process. Consequently, we face competition from traditional surgical procedures and topical agents. However, based on our clinical trials, we believe that NexoBrid has a sustainable competitive advantage over the current non‑surgical alternatives and is less invasive than surgery in removing eschar in patients with burn wounds. See “—NexoBrid“-NexoBrid and Our Clinical History” for the results of our clinical trials. Although we are in the clinical and preclinical phases for our pipeline product candidates for debridement of chronic and other hard‑to‑heal wounds and treatment of low risklow-risk basal cell carcinoma and connective tissue disorders and other indications, respectively, if one of our pipeline product candidates receives approval in the future, we would compete with traditional surgery and existing non‑surgical and other treatments. In chronic and other hard‑to‑heal wounds, we expect to face competition from current standard of care for debridement by sharp debridement or from the current non-surgical standard of care, either enzymatic debridement, primarily Smith & Nephew Plc’s Santyl,SANTYL, a collagenase-based product indicatedointment, approved by the FDA for debriding chronic dermal ulcers and severely burned areas or autolytic debridement. The current standard of care for treatment of low risk basal cell carcinoma,ulcers. SANTYL is surgical excision. In superficial basal cell carcinoma and inoperable nodular basal cell carcinoma, we expect to face competition from current topical applications such as imiquimod and 5FU.currently the market-leading enzymatic debridement product, with more than $360 million in estimated annual sales in the United States.
In addition to the currently available products, other products may be introduced to debride chronic and other hard‑to‑heal wounds or treat superficial and nodular basal cell carcinoma and connective tissue disorders during the time that we engage in necessary development. Accordingly, if one of our pipeline product candidates is approved, our main challenge in the market would be to educate physicians seeking alternatives to surgery or less effective non-surgical methods to use our product instead of already existing treatments. While we are still in the development stages, based on our studies, we believe that our pipeline product candidates will be more effective than the current non‑surgical alternatives and less invasive than surgery in removing escharnon-viable material in chronic and other hard‑to‑heal wounds or in tumor resection and may be comparable or perhaps better than currently available treatments for connective tissue disorders. NexoBrid received orphan drug status in the United States on August 20, 2003 for debridement of deep partial‑ and full‑thickness burns in hospitalized patients. In the United States, a sponsor that develops an orphan drug has marketing exclusivity for seven years post‑approval by the FDA. The exclusive marketing rights in both regions are subject to certain exceptions, including the development of a clinically significant benefit over the prevalent SOC. Once the market exclusivity for our orphan indication expires in a given jurisdiction, subject to other protections such as patents, we could face competition from other companies that may attempt to develop other products for the same indication.
Government Legislation and Regulation Our business is subject to extensive government regulation. Regulation by governmental authorities in the United States, the European Union and other jurisdictions is a significant factor in the development, manufacture and marketing of NexoBrid and in ongoing research and development activities.
European Union The approval process of medicinal products in the European Union generally involves satisfactorily completing each of the following: laboratory tests, animal studies and formulation studies all performed in accordance with the applicable E.U. GLP or GMP regulations;
submission to the relevant authorities of a clinical trial application (“CTA”), which must be approved before human clinical trials may begin;
performance of adequate and well‑controlled clinical trials to establish the safety and efficacy of the product for each proposed indication;
submission to the relevant competent authorities of a marketing authorization application (“MAA”), which includes the data supporting preclinical and clinical safety and efficacy as well as detailed information on the manufacture and composition and control of the product development and proposed labeling as well as other information;
inspection by the relevant national authorities of the manufacturing facility or facilities and quality systems (including those of third parties) at which the product is produced, to assess compliance with strictly enforced GMP;
potential audits of the non‑clinical and clinical trial sites that generated the data in support of the MAA; and
review and approval by the relevant competent authority of the MAA before any commercial marketing, sale or shipment of the product.
| • | laboratory tests, animal studies and formulation studies all performed in accordance with the applicable EU GLP or GMP regulations; | | • | submission to the relevant authorities of a CTA, which must be approved before human clinical trials may begin; | | • | performance of adequate and well‑controlled clinical trials to establish the safety and efficacy of the product for each proposed indication; | | • | submission to the relevant competent authorities of a marketing authorization application (“MAA”), which includes the data supporting preclinical and clinical safety and efficacy as well as detailed information on the manufacture and composition and control of the product development and proposed labeling as well as other information; | | • | inspection by the relevant national authorities of the manufacturing facility or facilities and quality systems (including those of third parties) at which the product is produced, to assess compliance with strictly enforced GMP; | | • | potential audits of the non‑clinical and clinical trial sites that generated the data in support of the MAA; and | | • | review and approval by the relevant competent authority of the MAA before any commercial marketing, sale or shipment of the product. |
Quality/preclinical studies In order to assess the potential safety and efficacy of a product, tests include laboratory evaluations of product characterization, analytical tests and controls, as well as studies to evaluate toxicity and pharmacological effects in animal studies. The conduct of the preclinical tests and formulation of the compounds for testing must comply with the relevant E.U. regulations and requirements. The results of such tests, together with relevant manufacturing control information and analytical data, are submitted as part of the CTA. Non-clinical studies are performed to demonstrate the health or environmental safety of new biological substances. Non-clinical studies must be conducted in compliance with the principles of good laboratory practice (“GLP”)GLP as set forth in EU Directive 2004/10/EC. In particular, non-clinical studies, both in vitro and in vivo, must be planned, performed, monitored, recorded, reported and archived in accordance with the GLP principles, which define a set of rules and criteria for a quality system for the organizational process and the conditions for non-clinical studies. These GLP standards reflect the Organization for Economic Co-operation and Development requirements.
Clinical trial approval Clinical drug development is often described as consisting of four temporal phases (Phase 1‑4)I‑IV). See, for example, the EMA’s note for guidance on general considerations for clinical trials (CPMP/ICH/291/95). Phase 1 (Most typical kind of study: Human Pharmacology);
Phase 2 (Most typical kind of study: Therapeutic Exploratory);
Phase 3 (Most typical kind of study: Therapeutic Confirmatory); and
Phase 4 (Variety of Studies: Therapeutic Use).
| • | Phase I (Most typical kind of study: Human Pharmacology); | | • | Phase II (Most typical kind of study: Therapeutic Exploratory); | | • | Phase III (Most typical kind of study: Therapeutic Confirmatory); and |
| • | Phase IV (Variety of Studies: Therapeutic Use). |
Studies in Phase 4IV are all studies other than routine surveillance performed after drug approval and are related to the approved indication. The phase of development provides an inadequate basis for classification of clinical trials because one type of trial may occur in several phases. The phase concept is a description, not a set of requirements. The temporal phases do not imply a fixed order of studies since for some drugs in a development plan the typical sequence will not be appropriate or necessary. Clinical trials of medicinal products in the EU must be conducted in accordance with EU and national regulations and the International Conference on Harmonization (“ICH”) guidelines on good clinical practices (“GCP”)GCP as well as the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki. If the sponsor of the clinical trial is not established within the EU, it must appoint an EU entity to act as its legal representative. The sponsor must take out a clinical trial insurance policy, and in most EU member states, the sponsor is liable to provide ‘no fault’ compensation to any study subject injured in the clinical trial. The regulatory landscape related to clinical trials in the EU has been subject to recent changes. The EU Clinical Trials Regulation (“CTR”) which was adopted in April 2014 and repeals the EU Clinical Trials Directive, became applicable on January 31, 2022. Unlike directives, the CTR is directly applicable in all EU member states without the need for member states to further implement it into national law. The CTR notably harmonizes the assessment and supervision processes for clinical trials throughout the EU via a Clinical Trials Information System, which contains a centralized EU portal and database. While the Clinical Trials Directive required a separate CTA to be submitted in each member state, to both the competent national health authority and an independent ethics committee, much like the FDA and IRB respectively, the CTR introduces a centralized process and only requires the submission of a single application to all member states concerned. The CTR allows sponsors to make a single submission to both the competent authority and an ethics committee in each member state, leading to a single decision per member state. The CTA must include, among other things, a copy of the trial protocol and an investigational medicinal product dossier containing information about the manufacture and quality of the medicinal product under investigation. The assessment procedure of the CTA has been harmonized as well, including a joint assessment by all member states concerned, and a separate assessment by each member state with respect to specific requirements related to its own territory, including ethics rules. Each member state’s decision is communicated to the sponsor via the centralized EU portal. Once the CTA is approved, clinical study development may proceed. The CTR foresees a three-year transition period. The extent to which ongoing and new clinical trials will be governed by the CTR varies. For clinicalClinical trials whose CTAfor which an application was madesubmitted (i) prior to January 31, 2022 under the Clinical Trials Directive, beforeor (ii) between January 31, 2022 and January 31, 2023 and for which the sponsor has opted for the application of the Clinical Trials Directive will continue to apply on a transitional basis for three years. Additionally, sponsors may still choose to submit a CTA under either the Clinical Trials Directive or the CTR until January 31, 2023 and, if authorized, those will beremain governed by the Clinical Trialssaid Directive until January 31, 2025. By thatAfter this date, all ongoingclinical trials (including those which are ongoing) will become subject to the provisions of the CTR. Medicines used in clinical trials must be manufactured in accordance with GMP. Other national and EU-wide regulatory requirements may also apply.
Pediatric investigation plan (“PIP”) We initiated a PIP study in November 2014. On January 26, 2007, Regulation (EC) 1901/2006 came into force with its primary purpose being the improvement of the health of children without subjecting children to unnecessary trials, or delaying the authorization of medicinal products for use in adults. The regulation established the Pediatric Committee (“PDCO”), which is responsible for coordinating the EMA’s activities regarding pharmaceutical drugsmedicinal products for children. The PDCO’s main role is to determine which studies the applicant needs to perform in the pediatric population as part of the PIP. All applications for marketing authorization for new pharmaceuticalmedicinal products that were not authorized in the EU prior to January 26, 2007 must include the results of studies carried out in children of different ages. The PDCO determines the requirements and procedures of such studies, describing them in a PIP. This requirement also applies when a company wants to add a new indication, pharmaceutical form or route of administration for a medicine that is already authorized. The PDCO can grant deferrals for some medicines, allowing a company to delay development of the medicine in children until there is enough information to demonstrate its effectiveness and safety in adults. The PDCO can also grant waivers when development of a medicine in children is not needed or is not appropriate, such asbecause the product is likely to be ineffective or unsafe in children, the disease or condition for diseases thatwhich the product is intended occurs only affectin adult populations, or when the elderly population. product does not represent a significant therapeutic benefit over existing treatments for pediatric patients. Before an MAA can be filed, or an existing marketing authorization can be amended, the EMA confirms that the applicant complied with the studies’ requirements and measures listed in the PIP. Since the regulation became effective, several incentives for the development of medicines for children become available in the European Union, including: medicines that have been authorized for marketing in the EU with the results of PIP studies included in the product information are eligible for an extension of their supplementary protection certificate extension (if any is in effect at the time of approval) by six months. This is the case even when the studies’ results are negative;
| • | medicines that have been authorized for marketing in the EU with the results of PIP studies included in the product information are eligible for an extension of their supplementary protection certificate extension (if any is in effect at the time of approval) by six months. This is the case even when the studies’ results are negative; | | • | for orphan medicines, such as NexoBrid, the incentive is an additional two years of market exclusivity instead of one; | | • | scientific advice and protocol assistance at the EMA are free of charge for questions relating to the development of medicines for children; and | | • | medicines developed specifically for children that are already authorized, but are not protected by a patent or supplementary protection certificate, can apply for a pediatric use marketing authorization (“PUMA”). If a PUMA is granted, the product will benefit from 10 years of market protection as an incentive. |
for orphan medicines, such as NexoBrid, the incentive is an additional two years of market exclusivity instead of one;
scientific advice and protocol assistance at the EMA are free of charge for questions relating to the development of medicines for children; and
medicines developed specifically for children that are already authorized, but are not protected by a patent or supplementary protection certificate, can apply for a pediatric use marketing authorization (“PUMA”). If a PUMA is granted, the product will benefit from 10 years of market protection as an incentive.
In November 2021,December 2023 we received positive scientific advice from the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) related to the pediatric label extension for NexoBrid. EMA’s CHMP agreed to assess a potential pediatric label extension on NexoBridCommission approval for the treatmentremoval of eschar in deep partial- and full-thickness thermal burns based on the available safety and efficacy results of the pivotal Phase 3 pediatric clinical study with its 12-month follow-up, and that the long-term follow up data are likely to be supportive data. Based on the feedback, the Company anticipates submitting a pediatric label extension request in the first half of 2022.for all ages.
Marketing authorization Authorization to market a product in the EU member states proceeds under one of four procedures: a centralized authorization procedure, a mutual recognition procedure, a decentralized procedure or a national procedure. Marketing authorization may be granted only to an applicant established in the European Union. Through our wholly‑owned German subsidiary, we received approval for NexoBrid pursuant to the centralized authorization procedure.
The centralized procedure provides for the grant of a single marketing authorization, issued by the European Commission based on the opinion of the EMA’s Committee for Medicinal Products for Human Use (“CHMP”), that is valid throughout the EU and the European Economic Area (“EEA”)EEA countries, and including Norway, Iceland and Lichtenstein. The centralized procedure is compulsory for medicines produced by certain biotechnological processes, products designated as orphan medicinal products, advanced therapy medicinal products (“ATMPs”) and products with a new active substance indicated for the treatment of certain diseases, and is optional for products which constitute a significant therapeutic, scientific, or technical innovation or for which a centralized process is in the interest of patients. Products that have received orphan designation in the EU, such as NexoBrid, will qualify for this centralized procedure, under which each product’s MAA is submitted to the EMA. Under the centralized procedure in the European Union, the maximum time frame for the evaluation of an MAA by the EMA is 210 days (excluding clock stops, when additional written or oral information is to be provided by the applicant in response to questions asked by the Committee of Medicinal Products for Human Use). In general, if the centralized procedure is not followed, there are three alternative procedures where applications are filed with one or more members state medicines regulators, each of which will grant a national marketing authorization:
| • | Mutual recognition procedure. If an authorization has been granted by one-member state, or the Reference Member State, an application may be made for mutual recognition in one or more other member states, or the Concerned Member State(s). |
| • | Decentralized procedure. The decentralized procedure may be used to obtain a marketing authorization in several European member states when the applicant does not yet have a marketing authorization in any country. |
| • | National procedure. Applicants following the national procedure will be granted a marketing authorization that is valid only in a single member state. Furthermore, this marketing authorization is not based on recognition of another marketing authorization for the same product awarded by an assessment authority of another member state. If marketing authorization in only one-member state is preferred, an application can be filed with the national competent authority of a member state. The national procedure can also serve as the first phase of a mutual recognition procedure. |
It is not always possible for applicants to follow the national procedure. In the case of medicinal products in the category for which the centralized authorization procedure is compulsory, that procedure must be followed. In addition, the national procedure is not available in the case of medicinal product dossiers where the same applicant has already obtained marketing authorization in one of the other European Union member state or has already submitted an application for marketing authorization in another member state and the application is under consideration. In the latter case, applicants must follow a mutual recognition procedure. After a drug has been authorized and launched, it is a condition of maintaining the marketing authorization that all aspects relating to its quality, safety and efficacy must be kept under review. Sanctions may be imposed for failure to adhere to the conditions of the marketing authorization. In extreme cases, the authorization may be revoked, resulting in withdrawal of the product from sale.
Period of authorization and renewals Under the above-described procedures, in order to grant the marketing authorization, the EMA or the competent authorities of the EU member states make an assessment of the risk benefit balance of the product on the basis of scientific criteria concerning its quality, safety and efficacy. Marketing authorization is valid for an initial five‑year period and may be renewed thereafter on the basis of a re‑evaluation of the risk‑benefit balance by the EMA or by the competent authority of the authorizing member state. To this end, the marketing authorization holder shall provide the EMA or other applicable competent authority a consolidated version of the file in respect of quality, safety and efficacy, including all variations introduced since the marketing authorization was granted, at least six months before the end of the initial five‑year period. Once renewed, the marketing authorization is valid for an unlimited period, unless the EMA or other applicable competent authority decides, on justified grounds relating to pharmacovigilance, to proceed with one additional five‑year renewal. Any authorization which is not followed by the actual placing of the drug on the E.U. market (in case of centralized procedure) or on the market of the authorizing member state within three years after authorization shall cease to be valid. On November 2017, the European Commission granted a five‑year renewal of our NexoBrid marketing authorization, and we plan to file for renewal during 2022.
Orphan designation62
On July 31, 2002, NexoBrid received orphan drug status in the European Union, and on December 20, 2012, the EMA confirmed NexoBrid’s designation as an orphan drug for marketing authorization.
In the EU, the Committee for Orphan Medicinal Products assesses orphan drug designation. The criteria for designating an “orphan medicinal product” in the EU are similar in principle to those in the United States. A medicinal product can be designated as an orphan if its sponsor can establish that (1) the product is intended for the diagnosis, prevention or treatment of a life threatening or chronically debilitating condition; (2) either (a) such condition affects not more than five in 10,000 persons in the EU when the application is made, or (b) the product, without the benefits derived from the orphan status, would not generate sufficient return in the EU to justify the necessary investment; and (3) there exists no satisfactory method of diagnosis, prevention or treatment of the condition in question that has been authorized for marketing in the EU or, if such method exists, the product will be of significant benefit to those affected by that condition.
In the EU, orphan drug designation also entitles a party to financial incentives such as reduction of fees or fee waivers and ten years of market exclusivity following drug or biological product approval. This period may be reduced to six years if, at the end of the fifth year, the orphan drug designation criteria are no longer met, including where it is shown that the product is sufficiently profitable not to justify maintenance of market exclusivity or a safer, more effective or otherwise clinically superior product is available. Granting of an authorization for another similar orphan medicinal product where another product has market exclusivity can happen at any time if: (i) the second applicant can establish that its product, although similar to the authorized product, is safer, more effective or otherwise clinically superior, (ii) inability of the applicant to supply sufficient quantities of the orphan medicinal product or (iii) where the applicant consents to a second orphan medicinal product application. A company may voluntarily remove a product from the orphan register.
Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process.
Regulatory data protection Without prejudice to the law on the protection of industrial and commercial property, some marketing authorizations benefit from an “8+2(+1)” year period of regulatory protection. During the first eight years from the grant of the innovator company’s marketing authorization, data exclusivity applies. If granted, the data exclusivity period prevents generic or biosimilar applicants from relying on the pre-clinical and clinical trial data contained in the dossier of the reference product when applying for a generic or biosimilar MA in the EU during a period of eight years from the date on which the reference product was first authorized in the EU. After the eight years have expired, a generic company can make use of the preclinical and clinical trial data of the originator in their regulatory applications but still cannot market their product until the end of the 10 years.year market exclusivity period. An additional one year of market exclusivity can be obtained if, during the first eight years of those 10 years, the marketing approvalauthorization holder obtains an approval for one or more new therapeutic indications which, during the scientific evaluation prior to their approval, are determined to bring a significant clinical benefit in comparison with existing therapies. However, there is no guarantee that a product will be considered by the EU’s regulatory authorities to be a new chemical or biological entity, and products may not qualify for data exclusivity. Under the current rules, a third party may reference the preclinical and clinical data of the reference product beginning eight years after first approval, but the third party may market a generic version only after 10 (or 11) years have lapsed. 58There is a special regime for biosimilars, or biological medicinal products that are similar to a reference medicinal product but that do not meet the definition of a generic medicinal product, for example, because of differences in raw materials or manufacturing processes. For such products, the results of appropriate preclinical or clinical trials must be provided, and guidelines from the EMA detail the type of quantity of supplementary data to be provided for different types of biological product. There are no such guidelines for complex biological products, such as gene or cell therapy medicinal products, and so it is unlikely that biosimilars of those products will currently be approved in the EU. However, guidance from the EMA states that they will be considered in the future in light of the scientific knowledge and regulatory experience gained at the time.
Additional data protection can be applied for when an applicant has complied with all requirements as set forth in an approved PIP. Post-Approval Requirements Similar to the United States, both marketing authorization holders and manufacturers of medicinal products are subject to comprehensive regulatory oversight by the EMA, the European Commission and/or the competent regulatory authorities of the member states. The holder of a marketing authorization must establish and maintain a pharmacovigilance system and appoint an individual qualified person for pharmacovigilance who is responsible for oversight of that system. Key obligations include expedited reporting of suspected serious adverse reactions and submission of periodic safety update reports (“PSURs”). All new MAA must include a risk management plan (“RMP”) describing the risk management system that the company will put in place and documenting measures to prevent or minimize the risks associated with the product. The regulatory authorities may also impose specific obligations as a condition of the marketing authorization. Such risk-minimization measures or post-authorization obligations may include additional safety monitoring, more frequent submission of PSURs, or the conduct of additional clinical trials or post-authorization safety studies. Failure to comply with the aforementioned EU and member state laws may result in administrative, civil or criminal penalties. These penalties could include delays or refusal to authorize the conduct of clinical trials, or to grant marketing authorization, product withdrawals and recalls, product seizures, suspension, withdrawal or variation of the marketing authorization, total or partial suspension of production, distribution, manufacturing or clinical trials, operating restrictions, injunctions, suspension of licenses, fines and criminal penalties. These penalties could include delays or refusal to authorize the conduct of clinical trials, or to grant the marketing authorization, product withdrawals and recalls, product seizures, suspension, withdrawal or variation of the marketing authorization, total or partial suspension of production, distribution, manufacturing or clinical trials, operating restrictions, injunctions, suspension of licenses, fines and criminal penalties. The aforementioned EU rules are generally applicable in the EEA. Brexit The United Kingdom (“UK”)UK left the EU on January 31, 2020, following which existing EU medicinal product legislation continued to apply in the United Kingdom during the transition period under the terms of the EU-UK Withdrawal Agreement. The transition period, which ended on December 31, 2020, maintained access to the EU single market and to the global trade deals negotiated by the EU on behalf of its members. The transition period provided time for the UK and EU to negotiate a framework for partnership for the future, which was then crystallized in the Trade and Cooperation Agreement (“TCA”) and became effective on the January 1, 2021. The TCA includes specific provisions concerning pharmaceuticals, which include the mutual recognition of GMP inspections of manufacturing facilities for medicinal products and GMP documents issued but does not foresee wholesale mutual recognition of UK and EU pharmaceutical regulations.
EU laws which have been transposed into UK law through secondary legislation continue to be applicable as “retained EU law”. However, under the Retained EU Law (Revocation and Reform) Bill 2022, which is currently before the UK parliament, any retained EU law not expressly preserved and “assimilated” into domestic law or extended by ministerial regulations (to no later than 23 June 2026) will automatically expire and be revoked by December 31, 2023. In addition, new legislation such as the EU CTR or in relation to orphan medicines will not be applicable. The UK government has passed a new Medicines and Medical Devices Act 2021, which introduces delegated powers in favourfavor of the Secretary of State or an ‘appropriate authority’ to amend or supplement existing regulations in the area of medicinal products and medical devices. This allows new rules to be introduced in the future by way of secondary legislation, which aims to allow flexibility in addressing regulatory gaps and future changes in the fields of human medicines, clinical trials and medical devices. As of January 1, 2021, the Medicines and Healthcare productsProducts Regulatory Agency (“MHRA”) is the UK’s standalone medicines and medical devices regulator. As a result of the Northern Ireland protocol, different rules will apply in Northern Ireland than in England, Wales, and Scotland, together, Great Britain (“GB”); broadly, Northern Ireland will continue to follow the EU regulatory regime, but its national competent authority will remain the MHRA. The MHRA has published a guidance on how various aspects of the UK regulatory regime for medicines will operate in GB and in Northern Ireland following the expiry of the Brexit transition period on December 31, 2020. The guidance includes clinical trials, importing, exporting, and pharmacovigilance and is relevant to any business involved in the research, development, or commercialization of medicines in the UK. The new guidance was given effect via the Human Medicines Regulations (Amendment etc.) (EU Exit) Regulations 2019 (the “Exit Regulations”). The MHRA has introduced changes to national licensing procedures, including procedures to prioritize access to new medicines that will benefit patients, including a 150-day assessment and a rolling review procedure. All existing EU MAs for centrally authorized products were automatically converted or grandfathered into UK MAs, effective in GB (only), free of charge on January 1, 2021, unless the MA holder chooseschose to opt-out. After Brexit, companies established in the UK cannot use the centralized procedure and instead must follow one of the UK national authorization procedures or one of the remaining post-Brexit international cooperation procedures to obtain an MA to commercialize products in the UK. TheFor a period of three years from January 1, 2021, the MHRA may rely on a decision taken by the an Commission on the approval of a new (centralized procedure) MA when determining an application for a GB authorization; or use the MHRA’s decentralized or mutual recognition procedures which enable MAs approved in EU member states (or Iceland, Liechtenstein, Norway) to be granted in GB. There will be no pre-MA orphan designation. Instead, the MHRA will review applications for orphan designation in parallel to the corresponding MA application. The criteria are essentially the same, but have been tailored for the market, i.e., the prevalence of the condition in GB, rather than the EU, must not be more than five in 10,000. Should an orphan designation be granted, the period or market exclusivity will be set from the date of first approval of the product in GB. Data Privacy and Security Laws Numerous state, federal and foreign laws, regulations, and standards govern the collection, use, access to, confidentiality and security of health-related and other personal information, and could apply now or in the future to our operations or the operations of our partners. In the United States, numerous federal and state laws and regulations, including data breach notification laws, health information privacy and security laws and consumer protection laws and regulations govern the collection, use, disclosure, and protection of health-related and other personal information. In addition, certain laws govern the privacy and security of personal data, including health-related data in the EU/EEA and in other foreign jurisdictions. For example, the GDPR imposes strict requirements for processing the personal data of individuals within the EEA. Companies that must comply with the GDPR face increased compliance obligations and risk, including more robust regulatory enforcement of data protection requirements and potential fines for noncompliance of up to €20 million or 4% of the annual global revenues of the noncompliant company, whichever is greater. Further, from January 1, 2021, companies have had to comply with the GDPR and also the UK GDPR, which, together with the amended UK Data Protection Act 2018, retains the GDPR in UK national law. The UK GDPR mirrors the fines under the GDPR, i.e., fines up to the greater of €20 million (£17.5 million) or 4% of global turnover. Privacy and security laws, regulations, and other obligations are constantly evolving, may conflict with each other to complicate compliance efforts, and can result in investigations, proceedings, or actions that lead to significant civil and/or criminal penalties and restrictions on data processing. Manufacturing The manufacturing of authorized drugs, for which a separate manufacturer’s license is mandatory, must be conducted in strict compliance with the EMA’s cGMP requirements and comparable requirements of other regulatory bodies, which mandate the methods, facilities and controls used in manufacturing, processing and packing of drugs to assure their safety and proper identification. The EMA monitors compliance with its GMP requirements through mandatory registration of facilities and inspections of those facilities. The EMA may have a coordinating role for these inspections while the responsibility for carrying them out rests with the competent authority of the member state under whose responsibility the manufacturer falls. Failure to comply with these requirements could interrupt supply and result in delays, unanticipated costs and lost revenues, and could subject the applicant to potential legal or regulatory action, including but not limited to warning letters, suspension of manufacturing, seizure of product, injunctive action or possible civil and criminal penalties. In January 2013, the EU and Israel signed the Protocol on Conformity Assessment and Acceptance of Industrial Products (the “ACAA”), which covers medicinal products. The ACAA provides for mutual recognition of the conclusions of inspections of compliance of manufacturers and importers with the principles and guidelines of EU GMP and equivalent Israeli cGMP. Certification of the conformity of each batch to its specifications by either the importer or the manufacturer established in Israel or in the EU shall be recognized by the other party without re‑control at import from one party to the other.
Marketing and promotion The marketing and promotion of authorized drugs,medicinal products, including industry‑sponsored continuing medical education and advertising directed toward the prescribers of drugs and/or the general public, are strictly regulated in the European Union, notably under Directive 2001/83 and subject to laws concerning promotion of medicinal products, interactions with physicians, misleading and comparative advertising and unfair commercial practices. The applicable legislation aims to ensure that information provided by holders of marketing authorizations regarding their products is truthful, balanced and accurately reflects the safety and efficacy claims authorized by the EMA or by the applicable national authority of the authorizing member state. All advertising and promotional activities for the product must be consistent with the approved summary of product characteristics, and therefore all off-label promotion is prohibited. Direct-to-consumer advertising of prescription medicines is also prohibited in the EU. Although general requirements for advertising and promotion of medicinal products are established under EU directives, the details are governed by regulations in each member state and can differ from one country to another. Failure to comply with these requirements can result in adverse publicity, warning letters, mandated corrective advertising and potential civil and criminal penalties. United States Review and approval of biologics In addition to E.U. regulations, NexoBrid is an investigational drugmarketed as a biologic product in the United States for eschar removal in adults with deep partial thickness and/or full thickness thermal burns and is therefore subject to various U.S. regulations. In the United States, the FDA regulates biologics under the Federal, Food, Drug and Cosmetic Act (“FDCA”), the Public Health Service Act, and their respective implementation regulations. On March 24, 2011, the FDA classified NexoBrid as a biological product. Biologics require the submission of a BLA and licensure by the FDA prior to being marketed in the United States. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations requires the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval may subject an applicant to a variety of administrative or judicial sanctions as well as enforcement actions brought by the FDA, the U.S. Department of Justice or other governmental entities. Possible sanctions may include the FDA’s refusal to approve pending BLAs or supplements, withdrawal of an approval, imposition of a clinical hold, issuance of warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement and civil or criminal penalties.
The process required by the FDA prior to marketing and distributing a biologic in the United States generally involves the following: completion of laboratory tests, animal studies and formulation studies in compliance with the FDA’s GLP and GMP regulations, as applicable;
submission to the FDA of an investigational new drug application (“IND”), which must become effective before clinical trials may begin;
approval by an independent institutional review board (“IRB”) at each clinical site before each trial may be initiated;
performance of adequate and well‑controlled clinical trials in accordance with GCP to establish the safety and efficacy of the product for each indication;
preparation and submission to the FDA of a BLA;
satisfactory completion of an FDA advisory committee review, if applicable;
satisfactory completion of one or more FDA inspections of the manufacturing facility or facilities at which the product, or components thereof, are produced to assess compliance with cGMP requirements, and to assure that the facilities, methods and controls are adequate to preserve the product’s safety, purity and potency, and of selected clinical investigation sites to assess compliance with GCP; and
payment of user fees and FDA review and approval of the BLA to permit commercial marketing of the product for particular indications for use in the United States.
| • | completion of laboratory tests, animal studies and formulation studies in compliance with the FDA’s GLP and GMP regulations, as applicable; | | • | submission to the FDA of an investigational new drug application (“IND”), which must become effective before clinical trials may begin; | | • | approval by an independent institutional review board (“IRB”) at each clinical site before each trial may be initiated; | | • | performance of adequate and well‑controlled clinical trials in accordance with GCP to establish the safety and efficacy of the product for each indication; | | • | preparation and submission to the FDA of a BLA; | | • | satisfactory completion of an FDA advisory committee review, if applicable; | | • | satisfactory completion of one or more FDA inspections of the manufacturing facility or facilities at which the product, or components thereof, are produced to assess compliance with cGMP requirements, and to assure that the facilities, methods and controls are adequate to preserve the product’s safety, purity and potency, and of selected clinical investigation sites to assess compliance with GCP; and | | • | payment of user fees and FDA review and approval of the BLA to permit commercial marketing of the product for particular indications for use in the United States. |
Preclinical studies Preclinical studies include laboratory evaluation of product chemistry, toxicity and formulation, as well as animal studies to assess the potential safety and efficacy of the product candidate. Preclinical safety tests must be conducted in compliance with FDA regulations regarding good laboratory practices. The results of the preclinical tests, together with manufacturing information and analytical data, are submitted to the FDA as part of an IND which must become effective before clinical trials may commence. Some preclinical testing may continue even after the IND is submitted. Clinical trials in support of a BLA Clinical trials involve the administration of an investigational product to human subjects under the supervision of qualified investigators in accordance with GCP requirements, which include, among other things, the requirement that all research subjects provide their informed consent in writing before their participation in any clinical trial. Clinical trials are conducted under written study protocols detailing, among other things, the objectives of the study, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. A protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA as part of the IND. An IND automatically becomes effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions related to a proposed clinical trial and places the trial on clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. In addition, an IRB representing each institution participating in the clinical trial must review and approve the plan for any clinical trial before it commences at that institution, and the IRB must conduct continuing review and reapprove the study at least annually. The IRB must review and approve, among other things, the study protocol and informed consent information to be provided to study subjects. An IRB must operate in compliance with FDA regulations. Information about certain clinical trials must be submitted within specific timeframes to the National Institutes of Health for public dissemination on their website, ClinicalTrials.gov.
For purposes of BLA approval, clinical trials are typically conducted in three sequential phases, which may overlap or be combined. In the United States, the three phases are generally described as follows:
| Phase 1:I: | The investigational product is initially introduced into healthy human subjects or patients with the target disease or condition and tested for safety, dosage tolerance, absorption, metabolism, distribution, excretion and, if possible, to gain an early indication of its effectiveness and to determine optimal dosage. |
| Phase 2:II: | The investigational product is administered to a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage. |
| Phase 3: | Phase III: | The investigational product is administered to an expanded patient population, generally at geographically dispersed clinical trial sites, in well‑controlled clinical trials to generate enough data to statistically evaluate the efficacy and safety of the product for approval, to establish the overall risk‑benefit profile of the product, and to provide adequate information for the labeling of the product. |
In some cases, the FDA may require, or companies may voluntarily pursue, additional clinical trials after a product is approved to gain more information about the product. These so-called Phase 4IV studies may be made a condition to approval of the BLA. Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and more frequently if serious adverse events occur. Phase 1,I, Phase 2II and Phase 3III clinical trials may not be completed successfully within any specified period, or at all. Furthermore, the FDA or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients.
Submission of a BLA to the FDA The results of the preclinical studies and clinical trials, together with other detailed information, including information on the manufacture, control and composition of the product, are submitted to the FDA as part of a BLA requesting approval to market the product candidate for a proposed indication. Under the Prescription Drug User Fee Act (PDUFA), as amended, applicants are required to pay user fees to the FDA for reviewing a BLA. These user fees, as well as the annual program fees required for approved products, can be substantial. Each BLA submitted to the FDA for approval is typically reviewed for administrative completeness and reviewability within 60 days following submission of the application. If found complete, the FDA will “file” the BLA, which triggers a full review of the application. The FDA may refuse to file any BLA that it deems incomplete or not properly reviewable at the time of submission. The FDA’s established goals are to review and act on standard applications within ten months after it accepts the application for filing, or, if the application qualifies for priority review, six months after the FDA accepts the application for filing. In both standard and priority reviews, the review process is often significantly extended by FDA requests for additional information or clarification. The FDA reviews a BLA to determine, among other things, whether a product is safe, pure and potent and the facility in which it is manufactured, processed, packed, or held meets standards designed to assure the product’s continued safety, purity and potency. The FDA may convene an advisory committee to provide clinical insight on application review questions. Before approving a BLA, the FDA generally inspects the facilities at which the product is manufactured or facilities that are significantly involved in the product development and distribution process, and will not approve the product unless cGMP compliance is satisfactory. Additionally, before approving a BLA, the FDA will typically inspect one or more clinical sites to assure compliance with GCP. If the FDA determines that the application, manufacturing process or manufacturing facilities are not acceptable, it will outline the deficiencies in the submission and often will request additional testing or information. Notwithstanding the submission of any requested additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval. After the FDA evaluates a BLA and conducts inspections of manufacturing facilities where the investigational product will be produced, the FDA may issue an approval letter or a Complete Response letter. An approval letter authorizes commercial marketing of the product with specific prescribing information for specific indications. A Complete Response letter will describe all of the deficiencies that the FDA has identified in the BLA, except that where the FDA determines that the data supporting the application are inadequate to support approval, the FDA may issue the Complete Response letter without first conducting required inspections, testing submitted product lots, and/or reviewing proposed labeling. In issuing the Complete Response letter, the FDA may recommend actions that the applicant might take to place the BLA in condition for approval, including requests for additional information or clarification.
The FDA may deny approval of a BLA if applicable statutory or regulatory criteria are not satisfied, or may require additional testing or information, which can delay the approval process. FDA approval of any application may include many delays or may never be granted. If a product is approved, the approval will impose limitations on the indicated uses for which the product may be marketed, will require that warning statements be included in the product labeling, may impose additional warnings to be specifically highlighted in the labeling (e.g., a Black Box Warning), which can significantly affect promotion and sales of the product, may require that additional studies be conducted following approval as a condition of the approval and may impose restrictions and conditions on product distribution, prescribing or dispensing. For example, the FDA may approve the BLA with a Risk Evaluation and Mitigation Strategy, or REMS, to ensure the benefits of the product outweigh its risks. A REMS is a safety strategy to manage a known or potential serious risk associated with a product and to enable patients to have continued access to such medicines by managing their safe use. A REMS program may be required to include various elements, such as a medication guide or patient package insert, a communication plan to educate healthcare providers of the drug’s risks, or other elements to assure safe use, such as limitations on who may prescribe or dispense the drug, dispensing only under certain circumstances, special monitoring and the use of patient registries. Once a product is approved, marketing the product for other indicated uses or making certain manufacturing or other changes requires FDA review and approval of a supplemental BLA or a new BLA, which may require additional clinical data. In addition, further post‑marketing testing and surveillance to monitor the safety or efficacy of a product may be required. Also, product approvals may be withdrawn if compliance with regulatory standards is not maintained or if safety or manufacturing problems occur following initial marketing. In addition, new government requirements may be established that could delay or prevent regulatory approval of our product candidates under development. Post‑approval requirements Any biologic products for which we receive FDA approvals are subject to pervasive continuing regulation by the FDA. Certain requirements include, among other things, record‑keeping requirements, reporting adverse experiences with the product, providing the FDA with updated safety and efficacy information annually or more frequently for specific events, product sampling and distribution requirements, complying with certain electronic records and signature requirements and complying with FDA promotion and advertising requirements. These promotion and advertising requirements include, among others, standards for direct‑to‑consumer advertising, prohibitions against promoting drugs for uses or in patient populations that are not described in the drug’s approved labeling, known as “off‑label use,” and other promotional activities, such as those considered to be false or misleading. Failure to comply with FDA requirements can have negative consequences, including the immediate discontinuation of noncomplying materials, adverse publicity, enforcement letters from the FDA, mandated corrective advertising or communications with doctors, and civil or criminal penalties. Such enforcement may also lead to scrutiny and enforcement by other government and regulatory bodies. Although physicians may prescribe legally available drugs for off‑label uses, manufacturers may not encourage, market or promote such off‑label uses. As a result, “off‑label promotion” has formed the basis for litigation under the Federal False Claims Act, violations of which are subject to significant civil fines and penalties.
The manufacturing of NexoBrid, EscharEx and our pipeline product candidates are and will be required to comply with applicable FDA manufacturing requirements contained in the FDA’s cGMP regulations. NexoBrid is manufactured at our production plant in Yavne, Israel, which is cGMP certified. The FDA’s cGMP regulations require, among other things, quality control and quality assurance, as well as the corresponding maintenance of comprehensive records and documentation. Biologic manufacturers and other entities involved in the manufacture and distribution of approved drugs and biologics are also required to register their establishments and list any products they make with the FDA and to comply with related requirements in certain states. These entities are further subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP and other laws. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain cGMP compliance. In addition, a BLA holder must comply with post‑marketing requirements, such as reporting of certain adverse events. Such reports can present liability exposure, as well as increase regulatory scrutiny that could lead to additional inspections, labeling restrictions or other corrective action to minimize further patient risk. Discovery of problems with a product after approval may result in serious and extensive restrictions on the product, manufacturer or holder of an approved BLA, as well as lead to potential market disruptions. These restrictions may include recalls, fines, warning letters, or untitled letters, clinical holds on clinical studies, refusal of the FDA to approve pending applicants or supplements to approved applications, product seizure or detention, or refusal to permit the import or export of products, suspension or revocation of a product license approval until the FDA is assured that quality standards can be met, and continuing oversight of manufacturing by the FDA under a “consent decree,” which frequently includes the imposition of costs and continuing inspections over a period of many years, as well as possible withdrawal of the product from the market. In addition, changes to the manufacturing process generally require prior FDA approval before being implemented. Other types of changes to the approved product, such as adding new indications and additional labeling claims, are also subject to further FDA review and approval. The FDA also may impose a number of post‑approval requirements as a condition of approval of a BLA. For example, the FDA may require post‑marketing testing, or Phase 4IV testing, as well as REMS and/or surveillance to monitor the effects of an approved product or place other conditions on an approval that could otherwise restrict the distribution or use of NexoBrid.
Orphan designation and exclusivity On August 20, 2003, NexoBrid received orphan drug designation in the United States. Under the Orphan Drug Act, the FDA may designate a drug product as an “orphan drug” if it is intended to treat a rare disease or condition, meaning that it affects fewer than 200,000 individuals in the United States, or more in cases in which there is no reasonable expectation that the cost of developing and making a drug product available in the United States for treatment of the disease or condition will be recovered from sales of the product. A company must request orphan product designation before submitting a BLA. If the request is granted, the FDA will disclose the identity of the therapeutic agent and its potential use. Orphan drug designation does not convey any advantage in or shorten the duration of the regulatory review and approval process. If a product with orphan status receives the first FDA approval for the disease or condition for which it has such designation, the product will be entitled to orphan product exclusivity. Orphan product exclusivity means that FDA may not approve any other applications for the same product for the same disease or condition for seven years, except in certain limited circumstances, such as a showing of clinical superiority to the product with orphan drug exclusivity. Competitors may receive approval of different products for the indicationdisease or condition for which the orphan product has exclusivity and may obtain approval for the same product but for a different indication. If a drug or drug product designated as an orphan product ultimately receives marketing approvalauthorization for an indicationa disease or condition broader than that designated in its orphan product application, it may not be entitled to exclusivity. In addition, exclusive marketing rights in the United States may be lost if the FDA later determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantities of the product to meet the needs of patients with the rare disease or condition. Expedited Development and Review Programs The FDA offers a number of expedited development and review programs for qualifying product candidates. The fast track program is intended to expedite or facilitate the process for reviewing new products that meet certain criteria. Specifically, new products are eligible for fast track designation if they are intended to treat a serious or life-threatening disease or condition and demonstrate the potential to address unmet medical needs for the disease or condition. Fast track designation applies to the combination of the product and the specific indication for which it is being studied. The sponsor of a fast track product has opportunities for frequent interactions with the review team during product development and, once a BLA is submitted, the product may be eligible for priority review. A fast track product may also be eligible for rolling review, where the FDA may consider for review sections of the BLA on a rolling basis before the complete application is submitted, if the sponsor provides a schedule for the submission of the sections of the BLA, the FDA agrees to accept sections of the BLA and determines that the schedule is acceptable, and the sponsor pays any required user fees upon submission of the first section of the BLA. A product intended to treat a serious or life-threatening disease or condition may also be eligible for breakthrough therapy designation to expedite its development and review. A product can receive breakthrough therapy designation if preliminary clinical evidence indicates that the product may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. The designation includes all of the fast track program features, as well as more intensive FDA interaction and guidance beginning as early as Phase 1I and an organizational commitment to expedite the development and review of the product, including involvement of senior managers. Any marketing application for a biologic submitted to the FDA for approval, including a product with a fast track designation and/or breakthrough therapy designation, may be eligible for other types of FDA programs intended to expedite the FDA review and approval process, such as priority review and accelerated approval. A product is eligible for priority review if it has the potential to provide a significant improvement in the treatment, diagnosis or prevention of a serious disease or condition compared to marketed products. For products containing new molecular entities, priority review designation means the FDA’s goal is to take action on the marketing application within six months of the 60-day filing date, compared with ten months under standard review.
Additionally, products studied for their safety and effectiveness in treating serious or life-threatening diseases or conditions may receive accelerated approval upon a determination that the product has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. As a condition of accelerated approval, the FDA will generally require the sponsor to perform adequate and well-controlled post-marketing clinical studies to verify and describe the anticipated effect on irreversible morbidity or mortality or other clinical benefit. In addition, the FDA currently requires as a condition for accelerated approval pre-approval of promotional materials, which could adversely impact the timing of the commercial launch of the product. In 2017, FDA established a new regenerative medicine advanced therapy, or RMAT, designation as part of its implementation of the 21st Century Cures Act, which was signed into law in December 2016. To qualify for RMAT designation, the product candidate must meet the following criteria: (1) it qualifies as a RMAT, which is defined as a cell therapy, therapeutic tissue engineering product, human cell and tissue product, or any combination product using such therapies or products, with limited exceptions; (2) it is intended to treat, modify, reverse, or cure a serious or life-threatening disease or condition; and (3) preliminary clinical evidence indicates that the drug has the potential to address unmet medical needs for such a disease or condition. Like fast track and breakthrough therapy designation, RMAT designation provides potential benefits that include more frequent meetings with FDA to discuss the development plan for the product candidate and eligibility for rolling review and priority review. Products granted RMAT designation may also be eligible for accelerated approval on the basis of a surrogate or intermediate endpoint reasonably likely to predict long-term clinical benefit, or reliance upon data obtained from a meaningful number of sites, including through expansion to additional sites. Once approved, when appropriate, the FDA can permit fulfillment of post-approval requirements under accelerated approval through the submission of clinical evidence, clinical studies, patient registries, or other sources of real world evidence such as electronic health records; through the collection of larger confirmatory datasets; or through post-approval monitoring of all patients treated with the therapy prior to approval. Fast track designation, breakthrough therapy designation, priority review, accelerated approval, and RMAT designation do not change the standards for approval but may expedite the development or approval process. Pediatric studies and exclusivity Under the Pediatric Research Equity Act of 2003, a BLA or supplement thereto must contain data that are adequate to assess the safety and effectiveness of the drug product for the claimed indications in all relevant pediatric subpopulations, and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. Sponsors must also submit pediatric study plans prior to the assessment data. Those plans must contain an outline of the proposed pediatric study or studies the applicant plans to conduct, including study objectives and design, any deferral or waiver requests, and other information required by regulation. The applicant, the FDA, and the FDA’s internal review committee must then review the information submitted, consult with each other and agree upon a final plan. The FDA or the applicant may request an amendment to the plan at any time.
The FDA may, on its own initiative or at the request of the applicant, grant deferrals for submission of some or all pediatric data until after approval of the product for use in adults, or full or partial waivers from the pediatric data requirements. Additional requirements and procedures relating to deferral requests and requests for extension of deferrals are contained in the FDASIA. Unless otherwise required by regulation, the pediatric data requirements do not apply to products with orphan designation. Separately, in the event the FDA issues a Written Request for pediatric data relating to a product, a BLA sponsor who submits such data may be entitled to pediatric exclusivity. Pediatric exclusivity is another type of non‑patent marketing exclusivity in the United States which, if granted, provides for the attachment of an additional six months of marketing protection to the term of any existing exclusivity, including other non‑patent and orphan exclusivity. This six‑month exclusivity may be granted if a BLA sponsor submits pediatric data that fairly respond to the Written Request from the FDA for such data. The data do not need to show that the product is effective in the pediatric population studied; rather, if the clinical trial is deemed to fairly respond to the FDA’s request, the additional protection is granted. If reports of requested pediatric studies are submitted to and accepted by the FDA within the statutory time limits, whatever statutory or regulatory periods of exclusivity or patent protection cover the product are extended by six months. This is not a patent term extension, but it effectively extends the regulatory period during which the FDA cannot accept or approve another application.
The Animal Rule70
In the case of product candidates that are intended to treat certain rare life-threatening diseases, conducting controlled clinical trials to determine efficacy may be unethical or unfeasible. Under regulations issued by the FDA in 2002, often referred to as the “Animal Rule”, the approval of such products can be based on clinical data from trials in healthy human subjects that demonstrate adequate safety and efficacy data from adequate and well-controlled animal studies. Among other requirements, the animal studies must establish that the drug or biological product is reasonably likely to produce clinical benefits in humans. Because the FDA must agree that data derived from animal studies may be extrapolated to establish safety and effectiveness in humans, seeking approval under the Animal Rule may add significant time, complexity and uncertainty to the testing and approval process. In addition, products approved under the Animal Rule are subject to additional requirements including post-marketing study requirements, restrictions imposed on marketing or distribution or requirements to provide information to patients.
Patent term restoration and extension A patent claiming a new drug product may be eligible for a limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984 (the “Hatch‑Waxman Act”), which permits a patent restoration of up to five years for the patent term lost during product development and the FDA regulatory review. The restoration period granted is typically one‑half the time between the effective date of an IND and the submission date of a BLA, plus the time between the submission date of a BLA and the ultimate approval date. Patent term restoration cannot be used to extend the remaining term of a patent past a total of fourteen years from the product’s approval date. Only one patent applicable to an approved drug product is eligible for the extension, and the application for the extension must be submitted prior to the expiration of the patent in question. A patent that covers multiple drugs for which approval is sought can only be extended in connection with one of the approvals. The U.S. Patent and Trademark Office reviews and approves the application for any patent term extension or restoration in consultation with the FDA. Biosimilars and reference product exclusivity The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act (collectively, the “ACA”), which was signed into law in 2010, includes a subtitle called the Biologics Price Competition and Innovation Act of 2009 (“BPCIA”), which created an abbreviated approval pathway for biological products that are biosimilar to or interchangeable with an FDA-approved reference biological product. Biosimilarity, which requires that there be no clinically meaningful differences between the biological product and the reference product in terms of safety, purity, and potency, can be shown through analytical studies, animal studies, and a clinical study or studies. Interchangeability requires that a product is biosimilar to the reference product and the product must demonstrate that it can be expected to produce the same clinical results as the reference product in any given patient and, for products that are administered multiple times to an individual, the biologic and the reference biologic may be alternated or switched after one has been previously administered without increasing safety risks or risks of diminished efficacy relative to exclusive use of the reference biologic. However, complexities associated with the larger, and often more complex, structures of biological products, as well as the processes by which such products are manufactured, pose significant hurdles to implementation of the abbreviated approval pathway that are still being worked out by the FDA.
Under the BPCIA, an application for a biosimilar product may not be submitted to the FDA until four years following the date that the reference product was first licensed by the FDA. In addition, the approval of a biosimilar product may not be made effective by the FDA until 12 years from the date on which the reference product was first licensed. During this 12‑year period of exclusivity, another company may still market a competing version of the reference product if the FDA approves a full BLA for the competing product containing the sponsor’s own preclinical data and data from adequate and well‑controlled clinical trials to demonstrate the safety, purity and potency of their product. The BPCIA also created certain exclusivity periods for biosimilars approved as interchangeable products. At this juncture, it is unclear whether products deemed “interchangeable” by the FDA will, in fact, be readily substituted by pharmacies, which are governed by state pharmacy law. The BPCIA is complex and continues to be interpreted and implemented by the FDA. In addition, recent government proposals have sought to reduce the 12‑year reference product exclusivity period. Other aspects of the BPCIA, some of which may impact the BPCIA exclusivity provisions, have also been the subject of recent litigation. As a result, the ultimate impact, implementation, and meaning of the BPCIA remains subject to significant uncertainty.
Review and Approval of Drug Products Outside the European Union and the United States In addition to the above regulations, we must obtain approval of a product by the comparable regulatory authorities of foreign countries outside of the European Union and the United States before we can commence clinical trials or marketing of NexoBrid in those countries. The approval process varies from country to country and the time may be longer or shorter than that required for FDA or EMAEU approval. In addition, the requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country. In all cases, clinical trials are conducted in accordance with GCP and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki. If we fail to comply with applicable regulatory requirements, we may be subject to, among other things, fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution. Pharmaceutical Coverage, Pricing and Reimbursement Significant uncertainty exists as to the coverage and reimbursement status of any products for which we obtain regulatory approval. In the United States, EU and other markets, sales of any products for which we receive regulatory approval for commercial sale will depend to a large extent on the availability of reimbursement from third‑party payors. Third‑party payors include governments, government health administrative authorities, managed care providers, private health insurers and other organizations. The process for determining whether a payor will provide coverage for a drug product may be separate from the process for setting the price or reimbursement rate that the payor will pay for the drug product. Third‑party payors may limit coverage to specific drug products on an approved list, or formulary, which might not include all of the drug products approved for a particular indication by the FDA, European Commission or National Ministries of Health. Third‑party payors are increasingly challenging the price and examining the medical necessity and cost‑effectiveness of medical products and services, in addition to their safety and efficacy. We may need to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost‑effectiveness of NexoBrid, in addition to the costs required to obtain the FDA or other Ministry of Health approvals. Additionally, NexoBrid may not be considered medically necessary or cost‑effective. A payor’s decision to provide coverage for a drug product does not guarantee that an adequate reimbursement rate will be approved. Adequate third‑party reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development. In the United States, the ACA substantially changed the way healthcare is financed by both governmental and private insurers and significantly impacted the pharmaceutical industry. The ACA contains a number of provisions, including those governing enrollment in federal healthcare programs, reimbursement changes and fraud and abuse provisions, which will impact existing government healthcare programs and will result in the development of new programs, including Medicare payment for performance initiatives and improvements to the physician quality reporting system and feedback program. Additionally, the ACA: increases the minimum level of Medicaid rebates payable by manufacturers of brand‑name drugs from 15.1% to 23.1%;
| • | increases the minimum level of Medicaid rebates payable by manufacturers of brand‑name drugs from 15.1% to 23.1%; | | • | requires collection of rebates for drugs paid by Medicaid managed care organizations; and | | • | imposes a non‑deductible annual fee on pharmaceutical manufacturers or importers who sell certain “branded prescription drugs” to specified federal government programs. |
requires collection of rebates for drugs paid by Medicaid managed care organizations; and72
imposes a non‑deductible annual fee on pharmaceutical manufacturers or importers who sell certain “branded prescription drugs” to specified federal government programs.
Since its enactment, there have been judicial, executive and congressional challenges to certain aspects of the ACA. On June 17, 2021, the U.S. Supreme Court dismissed the most recent judicial challenge to the ACA brought by several states without specifically ruling on the constitutionality of the ACA. Thus, the ACA will remain in effect in its current form. Further, prior to the U.S. Supreme Court ruling, President Biden issued an executive order that initiated a special enrollment period for purposes of obtaining health insurance coverage through the ACA marketplace from February 15, 2021 through August 15, 2021. The executive order instructed certain governmental agencies to review and reconsider their existing policies and rules that limit access to healthcare, including among others, reexamining Medicaid demonstration projects and waiver programs that include work requirements, and policies that create unnecessary barriers to obtaining access to health insurance coverage through Medicaid or the ACA. There has been heightened governmental scrutiny recently over the manner in which drug manufacturers set prices for their marketed products, which have resulted in several Congressional inquiries and proposed bills designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drug products. . In March 2021, the American Rescue Plan Act of 2021 was signed into law, which eliminates the statutory Medicaid drug rebate cap, currently set at 100% of a drug’s average manufacturer price, for single source and innovator multiple source drugs, beginning January 1, 2024. In August 2022, the Inflation Reduction Act of 2022, or IRA, was signed into law. Among other things, the IRA requires manufacturers of certain drugs to engage in price negotiations with Medicare (beginning in 2026), with prices that can be negotiated subject to a cap; imposes rebates under Medicare Part B and Medicare Part D to penalize price increases that outpace inflation (first due in 2023); and replaces the Part D coverage gap discount program with a new discounting program (beginning in 2025). The IRA permits the Secretary of the Department of Health and Human Services to implement many of these provisions through guidance, as opposed to regulation, for the initial years. For that and other reasons, it is currently unclear how the IRA will be effectuated, or the impact of the IRA on our business. We expect that additional U.S. federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that the U.S. federal government will pay for healthcare products and services, which could result in reduced demand for our products or additional pricing pressures. At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. In the EU, pricing and reimbursement schemes vary widely from country to country and often within regions or provinces of countries. Some countries may limit the annual budget of coverage or request that the company participate in the cost above certain use levels or for treatments perceived as unsuccessful and impose monitoring processes on the use of the product. Some countries and hospitals may require inclusion into the hospital formulary for payment from the hospital budget. Some countries and hospitals may require the completion of additional studies that compare the cost‑effectiveness of a particular drug candidate to currently available therapies. For example, the EU provides options for its member states to restrict the range of drugmedicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. European UnionThis Health Technology Assessment (“HTA process”) which is currently governed by the national laws of the individual EU member states, is the procedure according to which the assessment of the public health impact, therapeutic impact and the economic and societal impact of use of a given medicinal product in the national healthcare systems of the individual country is conducted. The outcome of HTA regarding specific medicinal products will often influence the pricing and reimbursement status granted to these medicinal products by the competent authorities of individual EU Member States. On December 15, 2021, the Health Technology Regulation (“HTA Regulation”) was adopted. The HTA Regulation is intended to boost cooperation among EU member states in assessing health technologies, including new medicinal products, and providing the basis for cooperation at EU level for joint clinical assessments in these areas. When it enters into application in 2025, the HTA Regulation will be intended to harmonize the clinical benefit assessment of HTA across the EU. Further, EU member states may approve a specific price for a drug product or may instead adopt a system of direct or indirect controls on the profitability of the company placing the drug product on the market. Other member states allow companies to fix their own prices for drug products, but monitor and control company profits. The downward pressure on health care costs in general, particularly prescription drugs, has become intense. As a result, increasingly high barriers are being erected to the entry of new products. In addition, in some countries, cross‑border imports from low‑priced markets exert competitive pressure that may reduce pricing within a country. Any country that has price controls or reimbursement limitations for drug products may not allow favorable reimbursement and pricing arrangements. Historically, products launched in the EU do not follow price structures of the United States and generally prices tend to be significantly lower.
Healthcare Law and Regulation; Data Privacy and Security LawsRegulation Healthcare providers, physicians and third‑party payors play a primary role in the recommendation and prescription of drug products that are granted marketing approval.authorization. Arrangements with healthcare providers, third‑party payors and other customers are subject to broadly applicable fraud and abuse and other healthcare laws and regulations. Such restrictions under applicable federal, state and foreign healthcare laws and regulations, include the following: the federal healthcare Anti‑Kickback Statute prohibits, among other things, persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, order or recommendation of, any good or service for which payment may be made, in whole or in part, under a federal healthcare program such as Medicare and Medicaid;
the federal False Claims Act imposes civil penalties, and provides for civil whistleblower or qui tam actions, against individuals or entities for knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government;
HIPAA, imposes criminal and civil liability for executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters;
HIPAA, as amended by HITECH and its implementing regulations, also imposes obligations, including mandatory contractual terms, on covered entities and their respective business associates with respect to safeguarding the privacy, security and transmission of individually identifiable health information;
the federal false statements statute prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statement in connection with the delivery of or payment for healthcare benefits, items or services;
the federal physician payment transparency requirements under the Affordable Care Act require certain manufacturers of drugs, devices and medical supplies to report to Centers for Medicare & Medicaid Services information related to payments and other transfers of value to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors), certain other healthcare professionals, and teaching hospitals and physician ownership and investment interests;
analogous state and foreign laws and regulations, such as state anti‑kickback and false claims laws, may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non‑governmental third‑party payors, including private insurers; and
similar healthcare laws and regulations in the E.U. and other jurisdictions, including reporting requirements detailing interactions with and payments to healthcare providers and laws governing the privacy and security of personal data, including the General Data Protection Regulation (“GDPR”), which imposes obligations and restrictions on the collection and use of personal data relating to individuals located in the E.U. and EEA (including with regard to health data).
| • | the federal healthcare Anti‑Kickback Statute prohibits, among other things, persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, order or recommendation of, any good or service for which payment may be made, in whole or in part, under a federal healthcare program such as Medicare and Medicaid. A person or entity does not need to have actual knowledge of this statute or specific intent to violate it in order to have committed a violation; | | • | the federal False Claims Act imposes civil penalties, and provides for civil whistleblower or qui tam actions, against individuals or entities for knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government. In addition, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act; | | • | HIPAA, imposes criminal and civil liability for executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters; | | • | the federal false statements statute prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statement in connection with the delivery of or payment for healthcare benefits, items or services; | | • | the federal physician payment transparency requirements under the Affordable Care Act require certain manufacturers of drugs, devices and medical supplies to report to Centers for Medicare & Medicaid Services information related to payments and other transfers of value to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors), certain other non-physician practitioners such as physician assistants and nurse practitioners, and teaching hospitals and physician ownership and investment interests; | | • | analogous state and foreign laws and regulations, such as state anti‑kickback and false claims laws, may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non‑governmental third‑party payors, including private insurers. |
Violations of any of these laws or any other governmental laws and regulations that may apply include, without limitation, significant civil, criminal and administrative penalties, damages, fines, imprisonment, exclusion of products from government funded healthcare programs, such as Medicare and Medicaid, disgorgement, contractual damages, reputational harm, diminished profits and the curtailment or restructuring of our operations. Some state laws require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government in addition to requiring drug manufacturers to report information related to payments to physicians and other health care providers or marketing expenditures. Additionally, certain state and local laws require the registration of pharmaceutical sales representatives. State and foreign laws also govern the privacy and security of health information in some circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts. For example, the California Consumer Privacy Act, or CCPA, which went into effect on January 1, 2020, among other things, creates new data privacy obligations for covered companies and provides new privacy rights to California residents, including the right to opt out of certain disclosures of their information. The CCPA also creates a private right of action with statutory damages for certain data breaches, thereby potentially increasing risks associated with a data breach. Although the law includes limited exceptions, including for “protected health information” maintained by a covered entity or business associate, it may regulate or impact our processing of personal information depending on the context. Further, the California Privacy Rights Act (CPRA), recently passed in California. The CPRA will impose additional data protection obligations on covered businesses, including additional consumer rights processes, limitations on data uses, new audit requirements for higher risk data, and opt outs for certain uses of sensitive data. It will also create a new California data protection agency authorized to issue substantive regulations and could result in increased privacy and information security enforcement. The majority of the provisions will go into effect on January 1, 2023, and additional compliance investment and potential business process changes may be required.
Environmental, Health and Safety Matters We are subject to extensive environmental, health and safety laws and regulations in a number of jurisdictions, primarily Israel, governing, among other things: the use, storage, registration, handling, emission and disposal of chemicals, waste materials and sewage; chemicals, air, water and ground contamination; air emissions and the cleanup of contaminated sites, including any contamination that results from spills due to our failure to properly dispose of chemicals, waste materials and sewage. Our operations at our Yavne manufacturing facility use chemicals and produce waste materials and sewage. Our activities require permits from various governmental authorities including, local municipal authorities, the Ministry of Environmental Protection and the Ministry of Health. The Ministry of Environmental Protection and the Ministry of Health, local authorities and the municipal water and sewage company conduct periodic inspections in order to review and ensure our compliance with the various regulations. These laws, regulations and permits could potentially require the expenditure by us of significant amounts for compliance or remediation. If we fail to comply with such laws, regulations or permits, we may be subject to fines and other civil, administrative or criminal sanctions, including the revocation of permits and licenses necessary to continue our business activities. In addition, we may be required to pay damages or civil judgments in respect of third‑party claims, including those relating to personal injury (including exposure to hazardous substances we use, store, handle, transport, manufacture or dispose of), property damage or contribution claims. Some environmental, health and safety laws allow for strict, joint and several liability for remediation costs, regardless of comparative fault. We may be identified as a responsible party under such laws. Such developments could have a material adverse effect on our business, financial condition and results of operations. In addition, laws and regulations relating to environmental, health and safety matters are often subject to change. In the event of any changes or new laws or regulations, we could be subject to new compliance measures or to penalties for activities which were previously permitted. For instance, new Israeli regulations were promulgated in 2012 relating to the discharge of industrial sewage into the sewer system. These regulations establish new and potentially significant fines for discharging forbidden or irregular sewage into the sewage system. Properties Our principal executive offices are located at 42 Hayarkon Street, Yavne 8122745, Israel. We leasehave leased these facilities from our largest shareholder, Clal Life Sciences, L.P. (“CLS”), pursuant to a sub‑third party. The lease agreement as amended, that expireswill expire on October 30, 2025.2035, with an option for a further three year extension until 2038. The facilities consist of approximately 32,300 square feet of space, and the yearly lease fee is approximately $469,000.$648,000. These facilities house our administrative headquarters, our research and development laboratories and our manufacturing plant. During 2023 we initiated the expansion of the Company’s manufacturing facility to address the increasing global demand for NexoBrid. This expansion project is scheduled for completion by the end of 2024 and is projected to achieve 6-fold manufacturing capacity increase in 2025. In July 2023, we entered into a turnkey scale-up agreement with Biopharmax Group Ltd. To bolster our manufacturing infrastructure and support our long-term growth trajectory. The sub-leaseobjective of this agreement includesis to establish, commission, and validate a cutting-edge, sterile, and GMP-compliant manufacturing facility. The venture aims to increase our production capacity significantly, projected to expand to six times the current capacity, aligning with our strategic plan to meet the escalating global demand for NexoBrid. The new facility, equipped with fully operational clean rooms, will be exclusively designed for NexoBrid production. It will comply with stringent regulations from the GMP, FDA, EMA, Israeli Ministry of Health, and relevant Israeli regulatory bodies. An estimated $12.7 million will be invested in the project, set for completion by mid-2024, with full-scale manufacturing expected to commence in 2025. We have furthermore leased additional office space in Yavne, Israel, consisting of approximately 4,000 square feet, for a term of 2 years, commencing in January 1, 2024, with an option to extendfor a further three year extension until 2028. The yearly lease fee is approximately $120,000. For the initial two years of the lease period for additional 3 years atterm, the majority of our sole discretion.lease payments are being covered by the DOD.
C. Organizational Structure The legal name of our company is MediWound Ltd. and we are organized under the laws of the State of Israel. Our corporate structure consists of MediWound Ltd., our Israeli parent company, (i) MediWound Germany GmbH, our active wholly‑owned subsidiary, which was incorporated on April 16, 2013 under the laws of the Federal Republic of Germany (ii) MediWound US, Inc., which was incorporated on December 8, 2020 under the laws of the State of Delaware and (iii) MediWound UK Limited, our inactive wholly‑owned subsidiary, which was incorporated on July 26, 2004 under the laws of England. D. Property, Plants and Equipment See “ITEM 4.B. Business Overview—Properties”Overview-Properties”, “ITEM 4.B. Business Overview—Manufacturing,Overview-Manufacturing, Supply and Production” and “ITEM 4.B. Business Overview—Environmental,Overview-Environmental, Health and Safety Matters”. Item 4AItem 4A.. UNRESOLVED STAFF COMMENTS None. Item 5. OPERATING AND FINANCIAL REVIEW AND PROSPECTS The information contained in this section should be read in conjunction with our consolidated financial statements for the year ended December 31, 20212023 and related notes, and the information contained elsewhere in this annual report. Our financial statements have been prepared in accordance with IFRS, as issued by the IASB. Company Overview We are a biopharmaceutical company that develops, manufactures, and commercializes novel, cost effective, bio-therapeuticbio‑therapeutic, non-surgical solutions for tissue repair and regeneration. Our strategy is centered aroundleverages our validatedbreakthrough enzymatic technology platform technology,into diversified portfolio of biotherapeutics across multiple indications to pioneer solutions for unmet medical needs. Our current portfolio is focused on next-generation protein-based therapies for burn andcare, wound care and for tissue repair. Our first innovative biopharmaceutical product, NexoBrid,NexoBrid®, has received marketing authorization from the FDA and marketing authorization from the European CommissionMedicines Agency (the “EMA”) and the Israeli, Argentinean, South Korean, Russian, Ukrainian, Eurasian economic union (Armenia, Kyrgyzstan, Belarus, Kazakhstan), Peruvian, Chilean, Taiwanese and United Arab Emirates Ministries of Healthother international markets for removal of dead or damaged tissue, known as eschar, in adults with deep partial‑partial-thickness and full‑thicknessfull-thickness thermal burns, also referred to as severe burns. In September 2020, the FDA accepted for review our BLA, which was based on acute data, including primary, secondary and safety endpoints, as well as 12-month safety follow-up data derived from our Phase 3 pivotal study. In June 2021, we received a Complete Response letter from the FDA stating that our BLA was not approved. We had a Type A meeting with the FDA in October 2021 to discuss a path forward for resubmission, in which we gained clarity on a path forward for resubmission of the BLA, and we plan to resubmit our BLA for NexoBrid in mid-2022. NexoBrid, a concentrated mixtureconcentrate of proteolytic enzymes enriched in bromelain, represents a new paradigm in burn care management, and our clinical trials have demonstrated, with statistical significance, its ability to non‑surgically and rapidly remove the eschar, without harming viable tissues, earlier relative to existing standard of care upon patient admission, without harming viable tissues.care. We commercialize NexoBrid globally viathrough multiple sales channels. We sell NexoBrid to burn centers in Europethe European Union, United Kingdom and Israel, primarily through our direct sales force, focusing on leadingkey burn centers and key opinion leader management, and are establishing additional distribution channels in the European Union to extend the product's outreach. We have signed distribution agreements with local distributors in multiple international markets, which are responsible for obtaining local marketing authorization within the relevant territory.KOLs. In the United States, we entered into exclusive license and supply agreements with Vericel Corporation (Nasdaq: VCEL) to commercialize NexoBrid in North America upon FDA’s approval. For additional informationAmerica. We have established local distribution channels in multiple international markets, focusing on Asia Pacific, EMEA, CEE and LATAM, which local distributors are also responsible for obtaining local marketing authorization within the commercialization of NexoBrid See ITEM 4.B. “Information on the Company - Marketing, Sales and Distribution.”relevant territories. We are conducting an expanded access treatment protocol (NEXT)have been awarded two contracts with the U.S. Biomedical Advanced Research and Development Authority ("BARDA"), for NexoBrid to treat burn patients with deep partial-the advancement of the development and full-thickness burns inmanufacturing, as well as the U.S., which is funded by BARDA and which will continue to take place during the review of our upcoming BLA by the FDA. We are also conducting a pediatric study to broaden the approved indicationprocurement of NexoBrid which is also being funded byhas initiated on January 2020, as a medical countermeasure as part of BARDA in which we reported positive topline results in July 2021.preparedness for mass casualty events.
An additional product candidate
EscharEx, our next-generation enzymatic therapy under development, is EscharEx, a topical bioactivebiological drug candidate designed to enzymatically debridefor the debridement of chronic and other hard-to-heal wounds. Designed for the outpatient setting, EscharEx is an easy-to-use concentrate of proteolytic enzymes enriched in bromelain; having the same API as NexoBrid. In Januaryseveral Phase II trials, EscharEx was shown to be well-tolerated and demonstrated safety and efficacy in the debridement of various chronic and other hard-to-heal wounds with only few daily applications. EscharEx’s mechanism of action is mediated by the proteolytic enzymes that cleave and remove the necrotic tissue and prepare the wound bed for healing. On May 12, 2022, we announced positive topline results from our ongoingU.S. Phase 2II clinical study of EscharEx for the treatmentdebridement of VLUs. These topline results suggest that theThe study met its primary endpoint with a high degree of statistical significance, demonstrating that patients treated with EscharEx had a statistically significant higher incidence of complete debridement during the 14-day measurement period within up to 8 applications, compared to the gel vehicle (EscharEx: 63% (29/46) vs. gel vehicle: 30% (13/43), p-value=0.004). EscharEx efficacy superiority remained statistically significant after adjusting for pre-specified covariates ascribed to patient baseline characteristics, wound size, wound age and regions. The study met key secondary and exploratory endpoints. Patients treated with EscharEx had a p-valuestatistically significant higher incidence of 0.004.complete debridement, during the same 14-day measurement period, compared to patients treated by non-surgical standard-of-care ("NSSOC") (EscharEx: 63% (29/46) vs. NSSOC: 13% (4/30)) and the time to achieve complete debridement was significantly shorter. Estimated median time to complete debridement was 9 days for patients treated with EscharEx and 59 days for patients treated with NSSOC (p-value=0.016). On average, complete debridement was achieved after 3.6 applications of EscharEx compared to 12.8 applications with NSSOC. Patients treated with EscharEx demonstrated significantly higher incidence of at least 75% granulation tissue at the end of the treatment period compared to gel vehicle (p-value <0.0001). Favorable trends were observed in wound area reduction and reduction of pain compared to gel vehicle. In addition, the study showed that EscharEx was safe and well tolerated, and the overall safety was comparable between the arms as assessed by the data safety monitoring board. Importantly, there were no observed deleterious effects on wound closure and no material differences in reported adverse events. Estimated time to complete wound closure was 64 days for patients treated with EscharEx compared to 78 days for patients treated with NSSOC. EscharEx was also evaluated in a U.S. Phase II pharmacology study. The study was prospective, open label, single-arm and conducted at three U.S. clinical sites. On July 7, 2022, we announced positive results from this study. 70% of patients achieved complete debridement during the course of treatment within up to 8 applications. On average, complete debridement was achieved after 3.9 applications of EscharEx. Additionally, an average reduction of 35% in wound size was achieved by the end of the 2-week follow-up period. In all patients that were positive for biofilm at baseline, the biofilm was reduced substantially to single individual microorganisms or completely removed by the end of treatment. Seven patients had positive red fluorescence (indicative of bacteria) at baseline and average red fluorescence was reduced from 1.69 cm2 pre-treatment to 0.60 cm2 post treatment. Biomarker analysis from wound fluid safety data showed that EscharEx is safe and well-tolerated. Our third innovative product candidate, MW005, is a topically applied biological drug candidate for the treatment of non-melanoma skin cancers, based on the same active substance ofAPI as NexoBrid and EscharEx products, a concentrated mixture(a concentrate of proteolytic enzymes enriched in bromelain.bromelain). In July 2021, we initiated a phase I/II study of MW005 for the treatment of low-risk BCC. On July 11, 2022, we announced positive initial data from this study. In the first cohort, eleven patients with either superficial or nodular BCC were treated. Patients enrolled into the study received seven topical applications of MW005, once every other day. At the end of eight weeks post treatment period, all patients undergo complete excision, and the specimen is subject to an independent histological clearance examination. In July 2023, we again announced positive results in our U.S. Phase I/II study of MW005 for the treatment of basal cell carcinoma. Fifteen patients were treated with MW005 and completed the study. Results showed MW005 to be safe and well-tolerated, with a high level of patient compliance. Based on clinical assessments, eleven out of fifteen patients achieved complete clearance of their BCCs; the majority of these patients also had histologically confirmed complete clearance. Based on the data generated to date, MW005 is safe, well-tolerated and an effective treatment for BCC with a majority of patients who completed the study demonstrating a complete histological clearance of target lesions. We manufacture NexoBrid and our product candidates in our state‑of‑the‑art, EMA‑cGMP certified cGMP‑compliant, sterile pharmaceutical products manufacturing facility at our headquarters in Yavne, Israel. Our securities are listed for trading on Nasdaq since March 2014 following our Initial Public Offering. As of December 31, 2021,2023, we had cash and cash equivalents and short term and restricted bank deposits of $11.0$41.7 million. Our revenues were $21.8$18.7 million and $23.8 million$26.5 in 20202023 and 2021,2022, respectively. Our net operating loss was $8.8$15.3 million and $11.2 million$8.3 in 20202023 and 2021,2022, respectively. We had an accumulated deficit of $148.5$174.8 million as of December 31, 2021.2023. We expect to incur significant expenses and operating losses forin the foreseeable future,coming years, as research and development activities are central to our operations, which will offset by cash inflows from NexoBrid. We expect to continue to invest in our research and development efforts, including in respect of our NexoBrid ongoing clinical trials which are fully funded by BARDA, as well as the clinical development and trials of EscharEx, MW005 and our other pipeline product candidates. In addition, we expect to continue to advance NexoBrid as a standard of care, and expand its commercial reach in international markets, including for potential use as a medical countermeasure during mass casualty events.
Key Components of Statements of Operations Revenues Sources of revenues. We derive revenues from sales of NexoBrid to burn centers and hospitals burn units in USA, Europe and Israel as well as to local distributors in other countries in accordance with distribution agreements we have in place, which also include revenues from licenses. We generate revenues from BARDA procurement of NexoBrid for emergency stockpile pursuant to BARDA contract. We generate revenues from development services provided to BARDA. BARDA, and to DOD/MTEC. Our ability to generate additional, more significant revenues will depend on the successful commercialization of NexoBrid, which itself will be dependent in part upon receipt of approval from the FDA.NexoBrid. Cost of Revenues Our total cost of revenues includes expenses for the manufacturing of NexoBrid, including: the cost of raw materials; employee‑related expenses, including salaries, equity based‑compensation and other benefits and related expenses, lease payments, utility payments, depreciation, changes in inventory of finished products, royalties and other manufacturing expenses. These expenses are partially reduced by an allotment of manufacturing costs associated with research and development activities to research and development expenses. Cost of revenues also includes costs associated with the research and development services provided to BARDA and MTEC, including salaries and related expenses, clinical trials, sub‑contractors and external advisors. We expect that our cost of revenues from sale of products will continue to increase as we expand the sale of NexoBrid throughout the European Union, the United States and other international markets.
Operating Expenses Research and Development Expenses Research and development activities are central to our business model. Product candidates in later stages of clinical development generally have higher development costs than those in earlier stages of clinical development, primarily due to the increased size and duration of later‑stage clinical trials. We expect research and development costs to increase significantly for the foreseeable future as EscharEx progresses in its clinical program in the U.S. and our other pipeline product candidates' progress in clinical trials. However, we do not believe that it is possible at this time to accurately project total program‑specific expenses to reach commercialization. There are numerous factors associated with the successful development of any of our product candidates, including future trial design and various regulatory requirements, many of which cannot be determined with accuracy at this time based on our stage of development. Additionally, future commercial and regulatory factors beyond our control will affect our clinical development programs and plans. Our actual spending could differ as our plans change and we invest in other drugs or potentially reduce our anticipated funding on research for existing products. Research and development expenses consist primarily of compensation for employees engaged in research and development activities, including salaries, equity‑based compensation, benefits and related expenses, clinical trials, contract research organization sub‑contractors, development materials, external advisors and the allotted cost of our manufacturing facility for research and development purposes.
Selling and Marketing Expenses Selling and marketing expenses consist primarily of compensation expenses for personnel engaged in sales and marketing, including salaries, equity based‑compensation and benefits and related expenses, as well as promotion, marketing, market access, medical, and sales and distribution activities. These expenses also includeare primarily comprised of costs related to our subsidiary in Germany, which is focused primarily on marketing NexoBrid in E.U., and costcosts related to maintain marketing authorization. General and Administrative Expenses General and administrative expenses consist principally of compensation for employees in executive and administrative functions, including salaries, equity‑based compensation, benefits and other related expenses, professional consulting services, including legal and audit fees, as well as costs of office and overhead. We expect general and administrative expenses to remain stable. Financial Income/Financial Expense Financial income includes interest income, revaluation of financial instruments and exchange rate differences. Financial expense consists primarily of revaluation of financial instruments, financial expenses in respect of deferred revenue, revaluation of lease liabilities and exchange rate differences. The market interest due on government grants received from the IIA is also considered a financial expense, and is recognized beginning on the date we receive the grant until the date on which the grant is expected to be repaid as part of the revaluation to fair value of liabilities in respect of government grants. Discontinued Operation
Following the expiration of our PolyHeal license in 2013, we accounted for our operation related to PolyHeal as a discontinued operation in accordance with IFRS accounting standard 5, “Non‑current Assets Held for Sale and Discontinued Operations.” Accordingly, the results of any legal process profit or loss are reported separately as a discontinued operation in our statement of operations for the periods presented below.
Taxes on Income The standard corporate tax rate in Israel is 23%. We do not generate taxable income in Israel, as we have historically incurred operating losses resulting in carry forward tax losses and other temporarily differences from R&D expenses totaling approximately $148$172 million as of December 31, 2021.2023. We anticipate that we will be able to carry forward these tax losses indefinitely to future tax years. Accordingly, we do not expect to pay taxes in Israel until we have taxable income after the full utilization of our carry forward tax losses. Under the Law for the Encouragement of Capital Investments, 5719‑1959 (the “Investment Law”), we have been granted “Beneficiary Enterprise” status, which provides certain benefits, including tax exemptions and reduced corporate tax rates. Income not eligible for Beneficiary Enterprise benefits is taxed at the regular corporate tax rate. The benefit entitlement period starts from the first year that the Beneficiary Enterprise first earns taxable income, and is limited to 12 years from the year in which the company requested to have tax benefits apply.
Comparison of Period to Period Results of Operations We are providing within this section a supplemental discussion that compares our historical statement of operations data in accordance with IFRS, as issued by the IASB. The below table and the below discussion provides data for each of the years ended December 31, 20202023 and 2021.2022. The below discussion of our results of operations omits a comparison of our results for the years ended December 31, 20192022 and 2020.2021. In order to view that discussion, please see “Item 5. Operating and Financial Review and Prospects—A.Prospects-A. Operating Results—Results- Comparison of Period to Period Results of Operations—Operations- Year Ended December 31, 20192022 Compared to Year Ended December 31, 2020”2021” in our Annual Report on Form 20-F for the year ended December 31, 2020,2022, which we filed with the SEC on February 25, 2021.March 16, 2023. | | | Years Ended December 31, | | | Years Ended December 31, | | | | | 2020 | | | 2021 | | | 2023 | | | 2022 | |
| | (in thousands) | | | (in thousands) | | | condensed statements of operations data: | | | | | | | | | | | Condensed statements of operations data: | | | | | | | | | Revenues | | $ | 21,763 | | | $ | 23,763 | | | $ | 18,686 | | | $ | 26,496 | | | Cost of revenues | | | 14,218 | | | | 14,992 | | | | 15,108 | | | | 13,331 | | | Gross profit | | | 7,545 | | | | 8,771 | | | | 3,578 | | | | 13,165 | | | | | | | | | | | | | | Operating expenses: | | | | | | | | | | | | | | | | | | Research and development | | | 7,698 | | | | 10,256 | | | | 7,467 | | | | 10,181 | | | Selling and marketing | | | 3,228 | | | | 3,388 | | | | 4,844 | | | | 3,725 | | | General and administrative | | | 5,459 | | | | 6,348 | | | | 6,768 | | | | 6,920 | | | Other (income) expenses | | | | (211 | ) | | | 684 | | | Operating loss | | | (8,840 | ) | | | (11,221 | ) | | | (15,290 | ) | | | (8,345 | ) | | Financial expenses, net | | | (436 | ) | | | (2,303 | ) | | | Loss from continuing operations | | | (9,276 | ) | | | (13,524 | ) | | | Profit from discontinued operation | | | 80 | | | | - | | | | Tax expenses | | | - | | | | (27 | ) | | | | | | | | | | | | | | Financial income (expenses), net | | | | 8,759 | | | | (11,176 | ) | | Loss before taxes on income | | | | (6,531 | ) | | | (19,521 | ) | | | | | | | | | | | | | Taxes on income | | | | (185 | ) | | | (78 | ) | | | | | | | | | | | | | Net loss | | $ | (9,196 | ) | | $ | (13,551 | ) | | $ | (6,716 | ) | | $ | (19,599 | ) |
Year Ended December 31, 20202023 Compared to Year Ended December 31, 20212022 Revenues
| | | Years Ended December 31, | | | | | | 2020 | | | 2021 | | | Years Ended December 31, | | | | | (in thousands) | | | 2023 | | | 2022 | | | | | | | | (in thousands) | | | Revenues from sale of products | | $ | 7,445 | | | $ | 9,613 | | | $ | 6,261 | | | $ | 5,347 | | | | | | | | | | | | | | Revenues from development services | | | 13,935 | | | | 12,372 | | | | 12,265 | | | | 12,943 | | | Revenues from license agreements | | | 383 | | | | 1,778 | | | | | | | 21,763 | | | | 23,763 | | | | | | | | | | | Revenues from license agreements and royalties | | | | 160 | | | | 8,206 | | | | | | | | | | | | | | | | | | 18,686 | | | | 26,496 | |
We generated total revenues of approximately $23.8$18.7 million for the year ended December 31, 20212023 compared to approximately $21.8$26.5 million for the year ended December 31, 2020.2022. The increasedecrease is primarily attributed to the BLA approval millstone payment from Vericel that took place in total revenues was a result of an increase in sale of products of $2.1 mainly derived from BARDA emergency stockpile procurement of $1.6 million and an increase in license sales of $1.4 million, partially offset by a decrease in development services to BARDA of $1.6 million.2022. Revenues from sale of products Revenues from sales of products in 2021 increased $2.22023 were $6.3 million, or 29%,a 17% increase compared to $5.3 million in comparison2022 mainly attributed to 2020, primarily as a result of BARDA’s procurement of NexoBrid for emergency stockpile of approximately $5.5 million net in 2021, versus approximately $3.8 million net during 2020. Revenues from BARDA’s procurement were recognized net of Vericel’s share pursuant to gross profit split.new customers. Revenues from development services Revenues from development services decreased 11%by 5% from $13.9$12.9 million in 20202022 to $12.4$12.3 million in 2021, as a result of completion of NexoBrid clinical studies.2023. The decrease mainly driven from BARDA. Revenues from license agreementagreements and royalties In 2021, we recognized $1.82023, revenues from license agreements and royalties were $0.2 million of license revenues,compared to $8.2 million in 2022, the decrease mainly driven by new distribution agreements and achieving certain milestones with current distributors agreements, compared to $0.4the BLA approval milestone of $7.5 million from Vericel in 2020.2022. Our revenues, as reported in our consolidated financial statements, are based on the location of the customers, as shown in the below table: | | | Years Ended December 31, | | | Years Ended December 31, | | | | | 2020 | | | 2021 | | | 2023 | | | 2022 | | | | | (in thousands) | | | (in thousands) | | | International (excluding U.S.) | | $ | 3,733 | | | $ | 5,649 | | | $ | 5,608 | | | $ | 4,624 | | | U.S. | | | 18,030 | | | | 18,069 | | | | 13,078 | | | | 21,872 | | | | | | 21,763 | | | | 23,718 | | | | 18,686 | | | | 26,496 | |
BARDA contributed 83%56% and 76%51% of ourthe Company’s total revenues in 20202023 and 2021,2022 respectively. Vericel contributed 4% and 28% of the Company’s total revenues in 2023 and 2022 respectively. DoD/MTEC contributed 10% and 2.8% of the Company’s total revenues in 2023 and 2022 respectively.
Costs and Expenses Cost of revenues | | | Years Ended December 31, | | | | | 2023 | | | 2022 | | | | | (in thousands) | | | Cost of revenues from sales of products | | $ | 4,927 | | | $ | 3,184 | | | Cost of revenues from development services | | | 10,177 | | | | 9,829 | | | Cost of revenues from license agreements | | | 4 | | | | 318 | | | | | | 15,108 | | | | 13,331 | |
| | | Years Ended December 31, | | | | | 2020 | | | 2021 | | | | | (in thousands) | | | Cost of revenues from sales of products | | $ | 3,151 | | | $ | 4,983 | | | Cost of revenues from development services | | | 11,067 | | | | 9,907 | | | Cost of revenues from license agreements | | | - | | | | 102 | | | | | | 14,218 | | | | 14,992 | |
Cost of revenues as a percentage of total revenues decreasedincreased from 65%50% for 20202022 to 63%81% for 2021.2023. Cost of revenues from sales of products as a percentage of revenues from sales of products increased to approximately 52%79% for the year ended December 31, 20212023, from approximately 42%60% in the year ended December 31, 2020.2022. The increase of cost of revenues from sales of product is primarily driven by BARDA procurement for emergency response preparedness.revenue mix changes.
Cost of revenues from development services as a percentage of revenues from development services was approximately 80%83% in the year ended December 31, 20212023, compared to approximately 79%76% in the year ended December 31, 2020.2022, mainly attributed to BARDA. Cost of revenues from license agreements and royalties as a percentage of revenues from license agreements were 6%was 3% in the year ended December 31, 2021, due2023 compared to costs associated withapproximately 4% in the support of our distributors to achieve their marketing authorizations.year ended December 31,2022. Research and development expenses, Research and development expenses increaseddecreased by 34%27% from approximately $7.7$10.2 million in the year ended December 31, 20202022 to approximately $10.3$7.5 million in the year ended December 31, 2021. The increase was primarily related2023, attributed mainly to the completion of EscharEx clinical development program.Phase II study. Selling and marketing expenses Selling and marketing expenses increased by 6%30% in 20212023 compared to 2020,2022, from approximately $3.2$3.7 million in the year ended December 31, 20202022 to approximately $3.4$4.8 million in the year ended December 31, 2021.2023, mainly related to consulting services, and various marketing expenses. General and administrative expenses General and administrative expenses increased 15%decreased by 2% in 20212023 compared to 20202022 from approximately $5.5$6.9 million in the year ended December 31, 20202022 to approximately $6.3$6.8 million in the year ended December 31, 2021. The increase in general and administrative expenses was primarily due to rent and maintenance allocation and legal consultation.2023. | Financial income, net | | Years Ended December 31, | |
| | 2020 | | | 2021 | | | | | (in thousands) | | | Financial income | | $ | 843 | | | $ | 11 | | | Financial expenses | | | (1,279 | ) | | | (2,314 | ) | | | | | (436 | ) | | | (2,303 | ) |
Other (income) expenses 76
Other one-time incomes for the year ended December 31, 2023 were $(0.2) million associated with non-cash income resulted with termination of sub-lease agreement with CBI. Other one-time expenses for the year ended December 31, 2022 were $0.7 million associated with management replacement and Vericel milestone payment fee. | Financial income, net | | Years Ended December 31, | | | | | 2023 | | | 2022 | | | | | (in thousands) | | | Financial income | | $ | 10,651 | | | $ | 461 | | | Financial expenses | | | (1,892 | ) | | | (11,637 | ) | | | | | 8,759 | | | | (11,176 | ) |
Financial income Financial income decreasedincreased from $0.8approximately $0.5 million in the year ended December 31, 20202022 to $0approximately $10.7 million in the year ended December 31, 2021.2023. The decreaseincrease was primarily driven by an $8.3 million adjustment resulting from the Teva contingent liability revaluation of warrants and by $2.3 million of interest on deposits.income from short term deposits in 2023. Financial expense Financial expense increasedexpenses decreased from approximately $1.3$11.6 million in the year ended December 31, 20202022 to approximately $2.3$1.9 million in the year ended December 31, 2021.2023. The increasedecrease in financial expenses in 2021 was primarily driven by $9 million adjustment resulting from the Teva contingent liability revaluation described below under “Application of Critical Accounting Policies and Estimates - Contingent Consideration for Purchase of Shares”, the Israeli innovation authority grant interest, currency exchange fluctuations and lease revaluations.warrants in 2022. Profit from Discontinued operations82
Profit from discontinued operations was $0 million for the year ended December 31, 2021 compared with $0.1 million for the year ended December 31, 2020. The profit from discontinued operations in 2020 was as a result of the Polyheal settlement of the litigation with certain PolyHeal Ltd.'s ("PolyHeal") shareholders. See “ITEM 8.A. Consolidated Statements and Other Financial Information—Legal Proceedings”.
B. Liquidity and Capital Resources Our primary uses of cash are to fund working capital requirements, manufacturing costs, research and development expenses ofactivities related to EscharEx and other products candidates, capital expenditure requirements, as well as sales and marketing activities associated with the commercialization of NexoBrid in Europe. WeOn March 7, 2022, the Company completed an underwritten follow-ona public offering in total of 744,048 new ordinary shares which were issued in consideration to offering price of $13.44 per share. The net proceeds were $8,653, after deducting commissions and other offering expenses. In addition, on March 22, 2022 the underwriters exercised their options to purchase an additional 89,012 ordinary shares at the same public offering price. The net consideration to the Company, less underwriting discounts and commissions was at additional of $1,021.
On September 2017, whereby we issued and sold 5,037,66426, 2022, the Company completed a registered direct (the “RD”) offering in an aggregate amount of $13,257 represent a combine purchase price of $12.25 for issuance of 1,082,223 ordinary shares and received net proceeds1,082,223 warrants that become exercisable on November 28, 2022, at an exercise price of approximately $22.7 million (after deducting the underwriting discount and offering expenses payable by us), pursuant to our previous shelf registration statement on Form F‑3. We$13.475 per ordinary share which will continue to use theexpire in four years, The net proceeds from this offering in the amount of $11,698 have been received on September 28, 2022. The issuance expenses related to the non-current financial liability were recorded through profit and lost and the issuance expenses related to the issuance of shares recorded as a deduction from the proceed in equity. On October 6, 2022 we entered the PIPE Securities Purchase Agreement with the several purchasers listed on the signature pages thereto (the “PIPE Purchasers”), in connection with the offer and sale of securities offered by us pursuant1,407,583 pre-funded warrants to that follow-on offeringpurchase up to fund our research1,407,583 Ordinary Shares (the “Pre-Funded Warrants”) and development activities, primarily1,407,583 ordinary warrants to purchase up to 1,407,583 Ordinary Shares (the “PIPE Ordinary Warrants,” and together with the clinical developmentPre-Funded Warrants, the “PIPE Warrants”) (the “PIPE Offering”). The combined purchase price for one Pre-Funded Warrant and associated PIPE Ordinary Warrant was $12.243. The Pre-Funded Warrants have an exercise price of EscharEx,$0.007 per Ordinary Share and the remainder, if any, for working capitalPIPE Ordinary Warrants have an exercise price of $13.475 per Ordinary Share and other general corporate purposes.each become exercisable on November 28, 2022. The timing and amount of our actual expenditures will be basedPIPE Offering closed on many factors, including cash flowsOctober 6, 2022. The gross proceeds from operations and the anticipated growth of our business. Under our current shelf registration statement on Form F-3 declared effective by the SEC on April 22, 2019, we may offer from time to time up to $125 million in the aggregate of our ordinary shares, warrants and/or debt securities in one or more series or issuances. In February 2020, we entered into an Open Market Sales Agreement with Jefferies LLC to issue and sell our ordinary shares with gross sales proceeds of up to $15 million, from time to time, through an at the market offering under which Jefferies LLC will act as our sales agent. As of the date hereof, we have not issued or sold any ordinary shares pursuant to the Open Market Sales Agreement. Funding under the BARDA contracts is classified under cash use for continuing operating activities.
PIPE Offering were approximately $17.23 million. As of December 31, 2021, we had $11.0 million2022, all Pre-Funded Warrants have been exercised and none of cash, cash equivalents and short-term deposits. Our net operating loss was $8.8 million and $11.2 millionthe PIPE Ordinary Warrants have been exercised. H.C. Wainwright & Co., LLC (“Wainwright”) acted as the exclusive placement agent for the years ended December 31, 2020RD Offering and 2021, respectively. As of December 31, 2021, we had an accumulated deficit of $148.5 million. We expect to incur significant expenses and operating losses for the foreseeable future. The net losses we will incur may fluctuate from quarter to quarter. Our capital expenditures for fiscal years 2020 and 2021 amounted to $0.9 million and $0.5 million, respectively. Capital expenditures consist primarily of investments in manufacturing equipment and leasehold improvements.
In March 2022, we entered into an underwriting agreement with Oppenheimer & Co., Inc., a representativePIPE Offering (together, the “2022 Offerings”). Upon closing of the several underwriters (the “Underwriters”), relating toOfferings, we issued Wainwright (or its designees) the issuance and sale of an aggregate of 5,208,333 of our ordinary shares at a price per share equal to $1.92. Total gross proceeds of the offering was approximately $10.0 million. The offering closed on March 7, 2022 and we received approximately $8.7 million in net proceeds, after deducting underwriting discounts and commissions and estimated offering expenses. Certain entities affiliated with CBI purchased approximately $2.8 million of ordinary shares in the offering at the public offering price. The Underwriters received the same underwriting discount on the shares purchased by these entities as they will on any other shares sold to the public in this offering. The securities purchased by these entities are subject to lock-up agreements with the Underwriters. We also granted the underwriters a 30-day optionwarrants to purchase up to an additional 781,249124,491 ordinary shares at(the “Wainwright Warrants”). The warrants have substantially the publicsame terms as the RD Warrants and the Series A Warrants, except that the Wainwright Warrants have an exercise price equal to $15.3125 per share (which represents 125% of the offering price less underwriting discountsper Ordinary Share in the Offerings) and commissions.will expire four (4) years after November 28, 2022, but no more than five (5) years following the commencement of the sales pursuant to the RD Offering.
77 On February 3, 2023, we entered into a securities purchase agreement (the “2023 Securities Purchase Agreement”) with the purchasers listed on the signature pages thereto (the “2023 Purchasers”), in connection with the offer and sale of 1,964,286 ordinary shares (the “2023 Offering”). The purchase price per ordinary share was $14.00. The 2023 Offering closed on February 7, 2023. The gross proceeds from the Offering were approximately $27.5 million.
The Company believes that its existing cash and cash equivalents, short-term and restricted bank deposits of $41.7 million as of December 31, 2023, will be sufficient to fund its operations and capital expenditure for at least twelve months from the date of issuance of these consolidated financial statements. Our future capital requirements will depend on many factors, including our revenue growth, timing of milestone payments, the timing and extent of our spending on research and development efforts, and international expansion. We may also seek to invest in or acquire complementary businesses or technologies. To the extent that existing cash and cash from operations are insufficient to fund our future activities, we may need to raise additional funding through debt and equity financing. Additional funds may not be available on favorable terms or at all. We believe our existing cash, cash equivalents and short‑term bank deposits will be sufficient to satisfy our liquidity requirements for at least the next 24 months.all The accompanying consolidated financial statements have been prepared on a basis which assumes that the Company will continue as a going concern. From inception to December 31, 2023, the Company has incurred cash outflows from operations, losses from operations, and has an accumulated deficit of $174.8 million.
Cash Flows The following table summarizes our consolidated statement of cash flows for the periods presented. The below discussion beneath the table omits a description of our cash flows for the year ended December 31, 2019.2023. In order to view that discussion, please see “Item 5. Operating and Financial Review and Prospects—B.Prospects Liquidity and Capital Resources—Cash Flows”Resources” in our Annual Report on Form 20-F for the year ended December 31, 2020,2022, which we filed with the SEC on February 25, 2021:March 16, 2023: | | | Year Ended December 31, | | | | | 2020 | | | 2021 | | | | | | | | | | | Net cash provided by (used in): | | | | | | | | Continuing operating activities | | $ | (6,700 | ) | | $ | (8,916 | ) | | Continuing investing activities | | | 17,385 | | | | 3,548 | | | Continuing financing activities | | | (629 | ) | | | (1,050 | ) | | Discontinued operating activities | | | (195 | ) | | | - | |
| | | Year Ended December 31, | | | | | 2023 | | | 2022 | | | | | | | | | | | Net cash provided by (used in): | | | | | | | | Operating activities | | $ | (10,465 | ) | | $ | (11,885 | ) | | Investing activities | | | (34,321 | ) | | | (481 | ) | | Financing activities | | | 22,917 | | | | 35,764 | |
Net cash used in continuing operating activities Net cash used in all periods resulted primarily from our net loss adjusted for non‑cashnon-cash charges and measurements and changes in components of working capital. Adjustments for non‑cashnon-cash items include depreciation and amortization, equity‑basedequity-based compensation, revaluation of contingent liabilities warrants, and lease liability, and changes in assets and liabilities items. Net cash used in continuing operating activities increaseddecreased to approximately $8.9$10.5 million in the year ended December 31, 20212023 compared to net cash used by continuingin operating activities of approximately $6.7$11.9 million in the year ended December 31, 2020,2022, primarily as a result of theour operational net loss, partially offset by various non-cash items such as depreciation, shared based compensation, financial income, net and revaluation of contingent consideration for the purchase of shares.considerations and warrants. Net cash used in discontinued operatinginvesting activities Net cash used in discontinued operatinginvesting activities primarily derives from investment in short term banks deposits and from purchases of property and equipment mainly related to scaling up our production facility, offset by interest received from short term bank deposit. Net cash used in investing activities was $0$34.3 million in the year ended December 31, 2021,2023, compared to approximately $0.2 million in the year ended December 31, 2020. The cash used in 2020 was primarily attributable to the consideration paid to PolyHeal’s shareholders following the settlement of the litigation with certain PolyHeal's shareholders. See “ITEM 8.A. Consolidated Statements and Other Financial Information—Legal Proceedings”. Net cash provided by continuing investing activities
Net cash provided by continuing investing activities primarily resulted from proceeds of investments in short‑term banks deposits offset by purchases of property and equipment. Net cash provided by investing activities was $3.5 million in the year ended December 31, 2021, compared to $17.4$0.5 million provided during the year ended December 31, 2020.
Net cash used2022, primarily result of investment in continuing financing activitiesshort- term bank deposits and investments in property and equipment.
Net cash provided by continuing financing activities primarily resulted from payments of lease liabilities and repayment to IIA. Net cash used in continuingprovided by financing activities was $1.1$22.9 million duringin the year ended December 31, 20212023 compared to $0.6$35.8 million duringin the year ended December 31, 2020.2022. The decrease in net cash provided by financing activities was primarily a result of the proceeds from the company’s 2022 Offerings, net of issuance expenses. Israeli Corporate-Level Tax Considerations and Government Programs The following is a brief summary of the material Israeli tax laws applicable to us, and certain Israeli Government programs that benefit us and therefore impact our results of operations and financial condition. To the extent that the discussion is based on new tax legislation that has not yet been subject to judicial or administrative interpretation, we cannot assure you that the appropriate tax authorities or the courts will accept the views expressed in this discussion. The discussion below is subject to change, including due to amendments under Israeli law or changes to the applicable judicial or administrative interpretations of Israeli law, which change could affect the tax consequences described below. General Corporate Tax Structure in Israel Generally, Israeli companies are subject to a corporate tax on their taxable income. Effective January 1, 2018 and thereafter, the corporate tax rate is 23%. However, the effective tax rate payable by a company that derives income from an Approved Enterprise, a Beneficiary Enterprise, a Preferred Enterprise or Technology Enterprise (as discussed below) may be considerably less. Capital gains derived by an Israeli company are generally subject to the prevailing regular corporate tax rate. Law for the Encouragement of Industry (Taxes), 5729-1969 The Law for the Encouragement of Industry (Taxes), 5729-1969 (the “Industry Encouragement Law”), provides several tax benefits for “Industrial Companies.”
The Industry Encouragement Law defines an “Industrial Company” as an Israeli resident-company which was incorporated in Israel, of which 90% or more of its income in any tax year, other than income from certain government loans, is derived from an “Industrial Enterprise” owned by it and located in Israel.Israel or in the “Area”, in accordance with the definition under section 3A of the Israeli Income Tax Ordinance (New Version) 1961. An “Industrial Enterprise” is defined as an enterprise whose principal activity in a given tax year is industrial production. The following tax benefits, among others, are available to Industrial Companies: amortization of the cost of purchased a patent, rights to use a patent, and know-how, which are used for the development or advancement of the Industrial Enterprise, over an eight-year period, commencing on the year in which such rights were first exercised;
under limited conditions, an election to file consolidated tax returns with related Israeli Industrial Companies controlled by it; and
expenses related to a public offering are deductible in equal amounts over a three years period commencing on the year of the offering.
| • | amortization of the cost of purchased a patent, rights to use a patent, and know-how, which are used for the development or advancement of the Industrial Enterprise, over an eight-year period, commencing on the year in which such rights were first exercised; | | • | under limited conditions, an election to file consolidated tax returns with related Israeli Industrial Companies controlled by it; and | | • | expenses related to a public offering are deductible in equal amounts over a three years period commencing on the year of the offering. |
Eligibility for benefits under the Industry Encouragement Law is not contingent upon approval of any governmental authority. We believe that we currently qualify as an Industrial Company within the meaning of the Industry Encouragement Law. However, there can be no assurance that we will continue to qualify as an Industrial Company or that the benefits described above will be available in the future. Law for the Encouragement of Capital Investments, 5719-1959 The Investment Law provides certain incentives for capital investments in production facilities (or other eligible assets). The Investment Law was significantly amended several times during recent years, with the three most significant changes effective as of April 1, 2005 (the “2005 Amendment”), as of January 1, 2011 (the “2011 Amendment”), and as of January 1, 2017 (the “2017 Amendment”). Pursuant to the 2005 Amendment, tax benefits granted in accordance with the provisions of the Investment Law prior to its revision by the 2005 Amendment remain in force but any benefits granted subsequently are subject to the provisions of the amended Investment Law. Similarly, the 2011 Amendment introduced new benefits to replace those granted in accordance with the provisions of the Investment Law in effect prior to the 2011 Amendment. However, companies entitled to benefits under the Investment Law as in effect prior to January 1, 2011 were entitled to choose to continue to enjoy such benefits, provided that certain conditions are met, or elect instead, irrevocably, to forego such benefits and have the benefits of the 2011 Amendment apply. The 2017 Amendment introduces new benefits for Technological Enterprises, alongside the existing tax benefits. Prior to 2011, we did not utilize any of the benefits for which we were eligible under the Investment Law.
The following is a summary of the Investment Law subsequent to its amendments as well as the relevant changes contained in the new legislation. Tax Benefits Subsequent to the 2005 Amendment The 2005 Amendment applies to new investment programs and investment programs commencing after 2004, but does not apply to investment programs approved prior to April 1, 2005 (“Approved Enterprise”). The 2005 Amendment provides that terms and benefits included in any certificate of approval that was granted before the 2005 Amendment became effective (April 1, 2005) will remain subject to the provisions of the Investment Law as in effect on the date of such approval. Pursuant to the 2005 Amendment, the Israeli Authority for Investments and Development of the Israeli Ministry of Economy (the “Investment Center”) will continue to grant Approved Enterprise status to qualifying investments. The 2005 Amendment, however, limits the scope of enterprises that may be approved by the Investment Center by setting criteria for the approval of a facility as an Approved Enterprise.
The 2005 Amendment provides that Approved Enterprise status will only be necessary for receiving cash grants. As a result, it is no longer necessary for a company to obtain the advance approval of the Investment Center in order to receive the tax benefits previously available under the alternative benefits track. Rather, a company may claim the tax benefits offered by the Investment Law directly in its tax returns, provided that its facilities meet the criteria for tax benefits set forth in the 2005 Amendment. Companies or programs under the new provisions receiving these tax benefits are referred to as Beneficiary Enterprises. Companies that have a Beneficiary Enterprise, are entitled to approach the Israel Tax Authority for a pre‑ruling regarding their eligibility for tax benefits under the Investment Law, as amended. Tax benefits are available under the 2005 Amendment to production facilities (or other eligible facilities), which are generally required to derive more than 25% of their business income from export to specific markets with a population of at least 14 million in 2012 (such export criteria will further increase in the future by 1.4% per annum). In order to receive the tax benefits, the 2005 Amendment states that a company must make an investment which meets certain conditions, including exceeding a minimum investment amount specified in the Investment Law. Such investment allows a company to receive “Beneficiary Enterprise” status, and may be made over a period of no more than three years from the end ofending in the year in which the company chose to have the tax benefits apply to its Beneficiary Enterprise. Where the company requests to apply the tax benefits to an expansion of existing facilities, only the expansion will be considered to be a Beneficiary Enterprise and the company’s effective tax rate will be the weighted average of the applicable rates. In this case, the minimum investment required in order to qualify as a Beneficiary Enterprise is required to exceed a certain percentage of the value of the company’s production assets before the expansion. The extent of the tax benefits available under the 2005 Amendment to qualifying income of a Beneficiary Enterprise depends on, among other things, the geographic location in Israel of the Beneficiary Enterprise. The location will also determine the period for which tax benefits are available. Such tax benefits include an exemption from corporate tax on undistributed income for a period of between two to ten years, depending on the geographic location of the Beneficiary Enterprise in Israel, and a reduced corporate tax rate of between 10% to 25% for the remainder of the benefits period, depending on the level of foreign investment in the company in each year. A company qualifying for tax benefits under the 2005 Amendment which pays a dividend out of income attributed to its Beneficiary Enterprise during the tax exemption period will be subject to corporate tax in respect of the amount of the dividend distributed (grossed‑up to reflect the pre‑tax income that it would have had to earn in order to distribute the dividend) at the corporate tax rate that would have otherwise been applicable. Dividends paid to Israeli shareholders out of income attributed to a Beneficiary Enterprise (or out of dividends received from a company whose income is attributed to a Beneficiary Enterprise) are generally subject to withholding tax at source at the rate of 15% (in the case of non-Israeli shareholders - subject to the receipt in advance of a valid certificate from the ITA allowing for a reduced tax rate, 15% or such lower rate as may be provided in an applicable tax treaty, applicable to dividends and distributions out of income attributed to a Beneficiary Enterprise.Enterprise). The reduced rate of 15% is limited to dividends and distributions out of income attributed to a Beneficiary Enterprise during the benefits period and actually paid at any time up to 12 years thereafter, except with respect to a qualified Foreign Investment Company (as such term is defined in the Investment Law), in which case the 12‑year limit does not apply.
The benefits available to a Beneficiary Enterprise are subject to the fulfillment of conditions stipulated in the Investment Law and its regulations. If a company does not meet these conditions, it would be required to refund the amount of tax benefits, as adjusted by the Israeli consumer price index, and interest, or other monetary penalties. We currently have Beneficiary Enterprise programs under the Investment Law, which we believe will entitle us to certain tax benefits. The majority of any taxable income from our Beneficiary Enterprise programs (once generated) would be tax exempt for a period of ten years commencing in the year in which we will first earn taxable income relating to such enterprises, subject to the 12-year limitation from the year the company chose to have its tax benefits apply. Tax Benefits Under the 2011 Amendment The 2011 Amendment canceled the availability of the tax benefits granted under the Investment Law prior to 2011 and, instead, introduced new tax benefits for income generated by a “Preferred Company” through its “Preferred Enterprise” (as such terms are defined in the Investment Law) as of January 1, 2011. The definition of a Preferred Company includes a company incorporated in Israel that is not fully owned by a governmental entity, and that has, among other things, Preferred Enterprise status and is controlled and managed from Israel.
The tax benefits under the 2011 Amendment for a Preferred Company meeting the criteria of the law include, among others, a reduced corporate tax rate of 15% for preferred income attributed to a Preferred Enterprise in 2011 and 2012, unless the Preferred Enterprise was located in a specified development zone, in which case the rate was 10%. Under the 2011 Amendment, such corporate tax rate was reduced in 2013 from 15% and 10%, respectively, to 12.5% and 7%, respectively, and then increased to 16% and 9%, respectively, in 2014 and thereafter until 2016. Pursuant to the 2017 Amendment, in 2017 and thereafter, the corporate tax rate for Preferred Enterprise which is located in a specified development zone was decreased to 7.5%, while the reduced corporate tax rate for other development zones remains 16%. Income attributed to a Preferred Company from a “Special Preferred Enterprise” (as such term is defined in the Investment Law) would be entitled, during a benefits period of 10 years, to reduced tax rates of 8%, or 5% if the Special Preferred Enterprise is located in a certain development zone. As of January 1, 2017, the definition of “Special Preferred Enterprise” includes less stringent conditions. Dividends paid to Israeli shareholders out of preferred income attributed to a Preferred Enterprise or to a Special Preferred Enterprise are generally subject to withholding tax at source at the rate of 20% (in the case of non-Israeli shareholders - subject to the receipt in advance of a valid certificate from the ITA allowing for a reduced tax rate, 20%, or such lower rate as may be provided in an applicable tax treaty (subject to the receipt in advance of a valid certificate from the Israel Tax Authority allowing for a reduced tax rate)treaty(. However, if such dividends are paid to an Israeli company, no tax is required to be withheld (although, if such dividends are subsequently distributed to individuals or a non‑Israeli company, withholding tax at a rate of 20% or such lower rate as may be provided in an applicable tax treatythe aforesaid will apply). The 2011 Amendment also provided transitional provisions to address companies already enjoying existing tax benefits under the Investment Law. These transitional provisions provide, among other things, that: unless an irrevocable request is made to apply the provisions of the Investment Law as amended in 2011 with respect to income to be derived as of January 1, 2011, a Beneficiary Enterprise can elect to continue to benefit from the benefits provided to it before the 2011 Amendment came into effect, provided that certain conditions are met. We have examined the possible effect, if any, of these provisions of the 2011 Amendment on our financial statements and have decided, at this time, not to opt to apply the new benefits under the 2011 Amendment. There can be no assurance that we will comply with the conditions required to remain eligible for benefits under the Investment Law in the future or that we will be entitled to any additional benefits thereunder.
New Tax benefits under the 2017 Amendment that became effective on January 1, 2017. The 2017 Amendment was enacted as part of the Economic Efficiency Law that was published on December 29, 2016, and is effective as of January 1, 2017. The 2017 Amendment provides new tax benefits for two types of “Technology Enterprises,” as described below, and is in addition to the other existing tax beneficial programs under the Investment Law. The 2017 Amendment provides that a technology company satisfying certain conditions will qualify as a “Preferred Technology Enterprise” and will thereby enjoy a reduced corporate tax rate of 12% on income that qualifies as “Preferred Technology Income,” as defined in the Investment Law. The tax rate is further reduced to 7.5% for a Preferred Technology Enterprise located in development zone A. In addition, a Preferred Technology Company will enjoy a reduced corporate tax rate of 12% on capital gain derived from the sale of certain “Benefitted Intangible Assets” (as defined in the Investment Law) to a related foreign company if the Benefitted Intangible Assets were acquired from a foreign company on or after January 1, 2017 for at least NIS 200 million, and the sale receives prior approval from the Israeli Innovation Authority.Authority (the “IIA”).
The 2017 Amendment further provides that a technology company satisfying certain conditions will qualify as a “Special Preferred Technology Enterprise” and will thereby enjoy a reduced corporate tax rate of 6% on “Preferred Technology Income” regardless of the company’s geographic location within Israel. In addition, a Special Preferred Technology Enterprise will enjoy a reduced corporate tax rate of 6% on capital gain derived from the sale of certain “Benefitted Intangible Assets” to a related foreign company if the Benefitted Intangible Assets were either developed by Special Preferred Technology Enterprise or acquired from a foreign company on or after January 1, 2017, and the sale received prior approval from IIA. A Special Preferred Technology Enterprise that acquires Benefitted Intangible Assets from a foreign company for more than NIS 500 million will be eligible for these benefits for at least ten years, subject to certain approvals as specified in the Investment Law.
Dividends distributed by a Preferred Technology Enterprise or a Special Preferred Technology Enterprise, to Israeli shareholders paid out of Preferred Technology Income, are generally subject to withholding tax at source at the rate of 20% (in the case of non-Israeli shareholders - subject to the receipt in advance of a valid certificate from the ITA allowing for a reduced tax rate, 20%, or such lower rate as may be provided in an applicable tax treaty (subject to the recipient in advance of a valid certificate from the Israeli Tax Authority allowing for reduced tax rate).treaty. However, if such dividends are paid to an Israeli company, no tax is required to be withheld.withheld (although, if such dividends are subsequently distributed to individuals or a non-Israeli company, the aforesaid will apply). If such dividends are distributed to a foreign company that holds solely or together with other foreign companies 90% or more in the Israeli company and other conditions are met, the withholding tax rate will be 4% (or a lower under the tax treaty, if applicable, subject to the receipt in advance of a valid certificate from the Israeli Tax Authority allowing for a reduced tax rate).
C. Research and Development, Patents and Licenses, etc. Our research and development strategy is centered on developing our patented proteolytic enzyme technology, which underlies NexoBrid and EscharEx, into additional products for high‑high value indications. Our research and development team is located at our facilities in Yavne, Israel, and consists of 2526 employees as of December 31, 20212022 and is supported by highly experienced consultants in various research and development disciplines. We have received government grants (subject to our obligation to pay royalties) as part of the NexoBrid and EscharEx research and development programs approved by the IIA. The total gross amount of grants actually received by us from the IIA, including accrued LIBOR interest and net of royalties actually paid, totaled approximately $13.8 million as of December 31, 20212023 and the amortized cost (using the interest method) of the liability totaled approximately $7.3$7.8 million and $8.1$7.6 million as of December 31, 20202023 and 2021,2022, respectively. Because the repayment of IIA grants is in the form of future royalties, the balance of the commitments to the IIA is presented as an amortized liability on our balance sheet. As of December 31, 2021,2022, we had accrued and paid royalties to the IIA totaling $1.3$2 million. We received funds from BARDA in accordance with the terms of our BARDA contracts. As of December 31, 20212023 we had accrued $70$93 million of BARDA’s participation in NexoBrid’s research and development programs. For a description of our research and development policies for the last three years, see “ITEM 4.B. Business Overview—ResearchOverview-Research and Development.”
D. Trend Information The COVID-19 pandemic has impacted companies in IsraelWe continue to closely monitor macro-economic conditions, including the headwinds caused by supply chain problems, inflation, increased interest rates and around the world, and as its trajectory remains highly uncertain, we cannot predict the duration and severity of the outbreak, its containment measures or the nature, timing and strength of recovery from it. Further, we cannot predict impacts,other trends and uncertainties involving the pandemic’s effects onthat have been adversely impacting economic activity on a global scale. We have been assessing, on an ongoing basis, the sizeimplications of those global conditions for our operations, supply chain, liquidity, cash flow and product orders, and will act in an effort to mitigate adverse consequences as needed. To the extent inflation increases our costs and expenses, we could consider price increases to offset those cost pressures.
Specific developments that may potentially impact our operating performance in an adverse manner include:
| • | further actions taken by central banks in Europe and the U.S. to increase interest rates as a means to slow down inflation, which may worsen credit/financing conditions for our customers who purchase our products; |
| • | potential contraction of economic activities and recessionary conditions that could arise as a result of interest rate increases and a decrease in consumer demand; and |
| • | the continued depreciated value of the Euro relative to the U.S. dollar, which may have an adverse impact on the U.S.- denominated value of our European-derived revenues for purposes of our financial statements. |
We cannot provide any assurances as to the extent of our labor force, our third-party partners, our investmentsresilience to the adverse impact of these specific developments in marketable securities, and the extent to which our revenue, income, profitability, liquidity, or capital resources may be materially and adversely affected prospectively.future periods. See also “ITEM 3.D. –- Risk Factors – “The coronavirus (COVID-19) outbreak could adversely impact our business, financial condition and results of operations.” and – “We-“We depend on a sole supplier to obtain our intermediate drug substance, bromelain SP, which is necessary for the production of our products.” Other than the foregoing and as disclosed elsewhere in this annual report, we are not aware of any trends, uncertainties, demands, commitments or events for the period from January 1, 20212022 to the present time that are reasonably likely to have a material adverse effect on our net revenue, income, profitability, liquidity or capital resources, or that would cause the disclosed financial information to be not necessarily indicative of future operating results or financial condition. E. Critical Accounting Estimates Our consolidated financial statements are prepared in conformity with IFRS, as issued by the IASB. The preparation of these historical financial statements in conformity with IFRS requires management to make estimates, assumptions and judgments in certain circumstances that affect the reported amounts of assets, liabilities and contingencies as of the date of the financial statements and the reported amounts of revenue and expenses during the reporting periods. We evaluate our assumptions and estimates on an ongoing basis. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions. Our critical accounting estimates are described in NotesNote 2 and 3 to our consolidated financial statements included elsewhere in this annual report.
Item 6. DIRECTORS, SENIOR MANAGEMENT AND EMPLOYEES A. Directors and Senior Management The following table sets forth the name, age and position of each of our executive officers and directors as of March 15, 2022:2024:
Name | | Age | | Position | Executive Officers | | | | | Ofer Gonen | | 51 | | Chief Executive Officer | Shmulik Hess | | Age51 | | Position | Executive Officers | | | | | Sharon Malka | | 50 | | Chief ExecutiveOperating Officer | Boaz Gur-Lavie | | 48 | | & Chief FinancialCommercial Officer | Lior Rosenberg, M.D. | | 76 | | Chief Medical Technology Officer | Ety Klinger Ph.D. | | 6062 | | Chief Research and Development Officer | Hani Luxenburg | | 51 | | Chief Financial Officer | | Yaron Meyer | | 4345 | | Executive Vice President, General Counsel and Corporate Secretary | Robert J. Snyder
| | 74 | | Chief Medical Officer | | | | | | | Directors | |
| |
| Stephen WillsNachum (Homi) Shamir (1)(2)(3)(5) | | 6569 | | Executive Chairman of the Board of Directors | Ofer GonenVickie R. Driver (1)(4)(5) | | 70 | | Director | | David Fox (2)(3)(5) | | 66 | | Director | Shmuel (Milky) Rubinstein (4)(5) | | 84 | | Director | | Stephen T. Willis (1)(2)(4)(5) | | 49 | | Director | Assaf Segal | | 5067 | | Director | Vickie R. Driver, M.D(1)(3)
| | 68 | | Director | Nissim Mashiach(1)(2)(3)(4)
| | 61 | | Director | Sharon Kochan(1)(2)(3)(4)
| | 53 | | Director | Samuel Moed (2)(3)
| | 59 | | Director | David Fox(3)
| | 64 | | Director |
(1) (1) | Member of our audit committee. |
(2) | Member of our compensation committee. | (3) | Member of our nominating and governance committee. | (4) | Member of our research and development committee. |
(5) (3) | Independent director under the listing rules of the Nasdaq Stock Market. |
(4) | External director under the Companies Law.
|
Executive Officers
Sharon MalkaOfer Gonen has served as our Chief Executive Officer since May 2019. Prior to that time, heJuly 2022 and on our board of directors since September 2003. Mr. Gonen is also the Chief Executive Officer of Cactus Acquisition Corp. 1 (Nasdaq: CCTS) and served as ourthe Chief FinancialExecutive Officer of Call Biotechnology Industries (TASE: CBI) from February 2017 until June 2022. Mr. Gonen has more than 20 years of experience in managing life science investments and Operations Officer, beginningbusiness collaborations in April 2007. From 2002 to 2007,both the US and Israel. Mr. MalkaGonen serves as a board member of several private and publicly-traded portfolio companies of CBI, including Gamida Cell (Nasdaq: GMDA) and Cactus, as well as a managing partner at the Anatomy Medical Fund. Before joining CBI, Mr. Gonen was the General Manager of Biomedical Investments Ltd., a partner at Variance Economic Consulting Ltd., a multi‑disciplinary consulting boutique that specializes in financial and business services. Mr. Malka also servedArte Venture Group, as well as a Senior Managertechnology consultant to various Israeli venture capital funds. Mr. Gonen gained extensive experience in R&D and management of defense-oriented projects at Kesselman Corporate Finance, a divisionthe prestigious “Talpiot” program of PricewaterhouseCoopers Global Network, from 1998 to 2002. Mr. Malkathe Israeli Defense Forces. He holds a B.Sc. in Business AdministrationPhysics, Mathematics and Chemistry from the Business Management College in IsraelHebrew University of Jerusalem, and an M.B.A.M.A. in Economics and Finance from Bar IlanTel Aviv University, Israel.with distinction.
Boaz Gur-LavieShmulik Hess has served as our Chief FinancialOperating Officer since June 2019.December 2023. Dr. Hess has over two decades of extensive expertise in drug development and commercial operations in healthcare. Prior to joining MediWound, Mr. Gur-Lavie co-founded in 2015 the Center for Digital Innovation (CDI), a non-profit organization determined to improve the quality of lives by creating innovative new solutions for challenges in the space of healthy aging and digital health, while focusing on senior citizens. In early 2015, he also co-founded MDClone, which introduced the world’s first Healthcare Data Sandbox, unlocking healthcare data to enable exploration, discovery and collaboration. Previously, he served as the chief financial officer of the Nasdaq-listed company, PluristemChief Executive Officer at Tabby Therapeutics, a stem-cell development company, from 2013 to 2015. He also served as the chief financial officer of STARLIMS, a Nasdaq listed company, until it was acquired by Abbott Laboratories in 2010, after which he served as the chief financial officer of Abbott’s informatics division until 2013. Mr. Gur-Lavie is a certified public accountantEnlivex Therapeutics (Nasdaq: ENLV), and received his B.A. in economics and M.B.A. in finance from the Ben-Gurion University in Israel.
Lior Rosenberg is one of our co‑founders and has served as our Chief Medical Technology Officer since 2001 andValin Technologies. Formerly, Dr. Hess served as a memberglobal operations executive at SciGen Ltd (acquired by VBI Vaccines). Dr. Hess is the inventor of our boardmultiple patents and author of directorsnumerous publications in peer reviewed scientific journals. He received his Ph.D. in Pharmaceutical Science from 2001 to 2013. Since 2001, Dr. Rosenberg has headed the unit for Cleft Lip Palate and Craniofacial Deformities at Soroka University Medical Center and Meir Medical Centers in Beer Sheva and Kfar Saba, Israel, respectively. Since 1987, he has served as a Full Professor of plastic surgery at the Ben‑Gurion University Medical School in Beer Sheva, Israel. He also serves as the Chairman of the Burn Disaster Committee for the International Society of Burn Injuries and the Israeli Ministry of Health. From 1987 to 2012, Dr. Rosenberg served as the chairman of the Department of Plastic Surgery and Burn Unit at Soroka University Medical Center in Beer Sheva, Israel. He is a founding member of the Israeli Burn Association and the Mediterranean Burn Council, a member of the American Burn Association and a national representative at the European Burn Association. Dr. Rosenberg holds a M.D. degree from Tel‑AvivHebrew University, Israel and was a Professor of Plastic Surgery degree from the Ben Gurion University, Israel.research fellow at Harvard-MIT Health Sciences and Technology (HST).
Ety Klinger has served as our Chief Research and Development Officer since May 2014. Prior to joining MediWound, Dr. Klinger was Vice President of Research and Development at Proteologics Ltd since July 2011, where she was responsible for discovery projects in the ubiquitin system, conducted in collaboration with GlaxoSmithKline plc and Teva. Prior to this, Dr. Klinger served for 17 years in numerous leadership positions at Teva’s global innovative R&D division and served as Teva’s Board representative at various biotechnology companies. Dr. Klinger was a key member of the Copaxone®Copaxone® development team. As a project leader she led the chemistry, manufacture and control, preclinical, clinical and post‑marketing R&D activities of various innovative treatments for multiple sclerosis (MS), autoimmune and neurological diseases. From 2006 to 2011, as a Senior Director at Teva, Dr. Klinger was a member of Teva’s global innovative R&D management team. From 2006 to 2008, she served as the Head of MS and Autoimmune Diseases at Teva, and led the Life Cycle Management (LCM) of innovative R&D. Dr. Klinger holds a B.Sc. in Biology from the Hebrew University in Jerusalem, a M.S. and a Ph.D. in Biochemistry from Tel‑Aviv University and an MBA degree from Tel Aviv University and Northwestern University. Hani Luxenburg has served as our Chief Financial Officer since May 2023. Ms. Luxenberg has over two decades of progressive leadership experience managing financial and accounting operations. Prior joining MediWound, Ms. Luxenburg served as Chief Financial Officer at BIRD Aerosystems. Prior to that, she held senior finance roles at AstraZeneca, Alvarion Technologies Ltd., and Ernst & Young. Ms. Luxenburg is a certified public accountant and holds a Bachelor of Arts in Economics and Accounting from the University of Haifa, along with a Bachelor of Law from IDC Herzliya. In addition, she is a member of the Israel Bar Association. Yaron Meyer has served as our Executive Vice President since March 2019 and as our General Counsel and Corporate Secretary since December 2013. From April 2008 to November 2013, he served as the Corporate Secretary of Clal Biotechnology Industries Ltd. (CBI).CBI. From November 2010 to November 2013, he served as the General Counsel and Corporate Secretary of D‑Pharm Ltd. From April 2008 to May 2010, he served as a legal counsel of Clal Industries Ltd. From May 2005 to April 2008, he worked as an associate at Shibolet & Co. Advocates. Mr. Meyer holds an LL.B. degree from Haifa University, Israel. Dr. Robert Snyder has served as our Chief Medical Officer since January 2023. Dr. Robert J. Snyder (DPM, MSc, MBA, CWSP, FFPM RCPS) is Dean, Professor, Director of Clinical Research and Fellowship Director in Wound Care and Research at Barry University School of Podiatric Medicine. He is certified in foot and ankle surgery by the American Board of Podiatric Surgery and is also a board-certified wound specialist. Dr. Snyder is past-president of the Association for the Advancement of Wound Care and past-president of the American Board of Wound Management. Dr. Snyder has completed an MBA in Health Management from The George Washington University and the Global Clinical Scholars Research Training Program at Harvard Medical School. Dr. Snyder is a key opinion leader and sought-after speaker, lecturing extensively throughout the United States and abroad. He has published several book chapters and over 165 papers in peer reviewed and trade journals on wound care, and was the recipient of the Dr. Robert Warriner Memorial Award for excellence in wound management. Dr. Snyder serves as the Associate Editor for JAPMA and on the editorial advisory boards of Ostomy Wound Management, Wounds and as a periodic reviewer for the Lancet and NEJM. He has been a Principal Investigator on more than 65 randomized controlled trials for innovative wound healing modalities and products.
Directors Stephen T. WillsNachum (Homi) Shamir has served as Chairman of our board of directors since August 2022. Mr. Shamir most recently the Chairman, and Chief Executive Officer of Luminex Corporation from 2014 through its sale to DiaSorin S.p.A.(“DiaSorin”) in 2021. Mr. Shamir continued to serve as President of Luminex after its sale to DiaSorin pursuant to a transition agreement with DiaSorin until June 2022. Additionally, Mr. Shamir has served as President and Chief Executive Officer of Given Imaging from 2006 through its sale to Covidien (now Medtronic) in 2014. Mr. Shamir currently serves on the Board of Directors of IsoPlexis Corporation (Nasdaq: ISO) and Strata Skin Sciences (Nasdaq: SSKN); and as Chairman of the Board of Cactus Acquisition Corp. (Nasdaq: CCTS). Mr. Shamir holds a Bachelor of Science degree from the Hebrew University of Jerusalem and a Masters of Public Administration from Harvard University.
Vickie R. Driver has served as a member of our Board since May 2017. She is board certified in foot surgery by the American Board of Podiatric Surgery and is a Fellow at the American College of Foot and Ankle Surgeons, licensed in Virginia, Massachusetts, and Rhode Island. Dr Driver serves as Chair, Board of Directors for the Wound Care Collaborative Community, an important collaboration with the FDA, CMS and the NIH and has received a prestigious honor of receiving The Robert A. Warriner III, MD Memorial Award. She is System Wide Medical Director of the Wound Care and Hyperbaric Centers at INOVA Healthcare in the DC Metropolitan area and is Professor, University of Virginia, School of Medicine. She is also Fellow, Royal College of Physicians and Surgeons-Glasgow, PM and Inaugural Fellow, Assoc for the Advancement of Wound Care, FAAWC. She currently serves as Honorary Visiting Professor at Cardiff University (UK) in the Department of Medicine and Professor at Barry University (USA). She proudly serves as a member of the Wound Healing Society (WHS) Board of Directors and as member Board of Directors for the Critical Limb Ischemia (CLI) Global Society. She has completed her tenure as president for the Advancement of Wound Care Association (AAWC) and served for 9 years on the Board of Directors. Dr. Driver is a former Professor of Surgery in the Department of Orthopedics at Brown University (Clinical) and Associate Professor of Surgery at Boston University. She has also chaired the Wound Care Experts and U.S. Food and Drug Administration (“FDA”) Clinical Endpoints Project [WEF-CEP]. The project was successful in developing the research to expand the wound healing clinical endpoints considered by FDA. She and her team proposed a combined effort to develop the Wound-care Experts/FDA-Clinical Endpoints Project [WEF-CEP] to strategically identify clinically meaningful, evidence-based, and patient-centered wound care endpoints that are relevant for clinical research and trials. The goal was to collaboratively work with the FDA to expand the list of acceptable primary endpoints, recognizing that new and innovative treatments, devices, and drugs may not have complete healing as the focus. She has served as a senior investigator for more than 70 important multi‑center randomized clinical trials, as well as developed and supervised multiple research fellowship training programs. She has served and chaired multiple committees for large national and international pivotal clinical trials, has authored over 150 publications and abstracts and is former Director, Translational Medicine at Novartis Institute for BioMedical Research. Dr. Driver is credited with the development and directorship of multiple major multidisciplinary Limb Preservation- Wound Healing Centers of Excellence, including Military/VA, Hospital and University based programs. Dr. Driver served on the Inaugural Educational Committee at the American College of Wound Healing and Tissue Repair at University of Illinois School of Medicine and was Scientific Director, Colorado Prevention Center, Wound Care Laboratory at the University of Colorado. Dr. Driver has held several leadership, teaching, research and clinical positions at Academic Medical Centers, Veterans Administration Medical Centers, and Military Medical Centers. Dr. Driver received a Doctorate of Podiatric Medicine and Surgery from the California College of Podiatric Medicine and Surgery and a master’s degree in medical education from Samuel Merritt University. Mr. David Fox has served as a member of our board of directors since April 2020. Mr. Fox has served as a member of our board of directors since April 2020. Mr. Fox was most recently a partner at Kirkland & Ellis LLP and served as a member of its Global Executive Management Committee until 2019. Prior to joining Kirkland, Mr. Fox was a partner at Skadden, Arps, Slate, Meagher & Flom LLP, where he was a member of its governing committee. Mr. Fox is a member of the executive committee and board of directors of the Park Avenue Armory, which enables artists to create and audiences to experience epic, adventurous work while also offering arts education programs at no cost to public school students, and is chairman of the advisory board of New Alternatives for Children, an organization that provides support to families caring for medically fragile children. Mr. Fox is the principal of David Fox & Co. LLC an advisory business and CEO of Bald Productions LLC, a movie and television development and production company. He is also an advisor to Longacre Square Partners, a communications and special situations advisory firm and to Nardello & Co, a global investigations firm. In addition, Mr. Fox serves on the executive committee and the board of governors, and is an honorary fellow, of the Hebrew University, Jerusalem. He holds an LL.B. degree from Jerusalem University, Israel.
Shmuel "Milky" Rubinstein has served as a member of our board since August 2023. Mr. Rubinstein brings a distinguished record of leadership in the pharmaceutical and biotechnology sectors to our board. Currently serving as Chairman of Trima Pharma, Milky's expertise extends across various prominent board roles, including Strata Skin Sciences (SSKN), Medison Biotech, and Keystone Dental. Notably, he held the esteemed position of CEO at Taro Pharmaceuticals (TARO), overseeing its successful acquisition by SUN Pharma. Milky's extensive board engagements also encompass Kamada (KMDA), Exalenz (Acquired by VIVO), and Clal Biotechnology Industries (CBI). With a proven track record, Milky Rubinstein's insights are poised to contribute significantly to our company's strategic direction and growth. Stephen T. Wills has served as a member of our Board since May 2017, as Chairman of our boardBoard since October 2017 and as Executive Chairman of our board since May 2019. Mr. Wills serves as Chief Financial Officer (since 1997) and Chief Operating Officer (since 2011) of Palatin Technologies, Inc. (NYSE: PTN), a biopharmaceutical company developing targeted, receptor‑specific peptide therapeutics for the treatment of diseases with significant unmet medical need and commercial potential. He also serves as Chief Financial Officer of Cactus Acquisition Corp. 1 Limited (Nasdaq: CCTS), a special purpose acquisition company, since 2021 until February 2024, when majority ownership was acquired by a new Sponsor. Mr. Wills serveserves on the boards of Gamida Cell Ltd. (Nasdaq: GMDA), a leading cellular and immune therapeutics company since March 2019 (audit chairperson and compensation and finance committee member) and of Amryt Pharma, a biopharmaceutical company focused on developing and delivering treatments to help improve lives of patients with rare and orphan diseases since September 2019 (chairman of audit committee and member of the compensation and finance committee)until April 2023, when Amryt was acquired by Chiesi Farmaceutici. Mr. Wills also serves on the board of trustees and executive committee of The Hun School of Princeton, a college preparatory day and boarding school since 2013, and its chairman since June 2018.2018 until his retirement in June 2023. Mr. Wills served on the board of directors of Caliper Corporation, a psychological assessment and talent development company since March 2016 and as chairman sincefrom December 2016 dountil December 2019, when Caliper was acquired by PSI. Mr. Wills servesserved as executive chairman and interim principal executive officer of Derma Sciences Inc. a provider of advanced wound care product from December 2015 to February 2017, when Derma Sciences was acquired by Integra Lifesciences (Nasdaq: IART). Previously, Mr. Wills served on the Board of Derma Sciences as the lead director and chairman of the audit committee from June 2000 to December 2015. Mr. Wills served as the Chief Financial Officer of Derma Sciences from 1997 to 2000. Mr. Wills served as the president and Chief Operating Officer of Wills, Owens & Baker, P.C., a public accounting firm from 1991 to 2000. Mr. Wills, a certified public accountant, earned his Bachelor of Science in accounting from West Chester University, and a Master of Science in taxation from Temple University. There are no family relationships among any of our executive officers or directors.
Ofer Gonen has servedThere are no arrangements or understandings with major shareholders, customers, suppliers or others, pursuant to which any person referred to above was selected as a director or member of our board of directors since September 2003. Mr. Gonen is the Chief Executive Officer of Clal Biotechnology Industries Ltd. (TASE: CBI) and Cactus Acquisition Corp. 1 (Nasdaq: CCTS). Mr. Gonen has more than 20 years of experience in managing life science investments and business collaborations in both the US and Israel. Mr. Gonen serves as a board member of several private and publicly-traded portfolio companies of CBI, including Gamida Cell (Nasdaq: GMDA), MediWound (Nasdaq: MDWD) and Cactus (Nasdaq: CCTS), as well as a managing partner at the Anatomy Medical Fund. Before joining CBI, Mr. Gonen was the General Manager of Biomedical Investments Ltd., a partner at Arte Venture Group, as well as a technology consultant to various Israeli venture capital funds. Mr. Gonen gained extensive experience in R&D and management of defense-oriented projects at the prestigious “Talpiot” program of the Israeli Defense Forces. He holds a B.Sc. in Physics, Mathematics and Chemistry from the Hebrew University of Jerusalem, and an M.A. in Economics and Finance from Tel Aviv University, with distinction.senior management.
Assaf Segalhas served as a member of our board of directors since October 2017. Mr. Segal serves as a board member of several companies, including Biokine therapeutics Ltd., Campus Bio L.P., Clal Life Sciences L.P. and Clal Application Center Ltd. Prior to that time, Mr. Segal was a Partner at Variance Economic Consulting Ltd., from 2004 until June 2015, where he provided in‑depth consulting for international and local clients in a wide range of industries, including telecommunications, internet, biotech, heavy industry and financial sectors. Previously, he founded a start‑up software company. Mr. Segal also previously held a managerial position at PriceWaterhouseCoopers Corporate Finance and was an Economic Department manager at the North American division of Amdocs Inc. His experience also includes risk management and house account (“Nostro”) trading at the Union Bank of Israel, and serving as an economist for capital markets in the Research Department of the Bank of Israel. Mr. Segal also has many years of experience in economic consulting and company valuations, joint ventures and financial instruments for investments, M&A, and IPOs. He has 15 years of experience in economic consulting for international and local clients in the Bio‑Tech sector as well as in Hi‑Tech, financial and other sectors. He holds a B.A. in Economics and Statistics and an M.B.A. (Finance and Information Systems) from the Hebrew University of Jerusalem.
Vickie R. Driverhas served as a member of our board of directors since May 2017. Dr. Driver is board certified in foot surgery by the American Board of Podiatric Surgery and is a Fellow at the American College of Foot and Ankle Surgeons, licensed in Rhode Island. Her career as a podiatric physician and surgeon has included a special emphasis on limb preservation and wound healing in her medical practice, as well as, research and education. Dr. Driver has been a Professor of Surgery in the Department of Orthopedics at Brown University (Clinical) since 2014. She has served for 9 years on the Board of Directors for the Association for the Advancement of Wound Care (“AAWC”), and recently completed her tenure as President for this international organization. Dr. Driver is also the chair of Wound Care Experts and U.S. Food and Drug Administration (“FDA”) Clinical Endpoints Project. She has just been named to serve as member at large to the Board of Directors of the Wound Healing Society (“WHS”) and Board Member to the Critical Limb Ischemia (“CLI”) Global Society. In addition, she serves on multiple national and international clinical committees that focus on preventing limb loss and improving wound healing in the high‑risk population. She has served as an investigator for more than 70 important multi‑center randomized clinical trials, as well as developed and supervised multiple research fellowship training programs. She has served and chaired multiple committees for large national and international pivotal clinical trials and has authored over 120 publications and abstracts. Dr. Driver is credited with the development and directorship of multiple major multidisciplinary Limb Preservation– Wound Healing Centers of Excellence, including Military/VA, Hospital and University based programs. Since 2015, she has served as Director, Translational Medicine, Wound Healing at the Novartis Institute for Biomedical Research. From 2011 to 2014, she was Program Director, Inaugural Educational Committee at the American College of Wound Healing and Tissue Repair at University of Illinois School of Medicine. From 2011 to 2015, she was also Scientific Director, Colorado Prevention Center, Wound Care Laboratory at the University of Colorado. From 2012 to 2015, Dr. Driver held a number of positions at the Providence Veterans Administration Medical Center in Rhode Island, including Chief, Section of Podiatric Surgery and Director, Clinical Research, Limb Preservation and Wound Healing. Prior thereto, she held various positions at multiple major multidisciplinary Limb Preservation – Wound Healing Centers of Excellence. Dr. Driver received a Doctorate of Podiatric Medicine and Surgery from the California College of Podiatric Medicine and Surgery and a Masters in Medical Education from Samuel Merritt University.
Nissim Mashiach has served as a member of our board of directors since June 2017. Mr. Mashiach served as President and Chief Executive Officer of Macrocure Ltd., a Nasdaq‑listed biotechnology company focused on the treatment of chronic and other hard‑to‑heal wounds, from June 2012 to January 2017. From 2009 to 2012, he served as General Manager at Ethicon, a Johnson & Johnson company. Prior to Ethicon, he served as President and Chief Operating Officer at Omrix Biopharmaceuticals, Inc., which was acquired by Johnson & Johnson in 2008. Prior to Omrix, Mr. Mashiach held leadership positions at several pharmaceutical companies. He holds an MBA from the University of Manchester in Manchester, England, an MPharmSc from the Hebrew University in Jerusalem, Israel, and a B.Sc, Chemical Engineering from the Technion‑Israel Institute of Technology in Haifa, Israel.
Sharon Kochan has served as a member of our board of directors since June 2017. Mr. Kochan is the CEO of Padagis LLC. a leading specialty pharma company that was curved out of Perrigo Company PLC. On July 2021. Prior to such, Mr. Kochan has served as Executive Vice President & President Pharmaceuticals / International, for Perrigo Company Plc., a global, over-the-counter, consumer goods and specialty pharmaceutical company listed on the New York Stock Exchange, since 2012 and has been a member of the Perrigo Executive Committee since 2007. From March 2007 to July 2012, he served as Executive Vice President, General Manager of Prescription Pharmaceuticals for Perrigo and from 2005 to 2007, he was Senior Vice President of Business Development and Strategy for Perrigo. Mr. Kochan was Vice President, Business Development of Agis Industries (1983) Ltd. from 2001 until Perrigo acquired Agis in 2005. He completed the Senior Management Program at the Technion Institute of Management in Haifa, Israel, received a Master of Science in Operations Research & Management Science from Columbia University in New York City and received a Bachelor of Science in Industrial and Management Engineering from Tel-Aviv University in Tel-Aviv, Israel.
Mr. Samuel Moed has served as a member of our board of directors since April 2020. Prior to joining our board, Mr. Moed served as an executive at Bristol-Myers Squibb, a global biopharma company focused on innovative therapeutics. In his most recent capacity as Senior Vice President, Corporate Strategy, Mr. Moed led the strategic planning of the company in all major business activities worldwide. Previously, Mr. Moed oversaw strategy for BMS’ Worldwide Pharmaceuticals Group, encompassing a range of global strategic initiatives, and managed a global portfolio of strategic alliances. Among other positions, he served as President of U.S. Pharmaceuticals and as President of Worldwide Consumer Healthcare. Mr. Moed received a BA in history from Columbia University in New York City.
Mr. David Fox has served as a member of our board of directors since April 2020. Mr. Fox was most recently a partner at Kirkland & Ellis LLP and served as a member of its Global Executive Management Committee until 2019. Prior to joining Kirkland, Mr. Fox was partner with Skadden, Arps, Slate, Meagher & Flom LLP, where he was a member of its top governing committee. Mr. Fox is a director of Israel Discount Bank of New York, a member of the borad of directors at the Park Avenue Armory and a member of the advisory board of New Alternatives for Children, for which he provides support to families caring for medically fragile children. In addition, Mr. Fox serves on the board of governors, and is an honorary fellow of the Hebrew University, Jerusalem. He holds an LL.B. degree from Jerusalem University, Israel.
B. Compensation Compensation of Directors and Executive Officers The table below reflects the compensation granted to our five most highly compensated officers during or with respect to the year ended December 31, 2021.2023. All amounts reported in the table reflect the cost to the company, as recognized in our financial statements for the year ended December 31, 2021.2023. | Name and Position | | Salary &
Social
Benefits(1) | | | Bonus | | | Share‑Based
Payment(2) | | | Other Compensation(3) | | | Total | | | Salary &
Social
Benefits(1) | | | Bonus | | | Share-Based
Payment(2) | | | Other Compensation
(3) | | | Total | | | | | ( thousand U.S. dollars)(4) | | | (thousand U.S. dollars)(4) | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | Sharon Malka, Chief Executive Officer | | | 427 | | | | 65 | | | | 227 | | | | 5 | | | | 724 | | | Lior Rosenberg, M.D., Chief Medical Technology Officer | | | 334 | | | | 39 | | | | 81 | | | | 25 | | | | 479 | | | Ofer Gonen, Chief Executive Officer | | | | 480 | | | | 152 | | | | 484 | | | | 17 | | | | 1,133 | | Ety Klinger, Chief Research & Development Officer | | | 292 | | | | 32 | | | | 73 | | | | 20 | | | | 417 | | | | 297 | | | | 58 | | | | 102 | | | | 22 | | | | 479 | | Boaz Gur-Lavie, Chief Financial Officer | | | 256 | | | | 29 | | | | 76 | | | | | | | | 385 | | | Yaron Meyer, Executive Vice President, General Counsel & Corporate Secretary | | | 208 | (5) | | | 27 | | | | 61 | | | | 5 | | | | 301 | | | | 250 | | | | 44 | | | | 80 | | | | 5 | | | | 379 | | Hani Luxenburg, Chief Financial Officer (5) | | | | 166 | | | | 43 | | | | 9 | | | | 26 | | | | 244 | | Boaz Gur-Lavie, former Chief Financial Officer (6) | | | | 140 | | | | 22 | | | | 46 | | | | 16 | | | | 224 | |
(1) | Represents the officer’s gross salary plus payment of mandatory social benefits made by the company on behalf of such officer. Such benefits may include, to the extent applicable to the executive, payments, contributions and/or allocations for savings funds (e.g., Managers’ Life Insurance Policy), education funds (referred to in Hebrew as “keren hishtalmut”), pension, severance, risk insurances (e.g., life or work disability insurance) and payments for social security. |
| (2) | Represents the equity‑based compensation expenses recorded in the company’s consolidated financial statements for the year ended December 31, 20212023 based on the options’ grant date fair value in accordance with accounting guidance for equity‑based compensation. |
| (3) | Represents the other benefits to such officer, which includes either or both of (i) car expenses, including lease costs, gas and maintenance, provided to the officers, and (ii) vacation benefits. benefits and (iii) severance payment. |
| (4) | Converted (i) from NIS into U.S. dollars at the rate of NIS3.229NIS 3.69 = U.S$1, based on the average representative rate of exchange between the NIS and the U.S. dollar in the year ended December 31, 20212023 as reported by the Bank of Israel in the year ended December 31, 2021. 2023. |
| (5) | Represents only 8 months’ salary due to paternity leave.Ms. Luxenburg Joined the Company in May 2023. | | (6) | Mr. Gur Lavie worked in the Company until July 2023. |
The aggregate compensation paid and equity‑based compensation and other payments expensed by us and our subsidiaries to our directors and executive officers with respect to the year ended December 31, 20212023 was $3.5$3.8 million. As of December 31, 2021,2023, options to purchase 1,811,319467,146 ordinary shares, exercisable at a weighted average exercise price of $3.86$15.3 per share, and restricted share units (“RSUs”) that may be settled for 55,00244,067 ordinary shares, in each case granted to our directors and executive officers, were outstanding under our equity incentive plans. We do not have any written agreements with any director providing for benefits upon the termination of such director’s relationship with our company or its subsidiaries.
Employment Agreements with Executive Officers We have entered into written employment agreements with all of our executive officers, which include standard provisions for a company in our industry regarding non‑competition/solicitation, confidentiality of information and assignment of inventions. Except for Prof. Rosenberg,Ofer Gonen, our Chief MedicalExecutive Officer, our executive officers will not receive benefits upon the termination of their respective employment with us, other than payment of salary and benefits (and limited accrual of vacation days) during the required notice period for termination of their employment, which varies for each individual. Upon termination of his employment, Prof. RosenbergMr. Gonen is entitled to a one‑time termination payment of tensix months of salary. Directors’ Service Contracts Other than with respect to our directors that are also executive officers, there are no arrangements or understandings between us, on the one hand, and any of our directors, on the other hand, providing for benefits upon termination of their service as directors of our company.
2003 Israeli Share Option PlanIncentive Plans
In November 2003, we adopted our 2003 Israeli Share Option Plan (the “2003 Plan”). The 2003 Plan provides for the grant of options to our and our subsidiaries’ directors, employees, officers, consultants and service providers, among others.
The initial reserved pool under the 2003 Plan was 1,710,000 ordinary shares and subsequently increased to a total of 3,230,000 ordinary shares. The 2003 Plan expired on December 31, 2013. Options that remain outstanding under the 2003 Plan continue to be governed by the terms of the plan, notwithstanding that expiration. The 2003 Plan is administered by our board of directors or a committee designated by our board of directors, which determines, subject to Israeli law, the grantees of options, the terms of the options, including exercise prices, vesting schedules, acceleration of vesting, the type of option and the other matters necessary or desirable for, or incidental to the administration of the 2003 Plan. The 2003 Plan provides for the issuance of options under various tax regimes including, without limitation, pursuant to Sections 102 and 3(i) of the Israeli Income Tax Ordinance (New Version) 1961 (the “Ordinance”).
Section 102 of the Ordinance allows employees, directors and officers who are not controlling shareholders and who are Israeli residents to receive favorable tax treatment for compensation in the form of shares or options. Section 102 of the Ordinance includes two alternatives for tax treatment involving the issuance of options or shares to a trustee for the benefit of the grantees and also includes an additional alternative for the issuance of options or shares directly to the grantee. Section 102(b)(2) of the Ordinance, which provides the most favorable tax treatment for grantees, permits the issuance to a trustee under the “capital gains track.” In order to comply with the terms of the capital gains track, all options granted under a specific plan and subject to the provisions of Section 102 of the Ordinance, as well as the shares issued upon exercise of such options and other shares received following any realization of rights with respect to such options, such as share dividends and share splits, must be registered in the name of a trustee selected by the board of directors and held in trust for the benefit of the relevant employee, director or officer. The trustee may not release these options or shares to the relevant grantee before the second anniversary of the registration of the options in the name of the trustee. However, under this track, we are not allowed to deduct an expense with respect to the issuance of the options or shares.
The 2003 Plan provides that options granted to our employees, directors and officers who are not controlling shareholders and who are considered Israeli residents are intended to qualify for special tax treatment under the “capital gains track” provisions of Section 102(b)(2) of the Ordinance. Our Israeli non‑employee service providers and controlling shareholders may only be granted options under Section 3(i) of the Ordinance, which does not provide for similar tax benefits.
Options granted under the 2003 Plan are subject to vesting schedules and generally expire ten years from approval of the option and vest over a four‑year period commencing on the date of grant, such that 25% of the granted options vest annually on each of the first, second, third and fourth anniversaries of the date of grant. Under the 2003 Plan, in the event of termination of employment or services for reasons of disability or death, the grantee, or in the case of death, his or her legal successor, may exercise options that have vested prior to termination within a period of six months after the date of termination. If a grantee’s employment or service is terminated for cause, all of the grantee’s vested and unvested options expire on the date of termination. If a grantee’s employment or service is terminated for any other reason, the grantee may exercise his or her vested options within 90 days after the date of termination. Any expired or unvested options are returned to the pool for reissuance.
The 2003 Plan provides that in the event of a merger or consolidation of our company or a sale of all, or substantially all, of our assets, the unexercised options outstanding may be assumed, or substituted for an appropriate number of shares of each class of shares or other securities as were distributed to our shareholders in connection with such transaction and the exercise price will be appropriately adjusted. If not so assumed or substituted, all non‑vested and non‑exercised options will expire upon the closing of the transaction. Our board of directors or its designated committee, as applicable, may provide in the option agreement that if the acquirer does not agree to assume or substitute the options, vesting of the options shall be accelerated so that any unvested option or any portion thereof will vest 10 days prior to the closing of the transaction. In the event that such consideration received in the transaction is not solely in the form of ordinary shares of another company, the board of directors or the designated committee, as applicable, may, with the approval of the acquirer, provide that in lieu of the assumption or substitution of the options, the options will be substituted by another type of asset or property, including cash.
2014 Equity Incentive Plan In March 2014, we adopted and obtained shareholder approval for our 2014 Equity Incentive Plan, which was amended as of December 18, 2018 (the “2014 Plan”). The 2014 Plan provides for the grant of options, restricted shares, RSUs and other share‑based awards to our and our subsidiaries’ and affiliates’ directors, employees, officers, consultants and advisors, among others and to any other person whose services are considered valuable to us or them, to continue as service providers, to increase their efforts on our behalf or behalf of a subsidiary or affiliate and to promote the success of our business. Following the approval of the 2014 Plan by the Israeli tax authorities, we are only granting options or other equity incentive awards under the 2014 Plan, although previously‑granted optionswhich shares will be rolled over to a new share incentive plan— the 2024 Share Incentive Plan— that we will adopted and awards will continue to be governed by our 2003 Plan and the shares underlying such options and awards will count against the reserved poolbring for the 2014 Plan. The initial reserved pool under the 2014 Plan was 3,032,742 ordinary shares, which will automatically increase on January 1 of each year by a number of ordinary shares equal to the lowest of (i) 2% of our outstanding shares, (ii) 600,000 shares and (iii) a number of shares determined by our board of directors, if so determined prior to January 1 of the year in which the increase will occur; provided that the pool of shares reserved under the Plan shall not exceed 15% (fifteen percent) of the then outstanding shares. Pursuant to an “evergreen” provision in the 2014 Plan, the reserved pool was increased by 431,006, 540,955, 543,577, 544,055 and 544,738 ordinary shares as of January 1, 2015, January 1, 2018, January 1, 2019, January 1, 2020 and January 1, 2021 , respectively, representing 2% of our outstanding shares as of each such date. We did not increase the reserved pool in 2016 or 2017.shareholder approval soon. The 2014 Plan is administered by our board of directors or by a committee designated by the board of directors, which determine, subject to Israeli law, the grantees of awards and the terms of the grant, including exercise prices, vesting schedules, acceleration of vesting and the other matters necessary in the administration of the 2014 Plan. The 2014 Plan enables us to issue awards under various tax regimes, including, without limitation, pursuant to Sections 102 and 3(i) of the Ordinance, as discussed under “—2003“-2003 Share Incentive Plan” above, and under Section 422 of the U.S. Internal Revenue Code of 1986, as amended (the “Code”). Options granted under the 2014 Plan to U.S. residents may qualify as “incentive stock options” within the meaning of Section 422 of the Code, or may be non‑qualified. The exercise price for “incentive stock options” must not be less than the fair market value on the date on which an option is granted, or 110% of the fair market value if the option holder holds more than 10% of our share capital. We currently intend to grant awards under the 2014204 Plan under the capital gains track of Section 102(b)(2) of the Ordinance only to our employees, directors and officers who are not controlling shareholders and are considered Israeli residents. Awards under the 2014 Plan may be granted until ten years from the date on which the 2014 Plan was approved by our board of directors.
Options granted under the 2014 Plan generally vest over three or four years commencing on the date of grant, such that 33% or 25%, respectively, vests annually on the anniversary of the date of grant. Options, other than certain incentive share options, that are not exercised within ten years from the grant date expire, unless otherwise determined by our board of directors or its designated committee, as applicable. Share options that qualify as “incentive stock options” and are granted to a person holding more than 10% of our voting power will expire within five years from the date of the grant. In the event of the death of a grantee while employed by or performing service for us or a subsidiary or within three months thereafter, or the termination of a grantee’s employment or services for reasons of disability, the grantee, or in the case of death, his or her legal successor, may exercise options that have vested prior to termination within a period of one year from the date of disability or death. If we terminate a grantee’s employment or service for cause, all of the grantee’s vested and unvested options will expire on the date of termination. If a grantee’s employment or service is terminated for any other reason, the grantee may exercise his or her vested options within three months of the date of termination. Any expired or unvested options return to the pool for reissuance. In the event of a merger or consolidation of our company or a sale of all, or substantially all, of our shares or assets or other transaction having a similar effect on us, then without the consent of the option holder, our board of directors or its designated committee, as applicable, may but is not required to (i) cause any outstanding award to be assumed or an equivalent award to be substituted by such successor corporation, or (ii) in case the successor corporation refuses to assume or substitute the award (a) provide the grantee with the option to exercise the award as to all or part of the shares or (b) cancel the options against payment in cash in an amount determined by the board of directors or the committee as fair in the circumstances. Notwithstanding the foregoing, our board of directors or its designated committee may upon such event amend or terminate the terms of any award, including conferring the right to purchase any other security or asset that the board of directors shall deem, in good faith, appropriate. Our board of directors or its designated committee may, in its discretion, approve that any awards granted under the 2014 Plan shall be subject to additional conditions in the case of a merger or a consolidation.
Restricted share awards are ordinary shares that are awarded to a participant subject to the satisfaction of the terms and conditions established by the board of directors or a committee designated by the board of directors. Until such time as the applicable restrictions lapse, restricted shares are subject to forfeiture and may not be sold, assigned, pledged or otherwise disposed of by the participant who holds those shares. Generally, if a grantee’s employment or service is terminated for any reason prior to the expiration of the time when the restrictions lapse, shares that are still restricted will be forfeited. Prospective 2024 Share Incentive Plan Because the 2014 Plan expires in March 2024, we expect to adopt a new share incentive plan—the 2024 Share Incentive Plan (the “2024 Plan”) - in the near future and bring it to our shareholders for approval. Outstanding grants under the 2014 Plan will remain subject to the 2014 Plan even after the expiration of that plan, but any ordinary shares available under the 2014 Plan as of the adoption of the 2024 Plan, or that subsequently become available under the 2014 Plan due to the expiration, cancellation, forfeiture or other surrender of outstanding grants under the 2014 Plan, will become available for new grants under the 2024 Plan. We will provide a description of the new 2024 Plan in the proxy statement for the general meeting of shareholders at which such plan will be presented for shareholder approval, and in subsequent documents filed with or furnished to, the SEC. The following table provides information regarding the outstanding options to purchase our ordinary shares, and RSUs held by each of our directors and executive officers who beneficially owns greater than 1% of our ordinary shares (after including shares underlying options or RSUs) as of March 15, 2022:2024:
| Name | | Number of
Options | | | Number of RSUs | | Grant Date | | Exercise
Price | | | Vested
Options/RSU's as
of March 15, 2022 | | Expiration Date | Sharon Malka, Chief Executive Officer | | | 121,600 | | | | | 12/24/2013 | | $ | 12.89 | | | | 121,600 | | 12/23/2023 | | | | | 50,000 | | | | | 12/23/2015 | | $ | 9.58 | | | | 50,000 | | 12/22/2025 | | | | | 135,000 | | | | | 12/31/2018 | | $ | 5.15 | | | | 101,250 | | 12/30/2028 | | | | | | | | | 45,000 | | 12/31/2018 | | | | | | | 33,750 | | | | | | | 40,000 | | | | | | 05/02/2019 | | $ | 4.92 | | | | 30,000 | | 5/1/2029 | | | | | | | | | 20,000 | | 05/02/2019 | | | | | | | 15,000 | | | | | | | 81,170 | | | | | | 06/29/2020 | | $ | 1.75 | | | | 20,292 | | 6/28/2030 | | | | | 45,692 | | | | | | 06/15/2021 | | $ | 5.36 | | | | - | | 6/14/2031 | | | | | | | | | 7,615 | | 06/15/2021 | | | | | | | - | | 6/14/3031 | | | | | | | | | | | | | | | | | | | | | Lior Rosenberg, Chief Medical Technology Officer | | | 76,000 | | | | | | 12/24/2013 | | $ | 12.89 | | | | 76,000 | | 12/23/2023 | | | | | 25,000 | | | | | | 12/23/2015 | | $ | 9.58 | | | | 25,000 | | 12/22/2025 | | | | | 20,000 | | | | | | 12/31/2018 | | $ | 5.15 | | | | 15,000 | | 12/30/2028 | | | | | | | | | 6,667 | | 12/31/2018 | | | | | | | 5,000 | | | | | | | 43,600 | | | | | | 04/23/2020 | | $ | 1.75 | | | | 10,900 | | 4/22/2030 | | | | | 27,953 | | | | | | 03/04/2021 | | $ | 5.36 | | | | - | | 3/3/2031 | | | | | | | | | 4,659 | | 03/04/2021 | | | | | | | 1,165 | | 3/3/2031 |
| Name | | Number of
Options | | Number of
RSUs | | Grant Date | | Exercise
Price - $ | | Vested
Options/RSU's | | Expiration Date | | Ofer Gonen | | 85,700 | | | | 07/06/2022 | | 14.42 | | 37,500 | | 06/06/2032 | | | | | | 20,089 | | 07/06/2022 | | 0 | | 15,625 | | 06/06/2032 | | | | 86,000 | | | | 31/05/2023 | | 11.89 | | 21,500 | | 30/05/2033 |
C. Board Practices Board of Directors Authorities Under the Israeli Companies Law, the management of our company is vested in our board of directors. Our board of directors may exercise all powers and may take all actions that are not specifically granted to our shareholders or to management. Our executive officers are responsible for our day‑to‑day management and have individual responsibilities established by our board of directors. Our Chief Executive Officer is appointed by, and serves at the discretion of, our board of directors, subject to the employment agreement that we have entered into with him. All other executive officers are also appointed by our board of directors, and are subject to the terms of any applicable employment agreements that we may enter into with them.
Composition Under our articles of association, our board of directors must consist of at least five and not more than nine directors, including, to the extent then required to appoint them, at least two external directors, who may be required to be appointed under the Israeli Companies Law. At any time the minimum number of directors (other than the external directors) shall not fall below three. Other than external directors, for whom special election requirements apply under the Israeli Companies Law when they are required to be elected, as detailed below, the Israeli Companies Law and our articles of association provide that directors are elected annually at the general meeting of our shareholders by a vote of the holders of a majority of the voting power represented present and voting, in person or by proxy, at that meeting. We have only one class of directors. In accordance with the exemption available to foreign private issuers under Nasdaq rules, we are not required to comply with the requirements of the Nasdaq rules with regard to having a majority of independent directors on our board of directors, as long as we follow Israeli law and practice, in accordance with which our board of directors includes at least two external directors. However, because we have elected under the Israeli Companies Law regulations to opt-out from compliance with Israeli law requirements related to appointment of external directors and audit and compensation committee composition (as described under “External Directors” below), we are not permitted to also exempt ourselves from the Nasdaq majority independent directors requirement. Our board of directors has determined that foursix of our sixeight current directors are independent under the Nasdaq Stock Market listing rules. The definition of “independent director” underrules such that we comply with the Nasdaq Stock Market listing rules and “external director” undermajority independence rule. Under the Israeli Companies Law, overlap to a significant degree such that we would generally expect the two directors that serve as external directors to qualify as independent under the Nasdaq Stock Market listing rules. However, it is possible for a director to qualify as an “external director” under the Israeli Companies Law without qualifying as an “independent director” under the Nasdaq Stock Market listing rules, or vice‑versa. The definition of external director under the Israeli Companies Law includes a set of statutory criteria that must be satisfied, including criteria whose aim is to ensure that there is no factor that would impair the ability of the external director to exercise independent judgment. The definition of independent director under the Nasdaq Stock Market listing rules specifies similar, although less stringent, requirements in addition to the requirement that theour board of directors must determine the minimum number of directors who are required to have accounting and financial expertise. In determining the number of directors required to have such expertise, our board of directors must consider, any factor which would impairamong other things, the abilitytype and size of the independent directorcompany and the scope and complexity of its operations. Our board of directors has determined that the minimum number of directors of our company who are required to exercise independent judgment. In addition, external directors serve for a period of three years pursuant to the requirements of the Israeli Companies Law. However, external directors must be elected by a special majority of shareholders while independent directors may be elected by an ordinary majority. See “—External Directors” for a description of the requirements under the Israeli Companies Law for a director to serve as an external director.have accounting and financial expertise is one. Nominations and Election In accordance with the exemption available to foreign private issuers under Nasdaq listing rules, we do not follow the requirementshave appointed a committee of the Nasdaq rules with regard to the process of nominating directors, and instead follow Israeli law and practice, in accordance with which our board of directors (or a committee thereof)that is authorized to recommend to our board, for nomination for election by our shareholders, director nomineesnominees. Please see “Committees of the Board of Directors— Nominating and Governance Committee” below for election.more information. Under the Israeli Companies Law and our articles of association, nominees for directors may also be proposed by any shareholder holding at least 1% of our outstanding voting power. However, any such shareholder may propose a nominee only if a written notice of such shareholder’s intent to propose a nominee has been given to our Secretary (or, if we have no such Secretary, our Chief Executive Officer). Pursuant to our Articles of Association, any such notice must include certain information, including, among other things, a description of all arrangements between the nominating shareholder and the proposed director nominee(s) and any other person pursuant to which the nomination(s) are to be made by the nominating shareholder, the consent of the proposed director nominee(s) to serve as our director(s) if elected and a declaration signed by the nominee(s) declaring that there is no limitation under the Israeli Companies Law preventing their election, and that all of the information that is required under the Israeli Companies Law to be provided to us in connection with such election has been provided. Under the Israeli Companies Law regulations, any such shareholder nomination must be delivered to our registered Israeli office within seven days after we publish notice of our upcoming annual general meeting of shareholders (or within 14 days after we publish a preliminary notification of an upcoming annual general meeting). In addition, our articles of association allow our board of directors to appoint directors to fill vacancies on our board of directors for a term of office equal to the remaining period of the term of office of the director(s) whose office(s) have been vacated. External directors are elected for an initial term of three years and may be elected for additional three‑year terms under the circumstances described below. External directors may be removed from office only under the limited circumstances set forth in the Israeli Companies Law. See “—External Directors.” Under the Israeli Companies Law, our board of directors must determine the minimum number of directors who are required to have accounting and financial expertise. See “—External Directors” below. In determining the number of directors required to have such expertise, our board of directors must consider, among other things, the type and size of the company and the scope and complexity of its operations. Our board of directors has determined that the minimum number of directors of our company who are required to have accounting and financial expertise is one.
We are not a party to, and are not aware of, any voting agreements among our shareholders. In addition, there are no family relationships among our executive officers and directors. Under regulations promulgated under the Israeli Companies Law, Israeli public companies whose shares are traded on certain U.S. stock exchanges, such as the Nasdaq Global Market, and that lack a controlling shareholder (as defined below) are exempt from the requirement to appoint external directors. Any such company is also exempt from the Israeli Companies Law requirements related to the composition of the audit and compensation committees of the Board. Eligibility for these exemptions is conditioned on compliance with U.S. stock exchange listing rules related to majority Board independence and the composition of the audit and compensation committees of the Board, as applicable to all listed domestic U.S. companies. Because we have a controlling shareholder (CBI), we are not eligible for these exemptions under these regulations.97
External Directors Under the Israeli Companies Law, our boardthe boards of directors isof companies, whose shares are publicly traded, including companies with shares traded in the United States, are generally required to include at least two members who qualify as external directors. Our current Under regulations promulgated under the Israeli Companies Law, Israeli public companies whose shares are traded on certain U.S. stock exchanges, such as the Nasdaq Global Market, and that lack a controlling shareholder (as defined below) are exempt from the requirement to appoint external directors. Any such company is also exempt from the Israeli Companies Law requirements related to the composition of the audit and compensation committees of the Board. Eligibility for these exemptions is conditioned on compliance with U.S. stock exchange listing rules related to majority Board independence and the composition of the audit and compensation committees of the Board, as applicable to all listed domestic U.S. companies. Eligibility is furthermore conditioned on our election of a female or male director at any time when we hold elections of directors and the Board is then composed of solely male or solely female members. On December 5, 2022, in light of our Board’s determination that Clal Biotechnology Industries Ltd. was no longer a “controlling shareholder” of our company under the Israeli Companies Law definition (provided further below), the Board elected, pursuant to the Israeli Companies Law regulations, to exempt our company from compliance with the (i) requirement to appoint external directors, are Nissim Mashiach and Sharon Kochan, each(ii) required composition of whom serves on ourthe audit committee and compensation committee. The provisionscommittees of the Board under the Israeli Companies Law. At the time that it made that election, our Board affirmatively determined that we meet the conditions for exemption from the external director requirement, including that a majority of the members of our Board, along with each of the members of the audit and compensation committees of the Board, are independent under the Nasdaq Listing Rules. Our Board has confirmed subsequently in an ongoing manner that we continue to fulfill those conditions for exemption from the Israeli Companies Law set forth special approval requirements related to (i) the appointment of external directors, and (ii) the composition of the audit committee and compensation committees of the Board.
Our election to exempt our company from compliance with the external director and related requirements can be reversed at any time by our Board, in which case we would need to hold a shareholder meeting to once again to appoint external directors, whose election would be for thea three-year term. The election of each external directors. External directors must be elected bydirector would require a majority vote of the shares present and voting at a shareholders meeting, of shareholders, provided that either: such• the majority voted in favor of election includes at least a majority of the shares held by allnon-controlling shareholders who are not controlling shareholders and do not have a personal interest in the election of the external director (other than a personal interest not deriving from a relationship with a controlling shareholder) that are voted at the meeting, excluding abstentions, to which we refer to as a disinterested majority; or
• the total number of shares votedheld by non‑controllingnon-controlling, disinterested shareholders and by shareholders who do not have a personal interest(as described in the election of the external directorprevious bullet-point) voted against the election of the external director does not exceed 2%two percent (2%) of the aggregate voting rights in the company. The term “controlling shareholder” as usedis defined in the Israeli Companies Law for purposes of all matters related to external directors and for certain other purposes (such as the requirements related to appointment to the audit committee or compensation committee, as described below), means a shareholder with the ability to direct the activities of the company, other than by virtue of being an office holder. A shareholder is presumed to be a controlling shareholder if the shareholder holds 50% or more of the voting rights in a company or has the right to appoint the majority of the directors of the company or its general manager. With respect to certain matters (various related party transactions)manager (i.e., a controlling shareholder is deemed to include a shareholder that holds 25% or more of the voting rights in a public company if no other shareholder holds more than 50% of the voting rights in the company, but excludes a shareholder whose power derives solely from his or her position as a director of the company or from any other position with the company.its CEO). The initial term of an external director is three years. Thereafter, an external director may be reelected by shareholders to serve in that capacity for up to two additional three‑year terms, provided that either:
| (i) | his or her service for each such additional term is recommended by one or more shareholders holding at least 1% of the company’s voting rights and is approved at a shareholders meeting by a disinterested majority, where the total number of shares held by non‑controlling, disinterested shareholders voting for such reelection exceeds 2% of the aggregate voting rights in the company, subject to additional restrictions set forth in the Israeli Companies Law with respect to affiliations of external director nominee; or
|
| (ii) | his or her service for each such additional term is recommended by the board of directors and is approved at a meeting of shareholders by the same majority required for the initial election of an external director (as described above).
|
The term of office for external directors for Israeli companies traded on certain foreign stock exchanges, including the Nasdaq Global Market, may be extended indefinitely in increments of additional three‑year terms, in each case provided that the audit committee and the board of directors of the company confirm that, in light of the external director’s expertise and special contribution to the work of the board of directors and its committees, the reelection for such additional period(s) is beneficial to the company, and provided that the external director is reelected subject to the same shareholder vote requirements (as described above regarding the reelection of external directors). Prior to the approval of the reelection of the external director at a general meeting of shareholders, the company’s shareholders must be informed of the term previously served by him or her and of the reasons why the board of directors and audit committee recommended the extension of his or her term.
External directors may be removed from office by a special general meeting of shareholders called by the board of directors, which approves such dismissal by the same shareholder vote percentage required for their election or by a court, in each case, only under limited circumstances, including ceasing to meet the statutory qualifications for appointment, or violating their duty of loyalty to the company.
If an external directorship becomes vacant and there are fewer than two external directors on the board of directors at the time, then the board of directors is required underFor further information concerning the Israeli Companies Law provisions related to call a shareholders’ meeting as soon as practicable to appoint a replacement external director. Each committee of the board of directors that exercises the powers of the board of directors must include at least one external director, except that the audit committee and the compensation committee must include all external directors, then servingplease see “Item 6. Directors, Senior Management and Employees- C. Board Practices- Board of Directors- External Directors” in our annual report on Form 20-F for the board of directors and an external director must serve as chair thereof. Underyear ended December 31, 2022, which we filed with the Israeli Companies Law, external directors of a company are prohibited from receiving, directly or indirectly, any compensation from the company other than for their services as external directors pursuant to the Israeli Companies Law and the regulations promulgated thereunder. Compensation of an external director is determined prior to his or her appointment and may not be changed during his or her term subject to certain exceptions.SEC on March 16, 2023.
The Israeli Companies Law provides that a person is not qualified to be appointed as an external director if (i) the person is a relative of a controlling shareholder of the company, or (ii) if that person or his or her relative, partner, employer, another person to whom he or she was directly or indirectly subordinate, or any entity under the person’s control, has or had, during the two years preceding the date of appointment as an external director: (a) any affiliation or other disqualifying relationship with the company, with any person or entity controlling the company or a relative of such person, or with any entity controlled by or under common control with the company; or (b) in the case of a company with no shareholder holding 25% or more of its voting rights, had at the date of appointment as an external director, any affiliation or other disqualifying relationship with a person then serving as chairman of the board or chief executive officer, a holder of 5% or more of the issued share capital or voting power in the company or the most senior financial officer.
The term “relative” is defined in the Israeli Companies Law as a spouse, sibling, parent, grandparent or descendant; spouse’s sibling, parent or descendant; and the spouse of each of the foregoing persons. Under the Israeli Companies Law, the term “affiliation” and the similar types of disqualifying relationships include (subject to certain exceptions):
an employment relationship;
a business or professional relationship even if not maintained on a regular basis (excluding insignificant relationships);
service as an office holder, excluding service as a director in a private company prior to the initial public offering of its shares if such director was appointed as a director of the private company in order to serve as an external director following the initial public offering.
The term “office holder” is defined in the Israeli Companies Law as a general manager (i.e., chief executive officer), chief business manager, deputy general manager, vice general manager, any other person assuming the responsibilities of any of these positions regardless of that person’s title, a director and any other manager directly subordinate to the general manager.
In addition, no person may serve as an external director if that person’s position or professional or other activities create, or may create, a conflict of interest with that person’s responsibilities as a director or otherwise interfere with that person’s ability to serve as an external director or if the person is an employee of the Israel Securities Authority of an Israeli stock exchange. A person may furthermore not continue to serve as an external director if he or she received direct or indirect compensation from the company including amounts paid pursuant to indemnification or exculpation contracts or commitments and insurance coverage for his or her service as an external director, other than as permitted by the Israeli Companies Law and the regulations promulgated thereunder.
Following the termination of an external director’s service on a board of directors, such former external director and his or her spouse and children may not be provided a direct or indirect benefit by the company, its controlling shareholder or any entity under its controlling shareholder’s control. This includes engagement as an office holder of the company or a company controlled by its controlling shareholder or employment by, or provision of services to, any such company for consideration, either directly or indirectly, including through a corporation controlled by the former external director. This restriction extends for a period of two years with regard to the former external director and his or her spouse or child and for one year with respect to other relatives of the former external director.
If at the time at which an external director is appointed all members of the board of directors who are not controlling shareholders or relatives of controlling shareholders of the company are of the same gender, the external director to be appointed must be of the other gender. A director of one company may not be appointed as an external director of another company if a director of the other company is acting as an external director of the first company at such time.
According to the Israeli Companies Law and regulations promulgated thereunder, a person may be appointed as an external director only if he or she has professional qualifications or if he or she has accounting and financial expertise (each, as defined below); provided that at least one of the external directors must be determined by our board of directors to have accounting and financial expertise. However, if at least one of our other directors (i) meets the independence requirements under the Exchange Act, (ii) meets the standards of the Nasdaq Stock Market listing rules for membership on the audit committee and (iii) has accounting and financial expertise as defined under the Israeli Companies Law, then neither of our external directors is required to possess accounting and financial expertise as long as each possesses the requisite professional qualifications.
A director with accounting and financial expertise is a director who, due to his or her education, experience and skills, possesses an expertise in, and an understanding of, financial and accounting matters and financial statements, such that he or she is able to understand the financial statements of the company and initiate a discussion about the presentation of financial data. A director is deemed to have professional qualifications if he or she has any of (i) an academic degree in economics, business management, accounting, law or public administration, (ii) an academic degree or has completed another form of higher education in the primary field of business of the company or in a field which is relevant to his/her position in the company or (iii) at least five years of experience serving in one of the following capacities, or at least five years of cumulative experience serving in two or more of the following capacities: (a) a senior business management position in a company with a significant volume of business, (b) a senior position in the company’s primary field of business or (c) a senior position in public administration or service. The board of directors is charged with determining whether a director possesses financial and accounting expertise or professional qualifications.
Our board of directors has determined that Sharon Kochan has accounting and financial expertise and possesses professional qualifications as required under the Israeli Companies Law, while Nissim Mashiach possesses professional qualifications.
Leadership Structure of the Board In accordance with the Israeli Companies Law and our articles of association, our board of directors is required to appoint one of its members to serve as chairman of the board of directors. Our board of directors has appointed Stephen T. WillsHomi Shamir to serve as executive chairman of the board of directors.
Committees of the Board of Directors Audit Committee Israeli Companies Law composition requirements Under the Israeli Companies Law, we are required to have an audit committee comprised of at least three directors. To the extent we are then required to appoint external directors, includingthis committee must include all of the external directors, one of whom must serve as chairman of the committee. The audit committee may not include the chairman of the board, a controlling shareholder of the company, a relative of a controlling shareholder, a director employed by or providing services on a regular basisThere are additional requirements as to the company, to a controlling shareholder or to an entity controlled by a controlling shareholder, or a director who derives most of his or her income from a controlling shareholder. In addition, under the Israeli Companies Law, the audit committee of a publicly traded company must consist of a majority of unaffiliated directors. In general, an “unaffiliated director’’ under the Israeli Companies Law is defined as either an external director or as a director who meets the following criteria: he or she meets the qualifications for being appointed as an external director, except for the requirement (i) that the director be an Israeli resident (which does not apply to companies such as ours whose securities have been offered outside of Israel or are listed for trading outside of Israel) and (ii) for accounting and financial expertise or professional qualifications; and
he or she has not served as a director of the company for a period exceeding nine consecutive years. For this purpose, a break of less than two years in the service shall not be deemed to interrupt the continuation of the service.
Each member of our audit committee (each, as identified in the second paragraph under the sub-heading “Nasdaq listing rules composition requirements” below) is an unaffiliated director under the Israeli Companies Law, thereby fulfilling the foregoing Israeli law requirement for the composition of the audit committee.committee under the Israeli Companies Law. However, when we elected to exempt our company from the external director requirement, we concurrently elected to exempt our company from all of such requirements (which exemption is conditioned on our fulfillment of all Nasdaq listing requirements related to the composition of the audit committee).
Nasdaq listing rules composition requirements Under the Nasdaq Stock Market listing rules, we are required to maintain an audit committee consisting of at least three independent directors, each of whom is financially literate and one of whom has accounting or related financial management expertise. If we choose to follow requirements under Israeli law in lieu of those Nasdaq requirements, we must disclose that fact in this annual report. Our audit committee consists of Sharon KochanStephen T. Wills (chairperson), Nissim MashiachVickie R. Driver and Vickie R Driver,Nachum Shamir, each of whom is an independent director in accordance with Rule 10A‑3(b)(1) under the Exchange Act and satisfies the independent director requirements under the Nasdaq Stock Market listing rules. All members of our audit committee meet the requirements for financial literacy under the applicable listing rules of the Nasdaq Stock Market. Our board of directors has determined that Sharon KochanStephen T Wills is an “audit committee financial expert,” as defined in the SEC regulations.regulations, and possesses accounting and financial expertise, as defined under the Israeli Companies Law. Audit committee role Our board of directors has adopted an audit committee charter that sets forth the responsibilities of the audit committee consistent with the rules and regulations of the SEC and the Nasdaq Stock Market listing rules, as well as the requirements for such committee under the Israeli Companies Law, including the following:
oversight of our independent registered public accounting firm and recommending the engagement, compensation or termination of engagement of our independent registered public accounting firm to the board of directors in accordance with Israeli law;
recommending the engagement or termination of the person filling the office of our internal auditor; and
recommending the terms of audit and non‑audit services provided by the independent registered public accounting firm for pre‑approval by our board of directors.
Our audit committee provides assistance to our board of directors in fulfilling its legal and fiduciary obligations in matters involving our accounting, auditing, financial reporting, internal control and legal compliance functions by pre‑approving the services performed by our independent accountants and reviewing their reports regarding our accounting practices and systems of internal control over financial reporting. Our audit committee also oversees the audit efforts of our independent accountants and takes those actions that it deems necessary to satisfy itself that the accountants are independent of management.
UnderOur board of directors has adopted an audit committee charter that sets forth the responsibilities of the audit committee consistent with the rules and regulations of the SEC and the Nasdaq Stock Market listing rules, as well as the requirements for such committee under the Israeli Companies Law, our audit committee is responsible for:including the following:
| • | overseeing our independent registered public accounting firm and recommending the engagement, compensation or termination of engagement of our independent registered public accounting firm to the board of directors in accordance with Israeli law; | | • | recommending the engagement or termination of the person filling the office of our internal auditor; | | • | recommending the terms of audit and non‑audit services provided by the independent registered public accounting firm for pre‑approval by our board of directors; | | • | pre-approving all audit, audit-related and all permitted non-audit services, and related fees and terms, to be provided to the Company by the independent auditor under applicable law and regulations; | | • | Establishing policies for hiring employees or former employees of the independent auditor in accordance with applicable law and regulations; | | • | reviewing periodically with management, the internal auditor and the independent auditor, the adequacy and effectiveness of the Company’s system of internal control over financial reporting; | | • | evaluating whether management is effectively communicating to employees and other persons retained by MediWound the importance of internal accounting and financial control effectiveness; | | • | reviewing with management and the independent auditor the annual and quarterly financial statements of MediWound prior to filing (or submission, as the case may be) with the SEC; | | • | discussing with management, and review prior to submission, any responses to SEC comments regarding the Company’s financial statements or financial reporting; | | • | discussing with management and MediWound’s independent auditors generally the types of financial information (including earnings guidance) to be disclosed in earnings press releases and earnings calls, as well as to analysts and rating agencies; | | • | reviewing and discussing with management and MediWound independent auditors MediWound’s earnings press releases, including the use of any pro forma, adjusted or other non-GAAP financial information, before their release to the public; | | • | reviewing with the MediWound’s general counsel and/or external counsel legal and regulatory matters that could have a material impact on the financial statements;
| | • | establishing procedures for (i) the receipt, retention, and treatment of complaints received by MediWound regarding accounting, internal accounting controls or auditing matters; and (ii) the confidential, anonymous submission by employees of MediWound of concerns regarding questionable accounting or auditing matters, and review any complaints or concerns received pursuant to such procedures; | | • | reviewing with management and the independent auditor risks of material misstatements due to fraud, and the process and controls implemented by the Company to manage the risks; | | • | recommending to the Board the retention and termination of the internal auditor, and the internal auditor’s engagement fees and terms, in accordance with the Companies Law, approve the internal auditor’s work plan, and review and discuss the internal auditor’s work on a quarterly basis; | | • | reviewing and monitoring, as appropriate, (i) litigation or other legal matters that could have a significant impact on MediWound’s financial results; and (ii) significant findings of any examination by regulatory authorities or agencies, in the areas of securities, accounting or tax; | | • | receiving reports of suspected business irregularities and legal compliance issues through periodic and, when appropriate, immediate reporting by members of MediWound’s management, legal counsel, the independent or internal auditor or pursuant to any “whistleblower policy” adopted by the Committee. | | • | establishing procedures for handling complaints by MediWound’s employees with respect to deficiencies in its business operations, including the protection to be granted to such complaining employees; | | • | overseeing MediWound’s policies and procedures regarding compliance with applicable financial and accounting related standards, rules and regulations; | | • | reviewing and considering the approval of related party transactions, including transactions between MediWound and a controlling shareholder (as defined under the Israeli Companies Law) or a transaction with another person in which a controlling shareholder has a personal interest, and transactions involving an office holder of MediWound (as defined in the Israeli Companies Law) that may present a conflict of interest between the duties of such office holder to the Company and his or her personal interests, in each case in accordance with Nasdaq listing rules, the Israeli Companies Law or as referred by the Board; | | • | confirming that MediWound’s independent auditors are informed of the committee’s understanding of the Company’s related party transactions that are significant to the Company; and reviewing and discussing with the Company’s independent auditors the auditors’ evaluation of the Company’s identification of, accounting for, and disclosure of, its related party transactions, including any significant matters arising from the audit regarding MediWound’s related party transactions; | | • | discussing MediWound’s policies with respect to risk assessment and risk management, and reviewing contingent liabilities and risks that may be material to MediWound and relevant major legislative and regulatory developments that could materially impact MediWound’s contingent liabilities and risks; | | •
| reviewing periodically the Company’s major financial risk exposures and the Company’s policies for managing such risks; | | • | conducting or authorizing investigations into any matters within the committee’s scope of responsibilities; and | | • | reviewing and approving any material change or waiver in MediWound’s Code of Conduct regarding directors or executive officers, and disclosures made in the Company’s annual report. |
determining whether there are deficiencies in the business management practices of our company, including in consultation with our internal auditor or the independent auditor, and making recommendations to the board of directors to improve such practices;
determining whether to approve certain related party transactions (including transactions in which an office holder has a personal interest and whether such transaction is extraordinary or material under the Israeli Companies Law) (see “—Approval of Related Party Transactions Under Israeli Law”);
establishing the approval process (including, potentially, the approval of the audit committee and conducting a competitive procedure supervised by the audit committee) for certain transactions with a controlling shareholder or in which a controlling shareholder has a personal interest;
where the board of directors approves the working plan of the internal auditor, examining such working plan before its submission to the board of directors and proposing amendments thereto;
examining our internal audit controls and internal auditor’s performance, including whether the internal auditor has sufficient resources and tools to fulfill his responsibilities;
examining the scope of our auditor’s work and compensation and submitting a recommendation with respect thereto to our board of directors or shareholders, depending on which of them is considering the appointment of our auditor; and
establishing procedures for the handling of employees’ complaints as to the management of our business and the protection to be provided to such employees.
Our audit committee may not approve any actions requiring its approval (see “—Approval“-Approval of Related Party Transactions Under Israeli Law”), unless at the time of the approval a majority of the committee’s members are present, which majority consists of unaffiliated directors including at least one external director.director, to the extent we then have external directors serving on the Board).
Compensation Committee and Compensation Policy Israeli Companies Law compensation committee composition requirements Under the Israeli Companies Law, the board of directors of a public company must appoint a compensation committee. The compensationIf a company is required to appoint external directors, the committee generally (subject to certain exceptions that do not apply to our company) must be comprisedconsist of at least three directors,members, including all of the external directors, whoone of whom must constitute a majorityserve as chairman of the members of, and includecommittee. There are additional requirements as to the chairpersoncomposition of the compensation committee. Each compensation committee member who is not anunder the Companies Law. However, when we elected to exempt our company from the external director must be a director whose compensation does not exceed an amount that may be paidrequirement, we concurrently elected to an external director. The compensation committee is subject toexempt our company from all of such requirements (including the same Israelithree-member minimum). Our exemption under the Companies Law restrictions as the audit committee as to who may not be a memberis conditioned on our fulfillment of the compensation committee. Each member of our compensation committee (each, as identified in the second paragraph under the sub-heading “Nasdaqall Nasdaq listing rules compensation committee composition requirements” below) fulfills the foregoing Israeli law requirements related to the composition of the compensation committee. Israeli Companies Law committee duties The duties of the compensation committee include the recommendation to the company’s board of directors of a policy regarding the terms of engagement of office holders, which we refer to as a compensation policy. That policy must be adopted by the company’s board of directors, after considering the recommendations of the compensation committee, and must be approved by the company’s shareholders, which approval requires what we refer to as a Special Majority Approval for Compensation. A Special Majority Approval for Compensation requires shareholder approval by a majority vote of the shares present and voting at a meeting of shareholders called for such purpose, provided that either (a) such majority includes at least a majority of the shares held by all shareholders who are not controlling shareholders and do not have a conflict of interest (referred to under the Israeli Companies Law as a “personal interest”) in such compensation arrangement or (b) the total number of shares of non-controlling shareholders and shareholders who do not have a personal interest in the compensation arrangement and who vote against the arrangement does not exceed 2% of the company’s aggregate voting rights.
Compensation policy requirements
We have adopted a compensation policy, most recently at the extraordinary general meeting of shareholders held on September 23, 2019,November 28, 2022, which policy serves as the basis for decisions concerning the financial terms of employment or engagement of office holders, including exculpation, insurance, indemnification or any monetary payment or obligation of payment or other benefit in respect of employment or engagement. Under the Israeli Companies Law, the compensation policy must relate to certain factors, including advancement of the company’s objectives, the company’s business plan and its long-term strategy, and creation of appropriate incentives for office holders. It must also consider, among other things, the company’s risk management, size and the nature of its operations. The compensation policy must furthermore consider the following additional factors: the knowledge, skills, expertise and accomplishments of the relevant office holder;
the office holder’s roles and responsibilities and prior compensation agreements with him or her;
the relationship between the terms offered and the average compensation of the other employees of the company, including those employed through manpower companies;
the impact of disparities in salary upon work relationships in the company;
the possibility of reducing variable compensation at the discretion of the board of directors;
the possibility of setting a limit on the exercise value of non-cash variable equity-based compensation; and
as to severance compensation, the period of service of the office holder, the terms of his or her compensation during such service period, the company’s performance during that period of service, the person’s contribution towards the company’s achievement of its goals and the maximization of its profits, and the circumstances under which the person is leaving the company.
| • | the knowledge, skills, expertise and accomplishments of the relevant office holder; | | • | the office holder’s roles and responsibilities and prior compensation agreements with him or her; | | • | the relationship between the terms offered and the average compensation of the other employees of the company, including those employed through manpower companies; | | • | the impact of disparities in salary upon work relationships in the company; | | • | the possibility of reducing variable compensation at the discretion of the board of directors; | | • | the possibility of setting a limit on the exercise value of non-cash variable equity-based compensation; and | | • | as to severance compensation, the period of service of the office holder, the terms of his or her compensation during such service period, the company’s performance during that period of service, the person’s contribution towards the company’s achievement of its goals and the maximization of its profits, and the circumstances under which the person is leaving the company. |
The compensation policy must also include the following principles: the link between variable compensation and long-term performance, which variable compensation shall, other than office holder who report to the CEO, be primarily based on measurable criteria;
the relationship between variable and fixed compensation, and the ceiling for the value of variable compensation;
the conditions under which an office holder would be required to repay compensation paid to him or her if it was later shown that the data upon which such compensation was based was inaccurate and was required to be restated in the company’s financial statements;
the minimum holding or vesting period for variable, equity-based compensation; and
maximum limits for severance compensation.
| • | the link between variable compensation and long-term performance, which variable compensation shall, other than office holder who report to the CEO, be primarily based on measurable criteria; | | • | the relationship between variable and fixed compensation, and the ceiling for the value of variable compensation; | | • | the conditions under which an office holder would be required to repay compensation paid to him or her if it was later shown that the data upon which such compensation was based was inaccurate and was required to be restated in the company’s financial statements; | | • | the minimum holding or vesting period for variable, equity-based compensation; and | | • | maximum limits for severance compensation. |
The compensation committee is responsible for (a) recommending the compensation policy to the company’s board of directors for its approval (and subsequent approval by its shareholders) and (b) duties related to the compensation policy and to the compensation of a company’s office holders as well as functions previously fulfilled by a company’s audit committee with respect to matters related to approval of the terms of engagement of office holders, including: recommending whether a compensation policy should continue in effect, if the then-current policy has a term of greater than three years (approval of either a new compensation policy or the continuation of an existing compensation policy must in any case occur every three years, other than following a company’s initial public offering, in which case such approval must occur within 5 years of the initial public offering);
recommending to the board of directors periodic updates to the compensation policy and assessing implementation of the compensation policy;
approving compensation terms of executive officers, directors and employees that require approval of the compensation committee;
determining whether the compensation terms of a chief executive officer nominee, which were determined pursuant to the compensation policy, will be exempt from approval of the shareholders because such approval would harm the ability to engage with such nominee; and
determining, subject to the approval of the board and under special circumstances, whether to override a determination of the company’s shareholders regarding certain compensation related issues.
| • | recommending whether a compensation policy should continue in effect, if the then-current policy has a term of greater than three years (approval of either a new compensation policy or the continuation of an existing compensation policy must in any case occur every three years, other than following a company’s initial public offering, in which case such approval must occur within 5 years of the initial public offering); | | • | recommending to the board of directors periodic updates to the compensation policy and assessing implementation of the compensation policy; | | • | approving compensation terms of executive officers, directors and employees that require approval of the compensation committee; | | • | determining whether the compensation terms of a chief executive officer nominee, which were determined pursuant to the compensation policy, will be exempt from approval of the shareholders because such approval would harm the ability to engage with such nominee; and | | • | determining, subject to the approval of the board and under special circumstances, whether to override a determination of the company’s shareholders regarding certain compensation related issues. |
A copy of our current compensation policy serves as an exhibit to this annual report on Form 20-F.
Nasdaq listing rules compensation committee composition requirements Under Nasdaq corporate governance rules, we are required to maintain a wholly-independent compensation committee consisting of at least two independent directors or, if we choose to follow requirements under Israeli law, we must disclose that fact in this annual report. Each of the members of the compensation committee is required to be independent under the Nasdaq rules relating to compensation committee members and Rule 10C‑1(b)(1) under the Exchange Act, which are different than the general test for independence of board members. Our compensation committee consists of Nissim MashiachNachum Shamir (chairperson), Sharon KochanDavid Fox and Samuel Moed,Stephen T. Wills, each of whom is an independent director under the Nasdaq Stock Market listing rules and each of whom satisfies the above-described additional requirements for compensation committee members under the Nasdaq rules and Exchange Act. Compensation committee charter and role Our board of directors has adopted a compensation committee charter setting forth the responsibilities of the compensation committee, which include: | • | the responsibilities set forth in the compensation policy; | | • | reviewing and approving the granting of options and other incentive awards to the extent such authority is delegated by our board of directors; and | | • | reviewing, evaluating and making recommendations regarding the compensation and benefits for our non-employee directors. |
Nominating and Governance Committee reviewingUnder Nasdaq corporate governance rules, director nominees must either be selected, or recommended for the Board's selection, either by independent directors constituting a majority of the Board's independent directors in a vote in which only independent directors participate, or a nominations committee comprised solely of independent directors. If we choose to exempt ourselves from that requirement in accordance with our home country practices, we must disclose that fact in this annual report. Each of the members of the nominating committee is required to be independent under the Nasdaq rules.
Our Board has established a nominating and approving the grantinggovernance committee, whose members consist of optionsNachum Shamir and other incentive awards to the extent such authority is delegatedDavid Fox, each of whom has been determined by our Board to meet that Nasdaq independence requirement, with Mr. Fox serving as chair. Our board of directors;directors has adopted a nominating and governance committee charter setting forth the responsibilities of the committee, which include: | • | overseeing and assisting our board in reviewing and recommending nominees for election of directors; | | • | assessing the performance of the members of our board; and | | • | establishing and maintaining effective corporate governance policies and practices, including, but not limited to, developing and recommending to our board a set of corporate governance guidelines applicable to our business. |
reviewing, evaluatingResearch and making recommendations regarding the compensation and benefits for our non-employee directors.
Development Committee Our Board has established a research and development committee, which is composed of Vickie R Driver, Stephen T. Wills and Sharon Malka, with Dr. Driver serving as chairperson. The primary functions of the research and development committee include: 98
| • | overseeing the Company's scientific, technical, research and development strategy, and the implementation thereof; and | | • | advising our board of directors and management regarding program prioritization, clinical development strategy, regulatory strategy and interactions, and related matters. |
Board Diversity Matrix (As of March 1, 2024) | Country of Principal Executive Offices | Israel | | Foreign Private Issuer | Yes | | Disclosure Prohibited under Home Country Law | No | | Total Number of Directors | 5 |
| | | | | | | | | | Part I: Gender Identity | | | | | | | | | Directors | 1 | | 4 | | 0 | | 0 | | Part II: Demographic Background | | | | | | | | | Underrepresented Individual in Home Country Jurisdiction | * | | | | | | | | LGBTQ+ | * | | | | | | | | Did Not Disclose Demographic Background | * | | | | | | |
(*) There are no directors who fall into any demographic background category who have agreed to disclose that information publicly.
Internal Auditor
Under the Israeli Companies Law, the board of directors of an Israeli public company must appoint an internal auditor recommended by the audit committee. An internal auditor may not be: a person (or a relative of a person) who holds 5% or more of the company’s outstanding shares or voting rights;
a person (or a relative of a person) who has the power to appoint a director or the general manager of the company (i.e., the chief executive officer);
an office holder (including a director) of the company (or a relative thereof); or
a member of the company’s independent accounting firm, or anyone on its behalf.
| • | a person (or a relative of a person) who holds 5% or more of the company’s outstanding shares or voting rights; | | • | a person (or a relative of a person) who has the power to appoint a director or the general manager of the company (i.e., the chief executive officer); | | • | an office holder (including a director) of the company (or a relative thereof); or | | • | a member of the company’s independent accounting firm, or anyone on its behalf. |
The role of the internal auditor is to examine, among other things, our compliance with applicable law and orderly business procedures. The audit committee is required to oversee the activities and to assess the performance of the internal auditor as well as to review the internal auditor’s work plan. Our internal auditor is Mr. Yisrael Gewirtz.
Fiduciary Duties of Directors and Executive Officers
The Israeli Companies Law codifies the fiduciary duties that office holders owe to a company. Each person listed in the table under “—Executive“Executive Officers and Directors” is an office holder under the Israeli Companies Law. An office holder’s fiduciary duties consist of a duty of care and a duty of loyalty. The duty of care requires an office holder to act with the level of care with which a reasonable office holder in the same position would have acted under the same circumstances. The duty of loyalty requires that an office holder act in good faith and in the best interests of the company.
The duty of care includes a duty to use reasonable means to obtain: information on the advisability of a given action brought for his or her approval or performed by virtue of his or her position; and
all other important information pertaining to any such action.
| • | information on the advisability of a given action brought for his or her approval or performed by virtue of his or her position; and | | • | all other important information pertaining to any such action. |
The duty of loyalty includes a duty to: refrain from any conflict of interest between the performance of his or her duties to the company and his or her other duties or personal affairs;
refrain from any activity that is competitive with the business of the company;
refrain from exploiting any business opportunity of the company to receive a personal gain for himself or herself or others; and
disclose to the company any information or documents relating to the company’s affairs which the office holder received as a result of his or her position as an office holder.
| • | refrain from any conflict of interest between the performance of his or her duties to the company and his or her other duties or personal affairs; | | • | refrain from any activity that is competitive with the business of the company; | | • | refrain from exploiting any business opportunity of the company to receive a personal gain for himself or herself or others; and | | • | disclose to the company any information or documents relating to the company’s affairs which the office holder received as a result of his or her position as an office holder. |
Disclosure of personal interests of an office holder and approval of certain transactions The Israeli Companies Law requires that an office holder promptly disclose to the board of directors any personal interest that he or she may be aware of and all related material information or documents concerning any existing or proposed transaction with the company. An interested office holder’s disclosure must be made promptly and in any event no later than the first meeting of the board of directors at which the transaction is considered. A personal interest includes an interest of any person in an act or transaction of a company, including a personal interest of such person’s relative or of a corporate body in which such person or a relative of such person is a 5% or greater shareholder, director or general manager or in which he or she has the right to appoint at least one director or the general manager, but excluding a personal interest stemming from one’s ownership of shares in the company. A personal interest furthermore includes the personal interest of a person for whom the office holder holds a voting proxy or the personal interest of the office holder with respect to his or her vote on behalf of a person for whom he or she holds a proxy even if such shareholder has no personal interest in the matter. An office holder is not, however, obliged to disclose a personal interest if it derives solely from the personal interest of his or her relative in a transaction that is not considered an extraordinary transaction. Under the Israeli Companies Law, an extraordinary transaction is defined as any of the following: a transaction other than in the ordinary course of business;
a transaction that is not on market terms; or
| • | a transaction other than in the ordinary course of business; | | • | a transaction that is not on market terms; or | | • | a transaction that may have a material impact on a company’s profitability, assets or liabilities. |
If it is determined that an office holder has a personal interest in a transaction which is not an extraordinary transaction, approval by the board of directors is required for the transaction, unless the company’s articles of association provide for a different method of approval. Further, so long as an office holder has disclosed his or her personal interest in a transaction, the board of directors may approve an action by the office holder that would otherwise be deemed a breach of his or her duty of loyalty. However, a company may not approve a transaction or action that is not in the best interest of the company or that is not performed by the office holder in good faith. An extraordinary transaction in which an office holder has a personal interest requires approval first by the company’s audit committee and subsequently by the board of directors. The compensation of, or an undertaking to indemnify or insure, an office holder who is not a director requires approval first by the company’s compensation committee, then by the company’s board of directors. If such compensation arrangement or an undertaking to indemnify or insure is inconsistent with the company’s stated compensation policy, or if the office holder is the chief executive officer (apart from a number of specific exceptions), then such arrangement is further subject to a Special Majority Approval for Compensation. Arrangements regarding the compensation, indemnification or insurance of a director require the approval of the compensation committee, board of directors and shareholders by ordinary majority, in that order, and under certain circumstances, a Special Majority Approval for Compensation. Generally, a person who has a personal interest in a matter which is considered at a meeting of the board of directors or the audit committee may not be present at such a meeting or vote on that matter unless the chairman of the relevant committee or board of directors (as applicable) determines that he or she should be present in order to present the transaction that is subject to approval. If a majority of the members of the audit committee or the board of directors (as applicable) has a personal interest in the approval of a transaction, then all directors may participate in discussions of the audit committee or the board of directors (as applicable) on such transaction and the voting on approval thereof, but shareholder approval is also required for such transaction. Disclosure of personal interests of controlling shareholders and approval of certain transactions Pursuant to Israeli law, the disclosure requirements regarding personal interests that apply to directors and executive officers also apply to a controlling shareholder of a public company. In the context of a transaction involving a shareholder of the company, a controlling shareholder also includes a shareholder who holds 25% or more of the voting rights in the company if no other shareholder holds more than 50% of the voting rights in the company. For this purpose, the holdings of all shareholders who have a personal interest in the same transaction will be aggregated. The approval of the audit committee or the compensation committee, the board of directors and the shareholders of the company, in that order, is required for (a) extraordinary transactions with a controlling shareholder or in which a controlling shareholder has a personal interest, (b) the engagement with a controlling shareholder or his or her relative, directly or indirectly, including through a company under the control of the controlling shareholder, for the provision of services to the company, (c) the terms of engagement and compensation of a controlling shareholder or his or her relative who is an office holder or (d) the employment of a controlling shareholder or his or her relative by the company, other than as an office holder. In addition, the shareholder approval requires one of the following, which we refer to as a Special Majority: at least a majority of the shares held by all shareholders who do not have a personal interest in the transaction and who are present and voting at the meeting approves the transaction, excluding abstentions; or
the shares voted against the transaction by shareholders who have no personal interest in the transaction and who are present and voting at the meeting do not exceed 2% of the voting rights in the company.
| • | at least a majority of the shares held by all shareholders who do not have a personal interest in the transaction and who are present and voting at the meeting approves the transaction, excluding abstentions; or | | • | the shares voted against the transaction by shareholders who have no personal interest in the transaction and who are present and voting at the meeting do not exceed 2% of the voting rights in the company. |
To the extent that any such transaction with a controlling shareholder is for a period extending beyond three years, approval is required once every three years, unless, with respect to certain transactions, the audit committee determines that the duration of the transaction is reasonable given the circumstances related thereto. Arrangements regarding the compensation, indemnification or insurance of a controlling shareholder in his or her capacity as an office holder require the approval of the compensation committee, board of directors and shareholders by a Special Majority, in that order, and the terms thereof may not be inconsistent with the company’s stated compensation policy. Pursuant to regulations promulgated under the Israeli Companies Law, certain transactions with a controlling shareholder or his or her relative, or with directors, that would otherwise require approval of a company’s shareholders may be exempt from shareholder approval upon certain determinations of the audit committee and board of directors.
As of March 15, 2022, Clal Biotechnology Industries Ltd. beneficially owned or controlled, directly and indirectly, 33.8% of our issued and outstanding ordinary shares and (assuming that no other shareholder holds more than 50% of the voting rights in our company) should therefore be deemed a “controlling shareholder” for purposes of the approval of related party transactions under the Israeli Companies Law.105
Shareholder duties Pursuant to the Israeli Companies Law, a shareholder has a duty to act in good faith and in a customary manner toward the company and other shareholders and to refrain from abusing his or her power in the company, including, among other things, in voting at a general meeting and at shareholder class meetings with respect to the following matters: an amendment to the company’s articles of association;
an increase of the company’s authorized share capital;
the approval of related party transactions and acts of office holders that require shareholder approval.
| • | an amendment to the company’s articles of association; | | • | an increase of the company’s authorized share capital; | | • | a merger; or | | • | the approval of related party transactions and acts of office holders that require shareholder approval. |
A shareholder also has a general duty to refrain from discriminating against other shareholders. In addition, certain shareholders have a duty of fairness toward the company. These shareholders include any controlling shareholder, any shareholder who knows that he or she has the power to determine the outcome of a shareholder vote and any shareholder who has the power to appoint or to prevent the appointment of an office holder of the company or other power towards the company. The Israeli Companies Law does not define the substance of the duty of fairness, except to state that the remedies generally available upon a breach of contract will also apply in the event of a breach of the duty to act with fairness. Exculpation, Insurance and Indemnification of Directors and Officers Under the Israeli Companies Law, a company may not exculpate an office holder from liability for a breach of the duty of loyalty. An Israeli company may exculpate an office holder in advance from liability to the company, in whole or in part, for damages caused to the company as a result of a breach of duty of care but only if a provision authorizing such exculpation is included in its articles of association. Our articles of association include such a provision. A company may not exculpate in advance a director from liability arising out of a prohibited dividend or distribution to shareholders. Under the Israeli Companies Law, a company may indemnify an office holder in respect of the following liabilities and expenses incurred for acts performed by him or her as an office holder, either pursuant to an undertaking made in advance of an event or following an event, provided its articles of association include a provision authorizing such indemnification: financial liability imposed on him or her in favor of another person pursuant to a judgment, including a settlement or arbitrator’s award approved by a court. However, if an undertaking to indemnify an office holder with respect to such liability is provided in advance, then such an undertaking must be limited to events which, in the opinion of the board of directors, can be foreseen based on the company’s activities when the undertaking to indemnify is given, and to an amount or according to criteria determined by the board of directors as reasonable under the circumstances, and such undertaking shall detail the abovementioned foreseen events and amount or criteria;
reasonable litigation expenses, including attorneys’ fees, incurred by the office holder (1) as a result of an investigation or proceeding instituted against him or her by an authority authorized to conduct such investigation or proceeding, provided that (i) no indictment was filed against such office holder as a result of such investigation or proceeding, and (ii) no financial liability was imposed upon him or her as a substitute for the criminal proceeding as a result of such investigation or proceeding or, if such financial liability was imposed, it was imposed with respect to an offense that does not require proof of criminal intent; and (2) in connection with a monetary sanction; and
| • | financial liability imposed on him or her in favor of another person pursuant to a judgment, including a settlement or arbitrator’s award approved by a court. However, if an undertaking to indemnify an office holder with respect to such liability is provided in advance, then such an undertaking must be limited to events which, in the opinion of the board of directors, can be foreseen based on the company’s activities when the undertaking to indemnify is given, and to an amount or according to criteria determined by the board of directors as reasonable under the circumstances, and such undertaking shall detail the abovementioned foreseen events and amount or criteria; | | • | reasonable litigation expenses, including attorneys’ fees, incurred by the office holder (1) as a result of an investigation or proceeding instituted against him or her by an authority authorized to conduct such investigation or proceeding, provided that (i) no indictment was filed against such office holder as a result of such investigation or proceeding, and (ii) no financial liability was imposed upon him or her as a substitute for the criminal proceeding as a result of such investigation or proceeding or, if such financial liability was imposed, it was imposed with respect to an offense that does not require proof of criminal intent; and (2) in connection with a monetary sanction; and | | • | reasonable litigation expenses, including attorneys’ fees, incurred by the office holder or imposed by a court in proceedings instituted against him or her by the company, on its behalf, or by a third party, or in connection with criminal proceedings in which the office holder was acquitted, or as a result of a conviction for an offense that does not require proof of criminal intent. |
Under the Israeli Companies Law, a company may insure an office holder against the following liabilities incurred for acts performed by him or her as an office holder, if and to the extent provided in the company’s articles of association: a breach of the duty of loyalty to the company, provided that the office holder acted in good faith and had a reasonable basis to believe that the act would not harm the company;
a breach of duty of care to the company or to a third party, to the extent such a breach arises out of the negligent conduct of the office holder; and
a financial liability imposed on the office holder in favor of a third party.
| • | a breach of the duty of loyalty to the company, provided that the office holder acted in good faith and had a reasonable basis to believe that the act would not harm the company; | | • | a breach of duty of care to the company or to a third party, to the extent such a breach arises out of the negligent conduct of the office holder; and | | • | a financial liability imposed on the office holder in favor of a third party. |
Under the Israeli Companies Law, a company may not indemnify, exculpate or insure an office holder against any of the following: a breach of the duty of loyalty, except for indemnification and insurance for a breach of the duty of loyalty to the company to the extent that the office holder acted in good faith and had a reasonable basis to believe that the act would not harm the company;
a breach of duty of care committed intentionally or recklessly, excluding a breach arising out of the negligent conduct of the office holder;
an act or omission committed with intent to derive illegal personal benefit; or
a fine or forfeit levied against the office holder.
| • | a breach of the duty of loyalty, except for indemnification and insurance for a breach of the duty of loyalty to the company to the extent that the office holder acted in good faith and had a reasonable basis to believe that the act would not harm the company; | | • | a breach of duty of care committed intentionally or recklessly, excluding a breach arising out of the negligent conduct of the office holder; | | • | an act or omission committed with intent to derive illegal personal benefit; or | | • | a fine or forfeit levied against the office holder. |
Under the Israeli Companies Law, exculpation, indemnification and insurance of office holders in a public company must be approved by the compensation committee and the board of directors and, with respect to certain office holders or under certain circumstances, also by the shareholders. See “—Approval“-Approval of Related Party Transactions Under Israeli Law.” Our articles of association permit us to exculpate, indemnify and insure our office holders to the fullest extent permitted or to be permitted by the Israeli Companies Law. We have obtained directors’ and officers’ liability insurance for the benefit of our office holders and intend to continue to maintain such coverage and pay all premiums thereunder to the fullest extent permitted by the Israeli Companies Law. In addition, we have entered into agreements with each of our directors and executive officers exculpating them from liability to us for damages caused to us as a result of a breach of duty of care and undertaking to indemnify them, in each case, to the fullest extent permitted by our articles of association and Israeli Law. The maximum indemnification amount set forth in those agreements is limited to an amount equal to the greater of (i) 25% of our total shareholders’ equity based on our most recently financial statements of the time of the actual payment of the indemnification; (ii) $50 million; (iii) 40% of our total market cap (which shall mean the average closing price of the Company’s ordinary shares over the 30 trading days prior to the actual payment of indemnification multiplied by the total number of issued and outstanding shares of the Company as of the date of actual payment); and (iv) in connection with or arising out of a public offering of our securities, the aggregate amount of proceeds from the sale by us and/or any shareholder of ours securities in such offering. The maximum amount set forth in those agreements is in addition to amounts actually paid, if any, under insurance policies and/or by a third-party pursuant to an indemnification arrangement.
As of December 31, 2021,2023, we had 77100 employees, 6788 of whom were based in Israel and 1012 based throughout Europe and employed by our German subsidiary. The distribution of our employees according to main areas of activity is as follows: 914 employees in the administrative department, 2526 employees in the research and development department, 3348 employees in the manufacturing department and 1012 employees in the sales and marketing department. As of December 31, 2021,2023, we did not employ a significant number of temporary employees.
Israeli labor laws govern the length of the workday and workweek, minimum wages for employees, procedures for hiring and dismissing employees, determination of severance pay, annual leave, sick days, advance notice of termination, payments to the National Insurance Institute and other conditions of employment, and include equal opportunity and anti-discrimination laws. While none of our employees is party to any collective bargaining agreements, certain provisions of the collective bargaining agreements between the Histadrut (General Federation of Labor in Israel) and the Coordination Bureau of Economic Organizations (including the Industrialists’ Associations) are applicable to our employees in Israel by order of the Israeli Ministry of the Economy. These provisions primarily concern pension fund benefits for all employees, insurance for work-related accidents, recuperation pay and travel expenses. We generally provide our employees with benefits and working conditions beyond the required minimums. We have never experienced any employment-related work stoppages and believe our relationships with our employees are good. For information regarding the share ownership of our directors and executive officers, see “ITEM 6.B. Compensation—2014Compensation-2014 Equity Incentive Plan” and “ITEM 7.A. Major Shareholders.”
F. Disclosure of Any Action to Recover Erroneously Awarded Compensation
None.
Item 7. MAJOR SHAREHOLDERS AND RELATED PARTY TRANSACTIONS The following table sets forth information with respect to the beneficial ownership of our shares as of March 15, 20222024 by:
each person or entity known by us to own beneficially more than 5% of our outstanding shares;
each of our directors and executive officers individually; and
all of our executive officers and directors as a group.
| • | each person or entity known by us to own beneficially more than 5% of our outstanding shares; | | • | each of our directors and executive officers individually; and | | • | all of our executive officers and directors as a group. |
The beneficial ownership of ordinary shares is determined in accordance with the rules of the SEC and generally includes any ordinary shares over which a person exercises sole or shared voting or investment power. The percentage of shares beneficially owned is based on 32,509,5449,491,718 ordinary shares issued and outstanding as of March 15, 2022.2024. Ordinary shares that are issuable under stock options or RSUs that are currently exercisable or exercisable within 60 days of March 15, 20222024 are deemed to be outstanding and to be beneficially owned by the person holding the stock option for the purpose of computing the number of shares and percentage ownership of that person. Those shares are not deemed outstanding, however, for the purpose of computing the percentage ownership of any other person. The beneficial ownership does not include a 30 –day option to purchase up to an additional 781,249 ordinary shares that we gave to the Underwriters in relation with the public offering that we had in March 2022. All of our shareholders, including the shareholders listed below, have the same voting rights attached to their ordinary shares. See “ITEM 10.B. Articles of Association.” None of our principal shareholders nor our directors or executive officers possesses different or special voting rights with respect to their ordinary shares. Unless otherwise noted below, each shareholder’s address is c/o MediWound Ltd., 42 Hayarkon Street, Yavne 8122745, Israel.
A description of any material relationship that our principal shareholders have had with us or any of our predecessors or affiliates within the past three years is included under “ITEM 7.B. Related Party Transactions.” | Name of Beneficial Owner | | Number of Shares Beneficially Held | | | Percentage of Class | | | Number of
Shares
Beneficially
Held | | | Percentage of
Class | | | Directors and Executive Officers | | | | | | | | | | | | | Nachum (Homi) Shamir
| | | | * | | | | * | | Ofer Gonen
| | | | 98,730 | | | | 1.1 | % | | Vickie R. Driver | | | | * | | | | * | | David Fox
| | | | * | | | | * | | Shmuel (Milky) Rubinstein
| | | | * | | | | * | | | Stephen T. Wills | | | * | | | | * | | | | * | | | | * | | | Ofer Gonen | | | * | | | | * | | | | Assaf Segal | | | * | | | | * | | | | Vickie R. Driver | | | * | | | | * | | | | Nissim Mashiach | | | * | | | | * | | | | Sharon Kochan | | | * | | | | * | | | | David Fox | | | * | | | | * | | | | Samuel Moed | | | * | | | | * | | | | Sharon Malka | | | 385,219 | | | | 1.2 | % | | | Boaz Gur-Lavie | | | * | | | | * | | | Lior Rosenberg(1) | | | 1,983,637 | | | | 6.1 | % | | Shmulik Hess
| | | | * | | | | * | | | Ety Klinger | | | * | | | | * | | | | * | | | | * | | Hany Luxenburg
| | | | * | | | | * | | | Yaron Meyer | | | * | | | | * | | | | * | | | | * | | All executive officers and directors as a group (13 persons)( 2) | | | 3,085,968 | | | | 9.2 | % | | Robert Snyder
| | | | * | | | | * | | All executive officers and directors as a group (11 persons)(1)
| | | | | | | | 3.8 | % | | | | | | | | | | | | | Principal Shareholders (who are not Directors or Executive Officers) | | | | | | | | | | | | | | | | | Clal Biotechnology Industries Ltd.(3) | | | 10,980,805 | | | | 33.8 | % | | Clal Biotechnology Industries Ltd. and affiliates (2)
| | | | 1,481,522 | | | | 16.0 | % | Israel Biotech Fund II, L.P. and affiliates (3)
| | | | 959,652 | | | | 9.9 | % | Deep Insight Limited Partnership and affiliates (4)
| | | | 959.652 | | | | 9.9 | % | Point72 Associates, LLC and affiliates (5)
| | | | 821,500 | | | | 8.9 | % | Rosalind Advisors, Inc. and affiliates (6)
| | | | 643,255 | | | | 6.8 | % |
| (1) | Shares beneficially owned consist of: (i) 146,532 ordinary shares held directly by Prof. Rosenberg; (ii) 126,900 ordinary shares issuable upon exercise of outstanding options held directly by Prof. Rosenberg that are currently exercisable or exercisable within 60 days of March 15, 2022; and (iii) 1,710,205 ordinary shares held by L.R. Research and Development Ltd. in trust for the benefit of Prof. Rosenberg. Prof. Rosenberg is the sole shareholder of L.R. Research and Development Ltd. |
(2) | Shares beneficially owned consist of 1,944,85679,068 ordinary shares held directly or indirectly by such executive officers and directors and 1,141,113267,058 ordinary shares issuable upon exercise of outstanding options and RSU’s that are currently exercisable or exercisable within 60 days of March 15, 2022.2024.
|
(3)(2) | Shares beneficially owned consist of: (i) 8,208,973Based solely on Schedule 13D/A filed on September 1, 2023, Clal Biotechnology Industries Ltd. (“CBI”) owns directly 308,811 ordinary shares, heldand may be deemed to share voting and investment power over the 1,172,710 ordinary shares owned directly by Clal Life Sciences LP, whose managingL.P. (“CLS”), the general partner isof which, Clal Application Center Ltd., a wholly-owned subsidiaryis wholly owned by CBI. Each of CBI; (ii) 2,682,665 ordinary shares held by CBI and (iii) 89,167 ordinary shares issuable upon exercise of outstanding options held directly by CBI that are currently exercisable or exercisable within 60 days of March 15, 2022. As reported on a Schedule 13G/A filed on February 14, 2019 by Access Industries Holdings LLC (“AIH”), Access Industries, Holdings LLC indirectly owns 100%(“Access LLC”), Access Industries Management, LLC (“AIM”), Clal Industries Ltd. (“Clal Industries”) and Mr. Blavatnik may be deemed to share voting and investment power over the ordinary shares owned directly by CBI and CLS because (i) Len Blavatnik controls AIM, AIH, Access LLC and AI International GP Limited (the general partner of AI SMS, as defined below), (ii) AIM controls Access LLC and AIH, (iii) Access LLC controls a majority of the outstanding sharesvoting interests in AIH, (iv) AIH owns a majority of Clal Industriesthe equity of AI SMS L.P. (“AI SMS”), (v) AI SMS controls AI Diversified Holdings Ltd. (“Holdings Limited”), (vi) Holdings Limited owns AI Diversified Parent S.à r.l., which owns 47.17%AI Diversified Holdings S.à r.l., which owns Access AI Ltd (“Access AI”), (vii) Access AI wholly owns Clal Industries, (viii) Clal Industries is the controlling shareholder of CBI, and (ix) CBI is the outstanding sharessole shareholder of CBI.Clal Application Center Ltd. The Reporting Persons, other than CBI and CLS, and each of their affiliated entities and the officers, partners, members and managers thereof, disclaims beneficial ownership of these securities. The address of Clal Industries Ltd. is the Triangular Tower, 3 Azrieli Center, Tel Aviv 67023, Israel and the address of Access Industries Holdings LLC is c/o Access Industries Inc., 40 West 57th Street, New York, New York 10019, United States. |
| (3) | Based solely on Schedule 13G/A filed on January 8, 2024, the 959,652 ordinary shares include 408,397 ordinary shares that are issuable upon the exercise of warrants held directly by Israel Biotech Fund II, L.P. (“IBF II”). Israel Biotech Fund GP Partners II, L.P. (“IBF GP”) is the sole general partner of IBF II, and I.B.F Management Ltd. (“IBF Management”) is the sole general partner of IBF GP. IBF GP and IBF Management may be deemed to share voting and dispositive power with respect to the ordinary shares that are beneficially owned by IBF II. The address IBF Management is HaOgen Tower, 4 Oppenheimer St., Rehovot 7670104, Israel and the address of the other reporting persons is 75 Fort Street, Clifton House, PO Box, 1350, KY1-1108, Grand Cayman. |
| (4) | Based solely on Schedule 13G/A filed on January 8, 2024, the 959,652 ordinary shares include 408,397 ordinary shares that are issuable upon the exercise of warrants held directly by Deep Insight Limited Partnership (“Deep Insight”). Deep Insight Fund GP Limited Partnership (“Deep Insight GP LP”) is the sole general partner of Deep Insight, Deep Insight GP Ltd. (“Deep Insight GP Company”) is the sole general partner of Deep Insight GP LP, Deep Insight Management Ltd. (“Deep Insight Management”) is the management company of Deep Insight GP LP and each of Barak Ben-Eliezer and Dr. Eyal Kishon hold 50% of the outstanding shares of Deep Insight GP Company and Deep Insight Management. Deep Insight GP LP, Deep Insight GP Company, Deep Insight Management, Barak Ben-Eliezer and Dr. Eyal Kishon may be deemed to share voting and dispositive power with respect to the Ordinary Shares that are beneficially owned by Deep Insight. Barak Ben-Eliezer and Dr. Eyal Kishon disclaim beneficial ownership of the Ordinary Shares reported by Deep Insight herein. The address of each of the reporting persons is 2 Rachel Imeinu St., Modiin, Israel 7177190. |
| (5) | Based solely on Schedule 13G filed on January 8, 2024, Point72 Associates, LLC (“Point72 Associates”) owns 821,500 ordinary shares. Pursuant to an investment management agreement, Point72 Asset Management, L.P. (“Point72 Asset Management”) maintains investment and voting power with respect to the securities held by Point72 Associates. Point72 Capital Advisors Inc. is the general partner of Point72 Asset Management. Steven Cohen controls each of Point72 Asset Management and Point72 Capital Advisors Inc. The address of the reporting persons is 72 Cummings Point Road, Stamford, CT 06902. |
| (6) | Based solely on Schedule 13G/A filed on February 14, 2024, Rosalind Master Fund L.P. (“RMF”) is the record owner of 479,990 shares of ordinary shares and 163,265 shares of ordinary shares issuable upon exercise of warrants. Rosalind Advisors, Inc. is the investment advisor to RMF and may be deemed to be the beneficial owner of shares held by RMF. Steven Salamon is the portfolio manager of Rosalind Advisors, Inc. and may be deemed to be the beneficial owner of shares held by RMF. Gilad Aharon is the portfolio manager and member of the Advisor which advises RMF. Notwithstanding the foregoing, Rosalind Advisors, Inc. and Mr. Salamon disclaim beneficial ownership of the shares. The address of RMF is P.O. Box 309 Ugland House, Grand Cayman KY1-1104, Cayman Islands, and the address of the rest of the reporting persons is 15 Wellesley Street West, Suite 326, Toronto, Ontario M4Y 0G7 Canada. |
Changes in Ownership of Major Shareholders
To our knowledge, other than as disclosed in the table above, our other filings with the SEC and this Annual Report, there has been no significant change in the percentage ownership held by any major shareholder since January 1, 2019. 2021, except as follows: as reported in their filings with the SEC, during 2023, CBI and its affiliates have decreased their holdings from approximately 34% to 16.1% as a result of a dilution from the 2023 Offering t as described in Item 5. “Operating and Financial Review and Prospects-Liquidity and Capital Resources. Voting Rights The major shareholders listed above do not have voting rights with respect to their ordinary shares that are different from the voting rights of other holders of our ordinary shares.
Controlling Shareholder110
Because CBI (and its affiliates) beneficially owned or controlled, directly and indirectly, 34.6% of our issued and outstanding ordinary shares as of December 31, 2021, it is considered a “controlled shareholder” under the Israeli Companies Law.Change in Control Arrangements
We are not aware of any arrangement that may at a subsequent date, result in a change of control of the Company. Registered Holders As of March 15, 2022,2024, we had one holder of record of our ordinary shares in the United States, which is Cede & Co., the nominee of The Depository Trust Company. This shareholder held in the aggregate 64%82.8% of the 32,481,2139,256,368 ordinary shares issued and outstanding as of March 15, 2022.2024. The number of record holders in the United States is not representative of the number of beneficial holders nor is it representative of where such beneficial holders are resident since many of these ordinary shares were held by brokers or other nominees.
| B. | Related Party Transactions |
Information Rights Agreement We have entered into an information rights agreement with CBI, which provides CBI with certain information rights relating to our financial information of the company and certain other information necessary for CBI to meet Israeli Securities Law requirements. CBI is not required to reimburse us for expenses we incur in providing such information. 2021 Registration Rights Agreement We are party to an amended and restated registration rights agreement, dated April 6, 2021, with certain of our shareholders (the “Registration“2021 Registration Rights Agreement”). The 2021 Registration Rights Agreement, which was approved by our shareholders at our 2021 annual general meeting of shareholders, replaced the registration rights agreement, dated March 3, 2014 (the “Original Registration Rights Agreement”), that we had entered into in connection with our initial public offering with certain of our pre-IPO shareholders, which expired by its own terms on its seven-year anniversary. The ordinary shares held by most of our pre-IPO shareholders who were party to the Original Registration Rights Agreement were no longer entitled to registration rights under that agreement as of the time that it expired, given their ability to freely sell their shares in the open market under Rule 144 of the Securities Act. However, each of CBI and Professor Lior Rosenberg, and their affiliated entities that hold ordinary shares (consisting of Clal Life Sciences LP and L.R. Research & Development Ltd., respectively) remained entitled to registration rights as of the time of the expiration of the Original Registration Rights Agreement, and we therefore entered into the Registration Rights Agreement with them as a means of extending those rights. . The 2021 Registration Rights Agreement provides to the holders of our ordinary shares that are party to the agreement the right to demand that we file a registration statement or request that their ordinary shares be covered by a registration statement that we are otherwise filing. In March 2019,May 2022, we filed, and the SEC declared effective, on April 22, 2019,June 3, 2022, a shelf registration statement on Form F-3 that registered the resale of the 11,240,8271,819,780 shares that were then entitled to registration rights under the Original2021 Registration Rights Agreement. That registration statement remains in effect as of the date of this Annual Report. The registration rights under the 2021 Registration Rights Agreement are described in more detail under “ITEM 10.B. Articles of Association” and in Exhibit 2.1 to this Annual Report, which is incorporated by reference in that ITEM 10.B. Articles of Association. Founders’ and Shareholders’ Agreement In January 2001, we entered into a founders’ and shareholders’ agreement (the “Founders Agreement”), with CBI, Prof. Lior Rosenberg, our Chief Medical Technology Officer, and LR, a private company which is wholly-owned by Prof. Rosenberg. The Founders Agreement was amended in 2006. Pursuant to the Founders Agreement, in exchange for the issuance of ordinary shares and certain rights thereunder and the payment of certain fixed amounts, Prof. Rosenberg granted to us a perpetual, exclusive, non-revocable, royalty-free, sub-licensable, worldwide license for intellectual property relating to debridement using products based on our proteolytic enzyme technology. As of the date hereof, all of the payments under the Founders Agreement were paid by us to Prof. Rosenberg in accordance with the Founders Agreement. The Founders Agreement also provided for anti-dilution, pre-emptive rights, a right of first refusal on the sale of our ordinary shares and bring-along rights, all of which were subsequently terminated. Sub-Lease Agreement
In January 2018,September 2022 we entered into a sub-leasean additional license agreement (the “Sub-Lease Agreement”), with Clal Life Sciences, L.P. (“CLS”), a subsidiary of CBI, our controlling shareholder, which was amended in February 2019. PursuantLR for intellectual property rights related to the Sub-Lease Agreement, we currently sublease approximately 32,300 square feetdevelopment of laboratory, officea synthetic hyaluronic acid polyurethane dressing for debrided and clean room space from CLS and our yearly rent is $0.4 million. The Sub-Lease Agreement is scheduled to expire on October 30, 2025.non-debrided burns. Under this license LR received an upfront payment of $150,000.
Agreements with Directors and Officers Employment Agreements We have entered into employment agreements with each of our executive officers, which include standard provisions for a company in our industry regarding non-competition/solicitation, confidentiality of information and assignment of inventions. However, the enforceability of the non-competition provisions may be limited under applicable law. Our executive officers, other than our CEO, will not receive benefits upon the termination of their respective employment with us, other than payment of salary and benefits (and limited accrual of vacation days) during the required notice period for termination of their employment, which varies for each individual. Our CEO, is entitled upon termination of employment (other than a termination by the Company for Cause, as defined below), to receive a single lump-sum payment equal 6 times his salary as of the last day of employment, less deductions and withholdings under applicable law, subject him signing a separation agreement and release of known and unknown claims in a customary form provided by the Company. Options Since our inception, we have granted options to purchase our ordinary shares to our directors and executive officers. Such option agreements may contain acceleration provisions upon certain merger, acquisition or change of control transactions. We describe our option plans under “ITEM 6.B. Compensation—2003Compensation-2003 Israeli Share Option Plan” and “ITEM 6.B. Compensation—2014Compensation-2014 Equity Incentive Plan.” If an executive officer is involuntarily terminated without cause or the executive officer voluntarily terminates his employment for good reason (as defined in the employment agreement), all options will immediately vest. Upon the consummation of a merger or acquisition transaction, an executive officer’s options will be assumed or substituted by the surviving company, if applicable, or, in the compensation committee’s sole discretion, will vest immediately or be amended, modified or terminated. Our compensation committee approved accelerated vesting in the case of a merger or an acquisition transaction for certain of our directors and executive officers with respect to the option agreements dated December 23, 2015, June 22, 2017, January 16, 2018, December 31, 2018, May 2, 2019, April 23, 2020, and March 4, 2021.2021 and February 15, 2023, April 3, 2023, August 15, 2023, December 1, 2023 and February 26, 2024. RSUs Under the 2014 Plan, we have granted RSUs to our executive officers and our chairman of the board. The RSU agreements generally provide for vesting of RSUs over a four-year period of continuous employment or service, with 25% of the RSUs vesting at the lapse of one year following the vesting commencement date, and the remaining 75% of the RSUs vesting in three equal installments, at the lapse of each of the following three years. Absent a specific acceleration provision, if a grantee’s service is terminated for any reason, all RSUs that have not vested will immediately terminate. RSUs that have vested but have not been settled yet for underlying ordinary shares may generally be settled within the three months following the termination of the service of the grantee, other than in the case of termination due to death or disability (in which case the grantee or his/her estate will have one year to settle the vested RSUs for underlying ordinary shares) or termination for cause (in which case all unsettled RSUs will immediately terminate). Upon the consummation of a merger or acquisition transaction, an executive officer’s or the chairman’s RSUs will be assumed or substituted by the surviving company, if applicable, or, in the compensation committee’s sole discretion, will vest immediately or be amended, modified or terminated. The RSUs that we grant may contain acceleration provisions upon certain merger, acquisition or change of control transactions, if approved by our board of directors with respect to a specific grant. The RSUs are generally subject to the further terms of the 2014 Plan, which we describe under “ITEM 6.B. Compensation—2014Compensation-2014 Equity Incentive Plan.” Exculpation, indemnification and insurance Our articles of association permit us to exculpate, indemnify and insure each of our directors and office holders to the fullest extent permitted by the Israeli Companies Law. Additionally, we have entered into indemnification agreements with each of our directors and executive officers, undertaking to indemnify them to the fullest extent permitted by Israeli law, including with respect to liabilities resulting from a public offering of our shares, to the extent that these liabilities are not covered by insurance. We have also obtained Directors and Officers insurance for each of our executive officers and directors. See “ITEM 6.C. Board Practices—Exculpation,Practices-Exculpation, Insurance and Indemnification of Directors and Officers.”
| C. | Interests of Experts and Counsel |
Not applicable.None.
Item 8. | FINANCIAL INFORMATION |
| A. | Consolidated Statements and Other Financial Information |
Consolidated Financial Statements See Item 18. “Financial Statements”. Legal and Arbitration Proceedings From time to time, we may be party to litigation or subject to claims incident to the ordinary course of business. Settlement of Litigation Involving Our Company PolyHeal Shareholders and Teva In March 2019, we entered into settlement agreements and mutual general releases with respect to our previously-reported litigation arising underUnder a series of agreements among PolyHeal, Teva and our company that we entered into in 2010 (collectively, the “2010 PolyHeal Agreements”). For a description of the history of the proceedings related to the 2010 PolyHeal Agreements and a dispute related to a collaboration agreement between Teva and our company that we entered into in 2007 (the "2007 Teva Agreement,") please see “ITEM 8. Financial Information— A. Consolidated Statements and Other Financial Information— Legal Proceedings” in our annual report on Form 20-F for the year ended December 31, 2018, filed with the SEC on March 25, 2019 (the “2018 Form 20-F”).
Aspreviously reported in the 2018 Form 20-F, on March 24, 2019, we entered into an initial settlement with the plaintiffs— certain shareholders of PolyHeal — which settlement was subsequently approved by the Israeli Supreme Court, which settled any and all debts, obligations or liabilities that we and the plaintiffs had to one another in connection with the transactions under the 2010 PolyHeal Agreements. Pursuant to the terms of this settlement agreement, the plaintiffsas amended, we were to repay a non-material portion of the amount that was ruled in their favor under a November 2017 ruling, and the Israeli Supreme Court was to approve and accept the appeal that was filed by us in December, 2017, cancel the 2017 ruling that was issued by the Israeli District Court against us, and reject the PolyHeal shareholders’ cross-appeal.
Also as reported in the 2018 Form 20-F, on March 24, 2019, we entered into a settlement agreement and mutual general release with Teva, which was contingent upon the Supreme Court’s approval of the settlement with the PolyHeal plaintiffs (which approval was received), which settled any and all debts, obligations or liabilities that each party or any of its controlled affiliates had to the other party or any of its controlled affiliates in connection with certain transactions and collaboration agreements entered into between us and Teva from 2007 to 2012, which had terminated effective as of December 31, 2012 and September 2, 2013, as applicable, and which had related to NexoBrid and PolyHeal, including a milestone payment to PolyHeal and certain additional payments, which were primarily intended to serve as reimbursement for development and manufacturing costs, which we had believed were to be borne by Teva through the effective date of termination of those collaboration agreements in December 2012.
Pursuant to the terms of the Teva settlement agreement, Teva agreed to pay us $4.0 million in cash, and to reduce the contingent consideration that is payable to Teva pursuant to our repurchase of our shares from Teva in 2013, so that we are obligated to pay Teva annual payments at a reduced rate of 15% of its recognized revenues from the sale or license of NexoBrid after January 1, 2019, up to a reduced aggregate amount of $10.2 million. In addition, we also agreed to indemnify Teva and its controlled affiliates from and against claims relating to a certain milestone related to PolyHeal under an agreement associated with our collaboration agreements with Teva, for up to an amount of $10.2 million, if a notice of such claim has been received by us prior to December 31, 2023.
On December 13, 2020, we signed an amendment to the Teva settlement agreement that replaces the revenue-based payment mechanism with a fixed payment schedule. The aggregate amount paid to Teva of up to $10.2 million and the other terms, including with respect to our indemnification obligations, in the Teva settlement agreement are unchanged. Out of the $3 million already due to Teva we paid $1 million of the on December 2020 and the balance will be paid inwhich twelve quarterly equal installments during the period commencing onwere paid from January 1, 2021 and ending onuntil December 31, 2023. In addition, commencing on January 1, 2021, we have agreed to pay Teva an aggregate annual amount of $1 million in four quarterly equal installments, unless we do not recognize any revenues generated from the sale or license of NexoBrid in any such quarter, up to an aggregate amount equal to $7.2 million regardless of the number of quarters required for purposes of the payment of such aggregate amount.
In September 2019, we entered into We also agreed to indemnify Teva and its controlled affiliates from and against claims relating to a series of additional settlementcertain milestone related to PolyHeal under an agreement associated with our collaboration agreements and mutual general releases with certain shareholders of PolyHeal, including Clal Biotechnology Industries Ltd. (CBI), our controlling shareholder, which together constitute the majority of PolyHeal's shareholders. Those additional settlement agreements settle any and all debts, obligations or liabilities that each party or any of its affiliates had or hasTeva, for up to the other party or any of its affiliates, in connection with or arising out of the series of 2010 PolyHeal Agreements. Pursuant to these settlement agreements, we paid an aggregate amount of approximately $2.8$10.2 million, if a notice of such claim had been received by us prior to December 31, 2023, however, no notice of a claim was received by such date and received 14,473 shares of PolyHeal.therefore the indemnification rights under this settlement agreement expired.
Dividend Policy We have never declared or paid cash dividends to our shareholders and we do not intend to pay cash dividends in the foreseeable future. We intend to reinvest any earnings in developing and expanding our business. Any future determination relating to our dividend policy will be at the discretion of our board of directors and will depend on a number of factors, including future earnings, our financial condition, operating results, contractual restrictions, capital requirements, business prospects, our strategic goals and plans to expand our business, applicable law and other factors that our board of directors may deem relevant. No significant changes have occurred since December 31, 2021,2022, except as otherwise disclosed in this annual report.
Item 9. | THE OFFER AND LISTING |
Our ordinary shares trade on the Nasdaq Global Market under the symbol “MDWD”.
Not applicable. See “—Listing“-Listing Details” above. Not applicable. Not applicable.
Not applicable. | Item 10. ADDITIONAL INFORMATION |
Not applicable. Item 10. | ADDITIONAL INFORMATION |
Not applicable.
| B. | Articles of Association |
A copy of our amended and restated articles of association is attached as Exhibit 1.1 to this Annual Report. Other than as disclosed below, the information called for by this Item is set forth in Exhibit 2.1 to ourthis Annual Report on Form 20-F, for the year ended December 31, 2019 andwhich information is incorporated by reference into this Annual Report. ElectionItem 10.B. of directors
Our ordinary shares do not have cumulative voting rights for the election of directors. As a result, the holders of a majority of the voting power represented at a meeting of shareholders have the power to elect each of our directors, subject to the special approval requirements for external directors described under “ITEM 6.C. Board Practices—External Directors.” Under our articles of association, our board of directors must consist of at least five and not more than nine directors, including at least two external directors required to be appointed under the Israeli Companies Law. At any time the minimum number of directors (other than the external directors) shall not fall below three. Pursuant to our articles of association, each of our directors, other than the external directors, for whom special election requirements apply under the Israeli Companies Law, will be appointed by a simple majority vote of holders of our voting shares, participating and voting at anthis annual general meeting of our shareholders. Each director will serve until his or her successor is duly elected and qualified or until his or her earlier death, resignation or removal by a vote of the majority voting power of our shareholders at a general meeting of our shareholders or until his or her office expires by operation of law, in accordance with the Israeli Companies Law. Our articles of association allow our board of directors to appoint directors to fill vacancies on the board of directors to serve until the next annual general meeting of shareholders. External directors are elected for an initial term of three years, may be elected for additional terms of three years each under certain circumstances, and may be removed from office pursuant to the terms of the Israeli Companies Law. Under regulations promulgated under the Israeli Companies Law, Israeli public companies whose shares are traded on certain U.S. stock exchanges, such as the Nasdaq Global Market and that lack a controlling shareholder are exempt from the requirement to appoint external directors. See “ITEM 6.C. Board Practices—Board of Directors and External Directors.”report.
"Except described below or otherwise described in this annual report in Item 4.A “History and Development of the Company”, Item 4.B “Business Overview”, “Item 5.B 'Operating and Financial Review and Prospects—Liquidity and Capital Resources”, Item 6.C “Board Practices” and Item 7.B “Related Party Transactions”, we are not currently, nor have we been for the two years immediately preceding the date of this Annual Report, party to any material contract other than contracts entered into in the ordinary course of business."
For a description of the registration rights that are subject to our 2021 Registration Rights Agreement, see “ITEM“Item 7.B. Related Party Transactions—Transactions – 2021 Registration Rights Agreement.” For a description of our contract with the U.S. Biomedical Advanced Research and Development Authority, see “ITEM 4.B. Our Focus—Burn Care—BARDA Contract.”
For a description of our exclusive license and supply agreements with Vericel, see “ITEM“Item 4.B. Business Overview—Overview- Marketing, Sales and Distribution—Distribution- Vericel License and Supply Agreements.”
For a description of our license agreement with Mark Klein, see “ITEM“Item 4.B. Business Overview—Intellectual Property—KleinOverview-Intellectual Property-Klein License Agreement.” We have entered into an agreement with Challenge Bioproducts Corporation Ltd. (“CBC”), a corporation organized and existing under the laws of the Republic of China, dated January 11, 2001, as amended on February 28, 2010, pursuant to which CBC uses proprietary methods to manufacture bromelain SP and supplies us with this intermediate drug substance in bulk quantities. According to the terms of the agreement, CBC shall not, and shall not permit related companies or a third party to, manufacture, use, supply or sell the raw materials for the use or production of a product directly or indirectly competing with any of our products. Our supply agreement with CBC has no fixed expiration date and can be voluntarily terminated by us, with at least six months’ advance written notice, or by CBC, with at least 24 months’ advance written notice. 109For a description of the main lease agreement, see “Item 4. Properties.”
For a description of the turnkey scale-up agreement with Biopharmax Group Ltd., see “Item 4. Properties.” 2022 Registration Rights Agreement In connection with the PIPE Offering (as described in “ITEM 5. Operating and Financial Review and Prospects— B. Liquidity and Capital Resources”), in October 2022, we entered into a registration rights agreement with the several investors named in the PIPE Securities Purchase Agreement (the “2022 Registration Rights Agreement”). Pursuant to the 2022 Registration Rights Agreement, within 45 calendar days of the date of the closing of the PIPE, we were required to file a registration statement to register for resale of (i) Pre-Funded Warrant Shares, (ii) the PIPE Ordinary Warrant Shares and (iii) the RD Ordinary Share Warrant (together, the “Registrable Securities”). Pursuant to the 2022 Registration Rights Agreement, we agreed to cause such registration statement to be declared effective under the Securities Act of 1933, as amended (the “Securities Act”), as promptly as possible after the filing thereof, but in any event no later 75 days, or in the event of a full review by the SEC, 110 days, after the closing date of the PIPE. We further agreed to use our best efforts to keep such registration statement continuously effective under the Securities Act until the date that all Registrable Securities covered by such registration statement have been sold or otherwise may be sold pursuant to Rule 144 under the Securities Act. On November 10, 202, we filed a registration statement on Form F-1 pursuant to our obligations under the 2022 Registration Rights Agreement, which became effective on November 25, 2022.
In 1998, Israeli currency control regulations were liberalized significantly, so that Israeli residents generally may freely deal in foreign currency and foreign assets, and non-residents may freely deal in Israeli currency and Israeli assets. There are currently no Israeli currency control restrictions on remittances of dividends on the ordinary shares or the proceeds from the sale of the shares provided that all taxes were paid or withheld; however, legislation remains in effect pursuant to which currency controls can be imposed by administrative action at any time.
Non-residents of Israel may freely hold and trade our securities. Neither our articles of association nor the laws of the State of Israel restrict in any way the ownership or voting of ordinary shares by non-residents, except that such restrictions may exist with respect to citizens of countries which are in a state of war with Israel. Israeli residents are allowed to purchase our ordinary shares.
The following description is not intended to constitute a complete analysis of all tax consequences relating to the acquisition, ownership and disposition of our ordinary shares. You should consult your own tax advisor concerning the tax consequences of your particular situation, as well as any tax consequences that may arise under the laws of any state, local, foreign or other taxing jurisdiction.
Israeli Tax Considerations for Our Shareholders Capital gains taxes applicable to non‑Israeli resident shareholders A non‑Israeli resident (whether an individual or a corporation) who derives capital gains from the sale of shares in an Israeli resident company that were purchased after the company was listed for trading on the Tel Aviv Stock Exchange or on a recognized stock exchange outside of Israel, will generally be exempt from Israeli capital gain tax so long as the shares were not held through a permanent establishment that the non‑resident maintains in Israel (and with respect to shares listed on a recognized stock exchange outside of Israel, so long as the particular capital gain is otherwise subject to the Israeli Income Tax Law (Inflationary Adjustments) 5745‑1985. These provisions dealing with capital gain are not applicable to a person whose gains from selling or otherwise disposing of the shares are deemed to be business income. However, non‑Israeli corporations will not be entitled to the foregoing exemption if Israeli residents (i) have a controlling interest of more than 25% in such non‑Israeli corporation or (ii) are the beneficiaries of, or are entitled to, 25% or more of the revenues or profits of such non‑Israeli corporation, whether directly or indirectly. If not exempt, a non-Israeli resident shareholder would generally be subject to tax on capital gain at the ordinary corporate tax rate (23% in 2023), if generated by a company, or at the rate of 25%, if generated by an individual, or 30%, if generated by an individual who is a “substantial shareholder” (as defined under the Tax Ordinance), at the time of sale or at any time during the preceding 12-month period (or if the shareholder claims a deduction for interest and linkage differences expenses in connection with the purchase and holding of such shares). A “substantial shareholder” is generally a person who alone or together with such person’s relative or another person who collaborates with such person on a permanent basis, holds, directly or indirectly, at least 10% of any of the “means of control” of the corporation. “Means of control” generally include, among others, the right to vote, receive profits, nominate a director or an executive officer, receive assets upon liquidation, or order someone who holds any of the aforesaid rights how to act, regardless of the source of such right. Individual and corporate shareholders dealing in securities in Israel are taxed at the tax rates applicable to business income (a corporate tax rate for a corporation (23% in 2023) and a marginal tax rate of up to 47% for an individual in 2023 (excluding excess tax as discussed below)) unless contrary provisions in a relevant tax treaty apply. Additionally, a sale of shares by a non‑Israeli resident may also be exempt from Israeli capital gains tax under the provisions of an applicable tax treaty. For example, under the Convention Between the Government of the United States of America and the Government of the State of Israel with respect to Taxes on Income, as amended (the “United States‑Israel Tax Treaty”), the sale, exchange or other disposition of shares by a shareholder who is a United States resident (for purposes of the United States‑Israel Tax Treaty) holding the shares as a capital asset and is entitled to claim the benefits afforded to such a resident by the United States‑Israel Tax Treaty (a “Treaty U.S. Resident”) is generally exempt from Israeli capital gains tax unless: (i) the capital gain arising from such sale, exchange or disposition is attributed to real estate located in Israel; (ii) the capital gain arising from such sale, exchange or disposition is attributed to royalties; (iii) the capital gain arising from the such sale, exchange or disposition can be attributable to a permanent establishment of the shareholder maintained in Israel, under certain terms; (iv) such Treaty U.S. Resident holds, directly or indirectly, shares representing 10% or more of the voting capital of a company during any part of the 12‑month period preceding such sale, exchange or disposition, subject to certain conditions; or (v) such Treaty U.S. Resident is an individual and was present in Israel for a period or periods aggregating to 183 days or more during the relevant taxable year. In each case, the sale, exchange or disposition of our ordinary shares would be subject to such Israeli tax, to the extent applicable; However, under the United States‑Israel Tax Treaty, such Treaty U.S. Resident would be permitted to claim a credit for such taxes against the U.S. federal income tax imposed with respect to such sale, exchange or disposition, subject to the limitations in U.S. laws applicable to foreign tax credits. The United States-Israel Tax Treaty does not provide such credit against any U.S. state or local taxes. In some instances where our shareholders may be liable for Israeli tax on the sale of their ordinary shares, the payment of the consideration may be subject to the withholding of Israeli tax at source. Shareholders may be required to demonstrate that they are exempt from tax on their capital gains in order to avoid withholding at source at the time of sale. Specifically, in transactions involving a sale of all of the shares of an Israeli resident company, in the form of a merger or otherwise, the Israel Tax Authority may require from shareholders who are not liable for Israeli tax to sign declarations in forms specified by this authority or obtain a specific exemption from the Israel Tax Authority to confirm their status as non‑Israeli resident, and, in the absence of such declarations or exemptions, may require the purchaser of the shares to withhold taxes at source.
Taxation of non‑Israeli shareholders on receipt of dividends Non‑Israeli residents (whether individuals or corporations) are generally subject to Israeli income tax on the receipt of dividends paid on our ordinary shares at the rate of 25% unless a relief is provided in a treaty between Israel and a shareholder's country of residence (provided that a valid certificate from the Israeli Tax Authority allowing for a reduced withholding tax rate is obtained in advance). With respect to a person who is a “substantial shareholder” at the time of receiving the dividend or on any time during the preceding 12 months, the applicable tax rate is 30%. A “substantial shareholder” is generally a person who alone or together with such person’s relative or another person who collaborates with such person on a permanent basis, holds, directly or indirectly, at least 10% of any of the “means of control” of the corporation. “Means of control” generally include the right to vote, receive profits, nominate a director or an executive officer, receive assets upon liquidation, or order someone who holds any of the aforesaid rights how to act, regardless of the source of such right. Such dividends are generally subject to Israeli withholding tax at a rate of 25% so long as the shares are registered with a nominee company (whether or not the recipient is a substantial shareholder), unless relief is provided in a treaty between Israel and the shareholder’s country of residence and provided that a valid certificate from the Israel Tax Authority allowing for a reduced withholding tax rate is obtained in advance. However, a distribution of dividends to non‑Israeli residents is generally subject to withholding tax at source at a rate of 15% if the dividend is distributed from income attributed to a Beneficiary Enterprise, unlessor such a reduced tax rate isas may be provided under an applicable tax treaty and provided(provided that a valid certificate from the Israel Tax Authority allowing for a reduced withholding tax rate or such lower tax rate as may be provided in an applicable tax treaty is obtained in advance.advance). For example, under the United States‑Israel Tax Treaty, the maximum rate of tax withheld at source in Israel on dividends paid to a holder of our ordinary shares who is a Treaty U.S. Resident is 25%. However, generally, the maximum rate of withholding tax on dividends, not generated by an Approved Enterprise or Beneficiary Enterprise, that are paid to a U.S. corporation holding 10% or more of the outstanding voting capital throughout the tax year in which the dividend is distributed as well as during the previous tax year, is 12.5%, provided that not more than 25% of the gross income for such preceding year consists of certain types of dividends and interest. Notwithstanding the foregoing, dividends distributed from income attributed to an Approved Enterprise or Beneficiary Enterprise are not entitled to such reduction under the tax treaty but are subject to a withholding tax rate of 15% for such a U.S. corporation, provided that the condition related to our gross income for the previous year (as set forth in the previous sentence) is met. The aforementioned rates under the United States-Israel Tax Treaty would not apply if the dividend income is derived through a permanent establishment of the U.S. resident in Israel. If the dividend is attributable partly to income derived from an Approved Enterprise, Beneficiary Enterprise or Preferred Enterprise, and partly to other sources of income, the withholding rate will be a blended rate reflecting the relative portions of the two types of income. We cannot assure you that we will designate the profits that we may distribute in a way that will reduce shareholders’ tax liability. A non‑Israeli resident who receives dividends from which tax was withheld, is generally exempt from the obligation to file tax returns in Israel with respect to such income, provided that (i) such income was not derived from a business conducted in Israel by the taxpayer, (ii) the taxpayer has no other taxable sources of income in Israel with respect to which a tax return is required to be filed and (iii) the tax payer is not obligated to pay the excess tax (as further explained below). Excess Tax Individuals who are subject to tax in Israel (whether any such individual is an Israeli resident or non-Israeli resident) are also subject to an additional tax at a rate of 3% on annual income exceeding a certain level, which amount is linked to the annual change in the Israeli consumer price index, including but not limited to, dividends, interest and capital gain. In 2021,2023, the additional tax was at a rate of 3% on annual income exceeding NIS 647,640.698,280.
United States Federal Income Taxation The following is a description of the material U.S. federal income tax consequences of the ownership and disposition of our ordinary shares by a U.S. Holder that holds the ordinary shares as capital assets. This description does not address tax considerations applicable to holders that may be subject to special tax rules, including, without limitation: banks, financial institutions or insurance companies;
real estate investment trusts, regulated investment companies or grantor trusts;
dealers or traders in securities, commodities or currencies;
tax‑exempt entities or organizations, including an “individual retirement account” or “Roth IRA” as defined in Section 408 or 408A of the Code, respectively;
certain former citizens or long‑term residents of the United States;
persons that received our shares as compensation for the performance of services;
persons that holds our shares as part of a “hedging,” “integrated” or “conversion” transaction or as a position in a “straddle” for U.S. federal income tax purposes;
partnerships (including entities classified as partnerships for U.S. federal income tax purposes) or other pass‑through entities, or holders that will hold our shares through such an entity;
holders that acquired ordinary shares as a result of holding or owning our preferred shares;
U.S. Holders (as defined below) whose “functional currency” is not the U.S. dollar;
persons that are residents of ordinarily resident in or have a permanent establishment in a jurisdiction outside the United States; or
holders that own directly, indirectly or through attribution 10.0% or more of the voting power or value of our shares.
| • | banks, financial institutions or insurance companies; | | • | real estate investment trusts, regulated investment companies or grantor trusts; | | • | dealers or traders in securities, commodities or currencies; | | • | tax‑exempt entities or organizations, including an “individual retirement account” or “Roth IRA” as defined in Section 408 or 408A of the Code, respectively; | | • | certain former citizens or long‑term residents of the United States; | | • | persons that received our shares as compensation for the performance of services; | | • | persons that holds our shares as part of a “hedging,” “integrated” or “conversion” transaction or as a position in a “straddle” for U.S. federal income tax purposes; | | • | partnerships (including entities classified as partnerships for U.S. federal income tax purposes) or other pass‑through entities, or holders that will hold our shares through such an entity; | | • | S corporations; | | • | holders that acquired ordinary shares as a result of holding or owning our preferred shares; | | • | U.S. Holders (as defined below) whose “functional currency” is not the U.S. dollar; | | • | persons that are residents of ordinarily resident in or have a permanent establishment in a jurisdiction outside the United States; or | | • | holders that own directly, indirectly or through attribution 10.0% or more of the voting power or value of our shares. |
Moreover, this description does not address the U.S. federal estate, gift or alternative minimum tax consequences, Medicare consequences, or any state, local or foreign tax consequences, of the ownership and disposition of our ordinary shares.
This summary is based on the Internal Revenue Code of 1986, as amended (the “Code”), administrative pronouncements, judicial decisions and final, temporary and proposed Treasury regulations, all as currently in effect and available. These authorities are subject to change or differing interpretation, possibly with retroactive effect. U.S. Holders should consult their tax advisors concerning the U.S. federal, state, local and foreign tax consequences of owning and disposing of our ordinary shares in their particular circumstances. For purposes of this summary, a “U.S. Holder” is a beneficial owner of our ordinary shares who is, for U.S. federal income tax purposes: an individual who is a citizen or individual resident of the United States;
a corporation, or other entity taxable as a corporation for U.S. federal income tax purposes, created or organized in or under the laws of the United States, any state thereof, or the District of Columbia;
an estate, the income of which is subject to U.S. federal income taxation regardless of its source; or
a trust that (1) is subject to the primary supervision of a U.S. Court and one or more U.S. persons that have the authority to control all substantial decisions of the trust or (2) has a valid election in effect under applicable Treasury regulations to be treated as a U.S. person.
| • | an individual who is a citizen or individual resident of the United States; | | • | a corporation, or other entity taxable as a corporation for U.S. federal income tax purposes, created or organized in or under the laws of the United States, any state thereof, or the District of Columbia; | | • | an estate, the income of which is subject to U.S. federal income taxation regardless of its source; or | | • | a trust that (1) is subject to the primary supervision of a U.S. Court and one or more U.S. persons that have the authority to control all substantial decisions of the trust or (2) has a valid election in effect under applicable Treasury regulations to be treated as a U.S. person. |
If a partnership (or other entity treated as a partnership for U.S. federal income tax purposes) holds our ordinary shares, the tax treatment of a partner in such partnership generally will depend upon the status of the partner and upon the activities of the partnership. Investors who are partners in a partnership should consult their tax advisors as to the particular U.S. federal income tax consequences of owning and disposing of our ordinary shares in their particular circumstances. A “Non‑U.S. Holder” is a beneficial owner of our ordinary shares that is neither a U.S. Holder nor a partnership for U.S. federal income tax purposes. Unless otherwise indicated, this discussion assumes that the company is not, and will not become, a “passive foreign investment company,” or a PFIC, for U.S. federal income tax purposes. See “ITEM 10.E. Taxation—UnitedTaxation-United States Federal Income Taxation—PassiveTaxation-Passive Foreign Investment Company Considerations” below. Further, this summary does not address the U.S. federal estate and gift, state, local or non‑U.S. tax consequences to U.S. Holders of owning and disposing of our ordinary shares. Investors should consult their own tax advisors regarding the U.S. federal, state and local, as well as non‑U.S. income and other tax consequences of owning and disposing of our ordinary shares in their particular circumstances.
Distributions If you are a U.S. Holder, the gross amount of any distribution made to you with respect to our ordinary shares before reduction for any Israeli taxes withheld therefrom, other than certain distributions, if any, of our ordinary shares distributed pro rata to all our shareholders, generally will be includible in your income as dividend income to the extent such distribution is paid out of our current or accumulated earnings and profits as determined under U.S. federal income tax principles. We do not expect to maintain calculations of our earnings and profits under U.S. federal income tax principles. Therefore, if you are a U.S. Holder you should expect that the entire amount of any distribution generally will be taxable as dividend income to you. Non‑corporate U.S. Holders may qualify for the lower rates of taxation with respect to dividends on ordinary shares applicable to long‑term capital gains (i.e., gains from the sale of capital assets held for more than one year), provided that certain conditions are met, including certain holding period requirements and the absence of certain risk reduction transactions. However, such dividends will not be eligible for the dividends received deduction generally allowed to corporate U.S. Holders. If you are a U.S. Holder, dividends paid to you with respect to our ordinary shares will generally be treated as foreign source income, which may be relevant in calculating your foreign tax credit limitation. Subject to certain conditions and limitations, Israeli tax withheld on dividends may be deducted from your taxable income or credited against your U.S. federal income tax liability. The limitation on foreign taxes eligible for credit is calculated separately with respect to specific classes of income. For this purpose, dividends that we distribute generally should constitute “passive category income.” A foreign tax credit for foreign taxes imposed on distributions may be denied if you do not satisfy certain minimum holding period requirements. Applicable U.S. Treasury Regulations have imposed additional requirements that must be met for a foreign tax to be creditable, depending on the nature of such foreign tax, although temporary relief from the application of certain aspects of these Regulations has been provided until new guidance or regulations are issued. The rules relating to the determination of the foreign tax credit are complex, and you should consult your tax advisor to determine whether and to what extent you will be entitled to this credit. Subject to the discussion below under “—Backup“-Backup Withholding Tax and Information Reporting Requirements,” if you are a Non‑U.S. Holder, you generally will not be subject to U.S. federal income (or withholding) tax on dividends received by you on your ordinary shares, unless you conduct a trade or business in the United States and such income is effectively connected with that trade or business (or, if required by an applicable income tax treaty, the dividends are attributable to a permanent establishment or fixed base that such holder maintains in the United States).
Sale, Exchange or Other Taxable Disposition of Ordinary Shares If you are a U.S. Holder, you generally will recognize gain or loss on the sale, exchange or other taxable disposition of our ordinary shares equal to the difference between the amount realized on such sale, exchange or other taxable disposition and your adjusted tax basis in our ordinary shares, and such gain or loss will be capital gain or loss. The initial tax basis in an ordinary share generally will be equal to the cost of such ordinary share. Except with respect to foreign currency gain or loss, if you are a non‑corporate U.S. Holder, capital gain from the sale, exchange or other taxable disposition of ordinary shares is generally eligible for a preferential rate of taxation applicable to capital gains, if your holding period for such ordinary shares exceeds one year (i.e., such gain is long‑term capital gain). The deductibility of capital losses for U.S. federal income tax purposes is subject to limitations under the Code. Any such gain or loss that a U.S. Holder recognizes generally will be treated as U.S. source income or loss for foreign tax credit limitation purposes. Because a U.S. Holder may use foreign tax credits against only the portion of United States federal income tax liability that is attributed to foreign source income in the same category, a U.S. Holder’s ability to utilize a foreign tax credit with respect to the foreign tax imposed on any such sale or other taxable disposition, if any, may be significantly limited.Applicable U.S. Treasury Regulations further restrict the availability of any such credit. However, a recent notice from the IRS indicates that the U.S. Department of the Treasury and the IRS are considering proposing amendments to such Regulations and allows, subject to certain conditions, taxpayers to defer the application of many aspects of such Regulations for taxable years beginning on or after December 28, 2021 and ending before the date that a notice or other guidance withdrawing or modifying the temporary relief is issued (or any later date specified in such notice or other guidance). Subject to the discussion below under “—Backup“-Backup Withholding Tax and Information Reporting Requirements,” if you are a Non‑U.S. Holder, you generally will not be subject to U.S. federal income or withholding tax on any gain realized on the sale or exchange of such ordinary shares unless: such gain is effectively connected with your conduct of a trade or business in the United States (or, if required by an applicable income tax treaty, the gain is attributable to a permanent establishment or fixed base that such holder maintains in the United States); or
| • | such gain is effectively connected with your conduct of a trade or business in the United States (or, if required by an applicable income tax treaty, the gain is attributable to a permanent establishment or fixed base that such holder maintains in the United States); or | | • | you are an individual and have been present in the United States for 183 days or more in the taxable year of such sale or exchange and certain other conditions are met. |
you are an individual and have been present in the United States for 183 days or more in the taxable year of such sale or exchange and certain other conditions are met.
Passive Foreign Investment Company Considerations If we were to be classified as a “passive foreign investment company,” or “PFIC,” in any taxable year, a U.S. Holder would be subject to special rules generally intended to reduce or eliminate any benefits from the deferral of U.S. federal income tax that a U.S. Holder could derive from investing in a non‑U.S. company that does not distribute all of its earnings on a current basis. A non‑U.S. corporation will be classified as a PFIC for federal income tax purposes in any taxable year in which, after applying certain look‑through rules with respect to the income and assets of subsidiaries, either: at least 75% of its gross income is “passive income”; or
at least 50% of the average quarterly value of its total gross assets (which may be determined in part by the market value of our ordinary shares, which is subject to change) is attributable to assets that produce “passive income” or are held for the production of passive income.
| • | at least 75% of its gross income is “passive income”; or | | • | at least 50% of the average quarterly value of its total gross assets (which may be determined in part by the market value of our ordinary shares, which is subject to change) is attributable to assets that produce “passive income” or are held for the production of passive income. |
Passive income for this purpose generally includes dividends, interest, royalties, rents, gains from commodities and securities transactions, the excess of gains over losses from the disposition of assets which produce passive income, and includes amounts derived by reason of the temporary investment of funds raised in offerings of our ordinary shares. If a non‑U.S. corporation owns at least 25% by value of the stock of another corporation, the non‑U.S. corporation is treated for purposes of the PFIC tests as owning its proportionate share of the assets of the other corporation and as receiving directly its proportionate share of the other corporation’s income. If we are classified as a PFIC in any year with respect to which a U.S. Holder owns our ordinary shares, we will continue to be treated as a PFIC with respect to such U.S. Holder in all succeeding years during which the U.S. Holder owns our ordinary shares unless we cease to be a PFIC and the U.S. holder has made a “deemed sale” election under the PFIC rules.
Based on our current estimates of our gross income and the estimated fair market value of our gross assets and the nature of our business, we do not believe we were classified as a PFIC for the taxable year ending December 31, 2021.2023. However, we must determine our PFIC status annually based on tests which are factual in nature, and our status in future years will depend on our income, assets and activities in those years. Further, because the value of our gross assets is likely to be determined in large part by reference to our market capitalization, a decline in the value of our ordinary shares or an increase in the value of our passive assets (including cash and short term investments) may result in our becoming a PFIC. There can be no assurance that we will not be considered a PFIC for any taxable year. If we were a PFIC and you are a U.S. Holder, then unless you make one of the elections described below, a special tax regime will apply to both (a) any “excess distribution” by us to you (generally, your ratable portion of distributions in any year which are greater than 125% of the average annual distribution received by you in the shorter of the three preceding years or your holding period for our ordinary shares) and (b) any gain realized on the sale or other disposition of the ordinary shares. Under this regime, any excess distribution and realized gain will be treated as ordinary income and will be subject to tax as if (a) the excess distribution or gain had been realized ratably over your holding period, (b) the amount deemed realized in each year had been subject to tax in each year of that holding period at the highest marginal rate for such year (other than income allocated to the current period or any taxable period before we became a PFIC, which would be subject to tax at the U.S. Holder’s regular ordinary income rate for the current year and would not be subject to the interest charge discussed below) and (c) the interest charge generally applicable to underpayments of tax had been imposed on the taxes deemed to have been payable in those years. In addition, dividend distributions made to you will not qualify for the lower rates of taxation applicable to long‑term capital gains discussed above under “Distributions.” Certain elections may be available that would result in an alternative treatment (such as mark‑to‑market treatment) of our ordinary shares.
If a U.S. Holder makes a valid mark‑to‑market election for the first tax year in which such U.S. Holder holds (or is deemed to hold) ordinary shares in a corporation and for which such corporation is determined to be a PFIC, the U.S. Holder generally will recognize as ordinary income any excess of the fair market value of the ordinary shares at the end of each taxable year over their adjusted tax basis, and will recognize an ordinary loss in respect of any excess of the adjusted tax basis of the ordinary shares over their fair market value at the end of the taxable year (but only to the extent of the net amount of income previously included as a result of the mark‑to‑market election). If a U.S. Holder makes the election, the U.S. Holder’s tax basis in the ordinary shares will be adjusted to reflect these income or loss amounts. Any gain recognized on the sale or other disposition of ordinary shares in a year when we are a PFIC will be treated as ordinary income and any loss will be treated as an ordinary loss (but only to the extent of the net amount of income previously included as a result of the mark‑to‑market election). The mark‑to‑market election is available only if we are a PFIC and our ordinary shares are “regularly traded” on a “qualified exchange.” Our ordinary shares will be treated as “regularly traded” in any calendar year in which more than a de minimis quantity of the ordinary shares, are traded on a qualified exchange on at least 15 days during each calendar quarter. Nasdaq is a qualified exchange for this purpose and, consequently, if the ordinary shares are regularly traded, the mark‑to‑market election will be available to a U.S. Holder.
If we are a PFIC, the general tax treatment for U.S. Holders described in this section would apply to indirect distributions and gains deemed to be realized by U.S. Holders in respect of any entity in which we hold equity that is also a PFIC (a "lower tier PFIC"). Because a mark‑to‑market election generally would not be available with respect to any lower‑tier PFICs, a U.S. Holder may continue to be subject to the PFIC rules with respect to such holder’s indirect interest in any investments held by us that are treated as an equity interest in such lower-tiers PFICs. We do not intend to provide the information necessary for U.S. Holders to make qualified electing fund elections if we are classified as a PFIC. U.S. Holders should consult their tax advisors to determine whether any of these elections would be available and if so, what the consequences of the alternative treatments would be in their particular circumstances. If a U.S. Holder owns ordinary shares during any year in which we are a PFIC, the U.S. Holder generally will be required to file an IRS Form 8621 (Information Return by a Shareholder of a Passive Foreign Investment Company or Qualified Electing Fund) or successor form with respect to the company, generally with the U.S. Holder’s federal income tax return for that year. If the company was a PFIC for a given taxable year, then you should consult your tax advisor concerning your annual filing requirements. U.S. Holders should consult their tax advisors regarding whether we are a PFIC and the potential application of the PFIC rules.
Backup Withholding Tax and Information Reporting Requirements U.S. backup withholding tax and information reporting requirements may apply to certain payments to certain holders of stock. Information reporting generally will apply to payments of dividendsdistributions on, and to proceeds from the sale, exchange or redemption of, our ordinary shares made within the United States, or by a United States payor or United States middleman, to a holder of our ordinary shares, other than an exempt recipient (including a payee that is not a United States person that provides an appropriate certification and certain other persons). Payments made (and sales or other dispositions effected at an office) outside the U.S. will be subject to information reporting in limited circumstances. A payor will be required to withhold backup withholding tax from any payments of dividends on, or the proceeds from the sale or redemption of, ordinary shares within the United States, or by a United States payor or United States middleman, to a holder, other than an exempt recipient, if such holder fails to furnish its correct taxpayer identification number or otherwise fails to comply with, or establish an exemption from, such backup withholding tax requirements, or to report dividends required to be shown on the holder’s U.S. federal income tax returns. Back up withholding is not an additional tax. Any amounts withheld under the backup withholding rules will be allowed as a credit against the beneficial owner’s U.S. federal income tax liability, if any, and any excess amounts withheld under the backup withholding rules may be refunded, provided that the required information is timely furnished to the IRS.
Foreign Asset Reporting Certain U.S. Holders who are individuals and certain entities may be required to report information relating to an interest in our ordinary shares, subject to certain exceptions (including an exception for shares held in accounts maintained by certain financial institutions) by filing IRS Form 8938 (Statement of Specified Foreign Financial Assets) with their federal income tax return. U.S. Holders are urged to consult their tax advisors regarding their information reporting obligations, if any, with respect to their ownership and disposition of our ordinary shares.
F. Dividends and Paying Agents Not applicable. G. Statement by Experts Not applicable. H. Documents on Display We are required to make certain filings with the SEC. The SEC maintains an internet website that contains reports, proxy statements and other information about issuers, like us, that file electronically with the SEC. The address of that site is www.sec.gov. We also make available on our website, free of charge, our annual reports on Form 20-F and the text of our reports on Form 6-K, including any amendments to these reports, as well as certain other SEC filings, as soon as reasonably practicable after they are electronically filed with or furnished to the SEC. Our website address is www.mediwound.com. The information contained on our website is not incorporated by reference in this document. I. Subsidiary Information Not applicable. J. Annual Report to Security Holders Not Applicable.
Item 11. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK We are exposed to a variety of risks, including foreign currency exchange fluctuations, changes in interest rates and inflation. We regularly assess currency, interest rate and inflation risks to minimize any adverse effects on our business as a result of those factors. Foreign Currency Risk The U.S. dollar is our functional and reporting currency. A significant portion of our operating expenses are denominated in Israeli shekels, accounting for approximately 40%45%, 44%45% and 45%44% of our operating expenses in the years ended December 31, 2019, 20202023, 2022 and 2021, respectively. We also have expenses in other non‑dollar currencies, in particular the Euro, and for the next few years, we expect that a substantial portion of our revenues will be denominated in U.S dollar. A devaluation of the shekel in relation to the U.S. dollar has the effect of reducing the U.S. dollar amount of our expenses or payables that are payable in shekels, unless those expenses or payables are linked to the U.S. dollar. Conversely, any increase in the value of the shekel in relation to the U.S. dollar has the effect of increasing the U.S. dollar value of our unlinked shekel expenses, which would have a negative impact on our profit margins.
Because exchange rates between the U.S. dollar and both the shekel and the Euro (as well as between the U.S. dollar and other currencies) fluctuate continuously, such fluctuations have an impact on our results and period‑to‑period comparisons of our results. The effects of foreign currency re‑measurements are reported in our consolidated financial statements of operations. The following table presents information about the changes in the exchange rates of the shekel against the U.S. dollar and changes in the exchange rates of the Euro against the U.S. dollar:
| | | Appreciation (Devaluation) of | | | Appreciation (Devaluation) of | | | Period | | Shekel against the U.S. dollar
(%) | | | Euro
against the U.S. dollar
(%) | | | Shekel against the U.S. dollar
(%) | | | Euro
against the U.S. dollar
(%) | | | | | | | | | | | | | | | | | 2019 | | | 7.8 | | | | (2.0 | ) | | | 2020 | | | 7.0 | | | | 8.0 | | | | 2021 | | | 3.3 | | | | (6.9 | ) | | | 3.3 | | | | 7.7 | | | 2022 | | | | (13.2 | ) | | | 5.8 | | | 2023 | | | | (3.1 | ) | | | (3.7 | ) |
A 10% increase (decrease) in the value of the NIS and Euro against the U.S. dollar would have increased (decreased) our net profit by (loss) approximately $0.74$1.54 million for the year ended December 31, 2021.2023. As we are marketing and selling NexoBrid in Europe and conducting clinical trials of outside the United States, we will continue to monitor exposure to currency fluctuations. We do not currently engage in currency hedging activities in order to reduce this currency exposure, but we may begin to do so in the future. Instruments that may be used to hedge future risks may include foreign currency forward and swap contracts. These instruments may be used to selectively manage risks, but there can be no assurance that we will be fully protected against material foreign currency fluctuations. Other Market Risks We do not believe that we have material exposure to interest rate risk due to the fact that we have no long‑term debt. We do not believe that we have any material exposure to inflationary risks. We do not believe that the rate of inflation in Israel has had a material impact on our business to date. However, our costs in Israel will increase if inflation in Israel exceeds the devaluation of the shekel against the U.S. dollar (to the extent that it devalues at all) or if the timing of such devaluation lags behind inflation in Israel.
Item 12. DESCRIPTION OF SECURITIES OTHER THAN EQUITY SECURITIES Not applicable.
PART II Item 13. DEFAULTS, DIVIDEND ARREARAGES AND DELINQUENCIES None. Item 14. MATERIAL MODIFICATIONS TO THE RIGHTS OF SECURITY HOLDERS AND USE OF PROCEEDS None. Item 15. CONTROLS AND PROCEDURES (a) Disclosure Controls and Procedures Our management, including our Chief Executive Officer and Chief Financial Officer, has evaluated the effectiveness of our disclosure controls and procedures (as such term is defined in Rules 13a‑15(e) and 15d‑15(e) under the Exchange Act) as of December 31, 2021.2023. Based on such evaluation, our Chief Executive Officer and Chief Financial Officer have concluded that, as of December 31, 2021,2023, our disclosure controls and procedures were effective. (b) Management Annual Report on Internal Control over Financial Reporting Our management, under the supervision of our Chief Executive Officer and Chief Financial Officer, is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rules 13a‑15(f) and 15d‑15(f) under the Exchange Act. Our management, including our Chief Executive Officer and Chief Financial Officer, assessed the effectiveness of our internal control over financial reporting as of December 31, 2021.2023. In making this assessment, our management used the criteria established in Internal Control—IntegratedControl-Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Our management has concluded, based on its assessment, that our internal control over financial reporting was effective as of December 31, 2021.
(c) Attestation report of the registered public accounting firm Somekh Chaikin, a member firm of KPMG International, an independent registered public accounting firm, to which we refer as KPMG Israel, which audited the financial statements included in this annual report containing the disclosure required by this Item 15 has issued an attestation report regarding the effectiveness of our internal control over financial reporting. That report is included in “Item 18. Financial Statements” on page F-[1] of this annual report, which attestation report is incorporated by reference in this Item 15(c). (d) Changes in Internal Control over Financial Reporting There were no changes in our internal control over financial reporting (as such term is defined in Rules 13a‑15(f) and 15d‑15(f) under the Exchange Act) that occurred during the period covered by this annual report that have materially affected, or that are reasonably likely to materially affect, our internal control over financial reporting. Item 16. [Reserved] Item 16A. AUDIT COMMITTEE FINANCIAL EXPERT Our board of directors has determined that Sharon KochanStephen Wills qualifies as an “audit committee financial expert,” as defined under the U.S. federal securities laws and has the requisite financial experience defined by the Nasdaq Marketplace Rules. In addition, Sharon KochanStephen Wills is independent as such term is defined in Rule 10A‑3(b)(1) under the Exchange Act and under the listing standards of the Nasdaq Global Market.
Item 16BItem 16B.. CODE OF ETHICS We have adopted a code of business conduct and ethics applicable to our executive officers, directors and all other employees. A copy of the code is delivered to every employee of MediWound Ltd. and its subsidiaries and is available to our investors and others on our website http://ir.mediwound.com/ or by contacting our investor relations department. Information contained on, or that can be accessed through, our website does not constitute a part of this annual report and is not incorporated by reference herein. Any waivers of this code for executive officers or directors will be disclosed through the filing of a Form 6‑K or on our website. We granted no waivers under our code of ethics in 2021.2023. Item 16C. PRINCIPAL ACCOUNTANT FEES AND SERVICES Principal Accountant Fees and Services
We paidThe cost for the following fees for professional services rendered by Kost Forer Gabbay & Kasierer, a member of Ernst & Young Global, who was our independent registered public accounting firm until the April 28, 2021, and by Somekh Chaikin, a member firm of KPMG International Haifa, Israel, Auditor firm ID: 1057, who became our independent registered public accounting firm on June 15, 2021 for the year ended December 31, 2021:2023 and 2022:
| | | 2020 | | | 2021 | | | 2023 | | | 2022 | | | Audit Fees | | $ | 170,000 | | | $ | 245,000 | | | $ | 375,000 | | | $ | 270,000 | | | Audit‑Related Fees | | | 33,500 | | | | — | | | | Tax Fees | | | — | | | | 15,000 | | | | 23,395 | | | | 28,549 | | | Total | | $ | 203,500 | | | $ | 260,000 | | | $ | 398,395 | | | $ | 298,549 | |
“Audit fees” are the aggregate fees paid for the audit of our annual financial statements for the year 2021.years 2023 and 2022. This category also includes services that generally the independent accountant provides, such as consents and assistance with and review of documents filed with the SEC. “Audit‑related fees” are the aggregate fees paid for assurance and related services that are reasonably related to the performance of the audit and are not reported under audit fees. These fees primarily include accounting consultations regarding the accounting treatment of matters that occur in the regular course of business, implications of new accounting pronouncements and other accounting issues that occur from time to time.
“Tax fees” include fees for professional services rendered by our independent registered public accounting firm for tax compliance, transfer pricing and tax advice on actual or contemplated transactions. The Audit Committee pre‑approves all audit and non‑audit services provided by the independent registered public accounting firm. Item 16D. EXEMPTIONS FROM THE LISTING STANDARDS FOR AUDIT COMMITTEES Not applicable. Item 16E. PURCHASES OF EQUITY SECURITIES BY THE ISSUER AND AFFILIATED PURCHASERS Not applicable. Item 16F. CHANGE IN REGISTRANT’S CERTIFYING ACCOUNTANT The information required by this Item 16F was previously reported in our report of foreign private issuer on Form 6‑K (File No. 001‑36349) filed with the SEC on March 2, 2022.Not applicable.
Item 16G. CORPORATE GOVERNANCE As a foreign private issuer, we are permitted to comply with Israeli corporate governance practices instead of the Nasdaq Stock Market requirements, provided that we disclose those Nasdaq Stock Market requirements with which we do not comply and the equivalent Israeli requirement that we follow instead. We currently rely on this “foreign private issuer exemption” with respect to the following requirements:
| • | Quorum. As permitted under the Israeli Companies Law pursuant to our articles of association, the quorum required for an ordinary meeting of shareholders will consist of at least two shareholders present in person, by proxy or by other voting instrument in accordance with the Israeli Companies Law, who hold at least 25% of the voting power of our shares (and in an adjourned meeting, with some exceptions, at least two shareholders), instead of 33 1/3% of the issued share capital required under the Nasdaq Stock Market listing rules. |
| • | Nomination of directors. With the exception of external directors and directors elected by our board of directors due to vacancy, our directors are elected by an annual meeting of our shareholders to hold office until the next annual meeting following one year from his or her election. The nominations for directors, which are presented to our shareholders by our board of directors, are generally made by the entire board of directors itself, in accordance with the provisions of our articles of association and the Israeli Companies Law. Nominations need not be made by a nominating committee of our board of directors consisting solely of independent directors or otherwise, as required under the Nasdaq Stock Market listing rules.•
|
| • | Majority of independent directors. Under the Israeli Companies Law, we are only required to appoint at least two external directors, within the meaning of the Israeli Companies Law, to our board of directors. Currently, four of our directors (of whom two are external directors, within the meaning of the Israeli Companies Law) qualify as independent directors under the rules of the U.S. federal securities laws and the Nasdaq Stock Market listing rules. If at any time we no longer have a controlling shareholder, we will no longer be required to have external directors, provided that we comply with the majority Board independence requirements and the audit and compensation committee composition requirements of the Nasdaq Stock Market.
|
| • | Shareholder approval. We do not intend to follow Nasdaq Stock Market rules which require shareholder approval in order to enter into any transaction, other than a public offering, involving the sale, issuance or potential issuance by the Company of ordinary shares (or securities convertible into or exercisable for ordinary shares) equal to 20% or more of the outstanding share capital of the Company or 20% or more of the voting power outstanding before the issuance for less than the greater of book or market value of the ordinary shares. We will follow Israeli law with respect to any requirement to obtain shareholder approval in connection with any private placements of equity securities. |
Item 16H. MINE SAFETY DISCLOSURE Not applicable.
Item 16I. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections.DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS. [Not applicable.]
Item 16J. DISCLOSURE REGARDING INSIDER TRADING POLICIES. The disclosure under this item is not yet required in this annual report. Item 16K. CYBERSECURITY Cybersecurity Risk Management and Strategy We have adopted a risk-based approach to protecting our information technology systems and confidential information through the adoption of certain technical and administrative safeguards, including certain information technology policies intended to protect the confidentiality, integrity, and availability of our critical systems and information. Key elements of our cybersecurity risk management program include: technical controls deployed to protect more critical systems and data, such as multi-factor authentication, firewalls, network segregation, and secure file transfer protocols; 120disaster recovery and business continuity plan;
monitoring of our information technology systems for information security risks; based on their level of risk, cybersecurity assessments of third-party software / platforms; and regular cybersecurity awareness training of our employees, designed to help identify any new threats and to address security incidents.
We have not identified risks from known cybersecurity threats, including as a result of any prior cybersecurity incidents, that have materially affected or are reasonably likely to materially affect us, including our operations, business strategy, results of operations, or financial condition. See “Risk Factors—Risks Related to Healthcare Laws and Other Legal Compliance Matters—Our business and operations may suffer in the event of information technology system failures, cyberattacks or deficiencies in our cybersecurity.”
Cybersecurity Governance Our Board considers cybersecurity risk as part of its overall risk oversight function and oversees management’s implementation of our cybersecurity risk management program. The Committee receives periodic reports from management on our cybersecurity risks. In addition, management updates the Committee, as necessary, regarding any material cybersecurity incidents, as well as any incidents with lesser impact potential. The full Board also receives briefings from management on our cyber risk management program. Board members receive presentations on cybersecurity topics from our Chief Operations Officer (CISO), internal security staff or external experts as part of the Board’s continuing education on topics that impact public companies. The Committee reports to the full Board regarding its activities, including those related to cybersecurity. The full Board also receives periodic briefings from management on our cyber risk management program, as well as from internal security staff or external experts engaged from time to time as part of the Board’s continuing education on topics that impact public companies. Our management team, including Chief Operations Officer, is responsible for assessing and managing our material risks from cybersecurity threats. The team has primary responsibility for our overall cybersecurity risk management program and supervises both our internal cybersecurity personnel and any retained external cybersecurity consultants which are engaged as required. Our management team’s combined experience includes over 10 years of organizational risk management experience. Our management team supervises efforts to prevent, detect, mitigate, and remediate cybersecurity risks and incidents through various means, which may include briefings from internal security personnel; threat intelligence and other information obtained from governmental, public or private sources, including external consultants engaged by us; and alerts and reports produced by security tools deployed in the IT environment.
PART III Item 17. FINANCIAL STATEMENTS Not applicable. Item 18. FINANCIAL STATEMENTS See pages F‑1 through F-49F-47 of this annual report. Item 19. EXHIBITS
| 101.INS | Inline XBRL Instance Document | | | | 101.SCH | Inline XBRL Taxonomy Extension Schema Document | | | | 101.CAL | Inline XBRL Taxonomy Extension Calculation Linkbase Document | | | | 101.DEF | Inline XBRL Taxonomy Definition Linkbase Document | | | | 101.LAB | Inline XBRL Taxonomy Extension Label Linkbase Document | | | | 101.PRE | Inline XBRL Taxonomy Extension Presentation Linkbase Document | | | | 104 | Cover Page Interactive Data File (the cover page iXBRL tags are embedded within the Inline XBRL document) |
129
† | Portions of this exhibit have been omitted pursuant to Instruction 4(a) to Exhibits to Form 20-F because they are both (i) not material and (ii) the type that the Registrant treats as private or confidential. |
| * | Indicates management contract or compensatory plan or arrangement. | (1) (1) | Previously filed with the SEC on March 3, 2014 pursuant to the Registrant’s registration statement on Form F‑1 (File No. 333‑193856) and incorporated by reference herein. |
(2) (2) | Previously furnished to the SEC on May 5, 2021 as Appendix B to the Registrant’s proxy statement for its 2021 annual general meeting of shareholders held on June 15, 2021, attached as Exhibit 99.1 to the Registrant’s report of foreign private issuer on Form 6‑K (File No. 001‑36349) and incorporated by reference herein. |
| (3) (3) | Previously filed with the SEC on February 10, 2014 pursuant to the Registrant’s registration statement on Form F‑1 (File No. 333‑193856) and incorporated by reference herein. |
| (4) (4) | Previously filed with the SEC on February 25, 2020 pursuant to the Registrant’s annual report on Form 20-F for the year ended December 31, 2019 (File No. 001-36349) and incorporated by reference herein. |
| (5) (5) | Previously furnished to the SEC on August 14, 2019October 21, 2022 as Appendix A to the Registrant’s proxy statement for its extraordinary general meeting of shareholders held on September 23, 2019,November 28, 2022, attached as Exhibit 99.1 to the Registrant’s report of foreign private issuer on Form 6‑K (File No. 001‑36349) and incorporated by reference herein.
|
| (6) (6) | Previously filed with the SEC on February 25, 2021 pursuant to the Registrant’s annual report on Form 20-F for the year ended December 31, 2020 (File No. 001-36349) and incorporated by reference herein. |
| (7) (7) | Previously filed with the SEC on January 25, 2016 as Exhibit 4.14 to the Registrant’s annual report on Form 20‑F for the year ended December 31, 2015 (File No. 001‑36349) and incorporated by reference herein. |
| (8) (8) | Previously filed with the SEC on February 21, 2017 as Exhibit 4.15 to the Registrant’s annual report on Form 20‑F for the year ended December 31, 2016 (File No. 001‑36349) and incorporated by reference herein. |
| (9) (9) | Previously filed with the SEC on March 19, 2018 as Exhibit 4.16 to the Registrant’s annual report on Form 20‑F for the year ended December 31, 2017 (File No. 001‑36349) and incorporated by reference herein. |
| (10) (10) | Previously filed with the SEC on March 25, 2019 as Exhibit 4.17 to the Registrant’s annual report on Form 20‑F for the year ended December 31, 2018 (File No. 001‑36349) and incorporated by reference herein. |
| (11) (11) | Previously filed with the SEC on March 19, 2018 as Exhibit 4.17 to the Registrant’s annual report on Form 20‑F for the year ended December 31, 2017 (File No. 001‑36349) and incorporated by reference herein. |
| (12) (12) | Previously filed with the SEC on March 25, 2019 as Exhibit 4.20 to the Registrant’s annual report on Form 20‑F for the year ended December 31, 2018 (File No. 001‑36349) and incorporated by reference herein. |
| (13) (13) | Previously filed with the SEC on March 25, 2019 as Exhibit 4.21 to the Registrant’s annual report on Form 20‑F for the year ended December 31, 2018 (File No. 001‑36349) and incorporated by reference herein. |
(14) (14) | Previously filed with the SEC by Vericel Corporation on August 6, 2019 as Exhibit 10.9 to its quarterly report on Form 10-Q for the quarter ended June 30, 2019 (File No. 001‑35280) and incorporated by reference herein. |
| (15) (15) | Previously filed with the SEC by Vericel Corporation on August 6, 2019 as Exhibit 10.10 to its quarterly report on Form 10-Q for the quarter ended June 30, 2019 (File No. 001‑35280) and incorporated by reference hereinherein. | (16) | Previously furnished to the SEC on June 9, 2022 as Appendix A to the Registrant’s proxy statement for its 2022 annual general meeting of shareholders held on July 19, 2022, attached as Exhibit 99.1 to the Registrant’s report of foreign private issuer on Form 6‑K (File No. 001‑36349) and incorporated by reference herein.
| (17) | Previously filed with the SEC on March 17, 2022 as Exhibit 4.11.7 to the Registrant’s annual report on Form 20‑F for the year ended December 31, 2021 (File No. 001‑36349) and incorporated by reference herein. | (18) | Previously furnished to the SEC on September 26, 2022 as Exhibit 4.1 to the Registrant’s report of foreign private issuer on Form 6‑K (File No. 001‑36349) and incorporated by reference herein. | (19) | Previously furnished to the SEC on September 26, 2022 as Exhibit 4.2 to the Registrant’s report of foreign private issuer on Form 6‑K (File No. 001‑36349) and incorporated by reference herein. | (20) | Previously furnished to the SEC on September 26, 2022 as Exhibit 4.4 to the Registrant’s report of foreign private issuer on Form 6‑K (File No. 001‑36349) and incorporated by reference herein.
| (21) | Previously furnished to the SEC on September 26, 2022 as Exhibit 10.3 to the Registrant’s report of foreign private issuer on Form 6‑K (File No. 001‑36349) and incorporated by reference herein. |
130
123
SIGNATURES The registrant hereby certifies that it meets all of the requirements for filing on Form 20‑F and that it has duly caused and authorized the undersigned to sign this annual report on its behalf. | | | | | MediWound Ltd. | | | | | Date: March 17, 202221, 2024 | By: /s/ Boaz Gur-LavieHani Luxenburg
| | | Boaz Gur-Lavie | | | Hani Luxenburg Chief Financial Officer | |
131
MEDIWOUND LTD. AND ITS SUBSIDIARIES CONSOLIDATED FINANCIAL STATEMENTS CONSOLIDATED FINANCIAL STATEMENTSDECEMBER 31, 2023
DECEMBER 31, 2021INDEX
INDEX
| | | | | | | F-2 – F-5F3 | (Firm Name: KPMG (Somekh Chaikin) / PCAOB ID No. 1057) | | (Firm Name: KOST FORER GABBAY & KASIERER / PCAOB ID No. 1281)
| | | | | | F-6F-4
| | | | | F -7F-5
| | | | | F-8F-6
| | | | | F-9F-7 - F-10F-8
| | | | | F-11F-9 - F-49F-47
|
- - - - - - - - - - - - - - - - - - - - -
MEDIWOUND LTD. AND ITS SUBSIDIARIES Report of Independent Registered Public Accounting Firm To the Shareholders and Board of Directors
MediWound Ltd.:
OpinionOpinions on the Consolidated Financial Statements and Internal Control Over Financial Reporting
We have audited the accompanying consolidated statementstatements of financial position of MediWound Ltd. and its subsidiaries (hereinafter – “the Company”)(the Company) as of December 31, 2021,2023 and 2022, the related consolidated statements of profit or loss and other comprehensive income (loss), changes in shareholders’ equity (deficit), and cash flows for each of the years in the three‑year thenperiod ended December 31, 2023, and the related notes (collectively, “thethe consolidated financial statements”)statements). We also have audited the Company’s internal control over financial reporting as of December 31, 2023, based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission. In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of the Company as of December 31, 2021,2023 and 2022, and the results of its operations and its cash flows for each of the year thenyears in the three-year period ended December 31, 2023, in conformity with International Financial Reporting Standards as issued by the International Accounting Standards Board. Also in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2023 based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission. Basis for OpinionOpinions TheseThe Company’s management is responsible for these consolidated financial statements, are the responsibilityfor maintaining effective internal control over financial reporting, and for its assessment of the Company’s management.effectiveness of internal control over financial reporting, included in the accompanying Management’s Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on thesethe Company’s consolidated financial statements and an opinion on the Company’s internal control over financial reporting based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our auditaudits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the auditaudits to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audit, we are required to obtain an understanding offraud, and whether effective internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.was maintained in all material respects. Our auditaudits of the consolidated financial statements included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our auditaudits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audit providesaudits provide a reasonable basis for our opinion.opinions. Definition and Limitations of Internal Control Over Financial Reporting A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements. MEDIWOUND LTD. AND ITS SUBSIDIARIES Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate. Critical Audit Matter The critical audit matter communicated below is a matter arising from the current period audit of the consolidated financial statements that was communicated or required to be communicated to the audit committee and that: (1) relates to accounts or disclosures that are material to the consolidated financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of a critical audit matter does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to which it relates. Israeli Innovation Authority grant liability As discussed in Notes 3e2c, 3d and 14 to the consolidated financial statements, in previous years the Company received grants from the Israeli Innovation Authority (“IIA”) to finance its research and development efforts. These grants were recognized as a liability to the extent the Company expected to refund them through royalties on its revenues derived from sales of products or services developed in whole or in part using the grants. The amount of the liability is reexamined each period using the Company’s updated future revenue forecasts discounted to their present value. Any changes in the IIA grant liability are recognized in profit or loss. The IIA grant liability was $8,105$7,803 thousand as of December 31, 2021.2023. We identified the evaluation of the subsequent period end measurement of the IIA grant liability as a critical audit matter. Specifically, a high degree of subjective auditor judgment was involved in evaluating certain significant assumptions used by the Company to develop its future revenue forecasts, including the likelihood and timing of achievement of regulatory approvals and potential market demand and market share for the Company’s products, which were based on external market research. These significant assumptions were forward-looking and could be affected by future economic and market conditions. The following are the primary procedures we performed to address this critical audit matter. We evaluated the design and effectiveness of certain internal controls related to the Company’s process for measuring the IIA grant liability, including controls related to the determination of the above referenced significant assumptions used to develop future revenue forecasts. We compared the Company’s assumption of the likelihood and timing for obtaining regulatory approvals for its products, based on the specific phases of their development, to relevant data in industry research reports. We evaluated the Company’s assumption of potential market demand and market share by evaluating the relevance and reliability of the external market research upon which the Company based its future revenue forecasts. We performed sensitivity analyses over these significant assumptions to assess the impact of changes in the assumptions on the period end IIA grant liability. Somekh Chaikin Member Firm of KPMG International We have served as the Company’s auditor since 2021. Haifa, Israel March 17, 202221, 2024 MEDIWOUND LTD. AND ITS SUBSIDIARIES | Kost Forer Gabbay & Kasierer
144 Menachem Begin Rd.
Tel-Aviv 6492102, Israel
| Tel: +972-3-6232525
Fax: +972-3-5622555
ey.com
|
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Shareholders and
Board of Directors of
MEDIWOUND LTD. AND ITS SUBSIDIARIES
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheet of MediWound Ltd and subsidiaries (the "Company") as of December 31, 2020, the related consolidated statements of comprehensive or loss, shareholders' equity and cash flows for each of the two years in the period ended December 31, 2020, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2020, and the results of its operations and its cash flows for each of the two years in the period ended December 31, 2020, in conformity with International Financial Reporting Standards as issued by the International Accounting Standards Board.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB. We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matter
The critical audit matter communicated below is a matter arising from the current period audit of the financial statements that was communicated or required to be communicated to the audit committee and that: (1) relates to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective or complex judgments. The communication of the critical audit matter does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the account or disclosure to which it relates.
| | Israel Innovation Authority (IIA) grant liability
| | | | Description of the matter
| | As described in Notes 3 and 17b to the consolidated financial statements, the Company’s research and development efforts have been financed in part through grants from the Israeli Innovation Authority (“IIA”). Grants received from the IIA are recognized as a liability if future economic benefits are expected from the research and development activity that will result in royalty-bearing sales. The Company undertook to pay royalties of 3% on the revenues derived from sales of products or services developed in whole or in part using IIA grants, up to the amount of total grants received, plus LIBOR interest. The liability to the IIA is measured at amortized cost using the effective interest method and amounted as of December 31, 2020 to $7,529 thousands.
Auditing the Company's IIA liability involved a high degree of subjectivity as it is based on assumptions about future revenue forecasts, such as long-term demand for the Company’s products and licenses and revenue growth rates. These significant assumptions are forward-looking and could be affected by future economic and market conditions.
|
How we addressed the matter in our audit
| | Our substantive audit procedures included, among others, evaluating the significant assumptions and operating data used by management. For example, we compared the significant assumptions and operating data used by management to historical trends, we performed look-back analyses by comparing the Company's historical financial forecasted revenues with the actual results and we agreed future revenues to approved budgets. In addition, we considered the phases of development of the Company's products and the Company’s ability of obtaining regulatory approvals. We also tested the completeness and accuracy of the relevant data used in management's calculation, tested the mathematical accuracy of management’s calculations and performed sensitivity analyses over significant assumptions used by management related to revenue growth rates.
|
| /s/ KOST FORER GABBAY & KASIERER | Tel-Aviv, Israel | KOST FORER GABBAY & KASIERER | February 25, 2021 | A Member of Ernst & Young Global |
We have served as the Company's auditor since 2001 to 2020
MEDIWOUND LTD. AND ITS SUBSIDIARIES
Consolidated Statements of Financial PositionsPosition
U.S. dollars in thousands | | | | | | As of December 31, | | | | | Note | | | 2023 | | | 2022 | | | | | | | | | | | | | Cash and cash equivalents | | 4 | | | | 11,866 | | | | 33,895 | | Short-term and restricted bank deposits | | 5 | | | | 29,842 | | | | - | | Trade receivables | | 6 | | | | 3,700 | | | | 9,332 | | Inventories | | 7 | | | | 2,846 | | | | 1,963 | | Other receivables | | 8 | | | | 1,441 | | | | 650 | | Total current assets | | | | | | 49,695 | | | | 45,840 | | | | | | | | | | | | | | | Other receivables | | 9 | | | | 233 | | | | 180 | | Long-term restricted bank deposits | | | | | | 440 | | | | 184 | | Property, plant and equipment, net | | 10 | | | | 9,228 | | | | 2,366 | | Right-of-use assets, net | | 11 | | | | 6,698 | | | | 1,215 | | Intangible assets, net | | 12 | | | | 165 | | | | 231 | | Total non-current assets | | | | | | 16,764 | | | | 4,176 | | | | | | | | | | | | | | | Total assets | | | | | | 66,459 | | | | 50,016 | | | | | | | | | | | | | | | Current maturities of long-term liabilities | | | | | | 1,410 | | | | 2,242 | | Trade payables and accrued expenses | | | | | | 5,528 | | | | 5,656 | | Other payables | | 13 | | | | 3,891 | | | | 4,159 | | Total current liabilities | | | | | | 10,829 | | | | 12,057 | | | | | | | | | | | | | | | Warrants, net | | 19c | | | | 7,296 | | | | 15,606 | | Liabilities in respect of IIA grants | | 14, 17b | | | | 7,677 | | | | 7,445 | | Liabilities in respect of TEVA | | 17c | | | | 2,256 | | | | 2,788 | | Lease liabilities | | 11 | | | | 6,350 | | | | 846 | | Severance pay liability, net | | 16 | | | | 456 | | | | 360 | | Total non-current liabilities | | | | | | 24,035 | | | | 27,045 | | | | | | | | | | | | | | | Total liabilities | | | | | | 34,864 | | | | 39,102 | | | | | | | | | | | | | | | Shareholders' equity: | | 19 | | | | | | | | | | Ordinary shares of NIS 0.07 par value: | | | | | | | | | | | | Authorized 20,000,000 shares as of December 31, 2023 and 12,857,143
shares as of December 31, 2022; Issued and Outstanding 9,221,764
shares as of December 31, 2023 and 7,240,020 shares as of December 31, 2022 | | | | | | 184 | | | | 143 | | Share premium | | | | | | 206,251 | | | | 178,882 | | Foreign currency translation reserve | | | | | | (18 | ) | | | (5 | ) | Accumulated deficit | | | | | | (174,822 | ) | | | (168,106 | ) | Total equity | | | | | | 31,595 | | | | 10,914 | | | | | | | | | | | | | | | Total liabilities and equity | | | | | | 66,459 | | | | 50,016 | |
| | | | | | As of December 31 | | | | | Note | | | 2021 | | | 2020 | | | | | | | | | | | | | Cash and cash equivalents | | 4 | | | | 11,046 | | | | 17,376 | | Restricted deposits | | 5 | | | | 0 | | | | 184 | | Short-term bank deposits | | 5 | | | | 0 | | | | 4,024 | | Trade receivables | | 6 | | | | 1,779 | | | | 2,767 | | Inventories | | 7 | | | | 1,200 | | | | 1,380 | | Other receivables | | 8, 25 | | | | 927 | | | | 462 | | Total current assets | | | | | | 14,952 | | | | 26,193 | | | | | | | | | | | | | | | Other receivables | | 9 | | | | 469 | | | | 0 | | Property, plant and equipment, net | | 10 | | | | 2,478 | | | | 2,630 | | Right-of-use assets, net | | 11 | | | | 1,548 | | | | 1,884 | | Intangible assets, net | | 12 | | | | 297 | | | | 363 | | Total non-current assets | | | | | | 4,792 | | | | 4,877 | | | | | | | | | | | | | | | Total assets | | | | | | 19,744 | | | | 31,070 | | | | | | | | | | | | | | | Current maturities of long-term liabilities | | | | | | 2,408 | | | | 2,417 | | Trade payables and accrued expenses | | | | | | 4,693 | | | | 2,992 | | Other payables | | 13, 25 | | | | 3,620 | | | | 2,857 | | Total current liabilities | | | | | | 10,721 | | | | 8,266 | | | | | | | | | | | | | | | Deferred revenues | | | | | | 119 | | | | 1,234 | | Liabilities in respect of IIA grants | | 14, 17b | | | | 7,885 | | | | 7,267 | | Liabilities in respect of purchase of shares | | 17c | | | | 3,922 | | | | 4,998 | | Lease liabilities | | 11 | | | | 1,391 | | | | 1,741 | | Severance pay liability, net | | 16 | | | | 288 | | | | 292 | | Total non-current liabilities | | | | | | 13,605 | | | | 15,532 | | | | | | | | | | | | | | | Total liabilities | | | | | | 24,326 | | | | 23,798 | | | | | | | | | | | | | | | Shareholders' equity: | | 19 | | | | | | | | | | Ordinary shares of NIS 0.01 par value: | | | | | | | | | | | | Authorized: 50,000,000 shares as of December 31, 2021 and December 31, 2020; Issued and Outstanding 27,272,818 shares as of December 31, 2021 and 27,236,752 shares as of December 31, 2020 | | | | | | 75 | | | | 75 | | Share premium | | | | | | 143,869 | | | | 142,193 | | Foreign currency translation reserve | | | | | | (19 | ) | | | (40 | ) | Accumulated deficit | | | | | | (148,507 | ) | | | (134,956 | ) | Total equity (deficit) | | | | | | (4,582 | ) | | | 7,272 | | | | | | | | | | | | | | | Total liabilities and equity | | | | | | 19,744 | | | | 31,070 | | | | | | | | | | | | | | |
The accompanying notes are an integral part of the consolidated financial statements. MEDIWOUND LTD. AND ITS SUBSIDIARIES Consolidated Statements of Profit or Loss and Other Comprehensive Income or Loss
U.S. dollars in thousands (except of share and per share data) | | | | | | | | | | | | | | Year Ended December 31 | | | | | Note | | | 2021 | | | 2020 | | | 2019 | | Revenues from sale of products | | | | | | 9,613 | | | | 7,445 | | | | 3,393 | | Revenues from development services | | | | | | 12,372 | | | | 13,935 | | | | 10,678 | | Revenues from license agreements | | | | | | 1,778 | | | | 383 | | | | 17,718 | | Total revenues | | 23a | | | | 23,763 | | | | 21,763 | | | | 31,789 | | | | | | | | | | | | | | | | | | | Cost of revenues | | 23b | | | | 14,992 | | | | 14,218 | | | | 11,849 | | | | | | | | | | | | | | | | | | | Gross profit | | | | | | 8,771 | | | | 7,545 | | | | 19,940 | | | | | | | | | | | | | | | | | | | Research and development, net of participations | | 23c | | | | 10,256 | | | | 7,698 | | | | 4,969 | | Selling and marketing | | 23d | | | | 3,388 | | | | 3,228 | | | | 4,064 | | General and administrative | | 23e | | | | 6,348 | | | | 5,459 | | | | 5,242 | | Other expenses | | 23f | | | | 0 | | | | 0 | | | | 1,172 | | Total operating expenses | | | | | | 19,992 | | | | 16,385 | | | | 15,447 | | | | | | | | | | | | | | | | | | | Operating profit (loss) | | | | | | (11,221 | ) | | | (8,840 | ) | | | 4,493 | | | | | | | | | | | | | | | | | | | Financial income | | 23g | | | | 11 | | | | 843 | | | | 556 | | Financial expense | | 23g | | | | (2,314 | ) | | | (1,279 | ) | | | (2,983 | ) | Financing expenses, net | | | | | | (2,303 | ) | | | (436 | ) | | | (2,427 | ) | | | | | | | | | | | | | | | | | | Profit (loss) before taxes on income | | | | | | (13,524 | ) | | | (9,276 | ) | | | 2,066 | | | | | | | | | | | | | | | | | | | Taxes on income | | | | | | (27 | ) | | | 0 | | | | 0 | | | | | | | | | | | | | | | | | | | Profit (loss) from continuing operations | | | | | | (13,551 | ) | | | (9,276 | ) | | | 2,066 | | Profit from discontinued operations | | 17c,22 | | | | 0 | | | | 80 | | | | 2,889 | | | | | | | | | | | | | | | | | | | Net profit (loss) for the year | | | | | | (13,551 | ) | | | (9,196 | ) | | | 4,955 | | | | | | | | | | | | | | | | | | | Other comprehensive income (loss): | | | | | | | | | | | | | | | | Foreign currency translation adjustments | | | | | | 21 | | | | (23 | ) | | | 8 | | Total comprehensive income (loss) | | | | | | (13,530 | ) | | | (9,219 | ) | | | 4,963 | | | | | | | | | | | | | | | | | | | Earning (loss) per share data | | 24 | | | | | | | | | | | | | | Basic and diluted net profit (loss) per share from continuing operations | | | | | | (0.50 | ) | | | (0.34 | ) | | | 0.08 | | Basic and diluted net profit per share from discontinued operations | | | | | | 0 | | | | 0 | | | | 0.10 | | Total Basic and diluted net profit (loss) per share - USD | | | | | | (0.50 | ) | | | (0.34 | ) | | | 0.18 | | | | | | | | | | | | | | | | | | | Number of shares used in calculating basic and diluted profit (loss) per share | | | | | | 27,244,475 | | | | 27,209,878 | | | | 27,178,839 | |
| | | | | | Year Ended December 31 | | | | | Note | | | 2023 | | | 2022 | | | 2021 | | Revenues from sale of products | | | | | | 6,261 | | | | 5,347 | | | | 9,613 | | Revenues from development services | | | | | | 12,265 | | | | 12,943 | | | | 12,372 | | Revenues from license agreements and royalties | | | | | | 160 | | | | 8,206 | | | | 1,778 | | Total revenues | | 22a | | | | 18,686 | | | | 26,496 | | | | 23,763 | | | | | | | | | | | | | | | | | | | Cost of revenues from sale of products | | | | | | 4,927 | | | | 3,184 | | | | 4,983 | | Cost of revenues from development services | | | | | | 10,177 | | | | 9,829 | | | | 9,907 | | Cost of revenues from license agreements | | | | | | 4 | | | | 318 | | | | 102 | | Total cost of revenues | | 22b | | | | 15,108 | | | | 13,331 | | | | 14,992 | | | | | | | | | | | | | | | | | | | Gross profit | | | | | | 3,578 | | | | 13,165 | | | | 8,771 | | | | | | | | | | | | | | | | | | | Research and development | | 22c | | | | 7,467 | | | | 10,181 | | | | 10,256 | | Selling and marketing | | 22d | | | | 4,844 | | | | 3,725 | | | | 3,388 | | General and administrative | | 22e | | | | 6,768 | | | | 6,920 | | | | 6,348 | | Other (income) expenses | | 22f | | | | (211 | ) | | | 684 | | | | - | | Total operating expenses | | | | | | 18,868 | | | | 21,510 | | | | 19,992 | | | | | | | | | | | | | | | | | | | Operating loss | | | | | | (15,290 | ) | | | (8,345 | ) | | | (11,221 | ) | | | | | | | | | | | | | | | | | | Financial income | | | | | | 10,651 | | | | 461 | | | | 11 | | Financial expense | | | | | | (1,892 | ) | | | (11,637 | ) | | | (2,314 | ) | Financing income (expenses), net | | 22g | | | | 8,759 | | | | (11,176 | ) | | | (2,303 | ) | | | | | | | | | | | | | | | | | | Loss before taxes on income | | | | | | (6,531 | ) | | | (19,521 | ) | | | (13,524 | ) | | | | | | | | | | | | | | | | | | Taxes on income | | | | | | (185 | ) | | | (78 | ) | | | (27 | ) | | | | | | | | | | | | | | | | | | Net loss | | | | | | (6,716 | ) | | | (19,599 | ) | | | (13,551 | ) | | | | | | | | | | | | | | | | | | Other comprehensive income (loss): | | | | | | | | | | | | | | | | Foreign currency translation adjustments | | | | | | (13 | ) | | | 14 | | | | 21 | | Total comprehensive loss | | | | | | (6,729 | ) | | | (19,585 | ) | | | (13,530 | ) | | | | | | | | | | | | | | | | | | Loss per share data: | | 23,19 | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | Basic and diluted net loss per share - USD | | | | | | (0.75 | ) | | | (3.93 | ) | | | (3.48 | ) | | | | | | | | | | | | | | | | | | Number of shares used in calculating basic and diluted loss per share | | | | | | 9,013,144 | | | | 4,987,069 | | | | 3,892,068 | |
The accompanying notes are an integral part of the consolidated financial statements. MEDIWOUND LTD. AND ITS SUBSIDIARIES Consolidated Statements of Changes in Shareholders’ Equity (Deficit)
U.S. dollars in thousands | | Share capital | | | Share premium | | | Foreign currency translation reserve | | | Accumulated deficit | | | Total equity (deficit) | | | | | | | | | | | | | | | | | | | | | | | Balance as of January 1, 2021 | | | 75 | | | | 142,193 | | | | (40 | ) | | | (134,956 | ) | | | 7,272 | | | | | | | | | | | | | | | | | | | | | | | Net loss | | | 0 | | | | 0 | | | | 0 | | | | (13,551 | ) | | | (13,551 | ) | Other comprehensive income | | | 0 | | | | 0 | | | | 21 | | | | 0 | | | | 21 | | Total comprehensive loss | | | 0 | | | | 0 | | | | 21 | | | | (13,551 | ) | | | (13,530 | ) | Exercise of options | | | (* | ) | | | 3 | | | | 0 | | | | 0 | | | | 3 | | Share-based compensation | | | 0 | | | | 1,673 | | | | 0 | | | | 0 | | | | 1,673 | | | | | | | | | | | | | | | | | | | | | | | Balance as of December 31, 2021 | | | 75 | | | | 143,869 | | | | (19 | ) | | | (148,507 | ) | | | (4,582 | ) | | | | | | | | | | | | | | | | | | | | | | Balance as of January 1, 2020 | | | 75 | | | | 140,871 | | | | (17 | ) | | | (125,760 | ) | | | 15,169 | | | | | | | | | | | | | | | | | | | | | | | Net loss | | | 0 | | | | 0 | | | | 0 | | | | (9,196 | ) | | | (9,196 | ) | Other comprehensive loss | | | 0 | | | | 0 | | | | (23 | ) | | | 0 | | | | (23 | ) | Total comprehensive loss | | | 0 | | | | 0 | | | | (23 | ) | | | (9,196 | ) | | | (9,219 | ) | Exercise of options | | | (* | ) | | | (* | ) | | | 0 | | | | 0 | | | | (* | ) | | | Share-based compensation | | | 0 | | | | 1,322 | | | | 0 | | | | 0 | | | | 1,322 | | Balance as of December 31, 2020 | | | 75 | | | | 142,193 | | | | (40 | ) | | | (134,956 | ) | | | 7,272 | | | | | | | | | | | | | | | | | | | | | | | Balance as of January 1, 2019 | | | 75 | | | | 139,637 | | | | (25 | ) | | | (130,715 | ) | | | 8,972 | | | | | | | | | | | | | | | | | | | | | | | Net profit | | | 0 | | | | 0 | | | | 0 | | | | 4,955 | | | | 4,955 | | Other comprehensive income | | | 0 | | | | 0 | | | | 8 | | | | 0 | | | | 8 | | Total comprehensive income | | | 0 | | | | 0 | | | | 8 | | | | 4,955 | | | | 4,963 | | Exercise of options | | | (* | ) | | | (* | ) | | | 0 | | | | 0 | | | | (* | ) | | Share-based compensation | | | 0 | | | | 1,234 | | | | 0 | | | | 0 | | | | 1,234 | | Balance as of December 31, 2019 | | | 75 | | | | 140,871 | | | | (17 | ) | | | (125,760 | ) | | | 15,169 | |
| | | Share capital | | | Share premium | | | Foreign currency translation reserve | | | Accumulated deficit | | | Total equity (deficit) | | | | | | | | | | | | | | | | | | | Balance as of January 1, 2023 | | | 143 | | | | 178,882 | | | | (5 | ) | | | (168,106 | ) | | | 10,914 | | | | | | | | | | | | | | | | | | | | | | | | Net loss | | | - | | | | - | | | | - | | | | (6,716 | ) | | | (6,716 | ) | Other comprehensive loss | | | - | | | | - | | | | (13 | ) | | | - | | | | (13 | ) | Total comprehensive loss | | | - | | | | - | | | | (13 | ) | | | (6,716 | ) | | | (6,729 | ) | Exercise of RSU | | | 1 | | | | - | | | | - | | | | - | | | | 1 | | Issuance of ordinary shares, net of issuance expenses (see note 19c) | | | 40 | | | | 25,429 | | | | - | | | | - | | | | 25,469 | | Share-based compensation | | | - | | | | 1,940 | | | | - | | | | - | | | | 1,940 | | Balance as of December 31, 2023 | | | 184 | | | | 206,251 | | | | (18 | ) | | | (174,822 | ) | | | 31,595 | | | | | | | | | | | | | | | | | | | | | | | | Balance as of January 1, 2022 | | | 75 | | | | 143,869 | | | | (19 | ) | | | (148,507 | ) | | | (4,582 | ) | | | | | | | | | | | | | | | | | | | | | | | Net loss | | | - | | | | - | | | | - | | | | (19,599 | ) | | | (19,599 | ) | Other comprehensive income | | | - | | | | - | | | | 14 | | | | - | | | | 14 | | Total comprehensive loss | | | - | | | | - | | | | 14 | | | | (19,599 | ) | | | (19,585 | ) | Exercise of options | | | (* | ) | | | (* | ) | | | - | | | | - | | | | (* | ) | Issuance of ordinary shares, net of issuance expenses (see note 19c) | | | 40 | | | | 17,389 | | | | - | | | | - | | | | 17,429 | | Exercise of pre-funded warrants (see note 19c) | | | 28 | | | | 15,678 | | | | - | | | | - | | | | 15,706 | | Share-based compensation | | | - | | | | 1,946 | | | | - | | | | - | | | | 1,946 | | Balance as of December 31, 2022 | | | 143 | | | | 178,882 | | | | (5 | ) | | | (168,106 | ) | | | 10,914 | | | | | | | | | | | | | | | | | | | | | | | | Balance as of January 1, 2021 | | | 75 | | | | 142,193 | | | | (40 | ) | | | (134,956 | ) | | | 7,272 | | | | | | | | | | | | | | | | | | | | | | | | Net loss | | | - | | | | - | | | | - | | | | (13,551 | ) | | | (13,551 | ) | Other comprehensive income | | | - | | | | - | | | | 21 | | | | - | | | | 21 | | Total comprehensive loss | | | - | | | | - | | | | 21 | | | | (13,551 | ) | | | (13,530 | ) | Exercise of options | | | (* | ) | | | 3 | | | | - | | | | - | | | | 3 | | Share-based compensation | | | - | | | | 1,673 | | | | - | | | | - | | | | 1,673 | | | | | | | | | | | | | | | | | | | | | | | | Balance as of December 31, 2021 | | | 75 | | | | 143,869 | | | | (19 | ) | | | (148,507 | ) | | | (4,582 | ) |
* Represents an amount lower than $1.
The accompanying notes are an integral part of the consolidated financial statements. MEDIWOUND LTD. AND ITS SUBSIDIARIES Consolidated Statements of Cash Flows
U.S. dollars in thousands | | Year ended December 31, | | | | 2021 | | | 2020 | | | 2019 | | | | | | | | | | | | | | | Cash flows from operating activities: | | | | | | | | | | | | | Profit (loss) for the year | | | (13,551 | ) | | | (9,196 | ) | | | 4,955 | | | | | | | | | | | | | | | Adjustments to reconcile net profit (loss) to net cash provided by (used in) continuing operating activities: | | | | | | | | | | | | | | | | | | | | | | | | | | Adjustments to profit and loss items: | | | | | | | | | | | | | Profit from discontinued operation | | | 0 | | | | (80 | ) | | | (2,889 | ) | Depreciation and amortization | | | 1,238 | | | | 1,090 | | | | 1,149 | | Share-based compensation | | | 1,673 | | | | 1,322 | | | | 1,234 | | Revaluation of liabilities in respect of IIA grants | | | 919 | | | | 828 | | | | (392 | ) | Revaluation of liabilities in respect of purchase of shares | | | 590 | | | | (433 | ) | | | 1,690 | | Revaluation of lease liabilities | | | 188 | | | | 305 | | | | 340 | | Increase (decrease) in severance pay liability, net | | | 13 | | | | 33 | | | | (105 | ) | Net financing income | | | (11 | ) | | | (297 | ) | | | (434 | ) | Un-realized foreign currency gain | | | (137 | ) | | | (211 | ) | | | (152 | ) | | | | | | | | | | | | | | | | | 4,473 | | | | 2,557 | | | | 441 | | Changes in asset and liability items: | | | | | | | | | | | | | Decrease (increase) in trade receivables | | | 929 | | | | 1,386 | | | | (3,553 | ) | Decrease in inventories | | | 257 | | | | 141 | | | | 67 | | Decrease (increase) in other receivables | | | (763 | ) | | | (13 | ) | | | 6,376 | | Increase (decrease) in trade payables and accrued expenses | | | 1,723 | | | | (1,096 | ) | | | 1,355 | | Increase (decrease) in other payables and deferred revenues | | | (1,984 | ) | | | (479 | ) | | | 247 | | | | | | | | | | | | | | | | | | 162 | | | | (61 | ) | | | 4,492 | | | | | | | | | | | | | | | Net cash provided by (used in) continuing operating activities | | | (8,916 | ) | | | (6,700 | ) | | | 9,888 | | | | | | | | | | | | | | | Net cash used in discontinued operating activities | | | 0 | | | | (195 | ) | | | (1,599 | ) | | | | | | | | | | | | | | Net cash provided by (used in) operating activities | | | (8,916 | ) | | | (6,895 | ) | | | 8,289 | |
| | | Year ended December 31, | | | | | 2023 | | | 2022 | | | 2021 | | Cash flows from operating activities: | | | | | | | | | | Net loss | | | (6,716 | ) | | | (19,599 | ) | | | (13,551 | ) | | | | | | | | | | | | | | | Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | | | | | | | | | | | | | | | | | | | | | | Adjustments to profit and loss items: | | | | | | | | | | | | | Depreciation and amortization | | | 1,303 | | | | 1,272 | | | | 1,238 | | Share-based compensation | | | 1,940 | | | | 1,946 | | | | 1,673 | | Revaluation of warrants accounted at fair value | | | (8,310 | ) | | | 8,977 | | | | - | | Issuance expenses of warrants through profit and loss | | | - | | | | 1,911 | | | | - | | Revaluation of liabilities in respect of IIA grants | | | 427 | | | | (132 | ) | | | 919 | | Revaluation of liabilities in respect of TEVA | | | 468 | | | | 533 | | | | 590 | | Financing income and exchange differences of lease liability | | | 257 | | | | (109 | ) | | | 188 | | Increase in severance pay liability, net | | | 83 | | | | 109 | | | | 13 | | Other income | | | (211 | ) | | | - | | | | - | | Financial income, net | | | (2,231 | ) | | | (74 | ) | | | (11 | ) | Un-realized foreign currency loss (gain) | | | 189 | | | | 525 | | | | (137 | ) | | | | | | | | | | | | | | | | | | | (6,085 | ) | | | 14,958 | | | | 4,473 | | Changes in asset and liability items: | | | | | | | | | | | | | Decrease (increase) in trade receivables | | | 5,658 | | | | (7,582 | ) | | | 929 | | Decrease (increase) in inventories | | | (906 | ) | | | (721 | ) | | | 257 | | Decrease (increase) in other receivables | | | (894 | ) | | | 364 | | | | (763 | ) | Increase (decrease) in trade payables and accrued expenses | | | (594 | ) | | | 414 | | | | 1,723 | | Increase (decrease) in other payables | | | (928 | ) | | | 281 | | | | (1,984 | ) | | | | | | | | | | | | | | | | | | | 2,336 | | | | (7,244 | ) | | | 162 | | | | | | | | | | | | | | | | Net cash used in operating activities | | | (10,465 | ) | | | (11,885 | ) | | | (8,916 | ) |
The accompanying notes are an integral part of the consolidated financial statements. MEDIWOUND LTD. AND ITS SUBSIDIARIES Consolidated Statements of Cash Flows
U.S. dollars in thousands | | Year ended December 31, | | | | 2021 | | | 2020 | | | 2019 | | Cash flows from investing activities: | | | | | | | | | | | | | | | | | | | | | | | | | | Purchase of property and equipment | | | (489 | ) | | | (923 | ) | | | (792 | ) | Interest received | | | 35 | | | | 274 | | | | 184 | | Proceeds from (investments in) short term bank deposits, net | | | 4,002 | | | | 18,034 | | | | (5,050 | ) | | | | | | | | | | | | | | Net cash provided by (used in) continuing investing activities | | | 3,548 | | | | 17,385 | | | | (5,658 | ) | | | | | | | | | | | | | | Net cash used in discontinued investing activities | | | 0 | | | | 0 | | | | (1,239 | ) | | | | | | | | | | | | | | Net cash provided by (used in) investing activities | | | 3,548 | | | | 17,385 | | | | (6,897 | ) | | | | | | | | | | | | | | Cash flows from financing activities: | | | | | | | | | | | | | | | | | | | | | | | | | | Repayment of leases liabilities | | | (693 | ) | | | (508 | ) | | | (630 | ) | Proceeds from exercise of options | | | 3 | | | | (* | ) | | | (* | ) | Repayment of IIA grants, net | | | (360 | ) | | | (121 | ) | | | (376 | ) | | | | | | | | | | | | | | Net cash used in continuing financing activities | | | (1,050 | ) | | | (629 | ) | | | (1,006 | ) | | | | | | | | | | | | | | Exchange rate differences on cash and cash equivalent balances | | | 88 | | | | 273 | | | | 140 | | | | | | | | | | | | | | | Increase (decrease) in cash and cash equivalents from continuing activities | | | (6,330 | ) | | | 10,329 | | | | 3,364 | | Decrease in cash and cash equivalents from discontinued activities | | | 0 | | | | (195 | ) | | | (2,838 | ) | Balance of cash and cash equivalents at the beginning of the year | | | 17,376 | | | | 7,242 | | | | 6,716 | | | | | | | | | | | | | | | Balance of cash and cash equivalents at the end of the year | | | 11,046 | | | | 17,376 | | | | 7,242 | | | | | | | | | | | | | | | Supplement disclosure of Non-cash transactions: | | | | | | | | | | | | | ROU asset, net recognized with corresponding lease liability | | | 155 | | | | 261 | | | | 209 | | Exercise of RSU’s | | | 147 | | | | 147 | | | | 97 | |
| | | Year ended December 31, | | | | | 2023 | | | 2022 | | | 2021 | | Cash flows from investing activities: | | | | | | | | | | | | | | | | | | | | | Purchase of property and equipment | | | (6,464 | ) | | | (555 | ) | | | (489 | ) | Interest received | | | 1,947 | | | | 74 | | | | 35 | | Proceeds from (investment in) short term bank deposits, net | | | (29,804 | ) | | | - | | | | 4,002 | | | | | | | | | | | | | | | | Net cash (used in) provided by investing activities | | | (34,321 | ) | | | (481 | ) | | | 3,548 | | | | | | | | | | | | | | | | Cash flows from financing activities: | | | | | | | | | | | | | | | | | | | | | | | | | | | Repayment of leases liabilities | | | (778 | ) | | | (701 | ) | | | (693 | ) | Proceeds from exercise of options | | | - | | | | (* | ) | | | 3 | | Proceeds from exercise of pre-funded warrants | | | - | | | | 10 | | | | - | | Proceeds from issuance of shares and warrants, net | | | 24,909 | | | | 38,380 | | | | - | | Repayment of IIA grants, net | | | (380 | ) | | | (258 | ) | | | (360 | ) | Repayment of liabilities in respect of TEVA | | | (834 | ) | | | (1,667 | ) | | | - | | | | | | | | | | | | | | | | Net cash provided by (used in) financing activities | | | 22,917 | | | | 35,764 | | | | (1,050 | ) | | | | | | | | | | | | | | | Exchange rate differences on cash and cash equivalent balances | | | (160 | ) | | | (549 | ) | | | 88 | | | | | | | | | | | | | | | | Increase (decrease) in cash and cash equivalents | | | (22,029 | ) | | | 22,849 | | | | (6,330 | ) | | | | | | | | | | | | | | | Balance of cash and cash equivalents at the beginning of the year | | | 33,895 | | | | 11,046 | | | | 17,376 | | | | | | | | | | | | | | | | Balance of cash and cash equivalents at the end of the year | | | 11,866 | | | | 33,895 | | | | 11,046 | | | | | | | | | | | | | | | | Supplement disclosure of non-cash transactions: | | | | | | | | | | | | | ROU asset, net recognized with corresponding lease liability | | | 6,825 | | | | 117 | | | | 155 | | Purchase of property and equipment | | | (1,011 | ) | | | - | | | | - | |
* Represents an amount lower than $1.
The accompanying notes are an integral part of the consolidated financial statements. MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) | | a. | Description of the Company and its operations: |
MediWound Ltd. Was incorporated in Israel.Israel in January 2000. The Company which is located in Yavne, Israel (The "Company" or "MediWound"), is biopharmaceutical company that develops, manufactures and commercializes novel, cost effective, bio-therapeutic, non-surgical solutions for tissue repair and regeneration. The Company’s strategy leverages its breakthrough enzymatic technology platform into diversified portfolio of biotherapeutics across multiple indications to pioneer solutions for unmet medical needs. The Company’s current portfolio is focused on next-generation bio-activeprotein-based therapies for burn andcare, wound care and tissue repair. The Company's first innovative biopharmaceutical product, NexoBrid,Nexobrid, has received in December 2022, an approval from the U.S. Food and Drug Administration (“FDA”) and marketing approval in each of India, Switzerland and Japan. In addition, it has a marketing authorization from the European Medicines Agency ("EMA"(“EMA”) as well as the Israeli, Argentinean, South-Korean, Russian, Taiwanese, Ukrainian, United Arab Emirates, Chilean and Peruvian Ministries of Health,regulatory agencies in other international markets for removal of dead or damaged tissue, known as eschar, in adults with deep partial and full thickness thermal burns. The Company commercialize Nexobrid globally through multiple sales channels. The Company sells NexoBridNexobrid to burn centers in the European Union, United Kingdom Norway, Switzerland and Israel, primarily through its commercial organizations while establishing additionalorganizations. The Company has established local distribution channels to extend its outreach in the European Union. In othermultiple international markets, the Company sells NexoBrid throughfocusing on Asia Pacific, EMEA, CEE and LATAM, which local distributors which are also responsible for obtaining the local marketing authorization within the relevant territory. territories. In the United States, the Company entered into an exclusive license and supply agreements with Vericel Corporation (“Vericel”) to commercialize NexoBridNexobrid in North America upon FDA'sFDA approval. On September 21, 2023, the Company announced the U.S. commercial availability of Nexobrid® for the removal of eschar in adults with deep partial- and/or full-thickness thermal burns. On September 20, 2022, The Company announced that EMA has validated for review the Type II Variation to expand the currently approved indication for Nexobrid (removal of eschar in adults with deep partial-and full-thickness thermal burn wounds) into the pediatric population. The Company’s second investigational innovativenext-generation enzymatic therapy product, EscharEx, a topical biological drug being developed for debridement of chronic and other hard-to-heal wounds, is currently under a U.S. phase 2 study and in January 2022, a positive topline results were announced from this study. Patient follow-up is ongoing and additional data, including secondary and exploratory endpoints as well as additional safety measurements, will allow further evaluation of clinical benefits, in the second quarter of 2022. The third clinical-stage innovative product candidate, MW005,On November 21, 2023 the Company announced in relate to EscharEx clinical trial that an updated Phase III protocol will be submitted during first half of 2024 in accordance with recent discussions with the U.S. FDA and EMA. Study initiation is a topical biological drug candidate forexpected to commence in the treatmentsecond half of non-melanoma skin cancers. A U.S. phase 1/2 study of MW005 for the treatment of low-risk basal cell carcinoma (BCC) was initiated in July 2021, and an investigator-initiated phase II trial of MW005 in non-melanoma skin cancer is being conducted in parallel in Israel.2024.
MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) | | b. | The Company's securities are listed for trading on NASDAQ since March 2014. In March, 2022, the Company completed a follow-on public offering. A total of 5,208,333 new ordinary shares were issued at a public offering price of $1.92 per share . The gross proceeds before deducting underwriting discounts and commissions and offering expenses, were approximately $10 million. (see also Note 26). |
On November 28, 2022, the Company’s shareholders general meeting approved a reverse stock split. Following that approval, on December 5, 2022, the Company’s board of directors approved a reverse stock split, in a range of 1-for-7 ratio. The reverse split went effective on December 20, 2022. (see also Note 19b). During March, September and October 2022, the Company completed a series of capital public and private offerings. The gross proceeds before deducting underwriting discounts and commissions and offering expenses, were approximately $41,700. The net proceeds were approximately $37,292 (see also Note 19). On February 7, 2023, the Company completed a registered direct offering. A total of 1,964,286 new ordinary shares were issued in consideration to offering price of $14 per share. The gross proceeds were $27,500, before deducting commissions and other offering expenses. The net proceeds were approximately $25,469. | | c. | The Company has three wholly owned subsidiaries: MediWound Germany GmbH, acting as Europe (“EU”) marketing authorization holder and EU sales and marketing arm, and MediWound UK Limited and MediWound US, Inc. which are currently inactive companies. |
| d. | In October 2023, Israel was attacked by a terrorist organization and entered a state of war. As of the date of these consolidated financial statements, the war in Israel is ongoing and continues to evolve. The company’s head quarter, manufacturing and R&D facilities are located in Israel. Currently, such activities in Israel remain largely unaffected. During the year ended December 31, 2023, the impact of this war on the company’s results of operations and financial condition was immaterial. |
| e. | The accompanying consolidated financial statements have been prepared on a basis which assumes that the Company will continue as a going concern. From inception to December 31, 2023, the Company has incurred cash outflows from operations, losses from operations, and has an accumulated deficit of $174.8 million. |
The Company believes that its existing cash and cash equivalents, short-term and restricted bank deposits of $41.7 million as of December 31, 2023, will be sufficient to fund its operations and capital expenditure for at least twelve months from the date of issuance of these consolidated financial statements. MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) The Company awarded two contracts with the U.S. Biomedical Advanced Research and Development Authority ("BARDA") valued at up to $168,000 for the advancement of the development, manufacturing and emergency readiness for NexoBrid deployment as well as the procurement of NexoBrid as a medical countermeasure as part of BARDA preparedness for mass casualty events. In February 2022 BARDA has expanded its awarded contract providing supplemental funding of approxemently $9,000 to support the NexoBrid BLA resubmission to the FDA and the continuous expanded access program.
| d. | On June 29, 2021, the Company received a Complete Response Letter (CRL) from the U.S. Food and Drug Administration (FDA) regarding its Biologics License Application (BLA) seeking approval of NexoBrid for eschar removal (debridement) in adults with deep partial-thickness and/or full-thickness thermal burns.
|
The FDA communicated that it had completed its review of the BLA, as amended, and has determined that the application cannot be approved in its present form. The FDA identified issues related to the Chemistry, Manufacturing and Controls (“CMC”) section of the BLA and requested additional CMC information. The FDA acknowledged receipt of several CMC amendments, submitted by the Company in response to the CMC information requests, which were not reviewed yet by the FDA.
The FDA also stated that an inspection of NexoBrid's manufacturing facilities in Israel and Taiwan, are required before the FDA can approve the BLA, but it was unable to conduct the required inspections during the current review cycle due to COVID-19 related travel restrictions. The FDA stated that it will continue to monitor the public health situation as well as travel restrictions and is actively working to define an approach for scheduling outstanding inspections. In addition, the CRL cited certain observations identified during good clinical practice (GCP) inspections related to the U.S. Phase 3 study (DETECT), and requested the Company to provide its perspective on the potential impact, if any, of these observations on the efficacy findings in the study. The FDA also requested to provide a safety update as part of its BLA resubmission, although there were no safety issues raised in the CRL.
Following a productive Type A meeting with the FDA, the Company gained clarity on a path forward for resubmission of its NexoBrid BLA, which is anticipated in mid-2022. In addition, the FDA’s facility inspection schedule which has been affected by COVID-19-related travel restrictions, is required before the FDA can approve the NexoBrid BLA.
Consequently, the Company expects the timing of the potential approval of NexoBrid to be impacted.
| e. | The Company addressed the challenges associated with the ongoing COVID-19 pandemic during the year ended 2020 and 2021, while prioritizing the health and safety of its workforce and maintaining operational efficiency and flexibility.
|
MEDIWOUND LTD. AND ITS SUBSIDIARIES| Note 2: | Basis of Preparation of the Consolidated Financial Statements |
Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data)
Note 2: Basis of Preparation of the Consolidated Financial Statements
| | a. | Statement of compliance with International Financial Reporting Standards |
These financial statements have been prepared in accordance with International Financial Reporting Standards ("IFRS") as issued by the International Accounting Standards Board ("IASB"). These consolidated financial statements were approved by the board of directors on March 17, 2022.21, 2024. | | b. | Functional currency, reporting currency and foreign currency: |
| | 1. | Functional currency and reporting currency: |
The reporting currency of the financial statements is the U.S. dollar. The Company determines the functional currency based on the currency in which it primarily generates and expends cash. The Company determined that its functional currency is the U.S. dollar since most of the Company's expensesrevenues are in U.S. dollars and the economic environment in which the Company operates in and performs its transactions is mostly affected by the U.S dollar. A certain portion of the Company's costs are denominated in NIS mainly due to payroll and related benefit costs incurred in Israel. To further support the Company's determination, the Company has analyzed the currency in which funds from financing activities are generated or held and the currency in which receipts from operating activities are usually retained. In this respect, funds from financing activities were principally derived from significant funds raising in U.S. dollars and U.S governmental funds. The Company operates and plans its activities in U.S. dollars and accordingly its periodic budgets and internal management reports are prepared and monitored using the U.S. dollar as the primary currency and provides the basis for the determination of share-based compensation. The functional currency of the Company's subsidiary in Germany has been determined to be its local currency - the EURO. Assets and liabilities of this subsidiary are translated at year end exchange rates and its statement of operations items are translated using the averegaeaverage exchange rates at the quarter of which those items are recognized. Such translation adjustments are recorded as a separate component of accumulated other comprehensive income (loss) in shareholders' equity (deficit). | | 2. | Transactions, assets and liabilities in foreign currency: |
Transactions denominated in foreign currency are recorded upon initial recognition at the exchange rate on the date of the transaction. After initial recognition, monetary assets and liabilities denominated in foreign currency are translated at the end of each reporting period into the functional currency at the exchange rate at that date. Exchange differences are recognized in profit or loss. MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) Note 2: Basis of Preparation of the Consolidated Financial Statements (Cont.)
| Note 2: | Basis of Preparation of the Consolidated Financial Statements (Cont.) |
| | c. | Use of estimates and judgments |
The preparation of the financial statements in conformity with IFRSs requires management to make judgments, estimates and assumptions that have an effect onaffect the application of the accounting policies and on the reported amounts of assets, liabilities, income and expenses. Actual results may differ from these estimates. Discussed below are the key assumptions made in the financial statements concerning uncertainties at the end of the reporting period and the critical estimates computed by the Company that may result in a material adjustment to the carrying amounts of assets and liabilities within the next financial year. | | • | Determining the fair value of share basedshare-based compensation to employees and directors: |
The fair value of share basedshare-based compensation to employees and directors is determined using the binomial option pricing models.Themodels. The assumptions used in the models include the expected volatility, early exercise factor, expected dividend and risk-free interest rate. | | • | Liabilities in respect to IIA grants: |
Government grants received from the IIA are recognized as a liability if future economic benefits are expected from the research and development activity that will result in royalty‑bearing sales. As the contingent liability is calculated based on future royalty-bearing sales, there is uncertainty regarding the estimated future cash flows and the estimated discount rate used to measure the amortized cost of the liability. | • | Fair value estimations of warrants |
The Company completed financing transactions in which it issued shares and warrants to purchase additional shares. The fair value of the warrants, which are not traded on an active market, is determined by using valuation techniques. These valuation techniques maximize the use of observable market data where it is available and rely as little as possible on entity specific estimates. MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) | Note 3: | SignificantMaterial Accounting Policies
|
The accounting policies set out below have been consistently applied for all periods presented in these consolidated financial statements: | | a. | Basis of consolidation: |
Consolidated financial statements include the financial statements of companies that the Company controls (subsidiaries). Control is achieved when the Company is exposed, or has rights, to variable returns from its investment with the investee and has the ability to affect those returns through its power over the investee. The financial statements of the Company and its subsidiaries are prepared as of the same dates and periods. The consolidated financial statements are prepared using uniform accounting policies by all entities in the Group. Significant intercompany balances and transactions and gains or losses resulting from intercompany transactions are eliminated in full in the consolidated financial statements. Cash equivalents are considered as highly liquid investments, including unrestricted short‑term bank deposits with an original maturity of three months or less from the date of deposit.
MEDIWOUND LTD. AND ITS SUBSIDIARIES
Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data)
Note 3: | Significant Accounting Policies (Cont.)
|
| c. | Short-term bank deposits:
|
Short-term bank deposits have a maturity of more than three months, but less than one year, from the deposit date.
Inventories are measured at the lower of cost and net realizable value. Net realizable value is the estimated selling price in the ordinary course of business less the estimated costs of completion and the estimated selling costs. The Company periodically evaluates the condition and age of inventories and makes provisions for slow moving inventories accordingly. Cost of inventories is determined as follows: Raw materials | - | At cost of purchase using the first-in, first-out method. | Finished goods | - | On the basis of average standard costs (which approximates actual cost on a weighted average basis) including materials, labor and other direct and indirect manufacturing costs based on practical capacity. |
| | c. | Property, plant and equipment, net: |
Property, plant and equipment are measured at cost, including directly attributable costs, less accumulated depreciation and accumulated impairment losses. Cost includes spare parts and auxiliary equipment that are used in connection with the plant and equipment. Depreciation is calculated on a straight‑line basis over the useful life of the assets at annual rates as follows: | | e.%
| Office furniture | | 7-10 | Manufacturing machinery and lab equipment | | 15-33 | Computers | | 33 | Leasehold improvements | | See below |
MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) | Note 3: | Material Accounting Policies (Cont.) |
Leasehold improvements are depreciated on a straight‑line basis over the shorter of the lease term (including the renewal option held by the Company which is expected to be exercised) and the expected life of the improvement. The useful life, depreciation method and residual value of an asset are reviewed at least each year-end and any changes are accounted for prospectively as a change in accounting estimate. | d. | Liability in respect of Israeli Innovation Authority ("IIA"): |
Grants from the IIA in respect of research and development projects are accounted for as forgivable loans according to IAS 20. Grants received from the IIA are recognized as a liability according to their fair value on the date of their receipt, unless on that date it is reasonably certain that the amount received will not be refunded. If future economic benefits are expected from the project that will result in royalty-bearing revenues from sale of products it will be treated as a contingent liability. At the end of each reporting period, the Company evaluates whether there is reasonable assurance that the liability recognized, in whole or in part, will not be repaid based on its best estimate of future sales and any changes in the present value of the cash flows discounted at the original interest rate of the grant are recognized in profit or loss. The difference between the amount received and the fair value on the date of receiving the grant is recognized as a deduction of research and development expenses. MEDIWOUND LTD. AND ITS SUBSIDIARIES
Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data)
Note 3: | Significant Accounting Policies (Cont.)
|
The Company accounts for a contract as a lease when the contract terms convey the right to control the use of an identified asset for a period of time in exchange for consideration. For leases in which the Company is the lessee, the Company recognizes on the commencement date of the lease a right-of-use (“ROU”) asset and a lease liability, excluding leases whose term is up to 12 months and leases for which the underlying asset is of low value. For these excluded leases, the Company has elected to recognize the lease payments as an expense in profit or loss on a straight-line basis over the lease term. In measuring the lease liability, the Company has elected to apply the practical expedient in the Standard and does not separate the lease components from the non-lease components (such as management and maintenance services, etc.) included in a single contract.
Following are the amortization periods of the ROU assets by class of underlying asset: | | | Years | | Motor vehicles | | 3 | | Buildings and equipment | | 5-86-15
| |
MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) | Note 3: | | | Material Accounting Policies (Cont.) |
The Company tests for impairment of the ROU asset whenever there are indications of impairment pursuant to the provisions of IAS 36. | | • | Variable lease payments that depend on an index: |
On the commencement date, the Company uses the index rate prevailing on the commencement date to calculate the future lease payments. For leases in which the Company is the lessee, the aggregate changes in future lease payments resulting from a change in the index are discounted (without a change in the discount rate applicable to the lease liability) and recorded as an adjustment of the lease liability and the ROU assets, only when there is a change in the cash flows resulting from the change in the index (that is, when the adjustment to the lease payments takes effect). | | • | Lease extension and termination options: |
A non-cancelable lease term includes both the periods covered by an option to extend the lease when it is reasonably certain that the extension option will be exercised and the periods covered by a lease termination option when it is reasonably certain that the termination option will not be exercised. In the event of any change in the expected exercise of the lease extension option or in the expected non-exercise of the lease termination option, the Company remeasures the lease liability based on the revised lease term using a revised discount rate as of the date of the change in expectations. The total change is recognized in the carrying amount of the ROU asset until it is reduced to zero, and any further reductions are recognized in profit or loss.
MEDIWOUND LTD. AND ITS SUBSIDIARIES
Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data)
Note 3: | Significant Accounting Policies (Cont.)
|
If a lease modification does not reduce the scope of the lease and does not result in a separate lease, the Company remeasures the lease liability based on the modified lease terms using a revised discount rate as of the modification date and records the change in the lease liability as an adjustment to the ROU asset.
If a lease modification reduces the scope of the lease, the Company recognizes a gain or loss arising from the partial or full reduction of the carrying amount of the ROU asset and the lease liability. The Company subsequently remeasures the carrying amount of the lease liability according to the revised lease terms, at the revised discount rate as of the modification date and records the change in the lease liability as an adjustment to the ROU asset.
Operating leases:
Leases in which substantially all the risks and rewards of ownership of the leased asset are not transferred to the Group are classified as operating leases. Lease payments are recognized as an expense in profit or loss on a straight-line basis over the lease term.
| g. | Property, plant and equipment, net:
|
Property, plant and equipment are measured at cost, including directly attributable costs, less accumulated depreciation, accumulated impairment losses and excluding day-to-day servicing expenses. Cost includes spare parts and auxiliary equipment that are used in connection with the plant and equipment.
Depreciation is calculated on a straight‑line basis over the useful life of the assets at annual rates as follows:
| | %
| Office furniture
| | 6-15
| Manufacturing machinery and lab equipment
| | 15-33
| Computers
| | 33
| Leasehold improvements
| | See below
|
Leasehold improvements are depreciated on a straight‑line basis over the shorter of the lease term (including the renewal option held by the Company which is expected to be exercised) and the expected life of the improvement.
The useful life, depreciation method and residual value of an asset are reviewed at least each year-end and any changes are accounted for prospectively as a change in accounting estimate.
| | h. | Intangible assets, net:
|
Separately acquired intangible assets with finite useful life are measured on initial recognition at cost.
MEDIWOUND LTD. AND ITS SUBSIDIARIES
Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data)
Note 3: | Significant Accounting Policies (Cont.)
|
Intangible assets are amortized over their useful life using the straight‑line method beginning in the period in which the intangible assets generates net cash inflows to the Company. The useful life is over the length of the patent or knowledge life. The intangible assets are reviewed for impairment at each reporting date until they begin generating net cash inflows and subsequently whenever there is an indication that the asset may be impaired.
| i.f. | Revenues recognition: |
The Company recognizes revenue when the customer obtains control over the promised goods or services. The revenue is measured according to the amount of the consideration to which the Company expects to be entitled in exchange for the goods or services promised to the customer, other than amounts collected for third parties. To determine revenue recognition for arrangements the Company evaluates the following criteria’s, which are within the scope of IFRS 15, it performs the following five steps: (i) identify the contract(s) with a customer, (ii) identify the performance obligations in the contract, (iii) determine the transaction price, (iv) allocate the transaction price to the performance obligations within the contract and (v) recognize revenue when (or as) the Company satisfies a performance obligation. The Company only applies the five-step model to contracts when it determines that it is probable it will collect the consideration it is entitled to in exchange for the goods or services it transfers to the customer.
Performance obligations are promises commitments in a contract to transfer a distinct good or service to the customer that (i) the customer can benefit from on its own or together with other readily available resources, and (ii) is separately identifiable from other promises commitments in the contract. Goods or services that are not individually distinct performance obligations are combined with other promised commitment goods or services until such combined group of promises commitments meet the requirements of a performance obligation.
The Company determines transaction price based on the amount of consideration the Company expects to receive for transferring the promised goods or services in the contract. Consideration may be fixed, variable, or a combination of both. At contract inception for arrangements that include variable consideration, the Company estimates the probability and extent of consideration it expects to receive under the contract utilizing either the most likely amount method or expected amount method, whichever best estimates the amount expected to be received. The Company then considers any constraints on the variable consideration and includes in the transaction price variable consideration to the extent it is probable that a significant reversal in the amount of cumulative revenue recognized will not occur when the uncertainty associated with the variable consideration is subsequently resolved. The Company then allocates the transaction price to each performance obligation based on the relative standalone selling price and recognizes as revenue the amount of the transaction price that is allocated to the respective performance obligation when (or as) control is transferred to the customer and the performance obligation is satisfied. MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) | Note 3: | SignificantMaterial Accounting Policies (Cont.)
|
The Company records amounts as accounts receivable when the right to consideration is deemed unconditional. Amounts received, or that are unconditionally due, from a customer prior to transferring goods or services to the customer under the terms of a contract are recognized as deferred revenue. Amounts expected to be recognized as revenue within the 12 months following the balance sheet date are classified as the current portion of deferred revenue. Amounts not expected to be recognized as revenue within the 12 months following the balance sheet date are classified as deferred revenue, net of current portion. The Company’s revenue generatingrevenue-generating arrangements typically include licensing arrangements, which comprise of upfront license fees, milestone payments and/or royalties and products sale arrangements. The promised goods or services in the Company’s licensing arrangements typically consist of a license to the Company’s intellectual property and/or research and development services. The Company may provide customers with options to additional items in such arrangements, which are accounted for separately when the customer elects to exercise such options, unless the option provides a material right to the customer. If a license is determined to be distinct from the other performance obligations identified in the arrangement, the Company recognizes revenue from nonrefundable, up-front fees allocated to the license when the license is transferred to the licensee and the licensee is able to use and benefit from the license. For performance obligations which consist of licenses and other promises, the Company utilizes judgment to assess the nature of the combined performance obligation to determine whether the combined performance obligation is satisfied over time or at a point in time and, if over time, the appropriate method of measuring progress. The Company evaluates the measure of progress each reporting period and, if necessary, adjusts the measure of performance and related revenue recognition. For arrangements that include sales-based royalties, including milestone payments based on the level of sales, where the license is deemed to be the predominant item to which the royalties relate, the Company will recognize revenue at the later of (i) when the related sales occur, or (ii) when the performance obligation to which some or all of the royalty has been allocated has been satisfied (or partially satisfied). In 2019, the Company entered into exclusive license and supply agreements with Vericel to commercialize NexoBridNexobrid in North America (the “Collaboration Agreements”) (see Note 19b)22). The Collaboration Agreements have multiple performance obligations, due to the contract covering multiple phases of the product lifecycle. Under the Vericel license and supply agreements, the Company identified three distinct performance obligations: (1) license rights (2) development services for BLA approval and (3) manufacturing and supply of NexoBrid.Nexobrid. As of the closing date of the agreement the manufacturing and development services were at market value, therefore the upfront payment was fully attributed to the license performance obligation and as such revenues are recognized at the point in time that control of the license is transferred to the customer. MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) | Note 3: | SignificantMaterial Accounting Policies (Cont.)
|
The Company allocated the Collaboration Agreements transaction price to each performance obligation using the best estimate of the standalone selling price of each distinct good or service in the contract.
The Company determined the license to the Intellectual Property ("IP") to be a right to use the IP, which has significant standalone functionality. Since Vericel has sublicensing rights, effective control over the development strategy in the Territory and is also entitled to generate revenues from BARDA procurement prior to BLA approval, the license is a distinct performance obligation and as such revenues are recognized at the point in time that control of the license is transferred to the customer. Since the manufacturing and development services are at market value, then the upfront payment was fully attributed to the license performance obligation. Future milestone payments are considered variable consideration and are subject to the variable consideration constraint, (i.e.i.e. will be recognized once concluded that it is “probable” that a significant reversal of the cumulative revenues recognized under the contract will not occur in future periods when the uncertainty related to the variable considerations are resolved)resolved. (see Note 18b). Therefore, as the milestone payments are not probable, revenues were not recognized in respect to such milestone payments.
AsRevenues from royalties under this agreement arewill be payable based on future commercial sales, which did not occur as of the financial statements date, the Company did not recognize any revenues from royalties.up on an occurrence.
Revenues from the sale of products to Vericel will be recognized when all the significant risks and rewards of ownership of the products have passed to the buyer and the seller no longer retains continuing managerial involvement. The delivery date of the products is usually the date of which ownership passes. Revenues from distribution licensing arrangements: The Company accounts for the bundled license provided to the distributers and related high specialized services as a single performance obligation and consequently recognize revenue using the cost-to-cost method, where the extent of progress towards completion is measured based on the ratio of actual costs incurred to the total estimated costs expected to be incurred upon satisfying such single performance obligation. The revenues from such bundled performance obligation are included within “Revenues from license agreements”. Significant finance components related to such arrangements are recognized as finance expense. Revenues from development services: Revenues from development services are recognized over time, during the period the customer receives and consumes the benefits provided by the Company's performance (see Note 3k).performance. MEDIWOUND LTD. AND ITS SUBSIDIARIES
Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data)
Note 3: | Significant Accounting Policies (Cont.)
|
Revenues from the sale of products: The Company generates revenues from sales of its innovative biopharmaceutical product, NexoBrid,Nexobrid, to burn centers and hospital burn units in Europe, U.S Israel and local distributors in international markets.markets through its commercial organizations and local distributors. Revenues from sale of goods is recognized in profit or loss at the point in time when the control of the goods is transferred to the customer, generally upon delivery of the goods to the customer. The transaction price is the amount of the consideration that is expected to be received based on the contract terms, excluding amounts collected on behalf of third parties (such as taxes).terms. MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) | Note 3: | Material Accounting Policies (Cont.) |
| | j.g. | Research and development expenses: |
Research and development expenses are recognized in profit or loss when incurred. An intangible asset arising from a development project or from the development phase of an internal project is recognized if the Company can demonstrate the technical feasibility of completing the intangible asset so that it will be available for use or sale; the Company's intention to complete the intangible asset and use or sell it; the Company's ability to use or sell the intangible asset; how the intangible asset will generate future economic benefits; the availability of adequate technical, financial and other resources to complete the intangible asset; and the Company's ability to measure reliably the expenditure attributable to the intangible asset during its development. Since the Company's research and development projects are often subject to regulatory approval procedures and other uncertainties, the conditions for the capitalization of costs incurred before receipt of approvals are not normally satisfied and, therefore, research and development expenses are recognized in profit or loss when incurred. Non-royalty bearing funds from BARDA for funding research and development projects were recognized at the time the Company was entitled to such grants on the basis of the related costs incurred.
The participation by BARDA was classified as reimbursement (deduction) of research and development expenses. Starting May 2019, following entrance into the Vericel license and supply agreements, in which Vericel has assumed the effective control over the BARDA contracts, funding by BARDA was classified as Revenues from development services.
| l. | Impairment of non-financial assets:
|
The Company evaluates the need to record an impairment of the carrying amount of non-financial assets whenever events or changes in circumstances indicate that the carrying amount is not recoverable. If the carrying amount of non‑financial assets exceeds their recoverable amount, the assets are reduced to their recoverable amount. The recoverable amount of an asset that does not generate independent cash flows is determined for the cash‑generating unit to which the asset belongs, and is calculated based on the projected cash flows that will be generated by the cash generating unit.
MEDIWOUND LTD. AND ITS SUBSIDIARIES
Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data)
Note 3: | Significant Accounting Policies (Cont.)
|
An impairment loss of an asset, is reversed only if there have been changes in the estimates used to determine the asset's recoverable amount since the last impairment loss was recognized. Reversal of an impairment loss, as above, may not increase the value above the lower of (i) the carrying amount that would have been determined (net of depreciation or amortization) had no impairment loss been recognized for the asset in prior years, and (ii) its recoverable amount.
| m.h. | Financial instruments: |
The accounting policy for financial instruments in accordance with IFRS 9, "Financial Instruments" ("the Standard") is as follows: Financial assets are measured upon initial recognition at fair value plus transaction costs that are directly attributable to the acquisition of the financial assets, except for financial assets measured at fair value through profit or loss in respect of which transaction costs are recorded in profit or loss.
The Company classifies and measures debt instruments in the financial statements based on the following criteria:
| - | The Company's business model for managing financial assets; and
|
| - | The contractual cash flow terms of the financial asset.
|
Impairment of financial assets:
The Company evaluates at the end of each reporting period the loss allowance for financial debt instruments which are not measured at fair value through profit or loss.
The Company has short-term financial assets such as trade receivables in respect of which the Company applies a simplified approach and measures the loss allowance in an amount equal to the lifetime expected credit losses.
An impairment loss on debt instruments measured at amortized cost is recognized in profit or loss with a corresponding loss allowance that is offset from the carrying amount of the financial asset.
| | a) | Financial liabilities measured at amortized cost: |
Financial liabilities are initially recognized at fair value less transaction costs that are directly attributable to the issue of the financial liability. MEDIWOUND LTD. AND ITS SUBSIDIARIES
Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data)
Note 3: | Significant Accounting Policies (Cont.)
|
After initial recognition, the accounting treatment of financial liabilities is based on their classification as follows:
After initial recognition, the Company measures all financial liabilities at amortized cost using the effective interest rate method, except for Financialfinancial liabilities at fair value through profit or loss such as derivatives; | | b) | Financial liabilities measured at fair value through profit or loss: |
At initial recognition, the Company measures financial liabilities that are not measured at amortized cost at fair value. Transaction costs are recognized in profit or loss. After initial recognition, changes in fair value are recognized in profit or loss. MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) | Note 3: | Material Accounting Policies (Cont.) |
Fair value is the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. Fair value measurement is based on the assumption that the transaction will take place in the asset's or the liability's principal market, or in the absence of a principal market, in the most advantageous market. The fair value of an asset or a liability is measured using the assumptions that market participants would use when pricing the asset or liability, assuming that market participants act in their economic best interest. A fair value measurement of a non-financial asset takes into account a market participant's ability to generate economic benefits by using the asset in its highest and best use or by selling it to another market participant that would use the asset in its highest and best use. The Company uses valuation techniques that are appropriate in the circumstances and for which sufficient data are available to measure fair value, maximizing the use of relevant observable inputs and minimizing the use of unobservable inputs. MEDIWOUND LTD. AND ITS SUBSIDIARIES
Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data)
Note 3: | Significant Accounting Policies (Cont.)
|
| | 4.i. | Classification of financial instruments by fair value hierarchy:Warrants:
|
All assets and liabilitiesReceipts in respect of warrants are classified as equity to the extent that they confer the right to purchase a fixed number of shares for whicha fixed exercise price. In the event that the exercise price or the numbers of shares to be issued are not deemed to be fixed (for example, in case of net share settlement provision), or warrants redemption in cash on the occurrence of Fundamental Transaction the warrants are classified as a non-current derivative financial liability. This liability is initially recognized at its fair value on the date the contract is measured or disclosed in the financial statements are categorized within theentered into and subsequently accounted for at fair value hierarchy, described as follows, basedat each reporting date. The fair value changes are charged to non-operating income and expense on the lowest level input that is significant to the fair value measurement as a whole:
Level 1
| -
| quoted prices (unadjusted) in active markets for identical assets or liabilities.
| | | | Level 2
| -
| inputs other than quoted prices included within level 1 that are observable either directly or indirectly.
| | | | Level 3
| - | inputs that are not based on observable market data (valuation techniques which use inputs that are not based on observable market data).
|
| 5. | Offsetting financial instruments:
|
Financial assets and financial liabilities are offset and the net amount is reported in the consolidated statement of financial position if there is a currently enforceable legal rightcomprehensive income or loss. Issuance costs allocable to offsetwarrants are also recorded as non-operating expense on the recognised amounts and there is an intention to settle on a net basis, to realise the assets and settle the liabilities simultaneously.statement of comprehensive income or loss.
A provision in accordance with IAS 37 is recognized when the Company has a present (legal or constructive) obligation as a result of a past event, it is expected to require the use of economic resources to clear the obligation and a reliable estimate has been made. | o. | Short-term employee benefits and severance pay liability, net:
|
The Company has several employee benefit plans:
| 1. | Short-term employee benefits:
|
Short-term employee benefits include salaries, paid annual leave, paid sick leave, recreation and social security contributions and are recognized as expenses as the services are rendered. A liability in respect of a cash bonus is recognized when the Company has a legal or constructive obligation to make such payment as a result of past service rendered by an employee and a reliable estimate of the amount can be made.
| 2. | Post-employment benefits:
|
The Company has liabilities for severance pay for its employees in several of jurisdictions and in Israel.
Post-employment benefit plans in Israel are normally financed by contributions to insurance companies and classified as defined contribution plans or as defined benefit plans. The Company has defined contribution plans for Israeli employees pursuant to the Severance Pay Law into which the Company pays fixed contributions and has no legal or constructive obligation to pay further contributions on account of severance pay if the fund does not hold sufficient amounts to pay all employee benefits relating to employee service in current and prior periods.
MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) | Note 3: | SignificantMaterial Accounting Policies (Cont.)
|
| k. | Short-term employee benefits and severance pay liability, net: |
The Company has several employee benefit plans: | 1. | Short-term employee benefits: |
Short-term employee benefits include salaries, paid annual leave, recreation and social security contributions are recognized as expenses as the services are rendered. | 2. | Post-employment benefits: |
The Company has liabilities for severance pay for its employees in several of jurisdictions and in Israel. The Company recognizes liability for severance pay mainly due to its employees in EU in accordancewithaccordance with local laws. | | p.l. | Share-based compensation: |
Certain Company employees and directors are entitled to remuneration in the form of equity-settled share-based compensation. Equity-settled transactions The cost of equity-settled transactions with employees is measured at the fair value of their equity instruments granted at grant date. The fair value is determined using the binomial option pricing model. The cost of equity-settled transactions is recognized in profit or loss, together with a corresponding increase in equity, during the period which the performance or service conditions are to be satisfied, ending on the date on which the relevant employees become fully entitled to the award. MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) | Note 3: | Material Accounting Policies (Cont.) |
| | q. | Discontinued operation:
|
A discontinued operation is a component of the Company that either has been disposed of or is classified as held for sale. Disposal group to be abandoned meets the criteria for being a discontinued operation at the date of which it ceases to be used. The operating results relating to the discontinued operation are separately presented in the consolidated statements of comprehensive income or loss.
| r.m. | Profit / Loss per share: |
Profit/lossThe Company presents basic and diluted earnings per share (EPS) data for its ordinary shares. Basic EPS is calculated by dividing the profit/profit or loss attributable to ordinary shareholders of the Company shareholders by the weighted average number of outstanding ordinary shares outstanding during the period. Potentialyear, adjusted for treasury shares. Diluted EPS is determined by adjusting the profit or loss attributable to ordinary shareholders of the Company and the weighted average number of ordinary shares are only included when theiroutstanding, after adjustment for treasury shares, for the effects of all dilutive potential ordinary shares, which comprise of warrants, share options and share options granted to employees.
| n. | New standards, amendments to standards and interpretations not yet adopted: |
Amendment to IAS 1, Presentation of Financial Statements: "Disclosure of Accounting Policies” As a result of applying the Amendment, the extent of the accounting policy disclosure provided in the financial statements for 2023 was reduced and adjusted according to the Company’s specific circumstances. | o. | Presentation of Financial Statements: Classification of Liabilities as Current or Non-Current (amendment to IAS 1) |
As a result of applying the Amendment the warrants presented in these financial statements (see note 19c), will be classified as a current liability pursuant to the conversion decreases incomeoption. | Note 4: | Cash and Cash Equivalents |
| | | December 31 | | | | | 2023 | | | 2022 | | | | | | | | | | | Balance in USD | | | 4,151 | | | | 24,475 | | | Balance in other currencies | | | 7,715 | | | | 9,420 | | | | | | | | | | | | | | | | 11,866 | | | | 33,895 |
| Note 5: | Short-term and restricted bank deposits |
| | | December 31 | | | | | 2023 | | | 2022 | | | | | | | | | | | Restricted bank deposits (1) | | | 167 | | | | - | | | USD Bank deposits (2) | | | 29,675 | | | | - | | | | | | | | | | | | | | | | 29,842 | | | | - | |
| (1) | Restricted bank deposits which are primarily used as security for the Company’s office leases. |
| (2) | The USD deposits bear annual interest of 6.26%-6.55% for the period of 91-365 days for 2023. |
MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share or increases lossdata) | | | December 31 | | | | | 2023 | | | 2022 | | | | | | | | | | | Vericel (Note 18b) | | | 236 | | | | 7,500 | | | BARDA (Note 18a) | | | 1,245 | | | | 701 | | | MTEC (Note 18c) | | | 617 | | | | 362 | | | Other trade receivables | | | 1,608 | | | | 771 | | | Less provision for impairment | | | (6 | ) | | | (2 | ) | | | | | | | | | | | | | | | 3,700 | | | | 9,332 | |
| | | December 31, | | | | | 2023 | | | 2022 | | | | | | | | | | Raw materials | | | 995 | | | | 913 | | Finished goods | | | *1,851 | | | | 1,050 | | | | | | | | | | | | | | | | 2,846 | | | | 1,963 | |
*Including write-down of $326 | Note 8: | Other Receivables- Short Term |
| | | December 31, | | | | | 2023 | | | 2022 | | | | | | | | | | | Government authorities | | | 322 | | | | 193 | | | Income receivables | | | 397 | | | | 170 | | | Prepaid expenses and other | | | 722 | | | | 287 | | | | | | | | | | | | | | | | 1,441 | | | | 650 | |
MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) | Note 9: | Other Receivables- Long Term |
| | | December 31, | | | | | 2023 | | | 2022 | | | | | | | | | | | Income receivables | | | 50 | | | | 180 | | | Prepaid expenses | | | 183 | | | | - | | | | | | | | | | | | | | | | 233 | | | | 180 | |
| Note 10: | Property, Plant and Equipment, Net |
| | | Office furniture | | | Manufacturing machinery and lab equipment | | | Computers | | | Leasehold improvements | | | Total | | | Cost | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | Balance as of January 1, 2023 | | | 293 | | | | 5,091 | | | | 166 | | | | 3,279 | | | | 8,829 | | | Additions | | | 25 | | | | 7,312 | | | | 101 | | | | 37 | | | | 7,475 | | | Capitalization of depreciation of ROU assets | | | - | | | | - | | | | - | | | | 59 | | | | 59 | | | Foreign currency translation | | | 1 | | | | - | | | | - | | | | - | | | | 1 | | | | | | | | | | | | | | | | | | | | | | | | | Balance as of December 31, 2023 | | | 319 | | | | 12,403 | | | | 267 | | | | 3,375 | | | | 16,364 | | | | | | | | | | | | | | | | | | | | | | | | | Balance as of January 1, 2022 | | | 257 | | | | 4,764 | | | | 176 | | | | 3,137 | | | | 8,334 | | | Additions | | | 37 | | | | 327 | | | | 49 | | | | 142 | | | | 555 | | | Disposals | | | - | | | | - | | | | (59 | ) | | | - | | | | (59 | ) | | Foreign currency translation | | | (1 | ) | | | - | | | | - | | | | - | | | | (1 | ) | | | | | | | | | | | | | | | | | | | | | | | | Balance as of December 31, 2022 | | | 293 | | | | 5,091 | | | | 166 | | | | 3,279 | | | | 8,829 | | | | | | | | | | | | | | | | | | | | | | | | | Accumulated Depreciation | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | Balance as of January 1, 2023 | | | 157 | | | | 3,818 | | | | 97 | | | | 2,391 | | | | 6,463 | | | Additions | | | 26 | | | | 448 | | | | 58 | | | | 140 | | | | 672 | | | Foreign currency translation | | | 1 | | | | - | | | | - | | | | - | | | | 1 | | | | | | | | | | | | | | | | | | | | | | | | | Balance as of December 31, 2023 | | | 184 | | | | 4,266 | | | | 155 | | | | 2,531 | | | | 7,136 | | | | | | | | | | | | | | | | | | | | | | | | | Balance as of January 1, 2022 | | | 133 | | | | 3,370 | | | | 94 | | | | 2,259 | | | | 5,856 | | | Additions | | | 25 | | | | 448 | | | | 62 | | | | 132 | | | | 667 | | | Disposals | | | - | | | | - | | | | (59 | ) | | | - | | | | (59 | ) | | Foreign currency translation | | | (1 | ) | | | - | | | | - | | | | - | | | | (1 | ) | | | | | | | | | | | | | | | | | | | | | | | | Balance as of December 31, 2022 | | | 157 | | | | 3,818 | | | | 97 | | | | 2,391 | | | | 6,463 | | | | | | | | | | | | | | | | | | | | | | | | | Carrying amounts of all fixed asset items | | | | | | | | | | | | | | | | | | | | | | December 31, 2023 | | | 135 | | | | 8,137 | | | | 112 | | | | 844 | | | | 9,228 | | | December 31, 2022 | | | 136 | | | | 1,273 | | | | 69 | | | | 888 | | | | 2,366 |
MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) The Company's offices and its production facility in Israel are located in a building that the Company leased from continuing operation.its Related party (see Note 24a), in accordance with a sub-lease agreement. The Company subleased approximately 3,000 square meters of laboratory, office and clean rooms space at a monthly rent fee NIS 125 (approximately $35). This sub-lease agreement was amended in October 2021, to extend the period up to October 2025 which was included in the calculation of the lease liability and RoU asset. Furthermore, potential ordinary shares converted duringIn July 2023, the period are includedcompany signed a termination agreement for the sub-lease agreement and signed on a new lease agreement for the same area of approximately 3,000 square meters in diluted loss per share only until the conversion date and from that date in basic loss per share.same building with the owner of the building at a monthly rent fee of NIS 195 (approximately $54). The lease agreement is for 12 years with an option for an additional 3 years. The company estimation is to use the 3 years option
In addition, the Company and its subsidiary have lease agreements for 16 vehicles for the remaining period of 1.75 years on average. | | s.b.
| ReclassificationAmounts recognized in profit or loss and in the statement of cash flows
|
| | | Year ended December 31, | | | | | 2023 | | | 2022 | | | | | | | | | | Interest expense on lease liabilities | | | 274 | | | | 102 | | Depreciation expenses | | | 565 | | | | 537 | | Cash outflow for leases | | | 778 | | | | 701 | |
Certain amounts previously reportedThe Company determined the appropriate interest rate for discounting leases, with the assistance of a third party. The valuation was based on: credit risk, the weighted average term of the leases and other economic variables. A weighted average interest rate in a range of 1% to 8.47% was used to discount future lease payments in the consolidated financial statements have been reclassifiedcalculation of the lease liability on the date of initial application of the standard (IFRS 16).
MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to conform to current year presentation. Such reclassifications did not affect net loss, Changesthe Consolidated Financial Statements
U.S. dollars in Stockholders' Equity or cash flows.thousands (except of share and per share data) | c. | Disclosures in respect of Right- of- Use assets: |
| | | Buildings | | | Motor vehicles | | | Total | | Cost | | | | | | | | | | Balance as of January 1, 2023 | | | 2,341 | | | | 550 | | | | 2,891 | | New leases | | | 6,460 | | | | 407 | | | | 6,867 | | Adjustments for indexation | | | 78 | | | | 3 | | | | 81 | | Disposals | | | (2,368 | ) | | | (308 | ) | | | (2,676 | ) | | | | | | | | | | | | | | | Balance as of December 31, 2023 | | | 6,511 | | | | 652 | | | | 7,163 | | | | | | | | | | | | | | | | Accumulated depreciation | | | | | | | | | | | | | Balance as of January 1, 2023 | | | 1,342 | | | | 334 | | | | 1,676 | | Depreciation and amortization | | | 334 | | | | 231 | | | | 565 | | Capitalized to Leasehold improvements | | | 59 | | | | - | | | | 59 | | Disposals | | | (1,527 | ) | | | (308 | ) | | | (1,835 | ) | | | | | | | | | | | | | | | Balance as of December 31, 2023 | | | 208 | | | | 257 | | | | 465 | | | | | | | | | | | | | | | | Depreciated cost | | | | | | | | | | | | | Balance as of December 31, 2023 | | | 6,303 | | | | 395 | | | | 6,698 | |
| | | Buildings | | | Motor vehicles | | | Total | | Cost | | | | | | | | | | Balance as of January 1, 2022 | | | 2,267 | | | | 654 | | | | 2,921 | | New leases | | | - | | | | 125 | | | | 125 | | Adjustments for indexation | | | 74 | | | | 9 | | | | 83 | | Disposals | | | - | | | | (238 | ) | | | (238 | ) | | | | | | | | | | | | | | | Balance as of December 31, 2022 | | | 2,341 | | | | 550 | | | | 2,891 | | | | | | | | | | | | | | | | Accumulated depreciation | | | | | | | | | | | | | Balance as of January 1, 2022 | | | 1,028 | | | | 345 | | | | 1,373 | | Depreciation and amortization | | | 314 | | | | 225 | | | | 539 | | Disposals | | | - | | | | (236 | ) | | | (236 | ) | | | | | | | | | | | | | | | Balance as of December 31, 2022 | | | 1,342 | | | | 334 | | | | 1,676 | | | | | | | | | | | | | | | | Depreciated cost | | | | | | | | | | | | | Balance as of December 31, 2022 | | | 999 | | | | 216 | | | | 1,215 | |
MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) Note 4: Cash and Cash Equivalents| | | December 31 | | | | | 2021 | | | 2020 | | | | | | | | | | Balance in USD | | | 7,735 | | | | 13,067 | | Balance in other currencies | | | 3,311 | | | | 4,309 | | | | | | | | | | | | | | | | 11,046 | | | | 17,376 | |
Note 5: Short-Term Bank Deposits
| | | December 31, | | | | | 2021 | | | 2020 | | | | | | | | | | USD bank deposits (1) | | | 0 | | | | 4,024 | | Restricted bank deposits (2) | | | 0 | | | | 184 | | | | | | | | | | | | | | | | 0 | | | | 4,208 | |
(1) The USD deposits bear annual interest of 1.12% for the period of 282 days for 2020.
(2)Restricted bank deposits which are primarily used as security for the Company’s office leases.
Note 6: Trade Receivables
| | | December 31 | | | | | 2021 | | | 2020 | | | | | | | | | | BARDA (see also Note 18a) | | | 1,085 | | | | 2,189 | | | | | | | | | | | | Other trade receivables | | | 696 | | | | 578 | | Less provision for impairment | | | (2 | ) | | | 0 | | | | | | 694 | | | | 578 | | | | | | | | | | | | | | | | 1,779 | | | | 2,767 | |
| | December 31,
| | | | 2021
| | | 2020
| | | | | | | | | Raw materials | | | 694
| | | | 631 | | Finished goods | | | 506
| | | | 749 | | | | | | | | | | | | | | 1,200
| | | | 1,380 | |
Note 8: Other Receivables- Short Term| | d. | December 31,Disclosures of the Company's lease liabilities:
| | | | 2021
| | | 2020
| | | | | | | | | Government authorities | | | 141
| | | | 73 | | Contract asset related to BARDA
| | | 347
| | | | 0 | | Prepaid expenses and other | | | 439
| | | | 389 | | | | | | | | | | | | | | 927
| | | | 462 | |
| | | Buildings | | | Motor vehicles | | | Total | | | | | | | | | | | | | Balance as of January 1, 2023 | | | 1,199 | | | | 179 | | | | 1,378 | | Repayment of leases liabilities | | | (588 | ) | | | (190 | ) | | | (778 | ) | Effect of changes in exchange rates | | | (19 | ) | | | 2 | | | | (17 | ) | New finance lease obligation recognized | | | 6,460 | | | | 365 | | | | 6,825 | | Adjustments for indexation | | | 78 | | | | 3 | | | | 81 | | Financial expenses | | | 257 | | | | 17 | | | | 274 | | Disposals-Termination of leases | | | (1,041 | ) | | | (11 | ) | | | (1,052 | ) | Balance as of December 31, 2023 | | | 6,346 | | | | 365 | | | | 6,711 | | | | | | | | | | | | | | | | Current maturities of long-term leases | | | (180 | ) | | | (181 | ) | | | (361 | ) | Lease liability Balance as of December 31, 2023 | | | 6,166 | | | | 184 | | | | 6,350 | |
| | | Buildings | | | Motor vehicles | | | Total | | | | | | | | | | | | | Balance as of January 1, 2022 | | | 1,691 | | | | 299 | | | | 1,990 | | Repayment of leases liabilities | | | (477 | ) | | | (224 | ) | | | (701 | ) | Effect of changes in exchange rates | | | (182 | ) | | | (28 | ) | | | (210 | ) | New finance lease obligation recognized | | | - | | | | 117 | | | | 117 | | Adjustments for indexation | | | 74 | | | | 9 | | | | 83 | | Financial expenses | | | 93 | | | | 8 | | | | 101 | | Disposals-Termination of leases | | | - | | | | (2 | ) | | | (2 | ) | Balance as of December 31, 2022 | | | 1,199 | | | | 179 | | | | 1,378 | | | | | | | | | | | | | | | | Current maturities of long-term leases | | | (399 | ) | | | (133 | ) | | | (532 | ) | Lease liability Balance as of December 31, 2022 | | | 800 | | | | 46 | | | | 846 | |
MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) Note 12: | Intangible Assets, Net |
| | | License and Knowhow | | | | | 2023 | | | 2022 | | Cost | | | | | | | Balance as of January 1, | | | 1,538 | | | | 1,538 | | Additions | | | - | | | | - | | Balance as of December 31, | | | 1,538 | | | | 1,538 | | Accumulated Amortization | | | | | | | | | Balance as of January 1, | | | 1,307 | | | | 1,241 | | Additions | | | 66 | | | | 66 | | Balance as of December 31, | | | 1,373 | | | | 1,307 | | Amortized cost | | | | | | | | | Balance as of December 31, | | | 165 | | | | 231 | |
Intangible assets include exclusive licenses to use patents, know-how and intellectual property for the development, manufacturing and marketing of products related to burn treatments and other products in the field of wound care. These licenses were purchased from third parties and from one of the Company's shareholders. Note 9:13: | Other Receivables- Long Term Payables |
| | | December 31, | | | | | 2021 | | | 2020 | | | | | | | | | | | Income receivables | | | 280 | | | | 0 | | | Restricted bank deposits (1) | | | 189 | | | | 0 | | | | | | | | | | | | | | | | 469 | | | | 0 | |
| | | December 31 | | | | | 2023 | | | 2022 | | | | | | | | | | Employees and payroll accruals | | | 2,109 | | | | 2,471 | | Liability in respect of TEVA (see Note 17c) | | | 1,250 | | | | 417 | | Related parties | | | 83 | | | | 307 | | Deferred revenues | | | 24 | | | | 63 | | Other | | | 425 | | | | 901 | | | | | | 3,891 | | | | 4,159 | |
| (1)Note 14: | Restricted bank deposits which are primarily used as security for the Company’s office leases.Liabilities in Respect of IIA Grants
|
Note 10: Property, Plant And Equipment, Net
| | | December 31 | | | | | 2023 | | | 2022 | | | | | | | | | | Balance as of January 1, | | | 7,566 | | | | 8,105 | | Royalties | | | (190 | ) | | | (407 | ) | Amounts carried to Profit or Loss | | | 427 | | | | (132 | ) | Balance as of December 31, | | | 7,803 | | | | 7,566 | | | | | | | | | | | | Current maturities | | | (126 | ) | | | (121 | ) | Long term liabilities in respect of IIA grants | | | 7,677 | | | | 7,445 | |
The Company is committed to pay royalties to the IIA up to the total grants received plus the applicable accrued interest. The total amount of grants received from IIA including accrued interest, net of royalties as of December 31, 2023 is approximately $13,783, while the amortized cost of this liability as of that date is $ 7,803, using the interest method. | | Office
furniture | | | Manufacturing machinery and lab equipment | | | Computers | | | Leasehold
improvements | | | Total | | Cost | | | | | | | | | | | | | | | | | | | | | Balance as of January 1, 2021 | | | 332 | | | | 4,775 | | | | 169 | | | | 2,904 | | | | 8,180 | | Additions | | | 18 | | | | 193 | | | | 45 | | | | 233 | | | | 489 | | Disposals | | | (89 | ) | | | (205 | ) | | | (36 | ) | | | 0 | | | | (330 | ) | Foreign currency translation | | | (4 | ) | | | 1 | | | | (2 | ) | | | 0 | | | | (5 | ) | | | | | | | | | | | | | | | | | | | | | | Balance as of December 31, 2021 | | | 257 | | | | 4,764 | | | | 176 | | | | 3,137 | | | | 8,334 | | | | | | | | | | | | | | | | | | | | | | | Balance as of January 1, 2020 | | | 301 | | | | 4,534 | | | | 124 | | | | 2,315 | | | | 7,274 | | Additions | | | 20 | | | | 241 | | | | 73 | | | | 445 | | | | 779 | | Disposals | | | 0 | | | | 0 | | | | (29 | ) | | | 0 | | | | (29 | ) | Re-classified from RSU assets | | | 0 | | | | 0 | | | | 0 | | | | 144 | | | | 144 | | Foreign currency translation | | | 11 | | | | 0 | | | | 1 | | | | 0 | | | | 12 | | | | | | | | | | | | | | | | | | | | | | | Balance as of December 31, 2020 | | | 332 | | | | 4,775 | | | | 169 | | | | 2,904 | | | | 8,180 | | | | | | | | | | | | | | | | | | | | | | | Accumulated Depreciation | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | Balance as of January 1, 2021 | | | 204 | | | | 3,092 | | | | 76 | | | | 2,178 | | | | 5,550 | | Additions | | | 22 | | | | 483 | | | | 55 | | | | 81 | | | | 641 | | Disposals | | | (89 | ) | | | (204 | ) | | | (35 | ) | | | 0 | | | | (328 | ) | Foreign currency translation | | | (4 | ) | | | (1 | ) | | | (2 | ) | | | 0 | | | | (7 | ) | | | | | | | | | | | | | | | | | | | | | | Balance as of December 31, 2021 | | | 133 | | | | 3,370 | | | | 94 | | | | 2,259 | | | | 5,856 | | | | | | | | | | | | | | | | | | | | | | | Balance as of January 1, 2020 | | | 175 | | | | 2,606 | | | | 60 | | | | 2,129 | | | | 4,970 | | Additions | | | 18 | | | | 486 | | | | 44 | | | | 49 | | | | 597 | | Disposals | | | 0 | | | | 0 | | | | (29 | ) | | | 0 | | | | (29 | ) | Foreign currency translation | | | 11 | | | | 0 | | | | 1 | | | | 0 | | | | 12 | | | | | | | | | | | | | | | | | | | | | | | Balance as of December 31, 2020 | | | 204 | | | | 3,092 | | | | 76 | | | | 2,178 | | | | 5,550 | | | | | | | | | | | | | | | | | | | | | | | Carrying amounts of all fixed asset items | | | | | | | | | | | | | | | | | | | | | December 31, 2021 | | | 124 | | | | 1,394 | | | | 82 | | | | 878 | | | | 2,478 | | December 31, 2020 | | | 128 | | | | 1,683 | | | | 93 | | | | 726 | | | | 2,630 | |
MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) a. Lease Agreements:
The Company's offices and its production facility in Israel are located in a building that the Company leases from its Parent Company (see Note 25a), in accordance with a sub-lease agreement. The Company subleases approximately 3,000 square meters of laboratory, office and clean room space at a monthly rent fee of NIS 119 (approximately $38) and NIS 125 starting November 2022 (approximately $40). This sub-lease agreement was amended on October 2021, to extand the priod up to October 2025 which was included in the calculation of the lease liability and RoU asset.
In addition the Company and its subsidiary have lease agreements for 13 vehicles for a period of three years.b.Amounts recognized in profit or loss and in the statement of cash flows
| | Year ended December 31,
| | | | 2021
| | | 2020
| | | | | | | | | Interest expense on lease liabilities
| | | 120
| | | | 144
| | Depreciation expenses relating to short-term leases
| | | 531
| | | | 427
| | Cash outflow for leases (1)
| | | 693
| | | | 652
| |
(1) For the year ended December 31,2020 the cash flow for leases includes $144 which were capitalized to Leasehold improvements.
The Company was assisted by external third party valuation expert in determining the appropriate interest rate for discounting its leases based on: credit risk, the weighted average term of the leases and other economic variables. A weighted average incremental borrowing in a range of 1% to 6.7% was used to discount future lease payments in the calculation of the lease liability on the date of initial application of the standard (IFRS 16).MEDIWOUND LTD. AND ITS SUBSIDIARIES
Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data)
c.Disclosures in respect of Right- of- Use assets:
| | Buildings
| | | Motor vehicles
| | | Total
| | Cost
| | | | | | | | | | | | | Balance as of January 1, 2021
| | | 2,225 | | | | 512 | | | | 2,737 | | | | | | | | | | | | | | | New leases
| | | 0 | | | | 162 | | | | 162 | | Adjustments for indexation
| | | 42 | | | | 7 | | | | 49 | | Disposals
| | | 0 | | | | (27 | ) | | | (27 | ) | | | | | | | | | | | | | | Balance as of December 31, 2021
| | | 2,267 | | | | 654 | | | | 2,921 | | | | | | | | | | | | | | | Accumulated depreciation
| | | | | | | | | | | | | Balance as of January 1, 2021
| | | 698 | | | | 155 | | | | 853 | | Depreciation and amortization
| | | 330 | | | | 201 | | | | 531 | | Disposals
| | | 0 | | | | (11 | ) | | | (11 | ) | | | | | | | | | | | | | | Balance as of December 31, 2021
| | | 1,028 | | | | 345 | | | | 1,373 | | | | | | | | | | | | | | | Depreciated cost
| | | | | | | | | | | | | Balance as of December 31, 2021
| | | 1,239 | | | | 309 | | | | 1,548 | |
| | Buildings | | | Motor vehicles | | | Total | | Cost | | | | | | | | | | | | | Balance as of January 1, 2020 | | | 2,362 | | | | 442 | | | | 2,804 | | New leases | | | 0 | | | | 305 | | | | 305 | | Adjustments for indexation | | | (17 | ) | | | (18 | ) | | | (35 | ) | Disposals | | | (76 | ) | | | (217 | ) | | | (293 | ) | Termination of leases | | | (44 | ) | | | 0 | | | | (44 | ) | | | | | | | | | | | | | | Balance as of December 31, 2020 | | | 2,225 | | | | 512 | | | | 2,737 | | | | | | | | | | | | | | | Accumulated depreciation | | | | | | | | | | | | | Balance as of January 1, 2020 | | | 381 | | | | 194 | | | | 575 | | Depreciation and amortization | | | 249 | | | | 178 | | | | 427 | | Capitalized to Leasehold improvements (1) | | | 144 | | | | 0 | | | | 144 | | Disposals | | | (76 | ) | | | (217 | ) | | | (293 | ) | | | | | | | | | | | | | | Balance as of December 31, 2020 | | | 698 | | | | 155 | | | | 853 | | | | | | | | | | | | | | | Depreciated cost | | | | | | | | | | | | | Balance as of December 31, 2020 | | | 1,527 | | | | 357 | | | | 1,884 | |
(1) As of the year ended December 31,2020 the cash flow for leases includes $144 which were capitalized to Leasehold improvements.
MEDIWOUND LTD. AND ITS SUBSIDIARIES
Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data)
d. Disclosures of the Company's lease liabilities :
| | Buildings
| | | Motor vehicles
| | | Total
| | | | | | | | | | | | | | | Balance as of January 1, 2021
| | | 1,953 | | | | 354 | | | | 2,307 | | Repayment of leases liabilities
| | | (477 | ) | | | (216 | ) | | | (693 | ) | Effect of changes in exchange rates
| | | 55 | | | | 13 | | | | 68 | | New finance lease obligation recognized
| | | 0 | | | | 155 | | | | 155 | | Adjustments for indexation
| | | 42 | | | | 7 | | | | 49 | | Interest
| | | 118 | | | | 2 | | | | 120 | | Disposals-Termination of leases
| | | 0 | | | | (16 | ) | | | (16 | ) | Balance as of December 31, 2021
| | | 1,691 | | | | 299 | | | | 1,990 | | | | | | | | | | | | | | | Current maturities of long-term leases
| | | (403 | ) | | | (196 | ) | | | (599 | ) | Lease liability Balance as of December 31, 2021
| | | 1,288 | | | | 103 | | | | 1,391 | |
| | Buildings | | | Motor vehicles | | | Total | | | | | | | | | | | | | | | Balance as of January 1, 2020 | | | 2,225 | | | | 225 | | | | 2,450 | | Repayment of leases liabilities | | | (479 | ) | | | (173 | ) | | | (652 | ) | Effect of changes in exchange rates | | | 134 | | | | 28 | | | | 162 | | New finance lease obligation recognized | | | 0 | | | | 283 | | | | 283 | | Adjustments for indexation | | | (17 | ) | | | (18 | ) | | | (35 | ) | Interest | | | 134 | | | | 10 | | | | 144 | | Disposals-Termination of leases | | | (44 | ) | | | (1 | ) | | | (45 | ) | Balance as of December 31, 2020 | | | 1,953 | | | | 354 | | | | 2,307 | | | | | | | | | | | | | | | Current maturities of long-term leases | | | (396 | ) | | | (170 | ) | | | (566 | ) | Lease liability Balance as of December 31, 2020 | | | 1,557 | | | | 184 | | | | 1,741 | |
MEDIWOUND LTD. AND ITS SUBSIDIARIES
Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data)
Note 12: Intangible Assets, Net
| | | License and Knowhow | | | | | 2021 | | | 2020 | | Cost | | | | | | | Balance as of January 1, | | | 1,538 | | | | 1,538 | | Additions | | | 0 | | | | 0 | | Balance as of December 31, | | | 1,538 | | | | 1,538 | | Accumulated Amortization | | | | | | | | | Balance as of January 1, | | | 1,175 | | | | 1,109 | | Additions | | | 66 | | | | 66 | | Balance as of December 31, | | | 1,241 | | | | 1,175 | | Amortized cost | | | | | | | | | Balance as of December 31, | | | 297 | | | | 363 | |
Intangible assets include exclusive licenses to use patents, know-how and intellectual property for the development, manufacturing and marketing of products related to burn treatments and other products in the field of wound care. These licenses were purchased from third parties and from one of the Company's shareholders.
|
| | | December 31 | | | | | 2021 | | | 2020 | | | | | | | | | | Employees and payroll accruals | | | 1,639 | | | | 1,910 | | Liability in respect of purchase of shares (see Note 17c)* | | | 417 | | | | 0 | | Related parties | | | 241 | | | | 225 | | Deferred revenues | | | 543 | | | | 462 | | Other | | | 780 | | | | 260 | | | | | | | | | | | | | | | | 3,620 | | | | 2,857 | |
• | An amount of $667 was classified from Liability in respect of purchase of shares to current maturities for the year ended 31, December 2020.
|
Note 14: Liabilities in Respect of IIA Grants
| | | December 31 | | | | | 2021 | | | 2020 | | | | | | | | | | Balance as of January 1, | | | 7,528 | | | | 6,935 | | Royalties | | | (342 | ) | | | (235 | ) | Amounts carried to Profit or Loss | | | 919 | | | | 828 | | Balance as of December 31, | | | 8,105 | | | | 7,528 | | | | | | | | | | | | Current maturities | | | (220 | ) | | | (261 | ) | Long term liabilities in respect of IIA grants | | | 7,885 | | | | 7,267 | |
The Company is committed to pay royalties to the IIA up to the total grants received plus the applicable accrued interest. The total amount of grants received from IIA including accrued LIBOR interest, net of royalties as of December 31, 2021 is approximately $13,681, while the amortized cost of this liability as of that date is $ 8,105, using the interest method.
MEDIWOUND LTD. AND ITS SUBSIDIARIES
Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data)
| Note 15: | Financial Instruments |
The Board of Directors has overall responsibility for the establishment and oversight of the Company’s risk management framework. The Company’s risk management practice was formulated to identify and analyze the risks that the Company faces, to set appropriate limits for the risks and controls, and to monitor the risks and their compliance within the limits. The risk policy and risk management methods are reviewed regularly to reflect changes in market conditions and in the Company’s operations. The Company Audit Committee oversees how management monitors compliance with the Company’s risk management policies and procedures and reviews the adequacy of the risk management framework in relation to the risks faced by the Company. The Company Audit Committee is assisted in its oversight role by Internal Audit. Internal Audit undertakes both regular and ad hoc reviews of risk management controls and procedures, the results of which are reported to the Audit Committee. The Company's activities expose it to various financial market risks, mainly foreign currency risk, interest rate risk and liquidity risk. The Company operates primarily in an international environment and is exposed to foreign exchange risk resulting from the fact that a certain portion of the Company's costs are denominated in NIS and EURO, mainly due to payroll and related benefit costs incurred in Israel and additionally due to marketing expenses incurred in Europe. | | 2. | Sensitivity tests relating to changes in market factors: |
The Company operates in an international environment and is exposed to foreign exchange risk resulting from the exposure to different currencies, mainly NIS and EURO. Foreign exchange risks arise from recognized assets and liabilities denominated in a foreign currency other than the functional currency. | | | December 31 | | | December 31 | | | | | 2021 | | | 2020 | | | 2023 | | | 2022 | | Gain (loss) from change: | | | | | | | | | | | | | 5% increase in NIS and EURO exchange rate | | $ | 3 | | | $ | 76 | | | $ | 176 | | | $ | 257 | | 5% decrease in NIS and EURO exchange rate | | $ | (3 | ) | | $ | (76 | ) | | $ | (176 | ) | | $ | (257 | ) |
The Company has performed sensitivity tests of principal market risk factors that may affect its reported operating results or financial position. The sensitivity tests present the profit or loss for the relevant risk variables chosen as of each reporting date. MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) | Note 15: | Financial Instruments (Cont.) |
Liquidity risk is the risk that the Company will not be able to meet its financial obligations as they fall due. The Company’s approach to managing liquidity is to ensure, as far as possible, that it will always have sufficient liquidity to timely meet its liabilities, under both normal and stressed conditions, without incurring unwanted losses. The Company manages the liquidity risk by holding cash balances, short-term deposits and secured bank credit facilities. | | | December 31, 2021 | | | | | Carrying | | | 12 months | | | | | | | | | | | | | | | | | | | | | | Non-derivative financial liabilities | | | | | | | | | | | | | Current liabilities | | | | | | | | | | | | | Current maturities of long-term liabilities | | | 3,024 | | | | 3,024 | | | | | | | | Trade payables and accrued expenses | | | 4,693 | | | | 4,693 | | | | | | | | Other payables | | | 3,620 | | | | 3,620 | | | | | | | | Non-current liabilities | | | | | | | | | | | | | | | Liabilities in respect of IIA grants | | | 15,286 | | | | | | | | 369 | | | | 14,917 | | Liabilities in respect of purchase of shares | | | 5,867 | | | | | | | | 1,667 | | | | 4,200 | | Lease liabilities | | | 1,522 | | | | | | | | 589 | | | | 933 | |
| | | December 31, 2023 | | | | | Carrying | | | 12 months | | | | | | | | | | | | | | | | | | | | | | Non-derivative financial liabilities | | | | | | | | | | | | | Current liabilities | | | | | | | | | | | | | Current maturities of long-term liabilities | | | 3,249 | | | | 3,249 | | | | - | | | | - | | Trade payables and accrued expenses | | | 5,528 | | | | 5,528 | | | | - | | | | - | | Other payables | | | 3,891 | | | | 3,891 | | | | - | | | | - | | Non-current liabilities | | | | | | | | | | | | | | | | | Liabilities in respect of IIA grants | | | 15,927 | | | | - | | | | 359 | | | | 15,568 | | Liabilities in respect of TEVA | | | 3,200 | | | | - | | | | 1,000 | | | | 2,200 | | Lease liabilities | | | 10,189 | | | | - | | | | 813 | | | | 9,376 | |
| | | December 31, 2022 | | | | | Carrying | | | 12 months | | | | | | | | | | | | | | | | | | | | | | Non-derivative financial liabilities | | | | | | | | | | | | | Current liabilities | | | | | | | | | | | | | Current maturities of long-term liabilities | | | 2,814 | | | | 2,814 | | | | - | | | | - | | Trade payables and accrued expenses | | | 5,656 | | | | 5,656 | | | | - | | | | - | | Other payables | | | 4,159 | | | | 4,159 | | | | - | | | | - | | Non-current liabilities | | | | | | | | | | | | | | | | | Liabilities in respect of IIA grants | | | 15,464 | | | | - | | | | 312 | | | | 15,152 | | Liabilities in respect of TEVA | | | 4,200 | | | | - | | | | 1,000 | | | | 3,200 | | Lease liabilities | | | 902 | | | | - | | | | 508 | | | | 394 | |
MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) | Note 15: | Financial Instruments (Cont.) |
| (1) | Financial instruments measured at fair value for disclosure purposes only. |
The carrying amountamounts of certain financial assets and liabilities, including cash, and cash equivalents, short‑term banktrade receivables, other receivables, deposits, trade and other receivables and trade and other payables approximatesare the same as or approximate to their fair value. | (2) | Fair value hierarchy of financial instruments measured at fair value. |
The financial instruments measured at fair value dueon a temporal basis, use valuation methodology in accordance with the fair value hierarchy levels when determining the fair value of an asset or liability, the Company uses observable market data as much as possible. Details regarding fair value measurement at Level 2 The fair value of the warrants which are classified as non-currents liabilities was measured by using Black and Scholes model. The following inputs were used to determine the short‑term maturitiesfair value: Expected period of such instruments.warrants– 2.91 years. Expected volatility – 67.1% Risk-free interest rate (3) – 4.03% Expected dividend yield – 0%. The fair value of liabilities in respect to IIA grants with fixed interest is based on a calculation of the present value of the cash flows at the interest rate for a loan with similar terms. The Company used a discount rate of 12% based in part of the Company'sCompany’s estimation at the time of the Company'sCompany’s recognition of the IIA grants which approximates the fair value at the respective balance sheet date. The fair valueliability in respect of the contingent consideration for purchase of sharesTEVA as presented in balance sheet which is approximate its fair value, based on a calculation of the present value of future payments. The expected cash flows already reflect assumptions about the uncertainty in future defaults, and therefore the Company used a discount rate of 14% that is commensurate with the risk inherent in the expected cash flows.flows (Note 17c). | Note 16: | Severance Pay Liability, Net |
Note 16: Severance Pay Liabilty, Net
The Company has liabilities for severance pay for its employees in Israel and in several EU jurisdictions. The Company'sCompany’s liability for employee benefits is based on local laws, valid labor agreements, the employee'semployee’s salary and the applicable terms of employment, which together generate a right to severance compensation. Post‑employment employee benefits are partially financed by deposits with defined contribution plans, as detailed below. The Israeli Severance Pay Law, 1963 ("(“Severance Pay Law"Law”), specifies that Israeli employees are entitled to severance payment, following the termination of their employment. Under the Severance Pay Law, the severance payment is calculated as one month salary for each year of employment, or a portion thereof. Under Section 14 of the Severance Pay Law ("(“Section 14"14”), employees are entitled to have monthly deposits, at a rate of 8.33% of their monthly salary, made on their behalf to their insurance funds. Payments in accordance with Section 14 release the Company from the liability for any future severance payments in respect of those employees. The majority of the Company'sCompany’s liability for severance pay is covered by Section 14. Acordingly,Accordingly, the Company does not recognize any liability for severance pay due to these employees and the deposits under Section 14 are not recorded as an asset in the Company'sCompany’s balance sheet. These contributions for compensation represent defined contribution plans. The Company recognizes liability for severance pay due to its employees in EU in accordance with local laws and its Israeli employees which are not under Section 14. MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) Note 17: Contingent Liabilities and Commitments
| Note 17: | Liabilities and Commitments |
| | a. | In 2000, the Company signed an exclusive license agreement (as amended in 2007) with a third party with regard to its patents and intellectual property. Pursuant to the agreement, the Company received an exclusive license to use the third party'sparty’s patents and intellectual property, for the purpose of developing, manufacturing, marketing, and commercializing products for treatment of burns and other wounds. |
In consideration for this exclusive license, theThe Company paid an aggregate amount of $ 950 following the achievement of certain development milestones as set forth in the agreement. In addition, the Companyand undertook to pay royalties of 1.5% to 2.5% from future revenues from sales of products which are based on this patent for a period ranging between 10 to 15of 12 years from the first commercial delivery in a major country, and thereafter the Company will have a fully paid-up royalty-free license for these patents. In addition, royalties will be paid at the rate of 10% - 20% from sub-licensing of such patents and for lump sum amounts paid to the Company by a third party, the Company will pay 2% of the proceeds up to $1,000 and 4% of the proceeds above this amount. Moreover, the Company agreed to pay a one-time lump-sum amount of $ 1,500 when the aggregate revenues based on these patents reach $ 100,000. The amount of royalty payments for the years 20202021, 2022 and 20212023 amounted to $42$149, $363 and $149$98 respectively.
| | b. | Under the Research and Development Law, (the "R“R&D Law"Law”) the Company undertook to pay royalties of 3% on the revenues derived from sales of products or services developed in whole or in part using IIA grants. The maximum aggregate royalties paid cannot exceed 100% of the grants received by the Company, plus annual interest equal to the 12-month LIBORinterest applicable to dollar deposits, as published on the first business day of each calendar year. (see also Note 14).TheThe total royalties amount paid as of December 31, 20212023 is $1,303.$1,942. (Note 14). |
| | c. | Beginning in 2007,In December 2020, Teva has agreed to revise the Company entered into a numberSettlement Agreement from March 2019, which was comprised of agreements with Teva Pharmaceutical Industries Limited (“Teva”) related topast agreement for collaboration in the development, manufacturing and commercialization of solutions for the burn and chronic wound care markets. In consideration for these agreements, Teva made investmentsmarkets, as well as the Company’s repurchase of shares from Teva. Under the new settlement the Company paid $1,000 in the Company's ordinary sharescash and agreedbecame obligated to fund certain researchpay an amount of $2,000 over three years and development expenses and manufacturing costs and perform all marketing activities for both NexoBrid, under the 2007 Teva Agreement, and the PolyHeal Product, under the 2010 PolyHeal Agreements (see also Note 22). Asan addition amount of $7,200 in quarterly fixed payments starting 2021 as long as there are revenues generated from sales of Nexobrid.
Total liabilities recorded as of December 31, 2012, all2023 and 2022 were approximately $4,428 and $4,794 respectively, and financial expenses of these agreements$468 and $533, respectively, were terminated.recorded in profit or loss within financial expenses. |
On September 2, 2013, in accordance with the terms of the Teva Shareholders’ Rights Agreement, the Company exercised its rights to repurchase all of its shares held by Teva, and purchased 755,492 ordinary shares, in consideration for an obligation to pay Teva future royalty payments.
Pursuant to a Settlement Agreement signed on March 2019, Teva agreed to reduce the contingent consideration that is payable to Teva pursuant to the Company's repurchase of its shares from Teva in 2013 and to paid the Company $4,000 in cash. As a result, the Company is obligated to pay Teva annual payments at a reduced rate of 15% of its recognized revenues from the sale or license of NexoBrid after January 1, 2019, up to a reduced aggregate amount of $10,200. MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) Note 17:18: | Contingent Liabilities and Commitments (Cont.)Materials Agreements
|
In addition, the Company also agreed to indemnify, defend and hold harmless Teva and its directors, officers, agents and employees from and against claims relating to a certain milestone related to PolyHeal under an agreement associated with the Collaboration Agreements, up to an amount of $10,200, if a notice of such claim has been received by the Company prior to December 31, 2023. In December 2020, Teva has agreed to revise the Settlement Agreement from March 2019. Under the new settlement the Company paid $1,000 in cash and became obligated to pay an amount of $2,000 over three years, in addition to a modified contingent consideration up to the amount of $7,200 in quarterly fixed payments starting 2021 subject to revenues generated from sales of NexoBrid. Total liabilities recorded as of December 31, 2021 and 2020 were approximately$ 5,928 and $6,587, respectively, and financial expenses (income) of $590 and ($433), respectively, were recorded in profit or loss within financial income of financial expenses.
Note 18: Materials Agreements
a.BARDAContracts
In September 2015, the Company was awarded the First BARDA Contract for treatment of thermal burn injuries, whichinjuries. This contract was valued atamended multiple times to extend its term until September 2024 (the Company is pursuing an extension of the contract) and its total value, up to $112,000. In July 2017 and in May 2019, BARDA expanded its commitment by an aggregate supplementala total amount of $41,000. $165,000 as of the end of 2022.
In March 2020,May 2023 BARDA further expanded its commitment byhas awarded an additional $5,500approximately $10,000 to the Company. The total amount of the contract is comprised of $110,000 to support emergency readiness for NexoBrid deployment upon request of use of NexoBrid in mass casualty situationsresearch and in February 2022 BARDA has expanded its awarded contract providing supplemental funding of $9,000 to support the NexoBrid BLA resubmission to the FDAdevelopment activities and the continuous expanded access program (collectively the "First BARDA Contract").Under the First BARDA Contract, BARDA provided technical assistance and a total of up to $91,000$65,000 to procure Nexobrid for U.S. emergency preparedness (which will be split between the Company and Vericel following Vericel agreement (see Note 18b)).
As of December 31, 2023, the Company has received approximately $88,282 in funding for NexoBrid development activities needed to request U.S. marketing approvalin the aggregate, from BARDA under the FDA. In January 2020, BARDA committedfirst contract, and an additional $16,500 to procure NexoBrid as partfor procurement of Nexobrid for U.S. emergency preparedness, which were recorded at the HHS mission to build national preparednessnet amount of approximately $10,500 following the split of gross profit agreement with Vericel for public health medical emergencies. The contract further includes a $10,000 option to fund development of other potential NexoBrid indications and an option to procure additional NexoBrid valued at up to $50,000.the initial BARDA procurement. In September 2018 the Company werewas awarded the second BARDA contract, (the "Second BARDA Contract"), which is an additional, separate contract to develop NexoBridNexobrid for the treatment of Sulfur Mustard injuries as part of BARDA’s preparedness for mass casualty events. The Second BARDA Contractcontract provides approximatelyup to $12,000 of funding to support research and development activities up to pivotal studies in animals under the U.S. FDA Animal Rule and contains options for BARDA to provide additional funding of up to $31,000$29,000 for additional development activities, animal pivotal studies, and the BLA submission for licensure of NexoBridNexobrid for the treatment of Sulfur Mustard injuries. The second BARDA contract expired in 2023. As of December 31, 2023, the Company has received approximately $4,368 of funding from the second BARDA contract. MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) | Note 18: | Materials Agreements (Cont.) |
Note 18: Materials Agreements (Cont.)The total aggregate value awarded by BARDA Contracts is up to $211,000 comprised of $144,500 to support research and development activities and $66,500 to procure NexoBrid for U.S. emergency preparedness (which will be splited between the Company and Vericel following Vericel agreement (see Note 18b)).
As of December 31, 2021, the Company has received approximately $69,400 in funding in the aggregate, from BARDA under the two contracts, and an additional of approximately $14,600 for procurement of NexoBrid for U.S. emergency preparedness which were recorded at the net amount of approximately $9,300 following the split of gross profit agreement with Vericel for the initial BARDA procurement.
b. Vericel Agreement:
On May 6, 2019, the Company entered into exclusive license and supply agreements with Vericel to commercialize NexoBridNexobrid in North America (the “Collaboration Agreements”). Pursuant to the Collaboration Agreements, Vericel will obtain the authority over and control of the development, regulatory approval and commercialization of licensed products in the North America territory. MediWound will be responsible for the development of the product through BLA approval, supported and funded by BARDA, as well as the manufacture and supply of NexoBrid.Nexobrid. In addition, MediWound retains the commercial rights to NexoBridNexobrid in non-North American territory.territory. Under the terms of the license agreement, Vericel has made an upfront payment to MediWound of $17,500 which was recorded as revenues from license agreements in 2019 and agreed to makeas well as an additional milestone payment of $7,500 payment contingentrecorded as revenues from license agreements in 2022 upon BLA approval andreceived in December 2022. Furthermore, Vericel has also agreed to pay MediWound up to $125,000 in payments contingent upon meeting certain annual sales milestones. Vericel has also agreed to pay MediWoundmilestones, tiered royalties on net sales ranging from high single-digit to teen-digit percentages, a split of gross profit on committed BARDA procurement orders and a teen-digits royalty on any additional future BARDA purchases of NexoBrid.Nexobrid. Under the terms of the supply agreement, Vericel will procure NexoBridNexobrid from MediWound at a fixed transfer price of cost plus a fixed margin percentage. price. AsTotal revenue from royalties as of the financial statements date,December 31, 2023 was approximately $82.
| c. | DOD and MTEC contracts: |
On February 17, 2022, the Company did not recognize any revenueswas entered into a contract with the U.S. Department of Defense (DoD), through the Medical Technology Enterprise Consortium (MTEC), to develop Nexobrid as a non-surgical solution for field-care burn treatment for the U.S. Army. The contract provides funding up to $2,727. During 2023, the DOD through MTEC awarded the Company additional funding of $9,117 in addition, the company awarded directly through MTEC funding of $1,190, to advance the development of a new temperature stable formulation of Nexobrid. The total additional fund from royalties.the DOD and MTEC during 2023 was $10,307. | d. | Biopharmax Group Ltd. contract: |
On July 17, 2023 thecompany signed a turnkey scale-up agreement with Biopharmax Group Ltd. This strategic agreement is designed to bolster our manufacturing infrastructure to support our long-term growth trajectory. The objective of this agreement is to establish, commission, and validate a cutting-edge, sterile, and GMP-compliant manufacturing facility. The venture aims to increase the company’s production capacity significantly, to expand up to six times the current capacity. The new facility, equipped with fully operational clean rooms, will be exclusively designed for Nexobrid production. It will comply with stringent regulations from the GMP, FDA, EMA, Israeli Ministry of Health, and relevant Israeli regulatory bodies. An estimated $12.7 million will be invested in the project, set for constructions completion by mid-2024, with full-scale manufacturing expected to commence in 2025 (see also note 10). MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) | | | December 31 | | | | | 2021 | | | 2020 | | | | | | | | | | Authorized number of shares | | | 50,000,000 | | | | 50,000,000 | | Issued and outstanding number of shares | | | 27,272,818 | | | | 27,236,752 | |
b.Rights attached to shares:
| | | December 31 | | | | | 2023 | | | 2022 | | | | | | | | | | Authorized number of shares | | | 20,000,000 | | | | 12,857,143 | | Issued and outstanding number of shares | | | 9,221,764 | | | | 7,240,020 | |
An ordinary share confers upon its holder(s) a right to vote at the general meeting, a right to participate in distribution of dividends, and a right to participate in the distribution of surplus assets upon liquidation of the Company. c.Movement in share capital:
| b. | Movement in share capital: | | | |
| 1. | During 2021, 2022 and 2023 the Company issued additional 4,852, 41,395 and 17,458 ordinary shares for each year upon vesting of outstanding RSU’s, respectively. |
| 2. | On November 28, 2022, at the Company’s extraordinary general meeting of shareholders, its shareholders approved: |
| (a) | An increase of the Company’s authorized share capital from NIS 500,000, consisting of 50,000,000 ordinary shares, per value NIS 0.01 per share to NIS 900,000 consisting of 90,000,000 ordinary shares, per value NIS 0.01 per share. |
| (b) | On December 5, 2022, the Company’s board of directors approved a reverse split of 1-for-7 ratio. The reverse split went effective on December 20, 2022. |
No fractional shares were issued as a result of the reverse share split. Instead, such shares were rounded up to the next whole number of shares. The reverse share split affected all shareholders uniformly and did not alter any person’s percentage interest in our outstanding ordinary shares, except for negligible adjustments that may have resulted from the treatment of fractional shares. ��During 2019,In connection with the reverse share split, The Company also amended and reduced the authorized number of shares was increased by 12,755,492 shares which has a nominal value of $40.•On December 31, 2019, the Company issued additional 23,956 ordinary shares upon vestingfrom 90,000,000 to 12,857,143, which reflected a reduction at the same 1-for-7 ratio as the reduction to the number of outstanding RSU’s.
•During 2020issued and 2021 the Company issued additional 33,958 ordinary shares for each year upon vesting of outstanding RSU’s.
Note 20: Share‑Based Compensation
a.Expense recognized in the financial statements:
The expenses recognized for services received from employees and directors is as follows:
| | | Year ended December 31 | | | | | 2021 | | | 2020 | | | 2019 | | Cost of revenues | | | 153 | | | | 115 | | | | 226 | | Research and development | | | 333 | | | | 179 | | | | 375 | | Selling and marketing | | | 0 | | | | 3 | | | | 40 | | General and administrative | | | 1,187 | | | | 1,025 | | | | 593 | | | | | | | | | | | | | | | | Total share-based compensation | | | 1,673 | | | | 1,322 | | | | 1,234 | |
b. Share-based payment plan for employees and directors:The Company has granted options and restricted stock units ("RSUs") for total of 3,863,089 ordinary shares.
Concurrently, the par value of the Company’s ordinary shares was increased proportionately, from NIS 0.01 per share to NIS 0.07 per share, in order to maintain the same overall authorized share capital under our Amended and Restated Articles of Association. On May 31, 2023 the Shareholders of the Company approved an amendment to Article 6 of the Company’s Amended and Restated Articles of Association, which increased the Company’s authorized share capital from 900,000 NIS consisting of 12,857,143 ordinary shares par value NIS 0.07 to NIS 1,400,000, consisting of 20,000,000 ordinary shares, par value NIS 0.07 per share.
MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) | c. | Financial transactions: |
| 1. | On March 7, 2022, the Company completed a public offering in total of 744,048 new ordinary shares which were issued in consideration to offering price of $13.44 per share. The net proceeds were $8,653, after deducting commissions and other offering expenses. In addition, on March 22, 2022 the underwriters exercised their options to purchase an additional 89,012 ordinary shares at the same public offering price. The net consideration to the Company, less underwriting discounts and commissions was at additional of $1,021. |
As part of the above- mentioned public offering, certain entities affiliated with CBI purchased 208,334 of ordinary shares at the public offering price. | 2. | On September 26, 2022, the Company completed a registered direct (the “RD”) offering in an aggregate amount of $13,257 represent a combine purchase price of $12.25 for issuance of 1,082,223 ordinary shares and 1,082,223 warrants that become exercisable on November 28, 2022, at an exercise price of $13.475 per ordinary share which will expire in four years (see also note 25.3). |
The warrants issued have been classified as a non-current financial liability due to a net share settlement provision and as they can be settled in cash on the occurrence of Fundamental Transaction as determined in the agreement. This liability was initially recognized at its fair value on the date the contract was entered into and is subsequently accounted for fair value at each balance sheet date and recorded through profit and loss. The fair value of the warrants has been evaluated with the assistance of external independent valuator and was computed based on then current price of the shares, a risk-free interest rate of 4.37% and an average standard deviation of 68%. The net proceeds from this offering in the amount of $11,698 have been received on September 28, 2022. The issuance expenses related to the non-current financial liability were recorded through profit and loss and the issuance expenses related to the issuance of shares recorded as a deduction from the proceeds in equity. | 3. | Concurrently, on October 6, 2022, the Company entered into a Private Issuance Purchase Equity agreement (the “PIPE”) with several purchasers, in connection with the offering of 1,407,583 unregistered pre-funded warrants to purchase up to 1,407,583 ordinary shares and 1,407,583 warrants to purchase up to 1,407,583 ordinary shares. Pre-Funded warrants become exercisable on November 28, 2022, at an exercise price of $0.007 per ordinary share and the warrants would be also exercisable upon the Authorized Share Increase Date at an exercise price of $13.475 per ordinary share and expire in four years. |
The Pre-Funded warrants and warrants issued have been classified as a non-current financial liabilities due to a net share settlement provision and they can be redeemed in cash on the occurrence of Fundamental Transaction as determined in the agreement. The initial fair value of the financial liabilities issued in the transaction was approximately $20,788, which comprised of: 1. The warrants which were valuated by Black and Sholtes model based on the current price of the shares and a risk-free interest rate of 4.26%, and 2. The pre-funded warrants which were valued in an amount which is approximate its share price upon their issuance. MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) The consideration received from this transaction was $17,233. As the fair value on initial recognition of the warrants differs from the transaction price, the difference, represents the First day loss at the amount of $3,555, and has been allocated to the warrants with respect to this transaction and is amortized on a straight line basis over the term of the warrants. The net proceeds from this offering amounted to approximately $15,920. The issuance expenses were recorded through profit and loss. During December 2022, the 1,407,583 pre-funded warrants were exercised into ordinary shares. | 4. | Upon closing of the RD and PIPE Offerings, the Company also issued the placement agent up to 124,491 warrants to purchase up to 124,491 ordinary Shares. The warrants have substantially the same terms as the RD and PIPE Warrants, except that the placement agent’s warrants have an exercise price equal to $15.312 per share (which represents 125% of the offering price per ordinary Share in the offerings). The Fair value of the placement agent’s options was recorded as an issuance expenses through profit and loss and as a deduction from proceed in equity based on the financial treatment of both RD and PIPE offering and in accordance with their part. |
| 5. | On February 7, 2023, the Company completed a registered direct (the “RD”) offering of 1,964,286 new ordinary shares which were issued in consideration to offering price of $14.0 per share. The gross proceeds were $27,500, before deducting commissions and other offering expenses in the amount of $2,031. |
| Note 20: | Share‑Based Compensation |
| a. | Expense recognized in the financial statements: |
The expenses recognized for services received from employees and directors is as follows: | | | Year ended December 31 | | | | | 2023 | | | 2022 | | | 2021 | | Cost of revenues | | | 271 | | | | 184 | | | | 153 | | Research and development | | | 485 | | | | 406 | | | | 333 | | Selling and marketing | | | 87 | | | | 42 | | | | - | | General and administrative | | | 1,097 | | | | 1,314 | | | | 1,187 | | | | | | | | | | | | | | | | Total share-based compensation | | | 1,940 | | | | 1,946 | | | | 1,673 | |
MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) | Note 20: | Share‑Based Compensation (Cont.)
|
| b. | Share-based payment plan for employees and directors: |
The Company has granted options and restricted stock units ("RSUs") for a total of 990,201 ordinary shares. As of December 31, 2021, 490,9272023, 861,255 ordinary shares of the Company were still available for a future grant. Any options or RSUs, which are forfeited or not exercised before expiration, become available for future grants.
In March 2014, the Company adopted and obtained shareholder approval for its 2014 Equity Incentive Plan (the “2014 Plan”). Options and RSU's granted under the Company's 2014 Plan are exercisable in accordance with the terms of the Plan. Options are exercisable within 5-10 years from the date of grant, against payment of an exercise price or cashless exercise and share units are granted immediately upon vesting of the RSU's. The options and the RSU's generally vest over a period of 1-4 years.
c.Share options activity:
| c. | Share options activity: |
The following table lists the number of share options, the weighted average exercise prices of share options and changes that were made in the option plan to employees and directors
| | | 2021 | | | 2020 | | | 2019 | | | | | Number of options | | | Weighted Average Exercise price | | | Number of options | | | Weighted Average Exercise price | | | Number of options | | | Weighted Average Exercise price | | | | | | | | | | | | | | | | | | | | | | Outstanding Options at beginning of year | | | 3,597,811 | | | | 6.55 | | | | 2,334,432 | | | | 9.18 | | | | 2,313,249 | | | | 9.31 | | Options Granted | | | 377,790 | | | | 5.36 | | | | 1,274,379 | | | | 1.43 | | | | 95,000 | | | | 4.45 | | Options Exercised | | | (3,750 | ) | | | 2.88 | | | | 0 | | | | 0 | | | | 0 | | | | 0 | | Options Forfeited and/or expired | | | (210,833 | ) | | | 8.13 | | | | (11,000 | ) | | | 7.19 | | | | (73,817 | ) | | | 5.17 | | | | | | | | | | | | | | | | | | | | | | | | | | | | Outstanding options and at end of year | | | 3,761,018 | | | | 6.35 | | | | 3,597,811 | | | | 6.55 | | | | 2,334,432 | | | | 9.18 | | | | | | | | | | | | | | | | | | | | | | | | | | | | Option's Exercisable at end of year | | | 2,335,325 | | | | 8.34 | | | | 1,952,014 | | | | 9.98 | | | | 1,753,803 | | | | 4.76 | |
| | | 2023 | | | 2022 | | | 2021 | | | | | Number of options | | | Weighted Average Exercise price | | | Number of options | | | Weighted Average Exercise price | | | Number of options | | | Weighted Average Exercise price | | | | | | | | | | | | | | | | | | | | | | Outstanding Options at beginning of year | | | 764,767 | | | | 30.44 | | | | 537,288 | | | | 44.45 | | | | 513,973 | | | | 45.85 | | Options Granted | | | 346,950 | | | | 11.87 | | | | 320,775 | | | | 14.25 | | | | 53,970 | | | | 37.52 | | Options Exercised | | | - | | | | - | | | | (807 | ) | | | 12.23 | | | | (536 | ) | | | 20.16 | | Options Forfeited and/or expired | | | (154,230 | ) | | | 57.55 | | | | (92,489 | ) | | | 55.58 | | | | (30,119 | ) | | | 56.91 | | | | | | | | | | | | | | | | | | | | | | | | | | | | Outstanding options at end of year | | | 957,487 | | | | 19.34 | | | | 764,767 | | | | 30.44 | | | | 537,288 | | | | 44.45 | | | | | | | | | | | | | | | | | | | | | | | | | | | | Option's Exercisable at end of year | | | 462,045 | | | | 26.25 | | | | 373,681 | | | | 46.18 | | | | 333,618 | | | | 58.38 | |
MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) | Note 20: | Share‑Based Compensation (Cont.) |
The following table summarizes information about share options outstanding: | | | Options outstanding as of December 31, 2023 | | Range of exercise prices ($ ) | | Number of options | | | Weighted Average Remaining contractual life | | | Weighted average exercise price | | | | | | | | | | | | | 8.26-9.64 | | | 55,950 | | | | 9.85 | | | | 8.44 | | 11.89-14.42 | | | 711,124 | | | | 7.89 | | | | 13.20 | | 26.88-37.52 | | | 125,535 | | | | 5.62 | | | | 35.99 | | 42.14-67.06 | | | 64,878 | | | | 1.85 | | | | 63.84 | | Total | | | 957,487 | | | | 7.29 | | | | 19.34 | |
| | | Options outstanding as of December 31, 2022 | | Range of exercise prices ($ ) | | Number of options | | | Weighted Average Remaining contractual life | | | Weighted average exercise price | | | | | | | | | | | | | 12.25-14.42 | | | 470,693 | | | | 7.84 | | | | 13.60 | | 26.88-37.52 | | | 145,354 | | | | 6.43 | | | | 35.95 | | 42.14-67.06 | | | 68,662 | | | | 2.85 | | | | 63.94 | | 90.23-96.32 | | | 80,058 | | | | 0.91 | | | | 90.65 | | Total | | | 764,767 | | | | 6.40 | | | | 30.44 | |
The following table summarizes information about RSU's outstanding:
| | | RSU's 2023 | | | RSU's 2022 | | | RSU's 2021 | | | | | | | | | | | | | Outstanding at beginning of year | | | 42,013 | | | | 14,581 | | | | 10,655 | | Granted | | | 9,100 | | | | 39,286 | | | | 8,992 | | Forfeited | | | (941 | ) | | | (27 | ) | | | (215 | ) | Vested | | | (17,458 | ) | | | (11,827 | ) | | | (4,851 | ) | | | | | | | | | | | | | | | Outstanding at the end of the period | | | 32,714 | | | | 42,013 | | | | 14,581 | |
The fair value of the options and RSU's granted to employees and directors at the grant date for the years ended December 31, 2023, 2022 and 2021 was $2,320, $2,970 and $1,392 respectively. The options and RSU’s of the Company are managed by a trustee. MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) | Note 20: | Share‑Based Compensation (Cont.) |
| 1. | On March 4, 2021, the Company's Board of Directors approved the grant of: (a) 34,013 options to purchase ordinary shares and 5,669 RSU's under the "2014 Share Incentive Plan" to its CEO, officers and board members of the Company at a fair value of $663 and $196, respectively, and (b) 19,958 options to purchase ordinary shares and 3,324 RSU's under the "2014 Share Incentive Plan" to its employees at a fair value of $417 and $116, respectively. The options are exercisable for an exercise price of $ 37.52 per share. |
| 2. | Over the second quarter of 2022, the Company’s Board of Directors approved the grant of 292,203 options to purchase the Company’s ordinary shares, for an exercise price of $ 14.42 per share as well as 39,286 restricted share units (“RSU’s”) to its CEO, officers and employees. The fair value of the options and RSU’s as of the grant date, was estimated at $2,314 and $498 respectively. The above-mentioned grant includes the grant of 151,786 options to purchase the Company’s ordinary shares and 39,286 restricted share units (“RSU’s”) to the directors and the CEO of the Company which are required to be approved by the Company’s General meeting as well. The fair value of the options and RSU’s, as of the approval date, was estimated at approximately $1,171 and $498, respectively. |
| 3. | On July 19, 2022, the Company’s Shareholders General meeting approved the abovementioned grants (Note 19) to the directors and the CEO, the compensation terms of Mr. Ofer Gonen as the Company’s new Chief Executive Officer, which terms will be effective as of July 1, 2022 and the termination terms for the previous CEO. |
| 4. | On August 18, 2022, the Company’s Shareholders General meeting approved the compensation terms and grant of 28,572 options to the Chairman of the Board of Directors which approved earlier by the board. The fair value of the options as of the grant date was estimated at $284. |
| 5. | On February 15, 2023, the Company's Board of Directors approved the grant of 130,600 options to purchase ordinary shares and 9,100 RSU's under the "2014 Share Incentive Plan" toemployees, officers, board members, CEO and some consultants at fair value of $1,012 and $117, respectively. The share options vest over a period of 1-4 years and the options are exercisable for an exercise price of $ 13.32 per share. |
| 6. | On April 3, 2023, the Company's Board of Directors approved the grant of 160,400 options to purchase ordinary shares under the "2014 Share Incentive Plan", for an exercise price of $11.89 and $11.91 per share to management and board members of the Company. The share options vest over a period of 1-4 years. The fair value of the options granted, as of the grant date, was estimated at approximately $884. |
| 7. | On May 31, 2023 the Shareholders of the Company approved the increase by 1,000,000 in the number of ordinary shares available for issuance under the Company’s 2014 Equity Incentive Plan. |
| 8. | On May 31, 2023 the Shareholders of the Company approved the extension to the exercise period of options which were granted to certain of the company’s directors on April 23, 2020 for an additional five years, until April 23, 2030. According to this extension, an expense of $146 was recognized. |
MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) | Note 20: | Share‑Based Compensation (Cont.)
|
The following table summarizes information about share options outstanding as of December 31, 2021:
| | | Options outstanding as of
December 31, 2021
| | Range of exercise prices ($ )
| | Number of
options
| | | Weighted
Average
Remaining
contractual
life
| | | Weighted
average exercise
price
| | | | | | | | | | | | | | | 2,307,469
| | | | 6.19
| | | | 3.29
| | 6.02- 9.58
| | | 642,249
| | | | 3.68
| | | | 9.01
| | 12.89 - 13.76
| | | 811,300
| | | | 1.92
| | | | 12.93
| | Total
| | | 3,761,018
| | | | 4.84
| | | | 6.35
| |
The following table summarizes information about RSU's outstanding:
| | | RSU's 2021 | | | RSU's 2020 | | | RSU's 2019 | | | | | | | | | | | | | Outstanding at beginning of year | | | 74,587 | | | | 108,544 | | | | 95,833 | | Granted | | | 62,947 | | | | 0 | | | | 36,667 | | Forfeited | | | (1,505 | ) | | | 0 | | | | 0 | | Vested | | | (33,958 | ) | | | (33,958 | ) | | | (23,956 | ) | | | | | | | | | | | | | | | Outstanding at the end of the period | | | 102,071 | | | | 74,587 | | | | 108,544 | |
The fair value of the options and RSU's granted to employees and directors at the grant date for the years ended December 31, 2019, 2020 and 2021 was $441 ,$1,819 and $1,392 respectively.
The options and RSU’s of the Company are managed by a trustee.
| 1.9. | On March 24, 2019, the Company granted to its incoming CEO and chairman of the board 60,000 options (40,000 and 20,000 respectively) to purchase ordinary shares, for an exercise price of $ 4.92 per share, and 30,000 RSU's (20,000 and 10,000 respectively), under the "2014 Share Incentive Plan". The options are exercisable in accordance with the terms of the plan and will vest over three-four years. The fair value of the options and RSU's granted, as of the grant date, was estimated at approximately $164 and $158, respectively. On May 2, 2019, the general meeting of the Company approved the abovementioned grants. |
| 2. | On June 6, 2019, the Company granted to its incoming CFO 40,000 options to purchase ordinary shares, for an exercise price of $ 3.84 per share, and 6,667 RSU's, under the "2014 Share Incentive Plan". The options are exercisable in accordance with the terms of the plan and will vest over four years. The fair value of the options and RSU's granted, as of the grant date, was estimated at approximately $93 and $26, respectively.
|
MEDIWOUND LTD. AND ITS SUBSIDIARIES
Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data)
Note 20: Share‑Based Compensation (Cont.) |
| 3. | On April 23, 2020,August 15, 2023, the Company's Board of Directors approved the grant of 1,274,3797,200 options to purchase ordinary shares under the "2014 Share Incentive Plan", for an exercise price of $ 1.759.63 per share to its employees, managments anda board membersmember of the Company. The share options vest over a 1 year. The fair value of the options granted, as of the grant date, was estimated at approximately $1,819.
$42. |
| | 4.10. | On March 4, 2021,November 20, 2023, the Company's Board of Directors approved the grant of: (a) 238,090of 48,750 options to purchase ordinary shares and 39,682 RSU's under the "2014 Share Incentive Plan" to its CEO, officers and board members of the Company at a fair value of $663 and $196, respectively, and (b) 139,700 options to purchase ordinary shares and 23,265 RSU's under the "2014 Share Incentive Plan" to its employees at a fair value of $417 and $116, respectively. The options are exercisable, for an exercise price of $ 5.368.78 and $8.13 per share.share to an officer and a consultant of the Company. The share options vest over a period of 1-4 years. The fair value of the options granted, as of the grant date, was estimated at approximately $265. |
| a. | The fair value of the Company's share options granted to employees and directors for the years ended December 31, 2023, 2022 and 2021 was estimated using the binomial option pricing models using the following assumptions: |
d. The fair value of the Company's share options granted to employees and directors for the years ended December 31, 2019, 2020 and 2021 was estimated using the binomial option pricing models using the following assumptions:
| | | December 31 | | | | | 2021 | | | 2020 | | | 2019 | | | | | | | | | | | | | Dividend yield (%) | | | 0 | | | | 0 | | | | 0 | | Expected volatility of the share prices (%) | | | 55-78 | | | | 51-71 | | | | 41-53 | | Risk-free interest rate (%) | | | 0.1-1.5 | | | | 0.2-0.9 | | | | 1.85-2.45 | | Early exercise factor (%) | | | 100-150 | | | | 100-150 | | | | 150 | | Weighted average share prices (Dollar) | | | 2.88 | | | | 2.43 | | | | 4.83 | |
| | | December 31 | | | | | 2023 | | | 2022 | | | 2021 | | Dividend yield (%) | | | 0 | | | | 0 | | | | 0 | | Expected volatility of the share prices (%) | | | 61-77 | | | | 59-77 | | | | 55-78 | | Risk‑free interest rate (%) | | | 2.1-5.36 | | | | 2.1-5.2 | | | | 0.1-1.5 | | Early exercise factor (%) | | | 100-150 | | | | 100-150 | | | | 100-150 | | Weighted average share prices (Dollar) | | | 10.71 | | | | 13.22 | | | | 36.33 | |
Measurement inputs include the share price on the measurement date, the exercise price of the instrument, expected volatility (based on the weighted average volatility of the Company’s shares, over the expected term of the options), expected term of the options (based on general option holder behavior and expected share price), expected dividends, and the risk-free interest rate (based on government debentures). | a. | The Company operates in two main tax jurisdictions: Israel and Germany. As such, the Company is subject to the applicable tax rates in the jurisdictions in which it conducts its business. |
| b. | Corporate tax rate in Israel: |
Note 21: Income Tax
a.The Company operates in two main tax jurisdictions: Israel and Germany. As such, the Company is subject to the applicable tax rates in the jurisdictions in which it conducts its business..
b. Corporatestandard tax rate in Israel:the years 2021-2023 is 23%.
The tax rates relevant to the Company in the years 2019-2021 is 23%. | c. | Benefits under the Law for the Encouragement of Capital Investments: |
c. Benefits under the Law for the Encouragement of Capital Investments:
Tax benefits under the Israel Law for the Encouragement of Capital Investments, 1959 (the "Investment Law"): MEDIWOUND LTD. AND ITS SUBSIDIARIES
Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data)
Note 21: Income Tax (Cont.)
Under the Investment Law, the Company has been granted "Beneficiary Enterprise" status which provides certain benefits, including tax exemptions and reduced tax rates. Income not eligible for Beneficiary Enterprise benefits is taxed at a regular rate. MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) | Note 21: | Income Tax (Cont.) |
During the benefit period, the Company will be tax exempt in the first two years of the benefit period and subject to tax at the reduced rate of 10%- 25% for an additional period of five to eight years (depending on the percentage of foreign investments in the Company) of the benefit period. The benefit entitlement period starts from the first year that the Beneficiary Enterprise first earned taxable income and is limited to 12 years from the year in which the Company requested to have tax benefits apply. In the event of distribution of dividends from the said tax exempttax-exempt income, the amount distributed will be subject to corporate tax at the reduced rate ordinarily applicable to the Beneficiary Enterprise's income. Tax exempt income generated under the Company's "Beneficiary Enterprise" program will be subject to taxes upon dividend distribution or complete liquidation. The entitlement to the above benefits is conditional upon the Company's fulfilling the conditions stipulated by the Investment Law and regulations published thereunder. Should the Company fail to meet such requirements in the future, income attributable to its Beneficiary Enterprise programs could be subject to the statutory Israeli corporate tax rate and the Company could be required to refund a portion of the tax benefits already received, with respect to such programs. | d. | The principal tax rates applicable to the subsidiary whose place of incorporation is outside of Israel is: |
d.The principal tax rates applicable to the subsidiary whose place of incorporation is outside of Israel is:
The statutory corporate tax rate in Germany was 29.79% in 2021, 20202023, 2022 and 2019.2021.
e.Final tax assessments:
The Company has finalized its tax assessments through the 20152016 tax year. The Company's subsidiary has not received athe final tax assessment since its incorporation.for the year 2021. | f. | f.Net operating carryforward losses for tax purposes and other temporary differences:
|
As of December 31, 2021,2023, the Company had carryforward losses and other temporary differences mainly from R&D expenses together amounting to approximately $148,000.$171,757. The Company did not recognize deferred tax assets for temporary differences at the amount of approximately $9,500 because their utilization in the foreseeable future is not probable. | h. | h.Current taxes on income:
|
The Company did not record any current taxes for MediWound Ltd. in Israel for the years ended December 31, 2019, 20202023, 2022 and 2021 as a result of its carryforward losses. The current tax expenses are in respect of taxes charged outside of Israel. MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) | Note 22: | Supplementary Information to the Consolidated Statements of Profit or Loss and Other Comprehensive Income or Loss |
| a. | Additional information on Revenues: |
Major customers: BARDA contributed 56%, 51% and 76% of the Company’s total revenues in 2023, 2022 and 2021respectively. Vericel contributed 4% and 28% of the Company’s total revenues in 2023 and 2022 respectively. MTEC contributed 10% and 2.8% of the Company’s total revenues in 2023 and 2022 respectively (see also Note 18). No other customer contributed 10% or more of the Company’s revenues in 2023, 2022 and 2021. Geographic information: The revenues reported in the financial statements are based on the location of the customers, as follows: | | | Year ended December 31 | | | | | 2023 | | | 2022 | | | 2021 | | | | | | | USA (see also Note 18a, 18b, 18c) | | | 13,078 | | | | 21,872 | | | | 18,102 | | EU and other international markets | | | 5,608 | | | | 4,624 | | | | 5,661 | | | | | | | | | | | | | | | | | | | | 18,686 | | | | 26,496 | | | | 23,763 | |
| 1. | Cost of Revenues from sale of products |
| | | Year ended December 31 | | | | | 2023 | | | 2022 | | | 2021 | | | | | | | | | | | | | Salary and benefits (including share-based compensation) | | | 2,703 | | | | 1,828 | | | | 2,047 | | Subcontractors | | | 230 | | | | 58 | | | | 190 | | Depreciation and amortization | | | 672 | | | | 426 | | | | 559 | | Cost of materials | | | 916 | | | | 636 | | | | 1,039 | | Other manufacturing expenses | | | 1,207 | | | | 779 | | | | 906 | | Decrease (increase) in inventory of finished products | | | (801 | ) | | | (543 | ) | | | 242 | | | | | | | | | | | | | | | | | | | | 4,927 | | | | 3,184 | | | | 4,983 | |
| 2. | Cost of Revenues from development services |
| | | Year ended December 31 | | | | | 2023 | | | 2022 | | | 2021 | | | | | | | | | | | | | Salary and benefits | | | 1,995 | | | | 1,691 | | | | 2,003 | | Subcontractors | | | 8,182 | | | | 8,138 | | | | 7,904 | | | | | | | | | | | | | | | | | | | | 10,177 | | | | 9,829 | | | | 9,907 | |
MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) | Note 22: | Supplementary Information to the Consolidated Statements of Profit or Loss and Other Comprehensive Income or Loss (Cont.) |
Note 21: Income Tax (Cont.)
| 3. | Cost of Revenues from license agreements |
i. Theoretical tax:
The reconciliation between the tax expense, assuming that all the income and expenses, gains and losses in the statement of income were taxed at the statutory tax rate and the taxes on income recorded in profit or loss, does not provide significant information and therefore was not presented (the main reconciliation item is due to operating losses and other temporary differences for which deferred tax assets were not recognized).
| | | Year ended December 31 | | | | | 2023 | | | 2022 | | | 2021 | | | | | | | | | | | | | Salary and benefits | | | 4 | | | | 38 | | | | 102 | | Royalties payments | | | - | | | | 280 | | | | - | | | | | | | | | | | | | | | | | | | | 4 | | | | 318 | | | | 102 | | | | | | | | | | | | | | | | Total Cost of Revenues | | | 15,108 | | | | 13,331 | | | | 14,992 | |
Note 22: Discontinued Operation
On September 15, 2014, a Statement of Claim was filed against the Company by some shareholders of Polyheal (the "Plaintiffs") related to '2010 PolyHeal Agreement' in which PolyHeal granted the Company an exclusive global license to manufacture, develop and commercialize all the Polyheal Products in consideration for royalty payments. | c. | Research and development expenses: |
During December 2017, following the Tel- Aviv District Court Ruling, the Company paid the Plaintiffs approximately $1,497 in consideration for PolyHeal's shares and recorded a full provision of $6,003 which represents the purchase price for the residual number of shares that the 2010 PolyHeal Agreements contemplate would be acquired by the Company from the shareholders of PolyHeal (the “Provision”).| | | Year ended December 31 | | | | | 2023 | | | 2022 | | | 2021 | | | | | | | | | | | | | Salary and benefits (including share-based compensation) (1) | | | 3,148 | | | | 4,494 | | | | 2,864 | | Subcontractors | | | 2,833 | | | | 4,054 | | | | 6,323 | | Depreciation and amortization | | | 396 | | | | 571 | | | | 396 | | Cost of materials | | | 785 | | | | 572 | | | | 347 | | Other research and development expenses | | | 305 | | | | 490 | | | | 326 | | | | | | | | | | | | | | | | Total Research and development | | | 7,467 | | | | 10,181 | | | | 10,256 | |
| (1) | The salary costs for the year ended December 31,2022 includes one-time payment of $205 derived from termination agreement with the CMO of the company. |
On March 24, 2019, the Company entered into a settlement agreement and mutual general release with the Plaintiffs (the "Polyheal Settlement Agreement"). Pursuant to the terms of Polyheal Settlement Agreement, the Plaintiffs repaid to MediWound a portion of the amount that was ruled in their favor under the Tel Aviv District Court Ruling, and it resulted the cancellation of the 2017 Ruling that was issued by the District Court against MediWound. | d. | Selling and marketing expenses: |
| | | Year ended December 31 | | | | | 2023 | | | 2022 | | | 2021 | | | | | | | | | | | | | Salary and benefits (including share-based compensation) | | | 1,991 | | | | 1,637 | | | | 1,643 | | Marketing and consulting | | | 1,637 | | | | 1,152 | | | | 627 | | Depreciation and amortization | | | 50 | | | | 49 | | | | 44 | | Shipping and delivery | | | 356 | | | | 385 | | | | 490 | | Registration and marketing license fees | | | 810 | | | | 502 | | | | 584 | | | | | | | | | | | | | | | | | | | | 4,844 | | | | 3,725 | | | | 3,388 | |
In September 2019, the Company entered a new series of settlement agreements (the "New PolyHeal Settlement Agreements") with the majority of the shareholders of Polyheal, including Clal Biotechnology Industries Ltd., its controlling shareholder. Pursuant to the terms of New PolyHeal Settlement Agreements, the Company paid an aggregate amount of approximately $2,800 and received 14,473 shares of PolyHeal, which was classified as royalty rights arising from the Company’s ownership of shares of Polyheal.
As a result of the New PolyHeal Settlement Agreements, the Company recognized one-time profit from discontinued operation of $2,889, following the decrease of the provision which was offset by an impairment of the royalty rights and settlement fees.
In 2020 the Company finalized PolyHeal Settlement Agreements and paid $195 for 1,558 shares of PolyHeal. As of December 31, 2020, the provision for liability in respect of discontinued operation, was fully offset.
MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) Note 23: Supplementary Information to the Statements of Comprehensive Profit or Loss
a. Additional information on Revenues:
| Note 22: | Supplementary Information to the Consolidated Statements of Profit or Loss and Other Comprehensive Income or Loss (Cont.) |
| e. | General and administrative expenses: |
| | | Year ended December 31 | | | | | 2023 | | | 2022 | | | 2021 | | Salary and benefits (including share‑based compensation) | | | 3,521 | | | | 3,344 | | | | 2,905 | | Professional fees | | | 2,189 | | | | 2,589 | | | | 2,480 | | Depreciation and amortization | | | 185 | | | | 225 | | | | 239 | | Other | | | 873 | | | | 762 | | | | 724 | | | | | | | | | | | | | | | | | | | | 6,768 | | | | 6,920 | | | | 6,348 | |
| f. | Other (income) expenses: |
Major customers:
BARDA contributed 76% of the Company’s total revenues in 2021, 83% in 2020, and 34% in 2019. Verical contributed 55% in 2019. (see also Note 18b).
NoThe other customer contributed 10% or more of the Company’s revenues in 2021, 2020 and 2019.
Revenue Re-classification:
Revenues in the amount of $383 from distributions agreements were classified from revenues from sale of productsone-time income amounted $211 for the year ended December 31, December 2020.
Geographic information:,
The revenues reported in2023, is associated with the financial statements are based on the locationtermination of the customers, as follows:
| | | Year ended December 31 | | | | | 2021 | | | 2020 | | | 2019 | | | | | | | | | | | | | USA ( see also Note 18a, 18b) | | | 18,102 | | | | 18,030 | | | | 28,504 | | EU and other international markets | | | 5,661 | | | | 3,733 | | | | 3,285 | | | | | | | | | | | | | | | | | | | | 23,763 | | | | 21,763 | | | | 31,789 | |
1. Cost of Revenues from sale of productssub-lease agreement (see note 11a).
| | | Year ended December 31 | | | | | 2021 | | | 2020 | | | 2019 | | | | | | | | | | | | | Salary and benefits (including share-based compensation) | | | 2,201 | | | | 2,139 | | | | 1,916 | | Subcontractors | | | 204 | | | | 153 | | | | 89 | | Depreciation and amortization | | | 603 | | | | 554 | | | | 512 | | Cost of materials | | | 1,091 | | | | 704 | | | | 456 | | Other manufacturing expenses | | | 953 | | | | 840 | | | | 657 | | Decrease in inventory of finished products | | | 242 | | | | 155 | | | | 344 | | Allotment of manufacturing costs to R&D | | | (311 | ) | | | (1,394 | ) | | | (1,621 | ) | | | | | | | | | | | | | | | | | | | 4,983 | | | | 3,151 | | | | 2,353 | |
2. Cost of Revenues from development servicesThe other one-time expenses amounted $684 for the year ended December 31, 2022, are associated with the management changes and FDA milestone payment fee (see Note 18b).
| | | Year ended December 31 | | | | | 2021 | | | 2020 | | | 2019 | | | | | | | | | | | | | Salary and benefits | | | 2,003 | | | | 2,320 | | | | 1,404 | | Subcontractors | | | 7,904 | | | | 8,747 | | | | 7,412 | | | | | | | | | | | | | | | | | | | | 9,907 | | | | 11,067 | | | | 8,816 | |
| g. | Financial income and expense: |
| | | Year ended December 31 | | | | | 2023 | | | 2022 | | | 2021 | | Financial income: | | | | | | | | | | | | | | | | | | | | | Interest income | | | 2,341 | | | | 270 | | | | 11 | | Revaluation of liabilities in respect of IIA grants | | | - | | | | 132 | | | | - | | Revaluation of Warrants | | | 8,310 | | | | - | | | | - | | Exchange differences, net | | | - | | | | 59 | | | | - | | | | | | | | | | | | | | | | | | | | 10,651 | | | | 461 | | | | 11 | | Financial expense: | | | | | | | | | | | | | | | | | | | | | | | | | | | Revaluation of liabilities in respect of IIA grants | | | 427 | | | | - | | | | 903 | | Financing income on net investment in lease | | | 274 | | | | 102 | | | | 120 | | Finance expenses in respect of deferred revenues | | | 8 | | | | 54 | | | | 143 | | Revaluation of liabilities in respect of TEVA | | | 468 | | | | 533 | | | | 590 | | Exchange differences, net | | | 654 | | | | - | | | | 511 | | Revaluation of Warrants | | | - | | | | 8,977 | | | | - | | Issuance expenses of warrants through profit and loss | | | - | | | | 1,911 | | | | - | | Other | | | 61 | | | | 60 | | | | 47 | | | | | | | | | | | | | | | | | | | | 1,892 | | | | 11,637 | | | | 2,314 | | | | | | | | | | | | | | | | Financial income (expenses), net | | | 8,759 | | | | (11,176 | ) | | | (2,303 | ) |
MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) | Note 23: | Net Loss Per Share |
| a. | Details of the number of shares and loss used in the computation of loss per share: |
Year ended December 31 | | 2023 | | | 2022 | | | 2021 | | Weighted average number of shares | | | Loss | | | Weighted average number of shares | | | Loss | | | Weighted average number of shares | | | Loss | | | | | | | | | | | | | | | | | | | | | | 9,013,144 | | | | (6,716 | ) | | | 4,987,069 | | | | (19,599 | ) | | | 3,892,068 | | | | (13,551 | ) |
| | | Year ended December 31 | | | | | 2023 | | | 2022 | | | 2021 | | | | | | | | | | | | | Basic and diluted loss per share: | | | (0.75 | ) | | | (3.93 | ) | | | (3.48 | ) |
In 2023, 2,614,297 warrants, 957,487 options and 32,714 RSU’s were excluded from the diluted weighted average number of Ordinary Shares calculation as their effect would have been anti-dilutive. In 2022, 2,614,297 warrants, 764,767 options and 42,013 RSU’s were excluded from the diluted weighted average number of Ordinary Shares calculation as their effect would have been anti-dilutive. In addition, the impact of 1,407,583 pre-funded warrants which were exercised in December 2022, have not taken in the diluted weighted average number of Ordinary Shares calculation as their effect would have been anti-dilutive as well. Note 23: Supplementary Information to the Statements of Comprehensive Profit or Loss (Cont.)
3. Cost of Revenues from license agreements
| | | Year ended December 31 | | | | | 2021 | | | 2020 | | | 2019 | | | | | | | | | | | | | | Salary and benefits | | | 102 | | | | 0 | | | | 0 | | | Royalties payments | | | 0 | | | | 0 | | | | 680 | | | | | | | | | | | | | | | | | | | | 102 | | | | 0 | | | | 680 | | | | | | | | | | | | | | | | | Total Cost of Revenues | | | 14,992 | | | | 14,218 | | | | 11,849 | |
c. Research and development expenses, net of participations:
| Note 24: | Balances and Transactions with Related Parties and Key Officers |
| a. | Related parties consist of: |
| • | Clal Biotechnologies Industries Ltd.- Related party (see also note 11) |
| • | Directors of the Company. |
| 1. | Balances with related parties: |
| | | Year ended December 31 | | | | | 2021 | | | 2020 | | | 2019 | | | | | | | | | | | | | Salary and benefits (including share-based compensation) | | | 2,811 | | | | 2,094 | | | | 2,965 | | Subcontractors | | | 6,309 | | | | 3,173 | | | | 4,694 | | Depreciation and amortization | | | 352 | | | | 346 | | | | 342 | | Cost of materials | | | 295 | | | | 517 | | | | 311 | | Allotment of manufacturing costs | | | 209 | | | | 1,394 | | | | 1,621 | | Other research and development expenses | | | 280 | | | | 174 | | | | 137 | | | | | | | | | | | | | | | | Research and development, gross | | | 10,256 | | | | 7,698 | | | | 10,070 | | | | | | | | | | | | | | | | Participations: | | | | | | | | | | | | | BARDA funds | | | 0 | | | | 0 | | | | (3,785 | ) | Revaluation of liabilities in respect of IIA grants | | | 0 | | | | 0 | | | | (1,316 | ) | | | | | | | | | | | | | | | | | | | 10,256 | | | | 7,698 | | | | 4,969 | |
| | Other Payables | | Related Party: | | | | | As of December 31, 2023 | | | - | | | As of December 31, 2022 | | | 177 | | | | | | | | Directors: | | | | | | As of December 31, 2023 | | | 83 | | | As of December 31, 2022 | | | 130 | |
d. Selling and marketing expenses:
| | | Year ended December 31 | | | | | 2021 | | | 2020 | | | 2019 | | | | | | | | | | | | | Salary and benefits (including share based compensation) (1) | | | 1,643 | | | | 1,700 | | | | 2,028 | | Marketing and medical support | | | 627 | | | | 740 | | | | 1,298 | | Depreciation and amortization | | | 44 | | | | 82 | | | | 49 | | Shipping and delivery | | | 490 | | | | 282 | | | | 200 | | Registration and marketing license fees | | | 584 | | | | 424 | | | | 489 | | | | | | | | | | | | | | | | | | | | 3,388 | | | | 3,228 | | | | 4,064 | |
(1) The salary costs for the year ended December 31,2020 includes one time payment of $243 derived from restructuring astrategy at the EU Subsidiary.
MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) Note 23: Supplementary Information to the Statements of Comprehensive Profit or Loss (Cont.)
e. General and administrative expenses:
| Note 24: | Balances and Transactions with Related Parties and Key Officers (Cont.) |
| | | Year ended December 31 | | | | | 2021 | | | 2020 | | | 2019 | | | | | | | | | | | | | Salary and benefits (including share-based compensation) | | | 2,905 | | | | 2,784 | | | | 2,621 | | Professional fees | | | 2,480 | | | | 2,267 | | | | 1,628 | | Depreciation and amortization | | | 239 | | | | 108 | | | | 247 | | Other | | | 724 | | | | 300 | | | | 746 | | | | | | | | | | | | | | | | | | | | 6,348 | | | | 5,459 | | | | 5,242 | |
| 2. | Transactions with related parties: |
The other one-time expenses amounted $1,172 for the year ended December 31, 2019, are associated with the review and assessment of the strategic deal with Vericel ( see Note 18b).Rental fee:
| | | Year ended December 31 | | | | | 2023 | | | 2022 | | | 2021 | | Related party | | | 267 | | | | 457 | | | | 469 | |
g. Financial income and expense:Professional fee *:
| | | Year ended December 31 | | | | | 2023 | | | 2022 | | | 2021 | | | | | | | | | | | | | Directors | | | 502 | | | | 484 | | | | 375 | | Related party | | | 25 | | | | 63 | | | | 85 | | | | | | 527 | | | | 547 | | | | 460 | | Number of Directors | | | ***9 | | | | **10 | | | | 8 | |
| * | Not included share- based compensation detailed in Note 20. |
| ** | During 2022 two members of the board of directors were replaced. |
| *** | During 2023 three members of the board have left and one member of the board was replaced. At the end of 2023, the total board member consisted of five members. |
| 1. | Balances with Key Officers of the Company |
| | Other Payables | Key Officers of the Company | | | | | | | | As of December 31, 2023 | | | 518 | | | As of December 31, 2022 | | | 754 | |
| | | Year ended December 31 | | | | | 2021 | | | 2020 | | | 2019 | | | Financial income: | | | | | | | | | | | | | | | | | | | | | | | | | Interest income | | | 11 | | | | 297 | | | | 434 | | | Revaluation of liabilities in respect of the purchase of shares | | | 0 | | | | 433 | | | | 0 | | | Exchange differences, net | | | 0 | | | | 113 | | | | 122 | | | | | | | | | | | | | | | | | | | | 11 | | | | 843 | | | | 556 | | | Financial expense: | | | | | | | | | | | | | | | | | | | | | | | | | | | | Interest in respect of IIA grants | | | 903 | | | | 832 | | | | 925 | | | Revaluation of liabilities in respect of IFRS16 | | | 120 | | | | 144 | | | | 140 | | | Finance expenses in respect of deferred revenues | | | 143 | | | | 247 | | | | 161 | | | Revaluation of liabilities in respect of the purchase of shares | | | 590 | | | | 0 | | | | 1,690 | | | Exchange differences, net | | | 511 | | | | 0 | | | | 0 | | | Other | | | 47 | | | | 56 | | | | 67 | | | | | | | | | | | | | | | | | | | | 2,314 | | | | 1,279 | | | | 2,983 | | | | | | | | | | | | | | | | | Financial expenses, net | | | (2,303 | ) | | | (436 | ) | | | (2,427 | ) |
MEDIWOUND LTD. AND ITS SUBSIDIARIES Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data) Note 24: Net Profit (Loss) Per Share
a. Details of the number of shares and loss used in the computation of loss per share from continuing operations:
| Year ended December 31 | | | 2021 | | | 2020 | | | 2019 | | Weighted average number of shares | | | Loss | | | Weighted average number of shares | | | Loss | | | Weighted average number of shares | | | Profit | | | | | | | | | | | | | | | | | | | | | | 27,244,475 | | | | (13,551 | ) | | | 27,209,878 | | | | (9,276 | ) | | | 27,178,839 | | | | 2,066 | |
b. Details of the number of shares and profit (loss) used in the computation of profit or (loss) per share from discontinued operation:
| Year ended December 31 | | | 2021 | | | 2020 | | | 2019 | | Weighted average number of shares | | | Profit | | | Weighted average number of shares | | | Profit | | | Weighted average number of shares | | | Profit | | | | | | | | | | | | | | | | | | | | | | 0 | | | | 0 | | | | 27,209,878 | | | | 80 | | | | 27,178,839 | | | | 2,889 | |
c. Net profit (loss) per share from continuing and discontinued operations:
| | | Year ended December 31 | | | | | 2021 | | | 2020 | | | 2019 | | Basic and Diluted loss per share: | | | | | | | | | | Profit (loss) from from continuing operations | | | (0.50 | ) | | | (0.34 | ) | | | 0.08 | | | | | | | | | | | | | | | | Profit from discontinued operation | | | 0 | | | | 0 | | | | 0.10 | | | | | | | | | | | | | | | | Profit (loss) per share | | | (0.50 | ) | | | (0.34 | ) | | | 0.18 | |
Note 25: Balances and Transactions With Related Parties and Key Officer
a. Related parties consist of:
• Clal Biotechnologies Industries Ltd.- Parent Company.
•Directors of the Company.
•CureTech Ltd.-Sister Company.
MEDIWOUND LTD. AND ITS SUBSIDIARIES
Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data)
Note 24: 25: | Balances and Transactions Withwith Related Parties and Key Officers(Cont.) |
1. Balances of related parties:
| | Other Payables
| | Parent Company (1):
| | | | | | | | As of December 31, 2020
| | | 138
| | As of December 31, 2021
| | | 144
| | | | | | | | | | | | | | | | | As of December 31, 2020
| | | 86
| | As of December 31, 2021
| | | 96
| |
2. Transactions with related parties:
Rental fee:
| | | Year ended December 31 | | | | | 2021 | | | 2020 | | | 2019 | | | | | | | | | | | | | Parent Company | | | 469 | | | | 446 | | | | 415 | | Sister Company | | | 0 | | | | 0 | | | | (59 | ) |
Professional fee:
| | | Year ended December 31 | | | | | 2021 | | | 2020 | | | 2019 | | | | | | | | | | | | | Directors | | | 375 | | | | 272 | | | | 249 | | Parent Company | | | 85 | | | | 54 | | | | 52 | | | | | | 460 | | | | 326 | | | | 301 | | Number of Directors | | | 8 | | | | 8 | | | | 6 | |
| • Not included share based compensation detailed in Note 20.
|
MEDIWOUND LTD. AND ITS SUBSIDIARIES
Notes to the Consolidated Financial Statements
U.S. dollars in thousands (except of share and per share data)
Note 25: Balances and Transactions With Related Parties and Key Officers (Cont.)
| 2. | 1. | BalancesCompensation of Key Officers of the CompanyCompany: |
| | Other Payables | | Key Officers of the Company | | | | | | | | As of December 31, 2021 | | | 353
| |
Represents the officer’s gross salary, bonuses and vacation provisions.
2. Compensation of Key Officers of the Company:
The following amounts disclosed in the table are recognized as an expense during the reporting period related to officers: | | | Year ended December 31 | | | | | 2021 | | | 2020 | | | 2019 | | Short-term employee benefits (*) | | | 1,788 | | | | 1,993 | | | | 2,533 | | Share-based compensation | | | 518 | | | | 467 | | | | 565 | | | | | | | | | | | | | | | | | | | | 2,306 | | | | 2,460 | | | | 3,098 | | Number of officers | | | 5 | | | | 5 | | | | 7 | |
| | | Year ended December 31 | | | | | 2023 | | | | 2022 | | | | 2021 | Short-term employee benefits (*)(**) | | | 2,084 | | | | 2,880 | | | | 1,788 | | Share-based compensation | | | 757 | | | | 797 | | | | 518 | | | | | | | | | | | | | | | | | | | | 2,841 | | | | 3,677 | | | | 2,306 | | Number of officers (***) | | | 8 | | | | 7 | | | | 5 | |
(*) One-time expenses amounted $309 for the year ended December 31, 2022, are associated with the management changes. (**) The amount for 2019 includes one-time payments for previous-CEO on the amount of $196. In December 2007, the Company's board of directors approved one‑time bonus payments to the Chief Medical Officer in the amounts of $ 120, to be paidwhich was recorded in profit and loss in December 2022 upon achieving marketing approval in the United States.
(***) During 2023 two key officers were replaced. At the end of 2023 the total key officers consist of six officers.
Note 26:Subsiquents| Note 25: | Subsequent events: |
On March 7, 2022, the Company completed a follow-on public offering. A total of 5,208,333 new ordinary shares were issued in consideration to offering price of $1.92 per share. The net proceeds, were $8,662, after deducting commissions and other offering expenses. MediWound also granted the underwriters a 30-day option to purchase up to an additional 781,249 ordinary shares at the public offering price, less underwriting discounts and commissions at an additional net proceeds of $1,388. | 1. | On January 1, 2024, the company entered into a new lease agreement for 380.21 square meters of office, in Yavne, Israel, close to the main office, the agreement is for two years with options for additional three years. The additional leased area is needed to support the company in extending its activity. The annual fee for this agreement is ILS 437 (approximately $120).In addition, certain entities affiliated with CBI purchased 1,458,333 of ordinary shares in the obove-mentioned offering at the public offering price.
|
| 2. | On February 26, 2024, the Company granted to employees, officers, board members, CEO and some consultants 311,022 share options for an exercise price of $12.73 per share and 30,482 RSUs. The share options vest over a period of 1-4 years. The grant to the directors and CEO is subject to the approval of the next shareholders annual meeting. |
| 3. | On March 6, 2024, 37,106 warrants were exercised to the Company shares for exercise price of $13.475 per ordinary share, in accordance with the terms of the warrants (See also note 19 C.2). |
F - 4947
| | | |
|