UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 20-F
(Mark One)
| ¨ | REGISTRATION STATEMENT PURSUANT TO SECTION 12(b) OR (g) OF THE SECURITIES EXCHANGE ACT OF 1934 |
OR
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 20142015
OR
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
OR
| ¨ | SHELL COMPANY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Date of event requiring this shell company report
Commission file number 001-36153File Number 001-36697
DBV TECHNOLOGIES S.A.
(Exact name of Registrant as specified in its charter and translation of Registrant’s name into English)
France
(Jurisdiction of incorporation or organization)
Green Square-Bâtiment D177-181 avenue Pierre Brossolette
80/84 rue des Meuniers
92220 Bagneux92120 Montrouge France
(Address of principal executive offices)
Dr. Pierre-Henri Benhamou
Chairman and Chief Executive Officer
DBV Technologies S.A.
Green Square-Bâtiment D177-181 avenue Pierre Brossolette
80/84 rue des Meuniers
92220 Bagneux92120 Montrouge France
Tel: +33 1 55 42 78 78 Fax: +33 1 43 26 10 83
(Name, Telephone, E-mail and/or Facsimile number and Address of Company Contact Person)
Securities registered or to be registered pursuant to Section 12(b) of the Act.
Title of each class | Name of each exchange on which registered | |
| American Depositary Shares, each representing one-half of one ordinary share, nominal value €0.10 per share | The Nasdaq Stock Market LLC | |
| Ordinary shares, nominal value €0.10 per share* | The Nasdaq Stock Market LLC* |
| * | Not for trading, but only in connection with the registration of the American Depositary Shares. |
Securities registered or to be registered pursuant to Section 12(g) of the Act. None
Securities for which there is a reporting obligation pursuant to Section 15(d) of the Act. None
Indicate the number of outstanding shares of each of the issuer’s classes of capital or common stock as of the close of the period covered by the Annual Report.annual report.
Ordinary shares, nominal value €0.10 per share: 19,160,66124,205,129 as of December 31, 20142015
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. ¨ Yes x No
If this report is an annual or transition report, indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934. ¨ Yes x No
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. x Yes ¨ No
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). ¨ Yes x No
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, or a non-accelerated filer. See definition of “accelerated filer and large accelerated filer” in Rule 12b-2 of the Exchange Act. (Check one):
Large accelerated filer ¨x Accelerated filer ¨ Non-accelerated filer x¨
Indicate by check mark which basis of accounting the registrant has used to prepare the financial statements included in this filing:
U.S. GAAP ¨ | International Financial Reporting Standards as issued by the International Accounting Standards Board x | Other ¨ |
If “Other” has been checked in response to the previous question, indicate by check mark which financial statement item the registrant has elected to follow. ¨ Item 17 ¨ Item 18
If this is an Annual Report,annual report, indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). ¨ Yes x No
| PAGE | ||||||
| INTRODUCTION | ||||||
| PART I | ||||||
| Item 1. | 4 | |||||
| Item 2. | 4 | |||||
| Item 3. | 4 | |||||
| 4 | ||||||
| 6 | ||||||
| 6 | ||||||
| 6 | ||||||
| Item 4. | ||||||
| Item 4A. | ||||||
| Item 5. | ||||||
| Item 6. | ||||||
| Item 7. | ||||||
| Item 8. | ||||||
i
| Item 9. | ||||||
| Item 10. | ||||||
| Item 11. | ||||||
| Item 12. | ||||||
| PART II | ||||||
| Item 13. | ||||||
| Item 14. | Material Modifications to the Rights of Security Holders and Use of Proceeds | |||||
| Item 15. | ||||||
| Item 16A. | ||||||
| Item 16B. | ||||||
| Item 16C. | ||||||
| Item 16D. | ||||||
| Item 16E. | Purchases of Equity Securities by the Issuer and Affiliated Purchasers | |||||
| Item 16F. | ||||||
| Item 16G. | ||||||
| Item 16H. | ||||||
| PART III | ||||||
| Item 17. | ||||||
| Item 18. | ||||||
| Item 19. | ||||||
ii
Unless otherwise indicated, “DBV,” “the company,” “our company,” “we,” “us” and “our” refer to DBV Technologies S.A. and its consolidated subsidiary.
We own various trademark registrations and applications, and unregistered trademarks and servicemarks, including “Diallertest®“Diallertest®,” “Viaskin®“Viaskin®,” “EPIT™,” “DBV Technologies®Technologies®” and our corporate logo. All other trademarks or trade names referred to in this Annual Report on Form 20-F are the property of their respective owners. Trade names, trademarks and service marks of other companies appearing in this Annual Report on Form 20-F are the property of their respective holders. Solely for convenience, the trademarks and trade names in this Annual Report on Form 20-F may be referred to without the® and ™ symbols, but such references should not be construed as any indicator that their respective owners will not assert, to the fullest extent under applicable law, their rights thereto. We do not intend to use or display other companies’ trademarks and trade names to imply a relationship with, or endorsement or sponsorship of us by, any other companies.
Our audited consolidated financial statements have been prepared in accordance with International Financial Reporting Standards, or IFRS, as issued by the International Accounting Standards Board, or IASB. Our consolidated financial statements are presented in euros. All references in this Annual Report on Form 20-F to “$,” “US$,” “U.S.$,” “U.S. dollars,” “dollars” and “USD” mean U.S. dollars and all references to “€” and “euros” mean euros, unless otherwise noted. Throughout this Annual Report on Form 20-F, references to ADSs mean ADSs or ordinary shares represented by ADSs, as the case may be.
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 20-F contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended, that are based on our management’s beliefs and assumptions and on information currently available to our management. All statements other than present and historical facts and conditions contained in this Annual Report on Form 20-F, including statements regarding our future results of operations and financial positions, business strategy, plans and our objectives for future operations, are forward-looking statements. When used in this Annual Report on Form 20-F, the words “anticipate,” “believe,” “can,” “could,” “estimate,” “expect,” “intend,” “is designed to,” “may,” “might,” “plan,” “potential,” “predict,” “objective,” “should,” or the negative of these and similar expressions identify forward-looking statements. Forward-looking statements include, but are not limited to, statements about:
You should refer to the section of this Annual Report on Form 20-F titled “Item 3.D—Risk Factors” for a discussion of important factors that may cause our actual results to differ materially from those expressed or implied by our forward-looking statements. As a result of these factors, we cannot assure you that the forward-looking statements in this Annual Report on Form 20-F will prove to be accurate. Furthermore, if our forward-looking statements prove to be inaccurate, the inaccuracy may be material. In light of the significant uncertainties in these forward-looking statements, you should not regard these statements as a representation or warranty by us or any other person that we will achieve our objectives and plans in any specified time frame or at all. We undertake no obligation to publicly update any forward-looking statements, whether as a result of new information, future events or otherwise, except as required by law.
You should read this Annual Report on Form 20-F and the documents that we reference in this Annual Report on Form 20-F and have filed as exhibits to this Annual Report on Form 20-F completely and with the understanding that our actual future results may be materially different from what we expect. We qualify all of our forward-looking statements by these cautionary statements.
This Annual Report on Form 20-F contains market data and industry forecasts that were obtained from industry publications. These data involve a number of assumptions and limitations, and you are cautioned not to give undue weight to such estimates. We have not independently verified any third-party information. While we believe the market position, market opportunity and market size information included in this Annual Report on Form 20-F is generally reliable, such information is inherently imprecise.
| Item 1. |
Not applicable.
| Item 2. | Offer Statistics and Expected Timetable. |
Not applicable.
| Item 3. | Key Information. |
| A. | Selected Financial Data |
Our consolidated audited financial statements have been prepared in accordance with IFRS, as issued by the IASB. We derived the selected statements of consolidated income data for the years ended December 31, 2012, 2013, 2014 and 20142015 and selected statements of consolidated financial position data as of December 31, 2012, 2013, 2014 and 20142015 from our consolidated audited financial statements included elsewhere in this Annual Report on Form 20-F. The selected consolidated statement of income data for the year ended December 31, 2012 and the selected consolidated financial position data as of December 31, 2012 have been derived from our audited consolidated financial statements and notes thereto which are not included in this Annual Report on Form 20-F. This data should be read together with, and is qualified in its entirety by reference to, “Item 5. Operating and Financial Review and Prospects” as well as our financial statements and notes thereto appearing elsewhere in this Annual Report on Form 20-F. Our historical results are not necessarily indicative of the results to be expected in the future.
Statement of Income (Loss) Data:Data (in thousands, except share and per share data):
| 2012 (3) | 2013 (3) | 2014 | Year Ended December 31, | |||||||||||||||||||||||||||||||||
| Euro | Euro | Euro | US$(1) | 2012(1) | 2013(1) | 2014 | 2015 | |||||||||||||||||||||||||||||
| Euros | Euros | Euros | Euros | US$(2) | ||||||||||||||||||||||||||||||||
Operating income | € | 2,776,588 | € | 3,826,313 | € | 4,761,522 | $ | 5,761,918 | 2,777 | 3,826 | 4,762 | 6,166 | 6,696 | |||||||||||||||||||||||
Operating expenses: | ||||||||||||||||||||||||||||||||||||
Cost of goods sold | 82,958 | 102,366 | 136,296 | 164,932 | 83 | 102 | 136 | 128 | 139 | |||||||||||||||||||||||||||
Research and development | 11,499,368 | 17,366,538 | 21,143,442 | 25,585,679 | 11,499 | 17,366 | 21,143 | 34,234 | 37,175 | |||||||||||||||||||||||||||
General and administration | 4,598,699 | 6,309,750 | 8,117,664 | 9,823,185 | ||||||||||||||||||||||||||||||||
Selling, general and administration | 4,599 | 6,310 | 8,118 | 17,350 | 18,840 | |||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
| ||||||||||||||||||||||||||||
Total operating expenses | 16,181,025 | 23,778,654 | 29,397,402 | 35,573,796 | 16,181 | 23,779 | 29,397 | 51,712 | 56,154 | |||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
| ||||||||||||||||||||||||||||
Operating profit (loss) | (13,404,437 | ) | (19,952,340 | ) | (24,635,880 | ) | (29,811,878 | ) | (13,404 | ) | (19,952 | ) | (24,636 | ) | (45,546 | ) | (49,458 | ) | ||||||||||||||||||
|
|
|
|
|
|
|
|
| ||||||||||||||||||||||||||||
Financial profit (loss) | 492,337 | 645,925 | 624,000 | 755,102 | 492 | 646 | 624 | 871 | 946 | |||||||||||||||||||||||||||
Net profit (loss) | € | (12,912,100 | ) | € | (19,306,416 | ) | € | (24,011,880 | ) | $ | (29,056,776 | ) | (12,912 | ) | (19,306 | ) | (24,012 | ) | (44,674 | ) | (48,513 | ) | ||||||||||||||
|
|
|
|
|
|
|
|
| ||||||||||||||||||||||||||||
Earnings (losses) per share | ||||||||||||||||||||||||||||||||||||
Basic | € | (1.05 | ) | € | (1.42 | ) | € | (1.49 | ) | $ | (1.81 | ) | € | (1.05 | ) | € | (1.42 | ) | € | (1.49 | ) | € | (2.08 | ) | $ | (2.26 | ) | |||||||||
|
|
|
|
|
|
|
|
| ||||||||||||||||||||||||||||
Diluted | € | (1.05 | ) | € | (1.42 | ) | € | (1.49 | ) | $ | (1.81 | ) | € | (1.05 | ) | € | (1.42 | ) | € | (1.49 | ) | € | (2.08 | ) | $ | (2.26 | ) | |||||||||
|
|
|
|
|
|
|
|
| ||||||||||||||||||||||||||||
Number of shares used for computing | ||||||||||||||||||||||||||||||||||||
Basic | 12,326,779 | 13,604,687 | 16,086,247 | 16,086,247 | 12,326,779 | 13,604,687 | 16,086,247 | 21,522,342 | 21,522,342 | |||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
| ||||||||||||||||||||||||||||
Diluted | 12,326,779 | 13,604,687 | 16,086,247 | 16,086,247 | 12,326,779 | 13,604,687 | 16,086,247 | 21,522,342 | 21,522,342 | |||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
| ||||||||||||||||||||||||||||
| (1) | The statement of consolidated income (loss) as of December 31, 2013 and 2012 corresponds solely to DBV Technologies S.A., as the company had no consolidated subsidiary as of this date. |
| (2) | Translated solely for convenience into dollars at the noon buying rate of |
| See Note 22 to our financial statements for further details on the calculation of basic and diluted loss per ordinary share. |
Statement of Financial Position Data (in thousands, except share and per share data):
| As of December 31, | ||||||||||||||||||||
| 2012 (1) | 2013 (1) | 2014 | 2015 | |||||||||||||||||
| Euros | Euros | Euros | Euros | US$(2) | ||||||||||||||||
Cash and cash equivalents | 38,348 | 39,403 | 114,583 | 323,381 | 351,159 | |||||||||||||||
Total assets | 42,975 | 46,236 | 125,416 | 343,280 | 372,768 | |||||||||||||||
Total shareholders’ equity | 39,173 | 40,395 | 115,445 | 322,076 | 349,742 | |||||||||||||||
Trade non-current liabilities | 632 | 1,607 | 4,419 | 5,183 | 5,628 | |||||||||||||||
Total current liabilities | 3,170 | 4,234 | 5,552 | 16,021 | 17,397 | |||||||||||||||
Total liabilities | 3,802 | 5,841 | 9,971 | 21,204 | 23,025 | |||||||||||||||
Total liabilities and shareholders’ equity | 42,975 | 46,236 | 125,416 | 343,280 | 372,768 | |||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
| The statement of consolidated |
Statement of Financial Position Data:
| As of December 31, | ||||||||||||||||
| 2012 (2) | 2013 (2) | 2014 | ||||||||||||||
| Euro | Euro | Euro | US$(1) | |||||||||||||
Cash and cash equivalents | € | 38,348,130 | € | 39,402,761 | € | 114,583,141 | $ | 138,657,059 | ||||||||
Total assets | 42,974,817 | 46,236,009 | 125,415,511 | 151,765,310 | ||||||||||||
Total shareholders’ equity | 39,173,135 | 40,394,685 | 115,444,959 | 139,699,945 | ||||||||||||
Trade non-current liabilities | 631,592 | 1,607,228 | 4,418,902 | 5,347,313 | ||||||||||||
Total current liabilities | 3,170,090 | 4,234,096 | 5,551,650 | 6,718,052 | ||||||||||||
Total liabilities | 3,801,682 | 5,841,324 | 9,970,552 | 12,065,365 | ||||||||||||
Total liabilities and shareholders’ equity | 42,974,817 | 46,236,009 | 125,415,511 | 151,765,310 | ||||||||||||
|
|
|
|
|
|
|
| |||||||||
| Translated solely for convenience into dollars at the noon buying rate of |
Exchange Rate Information
In this Annual Report on Form 20-F, for convenience only, we have translated certain euro amounts reflected in our financial statements as of and for the year ended December 31, 20142015 into U.S. dollars at the rate of €1.00$1.00 = US$1.2101,€1.0859, the noon buying rate of the Federal Reserve Bank of New York for euros onat December 31, 2014.2015. You should not assume that, on that or on any other date, one could have converted these amounts of euros into U.S. dollars at that or any other exchange rate.
The following table sets forth, for each period indicated, the low and high exchange rates for euros expressed in U.S. dollars, the exchange rate at the end of such period and the average of such exchange rates on the last day of each month during such period, based on the noon buying rate inof the CityFederal Reserve Bank of New York for the euro.euros into dollars. As used in this document,Annual Report, the term “noon buying rate” refers to the rate of exchange for the euro, expressed in U.S. dollars per euro, as certified by the Federal Reserve Bank of New York for customs purposes.
| Year Ended December 31, | ||||||||||||||||||||
| 2010 | 2011 | 2012 | 2013 | 2014 | ||||||||||||||||
High | 1.4536 | 1.4875 | 1.3463 | 1.3816 | 1.3927 | |||||||||||||||
Low | 1.1959 | 1.2926 | 1.2062 | 1.2774 | 1.2101 | |||||||||||||||
Rate at end of period | 1.3269 | 1.2973 | 1.3187 | 1.3779 | 1.2101 | |||||||||||||||
Average rate per period | 1.3216 | 1.4002 | 1.2909 | 1.3303 | 1.3297 | |||||||||||||||
High Low Rate at end of period Average rate per period Year Ended December 31, 2011 2012 2013 2014 2015 1.4875 1.3463 1.3816 1.3927 1.2015 1.2926 1.2062 1.2774 1.2101 1.0524 1.2973 1.3186 1.3779 1.2101 1.0859 1.3931 1.2854 1.3275 1.3306 1.1098
The following table sets forth, for each of the last six months, the low and high exchange rates for euros expressed in U.S. dollars and the exchange rate at the end of the monthsuch period, based on the noon buying rate as described above.
| October 2014 | November 2014 | December 2014 | January 2015 | February 2015 | March 2015 | |||||||||||||||||||
High | 1.2812 | 1.2554 | 1.2504 | 1.2015 | 1.1462 | 1.1212 | ||||||||||||||||||
Low | 1.2517 | 1.2394 | 1.2101 | 1.1279 | 1.1197 | 1.0524 | ||||||||||||||||||
Rate at end of period | 1.2530 | 1.2438 | 1.2101 | 1.1290 | 1.1197 | 1.0741 | ||||||||||||||||||
On December 31, 2014, the noon buying rate of the Federal Reserve Bank of New York for the euro was €1.00 = US$1.2101. Unless otherwise indicated, currency translations in this Annual Report on Form 20-F reflect theeuro.
| October 2015 | November 2015 | December 2015 | January 2016 | February 2016 | March 2016 | |||||||||||||||||||
High | 1.1437 | 1.1026 | 1.1025 | 1.0964 | 1.1362 | 1.1390 | ||||||||||||||||||
Low | 1.0963 | 1.0562 | 1.0573 | 1.0743 | 1.0868 | 1.0845 | ||||||||||||||||||
Rate at end of period | 1.1042 | 1.0562 | 1.0859 | 1.0832 | 1.0868 | 1.1390 | ||||||||||||||||||
On December 31, 2014 exchange rate.
On April 24, 2015, the noon buying rate of the Federal Reserve Bank of New York for the euro was €1.00$1.00 = $1.0876.€1.0859. Unless otherwise indicated, currency translations in this Annual Report on Form 20-F reflect the December 31, 2015 exchange rate.
On April 15, 2016, the noon buying rate of the Federal Reserve Bank of New York for the euro was $1.00 = €1.1295.
| B. | Capitalization and Indebtedness |
Not applicable.
| C. | Reasons for the Offer and Use of Proceeds |
Not applicable.
| D. | Risk Factors |
Our business faces significant risks. You should carefully consider all of the information set forth in this annual reportAnnual Report and in our other filings with the United States Securities and Exchange Commission, or the SEC, including the following risk factors which we face and which are faced by our industry. Our business, financial condition or results of operations could be materially adversely affected by any of these risks. This report also contains forward-looking statements that involve risks and uncertainties. Our results could materially differ from those anticipated in these forward-looking statements, as a result of certain factors including the risks described below and elsewhere in this reportAnnual Report and our other SEC filings. See “Special Note Regarding Forward-Looking Statements” above.
Risks Related to Our BusinessFinancial Condition and IndustryCapital Requirements
We Have Incurred Significant Losses Since Our Inception And Anticipate That We Will Continue To Incur Significant Losses For The Foreseeable Future.
We are a clinical-stage biopharmaceutical company, and we have not yet generated significant income, with the exception of the French research tax credit (crédit d’impôt recherche), or CIR, which is classified as other income in our statement of income (loss). We have incurred net losses in each year since our inception in 2002, including
net losses of €12.9 million, €19.3 million, €24.0 million and €24.0€44.7 million for the years ended December 31, 2012, 2013, 2014 and 2014,2015, respectively. Although we have historically generated non-meaningful revenues through the sale of our Diallertest Milk product in France, we discontinued our commercial partnership with respect to the product and ceased selling Diallertest Milk during the second half of 2015. We do not expect these sales to be a pointderive any revenues in 2016 from the sale of strategic focus for our company in the future and we may even discontinue these sales in the future.Diallertest Milk. As of December 31, 2014,2015, we had an accumulated deficit of €54.8€99.4 million.
We have devoted most of our financial resources to research and development, including our clinical and pre-clinical development activities. To date, we have financed our operations primarily through the sale of equity securities, obtaining public assistance in support of innovation, such as conditional advances from OSEO Innovation, or OSEO, and reimbursements of research tax credit claims. The amount of our future net losses will depend, in part, on the pace and amount of our future expenditures and our ability to obtain funding through equity or debt financings, strategic collaborations or additional grants or tax credits. WeWhile we have initiated a pivotal Phase III trial to evaluate the safety and efficacy of Viaskin Peanut, we have not yet completed pivotal clinical trials for any of our lead product candidates and it will be several years, if ever, before we have a product candidate ready for
commercialization, if at all. Even if we obtain regulatory approval to market a product candidate, our future revenues will depend upon the size of any markets in which our product candidates have received approval, and our ability to achieve sufficient market acceptance, reimbursement from third-party payors and adequate market share for our product candidates in those markets.
We expect to continue to incur significant expenses and increasing operating losses for the foreseeable future. We anticipate that our expenses will increase substantially if and as we:
The net losses we incur may fluctuate significantly from year to year, such that a period-to-period comparison of our results of operations may not be a good indication of our future performance. In any particular period or periods, our operating results could be below the expectations of securities analysts or investors, which could cause the price of theour ADSs or ordinary shares to decline.
We May Need To Raise Additional Funding, Which May Not Be Available On Acceptable Terms, Or At All. Failure To Obtain This Necessary Capital When Needed May Force Us To Delay, Limit Or Terminate Our Product Development Efforts Or Other Operations.
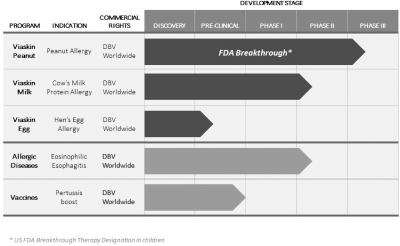
We are currently advancing our product candidates through pre-clinical and clinical development. Developing product candidates is expensive, lengthy and risky, and we expect our research and development expenses to increase substantially in connection with our ongoing activities, particularly as we advance Viaskin Peanut and Viaskin Milk through clinical development. We initiated the Peanut EPIT Efficacy and Safety Study, or PEPITES, a pivotal Phase III trial, of Viaskin Peanut in December 2015.
As of December 31, 2014,2015, our cash and cash equivalents were €114.6€323.4 million. We expect that our existing cash will be sufficient to fund our current operations until the end of 2016.2017. However, our operating plan may change as a result of many factors currently unknown to us, and we may need to seek additional funds sooner than planned, through public or private equity or debt financings, government or other third-party funding, marketing and distribution arrangements and other collaborations, strategic alliances and licensing arrangements or a combination of these approaches. In any event, we will require additional capital to pursue pre-clinical and clinical activities, pursue regulatory approval for, and to commercialize, our product candidates. Raising funds in the current economic environment may present additional challenges. Even if we believe we have sufficient funds for our current or future operating plans, we may seek additional capital if market conditions are favorable or if we have specific strategic considerations.
Any additional fundraising efforts may divert our management from their day-to-day activities, which may adversely affect our ability to develop and commercialize our product candidates. In addition, we cannot guarantee that future financing will be available in sufficient amounts or on terms acceptable to us, if at all. Moreover, the terms of any financing may adversely affect the holdings or the rights of our shareholders and the issuance of additional securities, whether equity or debt, by us, or the possibility of such issuance, may cause the market price of theour ADSs or ordinary shares to decline. The sale of additional equity or convertible securities would dilute all of our shareholders. The incurrence of indebtedness would result in increased fixed payment obligations and we may be required to agree to certain restrictive covenants, such as limitations on our ability to incur additional debt, limitations on our ability to acquire, sell or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business. We could also be required to seek funds through arrangements with collaborative partners or otherwise at an earlier stage than otherwise would be desirable and we may be required to relinquish rights to some of our technologies or product candidate or otherwise agree to terms unfavorable to us, any of which may have a material adverse effect on our business, operating results and prospects.
If we are unable to obtain funding on a timely basis, we may be required to significantly curtail, delay or discontinue one or more of our research or development programs or the commercialization of any product candidate or be unable to expand our operations or otherwise capitalize on our business opportunities, as desired, which could materially affect our business, financial condition and results of operations.
We Are Limited In Our Ability To Raise Additional Share Capital, Which May Make It Difficult For Us To Raise Capital To Fund Our Operations.
Under French law, our share capital may be increased only with shareholders’ approval at an extraordinary general shareholders’ meeting following the recommendation of our board of directors. The shareholders may delegate to our board of directors either the authority (délégation de compétence) or the power (délégation de pouvoir) to carry out any increase in share capital. SeeAs discussed further under “Item 10. B—Memorandum and Articles of Association”,Association,” our board may be precluded from issuing additional ordinary shares without first obtaining shareholders’ approval.
In addition, the French Commercial Code imposes certain limitations on our ability to price any offering of our share capital without preferential subscription right (sans droit préférentiel de souscription), which limitation may prevent us from successfully completing any such offering. Specifically, under the French Commercial Code, unless the offering is less than 10% of issued share capital, securities cannot be sold in an offering if it is not possible to fix the per share price of the shares at a level at least equal to the volume weighted average trading price on Euronext Paris over the last three trading days preceding the commencement of the marketing of the transaction, referred to as the “book building” process, less a maximum discount of 5%.
We Have Identified A Material Weakness In Our Internal Control Over Financial Reporting For The Year Ended December 31, 2015 And May Identify Additional Material Weaknesses In The Future Or Otherwise Fail To Maintain An Effective System Of Internal Controls, Which May Result In Material Misstatements Of Our Financial Statements Or Could Have A Material Adverse Effect On Our Business And Trading Price Of Our Securities.
As of the end of our second quarter of fiscal 2015, the market value of our common stock held by non-affiliates exceeded $700 million and we ceased to be an “emerging growth company” at the conclusion of fiscal 2015. Pursuant to Section 404 of the Sarbanes-Oxley Act of 2002, or Section 404, we are required to furnish a report by our management on our internal control over financial reporting, and we are required to include an attestation report on internal control over financial reporting issued by our independent registered public accounting firm.
In connection with the audit of our consolidated financial statements as of and for the year ended December 31, 2015 and our management’s assessment of our internal control over financial reporting, we identified a material weakness in our internal control over financial reporting. A material weakness is a deficiency, or combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of our annual or interim financial statements will not be prevented or detected on a timely basis. The material weakness that we identified is related to a lack of adequate segregation of duties given the size of our finance and accounting team to allow for appropriate monitoring of financial reporting matters and internal control over financial reporting.
This material weakness did not result in any adjustments or restatements of our audited consolidated financial statements or disclosures for any prior period previously reported by the company. However, there is a reasonable possibility that a material misstatement of the consolidated financial statements would not have been prevented or detected on a timely basis due to the failure in designing and implementing appropriate controls over the segregation of duties, and therefore, it has been evaluated as a material weakness. However, if the material weakness is not remediated, then it could result in material financial misstatements in the future.
We have limited accounting and financial reporting personnel and other resources with which to address our internal controls and procedures. We are currently designing and implementing new procedures and controls intended to remediate the material weakness described above. We have hired additional accounting and finance staff, who have significant external reporting experience and experience with establishing appropriate financial reporting policies and procedures, and engaged a professional advisor with sufficient technical accounting expertise to assist us in the implementation of internal controls over financial reporting and segregating duties amongst accounting personnel.
We expect to incur additional costs to remediate this weakness, primarily personnel costs and external consulting fees. We cannot assure you that the measures we have taken to date, together with any measures we may take in the future, will be sufficient to remediate the control deficiencies that led to the material weakness in our internal control over financial reporting or to avoid potential future material weaknesses.
If we are unable to successfully remediate our existing or any future material weakness in our internal control over financial reporting, or if we identify any additional material weaknesses, the accuracy and timing of our financial reporting may be adversely affected. If we are unable to maintain effective internal controls, we may not have adequate, accurate or timely financial information, and we may be unable to meet our reporting obligations as a U.S. public company or comply with the requirements of the SEC or Section 404. This could result in a restatement of our financial statements, the imposition of civil or criminal sanctions, the inability of registered broker dealers to make a market in our ADSs or ordinary shares, or an investigation by regulatory authorities. Any such action or other negative results caused by our inability to meet our reporting requirements or comply with legal and regulatory requirements or by disclosure of an accounting, reporting or control issue could adversely affect the trading price of our securities and our business. Our reporting and compliance obligations may place a significant strain on our management, operational and financial resources and systems for the foreseeable future. Failure to remedy any material weakness in our internal control over financial reporting, or to implement or maintain other effective control systems required of U.S. public companies, could also restrict our future access to the capital markets.
If We Do Not Obtain The Capital Necessary To Fund Our Operations, We Will Be Unable To Successfully Develop, Pursue Regulatory Approval For, And Commercialize, Our Biopharmaceutical Products.
The development of biopharmaceutical products is capital-intensive. We anticipate we may require additional financing to continue to fund our operations. Our future capital requirements will depend on, and could increase significantly as a result of, many factors including:
Until we can generate significant continuing revenues, we expect to satisfy our future cash needs through collaboration arrangements, sales of our securities, debt financings, obtaining public assistance in support of innovation, such as conditional advances from OSEO, and reimbursements of research tax credit claims, or by licensing one or more of our future product candidates. Dislocations in the financial markets have generally made equity and debt financing more difficult to obtain, and may have a material adverse effect on our ability to meet our future fundraising needs. We cannot be certain that additional funding will be available to us on acceptable terms, if at all. If funds are not available, we may be required to delay, reduce the scope of, or eliminate one or more of our research or development programs or our commercialization efforts. Additional funding, if obtained, may significantly dilute existing shareholders if that financing is obtained through issuing equity or instruments convertible into equity.
Our Product Development Programs For Candidates May Require Substantial Financial Resources And May Ultimately Be Unsuccessful.
In addition to the development of our lead product candidates, we may pursue development of our other early-stage development programs. In November 2015, we announced the enrollment of the first patient in theStudy of efficacy and safety of Viaskin Milk in Milk-Induced Eosinophilic Esophagitis, or SMILEE, a Phase IIa clinical trial assessing the safety and efficacy of Viaskin Milk for the treatment of milk-induced eosinophilic esophagitis. Our other earlier stage product development programs include a booster vaccine for pertussis, as well as potential treatments for Crohn’s disease and respiratory syncytial virus. Our current early-stage development programs are still in the pre-clinical proof-of-concept phase and may not result in product candidates we can advance to the clinical development phase. None of our other potential product candidates has commenced clinical trials, and there are a number of U.S. Food and Drug Administration, or FDA, and European Medicines Agency, or EMA, regulatory requirements that we must satisfy before we can commence these clinical trials, if at all. Satisfaction of these requirements will entail substantial time, effort and financial resources. We may never satisfy these requirements. Any time, effort and financial resources we expend on our other early-stage development programs may adversely affect our ability to continue development and commercialization of Viaskin patch product candidates based on our Viaskin technology platform, and we may never commence clinical trials of such development programs despite expending significant resources in pursuit of their development. Even if we do commence clinical trials of our other potential product candidates, such product candidates may never be approved by the FDA or the EMA.
The Requirements Of Being A U.S. Public Company May Strain Our Resources, Divert Management’s Attention And Affect Our Ability To Attract And Retain Executive Management And Qualified Board Members.
As a U.S. public company, we arehave incurred and will continue to incur significant legal, accounting, and other expenses that we did not previously incur. We are subject to the reporting requirements of the Securities Exchange Act of 1934, or the Exchange Act, the Sarbanes-Oxley Act, the Dodd-Frank Wall Street Reform and Consumer Protection Act, the Nasdaq listing requirements and other applicable securities rules and regulations. Compliance with these rules and regulations will continue to increase our legal and financial compliance costs, make some activities more difficult, time-consuming or costly and increase demand on our systems and resources, particularly after we are no longer an “emerging growth company” and/orqualify as a foreign private issuer. The Exchange Act requires that, as a public company, we file annual, semi-annual and current reports with respect to our business, financial condition and result of operations. However, as a foreign private issuer, we are not required to file quarterly reports with respect to our business, financial condition and results of operations. We currently make annual and semi-annual filings with respect to our listing on Euronext Paris. Unless otherwise required by the Exchange Act or the listing rules of the Nasdaq Global Select Market, we do not expect to file quarterly financial reports and will continue to file financial reports on an annual and semi-annual basis. Pursuant to Section 404 of the Sarbanes-Oxley Act of 2002, or Section 404, we are required to furnish a report by our management on our internal control over financial reporting, including an attestation report on internal control over financial reporting issued by our independent registered public accounting firm. However, while we remain an emerging growth company, we are not required to include an attestation report on internal control over financial reporting issued by our independent registered public accounting firm. To achieve compliance with Section 404 within the prescribed period, we have been and continue to be engaged in a process to document and evaluate our internal control over financial reporting, which is both costly and challenging. In this regard, we need to continue to dedicate internal resources, potentially engage outside consultants and adopt a detailed work plan to assess and document the adequacy of internal control over financial reporting, continue steps to improve control processes as appropriate, validate through testing that controls are functioning as documented and implement a continuous reporting and improvement process for internal control over financial reporting. As a result of being a U.S. public company, management’s attention may be diverted from other business concerns, which could adversely affect our business and results of operations. We may need to hire more employees in the future or engage outside consultants to comply with these requirements, which will increase our cost and expense. Despite our efforts, there is a risk that neither we nor our independent registered public accounting firm will be able to conclude within the prescribed timeframe that our internal control over financial reporting is effective as required by Section 404. This could result in an adverse reaction in the financial markets due to a loss of confidence in the reliability of our financial statements.
In addition, changing laws, regulations and standards relating to corporate governance and public disclosure are creating uncertainty for public companies, increasing legal and financial compliance costs and making some activities more time-consuming. These laws, regulations and standards are subject to varying interpretations, in many cases due to their lack of specificity, and, as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies. This could result in continuing uncertainty regarding compliance matters and higher costs necessitated by ongoing revisions to disclosure and governance practices. We intend to invest resources to comply with evolving laws, regulations and standards, and this investment may result in increased general and administrative expense and a diversion of management’s time and attention from revenue-generating activities to compliance activities. If our efforts to comply with new laws, regulations and standards differ from the activities intended by regulatory or governing bodies due to ambiguities related to their application and practice, regulatory authorities may initiate legal proceedings against us and our business may be adversely affected.
As a U.S. public company that is subject to these rules and regulations, we may find it is more expensive for us to obtain director and officer liability insurance, and we may be required to accept reduced coverage or incur substantially higher costs to obtain coverage. These factors could also make it more difficult for us to attract and retain qualified members of our board of directors, particularly to serve on our audit committee and compensation committee, and qualified executive officers.
As a result of disclosure of information in filings required of a U.S. public company, our business and financial condition will become more visible, which we believe may result in threatened or actual litigation, including by
competitors and other third parties. If such claims are successful, our business and results of operations could be adversely affected, and even if the claims do not result in litigation or are resolved in our favor, these claims, and the time and resources necessary to resolve them, could divert the resources of our management and adversely affect our business and results of operations.
Further, being a U.S. public company and a French public company has an impact on disclosure of information and compliance with two sets of applicable rules. This could result in continuing uncertainty regarding compliance matters and higher costs necessitated by ongoing revisions to disclosure and governance practices.
Risks Related to Product Development, Regulatory Approval and Commercialization
We Depend Almost Entirely On The Successful Development Of Our Novel Viaskin Technology. We Cannot Be Certain That We Will Be Able To Obtain Regulatory Approval For, Or Successfully Commercialize, Viaskin Products.
We currently have two lead Viaskin technology-based product candidates, Viaskin Peanut and Viaskin Milk, in clinical development, and our business depends almost entirely on their successful clinical development, regulatory approval and commercialization. We currently have no drug or biological product approved for sale and may never be able to develop a marketable drug or biological product. Viaskin Peanut and Viaskin Milk will require substantial additional clinical development, testing, and regulatory approval before we are permitted to commence their commercialization. Our other product candidates, such as Viaskin HDM,Egg, are still in pre-clinical development. The clinical trials of our product candidates are, and the manufacturing and marketing of our product candidates will be, subject to extensive and rigorous review and regulation by numerous government authorities in the United States and in other countries where we intend to test and, if approved, market any product candidate. Before obtaining regulatory approvals for the commercial sale of any product candidate, we must demonstrate through pre-clinical testing and clinical trials that, among other things, the product candidate is safe and effective for use in each target indication. This process can take many years and may include post-marketingpost- marketing studies and surveillance, which will require the expenditure of substantial resources. Of the large number of drugs in development in the United States, only a small percentage successfully completes the FDA regulatory approval process and is commercialized. Accordingly, even if we are able to obtain the requisite financing to continue to fund our development and clinical programs, we cannot assure you that Viaskin Peanut, Viaskin Milk or any other of our product candidates will be successfully developed or commercialized.
We are not permitted to market Viaskin Peanut or Viaskin Milk in the United States until we receive approval of a Biologic License Application, or a BLA, from the FDA, or in any other countries until we receive the requisite approval from such countries. Obtaining approval of a BLA, or requisite approval in other countries, is a complex, lengthy, expensive and uncertain process, and the FDA may delay, limit or deny approval of Viaskin Peanut and Viaskin Milk for many reasons, including, among others:
Any of these factors, many of which are beyond our control, could jeopardize our ability to obtain regulatory approval for and successfully market any of our product candidates based on our Viaskin patch product candidates.technology platform. Moreover, because our business is almost entirely dependent upon Viaskin technology, any such setback in our pursuit of regulatory approval would have a material adverse effect on our business and prospects.
Our Product Candidates Are Expected To Undergo Clinical Trials That Are Time-consumingTime-Consuming And Expensive, The Outcomes Of Which Are Unpredictable, And For Which There Is A High Risk Of Failure. If Clinical Trials Of Our Product Candidates Fail To Satisfactorily Demonstrate Safety And Efficacy To The FDA And Other Regulators, We, Or Our Collaborators, May Incur Additional Costs Or Experience Delays In Completing, Or Ultimately Be Unable To Complete, The Development And Commercialization Of These Product Candidates.
Pre-clinical testing and clinical trials are long, expensive and unpredictable processes that can be subject to extensive delays. We cannot guarantee that any clinical trials will be conducted as planned or completed on schedule, if at all. It may take several years to complete the pre-clinical testing and clinical development necessary to commercialize a drug or biologic, and delays or failure can occur at any stage. Interim results of clinical trials do not necessarily predict final results, and success in pre-clinical testing and early clinical trials does not ensure that later clinical trials will be successful. A number of companies in the pharmaceutical, biopharmaceutical and biotechnology industries have suffered significant setbacks in advanced clinical trials even after promising results in earlier trials, and we cannot be certain that we will not face similar setbacks. The design of a clinical trial can determine whether its results will support approval of a product, and flaws in the design of a clinical trial may not become apparent until the clinical trial is well advanced. An unfavorable outcome in one or more trials would be a major setback for our product candidates and for us. Due to our limited financial resources, an unfavorable outcome in one or more trials may require us to delay, reduce the scope of, or eliminate one or more product development programs, which could have a material adverse effect on our business and financial condition and on the value of the ADSs.our ADSs and ordinary shares.
In connection with clinical testing and trials, we face a number of risks, including:
The results of pre-clinical studies do not necessarily predict clinical success, and larger and later-stage clinical trials may not produce the same results as earlier-stage clinical trials. The prior clinical trials of Viaskin patchour product candidates based on our Viaskin technology platform showed favorable safety and efficacy data; however, we may have different enrollment criteria in our future clinical trials. As a result, we may not observe a similarly favorable safety and efficacy profile as our prior clinical trials. In addition, we cannot assure you that in the course of potential widespread use in future, some drawbacks would not appear in maintaining production quality, protein stability or allergenic strength. Frequently, product candidates developed by pharmaceutical, biopharmaceutical and biotechnology companies have shown promising results in early pre-clinical studies or clinical trials, but have subsequently suffered significant setbacks or failed in later clinical trials. In addition, clinical trials of potential products often reveal that it is not possible or practical to continue development efforts for these product candidates.
If we do not successfully complete pre-clinical and clinical development, we will be unable to market and sell our product candidates and generate revenues. Even if we do successfully complete clinical trials, those results are not necessarily predictive of results of additional trials that may be needed before a BLA may be submitted to the FDA. Although there are a large number of drugs and biologics in development in the United States and other countries, only a small percentage result in the submission of a BLA to the FDA, even fewer are approved for commercialization, and only a small number achieve widespread physician and consumer acceptance following regulatory approval. If our clinical trials are substantially delayed or fail to prove the safety and effectiveness of our product candidates in development, we may not receive regulatory approval of any of these product candidates and our business and financial condition will be materially harmed.
In Our Clinical Trials, We Utilize An Oral Food Challenge Procedure Intentionally Designed To Trigger An Allergic Reaction, Which Could Be Severe Or Life-Threatening.
In accordance with our food allergy clinical trial protocols, we utilize a double-blind, placebo-controlled food challenge procedure. This consists of giving the offending food protein to patients in order to assess the sensitivity of their food allergy, and thus the safety and efficacy of our product candidates versus placebo. The food challenge protocol is meant to induce objective symptoms of an allergic reaction. These oral food challenge procedures can potentially trigger anaphylaxis or potentially life-threatening systemic allergic reactions. Even though these procedures are well-controlled, standardized and performed in highly specialized centers with intensive care units, there are inherent risks in conducting a trial of this nature. An uncontrolled allergic reaction could potentially lead to serious or even fatal reactions. Any such serious clinical event could potentially adversely affect our clinical development timelines, including a complete clinical hold on our food allergy clinical trials. We may also become liable to subjects who participate in our clinical trials and experience any such serious or fatal reactions. Any of the foregoing could have a material adverse effect on our business, prospects, stock price or financial condition.
Delays, Suspensions And Terminations In Our Clinical Trials Could Result In Increased Costs To Us And Delay Or Prevent Our Ability To Generate Revenues.
Human clinical trials are very expensive, time-consuming, and difficult to design, implement and complete. The completion of trials for Viaskin Peanut, Viaskin Milk or our other product candidates may be delayed for a variety of reasons, including delays in:
The commencement and completion of clinical trials for our product candidates may be delayed, suspended or terminated due to a number of factors, including:
Many of these factors may also ultimately lead to denial of our BLA for our product candidate. If we experience delay, suspensions or terminations in a clinical trial, the commercial prospects for the related product candidate will be harmed, and our ability to generate product revenues will be delayed or such revenues could be reduced or fail to materialize.
In addition, we may encounter delays or product candidate rejections based on new governmental regulations, future legislative or administrative actions, or changes in FDA or other similar foreign regulatory agency policy or interpretation during the period of product development. If we obtain required regulatory approvals, such approvals may later be withdrawn. Delays or failures in obtaining regulatory approvals may result in:
Furthermore, if we fail to comply with applicable FDA and other regulatory requirements at any stage during this regulatory process, we may encounter or be subject to:
If Our Product Candidates Are Not Approved By The FDA, We Will Be Unable To Commercialize Them In The United States.
The FDA must approve any new drug or biologic before it can be commercialized, marketed, promoted or sold in the United States. We must provide the FDA with data from pre-clinical studies and clinical trials that demonstrate that, among other things, our product candidates are safe and effective for a defined indication before they can be approved for commercial distribution. Clinical testing is expensive, difficult to design and implement, can take many years to complete and is inherently uncertain as to outcome. We must provide data to ensure the identity, strength, quality and purity of the drug substance and drug product. Also, we must assure the FDA that the characteristics and performance of the clinical batches will be replicated consistently in the commercial batches. We will not obtain approval for a product candidate unless and until the FDA approves a BLA, if at all. The processes by which regulatory approvals are obtained from the FDA to market and sell a new or repositioned product are complex, require a number of years and involve the expenditure of substantial resources. We cannot assure you that any of our product candidates will receive FDA approval in the future, and the time for receipt of any such approval is currently incapable of estimation.
A Fast Track Designation By The FDA May Not Actually Lead To A Faster Development Or Regulatory Review Or Approval Process.Process, And It Does Not Increase The Likelihood That Our Product Candidates Will Receive Marketing Approval.
We have obtained fast track designation from the FDA for Viaskin Peanut, and we may pursue that designation for other product candidates as well. If a product is intended for the treatment of a serious or life-threatening condition and nonclinical or clinical data demonstrate the potential to address unmet medical needs for this condition, the sponsor may apply for FDA fast track designation. The FDA has broad discretion whether or not to grant this designation, and even if we believe our product candidates are eligible for this designation, we cannot be sure that the FDA would decide to grant it. Even if we do have fast track designation, we may not experience a faster development process, review or approval compared to conventional FDA procedures. The FDA may withdraw fast track designation if it believes that the designation is no longer supported by data from our clinical development program.
A Breakthrough Therapy Designation By The FDA For Our Product Candidates May Not Lead To A Faster Development Or Regulatory Review Or Approval Process, And It Does Not Increase The Likelihood That Our Product Candidates Will Receive Marketing Approval.
We have obtained breakthrough therapy designation for Viaskin Peanut in children, 6 to 11 years of age, and we may pursue that designation for other product candidates as well. A breakthrough therapy is defined as a product that is intended, alone or in combination with one or more other drugs, to treat a serious or life-threateninglife- threatening condition, and preliminary clinical evidence indicates that the product may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints. For product candidates that have been designated as breakthrough therapies, interaction and communication between the FDA and the sponsor of the trial can help identify the most efficient path for clinical development while minimizing the number of patients placed in ineffective control regimens. Such designation also offers an intensive and efficient review involving FDA senior managers and experienced review and regulatory health project management staff across disciplines. A breakthrough therapy designation affords the possibility of rolling review, enabling the agency to review portions of the marketing application before submission of a complete application, and priority review if supported by clinical data at the time of our BLA submission.
Designation as a breakthrough therapy is within the discretion of the FDA. Accordingly, even if we believe that our product candidates, in addition to Viaskin Peanut, meet the criteria for designation as a breakthrough therapy, the FDA may disagree and instead determine not to make such designation. In any event, the receipt of a breakthrough therapy designation for a product candidate may not result in a faster development process, review or approval compared to products considered for approval under conventional FDA procedures and does not assure ultimate approval by the FDA. In addition, even if one or more of our product candidates qualify as breakthrough therapies, the FDA may later decide that the products no longer meet the conditions for qualification.
The Approval Process Outside The United States Varies Among Countries And May Limit Our Ability To Develop, Manufacture And Sell Our Products Internationally. Failure To Obtain Marketing Approval In International Jurisdictions Would Prevent Our Product Candidates From Being Marketed Abroad.
In order to market and sell our product candidates in the European Union and many other jurisdictions, we, and our collaborators, must obtain separate marketing approvals and comply with numerous and varying regulatory requirements. The approval procedure varies among countries and may involve additional testing. We may conduct clinical trials for, and seek regulatory approval to market, our product candidates in countries other than the United States. Depending on the results of clinical trials and the process for obtaining regulatory approvals in other countries, we may decide to first seek regulatory approvals of a product candidate in countries other than the United States,
or we may simultaneously seek regulatory approvals in the United States and other countries. If we or our collaborators seek marketing approvals for a product candidate outside the United States, we will be subject to the regulatory requirements of health authorities in each country in which we seek approvals. With respect to marketing authorizations in Europe, we will be required to submit a European marketing authorization application, or MAA, to the EMA which conducts a validation and scientific approval process in evaluating a product for safety and efficacy. The approval procedure varies among regions and countries and may involve additional testing, and the time required to obtain approvals may differ from that required to obtain FDA approval.
Pursuing regulatory approvals from health authorities in countries outside the United States is likely to subject us to all of the risks associated with pursuing FDA approval described above. In addition, marketing approval by the FDA does not ensure approval by the health authorities of any other country, and approval by foreign health authorities does not ensure marketing approval by the FDA.
Even If We, Or Our Collaborators, Obtain Marketing Approvals For Our Product Candidates, The Terms Of Approvals And Ongoing Regulation Of Our Products May Limit How We Or They Market Our Products, Which Could Materially Impair Our Ability To Generate Revenue.
Even if we receive regulatory approval for a product candidate, this approval may carry conditions that limit the market for the product or put the product at a competitive disadvantage relative to alternative therapies. For instance, a regulatory approval may limit the indicated uses for which we can market a product or the patient population that may utilize the product, or may be required to carry a warning in its labeling and on its packaging. Products with boxed warnings are subject to more restrictive advertising regulations than products without such warnings. These restrictions could make it more difficult to market any product candidate effectively. Accordingly, assuming we, or our collaborators, receive marketing approval for one or more of our product candidates, we, and our collaborators will continue to expend time, money and effort in all areas of regulatory compliance.
Any Of Our Product Candidates For Which We, Or Our Collaborators, Obtain Marketing Approval In The Future Could Be Subject To Post-marketing Restrictions Or Withdrawal From The Market And We, And Our Collaborators, May Be Subject To Substantial Penalties If We, Or They, Fail To Comply With Regulatory Requirements Or If We, Or They, Experience Unanticipated Problems With Our Products Following Approval.
Any of our product candidates for which we, or our collaborators, obtain marketing approval in the future, as well as the manufacturing processes, post-approval studies and measures, labeling, advertising and promotional activities for such products, among other things, will be subject to continual requirements of and review by the FDA and other regulatory authorities. These requirements include submissions of safety and other post-marketing information and reports, registration and listing requirements, requirements relating to manufacturing, quality control, quality assurance and corresponding maintenance of records and documents, requirements regarding the distribution of samples to physicians and recordkeeping. Even if marketing approval of a product candidate is granted, the approval may be subject to limitations on the indicated uses for which the product may be marketed or to the conditions of approval, including the FDA requirement to implement a REMS to ensure that the benefits of a drug or biological product outweigh its risks.
The FDA may also impose requirements for costly post-marketing studies or clinical trials and surveillance to monitor the safety or efficacy of a product, such as long term observational studies on natural exposure. The FDA and other agencies, including the Department of Justice, closely regulate and monitor the post-approval marketing and promotion of products to ensure that they are manufactured, marketed and distributed only for the approved indications and in accordance with the provisions of the approved labeling. The FDA imposes stringent restrictions on manufacturers’ communications regarding off-label use and if we, or our collaborators, market any of our product candidates for which we, or they, receive marketing approval for treatment other than their approved
indications, we, or they, may be subject to warnings or enforcement action for off-label marketing. Violation of the FDCA and other statutes, including the False Claims Act, relating to the promotion and advertising of prescription drugs may lead to investigations or allegations of violations of federal and state health care fraud and abuse laws and state consumer protection laws.
If We Do Not Achieve Our Projected Development And Commercialization Goals In The Timeframes We Announce And Expect, The Commercialization Of Our Product Candidates May Be Delayed, And Our Business Will Be Harmed.
We sometimes estimate for planning purposes the timing of the accomplishment of various scientific, clinical, regulatory and other product development objectives.objectives for planning purposes. These milestones may include our expectations regarding the commencement or completion of scientific studies, clinical trials, the submission of regulatory filings, or commercialization objectives. From time to time, we may publicly announce the expected timing of some of these milestones, such as the completion of an ongoing clinical trial, the initiation of other clinical programs, receipt of marketing approval, or a commercial launch of a product. The achievement of many of these milestones may be outside of our control. All of these milestones are based on a variety of assumptions which may cause the timing of achievement of the milestones to vary considerably from our estimates, including:
If we fail to achieve announced milestones in the timeframes we expect, the commercialization of our product candidates may be delayed, our business and results of operations may be harmed, the trading price of the ADSs or ordinary shares may decline.
Access To Raw Materials And Products Necessary For The Conduct Of Clinical Trials And Manufacturing Of Our Product Candidates Is Not Guaranteed.
We are dependent on third parties for the supply of various materials, chemical or biological products that are necessary to produce patches for our clinical trials or diagnosis patches. The supply of these materials could be reduced or interrupted at any time. In such case, we may not be able to find other suppliers of acceptable materials in appropriate quantities at an acceptable cost. If key suppliers or manufacturers are lost or the supply of materials is diminished or discontinued, we may not be able to continue to develop, manufacture and market our product candidates or products in a timely and competitive manner. In addition, these materials are subject to stringent manufacturing processes and rigorous testing. Delays in the completion and validation of facilities and manufacturing processes of these materials could adversely affect our ability to complete trials and commercialize our products in a cost-effective and timely manner. To prevent such situations, we intend to diversify our supply
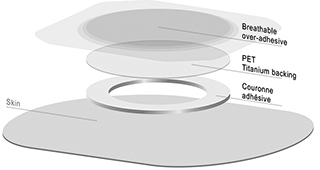
sources by identifying at a minimum a second source of supply for critical raw materials and materials, such as natural protein and polymer film with a titanium coating. If we encounter difficulties in the supply of these materials, chemicals or biological products, if we were not able to maintain our supply agreements or establish new agreements to develop and manufacture our products in the future, our business, prospects, financial condition, results and development could be significantly affected.
Relying On Third-partyThird-Party Manufacturers May Result In Delays In Our Clinical Trials And Product Introductions. We Or The Third Parties Upon Whom We Depend May Be Adversely Affected By Earthquakes Or Other Natural Disasters And Our Business Continuity And Disaster Recovery Plans May Not Adequately Protect Us From A Serious Disaster.
Developing and commercializing new medicines entails significant risks and expenses. Our clinical trials may be delayed if third-party manufacturers are unable to assure a sufficient quantity of the drug product to meet our study needs. Currently, we have only one manufacturer, for peanut protein extract, anSanofi S.A., or Sanofi, of the active pharmaceutical ingredientingredients used in our Viaskin Peanut clinical trials.product candidates, such peanut protein extract. If such manufacturerSanofi cannot manufacture the peanut protein extractactive pharmaceutical ingredients as required by us in a timely manner, we may not be able to find a substitute manufacturer on a timely basis and our clinic trials may be delayed. If our clinical trials are delayed, our commercialization efforts may be impeded, or our costs may increase. Further, we are aware that Sanofi has entered into licensing agreements of discovery platforms in selected food allergies, notably with Immune Design Corp. and Selecta Biosciences Inc. This potential competitive dynamic may make Sanofi less inclined to continue or renew their manufacturing arrangement with us on commercially reasonable terms or at all and, notwithstanding contractual protections, Sanofi may be able to utilize knowledge gained through their relationship with us in furtherance of their development of competitive therapies.
Once regulatory approval is obtained, a marketed product and its manufacturer are subject to continual review. The discovery of previously unknown problems with a product or manufacturer may result in restrictions on the product, manufacturer or manufacturing facility, including withdrawal of the product from the market. Any manufacturers with which we contract are required to operate in accordance with FDA-mandated current good manufacturing practices, or cGMPs. A failure of any of our contract manufacturers to establish and follow cGMPs and to document their adherence to such practices may lead to significant delays in the launch of products based on our product candidates into the market. Moreover, the constituent parts of a combination product retain their regulatory status (as a biologic or device, for example) and, as such, we or our contract manufacturers may be subject to additional requirements in the Quality System Regulation, or QSR, applicable to medical devices, such as design controls, purchasing controls, and corrective and preventive action. Failure by third-party manufacturers to comply with applicable regulations could result in sanctions being imposed on us, including fines, injunctions, civil penalties, revocation or suspension of marketing approval for any products granted pre-market approvals, seizures or recalls of products, operating restrictions, and criminal prosecutions.
Earthquakes or other natural disasters could severely disrupt our operations, and have a material adverse effect on our business, results of operations, financial condition and prospects. If a natural disaster, power outage or other event occurred that prevented us from using all or a significant portion of our headquarters, that damaged critical infrastructure, such as the manufacturing facilities of our third-party contract manufacturers, or that otherwise disrupted operations, it may be difficult or, in certain cases, impossible for us to continue our business for a substantial period of time. The disaster recovery and business continuity plans we have in place may prove inadequate in the event of a serious disaster or similar event. We may incur substantial expenses as a result of the limited nature of our disaster recovery and business continuity plans, which, particularly when taken together with our lack of earthquake insurance, could have a material adverse effect on our business.
We Rely, And Will Rely In The Future, On Third Parties To Conduct Our Clinical Trials And Perform Data Collection And Analysis, Which May Result In Costs And Delays That Prevent Us From Successfully Commercializing Product Candidates.
We rely, and will rely in the future, on medical institutions, clinical investigators, CROs, contract laboratories, and collaborators to perform data collection and analysis and others to carry out our clinical trials. Our development activities or clinical trials conducted in reliance on third parties may be delayed, suspended, or terminated if:
Third party performance failures may increase our development costs, delay our ability to obtain regulatory approval, and delay or prevent the commercialization of our product candidates. While we believe that there are numerous alternative sources to provide these services, in the event that we seek such alternative sources, we may not be able to enter into replacement arrangements without incurring delays or additional costs.
Even If Collaborators With Which We Contract In The Future Successfully Complete Clinical Trials Of Our Product Candidates, Those Candidates May Not Be Commercialized Successfully For Other Reasons.
Even if we contract with collaborators that successfully complete clinical trials for one or more of our product candidates, those candidates may not be commercialized for other reasons, including:
Our Viaskin Product Candidates May Not Be Able To Be Manufactured Profitably On A Large Enough Scale To Support Commercialization.
To date, our Viaskin product candidates have only been manufactured at a scale which is adequate to supply our research activities and clinical trials. There can be no assurance that the procedures currently used to manufacture our product candidates will work at a scale which is adequate for commercial needs and we may encounter difficulties in the production of Viaskin patches due to our or our partners’ manufacturing capabilities. We have not built commercial-scale manufacturing facilities, and we have limited manufacturing experience with Viaskin patches. We are working to develop a commercial-scale version of our electrospray manufacturing tool, but cannot predict or control issues that may arise with its development. These difficulties could delay the commercialization of our product candidates, reduce sales of our products, if approved, or increase our costs, any of which could harm our business.
We rely on a single supplier to produce, or contract for the production of, active ingredients for our clinical trials and for our commercial supplies of any future approved products. Even if we were to obtain access to quantities of
active ingredients sufficient to allow us otherwise to expand our Viaskin manufacturing capabilities, we may not be able to produce sufficient quantities of the product at an acceptable cost, or at all. In the event our Viaskin product candidates cannot be manufactured in sufficient quantities for commercialization, our future prospects could be significantly impacted and our financial prospects would be materially harmed.
We May Enter Into Agreements With Third Parties To Sell And Market Any Products We Develop And For Which We Obtain Regulatory Approvals, Which May Affect The Sales Of Our Products And Our Ability To Generate Revenues.
Given our development stage, we have limited experience in sales, marketing and distribution of biopharmaceutical products. However, if our product candidates obtain marketing approval, we intend to develop sales and marketing capacity, either alone or with strategic partners by contracting with, or licensing, them to market any of our products. Outsourcing sales and marketing in this manner may subject us to a variety of risks, including:
If we are unable to partner with a third party that has adequate sales, marketing, and distribution capabilities, we may have difficulty commercializing our product candidates, which would adversely affect our business, financial condition, and ability to generate product revenues.
Our Product Candidates Are Regulated As Biological Products, Or Biologics, Which May Subject Them To Competition Sooner Than Anticipated.
The Biologics Price Competition and Innovation Act of 2009, or BPCIA, was enacted as part of the 2010 enactments of the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, or collectively, the ACA, to establish an abbreviated licensure pathway for biological products shown to be biosimilar to, or interchangeable with, an FDA-licensed biological reference product. “Biosimilarity” means that the biological product is highly similar to the reference product notwithstanding minor differences in clinically inactive components and there are no clinically meaningful differences between the biological product and the reference product in terms of safety, purity, and potency of the product. To meet the higher standard of “interchangeability,” an applicant must provide sufficient information to show biosimilarity and demonstrate that the biological product can be expected to produce the same clinical result as the reference product in any given patient and, if the biological product is administrated more than once to an individual, the risk in terms of safety or diminished efficacy of alternating or switching between the use of the biological product and the reference product is not greater than the risk of using the reference product without such alternation or switch.
Under the BPCIA, an application for a biosimilar or interchangeable product cannot be approved by the FDA until 12 years after the reference product was first licensed, and the FDA will not even accept an application for review until four years after the date of first
licensure. The law is evolving, complex and is still being interpreted and implemented by the FDA. As a result, its ultimate impact, implementation, and meaning are subject to uncertainty. While it is uncertain when such processes intended to implement BPCIA may be fully adopted by the FDA, any such processes could have a material adverse effect on the future commercial prospects for our biological products.
We believe that any of our product candidates approved as a biological product under a BLA should qualify for the 12-year period of exclusivity. However, there is a risk that this exclusivity could be shortened due to congressional action or otherwise, potentially creating the opportunity for biosimilar or interchangeable competition sooner than anticipated. Moreover, the process by which an interchangeable product, once approved, will be substituted for any one of our reference products in a way that is similar to traditional generic substitution for non-biological products (i.e., drugs) is not yet clear, and will depend on a number of marketplace and regulatory factors that are still developing and subject to interpretation.
Even If Any Of Our Product Candidates Are Commercialized, They May Not Be Accepted By Physicians, Patients, Or The Medical Community In General. Even If We, Or Our Collaborators, Are Able To Commercialize Our Product Candidates, The Products May Become Subject To Market Conditions That Could Harm Our Business.
Even if the medical community accepts a product as safe and efficacious for its indicated use, physicians may choose to restrict the use of the product if we or any collaborator is unable to demonstrate that, based on experience, clinical data, side-effect profiles and other factors, our product is preferable to any existing drugs or treatments. We cannot predict the degree of market acceptance of any product candidate that receives marketing approval, which will depend on a number of factors, including, but not limited to:
We Are Dependent On A Single Exclusive Distributor For The Marketing Of Our Diallertest Milk Diagnostic Product. We May Discontinue Our Diallertest Milk Program.
In 2004, we introduced to the French market Diallertest Milk, the first ready-to-use patch test for detecting CMPA in young children. Diallertest Milk is the only product that we market to date, and is exclusively distributed in France though a distributor.
Diallertest Milk is currently available on the French market with a temporary exception status. Regulatory authorities are requesting a pivotal Phase III clinical trial to complete the marketing authorization file. We are examining the relevance of carrying out this clinical protocol, and are evaluating potential marketing and/or distribution relationships to market this product in Europe in the field of pediatrics. We may also elect, or may be required at the request of regulatory authorities, to stop the marketing of Diallertest Milk. If we were to stop the marketing of Diallertest Milk, or if our current or future distributors struggle to market this product in European markets, our ability to generate revenue from these product sales would be materially adversely effected. We cannot assure you that these product revenues will continue in the future and in any event we do not currently expect these product revenues to have a materially positive impact on our business and financial condition in future periods.
We Face Substantial Competition From Companies With Considerably More Resources And Experience Than We Have, Which May Result In Others Discovering, Developing, Receiving Approval For, Or Commercializing Products Before Or More Successfully Than Us.
The biopharmaceuticals industry is highly competitive. Numerous biopharmaceutical laboratories, biotechnology companies, institutions, universities and other research entities are actively involved in the discovery, research, development and marketing of therapeutic responses to treat allergies making it a highly competitive field. We have competitors in a number of jurisdictions, many of which have substantially greater name recognition, commercial infrastructures and financial, technical and personnel resources than we have. Although we believe we are currently in a unique position with respect to the testing and treatment of food allergies in young children, established competitors may invest heavily to quickly discover and develop novel compounds that could make the Viaskin patch products obsolete or uneconomical. Any new product that competes with an approved product may need to demonstrate compelling advantages in efficacy, convenience, tolerability and safety to be commercially successful. Other competitive factors, including generic competition, could force us to lower prices or could result in reduced sales. In addition, new products developed by others could emerge as competitors to Viaskin patch products. If we are not able to compete effectively against our current and future competitors, our business will not grow and our financial condition and operations will suffer.
In the case of food allergies, we are aware of several academic studies that are currently being conducted in major centers and hospitals worldwide. These studies are evaluating sublingual, subcutaneous, intranasal or other forms of desensitization or products using synthetic allergens, denatured allergens or combinations of medicines or methods, or medicines using traditional methods such as Chinese herbs. We are not aware of any pharmaceutical development in conjunction with these academic efforts at this time.
We expect studies combining other methods of immunotherapy, such as oral immunotherapy, or OIT, with anti-IgE treatments will be conducted. These types of co-administrations may significantly improve the safety of specific immunotherapies administered orally or subcutaneously, and may become significant competitors with our products.
To our knowledge, other pharmaceutical and biotechnology companies are also seeking to develop food allergy treatments, although many are in the discovery or pre-clinical stages. For example, Aimmune Therapeutics, Inc., formerly known as Allergen Research Corporation, is currently evaluatingenrolling patients in a Phase II clinical trialsIII trial to evaluate the safety and efficacy of its OIT product candidate, AR101, in peanut allergic patients. To our knowledge, the company uses a formulation of peanut flour for oral administration intended for oral desensitization.desensitization to peanut. We are also aware of other companies that are working on recombinant peanut proteins capable of initiating an attenuated immune response of using subcutaneous administration. We are also aware that Sanofi S.A. has entered into licensing agreements of discovery platforms in selected food allergies, notably with Immune Design Corp. and Selecta Biosciences Inc. and may pose a competitive risk to our products in the future.
Government Restrictions On Pricing And Reimbursement, As Well As Other Healthcare Payor Cost-containment Initiatives, May Negatively Impact Our Ability To Generate Revenues If We Obtain Regulatory Approval To Market A Product.
The continuing efforts of the government, insurance companies, managed care organizations and other payors of healthcare costs to contain or reduce costs of healthcare may adversely affect one or more of the following:
Sales of our products, when and if approved for marketing, will depend, in part, on the extent to which our products will be covered by third-party payors, such as federal, state, and foreign government health care programs, commercial insurance and managed healthcare organizations. There may be significant delays in obtaining coverage and reimbursement for newly approved products, and coverage may be more limited than the purposes for which the drug is approved by the FDA or comparable foreign regulatory authorities. Moreover, eligibility for coverage and reimbursement does not imply that a product will be paid for in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution. Third-party payors are increasingly reducing reimbursements for medical products, drugs and services. In addition, the U.S. government, state legislatures and foreign governments have continued implementing cost containment programs, including price controls, restrictions on coverage and reimbursement and requirements for substitution of generic products. Adoption of price controls and cost containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit our net revenue and results. Limited third-party reimbursement for our product candidates or a decision by a third-party payor not to cover our product candidates could reduce physician usage of our products once approved and have a material adverse effect on our sales, results of operations and financial condition.
The ACA is expected to significantly impact the provision of, and payment for, health care in the United States. Various provisions of the ACA take effect over the next several years, and are designed to expand Medicaid eligibility, subsidize insurance premiums, provide incentives for businesses to provide health care benefits, prohibit denials of coverage due to pre-existing conditions, establish
health insurance exchanges, and provide additional support for medical research. With regard to biopharmaceutical products, among other things, the ACA expanded and increased industry rebates for drugs covered under Medicaid programs and made changes to the coverage requirements under the Medicare prescription drug benefit. More recently, both the Budget Control Act of 2011 and the American Taxpayer Relief Act of 2012, or the ATRA, include, among other things, mandatory reductions in Medicare payments to certain providers. Additional legislative proposals to reform healthcare and government insurance programs, along with the trend toward managed healthcare in the United States, could influence the purchase of medicines and reduce demand and prices for our products, if approved. This could harm our or our collaborators’ ability to market any products and generate revenues. Cost containment measures that healthcare payors and providers are instituting and the effect of further healthcare reform could significantly reduce potential revenues from the sale of any of our product candidates approved in the future, and could cause an increase in our compliance, manufacturing, or other operating expenses.
In some foreign countries, the proposed pricing for a drug must be approved before it may be lawfully marketed. In addition, in certain foreign markets, the pricing of prescription drugs is subject to government control and reimbursement may in some cases be unavailable. The requirements governing drug pricing vary widely from country to country. For example, the European Union provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. A member state may approve a specific price for the medicinal product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. There can be no assurance that any country that has price controls or reimbursement limitations for biopharmaceutical products will allow favorable reimbursement and pricing arrangements for any of our products. Historically, biopharmaceutical products launched in the European Union do not follow price structures of the United States and generally tend to have significantly lower prices.
We believe that pricing pressures at the federal and state levels in the United States, as well as internationally, will continue and may increase, which may make it difficult for us to sell our potential products that may be approved in the future at a price acceptable to us or any of our future collaborators.
Our Product Candidates May Cause Undesirable Side Effects That Could Delay Or Prevent Their Regulatory Approval, Limit The Commercial Profile Of An Approved Label, Or Result In Significant Negative Consequences Following Marketing Approval, If Any.
Our product candidates are being developed to address the needs of severely allergic patients, for some of whom coming into contact with even minute amounts of an allergen can have a profound and life-threatening adverse reaction. Accordingly, safety is of paramount importance in developing these product candidates. To date, four clinical trials of Viaskin Peanut and Viaskin Milk product candidates have been conducted both outside and inside of the United States in over 400 human subjects to evaluate the safety and efficacy of these product candidates for the treatment of peanut allergies and milk allergies. Adverse events observed in these clinical trials have primarily involved general disorders and administration site conditions, such as erythema, pruritus, edema, and urticaria. It is worth noting that, as a desensitization patch bringing the allergen into contact with the skin, Viaskin patch product candidates can, in severely allergic patients, cause some erythematous or eczema skin reactions, which are a source of itching and discomfort for the patient. This reaction is typically temporary in duration and fades after a few weeks of use. In addition, during daily administration of the patches during treatments, depending on the severity of the allergies and patient response to treatment, precautionary measures are necessary when handling the patches after use due to risk of contamination.
Undesirable side effects caused by our product candidates could cause us or regulatory authorities to interrupt, delay, halt or terminate clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA or other regulatory authorities. Further, if our Viaskin patch product candidates receive marketing approval and we or others identify undesirable side effects caused by the products (or any other similar products) after the approval, a number of potentially significant negative consequences could result, including:
Any of these events could prevent us from achieving or maintaining market acceptance of the affected products and could substantially increase the costs of commercializing our products and significantly impact our ability to successfully commercialize our products and generate revenues.
Our Future Growth Depends, In Part, On Our Ability To Penetrate Foreign Markets, Where We Would Be Subject To Additional Regulatory Burdens And Other Risks And Uncertainties.
Our future profitability will depend, in part, on our ability to commercialize Viaskin patch product candidates based on our Viaskin technology platform in markets within and without the United States and Europe. If we commercialize Viaskin patch product candidates based on our Viaskin technology platform in foreign markets, we would be subject to additional risks and uncertainties, including:
Foreign sales of Viaskin patch products could also be adversely affected by the imposition of governmental controls, political and economic instability, trade restrictions and changes in tariffs.
We Are Subject To Healthcare Laws And Regulations, Which Could Expose Us To Criminal Sanctions, Civil Penalties, Exclusion from Government Healthcare Programs, Contractual Damages, Reputational Harm And Diminished Profits And Future Earnings.Earnings, Among Other Consequences.
Healthcare providers, physicians and others will play a primary role in the recommendation and prescription of Viaskin patch products, if approved. Our arrangements with such persons and third-party payors will expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we research, market, sell and distribute Viaskin patch products, if we obtain marketing approval. Restrictions under applicable federal, state and foreign healthcare laws and regulations include but are not limited to the following:
Analogous state or foreign laws and regulations, such as state anti-kickback and false claims laws, which may apply to items or services reimbursed by any third-party payor, including commercial insurers, state marketing and/or transparency laws applicable to manufacturers that may be broader in scope than the federal requirements, state laws that require biopharmaceutical companies to comply with the biopharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance
promulgated by the federal government, and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect as HIPAA. |
Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available, it is possible that some of our current and/or future business activities could be subject to challenge under one or more of these laws. In addition, recent health care reform legislation has strengthened these laws. For example, the Health Care Reform Law, among other things, amends the intent requirement of the federal Anti-Kickback Statute and criminal healthcare fraud statutes, in that a person or entity no longer needs to have actual knowledge of the statute or specific intent to violate it in order to commit a violation. Moreover, the Health Care Reform Law provides that the government and state laws governingmay assert that a claim including items or services resulting from a violation of the privacy and securityfederal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect as HIPAA.U.S. civil False Claims Act.
Ensuring that our business arrangements with third parties comply with applicable healthcare laws and regulations could be costly. It is possible that governmental authorities will conclude that our business practices do not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations were found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, disgorgement, individual imprisonment, and exclusion from government funded healthcare programs, such as Medicare and Medicaid, contractual damages, reputational harm, diminished profits and future earnings, and curtailment of operations, any of which could substantially disrupt our operations. Defending against any such actions can be costly, time-consuming and may require significant financial and personnel resources. Therefore, even if we are successful in defending against any such actions that may be brought against us, our business may be impaired. If the physicians or other providers or entities with whom we expect to do business are found not to be in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusionsexclusion from government funded healthcare programs.
Changes In Regulatory Requirements, FDA Guidance Or Guidance From Certain European Regulatory Authorities Or Unanticipated Events During Our Clinical Trials Of Viaskin Patch Products May Occur, Which May Result In Changes To Clinical Trial Protocols Or Additional Clinical Trial Requirements, Which Could Result In Increased Costs To Us And Could Delay Our Development Timeline.
Changes in regulatory requirements, FDA guidance or guidance from certain European regulatory authorities or unanticipated events during our clinical trials may force us to amend clinical trial protocols or the FDA or certain European regulatory authorities may impose additional clinical trial requirements. These discussions have caused us to adjust certain trial protocols. Similar amendments to our clinical trial protocols would require resubmission to the FDA and IRBs for review and approval, which may adversely impact the cost, timing or successful completion of a clinical trial. If we experience delays completing, or if we terminate, any of our clinical trials, or if we are required to conduct additional clinical trials, the commercial prospects for the Viaskin patch product candidates, or any other product candidates, may be harmed and our ability to generate product revenue will be delayed.
The FDA And Other Regulatory Agencies Actively Enforce The Laws And Regulations Prohibiting The Promotion Of Off-label Uses. If We Are Found To Have Improperly Promoted Off-label Uses, We May Become Subject To Significant Liability.
The FDA and other regulatory agencies strictly regulate the promotional claims that may be made about prescription products, such as Viaskin patch products, if approved. In particular, a product may not be promoted for uses that are not approved by the FDA or such other regulatory agencies as reflected in the product’s approved labeling. If we receive marketing approval for Viaskin patch products as a treatment for a particular allergy, physicians, in their professional medical judgment, may nevertheless prescribe Viaskin patch products to their patients in a manner that is inconsistent with the approved label. If we are found to have promoted such off-label
uses, we may become subject to significant liability under the FDCA and other statutory authorities, such as laws prohibiting false claims for reimbursement. The federal government has levied large civil and criminal fines against companies for alleged improper promotion and has enjoined several companies from engaging in off-label promotion. The FDA has also requested that companies enter into consent decrees or permanent injunctions under which specified promotional conduct is changed or curtailed. If we cannot successfully manage the marketing of Viaskin patch products, if approved, by restricting off-label promotion, we could become subject to significant liability, which would materially adversely affect our business and financial condition.
We May Not Obtain Biopharmaceutical Company Status And Therefore Have To Rely On Contract Manufacturers Indefinitely.
To date, we do not have biopharmaceutical company status, or PCS, and therefore, cannot manufacture the product candidates that we develop in France. A company with premises located in France must submit an application to the French Drug and Health Products Safety Agency, or ANSM, in order to get PCS status. The ANSM grants PCS to a company upon evaluation and determination that such company’s premises has adequate personnel, procedure and organization. We intend to seek PCS when our Viaskin patch product candidates have received MAA and thereby have the ability to manufacture our product candidates without contracting third parties. There are two types of PCS: (1) distributor “exploitant” status, which permits medicines to be marketed directly in France by the company after demonstrating control of certain key functions such as pharmacovigilance, medical information and advertising, management of quality complaints and batch recall; and (2) manufacturer status, which permits the manufacturing and quality control of medicines after demonstrating adequate manufacturing and quality control premises that exhibit a quality assurance system that meets cGMP.
Failure to obtain PCS status would force us to revise our strategy. First, failure to obtain manufacturer status would force us to entrust the manufacturing and control of the therapeutic products to one or more specialized contract manufacturing organizations, or CMOs, as is the case with the current production of our clinical lots. Second, if distributor “exploitant” status was not obtained, we could not conduct a direct commercial approach to the French market and would therefore have to enter into marketing license agreements with other biopharmaceutical companies. Failure to obtain any of the two types of PCS status would affect the production and marketing of our product candidates, once approved, and could be detrimental to our business, earnings, financial conditions and growth prospects.
Our Product Development Programs For Candidates Other Than Viaskin Patch Products May Require Substantial Financial Resources And May Ultimately Be Unsuccessful.
The success of our business depends primarily upon our ability to identify, develop and commercialize products to treat common food and/or environmental allergies. In addition to the development of Viaskin Peanut and Viaskin Milk, we may pursue development of our other development programs, including Viaskin HDM.Egg. None of our other potential product candidates has commenced any clinical trials, and there are a number of FDA requirements that we must satisfy before we can commence clinical trials. Satisfaction of these requirements will entail substantial time, effort and financial resources. We may never satisfy these requirements. Any time, effort and financial resources we expend on our other development programs may adversely affect our ability to continue development and commercialization of Viaskin Peanut and Viaskin Milk, and we may never commence clinical trials of such development programs despite expending significant resources in pursuit of their development. If we do commence clinical trials of our other potential product candidates, such product candidates may never be approved by the FDA. If any of these events occur, we may be forced to abandon our development efforts for a program or programs, which would have a material adverse effect on our business and could potentially cause us to cease operations.
If We Do Not Secure Collaborations With Strategic Partners To Test, Commercialize And Manufacture Certain Product Candidates Outside Of Food Allergies, We May Not Be Able To Successfully Develop Products And Generate Meaningful Revenues.
A key aspect of our current strategy is to selectively enter into collaborations with third parties to conduct clinical testing, as well as to commercialize and manufacture product candidates outside food allergies. Our ability to generate revenues from these arrangements will depend on our collaborators’ abilities to successfully perform the functions assigned to them in these arrangements. We currently have multiple collaboration agreements in effect, including collaborations for the development of applications in the field of respiratory allergies or autoimmune disease, as well as other therapeutic domains, such as vaccines. Collaboration agreements typically call for milestone payments that depend on successful demonstration of efficacy and safety, obtaining regulatory approvals, and clinical trial results. Collaboration revenues are not guaranteed, even when efficacy and safety are demonstrated. The current economic environment may result in potential collaborators electing to reduce their external spending, which may prevent us from developing our product candidates.
Even if we succeed in securing collaborators, the collaborators may fail to develop or effectively commercialize products using our product candidates. Collaborations involving our product candidates pose a number of risks, including the following:
Thus, collaboration agreements may not lead to development or commercialization of product candidates in the most efficient manner or at all.
Collaboration agreements are generally terminable without cause on short notice. Once a collaboration agreement is signed, it may not lead to commercialization of a product candidate. We also face competition in seeking out collaborators. If we are unable to secure new collaborations that achieve the collaborator’s objectives and meet our expectations, we may be unable to advance our product candidates and may not generate meaningful revenues.
Intellectual Property Risks Related to Our Business
Our Ability To Compete May Decline If We Do Not Adequately Protect Our Proprietary Rights.
Our commercial success depends on obtaining and maintaining proprietary rights to our product candidates for the treatment of common food and/or environmental allergies, as well as successfully defending these rights against third-party challenges. We will only be able to protect our product candidates, and their uses from unauthorized use
by third parties to the extent that valid and enforceable patents, or effectively protected trade secrets, cover them. Our ability to obtain patent protection for our product candidates is uncertain due to a number of factors, including:
Even if we have or obtain patents covering our product candidates or compositions, we may still be barred from making, using and selling our product candidates or technologies because of the patent rights of others. Others may have filed, and in the future may file, patent applications covering compositions or products that are similar or identical to ours. There are many issued U.S. and foreign patents relating to chemical compounds and therapeutic products, and some of these relate to compounds we intend to commercialize. Numerous U.S. and foreign issued patents and pending patent applications owned by others exist in the allergy treatment field in which we are developing products. These could materially affect our ability to develop our product candidates or sell our products if approved. Because patent applications can take many years to issue, there may be currently pending applications unknown to us that may later result in issued patents that our product candidates or compositions may infringe. These patent applications may have priority over patent applications filed by us.
Obtaining and maintaining a patent portfolio entails significant expense and resources. Part of the expense includes periodic maintenance fees, renewal fees, annuity fees, various other governmental fees on patents and/or applications due in several stages over the lifetime of patents and/or applications, as well as the cost associated with complying with numerous procedural provisions during the patent application process. We may or may not choose to pursue or maintain protection for particular inventions. In addition, there
are situations in which failure to make certain payments or noncompliance with certain requirements in the patent process can result in abandonment or lapse of a patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. If we choose to forgo patent protection or allow a patent application or patent to lapse purposefully or inadvertently, our competitive position could suffer.
Legal actions to enforce our patent rights can be expensive and may involve the diversion of significant management time. In addition, these legal actions could be unsuccessful and could also result in the invalidation of our patents or a finding that they are unenforceable. We may or may not choose to pursue litigation or other actions
against those that have infringed on our patents, or used them without authorization, due to the associated expense and time commitment of monitoring these activities. If we fail to protect or to enforce our intellectual property rights successfully, our competitive position could suffer, which could harm our results of operations.
Biopharmaceutical Patents And Patent Applications Involve Highly Complex Legal And Factual Questions, Which, If Determined Adversely To Us, Could Negatively Impact Our Patent Position.
The patent positions of biopharmaceutical companies can be highly uncertain and involve complex legal and factual questions. The interpretation and breadth of claims allowed in some patents covering biopharmaceutical compositions may be uncertain and difficult to determine, and are often affected materially by the facts and circumstances that pertain to the patented compositions and the related patent claims. The standards of the United States Patent and Trademark Office, or USPTO, are sometimes uncertain and could change in the future. Consequently, the issuance and scope of patents cannot be predicted with certainty. Patents, if issued, may be challenged, invalidated or circumvented. U.S. patents and patent applications may also be subject to interference proceedings, and U.S. patents may be subject to reexamination proceedings, post-grant review and/or inter partes review in the USPTO. Foreign patents may be subject also to opposition or comparable proceedings in the corresponding foreign patent office, which could result in either loss of the patent or denial of the patent application or loss or reduction in the scope of one or more of the claims of the patent or patent application. In addition, such interference, reexamination, post-grant review, inter partes review and opposition proceedings may be costly. Accordingly, rights under any issued patents may not provide us with sufficient protection against competitive products or processes.
In addition, changes in or different interpretations of patent laws in the United States and foreign countries may permit others to use our discoveries or to develop and commercialize our technology and products without providing any compensation to us, or may limit the number of patents or claims we can obtain. The laws of some countries do not protect intellectual property rights to the same extent as U.S. laws and those countries may lack adequate rules and procedures for defending our intellectual property rights.
If we fail to obtain and maintain patent protection and trade secret protection of our product candidates, we could lose our competitive advantage and competition we face would increase, reducing any potential revenues and adversely affecting our ability to attain or maintain profitability.
Developments In Patent Law Could Have A Negative Impact On Our Business.
From time to time, the United States Supreme Court, or the Supreme Court, other federal courts, the United States Congress, the USPTO or similar foreign authorities may change the standards of patentability and any such changes could have a negative impact on our business.
In addition, the Leahy-Smith America Invents Act, or the America Invents Act, which was signed into law in 2011, includes a number of significant changes to U.S. patent law. These changes include a transition from a “first-to-invent” system to a “first-to-file” system, changes to the way issued patents are challenged, and changes to the way patent applications are disputed during the examination process. These changes may favor larger and more established companies that have greater resources to devote to patent application filing and prosecution. The USPTO has developed new and untested regulations and procedures to govern the full implementation of the America Invents Act, and many of the substantive changes to patent law associated with the America Invents Act, and, in particular, the first-to-file provisions, became effective on March 16, 2013. Substantive changes to patent law associated with the
America Invents Act may affect our ability to obtain patents, and if obtained, to enforce or defend them. Accordingly, it is not clear what, if any, impact the America Invents Act will have on the cost of prosecuting our patent applications, our ability to obtain patents based on our discoveries and our ability to enforce or defend any patents that may issue from our patent applications, all of which could have a material adverse effect on our business.
If We Are Unable To Protect The Confidentiality Of Our Trade Secrets, Our Business And Competitive Position Would Be Harmed.
In addition to patent protection, because we operate in the highly technical field of development of therapies, we rely in part on trade secret protection in order to protect our proprietary technology and processes. However, trade secrets are difficult to protect. We expect to enter into confidentiality and intellectual property assignment agreements with our employees, consultants, outside scientific collaborators, sponsored researchers, and other advisors. These agreements generally require that the other party keep confidential and not disclose to third parties all confidential information developed by the party or made known to the party by us during the course of the party’s relationship with us. These agreements also generally provide that inventions conceived by the party in the course of rendering services to us will be our exclusive property. However, these agreements may not be honored and may not effectively assign intellectual property rights to us.
In addition to contractual measures, we try to protect the confidential nature of our proprietary information using physical and technological security measures. Such measures may not, for example, in the case of misappropriation of a trade secret by an employee or third party with authorized access, provide adequate protection for our proprietary information. Our security measures may not prevent an employee or consultant from misappropriating our trade secrets and providing them to a competitor, and recourse we take against such misconduct may not provide an adequate remedy to protect our interests fully. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret can be difficult, expensive, and time-consuming, and the outcome is unpredictable. In addition, courts outside the United States may be less willing to protect trade secrets. Trade secrets may be independently developed by others in a manner that could prevent legal recourse by us. If any of our confidential or proprietary information, such as our trade secrets, were to be disclosed or misappropriated, or if any such information was independently developed by a competitor, our competitive position could be harmed.
We Will Not Seek To Protect Our Intellectual Property Rights In All Jurisdictions Throughout The World And We May Not Be Able To Adequately Enforce Our Intellectual Property Rights Even In The Jurisdictions Where We Seek Protection.
Filing, prosecuting and defending patents on our product candidates in all countries and jurisdictions throughout the world would be prohibitively expensive, and our intellectual property rights in some countries outside the United States could be less extensive than those in the United States, assuming that rights are obtained in the United States. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as federal and state laws in the United States. Consequently, we may not be able to prevent third parties from practicing our inventions in all countries outside the United States, or from selling or importing products made using our inventions in and into the United States or other jurisdictions. The statutory deadlines for pursuing patent protection in individual foreign jurisdictions are based on the priority dates of each of our patent applications.
Competitors may use our technologies in jurisdictions where we do not pursue and obtain patent protection to develop their own products and further, may export otherwise infringing products to territories where we have patent protection, but enforcement is not as strong as that in the United States. These products may compete with our products and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing. Even if we pursue and obtain issued patents in particular jurisdictions, our patent claims or other intellectual property rights may not be effective or sufficient to prevent third parties from so competing.
The laws of some foreign countries do not protect intellectual property rights to the same extent as the laws of the United States. Many companies have encountered significant problems in protecting and defending intellectual
property rights in certain foreign jurisdictions. The legal systems of some countries, particularly developing countries, do not favor the enforcement of patents and other
intellectual property protection, especially those relating to biopharmaceuticals or biotechnologies. This could make it difficult for us to stop the infringement of our patents, if obtained, or the misappropriation of our other intellectual property rights. For example, many foreign countries have compulsory licensing laws under which a patent owner must grant licenses to third parties. In addition, many countries limit the enforceability of patents against third parties, including government agencies or government contractors. In these countries, patents may provide limited or no benefit. Patent protection must ultimately be sought on a country-by-country basis, which is an expensive and time-consumingtime- consuming process with uncertain outcomes. Accordingly, we may choose not to seek patent protection in certain countries, and we will not have the benefit of patent protection in such countries.
Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly, could put our patent applications at risk of not issuing and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded, if any, may not be commercially meaningful. In addition, changes in the law and legal decisions by courts in the United States and foreign countries may affect our ability to obtain adequate protection for our technology and the enforcement of intellectual property. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
Third Parties May Assert Ownership Or Commercial Rights To Inventions We Develop.
Third parties may in the future make claims challenging the inventorship or ownership of our intellectual property. We have written agreements with collaborators that provide for the ownership of intellectual property arising from our collaborations. These agreements provide that we must negotiate certain commercial rights with collaborators with respect to joint inventions or inventions made by our collaborators that arise from the results of the collaboration. In some instances, there may not be adequate written provisions to address clearly the resolution of intellectual property rights that may arise from a collaboration. If we cannot successfully negotiate sufficient ownership and commercial rights to the inventions that result from our use of a third-party collaborator’s materials where required, or if disputes otherwise arise with respect to the intellectual property developed with the use of a collaborator’s samples, we may be limited in our ability to capitalize on the market potential of these inventions. In addition, we may face claims by third parties that our agreements with employees, contractors, or consultants obligating them to assign intellectual property to us are ineffective, or in conflict with prior or competing contractual obligations of assignment, which could result in ownership disputes regarding intellectual property we have developed or will develop and interfere with our ability to capture the commercial value of such inventions. Litigation may be necessary to resolve an ownership dispute, and if we are not successful, we may be precluded from using certain intellectual property, or may lose our exclusive rights in that intellectual property. Either outcome could have an adverse impact on our business.
Third Parties May Assert That Our Employees Or Consultants Have Wrongfully Used Or Disclosed Confidential Information Or Misappropriated Trade Secrets.
We employ individuals who were previously employed at universities or other biopharmaceutical companies, including our competitors or potential competitors. Although we try to ensure that our employees and consultants do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or our employees, consultants or independent contractors have inadvertently or otherwise used or disclosed intellectual property, including trade secrets or other proprietary information, of a former employer or other third parties. Litigation may be necessary to defend against these claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.
A Dispute Concerning The Infringement Or Misappropriation Of Our Proprietary Rights Or The Proprietary Rights Of Others Could Be Time Consuming And Costly, And An Unfavorable Outcome Could Harm Our Business.
There is significant litigation in the biopharmaceutical industry regarding patent and other intellectual property rights. While we are not currently subject to any pending intellectual property litigation, and are not aware of any such threatened litigation, we may be exposed to future litigation by third parties based on claims that our product candidates, technologies or activities infringe the intellectual property rights of others. If our development activities are found to infringe any such patents, we may have to pay significant damages or seek licenses to such patents. A patentee could prevent us from using the patented drugs or compositions. We may need to resort to litigation to enforce a patent issued to us, to protect our trade secrets, or to determine the scope and validity of third-party proprietary rights. From time to time, we may hire scientific personnel or consultants formerly employed by other companies involved in one or more areas similar to the activities conducted by us. Either we or these individuals may be subject to allegations of trade secret misappropriation or other similar claims as a result of prior affiliations. If we become involved in litigation, it could consume a substantial portion of our managerial and financial resources, regardless of whether we win or lose. We may not be able to afford the costs of litigation. Any adverse ruling or perception of an adverse ruling in defending ourselves against these claims could have a material adverse impact on our cash position and the price of the ADSs. Any legal action against us or our collaborators could lead to:
We May Infringe The Intellectual Property Rights Of Others, Which May Prevent Or Delay Our Product Development Efforts And Stop Us From Commercializing Or Increase The Costs Of Commercializing Our Product Candidates, If Approved.
Our success will depend in part on our ability to operate without infringing the intellectual property and proprietary rights of third parties. We cannot assure you that our business, products and methods do not or will not infringe the patents or other intellectual property rights of third parties.
The biopharmaceutical industry is characterized by extensive litigation regarding patents and other intellectual property rights. Other parties may allege that our product candidates or the use of our technologies infringes patent claims or other intellectual property rights held by them or that we are employing their proprietary technology without authorization. Patent and other types of intellectual property litigation can involve complex factual and legal questions, and their outcome is uncertain. Any claim relating to intellectual property infringement that is successfully asserted against us may require us to pay substantial damages, including treble damages and attorney’s fees if we are found to be willfully infringing another party’s patents, for past use of the asserted intellectual property and royalties and other consideration going forward if we are forced to take a license. In addition, if any such claim were successfully asserted against us and we could not obtain such a license, we may be forced to stop or delay developing, manufacturing, selling or otherwise commercializing Viaskin patch products.
Even if we are successful in these proceedings, we may incur substantial costs and divert management time and attention in pursuing these proceedings, which could have a material adverse effect on us. If we are unable to avoid infringing the patent rights of others, we may be required to seek a license, defend an infringement action or challenge the validity of the patents in court, or redesign our products. Patent litigation is costly and time consuming. We may not have sufficient resources to bring these actions to a successful conclusion. In addition, intellectual property litigation or claims could force us to do one or more of the following:
Any of these risks coming to fruition could have a material adverse effect on our business, results of operations, financial condition and prospects.
Issued Patents Covering Our Product Candidates Could Be Found Invalid Or Unenforceable If Challenged In Court.
If we or one of our licensing partners initiated legal proceedings against a third party to enforce a patent covering our product candidate, the defendant could counterclaim that the patent covering our product candidate is invalid and/or unenforceable. In patent litigation in the United States, defendant counterclaims alleging invalidity and/or unenforceability are commonplace. Grounds for a validity challenge include alleged failures to meet any of several statutory requirements, including lack of novelty, obviousness or non-enablement. Grounds for unenforceability assertions include allegations that someone connected with prosecution of the patent withheld relevant information from the USPTO, or made a misleading statement, during prosecution. Third parties may also raise similar claims before administrative bodies in the United States or abroad, even outside the context of litigation. Such mechanisms include re-examination, post grant review and equivalent proceedings in foreign jurisdictions, e.g., opposition proceedings. Such proceedings could result in revocation or amendment of our patents in such a way that they no longer cover our product candidates or competitive products. The outcome following legal assertions of invalidity and unenforceability is unpredictable. With respect to validity, for example, we cannot be certain that there is no invalidating prior art, of which we and the patent examiner were unaware during prosecution. If a defendant were to prevail on a legal assertion of invalidity and/or unenforceability, we would lose at least part, and perhaps all, of the patent protection on our product candidates. Such a loss of patent protection would have a material adverse impact on our business.
Risks Related to Our Organization, Structure and Operation
We Will Need To Develop And Expand Our Company, And We May Encounter Difficulties In Managing This Development And Expansion, Which Could Disrupt Our Operations.
As of December 31, 2014,2015, we had 5691 full-time employees and we expect to significantly increase our number of employees and the scope of our operations. To manage our anticipated development and expansion, including the commercialization of our product candidates in North America, we must continue to implement and improve our
managerial, operational and financial systems, expand our facilities and continue to recruit and train additional qualified personnel. Also, our management may need to divert a disproportionate amount of its attention away from itsour day-to-day activities and devote a substantial amount of time to managing these development activities. Due to our limited resources, we may not be able to effectively manage the expansion of our operations or recruit and train additional qualified personnel. This may result in weaknesses in our infrastructure, give rise to operational mistakes, loss of business opportunities, loss of employees and reduced productivity among remaining employees. The physical expansion of our operations may lead to significant costs and may divert financial resources from other projects, such as the development of our product candidates. If our management is unable to effectively manage our expected development and expansion, our expenses may increase more than expected, our ability to generate or increase our revenue could be reduced and we may not be able to implement our business strategy. Our future financial performance and our ability to commercialize our product candidates, if approved, and compete effectively will depend, in part, on our ability to effectively manage the future development and expansion of our company.
We Depend On Key Personnel And Attracting Qualified Management Personnel And Our Business Could Be Harmed If We Lose Key Personnel And Cannot Attract New Personnel.
Our success depends to a significant degree upon the technical and management skills of our officers and key personnel, including in particular those of Pierre-Henri Benhamou, our Chairman and Chief Executive Officer, and Bertrand Dupont, our Chief Technical Officer.Senior Executive Vice President, Technology. The loss of the services of any of these individuals would likely have a material adverse effect on us. Our success also will depend upon our ability to attract and retain additional qualified management, marketing, technical, and sales executives and personnel. The loss of any of our key executives, or the failure to attract, integrate, motivate, and retain additional key personnel could have a material adverse effect on our business.
We compete for such personnel against numerous companies, including larger, more established companies with significantly greater financial resources than we possess. There can be no assurance that we will be successful in attracting or retaining such personnel and the failure to do so could have a material adverse effect on our business, financial condition, and results of operations.
Our Employees May Engage In Misconduct Or Other Improper Activities, Including Violating Applicable Regulatory Standards And Requirements Or Engaging In Insider Trading, Which Could Significantly Harm Our Business.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include intentional failures to: comply with the regulations of the FDA and applicable non-U.S. regulators, provide accurate information to the FDA and applicable non-U.S. regulators, comply with fraud and abuse and other healthcare laws and regulations in the United States and abroad, report financial information or data accurately or disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealingself- dealing and other abusive practices. These laws and regulations restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Employee misconduct could also involve the improper use of, including trading on, information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. We have adopted a code of conduct, but it is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may be ineffective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
Product Liability And Other Lawsuits Could Divert Our Resources, Result In Substantial Liabilities And Reduce The Commercial Potential Of Our Product Candidates.
The risk that we may be sued on product liability claims is inherent in the development and commercialization of biopharmaceutical products. Side effects of, or manufacturing defects in, products that we develop could result in the deterioration of a patient’s condition, injury or even death. For example, product liability claims may be brought by patients participating in our clinical trials as a result of unexpected side effects from our product candidates. Once a product is approved for sale and commercialized, the likelihood of product liability lawsuits increases. Criminal or civil proceedings might be filed against us by patients, the regulatory authorities, biopharmaceutical companies and any other third party using or marketing our products. These actions could include claims resulting from acts by our partners, licensees and subcontractors, over which we have little or no control. These lawsuits may divert our management from pursuing our business strategy and may be costly to defend. In addition, if we are held liable in any of these lawsuits, we may incur substantial liabilities and may be forced to limit or forgo further commercialization of the affected products.
We may be subject to legal or administrative proceedings and litigation other than product liability lawsuits which may be costly to defend and could materially harm our business, financial condition and operations.
We maintain product liability insurance coverage for our clinical trials with an €11.0 million annual aggregate coverage limit. Nevertheless, our insurance coverage may be insufficient to reimburse us for any expenses or losses we may suffer. In addition, in the
future, we may not be able to obtain or maintain sufficient insurance coverage at an acceptable cost or to otherwise protect against potential product or other legal or administrative liability claims by us or our partners, licensees or subcontractors, which could prevent or inhibit the commercial production and sale of any of our product candidates that receive regulatory approval, which could adversely affect our business. Product liability claims could also harm our reputation, which may adversely affect our collaborators’ ability to commercialize our products successfully.
We Must Maintain Effective Internal Control Over Financial Reporting, And If We Are Unable To Do So, The Accuracy And Timeliness Of Our Financial Reporting May Be Adversely Affected, Which Could Have A Material Adverse Effect On Our Business, Investor Confidence And Market Price.
We must maintain effective internal control over financial reporting in order to accurately and timely report our results of operations and financial condition. In addition, as a public company, the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, requires, among other things, that we assess the effectiveness of our disclosure controls and procedures semi-annually and the effectiveness of our internal control over financial reporting at the end of each fiscal year. We anticipate being first required to issue management’s annual report on internal control over financial reporting, pursuant to Section 404 of the Sarbanes-Oxley Act, in connection with issuing our consolidated financial statements as of and for the year ending December 31, 2015.
The rules governing the standards that must be met for our management to assess our internal control over financial reporting pursuant to Section 404 of the Sarbanes-Oxley Act are complex and require significant documentation, testing and possible remediation. These stringent standards require that our audit committee be advised and regularly updated on management’s review of internal control over financial reporting. We are in the process of designing, implementing, and testing the internal control over financial reporting required to comply with this obligation. This process is time-consuming, costly, and complicated. In addition, our independent registered public accounting firm will be required to attest to the effectiveness of our internal controls over financial reporting beginning with our annual report following the date on which we are no longer an “emerging growth company”. We may cease to qualify as an emerging growth company as soon as December 31, 2015, as a result of which we would be required to comply with these requirements in our annual report for the year ended December 31, 2015. Our management may not be able to effectively and timely implement controls and procedures that adequately respond to the increased regulatory compliance and reporting requirements that will be applicable to us as a public company. If we fail to staff our accounting and finance function adequately or maintain internal control over financial reporting adequate to meet the demands that will be placed upon us as a public company, including the requirements of the Sarbanes-Oxley Act, our business and reputation may be harmed and the price of the ADSs may decline. Furthermore, investor perceptions of us may be adversely affected, which could cause a decline in the market price of our ordinary shares.
Our Failure To Maintain Certain Tax Benefits Applicable To French Technology Companies May Adversely Affect Our Results Of Operations.
As a French technology company, we have benefited from certain tax advantages, including, for example, the CIR. The CIR is a French tax credit aimed at stimulating research and development. The CIR can be offset against French corporate income tax due and the portion in excess (if any) may be refunded at the end of a three fiscal-year period. The CIR is calculated based on our claimed amount of eligible research and development expenditures in France and represented €2.5 million, €3.3 million, €4.3 million and €4.3€5.7 million as of December 31, 2012, 2013, 2014 and 2014,2015, respectively. The French tax authority with the assistance of the Research and Technology Ministry may audit each research and development program in respect of which a CIR benefit has been claimed and assess whether such program qualifies in its view for the CIR benefit. The French tax authorities may challenge our eligibility to, or our calculation of certain tax reductions and/or deductions in respect of our research and development activities and, should the French tax authorities be successful, we may be liable to additional corporate income tax, and penalties and interest related thereto, which could have a significant impact on our results of operations and future cash flows. Furthermore, if the French Parliament decides to eliminate, or reduce the scope or the rate of, the CIR benefit, either of which it could decide to do at any time, our results of operations could be adversely affected.
We May Be Forced To Repay Conditional Advances Prematurely If We Fail To Comply With Our Contractual Obligations Under The Applicable Innovation Grant Agreements.
Since inception, we have received multiple conditional advances totaling €5.7 million for innovation granted by OSEO, the French Agency for Innovation and part of theBanque Publique d’Investissement. If we fail to comply with our contractual obligations under the applicable innovation grant agreements, including if we lose our exclusive right to commercially develop our product candidates, we could be forced to repay the sums advanced
ahead of schedule. Such premature repayment could adversely affect our ability to finance itsour research and development projects. In addition, we cannot ensure that we will then have the additional financial means needed, the time or the ability to replace these financial resources with others.
We May Be Exposed To Significant Foreign Exchange Risk. Exchange Rate Fluctuations May Adversely Affect The Foreign Currency Value Of Our ADSs.
We incur portions of our expenses, and may in the future derive revenues, in currencies other than the euro, in particular, the U.S. dollar. As a result, we are exposed to foreign currency exchange risk as our results of operations and cash flows are subject to fluctuations in foreign currency exchange rates. We currently do not engage in hedging transactions to protect against uncertainty in future exchange rates between particular foreign currencies and the euro. Therefore, for example, an increase in the value of the euro against the U.S. dollar could be expected to have a negative impact on our revenue and earnings growth as U.S. dollar revenue and earnings, if any, would be translated into euros at a reduced value. We cannot predict the impact of foreign currency fluctuations, and foreign currency fluctuations in the future may adversely affect our financial condition, results of operations and cash flows. The ADSs are quoted in U.S. dollars on the Nasdaq Global Select Market and our ordinary shares are trading in euros on Euronext Paris. Our financial statements are prepared in euros. Fluctuations in the exchange rate between euros and the U.S. dollar will affect, among other matters, the U.S. dollar value and the euro value of our ordinary shares and ADSs.
We May Use Hazardous Chemicals And Biological Materials In Our Business. Any Claims Relating To Improper Handling, Storage Or Disposal Of These Materials Could Be Time Consuming And Costly.
Our research and development processes may involve the controlled use of hazardous materials, including chemicals and biological materials. We cannot eliminate the risk of accidental contamination or discharge and any resultant injury from these materials. For example, in production, the confinement of the electrospray function and the use of the allergen in liquid form make it possible to prevent the allergens from contaminating the environment. However, we cannot assure you that in case of malfunction during the handling, storage or production process, allergen would not be released into the atmosphere and sensitize the persons present in the environment. We may be sued for any injury or contamination that results from our use or the use by third parties of these materials, and our liability may exceed any insurance coverage and our total assets. Federal, state, local or foreign laws and regulations govern the use, manufacture, storage, handling and disposal of these hazardous materials and specified waste products, as well as the discharge of pollutants into the environment and human health and safety matters. Compliance with environmental laws and regulations may be expensive and may impair our research and development efforts. If we fail to comply with these requirements, we could incur substantial costs, including civil or criminal fines and penalties, clean-up costs or capital expenditures for control equipment or operational changes necessary to achieve and maintain compliance. In addition, we cannot predict the impact on our business of new or amended environmental laws or regulations or any changes in the way existing and future laws and regulations are interpreted and enforced.
Our Internal Computer Systems, Or Those Of Our Third-party Contractors Or Consultants, May Fail Or Suffer Security Breaches, Which Could Result In A Material Disruption Of Our Product Development Programs.
Despite the implementation of security measures, our internal computer systems and those of our third-party contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. While we do not believe that we have experienced any such system failure, accident, or security breach to date, if such an event were to occur and cause interruptions in our operations, it could result in a material disruption
of our programs. For example, the loss of clinical trial data for our product candidates could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach results in a loss of or damage to
our data or applications or other data or applications relating to our technology or product candidates, or inappropriate disclosure of confidential or proprietary information, we could incur liabilities and the further development of our product candidates could be delayed.
We May Acquire Businesses Or Products, Or Form Strategic Alliances, In The Future, And We May Not Realize The Benefits Of Such Acquisitions.
At this stage, our strategy does not involve plans to acquire companies or technologies facilitating or enabling us to access to new medicines, new research projects, or new geographical areas, or enabling us to express synergies with our existing operations. However, if such acquisitions were to become necessary in future, we may not be able to identify appropriate targets or make acquisitions under satisfactory conditions, in particular, satisfactory price conditions. In addition, we may unable to obtain the financing for these acquisitions under favorable conditions, and could be led to finance these acquisitions using cash that could be allocated to other purposes in the context of existing operations. If we acquire businesses with promising markets or technologies, we may not be able to realize the benefit of acquiring such businesses if we are unable to successfully integrate them with our existing operations and company culture. We may encounter numerous difficulties in developing, manufacturing and marketing any new products resulting from a strategic alliance or acquisition that delay or prevent us from realizing their expected benefits or enhancing our business. We cannot assure you that, following any such acquisition, we will achieve the expected synergies to justify the transaction, which could have a material adverse effect on our business, financial conditions, earnings and prospects.
Risks Related to Ownership of Our Ordinary Shares and ADSs
The Market Price For The ADSs May Be Volatile Or May Decline Regardless Of Our Operating Performance.
The trading price of theour ADSs and ordinary shares has fluctuated, and is likely to continue to fluctuate, substantially. The trading price of the ADSsour securities depends on a number of factors, including those described in this “Risk Factors” section, many of which are beyond our control and may not be related to our operating performance.
Since theOur ADSs were sold atin our initial public offering on Nasdaq in October 2014 at a price of $21.64 per share, and the price per ADS has ranged from as low as $20.26 and as high as $28.50 through April 24,$44.76 during 2015. During this same period, our ordinary share prices have ranged from as low as €37.57 to as high as €83.48. The market price of the ADSsour securities may fluctuate significantly in response to numerous factors, many of which are beyond our control, including:
These and other market and industry factors may cause the market price and demand for the ADSsour securities to fluctuate substantially, regardless of our actual operating performance, which may limit or prevent investors from readily selling their ADSs or ordinary shares and may otherwise negatively affect the liquidity of the ADSs.our ADSs and ordinary shares. In addition, the stock market in general, and biopharmaceutical companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies.
Share Ownership Is Concentrated In The Hands Of Our Principal Shareholders And Management, Who Will Continue To Be Able To Exercise A Direct Or Indirect Controlling Influence On Us.
Our executive officers, directors, current 5% or greater shareholders and affiliated entities, including Sofinnova Capital V FPCI, entities affiliated with Bpifrance andParticipations, entities affiliated with Baker Bros. Advisors LP and entities affiliated with FMR LLC, together beneficially own approximately 41.97%52% of our ordinary shares. As a result, these shareholders, acting together, will have significant influence over all matters that require approval by our shareholders, including the election of directors and approval of significant corporate transactions. Corporate action might be taken even if other shareholders oppose them. This concentration of ownership might also have the effect of delaying or preventing a change of control of our company that other shareholders may view as beneficial.
If Securities Or Industry Analysts Do Not Publish Research Or Publish Inaccurate Or Unfavorable Research About Our Business, The Price Of The ADSs And Trading Volume Could Decline.
The trading market for theour ADSs and ordinary shares depends in part on the research and reports that securities or industry analysts publish about us or our business. If no or few securities or industry analysts cover our company, the trading price for theour ADSs and ordinary shares would be negatively impacted. If one or more of the analysts who covers us downgrades theour ADSs or ordinary shares or publishes incorrect or unfavorable research about our business, the price of theour ADSs and ordinary shares would likely decline. If one or more of these analysts ceases coverage of our company or fails to publish reports on us regularly, or downgrades theour ADSs or ordinary shares, demand for the our ADSs and ordinary shares could decrease, which could cause the price of theour ADSs or ordinary shares or trading volume to decline.
We Do Not Currently Intend To Pay Dividends On Our Securities And, Consequently, Your Ability To Achieve A Return On Your Investment Will Depend On Appreciation In The Price Of The ADSs. In Addition, French Law May Limit The Amount Of Dividends We Are Able To Distribute.
We have never declared or paid any cash dividends on our ordinary shares and do not currently intend to do so for the foreseeable future. We currently intend to invest our future earnings, if any, to fund our growth. Therefore, you are not likely to receive any dividends on your ADSs for the foreseeable future and the success of an investment in ADSs will depend upon any future appreciation in its value. Consequently, investors may need to sell all or part of their holdings of ADSs after price appreciation, which may never occur, as the only way to realize any future gains on their investment. There is no guarantee that the ADSs will appreciate in value or even maintain the price at which our shareholders have purchased the ADSs. Investors seeking cash dividends should not purchase the ADSs.
Further, under French law, the determination of whether we have been sufficiently profitable to pay dividends is made on the basis of our statutory financial statements prepared and presented in accordance with IFRS. In addition, payment of dividends may subject us to additional taxes under French law. Please see the section of this Annual Report on Form 20-F titled “Item 10.B—Memorandum and Articles of Association” for further details on the limitations on our ability to declare and pay dividends and the taxes that may become payable by us if we elect to pay a dividend. Therefore, we may be more restricted in our ability to declare dividends than companies not based in France.
In addition, exchange rate fluctuations may affect the amount of euros that we are able to distribute, and the amount in U.S. dollars that our shareholders receive upon the payment of cash dividends or other distributions we declare and pay in euros, if any. These factors could harm the value of the ADSs, and, in turn, the U.S. dollar proceeds that holders receive from the sale of the ADSs.
Future Sales Of Ordinary Shares Or ADSs By Existing Shareholders Could Depress The Market Price Of The ADSs.
As of December 31 2015, 24,205,129 ordinary shares were issued and outstanding. Sales of a substantial number of shares of our ordinary shares or ADSs in the public market, or the perception that these sales might occur, could depress the market price of our securities and could impair our ability to raise capital through the sale of additional equity securities. A substantial number of our shares are now generally freely tradable, subject, in the case of sales by our affiliates, to the volume limitations and other provisions of Rule 144 under the Securities Act. If our existing shareholdersholders of these shares sell, or indicate an intent to sell, substantial amounts of ordinary shares or ADSsour securities in the public market, the trading price of the ADSsour securities could decline significantly. As of March 24, 2015, 19,372,486 shares were eligible for sale in the public market, 6,147,285 of which shares are held by directors, executive officers and other affiliates and will be subject to volume limitations under Rule 144 under the Securities Act of 1933, as amended, or the Securities Act.
In addition, we have filed a registration statement with the SEC to register the ordinary shares that may be issued under our equity incentive plans. The ordinary shares subject to outstanding options under our equity incentive plans, and theordinary shares reserved for future issuance under our equity incentive plans and ordinary shares subject to outstanding warrants will become eligible for sale in the public market in the future, subject to certain legal and contractual limitations. We have filed a registration statement with the SEC covering ordinary shares available for future issuance under our equity incentive plans. Sales of a large number of the shares issued under these plans in the public market could have an adverse effect on the market price of the ADSs.our securities.
Our By-Laws And French Corporate Law Contain Provisions That May Delay Or Discourage A Takeover Attempt.
Provisions contained in our by-laws and the corporate laws of France, the country in which we are incorporated, could make it more difficult for a third-party to acquire us, even if doing so might be beneficial to our shareholders. In addition, provisions of our by-laws impose various procedural and other requirements, which could make it more difficult for shareholders to effect certain corporate actions. These provisions include the following:
| • | our shareholders have preferential subscription rights on apro rata basis on the issuance by us of any additional securities for cash or a set-off of cash debts, which rights may only be waived by the extraordinary general meeting (by a two-thirds majority vote) of our shareholders or on an individual basis by each shareholder; |
You May Not Be Able To Exercise Your Right To Vote The Ordinary Shares Underlying Your ADSs.
Holders of ADSs may exercise voting rights with respect to the ordinary shares represented by the ADSs only in accordance with the provisions of the deposit agreement. The deposit agreement provides that, upon receipt of notice of any meeting of holders of our ordinary shares, the depositary will fix a record date for the determination of ADS holders who shall be entitled to give instructions for the exercise of voting rights. Upon timely receipt of
notice from us, if we so request, the depositary shall distribute to the holders as of the record date (1) the notice of the meeting or solicitation of consent or proxy sent by us and (2) a statement as to the manner in which instructions may be given by the holders.
You may instruct the depositary of your ADSs to vote the ordinary shares underlying your ADSs. Otherwise, you will not be able to exercise your right to vote, unless you withdraw the ordinary shares underlying the ADSs you hold. However, you may not know about the meeting far enough in advance to withdraw those ordinary shares. If we ask for your instructions, the depositary, upon timely notice from us, will notify you of the upcoming vote and arrange to deliver our voting materials to you. We cannot guarantee you that you will receive the voting materials in time to ensure that you can instruct the depositary to vote your ordinary shares or to
withdraw your ordinary shares so that you can vote them yourself. If the depositary does not receive timely voting instructions from you, it may give a proxy to a person designated by us to vote the ordinary shares underlying your ADSs. In addition, the depositary and its agents are not responsible for failing to carry out voting instructions or for the manner of carrying out voting instructions. This means that you may not be able to exercise your right to vote, and there may be nothing you can do if the ordinary shares underlying your ADSs are not voted as you requested.
Your Right As A Holder Of ADSs To Participate In Any Future Preferential Subscription Rights Or To Elect To Receive Dividends In Shares May Be Limited, Which May Cause Dilution To Your Holdings.
According to French Law,law, if we issue additional securities for cash, current shareholders will have preferential subscription rights for these securities on a pro rata basis unless they waive those rights at an extraordinary meeting of our shareholders (by a two-thirds majority vote) or individually by each shareholder. However, the ADS holders in the United States will not be entitled to exercise or sell such rights unless we register the rights and the securities to which the rights relate under the Securities Act or an exemption from the registration requirements is available. In addition, the deposit agreement provides that the depositary will not make rights available to you unless the distribution to ADS holders of both the rights and any related securities are either registered under the Securities Act or exempted from registration under the Securities Act. Further, if we offer holders of our ordinary shares the option to receive dividends in either cash or shares, under the deposit agreement the depositary may require satisfactory assurances from us that extending the offer to holders of ADSs does not require registration of any securities under the Securities Act before making the option available to holders of ADSs. We are under no obligation to file a registration statement with respect to any such rights or securities or to endeavor to cause such a registration statement to be declared effective. Moreover, we may not be able to establish an exemption from registration under the Securities Act. Accordingly, ADS holders may be unable to participate in our rights offerings or to elect to receive dividends in shares and may experience dilution in their holdings. In addition, if the depositary is unable to sell rights that are not exercised or not distributed or if the sale is not lawful or reasonably practicable, it will allow the rights to lapse, in which case you will receive no value for these rights.
You May Be Subject To Limitations On The Transfer Of Your ADSs And The Withdrawal Of The Underlying Ordinary Shares.
Your ADSs, which may be evidenced by ADRs, are transferable on the books of the depositary. However, the depositary may close its books at any time or from time to time when it deems expedient in connection with the performance of its duties. The depositary may refuse to deliver, transfer or register transfers of your ADSs generally when our books or the books of the depositary are closed, or at any time if we or the depositary think it is advisable to do so because of any requirement of law, government or governmental body, or under any provision of the deposit agreement, or for any other reason subject to your right to cancel your ADSs and withdraw the underlying ordinary shares. Temporary delays in the cancellation of your ADSs and withdrawal of the underlying ordinary shares may arise because the depositary has closed its transfer books or we have closed our transfer books, the transfer of ordinary shares is blocked to permit voting at a shareholders’ meeting or we are paying a dividend on our ordinary shares. In addition, you may not be able to cancel your ADSs and withdraw the underlying ordinary shares
when you owe money for fees, taxes and similar charges and when it is necessary to prohibit withdrawals in order to comply with any laws or governmental regulations that apply to ADSs or to the withdrawal of ordinary shares or other deposited securities.
We Are An “Emerging Growth Company” And Will Be Able To Avail Ourselves Of Reduced Disclosure Requirements Applicable To Emerging Growth Companies, Which Could Make Our ADSs Less Attractive To Investors.
We are an “emerging growth company,” as defined in the JOBS Act, and we have taken advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not “emerging growth companies” including not being required to comply with the auditor attestation requirements of Section 404(b) of the Sarbanes-Oxley Act, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. In addition, Section 107 of the JOBS Act also provides that an emerging growth company can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act for complying with new or revised accounting standards. In other words, an emerging growth company can delay the adoption of certain accounting standards until those
standards would otherwise apply to private companies. Some investors may find the ADSs less attractive because we rely on these exemptions and, as a result, there may be a less active trading market for the ADSs and the price of the ADSs may be more volatile. We may take advantage of these reporting exemptions until we are no longer an emerging growth company. We will remain an emerging growth company until the earliest of (i) the last day of the fiscal year in which we have total annual gross revenue of $1.0 billion or more; (ii) the last day of our fiscal year following the fifth anniversary of October 22, 2014, the date of our global offering; (iii) the date on which we have issued more than $1.0 billion in nonconvertible debt during the previous three years; and (iv) the date on which we are deemed to be a large accelerated filer under the rules of the SEC.
As A Foreign Private Issuer, We Are Exempt From A Number Of Rules Under The U.S. Securities Laws And Are Permitted To File Less Information With The SEC Than A U.S. Company. This May Limit The Information Available To Holders Of ADSs.
We are a “foreign private issuer,” as defined in the SEC’s rules and regulations and, consequently, we are not subject to all of the disclosure requirements applicable to public companies organized within the United States. For example, we are exempt from certain rules under the Exchange Act that regulate disclosure obligations and procedural requirements related to the solicitation of proxies, consents or authorizations applicable to a security registered under the Exchange Act, including the U.S. proxy rules under Section 14 of the Exchange Act. In addition, our officers and directors are exempt from the reporting and “short-swing” profit recovery provisions of Section 16 of the Exchange Act and related rules with respect to their purchases and sales of our securities. Moreover, while we currently make annual and semi-annual filings with respect to our listing on Euronext Paris and expect to file financial reports on an annual and semi-annual basis, we will not be required to file periodic reports and financial statements with the SEC as frequently or as promptly as U.S. domestic issuers and will not be required to file quarterly reports on Form 10-Q or current reports on Form 8-K under the Exchange Act. Accordingly, there will be less publicly available information concerning our company than there would be if we were a U.S. domestic issuer.
As A Foreign Private Issuer, We Are Permitted To Adopt Certain Home Country Practices In Relation To Corporate Governance Matters That Differ Significantly From Nasdaq Corporate Governance Listing Standards. These Practices May Afford Less Protection To Shareholders Than They Would Enjoy If We Complied Fully With Corporate Governance Listing Standards.
As a foreign private issuer listed on the Nasdaq Global Select Market, we will be subject to corporate governance listing standards. However, rules permit a foreign private issuer like us to follow the corporate governance practices of its home country. Certain corporate governance practices in France, which is our home country, may differ significantly from corporate governance listing standards. For example, neither the corporate laws of France nor our by-laws require a majority of our directors to be independent and we could include non-independent directors as members of our compensation committee, and our independent directors would not necessarily hold regularly scheduled meetings at which only independent directors are present. Currently, we intend to follow home country practicepractices to the maximum extent possible. Therefore, our shareholders may be afforded less protection than they otherwise would have under corporate governance listing standards applicable to U.S. domestic issuers.
We May Lose Our Foreign Private Issuer Status In The Future, Which Could Result In Significant Additional Cost And Expense.
While we currently qualify as a foreign private issuer, the determination of foreign private issuer status is made annually on the last business day of an issuer’s most recently completed second fiscal quarter and, accordingly, the next determination will be made with respect to us on June 30, 2015. In2016, which would require us to comply with all of the future, we wouldperiodic disclosure and current reporting requirements of the Exchange Act applicable to U.S. domestic issuers as of January 1, 2017. We could lose our foreign private issuer status in the future if we to fail to meet the requirements necessary to maintain our foreign private issuer status as of the relevant determination date. For example, ifIn order to maintain our current status as a foreign private issuer, either (a) a majority of our ordinary shares or ADSs must be either directly or indirectly owned of record by non-residents of the United States or (b)(i) a majority of our executive officers or directors may not be U.S. citizens or residents, (ii) more than 50% of our securities are held by U.S. residents and more than 50% of our executive officers or members of our board of directors are residents or citizens ofassets cannot be located in the United States and (iii) our business must not be administered principally inside the United States. If we could lose ourlost this status, we would be required to comply with the Exchange Act reporting and other requirements applicable to U.S. domestic issuers, which are more detailed and extensive than the requirements for foreign private issuer status.issuers. As of January 15,September 30, 2015, approximately 48.2%61% of our outstanding ordinary shares were held by U.S. residents.
The regulatory and compliance costs to us under U.S. securities laws as a U.S. domestic issuer may be significantly more than costs we currently incur as a foreign private issuer. If we are not a foreign private issuer, we will be
required to file periodic reports and registration statements on U.S. domestic issuer forms with the SEC, which are more detailed and extensive in certain respects than the forms
available to a foreign private issuer. We would be required under current SEC rules to prepare our financial statements in accordance with U.S. GAAP, rather than IFRS, in USU.S. dollars rather than euros and modify certain of our policies to comply with corporate governance practices associated with U.S. domestic issuers. Such conversion of our financial statements to U.S. GAAP will involve significant time and cost. In addition, we may lose our ability to rely upon exemptions from certain corporate governance requirements on U.S. stock exchanges that are available to foreign private issuers such as the ones described above and exemptions from procedural requirements related to the solicitation of proxies.
U.S. Investors May Have Difficulty Enforcing Civil Liabilities Against Our Company And Directors And Senior Management And The Experts Named In This Annual Report.
Certain members of our board of directors and senior management, and those of our subsidiary, are non-residents of the United States, and all or a substantial portion of our assets and the assets of such persons are located outside the United States. As a result, it may not be possible to serve process on such persons or us in the United States or to enforce judgments obtained in U.S. courts against them or us based on civil liability provisions of the securities laws of the United States. Additionally, it may be difficult to assert U.S. securities law claims in actions originally instituted outside of the United States. Foreign courts may refuse to hear a U.S. securities law claim because foreign courts may not be the most appropriate forums in which to bring such a claim. Even if a foreign court agrees to hear a claim, it may determine that the law of the jurisdiction in which the foreign court resides, and not U.S. law, is applicable to the claim. Further, if U.S. law is found to be applicable, the content of applicable U.S. law must be proved as a fact, which can be a time-consuming and costly process, and certain matters of procedure would still be governed by the law of the jurisdiction in which the foreign court resides. In particular, there is some doubt as to whether French courts would recognize and enforce certain civil liabilities under U.S. securities laws in original actions or judgments of U.S. courts based upon these civil liability provisions. In addition, awards of punitive damages in actions brought in the United States or elsewhere may be unenforceable in France. An award for monetary damages under the U.S. securities laws would be considered punitive if it does not seek to compensate the claimant for loss or damage suffered but is intended to punish the defendant. The enforceability of any judgment in France will depend on the particular facts of the case as well as the laws and treaties in effect at the time. The United States and France do not currently have a treaty providing for recognition and enforcement of judgments (other than arbitration awards) in civil and commercial matters.
The Rights Of Shareholders In Companies Subject To French Corporate Law Differ In Material Respects From The Rights Of Shareholders Of Corporations Incorporated In The United States.
We are a French company with limited liability. Our corporate affairs are governed by our by-laws and by the laws governing companies incorporated in France. The rights of shareholders and the responsibilities of members of our board of directors are in many ways different from the rights and obligations of shareholders in companies governed by the laws of U.S. jurisdictions. For example, in the performance of its duties, our board of directors is required by French law to consider the interests of our company, itsour shareholders, its employees and other stakeholders, rather than solely our shareholders and/or creditors. It is possible that some of these parties will have interests that are different from, or in addition to, your interests as a shareholder. See the sections of this Annual Report on Form 20-F titled “Item 10. B—Memorandum and Articles of Association” and “Item 16.G—Corporate Governance.”
We May Be At Risk Of Securities Class Action Litigation.
Historically, securities class action litigation has often been brought against a company following a decline in the market price of its securities. This risk is especially relevant for us because biotechnology and biopharmaceutical companies have experienced significant share price volatility in recent years. If we were to be sued, it could result in substantial costs and a diversion of management’s attention and resources, which could harm our business.
We Believe That We Were Classified As A Passive Foreign Investment Company For 2014, And Certain U.S. Holders Of Our ADSs May Suffer Adverse U.S. Federal Income Tax Consequences.
Generally, if, for any taxable year, at least 75% of our gross income is passive income, or at least 50% of the value of our assets is attributable to assets that produce passive income or are held for the production of passive income, including cash, we would be characterized as a passive foreign investment company, or PFIC, for U.S. federal income tax purposes. For purposes of these tests, passive income includes dividends, interest, and gains from the sale or exchange of investment property and rents and royalties other than rents and royalties which are received from unrelated parties in connection with the active conduct of a trade or business. If we are characterized as a PFIC, U.S. holders of the ADSs may suffer adverse tax consequences, including having gains realized on the sale of the ADSs treated as ordinary income, rather than capital gain, the loss of the preferential rate applicable to dividends received on the ADSs by individuals who are U.S. holders, and having interest charges apply to distributions by us and the proceeds of sales of the ADSs. See “Item 10.E—Taxation—Certain Material U.S. Federal Income Tax Considerations—Passive Foreign Investment Company Considerations.”
Our status as a PFIC will depend on the composition of our income and the composition and value of our assets (which, if we are not a “controlled foreign corporation” under Section 957(a) of the Code or we are publicly traded for the entire year being tested, may be determined in large part by reference to the market value of the ADSs and our ordinary shares, which may be volatile) from time to time. Our status may also depend, in part, on how quickly we utilize the cash proceeds from our global offering in our business. Although it is not free from doubt, based on the composition of our income for our 2014 taxable year, we believe that we were a PFIC for our 2014 taxable year.
| Item 4. | Information on the Company. |
| A. | History And Development Of The Company |
Our legal and commercial name is DBV Technologies S.A. We were incorporated as asociété par actions simplifiée (S.A.S.) under the laws of the French Republic on March 29, 2002 for a period of 99 years and subsequently converted on March 13, 2003 into asociété anonyme. We are registered at the Nanterre Commerce and Companies Register under the number 441 772 522. Our principal executive offices are located at Green Square-Bâtiment D, 80/84 rue des Meuniers 92220 Bagneux,177-181 avenue Pierre Brossolette, 92120 Montrouge, France, and our telephone number is +33 1 55 42 78 78. Our agent for service of process in the United States is CT Corporation System. We also maintain a website at www.dbv-technologies.com. The reference to our website is an inactive textual reference only and the information contained in, or that can be accessed through, our website is not a part of this Annual Report on Form 20-F.
Our actual capital expenditures for the years ended December 31, 2012, 2013, 2014 and 20142015 amounted to €0.4 million, €1.4 million, €1.1 million, and €1.1€5.3 million, respectively. These capital expenditures primarily consisted of the acquisition of laboratory equipment and industrial tools, the refurbishment of our research and development laboratories, our relocation of our headquarters to Montrouge as well as cash contributions to our liquidity contract. We expect our capital expenditures to increase in absolute terms in the near term as we continue to advance our research and development programs and grow our operations. We anticipate our capital expenditureexpenditures in 20152016 to be financed from the cash flows from operating activities and proceeds offrom our recent July 2015 public offering. In aFor the near future, these investments will mainly remain in France where our research and development facilities are currently located.
| B. | Business Overview |
We are a clinical-stage specialty biopharmaceutical company focused on changing the field of immunotherapy by developing a novel technology platform called Viaskin. Our therapeutic approach is based on epicutaneous immunotherapy, or EPIT, our proprietary method of delivering biologically active compounds to the immune system through intact skin using Viaskin. We have generated significant data demonstrating that Viaskin’s mechanism of action is novel and differentiated as it targets specific antigen-presenting immune cells in the skin, called Langerhans cells, which capture the antigen and migrate to the lymph node in order to activate the immune system without allowing passage of the antigen into the bloodstream. We are advancing this unique technology to treataddress areas of unmet medical need, including in patients including infants and children, suffering from severe food allergies, for whom safety is paramount sinceas the introduction of the offending allergen into their bloodstream can cause severe or life-threatening allergic reactions, such anas anaphylactic shock.
Our proprietary platform is based on our epicutaneous Viaskin patch. We have designed and developed this technology internally, for which we have scalable manufacturing capabilities. Viaskin is an electrostatic patch, which offers a convenient, self-administered, non-invasive immunotherapy to patients. Once applied on intact skin, Viaskin forms a condensation chamber, which hydrates the skin and solubilizes the antigen allowing it to penetrate the epidermis, where it is captured by Langerhans cells. Based on numerous scientific publications and our own research, we believe this unique mechanism of action is safehas a favorable safety profile and that it generates a strong immune response that results in tolerance towards the allergen.allergen desensitization. Our epicutaneous immunotherapy method allows us to address severe food allergies, as well as unmet medical needs in other immunotherapy indications.
According to an expert panel convened by the American Academy of Allergy Asthma and Immunology, or the AAAAI, epidemiological studies suggest that over half of Americans are sensitive to at least one allergen. Allergy is considered a “disease of the developed world” as its increasing incidence is proportional to higher living standards. Based on a paper published by the AAAAI, approximately 3% to 5% of Americans suffer from food allergies, with a number of recent studies suggesting that nearly 6 million or approximately 8% of children have some type of food allergy. Food allergies in particular can lead to extremely dangerous reactions while significantly
impairing daily quality of life. According to a paper published in the Immunology and Allergy Clinics of North America, food, mainly peanut, allergies, are responsible for 150 to 200 deaths and about 200,000 emergency room visits every year in the United States. These patients often also experience skin discomfort, asthma symptoms, impaired lung function and gastrointestinal complications, such as sustained bloating, nausea, vomiting and diarrhea. Food allergies can be particularly difficult for young children to manage, and due to their life-threatening nature, severe food allergies can often lead to psychological traumas.traumas and social anxiety. In some cases, these allergies can also cause chronic diseases such as failure to thrive in children and an allergic inflammatory condition of the esophagus called eosinophilic esophagitis, or EoE.
We are committed to finding a safe, effective and patient-friendly therapy for food and pediatric allergy patients, for whom there are no currently approved treatments. Compared to other allergy treatment approaches, we believe the safety profile of our EPIT method carried-out via the Viaskin patch may offer significant therapeutic and ease-of-use advantages to these patient populations. EPIT can be utilized as an allergy-specific immunotherapy commonly referred to as desensitization. Desensitization consists of repeated administration of small quantities of allergen to decrease allergen reactivity in patients. Currently studied desensitization methods include subcutaneous, sublingual, and oral immunotherapy, which often require frequent or prolonged administration in highly specialized centers. In academic settings some successful cases exist, but large-scale pharmaceutical development in this field has been limited due to both the safety concerns and the commercial viability of these desensitization approaches. These methods seem tomay be poorly designed for young children due to their safety profile or the inconvenient method of administration. Most importantly, some of these approaches are also known for triggering severe adverse events during treatment, such as anaphylaxis, thus risking the patient’s life during administration;administration. Further, some of these methods have been also associated with an increased risk of adverse long-term treatment effects, such as eosinophilic esophagitis.EoE. As a self-administered treatment with a good safety profile, we believe Viaskin has positioned us as the company with the most advanced clinical program in food allergies.
The following table summarizes our most advanced product candidates:
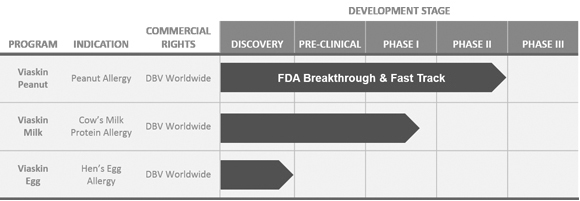
We are focused on becoming the leader in discovering, developing and commercializing food allergy products. Our pipeline development strategy is based on leveraging Viaskin’s scientific profile while taking into consideration a combination of target market characteristics, which include allergen prevalence, persistence and severity. We select our target productsproduct candidates with the aim to address the highestallergies that have high unmet medical needs.
Our lead product candidate, Viaskin Peanut, has obtained fast track designation and breakthrough therapy designation in children from the U.S. Food and Drug Administration, or FDA, which are designations intended to expedite or facilitate the process forof reviewing new drugs and biological products that are intended to treat a serious or life-threatening disease or condition and demonstrate the potential to address unmet medical needs for the disease or condition. The European Medicines Agency’s, or EMA, Pediatric Committee has also adopted a positive opinion with respect to our Pediatric Investigation Plan, or PIP, for Viaskin Peanut, which is a prerequisite for the filing of marketing authorization for any new medicinal product in Europe.
In September 2014, we announced positive topline results for our VIPES (ViaskinViaskin Peanut’s Efficacy and Safety)Safety, or VIPES, Phase IIb clinical trial of Viaskin Peanut, in peanut allergic patients, which was followed by a full study report presented at the 2015 AAAAI Annual Meeting in Houston, Texas. Pending consultation withFollowing results from our Phase IIb trial, we initiated the FDAPeanut EPIT Efficacy and Safety Study, or PEPITES, a pivotal Phase III trial, in December 2015. PEPITES is designed to evaluate the springsafety and efficacy of Viaskin Peanut in approximately 330 peanut allergic patients four to 11 years of age. Results from PEPITES are expected during the second half of 2017. In October 2015, we planannounced topline results from the first year of OLFUS-VIPES, an open-label extension trial of VIPES, in which we observed that 12 additional months of therapy with Viaskin Peanut 250 µg increased the number of patients benefiting from treatment to initiate70% in OLFUS-VIPES from 50% in VIPES, with 80% of children (ages six-11 at entry in VIPES) responding to therapy after 24 months.
We are developing our Phase III clinical trial in the last quarter of 2015 or first quarter of 2016.
Our second product candidate, Viaskin Milk, is being developed for the treatment of cow’s milk protein allergy, or CMPA, in the pediatric patient population.children two to 17 years of age. In November 2014, we initiated a 150-subject, multi-center, double-blind, placebo-controlled, randomized Phase I/II trial to study the safety and efficacy clinical trial of repeated doses of Viaskin Milk in approximately 176 patients with Immunoglobulin E, or IgE, mediated CMPA, which we refer to as the Milk Efficacy and Safety, or MILES, (MILkEfficacytrial. In June 2015, we announced the successful completion of Part A of the MILES study, or Phase I, andSafety). we launched Part B, or Phase II, in October 2015. Results from MILES are expected in the second half of 2017.
In February of 2015, we announced that our third food allergen product, Viaskin Egg, would addressseek to treat patients suffering from hen’s egg allergy. Preclinical development for Viaskin Egg commenced in the first half of 2015.
We have further usedalso use our Viaskin technology platform to advance other innovative product development programs that have the potential to address additional opportunitiestreat other immunological indications. In November 2015, we announced the enrollment of the first patient in immunology.theStudy of efficacy and safety of the Viaskin Milk in Milk-Induced Eosinophilic Esophagitis, or SMILEE, a Phase IIa investigator-sponsored clinical trial assessing the safety and efficacy of Viaskin Milk for the treatment of milk-induced EoE. Our other earlier stage product development programs include house dust mites allergy, eosinophilic esophagitis, pertusiss boosta booster vaccine for pertussis, as well as potential treatments for Crohn’s disease and birch pollen allergy,respiratory syncytial virus, none of which have resulted in a product candidate to date. We are also exploring earlier stage opportunities in respiratory syncytial virus vaccine, refractoryprograms for the treatment of hemophilia A, Crohn’s disease, Celiacceliac disease and type I diabetes.
We intend to commercialize our food allergy product candidates by ourselves in the United States and certain European countries. In other geographies and indications outside food allergies, we will explore selective partnerships with parties who have relevant clinical and commercial expertise in order to maximize shareholder value.
Our Strategy
Our goal is to become the leading global biopharmaceutical company focused on discovering, developing, manufacturing and commercializing treatments for severe allergies. Key elements of our strategy are:
| • | Rapidly Develop and Seek Marketing Approval for Viaskin Peanut—In December 2015, we initiated PEPITES, a pivotal Phase III trial in peanut allergic children ages four to 11, for which completion is expected in the second half of 2017. We also intend to explore additional indications for Viaskin Peanut in other patient populations as part of our clinical development strategy. Prior to launching our Phase III trial, we announced in September 2014 |
| • | Advance the Development of Our Viaskin Technology Platform into Other Areas of Unmet Medical Need in Food and Pediatric Allergies—We are advancing the clinical development of Viaskin Milk to address IgE mediated CMPA, which is |
| • | Become a Fully Integrated Biopharmaceutical Company Focused on the Commercialization of our Viaskin Food Allergy Product Candidates in the United States and Other Major Markets—We are utilizing our team’s unique food allergy expertise and knowledge to rapidly advance clinical development and approval of our product candidates. In anticipation of commercial launch, we continue to enhance our manufacturing and commercial production capabilities. Given the limited number and targeted nature of the prescribers in our target markets, we currently intend to launch and commercialize our food allergy product candidates with our own specialty sales force. |
| • | Maximize the Value of our Innovative Viaskin Technology Platform by Building a Broad Immunotherapy Product Pipeline—We believe that our Viaskin technology platform, for which we have worldwide commercialization rights, has the potential to support significant product opportunities beyond food allergies. We have commenced a Phase IIa trial of Viaskin Milk in patients suffering from milk-induced EoE, and we are also pursuing a number of pre-clinical collaborations, which could enable us to broaden our product pipeline, including collaborations for the development of applications in the field of |
Our Industry
Allergies are a Growing Global Health Problem
Allergy is considered a “disease of the developed world” as its increasing incidence is proportional to higher living standards. Epidemiological studies suggest that over half of Americans are sensitive to at least one allergen. Environmental and lifestyle changes, urbanization, pollution, dietary changes, development of sanitation standards and decrease in chronic bacterial infections all seem to be factors promoting the rapid increase in prevalence of allergies throughout the developed world.
Background on Allergic Reaction
An allergic reaction is the body’s inappropriate immune response to a foreign substance, or an allergen. While, for most people, exposure to an allergen is relatively harmless, for others, exposure to an allergen can provoke an allergic reaction of varying severity. An allergic reaction typically progresses in two stages.
In the first stage, the allergic immune response begins with allergen sensitization. The first time an allergen penetrates the body via the skin or the mucosa, for example, the eyes, respiratory or digestive tracts, the immune system identifies the foreign element as dangerous and begins to produce specific antibodies against it. Antibodies are substances produced by the immune system that recognize and destroy certain foreign elements to which the body is exposed. The immune system produces different types of antibodies targeted to specific allergens. For allergic people, this phenomenon is known as sensitization. In the second stage of an
allergic reaction, upon re-exposure to the allergen, the now sensitized immune system is ready to react. The antibody seeks to eliminate the allergen by triggering a collection of defense responses causing an allergic reaction. In various types of allergies, including food allergies, the antibody IgE plays an essential role in the development of the allergic disease. IgE is known for binding to allergens and triggering the release of cellular substances that can cause inflammation, thus triggering a cascade of allergic reactions. Allergic reactions range in severity and include hives, itching, swelling, shortness of breath, vomiting and cardiac arrhythmia. Reactions vary in duration, and allergy patients experience these symptoms frequently unless treated properly. The most severe allergic reaction is anaphylaxis, which if not treated quickly by epinephrine injection, may progress to anaphylactic shock causing a rapid drop in blood pressure, loss of consciousness and possibly death within a few minutes.
Current Challenges in the Treatment and Management of Allergy Patients
Symptomatic Allergy Treatments and their Limitations
For food allergies, there are no approved symptomatic or disease-modifying allergy treatments. By contrast, in the case of respiratory allergies, symptomatic allergy treatments, such as antihistamines, bronchodilators and corticosteroids, are among the most widely used treatments in the world. Non-sedating antihistamines such as histamine H1 inhibitors are the mainstay treatment for respiratory allergies. Allegra and Zyrtec are two leading antihistamines treatments. Another method of symptomatic treatment consists of blocking production of IgE, the allergy antibody.
However, all these treatments treat the symptoms of allergies, and are not intended to treat the underlying causes of the allergic reaction itself. As a result, when the treatment course is finished, the patient is once again susceptible to the original allergen and typically will suffer a similar allergic reaction if re-exposed to the original allergen.
Emergency Treatments and Their Limitations
Allergies can lead to severe reactions that require the use of treatments that have been designated to treat allergic symptoms during emergency situations, such as anaphylactic reactions. Epinephrine, also known as adrenaline, is the most widely used treatment for anaphylactic reactions, and it is usually administered by injection. The most commonly used type of epinephrine injections are Epipen Auto-Injectors, or Epipen,Epipens, which are indicated for the emergency treatment of severe allergic reactions including sudden anaphylaxis or for patients with a history of anaphylactic reactions to known triggers. Patients at risk of anaphylaxis are instructed by their physicians on how to recognize the symptoms of anaphylaxis and on when to use the Epipens. Epinephrine injections help relieve the symptoms of anaphylaxis, but they do not treat or help address the underlying causes of the allergic disease.
Desensitization Allergy Treatments and their Limitations
Another therapeutic approach for the treatment of allergies is through a type of immunotherapy, called desensitization therapy. Desensitization therapy consists of repeated administration of increasing quantities of allergen to decrease reactivity in allergic patients. It is currently recognized by the World Health Organization, or WHO, as the preferred therapeutic treatment for allergies. Desensitization therapy is widely used in respiratory allergies and allergies to insect bites. This treatment is traditionally performed by subcutaneous injections of increasing doses of the allergen at regular intervals in the hospital and under the supervision of a physician. Less invasive methods of administration, including oral drops and sublingual, or under the tongue, tablets, have also been developed to permit a simplified treatment that can be administered at home. For patients allergic to dust mites or pollen, desensitization by injection is the standard method of therapy.
However, while desensitization has shown potential in less severe allergies such as house dust mites or pollen, for food allergies and other severe allergies such as to peanut or milk proteins, existing desensitization therapies cannot beare not routinely used due to the high risk of anaphylactic shock, especially in young children. Subcutaneous methods of desensitization have been shown to cause significant side effects. Only limited academic studiesClinical trials have been performed using oral immunotherapy, andwhich consists in feeding small amounts of the offending allergen to the patient. While some of these studiestrials have not demonstrated an immune reaction deemed sufficiently consistent to supportshown a broadly applicable therapy. In some casesdesensitization effect, these therapies have been shown to trigger a high proportion of severe systemic reactions in certain cases, and we believe that this has limited their pharmaceutical development.development in the past.
Moreover, with current desensitization techniques, the achieved immunity may be short-lived; many patients are not able to tolerate the allergen permanently. A therapeutic approach that promotes tolerization to the allergen would be of particular clinical and societal benefit.
Food and Pediatric Allergies are a High Unmet Clinical Need
According to a paper published by the AAAAI, approximately 3% to 5% of Americans suffer from food allergies, with a number of recent studies suggesting that nearly 6 million or approximately 8% of children have some type of food allergy. Food allergies, in particular, can lead to extremely dangerous reactions and often lead to anaphylactic shock. According to a paper published in the Immunology and Allergy Clinics of North America, food, mainly peanut allergies, are responsible for 150 to 200 deaths every year in the United States. The U.S. Centers for Disease Control and Prevention reported that food allergies result in more than 300,000 ambulatory-care visits per year among children under the age of 18. Every three minutes a food allergy reaction sends someone to the emergency department, which is about 200,000 emergency department visits per year, and every six minutes the reaction is one of anaphylaxis. A recent U.S. study indicates an increase of 350% in the number of hospitalizations of children below age 18 for diagnosis of a food allergy for the period from 2004 to 2006 as compared to the period from 1998 to 2000. According to a paper published in the Immunology and Allergy Clinics of North America, the majority of fatal anaphylactic reactions in patients are caused by peanut allergy.
While anaphylactic shock is the most severe allergic reaction to food, patients also suffer from a poor quality of life. Symptoms tend to disappear within hours of exposure but, in some cases, can continue to affect patients for several days. Reactions can include, but are not limited to, skin discomfort, hay fever-like symptoms, impaired lung function and gastrointestinal complications, such as sustained bloating, nausea, vomiting and diarrhea. In some cases, food allergies can lead to chronic diseases such as failure to thrive in children and an allergic inflammatory condition of the esophagus called eosinophilic esophagitis, or EoE.
Recent studies suggest that patients with food allergies are especially at risk for experiencing significant disruption to their daily life. Food allergies are not only a physical disability; they are often associated with psychological traumas, including fear of eating, antisocial behavior and anxiety. In the case of pediatric patients, food allergies also have a significant impact on their caretakers. A recent study suggests that the quality of life in children with peanut allergy is more impaired than in children with insulin-dependent diabetes mellitus.
There Are No Approved Treatments Suitable for Food Allergies
Strict avoidance of food allergens and early recognition and management of allergic reactions to food are important measures to prevent serious health consequences. However, strict avoidance of food allergen is very difficult to achieve, especially for children. Some foods can contain hidden traces of allergens, labeling is often deceptive and contamination of allergen-free foods occurs regularly. For example, according to a paper published in the Journal of Allergy and Clinical Immunology, or JACI, it is estimated that accidental exposure to peanuts in peanut allergic patients occurs once every three to five years and the annual incidence of accidental ingestion is between 15% and 40%.
Treating Allergies Early in Life Can Modify the Disease, However, No Treatments Currently Exist for Young Children
Recent scientific studies suggest that treating allergies early in life could prevent disease progression or the development of polyallergies. A study of children desensitized to pollen and monitored for five years demonstrated that treating pollen allergy reduced the development of asthma. This early intervention, when the immune system is not mature, is referred to as the “window of opportunity.” Thus, research suggests that addressing allergies during this time in life is likely of critical clinical importance.
However, current techniques are poorly adapted to treating young allergy patients:
Due to these safety concerns, existing techniques are limited to children who are at least six years old. Given these limitations, it has been impossible to commercialize large-scale desensitization efforts for young children, even if medical research suggests that early allergy treatment during the “window of opportunity” is the best prophylactic and therapeutic management of the disease.
There is an Urgent Need for a Safe, Effective and Convenient Treatment for Food Allergy Patients
For all these reasons, food allergy patients, especially young children, their caregivers and their clinicians have long sought a safe, effective and convenient treatment. It is well understood that desensitization would be a desirable therapeutic approach as long as the procedure limits serious side effects, is convenient to apply and is effective. In particular, a therapeutic approach that promotes long-term therapeutic effect would be most desirable. To date, no such technique has been developed and approved.
Our Solution: Epicutaneous Immunotherapy (EPIT) Using Our Viaskin Technology Platform
Over the last decade, we have developed an innovative immunotherapy technology platform, with the potential for sustained therapeutic effect, by delivering biologically active compounds, including antigens, via intact skin. This technology platform, which we call Viaskin, is based on an electrostatic patch, which administers the antigen directly on the skin. Once administered, the antigen is concentrated in the superficial layers of the skin, where it activates the immune system by specifically targeting the LangheransLangerhans cells, without passage of the antigen into the bloodstream. We refer to this novel approach to immunotherapy as epicutaneous immunotherapy, or EPIT. Based on our trials and research, we believe that EPIT has the potential to provide all of the intended benefits of a disease-modifying treatment in allergy, while avoiding severe or life-threatening allergic reactions.
Viaskin—The First Epicutaneous Immunotherapy
Three important characteristics of our Viaskin technology platform contribute to its potential safety and efficacy:
Below is a diagram reflecting the primary components of the Viaskin patch:
|  |
The key elements of the Viaskin patch mechanism of action are the following:
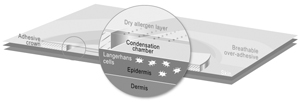  | Containing a dry layer of allergen in its center, the patch is positioned on intact skin, without prior preparation. |
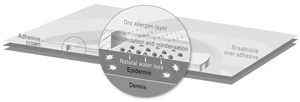  | The condensation chamber formed between the skin and the center of the patch creates hyperhydration of the skin and an accumulation of water. | |
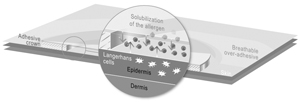  | The accumulation of water solubilizes the allergen. Due to this condensation chamber, the epidermis becomes more permeable allowing passage of the allergen into the epidermis. | |
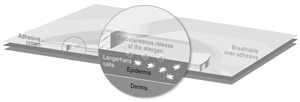  | Once in the epidermis, the allergen is captured by a population of highly specialized cells: Langerhans cells. These cells can take the protein at the surface of the skin, process it and present its epitopes to the lymphocytes in the lymph nodes. | |
Viaskin—Targeting the Unique Immunological Properties of Epicutaneous Langerhans Cells
The Viaskin’s effect on the immune system has been the subject of numerous scientific analyses and publications, which have been featured in major medical journals and allergology conferences. These epigenetic and mechanistic studies have helped us characterize the Viaskin’s novel mechanism of action.
Our mechanism of action is unique and differentiated as it targets specific epidermal dendritic cells, called Langerhans cells, which capture the antigen and migrate to the lymph node in order to activate the immune system without passage of the antigen into the bloodstream. After the antigen has been presented to the T cells in the lymph node, it activates the Tregs, the main factor in the down-regulation of Th2 response, while barely influencing the Th1 expression.expression, thus rebalancing the immune response.
Th2 cells are thought to play a role in allergic responses because allergies are known to be Th2 dominant conditions. An elevated Th2 response is ultimately responsible for the production of IgE, which can cause inflammation and trigger allergic reactions. Conversely, a normal, or non-allergic, immune response to an allergen is usually characterized by a well-balanced Th1/Th2 response.
We believe that EPIT can rebalance the immune reaction by decreasing, or down-regulating, the Th2 response to allergens, keeping Th1 and Th2 balanced and thus promoting long-term tolerance toward future allergen exposure. The first documentation of this mechanistic feature of our Viaskin patch in humans was presented at the 2016 AAAAI Annual Meeting in Los Angeles, California.
Viaskin—Compelling Clinical Benefits
We believe that our innovative approach to EPIT has the potential to offer compelling clinical benefits to patients suffering from severe allergies:
We believe that the Viaskin’s ability to induce epicutaneous immunological responses can also potentially be applied to other therapeutic areas, such as vaccination and treatment of inflammatory and autoimmune diseases.
Our Product Candidates
Our lead product candidate, Viaskin Peanut, is being developed for the treatment of peanut allergies in children, adolescents and adults. In December 2015, we initiated PEPITES, a randomized, placebo-controlled pivotal Phase III trial in children four to 11 years of age with Viaskin Peanut 250 µg. Results from PEPITES are expected in the second half of 2017. In September 2014, we announced topline results from our VIPES trial, a multi-center, double-blind, placebo-controlled, randomized Phase IIb clinical trial of Viaskin Peanut, which was followed by a full study report presented at the 2015 AAAAI Annual Meeting in Houston, Texas. The trial met its primary endpoint at the highest explored dose (Viaskin Peanut 250 µg), achieving statistical significance (p<0.01) in the percentage of treatment responders versus placebo. Our VIPES trial is the largest clinical trial in peanut allergy desensitization ever completed. Following the completion of VIPES, 171 patients enrolled in a two-year open label follow-up study, OLFUS-VIPES, which reported its first-year results in October 2015, followed by a complete study report in March 2016 during the AAAAI meeting in Los Angeles, California. Findings from the first twelve months of OLFUS-VIPES suggest that the treatment effect of Viaskin Peanut increases over time in children. In OLFUS-VIPES, we observed that 12 additional months of therapy with Viaskin Peanut 250 µg increased the number of patients benefiting from treatment to 70% in OLFUS-VIPES from 50% in VIPES, with 80% of children (ages six-11 at entry in VIPES) responding to therapy after 24 months. Viaskin Peanut has also been explored independently in two large academic trials, CoFAR6 and ARACHILD. CoFAR6, a Consortium of Food Allergy Research, or CoFAR, study sponsored by the National Institute of Allergy and Infectious Diseases, or NIAID, met its primary endpoint in cohorts treated with both Viaskin Peanut 100 µg (P=0.005) and Viaskin Peanut 250 µg (P=0.003). The 52-week results from CoFAR6 were presented at the 2016 AAAAI meeting. In June 2013, the Assistance Publique—Hôpitaux de Paris, or AP-HP, presented data from its ARACHILD pilot trial of Viaskin Peanut. In June 2012, we presented proof-of-concept data at the European Academy of Allergy and Clinical Immunology, or EAACI, Congress from a multi-center, double-blind, placebo-controlled, randomized Phase Ib clinical trial of Viaskin Peanut.
Our second product candidate, Viaskin Milk, is being developed for children (including infants) for the treatment of two indications,indications: IgE mediated CMPA and milk-induced EoE. Proof-of-concept data from a pilot clinical trial of Viaskin Milk was published in The Journal of Allergy and Clinical ImmunologyJACI in 2010. In November 2014, we initiated our MILES trial, a194 subject, multi-center, double-blind, placebo-controlled, randomized Phase I/II safety and efficacy MILES clinical trial of repeated doses of Viaskin Milk in patients with IgE mediated CMPA, and completion of its Part A (Phase I) occurred in June 2015. Part B (Phase II) is currently enrolling patients in the pediatric patient population with CMPA. InU.S. and Canada. Results from MILES are expected in the firstsecond half of 2017.
In November 2015, with our assistance,Dr. Jonathan Spergel from the Children’s Hospital of Philadelphia, intends to initiate aor CHOP, initiated an investigator-sponsored multi-center, double-blind, placebo-controlled, randomized trial to study safety and efficacy of Viaskin Milk in pediatric patient populations with milk-induced EoE. The trial is ongoing, and is expected to enroll approximately 20 patients four to 11 years of age.
We are also developing a third product candidate, Viaskin Egg, for the treatment of hen’s egg allergy. In the first half of 2015, we began pre-clinical work for this product candidate with the goal of initiating a clinical program if these studies are successful.
Viaskin Peanut
Background
Peanut allergy is one of the most common food allergies, and can cause severe, potentially fatal, allergic reactions, including anaphylaxis. Strict avoidance of peanut is essential, as even trace amounts of peanut can cause a severe allergic reactions. According to recent studies, food allergies, mainly peanut, are responsible for 150 to 200 deaths every year in the United States and about 200,000 emergency room visits. While anaphylactic shock is the most severe allergic reaction to peanuts, many patients also suffer from a poor quality of life. Peanut allergies have lifelong effects and are often associated with psychological traumas, including fear of eating, antisocial behavior and anxiety.
Allergy to peanuts appears to be on the rise and its prevalence has increased in the past 10 years. According to an article published in The Journal of Allergy and Clinical Immunology,JACI, a recent survey in the United States indicated that approximately 1% of the U.S. population, or more than three million people, are allergic to peanuts and/or nuts. Two recent studies conducted in the United States and the United Kingdom show that peanut allergy has doubled in five years in children below age five. A study funded by Food Allergy Research and Education, Inc., or FARE, indicates that the number of children in the United States with peanut allergy more than tripled between 1997 and 2008. Although some patients outgrow their peanut allergies, research indicates that only about 20% of individuals with peanut allergy outgrow it during a lifetime.
Development ProgramPhase III Clinical Trial — PEPITES
In December 2015, we initiated a pivotal Phase III trial designed to evaluate the safety and efficacy of Viaskin Peanut 250 µg in children four to 11 years of age suffering from peanut allergy. PEPITES is an international, randomized 2:1, double-blind, placebo-controlled Phase III study, in which pediatric peanut allergic patients will be treated with Viaskin Peanut 250 µg or placebo for 12 months. During the trial, patients’ peanut allergy will be assessed using a double-blind, placebo controlled food challenge, or DBPCFC, at baseline. The DBPCFC will be halted once the subject exhibits an objective symptom, as described on a pre-specified scale, thus establishing a subject’s peanut reactivity level, which is labeled as the patient’s eliciting dose, or ED. As in VIPES, subjects will receive a daily application of Viaskin Peanut or placebo over a 12-month treatment period. Each patch will be applied for 24 hours on the back of children.
Both the FDA and the EMA have agreed to a combined primary endpoint based on a responder analysis after 12 months of treatment. For patients with a baseline peanut protein ED equal to or less than 10 mg, a responder will be defined as a patient with a peanut protein ED equal to or greater than 300 mg of peanut protein after 12 months of treatment. For subjects with a baseline ED greater than 10 mg, a responder will be defined as a patient with a peanut protein eliciting dose equal to or greater than 1,000 mg of peanut protein after 12 months of treatment. Secondary endpoints will include the change from baseline of mean and median cumulative reactive dose of peanut protein, or CRD, which is used to establish the total quantity of peanut protein consumed during the DBPCFC. Serological markers will also be measured at baseline, three, six and 12 months in order to characterize the immunological changes in subjects.
PEPITES is expected to enroll approximately 330 patients in about 30 centers across North America (Canada and the U.S.), Europe, and Australia. Enrollment completion for PEPITES is expected in the third quarter of 2016. Based on this timeline, we expect study results to be available in the second half of 2017.
Phase IIb Clinical Trials—VIPES and VIPES OLFUSOLFUS-VIPES
VIPES (ViaskinPeanut’sEfficacy andSafety)
In August 2012, we initiated VIPES, a double-blind, placebo-controlled, multi-center Phase IIb clinical trial of Viaskin Peanut in 221 peanut allergic subjects with a well-documented medical history of systemic reactions after ingestion of peanut. Subjects completed their last food challenge visits after twelve months of treatment.
The VIPES trial was a multi-center clinical trial conducted at 22 sites in North America and Europe. In the trial, 221 peanut-allergic subjects were randomized into four treatment arms to evaluate three doses of Viaskin Peanut, specifically 50 µg, 100 µg and 250 µg peanut protein, compared to placebo. The trial was prospectively organized across the three dose levels with two patient strata, composed of three different patient age groups;groups: children (113 subjects, ages 6-11)six-11) for the first stratum and adolescents (73 subjects, ages 12-17) plus adults (35 subjects, ages 18-55) for the other stratum. Each patient underwent two double-blind, placebo-controlled food challenges, or DBPCFCs: one at initial screening and one at 12 months after initiation of treatment. The challenge was halted once the subject exhibited an objective symptom, thus establishing a subject’s peanut tolerance level.ED. Patients in VIPES received a daily application of the Viaskin Peanut patch over a 12-month treatment period. Each patch was applied for 24 hours, either on the upper arm for adults (age 18-55) and adolescents (age 12-17) or on the back of children (age 6-11)six-11).
Baseline peanut tolerance levels were established by measuring the peanut eliciting dose at which patients began to exhibit allergy symptoms, thus establishing the reactive baseline dose. The median baseline reactive dose in VIPES was 30mg30 mg for children and 100mg100 mg for adolescents and adults. The distribution of patients’ baseline reactive dose is summarized in the graph below.

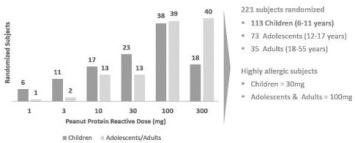
The primary efficacy endpoint in the trial was the percentage of treatment responders for each active treatment compared to placebo. Trial responders were defined as patients who, after 12 months of treatment with Viaskin Peanut and using a double-blind, placebo controlled food challenge,DBPCFC, started to react at a dose of peanut protein equal to or greater than 1,000 mg, or at least a 10-fold increase in the eliciting dose of peanut protein compared to baseline. As a secondary efficacy endpoint, Cumulative Reactive Dose, or CRD was also used to establish the total quantity of peanut protein that began triggering patient reactions at month 12 versus placebo. Serological markers were also measured as additional secondary endpoints at baseline, 3, 6three, six and 12 months in order to characterize the immunological changes in subjects. In terms of peanut consumption and immunological changes, a consistent dose effect was observed.
The principal coordinating investigator for VIPES in North America is Dr. Hugh Sampson, M.D., Chief of the Division of Allergy & Immunology in the Department of Pediatrics, Director of the Jaffe Food Allergy Institute, and Dean of Translational Biomedical Science at The Mount Sinai Medical Center in New York, United States. Dr. Sampson is also a member ofjoined us in June 2015 and was appointed as our Chief Scientific Advisory Board as well as principal investigator of the National Institutes of Health-sponsored Consortium of Food Allergy Research clinical trial with Viaskin Peanut (CoFAR6).Officer in November 2015.
The principal coordinating investigator for VIPES in Europe is Christophe Dupont, M.D., Ph.D., Head of the Pediatric-Gastroenterology Ambulatory Department at the Necker Hospital, (AP-HP). Heor AP-HP. Dr. Dupont is a member of the European Society for Pediatric Gastroenterology, Hepatology and Nutrition and of the Committee of Nutrition of the French Pediatric Society. Dr. Dupont is also the Chairman of our Scientific Advisory Board.
Results of VIPES Trial
In September 2014, we announced topline results for the VIPES trial, which was followed by a full study report at the 2015 AAAAI Annual Meeting in Houston, Texas, during an oral presentation by Dr. Sampson titled, “Epicutaneous Immunotherapy (EPIT) is effective and safe to treat Peanut Allergy: a multi-national, double-blind placebo-controlled randomized, phase IIb trial.” We discussed additional post-hoc analyses during a webcasted company-eventcompany event following the AAAAI meeting.
The primary efficacy endpoint was met with Viaskin 250mg, µg, with 50.0% responders vs 25.0% with placebo, p=0.0108 [Figure 1]. Moreover, children in the Viaskin 250mg µg arm (6-11(six-11 years) exhibited 53.6% responders vs 19.4% for placebo, p=0.0076 [Figure 2]. In children, the mean CRD showed a Viaskin Peanut dose-dependent response, with a change from baseline of +61 mg, +471 mg, +570 mg and +1121 mg for the placebo, 50mg, µg, 100mg, µg, and 250mg µg arms, respectively [Figure 3]. Children’s immunological responses were deemed to be robust. In the Viaskin 250mg µg arm, peanut-specific IgE exhibited a median increase³ 50 kUA/L at 3 months and decreased back to baseline at 12 months; median peanut-specific IgG4 at 12 months increased in a dose-dependent fashion: 1.3, 5.5-, 7.2- and 19.1-fold for each dose arm, respectively [Figure 4].
Additional post-hoc analyses on the children stratum demonstrated that on a peanut consumption basis, a significant amount of patients were strong responders to treatment. In a post-hoc analysis, VIPES shows that 32.1%, 26.9% and 17.9% of children subjects in the Viaskin 250mg, 100mg and 50mg arms, respectively, ingested both 10 times more peanut protein compared to baseline and at least 1,000 mg of peanut protein at month 12, compared to 0% in the placebo arm. We believe that this analysis confirms data presented by Dr. Sampson, which demonstrated both strong treatment and clear dose effect across the trial [Figure 5].
We are conducting additional analyses on the adolescents and adults age stratum. Due to a high placebo response rate, we believe these results need to be investigated further before determining our Viaskin Peanut development path in these patient populations [Figure 6]5]. After our End of Phase II Meeting with the FDA, we willWe intend to refine our development strategy for both peanut allergic adolescents and adults.adults in the next 12 months.
Patient compliance with daily Viaskin Peanut application was above 97%. The safety profile was confirmed across all active arms with no serious treatment-related adverse events reported or use of epinephrine related to treatment. Three separate Data Safety Monitoring Board, or DSMB, meetings concluded that VIPES did not have any safety concerns. In the trial, there were 20 serious adverse events, or SAEs, but none related to study drug. OutOf the 20 serious adverse events, or SAEs in VIPES, 1614 were anaphylaxes during the DBPCFC; twoDBPCFC, three were moderate anaphylaxes after accidental consumption of peanut containing foods outside of clinical trial site; theresite, one was alsoa reaction to fish consumption, one respiratory distress case;case and one psychiatric case. The trial drop-out rate was 6.4%, or 14 patients, which was below the 15% rate initially anticipated. Two out of the 14 drop-outs were related to the study drug due to dermatitis, one was due to uncontrolled asthma not related to treatment and the remaining 11 patients were drop-outs due to non-compliance, lost to follow-up or consent withdrawals. Furthermore, local cutaneous reactions, mostly mild and moderate, were observed in the majority of the active groups.
The following Figuresfigures summarize these results.
Figure 1: Summary of VIPES Responders
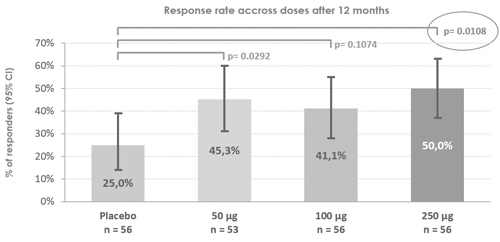
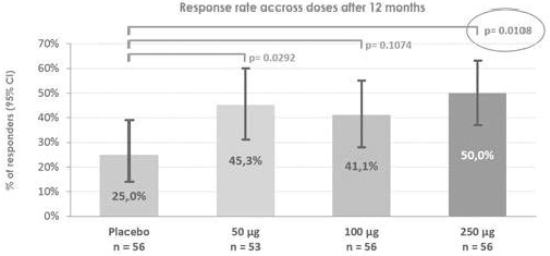
Figure 2: Summary of VIPES Responders: Children
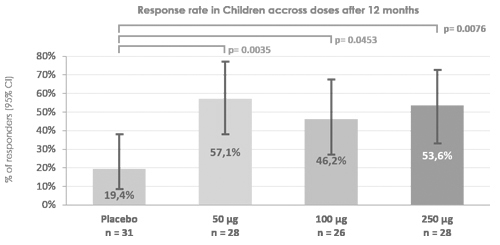
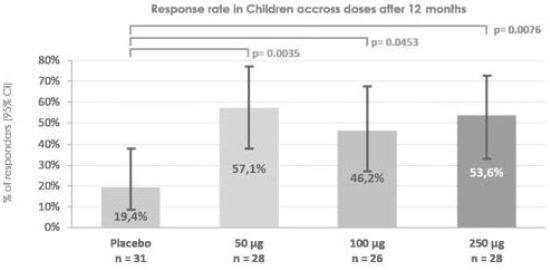
Figure 3: Summary of CRD Changes from Baseline in Children
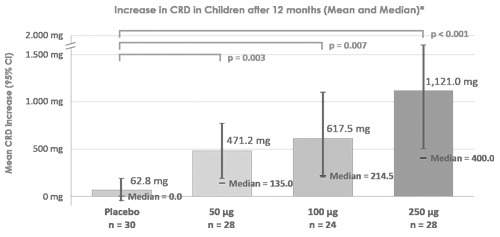
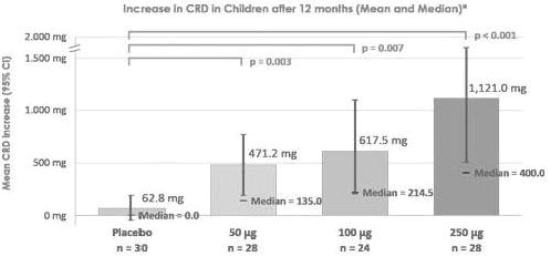
Figure 4: Summary of Immunological Responses in Children
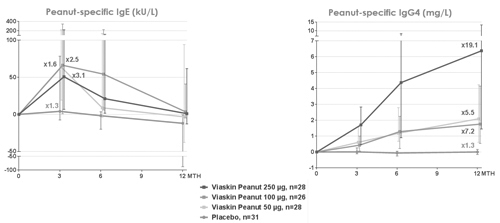
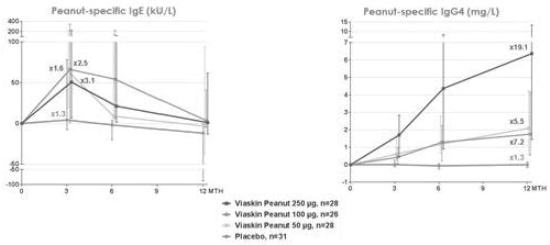
Figure 5: Summary of Children Ingesting 10 Times More Peanut Protein and at Least 1,000 mg of Peanut Protein at
Month 12
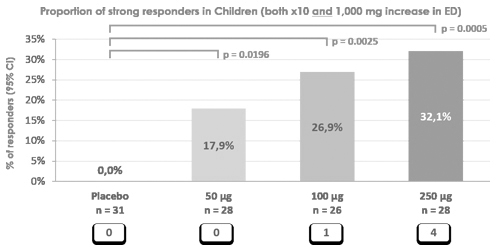
Figure 6: Summary of VIPES Responders: Adolescents and Adults
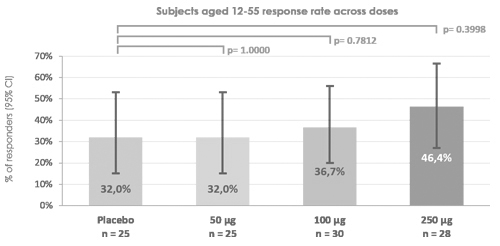
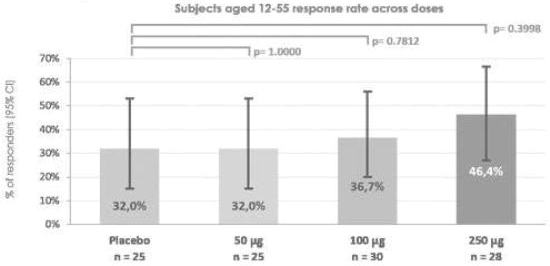
VIPES OLFUSOLFUS-VIPES (Open-LabelFollow-UpStudy)
In September 2013, we initiated an open-label follow-up Phase IIb clinical trial called VIPES OLFUSOLFUS-VIPES to assess the long-term efficacy and safety of Viaskin Peanut in subjects with peanut allergy. VIPES OLFUSOLFUS-VIPES is an extension trial for subjects having completed 12 months of the VIPES double-blind, placebo-controlled clinical trial, during which all subjects will be treatedare under active treatment with aViaskin Peanut 250 µg dose of Viaskin Peanut. VIPES OLFUSµg. OLFUS-VIPES includes 170171 subjects at 21 sites in North America and Europe, representing 82%83% of the patients who completed 12 months of therapy in the VIPES trial.
The objective of this trial is to assess the efficacy safety and sustained tolerancesafety of Viaskin Peanut after up to 36 months of epicutaneous immunotherapy in peanut allergic subjects.subjects, as well as sustained unresponsiveness of subjects to peanut protein after cessation of treatment.
Results
In October 2015, we announced topline results for OLFUS-VIPES, which show that the treatment effect with Viaskin Peanut seems to increase over time in children. Additional results were also presented by Dr. Sampson during the 2016 AAAAI meeting in Los Angeles, California. In the topline results for OLFUS-VIPES, we observed that 12 additional months of therapy with Viaskin Peanut 250 µg increased the number of patients benefiting from treatment to 70% in OLFUS from 50% in VIPES. No drug-related epinephrine use or SAEs due to Viaskin Peanut were reported. The study’s median compliance rate, which was maintained at 96%, was also consistent with previously reported results from VIPES.
Moreover, the response rate in children (ages six-11 at entry in VIPES) treated with the 250 µg dose for 24 months was observed to increase to 80% from 57% at OLFUS baseline; the average CRD for the this treatment group was 1,883 mg (1,440 mg median) of peanut protein compared to 84 mg (44 mg median) at baseline during VIPES entry [Figure 6]. Serological markers for this treatment group showed the strengthening of the immunological changes initially observed in VIPES. After 24 months, a median 40% decrease from the VIPES baseline value in peanut-specific IgE was observed, while the high median levels in immunoglobulin G4m, or IgG4, were maintained at an 800% increase from the VIPES baseline [Figure 7].
Children who received the placebo for one year in VIPES and received Viaskin Peanut for 12 months in OLFUS showed a 53.6% response rate compared to 17.2% at the OLFUS-VIPES baseline, which was consistent with previous findings from VIPES [Figure 8].
After 12 additional months of treatment with Viaskin Peanut, no additional significant clinical response was observed relative to the OLFUS baseline in the adolescents’ and adults’ treatment groups. Response rates consistent with the results observed in VIPES were shown in treatment-naïve adolescents and adults who received 12 months of therapy during OLFUS.
The following figures summarize these results.
Figure 6: Summary of Responders in OLFUS-VIPES - Children Treated for 24 Months with Viaskin 250 µg
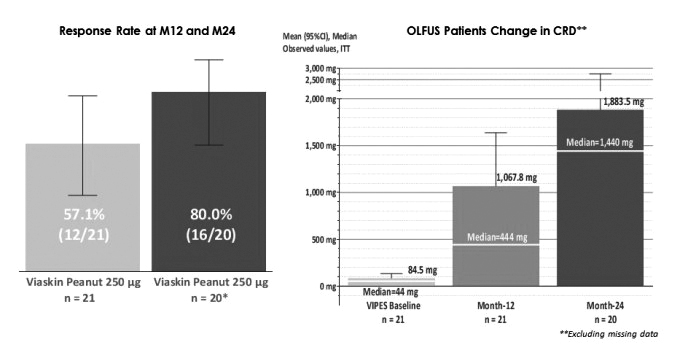
Figure 7: OLFUS-VIPES Serological Evolution in Children Treated for 24 Months with Viaskin 250 µg
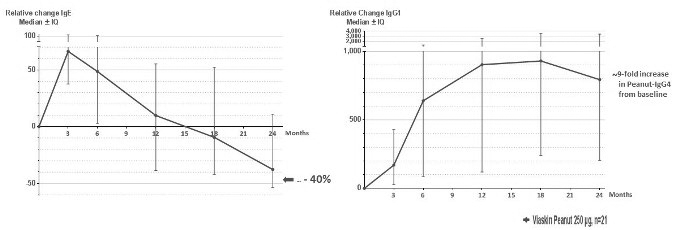
Figure 8: Summary of Responders in OLFUS-VIPES - Children Treated with Viaskin Peanut for 12 Months
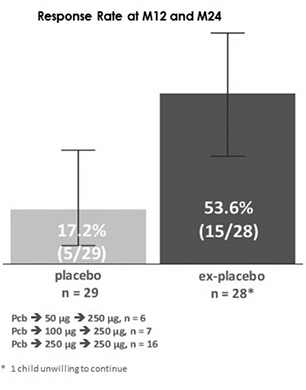
Phase Ib Clinical Trial
In July 2010, we initiated our first clinical trial of Viaskin Peanut in the United States, which was a Phase Ib trial to evaluate the safety and tolerability of repeated epicutaneous administration of Viaskin Peanut in patients allergic to peanuts. Results from this trial were published in February 2016 in JACI. In the trial, which was conducted at five leading centers in the United States, 100 subjects (initially adults, followed by adolescents and then children) allergic to peanuts, including 70 with a non-severe allergy and 30 with a severe allergy, were randomized and treated for two weeks with 20 µg to 500 µg of Viaskin Peanut or with placebo. Subjects with a history of severe anaphylactic reactions could be enrolled only after assessment of the safety of Viaskin Peanut in subjects with historical non-severe anaphylaxis. The primary endpoint of this clinical trial was safety, with the primary safety parameters of adverse events, physical examinations, vital signs, lab values, allergic reactions, any skin reactions, local or distant, echo-cardiogram, and Peak Expiratory Flow and spirometry (FEV1)(FEV1). Secondary endpoints included the proportion of subjects that experience systemic reactions such as urticaria, asthma and acute dyspnea, change in blood pressure, and digestive symptoms (vomiting, diarrhea)such as vomiting and diarrhea associated with Viaskin Peanut treatment versus placebo, the proportion of subjects requiring treatment for systemic reactions related to Viaskin Peanut treatment or placebo, and overall adherence to the clinical trial treatment.
In the overall population, the dose of 500 µg of Viaskin Peanut in adults and adolescents, and the dose of 250 µg of Viaskin Peanut in children, were each shown to be well-tolerated maximum doses regardless of the administration plan. Importantly, an excellent treatment compliance rate (>greater than 96%) was observed and the intermediate results suggested satisfactory usage safety of Viaskin Peanut in patients allergic to peanuts. The interim report was communicated to the FDA on December 15, 2011, and we released the complete results of this clinical trial at the EAACI Congress in June 2012.
Academic Trials
The lack of cure and approved treatments for food allergies has encouraged researchers and physicians to conduct several observational and mechanistic studies to further their understanding of these diseases. In the United States, for example, the National Institute of Allergy and Infectious Diseases, or NIAID of the United States National Institutes of Health has substantially increased its support for food allergy research since 2003, including the establishment of the Consortium for Food Allergy Research, or CoFAR in 2005.
As such, we have been approached by certain academic and research institutions interested in exploring Viaskin and EPIT’s mechanism of action and their impact on patients. In particular, both the AP-HP in France and CoFAR in the United States have initiated clinical trials to assess Viaskin Peanut’s efficacy: ARACHILD and CoFAR 6,CoFAR6, respectively. While not a sponsor of these trials, we have and will provide the doses of Viaskin Peanut needed to complete both of these trials.
CoFAR6 (Consortium forFoodAllergyResearch6)
In October 2013, CoFAR launched a multi-center, randomized, double-blind, placebo-controlled trial to evaluate Viaskin Peanut in children and adults allergic to peanuts. This trial is sponsored and funded by the NIAID and coordinated by Stacie M. Jones, MD, Professor Hugh Sampson in New York.of Pediatrics, University of Arkansas for Medical Sciences, Arkansas Children’s Hospital, Little Rock, Arkansas. The trial is being conducted in five hospitals in the United States and includes 75 patients, bothpatients; 54 children four to 11 years of age and 21 adolescents and adults and children. The recruitment12 to 25 years of age. In CoFAR6, ended in July 2014. Subjects will besubjects were randomized 1:1:1 to two doses of Viaskin Peanut (100 µg and 250 µg) or matched placebo and will undergo aplacebo. The primary outcome measure was the percent of patients desensitized to peanut protein during the peanut protein oral food challenge, or OFC, at week 52. ExpectedResponders were characterized as patients who successfully passed a 5044 mg OFC or who successfully consumed a dose ten times greater as compared to lastbaseline.
The 52-week CoFAR6 results were highlighted during two oral presentations at the 2016 AAAAI Annual Meeting in Los Angeles, California. Findings from this study were consistent with clinical data trends previously observed in
VIPES. In the CoFAR6 trial, Viaskin Peanut was observed to have a favorable safety and tolerability profile across treatment groups, with no SAEs or epinephrine use related to treatment observed. Treatment adherence was high (97.1%), dropouts were low (8%), and no withdrawals occurred in the 250 µg treatment group. Cohorts treated with both Viaskin Peanut 100 µg (P=0.005), and Viaskin Peanut 250 µg (P=0.003) met the primary efficacy endpoint in all populations. The treatment response was enhanced in children four to 11 years this trial will enable analysis of the effects of peanut desensitizationage, and also, with Viaskin Peanut over an initial period250 µg compared to Viaskin Peanut 100 µg. Based on additional analysis presented separately by us, we observed a greater response in children treated with the 250 µg dose (P=0.001).
CoFAR6 also explored mechanistic features of 12 months.Viaskin Peanut. Dr. Cecilia Berin, Associate Professor Pediatrics, Mount Sinai Hospital in New York, New York presented early findings from CoFAR6 at the 2016 AAAAI meeting, which supported Viaskin Peanut’s mechanistic features that have been observed at the preclinical level. Viaskin Peanut at the 250 µg dose showed a trend of decreased Th2 cell frequency without any increased trends in the Th1 response. In animal models, we have observed that Viaskin’s unique mechanism of action could rebalance the immune reaction by down-regulating the Th2 response to allergens while keeping Th1 responses balanced.
ARACHILD
The ARACHILD trial is a pilot trial conducted in France by the AP-HP. It is a DBPCFC trial to investigate the efficacy and safety of Viaskin Peanut in peanut allergic patients recruited from six centers. In the trial, 54 patients (35 children (age 5five to 11) and 19 adolescents (age 12 to 18)), were randomized into two treatment arms to evaluate a single dose of Viaskin Peanut, specifically 100 µg of peanut protein, compared to placebo. Patients in the placebo arm were crossed over at six months to Viaskin Peanut without unblinding the trial. Each patient underwent DBPCFCs at months 6,six, 12 and 18 after initiation of treatment. After the initialdouble-blind six-month treatment period, all patients went through an open-label period of 30 months. The primary endpoint of the trial was the proportion of patients who achieved at least a 10-fold increase in initial reactive dose or cumulative reactive dose, or CRD greater than 1,000 mg of peanut protein (about 4four peanuts). The secondary endpoints included significant immunological changes.
In June 2013, AP-HP reported the results from the initial six-month double-blind placebo-controlled phase of the trial and for the first 12 months of the open-label follow-up phase. In the active group (28 subjects), 6-six-, 12- and 18-month data showed 7.4%, 20% and 40% of subjects, respectively, consuming at least 10 times more peanut protein than tolerated at the beginning of the trial (versus 7.7% in the placebo arm before the crossover to Viaskin Peanut at month six, then 13% and 19% respectively after the crossover). Net trends of a specific sub-analysis of 19 adolescents (age 12 to 17) showed that despite a positive serological response of IgE, no adolescents qualified as responders at 6,six, 12 and 18 months. In an analysis of 35 children (age 5five to 11), we observed not only a positive serological response of IgE, but also that an immunological response is characteristic of an acquisition of tolerance leading to a continuous and progressive number of responders. For the children subgroup, 6-six-, 12- and 18- month data showed 12.5%, 33.3% and 66.7% of subjects, respectively, consuming at least 10 times more peanut protein than at the beginning of the trial (versus 10.5% in the placebo arm before the crossover to Viaskin Peanut at month six, then 16.7% and 23.5%, respectively, after the crossover). Viaskin Peanut also showed significant immunological changes (secondary efficacy endpoints) in the overall population, withclear-cut results in children. In treated children, peanut-specific IgE were increased by more than two-fold at 6-month, before decreasing and approaching toward initial levels at 18-month, while peanut-specific IgG4 (immunoglobulin G4) increased by more than eight-fold over 18-month of treatment.
Additional analyses of these data also suggest a linear relationship between body surface and response rate as well as onset of response. This analysis supports the belief that the 100 µg dose in Viaskin Peanut used in Arachild was potentially too low to generate a significant clinical outcome in patients with a higher body surface. In addition, these data also suggest that levels of the antibody IGg4 are potentially a good predictor of future patient response.
Pre-Clinical Studies
Prior to commencing our clinical trials of Viaskin Peanut, we completed a series of customary proof-of-concept and IND-enabling pre-clinical studies. These includedin vitropharmacokinetic/absorption studies,in vivopharmacology studies in a mouse model of peanut allergy, and toxicology studies, as well as ISO 10993-compliant biocompatibility studies for the device component.
Viaskin Milk
Background
Cow’s milk protein allergy, or CMPA is frequently the first allergy that appears during early childhood. CMPA is often missed in the primary care setting and can be a significant cause of infant distress when left undiagnosed. Symptoms can include gastrointestinal problems includingsuch as vomiting and diarrhea, skin rash, angioedema or rapid swelling of the skin, and anaphylaxis. The only option available for CMPA management is the avoidance of cow’s milk, which can lead to issues of dietary imbalance, failure to thrive and poor quality of life.
In addition, cow’s milk allergy is the primary causebelieved to be involved in two-thirdsa majority of thecases of EoE in children with EoE.and it is estimated that EoE impacts one in every 2,000 children. EoE is a debilitatingrecently recognized allergic inflammatory disease, involvingcharacterized by swelling of the esophagus. Typical symptoms include vomiting, abdominal pain, regurgitation, dysphagia and, in young children and infants, feeding difficulties and failure to thrive. Because the diverse and non-specific symptoms, EoE can be diagnosed only by esophageal dysfunction, duebiopsy. In addition to massivepresenting symptoms, acute and abnormal infiltration of eosinophil granulocytes in thechronic complications that may arise if EoE remains untreated include food impaction, esophageal stricture, narrow-caliber esophagus and the upper digestive tract,esophageal perforation. EoE is considered to be a chronic condition with inflammation of the esophagus.no currently approved treatments.
CMPA is the most common food allergy in infants and young children, affecting 2% to 3% of the general population. In approximately 80% of CMPA cases, the allergy to cow’s milk disappears after age 16. However, according to an expert panel convened by the AAAAI, approximately 35% of children with severe CMPA subsequently develop other food allergies or allergic respiratory diseases, such as asthma.
Development Program for Viaskin Milk
MILES (MILk Efficacy(MILkEfficacy and Safety)Safety)
In November 2014, we initiated our MILES trial, a multi-center, double-blind, placebo-controlled, randomized Phase I/II trial to study the safety and efficacy of Viaskin Milk in pediatric patient populations (age 2two to 17) suffering from CMPA with elevated IgE levels related to cow’s milk protein.mediated CMPA. We have an open IND for this trial. This trial is being conducted in select U.S. and Canadian clinical centers, including sites belonging to the CoFAR.centers. Up to 150194 subjects (18 subjects in Part A and 132176 subjects in Part B) will be randomized for treatment at approximately 10 to 1418 sites. Eligible subjects with confirmed IgE-mediated CMPA will perform a first food challenge at screening with escalating doses of cow’s milk proteins. Subjects who show the appearance of objective signs or symptoms to an eliciting dose of cow’s milk proteins£300 mg (approximately 9.4 mL of cow’s milk) will be randomized in the trial.
In June 2015, we announced results for Part A of MILES, which is equivalent to Phase I, will evaluatewhich evaluated the safety of repeated daily applications of three escalating dose-levels of Viaskin Milk (150 µg, 300 µg and 500 µg cow’s milk protein) versus placebo during three weeks. The DSMB for the study recommended that the study continue and expressed no safety concerns after evaluating the Part A safety data of subjects treated with the three doses of Viaskin® Milk.
Enrollment for Part B of the trial, which is equivalent to Phase II, was launched in October 2015, and it is expected to be completed in the third quarter of 2016, with study results expected in the second half of 2017. Part B is designed to evaluate the safety and efficacy of up to two selectedthree doses of Viaskin Milk (as determined from Part A), after a review of the safety data. After month(150 µg, 300 µg, 500 µg) compared to placebo for 12 all subjects from Parts A and B will continue treatment for another 12 months in anopen-label manner with Viaskin Milk, at the highest dose determined to be safe based on safety data from Part A.months. The primary efficacy endpoint will be the percentage of subjects who are treatment responders after 12 months, defined as subjects who meet at least one of the following criteria: a³10-fold(1) a10-fold or greater increase in the cumulative reactive dose, or CRD of cow’s milk proteins at the month 12 of the food challenge as compared to baseline value andin addition to reaching tolerance to at least 144 mg of cow’s milk proteinsprotein (approximately 4.5 mL of milk); or (2) a CRD of cow’s milk proteins³1444 greater than or equal to 1,444 mg (approximately 45 mL of milk) at the month 12 of the food challenge. Secondary efficacy endpoints include, among other things,others, the percentage of subjects who are treatment
responders at month 24, the mean and median CRD of cow’s milk proteins at months 12 and 24 andas well as the change in CRD from baseline, the change from baseline in the severity of symptoms elicited during the food challenge from baseline to months 12 and 24, and the change formfrom baseline in quality of life assessments at months 12 and 24.
SMILEE (Study of efficacy and safety of the ViaskinMILk in Milk-InducedEosinophilicEsophagitis)
SMILEE is a double-blind, placebo-controlled, randomized 3:1 trial designed to evaluate the safety and efficacy of Viaskin® Milk 500 µg for treating milk-induced EoE in children ages four to 11. The trial is being conducted by Dr. Jonathan Spergel at CHOP pursuant to an investigator-sponsored IND application that was accepted by the FDA in July 2015. Although we are providing assistance in the form of funding and trial supplies, this trial is being conducted by CHOP and supervised by Dr. Spergel.
Subjects with a documented medical history of EoE after ingestion of milk who currently adhere to a strict milk-free diet are being considered for participation in the trial. Approximately 20 subjects, 15 in the active treatment group and five in the placebo group, will be randomized and treated for nine months while remaining on a milk-free diet. The subjects will then continue their assigned treatment during a milk reintroduction period (1 week to 2 months), for a total of up to 11 months of treatment. The primary efficacy endpoint will evaluate the maximum esophageal eosinophil count in the active treatment group compared to placebo at the end of treatment. Secondary efficacy endpoints will include the change in symptoms score at the end of treatment compared to baseline and mean esophageal eosinophil count at the end of treatment. SMILEE began enrolling patients in November 2015.
Pilot Clinical Trial
ProfessorDr. Christophe Dupont and the AP-HP conducted a double-blind, placebo-controlled pilot clinical trial of EPIT in 2005 in children (age 3three months to 15 years) with high levels of specific IgE related to cow’s milk protein who were unable to consume more than 10 mL of cow’s milk. A publication discussing this trial’s results was published in the Journal of Allergy and Clinical ImmunologyJACI in 2010. In the trial, at the end of a three-month treatment, the mean cumulative tolerated dose increment was 12-fold in the active group versus 8% in the placebo group.
At the start of the clinical trial, out of the 19 patients included, some patients could not tolerate the equivalent of one drop of milk without having severe reactions. However, after three or six months of treatment, almost half of the Viaskin Milk treatment group was able to ingest milk in large quantities. In contrast, no patients treated during the first three months with a placebo (patch without active substance) showed meaningful improvement. These same non-responder patients were then treated with Viaskin Milk and after three or six months of treatment, 80% of them experienced an improvement in their tolerance of milk. There were no serious or unexpected adverse events in the trial nor premature withdrawal from the clinical trial. Although larger studies are needed to confirm the statistical efficacy, we believe the results of the pilot clinical trial provide proof-of-concept for specific immunotherapy via the epicutaneous route for this indication.
In addition, with assistance from us, The Children’s Hospital of Philadelphia, or CHOP, intends to file an IND for a trial that we call SMILEE in the first half of 2015, a double-blind, placebo-controlled, randomized trial to study efficacy and safety of the Viaskin milk for treating milk-induced eosinophilic esophagitis in children, we expect the SMILEE trial will be conducted as follows, subject to the applicable institutional review board, or IRB, and FDA approval. Twenty subjects aged from 4 to 17 years will be randomized into either Viaskin Milk or placebo arms. Treatment will start at randomization, during which subjects will maintain a milk-free diet. After nine months of treatment, subjects will be re-exposed to milk for up to two months, at which point biopsies will be performed to evaluate primary endpoints.
Although we are providing assistance in the form of funding and trial supplies, this trial will be conducted by CHOP and supervised by its staff member Jonathan Spergel, MD, PhD.
Pre-Clinical Studies
Prior to commencing our clinical trial of Viaskin Milk, we completed a series of customary proof-of-concept and IND-enablingpre-clinical studies. These includedin vitro pharmacology studies in a murine model of allergies, general safety studies in amilk-sensitized animal model, genetic and other toxicology studies with the milk protein extract, and local tolerance studies, as well as biocompatibility studies of the device component.
Viaskin Egg
Background
Hen’s egg allergy is the second most common food allergy in children. A 2011 study conducted in Australia estimated that up to 8.9% of infants react to raw egg. Several global studies suggest that egg allergy affects 1.5% to 3% of young children globally. However, most children seem to outgrow egg allergy before adolescence. A recent publication estimated that approximately 50% of children with egg allergy will become tolerant by six years of age, although resolution was highly correlated to lower egg-specific IgE levels and the absence of systemic reactions beyond topical sensitivity.
Egg-allergic reactions are mostly cutaneous in nature, including skin rashes and hives, and typically occur within 30 minutes of egg contact or ingestion. Gastrointestinal problems, such as vomiting, and respiratory complications, such as nasal congestion, are also common, but anaphylaxis is not often reported. Food allergy experts believe that about one thirdone-third of eczema patients react to food triggers, which can sometimes cause the eczema to worsen. The most common food allergen associated with eczema is egg.
Development Program for Viaskin Egg
We are developing Viaskin Egg as a treatment that we believe can reduce the clinical manifestations of hen’s egg allergy. Studies also suggest that the treatment of egg allergy in young children may have a significant impact on preventing the occurrence and development of eczema.
We recently begunbegan pre-clinical work for this product candidate with the goal of initiatingand plan to initiate a clinical program if these studies are successful.
Other Potential Viaskin Technology Applications
We believe that our broadly applicable technology platform, know-how and deep understanding of EPIT positions us well to develop product candidates in areas of unmet medical need in immunotherapy. We currently expect to selectively conduct product development programs outside of our core food allergy expertise, and will often seek to collaborate with companies or agencies that are experts in a particular field. To date, we have signed several collaboration agreements to broaden the number of indications we are pursuing with our Viaskin technology platform, while also developing other potential product candidates independently. These programs and collaboration agreements are in the early proof-of-concept phase, and we do not expect to provide regular updates unless and until we elect to move one of them forward in a meaningful way. These preclinical assets include the following:
| • | With the University of Geneva and Bionet Asia, we are developing a booster vaccine against pertussis, for which a first clinical trial is planned to start in 2016. In June 2015, scientific data demonstrating the potential application of the Viaskin technology to boost protective immunity against Bordetella pertussis (whooping cough) was published inVaccine. |
In addition, we are continuing to explore other cellular mechanisms modulated by EPIT, such as biomarkers, in collaboration with Mount Sinai Hospital in the United States, the Centre d’Immunologie de Marseille-Luminy, or CIML, in France and Commissariat à l’Énergie Atomique et aux Énergies Alternatives, or CEA, in France. We believe that with improved knowledge about the evolution of immunological biomarkers and epigenetic modulation, we may be able to determine the level of patient response earlier during treatment, ensure follow-up and measure tolerance maintained once treatment is completed.
Our First Marketing Experience: Diallertest Milk—A Ready-to-Use Patch Test For Detecting CMPA in Young Children
In 2004, we introduced to the French market Diallertest Milk, the first ready-to-use patch test for detecting CMPA in young children. Diallertest Milk is sold exclusively in France through a distribution partner. To date, sales of Diallertest Milk have been moderate, totaling approximately 25,000 kits per year on average over the past three years. We do not currently expect theseDuring the second half of 2015, we discontinued our commercial partnership with respect to the product revenues to have a material impact on our business and financial condition in future periods.ceased selling Diallertest Milk.
In June 2009, we received a letter from the Agence Française de Sécurité Sanitaire des Produits de Santé (French Agency for the Safety of Health Products), informing us that, in its view, Diallertest Milk, although previously marketed as a medical device in France, met the criteria for a drug product and accordingly would be required to complete a Phase III marketing authorization study in accordance with the regulations in France applicable to the marketing of drug products. However, in light of the safety profile and perceived clinical utility of this product, the agency did not require that we discontinue the marketing and sale of Diallertest Milk and instead indicated that they would review with us our progress towards completing a Phase III trial at a later date. As of the date of this Annual Report on Form 20-F we have not yet initiated this Phase III clinical trial, as we continue to evaluate the commercial rationale for doing so. We are also evaluating potential marketing and/or distribution relationships to market this product in Europe.Europe and in other major markets.
There can be no assurance that the French regulatory authorities will continue to permit us to market and sell Diallertest Milk prior to the completion of a Phase III clinical trial for this product. We may also elect to discontinue the marketing and sale of this product on our own. See “Risk Factors—We are dependent on a single exclusive distributor for the marketing of our Diallertest Milk diagnostic product. We may discontinue our Diallertest Milk program.”
Manufacturing and Supply
Our Proprietary Viaskin Technology
We have engineered a proprietary manufacturing technology for Viaskin patch, which is designed to comply with the most stringent pharmaceutical production standards, including those promulgated by the FDA, in order to enable Viaskin to deliver proteins via intact skin. This novel pharmaceutical process, which was fully developed by us, uses an electrospray to spray homogeneous, thin, dry protein layers onto the Viaskin patch.
  | This process sprays a liquid solution of electrically charged proteins onto the patch’s backing, which is then turned into a dry solid charged particle, which remains stuck onto the patch’s backing. It deposits very small and precise quantities of the active substance, devoid of adjuvants. The patch can then be stored at room temperature, providing a long shelf life. We believe this patented technology is highly scalable and complies with cGMP requirements. |
The principles of the Viaskin electrospray technology are the following:
 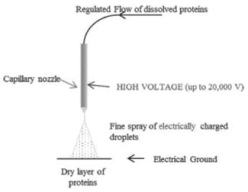 | When a liquid flows at a constant speed from a capillary and is subjected to a high voltage electric field (20,000 volts).
With our electrospray machine, we can transform these electrically charged liquid droplets into dry solid charged particles, and then drive them along the electric field lines onto the patch’s backing.
When the electric field lines are directed toward the grounded Viaskin patch, they force the dry particles to go directly to and only onto the patch. |
We have engineered the Viaskin patch with an electrically conductive backing in order to use an electrospray in its assembly. This conductive backing is placed under the machine’s cone at a specified distance; the patch is also grounded so that the electric field lines can be directed onto its surface. The dry particles from the electrospray follow these field lines and settle on the patch’s backing due to the attraction and conductivity produced by the electrostatic forces on the ground. Due to this process, the dry protein layers on the patch are homogenous and no loss of substance occurs during the spray. The electrostatic attraction between the particles and the medium keeps these particles attached on the patch.
With Viaskin manufacturing technology, we believe we can achieve:
Viaskin is a Highly Scalable Manufacturing Technology
Over the past five years, we have tailored our electrospray technology to conduct further clinical development of our Viaskin technology and for its subsequent commercialization.
We currently rely on a contract manufacturer, Sanofi, to manufacture the active pharmaceutical ingredients used in our Viaskin product candidates, such as peanut protein extract. Our manufacturing machine technology then uses an electrospray to deposit the active pharmaceutical ingredient onto the Viaskin patch. For our pre-clinical testing, we used two different prototypes. We then developed a third-generation machine in 2009 to manufacture patches for our clinical trials. For the Phase I and II of Viaskin Peanut and Viaskin Milk clinical trials, our electrospray machine, called GEN3.1, was able to produce 15,000 patches per batch, which was sufficient for our clinical needs. Overall, on a yearly basis, GEN3.1’s throughput is approximately 750,000 patches.
We have assembleddeveloped a new version of this tool, called GEN3.2, in 2014. This new generation manufacturing tool allows us to produce larger batch sizes of around 60,000 patches, which are compatible with our later-stage clinical development needs. Overall, on a yearly basis, GEN3.2’s throughput is expected to be approximately 2,250,000 patches.
We believe that a commercial-scale version of our electrospray manufacturing tool, called GEN4.0, will be completed in 2016.the second half of 2016 and efforts to increase our production capacity are currently ongoing. We anticipate that this tool will allow us to produce commercial batch sizes of around 500,000 patches, compatible with initial expected market demand. Overall, on a yearly basis, GEN4.0’s throughput is expected to reach approximately 20 to 30 million patches.
  | GEN3.1 (2009) 18 nozzles Used for Phase I and Phase II trials Batch size: 15,000 patches More than 200,000 produced since 2009 | |
  | GEN3.2 (2014) 54 nozzles Used for Phase III trials Batch size: 60,000 patches Improved electrospray process, forerunner of GEN4 | |
  | GEN4 (expected 2H 2016) Assembly ongoing Up to 300 nozzles Commercial products Used for commercialization Batch size: 500,000 patches |
We believe our proprietary Viaskin manufacturing technology creates high barriers to entry to our line of business, particularly in the engineering and manufacturing of our Viaskin product candidates. We design, develop and build our manufacturing tools, and contract third-party manufacturers to operate it. We are in the process of selecting third-party manufacturers to manage our manufacturing process after the approval and during the commercialization of our Viaskin product candidates.
Intellectual Property
Our patent portfolio includes pending patent applications and issued patents in the United States and in foreign countries. These patents and applications generally fall into four broad categories:
The term of a U.S. patent may be eligible for patent term extension under the Hatch-Waxman Act to account for at least some of the time the drug or device is under development and regulatory review after the patent is granted. With regard to a drug or device for which FDA approval is the first permitted marketing of the active ingredient, the Hatch-Waxman Act allows for extension of the term of one U.S. patent. The extended patent term cannot exceed the shorter of five years beyond the non-extended expiration of the patent or 14 years from the date of the FDA approval of the drug or device. Some foreign jurisdictions have analogous patent term extension provisions that allow for extension of the term of a patent that covers a device approved by the applicable foreign regulatory agency. In the future, if and when our Viaskin electrostatic patch receives FDA approval, we expect to apply for a patent term extension on the patent that we believe will provide the best exclusivity position if extended.
Co-Ownership Agreement
AP-HP and Université de Paris-Descartes
In January 7, 2009, we entered into an assignment, development and co-ownership agreement with Public Welfare-Hospitals of Paris (L’Assistance Publique—Hopitaux de Paris), referred to herein as AP-HP and Université Paris-Descartes, referred to herein asor UPD, by which we agreed to terms of co-ownership with AP-HP and UPD of certain U.S. and foreign patents and patent applications, referred to herein as the shared patents. We, and any licensees or
sublicensees that we designate, have the exclusive right to commercial uses of the shared patents. AP-HP and UPD agreed to use the shared patents only for internal research purposes and not to license the shared patents to any third party. Upon commercialization of any product covered by the shared patents, which we expect would include our Viaskin product candidates, we will be obligated to pay AP-HP and UPD a percentage of net sales as a royalty. This royalty is in the low single digits and varies depending on the particular patent used in the product and is in the low single digits.product. Additionally, if we license any of the shared patents to a third party and a licensee commercializes products covered by such shared patents, we will be obligated to pay AP-HP and UPD a percentage in the low single digits of the money that we receive from our licensee.
If we do not sell any of our product candidates covered by the shared patents within 30 months from the date we first market such product candidates, AP-HP may, upon six months’ notice and subject to certain exceptions, convert our exclusive right to the commercial use of the shared patents to a non-exclusive right.
Any party may terminate the license in the event of another party’s substantial breach which remains uncured after six months of receiving written notice of such breach. The agreement will also terminate in the event we cease operations or are subject to a dissolution or bankruptcy proceedings.
Absent early termination, the agreement will automatically terminate upon the expiration of the last shared patent. In the event the agreement is terminated, we would no longer have the exclusive right to commercial use of the shared patents, though we would retain
our shared ownership rights. In addition, our ownership stake in certain jointly made improvements covered by the shared patents would survive termination of the agreement. The longest lived patent rights licensed to us under the agreement are currently expected to expire in 2028.
Competition
The biotechnology and pharmaceutical industries are highly competitive and subject to significant and rapid technological change as researchers learn more about diseases and develop new technologies and treatments. Significant competitive factors in our industry include product efficacy and safety;safety: quality and breadth of an organization’s technology; skill of an organization’s employees and its ability to recruit and retain key employees; timing and scope of regulatory approvals; government reimbursement rates for, and the average selling price of, products; the availability of raw materials and qualified manufacturing capacity; manufacturing costs; intellectual property and patent rights and their protection; and sales and marketing capabilities.
We cannot assure you that any of our products that we successfully develop will be clinically superior or scientifically preferable to products developed or introduced by our competitors.
Our competitors may also succeed in obtaining FDA or other regulatory approvals for their product candidates more rapidly than we are able to do, which could place us at a significant competitive disadvantage or deny us marketing exclusivity rights. Market acceptance of our product candidates will depend on a number of factors, including: (1) potential advantages over existing or alternative therapies or tests,tests; (2) the actual or perceived safety of similar classes of products,products; (3) the effectiveness of sales, marketing, and distribution capabilities,capabilities; and (4) the scope of any approval provided by the FDA or foreign regulatory authorities.
Although we believe our product candidates possess attractive attributes, we cannot assure you that our product candidates will achieve regulatory or market acceptance, or that we will be able to compete effectively in the biopharmaceutical drug markets. If our product candidates fail to gain regulatory approvals and acceptance in their intended markets, we may not generate meaningful revenues or achieve profitability.
There are numerous competitors on the market for the therapeutic treatment of allergies. Numerous structures, pharmaceutical laboratories, biotechnology companies, institutions, universities and other research entities are
actively involved in the discovery, research, development and marketing of therapeutic responses to treat allergies. Many of our competitors have greater resources and experience in terms of clinical development, management, manufacturing, marketing and research than us.
In the case of food allergies, we are aware of several academic studies that are currently being conducted in major centers and hospitals worldwide. These studies are evaluating sublingual, subcutaneous, intranasal or other forms of desensitization or products using synthetic allergens, denatured allergens or combinations of medicines or methods, or medicines using traditional methods such as Chinese herbs. We are not aware of any pharmaceutical development in conjunction with these academic efforts at this time.
We expect studies combining other methods of immunotherapy, such as OIT, with anti-IgE treatments will be conducted. These types of co-administrations may significantly improve the safety of specific immunotherapies administered orally or subcutaneously, and may become significant competitors with our products.
To our knowledge, other pharmaceutical and biotechnology companies are also seeking to develop food allergy treatments, although many are in the discovery or preclinical stages. For example, Aimmune Therapeutics, Inc., formerly known as Allergen Research Corporation, is currently evaluatingenrolling patients in a Phase II clinical trialsIII trial to evaluate the safety and efficacy of its OIT product candidate, AR101, in peanut allergic patients. To our knowledge, the company uses a formulation of peanut flour for oral administration intended for oral desensitization.desensitization to peanut. We are also aware of other companies that are working on recombinant peanut proteins capable of initiating an attenuated immune response byof using subcutaneous administration. We are also aware that Sanofi S.A. has entered into licensing agreements of discovery platforms in selected food allergies, notably with Immune Design Corp. and Selecta Biosciences Inc. and may pose a competitive risk to our products in the future.
Government Regulation
Government authorities in the United States at the federal, state and local level and in other countries extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, post-approval monitoring and reporting, marketing and export and import of drug and biological products, or biologics, such as our product candidates. Generally, before a new drug or biologic can be marketed, considerable data demonstrating its quality, safety and efficacy must be obtained, organized into a format specific to each regulatory authority, submitted for review and approved by the regulatory authority.
U.S. Biological Product Development
In the United States, the FDA regulates biologics under the Federal Food, Drug, and Cosmetic Act, or FDCA, and the Public Health Service Act, or PHSA, and their implementing regulations. Biologics are also subject to other federal, state and local statutes and regulations. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval, may subject an applicant to administrative or judicial sanctions. These sanctions could include, among other actions, the FDA’s refusal to approve pending applications, withdrawal of an approval, a clinical hold, untitled or warning letters, product recalls or withdrawals from the market, product seizures, total or partial suspension of production or distribution injunctions, fines, refusals of government contracts, restitution, disgorgement, or civil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect on us.
Our product candidates must be approved by the FDA through the Biologics License Application, or BLA, process before they may be legally marketed in the United States. The process required by the FDA before a biologic may be marketed in the United States generally involves the following:
The data required to support a BLA is generated in two distinct development stages: pre-clinical and clinical. The pre-clinical development stage generally involves laboratory evaluations of drug chemistry, formulation and stability, as well as studies to evaluate toxicity in animals, which support subsequent clinical testing. The conduct of the pre-clinical studies must comply with federal regulations, including GLPs. The sponsor must submit the results of the pre-clinical studies, together with manufacturing information, analytical data, any available clinical data or literature and a proposed clinical protocol, to the FDA as part of the IND. An IND is a request for authorization from the FDA to administer an investigational drug product to humans. The central focus of an IND submission is on the general investigational plan and the protocol(s) for human trials. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA raises concerns or questions regarding the proposed clinical trials and places the IND on clinical hold within that 30-day time period. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. The FDA may also impose clinical holds on a product candidate at any time before or during clinical trials due to safety concerns or non-compliance. Accordingly, we cannot be sure that submission of an IND will result in the FDA allowing clinical trials to begin, or that, once begun, issues will not arise that could cause the trial to be suspended or terminated.
The clinical stage of development involves the administration of the product candidate to healthy volunteers or patients under the supervision of qualified investigators, generally physicians not employed by or under the trial sponsor’s control, in accordance with GCPs, which include the requirement that all research subjects provide their informed consent for their participation in any clinical trial. Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria, and the parameters to be used to monitor subject safety and assess efficacy. Each protocol, and any subsequent amendments to the protocol, must be submitted to the FDA as part of the IND. Further, each clinical trial must be reviewed and approved by an independent institutional review board, or IRB, at or servicing each institution at which the clinical trial will be conducted. An IRB is charged with protecting the welfare and rights of trial participants and considers such items as whether the risks to individuals participating in the clinical trials are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the informed consent form that must be provided to each clinical trial subject or his or her legal representative and must monitor the clinical trial until completed.
There are also requirements governing the reporting of ongoing clinical trials and completed clinical trial results to public registries. Sponsors of certain clinical trials of FDA-regulated products, including biologics, are required to register and disclose specified clinical trial information, which is publicly available at www.clinicaltrials.gov. Information related to the product, patient population, phase of investigation, study sites and investigators, and other aspects of the clinical trial is then made public as part of the registration. Sponsors are also obligated to discuss the results of their clinical trials after completion. Disclosure of the results of these trials can be delayed until the new product or new indication being studied has been approved. However, there are evolving rules and increasing requirements for publication of all trial-related information, and it is possible that data and other information from trials involving drugs and biologics that never garner approval could require disclosure in the future. In addition, publication policies of major medical journals mandate certain registration and disclosures as a pre-condition for potential publication, even if not currently mandated as a matter of law. Competitors may use this publicly available information to gain knowledge regarding the progress of development programs.
Clinical trials are generally conducted in three sequential phases that may overlap, known as Phase I, Phase II and Phase III clinical trials. Phase I clinical trials generally involve a small number of healthy volunteers who are initially exposed to a single dose and then multiple doses of the product candidate. The primary purpose of these clinical trials is to assess the metabolism, pharmacologic action, side effect tolerability and safety of the product candidate and, if possible, to gain early evidence on effectiveness. Phase II clinical trials typically involve studies in disease-affected patients to determine the dose required to produce the desired benefits. At the same time, safety and further pharmacokinetic and pharmacodynamic information is collected, as well as identification of possible adverse effects and safety risks and preliminary evaluation of efficacy. Phase III clinical trials generally involve large numbers of patients at multiple sites, in multiple countries (from several hundred to several thousand subjects) and are designed to provide the data necessary to demonstrate the efficacy of the product for its intended use, its safety in use, and to establish the overall benefit/risk relationship of the product and provide an adequate basis for product approval. Phase III clinical trials may include comparisons with placebo and/or other comparator treatments. The duration of treatment is often extended to mimic the actual use of a product during marketing. Generally, two adequate and well-controlled Phase III clinical trials are required by the FDA for approval of a BLA.
Post-approval trials, sometimes referred to as Phase IV clinical trials, may be conducted after initial marketing approval. These trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication. In certain instances, FDA may condition approval of a BLA on the sponsor’s agreement to conduct additional clinical trials to further assess the biologic’s safety and effectiveness after BLA approval.
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and written IND safety reports must be submitted to the FDA and the investigators for serious and unexpected suspected adverse, findings from other studies suggesting a significant risk to humans exposed to the drug, findings from animal orin vitro testing suggesting a significant risk to humans, and any clinically important rate increase of a serious suspected adverse reaction over that listed in the protocol or investigator brochure. Phase I, Phase II and Phase III clinical trials may not be completed successfully within any specified period, if at all. The FDA, the IRB, or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients. Additionally, some clinical trials are overseen by an independent group of qualified experts organized by the clinical trial sponsor, known as a data safety monitoring board or committee. This group provides authorization for whether or not a trial may move forward at designated intervals based on access to certain data
from the trial. We may also suspend or terminate a clinical trial based on evolving business objectives and/or competitive climate. Concurrent with clinical trials, companies usually complete additional animal studies and must also develop additional information about the
chemistry and physical characteristics of the product candidate as well as finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, must develop methods for testing the identity, strength, quality and purity of the final product. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life.
BLA and FDA Review Process
Following trial completion, trial data is analyzed to assess safety and efficacy. The results of pre-clinical studies and clinical trials are then submitted to the FDA as part of a BLA, along with proposed labeling for the product and information about the manufacturing process and facilities that will be used to ensure product quality, results of analytical testing conducted on the chemistry of the product candidate, and other relevant information. The BLA is a request for approval to market the biologic for one or more specified indications and must contain proof of safety, purity, potency and efficacy, which is demonstrated by extensive pre-clinical and clinical testing. The application includes both negative or ambiguous results of pre-clinical and clinical trials as well as positive findings. Data may come from company-sponsored clinical trials intended to test the safety and efficacy of a use of a product, or from a number of alternative sources, including studies initiated by investigators. To support marketing approval, the data submitted must be sufficient in quality and quantity to establish the safety and efficacy of the investigational product to the satisfaction of the FDA. FDA approval of a BLA must be obtained before a biologic may be marketed in the United States.
Under the Prescription Drug User Fee Act, or PDUFA, as amended, each BLA must be accompanied by a significant user fee, which is adjusted on an annual basis. PDUFA also imposes an annual product fee for human drugs and an annual establishment fee on facilities used to manufacture prescription drugs. Fee waivers or reductions are available in certain circumstances, including a waiver of the application fee for the first application filed by a small business.
Once a BLA has been accepted for filing, which occurs, if at all, sixty days after the BLA’s submission, the FDA’s goal is to review BLAs within ten months of the filing date for standard review or six months of the filing date for priority review, if the application is for a product intended for a serious or life-threatening condition and the product, if approved, would provide a significant improvement in safety or effectiveness. The review process is often significantly extended by FDA requests for additional information or clarification.
After the BLA submission is accepted for filing, the FDA reviews the BLA to determine, among other things, whether the proposed product candidate is safe and effective for its intended use, and whether the product candidate is being manufactured in accordance with cGMP to assure and preserve the product candidate’s identity, strength, quality, purity and potency. The FDA may refer applications for novel drug product candidates or drug product candidates which present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions. The FDA will likely re-analyze the clinical trial data, which could result in extensive discussions between the FDA and us during the review process. The review and evaluation of a BLA by the FDA is extensive and time consuming and may take longer than originally planned to complete, and we may not receive a timely approval, if at all.
Before approving a BLA, the FDA will conduct a pre-approval inspection of the manufacturing facilities for the new product to determine whether they comply with cGMPs. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. In addition, before approving a BLA,
the FDA may also audit data from clinical trials to ensure compliance with GCP requirements. After the FDA evaluates the application, manufacturing process and manufacturing facilities, it may issue an approval letter or a Complete Response Letter. An approval letter authorizes commercial marketing of the product with specific prescribing information for specific indications. A Complete Response Letter indicates that the review cycle of the application is complete and the application will not be approved in its present form. A Complete Response Letter usually describes all of the specific deficiencies in the BLA identified by the FDA. The Complete Response Letter may require additional clinical data and/or an additional pivotal Phase III clinical trial(s), and/or other significant and time-consuming requirements related to clinical trials, pre-clinical studies or manufacturing. If a Complete Response Letter is issued, the applicant may either resubmit the BLA, addressing all
of the deficiencies identified in the letter, or withdraw the application. Even if such data and information is submitted, the FDA may ultimately decide that the BLA does not satisfy the criteria for approval. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than we interpret the same data.
There is no assurance that the FDA will ultimately approve a product for marketing in the United States and we may encounter significant difficulties or costs during the review process. If a product receives marketing approval, the approval may be significantly limited to specific populations, severities of allergies, and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling or may condition the approval of the BLA on other changes to the proposed labeling, development of adequate controls and specifications, or a commitment to conduct post-market testing or clinical trials and surveillance to monitor the effects of approved products. For example, the FDA may require Phase IV testing which involves clinical trials designed to further assess the product’s safety and effectiveness and may require testing and surveillance programs to monitor the safety of approved products that have been commercialized. The FDA may also place other conditions on approvals including the requirement for a Risk Evaluation and Mitigation Strategy, or REMS, to assure the safe use of the product. If the FDA concludes a REMS is needed, the sponsor of the BLA must submit a proposed REMS. The FDA will not approve the NDA without an approved REMS, if required. A REMS could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. Any of these limitations on approval or marketing could restrict the commercial promotion, distribution, prescription or dispensing of products. Product approvals may be withdrawn for non-compliance with regulatory standards or if problems occur following initial marketing.
Review and Approval of Combination Products in the United States
Certain products may be comprised of components that would normally be regulated under different types of regulatory authorities, and frequently by different centers at the FDA. These products are known as combination products. Specifically, under regulations issued by the FDA, a combination product may be:
Our Viaskin product candidates are combination products comprising a device for delivery of a biologic. Under the FDCA, the FDA is charged with assigning a center with primary jurisdiction, or a lead center, for review of a combination product. That determination is based on the “primary mode of action” of the combination product, which means the mode of action expected to make the greatest contribution to the overall intended therapeutic effects. Thus, if the primary mode of action of a device-biologic combination product is attributable to the biologic product, that is, if it acts by means of a virus, therapeutic serum, toxin, antitoxin, vaccine, blood, blood component or derivative, allergenic product, or analogous product, the FDA center responsible for premarket review of the biologic product would have primary jurisdiction for the combination product.
Expedited Development and Review Programs
The FDA has a fast track program that is intended to expedite or facilitate the process for reviewing new drugs and biological products that meet certain criteria. Specifically, new drugs and biological products are eligible for fast track designation if they are intended to treat a serious or life-threatening condition and nonclinical or clinical data demonstrate the potential to address an unmet medical need. Fast track designation applies to the combination of the product and the specific indication for which it is being studied. The sponsor of a new drug or biologic may request the FDA to designate the drug or biologic as a fast track product concurrently with the submission of an IND or at any time before a pre-NDA meeting, and the FDA must determine if the product qualifies for fast track designation within 60 days of receipt of the sponsor’s request. Unique to a fast track product, the FDA may consider for review sections of the marketing application on a rolling basis before the complete application is submitted, if the sponsor provides a schedule for the submission of the sections of the application, the FDA agrees to accept sections of the application and determines that the schedule is acceptable, and the sponsor pays any required user fees upon submission of the first section of the application.
Any product submitted to the FDA for marketing, including under a fast track program, may be eligible for other types of FDA programs intended to expedite development and review, such as priority review and accelerated approval. Any product is eligible for priority review, or review within a six-month timeframe from the date a complete BLA is accepted for filing, if it treats a serious condition and has the potential to provide a significant improvement in safety or effectiveness. The FDA will attempt to direct additional resources to the evaluation of an application for a new drug or biological product designated for priority review in an effort to facilitate the review.
Additionally, a product may be eligible for accelerated approval. Drug or biological products studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit over existing treatments may receive accelerated approval, which means that they may be approved on the basis of adequate and well-controlled clinical trials establishing that the product has an effect on a surrogate endpoint that is reasonably likely to predict a clinical benefit, or on the basis of an effect on a clinical endpoint other than survival or irreversible morbidity. As a condition of approval, the FDA may require that a sponsor of a drug or biological product receiving accelerated approval perform adequate and well-controlled post-marketing clinical trials. If the FDA concludes that a drug shown to be effective can be safely used only if distribution or use is restricted, it will require such post-marketing restrictions as it deems necessary to assure safe use of the drug, such as:
The limitations imposed would be commensurate with the specific safety concerns presented by the product. In addition, the FDA currently requires as a condition for accelerated approval pre-approval of promotional materials, which could adversely impact the timing of the commercial launch of the product. Fast track designation, priority review and accelerated approval do not change the standards for approval but may expedite the development or approval process.
Breakthrough Designation
The Food and Drug Administration Safety and Innovation Act, or FDASIA, amended the FDCA to require the FDA to expedite the development and review of a breakthrough therapy. A product can be designated as a breakthrough therapy if it is intended to treat a serious or life-threatening condition and preliminary clinical evidence indicates that it may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints. A sponsor may request that a product candidate be designated as a breakthrough therapy concurrently with the submission of an IND or any time before an end-of-Phase-II meeting, and the FDA must determine if the product candidate qualifies for breakthrough therapy designation within 60 days of receipt of the sponsor’s request. If so designated, the FDA shall act to expedite the development and review of the product’s marketing application, including by meeting with the sponsor throughout the product’s development, providing timely advice to the sponsor to ensure that the development program to gather pre-clinical and clinical data is as efficient as practicable, involving senior managers and experienced review staff in a cross-disciplinary review, assigning a cross-disciplinary project lead for the FDA review team to facilitate an efficient review of the development program and to serve as a scientific liaison between the review team and the sponsor, and taking steps to ensure that the design of the clinical trials is as efficient as practicable.
Pediatric Trials
Under the Pediatric Research Equity Act, or PREA, a BLA or supplement to a BLA must contain data to assess the safety and efficacy of the product for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. FDASIA requires that a sponsor who is planning to submit a marketing application for a drug or biological product that includes a new active ingredient, new indication, new dosage form, new dosing regimen or new route of administration submit an initial Pediatric Study Plan, or PSP, within sixty days of an end-of-Phase II meeting or as may be agreed between the sponsor and FDA. The initial PSP must include an outline of the pediatric study or studies that the sponsor plans to conduct, including study objectives and design, age groups, relevant endpoints and statistical approach, or a justification for not including such detailed information, and any request for a deferral of pediatric assessments or a full or partial waiver of the requirement to provide data from pediatric studies along with supporting information. FDA and the sponsor must reach agreement on the PSP. A sponsor can submit amendments to an agreed-upon initial PSP at any time if changes to the pediatric plan need to be considered based on data collected from nonclinical studies, early phase clinical trials, and/or other clinical development programs. The FDA may, on its own initiative or at the request of the applicant, grant deferrals for submission of data or full or partial waivers.
Post-Marketing Requirements
Following approval of a new product, a manufacturer and the approved product are subject to continuing regulation by the FDA, including, among other things, monitoring and recordkeeping activities, reporting to the applicable regulatory authorities of adverse experiences with the product, providing the regulatory authorities with updated safety and efficacy information, product sampling and distribution requirements, and complying with promotion and advertising requirements, which include, among others, standards for direct-to-consumer advertising, restrictions on promoting products for uses or in patient populations that are not described in the product’s approved labeling (known as “off-label use”), limitations on industry-sponsored scientific and educational activities, and requirements for promotional activities involving the internet. Although physicians may prescribe legally available drugs and biologics for off-label uses, manufacturers may not market or promote such off-label uses. Modifications
or enhancements to the product or its labeling or changes of the site of manufacture are often subject to the approval of the FDA and other regulators, which may or may not be received or may result in a lengthy review process. Prescription drug promotional materials must be submitted to the FDA in conjunction with their first use. Any distribution of prescription drug products and pharmaceutical samples must comply with the U.S. Prescription Drug Marketing Act, or the PDMA, a part of the FDCA.
In the United States, once a product is approved, its manufacture is subject to comprehensive and continuing regulation by the FDA. The FDA regulations require that products be manufactured in specific approved facilities and in accordance with cGMP. Moreover, the constituent parts of a combination product retain their regulatory status, for example, as a biologic or device, and as such, we may be subject to additional requirements in the Quality System Regulation, or QSR, applicable to medical devices, such as design controls, purchasing controls, and corrective and preventive action. We rely, and expect to continue to rely, on third parties for the production of clinical and commercial quantities of our products in accordance with cGMP regulations. cGMP regulations require, among other things, quality control and quality assurance as well as the corresponding maintenance of records and documentation and the obligation to investigate and correct any deviations from cGMP. Manufacturers and other entities involved in the manufacture and distribution of approved products are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP and other laws. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain cGMP compliance. These regulations also impose certain organizational, procedural and documentation requirements with respect to manufacturing and quality assurance activities. BLA holders using contract manufacturers, laboratories or packagers are responsible for the selection and monitoring of qualified firms, and, in certain circumstances, qualified suppliers to these firms. These firms and, where applicable, their suppliers are subject to inspections by the FDA at any time, and the discovery of violative conditions, including failure to conform to cGMP, could result in enforcement actions that interrupt the operation of any such facilities or the ability to distribute products manufactured, processed or tested by them. Discovery of problems with a product after approval may result in restrictions on a product, manufacturer, or holder of an approved BLA, including, among other things, recall or withdrawal of the product from the market.
The FDA also may require post-approval testing, sometimes referred to as Phase IV testing, risk minimization action plans and post-marketing surveillance to monitor the effects of an approved product or place conditions on an approval that could restrict the distribution or use of the product. Discovery of previously unknown problems with a product or the failure to comply with applicable
FDA requirements can have negative consequences, including adverse publicity, judicial or administrative enforcement, warning letters from the FDA, mandated corrective advertising or communications with doctors, and civil or criminal penalties, among others. Newly discovered or developed safety or effectiveness data may require changes to a product’s approved labeling, including the addition of new warnings and contraindications, and also may require the implementation of other risk management measures. Also, new government requirements, including those resulting from new legislation, may be established, or the FDA’s policies may change, which could delay or prevent regulatory approval of our products under development.
Other Regulatory Matters
Manufacturing, sales, promotion and other activities following product approval are also subject to regulation by numerous regulatory authorities in addition to the FDA, including, the Centers for Medicare & Medicaid Services, or CMS, other divisions of the Department of Health and Human Services, the Drug Enforcement Administration, the Consumer Product Safety Commission, the Federal Trade Commission, the Occupational Safety & Health Administration, the Environmental Protection Agency and state and local governments. In the United States, sales, marketing and scientific/educational programs must also comply with state and federal fraud and abuse laws, data privacy and security laws, transparency laws, and pricing and reimbursement requirements in connection with
governmental payor programs, among others. The handling of any controlled substances must comply with the U.S. Controlled Substances Act and Controlled Substances Import and Export Act. Products must meet applicable child-resistant packaging requirements under the U.S. Poison Prevention Packaging Act. Manufacturing, sales, promotion and other activities are also potentially subject to federal and state consumer protection and unfair competition laws.
The distribution of pharmaceutical products is subject to additional requirements and regulations, including extensive record-keeping, licensing, storage and security requirements intended to prevent the unauthorized sale of pharmaceutical products.
The failure to comply with regulatory requirements subjects firms to possible legal or regulatory action. Depending on the circumstances, failure to meet applicable regulatory requirements can result in criminal prosecution, fines or other penalties, injunctions, recall or seizure of products, total or partial suspension of production, denial or withdrawal of product approvals, or refusal to allow a firm to enter into supply contracts, including government contracts. In addition, even if a firm complies with FDA and other requirements, new information regarding the safety or efficacy of a product could lead the FDA to modify or withdraw product approval. Prohibitions or restrictions on sales or withdrawal of future products marketed by us could materially affect our business in an adverse way.
Changes in regulations, statutes or the interpretation of existing regulations could impact our business in the future by requiring, for example: (i) changes to our manufacturing arrangements; (ii) additions or modifications to product labeling; (iii) the recall or discontinuation of our products; or (iv) additional record-keeping requirements. If any such changes were to be imposed, they could adversely affect the operation of our business.
U.S. Patent Term Restoration and Marketing Exclusivity
Depending upon the timing, duration and specifics of the FDA approval of our product candidates, some of our U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, commonly referred to as the Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term restoration period is generally one-half the time between the effective date of an IND and the submission date of a BLA plus the time between the submission date of a BLA and the approval of that application, except that the review period is reduced by any time during which the applicant failed to exercise due diligence. Only one patent applicable to an approved drug is eligible for the extension and the application for the extension must be submitted prior to the expiration of the patent. The U.S. PTO, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. In the future, we may apply for restoration of patent term for our currently owned or licensed patents to add patent life beyond its current expiration date, depending on the expected length of the clinical trials and other factors involved in the filing of the relevant BLA.
An abbreviated approval pathway for biological products shown to be similar to, or interchangeable with, an FDA-licensed reference biological product was created by the Biologics Price Competition and Innovation Act of 2009, or BPICA, which was part of the ACA. This amendment to the PHSA attempts to minimize duplicative testing. Biosimilarity, which requires that the biological product
be highly similar to the reference product notwithstanding minor differences in clinically inactive components and that there be no clinically meaningful differences between the product and the reference product in terms of safety, purity, and potency, can be shown through analytical studies, animal studies, and a clinical trial or trials. Interchangeability requires that a biological product be biosimilar to the reference product and the product can be expected to produce the same clinical results as the reference product in any given patient and, for products administered multiple times, the product and the reference product may be alternated or switched after one has been previously administered without increasing safety risks or
risks of diminished efficacy relative to exclusive use of the reference biological product. However, complexities associated with the larger, and often more complex, structure of biological products as compared to small molecule drugs, as well as the process by which such products are manufactured, pose significant hurdles to implementation that are still being worked out by the FDA.
A reference biological product is granted twelve years of exclusivity from the time of first licensure of the product, and the FDA will not accept an application for a biosimilar or interchangeable product based on the reference biological product until four years after the date of first licensure. “First licensure” typically means the initial date the particular product at issue was licensed in the United States. This does not include a supplement for the biological product or a subsequent application by the same sponsor or manufacturer of the biological product (or licensor, predecessor in interest, or other related entity) for a change that results in a new indication, route of administration, dosing schedule, dosage form, delivery system, delivery device, or strength, unless that change is a modification to the structure of the biological product and such modification changes its safety, purity, or potency. Whether a subsequent application, if approved, warrants exclusivity as the “first licensure” of a biological product is determined on a case-by-case basis with data submitted by the sponsor.
Pediatric exclusivity is another type of regulatory market exclusivity in the United States. Pediatric exclusivity, if granted, adds six months to existing exclusivity periods and patent terms. This six-month exclusivity, which runs from the end of other exclusivity protection or patent term, may be granted based on the voluntary completion of a pediatric trial in accordance with an FDA-issued “Written Request” for such a trial.
European Union Drug Development
In the European Union, our future product candidates may also be subject to extensive regulatory requirements. As in the United States, medicinal products can only be marketed if a marketing authorization from the competent regulatory agencies has been obtained.
Similar to the United States, the various phases of pre-clinical and clinical research in the European Union are subject to significant regulatory controls. Although the EU Clinical Trials Directive 2001/20/EC has sought to harmonize the European Union clinical trials regulatory framework, setting out common rules for the control and authorization of clinical trials in the European Union, the European Union Member States have transposed and applied the provisions of the Directive differently. This has led to significant variations in the Member State regimes. To improve the current system, a new Regulation No. 536/2014 on clinical trials on medicinal product candidates for human use, which repealed Directive 2001/20/EC, was adopted on April 16, 2014 and published in the European Official Journal on May 27, 2014. The new Regulation aims at harmonizing and streamlining the clinical trials authorization process, simplifying adverse event reporting procedures, improving the supervision of clinical trials, and increasing their transparency. The new Regulation entered into force on June 16, 2014 but will apply not earlier than May 28, 2016. Until then the Clinical Trials Directive 2001/20/EC will still apply. In addition, the transitory provisions of the new Regulation offer the sponsors the possibility to choose between the requirements of the Directive and the Regulation for one year from the entry into application of the Regulation.
Under the current regime, before a clinical trial can be initiated it must be approved in each of the European Union countries where the trial is to be conducted by two distinct bodies: the National Competent Authority, or NCA, and one or more Ethics Committees, or ECs. Under the current regime all suspected unexpected serious adverse reactions, or SUSARs, to the investigated drug that occur during the clinical trial have to be reported to the NCA and ECs of the Member State where they occurred.
European Union Drug Review and Approval
In the European Economic Area, or EEA (which is comprised of the 28 Member States of the European Union plus Norway, Iceland and Liechtenstein), medicinal products can only be commercialized after obtaining a Marketing Authorization, or MA. There are two types of marketing authorizations:
The Community MA, which is issued by the European Commission through the Centralized Procedure, based on the opinion of the Committee for Medicinal Products for Human Use, or CHMP, of the European Medicines Agency, or EMA, and which is valid throughout the entire territory of the EEA. The Centralized Procedure is mandatory for certain types of products, such as biotechnology medicinal products, orphan medicinal products, and medicinal products containing a new active substance indicated for the treatment of AIDS, cancer, neurodegenerative disorders, diabetes, auto-immune and viral diseases. The Centralized Procedure is optional for products containing a new active substance not yet authorized in the EEA, or for products that constitute a significant therapeutic, scientific or technical innovation or which are in the interest of public health in the European Union.
National MAs, which are issued by the competent authorities of the Member States of the EEA and only cover their respective territory, are available for products not falling within the mandatory scope of the Centralized Procedure. Where a product has already been authorized for marketing in a Member State of the EEA, this National MA can be recognized in another Member States through the Mutual Recognition Procedure. If the product has not received a National MA in any Member State at the time of application, it can be approved simultaneously in various Member States through the Decentralized Procedure. Under the Decentralized Procedure an identical dossier is submitted to the competent authorities of each of the Member States in which the MA is sought, one of which is selected by the applicant as the Reference Member State, or RMS. The competent authority of the RMS prepares a draft assessment report, a draft summary of the product characteristics, or SPC, and a draft of the labeling and package leaflet, which are sent to the other Member States (referred to as the Concerned Member States, or CMSs) for their approval. If the CMSs raise no objections, based on a potential serious risk to public health, to the assessment, SPC, labeling, or packaging proposed by the RMS, the product is subsequently granted a national MA in all the Member States (i.e. in the RMS and the CMSs).
Under the above described procedures, before granting the MA, the EMA or the competent authorities of the Member States of the EEA make an assessment of the risk-benefit balance of the product on the basis of scientific criteria concerning its quality, safety and efficacy.
Other Regulatory Matters
French Regulatory Framework
In the European Union, the regulation governing clinical trials is currently based on European Directive No. 2001/20/EC of April 4, 2001 relative to the implementation of good clinical practices in the conduct of clinical trials on medicinal products for human use. Each country of the European Union had to transpose this Directive into national law by eventually adapting it to its own regulatory framework.
In France, for example, Directive No. 2001/20/EC has been transposed by Act No. 2004-806 of August 9, 2004 relative to the public health policy and Decree No. 2006-477, April 26, 2006, modifying the title of the Code of Public Health dedicated to biomedical research. This regulation replaces the notification procedure arising from the Huriet-Sérusclat Act of December 20, 1988. Article L. 1121-4 of the Public Health Code, as amended by the Act of August 9, 2004, now establishes a system of prior authorization issued by the ANSM with the favorable opinion of a competent Research and Ethics Committee for the place where the investigator exercises his activity. On the basis of Article L. 1123-7 of the same code, the Committee shall deliver its opinion on the research’s conditions of validity, particularly with respect to participant protection, their information and how they collect informed consent,
as well as the project’s general relevance, the satisfactory nature of the assessment of benefits and risks and the adequacy between the objectives pursued and the means implemented. The ANSM, after submission of the complete file containing not only information on the clinical protocol, but also specific product data and its quality control, as well as results of pre-clinical studies may inform the sponsor that it objects to the implementation of the research. The sponsor can then modify the contents of his research project and submit this amended or supplemented request to the ANSM; this procedure may not, however, be applied more than once. If the sponsor does not alter the content of its request, the request is considered rejected. Under the terms of the Decree of April 26, 2006, the time limit for the examination of a request for authorization cannot exceed 60 days from the receipt of the complete file. Finally, under Article L. 1123-11, in the event of risk to public health or if the ANSM considers that the conditions in which the research is implemented no longer correspond to the conditions indicated in the request for authorization or does not comply with the provisions of the Public Health Code, it may at any time request changes to procedures for the realization of research, and suspend or ban this research. The decision of November 24, 2006 sets the rules for Good Clinical Practice for biomedical research on medicines for human use provided for in Article L. 1121-3 of the Public Health Code. The purpose of Good Clinical Practice, or GCP, is to ensure both the reliability of data arising from clinical trials and the protection of persons participating in these clinical trials. GCPs shall apply to all clinical trials, including pharmacokinetics, bioavailability and bioequivalence studies in healthy volunteers and Phase II to IV clinical trials.
Personal data collected during clinical trials should be declared in simplified form to the Commission Nationale Informatique et Liberté, or CNIL. Patients then have a right to access and correct this data pursuant to Act No. 78-17 of January 6, 1978, as amended by law No. 2004-801 of August 6, 2004, concerning computing, files and freedoms.
The main French regulatory texts concerning the conduct of clinical trials are as follows:
French Pharmaceutical Company Status
To date, we do not have the status of pharmaceutical establishment, and therefore, cannot either manufacture the product candidates we develop or directly consider their marketing. Obtaining the pharmaceutical establishment license, either as distributor “exploitant” or as manufacturer, requires the submission of a request file specific to each of the two qualifications with the ANSM, which only grants it after review of this file and evaluation, usually after verification that the company has adequate premises, the necessary personnel and an adapted structure with satisfactory procedures for carrying out the proposed pharmaceutical activities.
We currently entrust CMOs with the manufacturing of clinical batches and intend to continue relying on CMOs for the production of the first commercial batches. We may consider internalizing production once our first product candidate is approved by the regulatory authorities.
Reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of any product candidates for which we or our collaborators obtain regulatory approval. Sales of our products will depend, in part, on the extent to which our products, once approved, will be covered and reimbursed by third-party payors, such as government health programs, commercial insurance and managed healthcare organizations. These third-party payors are increasingly reducing reimbursements for medical products and services. The process for determining whether a third-party payor will provide coverage for a drug product typically is separate from the process for setting the price of a drug product or for establishing the reimbursement rate that a payor will pay for the drug product once coverage is approved. Third-party payors may limit coverage to specific drug products on an approved list, also known as a formulary, which might not include all of the FDA approved drugs for a particular indication.
In order to secure coverage and reimbursement for any product candidate that might be approved for sale, we may need to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost-effectiveness of the product candidate, in addition to the costs required to obtain FDA or other comparable regulatory approvals. Whether or not we conduct such studies, our product candidates may not be considered medically necessary or cost-effective. A third-party payor’s decision to provide coverage
for a drug product does not imply that an adequate reimbursement rate will be approved. Further, one payor’s determination to provide coverage for a product does not assure that other payors will also provide coverage, and adequate reimbursement, for the product. Third party reimbursement may not be sufficient to enable us to maintain price levels high enough to realize an appropriate return on our investment in product development.
The containment of healthcare costs has become a priority of federal and state governments, and the prices of drugs have been a focus in this effort. The U.S. government, state legislatures and foreign governments have shown significant interest in implementing cost-containment programs, including price controls, restrictions on reimbursement and requirements for substitution of generic products. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit our net revenue and results. Decreases in third-party reimbursement for our product candidate or a decision by a third-party payor to not cover our product candidate could reduce physician usage of the product candidate and have a material adverse effect on our sales, results of operations and financial condition.
For example, the ACA, enacted in March 2010, has already had, and is expected to continue to have, a significant impact on the health care industry. The ACA is expectedwas designed to expand coverage for the uninsured while at the same time containing overall healthcare costs. With regard to pharmaceuticalbiopharmaceutical products, among other things, the ACA is expected to expand and increase industry rebates for drugs covered under Medicaid programs and make changes to the coverage requirements under the Medicare Part D program. We cannot predict the full impact of the ACA on pharmaceuticalbiopharmaceutical companies as many of the ACA reforms require the promulgation of detailed regulations implementing the statutory provisions which has not yet occurred.
In addition, other legislative changes have been proposed and adopted in the United States since the ACA was enacted. On August 2, 2011, the Budget Control Act of 2011 among other things, created measures for spending reductions by Congress. AThe Joint Select Committee on Deficit Reduction was tasked with recommending to Congress proposals in spending reductions. Because they did not achieve a targeted deficit reduction of at least $1.2 trillion for thefiscal years 20132012 through 2021, was unable to reach required goals, thereby triggeringit triggered the legislation’s automatic reduction to several government
programs. This includes aggregate reductions to Medicare payments to providers of up to 2% per fiscal year, startedwhich went into effect in April 2013.2013 and will stay in effect through 2024 unless additional Congressional action is taken. On January 2, 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, or the ATRA, which delayed for another two months the budget cuts mandated by these sequestration provisions of the Budget Control Act of 2011. The ATRA, among other things, also reduced Medicare payments to several providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. We expect that additional federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, and in turn could significantly reduce the projected value of certain development projects and reduce our profitability.
In addition, in some foreign countries, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country. For example, the European Union provides options for its Member States to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. A Member State may approve a specific price for the medicinal product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. For example, in France, effective access to the market assumes that our future products will be supported by the hospital (through an agreement for local communities) or reimbursed by social security. The price of medications is negotiated with the Economic Committee for Health Products, or CEPS. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of our product candidates. Historically, products launched in the European Union do not follow price structures of the United States and generally tend to be significantly lower.
Other Healthcare Laws and Compliance Requirements
Our business operations in the United States and our arrangements with clinical investigators, healthcare providers, consultants, third-party payors and patients may expose us to broadly applicable federal, state, and stateforeign fraud and abuse and other healthcare laws. These laws may impact, among other things, our research, proposed sales, marketing and education programs of our product candidates that obtain marketing approval. The healthcare laws and regulations that may affect our ability to operate include, among others:
the federal Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, receiving, offering or paying remuneration (including any kickback, bribe or rebate), directly or indirectly, in cash or in kind,
to induce or reward, or in return for, either the referral of an individual for, or the purchase, lease, order, or recommendation of, an item, good, facility or service reimbursable under a federal healthcare program, such as the Medicare and Medicaid programs. Although there are a number of statutory exceptions and regulatory safe harbors protecting some common activities from prosecution, the exceptions and safe harbors are drawn narrowly. Practices that involve remuneration that may be alleged to be intended to induce prescribing, purchases or recommendations may be subject to scrutiny if they do not qualify for an exception or safe harbor. Several courts have interpreted the statute’s intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of federal healthcare covered business, the Anti-Kickback Statute has been violated;
|
The ACA broadened the reach of the federal fraud and abuse laws by, among other things, amending the intent requirement of the federal Anti-Kickback Statute and the applicable federal criminal healthcare fraud statutes. Pursuant to the statutory amendment, a person or entity no longer needs to have actual knowledge of this statute or specific intent to violate it in order to have committed a violation. In addition, the ACA provides that the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the civil False Claims Act or the civil monetary penalties statute.
Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws will involve substantial costs. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant administrative, civil, and/or criminal penalties, damages, fines, disgorgement, individual imprisonment, exclusion from government funded healthcare programs, such as Medicare and Medicaid, contractual damages, reputational harm, diminished profits and future earnings, and the curtailment or restructuring of our operations. If the physicians or other healthcare providers or entities with whom we expect to do business are found to be not in compliance with applicable laws, they may be subject to administrative, civil, and/or criminal sanctions, including exclusions from government funded healthcare programs.
Legal Proceedings
From time to time we may become involved in legal proceedings or be subject to claims arising in the ordinary course of our business. We are not presently a party to any legal proceedings that, if determined adversely to us, would individually or taken together have a material adverse effect on our business, results of operations, financial condition or cash flows. Regardless of the outcome, litigation can have an adverse impact on us because of defense and settlement costs, diversion of management resources and other factors.
The following diagram illustrates our corporate structure:


D. Property, Plants and Equipment.
We relocated our corporate headquarters to Montrouge, France in January 2016. Our principal offices occupy a 4,770 square meter facility consisting of office and laboratory space, pursuant to a lease agreement dated March 3, 2015, which expires on March 8, 2024. We made the decision to relocate to a larger facility to accommodate the growth of our office space,company. We also have a second facility in Bagneux, France, which was our former corporate headquarters. This facility consists of 1,479 square meters located in Bagneux, France. The lease for this facility expires on May 31, 2020. In order to accompany the growth of the company, we have concluded in January 2015 a lease agreement for 4,470 square meters of office and laboratory space locatedand is used by our industrial and production teams. This lease expires in Montrouge, France. May 31, 2020.
We intendalso sublease approximately 3,913 square feet of office space in New York, New York, which serves as temporary office space for our U.S. subsidiary. The sublease is for an initial period of 25 months and expires in June 1, 2017 and we expect to move and leaveinto a more permanent facility in the current premises later thisNew York area in the coming year.
Item 4A. Unresolved Staff Comments.
Not applicable.
Item 5.Operating and Financial Review and Prospects.
Overview
We are a clinical-stage specialty biopharmaceutical company focused on changing the field of immunotherapy by developing a novel technology platform called Viaskin. Our therapeutic approach is based on epicutaneous immunotherapy, or EPIT, our proprietary method of delivering biologically active compounds to the immune system through intact skin using Viaskin. We have generated significant data demonstrating that Viaskin’s mechanism of action is novel and differentiated, as it targets specific antigen-presenting immune cells in the skin, called Langerhans cells, that capture the antigen and migrate to the lymph node in order to activate the immune system without passage of the antigen into the bloodstream. We are advancing this unique technology to treat patients, including infants and children, suffering from severe food allergies, for whom safety is paramount, since the introduction of the offending allergen into their bloodstream can cause severe or life-threatening allergic reactions, such an anaphylactic shock.
We initially financed our operations through several private equity investments totaling €38.7 million. In 2012, we completed a €40.6 million initial public offering of our ordinary shares on Euronext Paris. In 2013, we completed a €29.9 million private investment in public equity, or PIPE, of which we received net proceeds of €15.1 million and our selling shareholders received net proceeds of €14.8 million. In 2014, we completed a €104.5 million global underwritten public offering of both ADSs on the Nasdaq Global Select Market, or Nasdaq, and ordinary shares on Euronext Paris, issuing an aggregate of 3,074 686 ordinary shares, from which we received net proceeds of €93.7 million. In July 2015, we completed a €255.3 million underwritten public offering of 4,140,000 ordinary shares in the form of 8,280,000 ADSs, from which we received net proceeds of €237.3 million. In connection with our 2015 public offering, our share capital increased by €414 thousand with a corresponding increase of €236.9 million in our share premium.
We have incurred net losses in each year since our inception. Substantially all of our net losses resulted from costs incurred in connection with our development programs and from general and administration expenses associated with our operations.
We expect to continue to incur significant expenses and increasing operating losses for the foreseeable future. We anticipate that our expenses will increase substantially in connection with our ongoing activities, as we:
We do not expect to generate material revenue from product sales unless and until we successfully complete development of, and obtain marketing approval for, one or more of our product candidates, which we expect will take a number of years and is subject to significant uncertainty. Accordingly, we anticipate that we will need to raise additional capital prior to completing clinical development of our product candidates. Until such time that we can generate substantial revenue from product sales, if ever, we expect to finance our operating activities through a combination of equity offerings, debt financings, government or other third-party funding and collaborations, and licensing arrangements. However, we may be unable to raise additional funds or enter into such arrangements when needed on favorable terms, or at all, which would have a negative impact on our financial condition and could force us to delay, limit, reduce or terminate our development programs or commercialization efforts or grant to others rights to develop or market product candidates that we would otherwise prefer to develop and market ourselves. Failure to receive additional funding could cause us to cease operations, in part or in full.
Our financial statements for 2012, 2013, 2014 and 20142015 have been prepared in accordance with International Financial Reporting Standards, or IFRS, as issued by the International Accounting Standards Board, or IASB.
Financial Operations Overview
Operating Income
Our operating income consistconsists of revenues and other income.
Revenues
We historically derive substantially all of our revenues from sales of Diallertest Milk, our diagnostic product for the detection of cow’s milk protein allergy, or CMPA, in children, which was sold exclusively in France through a distribution partner. To date, salesSales of Diallertest Milk have beenwere moderate, totaling approximately 25,000 kits per year on average over the past three years. Diallertest Milk is currently available onDuring the French marketsecond half of 2015, we discontinued our commercial partnership with a temporary exception status. Regulatory authorities are requesting a pivotal Phase III trialrespect to complete the marketing authorization file for this product. We are examining the relevance of carrying out this clinical protocol,product and are evaluating potential marketing and/or distribution relationships to market Diallertest Milk in Europe in the field of pediatrics. We may also elect, or may be required at the request of regulatory authorities, to stop the marketing ofceased selling Diallertest Milk. We do not currently expect these productto derive any revenues to have a material impact on our business and financial condition in future periods.2016 from the sale of Diallertest Milk.
Other Income
Government Assistance
Due to the innovative nature of our product candidate development programs, we have benefited from a certain number of sources of assistance from the central French government or local public authorities, intended to finance our research and development efforts or the recruitment of specific personnel. These funds are recognized as other income in our statement of income (loss) for the fiscal year that recorded the financed expenses or expenditures.
Research Tax Credits
The research tax credit (crédit d’impôt recherche), or CIR, is granted to companies by the French tax authorities in order to encourage them to conduct technical and scientific research. Companies demonstrating that they have expenditures that meet the required criteria, including research expenditures located in France or, since January 1, 2005, within the European Community or in another State that is a party to the Agreement on the European Economic Area that has concluded a tax treaty with France that contains an administrative assistance clause, receive a tax credit which can be used against the payment of the corporate tax due on the fiscal year in which the expenditures were made and during the next three fiscal years, or, as applicable, can be reimbursed for the excess portion. The expenditures taken into account for the calculation of the CIR only involve research expenses.
The main characteristics of the CIR are the following:
As a result, we have concluded that the CIR meets the definition of a government grant as defined in IAS 20 Accounting for Government Grants and Disclosure of Government Assistance and that the classification as other income within operating income in our statement of income (loss) is appropriate.
We received the reimbursement of the CIR for the 2014 fiscal year 2013 during the year 2014.in 2015. We have requested the reimbursement of the 20142015 fiscal year CIR under the Community taxapplicable rules for small and medium firms in compliance with the regulatory texts in effect for which we expect to be reimbursed in 2015. The CIR is presented under other income in our statement of income (loss). The CIR for the years 2008 and 2009 was subject to a tax audit in 2011. That audit, which ended on July 11, 2011, did not result in any significant adjustment.2016.
Cost of Goods Sold
Because we are not classified as a pharmaceutical laboratory, we contracthistorically contracted with a third party to manufacture our Diallertest Milk diagnostic patches according to current good manufacturing practices, or cGMPs. The cost of goods sold therefore includeshistorically included the costs of manufacture we incur through this service provider. We discontinued sales of Diallertest Milk the second half of 2015.
Operating Expenses
Since inception, our operating expenses have consisted primarily of research and development activities, and general and administration costs and sales and marketing costs.
Research and Development
We engage in substantial research and development efforts to develop innovative pharmaceutical product candidates. Research and development expense consists primarily of:
Our research and development expenses in the periods presented mainly relate to the following activities:
| • | Viaskin Peanut for the treatment of peanut allergies in |
| • | Viaskin Milk for the treatment of Immunoglobulin E, or IgE, mediated CMPA in children. We are currently enrolling patients in a Phase II, or Part B, clinical trial of Viaskin Milk in children called MILES (MilkEfficacy andSafety). The trial’s Phase I, or Part A, was completed in the third quarter of 2015. |
Our direct research and development expenses consist principally of external costs, such as startup fees paid to investigators, consultants, central laboratories, and CROs in connection with our clinical studies, and costs related to acquiring and manufacturing clinical study materials. We do not allocate personnel-related costs, costs associated with our general platform improvements, depreciation or other indirect costs to specific programs, as they are deployed across multiple projects under development and, as such, are separately classified as personnel and other expenses.
Research and development activities are central to our business. Product candidates in later stages of clinical development generally have higher development costs than those in earlier stages of clinical development, primarily due to the increased size and duration of later-stage clinical trials. We expect that our research and development expenses will continue to increase in the foreseeable future as we initiate clinical trials for certain product candidates and pursue later stages of clinical development of our product candidates.candidates, particularly our ongoing Phase III clinical trial of Viaskin Peanut.
We cannot determine with certainty the duration and completion costs of the current or future clinical trials of our product candidates or if, when, or to what extent we will generate revenue from the commercialization and sale of any of our product candidates that obtain regulatory approval. We may never succeed in achieving regulatory approval for any of our product candidates. The duration, costs and timing of clinical trials and development of our product candidates will depend on a variety of factors, including:
A change in the outcome of any of these variables with respect to the development of Viaskin Peanut or any other product candidate that we are developing could mean a significant change in the costs and timing associated with the development of Viaskin Peanut or such other product candidate. For example, if the FDA or other regulatory authority were to require us to conduct pre-clinical and clinical studies beyond those which we currently anticipate will be required for the completion of clinical development, or if we experience significant delays in enrollment in any clinical trials, we could be required to spend significant additional financial resources and time on the completion of the clinical development.
General and Administration
General and administration expense consists primarily of personnel costs and share-based compensation for personnel other than researchfinance, legal and developmentadministrative staff. General and administration expense also consists of fees for professional services, mainly related to audit, IT and legal services, real-estate leasing costs, insurance costs, investor relations costs and communication and travel costs, notably related to our being a public company in France.costs.
We anticipate that our general and administration expenses will increase in the future as we increase our headcount to support the expected growth in our research and development activities and the potential commercialization of our product candidates. We also anticipate continued increased expenses associated with being a public company in the United States, following the completion of our global public offering in October 2014, including costs relatedwith respect to audit, legal, regulatory and tax-related services associated with maintaining compliance with U.S. exchange listing and SEC requirements, director and officer insurance premiums, and investor relations costs.the Sarbanes-Oxley Act.
Sales and Marketing
IfSales and when we believe that a regulatory approvalmarketing expense consists primarily of the first product candidate appears likely, we anticipate an increase in payrollpersonnel costs and related expenses as a result of our preparationshare-based compensation for commercial operations, especially as it relates to the sales and marketing of our product candidates.staff, as well as consulting fees and travel costs.
Finance Income (Expense)
Our cash and cash equivalents have been deposited primarily in savings and deposit accounts with original maturities of three years or less, allowing the funds to be freely withdrawn at any time without notice or penalty. Savings and deposit accounts generate a modest amount of interest income. We expect to continue this investment philosophy.
Critical Accounting Policies and Estimates
Our financial statements are prepared in accordance with IFRS. Some of the accounting methods and policies used in preparing our financial statements under IFRS are based on complex and subjective assessments by our management or on estimates based on past experience and assumptions deemed realistic and reasonable based on the circumstances concerned. The actual value of our assets, liabilities and shareholders’ equity and of our earnings could differ from the value derived from these estimates if conditions changed and these changes had an impact on the assumptions adopted. We believe that the most significant management judgments and assumptions in the preparation of our financial statements are described below. See Note 3 to our financial statements for a description of our other significant accounting policies.
Conditional Advances
OSEO Innovation
Since inception we have received multiple interest-free conditional advances from OSEO Innovation, or OSEO, the French Agency for Innovation and part of theBanque Publique d’Investissement. OSEO’s mission is to provide assistance and financial support to emerging French enterprises by providing such enterprises with growth capital to facilitate the development and commercialization of innovative technologies. Each award of a conditional advance is made to help fund a specific development project. See also, Note 11 to our audited consolidated financial statements for the years ended December 31, 2012, 2013, 2014 and 2014.2015.
In the case of the conditional advances from OSEO, our obligation to repay these amounts is based on the technical and commercial success of the funded project, as determined by OSEO in its sole and subjective discretion. Once a project has been selected for funding by OSEO, both a payment and a repayment schedule are defined by contract. As the project advances, we provide OSEO with
one or more interim progress reports and a final report when the funded project ends. Based on these reports, OSEO makes a subjective determination, in its sole discretion, as to whether the project is a partial or total technical or commercial success, or a technical or commercial failure. In the event OSEO determines that the project is a failure, we are required by contract to repay a minimum amount. In the event OSEO determines that the project is a partial success, there is a specified repayment schedule, provided that the parties may renegotiate a different repayment schedule in good faith. In the event OSEO determines that the project is a complete success, we are obliged to repay 100% of the amount of the conditional advance.
In the case of each conditional advance, we assume that OSEO will determine the project to be a total technical or commercial success and thus the maximum amount repayable with respect to such project will become due. However, actual results related to the development of these programs may differ from these estimates in which case the financial liability reflected in our statement of financial statements for the conditional advances may be reduced. The current and non-current portions of the financial liability recognized in our statement of financial statements associated with these conditional advances are determined based on the applicable reimbursement schedules at the end of each reporting period. The portion of the conditional advances for terms longer than one year is classified as a non-current liability while the portion for terms of less than one year is classified as a current liability. In addition, in the case of each conditional advance, we treat the benefit resulting from the interest-free nature of the award as a subsidy and recognize this amount as other income over the applicable repayment period. We determine the amount of this deemed subsidy amount by applying a discount rate equal to the rate of fungible treasury bonds over the time period that corresponds to the time period of the repayment of the advances.
In addition to the conditional advances described above, since inception, we have received one non-refundable subsidy from OSEO in connection with our development of our legacy house dust mite product candidate. We refer to this development program as the ImmunaVia project. We account for this non-refundable subsidy as other income ratably over the duration of the funded project.
French Export Credit Insurance Company
In September 2007, we entered into an agreement with the French Export Credit Insurance Company, or COFACE, to support the promotion of our Diallertest Milk product internationally. COFACE offers and manages, on behalf and under the guarantee of the French State, public guarantees to support exportations and investments made by French companies abroad. Under the terms of this agreement, we must repay these advances at up to 7% of the revenues from the export of our Diallertest Milk until April 30, 2017. To date, our export revenues for Diallertest Milk have been modest. Diallertest Milk is currently availableDuring the second half of 2015, we discontinued our commercial partnership with a temporary exception status from French regulatory authorities. Regulatory authorities in France have requested a pivotal Phase III trialrespect to complete the marketing authorization file for this product. We are examining the relevance of carrying out this clinical protocol,product and are evaluating potential marketing and/or distribution relationships to market Diallertest Milk in Europe in the field of pediatrics. We may also elect, or may be required at the request of regulatory authorities, to stop the marketing ofceased selling Diallertest Milk. We do not currently expect these productto derive any 2016 revenues to have a material impact on our business and financial condition in future periods.from the sale of Diallertest Milk.
Share-Based Compensation
We have various share-based compensation plans for employees and non-employees. We account for share-based compensation in accordance with the authoritative guidance on share-based compensation. Under the fair value recognition provisions of this guidance, share-based compensation is measured at the grant date based on the fair value of the award and is recognized as expense, net of estimated forfeitures, over the requisite service period, which is generally the vesting period of the respective award.
Determining the fair value of share-based awards at the grant date requires judgment. We use the Black-Scholes option-pricing model to determine the fair value of share options. The determination of the grant date fair value of options using an option-pricing model is affected by assumptions regarding a number of complex and subjective variables. These variables include the expected term of the options, our share price volatility, risk-free interest rates, and expected dividends, which are estimated as follows:
Fair Value of Our Ordinary Shares. We established a policy of using the closing sales price per ordinary share as quoted on Euronext Paris on the date prior to the date of grant for purposes of determining the fair value of ordinary shares with a floor value of 95% of the average of the closing sales price per ordinary share for the 20 trading days preceding the grant.
Expected Term. The expected term represents the period that our share-based awards are expected to be outstanding. As we do not have sufficient historical experience for determining the expected term of the share option awards granted, we have based our expected term on the simplified method, which represents the average period from vesting to the expiration of the award.
Expected Volatility.We are using our volatility on Euronext Paris observed on a sufficient historical dataset.
Risk-Free Interest Rate. The risk-free interest rate is based on the yields of French government bonds with maturities similar to the expected term of the options for each option group.
Dividend Yield. We have never declared or paid any cash dividends and do not presently plan to pay cash dividends in the foreseeable future. Consequently, we used an expected dividend yield of zero.
If any of the assumptions used in the Black-Scholes model changes significantly, share-based compensation for future awards may differ materially compared with the awards granted previously.
The following table presents the weighted-average assumptions used to estimate the fair value of options granted during the periods presented:
| December 31 | December 31 | |||||||||||
| 2012 | 2013 | 2014 | 2013 | 2014 | 2015 | |||||||
Volatility | 40% | 40% | 40% | 40% | 40% | 40% | ||||||
Risk free interest rate | 1.21%-2.61% | 1.16%-1.72% | 0.71%-0.89% | 1.16%-1.72% | 0.71%-0.89% | 0.79%-0.81% | ||||||
Expected life (in years) | 5.5-7.0 | 7.0 | 5.0-6.0 | 7.0 | 5.0-6.0 | 7.0 | ||||||
Dividend yield | — | — | — | — | — | — | ||||||
For 2012, 2013, 2014 and 2014,2015, we recorded employee share-based compensation expense of €3.2 million, €5.0 million, €4.6 million and €4.6€10.4 million, respectively.
| A. | Operating Results |
ComparisonsComparison for the Years Ended December 31, 20122013 and 20132014
Operating Income
We generated operating income of €2.8 million in 2012 and €3.8 million in 2013 and €4.8 million in 2014, an increase of 37.9%24.4%. These incomes were mainly generated by our CIR, and more marginally, by Diallertest Milk sales, and by subsidies received for research projects conducted by us.
| December 31 | December 31, | |||||||||||||||
| 2012 | 2013 | 2013 | 2014 | |||||||||||||
| € | € | (Amounts in thousands of Euros) | ||||||||||||||
Revenues | 174,360 | 181,800 | 182 | 211 | ||||||||||||
Other income | 2,602,228 | 3,644,513 | 3,644 | 4,551 | ||||||||||||
o/w CIR | 2,522,399 | 3,312,462 | 3,312 | 4,340 | ||||||||||||
o/w subsidies | 79,829 | 332,051 | 332 | 211 | ||||||||||||
Total income | 2,776,588 | 3,826,313 | 3,826 | 4,762 | ||||||||||||
|
| |||||||||||||||
As no research and development expenditure is capitalized before obtaining a marketing authorization, the CIR related to such research programs is entirely recorded as operating income. The grants we received during the period were deducted from the calculation of the CIR base.
For the year ended December 31, 2013,2014, we recorded other income related to CIR of €3.3€4.3 million, which we requested forreceived reimbursement in 2014.2015. In 2013,2014, we received the reimbursement of €2.5€3.3 million for the 20122013 CIR under the Communitycommunity small and medium business scheme.
The increase of €790,063,€1.0 million, or 31.3%31.0%, in the CIR recorded in 20132014 reflects the acceleration of our various development programs in 2013.2014.
The revenues generated by Diallertest Milk, which iswas only marketed in France through a distributor, increased by 4.3%15.9% during the last financial year, from €174,360 in 2012 to €181,800 in 2013. We do not currently expect these product revenues2013 to have a material impact on our business and financial condition€210,759 in future periods.2014.
The increase of €252,222 in subsidies recorded in 2013 was due to the grant of the fourth OSEO advance.
Cost of Goods Sold
Because we are not classified as a pharmaceutical laboratory, we contractcontracted with a third party to manufacture our Diallertest Milk diagnostic patches according to the cGMP. The cost of goods sold therefore includes the costs of manufacture through this service provider. The cost of goods represented 47.6%56.3% and 56.3%64.7% of our sales revenues in 20122013 and 2013,2014, respectively.
| December 31 | ||||||||
| 2012 | 2013 | |||||||
| € | € | |||||||
Cost of goods sold | 82,958 | 102,366 | ||||||
| December 31, | ||||||||
| 2013 | 2014 | |||||||
| (Amounts in thousands of Euros) | ||||||||
Cost of goods sold | 102 | 136 | ||||||
|
|
|
| |||||
Research and Development Expenditures
From 20122013 to 2013,2014, the total amount spent by us for research and development activity increased from €11.5€17.4 million to €17.4€21.1 million, or an increase of 51.0%21.7%.
Our research and development expenses for the periods presented mainly relate to the following activities:
Our direct research and development expenses consist principally of external costs, such as startup fees paid to investigators, consultants, central laboratories and CROs in connection with our clinical studies, and costs related to acquiring and manufacturing clinical study materials. We do not allocate personnel-related costs, costs associated with our general platform improvements, depreciation or other indirect costs to specific programs, as they are deployed across multiple projects under development and, as such, are separately classified as personnel and other expenses as described in the table below:
| December 31 | ||||||||
| 2012 | 2013 | |||||||
| € | € | |||||||
Viaskin Peanut(1) | — | 6,503,562 | ||||||
Viaskin Milk | — | 957,714 | ||||||
Total direct research and development expenses(1) | — | 7,461,276 | ||||||
Personnel expenses | 4,800,518 | 7,194,722 | ||||||
Other Viaskin- & EPIT-related expenses(1) | 5,922,763 | 1,690,826 | ||||||
Facility expenses | 259,224 | 263,438 | ||||||
Conferences, travel expenses | 324,123 | 465,871 | ||||||
Depreciation, amortization and provisions | 192,740 | 290,406 | ||||||
Personnel and other expenses(1) | 11,499,368 | 9,905,262 | ||||||
Total R&D expenses | 11,499,368 | 17,366,538 | ||||||
| December 31, | ||||||||
| 2013 | 2014 | |||||||
| (Amounts in thousands of Euros) | ||||||||
Viaskin Peanut | 6,503 | 6,398 | ||||||
Viaskin Milk | 958 | 2,555 | ||||||
Total direct research and development expenses | 7,461 | 8,953 | ||||||
|
|
|
| |||||
Personnel expenses | 7,195 | 7,703 | ||||||
Other Viaskin- & EPIT-related expenses | 1,691 | 3,100 | ||||||
Facility expenses | 263 | 255 | ||||||
Conferences, travel expenses | 466 | 665 | ||||||
Depreciation, amortization and provisions | 290 | 466 | ||||||
Personnel and other expenses | 9,905 | 12,160 | ||||||
|
|
|
| |||||
Total R&D expenses | 17,367 | 21,143 | ||||||
|
|
|
| |||||
The increased expenditures from year to year resulted from the costs associated with both the VIPES Phase IIb and OLFUS-VIPES follow up trials of the Viaskin Peanut, the MILES Phase I/II trial of the Viaskin Peanut that began during the summer of 2012,Milk initiated, as well as a substantial strengthening of research and development teams due to the increasing number of active programs.
In particular, we have incurred:
General and Administration Expenses
During the period presented, our general and administration expenses increased from €4.6€6.3 million to €6.3€8.1 million, or an increase of 37.2%28.7%.
Our general and administration expenses break down as follows:
| December 31 | December 31, | |||||||||||||||
| 2012 | 2013 | 2013 | 2014 | |||||||||||||
| € | € | (Amounts in thousands of Euros) | ||||||||||||||
Personnel expenses | 3,107,246 | 4,698,848 | 4,699 | 5,109 | ||||||||||||
Fees | 512,709 | 586,638 | 587 | 1,166 | ||||||||||||
Real estate leasing | 157,467 | 111,232 | 111 | 204 | ||||||||||||
Insurance | 56,054 | 105,018 | 105 | 230 | ||||||||||||
Communication and travel expenses | 480,999 | 450,701 | 451 | 645 | ||||||||||||
Telecommunication expenses | 86,831 | 65,350 | 65 | 76 | ||||||||||||
Administrative costs and rental of personal property | 65,867 | 97,131 | 97 | 104 | ||||||||||||
Other | 131,526 | 194,832 | 195 | 583 | ||||||||||||
Total G&A expenses | 4,598,699 | 6,309,750 | 6,310 | 8,118 | ||||||||||||
|
| |||||||||||||||
General and administration expenses for the year ended December 31, 2012 were €4.6 million, compared to €6.3 million for the year ended December 31, 2013. The increase of €1.7€1.8 million in general and administration expenses was primarily due to:
Sales & Marketing Expenses
Sales and marketing expenses were immaterial for the years ended December 31, 2013 and 2014.
Financial Profit (Loss)
Our net financial profit increaseddecreased to €645,925€0.62 million in 2014 from €0.64 million in 2013, from €492,337 in 2012, an increasea decrease of 31.2%3.4%. The change in our financial profit in 20132014 is mainly explained by the cash investment income we received, notably as part of capital increases completed in March 2012November 2013 and November 2013,in October 2014, the financial revenues having increased from €517,540€0.67 million in 20122013 to €670,234€0.73 million in 2014. This was partly offset by a net foreign exchange loss of €0.02 million, compared to a net gain of €0.01 million in 2013.
Comparisons for the Years Ended December 31, 20132014 and 20142015
Operating Income
We generated operating income of €3.8 million in 2013 and €4.8 million in 2014 and €6.2 million in 2015, an increase of 24.4%29.5%. These incomes were mainly generated by our CIR, and more marginally by Diallertest Milk sales, and by subsidies received for research projects conducted by us.
| December 31 | December 31, | |||||||||||||||
| 2013 | 2014 | 2014 | 2015 | |||||||||||||
| € | € | (Amounts in thousands of Euros) | ||||||||||||||
Revenues | 181,800 | 210,759 | 211 | 202 | ||||||||||||
Other income | 3,644,513 | 4,550,763 | 4,551 | 5,964 | ||||||||||||
o/w CIR | 3,312,462 | 4,339,620 | 4,340 | 5,685 | ||||||||||||
o/w subsidies | 332,051 | 211,143 | 211 | 279 | ||||||||||||
Total income | 3,826,313 | 4,761,522 | 4,762 | 6,166 | ||||||||||||
|
| |||||||||||||||
As no research and development expenditure is capitalized before obtaining a marketing authorization, the CIR related to such research programs is entirely recorded as operating income. The grants we received during the period were deducted from the calculation of the CIR base.
For the year ended December 31, 2014,2015, we recorded other income related to CIR of €4.3€5.7 million, which we requested for reimbursement in 2015.2016. In 2014,2015, we received the reimbursement of €3.3€4.3 million for the 20132014 CIR under the Communitycommunity small and medium business scheme.
The increase of €1.0€1.3 million, or 31.0%, in the CIR recorded in 20142015 reflects the acceleration of our various development programs in 2014.2015.
The revenues generated by Diallertest Milk, which iswas only marketed in France through a distributor, increaseddecreased by 15.9%4.1% during the last financial year, from €181,800€211 thousand in 20132014 to €210,759€202 thousand in 2014. We do not currently expect these2015. During the second half of 2015, we discontinued our commercial partnership with respect to the product revenues to have a material impact on our business and financial condition in future periods.ceased selling Diallertest Milk.
Cost of Goods Sold
Because we are not classified as a pharmaceutical laboratory, we contractcontracted with a third party to manufacture our Diallertest Milk diagnostic patches according to the cGMP. The cost of goods sold therefore includes the costs of manufacture through this service provider. The cost of goods represented 56.3%64.7% and 64.7%63.3% of our sales revenues in 20132014 and 2014,2015, respectively.
| December 31 | ||||||||
| 2013 | 2014 | |||||||
| € | € | |||||||
Cost of goods sold | 102,366 | 136,296 | ||||||
Cost of goods sold December 31, 2014 2015 (Amounts in thousands of Euros) 136 128
Research and Development Expenditures
From 20132014 to 2014,2015, the total amount spent by us for research and development activity increased from €17.4€21.1 million to €21.1€34.2 million, or an increase of 21.7%61.9%.
Our research and development expenses for the periods presented mainly relate to the following activities:
Our direct research and development expenses consist principally of external costs, such as startup fees paid to investigators, consultants, central laboratories and CROs in connection with our clinical studies, and costs related to acquiring and manufacturing clinical study materials. We do not allocate personnel-related costs, costs associated with our general platform improvements, depreciation or other indirect costs to specific programs, as they are deployed across multiple projects under development and, as such, are separately classified as personnel and other expenses as described in the table below:
| December 31 | ||||||||
| 2013 | 2014 | |||||||
| € | € | |||||||
Viaskin Peanut | 6,503,562 | 6,398,226 | ||||||
Viaskin Milk | 957,714 | 2,555,090 | ||||||
Total direct research and development expenses | 7,461,276 | 8,953,316 | ||||||
Personnel expenses | 7,194,722 | 7,703,057 | ||||||
Other Viaskin- & EPIT-related expenses | 1,690,826 | 3,100,554 | ||||||
Facility expenses | 263,438 | 254,923 | ||||||
Conferences, travel expenses | 465,871 | 665,420 | ||||||
Depreciation, amortization and provisions | 290,406 | 466,172 | ||||||
Personnel and other expenses | 9,905,262 | 12,160,126 | ||||||
Total R&D expenses | 17,366,538 | 21,143,442 | ||||||
| December 31, | ||||||||
| 2014 | 2015 | |||||||
| (Amounts in thousands of Euros) | ||||||||
Personnel expenses | 7,703 | 13,268 | ||||||
Sub-contracting, collaboration and consultants | 10,703 | 15,325 | ||||||
Research supplies | 937 | 911 | ||||||
Purchases | 249 | 795 | ||||||
Real estate property rental | 255 | 1,094 | ||||||
Conferences and travel expenses | 665 | 1,233 | ||||||
Allowances for provisions and amortization and depreciation | 466 | 1,000 | ||||||
Others | 165 | 608 | ||||||
Total R&D expenses | 21,143 | 34,234 | ||||||
|
|
|
| |||||
The increased expenditures from year to year resulted from the costs associated with both the VIPESPEPITES Phase IIbIII and OLFUS-VIPES follow up trials of the Viaskin Peanut, running alongsidewhich were being conducted simultaneously in 2014,2015, the ongoing MILES Phase I/II trial of the Viaskin Milk initiated in November 2014, as well as a substantial strengthening ofincrease in research and development teams duepersonnel in order to thesupport our increasing numbernumbers of active development programs.
In particular, we have incurred:
General and Administration Expenses
During the period presented, our general and administration expenses increased from €6.3€8.1 million to €8.1€16.9 million, or an increase of 28.7%108%.
Our general and administration expenses break downare as follows:
| December 31 | ||||||||
| 2013 | 2014 | |||||||
| € | € | |||||||
Personnel expenses | 4,698,848 | 5,109,057 | ||||||
Fees | 586,638 | 1,165,989 | ||||||
Real estate leasing | 111,232 | 203,899 | ||||||
Insurance | 105,018 | 230,495 | ||||||
Communication and travel expenses | 450,701 | 645,175 | ||||||
Telecommunication expenses | 65,350 | 75,913 | ||||||
Administrative costs and rental of personal property | 97,131 | 104,374 | ||||||
Other | 194,832 | 582,762 | ||||||
Total G&A expenses | 6,309,750 | 8,117,664 | ||||||
| December 31, | ||||||||
| 2014 | 2015 | |||||||
| (Amounts in thousands of Euros) | ||||||||
Personnel expenses | 5,109 | 8,768 | ||||||
Fees | 1,166 | 4,234 | ||||||
Real estate property rental | 204 | 305 | ||||||
Maintenance | 140 | 356 | ||||||
Insurance policies | 230 | 1,239 | ||||||
Communication and travel expenses | 633 | 1,010 | ||||||
Postal and telecommunications expenses | 76 | 36 | ||||||
Administrative supplies and rentals of personal property | 104 | 162 | ||||||
Allowances for provisions and amortization and depreciation | 111 | 74 | ||||||
Others | 332 | 676 | ||||||
Total G&A expenses | 8,105 | 16,859 | ||||||
|
|
|
| |||||
General and administration expenses for the year ended December 31, 2013 were €6.3 million, compared to €8.1 million for the year ended December 31, 2014. The increase of €1.8€8.8 million in general and administration expenses was primarily due to:
Sales & Marketing Expenses
During the Nasdaq.second half of 2015, we hired our first employees to support the launch and commercialization of Viaskin Peanut in North America, if approved.
| December 31, | ||||||||
| 2014 | 2015 | |||||||
| (Amounts in thousands of Euros) | ||||||||
Personnel expenses | — | 133 | ||||||
Fees | — | 339 | ||||||
Communication and travel expenses | 13 | 20 | ||||||
Total S&M expenses | 13 | 491 | ||||||
|
|
|
| |||||
Financial Profit (Loss)
Our net financial profit decreasedincreased to €624,000€0.9 million in 20142015 from €645,925€0.6 million in 2013,2014, a decrease of 3.4%39.6%. The change in our financial profit in 20142015 is mainly explained by the cash investment income we received, notably as part of capital increases completed in November 2013October 2014 and in October 2014,July 2015, the financial revenues having increased from €670,234€0.7 million in 20132014 to €727,239€1.0 million in 2014. This was partly offset by a net foreign exchange loss of €22,887, compared to a net gain of €10,289 in 2013.2015.
| B. | Liquidity and Capital Resources |
We have financed our operations since inception through several private placements of equity securities totaling €38.7 million, a €40.6 million initial public offering of our ordinary shares on Euronext Paris in 2012, a €29.9 million PIPE in 2013, of which we received net proceeds of €15.1 million and our shareholders received net proceeds of €14.8 million, and a €104.5 million global
offering of both ADSs on the Nasdaq and ordinary shares on Euronext Paris, of which we received net proceeds of €93.7 million. In July 2015, we completed a €255.3 million underwritten public offering of 4,140,000 ordinary shares in the form of 8,280,000 ADSs from which we received net proceeds of €237.3 million. In connection with our 2015 public offering, our share capital increased by €414 thousand with a corresponding increase of €236.9 million in our share premium.
The table below summarizes our sources and uses of cash for the years ended December 31, 2012, 2013, 2014 and 2014.2015.
| December 31 | December 31, | |||||||||||||||||||||||
| 2012 | 2013 | 2014 | 2013 | 2014 | 2015 | |||||||||||||||||||
| € | € | € | (Amounts in thousands of Euros) | |||||||||||||||||||||
Cash used in operating activities | (10,432,549 | ) | (13,253,215 | ) | (20,559,652 | ) | (13,253 | ) | (20,560 | ) | (26,763 | ) | ||||||||||||
Cash used in investing activities | (368,760 | ) | (1,408,425 | ) | (1,096,231 | ) | (1,408 | ) | (1,096 | ) | (5,347 | ) | ||||||||||||
Cash flows provided by financing activities | 37,098,822 | 16,235,770 | 96,808,306 | 16,236 | 96,808 | 241,014 | ||||||||||||||||||
Net increase in cash and cash equivalents | 26,297,514 | 1,574,130 | 75,152,424 | �� | 1,574 | 75,152 | 208,904 | |||||||||||||||||
Our net cash flows used fromin operational activities was at €26.8 million, €20.6 million and €13.3 million for 2015, 2014 and €10.4 million for 2014, 2013, and 2012, respectively.
During 2013, our net cash flows used from operationalin operating activities increased due to our growingincreasing efforts in advancing our research and development programs, mainly Viaskin Peanut, thatwhich progressed through itsa Phase IIb clinical trial.trial in this period. This increase was partially offset by a positive change in working capital of €574,252 over the period.
During 2014, our net cash flows used from operationalin operating activities increased due to further growing effortsadvances in advancingour research and development programs, mainly due to the completion of our Phase IIb clinical trial of Viaskin Peanut, as well as the continuation into the study’s open-label extension trial. Further development of Viaskin Milk also contributed to the cash flow increase in this period.
During 2015, our net cash flows used in operating activities increased due to our advances in our research and development programs, mainly Viaskin Peanut, that runned both itswhich progressed into a Phase IIbIII clinical trial during this period, continuation of the OLFUS-VIPES trial, and follow up clinical trials, and Viaskin Milk that initiated its Phase I/II clinicalcontinuation of the MILES trial. This increase was partially offset by a positive change in working capital of €6.0 million over the period.
Our net cash flows used fromin investing activities stood at €1.4 million, €1.1 million €1.4and €5.3 million in 2013, 2014 and €368,760 in 2014, 2013 and 2012,2015, respectively.
This significant increase bothThe cash used in investing activities in 2013 and 2014 reflects the acquisition of industrial and laboratory equipment required to conduct our development programs, as well as the refurbishment of our research and development premises. The increase in 2015 resulted from the relocation of our headquarters to Montrouge (France), as well as purchasing materials for designing manufacturing tools for our product candidates.
Our net cash flows provided from financing activities declined to €16.2 million in 2013 from €37.1 million in 2012, mainly due to the fact that we only raised €15.2 million in 2013.
Our net cash flows providedresulting from financing activities increased to €241.0 million in 2015 from €96.8 million in 2014 and from €16.2 million in 2013, mainly as a result of our €104.5 million global offeringunderwritten public offerings in each of both ADSs on the Nasdaq2015 and ordinary shares on Euronext Paris, of which we received net proceeds of €93.7 million, partially offset by an additional €900,000 to our liquidity contract.2014.
Consistent with customary practice in the French securities market, we entered into a liquidity agreement (contrat de liquidité) with Natixis datedon April 13, 2012. The liquidity agreement complies with applicable laws and regulations in France. The liquidity agreement authorizes Natixis to carry out market purchases and sales of DBVour shares on Euronext Paris. As of December 31, 2014,2015, we have contributed an aggregate of €1.2€1.4 million to the liquidity account. The amount is classified in other non-current financial assets in our statement of financial position. At December 31, 2014, 8,0542015, 3,898 shares and €1,111,227€1.4 million were in the liquidity account. The liquidity agreement has a term of one year and will renew automatically unless otherwise terminated by either party.
Cash and Funding Sources
During 2012, 2013, 2014 and 2014,2015, we obtained new financing on the public markets by issuance of securities.
| Equity capital | Bank Loans | Other debt | Total | Equity capital | Bank Loans | Other debt | Total | |||||||||||||||||||||||||
2012 | 40,626,662 | — | — | 40,626,662 | ||||||||||||||||||||||||||||
| (Amounts in thousands of Euros) | ||||||||||||||||||||||||||||||||
2013 | 15,128,873 | — | 1,159,500 | 16,288,373 | 15,129 | — | 1,159 | 16,288 | ||||||||||||||||||||||||
2014 | 93,711,030 | — | 3,128,000 | 96,839,030 | 93,711 | — | 3,128 | 96,839 | ||||||||||||||||||||||||
2015 | 237,323 | — | 865 | 238,188 | ||||||||||||||||||||||||||||
Total | 149,466,565 | — | 4,287,500 | 153,754,065 | 346,163 | — | 5,152 | 351,315 | ||||||||||||||||||||||||
We have incurred net losses in each year since our inception. Substantially all of our net losses resulted from costs incurred in connection with our development programs and from general and administrative expenses associated with our operations.
We have not incurred any bank debt. Other debt is comprised of Conditionalconditional advances which are detailed as follows.
As of December 31, 2014,2015, we benefited from multiple conditional advances from OSEO, which advances do not accrue interest and are repayable at 100% in the event of technical and/or commercial success of our product, as determined solely and subjectively by OSEO, a non-refundable subsidy from OSEO, a conditional advance from COFACE and an interest-free loan by Bpifrance.
| • | 2nd OSEO advance: On January 10, 2005, we obtained a conditional advance of €600,000 from OSEO for a project to design a high-speed prototype machine to produce patches and to develop second-generation patches in particular intended for the detection of various allergies. The entire sum had been received as of December 31, 2010. The repayment of this grant was made in accordance with the initial schedule, with the final repayment made on April 2, 2013. |
| • | 3rd OSEO advance: In 2011, we obtained a conditional advance by OSEO for a total amount of €640,000 to finance the development of our programs to treat CMPA. This amount has been fully received, with a first payment of €256,000 in December 2011, a second payment of €256,000 in June 2013 and remaining €128,000 balance paid in January 2014. If the program is deemed to be technically or commercially successful, as determined by OSEO in its sole and subjective discretion it will be repaid in 16 quarterly installments defined as follows: four payments of €64,000 starting on September 30, 2014, then 12 payments of €32,000 starting on September 30, 2015, until June 30, 2018. If this project is deemed to be a technical failure, we will still be obligated to repay OSEO the amount of €256,000. |
| • | 4th OSEO advance: In 2013, we obtained a conditional advance by OSEO for a total amount of €3.2 million in the context of a research and clinical development collaborative project in the field of |
The activity for the conditional advances recorded during 2012, 2013, 2014 and 20142015 is summarized in the table below:
| 2nd OSEO advance | 3rd OSEO advance | 4th OSEO advance | Bpi France interest-free loan | COFACE | Total | 2nd OSEO advance | 3rd OSEO advance | 4th OSEO advance | Bpi France interest-free loan | COFACE | Total | |||||||||||||||||||||||||||||||||||||
| € | € | € | € | € | € | (Amounts in thousands of Euros) | ||||||||||||||||||||||||||||||||||||||||||
Balance debt at 01/01/2012 | 450,713 | 246,238 | — | — | 122,501 | 819,452 | ||||||||||||||||||||||||||||||||||||||||||
+ receipts | — | — | — | — | — | — | ||||||||||||||||||||||||||||||||||||||||||
- repayments | (200,000 | ) | — | — | — | — | (200,000 | ) | ||||||||||||||||||||||||||||||||||||||||
+/- other transactions(1) | 6,701 | 3,661 | — | — | 4,251 | 14,613 | ||||||||||||||||||||||||||||||||||||||||||
Balance debt at 12/31/2012 | 257,414 | 249,899 | — | — | 126,752 | 634,065 | ||||||||||||||||||||||||||||||||||||||||||
Balance debt at 01/01/2013 | 257 | 250 | — | — | 127 | 634 | ||||||||||||||||||||||||||||||||||||||||||
+ receipts | — | 256,000 | 903,500 | — | — | 1,159,500 | — | 256 | 903.5 | — | — | 1,159.5 | ||||||||||||||||||||||||||||||||||||
- repayments | (260,000 | ) | — | — | — | — | (260,000 | ) | (260 | ) | — | — | — | — | (260 | ) | ||||||||||||||||||||||||||||||||
+/- other transactions(1) | 2,586 | (1,579 | ) | (111,047 | ) | — | 19,300 | (90,740 | ) | 3 | (2 | ) | (111 | ) | — | 19 | (91 | ) | ||||||||||||||||||||||||||||||
Balance debt at 12/31/2013 | — | 504,320 | 792,453 | — | 146,052 | 1,442,825 | — | 504 | 792 | — | 146 | 1,443 | ||||||||||||||||||||||||||||||||||||
+ receipts | — | 128,000 | — | 3,000,000 | — | 3,128,000 | — | 128 | — | 3,000 | — | 3,128 | ||||||||||||||||||||||||||||||||||||
- repayments | — | (128,000 | ) | — | — | — | (128,000 | ) | — | (128 | ) | — | — | — | (128 | ) | ||||||||||||||||||||||||||||||||
+/- other transactions(1) | — | 2,276 | 12,932 | (416,361 | ) | 4,994 | (396,159 | ) | — | 2 | 13 | (416 | ) | 5 | (396 | ) | ||||||||||||||||||||||||||||||||
Balance debt at 12/31/2014 | — | 506,596 | 805,385 | 2,583,639 | 151,046 | 4,046,666 | — | 506 | 805 | 2,584 | 151 | 4,047 | ||||||||||||||||||||||||||||||||||||
+ receipts | — | — | 865 | — | — | 865 | ||||||||||||||||||||||||||||||||||||||||||
- repayments | — | (192 | ) | — | — | — | (192 | ) | ||||||||||||||||||||||||||||||||||||||||
+/- other transactions(1) | — | 3 | (2 | ) | 82 | 5 | 89 | |||||||||||||||||||||||||||||||||||||||||
Balance debt at 12/31/2015 | — | 318 | 1,669 | 2,666 | 156 | 4,809 | ||||||||||||||||||||||||||||||||||||||||||
| (1) | The changes in “other transactions” are comprised of the effect of discontinuing conditional advances. |
Operating Capital Requirements
We believe that our existing cash and cash equivalents will enable us to fund our operating expenses and capital expenditure requirements until the end of 2016.2017. We have based this estimate on assumptions that may prove to be wrong, and we could use our capital resources sooner than we currently expect. In any event, we will require additional capital to pursue pre-clinical and clinical activities, obtain regulatory approval for, and to commercialize our product candidates.
Until we can generate a sufficient amount of revenue from our product candidates, if ever, we expect to finance our operating activities through a combination of equity offerings, debt financings, government or other third-party funding and collaborations. Additional capital may not be available on reasonable terms, if at all. If we are unable to raise additional capital in sufficient amounts or on terms acceptable to us, we may have to significantly delay, scale back or discontinue the development or commercialization of one or more of our product candidates. If we raise additional funds through the issuance of additional debt or equity securities, it could result in dilution to our existing shareholders, increased fixed payment obligations and these securities may have rights senior to those of our ordinary shares. If we incur indebtedness, we could become subject to covenants that would restrict our operations and potentially impair our competitiveness, such as limitations on our ability to incur additional debt, limitations on our ability to acquire, sell or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business. Any of these events could significantly harm our business, financial condition and prospects.
Our present and future funding requirements will depend on many factors, including, among other things:
For more information as to the risks associated with our future funding needs, see the section entitled “Item 3.D—Risk Factors.”
Capital Expenditures
As all the clinical research and development expenditures are posted to the accounts as expenses until marketing authorizations are obtained, the principal investments made over 2012, 2013, 2014 and 20142015 have been related primarily to the acquisition of laboratory equipment and, secondarily, to the acquisition of computer and office equipment.
| December 31 | ||||||||||||
| 2012 | 2013 | 2014 | ||||||||||
| € | € | € | ||||||||||
Long-term intangible assets | 21,024 | 81,385 | 30,695 | |||||||||
Property, plant, and equipment | 340,411 | 1,089,902 | 941,301 | |||||||||
Long-term financial assets | 7,325 | 237,138 | 124,235 | |||||||||
Total | 368,760 | 1,408,425 | 1,096,231 | |||||||||
In 2012, we extended and refurbished our research and industrial development laboratories, for which we accounted for investments in property, plant and equipment of €164,000, of which €105,000 were spent on the acquisition of laboratory equipment, and €72,000 on the acquisition of our computer and office equipment.
| December 31, | ||||||||||||
| 2013 | 2014 | 2015 | ||||||||||
| (Amounts in thousands of Euros) | ||||||||||||
Long-term intangible assets | 81 | 31 | 148 | |||||||||
Property, plant, and equipment | 1,090 | 941 | 4,360 | |||||||||
Long-term financial assets | 237 | 124 | 839 | |||||||||
Total | 1,408 | 1,096 | 5,347 | |||||||||
In 2013, we have purchased tools and equipment for the design, the development and manufacturemanufacturing of industrial prototypes and tools for €399,000€0.4 million and continued to expand and refurbish our research and industrial development laboratories for €292,000,at a cost of €0.3 million, of which €192,000 were€0.19 million was spent on the acquisition of laboratory equipment and €157,000€0.15 million was spent on the acquisition of computers and office equipment. Also, €81,000 was spent on acquisition of software packages, notably in the context of updating the accounting and management software.
In 2014, we have purchased tools and equipment for the design, the development and manufacturemanufacturing of industrial prototypes and tools for €608,000, €277,000 were€0.6 million and we spent €0.3 million on the acquisition of laboratory equipment. Also, €848,000 were€0.8 million was allocated to additional funding of our liquidity contract and €124,000€0.1 million was spent as a guarantee deposit in the context of expending our facilities.
JOBS Act Exemptions
We qualify as an “emerging growth company” as defined inIn 2015, we purchased tools and equipment for the JOBS Act. As an emerging growth company,design, the development and manufacturing of industrial prototypes and tools for €2.2 million and we may take advantage of specified reduced disclosure and other requirements that are otherwise applicable generally to public companies. These provisions include:
We may take advantage of these exemptions for up to five years or such earlier time that we are no longer an emerging growth company. We would cease to be an emerging growth company upon the earliest to occur of (1) the last day of the fiscal year in which we have more than $1.0 billion in annual revenue; (2) the date we qualify as a “large accelerated filer,��� with at least $700 million of equity securities; (3) the issuance, in any three-year period, by our company of more than $1.0 billion in non-convertible debt securities held by non-affiliates; and (4) the last day of the fiscal year ending after the fifth anniversaryrelocation of our initial public offering. We may choosecorporate headquarters to take advantage of some but not all of these exemptions. For example, Section 107 of the JOBS Act provides that an emerging growth company can use the extended transition period provided in Section 7(a)(2)(B) of the Securities Act for complying with new or revised accounting standards. Given that we currently report and expect to continue to report under IFRS as issued by the IASB, we have irrevocably elected not to avail ourselves of this extended transition period and, as a result, we will adopt new or revised accounting standards on the relevant dates on which adoption of such standards is required by the IASB. We have taken advantage of reduced reporting requirements in this Annual Report on Form 20-F. Accordingly, the information contained herein may be different than the information you receive from other public companies in which you hold equity securities.Montrouge (France).
For a discussion of our research and development activities, see “Item 4.B—Business Overview” and “Item 5.A—Operating Results.”
For a discussion of trends, see “Item 4.B—Business Overview,” “Item 5.A—Operating Results” and “Item 5.B—Liquidity and Capital Resources.”
E. Off-Balance Sheet Arrangements
During the periods presented, we did not and do not currently have any off-balance sheet arrangements as defined under SEC rules, such as relationships with unconsolidated entities or financial partnerships, which are often referred to as structured finance or special purpose entities, established for the purpose of facilitating financing transactions that are not required to be reflected on our balance sheets.
F. Tabular Disclosure of Contractual Obligations
The following table discloses aggregate information about material contractual obligations and periods in which payments were due as of December 31, 2014.2015. Future events could cause actual payments to differ from these estimates.
| Less than 1 year | 1 to 3 years | 3 to 5 years | More than 5 years | Total | Less than 1 year | 1 to 3 years | 3 to 5 years | More than 5 years | Total | |||||||||||||||||||||||||||||||
| € | € | € | € | € | (Amounts in thousands of Euros) | |||||||||||||||||||||||||||||||||||
Long-term debt obligations | 212,653 | 437,238 | 1,536,666 | 1,914,266 | 4,100,823 | 149 | 1,324 | 1,066 | 2,303 | 4,842 | ||||||||||||||||||||||||||||||
Capital (finance) lease obligations | — | — | — | — | — | — | — | — | — | — | ||||||||||||||||||||||||||||||
Operating lease obligations | — | — | — | — | — | 2,013 | 3,448 | 3,143 | 4,877 | 13,481 | ||||||||||||||||||||||||||||||
Purchase obligations | — | — | — | — | — | — | — | — | — | — | ||||||||||||||||||||||||||||||
Other long-term liabilities | — | — | — | — | — | — | — | — | — | — | ||||||||||||||||||||||||||||||
Total | 212,653 | 437,238 | 1,536,666 | 1,914,266 | 4,100,823 | 2,162 | 4,772 | 4,209 | 7,180 | 18,323 | ||||||||||||||||||||||||||||||
The commitment amounts in the table above are associated with contracts that are enforceable and legally binding and that specify all significant terms, including interest on long-term debt, fixed or minimum services to be used, fixed, minimum or variable price provisions, and the approximate timing of the actions under the contracts. The table does not include obligations under agreements that we can cancel without a significant penalty. In March 2015, we signedentered into a new lease arrangement in relation to oura new premisesfacility located in Montrouge, France and expectFrance. We relocated our corporate headquarters to relocate our current siteMontrouge in Bagneux later in 2015.January 2016.
This Annual Report on Form 20-F contains forward-looking statements within the meaning of Section 27A of the Securities Act and Section 21E of the Exchange Act and as defined in the Private Securities Litigation Reform Act of 1995. See “Special Note Regarding Forward-Looking Statements.”
Item 6. Directors, Senior Management and Employee.Employees.
A. Directors and Senior Management.
The following table sets forth information regarding our executive officers and directors, including their ages, as of March 31, 2015.April 29, 2016. Unless otherwise stated, the address for our executive officers and directors is Green Square-Bâtiment D, 80/84 rue des Meuniers, 92220 Bagneux,177-181 avenue Pierre Brossolette, 92120 Montrouge, France.
| Name | Age | Position(s) | ||||
Executive Officers: | ||||||
Dr. Pierre-Henri Benhamou | Chief Executive Officer, Chairman of the | |||||
Bertrand Dupont | ||||||
David Schilansky | Chief Operating Officer | |||||
Charles Ruban | Chief Commercial Officer | |||||
Laurent Martin | 49 | Chief Development Officer | ||||
Non-Employee Directors: | ||||||
Dr. Torbjörn Bjerke(1)(2) | Director | |||||
| Director | |||||
George Horner III(1)(2) | ||||||
| Director | |||||
Chahra Louafi | Director | |||||
| Director | |||||
| (1) | Member of the audit committee. |
| (2) | Member of the compensation committee. |
Dr. Pierre-Henri Benhamou, co-founded DBVour company in 2002, and has served as our Chief Executive Officer from 2002 to the present (except from 2006 to 2010, when he was Chief Scientific Officer) and Chairman of our board of directors since our initial public offering on Euronext Paris in March 2012. With Dr. Christophe Dupont, he described for the first time and patented the epicutaneous method of immunotherapy and explored with our scientific team the wide range of applications of the method. Dr. Benhamou is a physician, specializing in pediatric gastroenterology. He has held numerous clinical and academic positions. He received the Altran Foundation Prize for Innovation in 2003 for his work on the development of diagnostic patch tests. Dr. Benhamou has published numerous papers and originated key scientific collaborations for us and has been instrumental in forming our Scientific Advisory Board. Dr. Benhamou holds an M.D. from Faculté de Médecine de Paris in 1984 and his specialization degree in Pediatrics in 1987. The board of directors believes that Dr. Benhamou’s leadership, deep knowledge of our company and scientific experience will allow him to drive us to the success of our objectives.
Bertrand Dupont, one of our founders, has served as our Senior Executive Vice President, Technology since January 2015, and served as our Chief Technical Officer since 2002.from 2002 to 2015. He is a member of our Executive Committee. From 1977 to 2002, Mr. Bertrand has served as a professor at the high school and at the university, teaching mechanics and robotics. In 1996,As our Senior Executive Vice President, Technology, Mr. Dupont began to put his skills and expertise to use in biomedical research. Since 2000, he has been atoversees the core of the development of the Viaskin patches and applications. As our Chief Industrial Officer, Mr. Dupont is a key figure in the development of the Viaskin technology and the application systems. He is responsible for every technical development around the Viaskin technology: design of the Viaskin patch, in-house industrial patch filling processes, design and building of the machines which are developed by DBV. He is also responsible for the production of the finished product in the facilities of the CMO DBV partners.technology. Mr. Dupont received an engineering degree from the School of Arts et Métiers of Paris in 1974 and an Aggregationaggregation in mechanical engineering in 1987.
David Schilansky has served as our Chief Operating Officer since January 2015. Mr. Schilansky was our Chief Financial Officer from December 2011 to January 2015. He supervises all of our financial work, human resources, general secretary, communication, investor relations as well as our partnership and business development activities. He is a member of our Executive Committee. From 2006 to 2011, Mr. Schilansky held various important positions at the Ipsen Group, or Ipsen, including Interim Chief Financial Officer, Deputy Chief Financial Officer, member of Ipsen’s Executive Committee and other positions in the administration and finance department and participated in various external growth operations and creation of Ipsen’s investor relations function. From 2003 to 2006, Mr. Schilansky spent three years at Thomson Inc. (now Technicolor S.A.) as co-head of investor relations. From 1999 to 2002, he spent three years at Warburg Dillon Read (now UBS Investment Bank) in the field of mergers and acquisitions. Mr. Schilansky received a Master degree from Université de Paris Dauphine and a Master degree from Imperial College in London.
Charles Ruban has served as our Chief DevelopmentCommercial Officer since January 2016 and has held various positions since joining us in June 2012, including serving as our Senior Executive Vice President of Clinical Development & North American Operations and Chief Development Officer. He currently oversees all of our development activities from manufacturing and quality control to clinical trials and regulatory affairs. Heaffairs and is a member of our Executive Committee. From 2003 to 2012, Mr. Ruban held various executive positions at Stallergènes S.A., including Senior Vice President of Product Development, member of the Executive Committee, Director of Research & Development Program and Director of Supply Chain. From 1994 to 2003, Mr. Ruban spent nine years at Eurogroup Consulting Holding as a management consultant. Mr. Ruban received an Engineering degree from the Ecole Centrale de Lyon, trained at Harvard-MIT Division of Health Sciences and Technology for his Master of Science in Biomedical Engineering and graduated with an Executive MBA from INSEAD.
Laurent Martin has served as our Chief Development Officer since January 2016 and has held various positions since joining us in 2007, including serving as our Senior Executive Vice President, Product Strategy & Regulatory Affairs and Director of Regulatory Affairs and Quality. He acquired his expertise in regulatory affairs through various pharmaceutical companies, such as Galderma, Guerbet, and finally, Orphan Europe, a company specialized in the development and marketing of orphan drugs, in which his last position was as Interim Responsible Pharmacist (Qualified Person), Manager of Pharmaceutical and Pre-Clinical Development, and Quality Assurance Manager. He received his PharmD from the Université René Descartes in Paris with an M.B.A. from IAE Paris Sorbonne and a Master of Law in Public Health from the faculty of Sceaux.
Dr. Torbjörn Bjerke has served as a member of our board of directors since 2006. Dr. Bjerke is the Chief Executive Officer of Karolinska Development AB. Previously, Dr. Bjerke was the President and Chief Executive Officer of Orexo AB, a position he held from 2007 until January 2011, President and Chief Executive Officer of Biolipox AB and Director of Pharmacology at AstraZeneca. Dr. Bjerke holds a Ph.D. in Medicine from Aarhus Universitet. The board of directors believes that Dr. Bjerke’s experience in the pharmaceutical industry, particularly his extensive experience in allergy treatment field, and his years of business and leadership experience allow him to make valuable contributions to the board of directors.
Michael J. Goller has served as a member of our board of directors since 2015. Mr. Goller serves as a Managing Director of Baker Brothers Investments, a fund management company focused on long-term investments in life-sciences companies. Prior to joining Baker Brothers in 2005, Mr. Goller was an Associate of JPMorgan Partners, LLC where he focused on venture investments in the life sciences sector from 1999 to 2003. Mr. Goller began his career as an investment banker with Merrill Lynch and Co. from 1997 to 1999. Mr. Goller holds a B.S. in Molecular and Cell Biology from The Pennsylvania State University, and a Master’s in both Biotechnology and Business Administration from the University of Pennsylvania. The board of directors believes that Mr. Goller’s experience in the life sciences industry and his knowledge of corporate development matters allows him to make valuable contributions to the board of directors.
George Horner III has served as a member of our board of directors since 2010. Mr. Horner has over 40 years of experience as a pharmaceutical executive. Since 2009, Mr. Horner has served as a biotech executive consultant for several private companies in the United States and Europe. Before that, from 2006 to 2008, Mr. Horner was the Chief Executive Officer of Prestwick Pharmaceuticals, Inc., or Prestwick, and under his leadership, Prestwick obtained FDA approval for Tetrabenazine (TBZ), the first drug ever licensed in the United States to treat Huntington’s disease patients. From 1996 to 2005, Mr. Horner was the Chief Executive Officer of Vicuron Pharmaceuticals, Inc. (previously known as Versicor), an anti-infective company that he grew from a market value of $12.8 million to a sale to Pfizer for $1.9 billion. He previously held numerous executive, general management, business development and marketing and sales positions at Abbott Laboratories and Bristol-Myers Squibb Company across four continents. Mr. Horner holds a AB of English History from Belmont Abbey College. From 2010 until its sale to AstraZeneca plc in July 2013, Mr. Horner was the chairman of the board of directors of Omthera Pharmaceuticals, Inc. The board of directors believes that Mr. Horner’s extensive executive and management experience in the pharmaceutical industry worldwide allows him to make valuable contributions to the board of directors.
Peter Barton Hutthas served as a member of our board of directors since 2009. He brings extensive knowledge and first-hand experience of U.S. food and drug legislation to our company. Mr. Hutt is currently a senior counsel at Covington & Burling LLP, a Washington DC law firm, specializing in food and drug law, where he has been practicing law since 1975. Mr. Hutt also teaches food and drug law at Harvard Law School. He has been a member of the U.S. Institute of Medicine since its establishment in 1971 and was former Chief Counsel for the FDA from 1971 to 1975. Mr. Hutt serves on the boards of directors of Momenta Pharmaceuticals, Inc., Xoma Corp., Concert Pharmceuticals Inc. and BIND Therapeutics, Inc. From 2008 to 2011, he served on the board of directors of Celera Corp. and from 2002 to 2012, he served on the board of directors of Ista Pharmaceuticals, Inc., Mr. Hutt holds a B.A. from Yale University, a LL.B. from Harvard Law School and a LL.M. in Food and Drug Law from New York University School of Law. The board of directors believes that Mr. Hutt’s extensive experience in the food and drug law and FDA matters allows him to make valuable contributions to the board of directors.
Chahra Louafi has served as a member of our board of directors since December 2010. Ms. Louafi is the Investment Director at Bpifrance Investissement, the fund manager for InnoBio FCPR, where she joined in 2001. Since October 2009, she has served the management team of InnoBio, a fund dedicated to biotech companies, managed by Bpifrance Investissement and invested by the pharmaceutical industry. Before that, she was at Mendel Partner in charge of initiating and implementing projects, as well as creating private business incubator specialized in biotechnology. Ms. Louafi serves on the boards of directors of Cysogene SAS and Pixium Vision SA. In addition, Ms. Louafi is the Chairman of the supervisory board of Inserm Transfert Initiative and a member of the
supervisory board of Cap Décisif Management. Ms. Louafi graduated from Paris Dauphine University with a Masters of Technology and Innovation Management, from Paris X Nanterre University with a Masters of Corporate Finance, and from Institut National Agronomique de Paris—Grignon with a Masters of Microbiology and Enzymatic Engineering. The board of directors believes that Ms. Louafi’s extensive investment experience in biotechnology industry allows her to make valuable contributions to the board of directors.
Rafaèle Tordjman, MD, Ph.D., has served as a member of our board of directors since 2005. She joined the French venture capital firm Sofinnova Partners in 2001 and is a Managing Partner specializing in life sciences investments. Prior to this, she worked as a research scientist at the Institut National de la Recherche Médicale (INSERM) in Cochin Hospital, Paris. Before joining INSERM, she was a medical doctor specializing in clinical haematology and internal medicine. She obtained her PhD, with high honors, in haematopoiesis and angiogenesis from the University Paris VII followed by a post-doctoral fellowship in immunology. Rafaèle obtained her medical degree and her specialization in Haematology and Internal Medicine as a five-year fellow in Paris University Hospitals. She also participated in the “Young Manager Program” at INSEAD (France, in 2002). She a member of the board of directors of Ascendis Pharma GmbH, Flexion Therapeutics, Inc., Nucana Biomed, MedDay and ObsEva. She was also on the board of Corevalve, Endoart before it was successfully sold to Allergan Inc., of Preglem before the latter was successfully sold to Gedeon Richter and of HBI Ltd before being acquired by Meda. The board of directors believes that Dr. Tordjman’s extensive clinical and research experience and pharmaceutical industry experience allow her to make valuable contributions to the board of directors.
Daniel Soland has served as a member of our board of directors since 2015. Mr. Soland most recently served as Senior Vice President and Chief Operating Officer of Viropharma, and currently serves on the Board of Tarsa Therapeutics. In addition to his role at Viropharma, where he helped build the organizational and commercial infrastructure that resulted in an 11-fold increase in Viropharma’s share price during his tenure, Mr. Soland previously served as President of Chiron Vaccines, and helped engineer a turnaround that contributed to Chiron’s acquisition by Novartis. Earlier, he was President and CEO of Epigenesis Pharmaceuticals. At GlaxoSmithKline Biologicals, Mr. Soland served as Vice President and Director, Worldwide Marketing Operations. Earlier in his career, Mr. Soland held positions of increasing responsibility in sales and product management at Pasteur-Merieux’s Connaught Laboratories. He holds a BS in Pharmacy degree from the University of Iowa. The board of directors believes that Mr. Horner’s extensive executive and management experience in the pharmaceutical industry worldwide, notably at various senior commercial operations positions, allows him to make valuable contributions to the board of directors.
Family Relationships
There are no family relationships among any of our executive officers or directors.
The aggregate compensation paid and benefits in kind granted by us to our current executive officers and directors, including share-based compensation, for the year ended December 31, 2014,2015, was €4.3€7.2 million. For the year ended December 31, 2014, €47,9922015, €0.2 million of the amounts set aside or accrued to provide pension, retirement or similar benefits to our employees was attributable to our executive officers.
Director Compensation
Our shareholders at theour ordinary shareholders’ general meeting ofheld on June 6, 201223, 2015 set the total annual attendance fees to be distributed among non-employee directors, except those who are affiliated with one of our significant shareholders, at €100,000.€350,000. This authorization is automatically renewed each year, unless otherwise decided by our shareholders at an ordinary shareholders’ general meeting. On September 25, 2012,March 24, 2015, upon recommendation of our compensation committee, our board of directors set attendance feefees for theseour non-employee directors at €2,500a fixed annual retainer of €30,000 per meeting.year, regardless of whether or not the director is independent. The members of our audit committee and compensation committee, regardless of whether or not the director is independent, are each entitled to an additional retainer of €5,000 per year. This amount will be increased to €10,000 per year for the chairman of said committees. The following table sets forth information regarding the compensation earned by our non-employee directors who are not executive officers or affiliated with one of our significant shareholders for service on our board of directors during the year ended December 31, 2014.2015. Dr. Benhamou, our Chief Executive Officer, Chairman and Co-Founder, is a director but does not receive any additional compensation for his services as a director.
Name | Fees Earned (€) | Warrants(1) (€) | Total (€) | |||||||||
Torbjörn Bjerke | 10,000 | 12,450 | 22,450 | |||||||||
Didier Hoch (2) | 10,000 | 12,450 | 22,450 | |||||||||
George Horner III | 10,000 | 12,450 | 22,450 | |||||||||
Peter Barton Hutt | 10,000 | 12,450 | 22,450 | |||||||||
Name Torbjörn Bjerke Michael J. Goller(2) George Horner III Peter Barton Hutt(3) Chahra Louafi Daniel Soland(4) Rafaèle Tordjman(5) Fees Earned Warrants(1) Total (Amounts in thousands of Euros) 45 — 45 — — — 45 169.5 214.5 15 — 15 30 — 30 40 268.5 308.5 20 — 20
| (1) |
| (2) |
| (3) | Resigned from the board of directors as of June 23, 2015. |
| (4) | Appointed on March 6, 2015 in replacement of Didier Hoch who had resigned. |
| (5) | Resigned from the board of directors as of October 21, 2015. |
Our other directors receive no compensation for their service as directors but are reimbursed for reasonable expenses incurred in connection with attending board and committee meetings.
CEO and COO Compensation
The following table sets forth information regarding compensation earned by Dr. Benhamou, our Chief Executive Officer, Chairman and Co-Founder, and Mr. Schilansky, our Chief Operating Officer, during the year ended December 31, 2014.2015.
Name and Principal Position | Salary (€) | Bonus (€) | Equity Awards (€) | Non-Equity Incentive Plan Compensation (€) | All Other Compensation (€) | Total (€) | Salary € | Bonus € | Equity Awards € | Non-Equity Incentive Plan Compensation € | All Other Compensation € | Total € | ||||||||||||||||||||||||||||||||||||
Pierre-Henri Benhamou | 318,500 | 109,200 | (1) | 1,140,600 | (2)(3) | — | — | 1,573,760 | 338,100 | 114,660 | (1) | 7,558,800 | (2)(3) | — | — | 8,011,560 | ||||||||||||||||||||||||||||||||
Chief Executive Officer, Chairman andCo-Founder | ||||||||||||||||||||||||||||||||||||||||||||||||
Chief Executive Officer, | ||||||||||||||||||||||||||||||||||||||||||||||||
Chairman and Co-Founder | ||||||||||||||||||||||||||||||||||||||||||||||||
David Schilansky | 253,575 | 85,995 | (4) | 5,039,200 | (2)(3) | — | — | 5,378,770 | ||||||||||||||||||||||||||||||||||||||||
Chief Operating Officer | ||||||||||||||||||||||||||||||||||||||||||||||||
| (1) | The bonus awarded to Dr. Benhamou for |
| (2) | This column reflects the full grant date fair value for equity awards granted during the year as measured pursuant to IFRS 2—Share-Based Payment as share-based compensation in our financial statements. Unlike the calculations contained in our financial statements, this calculation does not give effect to any estimate of forfeitures related to service-based vesting, but assumes that the executive officer will perform the requisite service for the award to vest in full. The assumptions we used in valuing options are described in Note 17 to our financial statements included in this Annual Report on Form 20-F. |
| (3) | The acquisition of the free shares is subject to the achievement of |
| (4) | The bonus awarded to Mr. Schilansky for 2015 amounted to €164,824 and |
Executive Compensation Arrangements
For a discussion of our employment arrangements with our executive officers, see “Item 7.C—Related Party Transactions—Arrangements with Our Directors and Executive Officers.” Except the arrangements described in “Item 7.C—Related Party Transactions—Arrangements with Our Directors and Executive Officers,” there are no arrangements or understanding between us and any of our other executive officers providing for benefits upon termination of their employment, other than as required by applicable law.
Limitations on Liability and Indemnification Matters
Under French law, provisions of by-laws that limit the liability of directors are prohibited. However, French law allowssociétés anonymes to contract for and maintain liability insurance against civil liabilities incurred by any of their directors and officers involved in a third-party action, provided that they acted in good faith and within their capacities as directors or officers of the company. Criminal liability cannot be indemnified under French law, whether directly by the company or through liability insurance.
We maintain liability insurance for our directors and officers, including insurance against liability under the Securities Act and we intend to enter into agreements with our directors and executive officers to provide contractual indemnification. With certain exceptions and subject to limitations on indemnification under French law, these agreements will provide for indemnification for damages and expenses including, among other things, attorneys’ fees, judgments, fines and settlement amounts incurred by any of these individuals in any action or proceeding arising out of his or her actions in that capacity. We believe that this insurance and these agreements are necessary to attract qualified directors and executive officers.
These agreements may discourage shareholders from bringing a lawsuit against our directors and executive officers for breach of their fiduciary duty. These provisions also may have the effect of reducing the likelihood of derivative litigation against directors and executive officers, even though such an action, if successful, might otherwise benefit us and our shareholders. Furthermore, a shareholder’s investment may be adversely affected to the extent we pay the costs of settlement and damage awards against directors and officers pursuant to these insurance agreements.
Certain of our non-employee directors may, through their relationships with their employers or partnerships, be insured against certain liabilities in their capacity as members of our board of directors.
Equity Incentives
We believe that our ability to grant incentive awards is a valuable and necessary compensation tool that allows us to attract and retain the best available personnel for positions of substantial responsibility, provides additional incentives to employees and promotes the success of our business. Due to French corporate law and tax considerations, historically, we have granted several different equity incentive instruments to our directors, executive officers, employees and other service providers. These are:
| • | employee warrants (otherwise known asbons de souscription de parts de créateurs d’entreprise, or BSPCE), granted to our officers and employees; |
| • | non-employee warrants (otherwise known asbons de souscription d’actions, or BSA), historically typically granted only to non-employee directors, members of our Scientific Advisory Board and other service providers not eligible for either employee warrants or employee share options; |
| • | employee share options (otherwise known asoptions de souscription d’actions, or OSA), granted to our officers and employees; and |
| • | free shares (otherwise known asactions gratuites). |
Our board of directors’ authority to grant these equity incentive instruments and the aggregate amount authorized to be granted must be approved by a two-thirds majority of the votes held by our shareholders present, represented or voting by authorized means at the relevant extraordinary shareholders’ meeting. Once approved by our shareholders, our board of directors can continue to grant such awards for 18 months for employee warrants and non-employee warrants authorized by the shareholders and 38 months for employee share options and free shares authorized by the shareholders.
We are no longer eligible to issue employee warrants since completion of our initial public offering on Euronext Paris in 2012.
In general, employee warrants, employee share options and non-employee warrants no longer continue to vest following termination of the employment, office or service of the holder and all vested shares must be exercised within post-termination exercise periods set forth in the grant documents. In the event of certain changes in our share capital structure, such as a consolidation or share split or dividend, French law and applicable grant documentation provides for appropriate adjustments of the numbers of shares issuable and/or the exercise price of the outstanding warrants or share options.
As of December 31, 2014,2015, employee warrants, non-employee warrants, employee share options and free share were allowing for the purchase of an aggregate of 2,207,5302,336,224 ordinary shares at a weighted average exercise price of €6.68€20.72 per share (not including the 641,3601,008,329 ordinary shares issuable upon the vesting of outstanding free shares that may be issued for free with no exercise price being paid).
Employee Warrants (BSPCE)
Employee warrants were granted only to our employees who are French tax residents as they carry favorable tax and social security treatment for French tax residents. Employee warrants may also be granted to our chairman and general manager and to our deputy general managers. Similar to options, they entitle a holder to exercise the warrant for the underlying vested shares at an exercise price per share determined by our board of directors and at least equal to the fair market value of an ordinary share on the date of grant. Employee warrants may only be issued by growth companies meeting certain criteria, which we will not meet following the completion of the offering. There is no legal limitation to the size of the employee warrant pool under French law.
We have issued three types of employee warrants as follows:
Plan title | BSPCE 4 | BSPCE X | BSPCE 2010 | BSPCE 4 | BSPCE X | BSPCE 2010 | ||||||||||||||||||||||||||
Meeting date | 1/21/2009 | 1/21/2009 | 12/16/2010 | 1/21/2009 | 1/21/2009 | 12/16/2010 | ||||||||||||||||||||||||||
Date of allocation by the Board of Directors | 1/21/2009 | 1/21/2009 | 6/24/2011 | 11/22/2011 | 1/21/2009 | 1/21/2009 | 6/24/2011 | 11/22/2011 | ||||||||||||||||||||||||
Total number of BSPCE authorized | 5,358 | 10,858 | 59,405 | 59,405 | 5,358 | 10,858 | 59,405 | 59,405 | ||||||||||||||||||||||||
Total number of BSPCEs granted | 5,358 | 2,296 | 24,000 | 10,039 | 5,358 | 2,296 | 24,000 | 10,039 | ||||||||||||||||||||||||
including those granted to Pierre-Henri Benhamou | — | — | 10,000 | — | — | — | 10,000 | — | ||||||||||||||||||||||||
David Schilansky | — | — | — | 10,039 | ||||||||||||||||||||||||||||
Start date for the exercise of the BSPCEs | 1/21/2009 | 1/21/2010 | 12/23/2011 | 11/22/2012 | 1/21/2009 | 1/21/2010 | 12/23/2011 | 11/22/2012 | ||||||||||||||||||||||||
BSPCE expiry date | 1/21/2019 | 1/21/2019 | 6/24/2021 | 11/22/2021 | 1/21/2019 | 1/21/2019 | 6/24/2021 | 11/22/2021 | ||||||||||||||||||||||||
BSPCE exercise price(1) | € | 4.33 | € | 4.67 | € | 5.13 | € | 5.13 | € | 4.33 | € | 4.67 | € | 5.13 | € | 5.13 | ||||||||||||||||
Number of shares subscribed as of December 31, 2014(1) | — | 34,440 | 46,005 | 15,000 | ||||||||||||||||||||||||||||
Total number of BCPCEs canceled or obsolete as of December 31, 2014 | — | — | — | — | ||||||||||||||||||||||||||||
Total number of BCPCEs outstanding as of December 31, 2014 | 5,358 | — | 20,933 | 9,039 | ||||||||||||||||||||||||||||
Total number of shares available for subscription as of December 31, 2014(1) | 80,370 | — | 313,995 | 135,585 | ||||||||||||||||||||||||||||
Number of shares subscribed as of December 31, 2015(1) | 40,005 | 34,440 | 158,025 | 112,950 | ||||||||||||||||||||||||||||
Total number of BCPCEs canceled or obsolete as of December 31, 2015 | — | — | — | — | ||||||||||||||||||||||||||||
Total number of BCPCEs outstanding as of December 31, 2015 | 2,691 | — | 13,465 | 2,509 | ||||||||||||||||||||||||||||
Total number of shares available for subscription as of December 31, 2015(1) | 40,365 | — | 201,975 | 37,635 | ||||||||||||||||||||||||||||
| (1) | The number of shares reflects an adjusted exercise parity of the division by 15 of the nominal value of the shares decided by the General Meeting of Shareholders held on December 9, 2011, namely that each BPSCE is now entitled to a subscription right to 15 new shares instead of 1 new share. For the same reason, the exercise price of each BPSCE plan has been adjusted accordingly and equals 1/15 of the price initially determined by the general meeting of shareholders having authorized each of the plans. |
All BPSCE 4, BPSCE X and BSPCE 2010 granted on June 2011 are exercisable. Except for 2,509 BPSCE 2010 which shall become exercisable on November 22, 2015, alland BSPCE 2010 granted on November 2011 are exercisable.
Administration. Pursuant to delegations granted by our shareholders, our board of directors determined the recipients, dates of grant and exercise price of employee warrants, the number of employee warrants to be granted and the terms and conditions of the employee warrants, including the period of their exercisability and their vesting schedule. The board of directors has the authority to extend the post-termination exercise period of employee warrants after the termination of the employment agreement.
Employee warrants are not transferable and may not be sold, pledged, assigned, hypothecated, transferred or disposed of in any manner other than by will or by laws of descent or distribution and may be exercised, during the lifetime of the beneficiary, only by the beneficiary.
Non-Employee Warrants (BSA)
Historically, non-employee warrants were typically granted by our board of directors to non-employee directors, members of our Scientific Advisory Board and other service providers not eligible for either employee warrants or employee share options. In addition to any exercise price payable by a holder upon the exercise of any non-employee warrant, non-employee warrants need to be subscribed for a price which is determined by the board on the date of grant. There is no legal limitation to the size of the non-employee warrant pool.
We have issued sixeight types of non-employee warrants (BSA) as follows(BSAs) as of December 31, 2014:2015, the terms of which are set forth in the chart below:
Plan title | BSA | BSA 2 | BSA X | BSA 2010 | BSA 2012 | BSA 2013 | BSA 2014 | BSA | BSA 2 | BSA X | BSA 2010 | BSA 2012 | BSA 2013 | BSA 2014 | BSA 2015 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Meeting date | 6/14/2007 | 1/21/2009 | 1/21/2009 | 12/16/2010 | 12/9/2011 | 6/4/2013 | 6/3/2014 | 6/14/2007 | 1/21/2009 | 1/21/2009 | 12/16/2010 | 12/9/2011 | 6/4/2013 | 6/3/2014 | 06/03/14 | 06/23/15 | 06/23/15 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Date of grant by the Board of Directors | 12/7/2007 | 1/21/2009 | 1/21/2009 | 6/25/2010 | 1/28/2011 | 6/24/2011 | 11/22/2011 | 1/17/2012 | 9/25/2012 | 7/25/2013 | 6/3/2014 | 12/7/2007 | 1/21/2009 | 1/21/2009 | 6/25/2010 | 1/28/2011 | 6/24/2011 | 11/22/2011 | 1/17/2012 | 9/25/2012 | 7/25/2013 | 6/3/2014 | 03/24/15 | 11/19/15 | 12/15/15 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Total number of BSAs authorized | 4,395 | 10,716 | 10,858 | 10,858 | 59,405 | 59,405 | 59,405 | 59,405 | 300,000 | 100,000 | 307,468 | 4,395 | 10,716 | 10,858 | 10,858 | 59,405 | 59,405 | 59,405 | 59,405 | 300,000 | 100,000 | 307,468 | 10,000 | 22,500 | 90,000 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Total number of BSAs granted | 1,717 | 10,716 | 306 | 1,825 | 10,039 | 8,000 | 1,338 | 89,835 | 30,000 | 73,000 | 10,000 | 1,717 | 10,716 | 306 | 1,825 | 10,039 | 8,000 | 1,338 | 89,835 | 30,000 | 73,000 | 10,000 | — | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Including those granted to Pierre-Henri Benhamou | — | 5,358 | — | — | — | — | — | — | — | — | — | — | 5,358 | — | — | — | — | — | — | — | — | — | — | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Start date for the exercise of the BSAs | 12/7/2008 | 1/21/2009 | 1/21/2010 | 6/25/2011 | 12/23/2011 | 12/23/2011 | 11/22/2012 | 1/17/2016 | 9/25/2013 | 7/25/2013 | 6/3/2014 | 12/7/2008 | 1/21/2009 | 1/21/2010 | 6/25/2011 | 12/23/2011 | 12/23/2011 | 11/22/2012 | 1/17/2016 | 9/25/2013 | 7/25/2013 | 6/3/2014 | — | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
BSA expiry date | 12/7/2015 | 1/21/2019 | 1/21/2019 | 6/25/2020 | 1/28/2021 | 6/24/2021 | 11/22/2021 | 1/17/2022 | 9/25/2022 | 7/25/2023 | 6/3/2024 | 12/7/2015 | 1/21/2019 | 1/21/2019 | 6/25/2020 | 1/28/2021 | 6/24/2021 | 11/22/2021 | 1/17/2022 | 9/25/2022 | 7/25/2023 | 6/3/2024 | 03/24/25 | 11/19/25 | 12/15/25 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
BSA exercise price | € | 4.33 | € | 4.33 | € | 4.33 | € | 4.33 | € | 5.13 | € | 5.13 | € | 5.13 | € | 5.13 | € | 8.59 | € | 8.10 | € | 18.79 | € | 4.33 | € | 4.33 | € | 4.33 | € | 4.33 | € | 5.13 | € | 5.13 | € | 5.13 | € | 5.13 | € | 8.59 | € | 8.10 | € | 18.79 | € | 43.00 | € | 66.06 | € | 64.14 | ||||||||||||||||||||||||||||||||||||||||||||||||||
Number of shares subscribed as of December 31, 2014 | — | 40,005 | — | — | 37,650 | — | 15,060 | — | 5,000 | 2,500 | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Total number of BSAs canceled or obsolete as of December 31, 2014 | 572 | — | — | — | — | — | — | — | — | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Total number of BSAs remaining as of December 31, 2014 | 1,145 | 8,049 | 306 | 1,825 | — | 8,000 | 334 | 89,835 | 25,000 | 70,500 | 10,000 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Total number of shares available for subscription as of December 31, 2014(1) | 17,175 | 120,735 | 4,590 | 27,375 | — | 120,000 | 5,010 | 89,835 | 25,000 | 70,500 | 10,000 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Number of shares subscribed as of December 31, 2015 | 4,290 | 160,740 | 16,425 | 37,650 | 109,350 | 15,060 | — | 20,000 | 60,000 | 5,000 | — | — | (3) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Total number of BSAs canceled or obsolete as of December 31, 2015 | 572 | — | — | — | — | — | — | — | — | — | — | — | 7,500 | (3) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Total number of BSAs remaining as of December 31, 2015 | 859 | — | 306 | 730 | — | 710 | 334 | 89,835 | 10,000 | 13,000 | 5,000 | 10,000 | 15,000 | (3) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Total number of shares available for subscription as of December 31, 2015(1) | 12,885 | — | 4,590 | 10,950 | — | 10,650 | 5,010 | 89,835 | 10,000 | 13,000 | 5,000 | 10,000 | 15,000 | (3) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| (1) | The number of shares reflects an adjusted exercise parity of the division by 15 of the nominal value of shares. Namely, each BSA is now entitled to a subscription right to 15 new shares instead of 1 new share. For the same reason, the exercise price of each BSA plan has been adjusted accordingly and equals 1/15 of the price initially determined. |
| (2) | The overall nominal amount of the shares to which the warrants issued are likely to give entitlement may not exceed €100,000. |
| (3) | The final subscription date for the BSAs issued in December 2015 was February 15, 2016; none of these BSAs were subscribed as of December 31, 2015. At February 15, 2016, 73,500 BSAs were subscribed. |
All BSA, BSA2 andBSA 2, BSA X are exercisable. Alland BSA 2010 are exercisable except for:
currently exercisable. All BSA 2012, 2013, 2014 2015 and 2014BSA X 2015 are exercisable subject to continuous membership of our boardBoard or Scientific Advisory Board (as the case may be) and subject to applicable insiders’ rules.
Administration. Pursuant to delegations granted by our shareholders, our board of directors determined the recipients, dates of grant and exercise price of non-employee warrants, the number of non-employee warrants to be granted and the terms and conditions of the non-employee warrants, including the period of their exercisability and their vesting schedule. The board of directors has the authority to extend the post-termination exercise period of non-employee warrants after the end of the term of office.
Non-employee warrants may be transferred to any person and may be exercised by their holder at any time subject to vesting.
Share Options (OSA)
We have granted share options to our employees and our officers pursuant to our 2013 Share Option Plan, or 2013 Plan, and our 2014 Share Option Plan, or 2014 Plan, and our 2015 Share Option Plan, or 2015 Plan. Our current plan, the 20142015 Plan, was adopted by our board of directors on June 3, 2014.September 21, 2015.
Share options may be granted to any individual employed by us or by any affiliated company under the terms and conditions of an employment contract. Employee share options may also be granted to our chairman and general manager and to our deputy general managers.
The maximum number of our ordinary shares that may be issued pursuant to share options granted under the 2013 Plan, 2014 Plan and 20142015 Plan are 518,000, 322,405 and 322,405,315,000, respectively. In addition, under French law, the maximum number of shares issuable upon exercise of outstanding employee share options may not exceed one-third of the outstanding share capital on a non-diluted basis as at the date of grant.
Plan title | SO 2013 | SO 2014 | SO 2013 | SO 2014 | SO 2015 | |||||||||||||||||||
Meeting date | 12/9/2011 | 6/3/2014 | 12/9/2011 | 6/3/2014 | 6/3/14 | 6/3/14 | ||||||||||||||||||
Date of allocation by the Board of Directors | 9/18/2013 | 6/3/2014 | ||||||||||||||||||||||
Total number of options authorized | 1,968,528 | 922,405 | ||||||||||||||||||||||
Date of allocation by the board of directors | 9/18/2013 | 6/3/2014 | 6/23/15 | 11/19/15 | ||||||||||||||||||||
Total number of options granted | 518,000 | 75,000 | 518,000 | 75,000 | 120,000 | 195,000 | ||||||||||||||||||
Including those granted to Pierre-Henri Benhamou | 129,000 | — | 129,000 | — | — | — | ||||||||||||||||||
Start date for the exercise of options(1) | 9/19/2017 | 6/4/2016 | 9/19/2017 | 6/4/2016 | 6/24/2016 | 9/30/2016 | ||||||||||||||||||
Options expiry date | 9/18/2023 | 6/3/2024 | 9/18/2023 | 6/3/2024 | 6/24/2026 | 9/30/2026 | ||||||||||||||||||
Options exercise price | € | 7.57 | € | 19.01 | € | 7.57 | € | 19.01 | € | 48.90 | € | 66.06 | ||||||||||||
Number of shares subscribed as of December 31, 2014 | — | — | ||||||||||||||||||||||
Total number of options canceled or obsolete as of December 31, 2014 | 47,000 | — | ||||||||||||||||||||||
Total number of options remaining as of December 31, 2014 | 471,000 | 75,000 | ||||||||||||||||||||||
Total number of shares available for subscription as of December 31, 2014 | 471,000 | 75,000 | ||||||||||||||||||||||
Number of shares subscribed as of December 31, 2015 | — | — | — | — | ||||||||||||||||||||
Total number of options canceled or obsolete as of December 31, 2015 | 47,000 | — | — | — | ||||||||||||||||||||
Total number of options remaining as of December 31, 2015 | 471,000 | 75,000 | 120,000 | 195,000 | ||||||||||||||||||||
Total number of shares available for subscription as of December 31, 2015 | 471,000 | 75,000 | 120,000 | 195,000 | ||||||||||||||||||||
| (1) | By way of exception, in the event of a change in control (as defined in Article L.233-3 of the French Commercial Code) occurring prior to September 19, 2017, or June 4, 2016, as applicable, all of the options could be exercised in advance. |
Our board of directors has set at 10% the number of acquired shares that must be kept by Dr. Pierre-Henri Benhamou in registered form until the cessation of his duties.
Administration. Our board of directors has the authority to administer the 2013 Plan, the 2014 Plan and 2014 Plan.the 2015 Plan, or collectively, the Plans. Subject to the terms of each of the 2013 Plan or 2014 Plan,Plans, our board of directors determines recipients, dates of grant, exercise price of share options, the number of share options to be granted and the terms and conditions of the share options, including the period of their exercisability and their vesting schedule.
The board of directors has the authority to modify awards outstanding under the 2013 Plan or 2014 PlanPlans subject to the consent of the optionee if such modification is detrimental to him/her, including in particular the authority to extend the post-termination exercise period after the termination of the employment.
The term of each share option is ten years from the date of grant or, in the case of death or disability of the optionee during such ten-year period, six months from the death or disability of the optionee in accordance with French law. In the event of the death of an optionee during the term of the options, unless otherwise resolved by the board of directors, the vested options may be exercised at any time within six months following the date of death, by the optionee’s estate or by a person who acquired the right to exercise the option by bequest or inheritance.
Share options are not transferable and may not be sold, pledged, assigned, hypothecated, transferred or disposed of in any manner other than by will or by laws of descent or distribution and may be exercised, during the lifetime of the optionee, only by the optionee.
Amendment and Termination. Our board of directors has the authority to amend, alter, suspend, or terminate the 2013 Plan and 2014 Plan,Plans, provided that such action does not impair the rights of any optionee without such optionee’s consent. We shall obtain shareholder approval of any amendment to the extent necessary and desirable to comply with applicable laws.
Free Shares
Under our 2012, 2013, 2014 and 20142015 Free Share Plans, we have granted free shares to our employees and officers. OurWe have two current plan,free share plans, the 20142015 Free Share Plan, wasPlans, which were adopted by our board of directors on June 3, 2014.September 30, 2015 and December 15, 2015, respectively.
Free shares may be granted to any individual employed by us or by any affiliated company under the terms and conditions of an employment contract. Free shares may also be granted to our Chairman, our general manager and to our deputy general managers. However, no free share may be granted to a beneficiary holding more than 10% of our share capital or to a beneficiary who would hold more than 10% of our share capital as a result of such grant.
Share Reserve. The maximum number of our ordinary shares that may be issued, in aggregate, under the 2012, 2013, 2014 and 20142015 Free Share Plans is 641,360.1,008,329. In addition, under French law, the number of free shares may not exceed 10% of the outstanding share capital on a non-diluted basis as at the date of grant.
The details of the grantgrants under the 2012, 2013, 2014 and 2015 Free Share Plans as of December 31, 20142015 are as follows:
| INFORMATION REGARDING FREE SHARES | INFORMATION REGARDING FREE SHARES | |||||||||||||||||||||||
Meeting date | December 09, 2011 | December 09, 2011 | December 09, 2011 | December 09, 2011 | June 3, 2014 | December 9, 2011 | December 9, 2011 | December 9, 2011 | December 9, 2011 | June 3, 2014 | September 21, 2015 | December 15, 2015 | ||||||||||||
Date of the Board of Directors’ meeting | April 02, 2012 | July 25, 2012 | November 28, 2012 | July 25, 2013 September 12, 2013 | June 3, 2014 | |||||||||||||||||||
Date(s) of the meeting of the board of directors | April 2, 2012 | July 25, 2012 | November 28, 2012 | July 25, 2013 and September 12, 2013 | June 3, 2014 | September 30, 2015 | December 15, 2015 | |||||||||||||||||
Total number of free shares granted | 669,796 | 134,081 | 35,360 | 501,500 | 186,000 | 669,796 | 134,081 | 35,360 | 501,500 | 186,000 | 708,500 | 42,000 | ||||||||||||
Number of shares granted to Pierre-Henri Benhamou | 304,461 | — | — | 58,500 | 60,000 | 304,461 | — | — | 58,500 | 60,000 | 120,000 | — | ||||||||||||
David Schilansky | — | — | — | — | — | 80,000 | — | |||||||||||||||||
Date of definitive acquisition of free shares (subject to the conditions of acquisition)(1) | April 2, 2014(2)(3) | July 25, 2014(2)(3) | November 28, 2014 | July 25, 2015(2)(4) | June 3, 2016(2)(5) | April 2, 2014(2)(3) | July 25, 2014(2)(3) | November 28, 2014 | July 25, 2015(2)(4) | June 3, 2016(2)(5) | September 30, 2017(6) | December 15, 2017(6) | ||||||||||||
End date of retention period | April 02, 2016(2) | July 25, 2016(2) | November 28, 2016 | July 25, 2017(2) | June 3, 2018(2) | April 2, 2016(2) | July 25, 2016(2) | November 28, 2016 | July 25, 2017(2) | June 3, 2018(2) | September 30, 2017 | December 15, 2017 | ||||||||||||
Number of shares acquired definitively as of December 31, 2014 | 667,936 | 134,081 | 35,360 | — | — | |||||||||||||||||||
Cumulative number of free shares canceled or lapsed as of December 31, 2014 | 1,860 | — | — | 81,500 | — | |||||||||||||||||||
Shares acquired free of charge remaining as of December 31, 2014 (in acquisition period) | — | — | — | 420,000 | 186,000 | |||||||||||||||||||
Number of shares acquired definitively as of December 31, 2015 | 667,936 | 134,081 | 35,360 | 286,338 | — | — | — | |||||||||||||||||
Cumulative number of free shares canceled or lapsed as of December 31, 2015 | 1,860 | — | — | 31,833 | 30,000 | — | — | |||||||||||||||||
Shares acquired free of charge remaining as of December 31, 2015 (in acquisition period) | — | — | — | 101,829 | 156,000 | 708,500 | 42,000 | |||||||||||||||||
| (1) | In the event of incapacity of a beneficiary as defined in Article L. 225-197-1, I of the French Commercial Code during the vesting period, said beneficiary may request the allocation of the shares within a period of six months from the event that led to the incapacity. In the event of the death of a beneficiary during the vesting period, his heirs may request the free allocation of shares within a period of six months from the death. |
| (2) | The acquisition and retention period end date start on achievement of performance criteria for executive officers. See (3), (4) or (5) below. |
| (3) | The acquisition of free shares is conditional for executive officers, including Dr. Benhamou, to the achievement of the three performance criteria below: |
| • | One-third of the shares allocated to executive officers will only be acquired from the later of the two following |
| (4) | The acquisition of free shares is conditional for executive officers, including Dr. Benhamou, to the achievement of the three performance criteria below: |
| • | One-third of the shares allocated to executive officers will only be acquired from the later of the two following |
| It is specified that in the event of a change of control (as defined in Article L. 233-3 of the French Commercial Code), the performance criteria will be considered as definitively achieved. |
| (5) | The acquisition of free shares is conditional for executive officers, including Dr. Benhamou, to the achievement of the three performance criteria below: |
| • | 50% of the shares allocated to executive officers will only be acquired from the later of the two following |
| Dr. |
| (6) | The acquisition of free shares is conditional for executive officers, including Dr. Benhamou, to the achievement of the three performance criteria below: |
Unless stated otherwise, the acquisition of the free shares allocated to Companycompany employees are not subject to the achievement of performance criteria.
Administration. Our board of directors has the authority to administer the 2012, 2013, 2014 and 20142015 Free Share Plans. Subject to the terms of the plans, our board of directors determines recipients, dates of grant, the number of free shares to be granted and the terms and conditions of the free shares, including the length of their acquisition period (period starting on the date of grant during which the beneficiary holds a right to acquire shares for free but not any shares yet) and holding period (period starting at the end of the acquisition period when the shares are issued and definitively acquired and issued but may not be transferred) within the limit determined by the shareholders (in particular the acquisition period is at least two years from the date of grant and the holding period two years from the end of the acquisition period, it being specified that no holding period will be applicable to the beneficiaries for whom the acquisition period is at least 4 years)four years for the 2012, 2013 and 2014 Free Share Plans, no additional holding period is applicable for the 2015 Free Share Plans).
The board of directors has the authority to modify awards outstanding under our 2012, 2013, 2014 and 20142015 Free Share Plans subject to the consent of the beneficiary if such modification is detrimental to him/her, including in particular the authority to release a beneficiary from the continued service condition during the acquisition period after the termination of the employment.
Free Shares. The free shares granted under our 2012, 2013, 2014 and 20142015 Free Share Plans will be definitively acquired at the end of the acquisition period as set by our board of directors (of a(a minimum of two years) subject to continued service during the acquisition period, except if the board releases a given beneficiary from this condition upon termination of his/her employment contract. At the end of the acquisition period, the beneficiary will be the owner of the shares. However, during the holding period under the 2012, 2013 and 2014 Free Share Plans (as set by our board of directors with a minimum of two years except if the acquisition period is at least equal to four years), the shares may not be sold, transferred or pledged. Under the 2015 Free Share Plans, the granted free shares may be sold, subject to the regulations governing companies whose shares are traded on a regulated market, as soon as the shares vest. The free shares allocated to the beneficiaries will be new ordinary shares and will immediately have the same rights as the existing company shares.
In the event of disability before the end of the acquisition period, the free shares shall be definitively acquired by the beneficiary on the date of disability. In the event the beneficiary dies during the acquisition period, the free shares shall be definitively acquired at the date of the request of allocation made by his or her beneficiaries in the framework of the inheritance provided that such request is made within six months from the date of death.
Under the 2015 Free Share Plans, in the event of a change of control of the company, the beneficiaries will remain eligible for the allocation at the end of each respective vesting period, even if the beneficiary’s employment contract and/or corporate mandate is terminated for any reason, between the date of the takeover and the last day of the vesting period. In this scenario, the shares will automatically vest and are not subject to the achievement of performance criteria.
Amendment and Termination. Our board of directors has the authority to amend, alter, suspend, or terminate our 2012, 2013, 2014 and 20142015 Free Share Plans, provided that such action does not impair the rights of any beneficiary without such beneficiary’s consent. The company shall obtain shareholder approval of any amendment to the extent necessary and desirable to comply with applicable laws.
We currently have seven directors, less than a majority of whom are citizens or residents of the United States. As permitted by French law, one of our directors is a legal entity. The entity has designated an individual to represent it and to act on its behalf at meetings of our board or directors. This representative has the same responsibilities to us and to our shareholders as she would have if she had been elected to our board of directors in her individual capacity. While the currently designated representative has served in that capacity since the entity was elected to our board of directors, the entity director retains the right to appoint a different representative at any time and there can be no assurance that the individual currently serving as the representative will continue in that capacity.
Under French law and our by-laws, our board of directors must be composed of between three and 18 members. Within this limit, the number of directors is determined by our shareholders. Directors are elected, re-elected and may be removed at a shareholders’ general meeting with a simple majority vote of our shareholders. Pursuant to our by-laws, our directors are elected for two-year terms. In accordance with French law, our by-laws also provide that our directors may be removed with or without cause by the affirmative vote of the holders of at least a majority of the votes of the shareholders present, represented by a proxy or voting by mail at the relevant ordinary shareholders’ meeting, and that any vacancy on our board of directors resulting from the death or resignation of a director, provided there are at least three directors remaining, may be filled by vote of a majority of our directors then in office provided that there has been no shareholders meeting since such death or resignation. Directors chosen or appointed to fill a vacancy shall be elected by the board of directors for the remaining duration of the current term of the replaced director. The appointment must then be ratified at the next shareholders’ general meeting. In the event the board of directors would be composed of less than three directors as a result of a vacancy, the remaining directors shall immediately convene a shareholders’ general meeting to elect one or several new directors so there are at least three directors serving on the board of directors, in accordance with French law.
The following table sets forth the names of our directors, the years of their initial appointment as directors and the expiration dates of their current term.
Name | Current Position | Year of Initial | Term | |||||||||
| Pierre-Henri Benhamou | Chairman | 2005 | 2016 | |||||||||
| Torbjörn Bjerke | Director | 2006 | 2016 | |||||||||
| Daniel Soland(3) | Director | 2014 | 2016 | |||||||||
| George Horner III | Director | 2010 | 2016 | |||||||||
| Peter Barton Hutt | Director | 2009 | 2016 | |||||||||
| Chahra Louafi(1) | Director | 2010 | 2016 | |||||||||
| Rafaèle Tordjman(2) | Director | 2005 | 2016 | |||||||||
Name Pierre-Henri Benhamou Torbjörn Bjerke Michael J. Goller(1) George Horner III Chahra Louafi(2) Daniel Soland(3) Current Position Year of Initial
Appointment Term
Expiration Year Chairman 2005 2016 Director 2006 2016 Director 2015 2016 Director 2010 2016 Director 2010 2016 Director 2015 2016
| (1) | Appointed on October 21, 2015, in replacement of Rafaèle Tordjman from Sofinnova Partners who resigned from the board of directors as of October 21, 2015. |
| (2) | From December 2010 to July 2014, Ms. Louafi served on our board of directors as representative of Bpifrance Investissement, the legal entity that previously held this board seat. Since July 2014, Ms. Louafi has served as a member of our board of |
| (3) | Appointed on March 6, 2015 in replacement of Didier Hoch who had resigned. |
In addition, Ms. Maïlys Ferrère was appointed as a non-voting observer pursuant to a shareholders’ agreement and her current term expires in 2016.
Director Independence
As a foreign private issuer, under the listing requirements and rules of Nasdaq, we are not required to have independent directors on our board of directors, except to the extent that our audit committee is required to consistent of independent requirements, subject to certain phase-in schedules. However, our board of directors has determined that, under current listing requirements and rules of Nasdaq (which we are not subject to) and taking account any applicable committee independence standards, Torbjörn Bjerke, George Horner III and Daniel Soland and Peter Barton Hutt are “independent directors.” In making such determination, our board of directors considered the relationships that each non-employee director has with us and all other facts and circumstances our board of directors deemed relevant in determining the director’s independence, including the number of ordinary shares beneficially owned by the director and his or her affiliated entities (if any).
Role of the Board in Risk Oversight
Our board of directors is primarily responsible for the oversight of our risk management activities and has delegated to the audit committee the responsibility to assist our board in this task. While our board oversees our risk management, our management is responsible for day-to-day risk management processes. Our board of directors expects our management to consider risk and risk management in each business decision, to proactively develop and monitor risk management strategies and processes for day-to-day activities and to effectively implement risk management strategies adopted by the board of directors. We believe this division of responsibilities is the most effective approach for addressing the risks we face.
Corporate Governance Practices
As a French société anonyme, we are subject to various corporate governance requirements under French law. In addition, as a foreign private issuer listed on the Nasdaq Global Select Market, we will beare subject to the Nasdaq corporate governance listing standards. However, the Nasdaq Global Select Market’s listing standards provide that foreign private issuers are permitted to follow home country corporate governance practices in lieu of the Nasdaq rules, with certain exceptions. We intend tocurrently rely on the certain exemptions for foreign private issuers and follow French corporate governance practices in lieu of the Nasdaq corporate governance rules, including Nasdaq corporate governance rules that state (1) a majority of the board of directors consists of independent directors; (2) establishing a nominating and corporate governance committee; (3) the compensation committee be composed entirely of independent directors; and (4) separate executive sessions of independent directors and non-management directors held by the company at least twice per year.
As a foreign private issuer, we are required to comply with Rule 10A-3 of the Exchange Act relating to audit committee composition and responsibilities. Rule 10A-3 provides that the audit committee must have direct responsibility for the nomination, compensation and choice of our auditors, as well as control over the performance of their duties, management of complaints made, and selection of consultants. However, if the laws of a foreign private issuer’s home country require that any such matter be approved by the board of directors or the shareholders, the audit committee’s responsibilities or powers with respect to such matter may instead be advisory. Under French law, the audit committee may only have an advisory role and appointment of our statutory auditors, in particular, must be decided by the shareholders at our annualgeneral shareholders’ meeting.
In addition, Nasdaq rules require that a listed company specify that the quorum for any meeting of the holders of common stock be at least 33 1/3% of the outstanding shares of the company’s common voting stock. Consistent with French Law,law, our by-laws provide that a quorum requires the presence of shareholders having at least (1) 20% of the shares entitled to vote in the case of an ordinary shareholders’ general meeting or at an extraordinary shareholders’ general meeting where shareholders are voting on a capital increase by capitalization of reserves, profits or share premium, or (2) 25% of the shares entitled to vote in the case of any other extraordinary shareholders’ general meeting. If a quorum is not present, the meeting is adjourned. There is no quorum requirement when an ordinary general meeting is reconvened, but the reconvened meeting may consider only questions which were on the agenda of the adjourned meeting. When an extraordinary general meeting is reconvened, the quorum required is 20% of the shares entitled to vote, except where the reconvened meeting is considering capital increases through capitalization of reserves, profits or share premium. For these matters, no quorum is required at the reconvened meeting. If a quorum is not present at a reconvened meeting requiring a quorum, then the meeting may be adjourned for a maximum of two months. See “Item 10.B—Memorandum and Articles of Association.”
Board Committees
The board of directors has established an audit committee and a compensation committee, which operate pursuant to a unique charter adopted by our board of directors. The composition and functioning of all of our committees will comply with all applicable requirements of the French Commercial Code, the Exchange Act, the exchange on which the ADSs are listed, and SEC rules and regulations.
In accordance with French law, committees of our board of directors will only have an advisory role and can only make recommendations to our board of directors. As a result, decisions will be made by our board of directors taking into account non-binding recommendations of the relevant board committee.
Audit Committee. Our audit committee reviews our internal accounting procedures, consults with and reviews the services provided by our independent registered public accountants and assists our board of directors in its oversight of our corporate accounting and financial reporting. Dr. Bjerke Mr.and Messrs. Horner and Mr. Soland currently serve on our audit committee. Dr. Bjerke is the chairperson of our audit committee. Our board has determined that each of Dr. Bjerke, Mr.Messrs. Horner and Mr. Soland is independent within the meaning of the applicable listing rules and the independence requirements contemplated by Rule 10A-3 under the Exchange Act. Our board of directors has further determined that Dr. Bjerke is an “audit committee financial expert” as defined by SEC rules and regulations and that each of Mr.Messrs. Horner and Mr. Soland qualifies as financially sophisticated under the applicable exchange listing rules. We intend to comply with the applicable independence requirements with respect to our audit committee within the applicable time frame under the applicable transition rules of the SEC. The principal duties and responsibilities of our audit committee include (1) analyzing economic and financial information, and (2) ensuring the accuracy and honesty of the our company’s financial statements, as well as the quality of the information provided.provided, and (3) monitoring the existence and effectiveness of our company’s accounting, internal controls and auditing matters.
Our board of directors has specifically assigned the following duties to the audit committee:
Compensation Committee. Our compensation committee assists our board of directors in reviewing and making recommendations to our board of directors with respect to the compensation of our executive officers and directors. Mr. Horner, Dr. Bjerke and Dr. TordjmanMessrs. Horner and Soland currently serve on the compensation committee. Mr. Horner is the chairperson of our compensation committee. The principal duties and responsibilities of our compensation committee include:
| D. | Employees. |
As of December 31, 2014,2015, we had 5691 employees. We consider our labor relations to be good. At each date shown, we had the following employees, broken out by department and geography:
| At December 31, | At December 31, | |||||||||||||||||||||||
| 2012 | 2013 | 2014 | 2013 | 2014 | 2015 | |||||||||||||||||||
Function: | ||||||||||||||||||||||||
Pre-clinical development and regulatory affairs | 4 | 5 | 6 | 5 | 6 | 14 | ||||||||||||||||||
Clinical development | 4 | 4 | 5 | 4 | �� | 5 | 8 | |||||||||||||||||
Research | 13 | 18 | 24 | 18 | 24 | 36 | ||||||||||||||||||
Engineering and production | 5 | 6 | 7 | 6 | 7 | 14 | ||||||||||||||||||
Management and administration | 8 | 11 | 14 | 11 | 14 | 18 | ||||||||||||||||||
U.S. marketing | — | — | 1 | |||||||||||||||||||||
Total | 34 | 44 | 56 | 44 | 56 | 91 | ||||||||||||||||||
Geography: | ||||||||||||||||||||||||
France | 34 | 44 | 55 | 44 | 55 | 86 | ||||||||||||||||||
United States | — | — | 1 | — | 1 | 5 | ||||||||||||||||||
Total | 34 | 44 | 56 | 44 | 56 | 91 | ||||||||||||||||||
| E. | Share Ownership. |
For information regarding the share ownership of our directors and executive officers, see “Item 6.B—Compensation” and “Item 7.A—Major Shareholders.”
| Item 7. | Major Shareholders and Related Party Transactions |
| A. | Major shareholders. |
The following table sets forth information with respect to the beneficial ownership of our ordinary shares as of March 24, 2015April 6, 2016 for:
Beneficial ownership is determined in accordance with the rules of the SEC. These rules generally attribute beneficial ownership of securities to persons who possess sole or shared voting power or investment power with respect to those securities and include ordinary shares that can be acquired within 60 days of March 24, 2015.April 6, 2016. The percentage ownership information shown in the table is based upon 19,372,48624,205,129 ordinary shares outstanding as of March 24, 2015.April 6, 2016.
Except as otherwise indicated, all of the shares reflected in the table are ordinary shares and all persons listed below have sole voting and investment power with respect to the shares beneficially owned by them, subject to applicable community property laws. The information is not necessarily indicative of beneficial ownership for any other purpose.
In computing the number of ordinary shares beneficially owned by a person and the percentage ownership of that person, we deemed outstanding ordinary shares subject to options and warrants held by that person that are immediately exercisable or exercisable within 60 days of March 24, 2015.April 6, 2016. We did not deem these shares outstanding, however, for the purpose of computing the percentage ownership of any other person. Beneficial ownership representing less than 1% is denoted with an asterisk (*). The information in the table below is based on information known to us or ascertained by us from public filings made by the shareholders. Except as otherwise indicated in the table below, addresses of the directors, executive officers and named beneficial owners are in care of DBV Technologies S.A., Green Square-Bâtiment D, 80/84 rue des Meuniers, 92220 Bagneux,177-181 avenue Pierre Brossolette, 92120 Montrouge, France.
| Shares Beneficially Owned | ||||||||
| Number | Percentage | |||||||
5% Shareholders: | ||||||||
Sofinnova Capital V FPCI(1) | 2,269,254 | 11.71 | % | |||||
Entities Affiliated with Bpifrance(2) | 2,516,186 | 12.99 | ||||||
Entities Affiliated with Baker Bros. Advisors LP(3) | 1,885,780 | 9.73 | ||||||
Directors and Executive Officers: | ||||||||
Pierre-Henri Benhamou(4) | 705,956 | 3.64 | ||||||
Torbjörn Bjerke(5) | 35,925 | * | ||||||
Daniel Soland(6) | — | * | ||||||
George Horner III(7) | 45,150 | * | ||||||
Peter Hutt(8) | 23,925 | * | ||||||
Chahra Louafi(9) | 2,516,186 | 12.99 | ||||||
Rafaèle Tordjman(10) | 2,269,254 | 11.71 | ||||||
All directors and executive officers as a group (10 persons)(11) | 6,245,705 | 32.24 | % | |||||
| Shares Beneficially Owned | ||||||||
| Number | Percentage | |||||||
5% Shareholders: | ||||||||
Entities affiliated with Bpifrance(1) | 2,212,259 | 9.14 | % | |||||
Entities affiliated with Baker Bros. Advisors LP(2) | 3,631,589 | 15.00 | ||||||
Entities affiliated with FMR LLC(3) | 2,412,324 | 9.97 | ||||||
Entities affiliated with Morgan Stanley(4) | 1,342,093 | 5.54 | ||||||
Janus Capital Management LLC(5) | 1,534,932 | 6.34 | ||||||
Directors and Executive Officers: | ||||||||
Pierre-Henri Benhamou(6) | 513,191 | 2.11 | ||||||
David Schilansky(7) | 198,510 | * | ||||||
Torbjörn Bjerke(8) | 35,925 | * | ||||||
Michael J. Goller(9) | 7,500 | * | ||||||
George Horner III(10) | 52,650 | * | ||||||
Chahra Louafi(11) | 2,212,259 | 9.14 | ||||||
Daniel Soland(12) | 20,000 | * | ||||||
All directors and executive officers as a group (10 persons)(13) | 3,761,896 | 15.31 | % | |||||
| (1) |
| The information is based solely on a Schedule 13D/A filed by Baker Bros. Advisors LP, Baker Bros. Advisors (GP) LLC, Felix J. Baker and Julian C. Baker on February 16, 2016. Consists of (a) |
| (3) | This information is based solely on a Schedule 13G filed by |
| (4) | This information is based solely on a Schedule 13G jointly filed by Morgan Stanley and Morgan Stanley & Co. International plc on February 12, 2016. Morgan Stanley beneficially owns 1,342,093 shares. It has sole voting power with respect to 1,334,502 of such shares, shared voting power with respect to 7,429 of such shares and shared dispositive power with respect to all 1,342,093 shares. Morgan Stanley & Co. International plc beneficially owns 1,287,265 shares. It has sole voting power with respect to 1,287,265 of such shares and shared dispositive power with respect to all 1,287,265 shares. The securities being reported on by Morgan Stanley as a parent holding company are owned, or may be deemed to be beneficially owned, by Morgan Stanley & Co. International plc, a broker dealer registered under Section 15 of the Securities Exchange Act of 1934, as amended. Morgan Stanley & Co. International plc is a wholly-owned subsidiary of Morgan Stanley. The principal business address for Morgan Stanley is 1585 Broadway, New York, New York 10036. The principal business address for Morgan Stanley & Co. International plc is 25 Cabot Square, Canary Wharf, London E14 4QA, England. |
| (5) | This information is based solely on a Schedule 13G filed by Janus Capital Management LLC, or Janus Capital, on February 16, 2016. Janus Capital has sole dispositive power with respect to an aggregate of 1,534,932 ordinary shares in its capacity as an investment adviser in accordance with Rule 13d-1(b)(ii)(E) under the Exchange Act and as a parent holding company or control person in accordance with Rule 13d-1(b)(ii)(G) under the Exchange Act. The principal business address for Janus Capital is 151 Detroit Street, Denver, Colorado 80206. |
| (6) | Consists of |
| Consists of |
| Consists of |
| Consists of |
| (10) | Consists of (a) 45,150 shares held and (b) 7,500 shares issuable upon the exercise of options and warrants that are immediately exercisable or exercisable within 60 days of |
| (13) | Includes 360,905 shares issuable upon the exercise of options and warrants that are immediately exercisable or exercisable within 60 days of April 6, 2016, subject to French law. |
The significant changes in the percentage ownership held by our principal shareholders since January 1, 20122013 are as a result of the transactions described in our prospectusprospectuses dated October 22, 2014 and July 15, 2015, each filed with the SEC pursuant to Rule 424(b), under the heading “Related Party Transactions—Transactions with Our Principal Shareholders” and the dilution resulting from our recentunderwritten public offering.offerings in October 2014 and July 2015. None of our principal shareholders have voting rights different than our other shareholders.
As of January 15,September 30, 2015, assuming that all of our ordinary shares represented by ADSs are held by residents of the United States, we estimate that approximately 48.21%61.0% of our outstanding ordinary shares (including ordinary shares in the form of ADSs) were held in the United States by 3257 holders of record. At such date, there were outstanding 6,487,292 ADSs, each representing one half of one ordinary share, and in the aggregate representing 16.89% of our outstanding ordinary shares. The actual number of holders is greater than these numbers of record holders, and includes beneficial owners whose ordinary shares or ADSs are held in street name by brokers and other nominees. This number of holders of record also does not include holders whose shares may be held in trust by other entities.
B. Related Party Transactions.
Since January 1, 2014,2015, we have engaged in the following transactions with our directors, executive officers and holders of more than five percent (5%) of our outstanding voting securities and their affiliates, which we refer to as our related-parties.
Shareholders’ Agreement
On March 9, 2012, Dr. Benhamou, PHYS Participations, Mr. Bertrand Dupont, DBCS Participations, Bpifrance Participations (formerlyFonds Strategique d’Investissement)d’Investissement) and our company entered into a shareholders’ agreement pursuant to which,which: (1) the parties entered into certain lock-up undertakings, which expired at the end of March 2016; (2) Bpifrance Participations is entitled to propose the appointment of one member of the board of directors (Dr. Didier Hoch was appointed, but resigned on February 16, 2015), which member shall serve on one of the board’s committees, (3) Bpifrance Participations is entitled to propose the appointment of one non-voting observer (currently, Ms. Maïlys Ferrère), and (4) Bpifrance Participations is entitled to certain information rights, subject to applicable laws and regulation. This shareholders’ agreement has a 10-year term and will automatically terminate if Bpifrance Participations ceases to hold at least 50% of the shares it held upon the first listing of the company on Euronext Paris.
Agreements with Our Directors and Executive Officers
Employment Arrangements
Pierre-Henri Benhamou
Dr. Benhamou, our Chief Executive Officer, does not have an employment agreement with the company. His compensation is determined annually by the board of directors upon recommendation of the compensation committee. On September 25, 2012, our board of directors resolved thatto provide certain benefits to Dr. Benhamou, which were subsequently amended in casesApril 6, 2016 and are currently set as follows: in the case of (1) termination of Dr. Benhamou’s term as Chief Executive Officer of the company, not due tofor any reason whatsoever, except in cases of termination or non-renewal of Dr. Benhamou’s employment against his will resulting from a violation of the law or our By-laws or gross or severe negligence, or (2) non-renewal of Dr. Benhamou’s term against his will, and not due to a violation of the law or the our by-laws or gross or severe negligence, the company mayagrees to pay him severance, the gross amount of which will be equal to the sum of the gross compensation he received from the company, of any kind whatsoever, over the 18 months preceding his termination if at least two of the following three criteria are met as of the date of his termination: (a) a management structure is in place permitting sale or collaboration involving Viaskin Peanut, with this criteria being considered as met if, on the date of his termination, all of the following positions are actually filled: technical director, director of development, financial director, head of strategic marketing and head of research; (b) stock market capitalization of the company equals to at least €80 million; (c) at least three Viaskin projects are in the process of development. In accordance with article L225-42-1 of the French Commercial Code, this commitment of severance shall be submitted for shareholder approval at our ordinary shareholders’ general meeting to be held on June 21, 2016 within the terms of a specific resolution.
We have entered into employment agreements with the following executive officers:
Bertrand Dupont
In January 2003, we entered into an employment agreement with Mr. Dupont, our Chief Technical Officer,current Senior Executive Vice President, Technology, which was amended as of January 1, 2006. Mr. DupontHe is entitled to an annual base salary. Mr. Dupont is also eligible to receive equity grants as our board may determine and to participate in our bonus plan. In addition, Mr. Dupont’shis employment agreement provides for restrictions on certain competitive activities during the one-year period following the date of termination of his employment subject to the payment by us of a monthly compensation equal to 33% of the monthly gross salary paid to Mr. Dupont prior to his termination.
David Schilansky
In September 2011, we entered into an employment agreement with Mr. Schilansky, our Chief Operating Officer, with an effective date as of September 30, 2011. Mr. SchilanskyHe is entitled to an annual base salary. Mr. Schilansky is also eligible to receive equity grants as our board may determine and to participate in our bonus plan.
Charles Ruban
In May 2012, we entered into an employment agreement with Mr. Ruban, our then Chief Development Officer and current Chief Commercial Officer, with an effective date as of May 30, 2012. In July 2015, he entered into an employment agreement with our U.S.
subsidiary, with an effective date of August 31, 2015. Mr. RubanRuban’s French employment agreement has been suspended during the term of his U.S. employment agreement. He is entitled to an annual base salary. Mr. Ruban is also eligible to receive equity grants as our board may determine and to participate in our bonus plan. He also received a retention bonus and reimbursement of certain relocation expenses, and is eligible for reimbursement or payment of certain housing, travel and tuition costs. In the event of Mr. Ruban’s termination without “cause” or relocation back to France, he is eligible for certain additional benefits.
Laurent Martin
In July 2007, we entered into an employment agreement with Mr. Martin, our then Senior Executive Vice President, Product Strategy & Regulatory Affairs and current Chief Development Officer. Mr. Martin is entitled to an annual base salary. Mr. Martin is also eligible to receive equity grants as our board may determine and to participate in our bonus plan.
Director and Executive Officer Compensation
See “Item 6.B—Compensation of Directors and Executive Officers” for information regarding compensation of directors and executive officers.
Equity Awards
Since January 1, 2014,On March 24, 2015, we haveissued 10,000 non-employee warrants (BSAs), which were granted equity awards to certainMr. Soland. Each BSA has been issued at a purchase price per BSA of our directors€4.30, and executive officers.gives the right to Mr. Soland to subscribe for one ordinary share for a purchase price per share of €43.00.
On June 3, 2014,23, 2015, we issued 186,000120,000 option shares, which were granted to employees.
On September 30, 2015 we issued 708,500 free shares, including 60,000120,000 free shares granted to Dr. Benhamou and 90,000170,000 free shares granted to other executive officers.
On June 3, 2014,November 19, 2015, we issued 10,000195,000 option shares, including 120,000 option shares granted to executive officers.
On November 19, 2015, we issued 15,000 non-employee warrants (BSA) as follows: (1) 2,500 BSA(BSAs), which were granted to Dr. Bjerke, (2) 2,500 BSA granted to Mr. Horner, (3) 2,500 BSA granted to Mr. HuttMessrs. Soland and (4) 2,500 BSA granted to Dr. Hoch.Horner. Each BSA has been issued at a purchase price per BSA of €1.88,€6.60, and gives the holder the right to subscribe for one ordinary share for a purchase price per share of €18.79.€66.06.
On December 15, 2015, we issued 42,000 free shares, which were granted to employees.
On December 15, 2015, we issued 90,000 non-employee warrants (BSAs), which included 7,500 BSAs granted to Mr. Goller. Each BSA has been issued at a purchase price per BSA of €6.41, and gives the holder the right to subscribe for one ordinary share for a purchase price per share of €64.14.
See “Item. 7A—Major Shareholders” for information regarding equity awards to our executive officers.
Bonus Plans
All our executive officers are entitled to a bonus ranging between 40% and 50% based on yearly objectives determined by our board of directors upon recommendation of our compensation committee.
Indemnification Agreements
We entered into indemnification agreements with each of our directors and executive officers. See “Item. 6B—Limitations on Liability and Indemnification Matters.”
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers or persons controlling us pursuant to the foregoing provisions, we have been informed that in the opinion of the SEC such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable.
Related-Party Transactions Policy
We have adopted a related-party transaction policy that sets forth our procedures for the identification, review, consideration and approval or ratification of related-party transactions. For purposes of our policy only, a related-party transaction is a transaction, arrangement or relationship, or any series of similar transactions, arrangements or relationships, in which we and any related parties are, were or will be participants, which are not (1) in the ordinary course of business, (2) at arms’ length and (3) in which the amount involved exceeds $120,000. Transactions involving compensation for services provided to us as an employee or director are not covered by this policy. For purposes of this policy, a related party is any executive officer, director (or nominee for director) or beneficial owner of more than five percent (5%) of any class of our voting securities, including any of their immediate family members and any entity owned or controlled by such persons.
Under the policy, if a transaction has been identified as a related-party transaction, including any transaction that was not a related-party transaction when originally consummated or any transaction that was not initially identified as a related-party transaction prior to consummation, our management must present information regarding the related-party transaction to our board of directors for review, consideration and approval. The presentation must include a description of, among other things, the material facts, the interests, direct and indirect, of the related parties, the benefits to us of the transaction and whether the transaction is on terms that are comparable to the terms available to or from, as the case may be, an unrelated third-party or to or from employees generally. Under the policy, we will collect information that we deem reasonably necessary from each director, executive officer and, to the extent feasible, significant shareholder to enable us to identify any existing or potential related-party transactions and to effectuate the terms of the policy. In addition, under our Code of Business Conduct and Ethics, our employees and directors have an affirmative responsibility to disclose any transaction or relationship that reasonably could be expected to give rise to a conflict of interest. In considering related-party transactions, our board, or to the extent permitted by applicable law an independent body of our board, will take into account the relevant available facts and circumstances including, but not limited to:
The policy requires that, in determining whether to approve, ratify or reject a related-party transaction, our board of directors, or if permitted by applicable law an independent body of our board of directors, must consider, in light of known circumstances, whether the transaction is in, or is not inconsistent with, our best interests and those of our shareholders, as our board of directors, or if permitted by applicable law an independent body of our board of directors, determines in the good faith exercise of its discretion.
C. Interests of Experts and Counsel.
Not applicable.
| Item 8. | Financial Information |
| A. | Consolidated Statements and Other Financial Information. |
Consolidated Financial Statements
Our consolidated financial statements are appended at the end of this Annual Report, starting at page F-1, and incorporated herein by reference.
Dividend Distribution Policy
We have never declared or paid any cash dividends on our ordinary shares. We do not anticipate paying cash dividends on our equity securities in the foreseeable future and intend to retain all available funds and any future earnings for use in the operation and expansion of our business.
Subject to the requirements of French law and our by-laws, dividends may only be distributed from our distributable profits, plus any amounts held in our reserves other than those reserves that are specifically required by law. See “Item 10. B—Memorandum and Articles of Association” for further details on the limitations on our ability to declare and pay dividends. Dividend distributions, if any, will be made in euros and converted into U.S. dollars with respect to the ADSs, as provided in the deposit agreement.
Legal Proceedings
From time to time we may become involved in legal proceedings or be subject to claims arising in the ordinary course of our business. We are not presently a party to any legal proceedings that, if determined adversely to us, would individually or taken together have a material adverse effect on our business, results of operations, financial condition or cash flows. Regardless of the outcome, litigation can have an adverse impact on us because of defense and settlement costs, diversion of management resources and other factors.
Dividend Distribution Policy
We have never declared or paid any cash dividends on our ordinary shares. We do not anticipate paying cash dividends on our equity securities in the foreseeable future and intend to retain all available funds and any future earnings for use in the operation and expansion of our business.
Subject to the requirements of French law and our by-laws, dividends may only be distributed from our distributable profits, plus any amounts held in our reserves other than those reserves that are specifically required by law. See “Item 10. B—Memorandum and Articles of Association” for further details on the limitations on our ability to declare and pay dividends. Dividend distributions, if any, will be made in euros and converted into U.S. dollars with respect to the ADSs, as provided in the deposit agreement.
| B. | Significant Changes. |
On April 29, 2015, we reported that for the three months ended March 31, 2015, our total income was €1,453,706, up from €1,277,349 for the same period in 2014. This 13.8% increase partly resulted from an increase in Research Tax Credit, amounting to €1,275,595 over the period, compared to €1,227,140 in 2014, and also from the sales of Diallertest, which amounted to €107,520 for three months ended March 31, 2015, whilst no revenue was booked for the sales of Diallertest over the same period in 2014. We also reported on April 29, 2015 that as of March 31, 2015, our cash and cash equivalents amounted to €109.7 million, as compared with €114.6 million as of December 31, 2014. These amounts are preliminary in nature, have not been audited and are subject to change upon completion of audit for the full fiscal year 2015. Additional information and disclosures would be required for a more complete understanding of our financial position and results of operations for these periods.Not applicable.
| Item 9. | The Offer and Listing. |
| A. | Offer and Listing Details. |
The ADS have been listed on Nasdaq Global Select Market under the symbol “DBVT” since October 22, 2014. Prior to that date, there was no public trading market for ADSs. Our ordinary shares have been trading on Euronext Paris under the symbol “DBV” since March 28, 2012. Prior to that date, there was no public trading market for ADSs or our ordinary shares. Our global offering in October 2014 was priced at $21.64 per ADS or €34.00 per ordinary share on October 22, 2014. The following tables set forth for the periods indicated the reported high and low sale prices per ADS on Nasdaq Global Select Market in U.S. dollars and per ordinary share on Euronext Paris in euros.
Nasdaq Global Select Market
| Per ADS | ||||||||
| High | Low | |||||||
Year Ended December 31, 2014: | ||||||||
Fourth Quarter (beginning October 22) | $ | 28.50 | $ | 22.20 | ||||
Year Ended December 31, 2015: | ||||||||
First Quarter | $ | 28.40 | $ | 20.26 | ||||
Month Ended: | ||||||||
October 2014 (beginning October 22) | $ | 28.50 | $ | 22.20 | ||||
November 2014 | $ | 25.98 | $ | 23.20 | ||||
December 2014 | $ | 27.59 | $ | 25.16 | ||||
January 2015 | $ | 28.40 | $ | 24.00 | ||||
February 2015 | $ | 26.00 | $ | 21.50 | ||||
March 2015 | $ | 24.07 | $ | 20.26 | ||||
April 2015 (through April 24, 2015) | $ | 27.99 | $ | 23.02 | ||||
| Per ADS | ||||||||
| High | Low | |||||||
Annual | ||||||||
2014 (beginning October 22, 2014) | $ | 28.50 | $ | 22.20 | ||||
2015 | $ | 44.76 | $ | 20.26 | ||||
Quarterly | ||||||||
Fourth Quarter 2014 (beginning October 22, 2014) | $ | 28.50 | $ | 22.20 | ||||
First Quarter 2015 | $ | 28.40 | $ | 20.26 | ||||
Second Quarter 2015 | $ | 30.04 | $ | 23.02 | ||||
Third Quarter 2015 | $ | 44.76 | $ | 29.54 | ||||
Fourth Quarter 2015 | $ | 39.84 | $ | 32.21 | ||||
First Quarter 2016 | $ | 36.05 | $ | 22.55 | ||||
Month Ended: | ||||||||
November 2015 | $ | 37.30 | $ | 33.42 | ||||
December 2015 | $ | 36.71 | $ | 32.26 | ||||
January 2016 | $ | 36.05 | $ | 24.23 | ||||
February 2016 | $ | 27.29 | $ | 22.55 | ||||
March 2016 | $ | 32.94 | $ | 24.94 | ||||
April 2016 (through April 22, 2016) | $ | 37.11 | $ | 32.37 | ||||
Euronext Paris
Period | High | Low | ||||||
Annual | ||||||||
2012 | € | 9.74 | € | 7.19 | ||||
2013 | € | 12.50 | € | 7.51 | ||||
2014 | € | 44.88 | € | 10.78 | ||||
Quarterly | ||||||||
First Quarter 2013 | € | 9.75 | € | 7.90 | ||||
Second Quarter 2013 | € | 8.79 | € | 8.00 | ||||
Third Quarter 2013 | € | 8.28 | € | 7.51 | ||||
Fourth Quarter 2013 | € | 12.50 | € | 7.66 | ||||
First Quarter 2014 | € | 23.50 | € | 10.78 | ||||
Second Quarter 2014 | € | 21.20 | € | 14.61 | ||||
Third Quarter 2014 | € | 37.27 | € | 18.37 | ||||
Fourth Quarter 2014 | € | 44.88 | € | 32.01 | ||||
First Quarter 2015 | € | 47.23 | € | 37.57 | ||||
Month Ended | ||||||||
October 2014 | € | 41.43 | € | 32.01 | ||||
November 2014 | € | 41.35 | € | 37.28 | ||||
December 2014 | € | 44.88 | € | 40.26 | ||||
January 2015 | € | 47.23 | € | 41.40 | ||||
February 2015 | € | 42.85 | € | 37.57 | ||||
March 2015 | € | 43.70 | € | 37.65 | ||||
April 2015 (through April 24, 2015) | € | 53.47 | € | 42.07 | ||||
Period | High | Low | ||||||
Annual | ||||||||
2012 (beginning March 28, 2012) | € | 9.74 | € | 7.19 | ||||
2013 | € | 12.50 | € | 7.51 | ||||
2014 | € | 44.88 | € | 10.78 | ||||
2015 | € | 83.48 | € | 37.57 | ||||
Quarterly | ||||||||
First Quarter 2014 | € | 23.50 | € | 10.78 | ||||
Second Quarter 2014 | € | 21.20 | € | 14.61 | ||||
Third Quarter 2014 | € | 37.27 | € | 18.37 | ||||
Fourth Quarter 2014 | € | 44.88 | € | 32.01 | ||||
First Quarter 2015 | € | 47.23 | € | 37.57 | ||||
Second Quarter 2015 | € | 53.47 | € | 42.01 | ||||
Third Quarter 2015 | € | 83.48 | € | 52.15 | ||||
Fourth Quarter 2015 | € | 73.00 | € | 57.27 | ||||
First Quarter 2016 | € | 66.79 | € | 38.69 | ||||
Month Ended | ||||||||
November 2015 | € | 69.80 | € | 62.09 | ||||
December 2015 | € | 70.99 | € | 58.26 | ||||
January 2016 | € | 66.79 | € | 42.61 | ||||
February 2016 | € | 49.89 | € | 38.69 | ||||
March 2016 | € | 57.99 | € | 45.58 | ||||
April 2016 (through April 22, 2016) | € | 64.89 | € | 56.82 | ||||
On April 24, 2015,22, 2016, the last reported saleclosing price of the ADSs on Nasdaq was $26.29$35.10 per ADS, and the last reported saleclosing price of the ordinary shares on Euronext Paris was €47.99€62.48 per share.
| B. | Plan of Distribution. |
Not applicable.
| C. | Markets. |
The ADS have been listed on Nasdaq under the symbol “DBVT” since October 22, 2014 and our ordinary shares have been listed on the Euronext Paris under the symbol “DBV” since March 28, 2012.
| D. | Selling Shareholders. |
Not applicable.
| E. | Dilution. |
Not applicable.
| F. | Expenses of the Issue. |
Not applicable.
| Item 10. | Additional Information. |
| A. | Share Capital. |
Not applicable.
| B. | Memorandum and Articles of Association. |
The information set forth in our prospectus dated October 22, 2014,July 15, 2015, filed with the SEC pursuant to Rule 424(b), under the headings “Description of Share Capital—Key Provisions of Our By-laws and French Law Affecting Our Ordinary Shares,” “Description of Share Capital—Differences in Corporate Law” and “Limitations Affecting Shareholders of a French Company” is incorporated herein by reference.
| C. | Material Contracts. |
Underwriting Agreements
We entered into an underwriting agreement among Citigroup Global Markets Inc. and Leerink Partners LLC, as representatives of the underwriters, on October 22, 2014, with respect to the ADSs and ordinary shares sold in our October 2014 global offering. We have agreed to indemnify the underwriters against certain liabilities, including liabilities under the Securities Act, and to contribute to payments the underwriters may be required to make in respect of such liabilities.
We entered into an underwriting agreement among Citigroup Global Markets, Inc., Morgan Stanley & Co. LLC, Barclays Capital Inc. and Leerink Partners LLC, as representatives of the underwriters, on July 14, 2015, with respect to the ADSs sold in our July 2015 underwritten public offering. We have agreed to indemnify the underwriters against certain liabilities, including liabilities under the Securities Act, and to contribute to payments the underwriters may be required to make in respect of such liabilities.
For additional information on our material contracts, please see “Item 4. Information on the Company,” Item 46. Directors, Senior Management and Item 6Employees,” and “Item 7.B. Related Party Transactions” of this Annual Report on 20-F.
| D. | Exchange Controls. |
Under current French foreign exchange control regulations there are no limitations on the amount of cash payments that we may remit to residents of foreign countries. Laws and regulations concerning foreign exchange controls do, however, require that all payments or transfers of funds made by a French resident to a non-resident such as dividend payments be handled by an accredited intermediary. All registered banks and substantially all credit institutions in France are accredited intermediaries.
| E. | Taxation. |
Certain Material U.S. Federal Income Tax Considerations
The following is a summary of certain material U.S. federal income tax considerations relating to the ownership and disposition of ADSs by a U.S. holder (as defined below). This summary addresses only the U.S. federal income tax considerations for U.S. holders of the ADSs that will hold such ADSs as capital assets. This summary does not address all U.S. federal income tax matters that may be relevant to a particular U.S. holder. This summary does not address tax considerations applicable to a holder of ADSs that may be subject to special tax rules including, without limitation, the following:
Further, this summary does not address the U.S. federal estate, gift, or alternative minimum tax considerations, or any U.S. state, local, or non-U.S. tax considerations of the ownership and disposition of the ADSs.
This description is based on the U.S. Internal Revenue Code of 1986, as amended (the “Code”), existing, proposed and temporary U.S. Treasury Regulations promulgated thereunder and administrative and judicial interpretations thereof, in each case as in effect and available on the date hereof. All the foregoing is subject to change, which change could apply retroactively, and to differing interpretations, all of which could affect the tax considerations described below. There can be no assurances that the U.S. Internal Revenue Service (the “IRS”) will not take a position concerning the tax consequences of the ownership and disposition of the ADSs or that such a position would not be sustained. Holders should consult their own tax advisers concerning the U.S. federal, state, local and non-U.S. tax consequences of owning and disposing of the ADSs in their particular circumstances.
For the purposes of this summary, a “U.S. holder” is a beneficial owner of ADSs that is (or is treated as), for U.S. federal income tax purposes:
If a partnership (or any other entity treated as a partnership for U.S. federal income tax purposes) holds ADSs, the U.S. federal income tax consequences relating to an investment in the ADSs will depend in part upon the status of the partner and the activities of the partnership. Such a partner or partnership should consult its tax advisor regarding the U.S. federal income tax considerations of owning and disposing of the ADSs in its particular circumstances.
As indicated below, this discussion is subject to U.S. federal income tax rules applicable to a “passive foreign investment company,” or a PFIC.
Persons considering an investment in the ADSs should consult their own tax advisors as to the particular tax consequences applicable to them relating to the ownership and disposition of the ADSs, including the applicability of U.S. federal, state and local tax laws and non-U.S. tax laws.
Distributions. Subject to the discussion under “Passive Foreign Investment Company Considerations,” below, the gross amount of any distribution (before reduction for any amounts withheld in respect of French withholding tax) actually or constructively received by a U.S. holder with respect to ADSs will be taxable to the U.S. holder as a dividend to the extent of the U.S. holder’s pro rata share of our current and accumulated earnings and profits as determined under U.S. federal income tax principles. Distributions in excess of earnings and profits will be non-taxable to the U.S. holder to the extent of, and will be applied against and reduce, the U.S. holder’s adjusted tax basis in the ADSs. Distributions in excess of earnings and profits and such adjusted tax basis will generally be taxable to the U.S. holder as either long-term or short-term capital gain depending upon whether the U.S. holder has held the ADSs for more
than one year as of the time such distribution is received. However, since we do not calculate our earnings and profits under U.S. federal income tax principles, it is expected that any distribution will be reported as a dividend, even if that distribution would otherwise be treated as a non-taxable return of capital or as capital gain under the rules described above. Non-corporate U.S. holders may qualify for the preferential rates of taxation with respect to dividends on ADSs applicable to long-term capital gains (i.e., gains from the sale of capital assets held for more than one year) applicable to qualified dividend income (as discussed below) if we are a “qualified foreign corporation” and certain other requirements (discussed below) are met. A non-United States corporation (other than a corporation that is classified as a PFIC for the taxable year in which the dividend is paid or the preceding taxable year) generally will be considered to be a qualified foreign corporation (a) if it is eligible for the benefits of a comprehensive tax treaty with the United States which the Secretary of Treasury of the United States determines is satisfactory for purposes of this provision and which includes an exchange of information provision, or (b) with respect to any dividend it pays on ADSs which are readily tradable on an established securities market in the United States. The ADSs are listed on the Nasdaq Global Select Market, which is an established securities market in the United States, and we expect the ADSs to be readily tradable on the Nasdaq Global Select Market. However, there can be no assurance that the ADSs will be considered readily tradable on an established securities market in the United States in the current taxable year or later years. The company, which is incorporated under the laws of France, believes that it qualifies as a resident of France for purposes of, and is eligible for the benefits of, the Convention between the Government of the United States of America and the Government of the French Republic for the Avoidance of Double Taxation and the Prevention of Fiscal Evasion with Respect to Taxes on Income and Capital, signed on August 31, 1994, as amended and currently in force, or the U.S.-France Tax Treaty, although there can be no assurance in this regard. Further, the IRS has determined that the U.S.-France Tax Treaty is
satisfactory for purposes of the qualified dividend rules and that it includes an exchange-of-information program. Therefore, subject to the discussion under “Passive Foreign Investment Company Considerations,” below, such dividends will generally be “qualified dividend income” in the hands of individual U.S. holders, provided that a holding period requirement (more than 60 days of ownership, without protection from the risk of loss, during the 121-day period beginning 60 days before the ex-dividend date) and certain other requirements are met. The dividends will not be eligible for the dividends-received deduction generally allowed to corporate U.S. holders.
A U.S. holder generally may claim the amount of any French withholding tax as either a deduction from gross income or a credit against U.S. federal income tax liability. However, the foreign tax credit is subject to numerous complex limitations that must be determined and applied on an individual basis. Generally, the credit cannot exceed the proportionate share of a U.S. holder’s U.S. federal income tax liability that such U.S. holder’s taxable income bears to such U.S. holder’s worldwide taxable income. In applying this limitation, a U.S. holder’s various items of income and deduction must be classified, under complex rules, as either “foreign source” or “U.S. source.” In addition, this limitation is calculated separately with respect to specific categories of income. The amount of a distribution with respect to the ADSs that is treated as a “dividend” may be lower for U.S. federal income tax purposes than it is for French income tax purposes, potentially resulting in a reduced foreign tax credit for the U.S. holder. Each U.S. holder should consult its own tax advisors regarding the foreign tax credit rules.
In general, the amount of a distribution paid to a U.S. holder in a foreign currency will be the dollar value of the foreign currency calculated by reference to the spot exchange rate on the day the U.S. holder receives the distribution, regardless of whether the foreign currency is converted into U.S. dollars at that time. Any foreign currency gain or loss a U.S. holder realizes on a subsequent conversion of foreign currency into U.S. dollars will be U.S. source ordinary income or loss. If dividends received in a foreign currency are converted into U.S. dollars on the day they are received, a U.S. holder should not be required to recognize foreign currency gain or loss in respect of the dividend.
Sale, Exchange or Other Taxable Disposition of the ADSs. A U.S. holder will generally recognize gain or loss for U.S. federal income tax purposes upon the sale, exchange or other taxable disposition of ADSs in an amount equal to the difference between the U.S. dollar value of the amount realized from such sale or exchange and the U.S. holder’s tax basis for those ADSs. Subject to the discussion under “Passive Foreign Investment Company Considerations” below, this gain or loss will generally be a capital gain or loss. The adjusted tax basis in the ADSs generally will be equal to the cost of such ADSs. Capital gain from the sale, exchange or other taxable disposition of ADSs of a non-corporate U.S. holder is generally eligible for a preferential rate of taxation applicable to capital gains, if the non-corporate U.S. holder’s holding period determined at the time of such sale, exchange or other taxable disposition for such ADSs exceeds one year (i.e., such gain is long-term taxable gain). The deductibility of capital losses for U.S. federal income tax purposes is subject to limitations under the Code. Any such gain or loss that a U.S. holder recognizes generally will be treated as U.S. source income or loss for foreign tax credit limitation purposes.
For a cash basis taxpayer, units of foreign currency paid or received are translated into U.S. dollars at the spot rate on the settlement date of the purchase or sale. In that case, no foreign currency exchange gain or loss will result from currency fluctuations between the trade date and the settlement date of such a purchase or sale. An accrual basis taxpayer, however, may elect the same treatment
required of cash basis taxpayers with respect to purchases and sales of the ADSs that are traded on an established securities market, provided the election is applied consistently from year to year. Such election may not be changed without the consent of the IRS. For an accrual basis taxpayer who does not make such election, units of foreign currency paid or received are translated into U.S. dollars at the spot rate on the trade date of the purchase or sale. Such an accrual basis taxpayer may recognize exchange gain or loss based on currency fluctuations between the trade date and the settlement date. Any foreign currency gain or loss a U.S. Holder realizes will be U.S. source ordinary income or loss.
Medicare Tax. Certain U.S. holders that are individuals, estates or trusts are subject to a 3.8% tax on all or a portion of their “net investment income,” which may include all or a portion of their dividend income and net gains from the disposition of ADSs. Each U.S. holder that is an individual, estate or trust is urged to consult its tax advisors regarding the applicability of the Medicare tax to its income and gains in respect of its investment in the ADSs.
Passive Foreign Investment Company Considerations. If we are classified as a passive foreign investment company (“PFIC”) in any taxable year, a U.S. holder would be subject to special rules generally intended to reduce or eliminate any benefits from the deferral of U.S. federal income tax that a U.S. holder could derive from investing in a non-U.S. company that does not distribute all of its earnings on a current basis.
A corporation organized outside the United States generally will be classified as a PFIC for U.S. federal income tax purposes in any taxable year in which, after applying certain look-through rules with respect to the income and assets of its subsidiaries, either: (i) at least 75% of its gross income is “passive income” or (ii) at least 50% of the average quarterly value of its total gross assets (which, if we are not a controlled foreign corporation or are publicly traded for the entire year being tested, would be measured by fair market value of our assets, and for which purpose the total value of our assets may be determined in part by the market value of the ADSs and our ordinary shares, which are subject to change) is attributable to assets that produce “passive income” or are held for the production of “passive income.”
Passive income for this purpose generally includes dividends, interest, royalties, rents, gains from commodities and securities transactions, the excess of gains over losses from the disposition of assets which produce passive income, and includes amounts derived by reason of the temporary investment of funds raised in offerings of the ADSs. If a non-U.S. corporation owns directly or indirectly at least 25% by value of the stock of another corporation, the non-U.S. corporation is treated for purposes of the PFIC tests as owning its proportionate share of the assets of the other corporation and as receiving directly its proportionate share of the other corporation’s income. If we are classified as a PFIC in any year with respect to which a U.S. holder owns the ADSs, we will continue to be treated as a PFIC with respect to such U.S. holder in all succeeding years during which the U.S. holder owns the ADSs, regardless of whether we continue to meet the tests described above.
The market value of our assets may be determined in large part by reference to the market price of the ADSs and our ordinary shares, which is likely to fluctuate after the offering. In addition, the composition of our income and assets will be affected by how, and how quickly, we use the cash proceeds from the global offering in our business. Whether we are a PFIC for any taxable year will depend on our assets and income in each year, and because this is a factual determination made annually after the end of each taxable year, there can be no assurance that we will not be considered a PFIC in any taxable year. Although it is not free from doubt, based on the composition of our income for our 20142015 taxable year, we believe that we were not a PFIC for our 20142015 taxable year and do not expect to be a PFIC for our 2016 taxable year.
If we are a PFIC, and you are a U.S. holder, then unless you make one of the elections described below, a special tax regime will apply to both (a) any “excess distribution” by us to you (generally, your ratable portion of distributions in any year which are greater than 125% of the average annual distribution received by you in the shorter of the three preceding years or your holding period for the ADSs) and (b) any gain realized on the sale or other disposition of the ADSs. Under this regime, any excess distribution and realized gain will be treated as ordinary income and will be subject to tax as if (a) the excess distribution or gain had been realized ratably over your holding period, (b) the amount deemed realized in each year had been subject to tax in each year of that holding period at the highest marginal rate for such year (other than income allocated to the current period or any taxable period before we became a PFIC, which would be subject to tax at the U.S. holder’s regular ordinary income rate for the current year and would not be subject to the interest charge discussed below), and (c) the interest charge generally applicable to underpayments of tax had been imposed on the taxes deemed to have been payable in those years. In addition, dividend distributions made to you will not qualify for the lower rates of taxation applicable to long-term capital gains discussed above under “Distributions.”
Certain elections exist that may alleviate some of the adverse consequences of PFIC status and would result in an alternative treatment (such as mark-to-market treatment) of the ADSs. If a U.S. holder makes the mark-to-market election, the U.S. holder generally will recognize as ordinary income any excess of the fair market value of the ADSs at the end of each taxable year over their adjusted tax basis, and will recognize an ordinary loss in respect of any excess of the adjusted tax basis of the ADSs over their fair market value at
the end of the taxable year (but only to the extent of the net amount of income previously included as a result of the mark-to-market election). If a U.S. holder makes the election, the U.S. holder’s tax basis in the ADSs will be adjusted to reflect these income or loss amounts. Any gain recognized on the sale or other disposition of ADSs in a year when we are a PFIC will be treated as ordinary income and any loss will be treated as an ordinary loss (but only to the extent of the net amount of income previously included as a result of the mark-to-market election). The mark-to-market election is available only if we are a PFIC and the ADSs are “regularly traded” on a “qualified exchange.” The ADSs will be treated as “regularly traded” in any calendar year in which more than a de minimis quantity of the ADSs are traded on a qualified exchange on at least 15 days during each calendar quarter (subject to the rule that trades that have as one of their principal purposes the meeting of the trading requirement as disregarded). The Nasdaq Global Select Market is a qualified exchange for this purpose and, consequently, if the ADSs are regularly traded, the mark-to-market election will be available to a U.S. holder.
We do not currently intend to provide the information necessary for U.S. holders to make qualified electing fund elections if we wereare treated as a PFIC for any taxable year. U.S. Holders should consult their tax advisors to determine whether any of these elections would be available and if so, what the consequences of the alternative treatments would be in their particular circumstances.
If we are determined to be a PFIC, the general tax treatment for U.S. Holders described in this section would apply to indirect distributions and gains deemed to be realized by U.S. Holders in respect of any of our subsidiaries that also may be determined to be PFICs.
If a U.S. holder owns ADSs during any taxable year in which we are a PFIC, the U.S. holder generally will be required to file an IRS Form 8621 (Information Return by a Shareholder of a Passive Foreign Investment Company or Qualified Electing Fund) with respect to the company, generally with the U.S. holder’s federal income tax return for that year. If our company were a PFIC for a given taxable year, then you should consult your tax advisor concerning your annual filing requirements.
The U.S. federal income tax rules relating to PFICs are complex. Prospective U.S. investors are urged to consult their own tax advisers with respect to the ownership and disposition of the ADSs, the consequences to them of an investment in a PFIC, any elections available with respect to the ADSs and the IRS information reporting obligations with respect to the ownership and disposition of the ADSs.
Backup Withholding and Information Reporting. U.S. holders generally will be subject to information reporting requirements with respect to dividends on ADSs and on the proceeds from the sale, exchange or disposition of ADSs that are paid within the United States or through U.S.-related financial intermediaries, unless the U.S. holder is an “exempt recipient.” In addition, U.S. holders may be subject to backup withholding on such payments, unless the U.S. holder provides a taxpayer identification number and a duly executed IRS Form W-9 or otherwise establishes an exemption. Backup withholding is not an additional tax, and the amount of any backup withholding will be allowed as a credit against a U.S. holder’s U.S. federal income tax liability and may entitle such holder to a refund, provided that the required information is timely furnished to the IRS.
Foreign Asset Reporting. Certain U.S. holders who are individuals are required to report information relating to an interest in the ADSs, subject to certain exceptions (including an exception for shares held in accounts maintained by U.S. financial institutions) by filing IRS Form 8938 (Statement of Specified Foreign Financial Assets) with their federal income tax return. U.S. holders are urged to consult their tax advisors regarding their information reporting obligations, if any, with respect to their ownership and disposition of the ADSs.
THE DISCUSSION ABOVE IS A GENERAL SUMMARY. IT DOES NOT COVER ALL TAX MATTERS THAT MAY BE OF IMPORTANCE TO A PROSPECTIVE INVESTOR. EACH PROSPECTIVE INVESTOR IS URGED TO CONSULT ITS OWN TAX ADVISOR ABOUT THE TAX CONSEQUENCES TO IT OF AN INVESTMENT IN ADSS IN LIGHT OF THE INVESTOR’S OWN CIRCUMSTANCES.
French Tax Consequences
The following describes the material French income tax consequences to U.S. Holders (as defined below) of purchasing, owning and disposing of our ADSs and, unless otherwise noted, this discussion is the opinion of Gide Loyrette Nouel A.A.R.P.I, our French tax counsel, insofar as it relates to matters of French tax law and legal conclusions with respect to those matters.
This discussion does not purport to be a complete analysis or listing of all potential tax effects of the acquisition, ownership or disposition of our ADSs to any particular investor, and does not discuss tax considerations that arise from rules of general application or that are generally assumed to be known by investors. All of the following is subject to change. Such changes could apply retroactively and could affect the consequences described below.
France has recently introduced a comprehensive set of new tax rules applicable to French assets that are held by or in foreign trusts. These rules provide inter alia for the inclusion of trust assets in the settlor’s net assets for purpose of applying the French wealth tax, for the application of French gift and death duties to French assets held in trust, for a specific tax on capital on the French assets of foreign trusts not already subject to the French wealth tax and for a number of French tax reporting and disclosure obligations. The following discussion does not address the French tax consequences applicable to ADSs held in trusts. If ADSs are held in trust, the grantor, trustee and beneficiary are urged to consult their own tax adviser regarding the specific tax consequences of acquiring, owning and disposing of securities.
The description of the French income tax and wealth tax consequences set forth below is based on the Convention Between the Government of the United States of America and the Government of the French Republic for the Avoidance of Double Taxation and the Prevention of Fiscal Evasion with Respect to Taxes on Income and Capital of August 31, 1994, or the Treaty, which came into force on December 30, 1995 (as amended by any subsequent protocols, including the protocol of January 13, 2009), and the tax guidelines issued by the French tax authorities in force as of the date of this Annual Report on Form 20-F (the “Treaty”).
For the purposes of this discussion, a “U.S. holder” is a beneficial owner of ADSs that is (or is treated as), for U.S. federal income tax purposes: (1) an individual who is a citizen or resident of the United States; (2) a corporation, or other entity that is treated as a corporation for U.S. federal income tax purposes, created or organized in or under the laws of the United States, any state thereof, or the District of Columbia; (3) an estate, the income of which is subject to U.S. federal income taxation regardless of its source; or (4) a trust, if a court within the United States is able to exercise primary supervision over its administration and one or more U.S. persons have the authority to control all of the substantial decisions of such trust or has a valid election in effect under applicable U.S. Treasury Regulations to be treated as a United States person.
If a partnership (or any other entity treated as partnership for U.S. federal income tax purposes) holds ADSs, the tax treatment of the partnership and a partner in such partnership generally will depend on the status of the partner and the activities of the partnership. Such partner or partnership is urged to consult its own tax adviser regarding the specific tax consequences of acquiring, owning and disposing of securities.
This discussion applies only to investors that hold ADS as capital assets that have the U.S. dollar as their functional currency, that are entitled to Treaty benefits under the ‘‘Limitation on Benefits’’ provision contained in the Treaty, and whose ownership of the ADS is not effectively connected to a permanent establishment or a fixed base in France. Certain U.S. Holders (including, but not limited to, U.S. expatriates, partnerships or other entities classified as partnerships for U.S. federal income tax purposes, banks, insurance companies, regulated investment companies, tax-exempt organizations, financial institutions, persons subject to the alternative minimum tax, persons who acquired the securities pursuant to the exercise of employee share options or otherwise as compensation, persons that own (directly, indirectly or by attribution) 5% or more of our voting stock or 5% or more of our outstanding share capital, dealers in securities or currencies, persons that elect to mark their securities to market for U.S. federal income tax purposes and persons holding securities as a position in a synthetic security, straddle or conversion transaction) may be subject to special rules not discussed below.
U.S. Holders are urged to consult their own tax advisers regarding the tax consequences of the purchase, ownership and disposition of securities in light of their particular circumstances, especially with regard to the ‘‘Limitations on Benefits’’ provision.
Estate and Gift Taxes and Transfer Taxes
In general, a transfer of securities by gift or by reason of death of a U.S. Holder that would otherwise be subject to French gift or inheritance tax, respectively, will not be subject to such French tax by reason of the Convention between the Government of the United States of America and the Government of the French Republic for the Avoidance of Double Taxation and the Prevention of
Fiscal Evasion with Respect to Taxes on Estates, Inheritances and Gifts, dated November 24, 1978, unless the donor or the transferor is domiciled in France at the time of making the gift or at the time of his or her death, or the securities were used in, or held for use in, the conduct of a business through a permanent establishment or a fixed base in France.
Pursuant to Article 235 ter ZD of theCode général des impôts (French Tax Code, or FTC), purchases of shares or ADSs of a French company listed on a regulated market of the European Union or an exchange formally acknowledged by the French Financial Market Authority (AMF) are subject to a 0.2% French tax on financial transactions provided that the issuer’s market capitalization exceeds 1 billion euros as of December 1 of the year preceding the taxation year. Nasdaq is not currently acknowledged by the French AMF but this may change in the future. A list of French relevant companies whose market capitalization exceeds 1 billion euros as of December 1 of the year preceding the taxation year is published annually by the French State.
Purchases of our securities may be subject to such tax provided that its market capitalization exceeds 1 billion euros and that Nasdaq is acknowledged by the French AMF.
In the case where Article 235 ter ZD of the FTC is not applicable, transfers of shares issued by a listed French company will be subject to uncapped registration duties at the rate of 0.1% if the transfer is evidenced by a written statement (“acte”) executed either in France or outside France.
Wealth Tax
The French wealth tax (impôt de solidarité sur la fortune) applies only to individuals and does not generally apply to securities held by an eligible U.S. Holder who is a U.S. resident, as defined pursuant to the provisions of the Treaty, provided that such U.S. Holder does not own directly or indirectly more than 25% of the issuer’s financial rights.
Taxation of Dividends
Dividends paid by a French corporation to non-residents of France are generally subject to French withholding tax at a rate of 30%. Dividends paid by a French corporation in a non-cooperative State or territory, as defined in Article 238-0 A of the FTC, will generally be subject to French withholding tax at a rate of 75%. However, eligible U.S. Holders entitled to Treaty benefits under the ‘‘Limitation on Benefits’’ provision contained in the Treaty who are U.S. residents, as defined pursuant to the provisions of the Treaty, will not be subject to this 30% or 75% withholding tax rate, but may be subject to the withholding tax at a reduced rate (as described below).
Under the Treaty, the rate of French withholding tax on dividends paid to an eligible U.S. Holder who is a U.S. resident as defined pursuant to the provisions of the Treaty and whose ownership of the ordinary shares or ADSs is not effectively connected with a permanent establishment or fixed base that such U.S. Holder has in France, is generally reduced to 15%, or to 5% if such U.S. Holder is a corporation and owns directly or indirectly at least 10% of the share capital of the issuer; such U.S. Holder may claim a refund from the French tax authorities of the amount withheld in excess of the Treaty rates of 15% or 5%, if any.
For U.S. Holders that are not individuals but are U.S. residents, as defined pursuant to the provisions of the Treaty, the requirements for eligibility for Treaty benefits, including the reduced 5% or 15% withholding tax rates contained in the ‘‘Limitation on Benefits’’ provision of the Treaty, are complicated, and certain technical changes were made to these requirements by the protocol of January 13, 2009. U.S. Holders are advised to consult their own tax advisers regarding their eligibility for Treaty benefits in light of their own particular circumstances.
Dividends paid to an eligible U.S. Holder may immediately be subject to the reduced rates of 5% or 15% provided that such holder establishes before the date of payment that it is a U.S. resident under the Treaty by completing and providing the depositary with a treaty form (Form 5000). Dividends paid to a U.S. Holder that has not filed the Form 5000 before the dividend payment date will be subject to French withholding tax at the rate of 30%, or 75% if paid in a non-cooperative State or territory (as defined in Article 238-0 A of the FTC), and then reduced at a later date to 5% or 15%, provided that such holder duly completes and provides the French tax authorities with the treaty forms Form 5000 and Form 5001 before December 31 of the second calendar year following the year during which the dividend is paid. Certain qualifying pension funds and certain other tax-exempt entities are subject to the same general filing requirements as other U.S. Holders except that they may have to supply additional documentation evidencing their entitlement to these benefits.
Form 5000 and Form 5001, together with instructions, will be provided by the depositary to all U.S. Holders registered with the depositary. The depositary will arrange for the filing with the French Tax authorities of all such forms properly completed and executed by U.S. Holders of ordinary shares or ADSs and returned to the depositary in sufficient time so that they may be filed with the French tax authorities before the distribution in order to obtain immediately a reduced withholding tax rate.
The withholding tax refund, if any, ordinarily occurs within 12 months from filing the applicable French Treasury Form, but not before January 15 of the year following the calendar year in which the related dividend was paid.
Tax on Sale or Other Disposition
In general, under the Treaty, a U.S. Holder who is a U.S. resident for purposes of the Treaty will not be subject to French tax on any capital gain from the redemption (other than redemption proceeds characterized as dividends under French domestic tax law or administrative guidelines), sale or exchange of ordinary shares or ADSs unless the ordinary shares or the ADSs form part of the business property of a permanent establishment or fixed base that the U.S. Holder has in France. Special rules apply to U.S. Holders who are residents of more than one country.
| F. | Dividends and Paying Agents. |
Not applicable.
| G. | Statement by Experts. |
Not applicable.
| H. | Documents on Display. |
We are subject to the information reporting requirements of the Exchange Act applicable to foreign private issuers and under those requirements will file reports with the SEC. Those reports may be inspected without charge at the locations described below. As a foreign private issuer, we are exempt from the rules under the Exchange Act related to the furnishing and content of proxy statements, and our officers, directors and principal shareholders are exempt from the reporting and short-swing profit recovery provisions contained in Section 16 of the Exchange Act. In addition, we are not required under the Exchange Act to file periodic reports and financial statements with the SEC as frequently or as promptly as United States companies whose securities are registered under the Exchange Act. Nevertheless, we will file with the SEC an Annual Report on Form 20-F containing financial statements that have been examined and reported on, with and opinion expressed by an independent registered public accounting firm.
We maintain a corporate website atwww.dbv-technologies.com. We intend to post our Annual Report on Form 20-F on our website promptly following it being filed with the SEC. Information contained on, or that can be accessed through, our website does not constitute a part of this Annual Report on Form 20-F. We have included our website address in this Annual Report on Form 20-F solely as an inactive textual reference.
You may also review a copy of this Annual Report on Form 20-F, including exhibits and any schedule filed herewith, and obtain copies of such materials at prescribed rates, at the SEC’s Public Reference Room in Room 1580, 100 F Street, NE, Washington, D.C. 20549-0102. You may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. The Securities and Exchange Commission maintains a website (www.sec.gov) that contains reports, proxy and information statements and other information regarding registrants, such as DBV Technologies, that file electronically with the SEC.
With respect to references made in this Annual Report on Form 20-F to any contract or other document of DBV Technologies,our company, such references are not necessarily complete and you should refer to the exhibits attached or incorporated by reference to this Annual Report on Form 20-F for copies of the actual contract or document.
| I. | Subsidiary Information. |
Not applicable.required.
| Item 11. | Quantitative and Qualitative Disclosures About Market Risk. |
Interest Rate Risk
We seek to engage in prudent management of our cash and cash equivalents, mainly cash on hand and common financial instruments (typically short- and mid-term deposits). Furthermore, the interest rate risk related to cash, cash equivalents and common financial instruments is not significant based on the quality of the financial institutions with which we work.
Foreign Currency Exchange Risk
We are exposed to foreign exchange risk inherent in some of our supplies obtained in the United States, which have been invoiced in U.S. dollars. As of this date, we do not have revenues in dollars nor in any other currency other than the euro. Due to the relatively low level of these expenditures we believe our exposure to foreign exchange risk is unlikely to have a material adverse impact on our results of operations or financial position. Our exposure to currencies other than the U.S. dollar is negligible.
For 2012, 2013, 2014 and 2014,2015, less than 11%10%, 10%7% and 7%, respectively, of our purchases and other external expenses have been made in U.S. dollars, generating a negligible net annual foreign exchange loss of €1,502, €2,831€3 thousands, €24 thousands and €24,337,€7 thousands, respectively, for those periods. In light of these insignificant amounts, we have not adopted, at this stage, a hedging mechanism in order to protect our business activity against fluctuations in exchange rates. We cannot rule out the possibility that a significant increase in our business, particularly in the United States, may result in greater exposure to exchange rate risk and would then consider adopting an appropriate policy for hedging against these risks.
Liquidity Risk
As of this date, we do not believe that we are exposed to a short-term (12 months) liquidity risk, considering the cash and cash equivalents that we have available as of December 31, 20142015 of €114.6€323.4 million, which are mainly composed of cash and term deposits that are convertible into cash immediately without penalties in case of a need for cash.
Moreover, we believe that the net proceeds of the globalour July 2015 underwritten public offering, together with our existing cash and cash equivalents, will enable us to fund our operating expenses and capital expenditure requirements until the end of 2016.2017.
Inflation Risk
We do not believe that inflation has had a material effect on our business, financial condition or results of operations. If our costs were to become subject to significant inflationary pressures, we may not be able to fully offset such higher costs through price increases. Our inability or failure to do so could harm our business, financial condition and results of operations.
| Item 12. | Description of Securities Other than Equity Securities. |
| A. | Debt Securities. |
Not applicable.
| B. | Warrants and Rights. |
Not applicable.
| C. | Other Securities. |
Not applicable.
| D. | American Depositary Shares. |
Citibank, N.A., as depositary, registers and delivers American Depositary Shares, also referred to as ADSs. Each ADS represents one-half of one ordinary share (or a right to receive one-half of one ordinary share) deposited with Citibank International Plc, located at EGSP 186, 1 North Wall Quay, Dublin 1 Ireland or any successor, as custodian for the depositary. Each ADS will also represent any other securities, cash or other property which may be held by the depositary in respect of the depositary facility. The depositary’s corporate trust office at which the ADSs will be administered is located at 388 Greenwich Street, New York, New York 10013.
A deposit agreement among us, the depositary and the ADS holders sets out the ADS holder rights as well as the rights and obligations of the depositary. New York law governs the deposit agreement and the ADSs. A copy of the Agreementdeposit agreement is incorporated by reference as an exhibit to this Annual Report on Form 20-F.
Fees and Charges
Pursuant to the terms of the deposit agreement, the holders of ADSs will be required to pay the following fees:
| Service | Fees | |
• Issuance of ADSs | Up to U.S. $0.05 per ADS issued | |
• Cancellation of ADSs | Up to U.S. $0.05 per ADS canceled | |
• Distribution of cash dividends or other cash distributions | Up to U.S. $0.05 per ADS held | |
• Distribution of ADSs pursuant to stock dividends, free stock distributions or exercise of rights. | Up to U.S. $0.05 per ADS held | |
• Distribution of securities other than ADSs or rights to purchase additional ADSs | Up to U.S. $0.05 per ADS held | |
• ADS Services | Up to U.S. $0.05 per ADS held on the applicable record date(s) established by the depositary | |
The holders of ADSs will also be responsible to pay certain fees and expenses incurred by the depositary and certain taxes and governmental charges such as:
| • | fees for the transfer and registration of ordinary shares charged by the registrar and transfer agent for the ordinary shares in France (i.e., upon deposit and withdrawal of ordinary shares); |
| • | taxes and duties upon the transfer of securities (i.e., when ordinary shares are deposited or withdrawn from deposit); and |
Depositary fees payable upon the issuance and cancellation of ADSs are typically paid to the depositary by the brokers (on behalf of their clients) receiving the newly issued ADSs from the depositary and by the brokers (on behalf of their clients) delivering the ADSs to the depositary for cancellation. The brokers in turn charge these fees to their clients. Depositary fees payable in connection with distributions of cash or securities to ADS holders and the depositary services fee are charged by the depositary to the holders of record of ADSs as of the applicable ADS record date.
The depositary fees payable for cash distributions are generally deducted from the cash being distributed. In the case of distributions other than cash (i.e., stock dividend, rights), the depositary charges the applicable fee to the ADS record date holders concurrent with the distribution. In the case of ADSs registered in the name of the investor (whether certificated or uncertificated in direct registration), the depositary sends invoices to the applicable record date ADS holders. In the case of ADSs held in brokerage and custodian accounts (via DTC), the depositary generally collects its fees through the systems provided by DTC (whose nominee is the registered holder of the ADSs held in DTC) from the brokers and custodians holding ADSs in their DTC accounts. The brokers and custodians who hold their clients’ ADSs in DTC accounts in turn charge their clients’ accounts the amount of the fees paid to the depositary.
In the event of refusal to pay the depositary fees, the depositary may, under the terms of the deposit agreement, refuse the requested service until payment is received or may set off the amount of the depositary fees from any distribution to be made to the ADS holder.
Note that the fees and charges the holders of ADSs may be required to pay may vary over time and may be changed by us and by the depositary. The holders of ADSs will receive prior notice of such changes.
The depositary may reimburse us for certain expenses incurred by us in respect of the ADR program established pursuant to the deposit agreement, by making available a portion of the depositary fees charged in respect of the ADR program or otherwise, upon such terms and conditions as we and the depositary may agree from time to time.
| Item 13. | Defaults, Dividend Arrearages and Delinquencies. |
Not applicable.
| Item 14. | Material Modifications to the Rights of Security Holders and Use of Proceeds. |
Global Offering
In October 2014, we sold 4,919,498 ADSs, each representing one-half of one ordinary share, nominal value €0.10, and 614,923 ordinary shares, in our global offering at a price of $21.64 per ADS and €34.00 per share, for aggregate gross proceeds to us of approximately €104.5 million. The net offering proceeds to us, after deducting underwriting discounts and commissions and offering expenses, werewas approximately €93.7 million. The offering commenced on October 15, 2014 and did not terminate before all of the securities registered in the registration statement were sold. The effective date of the registration statement, File No. 333-198870, for our global offering was October 21, 2014. Citigroup Global Markets Inc., Leerink Partners LLC and Bryan, Garnier & Co. are actingacted as joint global coordinators and joint book-running managers of the global offering.
A portionAll of the net proceeds from our global offering was used for general corporate purposes. The balance is held in cashhave been applied as of the date of this Annual Report and cash equivalents and is intended to also be used for general corporate purposes. None of the net proceeds of our global offering were paid directly or indirectly to any director, officer, general partner of ours or to their associates, persons owning ten percent or more of any class of our equity securities, or to any of our affiliates.
| Item 15. | Controls and Procedures. |
OurDisclosure Controls and Procedures
We maintain “disclosure controls and procedures,” as such term is defined in Rules 13(a) - 15(e) and 15(d) - 15(e) under the Exchange Act, that are designed to ensure that information required to be disclosed in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the rules and forms of the Securities and Exchange Commission. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed in our reports filed or submitted under the Exchange Act is accumulated and communicated to management, including our chief executive officer (principal executive officer) and chief operating officer (principal financial officer), after evaluatingas appropriate, to allow timely decisions regarding required disclosure.
Our management, with the participation of our chief executive officer (principal executive officer) and chief operating officer (principal financial officer), carried out an evaluation of the effectiveness of our disclosure controls and procedures (as defined in Rule 13a-15(e) of the Exchange Act) as of December 31, 2014, have2015. Based on that evaluation, management has identified a material weakness associated with the segregation of duties and therefore, our chief executive officer (principal executive officer) and chief operating officer (principal financial officer) concluded that as of such date, ourthe company’s disclosure controls and procedures were effectiveineffective as of December 31, 2015.
Management’s Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and ensured that information required to be disclosed by usmaintaining adequate internal controls over financial reporting (as defined in reports that we file or submitRules 13a-15(f) and 15d-15(f) under the Exchange ActAct) and for the assessment of the effectiveness of our internal control over financial reporting. Under the supervision and with the participation of our chief executive officer (principal executive officer) and chief operating officer (principal financial officer), management assessed our internal control over financial reporting based upon the framework in Internal Control — Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Based on this assessment, we concluded that we had a material weakness because we did not maintain adequate segregation of duties given the size of our finance and accounting team to allow for appropriate monitoring of financial reporting matters and internal control over financial reporting. We have limited accounting and financial reporting personnel and other resources with which to address our internal controls and procedures. The material weakness was partially remediated at December 31, 2015, but these new controls were not effective for almost all of 2015. Consequently, our chief executive officer (principal executive officer) and chief operating officer (principal financial officer) concluded that, due to this material weakness, we did not maintain effective internal control over financial reporting as of December 31, 2015.
A material weakness is accumulateda deficiency, or combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of our annual or interim financial statements will not be prevented or detected on a timely basis.
This material weakness did not result in any adjustments or restatements of our audited consolidated financial statements or disclosures for any prior period previously reported by the company. However, there is a reasonable possibility that a material misstatement of the consolidated financial statements would not have been prevented or detected on a timely basis due to the failure in designing and communicated toimplementing appropriate controls over the segregation of duties, and therefore, it has been evaluated as a material weakness.
Notwithstanding this material weakness and management’s assessment that internal control over financial reporting was ineffective as of December 31, 2015, our management, including our chief executive officer (principal executive officer) and chief operating officer (principal financial officer), believes that the consolidated financial statements contained in this Annual Report on Form 20-F present fairly, in all material respects, our financial position, results of operations and cash flows for the periods presented in conformity with IFRS.
Management’s Plan for Remediation
With the oversight of senior management and our audit committee, we continue to allow timely decisions regarding required disclosure and is recorded, processed, summarized and reported within the time periods specified by the SEC’s rules and forms.
This annual report does not include a report of management’s assessment regardingevaluate our internal control over financial reporting orand are taking several remedial actions to address the material weakness that has been identified:
Attestation Report of the company’sRegistered Public Accounting Firm
Our independent registered public accounting firm, due to a transition period establishedDeloitte & Associés, has audited the consolidated financial statements included in this Annual Report on Form 20-F, and as part of its audit, has issued its audit report on the effectiveness of our internal control over financial reporting. This report is included on page F-3 of this Form 20-F and is incorporated herein by rules ofreference.
Changes in Internal Control Over Financial Reporting
Other than the SEC for newly public companies.
Therematerial weakness and remediation activities described above, there were no changes in our internal control over financial reporting that occurred during the period covered by this annual reportAnnual Report that have materially affected, or that are reasonably likely to materially affect, our internal control over financial reporting.
| Item 15T. | Controls and Procedures. |
Not applicable.
| Item 16. | Reserved. |
Not applicable.
| Item 16A. | Audit Committees Financial Expert. |
Our board of directors has determined that Dr. Bjerke is an audit committee financial expert as defined by SEC rules and has the requisite financial sophistication under the applicable rules and regulations of the Nasdaq Stock Market. Dr. Bjerke is independent as such term is defined in Rule 10A-3 under the Exchange Act and under the listing standards of the Nasdaq Stock Market.
| Item 16B. | Code of Business Conduct and Ethics. |
We have adopted a Code of Business Conduct and Ethics, or the Code of Conduct, that is applicable to all of our employees, executive officers and directors. TheA copy of the Code of Conduct is available on our website atwww.dbv-technologies.com. The audit committee of our board of directors will be responsible for overseeing the Code of Conduct and will be required to approve any waivers of the Code of Conduct for employees, executive officers and directors. We expect that any amendments to the Code of Conduct, or any waivers of its requirements, will be disclosed on our website.
| Item 16C. | Principal Accountant Fees and Services. |
Deloitte & Associés, or Deloitte, has served as our independent registered public accounting firm for 20132014 and 2014.2015. Our accountants billed the following fees to us for professional services in each of those fiscal years:
| Year Ended December 31, | Year Ended December 31, | |||||||||||||||
| 2014 | 2013 | 2014 | 2015 | |||||||||||||
| (euro in thousands) | (Amount in thousands of Euros) | |||||||||||||||
Audit Fees | € | 884 | € | 192 | € | 884 | € | 686 | ||||||||
Audit-Related Fees | — | — | — | — | ||||||||||||
Tax Fees | — | — | — | — | ||||||||||||
Other Fees | — | — | — | — | ||||||||||||
|
|
|
| |||||||||||||
Total | € | 884 | € | 192 | € | 884 | € | 686 | ||||||||
|
|
|
| |||||||||||||
“Audit Fees”Fees” are the aggregate fees billed for the audit of our annual financial statements. This category also includes services that generally the independent accountantDeloitte provides, such as consents and assistance with and review of documents filed with the SEC. In 2014,2015, “Audit Fees” also include fees billed for assurance and related services regarding our globalJuly 2015 underwritten public offering.
“Audit-Related Fees”Fees” are the aggregate fees billed for assurance and related services that are reasonably related to the performance of the audit and are not reported under Audit Fees.
“Tax Fees”Fees” are the aggregate fees billed for professional services rendered by the principal accountantDeloitte for tax compliance, tax advice and tax planning related services.
“Other Fees”Fees” are any additional amounts billed for products and services provided by the principal accountant.Deloitte.
There were no “Audit“Audit Related Fees” “Tax Fees”or “Tax Fees” or billed or paid during 20132014 or 2014.2015.
Audit and Non-Audit Services Pre-Approval Policy
The audit committee has responsibility for appointing, setting compensation of and overseeing the work of the independent registered public accounting firm. In recognition of this responsibility, the audit committee has adopted a policy governing the pre-approval of
all audit and permitted non-audit services performed by our independent registered public accounting firm to ensure that the provision of such services does not impair the independent registered public accounting firm’s independence from us and our management. Unless a type of service to be provided by our independent registered public accounting firm has received general pre-approval from the audit committee, it requires specific pre-approval by the audit committee. The payment for any proposed services in excess of pre-approved cost levels requires specific pre-approval by the audit committee.
Pursuant to its pre-approval policy, the audit committee may delegate its authority to pre-approve services to the chairperson of the Audit Committee.audit committee. The decisions of the chairperson to grant pre-approvals must be presented to the full audit committee at its next scheduled meeting. The audit committee may not delegate its responsibilities to pre-approve services to the management.
The audit committee has considered the non-audit services provided by Deloitte & Associés as described above and believes that they are compatible with maintaining Deloitte & Associés’sDeloitte’s independence as our independent registered public accounting firm.
| Item 16D. | Exemptions from the Listing Standards for Audit Committees. |
Not applicable.
| Item 16E. | Purchases of Equity Securities by the Issuer and Affiliated Purchasers. |
Not applicable.
| Item 16F. | Change in Registrant’s Certifying Accountant. |
Not applicable.
| Item 16G. | Corporate Governance. |
As a Frenchsociété anonyme, we are subject to various corporate governance requirements under French law. In addition, as a foreign private issuer listed on the Nasdaq Global Select Market, we will beare subject to the Nasdaq corporate governance listing standards. However, the Nasdaq Global Select Market’s listing standards provide that foreign private issuers are permitted to follow home country corporate governance practices in lieu of the Nasdaq rules, with certain exceptions. We intend tocurrently rely on the certain exemptions for foreign private issuers and follow French corporate governance practices in lieu of the Nasdaq corporate governance rules, including Nasdaq corporate governance rules that state (1) a majority of the board of directors consists of independent directors; (2) establishing a nominating and corporate governance committee; (3) the compensation committee be composed entirely of independent directors; and (4) separate executive sessions of independent directors and non-management directors held by the company at least twice per year.
The following are the significant ways in which our corporate governance practices differ from those required for U.S. companies listed on Nasdaq:
As a foreign private issuer, we are required to comply with Rule 10A-3 of the U.S. Securities Exchange Act of 1934, as amended, or the Exchange Act relating to audit committee composition and responsibilities. Rule 10A-3 provides that the audit committee must have direct responsibility for the nomination, compensation and choice of our auditors, as well as control over the performance of their duties, management of complaints made, and selection of consultants. However, if the laws of a foreign private issuer’s home country require that any such matter be approved by the board of directors or the shareholders, the audit committee’s responsibilities or powers with respect to such matter may instead be advisory. Under French law, the audit committee may only have an advisory role and appointment of our statutory auditors, in particular, must be decided by the shareholders at our annualgeneral shareholders’ meeting.
In addition, Nasdaq rules require that a listed company specify that the quorum for any meeting of the holders of common stock be at least 33 1/3% of the outstanding shares of the company’s common voting stock. Consistent with French Law,law, our by-laws provide that a quorum requires the presence of shareholders having at least (1) 20% of the shares entitled to vote in the case of an ordinary shareholders’ general meeting or at an extraordinary shareholders’ general meeting where shareholders are voting on a capital increase by capitalization of reserves, profits or share premium, or (2) 25% of the shares entitled to vote in the case of any other extraordinary shareholders’ general meeting. If a quorum is not present, the meeting is adjourned. There is no quorum requirement when an ordinary general meeting is reconvened, but the reconvened meeting may consider only questions which were on the agenda of the adjourned meeting. When an extraordinary general meeting is reconvened, the quorum required is 20% of the shares entitled to vote, except where the reconvened meeting is considering capital increases through capitalization of reserves, profits or share premium. For these matters, no quorum is required at the reconvened meeting. If a quorum is not present at a reconvened meeting requiring a quorum, then the meeting may be adjourned for a maximum of two months.
| Item 16H. | Mine Safety Disclosure. |
Not applicable.
| Item 17. | Financial Statements. |
See pages F-1 through F-53F-52 of this Annual Report on Form 20-F.
| Item 18. | Financial Statements. |
Not applicable.
| Item 19. | Exhibits. |
The Exhibits listed in the Exhibit Index at the end of this Annual Report on Form 20-F are filed as Exhibits to this Annual Report on Form 20-F.
| Page | ||||
Annual Financial Statements for the Years Ended December 31, | ||||
Report of Deloitte & Associés, Independent Registered Public Accounting Firm | F-2 | |||
| F-3 | ||||
| F-4 | ||||
| F-5 | ||||
| F-6 | ||||
| F-7 | ||||
| F-8 | ||||
| Notes to the Consolidated Financial Statements | F-9 | |||
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Shareholders of DBV Technologies SA
Paris, France
We have audited the accompanying consolidated statements of consolidated financial position of DBV Technologies SA and subsidiariessubsidiary (the “Company”) as of December 31, 2012, 2013, 2014 and 2014,2015, and the related consolidated statements of consolidated income (loss), consolidated comprehensive income (loss), changes in consolidated shareholders’ equity, and consolidated cash flows for each of the three years in the period ended December 31, 2014.2015. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements,statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, suchthe consolidated financial statements present fairly, in all material respects, the consolidated financial position of DBV Technologies SA and subsidiariesthe Company as of December 31, 2012, 2013, 2014 and 2014,2015, and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2014,2015, in conformity with International Financial Reporting Standards (“IFRS”) as issued by the International Accounting Standards Board.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the Company’s internal control over financial reporting as of December 31, 2015, based on the criteria established in Internal Control — Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated April 28, 2016 expressed an adverse opinion on the Company’s internal control over financial reporting.
/s/ Deloitte & Associés
Represented by Fabien BrovedaniJulien Razungles
Neuilly-sur-Seine, France
April 29, 201528, 2016
Report of Independent Registered Public Accounting Firm
Internal Control Over Financial Reporting
To the Board of Directors and Shareholders of DBV Technologies SA
Paris, France
We have audited DBV Technologies SA and its subsidiary’s (the “Company’s”) internal control over financial reporting as of December 31, 2015, based on criteria established in Internal Control — Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission. The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Annual Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We do not express an opinion or any other form of assurance on management’s statements included in the Management’s Plan for Remediation section. We do not express an opinion or any other form of assurance on management’s statements included in the Management’s Plan for Remediation section.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on that risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed by, or under the supervision of, the company’s principal executive and principal financial officers, or persons performing similar functions, and effected by the company’s board of directors, management, and other personnel to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with International Financial Reporting Standards (“IFRS”) as issued by the International Accounting Standards Board, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of the inherent limitations of internal control over financial reporting, including the possibility of collusion or improper management override of controls, material misstatements due to error or fraud may not be prevented or detected on a timely basis. Also, projections of any evaluation of the effectiveness of the internal control over financial reporting to future periods are subject to the risk that the controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of the company’s annual or interim financial statements will not be prevented or detected on a timely basis. The following material weakness has been identified and included in management’s assessment: the Company did not maintain adequate segregation of duties given the size of its finance and accounting team to allow for appropriate monitoring of financial reporting matters and internal control over financial reporting. The Company has limited accounting and financial reporting personnel and other resources with which to address its internal controls and procedures. This material weakness was considered in determining the nature, timing, and extent of audit tests applied in our audit of the consolidated financial statements as of and for the year ended December 31, 2015, of the Company and this report does not affect our audit report on such financial statements.
In our opinion, because of the effect of the material weakness identified above on the achievement of the objectives of the control criteria, the Company has not maintained effective internal control over financial reporting as of December 31, 2015, based on the criteria established in Internal Control — Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated financial statements as of and for the year ended December 31, 2015, of the Company and our report dated April 28, 2016 expressed an unqualified opinion on those financial statements.
/s/ Deloitte & Associés
Represented by Julien Razungles
Neuilly-sur-Seine, France
April 28, 2016
CONSOLIDATED STATEMENTS OF CONSOLIDATED FINANCIAL POSITION
| Notes | Year Ended December 31, | |||||||||||||
| 2012 (1) | 2013 (1) | 2014 | ||||||||||||
| € | € | € | ||||||||||||
| ASSETS | ||||||||||||||
Non-current assets | ||||||||||||||
Intangible assets | 4 | 14,012 | 63,007 | 28,835 | ||||||||||
Property, plant, and equipment | 5 | 988,283 | 1,734,149 | 2,224,928 | ||||||||||
Other non-current financial assets | 6 | 384,357 | 623,829 | 1,595,861 | ||||||||||
Total non-current assets | 1,386,652 | 2,420,985 | 3,849,624 | |||||||||||
Current assets | ||||||||||||||
Inventories and work in progress | 7 | 29,673 | 6,568 | 124,071 | ||||||||||
Customer accounts receivable and related receivables | 8 | 92,875 | 182,900 | 136,112 | ||||||||||
Other current assets | 8 | 3,117,487 | 4,222,796 | 6,722,563 | ||||||||||
Cash and cash equivalents | 9 | 38,348,130 | 39,402,761 | 114,583,141 | ||||||||||
Total current assets | 41,588,165 | 43,815,024 | 121,565,887 | |||||||||||
TOTAL ASSETS | 42,974,817 | 46,236,009 | 125,415,511 | |||||||||||
| Notes | Year Ended December 31, | |||||||||||||||||||||||||||
| 2013 (1) | 2014 | 2015 | ||||||||||||||||||||||||||
| K€ | K€ | K€ | ||||||||||||||||||||||||||
| ASSETS | ||||||||||||||||||||||||||||
Non-current assets | ||||||||||||||||||||||||||||
Intangible assets | 4 | 63 | 29 | 94 | ||||||||||||||||||||||||
Property, plant, and equipment | 5 | 1,734 | 2,225 | 5,581 | ||||||||||||||||||||||||
Other non-current financial assets | 6 | 624 | 1,596 | 2,711 | ||||||||||||||||||||||||
Total non-current assets | 2,421 | 3,850 | 8,387 | |||||||||||||||||||||||||
Current assets | ||||||||||||||||||||||||||||
Inventories and work in progress | 7 | 7 | 124 | — | ||||||||||||||||||||||||
Customer accounts receivable and related receivables | 8 | 183 | 136 | — | ||||||||||||||||||||||||
Other current assets | 8 | 4,223 | 6,723 | 11,512 | ||||||||||||||||||||||||
Cash and cash equivalents | 9 | 39,403 | 114,583 | 323,381 | ||||||||||||||||||||||||
Total current assets | 43,815 | 121,566 | 334,893 | |||||||||||||||||||||||||
TOTAL ASSETS | 46,236 | 125,416 | 343,280 | |||||||||||||||||||||||||
| Notes | Year Ended December 31, | Notes | Year Ended December 31st | |||||||||||||||||||||||||
| 2012 (1) | 2013 (1) | 2014 | 2013 (1) | 2014 | 2015 | |||||||||||||||||||||||
| € | € | € | K€ | K€ | K€ | |||||||||||||||||||||||
| LIABILITIES | ||||||||||||||||||||||||||||
Shareholders’ equity | ||||||||||||||||||||||||||||
Share capital | 10 | 1,340,815 | 1,508,830 | 1,916,066 | 10 | 1,509 | 1,916 | 2,421 | ||||||||||||||||||||
Premiums related to the share capital | 54,612,601 | 69,640,899 | 163,876,789 | 69,641 | 163,877 | 403,910 | ||||||||||||||||||||||
Reserves | (3,868,181 | ) | (11,448,627 | ) | (26,336,016 | ) | (11,449 | ) | (26,336 | ) | (39,580 | ) | ||||||||||||||||
Net profit (loss) | (12,912,100 | ) | (19,306,416 | ) | (24,011,880 | ) | ||||||||||||||||||||||
Net (loss) | (19,306 | ) | (24,012 | ) | (44,674 | ) | ||||||||||||||||||||||
Total shareholders’ equity | 39,173,135 | 40,394,685 | 115,444,959 | 40,395 | 115,445 | 322,076 | ||||||||||||||||||||||
Non-current liabilities | ||||||||||||||||||||||||||||
Long-term financial debt | 11 | 376,651 | 1,316,533 | 3,888,170 | 11 | 1,317 | 3,888 | 4,693 | ||||||||||||||||||||
Non-current provisions | 12 | 254,941 | 290,695 | 530,732 | 12 | 291 | 531 | 490 | ||||||||||||||||||||
Total non-current liabilities | 631,592 | 1,607,228 | 4,418,902 | 1,607 | 4,419 | 5,183 | ||||||||||||||||||||||
Current liabilities | ||||||||||||||||||||||||||||
Short-term financial debt | 11 | 257,414 | 126,292 | 212,736 | 11 | 126 | 213 | 149 | ||||||||||||||||||||
Bank overdrafts | 519,499 | — | 27,956 | — | 28 | — | ||||||||||||||||||||||
Supplier accounts payable and related payables | 13 | 977,724 | 1,497,289 | 1,874,629 | 13 | 1,497 | 1,875 | 10,034 | ||||||||||||||||||||
Other current liabilities | 13 | 1,415,453 | 2,610,515 | 3,436,329 | 13 | 2,611 | 3,436 | 5,838 | ||||||||||||||||||||
Total current liabilities | 3,170,090 | 4,234,096 | 5,551,650 | 4,234 | 5,552 | 16,021 | ||||||||||||||||||||||
TOTAL LIABILITIES AND SHAREHOLDERS’ EQUITY | 42,974,817 | 46,236,009 | 125,415,511 | 46,236 | 125,416 | 343,280 | ||||||||||||||||||||||
| (1) | The consolidated statement of |
The accompanying notes form an integral part of these consolidated financial statements.
CONSOLIDATED STATEMENTS OF CONSOLIDATED INCOME (LOSS)
| Notes | Year Ended December 31, | Notes | Year Ended December 31st | |||||||||||||||||||||||||
| 2012 (1) | 2013 (1) | 2014 | 2013 (1) | 2014 | 2015 | |||||||||||||||||||||||
| € | € | € | K€ | K€ | K€ | |||||||||||||||||||||||
Operating income | ||||||||||||||||||||||||||||
Revenues | 15 | 174,360 | 181,800 | 210,759 | 15 | 182 | 211 | 202 | ||||||||||||||||||||
Other income | 15 | 2,602,228 | 3,644,513 | 4,550,763 | 15 | 3,645 | 4,551 | 5,964 | ||||||||||||||||||||
Total income | 2,776,588 | 3,826,313 | 4,761,522 | 3,826 | 4,762 | 6,166 | ||||||||||||||||||||||
Operating expenses | ||||||||||||||||||||||||||||
Cost of goods sold | 82,958 | 102,366 | 136,296 | (102 | ) | (136 | ) | (128 | ) | |||||||||||||||||||
Research and development | 16/17 | 11,499,368 | 17,366,538 | 21,143,442 | 16/17 | (17,366 | ) | (21,143 | ) | (34,234 | ) | |||||||||||||||||
Selling and marketing | 16/17 | (13 | ) | (491 | ) | |||||||||||||||||||||||
General and administration | 16/17 | 4,598,699 | 6,309,750 | 8,117,664 | 16/17 | (6,310 | ) | (8,105 | ) | (16,859 | ) | |||||||||||||||||
Total Expenses | 16,181,025 | 23,778,654 | 29,397,402 | (23,779 | ) | (29,397 | ) | (51,712 | ) | |||||||||||||||||||
Operating profit (loss) | (13,404,437 | ) | (19,952,340 | ) | (24,635,880 | ) | ||||||||||||||||||||||
Operating (loss) | (19,952 | ) | (24,636 | ) | (45,546 | ) | ||||||||||||||||||||||
Financial revenues | 18 | 517,54 | 670,234 | 727,239 | 18 | 670 | 727 | 1,018 | ||||||||||||||||||||
Financial expenses | 18 | (25,208 | ) | (24,310 | ) | (103,239 | ) | 18 | (24 | ) | (103 | ) | (146 | ) | ||||||||||||||
Financial Profit (loss) | 492,337 | 645,925 | 624,000 | |||||||||||||||||||||||||
Financial Profit | 646 | 624 | 871 | |||||||||||||||||||||||||
Income tax | 19 | — | — | — | 19 | — | — | — | ||||||||||||||||||||
Net profit (loss) | (12,912,100 | ) | (19,306,416 | ) | (24,011,880 | ) | ||||||||||||||||||||||
Basic / Diluted earnings per share (€/share) | 22 | (1.05 | ) | (1.42 | ) | (1.49 | ) | |||||||||||||||||||||
Net (loss) | (19,306 | ) | (24,012 | ) | (44,674 | ) | ||||||||||||||||||||||
Basic / Diluted (loss) per share (€/share) | 22 | (1.42 | ) | (1.49 | ) | (2.08 | ) | |||||||||||||||||||||
| (1) | The |
The accompanying notes form an integral part of these consolidated financial statements.
CONSOLIDATED STATEMENTS OF CONSOLIDATED COMPREHENSIVE INCOME (LOSS)
| Year Ended December 31 | ||||||||||||
| 2012 (1) | 2013 (1) | 2014 | ||||||||||
| € | € | € | ||||||||||
Net profit (loss) | (12,912,100 | ) | (19,306,416 | ) | (24,011,880 | ) | ||||||
Actuarial gains and losses on employee benefits, net of corporate tax | (99,900 | ) | 53,266 | (153,256 | ) | |||||||
Profit (loss) directly recognised in shareholders’ equity | (99,900 | ) | 53,266 | (153,256 | ) | |||||||
Other items in the total profit (loss) to be recycled subsequently to the net profit (loss) | — | — | (25,941 | ) | ||||||||
Total Comprehensive Income/ (loss) | (13,012,000 | ) | (19,253,150 | ) | (24,191,077 | ) | ||||||
| Year Ended December 31 | ||||||||||||
| 2013 (1) | 2014 | 2015 | ||||||||||
| K€ | K€ | K€ | ||||||||||
Net (loss) | (19,306 | ) | (24,012 | ) | (44,674 | ) | ||||||
Actuarial gains and losses on employee benefits, net of corporate tax | 53 | (153 | ) | 166 | ||||||||
Profit (loss) directly recognised in shareholders’ equity | 53 | (153 | ) | 166 | ||||||||
Other items in the total (loss) to be recycled subsequently to the net (loss) | — | (26 | ) | (90 | ) | |||||||
Total Comprehensive (loss) | (19,253 | ) | (24,191 | ) | (44,598 | ) | ||||||
| (1) | The consolidated statement of |
In accordance with IAS 1Presentation of Financial Statements (2007) (IAS 1), the Group, as defined in Note 1,2, presents a combined statement of other elements of comprehensive income (loss).
The Group does not hold any financial assets available for sale and non-current financial assets are measured at historical cost which approximates fair value; therefore, no change in fair value is reflected in the consolidated statement of consolidated comprehensive income (loss).
The accompanying notes form an integral part of these consolidated financial statements.
CONSOLIDATED STATEMENTS OF CONSOLIDATED CASH FLOWS
| Notes | 2012 (1) | 2013 (1) | 2014 | Notes | 2013 (1) | 2014 | 2015 | |||||||||||||||||||||
| € | € | € | K€ | K€ | K€ | |||||||||||||||||||||||
Cash flows from operating activities | ||||||||||||||||||||||||||||
Net profit (loss) for the period | (12,912,100 | ) | (19,306,416 | ) | (24,011,880 | ) | ||||||||||||||||||||||
Reconciliation of net profit (or loss) and of the cash used for operating activities: | ||||||||||||||||||||||||||||
Net (loss) for the period | (19,306 | ) | (24,012 | ) | (44,674 | ) | ||||||||||||||||||||||
Reconciliation of net ( loss) and of the cash used for operating activities: | ||||||||||||||||||||||||||||
Amortization and depreciation | 281,543 | 341,176 | 515,389 | 341 | 515 | 1,073 | ||||||||||||||||||||||
Retirement pension obligations | 36,495 | 89,572 | 86,781 | 90 | 86 | 125 | ||||||||||||||||||||||
Expenses related to share-based payments | 3,194,308 | 5,048,201 | 4,639,403 | 5,048 | 4,639 | 10,419 | ||||||||||||||||||||||
Other elements | — | — | 296 | |||||||||||||||||||||||||
Operating cash flows before change in working capital | (9,399,754 | ) | (13,827,467 | ) | (18,770,307 | ) | (13,827 | ) | (18,770 | ) | (32,761 | ) | ||||||||||||||||
Inventories and work in progress | 4,776 | 23,105 | (117,503 | ) | 23 | (117 | ) | 124 | ||||||||||||||||||||
Customer accounts receivable | (124,450 | ) | (57,675 | ) | (124,664 | ) | (58 | ) | (125 | ) | 136 | |||||||||||||||||
Other receivables | (230,647 | ) | (1,105,309 | ) | (1,701,831 | ) | (1,105 | ) | (1,702 | ) | (4,870 | ) | ||||||||||||||||
Supplier accounts payable | (1,226,754 | ) | 519,565 | (424,204 | ) | 520 | (424 | ) | 8,236 | |||||||||||||||||||
Other current liabilities | 544,280 | 1,194,565 | 578,857 | 1,195 | 578 | 2,372 | ||||||||||||||||||||||
Change in the working capital requirement | (1,032,794 | ) | 574,252 | (1,789,345 | ) | 574 | (1,789 | ) | 5,998 | |||||||||||||||||||
Net cash flow from operating activities | (10,432,549 | ) | (13,253,215 | ) | (20,559,652 | ) | (13,253 | ) | (20,560 | ) | (26,763 | ) | ||||||||||||||||
Cash flows from investment activities | ||||||||||||||||||||||||||||
Acquisitions of property, plant, and equipment | 5 | (340,411 | ) | (1,089,902 | ) | (941,301 | ) | 5 | (1,090 | ) | (941 | ) | (4,360 | ) | ||||||||||||||
Acquisitions of intangible assets | 4 | (21,024 | ) | (81,385 | ) | (30,695 | ) | 4 | (81 | ) | (31 | ) | (148 | ) | ||||||||||||||
Acquisitions of non-current financial assets | (33,685 | ) | (237,138 | ) | (124,235 | ) | (237 | ) | (124 | ) | (839 | ) | ||||||||||||||||
Other cash flows related to investment transactions | 26,360 | |||||||||||||||||||||||||||
Net cash flows from investment activities | (368,760 | ) | (1,408,425 | ) | (1,096,231 | ) | (1,408 | ) | (1,096 | ) | (5,347 | ) | ||||||||||||||||
Cash flows from financing activities: | ||||||||||||||||||||||||||||
Increase in conditional advances | 11 | 14,613 | 1,068,760 | 3,128,000 | 11 | 1,069 | 3,128 | 865 | ||||||||||||||||||||
(decrease) in conditional advances | 11 | (200,000 | ) | (260,000 | ) | (128,000 | ) | |||||||||||||||||||||
(Decrease) in conditional advances | 11 | (260 | ) | (128 | ) | (192 | ) | |||||||||||||||||||||
Treasury shares | (278,291 | ) | 230,697 | (888,977 | ) | 231 | (889 | ) | (175 | ) | ||||||||||||||||||
Capital increases, net of transaction costs | 10 | 37,562,500 | 15,196,313 | 94,643,126 | 10 | 15,196 | 94,643 | 240,538 | ||||||||||||||||||||
Other cash flows related to financing activities | 54,157 | 54 | (21 | ) | ||||||||||||||||||||||||
Net cash flows from financing activities: | 37,098,822 | 16,235,770 | 96,808,306 | 16,236 | 96,808 | 241,014 | ||||||||||||||||||||||
(Decrease)/Increase in cash | 26,297,514 | 1,574,130 | 75,152,424 | 1,574 | 75,152 | 208,904 | ||||||||||||||||||||||
Net Cash and cash equivalents at the beginning of the period | 11,531,117 | 37,828,631 | 39,402,761 | 37,829 | 39,403 | 114,555 | ||||||||||||||||||||||
Impact of exchange rate fluctuations | — | — | (78 | ) | ||||||||||||||||||||||||
Net Cash and cash equivalents at the close of the period | 9 | 37,828,631 | 39,402,761 | 114,555,185 | 9 | 39,403 | 114,555 | 323,381 | ||||||||||||||||||||
| (1) | The consolidated statement of |
The accompanying notes form an integral part of these consolidated financial statements.
CONSOLIDATED STATEMENTS OF CHANGES IN CONSOLIDATED SHAREHOLDERS’ EQUITY
(Amounts in thousands of Euros)
| Share Capital Ordinary Shares | Share Capital Ordinary Shares | |||||||||||||||||||||||||||||||||||||||||||||||
| Number of shares | Amount | Premiums Related to the Share Capital | Reserves | Profit (loss) | Total Shareholders’ Equity | Number of shares | Amount | Premiums Related to the Share Capital | Reserves | Profit (loss) | Total Shareholders’ Equity | |||||||||||||||||||||||||||||||||||||
At January 1, 2012 (1) | 8,822,745 | 882,275 | 17,508,641 | 553,964 | (7,238,262 | ) | 11,706,617 | |||||||||||||||||||||||||||||||||||||||||
Net profit (loss) | (12,912,100 | ) | (12,912,100 | ) | ||||||||||||||||||||||||||||||||||||||||||||
Profit (loss) directly recognized in shareholders’ equity | (99,900 | ) | (99,900 | ) | ||||||||||||||||||||||||||||||||||||||||||||
At January 1, 2013 (1) | 13,408,147 | 1,341 | 54,613 | (3,868 | ) | (12,912 | ) | 39,173 | ||||||||||||||||||||||||||||||||||||||||
Net (loss) | (19,306 | ) | (19,306 | ) | ||||||||||||||||||||||||||||||||||||||||||||
Profit directly recognized in shareholders’ equity | 53 | 53 | ||||||||||||||||||||||||||||||||||||||||||||||
Total profit (loss) directly recognized in shareholders’ equity | (99,900 | ) | (12,912,100 | ) | (13,012,000 | ) | 53 | (19,306 | ) | (19,253 | ) | |||||||||||||||||||||||||||||||||||||
Allocation of prior period profit (loss) | (7,238,262 | ) | 7,238,262 | |||||||||||||||||||||||||||||||||||||||||||||
Allocation of prior period (loss) | (12,912 | ) | 12,912 | |||||||||||||||||||||||||||||||||||||||||||||
Increase in capital | 1,680 | 168 | 14,961 | 15,129 | ||||||||||||||||||||||||||||||||||||||||||||
Treasury shares | 231 | 231 | ||||||||||||||||||||||||||||||||||||||||||||||
Issue of share warrants | 67 | 67 | ||||||||||||||||||||||||||||||||||||||||||||||
Foreign exchange translation | (1 | ) | (1 | ) | ||||||||||||||||||||||||||||||||||||||||||||
Share-based payments | 5,048 | 5,048 | ||||||||||||||||||||||||||||||||||||||||||||||
At December 31, 2013 | 15,088,298 | 1,509 | 69,641 | (11,449 | ) | (19,306 | ) | 40,395 | ||||||||||||||||||||||||||||||||||||||||
Net (loss) | (24,012 | ) | (24,012 | ) | ||||||||||||||||||||||||||||||||||||||||||||
(Loss) directly recognized in shareholders’ equity | (179 | ) | (179 | ) | ||||||||||||||||||||||||||||||||||||||||||||
Total (loss) directly recognized in shareholders’ equity | (179 | ) | (24,012 | ) | (24,191 | ) | ||||||||||||||||||||||||||||||||||||||||||
Allocation of prior period (loss) | (19,306 | ) | 19,306 | — | ||||||||||||||||||||||||||||||||||||||||||||
Increase in capital | 4,585,402 | 458,54 | 37,095,400 | 37,553,940 | 4,072,363 | 407 | 94,204 | 94,611 | ||||||||||||||||||||||||||||||||||||||||
Treasury shares | — | (278,291 | ) | (278,291 | ) | — | (41 | ) | (41 | ) | ||||||||||||||||||||||||||||||||||||||
Issue of share warrants | 8,560 | 8,560 | 32 | 32 | ||||||||||||||||||||||||||||||||||||||||||||
Share-based payments | 3,194,308 | 3,194,308 | 4,639 | 4,639 | ||||||||||||||||||||||||||||||||||||||||||||
At December 31, 2012 (1) | 13,408,147 | 1,340,815 | 54,612,601 | (3,868,181 | ) | (12,912,100 | ) | 39,173,135 | ||||||||||||||||||||||||||||||||||||||||
Net profit (loss) | (19,306,416 | ) | (19,306,416 | ) | ||||||||||||||||||||||||||||||||||||||||||||
Profit (loss) directly recognized in shareholders’ equity | 53,266 | 53,266 | ||||||||||||||||||||||||||||||||||||||||||||||
At December 31, 2014 | 19,160,661 | 1,916 | 163,877 | (26,336 | ) | (24,012 | ) | 115,445 | ||||||||||||||||||||||||||||||||||||||||
Net (loss) | (44,674 | ) | (44,674 | ) | ||||||||||||||||||||||||||||||||||||||||||||
Foreign exchange translation | (90 | ) | (90 | ) | ||||||||||||||||||||||||||||||||||||||||||||
Profit directly recognized in shareholders’ equity | 166 | 166 | ||||||||||||||||||||||||||||||||||||||||||||||
Total profit (loss) directly recognized in shareholders’ equity | 53,266 | (19,306,416 | ) | (19,253,150 | ) | 76 | (44,674 | ) | (44,598 | ) | ||||||||||||||||||||||||||||||||||||||
Allocation of prior period profit (loss) | (12,912,100 | ) | 12,912,100 | |||||||||||||||||||||||||||||||||||||||||||||
Increase in capital | 1,680,151 | 168,015 | 14,960,858 | 15,128,873 | ||||||||||||||||||||||||||||||||||||||||||||
Treasury shares | — | 230,697 | 230,697 | |||||||||||||||||||||||||||||||||||||||||||||
Foreign exchange translation | (511 | ) | (511 | ) | ||||||||||||||||||||||||||||||||||||||||||||
Issue of share warrants | 67,440 | 67,440 | ||||||||||||||||||||||||||||||||||||||||||||||
Share-based payments | 5,048,201 | 5,048,201 | ||||||||||||||||||||||||||||||||||||||||||||||
At December 31, 2013 (1) | 15,088,298 | 1,508,830 | 69,640,898 | (11,448,627 | ) | (19,306,416 | ) | 40,394,685 | ||||||||||||||||||||||||||||||||||||||||
Net profit (loss) | (24,011,880 | ) | (24,011,880 | ) | ||||||||||||||||||||||||||||||||||||||||||||
Profit (loss) directly recognized in shareholders’ equity | (179,197 | ) | (179,197 | ) | ||||||||||||||||||||||||||||||||||||||||||||
Total profit (loss) directly recognized in shareholders’ equity | (179,197 | ) | (24,011,880 | ) | (24,191,077 | ) | ||||||||||||||||||||||||||||||||||||||||||
Allocation of prior period Income (Loss) | (19,306,416 | ) | 19,306,416 | |||||||||||||||||||||||||||||||||||||||||||||
Allocation of prior period (loss) | (24,012 | ) | 24,012 | |||||||||||||||||||||||||||||||||||||||||||||
Increase in capital | 4,072,363 | 407,236 | 94,203,624 | 94,610,860 | 5,044,468 | 504 | 239,891 | 240,396 | ||||||||||||||||||||||||||||||||||||||||
Treasury shares | (41,179 | ) | (41,179 | ) | — | 273 | 273 | |||||||||||||||||||||||||||||||||||||||||
Issue of share warrants | 32,267 | 32,267 | 142 | 142 | ||||||||||||||||||||||||||||||||||||||||||||
Share-based payments | 4,639,403 | 4,639,403 | 10,419 | 10,419 | ||||||||||||||||||||||||||||||||||||||||||||
At December 31, 2014 | 19,160,661 | 1,916,066 | 163,876,789 | (26,336,016 | ) | (24,011,880 | ) | 115,444,959 | ||||||||||||||||||||||||||||||||||||||||
At December 31, 2015 | 24,205,129 | 2,421 | 403,910 | (39,580 | ) | (44,674 | ) | 322,076 | ||||||||||||||||||||||||||||||||||||||||
| (1) | The consolidated statement of changes in |
The accompanying notes form an integral part of these consolidated financial statements.
NOTES TO THE FINANCIAL STATEMENTS
Note 1: The Company
Incorporated in 2002 under the laws of France, DBV Technologies S.A. (“DBV Technologies,” or the “Company”) is a clinical-stage specialty biopharmaceutical company focused on changing the field of immunotherapy, specifically in young children.
The Company historically marketed a ready-to-use diagnostic product to detect t cow’s milk protein allergy (“CMPA”) in children called Diallertest® Milk, which was launched in France in 2004. This product was distributed in France only, by a commercial partner, under a temporary exception status from French regulatory authorities which, without such temporary exception, marketing of the product would not be allowed. During the second half of 2015, the Company discontinued its commercial partnership with respect to the product and ceased selling Diallertest Milk. The Company does not expect to derive any revenues in 2016 from the sale of Diallertest Milk. DBV Technologies is also developing a novel technology platform called Viaskin.Viaskin® . The Company’s therapeutic approach is based on epicutaneous immunotherapy, or EPIT, a proprietary method of delivering biologically active compounds to the immune system through intact skin using Viaskin.
The Company marketsViaskin® Peanut is the first specific immunotherapy product developed by DBV Technologies. Solid preclinical data have already been published. Pharmacological development was achieved through a ready-to-use diagnostic productvast network of collaborative efforts in the United States and in Europe. A tolerance study (Phase Ib) conducted in the United States demonstrated the safety and high level of tolerance of Viaskin® Peanut in patients with peanut allergies, and the FDA granted a Fast Track designation to detect the allergy to cow’s milk protein in children called Diallertest Milk, whichproduct. In France, the French Health Product Safety Agency (Agence française de sécurité sanitaire des produits de santé, AFSSAPS) authorized an efficacy study sponsored by the Paris Region Public Hospitals (Assistance Publique – Hôpitaux de Paris, AP/HP). In 2012, an efficacy study (Phase IIb) was launched in Francethe United States and Europe. The topline results for the studies were published during the second 2014 semester. A Phase III clinical study began during the last 2015 quarter.
Viaskin® Milk is the second product developed in 2004. This product is currently available withspecific immunotherapy. A Phase II pilot study published by Dupont et al. (JACI 2010) demonstrated the safety and effectiveness of Viaskin® Milk in children (JACI 2010). In 2014, a temporary exception status from French regulatory authorities. Regulatory authorities have requested a pivotal phase IIIclinical efficacy study to complete the marketing authorization file. The Company is currently assessing the relevance of conducting such a study and might decide, if necessary, to stop marketing Diallertest Milk.using Viaskin® Milk was launched.
Main events in 20142015
On September 22, 2014, we announced toplineIn February 2015, DBV Technologies published the totality of the results for ourthe VIPES study at the World Allergy Congress which was held in Houston, Texas. The detailed results of VIPES show the magnitude of Viaskin®’s therapeutic effect, which allow the Company to launch its third food allergy program, specifically on allergies to egg (Viaskin Peanut’s Efficacy® Egg).
In April 2015, DBV Technologies announced that it had obtained breakthrough therapy designation from the U.S. Food and Safety) phase IIb clinical trial ofDrug Administration (“FDA”) for Viaskin Peanut in children. 6 to 11 years of age A breakthrough therapy is defined as a product that is intended, alone or in combination with one or more other drugs, to treat a serious or life- threatening condition, and preliminary clinical evidence indicates that the product may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints.
In June 2015, DBV Technologies announced the positive opinion of the Pediatric Committee of the European Medicines Agency (“EMA”) regarding the Pediatric Investigation Plan (“PIP”) of Viaskin® Peanut for the treatment of children with a peanut allergicallergy. An accepted PIP is a prerequisite for the filing for marketing authorization for any new medicinal product in Europe.
In June 2015, subsequent to the end of Phase II, DBV Technologies also detailed the results of the meeting held with the FDA. The results of this meeting, which was regarding the plan to clinically develop Viaskin® Peanut, are consistent with the positive opinion previously given by the EMA regarding the Pediatric Investigation Plan (PIP) of Viaskin® Peanut. On the basis of the compulsory consultations, in the fourth trimester of 2015, DBV Technologies plans to launch a worldwide Phase II clinical study on Viaskin® Peanut for the treatment of peanut allergies in children from 4 to 11 years of age (Peanut EPIT Efficacy and Safety Study PEPITES). On the basis of these consultations, complementary developmental plans will be discussed with the FDA during the second semester of 2015 for Viaskin® Peanut in both younger and older patients.
In June 2015, Professor Hugh Sampson joined the Company as Scientific Director. As part of his duties, and in partnership with Professor Bertrand Dupont, President of the Scientific Advisory Board, Professor Sampson supervises DBV’s research team and drives the development of new applications for Viaskin® in the treatment of food allergies, while giving support to the Company’s clinical development teams.
In June 2015, the Company announced the end of Part A of the Viaskin® Milk Efficacy and Safety Phase I/II study (MILES). The Data Safety Monitoring Board (DSMB) for the study recommended that the study continue and expressed no safety concerns after evaluating the Part A safety data of subjects treated with a 150 µg, 300 µg and 500 µg doses of Viaskin® Milk.
In July 2015, the Company filed a registration statement on Form F-1 with the U.S Securities and Exchange Commission (“SEC”) for a proposed underwriter public offering of ordinary shares in the form of American Depositary Shares (“ADS”), as well as the registration of its Reference Document with the French Financial Markets Authority (“AMF”). On July 20, 2015, the Company consummated the ADS offering and increased its capital in the amount of 281.5 million US Dollars. The ADS were sold to the public at a price of 34 US dollars per ADS, which resulted in a subscription price of 61.68 Euros per new ordinary share (of which a share premium of 61.58 Euros). Each ADS gives the right to receive half of an ordinary share.
In July 2015, the FDA accepted an investigator-sponsored investigational new drug (“IND”) application for a Phase IIA clinical trial metfor the potential treatment of milk-induced EoE in children using the Company’s Viaskin® Milk. This IND acceptance enabled Dr. Jonathan Spergel at The Children’s Hospital of Philadelphia to initiate SMILEE (Study Efficacy and Safety of the Viaskin® Milk in milk-induced Eosinophilic Esophagitis), a clinical trial designed to assess the efficacy and safety of Viaskin® Milk in children from four to 17 years of age suffering from milk-induced EoE. The SMILEE trial was initiated in November 2015.
In October 2015, the Company presented its primary endpoint attopline findings from the highest explored dose (Viaskin Peanut 250 µg), achieving statistical significance (p=0.0108) in desensitizing a higher proportion of patients versus placebo afterfirst 12 months of Epicutaneous Immunotherapy (EPIT). Patients treatedthe OLFUS-VIPES study, which follows the Phase IIb VIPES clinical study. The Company announced that t it observed that 12 additional months of therapy with Viaskin Peanut 250 µg also showed statistically significant changesincreased the number of patients benefiting from treatment to 70% in measured serological markers while placebo patients did not exhibit material differences. The safety profile was confirmed across all active arms with no serious treatment-related adverse events reported, and patient compliance with dailyOLFUS-VIPES from 50% in VIPES.
In November 2015, the Company presented pre-clinical findings evaluating Viaskin Peanut application was above 97%. The trial drop-out rate was 6.4%, below® technology in the 15% rate initially anticipated. The VIPES trial iscontext of vaccinations against respiratory syncytial virus (“RSV”) at the largestRSV Vaccines for the World 2015 meeting in La Jolla, California.
In December 2015, the Company announced the launch of a pivot Phase III clinical trial which will evaluate the safety and efficiency of Viaskin® Peanut 250µg in peanut allergy desensitization ever completed,children aged from four to 11 years of age who are allergic to peanuts. PEPITES (Peanut EPIT® Efficiency and fullSafety Study) is an international, double-blind, placebo-controlled Phase III study.
In December 2015, the Company presented experimental results on the use of efficacy and safetyEpicutaneous Immunotherapy (EPIT®) in a model of Crohn’s disease. These will be presented at future scientific meetings.the Crohn’s & Colitis Foundation of America Advances in Inflammatory Bowel Diseases (“AIBD”).
Note 2: General Information and Statement of Compliance
Preliminary remarks
The company DBV Technologies Inc. was established on April 7, 2014.2014 (the “US subsidiary”). The share capital of this US subsidiary is 100% owned by DBV Technologies SA. These financial statements are the first annual consolidated financial statements of the group thus formed.SA (“DBV Technologies”).
The financial information as at December 31, 2012 and 2013 corresponds to the information previously published and includes only the transactions related to the parent company DBV Technologies SA, which had no equity interest in a subsidiary over the periodsperiod in question.
General principles
The accompanying consolidated financial statements and related notes (the “Financial Statements”) present the operations of DBV Technologies SA and its US subsidiary (the Group)“Group”) as of December 31, 2014.2015. The Company is a corporate venture under French law (société anonyme) and has its registered office is located at 80/84 rue des Meuniers, 92220 Bagneux.Bagneux at December 31, 2015. The Company moved its headquarters to 177-181 avenue Pierre Brossolette 92120 Montrouge on January 6, 2016.
Our Financial Statements as of December 31, 20142015 have been prepared under the responsibility of DBV Technologies’ management. The Financial Statements were approved by the Board of Directors of DBV Technologies on March 24, 2015.April 6th, 2016.
All amounts are expressed in thousands of euros, unless stated otherwise.
For consolidation purposes, both DBV Technologies SA and its subsidiary DBV Technologies Inc. have prepared individual financial statements for the period ended December 31, 2014.2014 and December 31, 2015.
Statement of Compliance
Our Consolidated Financial Statements have been prepared in accordance with International Financial Reporting Standards (“IFRS”) as issued by the International Accounting Standard Board (“IASB”) and whose application is mandatory for the year ended December 31, 2014.2015. Comparative figures are presented for December 31, 20122013 and 2013.2014.
Due to the listing of DBV Technologies S.A. ordinary shares on the Euronext Paris and in accordance with the European Union’s regulation No. 1606/2002 of July 19, 2002, the consolidated financial statements of the Group are also prepared in accordance with IFRS, as adopted by the European Union (EU)(“EU”).
As of December 31, 2014, all2015, there are no differences in the IFRS thatpublished and mandated by the IASB had published and that are mandatory are the same as those endorsed by the EU and mandatory in the EU, with the exception of IAS 39Financial Instruments: Recognition and Measurement (revised December 2003),the following specific accounting principles which the EU only partially adopted. The parthas not yet adopted byat the EUend of 2015:
This amendment has nonot had any impact on the Consolidated Financial Statements. Statements as of December 31, 2015.
As a result, the Consolidated Financial Statements comply with International Financial Reporting StandardsIFRS as published by the IASB and as adopted by the EU.
IFRS include International Financial Reporting Standards (“IFRS”), International Accounting Standards (“the IAS”), as well as the interpretations issued by the Standing Interpretations Committee (“the SIC”), and the International Financial Reporting Interpretations Committee (“IFRIC”). The main accounting methods used to prepare the Consolidated Financial Statements are described below. These methods were used for all years presented.
The followingThere are no new standards, and amendments to standards, or interpretations applicable with effect on the period ending December 31, 2015 that have been adopted by DBV Technologies from January 1, 2014 but have had noan impact on the Consolidated Financial Statements:Statements or on their presentation.
New and revised standards and amendments that may be relevant to the Company’s operations but are not yet effective. effective:
Management has not yet completed its assessmentis in the process of evaluating the impact of these standards and amendments and is therefore, not currently able to estimate reliably the impact of their adoption on the Company’s results on financial position or cash flows.
The accounting policies and measurement principles adopted for the consolidated financial statementsFinancial Statements as of and for the year ended December 31, 20142015 are the same as those used as of and for the years ended December 31, 20122013 and 2013.2014.
Note 3: Accounting Principles
Methods of consolidation
The consolidated financial statementsFinancial Statements incorporate the standalone financial statements of the CompanyDBV Technologies S.A and entitiesDBV Technologies Inc. which is controlled by the Company. Control is achieved when the Company:
The Company reassesses whether or not it controls a subsidiary if facts and circumstances indicate that there are changes to one or more of the three elements of control listed above.
Consolidation of subsidiaries begins when the Company obtains control over the subsidiary and ceases when the Company loses control over the subsidiary.
Profit or loss and each component of other comprehensive income are attributed to the owners of the Company and to the non-controlling interests. Total comprehensive income of subsidiaries is attributed to the owners of the Company and to the non-controlling interests even if this results in the non-controlling interests having a deficit balance.
When necessary, adjustments are made to the financial statements of subsidiaries to align their accounting policies with the Group’s accounting policies.
All intragroup assets and liabilities, equity, income, expenses and cash flows relating to transactions between members of the Group are neutralized in consolidation.
Translation of financial statements in foreign currencies
In preparing the financial statements of each individual group entity, transactions in currencies other than the entity’s functionnalfunctional currency are recognisedrecognized at the rates of exchange prevailing at the dates of the transactions. At the end of each reporting period, monetary items denominated in foreign currencies are retranslated at the rates prevailing at that date. Non-monetary items carried at fair value that are denominated in foreign currencies are retranslated at the rates prevailing at the date when the fair value was determined.
For the purpose of presenting these consolidated financial statements, the assets and liabilities of the Group’s foreign operations are translated into Currency Units using exchange rates prevailing at the end of each reporting period. Income and expense items are translated at the average exchange rates at the dates of the transactions. Exchange differences arising, if any, are recognized in other comprehensive income and accumulated in equity.
3.1 Intangible Assets
In application of the provisions in IAS 38Intangible Assets(“IAS 38”), intangible assets acquired are postedrecorded as assets on the statementConsolidated Statements of financial positionFinancial Position at their acquisition cost.
Research and Development Expenses
Research expenses are recorded in the financial statementsFinancial Statements as expenses.
In accordance with IAS 38, development expenses are recorded in the consolidated financial statementsFinancial Statements as intangible assets only if all the following criteria are met:
| (a) | technical feasibility necessary for the completion of the development project; |
| (b) | intention on the part of the Company to complete the project and to utilize it; |
| (c) | capacity to utilize the intangible asset; |
| (d) | proof of the probability of future economic benefits associated with the asset; |
| (e) | availability of the technical, financial, and other resources for completing the project; and |
| (f) | reliable evaluation of the development expenses. |
Because of the risks and uncertainties related to regulatory authorizations and to the research and development process, the Company believes that the six criteria stipulated by IAS 38 are only fulfilled once the MarketingMarket Access Authorization has been obtained from the competent authorities.
The application of this principle has resulted in all development costs being expensed as incurred.
Software
The costs related to the acquisition of licenses to software are posted to assets on the basis of the costs incurred to acquire and to implement the software in question.
They are amortized using the straight-line method over a period of one to three years depending on the anticipated period of use.
3.2 Property, Plant, and Equipment
Property, plant, and equipment are postedrecorded at their acquisition cost or, if applicable, at their production cost.
Property, plant, and equipment are depreciated on the basis of the straight-line method over the estimated use period of the property. The fixtures of property rented are depreciated over the term of their own lifetime or of the term of the rental agreement, whichever is shorter.
The depreciation periods used are the following:
PROPERTY, PLANT,AND EQUIPMENT ITEM | DEPRECIATION P | |||
Fixtures and improvements in structures | 9 years | |||
Research and development / production tools | 5 years | |||
Research equipment and technical facilities | 5 years | |||
Computer equipment | 3 years | |||
Office equipment and furniture | ||||
3.3 Financial Assets
Financial assets include assets available for sale, assets owned until their maturity, loans and accounts receivable, and cash and cash equivalents.
The valuation and the accounting treatment of the financial assets and liabilities are defined byIn accordance with IAS 39Financial (Financial Instruments: Recognition and Measurement (“Measurement) and IAS 39”).32 (Financial Instruments: Presentation), the Company has adopted the following classification for non-derivative financial assets, based on the type of asset and on management intention at the date of initial recognition. The designation and classification of such financial assets are subsequently reassessed at the end of each reporting period.
Non-derivative financial assets are recognized on the date when the Company becomes party to the contractual terms of the asset. On initial recognition, financial assets are measured at fair value, plus direct transaction costs in the case of financial assets not classified as fair value through profit or loss.
Classification, presentation and subsequent measurement of non-derivative financial assets are as follows:
Assets Owned Until Their Maturity
These securities are exclusively fixed income or determinable income and have fixed maturities, other than loans and accounts receivable, that the company has the intention and the ability to keep until maturity. After their initial posting at their fair value, they are valued and posted to the accounts at the cost amortized on the basis of the effective interest rate (“EIR”) method.
The assets owned until their maturity are monitored for any objective indication of impairment. A financial asset is impaired if its book value is greater than its recoverable amount as estimated during impairment tests. The impairment is postedrecorded to the income statement.
Loans and Receivables
This category includes otherLoans and receivables are non-derivative financial assets with fixed or determinable payments that are not quoted in an active market. They are presented in current assets with loans with a maturity of less than 12 months are presented within “other current assets” and with trade receivables presented within “accounts receivable.”. Loans with a maturity of more than 12 months are presented as “long-term loans and accounts receivable and commercial receivables.
These instrumentsadvances” within “other non-current assets.” Those financial assets are initially posted to the accountsmeasured at their fair value and then at the amortized cost calculated with the EIR method. The short-term receivables without an interest rate are valued at the amount of the original invoice unless the application of an implicit interest rate has a significant effect. For the loans and variable-rate accounts receivable, a periodic re-estimation of the cash flows, in order to reflect the change in the market interest rate, modifiesusing the effective interest rate and therefore the valuation of the loan or of the receivable.method.
The loans and receivables are monitored for any objective indication of impairment. A financial asset is impaired if its book value is greater than its recoverable amount as estimated during impairment tests. The impairment is posted torecorded in the income statement.Consolidated Statements of (Loss).
The loans and receivables also include the deposits and guarantees, which are classified under “Non-current financial assets” onin the statementConsolidated Statements of financial position.Financial Position.
Assets at Fair Value Per the Income StatementConsolidated Statements of (Loss)
TheThese assets considered to be held for trading purposes includeare classified on the assets that the Company intends to resellbalance sheet in the near future in order to realize a capital gain, which is part of a portfolio ofline items Other non-current assets, Current financial instruments managed together for which there exists a practice of selling in the short term. Theassets and Cash and cash equivalents.
Financial assets at fair value through profit or loss comprise assets held for trading may also include(financial assets voluntarily classifiedacquired principally for the purpose of reselling them in this category,the near term, usually within less than 12 months), and financial instruments designated as fair value through profit and loss on initial recognition in a manner that is independentaccordance with the conditions for application of the criteria listed above (“fair value” option).value option.
Realized and unrealized foreign exchange gains and losses on financial assets in currencies other than the euro are recognized in the income statement in Financial income or Financial expenses.
Assets Available for Sale
The assets available for sale include, primarily, securities that do not meet the criteria of the definition of the other categories of financial assets. They are valuedmeasured at their fair value, and the changes in value are posted to shareholders’ equity.recorded in the Consolidated Statements of Changes in Shareholders’ Equity.
The fair value corresponds to the market price for those securities that are listed on the stock exchange or to an estimate of the use value for unlisted securities, determined on the basis of the financial criteria most appropriate for the specific situation of each security. When there is an objective indication of the impairment of these securities, the accumulated impairment that has been posted to shareholders’ equityrecorded in the Consolidated Statements of Changes in Shareholders’ Equity is recognized in the income statement.Consolidated Statement (Loss).
3.4 Recoverable Amount of the Intangible Assets and Property, Plant, and Equipment
The property, plant, and equipment and intangible assets that have an established lifetime are subjected to an impairment test when the recoverability of their book value is called into question by the existence of indications of impairment. An impairment is posted to the accounts up to the amount of the excess of the book value over the recoverable value of the asset. The recoverable value of an asset corresponds to its fair value minus the costs of sale or its use value, whichever is higher.
3.5 Inventories and Work in Progress
Inventories are posted to the accountsrecorded at their cost or at their net liquidation value, if the latter is lower. In the latter case, the impairment is posted to income or loss. The inventories are valued on the basis of the “first-in, first-out” (“FIFO”) method.
3.6 Cash and Cash Equivalents
Cash equivalents are owned for the purpose of meeting short-term cash commitments rather than for the objective of investment or for other purposes. They are readily convertible into a known amount of cash and are subject to a negligible risk of change in value. Cash and cash equivalents are constituted by liquid assets that are available immediately, long-term investments that can be liquidated immediately without a penalty, and investment securities. They are valued on the basis of the IAS 39 categories under which they fall.
Investment securities are readily convertible into a known amount of cash and are subject to a negligible risk of change in value. They are valued at their fair value, and the changes in value are posted to the financial income or loss. Given the nature of these assets, their fair value is generally close to their net carrying value.
3.7 Share Capital
Common shares are classified under shareholders’ equity.Shareholders’ Equity. The costs of share capital transactions that are directly attributable to the issue of new shares or options are posted torecorded in the books under shareholders’ equityFinancial Statements in Shareholders’ Equity as a deduction from the revenue from the issue, net of tax.
3.83.7 Payments in Shares
Since its formation,incorporation, the Company has established several plans for compensation paid in equity instruments in the form of employee warrants (bons de souscription de parts de créateur d’entrepriseor “BSPCEs”) granted to employees and/or executives and in the form of “share warrants” (bons de souscription d’actions or “BSAs”) granted to non-employee members of the Board of Directors and scientific consultants.
Pursuant to IFRS 2Share-based payment (“IFRS 2”), the cost of the transactions paid with equity instruments is posted to the accounts as an expense in exchange for an increase in the shareholders’ equityShareholders’ Equity for the period during which the rights to be enjoyedbenefited from the equity instruments are acquired.
The Company has applied IFRS 2 to all equity instruments granted since 2002 to its employees, members of the Board of Directors, other individuals, or to companies.
The options are not subject to any market conditions. The characteristics of the options are presented in Note 17.
3.9 Recognition and measurement of Financial Liabilities
Financial Liabilities at Amortized Cost
Borrowings and other financial liabilities are valuatedvalued initially at their fair value and then at the amortized cost, calculated on the basis of the effective interest rate (“EIR”)EIR method.
The transaction expenses that are directly attributable to the acquisition or to the issue of a financial liability reduce that financial liability. These expenses are then amortized over the lifetime of the liability, on the basis of the EIR.
The EIR is the rate that equalizes the anticipated flow of future cash outflows with the current net book value of the financial liability in order to deduct its amortized cost therefrom.
Liabilities at Fair Value per the Income StatementConsolidated Statements of (Loss)
The liabilities at fair value per the income statementConsolidated Statements of (Loss) are valued at their fair value.
3.10 Subsidies and Conditional Advances
Subsidies
The Company receives from time to time assistance in the form of subsidies, which are grants that are not repayable by the Company. The subsidies are recognized when there is reasonable assurance that:
Subsidies that are upfront non-refundable payments are presented as deferred revenue and recognized ratably through income over the duration of the research program to which the subsidy relates.
A public subsidy that is to be received (e.g. from OSEO, the French Agency for Innovation) either as compensation for expenses or for losses already incurred, or for immediate financial support of the Company without associated expenses or losses, is recognized as other income ratably over the duration of the funded project.
Conditional advances
The Company also receives from time to time assistance in the form of conditional advances, which are advances repayable in whole or in part based upon acknowledgment by the funder of a technical or commercial success of the related project by the funding entity. The details concerning the conditional advances are provided in Note 11.
The amount resulting from the deemed benefit of the interest-free nature of the award is considered a subsidy for accounting purposes. This deemed benefit is determined by applying a discount rate equal to the rate of fungible treasury bonds over the time period that corresponds to the time period of the repayment of the advances.
In the event of a change in payment schedule of the stipulated repayments of the conditional advances, the Company makes a new calculation of the net book value of the debt resulting from the discounting of the expected new future cash flows. The adjustment that results therefrom is recognized in the income statement for the fiscal year during which the modification is recognized.
The conditional advances that can be subject to this type of modification are the COFACEwith Compagnie Française d’Assurance pour le Commerce Extérieur (“COFACE”) advances presented in Note 11.1.
3.11 Provisions
Provisions for Risks and Expenses
The provisions for risks and lawsuits correspond to the commitments resulting from lawsuits and various risks whose due dates and amounts are uncertain.
A provision is posted to the accounts when the company has a legal or implicit obligation to a third party resulting from a past event, concerning which it is likely or certain that it will cause an outflow of resources to that third party, without consideration that is anticipated to be at least equivalent to the latter, and that the future outflows of liquid assets can be estimated reliably.
The amount posted torecorded in the accounts as a provision is the best estimation of the expenses necessary to extinguish the obligation.
Pension Retirement Obligations
The employees of the Company receive the retirement benefits stipulated by law in France:
For the defined-benefit plans, the costs of the retirement benefits are estimated by using the projected credit unit method. According to this method, the cost of the retirement pensions is recognized in the income statementConsolidated Statement of (Loss) so that it is distributed uniformly over the term of the services of the employees. The retirement benefit commitments are valued at the current value of the future payments estimated using, for the discounting, the market rate based on the long-term obligations of the first-category companies with a term that corresponds to that estimated for the payment of the services provided.
The Company relies on external actuaries to conduct an annual review of the valuation of these plans.
The difference between the amount of the provision at the beginning of a fiscal year and at the close of that year is recognized through profit or loss for the portion representing the costs of services rendered and through other comprehensive income for the portion representing the actual gains and losses.
The Company’s payments for the defined-contribution plans are recognized as expenses on the income statement of the period with which they are associated.
3.12 Revenue
The sales revenue of the Company results mainly from the sale of the productDiallertest, a kit for diagnosing the allergy to proteins in cow’s milk.
The Company recognizes revenue when the amount can be measured reliably, when it is likely that the future economic advantages will benefit the Company, and when the specific criteria are met for the business activity of the Company. For the product sales, the sales revenue is recognized upon delivery.
3.13 Other Income
Subsidies
Since it was formed, because of its innovative character, the Company has received a certain number of sources of assistance or subsidies from the central government or from local public authorities such as OSEO or the Banque Publique d’Investissement, intended to finance its operations or the recruitment of specific personnel.
TheseWhen the obtention of the subsidy is reasonably certain, these subsidies are recognized as “Other income” forduring the fiscalcalendar year that recordedover which the corresponding expenses or expenditures when obtaining the subsidy is reasonably certain.were recorded.
Research Tax Credit
The Research Tax Credit (Crédit d’Impôt Recherche,CIR) is granted to companies by the French tax authorities in order to encourage them to conduct technical and scientific research. Companies that prove that they have expenditures that meet the required criteria (research expenditures located in France or, since January 1, 2005, within the European Community or in another State that is a party to the Agreement on the European Economic Area that has concluded a tax treaty with France that contains an administrative assistance clause) receive a tax credit that can be used for the payment of the corporate tax due for the fiscal year in which the expenditures were made and the next three fiscal years, or, as applicable, can be reimbursed for the excess portion. The expenditures taken into account for the calculation of the research tax credit involve only research expenses.
The Company has received the Research Tax Credit since it was formed.
The Company received the reimbursement of the Research Tax Credit for the year 2012 for an amount of €2.5 million during the year 2013.
The Company received the reimbursement of the Research Tax Credit for the year 2013 for an amount of €3.3 million during the year 2014. It
The Company received the reimbursement of the Research Tax Credit for the year 2014 for an amount of €4.3 million during the year 2015. The Company will request the reimbursement of the 20142015 Research Tax Credit for an amount of €5.7 million during the year 2016, under the Community tax rules for small and medium firms in compliance with the regulatory texts in effect.applicable.
3.14 Rental Agreements
The rental agreements involving property, plant, and equipment are classified as finance lease agreements when the Company bears a substantial portion of all the benefits and risks inherent in the ownership of the property. The assets that are covered under financing
lease agreements are capitalized as of the beginning date of the rental agreement on the basis of the fair value of the rented asset or the discounted values of the future minimum payments, whichever is lower. Each rental payment is distributed between the debt and the financial cost in such a manner as to determine a constant interest rate on the principal that remains due. The corresponding rental obligations, net of the financial expenses, are classified as financial liabilities. The portion of the financial expense that corresponds to the interest is recognized as an expense over the term of the agreement. The property, plant, or equipment acquired within the framework of a finance lease agreement is amortized over the use period or the term of the lease agreement, whichever is shorter.
The rental agreements for which a significant portion of the risks and advantages is preserved by the lessor are classified as ordinary rental agreements. The payments made for these ordinary rental agreements, net of any incentive measures, are recognized as expenses on the income statement in a linear manner over the term of the agreement.
3.15 Taxes
Income Tax
Deferred taxes are recognized for all the temporary differences arising from the difference between the tax basis and the accounting basis of the assets and liabilities that appear in the financial statements. The primary temporary differences are related to the tax losses that can be carried forward or backward. The tax rates that have been ratified by a legal text as of the closing date are utilized to determine the deferred taxes.
The deferred tax assets are posted torecorded in the accounts only to the extent that it is likely that the future profits will be sufficient to absorb the losses that can be carried forward or backward. Considering its stage of development, which does not allow income projections judged to be sufficiently reliable to be made, the Company has not recognized deferred tax assets in relation to tax losses carryforward in the statementConsolidated Statements of financial position.Financial Position.
3.16 Segment Information
The Company operates in a single operating segment: the conducting of research and development of epicutaneous immunotherapy products in order to market them in the future. The assets, liabilities, and operating loss realizedlosses recognized are primarily located in France.
3.17 Other Items in the Comprehensive Profit (or Loss)
The revenue and expense items for the period that are not posted toin the income statementConsolidated Statements of (Loss) as stipulated by the applicable standards are presented, as necessary, under the rubric “Other items in the comprehensive profit (or loss).”
3.18 Use of Estimates
Our Financial Statements are prepared in accordance with IFRS. The preparation of our Financial Statements requires usthe management to make estimates, assumptions and judgments that affect the reported amounts of assets, liabilities, revenue and expenses. We base our estimatesEstimates and assumptions are based on historical experience and other factors that we believemanagement believes to be reasonable under the circumstances. We evaluate our estimatesEstimates and assumptions are measured on an ongoing basis. Our actualActual results may differ from these estimates.
These estimates and judgments involve mainly:mainly involve:
3.19 Presentation of Financial Assets and Financial Liabilities Measured at Fair Value
In accordance with the amendments to IFRS 7, financial instruments are presented in three categories based on a hierarchical method used to determine their fair value:
3.20 Events After the Close of the Fiscal Year
The statementConsolidated Statements of financial positionFinancial Position and the income statementConsolidated Statements of (Loss) of the Company are adjusted to reflect the subsequent events that alter the amounts related to the situations that exist as of the closing date. The adjustments are made until the date the financial statements are approved and authorized for issuance by the Board of Directors.
Note 4: Intangible Assets
The intangible assets are broken down as follows:
| December 31 | ||||||||||||||||||||||||
| 2012 | 2013 | 2014 | 2013 | 2014 | 2015 | |||||||||||||||||||
| (Amounts in Euros) | (Amounts in thousands of Euros) | |||||||||||||||||||||||
Patents, licenses, trademarks | 29,848 | 31,080 | 45,793 | 31 | 46 | 54 | ||||||||||||||||||
Software | 66,172 | 146,325 | 162,307 | 146 | 162 | 302 | ||||||||||||||||||
Total historical cost | 96,020 | 177,405 | 208,100 | 177 | 208 | 356 | ||||||||||||||||||
Accumulated amort. of patents, licenses, and trademarks | 29,848 | 30,020 | 38,624 | |||||||||||||||||||||
Accumulated amort.of patents, licenses, and trademarks | 30 | 38 | 53 | |||||||||||||||||||||
Accumulated depreciation of software packages | 52,160 | 84,378 | 140,641 | 84 | 141 | 210 | ||||||||||||||||||
Accumulated amortization and depreciation | 82,008 | 114,398 | 179,265 | 114 | 179 | 262 | ||||||||||||||||||
Net total | 14,012 | 63,007 | 28,835 | 63 | 29 | 94 | ||||||||||||||||||
There has been no recognition of impairment losses in application of IAS 36Impairment of Assetsover for the fiscalcalendar years presented.
Note 5: Property, Plant, and Equipment
| 01/01/2012 | Increase | Decrease | 12/31/2012 | 01/01/2013 | Increase | Decrease | 12/31/2013 | |||||||||||||||||||||||||
| (Amounts in Euros) | (Amounts in thousands of Euros) | |||||||||||||||||||||||||||||||
Laboratory equipment | 676,795 | 104,507 | — | 781,302 | 781 | 590 | — | 1,372 | ||||||||||||||||||||||||
Building fixtures | 466,109 | 164,227 | — | 630,336 | 630 | 292 | — | 922 | ||||||||||||||||||||||||
Office equipment | 116,962 | 14,996 | — | 131,958 | 132 | 83 | — | 215 | ||||||||||||||||||||||||
Computer equipment | 143,334 | 56,681 | — | 200,015 | 200 | 74 | — | 274 | ||||||||||||||||||||||||
Other property, plant, and equipment | 48 | — | — | 48 | ||||||||||||||||||||||||||||
Property, plant and equipment in process | — | 51 | — | 51 | ||||||||||||||||||||||||||||
|
|
|
|
|
|
|
| |||||||||||||||||||||||||
Total, gross | 1,403,247 | 340,411 | — | 1,743,658 | 1,744 | 1,090 | — | 2,834 | ||||||||||||||||||||||||
|
|
|
|
|
|
|
| |||||||||||||||||||||||||
Accumulated depreciation of laboratory equipment | 403,262 | 102,974 | — | 506,236 | 506 | 164 | — | 670 | ||||||||||||||||||||||||
Accumulated depreciation of the building fixtures | 21,447 | 57,030 | — | 78,477 | 78 | 100 | — | 179 | ||||||||||||||||||||||||
Accumulated depreciation of office equipment | 43,143 | 15,888 | — | 59,031 | 59 | 33 | — | 92 | ||||||||||||||||||||||||
Accumulated depreciation of computer equipment | 86,156 | 25,427 | — | 111,583 | 112 | 47 | — | 159 | ||||||||||||||||||||||||
Accumulated depreciation of other property, plant, and equipment | 48 | — | — | 48 | ||||||||||||||||||||||||||||
Accumulated depreciation of Property, plant and equipment in progress | — | — | — | — | ||||||||||||||||||||||||||||
|
|
|
|
|
|
|
| |||||||||||||||||||||||||
Total accumulated amortization and depreciation | 554,056 | 201,319 | — | 755,375 | 755 | 344 | — | 1,099 | ||||||||||||||||||||||||
|
|
|
|
|
|
|
| |||||||||||||||||||||||||
Total, net | 849,191 | 139,093 | — | 988,284 | 988 | 746 | — | 1,734 | ||||||||||||||||||||||||
|
|
|
|
|
|
|
| |||||||||||||||||||||||||
Laboratory equipment Building fixtures Office equipment Computer equipment Other property, plant, and equipment Total, gross Accumulated depreciation of laboratory equipment Accumulated depreciation of the building fixtures Accumulated depreciation of office equipment Accumulated depreciation of computer equipment Accumulated depreciation of other property, plant, and equipment Total accumulated amortization and depreciation Total, net 01/01/2013 Increase Decrease 12/31/2013 (Amounts in Euros) 781,302 590,306 — 1,371,607 630,336 291,890 — 922,226 131,958 83,010 — 214,968 200,015 74,127 — 274,141 48 50,570 — 50,618 1,743,659 1,089,902 — 2,833,560 506,236 163,717 — 669,953 78,477 100,044 — 178,520 59,031 33,066 — 92,097 111,583 47,195 — 158,779 48 — — 48 755,375 344,023 — 1,099,397 988,284 745,879 — 1,734,163
| 01/01/2014 | Increase | Decrease | 12/31/2014 | |||||||||||||
| (Amounts in Euros) | ||||||||||||||||
Laboratory equipment | 1,371,607 | 885,680 | — | 2,257,287 | ||||||||||||
Building fixtures | 922,226 | 7,702 | — | 929,928 | ||||||||||||
Office equipment | 214,968 | — | 214,968 | |||||||||||||
Computer equipment | 274,141 | 47,919 | — | 322,060 | ||||||||||||
Other property, plant, and equipment | 50 618 | — | 48 | 50,570 | ||||||||||||
|
|
|
|
|
|
|
| |||||||||
Total, gross | 2,833,560 | 941,301 | 48 | 3,774,813 | ||||||||||||
|
|
|
|
|
|
|
| |||||||||
Accumulated depreciation of laboratory equipment | 669,953 | 245,716 | — | 915,669 | ||||||||||||
Accumulated depreciation of the building fixtures | 178,520 | 107,367 | — | 285,887 | ||||||||||||
Accumulated depreciation of office equipment | 92,097 | 33,806 | — | 125,903 | ||||||||||||
Accumulated depreciation of computer equipment | 158,779 | 63,647 | — | 222,426 | ||||||||||||
Accumulated depreciation of other property, plant, and equipment | 48 | — | 48 | — | ||||||||||||
|
|
|
|
|
|
|
| |||||||||
Total accumulated amortization and depreciation | 1,099,397 | 450,522 | 48 | 1,549,885 | ||||||||||||
|
|
|
|
|
|
|
| |||||||||
Total, net | 1,734,163 | 490,779 | — | 2,224,928 | ||||||||||||
|
|
|
|
|
|
|
| |||||||||
Laboratory equipment Building fixtures Office equipment Computer equipment Property, plant and equipment in process Total, gross Accumulated depreciation of laboratory equipment Accumulated depreciation of the building fixtures Accumulated depreciation of office equipment Accumulated depreciation of computer equipment Accumulated depreciation of Property, plant and equipment in progress Total accumulated amortization and depreciation Total, net Laboratory equipment Building fixtures Office equipment Computer equipment Property, plant and equipment in process Total, gross Accumulated depreciation of laboratory equipment Accumulated depreciation of the building fixtures Accumulated depreciation of office equipment Accumulated depreciation of computer equipment Accumulated depreciation of Property, plant and equipment in progress Total accumulated amortization and depreciation Total, net 01/01/2014 Increase Decrease 12/31/2014 (Amounts in thousands of Euros) 1,372 886 — 2,257 922 8 — 930 215 — — 215 274 48 — 322 51 — — 51 2,834 941 — 3,775 670 246 — 916 179 107 — 286 92 34 — 126 159 64 — 222 — — — — 1,099 451 — 1,550 1,734 491 — 2,225 01/01/2015 Increase Decrease 12/31/2015 (Amounts in thousands of Euros) 2,257 467 (368 ) 2,357 930 8 — 938 215 24 (30 ) 209 322 244 (95 ) 470 51 3,622 — 3,672 3,775 4,360 (489 ) 7,646 916 382 (349 ) 948 286 496 — 782 126 35 (30 ) 131 222 78 (95 ) 205 — — — — 1,550 990 (475 ) 2,065 2,225 3,370 (14 ) 5,581
Over the two fiscalthree years presented, the acquisitions correspond primarily to building fixtures and to laboratory and production equipment and material. The increase in the building fixturesproperty, plant and equipment in progress item is related to the improvements made infitting out of the Company’s new premises based in Bagneux, France.head office of the Company located at Montrouge, as well as the purchase of materials for the conception and tuning of the new prototypes (Gen 4.0 and Cut Pack for example).
Note 6: Non-Current Financial Assets
| December 31 | ||||||||||||||||||||||||
| 2012 | 2013 | 2014 | 2013 | 2014 | 2015 | |||||||||||||||||||
| (Amounts in Euros) | (Amounts in thousands of Euros) | |||||||||||||||||||||||
Deposits | 82,999 | 82,342 | 99,825 | 82 | 100 | 716 | ||||||||||||||||||
Pledged securities | 275,510 | 278,057 | 384,809 | 278 | 385 | 611 | ||||||||||||||||||
Liquidity contract | 25,848 | 263,430 | 1,111,227 | 263 | 1,111 | 1,384 | ||||||||||||||||||
Total non-current financial assets | 384,357 | 623,829 | 1,595,861 | 624 | 1,596 | 2,711 | ||||||||||||||||||
The non-current financial assets are composed of security deposits paid to the lessor and of open-ended mutual funds (sociétés d’investissement à capital variable “SICAVs”) pledged as guarantees of the ordinary rental agreements and the liquidity contract.
Under the liquidity contract, 8,0543,898 treasury shares were allocated for the reduction of shareholders’ equityShareholders’ Equity as at December 31, 20142015 with the cash balance being maintained in financial assets. The share capital is divided in 19,160,66124,205,129 shares including these 8,0543,898 treasury shares.
On March 31, 2014, DBV Technologies announced an additional contribution of €300,000 to the liquidity agreement held by Natixis in accordance with the Charter of Ethics established by the AMAFI of March 8, 2011 and approved by the Autorité des Marchés Financiers.
On December 15, 2014, DBV Technologies announced an additional contribution of €600,000 to the liquidity agreement held by Natixis in accordance with the Charter of Ethics established by the AMAFI of March 8, 2011 and approved by the Autorité des Marchés Financiers.
Note 7: Inventories and Work in Progress
| December 31 | ||||||||||||||||||||||||
| 2012 | 2013 | 2014 | 2013 | 2014 | 2015 | |||||||||||||||||||
| (Amounts in Euros) | (Amounts in thousands of Euros) | |||||||||||||||||||||||
Inventories of raw materials | 28,023 | 6,568 | 124,071 | 7 | 124 | 69 | ||||||||||||||||||
Depreciation of inventories and work in progress | 1,650 | — | — | — | — | (69 | ) | |||||||||||||||||
Total net value of the inventories and work in progress | 29,673 | 6,568 | 124,071 | 7 | 124 | — | ||||||||||||||||||
The inventories and work in progress involve the Diallertest product.® product which commercial partnership was settled on the 1st of September 2015.
Note 8: Customer Accounts Receivable and Other Current Assets
8.1 Customer Accounts Receivable and Related Receivables
| 2012 | 2013 | 2014 | ||||||||||
| (Amounts in Euros) | ||||||||||||
Accounts receivable and related receivables | 138,322 | 195,997 | 149,209 | |||||||||
Valuation allowance (charged to income statement) | (45,447 | ) | (13,097 | ) | (13,097 | ) | ||||||
Total net value of accounts receivable | 92,875 | 182,900 | 136,112 | |||||||||
Accounts receivable and related receivables Valuation allowance (charged to Consolidated Statements of (Loss)) Total net value of accounts receivable December 31 2013 2014 2015 (Amounts in thousands of Euros) 196 149 13 (13 ) (13 ) (13 ) 183 136 —
All the customer accounts receivable have payment terms of less than one year.
Accounts receivable and related receivables relate primarily to the sales of Diallertest.Diallertest®
| Additions | ||||||||||||||||||||||||||||||||||||||||
| (in thousands of euros). | Balance at beginning of period | Charged to | Charged to | Balance at end of period | ||||||||||||||||||||||||||||||||||||
Description—2015 | Income statement | Other accounts | Reversal | |||||||||||||||||||||||||||||||||||||
Valuation allowance deducted from account receivables | 13 | — | — | — | 13 | |||||||||||||||||||||||||||||||||||
| Additions | Additions | |||||||||||||||||||||||||||||||||||||||
| Balance at beginning of period | Charged to | Charged to | Balance at end of period | Balance at beginning of period | Charged to | Charged to | Balance at end of period | |||||||||||||||||||||||||||||||||
Description— 2014 | Income statement | Other accounts | Reversal | |||||||||||||||||||||||||||||||||||||
Description—2014 | Balance at beginning of period | Income statement | Other accounts | Reversal | Balance at end of period | |||||||||||||||||||||||||||||||||||
Valuation allowance deducted from account receivables | 13,097 | — | — | — | 13,097 | — | — | — | ||||||||||||||||||||||||||||||||
| Additions | ||||||||||||||||||||||||||||||||||||||||
| Balance at beginning of period | Charged to | Charged to | Balance at end of period | |||||||||||||||||||||||||||||||||||||
Description—2013 | Income statement | Other accounts | Reversal | |||||||||||||||||||||||||||||||||||||
Valuation allowance deducted from Account receivables | 45 | — | — | 32 | 13 | |||||||||||||||||||||||||||||||||||
| Additions | ||||||||||||||||||||
| Balance at beginning of period | Charged to | �� | Charged to | Balance at end of period | ||||||||||||||||
Description—2013 | Income statement | Other accounts | Reversal | |||||||||||||||||
Valuation allowance deducted from Account receivables | 45,447 | — | — | 32,350 | 13,097 | |||||||||||||||
| Additions | ||||||||||||||||||||
| Balance at beginning of period | Charged to | Charged to | Balance at end of period | |||||||||||||||||
Description—2012 | costs and expenses | Other accounts | Reversal | |||||||||||||||||
Valuation allowance deducted from Account receivables | 13,097 | 32,350 | — | — | 45,447 | |||||||||||||||
The valuation allowance of €32,350 posted in 2012 has been reversed in 2013 (and classified as a deduction of general and administrative expenses) following receipt of the corresponding payment. It explains the decrease in depreciation of accounts receivable in 2013.
8.2 Other current assets
The other current assets are broken down as follows:
| December 31 | ||||||||||||||||||||||||
| 2012 | 2013 | 2014 | 2013 | 2014 | 2015 | |||||||||||||||||||
| (Amounts in Euros) | (Amounts in thousands of Euros) | |||||||||||||||||||||||
Research tax credit | 2,522,399 | 3,312,462 | 4,339,620 | 3,312 | 4,340 | 5,702 | ||||||||||||||||||
Other tax claims | 355,728 | 594,723 | 1 022,563 | 595 | 1,023 | 2,550 | ||||||||||||||||||
Other receivables | 45,664 | — | 422,516 | — | 423 | 1,409 | ||||||||||||||||||
Prepaid expenses | 193,696 | 315,611 | 937,864 | 316 | 938 | 1,850 | ||||||||||||||||||
Total | 3,117,487 | 4,222,796 | 6,722,563 | 4,223 | 6,723 | 11,512 | ||||||||||||||||||
The other tax debt claims are primarily related to the deductible VAT as well as the reimbursement of VAT that has been requested.
As at December 31, 2015, 2014 and 2013, prepaid expenses are comprised primarily of rental and insurance expenses, as well as legal and scientific consulting fees.
As at December 31, Their variation is linked to an increase in insurance premiums following the Company’s public offering on the Nasdaq stock exchange in 2014 prepaid expenses are comprised primarily of rental and insurance expenses, as well as legal and scientific consulting fees.the July 2015 fundraising.
Research Tax Credit
The companyCompany benefits from the provisions in Articles 244quater B and 49septies F of the French Tax Code related to the Research Tax Credit. In compliance with the principles described in Note 3.14,3.13, the Research Tax Credit is posted to the accounts as “other income”“Other Income” during the year with which the eligible research expenditures are associated.
The changes in this Research Tax Credit over the last three fiscal years are presented as follows:
| Amounts in | ||||
| ||||
| ||||
| ||||
| ||||
| ||||
Opening balance sheet receivable as of January 1, 2013 | ||||
+ Operating revenue | ||||
- Payment received | ( | ) | ||
- Adjustment | ( | ) | ||
|
| |||
Closing balance sheet receivable as of December 31, 2013 | ||||
|
| |||
| Amounts in thousands of Euros | |||||||
Opening Balance Sheet Receivable as of January 1, 2014 | |||||||
+ Operating revenue | |||||||
- Payment received | ( | ) | |||||
|
| ||||||
Closing Balance Sheet Receivable as of December 31, 2014 | |||||||
| Amounts in thousands of Euros | |||||||
Opening Balance Sheet Receivable as of January 1, 2015 | 4,340 | ||||||
+ Operating revenue | 5,702 | ||||||
- Payment received | (4,322 | ) | |||||
- Adjustment | (18 | ) | |||||
Closing Balance Sheet Receivable as of December 31, 2014 | 5,702 | ||||||
|
| ||||||
The adjustments to the amount of the receivable reflect the penalties related to the fiscal verification of the Research Tax Credit 2012 that occurred in 2013.2015.
Note 9: Cash and Cash Equivalents
The cash and cash equivalents item is broken down as follows (in thousands of Euros):
| 2012 | 2013 | 2014 | ||||||||||
| (Amounts in Euros) | ||||||||||||
Cash | 1,085 | 826,154 | 107,690 | |||||||||
Cash equivalents: term deposits | 38,347,045 | 38,576,607 | 114,475,451 | |||||||||
Total cash and cash equivalent as reported in statement of financial position | 38,348,130 | 39,402,761 | 114,583,141 | |||||||||
Bank overdrafts | (519,499 | ) | — | (27,956 | ) | |||||||
Total net cash and cash equivalents as reported in the statement of cash flow | 37,828,631 | 39,402,761 | 114,555,185 | |||||||||
| December 31 | ||||||||||||
| 2013 | 2014 | 2015 | ||||||||||
| (Amounts in thousands of Euros) | ||||||||||||
Cash | 826 | 108 | 178,895 | |||||||||
Cash equivalents | 38,577 | 114,475 | 144,486 | |||||||||
Total cash and cash equivalent as reported in statement of financial position | 39,403 | 114,583 | 323,381 | |||||||||
Bank overdrafts | — | (28 | ) | — | ||||||||
Total net cash and cash equivalents as reported in the statement of cash flow | 39,403 | 114,555 | 323,381 | |||||||||
Term depositsCash equivalents are immediately convertible into cash at no cost. They are measured using level 1 fair value measurements. The increase in cash and cash equivalents mainly relates to the net proceeds received as part of our initial public offering that was completed in October 2014.July 2015 fundraising.
Note 10: Capital
10.1 Share Capital Issued
The share capital, as of December 31, 2014,2015, is set at the sum of €1,916,066.10.€2,420,512.90. It is divided into 19,160,66124,205,129 fully authorized, subscribed and paid-up shares with a nominal value of €0.10.
This number does not include share warrants (“BSA”), employee warrants (“BSPCE”), stock-options (“SO”) and performance shares (“AGA”) granted to certain investors and to certain natural persons, both employees and non-employees of the Company.
All the shares give their owners the right to a proportional share of the income and the net assets of the Company.
The table below presents the historical changes in the share capital of the Company as at December 31 2012,of December 31, 2013, 2014 and 2014:2015:
Date | Nature of the Transactions | Share Capital | Share premium | Number of Shares | Nominal value | |||||||||||||||||||||||||||||||
Balance as of January 1, 2012 | € | 882,274.50 | € | 17,508,641.11 | 8,822,745 | € | 0.10 | |||||||||||||||||||||||||||||
01/17/2012 and 09/25/2012 | Issue of share subscription warrants | € | 8,560 | |||||||||||||||||||||||||||||||||
03/28/2012 | Capital increase by issuance of common shares | € | 457,317.10 | € | 36,988,256.33 | 4,573,171 | ||||||||||||||||||||||||||||||
04/26/2012 | Capital increase by issuance of common shares | € | 1,223.10 | € | 107,143.56 | 12,231 | ||||||||||||||||||||||||||||||
|
|
|
| |||||||||||||||||||||||||||||||||
Balance as of December 31, 2012 | € | 1,340,814.70 | € | 54,612,601.00 | 13,408,147 | € | 0.10 | |||||||||||||||||||||||||||||
Date | Nature of the Transactions | Share Capital | Share premium | Number of Shares | Nominal value | Nature of the Transactions | Share Capital | Share premium | Number of Shares | Nominal value | ||||||||||||||||||||||||||
Balance as of January 1, 2013 | € | 1 340,814.70 | € | 54,612,601.00 | 13,408,147 | € | 0.10 | Balance as of January 1, 2013 | K€ | 1,340.8 | K€ | 54,612.6 | 13,408,147 | € | 0.10 | |||||||||||||||||||||
07/25/2013 | Issue of share subscription warrants | € | 67,440.00 | Issue of share subscription warrants | K€ | 67.4 | ||||||||||||||||||||||||||||||
11/14/2013 | Capital increase by issuance of common shares | € | 168,015.10 | € | 14,960,857.70 | 1,680,151 | Capital increase by issuance of common shares | K€ | 168 | K€ | 14,960.9 | 1,680,151 | € | 0.10 | ||||||||||||||||||||||
|
|
|
|
|
|
|
| |||||||||||||||||||||||||||||
Balance as of December 31, 2013 | € | 1,508,829.80 | € | 69,640,898.70 | 15,088,298 | € | 0.10 | Balance as of December 31, 2013 | K€ | 1,508.8 | K€ | 69,640.9 | 15,088,298 | € | 0.10 | |||||||||||||||||||||
Date | Nature of the Transactions | Share Capital | Share premium | Number of Shares | Nominal value | Nature of the Transactions | Share Capital | Share premium | Number of Shares | Nominal value | ||||||||||||||||||||||||||
Balance as of January 1, 2014 | € | 1,508,829.80 | € | 69,640,898.70 | 15,088,298 | € | 0.10 | Balance as of January 1, 2014 | K€ | 1,508.8 | K€ | 69,640.9 | 15,088,298 | € | 0.10 | |||||||||||||||||||||
01/23/2014 | Capital increase by issuance of common shares | € | 3,765.00 | € | 189,379.50 | 37,650 | Capital increase by issuance of common shares | K€ | 3.8 | K€ | 189.4 | 37,650 | € | 0.10 | ||||||||||||||||||||||
02/11/2014 | Capital increase by issuance of common shares | € | 500.00 | € | 41,225.00 | 5,000 | Capital increase by issuance of common shares | K€ | 0.5 | K€ | 41.2 | 5,000 | € | 0.10 | ||||||||||||||||||||||
04/02/2014 | Capital increase by incorporation of reserve | € | 24,248.40 | € | (24,248.40 | ) | 242,484 | Capital increase by incorporation of reserve | K€ | 24.2 | K€ | (24.2 | ) | 242,484 | € | 0.10 | ||||||||||||||||||||
06/11/2014 | Capital increase by issuance of common shares | € | 4,500.00 | € | 226,350.00 | 45,000 | Capital increase by issuance of common shares | K€ | 4.5 | K€ | 226.4 | 45,000 | € | 0.10 | ||||||||||||||||||||||
06/13/2014 | Capital increase by issuance of common shares | € | 4,000.50 | € | 169,221.15 | 40,005 | Capital increase by issuance of common shares | K€ | 4.0 | K€ | 169.2 | 40,005 | € | 0.10 | ||||||||||||||||||||||
06/18/2014 | Capital increase by issuance of common shares | € | 975.00 | € | 44,557.50 | 9,750 | Capital increase by issuance of common shares | K€ | 1.0 | K€ | 44.6 | 9,750 | € | 0.10 | ||||||||||||||||||||||
06/19/2014 | Capital increase by issuance of common shares | € | 100.50 | € | 5,055.15 | 1,005 | Capital increase by issuance of common shares | K€ | 0.1 | K€ | 5.1 | 1,005 | € | 0.10 | ||||||||||||||||||||||
07/25/2014 | Capital increase by incorporation of reserve | € | 4,469.30 | € | (4,469.30 | ) | 44,693 | Capital increase by incorporation of reserve | K€ | 4.5 | K€ | (4.5 | ) | 44,693 | € | 0.10 | ||||||||||||||||||||
09/19/2014 | Capital increase by incorporation of reserve | € | 25,741.80 | € | (25,741.80 | ) | 257,418 | Capital increase by incorporation of reserve | K€ | 25.7 | K€ | (25.7 | ) | 257,418 | € | 0.10 | ||||||||||||||||||||
10/03/2014 | Capital increase by issuance of common shares | € | 2,296.50 | € | 104,950.05 | 22,965 | Capital increase by issuance of common shares | K€ | 2.3 | K€ | 105 | 22,965 | € | 0.10 | ||||||||||||||||||||||
10/22/2014 | Capital increase by issuance of common shares | € | 307,468.60 | € | 104,231,855.40 | 3,074,686 | Capital increase by issuance of common shares | K€ | 307.5 | K€ | 104,231.9 | 3,074,686 | € | 0.10 | ||||||||||||||||||||||
11/01/2014 | Capital increase by incorporation of reserve | € | 25,742.20 | € | (25,742.20 | ) | 257,422 | Capital increase by incorporation of reserve | K€ | 25.7 | K€ | (25.7 | ) | 257,422 | € | 0.10 | ||||||||||||||||||||
11/30/2014 | Capital increase by issuance of common shares | € | 1,756.00 | € | 96,976.80 | 17,560 | Capital increase by issuance of common shares | K€ | 1.8 | K€ | 97 | 17,560 | € | 0.10 | ||||||||||||||||||||||
11/30/2014 | Capital increase by issuance of common shares | € | 1,672.50 | € | 83,333.25 | 16,725 | Capital increase by issuance of common shares | K€ | 1.7 | K€ | 83.3 | 16,725 | € | 0.10 | ||||||||||||||||||||||
12/31/2014 | Issue of share subscription warrants | € | 32, 266.55 | Issue of share subscription warrants | K€ | 32.3 | € | 0.10 | ||||||||||||||||||||||||||||
12/31/2014 | Fees charged to share premium 2014 | € | (10,909,078.52 | ) | Fees charged to share premium 2014 | K€ | (10,909.1 | ) | ||||||||||||||||||||||||||||
|
|
|
|
|
|
|
| |||||||||||||||||||||||||||||
Balance as of December 31, 2014 | € | 1,916,066.10 | € | 163,876,788.83 | 19,160,661 | € | 0.10 | Balance as of December 31, 2014 | K€ | 1,916.1 | K€ | 163,876.8 | 19,160,661 | € | 0.10 | |||||||||||||||||||||
Date | Nature of the Transactions | Share Capital | Share premium | Number of Shares | Nominal value | |||||||||||||||||||||||||||||||
Balance as of January 1, 2015 | K€ | 1,916.1 | K€ | 163,876.8 | 19,160,661 | € | 0.10 | |||||||||||||||||||||||||||||
01/06/2015 | Capital increase by issuance of common shares | K€ | 0.4 | K€ | 18.9 | 3,750 | € | 0.10 | ||||||||||||||||||||||||||||
01/07/2015 | Capital increase by issuance of common shares | K€ | 1.0 | K€ | 59.0 | 10,000 | € | 0.10 | ||||||||||||||||||||||||||||
01/22/2015 | Capital increase by incorporation of reserve | K€ | 3.5 | K€ | (3.5 | ) | 35,360 | € | 0.10 | |||||||||||||||||||||||||||
01/30/2015 | Capital increase by issuance of common shares | K€ | 9.8 | K€ | 476.8 | 97,720 | € | 0.10 | ||||||||||||||||||||||||||||
02/03/2015 | Capital increase by issuance of common shares | K€ | 0.8 | K€ | 37.7 | 7,500 | € | 0.10 | ||||||||||||||||||||||||||||
02/13/2015 | Capital increase by issuance of common shares | K€ | 3.8 | K€ | 188.6 | 37,500 | € | 0.10 | ||||||||||||||||||||||||||||
02/17/2015 | Capital increase by issuance of common shares | K€ | 2.0 | K€ | 100.6 | 19,995 | € | 0.10 | ||||||||||||||||||||||||||||
03/04/2015 | Capital increase by issuance of common shares | K€ | 1.0 | K€ | 59 | 10,000 | € | 0.10 | ||||||||||||||||||||||||||||
03/26/2015 | Capital increase by issuance of common shares | K€ | 5.0 | K€ | 400 | 50,000 | € | 0.10 | ||||||||||||||||||||||||||||
04/07/2015 | Capital increase by issuance of common shares | K€ | 4.0 | K€ | 169.2 | 40,005 | € | 0.10 | ||||||||||||||||||||||||||||
04/09/2015 | Capital increase by issuance of common shares | K€ | 15.0 | K€ | 692.1 | 150,375 | € | 0.10 | ||||||||||||||||||||||||||||
04/28/2015 | Capital increase by issuance of common shares | K€ | 1.4 | K€ | 77.1 | 14,290 | € | 0.10 | ||||||||||||||||||||||||||||
05/05/2015 | Issue of share subscription warrants | K€ | K€ | 43 | ||||||||||||||||||||||||||||||||
05/11/2015 | Capital increase by issuance of common shares | K€ | 2.0 | K€ | 100.7 | 20,010 | € | 0.10 | ||||||||||||||||||||||||||||
06/23/2015 | Capital increase by issuance of common shares | K€ | 59.1 | K€ | 27,029.4 | 590,543 | € | 0.10 | ||||||||||||||||||||||||||||
06/23/2015 | Capital Decrease | K€ | (58.6 | ) | K€ | (26,823.4 | ) | (586,048 | ) | € | 0.10 | |||||||||||||||||||||||||
07/20/2015 | Capital increase by issuance of common shares | K€ | 414.0 | K€ | 254,932.9 | 4,140,000 | € | 0.10 | ||||||||||||||||||||||||||||
07/24/2015 | Capital increase by issuance of common shares | K€ | 1.6 | K€ | 79.2 | 15,750 | € | 0.10 | ||||||||||||||||||||||||||||
07/25/2015 | Capital increase by incorporation of reserve | K€ | 28.6 | K€ | (28.6 | ) | 286,338 | € | 0.10 | |||||||||||||||||||||||||||
07/28/2015 | Capital increase by issuance of common shares | K€ | 1.7 | K€ | 91.5 | 17,000 | € | 0.10 | ||||||||||||||||||||||||||||
09/18/2015 | Capital increase by issuance of common shares | K€ | 0.3 | K€ | 46.7 | 2,500 | € | 0.10 | ||||||||||||||||||||||||||||
10/07/2015 | Capital increase by issuance of common shares | K€ | 4.8 | K€ | 241.2 | 47,955 | € | 0.10 | ||||||||||||||||||||||||||||
12/10/2015 | Capital increase by issuance of common shares | K€ | 1.8 | K€ | 146.9 | 17,500 | € | 0.10 | ||||||||||||||||||||||||||||
12/10/2015 | Issue of share subscription warrants | K€ | K€ | 99.2 | ||||||||||||||||||||||||||||||||
12/14/2015 | Capital increase by issuance of common shares | K€ | 1.6 | K€ | 69.5 | 16,425 | € | 0.10 | ||||||||||||||||||||||||||||
12/31/2015 | Fees charged to share premium | K€ | K€ | (18,269.7 | ) | |||||||||||||||||||||||||||||||
|
|
|
| |||||||||||||||||||||||||||||||||
Balance as of December 31, 2015 | K€ | 2,420.5 | K€ | 403,910.4 | 24,205,129 | € | 0.10 | |||||||||||||||||||||||||||||
The fees and banks commissions related to share capital increases in 2015 were posted in deduction of the share premium, amounting to €10,909,078.52€18.3 million.
On July 20, 2015, the Company announced the closing of its underwritten public offering of 4,140,000 ordinary shares in the form of 8,280,000 ADSs, at a public price of $34.00 per ADS, before underwriting discounts and commissions, which included an additional 1,080,000 ADSs sold pursuant to the full exercise of the underwriters’ option to purchase additional ADSs. Each ADS represents the right to receive one-half of one ordinary share.
As part of the public offering completed in July 2015, share capital increased by the issuance of 4,140,000 shares (K€414) with a corresponding increase of €236.9 million in share premium (€254.9 million gross, or €236.9 million net after deduction of fees and expenses of €18.0 million).
Initial public offering on the NASDAQ Global Market
On October 22, 2014, DBV Technologies announced the pricing of its global offering of 2,673,641 common shares, of which 2,138,913 ordinary shares represented by 4,277,826 American Depositary Shares (ADS)ADS at the subscription price of $ 21.64 per ADS, as part of a public offering conducted in the United States, Canada and some countries outside of France, and 534,728 common shares at a price of € 34€34 per share, under a concurrent private placement conducted by leaders banks and international sales agents in France and in some countries outside the United States and Canada.
As part of the initial public offering completed in October 2014, share capital increased by the issuance of 3,074,686 shares €(307,468.60)(€307,469) with a corresponding increase of €93,403,561.17€93,403,561 in share premium (€104,231,855.40104,231,855 gross, or €93,403,561.17€93,403,561 net after deduction of fees and expenses for €10,828,294.23)€10,828,294).
10.2 Share Warrants and Employee Warrants
The company has issued share warrants (BSAs), employee warrants (BSPCEs), performance shares (AGAs) and stock-options (SO) as follows:
Date | Type | Number of warrants issued as of 12/31/2012 | Number of warrants null and void as of 12/31/2012 | Number of warrants null and outstanding as of 12/31/2012 | Maximum number of shares to be issued | Strike price per share | ||||||||||||||||
12/23/2005 | BSA/BSPCE | 17,115 | 17,115 | — | — | € | — | |||||||||||||||
12/07/2007 | BSA | 1,717 | 572 | 1,145 | 17,175 | € | 4.33 | |||||||||||||||
01/21/2009 | BSA/BSPCE | 16,380 | — | 16,380 | 245,700 | € | 4.33 | |||||||||||||||
01/21/2009 | BSPCE | 2,296 | — | 2,296 | 34,440 | € | 4.33 | |||||||||||||||
06/25/2010 | BSA | 1,825 | — | 1,825 | 27,375 | € | 4.33 | |||||||||||||||
01/28/2011 | BSA | 10,039 | 7,529 | 2,510 | 37,650 | € | 5.13 | |||||||||||||||
06/24/2011 | BSA/BSPCE | 32,000 | — | 32,000 | 480,000 | € | 5.13 | |||||||||||||||
11/22/2011 | BSA/BSPCE | 11,377 | — | 11,377 | 170,655 | € | 5.13 | |||||||||||||||
01/17/2012 | BSA | 89,835 | — | 89,835 | 89,835 | € | 5.13 | |||||||||||||||
04/02/2012 | AGA | 669,796 | — | 669,796 | 669,796 | € | — | |||||||||||||||
07/25/2012 | AGA | 134,081 | — | 134,081 | 134,081 | € | — | |||||||||||||||
09/25/2012 | BSA | 30,000 | — | 30,000 | 30,000 | € | 8.59 | |||||||||||||||
11/28/2012 | AGA | 35,360 | — | 35,360 | 35,360 | € | — | |||||||||||||||
|
|
|
|
|
|
|
| |||||||||||||||
Total | 1,051,821 | 25,216 | 1,026,605 | 1,972,067 | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||||
Date | Type | Number of warrants issued as of 12/31/2013 | Number of warrants null and void as of 12/31/2013 | Number of warrants outstanding as of 12/31/2013 | Maximum number of shares to be issued | Strike price per share | Type | Number of warrants issued as of 12/31/2013 | Number of warrants null and void as of 12/31/2013 | Number of warrants outstanding as of 12/31/2013 | Maximum number of shares to be issued | Strike price per share | ||||||||||||||||||||||||||||||||
12/07/2007 | BSA | 1,717 | 572 | 1,145 | 17,175 | € | 4.33 | BSA | 1,717 | 572 | 1,145 | 17,175 | € | 4.33 | ||||||||||||||||||||||||||||||
01/21/2009 | BSA/BSPCE | 16,380 | — | 16,380 | 245,700 | € | 4.33 | BSA/BSPCE | 16,380 | — | 16,380 | 245,700 | € | 4.33 | ||||||||||||||||||||||||||||||
01/21/2009 | BSPCE | 2,296 | — | 2,296 | 34,440 | € | 4.33 | BSPCE | 2,296 | — | 2,296 | 34,440 | € | 4.33 | ||||||||||||||||||||||||||||||
06/25/2010 | BSA | 1,825 | — | 1,825 | 27,375 | € | 4.33 | BSA | 1,825 | — | 1,825 | 27,375 | € | 4.33 | ||||||||||||||||||||||||||||||
01/28/2011 | BSA | 10,039 | 7,529 | 2,510 | 37,650 | € | 5.13 | BSA | 10,039 | 7,529 | 2,510 | 37,650 | € | 5.13 | ||||||||||||||||||||||||||||||
06/24/2011 | BSA/BSPCE | 32,000 | — | 32,000 | 480,000 | € | 5.13 | BSA/BSPCE | 32,000 | — | 32,000 | 480,000 | € | 5.13 | ||||||||||||||||||||||||||||||
11/22/2011 | BSA/BSPCE | 11,377 | — | 11,377 | 170,655 | € | 5.13 | BSA/BSPCE | 11,377 | — | 11,377 | 170,655 | € | 5.13 | ||||||||||||||||||||||||||||||
01/17/2012 | BSA | 89,835 | — | 89,835 | 89,835 | € | 5.13 | BSA | 89,835 | — | 89,835 | 89,835 | € | 5.13 | ||||||||||||||||||||||||||||||
04/02/2012 | AGA | 669,796 | — | 669,796 | 669,796 | € | — | AGA | 669,796 | — | 669,796 | 669,796 | € | — | ||||||||||||||||||||||||||||||
07/25/2012 | AGA | 134,081 | — | 134,081 | 134,081 | € | — | AGA | 134,081 | — | 134,081 | 134,081 | € | — | ||||||||||||||||||||||||||||||
09/25/2012 | BSA | 30,000 | — | 30,000 | 30,000 | € | 8.59 | BSA | 30,000 | — | 30,000 | 30,000 | € | 8.59 | ||||||||||||||||||||||||||||||
11/28/2012 | AGA | 35,360 | — | 35,360 | 35,360 | € | — | AGA | 35,360 | — | 35,360 | 35,360 | € | — | ||||||||||||||||||||||||||||||
07/25/2013 | BSA | 73,000 | — | 73,000 | 73,000 | € | 8.10 | BSA | 73,000 | — | 73,000 | 73,000 | € | 8.10 | ||||||||||||||||||||||||||||||
09/12/2013 | AGA | 501,500 | — | 501,500 | 501,500 | € | — | AGA | 501,500 | — | 501,500 | 501,500 | € | — | ||||||||||||||||||||||||||||||
09/18/2013 | SO | 518,000 | — | 518,000 | 518,000 | € | 7.57 | SO | 518,000 | — | 518,000 | 518,000 | € | 7.57 | ||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
| |||||||||||||||||||||||||||||||||||||
Total | 2,127,206 | 8,101 | 2,119,105 | 3,064,567 | 2,127,206 | 8,101 | 2,119,105 | 3,064,567 | ||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||||||||||||||||||||||||||
Date | Type | Number of warrants issued as of 12/31/2014 | Number of warrants null and void as of 12/31/2014 | Number of warrants null and outstanding as of 12/31/2014 | Maximum number of shares to be issued | Strike price per share | ||||||||||||||||||||||||||||||||||||||
12/07/2007 | BSA | 1,717 | 572 | 1,145 | 17,175 | € | 4.33 | |||||||||||||||||||||||||||||||||||||
01/21/2009 | BSA/BSPCE | 16,380 | — | 13,713 | 205,695 | € | 4.33 | |||||||||||||||||||||||||||||||||||||
01/21/2009 | BSPCE | 2,296 | — | — | — | € | 4.33 | |||||||||||||||||||||||||||||||||||||
06/25/2010 | BSA | 1,825 | — | 1,825 | 27,375 | € | 4.33 | |||||||||||||||||||||||||||||||||||||
01/28/2011 | BSA | 10,039 | 7,529 | — | — | € | 5.13 | |||||||||||||||||||||||||||||||||||||
06/24/2011 | BSA/BSPCE | 32,000 | — | 28,933 | 433,995 | € | 5.13 | |||||||||||||||||||||||||||||||||||||
11/22/2011 | BSA/BSPCE | 11,377 | — | 9,373 | 140,595 | € | 5.13 | |||||||||||||||||||||||||||||||||||||
01/17/2012 | BSA | 89,835 | — | 89,835 | 89,835 | € | 5.13 | |||||||||||||||||||||||||||||||||||||
04/02/2012 | AGA | 669,796 | — | — | — | € | — | |||||||||||||||||||||||||||||||||||||
07/25/2012 | AGA | 134,081 | — | — | — | € | — | |||||||||||||||||||||||||||||||||||||
09/25/2012 | BSA | 30,000 | — | 25,000 | 25,000 | € | 8.59 | |||||||||||||||||||||||||||||||||||||
11/28/2012 | AGA | 35,360 | — | 35,360 | 35,360 | € | — | |||||||||||||||||||||||||||||||||||||
07/25/2013 | BSA | 73,000 | — | 70,500 | 70,500 | € | 8.10 | |||||||||||||||||||||||||||||||||||||
09/12/2013 | AGA | 501,500 | — | 420,000 | 420,000 | € | — | |||||||||||||||||||||||||||||||||||||
09/18/2013 | SO | 518,000 | — | 471,000 | 471,000 | € | 7.57 | |||||||||||||||||||||||||||||||||||||
06/03/2014 | BSA | 10,000 | — | 10,000 | 10,000 | € | 18.79 | |||||||||||||||||||||||||||||||||||||
06/03/2014 | AGA | 186,000 | — | 186,000 | 186,000 | € | — | |||||||||||||||||||||||||||||||||||||
06/03/2014 | SO | 75,000 | — | 75,000 | 75,000 | € | 19.01 | |||||||||||||||||||||||||||||||||||||
|
|
|
| |||||||||||||||||||||||||||||||||||||||||
Total | 2,398,206 | 8,101 | 1,437,684 | 2,207,530 | ||||||||||||||||||||||||||||||||||||||||
|
|
|
|
| ||||||||||||||||||||||||||||||||||||||||
Date | Type | Number of warrants issued as of 12/31/2014 | Number of warrants null and void as of 12/31/2014 | Number of warrants null and outstanding as of 12/31/2014 | Maximum number of shares to be issued | Strike price per share | ||||||||||||||||
12/07/2007 | BSA | 1,717 | 572 | 1,145 | 17,175 | € | 4.33 | |||||||||||||||
01/21/2009 | BSA/BSPCE | 16,380 | — | 13,713 | 205,695 | € | 4.33 | |||||||||||||||
01/21/2009 | BSPCE | 2,296 | — | — | — | € | 4.33 | |||||||||||||||
06/25/2010 | BSA | 1,825 | — | 1,825 | 27,375 | € | 4.33 | |||||||||||||||
01/28/2011 | BSA | 10,039 | 7,529 | — | — | € | 5.13 | |||||||||||||||
06/24/2011 | BSA/BSPCE | 32,000 | — | 28,933 | 433,995 | € | 5.13 | |||||||||||||||
11/22/2011 | BSA/BSPCE | 11,377 | — | 9,373 | 140,595 | € | 5.13 | |||||||||||||||
01/17/2012 | BSA | 89,835 | — | 89,835 | 89,835 | € | 5.13 | |||||||||||||||
04/02/2012 | AGA | 669,796 | — | — | — | € | — | |||||||||||||||
07/25/2012 | AGA | 134,081 | — | — | — | € | — | |||||||||||||||
09/25/2012 | BSA | 30,000 | — | 25,000 | 25,000 | € | 8.59 | |||||||||||||||
11/28/2012 | AGA | 35,360 | — | 35,360 | 35,360 | € | — | |||||||||||||||
07/25/2013 | BSA | 73,000 | — | 70,500 | 70,500 | € | 8.10 | |||||||||||||||
09/12/2013 | AGA | 501,500 | — | 420,000 | 420,000 | € | — | |||||||||||||||
09/18/2013 | SO | 518,000 | — | 471,000 | 471,000 | € | 7.57 | |||||||||||||||
06/03/2014 | BSA | 10,000 | — | 10,000 | 10,000 | € | 18.79 | |||||||||||||||
06/03/2014 | AGA | 186,000 | — | 186,000 | 186,000 | € | — | |||||||||||||||
06/03/2014 | SO | 75,000 | — | 75,000 | 75,000 | € | 19.01 | |||||||||||||||
|
|
|
|
|
|
|
| |||||||||||||||
Total | 2,398,206 | 8,101 | 1,437,684 | 2,207,530 | ||||||||||||||||||
|
| �� |
|
|
|
|
|
|
|
| ||||||||||||
Date 12/07/2007 01/21/2009 01/21/2009 06/25/2010 01/28/2011 06/24/2011 11/22/2011 01/17/2012 04/02/2012 07/25/2012 09/25/2012 11/28/2012 07/25/2013 09/12/2013 09/18/2013 06/03/2014 06/03/2014 06/03/2014 03/24/2015 06/23/2015 09/30/2015 11/19/2015 11/19/2015 12/15/2015 Total Type Number of
warrants
issued as of
12/31/2015 Number of
warrants
null and
void as of
12/31/2015 Number of
warrants
null and
outstanding
as of
12/31/2015 Maximum
number of
shares to
be issued Strike price
per share BSA 1,717 572 859 12,885 € 4.33 BSA/BSPCE 16,380 — 4,041 60,615 € 4.33 BSPCE 2,296 — — — € 4.33 BSA 1,825 — 730 10,950 € 4.33 BSA 10,039 7,529 — — € 5.13 BSA/BSPCE 32,000 — 13,465 201,975 € 5.13 BSA/BSPCE 11,377 — 2,509 37,635 € 5.13 BSA 89,835 — 89,835 89,835 € 5.13 AGA 669,796 1,860 — — € — AGA 134,081 — — — € — BSA 30,000 — 10,000 10,000 € 8.59 AGA 35,360 — — — € — BSA 73,000 6 13,000 13,000 € 8.10 AGA 501,500 113,333 101,829 101,829 € — SO 518,000 47,000 471,000 471,000 € 7.57 BSA 10,000 — 5,000 5,000 € 18.79 AGA 186,000 30,000 156,000 156,000 € — SO 75,000 — 75,000 75,000 € 19.01 BSA 10,000 — 10,000 10,000 € 43.00 SO 120,000 — 120,000 120,000 € 48.90 AGA 708,500 — 708,500 708,500 € — SO 195,000 — 195,000 195,000 € 66.06 BSA 15,000 — 15,000 15,000 € 66.06 AGA 42,000 — 42,000 42,000 € — 3,488,706 200,300 2,033,768 2,336,224
The totaltotals presented above doesdo not include the warrants cancelled prior to December 31, 2009.
As part of the initial public offering on Euronext, the nominal value of the shares underwent a fifteen-for-one share split following the decision of the Combined General Meeting of December 9, 2011.
The impact of the share-based payments on the net income (or loss)Consolidated Statements of (Loss) is presented in Note 17.
Note 11: Financial Liabilities
11.1 Conditional Advances
The conditional advances from public institutions are subject to contracts with OSEO and COFACE.
As of December 31, 2012,2015, the Company had two advance contracts with OSEO Innovation, and a contract with COFACE. As of December 31, 2013, the Company had three advance contracts with OSEO Innovation and a contract with COFACE. As of December 31, 2014, the Company had two advance contracts with OSEO Innovation and a contract with COFACE.
These advances do not bear interest and are 100% repayable at their nominal value in the event of technical and/or commercial success. The 3rd OSEO advance and the COFACE advance do not bear interest.
The Company also benefited from a third grant from BpiFrance Financement in November 2014.
The portion of the conditional advances for terms longer than one year is classified as non-current liabilities, while the portion for terms of less than one year is classified as current liabilities.
The table below presents the details of the debts recorded on the statement of financial position by the type of conditional advance (amounts in Euros):advance:
| 2nd OSEO advance | 3rd OSEO advance | 4th OSEO advance | COFACE | Total | ||||||||||||||||||||||||||||||||||||
Opening Balance Sheet Debt as of 1/1/2012 | 450,713 | 246,238 | — | 122,501 | 819,452 | |||||||||||||||||||||||||||||||||||
| (amounts in thousands of Euros) | 2nd OSEO advance | 3rd OSEO advance | 4th OSEO advance | COFACE | Total | |||||||||||||||||||||||||||||||||||
Opening Balance Sheet Debt as of 1/1/2013 | 257 | 250 | — | 127 | 634 | |||||||||||||||||||||||||||||||||||
+ receipts | — | — | — | — | — | — | 256 | 903.5 | — | 1,159.5 | ||||||||||||||||||||||||||||||
- repayments | (200,000 | ) | — | — | — | (200,000 | ) | (260 | ) | — | — | — | (260 | ) | ||||||||||||||||||||||||||
+/- other transactions | 6,701 | 3,661 | — | 4,251 | 14,613 | 3 | (2 | ) | (111 | ) | 19 | (91 | ) | |||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||||||||||||||||||||||
Opening Balance Sheet Debt as of 12/31/2012 | 257,414 | 249,899 | — | 126,752 | 634,065 | |||||||||||||||||||||||||||||||||||
Opening Balance Sheet Debt as of 12/31/2013 | 504 | 792 | 146 | 1,443 | ||||||||||||||||||||||||||||||||||||
Of which: | ||||||||||||||||||||||||||||||||||||||||
Non-current portion | 376,651 | 1,317 | ||||||||||||||||||||||||||||||||||||||
Current portion | 257,414 | 126 | ||||||||||||||||||||||||||||||||||||||
| 3rd OSEO advance | 4th OSEO advance | BPI advance | COFACE | Total | ||||||||||||||||||||||||||||||||||||
Opening Balance Sheet Debt as of 1/1/2014 | 504 | 792 | — | 146 | 1,443 | |||||||||||||||||||||||||||||||||||
+ receipts | 128 | — | 3,000 | — | 3,128 | |||||||||||||||||||||||||||||||||||
- repayments | (128 | ) | — | — | — | (128 | ) | |||||||||||||||||||||||||||||||||
+/- other transactions | 2 | 13 | (416 | ) | 5 | (396 | ) | |||||||||||||||||||||||||||||||||
|
|
|
|
| ||||||||||||||||||||||||||||||||||||
Opening Balance Sheet Debt as of 12/31/2014 | 507 | 805 | 2,584 | 151 | 4,047 | |||||||||||||||||||||||||||||||||||
Of which: | ||||||||||||||||||||||||||||||||||||||||
Non-current portion | 3,855 | |||||||||||||||||||||||||||||||||||||||
Current portion | 192 | |||||||||||||||||||||||||||||||||||||||
Stated interest rate | None | 2.05 | % | None | None | |||||||||||||||||||||||||||||||||||
|
|
|
|
| ||||||||||||||||||||||||||||||||||||
Discount rate | 0.4%-1.9 | % | 1.5%-1.8 | % | 3.2 | % | 4.25 | % | ||||||||||||||||||||||||||||||||
|
|
|
|
| ||||||||||||||||||||||||||||||||||||
Maturity (in years) | 0-3 | 7-9 | 2-7 | — | ||||||||||||||||||||||||||||||||||||
|
|
|
|
| ||||||||||||||||||||||||||||||||||||
| 3rd OSEO advance | 4th OSEO advance | BPI advance | COFACE | Total | ||||||||||||||||||||||||||||||||||||
Opening Balance Sheet Debt as of 01/01/2015 | 506 | 805 | 2,584 | 151 | 4,047 | |||||||||||||||||||||||||||||||||||
+ receipts | — | 865 | — | — | 865 | |||||||||||||||||||||||||||||||||||
- repayments | (192 | ) | — | — | — | (192 | ) | |||||||||||||||||||||||||||||||||
+/- other transactions | 3 | (2 | ) | 82 | 5 | 89 | ||||||||||||||||||||||||||||||||||
|
|
|
|
| ||||||||||||||||||||||||||||||||||||
Opening Balance Sheet Debt as of 12/31/2015 | 318 | 1,669 | 2,666 | 156 | 4,809 | |||||||||||||||||||||||||||||||||||
Of which: | ||||||||||||||||||||||||||||||||||||||||
Non-current portion | 4,681 | |||||||||||||||||||||||||||||||||||||||
Current portion | 128 | |||||||||||||||||||||||||||||||||||||||
Stated interest rate | None | 2.05 | % | None | None | |||||||||||||||||||||||||||||||||||
|
|
|
|
| ||||||||||||||||||||||||||||||||||||
Discount rate | 0.4%-1.9 | % | 1.5%-1.8 | % | 3.2 | % | 4.25 | % | ||||||||||||||||||||||||||||||||
|
|
|
|
| ||||||||||||||||||||||||||||||||||||
Maturity (in years) | 0-3 | 7-9 | 2-7 | — | ||||||||||||||||||||||||||||||||||||
|
|
|
|
| ||||||||||||||||||||||||||||||||||||
Opening Balance Sheet Debt as of 1/1/2013 + receipts - repayments +/- other transactions Opening Balance Sheet Debt as of 12/31/2013 Of which: Non-current portion Current portion 2nd OSEO
advance 3rd OSEO
advance 4th OSEO
advance COFACE Total 257,414 249,899 — 126,752 634,065 — 256,000 903,500 — 1,159,500 (260,000 ) — — — (260,000 ) 2,586 (1,579 ) (111,047 ) 19,300 (90,740 ) 504,320 792,453 146,052 1,442,825 1,316,533 126,292
| 3nd OSEO advance | 4th OSEO advance | BPI advance | COFACE | Total | ||||||||||||||||
Opening Balance Sheet Debt as of 1/1/2014 | 504,320 | 792,453 | — | 146,052 | 1,442,825 | |||||||||||||||
+ receipts | 128,000 | — | 3,000,000 | — | 3,128,000 | |||||||||||||||
- repayments | (128,000 | ) | — | — | — | (128,000 | ) | |||||||||||||
+/- other transactions | 2,276 | 12,932 | (416,361 | ) | 4,994 | (396,159 | ) | |||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Opening Balance Sheet Debt as of 12/31/2014 | 506,596 | 805,385 | 2,583,639 | 151,046 | 4,046,666 | |||||||||||||||
Of which: | ||||||||||||||||||||
Non-current portion | 3,854,666 | |||||||||||||||||||
Current portion | 192,000 | |||||||||||||||||||
Stated interest rate | None | 2.05 | % | None | None | |||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Discount rate | 0.4%-1.9 | % | 1.5%-1.8 | % | 3.2 | % | 4.25 | % | ||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Maturity (in years) | 0-3 | 7-9 | 2-7 | — | ||||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
The changes appearing in “Other transactions” are comprised of the effect of discounting conditional advances.
Second OSEO Advance
On January 10, 2005, DBV Technologies obtained from OSEO a repayable financial assistance for innovation in the amount of €600,000 for a project to design a high-speed prototype machine for the production and development of second-generation patches intended for the detection of various allergies. The principal steps of this advance are the following:
The terms of repayment are the following:
Third OSEO Advance
In 2011, the Company was notified by OseoOSEO Innovation of a new grant in the form of a conditional advance of up to €640,000 to finance the development of its program of treatment of the allergy to proteins in cow’s milk.
The amount of the assistance will bewas paid as follows:
The first payment of €256,000 was received in 2011.
The second payment of €256,000 was received in 2013.
The final balance of €128,000 has been received in 2014.
In the event of technical or commercial success of the program, the repayment schedule will be the following:
Regardless of the outcome of the development program, a fixed sum of €256,000 must be repaid in four quarterly instalments of €64,000 beginning on September 30, 2014.
Fourth OSEO Advance
In 2013, OSEO has provided assistance in the form of conditional advances for €3,206,162 to DBV Technologies as part of a collaborative research and clinical development in mite allergy in young children. ImmunaVia, the program, will be funded according to the following schedule, subject to the progress of the program:
The funds of October 2014, were finally received on January 22, 2015 for an amount of €864,989.
Such conditional advance bears interest at an annual rate of 2.05%. In case of technical or commercial success of the project, the repayment schedule, for a total amount of €3,750,000 (including interest), is as follows:
Furthermore, the financing program includes additional payment by OSEO to the company for a total of €1,919,056 in non-refundable subsidies.
BpiFrance Financement Interest-Free Loan
In 2014, BpiFrance Financement granted an interest-free Innovation loan of €3,000,000 to DBV Technologies to help financing the pharmaceutical development of Viaskin Milk. This amount was received in a single disbursement on November 27, 2014.
The planned repayment schedule is as follows:
COFACE Advance
On September 6, 2007, DBV Technologies signed a prospecting insurance contract with Compagnie Française d’Assurance pour le Commerce Extérieur (“COFACE”) in order to promote its Diallertest product internationally. Under the terms of that contract, the Company received conditional advances of up to €147,534.€147 thousands. DBV Technologies must repay these advances in amounts of up to 7% of its revenue from the export sales of its Diallertest product, until April 30, 2017.
As of December 31, 2014,2015, the remained amount that remainedwas €156 thousands compared to be repaid under this advance amounted to €145,760 (€146,040 as of€151 thousands on December 31 2013).st 2014.
The accounting treatment resulting from any changes in the anticipated flow of repayments of this advance is described in Note 3.11.3.10.
11.2 Due Dates of the Financial Liabilities
Due dates of the financial liabilities posted as of December 31, 2012
| Gross Amount | Less than One Year | One to Five Years | More than Five Years | |||||||||||||
| (Amounts in Euros) | ||||||||||||||||
Financial liabilities | ||||||||||||||||
Non-current conditional advances | 376,651 | — | 376,651 | — | ||||||||||||
Current conditional advances | 257,414 | 257,414 | — | — | ||||||||||||
Bank overdrafts | 519,499 | 519,499 | — | — | ||||||||||||
Supplier accounts payable and related payables | 977,724 | 977,724 | — | — | ||||||||||||
|
|
|
|
|
|
|
| |||||||||
Total financial liabilities | 2,131,288 | 1,754,637 | 376,651 | — | ||||||||||||
|
|
|
|
|
|
|
| |||||||||
Due dates of the financial liabilities recognized as of December 31, 2013
| Gross Amount | Less than One Year | One to Five Years | More than Five Years | Gross Amount | Less than One Year | One to Five Years | More than Five Years | |||||||||||||||||||||||||
| (Amounts in Euros) | (Amounts in thousands of Euros) | |||||||||||||||||||||||||||||||
Financial liabilities | ||||||||||||||||||||||||||||||||
Non-current conditional advances | 1,316,531 | — | 524,080 | 792,453 | 1,317 | — | 524 | 792 | ||||||||||||||||||||||||
Current conditional advances | 126,292 | 126,292 | — | — | 126 | 126 | — | — | ||||||||||||||||||||||||
Supplier accounts payable and related payables | 1,497,289 | 1,497,289 | 1,497 | 1,497 | ||||||||||||||||||||||||||||
|
|
|
|
|
|
|
| |||||||||||||||||||||||||
Total financial liabilities | 2,940,114 | 1,623,581 | 524,080 | 792,453 | 2,940 | 1,624 | 524 | 792 | ||||||||||||||||||||||||
|
|
|
|
|
|
|
| |||||||||||||||||||||||||
As detailed in Note 13, other current liabilities are primarily social security and tax liabilities and are mainly due in less than one year from the reporting date.
Due dates of the financial liabilities recognized as of December 31, 2014
| Gross Amount | Less than One Year | One to Five Years | More than Five Years | Gross Amount | Less than One Year | One to Five Years | More than Five Years | |||||||||||||||||||||||||
| (Amounts in Euros) | (Amounts in thousands of Euros) | |||||||||||||||||||||||||||||||
Financial liabilities | ||||||||||||||||||||||||||||||||
Non-current conditional advances | 3,854,666 | — | 1,940,400 | 1,914,266 | 3,855 | — | 1,940 | 1,914 | ||||||||||||||||||||||||
Non-current financial rent debts | 33,504 | 33,504 | 33 | 33 | ||||||||||||||||||||||||||||
Current conditional advances | 192,000 | 192,000 | — | — | 192 | 192 | — | — | ||||||||||||||||||||||||
Current financial rent debts | 20,653 | 20,653 | 21 | 21 | ||||||||||||||||||||||||||||
Accrued interest | 83 | 83 | ||||||||||||||||||||||||||||||
Supplier accounts payable and related payables | 1,874,629 | 1,874,629 | 1,875 | 1,875 | ||||||||||||||||||||||||||||
|
|
|
|
|
|
|
| |||||||||||||||||||||||||
Total financial liabilities | 5,975,535 | 2,087,365 | 1,973,904 | 1,914,266 | 5,976 | 2,087 | 1,974 | 1,914 | ||||||||||||||||||||||||
|
|
|
|
|
|
|
| |||||||||||||||||||||||||
Due dates of the financial liabilities recognized as of December 31, 2015
| Gross Amount | Less than One Year | One to Five Years | More than Five Years | |||||||||||||
| (Amounts in thousands of Euros) | ||||||||||||||||
Non-current conditional advances | 4,681 | — | 2,378 | 2,302 | ||||||||||||
Non-current financial rent debts | 12 | 12 | ||||||||||||||
Current conditional advances | 128 | 128 | — | — | ||||||||||||
Current financial rent debts | 21 | 21 | ||||||||||||||
Supplier accounts payable and related payables | 10,034 | 10,034 | ||||||||||||||
|
|
|
|
|
|
|
| |||||||||
Total financial liabilities | 14,876 | 10,183 | 2,391 | 2,302 | ||||||||||||
|
|
|
|
|
|
|
| |||||||||
As detailed in Note 13, other current liabilities are primarily social security and tax liabilities and are mainly due in less than one year from the reporting date.
Note 12: Non-Current Provisions
| December 31 | ||||||||||||||||||||||||
| 2012 | 2013 | 2014 | 2013 | 2014 | 2015 | |||||||||||||||||||
| (Amounts in Euros) | (Amounts in thousands of Euros) | |||||||||||||||||||||||
Pension retirement obligations | 254,389 | 290,695 | 530,732 | 291 | 531 | 490 | ||||||||||||||||||
Total | 254,941 | 290,695 | 530,732 | 291 | 531 | 490 | ||||||||||||||||||
Commitments for Compensation Payable to Employees Upon Their Retirement
Amounts in thousands of Euros | ||||
As of January 1, | ( | ) | ||
Costs of services rendered (operating expense) | ( | ) | ||
Interest expense (finance expense) | ( | ) | ||
Benefit paid | — | |||
Actuarial losses | ||||
| ||||
| ||||
| ||||
| ||||
| ||||
| ||||
|
| |||
As of December 31, 2013 | ( | ) | ||
Costs of services rendered (operating expense) | ( | ) | ||
Interest expense (finance expense) | ( | ) | ||
Benefit paid | — | |||
Actuarial gains | ( | ) | ||
|
| |||
As of December 31, 2014 | ( | ) | ||
Costs of services rendered (operating expense) | (116 | ) | ||
Interest expense (finance expense) | (9 | ) | ||
Benefit paid | — | |||
Actuarial gains | 166 | |||
As of December 31, 2015 | (490 | ) | ||
|
| |||
As part of the estimation of the retirement commitments, the following assumptions were used for all categories of employees:
| 2012 | 2013 | 2014 | 2013 | 2014 | 2015 | |||||||||||||||||||
% social security contributions | 50.0 | % | 50.0 | % | 50.0 | % | 50.0 | % | 50.0 | % | 50.0 | % | ||||||||||||
Salary increases | 3.3 | % | 4.0 | % | 4.0 | % | 4.0 | % | 4.0 | % | 2.0 | % | ||||||||||||
Discount rate | 2.90 | % | 3.16 | % | 1.30 | % | 3.16 | % | 1.30 | % | 2.08 | % | ||||||||||||
The discount rates come from the corporate AA zero coupon yield curve.
No employee has retired during the last three fiscal years presented.
Note 13: Supplier Accounts Receivable and Other Current Liabilities
13.1 Supplier Accounts Payable and Related Payables
No discounting was performed on the supplier accounts payable and related payables to the extent that the amounts did not present payment terms longer than one year at the end of each fiscal year presented.
13.2 Other Current Liabilities
| December 31 | ||||||||||||||||||||||||
| 2012 | 2013 | 2014 | 2013 | 2014 | 2015 | |||||||||||||||||||
| (Amounts in Euros) | (Amounts in thousands of Euros) | |||||||||||||||||||||||
Social security contribution liabilities | 1,158,362 | 1,708,526 | 2,160,437 | 1,709 | 2,160 | 4,464 | ||||||||||||||||||
Tax liabilities | 62,793 | 56,062 | 114,313 | 56 | 114 | 388 | ||||||||||||||||||
Other debts | 67,000 | 52,207 | 110,052 | 52 | 110 | 195 | ||||||||||||||||||
Deferred revenues from subsidies | 127,298 | 793,72 | 1,051,528 | 794 | 1,052 | 791 | ||||||||||||||||||
Total | 1,415,453 | 2,610,515 | 3,436,329 | 2,611 | 3,436 | 5,838 | ||||||||||||||||||
The other liabilities include the short-term debts to employees, as well as social wellfarewelfare and tax agencies. Deferred revenues include subsidies and conditional advances.
Note 14: Financial Instruments Recognized in the StatementConsolidated Statements of Financial Position and Related Effect on the Income StatementConsolidated Statements of (Loss)
2012 | Book value on the statement of financial position | Fair value through profit and loss(3) | Loans and receivables(1) | Debt at amortized cost(2) | Fair Value | |||||||||||||||
| (Amounts in Euros) | ||||||||||||||||||||
Financial assets | ||||||||||||||||||||
Non-current financial assets | 384,357 | 301,358 | 82,999 | — | 384,357 | |||||||||||||||
Customer accounts receivable and related receivables | 92,875 | — | 92,875 | — | 92,875 | |||||||||||||||
Cash and cash equivalents | 38,348,130 | 38,348,130 | — | — | 38,348,130 | |||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Total financial assets | 38,825,362 | 38,649,488 | 175,874 | — | 38,825,362 | |||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Financial liabilities | ||||||||||||||||||||
Conditional advances (non-current portion) | 376,651 | — | — | 376,651 | 376,651 | |||||||||||||||
Conditional advances (current portion) | 257,414 | — | — | 257,414 | 257,414 | |||||||||||||||
Supplier accounts payable and related payables | 977,724 | — | — | 977,724 | 977,724 | |||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Total financial liabilities | 1,611,789 | — | — | 1,611,789 | 1,611,789 | |||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
2013 | Book value on the statement of financial position | Fair value through profit and loss(3) | Loans and receivables(1) | Debt at amortized cost(2) | Fair Value | Book value in the Consolidated Statements of Financial Position | Fair value through Consolidated Statements of (Loss)(3) | Loans and receivables(1) | Debt at amortized cost(2) | Fair Value | ||||||||||||||||||||||||||||||
| (Amounts in Euros) | (Amounts in thousands of Euros) | |||||||||||||||||||||||||||||||||||||||
Financial assets | ||||||||||||||||||||||||||||||||||||||||
Non-current financial assets | 623,829 | 541,487 | 82,342 | — | 623,829 | 624 | 541 | 82 | — | 624 | ||||||||||||||||||||||||||||||
Customer accounts receivable and related receivables | 182,900 | — | 182,900 | — | 182,900 | 183 | — | 183 | — | 183 | ||||||||||||||||||||||||||||||
Cash and cash equivalents | 39,402,761 | 39,402,761 | — | — | 39,402,761 | 39,403 | 39,403 | — | — | 39,403 | ||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||||||||||||||||||||||
Total financial assets | 40,209,490 | 39,944,248 | 265,242 | — | 40,209,490 | 40,209 | 39,944 | 265 | — | 40,209 | ||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||||||||||||||||||||||
Financial liabilities | ||||||||||||||||||||||||||||||||||||||||
Conditional advances (non-current portion) | 1,316,533 | — | — | 1,316,533 | 1,316,533 | 1,317 | — | — | 1,317 | 1,317 | ||||||||||||||||||||||||||||||
Conditional advances (current portion) | 126,292 | — | — | 126,292 | 126,292 | 126 | — | — | 126 | 126 | ||||||||||||||||||||||||||||||
Supplier accounts payable and related payables | 4,107,804 | — | — | 4,107,804 | 4,107,804 | 4,108 | — | — | 4,108 | 4,108 | ||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||||||||||||||||||||||
Total financial liabilities | 5,550,629 | — | — | 5,550,629 | 5,550,629 | 5,551 | — | — | 5,551 | 5,551 | ||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||||||||||||||||||||||
2014 | Book value in the Consolidated Statements of Financial Position | Fair value through Consolidated Statements of (Loss)(3) | Loans and receivables(1) | Debt at amortized cost(2) | Fair Value | |||||||||||||||||||||||||||||||||||
| (Amounts in thousands of Euros) | ||||||||||||||||||||||||||||||||||||||||
Financial assets | ||||||||||||||||||||||||||||||||||||||||
Long-term financial assets | 1,596 | 1,496 | 100 | — | 1,596 | |||||||||||||||||||||||||||||||||||
Customer accounts receivable and related receivables | 136 | — | 136 | — | 136 | |||||||||||||||||||||||||||||||||||
Other current financial assets | 241 | — | 241 | — | 241 | |||||||||||||||||||||||||||||||||||
Cash and cash equivalents | 114,583 | 114,583 | — | — | 114,583 | |||||||||||||||||||||||||||||||||||
|
|
|
|
| ||||||||||||||||||||||||||||||||||||
Total financial assets | 116,554 | 116,079 | 477 | — | 116,554 | |||||||||||||||||||||||||||||||||||
|
|
|
|
| ||||||||||||||||||||||||||||||||||||
Financial liabilities | ||||||||||||||||||||||||||||||||||||||||
Long-term conditional advances | 3,855 | — | — | 3,855 | 3,855 | |||||||||||||||||||||||||||||||||||
Long term financial rent debt | 33 | — | �� | 33 | 33 | |||||||||||||||||||||||||||||||||||
Short-term conditional advances | 192 | — | — | 192 | 192 | |||||||||||||||||||||||||||||||||||
Short term financial rent debt | 21 | — | 21 | 21 | ||||||||||||||||||||||||||||||||||||
Accounts payable and other liabilities | 5,311 | — | — | 5,311 | 5,311 | |||||||||||||||||||||||||||||||||||
|
|
|
|
| ||||||||||||||||||||||||||||||||||||
Total financial liabilities | 9,412 | — | — | 9,412 | 9,412 | |||||||||||||||||||||||||||||||||||
|
|
|
|
| ||||||||||||||||||||||||||||||||||||
2014 | Book value on the statement of financial position | Fair value(3) | Loans and receivables(1) | Debt at amortized cost(2) | Fair Value | |||||||||||||||||||||||||||||||||||
2015 | Book value in the Consolidated Statements of Financial Position | Fair value through Consolidated Statements of (Loss)(3) | Loans and receivables(1) | Debt at amortized cost(2) | Fair Value | |||||||||||||||||||||||||||||||||||
| (Amounts in Euros) | (Amounts in thousands of Euros) | |||||||||||||||||||||||||||||||||||||||
Financial assets | ||||||||||||||||||||||||||||||||||||||||
Long-term financial assets | 1,595,861 | 1,496,036 | 99,825 | — | 1,595,861 | 2,711 | 1,365 | 1,346 | — | 2,711 | ||||||||||||||||||||||||||||||
Customer accounts receivable and related receivables | 136,112 | — | 136,112 | — | 136,112 | — | — | — | — | — | ||||||||||||||||||||||||||||||
Other current financial assets | 240,609 | 240,609 | 240,609 | 333 | — | 333 | — | 333 | ||||||||||||||||||||||||||||||||
Cash and cash equivalents | 114,583,141 | 114,583,141 | — | — | 114,583,141 | 323,381 | 323,381 | — | _ | 323,381 | ||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||||||||||||||||||||||
Total financial assets | 116,553,723 | 116,079,177 | 476,546 | — | 116,553,723 | 326,425 | 324,746 | 1,679 | _ | 326,425 | ||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||||||||||||||||||||||
Financial liabilities | ||||||||||||||||||||||||||||||||||||||||
Long-term conditional advances | 3,854,666 | — | — | 3,854,666 | 3,854,666 | 4,681 | — | — | 4,681 | 4,681 | ||||||||||||||||||||||||||||||
Long term financial rent debt | 33,504 | 33,504 | 33,504 | 12 | — | 12 | 12 | |||||||||||||||||||||||||||||||||
Short-term conditional advances | 192,000 | — | — | 192,000 | 192,000 | 128 | — | — | 128 | 128 | ||||||||||||||||||||||||||||||
Short term financial rent debt | 20,653 | 20,653 | 20,653 | 21 | — | 21 | 21 | |||||||||||||||||||||||||||||||||
Accrued interest | 83 | 83 | 83 | |||||||||||||||||||||||||||||||||||||
Accounts payable and other liabilities | 5,310,958 | — | — | 5,310,958 | 5,310,958 | 15,872 | — | — | 15,872 | 15,872 | ||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||||||||||||||||||||||
Total financial liabilities | 9,411,864 | — | — | 9,411,864 | 9,411,864 | 20,714 | — | — | 20,714 | 20,714 | ||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||||||||||||||||||||||
| (1) | The fair value of “loans and receivables” corresponds to the value reported in the |
| (2) | The book amount of financial liabilities measured at amortized cost was deemed to be a reasonable estimation of fair value. |
| (3) | The fair value of financial assets held for trading is determined based on Level 1 fair value measurements and corresponds to the market value of the assets. |
Note 15: Operating Income
The operating income is broken down in the following manner:
| December 31 | ||||||||||||||||||||||||
| 2012 | 2013 | 2014 | 2013 | 2014 | 2015 | |||||||||||||||||||
| (Amounts in Euros) | (Amounts in thousands of Euros) | |||||||||||||||||||||||
Revenues | 174,360 | 181,800 | 210,759 | 182 | 211 | 202 | ||||||||||||||||||
Research Tax Credit | 2,522,399 | 3,312,462 | 4,339,620 | 3 | 4,340 | 5,685 | ||||||||||||||||||
Subsidies | 79,829 | 332,051 | 211,143 | 332 | 211 | 279 | ||||||||||||||||||
Total | 4,761,372 | 3,826,313 | 4,761,522 | 3,826 | 4,762 | 6,166 | ||||||||||||||||||
The revenues of the Company are composed of the sales ofDiallertest® products.products whose commercialization ended on September 1st, 2015.
Note 16: Operating Expenses
The research and development expenses are broken down as follows:
| December 31 | December 31 | |||||||||||||||||||||||
| R&D expenses | 2012 | 2013 | 2014 | 2013 | 2014 | 2015 | ||||||||||||||||||
| (Amounts in Euros) | (Amounts in thousands of Euros) | |||||||||||||||||||||||
Personnel expenses | 4,800,518 | 7,194,722 | 7,703,057 | 7,195 | 7,703 | 13,268 | ||||||||||||||||||
Sub-contracting, collaboration and consultants | 5,229,379 | 8,212,083 | 10,703,130 | 8,212 | 10,703 | 15,325 | ||||||||||||||||||
Research supplies | 598,216 | 555,009 | 937,316 | 555 | 937 | 911 | ||||||||||||||||||
Purchases | — | 249 | 795 | |||||||||||||||||||||
Real estate property rental | 259,224 | 263,438 | 254,923 | 263 | 255 | 1,094 | ||||||||||||||||||
Conferences and travel expenses | 324,123 | 465,871 | 665,420 | 466 | 665 | 1,233 | ||||||||||||||||||
Allowances for provisions and amortization and depreciation | 192,740 | 290,406 | 466,172 | 290 | 466 | 1,000 | ||||||||||||||||||
Others | 95,168 | 385,009 | 413,424 | 385 | 165 | 608 | ||||||||||||||||||
Total R&D expenses | 11,499,368 | 17,366,538 | 21,143,442 | 17,367 | 21,143 | 34,234 | ||||||||||||||||||
The sales and marketing expenses are broken down as follows:
| December 31 | ||||||||
| S&M expenses | 2014 | 2015 | ||||||
| (Amounts in thousands of Euros) | ||||||||
Personnel expenses | 133 | |||||||
Fees | 339 | |||||||
Communication and travel expenses | 13 | 20 | ||||||
Total S&M expenses | 13 | 491 | ||||||
By nature, the breakdown of general and administrative expenses is as follows:
| December 31 | December 31 | |||||||||||||||||||||||
| G&A Expenses | 2012 | 2013 | 2014 | 2013 | 2014 | 2015 | ||||||||||||||||||
| (Amounts in Euros) | (Amounts in thousands of Euros) | |||||||||||||||||||||||
Personnel expenses | 3,107,246 | 4,698,848 | 5,109,057 | 4,699 | 5,109 | 8,768 | ||||||||||||||||||
Fees | 512,709 | 586,638 | 1,165,989 | 587 | 1,166 | 4,234 | ||||||||||||||||||
Real estate property rental | 157,467 | 111,232 | 203,899 | 111 | 204 | 305 | ||||||||||||||||||
Insurance policies | 56,054 | 105,018 | 230,495 | 105 | 230 | 1,239 | ||||||||||||||||||
Communication and travel expenses | 480,999 | 450,701 | 645,175 | 451 | 633 | 1,010 | ||||||||||||||||||
Maintenance | — | 140 | 356 | |||||||||||||||||||||
Postal and telecommunications expenses | 86,831 | 65,350 | 75,913 | 65 | 76 | 36 | ||||||||||||||||||
Administrative supplies and rentals of personal property | 65,867 | 97,131 | 104,374 | 97 | 104 | 162 | ||||||||||||||||||
Allowances for provisions and amortization and depreciation | 111 | 74 | ||||||||||||||||||||||
Others | 131,526 | 194,832 | 582,762 | 195 | 332 | 676 | ||||||||||||||||||
Total G&A expenses | 4,598,699 | 6,309,750 | 8,117,664 | 6,310 | 8,105 | 16,859 | ||||||||||||||||||
Personnel Expenses
The Company employed 5691 people as ofat December 31, 2015, in comparison with 56 as at December 31, 2014 in comparison withand 44 as ofat December 31, 2013.
The personnel expenses are broken down as follows (in Euros):follows:
| December 31 | ||||||||||||||||||||||||
| 2013 | 2014 | 2015 | ||||||||||||||||||||||
| 2012 | 2013 | 2014 | (Amounts in thousands of Euros) | |||||||||||||||||||||
Wages and salaries | 2,376,638 | 3,607,544 | 4,883,410 | 3,608 | 4,883 | 7,243 | ||||||||||||||||||
Social security contributions | 2,300,323 | 3,148,253 | 3,202,520 | 3,148 | 3,203 | 4,391 | ||||||||||||||||||
Expenses for pension commitments | 36,495 | 89,572 | 86,781 | 90 | 87 | 116 | ||||||||||||||||||
Share-based payments | 3,194,308 | 5,048,201 | 4,639,403 | 5,048 | 4,639 | 10,419 | ||||||||||||||||||
Total | 7,907,764 | 11,893,570 | 12,812,114 | 11,894 | 12,812 | 22,168 | ||||||||||||||||||
The increase in personnel charges is partly due to, on the one hand, the increased number of employees of the Company from one year to the next, and in part to the increasecharge to share-based payments linked to the global plan put in place during the second semester of 2015 (see Note 17).
Note 17: Share-Based Payments
The Board of Directors has been authorized by the general meeting of the shareholders to grant employee warrants (Bons de Souscription de Parts de Créateur d’Entreprise or “BSPCE”) and (Bons de Souscription d’Actions or “BSA”), Free shares and to implement share options plans as follows:
17.1 BCEX
The BCEX may be exercised by the beneficiary on the basis of the following vesting schedule:
By a decision by the Board of Directors meeting held on November 22, 2011, the BCEX warrants became exercisable beginning on the date of the first quotation of the shares of the Company on the Euronext regulated market in Paris.
Details of BCEX
Date of grant (Board of Directors) | 01/21/2009 | 01/21/2009 | 01/21/2009 | 01/21/2009 | ||||||||||||
Vesting period (years) | 1 | 2 | 3 | 4 | ||||||||||||
Plan expiration date | 01/20/2019 | 01/20/2019 | 01/20/2019 | 01/20/2019 | ||||||||||||
Number of BCEX granted | 574 | 574 | 574 | 574 | ||||||||||||
Share entitlement per BCEX(1) | 15 | 15 | 15 | 15 | ||||||||||||
Exercise price | 70 | 70 | 70 | 70 | ||||||||||||
Valuation method used | Black and Scholes | |||||||||||||||
Grant date share fair value | 70 | 70 | 70 | 70 | ||||||||||||
Expected volatility | 40 | % | 40 | % | 40 | % | 40 | % | ||||||||
Average life of BCEX | 5.5 | 6.0 | 6.5 | 7.0 | ||||||||||||
Discount rate(2) | 2.71 | % | 2.98 | % | 2.98 | % | 3.11 | % | ||||||||
Expected dividends | 0 | % | 0 | % | 0 | % | 0 | % | ||||||||
Performance conditions | NA | NA | NA | NA | ||||||||||||
Fair value per BCEX | 28.64 | 30.25 | 31.46 | 31.87 | ||||||||||||
| (1) | The number of shares takes into account an exercise parity adjusted by the division by 15 of the nominal value of the shares decided by the general meeting on December 9, 2011; each BCEX warrant now gives the right to subscribe to 15 new shares instead of 1 new share. For the same reason, the exercise price of each BCE plan was adjusted as a result and is thus equal to 1/15th of the price initially determined by the general meeting that authorized each of the plans. |
| (2) | Based on French government bonds (GFRN) with a maturity corresponding to the maturity of BCEX. |
Change in Number of BCEX Outstanding
| Year ended December 31, | December 31 | |||||||||||||||||||||||
| Number of BCEX | 2012 | 2013 | 2014 | 2013 | 2014 | 2015 | ||||||||||||||||||
Balance at beginning of period | 2,296 | 2,296 | 2,296 | 2,296 | 2,296 | — | ||||||||||||||||||
Granted during the period | — | — | — | — | — | — | ||||||||||||||||||
Forfeited during the period | — | — | — | — | — | — | ||||||||||||||||||
Exercised during the period | — | — | 2,296 | — | 2,296 | — | ||||||||||||||||||
Expired during the period | — | — | — | — | — | — | ||||||||||||||||||
Balance at end of period | 2,296 | 2,296 | — | 2,296 | — | — | ||||||||||||||||||
Breakdown of the Closing Balance
| Year ended December 31, | December 31 | |||||||||||||||||||||||||||||||||||||||||||||||
| 2012 | 2013 | 2014 | 2013 | 2014 | 2015 | |||||||||||||||||||||||||||||||||||||||||||
| Number of BCEX | Outstanding | Exercisable | Outstanding | Exercisable | Outstanding | Exercisable | Outstanding | Exercisable | Outstanding | Exercisable | Outstanding | Exercisable | ||||||||||||||||||||||||||||||||||||
BCEX with exercise price of €70 | 2,296 | 2,296 | 2,296 | 2,296 | — | — | 2,296 | 2,296 | — | — | — | — | ||||||||||||||||||||||||||||||||||||
Total | 2,296 | 2,296 | 2,296 | 2,296 | — | — | 2,296 | 2,296 | — | — | — | — | ||||||||||||||||||||||||||||||||||||
17.2 BSA
Date of Grant 12/07/2007
The BSA may be exercised by the beneficiary on the basis of the following vesting schedule:
Date of Grant 01/17/2012
The BSA may be exercised by the beneficiary on the basis of the following vesting schedule:
Date of Grant 09/25/2012
The BSA may be exercised by the beneficiary on the basis of the following vesting schedule:
Date of Grant 07/25/2013
The BSA may be exercised by the beneficiary on the basis of the following vesting schedule:
Date of Grant 06/03/2014
The BSA may be exercised by the beneficiary on the basis of the following vesting schedule:
Date of Grant 24/03/2015
The BSA may be exercised by the beneficiary on the basis of the following vesting schedule:
Date of Grant 19/11/2015
The BSA may be exercised by the beneficiary on the basis of the following vesting schedule:
Details of BSA
Date of grant (Board of Directors) | 12/07/2007 | 12/07/2007 | 12/07/2007 | 12/07/2007 | 01/17/2012 | 01/17/2012 | 01/17/2012 | 01/17/2012 | 12/07/2007 | 12/07/2007 | 12/07/2007 | 12/07/2007 | 01/17/2012 | 01/17/2012 | 01/17/2012 | 01/17/2012 | ||||||||||||||||||||||||||||||||||||||||||||||||
Vesting period (years) | 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | ||||||||||||||||||||||||||||||||||||||||||||||||
Plan expiration date | 12/06/2015 | 12/06/2015 | 12/06/2015 | 12/06/2015 | 01/17/2022 | 01/17/2022 | 01/17/2022 | 01/17/2022 | 12/06/2015 | 12/06/2015 | 12/06/2015 | 12/06/2015 | 01/17/2022 | 01/17/2022 | 01/17/2022 | 01/17/2022 | ||||||||||||||||||||||||||||||||||||||||||||||||
Number of BSA granted | 431 | 431 | 428 | 427 | 22,459 | 22,459 | 22,459 | 22,458 | 431 | 431 | 428 | 427 | 22,459 | 22,459 | 22,459 | 22,458 | ||||||||||||||||||||||||||||||||||||||||||||||||
Share entitlement per | 15 | 15 | 15 | 15 | 1 | 1 | 1 | 1 | 15 | 15 | 15 | 15 | 1 | 1 | 1 | 1 | ||||||||||||||||||||||||||||||||||||||||||||||||
Exercise price | 65 | 65 | 65 | 65 | 5.13 | 5.13 | 5.13 | 5.13 | 65 | 65 | 65 | 65 | 5.13 | 5.13 | 5.13 | 5.13 | ||||||||||||||||||||||||||||||||||||||||||||||||
Valuation method used | Black and Scholes | Black and Scholes | Black and Scholes | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Grant date share fair value | 65 | 65 | 65 | 65 | 5.13 | 5.13 | 5.13 | 5.13 | 65 | 65 | 65 | 65 | 5.13 | 5.13 | 5.13 | 5.13 | ||||||||||||||||||||||||||||||||||||||||||||||||
Expected volatility | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | ||||||||||||||||||||||||||||||||
Average life of BSA | 4.5 | 5.0 | 5.5 | 6.0 | 5.5 | 6.0 | 6.5 | 7.0 | 4.5 | 5.0 | 5.5 | 6.0 | 5.5 | 6.0 | 6.5 | 7.0 | ||||||||||||||||||||||||||||||||||||||||||||||||
Discount rate(2) | 4.06 | % | 4.09 | % | 4.09 | % | 4.10 | % | 2.33 | % | 2.33 | % | 2.61 | % | 2.61 | % | 4.06 | % | 4.09 | % | 4.09 | % | 4.10 | % | 2.33 | % | 2.33 | % | 2.61 | % | 2.61 | % | ||||||||||||||||||||||||||||||||
Expected dividends | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | ||||||||||||||||||||||||||||||||
Performance conditions | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | ||||||||||||||||||||||||||||||||||||||||||||||||
Fair value per BSA | 22.18 | 23.62 | 24.95 | 26.22 | 2.05 | 2.14 | 2.26 | 2.34 | 22.18 | 23.62 | 24.95 | 26.22 | 2.05 | 2.14 | 2.26 | 2.34 | ||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||||||||||||||||||||||||||||||||||||||||||||
| (1) | The number of shares takes into account an exercise parity adjusted by the division by 15 of the nominal value of the shares decided by the general meeting on December 9, 2011; each BSA warrant now gives the right to subscribe to 15 new shares instead of 1 new share. For the same reason, the exercise price of each BSA plan was adjusted as a result and is thus equal to 1/15th of the price initially determined by the general meeting that authorized each of the plans. |
| (2) | Based on French government bonds (GFRN) with a maturity corresponding to the maturity of BSA. |
Date of grant (Board of Directors) | 09/25/2012 | 09/25/2012 | 09/25/2012 | 09/25/2012 | 07/25/2013 | 06/03/2014 | ||||||||||||||||||
Vesting period (years) | 1 | 2 | 3 | 4 | 0 | 0 | ||||||||||||||||||
Plan expiration date | 09/25/2022 | 09/25/2022 | 09/25/2022 | 09/25/2022 | 07/25/2023 | 06/03/2024 | ||||||||||||||||||
Number of BSA granted | 7,500 | 7,500 | 7,500 | 7,500 | 73,000 | 10,000 | ||||||||||||||||||
Share entitlement per BSA(1) | 1 | 1 | 1 | 1 | 1 | 1 | ||||||||||||||||||
Exercise price | 8.59 | 8.59 | 8.59 | 8.59 | 8.1 | 18.79 | ||||||||||||||||||
Valuation method used | Black and Scholes | |||||||||||||||||||||||
Grant date share fair value | 8.4 | 8.4 | 8.4 | 8.4 | 8.15 | 19.01 | ||||||||||||||||||
Expected volatility | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | ||||||||||||
Average life of BSA | 5.5 | 6.0 | 6.5 | 7.0 | 5.0 | 5.0 | ||||||||||||||||||
Discount rate(2) | 1.21 | % | 1.21 | % | 1.53 | % | 1.53 | % | 1.16 | % | 0.71 | % | ||||||||||||
Expected dividends | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | ||||||||||||
Performance conditions | NA | NA | NA | NA | NA | NA | ||||||||||||||||||
Fair value per BSA | 2.29 | 2.43 | 2.61 | 2.74 | 2.18 | 4.98 | ||||||||||||||||||
| (1) | The number of shares takes into account an exercise parity adjusted by the division by 15 of the nominal value of the shares decided by the general meeting on December 9, 2011; each BSA warrant now gives the right to subscribe to 15 new shares instead of 1 new share. For the same reason, the exercise price of each BSA plan was adjusted as a result and is thus equal to 1/15th of the price initially determined by the general meeting that authorized each of the plans. |
| (2) | Based on French government bonds (GFRN) with a maturity corresponding to the maturity of BSA. |
Date of grant (Board of Directors) | 09/25/2012 | 09/25/2012 | 09/25/2012 | 09/25/2012 | 07/25/2013 | 06/03/2014 | ||||||||||||||||||
Vesting period (years) | 1 | 2 | 3 | 4 | 0 | 0 | ||||||||||||||||||
Plan expiration date | 09/25/2022 | 09/25/2022 | 09/25/2022 | 09/25/2022 | 07/25/2023 | 06/03/2024 | ||||||||||||||||||
Number of BSA granted | 7 500 | 7 500 | 7 500 | 7 500 | 73 000 | 10 000 | ||||||||||||||||||
Share entitlement per BSA(1) | 1 | 1 | 1 | 1 | 1 | 1 | ||||||||||||||||||
Exercise price | 8.59 | 8.59 | 8.59 | 8.59 | 8.1 | 18.79 | ||||||||||||||||||
Valuation method used | Black and Scholes | |||||||||||||||||||||||
Grant date share fair value | 8.4 | 8.4 | 8.4 | 8.4 | 8.15 | 19.01 | ||||||||||||||||||
Expected volatility | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | ||||||||||||
Average life of BSA | 5.5 | 6.0 | 6.5 | 7.0 | 5.0 | 5.0 | ||||||||||||||||||
Discount rate(2) | 1.21 | % | 1.21 | % | 1.53 | % | 1.53 | % | 1.16 | % | 0.71 | % | ||||||||||||
Expected dividends | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | ||||||||||||
Performance conditions | NA | NA | NA | NA | NA | NA | ||||||||||||||||||
Fair value per BSA | 2.29 | 2.43 | 2.61 | 2.74 | 2.18 | 4.98 | ||||||||||||||||||
Date of grant (Board of Directors) | 03/24/2015 | 11/19/2015 | ||||||
Vesting period (years) | — | — | ||||||
Plan expiration date | 03/24/2025 | 11/19/2025 | ||||||
Number of BSA granted | 10,000 | 15,000 | ||||||
Share entitlement per BSA(1) | 1 | 1 | ||||||
Exercise price | 43.00 | 66.06 | ||||||
Valuation method used | Black and Scholes | |||||||
Grant date share fair value | 43.00 | 66.06 | ||||||
Expected volatility | 36 | % | 50.91 | % | ||||
Average life of BSA | 5.0 | 5.0 | ||||||
Discount rate(2) | 0.68 | % | 0.81 | % | ||||
Expected dividends | 0 | % | 0 | % | ||||
Performance conditions | N/A | N/A | ||||||
Fair value per BSA | 9.90 | 22.60 | ||||||
Change in Number of BSA Outstanding
| Year ended December 31, | December 31 | |||||||||||||||||||||||
| Number of BSA | 2012 | 2013 | 2014 | 2013 | 2014 | 2015 | ||||||||||||||||||
Balance at beginning of period | 1,145 | 120,980 | 193,980 | 120,980 | 193,980 | 196,480 | ||||||||||||||||||
Granted during the period | 119,835 | 73,000 | 10,000 | 73,000 | 10,000 | 25,000 | ||||||||||||||||||
Forfeited during the period | — | — | — | — | — | — | ||||||||||||||||||
Exercised during the period | — | — | 7,500 | — | 7,500 | 77,786 | ||||||||||||||||||
Expired during the period | — | — | — | — | — | — | ||||||||||||||||||
Balance at end of period | 120,980 | 193,980 | 196,480 | 193,980 | 196,480 | 143,694 | ||||||||||||||||||
Breakdown of the Closing Balance
| Year ended December 31, | ||||||||||||||||||||||||
| 2012 | 2013 | 2014 | ||||||||||||||||||||||
| Number of BSA | Outstanding | Exercisable | Outstanding | Exercisable | Outstanding | Exercisable | ||||||||||||||||||
BSA with exercise price of €65 | 1,145 | 1,145 | 1,145 | 1,145 | 1,145 | 1,145 | ||||||||||||||||||
BSA with exercise price of €5.13 | 89,835 | — | 89,835 | — | 89,835 | — | ||||||||||||||||||
BSA with exercise price of €8.59 | 30,000 | — | 30,000 | 30,000 | 25,000 | 25,000 | ||||||||||||||||||
BSA with exercise price of €8.1 | — | — | 73,000 | 73,000 | 70,500 | 70,500 | ||||||||||||||||||
BSA with exercise price of €18.79 | — | — | — | — | 10,000 | 10,000 | ||||||||||||||||||
Total | 120,980 | 1,145 | 193,980 | 104,145 | 196,480 | 106,645 | ||||||||||||||||||
BSA2010 with exercise price of €65 BSA2010 with exercise price of €5.13 BSA2010 with exercise price of €8.59 BSA2010 with exercise price of €8.1 BSA2010 with exercise price of €18.79 BSA with exercise price of €43.00 BSA with exercise price of €66.06 Total December 31 2013 2014 2015 Number of BSA Outstanding Exercisable Outstanding Exercisable Outstanding Exercisable 1,145 1,145 1,145 1,145 859 859 89,835 — 89,835 — 89,835 — 30,000 30,000 25,000 25,000 10,000 10,000 73,000 73,000 70,500 70,500 13,000 13,000 — — 10,000 10,000 5,000 5,000 — — 10,000 10,000 — — 15,000 15,000 193,980 104,145 196,480 106,645 143,694 53,859
17.3 BSA 2
The BSA may be exercised by the beneficiary on the basis of the following vesting schedule:
By a decision by the Board of Directors meeting held on November 22, 2011, the BSA2 warrants became exercisable beginning on the date of the first quotation of the shares of the Company on the Euronext regulated market in Paris.
Details of BSA2
Date of grant (Board of Directors) | 01/21/2009 | 01/21/2009 | 01/21/2009 | 01/21/2009 | 01/21/2009 | |||||||||||||||
Vesting period (years) | 0 | 1 | 2 | 3 | 4 | |||||||||||||||
Plan expiration date | 01/20/2019 | 01/20/2019 | 01/20/2019 | 01/20/2019 | 01/20/2019 | |||||||||||||||
Number of BSA2 granted | 4,822 | 2,680 | 1,072 | 1,072 | 1,070 | |||||||||||||||
Share entitlement per BSA2(1) | 15 | 15 | 15 | 15 | 15 | |||||||||||||||
Exercise price | 65 | 65 | 65 | 65 | 65 | |||||||||||||||
Valuation method used | Black and Scholes | |||||||||||||||||||
Grant date share fair value | 70 | 70 | 70 | 70 | 70 | |||||||||||||||
Expected volatility | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | ||||||||||
Average life of BSA2 | 5.0 | 5.5 | 6.0 | 6.5 | 7.0 | |||||||||||||||
Discount rate(2) | 2.71 | % | 2.71 | % | 2.98 | % | 2.98 | % | 3.11 | % | ||||||||||
Expected dividends | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | ||||||||||
Performance conditions | NA | NA | NA | NA | NA | |||||||||||||||
Fair value per BSA2 | 29.05 | 30.32 | 31.89 | 33.05 | 33.45 | |||||||||||||||
| (1) | The number of shares takes into account an exercise parity adjusted by the division by 15 of the nominal value of the shares decided by the general meeting on December 9, 2011; each BSA2 warrant now gives the right to subscribe to 15 new shares instead of 1 new share. For the same reason, the exercise price of each BSA2 plan was adjusted as a result and is thus equal to 1/15th of the price initially determined by the general meeting that authorized each of the plans. |
| (2) | Based on French government bonds (GFRN) with a maturity corresponding to the maturity of BSA2. |
Change in Number of BSA 2 Outstanding
| Year ended December 31, | ||||||||||||
| Number of BSA2 | 2012 | 2013 | 2014 | |||||||||
Balance at beginning of period | 10,716 | 10,716 | 10,716 | |||||||||
Granted during the period | — | — | — | |||||||||
Forfeited during the period | — | — | — | |||||||||
Exercised during the period | — | — | 2,667 | |||||||||
Expired during the period | — | — | — | |||||||||
Balance at end of period | 10,716 | 10,716 | 8,049 | |||||||||
Balance at beginning of period Granted during the period Forfeited during the period Exercised during the period Expired during the period Balance at end of period December 31 Number of BSA2 2013 2014 2015 10,716 10,716 8,049 — — — — — — — 2,667 8,049 — — — 10,716 8,049 —
Breakdown of the Closing Balance
| Year ended December 31, | December 31 | |||||||||||||||||||||||||||||||||||||||||||||||
| 2012 | 2013 | 2014 | 2013 | 2014 | 2015 | |||||||||||||||||||||||||||||||||||||||||||
| Number of BSA2 | Outstanding | Exercisable | Outstanding | Exercisable | Outstanding | Exercisable | Outstanding | Exercisable | Outstanding | Exercisable | Outstanding | Exercisable | ||||||||||||||||||||||||||||||||||||
BSA2 with exercise price of €65 | 10,716 | 10,716 | 10,716 | 10,716 | 8,049 | 8,049 | 10,716 | 10,716 | 8,049 | 8,049 | — | — | ||||||||||||||||||||||||||||||||||||
Total | 10,716 | 10,716 | 10,716 | 10,716 | 8,049 | 8,049 | 10,716 | 10,716 | 8,049 | 8,049 | — | — | ||||||||||||||||||||||||||||||||||||
17.4 BCE 4
The BCE4 may be exercised by the beneficiary on the basis of the following vesting schedule:
By a decision by the Board of Directors meeting held on November 22, 2011, the BCE4 warrants became exercisable beginning on the date of the first quotation of the shares of the Company on the Euronext regulated market in Paris.
Details of BCE4
Date of grant (Board of Directors) | 01/21/2009 | 01/21/2009 | 01/21/2009 | 01/21/2009 | 01/21/2009 | |||||||||||||||
Vesting period (years) | 0 | 1 | 2 | 3 | 4 | |||||||||||||||
Plan expiration date | 01/20/2019 | 01/20/2019 | 01/20/2019 | 01/20/2019 | 01/20/2019 | |||||||||||||||
Number of BCE4 granted | 2,411 | 1,340 | 536 | 536 | 535 | |||||||||||||||
Share entitlement per BCE4(1) | 15 | 15 | 15 | 15 | 15 | |||||||||||||||
Exercise price | 65 | 65 | 65 | 65 | 65 | |||||||||||||||
Valuation method used | Black and Scholes | |||||||||||||||||||
Grant date share fair value | 70 | 70 | 70 | 70 | 70 | |||||||||||||||
Expected volatility | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | ||||||||||
Average life of BCE4 | 5.0 | 5.5 | 6.0 | 6.5 | 7.0 | |||||||||||||||
Discount rate(2) | 2.71 | % | 2.71 | % | 2.98 | % | 2.98 | % | 3.11 | % | ||||||||||
Expected dividends | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | ||||||||||
Performance conditions | NA | NA | NA | NA | NA | |||||||||||||||
Fair value per BCE4 | 29.06 | 30.33 | 31.90 | 33.06 | 34.35 | |||||||||||||||
| (1) | The number of shares takes into account an exercise parity adjusted by the division by 15 of the nominal value of the shares decided by the general meeting on December 9, 2011; each BCE4 warrant now gives the right to subscribe to 15 new shares instead of 1 new share. For the same reason, the exercise price of each BCE4 plan was adjusted as a result and is thus equal to 1/15th of the price initially determined by the general meeting that authorized each of the plans. |
| (2) | Based on French government bonds (GFRN) with a maturity corresponding to the maturity of BCE4. |
Change in Number of BCE4 Outstanding
| Year ended December 31, | December 31 | |||||||||||||||||||||||
| Number of BCE4 | 2012 | 2013 | 2014 | 2013 | 2014 | 2015 | ||||||||||||||||||
Balance at beginning of period | 5,358 | 5,358 | 5,358 | 5,358 | 5,358 | 5,358 | ||||||||||||||||||
Granted during the period | — | — | — | — | — | — | ||||||||||||||||||
Forfeited during the period | — | — | — | — | — | — | ||||||||||||||||||
Exercised during the period | — | — | — | — | — | 2,667 | ||||||||||||||||||
Expired during the period | — | — | — | — | — | — | ||||||||||||||||||
Balance at end of period | 5,358 | 5,358 | 5,358 | 5,358 | 5,358 | 2,691 | ||||||||||||||||||
Breakdown of the Closing Balance
| Year ended December 31, | December 31 | |||||||||||||||||||||||||||||||||||||||||||||||
| 2012 | 2013 | 2014 | 2013 | 2014 | 2015 | |||||||||||||||||||||||||||||||||||||||||||
| Number of BCE4 | Outstanding | Exercisable | Outstanding | Exercisable | Outstanding | Exercisable | Outstanding | Exercisable | Outstanding | Exercisable | Outstanding | Exercisable | ||||||||||||||||||||||||||||||||||||
BCE4 with exercise price of €65 | 5,358 | 5,358 | 5,358 | 5,358 | 5,358 | 5,358 | ||||||||||||||||||||||||||||||||||||||||||
BCE4 with exercise price of €65²² | 5,358 | 5,358 | 5,358 | 5,358 | 2,691 | 2,691 | ||||||||||||||||||||||||||||||||||||||||||
Total | 5,358 | 5,358 | 5,358 | 5,358 | 5,358 | 5,358 | 5,358 | 5,358 | 5,358 | 5,358 | 2,691 | 2,691 | ||||||||||||||||||||||||||||||||||||
17.5 BSA2010
Date of Grant 01/28/2011
The BSA may be exercised by the beneficiary on the basis of the following vesting schedule:
Date of Grant 06/24/2011
The BSA may be exercised by the beneficiary on the basis of the following vesting schedule:
Date of Grant 11/22/2011
The BSA may be exercised by the beneficiary on the basis of the following vesting schedule:
Details of BSA2010
Date of grant (Board of Directors) | 01/28/2011 | 01/28/2011 | 01/28/2011 | 01/28/2011 | 06/24/2011 | 06/24/2011 | 06/24/2011 | 06/24/2011 | 01/28/2011 | 01/28/2011 | 01/28/2011 | 01/28/2011 | 06/24/2011 | 06/24/2011 | 06/24/2011 | 06/24/2011 | ||||||||||||||||||||||||||||||||||||||||||||||||
Vesting period (years) | 0.9 | 1.9 | 2.9 | 3.9 | 0.5 | 1.5 | 2.5 | 3.5 | 0.9 | 1.9 | 2.9 | 3.9 | 0.5 | 1.5 | 2.5 | 3.5 | ||||||||||||||||||||||||||||||||||||||||||||||||
Plan expiration date | 01/27/2021 | 01/27/2021 | 01/27/2021 | 01/27/2021 | 11/22/2021 | 11/22/2021 | 11/22/2021 | 11/22/2021 | 01/27/2021 | 01/27/2021 | 01/27/2021 | 01/27/2021 | 11/22/2021 | 11/22/2021 | 11/22/2021 | 11/22/2021 | ||||||||||||||||||||||||||||||||||||||||||||||||
Number of BSA2010 granted | 2,510 | 2,510 | 2,510 | 2,509 | 2,000 | 2,000 | 2,000 | 2,000 | 2,510 | 2,510 | 2,510 | 2,509 | 2,000 | 2,000 | 2,000 | 2,000 | ||||||||||||||||||||||||||||||||||||||||||||||||
Share entitlement per BSA2010(1) | 15 | 15 | 15 | 15 | 15 | 15 | 15 | 15 | 15 | 15 | 15 | 15 | 15 | 15 | 15 | 15 | ||||||||||||||||||||||||||||||||||||||||||||||||
Exercise price | 77 | 77 | 77 | 77 | 77 | 77 | 77 | 77 | 77 | 77 | 77 | 77 | 77 | 77 | 77 | 77 | ||||||||||||||||||||||||||||||||||||||||||||||||
Valuation method used | Black and Scholes | Black and Scholes | Black and Scholes | Black and Scholes | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Grant date share fair value | 77 | 77 | 77 | 77 | 77 | 77 | 77 | 77 | 77 | 77 | 77 | 77 | 77 | 77 | 77 | 77 | ||||||||||||||||||||||||||||||||||||||||||||||||
Expected volatility | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | ||||||||||||||||||||||||||||||||
Average life of BSA2010 | 5.5 | 6.0 | 6.5 | 7.0 | 5.5 | 6.0 | 6.5 | 7.0 | 5.5 | 6.0 | 6.5 | 7.0 | 5.5 | 6.0 | 6.5 | �� | 7.0 | |||||||||||||||||||||||||||||||||||||||||||||||
Discount rate(2) | 2.70 | % | 2.82 | % | 2.82 | % | 3.04 | % | 2.55 | % | 2.68 | % | 2.68 | % | 2.87 | % | 2.70 | % | 2.82 | % | 2.82 | % | 3.04 | % | 2.55 | % | 2.68 | % | 2.68 | % | 2.87 | % | ||||||||||||||||||||||||||||||||
Expected dividends | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | ||||||||||||||||||||||||||||||||
Performance conditions | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | ||||||||||||||||||||||||||||||||||||||||||||||||
Fair value per BSA2010 | 31.33 | 32.90 | 34.23 | 35.84 | 31.15 | 32.70 | 34.02 | 35.57 | 31.33 | 32.90 | 34.23 | 35.84 | 31.15 | 32.70 | 34.02 | 35.57 | ||||||||||||||||||||||||||||||||||||||||||||||||
| (1) | The number of shares takes into account an exercise parity adjusted by the division by 15 of the nominal value of the shares decided by the general meeting on December 9, 2011; each BSA2010 warrant now gives the right to subscribe to 15 new shares instead of 1 new share. For the same reason, the exercise price of each BSA2010 plan was adjusted as a result and is thus equal to 1/15th of the price initially determined by the general meeting that authorized each of the plans. |
| (2) | Based on French government bonds (GFRN) with a maturity corresponding to the maturity of BSA2010. |
Date of grant (Board of Directors) | 11/22/2011 | 11/22/2011 | 11/22/2011 | 11/22/2011 | ||||||||||||
Vesting period (years) | 1.0 | 2.0 | 3.0 | 4.0 | ||||||||||||
Plan expiration date | 11/22/2021 | 11/22/2021 | 11/22/2021 | 11/22/2021 | ||||||||||||
Number of BSA2010 granted | 335 | 335 | 334 | 334 | ||||||||||||
Share entitlement per BSA(1) | 15 | 15 | 15 | 15 | ||||||||||||
Exercise price | 77 | 77 | 77 | 77 | ||||||||||||
Valuation method used | Black and Scholes | |||||||||||||||
Grant date share fair value | 77 | 77 | 77 | 77 | ||||||||||||
Expected volatility | 40 | % | 40 | % | 40 | % | 40 | % | ||||||||
Average life of BSA | 5.5 | 6.0 | 6.5 | 7.0 | ||||||||||||
Discount rate(2) | 2.23 | % | 2.60 | % | 2.60 | % | 2.85 | % | ||||||||
Expected dividends | 0 | % | 0 | % | 0 | % | 0 | % | ||||||||
Performance conditions | NA | NA | NA | NA | ||||||||||||
Fair value per BSA | 30.70 | 32.58 | 33.89 | 35.54 | ||||||||||||
| (1) | The number of shares takes into account an exercise parity adjusted by the division by 15 of the nominal value of the shares decided by the general meeting on December 9, 2011; each BSA2010 warrant now gives the right to subscribe to 15 new shares instead of 1 new share. For the same reason, the exercise price of each BSA2010 plan was adjusted as a result and is thus equal to 1/15th of the price initially determined by the general meeting that authorized each of the plans. |
| (2) | Based on French government bonds (GFRN) with a maturity corresponding to the maturity of BSA2010. |
Change in Number of BSA2010 Outstanding
| Year ended December 31, | December 31 | |||||||||||||||||||||||
| Number of BSA | 2012 | 2013 | 2014 | 2013 | 2014 | 2015 | ||||||||||||||||||
Balance at beginning of period | 19,377 | 11,848 | 11,848 | 11,848 | 11,848 | 8,334 | ||||||||||||||||||
Granted during the period | — | — | — | — | — | — | ||||||||||||||||||
Forfeited during the period | 7,529 | — | — | — | — | — | ||||||||||||||||||
Exercised during the period | — | — | 3,514 | — | 3,514 | 7,290 | ||||||||||||||||||
Expired during the period | — | — | — | — | — | — | ||||||||||||||||||
Balance at end of period | 11,848 | 11,848 | 8,334 | 11,848 | 8,334 | 1,044 | ||||||||||||||||||
Breakdown of the Closing Balance
| Year ended December 31, | December 31 | |||||||||||||||||||||||||||||||||||||||||||||||
| 2012 | 2013 | 2014 | 2013 | 2014 | 2015 | |||||||||||||||||||||||||||||||||||||||||||
| Number of BSA2010 | Outstanding | Exercisable | Outstanding | Exercisable | Outstanding | Exercisable | Outstanding | Exercisable | Outstanding | Exercisable | Outstanding | Exercisable | ||||||||||||||||||||||||||||||||||||
BSA2010 with exercise price of €77 | 11,848 | 6,845 | 11,848 | 9,180 | 8,334 | 8,000 | 11,848 | 9,180 | 8,334 | 8,000 | 1,044 | 1,044 | ||||||||||||||||||||||||||||||||||||
Total | 11,848 | 6,845 | 11,848 | 9,180 | 8,334 | 8,000 | 11,848 | 9,180 | 8,334 | 8,000 | 1,044 | 1,044 | ||||||||||||||||||||||||||||||||||||
17.6 BSAX
Date of Grant 01/21/2009 and 06/25/2010
The BSAX may be exercised by the beneficiary on the basis of the following vesting schedule:
By a decision by the Board of Directors meeting held on November 22, 2011, the BSAX warrants became exercisable beginning on the date of the first quotation of the shares of the Company on the Euronext regulated market in Paris.
Details of BSAX
Date of grant (Board of Directors) | 01/21/2009 | 01/21/2009 | 01/21/2009 | 21/01/2009 | 06/25/2010 | 06/25/2010 | 06/25/2010 | 06/25/2010 | ||||||||||||||||||||||||
Vesting period (years) | 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | ||||||||||||||||||||||||
Plan expiration date | 01/20/2019 | 01/20/2019 | 01/20/2019 | 01/20/2019 | 06/24/2020 | 06/24/2020 | 06/24/2020 | 06/24/2020 | ||||||||||||||||||||||||
Number of BSAX granted | 77 | 77 | 77 | 75 | 457 | 457 | 456 | 455 | ||||||||||||||||||||||||
Share entitlement per BSAX(1) | 15 | 15 | 15 | 15 | 15 | 15 | 15 | 15 | ||||||||||||||||||||||||
Exercise price | 65 | 65 | 65 | 65 | 65 | 65 | 65 | 65 | ||||||||||||||||||||||||
Valuation method used | Black and Scholes | Black and Scholes | ||||||||||||||||||||||||||||||
Grant date share fair value | 70 | 70 | 70 | 70 | 70 | 70 | 70 | 70 | ||||||||||||||||||||||||
Expected volatility | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | ||||||||||||||||
Average life of BSAX | 5.5 | 6.0 | 6.5 | 7.0 | 5.5 | 6.0 | 6.5 | 7.0 | ||||||||||||||||||||||||
Discount rate(2) | 2.71 | % | 2.98 | % | 2.98 | % | 3.11 | % | 2.04 | % | 2.23 | % | 2.23 | % | 2.50 | % | ||||||||||||||||
Expected dividends | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | ||||||||||||||||
Performance conditions | NA | NA | NA | NA | NA | NA | NA | NA | ||||||||||||||||||||||||
Fair value per BSAX | 30.32 | 31.89 | 33.05 | 33.45 | 29.47 | 30.88 | 31.99 | 33.44 | ||||||||||||||||||||||||
| (1) | The number of shares takes into account an exercise parity adjusted by the division by 15 of the nominal value of the shares decided by the general meeting on December 9, 2011; each BSAX warrant now gives the right to subscribe to 15 new shares instead of 1 new share. For the same reason, the exercise price of each BSAX plan was adjusted as a result and is thus equal to 1/15th of the price initially determined by the general meeting that authorized each of the plans. |
| (2) | Based on French government bonds (GFRN) with a maturity corresponding to the maturity of BSAX. |
Change in Number of BSAX Outstanding
| Year ended December 31, | December 31 | |||||||||||||||||||||||
| Number of BSAX | 2012 | 2013 | 2014 | 2013 | 2014 | 2015 | ||||||||||||||||||
Balance at beginning of period | 2,131 | 2,131 | 2,131 | 2,131 | 2,131 | 2,131 | ||||||||||||||||||
Granted during the period | — | — | — | — | — | — | ||||||||||||||||||
Forfeited during the period | — | — | — | — | — | — | ||||||||||||||||||
Exercised during the period | — | — | — | — | — | 1,095 | ||||||||||||||||||
Expired during the period | — | — | — | — | — | — | ||||||||||||||||||
Balance at end of period | 2,131 | 2,131 | 2,131 | 2,131 | 2,131 | 1,036 | ||||||||||||||||||
Breakdown of the Closing Balance
| Year ended December 31, | December 31 | |||||||||||||||||||||||||||||||||||||||||||||||
| 2012 | 2013 | 2014 | 2013 | 2014 | 2015 | |||||||||||||||||||||||||||||||||||||||||||
| Number of BSAX | Outstanding | Exercisable | Outstanding | Exercisable | Outstanding | Exercisable | Outstanding | Exercisable | Outstanding | Exercisable | Outstanding | Exercisable | ||||||||||||||||||||||||||||||||||||
BSAX with exercise price of €65 | 2,131 | 2,131 | 2,131 | 2,131 | 2,131 | 2,131 | 2,131 | 2,131 | 2,131 | 2,131 | 1,036 | 1,036 | ||||||||||||||||||||||||||||||||||||
Total | 2,131 | 2,131 | 2,131 | 2,131 | 2,131 | 2,131 | 2,131 | 2,131 | 2,131 | 2,131 | 1,036 | 1,036 | ||||||||||||||||||||||||||||||||||||
17.7 BCE2010
Date of Grant 06/24/2011
The BCE may be exercised by the beneficiary on the basis of the following vesting schedule:
Date of Grant 11/22/2011
The BSPCE may be exercised by the beneficiary on the basis of the following vesting schedule:
Details of BCE2010
Date of grant (Board of Directors) | 06/24/2011 | 06/24/2011 | 06/24/2011 | 06/24/2011 | 11/22/2011 | 11/22/2011 | 11/22/2011 | 11/22/2011 | ||||||||||||||||||||||||
Vesting period (years) | 0.5 | 1.5 | 2.5 | 3.5 | 1 | 2 | 3 | 4 | ||||||||||||||||||||||||
Plan expiration date | 11/22/2021 | 11/22/2021 | 11/22/2021 | 11/22/2021 | 11/22/2021 | 11/22/2021 | 11/22/2021 | 11/22/2021 | ||||||||||||||||||||||||
Number of BCE2010 granted | 6,000 | 6,000 | 6,000 | 6,000 | 2,510 | 2,510 | 2,510 | 2,509 | ||||||||||||||||||||||||
Share entitlement per BCE2010(1) | 15 | 15 | 15 | 15 | 15 | 15 | 15 | 15 | ||||||||||||||||||||||||
Exercise price | 77 | 77 | 77 | 77 | 77 | 77 | 77 | 77 | ||||||||||||||||||||||||
Valuation method used | Black and Scholes | Black and Scholes | ||||||||||||||||||||||||||||||
Grant date share fair value | 77 | 77 | 77 | 77 | 77 | 77 | 77 | 77 | ||||||||||||||||||||||||
Expected volatility | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | 40 | % | ||||||||||||||||
Average life of BCE2010 | 5.5 | 6.0 | 6.5 | 7.0 | 5.4 | 5.9 | 6.4 | 6.9 | ||||||||||||||||||||||||
Discount rate(2) | 2.55 | % | 2.68 | % | 2.68 | % | 2.87 | % | 2.05 | % | 2.42 | % | 2.42 | % | 2.66 | % | ||||||||||||||||
Expected dividends | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | ||||||||||||||||
Performance conditions | NA | NA | NA | NA | NA | NA | NA | NA | ||||||||||||||||||||||||
Fair value per BCE2010 | 31.16 | 32.71 | 34.03 | 35.58 | 30.42 | 32.29 | 33.58 | 35.2 | ||||||||||||||||||||||||
| (1) | The number of shares takes into account an exercise parity adjusted by the division by 15 of the nominal value of the shares decided by the general meeting on December 9, 2011; each BCE2010 warrant now gives the right to subscribe to 15 new shares instead of 1 new share. For the same reason, the exercise price of each BCE2010 plan was adjusted as a result and is thus equal to 1/15th of the price initially determined by the general meeting that authorized each of the plans. |
| (2) | Based on French government bonds (GFRN) with a maturity corresponding to the maturity of BCE2010. |
Change in Number of BCE2010 Outstanding
| Year ended December 31, | December 31 | |||||||||||||||||||||||
| Number of BCE2010 | 2012 | 2013 | 2014 | 2013 | 2014 | 2015 | ||||||||||||||||||
Balance at beginning of period | 34,039 | 34,039 | 34,039 | 34,039 | 34,039 | 29,972 | ||||||||||||||||||
Granted during the period | — | — | — | — | — | — | ||||||||||||||||||
Forfeited during the period | — | — | — | — | — | — | ||||||||||||||||||
Exercised during the period | — | — | 4,067 | — | 4,067 | 13,998 | ||||||||||||||||||
Expired during the period | — | — | — | — | — | — | ||||||||||||||||||
Balance at end of period | 34,039 | 34,039 | 29,972 | 34,039 | 29,972 | 15,974 | ||||||||||||||||||
Breakdown of the Closing Balance
| Year ended December 31, | December 31 | |||||||||||||||||||||||||||||||||||||||||||||||
| 2012 | 2013 | 2014 | 2013 | 2014 | 2015 | |||||||||||||||||||||||||||||||||||||||||||
| Number of BCE2010 | Outstanding | Exercisable | Outstanding | Exercisable | Outstanding | Exercisable | Outstanding | Exercisable | Outstanding | Exercisable | Outstanding | Exercisable | ||||||||||||||||||||||||||||||||||||
BCE2010 with exercise price of €77 | 34,039 | 14,510 | 34,039 | 23,020 | 29,972 | 27,463 | ||||||||||||||||||||||||||||||||||||||||||
BCE2010 with exercise price of €77.00 | 34,039 | 23,020 | 29,972 | 27,463 | 15,974 | 15,974 | ||||||||||||||||||||||||||||||||||||||||||
Total | 34,039 | 14,510 | 34,039 | 23,020 | 29,972 | 27,463 | 34,039 | 23,020 | 29,972 | 27,463 | 15,974 | 15,974 | ||||||||||||||||||||||||||||||||||||
17.8 OPTIONS
Grant of 09/18/2013
The share options may be exercised by the beneficiary on the basis of the following vesting schedule:
Grant of 06/03/2014
The share options may be exercised by the beneficiary on the basis of the following vesting schedule:
Details of SO
Date of grant (Board of Directors) | 09/18/2013 | 06/03/2014 | 09/18/2013 | 06/03/2014 | 06/23/2015 | 11/19/2015 | ||||||||||||||||||
Vesting period (years) | 4 | 2 | 4 | 2 | 4 | 4 | ||||||||||||||||||
Plan expiration date | 09/18/2023 | 06/03/2024 | 09/18/2023 | 06/03/2024 | 06/23/2025 | 11/19/2015 | ||||||||||||||||||
Number of SO granted | 518,000 | 75,000 | 518,000 | 75,000 | 120,000 | 195,000 | ||||||||||||||||||
Share entitlement per SO(1) | 1 | 1 | 1 | 1 | 1 | 1 | ||||||||||||||||||
Exercise price | 7.57 | 19.01 | 7.57 | 19.01 | 48.90 | 66.06 | ||||||||||||||||||
Valuation method used | Black and Scholes | Black and Scholes | ||||||||||||||||||||||
Grant date share fair value | 7.9 | 19.01 | 7.9 | 19.01 | 48.9 | 66.06 | ||||||||||||||||||
Expected volatility | 40 | % | 40 | % | 40 | % | 40 | % | 51 | % | 51 | % | ||||||||||||
Average life of SO | 7.0 | 6.0 | 7.0 | 6.0 | 7.0 | 7.0 | ||||||||||||||||||
Discount rate(2) | 1.72 | % | 0.89 | % | 1.72 | % | 0.89 | % | 0.89 | % | 0.81 | % | ||||||||||||
Expected dividends | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | ||||||||||||
Performance conditions | NA | NA | NA | NA | NA | NA | ||||||||||||||||||
Fair value per SO | 3.57 | 7.46 | 3.57 | 7.46 | 25.28 | 34.05 | ||||||||||||||||||
| (1) | The number of shares takes into account an exercise parity adjusted by the division by 15 of the nominal value of the shares decided by the general meeting on December 9, 2011; each SO now gives the right to subscribe to 15 new shares instead of 1 new share. For the same reason, the exercise price of each SO plan was adjusted as a result and is thus equal to 1/15th of the price initially determined by the general meeting that authorized each of the plans. |
| (2) | Based on French government bonds (GFRN) with a maturity corresponding to the maturity of SO. |
Change in Number of SO Outstanding
| Year ended December 31, | December 31 | |||||||||||||||||||||||
| Number of SO | 2012 | 2013 | 2014 | 2013 | 2014 | 2015 | ||||||||||||||||||
Balance at beginning of period | — | — | 518,000 | — | 518,000 | 546,000 | ||||||||||||||||||
Granted during the period | — | 518,000 | 75,000 | 518,000 | 75,000 | 315,000 | ||||||||||||||||||
Forfeited during the period | — | — | 47,000 | — | 47,000 | — | ||||||||||||||||||
Exercised during the period | — | — | — | — | — | — | ||||||||||||||||||
Expired during the period | — | — | — | — | — | — | ||||||||||||||||||
Balance at end of period | — | 518,000 | 546,000 | 518,000 | 546,000 | 861,000 | ||||||||||||||||||
Breakdown of the Closing Balance
| Year ended December 31, | ||||||||||||||||||||||||
| 2012 | 2013 | 2014 | ||||||||||||||||||||||
| Number of SO | Outstanding | Exercisable | Outstanding | Exercisable | Outstanding | Exercisable | ||||||||||||||||||
SO with exercise price of €7.57 | — | — | 518,000 | — | 471,000 | — | ||||||||||||||||||
SO with exercise price of €19.01 | — | — | — | — | 75,000 | — | ||||||||||||||||||
Total | — | — | 518,000 | — | 546,000 | — | ||||||||||||||||||
SO with exercise price of €7.57 SO with exercise price of €19.01 SO with exercise price of €48,90 SO with exercise price of €66,06 Total December 31 2013 2014 2015 Number of SO Outstanding Exercisable Outstanding Exercisable Outstanding Exercisable 518,000 — 471,000 — 471,000 — — — 75,000 — 75,000 — — — — — 120,000 — — — — — 195,000 — 518,000 — 546,000 — 861,000 —
The exercise prices, anticipated lifetime, and fair value of the underlying shares based on the share price on the Euronext market on the grant date of the warrants were used for the valuation of each category of compensation in shares.
17.9 FREE SHARES
Dates of Grant 04/02/2012, 07/25/2012, 11/28/2012, 09/12/2013 and 06/03/2014, 09/30/2015 and 12/15/2015
The free shares are subject to a two-year vesting period.
Details of Free Shares
Date of grant (Board of Directors) | 04/02/2012 | 07/25/2012 | 11/28/2012 | 07/25/2013&09/12/2013 | 06/03/2014 | 04/02/2012 | 07/25/2012 | 11/28/2012 | 07/25/2013&09/12/2013 | 06/03/2014 | ||||||||||||||||||||||||||||||
Vesting period (years) | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | ||||||||||||||||||||||||||||||
Number of free shares granted | 669,796 | 134,081 | 35,360 | 501,500 | 186,000 | 669,796 | 134,081 | 35,360 | 501,500 | 186,000 | ||||||||||||||||||||||||||||||
Share entitlement per free share (1) | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ||||||||||||||||||||||||||||||
Grant date share fair value | 8.86 | 8.20 | 8.70 | 7.96 | 19.01 | 8.86 | 8.20 | 8.70 | 7.96 | 19.01 | ||||||||||||||||||||||||||||||
Expected dividends | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | ||||||||||||||||||||
Performance conditions | Yes | (1) | Yes | (1) | NO | Yes | (2) | Yes | (3) | Yes | (1) | Yes | (1) | No | Yes | (2) | Yes | (3) | ||||||||||||||||||||||
Expected turnover during the vesting period | 1 | % | 1 | % | 1 | % | 1 | % | 1 | % | 1 | % | 1 | % | 1 | % | 1 | % | 1 | % | ||||||||||||||||||||
Date of grant (Board of Directors) | 09/30/2015 | 12/15/2015 | ||||||||||||||||||||||||||||||||||||||
Vesting period (years) | 2 | 2 | ||||||||||||||||||||||||||||||||||||||
Number of free shares granted | 708,500 | 42,000 | ||||||||||||||||||||||||||||||||||||||
Share entitlement per free share (1) | 1 | 1 | ||||||||||||||||||||||||||||||||||||||
Grant date share fair value | 62.99 | 64.14 | ||||||||||||||||||||||||||||||||||||||
Expected dividends | 0 | % | 0 | % | ||||||||||||||||||||||||||||||||||||
Performance conditions | Yes | (4) | Yes | (4) | ||||||||||||||||||||||||||||||||||||
Expected turnover during the vesting period | 0 | % | 0 | % | ||||||||||||||||||||||||||||||||||||
| (1) | The acquisition of free shares is contingent for certain individuals (the “Key Managers”), including Dr. Benhamou, upon the achievement of the three performance criteria below: |
| • | One-third of the shares allocated to Key Managers will only be acquired from the later of the two following dates (i) expiry of a period of two years from the date of allocation and (ii) inclusion of the 100th patient in the VIPES phase II study. |
| (2) | The acquisition of free shares is contingent for the Key Managers, including Dr. Benhamou, upon the achievement of the three performance criteria below: |
| • | One-third of the shares allocated to Key Managers will only be acquired from the later of the two following dates (i) expiry of a period of two years from the date of allocation and (ii) inclusion of the 100th patient in the Viaskin Peanut phase III study a maximum of twelve (12) months after the inclusion of the first patient in the study. |
It is specified that in the event of a change of control of the Company (as defined in Article L. 233-3 of the Commercial Code), the performance criteria will be considered as definitively achieved.
| (3) | The acquisition of free shares is contingent for the Key Managers, including Dr. Benhamou, upon the achievement of the two performance criteria below: |
| • | Half of the shares allocated to Key Managers will only be acquired from the later of the two following dates (i) expiry of a period of two years from the date of allocation and (ii) inclusion of the 100th patient in the Viaskin Peanut phase III study a maximum of twelve (12) months after the inclusion of the first patient in the study. |
| (4) | The acquisition of free shares is contingent for all the employees, including Dr. Benhamou, upon the achievement of the three performance criteria below: |
Performance conditions other than market conditions, which are taken into account by adjusting the number of equity instruments included in the measurement of the transaction amount, but are not taken into account when estimating the fair value of the shares.
Change in Number of Free Shares Outstanding
| Year ended December 31, | December 31 | |||||||||||||||||||||||
| Number of Free shares | 2012 | 2013 | 2014 | 2013 | 2014 | 2015 | ||||||||||||||||||
Balance at beginning of period | — | 839,237 | 1,340,737 | 839,237 | 1,340,737 | 641,360 | ||||||||||||||||||
Granted during the period | 839,237 | 501,500 | 186,000 | 501,500 | 186,000 | 750,500 | ||||||||||||||||||
Forfeited during the period | — | — | 83,360 | — | 83,360 | 61,833 | ||||||||||||||||||
Exercised during the period | — | — | 802,017 | — | 802,017 | 321,698 | ||||||||||||||||||
Expired during the period | — | — | — | — | — | — | ||||||||||||||||||
Balance at end of period | 839,237 | 1,340,737 | 641,360 | 1,340,737 | 641,360 | 1,008,329 | ||||||||||||||||||
Note 18: Financial Revenue and Expenses
The financial income and expenses are broken down as follows (in thousands of Euros):
| December 31 | ||||||||||||||||||||||||
| 2013 | 2014 | 2015 | ||||||||||||||||||||||
| 2012 | 2013 | 2014 | (in thousands of euros) | |||||||||||||||||||||
Financial revenues | 517,540 | 670,234 | 727,239 | 670 | 727 | 1,018 | ||||||||||||||||||
Financial expenses | (25,203 | ) | (24,310 | ) | (103,239 | ) | (24 | ) | (103 | ) | (146 | ) | ||||||||||||
Total | 492,337 | 645,925 | 624,000 | 646 | 624 | 871 | ||||||||||||||||||
The financial income is primarily comprised of capital gains on the disposals of investment securities. The foreign exchange losses and the expenses related to the accretion of the OSEO, BpiFrance and COFACE advances are classified as financial expenses in financial expenses.the Consolidated Statements of (Loss).
Note 19: Income Tax Expense
As mentioned in Note 3.13—Accounting Principles—Other Income, the French Research Tax Credit is not included in the line item “Income taxes” but included in the line item “Other Income.”
Reconciliation Betweenbetween the Effective and Nominal Income Tax Expense
The following table shows the reconciliation between the effective and nominal tax expense at the nominal standard French rate of 33.33% (excluding additional contributions):
| Year Ended December 31, | December 31 | |||||||||||||||||||||||
| 2012 | 2013 | 2014 | 2013 | 2014 | 2015 | |||||||||||||||||||
| (in thousands of euros) | (in thousands of euros) | |||||||||||||||||||||||
Income (loss) before taxes | (12,912 | ) | (19,306 | ) | (24,012 | ) | ||||||||||||||||||
(Loss) before taxes | (19,306 | ) | (24,012 | ) | (44,674 | ) | ||||||||||||||||||
Theoretical group tax rate | 33.33 | % | 33.33 | % | 33.33 | % | 33.33 | % | 33.33 | % | 33.33 | % | ||||||||||||
Nominal tax expense | 4,304 | 6,435 | 8,003 | 6,435 | 8,003 | 14,890 | ||||||||||||||||||
Increase/decrease in tax expense arising from: | ||||||||||||||||||||||||
Permanent differences(1) | 1,024 | 619 | 3,636 | 619 | 3,636 | 6,089 | ||||||||||||||||||
Research tax credit | 838 | 1,096 | 1,446 | 1,096 | 1,446 | 1,895 | ||||||||||||||||||
Share-based compensation | (1,065 | ) | (1,682 | ) | (1,546 | ) | (1,682 | ) | (1,546 | ) | (3,473 | ) | ||||||||||||
Non recognition of deferred tax assets related to tax losses and temporary differences | (5,088 | ) | (6,438 | ) | (11,458 | ) | (6,438 | ) | (11,458 | ) | (19,211 | ) | ||||||||||||
Other differences | (12 | ) | (30 | ) | (81 | ) | (30 | ) | (81 | ) | (190 | ) | ||||||||||||
Effective tax expense | 0 | 0 | 0 | 0 | 0 | 0 | ||||||||||||||||||
Effective tax rate | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | 0 | % | ||||||||||||
| (1) | The significant balance of permanent differences is mainly affected by transaction costs on capital increases occurred in |
Deferred Tax Assets and Liabilities
As mentioned in Note 3.15, the Company has not recognized deferred tax assets in the statementConsolidated Statements of financial position.Financial Position. The amount of the deficit carried forward at the end of December 2015 is €159 million including €156 million for DBV Technologie SA.
Note 20: Commitments
Obligations Under the Terms of the Ordinary Rental Agreements
On April 28, 2011, the Company signed with the company SELECTINVEST a lease for its premises.
On December 1, 2014, an expansion lease was signed between DBV and Nexity (formerly SELECTINVEST1).
On March 9, 2015 the Company signed a lease for the combined premises of offices and laboratories at Montrouge, in France, and moved its headquarters and its laboratories there on January 4, 2016. The Bagneux premises has however been kept pour the industrial teams.
On April 25, 2015, the Company signed a sub-lease for its premises for its American subsidiary company in New York.
The amount of future rents and charges in that capacity was breaks down as follows at December 31, 2014:2015:
| (Amounts in Euros) | 12/31/2014 | |||||||
| (Amounts in thousands of Euros) | 12/31/2015 | |||||||
2015 | 386,766 | |||||||
2016 | 427,565 | 2,013 | ||||||
2017 | 427,565 | 1,790 | ||||||
2018 | 427,565 | 1,658 | ||||||
2019 | 427,565 | 1,658 | ||||||
2020 | 178,152 | 1,485 | ||||||
2021 | 1,361 | |||||||
2022 | 1,361 | |||||||
2023 | 1,361 | |||||||
2024 | 794 | |||||||
|
| |||||||
Total | 2,275,178 | 13,481 | ||||||
|
| |||||||
The
In July 2014, the Company signed in July 2014 a lease agreement with EVOSCIENCES to lease laboratory equipment. The future rental payments as at December 31, 20142015 are as follows:
The company has signed various ordinary rental agreements for office equipment and vehicles. The future rental payments as at December 31, 20142015 are as follows:
Obligations Under the Terms of Other Agreements
The companyCompany signed with its bank CIC an acquisition contract of monetary market fund “SICAV CM-CIC” pledged as a guarantee for the ordinary rental agreements of the premises of Bagneux for an amount of €384,809.€0.4 million.
The Company also signed a letter of credit to ensure the sub-lease of its premises of its New York subsidiary company for $164 thousand due on March 17, 2016. This credit note has been extended for an additional year.
In addition, the Company took a term deposit for a sum of €227 thousand over 3 years.
As it has sub-contracted several important functions, the Company has been required to conclude, within the framework of its current operations, sub-contracting contracts or short- or medium-term delegation contracts with various third parties, in France and abroad, which include various obligations that are usual in these circumstances.
In 2011,On November 18, 2015, the Company signed a subcontracting agreement with a CROcontract research organization (CRO) within the context of launching its Phase IIIII clinical study for the Viaskin Peanut product. This study amounts to €5,390,637. €9.3 million.
As of December 31, 2014,2015, the amount remaining to pay as part of this contract for years 20152016, 2017 and 2018 was €323,123.€7.4 million.
The Company signed a subcontracting agreement with the same CRO within the context of launching its follow-up clinical study OLFUS for the Viaskin Peanut product. This study amounts to €6,800,000.€6.4 million.
As of December 31, 2014,2015, the amount remaining to pay as part of this contract for years 2015, 2016 and 2017 was €3,402,647.€2.2 million.
The Company signed a subcontracting agreement with a second CRO within the context of launching its clinical study for the Viaskin MolikMilk product. This study amounts to €7,050,758.€10.4 million.
As of December 31, 2014,2015, the amount remaining to pay as part of this contract for years 2015, 2016, 2017 and 2018 was €5,856,163.€7.7 million.
InOn January 7th 2009, the Company entered into an assignment, development and co-ownership agreement with PublicWelfare-Hospitals of Paris (L’Assistance Publique—Hopitaux de Paris), or AP-HP, and Université Paris-Descartes, or UPD, by which the Company agreed to terms of co-ownership with AP-HP and UPD of certain U.S. and foreign patents and patent applications, referred to herein as the shared patents. The Company, and any licensees or sublicensees the Company designates, have the exclusive right to commercial uses of the shared patents. AP-HP and UPD agreed to use the shared patents only for internal research purposes and not to license the shared patents to any third party. Upon commercialization of any product covered by the shared patents, which the Company expects would include its Viaskin product candidates, the Company will be obligated to pay AP-HP and UPD a percentage of net sales as a royalty. This royalty varies depending on the particular patent used in the product and is in the low single digits. Additionally, if the Company licenses any of the shared patents to a third party and a licensee commercializes products covered by such shared patents, the Company will be obligated to pay AP-HP and UPD a percentage in the low single digits of the money it receives from its licensee. If the Company does not sell any of its product candidates covered by the shared patents within 30 months from the date it first markets such product candidates, AP-HP may, upon six months’ notice and subject to certain exceptions, convert its exclusive right to the commercial use of the shared patents to a non-exclusive right. Any party may terminate the license in the event of another party’s substantial breach which remains uncured after six months of receiving written notice of such breach. The agreement will also terminate in the event the Company ceases operations or is subject to a dissolution or bankruptcy proceedings. Absent early termination, the agreement will automatically terminate upon the expiration of the last shared patent. In the event the agreement is terminated, the Company would no longer have the exclusive right to commercial use of the shared patents, though it would retain its shared ownership rights. In addition, its ownership stake in certain jointly made improvements covered by the shared patents would survive termination of the agreement. The longest lived patent rights licensed to the Company under the agreement are currently expected to expire in 2028. To date, this agreement has not had an impact on the Company’s financial statements.
Note 21: Relationships with Related Parties
The compensation amounts for 2015 presented below, which were awarded to the members of the Board of Directors and the Executive Committee of the Company were posted tototals 7.2 million euros.
Following the accounts as expenses during the coursereorganization of the fiscal years presented (in Euros):Company at the beginning of 2015, the Company henceforth considered the members of the Executive Committee to be related parties.
Amounts in thousands of euros:
| December 31 | ||||||||||||||||||||||||
| 2013 | 2014 | 2015 | ||||||||||||||||||||||
| 2012 | 2013 | 2014 | (Amounts in thousands of Euros) | |||||||||||||||||||||
Members of the Board of Directors | 203,450 | 380,800 | 433,160 | 381 | 433 | 605 | ||||||||||||||||||
Executive Committee | 686 | 887 | 1,768 | |||||||||||||||||||||
Directors’ fees | 45,000 | 40,000 | 40,000 | 40 | 40 | 195 | ||||||||||||||||||
Share-based payments to members of the Board of Directors | 1,211,454 | 1,612,191 | 1,117,538 | 3,530 | 2,771 | 4,637 | ||||||||||||||||||
Fees paid to SCP Benhamou Vannrom | 164,513 | — | — | |||||||||||||||||||||
Total | 1,624,417 | 2,032,991 | 1,590,698 | 4,637 | 4,131 | 7,205 | ||||||||||||||||||
The methods for the valuation of the benefit related to share-based payments are presented in Note 17.
StatementA schedule of the debtsamounts payable to related parties as of December 31:31, in thousands of euros:
| December 31 | ||||||||||||||||||||||||
| 2013 | 2014 | 2015 | ||||||||||||||||||||||
| 2012 | 2013 | 2014 | (Amounts in thousands of Euros) | |||||||||||||||||||||
Exceptional compensation | 75,600 | 109,200 | 114,660 | 109 | 345 | 674 | ||||||||||||||||||
Directors’ fees | 67,000 | 36,500 | 40,000 | 36 | 40 | 195 | ||||||||||||||||||
Pension obligations | 22,845 | — | — | 159 | 233 | |||||||||||||||||||
Total | 165,085 | 145,700 | 154,660 | 146 | 544 | 1,102 | ||||||||||||||||||
Note 22: Earnings Per Share
The basic earnings per share is calculated by dividing the net income going to the shareholders of the Company by the weighted average number of common shares outstanding during the course of the fiscal year. The weighted average number of shares was 13,604,687 in 2013. The weighted average number of shares was 16,086,247 in 2014. Taking into account the division of the nominal value of shares of the Company by 15, which was decided by the annual general meeting on December 9, 2011 the amount of shares is adjusted, and multiplying it by 15, for all the outstanding shares presented. The weighted average number of shares was 21,522,342 in 2015.
The instruments that entitle their holders to a portion of the share capital on a deferred basis (BSAs, BSPCEs) are considered to be anti-dilutive (1,437,684(2,119,109 instruments in 2013, 1,437,684 instruments in 2014 and 2,119,1092,033,768 instruments in 2013)2015). These instruments are presented in detail in Note 17. Therefore, the diluted earnings per share are identical to the basic earnings per share.
| As of December 31, | ||||||||||||
| 2012 | 2013 | 2014 | ||||||||||
Net income of the reporting period | (12,912,100 | ) | (19,306,416 | ) | (24,011,880 | ) | ||||||
Adjusted weighted average number of outstanding shares | 12,326,779 | 13,604,687 | 16,086,247 | |||||||||
Basic / Diluted earnings per share (€/share) | (1.05 | ) | (1.42 | ) | (1.49 | ) | ||||||
| December 31, | ||||||||||||
| 2013 | 2014 | 2015 | ||||||||||
| (Amounts in thousands of Euros) | ||||||||||||
Net income of the reporting period | (19,306 | ) | (24,012 | ) | (44,674 | ) | ||||||
Adjusted weighted average number of outstanding shares | 13,604,687 | 16,086,247 | 21,522,342 | |||||||||
Basic / Diluted earnings per share (€/share) | (1.42 | ) | (1.49 | ) | (2.08 | ) | ||||||
Note 23: Management of Financial Risks
The principal financial instruments of the Company are comprised of financial assets, cash, and investment securities. The purpose of managing these instruments is to allow the business activities of the Company to be financed. It is not the Company’s policy to subscribe to financial instruments for speculative purposes. The Company does not utilize derivatives.
The principal risks to which the Company is exposed are liquidity risk, interest rate risk and credit risk.
Liquidity Risk
The Company could need to strengthen its shareholders’ equity or rely on additional financing in order to ensure its development.
Since it was formed, the Company has financed its growth by reinforcing its shareholders’ equity through a succession of increases in the share capital, obtaining public assistance in support of innovation, and reimbursements of Research Tax Credit claims, but has never utilized bank loans. Therefore, the Company is not exposed to a liquidity risk resulting from the implementation of any early repayment clauses in loan agreements for such borrowings.
As of this date, the Company believes that it is not exposed to any short-term liquidity risk considering the cash and cash equivalents available as of December 31, 2014,2015, that is, €114,583,141.€323 thousand.
Significant research and development efforts and expenditures related to clinical studies have been initiated since the start-up of the Company’s business, which has thus far generated negative operating cash flows.
The Company will continue to have significant financing requirements in the future for the development of its technology, the continuation of its clinical development program, and the equipment for its own pharmaceutical laboratory, as well as for the production and marketing of its products in the future. It is possible that the company will find itself unable to self-finance its growth, which would compel it to seek other sources of financing, particularly through new increases in share capital.
The level of the financing requirements of the Company and how they are phased out over time depend on factors that are largely beyond the control of the Company such as:
| • | costs for responding to changes in the Viaskin® technology and for conducting the manufacturing and marketing on some or all of its products; and |
It is possible that the Company will be unable to obtain additional capital when it needs it, or that such capital may not be available on financial terms that are acceptable to the Company. If the necessary funds are not available, the Company could have to:
In addition, to the extent that the Company raises capital by issuing new shares, the investment of its shareholders could be diluted. Furthermore, financing by debt, to the extent that it is available, could also include restrictive conditions for the Company and its shareholders.
The occurrence of one or more of these risks could have a material adverse effect on the Company, its business, its financial position, its earnings, its development, and its prospects.
Interest Rate Risk
The Company’s exposure to interest-rate risk primarily involves investment securities. These are composed of money market funds and time deposit accounts. Changes in interest rates have a direct impact on the rate of return on these investments and the cash flows generated.
The Company has no variable rate debt. The repayment flows of its debts are not subject to interest rate risk.
The repayment of the conditional advances may vary depending on whether or not objectives are attained. The change in the flows of the anticipated repayments is treated in the income statement (Note 3.11)3.10).
As of this date, the Company has not contracted borrowings from credit institutions and, therefore, has only very low exposure to interest rate risk.
Credit Risk
The credit risk related to the cash, the cash equivalents, and the current financial instruments is not significant in light of the quality of the co-contracting financial institutions.
Fair Value
The fair value of financial instruments traded on an active market, such as the securities available for sale, is based on the market rate as of the closing date. The market prices used for the financial assets owned by the Company are the bid prices in effect on the market as of the valuation date.
The nominal value, less the provisions for depreciation, of the accounts receivable and current debts, is presumed to approximate the fair value of those items.
Foreign Exchange Risk
The Company is exposed to a very insignificant foreign exchange risk inherent in some of its supplies obtained in the United States, which have been invoiced in U.S.US dollars. As of this date, the company does not make sales revenue in dollars or in any other currency other than the euro; the Company does not receive any full or partial mechanical endorsement. The exposure to currencies other than the U.S. dollar is negligible.
For the fiscal years 2014 and 2013,2015, less than 7% and 10% respectively of the purchases and other external expenses had been made in U.S. dollars, generating a net annual foreign exchange gain of €79 thousand in 2015 in comparison with a net loss of €24,337 and €2,831 respectively for those periods.€24 thousand in 2014.
In light of these insignificant amounts, the Company has not adopted, at this stage, a hedging mechanism in order to protect its business activity against fluctuations in exchange rates. The Company cannot rule out the possibility that a significant increase in its business, particularly in the United States, may result in greater exposure to exchange rate risk and should thus consider adopting an appropriate policy for hedging against these risks.
Note 24: Events After the Close of the Fiscal Year
The second disbursement relatedNo event occurred between the closing date and the date of the Board of Directors that change or make necessary a mention in the Notes to the OSEO Immunavia advance expected in October 2014 was received on January 22, 2015. Given the expenditure declared, the amount received was €864,989.
In 2015, the Company has signed a new lease arrangement in relation to its new premises in Montrouge, France and expects to relocate its current site in Bagneux later in 2015. The maximum costconsolidated accounts of leaving the premises in Bagneux is estimated at €415k.DBV Technologies by not having been taken into consideration.
SIGNATURES
The registrant hereby certifies that it meets all of the requirements for filing on Form 20-F and that it has duly caused and authorized the undersigned to sign this annual report on its behalf.
| DBV Technologies S.A. | ||
/s/ Dr. Pierre-Henri Benhamou | ||
| By: | Dr. Pierre-Henri Benhamou | |
| Title: | Chief Executive Officer (Principal Executive | |
| Officer) | ||
Date: April 29, 201528, 2016
EXHIBIT INDEX
Incorporated by Reference | Incorporated by Reference | |||||||||||||||||||
Exhibit | Description | Schedule/ Form | File Number | Exhibit | File Date | Description | Schedule/ Form | File Number | Exhibit | File Date | ||||||||||
| 1.1 | By-laws (statuts) of the registrant (English translation) | Form F-1 | 333-198870 | 3.1 | 09/22/14 | By-laws (statuts) of the registrant (English translation) | Form F-1 | 333-198870 | 3.1 | 09/22/14 | ||||||||||
| 2.1 | Form of Deposit Agreement | Form F-1/A | 333-198870 | 4.1 | 10/15/14 | Form of Deposit Agreement | Form F-1/A | 333-198870 | 4.1 | 10/15/14 | ||||||||||
| 2.2 | Form of American Depositary Receipt | Form F-1/A | 333-198870 | 4.2 | 10/15/14 | Form of American Depositary Receipt | Form F-1/A | 333-198870 | 4.2 | 10/15/14 | ||||||||||
| 4.1 | Shareholders’ Agreement among the registrant and certain shareholders signatory thereto, dated March 9, 2012 (English translation) | Form F-1 | 333-198870 | 4.3 | 09/22/14 | Shareholders’ Agreement among the registrant and certain shareholders signatory thereto, dated March 9, 2012 (English translation) | Form F-1 | 333-198870 | 4.3 | 09/22/14 | ||||||||||
| 4.2# | Office Lease between the registrant and GENERALI VIE, dated March 3, 2015 (English translation) | |||||||||||||||||||
| 4.2 | Office Lease between the registrant and GENERALI VIE, dated March 3, 2015 (English translation) | Form 20-F | 001-36697 | 4.2 | 04/29/15 | |||||||||||||||
| 4.3 | Commercial Lease between the registrant and SELECTINVEST 1, dated April 28, 2011 (English translation) | Form F-1 | 333-198870 | 10.1 | 09/22/14 | Commercial Lease between the registrant and SELECTINVEST 1, dated April 28, 2011 (English translation) | Form F-1 | 333-198870 | 10.1 | 09/22/14 | ||||||||||
| 4.4 | Assignment, Development and Co-Ownership Agreement among the registrant, L’Assistance Publique—Hopitaux de Paris and Université Paris Descartes, date January 7, 2009 (English translation) | Form F-1 | 333-198870 | 10.2 | 09/22/14 | Assignment, Development and Co-Ownership Agreement among the registrant, L’Assistance Publique—Hopitaux de Paris and Université Paris Descartes, dated January 7, 2009 (English translation) | Form F-1 | 333-198870 | 10.2 | 09/22/14 | ||||||||||
| 4.5† | Form of Indemnification Agreement between the registrant and each of its executive officers and directors | Form F-1/A | 333-198870 | 10.3 | 10/15/14 | Form of Indemnification Agreement between the registrant and each of its executive officers and directors | Form F-1/A | 333-198870 | 10.3 | 10/15/14 | ||||||||||
| 4.6† | 2013 and 2014 Share Option Plans (English translation) | Form F-1 | 333-198870 | 10.4 | 09/22/14 | 2013 and 2014 Share Option Plans (English translation) | Form F-1 | 333-198870 | 10.4 | 09/22/14 | ||||||||||
| 4.7† | 2012, 2013 and 2014 Free Share Plans (English translation) | Form F-1 | 333-198870 | 10.5 | 09/22/14 | 2012, 2013 and 2014 Free Share Plans (English translation) | Form F-1 | 333-198870 | 10.5 | 09/22/14 | ||||||||||
| 4.8† | Summary of BSA | Form F-1 | 333-198870 | 10.6 | 09/22/14 | Summary of BSA | Form F-1 | 333-198870 | 10.6 | 09/22/14 | ||||||||||
| 4.9† | Summary of BSPCE | Form F-1 | 333-198870 | 10.7 | 09/22/14 | Summary of BSPCE | Form F-1 | 333-198870 | 10.7 | 09/22/14 | ||||||||||
| 4.10†# | 2015 Share Option Plan (English translation) | |||||||||||||||||||
| 4.11†# | 2015 Free Share Plans (English translation) | |||||||||||||||||||
| 8.1# | List of subsidiaries of the registrant | List of subsidiaries of the registrant | ||||||||||||||||||
| 12.1# | Certification by the Principal Executive Officer pursuant to Securities Exchange Act Rules 13a-14(a) and 15d-14(a) as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | Certification by the Principal Executive Officer pursuant to Securities Exchange Act Rules 13a-14(a) and 15d-14(a) as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | ||||||||||||||||||
| 12.2# | Certification by the Principal Financial Officer pursuant to Securities Exchange Act Rules 13a-14(a) and 15d-14(a) as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | Certification by the Principal Financial Officer pursuant to Securities Exchange Act Rules 13a-14(a) and 15d-14(a) as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | ||||||||||||||||||
| 13.1* | Certification by the Principal Executive Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | Certification by the Principal Executive Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | ||||||||||||||||||
| 13.2* | Certification by the Principal Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | Certification by the Principal Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | ||||||||||||||||||
| 15.1# | Consent of Deloitte & Associés | Consent of Deloitte & Associés | ||||||||||||||||||
| # | Filed herewith. |
| * | Furnished herewith. |
| † | Indicates a management contract or any compensatory plan, contract or arrangement. |